

Keywords
Abstract
The review summarizes the information accumulated to date on NH⋯π interactions, which represent an unconventional type of hydrogen bonding between the NH-donor (most often amines, carboxamides or NH heterocycles) and a proton acceptor (usually an electron-rich aromatic or heteroaromatic rings). The importance of NH⋯π interactions stems mainly from their involvement in the structuring of proteins and some other biomolecules, as well as in molecular recognition processes. The growth of knowledge on NH⋯π interactions contributes to a better understanding of complex biochemical processes, stimulates the development of new drugs and improved methods of their targeted delivery. Since the study of NH⋯π interactions on living tissues is rather difficult, their modelling on various synthetic objects has become widespread. In the review, various types of such models are systematized and compared in regard of their geometry, stereo dynamics, and NH⋯π binding energy. Examples of reactions driven by NH⋯π interaction are given, as well as examples of practical use. A rational classification of existing models is proposed, which facilitates their convenient consideration.The bibliography includes 196 references.
1. Introduction
One of the most important discoveries of the 20th century in the field of chemistry and molecular biology was the establishment of the structure of proteins, their functions in the body, the mechanism of biosynthesis and methods of synthetic production.1-3 Proteins are polymer molecules formed during the sequential condensation of α-amino acids, which is accompanied by elimination of water. In nature, this process, which involves 20 so-called proteinogenic acids, is controlled by genes. A variety of genes and countless amino acid sequences determine the intra- and interspecies differences in living organisms. The nature of proteins as linear polyamides (polypeptides) was established in 1902 by E.Fisher, and the specific sequence of amino acid residues was called the primary structure of proteins (Fig. 1).
![[{"id":"vpPhSGpcm0","type":"paragraph","data":{"text":"Fragment of polypeptide chain (primary protein structure) with amino acid residues containing different side groups R<sup>1</sup> - R<sup>4</sup>."}}]](/storage/images/resized/SaaGJmZUbwqiNckZZnj5JxxOrh36a3NkOPQnBJ0D_xl.webp)
Half a century later, based on the data of X-ray diffraction analysis (XRD), Pauling and Corey4,5 came to the conclusion that a highly flexible amino acid chain in proteins is capable of self-organizing into a right-handed α-helix, which is supported by NH…O=C hydrogen bonds (Fig. 2a). In the original paper, the energy of one such bond was estimated to be ≈8 kcal mol–1 (see Ref. 4), which was subsequently confirmed by precise quantumchemical calculations (ab initio, MP2 level).6 Hydrogen bonds (HB) in the protein helix are directed approximately parallel to its main axis. Their length (N…O distance) lies near 2.72 Å, and the NHO angle generally exceeds 150°. Each turn of the helix includes on average 3.6 amino acids, and the length of the turn along the main axis is 5.4 Å. The α-helix, along with the less common form of a corrugated β-sheet (Fig. 2b), is classified as a secondary structure of the protein.4,5 It should be noted that in many proteins, helical regions alternate with linear and β-sheets. Such sites are called domains.
![[{"id":"FW9cbQX1te","type":"paragraph","data":{"text":" Schematic representation of the two main types of secondary structure in proteins: a-helix (<i>a</i>), β-sheet (<i>b</i>)."}}]](/storage/images/resized/pxmoHR9Go9RgDKQotNwkS28tCMDz2pryqi1LYyZA_xl.webp)
It should also be noted that the very possibility of forming a-helices, β-sheets and other domains, as well as the overall complexity and diversity of protein architecture, stems from the possibility of the free rotation around C–C and C–N bonds in the protein backbone [excluding the amide C(O)–N bonds which have a considerable π-component]. This determines the general direction of the α-helix and the formation of so-called turns and loops. The properties, significance and classification of the latter constitute a separate chapter of structural biology. Discussion of this and related issues is beyond the scope of this review. More information on this subject can be found in studies.7,8
Over time, having more and more accurate XRD data, scientists came to the conclusion that in addition to NH…O=C bonds, other types of non-covalent interactions are also involved in the stabilization of the helical structure, such as salt, dipole--dipole, ion--dipole, hydrophobic, as well as unconventional hydrogen bonds of the XH…π type, where X=O, N and less often S or C.9--15 The latter interactions are formed between the proton-donor groups N–H, O–H, S–H, C–H and π-systems of benzene and indole, which are part of the side substituents of three main aromatic amino acids ̶ phenylalanine, tyrosine and tryptophan (Fig. 3). Another aromatic amino acid, histidine, occupies a special place. In living tissues, the imidazole ring of histidine is most often protonated and exists in the form of an imidazolium cation, largely losing its π-donor ability and becoming π-acceptor. At the same time, both in a neutral and in a protonated form, the imidazole ring can be relatively strong N–H donor in the formation of NH…π bonds.11,16 The amino acids arginine, lysine, asparagine, and glutamine are carriers of other NH-donor groups in the side chain (see Fig. 3). In contrast to arginine and lysine, the amino groups in asparagine and glutamine belong to the amide type and, as a rule, display their NH-activity in a neutral form. Many cases have been recorded when carboxamide groups of the protein backbone also act as NH donors in the formation of NH…π bonds.11,16 On average, one protein structure contains 519 NH…π bonds, or one per 20-25 amino acid residues. Of these, a little more than 50% of the NH bonds are donated by the side groups and somewhat less by the amide groups in the polypeptide backbone.11
![[{"id":"zudWMJWwoC","type":"paragraph","data":{"text":"Amino acids most often involved in NH...π and stacking interactions."}}]](/storage/images/resized/xMAjFgQBtSlmAzEXmV2bCy17UA7iqemHm5HV718R_xl.webp)
As for aromatic π-donor groups, XRD analysis of thousands of protein molecules showed that at least 15% of their total number are involved in XH…π interactions. The proportion of such interactions for tryptophan, tyrosine, and phenylalanine is 5:1.5:1, respectively.11 It is believed that this sequence is determined by a decrease in the surface of the π-system in the given series and, consequently, by an increase in its first ionization potential (IP1).
One of the key issues concerns parameters of the XH…π bond and the directionality of the X–H vector to the aromatic ring face. As applied to NH donors, these parameters include the NH…M and N…M distances, where M is the centroid of the ring, the linearity of the HB (NHM angle), and the angle ω between the perfectly linear HB and the normal to the ring plane (Fig. 4). In cases where the N–H vector is more oriented to one of the ring Ci atoms or to middle of the ring C–C bond than to the centroid, the corresponding N…Ci and NH…Ci distances are also considered.
![[{"id":"R8jooMC9Fn","type":"paragraph","data":{"text":"Schematic representation showing orientation of N-H vector to the aromatic ring centroid (M) and the nearest carbon atom(C<sub>i</sub>). The figure was created by the authors using data of Ref.11."}}]](/storage/images/resized/3tQ5Nr854srIlp2uUBA3zjn2n0e0WAuFSqpxQXUJ_xl.webp)
In reality, the NH…π bond is rarely linear and the NHM angle is almost always less than 180°. This is explained by the fact that the NH proton is also attracted to other nearby electronegative atoms, primarily to the oxygen of the OH, C = O groups or water molecules (Fig. 5a). Another common factor distorting the geometry of NH…π bonds is the pronounced tendency of the aromatic ring planes to stack with NH-donor groups.
![[{"id":"z1oy-PZv1j","type":"paragraph","data":{"text":"NH<sup>...</sup>π bond (<i>a</i>) and stacking (<i>b</i>) with participation of carboxamide group.The figurewas created by the authors using data of Refs 11,17."}}]](/storage/images/resized/WZPJuBlX2DXz1prqavuZZjwXwTvwlKbcWhaET5hh_xl.webp)
This type of interaction is especially characteristic of planar NH donors with a sp2-hybridized nitrogen atom and a sufficiently large surface.11,17 These include the carboxamide group in asparagine and glutamine (Fig. 5b), the indole system in tryptophan, as well as guanidinium and imidazolium cations in protonated arginine and histidine. Analysis of a large number of XRD protein structures showed that the border between stacking and NH…π interaction is rather blurred. Conventionally, the latter include clusters with an angle ω<25°, while stacking occurs with ω near 90° and slightly lower.11,17 For the absolute majority of the studied proteins, the ω value lies in the range of 45-60°. In fact, stacking and NH…π interactions are superimposed on each other, with the former dominating in a ratio of ≈2.5:1.11 It is believed that due to stacking, the potential of NH bonds is partially released for interaction with other partners, including aromatic nuclei, which gives an additional gain in energy.11,17 As Table 1 shows, for uncharged NH donors (carboxamide groups), the average interaction energy is about 4.4 kcal mol–1, while for the positively charged guanidinium and imidazolium systems of arginine and histidine, it is almost twice as large (8.0 kcal mol–1). These values include both the electrostatic and dispersion components, which can be separated in the calculation.17 As for the NH…M and N…M distances in protein clusters, in those cases when the NH…π interaction prevails over stacking, they amount to 3.2-3.8 and <4.3 Å, respectively. If stacking dominates, the NH…M values noticeably increase, falling within the range of 3.8-5.0 Å.11
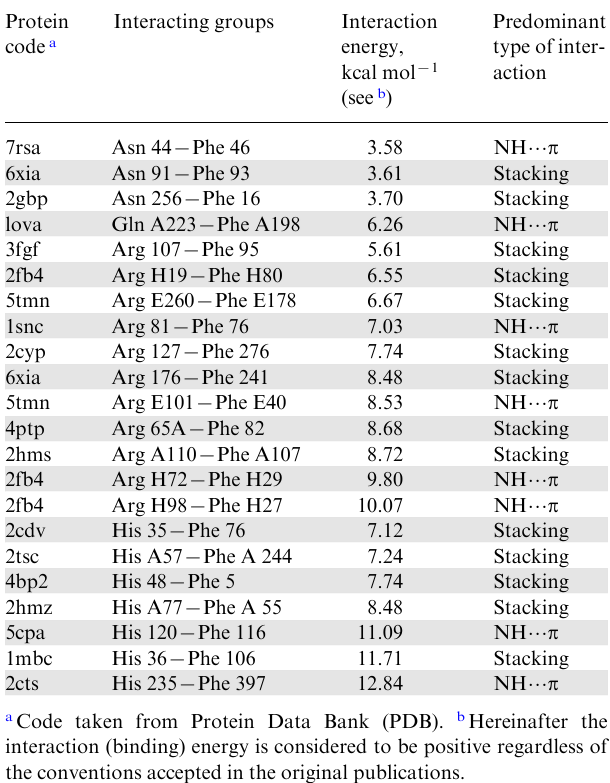
Now, let us clarify the question of where in the polypeptide chain amino acid residues can be located to form an NH…π bond between them, and in what terms it is usually expressed. It is known that the polypeptide chain is usually read from the N-terminal amino acid residue (see Fig. 1). Accordingly, the NH-donor moiety is denoted by the symbol i (sometimes n), and the following amino acid residues by the symbols (i+1), (i+2), etc. If the NH…π bond is formed with the moiety following the NH-donor and bearing the aromatic moiety, then this interaction is characterized as i→ (i+1). In the case when the aromatic residue precedes the NH-donor, the interaction is designated as i→ (i–1). The most common interactions in proteins are of the type i→ (i–2), followed by i→ (i+3) with a large margin. However, there are also many interactions between highly distant amino acid residues (see, e.g., Table 1). As a rule, this occurs in globular proteins in which the amino acid helix strand folds, most often due to the formation of sufficiently strong disulfide bonds between cysteine residues removed in the amino acid sequence (ES–S=55 kcal mol–1). The resulting folded form, which is referred to as the tertiary proteins structure, provides closeness of the initially distant NH-donor and NH-acceptor (π-donor) and the formation of NH…π bond between them.
XH…π bonds are often called soft ones. This means both their relatively low energy and a pronounced tendency to change geometry under the influence of various factors. The softness of XH…π bonds allows them to participate in the fine tuning of protein conformations, which is necessary for the implementation of the corresponding biological functions.
Along with intramolecular, intermolecular XH…π interactions are also very important. Most often, they manifest themselves when receptors of membrane proteins recognize signalling and exogenous molecules, for example, hormones, neurotransmitters, oligopeptides, drugs, etc.13,14,18 In addition, they are responsible for the formation of quaternary protein structures, which include nucleoprotein complexes and complex proteins like hemoglobin, consisting of several interconnected polypeptide strands.
The above, along with the imposition of other non-covalent forces on the XH…π interactions, makes their study directly on living tissues very difficult. Therefore, soon after the discovery of XH…π interactions in proteins, studies were launched on their modelling on simpler synthetic objects. This review summarizes the information accumulated since then on this issue. We focused mainly on NH…π interactions, given their high prevalence and, in part, our current research interests.19,20
2. Research methods
The choice of research methods for model NH…π clusters is primarily determined by their stability. Thus, in the case of simple and especially intermolecular models with the participation of benzene rings and neutral amino groups, the dissociation energy of NH…π bonds does not exceed 1-3 kcal mol–1. This greatly impedes XRD and solution studies. Therefore, most of the information concerning such models was obtained based on quantumchemical calculations, as well as gas-phase spectral measurements under conditions of a supersonic jet, which make it possible to strongly cool the sample.
The most commonly used laser spectroscopic method in the gas phase is the so-called double resonance IR/UV spectroscopy, based on a double optical excitation of jet-cooled neutral molecules, coupled to a fluorescence or mass-spectrometric detection. UV spectroscopy enables spectroscopists to selectively detect conformations or tautomers. IR/UV spectroscopy provides single-conformation IR spectra, which are then assigned by comparison with quantum chemistry calculations.
Individual components and variations of this method are also called laser-induced fluorescence excitation (LIF), dispersed fluorescence (DF), mass-resolved one-colour resonance enhanced two-photon ionization (RE2PI). Furthermore, various types of IR spectroscopy are often used, including combined IR/UV spectroscopy. A detailed description of all this techniques can be found in the experimental part of some relevant works.8,21
To simplify the experimental procedures and improve the reliability of the data, researchers seek to increase the stability of NH…π complexes. This is especially facilitated by the creation of preorganized models and the use of positively charged proton donors, e.g., ammonium salts. The advantage of preorganized structures is the proximity and optimal fixation of the NH-donor and NH-acceptor relative to each other. The resulting stabilization of NH…π clusters makes it possible to carry out their comprehensive studies both in solid form and in solution.
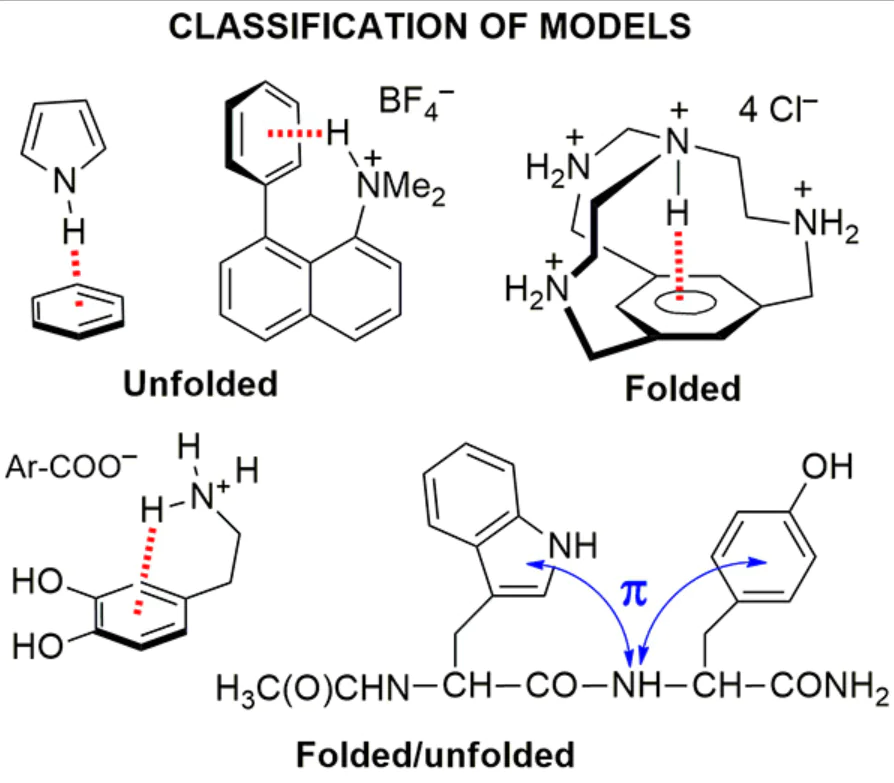
One of the main features of the majority of biologically significant molecules is their high flexibility and, as a consequence, the existence in the form of an equilibrium mixture of many conformers. The latter usually have a crumpled or folded shape, although in some cases unfolded conformations may also be more advantageous. Following this circumstance, we will further consider the existing models of NH…π clusters based on this principle.
3. Unfolded models
3.1. NH…π complexes of benzene with ammonia, amines and ammonium ions
Although the benzene molecule is often regarded as non-polar because of its symmetry consistent with zero dipole moment, this is not strictly the case. The point is that carbon atoms in benzene, being sp2 hybridized, have a slightly higher electronegativity than hydrogen atoms. As a result, the hydrogen edge of the ring acquires a partial positive charge, while the carbon rim with its π-electron sextet is negative (Fig. 6a). Such polarization makes benzene one of the strongest organic quadrupoles (Fig. 6b), the π-system of which can interact with numerous Lewis acids, including a proton, metal ions or ammonium cations.22--24 Let us first consider the π-complexes of benzene with ammonia and other simple NH-donors.
![[{"id":"W9YqjlE8T_","type":"paragraph","data":{"text":"Polarization of C H bonds (<i>a</i>) and schematic representation of quadrupole nature of the benzene ring (<i>b</i>). The figure was created by the authors using data of Ref. 22."}}]](/storage/images/resized/fYZVjUCOK4h44gLims9ZTYvQQ7bcCoT6agufMwtn_xl.webp)
Apparently, the first report on the existence of the C6H6•NH3 complex, in which ammonia acts as a proton donor for the benzene π-system, appeared in Nature in 1993 (Ref. 25) (see also the review26). The evidences for this were obtained for supersonic jet conditions and were based on RE2PI method and high-resolution IR spectroscopy. In addition, they were supported by quantum chemical calculations (ab initio, MP2/6-31G**). The results indicated that in vapours with a defficiency of ammonia (the latter is necessary to minimize the formation of a stronger HB between NH3 molecules), monodentate complex 1 is formed, in which only one N–H bond is oriented to the centroid of the benzene ring (Fig. 7). In this case, the rotational C3 axis of the NH3 molecule is inclined to the C6 axis of the ring at an angle of 58°. Simultaneously, the NH3 molecule rapidly rotates around the N–H bond vector, which makes it possible for two other NH atoms to be attracted to the carbon edge of the ring. The minimum N…M distance for the theoretically optimized structure 1 is 3.6 Å.26 The experimental value of the NH…π bond energy in the C6H6•NH3 complex was estimated as 1.4 kcal mol–1 (see Ref. 27) (1.84±0.12 kcal mol–1 according to other data28,29) while quantumchemical calculations, depending on their level, ranged from 0.1 to 2.4 kcal mol–1.
![[{"id":"YEmIsDUU0G","type":"paragraph","data":{"text":"Mono-, bi- and tridentate benzene-ammonia complexes. The figure was created by the authors using data of Refs 25,26."}}]](/storage/images/resized/nMj7ECkJ1VRQd0Bp79k8N8dHaaX40ukVEvQjzVQR_xl.webp)
It is believed that the NH3 molecule, known for the ease of pyramidal inversion, when complexed with benzene, does not undergo such inversion. The first ionization potential (IP1) of complex 1 is 9.07±0.03 eV,27 which is slightly lower than IP1 of benzene itself (9.21 eV). It was suggested that the NH…π bond in complex 1 is predominantly electrostatic30 with a certain contribution from the dispersion component.26,30
Di- and tridentate structures 2 and 3 were also considered, which, according to theoretical calculations, should be slightly less stable.25,26 At the same time, the analysis of the NH stretching vibrations in IR spectra and a number of other measurements testified to the existence of bidentate complex 2.31
As expected, based on the much higher proton-donating ability of the ammonium ion compared to that of ammonia, the C6H6•NH4+ complex is about an order of magnitude more stable than C6H6•NH3. Thus, according to MP2 calculations of mono-, bi-, and tridentate complexes 4-6 (Fig. 8), their energy (without BSSE correction) in the gas phase is 21.7, 22.4, and 20.0 kcal mol–1, respectively. At the same time, the N…M distance decreases from 3.6 Å in C6H6•NH3 down to 2.9 Å in C6H6•NH4+.32
![[{"id":"otIqGKof9y","type":"paragraph","data":{"text":" Mono-, bi- and tridentate benzene-ammonium ion complexes. The figurewas createdby the authors using data of Ref. 32"}}]](/storage/images/resized/c0A57HafCVHF6yYrPzUBr1vvZpqzzqSPYejCZPac_xl.webp)
Of interest are NH…π complexes 7a,b of benzene and toluene with the Lewis adduct of ammonia with tris(pentafluorophenyl)borane NH3•B(C6F5)3 (Fig. 9a), which were successfully characterized by XRD and IR spectroscopy.33 As can be seen from Fig. 9b, the nitrogen atom is located approximately above the centre of the benzene ring, while the proton participating in the formation of the NH…π bond is slightly deflected from it. The N…M distances in solid complexes 7a,b are 3.28 and 3.21 Å, while the NH…M distances are 2.64 and 2.48 Å, respectively. These values are intermediate between the analogous distances in the complexes C6H6•NH3 and C6H6•NH4+ (only theoretically calculated data are available for the latter two). As might be expected, the maxima of symmetric and asymmetric NH stretching vibrations in the IR spectra of complexes 7a are shifted towards lower frequencies by 30 cm–1 (Fig. 10). Although accurate data on the NH…π hydrogen bond energy in complexes 7a,b are lacking, there are some indications on their insufficient stability. Thus, when dried under reduced pressure, 7a lost a benzene molecule. Similarly, 1H NMR spectrum of 7a in CDCl3 represents an overlay of the spectra of benzene and the (C6F5)3B•NH3 complex, which indicates the displacement of the benzene molecule by deuterochloroform.
![[{"id":"UCEyt78sJZ","type":"paragraph","data":{"text":"NH<sup>...</sup>π Complexes of tris(pentafluorophenyl)borane-ammonia with benzene and toluene (<b>7a,b</b>) (<i>a</i>) and molecular structure of <b>7a</b> with selected key distances in Å (<i>b</i>) (CCDC refcode UBUBAW, T=90 K). The figure was created by the authors using data of Ref. 33."}}]](/storage/images/resized/MtVOu6IOF8rXjgsQDKImm1Rvlf8TGaIjRfdBh8Wg_xl.webp)
![[{"id":"K_huB4b4pa","type":"paragraph","data":{"text":"Region of stretching vibrations of N-H bonds in IR spectra (nujol) of (C<sub>6</sub>F<sub>5</sub>)<sub>3</sub>B•NH<sub>3 </sub>(<i>a</i>) and complex <b>7a</b> (<i>b</i>). The figure was created by the authors using data of Ref. 33."}}]](/storage/images/resized/lULoYQaDIU49FZ7bUNTOTrq7U5eU5IGVd4DuRNpA_xl.webp)
There are few examples of NH…π complexes between benzene and amines. One of them concerns the 2-amino-4,5-dihydrooxazole derivative 8, which was isolated as a solvate with a benzene molecule.34 XRD study revealed the presence of a distinct and moderately short (2.66 Å) NH…π bond between one of the hydrogen atoms of the NH2 group and the benzene ring centroid in the crystal lattice of 8 (Fig. 11). The N…M distance is 3.28 Å, while the second amine proton forms a 2.11 Å long NH…O bond with the solvate water molecule. Notably, that although the OH…π interaction is usually stronger than NH…π, the 4-OH group in 8 does not participate in it, what can be explained by its spatial shielding. Other complexes of this kind between 2aminobenzothiazole and phenylacetic acid have been also documented.35 A specific cluster of benzene with dimethylammonium cation is described in Section 4.1 (structure 86).
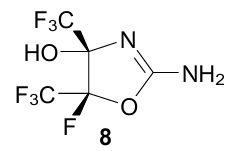
![[{"id":"E7wqn1TM2W","type":"paragraph","data":{"text":" NH<sup>...</sup>π and NH<sup>...</sup>O interactions in benzene solvate hemihydrate of amine <b>8</b> (NAJPIZ, 100 K). The figure was created by the authors using data of Ref. 34."}}]](/storage/images/resized/pQPd2x6JAbLygXiH2vPR9mqUgWpontv9ni0mfhcw_xl.webp)
3.2. NH…π complexes of simple benzene derivatives
The complexity of the study of NH…π complexes of benzene derivatives, in addition to their instability, lower symmetry, and mutual influence of the benzene ring and the substituent, lies in the possibility of the formation of HBs not only with the ring π-system, but also with a substituent (especially when it contains lone electron pairs). In the first case, we will consider such a cluster as a π-complex, and in the second, as a σ-complex. In this regard, we'll discuss a comparative study of the interaction in the gas phase of ammonia with benzene, toluene, chlorobenzene, fluorobenzene and 1,4-difluorobenzene.31 For this, the two-photon resonance ionization (R2PI), microwave and predissociation IR spectroscopy in the region of stretching vibrations of N–H bonds were employed. The experimental results were compared with ab initio calculations (MP2 theory level). Both approaches were in good agreement with each other and showed the formation of the complexes depicted in Fig. 12.
![[{"id":"ffHD5-D-rr","type":"paragraph","data":{"text":" Theoretically optimized structures of σ-, π- and , π,σ-complexes of ammonia with benzene and its fluoro-, chloro-, difluoro- and methyl derivatives. Reproduced from Ref. 31 with permission of the American Chemical Society."}}]](/storage/images/resized/TF2TjcfMxdPH91OYWcaXVqzUX7yQQVB9iqQ2DS03_xl.webp)
In agreement with the data of the aforementioned study,25 it was found that benzene forms only a monodentate π-complex in the gas phase. In the case of toluene, the monodentate π-complex, existing in the form of two rotational conformers, was also identified. In one of them, all three N–H bonds are directed towards the methyl group, while in the other they point to the opposite direction. In the case of fluoro- and chlorobenzenes, the data were interpreted in favour of the formation of three complexes. The σ-structure with NH…F(Cl) hydrogen bond was attributed to one of them, to the second, the structure of the π-complex was assigned, while the third one was determined as a σ,π-cluster of mixed type. For 1,4-difluorobenzene, two monodentate complexes with NH…π and NH…F bonds were detected. However, later, based on the rotational spectra, an unambiguous conclusion was made that the only stable 1,4-difluorobenzene-NH3 σ-complex can exist.36,37
The average value of the calculated binding energies for the considered π-complexes was estimated at 3-3.5 kcal mol–1 without taking zero-point energy (ZPE) into account and about 2.5 kcal mol–1 with ZPE. The smallest (3.39 Å) NH…M distance for the π-complexes and the largest stability were recorded for the toluene-NH3 complex. It was also noted that the structure of ammonia πcomplexes of benzene compounds is largely determined by dispersion forces. In another study of this kind, based on the data of resonance-enhanced multiphoton ionization spectroscopy (REMPI) and quantumchemical calculations (RICC2 method), Gosling et al.38 concluded that fluorobenzene forms only σ-complex with ammonia.
For 4-fluorotoluene-ammonia complex, two-photon ionization spectra featured signals of two forms assigned to the NH…F σ-complex 9a and the π-complex with the NH…π hydrogen bond 9b (Fig. 13). In this case, the signals in the low-frequency region of the spectrum were assigned to the π-complex, and in the high-frequency region, to the σ-complex. A quantumchemical calculation gave close values of bond energies in two forms.38 Similar conclusions based on the study of IR/UV double resonance spectra were also reached by Cocket et al.40 with the difference that the assignment of signals in the high-frequency and low-frequency regions attributed to N–H bonds was inverse.
![[{"id":"9UJFipp2wW","type":"paragraph","data":{"text":" Structures suggested for complexes 4-fluorotoluene-NH<sub>3</sub> (<b>9a,b</b>) and anisole-NH<sub>3</sub> (<b>10</b>). The figure was created by the authors using data of Refs 38, 39."}}]](/storage/images/resized/kiAgRizaDjxoIW55rAwS01AF3GDu4sIyeBOQLTaJ_xl.webp)
From the set of experimental and calculated data, anisole-ammonia complex was assigned structure 10, in which the ammonia molecule is bound to anisole by three hydrogen bonds of NH…O, N…HC and NH…π types (see Fig. 13).39 In a study,41 the length of the NH…π bond in the anisole•NH3 complex was estimated at 3.02 Å, and its dissociation energy at 3.82 kcal mol–1, which looks quite realistic.
The interaction of phenylacetylene with ammonia, methylamine, water and methanol as proton donors has been studied.42,43 Due to more extended π-system and a lower ionization potential than that of benzene (8.82 eV vs 9.24 eV), phenylacetylene should exhibit a greater tendency to form NH…π complexes. Fortunately, such complexes are quite informative because of the presence of easily detectable C≡C and Csp–H bonds. On the other hand, methylamine, like ammonia, has a fairly high basicity and very low NH-acidity, which is why, when forming hydrogen bonds, they tend to behave more like proton acceptors than proton donors. As research methods, the double IR/UV spectroscopy and a wide range of quantumchemical methods (mainly ab initio with the MP2 level of theory) were employed.
As can be seen from Fig. 14, the IR spectrum of phenylacetylene contains three bands of stretching vibrations of the Csp–H bond, namely, two intense ones at 3325 and 3343 cm–1 and one of low intensity at 3334 cm–1, located strictly between the first two (±9 cm–1). The latter is attributed to the vibrations of the unperturbed C–H bond, while the other two are considered as a consequence of the so-called Fermi resonance, reflecting the resonance of the νC–H vibrations with two out-of-plane vibrations of the C≡C bond. It is known that the Fermi resonance disappears when the C≡C triple bond enters into any interactions, for example, of the π-type.
![[{"id":"TLC7sD3UVO","type":"paragraph","data":{"text":" Phenylacetylene and its complexes with NH<sub>3</sub> and MeNH<sub>2</sub>. The figure was created by the authors using data of Refs 42, 43."}}]](/storage/images/resized/lbSRa3udANWqwk0sk2QDO33B8k7MYbXSrnxMii3a_xl.webp)
Experimental and theoretical data clearly indicate that, upon the interaction of phenylacetylene with ammonia, only structure 11 with the CspH…N hydrogen bond is realized, in which ammonia behaves as a proton acceptor.42 This is evidenced by a strong displacement (by 103 cm–1) of the ν(Csp–H) bond to the low-frequency region. The stabilization of this complex and the absence of NH…π interaction is not surprising, since the acidity of the CH bond (pKa 23.2) in phenylacetylene is 10 orders of magnitude higher than the NH-acidity of ammonia (pKa 33). It is assumed that electrostatic forces dominate here.43
Surprisingly, the phenylacetylene--methylamine complex, according to its IR spectrum, has a completely different structure, although the basicity of MeNH2 (pKa 10.6) is almost 1.5 orders of magnitude higher than that of NH3 (pKa 9.2). Indeed, the ν(Csp–H) band in this complex, just like in phenylacetylene itself, lies at 3333 cm–1, simultaneously losing the triplet structure characteristic of the Fermi resonance. Based on quantumchemical calculations of the PhC≡CH•MeNH2 complex, three optimized structures 12 and 13a,b were revealed, which correspond to potential energy minima (see Fig. 14).43 Structure 12 fits a global minimum, while structures 13a and 13b are local ones. Based on the spectral data, preference was given to structure 13a, in which methylamine forms two π-bonds, acting as a CH-donor with respect to the C≡C bond and as an NH-donor with respect to the benzene ring. The abnormal behaviour of methylamine in comparison with ammonia was explained by a more extensive bond system in the former, which highlights not electrostatic, but dispersion interactions with the participation of both components of the complex.
It was also suggested43 that complex 13a is the primary kinetic product of the reaction, while structure 13b is a thermodynamic product into which 13a transforms over time. To the naturally arising question of why complex 13b is thermodynamic and not 12, it was noted that, firstly, the energy difference between both types of clusters is very small and, secondly, the corresponding ΔG values substantially depend on the chosen calculation method. Under these circumstances, it cannot be ruled out that complex 13a may also be more stable, as evidenced by the IR spectroscopic data.
An IR/UV study of the interaction of phenylacetylene with triethylamine was also reported.42 Despite the even greater basicity of Et3N (pKa 10.9) as compared to NH3, it, like methylamine, does not form σ-cluster of the PhC≡C–H…NEt3 type. In contrast to methylamine, the IR spectrum of the complex with Et3N retains the Fermi resonance for the ν(Csp–H) band. Taken together, these data indicate that the interaction of triethylamine with phenylacetylene does not affect the C≡C bond and has dispersion nature touching on mostly the benzene ring π-system.
3.3. NH…π complexes of condensed arenes
There are few reports on this topic. The 1-naphthol-NH3 complex was studied by rotational spectroscopy and two-photon resonance ionization, which was supplemented by quantumchemical calculations.44 Using the ab initio method (MP2 level of theory), the stability of two 1-naphthol-NH3 isomeric structures with a composition of 1:2 was compared, one of which (14a) contains only the conventional OH…N and NH…N intermolecular HBs, and the other (14b) also contains an NH…π bond (Fig. 15). Although both forms have similar energies, the rotational spectroscopic data are more consistent with structure 14b. The latter differs by the smallest distance (2.568 Å) between NH proton and the centroid of the nearest benzene ring.
![[{"id":"eB7fe-kI4h","type":"paragraph","data":{"text":"Proposed structures of isomeric 1:2 1-naphthol-NH<sub>3</sub> complexes. The figurewas createdby the authors using data of Ref. 44."}}]](/storage/images/resized/DrCOcykYdJh0sbLFemfx3lGDnWYpPOBE2TxerlqI_xl.webp)
An anthracene-ammonia complex was examined using fluorescence spectra and a number of other methods.45 According to the data obtained, the ammonia molecule was proposed to be located above the central benzene ring with a distance N…M of 3.6 Å (Fig. 16), which is typical of HBs in other ammonia complexes of arenes. Based on the calculation results, it was suggested that the internal rotation of the NH3 molecule in complex 15 relative to the centre of mass of ammonia and anthracene is hindered due to the anisotropy of the π-system.
![[{"id":"vVsK4MjpVK","type":"paragraph","data":{"text":"Supposed structure of anthracene-NH<sub>3</sub> complex. The figure was created by the authors using data of Ref. 45."}}]](/storage/images/resized/UQfLeGiUu8uy0yJ9fdzNJFXon8YzeuxyPxzJkb72_xl.webp)
Pandith et al.46 created a highly sensitive fluorescent sensor based on imidazole-containing perylene 16 for determining the drug 5-aminosalicylic acid (5-ASA). The action of the sensor is based on the formation of a stable 1:1 complex with 5-ASA, in which, in addition to stacking and H-bonding between the imidazole ring and the carboxyl group, the components are connected also by NH…π bonds (3.55 Å) between the NH2 group of 5-ASA and the B fragment of the perylene system (Fig. 17).
![[{"id":"2K5tok0AOX","type":"paragraph","data":{"text":" Fluorescent reagent <b>16</b> for the determination of 5-aminosalicylic acid (<b>5-ASA</b>). The figure was created by the authors using dataof Ref. 46."}}]](/storage/images/resized/S9GwaoV6YOa0gsWCYkAFIAsTwCFl7HKRn8pO7fBQ_xl.webp)
3.4. NH…π complexes of arenes with (thio)carboxamides
The importance of such complexes is determined by the fact that they are especially often found in proteins11,16 and some other natural compounds, for example, vitamin B12 (see Section 7.3). Apparently, the Nikolic's group pioneered in considering the issue of modelling the corresponding NH…π interactions using benzene and its alkyl derivatives as π-donors and N-alkylcarboxamides as proton donors.47--50 The frequencies of stretching vibrations νNH in N-methyl-, N-ethyl-, N-butyl- and N-cyclohexyl derivatives of formamide, acetamide and propionamide were measured, using a 10-100 molar excess of arene as a solvent (Table 2). It was noted that, in comparison with solutions of carboxamides in CCl4 (νNH=3455 cm–1) in arene media, this band noticeably shifts to the low-frequency region. This observation was attributed to the formation of an NH…π hydrogen bond between the NH proton and the benzene ring. This was also supported by the fact that the displacement values, ΔνNH, (as well as the integral intensity of the band and the stability constants of the complexes) increased upon the introduction of alkyl substituents into the aromatic ring (see Table 2).
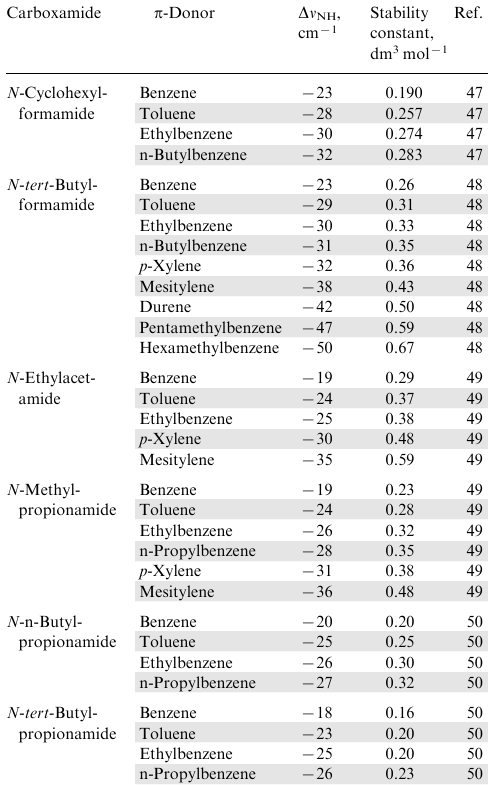
Cheng et al.51 significantly contributed to these studies by estimating geometric and energetic parameters of arene-carboxamide complexes. As a model, a benzene--N-methylformamide complex was used, for which quantumchemical calculations were performed using the MP2 method. It was found that the N…M distance, depending on parameters of the calculation, is in the range 3.2-3.6Å, while the HB energy for the optimized structure is 4.37 kcal mol–1 in average. This is almost twice as much as for the C6H6•NH3 complex, which can be due to the increased NH-acidity of carboxamides (pKa≈14-16) in comparison with ammonia and alkylamines (pKa≈33-35).52
In compounds in which the arene and amide components are part of one molecule, the situation is more complicated, and, in the solid form, the amide hydrogen bonds NH…O= C usually prevail over NH…π ones. Examples are acetamidonaphthalenes 1753 and 18.54 Thus, when considering the XRD structure of each individual molecule 17 (Fig. 18a), one might get the impression that NH…π and CH…π interactions between the acetamido group and the phenyl ring at position 8 dominate. This is supported by the very short distance (2.26 Å) between the NH proton and the Ci atom of the phenyl substituent. At the same time, attention should be drawn to the poor orientation of the N–H vector to the face of the 8-Ph group: the dihedral angle between this plane and the H–N-C =O plane of the amide group is only 32.1°. The reason for this becomes clear when considering the crystal lattice of amide 17 (Fig. 18b). It consists of chains in which the naphthalene rings of neighbouring molecules are located approximately parallel to each other, and the C= O and N–H bonds are directed towards each other, forming amide hydrogen bonds N–H…O=C. The length of these bonds (2.06-2.08 Å) is much shorter than those of N–H…M and N–H…Ci bonds (see Fig. 18a). In fact, carbonyl oxygen in solid amide 17 forms bifurcated HBs such as a relatively weaker intramolecular NH…π and a stronger intermolecular NH…O = C bonds.
![[{"id":"reMU6FZom6","type":"paragraph","data":{"text":"Molecular structure of one of three independent molecules of <b>17</b> (FAGTAK, 283-303 K) (<i>a</i>) and a fragment of crystal packing of <b>17</b> (<i>b</i>). The figure was created by the authors using data of Ref. 53."}}]](/storage/images/resized/P8P3T2Mx5S2HmqcDK1Wylq50jpeEj238y9kAkqaW_xl.webp)
In binaphthyl amide 18, two naphthalene systems are nearly orthogonal, thus making it possible for the amide NH-group to enter NH…π interaction with the benzene ring of another naphthalene moiety, which is activated by the OH group (Fig. 19). In this case, the NH…M distance (2.78 Å) is slightly less than the sum of the van der Waals (VDW) radii of hydrogen atom (1.2 Å) and the half-width of the benzene ring (≈1.7 Å) (2.513 Å). The NH-proton is even closer to the Ci atom of the same benzene ring, i.e., the NH…π interaction in 18 is quite efficient.
![[{"id":"sziV55Omju","type":"paragraph","data":{"text":"Fragment of the molecular structure of two H-linked molecules of amide <b>18</b> (BEFHIE, 150 K). The figure was created by the authors using data Ref. 54."}}]](/storage/images/resized/L9xq5pFycLzaNMvZruGUkip6jL2dJHBkwOI7YzsD_xl.webp)
Probably, in compound 18 there is also a relatively weak interaction of the NH-proton with the oxygen atom of the OH group, to which, along with electrostatic, dispersion forces make some contribution. Neighbouring carbonyl and hydroxyl groups are involved in the formation of the crystal lattice of 18: the length of C = O…HO hydrogen bonds is only 1.80 Å (see Fig. 19). For additional information on the involvement of carboxamide groups in NH…π interactions see Sections 3.8, 4, 5, 7.2, 8 and reviews.11,16
The data of the XRD study of 1-(1-naphthyl)-2-thiourea 19 revealed the presence of NH…π interactions between the NH2 group and the π-system of the benzene ring linked to the thioamide function (Fig. 20).55 To provide this interaction, the thioamide group rotates relative to the plane of the aromatic system at angle of 79.6°.
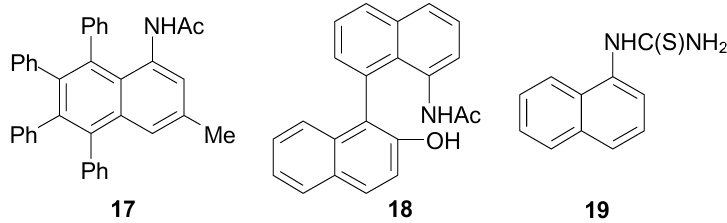
![[{"id":"xeK8Z-xwYv","type":"paragraph","data":{"text":"Molecular structure of 1-(1-naphthyl)-2-thiourea <b>19</b> (MOJDAS, 100 K). The figure was created by the authors using data of Ref. 55."}}]](/storage/images/resized/vZ6h1uk6BKMYAnA4SBCmPQmEux3Xr8i017YgxNUM_xl.webp)
3.5. NH…π complexes with sulfonamides
Recently, a simulation of NH…π interactions with the participation of an aromatic sulfonamide group has been carried out.56 As objects of study, 2,6-diarylbenzenesulfonamides 20 were taken, containing a number of donor and acceptor substituents in the para-positions of the flanking phenyl groups. XRD measurements of difluoride 20 (X=F) established the formation of an NH…π bond with one of the benzene rings with an NH…M distance of 2.68 Å and an NHM angle of 134.2°. Along with the fairly clear direction of the NH-bond vector towards the centroid of the ortho-phenyl group, there are two other characteristic features of the NH…π binding. One of them is a larger angle of rotation around the CAr–CAr bond of the phenyl ring facing the NH-proton. For difluoride 20, it is 63.9° vs 57.5° for another phenyl group. Even more revealing is the sharp flattening of the NH2 group in 20 (X=F) (ΣN=347.5°) in comparison with unsubstituted (parent) benzenesulfonamide (ΣN=334.7°). Obviously, this is due to an increase in the s-component of the valence orbitals of the more planar nitrogen atom, which enhances NH-acidity and, consequently, the NH…π interaction. An increase in the proton acceptor properties of the flanking phenyl nuclei, when electron-donor substituents are placed into them, leads to the same result.
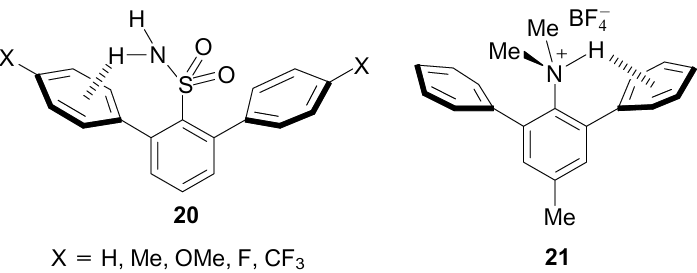
Based on the XRD studies of hydrazinylsulfonamides57 and hydrazincarbodithioic acid esters, NH…π interactions in their crystal lattices were also revealed.58
Strategy of using the flanking phenyl groups to model NH…π interactions turn out to be quite successful in the case of the dimethylammonium group. Thus, in the protonated form of N,N-dimethyl-2,6-diphenyl-p-toluidine 21, despite the imperfect orthogonality of the 2-Ph group and the central benzene ring and strong bifurcation of NH-hydrogen with the participation of the counterion (XRD data, Fig. 21), the NH…Ci distance is just about 2.31 Å, while the shortest (Me)CH…Ci distances characterizing CH…π interactions on the left side of the molecule are at the level of 2.60 Å.59 However, looking ahead, it should be said that the placement of dimethylammonium and phenyl groups in the peri-positions is more effective comprising the next progressive step in modelling NH…π interactions (see Section 3.8).
![[{"id":"UzYsDlGwpr","type":"paragraph","data":{"text":"Molecular structure of salt <b>21</b>. Selected parameters: φ<sub>AB</sub>=58.7°; φ<sub>AC</sub>=84.9°; ΣCNC=338.7°; NH<sup>...</sup>FBF<sub>3</sub><sup>-</sup>=2.13 Å"}},{"id":"R2j8bMpV-E","type":"paragraph","data":{"text":" (YUWWOF, 100 K). The figure was created by the authors using data of Ref. 59."}}]](/storage/images/resized/tzhAVKtLuBcr7aoAQNAm4c7YdYbSauY0YBwS6OgP_xl.webp)
3.6. NH…π complexes of arenes with NH-heterocycles
Considering the great importance of nitrogen heterocycles in biochemical systems, for example, histidine and tryptophan in proteins, purines and pyrimidines in nucleic acids and ATP, pyridines, pyridinium and imidazolium salts in enzymes, pyrrole in heme and chlorophyll,60 they have become very attractive objects for modelling NH…π interactions as both proton donors and proton acceptors. In this regard, it is logical to start this Section with pyrrole and pyrrole-based systems.
XRD analysis of unsubstituted pyrrole showed that T-oriented dimers 22 are formed in its crystals, in which one molecule acts as an NH-donor and the other as a proton acceptor (Fig. 22).61 In this case, the NH-proton is oriented not to the ring centroid, but to Cβ atoms, on which, as is known,63 the largest negative π-charge is concentrated. In the case of 1-(2-hydroxyphenethyl-2)pyrrole dimer 23, the OH groups are bonded to Cα atoms,64 which is apparently due to the greater stability of the ten-membered ring architecture in comparison with that which would arise at the OH…π binding at pyrrole Cβ positions. The NH…Cβ and OH…Cα distances in dimers 22 and 23 are 2.57 and 2.52 Å, respectively.
![[{"id":"dX0U9OIcl3","type":"paragraph","data":{"text":"Schematic representation of structures of pyrrole and indole dimers. Definition of intermolecular parameters, defining the mutual orientation of the units within the T-shaped indole dimer (<b>24a</b>). The figure was created by the authors using data of Refs 61, 62."}}]](/storage/images/resized/eTp7HBlVzuQS34bztTpuZRZhUnV6SdWXf09UiDZV_xl.webp)
Self-association of pyrrole was also registered in solution.65 Thus, according to quantumchemical calculations [B3LYP/6-31++G(d,p)] and IR spectra, pyrrole in CCl4 forms NH…π dimers with the T-configuration. In them, the centres of mass of monomer units are separated from each other by 4.52 Å, the interplanar angle is 73.0°, and the NH…π distance is 2.48 Å, which is in good agreement with XRD data. In the Fourier-IR spectrum of pyrrole in a dilute CCl4 solution, the νNH band has the form of a sharp peak at 3495 cm–1. In more concentrated solutions, a broadened band appears with a centre at 3408 cm-1, assigned to the NH…π bond in the dimer. Thus, the red shift value, ΔνNH, is –87 cm–1 (see also Ref. 66) which almost coincides with the calculated data (–84 cm–1).
Based on theoretical calculations and IR spectra, indole, similarly to pyrrole, forms T-dimer 23 (Fig. 22).62 However, structure 23, optimized using the B3LYP/6-31++G(d,p) method, indicates that the formation of NH…π bond occurs at the benzene rather than at the pyrrole ring. The interplanar angle α (see Fig. 22, structure 24a) is almost right (89.4°). The distance between the centres of mass of two molecules, RCM, and the length of the NH…π bond are 6.21 and 2.71 Å, respectively, while the angle φ between the straight line connecting the centres of mass and the normal is 51.5°. Noteworthy is the somewhat longer NH…π bond length in indole dimer 23 compared to pyrrole dimer 21, which indicates its lower stability. The calculated NH…π bond energy for dimer 23 is only 2.15 kcal mol–1. Returning to the orientation of the NH…π bond vector in the indole dimer to the benzene ring, it is interesting to note that in the case of tryptophan residues in protein structures this orientation is approximately equally distributed between the benzene and pyrrole rings.11
Using quantumchemical calculations (DFT-D density functional, SCS-MP2 and SCS-CC2 level of theory), as well as IR/UV double resonance spectroscopy under the supersonic-jet-cooled conditions the complexes of pyrrole with benzene and benzene-d6 were examined.67 The results of the study indicate their T-shaped structure 25a, in which the pyrrole ring is slightly inclined (by ±13°) to the normal drawn to the plane of the benzene ring. The distance N…M to the centroid of the benzene ring is 3.13 Å, and the red shift of the νNH band relative to free pyrrole (νNH=3531 cm–1) is –59 cm-1, which indicates a weaker NH…π interaction than in dimer 22 of pyrrole itself (–87 cm–1). Similar study of indole--benzene dimer using IR/UV double resonance spectroscopy revealed that in gas phase it also exists in T-form 25b with NH…π hydrogen bond.68 This is supported by the red shift (νNH=3479 cm–1) of the indole N–H stretching frequency (νNH=3525 cm–1).
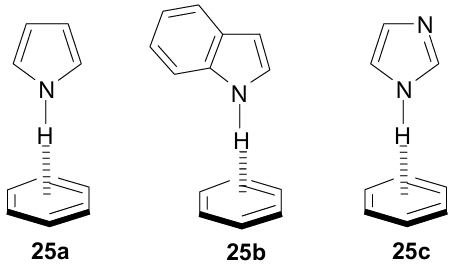
The vibration frequencies of benzene C–H bonds in the IR spectrum of complexes 25a,b practically do not change compared to those of benzene itself, which excludes CH…π interactions and confirms the acceptor action of the benzene ring with respect to the pyrrole NH-group. In the UV spectrum of 25a, a red shift of the long-wavelength band of benzene by 58 cm–1 relative to pure benzene was recordered. This was attributed to the violation of the symmetry of the π-electron system of benzene due to somewhat distorted geometry of the NH…π hydrogen bond.
Crystallographic studies of proteins showed that π-interactions involving such an important amino acid as histidine are very common in them.11 It is estimated that ≈80% of the contacts between the imidazole ring of histidine and the aromatic rings of phenylalanine, tyrosine, and tryptophan are CH…π and only are ≈20% NH…π interactions. In both cases, the imidazole ring behaves as a proton donor. Trachsel et al.69 simulated these interactions by studying the IR and UV spectra of a deeply cooled imidazole–benzene complex. DFT calculations of the ground and excited states using dispersion corrections and correlation methods SCS-MP2 and SCS–CC2 showed that complex 25c has a Cs-symmetric T-shaped structure with an NH…π bond vector directed to the benzene ring. Also, the NH-bond is inclined by 12° to the benzene C6 symmetry axis. IR spectra did not contradict this geometry. In particular, the νNH band in the IR spectrum of complex 25c is shifted by –73 cm–1 towards lower frequencies relative to spectrum of pure imidazole (νNH=3518 cm–1). The dissociation energy of the NH…π bond for the ground state of 25c in the gas phase was estimated as moderately strong (5.43 kcal mol–1). The authors did not find experimental evidence for the existence of a CH…π hydrogen bond in the ground state of 25c, which is not entirely consistent with the predominance of this geometry in proteins. In this regard, it was suggested that CH…π contacts in protein structures arise not so much as a result of energetically more favourable binding, but as a result of crystalline packing and folding of the polypeptide chain.
Other attractive pyrrole-based objects for studying NH…π interactions are 9H-pyrido[3,4-b]indoles 26a,b, also known as β-carbolines.70--72 They possess a rather extended π-system, which includes N-heteroatoms of the pyrrole and pyridine types. This makes them both potential NH- and π-donors, as well as proton acceptors, which expands the possibility of their binding to biochemical receptors through various types of non-covalent interactions. Perhaps for this reason, a number of medicines have been created on their basis, and some of their representatives are found in nature and are considered as a type of indole alkaloids.
Moreover, the behaviour of β-carbolines 26a,b towards benzene, naphthalene, phenanthrene and some azaarenes was also examined (Table 3).72 Using FTIR spectroscopy and quantumchemical calculations (AM1/MOPAC) it was shown that the νNH stretching vibration band of 26a in CCl4 decreases its intensity and, at the same time, a new band appears in the lower-frequency region, the intensity of which increases as the number of π-electrons in the arene grows. These changes were attributed to the formation of T-shaped NH…π clusters with a composition of 1:1, which, according to the results of theoretical calculations, corresponded to minima in the potential energy curves.
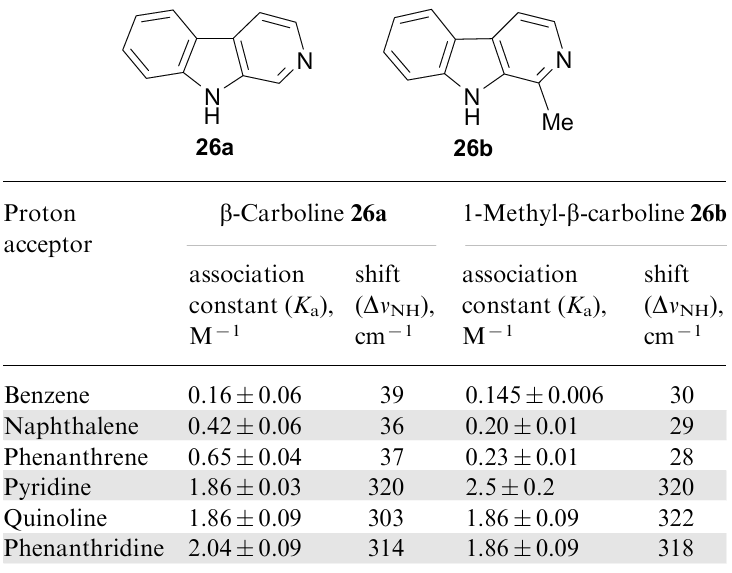
Analysis of the change in the intensity of the low-frequency band made it possible to calculate the association constants of the complexes (see Table 3), which increase with the expansion of the arene π-system, and, consequently, a decrease in its first ionization potential (growth of π-donor ability). In this context, however, it is not clear why the ΔνNH values themselves in the indicated complexes practically do not change in the series benzene–naphthalene–phenanthrene. Moving from 26a to its 1-methyl derivative (harmane) 26b, the association constants and ΔνNH values are noticeably decreased, which can be explained by steric reasons (especially in the case of naphthalene and phenanthrene) that hinders realization of the optimal geometry for the complex.
When arenes are replaced by azaheterocycles such as pyridine, quinoline, and phenanthridine, the structure of the resulting complexes changes dramatically. This is evidenced by a strong increase in their stability constants, as well as the value of ΔνNH (more than an order of magnitude! See Table 3). It is obvious that in these cases the nature of the bond between the components of the complex changes from NH…π mainly to NH…N.
Tsuzuki et al.73 carried out a detailed quantumchemical study (ab initio methods, 6-31G* and 6-311G**, MP2 level of theory) of benzene complexes with pyridine, pyridinium and N-methylpyridinium cations. Their attention was focused on the geometry of the complexes and the interaction energy (Eint) of their components. It was shown that in the weakest benzene-pyridine complex 27 both rings are located in face-to-face manner, which is typical of π, π-stacking (Fig. 23); the value of Eint in this case was equal to 3.04 kcal mol–1.
![[{"id":"oVZ3q4zTY4","type":"paragraph","data":{"text":"Theoretically optimized and alternative structures of complexes of benzene with pyridine, pyridinium and N-methyl pyridinium cations (E<sub>int.</sub> in kcal mol<sup>-1</sup>). The figure was created by the authors using data of Ref. 73."}}]](/storage/images/resized/UJuWbTHDYTtgRDUXQipYq1KAcN693EsUwXPA56Zd_xl.webp)
It is believed that the interaction of the rings in 27 is predominantly dispersive. A similar stacking is realized in benzene--N-methylpyridinium complex 30, in which the interaction energy is almost three times higher than in 27. The strongest interaction (14.77 kcal mol–1), related to the NH…π type, takes place in the benzene–pyridinium complex 28. The interaction in the alternative structure 29 with displaced π-stacking is almost half of that of 28. In the benzene-N-methylpyridinium pair, the lowest energy has a structure with π, π-stacking 30, while forms 31 with CH…π bonds and 32 with displaced π, π-stacking are less stable. Based on the large difference in the stability of benzene--pyridine 27 and benzene-pyridinium complexes 28 and 29, it was concluded that in the latter two species the attraction is determined not so much by dispersion as in 27, as by electrostatic and induction forces.
The amide group plays an important role in structural biology not only in an open-chain form, as in polypeptides and proteins, but also in a cyclic form, for example, in nucleobases (guanine, uracil, thymine, cytosine). To simulate NH…π interactions with the participation of such compounds, the investigation of complexes of 2- and 4-pyridones (2PY and 4PY) with benzene74 and various fluorobenzenes was performed.75--78 The latter are denoted below as 1FB, 2FB, 3FB, 4FB, 5FB and 6FB depending on the number of fluorine atoms.
Along with quantumchemical calculations (MP2 and SCS-MP2), double resonance IR/UV spectroscopy of strongly cooled dimers was used. As shown by the results of theoretical calculations, for the 2PY.C6H6 complex, the T-shaped structure 33 with NH…π hydrogen bond is the most stable (Fig. 24). In it, the pyridone ring is slightly inclined (by 12°) to the benzene ring, which, as in other similar cases (cf. Fig. 5), reflects a compromise between the perfectly orthogonal T-form and the plane-parallel form with π, π-stacking. The energy of the NH…π bond in 33 is estimated at 6.02 kcal mol–1, which is similar to that of the O–H…O hydrogen bond in the water dimer. Frequencies of stretching vibrations of N–H and C = O bonds in 2PY.C6H6 complex relative to the spectrum of pure 2-pyridone are reduced by 56 and 10 cm–1, respectively, which also indicates a rather strong NH…π interaction and a relatively weak participation of the C = O group in it. At the same time, the stretching vibrations of the C–H benzene bonds are almost unchanged (≈2 cm–1) compared to benzene itself, suggesting that they are not involved in the binding with 2-pyridone.
![[{"id":"csMm-d_ybq","type":"paragraph","data":{"text":"Structural motives in complexes of 2-pyridone with benzene and fluorobenzenes. The figure was created by the authors using data of Refs 75-78."}}]](/storage/images/resized/neWm11LlkCl88YmgtSgmwc3Eoir0VXqfU7UwWUoc_xl.webp)
For complexes of 2-pyridone with fluorobenzenes, the picture is much more complicated.75,77,78 Complexes 2PY.nFB, where n=1-4, are formed by two HBs, namely, C–H…O = C and NH…F–C (structure 34). In this case, with an increase in the number of fluorine atoms, the C–H…O = C bond becomes stronger than NH…F-C (for 2PY•4FB, by 4.11 kcal mol–1).
Unlike fluorobenzenes having at least one C–H bond, hexafluorobenzene is stabilized mainly due to π, π-stacking (structure 35) with an interaction energy of 5.81 kcal mol–1. In structure 35, a certain contribution of NH…F bonding is also allowed (Eint=2.32 kcal mol–1), which, in addition to the calculated data, is evidenced by the slight inclination of the plane of the pyridone molecule to the C6F6 plane, thus promoting the approach of the NH-group to one of the fluorine atoms. Table 4 summarizes the data on the interaction energies in all six 2PY•nFB dimers. As can be seen, a significant contribution of the π, π-stacking component commensurate with the H-bonding also occurs in the complexes of 1,2,4,5-tetrafluoro- and pentafluorobenzenes. The specificity of hexafluorobenzene in this series is explained as follows. The C6F6 molecule is also a quadrupole, but the direction of the quadrupole moment in it is opposite to that of benzene. Since fluorine is more electronegative than carbon, the edge of the ring in C6F6 is negatively charged, while a positive electrostatic potential forms over the ring itself. This circumstance prevents the formation of the NH…π bond, while the stacking with π-donors becomes much more preferable.
3.7. NH…π self-association of aniline and NH-heterocycles
Aniline is of interest as a model containing two proton-donor N–H bonds and two proton-acceptor centres (nitrogen atom and the ring π-system). Such structure suggests the possibility of self-association of aniline to form dimeric clusters of the head-to-head 36 or head-to-tail 37 types (Fig. 25). Yeh et al.79 calculated the energies of both forms in the gas phase using the methods of molecular mechanics (MM) and molecular dynamics (MD). In MD simulation, the head-to-head shape 36 with two parallel phenyl rings turned out to be more stable. On the contrary, the MM method gave preference to the head-to-tail conformation,44 in which each NH2 group of one molecule forms an NH…π bond with the benzene ring of the other with antiparallel orientation of the phenyl groups.
![[{"id":"LR65k3hHs5","type":"paragraph","data":{"text":"Conformations of aniline dimer calculated for the gas phase. The figure was created by the authors using data of Ref. 79."}}]](/storage/images/resized/oktIqzQiF9H5wSmTgOxaBRi4PBBV8oJEjjFgY04V_xl.webp)
Apart the calculations, the resonance two-photon spectroscopy R2PI has been used, according to which structure 36 prevails.79 However, in the same 1996, studying aniline dimers by infrared and laser mass spectroscopy, Sugawara et al.80 came to a little different conclusion. They completely ruled out the head-to-head structure 36 in favour of 37. Considering the inconsistency of these conclusions, Yamamoto et al.81 later repeated these experiments and performed ab initio calculations with the MP2/cc-pVDZ level of theory. Based on the results obtained, they concluded that both structures, 36 and 37, correspond to potential energy minima (Eint=3.36 and 4.54 kcal mol–1), but structure 37 with two NH…π bonds is 1.18 kcal mol–1 more stable.
XRD analysis of aniline at 100K also showed that NH…π and NH…N hydrogen bonds are the strongest in the crystal lattice, being realized inside the herringbone stacks that form layers.82 The energy of interaction of molecular pairs from adjacent layers due to H-bonds of two types are estimated at 2.2-3.8 kcal mol–1, and according to the so-called PIXSEL calculations, NH…π interactions make greater contribution. In addition, CH…π contacts are also involved in the formation of the crystal lattice, due to which the total interaction energy of molecular pairs can reach 5 kcal mol–1. As can be seen from Fig. 26, the distance between the NH-proton and the centroid (≈2.62 Å) and the nitrogen atom in the neighbouring molecule (2.36 Å) is noticeably less than the sum of the VDW radii of the hydrogen atom (1.2 Å) and the half-width of the benzene ring (1.7 Å).
![[{"id":"sIc3-2GJRF","type":"paragraph","data":{"text":" Fragment of aniline crystal structure (BAZGOY01, 100 K). The figure was created by the authors using data of Ref. 82."}}]](/storage/images/resized/lRs2hpz5pUbyT7vDZB5nqCi88OfSkagVl1cOSA4V_xl.webp)
In Section 3.6, we have already mentioned the self-association of pyrrole and indole. Recall that pyrrole in the solid state gives T-type NH…π dimers 22, in which the NH-proton of one molecule is directed to the Cβ atoms of the other. It is noteworthy, however, that along with the NH…C3(4) distances (2.57 Å), the NH…M distance (2.60 Å) is also rather short (Fig. 27a). Surprisingly, there are still no reliable XRD studies on unsubstituted indole (see, e.g., Ref. 85). Nevertheless, some indirect data, for example, obtained for a co-crystal of N,N’-di-n-butyl-3,6-bis(phenylethynyl)pyromellitic di-imide bis(indole) indicate the presence of NH…π interactions between the NH group of one indole molecule and the benzene ring of another one (Fig. 28).86 According to quantumchemical calculations and IR spectra,66 self-association of indole also proceeds via the NH…π binding with participation of the benzene ring (see Fig. 22, structure 24).
![[{"id":"fRDpDR3zC0","type":"paragraph","data":{"text":"NH<sup>...</sup>π binding in pyrrole<sup>61</sup> (RUVQII, 103 K, <i>a</i>) and carbazole<sup>83</sup> (CRBZOL04, 168 K, <i>b</i>) and dimer clusters of 3,6-di(4-alkoxyphenyl)carbazoles<sup>84</sup> (UHUJUC, c, UHUJOW, 283-303 K, <i>d</i>) (XRD data). The figure was created by the authors using data of Refs 61, 83, 84."}}]](/storage/images/resized/G9YJ2FJ98gU1DhlzTgTws0ATi4c7kTZqMU37npcw_xl.webp)
![[{"id":"av42mLnxRm","type":"paragraph","data":{"text":"Self-association of indole in a co-crystal of N,N'-di-n-butyl-3,6-bis(phenylethynyl)pyromellitic diimide bis(indole) (NOCTUV, 123 K). The figure was created by the authors using data of Ref. 86."}}]](/storage/images/resized/WDhvnKvQB35y585uzwo7OtkSMe8ccRAgNQQJfaJI_xl.webp)
Information on the self-association of multinuclear analogues of pyrrole and indole is no less interesting. XRD studies of carbazole83 showed that its crystallization resulted in the formation of T-type NH…π poly-associates similar to pyrrole (Fig. 27b). Indeed, the N–H bond in carbazole is symmetrically oriented to the β-carbon atoms of another molecule with NH…Cβ distances of 2.59 Å (for pyrrole, 2.57Å). In this case, similar to pyrrole, the molecules are antiparallel to each other, and the interplanar angle is 56.3°, which is noticeably less than in the pyrrole dimer (70.1°). The latter indicates the presence of other non-covalent interactions in carbazole crystals due to its more extended π-system, such as CH…π or π, π-stacking; notably, the N…M distance is 3.372 Å, which almost coincides with the sum of the VDW radii of the N atom and the half-width of the aromatic ring.
The dependence of the geometry of NH…π aggregates on the extension of the π-system is also traced in 3,6-di(4alkoxyphenyl)carbazoles.84 Thus, in dimers with two p-anisyl and 4-propoxyphenyl groups, the interplanar angle decreases to 47.7° and 41.5°, respectively (Fig. 27c,d). It is also noteworthy that in Fig. 27d the proton donor group behaves specifically with its N–H vector directed to the benzene, not pyrrole, ring of the carbazole system as in Fig. 27a-c. Apparently, this is due to an increase in steric factors when replacing methoxy with bulkier propoxy groups, which also affects the crystal packing. This explanation is supported by a noticeable increase in the banana-like curvature of the molecules upon transition to the propoxy-substituted compound (Fig. 27d).
A series of indolocarbazoles 38-41 with semiconducting properties was synthesized.87 XRD studies and DFT calculations disclosed the presence of NH…π dimer clusters in them, which enhance their conductivity. The energy of NH…π interactions is estimated at about 2.4 kcal mol–1, which is comparable to the energy of CH…π interactions also observed in these dimers (2.2 kcal mol–1). Accordingly, it was found that dimers of compounds 38 and 39 with the syn-oriented N–H bonds are more stable and thus encouraging for their examination as semi-conductors.
Analysis of the XRD data for the salt formed by 2-aminothiazolium cation with (2,4-dichlorophenoxy)-acetate anion showed self-association of two cations with participation of NH2 group of one unit and thiazolium ring centroid of the other one in its crystal lattice.88
3.8. peri-Disubstituted naphthalenes as NH…π models
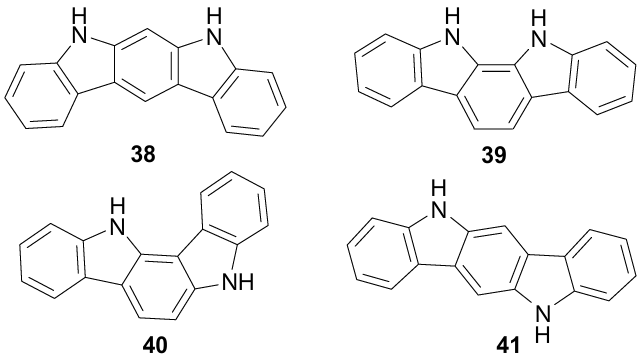
It is known that due to the 'proximity effect' of peri-substituents, 1,8-disubstituted naphthalenes are often used to study strong short hydrogen bonds,89,90 to simulate transition states,91 to stabilize atropoisomers,92,93 to conduct various cyclizations,62 to synthesize elementochelates,94 etc.95 Taking this into account, we recently proposed 1,8-disubstituted naphthalenes 42-49 (Ref. 19, 20) as models for studying NH…π interactions. They have the advantage of synthetic availability and stability, allowing XRD and practically any spectral measurements, providing information for the solid state, solution and the gas phase. Phenyl, 4-methoxyphenyl, 4-hydroxyphenyl, 2-naphthyl and 1-pyrrolyl groups were used as the π-donor components, and positively charged Me2NH+ and NH-pyridinium along with neutral NH2 and NHAc groups served as proton donors. In model salts 42-47, low-nucleophilic tetrafluoroborate was the counterion (Fig. 29).
![[{"id":"kJy1ZmZyJ4","type":"paragraph","data":{"text":"1,8-Disubstituted naphthalenes used studying NH<sup>...</sup>π for interactions. The figure was created by the authors using data of Ref. 20."}}]](/storage/images/resized/fpIFdycmjBTTjs9u9KCFYchM63Gza3eegRHwsN8f_xl.webp)
Two above-mentioned examples of neutral 1,8-disubstituted naphthalene 17 and 18 should be pointed out, in which the acetamido group acted as a proton donor. However, these sterically congested compounds were not specially taken as models of NH…π interactions and the latter were considerably masked by other types of hydrogen bonds.
Some important parameters that shed light on the nature of NH…π interactions in compounds 42--49 are summarized in Table 5. The first thing that attracts attention is the rather strict orientation of the NH bond vector in compounds 42--46 to the plane of the phenyl group in the position 8 [compare the values of the dihedral angles ∠HNC(1)C(2), which are close to 180°]. Simultaneously, the 8-phenyl group turns around the C(8)–C(1’) bond almost perpendicular to the naphthalene ring (φ=79-88°).
![Selected bond lengths, distances (Å) and angles (deg) in compounds <b>42-49</b> [all XRD measurements were conducted at 120 K except for salt <b>45</b> (100 K)].<sup>20</sup>](/storage/images/resized/0WFCiJ0bXPJ4WDrm1K85w9EO9LNbonHsPeSi3LDx_xl.webp)
Due to the specific arrangement of peri-substituents in salts 42-46, the NH-proton is much closer to the Ci atom of the phenyl group (2.05-2.08 Å) than to the centroid (NH…M=2.39-2.50 Å). The only exception, which should be discussed in more detail, is salt 46 with a 2,5-dimethyl-1-pyrrolyl substituent. In this compound, the distances of the NH proton from the ring centroid and the pyrrole N-atom are exactly the same (2.07 Å), and the first of them (NH…M) is the shortest among all currently known models with NH…π interaction.
Taking into account the short length of the NH…N bond in 46, the question arises whether this circumstance can be interpreted as the direct participation of the lone electron pair of the pyrrole nitrogen atom in the formation of HB. We tend to give a definitely negative answer to it, since the sum of the bond angles at the pyrrole N-atom at 46 is 359°, i.e., the heteroatom retains the planarity inherent in the sp2-state and, in fact, is fully involved in the 6π-electron aromatic system. It seems reasonable to classify such a hydrogen bond as NH…N(π).19
Meanwhile, pyrrole-containing systems have also been described in which the N-heteroatom is markedly pyramidal under similar circumstances, although its participation in cyclic conjugation is not in doubt. It is logical to denote such bonds as NH…N(n, π).19 They are realized, for example, in the protic salts of 1-methyl-9-dimethylaminobenzo[g]indole 50-52. The participation of not only the ring π-system, but also the electron pair of the heteroatom in hydrogen bonding, in addition to the loss of planarity by the latter (Fig. 30, parameter ΣN1), is evidenced by a noticeable deviation of the N(1)–Me group from the mean plane of the molecule.
![[{"id":"DII9JeoN-F","type":"paragraph","data":{"text":"Molecular structures and selected geometrical parameters in tetrafluoroborates of protonated 9-dimethylaminobenzo[g]indoles <b>51-53</b> (XOBRAK, XOBREO, XOBRIS, 120 K; XRD data). The figurewas created by the authors using data of Ref. 19"}}]](/storage/images/resized/wChYb2qBHc7LwBValFpfPESx3ykp1UP10S8hE3Ak_xl.webp)
The degree of pyramidalization of the N1 atom and, therefore, the NH…N(n, π) HB energy in salts 50-52 is noticeably influenced by the substituent at position 2 of the pyrrole ring. The HB is more stable in benzo[g]indoles 51, 52 bearing 2-aryl groups. Apparently, 2-aryl substituents sterically facilitate thedeviation of the N(1)–Me group from the heterocyclic ring plane, which moves it further away from the NH-proton. This, in turn, weakens the bifurcation of the latter with the BF4- anion [see Fig. 30, compare the tendency for the parameters NH…N(1), N(1) …N(2) and NH…FBF3-]. Thus, the distance from the NH-proton to the nearest fluorine atom of the BF4- anion decreases in the series 52 (2.40 Å)>51 (2.25 Å)>50 (2.17 Å). Among salts 42--47, the HB bifurcation is especially pronounced in benzo[h]quinolinium salt 47 (2.19Å). As a result, the NH…π interaction is sharply weakened, as evidenced by a decrease in the values of φ and NHM angles and an increase in the NH…M and NH…C(1’) distances (see Table 5).
The influence of the counterion on NH…π interactions in salts 42-47 and 50-52 does not allow adequate assessment of their energy (EHB), due to its strong and uneven underestimation.19 To exclude this complication, theoretical calculations of the EHB values were carried out for `naked' cations 42-44, 46 and 50-52 in the gas phase and in a solution of MeCN (Table 6). As expected, they arranged the first group of cations both for the gas phase and the solution in the series 46>43>44>42, which closely coincides with a decrease in the proton-acceptor ability of the π-donor substituent at position 8. In benzo[g]indole cations 50-52, the EHB values turned out to be on average 3 kcal mol–1 lower than in 42-44, 46, apparently due to the less favourable arrangement of the pyrrole ring relative to the Me2NH+ group.
![Calculated energies of NH<sup>...</sup>π hydrogen bonds in 'naked' cations <b>42-44</b>,<b> 46</b> (Ref.20) and <b>50-52</b> (Ref.19) [B3LYP/6-311++G(d,p)].<sup>a</sup>](/storage/images/resized/XglPJJ311lPfbqIMVLTqOOKTUp4ACdq0tV2ZC8hs_xl.webp)
Let us now turn to neutral models 48 and 49 with NH2 and NHAc groups as proton donors. The first thing that attracts attention in the structure of amine 48 (Fig. 31) is the strong pyramidalization of the nitrogen atom (ΣN=338.3°), which sharply distinguishes it from the completely flat NH2 group in 1-aminonaphthalene , which is coplanar with the ring, and benzene complex of 2aminooxazoline 8 (ΣN=360.0°).34 The second feature of the NH2 group in 48 is the striking difference in the lengths of the internal (0.85 Å) and external (1.07 Å) N–H bonds. While the length of N–H bond directed to the π-system of the benzene ring is close to the common values for aniline NH2 groups (0.84-0.90 Å), the external N–H bond is strongly stretched (1.07 Å). It is known that the proton-donor X–H bonds involved in HB formation are usually elongated. Stretching of the external N–H bond in amine 48 suggests that it is also involved in strong conventional H-bonding. This is confirmed by considering the crystal lattice of 48. As can be seen from Fig. 31b, the external N–H bonds in each crystallographic unit of 48 are linked by rather short HB with the amine nitrogen of the neighbouring molecule (NH…N=2.5 Å).
![[{"id":"JuptECGtoB","type":"paragraph","data":{"text":"Molecular structure (<i>a</i>) and fragment of the crystal lattice (<i>b</i>) of amine <b>48</b> (JUXFEQ, 120 K). The figure was created by the authors using data of Ref. 20."}}]](/storage/images/resized/a8Vz30M4IDjstCcNNn1PYvxOFO6Qzp3bgbKjnrpn_xl.webp)
In contrast to NH…N hydrogen bonds, the stretching of N–H bonds in the case of noticeably weaker NH…π interactions cannot be so significant. Indeed, the internal N–H bond (0.84 Å) in the benzene complex of 2-aminooxazoline 8 is only 0.01 Å longer than the external one (0.83 Å).34 In amine 48, the internal N–H bond has approximately the same length (0.85 Å) as in 8. It is possible that the slight increase in length in 48 is caused by differences in the hybridization of two nitrogen atoms or by the orientation of the NH-vector at 48 not to the centroid or the ring C(1’) atom, but to the middle of the C(1’)–C(2’) bond. In any case, however, there is little doubt that a weak NH…π bond exists in amine 48, as evidenced, for example, by a significant facing of the p-anisyl group towards the N–H vector.
Considering the increased NH-acidity of carboxamides in comparison with that of arylamines,52 one could expect that NH…π binding in compound 49 would be very noticeable. However, XRD measurements showed that the crystal lattice of 49 is formed exclusively by intermolecular amide bonds NH…O-C (Fig. 32b).20 This distinguishes compound 49 from its more complex naphthalene analogues 17 and 18, in which NH…π interactions are superimposed with other types of non-covalent bonds.
![[{"id":"K4HRy2fAWt","type":"paragraph","data":{"text":"Molecular structure (<i>a</i>) and a chain of H-bonded associates along the <i>c</i> axis (<i>b</i>) in the crystal lattice of amide <b>49</b> (JUXDUE, 120 K). The figure was created by the authors using data of Ref. 20."}}]](/storage/images/resized/7qNP814En1S5cRG26wekROqRFnPResZTW2HEIaAJ_xl.webp)
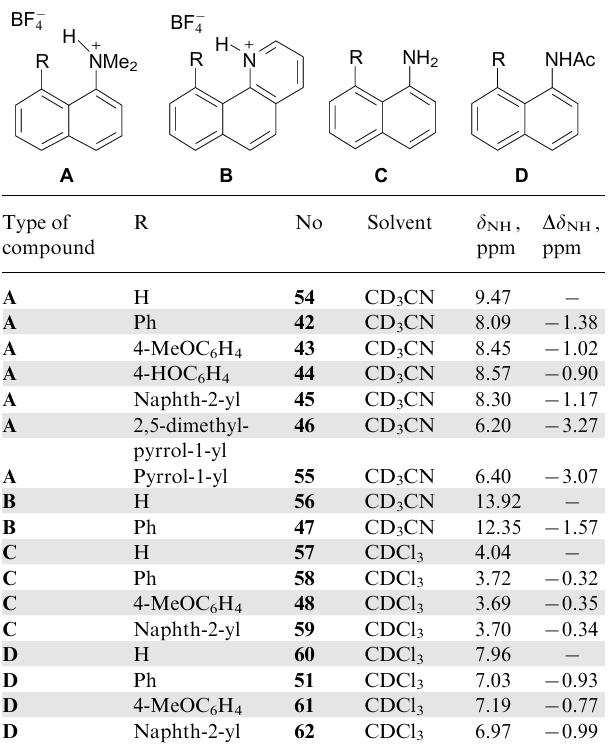
One of the significant advantages of peri-disubstituted naphthalene is an opportunity to systematically study NH…π interactions in its solutions using 1H NMR spectra. Here one can draw an analogy with the classic example of the CH…π hydrogen bond between chloroform and benzene (Fig. 33), which manifests itself in a noticeable shielding (ΔδCH=–1.56 ppm) of the CHCl3 proton.96 The existence of complex 53 in the solid state both inside and outside the supramolecular container was also confirmed using the XRD method;97--99 it is noteworthy that the CH…π distance in this case strongly depends on the external environment, varying from 2.103 to 2.826 Å. It is natural to believe that such an effect can also be useful for evaluating the NH…π interaction (see, e.g., Ref. 100). The corresponding values of chemical shifts, δNH, and paramagnetic displacement, ΔδNH, for some peri-disubstituted naphthalenes relative to the reference compounds, are represented in Table 7. As the reference compounds bearing no aromatic substituent in the adjacent peri-position, 1-aminonaphthalene 57, 1-acetamidonaphthalene 60, and dimethyl(naphth-1-yl)ammonium 54 and benzo[h]quinolinium tetrafluoroborates 56 were chosen.
![[{"id":"U8RugZo6jE","type":"paragraph","data":{"text":"Chemical shifts of the chloroform CH-proton in cyclohexane and benzene solutions. The figure was created by the authors using data of Ref. 96."}}]](/storage/images/resized/Efjxi6C4fY5rtyFDIGtZwZXWDJgHdGem8vyqLqLn_xl.webp)
It follows from Table 7 that the largest paramagnetic shifts, ΔδNH (–3.27 and -3.07 ppm), are observed for salts 46 and 55 containing 1-pyrrolyl groups as proton acceptors, while for their aryl analogues 42--45 ΔδNH values are 2.5-3 times less (–0.90...–1.38 ppm), and for neutral amines 48, 58 and 59 they are only –0.32...–0.35 ppm. It was then shown that in the case of salts 42--46 and 55, which do not have pronounced bifurcate interactions of the NH-proton with the BF4- anion, there is a satisfactory linear correlation between the ΔδNH values and the NH…M distances.20
Apparently, two factors mainly determine the magnitude of the paramagnetic shift of the NH proton signal in the NMR spectra of compounds A–D. Both act in opposite directions, with the result that ΔδNH reflects their compromise. The first factor is the magnetic anisotropy of the aromatic ring and the presence of a ring current, the paramagnetic component of which causes shielding of the NH proton. The second factor is the deshielding of the proton involved in the formation of a hydrogen bond. The first factor dominates especially clearly in the case of group A compounds.
As for amine 48, all data indicate that, in contrast to the solid state, there is no intramolecular NH…π binding in solution. Indeed, in the 1H NMR spectrum of 48 in CDCl3, NH2 protons appear as a two-proton singlet at δ 3.69 ppm. Their equivalence is retained even at –90 °C, indicating fast rotation of the amino group in the NMR time scale. Some paramagnetic shift (ΔδNH=–0.35 ppm) of the NH2-signal relative to that in 1-aminonaphthalene 57, as in the spectra of amines 58 and 59, most likely reflects the alternate short-term stay of each NH-proton in the paramagnetic region of the ring current of the neighbouring aromatic rings.
The absence of stable NH…π HB in 48 is also confirmed by IR spectra. Thus, the bands of symmetric and antisymmetric stretching vibrations of the NH2 group in the solution of 48 in CCl4 (νs=3401 cm–1, νas=3495 cm-1) in comparison with amine 57 (νs=3395 cm–1; νas=3476 cm-1) undergo a blue shift, rather than a red shift as usual during the formation of the HB, including NH…π bond.67
The situation for solutions of carboxamides 49, 61 and 62 looks ambiguous. Their 1H NMR spectra in CDCl3 demonstrate a moderate paramagnetic shift of the NH signal (ΔδNH=–0.77...–0.99 ppm) relative to that in spectrum of 1-acetamidonaphthalene 60 (see Table 7). At the same time, in the IR spectra of 49 and 61, the νNH band is strongly shifted to the high-frequency region (Table 8). The IR spectral data, which indicate the absence of NH…π interactions in amides 49 and 61 in solution, seem to be more reliable. The blue shift of the νNH band, along with an increase in the νCO frequency and a paratropic shift of the NH-signal in the 1H NMR spectrum, can be interpreted as a result of the destruction of carboxamide associates.

4. Folded and caged models
The main disadvantage of unfolded and predominantly intermolecular models is that they are poorly preorganized for effective NH…π interaction. This circumstance (in thermodynamic terms, the entropy factor) seriously reduces the stability of the complexes. As a result, their research requires special methods and conditions (see Section 2), which quite often leads to contradictory conclusions. The only exceptions are peri-disubstituted naphthalenes (see the previous Section), in which the NH…π binding is provided by the `proximity effect' of the proton-donor and proton-acceptor units.
Since the NH…π interactions in proteins and other biomolecules are commonly realized within folded and sufficiently preorganized structures, their similar intramolecular models seem to be very attractive. Over the past quarter century, several approaches have been proposed to make progress in this direction. Most of them use motifs of supramolecular chemistry, when the model is based on the derivatives of cyclophanes, cryptands, podands, calixarenes, chelated metal complexes, etc. In their molecules, both interacting components are either part of a single structural unit, or they are initially separated, but one of them, under suitable conditions, is encapsulated inside the cavity of the other.
The preorganized effect can be also achieved largerly in ammonium salts if the π-donating arene or hetarene is a part of a relatively complex anion. In this case, the very electrostatic attraction of counterions becomes a preorganizing factor. Typical examples of such models are summarized in Table 9. It is convenient to divide them into three general types:
(I) One-component systems based on a neutral molecule, cation or salt.
(II) Two-component systems in which an NH-donor guest is encapsulated into host framework.
(III) Two-component systems in which arene or hetarene guest molecule (e.g., benzene, pyrrole, imidazole, etc.) is encapsulated into an NH-containing cavity.
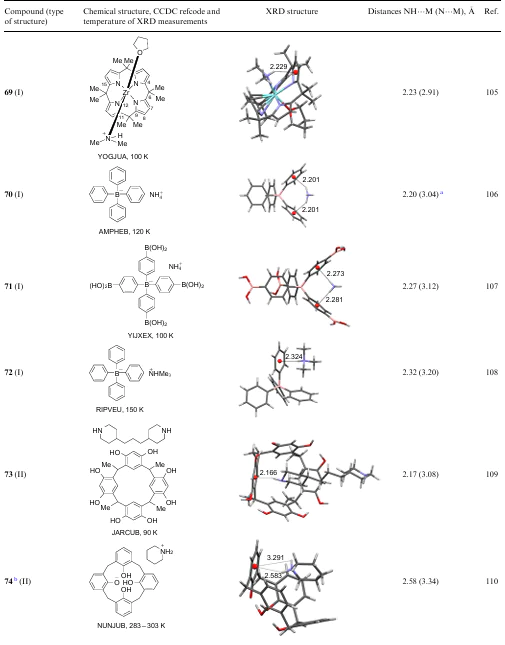
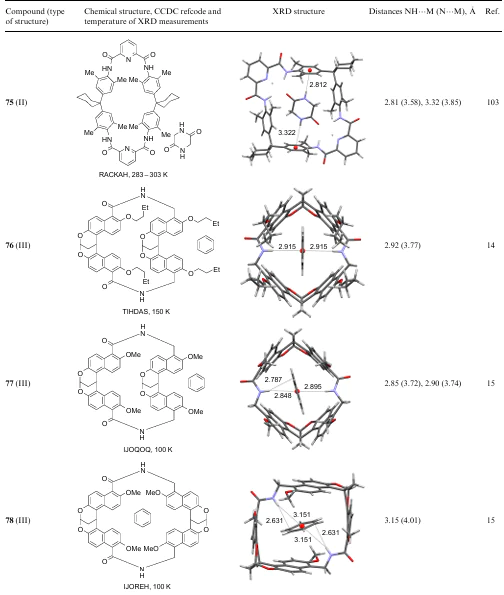
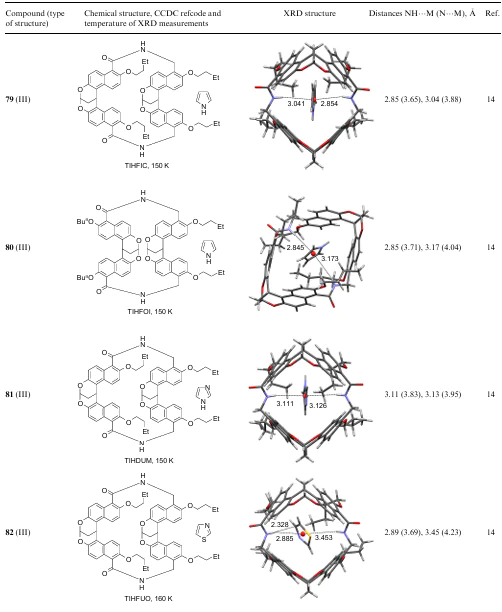
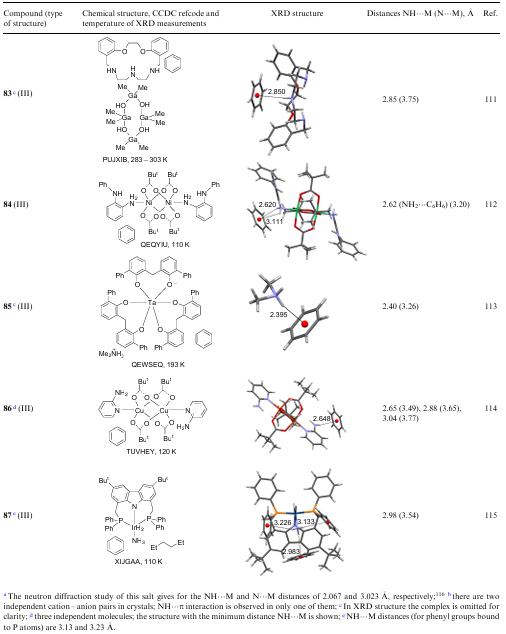
4.1. One-component NH…π systems
A typical representative of the first group of models is compact tetraazacyclophane 63 (Scheme 1), in the inner space of which no other Lewis acid except for a proton can be placed.101 The pKa values of this tetraacid base determined by potentiometric titration in an aqueous solution are: pK1=10.24; pK2=8.5; pK3=7.5; pK4=2.6. The first of these values characterizes the thermodynamic basicity of the bridging tertiary N-atom. Indeed, as shown by DFT calculations, monoprotonation of 63 in the gas phase, leading to the formation of cation 64, is 14 kcal mol-1 more favourable than that of the next secondary Nthe values N-atom. This is explained by the formation of the NH…π bond between the encapsulated NH-proton and the benzene ring, which can be considered as the internal solvation of monocation 64. In similar calculations for a solvating (aqueous) medium, the mentioned difference in monoprotonation energies decreases to 7 kcal mol–1 due to the possibility of external solvation of the NH2+ groups. Nevertheless, monocation 64 again remains the most stable species under these conditions. At the same time, kinetic basicity, i.e., the rate of attachment of a proton to secondary nitrogen atoms in 63, due to their lower steric shielding, is probably higher. This assumption allows understanding why the authors of the discussed work failed to obtain monocation 64 in an individual state, because of which they had to deal only with tetracation 65 (as tetrachloride).
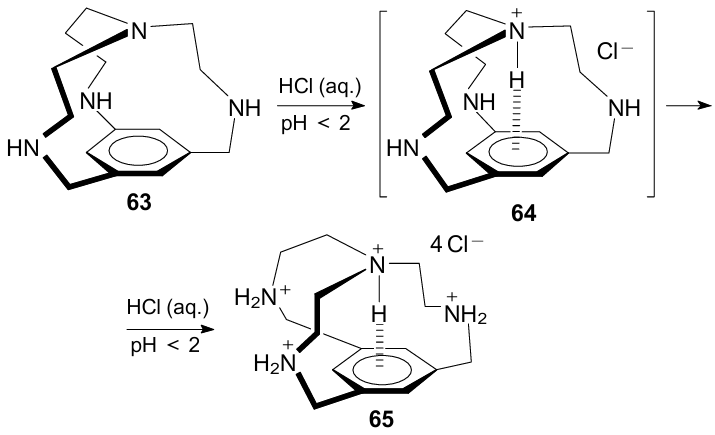
XRD measurements showed that the internal N–H vector in 65 is almost perfectly directed to the benzene ring centroid (NHM angle is 179°) with NH…M and N…M distances of 2.15 and 2.97 Å, respectively. Both latter values lie very close to the lower bounds of these distances measured for a wide range of compounds with NH…π interactions. Unfortunately, in the 1H NMR spectrum of salt 65, it was not possible to register the signal of the internal NH-proton due to its fast exchange (in the NMR time scale) with the protons of the NH2+ groups.
Compounds 66-69 belong to other members of the models of the first group (see Table 9). It is easy to see that they are not as perfectly preorganized as tetracation 65, which manifests itself in somewhat larger NH…M and N…M distances. Worth noting is cyclobis(phosphazene) 68, in which there are two identical NH…π interactions, strongly symmetrizing this complex structure. Changing the bulkiness of substituents present in the four-membered rings allows modulating the geometric characteristics of the entire system. Compound 69 is of interest, since the pyrrole system is involved as a π-donor in the formation of NH…π hydrogen bond instead of the more traditional benzene ring (cf. compounds 46 and 55).
Despite the non-framework architecture of ammonium tetraphenylborate salts 70 and 71, the NH…π interaction in them is very effective. Apparently, this is a consequence of both the strong electrostatic attraction of the cation and anion, and the polydentate nature of both counterions. Indeed, NH…π bonding in these salts involves four benzene rings and all N–H bonds of the NH4+ cation (Fig. 34). Due to this, chains are formed in the crystal lattice, including completely identical NH…π bonds with lengths of 2.20 and 2.27 Å for 70 and 71, respectively. The N…M distances (3.04 and 3.12 Å) are also quite short, which is lower than the sum (3.3 Å) of the VDW radii of the nitrogen atom and the half-thickness of the benzene ring. Against this background, the NH…π interaction of the tetraphenylborate anion with the monodentate cation of trimethylammonium in salt 72 looks expectedly weaker (see Table 9). It is noteworthy, however, that in 72 there is also a weak CH…π interaction between one of the Me groups of the proton donor and the nearest benzene ring of the tetraphenylborate anion (Fig. 35).
![[{"id":"BGPPK5JpkO","type":"paragraph","data":{"text":"Fragments of the crystal lattice of ammonium tetraphenylborates <b>70</b> (<i>a</i>) and <b>71</b> (<i>b</i>). The figure was created by the authors using data of Refs 106, 107."}}]](/storage/images/resized/ZlPFDPIkJjwvrxKtHfLmBR34GQIQXNO8G5ymLgrA_xl.webp)
![[{"id":"CWX61oTftK","type":"paragraph","data":{"text":"NH<sup>...</sup>π and CH<sup>...</sup>π contacts in salt <b>72</b>. The figure was created by the authors using data of Ref. 108."}}]](/storage/images/resized/hDCLSYmaOTe8WQmPuKTSlu7Gawl5tnyPF7Oh02ne_xl.webp)
4.2. Two-component NH…π host--guest systems
4.2.1. NH-Donor as a guest
The Table 9 shows data for three models of type II (compounds 73-75).103,109,110 The π-donor component in them is a calixarene or a macrocyclic framework structure, and the proton donor is a neutral diamine, 1,4-di(piperidin-4-yl)propane, piperidinium cation and glycine-based diketopiperazine, respectively. Despite the significantly higher NH acidity of the piperidinium cation in 74, judging by the NH…M and N…M parameters, the efficiency of the NH…π interaction is noticeably higher in model 73 with neutral amino groups. A reasonable explanation for this may be the rod-like form of 1,4-di(piperidin-4-yl)propane, which allows its deeper penetration into calixarene cavity. XRD patterns are consistent with such explanation. In the case of model 75, the internal cavity of the macrocyclic receptor is somewhat large for the guest molecule, as evidenced by the increased NH…π distance (2.81 Å), and for the second NH group it reaches 3.32 Å.
4.2.2. Benzene and other aromatics as guests
In NH…π clusters of the group III (see Table 9), the guest molecule is benzene (76-78 and 83-87)14,15,112--115 or five-membered heterocycle such as pyrrole, imidazole, and thiazole (79-82),14 while the proton donors in the host molecule more often are part of carboxamide (76-82) and, less often, amino groups of the alkyl (83) or aniline type (84, 86); in one case (87), the proton donor is an ammonia molecule coordinated with an iridium atom. Representatives of the NH…π models of group III are distinguished by even longer NH…M and N…M distances, which are in the ranges of 2.7-3.0 and 3.2-4.2 Å, respectively. Apparently, the main reason for this is the entropy factor caused by the disorder and strong anisotropy of the free benzene or hetarene molecule, which requires additional energy consumption for its proper fixation. Interestingly, imidazole and thiazole in structures 81 and 82 form NH…π rather than NH…σ clusters. At the same time, in the case of thiazole, attention is drawn to a very short contact (2.33 Å) between the NH proton and the C(4) atom of the heterocyclic ring.
In addition to the structures represented in Table 9, cluster C6H6•[HNMe2]+ (85) should be mentioned. It was rather unexpectedly isolated in the reaction of 2,2’-methylene-bis(6-phenylphenol) 88 with pentakis(dimethylamino) tantalum 89 (Scheme 2). The cluster is a salt in which the tantalum-containing chelate exists in the form of an anion, and dimethylamine, which is eliminated during the reaction, is in the form of a dimethylammonium cation. The sample that was subjected to XRD analysis contained crystallization benzene (2.3 C6H6 molecules per one metal complex species). Surprisingly, despite the electrostatic attraction of the anion and the cation and the disorder of the Me2NH+ cation in the crystal, the latter donates the NH proton not to the benzene rings of the macrocyclic ligand, but to one of the C6H6 solvate molecules. The resulting complex 85 is the simplest example of benzene NH…π clusters known to date, for which an XRD study was carried out (see Fig. 9). Interestingly, the NH…π distance in this cluster is rather short and amounts to 2.40 Å.
Concluding this Section, it should be emphasized that caged and folded models, along with their preorganization and a certain simulation of biological structures, have their drawbacks. They lie mainly in a tedious preparation of most of them, as well as in the difficulty of creating a space without too large voids and at the same time not being too cramped for the chosen molecular guest.
4.3. Zip-complexes
In the so-called zip complexes, two usually horseshoe-shaped components, formed by a certain number of covalently linked functional groups, are linked to each other through multiple non-covalent bonds. Thus, Adams et al.117 synthesized complex 90, in two parts of which (on the left and in the centre), adhesion is provided by NH…O = C hydrogen bonds, while on the right due to NH…π interaction between the pyrrolic NH group and the benzene ring. Comparing the stability constant of this complex in chloroform solution with that of a related complex devoid a pyrrole group, the NH…π bond energy in 90 was estimated to be 1.1±0.1 kcal mol–1. It is noteworthy that, as expected, the resonance of pyrrole hydrogen NH in the 1H NMR spectrum of 90 was paramagnetically shifted (–1.5 ppm). At the same time, the resonances of the NH amide protons were shifted to a low field (+1.4...2.1 ppm).
5. Folded/unfolded models
The models of this group directly simulate the high flexibility of proteins and some other biologically important compounds and their ability to change easily their conformation from folded to unfolded and vice versa.
5.1. Short peptide and diketopiperazine models
In the context of the above material, the idea of modelling XH…π interactions using short peptides seems pretty obvious. Studies of this kind started around the turn of the century. To exclude the formation of zwitterionic structures, modified analogue of classical di-, tri-, and (much less often) tetrapeptides containing protective groups such as NHCOCH3 and CONH2 at their termini are usually tested. The main goal of such simulations consisted in clarification of local backbone/chain and intra-backbone (normally amide one) interactions and their influence on conformational mobility and the character of protein folding.
A great majority of such experiments were conducted in the gas phase using double IR/UV resonant laser spectroscopy at a temperature close to zero degrees Kelvin (supersonic jet expansion). Quite narrow stretching vibration peaks at 3370-3450 were commonly assigned to NH…π bonds. Their number and νNH frequencies indicated the number and possible nature of the conformers. Additional information was provided by UV spectroscopy, since the change in the position of absorption bands of the chromophore (phenyl or indole residue) testified to its involvement in the XH…π interaction. The final conclusion about structure of each conformational isomer was obtained on the basis of the high-level theoretical calculations. Both the methods of these investigations and the corresponding results have been recently generalized in three excellent reviews.8,118,119 Given this circumstance, below we only briefly highlight the most general trends and results. Some of them are presented in Table 14.

One of the main problems in modelling non-covalent protein interactions using amino acids and peptides is the high flexibility of the latter, making it rather difficult to conduct fairly accurate measurements. A typical example is the study in which the conformational activity of tripeptide glycyl-phenylalanyl-alanine was examined.127 The authors' approach consisted in evaluation of surface free energy of various Gly-Phe-Ala conformers. Theoretical calculations (molecular dynamics, meta-dynamics and high-level ab initio) combined with the measurement of IR and two-photon resonance ionization spectra (R2PI), revealed 16 conformations for the gas phase, which differed in the shape of the peptide backbone. The differences in the corresponding potential energy minima did not exceed 3 kcal mol–1. Conformation 91 with an NH…π bond of the i→ (i–1) type turned out to be the most stable (Fig. 36).
![[{"id":"FSjSOsGccA","type":"paragraph","data":{"text":"NH<sup>...</sup>π interaction of type i → (i -- 1) for the most stable conformation of tripeptide Gly-Phe-Ala (gas-phase calculation). The figure was created by the authors using data of Ref. 127."}}]](/storage/images/resized/hSTsAmrFDtx0VieHpX5ouVQ2QdicddB0quwUy41a_xl.webp)
Paradoxically as it may seem at first glance, a significant number of cases have been reported where the complication of the peptide structure leads to a decrease in the number of conformers. This is due to an increase in the rigidity of their molecular structure, which limits the number of degrees of freedom. Thus, while seven different conformers were identified for tyrosine in the gas phase (see also similar data for phenylalanine128), judging by the number of νNH peaks in the IR spectrum, for di- and tripeptides, Tyr-Gly and Tyr-Gly-Gly, their number was reduced to four and three, respectively.122
As mentioned in the Introduction, the formation of the amide HBs is a key factor which stabilizes both the protein α-helix and βsheet. This is also manifested in oligopeptide models (Fig. 37). Formally, during the formation of NH…O = C bonds in the protein chain, rings of various sizes are closed, which, for example, for a five- and six-membered ring is denoted by the symbols C(5) and C(7), respectively. In some short peptides, even if they contain residues of aromatic amino acids, NH…π bonds are not detected at all. This depends on the nature of neighbouring amino acid residues and, to a large extent, on the character of specific chain turns (the classification of the latter is given in reviews8,118).
![[{"id":"sk5arBcNsU","type":"paragraph","data":{"text":"Selected examples of amide NH<sup>...</sup>O=C and NH<sup>...</sup>π hydrogen bonds in some peptide models. The figure was created by the authors using data of Refs 121, 123, 126"}}]](/storage/images/resized/FfVge0n9ayQOuJwC5r69JojrrI9mV3pPC0JKRoaK_xl.webp)
With the help of peptide models, it was found that the coexistence of several aromatic amino acids in them can lead to the formation of sandwich-like hydrogen bonds. In such cases one proton enters into NH…π interaction with two aromatic residues at once, for example, indole and p-hydroxyphenyl as in a tripeptide Ac-Trp-Tyr-NH (see Fig. 37). It is believed that in real proteins, such stabilization of domains with a high content of aromatic rings can improve their hydrophobicity.118,126 Interestingly, in the IR spectra of models with sandwich hydrogen bonds, the bands of the νNH stretching vibrations are in the region of 3370-3380 cm–1, i.e., they undergo a greater red shift compared to monodentate NH…π (3415-3450 cm–1) and especially free N–H bonds in simple amides (≈3495 cm–1). For comparison, the νNH values of NH…O = C bonds lie within 3300-3350 cm–1, depending on the degree of their stretching.
The possibility of structuring peptide chains through NH…π interactions in solution was also studied.129,130 For this, a series of oligomeric pseudopeptides with the inclusion of α-amino acids containing aromatic ring was synthesized. One of the model compounds of this kind 92, in which some of the N–H amide bonds are protected by a phthalimide group, is shown in Fig. 38. Using 1H NMR and FTIR spectroscopy, NH…π interaction of type i→ (i+2) between the amide N–H bond and phenylalanine residue, very rare for proteins, was found in 92. One evidence of this was the paramagnetic shift of the NH proton signal (Δδ=–0.8 ppm).
![[{"id":"lHJapkuFY5","type":"paragraph","data":{"text":"An example of a model pseudo peptide illustrating the possibility of structuring such compounds through NH...π interactions. The figure was created by the authors using data of Ref. 129"}}]](/storage/images/resized/bRqQcEcwoi5H79vHWyU15n1P52Hn4ornDFC2yuak_xl.webp)
More recently, a series of dipeptides and tripeptides of type X capable of assuming a stable folded conformation have been reported (Fig. 39a).131 The central fragment in them is an unnatural amino acid based on β-proline Y, abbreviated as Abh (7azabicyclo[2.2.1]heptane-2-carboxylic acid). By means of amide bonds, phenylalanine is attached to it as an N-terminal component, and glycine, alanine, leucine, etc., as a C-terminal one (Fig. 39b). The rigidity of the whole structure is provided by the proline component, which allows the benzene ring of phenylalanine and the NH group of the C-terminal amino acids to be close to each other, entering into NH…π interaction. Due to this, the latter can be registered using the NMR method both in weakly polar (CDCl3) and polar (DMSO-d6) solvents up to a temperature of 100 °C. As one can see, this is the i→ (i+2) type of interaction. Notably, that δ values of the amide proton NH in the NMR spectra of these compounds manifest a rather strong paramagnetic shift lying close to 6 ppm (cf. Table 7).
![[{"id":"kl94uz33DU","type":"paragraph","data":{"text":" Schematic representation of the rigid proline-containing tripeptides as a model for studying NH<sup>...</sup>π interactions in solution. The figure was created by the authors using data of Ref. 131."}}]](/storage/images/resized/Y5SS15ZeC9WXHnMwtJQX0jsw72T1hF0r2D42x3l2_xl.webp)
An attractive way to reduce the flexibility of a polypeptide chain, leading to a reduction in the number of its conformational isomers, is the modelling of the NH…π interactions using diketopiperazines (DKP) - products of dehydration of α-amino acids. The DKPs based on tryptophan, tyrosine, and phenylalanyl have been examined.21,132--134 Let us consider, as an example, linear phenylalanyl-phenylalanine (93, 94) and its DKP structures (95, 96), two diastereomers of which are shown in Fig. 40.
![[{"id":"WyyczY25o-","type":"paragraph","data":{"text":"Linear (<b>93, 94</b>) and diketopiperazine (<b>95 ,96</b>) structures of phenylalanyl-phenylalanine diastereomers. The figure was created by the authors using data of Ref. 132."}}]](/storage/images/resized/b9dejVbge4jLIcdPSh6Gx6grhHrtfeXwKAvGlwdy_xl.webp)
As can be seen from Fig. 41, in the solid form, DKP is asymmetric, with one benzyl group hanging over the piperazine ring, and the other turned outward from it.135 According to quantumchemical calculations [B3LYP-D3/6-311++g(d,p)], such a structure is also the most stable in the gas phase. Its stabilization is provided by a combination of CH…π and NH…π interactions. The former with the CH…π distance of 2.69 Å is manifested between the CH2 group of the outer benzyl group and the centroid of the inner benzene ring. The formation of the NH…π bond proceeds with the participation of the amide proton and the benzene ring of the unfolded benzyl. The shortest (2.65 Å) distance is NH…Ci, while the distance to the centroid is much greater (3.23 Å).
![[{"id":"D7ikZ1Arri","type":"paragraph","data":{"text":"Molecular structure of DKP form of SPhe-SPhe (DUZDUX, 283-303 K). The figure was created by the authors using data of Ref. 135."}}]](/storage/images/resized/f6xy4DPIOecmuKBCzetzq0Pi2um9cVO0vv5U6n2u_xl.webp)
Apparently, the DKP structure of this type is retained in solution, as evidenced by the paramagnetic shift of the signal of CH2 protons in the DMSO-d6 solution.136 Unfortunately, in this polar solvent, due to the fast exchange of the NH protons in the NMR time scale, it is difficult to determine the exact position of their resonances, while the use of non-polar media was hampered by the low solubility of polar DKPs.
The influence of the chirality of cyclodipeptides and their protonated forms on the conformation and the relative contribution of NH…π and CH…π interactions was also studied. Only a negligible dependence of the DKP structure on chirality was noted.24,132--134 In addition, whereas in the case of neutral DKPs, their structure is almost equally formed due to NH…π and CH…π interactions, when passing to cations in which the proton is attached to the oxygen atom, the OH…π bonds come to the fore.
5.2. Modelling neurotransmitters
Neurotransmitters (NTs) are relatively simple biological molecules that in living organisms provide communication between individual organs and tissues, on the one hand, and receptors of the autonomic or central nervous system, on the other hand. Such interactions serve as a signal to trigger certain biochemical processes.137 Many of the hundreds of known NTs contain a 2aminoethyl group attached to aromatic or heteroaromatic rings. Some of them, namely adrenaline, norepinephrine, dopamine, tyramine, serotonin and histamine, are shown in Fig. 42b,c.
![[{"id":"HjGjFSy2_G","type":"paragraph","data":{"text":" Examples of neurotransmitters (<i>b, c</i>) and related model compound 2-phenylethylamine (<i>a</i>)."}}]](/storage/images/resized/KwjP8nCfzE7oLJVzZsMBeNhROZteKJtksnRKY3nV_xl.webp)
In the structure of neurotransmitters, three main points attract attention: 1) the presence of a rather basic (pKa=9.8-10.2) primary amino group, 2) π-donor character of aryl and hetaryl substituents, provided by the presence of hydroxy groups or a pyrrole nitrogen atom in them, and 3) the presence of a CH2CH2 bridge, which makes the distance between the NH2 group and the aromatic ring approximately the same for all NTs. Taken together, all this leads to two consequences that seem to be especially important for understanding the biological activity of NTs and the diversity of their corresponding cell receptors. Firstly, the aliphatic amino group, even under conditions of low blood alkalinity (pH ≈7.4), should exist in tissues predominantly in the protonated form with a tail NH3+ group. Secondly, NTs are capable of taking both the expanded anti-form (97a) and the folded gauche-form (97b), which is stabilized by the intramolecular N…π interaction. Most of the studies attempting to shed light on these issues were performed using 2phenylethylamine 97 as a model (Fig. 42a). The conformational advantages of the gauche-structure have recently been elucidated in the study of the protonated phenylalkylamines of the general formula C6H5(CH2)nNH2 (n=1-4).138 The experiments, supported by quantumchemical calculations, were carried out in the gas phase using electrospray ionization with subsequent measurement of the infrared multiple photon dissociation (IRMPD) spectra of the ions formed in this case. It was found that the benzylammonium cation does not form a stable gauche-form, whereas in protonated 2-phenylethylamine this form is the most stable, differing the free energy of the anti-form by 5.0 kcal mol–1. The formation of the gauche-form is evidenced by the strong red shift of the νNH band (3143 cm–1), which is implicit for the bond that provides NH…π interaction. For comparison, similar bands of the other two N–H bonds are located at 3307 and 3337 cm–1. The closest (2.39 Å) NH proton in the gauche-form is directed to the C(ipso) atom of the phenyl ring. For phenylalkylamines with 3-4 methylene units, the number of conformations including folded one, increases significantly, but the latter become too stable to maintain the reversible equilibrium necessary for biological processes. This is evidenced by both the absence of νNH bands in the region of 3000-3500 cm-1 and quantum chemical calculations showing, in particular, a reduction in the NH…C(ipso) distances to 2.25 Å (for 3-phenylpropylamine). In a detailed work,139 in which numerous references can be found on this topic, very close results were obtained using similar methods for 2-phenylethylamine. It has also been argued that the stabilization of the gauche-form of 2-phenylethylamine 97b is partly due to London dispersion forces.
To some extent, 1-amino- and 2-aminoindanes can be considered analogues of 2-phenylethylamine as a model of neurotransmitters. Quantumchemical and spectral studies of their super-jet cooled samples in the gas phase led to the conclusion that 1-aminoindane under these conditions exists as two conformers,140 and 2-aminoindane141 as four conformers, of which 98 and 99 are the most stable, being stabilized by weak intramolecular NH…π bonds (Fig. 43). Their energies were estimated at 0.71.0 kcal mol–1 and 1.3-2.0 kcal mol–1 for 98 and 99, respectively, which is comparable to the C6H6•NH3 complex.
![[{"id":"OUnEusLR9R","type":"paragraph","data":{"text":" Preferred conformations of protonated 1-amino- and 2-aminoindanes. The figure was created by the authors using data of Refs 140, 141."}}]](/storage/images/resized/m2K1w0griceDVtLFAEQU9CECMaclLgjIzI0MAfr8_xl.webp)
One of the quantumchemical arguments in favour of the NH…π interaction in 98 was the somewhat longer (1.0189Å) N–Ha bond involved in the chelation as compared to the N–Hb bond (1.0172 Å) (for the further discussion of the effect of the NH…π interactions with the participation of NH2 groups on NH bond length see Ref. 139 and Section 3.8, Fig. 31).
By the example of protonated 2-(o-fluorophenyl)ethylamine, the effect of the ortho-substituent in the phenyl ring on the preferred geometry of similar NT analogues was studied.142 The study of the range of stretching vibrations of NH bonds revealed three low-energy gauche-conformers. The form 100 proved to be the most stable, being stabilized simultaneously by two moderately strong hydrogen bonds: NH+…π and NH+…F (Fig. 44). It was slightly inferior in stability to conformer 101, stabilized only by the NH+…F contact. The least stable form contains only the NH+…π bond. It was not detected experimentally, but its possible existence in low concentration followed from quantum chemical calculations.
![[{"id":"eUUSmK12rl","type":"paragraph","data":{"text":"The two most stable <i>gauche</i>-forms of 2-(o-fluorophenyl)ethylamine. The figure was created by the authors using data of Ref. 142."}}]](/storage/images/resized/CodIjmvD4rLqZICabGRhElAx7bRCxP7Fys7sb7tB_xl.webp)
The situation is less unambiguous for the solid NTs. According to the results of XRD analysis (Table 15), most of them exist in an expanded form even after protonation. This can be explained by the specificity of the crystal packing and the strong influence of the counterion. The important role of the latter factor is confirmed by short distances (1.91-2.37 Å) between the anion and the NH proton. In general, the implementation of the folded form with NH+…π binding is facilitated by low-nucleophilic anions with a strongly delocalized charge (e.g., picrate) and especially increased π-donor ability of the aromatic ring, as in the case of mescaline and tryptamine.
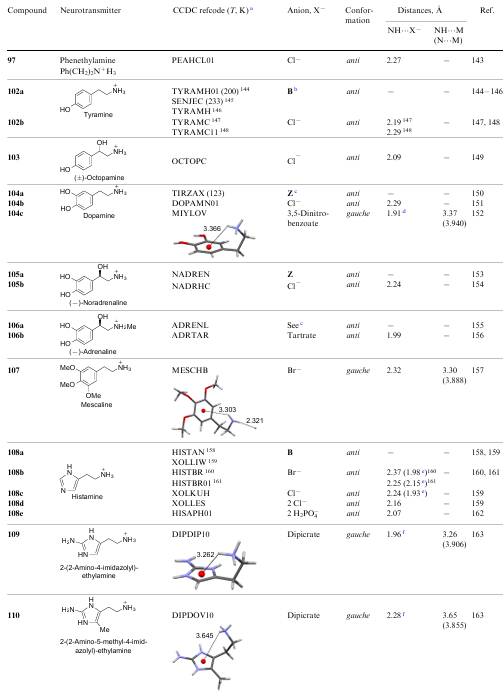
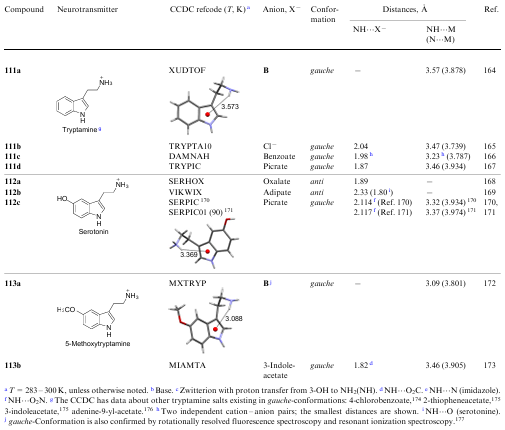
It is interesting to compare the NT discussed above with acetylcholine 114, one of the main representatives of such biomolecules (Fig. 45). Among many functions of 114, the regulation of muscle activity, participation in the parasympathetic nervous system, as well as in the processes associated with learning and memory are especially important. The molecule of 114 contains no groups that could enter into NH…π interaction, but there is a CH2CH2 chain, which once again emphasizes its important purpose.
![[{"id":"_PsiZbh00j","type":"paragraph","data":{"text":"Synthetic receptor<b> 115</b> which efficiently encapsulates acetylcholine <b>114</b>. The figure was created by the authors using data of Ref. 178."}}]](/storage/images/resized/iEM7HaCTus6pnmONsoHWthYALVcm9WPuYwT3IHvG_xl.webp)
Back in 1990, Dougherty and Stauffer178 reported the first evidence that the various receptors to which acetylcholine binds contain residues of aromatic amino acids such as phenylalanine, tyrosine, and tryptophan. One such evidence was the high affinity of acetylcholine for the synthetic receptor 115. The cluster 116 formed between them has a fairly high formation constant (Kd=50 mM), which is close to the binding energy of real biological complexes of the host-guest type. Compound 115 forms similar strong complexes with other ammonium salts. Accordingly, the forces promoting the incorporation of the ammonium cation into the cavity were characterized as a ‘cation-π’ interaction.23 Although living tissues contain many ions and molecules with lone electron pairs, their interaction with acetylcholine seems to play a secondary role due to the hydrophobicity of the inner cavity of the receptor.
Dougherty's work initiated many other studies along this line, including those related to NH…π interaction. In particular, Shishido et al.179 using pre-dissociative IR spectroscopy and mass spectrometry, tried to model molecular recognition of aromatic nuclei by acetylcholine in the gas phase. For this purpose, they simulated the formation of clusters of the trimethylammonium cation with different amounts (1-4 mol. equiv.) of benzene. Using natural bond orbital (NBO) calculations, it was shown that the positive charge in the [HNMe3]+ cation is almost completely dispersed over the methyl groups, as a result of which the interaction of this cation with benzene proceeds exclusively due to C…π, but not NH…π binding. Based on this, it was found more accurate to classify the forces responsible for molecular recognition of acetylcholine receptors as ‘activated CH…π interaction’. The review focused on cation-π and anion-π interactions has been published by Frontera et al.180
6. Reactions driven by NH…π interactions
One of the yet rare examples of such reactions is the base-catalyzed (preferably with DABCO) isomerization of 3-arylindenols 117 into 3-arylindanones 118 (Scheme 3).181 Being a variation of the well-known allyl rearrangement, this transformation is remarkable for its high enantioselectivity, which was used for the total synthesis of the muscarinic receptor antagonist (R)-tolterodine.182 Formally, the process represents a concerted suprafacial 1,3-hydrogen shift forbidden by the orbital symmetry rules.
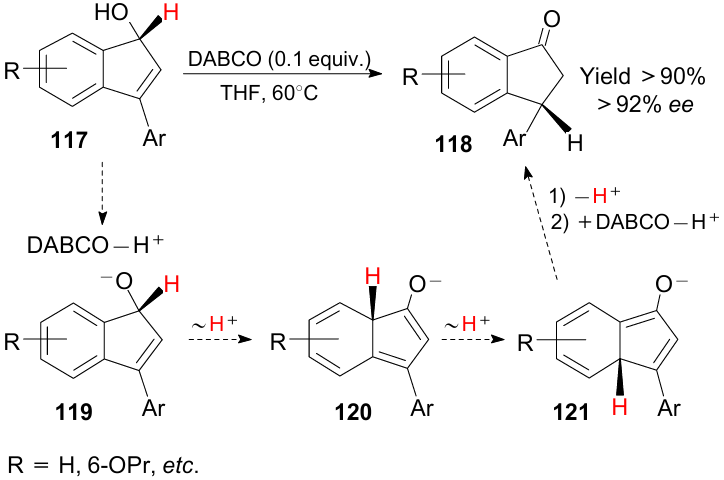
Initially, a stepwise proton transfer mechanism was proposed for it, shown in Scheme 3 and including intermediates 119-121. However, it did not explain two observations: why both enantiomers cannot be obtained at each stage, and how the high activation energy (>30 kcal mol–1) is compatible with high stereoselectivity. These issues were clarified in 2018 by Ascough et al.183 Based on theoretical calculations using molecular dynamics for condensed matter, a significantly different mechanism has been proposed, including only two stages of proton transfer (Scheme 4). Its principal feature is the deprotonation at the first stage of the C–H bond, which is geminal to the OH group, instead of the elimination of a proton from a seemingly more acidic hydroxyl. The authors proved that the acidity of this CH bond is at least commensurate with that of the secondary OH group. Indeed, the resulting carbanion 123 is simultaneously stabilized by allyl and benzyl resonances, as well as by conjugation with 3-aryl group. It was subsequently shown by theoretical calculations that the DABCO–H+ cation formed upon ionization of the C–H bond can enter into NH…π interaction with the formed rather extensive anionic π-system (structures 122→123) and move freely above it. As a result of such sliding over the surface of a five-membered ring, the proton is easily transferred to the C(3) atom (123→124→118a), which retains the stereochemical configuration without disturbing the aromaticity of the benzene ring as in the original mechanism (see structures 120 and 121). Another important thing is that the activation energy of the whole process, which is limited by deprotonation of the C–H bond, decreases by 15 kcal mol–1 with this mechanism. Note, that tautomerization of 118a to the final keto form 118 is determined by the Erlenmeyer-Eltekov rule.
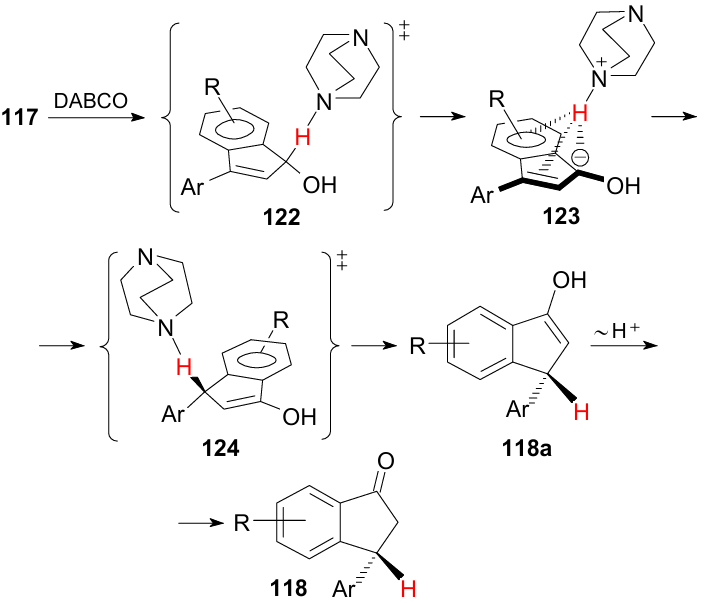
The second example of the influence of NH…π interactions on the reactivity was the first case of nucleophilic substitution of a hydrogen atom in an unactivated pyrrole ring, recently discovered in our laboratory (Scheme 5).184 It was found that the treatment of 1-dimethylamino-8-(pyrrol-1-yl)naphthalene 125 with an equimolar amount of HBF4 in MeCN produces tetrafluoroborate 55, which is sufficiently stable in solution for 2-3 h. This is confirmed by the complete similarity of 1H NMR spectra of 55 and its much more stable analogue 46 (see Table 7). As noted above (see Table 5), XRD measurements of salt 46 confirmed the formation of a pronounced intramolecular NH…N(π) hydrogen bond in its cation. The obvious presence of the similar HB in salt 55 makes it easy to explain its further behaviour. The point is that when the proton spectrum of 55 is measured again after 4 h, the clear signs of the presence of the second substance in the NMR tube appear. Its concentration slowly increases and after some time under ambient conditions it becomes the only product, which is a colourless crystalline pyrrolo[1,2-a]dihydroperimidinium tetrafluoroborate 128. Several circumstances are especially surprising in this transformation. First, this is the very fact of nucleophilic substitution of hydrogen in the electron-rich pyrrole ring. Secondly, the participation of such a weak nucleophile as the aniline NMe2 group in it. Third, the oxidation of intermediate 127, which is necessary for aromatization through the abstraction of the hydride ion, proceeds with the participation of atmospheric oxygen and does not require the addition of strong oxidants such as KMnO4. At last, the high selectivity and smoothness of the process are striking.
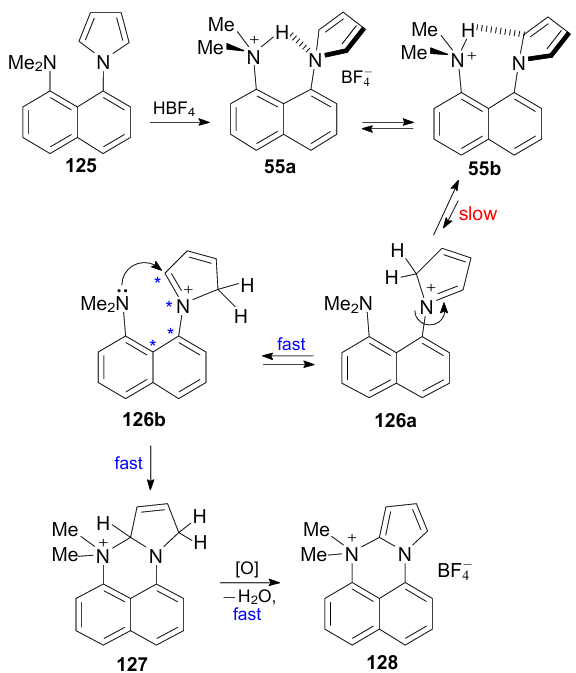
Based on quantum chemical calculations and a number of experiments, a putative mechanism was proposed for the reaction (see Scheme 5). There are good reasons to believe that the rate-determining stage is the formation of 2H-pyrrolium cation 126. Its activation energy is 14.5 and 17.1 kcal mol–1 for the gas phase and solution in acetonitrile, respectively. It is logical to assume that the formation of cation 126 occurs as a result of the slow sliding of the NH proton along the N–C(2) edge: 55a→55b→126. In general, the reaction becomes possible due to the proximity of the peri-substituents in 125 and the soft acid catalysis, which activates the pyrrole ring to the nucleophile attack.
7. Miscellaneous issues
7.1. NH…π interaction and recognition of α-amino acids
In an extensive collaborative study, Blasco et al.185 tried to figure out how biological receptors recognize hydrophilic and hydrophobic amino acids. The former were tested with histidine, aspartic and glutamic acids, and the latter with alanine, phenylalanine, tyrosine and tryptophan. Two pyridine-containing azacrown ethers with a pendant 2-aminoethyl (129) or 2-(naphth-1-yl)aminoethyl tail (130) served as receptor models. Potentiometric and calorimetric measurements in an aqueous medium, as well as high-level theoretical calculations, were used as research methods. It was assumed that the absence or presence of a 1-naphthyl group would provide a hydrophilic--hydrophobic balance when choosing the amino acids of ligand 129 or 130. It was found that the binding of the selected amino acids by the ligands is provided mainly by salt interactions (especially with the participation of the amino acid carboxylate anion) and hydrogen bonds. As for NH…π interactions, they appear only with ligand 129 upon protonation or hydration. One of the complexes formed between the tyrosine molecule and the doubly protonated ligand 129 is shown in Fig. 46b. A very short NH…π bond between the proton of the terminal ammonium group of the ligand and the centroid of the benzene ring of tyrosine deserves attention. To some extent, this interaction can be considered as a model for receptor binding of neurotransmitters carrying the terminal 2-aminoethyl group (see Section 5.2).
![[{"id":"dMW0NgRM3M","type":"paragraph","data":{"text":"Synthetic receptors for the recognition of hydrophilic and hydrophobic amino acids (<i>a</i>); theoretically calculated structure of the adduct of the doubly protonated receptor <b>129</b> with the tyrosine molecule lying in the potential minimum (<i>b</i>). Reproduced from Blasco et al.<sup>185</sup> with permission of the Royal Society of Chemistry."}}]](/storage/images/resized/gZJseNwWLCP4NGZ7BvWVmyiI4t0YODLDqlAKnXO6_xl.webp)
7.2. NH…π interaction and urea-assisted protein denaturation
Due to multiple non-covalent interactions, proteins commonly exist in a biologically active folded form. However, under the influence of various factors, for example, elevated temperature, mechanical impact or some chemicals, their molecules undergo denaturation, losing their secondary, tertiary and quaternary structure. One of the well-known denaturing agents is urea. The reasons for this have recently been specially studied using a number of experimental and computational methods.186
The main object of the study was a small tryptophan-containing protein. It was found that the mechanism of the denaturing action of urea is associated with its high affinity for aromatic nuclei and includes two types of interactions - stacking and the formation of NH…π hydrogen bonds, the ratio and strength of which is determined by the nature of the aromatic system. As follows from Fig. 47, which shows the calculated interaction energies for optimized structures, stacking is most efficient for the imidazolium ion, followed by indole, neutral imidazole, phenol, and benzene. As for NH…π interactions, they are stronger for indole and phenyl group. Decomposition of the obtained energies into components showed that the attraction of urea to aromatic nuclei is mainly due to dispersion forces (shown in parentheses in the top row), while the electrostatic interaction is slightly repulsive.
![[{"id":"ziSZ_nSmD8","type":"paragraph","data":{"text":"Calculated energies of interaction of urea with aromatic rings that make up amino acids. Reproduced from Goyal et al.<sup>186</sup> with permission of the American Chemical Society."}}]](/storage/images/resized/PZ2zKppyOkeuqSCfvx0m7BmK1tULZyptmmbotrmF_xl.webp)
7.3. NH…π interaction in vitamin B12 coenzyme
The significance of NH…π interactions is not limited to proteins and such biomolecules as neurotransmitters. Thus, Starikov and Steiner,187 using neutron diffraction on crystals of cobalamin 131 (a coenzyme of vitamin B12), found an NH…π bond in it with a length of 2.58 Å and an energy of 4.0 kcal mol–1. This bond is formed between the proton of one of the propioamide groups and the benzene ring of the 5,6-dimethylbenzimidazole moiety (Fig. 48).
![[{"id":"2AbnFnlnAL","type":"paragraph","data":{"text":"NH<sup>...</sup>π interaction in vitamin B<sub>12</sub> coenzyme. The figure was created by the authors using data of Ref. 187."}}]](/storage/images/resized/gUgQW1U2PmX4X0ycBcv8OHAmmLHdZNyt1esiZ61E_xl.webp)
8. Practical applications of NH…π interactions
Most of the research on the practical use of NH…π interactions is to some extent related to medicinal chemistry. Thus, it has been proposed to use carbon nanotubes [CNTs (5,0)] functionalized with a non-covalently bound amino acid, for example, alanine, for the targeted delivery of the anti-tuberculosis drug isoniazid (isonicotinic acid hydrazide).188 The success of such a task depends on the optimal binding energy of the drug with the carrier: it must be sufficiently high so that the drug is not desorbed along the way, but at the same time relatively low so that it is easily detached from the carrier upon reaching the receptor. The proposed combination seems to satisfy these requirements, since the binding energy of isoniazid to the CNT-alanine carrier, according to theoretical calculations, is 8.5 kcal mol–1. It is believed that the very binding of isoniazid is provided by a combination of stacking and NH…π interaction, which is facilitated by the planarity of the isoniazid molecule and the graphene-like structure of CNTs.
Another problem associated with the targeted drug delivery is the predominantly lipophilic nature of cell membranes, which hinders the penetration of polar substances into the cell. At the same time, receptors having a protein, i.e., very polar in nature, tend to better interact with polar molecules. To increase the membrane permeability with respect to both polar and low-polar drugs, it was proposed to use polyfunctional macrocyclic compounds with a molecular weight of at least 600-700 Da. It was assumed that due to the high conformational flexibility, macrocycles bearing suitable functional groups can more easily adapt to the environment, including tissue receptors. Such molecules were called 'chameleons'.
Based on this idea, Tyagi et al.189 synthesized macrocyclic polyamides 132a--e containing residues of lysine (132a), phenylalanine (132c), and other amino acids with aromatic nuclei (132b,d,e) (Fig. 49). They found that whereas in the 1H NMR spectrum of the lysine-containing macrocycle 132a in CDCl3 the NH proton resonates at δ 6.3 ppm, in the spectrum of compound 132c containing a phenylalanine residue, it undergoes a significant paramagnetic shift (ΔδNH≈-1.6 ppm), resonating at δ 4.7 ppm.
![[{"id":"Wb6HYEE34l","type":"paragraph","data":{"text":"Macrocyclic polyamides <b>132a-e</b> for studying NH<sup>...</sup>π interactions. The figure was created by the authors using data of Ref. 189."}}]](/storage/images/resized/UHs2SHMmubO2ezvgppXZzh5Lb3sSpTorO88sq5pI_xl.webp)
This indicates that an NH…π bond is formed between the phenyl ring of the benzyl group and the N(I)H proton, which was also confirmed by XRD analysis [structure 132c’; distances are: N(I)H…C1’=3.40 Å, N(I) …M=4.02 Å, N…C1’=3.45 Å]. As expected, the high-field NH proton shift in the spectra of compounds 132d and 132e, due to the absence of the benzyl CH2 group, is much lower (ΔδNH≈–0.4 ppm), and in spectrum of 132b the corresponding value is intermediate (–0.6 ppm) due to the greater distance of phenyl nuclei from the NH proton. Thus, the NH…π interaction in 132c and the realization of the folded conformation contribute to the shielding of the neighbouring C = O group and a decrease in its polarity. As a result, the lipophilicity of the whole compound increases and its penetration into the cell is facilitated.
As already noted above, NH…π interactions are involved in the recognition of various exogenous substances by biological receptors, including drugs. Here is another example on this topic, associated with the transmembrane protein neurokinin-1 (Fig. 50). The attachment to its receptors of a neurotransmitter polypeptide under code-name P, causes pain and neurogenic inflammation in animals. In search of an antidote that prevents the binding of the neurokinin receptor to P, a very effective drug was synthesized under the code CP-96345. It was possible to establish that it binds to the histidine residue (His-197) in the neurokinin receptor. Based on numerous experiments, it was concluded that the analgesic effect of CP-96345 is due to the presence of a benzhydryl group in its molecule, two benzene rings of which enter NH…π interaction with the rather acidic NH proton of the imidazole ring in neurokinin-1 (see Fig. 50).190
![[{"id":"4e7tjMQ7iP","type":"paragraph","data":{"text":" Schematic representation of NH<sup>...</sup>π interaction of the neurokinin receptor with the drug CP-96345. The figure was created by the authors using data of Ref. 190."}}]](/storage/images/resized/3GuveeS7NaXaxUxQz5FdObzBzdSp0UIKpStKTxYo_xl.webp)
In connection with the growing interest in molecular machines,191,192 Alfonso et al.193 synthesized 16- and 17-membered naphthocyclophanes 133a,b based on valine and phenylalanine (Fig. 51). Investigation of the rotation of the naphthalene ring as a rotor showed that in the case of phenylalanine it slows down significantly. This was attributed to the folding of the amide phenyl groups into a conformation stabilized by NH…π interaction.
![[{"id":"DSTQ5tUa7B","type":"paragraph","data":{"text":"Amino acid-based naphthocyclophane molecular rotors. The figure was created by the authors using data of Ref. 193."}}]](/storage/images/resized/jQSKNEg7z5U024Vwm9MXH9ZsuL5WPQGqrrkIL3PO_xl.webp)
The possibility of using NH…π interactions to improve the biocompatibility of surfactants and the quality of cosmetics, detergents, emulsifiers, etc., was investigated.194 For this, binary mixtures of anionic amphiphilic compounds based on amino acids were taken. It was found that when using mixtures of sodium salts of N-tetradecylalanine and N-tetradecylphenylalanine, the surface quality and the growth of micelle formation noticeably increase. This was attributed to binding amphiphilic components not only due to hydrophobic forces, but also as a result of NH…π and π…π binding (Fig. 52, structures 134 and 135).
![[{"id":"6Vg8Tg4Dr4","type":"paragraph","data":{"text":"Stabilizing micelle formation interaction of two amphiphilic molecules: NH<sup>...</sup>π interaction between alanyl-based and phenylalanyl-based surfactants (<b>134</b>); π<sup>...</sup>π interaction between two phenylalanyl-based surfactants (<b>135</b>). The figure was created by the authors using data of Ref. 194."}}]](/storage/images/resized/fB9bjRVEqOdzXcSPsiDepsUXZRqcqU6ebDJpON6I_xl.webp)
Along with other non-covalent forces, NH…π interactions play a role in the field of crystal engineering.195
Recently, the effect of NH…π interactions has been successfully used for the industrially important separation of a mixture of benzene, cyclohexene, and cyclohexane, which is formed by hydrogenation of benzene.15 Traditional separation methods of this mixture are based on extraction and azeotropic distillation processes and are extremely expensive. Yao et al.15 have developed a much more efficient adsorption method using amide naphthalene-containing nanotubes of the 76-82 types as a solid adsorbent (see Table 9). The method is based on the formation of very stable NH…π clusters by benzene and cyclohexene, and the process can be made highly selective by changing the conditions and choosing a suitable catalyst from this series.
Kodama et al.196 demonstrated that NH…π interactions can be helpful in chiral recognition processes. Thus, a procedure for the direct enantioseparation of axially chiral 1,1’-biaryl-2,2’-diols was developed. For this purpose, chiral amidines were proposed as resolving agents, for example, compound 137 synthesized from commercially available dehydroabietic acid. First, when mixing equimolar amounts of optically pure 137 and racemic 1,1’-binaphthyl-2,2’-diol 136, a mixture of easily separated diastereomeric amidinium salts was obtained as a result of a proton transfer from one of the OH groups on the amidine functionality. Their subsequent acidification and extraction resulted in the isolation of individual diol enantiomers with an optical purity ee >95-97%. The principle of chiral recognition is illustrated by XRD structure 138 (Fig. 53) of one of the obtained salts. It shows how the amidinium group holds two monoanions (R)-138 due to two hydrogen N–H…O and two NH…π bonds with modest NH…centroid distances.
![[{"id":"uU6JYK33s3","type":"paragraph","data":{"text":"Molecular structure of diastereomeric salt of 1,1'-binaphthyl-2,2'-diol with dehydroabietyl amidine <b>138</b> (ATOFIB, 150 K). The figure was created by the authors using data of Ref. 196."}}]](/storage/images/resized/6Hwljjx0fU3BVJdbbXS80ZcrNp6wbk0KnvfecW0D_xl.webp)
9. Conclusion
To summarize, the present survey shows that the history of NH…π interactions research is about 30 years old. During this time, more than 170 publications appeared, of which about 120 were published in the 2000s. Whereas in the first years after the emergence of interest in NH…π forces they were considered exclusively as a factor contributing to the structuring of proteins, then over time it became clear that they also play a significant role in the processes of molecular recognition. Accordingly, it turned out that NH…π interactions are found not only in proteins, but also in such types of biomolecules as neurotransmitters or vitamin B12. In recent years, they have been increasingly considered when solving problems of medicinal chemistry, creating new materials, and even for improving important production technologies. Synthetic reactions have been discovered whose stereochemistry and even the very possibility is determined by NH…π interactions. Although NH…π interactions are generally considered weak with energies in the range 0.5-5 kcal mol–1, in some cases they can reach 15-20 kcal mol–1, i.e., the level of sufficiently strong non-covalent forces. This is especially facilitated by the participation in NH…π clusters of positively charged NH donors, for example, ammonium salts and electron-rich π-donors such as pyrrole rings. Taking into account the difficulty of studying NH…π interactions on real living tissues, their modelling by creating simple and accessible synthetic compounds has become widespread. The most convenient and informative models are those in which the positions of the proton-donor and proton-acceptor groups are preliminary pre-organized. These include peri-disubstituted naphthalenes and folded or skeletal compounds. Their important advantage lies in the possibility of conducting research in solution and in the solid form. If the necessary conditions are met in the best NH…π models, the distance between the NH proton and the centroid of the aromatic ring M can reach only 2.07 Å, and the NHM angle approaches 180°.
The study was funded by the Russian Foundation for Basic Research (Project No 20-03-00112).
10. List of acronyms
5-ASA — 5-aminosalicylic acid,
ATP — adenosine triphosphate,
BSSE — basis set superposition error,
CCDC — Cambridge Crystallographic Data Centre,
CNT — carbon nanotube,
DABCO — 1,4-diazabicyclo[2.2.2]octane,
DF — dispersed fluorescence,
DFT — density functional theory,
DKP — diketopiperazine,
EHB — hydrogen bond energy,
Eint — interaction energy,
FTIR — Fourier-transform infrared spectroscopy,
HB — hydrogen bonding,
IMPT — intermolecular perturbation theory,
IP1 — first ionization potential,
IRMPD — infrared multiple photon dissociation,
IR/UV — infrared/ultraviolet spectroscopy,
LIF — laser-induced fluorescence excitation,
MD — molecular dynamics,
MeCN — acetonitrile,
NCI — non-covalent interaction,
NMR — nuclear magnetic resonance,
NT — neurotransmitter,
R2PI — resonant two-photon ionization,
RE2PI — mass-resolved one-color resonance enhanced two-photon ionization,
REMPI — resonance-enhanced multiphoton ionization,
UV-UV HB — ultraviolet--ultraviolet hole-burning,
VDW — van der Waals,
XRD — X-Ray diffraction,
ZPE — zero-point energy.