


Keywords
Abstract
The main challenge of modern polymer science is to search for ways of further development of polymer civilization, which obviously includes living organisms on the Earth, without harmful consequences for civilization and the planet in its entirety. The review considers approaches to handle the problem of environmental accumulation of plastic waste. Promising trends in the development of polymer technologies, which can significantly reduce the amount of waste produced, are highlighted. Separate Sections address original methods of additive manufacturing technologies, such as the extrusion printing technique to produce multilayer films, 3D printing by using high-temperature polyimide materials, new functional siloxane oligomers and hydrogels for medical uses. Much attention is paid to the development and applications of biodegradable materials in medicine, packaging industry and agriculture. An analysis of the European strategy for plastics and plastic disposal demonstrates that it has a number of limitations due to high energy requirements and changes in Earth's carbon balance. The modern approach to plastic waste management free from these shortcomings is briefly outlined.Bibliography — 1233 references.
1. Introduction
This review addresses the state-of-the-art research areas of polymer science. The year 2020 marked the 100th anniversary of Hermann Staudinger's discovery of the chain structure of polymers. This discovery is considered to be the beginning of the development of polymer science as a systematic approach to investigating this type of organic materials. The polymer science is currently a powerful interdisciplinary field. Polymers form the basis of human civilization and play a key role in its evolution. Over the past period, it has become clear that Staudinger's discovery is not only about providing a tool for the creation of polymer molecules but also, resulting from the development of these concepts, about realizing the undeniable fact that the variety of life on Earth is based on polymer materials, from proteins and polysaccharides (cellulose, chitin), covering the main forms of biomass, to DNAs and RNAs, which serve as carriers of the genetic information and play a key role in translating the genetic code in living things, providing their life stages and reproduction.
The idea of the life as a series of chemical transformations of organic matter was significantly improved and detailed in the past century, and the relationship between the organic and inorganic nature is clearly reflected in the so-called planetary cycles of carbon, oxygen, nitrogen, phosphorus, etc. Academician A.A.Berlin noted the key role of polymers in the carbon and oxygen cycles,1 attracting keen attention of researchers to the circulation of polymeric substances on Earth. In parallel with investigations of biological systems, numerous polymer-based synthetic materials were designed and chemically manufactured on the industrial scale. Today, a world without polymeric materials is almost unimaginable.
However, the mass production of polymers has created a new serious environmental problem associated with their disposal. This problem is equally or even more challenging than the design of new materials. Even a cursory acquaintance with the scale of this problem leads to understanding of the indisputable fact that the issues faced by mankind, which are caused by the plastic pollution of the natural environment, are crisis factors of growth and development but not fatal consequences of the progress in the manufacturing of plastics, according to many environmental experts. The goal of polymer science is not to minimize the production of plastics but to include them in the circulation of polymeric materials on the planet and to learn how to control it. Now it is clear that the mechanisms of waste disposal should be anticipated when designing the synthesis of polymers.
This review is intended to provide a tool of interdisciplinary analysis of the promising state-of-the-art research areas of polymer science and evaluate the challenges and prospects at the century mark of its development. The review considers the modern state of such important fields, as the development of technologies for producing multilayer film materials, enabling one not only to control the supramolecular structure and morphology of polymer films but also, during multiplication, to perform the mechanochemical synthesis of new copolymers, inaccessible by conventional synthetic approaches.2 Polymer aspects of 3D printing are considered in relation to the most complex and promising areas concerning the use of engineering plastics, silicone elastomers and hydrogels. Each of these areas is unique and the prospects of the application of these materials are truly limitless. Great attention is paid to prerequisites for the development of new polymer types, such as dense molecular brushes and unique materials based on these structures. Taking into account their high cost, it is supposed to impart new properties to these polymers, such as the ability not only to be organized to form more complex structures but also to be disassembled into building blocks for reuse. One Section is focused on the problems of biodegradable polymers and materials, which are considered by many experts as a mainstream approach for addressing issues associated with the plastic packaging as the major plastic waste. The processing and fabrication of plastic products are complicated by the specific nature of the rheology of polymer melts different from that of low-molecular-weight compounds, thereby making rheological studies an essential part of polymer science.
A separate Section is devoted to the analysis of approaches to plastic waste disposal. This issue is discussed in terms of the global carbon cycle, in which the synthesis and fabrication of plastics are considered as an alternative to the burning of hydrocarbons. An objective analysis of the approaches to plastic waste disposal, occurring in nature and used in modern civilizations, shows that, along with the fundamental differences, there are many features in common. A comparison of the efficiency of these approaches casts doubt on the accepted paradigm for processing plastic waste, and its constructive revision may solve these problems forever and rid the polymer science of social and political pressure. This, in turn, can bring the development of this research area back to natural logics, that is to the pathway to perfection.
The research areas included in this review were chosen by members of the Polymer Council of the Russian Academy of Sciences, as the most qualified experts on polymers in the Russian Federation. This choice is a matter of taste rather than a comprehensive systematic analysis of the leading research areas. It is equally important that the prospects of a particular research area were assessed based on the fundamental knowledge rather than consumer perceptions, which are commonly considered in specialized publications.
The unifying starting point of this review is the glance into the future, as seen by experts in different fields of polymer science. Here one should not expect futuristic projects and fantasies typical of non-fiction articles. The authors focused on the issues, which they considered to be of key importance for solving pressing current problems. The authors motto was `Polymers for the Future', which is used as the title of our mosaic review. We hope that the readers will get a feel for modern trends in polymer science and will use them in their research and that the mosaic canvas will get together into the clear and optimistic picture of the future of polymer science.
2. Materials based on molecular brushes
In the past decade, a parade of highly branched macromolecules, including dense molecular brushes and unique materials based on these polymers, was the most significant event in the synthesis of polymers, which actually overturned the traditional views on intermolecular interactions of macromolecules. In the studies,3--10 the authors demonstrated the unique high level of control over the properties of soft materials via controlling the architecture of bottlebrush polymer networks. The approach to the control of the three-dimensional structure by tailoring the properties through architecture is among the advanced techniques of controlling the properties of the polymer material during the synthesis (bottom-up method) and it provides an opportunity of using digital technologies in the design of materials.
As opposed to linear structures, the brush architecture offers significant advantages in the design of new materials with specified properties. First, the properties of the material can be changed in a wide range by varying only the architecture of brush blocks, which is determined by a set of parameters (nsc, ng, nx), where nsc, ng and nx are the degrees of polymerization of the side chains, the backbone between the grafted chains and the backbone between the crosslinks, respectively, with the chemical composition of the network remaining unchanged. For example, the modulus of elasticity (Young's modulus) can be changed by four orders of magnitude by varying the density of grafting and the length of side chains, as well as the distance between the crosslinks of the network (Fig. 1a,b).11–14 This significantly expands the potential of polymer technologies, in which the physical properties of the material are traditionally specified by its chemical composition.
![[{"id":"WQM-soHLi8","type":"paragraph","data":{"text":"Mechanical testing of polydimethylsiloxane (PDMS) bottlebrush elastomers (<i>a</i> is the true stress (σ<sub>true</sub>) versus the uniaxiale longation (λ); elastomers defined by different n<sub>g</sub> values at fixed n<sub>sc</sub> and n<sub>x</sub> demonstrate a wide range of mechanical properties, allowing the simultaneous increase in the stiffness (Young'smodulus, <i>E</i>) and elongation at break λ<sub>max</sub>;<sup>12</sup> <i>b</i>, the combined plot E(λ<sub>max</sub>) for a series of bottlebrush elastomers with different values of n<sub>sc</sub> and n<sub>x</sub>; E<sub>ent</sub> is the entanglement plateau modulus of linear polymers;<sup>12</sup> <i>c</i>, the plots σ<sub>true</sub>(λ) for physical networks self-assembled from linear-brush-linear block copolymers; such supersoft and strain-stiffening elastomers based on conventional PDMS (solid lines) make it possible to replicate the mechanics of biological tissues (empty squares); the numbers in square brackets correspond to the degree of polymerization of the brush backbone (printed in blue) and linear blocks (printed in red);<sup>12</sup> <i>d</i>, the softness (Young's modulus) of the network versus the firmness (defined as the ratio of the root-mean-square end-to-end distance to the square of the contour length of the polymer between crosslinks) for linear-brush-linear triblock copolymers with different ratios and densities of grafting of short and long side chains; each set of colour symbols corresponds to a series of samples with a variable degree of polymerization of the linear block at a constant length of the brush block (points are extrapolated by dashed lines for the sake of convenience). The grafting density and the fraction of long side chains increase from left to right. Therefore, the copolymerization of short and long macromonomers enables the precise independent tuning of the Young's modulus and the firmness of the network.<sup>13</sup> Figures <i>a-c</i> are reproduced with the permission of Springer Nature; Fig. <i>d</i>, with the permission of American Chemical Society."}}]](/storage/images/resized/gzkeOvduJJYjjJZ8kulg92XzRBaGnkmxPEr6XTou_xl.webp)
Second, having numerous independently controlled architecture parameters, one can program different physical properties (modulus of elasticity, elongation at break, melting point, degree of swelling) and, which is most important, change these properties independently of each other.12,15,16 This opens up intriguing possibilities for the fabrication of materials with a unique combination of usually incompatible properties, e.g., such as soft yet strong or elastic yet damping (Fig. 1c,d).13,17--21 A combination of such properties is characteristic only of biological tissues.
Third, brush macromolecules are highly reactive due to the presence of numerous potentially functional side chain ends. Depending on a particular application, a controlled fraction of side chain ends can be readily functionalized with either reactive or strongly associating groups that promote either permanent or dynamic crosslinking.20,22,23
Fourth, a more compact shape and low interpenetration of macromolecules due to a high grafting density lead to a decrease in the number of chain entanglements and, as a consequence, a decrease in the viscosity of the melt (Fig. 2). This facilitates the processing of polymers and injection molding, which is particularly important for additive manufacturing, such as 3D printing.20,23,24
![[{"id":"shH5Cl0hUj","type":"paragraph","data":{"text":" Frequency dependences o fthe complex viscosity (η*) of PDMS melts with similar molecular weights (Mn ≈ 500000) but with varying architecture (linear, bottlebrush and star-like bottlebrush) (<i>a</i>).<sup>20</sup> The 3D printing with a thermosensitive hydrogel based on linear-brush-linear block copolymers (<i>b</i>).<sup>23</sup> Figure <i>a</i> is reproduced with the permission of Springer Nature, Fig. <i>b</i>, with the permission of American Association for the Advancement of Science."}}]](/storage/images/resized/bk1dxYrRNaAKEpmoHdz4puUOg9bISFH61Fs6uz3p_xl.webp)
2.1. Reconfigurable polymer networks based on molecular brushes
The possibility of controlling the characteristics of polymer materials by changing the architecture with the chemical composition of the networks remaining unchanged open unique prospects for controlling their properties through the reconfiguration of the polymer network without the loss of integrity and even the shape of the material. This is achieved by the introduction of small molecules, serving as dormant crosslinkers or, on the contrary, as crosslinker scavengers, into polymer networks. These molecules can be activated at any time by an external stimulus (e.g., temperature or light) in order to introduce additional crosslinks or remove the existing ones (Fig. 3). In the former case, the elastomer or gel becomes firmer; in the latter case, softer up to the transition to the melt of bottlebrush mesoblocks. state. It is worth noting that for materials containing dormant crosslinks, as opposed to conventional stimuli-responsive materials, these changes are irreversible, i.e., in both states (before and after the activation), the materials remain stable indefinitely up to the next exposure to the corresponding external stimulus. This on-demand change in the properties may be useful in the development of upcycling technologies, allowing for the reuse of polymer materials to improve their properties.
![[{"id":"L6gKMVoufF","type":"paragraph","data":{"text":" Schematic views of the reconfiguration (<i>a</i>) and the reversible Diels-Alder reaction of maleimide with furan (dormant crosslinker) and the deactivation of crosslinker with (anthracen-9-yl)methanol (<i>b</i>)."}}]](/storage/images/resized/SeL2W4FxogKqsLN43rpZmydS4cwPXsASZHkHcdyQ_xl.webp)
Similar principles of architectural reconfiguration can be applied in technologies for closed-loop recycling of chemically cross-linked networks, which are most difficult objects for recycling (molecular modification). The disassembly is performed by removing crosslinks and transforming the networks to liquid melts consisting of bottlebrush mesoblocks. For this purpose, it is necessary to activate a dormant crosslinker scavengers, which will remove percolation bonds between the macromolecules. As mentioned above, materials remain stable before and after the reconfiguration, making it possible to use the polymer product up to the recycling and to store the raw material (melt of bottlebrush mesoblocks) over a long period of time for its subsequent use as the starting compound to reassemble the material possessing, depending on the goal, the same or new characteristics.
Therefore, the architectural reconfiguration can be used not only to assemble very complex structures with specified properties but to disassemble these structures for the reuse.
2.2. Degradation of molecular brushes
The disassembly of molecular brush-based networks can be accomplished in different ways, including the backbone degradation, the cleavage of side chains or the disintegration into bottlebrush mesoblocks. Correspondingly, different products, such as bottlebrush oligomers, gels swollen in cleaved side chains and melts of molecular brushes are produced.
The backbone degradation of molecular brushes can occur via acid25,26 or alkaline27--31 hydrolysis. The backbone can have different nature, for example, polynorbornene (PNB),25 polycarbonate (PC)26 or polyimide (PI).27--31
Shieh et al.25 described an original approach to perform mild acid hydrolysis of molecular brushes consisting of the PNB backbone with polyethylene glycol (PEG) side chains. The addition product of cyclopentadiene and maleimide bearing a nitrogen-attached PEG side chain was used as the main monomer. 2,2-Dialkyl-1,3-dioxa-2-silacyclooct-5-ene comonomer units were introduced into the backbone and were then subjected to hydrolysis with hydrochloric acid, providing the backbone degradation (Fig. 4). The acid hydrolysis was successfully applied to hydrolyze molecular brushes consisting of the PC backbone with PEG side chains.26 Under acidic condition, the backbone underwent complete degradation accompanied by carbon dioxide elimination (Fig. 5).
![[{"id":"roFUu5uKwh","type":"paragraph","data":{"text":"Schematic view of the hydrolysis of the copolynorbornene backbone at hydrolyzable moieties.<sup>25</sup> Reproduced with the permission of Springer Nature."}}]](/storage/images/resized/PHIBJZrnD6rmXJgTRTVQtw7AZdqjqt9iJVjP9cO7_xl.webp)
![[{"id":"qGBZBguMCg","type":"paragraph","data":{"text":"Schematic view of the acid hydrolysis of PI-graft-PEG molecular brushes.<sup>26</sup>"}}]](/storage/images/resized/pfiEMX3e7QTIppsF7zLOF9Mb7i9fHgDvZCnTUpCt_xl.webp)
In the studies,27--31 the complete alkaline hydrolysis of the PI backbone of molecular brushes with grafted poly(methyl methacrylate) (PMMA) chains (Fig. 6) and poly(tert-butyl methacrylate) (PtBMA) was performed in order to isolate and determine the molecular weight characteristics of the side chains. The reaction was carried out under the conditions, which did not cause, as proved by the authors, the saponification of side-chain ester groups (45 h at 65 °C in a 2% KOH solution in a 60:40 THF:methanol mixture).
![[{"id":"vbQ5T5ESbc","type":"paragraph","data":{"text":" Schematic view of the alkaline hydrolysis of the backbone of PI-graft-PMMA molecular brushes."}}]](/storage/images/resized/ixysBZk8JILXHNxTfNd2aUvUNp2PsbJU38WrFvHJ_xl.webp)
It should be noted that the acid hydrolysis of ester groups of PtBMA side chains in PI-graft-PtBMA molecular brushes afforded water-soluble PI-graft-PMAA molecular brushes with poly(methacrylic acid) (PMAA) side chains, which showed high efficiency as nanocontainers of cyanoporphyrazine agents in photodynamic therapy and diagnostics.29,31
Approaches associated with the depolymerization of the backbone of molecular brushes32,33 are of particular interest. Xiao et al.32 synthesized a series of molecular brushes by grafting azide-terminated polystyrene (PS) and PEG side chains onto the poly(benzyl ether) backbone bearing a triple bond at one end and a group initiating depolymerization at another end. The subsequent removal the latter group (Pd0 or F-) led to the irreversible degradation cascade from head to tail yielding individual side chains (Fig. 7). The depolymerization rate increased with increasing flexibility of the side chains in going from PS to PEG due to an increase in the conformational flexibility of the backbone, resulting in that the optimal orientation for the depolymerization of the terminal unit of the backbone was more rapidly reached (see Fig. 7).
![[{"id":"9Ug0bv_FFg","type":"paragraph","data":{"text":"Schematic view of the synthesis and depolymerization of molecular brushes.<sup>32</sup> Reproduced with the permission of American Chemical Society."}}]](/storage/images/resized/RLowCmYpHM4FBeske8zT8NheYH3Yq9oX4wuRvm2n_xl.webp)
Neary et al.33 performed the backbone degradation of molecular brushes consisting of the polycyclopentene backbone with PS side chains through ring-closing metathesis depolymerization (Fig. 8). It should be emphasized that the released side chains contain the cyclopentene end group, that is active in the addition to thiols. This can be used to prepare other polymers with a complex architecture, e.g., star-shaped (Fig. 9).
![[{"id":"OnLnvLeqQo","type":"paragraph","data":{"text":" Schematic view of the ring-closing metathesis depolyme rization.<sup>33</sup> Reproduced with the permission of American Chemical Society."}}]](/storage/images/resized/XJKTuFUHVnV35wyZOkDek4POiHg8BrvwuPjJTCMA_xl.webp)
![[{"id":"hLzQIDZCf1","type":"paragraph","data":{"text":"Schematic view of the sequential transformation of the polymer architecture from a linear macroinitiator (<i>1</i>) to a cylindrical molecular brush (<i>2</i>), a linear macromonomer (<i>3</i>) and a star-shaped polymer (<i>4</i>).<sup>33</sup> Reproduced with the permission of American Chemical Society."}}]](/storage/images/resized/kpvVlsytG8Ofdebt3tvwKOqWm7qgXd8JrvYijj5K_xl.webp)
It is also worth noting that the mechanical tension that occurs upon adsorption of molecular brushes onto the surface, can lead to the covalent bond scission in the backbone.34–37 The atomic force microscopy study showed that the avalanche-like backbone degradation of molecular brushes can occur once the bond tension approaches ≈1 nN (Fig. 10),35 the bond cleavage rate being sensitive to the substrate surface energy.37
![[{"id":"8kG1Z7_K79","type":"paragraph","data":{"text":" Schematic view of the appearance of a significant mechanical tension along the brush backbone due to steric repulsion between the densely grafted side chains and the spontaneous scission of carbon-carbon covalent bonds in the aliphatic backbone (<i>a</i>) and AFM images illustrating the progressive backbone degradation of molecular brushes upon spreading of a droplet of the brush polymer solution on asolid substrate with increasing distance from the droplet (<i>b</i>). f is the tensile force. The points of application of the force are represented by dots, the arrows indicate the force direction, x is the lateral size of the film; the random distribution of the breakage points of the backbone is seen in the magnified image in the lower part of Fig. <i>b</i>.<sup>35</sup> Reproduced with the permission of Royal Society of Chemistry."}}]](/storage/images/resized/1gzOEOBQGRiEZnC6KlajFHcSuiBak7m1uhLOTrfT_xl.webp)
Side chains of molecular brushes can also undergo different types of degradation, in particular hydrolysis. For example, Deshmukh et al.38 synthesized heterografted molecular brushes consisting of the PNB backbone with polylactide (PLA) side chains and liquid crystalline (LC) cyanobiphenyl mesogens (Fig. 11a), which self-assemble in an applied magnetic field to form cylindrical PLA domains (Fig. 11b).38 Then the PLA domains can be selectively removed by hydrolysis to produce nanoporous films.
![[{"id":"j8RfF145Sp","type":"paragraph","data":{"text":"Formula and schematic representation of the structure of a block copolymer brush consisting of the PNB backbone with PLA and cyanobiphenyl side chains (<i>a</i>) and an AFM image of the film produced from this brush blended with 4-(hexyloxy)-4-biphenylcarbonitrile in a ratio of 1:1 with respect to LC side chains, which demonstrates the self-assembly to form cylindrical domains (<i>b</i>).<sup>38</sup> Reproduced with the permission of American Chemical Society."}}]](/storage/images/resized/z2u85x9JDcZKZun1KEthP0n5sptAusfPsefxYFMc_xl.webp)
We decided to limit the description of conventional approaches, which are used to create materials based on polymer brushes, to this phenomenal research and focus the attention on the rapidly developing industrial techniques.
3. Multilayer coextrusion
The progress in studies of polymers is directly related to the state of science and technology, generating new ideas and approaches, as well as to an increasing demand of new materials. Thus, due to the development of electronic computing technique and software, in particular, computer-aided design systems, the additive manufacturing method, the most well-known variant of which is the 3D printing (layer-by-layer formation of products), has evolved from a revolutionary technology into a mainstream process used in a wide range of industries. This technology makes it possible, first, to fabricate items with complex geometry from several components or those, which do not require the assembly, and, second, enables the piece and small-scale production of components, in particular, those for the prototyping of products with individual design, individual medical products. Besides, the additive manufacturing is considered as an environmentally sustainable technology due to raw material savings and shortening of supply chains, resulting in the reduction of the environmental impact.39,40 The development of modern polymer materials includes the complication of molecular composites and the design of different hierarchical structures (polymer chains assembled into lamellae, fibrils, spherulites, etc.), which form higher levels of the spatial organization, such as superlattices and fractal structures.
Most of hierarchical materials are manufactured by bottom-up methods, from molecules to macrostructures. However, the top--down technology based on the nanostructuring of uniform components is often more practical, scalable and cost-effective. An increase in the percentage of materials manufactured by top-down methods is evidence that this technological path will replace the methods based on the self-assembly of materials, particularly in the mass production.
In the past decades, another additive technology, referred to as the forced assembly multilayer coextrusion, has attracted both scientific and industrial interest, as a method for manufacturing hierarchically structured polymer systems with a wide range of functional properties. This process can be used to fabricate polymer film products consisting of hundreds (and even thousands) of individual alternating micro- or nanolayers of two or three different polymers, provided that they can be processed from a melt.2
The schematic view of the multilayer coextrusion is shown in Fig. 12a. Two different melts of polymers (or a polymer composite) A and B are delivered from the corresponding extruders, pass through pumps and are then coextruded to form a multilayer laminar flow in a feedblock. By varying the performance of the extruders and pumps and the ratio of the flow rates of the melts from the extruders, one can change the ratio between the layer thicknesses of the two polymers A and B. Then the layered polymer melt flows through a layer multiplier. The presence of the latter is the distinguishing feature of a multilayer extrusion line, as opposed to conventional coextrusion setups, providing the fabrication of unique multilayer structures composed from different polymers.
![[{"id":"w2u3KabyRD","type":"paragraph","data":{"text":"Schematic view of the fabrication of multilayer films (a), the baker transformation and a multiflux layer multiplier setup; the arrow indicates the direction of the melt flow (<i>b</i>), the atomic force microscopy phase image of the cross-section of the structure.<sup>2</sup> Reproduced with the permission of Elsevier."}}]](/storage/images/resized/0SavgwCFGCXMf9rL8QrdEy7Pe0U3Hbyj5XWF16ZN_xl.webp)
In a layer multiplier, the polymer melt undergoes the baker's transformation, named after a kneading operation that bakers apply to dough. Due to the channel shape of the multiplier, the flow is spread, split into two layers and then recombined (see Fig. 12b). By repeating this process n times (combining several identical components; the case with n=2 is shown in Fig. 12b), one can fabricate a film with the desired number of layers.
Therefore, using several multipliers and providing the laminar flow of the melt, it is possible to fabricate films consisting of tens or even thousands of polymer layers alternating in the cross-section of the film (see Fig. 12b). The correctly chosen flow rates, temperature and channel geometry allow the fabrication of individual continuous layers with a thickness of up to 10 nm.2
There are several designs of layer multipliers. The schematic view of the most commonly used setup, so-called multiflux (see Fig. 12b),41 is based on Sluijters s static mixer.42 An alternative model of a serpentine-type layer multiplier (Fig. 13) was presented in the studies.43,44 This type of multipliers is topologically equivalent to a multiflux multiplier, but it is more compact and is simple to manufacture, particularly in the case of flow-rotating elements. Besides, in more compact multipliers, the contact of the melt with the walls is minimized and, as a consequence, the shear stress causing the disturbance of the laminar flow is decreased, resulting in a more uniform layer distribution.
![[{"id":"veJw0eBOX-","type":"paragraph","data":{"text":"Basic elements of a serpentine-like layer multiplier, performing the turn, mirroring, layering, rotation and addition (<i>a</i>)<sup>45</sup> and examples of serpentine-like layer multiplier setups<sup>43</sup> (<i>b</i>). The arrows indicate the direction of the melt flow. Reproduced with the permission of Wiley."}}]](/storage/images/resized/jozRmv4GlnvIRcnnrv50CgjAB1WgLtyIGxSjbDIs_xl.webp)
Guided by this principle, Neerincx and Peelen43 developed a mixer called PeelIncx. A two-layer flow is fed into the top part of the mixer (Fig. 14a) and is split into two flows, the left one (omitted in the Figure) and the right flow. Each flow enters a curved channel and then is split into six parallel flows, which are directed downwards. In the down part of the mixer, which is symmetrical to the top part (Fig. 14b), these six flows from the right channel are recombined in the narrowing channel and form twelve layers and then, at the exit, are recombined with twelve layers from the left channel, resulting in 24 layers. This flow can be fed into another element of the layer multiplier connected in series. Therefore, each multiplier increases the number of incoming layers by a factor of 12, and the total number of the fabricated layers is 2612n, where n is the number of connected multipliers.
![[{"id":"9ewZoFtiSY","type":"paragraph","data":{"text":"Top part (<i>a</i>) and the bottom part (<i>b</i>) of a PeelIncx layer multiplier.<sup>43</sup> Reproduced with the permission of Wiley."}}]](/storage/images/resized/YwbPwhlRX1t6ZXLDgDLwybcTRaCs4ixeVAuRoTyc_xl.webp)
It should be emphasized that the multilayer coextrusion technology makes it possible not only to assemble individual micro- and nanolayers to form simple sequences, such as (AB)n, (ACBC)n or (ABC)n, but also to arrange individual layers both parallel and perpendicular to the surface of the film product. The multilayer coextrusion was applied to produce polymeric fractal mixtures with a large interface.45--47 Fig. 15 presents photomicrographs of a tree-like structure manufactured by a combination of elements shown in Fig. 13. Wang et al.48 described the production of polycaprolactone nanofibres by multilayer coextrusion (Fig. 16). The process involved a combination of the multiplication and lamination of polycaprolactone (PCL) and polyethylene oxide (PEO) flows followed by the stretching and dissolution of PEO in water.
![[{"id":"3yA28nSfNn","type":"paragraph","data":{"text":"Fractal structure produced using a serpentine multiplier:<sup>47</sup> <i>a</i>, the general view; <i>b-e</i>, enlargements; <i>f</i>, an idealized structure, which can be produced in the absence of shear stresses. Reproduced with the permission of Wiley."}}]](/storage/images/resized/rDbpKJErKoCeen316jQPx6IvMBuOB45aAih3De41_xl.webp)
![[{"id":"kunZ4zypqr","type":"paragraph","data":{"text":"Schematics of the production of PCL nanofibres.<sup>48</sup> Reproduced with the permission of Elsevier."}}]](/storage/images/resized/wkJMoJgXGH4Q0XmGKbZNAOUXZ03Z6Cma5ZwIqpdU_xl.webp)
Multilayer film materials and fibres, produced using this technology, possess unique properties. The application of layered materials in electronic optical systems is of interest. The alternation of layers with different refractive indices and with a stepwise increasing thickness allows the creation of gradient refractive index lenses (GRIN lenses).49 Multilayer polymers with incorporated active nanoparticles and(or) organic molecules can be used to create 3D optical data storage systems.50 The precise control of the refractive index of multilayer polymer films at the nano- and microscales specifies the optical interference effects characteristic of these one-dimensional photonic crystal structures.51 For example, band-gap tunable lasers can be fabricated based on elastomers with strongly different refractive indices.52
The multilayer coextrusion of melts is a flexible continuous highly efficient technological process, which can not only be used for research purposes in laboratories but can be scaled up to industrial levels to manufacture unique film products and high-tech materials, with increasing importance of packaging industry.
3.1. Use of multilayer coextrusion technology in production of packaging materials
The packaging industry is the largest consumer of polymers. The main strategies for improving their barrier properties, mainly to water and oxygen, are based on the introduction of inorganic fillers and lamination with polymers with low gas permeability.53 High gas permeability of polymers is due to the presence of the free volume in the latter; therefore, amorphous regions have much higher permeability compared to crystalline regions. The barrier properties of commercially available polymers can be significantly improved by varying the crystal structure, including the alignment of crystallites. Thus, the formation of a layer of a crystalline polymer with a thickness of about tens of nanometers between two layers of another confining polymer results in the crystallization in a confined space,54 where crystalline lamellae are oriented parallel to the film surface, thereby significantly improving the barrier properties (Fig. 17). This effect was demonstrated in many studies, for example, for PS/PCL, poly(acrylic acid) (PAA)/PEO,55 PS/PEO,55 PS/PLA.
![[{"id":"9JVgutH5c-","type":"paragraph","data":{"text":" Oxygen permeability normalized to the corresponding permeability of the bulk film versus the thickness of single layers of multilayer films (<i>a</i>), and a schematic showing the gas diffusion pathway through a multilayer structure;<sup>55</sup> Fig. (<i>a</i>) was created by the authors on the basis of Ref. 55; (<i>b</i>) reproduced with the permission of American Association for the Advancement of Science."}}]](/storage/images/resized/hEGNpAPpEcFqlYYviLKPRwRg5t0nzROCZ8LcDCGw_xl.webp)
Multilayer coextrusion technologies for the fabrication of multilayer polymer films allow the creation of functional multilayer materials with characteristics required for application in a particular field. Thus, multilayer coextrusion can be used to manufacture multilayer packaging products with improved mechanical and gas-barrier properties.56--63 In the past years, the mechanical57,61,63 and transport56--60,62 properties of multilayer polymer films and the analysis of the layer structures, interlayer adhesion and interlayer diffusion58,62,64--69 have been extensively addressed in the literature. This interest is due to the fact that the changes in the macroscopic properties of coextruded films are largely associated with the effects at the interface between the layers and the dynamic constraints arising from the layered structure of the film. Multilayer polymer films are studied using both experimental methods and computer modeling.65
Let us consider in more detail the key factors responsible for the performance characteristics of coextruded multilayer films. The main factors are the composition of the layers and the degree of interlayer adhesion between the layers. The choice of a pair of polymers with good interfacial adhesion between the layers may significantly improve interactions between the layers, whereas poor adhesion increases the probability of film stratification in the presence of external deformations.63,68,70 As a consequence, the degree of interlayer adhesion is largely related to the stability of the final multilayer material.
The number and thickness of individual layers are also important factors, which significantly affect the properties of multilayer polymer films. As mentioned above, the increase in the number of layers in the films fabricated by multilayer coextrusion is related to a decrease in the layer thickness. In turn, the decrease in the thickness of individual layers may lead to the alignment of the polymer chains, a change in their mobility, confined crystallization and a change in the glass transition temperature.2,59,62,66,68,71,72 All these have a significant effect on the mechanical and transport properties of multilayer polymer films. Besides, it was shown72 that in the case of the layer thickness smaller than a certain critical value (generally, a few nanometers), the layers can degrade to nanoplates and nanoinclusions, facilitating an increase in the gas permeability of the film.
It is known that in multilayer films, there are dynamic constraints manifested in the changes in the mobility and organization of macromolecules in the layered system compared to the bulk material.2,73 For partially crystalline polymers, the dynamic constraints can promote confined crystallization with decreasing layer thickness;59,68,71,72 for amorphous polymers, they can lead to the alignment of the chains and a decrease in their mobility.62,66 The confined crystallization of partially crystalline polymers facilitates the sequential change in the crystal morphology with decreasing layer thickness from three-dimensional spherulites to one-dimensional crystalline lamellae with a unique orientation.54,62 There are two main types of the orientation of lamellae in the case of confined crystallization:
- polymer chains are aligned parallel to the plane of the layers in the film, while the lamellae are perpendicular to this plane (on-edge);
- lamellae are parallel to the plane of the layers, while the polymer chains are perpendicular to this plane (in-plane).54,56,59
Nassar et al.56 observed the alignment of PLA lamellae parallel to the plane of the layer (in-plane) in multilayer films composed of PLA layers with a thickness of 20 nm, which are confined between the layers of polycarbonate compatible with PLA, whereas the use of polystyrene incompatible with PLA as confining layers led to the mixed in-plane and on-edge orientation of PLA lamellae. It was suggested that the stronger interaction between confined partially crystalline polymers and confining amorphous polymers in multilayer films can facilitate the formation of crystalline lamellae in the layer plane of the film (in-plane). For multilayer polymer films composed of polycaprolactone and polystyrene, the confined crystallization of polycaprolactone was observed58 with decreasing layer thickness to the nanometer scale, resulting in the formation of lamellae aligned in the plane of the film.
It was shown that this phenomenon facilitated a significant improvement of the gas-barrier properties.58 Messin et al.62 demonstrated that a decrease in the water and gas permeability of polycarbonate/poly(m-xylene adipamide) (MXD6) multilayer films is related to the alignment of MXD6 chains and the reduction in the chain segment mobility due to dynamic constraints. In the case of composite films, dynamic constraints can promote the improvement of the dispersion of filler particles and their alignment in the plane of the layers upon coextrusion, which is also reflected in the mechanical and gas-transport properties of the material.57,61,63 Therefore, the confinement effect can significantly influence the structure of the multilayer film, as well as its mechanical properties and moisture- and gas-barrier properties.2,57,58,61--63,69,73
The interlayer diffusion is another important factor responsible for the properties of multilayer polymer films. The motion of polymer chains across the interface between the layers results in the mutual diffusion, which is reflected in the transport and mechanical properties of the films.2,64,67,74 The main driving force for interlayer diffusion is an increase in the entropy of the system caused by the interpenetration of the chains of the contacting layers.74,75 Once the contact between the layers occurs, the polymer chains at the interface have a distorted conformation as if reflected at this surface. Then the polymer chains relax towards the Gaussian conformation equilibrium in a melt, which maximizes the entropy of the system. However, because of topological entanglements, this relaxation process cannot take place through Fickian diffusion of the polymer chains, as was shown in the classical works by P.-G.de Gennes.76,77 The only mechanism of interlayer diffusion is the reptation through the motion of chain ends across the layer boundaries to the bulk.75
This mechanism of interlayer diffusion results in that its effect on the structure and properties of multilayer films fabricated by coextrusion is not negligible even in the case of immiscible polymers, particularly in the case of nano-thick layers.2,67 It was demonstrated64 that the coextrusion gives rise to numerous interfacial regions in the film with different layer thickness that depends on the contact time of the layers in the multiplier. In the general case, the fraction of interfacial regions in the film is determined by the degree of interaction between the alternating layers of polymers. This fraction increases with increasing contact time of the layers, the number of mixers and with decreasing thickness of individual layers.64,74 The degree of interlayer diffusion can differ for different layers in the film.64,74 The area of potential application of the fabricated multilayer materials depends on whether the layered structure of the film with different layer thickness is retained or destroyed due to interlayer diffusion.
Therefore, the main factors that affect the properties of multilayer films include the composition of the layers and the degree of interlayer adhesion between them, the number and thickness of the layers, the dynamic constraints and the degree of interlayer diffusion of the components. It is important to take into account the effects of these factors on the structure and performance properties of coextruded multilayer films when creating packaging.
Such materials, as polypropylene (PP), polyethylene terephthalate (PET), polyamide (PA), polyethylene (PE), ethylene vinyl alcohol (EVOH) and ethylene vinyl acetate, are currently most often used to manufacture packaging materials from multilayer polymer films. Layers of such polymers as EVOH and PA can provide enhanced gas-barrier properties of the films, whereas PP, PE and ethylene vinyl acetate are employed for protection from humidity;78 PP, PS and PET layers are often used to provide mechanical stability of the films.78
The issue of the safe disposal of multilayer films has attracted great attention. Therefore, the growing trend is to use biodegradable polymers. In particular, such biodegradable polymers as PLA, PCL, poly(butylene adipate terephthalate), poly(butylene succinate) (PBS), as well as different polyhydroxyalkanoates, can be used to create environmentally benign multilayer polymer films. These materials, taken alone, have drawbacks that limit their common use as packaging. However, their application in multilayer materials provides the possibilities for creating environmentally friendly packaging with altered characteristics. Thus, multilayer films with alternating PLA and PBS layers, which have improved moisture- and gas-barrier properties as compared to single-layer PLA and PBS films, were fabricated by multilayer coextrusion.60 Under the environmental conditions, films based on biodegradable polymers are most often degraded via enzymatic hydrolysis by microorganisms and also via chemical hydrolysis at a rate dependent on the temperature, humidity, pH of the medium and UV irradiation.79,80 These materials are degraded into water, carbon dioxide, methane, biomass and inorganic compounds, the degradation rate being hundred and thousand times higher than the rate of standard large-tonnage polymers.80
4. Modern rheology of polymer melts
With the development of state-of-the-art methods for the synthesis and processing of polymers, the conventional extrusion technologies based, in particular on the melt flow theory, did not lose the importance but even gained more significance. Actually, the main goal of modern technological processes for the manufacturing of goods from polymers and polymer composites is the formation of the desired structure of the material, which should provide the target functional performance. The notion structure is vague and requires the specification as applied to particular cases. The extrusion is one of the key technological processes for the fabrication of continuous goods with a specified profile, such as sheets, films, pipes, cable insulation, etc. The extrusion mechanics (i.e., the dynamics of the movement of the material in the extrusion channel and the profile extrusion die) was studied in sufficient detail and became the subject of university courses.41 Much more difficult is the analysis of physicochemical phenomena accompanying the extrusion of multicomponent polymer materials, which are largely determined by the rheological properties of polymers and their interfacial interactions. Therefore, these phenomena should be considered taking into account the complex stress states created in the extruder.
Before turning to the discussion of the conditions of polymer melt flow through multipliers and 3D printing heads, it is reasonable to briefly describe the main extrusion processes. The extrusion technology includes the preparation of a molten material of the basic polymer (or several polymers) containing different functional additives and the pushing of this material through the profile extrusion die with profile channels having a specified configuration.
In most cases, the functions of a single-screw extruder include the melting of the polymer composite based on thermoplastics and the creation of pressure for the uniform extrusion of the molten material through the profile extrusion die. Even in the case of extrusion of elastomers or thermoplastics, the chemical reactions in the extruder should be minimized. For this purpose, the operating elements of extruders (screws) are replaced by mixers with more complex geometry, the movement of the components to be mixed through the extruder playing a key role in the preparation of the mixture.
Screw extruders can be used as continuous flow reactors for the polymerization,81 the synthesis of thermosetting resins82 and polyurethanes,83 PET recycling,84 modification of polyolefins by grafting with maleic anhydride and other compounds,85,86 the synthesis of polyglycolides87 and many other reactions. This technique is successfully used to perform the reactions, which are not accompanied by the formation of side products.
The use of twin-screw extruders is particularly efficient for mixing polymers (both with and without fillers),88 which is reflected in the diversity of types of such extruders in the market, which are proved to be superior in different manufacturing processes (plastic and food industries, pharmaceutical applications, etc.).
Nevertheless, in any case, an extruder should produce a homogeneous material for the manufacturing of particular long-length products (tubes, rods, different profiles). The resulting structure (to be more precise, the morphology) of the material produced by the extruder depends on the degree of randomization on a particular scale. Along with the homogenization of the composition of a multicomponent system, the extrusion can give rise to regular morphological forms due not only to phase and relaxation transitions but also to the kinematics of the process and specific intercomponent interactions.
The step of the process, in which a particular three-dimensional structure can be produced under shear strain, is considered below. The formation of this structure is accompanied by physicochemical phenomena, which greatly depend on the rheological properties of the polymer composition.
4.1. Laminar flow
Homogeneous layers of two molten polymers (polymer composites) can be combined from two flows produced by two independent extruders. In the case of a flat slot die, the flow in the extruder producing the structure of the final material is laminar. In relation to the issue under discussion, the laminar flow theory was constructed for the first time for inelastic non-Newtonian fluids.89 Subsequently, this problem was considered in the general case for axially symmetric flows of polymer melts, the rheological properties of which are described by the power law.90
The fundamental problem in the discussion of stratified flows of polymer melts (like other fluids) is the interfacial instability (Fig. 18a). This problem is related to the classical Kelvin--Helmholtz instability caused by the density difference between two immiscible fluids. However, this mechanism of instability is generally of little significance for stratified flows of polymer fluids because the effects associated with the nonlinear properties and elasticity of polymer melts and solutions are much stronger than the density difference. The flow dynamics is most affected by the elasticity of polymer melts, which cause the interface deformation due to normal stresses (perpendicular to the flow direction).91 Besides, the presence or the absence of compatibility of polymers in interfacial layers also plays an important role, the compatibility facilitating the stabilization of the interface.92
![[{"id":"FyoH66-PDW","type":"paragraph","data":{"text":" Schematics illustrating the instability of the interface between two layers of immiscible fluids (<i>a</i>), the distortion of the flow profile in the laminar flow (<i>b</i>) and the distortion of the flow profile during the coextrusion of two layers in a cylindrical channel (<i>c</i>)."}}]](/storage/images/resized/wwCXq3VzQTg6FovE3IcsEJ5srrZubl1C8JSV0uZm_xl.webp)
The interfacial stability is particularly challenging in the fabrication of thin and superthin films using the modern technology for doubling coextruded polymers. The theoretical analysis of the dynamics of multilayer flows and the appearance of interfacial instability was performed in a number of studies.93 The complete solution of this problem requires using numerical methods and should rely on the analysis of very complex rheological equations (separately for each of the combined layers), which take into account the nonlinearity of the rheological properties of the fluids and their elasticity, giving rise to normal stresses.
The formulation of the boundary conditions at the contact interface between the layers plays a significant role in the consideration of the dynamics of multilayer fluids. Generally, it is assumed that the rates of two flows are equal; however, the difference in the stress at the interface due to different rheological properties of the layers can cause interfacial slip and breaks induced by periodic adhesion-deadhesion,94 the breaks being possible within the layers.95
Due to the specific rheological properties of the coextruded layers, the interface profile can be distorted not only with time but also along the flow length (Fig. 18b).
The situation is even more complicated in the case of coextruded layers when manufacturing products with a circular cross-section because the distortion of the flat interface between the layers results in that one of the layers is turned and pushed against the channel wall (Fig. 18c).
There are the following main areas of application of the multilayer extrusion technology:
- the direct manufacturing of multilayer long-profile products (in the simplest case, sheets);
- the use of multipliers of complex geometry (see above) to perform coextrusion of polymers, which are involved in chemical reactions under intense strain, resulting in the formation of an interfacial layer of intermediate composition.96 This process is similar to the process, which includes the addition of a reactive monomer and its copolymerization with a melt of high-molecular-weight polymer during coextrusion to form an aligned layer on the surface of the high-viscosity melt;97
- the multilayer extrusion can serve as the basis for the manufacturing of multiply doubled layers in an amount of 2n, where n can reach large values.
4.2. Phase separation in polymer blends
Generally, incompatible polymers are considered when discussing the phase separation in polymer blends. The tendency of such polymers to the separation is the natural thermodynamic driving force for the macrophase separation. However, the kinetic barriers hinder this process and often make the stratification impossible due to a very low diffusion rate because of high viscosity of the polymer components. Therefore, the phase separation can occur only under nonequilibrium conditions induced by external stimuli. Among these factors is the interaction of mechanical fields, i.e., shear or longitudinal strain. The following two situations are possible: the formation of morphological elements of one phase in the matrix of another component of the blend or the displacement of one component to the free surface to form a homogeneous layer. These phenomena are due to instability of the system induced by stresses, facilitating the disturbance of the quasi-equilibrium state.
Apart from the coextrusion of two (or more) polymers, of great interest is the specific morphology of the composites from a pseudohomogeneous blend of incompatible polymer melts produced in the profile extrusion die with a cylindrical channel. Actually, it is a polymer emulsion, in which droplets of the dispersed phase are deformed at the entrance to the channel and are converted to thin filaments at the exit (Fig. 19). This process for the fabrication of composites is particularly attractive when the dispersed phase is produced by a thermally stable fibre-forming polymer, in particular a LC thermoplastic, the aligned rigid macromolecules of which ensure high durability of furnished products.98 These systems are called in situ composites.
![[{"id":"78X37CcV97","type":"paragraph","data":{"text":" Schematic view of the formation of the fibril-reinforced morphology in a two-component flow of a blend of incompatible polymers in a capillary (profile extrusion die).<sup>99</sup> Reproduced with the permission of Wiley."}}]](/storage/images/resized/UKBckmhFtfTZPZWQorMpLHWEtfiokDjYUvWr20wK_xl.webp)
The problem of the spatial separation of polymer blends is no less interesting and is practically important. It is not just about the stretching of the droplets of the dispersed phase along the extrusion direction but also about the migration of this phase in the transversal direction to the periphery of the flow. This effect is most important in the case of high-viscosity blends of thermoplastics with LC polymers, the melts of which have a lower viscosity due to the anisotropy of the rheological properties. An example of the formation of a surface layer consisting of a LC polymer, which was fabricated by the extrusion of polysulfone (PSF) and liquid-crystalline copolyester (LC CPE), is presented in Fig. 20a.
![[{"id":"RdOSV4I3y9","type":"paragraph","data":{"text":"SEM image (1256) of an extrudate, which initially contained 70% PSF and 30% LC CPE Ultrax-4002, after the removal of PSF with dichloromethane (the LC matrix is indicated by an arrow) (<i>a</i>) and the plot of the viscosity of PSF-LC CPE blend melts versus the content of LC CPE Ultrax-4002 at different temperatures (<i>b</i>).<sup>99</sup> Reproduced with the permission of Wiley."}}]](/storage/images/resized/JsL3GAztjOm37kLHQOcf8RZV67gmUlKCBkwHlKhM_xl.webp)
It is seen that the morphological skeleton of LC CPE is similar to a wood or bamboo cut. Thus, the skeleton consists of concentric fibril-like layers and the outer shell (bark), which is responsible for the unique rheological properties of the melts of such composites. Due to the low-viscosity surface layer, acting as the lubricant, the viscosity of blends is much lower than that of its components (Fig. 20b), particularly at 280 °C.
Along with the presence of a low-viscosity peripheral layer, which causes the effect of slug flow during transport in channels, one should take into account the specificity of the velocity profile in the central zone of the flow in the presence of low-viscosity droplets of the dispersed phase in the polymer emulsion. Most likely, this is a polyparabolic profile, which should contain prominences corresponding to stretched droplets of the LC melt, resulting in the reduction in the viscosity of the flow and the formation of reinforced fibrils in the solid extrudate. In the presence of a LC component in the polymer blend, the specific features of the flow of composites with significantly different rheological properties of their components can be clearly tracked. This makes one think about the development of a rheological model describing the specific features of the extrusion of such systems.
The general and most significant effect of the phase separation in rotation flows is the formation of shear banding associated with the transition from a quasi-stable, but unstable, system to an organized structure. This fact is well-known for wormlike micelles100,101 and polymer solutions,102 as well as for polymer melt blends.103--105 The formation of a regular structure upon shearing is accompanied by the separation of a homogeneous mixture into clearly visible bands (generally, into two bands, but a much larger number of bands can also occur), which differ in the composition and, correspondingly, the rheological properties. In the case of a simple shear, shear rate jumps are observed at the band boundaries, determined from the velocity profiles. This effect is particularly pronounced in the case of the shear-banding flow between coaxial cylinders and a small gap between them (Fig. 21a) and also between the conical and flat surfaces at a small angle between the generatrix of the cone and the plane (Fig. 21b,c). The capillary flow serves as a model of the final step of extrusion, while the shear strain in a rotating setup models, to a large extent, the behaviour of polymer melts and solutions under conditions of worm rotation.
![[{"id":"rcrc-WOGk9","type":"paragraph","data":{"text":"Shear banding between coaxial cylinders that appear due to an inflection in the velocity profile (<i>a</i>), the schematics of the cone-and-plane setup (<i>b</i>) and the velocity profile in the gap between the conical and flat surfaces (<i>c</i>) for a homogeneous flow (<i>1</i>) and a shear banding flow (<i>2</i>). Ω is the rotation speed of the cone, γ̇ is the shear rate, ν<sub>cone</sub> is the maximum fluid flow velocity, ν is the fluid flow velocity at the point z. The Figure was prepared by the authors using original data from the studies.<sup>100-105</sup>"}}]](/storage/images/resized/MGM1zzG4gxJRNHPS3ZqwE7ZEF4PndXFENFcg8O1K_xl.webp)
It is most convenient to perform experimental studies of the appearance of shear banding in mixtures of polymer solutions and melts containing one liquid-crystalline component with a viscosity much lower than that of the isotropic component. As an example, Fig. 22 presents the evolution of the morphology of a model mixture composed of a 50% LC aqueous solution of hydroxypropyl cellulose (HPC) and a solution of polyisobutylene (PIB) in decalin at the same concentration in the 1:1 ratio.109 The viscosity of the HPC solution is approximately two orders of magnitude lower than that of the PIB solution.
![[{"id":"2vezO3riqE","type":"paragraph","data":{"text":" Photomicrographs (4×) illustrating the successive steps of the mixing of a coloured LC aqueous solution of HPC and a solution of PIB in decalin at a shear rate of 0.2 s<sup>-1</sup> (<i>a-c</i>) and 1.3 s<sup>-1</sup> (<i>d-f</i>) depending on the shear strain: γ=0 (<i>a</i>), 246 (<i>b</i>), 682 (<i>c</i>), 1600 (<i>d</i>), 2230 (<i>e</i>) and 3820 units (<i>f</i>).<sup>109</sup>"}}]](/storage/images/resized/X9TrHaU9GmUSz2CYnxLuuvqCgXXHhmMlp3admK9V_xl.webp)
A similar situation is observed for polymer melts, one of which is in the LC state. An example of the shear-induced texture development in a blend composed of high-viscosity isotropic PSF and low-viscosity LC CPE of polyethylene terephthalate with hydroxybenzoic acid is presented in Fig. 23.
![[{"id":"dDxyhuNpvM","type":"paragraph","data":{"text":" Photomicrograph (4×) illustrating the morphology of the PSF-LC CPE melt of polyethylene terephthalate with hydroxybenzoic acid produced at the cone-plane.<sup>106</sup>"}}]](/storage/images/resized/XiSMr6upOlu1oKRBcrQ6oq9el3o97bXZyX0HyWmV_xl.webp)
If we accept that the composite consists of a high-viscosity matrix with the viscosity ηm and droplets of a dispersed phase with the radius r0 and the viscosity ηd≪ηm, the problem of the flow stability and the formation of droplets can be considered as follows.99 The shear leads to the deformation of droplets, which adopt a stretched (cylindrical) shape with the length L and the radius r; the volume (V) of the droplet remains unchanged:
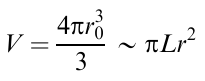
The stretching rate of the droplet is determined by dL(t)/dt. If the shear rate of the viscous matrix is γ̇, the difference between the stretching rate of the droplet and the shear rate of the matrix in the vicinity of the droplet is dL/dt– γ̇R. The dynamics of the droplet stretching is determined from the conditions of balance between the friction force and the capillary force
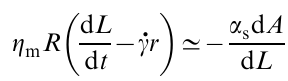
where αs is the surface tension and A≃2πrL≃√V̅L̅ is the surface area of the droplet. Ignoring the energy dissipation in the droplet and limiting only to the energy dissipation in the matrix, the stationary transverse radius of the droplet is
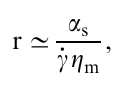
and the length of the stretched droplet is
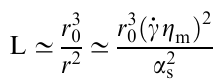
Therefore, the lateral size of the droplet increases with increasing volume and deformation rate of the droplets, and the latter can adopt a circular shape. The stability of such structures can be attributed to the normal stress.
Apart from the lateral deformation of large droplets, one should take into account the radial movement of small droplets, which is associated with the radial pressure gradient P(r) at the cone-plane and is determined by the first (N1) and second (N2) normal stress differences in a high-viscosity matrix,107
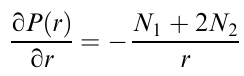
It is worth noting that small droplets can coalesce to form larger drops.
The formation of the circular morphology is accompanied by the appearance of maxima in the flow curves, the maxima being observed only in the rotation flow108 (Fig. 24).
![[{"id":"KKJ_vNIX8k","type":"paragraph","data":{"text":"Flow curves of PSF (<i>1</i>) and CPE (<i>2</i>) melts and their blends containing 25% (<i>3</i>), 50% (<i>4</i>) and 75% CPE (<i>5</i>). The dashed lines indicate the capillary rheometry data for composites <i>3</i> and <i>4</i>.<sup>108</sup> The spurt region <i>I</i>, the slip of the polymer melt along the interface. The region of negative slope in the flow curves after the maximum <i>II</i> corresponds to instability of the system manifested in the phase separation of components and, finally, in the region <i>III</i>, the LC polymer is responsible for the rheological behaviour of the blends; σ is voltage. Reproduced with the permission of Elsevier."}}]](/storage/images/resized/JYBKK3d6xOnI8Xff4hrT9xvV8QPfjccVHZ9Ousua_xl.webp)
The explanation of the observed effects is relied on the following points:99,108
1) at a shear rate of ≈1 s-1, the PSF melt undergoes the transition to a highly elastic state (so-called spurt or the flow stalling, i.e., a drastic increase in the shear rate at a constant shear stress occurs) and, therefore, it us out of the game and has not effect on the viscosity;
2) at shear rates higher than the maximum point, LC CPE is responsible for the rheological behaviour of the composite;
3) at higher shear rates, the viscosity of all blends approaches the viscosity of CPE, which may be indicative of the formation of the LC matrix.109
The concentration dependences of the viscosity of PSF/CPE composites at different shear stresses are shown in Fig. 25. As can be seen, in the vicinity of the maximum in the flow curve, the viscosity in the positive (2) and negative (2') branches differ by more than two orders of magnitude. In the capillary flow, the viscosity decreases more abruptly compared to the rotation flow, which may be due to the formation of low-viscosity flows and the matrix of the LC polymer. The situation in the rotating zone of screws and in the profile extrusion die can be attributed, to some extent, to a combination of the rotation and capillary flows. This mode of processing of such composites at different rates (i.e., in different branches of the flow curve) allows a significant increase in the performance of the product molding.
![[{"id":"ElZ5DplTe0","type":"paragraph","data":{"text":" Concentration dependences of the viscosity of PSF-CPE composites in the rotation flow at log σ (Pa)=2.0 (<i>1</i>) and 2.65 (2, <i>2'</i> ) and in the capillary flow at log σ (Pa)=4.0 (<i>3</i>).<sup>109</sup> Reproduced with the permission of Elsevier."}}]](/storage/images/resized/UgD8l3E6FbXF9CXPUKZAhDXsyRMqSjHg4WKu3VDN_xl.webp)
The shear banding can occur even in polydisperse polymer melts (blends of low- and high-molecular-weight polymers) due to the formation of zones (bands) with radically different molecular weights (Fig. 26). The high-viscosity (high-molecular-weight) fraction forms a nearly immobile layer at the stationary wall, and the rate at the movable wall remains almost constant, so that there is a region in the central part of the gap between the coaxial cylinders, in which the shear rate is abnormally high compared to the average shear rate.110 In this region, the concentration of different phases gradually changes, due to which the transition from one extreme flat profile to another one appears to be rather smooth.
![[{"id":"e7lmTWY1Za","type":"paragraph","data":{"text":"Shear rate (ν) profiles as a function of the position y in the gap between the coaxial cylinders d demonstrating the shear banding due to the separation of the sample into fractions according to the molecular weight. σ=const. The Figure was prepared by the authors using original data from the study.<sup>110</sup>"}}]](/storage/images/resized/R0THeIzyqDzmorGdmCadDrz0UKXDWjLgpz0BnXhA_xl.webp)
The interface between the layers can change in space and time,111 which is manifested, in particular, in auto-vibrations in the measurements of a local shear rate or stress.112,113 Meanwhile, the general cause of transition from random instability to a regular band structure is the possibility of the coexistence of two (or several) stable but different states of the fluid structure. This is reflected in the experimental data as a negative dependence of the shear stress on the shear rate or of the shear stress on the shear strain (see, e.g., Fig. 24). The flow curves with a branch of instability were observed also for many multicomponent polymer and colloidal systems, and generally this feature of the rheological properties of these systems was considered as a necessary condition for the appearance of shear banding.114,115 As shown above, such flow curves correspond to the evolution of different flow morphologies (see Fig. 22 and Fig. 23).
In some cases, in transient modes of shear strain, for example, in the pre-stationary region of flow at a specified shear rate, there is an area, in which the stress decreases with increasing strain (Fig. 27). It is this area, where the phase separation takes place regardless of whether there is a singularity in the steady-state flow curve.116 A similar situation was discussed above in terms of a significant decrease in the viscosity of composites due to phase separation (see Fig. 25 and Fig. 26). Subsequently, this concept was formulated as a general criterion for the formation of shear banding regardless of the equation of the rheological state of complex fluids.117
![[{"id":"CGZtP2Uosr","type":"paragraph","data":{"text":"Effect of the decrease in the shear stress σ with increasing shear strain γ at a constant shear rate, reflecting the existence of an instability region (dashed line) with two possible states for one polymer or for the components A and B in the polymer blend. The Figure was prepared by the authors using original data from the study.<sup>118</sup>"}}]](/storage/images/resized/A7jq3ojmTbQw1gD5LkFmNnBFxBIaS0UocoDi27AB_xl.webp)
The possibility of the coexistence of different states of polymers and colloidal liquids is explicitly reflected in the hysteresis of their rheological properties (viscosity or Young's modulus) in scanning measurements by increasing or decreasing the shear strain rate. This phenomenon can also lead to the structural self-assembly as shear banding.118
4.3. Filled polymers and nanocomposites
In the past decade, nanocomposites with nanosized fillers have attracted attention of researchers. Different aluminosilicates, carbon nanotubes, graphene, detonation nanodiamonds (the latter are an original Russian development119) are used as nanosized fillers. Abundant data demonstrating the effect of the composition on the rheological properties of the composites are available in the literature. However, the relationships between the rheological properties and the structure of the blends are rarely addressed. Meanwhile, the morphology, produced during a flow of nanocomposites in different dispersion media, is characterized by high sensitivity to the nature and properties of such media, in particular, to their elasticity. The evolution of the morphology can be visualized using an original technique based on a combination of a transparent mobile sphere and a transparent glass plate. The use of a spherical surface produces fields with different shear rates, allowing the observation of structural transitions that depend on the deformation mode.
In an aqueous medium, nanodiamonds form discrete rotating micro tornado, the long axis of which is perpendicular to the shear direction (Fig. 28). An increase in the shear rate leads to a series of transformations of micro tornado, resulting finally in its degradation (see Fig. 28f). These structures are similar to Taylor vortices, which appear in the case of predominance of inertial forces over viscous forces; their formation starts in the vicinity of the band and it develops towards the periphery of the flow, like the formation of the circular morphology (see above). The main difference is in the direction of loss of stability (lateral for circles and transversal for vortices).
![[{"id":"T5fFqJ4lqW","type":"paragraph","data":{"text":"Photomicrographs (4×) illustrating the morphology of a nanodiamond dispersion in an aqueous medium, which appears under a flow at a rate of 1 s<sup>-1</sup> and at a shear strain of 0 (<i>a</i>), 190 (<i>b</i>), 380 (<i>c</i>), 940 (<i>d</i>), 1480 (<i>e</i>) and 2000 units (<i>f</i>).<sup>120</sup>"}}]](/storage/images/resized/IbkQXdAbwRbpNXzoFpOwK6gtCANHFWRY38u4fsJj_xl.webp)
The addition of only 1% HPC to an aqueous dispersion medium leads to a radical change in the morphology of the dispersion. Thus, concentric circles or spiral elements are formed instead of radial rod-shaped vortices (Fig. 29). The transformation of a Newtonian fluid into a high-viscosity fluid induces another mechanism of the formation of regular structures by nanodiamond particles, where the elasticity is the driving force.121 This fact served as the basis for the detailed analysis of the role of elasticity in the formation of the circular morphology in rotation flows. The most telling in this respect are the studies,122,123 in which a new model was developed for the rheological behaviour of polymer systems, where the elastic reaction prevails over the dissipative one and the latter can be ignored. The development of this model was baws on the investigation of the circular morphology even in individual polymer melts produced by deformations at high shear rates (see Fig. 29). In the presence of filler particles in a melt, these particles are also ranked in concentric circles (Fig. 30).
![[{"id":"KTQbtk3RAk","type":"paragraph","data":{"text":"Photomicrographs (4×) illustrating the texturing of nanodiamond particles in a 1% aqueous HPC solution in the presence of Tween 80. Different steps of formation of a spiral structure with a time interval of 2 min at a shear rate of 1 s<sup>-1</sup> are shown.<sup>120</sup>"}}]](/storage/images/resized/KLCXWWoBi9QsaP81Csrx5Jg9z4m4umfwIPVr7qwp_xl.webp)
![[{"id":"sJvvV2r9vD","type":"paragraph","data":{"text":"Photomicrographs (4×) of the surface of a polycarbonate melt once the spurt mode is achieved (<i>a</i>) and the distribution of anisodiametric particles of the crushed mineral Na-sepiolite (aspect ratio is ≈100) in the PIB matrix under similar conditions (<i>b</i>).<sup>120</sup>"}}]](/storage/images/resized/WkMAZVXiZq1BixJ3HOsbAHfgOM75vuJ2kp13CUz6_xl.webp)
Sets of elastic spherical particles as units serve as the structural elements of the model. Elastic interactions arise from changes in the particle shape (from spheres to ellipsoids capable of aligning under deformation) or their collisions. The equation of motion of such elastic elements is defined by the Hamiltonian function, describing both elastic components of the system, the orientational interactions and contacts. Elastic deformations are described by a single parameter, namely, the modulus of elasticity E, which is a key parameter in the transformation of the particle shape and the alignment. The elastic energy density (w1) depends on the modulus of elasticity, the eccentricity and the angle between the long axes of the ellipsoids120
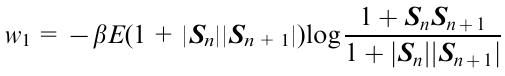
where SnSn+1 is the scalar product of vectors.
The second component of the Hamiltonian function (w2) describes the rotation caused by collisions of particles in the mechanical field
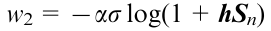
where s is the shear stress and h is the unit vector in the shear direction. In these equations, the numerical coefficients a and b are *1.
The full Hamiltonian Ĥ is the sum
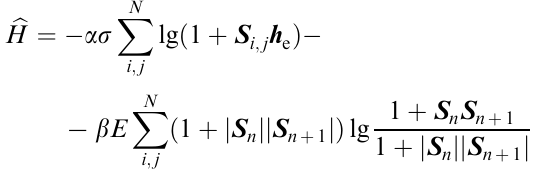
and the equation of motion of the elastic medium takes the form
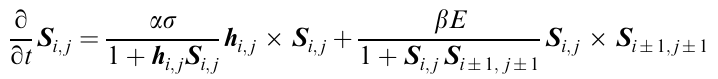
This is the third-order Schrödinger equation, which can be solved. The analysis of the solution demonstrated that this equation predicts bifuractions, giving rise to instability. The bifurcations are manifested in the formation of self-organized structures as spirals or other periodic structures. Therefore, the model under consideration describes the appearance of instability as actually observed structures (Fig. 31 and Fig. 32). The criterion M* serves as the quantitative measure of the transition from the flow state to elastic deformations.124
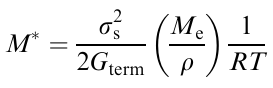
where ss is the stress, above which the forced rubber-like state occurs, Me is the molecular weight between the entanglements of macromolecules, Gterm is the modulus of elasticity in the final viscoelastic shear zone, ρ is the density, T is the absolute temperature, and R is the universal gas constant.
![[{"id":"8eCV-ByYoG","type":"paragraph","data":{"text":" Photomicrograph (8×) of the spiral texture formed in a PIB melt at the periphery of the flow. The arrows indicate the zones of evident bifurcations.<sup>122</sup> Reproduced with the permission of Springer Nature."}}]](/storage/images/resized/vIVud9kfQqSkhKEbPVtC98KKmx6KhxPL90u0VEiE_xl.webp)
![[{"id":"QUyyMtSMZc","type":"paragraph","data":{"text":"Formation of telescopic spirals in strong rotation flows (<i>a</i>) and the concentration of filler particles between these spirals (<i>b</i>).<sup>123</sup> Reproduced with the permission of Hindawi."}}]](/storage/images/resized/EsK3DCFIgeDBWZAu46DfbiIZI7iwmPM6RPfZoZaM_xl.webp)
Theoretical estimates demonstrated that the transition from a stable flow to unstable deformation regimes is accompanied by the formation of toroidal structures and their bifurcations. The tori have an inner telescopic structure, they consist of concentric cylinders with the surfaces of constant normal stresses. In the regions between the spiral-like tori, the normal stresses, related to the elasticity of the melt, are minimal. The experimental studies of the fine structures confirmed the bifurcations of spirals in strong flows (see Fig. 31). The spiral formation and the concentration of the filler in weak regions between them are shown in Fig. 32.
Apparently, this pattern of interactions between the polymer melt and the filler under the conditions of the rotation flow has a general significance for an understanding of the processes that occur in the zone of extrusion mixing and the transport of heterophase polymer systems. In the extrusion or spinning die, these processes are changed for the in situ formation of composites, i.e., single-dimensional self-reinforced products.
4.4. Phase transitions in the shear flow
The previous Section is concerned with strain-induced phase separation;125 however, the addressed phenomena are not related to phase transitions in terms of thermodynamic definition. Below we consider the appearance of a new phase that is not formed without a mechanical field. In this respect, of particular interest are multicomponent systems with dispersed components that are either initially anisometric or can be deformed and oriented under the action of flow-induced stresses. Such systems include rigid macromolecules, emulsions and micellar solutions containing highly anisometric worm-like micelles and suspensions of non-spherical particles.
A fundamental issue is whether (and, if yes, how) thermodynamic characteristics of the systems vary. Obviously, mechanical (rheological) characteristics do vary, as indicated by the stepwise change in the viscosity upon the formation of the LC phase in lyotropic systems.126 The formation of an LC phase can also be detected by optical and other physical methods (see, e.g.,127,128). As regards internal characteristics, heat release accompanying the shear flow of a solution of a rigid-chain polymer was detected at the qualitative level only in one study.129 It is noteworthy that in an initially isotropic solution heated up to the critical temperature, a LC structure is formed in the polymer-concentrated phase upon amorphous phase separation due to the lower critical solution temperature. As the two-phase system is cooled down, a homogeneous solution is formed again, and isotropization of the LC phase takes place, which is manifested as a viscosity step, reflecting heat absorption, i.e., this is caused by thermodynamic factors.
Strain-induced transitions from the isotropic state to the LC phase represent a separate class of phenomena. A necessary condition for the strain-induced formation of an LC structure in a multiphase system is the presence of anisometric elements, either rigid macromolecules or anisotropic rod-like or planar particles. Dispersions of natural clays in polymers also refer to such systems. The possibility of formation of various mesophase structures in clays was shown long ago.130 The discovery of strain-induced phase transitions in a system consisting of a lyotropic LC solution and layered silicate particles (since both components are structurally active) is the most interesting result of joint rheological and X-ray diffraction experiments.131 The design of this rheo-X-ray experiment is shown in Fig. 33.
![[{"id":"pBnvTF7Ca8","type":"paragraph","data":{"text":"Schematic diagram of the rheo-X-ray experiment.<sup>131</sup> The line shows the direction of an X-ray that passes through a dispersion of Na montmorillonite in an LC solution of HPC, which is subjected to shear in the annulus between the glass coaxial cylinders (capillaries). A video camera and a laser are used to align the working cell with the collimator and detector. The matrix is composed of aqueous solutions of HPC with a concentration of 50% (a mixture of LC and isotropic phases) and 60% (single LC phase) and 5% crystalline layered silicate as the dispersed phase. Published with permission from the American Chemical Society."}}]](/storage/images/resized/ujGVCVkTaf5Fyolnl6VnopHmSa02lku1jsR3lpEL_xl.webp)
It can be seen in the 2D X-ray diffraction patterns (Fig. 34a) that in the beginning of the experiment, reflections of the LC domains aligned along the flow in a two-phase solution of hydroxypropylcellulose (HPC) are located at the meridian, while clay particles arte transformed to the ordered state, with the corresponding reflections appearing at the equator. After 75 min of the intense shear (shear rate of 470 s–1), the clay reflection shift to the meridian (Fig. 34b). It can be stated that in the beginning of the process, clay particles form a columnar mesophase, which is converted to the discotic phase under prolonged shear. The crucial role of strain in the structurization of aluminosilicate particles was demonstrated in that study for the first time.131
![[{"id":"RfeiWjV9LA","type":"paragraph","data":{"text":" Evolution of X-ray diffraction patterns reflecting ordering of clay dispersions in a two-phase (<i>a, b</i>) and single plase LC solutions of HPC (<i>c, d</i>).<sup>131</sup> Published with permission from the American Chemical Society."}}]](/storage/images/resized/20j1h2hs7z5lZPg36FCKm706LXQ5XUrig6IA9cUb_xl.webp)
In the 100% LC matrix with HPC macromolecules aligned along the shear direction, a short-term shearing action gives rise to a four-point reflection of clay (Fig. 34c). This effect becomes more pronounced with time (Fig. 34d), which is possible only provided that that the clay particles in this phase form a combination of columnar and discotic mesophases.
Generally, during shearing, the situation develops as shown in Fig. 35. After injection of dispersions into the annulus between coaxial capillaries, the polymer phase is aligned along the capillary axis. The application of shear stress clearly reveals the type of orientation and the nature of the mesophases of HPC solution and Na montmorillonite: whereas for a solution, this is a nematic with the possible presence of elements of cholesteric mesophase typical of polysaccharides and the orientation occurs along the shear direction, in the case of clay, this is a combination of columnar and discotic mesophases.
![[{"id":"_Wf7LcI_qM","type":"paragraph","data":{"text":" Schematic diagram of the stages of formation of the mesophase structure of clay particles in anisotropic matrices upon intense shear.<sup>131</sup> Published with permission from the American Chemical Society."}}]](/storage/images/resized/UgOsgGs1F5QOZSlfRxNdJxDDX302EVaSuG1FZpUP_xl.webp)
Multicomponent polymer mixtures formed by incompatible polymers are, like colloids, thermodynamically non-equilibrium systems. The structure and, hence, the properties of such systems can considerably vary with time. Moreover, a moderate external mechanical impact is often sufficient to generate instability, which brings about fundamentally important consequences for the fate of the system. This largely refers to polymer solutions and melts in which mechanical impact also generates instability, giving rise to structural changes, which take place slowly, because the rate of propagation of new structural entities is restricted by diffusion processes in the highly viscous medium. Nevertheless, the development of instability in multicomponent polymer systems is determined by a number of contributing factors, the most interest and significant of which are as follows:
- instability at the interfaces preventing the formation of profiled parts with a specified geometry,
- macroscopic separation of mixtures into components, including separation according to molecular weights, which markedly changes the flow dynamics,
- phase transitions, including transitions from isotropic into anisotropic state. In the limiting case, this ends in the formation of the LC phase,
- formation of regular 3D structures as bands differing in the component concentration and/or phase composition; a spatial and temporal instability of the boundaries of these structures cannot be ruled out.
Instability description in dynamic problems is based on the key physical mechanism that determines this phenomenon. As applied to polymer systems, the driving force of instability is elasticity.
The understanding of the specific nature of the rheological processes involved in the formation and multiplication of layers is necessary for the development of not only multi-extruder techniques, but also various types of 3D printing.
5. 3D printing
A high-tech area traditionally related to polymer materials is additive manufacturing. In this respect, this is a widely used present.132 While speculating about the future of additive technologies, we decided to settle on three very complex, but simultaneously the most in-demand systems, particularly, thermally stable polymers, silicones and hydrogels. Development of research along each of these lines was accompanied by rather extensive modification of objects to make them suitable for 3D printing, and simultaneously by fundamental scientific advances, which provide an optimistic outlook.
5.1. 3D printing of thermally stable polymers
The progress of the computer methods for the creation of 3D images enabled the development of new approaches to the physical implementation of virtual 3D models in metals, ceramics or polymers and promoted improvement of manufacturing processes. The key role among the approaches belongs to layer-by-layer synthesis: computer modelling of the product and manufacturing of the product using a special equipment and various 3D printing techniques.133 A promising technique is the selective laser sintering (SLS).This technique implies formation of a 3D product according to the layer-by-layer principle. A fusible powdered material is supplied to the matrix and locally heated by a laser beam; as a result, the particles are sintered and fuse together. This produces an intricately shaped item that could not be manufactured by conventional moulding. Another method, fused deposition modelling (FDM), is based on extrusion deposition of the material. This method implies drawing of a fusible polymer filament through a heated extruder. On passing through the extruder, the filament transforms into the viscous-flow state and, as it comes out of the nozzle, it is deposited on the build platform where the product is thus formed. After the deposition, the polymer is rapidly solidified.
A type of FDM printing is direct printing using granules, which skips the filament preparation stage. 3D printing using granules differs from the conventional FDM printing in the design of the extruder, which additionally contains a screw conveyor. The advantages of this technique include lower cost of printing compared to the conventional FDM printing and a wide range of applicable materials.134 A drawback of the method is that products obtained in this way have lower mechanical characteristics than those obtained by conventional techniques. This is due to the presence of defects and pores caused by the layer-by-layer deposition of the polymer, insufficient interlayer strength and pronounced shrinkage of the material during printing.135 There are two key approaches for improving the mechanical characteristics:
- application of high-tech high-strength heat resistant polymers;
- modification of the used polymers by adding various fillers.136,137
The raw materials currently used for SLS and FDM technologies are polyamides such as polyamide 12,138--140 polyamide 11,141,142 polyamide 6143 and polystyrene and polycarbonate.144,145 Among high-tech heat resistant polymers, a group of poly(aryl ether ketones) are used most often for SLS printing, in particular poly(ether ketone)146,147 and poly(ether ether ketone) (PEEK);148,149 FDM printing is performed using also some other thermally stable polymers, e.g., polyphenylene sulfide,150 polyphenylene sulfone 151 and amorphous polyimide Ultem.136,152 Analysis of recent publications has demonstrated that there is relatively little information on the use of SLS and FDM techniques for processing partly crystalline polyimides. The cause is that most known and commercially available aromatic PIs are difficult to process using the fusion method due to high viscosity of the melt and high flow point, which in some cases may approach the temperature of the onset of polymer thermal destruction. Processing of these PIs into an end product (fibre, film or coating) is mainly performed using solutions of polyamide acids in high-boiling amide solvents and includes subsequent thermal imidization. The search for chemical structures of fusible heat resistant polymers resulted in the development of a large group of partially crystalline PIs with melting points (close to the flow points) being far below the thermal destruction temperatures.153–156 These PIs may become promising raw materials for SLS and FDM processing.
Studies of partially crystalline polymers attract considerable attention of researchers.157–159 The most well-known partially crystalline PIs are those manufactured under the brand names LaRC-CPI,153 LaRC-CPI2160 (developed by NASA) and New-TPI (developed by Mitsui Toatsu Chemicals)161 and some other partially crystalline polyimides based on 1,3-bis(4-aminophenoxy)benzene and diphenyltetracarboxylic acid 3,3’,4,4’-dianhydride developed at the Virginia Polytechnic Institute.155 Most of these PIs can crystallize only in the presence of a solvent and cannot recrystallize after being transformed to the molten state.
A group of partially crystalline PIs has been developed at the Institute of Macromolecular Compounds, Russian Academy of Sciences, on the basis of 3-bis(3,4-dicarboxyphenoxy)benzene dianhydride (R), 3,3’,4,4’-diphenyl oxide tetracarboxylic dianhydride (ODPA) and 4,4’-bis(4’’-aminophenoxy)biphenyl (diamine, BAPB) and on the basis of 3,3’,4,4’-benzophenonetetracarboxylic dianhydride (BZP) and 3,3’-diaminobenzophenone (diamine, 3,3’-BZP).162,163 These PIs (below referred to as synthetic PIs) are capable of controlled crystallization and reversible melting and can be transformed to the viscous flow state on heating.
5.1.1. Preparation of fusible polyimide powders
Development of the synthesis of partially crystalline PIs as fusible powders is highly important, as this expands the scope of engineering applications of PIs. Polyimide powders are obtained by chemical and thermal imidization methods. The first stage of the synthesis is common to both methods and includes polycondensation of a dianhydride with a diamine in an amide solvent to give polyamide acid (PAA). The thermal or chemical159,164,165 imidization of PAA affords PI powders (Fig. 36a). The developed syntheses furnished fusible polyimide powders: R-BAPB, ODPA-BAPB and BZP-3,3’-BZP.162,163,166 The chemical structures of the resulting polymers163 are shown in Fig. 36b.
![[{"id":"NL_-DllVm_","type":"paragraph","data":{"text":" General scheme of the synthesis of partially crystalline PIs as powders, which includes chemical and thermal PAA imidization methods (<i>a</i>) and chemical structures of obtained polymers (<i>b</i>)."}}]](/storage/images/resized/zRqL8aqjZpWlIOFt724ITMqxGEJr6U62MO0B5LOY_xl.webp)
The molecular structures of the PI powders were confirmed by IR spectroscopy. The course of imidization was monitored by appearance of bands at 1780 and 1720 cm–1 (symmetric and antisymmetric C=O modes of the imide ring) and at 1380 and 740 cm–1. As an example, Fig. 37 shows the IR spectra of the R-BAPB polyimide powders obtained by thermal (curve 1) and chemical (curve 2) imidization. In both cases, the limiting degree of imidization was attained.159,164,168
![[{"id":"ViqJqiVYZG","type":"paragraph","data":{"text":" IR spectra of R-BAPB PI-powders, synthesized by thermal (<i>1</i>) and chemical imidization methods (<i>2</i>).<sup>167</sup> Published with permission from Wiley."}}]](/storage/images/resized/s6hea5OcymwrXW5Z2qCalpZoq0tIxB0iB3nT0G9E_xl.webp)
Apart from the considered method for the preparation of PI powders by imidization in a PAA solution, there are other processes, e.g., an emulsion process.169 This method is based on the preparation of a poly(ether imide) solution in dichloromethane and emulsification of the solution by adding surfactants. Finally, spherical poly(ether imide) particles with a size from 10 to 100 m are formed. Grinding of poly(ether imide) granules is also used to obtain the powders.170
5.1.2. Structure and properties of polyimide powders
It is known171 that particle size and shape and the bulk density are important for the use of powdered polymers as 3D printing materials, especially for the SLS technique. The properties of partially crystalline R-BAPB, ODPA-BAPB and BZP-3,3’-BZP powders are summarized in Table 1. It can be seen, in relation to R-BAPB, that the powders obtained by chemical imidization have a higher density and a narrower particle size distribution than the thermal imidization products. In other words, by varying the imidization method, it is possible to obtain powders differing in the particle size and bulk density.159
Depending on the chemical structure of R-BAPB, ODPA-BAPB and BZP-3,3'BZP polyimides, the resulting chemically imidized powders can also differ in the particle size and shape (6 to 50 m). Furthermore, the bulk density is sufficiently high (at least 0.3 g mL–1). The structure of PI powders obtained by chemical imidization is depicted in Fig. 38.
![[{"id":"c8dbtfpFhl","type":"paragraph","data":{"text":" SEM images of PI powders obtained by chemical imidization: (<i>a</i>) R-BAPB, (<i>b</i>) ODPA-BAPB, (<i>c</i>) BZP-3,3'BZP.<sup>159 </sup>Published with permission from Springer Nature."}}]](/storage/images/resized/EjRgcve2GZCxsDRGeVlb2zcNRTPaI2kQcjdyC334_xl.webp)
All of the prepared PI powders show endotherms in the DSC curves, which correspond to melting of their crystalline phase (Fig. 39). In the case of BZP-3,3’BZP and ODPA-BAPB powders, the curves clearly show two melting peaks at different temperatures; therefore, additional heat treatment can influence the thermal stability of the finished products.163,172–174
![[{"id":"2VaNysrtW0","type":"paragraph","data":{"text":"DSC curves of PI powders obtained by chemical imidization. (<i>1</i>) R-BAPB, (<i>2</i>) BZP-3,3'BZP, (<i>3</i>) ODPA-BAPB. The figure was created by the authors using published data.<sup>163,172,173</sup>"}}]](/storage/images/resized/D16aY44Y61TbnrqgPoFSclCgODqCs2QV1v50VtcU_xl.webp)
According to DSC and thermogravimetric analysis (TGA) data, the synthesized powders possess a high level of thermal stability and heat resistance (Table 2). It can be seen that among the presented polymers, the highest thermal stability is inherent in the ODPA-BAPB polyimide: the glass transition temperature of this PI is 245 °C, and the melting point of the crystalline phase is above 370 °C; therefore, processing of the polymer by traditional methods such as injection moulding may be difficult.
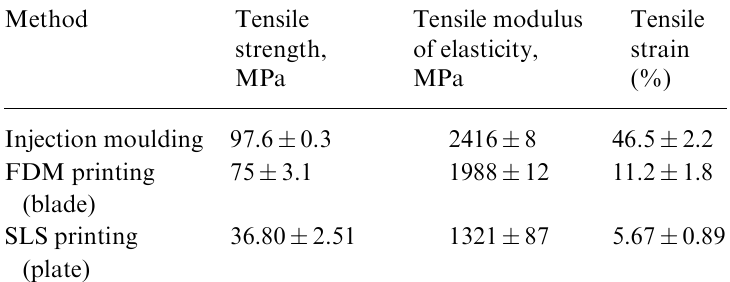
Apart from the above-noted thermal characteristics of the synthesized PI powders, the viscosity of melts is also highly important for the melt processing of polymer materials.159,175 The variation of the viscosity of melts of the PI powders with time is reflected in Fig. 40. For the R-BAPB and BZP-3,3’BZP systems (curves 1, 3), the viscosity of the melt changes little with time.174,176,177 In the case of ODPA-BAPB (curve 2), the viscosity sharply increases, which also complicates the injection moulding of this polymer. However, for the SLS technique in which fusion of powder particles takes fractions of a second, the value of viscosity at a definite laser power is more important than the stability of this value with time. For material processing by 3D printing (SLS and FDM techniques), the desired viscosity of the melt can be attained by varying the molecular weight of the polymer via a change in the molar ratio between the monomers. For example, PI powders that had a proper viscosity in the molten state were prepared by changing the stoichiometric ratio between the monomers (dianhydride:diamine) taken for the synthesis.172 As an example, 3D-printed samples were fabricated from the synthesized R-BAPB powders by SLS159 and FDM methods (Fig. 41).
![[{"id":"zN1hlYeBEY","type":"paragraph","data":{"text":"Time dependences of the viscosity of the melts of PI powders obtained by chemical imidization. (<i>1</i>) R-BAPB at 360 °C,<sup>176</sup> (<i>2</i>) ODPA-BAPB at 425 °C, (<i>3</i>) BZP-3,3'BZP at 330 °C.<sup>174</sup> The Figure was created by the authors using published data.<sup>174,176</sup>"}}]](/storage/images/resized/4oIv0nWtaWRVSvThc99McPZOHcOH5sCZUpoUuQWz_xl.webp)
![[{"id":"Q13h6Aum3-","type":"paragraph","data":{"text":"Photographs of the setup for SLS 3D printing of a chemically imidized R-BAPB powder (<i>a</i>), a three-dimensional sample formed as a plate (<i>b</i>) and a filament formed from this powder (<i>c</i>); FDM-printed blade (<i>d</i>).<sup>159,178</sup>"}}]](/storage/images/resized/WrOCu5PSqvrh1QmTw2hgpzNtdTxdOI1LHrHY0G3I_xl.webp)
Comparison of the mechanical properties of three-dimensional PI samples obtained by various methods (see Table 2) showed that the level of characteristics of samples obtained by FDM and SLS 3D printing is sufficient for manufacturing tough three-dimensional items. However, there is still unsolved problem of improving conditions of FDM and SLS processing in order to attain characteristics of the end products at the level attained by injection moulding. This especially refers to the strain behaviour, which determines the plastic properties of 3D-printed samples.
Preliminary experiments carried out for ODPA-BAPB polyimide powders with different molecular weights also demonstrated the possibility of SLS printing of 3D products.
5.2. 3D printing of silicone polymers
Silicones are highly important materials that have started to be widely used for polymer 3D printing. This is obviously related to unique properties of silicones such as very low glass transition temperature, high thermal and thermo-oxidative stability, high gas permeability, excellent dielectric characteristics, physiological inertness and biocompatibility and the possibility of easy functionalization.180--185 Owing to their unique properties, silicones are in high demand for soft robotics,18,186 manufacture of lithium batteries,187 anti-biofouling, anticorrosion, anti-icing188--190 and superhydrophobic coatings,191 biotechnology and medicine192,193 and other applications.194--197
The current state of 3D printing of silicones is covered in several review publications. Depending on the method of curing of siloxane polymers, the use of various 3D printing techniques to obtain various products, including special-purpose items, is considered. The most recent reviews on this subject appeared in 2018198 and 2020.199 A doctoral thesis200 addresses the silicone 3D printing by the thiol--ene addition mechanism to obtain preceramic materials. A review by Zhou et al.201 focuses on 3D techniques for manufacturing soft polymer materials in which silicones are in high demand as starting components. Analysis of 3D printing techniques using various polymers, including silicone elastomers, and evaluation of the properties of the resulting materials were reported by Herzberger et al.202 Some examples of silicone printing are given in a review203 dealing with the extrusion 3D printing. A short review by Luis et al.204 discusses silicone 3D printing techniques to fabricate items for medicine.
This Section briefly discusses the problems of production of silicone materials using additive manufacturing technologies in the context of chemical reactions used to form cross-linked polymers or obtain thermoplastics and evaluates the prospects for the development of these technologies taking account of the chemical processes underlying the 3D printing of silicones.
The following reactions that give cross-linked elastomers are most important as regards successful commercialization of the final products: hydrosilylation, which is usually conducted using platinum catalysts; condensation of silanol and alkoxy groups, which is most often catalyzed by tin; and radical cross-linking (Fig. 42).205
![[{"id":"LSqlvHXO-v","type":"paragraph","data":{"text":" Cross-linking of polysiloxanes containing vinyl and hydride functional groups by hydrosilylation reaction in the presence of a platinum catalyst (<i>a</i>); hydroxyl-terminated polydimethylsiloxanes with a tetrafunctional cross-linking agent, tetraalkoxysilane, in the presence of a tin-based catalyst (<i>b</i>); polydimethylsiloxane by a radical mechanism (<i>c</i>). Here and in other Figures, the wavy line indicates the polymer chain."}}]](/storage/images/resized/9jC4ee8fYQynPh7FJ6EalEBDq0iYXV26WCMAOogy_xl.webp)
The hydrosilylation reactions are widely used for the 3D printing of silicones, largely owing to the commercial availability of two-component kits (for example, Sylgard 184) containing a platinum catalyst in one of the components. This type of curing of polysiloxanes is compatible with 3D printing techniques such as direct ink writing (DIW)205--208 and inkjet printing.209,210 Using the DIW technique with silicones cured by hydrosilylation in the presence of platinum, porous polysiloxanes were printed for the first time.211 One more interesting technique that can make use of platinum-curable two-component silicone kits is freeform reversible embedding (FRE) technology: a hydrophobic silicone is cured in a support hydrogel based on a hydrophilic polymer.212,213 As a rule, after printing by these techniques, a long period of time at room temperature or several hours at 65--150 °C is required for complete curing of the end product. An original 3D printing technique based on two-photon three-dimensional microfabrication using photo-hydrosilylation reaction was proposed by Coenjarts and Ober.214 A similar process using UV radiation to activate the cross-linking via hydrosilylation reaction is discussed by Beach et al.215 One more method216 implies the formation of silicone microgranules that form a suspension with liquid PDMS and water. The resulting gel-like pulp can be extruded and then cured. This material can be printed both in air and in water. By embedded 3D printing217 using viscoelastic ink inside a silicone elastomer matrix, strain sensors were fabricated. This approach can be extended to obtaining high-tech products for wearable electronics, interfaces, soft robotics, etc. Luis et al.218 proposed one more original 3D printing technique involving commercial silicones cured by hydrosilylation. The authors designed their own 3D printer operating by the thermal extrusion method. As a result of optimization of 3D printing parameters and the mechanical properties of silicone, the authors were able to attain the shape and size accuracy of the product approaching those attained by moulding.
In a study by Porter et al.,219 the silicone precursor was extruded and thermally cured using an IR laser. In order to increase the IR absorption, the silicone was filled with a carbon black-based dye. This is one of the first studies demonstrating the influence of laser curing parameters in the near-IR range on the 3D printing of standard commercial high-viscosity silicone for medical applications. The same curing mechanism and the same silicone material were used220 for thermal curing of silicone; however, the authors used a 3D printer with a heated nozzle. Smith et al.221 considered 3D printing of the platinum-cured PDMS/PEEK composite. The material was extruded and then cured in a furnace at a temperature of up to 150 °C. This composite was proposed for printing products for biomedical applications.
The above examples demonstrate that hydrosilylation is one of the most demanded reactions for silicone 3D printing, which is largely due to the commercial availability of the starting materials and large amount of knowledge on this reaction concerning the synthesis of cross-linked silicone resins. An obvious benefit of this curing mechanism is the applicability of silicones with virtually any viscosity. A drawback is the necessity of the additional thermal curing stage for the printed product, which complicates and slows down the manufacturing process. In addition, from the standpoint of green chemistry principles, it is desirable to avoid the use of toxic compounds, for example, catalysts based on heavy metals.222
The UV-induced thiol-ene addition of the thiol SH group to the C=C double bond is considered more and more often as an alternative to hydrosilylation.223 This photoinduced reaction has been thoroughly studied for the formation of polymer networks. The thiol-ene addition reactions224 are characterized by high rates and high conversions, excellent solvent stability of the products, low susceptibility to oxygen inhibition and low volume shrinkage compared to these characteristics of acrylate and methacrylate systems (see below). The ready availability of vinyl-containing PDMS and polyfunctional mercaptans predetermined extensive use of 3D printing of silicones by the UV-initiated thiol-ene addition mechanism (Fig. 43).
![[{"id":"-dRHlyPO6B","type":"paragraph","data":{"text":"General scheme of the preparation of a cross-linked polymer based on vinyl-containing PDMS and trifunctional mercaptan by the UV-initiated thiol-ene addition reaction."}}]](/storage/images/resized/E5MiO3zExHBCYw9oCEnKj6yho2OMPuP4irCB8IQw_xl.webp)
The thiol-ene curing of polydimethylsiloxanes with vinyl groups is used in the DIW 3D printing,225,226 stereolithography (SLA)227--234 and digital light processing (DLP).235 Bryan et al.236 proposed an interesting 3D printing technique using commercial silicones, which are cured by the UV-initiated thiol-ene addition with a support organogel based on polystyrene block copolymers. This made it possible to fabricate micrometre-sized silicone items with a very high accuracy. It was shown237 that high-viscosity PDMS can be printed without the use of any support material via fast UV curing of compositions containing vinyl-terminated PDMS and PDMS with distributed mercaptopropyl groups reinforced with a mixture of fumed silica and MQ resins. Similar materials and a similar synthetic route (but without fillers) were used238 for the 3D printing of biocompatible silicone materials.
The abundance of publications devoted to 3D printing of silicone materials by the UV-initiated thiol-ene curing mechanism indicates that this approach obviously holds high promise. Materials for this technology are mainly commercially available. The processes using optical radiation have high speed and high accuracy. However, this approach has some disadvantages that affect its commercial promotion. The first one is the need to use a large amount of a photoinitiator, which impairs the properties of the products. The presence of the photoinitiator and a heteroatom such as sulfur in the end product may complicate secondary processing. Most often, 3D printing based on UV radiation requires expensive equipment and materials with a relatively low viscosity (<5000 Pas) for the fast formation of a thin layer with a smooth surface without air inclusions.239
Yet another important reaction widely used for curing of silicones is UV-initiated polymerization of acrylate groups (Fig. 44). Siloxanes containing acrylate groups are commercially available; however, original materials obtained by introducing acrylate groups into initial polysiloxanes are also used, and this naturally affects the properties of the final materials. The 3D printing of silicones containing acrylate groups is accomplished using various photopolymerization methods.240--243 Du et al.244 showed the possibility of DLP 3D printing of materials based on acrylate-terminated PDMS copolymers with thiourea. These copolymers have excellent elasticity and could potentially be used for the manufacture of medical items and for soft robotics.
![[{"id":"DqdEF3DaDr","type":"paragraph","data":{"text":"General scheme of UV radiation-induced curing of PDMS with acrylate functional groups."}}]](/storage/images/resized/Jowfz17EfAsvPNARnWrnJkmix1DCFl0NgAh12VKG_xl.webp)
The silicone 3D printing using UV-initiated polymerization of acrylate groups for curing has the same drawbacks as that using UV-initiated thiol-ene addition mechanism of curing: the presence and the adverse influence of a photoinitiator and the necessity to use expensive equipment.
Despite the existing drawbacks, some of the printing technologies described above are already utilized for the commercial production of silicone products. Apparently, one should expect further improvement of 3D printing techniques using the above curing reactions aimed at increasing the speed and accuracy of printing and eliminating the existing drawbacks.
The DIW printing technique was used to fabricate silicone elastomer products with a nano-sized silica filler as a modifier of rheological properties. As a result, silicones with various mechanical properties (e.g., superelastic materials stretchable to 2000%) become printable.245 The dynamic coordination bonds arising between the silanol groups of the filler and the polymer form a physical network, which breaks during extrusion, but is restored in the printed product, thus providing the desired viscosity.
An interesting work using an unusual method for cross-linking of silicones was performed by Brook s research group.246 It was shown that aqueous solutions of aliphatic aldehydes (glutaraldehyde, glyoxal or formaldehyde) rapidly react with siloxane polymers containing functional amino groups without catalysts. This curing composition can be used for 3D printing and for the development of adhesive and sealing formulations, as was demonstrated by the authors.
The 3D printing of the PDMS copolymer with pyrazole-urea moieties capable of forming dynamic (hydrogen) bonds was reported by Sun et al.247 A powder of this copolymer is applicable for SLS printing, and the end products based on this material are capable of self-healing.
In conclusion, we would like to note some achievements in the search for new effective methods of silicone cross-linking, which could also be applicable for the fabrication of 3D-printed silicone products. There is increasing number of publications on the introduction of azide and acetylene functional groups into polyorganosiloxanes and the post-polymerization modification by catalytic 1,3-dipolar addition.248--253 The possibility of cross-linking of functional PDMS by the non-catalyzed azide-alkyne cycloaddition mechanism was demonstrated.254 The copper-catalyzed 1,3-dipolar addition, which was first discovered as the Huisgen reaction255,256 and then, after discovery of copper(I) catalysis, was studied by Meldal's 257,258 and Sharpless' research groups 259-262 (Fig. 45), is one of the most often used click-reactions and has already proved to be efficient, in particular, for modification of polymers and preparation of materials based on them.257,263--265
![[{"id":"rINI_j2Q4k","type":"paragraph","data":{"text":" General scheme of the copper(I)-catalyzed azide-alkyne cycloaddition reaction."}}]](/storage/images/resized/vXk9BDGBWHYrZX61Q5Lr6OeVxMfojBFcMmbjWR1D_xl.webp)
Despite the absence of commercially available silicones for cross-linking via the azide-alkyne cycloaddition mechanism, this approach is quite promising for implementation into 3D printing technology. Note that apart from the obvious advantages inherent in an addition reaction requiring a cheap catalyst, the final cured structure contains a triazole moiety, which can improve the mechanical peformance of the end product.266
The Piers-Rubinsztajn reaction also holds high promise for silicone 3D-printing.267 This reactions proceeds as coupling between alkoxyl and silicon hydride groups catalyzed by B(C6F5)3 to give a siloxane bond and release an alkane. Reviews268--270 discuss the potential of this reaction for the generation of cross-linked silicone elastomers. Hydrosiloxanes can also be cured by α-diketones in the presence of a Lewis acid to give a Si–O–C bond.271 These materials are easily processed to give the starting monomers under mild conditions. The curing methods of hydrosiloxanes catalyzed by B(C6F5)3 are very convenient and environmentally benign; however, the evolution of gaseous products is a limiting factor hampering the implementation of these reactions in the 3D printing technology.
Thus, considering the global trend towards green technologies, it can be expected that the future development of silicone 3D in the future will also be related to the use of polyaddition reactions (in the ideal case, free from transition metal-based catalysts). Apparently, click-chemistry processes such as azide--alkyne cycloaddition, Diels-Alder reaction and other reactions would be used more and more often in 3D printing technology to obtain products with improved performance.
5.3. 3D printing of hydrogels
3D printing of polymer gels is a new area that has started to be developed in the last several years.201,272--328 Hydrogels are convenient materials to be used in 3D printing. They can serve to obtain soft products with a number of valuable properties: compatibility with biological tissues, high water content, porous structure that can accommodate various chemicals and biological objects, including living cells, etc. The gel products obtained by 3D printing can be used as adjustable valves,283,294 soft manipulators for robotics305,316,325 and personalized smart sensors that are glued to the skin to monitor the movement or concentration of various substances in the body and are capable of controlled release of required compounds (e.g., medications) in response to a change in their content.326,327 A new promising trend is 3D bioprinting,273,328 in which biocompatible molecules, biopolymers and even living cells are used as an ink. The major lines of research in this field are related to the design of artificial tissues and organs for the replacement of damaged parts in the human body.
Moreover, hydrogels are among the key objects for 4D printing. The concept of 4D printing in which the fourth dimension is time was first proposed by Tibbits in 2014.274 Since then, 4D printing has turned into a new, rapidly developing area, which attracts considerable research attention.275--282,316 The key feature of 4D printing is that the printed objects can interact with the environment.281 Under the action of external stimulus, they can change their shape in a pre-programmed way, thus performing certain functions. This is illustrated in Fig. 46 in relation to a soft actuator: it can be seen that at room temperature, the actuator can capture and trasport a vial, while at elevated temperature, the actuator tentacles are opened and the vial is released.
![[{"id":"_Z_azLOpxe","type":"paragraph","data":{"text":"Schematic comparison of 3D and 4D printing (<i>a</i>)<sup>280</sup> and photographs of a clamping unit fabricated by 4D printing (<i>b</i>), which grasps a vial in air at room temperature (<i>1</i>) and releases it into water at 70 °C (<i>2</i>).<sup>316</sup> Figure <i>a</i> is published with permission from Springer Nature, Fig. <i>b</i> is published with permission from Wiley."}}]](/storage/images/resized/3uayB0L6hNX92iljYh75BL0t4z4KQOpsZf4Yawhx_xl.webp)
The active gel materials for 4D printing include, in particular, shape memory hydrogels and hydrogels responsive to external stimuli.277,329--331 For 4D printing, hydrogels are usually combined with non-swelling polymers. When the printed product is placed into a solvent, hydrogel swells, while the second (inactive) polymer component does not absorb the solvent. This causes a change in the shape of the product. Instead of one active material, the ink for 4D printing may contain several active materials,288 which provides a finer control over the shape of the product and complicates the product architecture.
Hydrogels can be used in most of 3D printing techniques including laser (stereolithography, two-photon polymerization), extrusion and jet printing.328 Hydrogels are best suited for the layer-by-layer extrusion procedure, which is simple, inexpensive and provides high quality of printing. For successful high resolution extrusion printing, the materials should meet the following key requirements:
- polymer solutions (printing ink) should be characterized by shear thinning, i.e., they should markedly decrease their viscosity (which is necessary for solutions to be easily forced through the extruder and to be rapidly mixed), but nevertheless possess high elastic modulus and viscosity at rest (or yield stress), in order to retain the shape after extrusion up to the instant of fixation;289
- fixation that is performed by cross-linking of polymer chains into a network should proceed rapidly in order to provide a high speed of printing.290
The main environmentally friendly candidates for ink in the extrusion 3D printing are hydrogels based on biodegradable polysaccharides — alginate, hyaluronic acid, chitosan, methylcellulose, agarose and carrageenan.291,292 Quite a few studies devoted to the 3D printing of gels based on polysaccharide networks have been reported to date.201,285 However, printing of hydrogels with a single polymer network has considerable limitations. First, the viscoelastic properties of the solutions after extrusion are insufficient, which leads to spreading of the structure and decline of the 3D printing resolution. Second, the printed products have low mechanical strength and wear resistance. To overcome these drawbacks, it is necessary to increase the mechanical strength of the gels. This can be done by a number of methods such as the fabrication of double networks or nanocomposite gels.
The double networks293,295,332,333 are composed of two interpenetrating polymer networks with substantially different properties: the first network is usually formed by a tightly cross-linked and rigid polyelectrolyte, while the second one consists of a loosely cross-linked and flexible polymer (Fig. 47). Mechanical deformation leads to cleavage of so-called sacrificial bonds in the first network, which is accompanied by a pronounced energy dissipation in a rather large volume and prevents propagation of rupture and destruction of the whole gel. As this takes place, the second network undergoes elastic deformation and maintains the gel elasticity.334 The use of double networks results in a marked increase in the tensile strength, energy of rupture and strain at break.
![[{"id":"bZxFgu0Wbc","type":"paragraph","data":{"text":"Schematic image of a double polymer network.<sup>333</sup> Published with permission from the Royal Society of Chemistry."}}]](/storage/images/resized/CuIiqTmJXEtoxubKQmGDH7DciFG3cG21zEcceCnA_xl.webp)
There are several major approaches to the 3D printing of gel products based on double networks: those using a mixture of two monomers, a polymer and a monomer or two polymers as the ink. The monomer forms a network via polymerization, while the polymer network is formed upon cross-linking of the existing macromolecules. Network formation from the monomer usually takes place in the printed product, most often via photopolymerization. For 3D printing with a mixture of two monomers, a thickening agent should be added to the ink in order to produce a sufficiently high viscosity needed for printing.296 Polymer networks are formed, most often, using photo-cross-linkable macromolecules, ionic cross-links or thermoreversibly cross-linked polymers.328 The reversibility of cross-links between the macromolecules is often an important feature: cross-links are destroyed as the solution flows (e.g., during the extrusion printing), but subsequently they are rapidly recovered, with the shape of the printed object being retained.
The 3D printing of gel products based on a double network in which one component is a polysaccharide and the other one is a synthetic polymer has been reported (Fig. 48). In this case, the ink is based on a polymer mixture or a mixture of a polysaccharide and a monomer. For example, 3D printing was applied for double networks of κ-carrageenan and covalently cross-linked polyacrylamide.297 The ink contained κ-carrageenan, which formed the first network on cooling the ink below 40-45 °C at the outlet of the extruder, and acrylamide monomer and a photoinitiator, which were photopolymerized to generate the second network after formation of the product. The resulting hydrogels combined high mechanical strength and the ability to change electrical resistance upon deformation, which made them promising for the design of strain sensors. The preparation of biocompatible double networks of sodium alginate cross-linked by Ca2+ ions and photo-cross-linkable polyethylene glycol diacrylate (PEGDA) has been reported.298 For extrusion 3D printing, clay (laponite) nanoparticles were added to the ink, which provided a sufficiently high viscosity at rest and a reversible decrease in the ink viscosity during the flow. The PEGDA network was formed by photo-cross-linking after printing of the material. The double networks of PEGDA and gellan gum with inserted living cells were fabricated in a similar way.291 Gellan gum was cross-linked during printing on cooling from 37 to 25 °C. The capability for 3D printing was provided by the pronounced reversible decrease in the ink viscosity during flow, which is characteristic of gellan gum solutions, and the subsequent fixation of the structure by fast cross-linking of PEGDA. 3D-printed items were obtained from gels based on the double networks of chitosan cross-linked using sodium citrate and polyvinyl alcohol cross-linked woth PVA microcrystals; the networks were formed via freezing--thawing cycles.300 These networks have very high tensile strength (12.7 MPa) and elongation of break (300%).
![[{"id":"_S2YUSeGgT","type":"paragraph","data":{"text":"Formation of a double network of sodium alginate and acrylamide and acrylic acid (PAAm-PAA) copolymer and photographs of 3D-printed products. Each network is cross-linked by Fe<sup>3+</sup> ions.<sup>335</sup> Published with permission from the American Chemical Society."}}]](/storage/images/resized/gCxdQ52bbkELJIvYR0JmzYafjEkorRpGQs6CSTuc_xl.webp)
Recent publications describe the 3D printing of double networks based on two natural polymers. In this case, mixtures of polymers are used as ink; each polymer is cross-linked to form a network during or after the printing. These hydrogels are mainly considered as cell growth media to produce tissues and organs of a specified architecture, which is set by printing.279,301 For example 3D printed items were obtained using gelatin or agar gels cross-linked on cooling, a sodium alginate gel cross-linked with Ca2+ ions after 3D printing,302,303 and photo-cross-linked gelatin methacrylate and sodium alginate gel.304 The possibility of cell growth in the printed material was demonstrated. For example, smooth muscle cells were introduced into gelatin and alginate gels during the 3D printing for the subsequent formation of an artificial heart valve.302 Thus, 3D printing of gel items based on double networks is a new promising trend in the preparation of mechanically strong hydrogels.
Another approach for increasing the strength of polymer gels is to fill them with nanoparticles, which should also meet high requirements to environmental safety. Out of the whole diversity of nanofillers for traditional nanocomposite hydrogels, only only a few materials are considered as promising environmentally benign fillers for polysaccharide hydrogels meant for 3D printing, particularly, silica,306 graphene oxide321,336 and clay287,307--311,337 nanoparticles, cellulose crystals and nanofibrils,275,312--315,317,318 etc.
Filler particles can have various sizes and shapes and various surface functional groups, which can react with the polymer matrix. The interaction of nanoparticles with the polymer that forms the network structure may substantially improve the mechanical properties of the gel, since the nanoparticles act as an additional cross-linking agent for polymer chains. However, it is preferable to use nanoparticles that interact with polymer chains via non-covalent contacts or dynamic covalent bonds. In this situation, the lability of bonds between the system components would ensure a considerable decrease in the viscosity in the shear flow (during extrusion) and fast restoration of the mechanical properties of the gel at rest. Silica nanoparticles the surface of which is readily modified by functional groups are often used as fillers that actively interact with the matrix.306,338 For example, it was shown306 that the addition of spherical silica nanoparticles surface-modified with amino groups that form dynamic covalent bonds with the aldehyde groups of modified alginate results in a severalfold increase in the hydrogel yield point. A similar effect is exerted by spherical gold nanoparticles, which act as multifunctional dynamic cross-links for thiol-modified hyaluronic acid.320
Plate-like nanoparticles are promising fillers. In particular, plate-like graphene oxide nanoparticles with surface functional (carboxyl and hydroxyl) groups interacting with the polymer matrix provide higher mechanical performance of nanocomposites based on sodium alginate321,323 and chitosan322 hydrogels compared to the initial matrix.321--323,336 Plate-like clay (particularly laponite) nanoparticles are also widely used to improve the mechanical properties of polysaccharide hydrogels.287,307 Of particular interest is the case in which laponite particles present in a relatively high concentration in the nanocomposite system form a card house type structure due to the electrostatic attraction between the positively charged flat edges of nanoplates and the negatively charged surface. This structure shows an elastic (gel-like) response to minor mechanical impacts and a yield stress, and has shear thinning under high shear stress. After the stress is relieved, the structure is restored. Owing to these properties, laponite in a solution of polymer chains forms a gel-like structure suitable for extrusion 3D printing even in the absence of a cross-linking agent.307 After the end of 3D printing, the printed product is placed into a cross-linking agent solution to fix the structure and to increase the strength of the product.
Halloysite clay nanotubes proved to be good as a filler for reinforcement of hydrogels for 3D printing.308--311 High ratio between the particle thickness (50-100 nm) and length (100-1000 nm) and high particle strength provide a sharp enhancement of the mechanical properties of the polymer matrix, which was shown in relation to the chitosan and alginate hydrogel309,310 and gellan gum.311 Owing to the presence of the inner cavity (20-40 nm), nanotubes increase the matrix density to a minor extent and open up the possibility of encapsulating various components inside the cavity.
A recent trend is the use of fillers representing nanocrystals and nanofibrils based on cellulose and cellulose ethers (methylcellulose, carboxymethylcellulose), which have high mechanical properties and low thermal expansion coefficients and are biocompatible and structurally similar to the polysaccharide matrix. Therefore, they can be used to modify hydrogels for 3D printing.275,312,313,318 It has been shown that in sodium alginate hydrogels, cellulose nanocrystals are embedded into the matrix by forming hydrogen bonds. This makes the gels more stable, increases the elasticity of the matrix by an order of magnitude,313 and increases the resolution of 3D printing and the strength of the product.312 Note that as certain concentrations are attained, the nanocrystals and nanofibrils of cellulose and its derivatives can themselves form hydrogels with rheological properties appropriate for 3D printing;312,314,315 in combination with other polysaccharides, they are used to produce ink.318,319
Nanoparticles not only strengthen hydrogels, but can also endow them with special properties. For example, silver nanoparticles embedded into a sodium alginate hydrogel reinforced by cellulose nanocrystals endow the hydrogel with antimicrobial properties, with high mechanical characteristics being retained.324 Aluminosilicate nanotubes can encapsulate some additional components; a specific feature of this nanocontainer is prolonged release of the components, for example, antibacterial agents that protect the natural polysaccharide matrix from degradation.339 Carbon nanotubes endow the gels with conductivity.319
Thus, the specific feature of modern nanocomposite polysaccharide hydrogels applicable as ink for extrusion 3D printing is that the nanofillers not only introduce high mechanical properties, but also act as additional network structures alternative to cross-links or as nanocontainers. Moreover, they can additionally impart the gels with antimicrobial properties and conductivity. As a result, polysaccharide hydrogels acquire new functional properties and rheological characteristics required for extrusion 3D printing.
Meanwhile, it should be noted that studies on the use of double and nanocomposite networks in 3D printing technology are at their infancy, and the fundamental trends of the variation of the mechanical properties and structures of the gels in all stages of 3D printing providing fast printing of strong materials with a high spatial resolution are not ultimately clear as yet.
6. Universal methodology of processing and modification of polymer films, fibres and plastics
This section addresses the environmentally friendly and solvent-free methodology of processing and post-modification of films, fibres and plastics based on amorphous and semicrystalline polymers using two scientific and technological strategies, namely, orientational drawing in physically active liquid environments (PALEs) via the mechanism of environmental crazing and the use of labile and reactive precursors.
6.1. Environmental crazing
Environmental crazing is known to be a universal and scientifically justified strategy of structural-mechanical modification of polymers and offers ample opportunities for the development of innovative mesoporous and nanocomposite polymeric materials with unique functional task-oriented properties addressing the challenges of the 21st century.340--342
From the fundamental scientific viewpoint, environmental crazing constitutes a special mode of plastic deformation of polymers, which provides the formation of a highly ordered mesoporous structure with high volume porosity (up to 70%) and pore dimensions ranging from 1 to 50 nm.340,341,343--348 At the present time, two different modes of environmental crazing are identified: classical crazing for amorphous glassy polymers [PET, polyvinyl chloride (PC), PS] and intercrystallite crazing for semicrystalline polymers [PE, PP, polytetrafluoroethylene (PTFE), PLA, etc.],349--355 and controlled regimes of this process have been defined, thus allowing preparation of the materials with desired characteristics.
Classical crazing involves the nucleation and growth of separated crazes with their specific fibrillar-porous structure as discrete regions of localized plastic deformation.340,351,356 As the tensile strain increases, crazing proceeds via a gradual development of a highly developed surface (100-200 g cm–3), overall porosity increases to 60-70%, and nanoscale pores with dimensions below 15 nm are formed.343,357 In this case, PALEs, on the one hand, promote the development of crazing due to the Rehbinder phenomenon as adsorption-induced strength reduction,358 and, on the other hand, they stabilize the as-formed fibrillar-porous structure via adsorption interaction with the highly developed surface of nanosized fibrils.340,341,351 Mesoporous polymeric systems with high surface area and excess free surface energy are characterized by a high thermodynamic instability and, as typical colloidal systems, they tend to reduce the excess level of surface energy via collapse of fibrillar-porous structure provided by coagulation up to a complete monolithization of the system.340,351
The mechanism of intercrystallite environmental crazing is characteristic of semicrystallite polymers (PE, PP, PTFE, etc.). Necessary conditions for intercrystallite crazing are the high degree of crystallinity of polymers (>30%) and rubbery amorphous phase, thus providing an efficient separation of crystalline lamellae along the direction of the applied stress as well as cavitation and fibrillation via the Rayleigh-Taylor meniscus instability mechanism.349,359--363 Upon intercrystallite crazing, the development of porosity and specific fibrillar-porous structure takes place exclusively within the amorphous phase, whereas the crystalline lamellae serve as a load-bearing framework.349,353 In the case of intercrystallite crazing, volume porosity can achieve 70%, and pore dimensions are in the nanoscale range (below 15 nm).344,353,364 The role of the PALE is concerned with plasticization of the amorphous phase and development of highly porous structure as well as with stabilization of a highly developed fibrillar-porous structure of polymers.349 Noteworthy is that the above mesoporous polymeric materials are known to experience a low-temperature spontaneous strain recovery: upon unloading, the polymer fully restores its initial dimensions (strain recovery is 80-98%), 344,353,365,366 thus opening the avenue for the preparation of new mechanoresponsive hard-elastic materials based on a broad range of polymers.366 The efficient regime for stabilization of the highly porous structure of semicrystalline polymers is provided by annealing under isometric conditions at temperatures below the melting point of the polymer.367
Preparation of meso- and nanoporous polymeric materials is the challenging task of modern materials science.368--370 In this respect, key advantages of the scientific strategy of environmental crazing are concerned with the fact that this process can be applied for a broad range of amorphous and semicrystalline polymers, including high-tonnage polymers such as PE, PP, PTFE, PET, PVC, polyamides as films, fibres, ribbons, hollow fibres. Nowadays, from the viewpoint of ecological issues and environmental safety, of special interest are the switching from traditional synthetic to biodegradable polymers (PLA, PCL, etc.) and the development of related mesoporous and nanocomposite materials with task-oriented properties.352
Crazing can be implemented in a continuous technological regime using traditional technological equipment for orientational drawing of polymers with its minor modification or using the spot crazing protocol due to local straining of the polymer.371,372
Principal implications of environmental crazing as the technology for the preparation of mesoporous polymeric materials are primarily related to the use of high-cost, toxic and inflammable organic solvents as the PALEs.340,349 Nowadays, the transition to ecologically friendly regimes of environmental crazing dictates the search for new alternative PALEs with high efficiency comparable to the efficiency of traditional organic solvents. A new approach for the solution of this challenge is based on the use of oil-in-water emulsions of two thermodynamically incompatible liquids with high water content >95%.373 By their performance with respect to their potency in promoting crazing as estimated from the stress reduction and level of nanoscale porosity, emulsions containing micronic oil phase droplets (higher aliphatic alcohols, hydrocarbons, etc.) appear to be identical to pure organic solvents. This effect is provided by a selective adsorption of oil droplets by hydrophobic polymers (PE, PP, PET, PTFE) and the formation of a specific oil envelope on their surface, which serves as a pure organic solvent and allows an efficient crazing in the environmentally friendly mode.373
Hence, environmental crazing allows preparation of mesoporous polymeric materials with high mechanical characteristics, reduced density, unique heat-insulating properties, high MWTR and gas permeability, shape stability at elevated temperatures, materials with nanostructured surface and enhanced hydrophobicity, improved capillary characteristics, etc. (Fig. 49a).341,344,357,366,373,374 The as-prepared mesoporous polymeric materials can be efficiently used as breathable gas-permeable materials,374 sorbents, membranes, heat-insulating materials,341 water-proof materials,344,375 porous matrixes and substrates, packaging materials, sound-proof materials, mechanoresponsive materials,366,376 porous materials for gas storage, high-tech textile materials for comfort clothing, etc. (Fig. 49b).341,344,351,357,366
![[{"id":"7r0oiJ4ZRP","type":"paragraph","data":{"text":"Diagrams illustrating the performance of mesoporous polymeric materials after their mechanical structural modification via environmental crazing (<i>a</i>) and applied aspects of their practical use (<i>b</i>)."}}]](/storage/images/resized/hijfTFTCG6DMCzfETUszSSRvnt0mWemLcsRCZfub_xl.webp)
The strategy of environmental crazing offers new opportunities for the development of a new generation of nanocomposite polymeric materials with valuable functional properties, which are based on common high-tonnage polymers via controlled introduction of one or several target additives into mesoporous polymeric matrixes when the additives are uniformly distributed within the polymer volume in the highly dispersed nanoscale state. By now, efficient methods for the introduction of a broad range of target additives into polymers have been worked out and scientifically justified: forced impregnation and cyclic forced impregnation upon deformation in the PALE containing dissolved target additives or wet passive impregnation when the as-formed mesoporous matrix is impregnated with a solution or a melt of the target additive. Principal benefits of this approach are related to the possibilities of the incorporation of thermodynamically incompatible additives into polymers and their uniform distribution in the nanoscale state within the polymeric matrix, the controlled content of the additive in polymers, introduction of precursors into polymeric matrixes and subsequent in situ reactions (for example, reduction of metal salts to zero-valence state or polymerization of monomers).
Introduction of functional additives in the nanoscale state into polymers allows preparation of high-performance nanocomposite materials of new generation based on traditional high-tonnage and biodegradable polymers with task-oriented functional properties: flame-retardant materials with low content of antipyrenes,377 metal-polymer nanocomposites with metal nanoparticles,341,366,378,379 antibacterial materials against Gram-negative and Gram-positive bacteria, viruses and fungi,341,366,380 packaging materials, materials for medical applications, drug delivery systems, optochemical sensors with high spectral response,371,372,381--385 ‘smart’ materials as elements of ‘electronic nose’, electrically conductive polymeric materials, materials with controlled hydrophilic-lipophilic balance,373,375,386 gas separation materials, membranes, selective sorbents, polymer-polymer nanocomposites.387,388
6.2. Reactive precursors for processing and modification of plastics
The traditional route for the optimization of the performance of polymeric composites via introduction of additives and modifying agents is based on the centennial empirical experience accumulated by polymer engineering. However, as the knowledge on the behaviour of polymer melts progressed, more sensible and theoretically justified solutions came into play. This section addresses the examples of these meaningful solutions and analysis of the challenges related to processing of polymers in terms of classical approaches.
Processing and modification of bulk plastics can be achieved by the use of reactive precursors which, in general case, are polymer-monomer-initiator systems (PMI systems), among which the following types are identified:389–393
- I — polymerizates with incomplete conversion prepared by bulk polymerization;
- II — polymer solutions in their own and/or 'foreign' monomer;
- III — polymerization products in the reactor with conversion <100%.
Let us demonstrate how the above precursors work by considering the simplest Type I PMI system,394--397 namely, PMMA polymerizate with a conversion of 88% (or, in other words, containing 12% of the residual monomer and the initiator).
For comparison, let us consider the thermomechanical curves of bulk PMMA (Fig. 50a, curve 1) and bulk PMMA containing 12% of hydogenated monomer (curve 2).
![[{"id":"UD3nN7T6AF","type":"paragraph","data":{"text":"Thermomechanical curves of bulk PMMA (<i>1</i>), bulk PMMA plasticized by 12% of hydrogenated monomer (<i>2</i>) and PMMA polymerizate with conversion of 88% (<i>3</i>)<sup>395</sup> (<i>a</i>) and characteristic temperatures <i>versus</i> conversion of the polymerizate (<i>b</i>). Summarized data fromworks.<sup>389,390</sup>"}}]](/storage/images/resized/Lx1FnOE0FYUfLyI1IhtSBLtraB2wqZLvJcHUanXx_xl.webp)
The plasticizing effect of hydrogenated monomer is manifested as the depression of glass transition temperature by Tg(1) – Tg(2) and a comparable decrease in the flow temperature but the profile of the corresponding thermomechanical curve remains unchanged. In the reactive polymerizate with conversion of 88%, the residual monomer (concentration 12%) provides the similar plasticizing effect when the glass transition temperature of the polymerizate is Tg(2) (curve 3).
In this case, within the temperature range T1–Tmax, deformability of the polymerizate dramatically increases and achieves its maximum at Tmax when the system experiences the transition to the rubbery state. At the same time, the contribution from polymerization of the residual monomer to this process provides the vitrification of the system, and deformability of the material decreases down to the level of the initial bulk polymer within the temperature range Tmax–T2. Fig. 50b shows the correlation of the temperatures corresponding to the above temperature ranges and conversion of the polymerizate. As is seen in Fig. 50, for PMMA polymerizates with conversion of 80-95%, variations in the residual monomer concentrations have no effect on the characteristic temperatures Tmax and T2, whereas T1 linearly decreases. Let us mention that changes in Tmax are provided by variations in the concentration of the initiator.
To highlight the reactivity of the polymerizate with conversion of 88%, we consider the following data. Fig. 51 shows the temperature dependences of yield stress (σy.) for bulk PMMA (curve 1) and PMMA plasticized by 12% of hydrogenated monomer (curve 2). Plasticization of PMMA by hydrogenated monomer leads to the depression of glass transition temperature and to the concomitant shift of the linear temperature dependence of yield stress σy to lower temperatures (Fig. 51, transition from curve 1 to curve 2). At temperatures above 50 °C, 12% of residual monomer of the polymerizate with conversion of 88% provides the plasticization of the polymer similar to that of hydrogenated monomer. Within the temperature range of 50-80 °C, polymerization of residual monomer is accompanied by an increase in the yield stress of the material (Fig. 51, curve 3), and this value achieves the level of bulk PMMA (Fig. 51, curve 1). This evidence serves as the experimental basis for the development of low-cost technologies for processing and modification of plastics in the rubbery state, first of all, via blow molding and orientational drawing.
![[{"id":"odYH67HQo5","type":"paragraph","data":{"text":"Temperature dependences of yield stress for PMMA (<i>1</i>), PMMA plasticized by 12% of hydrogenated monomer (<i>2</i>) and polymerizate with conversion of 88% (<i>3</i>). Summarized data from works.<sup>394,395</sup>"}}]](/storage/images/resized/AfmYLA7vP0SvOgtQg9RwFrwCtKt4Eh32nHpdvUZ8_xl.webp)
Within the temperature range T1–Tmax the plasticizing effect of the residual monomer provides the low-temperature processing of the polymerizate, and subsequent polymerization of the monomer component of the system within the Tmax–T2 range ensures the thermal stabilization of the article and supramolecular structure, which was formed upon processing and modification. Combination of the above effects is coined as 'a temporary plasticization'. The position and width of the above temperature ranges can be varied by changing the concentrations of the monomer and the initiator. Let us mention that the technological procedure of polymerization of 'a temporary plasticizing agent' can easily solve the problem of exhaustion of the residual monomer.
Optimized processing of plastics in the flow state, mainly by extrusion and injection molding requires the reduced flow temperature of the precursor. As was mentioned above, the plasticizing effect of the monomer component in the PMI systems offers the solution for the above challenge. However, in this case, the use of far more complicated protocols and formulations is required when two (or more) initiators with different operating temperatures are involved, and the maximum temperature should be higher than the processing temperature. Heat setting is achieved by polymerization of the residual monomer via heating of the finished article after the processing stage. One of the options of this process is concerned with the use of the initiators based on actual initiators and photoinitiators. The latters provide photopolymerization of the residual monomer at lower temperatures.
Noteworthy is that, upon processing of plastics both in rubbery and flow states, crosslinking agents, for example, bifunctional monomers may be included in the formulations for the improvement of the performance and heat setting of the finished material.
Hence, the use of PMI precursors makes it possible to markedly reduce the polymer processing temperature (by 100-120°C) and, correspondingly, energy consumption. The reduced temperature also allows the processing of high-molecular-mass polymeric products, which is beyond the capabilities of traditional methods because, for such materials, the processing temperature often exceeds the temperature of thermal degradation.
Let us emphasize the key role of the PMI systems (Types II and III) in the preparation of plastics modified by nanoparticles.398 Controlled viscosity of the PMI precursor provides the sedimentation stability of the dispersions of nanoparticles in the polymerization system and stabilization of the fine dispersion of modifying agents in the finished article. We would also like to stress the benefits of the above types of the PMI systems as polymer reinforcing agents for the fabrication of a new class of protective structures with orthogonal arrangement of glass ceramic blocks.399
In addition to the above technological benefits, the use of the PMI systems offers a broader scope of polymeric articles based on a limited range of initial monomers. For example, as was shown for a small range of (meth)acrylate and vinyl monomers,389--393 this appraoch allows the production of high-impact and form-stable orientation-toughened glasses; polymeric glasses with a molecular weight of up to 107; layered polymer-polymer composites without physical interfaces between the layers; gradient composite materials; crosslinked and high-molecular-weight (>500000) extrusion glasses.
Summing up, we emphasize once again that the advanced methodology of processing and subsequent modification of polymer films, fibres and plastics offers ample opportunities for the development of environmentally friendly and low-cost production of a new generation of nanocomposite materials with valuable functional and engineering properties based on a limited scope of high-tonnage polymers and monomers.
Application of reactive PMI precursors allows an efficient solution to address the challenges of the production of plastics primarily related to high resource and energy costs and large amounts of waste. In addition to evident technological and economic benefits, this approach offers ample opportunities for the production of a wide range of plastics with a set of properties, which is almost unattainable when traditional technological regimes are used, as well as (nano)composites and polymeric adhesives.
7. Multiscale simulation concept in the studies of structure and properties of polymer materials
It was noted above (see Section 3.1) that computer simulation methods are in high demand for multi-layer extrusion processes used for the production of film materials. Of course, the significance of these methods is not exhausted by this special case. In this Section, we made an attempt to briefly characterize the most widely used methods for computer simulation of polymers and materials based on them.
Polymer nanocomposites (PNCs) are multiphase systems that are obtained by insertion of a nano-sized filler based on various nanoparticles (NPs) into the polymer matrix. The physicochemical properties of PNCs are strongly interrelated with the structural characteristics of the transient layer, that is, the interface between the polymer matrix and NPs. If the interfacial adhesion between the matrix and NPs is weak, the nanoparticles tend to agglomerate and behave as nanodefects, thus causing local stresses, which result in a considerable decrease in the strength of the material. The interaction between NPs and the polymer matrix can be controlled by modifying the NP surface. The development of new PNCs requires elaboration of versatile theoretical approaches to predicting the applicability of particular polymers for the manufacture of materials with desired properties. However, this is associated with considerable difficulties due to the necessity of taking into account a large number of parameters to characterize the properties of the polymer, nanoparticles and surface modification agent and the conditions of material use.
Owing to the sharp increase in the speed of numerical calculations and the rapid progress of computer hardware taking place in the recent decades, computer simulation turned into a separate method for accumulation of knowledge, existing on equal terms with theoretical and experimental studies. In some cases, computer models may replace laborious laboratory experiments requiring expensive facilities, reagents and methods for characterization of the products. The results derived from computations may fully reproduce the data that can be gained by EPR, NMR and IR spectroscopy, X-ray diffraction analysis, etc. This makes computer simulation an indispensable tool for preliminary prediction of the physicochemical properties of new materials, which is important for choosing the optimal strategies for their synthesis.
Currently, several levels of computer simulation are distinguished (Fig. 52) in terms of the space and time scales in which they are used. The most strict level of the study of molecular systems is provided by methods of quantum mechanics (QM). They can be applied to perform ab initio calculations in various approximations for solving the Schrödinger equation. This means that generation of a model requires only information on the compound structure. For example, for modelling of macromolecules, it is sufficient to know only the structural formulae of monomer units and the structure of polymer chain. QM methods provide accurate data about the energy characteristics, electronic spectra and optical, magnetic, electrostatic and other properties of molecular systems. The most balanced calculations (in terms of accuracy vs. time trade off) are implemented using the density functional theory (DFT). Depending on the used approximation, the level of QM simulations makes it possible to study molecular systems comprising several thousands of atoms in the time ranges from 10 to 100 ps.400
![[{"id":"0_KW4Ly-vP","type":"paragraph","data":{"text":"Hierarchy of computer simulation methods. The arrows indicate the transfer of information between the conventionally identified levels implemented in the framework of computer models. For abbreviations, see the text."}}]](/storage/images/resized/6nQWClUrvEGh43iTyOk6FM8nPgjaQb6rJzwSM2en_xl.webp)
The next simulation level combines the classical all-atom molecular mechanics, which includes various sorts of atomistic molecular dynamics (MD) and Monte Carlo (MC) calculations based on the laws of analytical mechanics and statistical physics. The so-called valence force field (VFF), the combination of phenomenological potentials composed of a set of analytical functions, is used as the interaction potential. In essence, VFF contains information about different types of intra- and intermolecular interactions. The intramolecular interactions include the deformation potentials of covalent bonds, bond angles and torsion angles (between the planes formed by groups of atoms) and other forces determining the structure of molecules. The intermolecular interactions usually include the van der Waals and electrostatic forces. The VFF parameters are selected by a cumbersome fitting procedure in such a way as to reproduce the chosen combination of thermodynamic properties of a set of known chemical compounds. Examples of VFF are the polymer consistent force field (PCFF)401 and condensed-phase optimized molecular potentials for atomistic simulation studies (COMPASS).402
Since the only external parameters used in all-atom MD and MC are VFF parameters, these methods can be regarded as macroscopic. Using these methods, it is possible to study both well-known and new polymers. In the latter case, if no adequate parameterization for the considered chemical compound in the chosen VFF procedure is available, it can be generated by QM calculations. The software implementation of the all-atom MD and MC methods can be used to study the properties of the models of materials composed of several millions of atoms over time ranges of a few microseconds.403
Currently, using MD, it is possible to predict rather accurately thermophysical, mechanical and other properties of PNCs404–406 and also to track visually (to an accuracy of a single atom) the structural evolution of a system. Therefore, MD can be regarded as a fairly accurate methodology for polymer characterization. For example, Komarov et al.406 reported the use of a large-scale MD simulation to study a self-organizing nanocomposite based on silica nanoparticles and thermoplastic silicone-urea block copolymer containing flexible non-polar PDMS blocks and rigid polar bis(4-isocyanatocyclohexyl)methane block. It was shown that urea blocks tend to be aligned parallel to one another owing to hydrogen bonding between them. The microphase separation, giving rise to a bi-continuous phase of flexible and rigid blocks, was observed in the simulation cell. Thus, the matrix itself can be considered as a self-organizing polymer-polymer nanocomposite. The urea blocks form the percolating reinforcing phase, which accounts for relatively high tensile strength inherent in these copolymers. After the introduction of a minor mass fraction of silica nanoparticles into the polymer, the thermophysical and mechanical properties of the system are corrected, with the reinforcing network of rigid urea blocks being retained.
The next simulation level (see Fig. 52) makes use of coarse-grained models based on Monte Carlo method and Langevin, Brownian and dissipative particle dynamics (DPD).407 The characteristic sample sizes of the model systems are 20 to 500 nm, while accessible time ranges reach in some cases 500 μs. Due to the use of intermediate scale, this simulation level is called mesoscopic, or mesoscale. In the models of molecular systems, exact chemical structure details are discarded, while collective variables are used instead. All molecular structures are replaced by an equivalent ensemble of centres of force — mesoscopic (or coarse-grained) particles. The interaction between the particles is assessed using coarse-grained types of VFF, which are restored by statistical processing of the evolution of representative prototypes of molecular systems at the atomistic level of simulation.
The mesoscale level models also use the self-consistent mean field method and related approaches, in particular the dynamic density functional theory (DDFT).408,409 Mention should also be made of the methods based on the integral equation theory. An example is the polymer reference interaction site model (PRISM).410 Coarse-grained simulation is usually employed to study relatively slow structurization processes in polymer systems, which are difficult to simulate in the framework of molecular mechanics.411–413
Consideration of PNCs at the macroscopic level (see Fig. 52) is performed using hydrodynamics, continuum mechanics and finite-element (FEM) methods. Model parameterization at this level is based on information on the physical properties calculated at the atomistic and mesoscale simulation levels. Examples are the elastic moduli of the polymer matrix and NPs, the structure and properties of the polymer/NP interface (they are determined at the atomistic level) and NP distribution in space (this is derived from mesoscale smulation data). Additionally, the equations of state, kinetic relations for a quantitative description of plasticity and fracture of the material, etc., can be used.414,415 The upper level in the hierarchy of the computer simulation methods depicted in Fig. 52 is the level of engineering and technological process design. At this level, the final products and methods for their production are designed. This is done using specific aproaches that are beyond the scope of this review.
Molecular mechanics, mesoscale simulation and finite element method require both preliminary parameterization of the force constants that determine the interaction between structural elements of the models and information on the filler distribution in the material bulk, which can be obtained experimentally. However, this is not always possible in the case of new materials. The problem of parameterization of computer models can be solved in terms of integrated calculation schemes based on the concept called multiscale simulation (MS).416–418
Currently, the MS concept is regarded as the most consistent approach to study of molecular systems requiring no external (beyond the calculation scheme) parameterization. Using this approach, it is possible design computer models proceeding from physical conditions of PNC application and PNC chemical structure and composition. The application of MS concept can be described by the followng chart
The MS strategy is based on identification of the most significant structural scales and relaxation processes in PNC followed by classification of their types. This makes it possible to establish the key degrees of freedom determining the properties of the system, with each space--time scale being described by a particular simulation method. The transition between the identified levels is accomplished by their successive parameterization according to the bottom-up principle. For example, quantum mechanical calculations are used to determine interaction parameters of both single atoms and groups of atoms, which is necessary for MD simulation. In the framework of molecular mechanics, the interaction between mesoscale particles can be parameterized and the properties of the polymer matrix, nanoparticles and the interface between them can be calculated. The latter is important for parameterization of the finite element method. In turn, mesoscale methods are used to explore the distribution of NPs in a polymer matrix, which is also important for the model parameterization in the finite element method (see Fig. 52).
Thus, the MS approach is an interrelated (via information exchange) hierarchical sequence of models aimed at elucidation of the structure→property relationships. The chosen space and time scales specify the level of detail for each of the used models. The opposite is also true, namely, the specified level of detailing dictates the choice of the simulation method regarding both the duration of processes and the scale of structures. In the case of PNCs, most implementations of the MS methods are reduced to the construction of two-scale schemes that implement transitions between all-atom and mesoscale levels.419–422
In some cases, simplifications based on the data on the key parameters of nanosystems can be used in the studies of PNC properties. One of such examples is considered below. It is known that many types of NPs are relatively large, and this complicates the direct use of atomistic MD. Meanwhile, it was shown that the physical properties of ultrathin polymer films sandwiched between two solid surfaces are similar to the properties of PNCs in which NPs have a high degree of ordering.423 This analogy is due to the fact that physical properties of hybrid systems are largely determined by the properties of the polymer/NP interface. This suggests that the formation of some physical properties in filled polymers and ultrathin polymer films on solid substrates is determined by the same mechanism. The computer simulation of PNCs can be simplified in this way. This model was employed424 to study the effect of the mass fraction of SiO2 NPs on the thermophysical properties of the polyimide matrix. The calculations were carried out using two methods: atomistic MD and QM. The model formed in this way was a two-scale calculation scheme. The inorganic filler was modelled by an infinite substrate (through periodic boundary conditions) based on the β-cristobalite (quartz polymorph) unit cell, which was multiplied along main crystallographic directions. The partial charges needed to calculate the electrostatic interactions were parameterized by QM calculations. Using the model they developed, the authors demonstrated a highly nonlinear variation of the temperature properties of the nanocomposite depending on the mass fraction of the filler. After the threshold content of SiO2 (20 mass%) was exceeded, the coefficient of linear expansion of the material sharply decrease, because of increasing contribution of the polymer/SiO2 boundary region in which the surface forces strongly influence the polymer chain dynamics, which is reflected in the properties of the whole system.
This simulation protocol is very convenient for studying both the polymer/NP interface structure and the properties of ultrathin films of organic liquids or melts supported on solid surfaces (or sandwiched between two solid surfaces).424–429 These models provide an answer to the question of how a change in the external conditions and the type of surface would affect the morphology of the near-surface layer and the properties of the whole material.
Examples of a more complex hierarchical simulation were described in a number of publications.430–437 All-atom simulations of samples of highly cross-linked polymer matrices were performed and multi-stage calculation schemes were proposed. The simulation of polymer matrices based on epoxide426,431 and phthalonitrile432,433 resins consists of the following sequence of stages: generation of a coarse-grain monomer model→mesoscale level simulation of the polymerization→reverse mapping→study of the properties of samples of the resulting material. The simulation of highly cross-linked polystyrene matrices and reactions of their synthesis in dichloroethane included the stages of quantum mechanical calculations of partial charges on atoms of polystyrene and dichloroethane, all-atom calculations of polystyrene solutions in dichloroethane at different polymer concentrations, forward and reverse data mapping and study of the macroscopic properties (Fig. 53).
![[{"id":"wm2ZJJEpqO","type":"paragraph","data":{"text":"Scheme of the multiscale simulation of the synthesis of highly cross-linked polystytrene."}}]](/storage/images/resized/rpYQEDMnhoukxkAcZugVEx1OfxhFdusU9udwQWgF_xl.webp)
Polystyrene cross-linking in dichloroethane was modelled in two ways. First, multiscale simulation in which the all-atom model of linear polystyrene macromolecules dissolved in dichloroethane was converted to a mesoscopic coarse-grained model, the model cross-linking was performed and then reverse mapping to the all-atom structure was done.434--436 Second, the reactive force field (ReaxFF) method was used: the reaction was conducted in a system including all atoms of polystyrene, cross-linking agent and catalyst.437 According to both calculation methods, highly cross-linked polystyrene is an inhomogeneous structure in which solvent-filled cavities coexist with areas with a high polymer concentration connected by bridges. This inhomogeneity is a consequence of the non-uniform distribution of cross-links both in the bulk and along the chain: some benzene rings have several cross-links, while other ones are completely free.
The calculated parameters, including specific and local sample densities, specific surface area, elastic modulus, pore size distribution and the maximum pore size, were in quantitative and qualitative agreement with experimental data.
It should be emphasized that a central idea underlying the procedure of construction of cross-linked polymer matrices related to the notion of mesoscale chemistry is described in a number of publications.426,431,432,438,439 According to this concept, the polymer matrix is generated by modelling of the chemical reaction using coarse-grained particles. The use of the mesoscale level is caused by the following factors. Chemical reactions can be modelled directly, for example, using QM methods440 or atomistic MD with the ReaxFF potential;441 however, in the case of large model samples of polymer materials sufficient for obtaining adequate physical characteristics (elastic modulus, coefficient of thermal expansion, glass transition temperature, etc.), this method requires very large computing resources. This is due to the fact that the kinetics of chemical reactions of curing of, e.g., epoxide resins is determined by the rates of monomer diffusion, and the reaction is sharply retarded as the polymer conversion increases.442 A study of epoxide matrices based on bisphenol A diglycidyl ether and diethyltoluenediamine (DGEBA-DETDA) monomers showed that for obtaining correct local structure of epoxy resin, the simulation cells should be as large as possible. For the DGEBA-DETDA matrix, the simulation cell volume should be at least ≈ (150 Å)3 (Ref. 414). Most publications ignore this circumstance. As a rule, the chosen size of MD-simulated systems is too small (for saving computing resources).443 In our opinion, simulation of the polymerization at the mesoscopic level is often the only possible way to model materials based on cross-linked polymer matrices, including nanocomposites.
One more example of using MS protocols444--447 is concerned with PLA, one of the most popular biodegradable polymers. The MS protocol includes all-atom calculations, forward and reverse mapping (FIg. 54a) and temperature-dependent renormalization of coarse-grained potentials; finally, the results of atomistic simulation of PLA melts were reproduced with high accuracy over a broad temperature range (FIg. 54b) and the effect of mechanical stretching on the structure and properties of polylactide was studied using coarse-grained calculations. Furthermore, good quantitative agreement was attained with considerable (by a factor of several tens) increase in the calculation speed.
![[{"id":"8YjFVheXvg","type":"paragraph","data":{"text":" Mapping of the polylactic acid structure (<i>a</i>) and temperature dependences of the specific volume of oligolactide determined by all-atom simulation (red line), coarse-grained simulation (yellow line) and simulation after the temperature-dependent (blue line) renormalization (<i>b</i>).<sup>446</sup> (<i>1</i>) atomistic, (<i>2</i>) coarse grained γ<sub>nb</sub>=0, γ<sub>b</sub>=0, (<i>3</i>) coarse-grained γ<sub>nb</sub>=-0.25, γ<sub>b</sub> =0.5. γ<sub>nb</sub> and γ<sub>b</sub> are the empirical temperature-dependent renormalization coefficients for non-bonding and bonding interactions, respectively. Reproduced with permission from Elsevier."}}]](/storage/images/resized/BApVwKAkauiBH1xLXzDwZqBUmsJwnfctD2tBSI6t_xl.webp)
The local structure of the PLA molecule depending on its isomeric composition can be taken into account in terms of a model in which quantum mechanical calculations of partial charges are applied to ordered helical conformations.447 This model provides structural and thermophysical characteristics for PLA containing different percentages of L- and D-isomers.
In conclusion, we would like to emphasize that this brief review does not cover all of the modern approaches used to predict the properties of polymer materials. Nevertheless, the presented examples demonstrate the enormous potential of computer simulation. At the same time, advanced calculation schemes suitable for modelling and investigation of PNCs and requiring no additional parameterization are implemented only using the concept of multiscale simulation. The success of application of this concept relies on the design of a sequence of interrelated models (based on identification of the most important degrees of freedom of the molecular systems) and gaining the required information for implementing each level of the calculation scheme. One more important issue is analysis of the experimental data, which often prompts the strategy of designing the calculation scheme.
8. Biodegradable polymeric and composite materials based on plant raw materials
8.1. Biodegradable polymers with improved properties as a promising alternative to polyolefins in packaging production
The concept Polymers for the Future implies the environmental friendliness and safety of the materials at all life cycle stages - from renewable sources of raw materials to controlled degradation to environmentally friendly products. Natural and synthetic biodegradable polymers are perceived as promising materials of the future because, after their use, they degrade down to carbon dioxide, water, and biomass under the action of environmental factors and microorganisms.448 Due to biocompatibility and biodegradability, these materials have long been successfully used in medicine and pharmaceutics, in the production of sutures, implants, scaffolds, targeted drug delivery release systems and prolong drug, etc.449,450
In the past two decades, due to the pressing problem of environmental pollution from plastic waste, biodegradable polymers have attracted the attention of manufacturers of packaging materials, diverse disposable products and materials for 3D printing.451,452 Polyolefin-based packaging materials are the main source of environmental pollution.
Additionally, a severe environmental problem is concerned with the accumulation of the so-called microplastics, which are the particles of high-tonnage polymers with dimensions ranging from several microns to several millimetres, and their source is the gradual breakdown of polymeric products (clothes, cosmetics, car tires) and partially biodegradable materials consisting of polyolefins and special catalysts or degradable fillers in landfills.453 In contrast to traditional polymeric waste, which can be collected and utilized, spreading of microplastics is uncontrolled and they contaminate groundwater, rivers and seas, and eventually can be traced in human and animal bodies.454 Therefore, the problem of contamination by polymer waste is pressing not only from the viewpoint of ecological issues but its solution also is crucial for human health control and protection.
Switching to biodegradable polymers for the production of both packaging materials and some other articles, including car spare parts, can mitigate the accumulation of polymer waste. Biodegradable polymers, like traditional polymers, should be collected and properly disposed, for example, by industrial composting or recycling. Main benefits of biodegradable polymers are concerned with their short-term degradation into harmless products when released to the natural environment. Noteworthy is that the above polymers can hardly be considered as fully environmentally safe if their biodegradability alone is taken into account. The source of raw materials and environmental hazard at the stage of production should also be accounted for.
According to the European Bioplastics e.V., the global annual production of biodegradable polymers was 1.23 million tons in 2020, and by 2025, the growth up to 1.8 million tons is planned.* More than a half of the global consumption of biodegradable polymers accounts for the packaging industry, whereas consumer products and agriculture take second and third places, respectively. The main triggers stimulating the use of biodegradable polymers are concerned with various restrictions and tax measures. The development of this particular area is limited by relatively high cost of biodegradable materials and their certain characteristics which are inferior to those of polyolefins.
To find a broader use, biodegradable materials should be more competitive in price and properties as compared with high-tonnage polymers, should retain the desired operation performance and undergo degradation within the scheduled time period. The development of sustainable 'green' materials, which can replace large-tonnage polymers in many areas, presents the challenging task for scientific community all over the world.
Biodegradable polymers are classified into natural polymers polymers and synthetic polymers, produced either by microorganisms or by chemical synthesis (Fig. 55). Synthetic polymers are produced from both natural and synthetic monomers (based on oil). Such polymers [for example, polybutylene adipate terephthalate (PBAT), poly(ε-caprolactone)] can hardly be considered as fully environmentally friendly because the source of their raw materials is not renewable. Currently, the most common biodegradable polymers are PLA and various starch-based blends; their share in the global market of biodegradable polymers is approximately equal (32% each), and the annual production volume of each is 400000 tons.** Less share belongs to polymers based on petroleum raw materials such as PBAT (23%), PBS (7%) and PHA synthesized by microorganisms (3%). Therefore, natural polymers (primarily, starch and its blends) are the most advantageous candidates for high-tonnage biodegradable polymers due to their availability and excellent biodegradability, this also concerns PLA due to its good physicomechanical characteristics; however, all the above polymers also suffer from certain drawbacks (see below). In order to develop the materials with improved properties, diverse approaches have been worked out.
![[{"id":"DvrhzXRFTQ","type":"paragraph","data":{"text":"Classification of biodegradable polymers (summarized from works<sup>465</sup> and F.Baghi, A.Gharsallaoui, E.Dumas, S.Ghnimi. <i>Advance ments in Biodegradable Active Films for Food Packaging: Effects of Nano/Microcapsule Incorporation</i>. Foods, <b>11</b>, 760 (2022)."}}]](/storage/images/resized/FpVY6qXuMJb7CO9mAuKfLY3yxqOKdCPRnIThd9LZ_xl.webp)
Starch is known to be the most common and accessible among natural polymers. Starch presents a mixture of two polysaccharides: amylose (10-20%) and amylopectin (80-90%). The source of starch is the renewable plant raw materials such as cereals, corn, potatoes, etc. Pure starch is an unstable brittle material and its processing is difficult due to the crystalline phase of branched amylopectin. Preparation of thermoplastic starch-based materials, pristine starch should be modified by plasticizers (glycerol, sorbitol) under heating and shear stresses. On the industrial scale, processing is performed using an extruder. Depending on the mode of modification, the elastic modulus and strength of the resultant amorphous thermoplastic starch are equal to 0.1-1 GPa and 20 MPa, respectively. After the modification, even though thermoplastic starch becomes processable, it can hardly be used for packaging production because of its high hydrophilicity. At the next stage, starch is blended with polyesters or vinyl-alcohol based polymers; also, various mineral additives are introduced.455 The most commonly used are the commercial starch-based biodegradable materials produced under the brand name 'Mater-Bi', which are mixtures of the thermoplastic starch with one of the following polymers: PBAT, poly(ε-caprolactone) or acetylcellulose. Hydrophilicity of the starch-based materials can be reduced and their barrier properties can be improved by the deposition of UV-crosslinked coatings based on the modified soybean oil. As a result of this modification, water permeability of the material decreases by a factor of 10.456 Alternative modification methods have also been developed, and some of them can be implemented on the industrial scale.457
Among all natural polymers, starch is credited as the most easily processable polymer but the feasibility of other polysaccharides (mostly, cellulose derivatives) is also being investigated.458 For example, recently, a new approach for the preparation of easily processable bioplastic from sawdust has been proposed,459 and the advantages of this approach are related to the facts that no costly extraction of cellulose and lignin is required, and the finished material is prepared as finely dispersed cellulose micro- and nanofibrils in the lignin matrix. As a result, the materials possessing this structure are characterized by high strength (128 MPa) and excellent water resistance. Due to their poor processability, polysaccharides are mostly considered as fillers in biodegradable composite materials. Natural composites (wood, crustacean exoskeletons, bones) are constructed in the same manner.460 The most promising functional fillers are nanocrystalline cellulose,458 chitin461 and chitosan.462,463 The introduction of the above fillers allows the development of biodegradable composites with improved performance (thermal, physicomechanical, barrier and antibacterial properties).
A promising representative of the family of synthetic biodegradable polymers is PLA which is an amorphous or semicrystalline polymer with an elastic modulus up to 2.5-3.5 GPa and a strength of 60 MPa. Under industrial composting, degradation of PLA via hydrolysis and enzymatic degradation is completed within few weeks. Being released into the environment, the degradation rate is controlled by environmental conditions (temperature, humidity, presence of microorganisms); usually the degradation period does not exceed 5 years. Degradability and the degradation time of PLA can be controlled in a wide range by varying the ratio of L- and D-lactic acid units in the polymer chain.464,465 Initially, PLA was exclusively used for the production of biodegradable implants for surgery, regenerative medicine, traumatology, orthopedics, etc. However, since 2000, the production of PLA was greatly increased, and its cost has plummeted by hundreds of times and almost approached the cost of polyolefins, thus offering the opportunity to use PLA for packaging materials, various consumer goods and even for the fabrication of automotive parts.466 For the synthesis of PLA, the source of raw material is starch, from which lactic acid is obtained; then, its cyclic dimer, lactide, is synthesized. Commercial PLA grades are copolymers of poly(L-lactide-co-D-lactide) with low content (2-10%) of D-lactide units, which provide an easier processing due to the reduced melting point. Different grades of PLA are characterized by different viscosity optimal for a particular mode of processing. On the industrial scale, high-molecular-weight PLA is synthesized in a melt by ring-opening polymerization of lactide in the presence of various catalysts. Traditional catalysts are tin compounds; however, after degradation of the material, they are retained in the environment; hence, the search for new effective catalysts based on lighter metals, for example, zinc467 or zirconium, is invited.468 Alternative areas of research address the elimination of drawbacks inherent in the PLA-based materials, which limits the expansion of their applications for the development of packaging materials and household goods. Principal disadvantages of PLA are their brittleness, low impact toughness, softening temperature <100 °C, poor barrier properties and high cost as well as the slow-rate degradation in certain natural environments.
Noteworthy is that, over the long history of their application in medicine, numerous methods have been developed for the modification and controlled degradation of lactide-based polymers, which proved to be successful in the elimination of nearly all of the above shortcomings. For instance, brittleness of the material can be reduced by the incorporation of only 5-10% of more flexible units of ε-caprolactone into the molecular structure of PLA at the stage of polymerization.469 Copolymerization of lactide and more hydrophilic glycolide allows the adjustable control over the degradation time ranging from a few months to several years. However, the potential of these approaches for producting packaging materials is limited as the production cost markedly increases. Basic approaches to the preparation of PLA with improved properties for packaging involve the modification of the supramolecular structure, blending with other polymers and introduction of various functional fillers, including the preparation of nanocomposites.
As was shown in the work,470 the main cause of embrittlement of PLA at room temperature is the rapid aging and crystallization leading to the formation of micronic spherulites. The above undesirable phenomena can be eliminated by fast deformation of the material upon heating, which changes the structure of the entanglement network. Structural modification of PLA via environmental crazing also provides an efficient tools for reducting the brittleness.471 Impact toughness of PLA can be improved by blending with various biodegradable polymers such as polycaprolactone, polyurethanes, polysaccharides, copolymers of glycidyl methacrylate with ethylene and butylene, etc.472,473
Other shortcomings of PLA such as the low degradation rate in seawater can also be eliminated by the preparation of polymer blends.448 When PLA is blended with more hydrophilic polymers (in particular, starch), water uptake increases, thus accelerating the degradation.474 Moreover, the cost of the finished products can be reduced due to the availability of starch. However, it should be noted that other characteristics of polymer blends or composites are compromised (for example, strength may be reduced due to poor adhesion between a hydrophobic polymer and a hydrophilic filler). Therefore, the search for efficient compatibilizers and various functional fillers is important for the improvement of the physicomechanical properties of final materials. The glass transition temperature of poly(L-lactide) is 60-65 °C; hence, at ≈70 °C, the PLA-based articles are softened and deformed, which means that this polymer can hardly be applicable for the production of articles, which are used at elevated temperatures. This challenge can be addressed by preparing the material based on the blend of poly(L-lactide) and poly(D-lactide) when co-crystallization provides the formation of a stereocomplex with a different crystal lattice. Such materials are thermally stable up to 150 °C.475,476 The nanocomposite materials based on PLA and organo-modified montmorillonite show thermal stability up to 112 °C.477 Moreover, due to the layered structure of the filler, such composites are characterized by a reduced (up to four times) oxygen permeability. Hence, the preparation of various composites based on PLA, available natural polymers and functional additives seems to be promising. The production of biodegradable polymers can be developed on the basis of complexes for deep processing of grain, among the products of which starch and various sugar syrups occupy a significant share.
* According to the data of European Bioplastics e.V. [European Bioplastics nova-Institute (2022); https//www.european-bioplastics.org/market] (accessible 21.12.2022).
** According to the data of European Bioplastics e.V. [European Bioplastics nova-Institute (2022); https//www.european-bioplastics.org/market] (accessible 21.12.2022).
8.2. Application of polylactide in coatings of controlled release fertilizers
Numerous studies of scientists in different countries are targeted to the improvement of the methods to achieve better technical and economic indicators of the biosynthesis of lactic acid, synthesis of PLA and preparation of the PLA-based composites. The final goal is to reduce PLA as a raw material and increase the scope of its application. In particular, PLA has great prospects in agriculture for the production of mulching films, seedling pots, controlled release fertilizers (nutrient mixtures), etc. This approach will significantly mitigate the environmental pollution by polymer waste (including microplastics) generated by the use of the polyolefin-based materials.478 The search of scientific and patent literature on the use of PLA as coatings for fertilizers shows that the number of works in this area significantly increased over the past 5 years.
The basic idea is that the PLA-based capsule provides a gradual release of one or several fertilizers in accordance with the growing season of plants. Of special interest is the fact that the PLA degradation products are not only environmentally friendly but also promote the flora conservation as they boost the concentration of chlorophyll and sprout formation rate, increase the digestibility of nutrients necessary for plant digestibility and protect plants from toxic impact of mineral salts at high concentrations. The technology of controlled nutrient release offers an efficient approach for plants, which provides a marked reduction in wastage and gradual delivery of nutrients to agricultural crops. Various additives are used as nutrient compounds: nitrogen-containing (urea, guanidine, hexamethylenetetramine, glycine, alanine),479--482 salts containing Cl, S, Na, K, Mg, Ca, Fe, Mn, Zn, Cu, Se, e.g., Na(NH4)2PO4, NaH2PO4, Na2SO4, (NH4)2SO4, NH4NO3, NaNO3, MgSO4, KH2PO4, CaO, FeSO4, NH4Cl, MnSO4, MnCl2, MnHPO4, ZnSO4, ZnO, CuSO4, K2SeO4, Na2SeO4, commercial fertilizers, for example, 'Nitrolusal',* boron compounds483--485 (see**), etc. as individual compounds or their mixtures. The final composition of the fertilizer depends on the type of plants for which a given fertilizer is intended, conditions of their use and type of soils.
Both inorganic (sulfur and clay) and organic coatings [PSF, PE and poly(acrylic acid-co-acrylamide)] as well as biopolymers are used to provide a prolonged release of micronutrients.486 The basic property of such coatings is their vapour permeability. In the work,487 polymer coatings were classified according to their water vapour permeability. For synthetic polymers, the gas permeability coefficient ranges from 70 to 3000 cm2 s–1 Pa–1; for unmodified polysaccharides, from 4000 to 12000 cm2 s–1 Pa–1.
Controlled release of fertilizers to soil is provided by the formation of the coating on the surface of granulated nutrient composites. Release of the nutrient components, on the one hand, should be retarded in order to protect the seedling from over-abundance and the fruiting plant from the deficiency of elements and, on the other hand, it should be fast enough to supply the plant with nutrients within the growing season.484 The optimal period for the release of target components should be 30-120 days. The studies have shown that, in order to provide a slow release of nutrients within 30 days, the thickness of the PLA shell of the granule should be 200-600 μm (in some cases, up to 2 mm), thus markedly increasing the cost of the final controlled release fertilizers product. The cost of the PLA-based coatings can be reduced and their performance can be improved by the preparation of the PLA-based composites with natural polymers or by the use of multilayered coatings.479,483,488--490 Polymers used in agriculture are referred to as agro-polymers. Agropolymers and additives include cellulose, starch, lignin, alginates, soy protein, chitin, chitosan, xylan, wax, clays, etc. In most cases, agropolymers are waste products of related industries such as wheat straw, coffee cake, biochar, peat, etc.
In addition to the methods for deposition of coatings, alternative approaches are used for the development of controlled release fertilizers, for example, micro(nano)cross-linked gels with incorporated nutrients491 or nanocomposites prepared by the extrusion of polymer blends with fertilizers.492 An active ingredient is incorporated into the polymer by adsorption, dispersion of an active ingredient in the polymer composite or by encapsulation; and the release of target components is provided by desorption, diffusion or breakdown of the polymer matrix (Fig. 56).
![[{"id":"mmy3Hom3Hi","type":"paragraph","data":{"text":"Scheme illustrating the mechanisms behind the release of active compounds from the matrix of the polymer composite."}}]](/storage/images/resized/cwE4wwdAQSclz7vc0Ng7MojVvgMEoNNCzveLMf2J_xl.webp)
The use of controlled release fertilizers and stabilized fertilizers is an indicator of good agricultural practice. Such fertilizers are in the greatest demand in agricultural holdings for the cutivation of ornamental plants as well as indoors and outdoors fruit and vegetable crops, and for the introduction of innovative farming systems, for example, no-till technology when large amounts of fertilizers should be applied in one hit. Improved methods for the preparation and use of 'smart' fertilizers make a significant contribution to the sustainable development of agriculture.
* See M.Oliveira, C.Mota, A.S.Abreu, J.M.Nobrega. J. Polym. Eng., 35 (4), 401 (2015).
** L.Xie, M.Liu, B.Ni, X.Zhang, Y.Wang. Chem. Eng. J., 167, 342 (2011).
9. Modification of biodegradable materials based on polysaccharides for the control of the surface properties and biodegradation performance
At the present time, the traditional field of research addressing control over hydrophilic or hydrophobic surface properties of the materials is reaching a new level.493--500 The contact (or wetting) angle serves as a quantitative measure of these properties (>90° for hydrophobic surfaces and ≤90° for hydrophilic surfaces). Of special interest are the limit states of wettability — superhydrophilicity (contact angle <15°) and superhydrophobicity (contact angle >150°), which provide a complete wettability or non-wettability, respectively.493,501--503
Tailored modification of the surface layers of the materials based on natural polymers offers new properties while retaining the performance of the material in whole.504--507 For example, in-depth research addresses the materials based on cellulose and chitosan as fibres, films, aerogels, hydrogels, hollow tubes, etc., which are characterized by biocompatibility and biodegradability after their modification by the compounds which control the lyophilic properties and degradation rate.508--517
9.1. Factors controlling bioadegradation of polymers
As was mentioned above, in contrast to common polymers, biodegradable materials can be degraded by bacteria and fungi in environmental conditions.518--520 Biodegradable polymers usually degrade in soil or in water within 6-9 months.521 Key parameters that control the resistance of polymers to biodegradation are the following: molecular weight, supramolecular structure (degree of crystallinity), the presence of functional groups and imperfect units.522,523 Oligomers serve as a source of carbon for microorganisms, whereas polymers with higher molecular weight are resistant to bacteria and fungi.524 Semicrystalline and crystalline polymers experience no swelling or limited swelling in water, thus hindering the penetration of enzymes from microorganisms into the polymer matrix and increasing the resistance of crystalline polymers to biodegradation.525--527
Surfaces can be functionalized by grafting of the compounds that interact with the reactive sites of polysaccaharides. Surface modification is generally performed using low-molecular-weight compounds containing highly reactive groups. Hydrophilicity can be improved by the treatment with acetic anhydride or acyl chloride in the presence of urea,528,529 as well as by grafting of hydrophilic copolymers based on glycidyl methacrylate with the subsequent formation of chelate complexes.530 Due to improved hydrophilicity, biodegradation of the as-treated materials is accelerated in the presence of moisture due to the presence of enzymes and ions, which control the kinetics of degradation and the rate of active reproduction of microorganisms in this environment.
The surface hydrophobization of the cellulose- and chitosan-based materials is traditionally used for the improvement in their performance for the production of paper, textiles, construction materials, etc. To this end, two principal methods of surface modification are used:
- adsorption of various compounds on the surface of cellulose-containing substrates;531--535
- electrostatic interaction and complexation of protonated amino groups of chitosan with negatively charged polyanions or low-molecular-weight compounds, for example, fluorine-containing surfactants.536
Among low-molecular-weight reagents, the most known are chlorosilanes,531,537 carbonyl compounds,538--541 acid halides542 and haloalkanes (C10-C18).543 However, materials modified with often toxic low-molecular-weight reagents do not show sufficiently stable hydrophobic or superhydrophobic properties in a humid environment. From this viewpoint, chemical modification of chitosan, cellulose and related materials with synthetic polymers seems to be more promising,544--546 as it provides the preparation of new materials with improved properties, in particular, with high strength, thermal stability, adsorption, antimicrobial and antioxidant properties.
9.2. Properties of cellulose and chitosan and approaches to their processing
Cellulose is known to be the most common plant raw material, which is used for the preparation of composite materials, textiles, drug delivery systems, personal care products, etc. This low-price, biodegradable and renewable resource has also attracted the attention of researchers due to its physical and chemical properties. However, cellulose lacks some universal performance properties inherent in synthetic polymers, and this aspect explains the ever increasing interest in its modification, preparation of cellulose-based composites and blends.
Cellulose is characterized by high reactivity due to the presence of three hydroxyl groups in each unit. From this standpoint, cellulose can be considered as a polymer polyatomic alcohol with elementary units containing one primary (at the sixth carbon atom) and two secondary (at the second and third carbon atoms) hydroxyl groups. These functional groups are responsible for the reactivity of cellulose. Cellulose is a fibre-forming compound with a high molecular weight, and its modification is often performed under heterogeneous conditions or, in other words, treatment with reagents takes place on the surface of the materials.547--549 In contrast to some synthetic polymers, cellulose cannot be melt processed because its melting point is close to the degradation temperature. Processing of cellulose through solutions is hindered because the use of traditional solvents is impossible due to the presence of inter- and intramolecular hydrogen bonds. To tackle this problem, special dissolving systems have been developed,550--553 which allow the production of cellulose materials and products as well as their modification. The examples of such systems are the following: water-alkaline solutions,554--556 melts of hydrated inorganic salts,557--561 concentrated aqueous solutions of inorganic acids,550,562--564 supercritical water.565--567 Dissolution of cellulose without chemical modification is a laborious process due to the rigid long main chain with strong inter- and intramolecular hydrogen bonds.551,568 Therefore, cellulose should be activated to make it more solvent-accessible. In the late 1970s, the studies were launched to develop new approaches to dissolve cellulose using mixtures of aprotic polar solvents, such as dimethylformamide, dimethyl sulfoxide, dimethylacetamide, ethyl acetate with nitrogen oxides, lithium chloride, paraformaldehyde, etc.556,569,570
Since the 1980s, solutions of cellulose in tertiary amine N-oxides were studied in the Institute of Macromolecular Compounds of the Russian Academy of Sciences in order to obtain the related cellulose-hydrate fibres.571 At the present time, some manufacturers, including those in Russia, have advanced the technologies for the preparation of cellulose-hydrate fibres with the performance, similar to that of cotton, using N-methylmorpholine-N-oxide solutions.572--574 However, challenges associated with the improvement of technological production lines and the search for new solvent systems remain pressing since, along with the benefits of fibres produced by new technologies, they suffer from certain drawbacks (in particular, fibre fibrillation upon spinning due to an increased crystallinity of cellulose).
In terms of availability and distribution, chitosan is an equally promising polysaccharide. Chitosan is the only cationic natural polysaccharide which is the product of deacetylation of chitin, a structure-forming component of the exoskeleton of crustaceans and insects. In this area, pioneering works by P.P.Shorygin were concerned with the study of acetylation, nitration and methylation of chitin.577 Chitosan is a copolymer consisting of N-glucosamine and N-acetylglucosamine monomer units, and their ratio (degree of deacetylation) depends on the method of its production.578 The structure of chitosan is similar to that of cellulose, and therefore, like cellulose, it has fibre- and film-forming properties.579 The presence of hydroxyl groups and uncharged amino groups is responsible for the ability of chitosan macromolecules to form strong intra- and intermolecular hydrogen bonds. In turn, linear aggregates and rigid crystalline domains are formed in chitosan. In a solution, individual chitosan chains are arranged into a helix with a repeat of 10.1-10.5 Å along the polymer chain.
Potential sources of chitin and, consequently chitosan, are widely distributed in nature.580 Their total reproduction in the world's oceans is estimated at 2.3 billion tons per year, providing the global potential for chitin and chitosan production equal to 750 thousand tons per year. From the viewpoint of industrial production, the most accessible sources of chitin and chitosan are the shells of commercial crustaceans;581 in Russia, the catch of the Kamchatka crab and strigun crab in the Far East is 80 thousand tons per year.*
Like cellulose, chistosan cannot be melt processed; however, dissolving systems appear to be ‘softer'. Due to the presence of amino groups, chitosan can be dissolved in dilute organic acids, for example, in aqueous solutions of acetic, succinic or lactic acids, thus allowing the formation of films, sponges and fibres.
* A.G.Dvoretskii, A.P.Balykin. Izv. Ros. Akad. Nauk. Ser. Geografiche skaya, 85 (3), 405 (2021); https://doi.org/10.31857/s2587556621030055
9.3. Modification of cellulose and chitosan
The presence of many functional groups in both cellulose and chitosan makes them unique candidates for subsequent modification. In most cases, the final goal of the modification of cellulose and chitosan is either their solubility in aqueous media,582,583 or their ability for water uptake due to get formation via crosslinking,584--586 or required lyophilic properties.587--589 Physicochemical methods for surface hydrophobization of biodegradable materials based on polysaccharides (Fig. 57) involve adsorption of the following compounds on their surface: complexes of natural polymers or copolymers based on unsaturated carboxylic acids, containing titanium, zinc, silicon dioxides,532--534 as well as vegetable oils535 and thiols.531 For this purpose, the materials are immersed and allowed to stay in a hydrophobizing solution, and then dried. In this case, the materials acquire superhydrophobic (contact angle of water is 150°) and superoleophilic properties (contact angle of oils is 0°). This procedure allows the preparation of oleophilic-hydrophobic materials, which are promising for the separation of water-organic emulsions and retain their ability for biodegradation in enzyme solutions for 15 days.
![[{"id":"SUgekCMQyi","type":"paragraph","data":{"text":"Scheme illustrating the modification of woven materials by adsorption for imparting superhydrophobic and oleophilic properties."}}]](/storage/images/resized/Q85BQ3uvTHAcnhzQGlC9lELhttghbPxInQ70v6Fs_xl.webp)
Materials with controlled surface wettability can be prepared by photolithography,590 electrodeposition,591 sol-gel deposition,592 chemical vapour deposition593 and electrospinning.594,595 However, the above methods are complicated, labour-intensive and high-cost. As a result, their practical application is limited. Moreover, the resultant materials are unable to switch their wettability as transition from superhydrophobicity to superhydrophilicity, and back. Therefore, the challenging task is to develop new materials with controlled lyophilicity and response to environmental impact (changes in temperature, electric field, light radiation, pH).596--602
Modification of polysaccharides by synthetic polymers via graft copolymerization presents a relatively new approach to the synthesis of graft copolymers. Graft copolymerization allows introduction of polymer side chains via free-radical,603 ionic604 and controlled radical polymerization605 giving new derivatives composed of natural and synthetic polymers.544
Grafting of monomers on functional groups of the backbone of polysaccharides makes it possible to prepare compounds with different structures, degrees of branching, and content of unmodified hydroxyl and/or amino groups of cellulose and chitosan.606 Combination of useful properties of synthetic polymers based on vinyl monomers (primarily, physicomechanical properties) and biopolymers (adhesion to mucous membranes of living organisms, biocompatibility, ability to adsorb biopharmaceuticals as well as the ability to flocculation and coagulation, the absence of toxicity) can be achieved by radical graft polymerization. The graft copolymers are mainly used as supports for tissue engineering, separation membranes, hydrogels, aerogels, etc.607--609
Graft copolymers of chitosan and cellulose are composed of polysaccharide macromolecular chains with randomly distributed branchings of grafted polymers. Performance of graft copolymers is markedly controlled by the chemical structure, length and number of grafted chains. Two approaches, namely, grafting from and grafting to/onto, have been used to obtain graft copolymers of chitosan and cellulose.493,494,610 The former approach involves polymerization of monomers by means of initiators anchored to the carrier surface. Active sites on polysaccharide macromolecules can be produced either at the stage prior to blending with a monomer or directly in the course of the reaction with a monomer. In the latter approach, end groups of the synthesized polymers react with the complementary functional groups on the to-be-modified surface to form graft copolymers.610,611 When polymers sensitive to external changes are grafted onto the surface of polysaccharides, this approach allows preparation of `smart' materials, which can serve as antibacterial surfaces,612 heat-sensitive materials, components of composite materials with improved adsorption properties. In particular, adsorption properties of chitosan can be improved by modification via grafting with polyglycidyl methacrylate (PGMA).613 As was shown,614 modification of cellulose with hydrophobic PGMA leads to a decrease in water uptake with increasing PGMA content, and post-modification with hydrochloric acid accompanied by epoxide ring-opening to form a hydrophilic material promising for biomedical applications. Glycidyl methacrylate interacts with chitosan and cellulose in the same manner,615,616 when double bonds at the second stage participate in free-radical polymerization with such monomers as methyl methacrylate615 and butyl methacrylate.617 The same approach has been applied for polymerization on hemicellulose of methacrylates — stearyl acrylate618 and lauryl methacrylate.619 After modification of film materials, the water contact angle increases to 114-123° as compared with 70° of the initial film; and this is the maximum achievable increase in hydrophobicity for smooth surfaces.
The use of glycidyl methacrylate and (fluoro)alkyl methacrylate copolymers for the modification of cellulose-containing materials620--622 relies on the same principles. The surface layer is composed of polysaccharide macromolecular chains with randomly distributed branchings of the graft copolymer.623 To provide interaction of hydroxyl and oxirane groups, acidic or basic catalysis is used,623,624 a noncatalytic process at 130-150 °C is also possible.625
The studies on physicochemical, mechanical and biological properties of the materials based on modified chitosan showed that they are promising for the development of biodegradable vascular prostheses.626 These materials act as a hollow skeleton. Endothelial cells are attached to the surface of chitosan, and they proliferate with the formation of new living tissues. In an ideal case, this framework can degrade with a certain rate, which depends on the chemical composition of surface functional groups which are responsible for lyophilic properties of the surface of the materials.
Physical characteristics of aerogels based on cellulose and chitosan modified by grafted synthetic polymers open up the opportunities for practical applications of these materials as catalyst carriers, adsorbents for oils627,628 and heavy metals,629,630 filtering materials631,632 for separation of water-organic emulsions. The above properties of the modified materials, which distinguish them from the properties of initial natural polymers, are very important because the high affinity for the organic phase, low water and moisture absorption, biodegradability as well as the possibility of repeated and long-term use are the main requirements for modern adsorbents for oils and petroleum products.
To conclude, surface chemical modification of the materials based on cellulose and chitosan involves interaction of reagents through hydroxyl and amino functional groups of polysaccharides. The resultant materials retain the key properties of polysaccharides, such as biocompatibility and biodegradability, but show improved physicochemical, physicomechanical and adsorption characteristics and adjustable lifetime.
10. Polymer waste management problem
At the first glance, the issues related to accumulation of polymer trash and pollution of the world ocean by plastic waste are not directly related to the polymer science, but refer to organizational and management tasks of particular countries, first of all, the most developed countries. These countries are characterized by the highest polymer consumption per capita, which is largely correlated with life standards and well-being of the society.633 However, the global character of the arising problem and the designed approaches to the problem solution adopted in particular countries or associations of countries directly affect the trends of development of polymer science and engineering and thus interfere with their natural progress. Examples of such solutions are the ban of the single-use plastic items* and reducing the consumption of lightweight plastic carrier bags** adopted by the European Parliament. One of the goals of the project Polymers of the Future is to elaborate a fundamental concept of this problem in order to decrease or mitigate the social and political impact on the progress polymer science.
In the European Union, a polymer waste management program has been developed and is consistently implemented (see below). In a review634 that analyzes the existing approaches to polymer waste management, three main trends are distinguished (Fig. 58):
- recycling, which implies thorough sorting and crushing;
- incineration, which is accompanied by release and utilization of a large amount of energy;
- landfilling.
![[{"id":"XYq-d7NIC7","type":"paragraph","data":{"text":" Life cycle of a polymer material.<sup>634</sup> Published with permission from Springer Nature."}}]](/storage/images/resized/GcvfsBLcZD3OyW4ujUYuDnEHaXuSZLneLynyDoi2_xl.webp)
The common practice is directed towards increasing the proportion of recycled waste. Adherents of this trend believe that this would decrease the consumption of hydrocarbons and energy expenditure for the synthesis of polymers. However, they usually neglect the fact that in 90% of cases, secondary polymers have a lower quality than primary ones, which naturally deteriorates the quality of polymer products. One more neglected fact is that the equipment cost and the energy consumption related to the sorting and crushing are added to the cost of the end products and thus generate a considerable energy and monetary imbalance in the operation of plants that use this approach.
Much more encouraging is incineration with energy recovery635 and energy use with a positive economic balance.636 A reverse side of this waste management method is the need to remove components containing organic chlorine (first of all, PVC) from the waste bulk and to construct powerful treating facilities for effluent gases, which markedly decreases the expected economic benefit. The percentage of incinerated waste permanently increases, despite one more large drawback: combustion gives off an enormous amount of carbon dioxide. In other words, this route of waste disposal directly contradicts the concept of decreasing the carbon footprint, which is a part of environmental agenda. This route is also incorrect in terms of D.I.Mendeleev s statement that burning oil is akin to firing up a kitchen stove with bank notes. Burning polymers differs from burning oil only by the fact that before being converted to CO2, polymers have already performed a useful function. However, this is a weak argument from the point of view of environmentalists.
The third option, waste landfilling,637 is least popular, because it is supposed to landfill only the most hazardous waste, which cannot be disposed of by the first or the second method. This approach is considered to be least favoured, as landfilling requires huge underground storages with unpredictable reclamation periods; also, there are concerns about possible contamination of groundwater and soil water with components of plastic degradation and withdrawal of large areas of deficient land from turnover. Nevertheless, landfilling is performed on a large scale, most often, in an unqualified manner, i.e., without preliminary sorting. In those cases where landfilling is performed in a qualified manner, the landfill sites are often found in some other region.
Unfortunately, the current approaches are up-to-the-minute and arbitrary. This can be easily found out by analyzing the available regulatory documents. The delay in waste management measures brings about the need to introduce bans and restrictions on the production of certain types of polymers, while from the point of view of efficiency and prospects, it is better to increase the capacity for waste collection and processing into safe construction materials, for example, for aquatic construction.
What is the focus of modern approaches to the processing of polymer waste? The commonly accepted hierarchy of polymer waste management establishes the priority trends of development of technologies according to the closed-loop economy concept, which must obey the so-called 3R s rule (Reduce, Re-use, Recycle).638
The Russian Federation law 89-FZ, dated June 24, 1998, On Production and Consumption Waste envisages improvement of the waste management system and establishes the state policy measures in the field of waste management. The fullest use of the raw substances and materials, increase in the service life of polymer products and waste treatment and disposal were supposed to be the factors preventing and reducing the amount of polymer waste.
In order to prevent waste formation, it is necessary to take measures before a polymer or an item become trash, such as withdrawal of single-use items (containers and packaging for non-medical purposes, plastic bags for retail stores).639 An alternative option is to integrate the packaging disposal into the value-added process flow. Also, manufacturers of packaging materials could themselves develop processes for disposal of their complex waste.***
A way to address the problem of reducing the adverse impact of polymer waste on the environment is the creation and industrial production of environmentally benign self-degrading compounds such as biodegradable polymers (see below).640
The longest service life of polymer materials and products other than packaging implies than they can be repaired and reused. First of all, this refers to the use of self-regenerating and self-healing materials, but not implies the transfer of second-hand principle to critical items made of polymers. Scaling up from laboratory research to the industrial production of these innovative materials is a primary challenge faced by researchers.
Most polymer products retain their technical characteristics after being converted to waste. Hence, it is necessary to find how they can be usefully employed. Polymer waste recycling as a production process of secondary polymers includes the following stages:641
- preparation: collection, sorting and purification;
- mechanical, chemical or biological processing;
- fine tuning of the secondary product: enhancement of performance characteristics, creation of marketable appearance (upgrading), etc.
In the vast majority of cases, the products based on polymer materials are complex compositions; apart from a polymer or a blend of several polymers, they contain modifiers, fillers, antioxidants and various functional additives. When the polymer materials and products turn into trash, they are usually contaminated, and their recycling requires considerable energy, material and labour expenditures for sorting and purification before the secondary processing.642 For this reason, recycling is economically reasonable at enterprises that can utilize waste generated by their own manufacturing processes.
Recycling is economically and environmentally justified only for relatively large flows with a uniform composition free from foreign polymer waste impurities. In this regard, the first stage comprising the collection, sorting and purification is the key stage in the whole polymer recycling process. One way to optimize this stage is to reduce the number of sorting processes and to use two-stream waste collection,643 which implies domestic separation of waste into two streams, one containing waste to be recycled and the other containing all other types of waste.
It is noteworthy that the separate waste collection is a long-standing problem. In particular, in the USSR, it was solved by existence of separate collection points for glass containers, waste paper, scrap metal and textiles. Perhaps, it would be expedient to revise an old approach to an old problem in a new way, that is, to arrange the separate collection of secondary raw materials adapted to new reality (by adding collection points for polymeric bottles, fuel cans, etc.).
One more possible example is to deliberately take away plastic trash, e.g., plastic parts of obsolete office equipment, from enterprises by companies that recycle waste polymers. As a rule, office equipment is manufactured using high-impact polystyrene and ABS plastics (copolymers of acrylonitrile, butadiene and styrene). This means that the same sorting is performed just on the spot, but not at a garbage recycling plant. The same is true for collection of waste single-use tableware from public catering facilities. Conversion of the above measures from special cases into common practice requires economic interest of the enterprises participating in the separate collection of polymer waste. Collection of single-type plastic trash, structurization of the flows of obsolete polymer products and materials is a socioeconomic task.
The polymer waste delivered to the processing plant is subjected to crushing (shredding), purification, washing and additional separation. This is done using sorting stations, crushing machines, washing equipment, drying units, separators, agglomerators and granulating units. The types, kinds and specific features of the equipment were described in a number of reviews devoted to polymer recycling (e.g.644,645). The resulting secondary polymer raw material as flakes or granules is routed to further processing, which consists in the conversion of waste into secondary products and can be performed by non-destructive (mechanical treatment: crushing, final purification of the melt, granulation; polymer blending, introduction of modifying agents and fillers and chemical modification) or destructive (hydrolysis, glycolysis, methanolysis, pyrolysis) methods.
Processing without the change in the chemical composition of polymers includes refinement (additional separation according to types), regranulation and reuse in the manufacture of new articles. In view of the fact that an increase in the number of processing stages usually deteriorates the properties of polymers because of the thermo-oxidative degradation, a relevant issue is the search for efficient stabilizers and antioxidants that would favour repeated recycling with retention of properties.
If the requirements to the mechanical properties of the secondary polymer raw material are relatively low, the waste mixture is recycled into final products (without additional separation according to types). Compatibilizers are added to improve blending and the quality of blends. Without compatibilizers, the properties of the obtained compositions, which often consist of incompatible polymers, are very poor.646
A method for optimization of polymer recycling is the targeted blending of polymer wastes to obtain materials of a higher quality or more environmentally benign (upcycling). An example is provided by the production of blends of poorly separable polymers such as PE and PP and composites, i.e., the addition of fillers (fibres, micro- or nano-sized particles) into polymer waste. The newly obtained materials with satisfactory properties are used in some fields of economy.647 Production of composites of secondary polymers involves solution of the same problems as the fabrication of composites based on primary polymers: enhancement of the compatibility (use of compatibilizers, surface modification of fillers, optimization of filler composition and concentration).
Meanwhile, the above methods of polymer waste recycling without change in the chemical composition reduce the amount of plastic waste, but do not solve the problem of waste accumulation. After the end of service life, they become waste or trash once again. The use of secondary polymers for manufacturing so-called non-critical items (buckets, garbage bags, plastic containers, bungalow furniture, etc.), which in some cases contain some primary polymers, only enhances the spread of short-lived products that are quickly thrown away, thus contributing to the growth of plastic trash piles.
Chemical modification changes the structure of recycled polymers.648,649 Most often, this involves addition of compounds for restoring the molecular weight of polymers, so-called chain extenders, which act as cross-linking agents.650
The method of thermochemical degradation of polymers to give low-molecular-weight products is based on the destruction processes induced by factors that cleave the chemical bonds of macromolecules.
During the thermal processing of polymer waste, the destruction of macromolecules proceeds at 400-450 °C to give low-molecular-weight hydrocarbons. The reaction can take place under either atmospheric or elevated pressure with or without a catalyst. In particular, polyolefin waste is processed to give liquid furnace oil and diesel fuel.651
The ways of chemical recycling of polymer waste include the following high-temperature processes: hydrogenation in a hydrogen atmosphere; cracking and hydrocracking; gasification in an oxygen, steam or carbon dioxide atmosphere and in their mixture to give synthesis gas (a mixture of carbon monoxide and hydrogen); pyrolysis in an oxygen-free atmosphere;652,653 hydrolysis based on cleavage of polymers with aqueous solutions of acids.654 A reaction similar to hydrolysis is glycolysis in which glycols are used for depolymerization.
According to reviews,655,656 chemical recycling of polymer waste is the most promising approach for the integration of polymers into closed-loop economy. Successful implementation of these plans requires new reactor designs and deeper understanding of processes that underlie chemical recycling. In order to optimize the chemical processing methods and adapt them to plastic waste flows, irrespective of the sorting quality, it is necessary to find experimental correlations between the feed composition, operating conditions, type of the reactor and the composition of reaction products and also to use mathematical modelling methods and model systems.
A prevalent opinion is that incineration of polymer waste with energy recovery is the most efficient way for polymer waste elimination.657,658 Polymer waste is incinerated using furnaces of various designs: hearth, rotary, burner-fired, fluidized bed and other furnaces. Complete conversion of polymer waste to CO2 and H2O at a relatively high temperature requires preliminary grinding.659 It is noteworthy that this method of polymer waste recycling does not solve the carbon footprint problem (for more details, see Section 12).
Self-degradation of polymers induced by bacteria, UV radiation and water also refers to elimination methods. Degradation of, for example, polyethylene or polystyrene under the action of UV radiation (290-320 nm wavelength) is activated by introduction of 2--5% vinyl ketone monomer into the polymer backbone. The low-molecular-weight fragments formed upon the photodegradation are ultimately taken up by soil microorganisms.
The photodegradation of common plastics can be accelerated, in particular, by using special degradation agents. Most often, these are transition metal compounds, which catalyze the polymer destruction on exposure to UV light. The addition of the degradation agents to polymers is complicated by the fact that the additive should accelerate the polymer destruction in the light and simultaneously do not interfere with long-term operation also in the light. In other words, the additive should trigger the degradation at a definite time point. This can be achieved by introducing encapsulated additives. The use of microcapsules containing a degradation agent makes it possible to predict the service life of the polymer, which would be limited by stability of the microcapsule material. Over time, the polymer photodegradation may be accompanied by the degradation induced by bacteria; therefore, these agents are often added to biodegradable polymer composites.
* See https://www.europarl.europa.eu/news/en/press-room/20190321IP-R32111/parliament-seals-ban-on-throwaway-plastics-by-2021 (accessible 20.12.2022)
** See https://www.eur-lex.europa.eu/legal content/EN/TXT/?uri=celex:32015L0720 (accessible 20.12.2022)
*** See https://www.tetrapak.com/sustainability/planet/environmental-I,pact/tetra-pak-operations (accessible 20.12.2022)
11. Processing of plastics using supercritical fluids
Most of the approaches to polymer waste processing considered above use common liquid solvents and various catalysts. Currently, supercritical fluids (SCFs) have been used more and more often as the most promising media for the destruction and modification of polymer waste. Below we consider processing of typical plastic waste of diverse nature and composition in SCFs of various polarity, including the destruction via solvolysis. The products thus formed and the possibility of their reuse are discussed. Primary attention is given to processes in sub- and supercritical water and in supercritical carbon dioxide, as these solvents comply, to the greatest extent, with the green chemistry concept.
11.1. Chemical recycling of plastics in sub- and supercritical solvents
11.1.1. Hydrolysis and destruction in water
In many respects, water appears to be a perfect reaction medium for chemical processing of polymers: it is a non-flammable, non-toxic and fully environmentally compatible solvent. Under atmospheric pressure and a temperature of 25 °C, water is a polar medium, but at high pressure and temperatures above 150 °C, it becomes less polar and suitable for dissolving non-ionic organic compounds. Near the critical point (374 °C and 217 bar), minor variations of temperature or pressure have a considerable effect on the physical or chemical parameters of water, which makes SC ОH2O a solvent with tunable properties. In addition to the fact that supercritical water can serve as a solvent for nonpolar organic compounds, while retaining certain dissolving capacity towards polar compounds, it can also act as the active medium in acid- and base-catalyzed reactions. In the vicinity of supercritical conditions, the oxidative ability and the overall reactivity of water considerably increase.
It is not surprising that particularly SC water is used most often in the studies devoted to the chemical recycling of polymers in SCFs. Degradation of polymers in water with the goal of recycling is the subject of a number of reviews (see, e.g.660--664). This part of our review deals with the approaches to processing of the main types of polymers and macromolecular systems in SC water. The considered materials include condensation polymers, addition polymers, cured macromolecular resins, cross-linked polymer compositions and carbon fibre- (CFRP) and glass fibre-reinforced plastics (GFRP).
11.1.1.1. Condensation polymers
Polyethylene terephthalate is a thermally, chemically and mechanically stable polymer, which is widely used in the packaging industry, particularly for the production of plastic water containers. Among the possible approaches to the chemical recycling of PET, Shojaei et al.665 distinguished hydrolysis, which can proceed in acidic, alkaline or neutral medium (usually, at temperature >200 °C and pressure >15 bar). The hydrolysis affords PET monomers: terephthalic acid (TPA) and ethylene glycol (EG).666,667 If a methylating agent is added to the reaction mixture, the reaction gives dimethyl terephthalate (DMT) together with EG.668 Kazaryan and Martirosyan669 showed that the hydrolysis of PET in water can be initiated even at ≈180 °C. Exposure of PET in steam at 200 °C and 16 bar also results in a decrease in the molecular weight.670 Campanelli et al.671 reported a kinetic study of PET hydrolysis in subcritical water at temperatures above the melting point of PET (230-245 °C). When the reaction was conducted in a dilute solution of ammonia, the optimal temperature of hydrolysis decreases to 220 °C.672 Fang et al.673 reported important data on the phase changes in the water/PET systems at temperatures of the onset of PET hydrolysis in subcritical and supercritical water: PET and water form a homogeneous phase at a temperature close to the melting point of PET, whereas TPA is a water-insoluble crystalline solid under these conditions. Dissolution of TPA in water takes place above 350 °C. Publications that describe hydrolysis of PET analogues and derivatives such as polyethylene naphthalate674,675 and polybutylene terephthalate 676 in hot and SC water are also available.
Hydrolysis in sub- and supercritical water was utilized to degrade bisphenol A polycarbonate to the monomer and to bisphenol A degradation products: isopropylphenol and isopropenylphenol.677,678 The transition from subcritical to supercritical water was accompanied by increase in the yield of reaction products from 38 to 89%. When alkali metal salts were used as catalysts, the yield of products increased over the whole investigated temperature range. Watanabe et al.679 found that the process carried out with high-pressure steam (79 bar and 300 °C) afforded bisphenol A, which, however, did not further decompose.
Nylon-6 obtained by ε-caprolactam casting accompanied by ring opening polycondensation is a polyamide very hard to mechanically process. Wang et al.680 investigated the hydrolytic degradation of this type of nylon in subcritical water at 90 bar at 345 °C. Under these conditions, monomer casting nylon completely decomposed into water-soluble monomer and oligomers after 45 min; the maximum yield of the products (89%) was observed after 75 min.680 The reaction can be accelerated by using zeolites and acid metal oxides as catalysts.681
The high potential of using SC water for polymer recycling was demonstrated for Okajima et al.,682 who performed decomposition of aromatic polyamide Kevlar® fibres. The authors studied the thermochemical decomposition of this mechanically and chemically relatively stable polymer in various media: supercritical water, subcritical water with and without the addition of NaOH. Hydrolysis gave the corresponding monomers: para-phenyldiamine and TPA; the reaction rate, which depended on the medium, increased in the following series of solvents: subcritical water<supercritical water<subcritical water with NaOH. For the identified optimal conditions (250 °C, 40 bar, NaOH:Kevlar® ratio of 1:5), the yield of the two monomers was 95% (Fig. 59).
![[{"id":"1r2QmFGZxm","type":"paragraph","data":{"text":"Images of Kevlar® fibres (<i>a</i>) and monomers resulting from Kevlar® fibre treatment in SC water: <i>para</i>-phenyldiamine (<i>b</i>) and TPA (<i>c</i>).<sup>682</sup> Published with permission from Elsevier."}}]](/storage/images/resized/Ti4SR2ZIcBD4lBoClUH1h3UOZlcjtFRkvSIkPoxM_xl.webp)
11.1.1.2. Addition polymers
In the first stage of the thermal decomposition of PVC in SC water, chlorine atoms are eliminated from the backbone by the dehydrohalogenation mechanism to give hydrochloric acid in the aqueous phase.683--687 Sato et al.684 presented the distributions of gaseous and liquid products of PVC hydrolysis in SC water under milder (370 bar and 450 °C) and more harsh (600 bar and 600 °C) conditions. At 600 °C, all liquid products of the reaction were aromatic hydrocarbons: phenol, benzene, naphthalene. If the reaction was carried out at 450 °C, acetic acid was also present among the products.
The PS degradation in SC water (370-420 °C and 240-320 bar) afforded a complex mixture of liquid products, including styrene monomers and dimers.688 The yield of styrene was higher for PS treatment in SC water than for PS pyrolysis.689 The product distribution considerably depended on the reaction conditions and on the use of a catalyst (e.g., NaOH).690 A complex mixture of products was also formed upon decomposition of polyolefins, PE691--694 and PP,695,696 in SC water. The studies on supercritical water gasification of PS and polyolefins have been considered in detail in a review.697 The processing of fluoroplastics such as polytetrafluoroethylene, polyvinyidene fluoride and copolymers based on them, deserves a special interest. An important advantage of SC water is the possibility to convert fluorine atoms into the anions, which are located in the aqueous phase, and this minimizes hazardous emissions of fluorinated compounds to the atmosphere.698,699
A number of recent studies are devoted to comparison of liquid organic products formed upon SC water treatment of chemically diverse plastics, including both polymerization and polycondensation products.700--702 The results of these studies confirmed once again that the use of SC water as a solvent is promising for the chemical recycling of polymers.
11.1.1.3. Thermosetting plastics, cross-linked polymers, composites and systems of a complex composition
Tagya et al.703 showed the possibility of decomposition of phenol resin model compounds and molding materials in supercritical water to the monomers. Cross-linked epoxy resins are also efficiently decomposed in SC water to give phenol derivatives as the products; this reaction can be catalyzed by adding Na2CO3.704--707 Apart from epoxy resins, decomposition in SC water was studied for thermally stable plastics such as poly(ether ether ketones),704,708 styrene copolymers,709 butadiene copolymers,710 cation exchange resins711 and polyurethanes.712
Chen et al.713 studied the thermal degradation of waste tyres and natural rubber in SCFs and compared the results obtained using supercritical water and CO2. In both cases, a considerable decrease in the molecular weight was noted; the use of SC water resulted in a more efficient degradation of both tyre rubber and natural rubber. Computer simulation data showed that the tyre decomposition in SC water follows a radical mechanism.714 The conversion of waste tyres into low-molecular-weight products can also be efficiently carried out in SC water with the addition of hydrogen peroxide.715 Of particular interest are gaseous reaction products, specifically, the formed hydrogen, the yield of which can be increased by adding catalysts [for example, Ba(OH)2, Ca(OH)2 or Mg(OH)2] as a result of steam gasification reaction.716 The destruction of waste tyres may be combined with crushing by conducting the process under high-pressure hot water jet (jet pressure of up to 2200 bar) (Fig. 60).717
![[{"id":"p76tzh4eHS","type":"paragraph","data":{"text":"Experimental facility for investigation of tyre destruction under a high-pressure hot water jet (<i>a</i>) and scheme of areal unit for tyre shredding and processing (<i>b</i>).<sup>717</sup> Published with permission from De Gruyter."}}]](/storage/images/resized/1bf1m4j2oHX3bqAAGGNdlcTJF4GmYXATAmI3c9J8_xl.webp)
Recycling of plastics used in electronic and computer equipment in SC water can be distinguished as a separate area (the rapid increase in the consumption of the products of electronic industry and annually increasing amounts of the corresponding waste is a more and more pressing environmental issue).718--720 This refers to printed circuit boards,721--726 integrated microcircuits,727 keyboards,728 liquid crystal display parts.729,730 It was shown that up to 99% of metallic gold and copper can be recovered from printed circuit boards722 via treatment in SC water at 400 °C and 250 bar. Furthermore, the listed items often contain brominated organic compounds as fire-retarding agents; in this case, supercritical water treatment results in efficient conversion of bromine to HBr, which occurs in the aqueous phase and can thus be separated from nonpolar degradation products, which form a separate phase.720,721,724,725,731--734
A widely adressed process of plastic waste recycling in SCFs is the processing of CFRPs with separation of the carbon and polymer phases; the key publications in this area were analyzed in a few reviews.735--737 The chemical nature of the polymer component of CFRPs treated in SC water may be different, including plastics based on epoxy resins,738--743 in particular in the presence of brominated fire-retarding agents,744 polyesters,745 PEEK,746 acrylonitrile, styrene and butadiene copolymer747 and polyamides.748 It was shown740,749 that the recovered carbon fibre fabric preserves the woven architecture and can be used again. Apart from CFRPs, examples of recycling using SC water are known for GFRPs.750 The discussion of CFRP recycling in this stage includes not only scientific aspects, but also economic751 and environmental752,753 considerations. A pilot unit for CFRP recycling using SC water with a reactor working volume of 2.9 m3 was put into operation by Panasonic Corporation in Tochigi (Japan).663
An important factor hampering the industrial implementation of plastic recycling processes discussed here is high corrosion activity of sub- and supercritical water, resulting in fast wear of equipment and requiring the use of expensive corrosion-resistant alloys for the manufacture of reactor equipment.754 This problem is aggravated by the fact that the reaction products may contain both corrosive low-molecular-weight compounds (salts, acids) and high-viscosity fractions, which makes handling of equipment more complicated. Nevertheless, in our opinion, the benefits of using SC water for plastic recycling can provide sufficient incentive to search for the required optimized engineering and design solutions.
11.1.2. Alcoholysis and glycolysis
The critical parameters of methanol (240 °C and 78 bar), ethanol (241 °C and 63 bar), 1-propanol (263 °C and 52 bar) and 1-butanol (289 °C and 45 bar) are much lower than those of water (374 °C and 217 bar); furthermore, the corrosion activity of SC alcohols is lower than that of SC water. Thus, plastic recycling using SC alcohols is more gentle for the operating equipment. In addition, the decomposition of plastics in SC alcohols may follow a different chemical pathway compared to that in SC water and give a different set of reaction products. In the case of condensation polymers, SC alcohol is not only a decomposition medium, but also a reactant involved in alcoholysis.
Studies of methanolysis of PET in supercritical fluids are widely represented in scientific literature.755--762 Depolymerization of PET in SC methanol gives DMT and EG,763 which can be easily purified and reused in PET synthesis. When other SC alcohols are used instead of methanol, the corresponding TPA esters are formed together with EG.764--768 Also, there are examples of PET depolymerization upon the glycolysis in SC EG to give bis(2-hydroxyethyl) terephthalate as the product.769 A number of studies are devoted to the mechanism and kinetics of PET methanolysis in SCF.770--773 Depolymerization of PET analogues in SC methanol has been studied.774--777 Mitsubishi Heavy Industries, Ltd. (Japan) launched a pilot line for the chemical processing of PET in SC methanol.778 Regarding condensation polymers, works dealing with decomposition of polycarbonates779--781 and polyamides782--784 in SC alcohols are worth mentioning.
As the above-considered thermal decomposition of polystyrene in SC water, the reaction in SC methanol gives styrene monomers and dimers among the reaction products.785 As the reaction time increases, the yield of dimers decreases, while other reaction products include an increased proportion of toluene. In the thermal decomposition of HDPE in SC ethanol, like in the decomposition in SC water, the liquid product fraction is a complex mixture of compounds.786
A potential advantage of SC alcohols over SC water is the possibility of recycling cross-linked polyolefins in which the cross-links are cleaved selectively without the pyrolytic degradation of the polymer backbone.787--790 Regarding the potential industrial implementation, an important factor (demonstrated in a number of studies) is the possibility of selective cleavage of the PE cross-links in a continuous flow process during extrusion of polymers in SC alcohols.791--795
Apart from cross-linked polyethylene, studies on the recycling in SC alcohols are focused on polyurethane foams,796,797 cured epoxy resins,798,799 rubber crumb 800 and plastic parts of light-emitting diode displays.801 As with SC water, a considerable part of studies are devoted to CFRP processing in SC alcohols.802--819 While comparing the liquid products and the solid carbon-containing products of CFRP processing in various SCFs (methanol, 1-propanol, 2-propanol, 1-butanol and acetone), Cheng et al.809 showed that at a particular temperature and pressure, the carbon filler of CFRPs is most efficiently recovered in SC 1-butanol. Recycling of CFRPs is also efficient in SC EG and a EG/water mixture in which 97% of the polymer component of the composite was removed.820
11.1.3. Processing in acetone
Supercritical acetone is widely used in research practice.821 The destruction of composite materials based on thermosetting polymers in acetone at elevated temperature and pressure attracts attention of specialists by the possibility of degrading epoxy resins reinforced by glass or carbon fibre fabric, recovery and reuse of the filler (this is especially important for expensive carbon fibre) and conversion of the cured resin to low-molecular-weight compounds, which can be reused. This type of destruction is accompanied by chemical transformations of the solvent via aldol reaction, aldol condensation, Michael reaction and other pathways to give diacetone alcohol, isomesityl and mesityl oxide, isophorone, phorone, mesitylene, its isomers and so on (Fig. 61).822 Similar reactions and condensations can also proceed in supercritical primary (secondary) alcohols provided that they are oxidized to aldehydes (ketones).
![[{"id":"1ORc40GoOc","type":"paragraph","data":{"text":"Possible chemical reactions of SC acetone.<sup>822</sup>"}}]](/storage/images/resized/LXSk4Ov66V2MIDa1cvo6fnafSQeQwrcVY0m3CdUJ_xl.webp)
The products formed upon transformations of SC acetone and polymer decomposition have a beneficial effect on the rate and degree of thermal destruction of reinforced composites.822 Apparently, these products promote the subsequent cleavage of ester bonds in the cross-linked epoxide material. In this case, it can be stated that SC acetone functions as not only a solvent (the term solvolysis is even used in the literature, see below).822 It appears possible to recover both the filler and a part of the starting reactants used to obtain the thermoset. It was shown that after repeated destruction cycles, unreacted acetone can be efficiently separated and reused.823
Okajima et al.736 compared the decomposition of carbon fibre-reinforced epoxy resin cross-linked with amines in a series of SC solvents, alcohols and ketones, including methanol, ethanol, propanols, butanols and acetone and methyl ethyl ketone with a higher critical temperature. For invariable reaction temperature of 320 °C and solvent concentration in the reactor of 3.64 mol L–1, different pressures (in the range from a few to 12 MPa) exceeding the critical pressure were set. The authors showed that in the initial reaction period, the decomposition rate was positively affected by solvents with smaller size of the molecules, while at the final stage, the dissolving capacity is more important, which must be taken into account in relation to appearance of carbonized reaction products. Indeed, the efficiency of SC acetone in elimination of the adverse effects of carbonization during the thermal destruction of polymers is well known.824 Among the tested solvents, particularly SC acetone demonstrated a combination of high rate and high degree of destruction of thermosetting composites. The conversion efficiency at an invariable temperature increased with increasing pressure (acetone density).825 The recovered carbon fibre sheets preserved the surface and strength characteristics. In experiments using acetone and water mixtures (80:20), temperature rise resulted in acceleration of the composite destruction, but a temperature above 340-360 °C was undesirable, because this deteriorated the properties of carbon fibre recovered from the composite making it more fluffy.826
A comparative study of the degradation of epoxide-based GRFPs and CFRPs in SC acetone and in subcritical water under the same conditions (pressure, temperature) showed that treatment in acetone is more gentle for the recovered glass fibre;827 in this case, the strength characteristics of this filler are better preserved. The fibre recovered from CFRPs retains high strength characteristics irrespective of the type of solvent used for the destruction.
In the case of recycling of hydrophobic polymers in SC acetone, destruction is also accelerated owing to better solubility compared to usual thermolysis and removal of the products under the same conditions, as was shown in relation to polystyrene828–830 and polyolefins.831
11.1.4. Solvolysis and destruction in ammonia
For supercritical ammonia (132 °C, 11 MPa), chemical transformations of molecules of the pure solvent are impossible. In this respect, SC ammonia resembles SC water and differs from supercritical ketones, aldehydes and some other organic solvents (see below). The dipole moment of the ammonia molecule (1.65 D) is comparable with that for water and methanol molecules (1.8 and 1.65 D, respectively); the presence of a dipole moment distinguishes these solvents from non-polar CO2, which has only a quadrupole moment. Decomposition of polymers with polar groups is expected to involve solvolysis, i.e., direct participation of the ammonia molecule in the induced chemical transformations of the macromolecules.
A typical process that can be mentioned as solvolysis in ammonia is the decomposition of PET. It is known that solvolysis of PET catalyzed by zinc acetate occurs in aqueous ammonia even at room temperature and ambient pressure.832 Under supercritical or nearly supercritical conditions, a high reaction rate is attained even without catalysts. Ammonia molecules actively participate in the reaction; therefore, unlike hydrolysis or methanolysis (see above), decomposition of PET in ammonia gives terephthalic acid diamide, instead of TPA or DMT (Fig. 62).833 Unlike esters or acids, these amides cannot be directly used for the two-step transesterification-condensation or one-step condensation reaction to give again the polymer.
![[{"id":"3a7Jhv8OQf","type":"paragraph","data":{"text":"Products of PET solvolysis in supercritical water, alcohols and ammonia.<sup>833</sup>"}}]](/storage/images/resized/R6UoVourABQHkyuKmI2MffshPhL10o6rpoP69tNa_xl.webp)
Polyurethane foams, traditionally produced by reactions of polyhydric alcohols (polyols) with isocyanates, can also be degraded in SC ammonia.834 Ammonia molecules actively participate in the foam destruction reactions. Consequently, apart from polyols, the recovered products contain amines (instead of the initial isocyanates) and amides (mainly ureas), resulting from transformations of the fragments of carboxylic acid derivatives.834 After the separation of products and conversion of amines to isocyanates, repeated synthesis of polyurethanes is possible.
In addition, the destruction of aliphatic835 and aromatic polycarbonates in SC ammonia makes it possible to obtain alcohols (phenols) and urea.833 In particular, this type of decomposition of polycarbonates based on bisphenol A allows recovery of bisphenol A for the subsequent use without the need for its additional transformation.833 This determines good prospects of polycarbonates (or related compounds) for the degradation in SC ammonia. Mormann and Frank836 investigated the chemical recycling of thermosetting polycyanurate based on bisphenol A, phenyl isocyanurate as a model of polyisocyanurates and cured bismaleimide resin. Particularly SC ammonia ensured conversion of the starting materials to low-molecular-weight products (bisphenol A and melamine; aniline and urea; and aniline and two methylenedianiline isomers, respectively) suitable for reuse. Later, analogous processes were successfully implemented at elevated temperature and pressure in aqueous (including dilute) solutions of ammonia.672,837,838
The high reactivity of ammonia may be fairly useful. Xiu et al.839 showed that treatment of plastic-containing wastes (in particular, recycled printed circuit boards) in aqueous ammonia under subcritical conditions gives nitrogen-containing products of degradation of the epoxy resin binder valuable for fine chemical industry, e.g., pyrazine and pyridine derivatives. A later publication of the same authors showed that the plasticizer and chlorine can be effectively removed from recycled PVC via reactions with ammonia molecules under similar conditions.687
11.1.5. Processing in miscellaneous solvents
Supercritical nonpolar hydrocarbons are a reasonable choice of solvent for the destruction of hydrophobic polymers, because of the expected good solubility of the starting materials and/or decomposition products in these solvents. In particular, efficient thermal destruction of polystyrene in supercritical hexane,840 cyclohexane and ethylcyclohexane841 has been carried out. The processes were more intense 841 than those in the experiment carried out at the same temperatures (300-400 °C) and pressures (2-5 MPa) under inert atmosphere (nitrogen) and in the presence of a catalyst. The process was also accelerated in supercritical pentane and toluene.842
Relatively high destruction rates were attained in some subcritical solvents with hydrogen donor properties including phenol.843 Phenol was successfully used to decompose not only polystyrene,844 but also polyolefins.845 Similar experiments were carried out using subcritical decalin.846
Dhawan et al.847 found dozens of detectable low-molecular-weight organic products upon depolymerization of natural rubber and vulcanized rubber waste in SC toluene and noted a cascade of transformations of the solvent. Reaction mechanisms were proposed for typical products (distribution of product yields depended on the particular reaction conditions); some of the mechanisms included cross-reactions of radicals formed upon thermal decomposition of the macromolecules and the solvent molecules. The average molecular weight of liquid products showed a tendency to decrease with increasing reaction temperature.848 Depolymerization processes of cis-polyisoprene in SC tetrahydrofuran were considered by Lee and Hong.849
Jie et al.50 visually observed the degradation of polystyrene in SC toluene in a reactor with an optical window. The observation indicated that the polymer first rapidly dissolved and then gradually decomposed. The mechanism of the decomposition starting with the end group was discussed.851 Among the tested SC solvents, toluene was found to be one of the most efficient for depolymerization of polystyrene.852 Sub- and supercritical D-limonene, which are good solvents for polystyrene, were successfully used to recycle cured ester-based glass fibre-reinforced thermosets. The fibres were recovered for the subsequent use, with their mechanical properties being retained.853
Subcritical acetic acid is used for efficient degradation of cured bismaleimide resin to recover the reinforcing carbon fibre.854 Cured bromine-containing epoxy resin from recycled printed circuit boards was successfully decomposed in this solvent to bisphenol A and phenol; bromine-containing fire-retarding agents decomposed to release HBr.855 The clear-cut beneficial effect of the chosen solvent on the process rate was attributed to a high degree of swelling of these thermosets in this solvent.
The degradation of polar polymers in nonpolar SC hydrocarbons may require the use of polar co-solvents. Efficient depolymerization of nylon was carried out in SC hydrocarbons in the presence of water, which was necessary just for the reaction to proceed.856
The operating parameters of reactions involved in polymer destruction in compressed gases often exceed characteristics (pressure, temperature) of the critical points of these gases; therefore, these processes can be classified as decomposition under SC conditions. Feng et al.857 reported decomposion of plastic waste (polyolefins) in nitrogen or hydrogen at high pressure; in the case of hydrogen, the pressure was above the critical point (SC hydrogen). However, no significant difference between the yields was detected. Under optimized conditions, a liquid low-molecular-weight fraction was formed as the major product.857 By varying the process conditions, it was possible to increase the yield of the gas fraction.858 Combining of plastic waste with coal for recycling gave a clear-cut positive synergistic effect,857 which was in particular found for mixtures of polyolefins with polystyrene.859 Similar processes were conducted to recycle plastic waste together with heavy oil residues.860 The enhanced degradation of mixed polyolefin plastics, specifically in compressed hydrogen, was attributed861,862 to the catalytic action of the added elemental sulfur. Other applicable catalysts include dispersed alumina-silica,857 zeolites863 and superacids.864,865 A procedure involving dechlorination stage was proposed for the hydrocracking of mixtures of polyolefins with PVC.866 It should be emphasized that, despite the fact that formally these processes take place under SC conditions, the densities of compressed gases in these experiments are moderate.867
11.2. Plastic recycling using supercritical carbon dioxide
The use of SCFs to control the properties of materials produced from polymer waste868 attracts considerable interest. Modification of these materials to impart specified properties to the final processing product may involve extraction, impregnation and foaming processes, which reflects the diversity of methods of treating the items depending on the goals and objectives of this treatment.
Studies of the possible plastic recycling methods using supercritical CO2 are widely (in comparison with other SCFs) presented in the literature. This is largely caused by the environmental friendliness of this solvent and relative ease of converting CO2 to the SC state (in comparison with water and other polar solvents, CO2 has relatively low critical parameters, namely, 31.1 °C and 7.39 MPa).
11.2.1. Supercritical fluid extraction
Supercritical fluid extraction (SFE) is a highly demanded and well-studied application of both SC CO2 and, in general, SCF for plastic recycling. The process is based on the dissolution and mass transfer of the substance impregnated into the polymer matrix into an external solution using SCF, which acts as the extracting solvent (eluent). Owing to their high density, SCFs have a pronounced dissolving capacity for many types of fillers. In addition, due to the absence of surface tension related effects, SCF can penetrate deep into the polymer matrices without hindrance, including matrices with a complex pore morphology, and can also freely leave them upon decompression. This favourably distinguishes SFE from the extraction methods based on the use of classic liquid solvents.869,870 Indeed, SCFs in the compressed state occupy the whole volume, like gases, while pressure release induces a uniform decrease in the fluid density. The action of these two factors eliminates the problem of (non)wettability of pore walls. The concentration gradient of the solute is the driving force for the diffusion extraction process. The characteristic times of diffusion in SCFs are much lower than these times in liquid media. In addition, by varying the extraction parameters (temperature and pressure), it is possible to smoothly change the dissolving capacity of SCFs and thus to carry out selective extraction.
As noted above, SC CO2, a cheap non-toxic gas, is used most often for SFE processes. Owing to ready availability and safety, it has traditionally attracted considerable interest for food industry and pharmaceutical applications related to the fluid extraction of valuable compounds (carotenoids, alkaloids, phenols, essential oils, etc.) from plant sources. The practical implementation of these approaches is described in numerous reviews.871--874 Supercritical CO2 is also widely investigated with regard to the use for sterilization, because it efficiently inactivates bacteria and viruses in relatively low-temperature (non-destructive and safe) processes.875--877 The sterilization may be an important stage of recycling of plastic waste of medical origin. In this part of the review, the attention is concentrated on SFE as applied to plastic materials and general-purpose waste.
Many polymers, especially amorphous polymers, have a clear-cut ability to swell in supercritical CO2; this unfreezes the mobility of macromolecules; therefore, SFE additionally accelerates the diffusion of compounds extracted from the polymer matrix.878,879 As a consequence, SFE can be used to remove the pollutants introduced into these polymers during operation and specially added dopants meant for enhancing the performance characteristics of the materials. The main compounds that should be recovered (in some cases, with the prospect of subsequent reuse) include plasticizers, dyes, antioxidants, UV light absorbers, fire retarding agents, stabilizers, modifies and additives, residual oligomers, monomers, catalysts, initiators and organic and other contaminants.880 It is important to emphasize that SFE can be performed using either one fluid (e.g., SC CO2) or also co-solvents (fluid mixtures) for increasing the dissolving capacity of the medium towards a wider range of compounds (for example, a typical expedient is to add a polar co-solvent to nonpolar CO2).881 The main classes of polymers purified by SFE and compounds recovered during this extraction are considered below.
Packaging plastics are the most abundant polymers that get to the trash basket. First of all, this refers to PP and PE. High-quality and efficient recycling of these thermoplastics, in particular for the subsequent reuse, includes purification, which can be accomplished by SFE methods. The recovered extraction products can also be valuable for the subsequent use. Antioxidants (BHT, Irganox, Irgafos, Hostanox, Topanol), UV light absorbers (Tinuvin, Cyasorb) and slip additives (e.g., erucamide) were extracted from PP matrices using SC carbon dioxide.882--885 Depending on the extractable compound, the extraction efficiency was 10 to 100%. The above antioxidants were also extracted upon the addition of various co-solvents (methanol, hexane, acetone) to pure SC carbon dioxide.886--888 A similar situation occurs for the supercritical fluid extraction from PE matrices: pure CO2 was used to recover the same sets of antioxidants, UV light absorbers and slip additives.884,885,889,890 Co-solvents (out of the series: methanol, xylene, toluene, chloroform, acetonitrile) were added to increase the efficiency of extraction of the same compounds.891,892 In addition, a number of publications address SFE of various organic pollutants (stearin, dilaurin, trilaurin and tripalmitin molecules were considered as models) from polyolefin polymers used for food packaging.893--895 It was proposed to use SFE to remove residual monomers and initiators from polyolefins.896 Finally, it was shown that SC CO2 can be applied for decontamintion of polyolefin containers from oil products897 and for the separation or purification of paraffins resulting from the chemical recycling of polyolefins.898
Among polymers that are successfully purified by SC- CO2 (in some cases, using co-solvents), mention should be made of PET,899--902 nylon,903--905 PVC,906--909 polystyrene,910--913 polyvinylidene fluoride (PVDF) and PTFE,914 polyurethane,915,916 polyvinylbutyral.917 Supercritical fluid extraction can also be used to extract bromine-containing agents present in fire-retardant coatings918--922 and printed circuit boards in electronics.922,923 In addition, the same approach is also suitable for recovering macromolecular components from printed circuit boards.924,925 When polar water or hydrogen peroxide were used as co-solvents for nonpolar SC CO2, it was possible to efficiently extract copper,926,927 cobalt,928 indium,929 palladium, silver930 and other 931 metals from waste printed circuit boards, batteries and LC displays for the subsequent use. A recently proposed and successfully tested technique for the extraction of organic electrolyte from spent lithium ion batteries using SC CO2 with the goal of subsequent regeneration is of special practical interest.932--935 Separation of the products of depolymerization of polycarbonates936 and isolation of low-molecular-weight destruction products by mechanical processing of polymers937 was described. An approach in which SC CO2 is used as the precipitation agent for the macromolecular fraction was elaborated. This approach can be implemented in the processing of polystyrene938 or extraction of fire-retarding agents from PS939,940 and for nylon precipitation during the recycling of carpet waste.941 The selectivity of SC CO2 as a precipitating agent for polymers (it readily dissolves many classes of low-molecular-weight organic compounds, but only few types of polymers) provides the purity of the precipitated macromolecular fractions.
11.2.2. Supercritical impregnation
It was noted above that SC CO2 may efficiently wash out various compounds from polymer matrices, owing to its diffusion and penetration properties. Meanwhile, it can also be used to solve the inverse problem: impregnation of polymer matrices. For this purpose, it is necessary to reverse the sign of the concentration gradient of the compound dissolved in SCF. This approach is used to tackle research problems such as insertion of the pharmaceutical agents into polymeric implants,942 textile dyeing,943 formation of diverse nanoparticles in matrices or on substrates for the fabrication of composites,944 addition of monomers and initiators to a polymer to form polymer blends945 and obtaining other functional materials.946,947 However, for waste recycling problems, SC impregnation of polymers with agents promoting degradation of matrices, e.g., cross-linked macromolecular systems, is most important. Supercritical impregnation is most widely used in devulcanization processes in which agents that cleave the sulfur cross-links in the polymer are introduced into rubber before recycling.948 When a devulcanizing agent (diphenyl disulfide is a typical example) is used in SC CO2, some of the dissolved molecules of the agent also penetrate into swollen rubber on heating and react with sulfur cross-links to cleave them.949 This approach, proposed by Kojima et al.,950,951 was further developed in subsequent studies.952,953 Owing to high penetration ability of the solvent and fast diffusion, it is possible to attain a high degree of devulcanization even in carbon black-filled rubber (this type of filling markedly impairs the diffusion for conventional liquid media, but this impairment is less pronounced for SCF).954 The recycled rubber markedly swells in SC CO2, which results in efficient devulcanization. This approach proved to be efficient for vulcanized rubbers based on various types of natural and synthetic rubbers.955--957
11.2.3. Foaming
The fact that numerous polymers (especially amorphous ones) swell in SC CO2 forms the basis for foaming processes widely used in research. These processes are implemented in the following way: the polymer is saturated with CO2 at constant elevated temperature and pressure, then depressurizing is carried out, and, hence, the polymer-SCF system leaves the thermodynamic single-phase region, i.e., phase separation takes place. In other words, the plasticized polymer matrix becomes supersaturated with CO2, and gas bubbles are formed; this accounts for the formation of a porous polymer structure, the morphology of which is fixed upon removal of the plasticizer, i.e. CO2 molecules.958 Foaming can be used in plastic processing for the fabrication of composite materials with inclusion of the products of vulcanized rubber recycling. Indeed, foaming of mechanically produced mixtures provides stirring and distribution of the inserted agents throughout the whole polymer matrix. Unlike impregnation, in this case, good solubility of the compounds in SC CO2 is not a necessary condition. Composite materials based on PP and PE filled with rubber destruction products were obtained.959--962 This increased the thermomechanical characteristics of the composites. Using SC foaming, it is possible to fabricate composites, e.g., with PET, the conventional recycling of which is an intricate process requiring a high degree of purity. Therefore, it is economically reasonable to reuse PET without high purification in the form of composites.963 The residual contaminants can act as functional additives that provide controllable properties and thus promote diversification of possible applications of the materials.964
11.2.4. Destruction
Supercritical CO2 can be used as a medium for polymers destruction. Currently, there is a moderate number of publications devoted to the use of SC CO2 for depolymerization and destruction of polymers. Chen and co-workers965,966 reported PET recycling via oxidative pyrolysis in SC CO2 to give a superhydrophobic carbon material. The use of this medium increased the efficiency of the process in comparison with conventional oxidative pyrolysis. Supercritical CO2 was proposed for the oxidative destruction of nylons.967 During this process, polymer matrices swelled, which allowed for more efficient penetration of the oxidant NO2 into the material. Gao and co-workers968,969 carried out the catalytic hydrolysis of PET in SC CO2, which also acted as the plasticizer and promoted more efficient penetration of the catalyst and water molecules into the polymer matrix. Elmanovich et al.970 carried out the oxidative decomposition of PP in SC CO2 and showed that this medium increases the efficiency of polymer destruction to obtain more selective and controllable distribution of the final products. It was shown971 that, depending on the ratio of the amounts of oxygen and the polymer, the thermochemical destruction of PE in oxygen-enriched SC CO2 affords either waxy oligomers or a liquid product with predominance of acetic acid (Fig. 63).
![[{"id":"mJROaMo8_c","type":"paragraph","data":{"text":"General scheme of the chemical processing of PE in oxygen-enriched SC CO<sub>2</sub>.<sup>971</sup> Published with permission from Elsevier."}}]](/storage/images/resized/MHzKQKzwm45U1ecVb8s1J2FLtUCPTGwLdkGCQDRk_xl.webp)
These examples provide the conclusion that SC CO2 is a promising medium for chemical degradation of polymers. The fact that the number of relevant publications is still moderate indicates that further studies along this line would be relevant.
Thus, the diversity of examples of recycling of plastic wastes in various types in supercritical fluids attests to versatility of this strategy, which has no competitors in the diversity of polymer waste composition and possibility of intensifying the degradation or modification processes. Particularly the versatility accounts for somewhat exagerated attention received by this approach in the literature. However, in view of the possible ecological costs, the method does not appear perfectly trouble-free, and this aspect still requires systematic analysis.
The studies dealing with plastic degradation in supercritical water, ammonia, alcohols, acetone, etc. implement an approach to chemical recycling in which the chemical structure of the polymer is broken and new low-molecular-weight compounds are formed. In most cases, supercritical fluids are direct participants of the reaction. For condensation polymers (especially for PET), solvolysis in SCFs gives EG and TPA and its derivatives, which can be reused as monomers to obtain PET. In the case of vinyl polymers (PE, PP, PVC, PS), decomposition in SCFs results in the formation of a multiphase product, the liquid phase of which consists of a variety of compounds. Meanwhile, chemically inert carbon dioxide, which is readily converted to the SC state, acts primarily as a mediator, a plasticizing agent for the matrix to be decomposed, a carrier of functional additives or a solvent for the products of the induced destruction.
Regarding SCF solvolysis of plastics, a strategy that seems quite promising is not the recycling of single pure polymers that selectively gives the monomer or a particular low-molecular-weight compound, but rather the chemical recycling of polymer composites (printed circuit boards, natural and vulcanized rubber items, carbon fibre- and glass fibre-reinforced plastics), which may lead to isolation of pure solid-phase components - metals or various fillers, including carbon and glass fibres. Recycling processes of polymer blends can be characterized by positive synergistic effect,972 which has not yet been studied in detail.
Despite the existence of significant scientific grounds for the chemical recycling of plastics in SCFs, there are still few economic calculations and commercial implementations. Apparently, the bottleneck is promotion of new engineering and design solutions. Therefore, the search for new approaches to plastic recycling in SCFs should be concentrated, first of all, on easily industrially scalable processes.
12. Alternative concept of polymer waste management
The material presented in the previous parts of the review indicates that currently there is no universal way for polymer waste management. Therefore, some ecologists put forward the idea of abandoning polymers in the most large-scale applications — packaging industry and construction — and there are appeals to return to the glass or paper packaging. However, they forget that paper is a polymer material the manufacture of which is associated with considerable environmental costs and pollution, and that the power consumption for the glass manufacture casts doubt on the possible increase in the scope of glass application. One more dangerous idea is to turn the manufacture of polymers into the production of biodegradable polymer materials, which is proposed as a panacea in view of the failure to collect and recycle polymer waste in an economically feasible and environmentally benign way.473,474,640,973,974 The adherents of this approach forget that the final products of destruction of biodegradable plastics include greenhouse gases, which ecologists try so hard to control. Nevertheless, this ideology is persistent and is steadily developing, thus diverting the efforts and resources of scientists and industrialists from the search for really effective solutions, i.e., political and economic practice begins to affect the prospects of polymer science and engineering.
The packaging market size increases every year; hence, the environmental pollution by plastic waste grows. This problem has no one common solution. It is necessary to simultaneously develop polymer waste collection and sorting, recycling and environmentally safe disposal. Switching to biodegradable polymers is just one of the tools to reduce pollution. Examination of the recent scientific achievements demonstrates that many drawbacks of natural and synthetic biodegradable polymers can be eliminated, which would expand the scope of their applicability. Meanwhile, for the fabrication of available and fully environmentally benign materials, it is necessary to continue the design of more efficient processes for obtaining raw materials (in particular, lactic acid), studies in the field of green polymerization (using new catalysts, reducing energy expenditure) and the synthesis of composites with enhanced properties (functional additives and compatibilizers).
However, ignoring the fundamental approaches to solving the problem of polymer waste recycling not only retards the production of the most massive types of polymers, but also influences the progress of polymer science and engineering. Indeed, one or two research areas are normally selected to be supported out of the whole possible range. However, it is evident that they just cannot meet with success, since they immediately generate another, no less important problem, namely, a pronounced increase in the carbon footprint. Analysis of publications devoted to various aspects of the concept of biodegradable polymers, starting from development of methods for their synthesis (including studies of composting kinetics) and ending with kinetics of the degradation products, demonstrated that the insufficiently scientifically proven choice of the development strategy leads to accelerating movement in a wrong direction.
We do not mean that the development of biodegradable polymers has no prospects. They are needed for drug delivery systems, matrices for cell structures, medical suture material and various types of implants and prostheses, i.e., it is necessary to develop the manufacture of these materials according to the needs of their unique applications. However, these materials should not be opposed to readily available, cheap and efficient polymers as a result of a biased comparison of their benefits and drawbacks. This opposition brings about unjustified distortions in the distribution of efforts and funds and does not help to approach the main goal: development of a strategy covering the collection, processing and disposal of polymer materials in economically and environmentally sound ways.
In the situations where empirical search leads to a dead end, it is reasonable to find out how a particular problem is solved by nature, and often this helps to find a right way. Unlike human society, nature has successfully and long ago solved the problem of disposal of polymer waste, which is present in the environment in markedly larger amount than has been newly synthesized. Strange as it may seem, we were unable to find a systematic analysis of this fact in the literature. Let us look into this issue ourselves. It was noted in the Introduction that the nature uses renewable polymer materials as a potent tool for the control of not only the carbon cycle, but also the oxygen cycle of the Earth. Thus, plants, which are largely represented by various types of cellulose-based polymer materials, absorb carbon dioxide to generate oxygen, but simultaneously they actively release carbon dioxide and consume oxygen during wood oxidation. The following constituents of the handling of wood in nature can be noted:
(1) large-scale combustion during forest fires;
(2) active secondary use of wood (after completion of the life cycle of plants) for the construction of houses and other stuff for the animal world, which range from birds making nests to beavers, who use wood extensively and skilfully;*
(3) successive transformation of wood under oxygen deficiency into peat, brown coal, hard coal and oil, i.e., into the stored carbon reserves against which 'environmentalists' actively argue for the reason of changing the Earth's carbon balance.**
Surprisingly, the above-described 'civilization' approaches to polymer waste magagement generally mimic the natural ways. However, there is a significant difference: civilization methods show increasing trend towards combustion, whereas the nature uses the option of combustion very sparingly. Recycling (including that for meeting people's needs), which removes wood from oxygen generation processes and from the combustion with evolution of carbon dioxide, makes a large contribution to the disposal of wood. As regards landfilling, the contribution of this type of disposal gradually decreases, with widespread swamp drainage, forest clearance and changes in the Earth s crust, which are far from being regular.
Despite the apparent spontaneity and independence of the three constituents of wood handling, the concentrations of oxygen and carbon dioxide on the Earth are maintained constant. The role of wood in establishing this equilibrium is not entirely clear, but coincidence of the oxygen content in the atmosphere and the oxygen index of wood prompts the idea that this fact is not occasional. This emphasizes once again the important role of wood in this equilibrium. The relationship between the three main constituents of wood handling in nature is sharply changed in particular periods, which leads to pronounced fluctuations of the carbon dioxide content in the atmosphere. Nevertheless, the nature always returns to the observed optimal levels.
It follows from the above brief superficial analysis that despite the similar structure of trends of the natural and civilization handling of polymer matter, they also have basic differences. Recycling of plant wood provides for the production of high-quality materials at minimum cost (except for paper manufacture), whereas the recycling of polymer waste is always accompanied by a quality loss. Incineration gives rise to carbon dioxide and some hazardous components in both cases. Perhaps, the civilization approach is more favourable in this respect, because the hazardous compounds formed upon waste incineration are trapped, while the exhaust products of fires and even burning of firewood are not treated. The natural landfilling and conversion of polymer matter occur over different time periods; therefore, the similarity is only formal. It cannot but be mentioned that landfilling in the natural version is most efficient, as huge amounts of biomass are withdrawn from the turnover and are finally converted to effective fuel.
Although the operations on polymer waste treatment are formally similar, the time frame and the scale factor preclude direct implementation of the biomimetic approach. The natural cycle of waste treatment may be highly non-uniform and may result in sharp fluctuations of the carbon dioxide content in the atmosphere. The human population may not withstand such fluctuations; therefore, it is vitally important to have mechanisms for controlling the carbon dioxide and oxygen concentrations. In this respect, the only objective biomimetic conclusion that follows from the presented comparison is that carbochain polymers should be considered as an important tool to control the carbon cycle. For this tool to operate, it is necessary to optimize two most promising constituents of polymer matter disposal, namely, incineration and landfilling, with the focus being placed on the latter. The efficiency of landfilling may be increased at the expense of the former route, i.e., recycling may consist in the conversion of waste into the form optimal for the environmentally benign landfilling.
* It should also be borne in mind that people use enormous amounts of wood for economic purposes (building of houses and manufacture of furniture, transport facilities and various homeware).
** We did not mention natural gas, which is also formed upon biomass transformation under anaerobic conditions and which is concentrated, in the best case, in underground cavities and, in the worst case, as gas hydrates on the bottom of northern seas in permafrost areas.
13. Environmental and medical applications of polymer materials
Whereas the previous parts of the review consider how to prevent the environmental pollution by polymer waste, this Section, conversely, addresses a number of examples in which polymers serve for environmental protection.
13.1. Application of biocompatible and biodegradable natural polymers as multifunctional binders and sorbents for nature protection purposes, environmental geology and agrochemistry
Soil is a highly valuable natural resource, the state of which is exceptionally important for the production of foodstuff and maintenance of balanced life activity of ecosystems as a whole.975,976 Unfortunately, assessment of the state of soil cover in the world indicates that soils are being degraded: the soil fertility decreases, the acid-base properties change and contamination with toxic compounds, mineralization, etc. take place.977,978 Most of the problems are caused by poor structural state of soil and low soil stability to erosion processes in which soil degrades under the action of external factors such as wind, water flows or mechanical treatment.979,980
The physicochemical properties of soils are enhanced by using chemical binders, usually Portland cement, hydrated lime and microsilica.981,982 The introduction of these agents enhances the strength characteristics of soil.982,983 However, the cementing binders are not environmentally benign: their production and application are accompanied by considerable atmospheric discharge of carbon dioxide and nitrogen oxides.984
These factors initiated the search for more environmentally acceptable and inexpensive binders. A solution was found in the use of polymer additives.985 Commercially available urea formaldehyde resins, poly(vinyl acetate) and its derivatives, poly(vinyl alcohol), polyacrylates, polyurethanes and latexes are used as polymer soil stabilizers (which were called non-traditional).986 Acrylamide and sodium acrylate copolymers, known under the general term polyacrylamide (PAM), which demonstrate stable erosion prevention properties, have occupied the top position in this list over time. The accumulated experience of the use of PAM in laboratory and field tests is most fully reflected in review publications.987,988 In particular, it was shown that the rate of PAM degradation in the environment is less than 10% per year. The high stability of PAM and the necessity of frequent PAM addition to maintain the erosion prevention properties lead to the accumulation of the polymer in soil and simultaneous accumulation of the residual acrylamide monomer, possessing neurotoxic properties even in very low concentrations.
In recent years, polymers of natural origin (biopolymers) and the products of their processing have been used more and more often as soil conditioners;989 these polymers include heterofunctional polysaccharides (starch, agar, xanthan gum, carboxymethyl cellulose, hyaluronic acid, pectin, chitosan, etc.) and derivatives of lignin or soil organic matter (lignosulfonates, lignohumates, humic acids, fulvic acids). These polymers are biocompatible; under the action of physical factors and soil microorganisms, they degrade to low-molecular-weight products. The use of biopolymers markedly reduces the environmental impact on soil. A brief review of the studies dealing with biopolymers used most often as construction binding materials and soil structure forming agents is presented below.
13.1.1. Lignosulfonates
Lignosulfonates (LGS) are sulfo derivatives of lignin formed as by-products of wood processing in the paper and woodworking industry.990 The macromolecules of industry-based LGS are usually branched and contain functional groups, which provide good solubility in water, that is, alcohol, phenol, carboxyl and sulfo groups.991 The molecular weight of LGS is in the range from 5 to 400 kDa; they are non-toxic, do not cause metal corrosion and do not affect the soil acidity.992
Many researchers observed improvement of geotechnical characteristics of soils of various textures (dry sand, silty sand, sandy loam and soft clay) after treatment with LGS.993,994 The application rate of LGS (mass fraction of the polymer in soil) depends on the type of soil and varies from 0.2-0.6 mass% for loamy soil substrates to 2-6 mass% for sandy soil substrates. Numerous advantages of LGS over the traditional cement for increasing the stability of loamy soil to erosion by water was demonstrated by Vinod et al.995
Shivashankar et al.996 used LGS to prevent the erosion of dip slopes of clayey soil. LGS were also used as stabilizers for industry-produced soil substrates; for example, they are successfully suppressed the wind erosion of red sand, a coarse sandy bauxite residue formed as a by-product of alumina production.997
The above examples attest to stabilizing ability of LGS for both compact and loose soils; currently the interest in this polymer binder continues to increase.
13.1.2. Xanthan gum
Xanthan gum (XG), an anionic branched polysaccharide, is formed via aerobic fermentation of sucrose or glucose by Xanthomomanas bacterium.998 Xanthan gum is soluble in hot and cold water; the solutions have high viscosity even at low concentrations of the biopolymer owing to the rigid structure of helical macromolecules and their capacity for intermolecular interactions.999,1000 This anionic biopolymer easily binds water molecules through hydrogen bonds and forms elastic hydrogels at high concentrations. As compared with solutions of other polysaccharides, solutions of XG are distinguished by long-term stability upon variation of ionic strength and pH of the solution, successive freezing--thawing cycles, heating--cooling cycles and applying mechanical stress.1001
Xanthan gum is used for strengthening of loose sandy soils 1002--1004 and soft clayey soils;1005--1007 for waste stabilization in the tailing dumps of mining and processing plants1008 and for increasing the fracture toughness of clay structures.1009 The optimal content of XG for increasing soil strength is 1-2.5 mass%.1010
Xanthan gum proved to be efficient for preventing water erosion of the sandy banks of water streams.1011 The addition of XG to the Korean red soil, which refers to silty loams, in a quantity of 0.5-1.0 mass% increased the resistance of soil to erosion by water by almost an order of magnitude.1012 Being in contact with water, XG swelled and retained water in soil for a long period of time, which was favourable for plant growth.
Numerous examples of application of XG as an environmentally benign binder for geotechnical purposes are summarized in reviews.1013,1014 Our analysis of the published data and our own data show that the mechanical characteristics of soils are determined by the following parameters: the amount of XG applied; the type, density, porosity and moisture content of the soil substrate; and method of mixing of soil with the polymer. Alkali and alkaline earth metal ions improve the elastic properties of XG and act as bridges between the anionic XG macromolecules and negatively charged soil particles, giving rise to polymer--soil composites, which behave similarly to solid plastics. The considerable decrease in the selling price of this biopolymer indicates that it may serve as a competitive modern polymer binder.
13.1.3. Alginic acid salts
Commercially available alginates are usually extracted from brown algae, for example, from Laminaria digitata and Ascophyllum nodosum, by treating the algae with aqueous solutions of alkalis.1015 Alginates are linear copolymers of β-D-mannuronic acid (M) and α-L-guluronic acid (G). Commercial alginates differ in the number and sequence of acid residues and consist of homopolymeric M and G blocks alternating with variable-structure MG blocks, the ratio of which varies depending on the source.1016 In solution, alginates behave as flexible helices. Divalent metal ions form ionic bonds with the anionic groups of the polymer, which causes a collapse of the macromolecules and, in the limiting case, leads to the loss of their solubility.1017
The critical parameters determining the properties of alginate materials are their composition and molecular weight. An increase in the proportion and length of G blocks in the polymer is accompanied by increasing strength characteristics of alginate films and hydrogels, whereas a greater proportion of M blocks promotes the flexibility and elasticity.1018
Alginates were among the first polymers used to stabilize soils; the application of alginates for soil stabilization started in the 1940s.1019 However, the efficiency of these plant polymers proved to be moderate: a robust stabilization effect could be attained by introducing several tons of the polymer material per hectare. Later, it was shown that at these consumption rates, the content of nitrogen taken up by plants in soil decreases to a very low level.1020
Currently, the interest in alginates is being revived, although studies reported in the literature are mainly limited to laboratory tests. According to Fatehi et al.,1021 the compressive strength of finely dispersed sand of shifting dunes linearly increases as the biopolymer content increases from 1 to 5 mass %. Similar results were reported1013 for the alginate-silty sand system; the authors noted a lower stabilizing capacity of alginate compared to that of XG. Alginate was used to fabricate a soil constructor, an artificial soil blend composed of a high plasticity silt clay and tyre-derived ground rubber1022 and to immobilize shifting sand and stimulate the vital activity of microorganisms with subsequent desert reclamation.1023 Alginates formed the hydrophilic interlayers between quartz particles, which retained water and promoted colonization and growth of cyanobacteria, which are able to survive under extreme conditions of arid climate and take up atmospheric nitrogen and carbon dioxide, which mitigates the greenhouse effect1024,1025 and leads to formation of a productive soil in a desert.1026
Generally, the studies demonstrated an acceptable stabilizing effect and water-retaining properties of alginates; the results attest to good prospects of using this biopolymer and its derivatives as soil conditioners.
13.1.4. Carboxylated cellulose soils
Sodium carboxymethyl cellulose (Na-CMC) is a water-soluble product of reaction of cellulose with monochloroacetic acid in the presence of alkali;1027 it is widely used as a viscosity modifier, an emulsion stabilizer, a binder and a water-retaining agent. The key properties of this salt — solubility and viscosity and gelation capacity of solutions — are closely related to the polymer molecular weight and degree of carboxymethylation, i.e., the degree of replacement of cellulose hydroxyl groups by carboxymethyl groups.1028
Modification of the slopes of sandy loam soil with the biopolymer gave rise to an effective erosion control barrier.1029,1030 A noticeable increase in the sand stability to intense water flow was observed at the Na-CMC application rate of 0.7 mass%; however, the increase in the water resistance was accompanied by a decrease in the rate of water penetration into soil,1031 which was due to polymer swelling and formation of a viscous gel upon polymer contact with water. Field tests with long-term (three-year) monitoring showed that the surface layer involving Na-CMC can effectively control erosion of sand slopes for long periods of time.1031
Agrochemists consider Na-CMC to be a promising agent for combating desertification of territories and for obtaining polymer materials with controlled release of biologically active compounds. For this purpose, so-called superabsorbent hydrogels able to retain considerable amounts of water in the bulk were obtained on the basis of Na-CMC. Cross-linked derivatives of Na-CMC improve the hydrophysical characteristics of coarse-grained sandy soils and increases several-fold the amount of water accessible for plants in the root zone, which makes it possible to increase the time between waterings and mitigate the abiotic stress in plants caused by the lack of water.1032--1034 Particles of carboxymethyl cellulose hydrogels can act as depots for pesticides and/or adsorbents for binding toxic heavy metal cations.1035,1036 The biodegradation rate of Na-CMC hydrogel depends on the composition of the soil substrate and soil humidity and, on average, amounts to six months.1037 The biodegradation rate can be varied by adding antiseptic agents, which suppress the microbial activity.
The above properties confirm the high promise of using Na-CMC for formulations capable of suppressing erosion processes in soil and create (maintain) conditions for plant growth, especially in arid areas with insufficient water supply.
13.1.5. Chitosan
Chitosan is a biocompatible material that slowly decomposes into harmless products (amino sugars), which are either absorbed by living organisms or excreted from organisms.1038 Owing to accessible free amino groups, chitosan reacts with negatively charged micro- and macro-sized species and forms complexes both with anionic low- and high-molecular-weight compounds and with polyvalent metal ions.1039 For this reason, chitosan is actively used in environmental protection technologies as a flocculant for wastewater treatment and an adsorbent for binding and deactivation of various pollutants: dyes, phenol derivatives, pesticides and metals.578
Chitosan was first used for geo-environmental purposes about 20 years ago as a binder to suppress erosion of loamy soils during irrigation. According to laboratory tests, it was only slightly inferior to polyacrylamide in efficiency.1040,1041 Wind tunnel tests of sandy clays treated with chitosan showed their high resistance to air erosion.1042 A sharp decrease in the hydraulic conductivity of sandy soil after treatment with chitosan was noted;1043,1044 this may provide grounds for the design of barrier filters and removal of toxic metal ions from moving contaminated groundwater plumes.1044 During field tests, coatings obtained by applying a chitosan solution (0.5 mass%) on a soil of coastal region with a high content of clay particles protected the soil from water erosion.1045 In a similar testing of a sandy soil with a lower content of clay,1046 the water resistance was acceptable only upon successive application of 0.2 mass% of an aqueous solution of an anionic polysaccharide, carrageenan, and then 0.2 mass% of an aqueous solution of cationic chitosan onto the substrate surface.
Biodegradation of films involving chitosan was studied by Makarios-Laham and Lee.1047 Under laboratory conditions, the bacteria Serratia marcescens and Pseudomonas oeruginosa and fungus Beauveria bassiana absorbed 80% of the biopolymer within eight weeks of incubation at room temperature. In the soil, 100% biopolymer degradation took place within six months.
Chitosan may enhance the vital activity of plants.1048,1049 It was proposed as an antifungal agent against diseases of horticultural crops caused by the fungus Fusarium.1050,1051 The biocidal properties of chitosan and film forming properties promote the formation of film coatings on seeds for protection against pathogens and stresses.1052 The formation of coatings with optimized structure and mechanical properties requires, in some cases, the introduction of a plasticizer and/or a cross-linking agent.1053
Thus, chitosan has a high potential as a geo-ecological and agrotechnical material. However, field tests of this biopolymer that would describe the integrated effect of chitosan inclusion on the physical, mechanical and microstructural state of the soil and the vital activity of the soil microbiota have not yet been carried out.
13.1.6. Interpolyelectrolyte complexes of biodegradable polymers
The improvement of properties, increase in the stability and generation of new functions of biopolymers are usually attained by chemical modification. As an alternative, it was proposed to modify biopolymers by incorporating them into interpolyelectrolyte complexes (IPECs) formed upon interaction of oppositely charged ionic polymers (Fig. 64a).
![[{"id":"FeeKLptrkZ","type":"paragraph","data":{"text":" IPEC structure (<i>a</i>) and binding to soil particles (<i>b</i>).<sup>1054</sup> Published with permission from Elsevier."}}]](/storage/images/resized/NYJbxkYz9ELBX0LPufwfu0vrTlQ6U5O5Ztucij4K_xl.webp)
More than 50 years have passed since the first publications reporting the IPEC preparation. During this period, the regularities of formation and properties of these compounds have been extensively studied and repeatedly described.1054--1060 A fundamental characteristic of the polyelectrolyte complexes is their composition, which is usually expressed as the ratio of the concentrations of ionic groups of both components — the polyanion and the polycation.1056 In the case of equal charge to charge ratio of the complexes, stoichiometric water-insoluble polycomplexes are formed; an excess of any of the components gives water-soluble non-stoichiometric IPECs. The polycomplexes can be considered as copolymers containing both hydrophilic and hydrophobic blocks (see Fig. 64a). Isolated parts of polymer chains act as hydrophilic blocks, and hydrophobic blocks are sequences consisting of oppositely charged units of both polymers.
An important benefit of IPECs is simplicity of their preparation. A traditional method is mixing of aqueous solutions of the cationic and anionic polymers in a certain volume ratio.1058 This makes it possible to change the hydrophilic--hydrophobic balance of IPECs in a specified manner and to obtain polycomplexes with different overall charges and thus to control the IPEC solubility and reactivity towards low-molecular-weight, colloidal and macro-sized objects.
The amphiphilic nature of IPECs allows them to efficiently bind soil particles.1054 The charged areas on the surface of soil particles bind to oppositely charged isolated IPEC blocks; the hydrophobic areas on the particle surface bind to the hydrophobic blocks of IPECs (Fig. 64b). These interactions together ensure aggregation of soil particles.
The interpolyelectrolyte complexes of synthetic polymers, e.g., anionic sodium polyacrylate and cationic polydiallyldimethylammonium chloride, surpass single ionic polymers in their structuring properties.1061--1064 These IPECs efficiently bind both light loose sandy soils and well-aggregated loamy soils. By varying the IPEC composition, it is possible to vary the polycomplex composition, taking account of the type of soil, and thus to affect the mechanical strength and erosion stability of the protective polymer-soil coatings. The use of biodegradable polymers for the synthesis of IPECs opens up new prospects in the development of production techniques for soil conditioners. IPECs made of biodegradable polymers possess the same benefits as the traditional IPECs obtained from synthetic polymers, namely, the possibility of adjusting the composition of the polycomplex to physicochemical properties and structural features of soil, the ability to form strong and simultaneously moisture- and air-permeable coatings with high water-retaining properties for reasonable cost. In addition, new IPECs are biocompatible, biodegradable and, as a consequence, environmentally benign. After performing the stabilizing function, IPECs degrade to small molecules (fragments) and are removed from soil together with subsoil water and/or are digested by soil microorganisms.
Examples of using IPECs composed of biodegradable polymers for solving environmental problems have been reported in the literature. The studies addressed both polycomplexes in which only one component (lignosulfonate) was capable of biodegradation 1065,1066 and polycomplexes fully composed of biodegradable polymers, e.g., a mixture of alginate (or Na-CMC) and chitosan.1067,1068 Treatment of sandy soils with such formulations resulted in the formation of 5-10 mm thick protective crusts, which resisted the action of wind and rain and increased the moisture permeability of soil. The works along this line are now mainly restricted to demonstration of the stabilizing properties of biodegradable IPECs. Much less attention is given to the relationship between the composition of IPECs and the properties of soil substrate, on the one hand, and the strength of the polymer--soil coatings and their stability to the wind and air erosion, on the other hand. Meanwhile, this correlation is necessary for understanding the mechanism of action of new IPECs and, what is more important, for the development of multiple-action polymer structures (soil conditioners) capable of performing a number of essential functions: suppress erosion processes in soil, improve the soil structure, retain water, supply nutrients, bind toxins, control pH and so on. Solving these issues would markedly expand the potential of biodegradable IPECs as a result of using new polymers for the preparation of polycomplexes and using new types of soil, in particular degraded and artificial soils.
Owing to the broad set of functional groups, biopolymers form multiple electrostatic, hydrophobic and hydrogen bonds with structurally diverse mineral particles of various nature, soil substrates and low-molecular-weight compounds. This provides conditions for efficient and safe solution of a number of geo-environmental and agrotechnical problems. The biodegradability of natural macromolecules can reduce the environmental pollution and minimize the consequences of the secondary soil contamination caused by uncontrolled physicochemical and biological transformations of the polymers put into soil.
Currently, biocompatible and biodegradable natural ionic polymers are considered to be promising environmentally benign additives able to enhance the mechanical and rheological characteristics of mineral soil, prevent dust transfer and immobilize quick sand. Biopolymers can be used as soil conditioners to reduce the degradation of agricultural lands, to bind organic carbon and to detoxify and reclaim contaminated soils. Binding of a biopolymer into an IPEC with an oppositely charged (bio)polymer provides controlled modification of the polymer binder and formation of environmentally benign protective coatings on the soil surface. There are virtually no published data on the optimal compositions of these polycomplexes and their behavior as combined-action soil conditioners, whereas the experience of using polycomplex binders made of synthetic polymers demonstrated obvious advantages of polycomplexes over single polymers.
13.1.7. Waterborne polyurethanes
Owing to the enhanced ability to form host-guest supramolecular compounds, waterborne polyurethanes can be used both to prevent water pollution and to selectively and efficiently remove organic pollutants contained in water even in minor amounts.
The synthesis of environmentally benign waterborne polyurethanes (WBPU) is a vigorously developing trend of polymer chemistry. These materials have already largely displaced the conventional organo-soluble polyurethanes. Waterborne polyurethanes are used more and more often as coatings,1069 adhesives,1070 targeted delivery systems for biologically active compounds, including medicinal drugs1071 and agrochemicals1072 and for water treatment systems.1073
Like in the case of conventional organo-soluble polyurethanes, WBPUs are mainly synthesized by the reaction of diisocyanates with low-molecular-weight and oligomer diols and with diamines (Fig. 65).
![[{"id":"nNOEWe7Vmw","type":"paragraph","data":{"text":"Scheme of the synthesis of aqueous dispersions of poly urethanes."}}]](/storage/images/resized/u0gLgYxstYGLEJ9NMi5nQQDmTCZDGbKYlSVQyH0P_xl.webp)
The isocyanate components used for the synthesis of WBPUs include aromatic1074 and aliphatic diisocyanates1070 and also isocyanurate oligomers, resulting from their polymerization.1075 Among the wide diversity of diisocyanates, isophorone diisocyanate is used most widely for WBPU synthesis. This is due to the possibility of controlling urethane formation process taking advantage of different reactivities of the isocyanate groups of this diisocyanate.1076,1077 In order to increase the environmental safety of WBPUs, they are often prepared using biodegradable oligodiols, particularly, polycaprolactonediol and poly(butylene adipate).1078 One more hydroxyl-containing compound used for this purpose is castor oil obtained from renewable sources of raw materials.1079
The synthesis of non-isocyanate WBPUs by the reaction of bifunctional cyclocarbonates with diamines has been actively developed in recent years (Fig. 66).1080–1082
![[{"id":"UCrDT9wR7i","type":"paragraph","data":{"text":" Scheme of the synthesis of aqueous dispersions of non-isocyanate polyurethanes.<sup>1082</sup> Published with permission from the Royal Society of Chemistry."}}]](/storage/images/resized/NZ3do1Ik3lTBEZZWyE39S2ZU2AqTuRz8wfToUTEN_xl.webp)
Thus, a large diversity of the raw materials for the preparation of WBPUs in combination with the possibility of controlling the process of synthesis make an effective tool for tuning the properties of the final material.
The stability of aqueous dispersions of polyurethanes is largely attained by introducing internal emulsifiers into polyurethanes. All hydrophilic agents used for the synthesis of WBPUs can be divided into three groups:
- anionic [e.g., 2,2-bis(hydroxymethyl)propionic acid];1083
- cationic (e.g., N-methyldiethanolamine);1079
- non-ionic (e.g., PEG).1084
It should be emphasized that cationic type internal emulsifiers (specifically, 3,3’-diamino-N-methyldipropylamine) are mainly used for the synthesis of non-isocyanate WBPUs. Ionic type internal emulsifiers are neutralized by tertiary amines or carboxylic acids to form quaternary ammonium bases, which is also favourable for stabilization of waterborne polyurethanes.
It follows from the above that WBPUs are amphiphilic polymers in which urethane moieties linked to ionic centres form the hydrophilic part, while the urethane moieties of oligoesters and low-molecular-weight diols (diamines) represent the hydrophobic part. As a result, WBPUs tend to self-organize and can form micelles or vesicles.1071,1078,1083 This is a key feature of WBPUs, which ensures the possibility of encapsulating low-polarity molecules of biologically active compounds.
All benefits of WBPUs are fully utilized in the design of water treatment systems and in prevention of water pollution, as indicated by the examples listed below.
For preventing pollution of water by pesticides, WBPUs are widely used as pesticide delivery systems in agrochemistry, as they provide a controlled release of pesticides owing to their amphiphilic nature.1072,1079,1085,1086 An increase in the efficiency of pesticide action is achieved by encapsulation of biologically active substances into the nonpolar WBPU core. The resulting WBPU nanoemulsions applied on plant leaves ensure the location of pesticides in the close contact with potential pests for a long period of time. Due to the low surface tension of these emulsions and the possibility of hydrogen bonding between the leaf surface and the polymer matrix, the efficiency of retention of pesticides encapsulated in WBPUs is much higher than that with the use of commercially available formulations as emulsifiable concentrates or powders.1085 The limiting content of the popular pesticides such as avermectin and lambda-cyhalothrin in WBPUs can reach 40% with encapsulation efficiency being>85%.1072,1085,1086 As expected, the rate of release of pesticides from the polymer matrix depends on the amount of encapsulated biologically active compound, pH and temperature.1086
Zhang et al.1079 analyzed the effect of the nature of ionic emulsifier on the efficiency of azadirachtin encapsulated in castor oil-based WBPU. The highest photochemical stability of the pesticide was attained by encapsulation in polyurethanes obtained using a cationic type internal emulsifier. In particular, it was shown that 7-day natural light irradiation of azadirachtin applied using conventional Tween-80 emulsifier leads to decomposition of almost 70% of the biologically active compound, whereas the photodegradation of the WBPU-encapsulated pesticide is only 30% over the same period of time. The efficiency of azadirachtin retention in WBPU after simulation of atmospheric precipitation was 87.9%, whereas this value obtained after wash-off of samples applied using traditional formulations was only 7.3%.
Due to their hydrophilicity and environmental safety, foams based on WBPU with various immobilized nanoparticles can act as effective filtering systems for the treatment of water to remove various sorts of lyophobic compounds,1087 heavy metal ions,1088 phenol1089 and bacteria such as E.coli.1074 It is noteworthy that all composite materials used in the experiments were obtained in situ in an aqueous medium.
For example, WBPU-based polyurethane foams with immobilized thermally expanded graphite particles were prepared1087 for effective separation of oil-in-water type emulsions under the action of gravity. The results of measurement of the contact angles and sorption for water and various oleophilic compounds (diesel fuel, toluene, silicone oil) showed that these composite materials possess both hydrophilic and oleophilic properties with respect to air environment. After swelling in water, the foams started to show clear-cut oleophobic behaviour (contact angle of >133°). Filtering systems for oil-in-water emulsions with a separation efficiency of 99.99% were developed on the basis of thermally expanded graphite immobilized in WBPU.
WBPU-based gels are often employed for aerobic-anaerobic removal of nitrogen from waste water. In particular, cross-linked WBPUs can serve as matrices for immobilization of sources of nitrifying bacteria such as active sludge.1084,1090,1091 WBPUs used to fabricate these composites are mainly obtained using oligoether diols (PEG), isophorone diisocyanate and 2-hydroxymethyl acrylate.1084 Nitrifying bacteria are immobilized in situ in the polymer matrix in water via three-dimensional radical polymerization of WBPU.
Composites based on active sludge immobilized in a cross-linked WBPU matrix markedly surpassed their analogues using poly(vinyl alcohols), sodium alginate, their mixture and carboxymethylcellulose as the carrier for nitrifying bacteria in all key characteristics such as mechanical strength, biological activity, tensile strength and swelling ratio.1092 Moreover, WBPU did not affect the growth of bacteria or the structure of the bacterial community, resulting in increase in the amount of biomass. The obtained experimental data attest to good prospects of using cross-linked WBPUs for treatment of waste water from industrial plants. This is also confirmed by the fact that a number of laboratory bioreactors using biomass immobilized in WBPU-based gels have already been constructed.1073,1093,1094 The efficiency of waste water treatment by this method may reach 99.9% for NH4+ removal. 1095
Thus, considering polyurethanes, it was shown that the use of common polymers is shifting towards the environmental protection. The progress of polymer chemistry is accompanied by the appearance of new materials that promote highly efficient water treatment for removal of hazardous organic compounds. New measures for preventing contamination of living nature with hazardous compounds are based on more efficient and careful application of materials using special polymer systems.
13.2. Promising polymer materials for water treatment and for preventing water pollution with organic compounds
Numerous natural and synthetic polymer systems that are described in the previous part of the review by no means exhaust the applications of polymer materials for environmental protection purposes. Ever increasing attention is drawn by the polymers specially designed for useful functions in this field. The design and tailoring of polymer systems for water treatment processes are described below.
The traditional treatment methods such as precipitation, flocculation, coagulation and activated carbon treatment do not ensure efficient removal of organic pollutants from water. Currently, the main efforts in the development of water treatment methods are directed towards the use of membrane technologies1096 for micro-, ultra- and nanofiltration.1097,1098 The elaboration of relevant membrane processes requires new advanced materials, among which mention should be made of metal-containing nanoparticles, carbon nanomaterials, zeolites and polymer materials. Water treatment membranes are now being developed on the basis of nanocellulose,1099--1101 carbon nanotubes in a polymer matrix,1102,1103 metal nanoparticles in a polymer matrix.1104,1105 Dendrimers and highly branched polymers,1106,1107 molecularly imprinted polymers (MIPs)1108--1110 and other materials 1111,1112 are also considered to be promising for water treatment.
13.2.1. Molecularly imprinted polymers
The concept of molecular imprinting for the design of water treatment materials to remove organic pollutants is based on formation of so-called imprints of the template molecules of pollutants. As a result, MIPs are able to specifically (complementarily), selectively and efficiently recognize and covalently or non-covalently bind the molecules of the imprinted organic pollutants (Fig. 67). The issue concerning the manufacture, properties and various applications of MIPs are considered in a number of reviews.1109,1113--1115 The progress in the development of MIPs can be evaluated by considering the number of publications retrieved from Web of Science in response to the query molecularly imprinted polymer: 269 papers were published in 2006, 828 papers appeared in 2015, and more than 1200 papers appeared in 2022.
![[{"id":"1jrpU9VUA4","type":"paragraph","data":{"text":"Conceptual chart of the preparation of molecular-imprinted polymers and their use to remove organic pollutants from water."}}]](/storage/images/resized/YpT31zYDY05cj4o4DO2b1FEuRlintOABLsGsafPX_xl.webp)
Non-covalent imprinting is most popular because of the relative ease of implementation and versatility. Here the key role belongs to the selection of the functional monomer that form associates with the functional groups of the template molecule via ionic or hydrogen bond, π–π contacts, hydrophobic interactions, etc. The most promising functional monomers are, for example, methacrylic, acrylic, itaconic, p-vinylbenzoic, 2-trifluoromethylacrylic and 2-acrylamido-2-methyl-1-propanesulfonic acids. Apart from these functional monomers, some monomers less prone to form associates are used. Among such monomers, most popular are 4-vinylpyridine, vinylimidazole, aminostyrene, N,N-diethylaminoethyl methacrylate, 2-hydroxyethyl methacrylate, N-vinylpyrrolidone and acrylamide. These monomers provide control over the thermodynamic compatibility of MIPs with the medium in which they are synthesized and used for water treatment to remove organic pollutants. Combining various functional monomers for the formation of MIPs based on non-covalent contacts leads to the most efficient recognition and binding of organic molecules, because the imprint is most complementary to the template molecule.
The efficiency of MIPs for water treatment to remove organic pollutants is due to the fact that the imprint is preserved with time both in the absence of external impacts and under the action of elevated temperature, mechanical stress, osmotic pressure, irradiation, etc. The key role in this aspect is played by the cross-linking monomer, which fixes the imprint of the template in the polymer during the synthesis of MIPs via the formation of a cross-linked structure, which ensures the mechanical strength and a decrease in the MIP ability for swelling, melting and deformation at temperatures above the glass transition temperature. However, a superfluous content of the cross-linking monomer leads to a decrease in the imprinting efficiency, since the number of formed imprints decreases. The optimal content of the cross-linking monomer usually corresponds to a 3-7-fold molar excess of the cross-linking monomer over the functional monomer.1116 A lower excess of the cross-linking monomer gives rise to MIPs in which the imprints of template molecules are located too closely to one another and can even overlap, which disrupts their complementarity and decreases the MIP selectivity and efficiency. Ethylene glycol dimethacrylate, trimethylolpropane trimethacrylate, divinylbenzene, N,N’-methylene-bis-acrylamide, etc. are widely used as cross-linking monomers.
The efficiency of MIPs is considerably affected by the medium in which they are synthesized. The solvent can influence, first, the morphology of the polymer matrix (the presence of meso- and macropores) and, second, the strength of interaction between the template molecule and functional monomers, thus changing both the shape of the imprint and functional group accessibility and proximity to the molecular imprint surface . The solvents used most often in the synthesis of MIPs include acetonitrile, chloroform, toluene, N,N-dimethylformamide, methanol, tetrahydrofuran and dichloroethane. The efficiency of prepared MIPs depends, first of all, on the solvent polarity. When water is treated to remove organic pollutants, the effect of water as a highly polar solvent is great; for example it can disturb the equilibrium between organic pollutant molecules bound to the imprint and pollutant free molecules in the aqueous solution. This fact should be borne in mind while choosing functional and cross-linking monomers to increase the efficiency of MIPs.
The most frequently used MIP production processes are free radical polymerization, ionic polymerization, polycondensation and polyaddition, electropolymerization and oxidative polymerization. The main requirement to these processes are high conversion of the monomers under conditions favourable for specific interactions between the functional monomer and the template molecule. Free radical polymerization is used most widely due to the wide choice of vinyl(idene) monomers, moderate requirements to the special purification of monomers and solvents and initiating systems that operate at room temperature (UV and visible light initiation,1117,1118 initiation by γ-radiation1119) or at moderate temperatures. Furthermore, an additional benefit of this method for obtaining MIPs is the development of reversible deactivation radical polymerization, which produces polymers with a narrow molecular-weight distribution and, in the case of copolymerization, gives compositionally uniform copolymers with fine-tuned macromolecular design,1120,1121 which is necessary for providing highly specific interaction between the imprint and an organic molecule. Meanwhile, copolymerization with cross-linking comonomers, which follows a clearly microheterogeneous mechanism under usual conditions, involves a lower degree of microsyneresis when occurs in the living polymerization mode; this allows the preparation of cross-linked polymers with a more uniform morphology at the microlevel.1122 This increases the capacity and efficiency of MIPs for the recognition of the molecules of organic pollutants.
Molecular imprinted polymers with an extensive specific surface area are developed for highly efficient removal of organic pollutants. The traditional approach involves the formation of porous and mesoporous polymers, which can be obtained in different ways,1123,1124 in particular, in the presence of non-polymerizable low-molecular-weight1125 and high-molecular-weight porogens.1126 Mesoporous MIPs can be utilized as monolithic membranes or microparticles obtained by grinding of monolithic samples. An alternative approach is based on the formation of polymer nano- and microspheres formed upon precipitation,1127 suspension,1128 emulsion or miniemulsion polymerization.1129 These nano- and microspheres of polymers are characterized by a narrow size distribution and can also have a mesoporous morphology. A promising approach is the fabrication of (nano)composite materials in which MIP are integrated.1130
As examples, consider several particular results on the development of MIPs for the recognition and binding of organic pollutants. Li et al.1131 obtained MIP based on oleic acid and divinylbenzene by inverse microemulsion polymerization; the maximum adsorption capacity of the product towards 2,4-dichlorophenol was 183.8 mg g–1. Reversible addition-fragmentation chain transfer precipitation polymerization was used1132 to obtain MIP imprinted with 2,4-dichlorophenoxyacetic acid, which showed 25-45% higher maximum capacity than non-impregnated analogues. High efficiency was found for MIPs based on acrylamide and glycidyl methacrylate copolymers;1133 in the formation of molecular imprints complementary to 2,4-dinitrophenol, they increased the maximum sorption capacity from 41.5 to 138.9 mg g–1. For the removal of polycyclic aromatic hydrocarbons (benzanthracene, benzopyrene, dibenzopyrene, etc.) using polymer nanoparticles based on methacrylic acid and trimethylolpropane trimethacrylate, the adsorption capacity increased after molecular imprinting from 119 to 687 mg g–1 (Ref. 1134). It is evident that molecular imprinting considerably increases the efficiency of polymer materials for removal of organic pollutants from water.
It is noteworthy that the development of MIPs is a multidisciplinary problem that should be addressed taking account of a lot of factors (Fig. 68). Solution of such a multidisciplinary problem requires a comprehensive approach including both fundamental and applied research.
![[{"id":"80COMIup-j","type":"paragraph","data":{"text":"Basic interrelated factors that should be considered to develop MIP for a particular imprinted organic pollutant."}}]](/storage/images/resized/gPxWFFtxqwSpQirmiq6RkGwpcQzFEFGInAHVtisC_xl.webp)
13.2.2. Microgels
By microgels are traditionally meant solvent-swollen polymer particles of a network architecture with sizes from tens of nanometres to a few micrometres.1135 These macromolecular entities combine the properties of polymers and colloidal particles. Owing to their structure, microgels swell in good solvents and thus become deformable (soft), porous and colloidally stable, which is typical of polymers in good solvents. Conversely, in poor solvents, microgels collapse, and in this state they displace most of the solvent from the bulk of the network and behave as solid colloids, including the capability for aggregation. Also, microgels are known for their high surface activity: upon adsorption at the interface, the networks are distorted and flattened, and the surface tension decreases. This points to an analogy between microgels and surfactants.1136 Of special interest are polymer particles sensitive to changes in the medium in which they dissolved. In particular, water-soluble microgels can change the degree of swelling (and, hence, the permeability and deformability) depending on external stimuli such as temperature and pH changes.1137 The above properties characterize microgels as unique high-molecular-weight compounds possessing a high potential for practical applications in various fields, one of the fields being the removal of organic compounds, metal ions and other pollutants from water.1138 In this case, benefits of microgels are the possibility of catalytic neutralization of the collected material and the possibility of reuse. This part of the review describes examples of successful application of stimulus-sensitive microgels as water-soluble sorbents.
13.2.2.1. Chemical structure
The responsiveness of a polymer network to external stimuli depends on the polymer composition (Fig. 69). The most frequently encountered type of microgels are temperature-responsive particles based on N-isopropylacrylamide (NIPA), which swell in water at temperatures below the volume phase transition temperature (VPTT), amounting to ≈33 °C.1139--1143 This feature is due to the presence of hydrophilic and hydrophobic groups in the network units: the hydrophilic groups are hydrogen-bonded to water, while hydrophobic interactions of insoluble groups increase with temperature rise. Other examples of temperature-responsive monomers are N-isopropylmethacrylamide (NIPMA) with a higher VPTT (≈45 °C)1144--1146 and a NIPA isomer, N-n-propylacrylamide (NNPA) with a lower VPTT (≈23 °C).1147,1148 The incorporation of comonomers with functional groups into microgels during the synthesis gives multiresponsive microgels. The swelling ratio and response to the main stimulus for this material differ from those for homopolymer networks. For example, VPTT of amphiphilic microgels decreases depending on the proportion of the hydrophobic comonomer.1145,1149 Conversely, an increase in VPTT can also be attained by incorporation of charged comonomers (anionic1139--1141,1144,1150 or cationic1143,1151). The electrostatic repulsion between the like charges would result in additional extension of the polymer network subchains, and the microgels would become responsive to solution pH and ionic strength. For example, the polymers containing acrylic acid (AA) monomers swell at pH≥5,1141 while those containing 1-vinylimidazole monomer swell at pH≤4.1143 Mention should also be made of the possibility of obtaining water-soluble particles containing biodegradable cellulose,1152 β-cyclodextrin1153 or chitosan1154 macromolecules. Finally, it is possible to incorporate inorganic metal nanoparticles,1147,1149,1155,1156 oxide nanoparticles1150,1151 or minerals1157,1158 into the polymer networks. This gives hybrid microgels possessing high capacity for absorption of metal ions, which serve as catalytic sites, or responding to an external magnetic field (apart from the main stimulus).1150,1151
![[{"id":"t6FdHL9_A5","type":"paragraph","data":{"text":"Wheel of functional monomers forming the basis of water-soluble stimuli-responsive microgels."}}]](/storage/images/resized/iHynOXuF7HSy78Nb0n2i0n3sJQ6BuR5YGdHFk5F9_xl.webp)
13.2.2.2. Removal of pollutants
The efficiency of application of microgels of a definite composition depends on the type of water pollutants, the main of which are heavy metal ions (mercury, lead, cadmium, zinc and their compounds), dyes (first of all, azo dyes) and other organic compounds dissociating in water (ionic surfactants, phenols, etc.).1138 The electrostatic repulsion between the ion or molecule and a charged group of the polymer network is the driving force for the pollutant removal. Anionic polymer microgels were reported to absorb Cu2+ (see1139,1145,1146,1153,1156), Pb2+ (see1145,1153,1159), Cd2+ (see1145,1153), Co2+ (see1146) and Cr3+ ions1145 and cationic dyes: safranin,1140 brilliant green,1140 brilliant cresyl blue,1140 methylene blue1144 and rhodamines.1144,1150 The networks with cationic groups are able to absorb AsO43- anions,1151 orange II dye molecules,1143 and also anionic surfactants,1160 nitrophenols1161 and humic acids.1162 The hybrid microgels containing nanoparticles of the mineral attapulgite can, in addition, collect Zn2+ and Ni2+ ions.1157,1158 The uptake of the networks directly depends on the content of charged groups (the higher the charge, the greater the amount of pollutant that can be captured),1140,1159 their chemical structure (e.g., microgels containing AA monomers capture lead ions more efficiently than ions of other metals)1145,1153,1157,1158 and pH of the medium (the anionic and cationic groups are activated at high and low pH, respectively).1143,1144 Another mechanism of absorption may be due to the hydrophobic interactions, which are enhanced in temperature-responsive microgels as the temperature is raised. This allows one to conduct absorption of charged molecules by both neutral networks1163 and likely charged networks. The latter fact was demonstrated for microgels based on NIPA and AA and also based on NIPMA and AA in the removal of anionic dyes, orange II1141,1142 and Congo red,1138 respectively. In addition, hydrophobic interactions are the key factors responsible for the absorption of oil and oil products by water-insoluble (and non-responsive) microgels.1164,1165
13.2.2.3. Multiple use
Apart from the absorption of pollutants, an important aspect is the collection of microgels after treatment and the possibility of reuse. The simplest method is flocculation, which takes place on heating of temperature-responsive networks1141,1142,1159 or spontaneously as the substance is being absorbed by polyelectrolyte networks.1151,1157,1158 In this case, the sorbent can be readily separated from the aqueous medium. In addition, magnetically responsive composite particles can be collected by applying an external magnetic field; therefore, they can be used in open water areas (however, it should be borne in mind that the uptake rate somewhat decreases compared to fully polymer analogues).1161,1162 A pollutant can be extracted from microgels by swelling in a solvent upon decrease in the temperature1142 or change in the pH,1151 while the networks can be reused. For example, multiple use (five absorption-release cycles) was demonstrated for both anionic microgels (using the orange II dye)1141,1142 and cationic microgels (using AsO43- ions).1151 Meanwhile, for composite networks with the mineral attapulgite nanoparticles, the efficiency of removal of Pb2+ and Cu2+ ions from water virtually did not change after 10 cycles; the networks containing Fe3O4 nanoparticles were shown to be applicable five times for removal of various phenols from water.1161
13.2.2.4. Catalytic neutralization
Apart from being used as sorbents, water-soluble microgels can be used for the manufacture of green catalytic systems 1166--1168 for the conversion of the absorbed pollutants. In most cases, these systems are aqueous solutions with composite particles made of responsive polymer networks and metal nanoparticles (mainly, of noble metals).1147--1149,1155,1156,1166,1169,1170 Examples of conversion of aromatic compounds using composite microgels include the following processes: reduction of 4-nitrophenol to 4-aminophenol with gold,1166,1170 silver,1147,1149,1170 platinum1156 and palladium1148 nanoparticles; reduction of nitrobenzene to aniline with silver nanoparticles;1149 oxidation of benzyl alcohol to benzaldehyde with gold nanoparticles.1169 Organic pollutants can also be degraded, which was shown for dyes: Congo red,1170 methylene blue,1170 methyl orange1155 and eosin H.1155 The catalytic activity correlates with the absorption capacity of the polymer network, which in turn depends on various factors (see above) and can be controlled by an external stimulus.
Thus, stimuli-responsive microgels are promising candidates for cleaning of water areas, which was confirmed by numerous laboratory studies. The extensive practical use of network particles could be facilitated by solving a number of relevant issues that currently exist. First of all, it is necessary to arrange a large-scale production of microgels with a controlled internal structure; one can hope that this would benefit from the active development of microfluidics.1171,1172 One more task is to provide multiple use of microgels for scavenging of oil and oil products, because repeated use of a non-responsive sorbent can be difficult.1165 This problem should be addressed by choosing networks readily soluble in oil (for example, networks based on N-vinylcaprolactam1166--1168,1173) and with water solubility varying in reponse to external stimuli. Finally, one more task is to collect the microgel sorbent with the absorbed pollutant on open water areas. This problem can be addressed by using composite particles responsive to external magnetic field.1161,1162 Generally, it can be expected that the high research interest in responsive colloidal networks and their undoubted practical potential would serve as the base for wide use of polymer microgels in various industrial branches to improve their environmental friendliness.
13.3. Biologically active water-soluble synthetic polymers for biomedical purposes
The progress in the field of water-soluble polymers also involves a highly important application of these systems, namely, the design of polymer materials for medical purposes. Pressing problems of biology and medicine initiated the appearance of the chemistry of biomedical polymers. Recent research in this area has been focused, first, on modification of known drug substances in order to improve their therapeutic properties, provide targeted delivery to the biotarget (organ), and, second, on the search for new macromolecules possessing their own biological activity and simulating the functioning of biopolymers.
The synthesis of structurally diverse macromolecules and in-depth study of their properties revealed a broad range of biological activity, manifested at the molecular and cellular levels and at the whole-body level.
Using the accumulated knowledge on the control of synthesis methods, molecular weight characteristics, chemical structure and supramolecular structure of macromolecules, it is possible to solve particular biomedical problems and fabricate polymer systems with targeted biological action. It should be noted that the biological activity of most synthetic polymers is caused by their macromolecular nature and exists only at the polymer level.1174 Water-soluble polymers provide unique opportunities for the design of polyfunctional biologically active systems and delivery systems for the targeted therapy and diagnosis.1175–1178
The current coronavirus pandemic stimulates the search for effective antiviral compounds that would not be toxic to the host cells. Since viral infection is often accompanied by a bacterial infection, macromolecular systems possessing combined (antimicrobial and antiviral) biological activity may be useful for prevention of viral and bacterial infections.
The most promising compounds that deserve close attention are membranotropic polymers, polyelectrolytes (charged macromolecular compounds — polycations and polyanions) and water-soluble macromolecular chelating agents.
13.3.1. Membrane-active polymers
Membrane-active polymers are water-soluble macromolecular systems that actively interact with biological membranes and change their barrier functions. Among these polymers, the most interesting macromolecules are cationic polyelectrolytes with positive charge both in the backbone and in the side chain. The main biological target of these polymers in a bacterial cell is the cytoplasmic membrane.1177,1179 The decisive factor is the electrostatic interaction of cationic polyelectrolytes with proteins and phospholipids in the cell membrane. This entails neutralization of the charge of the membrane and the whole cell. This changes the packing density of lipid chains, which results in increase in their mobility, decrease in the barrier functions, increase in the membrane permeability, escape of low-molecular-weight metabolites from the cell and cell death. Polycations have a broad-spectrum antimicrobial activity; they suppress the growth of gram-positive and gram-negative bacteria, viruses and fungi when present in concentrations of 5-100 μgmL–1 (Ref. 1177).
The molecular weight distribution of ionogenic groups along the chain and type of counter-ion do not affect the antimicrobial activity of polycations. It is determined by the content of ionogenic groups in the macromolecule.1180 Owing to the presence of membrane activity, cationic polyelectrolytes can be used to increase the cellular uptake of low-molecular-weight (e.g., antibiotics)1177 and high-molecular-weight (e.g., DNA)1181 target compounds, and in biotechnological processes, this can be used to improve the release of target compounds from cells.
The targeted delivery of the foreign genetic material to specified intact cells is a promising method for solving problems of biotechnology, molecular biology and gene therapy. The construction of gene vectors for the targeted delivery of genetic material is associated with the necessity of compacting the rigid DNA molecule bearing negatively charged phosphate groups. The gene vector must penetrate through cell membrane and act as a viral particle carrying modified DNA. Viral particles are not actively used due to immunological reactions and the risk of pathological cell transformation.
An alternative method is to form IPECs of DNA with synthetic polycations in which DNA is compacted and protected from protonation, the action of nucleases and other unfavourable factors of the environment. Interpolyelectrolyte complexes with DNA are obtained using various polycations: polylysine, polyethyleneimine, polyvinyl-polyallylamine, polyaminoacrylates1182--1184 and so on. Note that DNA-polycation complexes are stable, they have a lower cytotoxicity than the polycation and a higher degree of transfection (up to 90% and more) of eukaryotic cells. Hence, these complexes could serve as promising vectors for DNA transfection. Since polycations act on several targets in the cell (destabilize the cytoplasmic membrane and suppress the activity of bacterial enzymes located in the membrane), bacterial resistance to them is formed more rarely than the antibiotic resistance. Therefore, in recent years these polymers have attracted increasing attention1178,1185,1186 as potential agents for the prevention and treatment of diseases caused by bacterial and viral infections and as compounds with a broad spectrum of biological activity. Thus, polycations based on aminoacrylates were found to have desensitization and reparative effects on bone and soft tissues1187 and also antitumour and immunomodulatory activities.1188 In addition, water-soluble copolymers of dimethylaminoethyl methacrylate with N-methyl-N-vinylacetamide and methacryloylaminoglucose reduce Ag+ to Ag0 and form water-soluble dispersions of silver nanoparticles active against H1N1 influenza virus and against gram-negative (E.coli) and gram-positive (Staph. aureus) bacteria.1189
13.3.2. Macromolecular chelating agents and polyanions
Membrane activity is also inherent in water-soluble polymers containing carboxyl, phosphonic, sulfo and various chelating groups.1177,1190–1192 These polymers are able to bind metal ions not only in common solvents, but also in biological objects. For example, ethylenediaminetetraacetic acid (EDTA), a known low-molecular-weight chelating agent, binds up to 40% of Mg2+ ions and up to 80% of Ca2+ ions present in the bacterial cell wall and cytoplasmic membrane, thus increasing their permeability. A polymeric EDTA analogue, copolymer of N-vinylpyrrolidone and N-vinyliminodiacetic acid, also binds Ca2+, Sr2+, Zn2+, Mg2+ and other cations,1190 changes the cell membrane permeability and increases the sensitivity of antibiotic resistant bacterial strains to, for example, tetracycline, penicillins, etc.
Water-soluble methacryloylacetone-N-vinylpyrrolidone copolymers, in which more than 90% of β-dicarbonyl groups are enolyzed in aqueous solutions, form stable complexes with Ca2+, Y3+, Zn2+, Mg2+ and Cu2+ ions.1191 These copolymers are able to break the Mg2+ and Ca2+ complexes with the phospholipids, proteins and lipopolysaccharides of cell membranes; they increase the membrane permeability and promote, for example, the chlortetracycline transport across the cell membrane (the bacterial resistance to this antibiotic is caused by decreasing membrane permeability).1177
13.3.3. Interferon-inducing and antiviral activity of polyanions
The antiviral activity of polyanions was first discovered by Regelson1193 for ethylene copolymers with maleic anhydride. Later, it was ascertained that preliminary administration of solutions of N-vinylpyrrolidone copolymer with crotonic acid or with maleic anhydride prevents the death of mice infected with tick-borne encephalitis and louping ill viruses.1194,1195 Furthermore, these polymers protected animals from infection with Aleutian mink disease and influenza viruses. The antiviral activity was found for polyacrylic and polymethacrylic acids and copolymers of divinyl ether with maleic anhydride (DIVEMA). It was found that DIVEMA and N-vinylpyrrolidone-maleic anhydride copolymers increase the resistance of experimental animals to various types of viruses (in particular, rabies virus).
The antiviral action of the polyanions upon intraperitoneal administration is associated with induction of interferon synthesis (which usually takes place in response to a viral infection) in the animal body.1196
The antiviral and interferon-inducing activities was found for polymer derivatives of para-aminosalicylic acid (PAS), which is used as an antituberculosis drug.1197 The authors studied the effect of the chemical structure of the polymer derivatives of PAS such as the type of the polymer-PAS bond (azomethine, amide) on the degree of protection of mice infected with tick-borne encephalitis virus and on the interferon concentration in the blood after intraperitoneal administration of 1% aqueous solutions of polymers. The survival rate of animals reached 35-45%, and interferon was detected in blood for 48 h. High interferon-inducing activity was found for DIVEMA copolymer after subcutaneous, intraperitoneal and intragastric administration.1198 Irrespective of the way of administration, interferon appeared in blood after 6-12 h and reached the maximum concentration within 24 h to 4 days. Analogous results were obtained1199 when interferon synthesis was induced by complexes of acridone acetic acid and N-alkyl-vinylamine copolymer with N-vinylpyrrolidone. The highest level of interferon was observed 8 h after the intraperitoneal administration of the complexes. These data indicate that these polymers (unlike low-molecular-weight analogues) are capable of delayed and remote induction of interferon synthesis; hence, these compounds can be used for the prevention of diseases caused by viral infections.
13.3.4. Anti-adhesion and virucidal action of polyanions
A viral infection starts with non-specific adsorption of viral particles (virions) on the membrane of the host cell. This is followed by receptor-mediated adsorption, which leads to penetration of the virus into the cell. In the early stage, the key role is played by electrostatic interactions and effects related to the structure of viral receptors. The pH dependence of the adsorption of the virus (reovirus) on the charged membranes was demonstrated under model conditions, and it was shown that in neutral media the viral particles are negatively charged.1200 Detailed electron microscopic examination of the effect of the nature of synthetic polycation, polyanion and neutral polymer on the adsorption of some viruses dangerous for humans was used to study the adsorption of viral particles 1201 on the polymer layers deposited on a substrate. The chosen water-soluble polymers included macromolecules carrying an NH2 group (polyvinylallylamine), a tertiary amino group and a quaternary ammonium group (polydimethylaminoethyl methacrylate and its ammonium salts), a neutral polymer (polyvinylpyrrolidone) and a polyanion (polyacrylic acid). In sorption experiments, the virions of rotavirus, adenovirus, astrovirus and two types of caliciviruses were used.
Polyvinylpyrrolidone did not significantly affect the adsorption of these virions; polyacrylic acid substantially decreased the adsorption for all viruses, which attested to their negative charge (except for adenovirus); and all polycations efficiently adsorbed astroviruses, rotaviruses and caliciviruses. The viruses showed the highest affinity to polyallylamine, in which the NH2 group was more remote from the backbone. Zerda et al.1200 gave an explanation for the high antiviral activities of synthetic and natural sulfo-containing polymers: dextran sulfate, heparin, polyvinyl sulfonate, polystyrene sulfonate, polyvinyl alcohol sulfate, etc.1180,1202,1203 It is believed1204 that these polymers inhibit the interaction stage of viral particle with the glycosaminoglycan receptor of the target cell.
Among the variety of sulfo-containing polymers, considerable attention of researches is paid to polystyrene sulfonate, which has a broad range of biological action.1205 It is active against influenza, herpes and rabies viruses, coronavirus, HIV, etc. It also shows an antibacterial activity against gonococci, chlamydia, and some other bacteria and is considered as a promising prophylactic agent against pathogenic microorganisms that cause sexually transmitted diseases. However, homopolymers (sodium polystyrene sulfonate, sodium polyvinyl sulfonate, poly-2-acrylamido-2-methylpropanesulfonic acid) have relatively high cytotoxicity, with IC50 being 6-18 μg mL–1, and this restricts their application. Therefore, a number of sulfo-containing copolymers with hydrophilic monomers such as N-vinylpyrrolidone, acrylamide, etc. were synthesized and showed a lower toxicity.1206,1207
With the goal of extending the range of biological activity and obtaining a macromolecular system combining antiviral and antibacterial activities, carboxyl- and sulfo-containing polymer carriers, namely, copolymers of crotonic acid and sodium vinyl sulfonate with N-vinylpyrrolidone and 2-acrylamino-2-methylpropanesulfonic acid, were used to obtain complexes with cationic surfactants (Katapol bactericidal agent), antibiotic gentamycin and antiviral agent Arbidol. The products were active against the herpes and influenza viruses and had a broad spectrum of antimicrobial activity and low toxicity.1207,1208
The interaction of Katapol with some viruses was studied by transmission electron microscopy; it was found that Katapol nanoparticles (Fig. 70, Fig. 71) actively interact with the virion envelope. For 0.05-0.2% concentration of Katapol,100% destruction of influenza, parainfluenza, coronavirus and rotavirus virions was observed. Higher stability of coronavirus compared to influenza virus is probably attributable to the fact that the lipid envelope of the coronavirus is stabilized by special M-protein, which pierces the lipid bilayer. Adenoviruses are more resistant to Katapol. When the concentration of the bactericidal agent was 0.5%, up to 50% of the virions were damaged.
![[{"id":"tdneJuu1Ml","type":"paragraph","data":{"text":"TEM images of the interaction of Katapol nanoparticles with the virions of influenza virus (C=0.005%) (<i>a</i>), coronavirus (C=0.05%) (<i>b</i>), rotavirus (C=0.05%) (<i>c</i>). Figure a shows fusion of Katapol nanoparticles with the virion envelope (400000 magnification); Figureb depicts the destruction of surface receptors and disruption of the integrity of the virion envelope (250000 magnification). The photographs from the personal archive of E.F.Panarin were presented at the symposium Improvement of the Preparedness for Influenza Pandemic based on the Civilian Military Cooperation (A.K.Sirotkin, I.S.Kochetkova, E.F.Panarin. <i>Proceedings of the Symposium.</i> St. Petersburg, 2003, p. 59)."}}]](/storage/images/resized/eeY2WHSXWEp8pONaFL4gpcUfBENFd8klX7O08nQs_xl.webp)
![[{"id":"eWDfjLYyoT","type":"paragraph","data":{"text":"TEM images of the coronavirus (<i>a</i>) and rotavirus (<i>b</i>) virions (250000 magnification). The photographs from the personal archive of E.F.Panarin were presented at the symposium Improvement of the Preparedness for Influenza Pandemic based on the Civilian Military Cooperation (A.K.Sirotkin, I.S.Kochetkova, E.F.Panarin. <i>Proceedings of the Symposium</i>. St. Petersburg, 2003, p. 58)."}}]](/storage/images/resized/L8PULfa6f8qSz2i6uIAOIAum20nFLtllEgDR3707_xl.webp)
The synthesized polymers with polyfunctional biological activities are of interest for comprehensive investigation and for development of agents for preventing viral and bacterial infections.
13.3.5. Metallopolymer complexes for bioimaging. Luminescence imaging
Luminescence methods of imaging have long been used in biological and medical studies. Organic chromophores are mainly used as luminescent materials. However, they do not provide stable luminescence, show a broad (>100 nm) luminescence band and decolorize. Therefore, in the last decades, a lot of attention has been paid to luminescent lanthanide coordination complexes.1209,1210
Lanthanides (Eu, Tb, Sm, etc.) possess unique optical properties: monochromatic emission (5-10 nm-wide luminescence bands), stability of emission over time, invariable positions of luminescence bands irrespective of the solvent and ligand nature and long excited state lifetimes (≈1000 μs).1211 Lanthanide metallopolymer complexes are of particular interest for bioimaging; unlike low-molecular-weight complexes, they ensure a longer residence time and increased concentrations of the lanthanide in the animal body and are suitable for studying the pharmacokinetics and localization of biologically active compounds in tissues and cells. Metallopolymer complexes for bioimaging should be water-soluble, stable, non-toxic and compatible with fluids and tissues of the living body, and the polymers should have chelating groups in the molecule and provide the transfer of electron excitation energy from the triplet level to the resonance level of the lanthanide ion.
Macromolecular ligands appropriate for binding lanthanide ions are carboxyl-containing polymers such as homo- and copolymers of acrylic, methacrylic and maleic acids with hydrophilic monomers (N-vinylamides, vinyl saccharides), polymer derivatives of aminobenzoic and salicylic acids, β-dicarbonyl compounds, etc.1212,1213 The most widely used chelating agents are iminodiacetic acid derivatives: 1,4,7,10-tetraazacyclodecane-1,4,7,10-tetraacetate (DOTA) and diethylenetriaminepentaacetic acid (DTPA), which were approved for clinical use.1214 Using DOTA, intensely luminescent iridium complexes were synthesized and were incorporated into temperature-responsive poly(isopropyl methacrylamide).1215
The introduction of a relatively high amount of DOTA or its derivatives into a macromolecule is associated with certain experimental difficulties caused by steric restrictions generated by the structure of the acid. Therefore, research and development of methods for the synthesis of new macromolecular chelating agents with different structure of the chelating groups have been carried out in recent years. Salicylaldehyde-based macromolecular chelating agents, which formed stable complexes with Ga3+, were prepared using reactions in N-vinylpyrrolidone copolymer backbones.1216,1217 It was shown that water-soluble copolymers of N-vinylpyrrolidone with N-allyl-N-aminosalicylic acid with a low content of chelating units form stable complexes with Eu3+ in the presence of thenoyltrifluoroacetone as a low-molecular-weight co-ligand; the intensity of luminescence of these complexes in aqueous solutions was 1-1.5 orders of magnitude higher than the luminescence intensity of a single Eu3+ complex. The observed effect of the polymer is probably caused by hydrophobization of the macromolecular coil upon the replacement of water molecules in the solvate environment of the chelating group by Eu3+ ions. The metallopolymer complexes were stable in solutions simulating biological fluids.
DOTA analogues, water-soluble macromolecular chelating agents with a high content of chelating groups, were prepared by copolymerization of N-vinylpyrrolidone with a vinyl monomer, 2-methacryloyloxyethyliminodiacetic acid. Their reactions with Eu3+ and Gd3+ ions were studied in dilute aqueous solutions by luminescence-based techniques.1218 It was shown that transition from the monomer to polymer complex, and from monoligand to a biligand complex is accompanied by 25-fold enhancement of luminescence. Moreover, the formation of bimetallic complex upon partial replacement of Eu3+ ions by Gd3+ further enhances luminescence by up to two times, which is indicative of the efficiency of Gd3+→Eu3+ energy transfer. Thus, the formation of biligand and bimetallic complexes with variable composition and structure offers the possibility of controlling the luminescence intensity. Gadolinium is a paramagnetic metal used in contrast agents for magnetic resonance imaging; therefore, the Eu3+ and Gd3+ bimetallic polymer complexes are of interest as non-invasive agents for the synchronous optical and magnetic resonance imaging.
β-Dicarbonyl compounds can serve as promising ligands. Water-soluble polymers containing β-dicarbonyl groups can be prepared both by reactions in hydroxyl-containing polymer chains and by copolymerization of, for example, methacryloylacetone with hydrophilic comonomers.1176,1191
Nekrasova et al.1219 described the synthesis of a water-soluble terpolymer of methacryloylacetone with 2-deoxy-N-methacryloylaminoglucose and methacrylic acid and the formation conditions and luminescence of the polymer complex with Tb3+ ions. Using luminescence-based techniques, the complex formation was studied in aqueous solutions at low concentrations of reactants close to the concentrations used in nuclear medicine (≈10–5 mol.%). The luminescent Tb3+ polymer complexes were stable in the presence of NaCl and CaCl2; therefore, these complexes are promising for the preparation of luminescence agents for the therapy of cancer using radioactive 161Tb.
13.3.6. Metallopolymer complexes for magnetic resonance imaging
Magnetic resonance imaging (MRI) is a non-invasive diagnostic technique, which is highly important for medicine, as it allows 3D imaging of soft tissues with a submillimetre resolution in real time.1214 The method is based on the electromagnetic excitation of water molecules. The MRI signal is determined by longitudinal (T1) and transverse (T2) relaxation times of the hydrogen nucleus. Paramagnetic metals (Gd, Mn) are used to accelerate the proton relaxation and amplify the signal, in order to increase the resolution for the imaging of tissues with high contents of water. However, Gd3+ and Mn2+ are toxic; therefore, stable chelates of these metals are used in clinical practice.1220 However, due to relatively low molecular weight, these contrast agents are rapidly eliminated from the bloodstream and excreted from the body, and it is difficult to distinguish between the damaged and healthy tissues using these compounds. Presumably, this drawback could be overcome by inclusion of contrast agents into a macromolecular system. This results in increasing relaxivity, resolving power and circulation time and enables early diagnosis. Furthermore, polymers and nanostructured materials can penetrate into solid tumours and be retained there. An early example of application of water-soluble polymers as carriers for contrast agents was reported by Sieving et al,1221 who described the addition of DOTA and DTPA to polylysine by covalent bonds; the resulting conjugate was additionally modified with human serum albumin. The relaxivity of the final derivative was 2-3 times higher than that of the starting low-molecular-weight analogues (Gd-DOTA and Gd-DTPA). Modification of polyglutamic acid,1222 homo- and graft-copolymers of various amino acids and proteins was reported; no effect of the molecular weight on the relaxivity was noted, but the relaxivity changed upon variation of the distance between chelating units.
Apart from the modification of polypeptides, binding of Gd3+ complexes to polysaccharides was performed. For example, dextran was functionalized with the Gd-DTPA complex with a high binding efficiency (40-50%). The dextran derivative that showed a 1.5-2-fold increase in the relaxivity compared to Gd-DTPA was non-toxic and had some more advantages over other Gd-DTPA polymer derivatives for blood imaging.
A promising polymer carrier for Gd3+ complexes is PEG. This flexible-chain polyether possessing anti-aggregation properties serves as the base for the plasma substitute called Polyoxidin. Linear, graft and block copolymers of EG with DTPA and polyhistidine were synthesized.1223 These copolymers showed long circulation times in blood and liver accumulation. Of obvious interest are polyamides based on DTPA and diamines containing 4, 5, 10 and 12 methylene units between the NH2 groups.1198--1200 These polymers showed enhanced relaxivity, which increased with increasing length of the hydrophobic block.1224
Magnetic resonance imaging contrast agents were developed on the basis of water-soluble carbochain polymers. Polyhydroxypropyl methacrylate was modified with Gd-DTPA complex using reactions in polymer chains and the distribution of the product in rats was studied experimentally.1225 Monomeric methacrylamide DTPA derivatives and iminodiacetic acid analogues were synthesized and subjected to radical copolymerization, which resulted in the synthesis of water-soluble copolymers1226 with hydroxypropylmethacrylamide and N-methacryloyltyrosinamide, which were retained in the tissue of rats for more than 14 days. Modification of DOTA with N-vinylpyrrolidone, acrolein and vinylamine copolymers1227 yielded water-soluble polymers with increased content of Gd3+ and Mn2+ chelate units; relaxivity of these polymers considerably exceeded the relaxivity of clinically used low-molecular-weight analogues.
Dendrimers and dendronized polymers are also actively used, in addition to linear polymers, in the studies dealing with macromolecular contrast agents. The interest in dendrimers is due to the fact that quite a few chelate groups can be introduced into one macromolecule and, hence, the density of these groups and the gadolinium content can be considerably increased. For example, sixth generation polyamidoamine dendrimer (PAMAM) contained 170 chelate groups, demonstrated high relaxivity per Gd3+ ion and was considerably superior to commercial Magnevist (Gd-DTPA) regarding the blood circulation time of experimental animals.1228 Second to fourth generation dendrimers were completely renally excreted, fifth and sixth generation dendrimers were excreted by both the kidneys and the liver, and seventh to tenth generation dendrimers were excreted only by the liver.
Polylysine dendrimers functionalized with DTPA, chelates and additionally with vectors (galactose fragments) to make them specific to hepatocytes were synthesized.1229 The dendrimer containing galactose residues showed a higher signal intensity. Polymers in which two polylysine dendrons are linked by a polyethylene glycol bridge appear to be promising.1230
Along with dendrimers incorporating Gd-DTPA and Gd-DOTA chelates in the outer sphere, dendrimers containing gadolinium chelates in the middle of the molecule were prepared; this improved the relaxivity by substantial retardation of the random molecular rotation.1231,1232 A considerable improvement of the relaxivity was attained by adding dendrons loaded with Gd3+ and Mn2+ DOTA complexes to linear N-vinylpyrrolidone copolymers.1227
The above studies extended the views on MRI contrast agents. Methods for the synthesis of new macromolecular systems were developed and data on their safety and distribution in living organisms were obtained. The accumulated knowledge opens up the way for developing new effective pharmaceuticals based on hydrophilic polymers that will combine the properties of diagnostic and therapeutic agents and provide bioimaging based on luminescent, paramagnetic and radioactive ions.
14. Conclusion
To complete the screening of the promising trends in the development of polymer chemistry and the most important applications of polymers, although the choice of issues and areas was mosaic and arbitrary, we can state that polymer science is on the ascending branch of development and that the mosaic is evolving into a clear pattern. The superfluous politicization of the problem of Earth's pollution with polymer waste brought about certain bias in the choice of the most promising methods for polymer synthesis and processing; however, this could not divert the polymer science from the avenues such as making the synthesis more facile, minimizing the costs, reduction of energy consumption, focus on renewable raw materials, transition to water-soluble systems and generation of new ideas and approaches to the processing and recycling of polymer waste. The search for architecturally complex macromolecules continues, with the major line of research being shifted from the design of spectacular structures such as dense molecular brushes and dendrimers to the demonstration of unique properties inherent only in these structures and to increase in the efficiency of synthesis and also (as demanded by the present time) to the development of effective ways for their taking to pieces. Whereas earlier such studies were considered to be a chemical trick,1233 in our review this approach is followed as a promising trend in the field of macromolecular systems of a complex architecture.
The border between the extrusion of polymer blends and highly efficient methods for the synthesis of new materials is blurred. The multi-layer extrusion processing makes it possible to obtain new polymer systems based on a limited set of components manufactured on a large-scale. Preliminary results attest to enormous prospects of this, seemingly purely engineering solution unrelated to high technology or sophisticated synthetic approaches. The combinatorial prospects of this approach are just endless and involve almost no scaling problem, i.e., transition from laboratory experiments to industrial production does not require intermediate stages. The only limiting factor is the paradigm of polymer waste disposal that implies preliminary very costly waste separation. Here we cast doubt on this obsolete ideology. The development of a biomimetic approach to polymer waste management would not only give rise to more efficient recycling options, but also break down numerous barriers that stand in the way of this key trend.
3D printing processes are being actively developed as a pass to the future demonstrating waste minimization in the manufacture of finished products. This high-technology field is represented in the review by examples most complicated for implementation such as the production of high-temperature polyimides and silicone materials. The lack of waste is especially important for these classes of polymers and can counterbalance the use of more intricate and expensive equipment. The review demonstrates the obvious progress in the development of options for the manufacture of precursors and curing conditions during printing; this ensures a steady growth of the production of diverse materials for 3D printing.
The idea to replace all sorts of polymers produced on a large-scale by their biodegradable analogues, which has persisted in recent years, has also changed. Although this idea is utopian, it is convenient from the standpoint of consumer society, as there is no need to take care of collection and disposal of trash: the nature is supposed to do it by itself. However, a long service life and a long period of complete degradation are, most often, interrelated. Therefore, fine tuning of these characteristics is very costly and inefficient. A certain role in the perception of the low efficiency of self-degradation belongs to the fact that the most harmless degradation products of even efficiently decomposed polymer systems contain carbon dioxide, with all ensuing consequences for the environment.
Although slowly, manufacturers start to understand that the efforts should be concentrated on the design of collection and effective recycling of polymer waste rather than on the self-destruction. Therefore, the use of biodegradable systems is redirected from the manufacture of packaging to the applications where the collection of waste is just impossible. For example, biodegradable polymers are included in formulations used in agriculture, while in medicine these polymers are used to produce drug delivery systems and implantation materials based on various smart systems, including those with precisely controlled biodegradation time. Finally, most important is the polymer waste recycling to composites for the aquatic construction. In these composites, finely crushed polymer waste serves as an inert filler encapsulated in an inorganic or hybrid binder.
This idea seems promising regarding simplification of the processing and safe storage of polymer waste in the bottomless depth of the global ocean; moreover, this implies a useful function of construction of underwater structures and artificial islands. The research along this line, feasibility calculations and particular practical solutions are the obvious milestones of the near future, which would change the ideology of polymer waste management and, after this, would eliminate the bottlenecks in the main research avenues into polymers and environmentally benign polymer technologies.
This review was prepared in the framework of the program of large research projects of the Ministry of Education and Science and the Russian Academy of Sciences; the team of authors includes members of the Scientific Council for Macromolecular Compounds of RAS. The review was financially supported by the Ministry of Science and Higher Education of the Russian Federation (agreement no.075-15-2020-794).
15. List of abbreviations and symbols
The following abbreviations and symbols were used:
3,3’-BZP — 3,3’-diaminobenzophenone,
AA — acrylic acid,
ABS plastic — acrylonitrile, butadiene and styrene copolymer,
ATRP — atom transfer radical polymerization,
APTMAC — (3-acrylamidopropyl)trimethylammonium chloride,
BAPB — 4,4’-bis(4’-aminophenoxy)diphenyl,
BZP — 3,3’,4,4’-benzophenonetetracarboxylic acid dianhydride,
CFRP — carbon fibre reinforced plastic,
CPE — copolyester,
DFT — density functional theory,
DDFT — dynamic density functional theory,
DGEBA — diglycidyl ether of bisphenol A,
DETDA — diethyltoluenediamine,
DIVEMA — copolymer of maleic anhydride with divinyl ether,
DIW — direct ink writing,
DLP — digital light processing,
DMT — dimethyl terephthalate,
DOTA — 1,4,7,10-tetraazacyclodecane-1,4,7,10-tetraacetic acid,
DPD — dissipative particle dynamics,
DSC — differential scanning calorimetry,
DTPA — diethylenetriaminepentaacetic acid,
E — Young modulus,
Eent — entanglement plateau modulus of linear polymers,
EG — ethylene glycol,
EDTA — ethylenediaminetetraacetic acid.
EVOH — ethylene vinyl alcohol,
FDM — fused deposition modelling,
FEM — finite element method,
FRE — freedom reversible embedding,
Gterm — modulus of elasticity in the final viscoelastic shear zone,
GFRP — glass fibre reinforced plastic,
GRIN — gradient index,
h — unit vector in the shear direction,
HPC — hydroxypropyl cellulose,
IPEC — interpolyelectrolyte complex,
ItA — itaconic acid,
L — droplet length,
LC — liquid crystal,
LGS — lignosulfonates,
Me — molecular weight between the entanglements,
Mn — number-average molecular weight,
MAA — methacrylic acid,
MC — Monte Carlo method,
MD — molecular dynamics,
MIP — molecular imprinted polymer,
MRI — magnetic resonance imaging,
MS — multiscale simulation,
MXD6 — poly-meta-xylyleneadipamide,
nsc — side-chain degree of polymerization,
ng — degree of polymerization of the backbone between the grafted chains,
nx — degree of polymerization of the backbone between the cross-links,
Na-CMC — sodium carboxymethyl cellulose,
NIPA — N-isopropylacrylamide,
NIPMA — N-isopropylmethacrylamide,
NNPA — N-n-propylacrylamide,
NP — nanoparticle,
ODPA — 3,3’,4,4’-diphenyl oxide tetracarboxylic dianhydride,
Pbulk — bulk film permeability,
Pconfined layer — oxygen permeability,
P(r) — pressure gradient,
PA — polyamide,
PAA — polyacrylic acid,
PALE — physically active liquid environment,
PAM — polyacrylamide,
PAMAM — polyamidoamine dendrimer,
PAS — para- aminosalicylic acid,
PBAT — polybutylene adipate terephthalate,
PBS — polybutylene succinate,
PC — polycarbonate,
PCFF — polymer consistent force field,
PCL — polycaprolactone,
PDMS — polydimethylsiloxane,
PEEK — poly(ether ether ketone),
PEGDA — polyethyleneglycol diacrylate,
PEG — polyethylene glycol,
PE — polyethylene,
PEO — polyethylene oxide,
PET — polyethylene terephthalate,
PGMA — polyglycidyl methacrylate,
PHA — polyhydroxyalkanoate,
PIB — polyisobutylene,
PI — polyimide,
PLA — polylactide,
PMAA — polymethacrylic acid,
PMI systems — polymer--monomer--initiator systems,
PMMA — polymethyl methacrylate,
PNB — polynorbornene,
PNC — polymer nanocomposite,
PP — polypropylene,
PRISM — polymer reference interaction site model,
PS — polystyrene,
PSF — polysulfone,
PtBMA — poly(tert-butyl methacrylate),
PTFE — polytetrafluoroethylene,
PVA — polyvinyl alcohol,
PVC — polyvinyl chloride,
PVDF — polyvinylidene fluoride,
QM — quantum mechanics,
r — drop radius,
R — universal gas constant,
R — 1,3-bis(3,4-dicarboxyphenoxy)benzene dianhydride,
RCM — ring closing metathesis depolymerization,
ReaxFF — reactive force field,
SC — supercritical,
SCF — supercritical fluid,
SFE — supercritical fluid extraction,
SLA — stereolithography,
SLS — selective laser sintering,
T — absolute temperature,
Tg — glass transition temperature,
Tm — melting point.
TBAF — tetrabutylammonium,
TGA — thermogravimetric analysis,
TPA — terephthalic acid,
V — droplet volume,
VI — 1-vinylimidazole,
VFF — valence force field,
VPTT — volume phase transition temperature,
WBPU — waterborne polyurethane,
XG — xanthane gum,
αs — surface tension,
γ – deformation,
γ̇ — shear rate,
δ — gap between the coaxial cylinders,
λ — uniaxial elongation,
η* — complex viscosity,
η — viscosity,
ηm — matrix viscosity,
ηd — dispersed phase viscosity,
λmax — elongation at break,
ν — fluid flow velocity at point z,
ρ — density,
σ — shear stress,
σs — stress above which the forced rubber-like state occurs,
σtrue — true stress,
Ω — cone rotation speed.