


Keywords
Abstract
α-Amino carbonyl compounds are an important class of nitrogen-containing substances. This review highlights their synthesis using various strategies and considers mechanisms of the processes. The strategies are classified into subcategories based on the type of starting materials, chemical reactions and synthetic methods in use. In the literature survey, different types of reactions are discussed like oxidation, reduction, addition, coupling, C–H amination, oxidative cleavage and rearrangement, amidation, multicomponent cascade reaction, etc., for the synthesis of these compounds.
The bibliography includes 100 references
1. Introduction
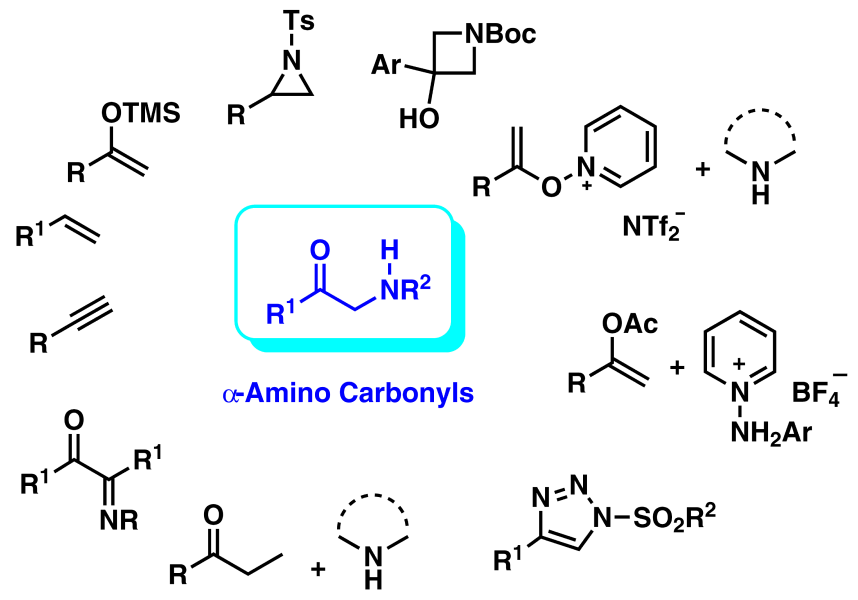
α-Amino carbonyl compounds or α-amino ketones are important building blocks for the synthesis of various natural products. Also, they are found in a large number of biologically and pharmacologically active natural products, so they play a significant role in pharmaceutical and medicinal chemistry.1--8 These nitrogen-containing skeletons are also important synthetic intermediates for the synthesis of 2-amino alcohols9--12 and various nitrogenous heterocycles due to their unique structures and reactivity.13--16 Commercial drugs such as mephedrone17 and bupropion18 (Fig. 1) contain α-amino carbonyl moieties. The Scheme 1 shows synthetic strategies to prepare some nitrogenous heterocycles (A, B, C) derived from α-amino carbonyl compounds.
![[{"id":"1y1CR_mSJg","type":"paragraph","data":{"text":"Examples of commercial α-amino carbonyl-containing drugs"}}]](/storage/images/resized/fQsmje5zyubOvZgbp2oGqWAAtecbzMOeDk3DMyMJ_xl.webp)
So, among nitrogen-containing heterocyclic scaffolds, the synthesis of those bearing an α-amino carbonyl unit has gained significant attention due to the importance of this structural motif. In view of this, several methods have been developed up to date based on various strategies.19--22 In the present review, publications concerning some recent strategies for the synthesis of this important scaffold are discussed. This review is divided into some subsections based on the type of starting materials, chemical reactions and synthetic methods of choice. Also, plausible mechanisms and limitations of the presented methods are considered. The collected data have been published mainly over the last two decades (2001--2022). In a review,20 synthetic approaches are described using natural α-amino acids as starting materials.20 We did not include these methods herein, since they are quite old. Although some works published before the year 2000 will be mentioned so as to illustrate the history of the synthesis of this moiety.
2. Synthetic strategies
2.1. Thermolysis of ethyl azidoformate in enol silanes
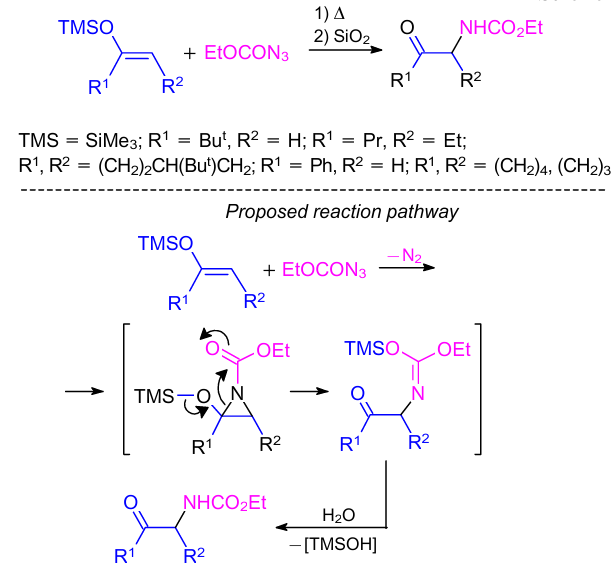
Initially, Lociuro et al.23 was the first to develop a one-step method for the synthesis of N-protected α-amino carbonyl derivatives by thermolysis of ethyl azidoformate in enol trimethylsilyl ethers. Various enol ethers derived from alkyl, cycloalkyl, and alkyl aryl ketones were successfully used in this reaction to provide high yields of the products. A reaction pathway was proposed as shown in Scheme 2. In a follow-up study,24 another approach to N-protected α-amino carbonyl derivatives from enamines and (ethoxycarbony1)nitrene was developed. The authors also described the outcome of the reactions between ketene silyl acetals and ethyl azidoformate.25
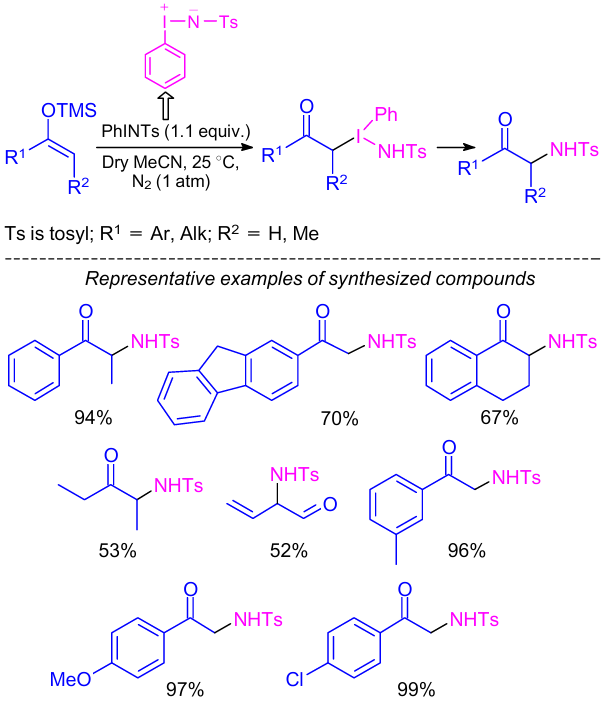
In 1994, Evans et al.26 reported the synthesis of α-amino ketones by the reaction between (N-(p-tolylsulfonyl)imino)phenyliodinane (PhI=NTs) and enol silanes in the presence of CuClO4. The product yields in the reaction the 53--75% based on the nitrene precursor.
Later, Lim and Ahn27 reported a convenient catalyst-free method for the synthesis of α-tosylamino carbonyl compounds from enol silanes using the same aminating reagent PhI=NTs (Scheme 3). Various aromatic enol silanes reacted with PhI=NTs to afford α-tosylamino carbonyls in good yields. Relatively lower yields were obtained when PhI=NTs reacted with aliphatic enol silanes such as 3-(trimethylsilyloxy)-2-pentene (53%) and 1-(trimethylsilyloxy)-1,3-butadiene (52%). Unfortunately, enol silanes such as 1-(trimethylsilyloxy)cyclohexene and 1-phenoxy-1-(trimethylsilyloxy)ethane did not react with PhI=NTs at all.
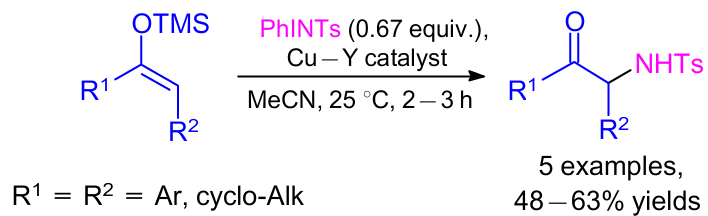
Phukan and Sudalai28 demonstrated the possibility to prepare α-amino carbonyl compounds from silyl enol ethers using heterogeneous catalyst such as Cu-exchanged Y-zeolite (Cu–Y) at room temperature in acetonitrile in good yields (Scheme 4). The catalyst was prepared by an ion exchange method in an aqueous solution of copper acetate. It was observed that the yields were lower in the case of cyclic substrates; but the rate of reaction was higher due to the ring strain effect.
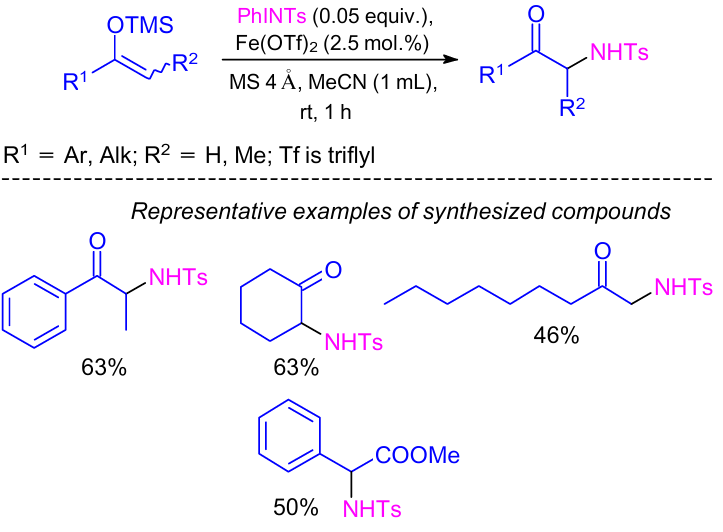
In 2008, Bolm and co-workers29 developed a method for aziridination of various enol silyl ethers at ambient temperature. The reaction was carried out using iron catalyst in acetonitrile to provide the desired products in moderate to good yields (Scheme 5).
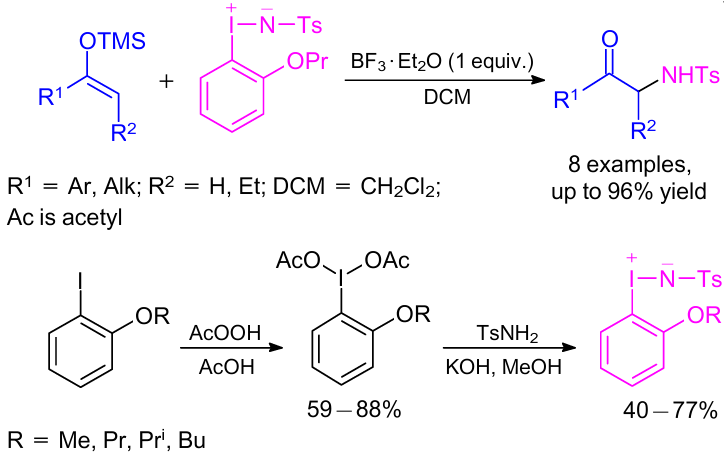
In 2011, Zhdankin and co-workers30 described a metal-free amination reaction using nitrene precursors derived from ortho-alkoxyiodobenzenes (Scheme 6). Various silyl enol ethers reacted smoothly and provided the desired products in moderate yields in the presence of boron trifluoride diethyl etherate in DCM in 0.2--24 h. Readily available 2-iodophenol ethers were used to prepare the desired o-alkoxyphenyliminoiodanes in two simple steps.
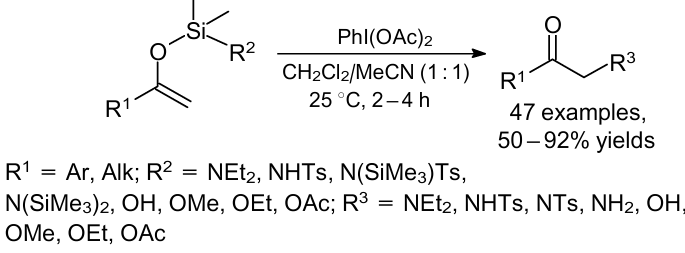
Mizar and Wirth31 carried out the synthesis of α-amino carbonyl derivatives as well as functionalization of carbonyl compounds at α-position through the umpolung reaction in the presence of hypervalent iodine, (diacetoxyiodo)benzene [PhI(OAc)2] (Scheme 7). This reaction proceeded smoothly at ambient temperature in the presence of phenyliodine(III) diacetate (PIDA) without using any Lewis acid. Various nucleophiles gave the desired products in moderate to good yields.
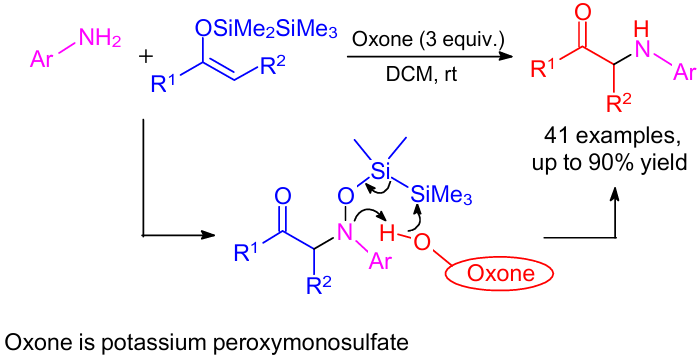
A metal-free nitroso aldol reaction providing α-amino ketones was carried out at room temperature by the direct cross-coupling of various anilines with silyl enol ethers (Scheme 8).32 For this purpose, only inexpensive and user-friendly Oxone was required, and the reaction demonstrated remarkable functional group tolerance. The reaction sequence comprised the following steps: one-pot tandem generation of nitroso compound, the selective C–N bond formation and the N–O bond cleavage.
2.2. Additions of acyl silanes to imines
A new base-mediated organocatalytic process for the synthesis of α-amino carbonyl derivatives is based on the addition of acylsilanes to N-diphenylphosphinoylimines (Scheme 9).33 Neutral carbenes (or zwitterions) generated in situ from readily available thiazolium salts served as catalysts. It was found that both alkyl and aryl acyl silanes and various substituted aryl imines reacted smoothly. A plausible reaction mechanism was proposed (see Scheme 9).
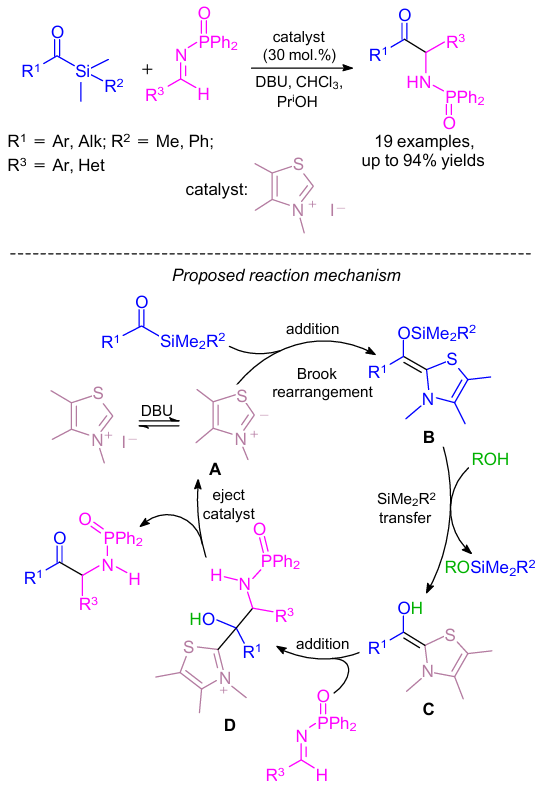
The formation of the heterocyclic carbene/zwitterion catalyst species A was initiated by the thiazolium salt.34--36 This species A underwent the Brook rearrangement by addition to the acyl silane to generate intermediate B.37 In the presence of 2-propanol, the newly formed enol silane was converted to C. The resulting reduced steric environment around the double bond initiated addition to the imine in the next step. Collapse of the tetrahedral intermediate (D) occurred in the final step and the desired product was obtained with concomitant regeneration of the catalyst.
2.3. Coupling of enol acetates with N-arylsulfonyl-1-aminopyridine salts
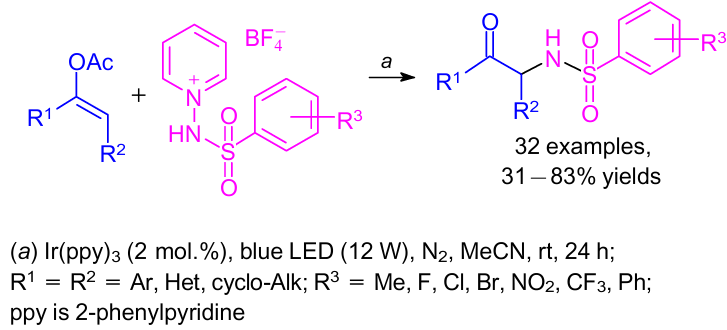
A visible-light-promoted photoredox-catalyzed synthesis of α-sulfonylamino ketones was reported.38 The reaction was carried out by coupling aryl enol acetates with N-arylsulfonyl-1-aminopyridine salts in up to 83% yields (Scheme 10). It was observed that catalytic amounts of Ir(ppy)3 (2 mol.%) (ppy is 2-phenylpyridine) accelerated the formation of N-centered radical intermediates from N-arylsulfonyl-1-aminopyridine salts upon irradiation with a blue LED (λ=425±15 nm) and these intermediates were directly involved in the coupling reaction.
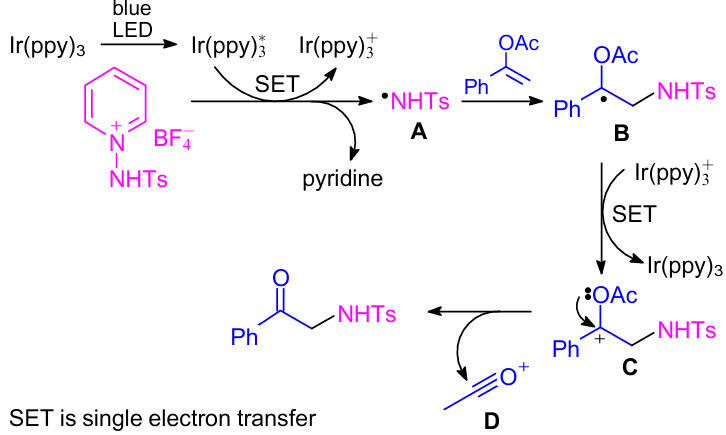
According to the proposed reaction mechanism (Scheme 11), the blue LED light excited the photocatalyst Ir(ppy)3 to Ir*(ppy)3. Next, the N-centered radical A was formed via the reduction of N-Ts-protected 1-aminopyridinium salt initiated by Ir*(ppy)3 through a single electron transfer (SET) process with simultaneous oxidation of Ir*(ppy)3 to Ir(ppy)+3. The radical intermediate B was produced by an attack of N-centered radical species A on the enol double bond. Cation C was generated from intermediate B by oxidation in the presence of Ir(ppy)+3 with simultaneous regeneration of Ir(ppy)3 to complete the catalytic cycle. Finally, the desired α-sulfonylamino ketone along with the acetyl cation D was formed from the resulting cation C. It is worth mentioning that existing nucleophilic species (e.g., pyridine) captured the acetyl cation D.
2.4. Reduction of α-imino carbonyl compounds
In 2001, Shimizu et al.39 offered a method for the synthesis of α-amino carbonyl derivatives by the reduction of α-imino carbonyl compounds using titanium tetraiodide (TiI4) as a reducing reagent (Scheme 12). Various types of imine substrate were reduced to the corresponding amino compounds in good to high yields.
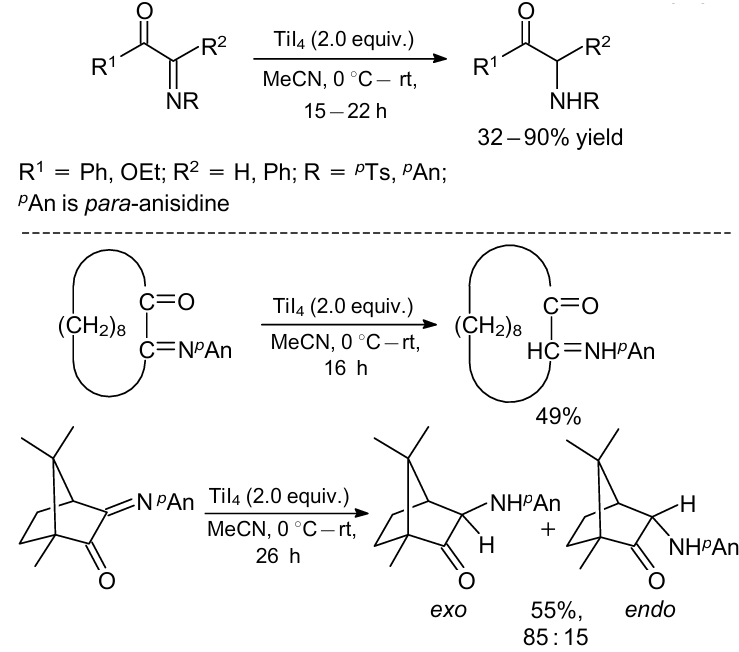
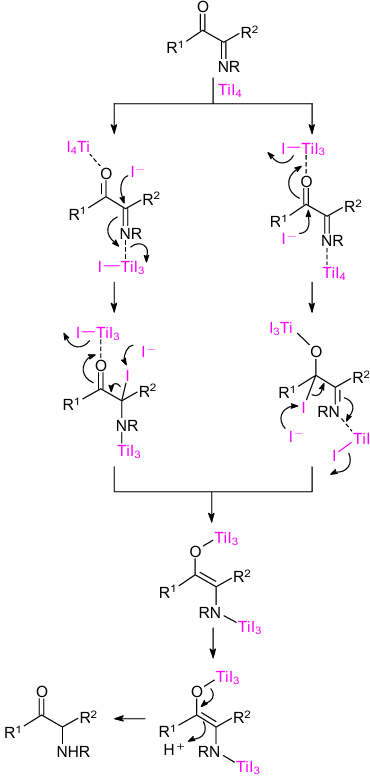
The authors proposed a reaction pathway for this process (Scheme 13). First, an initial attack of iodide anion occurs on the imino or carbonyl carbon followed by the reaction with another iodide anion to generate enolates species. The latter are protonated to give chemoselectively α-amino carbonyl compounds.
2.5. Oxidative cleavage of aziridines
In 2003, Rao and co-workers40 described a procedure for the oxidative cleavage of β-cyclodextrin (β-CD)-aziridine(epoxide) complexes with 2-iodoxybenzoic acid (IBX) to produce α-hydroxyketones and α-aminoketones in high yields (Scheme 14, method A). This reaction is mediated by β-CD and proceeds in water. Cyclodextrins are cyclic oligosaccharides, which have a cavity-like structures and can selectively bind substrates and catalyze chemical reactions. In the absence of cyclodextrin, the reaction does not proceed. This is the first example of a direct one-step synthesis of α-amino carbonyl derivatives from aziridines.
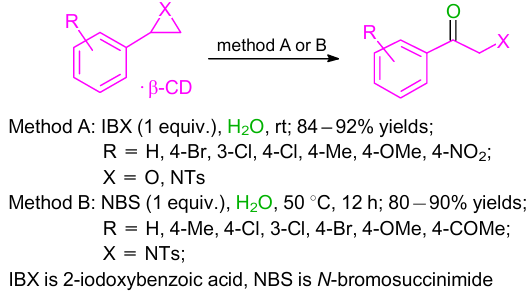
In a follow-up study,41 α-tosylamino carbonyl derivatives were effectively synthesized from aziridines at 50 °C in water (Scheme 14, method B). In this reaction, NBS (N-bromosuccinimide) was used for the first time for the oxidative cleavage of the in situ formed β-cyclodextrin-aziridine complexes. As a result, the corresponding α-tosylamino carbonyl derivatives were produced in high yields.
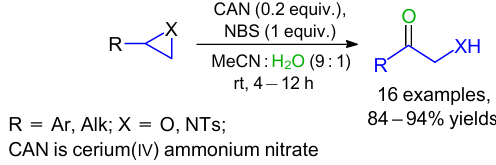
The authors developed one more procedure to prepare α-hydroxy ketones and α-amino ketones by oxidation of epoxides and aziridines under ambient conditions (Scheme 15).42 This reaction was carried out in the presence of cerium(IV) ammonium nitrate (CAN) and NBS in an acetonitrile-water mixture to give the corresponding products in high yields. In this case, the substrate scope was much wider as compared to the above-mentioned methods. Both aryl- and alkyl-substituted aziridines performed well under the reaction conditions.
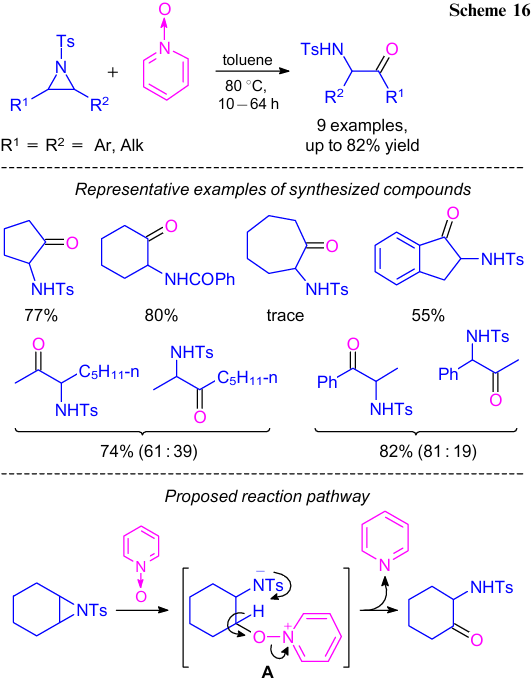
Luo et al.43 reported the synthesis of α-tosylamino carbonyl derivatives by the oxidative ring-opening of aziridines in presence of a stoichiometric amount of pyridine N-oxide (Scheme 16). The corresponding carbonyl derivatives were obtained in high yields. A reaction pathway was proposed (see Scheme 16). At first, intermediate A is formed by a nucleophilic attack of amine oxide on the aziridine. α-Amino ketones were produced in the next step by the intramolecular proton transfer and elimination of pyridine.
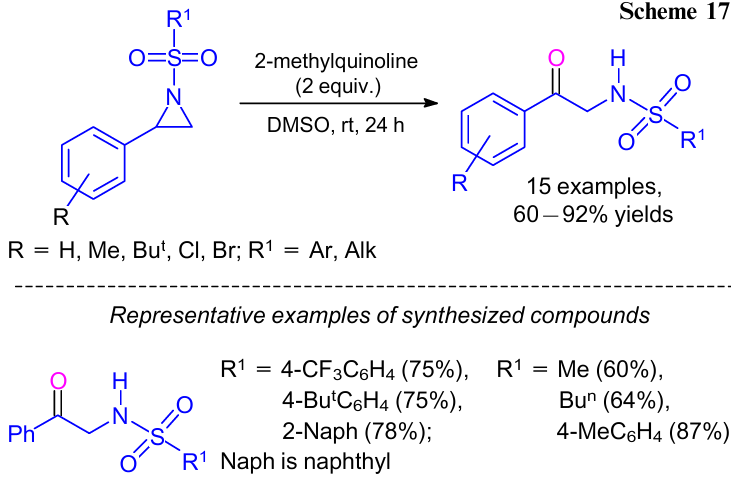
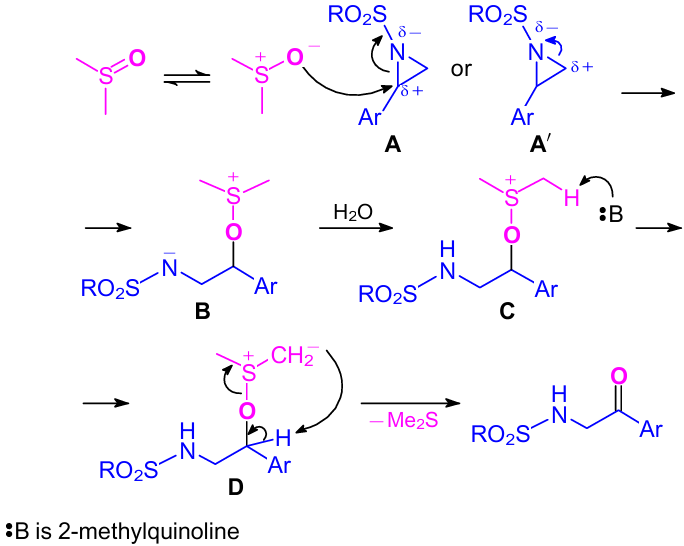
In 2016, Zhang et al.44 developed an efficient approach to α-amino aryl ketones via regiospecific oxidative ring-opening of N-sulfonyl aziridines with DMSO at ambient temperature (Scheme 17). Here, 2-methylquinoline was used as a ring-opening promoter to provide the corresponding α-amino ketones in high yields. Supposedly, it was due to the appropriate alkalinity of 2-methylquinoline.45,46 Although, the exact role of this 2-methylquinoline is still unclear. The reaction was also applicable to the gram-scale synthesis. A reaction mechanism was also proposed by the authors (Scheme 18). Given the stability of the polarized cations, aryl-substituted N-sulfonyl aziridines were more easily polarized to the intermediate A than to Aʹ. Hence, the oxygen atom (nucleophilic) of DMSO regiospecifically attacked the benzylic carbon of the N-sulfonyl aziridines and gave the zwitterionic intermediate B. Then, intermediate C (highly polarized) was formed by the protonation of intermediate B by water. Further, alkoxysulfonium ylide D was formed by deprotonation of the methyl group in the intermediate C by 2-methylquinoline. Upon intramolecular deprotonation, ylide D gave the desired α-sulfonylamino aryl ketones.
2.6. Oxidative deconstruction of azetidinols
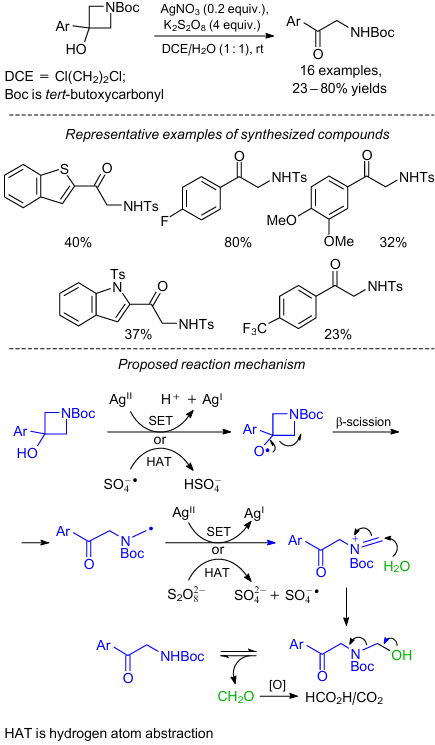
Quite recently, a silver-mediated oxidative deconstruction of azetidinols was reported, which provided α-amino carbonyl derivatives in up to 80% yields (Scheme 19).47 Relatively inexpensive oxidants were used for this scalable procedure and a wide range of aryl and heteroaryl substituents were compatible with the reaction conditions. In addition, the authors carried out some control experiments to understand the reaction pathway (see Scheme 19). It was suggested that the reaction proceeds through the sequence of β-scission of an alkoxy radical, followed by oxidation and C–N cleavage of the resulting α-amido radical.
2.7. Oxidation of alkenes
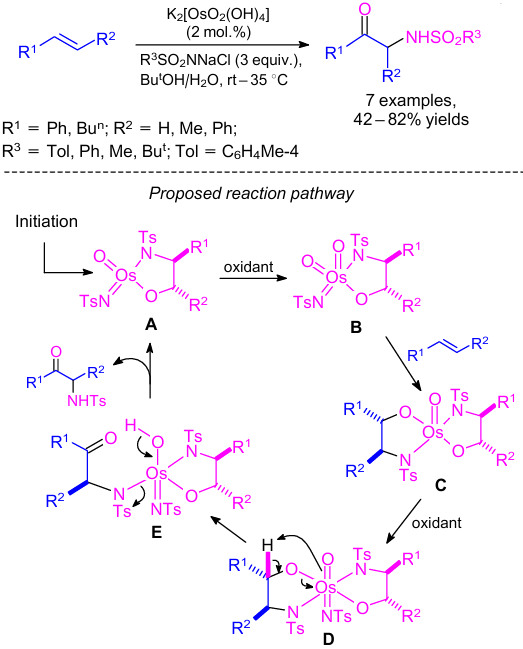
In 2005, MuХiz and co-workers48 described osmium-catalyzed oxidation of alkenes to α-amino carbonyls (Scheme 20). This reaction was carried out in the presence of osmium complex, K2[OsO2(OH)4] (2 mol.%) and 3 equiv. of chloramine T in a tert-butyl alcohol/water mixture. According to the proposed reaction pathway, osma(VI)azaglycolate A was formed after preliminary aminohydroxylation of alkene (see Scheme 20). Intermediate A was re-oxidized to osma(VIII)azaglycolate B. The absence of any cinchona alkaloid ligand slowed down hydrolysis of the amino alcohol leading to concomitant oxidation of the second alkene to provide bis(azaglycolate) C and, upon additional re-oxidation, osma(VIII)-bis(azaglycolate) D. Then mono-bound 2-amino ketone (E) was formed by the intramolecular oxidation of an amino alcohol ligand. This compound E was hydrolysed to redevelop complex A and complete the overall catalytic cycle.
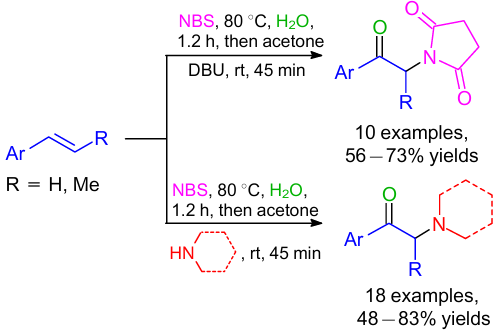
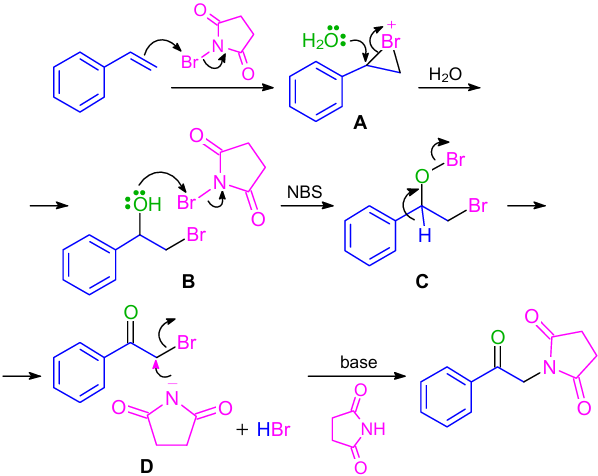
An NBS-promoted one-pot approach for the synthesis of α-amino carbonyl derivatives by the reaction with styrenes was developed (Scheme 21).49 NBS facilitated the bromonium ion formation, bromohydrin oxidation and also provided the nucleophilic nitrogen source. The reaction path could be easily switched between α-imido and α-amino carbonyl derivatives by a simple change of a base. It was observed that when DBU was used as a base, α-imido carbonyl derivatives were formed in the reaction with NBS; but with amines, α-amino ketones were isolated in good yields. The reaction pathway included the formation of bromohydrin (B) by the sequential NBS-mediated generation of a bromonium ion (A), followed by regioselective addition of water (Scheme 22). Bromohydrin (B) then reacted with NBS to give a hypobromite intermediate (C), which transformed to phenacyl bromide (D), which afforded the resulting α-imido ketone through the nucleophilic displacement with a succinimide anion in the presence of a base.50--52 Using an amine, α-amino ketone was produced because of its more nucleophilic and less basic nature and further underwent direct nucleophilic displacement with phenacyl bromide (D) generated in situ.
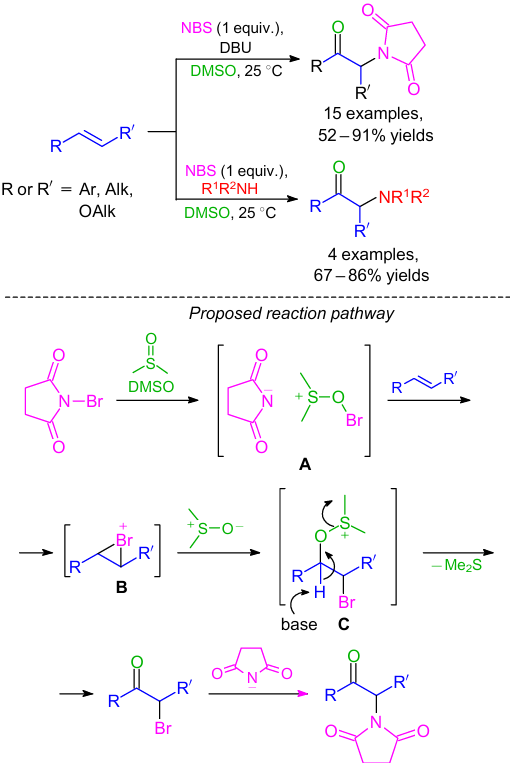
Another effective approach to the synthesis of the similar compounds by the regioselective oxo-amination of alkenes and enol ethers using NBS–DMSO–secondary amine oxidative system was developed (Scheme 23).53 In addition, a one-pot method for the synthesis of vicinal amino alcohols and α-ketoamides through the oxidative coupling of alkenes and secondary amines was also reported in this work. It is worth mentioning that the (bromooxy)-dimethylsulfonium ion generated by the interaction of NBS and DMSO was identified as a reactive species in the oxo-amination reaction (see Scheme 23).
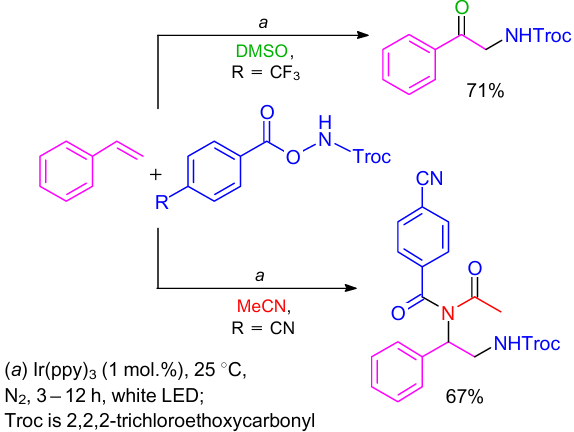
Yu and co-workers54 carried out deamidation and oxidative amidation of alkenes with O-acyl hydroxylamine derivatives under photoredox-catalysis conditions (Scheme 24). Here, O-acyl hydroxylamine derivatives generated N-centered radicals under visible light irradiation. Diamidation and oxidative amidation of alkenes can be achieved in MeCN and DMSO solvents, respectively.
2.8. Using N-sulfonyl-1,2,3-triazoles
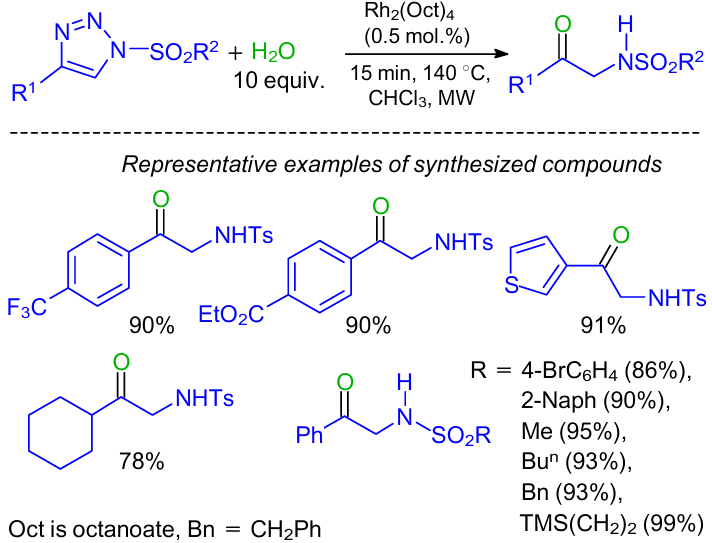
In 2011, Murakami and co-workers55 reported the rhodium-catalyzed synthesis of α-amino carbonyl derivatives from terminal alkynes via denitrogenative hydration of N-sulfonyl-1,2,3-triazoles (Scheme 25). Substrate scope of this reaction was broad. Not only an aryl sulfonyl group but also simple alkyl substituents such as methyl, n-butyl, benzyl, etc., were suitable for this reaction.
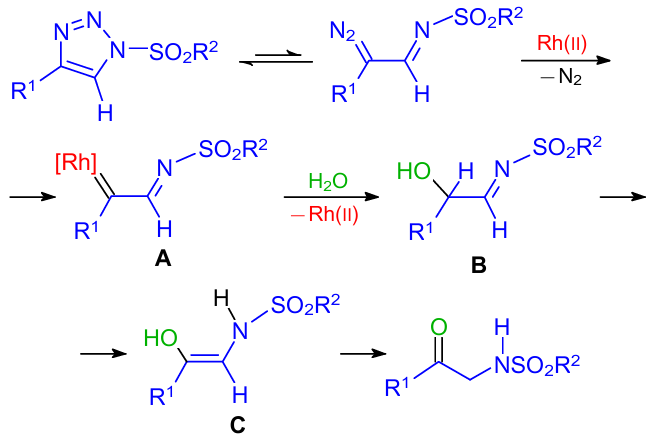
The proposed reaction mechanism is shown in Scheme 26. In the first step, the ring-chain tautomerization of 1,2,3-triazole led to the generation of an α-diazo imine,56,57 which then reacted with rhodium(II) to form α-imino rhodium carbenoid A by releasing molecular nitrogen. In the next step, α-imino alcohol B was formed by the insertion of A into the O–H bond of water,58,59 and the rhodium(II) catalyst was regenerated. Finally, α-amino enol C was produced by imine-enamine tautomerization; the following keto-enol tautomerization afforded the final product.
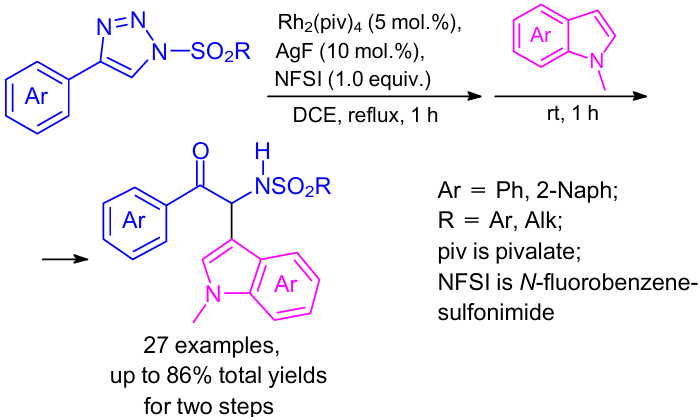
Another convenient one-pot two-step procedure to prepare α-amino carbonyl compounds was developed using intramolecular oxygen transfer from sulfonyl to carbenoid carbon as an extension of the reaction behaviour of the corresponding common N-sulfonyl-1,2,3-triazoles (Scheme 27).60 Various nucleophiles such as indoles and some other arenes were reacted with the key N-sulfinyl imine intermediate.
2.9. Multicomponent reactions
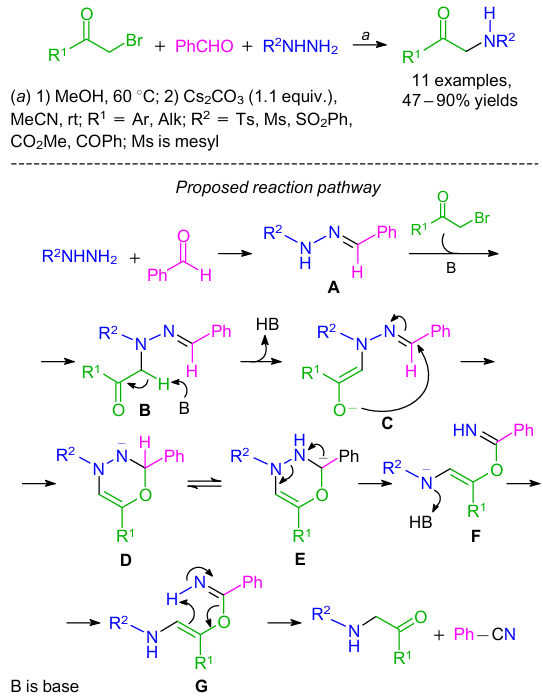
A Cs2CO3-promoted multicomponent synthesis of α-amino carbonyls was developed by the reaction between hydrazines, benzaldehyde, and α-haloketones (Scheme 28).61 The reaction proceeded through the cascade condensation, nucleophilic substitution and the N-N bond cleavage. α-Halo ketones and hydrazines bearing groups of different steric and electronic nature, reacted smoothly to afford α-amino carbonyl derivatives in satisfactory to high yields. It was observed that varying the structure of the starting aldehyde, a broad range of nitriles can be obtained. According to the authors, nucleophilic substitution product B was produced by the reaction between hydrazone A (obtained in situ from hydrazine and benzaldehyde) and αhaloketone under basic conditions (see Scheme 28). A six-membered ring intermediate D was generated through the transition state C from product B by enolization reaction/intramolecular cyclization. The intermediate G was yielded from an unstable D via the 1,2-hydrogen shift/N–N bond cleavage/protonation step. Finally, intermediate G gave the final products through the rapid retro-ene type fragmentation.62,63
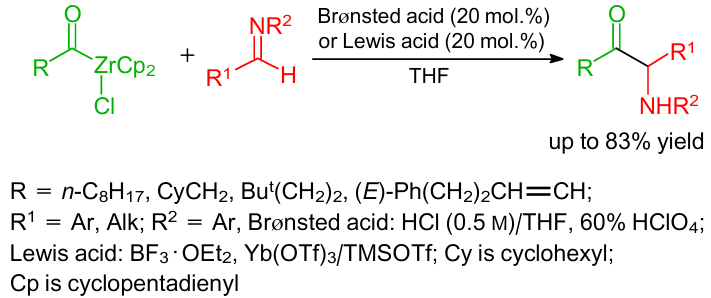
A catalyst-free method to produce α-amino carbonyls based on the three-component reaction between aldehydes, anilines, and acylzirconocene chlorides was developed.64 As an alternative, the same reaction was also carried out in the presence of Yb(OTf)3/TMSOTf as catalysts (Scheme 29).64 In this case, acylzirconocene chlorides reacted as "unmasked" acyl anion donors.
2.10. Alkyne amidation reactions
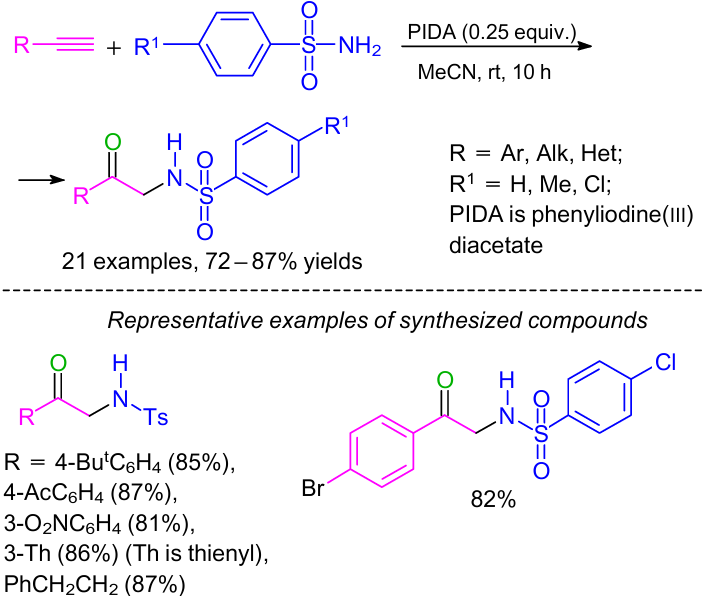
Recently, we reported65 quite a different approach to the synthesis of α-amino carbonyls using (diacetoxy)iodobenzene (phenyliodine(III) diacetate) (PIDA)66 as the oxidant under ambient conditions. It was found that α-amino carbonyl derivatives (α-sulfonylamino ketones) could be obtained in high yields by the reaction between terminal alkynes and benzenesulfonamide in the presence of PIDA (Scheme 30). The broad substrate scope included diversely substituted terminal alkynes and benzenesulfonamides. It was observed that α-acetoxy ketones were formed in the absence of sulfonamide.
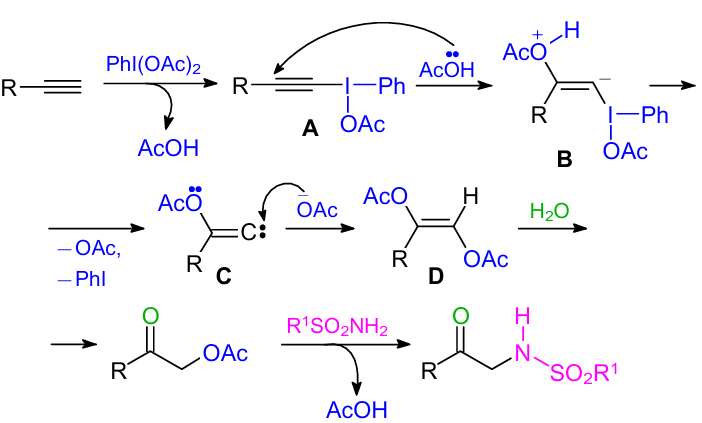
Based on the literature data,67--69 we proposed a plausible reaction pathway as shown in Scheme 31. Phenylalkynyl iodanyl acetate intermediate A is formed by the reaction of alkyne with PhI(OAc)2. Then, intermediate A undergoes Michael type addition of AcOH to provide intermediate B. The intermediate carbene C is generated via the expulsion of acetate anion and then reacts with an acetoxy nucleophile to produce diacetoxy alkene intermediate D. The latter species reacts with residual water to afford α-acetoxy ketone. Finally, α-sulfonylamino ketone is formed in the presence of sulphonamides.
2.11. Sulfur-mediated difunctionalization of internal alkynes
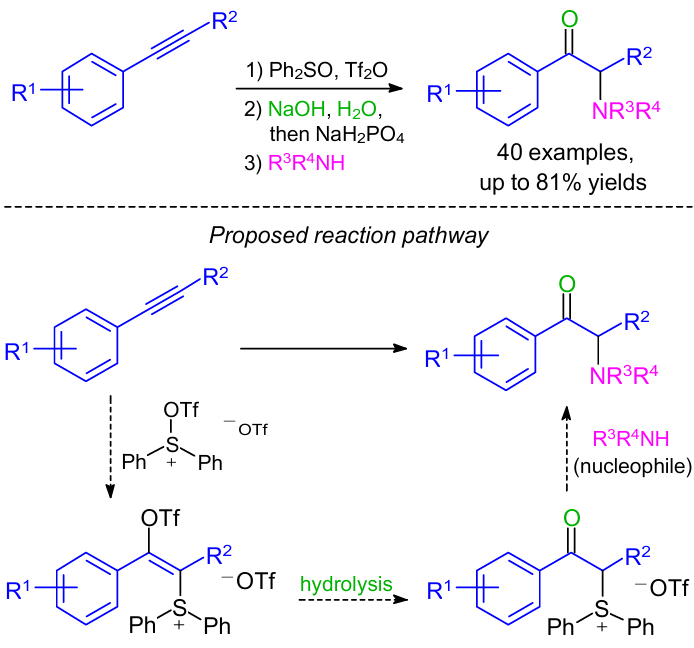
In 2019, Zhang et al.70 developed an efficient one-pot method for the synthesis of α-amino carbonyl compounds through the sulfur-mediated difunctionalization of internal alkynes (Scheme 32). It was observed that the reaction proceeded through the attack of the internal alkyne on triflic anhydride-activated diphenyl sulfoxide, which generated a sulfonium vinyl triflate intermediate, and was followed by hydrolysis to give an α-sulfonium ketone and finally substitution with various nucleophiles.
2.12. Oxidative rearrangement of enamines
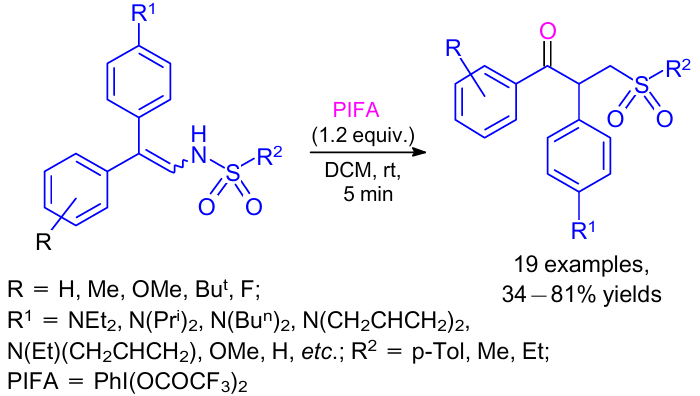
An efficient and fast chemoselective oxidative rearrangement of enamines was carried out by Yadagiri and Anbarasan.71 The reaction proceeded through the initial enamine oxidation to an α-acyloxyimine intermediate and simultaneous semipinacol rearrangement in the presence of PhI(OCOCF3)2 (PIFA). The reaction provided various α-amino carbonyl compounds in excellent yields using enamines bearing mesyl, tosyl and ethanesulfonyl substituents on the nitrogen atom (Scheme 33).
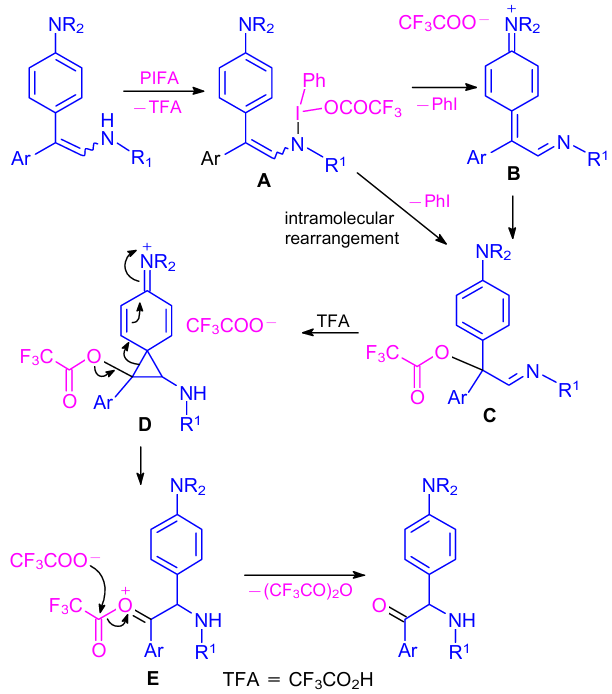
According to the proposed reaction pathway, the reaction between the enamine and PIFA starts from the substitution of CF3COO– with enamine to give the iodonium intermediate A (Scheme 34). Next, α-acyloxyimine intermediate C was obtained by the reaction sequence of enamine-assisted iodonium A reduction to phenyl iodide and cationic imine B, intermolecular trapping of B with the CF3COO–anion. Instead of this step, α-acyloxyimine C formation can also be expected from intermediate A through an intramolecular rearrangement. The formation of the final product from α-acyloxyimine C was envisioned in the next step via the acid-promoted semipinacol rearrangement.72,73 Thus, electrophilic C activation with TFA and subsequent stabilization by an electron-rich aryl group resulted in the intermediate phenonium ion D. Oxonium ion E was generated in the final step due to the 1,2-migration of electron-rich aryl group in D. The oxonium ion E hydrolysis provided the key product.
2.13. α-C–H amination of ketones
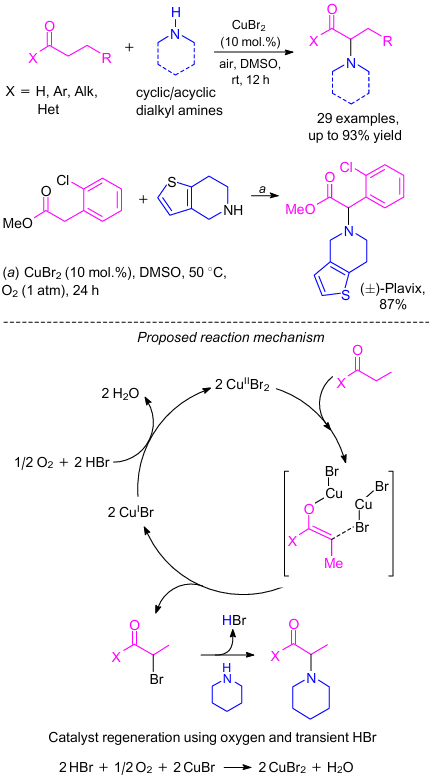
MacMillan and co-workers74 developed a copper(II)-catalyzed direct α-amination of ketones (Scheme 35). This method also works well for esters and aldehydes. The most highlighted application of this method is the one-step synthesis of two important pharmaceutical agents, plavix and amfepramone. The authors proposed a plausible reaction mechanism for this transformation (see Scheme 35). In the presence of catalytic copper(II) bromide, carbonyl substrate undergoes bromination at the α-position75 via a copper-bound enolate to produce an α-bromo carbonyl derivative along with an equivalent of HBr and two molecules of copper(I) bromide.76 Next, the facile nucleophilic displacement of α-C=O bromide functionality by a secondary amine delivered the key product along with the second equivalent of HBr. Copper(II) bromide catalyst was regenerated by reoxidation of copper(I) bromide in the presence of HBr. It should be noted that water was the only by-product.
The first example of oxidative imidation of N–H bonds in imides and C(sp3)–H bonds in simple ketones in the presence of TBHP as the oxidant and Bu4nNI as the catalyst was reported (Scheme 36).77 A number of imides such as succinimide, saccharin and phthalimide and various ketones including the simplest acetone were used and all performed well. During optimization, various common oxidants such as TBHP, K2S2O8, di-tert-butylperoxide (TBP), O2 and 30% H2O2 were used; among them, TBHP was the most effective peroxide for this conversion. In addition, catalysts other than Bu4nNI such as NaI, NH4I, Bu4nNBr, Bu4nNCl, I2, and NIS either decreased the yields or have no effect on the reaction at all.
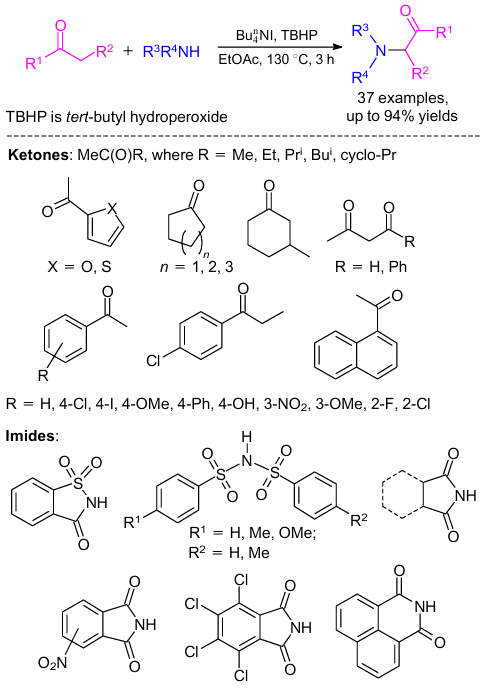
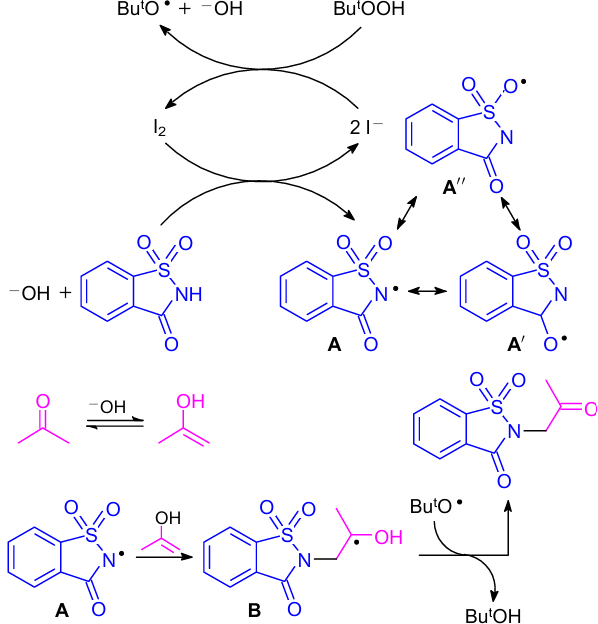
A plausible reaction pathway for this transformation is illustrated in Scheme 37. First, tert-butoxy radical and hydroxide anion were generated from TBHP in the presence of an iodide anion. After that, hydroxide reacted with imide (e.g., saccharin) to form an imidyl radical A in the presence of I2,78--83 which might be stabilized by its resonance structures Aʹ and Aʹʹ. In the next step, enol form of ketone underwent another reaction with radical intermediate A in the presence of hydroxide to generate radical B, which gave the final product upon oxidation.84
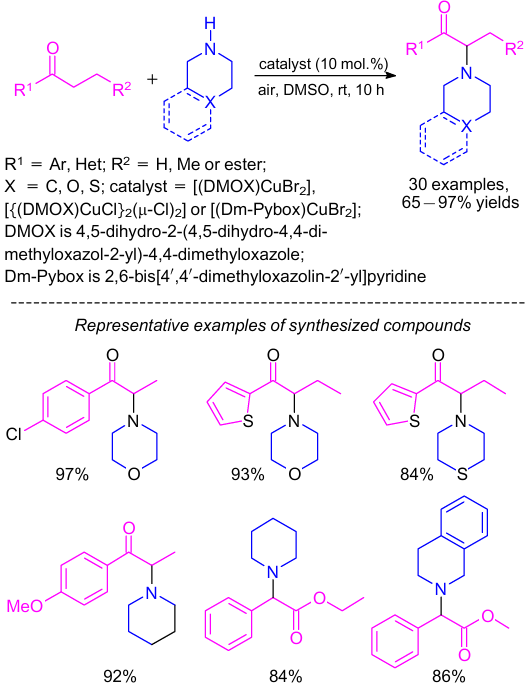
Jia et al.85 synthesized and characterized several Cu(II) mononuclear complexes and binuclear complexes by reacting CuX2 (X=Cl, Br) with DMOX and Dm-Pybox ligands. During these studies, these complexes were tested as catalysts in α-amination of ketones and esters through the formation of α-bromo carbonyl intermediates and proved to be quite promising (Scheme 38). A number of secondary amines (e.g., morpholine, thiomorpholine, piperidine and 1,2,3,4-tetrahydroisoquinoline) were reacted with various carbonyls in this amidation reaction to provide good to high yields of the corresponding α-amino carbonyl compounds.
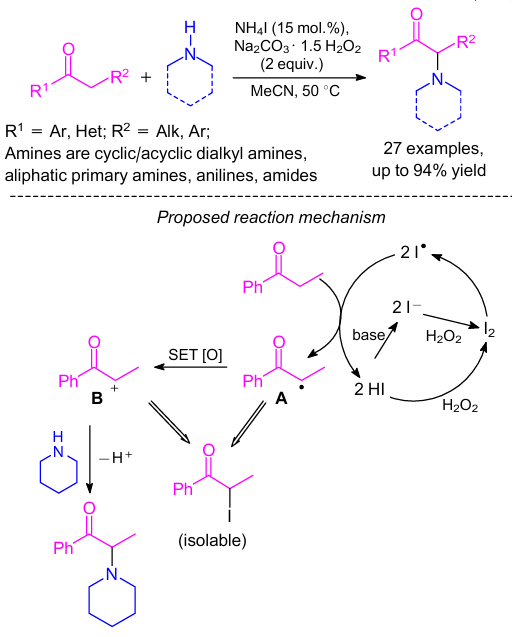
A transition-metal-free method for the oxidative α-C–H amination of ketones by the reaction with amines was developed (Scheme 39).86 Herein, sodium percarbonate was used as the oxidant. Various amines such as cyclic or acyclic dialkyl amines, aliphatic primary amines, anilines and amides can be used in this reaction. Interestingly, an appetite suppressant amfepramone87 was synthesized following this protocol by the reaction of propiophenone with diethylamine. Based on a number of control experiments, which revealed that the reaction follows the radical pathway, the authors suggested its plausible mechanism (see Scheme 39).86 First, molecular iodine was generated by iodide (I–) oxidation with hydrogen peroxide. The molecular iodine was again decomposed into iodine radical. Next, radical intermediate A was formed by the abstraction of α-hydrogen of the propiophenone by the iodine radical releasing the HI molecule, which was trapped by the base to regenerate iodide (I–). In addition, HI could also be reoxidized into molecular iodide in the presence of hydrogen peroxide. The carbon radical A was converted to the intermediate cation B through the subsequent single electron oxidation. The desired product was formed by the nucleophilic addition of amine to the intermediate B in the final step.
Liang et al.88 reported an electrochemical protocol for the synthesis of α-amino carbonyl compounds. The reaction was carried out by the coupling of ketones with secondary amines at ambient temperature in a simple undivided cell with NH4I as the redox catalyst and cheap graphite plate as the working electrode (Scheme 40). As for the mechanistic study, initial α-iodination of ketone followed by a nucleophilic substitution of amines gave the desired products.

Kumar et al.89 developed a straightforward strategy for the synthesis of α-amino carbonyl compounds by α-amination of the in situ generated deoxybenzoin in the presence of NBS (Scheme 41). It was observed that a wide range of functionalities in arylacetonitriles, boronic acids and amine reaction components were well tolerated under the reaction conditions.
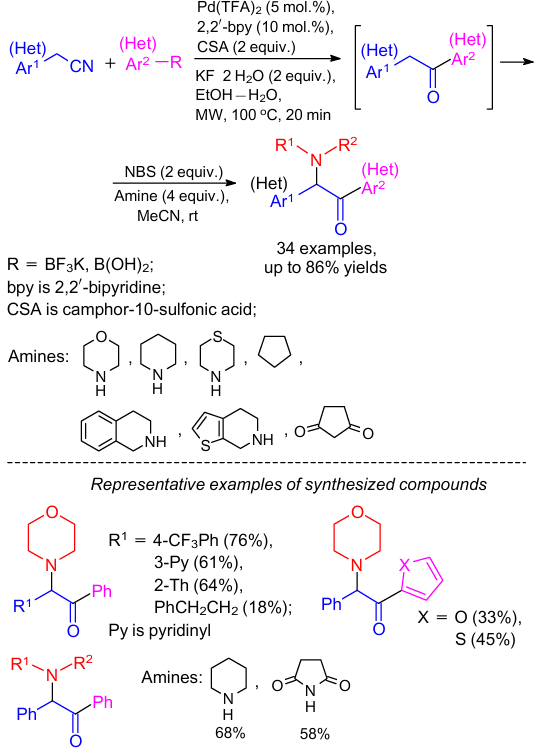
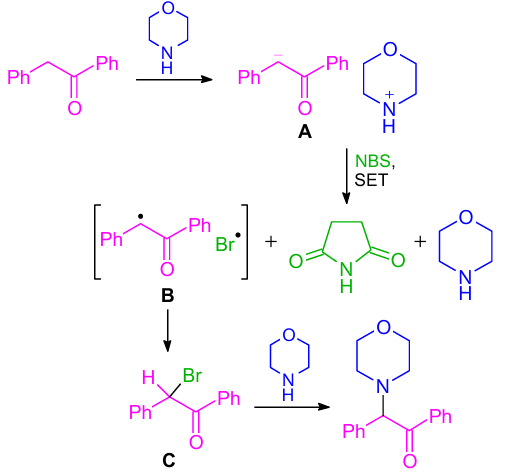
The authors proposed a plausible reaction mechanism as shown in Scheme 42. The reaction started with the formation of an enolate-anion A from deoxybenzoin in the excess of morpholine. Then, enolate-anion and NBS were involved in the SET90--92 reaction to generate benzylic radical B and bromine radical. Accordingly, the benzylic radical B could trap the in situ generated bromine radical thus affording intermediate C. The desired α-amino diaryl ketone was produced by the nucleophilic substitution of α-bromo ketone C with a nucleophile in the final step.
2.14. α-C–H amination of benzylic secondary alcohols
Sekar and co-workers93 described an efficient one-pot method for the synthesis of the above-mentioned compounds directly from benzylic alcohols using various primary and secondary amines. The reaction proceeded through sequential alcohol oxidation and ketone α-bromination/C–N bond formation in the presence of NBS (Scheme 43). Various amines such as piperidine, 1,2,3,4-tetrahydroisoquinoline and pyrrolidine were examined and the corresponding desired products were isolated in good to excellent yields. In addition, aliphatic and aromatic primary amines (e.g., para-toluidine, tert-butylamine, and para-anisidine) were also quite suitable and gave the desired products in moderate yields. This methodology also provided pharmaceutical agents like amfepramone and pyrovalerone in good yields.
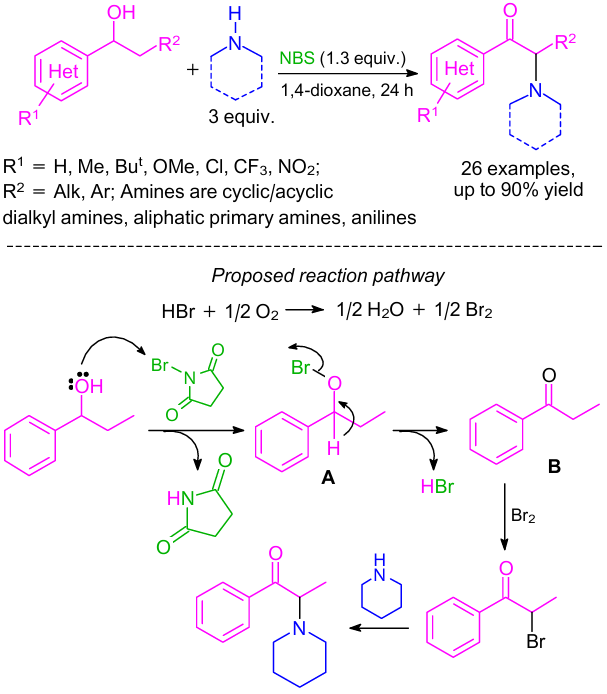
According to the plausible reaction mechanism, hypobromite intermediate A was first formed by the reaction between NBS and alcohol (see Scheme 43). Then, further oxidation of A gave ketone and HBr. It was assumed that the oxidation was very fast and exothermic, and the heat thus generated facilitated oxidation of HBr to Br2 in the presence of oxygen.94 The processes of α-bromination of ketone B by the in situ generated bromine and nucleophilic substitution of α-bromoketone with the amine affording α-amino ketone occurred consecutively.
2.15. Heyns rearrangement of α-hydroxyl ketones
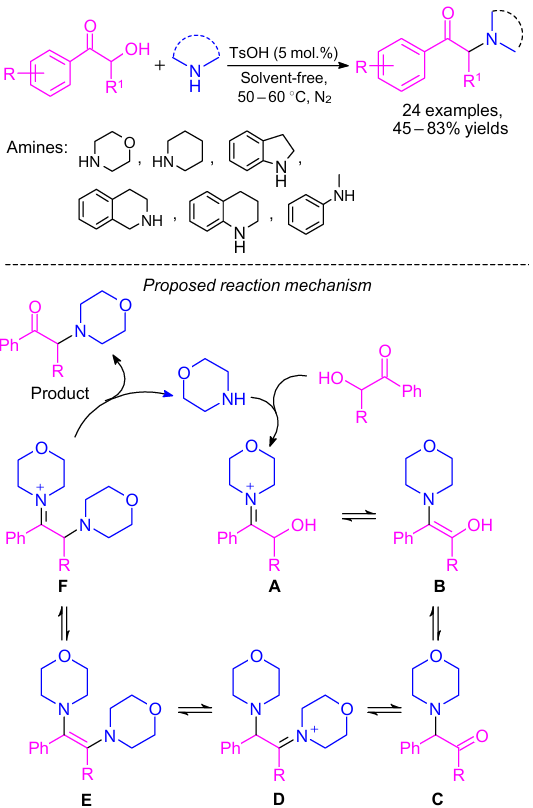
Tang and co-workers95 developed a method to produce α-amino ketones based on the Heyns rearrangement of α-hydroxyl ketones and secondary amines (Scheme 44). In this synthetic procedure, the solvent-free reaction was carried out using p-toluenesulfonic acid (PTSA) as the catalyst. In carbohydrate chemistry, the Heyns rearrangement has been in high demand over the decades.96 Various types of ketones with a primary or secondary α-hydroxy group and various amines including aliphatic and aromatic secondary amines were used in this reaction. The authors suggested the putative mechanism of the reaction by virtue of the Heyns rearrangement (see Scheme 44).
2.16. Amination of umpoled enolates
Xu and co-workers97 reported a convenient method for the synthesis of α-amino carbonyl derivatives using the umpolung strategy via an SN2ʹ pathway. In this method, umpoled enolates (e.g., N-alkenoxypyridinium salts) efficiently reacted with aromatic amines (Scheme 45). Both non-functionalized alkenoxypyridiniums and aliphatic N-alkenoxypyridiniums bearing several functional groups such as cyano, 1,3-ketoester, allyl, benzyl ether, benzoxy ester and phenoxy groups were well tolerated in this reaction.
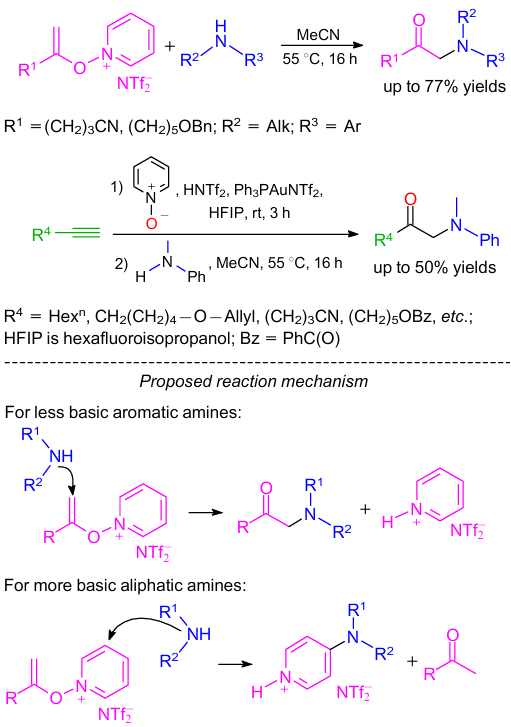
According to the proposed reaction mechanism (see Scheme 45), there are two electrophilic sites in N-alkenoxypyridinium. In the case of less basic aromatic amine, the amine nucleophile attacks the terminal carbon of the double bond yielding the desired product. At the same time, more nucleophilic aliphatic amine attacks the 4-position of pyridinium to furnish 4-aminopyridine.
2.17. Aza-benzoin reaction
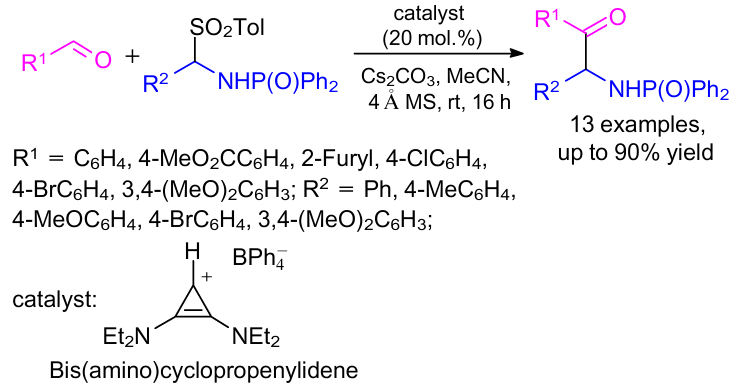
In 2014, Wilde and Gravel98 reported for the first time an aza-benzoin reaction between aldehydes and phosphinoyl imines in the presence of bis(amino)cyclopropenylidene (BAC) catalyst (Scheme 46). The reaction performed well for a variety of (hetero)aromatic aldehydes and a wide range of aromatic phosphinoyl imines to afford various α-amino carbonyl derivatives.
2.18. C(sp3)–H aroylation of amines
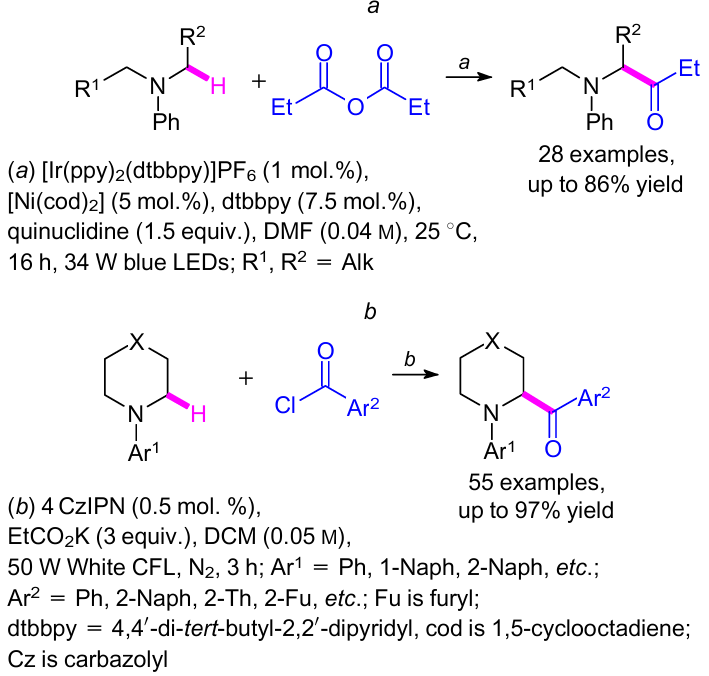
The strategy of C(sp3)–H aroylation of amines (see Scheme 45) gave rise to two different methods. For example, Joe and Doyle99 proposed a photoredox nickel-catalyzed C(sp3)–H coupling of acyl donors with N-aryl amines to afford α-amino carbonyl derivatives, in which saturated aza-heterocyclic moiety was substituted with a broad range of aliphatic acyl groups (Scheme 47, reaction a). Recently, G.-Q.Xu et al.100 reported a convenient method for the direct cross-coupling of α-amino and aroyl radicals using an organic photocatalyst, which allowed to prepare the target compounds (Scheme 47, reaction b).
3. Conclusion
In recent years, there has been a keen interest in the synthesis of α-amino carbonyl motif due to its presence in natural products, pharmaceuticals and photoluminescent materials. This review summarizes the advances in the synthesis of α-amino carbonyl derivatives from the viewpoint of substrates, reagents, catalysts, and mechanistic aspects of various versatile protocols developed over the last few decades. Several methods are discussed based on the starting materials in use such as enol silanes, acylsilanes, alkenes, alkynes, aziridines, etc. In addition, various types of reactions like oxidation, reduction, addition, coupling, C–H amination, oxidative cleavage and rearrangement, amidation, multicomponent cascade reaction etc., were carried out. However, it is worth mentioning that the synthetic methods are still in their infancy and need more attention from organic chemists. Besides, it is still essential to overcome several challenges to explore the major synthetic utility of α-amino carbonyls. The reaction mechanisms are also discussed to help the young researchers in academic circles. Moreover, these nitrogen-containing scaffolds are also important synthetic intermediates, which are used in organic and medicinal chemistry. Therefore, we believe that this review will provide new insights into medicinal chemistry for carrying out future research and will inspire synthetic chemistry practitioners.
This work was funded by the Ministry of Science and the Higher Education of the Russian Federation (Ref. No. 075-15-2022-1118, dated 29 June 2022 (Chapters 2.16--2.18)). Mukherjee and S.Santra are thankful to the Russian Science Foundation for funding (Grant No. 20-73-10205). A.Majee acknowledges financial support from the CSIR Major Research Project (Ref. No. 02(0383)/19/EMR-II). The authors are thankful to the DST-FIST and UGC-SAP program.
4. List of acronyms
Ac — acetyl,
atm — atmospheric pressure,
BAC — bis(amino)cyclopropenylidene,
Bn — benzyl,
Boc — tert-butyloxycarbonyl,
bpy — 2,2ʹ-bipyridine,
CAN — cerium(IV) ammonium nitrate,
CCE — constant current electrolysis,
CD — cyclodextrin,
CFL — compact fluorescent lamp,
Cp — cylopentadienyl,
cod — 1,5-cyclooctadiene,
CSA — camphor 10-sulfonic acid,
Cz — carbazolyl,
DBU — 1,8-diazabicyclo[5.4.0]undec-7-ene,
DCE — 1,2-dichlorethane,
DCM — dichloromethane,
DDQ — 2,3-dichloro-5,6-dicyano-1,4-benzoquinone,
DMF — N,N-dimethylformamide,
DMOX — 4,5-dihydro-2-(4,5-dihydro-4,4-dimethyloxazol-2-yl)-4,4-dimethyloxazole,
Dm-Pybox — 2,6-bis[4ʹ,4ʹ-dimethyloxazolin-2ʹ-yl]pyridine,
DMSO — dimethyl sulfoxide,
dtbbpy — 4,4ʹ-di-tert-butyl-2,2ʹ-dipyridyl,
Fu — furyl,
HAT — hydrogen atom abstraction,
Het — heteroaryl,
HFIP — hexafluoroisopropanol,
IBX — 2-iodoxybenzoic acid,
LED — light-emitting diode,
MS — molecular sieves,
Ms — mesyl (methanesulfonyl),
MW — microwave irradiation,
Naph — naphthyl,
NBS — N-bromosuccinimide,
NFSI — N-fluorobenzenesulfonimide,
Oct — octanoate,
PIDA — (diacetoxyiodo)benzene, or phenyliodine(III) diacetate,
PIFA — (bis(trifluoroacetoxy)iodo)benzene,
PhINTs — (N-(p-tolylsulfonyl)imino)phenyliodinane,
ppy — 2-phenylpyridine,
PTSA — p-toluenesulfonic acid,
Py — pyridinyl,
rt — room temperature,
SET — single electron transfer,
TBHP — tert-butyl hydroperoxide,
TBP — di-tert-butylperoxide,
Tf — triflyl,
TFA — trifluoroacetic acid,
Th — thienyl,
THF — tetrahydrofuran,
TMS — tetramethylsilane,
Tol — p-tolyl (p-methylphenyl),
Troc — 2,2,2-trichloroethoxycarbonyl,
Ts — tosyl (p-toluenesulfonyl).