


Keywords
Abstract
The review summarizes and critically analyzes the results of exploratory and basic studies carried out mainly in the last 10 years concerning the mechanism and role of hydrogen transfer reactions in hydrocarbon conversion processes. A discussion addresses the established views on the regularities of these reactions (mainly involving hydrogen–hydrogen, carbon–carbon and carbon–hydrogen bonds) in various systems containing cations of d-, p - and s-elements. The features of intermolecular hydrogen transfer reactions involved in the zeolite-catalyzed cracking of hydrocarbons are considered. Data on the development of compositions and investigation of properties of modern zeolite-based catalysts for hydrocarbon cracking are discussed. The attention is focused on the role of intermolecular hydrogen transfer in this process, especially in the transformations of heteroatomic compounds. Regarding zeolite compositions, the main attention is paid to the influence of the acid site distribution on the zeolite surface on the hydrogen transfer reactions and the role of extra-framework aluminium species and added modifier cations in these processes. The results of studies of hydrogen transfer by model calculations and by drawing analogies are presented. It is demonstrated that intermolecular hydrogen transfer reactions must be taken into account while addressing problems of improving the cracking catalysts and creating catalytic compositions for joint processing of sulfur, nitrogen and oxygen compounds together with hydrocarbons in the catalytic cracking.
The bibliography includes 249 references.
1. Introduction
At the current stage of oil refining technology, catalytic cracking provides for >40% of a modern refinery gasoline pool;1,2 catalytic cracking is also one of the main sources of propylene, butylenes and isobutane.3
Catalytic cracking is a thermocatalytic secondary oil refining process taking place in the presence of acid catalysts (currently, special aluminosilicate--zeolite compositions) in which hydrocarbon feedstock consisting of large molecules is converted to products with a lower molecular weight. The amount of hydrocarbon feedstock processed via catalytic cracking throughout the world in 2020 was >800 million tons,2,3 and the amount of the used cracking catalysts was greater than the amount of any other oil refining catalyst; their annual consumption in 2019 exceeded 650 thousand tons.*
The results of the first scientific studies of high-temperature non-catalytic transformations of crude oil were reported by A.A.Letniy back in 1875,3 while in 1891 V.G.Shukhov patented the first equipment design of this process.4 At about the same time (turn of the 19th and 20th centuries), first mentions of the catalytic cracking appeared. As a rule, they were associated with the conversion of oil fractions in the presence of anhydrous chlorides of some metals (so-called Friedel--Crafts catalysts).5 The first industrial implementation of cracking processes in the presence of such catalysts was done by the Gulf Refining Company (USA) in 1915 using anhydrous aluminium chloride.**
The first description of the conversion of liquid hydrocarbons (crude oil or heavy fractions of crude oil) in the presence of heterogeneous catalysts can apparently be found in a patent6 in which crushed brick was proposed as such a catalyst; somewhat later, the use of clay and synthetic alumina in various designs was proposed for this purpose.7 The Houdry process, licensed in 1936--1938, using the catalyst based on bentonite clay pretreated with concentrated sulfuric acid, is commonly considered to be the first industrial implementation of the heterogeneous catalytic cracking. The catalyst of the Houdry process was rapidly coked during the catalytic cracking; therefore, the process was conducted using several reactors, which alternately operated in the reaction and regenerated modes.8
The success of the Houdry process gave a new impetus to the studies of catalytic cracking. In the period from 1940 to 1970, the equipment design of the process considerably changed. The changes included approaches towards a continuous flow process (thermafor catalytic cracking, TCC) with a moving catalyst bed,9 the Standard Oil Dev. Co. process in a fluidized pulverized catalyst bed in a lift reactor10--12) and changes in the nature of the catalyst (transition from baked clay7,13 to synthetic amorphous aluminosilicate and microcrystalline zeolites — modern bi-zeolite catalytic compositions based on USY (ultrastable Y zeolite) and ZSM-5.13,14
It is important to note that the described generational change of catalytic compositions reflects the overall trend towards improvement of the most important characteristics of the process: increase in the degree of conversion of hydrocarbon feed and decrease in the coke formation (Fig. 1a) and also a change in the composition of the reaction products (Fig. 1b).2
![[{"id":"WxIAuP2ebr","type":"paragraph","data":{"text":"Diagrams reflecting the trend towards upgrading of heterogeneous cracking catalysts: from low activity and a high yield of coke to high activity and a low yield of coke (a) and to high selectivity to target products (b). T is thermal cracking (without a catalyst). The Figure was created by the authors using published data 2 and J.E.Naber, P.H.Barnes, M.Akbar. The Shell Residue Fluid Catalytic Cracking Process. (Tokyo: Japan Petroleum Institute, Petroleum Refining Conference, 1988)"}}]](/storage/images/resized/elAWkaaibVqWaOsswLRqCtlL0sDSOzI0hZYIHLvb_xl.webp)
The advancement of catalysts and equipment design of the cracking processes was accompanied by comprehensive studies of the conversion of hydrocarbon feedstock on the catalytic sites. The first reports15 on these processes catalyzed by homogeneous Friedel--Crafts catalysts date back to the end of the 19th and beginning of the 20th century. The idea that the activity of heterogeneous cracking catalysts is related to the acid--base nature of the functional groups located on the catalyst surface (active sites) appeared right after the commencement of the process (i.e., in the 1920s--1930s). As was shown later, a significant parameter determining the activity of a heterogeneous catalyst towards carbon--carbon bond scission is the Hammett acidity function (H0) of these sites. For natural clays, this value is in the range from +1.5 to –3.0; treatment with sulfuric acid may increase the value to H0=–5.6 to –8.2,16 and even higher acidity is inherent in synthetic amorphous (H0≤–9) and crystallite (H0≤–10) aluminosilicates (e.g., ZSM-5 zeolites, ferrierite).17,18
The introduction of the notions of protic and aprotic acids in 1923 brought the description of hydrocarbon conversions on acid--base catalysts, including catalytic cracking, to a new qualitative and quantitative level. The theory of formation and conversion of carbocations proposed by Whitmore19 became the main approach, which was further developed by Olah and Schlosberg,20 who reported superacid catalysts. It is important that the basic theory of carbocations reliably describes the reactions of compounds in solutions; however, in the absence of a liquid phase on heterogeneous catalysts, carbocations form intermediate compounds (adducts) with appropriate ions located on the catalyst surface. The most complete chart of transformations of various types of hydrocarbons during catalytic cracking was proposed by B.Voitsekhovskii21 in 1986. The modern mechanistic studies of the reactions of hydrocarbons on zeolite catalysts confirmed the formation of covalent alkoxy groups (–OR)22 (activation by Brønsted acid sites) or adsorbed complexes on metal ions23 (activation by Lewis acid sites) and showed that the catalytic cracking of hydrocarbons involves three fundamentally important steps (Fig. 2):
(1) formation of adsorbed carbocations (carbenium ions from alkenes and carbonium ions from alkanes) via the hydrocarbon adsorption on an acid site of the catalyst (OH group as a Brønsted acid site or metal cation with a coordination vacancy as a Lewis acid site);
(2) transformations of adsorbed carbocation: positional and skeletal isomerization, carbon--carbon bond scission (in α- or β-position relative to the C+ centre) giving hydrocarbons and adsorbed carbocations with lower molecular weights; their alkylation and cyclization followed by aromatization (considering the nature of the target products of cracking, hydrogen transfer steps play the crucial role in these transformations);
(3) destruction of carbocations, resulting in the formation of hydrocarbons and regeneration of acid sites, or formation of polycondensation products (coke) accompanied by blocking of the active sites and, as a consequence, catalyst deactivation.
![[{"id":"lDBwm93M-G","type":"paragraph","data":{"text":"Conversion of n-C<sub>30</sub>H<sub>62</sub> as an averaged hydrocarbon subjected to catalytic cracking on Brønsted (B) and Lewis (L) sites of a solid acid catalyst. For the steps of formation, transformation and destruction of the adsorbed carbocations, see text. The Figure was created by the authors using published data.2, 24"}}]](/storage/images/resized/WN7m6T8faNabGFgU3Vs6nkNSn31eMKSGKrlxnFm3_xl.webp)
While considering the hydrocarbon conversion mechanism during the catalytic cracking (see Fig. 2) in the presence of zeolite catalysts, one should take into account the following factors: the appearance of the molecular-sieve properties (i.e., confined space), which affect the possibility of bimolecular steps (intermolecular hydrogen transfer); potential possibility of the change in the type of acidity during the process (interconversion of the Lewis and Brønsted acid sites via dehydration and rehydration, respectively; this takes place most often in the hydrocarbon cracking in the presence of steam or in the cracking of oxygen-containing organic compounds); and introduction of various modifying elements into the zeolite.24--28
The above factors markedly influence the ratio of the rates of various processes such as
- protolytic cracking (intramolecular hydrogen transfer) leading subsequently to carbon--carbon bond scission (in the α-position to the C+ centre for a five-coordinate carbonium cation; and in the β-position to the C+ centre to a three-coordinate carbenium cation);
- intermolecular hydrogen transfer (hydride transfer from the hydrogen donor hydrocarbon to the C+ centre of another hydrocarbon molecule acting as the hydrogen acceptor), which markedly affects the composition of the products.
The introduction of a more acidic and narrow-pore crystalline aluminosilicate (e.g., Y zeolite) into amorphous aluminosilicate induces a sharp change in the content of unsaturated compounds in the obtained C2–C4 hydrocarbons and, in addition, leads to the formation of C6–C12 aromatic compounds and average molecular weight paraffins (gasoline and diesel fractions; fuel mode of catalytic cracking). This attests to increasing contribution of intermolecular hydrogen transfer reactions. The transfer of the process to the petrochemical mode, in which light olefins should be the target products, necessitates the introduction into the catalyst of a second zeolite (e.g., ZSM-5) with a higher acidity (strength of acid sites) and smaller pores compared to the Y zeolite.29 This increases (due to the increase in the site acidity) the activity of short-chain hydrocarbons in the protolytic cracking and hampers the occurrence of intermolecular hydrogen transfer reactions in the channels of this zeolite (due to steric restrictions, confined pore space).
Thus, the differences in the strength of acid sites and porosity of the components (zeolites and the amorphous aluminosilicate matrix) of the composite cracking catalyst are the key factors determining the contributions of carbon--carbon bond scission and hydrogen transfer reactions, which dictate the pathways of the successive transformations of hydrocarbons on catalyst components (Fig. 3).
![[{"id":"IOs6xqbmFU","type":"paragraph","data":{"text":"Hierarchical diagram of the catalytic cracking processes of hydrocarbons on components of a modern composite catalyst: (a) hydrocarbon cracking on a catalyst comprising three functions: (I) cracking by the matrix (amorphous aluminosilicate, alumina, clay), (II) cracking by Y zeolite. (III) cracking by ZSM-5 zeolite, FER is ferrierite; (b) conversion of fractions on catalyst components, (d ) pore diameter or channel size for zeolites"}}]](/storage/images/resized/V0Z6j9WGFyBokskfsq2vneZqbugEDcJ6v68NPQR4_xl.webp)
The attention of researchers to hydrogen transfer reactions involved in the cracking and to mechanisms of these reactions were intensified in the late 1960s, simultaneously with the start of implementation of zeolite-containing catalysts based on faujasite type zeolites [FAU, 0.74 nm size of pores (aperture)].30 These studies are still in progress. First of all, this is due to the fact that, as noted above, a change in the ratio between the rates of the protolytic cracking and hydrogen transfer reactions substantially affects the composition of the products. The use of zeolite-based catalysts rather than previous-generation catalysts based on amorphous aluminosilicates is also one of the primary causes of higher selectivity to gasoline. One more fact stimulating these studies is the intention to perform the catalytic cracking for oil fractions that were not preliminarily hydrotreated to remove heteroatomic organic compounds (most stable among them are cyclic aromatic heteroatomic sulfur and nitrogen compounds25,31--33) and to use hydrogen transfer reactions for the internal (proceeding during cracking) hydrogenolysis of these compounds.
Generally, issues concerning the mechanisms and role of hydrogen transfer reactions in the cracking processes were raised in reviews and monographs starting from the 1980s (see, for example34--36) and have been frequently discussed in the subsequent years (see, for example 22,24,37--43). The appearance of new-generation equipment and development of computational technologies gave an impetus to new original ways to address long-existing problems related, first of all, to functioning of the active sites of the cracking catalysts. These studies gave rise to a large body of new data requiring analysis and systematization. Meanwhile, the reviews of the last decade are focused on the applied aspect. A series of publications36,41--47 are mainly concerned with the involvement of renewable raw materials into cracking, increase in the catalyst resistance against the poisoning effect of heavy metals in the processing of oil residues, improving the contact between the feed and the catalyst and modelling of both the whole process and particular stages. Hydrogen transfer reactions are most often mentioned in these reviews among the general list of possible reactions, and, in our opinion, the role of these reactions in the transformation pathways of sulfur, nitrogen and oxygen compounds has not received adequate attention.
In the present review, we consider and analyze the results of exploratory and fundamental studies, carried out mainly in the last 10 years, devoted to the mechanisms of hydrogen transfer reactions in the hydrocarbon conversion processes, properties of modern zeolite-based catalysts of hydrocarbon cracking and development of compositions for these catalysts. A lot of attention is given to the role of intermolecular hydrogen transfer reactions, especially in the transformations of heteroatomic compounds.
The review is arranged according to the description and analysis of the following aspects of these studies:
- the existing views on the regularities of hydrogen transfer reactions taking place during the hydrocarbon conversions (mainly reactions involving C=C and H–R bonds, where R is any hydrocarbon moiety) in various systems (containing cations of d-, p- and s-elements) and on the specific features of these reactions in the presence of zeolite-based catalysts of hydrocarbon cracking, particularly, on the role of the structure and porosity of the zeolite surface; the role of extra-framework aluminium cations and added modifying cations in the activation of hydrocarbon molecules; and the use of model calculations and drawing analogies to study the hydrogen transfer reactions;
- components of the cracking catalysts and effect of the catalyst composition on the rates and pathways of hydrogen transfer reactions;
- significance of intermolecular hydrogen transfer reactions for the transformation of sulfur-, nitrogen- and oxygen-containing compounds in the presence of hydrocarbons during the catalytic cracking.
* Fluid Catalytic Cracking Market Size, Share & Trends Analysis Report by Application, Regional Outlook, Competitive Strategies, and Segment Forecasts, 2019 to 2025; https://www.grandviewresearch.com/industry-analysis/fluid-catalytic-cracking-fcc-market (accessed on February 27, 2023)
** See J.G.Speight. The Chemistry and Technology of Petroleum. (Boca Raton, FL: CRC Press, 2006).
2. Current views on the hydrogen transfer mechanisms
The transformations involving hydrogen atom redistribution between the reacting molecules without participation of molecular hydrogen supplied from the outside are commonly considered as hydrogen transfer reactions. The hydrogen transfer reactions that take place during hydrocarbon conversions have been known since the early 20th century. One of the first examples is the palladium-catalyzed conversion of cyclohexene derivatives to benzene and cyclohexane discovered by N.D.Zelinsky and N.L.Glinka48 in 1911. Currently, numerous experimental methods for detecting intermolecular hydrogen transfer have been developed and used. Systematic analysis of the results of studies in this area started apparently from a number of reviews49--51 and is still relevant (for example, Refs 52--55). The key approaches used to study these reactions are as follows:
- detection of the intermediate compounds56,57 and changes in the state of the catalyst;5859
- use of hydrogen and carbon isotopes;60,61
- experimental investigation of model systems62 and theoretical modelling.63
Three groups of the most frequently encountered mechanisms of hydrogen transfer can be distinguished:
- mechanisms involving the intermediate formation of molecular hydrogen;64
- mechanisms involving atomic (dissolved) hydrogen between molecules adsorbed on the catalyst surface (most typical of catalysts based on noble metal particles);64--67
- mechanisms involving protons and hydride ions (typical of catalysts containing acid and basic sites, including those formed by transition metal compounds).68--73
According to the above definition, hydrogen transfer reactions require hydrogen donor and hydrogen acceptor molecules. Hydrogen donors are, most often, compounds with high hydrogen contents (alkanes, cycloalkanes, e.g., n-hexadecane, methylcyclohexane,74 decalin, tetralin75) or compounds containing an active hydrogen atom (alcohols, acids, alkylarenes, etc., for example, isopropyl alcohol,76 formic acid77). Hydrogen acceptors are compounds with low contents of hydrogen (olefins, e.g., hex-1-ene78), heteroatomic compounds (e.g., thiophene,79 nitroarenes,78,80 aldehydes and ketones81) or carbon.82 For donors, this reaction is hydrogen-free (without formation of H2) dehydrogenation, while for acceptors, the reaction is hydrogen-free (i.e., without the use of H2) hydrogenation. However, hydrogen transfer on acid--base sites may be closely connected to the formation of intermediate compounds involving both protons and hydride ions, while molecular hydrogen can appear, for example, in reactions of alkanes with a strong acid site (α-scission of the C–H bond in five-coordinate carbonium cation); therefore, it is important to study the activation on these sites of not only C–C and C–H bonds, but also H–H bonds.
It was noted in the Introduction that the main goal of this review is to consider the role of hydrogen transfer reactions in cracking processes catalyzed by zeolite-based compositions; therefore, analysis of published data is focused on systems the catalytic activity of which is related to the presence of acid--base sites. Thus, below we consider and analyze the published data on catalytic systems operating via acid--base sites, namely, spatially separated, {Aδ+} and {–δB}, or spatially coincident (conjugated), {Aδ+…-δB}, sites; where δ is the effective charge of the site, Aδ+ and -δB are functional groups: Aδ+ either gives off a proton to the substrate or accepts a lone pair of electrons from the substrate, -δB either accepts a proton from the substrate or gives off its lone pair to the substrate. In this case, the substrate (more precisely, the reacting groups of the substrate) are H–H, C–H and C–C bonds. We decided to start our discussion with compositions that perform these reactions in liquid solutions (since for these compositions, more unambiguous conclusions about the structure and functioning mechanism of active sites are provided by physical methods and modelling approaches). This is followed by consideration of compositions in which the active sites are located on the surface of solids. To understand the catalytic action mechanism for these compositions, comparison with homogeneous analogues is often used apart from the physical methods.
2.1.Regularities of hydrogen transfer reactions in homogeneous liquid-phase systems
It should be noted that because of thermodynamic restrictions, some reactions that take place during cracking of hydrocarbons cannot be modelled by comparison with homogeneous analogues. Khoshbin and Karimzadeh83 carried out detailed analysis of the variation of Gibbs energy (ΔGT) as a function of temperature (T) for numerous n-hexane conversion reactions taking place during cracking. According to that publication, in the temperature range of 300--400 K (typical of homogeneous catalysis in solutions), hydrogenation of n-hexene
n-C6H12+H2 → n-C6H14
and hydrogenolysis of hexane
n-C6H14+H2 → 2C3H8
with ΔG<-40 kJmol–1 can be considered to be irreversible; while dehydrogenation
n-C6H14 → n-C6H12+H2
and dehydrocyclization
n-C6H14 → C6H6+4H2
with ΔG>+40 kJmol–1 are thermodynamically forbidden; and the reactions such as alkane addition to olefin (alkylation)
C3H8+C3H6 ↔ C6H14
and hydrogen exchange (conjugate hydrogenation--dehydrogenation)
C3H8+C2H4 ↔ C3H6+C2H6
with ΔG in the range from -40 to +40 kJ mol–1 are taken as conventionally equilibrium reactions, because at these ΔG values, the direction of the reaction either to the right or to the left can be changed by varying the partial pressures of the reactants.
Thus, for considering the influence of the nature of acid--base sites on the hydrogen transfer reactions using comparison with homogeneous analogues, it is reasonable to use published data for reactions such as hydrogenation of the C=C bond, addition of the H–R group to this bond, and transfer of two hydrogen atoms from the H–C-C–H moiety onto this bond.
Generally, the possibility and the pathway of heterolytic activation of the H–H and H–C bonds on the {Aδ+…-δB} site depend on the strength (hardness) of acid A and base B composing this site; this strength is determined by the types of acceptor and donor orbitals (s-, p- and d-contributions) and by the relative spatial positions of the orbitals. The Pearson's concept of hard and soft acids and bases (HSAB) should be used as a common theoretical platform for analysis of these issues.84 For example, in the catalysis by metal complexes, a transition metal ion in an electron configuration with a noticeable contribution of d orbitals (e.g., dsp2, d2sp3, dp3) is formally described as a soft Lewis acid (SLA) or a soft Lewis base (SLB), with the possibility of interconversion between SLA and SLB via electron pair exchange (2e–, two-electron oxidation--reduction). For example, d8 Pd2+ ion with a square coordination (dsp2) is a soft acid, whereas tetrahedral Pd0 in the d10 state (dp3) is a soft base.
It is reasonable to begin the consideration of hydrogen transfer reactions in the liquid phase with the catalytic compositions containing d element compounds. One of the mechanisms of olefin hydrogenation with molecular hydrogen catalyzed by L2PdII complexes is formally similar to the mechanism of non-catalytic ionic hydrogenation (Fig. 44): addition to the C=C double bond, first, of the hydride ligand (hydride ion) and then the proton, both resulting from the heterolytic activation of a hydrogen molecule on the {Pdδ+…-δX} site in which -δX is a hard base, namely, a water molecule (Fig. 55a). The same palladium complexes can also perform the simple hydrogen transfer from the C–H bond; a similar hydride--proton pair is formed when formic acid is used as the hydrogenating agent (hydrogen donor); in this case, the hydrogen atom of the C–H group is the source of the hydride ligand, while the HO group of formic acid is the proton source (see, e.g., Ref. 89) (Fig. 5b).
![[{"id":"pYv_ExtP52","type":"paragraph","data":{"text":"Ionic hydrogenation of 2-methylbut-2-ene: hydrogen-free hydrogenation without a catalyst proceeding via successive addition of H<sup>+</sup> and H<sup>-</sup> upon treatment of the substrate first with trifluoroacetic acid (proton donor) and then with triethylsilane (hydride ion donor).85, 86"}}]](/storage/images/resized/P3AaeLAFPAJejLoMQQDAYI8TBbO3EuRduI8BZUNs_xl.webp)
![[{"id":"ShKWxUbsCN","type":"paragraph","data":{"text":"Mechanisms of hydrogen transfer in the catalysis with metal complexes in the liquid phase: (a) basic scheme of the mecha\u0002nism of olefin hydrogenation (in relation to ethylene) via heterolysis of H<sub>2</sub> molecule and the successive addition (transfer) of the hydride ion and the proton to ethylene in the Pd<sup>II</sup> complex in aqueous trifluoroacetic acid, L = PPh<sub>3</sub>; 87 ± 89 (b) basic scheme of the mech\u0002anism of hydrogen transfer to the C=C moiety (in relation to ethylene) from the C-H bond of formic acid for Pd<sup>II</sup> complex in aqueous trifluoroacetic acid.90, 91"}}]](/storage/images/resized/BcIer0XLF6R1PilpDTZ3uM3mYabMLGvuKUjWUdY7_xl.webp)
The applicability of complexes of d elements as catalysts for low-temperature liquid-phase hydrogenation of the double bond via transfer of two hydrogen atoms from two C–H bonds, i.e., by conjugate hydrogenation-dehydrogenation
C(1) = C(2) + H-C(3)–C(4)– H → H-C(1)–C(2)-H + C(3)= C(4)
was demonstrated92,93 using RhI and IrI complexes, respectively. A dioxane molecule served as the hydrogen donor, being converted to a dioxene molecule. These issues are considered in detail in a review by Samec et al.94 A similar sequence of steps of formation of hydride complexes and hydride ligand transfer to the unsaturated carbon--carbon bond also takes place in the hydrocarbonation reaction (in a special case, known as alkylation): the addition of C–H group to unsaturated C=C bond
C=C + H–R → H-C–C-R
This reaction is catalyzed by low-valent metal (Ru0, RhI, IrI) complexes.95
The above examples demonstrate the ability of SLA-containing acid--base sites to catalyze the hydrogen transfer from donors such as H–H and C–H bonds to unsaturated C=C groups under ambient conditions. However, in these cases, the C=C bond is also activated, most often, because of the formation of π-complexes with SLA (due to the participation of d orbitals in the dative bonding). Usually this results in elongation of the C=C bond because of the change in the hybridization state of the carbon atoms of this bond.96 In any case, these intermediate complexes are relatively stable and can be isolated from the catalytic reaction medium (or specially synthesized) to comprehensively study their structure and properties.
Upon transition from d- to p-elements, the hardness of the central ion as a Lewis acid site increases. The properties of an acid--base site as a hard Lewis acid--Lewis base pair in hydrogen transfer reactions were studied in relation to B3+ and Al3+ compounds (Lewis acids) and N3– and P3- compounds (Lewis bases), which form so-called frustrated Lewis pairs (FLPs). An example of non-frustrated structure is H3NBH3 with a strong N–B bond formed by donor--acceptor interaction

(quenched unreactive bond, see Fig. 6a). However, if the nitrogen (phosphorus) and boron (aluminium) carry bulky substituents [C6F5, (H3C)3C, etc.], the steric repulsion between them does not allow the atoms to approach each other to a distance that would enable the formation of the donor--acceptor bond; this gives rise to FLP with an unquenched reactive donor--acceptor pocket (Fig. 6b).
Quenching of this pocket is energetically very favourable: both heterolytic cleavage of a hydrogen molecule (Fig. 6c) and binding of alkene molecule can take place in the pocket; hence, hydrogenation of alkenes is implemented (Fig. 6d).97,98 The detailed mechanism of activation of H2 molecule upon the interaction with compositionally and structurally diverse FLPs is analyzed in a number of reviews.99--102
![[{"id":"cu0R0eJZ5Y","type":"paragraph","data":{"text":"Formations, types of interaction and properties of Lewis acid - Lewis base pair: (a) interaction in the H<sub>3</sub>B-NH<sub>3</sub> system to form a strong donor-acceptor adduct: compound H<sub>3</sub>B-NH<sub>3</sub>; (b) interaction in the (C<sub>6</sub>F<sub>5</sub>)<sub>3</sub>B-P(C(CH<sub>3</sub>)<sub>3</sub>)<sub>3</sub> system to give FLP with an unquenched reactive donor-acceptor pocket; (c) heterolytic activation of the hydrogen molecule in the unquenched reactive donor-acceptor pocket to give hydride- [(C<sub>6</sub>F<sub>5</sub>)<sub>3</sub>B-H<sup>-</sup>] and proton-containing [<sup>+</sup>H-P(C(CH<sub>3</sub>)<sub>3</sub>)<sub>3</sub>] species, which then undergo ionic hydro\u0002genation; (d ) (1) H<sub>2</sub> heterolysis; (2) proton transfer; (3) hydride ion transfer (Figs a, b, see https://isonlab.wordpress.ncsu.edu/research/frustrated-lewis-pairs/(accessed on February 27, 2023; published with permission from the North Carolina State University; Fig. c, d, see https://nomadicchemist.wordpress.com/2014/10/19/frustration-to-a-good-end/ (accessed on February 27, 2023, published with permission from D.Palomas)"}}]](/storage/images/resized/wI9hHUxkrvhOAccWnRXVwiDluVKnQJytM5joOLyj_xl.webp)
The Lewis acid hardness of the central ion increases to even a higher extent on going from p- to s-elements. This was demonstrated, in particular, for compounds of alkaline earth metals (MAE): Ca, Sr and Ba. In this case, only hydrocarbons (mainly aromatic) are applicable as solvents; furthermore, they must not contain even traces of water or alcohols, as they irreversibly deactivate the precursors of catalytically active sites — MAEII(Nʹʹ)2 diimides, where Nʹʹ=N(SiMe3)2. According to published data,103 these compounds catalyze, even at room temperature, hydrogenation of styrene to ethylbenzene via hydrogen transfer from a cyclohexa-1,4-diene molecule, which is thus converted to benzene. At higher temperature (120 oC), the less reactive hex-1-ene molecule is hydrogenated in a similar way, while no migration of the C=C bond takes place in this case. It is assumed [the assumption was confirmed by density functional theory (DFT) data] that the Nʹʹ–MAEII–H hydride is the catalytically active intermediate of this hydrogenation reaction. This species transfers the hydride ion to the C=C bond of the olefin molecule (Fig. 7a). The formation of this hydride and its high activity in the hydrogenation of alkenes have been confirmed experimentally for MAEII=Ca.104 As an alternative, it was assumed that the coordinatively unsaturated [Nʹʹ–MAEII]+ cation (Fig. 7b), the possibility of formation of which was shown by Thum et al.,105 may also serve as the catalytic active site to transfer hydrogen from the cyclohexa-1,4-diene molecule to an olefin molecule. Being a Lewis acid, this cation can perform the hydride transfer by a mechanism similar to that operating for B(C6F5)3, which is a hard Lewis acid.106,107
![[{"id":"4p7UaTETSh","type":"paragraph","data":{"text":"Intermolecular hydrogen transfer on Lewis acid sites based on s-elements: (a) imide complexes containing coordinatively unsaturated alkaline earth metal cations (shown in red) as potential catalytic active sites for the intermolecular hydrogen transfer from the cyclohexa-1,4-diene to the olefin;105 (b) basic diagram of the hydrogenation of olefins with cyclohexa-1,4-diene catalyzed by alkaline earth metal diimides M<sup></sup><mark><sup>II</sup> <sub>AE</sub></mark><sub></sub>(Nʹʹ)<sub>2</sub> , where Nʹʹ = N(SiMe<sub>3</sub>)<sub>2</sub> .103 Published with permission from John Wiley & Sons"}}]](/storage/images/resized/zFwcfZE9mEBBkDDomojxLy1vV71x4unPYgwWyFgK_xl.webp)
The catalytic properties of s block elements of Group 1 are mainly studied in relation to lithium cations; this cation is of particular interest, as it is the second hardest Lewis acid (after H+). Studies along this line started relatively recently with development of the synthesis of lithium compounds that are soluble in hydrocarbons (e.g., in toluene) and generate a naked lithium ion upon dissociation in solutions.108 The naked ions can be obtained using lithium compounds with very large spherical supersoft anions such as [B((F3C)2C6H3)4]– and [CB11(CH3)12]–. The reactions catalyzed by naked lithium ions are associated with the activation of the C=O or C=C bonds in the Diels--Alder addition,108 polymerization106 and Friedel--Crafts alkylation109 (hydrocarbonation reaction involving a hydrogen transfer step).
From the above examples, one can see that in the presence of homogeneous catalysts, hydrogen transfer from the H–H and C-H bonds to the C=C bond occurs via heterolytic cleavage of the former bonds to give protons, which bind to Lewis bases, that is, solvent molecules or ligands present in the inner or outer coordination sphere of the Lewis acid site, and hydride (or alkyl) species directly bound to it. In the series of hybridization states of the Lewis acid site cation (dsp, sp, s), an increase in the hardness of the cation is accompanied by increasing contribution of the heterolysis of the H–H or C-H bonds to the activation energy of hydrogen transfer, while the role of the C=C bond activation (e.g., as a result of p-complex formation) decreases. For example, it was shown107 that the adduct of the Mg2+ ion with 1,1-diethylethylene is very unstable and has an asymmetry in the Mg...C distances, which may be attributed to the electrostatic nature of the ion--induced dipole interaction with a slight electron density transfer (Fig. 8). This sharply differs from the case where the π-complex is formed upon the olefin interaction with a Lewis acid site that has d electrons participating in the dative bonding, resulting in a considerable change in the reactivity of the olefin ligand.
![[{"id":"IcxH8PzcIp","type":"paragraph","data":{"text":"Schemes illustrating the nature of C=C bond activation in the π-complexes of olefins with a Lewis acid site that has d electrons (a) or only s electrons (b).110 Published with permission from John Wiley & Sons"}}]](/storage/images/resized/QtQPvgtfdrOaF7Ajn1ALTjixdgWBG1uWTsTzI13I_xl.webp)
In the limiting case of hardness, e.g. for H+ (this the hardest Lewis acid according to the HSAB concept), the π-complex of an olefin with H+ does not exist as a stable compound (because of weak soft base--hard acid interaction), but occurs in equilibrium with the starting olefin molecule and s-complex (carbocation); the latter further reacts with a hydrogen donor by eliminating a hydride ion from it.
To complete the discussion of hydrogen transfer in homogeneous liquid-phase systems, it is expedient to emphasize once again that the strength of a Lewis acid site affects both the rate and the pathway of hydrogen transfer, i.e., affects the selectivity of hydrogen addition if the substrate has several sites for hydrogen attachment. In this aspect, note a study111 in which it was shown that in ionic hydrogenation reactions (see Fig. 4), Lewis acids activate the substrate towards the addition of the hydride ion via the formation of a donor--acceptor complex with the substrate; this effect correlates with the affinity of the Lewis acid for the hydride ion (Fig. 9).
![[{"id":"SWyTB2N3WD","type":"paragraph","data":{"text":"Effect of the Lewis acid strength on the substrate activation towards hydride ion addition: (a) hydride ion transfer from the [BH<sub>4</sub>]<sup>-</sup> donor to the substrate activated by binding to a Lewis acid site; (b) general trend of the effect of the Lewis acid strength on the ease of hydride ion addition to the substrate. The Figure was created by the authors using published data.111"}}]](/storage/images/resized/GBLP4GmOEsJDGk7rZefwm0G3jj1E79zlMbO2eryN_xl.webp)
2.2. Hydrogen transfer reactions on the active sites located on the surface of solids
The use of the surface of solids in order to generate catalytic active sites for the desired chemical reactions is a promising trend of the science and practical application of catalysis. These active sites are generated using three key methods:
(1) heterogenization of the homogeneous compositions of metal complexes by grafting them in the molecular state to a support surface;
(2) deposition of metal complexes on a surface as a nanodispersed phase (islands);
(3) conduction of specific reactions involving atoms exposed on the solid crystallite surface, resulting in the formation of surface defects with a structure needed for the catalytic behaviour.
Currently, the last-mentioned research trend, which is called "molecular design of catalysts", has become very popular and efficient, which is reflected in frequent publication of scientific reviews (among the reviews, note Refs 112--115). In the case of the first approach, under conditions similar to the homogeneous reaction conditions, that is, in the presence of the liquid phase, a heterogenized metal complex demonstrates the properties of a homogeneous analogue. However, when the reactions are carried out without a solvent, the heterogenized complex, as a rule, loses ligands that stabilize its structure under the action of gas phase components (especially hydrogen), being thus converted to another surface compound with different catalytic properties (or without catalytic properties).
In the case of heterogeneous systems obtained by grafting of metal cations directly to surface OH groups (second approach), the use of hydrogen and hydrocarbons as substrates may lead to reduction of the transition metal ion to give metal particles; therefore, it is important that the metal ions present in the complex cannot thermodynamically be reduced with hydrogen to the metal (suitable cations are, e.g., titanium, zirconium, cerium, molybdenum, tantalum, tungsten and rhenium). For the use of catalytic systems obtained by the third approach, studying hydrogen transfer processes requires that the oxide of the element that forms the crystallite used to generate surface defects be stable against reductive destruction; examples of such materials are aluminium, silicon, titanium and gallium oxides.
In recent years, particular interest of specialists has been directed towards the synthesis and studying of systems corresponding to the second approach as related to FLPs. The achievements in this area are discussed in detail in a review by Stephan,116 who noted that heterogenization of FLP companions diversifies their nature and thus expands the options for heterolytic activation of hydrogen and for the subsequent steps of proton and hydride ion transfer to the substrate.
While turning to consideration of the achievements in studying the catalytic properties of systems obtained by the third approach, note that the possibility of heterolytic activation of H–H and C–H bonds on the oxide surface was demonstrated in relation to alumina (e.g., Ref. 117). Treatment of γ-Al2O3 with hydrogen at 25 °C or with methane at 150 °C gave rise to Al-H and Al–CH3 groups, respectively, on the alumina surface and resulted in the protonation of the surface oxygen ions. This behaviour of γ-Al2O3 was promoted by its heat pre-treatment at up to ~600 °C.118 Computational studies 119 provided the conclusion that the AlIII...OIII group located at the [110] face, with the Al3+ coordination number with respect to oxygen and O2– coordination number with respect to aluminium being both equal to three, is highly reactive towards C–H bond heterolysis (as estimated from the heat of dissociative addition to the centre of methane) (Fig. 10a). This group comprises closely spaced but not chemically bonded Al3+ cation and O2– anion; therefore, it can behave as FLP. Oxide systems having specially generated oxygen vacancies on the surface possess even a higher capacity for the heterolytic addition of hydrogen. These vacancies are generated, for example, upon the photoactivated hydroxylation of the In2O3 crystal,120,121 and they form groups behaving as FLPs (Fig. 10b).
![[{"id":"KEuiAsjsRr","type":"paragraph","data":{"text":"Structure of the sites (enclosed by dashed ovals) for heterolytic addition of hydrogen and methane at the [110] face of γ-Al<sub>2</sub>O<sub>3</sub> (a) (the Roman numerals indicate the coordination numbers of atoms) 117 and at the defective face of In<sub>2</sub>O<sub>3</sub><sub>-x</sub>(OH)<sub>y</sub> (b) (Ha and Hb show rupture of the hydrogen molecule into proton and hydride ion).122 Figure a is published with permission from the American Chemical Society, Fig. b is published with permission from John Wiley & Sons"}}]](/storage/images/resized/8nbYj9lALuT8IXpSWNTSy9Xxity0b2zYOVtN7nkq_xl.webp)
The FLP-forming oxygen vacancies are present in oxides obtained as nanocrystals with a curved and, hence, highly defective surface. In this regard, note a study by Zhang et al.,123 who found that CeO2 shaped as nano-sized rods exhibited high activity and selectivity in low-temperature gas-phase hydrogenation of the unsaturated C=C bond; furthermore, this feature markedly distinguishes this form of CeO2 from nano-sized cubes or polyhedra. Modification of these oxides with cations that are softer (e.g., Ni in ceria124) or harder (e.g., Ga in ceria125) than the oxide cations with respect to oxygen anion or introduction of Brønsted acid sites and d-element cations or clusters, apart from FLPs, provide more opportunities for the generation of surface FLPs and for controlling their behaviour in hydrogen transfer reactions.
Zeolites are the supports of choice for the design of such polyfunctional catalytic compositions. In this respect, running ahead of the story, we would like to mention a paper by Lee et al.,126 who were able to generate a separated H+...–H Lewis pair in the NaY zeolite by the deposition of Pt nanoparticles on the external surface of the zeolite crystallite and the subsequent hydrogen treatment. The hydride ion was stabilized via binding to the Na+ cation located in the zeolite cavity and acting as a Lewis acid (Fig. 11).
![[{"id":"msz1YWQoCS","type":"paragraph","data":{"text":"Formation (a) and structure (b) of (<mark>structure</mark>) FLP in the Y zeolite cavity; the H<sup>+</sup>...H<sup>-</sup> distance is 0.15 nm.126 Published with permission from John Wiley & Sons."}}]](/storage/images/resized/30I9MH2mvz3al8CO07bzXl3Yro88gImCu9PAcKfl_xl.webp)
2.3. Specific features of hydrogen transfer reactions in zeolite-based systems
Zeolites are crystalline aluminosilicates characterized by the presence of nano-sized 3D-ordered channels and cavities. Currently, there are >30 known natural zeolites and >200 synthetic zeolites with no natural analogues.* Only a minor part of the zeolites are used in practice. The modern industry employs zeolites to prepare catalysts for catalytic cracking. Most often, specially synthesized Y zeolite and additionally ZSM-5 zeolite are used for this purpose. Thus, in view of the subject of our review, the subsequent consideration of the features and methods for the control of hydrogen transfer reactions on zeolites is based, first of all, on publications addressing the use of Y and ZSM-5 zeolites.
The main feature that determines the unique properties of zeolites employed in catalysis is combination of a very high Brønsted acidity, pronounced molecular-sieve properties and extended surface, which can thus accommodate additional catalytic active sites of a different nature (Lewis, metal and so on). The Brønsted acidity of zeolites is similar to that of superacids. This is due to the general similarity between the zeolite and superacid structures, which can be described as a proton located on a very large, bulky anion, as indicated by the zeolite chemical formula Hx[AlxSiyO2(x+y)] and by the fact that the acid site strength tends to increase with increasing (x+y) accompanied by a decrease in x. Indeed, the composition of the aluminosilicate anion of the Y zeolite is [Al7Si17O48]7– where x/(x+y)=7/24. In the case of ZSM-5, the anion similar in nature has the composition [AlnSi96-nO192]n- (0<n<27) and, for example, for n=7, the ratio x/(x+y)=7/96, i.e., it is four times lower than that of Y zeolite. This accounts for the higher strength of the acid sites in ZSM-5. The (x+y) value and the x/(x+y) ratio determine the structure of the zeolite aluminosilicate framework, namely, the size and shape of channels and cavities and their architecture in the crystal. The primary structural units of zeolites are tetrahedra consisting of four oxygen anions, which surround a smaller silicon or aluminium cation (Fig. 12a). These tetrahedra are grouped in such a way that each of the four oxygen anions belongs to another tetrahedron (according to the empirical Lлwenstein rule,127 in this case, two aluminium atoms cannot share an oxygen atom); a certain number of the tetrahedra give rise to secondary structural units, cages (Fig. 12b), which further form a crystal (Fig. 12c).
![[{"id":"RDezpVP9MW","type":"paragraph","data":{"text":"Architecture of zeolite crystals: (a) SiO<sub>4</sub> and AlO<sub>4</sub> tetrahedra; (b) sodalite (for Y zeolite, above) and pentasil (for ZSM-5 zeolite, below) cages; (c) unit cell structures formed by the cages for Y (above) and ZSM-5 (below) zeolites"}}]](/storage/images/resized/CurtDikMOcsbXIdv61ywzdnzKq0gosNFRtSAeJw7_xl.webp)
Thus, only Si–O–Si and Si-O–Al bonds can be present in the aluminosilicate framework of the zeolite, while the Al–O–Al bonds are impossible. Various types of zeolite post-treatment (calcination, ultrastabilization, etc.) can give rise to extra-framework species, in particular, extra-framework aluminium, which provides for the possibility of Al–O–Al bonds. This and other factors of the zeolite synthesis and post-treatment affecting the zeolite properties, in particular the activity in hydrogen transfer reactions, are considered below. Here we would like to emphasize the following important feature of the synthesis of zeolite as a Brønsted acid: since the formation and the subsequent interactions of the silicon- and aluminium oxide precursors to give the aluminosilicate framework take place in basic media, the crystalline zeolite is formed as Mx[AlxSiyO2(x+y)], where M is usually Na; the subsequent ion exchange between Na+ and ammonium cations and the thermal decomposition of zeolite ammonium forms result in the formation of the proton (decationized) form Hx[AlxSiyO2(x+y)] (Fig. 13).
![[{"id":"T05Z2IkkzX","type":"paragraph","data":{"text":"Zeolite sodium form (<i>a</i>) and thermal decomposition of the ammonium form (<i>b</i>), which yields Brønsted acid sites."}}]](/storage/images/resized/ZI7vWhL46ohkpuxP4NpQjDJesd0vLS6GLN8SgDZK_xl.webp)
Note that special designations are used for the cationic forms of zeolites, e.g., NaY and NaZSM-5; however, for the proton forms (i.e., HY and HZSM-5), the symbol of hydrogen is often omitted, as this is done in the present review.
The introduction of other cations (e.g., Zn2+, Ga3+, La3+, Ce3+, Ti4+), apart from Na+, into the zeolite sharply changes its acidic properties.128--130 For example, the presence of Zn2+ leads to increasing concentration of the Lewis acid sites, while the presence of La3+ cations increases the concentration of Brønsted acid sites. It is important that cations can be introduced into the structure either via the conventional ion exchange (replacement of Na+) or during the synthesis at the stage of crystallization (e.g., introduction of Sn4+, Ti4+ and Ga3+). In the latter case, isomorphous replacement of silicon or aluminium atoms in the zeolite framework takes place (Fig. 14).131
![[{"id":"8XbMcn2ChN","type":"paragraph","data":{"text":"Formation of Lewis acid sites by ion exchange (<i>a</i>) or isomorphous replacement (<i>b</i>) and formation of Brønsted acid sites (<i>c</i>) in the zeolite upon modification with metal cations."}}]](/storage/images/resized/ZtydtETqUOJ3qXlQq2nGayJQDXhTZKrnbX894ohN_xl.webp)
Treatment of the zeolite at a temperature above 500 oC induces dehydroxylation of its surface (Fig. 15a),129 yielding structural groups such as AlO+, Al(OH)2+ and AlOH2+, which behave as Lewis acid sites.
Heat treatment of zeolites in a steam environment may induce hydrolysis of the Al–O bonds. This leads to zeolite dealumination (or ultrastabilization): escape of the aluminium cations from the zeolite framework and partial destruction of the framework. These reactions generate extra-framework aluminium (EFAL) oxide clusters and dealuminated ultrastable Y zeolite (USY zeolite) (Fig. 15b), which exhibit clear-cut Lewis acid properties.133 Hence, the acid--base characteristics of the zeolite substantially change. Thus, zeolite dealumination on treatment in steam can be considered as the transformation of a part of Brønsted acid sites to Lewis acid sites.
![[{"id":"1NZEqELKmD","type":"paragraph","data":{"text":"Dehydroxylation of the zeolite surface accompanied by transformation of Brønsted acid sites into Lewis acid sites (<i>a</i>) and simplified scheme of the process giving the AlO<sup>+</sup> EFAL moiety carrying a Lewis acid site (<i>b</i>).132 Published with permission from Elsevier"}}]](/storage/images/resized/G6qznQFbzDaetBBlrHnS0B2iDI6V3yTXNoqaVFNW_xl.webp)
The AlO+ groups arising in this process (see Fig. 15b) can form various oligomer structures (dimers, trimers, tetramers, etc.), that is, cationic aluminium oxide clusters. Theoretical DFT studies of the formation of this type of particles indicate the predominant formation of tri- and tetranuclear clusters, [Al3(OH)6]3+ and [Al4O6], respectively. These clusters are mainly located in small zeolite cavities and additionally stabilize the framework; they also give rise to Lewis acid sites in the close vicinity of the Brønsted acid sites retained in the framework.134,135 Zeolite dealumination can also be attained by treatment with organic acids136--138 or by impregnation with aluminium salts133,139,140 followed by heat treatment and desilylation, that is, removal of some of silicon from the zeolite framework, which gives EFAL.141
Not only alumina, but also oxides of other metals — calcium, barium, zinc, gallium, molybdenum, titanium and tin — can act as extra-framework species. These species can appear in zeolites, for example, upon hydrothermal treatment (accompanied by hydrolysis of M–O–Al and M–O–Si bonds) of either various cationic zeolite forms (e.g., calcium, barium)142 or zeolites containing other metal cations (titanium, gallium, tin) introduced into the framework by isomorphous replacement of the framework aluminium and silicon cations.143 The extra-framework oxide nanoparticles can also appear upon the deposition of compounds of the above-noted elements on the zeolite surface, in particular, by impregnation.144,145 This zeolite modification gives, as a rule, Lewis acid sites of a different elemental composition on the surface; as a result, modified zeolite acquires new unique properties, which are used for C–C and C–H bond activation and hydrogen transfer reactions.146
The nature of the modifying element (s-, p- or d-element) considerably affects the strength (hardness) of the Lewis acid site formed by this cation. Generally, the strength of Lewis acid sites varies similarly to that of metal complexes described above in terms of the HSAB concept. For example, using DFT calculations, it was shown147 that in the series of cation exchange MY zeolites, where M = Li+, Na+, K+, Cs+, Cu+ and Ag+, the interaction energy between the toluene molecule and the Lewis acid site M+ gradually increases on going from Li+ to Cs+ and then increases stepwise towards Cu+ and Ag+ (Fig. 16a). This trend fully corresponds to the expected one if one takes into account that toluene (π-conjugated system of the ring) is a soft Lewis base, while the alkali metal ions are Lewis acids the hardness of which decreases from Li+ to Cs+ (in terms of the HSAB concept, it should be expected that the interaction energy for a soft base should increase with decreasing hardness of the Lewis acid). It is noteworthy that an opposite trend is observed for the adsorption of water, since water as a hard Lewis base forms a stronger bond (aqua complex) with Li+ (harder Lewis acid). The stepwise (after Cs+) increase in the interaction energy between toluene and Cu+ and Ag+ cations can be attributed to higher softness (compared to Cs+) and d-character of the atomic orbitals of these cations, resulting in a different type of their binding to the toluene molecule (Fig. 16b).
![[{"id":"3SbkfJXNpS","type":"paragraph","data":{"text":"Diagram of variation of the interaction energy of toluene and water with MY zeolites, where M = Li<sup>+</sup>, Na<sup>+</sup>, K<sup>+</sup>, Cs<sup>+</sup>, Cu<sup>+</sup>, Ag<sup>+</sup> (<i>a</i>), and DFT models for the adducts (<i>b</i>).147 Published with permission from MDPI"}}]](/storage/images/resized/7CbPwv9gCuu3vgYfTDBUKYrw83r8iezbG7COddZ8_xl.webp)
The experimental study of the acid--base properties of Y zeolite carried out by Pang et al.148 showed that the introduction of Zn2+, V3+ and Cu2+ cations into this zeolite increases the content of Lewis acid sites 5--6-fold (according to IR spectroscopy data for adsorbed pyridine) compared to that in the unmodified zeolite in the H-form. Copper modification gives rise to stronger Lewis acid sites than modification with zinc; this is qualitatively in line with the position of Cu2+ and Zn2+ cations on the HSAB scale.
While turning to elucidation of the factors that influence the catalytic properties of zeolites in the reactions including hydrogen transfer, we would like to note that the pristine zeolite without modifying elements is catalytically active in the hydrogenation reactions. For example, it was noted149 that HZSM-5 (Si:Al=120, treatment with HCl followed by annealing at 500 °C) possessed a noticeable activity towards ethylene hydrogenation at 450 °C, and this was not caused by the iron impurity (~0.007 mass%). One of the explanations to this activity is the assumption that the bifunctional Brønsted--Lewis acid site participates in the activation of reactants (C2H4 and H2); this site catalyzes both the protonation of ethylene and the transfer of the hydride ion (resulting from heterolysis of hydrogen molecule on the Lewis Al–O site) to the ethyl carbocation. The possibility of formation of these tandem sites was considered in the studies of the effect of the Al3+ distribution in the framework on the catalytic properties of the zeolite. A detailed critical analysis of the results of such studies has been reported.150 Generally, only three probable distribution patterns of aluminium cations on the channel surface should reasonably be considered for zeolites with Si:Al>10:
(a) Al pairs (aluminium cations are linked by one OSiO group; Fig. 17a);
(b) close unpaired Al atoms (aluminium cations are bound by a sequence of two or three OSiO groups; Fig. 17b);
(c) single Al atoms (Fig. 17c).
![[{"id":"3BBSu75ruQ","type":"paragraph","data":{"text":"Schematic images of the types of distribution of aluminium cations (highlighted in red) on the channel surface.150 For <i>a-c</i>, see text. Published with permission from John Wiley & Sons."}}]](/storage/images/resized/v7AF4lrPZXf6yPxSCSRz57A5dfl4KRbiMnAbmeoZ_xl.webp)
These patterns of distribution of aluminium cations in the zeolite (especially a and c) can be distinguished using MAS 29Si NMR spectroscopy and especially various techniques of MAS 27Al NMR spectroscopy.150 The results of catalytic experiments are interpreted considering the fact that each framework aluminium cation possesses a Brønsted acid site, –O–Si–O(H)–Al–O–; therefore, the difference between sites a and c is not exhausted by the fact that site a has two closely spaced aluminium cations, but in addition, this site has two closely spaced OH groups. A study of the catalytic behaviour of HZSM-5 zeolites differing in the ratio of the concentrations of sites a and c in the cracking of but-1-ene demonstrated that sites c are more active towards cracking of butene and octene (as the butene dimerization product) and give light olefins; sites a promote, to a greater extent, olefin oligomerization and hydrogen transfer reactions, resulting in the formation of aromatic compounds (Fig. 18).
![[{"id":"_K2s1tIuKQ","type":"paragraph","data":{"text":"Diagram reflecting the effect of the distribution of aluminium cations in the HZSM-5 zeolite framework (Si : Al = 15) on the catalytic properties in the but-1-ene cracking (500 <sup>o</sup>C, 15 h<sup>-1</sup>, 20 min): (1) for a zeolite in which 84% of aluminium occurs as sites <i>c</i>; (2) for a zeolite in which 40% of aluminium occurs as sites c.150 Published with permission from John Wiley & Sons."}}]](/storage/images/resized/GsZe9GOxh5Df5zTs5ROIWYlhMA4s2zUY5KUsCNPQ_xl.webp)
The presence of so-called aluminium pairs (AlPs) is important for the preparation of zeolites containing metal cations (especially transition metal cations) incorporated in the surface. This markedly expands the scope of catalytic applications of zeolites up to the reactions traditionally catalyzed by metal complexes. As an example, consider a study by Deng et al.,151 who demonstrated that Y zeolite with incorporated Ni2+ cations (NiY) is an efficient catalyst for the selective hydrogenation of acetylene to ethylene. Detailed study of the structure of this catalyst provided conclusions about the nature of its active site involving AlP and about the probable mechanism of functioning of this site; according to this mechanism, the key (rate-determining) step is heterolytic activation of hydrogen on the Ni–O moiety (apparently acting as FLP) followed by addition of, first, a hydride ion and, second, a proton to the acetylene molecule activated with Ni2+ cation (Fig. 19), i.e., the mechanism is similar to the mechanism of ethylene hydrogenation in the presence of PdII complexes considered above (see Fig. 5a).
![[{"id":"5JqQvm-Oa5","type":"paragraph","data":{"text":"Characterization of Ni<sup>2+</sup>-containing Y zeolite as a catalyst for acetylene hydrogenation.151 Local environment of the Ni<sup>2+</sup> cation incorporated (<i>via</i> AlP) into zeolite framework from the data of XANES (X-ray absorption near edge structure) (<i>a</i>), EXAFS (extended X-ray absorption fine structure) (<i>b</i>) and DFT (<i>c, d</i>); (<i>e</i>) changes in the Gibbs energy of formation and structures of intermediates formed during hydrogenation according to DFT calculation data. Oxygen atoms are shown in red, silicon is yellow, aluminium is purple, nickel is grey-blue, carbon is grey and hydrogen is white. Published with permission from the Chinese Chemical Society"}}]](/storage/images/resized/FH0cBXSiFLwToG8tORBfJSlqkf4OE4m8fNuppVKj_xl.webp)
While returning to the issue of involvement of tandem sites in the catalytic processes, note that the relative arrangement and the concentration ratio of Lewis and Brønsted sites may produce, in some cases, a synergistic effect in the activation of C–H and C–C bonds. For example, study of a system containing an extra-framework Ga3+ cation in the hydrogen form of the MFI zeolite 131 revealed an interesting hydrogen transfer reaction: olefin hydrogenation with methane giving ethylene as a product of methane dehydrogenation. The mechanism of this unusual reaction was interpreted152 under the assumption that there are proximate Brønsted and Lewis sites that activate the olefin (H+ addition) and methane (hetrolytic addition to the Ga–O moiety), respectively, and this is followed by hydride ion transfer to the ethyl carbocation and coupling of methylene groups.
Generally, the steric restrictions generated by the pore system inside the crystal, which depends on the type of the zeolite, promote the formation of bifunctional C–C and C–H bond activation sites, facilitating the concerted proton and/or hydride ion transfer between different sites. Simultaneously, they hamper intermolecular hydrogen transfer reactions such as the reaction between the carbocation and a hydrogen donor hydrocarbon, which is accompanied by hydride ion transfer and gives a new hydrocarbon and a new carbocation. This effect was demonstrated by Meusinger and Corma,153 who compared the composition of n-heptane cracking products formed using Y and MFI zeolites. The paraffin:olefin ratio, which indirectly characterizes the contribution of intermolecular hydrogen transfer reactions (hydrogen transfer coefficient, see below), is markedly higher for Y zeolite, in which the channels are larger than in MFI.
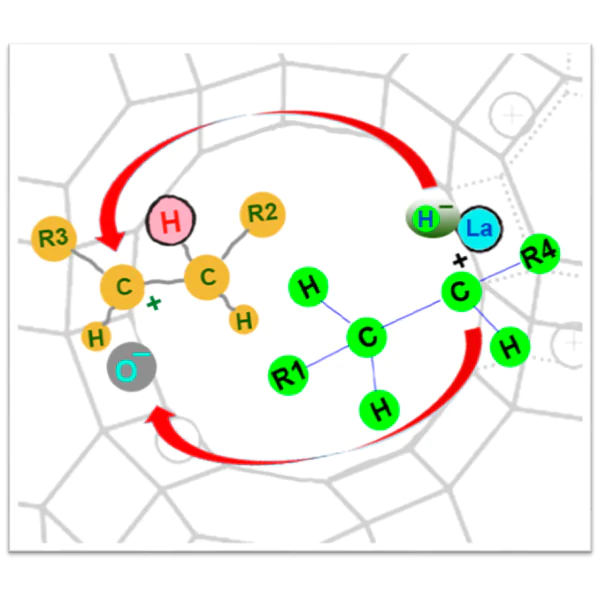
The contribution of intermolecular hydrogen transfer reactions to cracking processes was quantitatively estimated40 using deuterated compounds for the conversion of hex-1-ene and a naphthene (cyclohexane-d12) mixture and a series of zeolite-based catalysts differing in the composition of the zeolite component (cation Y zeolite; Y and ZSM-5 zeolites in different ratios) (Fig. 20a,b). Using gas chromatography--mass spectrometry, the authors showed that only one hydrogen (deuterium) atom comes from the donor molecule in the hydrogen transfer reaction, while the other hydrogen atom originates from the Brønsted acid site of the catalyst (see Fig. 20c). Meanwhile, the cracking of pure hydrogen donor (cyclohexane-d12) does not give deuterated hexanes (only deuterated C1–C5 hydrocarbons are present among the products). This indicates that deuterated hexanes appear only upon the transfer of the deuteride ion from the naphthene to the olefin, with the acid site of the catalyst being regenerated in the deuterated form (see Fig. 20c).
![[{"id":"Ii0ShcQguu","type":"paragraph","data":{"text":"Scheme of intermolecular hydrogen trans\u0002fer during the cracking of hydrocarbons: (<i>a</i>) components of the cracking catalysts; (<i>b</i>) amount of deuterium transferred to hexanes (hydrogen transfer products) <i>vs.</i> hydrogen transfer coefficient (ratio of the selectivities to butanes and butylenes) for joint conversion of hex-1-ene and deuterocyclohexane on various zeolite-based catalysts [500 <sup>o</sup>C, weight hourly space velocity (WHSV) of 10 h<sup>-1</sup>, equilibrium cracking catalysts, i.e., bi-zeolite (Y and ZSM-5) catalysts taken from the process]; (<i>c</i>) hypothesized mechanism of the intermolecular hydrogen (deuterium) transfer.40 Published with permission from Elsevier"}}]](/storage/images/resized/01c4U4xNub1tsvLxtXwOcC2E23rQ7lZSkVWrJjY4_xl.webp)
In general, it is worth noting that experimental studies of hydrogen transfer reactions during the cracking of hydrocarbons in the presence of zeolite systems are complicated by numerous reactions occurring in parallel (e.g., isomerization, alkylation, aromatization, etc.), non-uniformity of active sites on the catalyst surface, heteroatomic impurities and some other factors; this makes interpretation of the results of these studies a difficult task.
*See http://europe.iza-structure.org/IZA-SC/ftc_table.php (accessed on February 27, 2023).
3. Effect of the components of cracking catalysts on their behaviour in hydrogen transfer reactions
The modern cracking catalysts are complex multicomponent systems, with each component contributing to the catalyst behaviour. It is noteworthy that the contributions of components to the catalytic and physical properties of systems are not simply added; a pronounced role is played by the interaction between the components, resulting in the synergistic effects changing the catalyst activity and selectivity.
As the variable components of the cracking catalysts that form the catalyst structure, it is customary to distinguish the matrix (the first cracking function, see Fig. 3) and active components, zeolite Y and ZSM-5 (the second and third cracking functions, respectively).154 The practical use of other zeolites is considerably restricted by their high cost [e.g., ITQ-7 (Ref. 155)] or low thermal stability, which leads to fast catalyst deactivation under drastic conditions of the catalytic cracking (e.g., MCM, Beta).156--158 The catalyst matrix is a blend composed of aluminosilicates (amorphous natural clays) and alumina. The matrix has important functions for the industrial cracking process, in particular
- performs the primary cracking of the feedstock and efficiently utilizes the cracking potential of zeolite components;
- ensures the mechanical strength of the catalyst microsphere;
- withdraws the heat during the oxidative regeneration;
- reduces the pore diffusion resistance by creating a system of transport pores;
- forms the required bulk density;
- increases the thermal stability of the zeolite components, i.e., prevents the zeolite amorphization during the catalyst operation.
The preparation techniques of composite cracking catalysts are based on the following principles:
- the matrix and the zeolites are produced separately, with a specified ratio of crystallite size and desired crystallite shape, and the desired porosity and acidity of the matrix. Then the components are mixed to form a composition, while the possible interaction between the components is taken into account; this may increase the activity and stability of the components and of the catalyst as a whole;159,160
- the zeolite component is formed directly in the matrix (in situ process).161
Among the known production techniques of the composite cracking catalyst, the following three processes are used most often:154,162
1) in situ (used by BASF; the synthesis of Y zeolite directly in the precursor of the catalyst matrix);
2) sol process (used by Grace, BASF and Albemarle Sinopec; the matrix is based on alumina sols);
3) sol-gel process (used in public joint-stock company Gazpromneft; the matrix is based on amorphous aluminosilicate and alumina pseudo-sol).
The first and second processes afford cracking catalysts with a relatively high content of Y zeolite (~30-40 mass%)163,164 compared to the content in the catalyst prepared by the third technique (~20 mass%).154,162,165 The components of the composite catalyst show different activities towards hydrogen transfer reactions; therefore, a markedly different zeolite component:matrix ratio would result in a considerable change in the composition of products for similar degrees of conversion of the hydrocarbon feed. Therefore, this part of the review is focused on analysis of the causes for differences in the activities of the components of the cracking catalyst (including methods of synthesis and subsequent modification).
3.1. Y zeolite
Among the components of cracking catalysts, the highest activity in the intermolecular hydrogen transfer is inherent in Y zeolite.166 The pore aperture of this zeolite is ~0.74 nm, while the size of the large cavities is ~1.1 nm; hence, there are no steric hindrances for the formation of hydrogen acceptor--hydrogen donor adducts (transition states necessary for the intermolecular hydrogen transfer) on the acid sites of these cavities. This circumstance, together with high concentration of acid sites on this component (>1500 mmol g–1 according to the ammonia temperature-programmed desorption data), account for the very high specific activity of Y zeolite in the intermolecular hydrogen transfer reactions.
The ratio between the Lewis and Brønsted acid sites and the number and strength of acid sites, which affect the activity of zeolite, may considerably differ depending on the zeolite SiO2:Al2O3 molar ratio and the cationic composition of the framework and upon the formation of extra-framework species. For example, it was shown167--170 that an increase in the content of rare earth elements (La, Ce, Nd, Pr) in the Y zeolite from 0.5 to 10 mass% leads to increase in the overall concentration of acid sites and increase in the catalyst efficiency in the intermolecular hydrogen transfer, which is reflected by the hydrogen transfer coefficient (Fig. 21).
![[{"id":"jV061HBmEC","type":"paragraph","data":{"text":"Effect of the content of rare earth oxides in the cracking catalyst on the hydrogen transfer coefficient (found as the ratio of the integral selectivities to butanes and butylenes, experimental time of 30 s) for the conversion of coked gasoline and straight-run gasoline mixture.171 Published with permission from Springer"}}]](/storage/images/resized/nkg0vpnVUUH69E9zfXLPOJLBznioX9S5Wisj5qVi_xl.webp)
In the design of petrochemical catalysts, it is necessary to bear in mind that the activity of Y zeolite towards the intermolecular hydrogen transfer should be suppressed to the highest possible extent, i.e., in this case, the use of zeolite with a minimized content of rare earth (RE) oxides is preferred. At the same time, it should be remembered that Y zeolite is responsible for the transformation of the intermediates formed on the matrix (see Fig. 3) and provides extensive conversion of vacuum gas oil. The latter process requires the use of Y zeolite with a relatively high acidity (H0≈–9), which is, in turn, accompanied by increasing activity towards hydrogen transfer and decreasing yield of light olefins due to their hydrogenation.
The authors of a number of publications made attempts to model hydrogen transfer reactions on zeolites, in particular faujasite (Y zeolite). The first publications devoted to modelling of hydrogen transfer on zeolites mainly presented experimentally determined indirect characteristics that reflected the contribution of hydrogen transfer (such as the hydrogen transfer coefficient mentioned above; the paraffin to olefin molar ratio in the cracking products; and the content of isobutane, which is considered to be the major product of hydrogen transfer involving the tert-butyl carbocation) and determined the kinetic regularities of transformations of model hydrocarbons. Lukyanov et al.172 addressed hydrogen transfer reactions involved in the conversion of n-hexane (hydrogen donor) with C2-C5 olefins (hydrogen acceptors) adsorbed on Brønsted acid groups of the catalyst, ZSM-5. It was shown that the rate of butane formation upon the reaction of hexane with the butyl carbocation is ~70 times higher than the ethane formation rate from the ethyl carbocation.
Jiao et al.173 performed a theoretical study of the effect of the nature and content of RE cations in the zeolites on their acid--base properties and proposed a bimolecular hydrogen transfer model, which was based on the concept of `symbiosis' of the Lewis and Brønsted acid sites in this process. According to this model, the cerium(III) cation located in small cavities of Y zeolite promotes the adsorption of the hydrogen donor, elimination of the hydride ion and transfer of the hydride ion to the hydride ion acceptor, that is, the carbocation formed on the Brønsted acid site (Fig. 22). In the opinion of the authors, the presence of cerium-containing sites with a low (soft) Lewis acidity rather than the number and strength of the Brønsted acid sites plays the crucial role for the hydride transfer reactions. Blaszkowski et al.,174 who carried out a DFT study of hydrogen transfer on zeolites, showed that an increase in the zeolite acidity increases the contribution of hydrogen transfer to a markedly higher extent than it promotes other reactions (cracking, isomerization, etc.) that take place on the zeolite. Li et al.,175 who considered the butane conversion on H-BEA zeolite also using computational methods, showed that an increase in the acid site strength leads to a higher rate of the hydride transfer. It is of interest that a similar conclusion follows from studies of homogeneous analogues (see Section 2.1, Fig. 9): an increase in the strength of the acid site carrying the hydrogen acceptor facilitates the hydride transfer from the donor to the acceptor.
![[{"id":"TEh_cMs_Qk","type":"paragraph","data":{"text":"Hydrogen transfer from a hydrogen donor molecule (R<sup>1</sup>HH) to a hydrogen acceptor molecule in the FAU zeolite cavity bearing a cerium cation (Lewis acid) and protons (Brønsted acid) on the surface. The square designates a vacancy. The Figure was created by the authors using pub\u0002lished data.173"}}]](/storage/images/resized/6AW4gw0q1llwOv9hvkkmI5SWA6wkyhHEdjvZu8Q0_xl.webp)
Maihom et al.176 explored the conversion of n-hexane on the HY zeolite using 38T model (where T is AlO4 or SiO4 tetrahedron in the zeolite framework unit being calculated, 38 is the total number of the tetrahedra). The results of DFT calculations attested to occurrence of cracking reactions to give propane and propylene via the step of hexane adsorption on a Brønsted acid site of the zeolite giving hexane-3-carbonium intermediate. Mullen and Janik177 investigated the structures of intermediates formed upon the hydrogen transfer from ethane, propane and isobutane molecules to an alkoxide molecule (that is, ethoxide, propoxide or tert-butoxide molecule formed upon olefin adsorption on the zeolite OH group) adsorbed on mordenite (48T model) and ferrierite (36T model). According to DFT calculations, the hydride ion transfer proceeds via the formation of a common hydride ion--carbocation complex in the zeolite channels at the Lewis and Brønsted acid site position. It is of interest that on the basis of DFT data for ultrastable (dealuminated) Y zeolite in the close vicinity of the Brønsted acid site, Mota et al.178 drew the conclusion that the presence of a Lewis acid site (aluminium cation) leads to increasing strength of the Brønsted acid site.
It is noteworthy that the activity towards hydrogen transfer is more beneficial for increasing the yield of isobutane than butylenes. Butylenes and butane are both formed via the intermediacy of the butyl carbocation (C4H9+); the ratio of the rates of formation of these products is determined by both the nature of the hydrogen donor (paraffins, naphthenes) and the catalyst activity in the intermolecular hydrogen transfer.9,21,22,179 In the case of composite catalyst, this activity sharply increases with increasing content of Y zeolite in the catalyst; simultaneously, the yield of butylenes sharply decreases (so does the content of unsaturated compounds in the butane--butylene fraction), while the isobutane yield increases.
Other products formed via hydrogen transfer reactions are aromatic compounds; in the gasoline fraction, they are mainly C7–C9 alkylbenzenes. The increase in the catalyst activity towards the intermolecular hydrogen transfer upon increasing content of Y zeolite leads to higher yields of aromatic compounds in the heavy fraction of the cracked gasoline and lower content of olefins in the light fraction.
3.2. ZSM-5 zeolite
ZSM-5 is the basic component for the manufacture of cracking catalyst for petrochemical purposes.180,181 The ZSM-5 channels are narrower (0.55 nm) than those in Y zeolite;182 therefore, products of lower molecular weights are formed on ZSM-5. As a rule, the introduction of ZSM-5 zeolite leads to higher yield of propylene.
Despite the relatively small size of channels, the activity of pristine (unmodified) ZSM-5 in the H-form towards intermolecular hydrogen transfer is relatively high. In order to decrease this activity, the zeolite is modified with phosphorus compounds.183 This modification is accompanied by the interaction between the aluminium cations in the zeolite framework and PO4 groups; this gives extra-framework aluminium polyphosphates; however, inside the zeolite channels, first, the zeolite acidity decreases (this is especially true for the strong Brønsted acid sites) and, second, the size of the channels decreases due to the formation of aluminium phosphates. Generally, the modification with phosphorus compounds affects the catalytic properties of the zeolite in the same way as the increase in the silica to alumina ratio or the decrease in the size of channels. For example, the introduction of >7.5 mass% phosphorus into ZSM-5 leads to a 10-fold decrease in the hydrogen transfer coefficient.184
In addition to the change in the physicochemical and catalytic properties, phosphorus modification increases the thermal stability of the zeolite; this ensures longer retention of the catalyst activity in high-temperature cracking.185 The improvement of the stability is due to the increase in the silica to alumina ratio of the zeolite and to the appearance of extra-framework species.
The variation of the mass ratio of the modified Y zeolite (by introducing extra-framework species, changing the silica to alumina ratio and the cationic composition, first of all, the content of RE oxides) and ZSM-5 zeolite (by introducing phosphorus compounds and changing the silica to alumina ratio) in the composite cracking catalyst can be used to control the pathway of cracking reactions (carbon--carbon bond scission; skeletal isomerization; hydrogenation of hydrogen acceptor and dehydrogenation of hydrogen donor; and aromatization of hydrocarbons) and, finally, to control the ratio between the yields of propylene, butylene, isobutane and gasoline components. The increase in the fraction of phosphorus-modified ZSM-5 in the composite catalyst results in increasing propylene yield and decreasing gasoline yield, As the content of Y zeolite modified with RE cations in the catalyst increases, the yield of butylenes increases, while the high gasoline yield is retained.
Fu et al.186 used DFT calculations to compare the hydrogen transfer pathways involved in the transformations of n-octane on hydrogen forms of Y and ZSM-5 zeolites. The authors considered the reaction of adsorbed butyl carbocation (2-butyl alkoxide) as a hydrogen (hydride ion) acceptor with n-octane molecule as a hydride ion donor to give n-butane and adsorbed octyl carbocation (3-octyl oxide). In the presence of ZSM-5, this reaction mainly proceeds by a monomolecular mechanism, while in the case of Y zeolite, the mechanism is mainly bimolecular. The calculated energy barriers for the monomolecular and bimolecular reactions over ZSM-5 were 5.30 and 23.12 kcal mol–1, while in the case of Y zeolite, these values were 17.39 and 6.95 kcal mol–1. The results of calculations were in good agreement with the above data on the comparison of hydrocarbon conversion pathways over Y and ZSM-5 zeolites with substantially different channel sizes (0.74 and 0.55 nm, respectively), which attest to steric restrictions for fast bimolecular hydrogen transfer reactions in the case of narrower-pore ZSM-5 (in particular, C2–C4 olefins can be obtained in relatively high yields upon cracking of alkanes).
3.3. Matrix of the catalyst
The basic components of the matrix used to manufacture cracking catalysts are amorphous aluminosilicate, alumina and clays. Clays can be represented by kaolin (layered mineral consisting of a layer of SiO4 tetrahedra and a layer of AlO6 octahedra linked via an oxygen atom) or montmorillonite (layered mineral consisting of three layers: two layers of SiO4 tetrahedra and a layer of AlO6 octahedra sandwiched between them). Montmorillonite is more preferable, because it is less active towards intermolecular hydrogen transfer.
In relation to model hydrocarbons, Lipin187 showed that the activity of matrix components in both the carbon--carbon bond scission and hydrogen transfer reactions increases in the series
amorphous aluminosilicate>alumina>clay
While considering the catalyst composition as a whole, it is necessary to take into account the interaction between the components during the synthesis and pretreatment. An increase in the thermal stability of Y zeolite in the aluminosilicate matrix (due to redistribution of sodium cations) and ZSM-5 zeolite in the alumina--montmorillonite matrix188 and enhancement of interaction of amorphous aluminosilicate with alumina (with increasing content of alumina in amorphous aluminosilicate and, hence, increasing concentration of acid sites)189 may change the dependence of the catalyst activity towards hydrogen transfer on the catalyst composition. For example, an increase in the thermal stability of Y zeolite in the aluminosilicate matrix would result in a higher fraction of this zeolite being retained during the operation of the cracking catalyst relative to the zeolite content present after the synthesis. The increase in the fraction of the retained zeolite would increase the catalyst activity in the presence of the aluminosilicate matrix towards hydrogen transfer compared to the catalyst activity without this matrix, with the initially equal contents of Y zeolite in both catalyst compositions.
The ratio of Brønsted and Lewis acid sites in the cracking catalyst matrix is determined by the amorphous aluminosilicate:aluminium-containing component ratio (the aluminium-containing component is a mixture of montmorillonite and alumina in equal mass proportions). A decrease in the content of amorphous aluminosilicate leads to high proportion of Lewis acid sites. When the indicated ratio is ~0.9, the total yield of light olefins (C2–C4) reaches a maximum (Fig. 23).
![[{"id":"G6Sgc1_sqV","type":"paragraph","data":{"text":"Total yield of C<sub>2</sub>-C<sub>4</sub> olefins upon cracking of feed characterized by different degrees of hydrotreating on petrochemical cracking catalysts <i>vs.</i> composition of the matrix in which the aluminium-containing component is a mixture of montmorillonite and alumina. Designations: HVGO is hydrotreated vacuum gas oil, NHVGO is non-hydrotreated vacuum gas oil, HCR is hydrocracking residue. The activity was measured according to ASTM D 3907, 527 <sup>o</sup>C, the catalyst : feed ratio was 4. The Figure was created by the authors using published data.169"}}]](/storage/images/resized/OLx5Dm3UkYidArl0hIuJphOOuyniyV6Qgke7z6EE_xl.webp)
The presence of a maximum yield of light olefins at a certain ratio between amorphous aluminosilicate and aluminium-containing component is characteristic of each type of feed. The minimum total yield of olefins is observed when this ratio is 0.5. In this case, the Y zeolite stabilization and the catalyst activity towards hydrogen transfer are maximized. It is important that the highest content of olefins in the propane--propylene and butane--butylene fractions is attained when this ratio is low (~0.3), i.e., at the highest content of alumina.
Thus, for the design of catalysts with a specified activity towards hydrogen transfer meant to perform cracking along a chosen pathway, it is necessary to take into account both the properties of particular components, which are determined by their nature and modification conditions, and the interaction between the components within the catalyst (Table 1).

An example of targeted use of hydrogen transfer is hydrogen-free upgrading of thermally processed gasolines.189,190 The process consists of purification of low-grade gasoline fractions (such as delayed coker gasoline, visbreaker gasoline and heavy catalytically cracked gasoline) by decreasing the content of unsaturated (via hydrogenation) and sulfur (C–S bond hydrogenolysis to give H2S) compounds as a result of intermolecular hydrogen transfer reactions. Other non-demanded gasoline fractions with high content of paraffins and napthenes (e.g., 62--5 °C gasoline fraction) serve as the hydrogen donors, being thus converted to aromatic products. The hydrogen-free upgrading process may remove more than 95 and 90% of unsaturated and sulfur compounds, respectively, from low-grade gasoline fractions and change the type composition of hydrocarbons (increase the content of aromatic hydrocarbons and isoparaffins), thus increasing the octane number of the resulting overall gasoline by 5--10 points.
It was noted above that the hydrocarbon conversion on acid--base catalysts involves two types of reactions (see Fig. 2): cracking (carbon--carbon bond scission) to give products with a lower molecular weight and hydrogen transfer to give products with similar molecular weight, but differing in the composition or structure. The reactions of both types are not thermodynamically unfavoured in the temperature range of 450--650 °C, and the activation energy of cracking is much higher than that of hydrogen transfer.172,191 The latter accounts for the predominance of hydrogen transfer at temperatures of 450--500 °C and predominance of cracking at 520--650 °C. These trends were confirmed by experimental results.40,191--194 Thus Potapenko et al.192 comprehensively studied the effect of conditions and catalyst composition on the contribution of hydrogen transfer reactions to various types of the catalytic cracking of hydrocarbon feed. For the model cyclohexane--hex-1-ene--cyclohexane-d12 mixture, the authors estimated the deuterium transfer coefficient, which takes into account not only butane and butylene selectivity (S) ratio, but also the content of deuterium atoms in them (χD). The deuterium transfer coefficient (DTC) was found by the formula
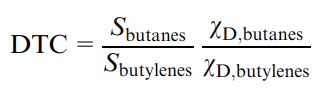
The transformations of the model system were carried out192 under conditions optimal for these reactions (classic catalytic cracking of vacuum gas oil, catalytic hydrogen-free upgrading, catalytic cracking of gasoline fractions). The process conditions and hydrogen and deuterium transfer coefficients are given in Fig. 24. In the catalytic conversion of the model mixture, the highest HTC and DTC values were observed under the conditions optimized for the catalytic hydrogen-free upgrading. The deuterium transfer coefficient was somewhat higher, which attests to the predominant transition of deuterium (as labelled hydrogen) to saturated C4 hydrocarbons. The catalytic cracking of gasoline fractions was characterized by minimized (nearly zero) HTC values, which attests to single (monomolecular) conversion of feed components and predominant accumulation of deuterium in unsaturated C4 products.
![[{"id":"d1X8WPGf3G","type":"paragraph","data":{"text":"Hydrogen (HTC) and deuterium (DTC) transfer coefficients for various processes in the conversion of a model cyclohexane - hex-1-ene - cyclohexane-d<sub>12</sub> mixture. The catalyst for vacuum gas oil cracking (process I) is based on Y zeolite with a reduced (<5 mass %) content of RE (La, Ce, Nd, Pr) oxides; the catalyst for hydrogen-free upgrading (process II) is based on Y zeolite with an increased (>7 mass %) content of RE oxide and ZSM-5 zeolite not modified with phosphorus compounds; the catalyst for cracking of gasoline fractions (process III) is based on phosphorus-modified (4% P) ZSM-5, exhibiting a minor activity towards hydrogen transfer.192 <i>P</i> is pressure, <i>C<sub>dil.</sub></i> is dilution of the reaction mixture by an inert component. Published with permission from Elsevier."}}]](/storage/images/resized/SCR2xbUqpnDtvr1f8c0if0sQe8oOWzudbi61EEhJ_xl.webp)
The classic cracking of vacuum gas oil occupies an intermediate position between hydrogen-free upgrading and cracking of light fractions. For this process, both types of reactions are important, that is, cracking (forming the essence of the process and determining the yield of propane--propylene and butane--butylene fractions and gasoline) and hydrogen transfer, which promotes the formation of isoalkanes and aromatic compounds.
4. Significance of the intermolecular hydrogen transfer for the conversion of
sulfur-, nitrogen- and oxygen-containing compounds in the presence of hydrocarbons on zeolite catalysts
The intermolecular hydrogen transfer on zeolite-based catalysts of catalytic cracking is especially important for co-processing of sulfur-, nitrogen- and oxygen-containing compounds in the presence of hydrocarbons, the major components of mixed feed. Note that the proportion of non-hydrotreated types of feed in the total amount of feed supplied to the catalytic cracking worldwide exceeds 70% (Table 2).

A non-hydrotreated mixed feed contains up to 3 mass% sulfur and up to 0.5 mass% nitrogen.195 The sulfur compounds are mainly thiophene derivatives, which account for ~70% of the whole amount of sulfur.196 Depending on the origin of the feed, it may contain both polycyclic and monocyclic sulfur derivatives with bulky alkyl groups. The rest 30% of sulfur are concentrated in non-aromatic sulfur compounds: mercaptans, sulfides and disulfides. Nitrogen compounds include amines, pyrrole and pyridine derivatives, polycyclic aromatic and non-aromatic nitrogen compounds and porphyrins. The total content of nitrogen compounds is, most often, an order of magnitude lower than the content of sulfur derivatives. The presence of sulfur and nitrogen compounds in the feed leads to faster deactivation of the cracking catalyst, with the poisoning action of nitrogen compounds being more pronounced.195
The involvement of renewable (green) types of feed into cracking requires joint processing of hydrocarbons and oxygen-containing compounds; the latter include, depending on the source of feed, fatty acid triglycerides (vegetable oils),197 aliphatic alcohols,198 phenol derivatives (bio-oil),199--201 furan derivatives,202 etc.
During the catalytic cracking of a mixed feed comprising hydrocarbons, together with the sulfur-, nitrogen- and oxygen-containing organic compounds, heteroatomic molecules can undergo the following main reactions:
- cracking of the C–C and C–E bonds, where E=S, N and O, to give the corresponding E hydrides and mixtures of unsaturated organic compounds with a lower molecular weight either containing heteroatoms or not; in addition, E-containing coke can be formed;
- hydrocracking of C–C and C–E bonds to give E hydrides and mixtures of saturated and aromatic hydrocarbons.
The second pathway may be accompanied by a considerable decrease in the content of heteroatomic compounds in the target products of cracking, but it requires a large contribution of hydrogen transfer reactions. Since the effect of the hydrocarbon (hydrogen donor) nature and process conditions (temperature) on these reactions has been addressed in the literature,203--205 in this review, the attention is focused on analysis of the possibilities of increasing the contribution of hydrogen transfer reactions by varying the nature of active sites and by introducing additional sites.
4.1. Sulfur compounds
The transformation of sulfur non-aromatic compounds on acid--base catalysts proceeds relatively easily.206 The cracking of mercaptans, sulfides and disulfides affords hydrogen sulfide and the corresponding unsaturated hydrocarbons. The reaction is so fast that the residual content of non-aromatic compounds in the liquid cracking products does not exceed 5% of the total amount (in terms of sulfur atoms) of all sulfur compounds, and these compounds are fully concentrated in the fraction with initial boiling point of 80 °C.
The conversion of mercaptans to give olefins and hydrogen sulfide (dehydrosulfidation reaction) is an analogue of the dehydration of alcohols. Hence, this reaction can be catalyzed by both Brønsted and Lewis acid sites; the presence of even very weak sites in the catalyst is sufficient (e.g., H0≈+4 for Brønsted sites).
Thiophene aromatic compounds are much more stable during catalytic cracking than mercaptans and sulfides. In the absence of hydrocarbon, the following transformation pathways can be considered for alkyl-substituted thiophene derivatives: alkylation, isomerization (change in the position of the alkyl substituent) and condensation. During the joint conversion of the hydrocarbon acting as a hydrogen donor and a thiophene compound acting as a hydrogen acceptor, hydrogen transfer reactions give first a cyclic non-aromatic sulfide (di- or tetrahydrothiophene derivative). The loss of aromaticity in the ring sharply decreases the stability of this compound, which leads to its fast conversion to hydrogen sulfide and hydrocarbon. The dehydrosulfidation of tetrahydrothiophene, like that of mercaptans (see above), does not require strong acid sites. The overall chart of transformations of thiophene compounds in the course of catalytic cracking and in the presence of hydrogen donor is depicted in Fig. 25.
![[{"id":"48DBXIIJtp","type":"paragraph","data":{"text":"Transformations of thiophene compounds,207 including isomerization (<i>1</i>), alkylation (<i>2</i>), dealkylation (<i>3</i>) and hydrogen transfer (<i>4</i>). [H] is hydrogen donor, R is alkyl substituent, R = is olefin. Published with permission from Springer"}}]](/storage/images/resized/OmJfOuEIfRqLVlqgpYJgIzKA6V0BCJDCMkTVfnGX_xl.webp)
Analysis of the product compositions and experiments with deuterated compounds make it possible to describe the hydrogen transfer from cycloalkane to thiophene compound by the scheme presented in Fig. 26a. It can be seen that the hydrogen transfer to thiophene compound from a hydrogen donor hydrocarbon requires the presence of both Lewis and Brønsted acid sites in the catalyst (Fig. 26b). Thus, modification of a catalytic system with the goal to increase the selectivity to hydrogen sulfide via hydrogen donation can be attained by controlling both the concentration ratio and the acidity level of the Brønsted and Lewis sites. Furthermore, the step shown in Fig. 26c suggests that the Lewis and Brønsted acid sites should be proximate to each other in such a way as to enable the hydride ion transfer through its bound states.
![[{"id":"pZOiaTXWAp","type":"paragraph","data":{"text":"Cracking reactions of thiophene derivatives to give hydrogen sulfide in the presence of cycloalkane. (<i>a</i>) Formation of the s-complex with a Brùnsted acid site and alkylthiophene (proton transfer); (<i>b</i>) formation of the carbenium ion and hydride ion upon the interaction of the hydrogen donor hydrocarbon and Lewis acid site; (<i>c</i>) hydride ion transfer to the s-complex and regeneration of Brønsted and Lewis acid sites"}}]](/storage/images/resized/gUWLAUJKVv57LDAzzMKtMajyMsZOa53sRILj4S4M_xl.webp)
Li et al.208 analyzed the transformation routes of thiophene only on the Brønsted acid sites without participation of other sites (i.e., via single site adsorption) and in the absence of hydrogen donor hydrocarbons in the system. According to DFT calculations, destruction of the thiophene ring proceeds in this case via disproportionation, which first gives 2-(2,5-dihydrothiophen-2-yl)thiophene, that is, hydrogenated thiophene dimer, bound to the acid site, and then leads to C-S bond scission in 2,5-dihydrothiophenyl radical as a result of hydrogen transfer within the dimer molecule. Mechanisms of destruction of thiophene derivatives to mercaptans (which are highly prone to dehydrosulfidation, as noted above) on the catalysts containing not only acid but (additionally) basic sites both in the absence and in the presence of hydrogen in the reaction system were also proposed on the basis of DFT calculations.209,210
In the former case, the process includes double-site adsorption of a thiophene molecule on a Brønsted acid site and a Lewis basic site followed by ring opening to give unsaturated vinyl-acetylene mercaptan (upon intramolecular hydrogen transfer from one carbon atom to another) (Fig. 27a). In the latter case (in the presence of hydrogen), the intramolecular hydrogen transfer step (occurring in the former case) is replaced by the transfer of proton and hydride ion (arising upon the heterolytic activation of hydrogen) to the adsorbed mercaptan intermediate, which gives rise to divinyl mercaptan (Fig. 27b).
![[{"id":"mCZnBu3oJ_","type":"paragraph","data":{"text":"Mechanisms of destruction of the thiophene ring to give mercaptans on an acid - base site in the absence (<i>a</i>) and in the presence (<i>b</i>) of hydrogen.209, 210 Published with permission from Elsevier"}}]](/storage/images/resized/Jy6qRoDdU5SJb9rTrsIn075E5x8Vxj8cDBFw4jdq_xl.webp)
Can et al.211 considered an unusual mechanism for the conversion of thiophene compounds. As the first (key) stage, the authors identified the hydrogen transfer from a hydrocarbon molecule to the molecule of a thiophene derivative to give tetrahydrothiophene; the mechanism of this transfer was not specified. In the authors opinion, the subsequent transformations of tetrahydrothiophene also deserve special attention; the destruction of tetrahydrothiophene molecule can be intensified by using a special additive containing both acid and basic sites, e.g., mixed magnesium aluminium oxides (Fig. 28).
![[{"id":"420QgSmbUr","type":"paragraph","data":{"text":"Mechanism of tetrahydrothiophene conversion on a basic alumina additive.211 Published with permission from Elsevier"}}]](/storage/images/resized/pM8GdkWdhTU3JmZrCS9PyGmQxgnYvXiFKsylRseo_xl.webp)
Corma et al.212 considered approaches to decreasing the sulfur content in gasoline in the course of catalytic cracking and studied the reactions of mercaptans and alkylthiophenes. In the latter case, dealkylation, condensation (cyclization followed by aromatization) and dehydrogenation are considered as the major pathways. Lefleive et al.213 discussed two ways of appearance of sulfur compounds in the gasoline fraction during the catalytic cracking of sulfur-containing hydrocarbon feed:
- dealkylation of thiophene compounds with large alkyl substituents;
- reactions of hydrogen sulfide and olefins accompanied by cyclization and aromatization.
Dupain et al.214 also pointed to the pronounced influence of dealkylation of alkylthiophenes, alkylbenzothiophenes and alkyldibenzothiophenes on the residual sulfur content in the liquid cracking products and noted that the resulting thiophene, benzothiophene, dibenzothiophene and their methyl derivatives are very stable during catalytic cracking (i.e., they are not cracked to give hydrogen sulfide).
A number of studies148,215--220 present a detailed analysis of various approaches to the modification of components of zeolite-based cracking catalysts directed towards increasing the contribution of reactions that reduce the sulfur content in the liquid products directly in the reactor without using other processes (hydrotreating, adsorption, etc.). The modification methods indicated in the cited studies include the increase in the activity and selectivity of reactions that give hydrogen sulfide ( internal hydrogenolysis of the carbon--sulfur bond; method 1) and alkyl thiophene derivatives (which are then condensed to give polyaromatic compounds and coke; method 2). Method 1 is preferred, because it decreases the duty of the diesel hydrotreating unit and reduces the amount of sulfur oxides discharged with the catalyst regeneration gases.
As a rule, modification of a catalytic system consists in an increase in the concentration of Lewis acid sites. Most often, this is done using Zn2+ (Refs 221, 222) and Al3+ ions.223 Modifying agents of other types (e.g., V3+, Cu2+, Cr3+, which are introduced into the zeolite by impregnation or ion exchange) are used much more rarely.222,224--226 The function of these sites as Lewis acids is to enhance the adsorption of sulfur compounds, which are Lewis bases, and to increase the catalytic activity towards hydrogen transfer (see Fig. 26).
Like in the conversion of hydrocarbons, the hydrogen transfer from hydrocarbons to thiophene compounds are markedly affected by the conditions of the process. A temperature rise, a pressure decrease and addition of a diluent decrease the contribution of hydrogen transfer and, as a consequence, decrease the selectivity to hydrogen sulfide and increase the sulfur content in the liquid cracking products (Fig. 29).
![[{"id":"A6fvol76de","type":"paragraph","data":{"text":"Diagram reflecting the effect of conditions (temperature, steam dilution) of the cracking of a cyclohexane - 2-methylthiophene mixture on the content of sulfur in liquid cracking products (fixed catalyst bed, WHSV = 2.5 h<sup>-1</sup>, 5000 ppm of sulfur).227 Published with permission from Elsevier."}}]](/storage/images/resized/AO53CMpRe7kh2dvFIul2e9yNeHwP67LVnKuImfyg_xl.webp)
The chemical nature of the used hydrocarbon also considerably influences the activity of the catalytic composition towards the conversion of thiophene compounds to give H2S. In this case, the most important characteristic is the [H]-donor ability of the hydrocarbon.* This value is maximized for hydrocarbons with relatively high hydrogen contents in the molecule provided that the reaction gives an aromatic compound that splits off a hydride ion to give the most stable carbocation. The effect of the type of hydrogen donor on the activity of the cracking catalyst towards the conversion of 2-methylthiophene to give hydrogen sulfide is illustrated in Fig. 30; an increase in the sulfur content in the liquid products attests to lower catalyst activity in this reaction. Note that increase in the [H]-donor ability in the series
arene<paraffin<naphthene
(established for model compounds) increases the hydrogen sulfide formation rate by more than two orders of magnitude.
![[{"id":"Gik62CgHgB","type":"paragraph","data":{"text":"Diagram reflecting the effect of the type of hydrogen donor on the content of sulfur compounds in the liquid products of hydrocarbon cracking (monozeolite cracking catalyst based on Y zeolite, 450 <sup>o</sup>C, 10,000 ppm of sulfur from 2-methylthiophene).40 Designations: DEC is decalin, MCH is methylcyclohexane, MCP is methylcyclopentane, I8 is isooctane, CH is cyclohexane, N8 is n-octane, XYL is xylene. Published with permission from Elsevier"}}]](/storage/images/resized/RdHfpwkFV6hCluDDZczz9oCLqpCKMGa2Mq2MH3Sq_xl.webp)
* By [H]-donor ability is meant an integrated characteristic of hydrocarbon that reflects its ability to act as a hydride ion donor in the reactions taking place on acid-base catalysts.207
4.2. Nitrogen compounds
Nitrogen-containing compounds present in the catalytic cracking feed are mainly aromatic. These compounds can be classified into two main groups: nitrogen bases and neutral nitrogen compounds.228 In the nitrogen bases, the lone pair of electrons of nitrogen is not involved in the formation of the (4n+2)-electron aromatic system; therefore, this nitrogen atom has Lewis base properties, and molecules containing this atom have a pronounced poisoning effect on the catalyst acid sites. In neutral nitrogen compounds, the lone pair participates in the formation of the (4n+2)-electron aromatic system, while the remaining electron is involved in the formation of the N-H bond, acting as a weak acid; therefore, compounds containing such a pyrrole nitrogen atom have a minor poisoning effect on the catalyst acid sites.229
Most publications that describe cracking in the presence of nitrogen compounds note that increase in the nitrogen basicity leads to higher poisoning effect. For example, for cracking of isopropylbenzene in the presence of nitrogen bases, the poisoning effect of the nitrogen bases was found230 to decrease in the series
quinoline>pyrrole>aniline
in parallel with the decrease in the basicity constants of these compounds. This conclusion is valid if the cracking feed contains a large amount of aromatic compounds, which, as indicated above, have a low [H]-donor ability. Bobkova et al.231--233 investigated how the type of poisoning nitrogen compounds affects the conversion of hydrocarbons with different [H]-donor abilities and demonstrated that conversion of a hydrocarbon with a high [H]-donor ability is accompanied by a change in the poisoning effect of aromatic and non-aromatic nitrogen compounds. This change is a consequence of increasing contribution of intermolecular hydrogen transfer, resulting in fast saturation of the aromatic ring and formation of a nitrogen compound with pronounced basic properties (Fig. 31 and Fig. 32).
![[{"id":"_IslJqvOel","type":"paragraph","data":{"text":"Diagram reflecting the effect of [H]-donor ability of hydrocarbons (low for cumene and high for n-undecane) on the poisoning effect of aromatic nitrogen compounds of various classes, basic (quinoline, pKa≈ 5) and neutral (indole, pKa ≈ 17; ~4500 ppm of nitrogen), in the cracking of cumene and n-undecane on the zeolite-based cracking catalyst (conversion temperature of 450 <sup>o</sup>C). The Figure was created by the authors using published data.233"}}]](/storage/images/resized/WwQHwb0yYC3y2rVai5wNUcbxYtBuUCCuhVVfqAPa_xl.webp)
![[{"id":"mR_BzXKJqk","type":"paragraph","data":{"text":"Scheme of indole conversion in the presence of a hydrogen donor to give dihydroindole, a compound with a high poisoning effect with respect to acid sites (<i>a</i>), and possible pathways for further transformations of the product.233 Published with permis\u0002sion from Springer."}}]](/storage/images/resized/sF0Kfmuq9hdhsqhf3fjqMCoKDttM5v5U7ZcC8gq0_xl.webp)
Meanwhile, by analogy with sulfur compounds, high catalyst activity towards hydrogen transfer combined with high [H]-donor ability of the medium enable hydrogenation (hydrogenolysis) to ammonia and thus reduce the poisoning effect (see Fig. 32b). For example, for a mixture of decalin and nitrogen compounds (exemplified by quinoline) in the presence of Y zeolite-based cracking catalyst, more than 50% of nitrogen present in the feed is converted to ammonia.
Note that the transformations of nitrogen compounds in the presence of hydrocarbons catalyzed by zeolite-based materials have been much less studied than those of sulfur compounds. This may be caused by two factors: first, the content of nitrogen compounds in the hydrocarbon feed is approximately an order of magnitude lower than the content of sulfur compounds, and, second, the number of possible transformation pathways is much greater for nitrogen compounds, and the obtained products are more diverse, which complicates the identification of products and interpretation of the identified trends.
4.3. Oxygen compounds
As noted above, the key studies in the field of catalytic cracking during the last five years have been focused on the transformations of alternative (green) types of feedstock. The major part of plant-based feedstock consists of oxygen-containing compounds — fatty acid triglycerides, alcohols, phenols, etc.
The conversion of oxygen-containing compounds in the first stage of catalytic cracking is, most often, accompanied by deoxygenation (dehydroxylation, decarbonylation, decarboxylation)234 to give carbon monoxide and dioxide, water and the corresponding olefin. The subsequent reactions can include both the transformations of olefins, which have very high reactivity, and joint transformations of olefins and hydrocarbons present as the major components of the feed (e.g., like in the co-processing of oxygen-containing compounds with petroleum feedstock). In the latter case, the intermolecular hydrogen transfer reactions have a decisive importance for the formation of the final products.
In the presence of a reactive olefin, a hydrogen acceptor, the hydrogen transfer reactions are intensified and the degree of conversion of petroleum hydrocarbons (hydrogen donors) increases. Therefore, co-processing of plant- and petroleum-based feedstock may considerably increase the efficiency of cracking. It was shown235 that the addition of up to 5 mass% vegetable oils considerably (by 5-10 mass%) increases the feed conversion and the yield of the gasoline fraction.
Detailed studies of the transformations of aliphatic alcohols containing 2 to 5 carbon atoms in the molecule were mainly carried out with ZSM-5 zeolites. The studies differed in the methods and conditions of modification, resulting in the change in the zeolite acidity.236,237 The acidity and, hence, the catalytic behaviour of ZSM-5 in the transformations of alcohols are markedly affected by silica to alumina ratio of the zeolite. As the SiO2:Al2O3 ratio increases, the number of acid sites decreases; in view of small size of pores, the zeolite activity towards the hydrogen transfer is suppressed. As a result, β-scission of C–C bonds predominates, and the selectivity to light olefins increases. In order to attain a high yield of aromatic hydrocarbons, which result from hydrogen transfer, it is necessary to decrease the silica to alumina ratio of the zeolite and thus to increase the concentrations of the Brønsted and Lewis acid sites.238 Modification of ZSM-5 by treatment with phosphorus compounds or with desilylating agents (e.g., sodium hydroxide solution) can change its physicochemical properties (concentration and strength of acid sites, porosity). In particular, this modification significantly affects the contents of silicon and aluminium in the zeolite; hence, this treatment can be considered as being equivalent to a change in the silica to alumina ratio. Altynkovich et al.184 investigated the physicochemical and catalytic properties of ZSM-5 zeolites subjected to alkaline treatment and phosphorus modification in the cracking of 3-methylbutan-1-ol. Treatment with alkali makes it possible to change (due to the partial removal of the zeolite framework) the porosity, thus increasing the surface area and the total pore volume due to the formation of mesopores. In contrast to alkaline treatment, modification with phosphorus causes a decrease (due to the formation of aluminium and phosphorus compounds) in the total acidity of the zeolite. According to the data of temperature-programmed desorption of ammonia, the number of strong acid sites decreases to a greater extent than the number of weak acid sites. The catalyst activity in intermolecular hydrogen transfer reactions changes correspondingly: the alkaline treatment (followed by ion exchange of Na+ cations with NH4+ and annealing) promotes hydrogen transfer reactions and the formation of aromatic compounds due to the increase in the concentration of acid sites and increase in the pore size, while treatment with phosphorus compounds suppresses the hydrogen transfer and promotes the formation of light olefins, because of the decrease in the acidity and decrease in the pore size (Fig. 33).
![[{"id":"6OaXQH8Nny","type":"paragraph","data":{"text":"Effect of ZSM-5 modified with ammonium hydrogen phosphate (<i>a</i>) and sodium hydroxide (<i>b</i>) on the hydrogen transfer coefficient during cracking of 3-methylbutan-1-ol (450 <sup>o</sup>C; WHSV = 2.5 h<sup>-1</sup>).184 Published with permission from Springer"}}]](/storage/images/resized/7JLUP6p6fz0CJ5YZl12FTbcnJ9fGTsvXV3zJmtvj_xl.webp)
5. Conclusion
In the 1960s, the most significant leap in the research into hydrogen transfer reactions on heterogeneous catalysts took place. This was mainly stimulated by the beginning of the wide use of zeolite-based catalysts in the cracking of hydrocarbons. Due to the need for continuous improvement of this important process, which provides deeper oil refining, the studies of the mechanisms of hydrogen transfer reactions and development of rational approaches to the control over the cracking pathways are still highly relevant.
The last decade witnessed an increase in the number of publications considering the patterns of joint conversion of heteroatomic organic compounds and hydrocarbons under cracking conditions, emphasizing the role of hydrogen transfer for these processes. A reason for the publication activity of the last 10 years is the emergence of a new generation of equipment, which makes it possible to perform experimental studies and theoretical calculations at a qualitatively higher level. In addition to solving long-existing problems concerning the nature of the catalytic action of acid--base catalysts, mainly zeolites, in the cracking of hydrocarbons (e.g., Ref. 239), it is possible to switch to studying more complex systems containing transition metal compounds, so-called hybrid polyfunctional catalytic compositions (e.g., Ref. 240).
Analysis of the data on the design and study of properties of modern zeolite-based catalysts for the cracking of hydrocarbons carried out in this review, with the accent being placed on the role of intermolecular hydrogen transfer reactions, demonstrated the importance of considering these reactions in the overall cracking process and the need to search for the scientific and engineering tools for the control of these reactions both during the choice and design of the catalyst and during the proper conversion of the hydrocarbon feedstock. The main approaches to the control over the contribution of hydrogen transfer reactions are summarized below.
1. This contribution is affected, most appreciably, by a change in the component (aluminosilicate) composition of the cracking catalyst. The highest activity towards hydrogen transfer in the course of hydrocarbon conversion in inherent in Y zeolite, which is due to both high concentration (>1500 μmol.g–1) of strong (H0≤–9) acid sites and the absence of steric restrictions for occurrence of bimolecular reactions between the hydrogen donor and hydrogen acceptor hydrocarbons. Other components of the catalyst such as ZSM-5, amorphous aluminosilicate, alumina and especially clay are significantly less active in hydrogen transfer reactions due to either confined pore space accommodating the active sites (ZSM-5) or lower acidity and concentration of acid sites (amorphous aluminosilicate).
2. The introduction of heteroatoms (p-, d- and f-elements), which have Lewis acidity, into the zeolite component provides additional activation of hydrogen donor molecules, which is selective to certain C–H bonds accepting the hydride ion (hydrogen acceptor molecules are mainly activated by Brønsted sites). According to recent studies, important factors are not only the type, strength and concentration of these Lewis sites, but also their position relative to the Brønsted sites. The presence of closely spaced strong Lewis and Brønsted acid sites is optimal for the bimolecular hydrogen transfer. Generation of the Lewis acid sites in zeolites and zeolite-like materials may give rise to frustrated Lewis pairs and transition metal complexes in the oxygen ligand environment formed by the aluminium pair of the zeolite framework; analogues of these structures exist in homogeneous catalytic systems. The adsorption and catalytic properties of these heteroatomic structures in zeolites can be explained and, in some cases, even predicted in terms of Pearson's HSAB concept. The formation of sites active towards hydrogen transfer in zeolite materials is possible if the zeolite framework contains both cation vacancies and extra-framework species. Both types of defects, being located in the pores of the zeolite framework, can greatly affect the hydrocarbon transformation pathways. Note that cationic (introduction of metal cations or protons) and anionic (alkaline treatment, introduction of phosphate ions, etc.) modification as well as the heat treatment (accompanied by dealumination) of zeolites are efficient and practically useful tools for the control of the physicochemical and catalytic properties of Y and ZSM-5 zeolites. In addition, increasing attention of researchers who intend to attain the control over the catalytic cracking is attracted by polyfunctional systems that are formed by creating platinum metal nanoparticles or nano-islands of Group 4--7 transition metal oxides on the surface or inside the pore space of the zeolite (e.g., see reviews241--244).
3. Along with the composition of the catalytic system, the contribution of hydrogen transfer reactions is affected by the conditions of hydrocarbon transformations. An increase in the process temperature, a pressure decrease, addition of a diluent (reducing the partial pressure of hydrocarbons) and a decrease in the catalyst:feed ratio decrease the contribution of these reactions.
4. In the catalytic cracking of hydrocarbons containing heteroatomic organic compounds using the same approaches to the control of the contribution of hydrogen transfer reactions as for the cracking of pure hydrocarbons, it should be borne in mind that intensification of hydrogen transfer may be undesirable in some cases. For example, in the conversion of sulfur compounds, a high contribution of hydrogen transfer increases the selectivity to hydrogen sulfide (internal C–S bond hydrogenolysis takes place) and, hence, decreases the amount of sulfur compounds in the liquid products (gasoline and diesel fractions formed upon the catalytic cracking of oil distillates on zeolite-based catalysts).
In the conversion of nitrogen compounds, the intensification of hydrogen transfer leads to an increase in the contribution of hydrogenation of aromatic molecules, resulting in increasing basicity of these compounds. This would enhance deactivation of both Brønsted and Lewis acid sites of the catalysts and would adversely affect their ability to conduct the subsequent hydrogenolysis of the C–N bond to give ammonia. Oxygen-containing compounds, which are thermally less stable than sulfur and nitrogen compounds, undergo most often thermocatalytic destruction (deoxygenation) and thus serve as sources of unsaturated compounds, which act as highly reactive hydrogen acceptors and may enhance the formation of coke.
In general, addressing the issues related to processing of crude hydrocarbon feedstock and combined mixed feedstock via catalytic cracking is currently acquiring particular interest, as evidenced by enhanced publication activity in both theoretical and applied aspects of this subject matter.242,245--249
This review was prepared with the financial support of the Ministry of Science and Higher Education of the Russian Federation within the framework of the State Assignment of the Federal Research Center, G.K.Boreskov Institute of Catalysis, Siberian Branch of the Russian Academy of Sciences (Project AAAA-A21-121011890074-4).
List of abbreviations and symbols
A — arenes,
CH — cyclohexane,
DEC — decalin,
DFT — density functional theory,
DTC — deuterium transfer coefficient,
EFAL — extra-framework aluminium,
FLP — frustrated Lewis pair,
HCR — hydrocracking residue,
HSAB — hard and soft acids and bases (theory),
HTC ---hydrogen transfer coefficient,
HVGO — hydrotreated vacuum gas oil,
I8 — isooctane,
MCH — methylcyclohexane,
MCP — methylcyclopentane,
N — naphthenes,
NHVGO — non-hydrotreated vacuum gas oil,
N8 — n-octane,
O — olefins,
P — paraffins,
RE — rare earth
SLA — soft Lewis acid,
SLB — soft Lewis base,
TCC --- thermafor catalytic cracking,
USY — ultrastable Y zeolite,
WHSV — weight hourly space velocity,
XYL — xylene.