


Keywords
Abstract
The chemistry of organic di- and polyazides has been extensively developed in recent years. Methods for the synthesis of 1,1- and 1,2-diazides are diverse and include stoichiometric, catalytic, photochemical and electrochemical approaches. These organic compounds are highly reactive and are involved in a wide range of reactions, from thermal transformations to click chemistry. Of particular attention are transformations, in which one azido group undergoes intramolecular cyclization, while another azido group remains intact and can participate in further chemical transformations. Tri- and polyazides are investigated as precursors for the production of high-energy materials and the synthesis of high-molecular-weight compounds. This review addresses the methods for the synthesis of organic compounds containing two or more azido groups and their chemical transformations. Aliphatic, aromatic and heteroaromatic diazides are considered. Specific transformations characteristic of geminal and vicinal diazides, which differ from the reactions of monoazides, are described. Examples of the practical application of di-, tri- and polyazides are highlighted.
The bibliography includes 355 references
1. Introduction
Due to unique properties, azides are widely used in various fields of human activity. For example, the well-known inorganic azide NaN3 is a gas-generating agent for air bags, since this compound decomposes under thermal or mechanical stress to release nitrogen gas.
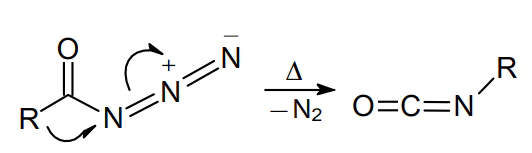
Organic azides have been known since the second half of the 19th century. Thus, in 1864 the German organic chemist Peter Griess1,2 synthesized phenyl azide. More than 25 years later, Theodor Curtius3,4 obtained the extremely unstable hydrogen azide and found that acyl azides are capable of rearranging to the corresponding isocyanates; this transformation became known as the Curtius rearrangement (Scheme 1).
However, after that organic azides were almost not studied until the middle of the last century, when a representative series of alkyl, aryl and acyl azides was obtained.5,6
At present, organic azides are widely used not only in the laboratory practice but also in the modern chemical industry. For example, polysulfonyl azides are employed as blowing agents to create foams and porous rubbers.7 The functional azide moiety, despite its potential danger, is present in the molecule of azidothymidine (zidovudine), the first antiretroviral drug used to treat human immunodeficiency syndrome.8 Azides are important building blocks in the construction of nitrogen-containing heterocycles, in particular triazoles and tetrazoles, and also serve as substrates in the Staudinger reaction9,10 and the Meldal--Sharpless click reaction of copper-catalyzed azide--alkyne cycloaddition (CuAAC).11,12 These reactions are widely used in materials science13--15 and biochemistry.16,17 It should be noted that during the preparation of this review, Morten Meldal and Barry Sharpless were awarded a 2022 Nobel Prize in Chemistry for the development of click chemistry and bioorthogonal chemistry.
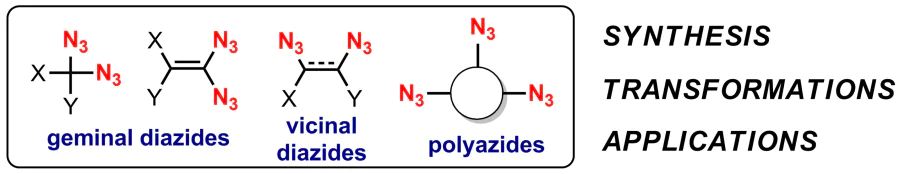
The presence of two and more azide groups in one organic molecule leads to the expansion of the pool of available chemical transformations and the diversification of possible applications of di- and polyazides while ensuring that increased instability of such compounds is taken into account. Note that in addition to Banert's monograph,18 several reviews and monographs have appeared in recent years devoted to azide-containing organic compounds.19--23 It is obvious that this trend got a second wind mainly due to the CuAAC reaction discovered at the beginning of the 3rd millennium.11,12,24 The aim of the present review is to comprehensively summarize the current state of chemistry of organic di- and polyazides with various mutual arrangement of azide groups. Diazides can be conditionally divided into the following three classes: geminal, vicinal and other azides, in which azide moieties are tethered to each other by at least two carbon atoms.
In this review, much attention is paid to the systematization of key synthetic approaches to geminal and vicinal diazides using a variety of substrates and azide sources. Besides, it summarizes chemical transformations typical of 1,1- and 1,2-diazide organic derivatives, in particular, thermal and photochemical reactions. Also, not only emphasis is given to modern publications but also the relationship of studies of the chemistry of such compounds in the historical development is shown. Therefore, this publication can be considered as a kind of guide to the methods of synthesis and chemical properties of organic diazides.
The attention is also paid to the synthesis and chemical transformations of organic compounds bearing three or more azide groups known to date. Such derivatives have been closely studied in recent years as target explosives and coupling agents in macromolecular chemistry.
2. Synthesis and transformations of geminal diazides
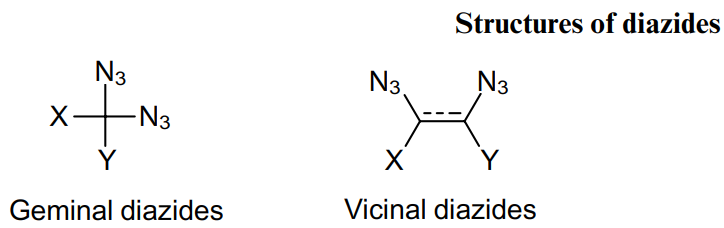
In geminal diazides, both azide moieties are attached to the same carbon atom, which can be bonded either to another carbon atom or to a heteroatom. The synthesis method and chemical properties of these compounds depend on which atoms are adjacent to the diazide centre. Structures 1-5 are the most common for geminal, or 1,1-substituted, diazides.
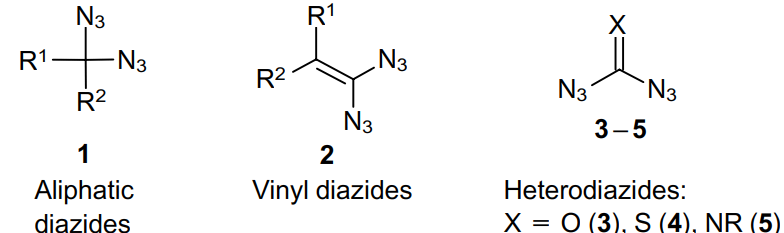
The chemistry of geminal diazides is discussed in depth in a review by Hйring and Kirsch.20 The authors focus the readers attention on the extremely explosive nature of such compounds.
2.1. Preparation of aliphatic 1,1-diazides
This Section systematically explores the literature data on the preparation of geminal aliphatic diazides, considers the nucleophilic substitution reactions of a wide range of substrates involving various azide sources and gives examples of diazidation of alcohols, ketones, alkenes, alkynes, etc., in the presence of various oxidants.
2.1.1. Substitution reactions
Sodium azide is the cheapest and most available source of the azide anion. This is why it is widely used in nucleophilic substitution reactions affording geminal diazides. The synthesis of geminal diazides involving sodium azide was first reported over 100 years ago. Thus, in 1908 Forster et al.25 for the first time isolated ethyl 2,2-diazidoacetate (7) in a low yield via the double substitution of ethyl dichloroacetate (6) in the presence of excess sodium azide (Scheme 2).
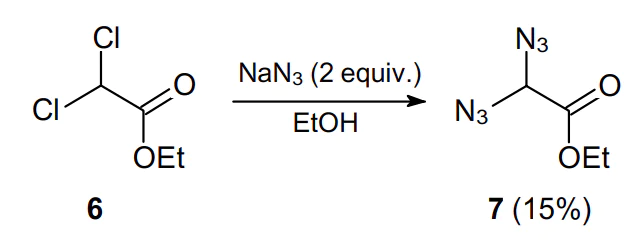
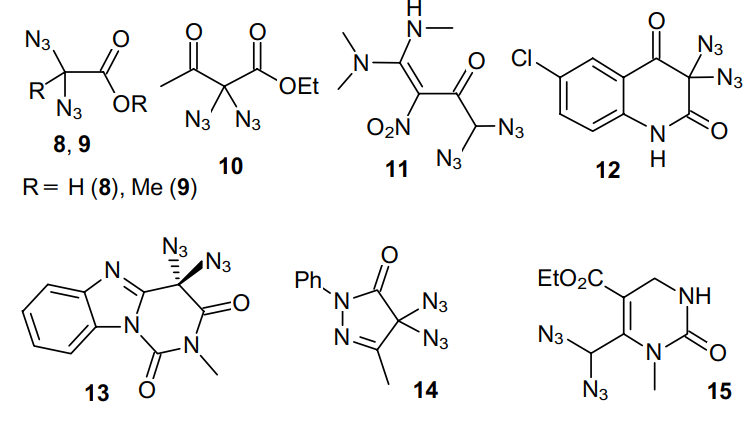
Since then, syntheses of geminal diazides from dihalides using NaN3 as an azide source have have gained popularity, and a large number of such compounds were obtained to date, in particular, a,a-diazido esters,2--27 a,a-diazidoketones,28 2,2-diazidomalonates,29 heterocyclic diazides,30--33 etc. (e.g., Structures 8-15).34,35
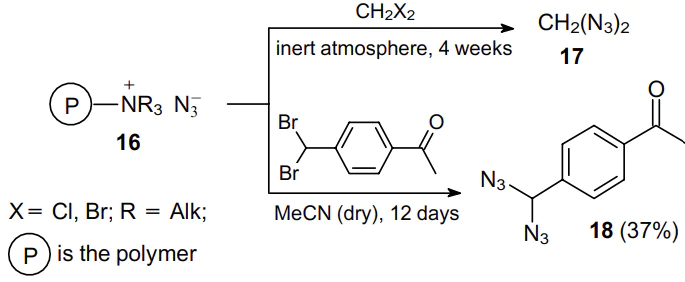
Recently, in addition to sodium azide, other reagents were also used for this purpose. An example is azidation polymer 16, which is an Amberlite IR-400 ion exchange resin with azide ions as counterions (Scheme 3).36,37 Such a polymer provides access to low-molecular-weight azides or polyazides, e.g., methyl azide, diazidomethane (17), triazidomethane (azidoform) and diazide 18. Removing the polymeric azidation reagent by filtration gives a solution of a nearly pure reaction product.
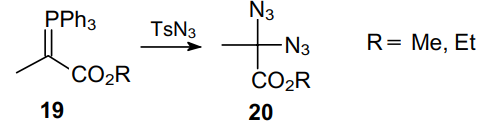
Geminal diazides can also be prepared using an electrophilic source of azide ions, namely, tosyl azide TsN3 (Ts is p-toluenesulfonyl). Thus, in 1972 Sohn et al.,26 expecting to obtain diazopropionate on treating phosphorus ylide 19 with diazopropionate, observed the formation of the corresponding diazide 20 (Scheme 4).
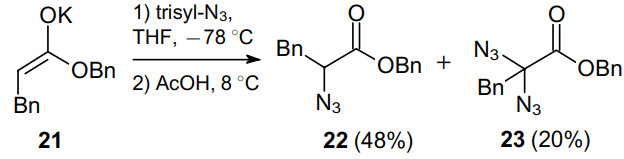
Besides, an attempt was made to perform the direct azidation using hydrocinnamic acid.38 The authors applied the reaction of potassium enolate 21, generated using potassium hexamethyldisilazide (KHMDS), with 2,4,6-triisopropylbenzenesulfonyl azide (trisyl-N3) followed by the reaction with acetic acid. Under these conditions, a mixture of monoazide 22 and diazide 23 as a minor product was obtained (Scheme 5). The reverse addition of enolate 21 to an electrophile improved the yield of azide 22 up to 72%.
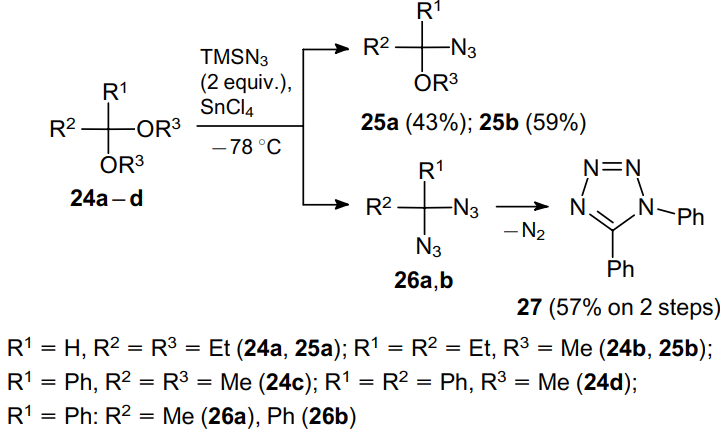
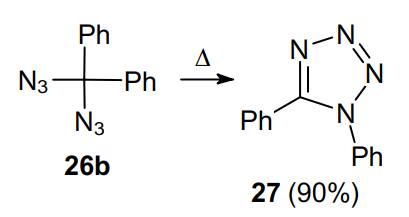
Geminal aliphatic diazides are produced by the reactions of acetals and ketals 24a--d with excess TMSN3 (TMS is trimethylsilyl) in the presence of strong Lewis acids, e.g., SnCl4 (Scheme 6).39 In this case, to form diazide, substrate 24 should bear at least one phenyl substituent; otherwise the reaction affords alkoxy azides 25. Geminal diazide 26a is produced in 49% yield, whereas its analogue 26b loses a nitrogen molecule to form tetrazole 27.
Ketones40,41 and aldehydes42,43 can also be used as starting materials to prepare geminal diazides; in this case, catalyst SnCl2 is preferable. This protocol is suitable not as much for aliphatic and aromatic aldehydes, as for their heterocyclic analogues.44 Also, it was shown that the addition of a combination of NaN3 with a crown ether significantly accelerates this process.42 It is also known that aldehydes can be directly converted into 1,1-diazides, being treated with NaN3 in the presence of AlCl3 or TiCl4.42,45
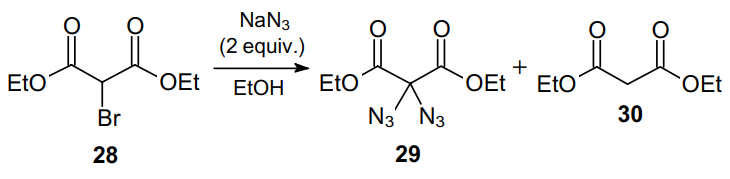
Another approach to 1,1-diazides involves the disproportionation of alkyl 2-bromomalonates 28, giving the corresponding diazidomalonate 29 (Scheme 7).46 However, this method is not very effective, since during the process, the starting compound is partially converted into a by-product, alkyl malonate 30. Nevertheless, this reaction provides pure 2,2-diazidomalonate 29, with no traces of monoazide detected.47,48
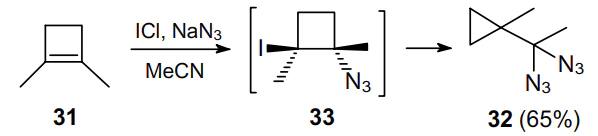
When studying the reactivity of 1,2-dimethylcyclobutene (31) by treating it with a mixture of excess iodide(I) chloride and sodium azide, O'Hare and Swern49 unexpectedly obtained geminal diazide 32 (Scheme 8). In this case, intermediate 33 undergoes four-membered ring contraction, and both azide moieties occur at the exocyclic carbon atom.
Bromine azide, prepared by the reaction of N-bromosuccinimide (NBS) with sodium azide, was used to convert nitro olefins 34 to geminal diazides 35 via the addition--elimination--addition sequence (Scheme 9).50

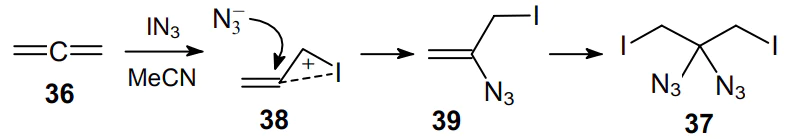
Allene (propa-1,2-diene) 36 reacts with iodine azide to give geminal diazide 37 through the intermediate formation of cyclic iodonium ion 38 and monoazide 39 (Scheme 10).51
2.1.2. Synthesis of aliphatic 1,1-diazides under oxidizing conditions
Modern techniques enable the direct diazidation of a number of organic compounds, namely, benzyl alcohols,52 ketones,52,53 diketones, alkenes,53 alkynes,54 and also malonates and b-ketoesters,55 under oxidizing conditions. In such cases, the reactions afford a-diazidocarbonyl compounds. Hydrogen peroxide,56 NaIO4, potassium 3-carboxy-4-iodylbenzenesulfonate (IBX-SO3K) and N-iodosuccinimide (NIS) are used as oxidants.
For example, the reaction of both linear and cyclic aryl ketones containing the a-methylene moiety with NaN3 in the presence of NaIO4 in a 1:4 AcOH--DMSO mixture at 75 °C furnishes a,a-diazidoketones 40 in 93--96% yields (Scheme 11).52 However, under these conditions, isobutyrophenone gives solely the a-monoazidation product in 35% yield.
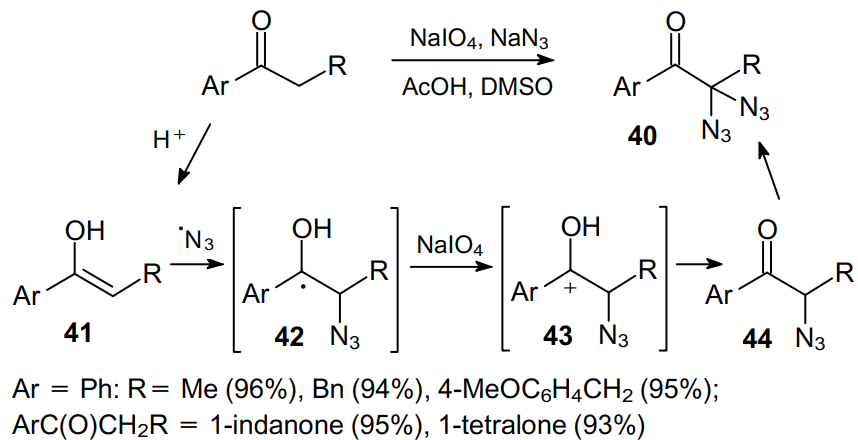
It is believed that the mechanism of this transformation comprises one-electron oxidation of an azide ion, which then adds to enol 41. The further oxidation of radical 42 generates cation 43, the abstraction of a proton from which leads to a-azidoketone 44, which is again involved in the oxidation cycle to provide a,a-diazidoketone 40. To confirm the formation of intermediate monoazidoketone 44, tetralone was diazidated52 using equimolar amounts of NaN3 and NaIO4 to give the corresponding a-diazidoketone in 28% yield. When individual 2-azido-1-phenylpropan-1-one was diazidated with a mixture of NaIO4 (1 equiv.) and NaN3 (2 equiv.), 2,2-diazido-1-phenylpropan-1-one proved to be the only product (92% yield). This fact indicates that the rate of diazidation of monoazidoketone is much higher than that of the starting aryl ketone.
Another approach to 2,2-diazide derivatives using the easy-to-handle and environmentally friendly oxidant IBX-SO3K (45) was developed by Kirsch's reseach group.55,57 This approach is characterized in that it makes available a number of tertiary 2,2-diazido-1,3-dicarbonyl products, which are pretty difficult to prepare using conventional procedures. This method provides an access to diazides starting from 1,3-dicarbonyl compounds, in particular, malonates and b-ketoesters (Scheme 12).
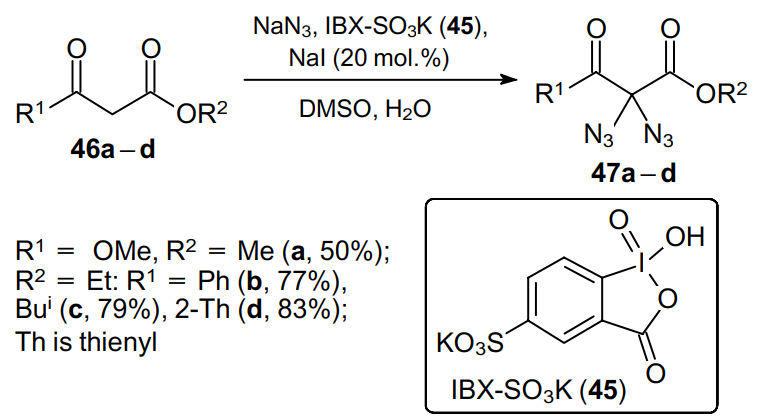
Being treated with a mixture of IBX-SO3K (3 equiv.), NaI (0.2 equiv.) and NaN3 in the DMSO--H2O system, at room temperature for 10 min, 1,3-dicarbonyl compounds 46a--d, devoid of an additional substituent in the 2 position, afford rather stable 2,2-diazido-1,3-dicarbonyl compounds 47a--d in good yields. An advantage of this protocol is the rapid incorporation of an azide-containing moiety, which can further be additionally functionalized via conventional reactions of 1,3-dipolar cycloaddition with terminal alkynes.
Later, this approach was improved and expanded, thereby providing a direct access to geminal diazides bearing one carbonyl moiety from a-azidoketones, a-iodoketones and olefins with an internal double bond (Scheme 13).53
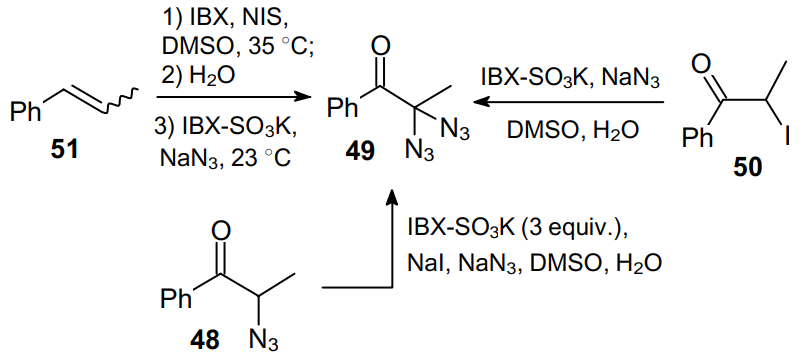
In all cases, NaN3 was employed as a cheap and easily available azide source, while a combination of IBX-SO3K with iodides proved to be a perfectly balanced oxidizing agent. Thus, in the presence of substoichiometric amounts of NaI, 2-azido-1-phenylpropanone 48 was converted into 2,2-diazido-1-phenylpropanone 49 in 82% yield.
2-Iodo-1-phenylpropanone 50 can be diazidated using IBX-SO3K and NaN3 in aqueous DMSO at room temperature in good yield (79%). The synthesis of diazide 49 from an internal olefin, propenylbenzene 51, comprises primarily the in situ transformation of the starting compound into iodomethyl ketone using 2-iodoxybenzoic acid (IBX) and NIS (see Scheme 13).
Diazides 47 can also be prepared from dicarbonyl compounds 46 using an alternative azidation system, I2--NaN3, in aqueous DMSO.58 This method performed well in diazidating monocarbonyl derivatives 52a--d. In this way, diazides 53a--d were produced in high yields (Scheme 14).59

In 2015, Okamoto et al.54 developed a rather productive approach to 1,1-diazides 54 based on the reaction of disubstituted arylacetylenes 55 with TMSN3 as an azide source (Scheme 15). The presence of a minor amount of atmospheric moisture and oxygen contributes to the successful outcome of the reaction, but the authors failed to determine accurately the role of water and oxygen.
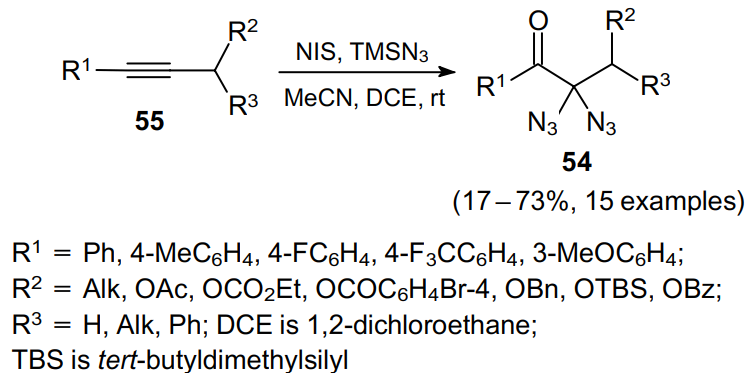
On the other hand, an excess of water in the reaction mixture was found to significantly reduce the product yield due to the rapid hydrolysis of TMSN3. The putative mechanism of the process as exemplified by the transformation of phenylacetylenes 56 is illustrated in Scheme 16.
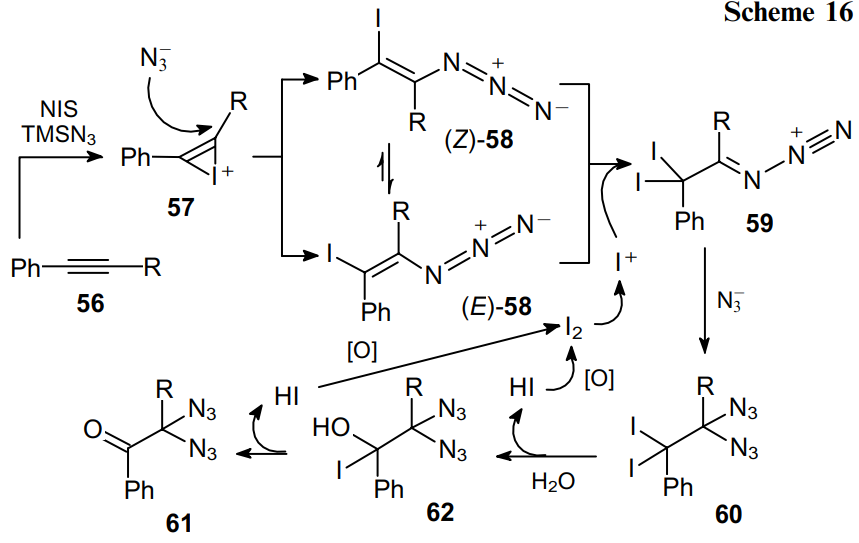
In the first step, alkyne 56 reacts with NIS and TMSN3 to form regioselectively, via the ring-opening of iodonium intermediate 57, a mixture of E and Z isomers of alkene 58. It was found60--62 that the regioselectivity of the addition of IN3 to arylalkyne is opposite to that predicted from the electronic effect and that the addition of IN3 results in the formation of two stereoisomers. Using 1H NMR spectroscopy, it was shown that the reaction between NIS and TMSN3 affords iodine azide IN3. The iodination of alkene 58 generates intermediate 59, and its subsequent reaction with the azide anion gives diazide 60, which is then hydrolyzed by atmospheric moisture to afford diazidoketone 61 via the formation of intermediate 62. In this case, oxygen oxidizes HI to I2, which binds to the remaining compound 58 in the form of I+.
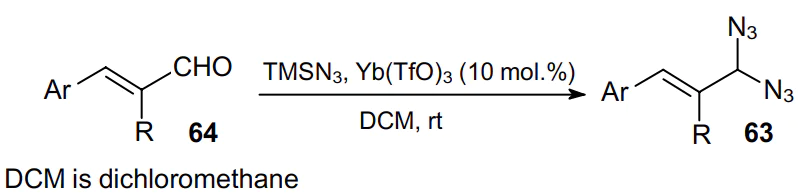
Particularly noteworthy is the recently developed synthetic approach to a,b-unsaturated geminal diazides 63 derived from unsaturated aldehydes 64 using TMSN3 as the azide source and Yb(TfO)3 as the catalyst (Scheme 17).63 This reaction proceeds in a one-step fashion under mild conditions to provide the product in moderate yield.
2.2. Reactions of geminal aliphatic diazides
This Section presents the fundamental reactions of geminal diazides, including thermolysis and photolysis, and also several examples of intermolecular reactions, most of which afford nitrogen-containing heterocycles of unique classes.
2.2.1. Thermal and photochemical reactions
In the early 20th century, it was found that the thermal decomposition of diazidodiphenylmethane 26b gives tetrazole 27 (Scheme 18).64,65
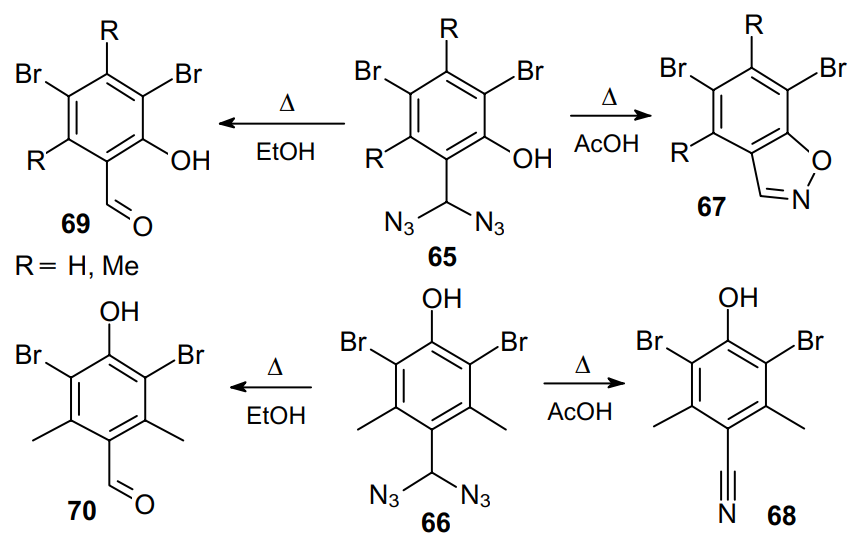
However, when studying the thermal reactions of ortho- (65) and para-hydroxy-disubstituted (66) benzyl diazides, Lindemann and Mмhlhaus66 revealed the formation of benzisoxazoles 67 and benzonitrile 68, respectively (Scheme 19). Besides, when carrying out the reaction in ethanol, benzaldehydes 69 and 70 were obtained. The formation of these compounds is probably attributed to the hydrolysis of diazides due to the presence of water in the solvent.
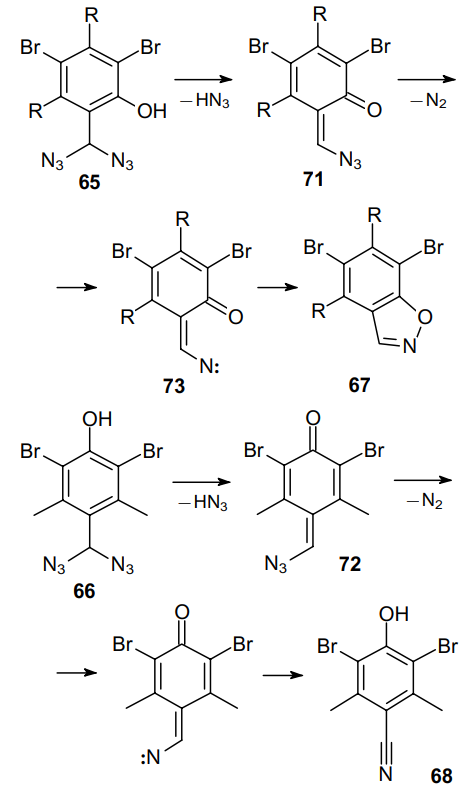
The mechanisms of formation of benzisoxazoles 67 and benzonitrile 68 are presented in Scheme 20. One might note the similarity of these mechanisms: in both cases, HN3 is first eliminated to afford quinoid derivative 71 or 72 followed by the loss of a nitrogen molecule. The difference occurs only in the last step. In the presence of the ortho-hydroxy group, intermediates 73 are stabilized via the formation of a new isoxazole ring. When the hydroxy group is in the para position, only the diazidomethyl moiety undergoes transformations, without affecting the hydroxy functionality. A number of more recent publications42,45,67--69 also reported the synthesis of benzonitriles using conventional reagents, such as NaN3 or TNSN3. In these reactions, benzyl diazides are often formed in situ as intermediates.
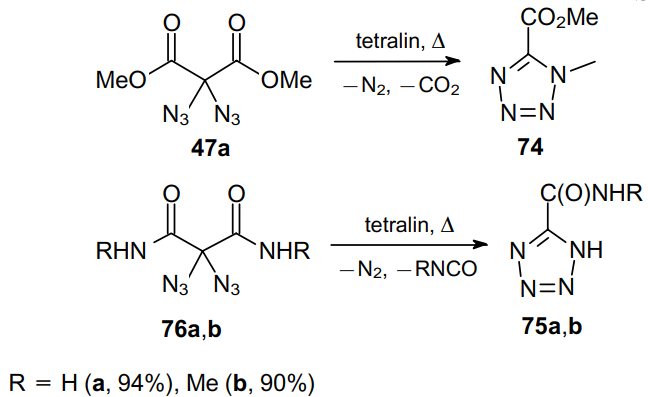
Thermal decomposition of geminal diazides also affords interesting organic compounds. Thus, tetrazoles 74 and 75 can be obtained by heating dimethyl 2,2-diazidomalonate 47a and diamides 76a,b, respectively, in nonpolar solvents, e.g., tetralin (Scheme 21).48
Thermal decomposition of glucopyranoside gem-diazide 77 produces tetrazole 78 in high yield. Meanwhile, the photochemical decomposition of diazide 77 gives a mixture of tetrazoles 78 and 79 (Scheme 22).70 Probably, tetrazole 79 is formed through the generation of intermediate azidonitrene followed by its insertion across the carbon--oxygen bond.
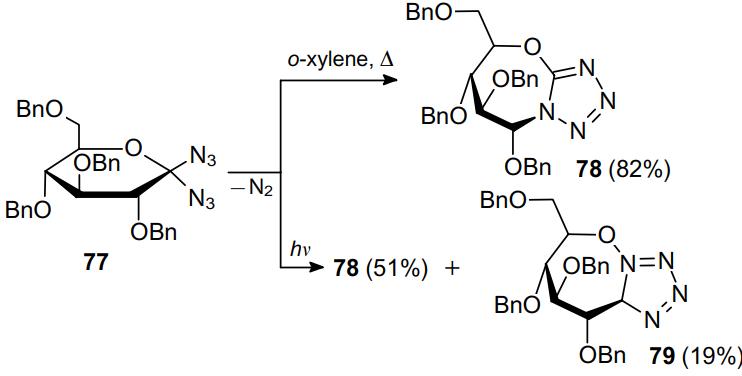
When studying the thermal decomposition of 3,3-diazido-2,4-dioxoquinolines 12 and 80, Landen and Moore30 suggested the formation of N-cyano derivatives 81. However, a careful study of the by-product structures indicated the formation of isomeric 3-diazoquinoline-2,4-diones 82, which have already been known for a long time and used to prepare azo dyes71 and their complexes with copper72 (Scheme 23). In addition to diazo compounds 82, the authors also observed the formation of tetrazoles 83. It was suggested that products 82 and 83 are formed via the common intermediate, spirocyclic tetrazole 84. Besides, in refluxing pyridine in the presence of copper, 3,3-diazido-2,4-dioxoquinoline 80 gave coordination compound 85 in high yield.
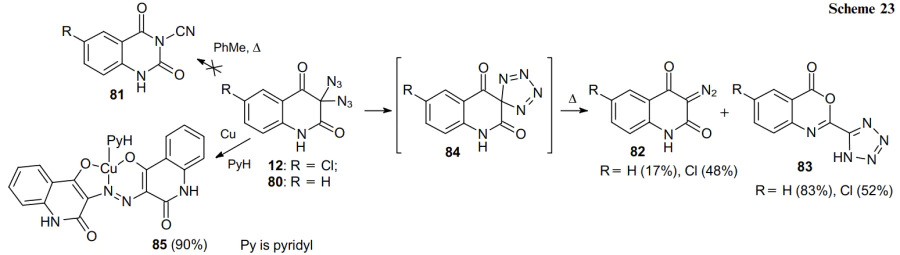
Microwave irradiation (MW) enables the cyclization of geminal diazides 86 to tetrazole derivatives 87 in high yields (Scheme 24). The same method was used to produce tetrazoles from the open-chain diazidoketones. However, under these conditions, no isomeric tetrazoles 88 are formed.73
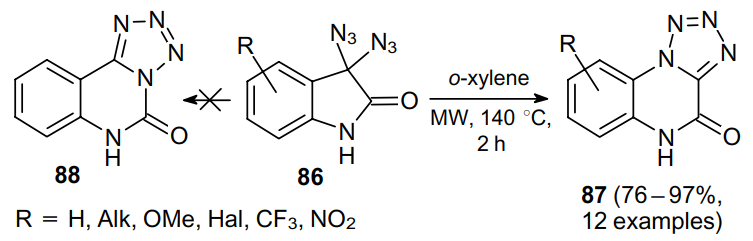
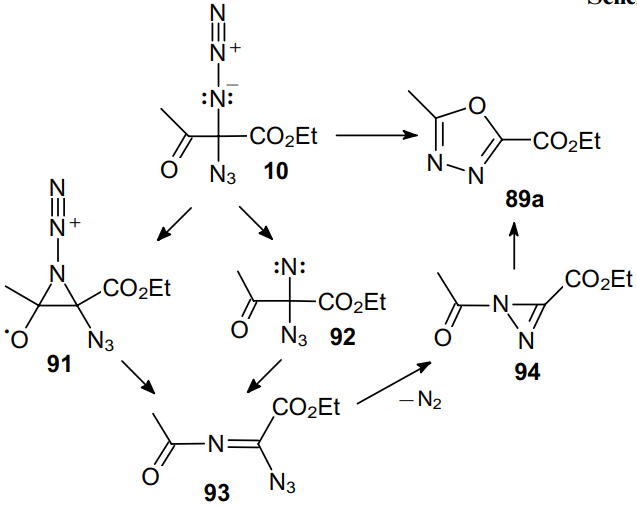
Ogilvie and Rank74 pioneered in the synthesis of 1,3,4-oxadiazoles 89a,b via heating a,a-diazido-b-ketoesters 10 and 47b (Scheme 25). The structure of product 89a was additionally confirmed by the independent synthesis from 5-methyltetrazole (90).
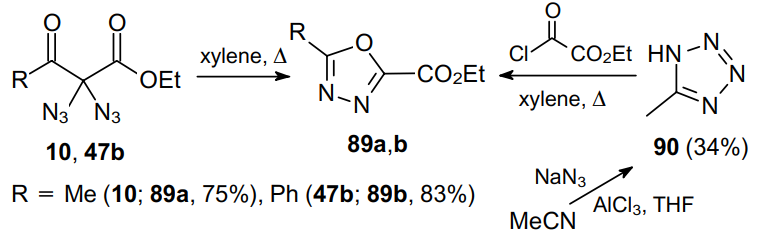
The authors believe that diazido compound 10 converts to aziridine 91 or loses a nitrogen molecule to afford nitrene 92. The ring opening or subsequent migration of the acyl group to the nitrogen atom leads to azido azomethine intermediate 93. The loss of a second nitrogen molecule directly from azido azomethine 93 gives diazirine 94, which is finally rearranged into 2,5-disubstituted 1,3,4-oxadiazole 89a (Scheme 26).
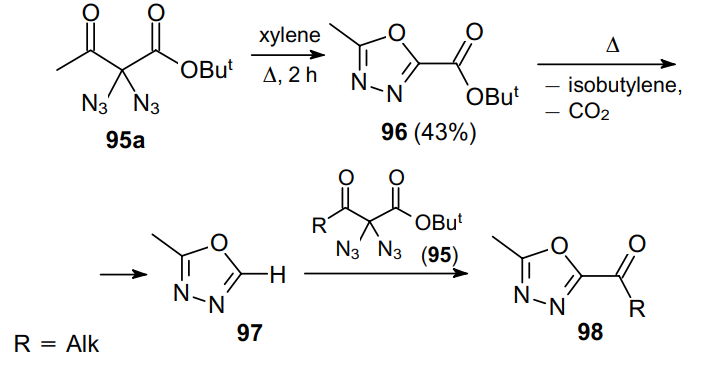
Almost 30 years later, Erhardt, Mohr and Kirsch75 continued to study this reaction and significantly expanded the scope of the resulting 1,3,4-oxadiazoles. It was also demonstrated that in the case of tert-butyl-substituted esters (e.g., compound 95a), further heating of product 96 led to the removal of the ester functionality (intermediate 97) accompanied by the possible transfer of a new acyl moiety from ester 95a giving compounds 98 (Scheme 27).
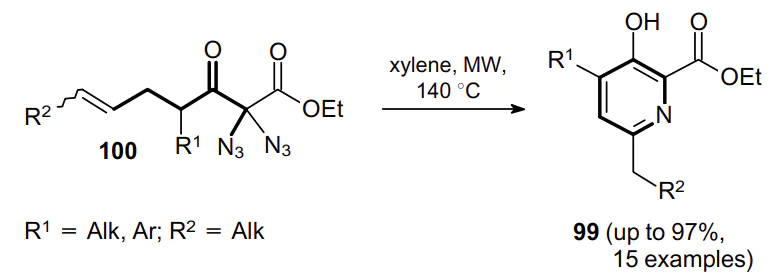
A similar approach can be applied to the synthesis of pyridine derivatives 99 from 1,1-diazides 100 under microwave irradiation (Scheme 28).76
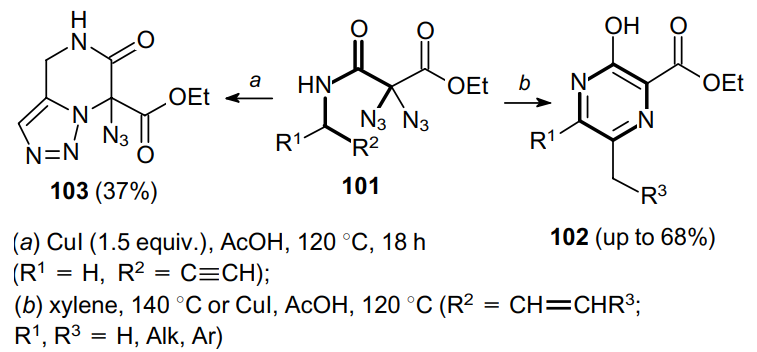
Heating of diazides 101 induces the cyclization to pyrazines 102 or the intramolecular cyclization onto one azido moiety leading to azidotriazole 103 (Scheme 29).77
Geminal diazides can be used as nitrene precursors. For example, C60 fullerene (104) was refluxed in xylene with ethyl 2,2-diazidomalonate (29) to produce functionalized bridged fulleroid 105 (Scheme 30).78
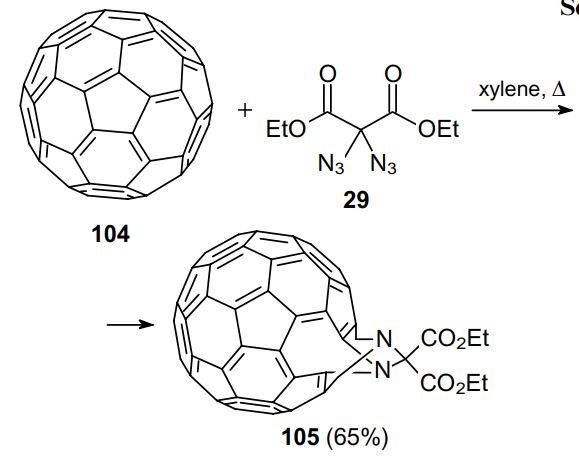
Over half a century ago, Moriarty et al.47 studied the photolysis of three geminal diazides, namely, methyl 2,2-diazidomalonate (47a), 2,2-diazidomalonimide (76a) and diazidodiphenylmethane (26b). As is seen in Scheme 31, the photolysis of diazides 47a, 76a produces mixtures of different products 106--109, suggesting the existence of several reaction pathways.
The authors presented some mechanistic aspects79 including the formation of nitrene intermediates, which was confirmed by EPR spectroscopy. However, the final mechanism that explains all observations on photolysis of diazides, is still unclear. From a synthetic point of view, tetrazole 107 is the major product that is formed upon irradiation of compound 47a. Its further irradiation results in a loss of a nitrogen molecule to give methyl 5-methoxy-1,2,4-oxadiazole-3-carboxylate 108, while irradiation of 2,2-diazidomalonamide 76a furnishes tetrazole 109.
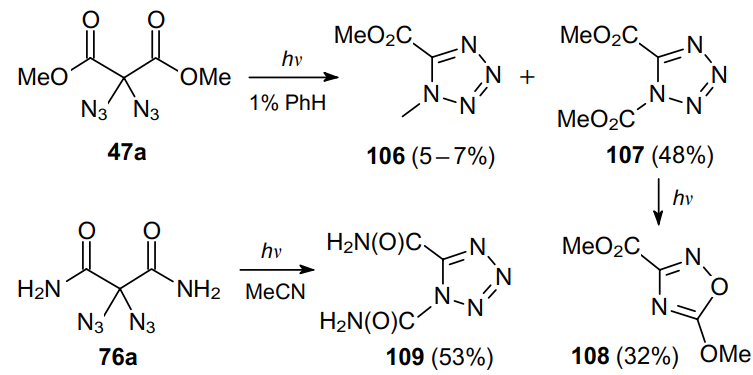
Meanwhile, diazidodiphenylmethane (26b) shows a completely different distribution of products 27, 110 and 111 (Scheme 32), which implies the reaction to proceed via intermediates 112--114.
More recent studies of the photochemistry of geminal benzyl azides using density functional theory (DFT) calculations and isotope labeling experiments also supported the hypothesis that there is more than one reaction pathway.37
Harter et al.80 synthesized salts of 2,2-diazidomalonate containing sodium, potassium, ammonium and guanidinium counterions. These compounds were tested for sensitivity towards impact, friction and electrostatic discharge in comparison with trinitrotoluene. It turned out that some of them are superior in terms of velocity of detonation to trinitrotoluene. Also, the toxicity of these compounds to aquatic organisms was studied using the bioluminescent marine bacterium Vibrio fischeri.80
2.2.2. Intermolecular reactions
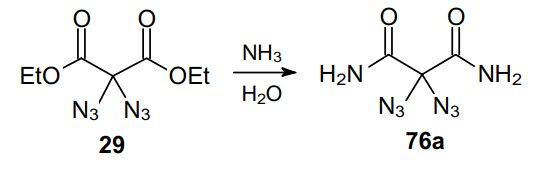
The conversion of geminal diazide 29 to 2,2-diazidomalonamide (76a) in the presence of ammonia has been known for more than 100 years (Scheme 33).29 Azide moieties are not directly involved in this reaction but appear to exert a strong activating effect since the formation of the corresponding amide from unsubstituted diethyl malonate (30) is much slower.
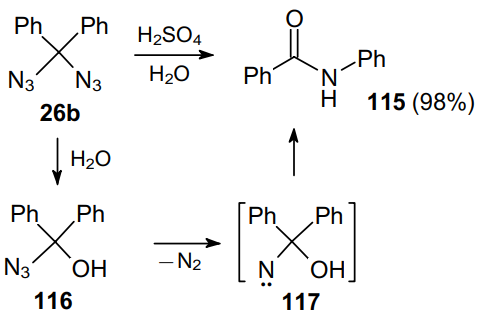
In 1931, the transformation of diazidodiphenylmethane 26b into N-phenylbenzamide 115 under the action of strong acids was reported.65 The hydrolysis of diazide 26b gives intermediate 116, which loses a nitrogen molecule to afford nitrene 117. The subsequent rearrangement of this intermediate yields N-phenylbenzamide 115 (Scheme 34).
It should be noted that because of the issues with the commercial availability and storage of gem-diazides, studies of the reactivity of such compounds were scarce for a long time in contrast to the studies of more stable monoazides. Nevertheless, significant progress has been made in this area over the past 5 years. The reaction between aromatic aldehydes with TMSN3 was used to produce gem-diazides 118, and their reactions with heterocyclic carbene 119 and triphenylphosphine giving products 120 and 121, respectively, were described (Scheme 35).81

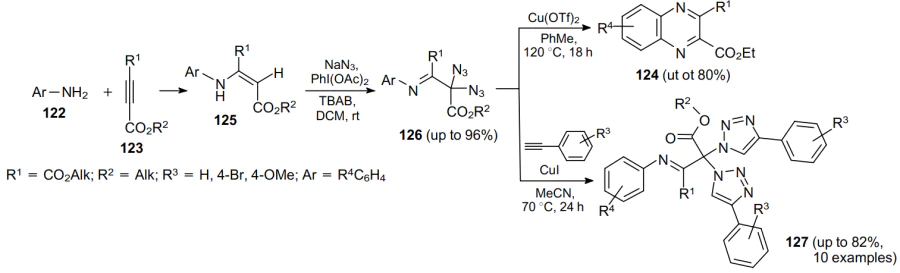
Recently, oxidative geminal diazidofunctionalization of alkynes was accomplished, in which tetra-n-butylammonium bromide (TBAB) was a precursor in the in situ preparation of tetra-n-butylammonium azide.82 Besides, based on the reaction of anilines 122 with acetylenedicarboxylates 123, the cycloamination strategy was first demonstrated yielding quinoxalines 124 (Scheme 36). The reaction sequence includes the formation of compounds 125 and 126. From the latter compound, product 127 was also obtained.
Considerable advances in intermolecular reactions of geminal diazides were achieved by Kirsch and co-workers83 (Scheme 37). Azido moieties in compound 29 can be hydrogenated over Pd/C to produce amines 128, in which an amino functionality is located between two carbonyls.84 This reaction can be applied to a variety of geminal dicarbonyl diazides and is an alternative to the conventional synthesis of such amines via the reduction of the appropriate oximes.85
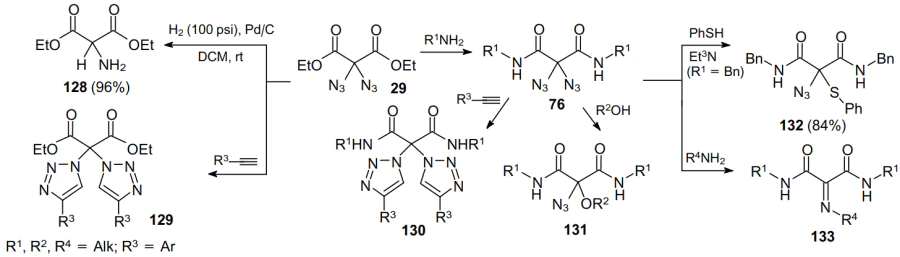
Diazide 29 undergoes the [3+2]-cycloaddition with terminal acetylenes to give bis(triazoles) 129.53,86 The reaction of this diazides with primary amines enables the preparation of diamide derivatives of geminal 2,2-diazides 76,58,59 which in turn can react with alkynes to afford bis(triazoles) 13087 and also react with alcohols accompanied by the substitution of one azido group to form monoazides 131. This transformation involving thiols leads to compounds 132.88 Finally, diazides 76 can be transformed into imines 133 using primary amines as nucleophiles.79
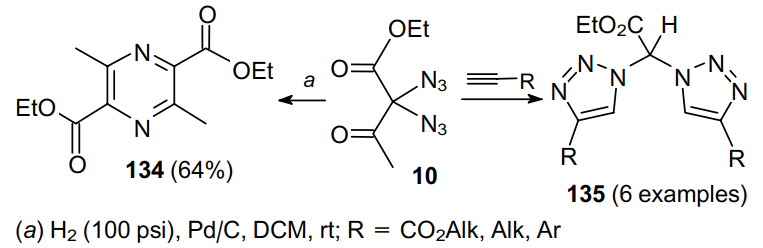
It is remarkable that the hydrogenation of substrate 10, in contrast to the hydrogenation of diazide 29 under similar conditions, leads to a kind of dimerization of diazide to furnish pyrazine 134 in moderate yield.79 Diazide 10 can also undergo the [3+2]-cycloaddition reaction catalyzed by the conventional CuSO4--sodium ascorbate system to give geminal bis(triazoles) 135 in yields up to 91% (Scheme 38).89
Kirsch and co-workers58,90,91 also developed a method for the oxidative cleavage of 1,3-diketones 136 to amides 137 upon the treatment with (BuN)N3 and iodine in the presence of an amine at room temperature. In most cases, 4-pyrrolidinopyridine (4-PPY) was used as the additive. According to the preliminary mechanistic studies, following the diazidation of the enol form, diazide 138 reacts with the amine through the attack on the carbonyl group, resulting in the cleavage of diazide 139, which also gives the final product via the formation of intermediates 140 and 141, (Scheme 39).
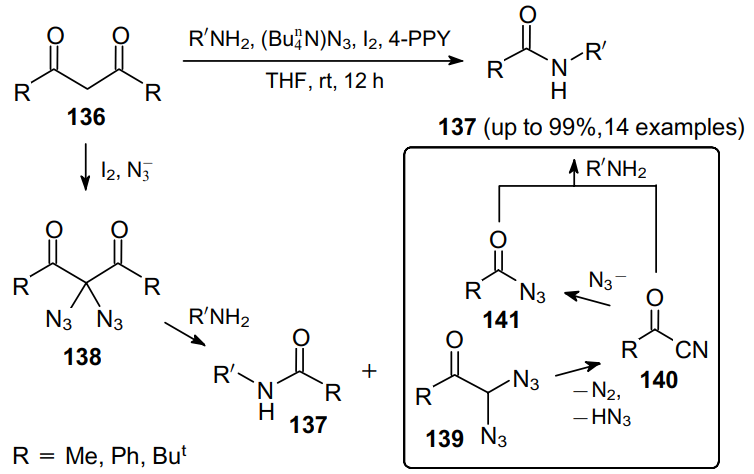
It was subsequently demonstrated that diazidoketones 53 can undergo a controlled fragmentation when treated with primary amines in the presence of caesium carbonate, thereby providing access to the corresponding amides 142 and benzonitriles 143 (Scheme 40).59
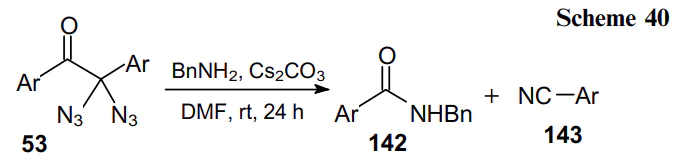
Diazidomalonate 29 was used as a monomer in the synthesis of polyamides 144 containing geminal diazido units within the polymer backbone. The post-modification of such diazido units using the CuAAC reactions allows for the design of highly functionalized polymeric structures (Scheme 41).87 This reaction and some other transformation typical of geminal aliphatic diazides, are presented in a mini-review by Тelik and Kirsch.19
2.3. Geminal vinyl diazides
2.3.1. Synthesis of vinyl diazides
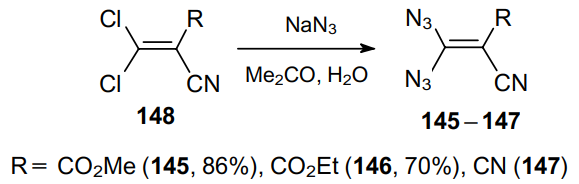
A significant number of publications addressing the synthesis of geminal vinyl diazides appeared in the 1980s.92--95 Diazides 145--147 are generally prepared by the simple replacement of chlorine atoms in compounds 148 with sodium azide in an acetone--water mixture at a temperature below 0 °C. The resulting methyl 3,3-diazido-2-cyanoacrylate (145) and ethyl 3,3-diazido-2-cyanoacrylate (146) are yellow crystalline compounds, stable in solution at 15 °C within several days but slowly decomposing at room temperature. Compounds 145 and 146 spontaneously detonate at 70 8C and 50 °C, respectively. 2-(Diazidomethylene)malononitrile 147 was described as an orange oily compound (Scheme 42). Diazides 145 and 146 were isolated by filtration, while in the case of product 147, the water-insoluble oily product was separated.
Research of the scientific group led by V.G.Nenajdenko96,97 also included the synthesis of geminal vinyl diazides from the apporpiate dihalo derivatives using sodium azide as the azide source. These works will be considered below in the Section on the reactivity of such diazides.
2.3.2. Transformations of vinyl diazides
Transformations involving geminal vinyl diazides are quite diverse.98 The reactions characteristic of such compounds can be divided into three main types. The first type includes the monomolecular decomposition, which can be both thermal and photochemical. Scheme 43 exemplifies a photochemical reaction, which occurs upon photoirradiation of a methanolic solution of methyl 3,3-diazido-2-cyanoacrylate (145). This reaction affords methyl 3-methoxy-2-cyanoacrylate (149).99--101
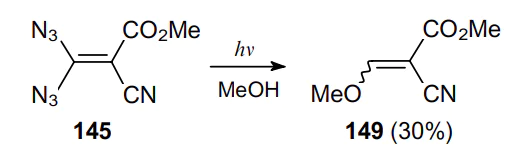
Compared to the photochemical decomposition, which has not yet been studied in detail, thermal reactions of geminal vinyl diazides were investigated more extensively (Scheme 44).102--106
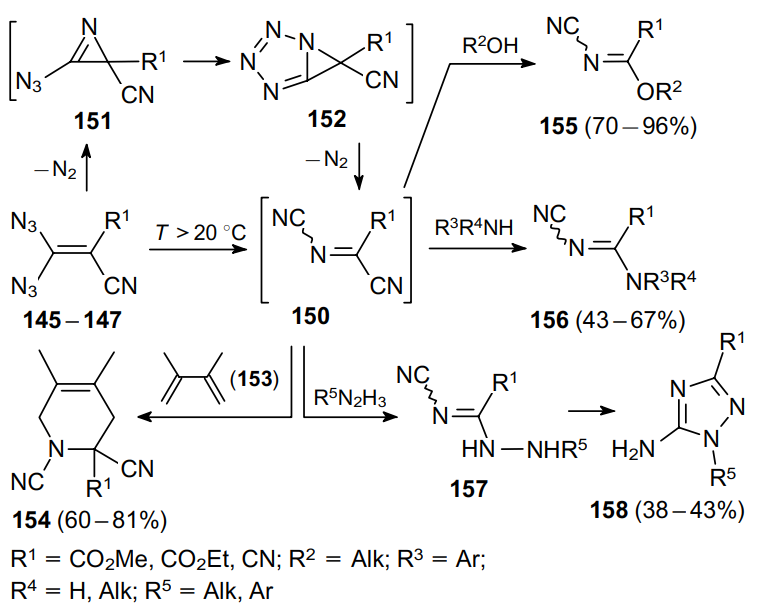
Electrophilic doubly acceptor-substituted N-cyanoimines 150 are key intermediates in such transformations.107 At temperatures >20 °C, vinyl azides 145--147 lose one nitrogen molecule to form azidoazirines 151 that spontaneously cyclize to tetrazoles 152.106 After releasing a second nitrogen molecule and subsequent rearrangement, the latter compounds give N-cyanoimines 150. Such intermediates were characterized only in solution94,106 since all attempts to isolate them in an individual state ended in polymerization.106,107 The existence of these intermediates was also confirmed by their cycloaddition to 2,3-dimethylbuta-1,3-diene (153) to afford Diels--Alder products 154.92--94,107,108 In the presence of alcohols102,103,107,108 or amines,102--106 N-cyanoimines 150 are instantly attacked at the electrophilic carbon atom to produce, after the release of hydrogen cyanide, the corresponding compounds 155 or 156 in good yields. The reaction of N-cyanoimines 150 with hydrazines furnishes compound 157, the product of the formal substitution of the cyanide moiety. A further cyclization yields 1,2,4-triazoles 158.108
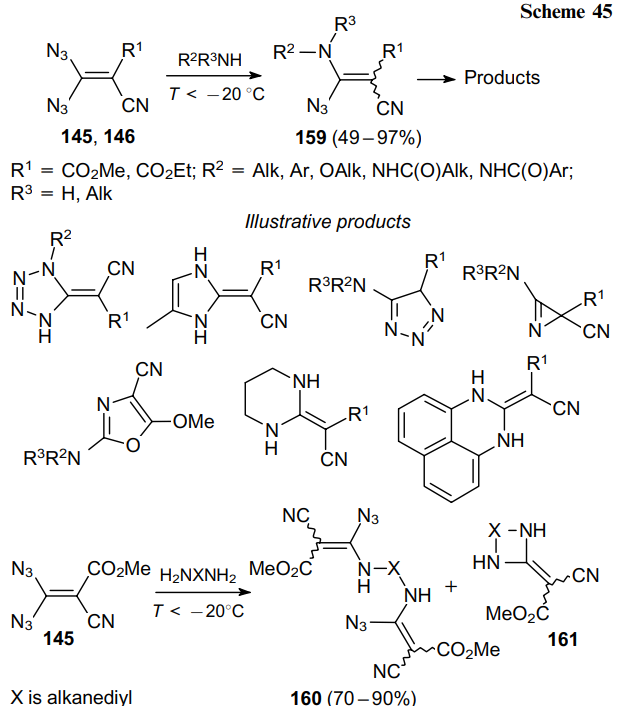
The nucleophilic substitution of an azide moiety is another fundamental reaction of geminal vinyl diazides 145 and 146 to produce intermediates 159 (Scheme 45). Formally, this process should be considered as an addition followed by elimination of hydrazoic acid. Due to thermal instability of the starting compound, this reaction should be carried out at a temperature below –20 °C. Otherwise, N-cyanoimine will be generated before the intermolecular reaction occurs. Primary95,106,108,109 and secondary amines,103,106 diamines104,106 and hydrazines95 were tested as nucleophiles. In the case of diamines, the substitution occurs either in two different molecules of diazide 145 to yield bis(vinyl azides) 160,104,106,108 or in the same molecule to afford cyclic products 161.104 In the reaction of simple amines with geminal vinyl azides, one azide functionality is substituted to give the corresponding vinyl azide 159. Further transformations of vinyl azide 159 provide access to a variety of nitrogen-containing heterocycles such as tetrazolylidenes,95 imidazolylidenes,109 oxazoles,104,109 1,2,3-triazoles,108 azirines, etc. The reaction outcome strongly depends on the substitution pattern in vinyl azide 159 and reaction conditions.
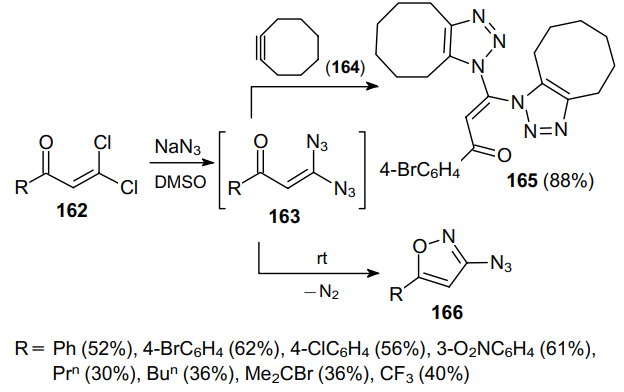
Noteworthy are the reactions of geminal vinyl diazides discovered in recent years, in which one azido group is involved in the intramolecular cyclization with any functional group, while another azide group remains intact and can be involved in further transformations of the newly formed cyclic azide.96,97 Thus, the reaction between geminal dichlorovinyl ketones 162 and sodium azide was studied.96 Using NMR spectroscopy and quantum chemical calculations, it was shown that this process produces the corresponding geminal 3,3-diazidoenones 163 (Scheme 46). These highly reactive compounds can be captured by cyclooctyne (164) to give bis(triazoles) 165 in high yields. In the absence of the capturing agent, 3,3-diazidoenones 163 spontaneously cyclize into 3-azidoisoxazoles 166. According to calculations, the intramolecular cyclization proceeds through the nucleophilic attack of the carbonyl oxygen on the nitrogen atom with the release of a N2 molecule.
Tsyrenova and Nenajdenko110 developed an effective method for the synthesis of 4-azido-1,2,3-triazoles 167 based on the treatment of dichlorodiazadienes 168 with an excess of NaN3 (Scheme 47). Geminal diazide 169 is an intermediate product of this transformation; its formation was confirmed by the reaction of dichlorodiazadiene with NaN3 in the presence of cyclooctyne (164), which produced bis(triazole) derivative 170.
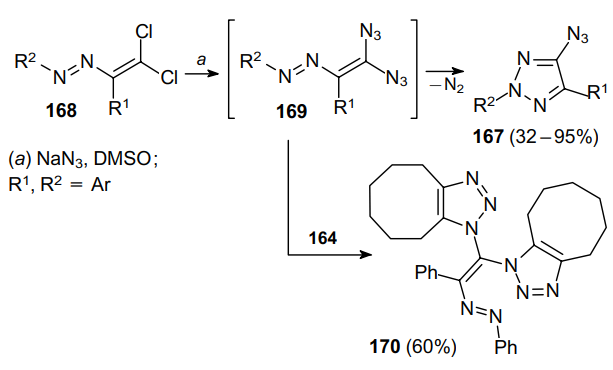
The introduction of an acetylene moiety in the ortho position of the aryl substituent at the C(3) carbon atom of dichlorodiazadienes 171 and their subsequent treatment with sodium azide lead first to diazides 172 and then to the corresponding acetylene-substituted 4-azido-1,2,3-triazoles 173, which can be transformed into various fused heterocyclic systems.110 In particular, the thermal intramolecular cyclization of o-propargyloxy-substituted 4-azido-1,2,3-triazoles 173 gives rise to a variety of oxazocine derivatives 174 in good yields (Scheme 48).
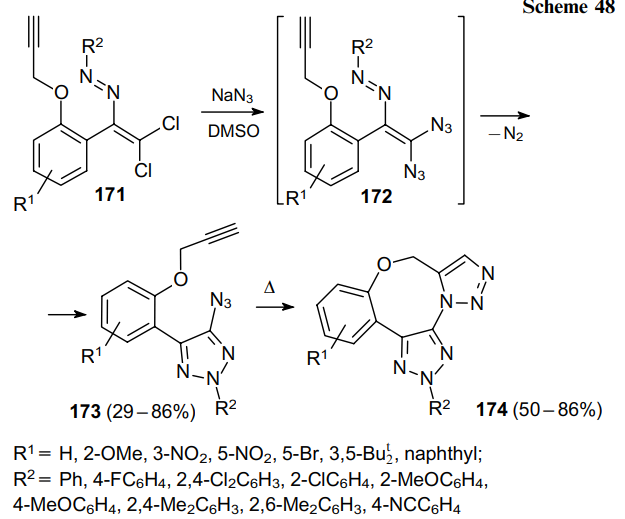
Dichlorodiazadienes 175, via the formation of vinyl diazides 176, also gave ortho-alkynyl-substituted 4-azido-1,2,3-triazoles 177, which were transformed into isoquinoline derivatives 178 when heated in toluene (Scheme 49).111
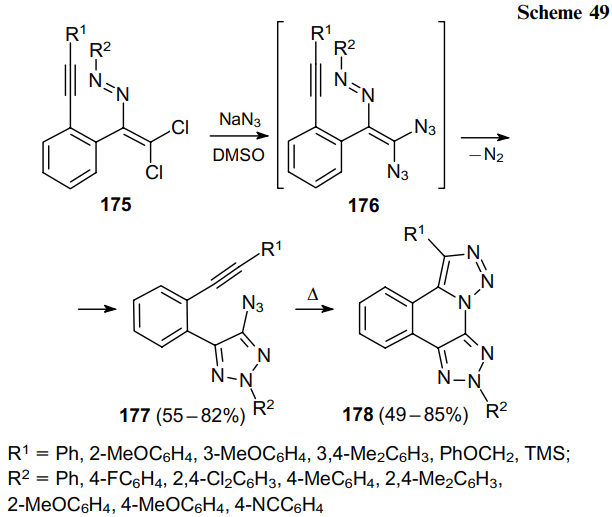
Finally, under similar conditions, pyridine-containing dichlorodiazenes 179 gave 2-pyridine-substituted diazides 180 and 4-azido-1,2,3-triazoles 181, which, when heated in o-xylene, furnished pyridopyrazole derivatives 182 with a highly polar N+–N– bond (Scheme 50).112
2.4. Geminal heterodiazides
2.4.1. Diazidophosgene
Recently, a large number of notable polyazides of Groups 15 (P, As, Sb)113--116 and 16 (Se, Te)117--122 of the Periodic table were synthesized and structurally characterized. As for carbon, Group 14 element, a few organic mono- and diazides are well known, and some of them are quite stable at room temperature and even commercially available. Meanwhile, one of the simplest carbon diazides, carbonyl diazide (or diazidophosgene, OC(N3)2 (3)), was poorly explored. Thus, the synthesis of this compound and its use in situ were reported in a few papers.123--125 In 2007, diazide 3 was mentioned in a publication126 as a hydrolysis product of diazidomethane, as evidenced by 13C and 15N NMR spectroscopy, but the authors did not isolate it in the pure form.
Three years later, a safe method for the synthesis and precautions for handling carbonyl diazide OC(N3)2 as a pure substance in small quantities were described.127 The physical properties of this compound were investigated and it was fully characterized by IR (gas-phase, Ar matrix) and Raman (solid-state) spectroscopy and X-ray crystallography. Carbonyl diazide was prepared by reacting NaN3 with an excess of FC(O)Cl in a sealed glass ampoule at room temperature for 4 days. Volatile products from three batches were collected and separated by repeated condensation at different temperatures. Thus, product 3 was trapped at –60 °C as a white solid. More volatile intermediate azide FC(O)N3 was trapped at –100 oC, while unreacted FC(O)Cl was trapped at –196 °C. Later, various routes for the generation of carbonyl diazide were described,128 including that from tetraazidomethane (183) (Scheme 51) but none of these reactions can be considered as an effective method for the synthesis of carbonyl diazide.

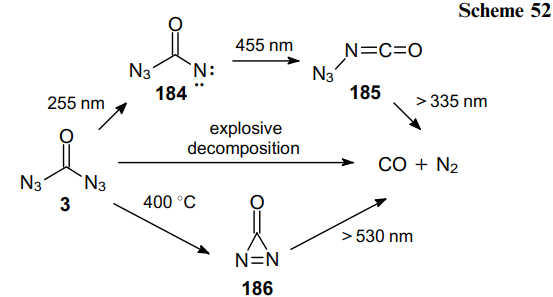
An advantage of FC(O)Cl as the starting material compared to C(O)Cl2 or C(O)F2 is that a stable intermediate product FC(O)N3 is generated in the first step via the rapid chlorine--azido substitution reaction. In the second step, this monoazide undergoes a slower fluorine--azide substitution reaction to afford OC(N3)2. In the condensed phase, diazide 3 is rather shock-sensitive; however, it demonstrates high thermal stability, as it did not decompose either in the gas phase or in the liquid and solid states. The solid melts at 16 °C and can be transferred from one vessel to another by vacuum sublimation. Although Nolan et al.128 did not indicate the yield of product 3, the very fact of the isolation of carbonyl diazide in an individual state is particularly noteworthy. During thermal and photochemical decomposition of compound 3, intermediates 184--186 can be generated (Scheme 52).
2.4.2. Carbamoyl diazides
In 1912, Darzens and Hebd129 carried out the reaction between bromocyan and sodium azide, assuming that they isolated cyanogen azide 187 as a product. Much later, it was revealed that the structure of the resulting compound was determined incorrectly and the reaction gave diazide 188 (Scheme 53), which is the first example of geminal diazide containing two azide groups on the unsaturated carbon atom.130--132
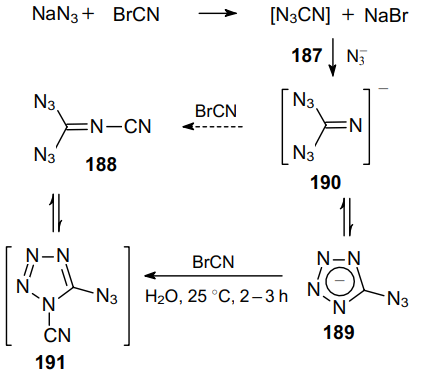
The initial fast reaction of sodium azide with cyanogen bromide generates unstable intermediate 187, which rapidly reacts with aqueous sodium azide to give tetrazole 189 in 80% yield. This tetrazol is believed to be formed through the attack of the azide ion on the electron-deficient carbon atom in intermediate 187, which affords hypothetical geminal diazide 190. The second, slower step involves the substitution yielding compound 191, which apparently undergo ring opening to give gem-diazide 188, which is typical of many tetrazoles.
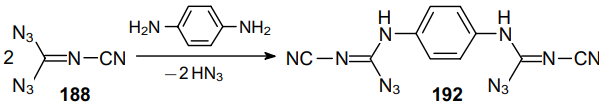
Norris and Henry133 reported the possible reaction of cyano-containing diazide 188 with p-phenylenediamine (Scheme 54); however, the authors did not provide the yield of product 192.
3. Synthesis and transformations of vicinal diazides
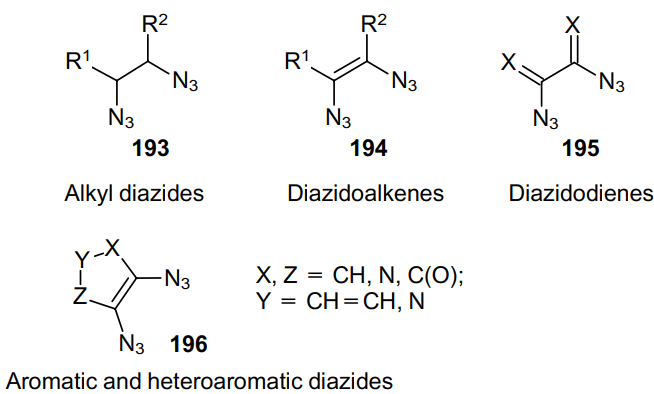
Vicinal diazides comprise azido moieties located at the neighboring carbon atoms (Structures 193-196). Derivatives of this class are commonly found in the literature as intermediates. In particular, they are precursors to vicinal diamines, the structural unit of which are found in many natural compounds and drugs, 134 and also play an important role in modern organic chemistry as ligands for catalysts for asymmetric synthesis.135
3.1. Methods for the synthesis of vicinal alkyl diazides
This Section considers the main methods for the preparation of aliphatic 1,2-diazides. The analysis of the literature data showed that the main approaches to the synthesis of such compounds include the nucleophilic substitution in dihalo, dihydroxy and sulfo derivatives of organic compounds, and also diazotization and diazo transfer reactions. Besides, vicinal alkyl diazides are derived from appropriate olefins.
3.1.1. Nucleophilic substitution reactions
The direct nucleophilic substitution of functional groups in derivatives 197 with an azide anion is the simplest and most reliable method for the synthesis of diazides 193 (Scheme 55).
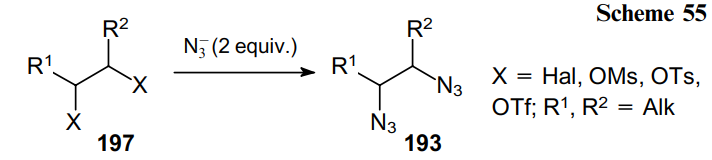
For example, 1,2-diazidoethane 198 can be obtained from dibromoethane via nucleophilic substitution under the action of sodium azide and then employed in the synthesis of thermally stable 1,2-bis(5-carboxyethyl-1H-tetrazolyl)ethane 199 (Scheme 56), a building block for the design of nitrogen-rich polymers.136
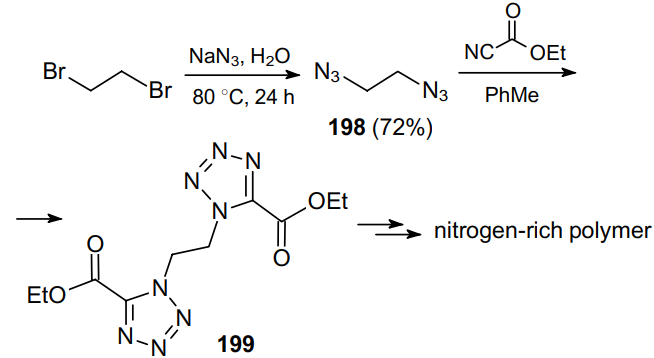
A number of publications137--141 describe the use of alkenes 200 in the preparation of vicinal diazides 201, which served as substrates in the synthesis of diamines 202 (Scheme 577). In this case, the diazotization of dibromides 203 in carried out using NaN3 to form the products in almost quantitative yields.
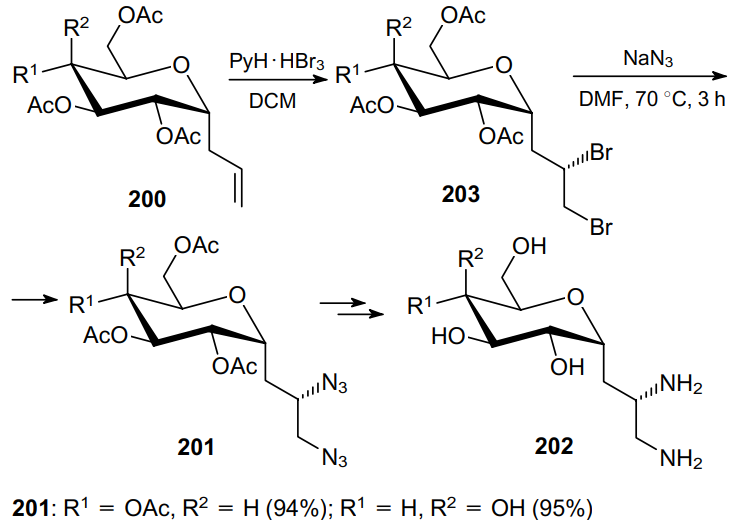
However, the reactivity of such alkali metal azides as NaN3 and LiN3 may be insufficient for the nucleophilic substitution of sterically hindered alkyl and alkenyl sulfonates. In this case, more reactive azide sources are generally employed, in particular, the reagent [Et2NH2]+N3-(DEAHN3), which can be prepared by mixing an ethereal solution of HN3 with diethylamine (DEA).67 Thus, DEAHN3 was successfully applied in the synthesis of diazides 205 from tosylates 204 (Scheme 58), and according to NMR spectroscopic data, the reaction occurred with almost complete inversion of both stereogenic centres.142
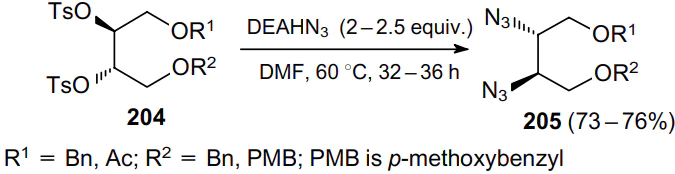
Another azidating agent, trimethylsilyl azide, proved effective in the nucleophilic substitution of OH moieties in uridine derivative 206 (Scheme 59).143 Here, the azidation occurs in the presence of tetra-n-butylammonium fluoride (TBAF), which makes it possible to remove the silyl protection and obtain vicinal diazide 207.
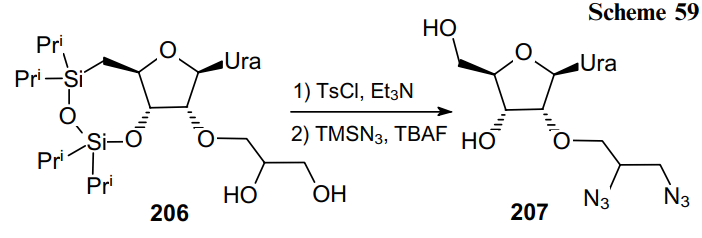
The azidation of diol 208 can also be accomplished in a two-step fashion to provide a high yield of the product (Scheme 60). First, the tosylation is carried out followed by the nucleophilic substitution with two equivalents of NaN3 in DMF. This process makes available acyclic nucleoside analogues of pyridothienopyrimidines 209 containing a vicinal diazide moiety.144--147
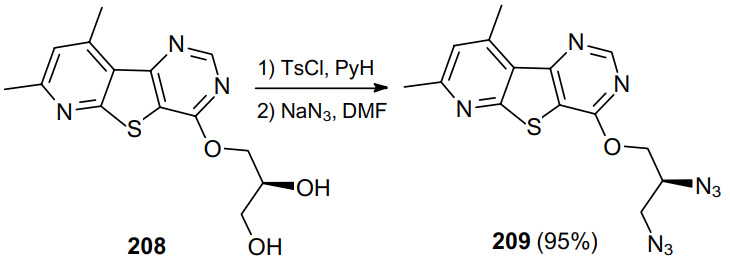
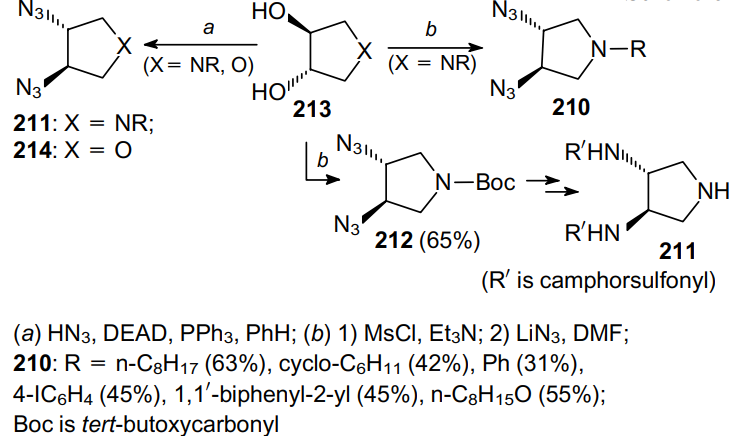
A similar strategy was used to prepare 3,4-diamino-1-substituted pyrrolidines.148 Thus, Marson and Melling149 synthesized diazides 210 in yields up to 63%, using LiN3 as the azidating agent (Scheme 61). This approach has proven to be suitable for the synthesis of sulfonamides 211, serving as promoters in the asymmetric addition of ZnEt2 to p-chloroacetophenone, via the formation of diazide 212.150--153 Reported also are successful attempts to carry out the Mitsunobu reaction154--156 for enantiomeric diols 213, which were transformed into diazides 211 and 214, when treated sequentially with HN3, PPh3 and diethyl azodicarboxylate (DEAD), in yields up to 80%.157
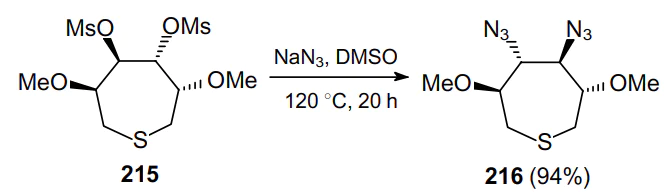
A simple and effective approach to 2,3-diaminoconduritols, promising compounds with high biological activity,158,159 comprises the azidation of 4,5-dimesyl-3,6-dimethoxythiepane 215 with sodium azide in DMSO. This led to an inversion of configuration at the C(4) and C(5) atoms when forming diazide 216 (Scheme 62).160,161
The two-step azidation of 3,4,5,6-tetra-O-benzyl-myo-inositol (217) was carried out under similar conditions in a DMF medium (Scheme 63). The yield of diazide 218 over two steps ranges from 54 to 79% according to various sources.162--164
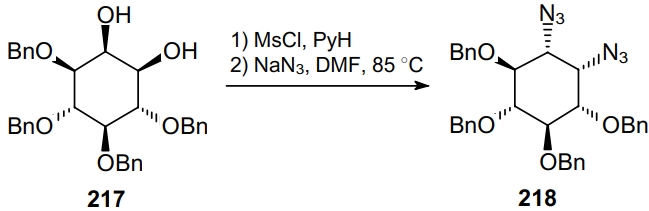
The mesylation of diol 219 followed by the treatment with NaN3 in the presence of catalytic amounts of 15-crown-5 provides diazide 220 in high yield (Scheme 64). This diazide 220 can be further reduced to diamine 221, which is a precursor to chelating agents based on polyaminocarboxylic acids.165,166
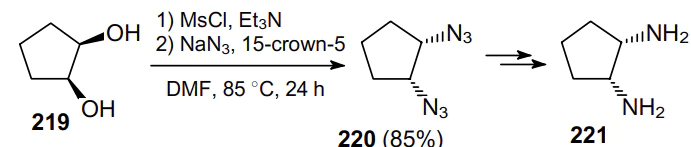
This approach has proven effective also for the synthesis of diazide-substituted terpenoids, e.g., from diol 222 (Scheme 65).167 Unfortunately, the authors failed to adequately justify the retention of stereoconfiguration of asymmetric centres in the synthesis of diazide 223.
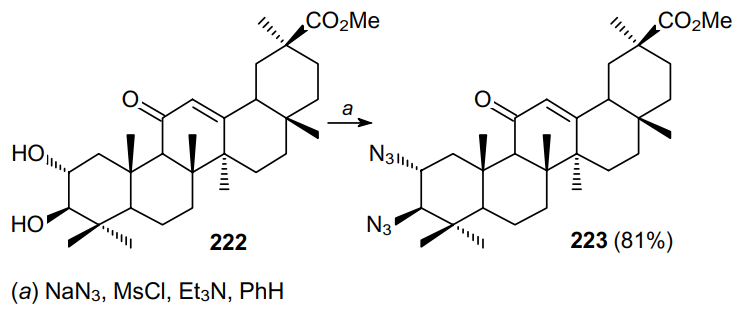
When ditriflates are used in the azidation, the resulting products can contain two good leaving groups and serve as valuable substrates for subsequent transformations.168 At room temperature, triflate 225, derived from protected glucoside 224, reacts with tetra-n-butylammonium azide in THF to give a mixture of mono- (226) and diazidation (227) products (Scheme 66).
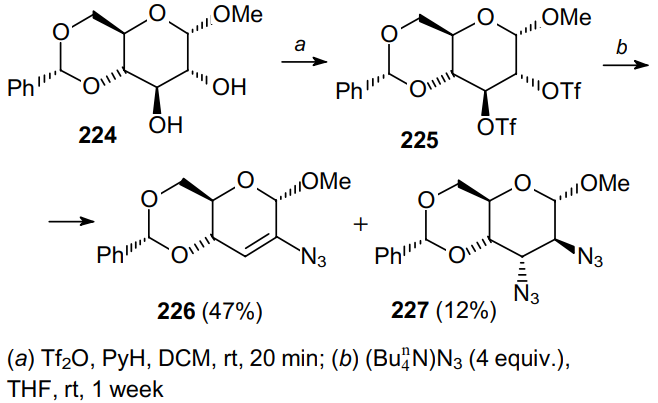
In a similar way, using trifluoromethanesulfonic anhydride, 2',3'-diamino-substituted uridines with xylo configuration can be produced from compound 228 (Scheme 67).169 In this case, the nucleophilic attack of an azide anion on the resonance-stabilized cation 229 occurs. As a result, two main products, diazide 230 and monoazide 231, are obtained. Evidently, the latter product is generated via the elimination of HN3 from diazide 230.
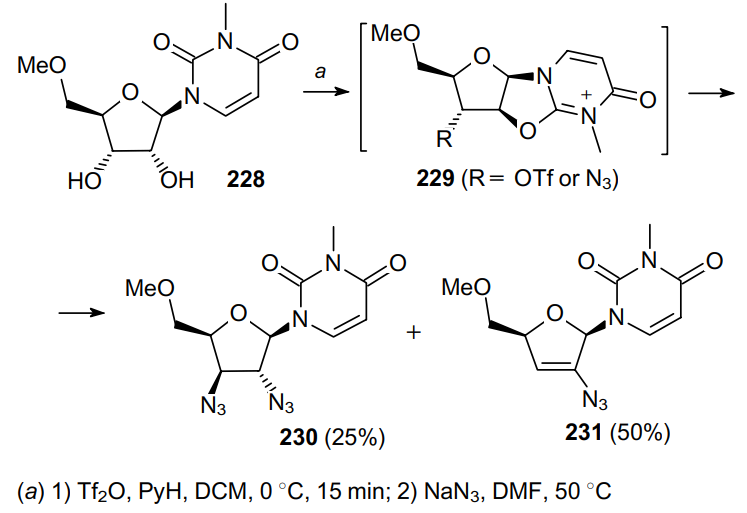
The diazo transfer reaction of 1,2-diaminocyclohexane (232) with triflyl azide afforded 1,2-diazidocyclohexane (233) in good yield (Scheme 68).170

3.1.2. Synthesis of alkyl-1,2-diazides from alkenes
3.1.2.1. Stoichiometric 1,2-diazidation of olefins
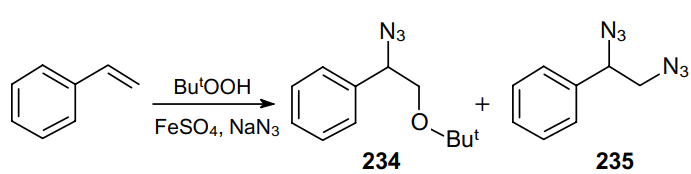
The synthesis of vicinal diazides was first described in 1962.171 The iron(II)-catalyzed decomposition of tert-butyl hydroperoxide in the presence of styrene and NaN3 produces a mixture of azidoether 234 and diazide 235 (Scheme 69). The authors provided no information on the ratio and yields of the products.
The mechanism of this reaction99 involves a stepwise radical diazidation, as exemplified by 2,3-dimethylbut-2-ene in Scheme 70, which leads to the generation of azidoalkyl radicals 236 and the formation of vicinal diazide 237.
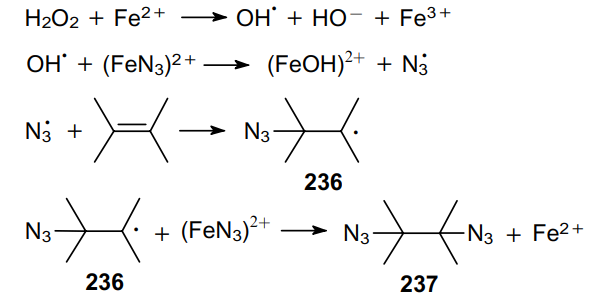
The pathway of the reaction between styrene and NaN3 is driven by the electrophilic character of the reactive oxygen radical.172 Due to its high electrophilicity, the OH• radical reacts with the azide ion coordinated to the iron salt rather than with the olefin. The electrophilic character of the newly formed azide radical N3• is not quite as pronounced as in the OH• radical. This reaction can be extended to conjugated dienes (butadiene, cyclopentadiene, etc.) and also applied to non-activated alkenes, such as pent-2-ene, hex-1-en, cyclohexene, etc. In the presence of electron-withdrawing groups attached to the double bond, such as in the case of acrylonitrile, the reaction proceeds less efficiently. There are example of using lead173 and thallium100 salts as catalysts for such processes; however the yields of diazides turned out to be low.
Alkenes can be converted into vicinal diazides 193 in moderate yields using an excess of NaN3 in the presence of Mn(OAc)3 (Scheme 71).174
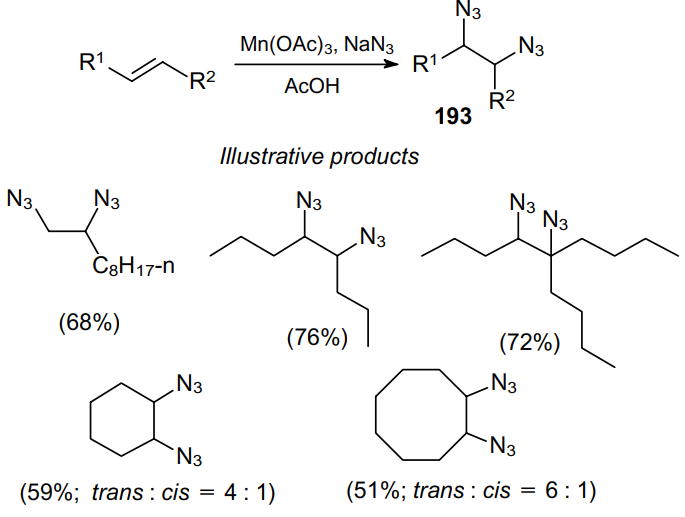
The mechanism of this reaction involves the generation of azidoalkyl radical 238, which reacts with the MnIII--N3 complex to afford vicinal diazide 193 (Scheme 72).
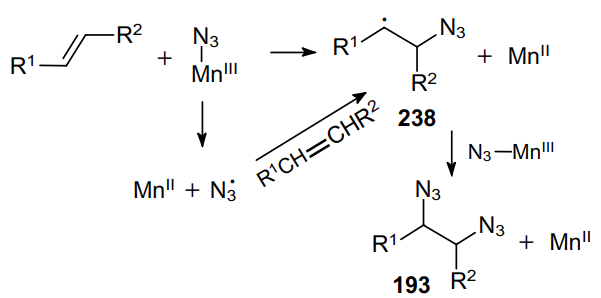
This approach was extended to the natural alkene a-pinene (239),175 which gave a mixture of isomeric diamines 240 and 241 (Scheme 73). Meanwhile, b-pinene (242) can be regioselectively converted into the corresponding 1,2-diazide 243 in high yield.176 The above procedure was modified and applied to other, more acid-sensitive alkenes.
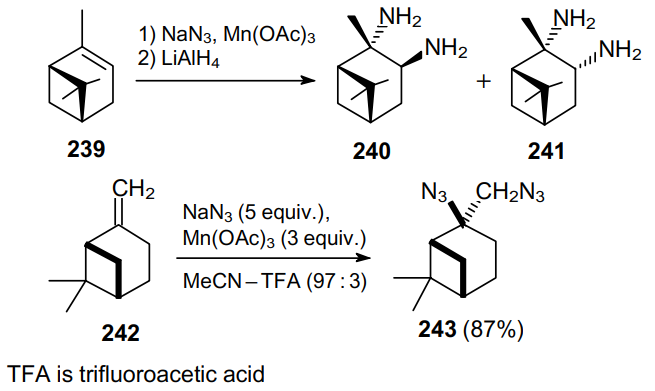
Model experiments with dihydropyran 244 (Scheme 74) demonstrated that the diazidation in a Mn(OAc)3--NaN3--AcOH system proceeds much slower than the protonation of the double bond. Therefore, the reaction gives tetrahydropyranol and tetrahydropyranyl azide as the major products. Carrying out this reaction at a low temperature in a 9:1 MeCN--TFA mixture significantly reduces the potential for protonation, thereby producing a mixture of trans- (245) and cis-diazides (246).
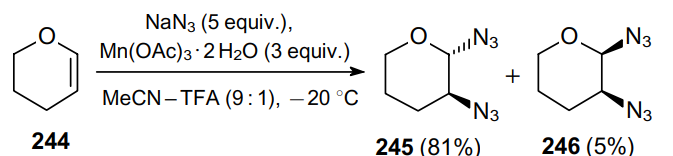
The same solvent system proved to be suitable for 1,2-diazidation using peracetylated or perbenzylated glycals as substrates. Thus, the reaction of 3,4,6-tri-O-acetyl- D -galactal (247) with a mixture of Mn(OAc)3•2H2O and NaN3 with the same ratio, in a 9:1 MeCN--TFA mixture at 25 °C for 2 h affords a mixture of diazides 248 and 249 (Scheme 75). Such 2-azido-2-deoxyglycopyranosyl azides can be used to produce glycosylated asparagines.177,178 The above examples sufficiently demonstrate the scope of the the addition reaction of azide with alkenes in the presence of Mn(OAc)3.
3.1.2.2. 1,2-Diazidation using hypervalent iodine
In recent years, reagents based on hypervalent iodine have gained great popularity in synthetic organic chemistry due to their low toxicity, ease of handling and commercial availability. Thus, an interesting approach to vicinal diazides is based on the reaction of alkenes with iodosobenzene (250) in the presence of sodium azide (Scheme 76). This method provides a simple and efficient route to 1,2-diazides 193, which can be prepared in yields up to 85%. Terminal alkenes and also disubstituted and cyclic olefins can be used in this process.179
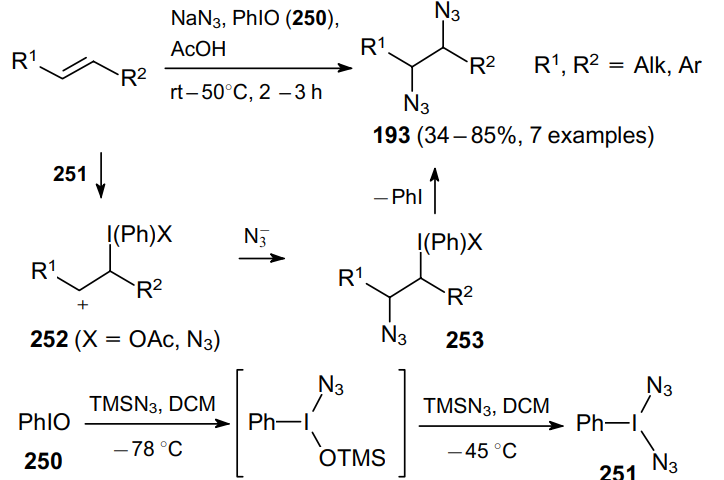
The reaction mechanism involves the electrophilic attack of the in situ formed diazide derivative of hypervalent iodine 251 on the alkene double bond. This generates carbocation 252, to which an azide anion then adds yielding intermediate 253. The subsequent reductive elimination of iodobenzene affords vicinal diazide 193. Alternatively, this step can be considered as the nucleophilic substitution of iodobenzene with an azide ion.
It was shown180 that diazide 251 can act as an azidating agent for triisopropylsilylated enols 254. Depending on the temperature conditions, the reaction can afford predominantly b-azide 255 or 1,2-diazide 256 (Scheme 77). Consequently, in this case there are two competing mechanisms; the authors suggested that the formation of diazide 256 occurs via the radical addition. The use of a stable organic radical (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO, 257) allows the synthesis of diazidation product 256 as the major product.
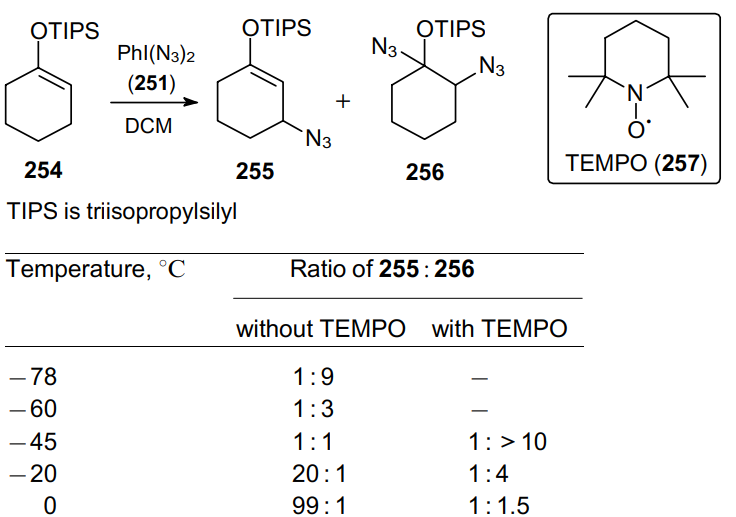
Under the optimized conditions, in the presence of TEMPO, TIPS-substituted enols 258a--g gave rise to a large number of diazides 259a--g in yields from 41 to 91%, with the products 259a--c forming a single diastereomer (Scheme 78).
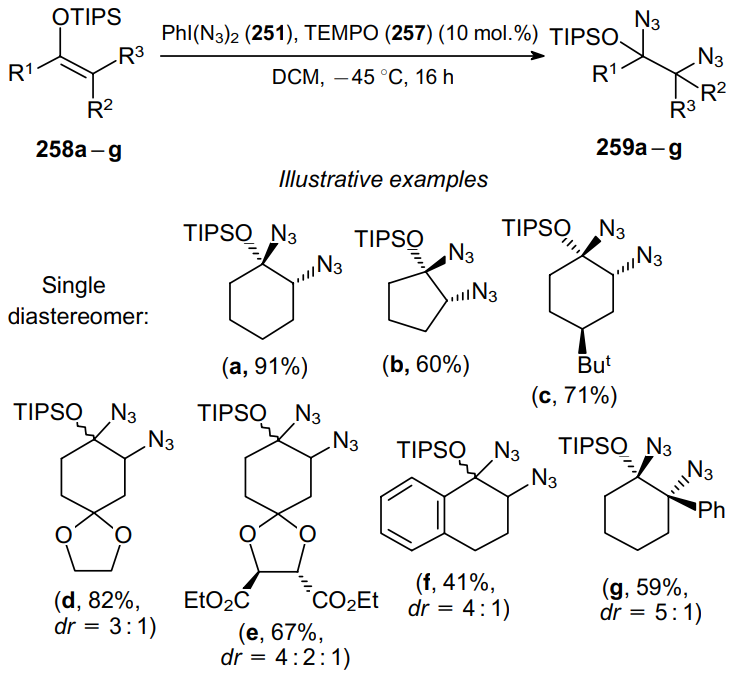
The reaction of b-substituted allylsilanes 260a--e with iodosobenzene (1.5 equiv.) and TMSN3 (2 equiv.) furnishes 1,2-diazides 261a--e in yields up to 86% (Scheme 79).181
It is noteworthy that 1,2-diazides can be prepared by the reaction of cyclic conjugated polyolefins with IN3 and an excess of NaN3 affording the corresponding cis isomers.182 Thus, the reaction of cyclooctatetraenes 262 with iodine azide, prepared in situ from an excess of sodium azide and iodine monochloride in acetonitrile, gives a cis product of the general formula 263 (Scheme 80), however, for X=CH= CH, the diazide is rapidly transformed into bicyclic product 264. Moreover, the reaction of cycloocta-1,3- and cycloocta-1,5-dienes under the above conditions produces monoazides, which can be further converted to the corresponding diazides when treated with NaN3 in DMF at 35--40ºC.
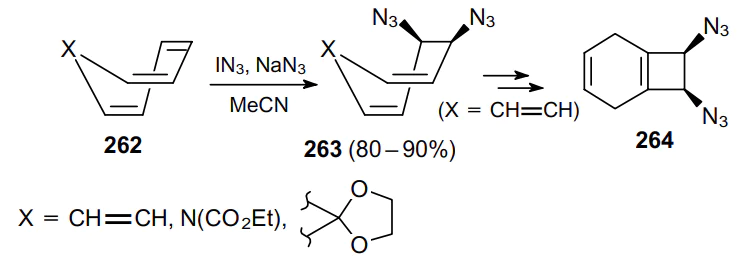
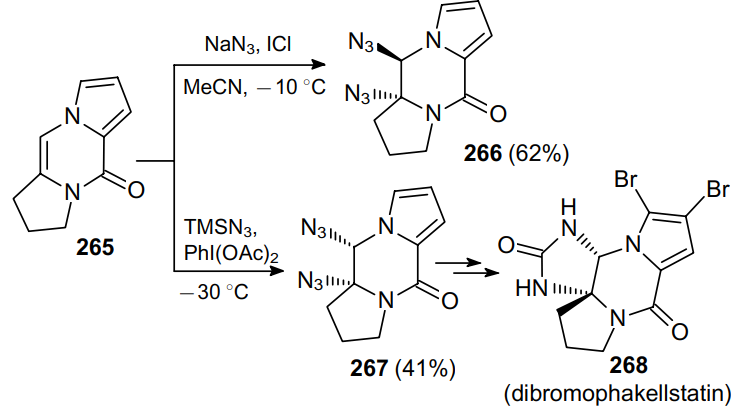
This approach was employed to azidate pyrazinone derivative 265 to form diazide 266 in moderate yield. When changing the reaction conditions, ketone 265 gave isomeric diazide 267, which was used in the synthesis of the tetracyclic pyrrole imidazole alkaloid (±)-dibromophakellstatin (268) isolated from the marine sponge Agelas (Scheme 81).183
Another variant of the direct diazidation based on the use of the sodium periodate--sodium azide--DMSO--AcOH system is notable for its efficiency and simplicity.52 By varying the stoichiometric ratio of reactants, the optimal combination was identified, consisting of 3 equiv. of NaN3 and 1 equiv. of NaIO4. Under these conditions, styrene derivatives and aliphatic olefins, including linear and cyclic alkenes, produce 1,2-diazides 193 in high yields (Scheme 82).
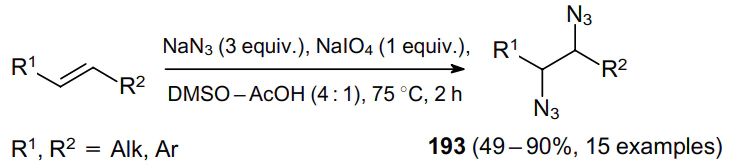
Internal olefins provide high yields of syn- and anti-isomeric products formed in a 1:1 ratio. However, no reaction occurs with a,b-unsaturated carbonyl compounds [e.g., cinnamic esters, (R)-(–)-carvone] and also sterically hindered alkenes (e.g., a-pinene). The process follows a radical pathway, since the diazidation of styrene in the presence of catalytic amounts of TEMPO resulted in a sharp decrease in the yield of the product (up to 10%) due to the radical quenching effect.
To diazidate double bonds, an approach can be employed based on the treatment of enamides 269 with a combination of bis(tert-butylcarbonyloxy)iodobenzene (270) and LiN3 in acetonitrile (Scheme 83).184 The corresponding 1,2-diazides 271 are produced in yields ranging from 18 to 61%; high degrees of stereoselectivity of diazidation (up to 90% of syn-isomer) are achieved.
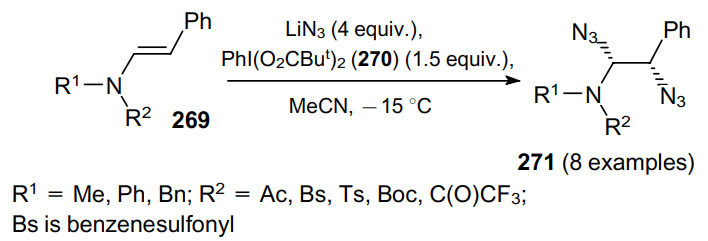
The diazidation of the double bond can also be performed using the bis(trifluoroacetoxy)iodobenzene (PIFA)--TMSN3 system in the presence TMSOTf as a catalyst.185 In particular, 1,2-unsaturated sugars 272--274 can be converted under these conditions to the corresponding vicinal diazides 275--277 in moderate yields (Scheme 84). While galactoses 272 produce the corresponding cis-1,2-diazide products 275, glucoses 273 form mixtures of a- and b-anomers in different ratios (given in parentheses after the yield of the product), with a-anomer dominating. Interestingly, the reaction of arabinose derivatives 274 affords vicinal diazides 277 in the 1C4 conformation.
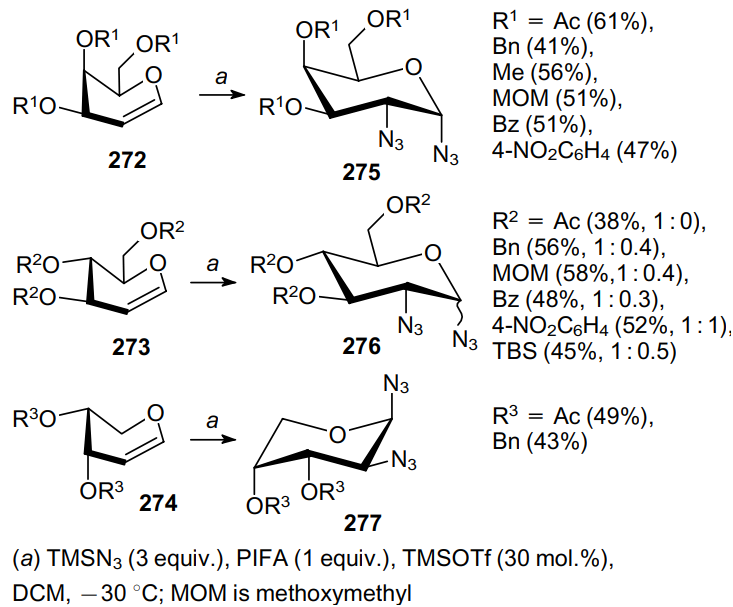
The mechanism of this reaction for derivatives 272 is illustrated in Scheme 85.185 Diazide PhI(N3)2 (251) generated in situ produces intermediate 279 upon p-interaction (complex 278) with the galactal double bond from the axial side. Then the azide moiety is transferred to the C(2) atom from the axial side in an SNi fashion. After this, due to steric hindrance caused by the substituents at the C(3), C(4) and C(5) atoms, the second azide moiety preferentially attacks the anomeric carbon atom of the galactose derivative from the axial side to form diazide 275.
3.1.2.3. Catalytic 1,2-diazidation of olefins
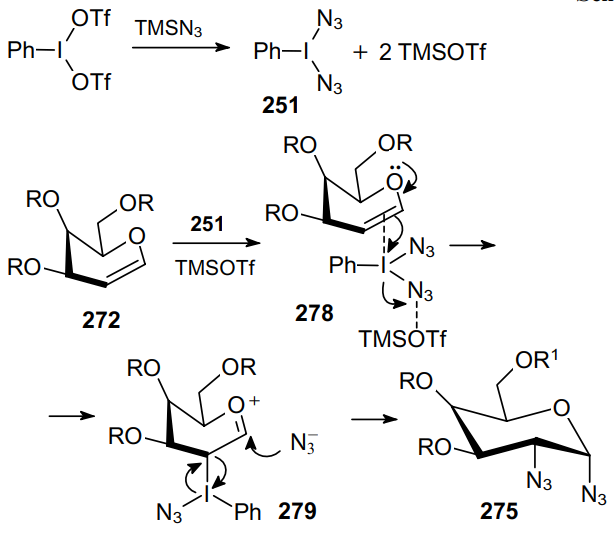
Transition metal catalysts are also used to prepare vicinal diazides. Thus, the copper(II)-catalyzed 2,3-diazidation of indoles of the general formula 280, involving azidotrimethylsilane as the azide source and iodobenzene acetate as the oxidant, provides a route to a series of 2,3-diazido-substituted indoles 281 in high yields (Scheme 86).186
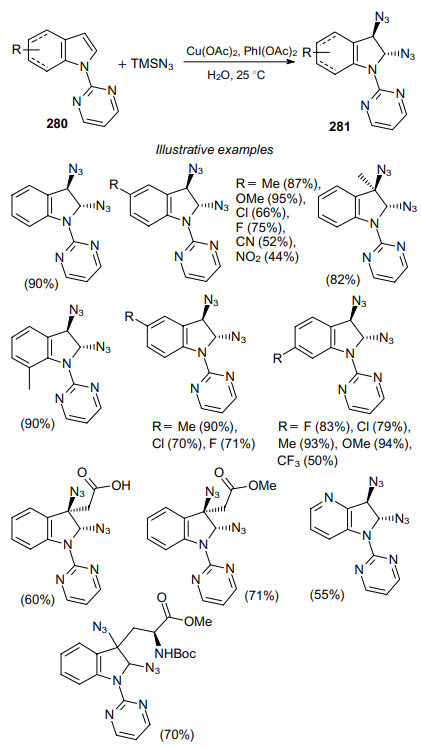
Indoles bearing electron-donating groups, such as Me and OMe, provide higher yields of the products compared to the substrates containing electron-withdrawing substituents. This reaction shows high diastereoselectivity and occurs under mild conditions in an aqueous medium.
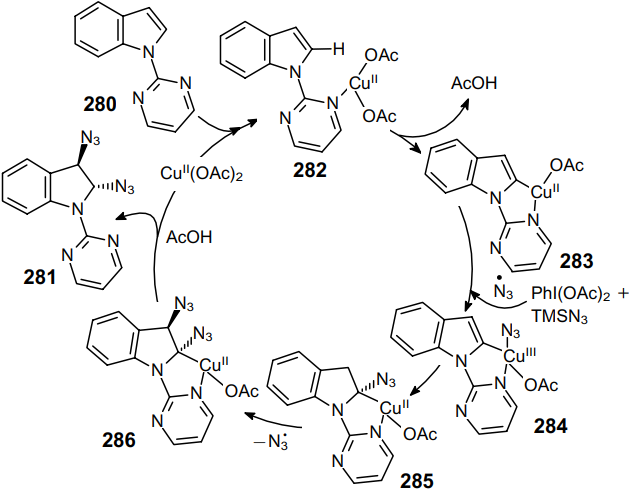
The proposed mechanism involves the coordination of Cu(OAc)2 by the pyrimidine ring of the substrate to afford complex 282 followed by the regioselective activation of the C–H bond at the C(2) position of indole, thereby yielding CuII complex 283. Then the N3 radical, that formed in the reaction of PhI(OAc)2 with TMSN3, is coordinated to the CuII ion to form complex 284 (Scheme 87). After this, the N3 group is rapidly inserted at the indole C(2) atom. This is accompanied by the dearomatization of the double bond at the C(2) and C(3) positions to give radical 285. The formation of trans-isomeric product 286 is due to the addition of the N3 radical from the more accessible side of complex 285.
Vicinal diazides 290 can be prepared by the addition of an azide group from Zhdankin reagent (1-azido-1,2-benziodoxol-3(1H)-one, 288) to the double bond of various styrenes 289 using the catalyst [Cu(287)2]Cl (Scheme 88).187 Under light irradiation, the azidation in a methanolic solution affords methoxyazides 291. On the contrary, in the absence of light, double azidation takes place to furnish 1,2-diazides 290. The reaction proceeds under mild conditions and performs well for a number of styrenes, 2-vinylnaphthalene and indene.
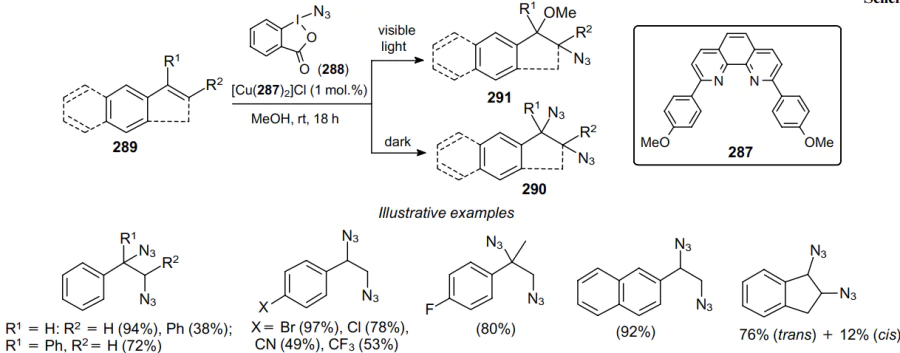
A similar procedure for the diazidation of styrenes 289 was developed by Lu et al.188 They carried out the reaction in DMSO in the presence of catalytic amounts of Cu(OTf)2. By changing the catalyst to CuI, the authors increased the yield of the product up to 82%. Various ortho-substituted substrates react well under these conditions to afford diazidation products 290 in 75--78% yields. It should be noted that substrates bearing electron-withdrawing substituents give 1,2-diazides in good yields (55--77%). The reactions of 1-vinylnaphthalene and 2-vinylthiophene with Zhdankin reagent also produce the corresponding products in moderate yields. Finally, dihydronaphthalene gives a diazide in a fair yield with moderate diastereoselectivity (Scheme 89).
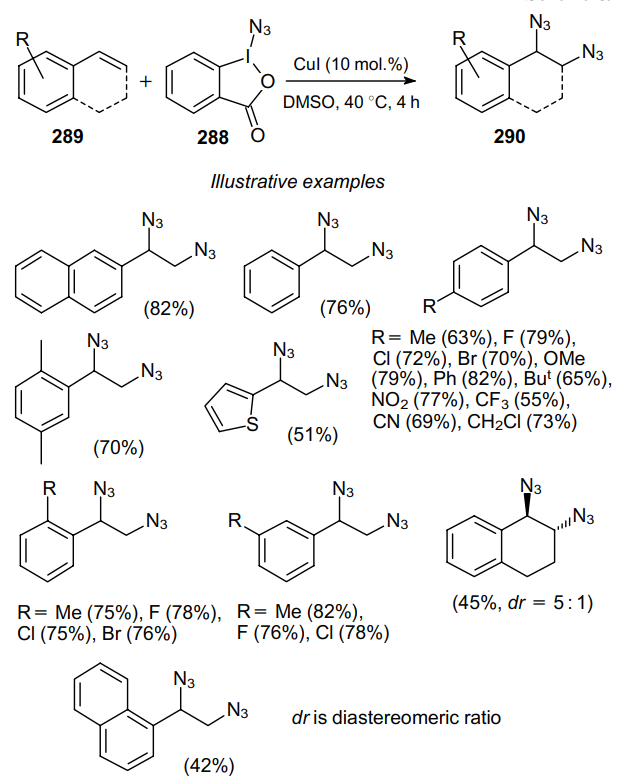
To investigate the mechanism of this transformation, the diazidation of 2-vinylnaphthalene (289a) was carried out in the presence of 3 equiv. of TEMPO or 2,6-di-tert-butyl-4-methylphenol (BHT, 292) (Scheme 90b). In both cases, a strong inhibition of the process was observed. Moreover, in the radical clock reaction, compound 293 gave cyclization product 294 in low yiels. These observations suggest the radical mechanism of the process.
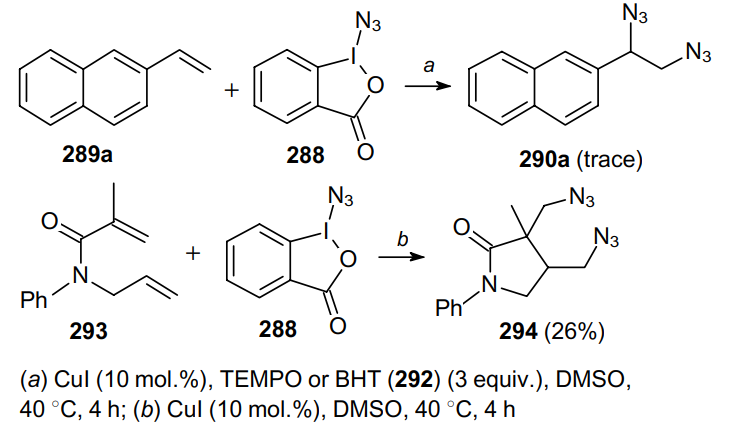
Zhou et al.189 proposed the environmentally friendly copper-catalyzed ligand-free diazidation of olefins including vinyl(het)arenes, unactivated alkenes, as well as allenes and dienes, under mild conditions using TMSN3 as the azide source (Scheme 91). This reaction occurs in the presence of tert-butyl peroxybenzoate (TBPB, 295a) for 6 h and is implemented in two systems, namely, in water (method a, the yield is given in Scheme 91 as the first value in parentheses) and in MeCN (method b). Substrates bearing various substituents in aromatic rings produce the desired products 296 in yields up to 94%. Interestingly, the reaction with cyclic olefins in various solvent systems affords diastereomers in different ratios. Diazides containing free hydroxy and carboxy functionalities are formed in trace amounts in an organic solvent; however, in water it is possible to obtain them in 63--67% yields. These reaction conditions are also suitable for dienes and allenes; under these conditions, the corresponding products were prepared in moderate yields.
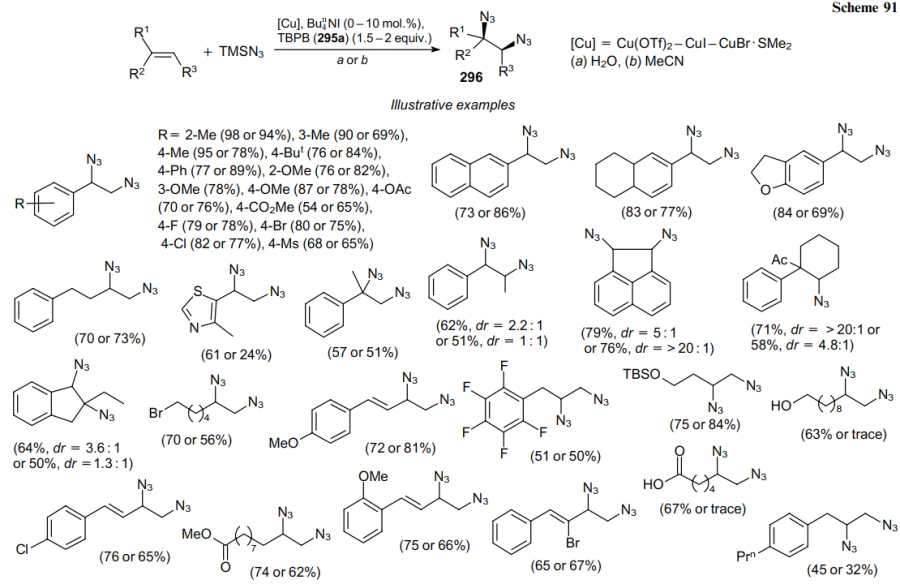
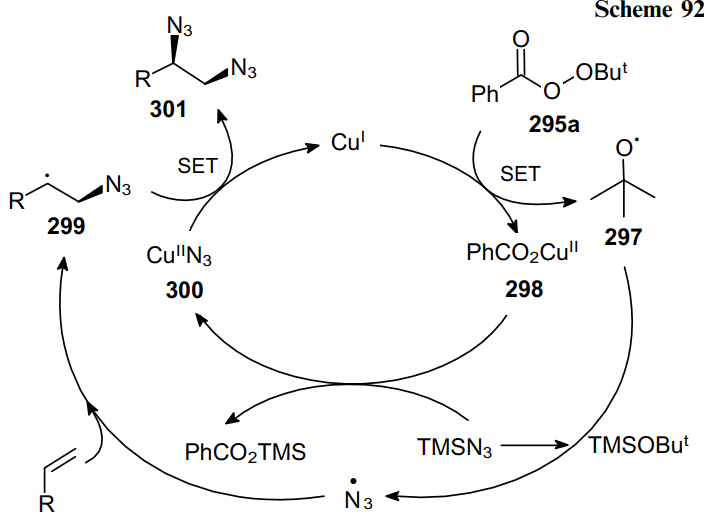
A radical catalytic cycle was proposed to explain the mechanism of this transformation. A single electron transfer (SET) between CuI and tert-butyl peroxybenzoate (295a) initiates the reaction of alkene RCH= CH2 to generate the tert-butoxy radical (297) and CuII species (298). Then a radical relay process occurs between TMSN3 and radical 297 yielding an azide radical, which adds to the starting olefin to give C-radical 299. The ligand exchange produces intermediate CuIIN3 (300), which reacts with internal radical 299 to produce the desired diazidation products 301 and regenerate CuI (Scheme 92).
Iron(II) salts in the presence of pyridine-bis(oxazoline) ligand 302 are another type of catalysts for diazidation.190,191 The use of trimethylsilyl azide--compound 303 catalytic system enables the diastereoselective diazidation of olefins at room temperature (Scheme 93).
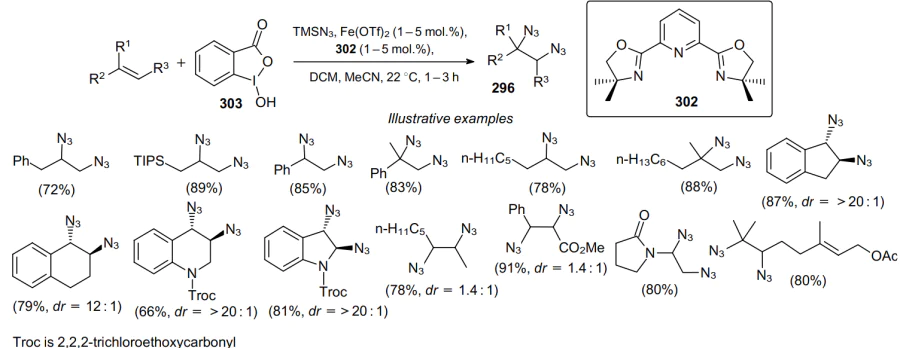
This reaction provides a convenient route to vicinal primary diamines, as well as other synthetically valuable nitrogen-containing building blocks that are otherwise not readily available. To assess the scope of this diazidation method, a broad range of the starting olefins were tested. The target diazides 296 were obtained in yields up to 89%.
The mechanism of this type of diazidation (Scheme 94) involves the formation of azides 288 via the reaction of TMSN3 with compound 303. Azide 288 can be additionally activiated by trimethylsilyl azide to afford diazide 304, which undergoes the I–N3 bond cleavage in the presence of a Fe catalyst and presumably generates high-valent iron species and an azide radical. Thus, the azide radical can react, e.g., with cis-1,2-diphenylethylene (305) to form radical 306, which is bound to the iron complex, and ester 307. Finally, ferric species can further oxidize radical 306 through the azide transfer to the inner sphere of the complex to yield diazide 308.
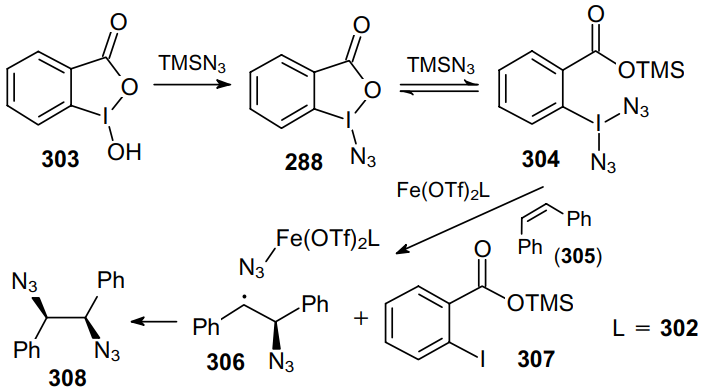
The reported iron-catalyzed direct diazidation192 is based on activation of stable peroxyesters 295 promoted by nitrogen-containing ligands (Scheme 95). In this case, nearly stoichiometric amounts of an oxidant and TMSN3 are sufficient to carry out an efficient diazidation of a broad range of olefins and N-heterocycles at room temperature in the presence of both tridentate (302) and bidentate (309) ligands. It is noteworthy that various substrates including allylic esters and carbamates, β-pinene, highly functionalized indoles, pyrroles (prone to rapid rearomatization), β,γ-unsaturated ketones enter this reaction to provide very high yields of the corresponding diazides.

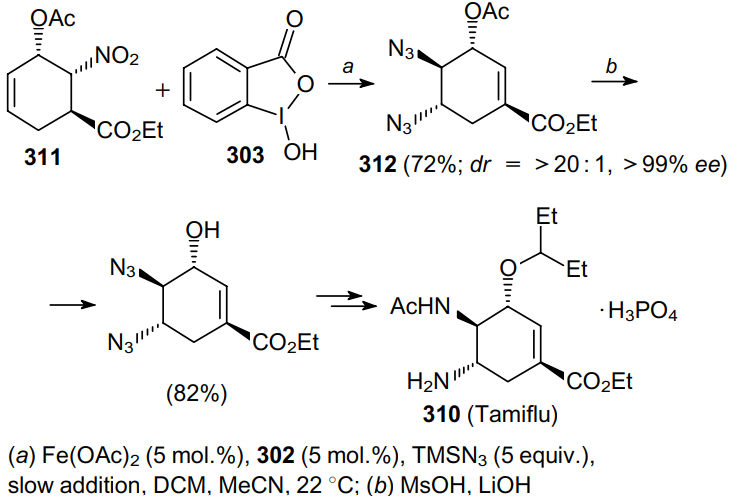
Li et al.193 carried out an enantioselective synthesis of the antiviral drug Tamiflu (310), in which the key trans-diamino moiety was formed via the iron(II)-catalyzed stereoselective diazidation of olefin 311 to give product 312 (Scheme 96). This method is efficient even for densely functionalized electronically deactivated substrates, for which the previously developed methods had limitations.
Other metals can also be used to deactivate alkenes. Thus, recently Peng et al.194 reported the oxidative vicinal diazidation of unsaturated compounds catalyzed by Pd(O2CCF3)2 bearing the bidentate ligand [4,7-bis(4-bromopheny)-1,10-phenanthroline (313)] in the presence of an azide source (TMSN3) and an oxidant, N-fluorobenzenesulfonimide (NFSI, 314) (Scheme 97). Styrenes 289b--f with various substituents at the para position proved to be suitable substrates to prepare the corresponding vicinal alkyl diazides 290b--f. In the case of unactivated terminal alkenes 315a,b, the addition of a small amount of p-nitrobenzoic acid and an increase in the amount of the catalyst to 5 mol.% are required, which improve the yields of products 316a,b up to 83%.
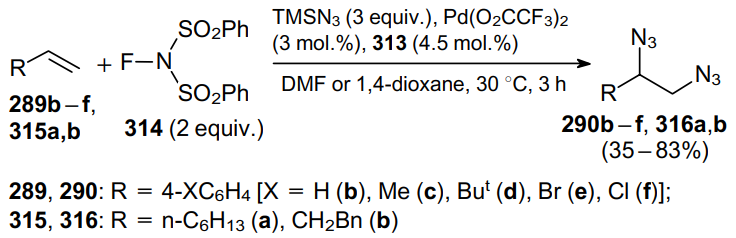
Using this approach, the corresponding trans-diazides 318a--h can be obtained from cyclic alkenes 317a--h (Scheme 98).
Originally, 1H-indene gave diazide 318a in 70% yield with a 2:1 diastereomeric ratio under standard conditions. However, the diastereoselectivity was improved to dr=10:1 by adding water as a cosolvent. A similar result was observed for 1,2-dihydronaphthalene 317b. It should be noted that reactions of 1,2-dihydroquinoline and 2H-chromene afforded trans-isomeric products 318c,d as single isomers in moderate yields. Interestingly, the reaction of α-phenylcyclopentene also showed an excellent diastereoselectivity, since it provides a single isomer 318e in 60% yield. Unactivated cyclic alkenes containing five--eight-membered rings were found to give products 318f--h in good yields with moderate diastereoselectivity dr=(3-5):1.
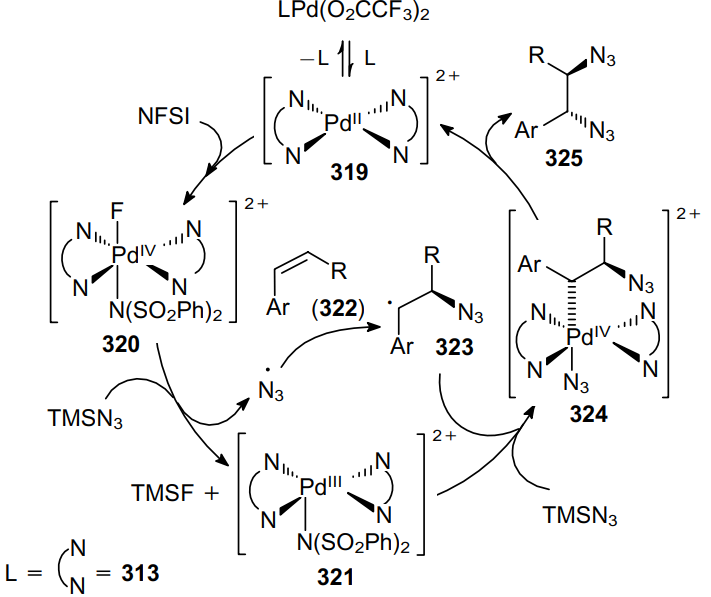
The mechanism of these transformations involves the oxidation of cationic complex 319 with N-fluorobenzenesulfonimide (314) to give complex 320. The latter oxidizes TMSN3 via a single electron transfer to generate an azide radical and complex 321. Then the azide radical adds to alkene 322 to yield C-radical 323, which reacts with intermediate 321 to afford PdIV compound 324. The subsequent ligand exchange and reductive elimination give rise to vicinal diazide 325 (Scheme 99).
3.1.2.4. Photochemical 1,2-diazidation of olefins
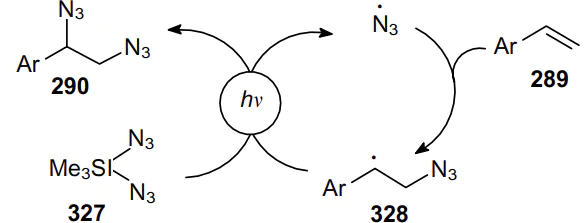
The double azidation of alkenes can be carried out photochemically without the use of photoredox or metal catalysts.195 The reaction proceeds stereospecifically under visible light irradiation and involves sulfonium bis(acetoxy)iodate(I) (Me3SI(OAc)2, 326) and sodium azide as the radical source. Various vinylarenes and unactivated alkenes readily undergo 1,2-diazidation. It is important to note that sterically hindered 2,4,6-trimethylstyrene forms a diazide in high yield. Under these conditions, the diazidation of 2-vinylpyridine and 2-vinylnaphthalene was successfully carried out (75--78% yields). Indene and 1,2-dihydronaphthalene are converted into the corresponding vicinal syn-1,2-diazides in 72--77% yields with good diastereoselectivity (dr=96:4) (Scheme 100).
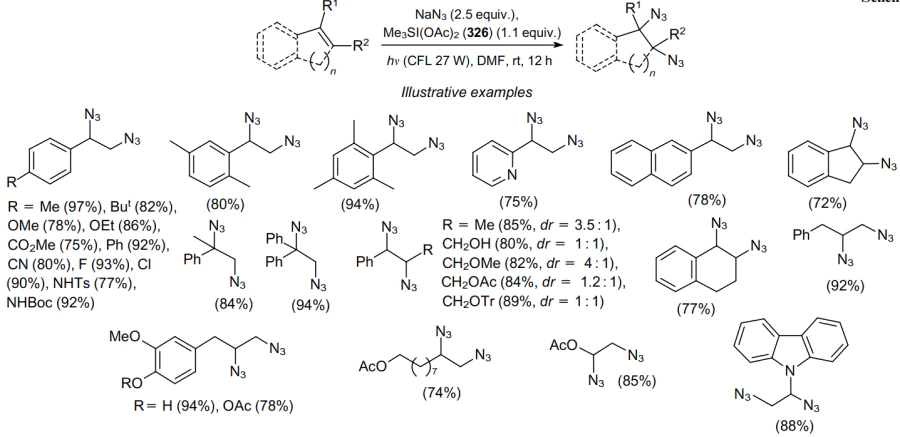
The mechanism of this transformation involves the in situ formation of Me3SI(N3)2 (327) and its subsequent visible light-induced homolysis. In this case, an electrophilic azidyl radical is generated, which attacks the C= C bond, e.g., of alkene 289 to give benzylic radical 328. Then the azide radical derived from an iodonium salt adds to radical 328 to yield diazide 290 and a free azide radical, which can reattack the alkene molecule (Scheme 101).
3.1.2.5. Electrochemical 1,2-diazidation of olefins
One of the electrochemical diazidation methods reported in the literature is a double azidation using a reticulated vitreous carbon anode in 0.1M LiClO4 solution in MeCN with an applied cell potential (Ecell) of 2.3 V. Manganese(II) bromide was used served as the catalyst; acetic acid, as the proton source.196,197 This protocol is applicable to diazidation of a broad range of alkenes (Scheme 102). For example, aryl-substituted acyclic alkenes give the corresponding 1,2-diazides in 66--95% yields.
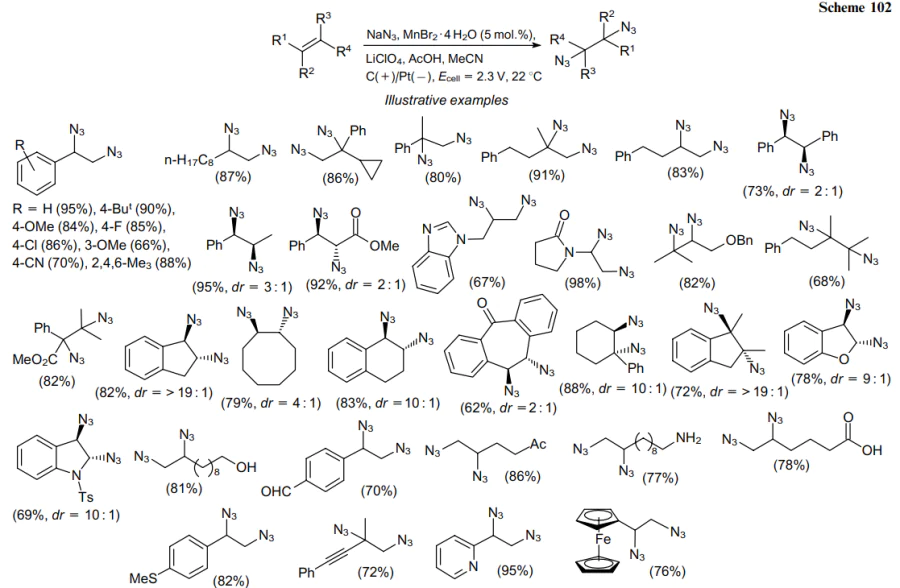
1,2-Diazides can be produced from terminal and also 1,1- and 1,2-disubstituted olefins under these conditions. Cyclic alkenes are also suitable substrates for this reaction, forming mainly trans-1,2-diazides in high yields with good diastereoselectivity. Electron-rich benzofuran and N-tosylindole were dearomatized to 1,2-diazides in yields of 78 and 69%, respectively. Substrates bearing hydroxy, carbonyl, carboxy, amine, sulfide or alkyne moieties, which react without undesired side reactions, are suitable for this reaction system.
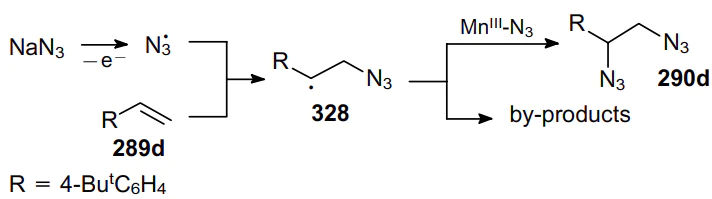
It is believed that the anodic generation of an azide radical followed by its addition to the double bond of, e.g., alkene 289d and to intermediate radical adduct 328 results in the formation of two new C–N bonds and gives rise to vicinal diazide 290d (Scheme 103). This catalytic system combines high reactivity and good chemoselectivity.198
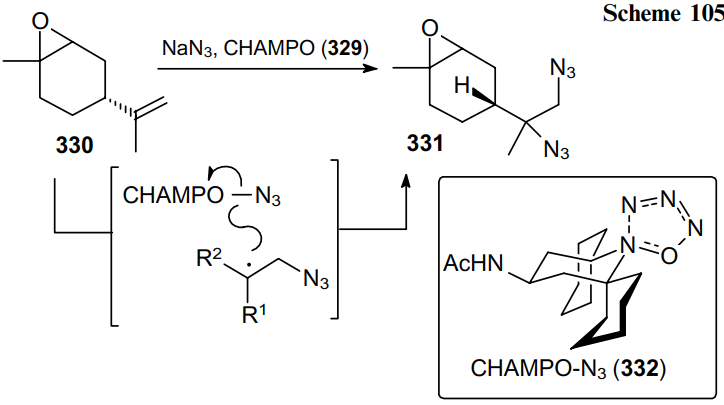
Recently, the aminoxyl catalyst CHAMPO (329) has been developed for the electrochemical diazidation of a wide range of alkenes to produce the corresponding diazides (Scheme 104).199
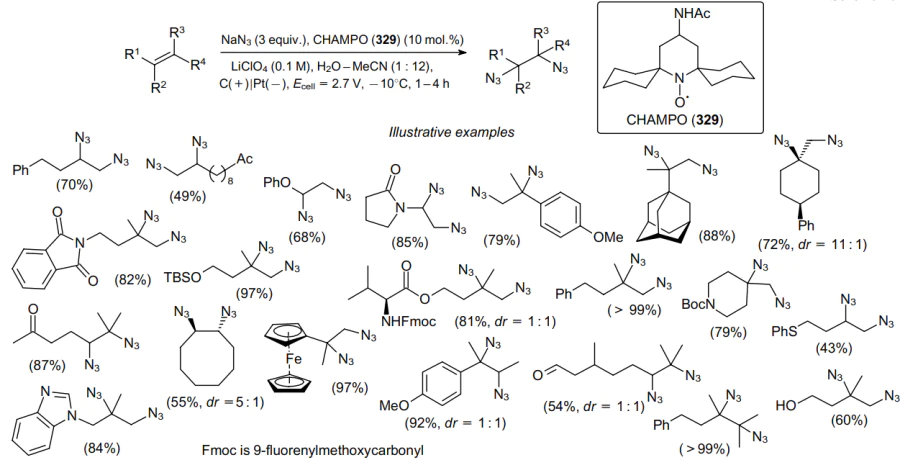
The radical diazidation of alkene 330 to product 331 by an anodically generated charge-transfer complex in the form of CHAMPO-N3 (332) was achieved without using transition metal complexes or oxidants (Scheme 105).199
3.2. Synthetic approaches to vicinal diazidoalkenes
Literature data on the preparation of 1,2-diazidoalkenes are limited to only two examples including the substitution of 1,2-diboronic esters involving CuII species and the treatment of propargyl azides with an alternative azide source.
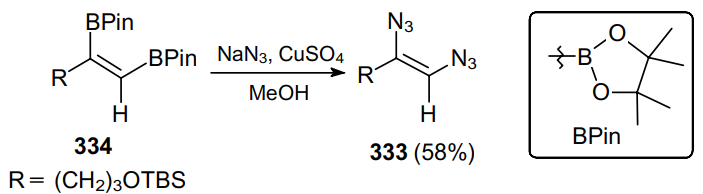
Mali et al.200 developed a versatile stereoselective route to the synthesis of (Z)-1,2-diazidoalkenes 333 from the appropriate 1,2-diboronic esters 334 via the copper-mediated reaction with sodium azide (Scheme 106). This transformation occurs with the retention of stereochemistry of the original double bond. The mechanism of this transformation was not considered.
Open-chain 1,2-diazidoethenes 335--338 were obtained from propargyl azides 339, 340, bearing an acceptor substituent at the triple bond by the reaction with tetramethylguanidinium azide (TMGA, 341) (Scheme 107).
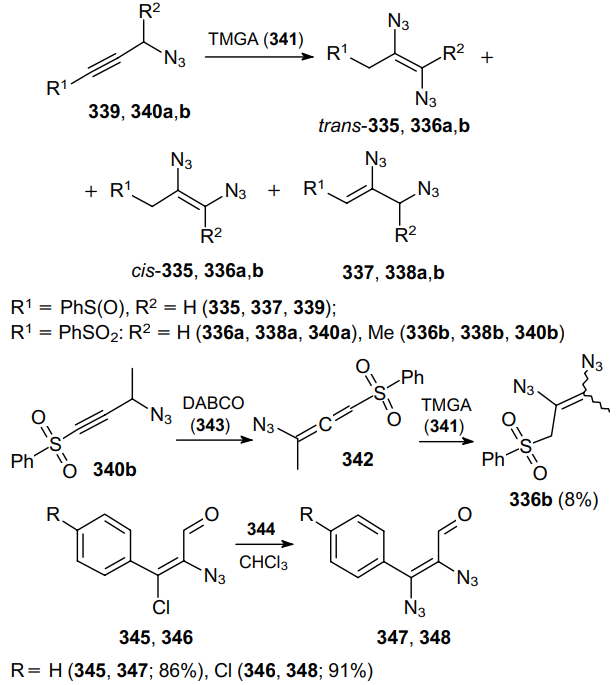
In some cases, such products are prepared via a two-step procedure. The transformation sequence begins with the in situ formation of the corresponding azidoallene 342 in the presence of 1,4-diazabicyclo[2.2.2]octane (DABCO, 343) followed by the addition of hydrazoic acid derived from TMGA.201 These diazides are extremely unstable; however, the introduction of electron-withdrawing substituents and the use of another N3 source, tri-n-butylhexadecylphosphonium azide (344), allows the synthesis of cis-diazide products 347, 348 from cinnamic aldehydes 345, 346 in high yields.
3.3. Synthesis of aromatic and heteroaromatic vicinal diazides
This Section considers approaches to the synthesis of aromatic and heteroaromatic (pyridinic, pyrrolic, etc.) 1,2-diazides via the nucleophilic substitution and diazo transfer reactions.
3.3.1. Synthesis of aromatic 1,2-diazides
3.3.1.1. Substitution reactions
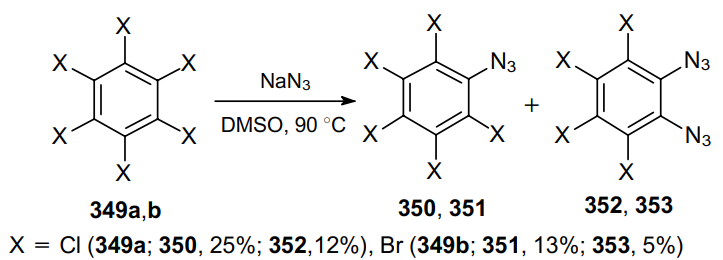
Perchloro- and perbromobenzenes 349a,b undergo selective dehalogenation when heated with an excess of sodium azide in dimethyl sulfoxide; the resulting mixture contains mainly monoazide (350, 351) and 1,2-diazide (352, 353) (Scheme 108).202
As part of the study on the reactivity of hexakis[4-(dimethyl)pyridinio]benzene hexakis(trifluoromethanesulfonate) (354) towards various nucleophiles, Weiss et al.203 treated a suspension of this compound in acetonitrile with azide ions and obtained salt 355 (Scheme 109).
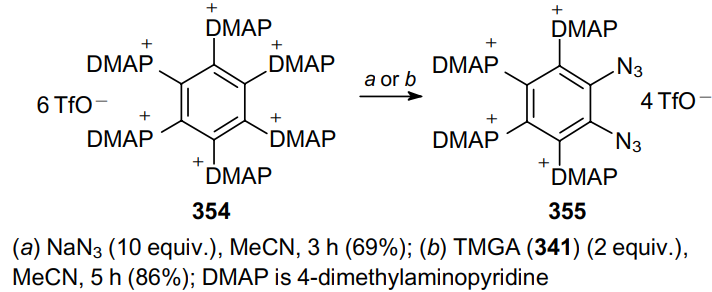
Sodium azide and TMGA (341) were used as azide sources. In the case of sodium azide, the amount of exchanged onio ligands can be affected by the presence of water, which reduces the nucleophilicity of the azide ions due to solvation. The use of TMGA as the azide source makes it possible to control the degree of onio exchange via stoichiometry. Reactions with NaN3 often produces mixtures of mono- and diazides, as well as sodium triflate, which is difficult to remove from the reaction mixture. Therefore, TMGA is a more preferable azide source.
Treating of 7-azide-8,9-dinitro-2,3,4,5-tetrahydrobenzo[b][1,4]-dioxocin (356) with an excess of NaN3 in DMSO results in the substitution of the nitro moiety to afford 7,8-diazide 357 in good yield (Scheme 110).204
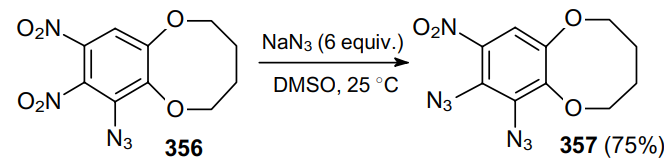
The reaction of benzo-1,2,3,4-tetrazine 1,3-dioxide 358 with sodium azide in aqueous acetone gives triazide 359 (88% yield).205 Its subsequent treatment with NaN3 (1 equiv.) in DMF produces the corresponding tetraazide 360 (Scheme 111).
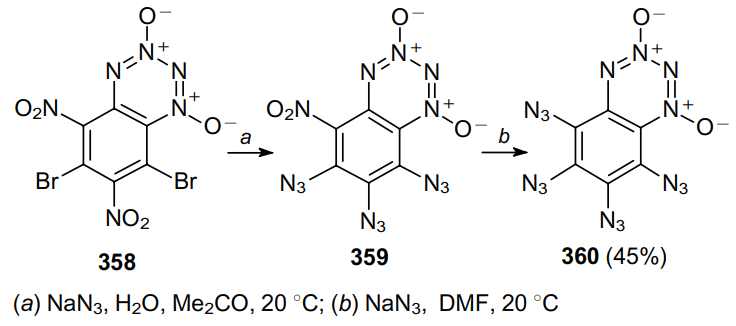
Adams and Moje206 used monoazide derivative 361 to prepare 2,3-diazido-1,4-naphthalenebenzenesulfonamide 362 in quantitative yield (Scheme 112).
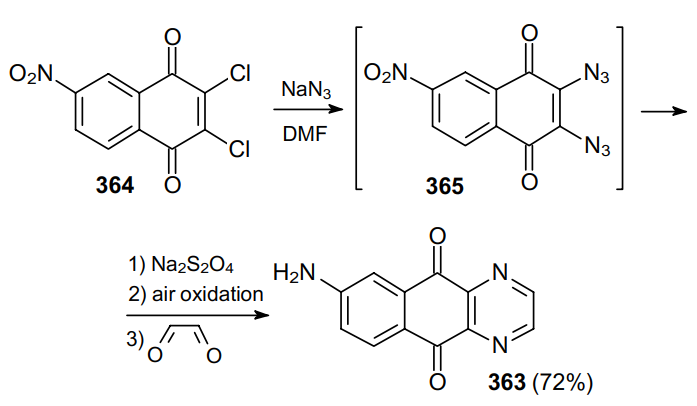
The synthesis of 6-amino-1,4-diazaanthraquinone 363 involves the nucleophilic substitution of dichloronaphthoquinone 364 to produce in situ 2,3-diazido-6-nitronaphthalene-1,4-dione (365) as an intermediate (Scheme 113).207 The resulting diazide is then reduced with sodium dithionite and oxidized with ambient oxygen to the corresponding diamino derivative, which reacts with glyoxal to afford diazaanthraquinone 363. The formation of diazide 365 was confirmed experimentally.208 A number of studies209--220 reported similar examples of the nucleophilic substitution involving dihalobenzoquinone and -naphthoquinone derivatives to afford vicinal diazides.
3.3.1.2. Diazotization and diazo transfer reactions
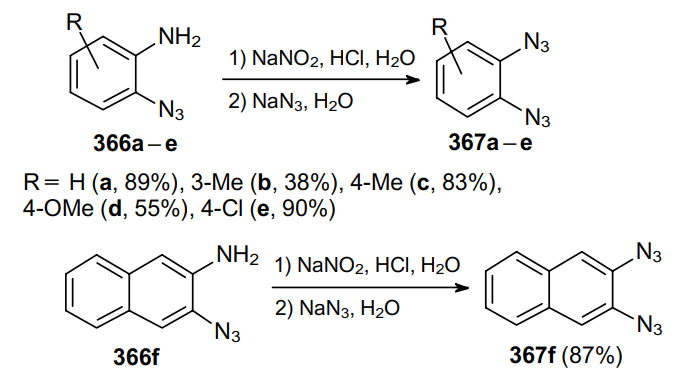
The diazotization of 2-azidoanilines 366a--e followed by the treatment of a diazonium salt solution with sodium azide forms the basis for the conventional synthesis of aromatic vicinal diazides, 1,2-diazidobenzenes 367a--e (Scheme 114).221--223 In a similar way, 2,3-diazidonaphthalene 367f can be prepared from aminoazide 366f in high yield.224
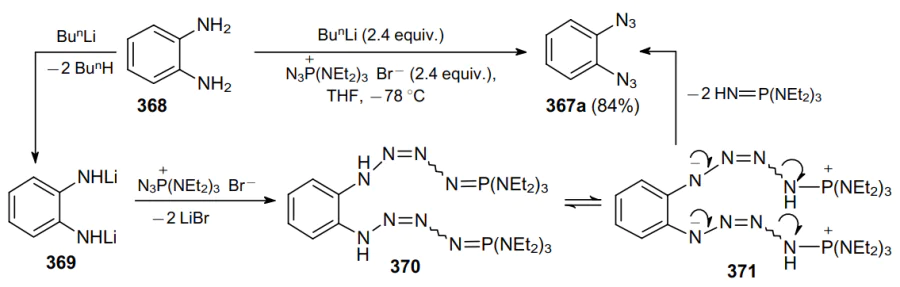
The double diazo transfer reaction was carried out to synthesize 1,2-diazidobenzene 367a from o-phenylenediamine 368 (Scheme 115).225,226 The mechanism of this transformation involves the deprotonation of the substrate giving dilithium derivative 369, which is then converted to intermediate 370 following the addition of azidotris(diethylamino)phosphonium bromide. Subsequently, via the formation of zwitterion 371, two tris(diethylamino)phosphorimine molecules are eliminated to give 1,2-diazide 367a in 84% yield.
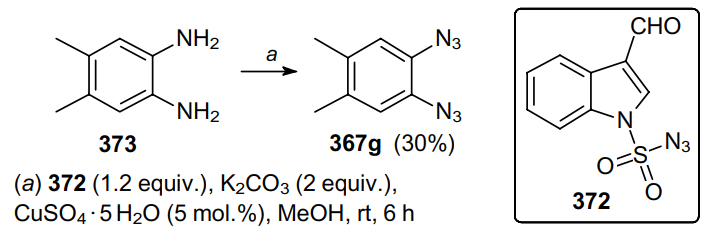
Mishra and Tiwari227 proposed another diazo-transfer reagent, namely, indole-3-carboxaldehyde-1-sulfonyl azide (372), which reacts with primary amines, including vicinal diamine 373, in the presence of CuSO4•5H2O and K2CO3 to yield diazide 367g (Scheme 116).
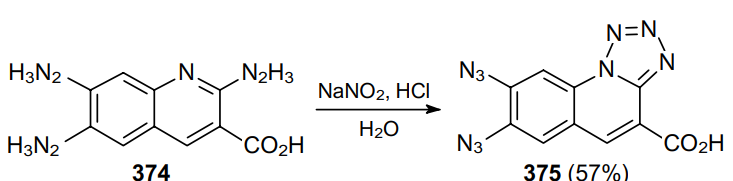
The treatment of 2,6,7-trihydrazinylquinoline-3-carboxylic acid (374) with nitrous acid affords tetrazoloquinoline 375 (Scheme 117).228
The effect of substituents at the positions 2 and 3 of quinoxaline 1,4-dioxides 376a,b on the reactivity of fluorine atoms towards various nucleophiles, including NaN3, was studied.229 Quinoxaline 1,4-dioxide 376a, when heated with an aqueous NaN3 solution (1:1 molar ratio) in acetonitrile, gives a product of monosubstitution of fluorine in 63% yield, while using 2 equiv. of NaN3, diazido derivative 377a is formed. However, the monosubstitution product was not isolated in the reaction of 2,3-dimethyl-substituted quinoxaline 1,4-dioxide 376b with NaN3. These results indicate that an azido functionality facilitates the further substitution of the fluorine atom at the position 6 and, consequently, the second fluorine atom is substituted more rapidly than the first one (Scheme 118).
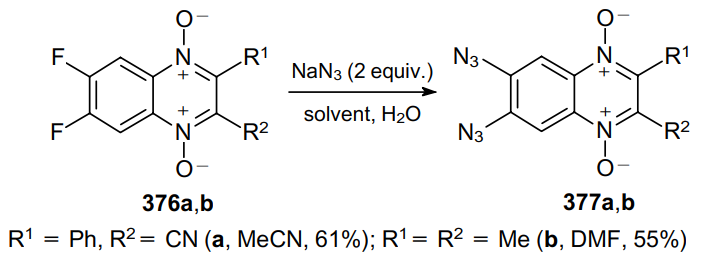
3.3.2. Synthesis of heteroaromatic 1,2-diazides. Substitution reactions
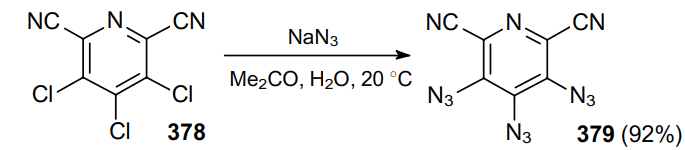
The reaction of trichloropyridine-2,6-dicarbonitrile 378 with an excess of azide in aqueous acetone at room temperature provides a high yield of triazidopyridine 379, a promising component of energetic materials (Scheme 119).230
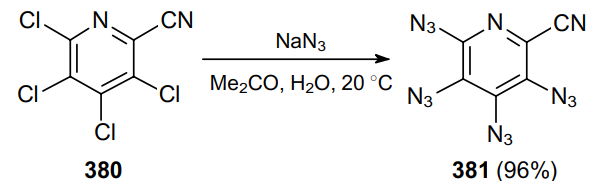
In 2009, the same research group231 prepared tetraazido-substituted pyridine 381 from substrate 380 under identical conditions in almost quantitative yield (Scheme 120).
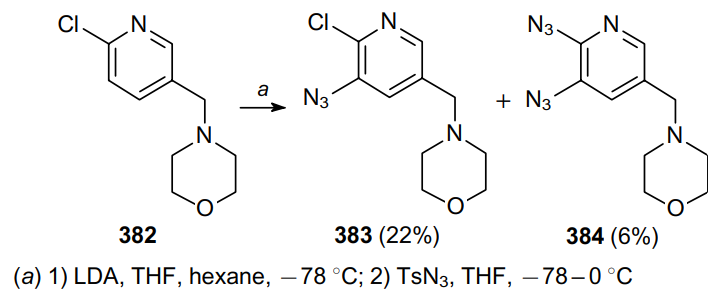
It is also possible to introduce an azide functionality into pyridine via metalation with the followed by the use of tosylamide as an electrophilic agent.232 The starting chloro-substituted pyridine 382 was first lithiated with lithium diisopropylamide (LDA) and then TsN3 was added to the reaction mixture. As a result, azidopyridine 383 and diazidopyridine 384, as a by-product, were obtained (Scheme 121). Diazide 384 is unstable and decomposes within 16 h at room temperature.
Under conventional diazidation conditions, N-aryldichloromaleimides 385 give N-arylmaleimidediazides 386 in high yields (Scheme 122).233,234
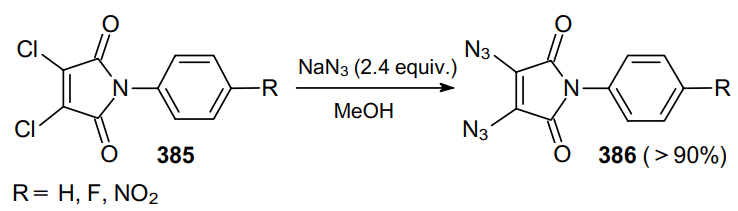
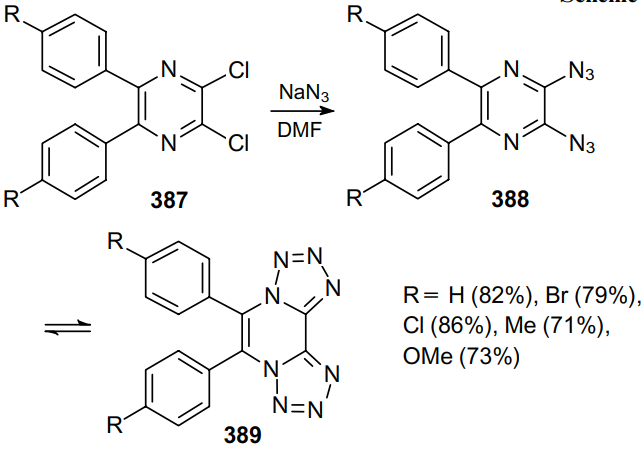
Dichloropyrazines 387 react with NaN3 in DMF to afford 2,3-diazidopyrazines 388 (Scheme 123).235,236 The IR spectra of these products show no signals of an azido moiety. This fact suggests that 2,3-diazidopyrazines exist in the tetrazole form (389) rather than in the diazide one both in solution and in the solid state.
3.4. Reaction of vicinal diazides
3.4.1. Thermal decomposition
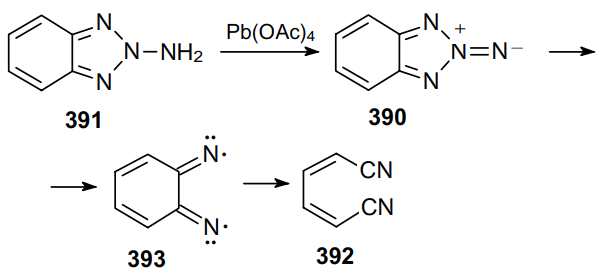
In 1965, Campbell and Rees237 postulated the existence of compound 390 upon the oxidation of 2-aminobenzotriazole (391) with lead tetraacetate. It was proved that it is this compound that is a possible precursor to cis,cis-1,4-dicyanobuta-1,3-diene 392. The N–N bonds of compound 390 are homolytically cleaved to generate diradical 393, which is then converted to dinitrile 392 due to a homolytic cleavage of the corresponding C–C bond. As a result, the configuration of the aromatic ring system is completely retained (Scheme 124).
In 1967, in view of these findings, Hall and Patterson221 developed an efficient method for the preparation of dicyano derivatives based on the thermal decomposition of 1,2-diazido-substituted compounds. Thus, when heated to reflux in decalin, 1,2-diazidobenzenes 367a--e produce cis,cis-1,4-dicyanobuta-1,3-diene (392) and its derivatives 394a--d in high yields (Scheme 125, the positions of the substituents in the product are indicated).
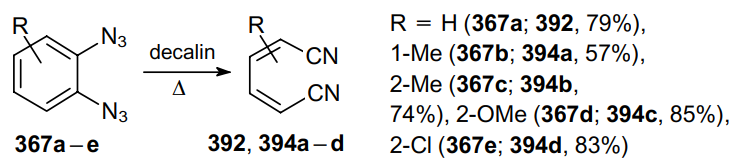
In turn, 1,2-diazidonaphthalene 395 in refluxing decalin gives cis-2-(2-cyanovinyl)benzonitrile (396) in moderate yield (Scheme 126).
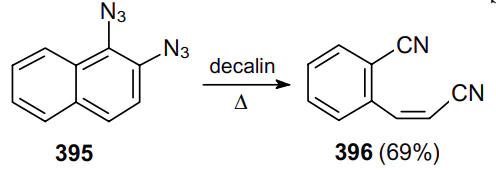
Later, this approach was implemented to synthesize 1,4-diacetoxy-1,4-dicyanobuta-1,3-dienes 397 via aromatic ring cleavage of 1,4-diacetoxy-2,3-diazidobenzenes 398 in refluxing o-dichlorobenzene (Scheme 127).210
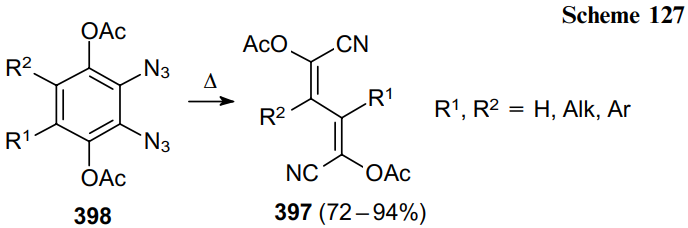
The thermal decomposition of 1,4-diacetoxy-2,3-diazidonaphthalene (399) affords intermediate quinodimethane 400 but the electronic and steric effects lead to electrocyclization of this compound, thereby providing a direct route to the benzocyclobutene ring system. This gives a mixture of isomeric benzocyclobutenes 401 and isoquinoline 402 (Scheme 128).210,238
Noteworthy are examples of the synthesis and transformations of pyrazines bearing two or more azide moieties, e.g., compounds 403 derived from appropriate dichloro derivatives 404. While cyclization of azidopyrazines to imidazoles is a well-studied transformation,239 the possible pyrolytic cyclization of 2,3-diazidopyrazines 403 (see240) into four-membered rings 405 was assumed (Scheme 129). Later,235 a representative series of such diazides were synthesized, and it was shown that their thermal decomposition affords only dicyanoimines 406.
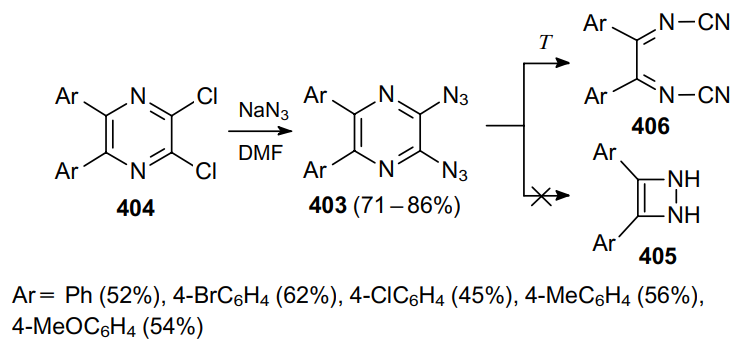
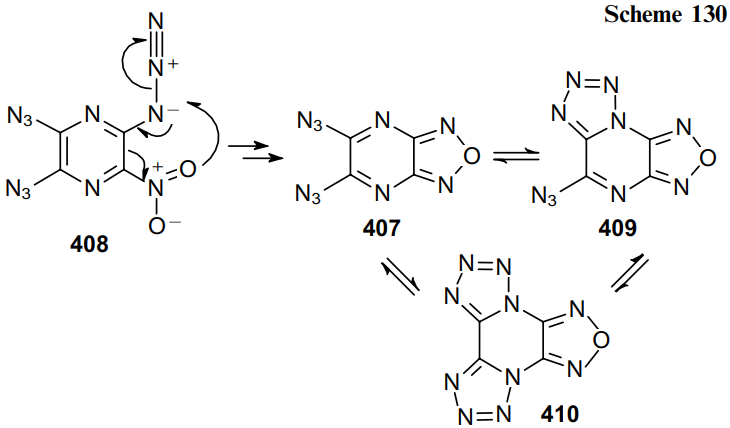
Guillou at al.241 reported the synthesis of diazidofurazanopyrazine 407 from triazidonitropiperazine 408 and the investigation of its tautomerism with forms 409 and 410 using 15N-labelled atoms (Scheme 130).
3.4.2. Photochemical reactions
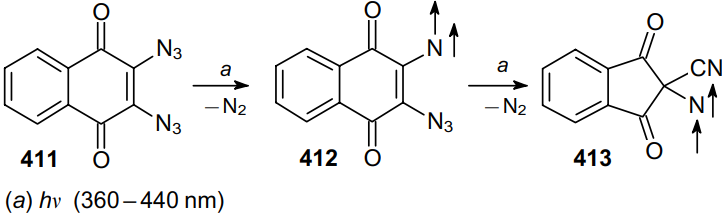
Using EPR and IR spectroscopy, it was shown that under cryogenic conditions, irradiation (with a wavelength of 360--440 nm) of 2,3-diazido-1,4-naphthoquinone (411) affords vinyl nitrene 412, which absorbs another photon to form triplet alkyl nitrene 413 (Scheme 131).211 This process is a striking example of photochemical transformations of 1,2-diazides. Thus, a new method for the preparation of triplet alkyl nitrenes was developed, which have a potential in high-spin assemblies.
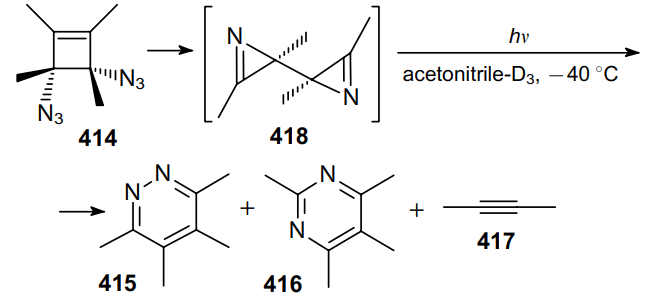
In 2001, Banert and Kлhler242 showed that the photolysis of cis- and trans-1,2-diazidocyclobutenes (e.g., compound 414) in CD3CN at –40 °C produces a mixture of products 415 and 416 along with a trace amount of alkyne 417. This process occurs via intermediate biazirinyl 418 (Scheme 132).
In 2008, it was demonstrated243 that 2-azido-2H-azirines 419 and 420 are intermediates in the photochemical transformation of open-chain 1,2-diazidoethenes 194 to cyano compounds 421 and 422. Intermediates 419 and 420 were identified by NMR spectroscopy at low temperature. Upon prolonged irradiation or heating, such heterocycles lose a second molecule of nitrogen to form two moieties of the corresponding cyano compounds (Scheme 133).

3.4.3. Reactions with organophosphorus compounds
Mao et al.233 studied the reaction of vicinal diazides of N-arylmaleimide 386 with triphenylphosphine (Scheme 134). At room temperature and above, triazole 423 is the major product of this reaction. Also, double Staudinger product 424 is formed in trace amounts, which can readily be obtained at lower temperatures.244 In the presence of methanol or ethanol, compound 423 undergoes a maleimide ring-opening to give the corresponding ester 425. Upon acid hydrolysis, compound 423 eliminates P(O)Ph3 and affords the corresponding amine 426.

In a similar way, triphenylphosphine reacts with naphthoquinone-2,3-diazide 411 derived from dichloride 427 to produce 2-iminophosphorane-1,2,3-triazole 428 in almost quantitative yield.245 In turn, in acidic medium, iminophosphorane 428 is hydrolyzed to free 2-amino-1,2,3-triazole 429. In the presence of moisture, the reaction of compound 411 with PBu3n also furnishes product 429 (38%). Diazide 411 also reacts smoothly with bis(diphenylphosphino)methane to form dimer 430 (Scheme 135). The mechanism of the formation of 2-iminophosphorane-1,2,3-triazole 428 involves the addition of a phosphine molecule to an azide function followed by the elimination of a nitrogen molecule from intermediate 431.223
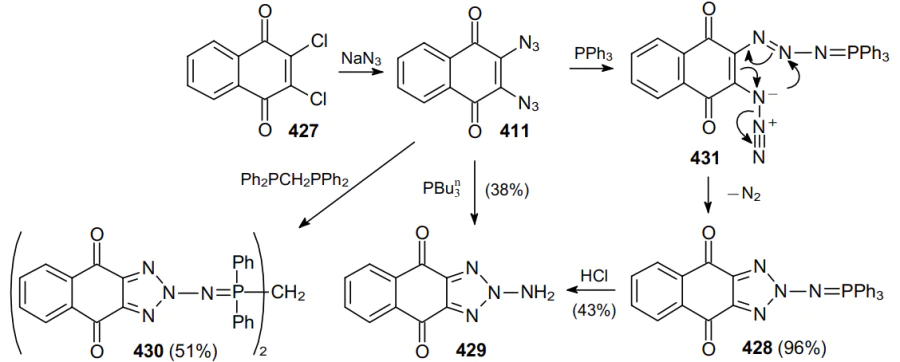
In a similar way, diazides 432 and 433 can provide an access to (iso)quinolinediones 434 and 35 (Scheme 136).246
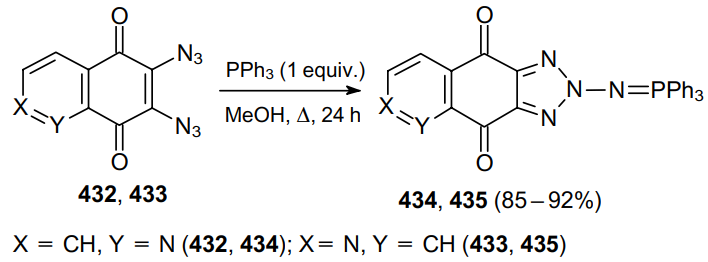
The reaction of an azide moiety with phosphine was used in the synthesis of N,N-ligands based on 2,3-diazido-2,3-dideoxy-α-D-mannopyranose.247 Diazide 436 reacted with dimethylphenylphosphine in toluene to afford iminophosphorane 437. The latter was converted in situ to diimine 438 through the reaction with benzaldehyde (Scheme 137).
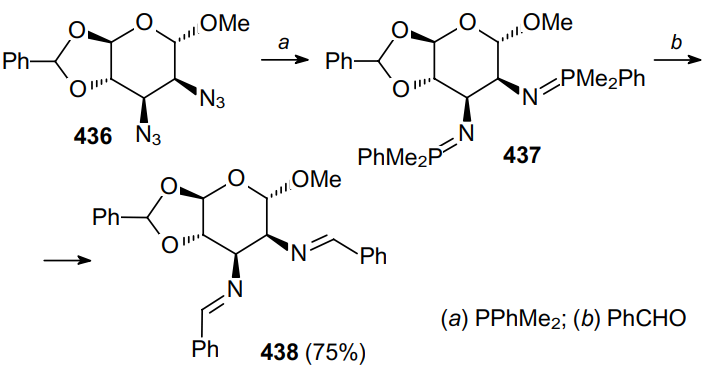
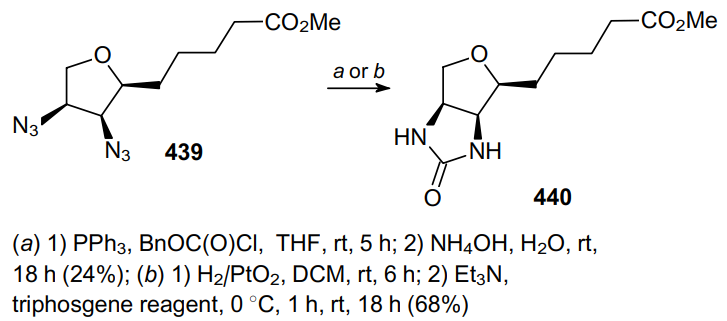
Diazide 439 was used as a precursor in the synthesis of (+)-oxybiotin (Scheme 138). When treated with PPh3 and benzyl chloroformate (method a), it gave the desired imidazolidinone 440 in low yield. It should be noted that the catalytic one-pot reduction of diazide 439 followed by the treatment with triphosgene (method b) significantly improves the yield of the product.248,249 Compound 440, when treated with barium hydroxide under the hydrolysis conditions, is converted to (+)-oxybiotin.
3.4.4. Reduction of vicinal diazides to diamines
In 1980, Saito et al.250 reported the synthesis of potential immunostimulants, hexahydronaphthimidazothiazoles, using cis- and trans-isomeric 1,2-diamino-1,2,3,4-tetrahydronaphthalenes 441 as key intermediates (Scheme 139). The reduction of azide moieties with LiAlH4 in diethyl ether makes it possible to convert vicinal diazides 442 to the corresponding diamines in almost quantitative yields.
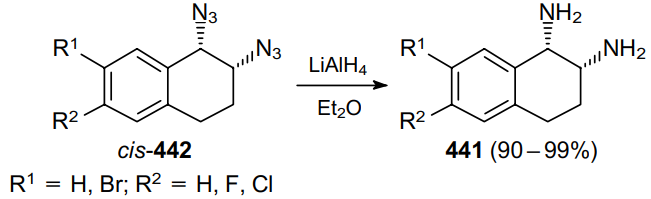
Lithium aluminium hydride was also employed in the final step of the synthesis of methyl-2,3-diamino-4,6-O-benzylidene-2,3-dideoxy-α-D-hexapyranoside ligands, in which diazide 443 was reduced to diamine 444 (Scheme 140).251--253 In a number of studies,254--256 the similar protocol was applied to reduce vicinal diazides.
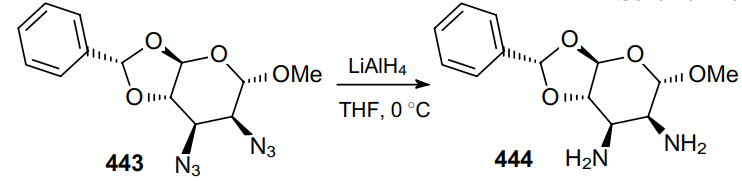
In 2017, (±)-trans-1,2-diazidoindane (445) was used to prepare the corresponding diamine.191 The reduction using the PPh3--H2O system and p-toluenesulfonic acid allowed the preparation of trans-indene diaminium salt 446 in 92% yield (Scheme 141).
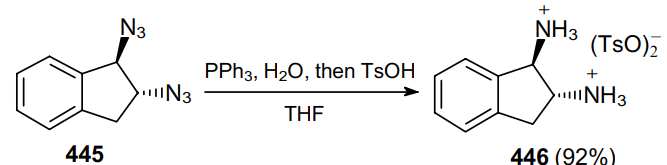
The catalytic hydrogenation of azide moieties in compound 447 in the presence of Adams catalyst (PtO2) in a H2 atmosphere gives bis(amino alcohol) 448 in nearly quantitative yield (Scheme 142).34,137,160,162,249
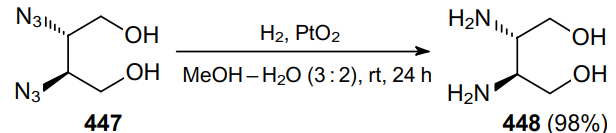
In this case, Adams catalyst is more preferable compared to such systems as Pd/C and Pearlman's catalyst [Pd(OH)2/C], which provide modest yields of the product (20--30%). The protection of hydroxy groups significantly affects the reaction outcome. Thus, O-benzylated diazide 449 is reduced over Pd/C to diamine 450 in quantitative yield (Scheme 143).257
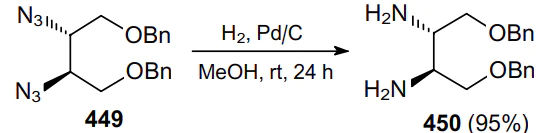
The catalytic reduction of (±)-trans-1,2-diazidoindane 445 with hydrogen in the presence of palladium on carbon in anhydrous ethanol gives (±)-trans-1,2-diaminoindane 451 in quantitative yield (Scheme 144).258 In general, this method of reduction of diazides is suitable for a wide range of diazides and its application is well documented in the literature (see, e.g., Refs 259--267).
3.4.5. Azide-alkyne [3+2]-cycloaddition. Bis(triazoles)
Using classical click chemistry conditions,268 a versatile synthetic approach has recently been developed to prepare bifunctional chelating agents 452 and 453, which are bis(triazole)-based polyaminocarboxylic acids. Thus, the azide--alkyne cycloaddition catalyzed by the Cu(OAc)2--sodium ascorbate system provides the synthesis of the target bis(triazoles) 452 and 453 in high yields from alkyne 454 and diazides 455 and 456, respectively (Scheme 145).
This catalytic system is more preferable than the CuI--diisopropylamine system. Instead of catalysts based on Cu(OAc)2, the CuSO4--sodium ascorbate system is often used.200,269--275 Thus, via the Cu-catalyzed double [3+2]-cycloaddition involving phenylacetylene, cis-diazide 457 gives 1,4-disubstituted cis-bis(triazole) 458 in 59% yield (Scheme 146).184 Diazide 457 can be converted to the corresponding bis(benzotriazole) 459 in 44% yield. The latter case is a non-catalytic version of the [3+2]-cycloaddition, where an alkyne component is formed in situ during the reaction. Similar procedures were described in the publication.186
It was demonstrated that 1,2-diazides 460 and 461, derived from glycals, can be subjected to [3+2]-cycloaddition. Thus, the corresponding bis(triazoles) 463 and 464 were obtained in high yields using phenylacetylene or carbohydrate 462 bearing a terminal alkyne moiety (Scheme 147).185
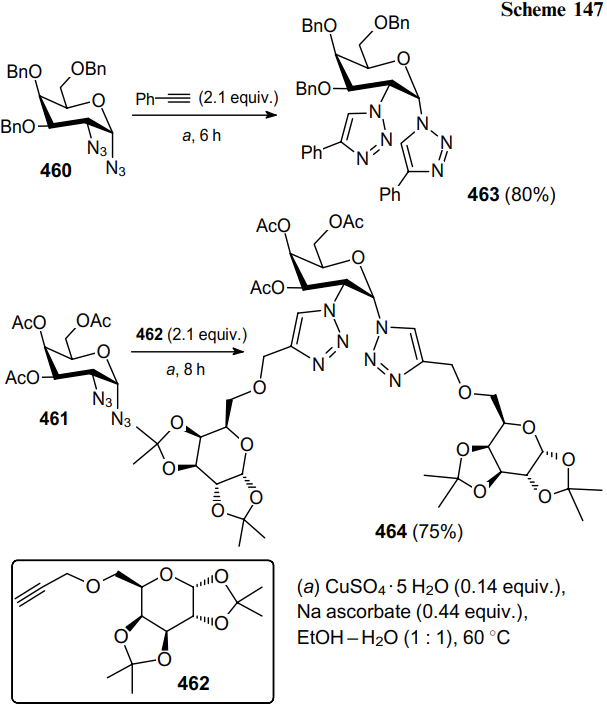
Mali et al.200 described the synthesis of vicinal cis-diazidoalkenes from 1,2-diboronic acid pinacol esters 465. Because of instability of such diazidoalkenes, they were generated in situ using sodium azide and the CuSO4--sodium ascorbate catalytic system. As a result, bis(triazoles) 466 were obtained (Scheme 148). It should be noted that modifying the reaction conditions and switching to another catalytic system provide a convenient route to bis(triazolo)pyrazines 467, a new class of fused heterocycles. This approach to bis(triazolo)pyrazines stimulates the formation of five bonds at once in a single reaction step while retaining all atoms present in the substrates.
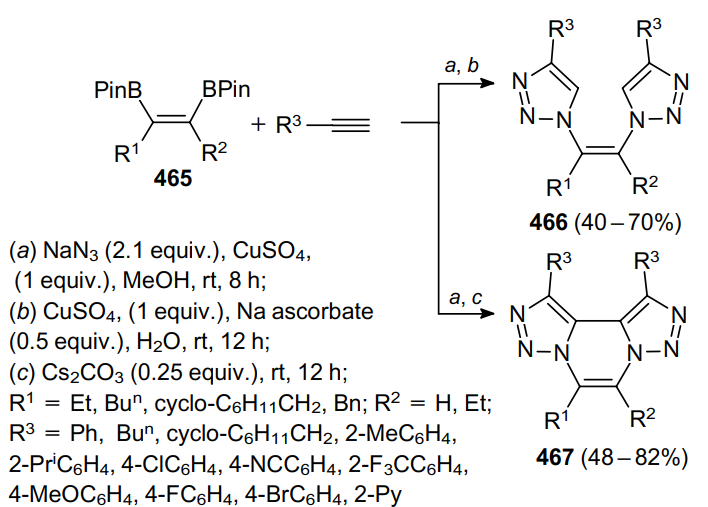
3.5. Vicinal diazidodienes and their heteroanalogues
The main classes of diazides related to compounds of this type can be represented by the general formula 468 (Structure 468). Examples of each class are considered below.
The synthesis of butadienes bearing azide functionalities at the positions 2 and 3 is mentioned in publications.276,277 1,4-Dibromobut-2-yne (469) was used as a starting compound, which reacted with an azide ion to give first bis(diazidomethyl)acetylene (470), being in equilibrium with allene 471, and then 2,3-diazidobuta-1,4-diene (472) (Scheme 149). Structural features of diazidobutadienes were studied in 1986, soon after the first report about the possibility of the synthesis of such compounds.278,279
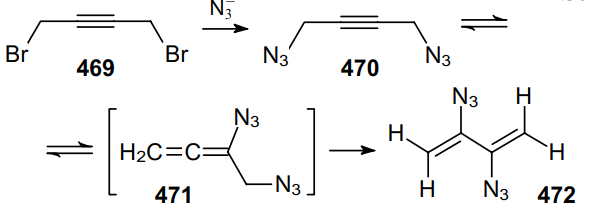
The [4+2]-cycloaddition of a variety of compounds acting as dienophiles to diene 472 was investigated by Banert.280,281 Scheme 150 illustrates the examples of transformations involving 4-phenyl-3H-1,2,4-triazole-3,5(4H)-dione and tetracyanoethylene, resulting in the formation of adducts 473 and 474, which eliminate two nitrogen molecules under photochemical or thermal conditions to furnish products 475 and 476.
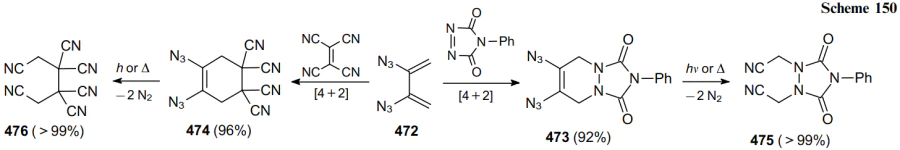
In the same publications, the reactivity of homologues of diazidobutadiene 477--481 in the [4+2]-cycloaddition reaction was studied.
In the case of isomers and homologues 477--479 (Structures 477-481), the [4+2]-cycloaddition to tetracyanoethylene occurs similar to that depicted in Scheme 150, while no reaction is observed for compound 480 (E,E isomer). Homologue 481 bearing four methyl groups is not involved in [4+2]-cycloaddition; however, under these conditions, partial intramolecular cyclization of diazidodiene into azirine 482 occurs. The synthesis of azirines from diazidobutadiene was first reported by Banert in 1985.282 Two years later, this type of reactions, typical of unsaturated azides, was described in details.283 Scheme 151 represents the general sequence of thermal or photochemical destruction of diazidodienes 472 and 481 derived from dihalides 469 and 483. As a result, mono- (482, 484) and diazirines (485, 486) were obtained.
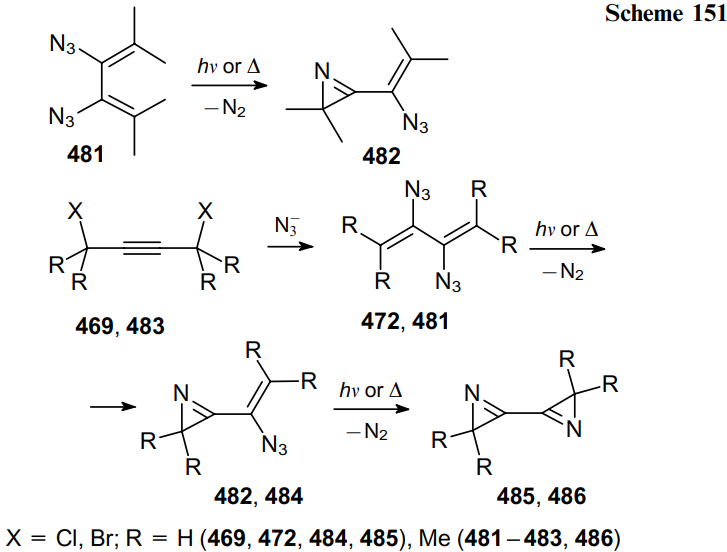
The synthesis of dicarbonyl diazide (oxalyl diazide) 488 from oxalyl dihydrazide 487 and its high explosive sensitivity were first described in 1964.284 In the same publication, selected chemical properties of compound 488 are considered, in particular its conversion to azidocarbonyl isocyanate 489 accompanied by the loss of one nitrogen molecule. The reaction of azide 489 with ethanol affords compound 490, with an azide moiety being intact (Scheme 152). More than 40 years later, diazide 488 was prepared by passing oxalyl chloride vapour through a tube filled with the freshly prepared silver azide.285 The product was not isolated but just identified by photoelectron spectroscopy. Harter et al.286 studied in detail the chemical properties of intermediate compound 489 formed via the thermal decomposition of oxalyl diazide 488. When carrying out this reaction in protic solvents, the newly formed isocyanate 489 reacts with moist air or solvent to give product 491 in high yield. The reaction with hydroxy-containing compounds can involve either one or both azide moieties to form the corresponding products 492 and 493. This strategy was extended to compounds bearing an amino functionality. For example, the reaction of azide 489 with aminotetrazole affords azide 494 though in low yield.
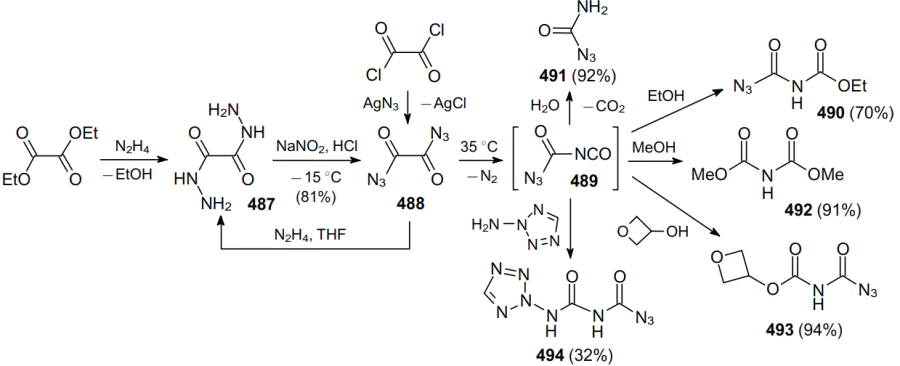
For oxalyl diazide 488, quantum chemical calculations were carried out (DFT, B3LYP/6-311++G(3df)) and the transition energies between its possible A--E conformers were calculated (Scheme 153; the relative energies of the conformers are given in parentheses under the structures; the energies of the transition states are given in parentheses above the arrows). According to the calculations, conformer A is the most energetically favourable, the energy of its transition to the D form is as low as 1.31 kcal mol–1.
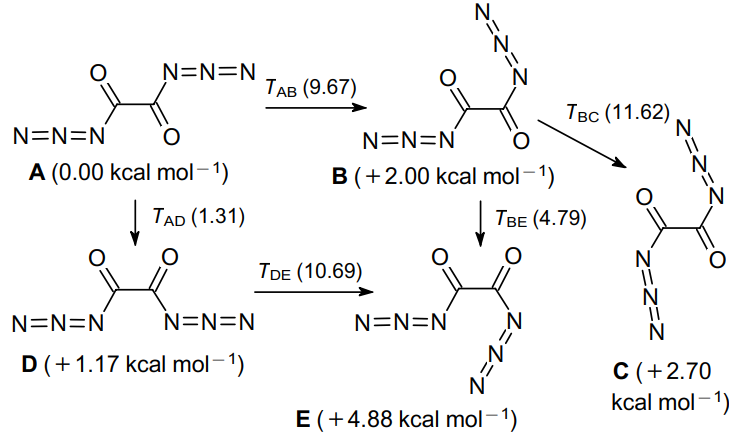
Also reported286 is the synthesis of diazide 495 containing the N3C(O)NHR structural motif (Scheme 154). The results of the study of chemical properties of this diazide were not considered, but the energies of possible tautomeric forms of this compound were assessed (given under the formulas in kcal mol–1), the stability of this compound was evaluated to be higher than that of oxalyl diazide 488.

Worthy of special attention are studies aimed at developing efficient and safe methods for the production of the TKX-50 explosive (Structure 496, Scheme 155). This compound is most commonly synthesized from glyoxal using multistep methods, which involve the preparation of diazidoglyoxime 497 as a key step. Chlorine atoms of compound 498 are very effectively replaced due to the presence of activating substituents. When studying this reaction, Tselinskii et al.287 observed the solvent effect. Thus, in acetone or acetonitrile, the diazide is formed in *50% yield; in aqueous ethanol, the yield is 76%; in DMF, it is nearly quantitative. Then diazide 497 undergoes tautomerization in an acidic medium to bis(hydroxytetrazole) 499, which can form salts with alkali and alkaline earth metals,288 ammonium289 or hydroxylamine to afford the target explosives.290,291 Patents292,293 address the possibility of combining two intermediate steps into one by using N-halogenated succinimides as halogenating agents for dioxime 500 (see Scheme 155). Besides DMF, DMSO and N-methyl-2-pyrrolidone are also suitable solvents for this transformation.

In 2018, the thermal stability of intermediates and TKX-50 was studied using thermogravimetric analysis and differential scanning calorimetry and also the corresponding quantum chemical calculations.294 The safety of the synthetic protocol for compound 496 can be enhanced by adding one more intermediate step, namely, the preliminary protection of hydroxyl groups of dihydroxyglyoxime immediately prior to the replacement of chlorine atoms by azide moieties. In most cases, the tetrahydropyranyl protection is used for this purpose.295--298 However, in 2021 it was proposed299 to protect OH functionalities with 2-methoxyprop-1-ene and to isolate intermediate diazide 501 (Scheme 156).
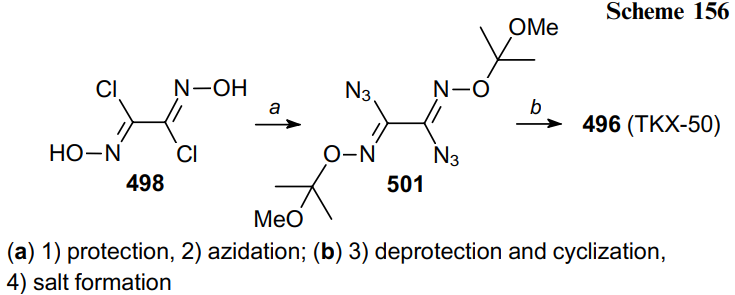
Diazidoglyoxime 497 can be acylated by the treatment with acetic anhydride or acetyl chloride.287 In the latter case, a mixture of diacetylated diazidoglyoxime 502 and bis(tetrazole) derivative 503 was obtained as a major reaction product (Scheme 157).
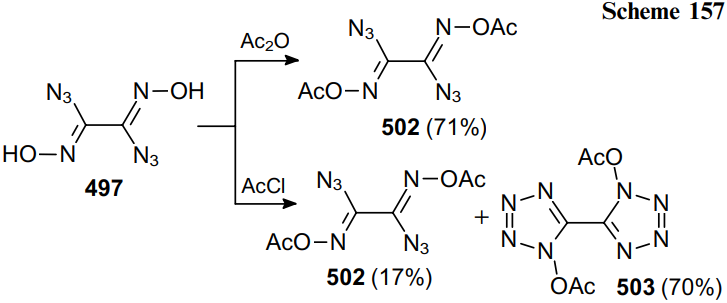
Over the past decade, publications have emerged on the synthesis of 1,1ʹ-dinitramino-5,5ʹ-bis(tetrazole) (DNABT, 504) (Scheme 158), its salts and derivatives, which are explosives.300--303 The complete scheme for the synthesis of diammonium salt of DNABT from dimethyl carbonate (505), hydrazine and glyoxal was reported in the study.301 Diazobutadiene 506 and diazide 507 were isolated as intermediates, the latter compound being cyclized into bis(tetrazole) derivative 508 in an acidic medium. Its nitration gives compound 509, which is transformed into product 504. A similar scheme is used to prepare dipotassium salt of DNABT.303
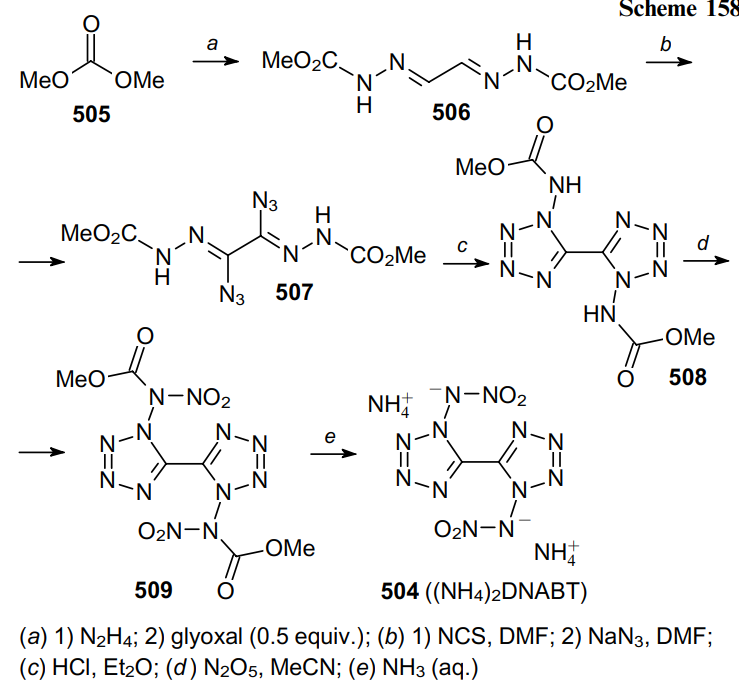
4. Organic tri-, tetra- and polyazides. Synthesis, properties and application
Cationic triazide C(N3)3+SbCl6- was first synthesized in 1966.304 Using IR spectroscopy, it was established that the newly formed triazidocarbenium cation C(N3)3+ has C3v-symmetry, with the central carbon atom lying in the same plane with three neighbouring nitrogen atoms. The cationic part of this compound is isoelectronic with diazide NC-N=C(N3)2, which can be prepared from sodium azide and cyanogen bromide (BrCN).130 The triazidocarbenium cation C(N3)3+, being highly endothermic, forms the corresponding energetic salts with such anions as N(NO2)2– and ClO4-. Also, the salt C(N3)3+BF4- was prepared.305 Electronic structure calculations carried out for the C(N3)3+ cation showed that in all cases the planar structure with C3v symmetry has the minimum energy. The heats of formation of C(N3)3+N(NO2)2– and C(N3)3+ClO4- were evaluated as endothermic with values of 252 and 218 kcal mol–1, respectively, which explains the high explosiveness of these compounds. Despite the fact that the sensitivity of triazidocarbenium salts to heat and impact is too high for practical use, such compounds represent a new class of materials with high-energy density and demonstrate that endothermicity up to 1 kcal g–1 is quite achievable for ionic solids.
As for organic tri-, tetra- and polyazides, aliphatic, alkenyl and aromatic representatives of this class of compounds are known to date.
4.1. Aliphatic tri-, tetra- and polyazides
Among alkyl derivatives of polyazides (Structures 183, 510-512), deserve attention.
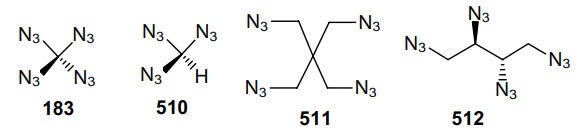
Triazidomethane (510) was first mentioned by Hassner et al.306 in 1990. The procedure for the preparation of this compound from bromoform involves the replacement of bromine atoms with azide groups using azide-containing polymer 513, which was prepared from the anion exchange resin Amberlite IR-400 and sodium azide (Scheme 159). It is important to note that the authors of the cited study did not use an individual compound but an ethereal solution of a mixture of the starting bromoform and supposedly triazidomethane, taking diethyl ether:CHBr3:510 in the 10:34:35 ratio. The formation of the product was confirmed by gas chromatography and NMR spectroscopy; the transfer of this solution into the ampoule may cause an explosion.
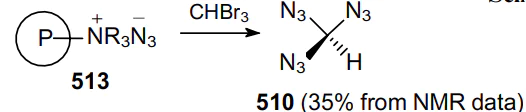
More than 25 years later, in 2007, Banert et al.126 reported the results of the synthesis and studies on the chemical properties of tetraazidomethane (183). A variety of the starting halogenated organic reagents were tested but the desired tetraazidomethane was isolated only using trichloroacetonitrile (Scheme 160). Unfortunately, the authors did not report the yield of tetraazide 183 but noted that the resulting compound is highly explosive. Nevertheless, this tetraazide was characterized by 15N NMR spectroscopy and was shown to have rich chemistry. Thus, it can give rise to products 3, 15N-183 and 514--521 (Scheme 161).
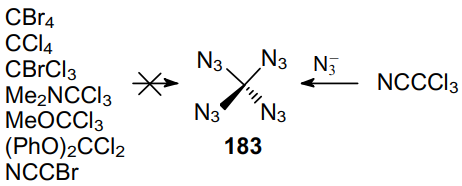
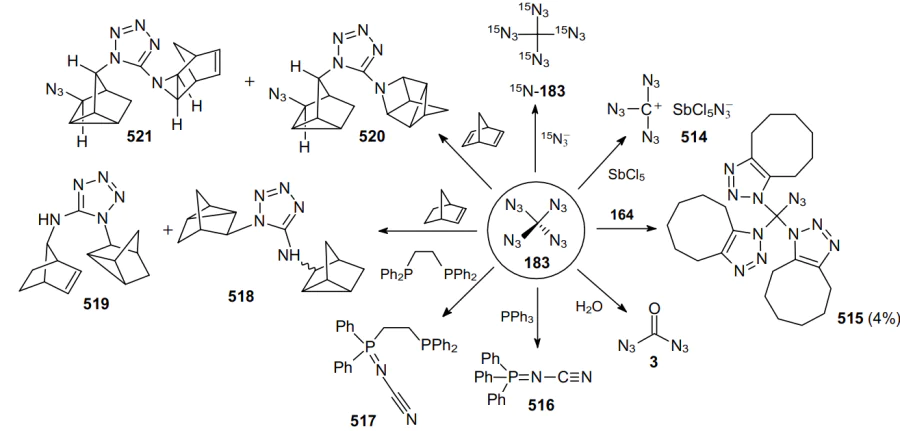
Tetraazide 183 reacts with antimony pentachloride to form salt 514, in which the above-mentioned triazidocarbenium cation acts as a cation. Also worthy of note are the hydrolysis of tetraazidomethane to diazidophosgene (3), its reactions with phosphorus(III) compounds to afford cyanophosphazenes 516 and 517, respectively, and also the cycloaddition involving norbornene and dicyclopentadiene to form disubstituted tetrazoles 518--521. Tetraazide 183 can be detected using cyclooctyne (164) as a molecular trap to yield stable tris(triazole) 515. The subsequent reaction of the latter compound with cyclooctyne (164) leading to the corresponding tetrakis(tetrazole) 522 (Scheme 162) is also possible.126
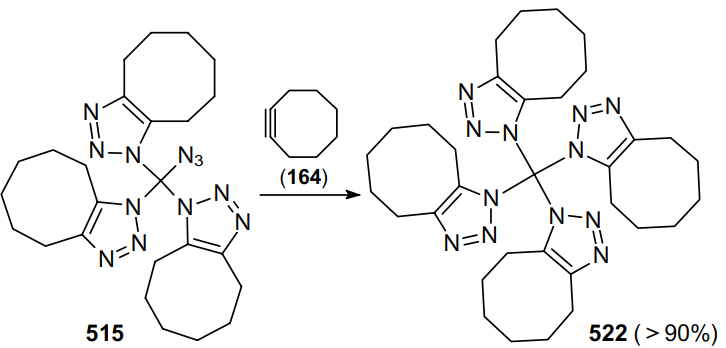
In turn, the reaction of tetraazidomethane with norbornene and dicyclopentadiene affords mixtures of aminotetrazole derivatives. Using the reaction with norbornene, the reaction pathway for chemical transformations occurring under these conditions was proposed (Scheme 163).126
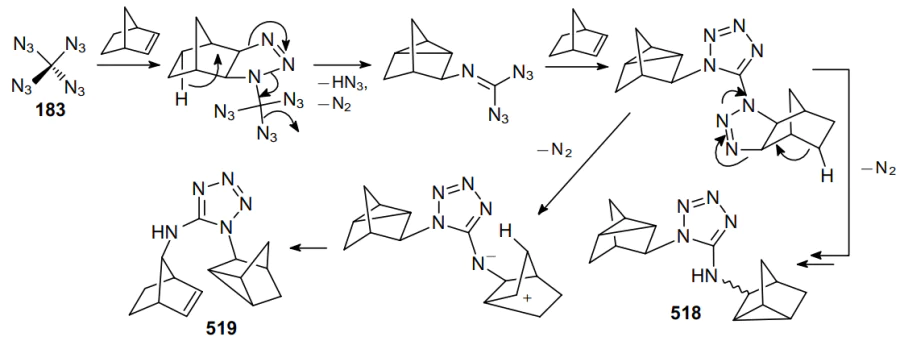
Lyssenko et al.307 studied the structural features of pentaerythrityl tetraazide (511). They showed that the formation of short contacts between azide moieties in crystals is not typical of this polyazide. Therefore, the assembling of molecules in the solid state for such rigid molecular framework gives rise to a structure with high impact sensitivity, since even small deformations of the crystal structure cause shortening of favourable intermolecular contacts. The synthesis of several azido-containing pentaerythritols is presented in a patent.308 Polynitramine compounds based on tetraazide 511 were produced. In particular, pentaerythrityl tetranitramine 526 was obtained via the formation of derivatives 523--525 (Scheme 164).309 Compound 526 has high density and thermally decomposes at 183 °C; its chemical, thermal and calculated detonation properties were described.309
Azide and nitrate derivatives of pentaerythrytol 527--530 were prepared in good yields based on the azide exchange of appropriate bromo derivatives. Tetraazide 511 was the most stable in this series.310 Thermal stability of organic nitrates is limited to 180 °C, while this tetraazide is stable up to 218 °C. The study showed that organic azides are potential candidates for use as novel initiating or melt cast explosives although their thermal or mechanical properties are generally insufficient for practical applications.Structures 511,527-530
This approach, involving a combination of different functional groups in one molecule, makes it possible to tune the chemical and physical properties of organic compounds. It can help, for example, in the development of novel explosives, the specifications of which are based on certain characteristics of the substance.311 Thus, an interesting pattern of changes in the properties of compounds 512, 531--534 (Structures 512,531-534) is given below.
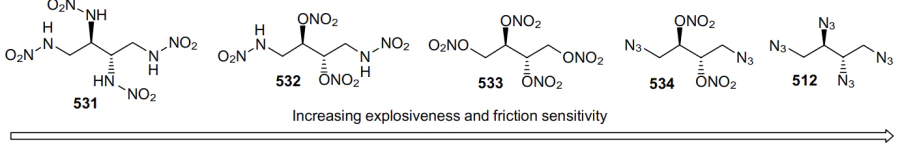
The synthesis of a number of nitro- and azido-functionalized compounds for their possible use as energetic plasticizers was described in the study.312 Tetraazide 535 can be prepared in four steps from glycolic acid; in the final step, the dimerization is carried out in the presence of triazine 536 (Scheme 165).
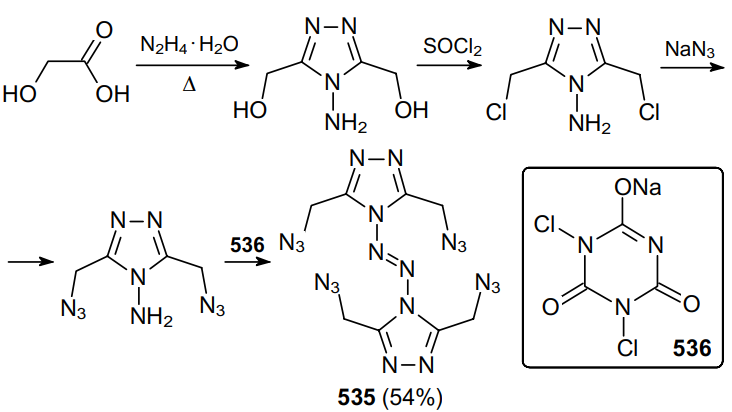
Four novel polyazidomethyl (Structures 537-540) based on heterocyclic compounds were prepared and characterized.313
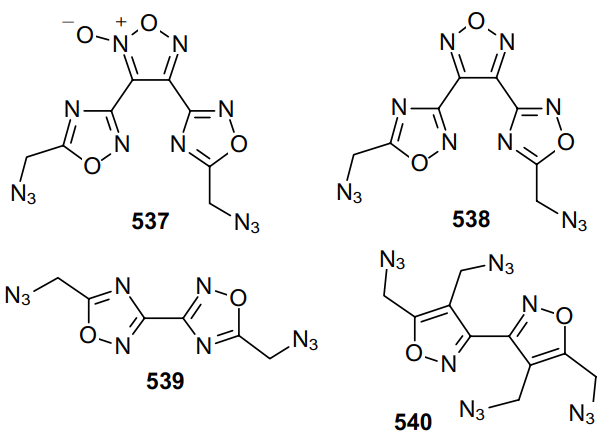
Compounds 537, 538 and 540 possess suitable properties as energetic plasticizers. The synthesis of tetraazide 540 from dichloroglyoxime deserves attention (Scheme 166). In the final step, chlorine atoms are replaced with azide moieties.
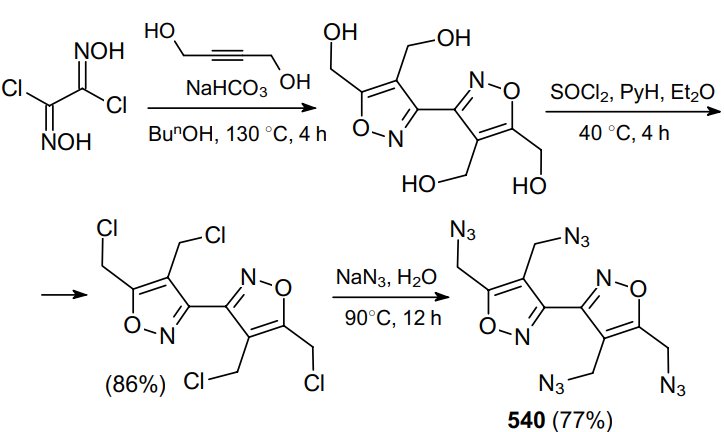
The synthesis from 1,2,4,5-tetramethylbenzene 541 through the formation of polybromide 542 afforded 1,2,4,5-tetrakis(diazidomethyl)benzene (543), which was studied by X-ray diffraction analysis (Scheme 167).314 This compound appeared to be more stable in the solid state than expected. It is of interest both for high-energy experiments and as a precursor for the synthesis of carbon nanotubes, nanospheres and high-nitrogen carbon nitrides with great potential in biological and technological applications.
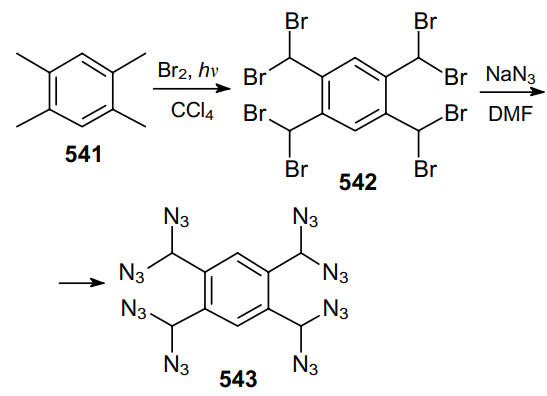
4.2. Alkenyl and carbonyl tri-, tetra- and polyazides
Alkenyl tri- and tetraazides 544--547 (Structures 544-547) were studied by quantum chemical calculations and their structures were optimized; however, these compounds were not synthesized.315
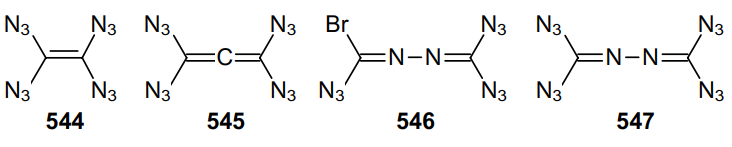
It was shown that triazide 548 can be synthesized using triaminoguanidinium chloride (549) as the starting compound (Scheme 168).316 The treatment of the latter compound with two equivalents of sodium nitrite under acidic conditions affords diazide 550, which undergoes spontaneous cyclization to 5-azido-1-aminotetrazole (551).317 The treatment with an aqueous solution of sodium hydroxide leads to the dimerization of salt 550.316 Therefore, dimer 552 bearing four azido moieties is a precursor to triazide 548. The structures of triazide 548 and the product of its reaction with HCl, compound 553, were confirmed by spectroscopic methods and X-ray diffraction analysis.
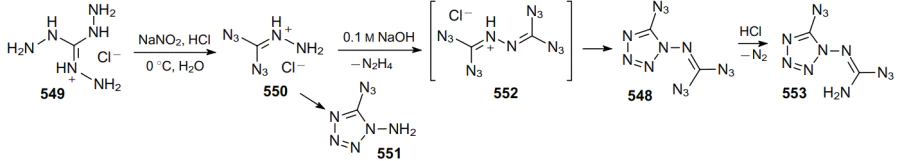
In 2013, Banert et al.318 reported the synthesis of triazide 548 from tetrabromide 554 according to a known procedure.319 Besides, the authors converted this triazide to tetrazole derivative 555 with the use of cyclooctyne (164) (Scheme 169). Triazide 548 unexpectedly turned out to be more reactive as compared to its tetraazide precursor 547.
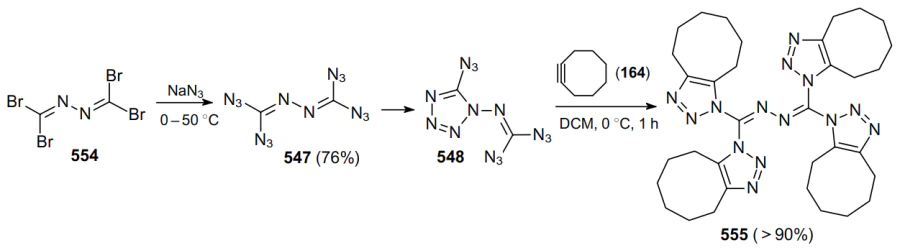
In 2014, a representative series of a novel class of geminal acyl triazides 556 with various substituents at the carbonyl centre was prepared (Scheme 170).320 Three simple approaches were proposed to synthesize such triazides, namely, starting from 3-oxocarboxylic acids (method a) iodomethyl ketones (method b) and terminal olefins (method c). Actually, all three proposed methods are variants of an oxidative azidation using a sulfonylated derivative of 2-iodoxybenzoic acid (45) and NaN3 as the azide source. The methods b and c have proven most effective and provided the synthesis of the target products in yields from 30 to 77% and from 32 to 48%, respectively. Despite a high nitrogen content, geminal triazides are easy to handle when carrying out the synthesis on a preparative scale and are potentially valuable building blocks for the synthesis of amino acids.
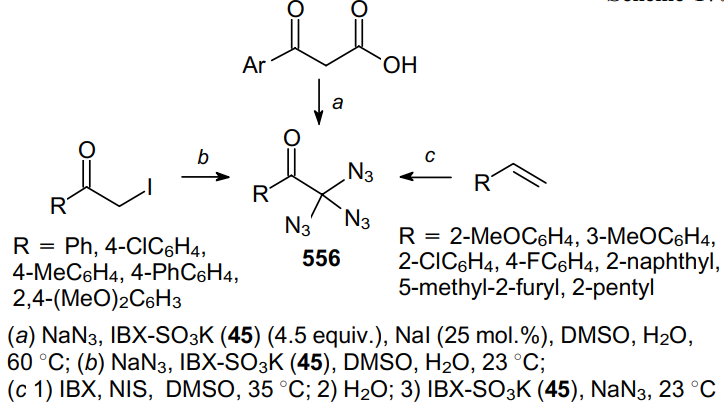
4.3. Aromatic and heterocyclic tri-, tetra- and polyazides
The preparation and chemical transformations of heterocyclic monoazides are discussed in detail in the review.22 The reviews by Chapyshev321,322 address the synthesis, chemical transformations and possible applications of six-membered aromatic azides 557--574 bearing three or more azide moieties in the ring. (Structures 557-578)
These compounds are widely used as the starting materials in organic synthesis and photochemical research and also as photoresistors in microelectronics and crosslinking agents in polymer chemistry. Some aromatic polyazides show high antitumour activity, while the other are of significant interest as energetic compounds and precursors to nanomaterials. The use of aromatic polyazides in click reactions can become a new trend in the development of various supramolecular systems with valuable chemical, physical and biological properties. A special attention is paid to chemical transformations of cyanuric triazide 557.
The synthesis of 1,3,5-triazido-2,4,6-trinitrobenzene (564) was first mentioned in a patent323 filed in 1927. A long chain of transformations begins with aniline, which gives 1,3,5-trichlorobenzene in three steps, followed by the nitration of the latter and the replacement of aromatic chlorine atoms with sodium azide in ethanol. In 1951, 1,3,5-triazido-2,4,6-trinitrobenzene (564) was synthesized through the substitution of chlorine atoms in the corresponding trichloro derivative 579 treated with NaN3 in wet acetone (Scheme 171).324
Bailey and Case325 synthesized this triazide using another approach based on the conversion of trichlorobenzene 580 to dinitrotriazidobenzene 563, which, in turn, is subjected to subsequent nitration. It is known that triazide 564 can form benzotrifuroxan 581 via a spontaneous intramolecular cyclization.326
Chapyshev and Chernyak327 reported the azidation of 1,3,5-trifluorobenzenes 582a--c (Scheme 172). When heated in DMSO with sodium azide, trifluorobenzenes 582a,b undergo rapid chemoselective defluorination to afford triazides 583 and 584 in high yields. Under the same conditions, the azidation of trifluorobenzene 582c leads to a mixture of triazides 585 and 586 in a 2:1 ratio. The equilibrium between products 585 and 586 can be shifted towards the predominant formation of the latter compound by carrying out the reaction in aqueous acetone. Compounds 583--585 are the first representatives of available 1,3,5-triazidobenzenes, which show promise for practical application as photoactive crosslinking agents in the polymer industry, as well as the starting materials for preparing organic materials with magnetic properties.
It should be noted that hexafluorobenzene (587) reacts with an excess of sodium azide in DMSO at room temperature to produce a mixture of diazide 588, tetraazide 589 and hexaazide 570 (Scheme 173). For this reason, the azidation of hexafluorobenzene 587 or its hexachloro- and hexabromo-substituted analogues cannot be considered as an approach to the preparation of 1,3,5-triazidobenzenes.328 Photolysis of halo- and cyano-containing 2,4,6-triazidopyridines in cryogenic matrices was also studied.329

2,4,6-Triazido-1,3,5-tricyanobenzene (590) was first mentioned in the literature in 1967.330 This triazide was synthesized (yield >95%) from appropriate cyanuric trichloride and sodium azide in refluxing acetonitrile. In 2012, Becker et al.331 developed an efficient route to triazide 590 starting from mesitylene (591), wherein tribromide 592 was reacted with sodium azide in the final step upon heating in acetonitrile (Scheme 174).

The preparation of 2,4,6-triazido-3,5-dichloropyridine (573) from tetrachloride 593 as well as some chemical transformation of this product were described by Ranjbar-Karimi et al.332 (Scheme 175). Also noteworthy is the possibility of the selective reaction of triazide 573 with norbornene at the 4-positioned azido moiety to give aziridine 594. In contrast, the reaction with dimethyl acetylenedicarboxylate produces cycloadduct 595, the formation of which involves 2- and 6-positioned azide moieties.
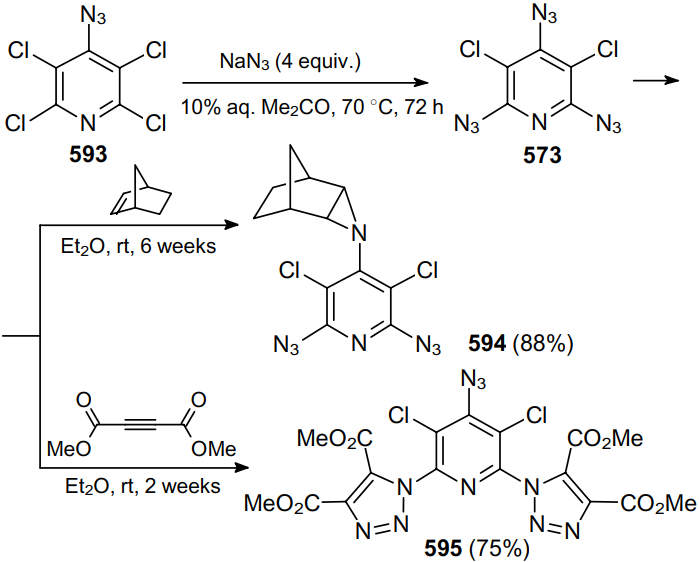
Cyanuric triazide (557) was first reported more than 90 years ago130 It is difficult to judge whether the compound the author dealt with was individual in view of the imperfect analytical base. After a long time, this triazide and relevant tetraazide 596 were mentioned in publications333,334 in 2004, devoted to studies of various polyazido high-nitrogen compounds. Scheme 176 shows the reaction sequence, as a result of which the chlorine atoms in tetrachloride 597 were first replaced with hydrazide moieties (intermediate 598) and then by azide ones. In the final step, hydrazide derivative 599 was converted to the corresponding aza compound 596.
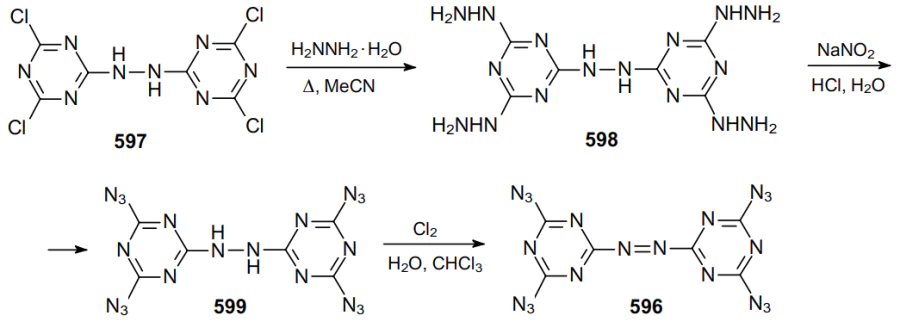
In 2004, Zheng et al.335 performed quantum chemical calculations of compounds 557, 596 and other heterocyclic polyazides and also predicted the azide--tetrazole tautomerism. The energy difference between the conformations A and B of triazide 557 is as low as 0.3 kcal mol–1, while the energy of the transition state is 11.7 kcal mol–1 (Scheme 177).336
The azide--tetrazole ring-chain tautomerism was studied for a wide range of aromatic di-, tri- and tetraazides 557, 596, 599--605 (Scheme 178).337 Ab initio quantum-chemical methods showed that the electron-donating amino group promotes the ring closure of tetrazoles, while the electron-withdrawing nitro group favours the retention of the azido form. It was predicted that diazidoazo- and hydrazotetrazines would readily cyclize to tetrazoles in polar solvents.338
An unambiguous assignment of signals in the 15N NMR spectra of 2,4,6-triazido-s-triazine, 2,4,6-triazidopyrimidine and six 2,4,6-triazidopyridines was made using computational and experimental methods. The different reactivity of a- and g-azido moieties of pyridines correlates well with the 15N chemical shifts in the NMR spectra. Among two non-equivalent azido functionalities of azines, an azido moiety with a more downfield shifted 15N signal is more electron-deficient and reactive towards electron-rich agents. In contrast, an azido moiety with the most upfield shifted 15N signal is the most reactive towards electron-poor reagents.339
In 2000, it was shown340 that the slow thermal decomposition of compound 557 in a high-pressure reactor produces nitrogen-rich carbon nitrides (Scheme 179). The authors suggested that in this case, the elimination of one nitrogen molecule affords intermediate 606 followed by its transformations.
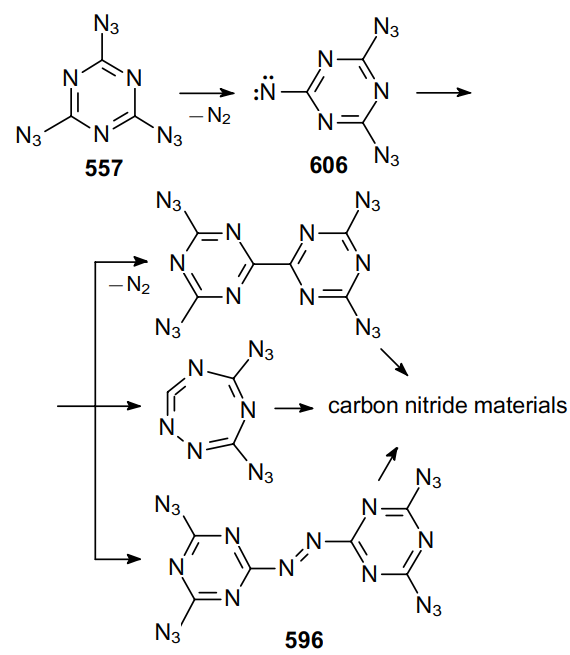
The resulting material is an amorphous bulk solid having a glassy microstructure with large pores and voids. Compound 557 can be used to prepare amorphous films exhibiting optical properties. This triazide can be employed to deposit carbon nitride coatings onto a wide range of thermally sensitive substrates.341 The recent advances in this field are discussed in a review342 published in 2021.
4.4. High-molecular-weight polyazides
Polyazides are being extensively studied as coupling agents for the construction of nitrogen-containing macromolecules. Highly branched structures, such as dendritic and star-shaped polymers, have been of great interest to researchers for a long time due to their compact structure and high functionality of the end groups.This is the subject of a review by Lammens and Du Prez.343 The synthesis of high-molecular-weight polymers involving di- and polyazides is generally based on a convenient azide--alkyne [3+2]-cycloaddition reaction. It provides controlled cross-linking of shorter chains, resulting in nodal nets of 1,2,3-triazoles.344 The CuAAC reaction is commonly used to design materials with architectural integrity due to the presence of stable triazole moieties in their structure. The reported examples include linear polymers, dendrimers, polymer networks, polymer nanoparticles and other high-molecular-weight structures.345
Brunner et al.346 developed dendritic siRNA nanostructures capable of penetrating even difficult-to-transfect cells such as neurons due to a special receptor ligand. The cell-penetrating siRNA dendrimers, constructed using click chemistry methodology, provide great opportunities for finding novel molecules that are able to treat, e.g., fatal illness caused by the rabies virus.
1,2,3-Triazole cross-linked polymers were synthesized as binders for solid rocket propellant systems by reacting the azide moieties of the polymer with the alkyne groups of a dipolarophile curing agents. A carbonyl functionality adjacent to the triple bond, which acts as an electron-withdrawing group, significantly accelerates the 1,3-dipolar cycloaddition reaction between azide and alkyne functionalities, allowing the preparation of rubbery materials with a high degree of cross-linking.347
The covalent synthesis of nanoscale frameworks is easily carried out in high yields using click chemistry methodology via the CuI-catalyzed cycloaddition of appropriate polyazides 607 and polyalkynes 608 in the presence of (EtO)3P.CuI as the catalyst to afford structures 609 (Scheme 180).348
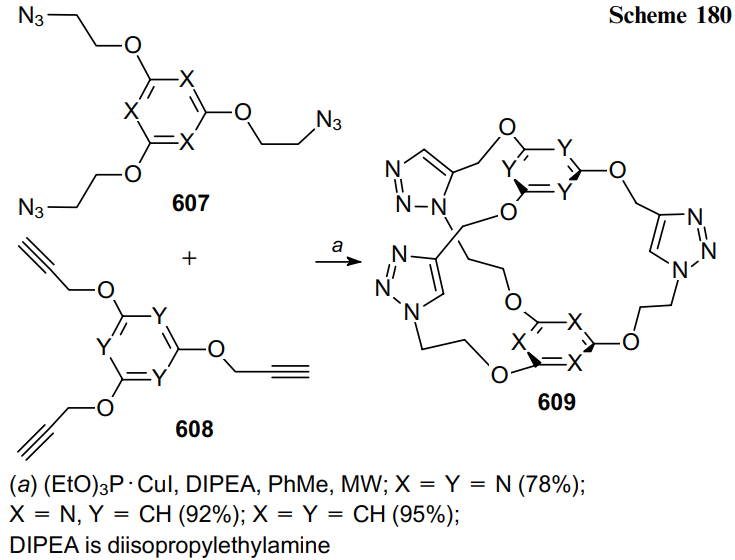
A similar approach was implemented to produce cured polymers based on commercial polyols.349 Compounds 610-617 Structures 610-617 were used as an azide component.
The click chemistry concept was employed to construct fullerene building blocks based on azide-containing fullerenes. Scheme 181 illustrates one such example, the conversion of polyazide 618 into polytetrazole 619. Current investigations in this field are summarized in a review.350
Hiba and Sreekumar351 presented the synthesis of dendrimer 620 from appropriate polyazide 621 (Scheme 182). In the resulting product, the dendritic cavity and surface are functionalized with reactive centres. This amino-functionalized dendronized polymer was used as a homogeneous organocatalyst for the synthesis of 4-substituted triazoles.
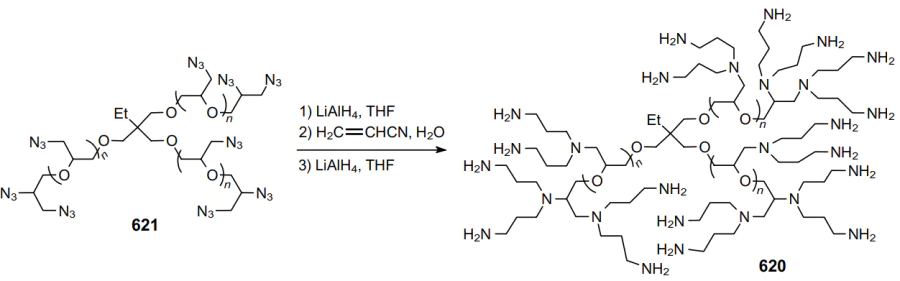
Conventional thermosetting polyurethane polymers are prepared by crosslinking a polyisocyanate with a polyol. An alternative approach to cross-linked polymers is possible using polyazides. Figure 1 shows a general strategy for obtaining novel promising polymer materials based on azide-containing polymers.352,353
The azide--alkyne cycloaddition allows the creation of novel structural motifs in DNA nanotechnologies and the introduction of new functional groups into DNA nanostructures,354 and also have proved to be effective in the synthesis of fluorinated surfactants for droplet microfluidics.355
Conclusion
To summarize, the chemistry of 1,1- and 1,2-diazides has attracted great interest of experts and has been extensively developed in recent years. Noteworthy is the importance of these classes of azides in modern organic chemistry. This review presents a large number of various methods for the synthesis of such compounds, including stoichiometric, catalytic, photochemical and even electrochemical approaches. It is important to highlight high reactivity of diazides and a variety of their reactions, from thermal transformations to click reactions. Of particular attention are processes, in which one azide functionalities undergoes intramolecular cyclization, while another azide moiety remains intact. This enables further chemical transformations involving the remaining azido moiety of the resulting heterocycles, in particular of azidoisoxazoles and azido-1,2,3-triazoles. Tri- and tetraazides are often investigated as precursors to explosives and monomers in macromolecular chemistry. In general, this line of research has so far been studied only fragmentary and there is a wide field of activity for organic chemists and researchers in related disciplines.
List of abbreviations and designations
The following abbreviations and designations are used in the review:
Boc — tert-butoxycarbonyl;
BHT — 2,6-di-tert-butyl-4-methylphenol;
Bs — benzenesulfonyl;
CuAAC — click reaction of copper-catalyzed azide--alkyne cycloaddition;
DABCO — 1,4-diazabicyclo[2.2.2]octane;
DCE — 1,2-dichloroethane;
DCM — dichloromethane;
DEA — diethylamine;
DEAD — diethyl diazodicarboxylate;
DFT ---density functional theory;
DIPEA — diisopropylethylamine;
DMAP — 4-dimethylaminopyridine;
DNABT — 1,1ʹ-dinitramino-5,5-bis(tetrazole);
dr — diastereomeric ratio;
Fmoc — 9-fluorenylmethoxycarbonyl;
IBX — 2-iodoxybenzoic acid;
IBX-SO3K — potassium 3-carboxy-4-iodylbenzenesulfonate;
KHMDS — potassium bis(trimethylsilyl)amide (potassium hexamethyldisilazide);
L — ligand;
LDA — lithium diisopropylamide;
MOM — methoxymethyl;
Ms — methanesulfonyl (mesyl);
MW — microwave irradiation;
NBS — N-bromosuccinimide;
NFSI — N-fluorobenzenesulfonimide;
NIS — N-iodosuccinimide;
PMB — p-methoxybenzyl;
PIFA — bis(trifluoroacetoxy)iodobenzene;
Py — pyridyl;
4-PPY — 4-pyrrolidinopyridine;
SET — single electron transfer;
SPAAC ---strain-promoted azide--alkyne cycloaddition;
TBAB — tetra-n-butylammonium bromide;
TBAF ---tetra-n-butylammonium fluoride;
TBPB — tert-butyl peroxybenzoate;
TBS — tert-butyldimethylsilyl;
TEMPO — (2,2,6,6-tetramethylpiperidin-1-yl)oxyl;
TFA — trifluoroacetic acid;
Tf — trifluoromethanesulfonyl (triflyl);
Th — thienyl;
TIPS — triisopropylsilyl;
TMGA — tetramethylguanidinium azide;
TMS — trimethylsilyl;
Troc — 2,2,2-trichloroethoxycarbonyl;
Ts — p-toluenesulfonyl (tosyl).