



Keywords
Abstract
This review analyzes literature on polymeric transition metal polychalcogenides of group IV–VII This review systematizes literature data on polymeric polychalcogenides of group IV–VII transition metals (TiS3, VS4, NbSe3, MoSx, ReS4, etc.). It covers the structural characteristics of crystalline compounds and the proposed structural models of amorphous phases. The latest advances in the synthesis of these low-dimensional materials at the nanoscale are presented. Their chemical properties are discussed, in particular, those arising from the presence of the dichalcogenide bonds. It is emphasized that the properties of the S–S (or Se–Se) groups largely determine the distinctive features of polychalcogenides and the unusual phenomena observed in them. In particular, these groups play an important role in the performance of electrode materials in metal-ion batteries, photo- and electrocatalysts for hydrogen evolution reaction, and mercury vapour sorbents based on transition metal polychalcogenides, which is also addressed in this review.
The bibliography includes 304 references.
1. Introduction
Metal polychalcogenides are inorganic compounds where anions are formed by interconnected chalcogen atoms of the (Q–Q)2– type, where Q=S, Se, Te. Such linkage is well known for elemental sulfur and selenium, the structure of which comprises stable chains and cycles (S8, S6, S4, Se4), and also for Li2Sx-like alkali metal ionic polysulfides (S82-, S62-, S42-, S22-),1--3 for organic4 and biological5 systems. Transition metal complexes containing Qx (x=2-6) moieties as chelate and bridging ligands (Fig. 1a,b) are also very numerous.
![[{"id":"oHL9X6PQJN","type":"paragraph","data":{"text":"Structure of crystalline transition metal polychalcogenides. Cluster anions: [Mo<sub>2</sub>S<sub>12</sub>]<sub>2</sub> (<i>a</i>),<sup>2-6</sup> [Mo<sub>3</sub>S<sub>13</sub>]<sub>2</sub> (<i>b</i>).<sup>2-7</sup> Chain structures: MS<sub>4</sub> (M=V,Nb) (<i>c</i>),8 M<sub>2</sub>Se<sub>9</sub> (M=V,Nb) (<i>d</i>);<sup>9,10</sup> an example of ternary polychalcogenide: polysulfide NaNbS<sub>6</sub> - the structure comprises <sub>∞</sub>{NbS<sub>6</sub>}<sup>-</sup> infinite chains, where Nb<sup>5+</sup> ions are in a coordination environment of (S<sub>2</sub>)<sub>3</sub>(S)<sub>3</sub> disulfide groups (<i>e</i>).<sup>11 </sup>Structures combining the properties of chain and layered compounds: NbS<sub>3</sub> (<i>f</i>),<sup>12</sup> MTe<sub>5</sub> (M=Zr,Hf)(<i>g</i>).<sup>13</sup> Here and below, transition metal atoms are marked in black; chalcogen atoms in an oxidation state of -2 are marked in light-grey; chalcogen atoms in an oxidation state of -1 (inQ-Q units) and higher (in longer chalcogen chains) are marked in yellow. Red colour indicates the “chalcogen-chalcogen“ bonds, the presence of which in the compound allows it to be assigned to the class of transition metal polychalcogenides."}}]](/storage/images/resized/mCHbtm5tj8hRDI9hnLs6nmpZvNHvih8e9iEwA8mP_xl.webp)
Key molecular representatives of polychalcogenides have been considered in detail in earlier reviews14--22 prepared by our scientific group and other researchers. A critical view on polychalcogenide anions is presented in the review.23
This review addresses low-dimensional group IV–VII transition metal polychalcogenides (TMPC) (TiS3, VS4, NbSe3, MoSx, ReS4, etc.), which are compounds with extended structures (quasi-1D, -2D). They are composed of covalently bonded chains or layers that interact with each other via van der Waals forces (Fig. 1c-g).
The family of low-dimensional group IV–VII metal polychalcogenides comprises crystalline MQ3 trichalcogenides (M=Ti, Zr, Hf, Nb, Ta; Q=S, Se, Te)12,24--83 (see Fig. 1f), MQ4 tetrachalcogenides (VS4,8,84--111 NbS4,112 NbTe4,113,114 TaTe4113,115) (see Fig. 1c), M2Se9 dimetal nonaselenides (V2Se9 (see 116,117) and Nb2Se9 (see 118)) (see Fig. 1d) and a variety of the MQ≈3-type amorphous phases (M=Cr, Mo, W; Q=S, Se)68,119--142 (Fig. 2), MoS4,142--148 TiS4,149--151 MoS5,142,152,153 WS5,143,154 MoS6,142,152 MoSe≈5, WSe≈6–7,155 NbS≈2.5,156 and also MoSe3S-type chalcogen-mixed or metal-mixed phases,157,158 Nb1+xV1-xS5,159 Zr1-xTixS3 (0<x<0.33),160,161 Ti1-xNbxS3,162,163 Nb1-xTaxS3.164 Moreover, (Qx)2– chains (x=2-6) are an important structural unit of such an interesting class of objects as MoSx-type chalcogenide gels,165--169 in which polychalcogenide chains are linked by metal ion bridges. Finally, there are ternary and quaternary polychalcogenides, whose structure is stabilized by halide ions or metal cations, most often, alkaline ones (see Fig. 1e).11,170
![[{"id":"7IvelYhNky","type":"paragraph","data":{"text":" Structure of amorphous transition metal polysulfides. Chain model for MoS<sub>3</sub> (<i>a</i>),<sup>123</sup> cluster model for MoS<sub>3</sub> (<i>b</i>),<sup>171</sup> CrS<sub>3</sub> (<i>c</i>),<sup>172</sup> ReS<sub>4</sub> (<i>d</i>),<sup>173</sup> Re<sub>2</sub>S<sub>7</sub> (<i>e</i>),<sup>173</sup> MoS<sub>5</sub> (<i>f</i>).<sup>174</sup> The Figure was prepared by the authors using original data from the cited publications."}}]](/storage/images/resized/vTZahiCMxZepN5bmys6adwH52n16i6Qb9BT988Ir_xl.webp)
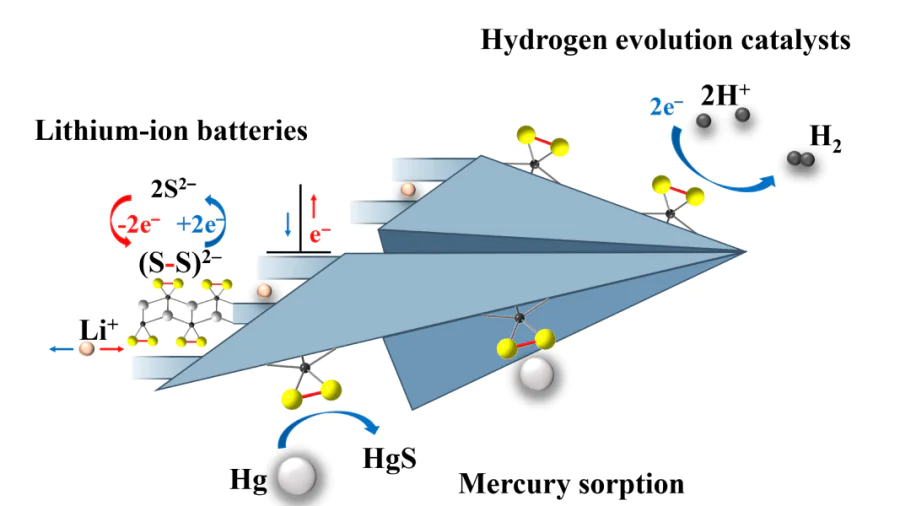
It seems timely to systematize and generalize the latest data on TMPCs accumulated in recent years, since in the 2000s, these studies have evolved to a new stage. A large number of works have appeared on the preparation of these compounds at the nanoscale. Structures of amorphous phases have been refined, and in 2020-2022, novel polychalcogenides were prepared, previously thought to be non-existent such as crystalline CrS3,175 NbS4112 and WS3.176 However, practically-oriented research develops especially fast. On the one hand, these are applications in the new-generation electronics and optoelectronics devices based on TiS3 and related materials,28,29,39,40,42,50--52,59,61,65,67,69,80 in which the physical properties of TMPCs are important. On the other hand, these are areas, where chemical processes play an important role: electrodes of high-capacity metal-ion batteries, catalysis of hydrogen evolution reaction, photocatalytic decomposition of organic molecules, and sorption of toxic vapours. To a large extent, these functional properties depend on the structure and chemical properties of materials, in particular, on transformations in di- or poly-chalcogenide anions. It is proved that (S–S)2–/(Se–Se)2– groups are involved in redox processes occurring in the electrodes of lithium-ion batteries68,90,105,108,109,140,142,169,177--180 or in electrocatalysts (Fig. 3).53,125,126,128,133,136,138,181,182
![[{"id":"hcZY-g0l32","type":"paragraph","data":{"text":"Schematic of several practically relevant processes, in which polysulfide materials are explored: cathode materials for lithium-ion batteries (<i>a</i>); catalysts for hydrogen evolution reactions (can act as photo-, electro- and photoelectrocatalysts) (<i>b</i>); mercury vapour sorbents (<i>c</i>). The (S-S)<sup>2-</sup> groups of the material, which are involved in reversible redox transformations, play a special role."}}]](/storage/images/resized/LzdQnOeGy7jY6KTgGB8f7WpceNvMnpfiVOxNmm0b_xl.webp)
Thus, during charge/discharge of a battery, an anionic disulfide group in the electrode material undergoes a reversible redox transformation: S22-+2e–↔2S2– (Fig. 3a).177 As a result, the involvement of both cations and anions in the redox processes on the electrode makes it possible to accomplish multielectron reactions and hence, to achieve higher capacities. At the same time, layered or chain structures of the electrode material can potentially contribute to stable cycling. Disulfide units (S–S)2- are also involved in electrocatalytical processes, e.g., in the hydrogen evolution reaction (HER), which is important from the practical point of view and is now being actively studied. Much evidence suggests that it is the (S–S)2– groups that act as active centres in the electro- and photocatalysts like FeS2, MoS3 and ZrS3 (Fig. 3b). Moreover, it is interesting that their important role in electrocatalysis is observed not only for polysulfide materials,53,181 where all sulfur exists as S22-, but also for the edge disulfide centres of nanosized MoS2, which is not a polysulfide but contains terminal sulfur dimers.7,183--185 Numerous proofs of this fact have been obtained using molecular models186 (clusters like [Mo2S12]2– (Ref. 6) and [Mo3S13]2– (Ref. 7), see Fig. 1a,b), which once again illustrate the intimate connection between the chemistry of molecular and non-molecular polychalcogenides. The above examples show that diverse structurally distinct polychalcogenides have in fact many common properties due to the presence of polysulfide groups Sn. Therefore, it is worth considering low-dimensional TMPCs as a separate class of compounds and to summarize recent related data accordingly.
Most of the reviews on polychalcogenides were written more than 20 years ago.24,170,187--192 Some recent reviews have focused on specific members of the TMPC family: in 2007-2022, papers were published regarding amorphous molybdenum polysulfides119--121 and group IV metal trichalcogenides (TiS3, ZrS3, etc.),26--29,82 however, they highlighted mainly electronic properties and prospects for applying such materials in electronics.
In this review, we consider a wide range of polychalcogenide materials trying to demonstrate their similarity, and emphasize more their chemical properties and the applications they define, in particular as electrocatalysts and electrode materials (the concept of their functioning we have recently summarized177). We will focus on the structural features of TMPCs, methods for the synthesis of their nanostructures, chemical properties, present latest advances in the use of these materials as electrodes in metal ion batteries, photo- and electrocatalysts, etc., and describe our views on the further development of their chemistry and applications. The review will largely discuss sulfides, since most of the results accumulated in the literature apply to them; at the same time, if possible, data on selenides and tellurides will be presented.
2. Structures of transition metal polychalcogenides
Since the family of low-dimensional transition metals polychalcogenides includes a large number of representatives with different structures and compositions, it is useful to distinguish several groups. For instance, pure polychalcogenides containing chalcogen atoms only as (Q2)2– groups such as VS4, MoS5, CrS3, NbS4, NbTe4, TaTe4, ZrTe5 can be considered separately. Mixed polychalcogenides, such as layered trichalcogenides MQ3, contain both dichalcogenide and monochalcogenide groups. It is possible to classify TMPCs in a different way and to consider separately crystalline materials, which structures have been determined, and purely amorphous materials, for which no crystalline analogues have been obtained, and whose structures have been determined using a combination of indirect data from various methods. Section 2 of this review is based on the latter classification. (1D/2D) layered-chain crystal structures characteristic of metal trichalcogenides MQ3, linear chain (1D) structures of VS4 and also structures of several amorphous metal polychalcogenides will be discussed briefly below.
2.1. Structures of crystalline transition metal polychalcogenides
Crystal structures of low-dimensional transition metal polychalcogenides are diverse (see Fig. 1), since chalcogens can serve as bridging and terminal ligands Q2– and (Q2)2–, and V–VII group metals can form metal–metal bonds (metal cluster complexes). The structures in question are defined by 1) the formation of strong dichalcogenide units with short Q–Q bond lengths (≈2.0-2.1, 2.3-2.9 and 2.8-3.0 Å for Q=S, Se and Te, respectively) and 2) the presence of weakly bonded layers or chains (van der Waals solids). Several structural types characteristic of low-dimensional transition metal polychalcogenides can be distinguished.
Trichalcogenides of IV and V group metals of general formula MQ3 (M=Ti, Zr, Hf, Nb, Ta; Q=S, Se, Te) make up a family of compounds with slightly different structures,12,24--81 which can be considered as derivatives of ZrSe3 structure (monoclinic syngony, P21/m space group). These structures are composed of the MQ6/2 trigonal prismatic columns, which in turn are made up of MQ6 elementary prisms sharing their triangular faces and extending along the b-axis (see Fig. 1f). In a triangular base of the prisms, two chalcogen atoms are bonded into dichalcogenide groups [e.g., dS–S=2.04 Å (TiS3), dSe–Se=2.34 Å (ZrSe3)], while one chalcogen ligand is a sulfide or selenide ion (S2– or Se2–). Since chalcogen atoms in MQ3 structures are in two electronic states, the formula for trichalcogenides can be represented as M4+Q2–Q22-. Shape and packing of prisms differ for various trichalcogenides. Polymorphism of NbS3 is addressed in a recent study.12 Covalently bonded columns in MQ3 structures determine their quasi-1D nature. On the other hand, there is also strong covalent bonding between the prisms from neighbouring columns, connecting the chains into layers (quasi-2D structure), which in turn are linked by van der Waals contacts. Thus, trichalcogenides are characterised by the combined properties of 1D/2D materials.
The recently synthesized crystalline chromium trisulfide175 (Fig. 4) differs significantly in structure from crystalline trisulfides of groups IV and V transition metals: its structure is three-dimensional, derived from the structure of marcasite FeS2. Crystalline CrS3 contains only disulfide sulfur: in its structure, Cr2S10 dimers are interconnected through disulfide bridges. In these dimers, two CrS6 octahedrons share an S–S edge.
![[{"id":"eDc9mcpWV9","type":"paragraph","data":{"text":" Crystal structure of CrS<sub>3</sub>.<sup>175</sup> Reprinted with the permission of the American Chemical Society."}}]](/storage/images/resized/NmWeEmgOsAV3AIwb664eaqA8tGXkovnakXCXknhd_xl.webp)
Vanadium tetrasulfide VS4 (V4+(S22-)2) (see Fig. 1c) crystallizes in the monoclinic space group C2/c, and its structure is a packing of infinite {VS4}∞ chains. The chains of V4+ ions run along the c axis, the vanadium atoms being linked in pairs by short V–V (d=2.84 Å) bonds, while the distances between V2 pairs are significantly longer (3.21 Å). Such structure is characteristic of a Pierels insulator, which is typical of d1 chain compounds. The distance between V . . . V chains is >6.1 Å, therefore, they interact weakly, which determines the 1D nature of the structure. Each vanadium atom is surrounded by eight sulfur atoms. All sulfur atoms are bonded into (S2)2– groups with short S–S distances (d=2.03-2.04 Å). Rectangular S4 planes perpendicular to the axis of the metal chains define the tetragonal-antiprismatic coordination of the vanadium atoms.8 A recently prepared crystalline niobium tetrasulfide NbS4112 is isostructural with VS4.
Binary tetraselenides MSe4 are unknown, but (MSe4)xAy ionic compounds with infinite chains {MSe4}∞ and halide ions or anionic halide complexes (A) have been obtained: (NbSe4)3I,193 (NbSe4)10I3,194 (NbSe4)4Br2,195 (TaSe4)2I,196 (TaSe4)2TaBr6.197 Similar ionic compounds were also reported for tellurium: (NbTe4)I, (TaTe4)I,198 (TaTe4)4I2TaI6 (Ref. 199) and (TaTe4)6I4TaI6,200 but simple binary tetratellurides NbTe4 and TaTe4 are also known. Unlike tetrasulfides, infinite MTe4 chains comprise clusters composed of three metal atoms. In NbTe4 chains,114 niobium atoms form Nb3 clusters with Nb–Nb bond lengths of 3.07--3.28 Å, separated by Nb . . . Nb distances of 3.90-3.93 Å. Another structural feature of MTe4 is the covalent Te–Te bonding between neighbouring {MTe4} chains (in NbTe4, distances are dTe–Te=2.91-2.94 Å).
Two representatives are known for the stoichiometric MQ5, both being tellurides — ZrTe5 and HfTe5 (M4+(Te22-)(Te32-), where M=Zr, Hf).13,201 Their crystal packings consist of infinite layers, which in turn are composed of trigonal-prismatic infinite columns cross-linked by polytelluride bridges (see Fig. 1g).
Infinite M2Se9 chains appear in V2Se9 and Nb2Se9 compounds. In these, binuclear {M2Se4} clusters alternate with polyselenide bridging Se5 ligands: ...–M2Se4–Se5–M2Se4–Se5–... (see Fig. 1d). In V2Se9 chains ((V5+)2(Se22-)4(Se2–)),117 the V-V bond lengths in V2Se4 clusters are 2.84 Å, and those between the clusters are dV...V=3.65 Å.9 Nb–Nb bond lengths in Nb2Se9 are 2.88(3) and 2.89(3) Å in Nb2Se4 clusters, while long Nb . . . Nb distances are equal to 3.76(3) Å.10
Electron-rich rhenium forms a crystalline polytelluride having an unusual structure: in a cage (3D) structure of Re6Te15 ((Re3+)6(Te2–)8(Te7)2–)), {Re6Te8}2+ cluster cores are bridged by polytelluride (Te7)2– groups.202
2.2. Structures of amorphous metal polychalcogenides
For a number of amorphous metal polysulfides and polyselenides crystalline analogues are unknown. Amorphous chalcogen-rich group VI TMPCs (MoS3, WS3, CrS3, MoSe3, CrSe3, MoS5–6, MoSe5–6, WSe5) are particularly diverse, but the structures of amorphous group IV (TiS4) and group VII (ReS4 and Re2S7) TMPCs are also being studied. The lack of translational symmetry hampers the elucidation of their structures. They were studied by extended X-ray absorption fine structure (EXAFS-spectroscopy), analysis of the radial distribution function, X-ray photoelecron spectroscopy (XPS), Raman spectroscopy, etc., and also by an indirect method of chemical excision of structural units in chemical reactions122 and the reverse Monte Carlo method.123 Various models for structures of such polychalcogenides were proposed, and discussions in the literature on this issue are ongoing (see Fig. 2a,b). For example, it is likely that the structures of MoS3 samples obtained by different methods may differ and there may be polymorphism/isomerism inherent in Mo/S fragments bearing sulfide and disulfide groups.119
The two most commonly discussed structural models for MoS3, WS3 and similar compositions are the chain and cluster models,123,139,171 variants of which are shown in Fig. 2a,b. The chain model (see Fig. 2a) suggests the formation of octahedral MoVS6 units sharing their trans-faces, with pair bonds between molybdenum atoms (Mo–Mo). In the cluster model (see Fig. 2b), molybdenum atoms are assembled in triangular structures Mo3 IV. Recent density functional theory (DFT) calculations show that different structures can coexist depending on MoS3 particle size.171 At smaller sizes, structures formed by triangular clusters are more energetically favourable, and as the size increases, chain structures begin to prevail; it is emphasised that these chains are bent.
The structural model of amorphous CrS3 is made up of chains, which are additionally linked together by the S–S groups (CN (Cr)=6) (see Fig. 2c).172 At the same time, the recently synthesized crystalline chromium trisulfide (see Fig. 4)175 differs significantly in structure from the amorphous CrS3: it has a three-dimensional structure, derived from the structure of marcasite FeS2. Therefore, the structure of this compound differs from that of both its amorphous counterpart and the other crystalline trichalcogenides (e.g., trichalcogenides of Nb, Ta, Ti, Zr, Hf, having a structure like the one depicted in Fig. 1f).
Sulfur-rich amorphous polychalcogenides such as MoS4.7, MoS5, MoS5.6 and MoS6, are obviously similar in structure to MoS3. New S–S bridges may incorporate into the chains shown in Fig. 2a. Since MoS3 is amorphous, there are no structural restrictions that could hamper such chain growth. In fact, one managed to prepare amorphous polysulfides MoSx with a wide range of compositions (x=3-6) and this not be the limit, as the S42- units are known and found in thiomolybdates.119, 152, 174 In polymeric materials MoSx, sulfur exists in different states such as bridging (S–S)2- groups, terminal S2–, S22- groups, etc.124--127,138,203 The large number of bridging (S–S)2– units between building blocks explains why sulfur-rich polysulfides exist only as amorphous phases and form in an unusual morphology. Thus, in MoS5, dimers M2S8 ([(M5+–M5+)(S–S)42-]2+) are linked by disulfide bridges into infinite chains, which bend like the classical polymeric chains and are arranged in such a way that the MoS5 and WS5 particles have a globular morphology, as shown experimentally and confirmed by simulations using DFT and DFT molecular dynamics methods.174 For this reason, sulfur-rich TMPCs can only be produced in an amorphous state. The corresponding amorphous selenides MoSe3,204 CrSe3, MoSe5–6 (Ref. 143) and WSe5 have also been reported.205
The data from the pair distribution function (PDF), XPS, Re LIII edge EXAFS and sulfur K-edge XANES for two related rhenium polysulfides ReS4 and Re2S7 also suggest their chain-type structure with a zig-zag arrangement of the Re–Re bonds173 (see Fig. 2d,e). Apart from bridging S2– and S22- in different ratios, both structures contain μ3-sulfur atoms.
For amorphous TiS4, the PDF analysis and DFT modelling data indicated a three-dimensional network-like structure in which titanium ions are linked by bridging polysulfide ions (S22-, S32-, etc.) rather than the chain structure. The average S–S and Ti–S bond lengths are 2.09 and 2.46 Å, respectively, and the average coordination number of Ti is 6.9. In this case, the structure switches into the chain one by the intercalation of lithium ions in the electrochemical process.150,151
Based on the literature data, it can be concluded that great progress in the study of amorphous TMPCs is due to the use of theoretical modelling, and it is clear that in the coming years it will be an integrated approach, including modern experimental and computational methods, that will allow further advances in the understanding of the structure of amorphous TMPCs.
2.3. Features of electronic structures
Features of crystal structure of TMPCs are associated with several interesting properties, which have been developed in studies on electrochemistry and the exploration of the electro-physical properties of one-dimensional systems. Most TMPCs are of the semiconductor or metallic conductivity type. For example, NbS3 of I and II structural types are semiconductors at room temperature, NbS3(III) is a semimetal, NbSe3, TaSe3 (rhombic and monoclinic) have a metallic conductivity, while (TaSe4)2I and (NbSe4)10I3 have a semi-metallic conductivity. When considering the electronic structure of TMPCs, the electronic structure of the conduction bands is of the greatest interest because it can explain the features of the electrophysical behaviour. This is important for many applications, particularly in the use of TMPCs in electrochemical and photocatalytic processes (see Section 5).
A description of the calculated electron densities of states of TMPCs can be found in the monograph206 or in later works. The contribution of s- and p-states of chalcogen, d-bands of metal atoms to the blocks of the energy spectrum, as well as their mutual location and overlapping near the Fermi level and the filling of bands with electrons constitute the principal features of the electronic structure of TMPCs. For example, first three bands below the Fermi level for NbS4 are represented by the following blocks (Fig. 5): from –0.5 to –1.0 eV, Nb4d states, responsible for the overlapping of Nb4d orbitals and the formation of Nb-Nb covalent bonds, prevail; the band from –1 to -3 eV corresponds to the overlapping of the S3p and Nb4d states and the formation of the Nb–S bonds; two bands from –3 to –3.5 and from –3.5 to –7 eV consist mainly of S3p states responsible for the formation of S2 covalent bonds, which coordinate covalently bound Nb2 pairs in the{NbS4}∞ infinite chains. The theoretical value of the band gap is 1.07 eV, which is in line with the experimental value of 1.1 eV derived from spectral data.112
![[{"id":"WVB5oJDmGh","type":"paragraph","data":{"text":"Calculation of the electron density of crystalline NbS<sub>4</sub> in DFT:<sup>112</sup> electronic band structure, total and selected partial density of states (DOS) (<i>a</i>). Total DOS is represented by a solid black line, Nb4d and S3p valence states are marked in blue and yellow, respectively; electron density difference map in the plane (010), passing through the NbS<sub>4</sub> molecular chain (<i>b</i>); phonon band structure (<i>c</i>). Reprinted with the permission of the American Chemical Society."}}]](/storage/images/resized/TnMQtA9HUmANZCDCdk16iyJ3Bs3tckw8Lcm14Hvb_xl.webp)
The current computational studies show that the electronic structure of TMPCs can be altered under the influence of various factors, namely, through bulk-to-nano transition, doping,207 tensile strain,208 exposure to high pressure.209,210 For example, the O-doping or S-vacancy in VS4 narrows the band gap (Fig. 6).211 Moving from crystalline VS4 to its nanowire, the valence band maximum and the conduction band minimum are formed by the overlapping of S2p orbitals and V3d levels, but compared to the crystal, S2p orbitals make a greater contribution, and vanadium contributes the dxy+dx2-y2 orbitals.212 Metal polychalcogenides are often referred to as electron reservoirs due to the presence of (Q2)27 dichalcogenide groups, which can serve both as an electron donor and an electron acceptor.189 In TMPCs, chalcogen p orbitals overlap with metal d orbitals to form hybrid d/p orbitals near the Fermi level, which allows the electron density transfer from metal atoms to the dichalcogenide group thus forming an electron reservoir.177
![[{"id":"W0jIwfXv-D","type":"paragraph","data":{"text":" Simulation models of electronic structure for crystalline VS<sub>4</sub> (<i>a</i>),VS<sub>4</sub> with O-doping (<i>b</i>),VS<sub>4</sub> with S-vacancies (<i>c</i>),V S<sub>4</sub> with O-doping and S-cacancies at the same time (<i>d</i>).<sup>211</sup> Reprinted with the permission of the American Chemical Society."}}]](/storage/images/resized/Tgb7Hn6gvRZUYzwvBTqWLlckeWeOhPKsEiXrVnf7_xl.webp)
When describing electrophysical properties by single-particle electron processes, TMPCs are seminconductors or metals. There is a number of publications devoted to the study of electrophysical properties of TMPCs, for which the relationship with their quasi-one-dimensional structure is clearly seen, and anomalies in electrical conductivity are discussed in terms of a collective state (electron-hole condensate movement) along a preferred direction. Compounds MS4, M2Se9, MTe5 belong to one-dimensional van der Waals structures, i.e., including infinite chains, which form van der Waals interactions with neighbouring chains, while MQ3, NbTe4, TaTe4 are quasi-one-dimensional: in their structures infinite chains of metal atoms can be distinguished, which assemble into infinite layers via interatomic bonds stronger than van der Waals forces.
Studies of electrophysical properties of trichalcogenides such as NbS3,213 NbSe3,214 TaS3,215 TaSe3,216 ionic compounds with infinite chains (NbSe4)10I3,217 (TaSe4)2I,218 and also tetrachalcogenides NbTe4 and TaTe4113 contributed much to the discussion on electrophysical properties of one-dimensional and quasi-one-dimensional compounds.
In the [MQ2-(Q2)2–]∞ infinite chains in MQ3 and [M(Q2)2]∞ infinite chains in MQ4, metal atoms can shift along the chains under certain conditions, according to the Peierls theorem on the instability of one-dimensional crystals consisting of evenly spaced atoms with one unpaired electron.219,220 In the Peierls transition, the chain under consideration converts to an insulating state. For example, the Nb . . . Nb distances of 3.04 and 3.69 Å alternate in the [NbS2–(S2)2-]∞ chains of compound NbS3(I), which characterizes an insulating state of these chains at room temperature. A large number of polymorphs are known for NbS3, but the difficulty of crystallizing most of them prevents their experimental study. In a theoretical work,12 models of electronic structure of seven stable polymorphs are compared, four of them being experimentally obtained ones and the three others being unreported ones which were refined in the structures of NbSe3, TiS3 and TaSe3. It was shown that structural differences of these polymorphs stem from the differences in their conductivities and electron transport12 (Fig. 7). Thus, for NbS3(I), linkages between infinite chains in pairs of chains of the same type are quite strong compared to other polymorphs. Further, NbS3(V), NbS3(II) and ʹNbSe3ʹ phases exhibit metallic properties with incommensurable charge density waves (CDW), which is associated with different partial occupation of the Nb dz2 band of the trigonal prismatic chains by half, one-third and one-quarter, respectively.
![[{"id":"5bVa-qT7ox","type":"paragraph","data":{"text":" Summary of electrophysical properties of NbS<sub>3</sub> polymorphs.<sup>12</sup> Reprinted with the permission of the American Chemical Society."}}]](/storage/images/resized/YCRgqPSCpGNlCf4XO1FEqQLtrbTRGWUhxBBDQg4Y_xl.webp)
In TMPC crystals, a nonohmic character of electrical conductivity is observed: anomalies in the dependences of electrical conductivity on temperature, pressure or exposure to electromagnetic radiation are accompanied by a periodic modulation of the electron density — charge density waves.221 The emergence and dynamics of CDW, the state of superconductivity, and related phenomena at TMPCs have been studied since the 1960s, as reflected in several monographs;206,222 the research has also continued in recent years.223
3. Methods for the synthesis of transition metal polychalcogenides
Synthetic approaches and theoretical foundation of preparing such materials were laid as early as the 1980-1990s189 and were detailed in earlier papers,24,27--29,187 so here we will discuss recent publications addressing their synthesis, with a focus on obtaining nanomaterials. Polychalcogenide crystals feature anisotropic morphology in the form of ribbons, rods or wires growing along the b axis, i.e. parallel to the infinite chains (more pronounced growth occurs along the a axis with increasing temperature41) and, often, one (or more) of the crystal sizes is in the nanoscale range.Therefore, many of the polychalcogenide samples discussed here can be micro-or nanocrystals (or amorphous particles). Theoretical calculations also supported the possibility of stabilization and predicted interesting physical properties of various nanostructures of polychalcogenides, including monolayers.37,62,65--67,71,77,79 The main synthetic approaches to various crystalline and amorphous TMPCs are listed in Table 1 and Table 2.
3.1. Synthesis from elements
Transition metal polychalcogenides are prepared from elements at moderately high temperatures: the upper limit of the reaction temperature is generally limited by the formation of more stable dichalcogenides. For the synthesis of monocrystalline samples the chemical vapour transport technique is used: a transport agent such as iodine, bromine or a chalcogen chloride is introduced into the reaction medium. Sulfur/selenium, taken in excess, can also act as transport agents, producing MQx intermediates responsible for mass transfer.26,41,187 In these cases, smaller crystals are obtained due to less active material transport.187 The procedure consists of three steps:26,41 evaporation of the solid reactants, transport of gaseous substances, condensation on a surface accompanied by the crystal growth.
For the synthesis of TiS3, the following mechanism is proposed:29,40,61 at a temperature of 450 °C, sulfur turns into a gaseous state, precipitates on the titanium surface and reacts with it to generate unstable gaseous intermediates such as TiSx. Next, TiSx reacts with S in the gaseous phase and the resulting stable TiS3 phase is again deposited on the titanium surface, becoming the centre of crystallisation and growth of TiS3 (nano)fibres from the gaseous phase through further interactions between TiSx and S. Probably, ZrS3 and HfS3 nanoribbons are also formed in a similar way.41 This mechanism is evidenced by the pointed shape of the ends of trichalcogenide nanocrystals.29 Thus, the direction of growth is determined by the competition among the growth rates of the different crystallographic faces. Growth will occur towards the edge with the highest free energy, i.e., the most reactive (the least stable) face.
In the process of the synthesis, parameters such as vapour pressure, heating and cooling rates, reaction time, reaction temperature and temperature gradient along the length of the ampoule can be varied. The growth temperature and temperature gradient have the most significant impact on the characteristics of the resulting samples.36,41,61,81 In the case of TiS3, crystals displayed a morphology of nanoribbons at 500 °C, nanosheets at 400 °C, and at 450 °C, a sample of a mixed morphology was formed.61 It was noted that titanium trisulfide microparticles are formed in a temperature range from 400 to 550 °C for 24 h, with the morphology of the resultant microparticles depending on the synthesis temperature — from broad thin ribbons (400 °C) to narrow ribbons (500-550 °C).81 The growth temperature for TiS3 should not exceed 632 °C, since TiS2 is stable at higher temperatures.28,61,81 Zirconium trisulfide is stable at even higher temperatures, and it was also noted83 that upon increasing the temperature of the ampoule synthesis from Zr and S from 350 to 650 °C, the morphology of the resultant ZrS3 changed from thin fibres to nano- and microribbons (Fig. 8).
![[{"id":"47yZEIa1xt","type":"paragraph","data":{"text":" Temperature dependence of the morphology of ZrS<sub>3</sub>, microcrystals obtained from elements in the range from 350 to 650 °C.<sup>83</sup> Reprinted with the permission of Elsevier."}}]](/storage/images/resized/jLqOntng6uRBpE0ROBgCCpEJ8FKEqadjpVPYQIyG_xl.webp)
Niobium triselenide in the form of nanoribbons and nanowires begins to grow from a stoichiometric mixture of elements at temperatures above 610 °C, with samples of the highest purity being obtained at 630-700 °C.36 The cross-section area of nanostructures increases with increasing synthesis temperature. A sample of NbSe3 fabricated at 700 °C, has a fibre morphology up to a few millimetres long and 20-700 nm wide.36,42,43 At temperatures beyond this range, i.e. both below 610 °C and above 700 °C, other niobium selenides, Nb2Se9 and NbSe2, are formed, respectively. Nanoribbons of ZrS3 and HfS3 prepared at 600 °C,41 and TaS3 prepared at 650 °C47 possessed a morphology and dimensions similar to those of NbSe3, however in the case of HfS3, a thin amorphous layer with a thickness of 2-20 nm and a significant content of TaS2 was also found on the surface of the particles, which leads the authors to classify these structures as the ʹcore--shellʹ architecture.
Careful control of experimental conditions allows the growth of nanostructures of transition metal trichalcogenides of very different and sometimes unusual morphology: in the form of ring-shaped structures (NbSe3242 and TaSe3 (Ref. 32)), Möлbius strips (TaSe332 and NbSe3 (Ref. 31)), ʹflowersʹ (TiS3)45 and other complex structures.25,26,31 Such structures are formed due to a temperature gradient.31 Under strongly non-equilibrium conditions, selenium simultaneously exists as vapour, aerosol and liquid droplets, which act as templates: growing trichalcogenide filamentary crystals are ʹwrappedʹ around them.
There are also reports of binary niobium and vanadium polyselenides of the stoichiometric composition M2Se9 (M=V or Nb) prepared from the elements. Divanadium nonaselenide V2Se9 is formed on heating a mixture of 2V+12.6Se (molar ratio) at a temperature of up to 330 °C,116 and Nb2Se9 — on heating a mixture of Nb+200Se (molar ratio) at a temperature of 500-600 °C in 3 days.118
Tetratellurides of niobium113 and tantalum113,115 MTe4 (M=Nb, Ta) can be produced on prolonged heating of a mixture of elements in an almost stoichiometric ratio at 900 °C. Zirconium and hafnium pentatellurides MTe5 (see Fig. 1g)13 were prepared from elements at a significantly lower temperature (500 °C) in 7 days.
The synthesis of trichalcogenides from elements can also give products with partial substitution of the metal or chalcogen atoms, similarly to the synthesis of transition metal dichalcogenides, which allows to adjust the electronic properties of the material.243 Thus, in a study,160 in the synthesis of crystalline ZrS3 nanolayers, zirconium was partially replaced by titanium. The (Ti,Zr)S3 solid solutions were obtained by the reaction of an Ti/Zr alloy and sulfur vapour at 800 °C. As a result, the samples with the morphology of microribbons were obtained, which is typical of transition metal trichalcogenides. The solid solutions had the ZrSe3 structural type (monoclinic). Interestingly, with molar ratios of zirconium and titanium Zr/Ti from 20/80 to 80/20, the stoichiometry of the resultant solid solutions was close to Zr0.8Ti0.2S3. The formation of solid solutions with close stoichiometry is probably influenced by the difference in stabilities of di- and trisulfides for titanium and zirconium: ZrS3 is stable up to 800 °C,161 whereas TiS3 decomposes to TiS2 and elemental sulfur at a temperature above 630 °C.244 At 800 °C, zirconium only formed trisulfides while titanium, when used in excess, formed TiS2. Nanolayers of substituted zirconium trisulfide were fabricated by scotch-tape exfoliation of the crystals, and their electrotransport properties were examined. The synthesis of a number of solid solutions Ti1-xNbxS3 (x=0.05, 0.07, 0.10) from stoichiometric quantities of metals and sulfur at 550 °C is reported;162 for the obtained materials, temperature-dependent electrical resistivity and the Seebeck coefficient value were studied.
3.2. Synthesis from lower chalcogenides or metal oxides
Not all polychalcogenides can be obtained from the elements in good yield — many amorphous polychalcogenides were prepared from metal compounds and various sulfurizing agents. The formation of crystalline VS4 and NbS4 tetrasulfides from the elements is kinetically hampered. The synthesis of VS4 from metal vanadium and sulfur takes 10 days at 400 °C, and NbS4 is formed from the elements in a very low yield. At a temperature >450 °C these tetrasulfides are unstable. In both cases, the yield of MS4 increases significantly using metal sulfide and excess sulfur as reactants. Thus, VS4 can be produced in quantitative yield by heating a mixture of V2S3+6S (molar ratio) in an evacuated sealed glass tube at 400 °C for 24 h;105 similarly, NbS4 is obtained from a mixture of Nb1.14S2+11.4S at 440 °C in 60 h in 85% yield.112 The study227 showed that ZrS3 nanoribbons are produced via the reaction of ZrS2 with sulfur in a nitrogen atmosphere at 550 °C in a sealed tube for 2 h. This conversion is probably facilitated by the higher pressure (due to the addition of nitrogen) compared to the vapour pressure of sulfur when synthesised from the elements. In the above instances, sulfide ions were oxidized with sulfur to S– along with the partial metal oxidation in the case of vanadium and niobium.
Carrying out the high-temperature synthesis also at elevated pressure allows to obtain polychalcogenides that differ in structure from the known analogues, as well as polychalcogenides with previously unknown metal–chalcogen combinations. For example, chromium trisulfide CrS3 was recently obtained for the first time as a crystalline product from Cr3S4 and sulfur at 800 °C and a pressure of 13 GPa (see Fig. 4)175 (CrS3 is also formed from Cr and S under similar conditions). The structure and physical properties (superconductivity) of TiS3 at pressures up to 100 GPa were theoretically predicted. At 20 GPa denser structures (layered, corresponding to the experimentally obtained phase II) and cubic (cage) structures at 80 GPa) are suggested.
An alternative way of activating the reaction mixture is to treat them mechanochemically in an inert atmosphere. It is commonly used to prepare amorphous polysulfides of titanium and niobium. Amorphous TiS344,225,226 and TiS4149,150,225,226,239 were obained from a stoichiometric mixture of TiS2 and S. During a rather short synthesis time (several hours to tens of hours), X-ray amorphous products were formed that, according to their XRD patterns, did not contain the pristine crystalline TiS2 and S. In a similar way, a number of amorphous niobium polysulfides were prepared such as NbS3, NbS4 and NbS5.228 The study151 showed that the Ti 2p XPS spectrum of amorphous TiS4 corresponds to Ti4+, and in the S 2p spectrum, sulfur atoms in three charge states were found, namely, S2–, S–, and S with a small negative charge due to inner position within the (S-S–S)2– bridging groups.
Interestingly, mechanochemical synthesis also makes available amorphous MoS3 and other sulfur-rich polysulfides starting from MoS2 and sulfur,142 which do not react on conventional, even prolonged, heating. A series of amorphous molybdenum polysulfides MoSx (x=3--7) is formed via the prolonged (180 h) mechanochemical synthesis from crystalline MoS2 and elemental sulfur in an argon atmosphere. These polysulfides were produced as a mixture with partially amorphized MoS2; resulting mixtures were then explored as cathode materials in solid-state sodium-ion batteries. Noteworthy that according to the XPS data for the obtained samples, the binding energy of the Mo 3d 5/2 electrons is in the narrow range of 228.9-229.0 eV, which is very close to MoS2 (229.1 eV), and it can be assumed that using this synthetic method, molybdenum retains its Mo4+ oxidation state. The S 2p 3/2 XPS data confirm the polysulfide nature of the obtained materials: there are both S2– (161.8-162.1 eV) and S22- (163.0-163.2 eV).
An example when one polychalcogenide is prepared from another is the transformation of niobium polyselenides. When Nb2Se9 crystals were heated in an evacuated sealed tube at 600-630 °C for 3--6 h, the formation of NbSe3 crystals was observed.242 Niobium triselenide is considered to be a stable phase in this temperature range, and this transition is in agreement with the Nb–Se phase diagram. A fascinating feature of this transformation was the organisation of the resulting crystals into perfect rings.
Apart from sulfides, oxides can also serve as starting materials to prepare certain TMPCs. Crystalline trisulfides of titanium and niobium as well as vanadium were derived from Ti (TiO2), V (V2O5), Nb (NbO, Nb2O5) oxides, which reacted with sulfur and bromine in the high-temperature ampoule process.224 The reactions are driven by the formation of thermodynamically favourable B2O3, with boron sulfides (B2S3 and BS2) generated in situ being sulfidation agents along with sulfur; metal in the process can be oxidized (NbO) or reduced (V2O5, Nb2O5). Using this procedure, polysulfides TiS3, VS4, NbS3 and various disulfides were obtained at 350-400 °C. Finally, the solvothermal reaction between WO3·0.33H2O and thioacetamide has recently been used to prepare the first microcrystalline WS3.176
3.3. Synthesis from salts and molecular precursors
Methods for obtaining IV-VII group metal polychalcogenides in solutions, as well as methods of vapour deposition using salts or complex metal compounds, have also been developed. As starting metal-containing compounds, one can use tetrachlorides (to prepare TiS3, ZrS3 and VS433,86), thiocomplexes (MoS3), carbonyls M(CO)6143,144,155 (polychalcogenides of Cr, Mo, W) and other coordination compounds. The products of such syntheses are generally X-ray amorphous.
The reaction of ZrCl4 with thiourea CS(NH2)2 under solvothermal conditions (160-230 °C) produces ZrS3 nanocrystallites.33 It is assumed that one stage in the production of ZrS3 is the formation of an intermediate complex via the coordination of the thiourea S atom with the ZrIV atom. In this regard, it was noted that the synthesis was successful only in solvents having no coordination ability towards zirconium atoms, such as n-hexane, toluene and cyclohexane, while pyridine or ethylenediamine, capable of complex formation with ZrIV, hampered the reaction with thiourea. When sulfur or carbon disulfide was used instead of thiourea, the reaction did not proceed.
As sulfurizing agents in the syntheses from halides at a temperature of ≤160 °C, bis(trimethyldisil)thiane, di-tert-butyl disulfide, di-tert-butyl sulfide and hydrogen sulfide are also used. The following equations describe the synthesis of amorphous VS486 and MoS3236,237 using bis(trimethyldisil)thiane:
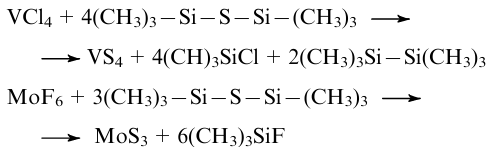
Sulfurization reaction using di-tert-butyl disulfide (CH3)3C–S–S–C(CH3)3 is outlined in a quite detailed study on the synthesis of amorphous and crystalline titanium sulfides.245 Thus, crystalline TiS3 was prepared by a two-step procedure: first, a precipitate of amorphous titanium sulfide was obtained by reacting TiCl4 with di-tert-butyl disulfide, then, it was heated with excess sulfur in an evacuated sealed ampoule; pure TiS3 was formed in a temperature range of 300-500 °C.
Reacting the same reactants, TiCl4 and di-tert-butyl disulfide, under chemical vapour deposition (CVD) conditions afforded amorphous titanium sulfide TiS3 at a temperature below 260 °C.246 Interestingly, the reaction of other sulfur sources with TiCl4 yielded TiS2.
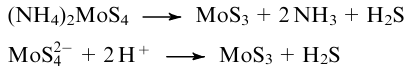
Preparation of amorphous VI group metal polysulfides MS3 (M=Cr, Mo, W), MoS4.7, WS5 can be achieved by solvent chemical methods and electrochemical deposition on various surfaces (see reviews119,121,191). A well-known procedure to prepare trisulfides MoS3124,138,203,232--235 and WS3,232--234, 238 and also MoSx136,139 comprises thermal decomposition of (NH4)2MS4 (M=Mo, W) or acidification of their aqueous solutions:119
Electrodeposition method allows to obtain MoS3 film samples of tens or hundreds of nanometres thickness from MoS42- solutions on various surfaces (indium--tin oxide (ITO), fluorine-doped tin oxide (FTO), glassy carbon).124,131,138 For example, 20-30 nm thick MoS3 layers were fabricated via oxidation of an aqueous solution of (NH4)2MoS4/KCl at a potential of +0.11 V (vs RHE) for 100 s:138
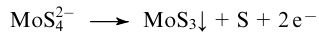
Similarly, it was shown173 that various amorphous ReS4 species can be produced by electrodeposition of a Et4NReS4 solution in acetonitrile (at 0.6 V vs AgCl/Ag) for 2 min, this reaction being reversible: at 7900 mV, the precipitated solid dissolved to give (ReS4)–. In the same study, amorphous ReS4 was prepared by oxidation of Et4NReS4 with iodine and ferrocenium salt (Cp2Fe)PF6 in acetonitrile. Based on a set of methods, ReS4 is shown to be a superposition of two states, ReV(S2–)(S22-)1.5 and ReVI(S2-)2(S22-)1. It was also found out that a commercial product Re2S7 (Aldrich), related to tetrasulfide ReS4, is a superposition of two states such as ReIV(S2–)0.5(S22-)1.5 and ReV(S2–)1.5(S22-)1. Therefore, the Re7+ ion in thioperrhenate ReVIIS4-, in which it is stabilized by four terminal sulfide groups, acting as strong π-donors, is reduced in these reactions allowing some of the sulfide groups to oxidize to disulfide ones.
Sulfur-rich molybdenum polysulfides MoS5, MoS5.6 and MoS6 in the form of amorphous solids were derived from solutions of (NH4)2Mo2S12 in various media (acetone, I2/DMF, aqueous HCl).145,146,152,240 Interestingly, under hydrothermal conditions, the species of non-stoichiometric polysulfide MoS5.6 grew in the original morphology of hollow spheres, which is probably due to the slow steps of nucleation and particle growth.152
Also, TMPCs derived from (NH4)2MoS4 were produced in the form of aerogels, which are fluffy porous structures.167 For this purpose, a solution of (NH4)2MoS4 in formamide/DMF was treated with iodine to afford amorphous molybdenum polysulfide as a paste. The precipitate was allowed to age for a month, washed repeatedly with a water-alcohol mixture and then with alcohol and dried in supercritical CO2 at 42 °C and a pressure of 1400 psi (≈95 atm). As a result, chalcogenide aerogel (chalcogel) MoSx (x≈3-4) was formed. According to PDF and XPS data, the aerogel is a network having a residual negative charge; its structure consists mainly of trinuclear [Mo3S13]2– clusters and the aerogel formula was determined as (NH4)0.2MoS4. The product features high specific surface area (up to 370 m2 g–1) and porosity, hence, exhibits good sorption capacity towards iodine, mercury and several gases. This study is a continuation of a cycle of publications on the synthesis and investigation of chalcogels of other transition metals.165
Several procedures to prepare molybdenum polychalcogenides from molybdenum(VI) oxygen compounds have been reported. For instance, MoS3 was prepared by reacting MoO3 with Na2S in an acidified aqueous solution.127 The study126 used ammonium heptamolybdate (NH4)6(Mo7O24)·4H2O and Na2S as a sulfur source to produce MoS3. An alternative approach is to use sulfur: heating a mixture of (NH4)6Mo7O24·4H2O+Sex in propylene carbonate furnished MoS≈3 and MoS≈4 nanoparticles of 15-29 nm size.148 Apparently, under these conditions, both sulfur and the solvent per se had reductive properties. Potato starch was used as a particle growth inhibitor. A mixed amorphous tetrachalcogenide MoSe3S was prepared by bubbling a mixture of hydrogen sulfide and hydrogen selenide through a solution of ammonium molybdate in aqueous ammonia.157
Sodium molybdate was also used in the reaction with elemental selenium to produce amorphous molybdenum triselenide MoSe3.204 Reactants were heated under hydrothermal conditions in 80% aqueous hydrazine at 180 °C for 12 h. The Se 3d XPS spectrum of amorphous MoSe3 showed peaks corresponding to Se2– and Se22- in a ratio of 1/1, which allowed the triselenide formula to be defined as Mo4+(Se2-)(Se22-). Heating of purified MoSe3 in a dynamic vacuum at 450 °C gave crystalline MoSe2, which demonstrates the similarity of the chemical behaviour of this substrate with other metal trichalcogenides.
Sulfur-rich amorphous polysulfides CrS3, MoS5 and WS5 and their polyselenide analogues are produced when treating carbonyls M(CO)6 with an excess of sulfur and selenium, respectively, in 1,2-dichlorobenzene at 160 °C (seeFig. 2f).143,144,155,174
3.4. Synthesis of nanosized transition metal polychalcogenides using templates and surfactants
It is of particular interest to develop synthetic approaches to produce TMPC materials on the nanoscale. Carbon templates are often used in the synthesis, which help to stabilize the metal polychalcogenides on the surface and also affect the composition of the product and sometimes even facilitate the formation of novel compounds in the form of nanostructures.
Recently, nanosized tellurides TaTe3,231 NbTe3, VTe3 and TiTe3,230 were prepared, whose crystal analogues were not previously reported and whose structures were not described. These phases were stabilized as thin crystals consisting of several {MTe3}∞ chains, with structures similar to that of the infinite chains in the NbSe3 structure (a member of the trichalcogenide family, such as NbS3, see Fig. 1f). The above compounds were synthesized within the cavity of multiwalled carbon nanotubes: they were formed on heating stoichiometric quantities of a metal and tellurium in the presence of iodine vapour and multiwalled end-opened carbon nanotubes at 520-625 °C. Single- and many-chain specimens of Ti, V, Nb tritellurides are probably stabilized by the carbon nanotube walls, which act as suitable nanoreactors.
Nanosized quasi-1D VS4 (Ref. 87) was grown using graphitic templates (graphene, graphene oxide, carbon nanotubes, perylene-3,4,9,10-tetracarboxylic dianhydride) in the presence of Na3VO4 and thioacetamide C2H5NS under hydrothermal conditions, which has been confirmed by numerous follow-up publications.88--90,92--95,99,101--103,110 In some cases, the method was modified, e.g., by the addition of a surfactant.109,111 Graphene oxide was found to contribute specifically to the formation of the VS4 phase.
However, the template-free synthesis of VS4 accomplished under hydrothermal conditions using the same reactants, vanadate Na3VO4 and thioacetamide C2H5NS, with careful control of experimental conditions was also reported.1,96,106 For example, it was shown that increasing the reaction time up to 48 h favours the formation of VS4, and to prevent oxidation of vanadium to its oxides, it is important to reduce the reaction temperature from 180 to 140-160 °C.91 The authors note that the change in pH of the aqueous solution from 5 to 12 has no significant effect on the crystallinity of VS4 but is a factor in controlling morphology of the resulting particles. The study96 also provides the method for coating VS4 microspheres with organic polymer films. Moreover, in the absence of a structural template, VS4 amorphous films can be produced via an atomic layer deposition onto aluminium oxide using vanadium(III) tris-(N,N|'|-diisopropylacetamidinate) (V(amd)3) and H2S.100 The process occurred at a temperature <200 °C; in the first half-cycle (the pulse of a vanadium precursor followed by nitrogen purging) the parent vanadium complex lost its organic ligands, while in the second half-cycle (the pulse of 4% H2S/N2 followed by nitrogen purging), hydrogen sulfide generated S22- disulfide moieties followed by oxidation of V3+ to V4+. At higher temperatures, V2S3 films were obtained.
Transition metal polychalcogenide nanomaterials are also accessible by solvent methods carried out in the presence of surfactants, as in the case with nanoparticle synthesis of many other compounds. For example, 2D nanoplates of ZrSe3 and HfSe3 were produced from ZrCl4 and HfCl4 and selenium in oleylamine, a high-boiling solvent with coordinating and stabilizing properties.229
MoS≈3 and MoS≈4 nanoparticles of 15-29 nm size were prepared by heating a solution of ammonium heptamolybdate with excess sulfur in propylene carbonate with potato starch added, which was used as a particle growth inhibitor.148 After the process is completed, the particles were heated to 400 °C in a flow of nitrogen or hydrogen to afford highly porous MoS2.
Nanoparticles of MoS3 and ReS≈5 of 8-110 nm size prepared from thiometallates were explored as friction modifiers for mineral lubricating oils. Several tetraalkylammonium thiometallates (Alk)4NMoS4 and (Alk)4NReS4247 were subjected to thermosolvolysis in DMF at 155-165°С in the presence of alkenyl succinimide.
3.5. Ultrasonic exfoliation of bulk to nanosized samples
Polychalcogenides in the form of nano-objects can also be produced via post-synthetic treatments, which enable the isolation of species of a certain dispersibility and/or possessing given properties. For example, this can be achieved through exfoliation of their bulk analogues by mechanical28,29,52,58,59,61,71,73,77 or physicochemical8,53--57,72,75,76,248 means or their combinations.69 However, it should be noted that high anisotropy of such materials (width/height ratio) makes mechanical exfoliation very difficult in practice.
Liquid-phase exfoliation results from overcoming the weak van der Waals interactions in the polychalcogenide structure and thereby separating the layers or chains that make up the bulk material. This group of methods is to a great extent based on a large body of literature data on exfoliation of other weakly bonded materials249--251 such as graphite/graphene,252 MoS2-like transition metal dichalcogenides253 and hexagonal boron nitride.254 Generally, a layered material is treated with solvents under ultrasonication (US); to remove unexfoliated bulk materials, centrifugation is applied. As a whole, US-stimulated interaction between the material and the solvent can be represented as a following sequence: 1) immersion into the solvent, 2) exfoliation and 3) stabilisation of the colloidal solution containing the nanoplates. The width of the resultant plates, their lateral dimensions, concentration and other characteristics depend on the experiment conditions. Ultrasonication parameters, the solvent nature and the composition of the liquid medium have the greatest impact on the properties of the resultant dispersions.
In this regard, of particular interest is liquid-phase chemical exfoliation of polychalcogenides. Some of the first experiments in this direction were carried out in our laboratory with VS4 and MQ3 (M=Ti, Zr, Nb, Ta).8,54--57,75,83,248 We have systematically explored the dispersion of polychalcogenides in liquid media and comprehensively characterised the resulting products by modern physicochemical methods. It was shown that under direct ultrasonication, bulk trichalcogenides NbS3, NbSe3,54,55,75 TaS3,56 ZrS3 (Ref. 83) and tetrachalcogenide VS48 are consistently dispersed in a series of organic solvents (Fig. 9).
![[{"id":"mK6dgIykmS","type":"paragraph","data":{"text":" Schematic illustrating the process of exfoliation crystal line samples to nanoscale by the example of VS<sub>4</sub> (<i>a</i>),<sup>8</sup> and TEM images of the products obtained in the form of VS<sub>4</sub> nanorods (<i>b, c</i>);<sup>8,98</sup> ZrS<sub>3</sub> nanosheets (<i>d</i>);<sup>53</sup> TiS<sub>3</sub> nanoribbons (<i>e</i>);<sup>76</sup> ZrS<sub>3</sub> particles (<i>f, g</i>).<sup>83</sup> Reprinted with the permission of Wiley, American Chemical Society, Royal Society of Chemistry, Springer, Elsevier."}}]](/storage/images/resized/4TUnBpBF0I9V50DNspRNrbymTGdRvzBGbSqsGffH_xl.webp)
The most stable and concentrated colloids (up to 0.4 g L–1 for NbS3 and NbSe3 and up to 0.3 mg L–1 for VS4) are formed in isopropanol and water-ethanol mixture, which distinguishes polychalcogenides from dichalcogenides and graphene, which are best dispersed in DMF and N-methylpyrrolidone. Transformation of TaS3 into colloid occurs only in acetonitrile and DMSO.56 According to the data of atomic force microscopy and photon correlation spectroscopy, colloidal NbS3 and NbSe3 contain thin, well-crystallized nanostructures with a high linear size ratio, which allows us to term them as ʹnanoribbonsʹ (50-200 nm length, 1-20 nm width).
Trichalcogenide nanoribbons obtained in colloids are thinner and shorter than nanoribbons synthesised from elements (the bottom up method, Section 3.1), where the thickness of NbSe3 species ranged from 20-30 nm to 95 nm.36,42 In the case of VS4, individual particles are shaped like rods or elongated plates several hundred nanometres long (Fig. 9b,c).8 Interestingly, TEM images show that VS4 nanorods align themselves in an end-to-end fashion, which is probably due to the features of their edges, e.g., dangling bonds and functional groups. Another indirect proof is that the colloidal nanostructures are charged (zeta potential is –44 mV (NbSe3/alcohol),54 –34 mV (VS4/alcohol),8 –12.6 mV (TaS3/acetonitrile) and +20.2 mV (TaS3/DMSO)56). Filtration or spraying of the colloids produces textured thin films with good conductivity, volt-ampere characteristics55 and other transport properties.57 For example, a colloidal solution of ZrS3 in isopropanol was used to sputter ZrS3 on the surface of perovskite CH3NH3PbBr3 as an electron-injection layer in an LED material.255
Besides the works of our scientific group, one of the few examples of obtaining polychalcogenide nanostructures in colloidal dispersions is the study53 proposing a two-step procedure to prepare 2D ZrS3 comprising an initial treatment of ZrS3 with n-propylamine in an autoclave at 120 °C for 3 days followed by dispersing the washed product in a mixture of ethanol and 1-cyclohexyl-2-pyrrolidinone under ultrasonication. The resulting nanostructures have proven efficient catalysts for oxygen evolution reaction. Finally, another study made use of a sequence of chemical liquid-phase dispersion in ethanol and mechanical exfoliation of TaSe3 to produce TaSe3 nanowires up to several millimetres long and lateral dimensions of ≈20-70 nm.69 The obtained species were then modified with hexagonal boron nitride (h-BN), which prevented oxidation of the edges of the TaSe3 nanostructures, and the resulting heterostructures were used in model electronic devices.
The question arises, how exactly does the transformation of the bulk MQ3 powder into a colloid occur? Theoretically, there are three possible cutting directions for a nanocrystal (Fig. 10): (1) parallel to the plane ab (direction 001) — exfoliating the layers from the bulk phase, (2) parallel to the plane bc (direction 100) — breakage of the interchain linkages and (3) parallel to the plane ac (direction 010) — ʹcutting' the chains length.
![[{"id":"-j_G4MYg09","type":"paragraph","data":{"text":"Schematic representation of NbS<sub>3</sub> crystal cutting potential directions during liquid phase exfoliation.<sup>55</sup> Reprinted with the permission of the Royal Society of Chemistry."}}]](/storage/images/resized/zTZ1QJa8WDKV92QzAtTWPmofTW7lYIN2CGeeLJmK_xl.webp)
As thin nanoribbons are found in the colloids, it is likely that exfoliation occurs parallel to the ab plane as expected, given that only weak van der Waals bonds between the layers need to be overcome to separate layers in this direction. Exfoliation and stabilization of NbS3, NbSe3, ZrS3, and also VS4 in the form of colloidal nanoribbons (nanorods for VS4) appear to be the result of solvent intercalation into the spaces between the columns, leading to material exfoliation and stabilization of the resulting colloids by solvent molecules with suitable surface energies. Bulk particles may also be cleaved in other directions, for example, nanoribbons may be ultrasonically cut perpendicular to the ribbon axis, breaking not only van der Waals but also stronger covalent bonds, which is most clearly observed for VS4.8
The question of which particular properties of a solvent render it capable or incapable of dispersing a layered material has been discussed from various standpoints. On the one hand, intermolecular interactions with solvent molecules, which do not provide covalent bonds but are strong enough to overcome the forces of attraction between nanosheets, are considered. On the other hand, recent studies on dichalcogenides MQ2 (M=Mo, W, etc.) show that interactions at the solvent–MQ2 interface may be of a more complex, chemical nature, determined in particular by the solvent's ability to form chemically active species during sonolysis.256--259 Probably, the same applies to polychalcogenide materials.
Depending on how the TMPC nanoparticles are prepared, different methods are used to determine the size of the particles, in particular microscopic methods (scanning and transmission electron microscopy, sometimes atomic force microscopy). As for colloidal solutions of TMPC nanoparticles, colloidal chemistry methods usual for sols of other nanoparticles are applied. For example, particle size distributions are obtained from photon correlation spectroscopy data, possibly followed by modelling of the distributions with an idealized particle shape (e.g., within the framework of Deryagin-Landau-Verwey-Overbeek theory). Since the above methods of particle shape and size characterisation are not specific to TMPCs, we did not focus on them in this review.
4. Chemical properties
Since polychalcogenide materials are very diverse, in this review we tried to summarize some of their characteristic chemical properties. Many of the interesting chemical properties of TMPCs stem from the presence of S–S fragments therein.
When heated in a vacuum, TMPCs lose some of their chalcogen atoms and eventually transform to dichalcogenides. In particular, trisulfides of Ti, Zr and Hf are directly converted to disulfides, amorphous pentasulfides first lose sulfur to give trisulfides, and then disulfides,260 whereas Nb2Se9 when heated in vacuo, transforms to NbSe3.242 The transformation of NbS3 nanoribbons into NbS2 nanosheets has recently been studied in detail by in situ electron microscopy (Fig. 11).261
![[{"id":"Ja5eV1ssEi","type":"paragraph","data":{"text":"Structure of pristine NbS<sub>3</sub> (<i>a</i>). When heated in vacuo to 1000 °C, the loss of sulfur occurs (marked in yellow), the bonds are then rearranged, and NbS<sub>2</sub> is formed; schematic representation of the topotactic transformation of quasi-1D NbS<sub>3</sub> to quasi-2D NbS<sub>2</sub> (<i>b</i>).<sup>261</sup> Reprinted with the permission of the American Chemical Society"}}]](/storage/images/resized/VE92jyvh6ychdO3Wx1bjDTbgpPo8uZitJno2IbEH_xl.webp)
It is likely that on heating to 500 °C, each (S2)2– loses one sulfur atom, less strongly bonded to niobium than S2– (Fig. 11a). The atoms then need to be rearranged so that the existing chains are transformed into layers (Fig. 11b). Bonds are formed between each Nb atom and two additional S atoms from neighbouring chains, which is promoted by pre-existing Nb . . . S interactions. As a result, a half of the new {NbS6} polyhedron of the NbS2 building block is constructed.
As these chemical changes occur, niobium sulfides ʹfuse' to form continuous sheets of NbS2 made up of {NbS6} polyhedrons sharing their edges.
Heating the trichalcogenide to a temperature sufficient to remove some S22- disulfide groups but not sufficient for the complete conversion of MX3 to MX2, is used to create targeted defects on the disulfide groups (M=Ti,262 Zr (Ref. 263)). For this purpose, annealing of ZrS3 (at 700 °C) or TiS3 (at 550 °C) in a sealed ampoule for 10--30 min is carried out. This produces no disulfide, since to obtain TiS2, temperature should be >600 °C,40 while for ZrS2 it should be 820 °C,39 but the S22- vacancies arise (the formula was determined as ZrSS2-x or TiS3-x), affecting the catalytic activity of the species in the reactions of photochemical hydrogen evolution262 or hydrogen peroxide synthesis.263
On heating sulfur-rich amorphous pentasulfides MoS5 and WS5, sulfur is lost in two steps: first, trisulfides MS3 (M=Mo (190 °C), W (240 °C)) are formed, then poorly crystalline MS2 disulfides of spherical morphology similar to the pristine MS5 (M=Mo (380 °C), W (300 °C)).260 The kinetics of their thermal decomposition was studied based on thermogravimetric analysis and it was shown that both steps are described by the Avrami-Erofeev model including random nucleation and subsequent nucleate growth. The rate-limiting step is the diffusion of sulfur from spherical MS5 particles. Theoretical modelling results suggest that the first step of thermolysis is depolymerization of MS5 chains to afford cluster units followed by desulfurization.
Amorphous molybdenum sulfides transform into cluster compounds through a number of chemical reactions.122,203,264--268 Thus, amorphous MoS3 reacts with an aqueous solution of sodium cyanide to give an anionic cluster complex [Mo3S4(CN)9]5– and a small amount of [Mo4S4(CN)12]8– (Refs 122, 203, 266), while the reaction of MoS3 with ammonia or aqueous KOH affords complex [Mo3S13]2– (Refs 203, 265, 267). At the same time, the cluster anion salt [Mo3S7Cl6]2– was isolated from the reaction of MoS3 with concentrated HCl.203 At first, these experiments were carried out to establish the structure of amorphous MoS3: it was assumed that cluster units are excised from the polycrystalline material and therefore, MoS3 is described by a cluster model (see Fig. 2b), rather than a chain one (see Fig. 2a). Nevertheless, there is an ongoing debate on this topic, since there may also be a sequential degradation of the chain structure and then — an assembly of cluster units in solution: for example, the fusion of two Mo2 dimers and monomer extraction may produce a Mo3 metal cluster.123 In any case, the findings once again confirm the close relationship between polychalcogenide structures and molecular clusters, at least in the case of the ʹMo–S' system.
We have recently described the formation of cluster complexes from the sulfur-rich amorphous pentasulfides MoS5 and WS5.268 These compounds react with molten thiuram disulfide to furnish triangular cluster complexes comprising {Mo3S7}4+ and {W3S4}4+ cores, and the reaction between MoS5 or WS5 and halogenating agents (CHCl3 and Br2) gives well-known coordination polymers M3S7Hal4 (M=Mo, W; Hal=Cl, Br). Assuming that the parent MS5 has a chain-like structure based on M2S8 dimers or M3S12 trimers,174 it can be stated that the rearrangement of clusters depends on the reaction conditions. Remarkably, molybdenum formed only clusters with {Mo3S7}4+ core, and tungsten formed two different cluster cores depending on conditions: {W3S4}4+ in dithiocarbamate complex and {W3S7}4+ in coordination thiohalide polymers.
Transition metal polysulfides, like many other layered and chain compounds, are characterized by metal ion intercalation reactions, primarily the electrochemical intercalation of Li+ in electrodes for lithium-ion batteries (LIBs), but also other metals such as Na+, Mg2+, Zn2+, Al3+ to create Na-ion,93,99,102,106,107,111 Mg-ion,109,269 Zn-ion270,271 and Al-ion110 batteries (see Section 5.1). In intercalation reactions, the electron density is redistributed between guest and host and in the case of polychalcogenides, this means electron transfer to disulfide groups and elongation or cleavage of the Q–Q bonds: (Q–Q)2–+2e–→2Q2–. In addition to intercalation, a conversion mechanism can also take place, where the reaction is completed to form (nano)transition metal species and lithium chalcogenide. The reversibility or irreversibility of the intercalation process depends on the specific compound and the amount of inserted lithium. Recently, using NbSe3 as an example, it was shown272 that a particular reaction pathway also depends on the size of the chalcogenide species (Fig. 12). During electrochemical intercalation of lithium cations into NbSe3 species with lateral sizes of several hundred nanometres, the lattice is capable of relaxation and accomodation of Li+ ions in the interchain/interlayer space. On the surface of the particles, however, the conversion process yielding Li2Se (NbSe3+6Li++6e–→Nb+3Li2Se) occurs. The same happens during lithiation of particles <100 nm in size, where a nanocomposite containing Nb nanoparticles on Li2Se is formed very rapidly.272 It is obvious that metal ion intercalation is of direct practical interest for LIB applications, which is the subject of Section 5.1 of this review.
![[{"id":"feod2oOzXo","type":"paragraph","data":{"text":"Different mechanisms of interaction of NbSe<sub>3</sub> with lithium cations depending on the particle size: large particle size favours intercalation, while small particle size favours conversion process (formation of Nb + Li<sub>2</sub>Se).<sup>272</sup> TMC - transition metal chalcogenides. Reprinted with the permission of the American Chemical Society."}}]](/storage/images/resized/0cyYW33osUs4vFkIErXnb8oUX4mo8xQkroUUb8T2_xl.webp)
In addition to electrochemical intercalation of metal ions into TMPCs, intercalations of hydrazine into TaSe3 (Ref. 273) and ammonia into NbS3 were reported.274 The intercalation of K+, Rb+, Cs+ and Ca2+ cations into ZrSe3 via a chemical process using alkali metal and calcium solutions in ammonia in an inert atmosphere at reduced temperatures has recently been described in detail.275 Intercalation of alkali metal ions from NH3 solution at –78 °C into ZrSe3 furnished intercalates K0.45(2)ZrSe3 (space group Cmc21), Rb0.41(1)ZrSe3 (space group Immm) and Cs0.42(1)ZrSe3 (space group Immm). Compared to ZrSe3, the interlayer spacing in the products increased by 16, 20 and 25%, respectively, as expected based on the large sizes of cesium and rubidium atoms. The Zr–Se distances (2.7654(2) Å for ZrSe3) showed no significant changes: for example, in potassium intercalates, this value was 2.789(7) Å, and the oxidation state of Zr remained at 4+. The main change affected the Se–Se distances due to their partial reduction: in ZrSe3, the Se–Se distance was 2.3441(6) Å, and in Rb0.41(1)ZrSe3 and Cs0.42(1)ZrSe3 it was 2.592(4) and 2.605(5) Å respectively. The intercalation with Ca2+ from the Ca/NH3 solution proceeds with co-intercalation of ammonia molecules, so that the product formula is expressed as Ca0.2(NH3)0.45(7)(NH2)0.15(7)ZrSe3 and the interlayer distance is 10 % larger than in the potassium intercalate. The reaction with metal might afford not only an intercalate but, under certain conditions, a material with selectively formed sulfide ligand vacancies (S2–), which was shown in a study263 for ZrS3, treated with lithium solution in ethylenediamine at 120 °C.
The surface properties of polychalcogenide particles differ from those of surfaces formed only by S2- atoms. Thus, we observed oxidative properties of (S–S)2– units in the reactions of VS4 and NbS3 with silver particles.98 The reduction of silver salts in the presence of ʹnormal' MoS2-type sulfides (devoid of the S–S bonds) stabilizes silver metal particles (Ag/MoS2) on the sulfide surface (in situ approach). Unlike MoS2, polysulfide materials under similar conditions favour formation of Ag2S nanoparticles. This process can proceed in two ways (Fig. 13): in a one-step (a) or two-step (b) fashion with the intermediate formation of silver metal nanoparticles. The (S22-) disulfide groups act as oxidant in both cases. The second path (two-step) is also accomplished when mixing a colloidal polychalcogenide (VS4, NbS3) with pre-synthesized Ag0 sol (ex situ approach).
![[{"id":"RHisFzyTSl","type":"paragraph","data":{"text":" Schematic of two possible pathways for the for mation of Ag<sub>2</sub>S nanoparticles anchored to VS<sub>4</sub> nanorods: the direct formation of Ag<sub>2</sub>S when a reducing agent is added to VS<sub>4</sub> colloid containing Ag<sup>+</sup> (<i>a</i>); a two-step process in which Ag<sup>+</sup> ions are first reduced by sodium citrate to form Ag<sup>0</sup> nanoparticles, and then oxidized to Ag<sub>2</sub>S by disulfide groups (<i>b</i>).<sup>98</sup> Reprinted with the permission of Wiley"}}]](/storage/images/resized/I9w1lm5fsG8MTsUh4SbOFzu6fYRpZ00E7pDUXJlL_xl.webp)
To summarize, ʹAg/VS4' or ʹAg/NbS3' species cannot be obtained: the silver is stabilized as a sulfide. Ag/MoS2, Ag2S/NbS3 and Ag2S/VS4 samples were characterized by a number of methods and it was shown that the unexpected formation of Ag2S instead of Ag on polysulfide surfaces is probably due to complex redox processes involving S-S disulfide units, which are absent in MoS2, but are present in VS4 or NbS3 structures. This observation is of interest both for a fundamental understanding of the properties of sulfur-rich surfaces and in terms of a new synthetic approach to Ag2S nanoparticles as part of composites for various applications.
Similar observations regarding the reactivity of (S–S)2– groups were described by Afanasiev et al.276 when studying the reactions of amorphous molybdenum polysulfides MoSx with H2. At room temperature, polysulfide groups irreversibly react with hydrogen, resulting in the cleavage of S–S bonds and the formation of labile –SH functional groups, which escape from the surface as H2S in the hydrogen flow:
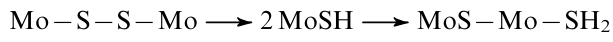
The amount of absorbed hydrogen directly depended on the number of S–S bonds, which varied over a wide range.
In the case of MoS3, it was shown that the surface bridging S-S groups are also able to reversibly bind water molecules, which was used to create moisture sensors.182
5. Potential applications
5.1. Metal-ion batteries
Ideas of using transition metal chalcogenides in lithium-ion batteries date back to the 1970-1980s, in particular to the works of M.S.Whittingham,277 who was awarded the 2019 Nobel Prize in chemistry. Both di- and trichalcogenides were investigated then, but since the technological breakthrough came with the use of LiCoO2-type oxide materials, chalcogenide materials faded into background for some time.
However, in the last 5-10 years interest in chalcogenide materials has renewed, and experimental evidence appeared that new generation electrodes with increased capacity can be developed on their basis.1,278 Moreover, use of TMPC-based electrodes allows successfully solving the problem preventing development of Li-S batteries, namely, an escape of polysulfide anions (polysulfide shuttle effect) into liquid medium, which lead to corrosion of anodes. Metal–sulfur covalent bonds in TMPCs inhibit this process. Many state-of-the-art works use nanosized TMPCs in metal-ion batteries, but at the same time, the effect of electrode particle size on battery performance is ambiguous since, on the one hand, diffusion of lithium (or other metal) ions in the nanomaterial is facilitated, but on the other hand, side `harmful' reactions of the electrode nanomaterial with the electrolyte are simultaneously accelerated, impairing cyclability and stability.
Among chalcogenides, both di- and polychalcogenides are studied, but it is noteworthy that when made into electrodes, they operate following remarkably different mechanisms. Under reversible intercalation of lithium ions, TiS2 dichalcogenides form LixTiS2 due to redox conversion at cationic centres: Ti4+/Ti3+, similarly to the way it occurs in classical oxide electrodes LiCoO2, LiMn2O4, LiFePO4 (Fig. 14a).
![[{"id":"6-SOEwDMoD","type":"paragraph","data":{"text":"Two different mechanisms of redox activity of electrode materials. Schematic representation of band structure of conventional oxide electrode materials characterized by higher position of 3d orbitals of the transition metal M in relation to p orbitals of O anions (<i>a</i>). Schematic representation of band structure of the transition metal polysulfude (<i>b</i>). In this case, p orbitals of less electronegative S lie higher (than in O) and can overlap with the metal 3d orbitals, thus enabling the intramolecular electron transfer and triggering the anionic redox process. EF is the Fermi level.<sup>177</sup> Reprinted with the permission of Wiley."}}]](/storage/images/resized/G88SnG5lZIY2h1SZBtquh49ITE1xUiDot8NxsQk9_xl.webp)
However, in the case of polysulfide electrodes other chemical processes are added, because anionic groups which traditionally were considered inactive are actively involved in electrochemical transformations, e.g., S22-+2e-=2S2– (anionic redox mechanism). Such behaviour of polysulfide electrodes is due to the fact that the 3p orbitals of S atoms largely overlap with the d orbitals of transition metal atoms (Fig. 14b), and hence electron density may transfer between them, and therefore, the anionic redox mechanism works, for example, in Li1-xTiS3 but not in Li1-xTiO2.177,278
From a practical point of view, it is important that due to the anionic redox mechanism, more lithium atoms can be involved in the reaction: 2Li+TiS3→Li2TiS3 (as opposed to Li+TiS2→LiTiS2), and therefore an increase in battery capacity with such electrodes can be achieved. Below we will take a closer look at recent data on the use of various transition metal polychalcogenides as electrode components for metal-ion batteries.
In the series of TMPCs, transition metal trichalcogenides MQ3 with quasi-layer structures were historically the first to be tested as LIB electrodes. In the 2000s, such compounds began to be re-explored both theoretically,269,279 and in practice, in particular TiS3,44,68,280 ZrS3,74 NbSe3,78 NbS3.228 The MQ3 interlayer spacing can accommodate up to three lithium ions per MQ3 formula unit, as exemplified by TiS3:

Density functional calculations show that the electron density from the absorbed lithium is largely transferred to the nearest sulfur atoms.269,279 The capacity of an electrode based on crystalline TiS3 in LIB is calculated to be 339 mA h g–1, while for a monolayer it is 260 mA h g–1.279 In practice, higher values are obtained, which is probably due to the use of amorphous materials: it has been observed that amorphization increases the availability of the structure for lithium ions and allows more anionic centres to be involved in the redox process.44,135,149,150,225,226,228,239 Thus, all-solid-state batteries with electrodes based on amorphous TiS3 (particles in the micron size range) reached capacities of 400 mA h g–1,44 while batteries with liquid organic electrolytes reached 350 mA h g–1.280 However, in the latter case, after a few initial cycles the capacity decreases rapidly and is set at around 50 mA h g-1.280 In the case of a solid-state battery, the results are slightly better — 300 mA h g–1 after 10 cycles.44 Maintaining a high capacity during cycling is challenging, since lithium intercalation into TiS3 is only partially reversible.
A significant part of the new research using TMPCs as electrode materials is devoted to sulfur-rich amorphous phases (MSx, where x≥4). Many such materials are produced by mechanochemical synthesis from a lower sulfide, and sulfur and in this process the reagents amorphize and enter the reaction, which is accompanied by a reduction in particle size.
In a series of amorphous niobium polysulfides NbS3, NbS4 and NbS5, the initial discharge capacities raised from 281 (NbS3) and 446 mA h g–1 (NbS4) to 596 mA h g–1 (NbS5), which corresponded to 2.0, 3.7 and 5.6 electrons per formula unit.228 It is assumed that both sulfur and niobium atoms are involved in the redox process, and these reactions depend on each other, since niobium and sulfur are covalently bound in NbSx (as in other TMPCs). However, attempts to achieve stable cycling for NbS5 have so far failed, and capacities drop significantly after the 10th cycle.
Amorphous titanium polysulfide TiS4 has been investigated not only in Li-ion,149--151 but also in Al- (Ref. 225) and Mg-ion batteries.226 When switching from crystalline TiS2 to amorphous TiS3 and TiS4, the capacity of LIB cathodes increased to over 500 mA h g–1 (TiS4) with reversibility over 10 cycles.149
A 4-electron process is assumed. In-depth studies on the mechanism of the TiS4-based electrode150 by PDF, DFT, Ti K-edge and S K-edge XANES techniques show that the typical products of the conversion process, Ti and Li2S, are not formed. Instead, TiS4 is discharged to Li4TiS4, which is accompanied by the cleavage and reduction of the disulfide S–S bonds with a simultaneous change in the coordination number of Ti. The structure turns from a ʹnetwork' type in TiS4 to a chain type in Li4TiS4 and back again.
More stable cycling of amorphous TMPC-based electrodes in all-solid-state batteries is observed: it was possible to optimize the composition of solid electrolytes and achieve stable operation of a battery with a high-capacity cathode based on TiS4 for 100-200 cycles (507.4 mA h g–1) after 100 cycles at a current density of 0.1 A g–1 and 350 mA h g–1 after 200 cycles at a current density of 1 A g–1.239
Promising results were obtained for batteries using TiS4-based cathodes and aluminium anodes: 206 mA h g-1 after 1000 cycles, which is one of the best results for Al-ion devices.225 Its mechanism involves the anionic oxidation-reduction of S22- ↔ 2S2–, lowering the CN of Ti atoms, breaking the S–S bonds and forming Al–S bonds.
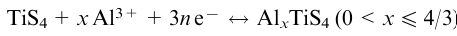
Mg-ion batteries with TiS4-based cathodes operate similarly, as shown by the same group of authors.226
In addition to amorphous titanium polysulfide, amorphous molybdenum polysulfides MoSx (x=3-7) are actively studied as electrode materials for Li- (Refs 68, 130, 135, 153, 281) and Na-ion batteries.140,142,179,180,281 Note281 that amorphous nanosized MoS3 (prepared from (NH4)2MoS4 in the presence of carbon nanotubes) has several advantages over TiS3, TiS4 and NbS3 when used as a LIB cathode with liquid electrolyte: it allows operation at higher potential and is more stable under cycling (Fig. 15a). In all-solid-state LIB,68,135 submicron-sized MoS3 particles prepared using mechanochemical process, showed a capacity of ≈670 mA h g–1 after 60 cycles, with the redox process affecting both sulfur and molybdenum atoms.
![[{"id":"OiMkS6Xbpv","type":"paragraph","data":{"text":"Stable cycling of MoS<sub>3</sub> (<i>a</i>)<sup>281</sup> and lithium intercalation/deintercalation mechanism in VS<sub>4</sub> on discharge/charge during the first and second cycles (<i>b</i>) in Li ion batteries;<sup>108</sup> reverse Mg<sup>2+</sup> insertion/extraction into/from VS<sub>4</sub> during discharge/charge in magnesium-ion batteries (<i>c</i>).<sup>109</sup> Reprinted with the permission of the United States National Academy of Sciences, Elsevier and Wiley"}}]](/storage/images/resized/KgzvrJzrochJeMnRMsLp2460j54U4u2OlREr8aBV_xl.webp)
Amorphous MoSx samples with sulfur content of x>3 have also been studied. For example, MoS5.7 showed a capacity of 746 mA h g–1 with 86% efficiency retained for 50 cycles in a liquid electrolyte-based battery.153 It was found that the electrode operates by an unusual mechanism, as Li2S2 is formed in a reversible process:
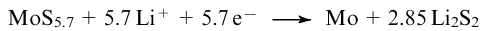
In the case of all-solid-state sodium batteries, the samples of MoSx (x=3-7) synthesized by two different methods such as mechanochemical process and thermolysis from molecular precursors were compared.142,179,180 It turned out that the samples obtained via thermolysis demonstrate higher content of disulfide groups and higher capacity of batteries using such electrodes. For example, MoS6 produced by mechanochemical method shows capacity of 510 mA h g–1 while maintaining efficiency of 80% after 30 cycles, and for MoS6 this value was 690 mA h g–1 while maintaining efficiency of 92%.180 This once again confirms the important role of the anionic redox processes.
The operation mechanism of a cathode based on another related material such as molybdenum polysulfide chalcogel MoS3.4 was studied in detail.169 This substance is a porous amorphous network where the Mo4+ metal centres are linked by polysulfide chains, but are themselves chemically inactive in the electrode process. The polysulfide chalcogel features high gravimetric capacity of 578 mA h g–1, maintained at 83% after 10 cycles and at 60% after 30 cycles, which is comparable to the performance of Li–S batteries. The results of XPS, EXAFS and PDF showed that upon the discharge of the battery, the S–S bonds in the polysulfide chains are cleaved and sulfur is reduced to S2–, and upon its charge, S2– is oxidised to form S–S bonds.169
Another polysulfide that has recently attracted great interest as an electrode material for metal-ion batteries is VS4 with a quasi-chain structure, which is one of the most investigated TMPCs for battery applications: a recent review282 is devoted entirely to electrodes based on this material. Vanadium tetrasulfide and its composites with graphene oxide were explored as electrode materials for Li-ion,87,89,90,94,96,102,105,108 Na-ion,93,99,102,106,107,111,283 Mg-ion,109,284,285 Zn-ion270,271 and Al-ion110,286 batteries. VS4-based electrodes (nanoparticles 50-500 nm) exhibit stable performance with high capacity: 727 mA h g–1 after 50 cycles even at currents up to ≈3 A.87,89 In continuation of the research by Xu et al.,89 Britto et al.90 carried out an in-depth study on complex and unusual redox processes occurring in VS4-based electrodes involving both anion and cation. The products on different cycling stages were examined by solid-state NMR (6Li and 51V), S K-edge XANES and PDF analysis. Upon lithiation, sulfur dimers (S-S)2– first degrade to give an intercalated phase with a stoichiometry close to Li3VS4. In this compounds, formal oxidation states of V and S are 5+ and 2- respectively. Therefore, the conversion of VS4 to Li3VS4 is characterized by an unusual redox reaction comprising an internal electron transfer from V4+ to (S–S)2– disulfide groups. As lithiation proceeds, once all the disulfide groups are reduced to sulfide ions, V5+ is partially reduced to V4+ in the Li3+xVS4 intercalate. Finally, V metal and Li2S are formed, and the reverse reaction occurs, as upon charging, Li3+xVS4 is gradually formed again followed by amorphous VS4. These interesting data show that the reversibility in this system is not exclusively due to the Li2S↔S reaction (which is similar to the process occurring in Li–S batteries), as previously assumed, but involves the metal V and at least partial conversion to the pristine phase.
The study108 notes that lithiation and delithiation of VS4 (nanoparticle size of <100 nm) in the first and subsequent cycles follow different mechanisms (see Fig. 15b). Lithium sulfide is only formed in the first cycle, after which it converts back into VS4, but now in an amorphous state, and then the following reaction occurs: VS4+yLi++ye–↔ LiyVS4 (0<y<8) with reversible oxidation-reduction of the S22- ↔2S2– disulfide groups. Thus, a mixed mechanism is confirmed, and not exclusively intercalation or conversion to V/Li2S.
In contrast to Li-ion batteries, in Mg-ion devices, magnesium intercalation into VS4 nanodendrites does not furnish alkaline-earth metal sulfide (MgS) and V metal (Fig. 15c).109 The reactions at the cathode can be represented as follows:
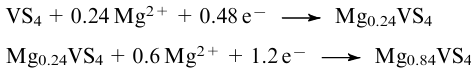
Upon discharge, S22- is partially reduced to S2–, and V4+ is partially oxidized to V5+. During cycling, the samples retain their morphology of nanodendrites (ʹsea urchins'), which affected the stable performance with a capacity of up to 74 mA h g–1 over 800 cycles at a high current density of 500 mA g–1. This example shows that optimizing the morphology and nanostructuring of VS4 and other TMPCs is a promising direction for the development of high-capacity and stably cyclable electrode materials. In addition, the use of novel electrodes requires optimization of electrolyte compositions and other constituents and battery operating conditions, as well as a better understanding of electrode mechanisms, but it can already be said that the potential for the use of TMPCs in this area is very high.
5.2. Electrocatalysis and photocatalysis
5.2.1. Electrocatalytic water decomposition
The hydrogen evolution reaction (HER) using various catalysts has been actively investigated in recent years,287 when the production of green fuels has become an urgent task. In electrolysis, the surfaces of the working electrodes are coated with a layer of catalyst, which reduces the applied potential and hence the energy consumption, and makes the process more economical. The first step in the catalytic process is the adsorption of hydrogen (Hads) onto the catalyst, after which different mechanisms are possible leading to the formation of H2 molecules, but, in any case, an effective catalyst must accelerate such adsorption processes. A plethora of materials have been studied for this purpose, in particular metal sulfides, among which various amorphous polysulfides of molybdenum (MoS3, etc. (see Fig. 2a,b and Fig. 3b)) and tungsten (WS3-x)137 showed promising results.120,121,124,126--128,131--134,136,138,182,288 For catalytic applications, TMPCs in the form of nanosized samples are often used because catalysis and adsorption proceed more efficiently on materials with a developed surface and with a large number of available active sites (in this case, disulfide groups). For example, Fig. 16 compares the characteristics of various MoSx samples. A high reaction rate can be judged by a lower initial potential and a lower overpotential with a more negative current degree.
![[{"id":"ZjBOhYGNmW","type":"paragraph","data":{"text":"Comparison of Tafel curves in HER processes for different MoS<sub>x</sub> samples.<sup>126</sup> Reprinted with the permission of the American Chemical Society."}}]](/storage/images/resized/8S2GJrfvvH7UrzERBksmjGBXXncnObxU4CiwItiQ_xl.webp)
A number of recent studies showed that the activity of MoSx nanomaterials is largely related not to molybdenum atoms,136 but to disulfide groups,127,128,133,134,138 although opinions differ as to whether these are terminal133 or bridging groups.128--138 Thus, direct evidence of the important role of the catalyst disulfide groups in the water electrolysis reaction was obtained from in situ X-ray absorption spectroscopy studies of MoSx film samples.133 It was shown that under catalytic reaction conditions (–0.3 eV vs reversible hydrogen electrode (RHE)) the sample is reduced to an amorphous MoS2 with a surface formed by terminal (η2-S22-) disulfide groups, which are reversibly cleaved and protonated during the catalytic process (Fig. 17).
![[{"id":"J2Xo_EIQ3k","type":"paragraph","data":{"text":"Proposed mechanism of the catalysis in the HER reaction involving (S-S)<sup>2-</sup> disulfide groups as active sites of H<sub>2</sub> formation. The Figure was prepared by the authors using original data from the cited publication.<sup>133</sup>"}}]](/storage/images/resized/78Fx3llwKQJJltaPPxWXW3gGBCaphvJezHDiJsIq_xl.webp)
It was also noted124,126,127,131 that the pristine MoS3 converts to an active phase (activation of the catalyst), which is probably an amorphous MoS2+x containing disulfide ligands. Based on Raman spectroscopy, H/D isotope exchange and DFT calculations, Yeo and co-workers136 provided direct evidence for the S–H bond formation during the reaction of electrocatalytically active MoSx films consisting of 20-30 nm particles. XPS data and density functional calculations of energy of H binding to various sites also support the involvement of disulfide centres in catalysis, although these data show that it is not the terminal, but the bridging disulfide groups that are active.138 The correlation between the turnover frequency of the MoSx electrocatalyst (amorphous films consisting of 20-30-nanometre particles) (Fig. 18a) in the hydrogen evolution reaction, i.e. its activity, and the content of S atoms therein with higher binding energies (163.8/165 eV) (Fig. 18b) based on the XPS data was pointed out.138 The bridging S22- groups and apical S2– groups fall in this region of binding energies. DFT calculations of binding energies of H atoms to these centres in various configurations were performed,134,136,138 and it was shown that apical S atoms (Fig. 18a) bind H atoms the weakest (Gibbs binding energies of hydrogen atom adsorption ΔGH>+1 eV) and, therefore, cannot be considered as active centres (Fig. 18b). At the same time, S atoms in terminal S22- ligands (see Fig. 2b) bind atomic H too strongly, causing passivation and consequently low HER catalytic activity of these centres. Optimal values close to ΔGH=0 were obtained for bridging S22- groups (see Fig. 2b), which appear to be the most catalytically active centres. A possible mechanism for such a catalyst is depicted in Fig. 17.
![[{"id":"zpsvVlPUFe","type":"paragraph","data":{"text":"Dependence of turnover frequencies (TOF) of the MoSx catalyst in the HER vs the proportion of S atoms with high binding energy in XPS spectra (<i>a</i>); the Gibbs energy (ΔG) of hydrogen evolution <i>via</i> the sorption of H atoms on various MoS<sub>x</sub> catalytic sites (<i>b</i>);<sup>138</sup> simulated models for the Gibbs energy calculations (<i>c</i>). Possible hydrogen binding sites are designated along the X axis as 1t and 2t (terminal), 1b and 2b (bridging) and apical. For the 1t and 1b centres, only one S atom is bonded to the corresponding Mo atom. For the 2t and 2b, another S atom is bonded to the same Mo atom. Reprinted with the permission of the American Chemical Society."}}]](/storage/images/resized/72re5xZzKiBo5VnONWX7aBQlW2endeXab4tkcO8b_xl.webp)
Specific synthetic approaches have been developed to increase precisely the content of catalytically active S22- groups. The preliminary reaction of ammonium tetrathiomolybdate with ammonium polysulfide (NH4)2Sx was shown not only to promote the formation of a bridging S configuration but also to increase the total S atom proportion in the samples. The S/Mo=3.5 ratio and bridging sulfur content in MoSx of 67% of the total content (normally in a-MoSx from (NH4)2MoS4 these values are close to 2.8 and 47% respectively) were achieved in this way.These samples demonstrated excellent performance in the HER reaction with low onset potential (–96 mV), overpotential (–0.118 V) and Tafel slope of the polarization curve (46 mV dec–1) at a current of 10 mA cm-2.235
A sample for electrocatalysis with even higher sulfur content, MoS6, was prepared by oxidation of ammonium thiodimolybdate (NH4)2Mo2S12 with iodine. The product represents amorphous particles with a diameter of ≈400-800 nm. The HER with this sample proceeded at an overpotential of 130 mV (current density 10 mA cm–2), while for the MoS4 control sample, this value was higher (178 mV). The authors also attribute the good results obtained for this material to the presence of increased amounts of disulfide groups.240
It should be noted that the important role of disulfide groups in electrocatalysis is observed not only for polysulfide materials, where all or considerable part of sulfur exists as S22-, but also for the extensively studied electrocatalyst MoS2, which is not a polysulfide, but whose edge fragments can be represented by disulfide groups.7,183--185
In addition to their application in the process of electrolytic hydrogen evolution reaction, TMPCs have also been investigated as electrocatalysts for the oxygen evolution reaction. Well-crystallized ultrathin ZrS3 nanosheets with in-plane dimensions at hundreds of nanometres have proven to be efficient and stable (over 1000 cycles) electrocatalysts for this process in both alkaline and neutral solutions.53 In strongly alkaline solutions (pH=14), the onset overpotential was as low as 244 mV (≈500 mV vs Ag/AgCl electrode) and a Tafel slope was 45 mV dec–1, and no side reactions, which often occur using other electrocatalysts, were observed. For crystalline ZrS3, the values of Tafel slopes of polarization curves were significantly higher (≈176 mV dec–1), thus indicating the importance of the dispersion degree of this material. Traditionally, transition metal cations have been considered to be catalytically active centres in water decomposition processes, but this point of view is currently under revision.289 Xie et al.53 emphasize that splitting of low-dimensional structures increases the active surface of ZrS3 nanosheets formed by the S–S disulfide groups, which are probably the active centres of electrocatalysts. Similar observations were also made for a number of pyrites FeS2, CoS2, NiS2, which were explored as catalysts for hydrogen evolution reactions.181
5.2.2. Photocatalytic and electrocatalytic water decomposition
Semiconducting polychalcogenide materials are also actively explored as catalysts for water decomposition reactions under visible light irradiation. In this process, an important role belongs to the light absorbing capacity of the material (i.e. band gap width) and a suitable arrangement of the energy bands (conduction and valence bands) according to the redox potentials of water.288,290--292 Based on half-reaction potentials of water oxidation and hydrogen formation, the bandgap width of a semiconductor photocatalyst should be >1.23 eV, which corresponds to near-IR wavelength λ≈1008 nm, and simultaneously, preferably, <3 eV (>400 nm) to provide the possibility of using not only UV radiation (4% of the solar spectrum) but also visible light (53% of the solar spectrum).292 In reality, however, energy losses are inevitable during electron transfer, leading to kinetic overpotentials and, accordingly, the bandgap of the photocatalyst must be ≈2 eV.288,290 On the other hand, the issue with narrow-gap semiconductors, which can absorb in the visible region, is the probability of e––h+ rapid recombination.88 The positions of the conduction and valence bands in relation to the redox potentials of water are also important:63 for efficient reduction and oxidation of water by photo-excited electrons and holes, the width of the band gap should overlap the values of the potentials of both reactions.291
According to the theory calculations,70 the transition metal trichalcogenides are characterized by the band gap widths in the range of 0.58-2.17 eV. For example, the calculated direct band gap for TiS3 is 1.35 eV and the calculated indirect band gap is 1.21 eV (for ZrS3, 2.16 and 2.06 eV, respectively). The results of the analysis of electronic absorption spectra showed that the absorption of light began at 0.9-1.63 eV, hence, such materials are of interest for application in photovoltaics and photocatalysis, the study of which was started back in the 1980s,293,294 however was actively continued only in the last few years.46,63,72,295
The experimental results proved that n-type semiconductors such as TiS3, ZrS3 and HfS3 can be used as photoanodes,46,63,72,293,294 in which photo-excited holes gather on the surface and are involved in oxidation reactions, while electrons move to the counter-electrode through an external circuit and participate in reduction reactions.292 Thus, TiS3 nanoribbons (width of ≈1-5 μm, length >100 μm, thickness of 50-200 nm) were explored as a photoanode in photoelectrochemical cell to generate hydrogen from 0.5M solution of Na2SO3 (pH=9) under visible light irradiation.63 According to the authors, the band gap (1.1 eV) and the positions of the valence and conducting bands are thermodynamically favorable only for the evolution of hydrogen, but not oxygen, which was also confirmed by the composition of the evolved gas. The quantified hydrogen flux was 1.8 μmol H2 min–1 at 0.3 V (Ag/AgCl) with a quantum efficiency of 7%. The Ti-coated pyrite FeS2 was also studied under similar conditions and showed 2 times lower efficiency compared to TiS3.296 In a follow-up study,72 the same research group tested the samples of TiS3, ZrS3 and HfS3, deposited from dispersions onto supports, as photoanodes. Compared to TiS3 nanoribbons grown on a Ti metal surface, the results were more modest and the recorded hydrogen flux was 50 times lower, probably due to insufficient electrical contact between the particles. However, this work was one of the first examples of the study of HfS3 as a photoanode. In their follow-up study, the authors compared catalytic performance (hydrogen evolution reaction) of four trisulfides with a morphology of nanoribbons such as pure trisulfides of titanium and niobium, and two ternary trisulfides, NbxTi1-xS3 and TixNb1-xS3.163 It was found that niobium-doped titanium trisulfide (<10% Nb) shows slightly higher hydrogen photogeneration than the others (Fig. 19).
![[{"id":"lZdAhNS0k0","type":"paragraph","data":{"text":"Generated hydrogen fluxes (<i>a</i>) and average hydrogen generation rate (<i>b</i>) obtained from various trisulfides deposited on metal plates, as photoanodes (mass-spectrometry data). The experiment was carried out in 0.5M Na<sub>2</sub>SO<sub>3</sub> aqueous solutions with pH=9.5 using a white light source. For all trisulfides, estimated band gap value was 1+/-0.05 eV. Comparative scheme of energy levels of the MS<sub>3</sub>/electrolyte interface in different sulfides (<i>c</i>).<sup>163</sup> Reprinted with the permission of Elsevier."}}]](/storage/images/resized/LQbSlXvdcwChlWOsKJ9Q1PBF5sXX4J0IZmrhcffr_xl.webp)
It was recently proposed to modify a traditional titanium dioxide (TiO2)-based photocatalyst with a TiS3 shell for the application in the photocatalytic water splitting. Such surface modification of a wide band gap semiconductor TiO2 (Eg≈3.2 eV) with a narrow band gap semiconductor TiS3 (Eg<1.2 eV) facilitates the shift of the absorption of the sample towards visible region.295 Indeed, according to the diffuse reflectance spectroscopy data, shift of the absorption onset from 3.14 eV to <2.6 eV and the absorption edge broadening are observed (Fig. 20).
![[{"id":"PtsUD09K8O","type":"paragraph","data":{"text":"Diffuse reflection absorbance spectra of pristine TiO<sub>2</sub> nanofibres (TiO<sub>2</sub> NF) and samples obtained by modifying TiO<sub>2</sub> nanofibres with different reagents: NaOH (Na - TiO<sub>2</sub> NF), thioacetamide (S - TiO<sub>2</sub> NF) and Na<sub>2</sub>S (Na/S - TiO<sub>2</sub> NF). Samples (S - TiO<sub>2</sub> NF and Na/S - TiO<sub>2</sub> NF) contain the TiS<sub>3</sub> phase, so there is a shift in absorption onset from 3.14 eV to <2.6 eV and a line broadening.<sup>295</sup> Reprinted with the permission of the Royal Society of Chemistry."}}]](/storage/images/resized/NCzl62uJvnxgoDhaStBrWhgNJwuylDOicsL3qyKg_xl.webp)
Vanadium tetrasulfide has also recently been investigated as a photocatalyst for the water splitting to produce hydrogen (theoretical band gap width of VS4 is ≈1 eV).88 It was used as a component of a nanocomposite with reduced graphene oxide, which, when applied in the optimal amount of 5 wt%, hampered the rapid e––h+ recombination, promoted the transfer of photo-excited electrons and their interaction with H+. The highest hydrogen evolution rate was 0.7 mmol L–1 h–1 in a photocatalytic reactor containing 500 mL of water. The study125 also used a composite containing polychalcogenide MoS3 deposited on CdS/CdSe nanorods for photocatalytic decomposition of water under visible light irradiation. High values of hydrogen evolution 100 mmol h–1 g–1 and efficacy ≈10% were achieved. An interesting embodiment of photocatalytic decomposition of water in gaseous phase using a combination of MoSx nanostructure with TiO2 was proposed.182 The methods is based on the use of the hygroscopic nature of MoSx: water vapour is sorbed on the material, and then decomposes with the release of hydrogen.
5.2.3. Photocatalytic decomposition of organic compounds
Polychalcogenide materials, particularly, patronite VS4 and its composites with graphene, were also explored as photocatalysts for decolorization of dyes such as methylene blue91,297 and methyl orange,97,101 Rhodamine B,297 and also phenol,97 which were used as model compounds for water purification as well as disinfection (elimination of E.coli).103 Since the band gap width of VS4 is only 0.8-1.2 eV, such samples are promising for use in the near-infrared101 and visible97 regions, not just in the ultraviolet.91,97 Vanadium tetrasulfide with a morphology of microspheres performed better in catalytic decolorization of methyl orange under near-infrared light irradiation than TiO2 (60% vs 9% within 2 h).101 The authors also studied the luminescence decay on these samples and obtained quite a long luminescence lifetime (1.99 μs), indicating relatively slow recombination, that is, efficient separation of electrons and holes in the material. Even greater efficiency can be achieved by using VS4 nanocomposites with various carbon materials,103 which are known for their ability to inhibit recombination of electrons and holes. Thus, nanocomposite ʹreduced graphene oxide/VS4' has proven more effective in decolorization of methylene blue under UV irradiation compared to pure VS4.91 It was emphasized that more active samples had a slightly higher specific surface area, which could lead to a greater availability of the active sites, but the specific surface area of these samples varied insignificantly (7-12 m2 g–1). In another study,97 5% C/VS4 composites (VS4 particle size of 50-100 nm) also had higher decolorization efficiency of methyl orange under visible light (98.8±0.9% in 30 min), than carbon-free samples of VSx (44.0±1.4%). When irradiated with UV light, the photocatalytic performance decreased to 67.0±3.9%, which is due to its narrow band gap width (1 eV). Under the action of sunlight, the photocatalyst decolorized 70.1±3.3% of the methyl orange. Compared to other semiconductor photocatalysts (graphene oxide/TiO2, CdS/TiO2), which provide similar decolorization efficiency under visible light, the C/VS4 composite has clear advantages such as short reaction time, low catalyst loading, high decolorization rate of methyl orange. For comparison, decolorization rate was 19.8 mg L–1 h–1 for 5% C/VS4, 4.8 mg L–1 h–1 for CdS/TiO2, and 1.4 mg L–1 h–1 for graphene oxide/TiO2. When irradiated with UV and sunlight, the VS4 composite is inferior in efficiency to samples such as Cu/ZnO, Co2TiO4 and Se/ZnS, but still outperforms them in decolorization rate. A systematic study of various factors affecting this process, such as pH, catalyst loading and dye concentration was conducted.97 Moreover, other polysulfides were also explored as photocatalysts for dye degradation, in particular nanostructured TiS3 in the photodegradation of methyl orange and methylene blue,298 ZrS3 microribbons in the degradation of methyl orange299 and MoS3 nanoparticles in photodecolorization of methyl red.300
Another example of photocatalytic properties of TMPCs includes their application in photogeneration of hydrogen peroxide.263 Surface-modified zirconium sulfide nanoribbons (average length, width and thickness were 24 μm, 840 and 38 nm, respectively, according to atomic-force microscopy) were employed to produce hydrogen peroxide, and were compared to the non-modified ZrS3. Samples with two kinds of modification were used — those with defects of S22- positions and those with defects of both sulfur positions, S22- and S2–. It was shown that in the presence of all zirconium trisulfide samples photogeneration of hydrogen peroxide occurs from an aqueous solution of benzylamine through which oxygen was bubbled (with benzylamine as a sacrificial agent oxidized to benzonitrile). It was found that the performance of ZrS3 as a photocatalyst increased with increasing concentration of sulfur point defects on the surface of trisulfide. The lifetime of photoinduced charge carriers increased with increasing defect concentration (from 0.3 (ZrS3) to 0.69 s (ZrSS2-x) and 0.82 s (ZrS1-xS2-x)). The electron concentration also increased, and its value was estimated by experiments on charge carrier dynamics using the Mott--Shottky method.
5.3. Other applications
Below are a few more examples of practically relevant processes in which the chemical properties of TMPC's polychalcogenide groups play a significant role.
The use of polychalcogenide materials in gas sensors is described. For example, MoSx (x=3.66) was employed as an air moisture sensor, and it turned out that water molecules reversibly bound to disulfide ligands of MoSx porous branched material.182 Based on TaS3 nanofibres, NO gas sensors were created.80
Ternary polychalcogenides in the form of porous chalcogels performed well in sorption of radioactive isotopes of iodine (129I and 131I),301 suggesting that this area will also be promising for other TMPCs. Moreover, aerogel (NH4)0.2[MoS4] was explored as adsorbent of gases (H2, CO2, CH4 and C2H6).167 Due to the polarizable internal surface of the mesoporous aerogel (estimated internal surface area was 370 m2 g–1), the aerogel was expected to adsorb polar molecules. The best results were obtained for C2H6 and CO2, with the aerogel exhibiting pressure-independent selectivity in C2H6/H2 and CO2/H2 pairs with fairly high selectivity factors.
Various metal chalcogenides are successfully used as sorbents of mercury vapour (see Fig. 3c),302 and it has been noted that it is polychalcogenides, such as MoS3 and MoSe3, that are extremely efficient at sorbing mercury vapour due to their S–S and Se–Se groups, respectively.167,204,303 Chalcogels MoSx captured ≈2 g of mercury per gram of the sorbent.167 The material reacted with Hg, and the S–S sites reacted with mercury to afford HgS. According to XPS data, oxidation of mercury physically adsorbed onto MoS3/TiO2 composites occurred on the S22- active sites.303 Diselenide ligands of MoSe3 amorphous nanosheets also exhibit excellent affinity towards mercury,204 with uptake capacity of this material of 1 g g–1, and the uptake rate of 240 mg g–1 min–1, which are record values compared to the performance of other sorbents, including chalcogenide ones.
To summarize, chalcogenide aerogels could be promising matrices for capturing iodine and mercury vapour, which is of interest to the nuclear power industry and environmental protection.
6. Conclusion
Low-dimensional transition metal polychalcogenides have attracted the attention of researchers following graphene and dichalcogenides. Although TMPCs have been known for a long time, in the last decade their study has reached a new level. Thus, new crystalline phases, previously thought to be non-existent, have been synthesised, as well as numerous TMPC nanomaterials. A plethora of novel amorphous phases have been obtained, and their structure is actively studied using a set of modern experimental methods and theoretical modelling. New applications have been found, and important steps have been taken to understand how these materials function.
This review shows the influence of the structure of low-dimensional TMPCs on their functional properties. The structure of these substances, which combines the properties of 1D and 2D materials, and features chalcogen--chalcogen bonds makes them efficient materials for battery electrodes, as well as electro- and photocatalysts, since it is S–S (or Se–Se) centres that show redox activity in intercalation of metal ions, catalytic activity in water splitting processes, etc. Lithium-, sodium-, magnesium-, and aluminium-ion batteries with promising characteristics have been designed using various, mainly amorphous, TMPCs. Meanwhile, the potential applications of TMPCs are not limited to batteries and catalysis, but new areas can be found. For example, S–S centres actively interact with mercury vapour, which has been used to create relevant sensors.
An important trend in the development of TMPC chemistry will be a further search for synthetic approaches to the preparation of new phases, both crystalline and amorphous. Thus, MQ4I-like ternary phases can be thought of as partially oxidized polychalcogenides, which could be hypothetically converted to binary polychalcogenide phases by careful reduction. Nanostructuring and design of morphology, defects and surface chemistry will also develop, as well as further study of the chemical properties of TMPCs in various processes. To obtain colloidal dispersions of polychalcogenides, it might also be promising to explore the intercalation approach widely used to obtain aqueous MoS2 colloids, when a LixMoS2 intercalate reacts with water to form negatively charged dispersed nanosheets.253,304 However, although intercalates of various polychalcogenides are well known, their use as precursors for colloidal nanostructures is not reported.
Thus, in the coming years we can expect an active development of TMPC research aimed both at the synthesis of new materials based thereon and at the creation of prototype energy storage and conversion devices based on a better understanding of the mechanisms of action of these materials.
This work was funded by the Ministry of Science and the Higher Education of the Russian Federation (Project No. 121031700321-3) and Russian Science Foundation (Project No. 21-13-00274, https://rscf.ru/project/21-13-00274/).
7. List of acronyms
1D — compound with one-dimensional (chain) structure,
2D — compound with two-dimensional (layered) structure,
amd — N,Nʹ-diisopropylacetamidinate,
CDW — charge-density wave,
CN — coordination number,
CVD — chemical vapour deposition,
d — interatomic spacing,
DFT — density functional theory,
DMSO — dimethylsulfoxide,
DMF — N,N-dimethylformamide,
DOS — density of states,
EXAFS — extended X-ray absorption fine structure,
HER — hydrogen evolution reaction,
LIB — lithium-ion batteries,
mV dec–1 — millivolts per decade - unit of measure of the Tafel slope of the polarization curve,
NF — nanofibers,
PDF — pair distribution function,
RHE — reversible hydrogen electrode,
TEM — transmission electron microscopy,
TMDC — transition metal dichalcogenides,
TMPC — transition metal polychalcogenides,
TOF - turnover frequency,
XANES — X-ray absorption near edge structure,
XPS — X-ray photoelectron spectroscopy,
US — ultrasonication.