


Keywords
Abstract
To date, nitro-containing compounds form one of the most important classes of organic compounds. The chemistry of these molecules attracts attention primarily due to their use as high-energy reagents and drugs. Also, the introduction of a nitro group is a popular synthetic strategy for constructing new organic molecules. The present review summarizes the latest research findings of compounds bearing a nitro group on a two-carbon moiety such as aminonitroethylenes, α,α-bis(alkylsulfanyl)nitroalkenes and their amino derivatives, α-nitroketones, alkyl nitroacetates and nitroacetonitrile. The literature data are systematized according to the type of chemical reactions such as reactions between nucleophiles and electrophiles, various cyclization reactions, reactions at the C–H bond, etc. The reactivities of the said nitro compounds and the conditions of chemical transformations are compared to assess the prospects of their application.
The bibliography includes 314 references.
1. Introduction
For over one hundred and fifty years ago, organic chemists all over the world work with nitro-containing compounds. Initially, such compounds were considered as intermediates in the production of azo dyes, and were directly used as explosive materials. Currently, the use of nitro-containing structures as high-energy compounds has also not lost its relevance,1 although modern studies of the properties of nitro derivatives are focused mostly on nitrogen-containing nitroheterocycles,2--5 rather than on polynitrobenzenes.6 For example, there are monographs and reviews on nitroazoles7--9 and nitroazines10 devoted to chemical properties and biological activities of the corresponding nitro compounds. Moreover, many nitro derivatives have found application as drugs.3,11--14 In 2019, Nepali et al.15 published a detailed review on nitro compounds used in medicine, focusing on the uniqueness of the biological activity of the nitro group.
Nevertheless, the chemistry of nitro compounds is the most studied research subject for synthetic chemists. Scientists such as Henry, Konovalov, Nef, Zinin, Meyer, etc., have studied organics since their university years. Thanks to them and other scientists, we know that nitro compounds are not only an aim in themselves for synthesis but are also key intermediates in the preparation of numerous other organic molecules. At the turn of XX-XXI centuries, the frequently cited book by Noboru Ono16 entitled The Nitro Group in Organic Synthesis was published, which details the methods for introducing a nitro group into molecules and also chemical properties of nitro compounds.
At the same time, modern reviews are often focused on specific classes of compounds that can be represented as nitro-containing synthetic equivalents (NSEs), e.g., nitroalkenes,17--23 nitrodienes,24 nitroalkanes,25 γ-nitrocarbonyl compounds,26 nitroaziridines27 and nitroalkynes.28 This is not surprising, since the introduction of the nitro group into molecules, not via direct nitration or oxidation, makes available products containing substituents that are sensitive to harsh reaction conditions. Consequently, understanding the chemical properties of such NSEs as well as their synthetic possibilities provides access to a various new approaches and compounds.
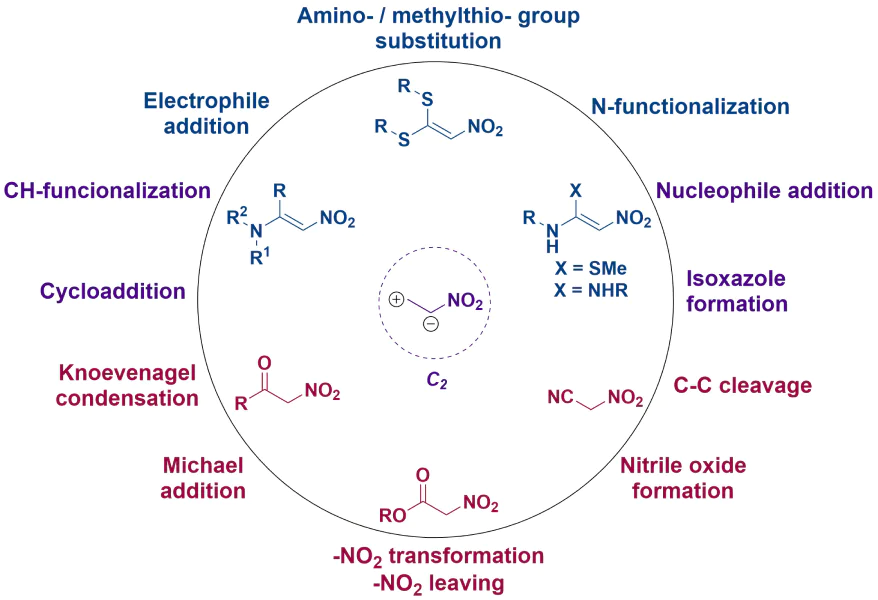
The present review considers groups of two-carbon NSEs such as α-aminonitroethylenes (1), α,α-di(alkylthio)nitroalkenes and their amino derivatives (3, 4), α-nitroketones (5), alkyl nitroacetates (6) and nitroacetonitrile (7) (Fig. 1), and shows their diverse synthetic opportunities. Particular attention is paid to similarities and differences in the chemical behaviour of these groups. As mentioned above, the study on the synthetic potential of such compounds is necessary, first of all, to understand how to implement the related synthetic strategies, since in some cases the simplest or the only approach to the target molecule requires the use of these NSEs. Accordingly, with new methods for the modification of low-molecular-weight nitro compounds, the analysis of modern trends in the chemistry of such structures is very relevant, especially considering that these compounds, until now, have never been explored within a single study.16,29--32
![[{"id":"hJ8Hg64v3A","type":"paragraph","data":{"text":" Structures of nitro-containing synthetic equivalents and possible transformations with their participation. The following abbreviations are used: ANE is aminonitroethylene, DiSAlk is α,α-di(alkylthio)nitroalkene, SMeNHR is (alkylthio)aminonitroalkene, DiNH is α,α-di(amino)nitroalkene, NK is α-nitroketone, ANA is alkyl nitroacetate, NAN is nitroacetonitrile."}}]](/storage/images/resized/b6zxVfnvECpCAzDrmakOiaZXrqduWLKV7MmzpGYH_xl.webp)
It should be noted that the definition two-carbon for a fragment of a molecule is not canonical. In this review, this term is proposed to be applied to those structures whose central fragment contains two carbon atoms linked by a single or double bond. One of these atoms contains an NO2 moiety, which causes a special distribution of electrons in the molecule.
2. Conjugated nitroalkenes
2.1. α-Aminoethylenes
α-Aminonitroethylenes, like nitroacetonitrile, form rather underexplored group of NSEs. Apparently, this is due to the few available methods for the synthesis of such push-pull structures. At the same time, α-aminonitroethylenes have great synthetic potential; the main routes for their modification are depicted in Fig. 2, and the most common methods for the synthesis of these compounds can be found in publications.33--35
![[{"id":"xEM08QLIAB","type":"paragraph","data":{"text":" Possible ways to modify α-aminonitroethylene"}}]](/storage/images/resized/viPgmISRVxbhoSZXIzJpchVKoLqkpnSAKfiz5aBb_xl.webp)
A key factor governing the chemical properties of ANEs and DiSAlk, is the possibility of imine--enamine tautomerism at R1=H. In this case, the carbon atom adjacent to the nitro group, and the NH moiety become available to the reaction with various electrophiles. Moreover, when using transition metal catalysis or oxidizing systems, the CH bond is modified.
2.1.1. Reactions with electrophiles
In 2020, Pilipecz et al.36 carried out nitrosation of (nitromethylene)pyrrolidin-2-yl 1a to study chemical properties of 2-[nitro(nitroso)methylene]pyrrolidine (8). The reaction is completed in just a few minutes. The authors also noted that product 8 decomposes in a day when stored at room temperature (rt), however, it can be stored in the refrigerator for over a week (Scheme 1).
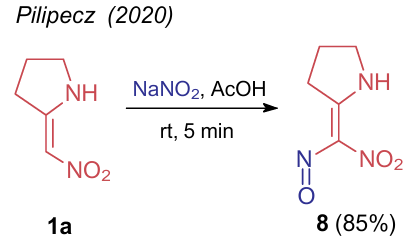
Alekszi-Kaszвs et al.37 studied the Mannich reaction of the same compound 1a with C-electrophiles. The use of a double excess of formaldehyde (as 35% formaline) and primary aromatic amine provided 4-nitro-1,2,3,5,6,7-hexahydropyrrolo[1,2-c]pyrimidines 9a--d (Scheme 2). Initially, it is the C-atom bound to the nitro group that is attacked by the electrophile. This was demonstrated by the reaction of NSE 1a with an alkylated benzotriazole yielding product 10 (see Scheme 2).
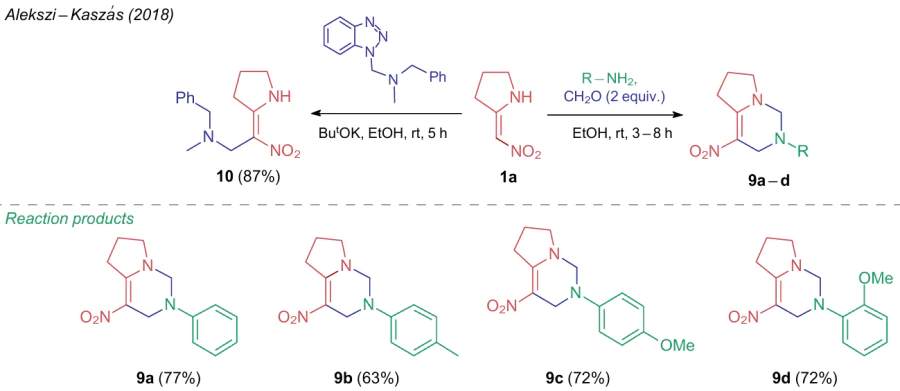
In 2019, Lyapustin et al.38,39 found that 1-morpholino-2-nitroalkenes 1 react with boron trifluoride etherate in alcohol to give the corresponding nitroalkynes 11 (Scheme 3). These products were reacted in situ with aminoazoles 12 to afford azolylenamines 13, capable of heterocyclization with aldehydes 14 when heated in the same reaction system. The resultant 4,7-dihydro-6-nitroazolo[1,5-a]pyrimidines 15 were also produced using a multicomponent procedure when refluxing the starting compounds in n-butanol in the presence of the same Lewis acid. The plausible reaction pathway includes the formation of morpholino-containing intermediates 16 and 17.
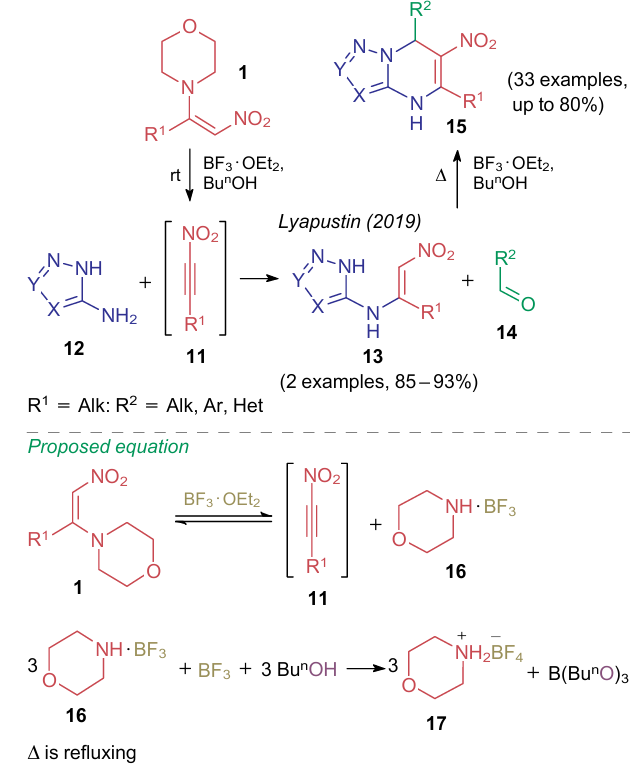
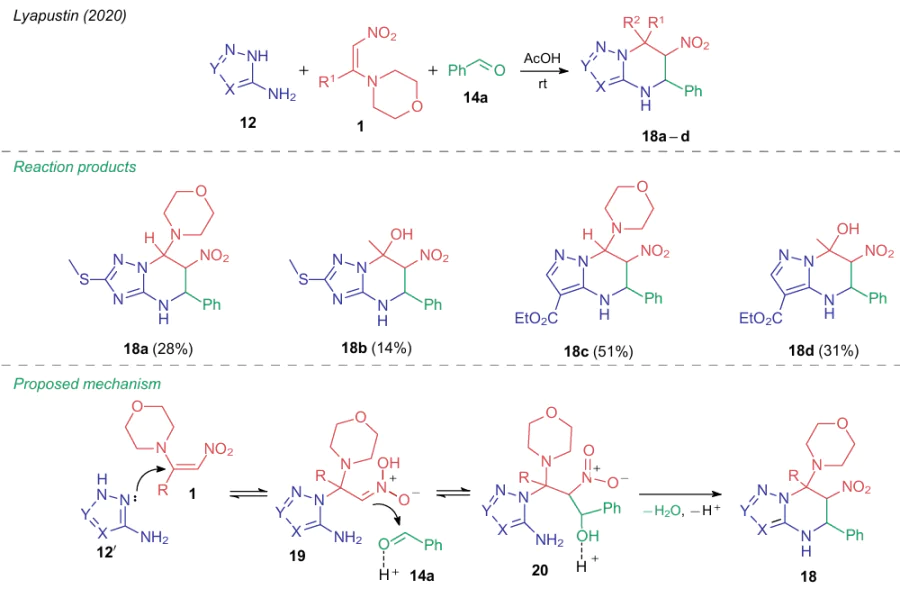
The same multicomponent reaction (MCR), when carried out in acetic acid, follows an alternative pathway. In 2020, the same research group40 found that acetic acid disfavours the formation of nitroalkyne 11, and the reaction can occur only if all three reactants are present. In this way, several 6-nitro-4,5,6,7-tetrahydroazolo[1,5-a]pyrimidines (18a--d) were obtained. The reaction pathway under the catalysis with Brуnsted acid through formation of intermediate compounds 19, 20 was proposed (Scheme 4).
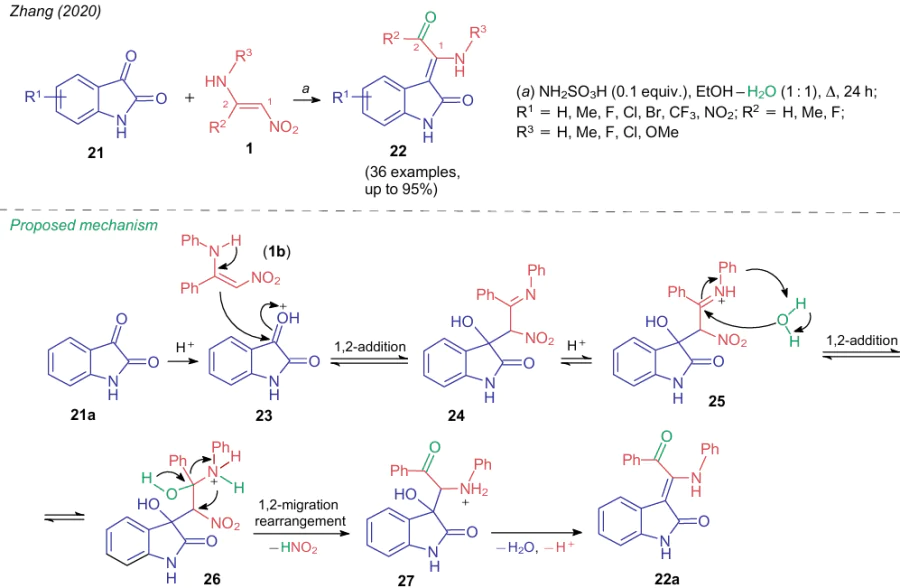
In 2020, Zhang et al.41 found that the reaction of aminonitroethylenes 1 with the carbonyl moiety of substituted isatins 21 under acidic conditions leads to the cleavage of both C–N bonds, unprecedented for this NSE group, and formation of products 22 (Scheme 5). The authors suggested that the reversible addition, e.g., of aminonitroethylene 1b to protonated isatin 23 catalyzed by sulfamic acid first occurs to give adduct 24. Further, under the action of an acid, a water molecule adds to the imine fragment of adduct 25. Then, intermediate 26 undergos a loses of a nitrous acid molecule and an 1,2-amine migration to give a rearrangement product 27. Finally, intermediate 27 is deprotonated and dehydrated to afford product 22.
In 2017, Siddaraju and Prabhu42 described the only example of the reaction of 1-dimethylamino-2-nitroethene (1c) with the in situ generated species containing the S–I bond. The authors argue that compound 1c reacts with 1-phenyltetrazole-5-thiol (28) when heated in DMSO in the presence of iodine with the formation of sulfide 29 (Scheme 6).

Based on the control experiments, the authors found that 1-phenyltetrazole-5-tiol (28) first reacts with iodine to give dimer 30 and HI. The resultant dimer then reacts with iodine to afford intermediate 31 containing the S-I bond. Nucleophilic displacement of an iodide group by enamine 1c affords product 29. Further, iodine is regenerated by the reaction of HI with DMSO and the catalytic cycle is repeated.
2.1.2. Reactions with nucleophiles
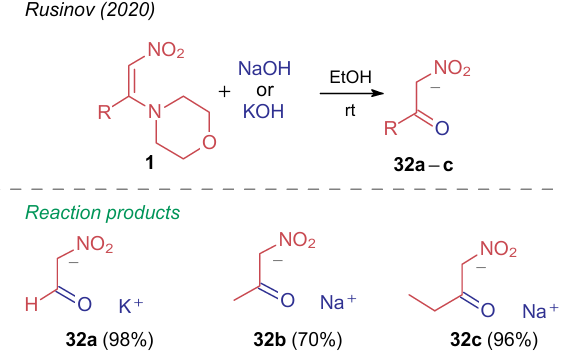
One reaction demonstrating the interconversion between aminonitroethylenes and α-nitroketones, is the reaction of the former compounds with alkalis. Rusinov et al.33 showed that 1-morpholino-2-nitroalkenes 1 react with alcoholic solutions of sodium or potassium hydroxides at room temperature to yield the salts of α-nitrocarbonyl compounds 32a-c (Scheme 7). Numerous methods for the preparation of such ketones are available in the literature; however, the synthesis from ANE is relatively recent.33 This way is among the easiest approaches to produce a stable form of nitroacetic aldehyde, though the disadvantage of this method is a limitation in structural modifications of compounds 1.
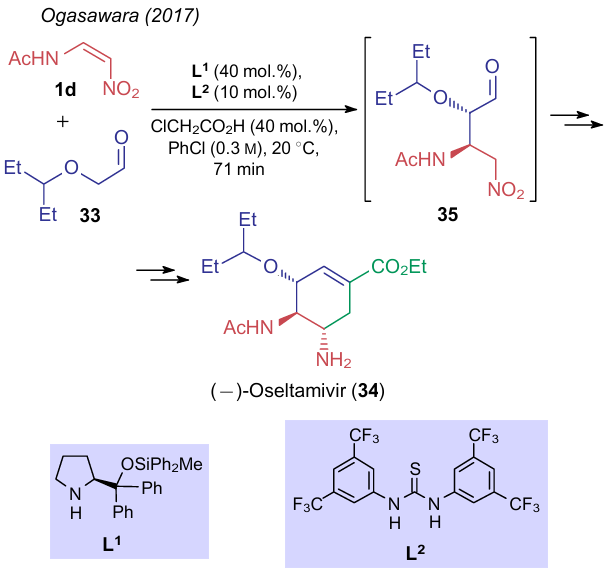
An electrophilic centre in aminonitroethylenes is prochiral, and nucleophile addition reactions imply the appearance of at least one chiral centre in the molecule. Therefore, investigations of such reactions often include the development of stereoselective synthetic approaches. For example, Ogasawara and Hayashi43 improved a method to produce the antiviral drug Tamiflu [(–)-Oseltamivir, 35] from substrates 1d and 33 in a flow reactor. Scheme 8 illustrates only the first step of this transformation, directly related to the subject of this review. Although all steps in the above scheme occur sequentially without isolation of intermediate products, the authors explored each reaction separately. It was found that the use of a modified Hayashi--Jуrgensen catalyst (L1) and 1,3-bis[3,5-bis(trifluoromethyl)phenyl]thiourea (L2) provides N-[(2R,3S)-1-nitro-4-oxo-3-(pentan-3-yloxy)butan-2-yl]acetamide (34) with a 97% enantiomeric excess in the first step.
The Michael reaction involving the α-CH-acidic atom of carbonyl compounds was also studied for preparing stereoselective adducts with optically active thiourea L3 (method A)44 as in the previous example, and pyrrolidine catalyst L4 (method B)45 (Scheme 9). 2-(2-Nitrovinyl)isoindoline-1,3-dione (36) reacts with aldehydes and ketones to afford, depending on the reaction conditions, different stereoisomers 37 (14 examples, yields up to 99%, diastereomeric ratio (dr) >20:1, enantiomeric excess (ee) (80-99%) and 38 (6 examples, yields up to 84%, dr from 3:1 to 80:1, 85-99% ee).
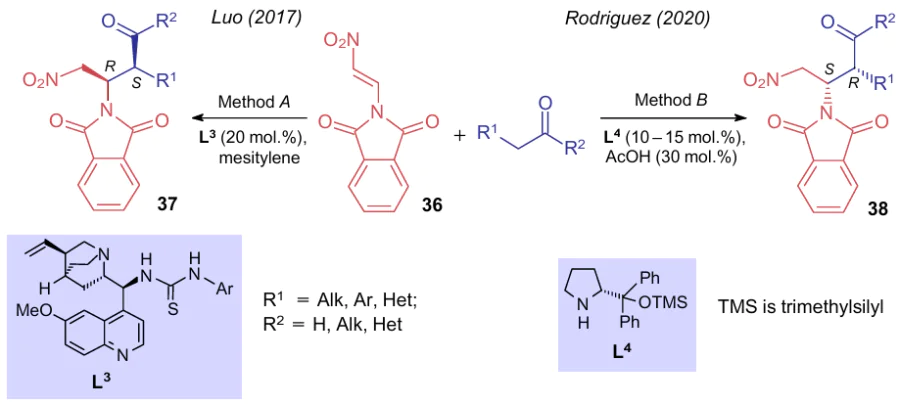
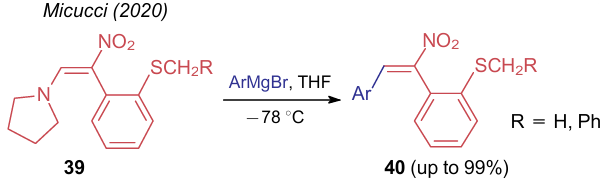
The reaction of compounds 39 with Grignard reagents in tetrahydrofurane is reported,46 in which an amino moiety is almost quantitatively displaced with a C-nucleophile to give 2-(1-nitro-2-arylvinyl)phenyl sulfides 40 (Scheme 10).
One of the common applications of ANEs is the introduction of a nitrovinyl group into indoles 41 for the further synthesis of the corresponding biologically active compounds from derivatives 42.47--50 Such procedure is mainly carried out in methylene chloride in the presence of trifluoroacetic acid (TFA) (Scheme 11).
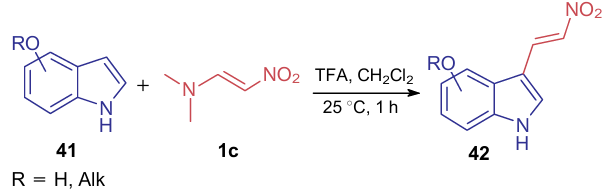
The role of trifluoroacetic acid in this reaction is likely to activate ANE. However, the cited publications did not provide any data on the use of other acids in this transformation or detailed studies on the mechanism of the process.
2.1.3. Reduction of the double bond
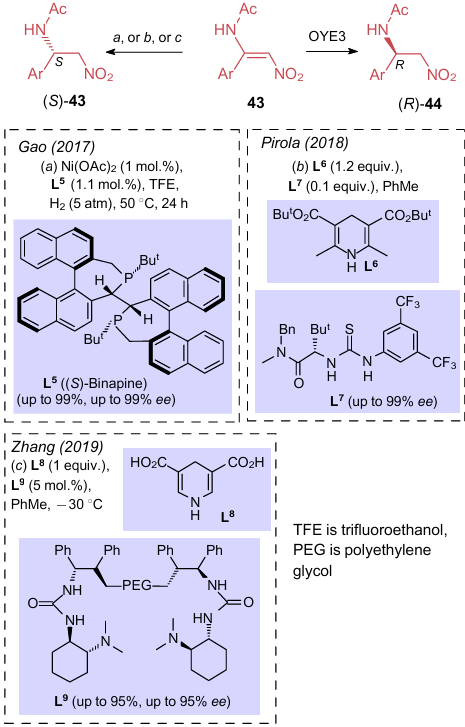
Hydrogenation of the double bond of aminonitroalkenes, similar to the addition of nucleophiles, results in the formation of a chiral centre. Accordingly, depending on the structure, this can produce both R- or S-isomers and also a racemate if there is no need for a stereoselective product. Thus, in 2017, Brenna et al.35 pioneered in bioreduction of ANE in the presence of an enzyme (reductase) in high yields (up to 88%) and with high enantioselectivity (up to 99%). When reducing the substrates 43 with Old Yellow Enzymes 3 (OYE3), isomers (R)-44 were the major products (Scheme 12). A few publications addressed the synthesis of their epimers (S)-44. For example, Gao et al.51 carried out hydrogenation with hydrogen gas using nickel acetate and (S)-Binapine ligand (L5). This procedure features excellent stereoselectivity and high conversion rates. Pirola et al.52 described in detail the use of a flow reactor to reduce ANE in the presence of an organocatayst based on dihydropyrimidine (L6) and thiourea (L7). The authors optimized the protocol and further reduced the nitro group under the same continuous-flow conditions. Zhang and Shi53 accomplished the reduction of nitroenamines in the presence of dihydropyrimidine (L8) and polyethylene glycol organocatalyst (L9). However, the authors evaluated the enantiomeric purity of the products without establishing the absolute configuration.
2.1.4. Cycloaddition reactions
In 2018, Chalyk et al.54 reported their findings on cycloaddition of ANEs to halooxime 45 to prepare 4-nitroisoxazoles 46 (Scheme 13). The plausible mechanism suggests the reaction of halooxime 45 with sodium hydrocarbonate to deliver nitrile oxide 47. Then, intermediate 47 enters the pericyclic reaction with 1-dimethylamino-2-nitroalkene 1c, which leads to the aromatization of the system via intermediate state 48 and intermediate 49.
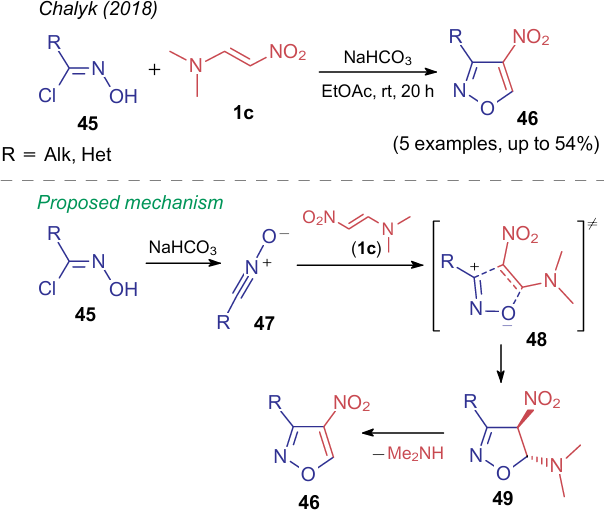
In the present review, the synthesis of isoxazoles from NSE is discussed below in the relevant sections, however, this example is the only one, in which the nitro group is not directly involved in the formation of the heterocycle.
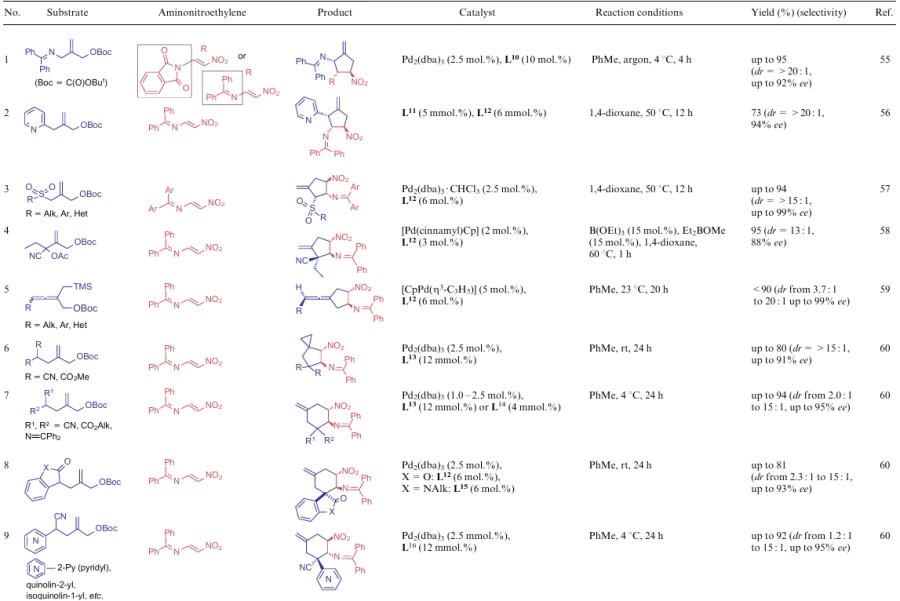
A series of works of the research group led by Trost55--60 concerns the synthesis of 2-amino-1-nitrocyclopentanes (Table 1, lines 1-6) and -cyclohexanes (see Table 1, lines 7-9) from the appropriate allyl (tert-butyl) carbonates and aminonitroethylenes. The authors mainly consider the variability of substrates and the effect of the catalyst on the stereoselectivity of the process. In all cases, palladium catalysts [e.g., in the form of complexes with dibenzylideneacetone (dba) or cyclopentadiene (Cp)] and phosphorus-containing ligands L10-L16 were used.
![[{"id":"LbdcIuF1om","type":"paragraph","data":{"text":"Structures L<sup>10</sup>-L<sup>16</sup>"}}]](/storage/images/resized/q6P2XtJzuduiF8oDYxhlRZzKrBFDlhfG7xTX3wrG_xl.webp)
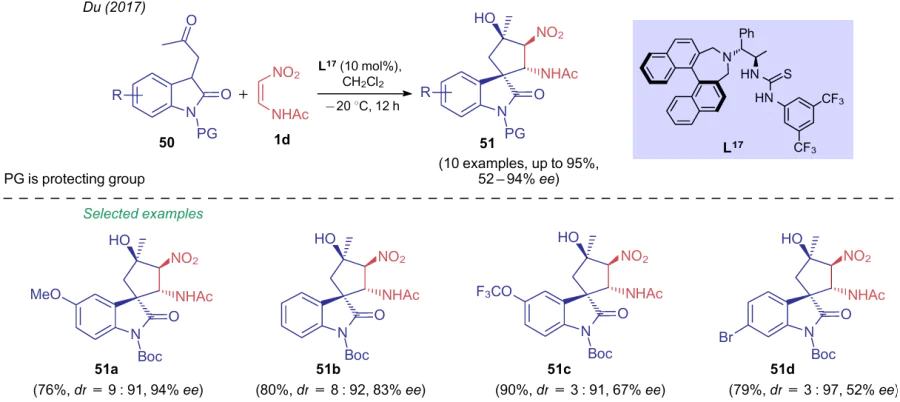
To prepare spiroindoles 51 from oxindoles 50 and aminonitroethylene 1d, Du et al.61 used an optically active catalyst based on thiourea L17. Depending on substrates in choice, enantioselectivity of the process varied over a wide range, while diastereoselectivity, on the contrary, was quite high (Scheme 14).
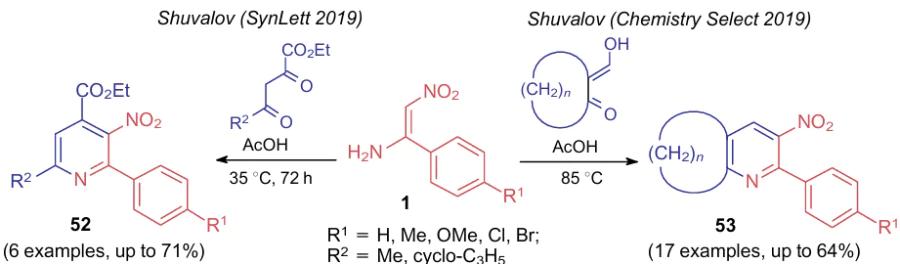
In 2019, two publications of the Shuvalov's group62,63 appeared on the reaction of ANEs with 1,3-dicarbonyl compounds furnishing 3-nitropyridines 52, 53 (Scheme 15). The study62 concerned only the reaction between nitroenamines 1 and hydroxymethylenecyclohexanone when heated without solvent, while in the publication,63 the scope of the starting carbonyl compounds was significantly expanded. The synthesis was carried out in acetic acid to improve the yield of the product. Moreover, the reaction of nitroenamines 1 with β,γ-diketocarboxylate was carried out.
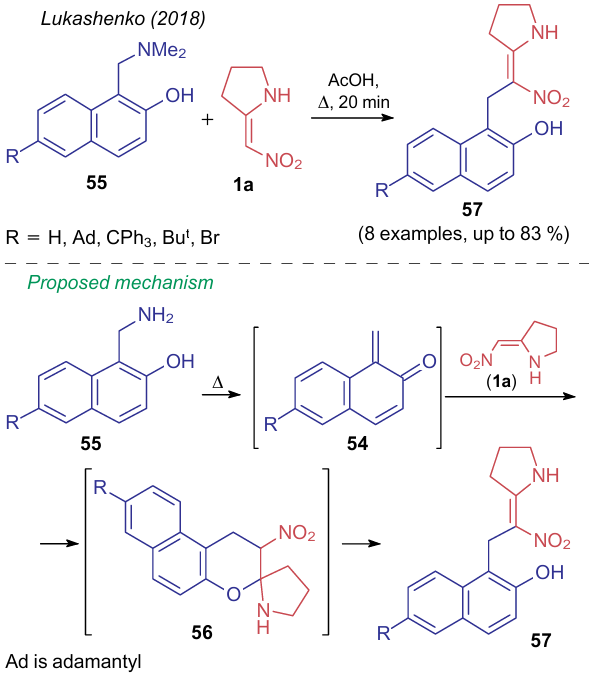
Lukashenko et al.64 described the reaction of compound 1a with an intermediate diene 54 generated by heating 1-[(dimethylamino)methyl]naphthalen-2-ols 55 in acetic acid (Scheme 16). According to the authors, the reaction follows the [4+2]-cycloaddition route, however, the resulting ring system 56 eventually undergoes ring-opening to give the Mannich product 57.
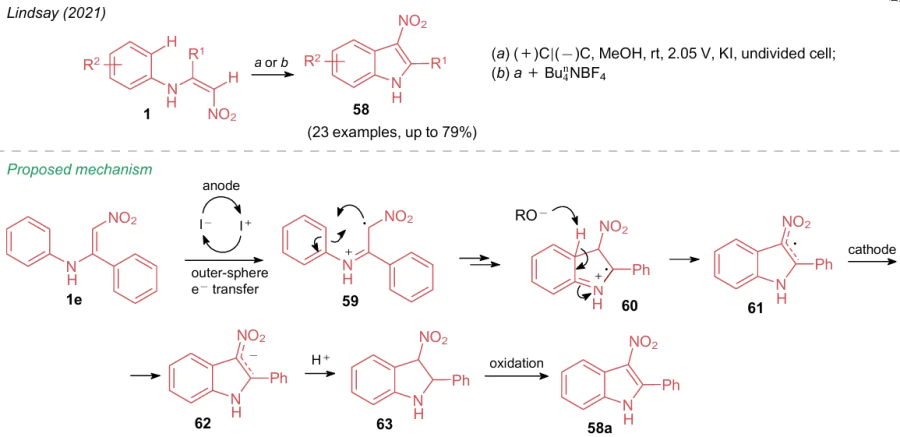
In 2021, Lindsay et al.65 showed that N-aryl-substituted aminonitroethylenes are prone to intramolecular cyclization under oxidative conditions. To carry out this reaction, the authors developed an oxidative system, in which arylaminonitroethylenes 1 produced 3-nitroindoles 58 in an electrochemical cell in the presence of KI catalyst (Scheme 17). Through a series of 13 control experiments, on the example of substrate 1e, the authors proposed a reaction mechanism, in which the process is initiated by electron transfer from nitroenamine to an iodine atom at the anode to generate a cation radical 59. Further, after the formation of a new C–C bond and deprotonation of a species 60, the radical intermediate 61 at the cathode transforms into the anionic form 62, which is protonated to give 3-nitro-2-phenyl-2,3-dihydroindole (63). Under electrochemical cell conditions, semiproduct 63 undergoes autoaromatization to afford product 58a.
2.1.5. Other reactions
This subsection include reactions that do not formally fall into the above-mentioned classifications, and can not be grouped in any way, since they are rarely mentioned in the literature. For example, Cheng et al.66 demonstrated the possibility of oxidative coupling of (E)-N,N-dimethyl-2-nitroetheneamine (1c) with 1,3-diphenylprop-1-ene (64) in dichloroethane (Scheme 18). The authors considered a combination of various EWG-alkenes (EWG is an electron-withdrawing group), however, the amino group hydrolyzed only in compound 1c, and the intermediate most likely oxidized to a carboxylic acid with further decarboxylation to product 65.

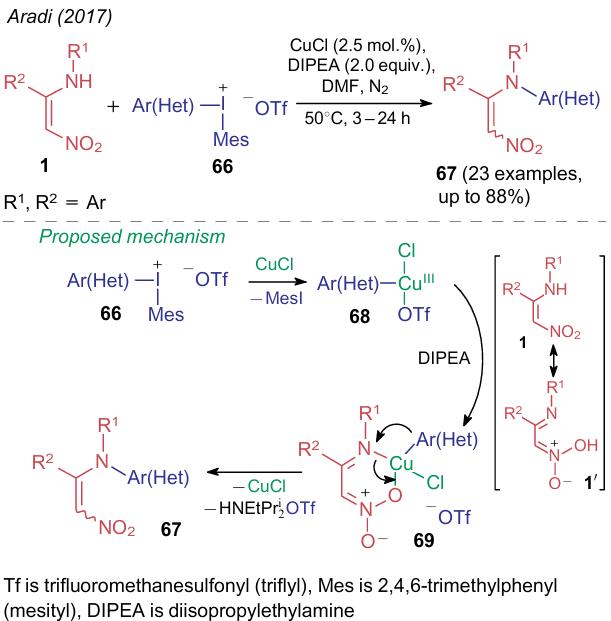
Compounds 1, bearing the monosubstituted amino group, can be arylated in dimethylformamide in the presence of 2 equiv. of a base and catalytic amounts of copper(I) chloride. Aradi et al.67 described the procedure of the reaction of ANE with diaryliodonium triflate 66, which might be formally considered as amination of iodine-containing (hetero)aromatic compounds. The proposed reaction pathway implies the formation of cis- and trans-isomers 67 in non-stoichiometric amounts via copper-containing intermediates 68 and 69 (Scheme 19).
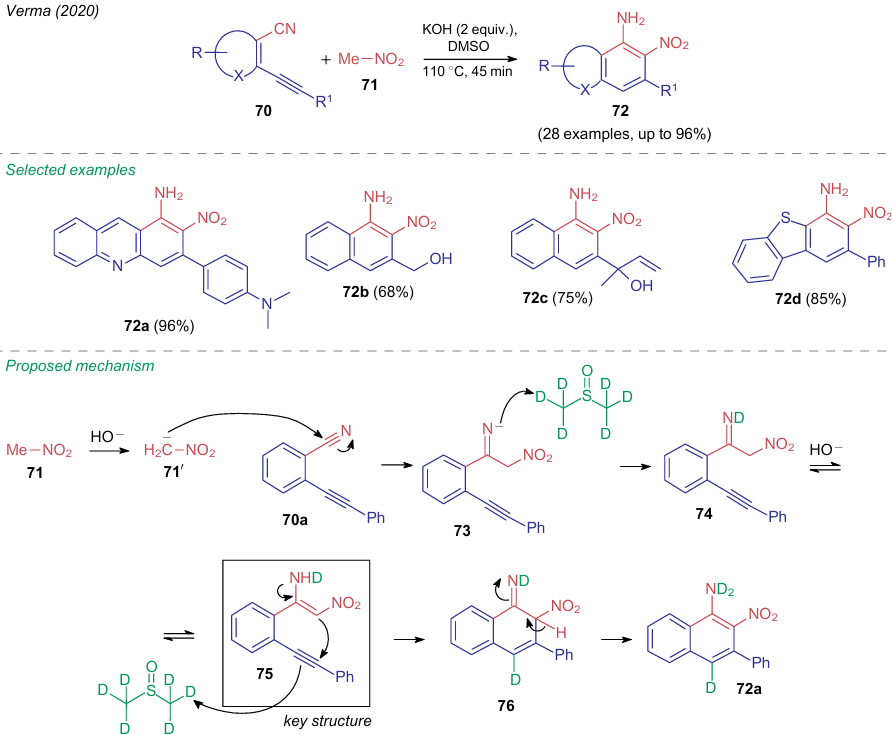
In 2020, Verma et al.68 developed an elegant atom-economical approach to the synthesis of various nitro-substituted aromatic and heteroaromatic compounds bearing sensitive functional groups. The method includes regioselective annulation of 2-alkenylbenzonitriles 70 with nitromethane 71 in dimethylsulfoxide in the presence of 2 equiv. of potassium hydroxide to afford fused systems 72 (Scheme 20). Based on control experiments and the findings on using a deuterated solvent, the authors proposed a reaction pathway as illustrated on the example of 2-(phenylethynyl)benzonitrile (70a). The aza-Henry addition of the nitromethane potassium salt 71ʹ to a nitrile group of the substrate gives anion 73, which reacts with deuterated DMSO to afford intermediate 74. The key structure of this mechanism is ANE 75, which cyclizes to compound 76 when reacting with a solvent.
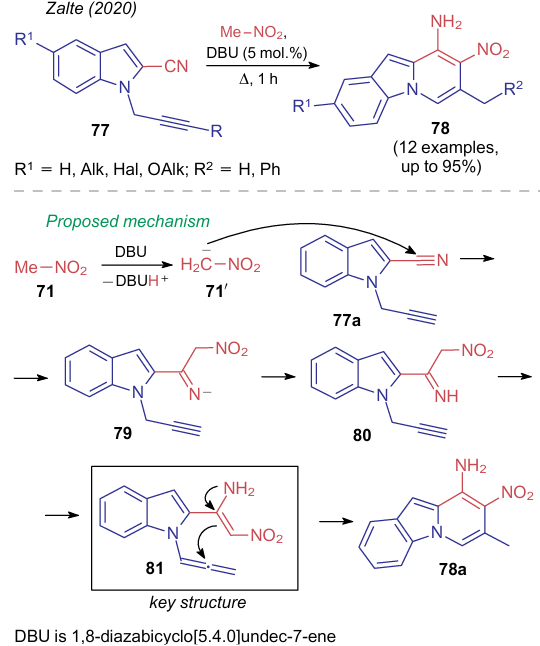
In the same year, Zalte et al.69 employed the similar reaction to prepare fused compounds, 9-aminopyrido[1,2-a]indoles 78, from indoles 77. As in the previous case, the reaction mechanism comprised the formation of a push--pull enamine (Scheme 21). However, in this example, the reaction began with the formation of anion 79 and was completed presumably via the alkyne-allene rearrangement 80→81 followed by an intramolecular cyclization.
When considering methods for the preparation of ANE 1, it can be concluded that over the past five years, the chemical properties of such NSEs have been enriched by a significant number of new and nontrivial examples. These transformations show good prospects for their application to the synthesis of a variety of hetero- and carbocyclic compounds, and other acyclic molecules necessary to construct more complex organic skeletons.
2.2. 1-Nitro-2,2-disubstituted ethylenes
The chemical behaviour and a pool of reactions of disubstituted nitroethylenes 2-4 are generally similar to those for the above-mentioned NSEs (Fig. 3). Significant differences are evident in that compounds 2-4 are much more likely to react with the leave of the nitro group. This is mainly due to the need to form an aromatic system during cyclization. As for NSEs 3, 4, they tend to form isoxazole rings through the transformation of the nitro group. Compound 2 and 3, 4 are noticeably different: since imine-enamine tautomerism is impossible for DiSAlk, such derivatives do not interact with electrophiles either. To functionalize the C-atom adjacent to the nitro group, palladium catalysts should be used (see Section 2.2.4). Therefore, various primary amines are used in the reactions involving compounds 2 to allow the electrophile to react with the in situ generated SMeNH. Interestingly, no studies are available on the chemical properties of (alkylthio)aminonitroalkenes substituted with a SAlk fragment and a secondary amino group. The most common methods for producing such compounds are available in the studies.70,71
![[{"id":"jwrRTCGks0","type":"paragraph","data":{"text":" Possible ways to modify compounds <b>2</b> - <b>4</b>"}}]](/storage/images/resized/HtW8VX2EjAZtlkdx1CgG9kv014AwGLgXrpluqEsg_xl.webp)
2.2.1. Cycloaddition reactions
The overwhelming majority of transformations described for NSEs 2-4 are heterocyclization reactions affording polysubstituted 3-nitropyridines (Table 2, lines 1-31), 3-nitropyranes (see Table 2, lines 32--45) (2, 32–45) or 5-nitropyrimidines (see Table 2, lines 46, 47). 3-Nitropyridines are accessible via the [3+3]-cycloaddition reaction involving NSEs and di-C-electrophiles, and also [3+2+1]-type reactions using NSE, C-electrophiles and various 1,2-bifunctional structures. In the case of 2,2-dimethylthio-1-nitroethylene (2a), mono- or diamines are added to the reaction mixture, which initially generate in situ mono- or diamino-1-nitroethylenes 3 or 4 to be cyclized. To produce nitropyranes, NSEs, C-electrophiles and bifunctionalized O,C-nucleophiles are mainly used in the [3+2+1]-cyclization or NSE, while NSEs and bifunctionalized compounds (O-nucleophile, C-electrophile) are employed in the [4+2]-addition reaction. In these cases, the methylthio group is displaced with an O-nucleophile, and the effect of the NH-alkyl group is noted only in the reaction pathway of tautomeric transformations. Nitropyrimidines are commonly produced via the [3+2+1]-type reaction of aminoazoles, aromatic aldehydes and 2-alkylamino-2-methylthio-1-nitroethylenes.
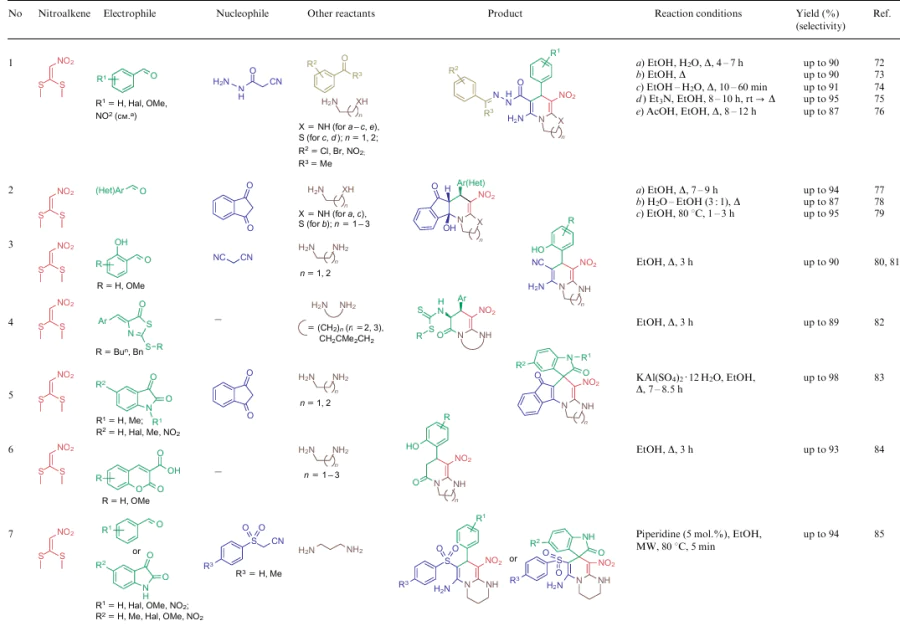
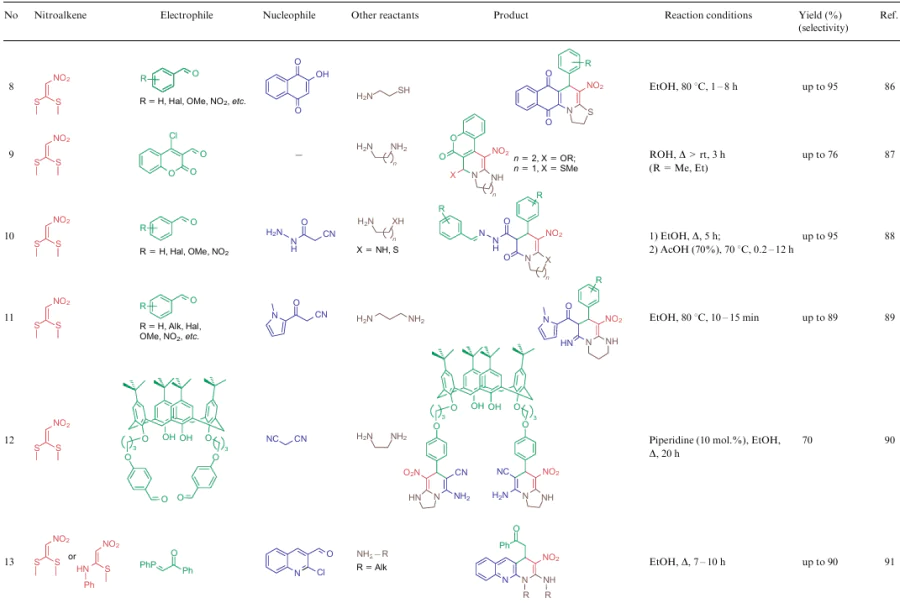
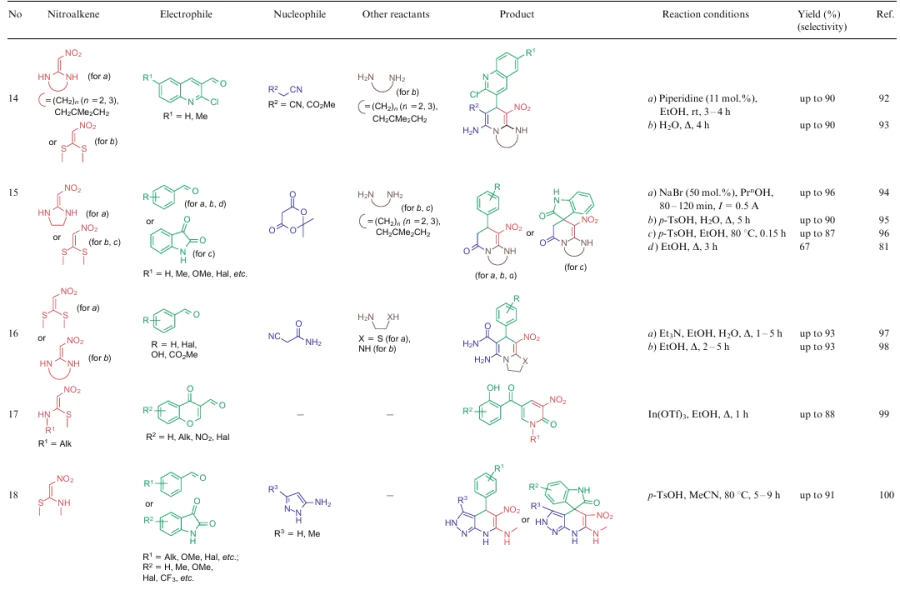
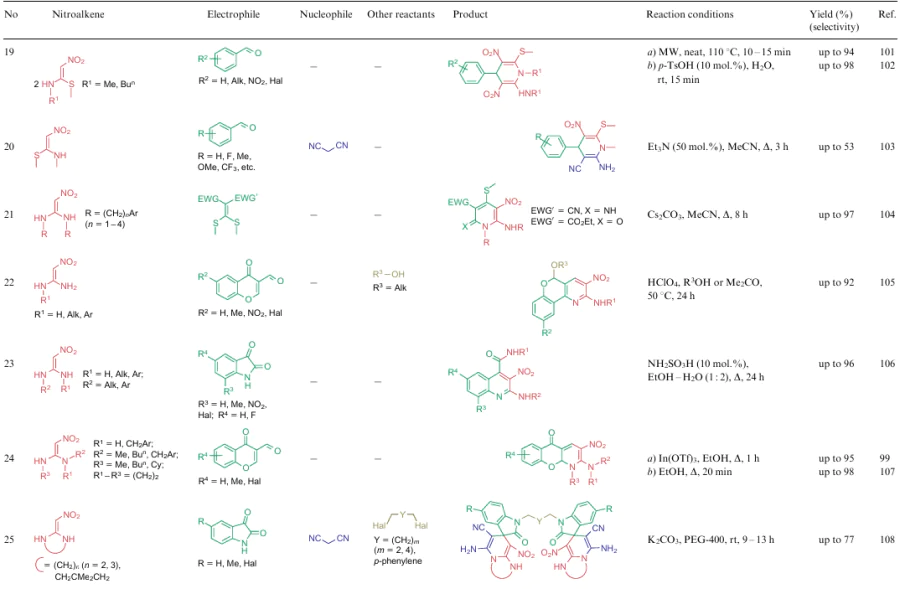
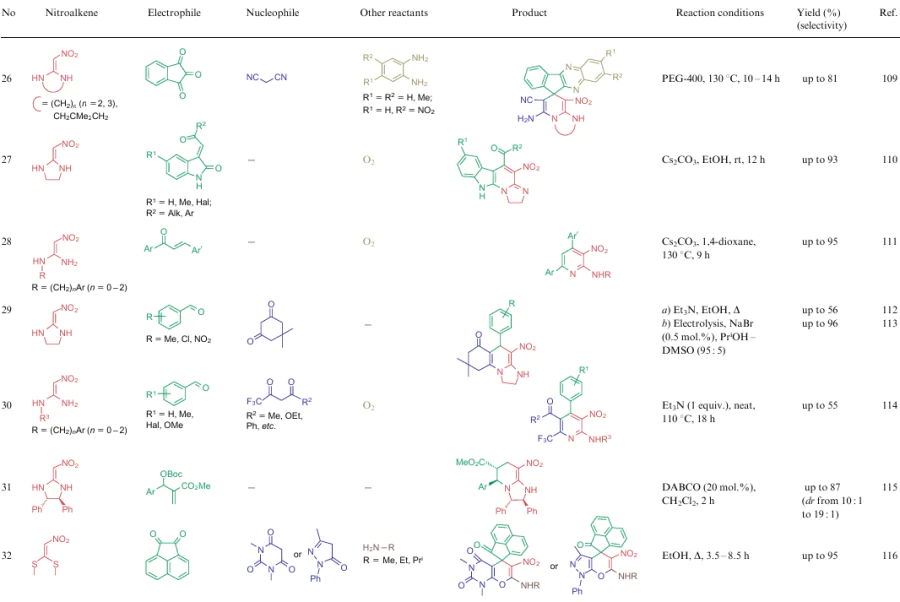
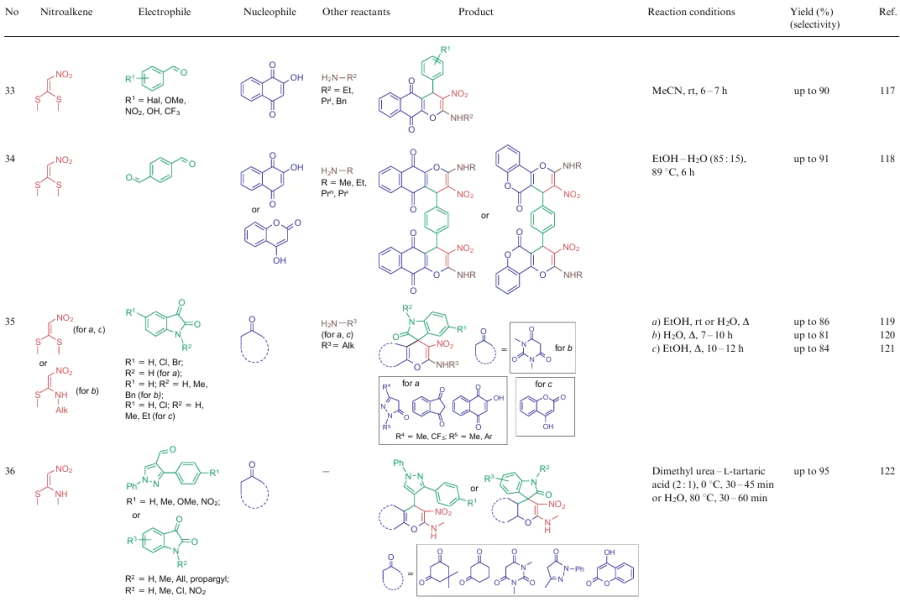
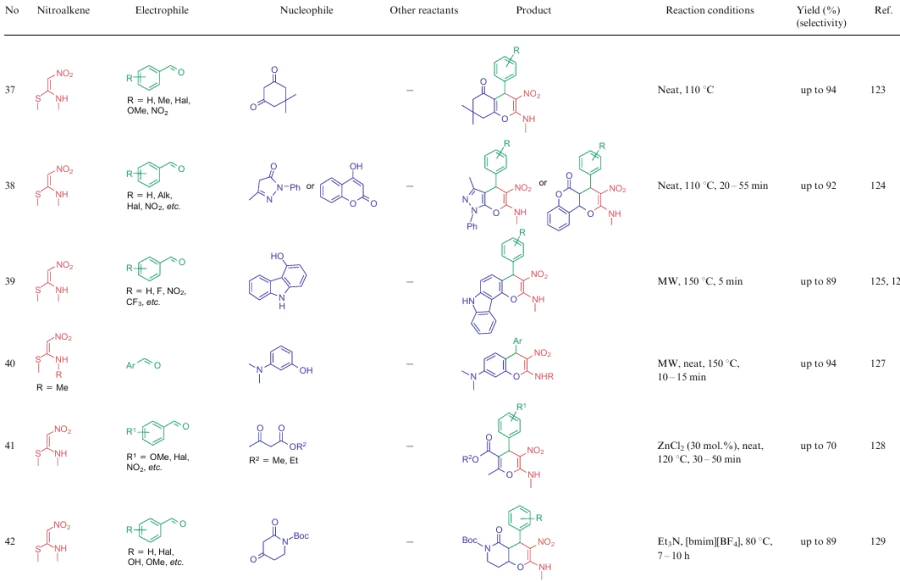
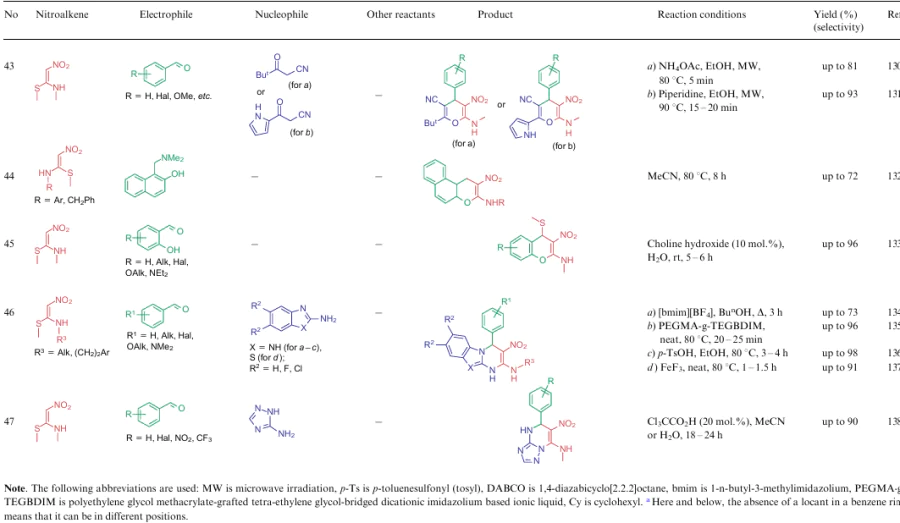
For all these transformations, no clear relationship between the cyclization type or the features of certain substrates and the preferred reaction conditions or the type of catalysis was revealed. Nevertheless, these multicomponent processes are carried out mainly in polar solvents in the presence of catalytic amounts of acid or base, or on heating without a catalyst. Fusion, microwave irradiation, ionic liquids or electrolysis conditions are less commonly used. The data on cycloaddition reactions producing six-membered rings are summarized in Table 2.
The authors often point out the beneficial properties of the resultant compounds, primarily their biological activities and photophysical characteristics, or use these structures as scaffolds to create practically valuable compounds. Note that in a review of 2019, Saigal et al.29 highlighted some transformations related to the reactions of 1-nitro-substituted 2-alkylthio-2-alkylaminoalkenes. However, in the present review, the chemical properties of this group of NSE are presented in more detail. Often there are cases of two-, three- and four-component reactions affording polysubstituted 3-nitropyrroles (Table 10). The process in general is the reaction between an 1,2-dielectrophile and nitroalkene 3 bearing at least one unsubstituted NH group. The dielectrophile can be both generated in the reaction and used in a finished form. In this process, as in the formation of six-membered rings, it was problematic to identify the general dependence of the reaction conditions on particular substrates. Nevertheless, in most cases, the reaction is carried out in polar solvents without any catalyst.
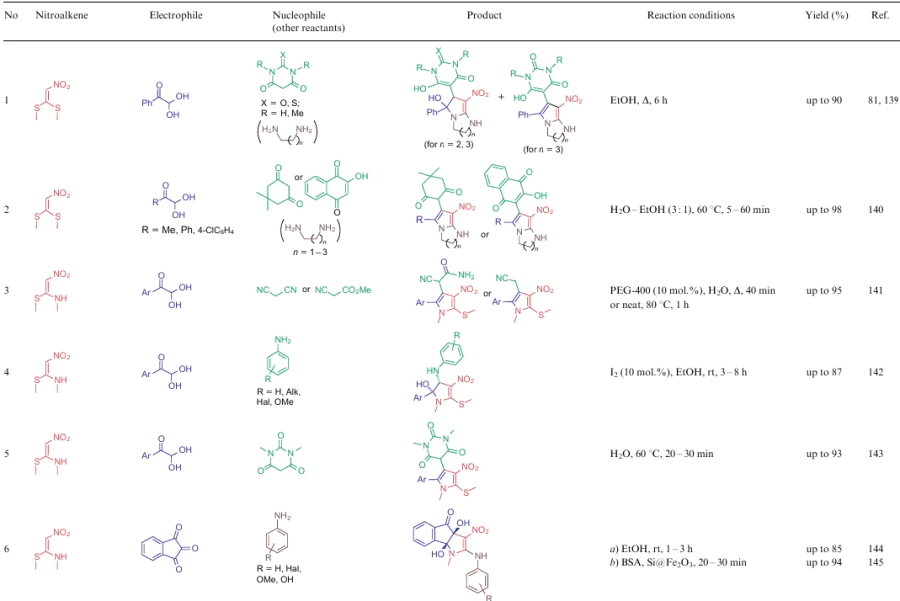
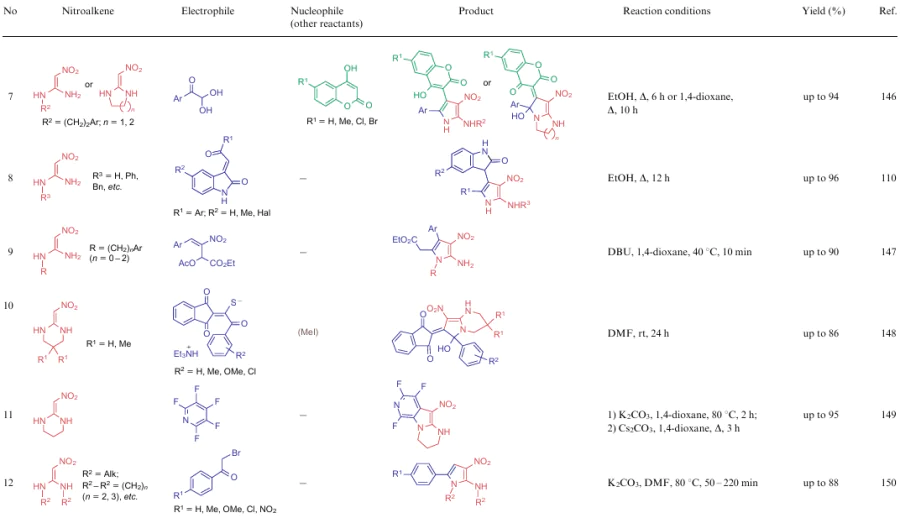
The cyclization ability of α, α-di(alkalkylthio)nitroalkenes 2 is not limited to the examples shown above. Based on such nitroenamines, other five-six-membered heterocycles as well as seven- and eight-membered compounds can also be produced. For example, Hasaninejad et al.151 carried out the three-component consequtive reaction involving compounds 2, hydrazine (82) and aldehydes 14 in ethanol at room temperature to obtain 4-nitro-2,3-dihydropyrazoles 83 in nearly quantitative yields (Scheme 22). First, methylthio moieties are displaced with hydrazine molecules, while an intermediate is converted to product 83 via the [4+1]-cyclization with a molecule of aldehyde 14.

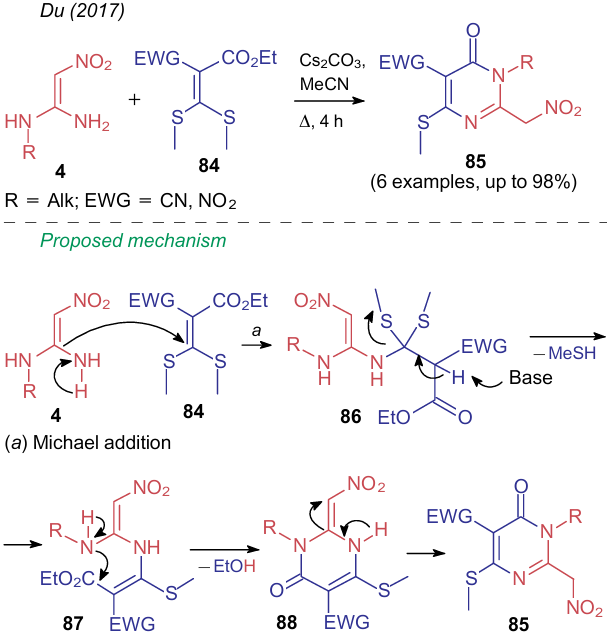
As was shown above, α, α-di(amino)nitroalkenes 4 react with 2,2-di(methylthio)ethylene 84, bearing two electron-withdrawing groups, in boiling acetonitrile in the presence of cesium carbonate to furnish nitropyridines (see Table 2, line 21). However, as noted in the cited publication,104 formally the same reaction but involving NSE 4, containing one unsubstituted amino group, and 1-EWG-1-ethoxycarbonyl-2,2-di(methylthio)ethylenes 84 affords the products of an alternative heterocycization, 5-EWG-substituted 6-methylthio-2-nitromethylpyrimidin-4-ones 85. The authors propose two different mechanisms for these processes (the second one implying the formation of structures 86--88), from which it can be concluded that regioselectivity is governed by the spatial availability of the unsubstituted amino group (Scheme 23).
The reaction of nitroaminoalkenes 3 or 4 with an aromatic amine [2,5-dimethylaniline (89)] and succinaldehyde (90) was used to synthesize compounds 91 based on the 3-nitroazepine core (Scheme 24).152 Note that in the absence of the aromatic amine, the reaction stops at the step of formation of azepine with a bridging oxygen atom, while the addition of 2,5-dimethylaniline gives an aza derivative 91.
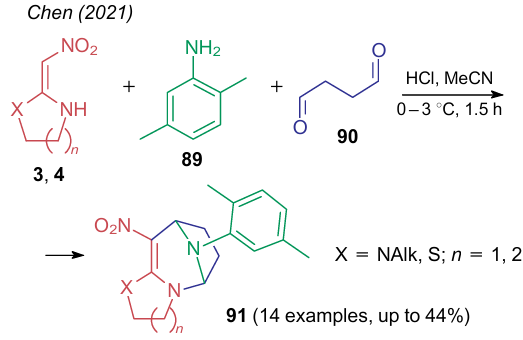
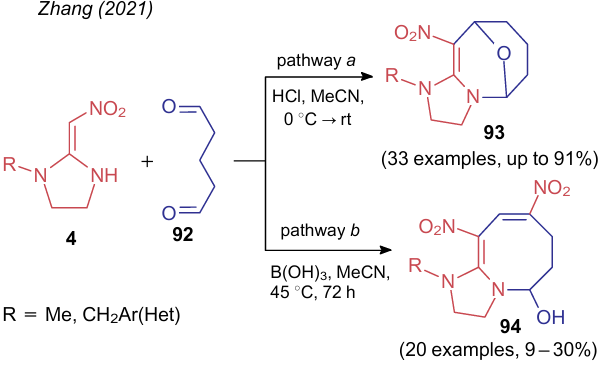
In 2021, Zhang et al.153,154 showed two pathways for the reaction of α, α-di(amino)nitroalkenes 4 with glutaraldehyde (92) depending on the type of acid catalyst (Scheme 25). In the first case (pathway a), oxabridged 10-nitro-5,9-epoxy-1,2,3,4,5,6,7,8-octahydroimidazo[1,2-a]-azocines 93 were formed.153 The reaction occurred in acetonitrile in the presence of Brуnsted acid catalyst (HCl) and was in fact the condensation of a dinucleophile with a dielectrophile. Catalysis with Lewis acid [B(OH)3] unexpectedly led to 8,10-dinitro-1,2,3,5,6,7-hexahydroimidazo[1,2-a]azocin-5-ol 94 (pathway b).154 The low yield of the product may indicate that the second nitro group is incorporated in a molecule 94 due to the destruction of the starting NSE 4. However, the authors did not proposed any reaction pathway.
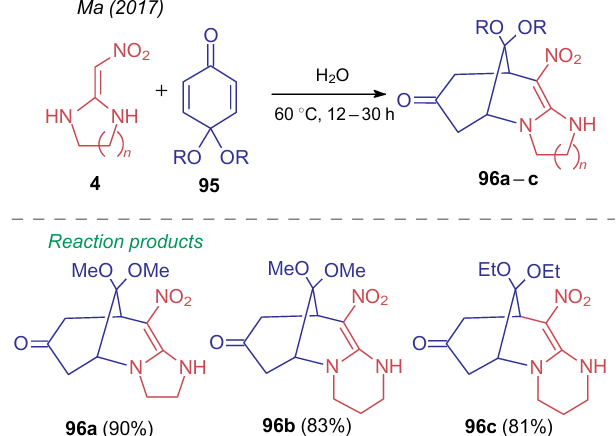
To construct an azocine ring, Ma et al.155 used quinone-based dielectrophiles 95. Thus, reactions of compounds 4 with ketones 95 provides 10-nitro-2,3,5,6,8,9-hexahydro-5,9-methanoimidazo[1,2-a]azocin-7-one (96a) and 11-nitro- 1,2,3,4,6,7,9,10-octahydro-6,10-methanopyrimido[1,2-a]azocin-8-ones 96b,c in high yields (Scheme 26). Noteworthy that unlike the previous similar examples, the process occurs in water under catalyst-free conditions, thus being consistent with the principles of green chemistry.
2.2.2. Synthesis of isoxazoles
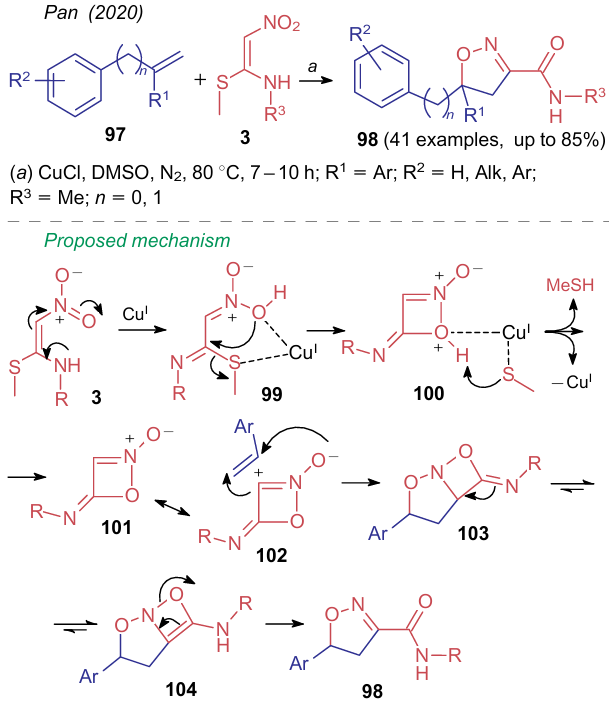
Recently, two approaches to prepare isoxazoles based on NSE 3 have been reported. In the first case, in 2020, Pan et al.156 carried out the reaction between 2-alkyl(aryl)amino-2-methylthio-1-nitroethylenes 3 and olefins 97 in dimethylsulfoxide in the presence of copper(I) chloride to obtain 3,5-disubstituted 4,5-dihydroisoxazoles 98 (Scheme 27). According to the authors, NSE activated by the catalyst first coordinates to the metal atom to generate species 99, and then, via intermediate 100, forms an oxazetine ring 101, which further undergoes cycloaddition to the starting olefin as a zwitterion 102. Intermediate isoxazolidinoxazetine bicycle 103 transforms into its tautomer 104, followed by the ring-opening of the four-membered heterocycle to afford 4,5-dihydroisoxazole 98. Simple procedure, availability of the catalyst and high regioselectivity of the process allow considering NSE 3 as a versatile synthone for the construction of such isoxazoles. An alternative approach had been used a few years before to prepare 3,4,5-trisubstituted isoxazoles 105.
Lei et al.157,158 carried out the reaction between 2,2-diamino-1-nitroalkenes 4 and aromatic aldehydes 14 in alcohol in the presence of potassium carbonate and L-proline (Scheme 28). In this case, two molecules of NSE, e.g., compound 4a, cross-link with a molecule of aldehyde 14 to generate intermediate 106, which then undergoes intramolecular nucleophilic substitution of the nitro group to afford finally isoxazoline-N-oxide 107. The following tautomeric transformations and dehydration yield polysubstituted isoxazoles 105.
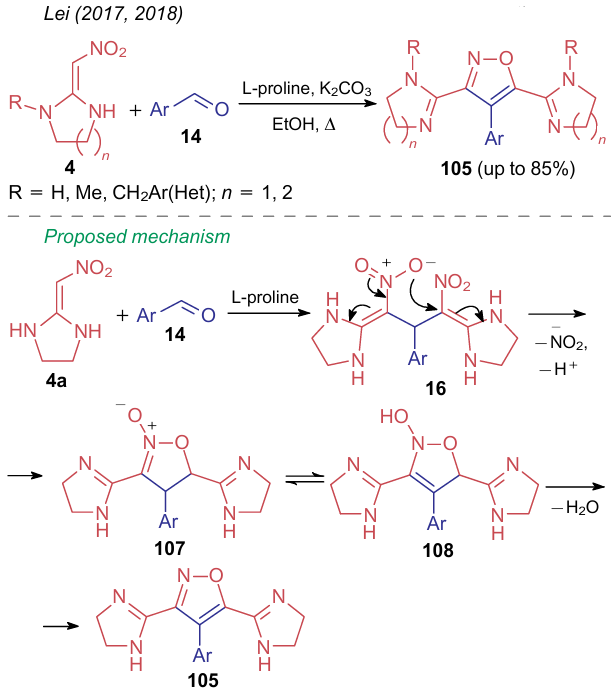
2.2.3. Reactions with the elimination of a nitro group
The structural features of a nitro group are not limited only to its ability to enter cycloaddition reactions. The nitro group can often serve as a leaving group, generally, as a nitrite ion or nitrous acid. However, under certain conditions, some multicomponent processes affording 3-nitropyridines (see Table 1) can follow an alternative reaction path. For example, in 2019, Du et al.114 studied the base-promoted reaction of 2-arylamino-substituted 2-amino-1-nitroalkenes 4, trifluoroacetoacetates 109 and aromatic aldehydes 14 (Table 2 and Scheme 29). When melting the starting compounds together with an equivalent amount of triethylamine, a mixture of 3-nitropyrimidine (see line 30) and pyridine 110 is obtained (see Scheme 29). Carrying out this reaction in propylene carbonate with an equivalent amount of piperidine provides the regioselective synthesis of pyridines 110. The plausible reaction pathway suggests the formation of intermediates 111-114 and implies aromatization of dihydronitropyridine 115 in two ways, namely, under the action of oxygen (see line 30) or by the loss of a nitrous acid molecule (see Scheme 29).
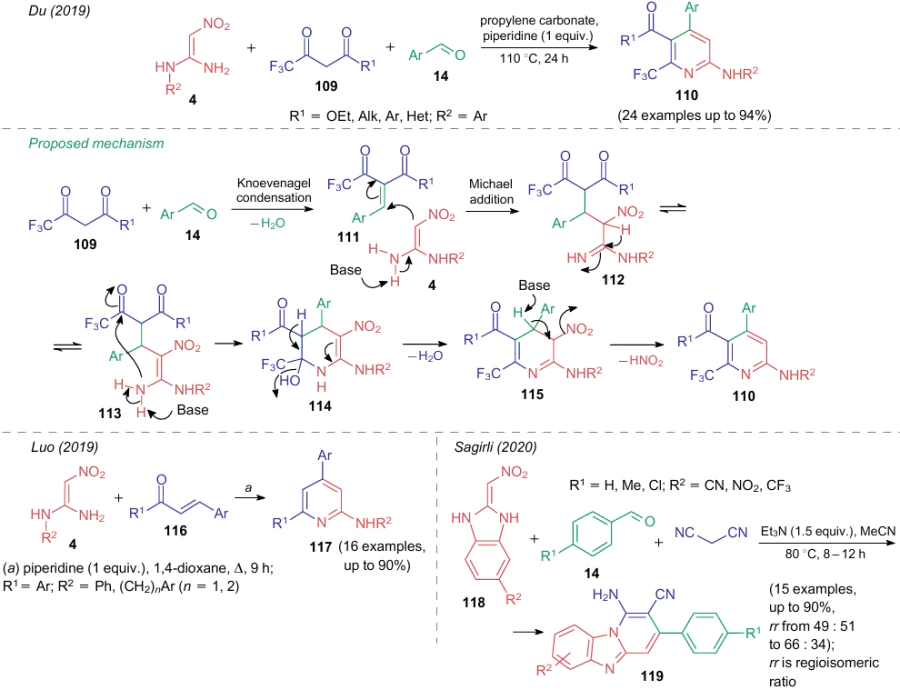
The same year, Luo et al.111 carried out a similar process involving α,β-unsubstituted ketones. The reaction mechanism provided in the cited publication111 is similar to that illustrated in Scheme 29, however, by varying the reaction conditions, the authors succeeded in obtaining two alternative products in the individual state. Refluxing 2-alkyl(aryl)amino-substituted 2-amino-1-nitroalkenes 4 with chalcones 116 in the presence of a strong inorganic base (Cs2CO3) promotes aromatization of the intermediate under the action of atmospheric oxygen and furnishes diaryl-substituted 2-amino-3-nitropyridines (see line 28 in Table 2). On the contrary, refluxing with piperidine leads to the loss of a molecule of nitrous acid and affords pyrimidines (see Scheme 29). Findings of Sagiri159 add to these examples: in this case, NSE 118, benzaldehydes and malononitrile construct the heterocyclic system 119.
An uncharacteristic transformation is observed when α, α-di(methylthio)nitroethylene (2a) reacts with the in situ formed diene 54a (Scheme 30). Lukashenko et al.132 carried out the synthesis of benzo[f]chromen-3-one 120, in which NSE served as an ‘acetylene’ source. When heated, 1-[(dimethylamino)methyl]-2-naphthol (55a) forms 1-methylenenaphthalen-2-one (54a), which enters the Diels-Alder reaction twice: first, it reacts with DiSAlk 2a and then with intermediate 121 releasing a molecule of methanethiol. Under the action of water present in the reaction mixture, symmetric dimer 122 underwent ring-opening, displacement of the methylthio group with a hydroxyl group and aromatization, accompanied by the loss of a molecule of nitrous acid from intermediate 123 to form product 120.

2.2.4. Reactions at the C–H bond
The C–H bond in NSEs is often modified through reactions with nucleophiles. However, such reactions do not always produce cyclic structures. For example, reactions of 2,2-diamino-1-nitroethylenes 4 and 124 with p-quinone methide 125 in acetone in the presence of cesium carbonate occur as an addition of NSE to a C-electrophile.160 The subsequent dehydrogenation lead to conjugated compounds 126 and 127, respectively (Scheme 31). The plausible reaction pathway (via the formation of intermediates 128 and 129) is provided for the second process.
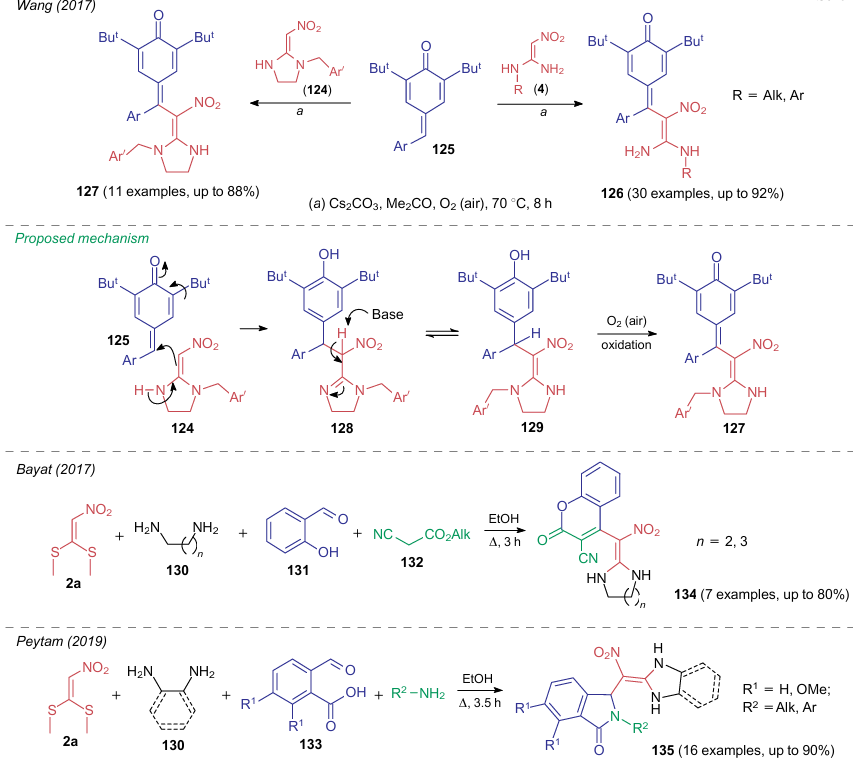
As mentioned in the beginning of this review, the key factor in all such processes is the possibility of enamine-imine tautomerism, which promotes further addition reactions. The research groups of Bayat80,81 and Peytam161 carried out multicomponent reactions, also involving 2,2-diamino-1-nitroethylenes derived from α,α-di(methylthio)nitroethylene (2a) and amines, and C-electrophiles as the key interacting intermediates. Although these reactions proceeded on heating in ethanol without a catalyst, they were completed mainly with dehydrogenation to afford the corresponding conjugated structures. Scheme 31 illustrates the examples of MCRs with compounds 130-133 to produce products 134, 135 (see Scheme 31).
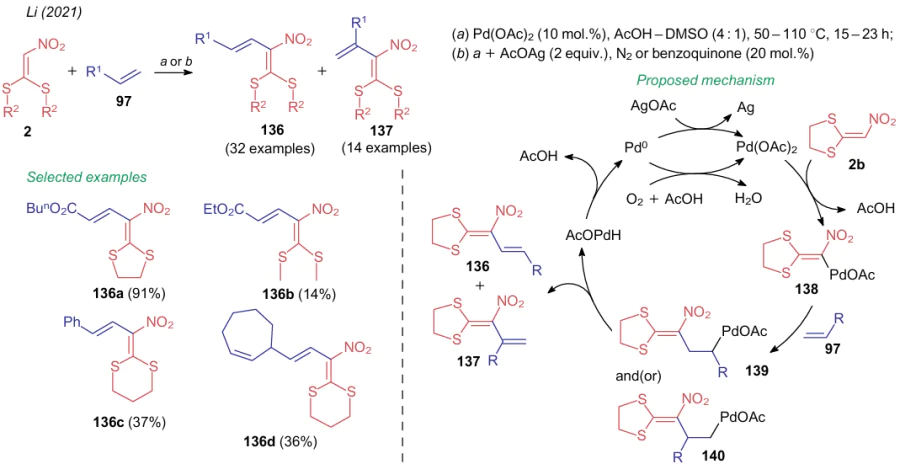
In turn, chemical properties of DiSAlk 2 differ somewhat from those of the above-mentioned compounds. Since tautomerism, which fundamentally changes the chemical properties of the C-atom adjacent to the nitro group, is not possible in such structures, the reaction with electrophiles is not typical for this NSEs. Therefore, in 2021, Li et al.162 developed a procedure for the preparation of regioisomers 136 and 137, which included the activation of the C--H bond of compounds 2 with palladium(II) acetate (Scheme 32). The mechanism of the reaction as illustrated on the example of substrate 2b suggests the divergent addition of a molecule of the terminal alkene to the activated complex 138 to afford Pd-derivatives 139 and(or) 140. Nevertheless, in practice, the process selectivity turns towards the formation of linear isomers 136 (ratio from 1.4:1 to 45:1 with a total yield of 11-92%).
2.2.5. Other reactions
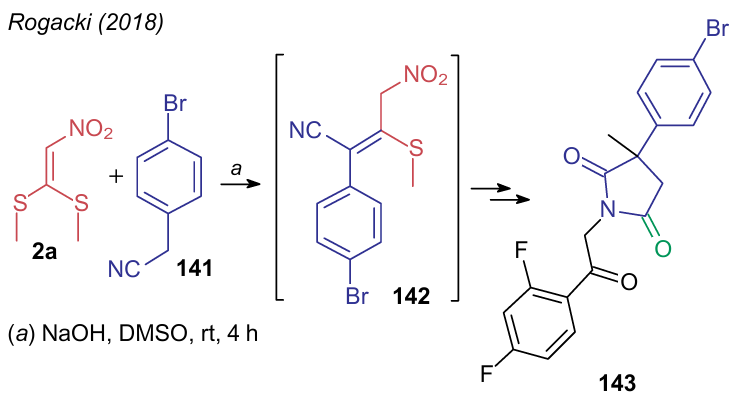
If the substitution of alkylthio groups in DiSAlk 2a by various primary amines is not a specific procedure and is used both directly and as part of multicomponent transformations, a similar substitution by C-nucleophiles is much less common. In 2018, Rogacki et al.163 developed a multistep procedure to prepare compounds having antituberculotic activity, which starts from the condensation of substrate 2a with the CH-acidic p-bromophenylacetonitrile (141) in the presence of sodium hydroxide in dimethyl sulfoxide (Scheme 33). The generated push-pull alkene 142 was further used in situ in the Nef reaction to produce product 143.
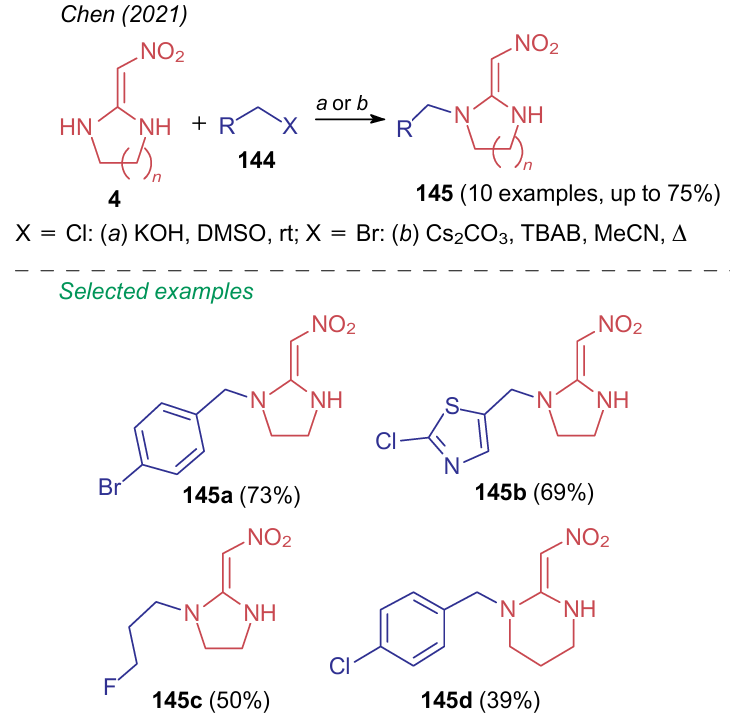
As with aminonitroethanes, DiNH 4, bearing the secondary amine group, enters the alkylation reaction. However, contrary to the example given in Scheme 19, in 2021, Chen et al.152 carried out alkylation with halides 144 to afford monoalkyl derivatives 145 under conditions close to standard for reactions of this type. When using alkyl chlorides, the reaction was conducted in dimethyl sulfoxide in the presence of potassium hydroxide at room temperature, while with alkyl bromides, cesium carbonate in acetonitrile was used with the addition of a phase transfer catalyst, tetra-n-butylammonium bromide (TBAB) (Scheme 34).
Thus, in contrast to the above-mentioned ANE 1, compounds 2-4 were explored much better. At the same time, the variability of their chemical properties is mostly limited to transformations resulting in the heterocycle formation. This is not surprising, since NSE 2-4 are strongly pronounced push--pull compounds and contain two leaving groups at once, which promote heterocyclization. Nevertheless, an in-depth study of methods for modifying the C–H bond attached to the nitro group creates prerequisites for the development of new synthetic approaches to complex organic molecules based on such compounds.
3. Structures of the O2N-CH2-EWG type
The next three groups of NSEs: α-nitroketones (5), alkyl nitroacetates (6) and nitroacetonitrile (7) have similar structural features, which indicates their similar chemical behaviour (Fig. 4). The main affinity element is the C(sp3)-hybridized atom conjugated with two electron-withdrawing groups and allowing these compounds to enter the Knoevenagel and Michael reactions. In addition, NSEs 5-7 are often used in the synthesis of isoxazoles bearing the appropriate electron-withdrawing substituents. Compounds 2–4, described in the previous Section, found application in such transformations much less frequently, while aminonitroethylenes were not used at all during the considered period. A common feature of NSEs 5-7 is also their participation in the cycloaddition reactions. Of these three groups of compounds, nitroacetonitrile (7) is the least represented in the literature, which is due to its low availability and low stability. Certain chemical properties described above for nitro derivatives 1-3, e.g., the reaction at the C–H bond, modification of the nitro group and transformations with its elimination are also typical of ANA and NK. Moreover, new properties appear such as the possibility to deliver nitrile oxides, and also the C–C bond cleavage, which give, due to the presence of an α-CH acidic atom and a carbonyl group, four-six-membered rings, which further undergo ring-opening. Note that nitroketones can enter reactions characteristic of carbonyl compounds, e.g., the addition of nucleophile and reduction. Moreover, NKs can eliminate not only nitro group but also a molecule of nitromethane, as discussed in detail below.
![[{"id":"E-ST6h0UDh","type":"paragraph","data":{"text":" Possible ways to modify compounds <b>5</b> - <b>7</b>"}}]](/storage/images/resized/8IxpRgAAHnoVbzyliz0dDuUF4sL6pJMuubuayGKN_xl.webp)
Considering similar trends in chemical behaviour of nitro derivatives 5-7, the examples of the Knoevenagel and Michael reactions, as well as the synthesis of isoxazoles for these compounds were combined and represented in the form of the corresponding Tables.
3.1. General reactions
3.1.1. Knoevenagel condensation
α-Nitroketones (5), alkyl nitroacetates (6) and nitroacetonitrile (7) are excellent examples of compounds having an active methylene centre to react with carbonyl compounds by the Knoevenagel reaction. Such transformations, found in the literature during the reporting period, are summarized in Table 12.
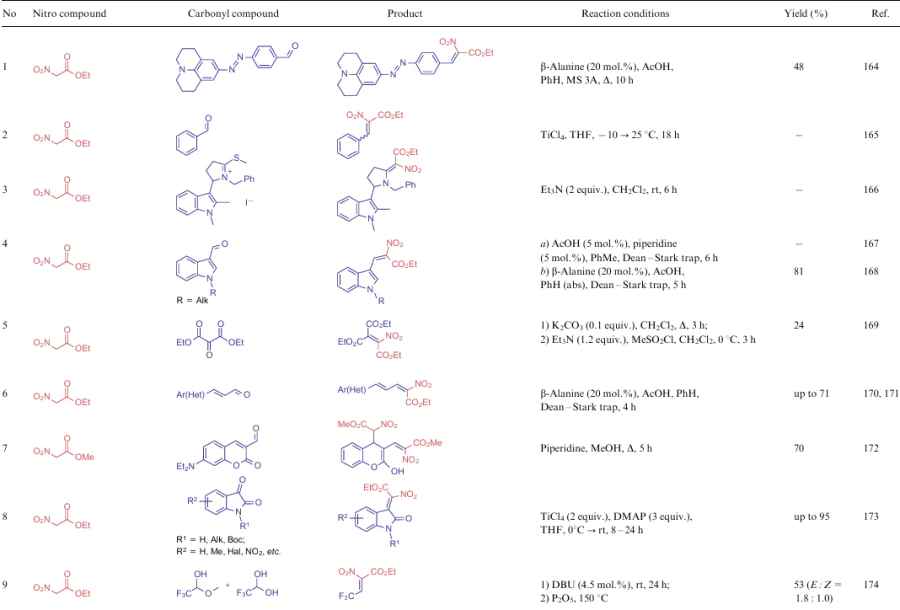
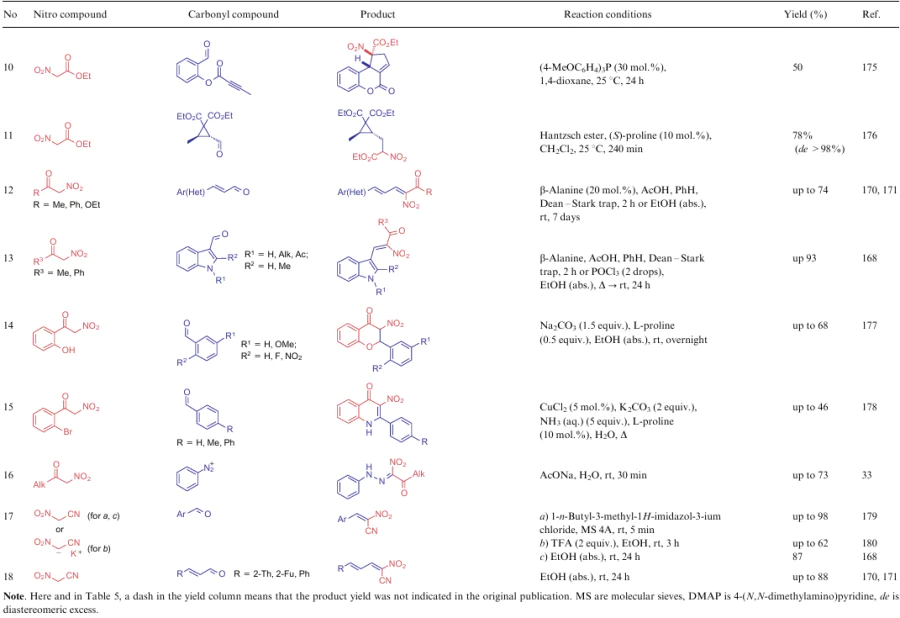
Despite the variety of protocols of the Knoevenagel reaction, due to numerous theoretical and practical studies for the NSEs of this group, several features of this transformation can be highlighted: the use of methyl thioimine as the electrophilic substrate instead of carbonyl compound (see Table 12, line 3), a two-step synthesis to produce an alkene bearing four electron-withdrawing substituents (see Table 12, line 5), and also an unusual catalyst in the reaction between ANA and isatin (see Table 12, line 8). Zhu et al.173 showed that the use of piperidine as the catalyst results in the replacement of a nitro group in the product by a piperidine moiety. Therefore, Lewis acids are recommended for use in such processes along with a non-nucleophilic organic base. Compound 7, as the strongest CH-acid of the three groups under consideration, can react with aldehydes without any catalyst in anhydrous ethanol (see Table 12, lines 17, 18).
3.1.2. Michael reaction
An active methylene centre of NSEs 5-7 is also often involved in the Michael reaction. Examples of such transformations are shown in Table 14. The addition of NSEs 5-7 is mainly catalyzed by bases (see Table 14, lines 1, 2, 4, 6, 15, 16), although the use of bacteria Aspergillus niger in the stereospesific reaction is also known (see Table 14, line 7). In addition to the biocatalyst, optically active catalysts L18 and L19 derived from diaminocyclobutenediones (see Table 14, lines 3, 5, 12 and 14) and pyrrolidine (see Table 14, line 11) can also be used for the stereoselective implementation of the process. An interesting case is the Michael reaction using a coordination compound Pd2L4 (L20) in combination with 18-crown-6 (see Table 14, lines 10 and 13). This catalytic system provides not only a significant improvement in the reaction rate, but also makes the process highly stereoselective.
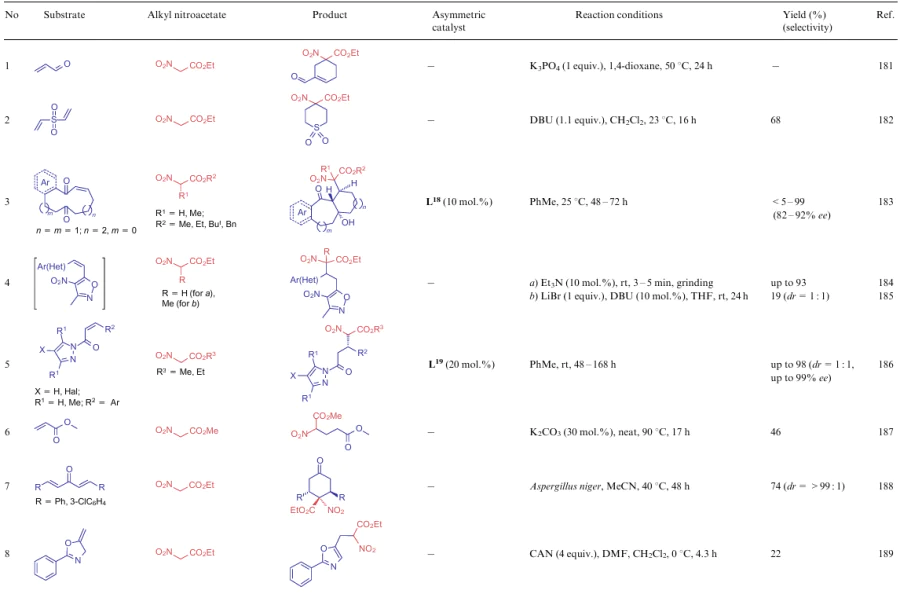
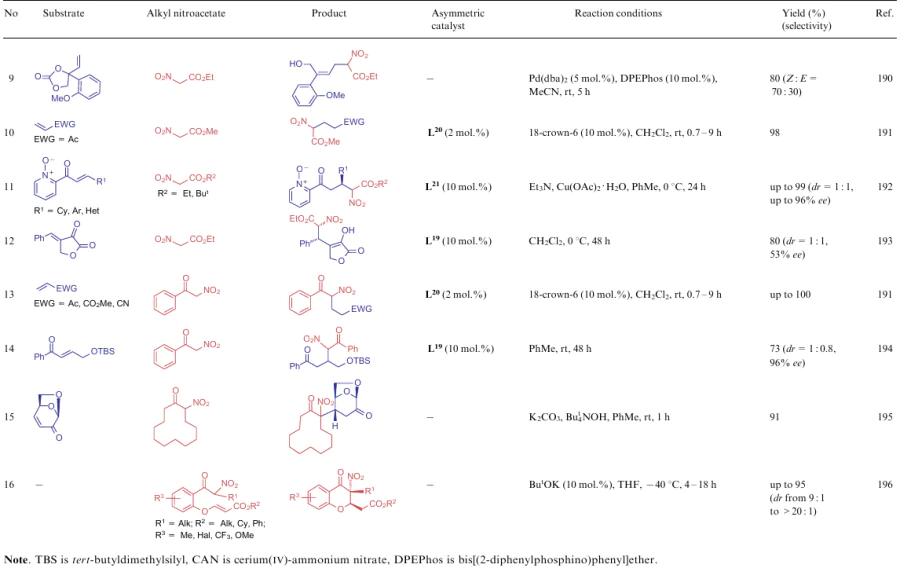
![[{"id":"DbHCPZ7pof","type":"paragraph","data":{"text":" Structures L<sup>18</sup>-L<sup>21</sup>"}}]](/storage/images/resized/s71ZacDblOjDaQYCnc62uKmAZrSQZ2IQJf6v8KcD_xl.webp)
3.1.3. Synthesis of isoxazoles
The most common applications of NSEs 5-7 also include their use as starting materials in the synthesis of various isoxazoles and isoxazole N-oxides. Both the active methylene centre of the subject compounds and the nitro group per se are involved in the construction of the five-membered ring. Various alkenes, alkynes and carbonyl compounds are typical substrates for such process (Table 16). Noteworthy that the current ideas about the formation of (dihydro)isoxazoles suggest the initial generation of the corresponding (dihydro)isoxazole N-oxides, which are either dehydrated under more harsh conditions or reduced in boiling trimethyl phosphite. The only reaction that occurs in the presence of an asymmetric catalyst L21 is shown in line 8 of Table 16 (conditions a).
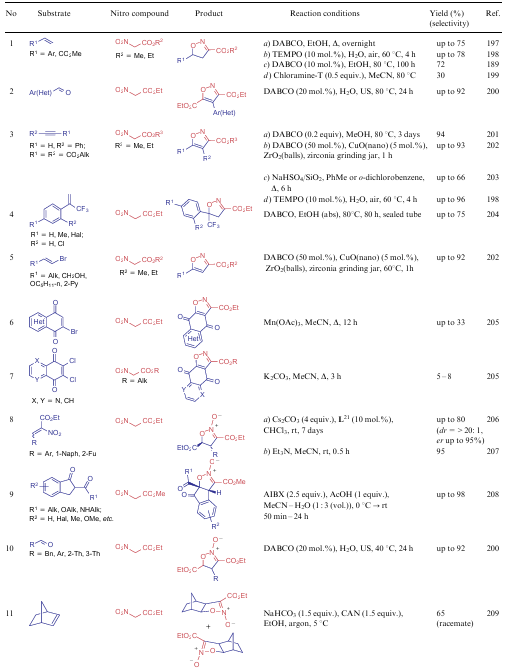
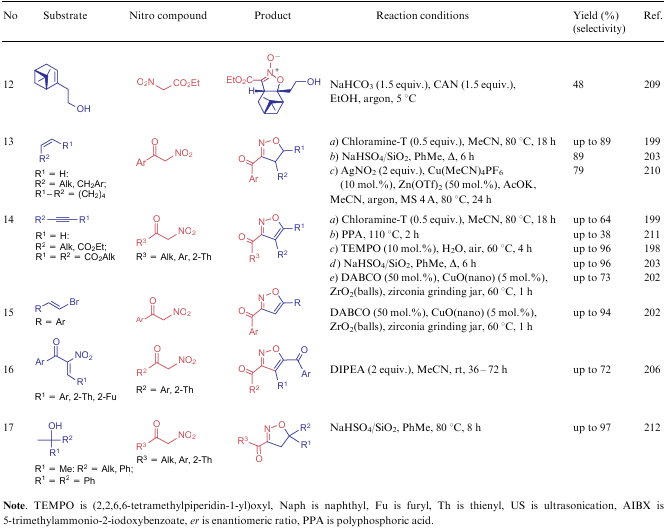
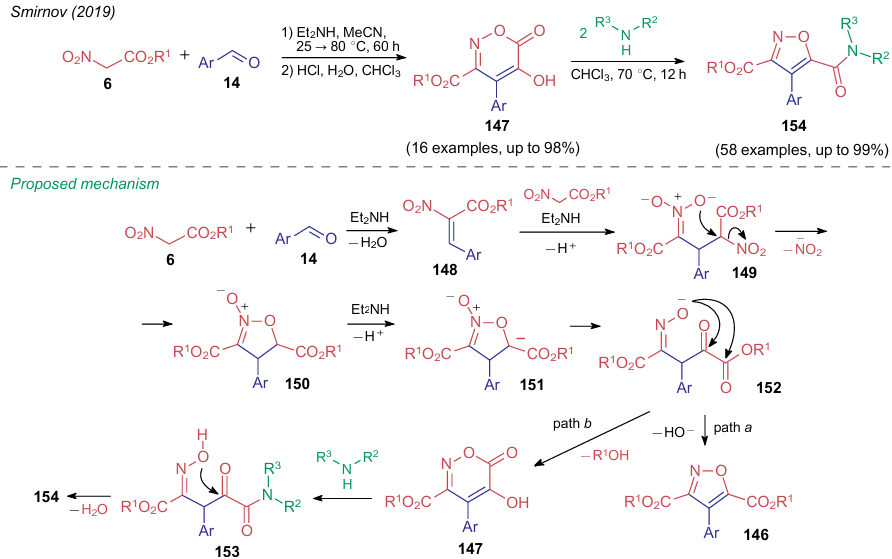
In 2019, Smirnov et al.213 revised the classical mechanism of the Dornow-Wiehler reaction. The authors found that aromatic aldehydes 14 can react with 2 equiv. of nitro compounds 6 to produce not only 3,5-alkoxy-4-arylisoxazoles 146 but also 4-aryl-5-hydroxy-6-oxo-6H-1,2-oxazine-3-carboxylates 147 (Scheme 35). However, the formation of six-membered rings directly depends on the reaction conditions: this requires the use of a polar aprotic solvent, as well as an unusual temperature mode. The starting compounds are first stirred at room temperature for two days, and then the reaction mixture is heated at 80 °C for 3 h. Using aliphatic aldehydes, a mixture of products 146 and 147 is obtained, and in the case of aromatic aldehydes with strong electron-withdrawing groups, only isoxazoles 146 are isolated. It is assumed that this process starts from the reaction of one NSE molecule with an aldehyde to give intermediate 148, which adds the second molecule of the nitro compound. 3,5-Dinitroglutaric ester 149 cyclizes into isoxazoline N-oxide 150. When treated with a base, intermediate 150 delivers the corresponding salt 151, which is ring-opened to form oxime 152 capable of cyclizing into products 146 and 147 (see Scheme 35, paths a and b, respectively). The authors213 was also showed that oxazines 147 react with various amines and water to afford intermediates 153, cyclizing into unsymmetrical isoxazoles 154, which is a significant contribution to the study of such structures.
3.2. Alkyl nitroacetates
The following are the most specific properties of alkyl nitroacetates. Quite often, these compounds are considered as latent analogues of glycine alkyl esters. This determines the ways of their modifications, which were discussed in detail in the review by Hervin et al.30 The methods for the synthesis of such compounds can be found in publications.214,215
3.2.1. Cycloaddition reactions
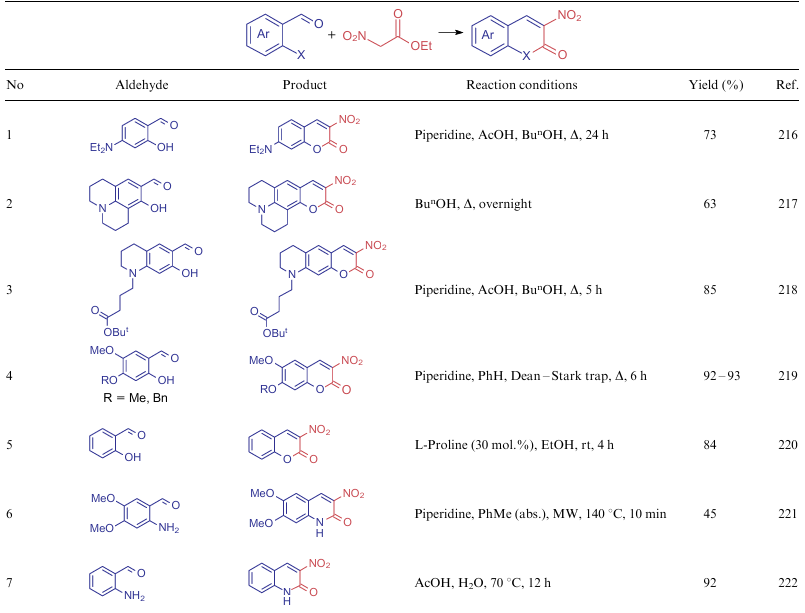
The presence of a nitro group, which significantly increases the acidic properties of the methylene carbon atom, as well as the ethoxycarbonyl group, turns ethyl nitroacetate (6a) into a pronounced bifunctional NSE, which is often used in the synthesis of six-membered rings. Several examples demonstrating the involvement of compound 6a in the construction of a ring of nitroquinolones and nitrocoumarines are presented in Table 18. In general, these transformations are a modification of the Friedlйnder reaction, which can involve both o-aminobenzaldehydes and salicylic aldehydes.216--222
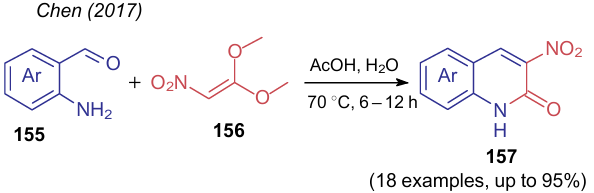
An interesting example is the use of 1,1-dimethoxy-2-nitroethylene (156) instead of alkyl nitroacetate in the reaction with o-aminobenzaldehydes 155 (Scheme 36). Chen et al.222 pointed out that such NSE was first developed so as to introduce the CH=CHNO2 fragment into molecules to design biologically active compounds. Despite the structural similarity of nitroalkene 156 with dimethyl carbonate, the authors failed to use this NSE as an alkylating agent, but this compound found application in the preparation of 3-nitroquinolin-2-ones 157 on heating in dilute acetic acid. Apparently, in the presence of water, 1,1-dimethoxy-2-nitroethylene (156) hydrolyzes to methyl nitroacetate, which is involved in the cycloaddition reaction by known mechanisms.
Of particular interest is the synthesis of 3-nitroazolo[5,1-c]triazines 15810,13,14,223--227 from ethyl nitroacetate (6a) and diazoles 159 generated in situ during diazotization of the corresponding amino azoles 12 (Scheme 37). The reaction tolerates a wide range of amino-substituted pyrazoles, imidazoles, triazoles and tetrazoles. Note that hydrazones 160 resulting from azo coupling can heterocyclize to products 158 without being isolated from the reaction mixture.225,227
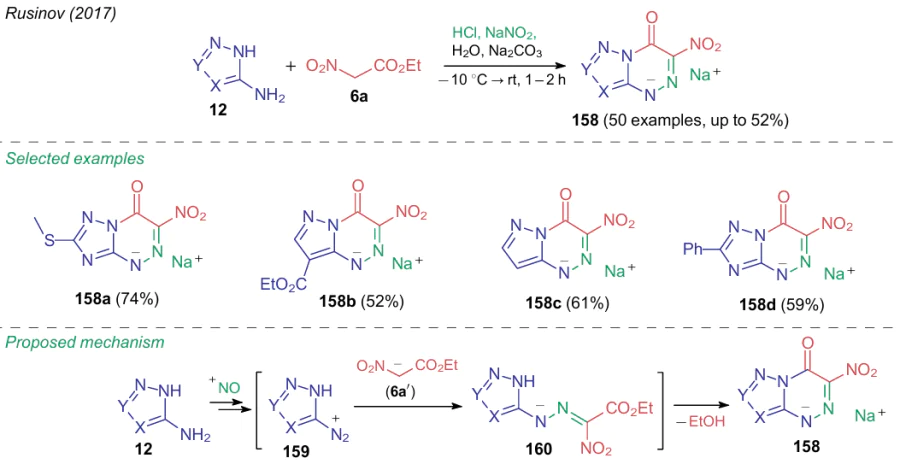
3-Nitroazolo[5,1-c][1,2,4]-triazin-4-ones 158 thus obtained form a new class of antiviral agents effective in the treatment and prevention of influenza, SARS, tick-borne encephalitis, as well as COVID-19 and a number of other viral infections. An antiviral drug Triazavirin (Riamilovir),228,229 6-methylthio-3-nitro[1,2,4]triazolo[5,1-c][1,2,4]triazin-4-one sodium salt dihydrate (158a), is produced in Russia on an industrial scale and are used in medical practice. Compounds 158 are also of interest as antidiabetic drugs.223,225 Moreover, the synthesis of Triazavirin labeled with several stable isotopes was reported, since incorporation of deuterium, 13C and 15N atoms into its structure provides unique opportunities for studying its metabolic processes and mechanism of action in the body.230
3.2.2. Transformations involving the elimination of the nitro group
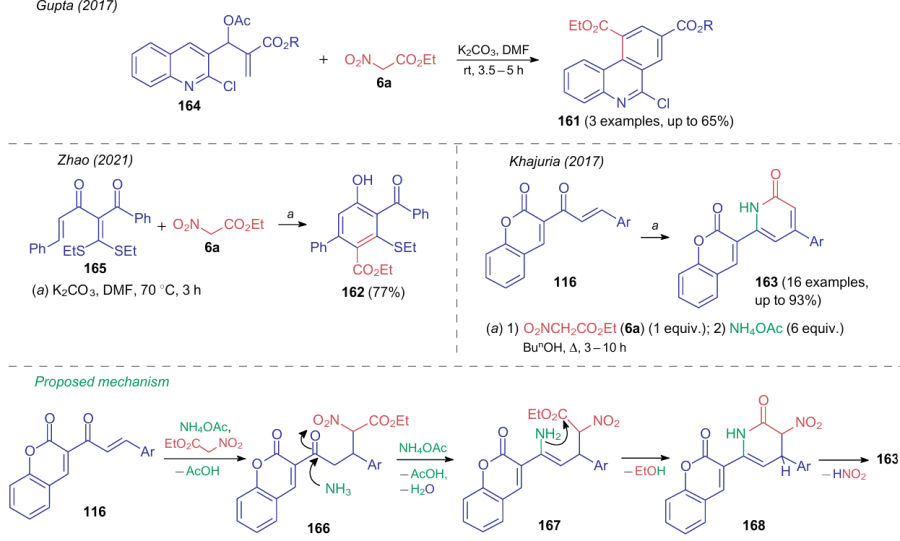
Similar to NSEs outlined in the previous Section, alkyl nitroacetates enter the cycloaddition reactions, which lead to aromatization via the expulsion of the nitrite anion. Thus, a number of publications231--233 deal with the synthesis of six-membered and heteroaromatic compounds 161--163 from substrates 116, 164, 165 and ethyl nitroacetate (6a) (Scheme 38). In general, such processes often requires the use of polar solvents and base catalysts. Despite the structural features of substrates 116, 164, 165, the cyclization follow the similar mechanisms. For example, for products 163, the formation of intermediates 166-168 is assumed.
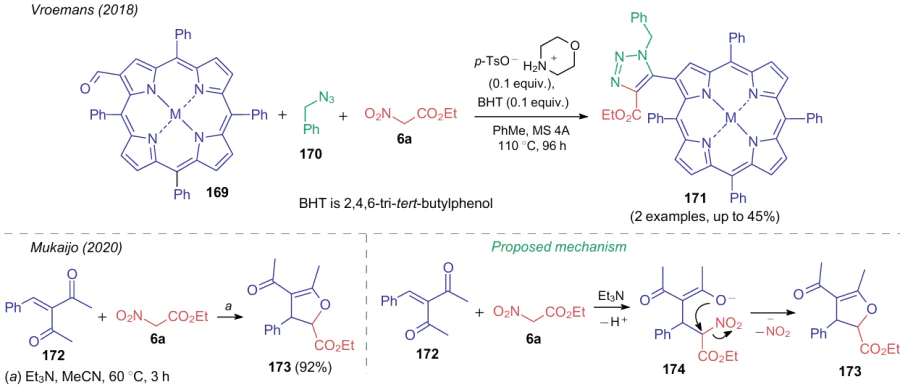
The synthesis of five-membered heterocycles using ANAs is also possible. Starting from complexes 169, benzyl azide (170) and ethyl nitroacetate (6a), triazole-containing porphyrins 171234 were obtained, whereas 3-benzylidenepentane-2,4-dione (172) and the nitro compound 6a afforded dihydrofurane 173.207 However, the mechanism of expulsion of the nitro group differs somewhat from that described in the previous examples. In reactions as illustrated in Scheme 39, the nitrite anion was not eliminated leading to aromatization of the molecule, but an intramolecular substitution occurs, e.g., in structure 174 to form a heterocycle.
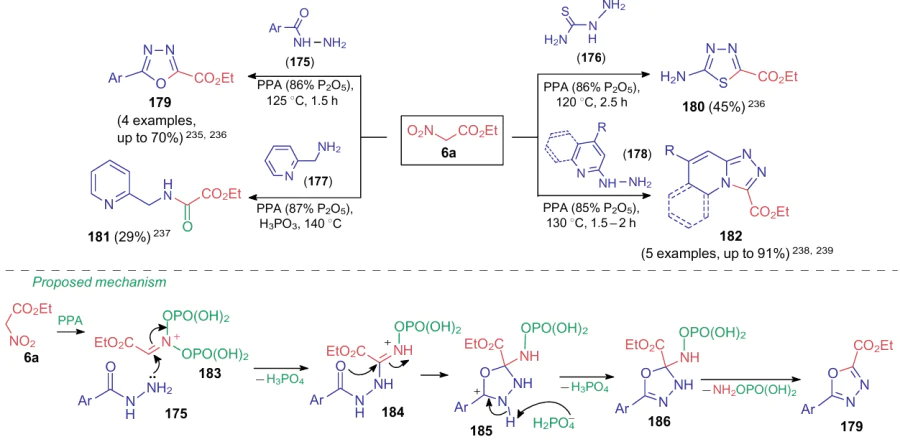
Aksenov et al.235--239 described a special type of nitro group substitution in ANAs. It was shown that the reaction between ethyl nitroacetate (6a) and polyphosphoric acid affords a diphosphorylated intermediate, which can further react with various N-nucleophiles, e.g., with compounds 175-178 (Scheme 40). As a result, products 179-182 are formed. The authors suggest that N-nucleophile (e.g., hydrazide 175) attacks diphosphorylated intermediate 183 to generate species 184, which further cyclizes into oxadiazolidine 185. In the final step, the nitrogen atom of the nitro group leaves intermediate 186 in the form of aminooxyphosphoric acid.
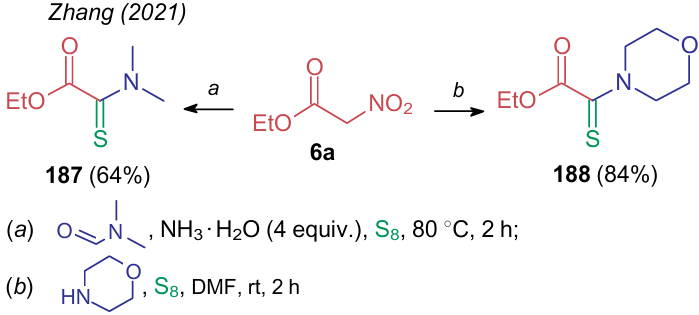
In 2021, the research group of Zhang240 carried out the simultaneous thionation of ethyl nitroacetate (6a) on the CH-acidic carbon atom and displacement of the nitro group with dimethylamine (compound 187) or morpholine (188) moieties (Scheme 41). (This process will be detailed in Section 3.3.5 on nitrocarbonyl compounds.)
3.2.3. Reactions at the C–H bond
This subsection covers the reactions of alkyl nitroacetates, in which an active methylene fragment is modified in ways other than those discussed above in the Knoevenagel and Michael reactions (Table 19). Among such processes, one can single out the addition of ethyl nitroacetate (6a) to heptene in the presence of ligand L22 and stereoselective aza-Henry conversion of imines and alkyl nitroacetates under the action of optically active quinoline catalyst L23 (see Table 19, line 4). Also known is the regioselective reaction of ANAs with alkynes catalyzed by palladium or rhodium complexes with (2-biphenyl)dicyclohexylphosphine (Cy-Johnphos) or ligand L24 (see Table 19, lines 5, 6), which is likely to occur via an intermediate allene structure. The reaction of ANA with alkylating agents containing various leaving groups (see Table 19, lines 7-13), such as bromide, tosyl, hydroxide, alkoxide, and amines, was studied. As ligands, compounds L25-L27 can be used. It is also worth noting that in the presence of metal complexes, the alkylation reaction prevails in the competitive processes of addition of ANAs to alkynes (see Table 19, line 11) and alkenes (see Table 19, line 12).
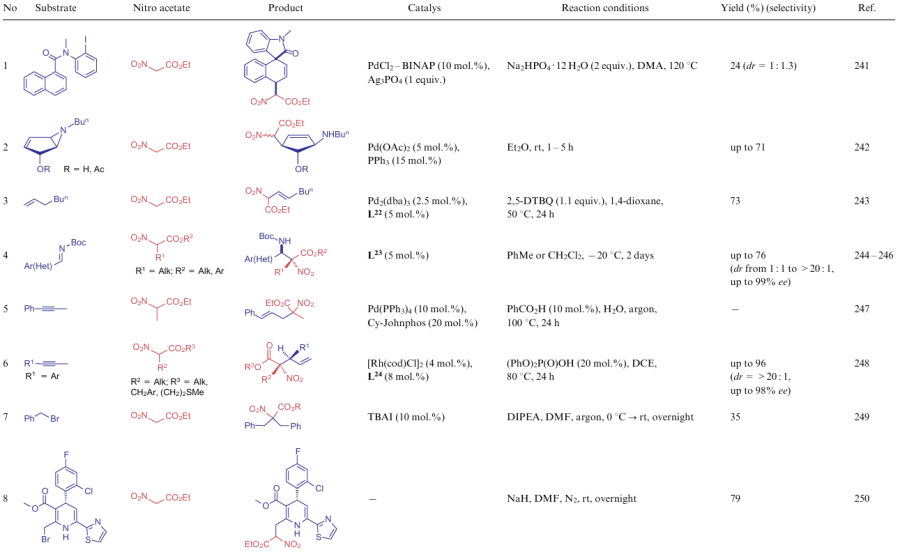
![[{"id":"0kkUIkn3Bn","type":"paragraph","data":{"text":" Structures L<sup>22</sup>-L<sup>27</sup>"}}]](/storage/images/resized/n4T7pQ4cG376hn75DbKGruaoxAYxdAs3sTayJQf0_xl.webp)
3.2.4. Other reactions
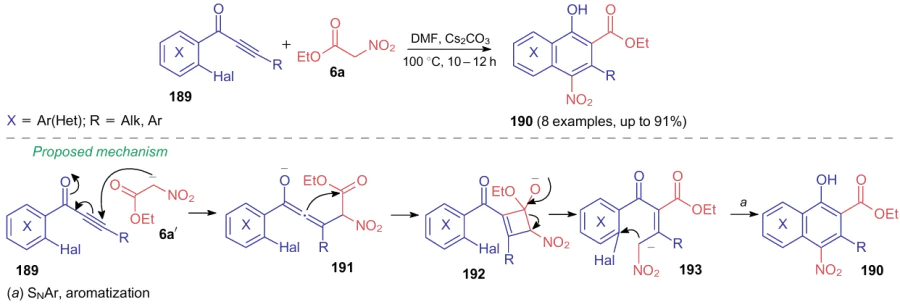
Ethyl nitroacetate (6a) indergoes an unusual C–C bond cleavage when reacting with ketoalkynes 189 in the presence of cesium carbonate at elevated temperatures to produce nitro(het)arenes 190.256,257 The authors note that activated NSE 6aʹ first adds across a triple bond of substrate 189 to yield allene 191, which then undergoes an intramolecular cyclization to afford nitro-containing cyclobutene 192 (Scheme 42). Further, the C–C bond, which previously belonged to the starting compound 6a, is cleaved in the intermediate 192 to generate anion 193. Given the halogen atom ortho-positioned to the alkynylcarbonyl substituent in substrate 189, the final reaction step is aromatization with nucleophilic aromatic substitution of that halogen atom.
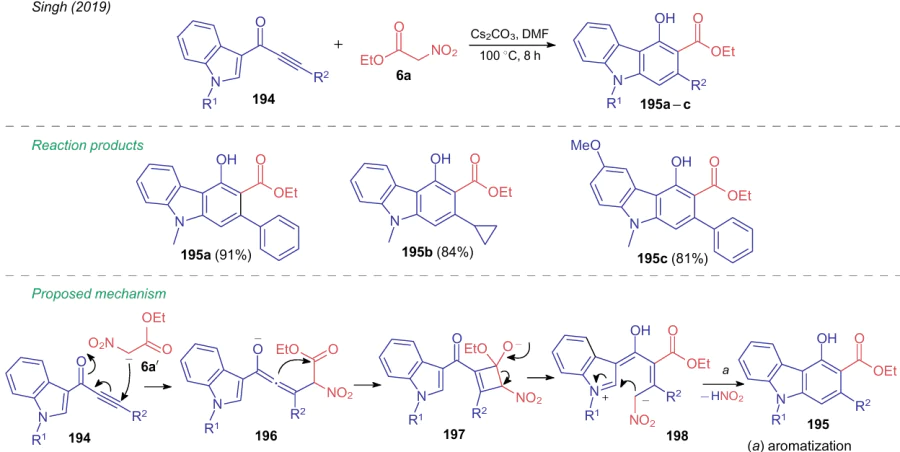
Noteworthy that similar reaction of 3-alkynyloxy-substituted indoles 194 devoid of the 2-positioned halogen atom delivers products 195a-c containing no nitro group.258 In this case, the generation of intermediates 196 and 197 is followed by the final aromatization of an intermediate anion 198 via the loss of a molecule of nitrous acid (Scheme 43).
In 2020, Roscales and Csвky259 described the protocol for a simultaneous reduction of a nitro group and its modification in alkyl nitroacetates. Thus, a prolonged heating of compounds 6 with arylboronic acids 199 and trialkyl phosphites 200 furnishes aminoacetic acid esters 201 in satisfactory-to-good yields (Scheme 44). The plausible mechanism suggests the formation of a four-membered ring intermediate 202, which attacks arylboronic acid 199. Next, two molecules of trialkyl phosphate are released sequentially from intermediate compounds 203 and 204. The newly formed boramide acid 205 is alkylated with trialkyl phosphate to give aminodisubstituted product 201.
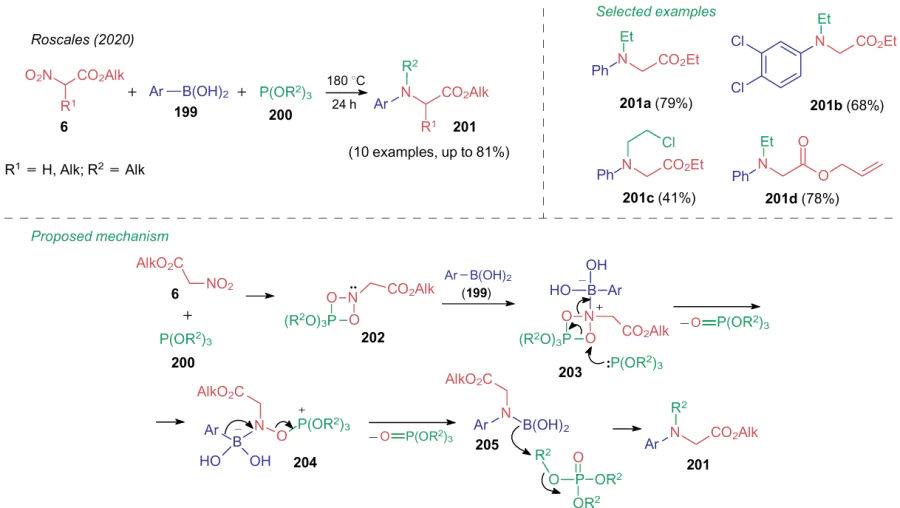
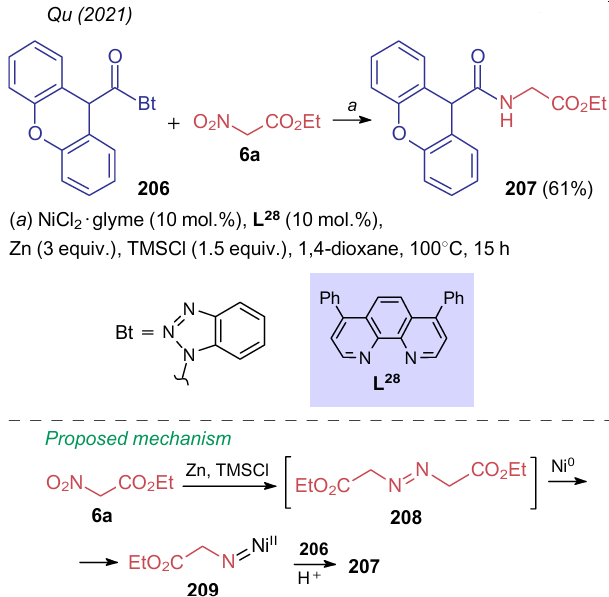
Alkylation and arylation are not the only reactions of the nitro group reductive modification. Qu et al.260 showed the possibility of acylating ethyl nitroacetate (6a) with benzotriazole 206 on reduction with zinc in the presence of phenanthroline L28, which delivered product 207 followed heating in dioxane (Scheme 45). Based on the control experiments, the author proposed the mechanism including the initial formation of azodicarboxylate 208, which further converts to a nitrene intermediate 209 under the action of Ni0.
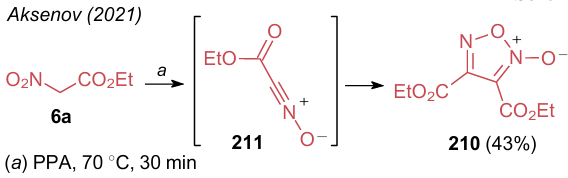
Cited above publications of Aksenov et al.235--239 describe a special influence of polyphosphoric acid on the reactions of alkyl nitroacetates 6 with nucleophiles (see Scheme 40). At the same time, under certain conditions,211 polyphosphoric acid reacts with ANAs as a conventional mineral acid, i.e., it does not provoke the expulsion of the nitro group, but promotes dehydration. Thus, compound 6a delivers disubstituted furoxan N-oxide 210 in the presence of polyphosphoric acid at 70 °C (Scheme 46). This reaction was carried out to study the influence of PPA on NSEs. Nitrile oxide 211 is proposed to be the key intermediate; however, it was not possible to isolate it because of its high reactivity.
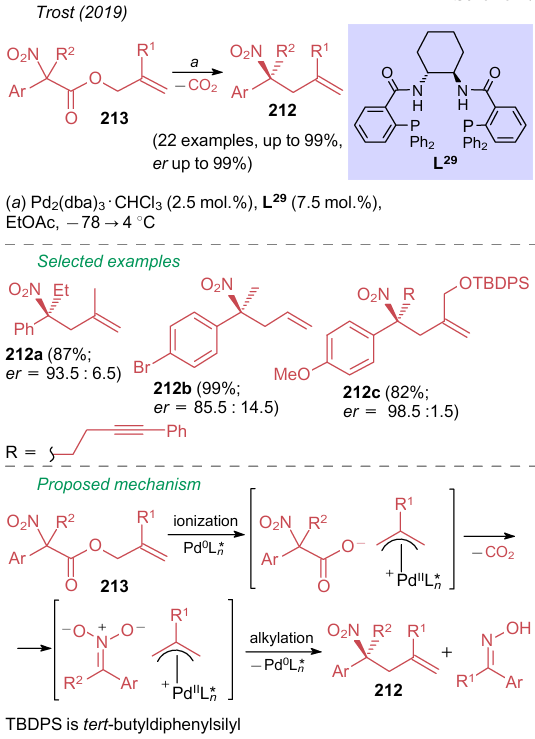
An interesting example of enantioselective palladium-catalyzed decarboxylation was reported by Trost et al.261 in 2019. The use of Pd2(dba)3·CHCl3 complex and optically active ligand L29 in ethyl acetate at low temperature makes available the synthesis of tetrasubstituted nitroalkanes 212 from allyl nitroacetates 213 (Scheme 47). The authors note that this synthetic approach to nitroalkanes 212, referred to as decarboxylative asymmetric allylic alkylation (DAAA), has several advantages over the alternative strategy involving allylation of disubstituted nitroalkanes. The main advantages of this reaction are high stereoselectivity and an insignificant effect of steric factors.
To summarize this Section, it can be said that the difference in chemical behaviour between conjugated NSEs and nitroalkane-type structures is already evident in the example of alkyl nitroacetates. The C-H bond in nitroacetates has a strong acidic character, providing a variety of simple transformations with the formation of a new C-C bond. Moreover, compounds 6 are much more likely to enter reactions with direct involvement of the nitro group, thereby providing an access to both new heterocyclic compounds and polysubstituted amines.
3.3. α-Nitrocarbonyl compounds
As mentioned above, α-nitroketones (5) and alkyl nitroacetates (6) have similar chemical behaviour patterns. Their most distinctive features are the reactions arising from the presence of a carbonyl rather than an ester moiety, such as addition of nucleophiles and reduction. In general, α-nitroketones are quite ubiquitous compounds and methods for their preparation are detailed in a monograph.16
3.3.1. Cycloaddition reactions
Similar to ANAs, α-nitroketones, due to their bifunctional nature, enter cyclization reactions. These can be cyclocondensations involving only nitroketones, as well as nitroketones and other bifunctional substrates with various carbonyl compounds or their latent forms (Table 21).
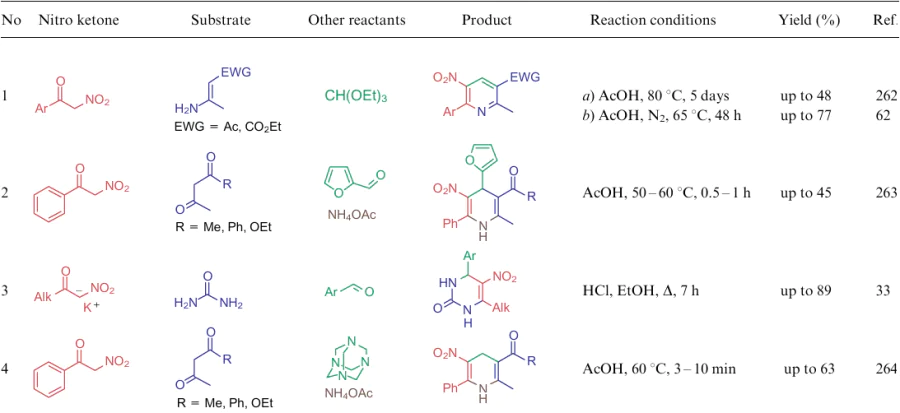
Like ethyl nitroacetate (6a), salts of nitroacetaldehyde and α-nitroketones 32 are versatile substrates for the construction of nitro-1,2-4-triazine core in the synthesis of 4-hydroxy-3-nitro-1,4-dihydroazolo[5,1-c]-1,2,4-triazines 214 (Scheme 48).265--267 This transformation, as that depicted in Scheme 37, suggests diazotization at –10 °C without isolation of an intermediate hydrazone.

3.3.2. Reactions with the cleavage of the C(NO2)–C(O) bond
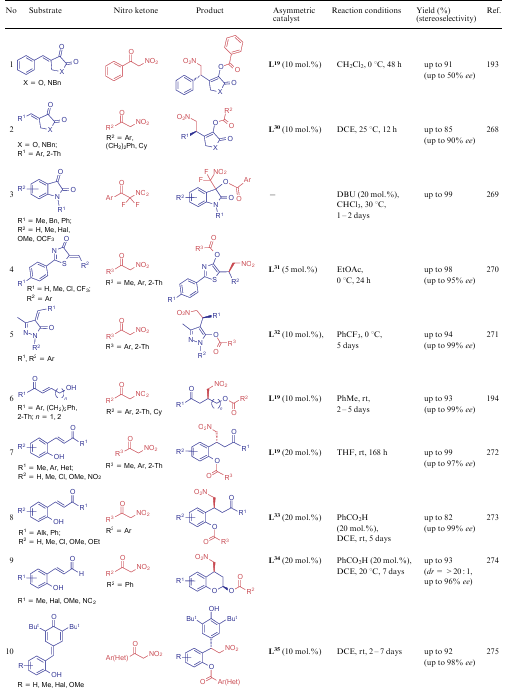
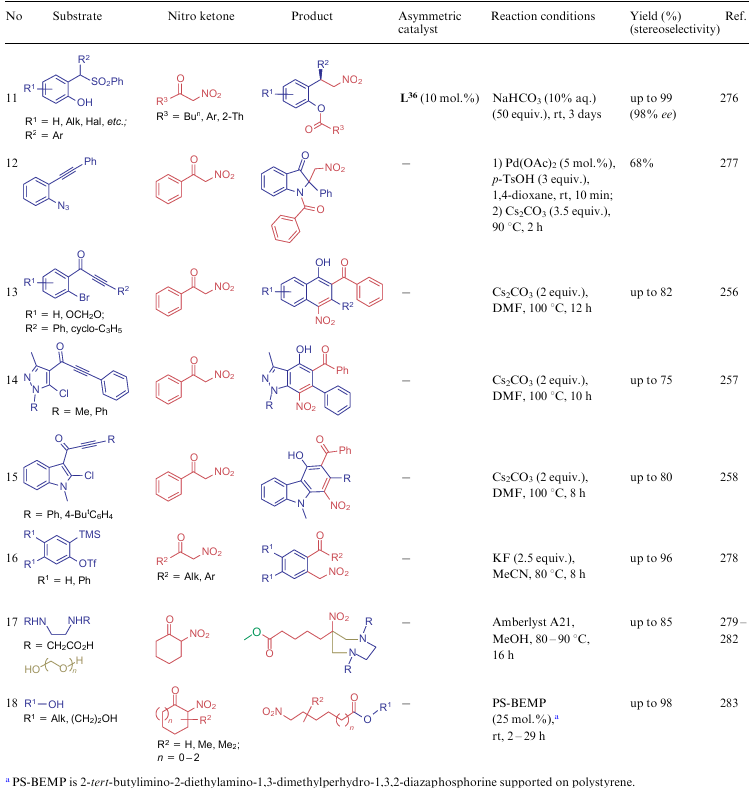
A distinctive feature of α-nitroketones is the C–C bond cleavage in reactions with various bifunctional compounds (Table 22). Several examples of such interaction were observed for ANAs (see Scheme 42, Scheme 43), however, for compounds 5, a much larger number of such reactions are known. Ligands L19 (see the structure above), L30-L36 were used as asymmetric catalysts.
![[{"id":"sCis5xiRMp","type":"paragraph","data":{"text":" Structures L<sup>30</sup>-L<sup>36</sup>"}}]](/storage/images/resized/HYZlHa4YXcEekfqNXOwdaEqxQlTTNARmnKK8B6mU_xl.webp)
3.3.3. Reactions with nucleophiles
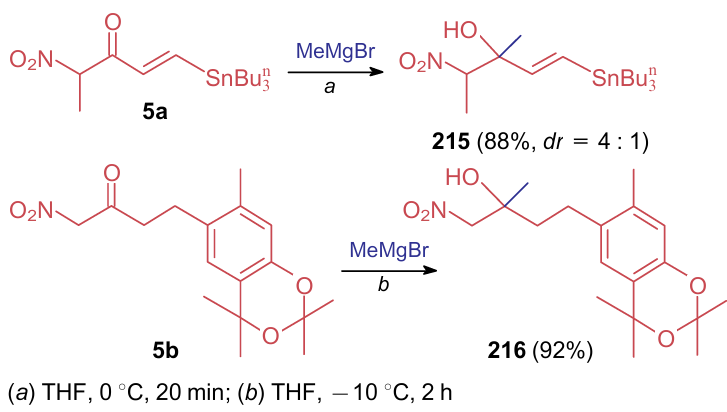
Reactions of nitrocarbonyl compounds with C- and N-nucleophiles generally affect the carbonyl moiety by the addition or ANE patterns (i.e., with the loss of a water molecule). Thus, examples of the reaction of nitroketones 5a,b with the Grignard reagent to afford nitro derivatives 215, 216 are known (Scheme 49).284,285
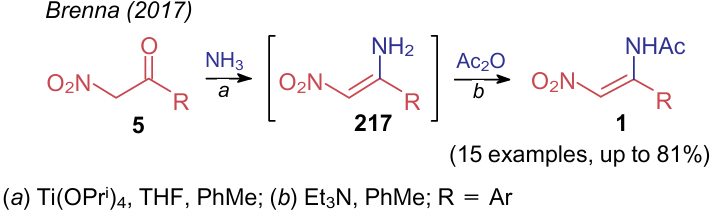
Brenna et al.35 described the preparation of N-acylated aminonitroethylenes 1 by treating nitroketones 5 with ammonia in the presence of titatium isopropoxide followed by acylation of intermediate 217 (Scheme 50).
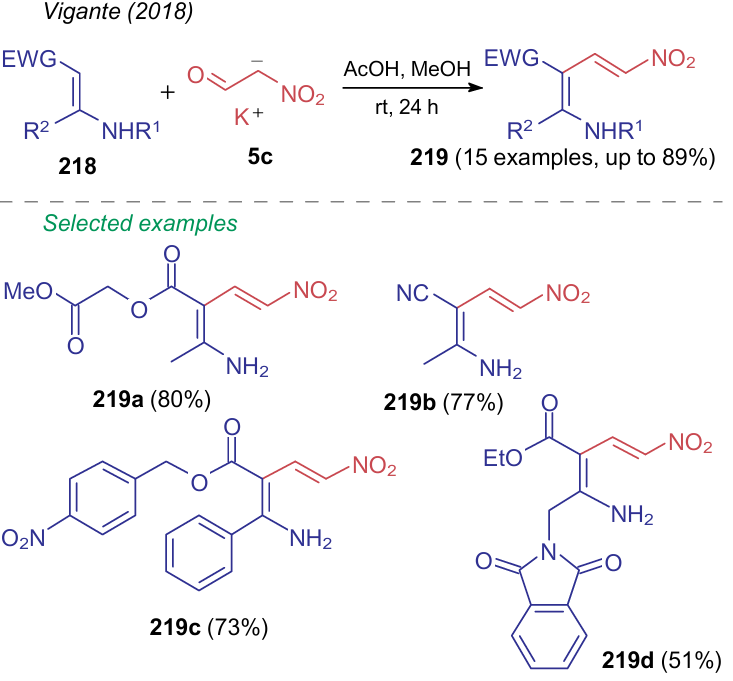
Potassium salt of nitroacetic aldehyde 5c can react with push--pull pre-nucleophiles 218 in a methanol--acetic acid mixture (Scheme 51).286 This process can also be considered as an incorporation of a nitrovinyl fragment into a molecule.
3.3.4. Reactions at the C–H bond
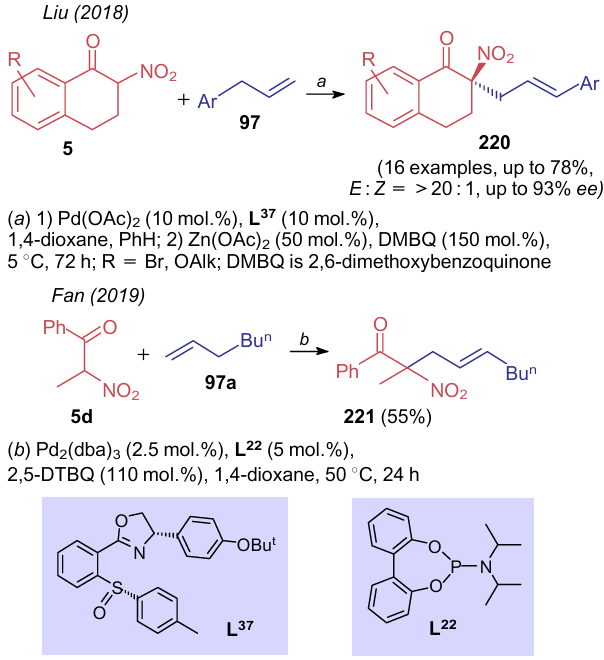
Modification of the C(sp3)-hybridized atom in nitro-containing carbonyl compounds is mainly represented by their reactions with unsaturated hydrocarbons. Thus, Liu et al.287 and Fan et al.243 reported alkylation of terminal olefins using palladium catalysts and appropriate ligands L37 and L22 (Scheme 52). Using aryl sulfoxide--oxazoline catalyst L37, (R)-2-(3-arylallyl)-3,4-dihydro-2-nitronaphthalen-1-ones 220 were produced with high enantioselectivity. The similar reaction involving nitroketone 5d and heptene 97a in the presence of ligand L22 furnished product 221 in moderate yield.
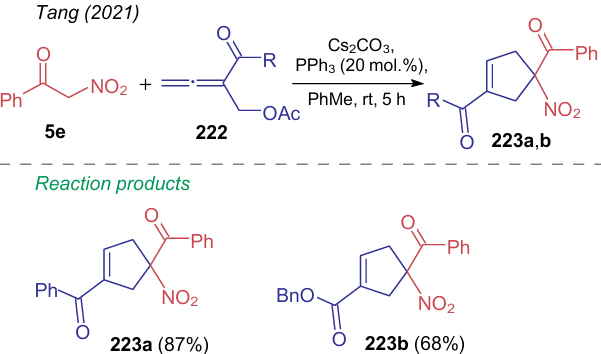
When treated with a base and triphenyl phosphine, nitroacetophenone (5e) reacts with allenes 222 to give 1-benzoyl-1-nitrocyclopent-3-ene-3-carboxylates 223a,b in high yields (Scheme 53).288
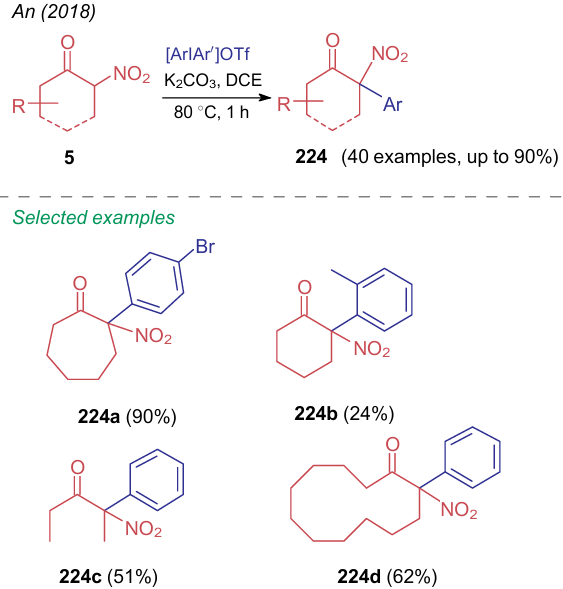
In 2018, An et al.289 pioneered in carrying out α-C-arylation of nitroketones 5 with diaryliodonium salts under metal-free conditions to afford a wide range of nitro compounds 224 (Scheme 54).
3.3.5. Transformations with elimination of the nitro group
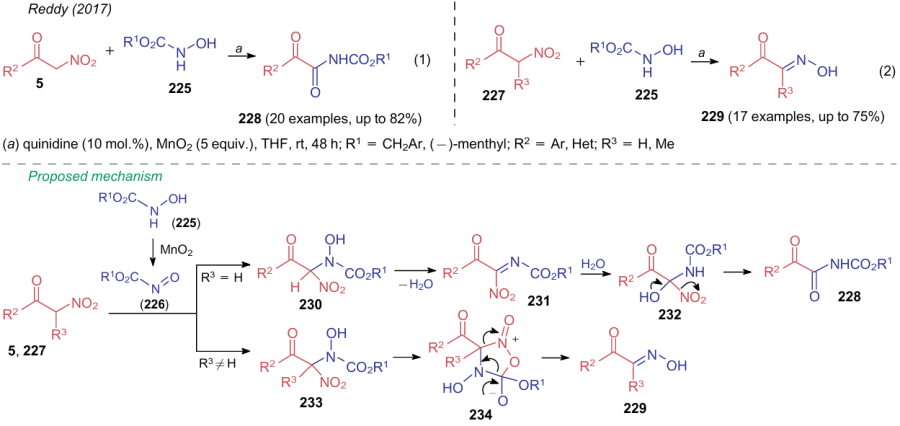
In 2017, Reddy et al.290 showed an example of the Henry reaction between nitroketones and nitrosocarboxylic acid esters 226 generated in situ from alkyl-N-hydroxycarbamates 225, which ends up with elimination of a nitrite ion (Scheme 55). The reaction pathway differs for unsubstituted α-nitroketones (5) and their analogues 227 substituted at the CH-acidic atom: in the first case, the reaction affords α-ketoamides 228 [see Scheme 55, reaction (1)], while substrates 227 deliver α-ketoximes 229 [see Scheme 55, reaction (2)]. The plausible mechanism of both processes include the formation of intermediates 230--234.
For nitro carbonyl compounds, a process was also described similar in conditions to the Willgerodt-Kindler reaction. Zhang et al.240 noted that thionation of nitroketones 5 with elemental sulfur in dimethylformamide in the presence of various amines 235 occurs with the displacement of a nitro group to give products 236 in good yields (Scheme 56). If ammonia is used as the amine, the products are dimethylamine derivatives of ketothioamides 236 (R1=R2=Me), since dimethylformamide 237 acts as a reactant under reaction conditions and decomposes into carbon monoxide and dimethylamine. The mechanism of this process suggests the initial thionation of the CH-acidic atom of the enol form 238 to afford adduct 239, which significantly facilitates the subsequent nucleophilic substitution of the nitro group in intermediate 240.

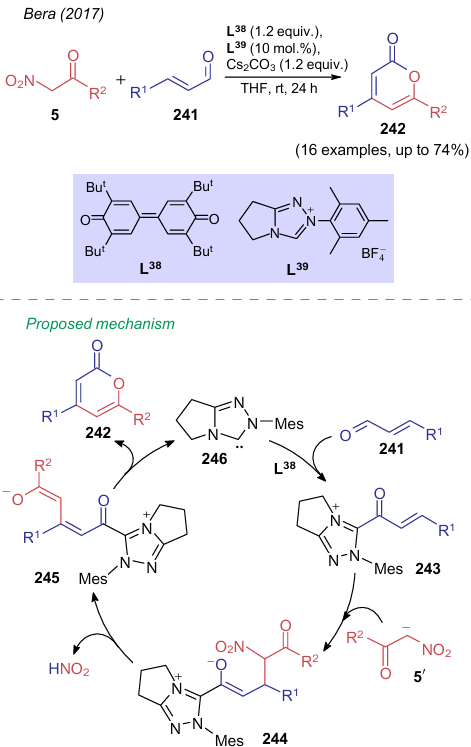
In nitrocarbonyl compounds as in alkyl nitroacetates, the nitro group often serves as a leaving group, leading to the formation of aromatic compounds. Thus, the reaction of β-arylcinnamaldehydes 241 with nitroketones 5 in the presence of oxidant L38 and triazolium salt L39 generating the carbene catalyst affords α-pyrones 242 (Scheme 57).291 According to the authors, NK 5 adds to the activated species 243 followed by the loss of nitrous acid by salt 244. Then, intermediate 245 undergoes lactonization to yield α-pyrone 242 and carbene 246, to complete the catalytic cycle.
Starting from compounds 247 prepared by the Morita-Baylis-Hillman reaction, Reddy et al.292 synthesized in 2020 a number of 2H-pyranes 248. The reaction with nitroketones 5 was carried out in THF using 3 mol.% of DABCO (Scheme 58). Based on quantum chemical calculations and isolation of intermediate 249 in the individual form, the authors proposed the mechanism of the process, in which DABCO initially adds to substrate 247, while cation 250 reacts, e.g., with the anion of nitroacetophenone 5e to form a novel C–C bond (see Scheme 58).
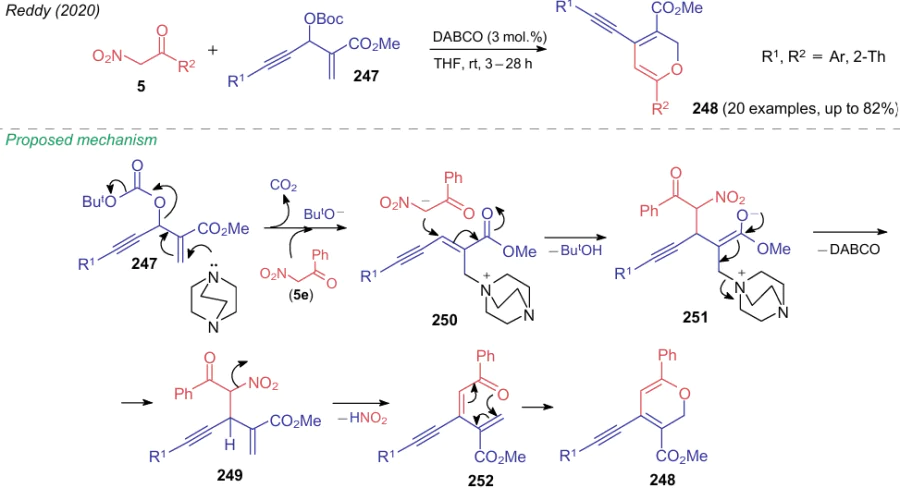
Elimination of DABCO from compound 251 gives intermediate 249, which was characterized by spectral methods. Under reaction conditions, the loss of a molecule of nitrous acid occurs, and then compound 252 cyclizes to 4-alkynylpyrane-3-carboxylate 248.
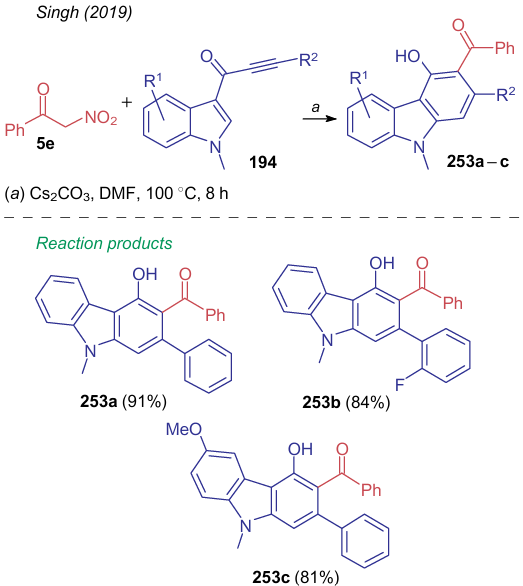
In addition to pyranes and other heterocycles, nitroacetophenone 5e can deliver compounds containing an aromatic benzene ring. For instance, heating of acylated N-methyl indole 194 with nitroketone 5e in DMF in the presence of cesium carbonate gives 4-hydroxy-9-methylcarbazol-3-ylphenylmethanones 253a-c in good yields (Scheme 59).258 The mechanism of this reaction was presented above (see Scheme 43).
Five-membered heterocycles were prepared by a procedure, which is a modification of the Nazarov reaction, denitrative imino-diaza-Nazarov cyclization (DIDAN) (Scheme 60). Aegurla et al.293 reacted nitroketones 5 with the in situ formed hydrazones 254 in the presence of iodine in ethanol to afford 3,5-disubstituted pyrazoles 255. It is assumed that the resulting enamine-imine 256 reacts with iodine to undergo, after tautomeric transformations, 4p-electrocyclization to generate diazaallyl cation 257, which is aromatized through the loss of a molecule of nitrous acid from intermediate 258.

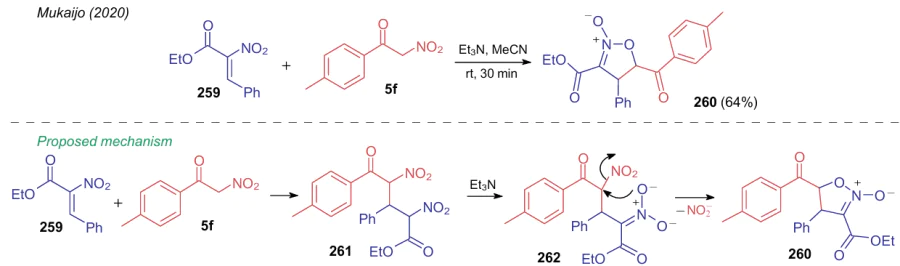
An interesting example was described by Mukaijo et al.,207 who studied the reaction of nitroketone 5f with 2-nitro-3-phenylacrylate 259 (Scheme 61). The major product of the reaction catalyzed by triethylamine is 5-(4-methylbenzoyl)-4-phenyl-3-ethoxycarbonylisoxazole-2-oxide (260), which includes a nitro group from acrylate 259. The authors note that adduct 261 formed in this reaction can cyclize in three ways, however, since the tolyl substituent lowers the nucleophilicity of the nitronate ion and increases the electrophilicity of the α-CH acidic atom compared to ethoxycarbonyl substituent, it is isoxazole 260 that is predominantly formed from intermediate 262.
3.3.6. Transformation with elimination of nitromethane
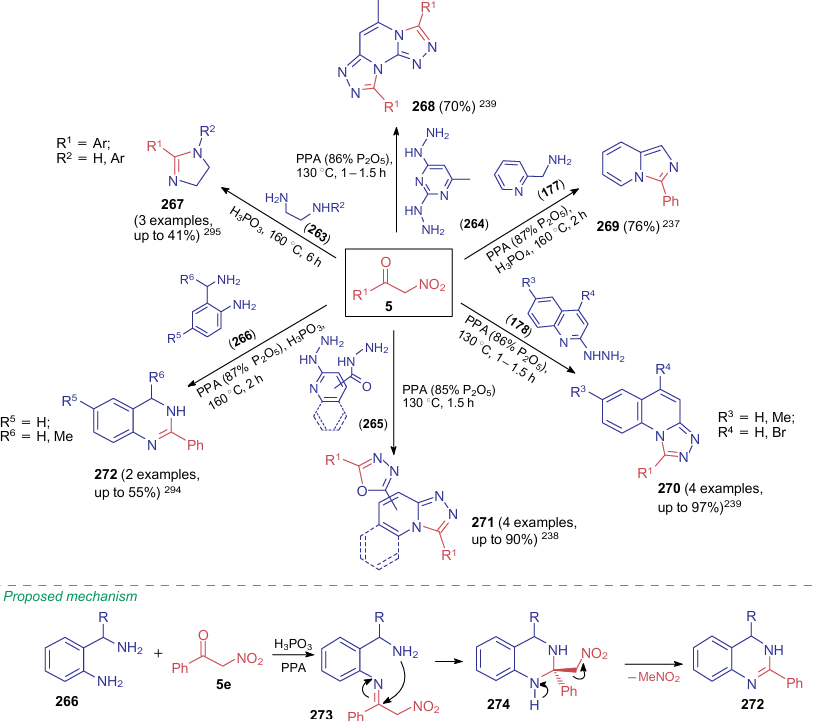
As noted above, the scientific group led by Aksenov237--239,294,295 studied the reactions of alkyl nitroacetates with various nucleophiles promoted by polyphosphoric acid (see Scheme 40). The same authors also explored the reaction of nitroketones 5 with dinucleophiles 177, 178, 263-266 under similar conditions, which delivered products 267-272 (Scheme 62). These reactions show a major difference between chemical behaviour of alkyl nitroacetates and nitroketones — in nitro-containing esters 6, aminooxyphosphoric acid acts as a leaving group, while in nitrocarbonyl compounds 5, the leaing group is a nitromethane molecule. The authors provide no explanation of eliminating just this small molecule. The mechanism suggesting the formation of intermediates 273 and 274 is exemplified by the reaction of nitroacetophenone (5e) with 2-(aminomethyl)anilines 266 in 87% PPA.
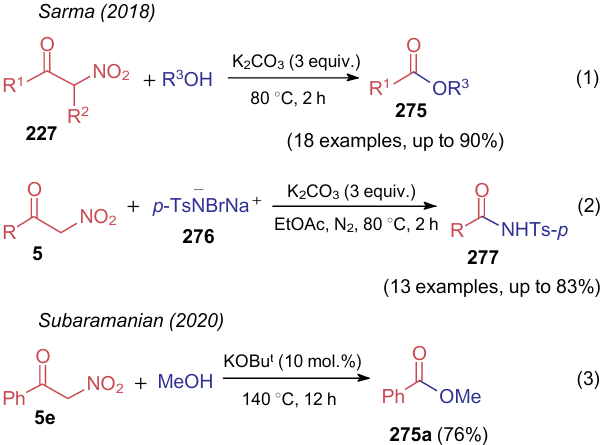
The C–C bond cleavage in nitroketones 5 and their substituted analogues 227 was also observed296,297 in their base-promoted reactions with alcohols affording esters 275 (Scheme 63). The reaction with alcohols is catalyzed with potassium carbonate (reaction a) or tert-butoxide (reaction c), while such acylation of free amines is considered impossible due to high nucleophilicity of the latter. Therefore, Sarma and Phukan296 carried out the reaction b with bromamine-T (276) to furnish tosyl amides 277.
3.3.7. Other reactions
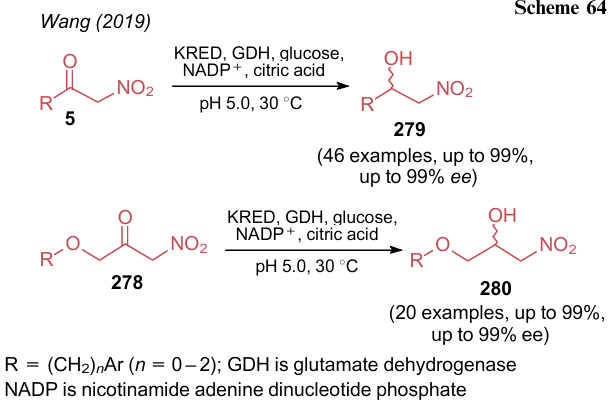
Wang et al.298 carried out a reaction characteristic of carbonyl compounds such as the reduction of a keto group in compounds 5 and 278 to the corresponding alcohols 279 and 280 (Scheme 64). The authors explored the stereoselective bioreduction of ketones with ketoreductases (KRED). At pH 5, the use of YGL039w afforded S-isomer, while in the presence of RasADH/SyADH, R-isomer was obtained. The yields of the products and enantioselectivity reached 99%.
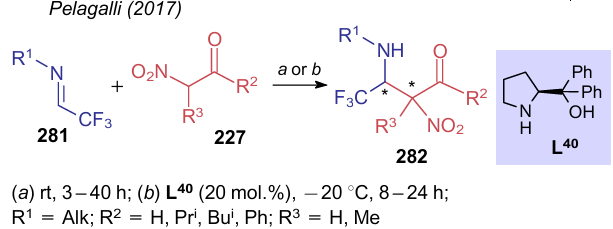
Stereoselectivity of the process was also studied299 on the example of the Mannich reaction between nitroketones 227 and trifluoromethyl aldimines 281 (Scheme 65). The authors found that structures of both substrates play a pivotal role in increasing stereoselectivity. With a certain substituent R1, it is possible to shift the direction of the reaction towards the formation of an anti-isomer 282. The use of (S)-α,α-diphenylprolinol (L40) also favours the formation of this isomer.
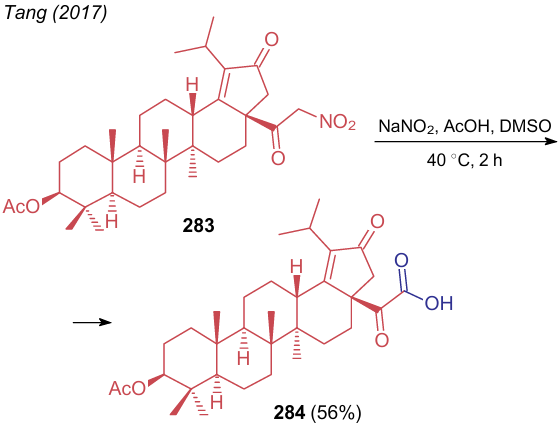
Tang et al.300 carried out the convertion of betulin derivative 283 to α-ketocarboxylic acid 284 (Scheme 66). Although this transformation resembles the Nef reaction, the intermediate compound seems to be nitrolic acid, which under the reaction conditions gives product 284 with releasing nitrous oxide.
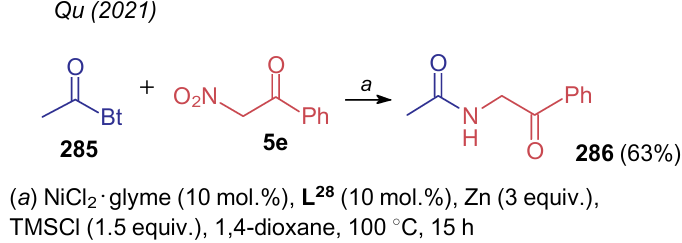
As with alkyl nitroacetates, Ni-catalyzed trans-amidation using acylated benzotriazole 285 is also possible260 for nitroacetophenone (5e) (Scheme 67). The mechanism of this process was illustrated above (see Scheme 45).
One more property combining nitroketones and alkyl nitroacetates is acid-catalyzed dimerization. Thus, Aksenov et al.211 developed a straightforward method for the synthesis of furoxan N-oxides 287 intended to elucidate the mechanism of the reaction between nitroketones 5 and polyphosphoric acid (Scheme 68). Similarly to the reaction in Scheme 46, nitrile oxide 288 acts as an intermediate.

Disubstituted furoxans 287 are not the only product that can be derived from intermediate nitrile oxides 288. Even though this intermediate has an explicit dipole structure, its modification should not necessarily lead to the formation of a ring structure. For example, in 2017, Aoyama et al.301 showed the possibility of carrying out the addition of alkyl halides 289 to intermediates 288 to furnish ketoximes 290 (Scheme 69). In this study, nitrile oxides per se were generated in situ in the presence of NaHSO4/SiO2.
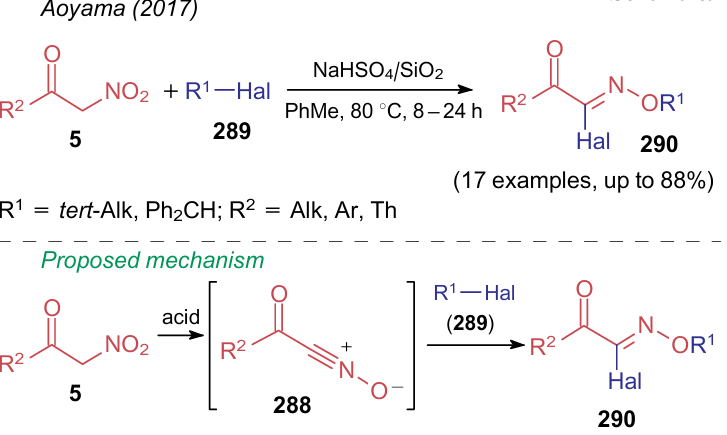
Thus, it can be noted that over the past five years, among all the considered NSEs, α-nitroketones have been studied in the most detail in terms of the diversity of their chemical properties.This is mainly due to the availability and high reactivity of these compounds. Their main difference from alkyl nitroacetates includes the reactions at the carbonyl group. Due to this, compounds 5 are characterized by reactions with nucleophiles, as well as the C-C bond cleavage reactions resulting in a wide range of cyclic and acyclic molecules.
3.4. Nitroacetonitrile
Nitroacetonitrile (7) is the least studied of the above-mentioned NSEs. The scope of its application relates primarily to energetic materials with low impact sensitivity and biologically active compounds. Despite the small number of studies related to it, two reviews31,32 have been devoted to nitroacetonitrile and its derivatives over the past few years. Moreover, methods for the synthesis of nitroacetonitrile can also be found in publications.302,303 This review discusses works on nitroacetonitrile published since 2016.
3.4.1. Cycloaddition reactions
As mentioned above, one of the main applications of compound 7 is the synthesis of nitrogen-containing high-energy heterocyclic compounds bearing a nitro group. These transformations are generally diazotization of various aminopyrazoles,304 triazoles226,305--309 and tetrazole310 followed by azo coupling with nitroacetonitrile. Scheme 70 and Table 24 illustrate the reactions described for the period under consideration. Generally, diazotization was carried out using hydrochloric or diluted sulfuric acids and sodium nitrite served as a nitrosating agent in almost all cases. Tang et al.307 employed methanolic hydrogen chloride for the diazotization of 3,5-diamino-1,2,4-triazole followed by the addition of tert-butyl nitrite. The azo coupling process first delivers hydrazone 291, which in some cases can be isolated and identified. The target azolo-1,2,4-triazines 292 can also be accessible via thermal cyclization.
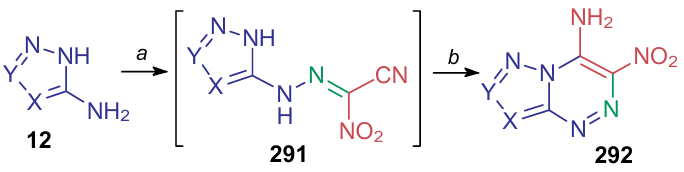

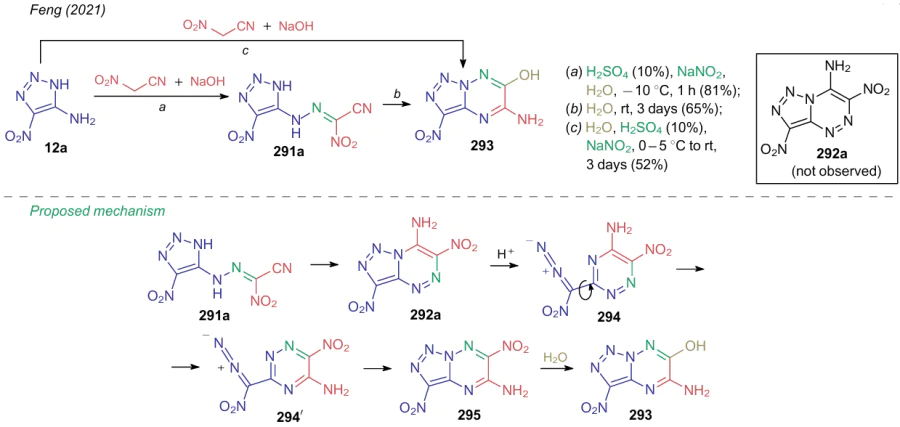
A curious example of similar azo coupling reaction was demonstrated by the Feng's research group311 in 2021. It was found that coupling of 5-amino-4-nitro-1,2,3-triazole 12a with compound 7 gives the corresponding hydrazone 291a, however, its further intramolecular cyclization furnishes 5-amino-3-nitro-1,2,3-triazolo-[1,5-b][1,2,4]triazin-6-ol (293) rather than 3,6-dinitro-1,2,3-triazolo[5,1-c][1,2,4]triazine-7-amine (292a) (Scheme 71). Nitro-containing product 292a can not be obtain either via the azo coupling-cyclization sequence or by cyclization of an intermediate hydrazone 291a in water. The authors argue that initially, cyclization of hydrazone 291a does afford nitrotriazolotriazine 292a, however, further it undergoes the Dimroth rearrangement via the triazole ring-opening to give diazo-intermediate 294, which converts to nitro compound 295, furnishing the observed product upon hydrolysis.
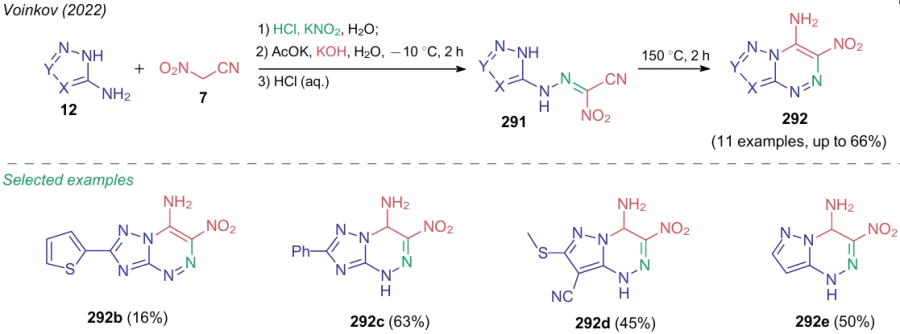
If in the above examples the resulting azolotriazines 292 were mainly considered as nitrogen-containing high-energy compounds, then by changing the azole component, Voinkov et al.312 prepared a number of heterocycles possessing antimicrobial activity. Versatile and environmentally friendly protocol has been proposed comprising isolation of hydrazones 291 and their thermal cyclization under solvent-free conditions (Scheme 72).
3.4.2. Reactions with a latent form of nitroacetonitrile
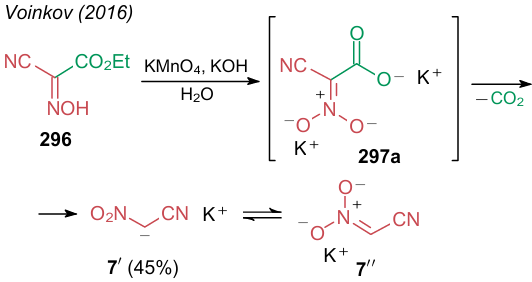
In 2016, Voinkov et al.180 developed a convenient synthetic procedure to obtain potassium salt of nitroacetonitrile (7ʹ) via oxidation of ethyl 2-hydroxyimino-2-cyanoacetate (296) with potassium permanganate in the presence of potassium hydroxide (Scheme 73). The intermediate under these conditions is the dipotassium salt of nitrocyanoacetic acid 297a, which undergoes decarboxylation to form NSE. Note the potassium salt 7ʹ is in an aci-nitro form 7ʹʹ, as confirmed by X-ray diffraction analysis.180
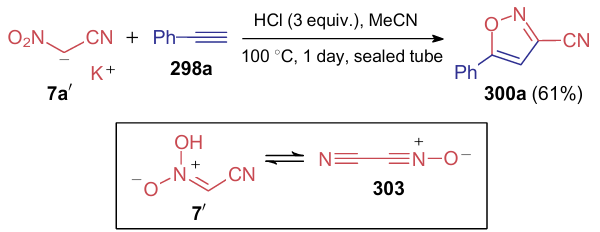
In the following publications, dianion 297 was used as a starting compound for various reactions, since the monopotassium salt 7ʹ is explosive. Nishiwaki et al.313 implemented the reaction of dipotassium salt 297a with alkynes (298) and alkenes (299) in an acetonitrile-water mixture to prepare 3-cyanoisoxazoles 300 and 3-cyano-4,5-dihydroisoxazoles 301, respectively. The process was carried out in the presence of 3 equiv. of hydrochloric acid on heating in a sealed tube (Scheme 74). The reaction with phenylacetylene (298a) is probably mediated by intermediate 302.

In the same work, the model reaction of phenyl acetylene (298a) with potassium salt 7ʹ was carried out to gain insight into the mechanism of the process (Scheme 75). Based on this reaction, the authors suggest that when treated with an acid, dipotassium salt 297a is decarboxylated to give nitroacetonitrile, which serves as a 1,3-dipole in the cycloaddition reaction with phenylacetylene. According to the authors, an excess hydrochloric acid can indicate the presence of cyanonitrile oxide 303 in the reaction mixture, and dipotassium salt 297a, like compound 7, can be considered as its masked form.
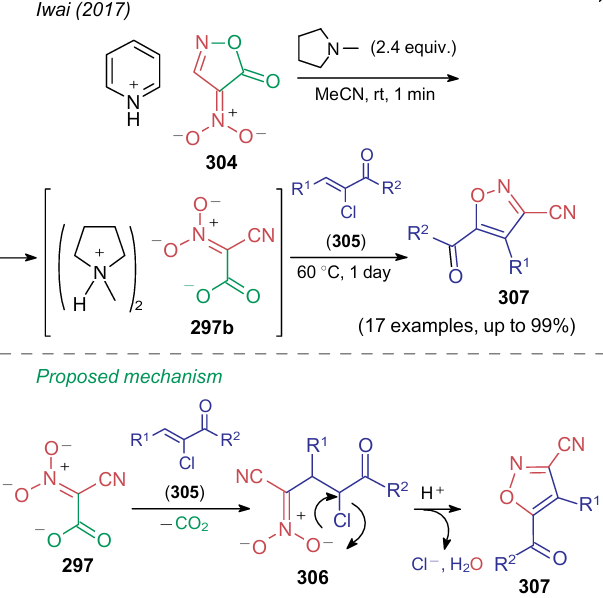
Di(N-methylpyrrolidinium) salt of nitrocyanoacetic acid 297b is an alternative to dipotassium salt 297a. Its main advantage over potassium salt 7ʹ and dipotassium salt 297a is its solubility in organic media. Thus, Iwai et al.314 reported the synthesis of dianion 297b from the salt of nitroisoxazolone 304 and its subsequent reaction with α-chloro-α, β-unsaturated ketones 305 (Scheme 76). The reaction pathway suggests decarboxylation of intermediate 297 followed by the Michael addition of substrate 305. The final stage of the process is an intramolecular cyclization of intermediate compound 306 to yield 5-acyl-3-cyanoisoxazoles 307.
It is obvious that the search for the methods to produce stable forms of nitroacetonitrile and the development of novel approaches to modify its latent forms should significantly increase the synthetic potential of this NSE, which will be reflected in an increase in the number of publications devoted to nitroacetonitrile over time.
4. Conclusion
Summing up the literature survey, it can be concluded that over the past five years, interest in the NSE chemistry has increased significantly. This is largely due to the search for new ways to functionalize such structures. For example, for nitro-containing alkenes, the functionalization of the C–H bond adjacent to the nitro group appears to be quite indicative but yet little-studied trend. For derivatives in which imine-enamine tautomerism is impossible, functionalization methods are just beginning to be developed, which, with due attention of scientists, can become very useful for the practical and theoretical study of this subject. This also includes reactions involving the cleavage of the C–C and(or) C–NO2 bonds, as well as the accompanying rearrangements. Some reactions of this type were described for the first time precisely in the period under consideration, which indicates the relevance of further study of these processes so as to obtain more accurate ideas about their capabilities. However, the bulk of the transformations associated with nitroalkenes falls on cycloaddition reactions in multicomponent and sequential fashions.This is not surprising, since the use of such NSEs offers access to a wide range of polysubstituted heterocycles, either of independent interest or intended for further modification, often due to the presence of a nitro group in the molecule.
In turn, nitroalkanes are also used mainly to construct heterocycles. On the other hand, in their case, the tendency to heterocyclization is more pronounced, resulting in the loss of nitrous acid or nitromethane molecules to form an aromatic structure. Undoubtedly, this indicates the uniqueness of such NSEs as synthetic units in the construction of complex organic structures. Accordingly, the use of the features of the reactivity of such compounds, as well as the elucidation of conditions allowing an easy change in the cyclization course, will become a relevant task in the near future. Moreover, for nitroalkanes, processes (for example, the Michael reaction) are increasingly being considered, which, depending on the type of catalyst, proceed stereoselectively or stereospecifically. Given that such NSEs are often used when creating medicinal drugs, it can be argued that the development of conditions for obtaining enantiomerically pure products using such substrates will also be a promising area of research.
The reactions discussed in this review are mostly implemented to prepare potential biologically active molecules and high-energy compounds. Of course, these are not the only fields of practical application of NSEs, but even they are enough to demonstrate the high relevance of the ongoing research. The authors of many of the cited works made a great contribution to the description of new reaction mechanisms, as well as to the addition of theoretical ideas about the course of known processes. To conclude, it should be noted that based on the discovery of new reactions involving low-molecular-weight NSEs, the chemical potential of these compounds is limited only by the imagination of synthetic chemists.
This work was funded by the Ministry of Science and the Higher Education of the Russian Federation [State contract No. FEUZ-2023-0021 (H687/42Б.325/23)].
5. List of acronyms
Following acronyms are used in the review:
Δ --- boiling point,
2,5-DTBQ --- 2,5-di-tert-butyl-p-hydroquinone,
Ad --- adamantyl,
AIBX --- 5-trimethylammonium-2-iodoxybenzoate,
ANA --- alkyl nitroacetate,
ANE --- aminonitroethylene,
BArF — tetrakis-3,5-bis(trifluoromethyl)phenyl borate,
BHT --- 2,4,6-tri-tert-butylphenol,
BINAP --- [2,2ʹ-bis(diphenylphosphino]-1,1ʹ-binaphthyl,
bmim --- 1-n-butyl-3-methylimidazolium,
Boc --- tert-butyloxycarbonyl,
CAN --- cerium(IV)-ammonium nitrate,
cod --- cycloocta-1,5-diene,
Cp --- cyclopentadiene,
Cy --- cyclohexyl,
Cy-Johnphos --- (2-biphenyl)dicyclohexylphosphine,
DAAA --- decarboxylative asymmetric allylic alkylation,
DABCO --- 1,4-diazabicyclo[2.2.2]octane,
dba --- dibenzylideneacetone,
DBU --- 1,8-diazabicyclo[5.4.0]undec-7-ene,
DCE --- 1,2-dichloroethane,
DDQ --- 2,3-dichloro-5,6-dicyano-1,4-benzoquinone,
DIPEA --- diisopropylethylamine,
DiNH --- α,α-di(amino)nitroalkene,
DiSAlk --- (α,α-di(alkylthio)nitroalkene,
DMA --- dimethylacetamide,
DMAP --- 4-(N,N-dimethylamino)pyridine,
DMBQ --- 2,6-dimethoxybenzoquinone,
DMS --- dimethyl sulfide,
DPEPhos --- bis[(2-diphenylphosphino)phenyl] ether,
de --- diastereomeric excess,
dr --- diastereomeric ratio,
er --- enantiomeric ratio,
EWG --- electron withdrawing group,
Fu --- furyl,
GDH — glutamate dehydrogenase,
KRED --- keto reductases,
MCR --- multicomponent reaction,
Mes --- 2,4,6-trimethylphenyl (mesityl),
MS --- molecular sieves,
MW --- microwave irradiation,
Naph --- naphthyl,
NADP — nicotinamide adenine dinucleotide phosphate,
NAN --- nitroacetonitrile,
NK --- α-nitroketone,
NSE --- nitro synthetic equivalents,
PEG --- polyethylene glycol,
PEGMA-g-TEGBDIM --- polyethylene glycol methacrylate-grafted tetraethylene glycol-bridged dicationic imidazolium based ionic liquid,
PG --- protecting group,
PPA --- polyphosphoric acid,
PS-BEMP --- 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine supported on polystyrene,
Py --- pyridyl,
p-Ts --- p-toluenesulfonyl (tosyl),
rt --- room temperature,
rr --- regioisomeric ratio,
SMeNHR --- (alkylthio)aminoalkene,
TBAB --- tetra-n-butylammonium bromide,
TBAI --- tetra-n-butylammonium iodide,
TBDPS --- tert-butyldiphenylsilyl,
TBS --- tert-butyldimethylsilyl,
TEMPO --- 2,2,6,6-tetramethylpiperidin-1-yl)oxyl,
TFA --- trifluoroacetic acid,
Tf --- trifluoromethanesulfonyl (triflyl),
TFE --- trifluoroethanol,
Th --- thienyl,
TMS --- trimethylsilyl,
US --- ultrasonication.