


Keywords
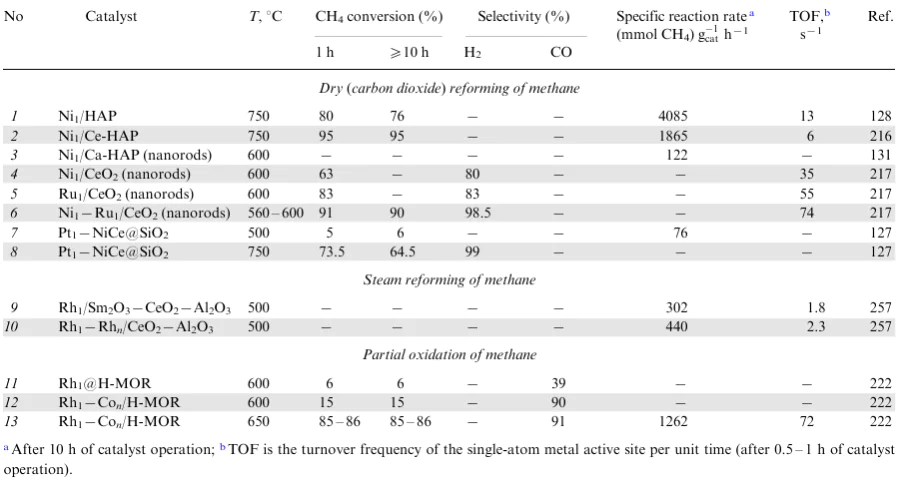
Abstract
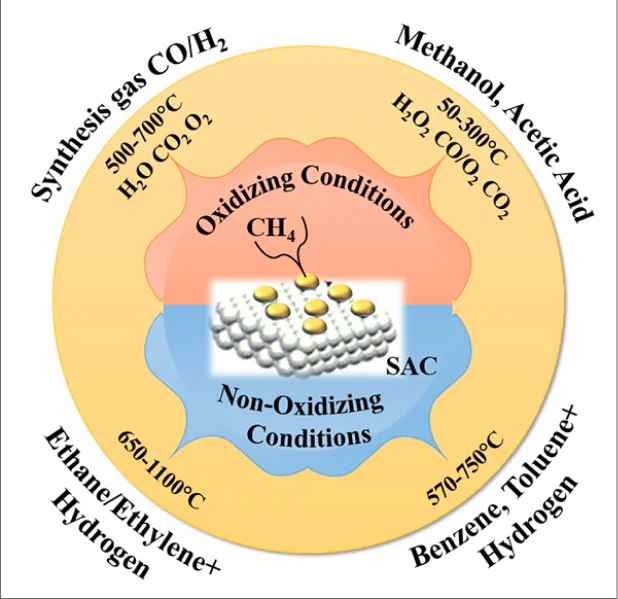
The review integrates and systematizes literature data on the use of single-atom catalysts in methane chemistry, with the emphasis on the most recent results. The single-atom catalysts are heterogeneous catalysts of the latest generation in which single metal atoms supported on an inorganic material act as the active sites. The features of CH4 activation on the surface of these catalysts are considered and compared with the behaviour of other known heterogeneous catalysts containing metal nanoclusters or active metal nanoparticles in relation to the direct oxidative and non-oxidative conversion of methane into various chemicals. The efficiency of application of single-atom catalysts of various compositions and types (supported single metal atoms, mono- and polymetallic monoatomic contacts, complex disperse compositions, etc.) is considered for reactions involving methane, including dry, steam and oxidative reforming, partial oxidation to methanol, oxidative carbonylation and carboxylation to acetic acid, non-oxidative and oxidative methane coupling to ethane/ethylene, methane dehydroaromatization, and methylation of benzene with methane. The new prospects opened up in methane chemistry by using single-atom catalysts are discussed.
The bibliography includes 307 references.
1. Introduction
Methane is the major component of natural gas and the most environmentally safe carbon resource for the production of high value-added chemical products.1--7 Catalytic1,2,8--14 and non-catalytic15--22 methods for the conversion of this simplest hydrocarbon to basic products (hydrogen, ammonia, synthesis gas, ethylene, acetylene, carbon black and nanofibrous carbon, methanol, motor fuel, etc.) have been practiced for several decades; as a result, gas chemistry is now a leading branch of world economy with numerous large-scale plants (with a total capacity of >1 million tons per year) and a total production output of various chemicals of ≈500 million tons per year.16
In recent years, the greatest attention of researchers has been directed towards development of processes for methane conversion to organic compounds that were previously commercially manufactured by petrochemical industry (oxygenates, olefins, aromatic hydrocarbons, polymers based on them, etc.).3--7,11,12,23--36 There are two principal pathways for the conversion of gas feedstock to these products: indirect and direct conversion.7,15,16,23--38 In the case of indirect conversion, CH4 is converted to synthesis gas (CO and H2 mixture), which is used for the industrial synthesis of oxygenates (methanol, dimethyl ether, acetic acid, methyl acetate) and other products.18--20,29,39,40 In the case of direct methane conversion under oxidative or non-oxidative conditions, these valuable chemicals are obtained in one step.4,6,21,36,37,41--46
In view of the difficulty of activation of the thermally stable CH4 molecule, until recently, the chemical reactions of methane were carried out only at high temperatures.1,2,23,24 However, the high-temperature processes are energy demanding and expensive, and in the case of catalytic processes that enable conversion of methane under milder conditions, the use of harsh reaction conditions leads to a low stability of the catalyst structure and active sites and hampers selective preparation of target products (due to the higher reactivity of any other organic compound compared to CH4).2,3,6
A variety of strategies have been attempted for decreasing the temperature of reactions involving methane.47 Some research teams are developing methods for the conversion of methane under mild conditions in the presence of homogeneous catalytic systems.31,48,49 However, the primary attention is devoted to the design of low-temperature (T=25-300 °C) selective heterogeneous catalytic processes, which are most promising for the subsequent industrial implementation.50 By analogy with known homogeneous catalysts, so-called quasi-homogeneous heterogeneous catalytic systems are developed.51 Procedures of doping of traditional oxide catalysts have been used;52,53 transition metal alloys54,55 and proton-conducting oxide membranes with the perovskite structure 56,57 have been proposed as catalysts. The most pronounced effect was attained on going to nano-sized catalytic systems.23,58--60 Meanwhile, special expectations of chemists are associated with the use of single-atom catalysts (SACs), which are able to activate methane even at room temperature.61--64
Single-atom catalysts are new-generation heterogeneous catalytic systems with monoatomic dispersion of metal active sites.63,65,66 Unlike highly dispersed nano-sized systems (nanoparticles, nanoclusters), these systems have no metal--metal bonds on the surface. They are single metal sites (usually cations) that are chemically isolated and supported on an inorganic substrate.67--70 All isolated metal atoms occur in direct chemical contact with the surface, which provides 100% accessibility for the reactants in chemical reactions. This configuration makes these heterogeneous catalysts somewhat similar to homogeneous catalysts71,72 and determines their unique properties: for most chemical reactions, SACs are effective under mild conditions and surpass in the activity, selectivity and stability other known heterogeneous catalytic systems, including those containing nanoparticles and nanoclusters of active metals.73--78
Single-atom catalysts differ from other catalytic systems in the electronic structure of the active sites.71 It is not a single metal atom that is considered to be the SAC active site, but an active region centred by a single metal atom and including neighbouring atoms and/or vacancies.63 These heterogeneous systems are characterized by strong interaction between the single metal site and chemical elements of the support (O, N, P, S etc.).79 By varying the chemical composition of the support, it is possible to affect the electronic state of the active metal and to tune its catalytic properties.79--81 The efficiency of a single-atom site is increased by using modification with organic ligands or doping with metals,73,75,81--84 variation of the functional groups of the support,66,79 and, in some cases, special formation of complex dispersion systems that include, apart from isolated atoms, also nano or atomic clusters, or nanoparticles of active metals.73,81,82
Quite a number of single-atom catalysts have now been prepared; they contain a broad range of noble and some non-noble transition metal atoms79,85,86 dispersed on supports of various chemical compositions and various natures.82 The supports used for the formation of single metal active sites include metal oxides and sulfides,87--90 metal and non-metal nitrides,91--94 graphene (G) and graphite and their derivatives,74,76,95,96 porous structured materials like zeolites and metal-organic frameworks.69,97,98 There are structurally diverse mono- and multimetallic heterogeneous contacts: catalysts with metal sites located on the support surface or embedded into the support crystalline framework79,85,99,100 or core@shell capsule catalysts.74,93--96 Nanostructured 2D systems (nanosheets, nanorods, etc.) have been obtained.99,101,102 New types of SACs such as surface organometallic compounds73,103,104 and single-atom metal alloys105--107 have been synthesized.
Each type of single-atom heterogeneous catalysts has its own benefits. For example zeolites or metal-organic frameworks are used to obtain the most uniform catalytic structures.97,98 Nanostructured 2D systems are characterized by the maximum exposure of active sites and, hence, the highest production rate.101 Single-atom alloys are distinguished by resistance to coking.99,107 Nano-heterogeneous systems of a complex size composition, in which synergistic effect occurs between single metal sites and other active species (atomic clusters or nanoparticles), exhibit unique catalytic properties in complex processes.65,76,81,108--110
The rapid development of single-atom catalysis started in 2013--2014111 when new-generation high-resolution instrumental equipment was implemented in the research practice. It became possible to determine the catalyst fine structure at the atomic level using high-resolution transmission electron microscopy (HRTEM) and X-ray absorption spectroscopy (XAS) as extended X-ray absorption fine structure (EXAFS) or X-ray absorption near edge structure (XANES) techniques.57,66,111 Particular research attention was attracted by the potential application of various types of SACs in redox reactions;104 these catalysts have already showed high performance and high energy efficiency in a series of thermo-, electro- and photocatalytic processes112--119 used in the production of organic compounds, energy engineering, environmental protection and biomedicine.104,120--122 However, the most impressive results were attained by the use of SACs in the chemistry of small molecules (CO, H2, N2, CO2, CH4),65,72,78,84,123,124 especially for the activation of strong chemical bonds (N≡N, O=C=O),79,96,98,110 including the C–H bonds of the thermally stable methane molecule.72,78,84
The studies dealing with SACs are rapidly developing, which is confirmed, in particular, by the apprarance of numerous reviews (>40);63,65--73,77--79,81--91,97--107,120--126 however, there are almost no studies specially devoted to the methane chemistry. There are only two review publications in international journals 125,126 that summarize the results of studies of methane oxidation in the presence of single-atom catalysts and there are also particular chapters on similar topics in several more general reviews.63,72,78,103 Recent publications report the successful use of single-atom heterogeneous catalysts in other reactions in which methane is selectively converted to the target C1 or C2+ products: dry, steam and oxidative reforming,127--131 methylation,132--135 oxidative carbonylation,136--138 carboxylation139--143 and oxidative144 and non-oxidative145--147 coupling. New publications on the application of SACs in methane chemistry continuously appear,64,147--150 but this rather extensive information has not yet been summarized.
This review analyzes, integrates and arranges the latest achievements in the application of SACs in the direct conversion of CH4 to valuable chemical products (synthesis gas, C1–C2 oxygenates, C2+ hydrocarbons) both under oxidative and under non-oxidative conditions. New prospects for the catalytic transformations of methane in the presence of SACs caused by the specific features of CH4 activation on single metal sites are demonstrated.
2. Features of methane activation by single-atom catalysts
The stable non-polar methane molecule is shaped like a regular tetrahedron and has a low polarizability and exceptionally high C-H bond dissociation energy (≈439 kJ mol–1). In most non-oxidative and oxidative methane conversion reactions, dissociation of the first C–H bond in the CH4 molecule is the rate-limiting step.32
Today, while developing low-temperature catalysts for reactions involving methane, chemists rely on the results of quantum chemical calculations,151--153 which predict a markedly lower energy barrier for heterolytic dissociation of the methane C–H bond, i.e., via polarization (CH4→:CH3δ-+Hδ+), than for the homolytic dissociation to give radicals (CH4→ •CH3+ •H). Therefore, preference is given to catalysts with charged active sites, which ensure the required polarization of the C–H bond.
The polarization of the CH4 molecule is attained on solid catalytic surfaces containing Lewis pairs (acid and base), e.g., on the Al2O3,154,155 ZnO@Au,156 boron nitride157 and other surfaces. Heterolytic cleavage of the C–H bond is provided upon modification of zeolites or metal-organic frameworks with transition metal cations or oxide clusters.158--164 Compounds considered to be promising for the heterolytic activation of methane include transition metal carbides and cationic boride clusters59,165--168 and charged vanadium, manganese and rhenium oxide clusters.169--171 Catalytic systems containing cationic heteronuclear oxide clusters ([Al2CuO5]+, [TiAlO4]+, [TiSiO2]+) are also used.130,131,172 The best results were achieved by using catalytic systems based on charged atomic clusters and ions (cations or anions) of noble and some non-noble transition metals: in the presence of these compounds, dissociation of methane molecule is possible even at room temperature.59,61,149,173--184
Using simplified models for transition metal cluster systems (Mn) in gaseous plasma, a number of researchers176,177,182,185--200 demonstrated that the activity towards dehydrogenation of the methane molecule
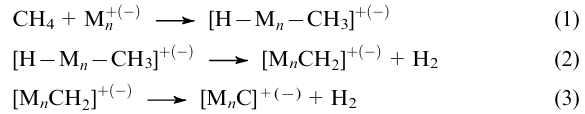
is inherent only in charged Mn+(-) clusters of moderate nuclearity (n=1--4). When n >2, the target process is, most often, accompanied by oligomerization of the [MnCH2]+(–) groups
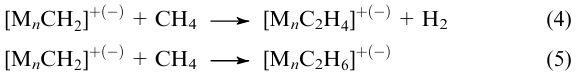
When n=1, the process can be retarded after elimination of only one hydrogen atom from the methane molecule to give relatively stable methyl hydride complex [reaction (1)].187,189,191 However, when a charged cluster with a complex chemical composition (e.g., [Rh1Al2O4]–, [Rh1TiO2]–)183,184 contains a transition metal ion M1+(-), the C–H bond in the methane molecule is cleaved via an alternative pathway, in which the methyl radical is attached to the noble metal atom site and the primary hydrogen atom is fixed on the heteroatom, e.g., as shown below184,201

The activity of these complex systems is several orders of magnitude higher than that of gas-phase transition metal ion.176,183,184
The model heteroatomic systems containing one active transition metal atom resemble real SACs in which the single-atom metal site (usually a cation) is supported on an inorganic material.32,144,202 The electrophilic chemical elements (O, N, S, etc.) present on the support surface and bound directly to the active metal cation can participate in the activation of the primary C–H bond of methane by trapping the eliminated hydrogen atom. The hydrogen chemisorption on the polar sites of the support facilitates the C-H bond polarizability and increases the probability of its heterolytic cleavage with a reduced energy barrier. As a result, the temperature of methane activation decreases, and the methyl σ-complex [CH3–M1]+ involving the active metal cation is formed.203 According to the energy profile of methane dehydrogenation (Fig. 1a), this complex has the lowest energy.144 This attests to the high stability of this intermediate, which rules out the subsequent dehydrogenation steps. The formation of a stable methyl complex was detected by spectral methods for various SACs [Rh1/ZrO2,144 Rh1/CeO2,202 Pd1/CeO2,203 H-Ta1/SiO2 (Ref. 204)] and the stability of such systems was confirmed by numerous quantum chemical calculations.144,178,179,202,203
![[{"id":"npINejxSdW","type":"paragraph","data":{"text":" Energy profile of methane dehydrogenation over the Rh<sub>1</sub><sup>3.6+</sup>/ZrO<sub>2</sub> single-atom catalyst (<i>a</i>) and CH<sub>4</sub> conversion pathways (<i>b</i>).<sup>144</sup> The asterisk refers to the excited state. Published with permission from the American Chemical Society (ACS)"}}]](/storage/images/resized/g14r8c998uoechw9QR4rv3KZUo72Ju7lUr7MWCQN_xl.webp)
Stabilization of the [CH3–M1]+ methyl intermediate is a key feature of CH4 activation by SACs, which determines their unique selectivity and the possibility of selective synthesis of organic products containing a methyl group (CH3OH, C2H6, CH3COOH, etc.) (Fig. 1b). This feature accounts for the differences between the SAC behaviour in the methane chemistry and the behaviour of low-nuclearity atomic metal clusters, which form the basis of the modern nano-sized heterogeneous systems. Note that the latter catalysts show high activity towards the stepwise dissociation of the CH4 molecule, which may result in complete dehydrogenation or be accompanied by oligomerization of the CH2 or CH groups. These properties are valuable for catalysts meant for methane reforming processes (to give CO+H2) or for CH4 coupling processes giving a C2+ carbon skeleton. Catalytic systems with a complex size composition containing simultaneously metal nanoclusters and single-atom metal sites may be of particular interest.
In the design of SACs, the choice of the support for the active site is an important factor, because adjustment of the strength of interaction between the metal cation and the support elements is a crucial aspect of tuning of the electronic state of the active site, which would influence its efficiency in the low-temperature methane conversion. For example, in the case of oxygen-containing supports (metal oxides, zeolites), the energy barrier for methane activation decreases with increasing negative charge on the substrate oxygen atom.151 A similar situation was observed for the graphene support doped with nitrogen atoms.205,206 When zeolites are used as supports, the energy barrier for methane activation can be further reduced (by more than 50%) due to the synergistic effect caused by the presence of active metal ions in the catalyst and Brуnsted acid sites in the zeolite framework.158,207
Over the last ≈5 years, >40 SACs based on noble (Rh, Pt, Pd) and some non-noble transition (Fe, Cu, Ni, Co, Zn, Cr, Ta, etc.) metal cations with various types of supports have been proposed for low-temperature methane activation (Table 1). The supports used for this purpose are mainly metal oxides with nanoparticle morphology (e.g., nanowires, nanorods and so on) or zeolite matrices (usually in the H-form) with different crystal structures: MFI, CHA, MOR or BEA (items 2--23 in Table 1). Single active metal atoms can also be supported on monolayer single crystals (items 25, 26, 39), fused with another metal (items 27--30) or embedded into the graphene matrix (items 31--36). Single-atom catalysts of unusual types have been designed. For example, there are single-atom alloys encapsulated into a silicate lattice or supported on metal oxide nanoparticles (items 29, 30). A more intricate system has also been proposed, that is, graphene matrix with attached single rhodium atoms, which are bound by strong covalent bonds to nickel single crystals (item 39). Molybdenum sulfide and porous carbon nitride, including its oligomeric polymorphs (items 37, 38, 40) are also considered as promising supports. Hexagonal boron nitride sheets possessing high thermal conductivity (item 41) are considered to hold good promise as a support material for single-atom catalysts.
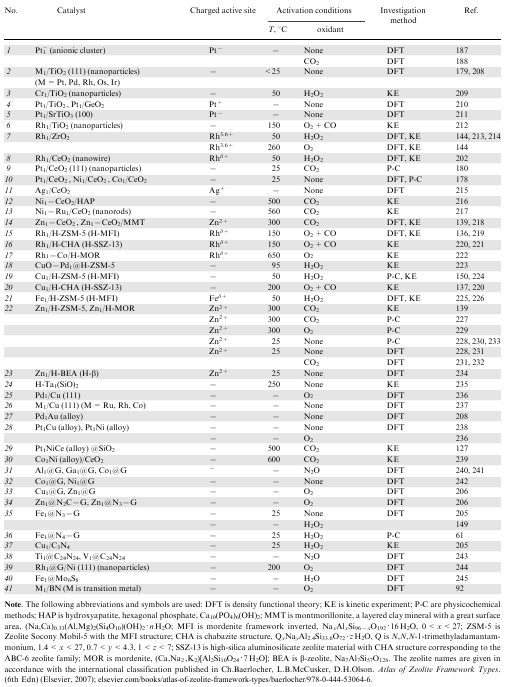
The efficiency in the activation of methane molecules for most of the proposed SACs has so far been investigated by theoretical computer simulation methods (mainly DFT). Some of these catalysts have already been tested in kinetic experiments in various oxidative and non-oxidative methane conversion reactions.
3. Reforming of methane into synthesis gas
Methane reforming is one of the few modern large-scale industrial processes for the oxidative processing of natural gas for the formation of synthesis gas.39,246--248 This gas mixture is used as a source of hydrogen for the production of ammonia and mineral nitrogen fertilizers247 and as an intermediate in large-scale industrial processes such as synthesis of methanol,249,250 acetic acid,24 formaldehyde, ethers,251 lower olefins 23 and synthetic fuel.252
The existing commercial processes of methane conversion to synthesis gas involving the use of oxidants (H2O, O2, CO2) at high temperatures (800--950 °C) catalyzed by heterogeneous nickel-containing oxide cermets253--255 are characterized by high energy consumption and fast catalyst deactivation, mainly as a result of carbon deposition on the catalyst surface.246,256
The hope for efficient elimination of the drawbacks of the industrial methane reforming processes is placed, in particular, on the replacement of the traditional heterogeneous bulk catalysts by single-atom catalytic systems:68,216 owing to the low-energy heterolytic cleavage of the primary C–H bond upon CH4 activation on SAC active sites, it is possible to decrease the reaction temperature, while stabilization of the methyl intermediate prevents methane pyrolysis and oligomerization, which are the major causes of coking and carbonization of the catalytic surface.
According to the results of quantum chemical calculations,68,216,217 the CH4 conversion to CO+H2 over SACs consists of a sequence of reactions of the methyl intermediate including oxidative dehydrogenation steps

This process is chemically complex and requires catalysts of definite chemical and phase compositions.
In a number of studies,181,183,184 mass spectrometry and photoelectron spectroscopy were used to study the behaviour of rhodium-containing anionic species of two types, those doped with one noble metal atom ([Rh1Al2O4]–, [Rh1TiO2]–)183,184 and those consisting of three rhodium atoms (Rh3-),181 in the methane reactions with oxidants (O2, CO2) proceeding under ideal thermal collision conditions (at temperatures close to room temperature). In relation to these models it was shown that single-atom systems can promote and low-nuclearity atomic clusters can catalyze the conversion of methane to synthesis gas under thermal conditions. In view of this, researchers attempt to use SACs in methane reforming both separately and as parts of complex dispersed catalytic systems containing single atoms of the active metal together with low-nuclearity nanoclusters (Table 2).
Most of the known single-atom heterogeneous catalysts for methane reforming are based on nickel or noble metals (Ru, Pt, Rh) supported on various materials.127--131,216,217,222,257 Using wet impregnation131,222,257 or strong electrostatic adsorption,128,216 the single-atom sites of active metals are introduced, in the form of cations, into zeolite matrices (MOR, BEA, MFI),222 cerium oxide217 or hydroxyapatite lattice.128,131,216 Atomically dispersed systems with strong bonds between the metal and the support can also be obtained by combustion of nitrogen-containing metal precursors mixed with the oxide support (lanthanum oxide). Apart from active metals, doping components such as calcium oxide,131 cobalt oxide222 or lanthanide oxides (CeO2, Sm2O3) are added to the catalysts.216,257 In all cases, during the catalyst formation, chemical elements of the support or the doping promoter [Brуnsted acid sites (BAS), Ca2+, Ce4+ cations and so on] are partially replaced by cations of the active metal (Mδ+). This gives rise to a new interfacial structure (such as Mδ+–O–Al,222 Mδ+–O–P,128 Mδ+–O–Ce216,217), which provides stability of the single-atom site capable of activating methane by the low-energy-consuming heterolytic mechanism, in which the hydrogen atom eliminated upon dissociation of the methane C–H bond can be chemisorbed on an oxygen atom of the support.
Apart from the conventional supported SACs, more complex heterogeneous catalysts have also been proposed for methane reforming, particularly, polymetallic single-atom systems (Ni1–Ru1/CeO2,217 Rh1–Co/H-MOR 222), single-atom alloys (Pt1-NiCe),127 encapsulated systems (Pt1-NiCe@SiO2),127 and systems of a mixed size composition, including simultaneously single-atom metal sites and metal nanoclusters (Rh1–Rhn/CeO2–Al2O3,257 Rh1–Con/H-MOR 222).
The efficiency of a particular catalyst design depends, first of all, on the oxidant (CO2, H2O or O2) used for CH4 conversion (see Table 2). However, in all cases, in the design of SACs for methane reforming, chemists have followed the strategy according to which CH4 and the oxidant are activated on different sites.
Single-atom catalysts for dry reforming (CO2 as the oxidant) are often bimetallic systems that comprise a single-atom metal site (Ni, Pt, Ru) where the CH4 molecules are activated and a basic site meant for the adsorption and dissociation of CO2 molecules (cerium oxide,217 calcium oxide,131 etc.). The synergistic effect caused by the presence of two sites for reactant activation is most pronounced in the case of the polymetallic catalyst with two heterometallic single-atom sites (nickel and ruthenium) that are directly bound to each other and are fixed on cerium oxide nanorods (Ni1–Ru1/CeO2).217 According to Tang et al.,217 the primary C–H bond of the CH4 molecule dissociates on the nickel site, and the resulting H atoms migrate to ruthenium (hydrogen spillover) where they are converted to molecular hydrogen. The CO2 molecule is adsorbed on the ruthenium atom, and its dissociation takes place with participation of cerium oxide defects
Thus, all components of the Ni1–Ru1/CeO2 catalytic composition, including the support, are involved in the reactant activation and CO and H2 formation.
A catalytic system effective for steam reforming of methane (with H2O as the oxidant) has a mixed size composition, including single-atom Rh1 sites and Rhn nanoclusters supported on cerium oxide-doped alumina (Rh1–Rhn/CeO2-Al2O3).257 In the opinion of Duarte et al.,257 methane activation takes place on rhodium single-atom sites, but they are inert towards water molecules; the H2O activation and CO formation occur on rhodium nanoclusters. The activation of water molecules involves both Rhn nanoclusters and CeO2-x promoter: the oxygen atoms formed on the rhodium sites migrate to CeO2-x vacancies via spillover. Thus, methane conversion to synthesis gas upon the reaction with water is provided by the synergistic effect of three components of the complex catalytic system: Rh1, Rhn and CeO2-x.
Different-size catalysts with a single-atom site and a metal nanocluster formed by different metals are also used for the partial oxidation of methane to synthesis gas (oxidative reforming, O2 as the oxidant). For example, the Rh1–Con/H-MOR bimetallic system has been proposed;222 in the authors opinion, methane molecules are activated on the single-atom rhodium site, while oxygen molecules are mainly activated on the cobalt nanoclusters. Cobalt is oxidized during the process, but is rapidly reduced to Co0 under the action of hydrogen, which results from methane dissociation on Rh1 site and migrates towards cobalt via spillover. The metallic components of this catalytic system were fixed in the zeolite matrix, which is not involved in the reforming of methane, but maintains the highly dispersed state of the active metal sites.257,258
The catalytic properties of single-atom systems towards methane reforming have been compared with the properties of heterogeneous catalysts of the same composition, but fully consisting of metal nanoclusters.127,128,216,257 The following pairs of systems were tested in the dry reforming of methane: Ni1/Ce-HAP and Nin/Ce-HAP,216 Pt1–NiCe@SiO2 and Ptn–NiCe@SiO2 127 and Ni1/HAP and Nin/HAP;128 also, Rh1/Al2O3 and Rhn/Al2O3 were tested in the steam reforming.257 According to the results, at any type of reforming, the starting activity was 2.5-4 times higher for single-atom systems than for nanoclusters, all other factors being equal. The catalytic properties of single-atom systems were virtually invariable for at least 50-100 h,216,222 whereas nanoclusters were rapidly coked and deactivated in the first 10-15 h.256 The above cited authors obtained experimental data, demonstrating advantages of single-atom catalysts over nanocluster systems such as higher starting activity, the possibility of reducing the reaction temperature down to 500-750 °C, resistance to coke formation and increase in the time of continuous operation of the catalyst before regeneration.
Apart from the single-atom metal active sites, the surface of SACs may carry metal nanoclusters of different size and nanoparticles (Fig. 2a). Akri et al.,216 who investigated dry reforming of methane on heterogeneous Ni1/Ce-HAP and Nin/Ce-HAP catalysts, showed that a heterogeneous catalyst composed of nanoclusters and a large amount of nanoparticles (10% Nin/Ce-HAP) is rapidly deactivated during the reforming, but the presence of a small amount of nanoclusters in the single-atom catalyst has a beneficial effect on the catalyst performance [activity and resistance to coking, cf. 0.5% Ni1/Ce-HAP and 2% (Ni1–Nin)/Ce-HAP in Fig. 2b,c].
![[{"id":"fpDQSecIOh","type":"paragraph","data":{"text":"HRTEM images of the 2% (Ni<sub>1</sub>-Ni<sub>n</sub>)/Ce-HAP sample (the circles mark single-atom sites and the squares mark nanoclusters) and distribution diagram of nickel-containing particles of various sizes on its surface (<i>a</i>); distribution of nickel single-atom sites, nanoclusters and nanoparticles on the surface of Ni/Ce-HAP catalyst with Ni content of 0.5, 2 and 10 mass% Ni (<i>b</i>) and CO<sub>2</sub> conversion in methane dry reforming over these catalysts (reaction conditions: T=750 °C, CH<sub>4</sub>:CO<sub>2</sub>:He=10:10:30, gas flow rate 50 mL min<sup>-1</sup> (60000 mL g<sub>cat</sub><sup>-1</sup> h<sup>-1</sup>) (<i>c</i>).<sup>216</sup> Published under the Creative Commons Attribution 4.0 International License"}}]](/storage/images/resized/pnzmZf7rQRv8FQKjnNiaV0IKVahJwM86Q5gMEn3w_xl.webp)
A number of highly active and stable catalytic compositions with single-atom metal sites suitable for the industrial implementation have been proposed to date. For example, the Rh1–Con/H-MOR system was developed for partial oxidation of methane to synthesis gas (H2:CO=2 molar ratio) at 650 °C,222 while the Ni1–Ru1/CeO2 system was designed for dry reforming at temperatures of 560--600 °C;217 in particular, it was tested in fuel cells at 500 °C.259 In the presence of these catalytic compositions, reforming proceeds with a selectivity of 90-99% (to CO and H2), with methane conversion being 85-90%. On each catalytic active site, 72--74 H2 molecules per second are formed without noticeable coke formation.
The studies on the development of SACs for methane reforming are still few in number, but the obtained results already attest to high potential of these innovative catalytic systems in industry with the possibility of reducing the reaction temperature by 200-250 °C and considerable increase in the operating time of the catalyst.
4. Direct oxidative conversion of methane into oxygenates
The prospects of direct one-step conversion of methane to oxygenates has long attracted the attention of a broad range of researchers3,4,25,48,260,261 as a possibility of decreasing the number of stages in the advanced industrial processing of natural gas. In recent years, the probability of practical solution of this problem has markedly increased owing to the targeted development of SACs for these reactions.
In view of the ability of these catalysts to stabilize the methyl intermediate upon the methane activation, chemists make attempts to prepare effective single-atom heterogeneous catalysts for the direct oxidative conversion of methane to organic compounds containing a CH3 group, that is, methanol and acetic acid, which are large-scale industrial products.125,126,138,262,263
Examples of SACs experimentally studied by various research groups in the direct methane oxidation to oxygenates with various oxidants such as hydrogen peroxide,223,262 oxygen and carbon monoxide mixture 138,212,263 and carbon dioxide139 are summarized in Table 3. The Table includes mono- and polymetallic heterogeneous catalysts with single-atom sites of noble metals, Rh,136,144,202,263 Pd,138,223 Ir,138 and systems based on some non-noble metals: Fe,61,64,148,226 Cu,137,150,264 Cr,209 Zn,139 Ni.262 The supports used in these systems are metal oxides [TiO2,190 ZrO2,144 CeO2 (Ref. 202)], zeolites (H-ZSM-5,64,150,223 H-MOR148 or H-SSZ-13137) and carbon materials usually functionalized with nitrogen (nitrogen-containing graphene149 and carbon nanotubes,262 porous carbon nitride264).
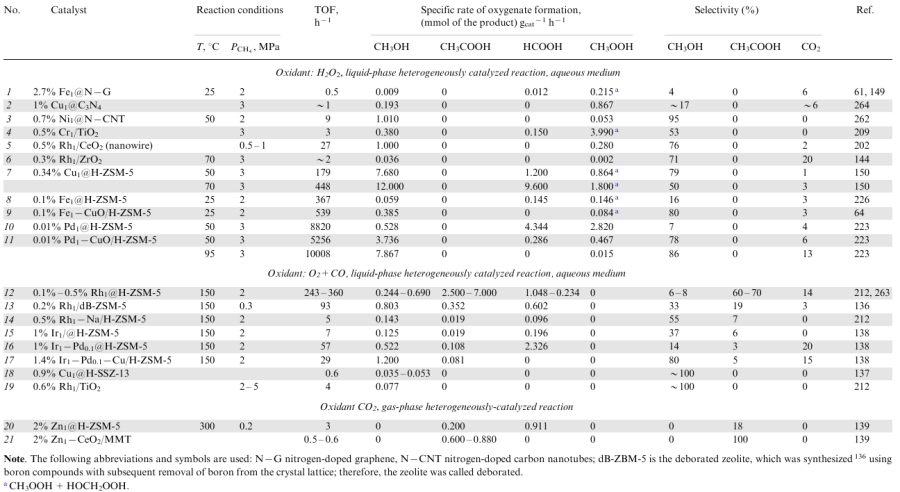
It can be seen from Table 3 that the oxidative conversion of methane in the presence of SACs is a low-temperature process selective to CH3-containing oxygenates, the proportion of which in the product reaches 70-100%. In the methane oxidation with hydrogen peroxide, high selectivity to methanol is attained in some cases (see Table 3, items 1--11); the reaction of CH4 with CO2 gives acetic acid (item 21) or acetic and formic acids (item 20), and the reaction of methane with oxygen in the presence of carbon monoxide may afford methanol and acetic and formic acids (items 12-19). Irrespective of the oxidant used, almost no deep oxidation products (CO2, CO) are formed in the presence of single-atom catalytic sites. However, when methane reacts with H2O2, the products can also contain organic peroxides (methyl peroxide, hydroxymethylene peroxide). Currently, the efforts of chemists are directed towards increasing the selectivity to the target oxygenate products.
Most studies are devoted to the development of SACs for the selective methane oxidation to methanol with hydrogen peroxide. This reaction occurs under the mildest conditions: at T=25-95 °C in water (see Table 3, items 1-11). Recent studies61,144,149,202,262,264 demonstrated that the formation of formic acid in this oxidative reaction can be minimized when the single-atom sites of active metal (Rh, Fe, Cu, Ni) are immobilized on nitrogen-doped carbon supports or attached to the surface of some metal oxides (ZrO2, CeO2). This result is attributable to the synergistic effect of the support and the active metal in the reactant activation.61,144,149,209 The support participates in the adsorption and spontaneous dissociation of hydrogen peroxide61,144,213
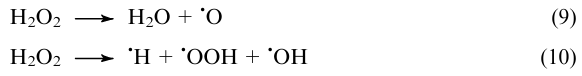
The resulting radicals are attached to the support surface, which prevents the excess oxidation of reactants. The adsorbed highly reactive •OOH (Ref. 144) or •O (Ref. 61) radicals react with the active metal atoms to give metal-oxo sites, which are highly active in the dissociation of the primary C–H bond of methane to give the •CH3 radical, which recombines with the •OH and •OOH radicals, giving rise to methanol (CH3OH) and its precursor, methyl peroxide (CH3OOH).213,265 Owing to stabilization of the •CH3 radical, the reaction selectivity to methyl-containing products (CH3OH+CH3OOH) exceeds 95%. However, methyl peroxide may dominate in the products over methanol, as a result of the general retardation of the process. It is believed225 that in the single-atom catalytic systems with an active support, the reactants (H2O2 and CH4) can compete for the same active sites; since H2O2 dissociation is energetically more favourable than the homolytic cleavage of the primary C–H bond of CH4 and its oxo functionalization, the overall efficiency of the methane oxidation remains low: the turnover frequency of the metal active sites per unit time is 0.5-27 h–1 (see Table 3, items 1-6).
A much higher (10-1000-fold) activity is inherent in SACs obtained by immobilization of active metals in micropores of a zeolite matrix [Cu1@H-ZSM-5,150 Fe1@H-ZSM-5,226 Fe1–CuO/H-ZSM-5,64 Pd1@H-ZSM-5 223 and Pd1–CuO/H-ZSM-5 (see Ref. 223 and Table 3, items 7-11)]. The exceptionally high activity of these systems is attributed to the high content of the cationic species of the active metal (Pd2+, Cu2+, Fe2+) located in the channels of a structured microcrystalline material;64,223 this creates conditions for the heterolytic cleavage of the C–H bond, which is more energy efficient than the homolytic dissociation of this bond. In these catalysts, the zeolite support does not participate in H2O2 activation and cannot scavenge radicals (which are inevitably generated in any oxidative process involving H2O2); therefore, the reaction in the presence of these catalysts affords a large amount of excessive oxidation products such as formic acid and methyl peroxide, the proportion of which can reach 93% (see Table 3, items 7, 10). In order to suppress the formation of these products, copper(II) oxide is added to the catalytic system as a radical scavenger.266,267 This results in the formation of a bimetallic catalyst selective to methanol (78-80%) [Fe1–CuO/H-ZSM-5,64 Pd1–CuO/H-ZSM-5 (see Ref. 223 and Table 3, items 9, 11)].
A high methanol selectivity (79%) was found for the monometallic copper single-atom catalyst Cu1@H-ZSM-5.150 Palladium doping of copper oxide deposited on the H-ZSM-5 zeolite surface (the noble metal content being at the impurity level, only 0.01 mass %) gave the most active catalyst — 0.01% Pd1-CuO/H-ZSM-5 (see Table 3, item 11). With this bimetallic system, the synthesis of methanol is carried out at 50 °C with a production rate of ≈4 mol kg cat-1h–1 at a selectivity of 78%. When the reaction temperature is increased to 95 °C, the rate of formation of the target product can be doubled (up to ≈8 mol kg cat-1h–1) and the selectivity increases up to 86%, while the formation of the side oxygen-containing products (HCOOH and CH3OOH) is almost suppressed.
A review78 and several papers144,202,264,268 reported comparison of the reactions between CH4 and H2O2 in the presence of single-atom catalysts and nanocluster systems of the same composition: Rh1@ZrO2 and Rhn/ZrO2,144 Rh1@CeO2 and Rhn/CeO2,202 Cu1@C3N4 and Cun/C3N4,264 and Fe1@H-ZSM-5 and Fen/H-ZSM-5.226,268 All authors noted pronounced increase in the reaction rate (5-8-fold) and selectivity to methanol (1.5-3-fold) on going from the nanoclusters to SACs. On the basis of the results of kinetic experiments and calculations, various reaction pathways for methane conversion, resulting in different products, were proposed. For example, as can be seen from the energy profiles of product formation during methane oxidation with hydrogen peroxide in the presence of Rh1@CeO2 and Rhn/CeO2 systems (Fig. 3), the energetically favourable pathway in the case of single-atom sites includes dissociation of the primary C–H bond of the methane molecule to give the •CH3 methyl intermediate followed by its oxidation to methanol or methyl peroxide265 (Fig. 3a). Meanwhile, in the presence of Rhn/CeO2, the probability of formation of the CH3 products is relatively low, but a more favourable pathway is the dissociation of the C-H bond in the methyl radical to give the •CH2 intermediate, which is then completely oxidized to CO2 and CO through HCOOH (Fig. 3b). Therefore, the major products obtained on single-atom sites are CH3OH and CH3OOH (where methanol predominates), which are formed at a fairly high rate, whereas over cluster systems (Rhn/CeO2), a lot of CO2 is generated.
![[{"id":"i9OM8Vq5px","type":"paragraph","data":{"text":"Energy profiles of product formation in the methane oxidation with hydrogen peroxide over the Rh1@CeO<sub>2</sub> single-atom rhodium catalyst (<i>a</i>) and Rh<sub>n</sub>/CeO<sub>2</sub> nanocluster (<i>b</i>). Green colour marks the lowest energy levels; red, blue and lilac colours designate various reaction paths.<sup>202</sup> Published under the Creative Commons Attribution 4.0 International License"}}]](/storage/images/resized/s8WZjT7M0P0Q804hJRq6FzNRu8unJINTrMApkRl9_xl.webp)
A similar trend holds for rhodium-containing compositions [e.g., Rh1@ZrO2 and Rhn/ZrO2 (Ref. 144)] and for copper- and iron-based catalysts [Cu1@C3N4 and Cun/C3N4,264 Fe1@ZSM-5 and Fen/H-ZSM-5 (Ref. 268)]. However, in the case of non-noble transition metals, a minor difference was noted, caused by their lower activity compared with the activities of rhodium systems: the reaction product formed on the single-atom sites (Cu1@C3N4) was dominated by methyl peroxide, and the process conducted over nanoclusters (Cun/C3N4) could be suppressed after the formation of HCOOH.
Thus, by varying the nature and the degree of dispersion of the active metal, it is possible to control the selectivity to oxygenates upon the methane oxidation with hydrogen peroxide, which can give either up to 95-100% CH3OH+CH3OOH on single-atom metal sites or up to 64-84% HCOOH over nanoclusters.264,268
Recent publications describe the selective formation of not only methanol or formic acid, but also acetic acid (C2 oxygenate) upon methane oxidation with hydrogen peroxide. This became possible owing to the addition of carbon monoxide to the reaction system [Rh1@H-ZSM-5 (Ref. 220) and Fe/H-ZSM-5 (Ref. 269) catalysts]. The reaction conditions in this case were almost as mild as those used to obtain C1 oxygenates (T=50-150 °C).
The benefits of the selective low-temperature oxidative conversion of CH4 with hydrogen peroxide to C1 and C2 oxygenates is beyond doubt, especially when SACs are used. Unfortunately, they are hardly suitable for the industrial implementation due to the high cost of H2O2. The search for less expensive oxidants and development of the appropriate single-atom catalytic systems are carried out using both kinetic and computational methods.
According to the results of DFT calculations, selective oxidation of methane to methanol under mild conditions is potentially possible with nitrogen(I) oxide as the oxidant on single-atom sites of a number of metals (Al,240 Ga,240 Co,241 Ti,243 V 243) embedded into porous graphene240,241 or carbon polynitride (C24N24) matrix.243 With these systems, N2O is expected to undergo spontaneous decomposition to N2 and an adsorbed oxygen atom (•O), which activates the methane molecule240,243
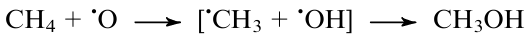
As regards the possibility of using air oxygen as a cheap and the most practically convenient oxidant for the reaction with methane, the results of DFT calculations270 indicate that the activation of the O=O double bond together with the methane activation on a single-atom active site may be difficult. According to estimates,244,270 the probability of this pathway increases when the adsorption sites of O2 and CH4 molecules are separated (for example, when the Pd2/MoCO2 diatomic palladium cluster270 or Rh1@G/Ni(111) complex bimetallic composition244 is used as the catalyst). The monolayer hexagonal boron nitride doped with a single transition metal atom is considered to be a promising catalyst for the partial methane oxidation:92 the adsorption of O2 molecule on this catalyst is likely to give peroxide, the O–O bond of which can be cleaved to give O– anions, that is, reactive oxygen species for activating CH4 molecule under mild conditions.
However, only one original method for the activation of oxygen molecule towards the reaction with methane has been practically implemented as yet, that is, a process involving addition of carbon monoxide to the reaction medium.136--138,212 This reaction, resulting in the formation of acetic acid and methanol, is called oxidative carbonylation of methane; conduction of this reaction over SACs is studied at several research centres.136--138,212,220,221,263,271--273
The reaction of CH4 with O2 in the presence of CO proceeds as a heterogeneously catalyzed liquid-phase reaction under mild conditions [150 °C, total pressure of 2.7-6.8 MPa, and composition of the reaction mixture, vol.%: CH4(2-5)+CO(0.5-1)+O2(0.2-0.8)] over single-atom catalysts dispersed in an aqueous medium (Rh, Ir, Pd or Cu atoms fixed in a zeolite matrix or on the surface of titanium oxide). This reaction yields oxygenates (methanol, acetic acid, formic acid) at an overall rate of up to 10000 μmol g cat-1h–1 with the selectivity to CH3-containing products ranging from 60 to 100%;212 the yield of enhanced oxidation products is low (0-14% CO2). In the absence of carbon monoxide, methane oxidation products are not formed on the single-atom catalytic sites.
In the opinion of Moteki et al.,220 carbon monoxide is a co-catalyst of the process (when occurs as a ligand bound to the active metal atom) and also acts as a reducing agent and a reactant. An original hypothetical scheme for the mechanism of methane oxidation with oxygen in the presence of carbon monoxide was proposed (Fig. 4). In relation to the reaction on a rhodium single-atom site, it was stated that the CO ligand located in the coordination sphere of the active metal atom reacts with an oxygen molecule (arriving from the gas phase) to be converted to CO2, and simultaneously surface metal-oxo compounds are formed. A CH4 molecule is activated by the oxo species to give Rh–CH3 and Rh–OH bonds, while the subsequent release of the methoxy form or CO insertion into the Rh–C bond results in the formation of CH3OH or CH3COOH, respectively.
![[{"id":"-fVjEPhqFA","type":"paragraph","data":{"text":"Hypothetical mechanism of methane oxidation with oxygen in the presence of CO on the rhodium single-atom site to form methanol or acetic acid. The blue and red colour marks different reaction products.<sup>220</sup>"}}]](/storage/images/resized/oSqlTULBf8U0s6Wb3yHtRqQHdb0dfVBtwUO1d5AQ_xl.webp)
According to DFT calculation data,219 the rate-limiting step of the reaction is the formation of the C–OH bond rather than the activation of CH4 (the C–OH bond formation is promoted by carbon monoxide, which draws the O2 molecule into the catalytic cycle). The proposed way of O2 activation involving CO cannot be called new: it completely agrees with the known facts of effective CO oxidation under mild conditions on the surface of SACs based on noble metals embedded in a zeolite matrix.274,275
Moteki et al.220 confirmed experimentally that CO is oxidized to CO2 with air oxygen over the ZSM-5 zeolite modified with noble metals (rhodium, ruthenium, etc.). However, the hypothetical mechanism of methane oxidation catalyzed by the Rh1@H-ZSM-5 type SAC (see Fig. 4) to give methanol or acetic acid proposed by the authors appears debatable, as this at variance with the low content of CO2 in the products. The authors themselves agree with this: in their opinion, further spectral studies and analysis of the state of the catalyst surface are required, in order to identify the active sites and intermediates and establish the details of the reaction mechanism.220
Meanwhile, according to the proposed scheme (see Fig. 4) and experimental results of other researchers (e.g., Refs 212 and 272), methanol and acetic acid are formed by independent pathways and on different sites, which creates prerequisites for the separate preparation of these products.
Systems containing rhodium immobilized in the H-ZSM-5 type zeolite matrix (with the SiO2:Al2O3 molar ratio of 30-300) were studied in most detail.212,260,263,272,273 It was shown that acetic acid is generated only on single-atom sites, namely, on metal cations possessing high carbonylating activity (e.g., Rh1+); only in the presence of BAS, it is formed on the heterogeneous catalytic surface.212,260,272 A number of chemists260,272 believe that the zeolite acid sites participate in the oxidation of methane, together with the single-atom rhodium sites: the secondary conversion of methanol (the primary hydrocarbon oxidation product) into acetic acid takes place on zeolite BAS.
According to a viewpoint,273,276 the zeolite acid sites affect the electronic state of the metal in SACs: the content of BAS in the zeolite influences the electron transfer from the metal (Rh) to the zeolite framework, which was detected by X-ray photoelectron spectroscopy.273 The fact of interaction between the metal cation and BAS located close to each other in the H-ZSM-5 zeolite matrix was ascertained by Wang et al.207 and Arzumanov et al.233 It was shown that this arrangement of Rh and BAS gives rise to a synergistic effect for the C–H bond activation in the methane molecule 207 and also promotes the formation of acetic acid upon the oxidative carbonylation of methane.277 In a number of studies,263,272,273 it was shown using X-ray absorption spectroscopy techniques (EXAFS or XANES) in combination with quantum chemical (DFT) data that single rhodium atoms are fixed in the internal micropores of ZSM-5 type zeolites (in the H-form, H-ZSM-5) via the coordination of the metal atom to five oxygen atoms of the zeolite matrix (Rh1O5@H-ZSM-5),244,272 but high activity towards the methane conversion to acetic acid is inherent only in the single-atom systems in which each rhodium atom is bound to five oxygen atoms one of which belongs to the hydroxyl group [Rh1O4(OH)]. These sites are mainly formed when the Rh atoms are anchored at channel intersections in the zeolite framework.273 The single-atom rhodium catalysts enable the preparation of acetic acid by oxidative carbonylation of methane (CH4+CO+O2) with a selectivity of 60-70% and a specific production rate of up to 420 (g CH3COOH) kg cat-1h–1 (Refs 212, 263). These catalysts are superior in performance to all other catalysts known for this reaction,24,271 including new single-site quasi-homogeneous heterogeneous systems such as Rh1–Cu/polyorganic polymer, which are active only upon the addition of a iodine-containing polymer.51
Methanol is the main by-product in the oxidative carbonylation of methane to acetic acid. A study of a series of 0.5% Rh1@H-ZSM-5 catalyst samples with different physicochemical characteristics prepared using impregnation methods with compositionally different polymers in combination with ultrasonic treatment and desilylation 272,273demonstrated that methanol, unlike acetic acid, can be formed on cluster metal sites: as the content of rhodium cluster species (Rh–Rh) on the catalyst surface increases, the methanol yield grows, while the yield of acetic acid decreases (Fig. 5a,b). The highest selectivity to methanol was observed for a mesoporous catalyst (Fig. 5c), since the location of rhodium in the zeolite mesopores is favourable for the clusterization of metal atoms. As the acidity of the zeolite framework decreases, the methanol selectivity of the 0.5% Rh1@H-ZSM-5 catalyst also increases (Fig. 5d).
![[{"id":"d3jvixUQ5h","type":"paragraph","data":{"text":" Dependence on the selectivity to acetic acid and methanol for the oxidative carbonylation of methane on the physicochemical characteristics of the 0.5% Rh@H-ZSM-5 single-atom catalyst: the Rh - Rh cluster particles on the surface of catalyst samples <b>1</b> - <b>4</b> with different EXAFS characteristics (<i>a, b</i>); the predominant location of the rhodium atom in the zeolite crystal lattice (<i>c</i>); zeolite acidity (<i>d</i>). For reaction conditions, see Table 3, item 12; <i>k</i> is the wave number of photoelectrons.<sup>272,273</sup> R is the distance to the atom emitting a photoelectron."}}]](/storage/images/resized/Do5wZh4GVe9XznuDkYn6HyXFUewvlMn7whTS9XWO_xl.webp)
A methanol-selective catalyst can be obtained by using a metal with a low carbonylating activity (Cu, Ir)137,138 or by using a narrow-pore zeolite as the support (with the size of the framework cavities corresponding to the kinetic diameter of the CH3OH molecule), e.g., with CHA220 or AEI221 topology (see*). Using both the single-atom Cu1@H-SSZ-13 and cluster Cun/H-SSZ-13 catalytic systems based on copper and the H-SSZ-13 zeolite (CHA topology), methanol is synthesized from CH4 and O2+CO with 100% selectivity, but the SAC activity is 1.5 times higher.137
The most efficient single-atom system for methanol synthesis was obtained138 on the basis of iridium promoted by palladium and copper (to increase the activity and the selectivity, respectively). Methanol is formed over the polymetallic heterogeneous 1.4% Ir1--0.1% Pd1--Cu/H-ZSM-5 catalyst upon the methane oxidation with air oxygen (in the presence of CO) at a temperature of 150 °C with a specific production rate of 1.2 (molCH3OH) kg cat-1h–1 at 80% selectivity, which is comparable with these characteristics attained in the synthesis of methanol from CH4 using expensive H2O2 as the oxidant (see Table 3, cf. item 17 with items 5 and 11). In other words, the design of the polymetallic Ir1–Pd1–Cu/H-ZSM-5 single-atom catalyst enables the efficient one-step catalytic synthesis of methanol by oxidation of methane with air oxygen (with carbon monoxide promoting the reaction). This process favourably compares with the recently widely advertised methods of partial oxidation of methane by air oxygen to methanol on zeolites modified with metal-oxo clusters:158,278,279 cluster catalysts are efficient at higher temperatures (not lower than 250 °C), and their performance is moderate; in addition, the chemical looping process design is used most often, that is, two successive stoichiometric reactions are combined.
Recent studies address the possibility of the catalytic oxidation of methane by carbon dioxide to oxygenates,4,140--143,280,281 including processes involving SACs based on Zn1,175,188 Zr1,281 Pt1188 and Ag1.141 The supports used for such systems are zeolites (most often, H-ZSM-5)139,232 or metal oxides (CeO2,139,141 In2O3,142 ZnO143). The reaction of CH4 with CO2 is carried out at moderate temperatures (>300 °C), i.e., under more drastic conditions than methane oxidation reactions with other oxidants (H2O2, O2+CO), which is due to high strength of the C=O bonds of the CO2 molecule; the activation energy of the carbon dioxide molecule (532 kJ mol–1) is higher than that of the methane molecule (435 kJ mol–1). The target product of the reaction is acetic acid. No methanol is formed, but formic acid may appear as a by-product (see Table 3, items 20, 21).
The most active single-atom catalyst for the reaction of CH4 with CO2, as well as with other oxidants, is formed using ZSM-5 type zeolite (in this case, Zn1@H-ZSM-5).139,232 However, the selectivity of this catalyst to acetic acid is very low (18%),139 while formic acid is the major reaction product (82% selectivity). It is believed 4,232 that this is caused by the difficulty of C–C bond formation on a monometallic site.
In a series of studies139--143,218 performed using mainly quantum chemical calculations, it was demonstrated that an efficient catalyst for the methane oxidation with carbon dioxide to acetic acid can be formed only provided that the activation sites of reactants (CH4 and CO2) are separated. On the basis of DFT calculation results,141 it was recommended to use binary catalytic systems consisting of a single-atom metal site (to activate the CH4 molecule) and a metal oxide (to activate the CO2 molecule as a frustrated Lewis pair). The frustrated pair (acid and base) Lewis sites can be formed spontaneously upon the deposition of single metal atoms onto the surface of some oxides [CeO2,141 In2O3 (Ref. 142)] owing to the formation of oxygen vacancies. High efficiency is predicted for the following binary systems: Ag1/CeO2,141 Zr1/In2O3,142 Fe1/ZnO.143 The efficiency of the Zn1/CeO2 system was confirmed by kinetic experiment.139,218 Using the catalyst obtained upon the deposition of this binary system on montmorillonite, acetic acid was prepared, with the specific production rate being 0.6-0.9 mol kg cat-1h–1 at 100% selectivity.139 The reaction of CH4 with CO2 was conducted as a gas-phase heterogeneously catalyzed process at an equimolar ratio of the reactants, a temperature of 300 °C and a total pressure of 0.2 MPa.
Thus, using SACs efficient in the direct oxidative conversion of methane and readily available oxidants (CO2, H2O2, O2+CO) can provide a low-temperature (or, at least, medium-temperature) selective synthesis of CH3-containing oxygenates (methanol, acetic acid). The highest activity was noted for the catalytic systems with the active metal immobilized into a zeolite framework. The bimetallic or polymetallic systems are more selective in the methane conversion to methanol (with H2O2 and O2+CO as oxidants) and in the methane carboxylation to acetic acid. In the oxidative carbonylation of methane to acetic acid on single-atom rhodium sites, only monometallic catalysts have been studied so far.
In the oxidative processes described above, the C2 product (acetic acid) is formed via the C–C bond formation involving carbon oxides (CO2 or CO). Meanwhile, there is another way for the formation of the carbon skeleton upon coupling of the CH4 molecules, which results in the formation of C2+ hydrocarbons.
* AEI is the crystalline framework code taken from the Atlas of Zeolite Framework Types, see Note to Table 1 of this review.
5. Direct conversion of methane into hydrocarbons
Direct conversion of methane under non-oxidative conditions results in the formation of ethane (ethylene) and aromatic hydrocarbons (benzene, toluene, naphthalene).5,6,45,46,147,282--285 The chemical conversion of CH4 is implemented via successive dehydrogenation of the methane molecule in combination with the condensation processes of carbon-containing products. This transformation includes the activation of the C–H bonds of the methane molecule on the catalyst surface to give the •CH3, •CH2 and •CH species, which then undergo recombination to give C–C bonds either in the gas phase23,284,285 or on the catalyst surface.5,282 By varying the reaction conditions and composition of the heterogeneous catalyst, it is possible to terminate the carbon chain growth at a definite stage and to direct the process along the desired pathway, thus providing the selective formation of the target hydrocarbon product: ethane (ethylene) or benzene (toluene). The difficulty of the successful solution of this problem is associated with the thermodynamic factors, which dictate that conversion of methane to polyaromatic compounds (naphthalene and coke structures) is more favourable than the conversion to C2, C6 or C7 hydrocarbons over the whole range of applicable temperatures (at least up to 950 °C).5,285
The good prospects for the application of SACs in these processes is obvious: stabilization of the methyl groups upon the activation of methane characteristic of these heterogeneous catalytic systems may retard the propagation of the carbon chain and, hence, the undesirable extensive condensation processes would be suppressed.
The data on SACs used in non-oxidative conversion of CH4 by methane dehydroaromatization (DHAM) to give benzene,286--291 benzene methylation with methane (MBM) to give toluene132--135 and non-oxidative coupling of methane (NOCM) to give C2 hydrocarbons (ethane or ethylene) are summarized in Table 4. In most of the known examples, the single-atom metal sites are stabilized by the zeolite framework with ten-membered (10MR) channels (ZSM-5,132--135,287--292 ZSM-11 135 and MCM-22 294,295 zeolites), which ensure the shape selectivity of the catalyst to hydrocarbons with up to 10-12 carbon atoms.
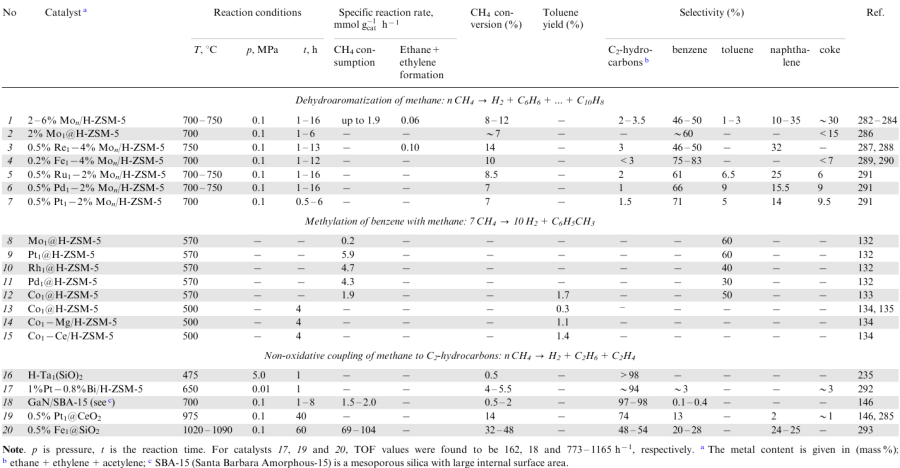
The catalytic systems for methane dehydroaromatization are obtained, as a rule, using molybdenum.295--297 Until recently, only heterogeneous catalysts containing metal clusters of low nuclearity, mainly Mon/H-ZSM-5 (n=4-6), were used in this reaction.282--284,288 It is believed 282,286 that C–H bond activation in the methane molecule and formation of the C–C bonds can occur simultaneously on the cluster species, while aromatization of the resulting fragments occurs in micropores in the zeolite matrix. Methane conversion to a number of aromatic compounds (a mixture of benzene, toluene, naphthalene and coke) takes place over the catalysts containing molybdenum nanoclusters at temperatures of 700-750 °C; the product also contains ethane and ethylene impurities. The benzene selectivity does not exceed 50%; polyaromatic compounds and coke are intensely formed and, hence, the catalyst is rapidly deactivated.
The behaviour of molybdenum catalysts with single metal atoms in the DHAM reaction has been studied.286--291 Two forms of heterogeneous catalysts have been tested, those with a single-atom molybdenum site286 and with a single-atom sites of other transition metals introduced into the molybdenum nanocluster precursor (M1–Mon/H-ZSM-5, where M is a transition metal).287--291 In all cases, switching from nanocluster systems to single-atom catalysts was accompanied by a considerable (≈2-6-fold) decrease in the coke formation rate and increase in the catalyst stability, selectivity and yield of benzene (the target product). However, the activity of the monometallic single-atom Mo1@H-ZSM-5 catalyst was noted to be lower than that of nanocluster analogues (cf. items 1 and 2 in Table 4).
The catalytic systems efficient in all respects were obtained as bimetallic species by doping the molybdenum cluster by single-atom sites of another transition metal (Re, Fe, Pt, Pd, Ru).287--291 The highest activity was found for bimetallic systems containing rhenium as the dopant — Re1–Mo4/H-ZSM-5.287,288 This was explained288 by acceleration of the C–C bond formation by the action of the Re-Mo metal pair. This is evidenced by the fact that the rate of ethane formation from CH3 fragments increases twofold when the initial molybdenum catalyst is modified with rhenium. The most selective and stable catalyst was obtained by doping the molybdenum system with single-atom iron.289,290 In the presence of the Fe1–Mon/H-ZSM-5 catalysts, benzene is formed at a high constant rate and with a selectivity of up to 83%; the methane conversion reaches equilibrium values (10-12%), while the catalyst operation time is at least 10 h. Unlike the iron-containing system, bimetallic molybdenum-containing catalysts modified with noble metal atoms (Pt, Pd, Ru) 291 are less stable, but in the presence of these catalysts, the formation of aromatic compounds from methane proceeds with high total selectivity (≈90%); apart from benzene (61-71%), the reaction products contain a considerable concentration (5-9%) of toluene, an important petrochemical hydrocarbon.
Katada and co-workers132--135 demonstrated that toluene can be synthesized by the MBM reaction at temperatures of 500--570 C using SACs based on some transition metals (Pt, Pd, Rh, Co) embedded into the H-ZSM-5 zeolite matrix. In these systems, the single-atom metal sites activate CH4 to give methyl species, while the zeolite acid sites participate in benzene activation. As can be seen from the data presented in Table 4 (items 8-15), the Mo1@H-ZSM-5 single-atom catalyst has virtually no activity in this reaction (this apparently accounts for the low content of toluene in the DHAM reaction products). Meanwhile, when SACs based on noble metals (Pt1@H-ZSM-5, Pd1@H-ZSM-5, Rh1@H-ZSM-5) are used, toluene is formed at a high initial rate, which is 2-3 times higher than the rate of the DHAM reaction. However, due to the agglomeration of metal particles, these catalytic systems are rapidly deactivated during the MBM reaction.
The stability of cobalt-based SACs is markedly higher. For example, in the presence of Co1@H-ZSM-5 at 500 °C, toluene is produced from CH4 and C6H6 at a constant rate (156 μmol g cat-1h–1) for at least 4 h, while the reaction rate in the presence of Pt1@H-ZSM-5 decreases over the same period from 414 to 30 μmol g cat-1h–1 (Refs 132, 133). The introduction of a second metal (M=Mg, Ca, Zn, Pb, Ni, Ce)134 into Co1@H-ZSM-5 additionally stabilizes the single-atom active sites — Co2+ cations;133,135 as a result, on going to bimetallic Co1–M/H-ZSM-5 systems, the yield of toluene can be increased 3-5-fold. An important role in the catalytic process is played by the shape selectivity of the zeolite support, which should match the kinetic diameter of benzene and toluene molecules. Considering this fact, effective catalysts are obtained on the basis of zeolites with MFI morphology (ZSM-5, ZSM-11),135 while immobilization of cobalt atoms into the MOR, BEA or FAU (fojasite crystal structure) crystalline framework results in absolutely inert systems.133
In the methane conversion under non-oxidative conditions, it is most difficult to obtain C2 hydrocarbons (ethane, ethylene) by arresting the condensation processes in the initial stage (NOMC reaction).298,299 However, exactly SACs can cope with this challenge.
A variety of SACs have been used for the NOMC reaction (see Table 4, items 16-20). It was reported that ethane is formed with >90% selectivity in the presence of H-Ti1(SiO)2 (Ref. 235) and Pt1–Bi/H-ZSM-5 systems (bimetallic catalyst)292 and that ethylene is formed selectively (97%) in the presence of the GaN/SBA-15 gallium nitride catalyst.145 Using these SACs, the reactions are carried out at moderate temperatures (475-700 °C); the catalyst activity is relatively high: the turnover frequency of the metal site is >160 h-1, and the reaction rate is not lower than the rate of methane dehydroaromatization or benzene methylation (cf. items 1, 12 and 18 in Table 4). However, the attained yield of the C2 products is moderate (0.5-5%), apparently, due to the severe thermodynamic limitations in this temperature range.146
The equilibrium values of methane conversion to C2 hydrocarbons acceptable for practical use (at ≈20% level) can be attained only at temperatures of >950 °C;285 therefore, high-temperature single-atom heterogeneous catalysts, Pt1@CeO2 (Refs 146, 285) and Fe1@SiO2,293,300--303 were developed for conducting the NOMC reaction under drastic conditions. The methane conversion over the Pt1@CeO2 platinum sites at 975 °C is ≈14%; the hydrocarbon products are dominated by C2 compounds (74%), but with a high percentage of acetylene (35% C2H2) and lower percentages of ethane and ethylene (6% C2H6, 33% C2H4).146,285 The single-atom Fe1@SiO2 iron sites are more efficient: the ethylene selectivity reaches 45-48% (the other products are benzene and naphthalene); at a temperature of 1090 °C, the conversion of methane is 48%.293 According to the results of DFT calculations,300 a more efficient catalyst can be produced by incorporating a tungsten atom, instead of iron, into the silica matrix. However, the W1@SiO2 system has not yet been tested experimentally.
Only the Fe1@SiO2 single-atom catalyst is of interest considering the potential commercialization: over this catalyst, ethylene is formed in 25% yield, the reaction is stable and the catalyst operation time exceeds 60 h. Currently, studies are carried out to test various designs of NOMC process with this catalytic system (in particular, using a membrane reactor with a hydrogen-permeable membrane 301 and an iron quartz reactor).302,303 Simultaneously, details of the reaction mechanism are being studied; the reaction is assumed to follow a radical mechanism.303--306 The isolated nature of the single active sites prevents the catalytic C–C coupling of the •CH3 and •CH2 species formed in the initial stage. At a high reaction temperature, these species rapidly diffuse from the catalytic surface and undergo recombination in the gas phase; this provides a high selectivity of the reaction to the C2 hydrocarbons.293 As it was shown,285,293 this effect cannot be attained using nanocluster-based catalysts: the use of heterogeneous Fen/SiO2 (T=1020 °C) and Ptn/CeO2 (T=975 °C) catalysts leads to the predominant (80--100% selectivity) formation of coke, resulting from enhanced condensation; the catalyst loses activity in the first minutes of the process, and the yields of C2 hydrocarbons do not exceed 0.3%.285,293
Thus, the use of single-atom catalysts in the NOMC reaction, like in the syntheses of aromatic hydrocarbons (DHAM, MBM), can be regarded as a key to the selective stable preparation of the target hydrocarbons from methane under non-oxidative conditions. Which of the products, ethane (ethylene) or benzene (toluene), would be formed in a larger amount, depends on the type of single-atom heterogeneous catalyst used and on the reaction temperature. For the synthesis of C2 hydrocarbons, it is most appropriate to combine the use of a single-atom monometallic site on an oxide support and a high (>950 °C) temperature of the reaction for fast removal of the primary intermediates formed on the single-atom metallic site from the catalyst surface; this makes it possible to terminate the coupling in an initial stage. The aromatic hydrocarbons are efficiently obtained at lower temperatures (570-750 °C) on bimetallic catalytic systems of a complex size composition. These systems contain single atoms of one metal, which activate CH4, and nanoclusters of another metal immobilized in the zeolite framework (usually H-ZSM-5 type), which provide the formation of the C2+ carbon skeleton needed for dehydroaromatization of the obtained hydrocarbon chain. The separate (selective) synthesis of these two groups of hydrocarbon products is possible, despite the common mechanism of activation of the primary C–H bond of methane, owing to the difference between the mechanisms of C–C bond formation: a radical gas-phase process gives ethane (ethylene), while the growth of the carbon skeleton on the heterogeneous catalytic surface involving nanocluster metal species leads to the production of benzene (toluene), provided that the subsequent dehydroaromatization of the resulting hydrocarbon chain takes place.
A common drawback of all processes of methane conversion to hydrocarbons under non-oxidative conditions is their exceptionally unfavourable thermodynamic characteristics at low and moderate temperatures. Unfortunately, the use of single-atom heterogeneous catalysts does not turn these processes into low-temperature ones. Meanwhile, a decrease in the reaction temperature may be attained by adding an oxidant (air oxygen) to the reactant mixture. For example, the oxidative coupling of methane is carried out at temperatures of 410-780 °C over nanostructured heterogeneous catalysts based on lanthanum oxide; this results in the formation of C2 hydrocarbons with 45-73% selectivity.23,58,307 The conceptual possibility was demonstrated for selective preparation of ethane from a CH4+O2 mixture (300:1 v/v) via a heterogeneously catalyzed gas-phase process over the Rh1@ZrO2 single-atom rhodium catalyst at a moderate temperature (260 °C) and an atmospheric pressure.144 In the case of nanoclusters of the same composition (Rhn/ZrO2) under similar conditions, only CO2 is formed. Hence, the use of SACs suppresses deep oxidation, i.e., this solves the key problem of the oxidative methane conversion, that is, high thermal lability of the primary products of CH4 oxidation. The above-cited publication144 is still the only one example of low-temperature methane coupling to C2 hydrocarbons over SACs; however, the results attained by the authors attest to good prospects of these catalytic systems in the oxidative coupling of methane.
6. Conclusion
The use of single-atom catalysts for the activation of stable CH4 molecule opens up new prospects in methane chemistry, namely:
- there appears the possibility of one-step heterogeneously catalyzed selective conversion of CH4 to oxygenates and hydrocarbons containing a methyl group (methanol, acetic acid, ethane);
- the inclusion of single-atom metal sites into heterogeneous catalytic systems may decrease the reaction temperature (by at least 200 °C) and thus suppress the undesirable side reactions that give polyaromatic compounds and coke and sharply increase the catalyst operation time in the methane reforming and dehydroaromatization processes;
- single-atom catalysts demonstrated a high potential virtually in all reactions of direct methane conversion; their appearance opened up the first real possibility for the design of low-temperature selective processes for single-stage methane conversion into a wide range of value-added chemicals (synthesis gas, methanol, acetic acid, ethane, ethylene, benzene, toluene).
On the basis of analysis of the published experimental data and the mechanistic details of the conversion of methane and co-reactants (O2, H2O, CO2, benzene) to the target products, it is possible to give some recommendations concerning the use of one or another type of single-atom catalyst in the particular methane conversion process. For example, systems of a complex size composition that include both SACs and nanoclusters (of the same or another transition metal) are needed in two cases:
- if apart from CH4 activation, the formation of C2+ carbon skeleton (methane dehydroaromatization) is required;
- if the co-reactant (H2O, O2) of methane is activated only on cluster metal sites, but is not activated on SACs (methane steam reforming and partial oxidation to synthesis gas).
Bimetallic systems with single-atom sites of two different metals are efficient for the reactions that involve CH4 and CO2, which are activated according to different principles (dry reforming, methane carboxylation into acetic acid). Meanwhile, in those cases where the catalytic cycle can be accomplished with only one metal site [methane oxidation and oxidative carbonylation, non-oxidative coupling to ethane (ethylene), or methylation of benzene with methane], a second metal can also be introduced into the catalytic composition in order to increase the stability of the single-atom sites of the active metal or to affect the catalyst selectivity.
It is also noteworthy that almost in cases, the most active and selective single-atom catalytic systems (both mono- and bimetallic) are obtained by immobilization of active metal sites in a zeolite matrix (mainly, H-ZSM-5 type): metal immobilization in the micropores of a structured support provides a uniform distribution of metal active sites over the catalytic surface; most often, they exist as cations acting as sites for the low-temperature activation of CH4.
Unfortunately, there are still few publications on the application of these promising catalytic systems in each particular reaction of CH4 (this is especially true for the methane reforming and methylation and for the reaction of CH4 with CO2 to give acetic acid). Hence, it is necessary to intensify the studies in this field in the near future.
The present review, devoted to a new promising area of chemistry, transformations of the stable methane molecule over single-atom catalysts, is meant for a wide circle of researchers engaged in the development of new efficient catalysts for reactions involving methane. We hope that this information would also be of interest for higher school teachers, post-graduates and students.
This work was performed with the financial support of the Russian Science Foundation (Grant No.21-73-20042).
7. List of abbreviations and symbols
BAS — Brуnsted acid sites,
DFT — density functional theory,
DHAM — dehydroaromatization of methane,
EXAFS — extended X-ray absorption fine structure,
G — graphene,
HAP — hydroxyapatite,
HRTEM ---high-resolution transmission electron microscopy,
MBM — methylation of benzene with methane,
NOMC — non-oxidative methane coupling,
OCM — oxidative coupling of methane,
R — the distance to the atom emitting a photoelectron
SACs — single atom catalysts,
SBA-15- — mesoporous crystalline silica (Santa Barbara Amorphous-15),
TOF — turnover frequency,
XAS — X-ray absorption spectroscopy,
XANES — X-ray absorption near edge structure,
ZSM-5 — Zeolite Socony Mobil-5,
ZSM-11 — Zeolile Socony Mobil-11 (MEL).