



Keywords
Abstract
This review analyzes the current state of the art in the direct synthesis ofalkoxysilanes (DSA). The main approaches, challenges and prospects of this process are highlighted. The plausible reaction mechanism is considered, as well as factors that have a significant influence on the process, including temperature, type and concentration of the catalyst, promoter additives, the method of carrying out the process, etc.
The bibliography includes 232 references.
1. Introduction
The practical application of silicones is over 80 years old. Today, it would be easier to indicate areas in which silicones are not used. Silicone applications include aircraft and spaceship engineering, medicine and pharmacology, construction of buildings and roads, textile and paper production.1 – 3 However, the opinion of professor F.S.Kipping, one of the most famous scientists working in silicone chemistry sounded like a sentence: ‘... the few [organosilicon compounds], which are known, are very limited in their reactions, the prospect of any immediate and important advances in this section of organic chemistry does not seem to be very hopeful’.4 Fortunately for silicones and for us, this opinion was quickly disproved by the first practical applications of silicones, which determined their fate for many years. In the late 1930s, K.A.Andrianov prepared ethylsiloxane resins and demonstrated their effectiveness as hydrophobisers.5–8
Since then, the industrial production of silicones has developed sustainably and continuously. The first methods of silicones production were based on the Grignard reaction, but soon E.Rochow9 made a revolutionary discovery. He developed a method for direct synthesis of organochlorosilanes (DSC) based on the reaction of elemental silicon with alkyl chloride in the presence of a copper catalyst at a temperature of 200 – 400 °C (Scheme 1).
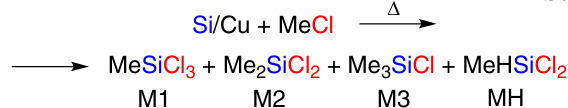
This process is implemented using methyl-, ethyl- and phenyl chlorides. The reaction gives mainly diorganodichlorosilane. Dimethyldichlorosilane (M2) is the most demanded among all organochlorosilanes, — truly the primary building block from which the magnificent building of silicones is constructed.4 This is due to the fact that the production volume of polydimethylsiloxane (PDMS), the precursor of which is M2, accounts for up to 90% of the total volume of silicone products. To be fair, it is also necessary to mention the German scientist R.Müller, who discovered the DSC process independently of Rochow, so in modern literature DSC is usually referred to as the Müller – Rochow process.4
Ironically, E.Rochow did not win the Nobel Prize, although he solved no less difficult and practically significant problem than the one solved by the prize winner V.Grignard. E.Rochow used a less reactive alkyl halide and much less active, compared to magnesium, silicon. In this way, his method for a long time pulled out the Grignard synthesis from the main paths of development of the chemistry of silicones. Unfortunately, the scientists of that time clearly underestimated the huge potential of E.Rochow’s discovery.
The direct synthesis has come a long way since its discovery. The optimum process conditions and reactor type were found out. A lot of work was done to find a suitable method for preparing the so-called contact mass (CM) — a mixture of silicon and copper. All this provided an opportunity to produce M2 with a selectivity of 85 – 95% and silicon conversion close to quantitative.10 In specialized plants, PDMS is produced from M2 by direct synthesis. This makes it possible to create virtually waste-free production, where M2 immediately enters the hydrolysis stage (Fig. 1).11
![[{"id":"yki-LD5BhK","type":"paragraph","data":{"text":"General scheme for the production of polydimethylsiloxanes.<sup>11</sup>"}}]](/storage/images/resized/OMFNE07msLm80pWebQA3lKZgiqO4Q0g7mxULY9Xh_xl.webp)
The obtained PDMS is sent for further processing (catalytic rearrangement, separation from cyclic products, etc.), and the HCl released in the reaction is used to produce the starting methyl chloride.12,13
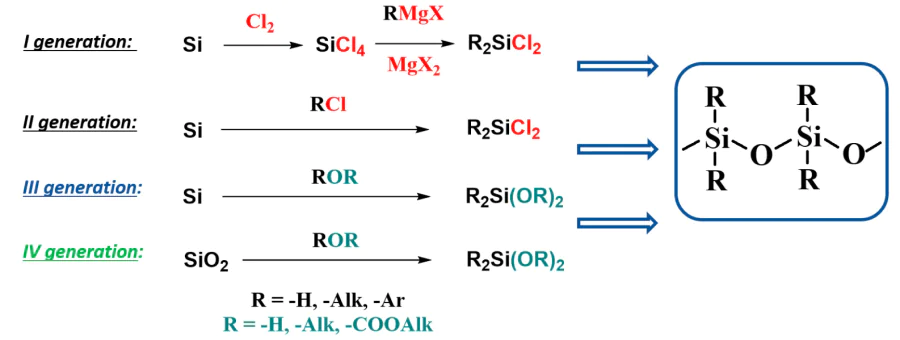
Like many other fields of technology, the production of silicones can be represented by its technological modes in the form of generations13 (Fig. 2):
![[{"id":"RWdS3lC2Mt","type":"paragraph","data":{"text":"Scheme for technological generations in silicones production."}}]](/storage/images/resized/JovYWT8RiMTJXqgQ3cfnKogN61H4r7vxHhjo5yUZ_xl.webp)
In fact, a generation is a set of basic technologies of silicones production that determine the production volume and cost (and hence availability) of silicone products.
The first generation includes technologies for the synthesis of tetrachlorosilane, the subsequent organomagnesium synthesis of organochlorosilanes and their further conversion into organosiloxanes (see Fig. 2, I). The transition to direct synthesis was revolutionary, an instant shift in the time scale. The second technological mode (see Fig. 2, II) in many variations became dominant at all production sites.4 The production of polyorganosiloxanes from organochlorosilanes also underwent modifications from generation to generation, but it was always the synthesis of starting organosilicon monomers that was the limiting step.
As for production sites, it should be noted that there were few of them.14 Until the breakup of the Soviet Union, only five countries were members of the ‘silicone club’ — i.e., they had a full cycle of silicone production, starting from copper-silicon alloys and mixtures and ending with a wide range of silicone products, including binders, liquids, rubbers and sealants. The world did not notice the Soviet Union’s withdrawal from the global silicone market, as powerful players such as South Korea and China emerged just a decade later. It is China that is now associated with the constant growth of global production of silicones, which are still produced by the proven Müller – Rochow method, perfected in all technological components. Suffice it to say that the individual unit capacity † of Chinese direct synthesis reactors exceeds that of the devices used in the Soviet Union by several times. At present, Chinese reactors with a single capacity of 150 000 tonnes per year are known.10
Nevertheless, the second-generation silicon production technology has already practically exhausted itself. Even the increase of single capacities cannot save this process from ‘generic’ disadvantages, the main of which is the so-called ‘chlorine cycle’ (Fig. 3), in which silicon is first reduced from silica,15 getting rid of silicon-oxygen bonds, then oxidized with chlorine, and then or simultaneously chlorine is replaced by carbon or oxygen in the final products. The cycle is completed by the reaction of HCl with MeOH to give the starting methyl chloride.
![[{"id":"8ssenJTPAB","type":"paragraph","data":{"text":" ‘Chlorine cycle’ of the second-generation technology"}}]](/storage/images/resized/QF6enL2Vh9TyrfN3IKYDo6KxkbkKKQS4uWuSgiAE_xl.webp)
Moreover, the ‘chlorine cycle’ is potentially very dangerous, since to produce a variety of silicones, chlorosilanes are used as monomers. The increase of single reactor capacities led to the fact that tens and hundreds of thousands of tonnes of these hazardous reagents are concentrated in one place. The storage, transport, and purification of chlorosilanes involve risks on a scale comparable to the use of phosgene during World War I.
Other disadvantages are economic. And while large companies deal with environmental aspects out of necessity, economic factors are strong arguments in favour of rejecting chlorosilanes. Processes with a large amount of hazardous waste and reagents require rather complex technological lines with a lot of recycling and other complex instrumental solutions to achieve waste-free production, and need significant financial resources. Given the same efficiency, the choice will always be in favour of simplicity and low cost. This is also true for the production of organosilicon compounds.
A silicones production plant based on the second-generation technology should have a chlorosilanes production facility, a unit for production of siloxanes on their basis, modules for synthesis and preparation of feedstock (see Fig. 1), not to mention silicon and CM preparation workshops and places for disposal of spent CM. It is extremely disadvantageous to produce CMs separately and transport them to the DSC plant. CMs oxidize rapidly in air and cannot be used.16, 17 Disposal of spent CMs also needs to be done locally due to their high explosion hazard.18 Organochlorosilanes must be used immediately, as their transport is quite dangerous. These highly reactive compounds hydrolyze very quickly in air, releasing enormous amounts of gaseous HCl into the atmosphere.
Finally, the most important economic factor is the separation of the target methylchlorosilanes. As already mentioned, M2 is the major product of DSC. In addition to M2, methyltrichlorosilane (M1) and trimethylchlorosilane (M3) are formed. The boiling points of these products are 70, 66 and 57 °C, respectively. It is very difficult to separate such a mixture; for this, several sequentially connected rectification columns more than 50 m high are used.19 Obviously, such columns are expensive to maintain and energy intensive.
So, can the ‘chlorine curse’ be overcome? Yes, it is. When developing the direct synthesis of organochlorosilanes, Rochow 20, 21 showed that chlorine-free synthesis of alkoxysilanes by the reaction of alcohol with silicon was possible. But at that time, the chlorine process was much more favourable from all points of view, except from the ecological one, which was then the last priority. The direct synthesis of monomers from alcohol and silicon is not just more environmentally friendly, it eliminates two chemical steps in the silicone synthesis, namely the introduction and then removal of chlorine. Due to this feature, this approach can be safely attributed to the third-generation technologies — chlorine-free processes of silicone synthesis.
In contrast to the revolutionary progress of the second generation, the third generation (direct synthesis of alkoxysilanes, see Fig. 2, III) evolves slowly and is in no hurry to displace its predecessor from the pedestal. Nevertheless, the work in this direction is ongoing and is drawing increasing attention. Unfortunately, until very recently, researchers have worked with tri- and tetraalkoxysilanes, which need at least one additional step prior to be converted into organosilicon derivatives containing Si – C bonds.22, 23 The synthesis of these compounds has been brought to industrial scale, but as an auxiliary option for utilization of silicon dust, a waste product of the DSC process. This third-generation sprout is discussed in detail below (see Section 2.2).
Third generation technologies will slowly grow out of the second generation, absorbing all the best and eventually dominating the global silicones market. The fact that the generational shift will take place in the next decade became obvious after reports of the successful chlorine-free synthesis of dimethyldimethoxysilane via the direct reaction of dimethyl carbonate with copper silicide.24 It is no surprise, this breakthrough was made by Dow Corning scientists, the main victims of the first generational shift in the forties of the last century. Their dominance in the silicone market was shaken for many years as direct synthesis was developed at rival General Electric. Since then, Dow Corning, having made the right conclusions, has been among the leaders in innovation, science funding, development of relations with universities and academia.
The progress of the third-generation technology in our country is especially relevant. This is due to the fact that after the breakup of the USSR all production based on the second generation was lost. At the same time, Russia retains its leading position in silicon production in the world.25 It makes no sense to recreate the second-generation production in Russia, as Soviet technologies are long outdated and will not be competitive in the global market.
In this context, one should not fail to mention the fourth-generation technology (see Fig. 2, IV) based on the direct production of alkoxysilanes from SiO2.26 Although it has not yet been possible to obtain organoalkoxysilanes by this method, there are several examples of deriving tetraalkoxysilanes from silica gel, rice hull ash and some minerals.27 – 36
Importantly, the entire technological interface for the transition to the third and, potentially, to the fourth generation has already been established.13 Chlorine-free chemistry based on organoalkoxysilanes is already working and very effectively used, marking a new milestone in controlling the properties of materials.37 – 43 In addition, there is no worthy alternative to alkoxysilanes in a number of reactions and processes (e.g., in the Piers – Rubinsztajn reaction 44 and sol – gel process 45). We should also mention technological processes based on alkoxysilanes, which were implemented in the USSR. The manufacturing processes for phenylsilsesquioxane resin F-9 and polydiethylsiloxanes (PES) were carried out on an industrial scale.46, 47 Undoubtedly, there are no less efficient technologies implemented in other world centres of silicones production, but such data belong to the industrial secrets of companies, which are usually not disclosed.
Therefore, the key element of the third-generation technology is the direct synthesis of organoalkoxysilanes, the realization of which on an industrial scale is the crucial target for modern organosilicon chemistry. Analyses of the available prerequisites for solving this issue, as well as an assessment of the prospects for the technological implementation of processes for the direct synthesis of organoalkoxysilanes are the subject of this review.
2. Direct synthesis of alkoxysilanes
Alkoxysilanes and alkylalkoxysilanes are currently a low-tonnage product of the chemical industry. They are widely used to produce a huge nomenclature of organosilicon materials that are applied in various fields.13
As mentioned above, the direct synthesis of organoalkoxysilanes (see Fig. 2, III) is a priority for researchers. In 2020, a successful approach to obtaining methyl methoxysilanes from silicon silicide was presented.24 Earlier, positive results were obtained in the synthesis of tri- and tetraalkoxysilanes based on the reaction of silicon with alcohol (Scheme 2).
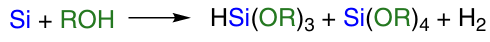
2.1. Valuable synthetic intermediates based on HSi(OR)3 and Si(OR)4
Tetraalkoxysilane (TAOS) is a major precursor for sol – gel processes,48 synthesis of carriers for catalysts, zeolites and adsorbents,49, 50 curing agent in siloxane compositions.51 In addition, TAOS is a starting compound for the preparation of phenyl and ethyl ethoxysilanes by organomagnesium synthesis (e.g., phenyltrialkoxysilane (PTAS) and diethyldialkoxysilane (DEDAS)).14, 46, 47 PTAS serves as a basic monomer for a wide range of various phenylsilsesquioxanes.52 Phenylsilsesquioxanes are used as bases for varnishes and resins, optical materials, and are part of many polymer compositions to give them improved physico-mechanical properties. DEDAS is the main monomer for obtaining a unique product — polyethylsiloxane liquids (such as PES liquid). These polymers have significant advantages over classical polydimethylsiloxanes such as improved compatibility with organic compounds and absence of crystallisation. Due to this, they are used as low-temperature heat carriers, in lubricating compositions, cosmetics.53
Trialkoxysilane is even more valuable. Firstly, it is a source of semiconductor silicon, without which no modern electrical device can be imagined. At present, the process for producing semiconductor-grade silicon, called the Siemens process, is based on the disproportionation of trichlorosilane.54 Obviously, switching to trialkoxysilane will seriously improve the environmental impact of this method. Secondly, the production of semiconductor silicon via a chlorine-free process is much more favourable in terms of energy consumption.54
Also, trialkoxysilane is a precursor to a number of organotrialkoxysilanes, which can be used, in particular, for modification of polymer systems and obtaining various types of hybrid materials: from sorbents for HPLC or adsorbents for the separation of various metals from solutions to the creation of new catalysts. Let us list some of the most important representatives of such trialkoxysilanes and their applications:
— 3-aminopropyltriethoxysilane, a coupling agent for phenolic, epoxy, polyamide and polycarbonate resins,55 used to bind copper salicylaldimine in silica gel;56
— vinyltriethoxysilane used as a binder for fillers or fibre-glass resins,57 in some cross-coupling reactions,58 in the preparation of some olefin copolymers;59
— 3-mercaptopropyltrimethoxysilane is a curing agent for some resins,55 widely used in surface treatments and adsorbents for highly efficient binding of heavy metals,60 immobilization of fluorescent labels on nanoparticle surfaces;61
— 3-azidopropyltrimethoxysilane is used in click chemistry for immobilization of discrete complexes 62 or poly-l-lysine onto the surface of mesoporous silica gel.63
The above data suggest that the development of the alcohol variant of the direct synthesis of alkoxysilanes will give a powerful impetus to the development of their industrial production and, in the long term, a number of practical applications of silicones.
2.2. Direct synthesis of tri- and tetraalkoxysilanes
The reaction of direct synthesis of tri- and tetraalkoxysilanes (see Scheme 2) was first discovered by E.Rochow.20, 21 This reaction proceeds at a temperature of 200 – 350 °C. Copper or its compounds are used as catalysts. The most commonly used alcohols are methanol and ethanol.
Scheme 2 illustrates the general scheme of the process. In fact, the main reaction looks as shown in Scheme 3, reaction 1.
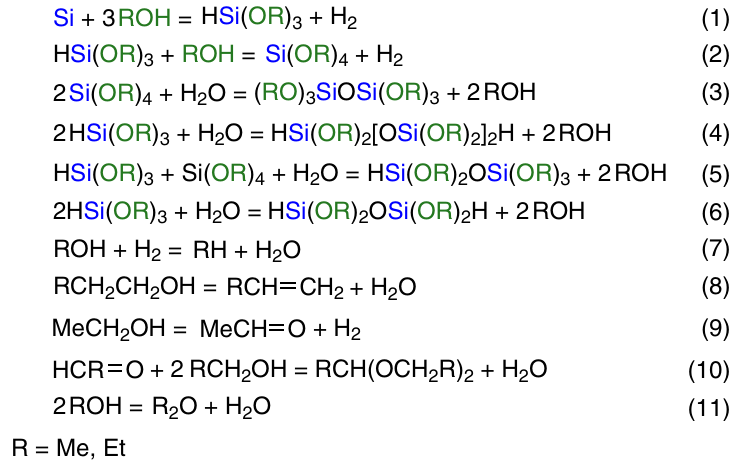
The main product is trialkoxysilane, which further reacts with alcohol to give tetraalkoxysilane (see Scheme 3, reaction 2). A number of other side processes may occur (see Scheme 3, reactions 3 – 11). Water formed in reactions 7, 8, 10, 11 leads to hydrolysis and condensation of the main products (see Scheme 3, reactions 3 – 6). Dehydrogenation of alcohol furnishes aldehyde (see Scheme 3, reaction 9). Its further reaction with alcohol affords acetals (see Scheme 3, reaction 10). By-products obtained in reactions 3 – 11 (see Scheme 3) were reported by a number of authors.64 – 67 All these reactions deteriorate the characteristics of the main process, such as speed, degree of silicon conversion, selectivity. The ratio of the rates of these reactions depends on process control factors such as temperature, pressure, reagent feed rate and reactor hydrodynamics. The method of CM preparation, silicon grade, type and concentration of copper catalyst, presence, amount and method of introduction of promoter additives, reactor type also play a role.
Almost all researchers note that initially, HSi(OR)3 with a small admixture of Si(OR)4 prevails, while the selectivity for trialkoxysilane decreases with increasing reaction time and silicon conversion. As an illustrative example, the dependence of the product mixture composition on silicon conversion obtained by the authors of the patent 68 for the reaction of activated silicon with methanol is given below (Fig. 4).
![[{"id":"RULiDzZ61A","type":"paragraph","data":{"text":"The product mixture composition of the reaction of Si with methanol vs Si conversion in a liquid-phase reactor. TMS is (MeO)<sub>3</sub>SiH, TMOS is (MeO)<sub>4</sub>Si.<sup>68</sup>"}}]](/storage/images/resized/v612bQuNYb28Ml9eHzQpG2iLS9S0U3PHZiYFmq4C_xl.webp)
Despite the complexity of the process, the proper choice of its conditions, namely, the type of catalyst, promoting additives and equipment allows to optimize the process and minimize the effect of side reactions.
2.3. Direct synthesis mechanism
Although the DSC and DSA were discovered in the middle of the last century, their exact mechanism satisfactorily describing all stages of the process has never been established. According to Newton and Rochow,64 these reaction pathways should be largely similar and consist in the polarization of the organic component (in the case of alkoxysilanes it is an alcohol) on the copper-silicon active site with subsequent desorption of the reaction products (Fig. 5).
![[{"id":"9shxYa6L9A","type":"paragraph","data":{"text":"Plausible mechanism of the DSA according to E.Rochow.<sup>64</sup>"}}]](/storage/images/resized/6wMV5JANLGo3DFV6FQQRhZlsowdYBwOrtUDcTT0r_xl.webp)
Subsequently, the mechanism of DSA has hardly been investigated. The choice of DSC as a basis for the development of industrial production of silicones focused the main attention of researchers on the mechanism of direct synthesis of chlorosilanes.
The mechanism of DSC, originally proposed by Hurd and Rochow 69 (Scheme 4, reactions 1 – 4), was subsequently refuted by numerous studies.
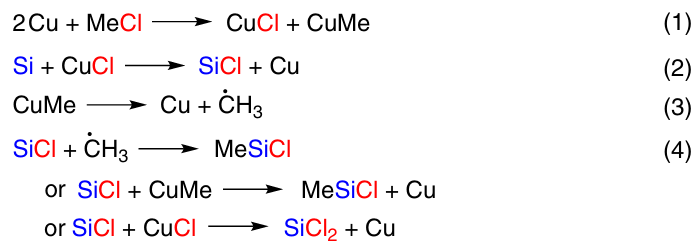
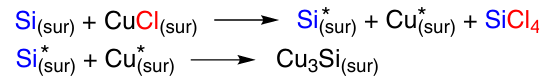
It was found that the reaction proceeds on separate sites on the CM surface. The density of the reaction sites is controlled by the nature of the surface oxide layer and depends on the number of surface defects. The reaction has an induction period, the duration of which depends on the method of the CM preparation, and affords surface copper – silicon intermetallic compounds CuxSi (x ≥ 3) and active Si – Cl sites, the latter being the active reaction sites.70 As silicon leaves the surface during the reaction, it diffuses from the bulk to the Cu3Si/Si interphase, forming a new η-phase that is involved in the further reaction. The role of the Cu3Si phase is to mediate the transport of silicon from the lower layers to the CM surface. Examination of the CM using scanning electron microscopy (SEM) before and during the reaction with methanol showed that the reaction starts around the Cu3Si localization area and proceeds in voids/pits on the silicon surface. The number of pits per unit of surface area characterizes the specific concentration of active sites. In the course of the reaction, these pits become larger and at 45% conversion they merge with each other.71, 72 Two main mechanisms for the formation of Cu silicides were presented in the literature. Both of them suggest the presence of a Cu – Si phase and neutral Cu and the removal of Cl in the form of SiCl4. In the first variant, at the physical contact point on the surface (sur), Si and CuCl interact to form active Si* and active Cu*, which react with each other to form Cu3Si (Scheme 5).73
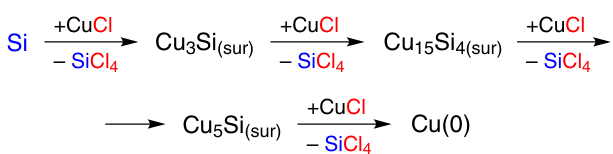
In the second variant, the Cu3Si phase is formed immediately from Si and CuCl at the point of contact and is further transformed into copper through consecutive reactions with CuCl (Scheme 6).74
Exploring the reaction in a liquid-phase reactor, Adonin et al.75 found that the process is more complex. The authors noted the topochemical nature of the reaction of CuCl with Si, which begins at the point of their contact and is accompanied by the distribution of chlorine onto the silicon surface. As a result, Cu nanoparticles partially stabilized by chlorine adsorbed on the surface are formed (Scheme 7, reaction 1), and a Cu – Si phase appears at the interface boundary between metallic Cu and silicon.
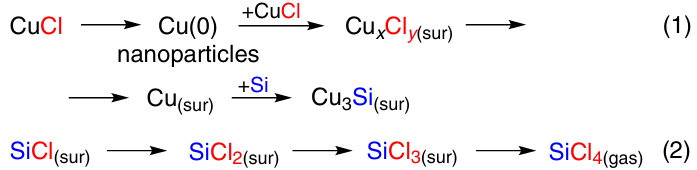
Chlorine is consumed during the diffusion over the silicon surface successively forming Si – Cl bonds (Scheme 7, reaction 2).
If one imagines a cross section of the active phase area of the process, the picture for the DSA reaction can be represented as follows (Fig. 6).
![[{"id":"xMK9j48_1Z","type":"paragraph","data":{"text":"Possible chemical forms of silicon in the CM and processes occurring in the catalytic DSA.<sup>25, 76</sup> 1 is Si polycrystalline lattice, 2 is Si interphase (crystal Si – Cu<sub>3</sub>Si alloy), 3 is a Si silicide (e.g., Cu<sub>3</sub>Si), 4 is interfacial Si transport (Cu<sub>3</sub>Si – Cu alloy – metal), 5 is a silylene on the copper crystallite surface, 6 is a Si – OR alkoxide form chemically absorbed onto the CM surface, 7 is a desorbed reaction product (alkoxysilane)"}}]](/storage/images/resized/2qrfeRkDCrVHL35sfTlWaNrFsyL2ji9xckuG3aP4_xl.webp)
Zone 1 comprises polycrystalline silicon particles ranging from 500 nm to 20 mm, zone 3 comprises silicide phases (e.g., Cu3Si) hundreds of nanometres thick with the silicon-to-copper concentration gradient decreasing towards the contact surface with zone 5, in zone 5, copper prevails and highly reactive silylene units occur, zone 7 is the zone of gas-phase diffusion of reagents and products. Silicon diffuses from the bulk to the active surface through the copper silicide layer, where it coordinates alkoxides and hydrogen absorbed on the CM surface, after which it moves into the gas phase in the form of reaction products.
The mechanism currently discussed in the literature, involving the participation of surface intermediate silylene particles in the reaction, was proposed by Andrianov et al.,77 who showed that in the case of MeCl, methylchlorosilanes Me2SiCl2 and MeSiCl3 are mainly formed on the CM surface through MeSiClsur and SiCl2sur (Scheme 8). Although the silylene particles are desorbed into the gas phase, their transformations in the gas phase are irrelevant to the direct synthesis. This approach was subsequently developed by other researchers.78 – 80
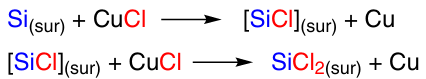
Yet another argument in favour of the participation of silylene particles in the reaction of silicon with methyl chloride is the production of silacyclopentenes along with methylchlorosilanes. In experiments carried out in the presence of butadiene, which is a good trap for silylenes, silacyclopentenes I – III were obtained in an overall yield of 25%.81, 78
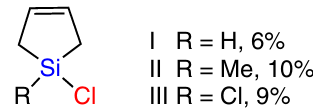
A similar mechanism was proposed by Okamoto et al.82 – 84 for the reaction of silicon with alcohols. According to the authors, the active sites on the CM surface are of silylene nature (particle 1 in Scheme 9).85
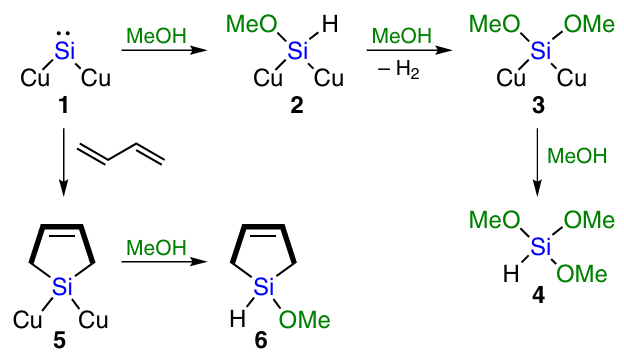
The active silylene site reacts with an alcohol molecule to form an intermediate compound 2. Reaction with the next alcohol molecule gives intermediate 3, and hydrogen gas is released. At the final step, reaction with the third alcohol molecule occurs to afford trialkoxysilane 4.
To prove the proposed mechanism, this reaction was carried out in the presence of carbene traps and their analogues. The addition of butadiene trap to the methanol stream leads to cyclic alkoxysilane 6 via intermediate 5. Similar results were obtained in the chlorine variant of the synthesis,78 – 80 thus supporting this mechanism. Despite this, the reaction using traps provides low yields. The selectivity for the obtained organoalkoxysilanes ranges from 3 to 33%.
The results of the study of cryogenic reactions between silicon and methanol in an argon matrix by IR spectroscopy can serve as an additional argument for the reaction to occur via the silylene intermediate. The experiments showed that the above reaction proceeds stepwise and involves the formation of various intermediates. At a ratio of reactants Si : MeOH : Ar = 1 : 1 : 500 at 15 K, the IR spectrum showed intense bands (1966, 1929 and 1917, 1911, 1849 cm–1) attributed to the insertion product of silicon into the methanol O – H bond, i.e., MeOSiH. It was formed much more favourable than the insertion product into the C – O bond, although very weak bands attributed to the MeSiOH fragment were also recorded in the IR spectrum. The results of DFT (density functional theory) calculations showed that the initially exothermically formed Me(H)O∙∙∙Si ( – 23.2 kcal mol–1) complex can give either silylene MeOSiH or silylene MeSiOH, with the energy barrier for the formation of the former (5.6 kcal mol–1) being much lower than that for the latter (26.1 kcal mol–1), with exothermity values of – 96.3 and – 119.3 kcal mol–1, respectively.86 Preparative experiments at 77 K showed that the major products are (MeO)2SiH2 and (MeO)2Si(H)Si(H2)(OMe), along with minor (MeO)SiH3, (MeO)3SiH and (MeO)4Si. The formation of disilane is possible via the insertion of the initially formed silylene MeOSiH into the Si – H bond of dimethoxysilane (Scheme 10).

The absence of silicon insertion into the alcoholic C – O bond is also characteristic for the surface of single-crystal silicon. Study by photoemission spectroscopy showed that methanol adsorbs on the Si(100) silicon surface with cleavage of the O – H bond and formation of the Si – O bond.87 Similar reactivity was also found for ethanol,88 phenol 89 and 1,4-butadiol.90 No fragmentation of alkoxy groups was also found for the Si(111) surface during adsorption of alcohols.
Not all experimentally observed details of the reaction can be explained by the mechanism of the direct process discussed in the literature. The mechanisms of influence of various activators and additives on the process remain unclear. Therefore, the reaction pathway of the direct synthesis certainly needs further clarification and development, which is understandable in view of the issues associated with the heterogeneous nature of the process and a number of factors that affect its course and have not yet been adequately described within the proposed mechanism.
Such factors include:
1) the type of silicon;
2) method of CM preparation;
3) type and concentration of the copper catalyst;
4) the presence, type, concentration and the way to introduce various promoters;
5) technological features of the process (reactor type, reagent injection rate, etc.);
6) temperature of the process.
Items 1 to 3 can be referred to the preparatory stage, i.e. the selection of starting materials and their preparation for the reaction. Items 5, 6 refer to the process per se and include the method in choice (reactor type) and the temperature mode. DSC is generally carried out in fluidized bed reactors.10 DSA is mainly carried out in a fixed bed reactor or in a high-temperature heat transfer fluid.25 The methods of carrying out DSA will be described in more detail in Section 3.7. Particular noteworthy is item 4, which can be classified in both the first and second categories. This is due to the fact that promoters can be added both at the preparation stage, i.e. by adding them to the contact mass, and directly during the reaction.91
From a simple enumeration of the factors and their classification into one or another stage of the process, it is clear that changing at least one of these items can affect all the others. Thus, for example, when exploring the effect of catalyst concentration on the main process characteristics (such as reaction rate, silicon conversion, product selectivity), it is necessary to keep all the others unchanged. For example, if the type of silicon is changed, the study has to be started from the beginning because of differences in the composition of impurities in the latter (see below).
Therefore, the process of direct synthesis of chloro- and alkoxysilanes is rather complicated. Despite the available studies of this reaction, there is no unambiguous understanding of its mechanism to date. This is primarily due to numerous factors that affect the reaction and, as a consequence, obscure its understanding. Another important circumstance, not mentioned before, is the applied importance of direct synthesis for silicon chemistry. Eventually, the results of research in this field are kept confidential in the form of patents and know-how, which inevitably affects the pace of development of knowledge about this process. Despite all these challenges, progress in the field of research on the mechanism of direct synthesis is evident. Let us consider sequentially the influence of the above factors on the DSA process.
3. Factors affecting the DSA process
Due to the poor literature data on DSA, this Section also presents the findings on DSC considering the impact of various factors on the synthesis stages, which are similar for DSA and DSC.
3.1. Type of silicon
Industrial DSC processes traditionally use technical-grade silicon. It can be produced by any method such as casting, aqueous granulation, atomization and acid treatment, etc. The presence of impurities of various metals can both promote the direct synthesis and slow down the process, as well as affect the selectivity for target products. Examples of using different silicon, from technical-grade silicon (purity ≤98%) to semiconductor-grade silicon, in DSA are known. Silicon is first ground and fractionated.
In the Soviet Union, technical-grade silicon with a purity of 98%, produced under the brand name KR-1, was usually employed for DSC.92 The remaining 2% of impurities included iron (0.7%), aluminium (0.7%) and calcium (0.6%). The use of technical-grade silicon is not only due to its lower price compared to semiconductor-grade silicon. It was shown 93 that the reaction of technical-grade silicon with ethanol (in the presence of EtCl as a promoter) is characterized by a shorter reaction induction period (2 vs 5 h) and a higher rate, compared to the same reaction using semiconductor-grade silicon. The main impurities (Fe, Al, Ca, Sn) present in technical silicon have a lower redox potential than silicon and are able to reduce CuCl to form active copper atoms, and the reduction ability of Al and Ca is higher than that of Si. This increases the number of active sites and, consequently, the reaction rate (perhaps impurities serve as Cu3Si nucleation centres). Another reason may be the promoting ability of the impurities, e.g., Al affects both the formation rate of the active Cu3Si intermetallide and its decomposition, while additional Ca and Fe impurities stabilize Cu3Si, reducing the influence of aluminium on the decomposition of the Cu3Si phase.94 Silicon of different grades may contain different impurities. For example, the typical composition of commercial silicon used by E.Rochow 95 for direct synthesis was as follows (wt.%): Si 98.53%, Fe 0.56%, Al 0.31%, Ca 0.12%, Mn 0.04%, Ti 0.02%, other metals 0.08%, oxygen 0.35%. Among the impurities, the lead content is the most critical; it should not exceed 0.001 wt.%.96 The formation of active copper dispersed onto the silicon surface can also occur through other alloys of silicon with metals present in the CM (Scheme 11).97
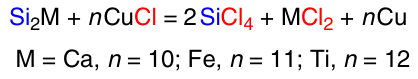
The use of ferrosilicon with iron content of up to 30% and calcium-silicon alloys with calcium content of up to 27% in the direct synthesis reaction has been patented.98 Silicides of iron, calcium, magnesium and copper prepared in advance can be used in the synthesis either as an individual phase or in a mixture with silicon.
The size of the silicon particles used can range from 1 to 500 μm,66 but typically the particle size is 40 to 200 μm. The smaller the particle size, the lower the tendency of the resulting dispersion to precipitate when the reaction is carried out in a liquid medium, the higher the reaction rate and the less erosion of the reactor. However, this silicon-containing fraction has a larger surface area and hence a larger surface area of the oxide layer.
3.2. Effect of the presence of an oxide layer on the contact mass (CM) surface on direct synthesis
Metallurgical-grade (technical-grade) silicon used in industrial processes for the production of organochlorosilanes is coated with an oxide layer that inhibits the direct synthesis reaction by preventing the contact of Cu with Si. The formation of copper silicides has not been reported even when annealed at 800 °C for 3 h given a 200 nm layer of SiO2.16 A 20 nm thickness is sufficient to completely inhibit the reaction of Si with MeCl.17 The native oxide layer possesses defects, which are holes, voids or pores, whose density on the surface decreases with increasing oxide layer thickness. Numerous studies showed that the rate of the reaction between Si and MeCl and selectivity for Me2SiCl2 formation raise with decreasing of oxide layer thickness. In the reaction of Si and CuCl powders, the process occurs upon heating in the regions of defects in the oxide layer to afford Cu3Si, Cu5Si, Cu15Si4 copper silicides, and also a liquid condensate consisting of SiCl4 (90%) and Si2Cl6 (10%) (according to GC-MS). In this case, more than 95% of Cl from CuCl is consumed to give silanes. The authors noted that at 300 °C this reaction occurs within seconds, whereas at 260 to 300 °C, the reaction is slower, but they provided no specific data.17 Therefore, factors favouring a decrease in the thickness of the oxide layer and an increase in the number of defects on the surface positively affect the reaction. Even mechanical damage of the silicon wafer surface with a diamond scribe leads to preferential deposition of copper salt on the scratch.17 Application of a monolayer of some elements (Au, Ag, Cu, W, Ni, Pt, Ti, Ge, Si, Mg, At) onto the SiO2/Si(100) surface favours the decomposition of the oxide layer during high vacuum anneal.99
In most publications devoted to DSA, the oxide film was removed from the silicon surface by the reaction with HF. For example, Suzuki et al.71 used silicon both washed and not washed with hydrofluoric acid. It was shown that in the case of washed silicon, it reacted with alcohol without an induction period, with a higher rate and greater selectivity for trimethoxysilane (> 98%) with almost complete conversion of Si. In this case, contact mass (CM) was obtained by mixing Si and CuCl powders followed by heating at 240 °C in an inert gas flow. Still unexplained are the results of experiments obtained by preheating a mixture of CuCl with silicon covered with a native oxide layer at 450 °C, showing that the reaction sites on the surface are formed even in the presence of an oxide layer, which does not prevent in this case the formation of intermetallic compound Cu3Si. The selectivity for trimethoxysilane decreases to 60%, the reaction rate decreases, and the silicon conversion is 70%.30, 72 The reaction order of silicon consumption at the reaction site and the activation energy of the reaction of silicon with methanol change when the mixture is heated at 220 °C and at 400 to 450 °C: first order and 88 kJ mol–1 to order 1.4 and 44 kJ mol–1, respectively. The authors believe that such a change in the kinetic parameters indicates that different temperatures of the mixture heating change the nature of reaction sites on the silicon surface.71 Perhaps, this is also due to the fact that at high temperature copper diffuses more actively into silicon in the areas of defects in the SiO2 layer.
Thus, the silicon grade and its pretreatment has a decisive impact on the main parameters of the direct synthesis reaction, such as reaction rate, selectivity for alkoxysilane formation and silicon conversion.
3.3. Method of contact mass pretreatment
Based on the analysis in the previous Section, it is obvious that the pretreatment of the contact mass is the most important factor significantly affecting the direct synthesis process. There are three approaches to CM pretreatment used in the industry:100
1) pressing of copper and silicon powders at pressure of 500 MPa at 1050 °C in a hydrogen atmosphere;
2) fusing silicon with copper at 1200 – 1400 °C in a hydrogen atmosphere;
3) chemical reaction of silicon with copper(i) chloride.
The latter method is the most convenient for laboratory research. This is probably why it is the most frequently encountered method in the DSA literature (Scheme 12).84
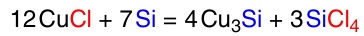
The CM pretreatment conditions significantly influence the subsequent reaction of direct synthesis. Suzuki and Ono 101 showed that in a reactor with a fixed bed of CM preheated at 260, 350, 400, 450, and 500 °C in a helium current for 3 h, the reaction rate with methanol at 240 °C raises significantly with increasing CM heating temperature, and the time dependence of the reaction rate passes through the maximum, then monotonically declines. The selectivity for trimethoxysilane reaches 98 – 99% for CM heated at 240 – 350 °C, whereas for CM heated at higher temperature the selectivity decreases to 77%. At the same time, an increase in the CM preheating temperature has a positive effect on the conversion of silicon: 98% in 3 h with preheating at 450 – 500 °C and 28% in 5 h at 350 °C.101 The same authors102 showed that the reaction at 270 °C with CM preheated at 350 °C in a helium current for 3 h achieved 98% selectivity for trimethoxysilane in 5 h with a 82% conversion of silicon.
In a follow-up study, slightly different data were presented (Fig. 7).72 Fig. 7 shows that at a reaction temperature of 240 °C and preheating temperature of 240 – 450 °C in a helium current for 1 h, the reaction started just after start of methanol feeding (see Fig. 7, line 1) when heated at 240 °C. At the same time, when the heating temperature was raised to 350 °C, an induction period appeared in the reaction (see Fig. 7, line 2). This decreased the reaction rate. An interesting effect was observed at the CM preheating temperature of 330 °C. Two maxima were observed on the DSA rate-time curve (see Fig. 7, line 3). The authors attribute this character of the rate of methoxysilane formation to the presence of two types of reaction sites in CM. Accordingly, at the heating temperature of 330 °C, both types of reaction centres are formed in CM. At T ≤ 330 °C, only active sites of the first type are formed, and at T ≥ 330 °C, only those of the second type are formed.
![[{"id":"RsvR8wMAPF","type":"paragraph","data":{"text":"Effect of the CM preheating temperature on the formation rate of methoxysilanes (HSi(OMe)<sub>3</sub> и Si(OMe)<sub>4</sub>) (solid line) and the selectivity (dashed line) for HSi(OMe)<sub>3</sub> with time. Preheating time is 1 h, Cu concentration is 10 wt.%. The reaction temperature is 240 °С.<sup>72</sup>"}}]](/storage/images/resized/POwU4Wcx2yA8lzWeoHLydL7ytUVRjExr9KGxczev_xl.webp)
Such a difference in the results of studies 101, 102 and study 72 is probably accounted for the different method of silicon preparation and its purity.
Chigondo et al.103 explored the reaction of the CM with ethanol in a packed bed flow tubular reactor. Thus, the formation rate of triethoxysilane is higher when the CM is preheated at 220 and 350 °С (up to 0.12 mol h–1). When heated above 500 °C, the rate of triethoxysilane formation decreases to the values obtained with the CM used without a preheating step (< 0.055 mol h–1).
The observed general trend that the selectivity for trimethoxysilane is higher when CM is heated at 200 – 240 °C than at 260 – 340 °C was confirmed by other researchers,104 with selectivity for tetramethoxysilane being higher than that for trimethoxysilane when CM is preheated at temperatures above 280 °C.
For the reaction with ethanol, a similar pattern was observed: at a preheating temperature of 500 °C for 2 h, the conversion of ethanol was 97%, the selectivity for tetraethoxysilane exceeded 92%, and for triethoxysilane it was 8%. When heating CM at 240 – 300 °C, triethoxysilane is formed with a selectivity of 84 – 97%.105 For other members of the homologous series, the following general trend is observed–silicon conversion and selectivity for trialkoxysilane formation decrease with increasing number of carbon atoms in the alcohol, which is attributed to a decrease in the reactivity of the corresponding alcohol. Thus, for 1-propanol and 1-butanol, the silicon conversion is 54 and 48%, respectively, and the selectivities are 91 and 88%, respectively.103
The duration of CM activation by preheating also has a noticeable effect on the DSA process. The optimal CM pre-activation time is 2.5 to 5 h, longer duration (15 and 20 h) decreases the rate of triethoxysilane formation.103 A high (98%) selectivity for trimethoxysilane in the reaction with CM heated at 330 °C in helium current for less than an hour was reported.72
The selectivity for trimethoxysilane decreases with longer CM activation, while the silicon conversion in the reaction at 240 °C for 5 h was 83, 91, 67, 75, 83 and 74% when CM was activated at 330 °C for 0, 0.17, 0.5, 1.0, 5.0 and 12.0 h, respectively.72 Fig. 8 shows plots of the rate of methoxysilane formation/selectivity for HSi(OMe)3 vs reaction time.
![[{"id":"P2XTrkbdoS","type":"paragraph","data":{"text":"Effect of the CM preheating time on the formation rate of methoxysilanes (HSi(OMe)<sub>3</sub> and Si(OMe)<sub>4</sub>) (solid line) and the selectivity (dashed line) for HSi(OMe)<sub>3</sub> with time. Preheating temperature is 330 °С, Cu concentration is 10 wt.%. The reaction temperature is 240 °С.<sup>72</sup>"}}]](/storage/images/resized/7FwHZitzk2frSRnJ4I2pcZSVMaQE6uz1dCKlnjjX_xl.webp)
Fig. 8 shows that the preheating time significantly affects the reaction course. When devoid of the CM preheating, the reaction starts immediately, and its rate rapidly decreases. At the same time, increasing the CM preheating time up to half an hour leads to the appearance of the induction period. The most interesting is the example for preheating for 1 h (see Fig. 8, line 4). The graph shows two maxima on the rate curve of methoxysilane formation. As with the effect of heating temperature, the authors attribute this to the formation of two types of active sites on the CM. Their ratio depends on the CM preheating time. Thus, at heating for up to 0.5 h, only the first type sites are formed, while at 1 h of heating, the second type sites appear in the CM. Heating for more than 1 h produces the second type sites in the CM.
The probable reason for the difference in DSA results at different CM preheating times is the formation of an active intermetallide Cu3Si. As mentioned above, in a number of studies on both DSC and DSA, this alloy is the active site of the direct synthesis reaction. Wang et al.104 showed that when preheating CM directly in the reactor prior to the direct synthesis, Cu3Si phase is formed only at T = 280 °C and above (Fig. 9).
![[{"id":"_Lqxe28B-a","type":"paragraph","data":{"text":"X-ray diffraction patterns of contact masses at different preheating temperatures.<sup>104</sup>"}}]](/storage/images/resized/X728qLRq1Pnv4uVduqnZ1E2v32JWtDVDu2WHsJyk_xl.webp)
Probably, this changes the rate of the reaction between silicon and methanol described in Refs 71, 72. As mentioned above, when pretreating CM at 450 °C, the reaction rate increased significantly, and the selectivity for trimethoxysilane decreased from 98 to 60%. The ability of copper silicides to weaken the adjacent Cu – Cu and Si – Si bonds has been known for quite a long time. This fact is detailed in the publication.106 For a long time, the h-phase (Cu3Si) was considered to be the main source of activity,107 however, over time it was understood that the process is much more complex and there are other silicides on the surface of the reacting silicon species, the presence of which can also be linked to the contact mass activity.70 Therefore, this led to an interest in looking more deeply into silicon-copper reactions.
Studies of the reaction of silicon and copper carried out both in bulk 108 and in thin films 109 showed that the direct reaction of silicon and copper affords Cu3Si. The temperature of this process depends on the initial state of the silicon and copper, as well as on the method of their mixing. In general, such systems are characterized by the formation of different silicon-copper phases under different (sometimes even insignificantly different) conditions. In addition to the initial ratio of reagents and annealing temperature, such factors as the nature of the substrate material, the stacking faults in the film 110 and even the presence of an applied magnetic field 111 may be important.
For example, when using extremely amorphous, as-deposited silicon, the temperature of Cu3Si formation is ~30 °C, while when using crystalline silicon Si(100) or Si(111), the reaction temperature increases to 200 °C.106, 109, 112 This difference may be due either to the higher activity of such destructured silicon, or to the complete absence of the surface oxide film, which can be found even on the purest samples of crystalline silicon. Stolt et al.112 are inclined to the second version.
However, there are other, more copper-enriched silicides. A phase diagram was constructed for the Cu – Si system, in which the temperatures and atomic ratios of copper and silicon were determined at which silicides of different compositions exist (Fig. 10).113
![[{"id":"Ww40UtvUUn","type":"paragraph","data":{"text":"Cu – Si phase diagram.<sup>113</sup>"}}]](/storage/images/resized/S9xK9m3j4VpjlA7RRTg9MXH5T49Mg3YlrynUer9r_xl.webp)
The phase diagram shown in Fig. 10 has a rather complex appearance. This is partly due to the differences in the relative solubility of copper and silicon. The solubility of copper in silicon is extremely low (0.5 at.% at 200 °C), while the solubility of silicon in copper can reach 15 at.%. The diffusion of silicon into copper is very sluggish, in contrast to that of copper into silicon.114
Stolt et al.112 explored the features of the formation of these phases. The authors studied the transformations in silicon layers ~ 100 nm thick, onto which copper was deposited in the amount necessary to obtain silicides of a certain composition. With this method of deposition of copper onto silicon, at 25 at.% of silicon, the reaction between silicon and copper affording Cu3Si occurs at 150 – 200 °C. At silicon content < 25 at.% and on reaching 300 °C, other phases are formed and begin to react with each other. As a result, the following conclusions were drawn:
— even at low silicon content (< 25 at.% required to produce Cu3Si), Cu3Si is the first compound formed (h'', see Fig. 10);
— at excess copper, the h''-phase reacts with the former to generate the g-phase (Cu5Si), which contains 17 – 18 at.% silicon;
— at ~20 at.% silicon, ε-phase (Cu15Si4) may appear, which is formed by the reaction of h''- and g-phases;
— the k-phase (Cu7Si) is derived from the g-phase only at temperatures > 555 °C and at 10 – 17 at.% silicon (see Fig. 10). Thus, the above described transformations can be expressed by the following scheme (Fig. 11).
![[{"id":"llpcVqZl7K","type":"paragraph","data":{"text":" Sequence of transformations during the solid-phase reaction of silicon with copper.<sup>112</sup>"}}]](/storage/images/resized/26WD6nrhmuP8X366QIvtGnJhJUC31KSmrUbQWPhA_xl.webp)
Later, the possibility of obtaining nanoporous silicide films by annealing and subsequent etching of the Si (50 nm) – Cu (200 nm) – Si (50 nm) trilayer system was studied, where the authors suggest a different sequence of transformations (Fig. 12).115
![[{"id":"BX7Jku4RBi","type":"paragraph","data":{"text":"An alternative sequence of transformations during the solid-phase reaction of silicon with copper.<sup>115</sup>"}}]](/storage/images/resized/srqHl3M5y6WzFkzCLnCphOQOE36aHDXPGDm7TvJK_xl.webp)
The right boundaries of the temperature ranges correspond to the temperature of the beginning of the subsequent phase formation rather than to the maximum conversion of the previous phase into the subsequent one. In other words, it corresponds to the accumulation of the previous phase in an amount sufficient for it to become a matrix for the formation of the subsequent phase, i.e., given 16% Si content and 84% Cu, the achievement a temperature of 450 °C does not mean that all Cu3Si was converted to Cu5Si. It means that at 450 °C and above, the Cu7Si phase can already start to form.
It should be noted that the data of Stolt et al.112 and Buchin et al.115 are not consistent. The reasons have already been discussed above: the studied systems are extremely sensitive to the reaction conditions. Some of the conditions are given above, and Ibrahim et al.114 additionally showed the dependence of the result on the sequence of layer deposition (Fig. 13a). Thus, it was found that when silicon is deposited onto copper, a sharp interface is formed (Fig. 13c) devoid of a surface concentration gradient. At such an interface, the appearance of Cu3Si is difficult and starts only at temperatures above 200 °C. When copper is deposited onto the silicon layer, a concentration gradient zone is formed, in which the nucleation of Cu3Si occurs much easier (Fig. 13b).
![[{"id":"UOOukDsln0","type":"paragraph","data":{"text":"SEM images: the effect of deposition order on the interface morphology.<sup>114</sup> General picture of layers (<i>a</i>): magnification of the corresponding zone showing the result of copper deposition onto silicon (formation of intermetallide) (<i>b</i>). The sequence was reversed in the c zone, therefore, no intermetallides were formed"}}]](/storage/images/resized/J8qxeRcyLvlZy7JfmLKT71eUIlxwjt5zHFSs9zEi_xl.webp)
Another illustrative example of the dependence of the result of the interaction of various silicides on the conditions was provided by Klementová et al.116 The reduction products of various silicon-containing compounds (SiH4, EtSiH3, BuSiH3) on Cu/Cu5Si precursors at 500 °C were studied both with and without added hydrogen. The authors found that depending on the process conditions, either h'-Cu3Si nanorods, h''-Cu3Si nanoribbons, or γ-Cu5Si in the form of nanowires were formed.
K.Su et al.117 obtained nearly pure Cu3Si and used it in the synthesis of trichlorosilane (TCS).
The intermetallide was prepared by mechanical grinding of finely dispersed silicon and copper in a mortar followed by calcination in an argon atmosphere at 1050 °C. Based on the results of photoelectron spectroscopy and X-ray phase analysis, it was concluded that Cu3Si is formed on the surface of silicon particles and converts into Cu6.69Si, which, according to the authors, is the active surface site of the reaction (Scheme 13).
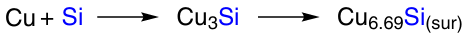
As proof, the authors cite the data of physico-chemical study of contact masses. In the samples taken at the moments of the highest CM activity (high silicon conversion and selectivity for TCS), the Cu6.69Si phase was found in a smal amount.
Another work of the same authors 118 is devoted to the direct synthesis of trimethoxysilane from methanol and silicon. In this case, a catalyst based on a mixture of CuCl and Cu2O was used. Comparing the contact masses prepared under different conditions using this catalyst, the authors deduced a slightly different chain of reactions leading to the formation of the Cu15Si4 active site (Scheme 14).
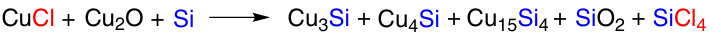
From our point of view, the cause-effect structures of the last two papers are not flawless. Based on the above, the more copper-rich phases are formed from Cu3Si, i.e., it is a kind of matrix in which other phases can develop. During the direct synthesis, elemental copper is released, which, when entering Cu3Si-rich regions, is able to react with the latter to form Cu-rich silicides. However, this does not mean that these copper-rich silicides should be responsible for the higher CM activity for a particular product. They may even have a lower activity, as was shown in a study 119, which will be discussed in detail in the next Section. This can also explain the authors’ noted decrease in the amount of free copper released in the reaction.
Thus, the conditions of CM preparation are crucial for the subsequent direct synthesis of alkoxysilanes.
The reaction of silicon with copper under different conditions can give silicides of different compositions. This additionally complicates the determination of the chemical composition and nature of the active sites of the reaction, although Cu3Si should still be considered as the main source of activity.
3.4. Effect of the type and concentration of the catalyst
The most commonly used catalyst for the direct synthesis of alkoxysilanes is copper(i) chloride. It was found 105, 119, 120 that its catalytic performance is affected by the particle size, crystallinity, and method of synthesis. Zhang et al.119 compared commercially available CuCl and CuCl freshly prepared by laboratory method. As a result, it was found that when using laboratory, structured (according to SEM data), finely dispersed, nanosized CuCl obtained by the wet method 121 (Scheme 15), the rate of the reaction between Si and EtOH is higher than when using commercial CuCl. The latter is obtained in industry by the dry method by burning copper in a chlorine atmosphere. The optimum temperature to produce CuCl by the wet method is 45 °C, and the obtained CuCl has particle size less than 80 nm, larger specific surface area, larger total and average pore size than CuCl obtained by the dry method.122 With 5 wt.% CuCl (obtained by the wet method) in CM, high selectivity (98%) for triethoxysilane was achieved, and silicon conversion was 95%. Increasing the amount of copper chloride up to 10% decreased the selectivity for triethoxysilane. The detection of CuCl on the surface of silicon particles after the reaction indicates that high concentrations of CuCl inhibits the formation of copper during the synthesis process, being located on the surface of silicon particles.
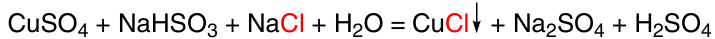
When carrying out the reaction in a fixed bed reactor, the selectivity for triethoxysilane decreased from ~ 100 to 94% on reducing the amount of catalyst from 5 to 2.5%. The silicon conversion was 83, 90, 95 and 93% at CuCl contents of 2.5, 5, 10 and 20%, respectively.123 The same trend – decrease in selectivity for trialkoxysilane formation and decrease in silicon conversion when reducing the amount of catalyst in the CM from 10 to 1% is observed for methanol.84
The literature presents contradictory data on this reaction rate. When increasing the amount of the catalyst, the number of active sites formed on the silicon surface increases and, consequently, the reaction rate increases. As the catalyst concentration increases from 5 to 15%, an increase in the CM surface area and an increase in the amount of methanol adsorbed on it were observed.124 As the amount of catalyst increases from 0.5 to 12%, a gradual increase in the reaction rate maximum was observed.93, 123
In other studies, the rate of triethoxysilane formation in a fixed-bed reactor for different CuCl contents (2.5, 5, 10 and 15%) was found to differ slightly, growing gradually over the course of 6 h.103
With 10% CuCl, an induction period in the direct synthesis was observed (Fig. 14), the occurrence of which was attributed to successive reactions during the reaction of CuCl with Si (Scheme 16).119
![[{"id":"U3LbTOeJp1","type":"paragraph","data":{"text":"Effect of CuCl concentration on the direct synthesis rate.<sup>119</sup>"}}]](/storage/images/resized/EeSeTRAQPbHJJwUfLHwaPaKAm72rDkPHTICWOgaX_xl.webp)

In the presence of excess CuCl, the η-phase Cu3Si is transformed into the less active Cu5Si, and the time of formation of the active site of the reaction increases. It should be noted that this explanation agrees with our interpretation (see the end of Section 3.3.) of the processes considered in works.117, 118
Cu(0) is released due to the deactivation of the active sites,93 as shown in Fig. 15.
![[{"id":"4MOVTy1Avp","type":"paragraph","data":{"text":"Scheme for the formation of inactive Cu(0).<sup>93</sup>"}}]](/storage/images/resized/CpPVHlirzK2av9UQgLp4r1LMfHksEhenbL2frS3L_xl.webp)
The use of CuCl as a catalyst entails the formation of chlorine-containing products that pollute the environment. To avoid their formation, other copper compounds were explored as catalysts for the direct process. Thus, it is shown that the catalytic activity decreases in the series of CuCl, Cu(OH)2, CuO, Cu2O, CuSO4, which affects the reaction rate (Fig. 16).103
![[{"id":"a3Ja52jDSp","type":"paragraph","data":{"text":"Comparison of rates of reactions catalyzed by different copper compounds.<sup>103</sup>"}}]](/storage/images/resized/Cwy5jWDw8wPXhx5Wy39KvJxrKoWt1UREaWOUpfkU_xl.webp)
When testing a number of copper(II) compounds, it was found that in the series of oxalate, formate, phthalate, oxide and acetate, the silicon conversion is 1, 11, 23, 50 and 82%, respectively, with selectivity for trimethoxysilane of 0, 9, 21, 57 and 8%, respectively. The best result was achieved for Cu2O with silicon conversion of 82% and selectivity of 19%. In comparison, when CuCl was used under these conditions, the conversion was 88% and the selectivity for trimethoxysilane was 98%. Other compounds such as copper(I) thiocyanate, copper(II) sulfate, copper(II) hydroxocarbonate, copper(ii) nitrate did not show any catalytic activity.125
In addition to the above examples, the use of a significant number of copper-based catalysts is reported in the patent literature.66, 67, 126, 127
Thus, the choice of the catalyst and its content in the contact mass significantly affects the course of the direct synthesis of alkoxysilanes. The most common catalyst is copper(I) chloride. However, different sources of CuCl, such as commercial or produced in the laboratory, sometimes have different effects on the reaction of silicon with alcohol. These data need to be taken into account when comparing other factors, such as temperature or process method, with each other.
3.5. Use of different cocatalysts. Presence of promoters in contact masses
State-of-the-art contact masses for the production of organochlorosilanes contain promoters. Two of them, Zn and Sn, are always present in CM. The mechanism of the effect of promoters on the DSC is not less complicated than the main pathway of the direct synthesis. The influence of promoter additives on DSC was highlighted in a number of publications 91, 128, 129 and is also detailed in the review.25 In the present review, we will only address the crucial features of the effect of promoters on DSC, which we believe may be a key to understanding the pathways for the formation of methylalkoxysilanes in the reaction of CM with an organic precursor.
The use of promoters in DSA is not as well covered in the literature as for DSC. Kareem et al.124 found that the addition of Al, Sn, ZnCl2 or CaCl2 has a beneficial impact on the main parameters of the direct synthesis process. The addition efficiency decreases from left to right in the above series. The authors of the patent 121 also achieved good results when adding aluminium to CM. Apparently, the role of additives of various metals and their compounds in CMs is quite similar to the above described effect of impurities in technical silicon. Nevertheless, comparison of the influence of the additives in this series is not quite correct. This is due to the different mechanism of action of different elements on the direct synthesis. For example, aluminium and calcium additives in CMs improve the selectivity of DSC for alkyl-containing chlorosilanes.100 Also, the mass ratio of promoters in CMs is of great importance. For example, zinc and aluminium are added in amounts of 100 – 5000 and 500 – 4000 ppm, respectively. The addition of tin above a certain amount (from 100 to several thousand ppm depending on the technology and method of the CM preparation) leads to CM poisoning.130 More precise data for optimal additive loadings are usually a commercial secret.
We will focus on the most important, from our point of view, promoters, namely, Zn and Sn. In the review,128 these promoters are referred to different types according to the principle of their action in the direct synthesis. Zinc is added in relatively large amounts (0.05 – 1 wt.%). Its addition reduces the activation energy of the reaction of CM and methyl chloride. At the same time, tin is added to CM in the amount of 300 – 1000 ppm. Its addition does not decrease the activation energy of DSC. Tin concentrates on CM at the reaction sites (active centres) of DSC. It is suggested that Sn migrates to grain boundaries in the Cu – Si phase and causes the formation of cracks at the interface, which leads to the appearance of additional active reaction sites.128 In this way, tin was detected only on the CM surface,131 i.e., it does not diffuse into bulk silicon. Accordingly, in large quantities, tin can mask the active sites of the reaction, which leads to CM poisoning. Another remarkable feature of tin is its ability to stabilize methyl radicals. It was shown that this decreases selectivity for SiH-containing products, which are formed in the DSC process along with methylchlorosilanes.132 Of greatest interest is the combined effect of promoters on the DSC reaction. Banholzer and Burrell 131 managed to demonstrate the synergistic effect of Zn and Sn in the reaction of CM and methyl chloride. The experimental results showed that the presence of Zn in CM promotes the formation of Cu3Si intermetallide. At the same time, Sn promotes the consumption of Cu3Si. Thus, when zinc and tin are used together (which is actually the case in industry), there is a balance between the formation and consumption of Cu3Si. This, in turn, favours the maintenance of high CM activity throughout the synthesis and the completion of the catalytic cycle.
3.6. Coreagents (promoters) introduced with the reagent stream during the reaction process
Besides adding promoters to CMs prior to reaction, it is also possible to introduce various organic compounds during direct synthesis. While the first option is more used for DSC, both activation options are being explored in studies on DSA.
Organohalides have an interesting effect on the DSA.93, 133, 134 Thus, preheating of CM in a MeCl stream increases the selectivity for trimethoxysilane from 56 to 84% compared to CM preheated without MeCl addition, while the silicon conversion also increases from 66 to 84%.133 The introduction of alkyl halides directly during the reaction has an extremely favourable effect on the selectivity for trialkoxysilane, increasing it to almost 100%. Okamoto et al.84 found that this effect is caused by the poisoning of Cu(0), which is the catalyst for the conversion of trialkoxysilane into tetraalkoxysilane (see Scheme 3, reaction 2) by an organohalide. Similar results were achieved with thiophene.82
Hydrofluoric acid introduced in a mixture with ethanol throughout the reaction has a remarkable effect on the direct synthesis. In particular, it was shown that under its influence the inactive zero-valent copper returns to the catalytic cycle (Fig. 17).93
![[{"id":"tp9KgHnJ5l","type":"paragraph","data":{"text":"Regeneration of deactivated copper by fluoride anion.<sup>93</sup>"}}]](/storage/images/resized/equOAFqufDAkwlgbafude4aWsavEGEuppXjzXIll_xl.webp)
Zero-valent copper is formed through deactivation of the active sites. This is the way to avoid reduction of the number of active sites in the contact mass, which noticeably improves the reaction rate and silicon conversion.
It appears from the foregoing that the introduction of various promoting additives can significantly affect the main parameters of the direct synthesis of alkoxysilanes. All additives can be divided into two categories: those introduced into the contact mass prior to the reaction and those introduced directly during the process. The role of the former is similar to that of impurities in technical silicon. The latter influence the catalyst by poisoning deactivated copper or returning it to the catalytic cycle.
The question of promoter additives may be a key issue for the preparation of organoalkoxysilanes. As will be shown below, promoter additives play a crucial role in the activation of the organic precursor necessary for the formation of the Si – C bond.
3.7. Technological features of the process
The remaining two points are the top of the ‘pyramid’ of factors affecting the process of direct synthesis of alkoxysilanes. That might account for the controversial situation around this issue, described below.
The methods of direct synthesis of alkoxysilanes can be divided into two main types: liquid-phase and gas-phase. Most of the studies reviewed above belong to the second type. This is primarily due to the fact that researchers prefer this particular method. For example, only one-sixth of the publications found in peer-reviewed journals describe the liquid-phase variant of direct synthesis. At the same time, the opposite situation is observed in the patent literature aimed at industrial applications. Among 25 found patents,66 – 68, 121, 126, 127, 133, 135 – 152 twenty four fall on the liquid-phase method, with seven of which providing the process flow diagram. For example, the US patent 67 uses a capacitive reactor (Fig. 18).
![[{"id":"9mCnbk2dxc","type":"paragraph","data":{"text":"Process flow diagram of liquid-phase process for direct synthesis of alkoxysilanes.67 Alcohol is supplied to the vessel reactor 5, equipped with a mechanical stirrer 7, by pump 2 to the evaporator 4, after which the alcohol enters the lower part of the reactor 5 containing contact mass dispersed in a high-boiling solvent in the presence of surfactants that control foaming. The reaction products, together with the unreacted alcohol, enter the distillation column 16. The alcohol separated from the mixture is returned to the reaction. 3 is flow meter, 6 is hopper, 8 is temperature controller, 9 is thermocouple, 10 are internal baffles, 11 are spargers, 12 is pressure gauge, 13 is pressure release safety valve, 14 is entrainment separator, 15 is valve, 17 is reboiler, 18 is reflux condenser, 19 are liquid reaction products, 20 is pump, 21 is flow of low-boiling by-products, 22 is portion of the liquid overhead stream, 23 is unreacted alcohol, 24 is vent gas stream."}}]](/storage/images/resized/Oqs15QKSSka5TzsGaNHBKEtxT1suZ1E5A7d6kMvv_xl.webp)
There are no fundamental difference between the described process flow diagram and the schemes presented in other patents. In general, solvents of Therminol 59, Therminol 60, Therminol 66, Dowtherm HT, Marlotherm S, Marlotherm L trademarks, which are alkylaromatic compounds, are used as high-boiling solvents. It should be noted that the presence of the solvent significantly complicates the process of obtaining alkoxysilanes, since following the direct synthesis, its regeneration with separation from silicate by-products is required.136, 153 The need to introduce surfactants to control foaming is another challenge. All these issues are absent when using the gas-phase method of carrying out the reaction. Findings of the studies describing this approach are not inferior, and often even superior to those using liquid-phase technology. Nevertheless, these methods cannot be compared correctly. The reason for this, as already mentioned, is a lot of factors at the base of the ‘pyramid’. A change in one factor involves a change in the whole process. It follows that a comparison of the two main ways of carrying out the direct synthesis of alkoxysilanes requires the other factors listed above to remain unchanged. In addition, some features of the described processes simply have not received sufficient attention. For example, the direct synthesis of organochlorosilanes is known to be an exothermic reaction (ΔH = –37.7 kcal mol–1 per one MeCl molecule).128 This is associated with great difficulties in the design of fluidized bed reactors in which DSC is carried out. When designing such an apparatus, heat removal must be ensured, and heat exchangers must not interfere with the hydrodynamics of the process. The DSA reaction is also exothermic, but calculated thermodynamic values are practically absent in the literature. Based on data found in a single source,154 the reaction of silicon with alcohols also has a significant heat effect (ΔH = –58.81 kcal mol–1).154
Accordingly, when the reaction is carried out in a heat carrier fluid, this factor is insignificant because the heat is removed by this fluid. At the same time, when the reaction is carried out in a fixed bed layer, the thermal effect can be decisive. The self-heating of the CM layer can provoke side processes such as pyrolysis of the organic precursor and condensation of alkoxysilanes. Also, there are problems with the reaction mode as well as the equipment aging when scaling up.
Probably, that is why in the patent literature, which is closer to the practical application of these processes, the liquid-phase method of trialkoxysilane synthesis is favoured. Indeed, in the liquid-phase approach, heat removal is provided by the dispersion medium, and poor performance of the apparatus is levelled by the relatively small demand for trialkoxysilane.
Temnikov et al.155 compared liquid-phase and gas-phase approaches to the DSA process. Other conditions being equal, the gas-phase process was characterized by a higher reaction rate, but the liquid-phase mode showed higher selectivity for trialkoxysilane and was characterized by a higher silicon conversion. Moreover, the liquid-phase process is preferable for scaling, since the demand for tri- and tetraalkoxysilanes is not comparable to the demand for dimethyldichlorosilane. Accordingly, it makes no sense to develop more intensive processes. However, the situation will change when implementing the direct synthesis of dimethyldialkoxysilane. Therefore, the path of development of the liquid-phase method with respect to the synthesis of alkoxy- and organoalkoxysilanes is most likely a dead end.
The development of the liquid-phase mode of the direct synthesis can be considered completed, but unpromising for the synthesis of dialkyldialkoxysilanes. Consequently, it is important to trace, how the disadvantages of the gas-phase method can be neutralized. We have already mentioned above that the introduction of alkyl chlorides improves the selectivity for trialkoxysilane. In addition, the proper apparatus design allows to control the intensity of DSA,103 but no examples of scaling-up the gas-phase method were found in the literature. Of the reviewed examples, only one paper used a fluidized bed reactor.93 The CM loading was ca. 60 g, but the authors did not provide technological aspects of the reaction. At the same time, in most of the works considered in the review, the CM loading ranges from hundreds of milligrams to a few grams.
3.8. Process temperature
Temperature is a critical factor affecting the process of direct synthesis of alkoxysilanes. However, it is rather difficult to separate the effect of temperature from the influence of other factors. For example, it can be assumed that the difference in the type of silicon and copper chloride used, as well as their pretreatment conditions, can make it difficult to accurately determine the influence of temperature.
This assumption is supported by the two studies described below, carrying out the experiment in a solvent-free manner (gas-phase variant). In the first study,102 it was shown that the reaction rate increases markedly with increasing temperature from 473 to 543 K (Fig. 19).
![[{"id":"AMowQeNSLL","type":"paragraph","data":{"text":"The dependence of the rate of for mation of methoxysilanes on temperature.<sup>102</sup>"}}]](/storage/images/resized/bsb66Gg6JmsQ6iYoKdnRPlUCLmbLw7nwgRohnZfn_xl.webp)
The same happens with silicon conversion (Fig. 20), which reaches its maximum value (~91%) at 543 K. It is worth noting that the selectivity for trimethoxysilane remains almost 100% throughout the process.
![[{"id":"9wgJcCUBKs","type":"paragraph","data":{"text":"The dependence of silicon conversion (<i>a</i>) and selectivity for trimethoxysilane (<i>b</i>) on temperature (K).<sup>102</sup>"}}]](/storage/images/resized/AyHmfK0iEiSOXscBIYBAS4veLo6KWUFvpkeu96y0_xl.webp)
The data for the second study 124 are given in Table 1.

In the whole temperature range, the selectivity for trimethoxysilane remains at 60%. At the same time, the content of silicon-containing products increases while changing process temperature from 250 to 350 °C. Apparently, the conclusions are inconsistent since other circumstances affecting the process were not considered.
The results of the only work 156 using the liquid-phase variant, in which the influence of temperature was considered, are presented in Table 2.
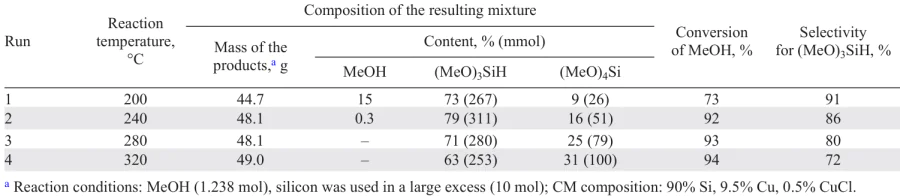
Table 2 shows that elevated temperature leads to an increase in the yield of silicon-containing products, while the selectivity for trimethoxysilane decreases. Most of the studies on the liquid-phase mode of the process are patents, so the data on the effect of temperature on direct synthesis are very conditional and are intended to cover the widest range in order to protect patent rights.
The influence of temperature on DSA was discussed in a publication,157 in which the synthesis was carried out in a high-pressure mechanochemical reactor. Fig. 21 shows the curves describing the dependence of hydrogen pressure (proportional to its amount) on time.
![[{"id":"I85C-9XKK5","type":"paragraph","data":{"text":"Time-dependent pressure curves of released hydrogen for DSA in a mechanochemical autoclave at different temperatures.<sup>157</sup>"}}]](/storage/images/resized/hWFAo67CA1Ocp0vASe3XjRulX2PJFy3eDSg9P1qH_xl.webp)
From the above curves it can be seen that at 150 °C the reaction proceeds in a stationary mode, while at 200 and 250 °C S-shaped curves are observed, indicating a sharp increase in the reaction rate after a certain period. By examining contact masses sampled at various stages of the process, the authors showed that the reaction can be carried out to almost 100% silicon conversion, with the bulk of the silicon reacting during the first 10 min after heating the reactor to 190 °C. At 150 °C, the silicon conversion was 80%. It is shown that the formation of Cu3Si in CM is observed only at 250 °C. The described method allows the reaction to be carried out both with liquid methanol and with gaseous and even supercritical methanol.
From all of the above, it can be concluded that, despite significant advances in the direct synthesis of tri- and tetraalkoxysilanes, the research on this method is far from describing the complete reaction mechanism and choosing the optimal conditions for its implementation. To date, it is impossible to choose even the optimal mode of the process (liquid-phase or gas-phase).
3.9. Methods of physical activation of DSA
Besides the use of various promoters at the CM pretreatment stage or the addition of various coreagents to the organic reagent, the effect of various physical effects such as radiation, mechanoactivation, and elevated pressure on the main reaction parameters of the direct synthesis of alkoxysilanes was explored.
3.9.1. Use of radiation
There are examples of the use of UV and microwave radiation in the DSA reaction. The reaction of silicon with ethanol at 190 °C in a liquid-phase reactor in Marlotherm SH medium under microwave irradiation (radiator power 335 – 1000 W) achieved the selectivity for triethoxysilane close to 100%, with silicon conversion up to 95%.145 However, the reaction must be carried out in the medium of a polar heat-transfer fluid interacting with the microwave field. Otherwise, the polarity of the medium would not be sufficient to heat the reactor to the required temperature. Therefore, it remains unclear how the authors managed to achieve the desired temperature (190 °C) in the reactor using non-polar Marlotherm SH.
An interesting example of the use of UV radiation is given in a study.155 The synthesis was carried out using the gas-phase approach to DSA using methanol. The formation of oligomeric siloxanes in addition to basic methoxysilanes was observed. The authors compared the 1H NMR spectra of these oligomers when carrying out DSA with and without UV irradiation. The results showed that under UV irradiation, the resulting oligomeric products contain ethyl groups (Fig. 22, spectrum 2), in contrast to the same products obtained in DSA devoid of UV irradiation (Fig. 22, spectrum 1).
![[{"id":"vWgJs9_1sF","type":"paragraph","data":{"text":"<sup>1</sup>Н NMR spectrum of oligomers formed in the direct synthesis without (<i>1</i>) and with (<i>2</i>) exposure to UV irradiation.<sup>155</sup>"}}]](/storage/images/resized/YjchFFdBIUzUTgveajelqwHi5VyXQrl1qa80eqs0_xl.webp)
The DSA process itself had a more stationary character under UV irradiation.
Nevertheless, to date, it is impossible to speak with certainty about the essential influence of electromagnetic radiation on the DSA process, which casts doubt on the prospects of its use for the direct synthesis of organoalkoxysilanes.
3.9.2. Mechanochemical approach
In contrast to electromagnetic radiation, mechanochemistry has proven to be a powerful tool to influence the direct synthesis process. Indeed, many examples of DSC show that the silicon dispersity has a significant effect on all the characteristics of the process. The larger the surface area of the starting silicon, the more intense and selective the DSC process. Obviously, fine grinding during the reaction itself will have a favourable effect on the characteristics of direct synthesis.
This thesis was successfully confirmed by studies,158 – 160 in which DSA was carried out in a mechanochemical reactor (Fig. 23).
![[{"id":"6pcA75vZbk","type":"paragraph","data":{"text":"Schematic diagram of the semi-continuous mechano-chemical synthesis unit.<sup>160</sup> 1 is reactor (1 L), 2 is electric heater, 3 is thermocouple, 4 is thermoregulator, 5 is inlet process connection, 6 is outlet process connection, 7, 8 are milling bodies d = 5, 10 mm, (2/3 of the reactor volume), 9 is vibration drive, 10 is dosage pump, 11 is descending condenser, 12 is receiver, 13 is sampler."}}]](/storage/images/resized/NR8MbL78yOZZPmfpBOvkIpATdNJTIyzaBPPpMuJl_xl.webp)
Alcohol was fed into reactor 1 heated to the required temperature. Inside the reactor, silicon was continuously ground by milling bodies of different dispersity 7, 8 due to vibration. The DSA products were discharged from the reactor through the outlet process connection 6. The milling bodies were brass balls. The last fact is especially important because brass mainly consists of zinc and copper – the main promoter and catalyst of direct synthesis. This allowed the reaction to be carried out without special introduction of copper, since the necessary amount of this metal was supplied to the reaction by abrasion of the brass balls. Large silicon particles (1 – 2 mm) covered with a thin oxide film were used in the reaction. As mentioned above, the oxide layer inhibits the reaction of silicon with alcohol. However, in the case of mechanoactivation, the initial specific surface area of silicon is low. In the case of mechanoactivation, the silicon is milled and its surface is constantly renewed, thus avoiding the negative influence of the SiO2 layer. With this gas-phase version of DSA, almost complete conversion of silicon is achieved. The selectivity is fully directed towards tetraalkoxysilane. An additional advantage of the method is the elimination of a number of preparation stages: fine grinding of silicon, its fractionation and purification from surface SiO2 are not required. The CM preparation stage is also not necessary.
3.9.3. High pressure
The study of the effect of high (i.e., exceeding atmospheric) pressure on the efficiency of direct synthesis of alkoxysilanes is due to the currently accepted mechanism of this reaction, which involves the interaction of silylene silicon sites on the surface of CMs. Considering the heterogeneity of this process, it is logical to assume that an increase in pressure will increase the concentration of the gaseous precursor on the CM surface. This should at least improve the reaction rate. This assumption is confirmed in the studies.65, 83 An increase in reaction rate with increasing pressure was also noted in the publication.161
Since the first stage of the reaction is the nucleophilic interaction of a lone pair of electrons of the O atom involved in the alcohol reaction with the free p-orbital of the silylene-like particle on the silicon surface with the initial formation of the intermediate complex Si∙∙∙O< , it is logical to consider the known results of studies of monomeric silylene SiH2 with oxygen-containing substrates in the gas phase. Thus, quantum chemical calculations of the interaction of SiH2 with alcohols showed (Scheme 17, reaction 1) that for the intermediate exothermically formed complex H2Si∙∙∙O(H)R in the case of methanol (R = Me, ΔE = –75.6 kJ mol–1) and ethanol (R = Et, ΔE = – 78.6 kJ mol–1), the formation of alkoxysilane H3Si – OR corresponding to the 1,2-hydrogen shift direction, passing through the energy barrier of 13 and 9 kJ mol–1, with exothermities of ΔE = –304 kJ mol–1 and ΔE = –306 kJ mol–1, respectively, is favoured.159 In the case of water (see Scheme 17, reaction 2), both the 1,2-hydrogen shift affording silanol (ΔE = –53.3 kJ mol–1) and hydrogen release affording hydroxysilylene (ΔE = –112 kJ mol–1) are energetically equally probable for the exothermically formed intermediate complex H2Si∙∙∙OH2 (ΔE = –53.3 kJ mol–1).
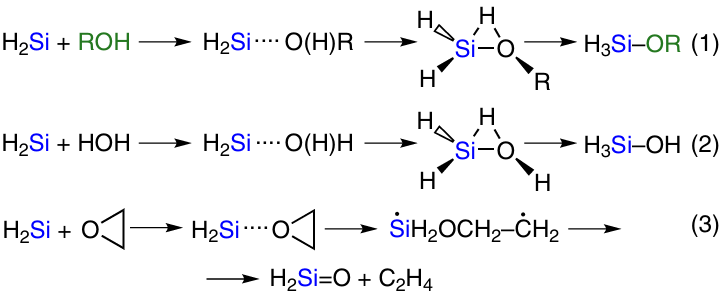
For the SiH2 + Me2O system, which has a relatively deep energy well for the initially generated associative complex H2Si∙∙∙O(Me)Me (ΔE = –84 kJ mol–1), the energy barrier for the migration of the Me group from O to Si is quite high (ΔE = – 193.3 kJ mol–1) and therefore, this process is unlikely.162 Experimental studies of reactions of silylene with dimethyl ether 163 and cyclic ethers, oxirane, oxetane, and tetrahydrofuran in the gas phase 164 showed that the rate constant of the reaction affording the Lewis acid — Lewis base complex depends on pressure and temperature, which agrees with quantum chemical calculations. The products detected in the reaction, however, are not the result of radical migration from O to Si. The reaction rate constant increases with increasing pressure. In the reaction of SiH2 with oxirane (see Scheme 17, reaction 3), the formation of ethylene was detected, which agrees with calculations showing that the reaction with the formation of H2Si = O and C2H4 is highly exothermic (ΔE = –61.8 kcal mol–1).165
An experimental study of the kinetics of reactions of silylenes (SiMe2, SiPh2, SiMes2) with alcohols revealed that the O – H insertion of dialkyl- and diarylsilylenes into alcohols proceeds to give first a silylene-alcohol complex (Lewis acid – Lewis base), which converts into alkoxysilane with proton transfer from O to Si catalyzed by the second alcohol molecule (Fig. 24a).166 The reaction of Ph2Si with the simplest alkoxysilane MeOSiMe3 produces in 54% yield the insertion product into the Me3Si – O bond, which is formed by a two-step mechanism via the initial formation of the complex Ph2Si∙∙∙OMeSiMe3 followed by the SiMe3 group migration (see Fig. 24b). The authors did not observed the insertion of a silicon atom into the C – O bond.
![[{"id":"ndBB9HZmGh","type":"paragraph","data":{"text":"Conversion of the initially formed Ph2Si∙∙∙OHR complex into alkoxysilane catalyzed by the second alcohol molecule (<i>а</i>), and the product of the reaction between photolytically generated Ph<sub>2</sub>Si and MeOSiMe<sub>3</sub> (<i>b</i>)"}}]](/storage/images/resized/d451CWAyyqnKOFzb9mH120gSko7fAsQyQGxPR26L_xl.webp)
Thus, experimental studies and theoretical calculations of reactions of silylenes with oxygen-containing substrates demonstrate the barrier-free formation of the associative complex and pressure dependence of the rate constant of the subsequent reaction.
Taking this into account and assuming that the mechanism of the reaction of silicon with alcohol is similar to that for silylenes, we should expect a similar dependence and acceleration of the reaction with increasing pressure.
To date, few papers have been published on the study of direct synthesis at elevated pressures. A method for the preparation of alkylalkoxysilanes based on the reaction of silicon with methanol in an autoclave at pressures of up to 60 atm and temperatures of 250 °C in the presence of catalytic amounts of alkali metal carboxylates was patented.167 This approach allows to obtain up to 9% of methyltrimethoxysilane per tetramethoxysilane with a total yield of 43%. Catalysis with alkali metal carboxylates was also favourable to produce tetraalkoxysilanes with selectivity of up to 95% in autoclave on heating up to 200 °C and autogenous pressure up to 22 atm 168, 169 with complete absence of the products with Si – C bonding. Union Carbide Corporation patented a process for the preparation of alkylsilicates 170 in an autoclave using a mixture of MeOH/EtOH alcohols and K2CO3 as the catalyst, but the silicon conversion did not exceed 40%. The formation of all five expected products was recorded by the GC techniques.
The reaction of silicon with alcohols in autoclave at autogenous pressure showed that the main released products in the reaction with primary alcohols ROH (R = Me, Et, Prn, Bun, Bui, EtOCH2CH2) are tetraalkoxysilanes with 59 – 86% yield at silicon conversion up to 90%.171 In the case of methanol, the main by-product is hexamethoxydisiloxane (MeO)3SiOSi(OMe)3 with selectivity up to 18%. For ethanol, besides the main product tetraethoxysilane (TEOS) with up to 90% content in the mixture, siloxanes with Si – H bond were detected, whereas for isopropanol the main isolated product is HSi(OPri)3 with up to 67% content in the mixture. For cyclohexanol and ButOH, only products of alcohol dehydration with small yields were identified. Newton and Rochow 64 previously showed that the reaction of silicon with PrnOH carried out at atmospheric pressure in a liquid-phase reactor gives HSi(OPrn)3 with a yield <11% based on the reacted alcohol, while BunOH and CH3OCH2CH2OH provided no products. Obtaining alkoxysilanes in good yields with good conversion of silicon in the autoclave indicates a positive effect of pressure on the reaction of silicon with alcohols.
Particular attention deserves the work of Muzafarov et al.,157 who described the direct synthesis of tetraalkoxysilanes in a high-pressure mechanochemical reactor, i.e., two activating factors were realized within one reactor. Such a method, on the one hand, makes it possible to utilize the advantages of the mechano-activation described above. On the other hand, due to the high pressure, the authors managed to carry out DSA at a record low (at the time of publication) temperature (100 °C). This confirms the really important role of both factors in the direct synthesis of alkoxysilanes. Later, this concept was adopted by Japanese researchers,172 who developed an approach to tetraalkoxysilane using a planetary mill under extremely mild conditions (40 °C, 4 bar). At a significant (31 mol.%) copper content in CM, the maximum yield of tetramethoxysilane (TMOS) was 51%.
3.9.4. Activation with hydrogen
Activation of CM by hydrogen flow prior to DSC has been known for a long time.11 However, there are not many examples of the use of CM activation by hydrogen in DSA. The most detailed study of the effect of CM activation by hydrogen was performed by Adonin et al.75 CM pretreatment was carried out through a number of consecutive stages (Scheme 18). Silicon and copper chloride powders were mixed in the required proportions, then milled (CM1). After that, hydrogen was passed through the CM mixture in a high-boiling solvent at a temperature of 240 – 250 °C (CM2). Further, the mixture was also heated in an argon stream (CM3).
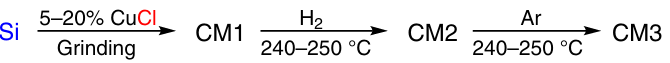
According to the authors, hydrogen is necessary to clean the silicon surface from oxygen. The latter can be in the form of adsorbed gas or be chemically bound to silicon. Hydrogen concentration at the interface of silicon crystallites leads to their destruction.
This leads to an increase in the specific surface area of silicon. However, adsorption of hydrogen onto the silicon surface has an adverse effect: hydrogen displaces copper from the surface phases, causing its segregation on the silicon surface and the formation of chlorine-coated metallic copper nanoparticles. Thus, sorbed hydrogen can slow down the surface diffusion of copper into silicon. Therefore, the stage of CM heating in argon is necessary because it helps to remove the sorbed hydrogen from silicon. The study is convincingly supported by data from techniques such as scanning electron microscope with energy dispersive X-ray spectroscopy (SEM-EDX), X-ray photoelectron spectroscopy (XPS) and X-ray phase analysis (XPA), as well as by studying the extended fine structure of the X-ray absorption spectrum (EXAFS).
3.10. Prospects of organoalkoxysilanes production
Thus, the DSA of tri- and tetraalkoxysilanes in general gives an idea of the nature of the process. In the very first approximation, the ideal precursor for the preparation of organoalkoxysilanes by direct synthesis is dimethyl ether (DME) (Scheme 19).

However, it is extremely difficult to realize such a synthesis. The first obvious reason for this is the inertness of an ether or any other organic precursor (OP) towards silicon. Indeed, when comparing alkyl chlorides with ethers, the latter are extremely inert chemically. Knyazev et al.154 carried out computer calculations of thermodynamic parameters of obtaining methylalkoxysilanes from various organic precursors.
The obtained data suggest that there are no thermodynamic obstacles for the formation of methylalkoxysilanes. The Gibbs energies are negative for reactions with both alcohols and simple ethers. This fact is not surprising, since the bonding energy of Si – O is much higher than the bonding energies in ethers. Accordingly, the main challenge for chemists in this field is to search for activators of C – O – C bonds, and such that the reaction takes place on the surface of CMs to provide surface particles with Si – O – C and Si – C bonds. Otherwise, a wide range of products can be formed during the transformation of ethers: from the simplest molecules (H2, H2O, CO, CO2 and CH4), lower olefins, methanol, ethanol, methyl acetate, to substituted mono- and polyaromatic hydrocarbons.173 – 176
However, significant results on the preparation of methyl methoxysilanes from DME and CM are not available to date. Further, few examples of works on this topic will be discussed. In addition, at the end of the review there is a section on possible methods of activation of the ether bond, with examples of obtaining various organic compounds from DME.
3.10.1. Use of ethers in DSA
The first attempts to exclude chlorine from the process of preparation of organoalkoxysilanes appeared almost immediately after the development of the process of direct synthesis of organochlorosilanes. The first experiments on this subject were carried out by Rochow,20 who pioneered in describing the reaction of silicon with methanol and ethanol. However, it quickly became clear that reactions with alcohols do not produce alkyl-containing products in appreciable amounts.64 Therefore, all subsequent efforts were directed to the search for other chlorine-free organic precursors and methods of their activation in direct synthesis reactions. Obviously, ethers are the most atom-economical and economically viable precursors.
The reaction of silicon with dialkyl ethers in the presence of HCl was also first studied and patented by Rochow.177 A similar process was presented by Turetskaya et al.178 However, this procedure yields organochlorosilanes, which made this method rather unpromising. According to the authors of the patent,179 organoalkoxysilanes in this case are not formed because of the following reaction (Scheme 20).

In 1951, patent 180 was published describing the reaction of alkyl and aryl ethers with silicon in a vertical tube at atmospheric pressure and temperature ~ 400 °C. Copper and silver were used as catalysts. This method would have been very attractive, but when it was reproduced 181 it was found that it did not provide organosilicon products.
In 1978, the US patent 98 was obtained describing an autoclave process in which the reaction proceeds at temperatures between 200 and 300 °C in the presence of catalytic amounts of chlorine- or bromine-containing compounds (metal halides, halogenohydrates, halogenosilanes and siloxanes, organohalides). The highest efficiency was observed for methyl bromide. This method can be represented by the following reaction sequence (Scheme 21).
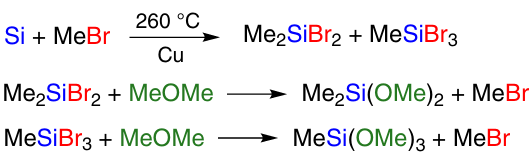
The main disadvantages of this method include the low reaction rate and the use of an autoclave, which allows only a batch process. Several years later, an attempt was made to intensify and convert the process to continuous type.179 However, despite notable improvements,182 it has not been commercialized. The main reason is the use of alkyl halide to obtain products in the form of cyclic or linear oligomeric organosiloxanes. In addition, the presence of appreciable amounts of halogen-containing impurities posed some issues.
Thus, the activation of an ether bond and further interaction of the ether with CM is a great challenge, as evidenced by the absence of a noticeable number of publications on this subject in the peer-reviewed journals. All data on the direct synthesis of methylalkoxysilanes are covered by patents, which indicates purely empirical approaches to solving the problem.
3.10.2. Other precursors
Hydrosilylation of ethylene with trichlorosilane in the presence of a platinum catalyst has been known since 1946.183 Accordingly, it was quite natural to combine this reaction with alcoholic direct synthesis. This was first realized in the patent 184 (Scheme 22).
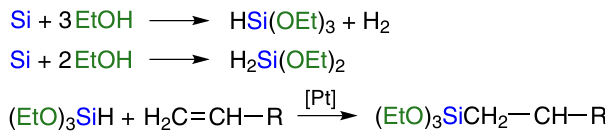
In addition to the platinum hydrosilylation catalyst, iron pentacarbonyl was also used. Besides the process described above, the same patent mentions other reactions with unsaturated compounds, such as with acetylene and allyl acetate under similar conditions, with benzene in the presence of boron trifluoride (Lewis acid).
In 1994, as part of a study on the direct synthesis of alkoxysilanes, Okamoto et al.84 proposed a mechanism for their formation via intermediate silylenes. In a follow-up publication,82 methyldimethoxysilane was obtained with a selectivity of 22% at a silicon conversion of ~40%. The authors suggested that the formation of MeSiH(OMe)2 is possible due to the interaction of methanol with metallic copper, during which the organometallic compound VI is generated (Fig. 25).
![[{"id":"8xU4SiYWiB","type":"paragraph","data":{"text":"Plausible mechanism for the formation of MeSiH(OMe)<sub>2</sub>.<sup>82</sup>"}}]](/storage/images/resized/0cvUHXLa4j31pizHjxqd0zGRHVowuXHTllsKTXDu_xl.webp)
Earlier, in Section 2.3, we have already mentioned studies,82 – 84 in which the silylene nature of active copper-silicon centres was assumed and reactions with carbene traps were carried out. The same authors studied the reaction of silicon with a methanol/ethylene mixture in a high-pressure flow reactor (Scheme 23).83 They managed to obtain ethyldimethoxysilane with a selectivity of 33% at a silicon conversion of ~60%.
Finally, an approach presented recently in the publication 24 and patent 185 comprises the use of dimethyl carbonate (DMC) as both a methylating and methoxylating agent.
Dimethyl carbonate is a ‘green’ analogue of organohalide, successfully used for alkylation and alkoxylation of organic compounds.186, 187
The reactions were carried out in a fixed bed reactor. Cu5Si copper silicide was used as a silicon source at 350 °C (Scheme 24).
Tin addition plays a pivotal role in the formation of methylalkoxysilanes. In the above-mentioned study,24 SnCl2 was used (Table 3).

The Table 3 show that the addition of tin provides the maximum selectivity for Me2Si(OMe)2.
According to the proposed mechanism, the process begins with the enrichment of the CM surface with tin (Fig. 26). Then, DMC dissociatively chemisorbs onto the tin-enriched surface to generate surface species with –Me and –OCOOMe bonds. Surface carboxyl esters are less stable than methyl esters, so they are rapidly decarboxylated to methoxy particles ( – OMe), releasing CO2 into the vapour phase. Once on the CM surface, Me and OMe groups can migrate onto Si to form silylene intermediates. When the reaction products are released, the CM surface loses silicon, the surface concentration of which is replenished due to its gradual diffusion from the bulk CM to the surface. However, due to the high temperature of this process, simultaneously with the formation of target products, decomposition of reagents and intermediate products occurs, which leads to gradual coking of the CM surface and loss of activity.
![[{"id":"EXjm1uuK1A","type":"paragraph","data":{"text":"Plausible mechanism for the direct synthesis using dimethylcarbonate and Cu<sub>5</sub>Si.<sup>24</sup>"}}]](/storage/images/resized/OF6tyUBwnyIqdFJr77Z4zPZZ7c2XmRN5u9MM90g7_xl.webp)
The described direction seems to be the most promising at present, despite a number of drawbacks inherent in this particular implementation. The research is at its infancy. Thus, publication24 provides no values of conversion of Si to Cu5Si, and according to the given data it is possible to calculate that as low as 1 – 2 mg of the product mixture was obtained. Furthermore, the use of this silicide also has no industrial prospects since it contains only 7 wt.% silicon. The main result of this study, which is of great importance for further research, lies precisely in the use of tin as an additive that determines the possibility of forming organoalkoxysilanes.
The reaction conditions of Cu5Si with DMC are also rather drastic (350 °C). While with methyl chloride such temperature is acceptable, for dimethyl carbonate it should be reduced at least to 300 °C. This is due to the fact atht at T > 300 °C, DMC spontaneously decomposes, resulting in a significant decrease in the process productivity and the appearance of a large number of by-products.
Another patent of the same authors 188 claims that similar products can be obtained by using a mixture of carbon dioxide with ethers on various contact masses and pure intermetallides. Unfortunately, the authors provide neither references nor any significant experimental evidence.
4. Synthesis of dimethylcarbonate
Based on the results described above, DMC appears to be the most suitable precursor for direct synthesis of methyl methoxysilanes in terms of its reactivity.
Currently, the global DMC market is estimated to be worth $1.1 billion with a forecast to grow to $1.6 billion by 2027.189 According to this forecast, DMC production in the Asia – Pacific region will exceed 200 000 tonnes by 2028.190 Methods of DMC synthesis are shown in Fig. 27.191
![[{"id":"8QrrK9Y-w-","type":"paragraph","data":{"text":"State-of-the-art methods for producing dimethylcarbonate.<sup>191</sup>"}}]](/storage/images/resized/V3xCuoiyG6jd0Dj9PtmyUuZEPqHjF00Z6gzXisZM_xl.webp)
Dialkyl carbonates are traditionally prepared by alkoxylation of phosgene.192 Thus, to obtain diethyl carbonate (DEC), gaseous phosgene is bubbled through boiling ethyl alcohol. The reaction yields DEC and hydrogen chloride. The process is carried out with a large excess of ethanol (2.5 – 4.0 equiv.) without any catalyst. To obtain a marketable product, the reaction mass is distillated with separation of unreacted ethanol and hydrogen chloride. The yield of DEC based on phosgene is close to quantitative (> 95%). In the case of phosgene methoxylation, the use of a catalyst (NaOH) and a more complicated instrumentation are required; therefore, DMC is often manufactured by transesterification of other carbonates (e.g., ethylene carbonate) or by oxidative carbonylation of methanol. Also, the direct synthesis of DMC from methanol and CO2 is a promising area of research in this field,193, 194 but this method has not yet been commercialized.
The method of oxidative carbonylation of methanol has been actively used in industry since 1984.195 This approach is considered to be more modern and environmentally benign compared to the traditional phosgene route; the process is carried out in the presence of homogeneous catalyst Cu2Cl2 at 120 – 130 °C and increased pressure (see Fig. 27). The reaction scheme is shown in Fig. 28. Under these conditions, methanol is almost quantitatively converted into DMC, while the selectivity of CO conversion tends to 80%; the by-products of the reaction are methyl chloride, DME and CO2. Further development of technology is connected with swiching to heterogeneous catalysts; the main types of developed systems are considered in the review.196 Most commonly used are various copper catalysts on different carriers such as FAU (FAU-type zeolite (faujasite)), SiO2 – TiO2, MFI (MFI-type zeolite (Mordenite Framework Inverted)), MOR (MOR-type zeolite (mordenite)), MWW (MWW-type zeolite); CuCl2 – PdCl2/AC (activated carbon) and others. The created catalysts are characterized by rather high activity and selectivity for DMC in oxidative carbonylation of methanol and allow to obtain DMC with yields of ~10 – 20% per pass in a continuous flow reactor.
![[{"id":"n0Oet4Sv0q","type":"paragraph","data":{"text":"Scheme for dimethylcarbonate production via oxidative carbonylation of methanol in the presence of Cu<sub>2</sub>Cl<sub>2</sub>."}}]](/storage/images/resized/GMXsSvkUXx7ILhz7Bp8nKJdGNLBExeYPFS3VHbL3_xl.webp)
The possibility of DMC production from DME deserves a separate mention. Li et al.197 demonstrated the principal possibility to derive DMC from DME and studied the thermodynamic parameters of the system to modell the reaction apparatuses. When studying the decarboxylation of DME in the presence of Zn-FAU, it was found that the decarboxylation is indeed reversible and can be used for chemical binding of CO2 to form DME, but its rate is much slower than that of DME decomposition under the same conditions.198 These circumstances are caused by extremely unfavourable thermodynamics of the reaction of CO2 with DME. At the same time, in a number of studies, the problem of unfavourable thermodynamics of the process is solved by successive transformation of the product into a more energetically favourable one. A striking example, in which thermodynamic constraints on the system are removed in this way, is the partial reduction of CO to formaldehyde:199, 200 under normal conditions, this reaction produces methanol and CH4, but when carried out in an aqueous medium, formaldehyde, which is a Bodenstein intermediate in the reduction route, reacts with water to form methylene glycol as a thermodynamically allowed adduct. As a result, commercial formalin can be obtained and gaseous formaldehyde can be synthesized via dehydration of methylene glycol (Scheme 25).
Considering the high activity of copper-containing catalytic systems in the process of DME production from methanol, CO and O2, it can be assumed that CM used for the direct synthesis of organoalkoxysilanes may possess some catalytic activity in carboxylation of DME to DMC. In this case, the activation pathway of DME producing more reactive OP may accelerate the formation of organoalkoxysilanes, which, in turn, requires a more detailed experimental study of the whole direction in the complex.
5. Activation of a molecule of dimethyl ether
Let us return to the ideal precursor: dimethyl ether is of the greatest interest in terms of its use as a feed in the direct synthesis of methylalkoxysilanes. The increased interest in this raw material is caused by the environmental challenges faced by mankind, which prompt to search for alternative sources of energy and raw materials for chemical technology to petroleum.201, 202 Firstly, syngas is used as a traditional raw material for DME production, and the process is carried out either directly from it or through the formation of methanol.203, 204 A promising approach is the process using CO2,205 which allows the use of various wastes and biomass as a raw material, while solving the problem of carbon dioxide emission and capture. Secondly, the physico-chemical properties of DME favour its application as an available and environmentally-friendly fuel for engines or a component thereof,203, 204 a promising liquid organic hydrogen carrier 206 and an efficient extraction agent.207, 208 The combustion of DME-based fuels produced from CO2 or biomass does not release additional amounts of CO2, making the carbon footprint of such fuels minimal.
Chemical properties of DME do not allow to consider it as a versatile precursor in organic chemistry, but the use of modern catalysts makes it possible to apply it successfully as a raw material in many petrochemical processes, mainly as an alkylating agent. Thus, the processes of obtaining olefins,209 – 211 motor fuels and individual hydrocarbons,174, 212 – 214 formaldehyde,215, 216 ethanol and methyl acetate,175, 176, 217 polyoxymethylene ethers 176, 218, 219 from DME are being actively investigated. Its transformation pathways depend significantly on the presence of additional reagents, composition and nature of the catalyst.
The most common catalysts for the activation of esters are acids, in particular heterogeneous acid catalysts (zeolites, heteropolyacids (HPAs), metal organic frameworks, etc.). During the interaction of DME with surface Brønsted acid sites (BASs) of such systems, the surface methoxide is generated (Scheme 26).176
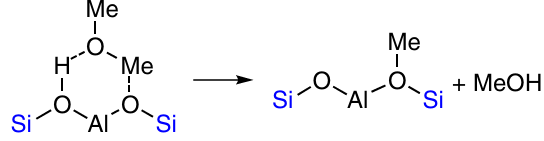
Further transformation of the surface intermediate occurs due to interaction with other particles following the Eley – Rideal mechanism. Depending on which particle acts as such a substrate, different products can be formed. Wang and Hunger 220 explored the transformation pathways of 13CH3OH on the surface of BASs of zeolites as the model substrate, the activation of which occurs similarly to the DME transformation. The obtained distribution of carbon labels in the products of interaction of methoxide with a variety of nucleophiles is shown in Fig. 29.
![[{"id":"UxLlzxnulQ","type":"paragraph","data":{"text":"Scheme for the conversion of a surface methoxy species into main products.<sup>220</sup>"}}]](/storage/images/resized/PxFIG4Gx5ZGbejQHtsTiur7sOqh8d1TmcWHc8abO_xl.webp)
It is worth noting that some acid catalysts are able to convert DME into certain products with extremely high selectivity. In particular, zeolite catalysts (based on MFI, FAU, BEA (beta polymorph A), MOR, FER (ferrite) and other zeolites) demonstrate high activity in DME conversion.173 – 176 Such reactions include conversion of DME into methyl acetate and ethanol.
DME reacts with CO according to the regularities of a well-studied process – carbonylation of methanol (Scheme 27), which has attracted and attracted researchers since the beginning of the 20th century due to the high demand for the reaction products (acetic acid, methyl acetate, etc.). As for DME, the process is generally used in industry to produce acetic anhydride or methyl acetate.221 The process is usually carried out in the presence of homogeneous metal complex catalysts (traditionally, [Rh(CO)2I2] – or [Ir(CO2)I2] – ) and HI as a promoter. Other platinum group metals can also be used. Thus, DME was shown to be successfully activated on Ru(acac)2 in combination with MeI in toluene; the reaction proceeds towards the formation of methyl acetate (MA).222

The research of Shikada et al.223, 224 should be attributed to the pioneer works in the field of heterogeneous-catalytic carbonylation of DME, in which the process was explored in the presence of nickel catalyst on activated carbon obtained by impregnating activated carbon with nickel acetate solution. Under optimum conditions, the yield of MA is 28.6%, and the reaction was carried out not in a batch apparatus but in a flow system at a pressure of 11 atm.223 When the pressure is raised to 40 atm, the yield of MA increases to 34%.224 The disadvantages of the process include the use of HI as a promoter. At the same time, the activity of other metals in this process was also studied, but their activity and selectivity turned to be low. For example, the use of Mo/AC as a catalyst for the carbonylation process in the case of DME conversion was inefficient (~ 5%, see Scheme 27, conditions b).226
An important milestone in the development of DME conversion technology was the discovery of the activity of superacid systems as active catalysts for carbonylation of methane, methyl halides, methanol, and DME. When studying the activity of HF – BF3 systems in the conversion of DME to MA, it was found that in 6 h of the reaction MA can be obtained in 66.2% yield (see Scheme 27, conditions c).225
Later, it turned out that heterogeneous acid catalysts are also characterized by extremely high activity and selectivity in carbonylation of methanol and DME. Wegman 227 studied the activity of HPAs of the type H(3 – n)Mn+W12PO40 (where Mn + = Ir3+, Rh3+, Pd2+, Mn2+, Co2+, Ni2+ and Fe3+), deposited on SiO2 during the carbonylation of methanol in CO at 225 °C and 1 atm pressure. All the above samples were active towards the dehydration of methanol in DME, however, in the presence of IrPW12O40 and RhPW12O40, the MA yields were 40 and 34%, respectively. This fact directly indicates the high activity of Ir and Rh derivatives of HPAs in DME carbonylation. Studies in this field led to the development of highly selective Rh – CsPW12O40 systems whose selectivity for MA tends to 96% with a maximum capacity of 190 gМА (Lcat h) – 1 (see Ref. 228), and Sardesai et al.229 proposed an IrPW12O40 system deposited on Grade 643 silica gel that retains high activity during 8 h of the reaction.
The general disadvantage of the systems with HPAs is the necessity to use noble metal salts to prepare active catalysts, while HPAs with transition metal cations are characterized by rather low activity in the target conversion of DME to MA. Zeolites can be an accessible and cheap alternative to HPA-based catalysts. Iglesia and co-workers 230, 231 first studied activity of zeolite catalysts in the carbonylation DME. Common and industrially available zeolites, such as H-MFI, H-FER and H-MOR were chosen as the object of study.230 It was found that in the temperature range of 150 – 190 °C all the samples are characterized by almost 100% selectivity for MA, while at higher temperature DME transformations lead to a mixture of hydrocarbons. The H-MOR sample (Si/Al = 10) showed the highest activity (to 0.5 gМА (gcat h) – 1) towards MA formation. A detailed study of the regularities of the reaction 231 allowed to expand the understanding of its mechanism (Fig. 30) and the possibility of its control. The carbonylation process has an induction period during which surface methoxide species from DME or methanol are formed. After the formation of sufficient methoxides, the main process is initiated. The first step involves the insertion of CO into the C – O bond of the methoxide, and then the formed acetate moiety interacts with a second DME molecule from the gas phase. As a result, the acetate fragment is methoxylated and the methyl substituent restores the structure of the active site.
![[{"id":"Aa9xlMrCt_","type":"paragraph","data":{"text":" Proposed mechanism for MA formation during DME carbonylation.<sup>231</sup>"}}]](/storage/images/resized/dNKmt44PXt3PoONqjhwxtcTPdQ4oLbsYxeiH2MUj_xl.webp)
In addition to MOR zeolites, FER zeolites show high activity in the DME carbonylation process. The unusual activity of both types of zeolites is due to the structure of their 8T-atomic pores (Fig. 31), which allows activating not only DME but also CO molecule during the process.232 At the same time, zeolites of other common types (MFI, FAU, BEA, etc.), which are not characterized by the presence of 8T-atomic pores, are practically inactive with respect to the formation of MA,230, 231 and produce mainly hydrocarbons and methanol as the reaction products.173, 174, 209 – 214, 230, 231 It is worth noting that by adding components active in hydrogenation (e.g., Cu) to H-MOR and hydrogen to the reaction mixture, it is possible to direct the process towards the formation of ethanol. The processes of carbonylation and hydroformylation of DME to form MA and ethanol are discussed in more detail in reviews.175, 176 Thus, by selecting an optimal catalyst, it is possible to direct the DME conversion process either towards the formation of active and reactive surface methoxides, the conversion of which is controlled by both reagents and catalyst, or towards the formation of more active OPs (MA and ethanol). DME derivatives promising for PSA also include DMC and trimethylorthoformate.
![[{"id":"4quHxkKg1b","type":"paragraph","data":{"text":"Framework structure of MOR zeolite.<sup>175</sup>"}}]](/storage/images/resized/jHh1M3HEkEqvzrKFZChrO7ECT5BvW4Uljj8vk249_xl.webp)
To summarize, DME is a promising precursor for the direct synthesis of methylalkoxysilanes due to its commercial availability and the existing knowledge of its activation routs under conditions suitable for direct synthesis.
6. Conclusion
Direct synthesis of alkylalkoxysilanes is a key process in transition to environmentally friendly production of silicones. Such a process can be much more economically and environmentally attractive compared to the second generation chlorine technology. This is due to the efficient separation of the methylalkoxysilanes mixture compared to their chlorine-containing counterparts, as well as the possibility of their safe storage and transportation. In addition, silicones can be recycled into organoalkoxysilanes rather than into chlorosilanes. Accordingly, direct synthesis of organoalkoxysilanes would close this production cycle and make silicones completely ‘green’.
To date, the synthesis of tri- and tetraalkoxysilanes has been most extensively studied. Analysis of the literature allows to conclude that the liquid-phase version of this process is the closest to industrial implementation, and probably already realized. Nevertheless, the DSA process is very complex. It is influenced by many factors. This complicates its study and, consequently, its application. However, the high practical and environmental significance of this process makes the work in this direction very relevant.
The most significant green synthesis of methylalkoxysilanes, apparently, is only at the first stage of its development. This is evidenced by a recent paper by Roberts et al.,24 in which methylmethoxysilanes were derived from copper silicide and DMC. This work can rightly be considered pioneering and extremely important for further development of the DSA process. Analysis of the literature suggests that researchers in this field have all the necessary means to achieve the goal. For example, it is known from chlorine synthesis technology that active contact masses should comprise Cu3Si intermetallide. In addition, there is much data regarding the influence of various additives, in particular, synergistic effects have been studied as a result of the combined use of tin and zinc in chlorine direct synthesis. The fact that such a necessary component of the emerging green process as tin is also one of the most effective additives in the chlorine process may indicate a significant similarity in the mechanisms of action of additives on these processes. Thus, there are assumptions about the necessity of the presence of Cu3Si intermetallide in the active CMs. This aspect has long been studied and, in particular, the appearance of this intermetallide is associated with the action of some additives, for example, Zn. At the same time, the mentioned synergistic interaction between zinc and tin, manifested in DSC, can also occur in the case of DSA, which will contribute to the activation of organic precursors.
All this inspires optimism about the prospects of this process. The reaction conditions stated by Roberts et al.24 can be made considerably milder, for example, by using mechano-activation of CM, successfully used for the synthesis of tetraalkoxysilanes. In addition, a search for promoters of methylalkoxysilanes formation, alternative to toxic tin is needed. On the other hand, its content in CM is extremely small (< 1000 ppm), so at this stage, the environmental benefits of switching from DSC to DSA undoubtedly outweigh the negative impact of tin on the environment.
Silicone production appears to be on the threshold of successful development of direct synthesis of organoalkoxysilanes on an industrial scale. Its successful realization will introduce silicone chemistry into a new, ‘green’ era and will give a strong impetus to the research and implementation of silicone materials in both existing and completely new areas of practical application.
Section 1 was prepared with the financial support of the Ministry of Science and Higher Education of the Russian Federation using the equipment of the Center for Molecular Composition Studies of INEOS RAS (Agreement No. 075-03-2023-642).
Section 2 was prepared with the financial support of the Government of the Tula Region (Resolution No. 899 of 30.12.2021) within the framework of the Agreement No. 11 of 07 September 2022.
Section 3 was prepared with the financial support of the Russian Science Foundation (Project No. 18-73-10153)
Sections 4 and 5 were prepared with the financial support of the Russian Science Foundation (Project No. 22-13-00279)
7. List of abbreviations
AC — activated carbon,
BAS — Brønsted acid sites,
BEA — BEA zeolite (beta polymorph A),
cat — catalyst,
CM — contact mass,
D4, D5 — polydimethylsiloxane rings (the figure denotes a number of units),
DEDAS — diethyldialkoxysilane,
DFT — density functional theory,
DMC — dimethylcarbonate,
DME — dimethyl ether,
DSA — direct synthesis of alkoxysilanes,
DSC — direct synthesis of chlorosilanes,
EXAFS — extended X-ray absorption fine structure,
FAU — FAU zeolite (faujasite),
FER — FER zeolite (ferrerite),
GC-LC — gas liquid chromatography,
HPA — heteropolyacids,
HPLC — high precision liquid chromatography,
МА — methyl acetate,
MFI — MFI zeolite (mordenite fraimwork inverted),
MOR — MOR zeolite (mordenite),
MWW — MWW zeolite,
OP — organic precursor,
PDMS — polydimethylsiloxane,
PTAS — phenyltrialkoxysilane,
SEM — scanning electron microscopy,
SEM-EDX — Scanning Electron microscopy with energy-dispersive X-ray spectroscopy,
sur — surface,
ТАOS — tetraalkoxysilane,
TCS — trichlorosilane,
TEOS — tetraethoxysilane,
ТМOS — tetramethoxysilane,
XРS — X-ray photoelectron spectroscopy.