

Keywords
Abstract
The potential wide use of environmentally friendly low-carbon gas fuel based on natural gas, hydrogen, their mixtures, and syngas in power generation and transport requires detailed information about the kinetics of ignition of these gases at temperatures below 1000 K, at which fuel ignition occurs in internal combustion engines (ICEs) and gas turbines. The same temperature range is also important for monitoring the storage and transportation conditions of these fuels. Although there are quite a few studies addressing the ignition of classical gas fuels such as methane or hydrogen, there is an obvious lack of works dealing with real natural gases and gas mixtures. Furthermore, even for methane and hydrogen, data on the ignition at high temperatures (T > 1000 K), which have been mainly gained by the shock-wave method for highly diluted mixtures, are at variance with the kinetic estimations for real conditions of operating with them or their use in ICEs. Considering the ignition characteristics at T < 1000 K is also important for syngas, the largest-scale base product of gas chemistry and the main industrial source of hydrogen. The pronounced discrepancies between the extrapolation of the results obtained for high-temperature ignition to lower temperatures and the results of kinetic modelling of these processes make it necessary to analyze their causes. This review addresses new experimental results on the ignition of methane–alkane and methane–hydrogen mixtures (real gas fuels) and kinetic modelling of these processes, which reveal significant changes in the ignition behaviour at T < 1000 K. These changes in the ignition process upon the variation of the temperature, pressure, and composition of the mixture are related to significant changes in the methane and hydrogen oxidation mechanisms in this temperature range. They are mainly caused by changes in the kinetics and, hence, the role of peroxide compounds and radicals in methane and hydrogen oxidation following temperature and pressure variation. The established features bring about the question of the adequacy of the existing criteria for assessing the knock resistance of gas engine fuels, primarily those containing hydrogen, when they are used in ICEs, and for assessing their explosiveness and measures taken for their safe handling. The review considers the possible methods for improving the knock characteristics of natural and associated gases to meet the requirements of power equipment manufacturers.
The bibliography includes 128 references.
1. Introduction
The current stage of development of the world economy is governed by two global trends: the apparent stabilization of oil production,1 which would inevitably decline in the future, making oil progressively less accessible, and the desire to reduce greenhouse gas emissions during power generation. The decrease in the greenhouse gas emission was announced as a goal of the Paris climate agreement,2 intended to stabilize the observed climatic processes or, at least, delay their consequences.2, 3 This is usually formulated as a demand to reduce the carbon footprint of the global energy. The most real way for reducing the carbon dioxide emission upon power generation is to more widely use natural fuels and secondary energy carriers with low carbon content, which include natural gas, syngas, and hydrogen.
Of the listed low-carbon gas fuels, only natural gas is a primary energy source. The natural gas resources in Earth’s crust, including unconventional resources, such as shale gas, coalbed methane, gas hydrates, and several other, are enormous and can supply the world’s energy production industry for quite a long period of time.4, 5 Syngas and hydrogen are secondary energy carriers, which are mainly produced from natural gas and coal. Since the production of secondary energy carriers is always associated with additional expenses and, furthermore, hydrogen and syngas have a significantly lower specific volumetric energy content than even methane and the problems of their transportation and long-term storage still lack a reasonable solution,6, 7 one can hardly expect that they will play a serious role in the global energy in the near future. However, their more extensive use for solving local environmental problems, particularly in the field of transport, by partial conversion of the traditional hydrocarbon fuels directly on-board the vehicle,8, 9 is quite probable.
The prospects for wide use of various gas fuels for energy production and transport dictate the need for thorough analysis and control of conditions of their ignition and combustion, first of all, in the pressure and temperature regions that most closely correspond to the conditions of their real application. Most often, this concerns moderate temperatures. For example, as shown in a recent study,10 the ignition of the fuel mixture in ICE takes place in the 500 – 900 K temperature range and, hence, it is determined by the kinetics of processes that occur at these temperatures. However, the vast majority of studies on the ignition of hydrocarbon gases were carried out in shock waves, which is most common for studying high-temperature processes,11 – 13 the lower temperature limit of which is only slightly below 1000 K. Therefore, the intricate and interesting details of ignition of methane and hydrogen at lower temperatures have not received due attention so far. One more considerable drawback of shock-wave experiments is that they are usually carried out under high dilution of the reactants with an inert gas, which inevitably influences the reaction kinetics.
Lower-temperature ignition is studied using rapid compression machines 14 and static bypass installations (high-pressure bombs).15, 16 However, these methods also have drawbacks and limitations. In particular, static bypass installations (SBIs) are unsuitable for investigation of very fast ignition of hydrogen and mixtures with high hydrogen contents. Therefore, kinetic modelling plays an important role in determining the features of ignition of methane, hydrogen, and mixtures containing these gases.
Today, quite reliable kinetic mechanisms describing the oxidation of light hydrocarbons and hydrogen at moderate temperatures (T < 1500 K), suitable for analyzing processes in this temperature range, have been reported in the literature. Our analysis of the known mechanisms 17 and the results of analysis of these mechanisms by other authors 18 made it possible to give preference to the group of mechanisms proposed at the National University of Ireland (NUI, Galway), in particular NUIGMech1.1, AramcoMech 3.0, and some other,19 which were used in most of our kinetic studies, in particular those considered here.
The present review addresses the experimental results obtained to date on the ignition of methane, hydrogen, and mixtures containing these gases at moderate temperatures (T < 1000 K) and the kinetic analysis of these processes. Out of the large array of studies on the ignition of considered gases at higher temperatures, we mention only those in which the temperature range was close to the temperature range of interest and the results of which illustrate, to some extent, the trends observed at lower temperatures.
2. Ignition of methane and other light alkanes
The ignition delay time is a highly important characteristic of ignition of any fuel, including hydrocarbons. Owing to very strong C – H bonds, methane is among the least reactive hydrocarbons 20, 21 characterized by long ignition delay and, correspondingly, high knock resistance. The ignition delay time of methane is much longer than those of other alkanes. As a rule, this value decreases with increasing number of carbon atoms in the molecule. However, even in an early study,11 in which ignition delay times of the first five alkanes C1 – C5 were determined in the reflected shock waves in the range of 1165 – 1900 K, the authors paid attention to an abnormal behaviour of ethane in this series: ignition delay time of ethane was not located between those of methane and other alkanes, but was noticeably shorter than the ignition delay times of n-propane and even n-butane. The effective activation energy of ignition delay for methane found in this study was approximately 50 kcal mol–1, which is much higher than the value of ~ 40 kcal mol–1, corresponding to ethane, propane, and butane. Only n-pentane had a markedly lower activation energy of ignition delay (~ 37 kcal mol–1). The authors suggested that the abnormal behaviour of ethane is due to the fact that it gives two methyl radicals upon dissociation. Except for the two first members of the series, methane and ethane, other gaseous alkanes are very similar in their ignition behaviour at elevated temperatures in stoichiometric mixtures with oxygen.
Similar values for the effective activation energy of ignition delay were found using a flow test rig at atmospheric pressure in the 925 < T < 1060 K temperature range for heavier homologues of methane, including propane, n-pentane, and aviation kerosene.22 These values were 38.2 kcal mol–1 for propane and 40.9 kcal mol–1 for kerosene.
Higher activation energies of ignition delay for C1 – C3 alkanes were found in reflected shock wave experiments for higher temperatures (1485 – 1900 K), pressures of 3 – 13 atm, and the equivalence ratio (fuel excess ratio) ϕ = 0.5 – 2 for alkanes diluted with 95 – 99 vol.% argon. The results were as follows: 54.5 kcal mol–1 for methane, 55.6 kcal mol–1 for ethane, and 56.9 kcal mol–1 for n-propane.23
Holton et al.24 investigated the ignition of methane, ethane, and n-propane under flow conditions at atmospheric pressure, ϕ = 0.5 – 1.25, and a temperature of 930 – 1140 K. The activation energy of methane oxidation amounted to 46.6 kcal mol–1. Ethane and propane had similar oxidation activation energies: 40.0 and 38.5 kcal mol–1, respectively. As the equivalence ratio increased, ignition delay time decreased for both pure fuels and methane, ethane, and propane mixtures.
An interesting study in the relevant temperature range from 785 to 935 K was carried out by Beerer and McDonell,25 who used a turbulent flow reactor at elevated pressure (7 to 15 atm). The authors determined the ignition delay times for both single alkanes and alkane mixtures under conditions simulating the conditions at the inlet to the fuel and air pre-mixing zone in modern gas turbines with lean fuel – air mixtures. The authors were also interested in the kinetics of processes in this temperature range in which ignition is controlled by reactions involving O2 and H2O2. The results were compared with the literature data on ignition at higher temperatures, which revealed a number of interesting differences between these temperature ranges. The resulting overall activation energies for methane, ethane, and propane were 18.4, 33.5, and 29.9 kcal mol–1, respectively, with the possible error being estimated as ± 2.4 kcal mol–1. In other words, these values were markedly lower than those reported in most other studies carried out at higher temperature. As a result, the authors concluded that the activation energy increases with temperature rise. A fact deserving attention is that the minimum effective activation energy of ignition delay was found for methane and the maximum value was inherent in ethane. The ignition delay time monotonically decreased with increasing pressure proportionally to P –1.0 ± 0.1. The effective activation energy of ignition delay also slightly decreased with increasing pressure. Experiments for alkane mixtures showed a similar pressure dependence. The equivalence ratio in the range ϕ = 0.4 – 0.6 did not influence the ignition delay.
The understanding of the ignition kinetics of light alkanes and the corresponding unsaturated hydrocarbons can be gained by using kinetic modelling of these processes. Unfortunately, most of the early attempts of this modelling utilized kinetic mechanisms developed to describe high-temperature combustion of light hydrocarbons such as GRI-Mech 3.0 (Ref. 26) and the like. These mechanisms do not include large blocks of elementary reactions necessary to describe the low-temperature processes occurring in the period of ignition delay of hydrocarbons at low initial temperatures. First of all, this refers to reactions involving peroxides and peroxyl radicals, which, as shown below, make a significant contribution to low-temperature oxidation processes 20, 21 and ignition of hydrogen and hydrocarbons. Therefore, although the kinetic description of high-temperature ignition of these mixtures is satisfactory, the modelling results in the temperature range of interest (T < 1000 K) have been repeatedly noted to deviate from the experimental results.
The emergence of new mechanisms meant for the kinetic description of lower-temperature oxidation processes involving light alkanes and corresponding unsaturated compounds, first of all, mechanisms developed by the NUI Galway team,19 enables a more adequate analysis of the oxidation and ignition of light hydrocarbons at lower temperatures. According to our analysis,17 these mechanisms are applicable even at a markedly lower temperature than it was initially assumed by their authors.
With direct participation of NUI Galway researchers, who proposed a group of kinetic mechanisms,19 experimental and theoretical studies of the ignition delay period of methane, ethane, and ethylene were carried out.27 The studies covered broad ranges of temperature (800 – 2000 K), pressure (1 – 80 bar), equivalence ratio (0.5 – 2.0), and inert gas dilution (75 – 90 vol.%). The authors demonstrated the possibility of reliable description of the kinetics of oxidative processes in terms of these mechanisms.19 However, their analysis was mainly based on shock-tube experiments, which require high dilution of the components with an inert gas. Probably, that is why, despite the broad temperature range, a number of interesting phenomena described by Arutyunov and co-workers 16 were left unnoticed in this analysis. Below these features are considered in more detail.
Arutyunov and co-workers,16, 28 who used a static bypass installation in the temperature range T = 523 – 1000 K, demonstrated a sharp difference between the ignition delay time of methane and those of its close homologues, C2 – C5, for which the observed ignition delay times under the same conditions were approximately equal (curve 1 in Fig. 1).
![[{"id":"YVSazeVlsR","type":"paragraph","data":{"text":"Ignition delay time of C<sub>1</sub> – C<sub>5</sub> alkane mixtures with air (<i>1</i>) and alkane (10 vol.%) – methane mixtures with air (<i>2</i>) <i>vs.</i> the number of carbon atoms N<sub>c</sub> in the added alkane molecule. T = 900 K, P = 1 atm, ϕ = 1; the dots are averaged results of two series of experiments.<sup>16</sup>"}}]](/storage/images/resized/SwyFTIcl3zZRoOLPc8pfWbh12Q6SVih4VJ2LaG5d_xl.webp)
The temperature dependence of the ignition delay time τ was relatively well described in all cases by the Arrhenius relation
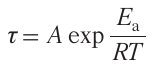
where Ea is the effective activation energy of ignition delay, А is the pre-exponential factor, R is the gas constant. Meanwhile, the ignition delay activation energy Ea of single C1 – C4 alkanes varied non-monotonically and had a clear maximum for ethane (Fig. 2). Although, according to the known views on the relative reactivities of these hydrocarbons,20, 21 it would be more reasonable to expect that the activation energy of ignition delay would be lower for ethane than for methane. However, as can be seen in Fig. 2, the effective activation energy even for n-propane is higher than that for methane, and only in the case of n-butane, it is close to that of methane. The absence of data for heavier alkanes, n-pentane and n-hexane, in Fig. 2 is due to the fact that the oxidation of these alkanes in this temperature range is substantially affected by the appearance of the negative temperature coefficient (NTC) of the reaction rate, which makes the choice of Ea for these alkanes in this temperature range almost arbitrary. This dependence of the effective activation energy for the first members of the alkane series on the number of carbon atoms NC is quite consistent with analogous dependence found for the same temperature range in a study 25 in which Ea was found to be much lower for methane (18.4 kcal mol–1) than for ethane (33.5 kcal mol–1) and even for n-propane (29.9 kcal mol–1).
![[{"id":"xMgBZrJd7S","type":"paragraph","data":{"text":"Effective activation energy E<sub>a</sub> of ignition delay of single C<sub>1 </sub>– C<sub>4</sub> alkanes <i>vs</i>. the number of carbon atoms N<sub>C</sub> in the alkane molecule (● and о correspond to two series of experiments). T = 900 K, P = 1 atm, ϕ = 1.0.<sup>16</sup>"}}]](/storage/images/resized/dsG43F4yBXZFc1l80naLSWszgXi0iArVqXewL4Ko_xl.webp)
This type of dependence of Ea on NC is a consequence of the unique mechanism of methane oxidation, which provides the possibility of efficient chain branching in the T ≤ 900 K range via the formation and the subsequent decay of methyl hydroperoxide CH3OOH,20 which is absent even for the close homologues of methane. The methyl radical resulting from methane oxidation is involved in the following sequence of transformations
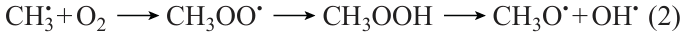
leading to degenerate chain branching. Meanwhile, for all higher homologues, the alkylperoxy radicals ROO• formed in a similar way have a high probability of isomerization followed by decay into the corresponding olefin Q= and unreactive HО2• radical, which results in chain termination
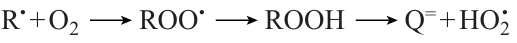
3. Effect of methane homologues and other components on the methane ignition delay
3.1. Effect of methane homologues on the methane ignition delay
Light gaseous C2 – C5 alkanes are native components of natural gas, always present in natural gas in some concentrations. Therefore, study of their influence on the ignition and, hence, on the knock resistance and other engine characteristics of gas fuels is of high practical importance. All the more so, because, as will be shown below, this influence is complex.
It is well known that even small amounts of heavier alkanes substantially reduce the ignition delay time of methane. According to Khalil and Karim,29 for the initial gas temperature of 800 and 650 K, the addition of only 0.5 vol.% n-heptane to methane leads to a decrease in the ignition delay time by 50% and 75%, respectively. However, when the concentration of heavier alkanes is above ~ 10 vol.%, further change in the ignition delay time becomes insignificant (Fig. 3). The greater part of the total ignition delay period falls to the processes that occur at low temperature, because as the ignition develops and a temperature above 1200 K is attained, fast branched-chain reactions involving methyl radicals start to predominate, which sharply accelerates the process.
![[{"id":"zhU3xKVX6U","type":"paragraph","data":{"text":"Calculated ignition delay time (dashed line) and overall combustion time (solid line) in an adiabatic constant volume reactor <i>vs</i>. n-heptane content in the fuel mixture with methane for two initial temperatures (T = 650 and 800 K). P = 2.8 MPa, ϕ = 1.0.<sup>29</sup>"}}]](/storage/images/resized/YaSIc1TI5fztYAAJ5Skrdd41Z79dn3Pld0oOTAru_xl.webp)
The addition of any of methane homologues has a promoting effect on the methane ignition. This promoting effect, in particular that of ethane and n-propane, at moderate temperatures (T < 1100 K) was originally attributed to the action of methylperoxy and methyl hydroperoxide compounds.30
Holton et al.24 reported experimental determination the ignition delay times for methane – ethane and methane – propane binary fuels and methane – ethane – propane and methane –ethane – CO2 ternary fuels under flow conditions at atmospheric pressure, ϕ = 0.5 – 1.25, and temperature of 930 – 1140 K. The addition of 5 – 10 vol.% ethane or n-propane decreased the ignition delay time of a methane-based binary fuel by 30 – 50%. Further addition of ethane or n-propane further decreased the ignition delay, but to a lower extent. The addition of either ethane or n-propane to methane decreased the activation energy of oxidation to approximately equal extents. The activation energy of oxidation of the methane – ethane binary fuel was 42.2 kcal mol–1, while that for methane – n-propane was 41.8 kcal mol–1. Like for single C1 – C3 alkanes, the ignition delay for methane, ethane, and n-propane mixtures decreased with increasing equivalence ratio.
The abnormal behaviour of ethane in the series of alkanes when added to methane was noted by Spadaccini and Colket III,31 who determined the ignition delay of pure methane, methane containing small amounts of ethane, n-propane, or n-butane, and a typical multicomponent natural gas by the shock wave method in the 1300 – 1900 K temperature range, 3.5 – 15 atm pressure range, and equivalence ratio ϕ = 0.45 – 1.25. In the correlation equations obtained for the calculation of the ignition delay time of methane with addition of various alkanes, the following effective activation energies (kcal mol–1) were taken: 45.0 for methane; 37.5 for methane + ethane; 43.1 for methane + n-propane; and 39.2 for methane + butanes. Thus, the lowest activation energy was taken for methane and ethane mixtures, although butanes were somewhat more efficient in reducing the ignition delay than ethane or n-propane. No noticeable difference between n-butane and isobutane was found.
The kinetic modelling based on the hydrocarbon oxidation mechanism 19 of the effect of H2, C2H6, and n-C3H8 added to methane demonstrated 32 that the addition of 10 vol.% ethane or n-propane decreased the ignition delay ~ 4 – 5-fold; C2H6 caused a more pronounced decrease in the ignition delay time than n-propane, while any of the alkanes had a more pronounced effect than hydrogen (Fig. 4). The ignition delay time was determined for both stoichiometric mixtures and mixtures with ϕ = 0.5.
![[{"id":"_diXruBR80","type":"paragraph","data":{"text":"Effect of addition of H<sub>2</sub>, C<sub>2</sub>H<sub>6</sub>, and n-C<sub>3</sub>H<sub>8</sub> on the ignition of methane.<sup>32</sup>"}}]](/storage/images/resized/ADc99ipGcZVwWPmuXyciYOSMvc8MpB1YVmmFq4pX_xl.webp)
At a temperature of 785 – 935 K and an elevated pressure (7 – 15 atm), which simulate the conditions at the gas turbine inlet, in a turbulent flow reactor, it was shown that an increase in the ethane or n-propane percentage in the mixture with methane leads to monotonic decrease in the ignition delay time and monotonic increase in the effective activation energy.25 Despite the fact that the general trends for methane mixtures with ethane and n-propane were similar, n-propane decreased the delay time to a greater extent than ethane, with their contents in the fuel being equal. Fuels containing 0.1 – 1 vol.% C4 – C6 alkanes (with 5 to 10 vol.% ethane and n-propane content) were also studied. The delay times for these fuels were approximately 1.5 times smaller than those for pure methane under similar testing conditions. In the authors’ opinion, this indicates that even trace amounts of these alkanes have a pronounced effect on the ignition delay.
A series of studies 15, 16, 28 carried out using a bypass installation in the range T = 523 – 1000 K at atmospheric pressure demonstrated that the addition of any of the C2 – C6 alkanes has virtually the same effect on the ignition of methane (Fig. 5).
![[{"id":"5Hp9gaJyLX","type":"paragraph","data":{"text":"Ignition delay time in air for binary methane – alkane mixtures <i>vs</i>. the concentration of the added alkane [C<sub>n</sub>].<sup>16</sup> T = 900 K, P = 1 atm, ϕ = 1."}}]](/storage/images/resized/RKsxFQRqU8KuMlqd9RcVBAhVw9wBCumzr6Y6oUPI_xl.webp)
When any of the C2 – C6 alkanes was added to methane in an amount of only 10 vol.%, the ignition delay time coincided with that for this alkane; therefore, the measurements were limited to mixtures containing less than 10 vol.% of these alkanes in methane. In the case of methane mixtures with n-hexane, this range was limited to 2.5 vol.% for operational reasons. It can be seen from Fig. 5 that the dependences of the ignition delay time for all binary methane – alkane mixtures in air on the concentration of the alkane additive are virtually identical. Moreover, despite the considerable differences between their carbon chain lengths and reactivities, the ignition delay times are fairly similar when the concentration of the added alkanes are equal. It should be noted that the addition of C2 – C6 alkanes even at the level of 1 vol.% decreases the methane ignition delay time by a factor of two to three, while the addition of 10 vol.% (see Figs 2 and 5) makes the ignition delay time of the mixture virtually indistinguishable from the ignition delay time of the hydrocarbon added; this is quite consistent with the results of Khalil and Karim.29
Despite the identical characters of the influence of various light alkanes on the ignition delay time of methane, there were also some differences. Contrary to expectations and the above-indicated order of variation of the ignition delay activation energy in the series of C2 – C4 hydrocarbons (see Fig. 2), the most pronounced decrease in the ignition delay time of methane in a binary mixture with alkane was induced by ethane (see Fig. 1, solid line). The decrease in the ignition delay time upon the addition of the same amounts of n-propane or n-butane was markedly smaller. Only upon the addition of pentane, was the ignition delay time somewhat shorter than that upon the addition of ethane. This behaviour holds over the whole range of concentrations of the added hydrocarbons (1 to 10 vol.%), and it is consistent with the results of some other papers (see, e.g., Ref. 32). The obtained non-monotonic dependence of the effect of C2 – C5 hydrocarbons on the ignition delay time of methane – air mixtures on the carbon chain length is non-trivial and calls for detailed analysis of the process kinetics. This is even more surprising in view of the fact that the ignition delay times observed for single C2 – C5 alkanes under the same conditions were approximately equal (see Fig. 1, dashed line).
Nevertheless, as follows from Fig 1, in the first approximation, the effects of addition of any of C2 – C5 alkanes on the ignition delay of the methane – alkane – air mixtures are quite similar, which suggests the possibility of their analytical description (see Section 7).
Troshin et al.16 also investigated the influence of the concentration of the C2 – C5 alkanes on the ignition delay of multicomponent methane – alkane mixtures simulating natural gas, with the goal of elucidating the applicability of the additivity principle for predicting the behaviour of these mixtures. This would substantially simplify the evaluation of knock resistance of complex gas mixtures directly from their composition and would increase the velocity of analysis. The authors investigated the ignition delays for ternary methane – alkane mixtures, with the overall concentration of the added C2+ alkanes being 10 vol.%, but different ratios between the components, and also more complex mixtures simulating the composition of the real associated gas, in which the sum of the heavy components Σ[CnH2n + 2] was ~ 10 vol.% (Fig. 6). For all mixtures containing 10 vol.% of heavier hydrocarbons in methane, the ignition delay times differed insignificantly, within the experimental error of their determination. Thus, the effect of the carbon chain length in the added hydrocarbons can be considered to be insignificant, in the first approximation. In view of the pronounced difference in the reactivities of light alkanes, this result is quite unexpected.
![[{"id":"NNXkVT4Kl7","type":"paragraph","data":{"text":"Ignition delay of methane – ethane – n-propane (●), methane – ethane – n-butane (■), and methane – ethane – n-pentane (▲) mixtures and multicomponent mixtures (♦) <i>vs</i>. ethane concentration in the mixtures.<sup>16</sup> [CH<sub>4</sub>] = 90 vol.%, Σ[C<sub>n</sub>H<sub>2n+2</sub>] = 10 vol.%; T = 900 K, P = 1 atm, ϕ = 1."}}]](/storage/images/resized/vtQ4eyDKq553KWrjgey4qP3YpD2VB4ifzFcXIm4Q_xl.webp)
The determined effective activation energies of ignition delay for all mixtures containing ~10 vol.% heavier alkanes added to methane also differ insignificantly and occur in the range of 40 ± 10 kcal mol–1 (Fig. 7), which is characteristic of the activation energy of ignition delay of ethane, n-propane, and n-butane.11 No regular trends were found for the dependence of the activation energy in complex mixtures on the concentration ratio of the alkanes added to methane (for invariable total alkane concentration).
![[{"id":"N8c1wDuoyL","type":"paragraph","data":{"text":"Effective activation energy for the ignition delay of methane – ethane – propane mixtures <i>vs</i>. ethane concentration in the mixtures.<sup>16</sup> P = 1 atm, ϕ = 1."}}]](/storage/images/resized/tElXdOBljYqY3J6qgAGLNsvW2La92Uz2S13NBOOv_xl.webp)
The influence of the added alkanes heavier than C5 on the methane oxidation and ignition and the influence of the methane addition on the ignition and behaviour of these mixtures in diesel engines is permanently in the focus of research attention. However, these studies can hardly add anything conceptually new to the understanding of the ignition kinetics of methane mixtures with alkanes. A study of the ignition of methane – n-heptane mixtures under conditions resembling engine conditions demonstrated 33 that the addition of n-heptane promotes the ignition of methane. In this case, the phenomena related to NTC of the reaction rate typical of these alkanes are clearly manifested. The higher the fraction of n-heptane in a mixture and the higher the equivalence ratio, the more pronounced these phenomena. Study of the product formation and consumption indicates that methane is ignited as a result of n-heptane combustion. However, the subsequent process is mainly controlled by the reactions related to methane oxidation, as the oxidation of n-heptane is completed very rapidly, and the greater part of the fuel is represented by methane. The n-heptane contribution to the ignition decreases with increasing initial temperature.
The results of oxidation experiments in the temperature range of interest (450 – 900 K) at a pressure of 21 and 100 atm of stoichiometric and lean mixtures of methane with n-heptane in a laminar flow reactor were described by Thorsen et al.34 Under any of the conditions, the n-heptane conversion started at T < 600 K and was completed below 900 K. At a pressure of 21 atm, a region of reaction rate NTC was clearly manifested. At a sufficiently high temperature, the presence of n-heptane promoted the methane oxidation by initiating its onset at the moment of complete conversion of n-heptane. It is of interest that, in comparison with the oxidation of pure n-heptane, the presence of methane, in turn, promoted n-heptane oxidation over the whole temperature range, probably, as a result of formation of methyl radicals.
3.2. Effect of unsaturated compounds on the methane ignition delay
The effect of unsaturated compounds on the ignition delay of methane is of special interest. Alkenes, and also cycloalkanes are widely used to increase the engine performance of liquid hydrocarbon fuels. As a rule, they have better engine performance than alkanes containing the same number of carbon atoms.35 Using experiments with a bypass installation and kinetic modelling, the ignition delays for stoichiometric methane – ethylene – air mixtures have been studied at an initial temperature in the range T = 800 – 1000 K at pressures P = 1 and 3 atm.36, 37 As the ethylene concentration in stoichiometric methane – ethylene – air mixtures increases, the ignition delay time monotonically decreases, but less sharply than the ignition delay times of the stoichiometric methane – air mixtures upon the addition of C2 – C6 alkanes. Whereas the addition of any C2 – C6 alkane in an amount of only 10 vol.% decreases the ignition delay time of methane in air to a value corresponding to the ignition delay of the added alkane itself (see Fig. 5), in the case of methane – ethylene – air mixtures, the ignition delay time characteristic of ethylene is attained only when the ethylene concentration is > 60 vol.%. However, the overall pattern of dependence of the ignition delay of stoichiometric methane – air mixtures on the concentration of the added hydrocarbon was quite similar for methane – ethylene – air and methane – ethane – air mixtures. An increase in the pressure shortened the ignition delay time,37 without changing the general pattern of its dependence on the ethylene concentration in the mixture.
It was established experimentally that the effective activation energy of the ignition delay increases with increasing concentration of ethylene in the mixture. A comparison with the ignition delay of methane – ethane – air mixtures provided the conclusion that the knock resistance of light C2 – C3 alkenes, in particular, in their mixtures with methane, does not exceed the knock resistance of the corresponding alkanes.
3.3. Effect of inert components on the methane ignition delay
Since it is known that the addition of inert components increases the knock resistance of fuels, it is of interest to study their effect on the ignition of methane fuel. Holton et al.24 studied the effect of CO2 addition on the ignition of a binary fuel mixture consisting of methane (75 vol.%) and ethane (25 vol.%). This mixture was diluted with CO2 in form mixtures containing 5 and 10 vol.% CO2. The resultant ignition delay of the mixtures containing CO2 did not differ significantly from the ignition of the mixture containing no CO2. For the highest temperature (1137 K) and ϕ = 0.5, the addition of 5 vol.% CO2 increased the ignition delay time by only 2%, but an increase in the CO2 concentration to 10 vol.% resulted in an increase in the ignition delay time by 46%. This is attributable to the fact that the efficiency of CO2 as the third body for collisions is an order of magnitude higher than that of N2.
Data on the effect of haloalkanes on the ignition delay of methane also deserve attention. Their addition narrows down the concentration range of ignition of methane – air mixtures more appreciably than the addition of inert gases.38 However, in the case of shock wave initiation in the 1100 – 1800 K temperature range, the ignition of a stoichiometric methane – oxygen mixture containing 1 – 3 vol.% haloalkanes is not suppressed, but, conversely, does take place with a reduced induction period; the authors attributed this result to the considerable difference between the concentrations of active centers and the gas heating regimes.
It is noteworthy that the above experimental data about the effect of methane homologues on the methane ignition delay can be described, not only qualitatively, but also quantitatively, in terms of the modern kinetic mechanisms of oxidation of light alkanes at moderate temperatures, e.g., the mechanism proposed in the NUI Galway publication.19 Fig. 8a shows a comparison of the experimental and theoretical temperature dependences of the ignition delay of stoichiometric methane and ethane mixtures with air and their binary mixtures, while Fig. 8b presents the ignition delay of the methane – n-pentane binary mixture as a function of n-pentane concentration. Considering the real experimental error, estimated to be approximately 30%,16 the agreement between the calculated and experimental results appears quite satisfactory.
![[{"id":"SE-Aniwttn","type":"paragraph","data":{"text":"Comparison of the experimental and calculated data on the ignition delay: temperature dependence of the ignition delay time in air for stoichiometric mixtures: methane, mixture of methane with 5 and 10 vol.% ethane, and ethane (<i>a</i>);<sup>16</sup> ignition delay time of a binary methane – pentane mixture vs. n-pentane concentration (<i>b</i>).<sup>39</sup> Initial temperature T = 990 K; initial pressure P = 1 atm. The symbols show the experimental data, and the lines correspond to the calculated results based on the NUI Galway mechanism. The error of experimental determination of the delay time τ does not exceed 30%."}}]](/storage/images/resized/63HmG44J080kzA79jvwQAfXIgFfLeLhJi5oFTOLs_xl.webp)
The calculations carried out by Arutynov et al.39 on the basis of the reported 19 kinetic model confirmed the experimentally elucidated trends, particularly the conclusion that the ignition delay time of stoichiometric methane – n-propane – air mixtures at a pressure of 1 atm and 800 ≤ T (K) ≤ 1000 may exceed the ignition delay time of a methane – ethane – air mixture, all other conditions being equal. Fig. 9 shows the calculated temperature dependence of the ignition delay time of stoichiometric methane – ethane – air and methane – n-propane – air mixtures at 800 ≤T (K) ≤ 1000 and atmospheric pressure; they indicate that the ignition delay time of the methane – ethane mixtures is higher at T = 800 K, which is in line with experimental results (see Fig. 1). This means that due to the difference between the effective activation energy of ignition delay, the relative effect of each of the C2 – C5 alkanes on methane ignition may differ for different temperature ranges. Thus, the relative influence of alkanes on the knock resistance of methane and, hence, the knock resistances of natural gas and associated petroleum gas may depend on the particular type and operation mode of the engine.
![[{"id":"1ExporgWNu","type":"paragraph","data":{"text":"Calculated dependence of the ignition delay time on the initial temperature of stoichiometric methane – ethane – air (dashed line) and methane – n-propane – air (solid line) mixtures at P = 1 and at 5 vol.% (<i>1</i>) and 10 vol.% (<i>2</i>) concentration of the alkane added to methane. The Figure was created by the authors using published data.<sup>39</sup>"}}]](/storage/images/resized/GC99P59kCdABl1I6tvP2yK0kU2z09O9lKAbNYObx_xl.webp)
Modelling also confirmed the assumption based on experimental results about the existence of a weak synergistic effect on the ignition delay time of methane mixed with two heavier alkanes. The minimum delay is attained when the concentrations of any two C2 – C5 alkanes added to methane are approximately equal.39
The results of modelling indicate that the currently attained level of theoretical description of the oxidation of light alkanes is quite adequate. This enables theoretical analysis of the behaviour of systems based on the oxidation of light alkanes under conditions for which no experimental data are now available or are very difficult to obtain. The agreement between the modelling and experimental results also makes it possible to substantiate the use of analytical dependences for fast assessment of the ignition delay for complex hydrocarbon mixtures.39 These dependences are considered in more detail in Section 7.
4. Ignition of methane mixtures with carbon monoxide
In addition to hydrocarbons and hydrogen, carbon monoxide is one of the major components of various industrial gas chemistry processes, related to the production of large-scale products such as ammonia, methanol, synthetic liquid hydrocarbons, carbonylation products, and some other. Therefore, study of the conditions of autoignition of carbon monoxide mixtures with hydrocarbons, first of all, methane is necessary to ensure safety of these processes. It is also important for fire safety of coal mines. The inevitable penetration of CO during fires into combustible mixtures that have not yet ignited may affect the ignition conditions and flame propagation. Experimental data about the ignition delay time of methane and carbon monoxide mixtures is also necessary to further develop the kinetic mechanisms of hydrocarbon oxidation, in which a noticeable role is played by reactions involving CO.
Characteristic features of carbon monoxide ignition have attracted attention back at the infancy of kinetics where CO has become a model object.40 A carbon monoxide – air mixture free from any gases containing hydrogen atoms does not ignite under standard conditions, due to the absence of chain carriers and, hence, the absence of branched-chain reactions needed for flame propagation. However, the presence of even a minor amount of hydrogen, e.g., as water vapour, provides conditions for the formation of chain carriers, with hydrogen and water vapour having approximately the same effect on the CO oxidation kinetics.
Carbon monoxide is one of the main combustion intermediates of virtually all hydrocarbons, which is already formed in the very early stages of the induction period, along with radicals such as H•, OH•, O••, HO2•, CH3•, etc. Carbon monoxide is involved in many important elementary reactions in the hydrocarbon oxidation mechanism. Its presence can substantially affect the ignition of hydrocarbons, especially in the low-temperature region where a pronounced contribution is made by the specific oxidation mechanism involving the formation of relatively stable peroxide compounds. The addition of CO to hydrocarbon fuel may also influence particular elementary reactions by changing the ratio between the forward and reverse reaction rates.
There are diverse experimental data on the ignition of both pure CO 41, 42 and CO – H2 mixtures (syngas),43 – 45 which are considered in more detail in Section 6. However, the information on the influence of carbon monoxide addition on the ignition delay of methane and other light alkanes is scarce. Our results on the effect of the composition of CH4 and CO mixtures on their ignition at ambient or elevated pressure at relatively low temperatures (T ≤ 1000 K)46 are presented below.
As expected,40 ignition of stoichiometric mixtures of carbon monoxide and air does not take place in the indicated temperature range in the bypass installation described earlier.15, 16 The ignition of stoichiometric methane – carbon monoxide mixtures in air containing 10 to 95 vol.% CO (as a mixture with methane) showed good agreement of the temperature dependence of the ignition delay time with the Arrhenius equation. However, the effect of carbon monoxide concentration on the methane ignition proved to be non-trivial. When present in a concentration of up to 60 vol.%, carbon monoxide had a weak promoting effect on the ignition of methane by slightly decreasing the ignition delay time. However, further increase in the CO concentration in the fuel leads to a sharper decrease in the ignition delay time (Fig. 10).
![[{"id":"jxG6wm2yy_","type":"paragraph","data":{"text":"Experimental and calculated dependence of the ignition delay time of CH<sub>4</sub> – CO – air mixtures for CO concentration in the fuel (mol.%): 10 (<i>1</i>), 40 (<i>2</i>), 80 (<i>3</i>), 95 (<i>4</i>). The symbols correspond to experimental data and the lines show the calculation results. ϕ = 1, P = 1 atm.<sup>46</sup>"}}]](/storage/images/resized/CaLBxuIyfuwoD1mEa6Vo6quUOw53hzaiAytUMLXC_xl.webp)
In this case, the effective activation energy of ignition delay (Eа) for the mixture increases almost fivefold (Fig. 11), which unambiguously attests to profound changes in the process mechanism. This increase in the effective activation energy is accompanied by an equally pronounced decrease in the pre-exponential factor A in the Arrhenius equation (see Fig. 11).
![[{"id":"JOge3kYGXL","type":"paragraph","data":{"text":"Experimental values of the effective activation energy E<sub>a</sub> (<i>1</i>) and pre-exponential factor A (<i>2</i>) <i>vs</i>. the concentration of carbon monoxide in its mixtures with methane during oxidation by air. ϕ = 1, P = 1 atm.<sup>46</sup>"}}]](/storage/images/resized/mbNjuK09AbW4OQLlnJMU3fvllo6vcXfXv11ujUWX_xl.webp)
This effect of CO concentration on the effective activation energy of ignition delay for CO mixtures with methane is very similar to the effect of hydrogen concentration on the effective activation energy for methane – hydrogen mixtures (Section 5). In a certain range of conditions, an increase in the hydrogen concentration also induces an approximately fivefold increase in the activation energy accompanied by a counterbalancing decrease in the pre-exponential factor. In both cases, this is related to pronounced changes in the oxidation mechanisms of both components,47 – 49 which are discussed below.
The results of kinetic modelling of self-ignition of carbon monoxide mixtures with methane of various compositions in air in terms of the proposed mechanism 19 in comparison with the experimental results obtained by Troshin et al.46 are presented in Fig. 10. With the effects of temperature and CO concentration on the methane ignition delay being qualitatively the same, it is noteworthy that, unlike experimental results, modelling follows the Arrhenius dependence less stricly, with an obvious trend towards a decrease in the activation energy with increasing temperature, which is especially pronounced in the case of high contents of carbon monoxide in the mixture. Generally, a quite satisfactory qualitative and even quantitative agreement between the modelling and experimental results can be noted.
The ignition delay times of stoichiometric CH4 – CO – air mixtures were calculated for the initial temperature range of 800 – 1000 K, initial pressures of 1, 5, 10, and 15 atm, and CO concentrations in the fuel of 20, 30, 80, and 95 vol.%.46 Fig. 12 shows the pressure dependence of the ignition delay time and the effective activation energy of ignition delay for three compositions of the mixture.
![[{"id":"UDPTByLyKr","type":"paragraph","data":{"text":"Calculated dependences of the ignition delay time for CH<sub>4</sub> – CO – air mixtures (<i>a</i>) and effective activation energy of ignition delay for these mixtures on the initial pressure P0 (<i>b</i>) for CO content in the fuel of 20 (<i>1</i>), 80 (<i>2</i>), and 95 vol.% (<i>3</i>). ϕ = 1, T = 950 K.<sup>46</sup>"}}]](/storage/images/resized/nOArUmoy4Co3D9KBhk3Vx8itW0R0b8aQhl7WXRGt_xl.webp)
The results of modelling indicate that the effects of pressure on the ignition, and even on its nature, differ considerably depending on the carbon monoxide content in the mixture. When the carbon monoxide content is low, a pressure increase induces a marked decrease in the ignition delay time (see Fig. 12а), but also a noticeable increase in the effective activation energy (see Fig. 12b). However, at higher pressure, the effect of CO becomes less pronounced. Probably, this accounts for the fact that a minor influence of 20 vol.% CO on methane ignition was noted in experiments carried out at pressures from 20 to 80 atm by the rapid compression method.50 When [CO] = 80%, the effect of pressure on the ignition delay time becomes less pronounced (see Fig. 12а). The activation energy of the ignition delay in the considered temperature range at this carbon monoxide concentration is nearly constant (see Fig. 12b).
For a mixture with suppressing concentration [CO] = 95 vol.%, or higher, the ignition delay time is maximized at a pressure of ~5 atm, and then smoothly decreases as pressure is further increased (see Fig. 12а). Meanwhile, the activation energy decreases rather significantly with pressure rise at this CO concentration (see Fig. 12b).
Special attention should be paid not only to the sharply different patterns of pressure dependence of the ignition delay time for CH4 – CO – air mixtures with different carbon monoxide contents (Fig. 12а), but also to equally sharp difference between the pressure dependences of the effective activation energy of ignition delay. When the carbon monoxide content is relatively low, Ea increases with pressure rise; when [CO] = 80 vol.%, it is barely pressure-dependent; and when the carbon monoxide concentration is very high ([CO] = 95 vol.%), it noticeably decreases with pressure rise (see Fig. 12b). As a result, when the temperature is above 900 K, the ignition delay for a mixture with [CO] = 20 vol.% becomes lower with increasng temperature than those for mixtures with higher CO contents. In other words, promotion of methane ignition by carbon monoxide at low temperature is replaced by inhibition of methane ignition by CO above 900 K. However, despite this highly different effects of pressure on the ignition delay time of CH4 – CO – air mixtures of different composition, the pressure effect on the maximum concentrations of the OH• radicals, which are major radicals that maintain the ignition process in this system, and other reactive radicals, such as Н•, О••, and НО2•, is virtually the same: their maximum concentrations attained at the moment of ignition monotonically decrease with pressure rise.
Fig. 13 shows the calculated variation of the concentration of the determining components (CO and OH•) during ignition of stoichiometric CH4 – CO – air mixtures at T = 950 K and CO content in the fuel of 5, 30, and 80 vol.% for three initial pressures P = 5, 10, and 15 atm. The kinetics of other reactive radicals (Н•, О••, and НО2•) are similar to that of OH•.
![[{"id":"H_c2Y2EwyI","type":"paragraph","data":{"text":" Kinetics of variation of the concentrations (X<sup>j</sup>, mole fraction) of carbon monoxide (solid line) and OH<sup>•</sup> radicals (dashed line) during ignition of stoichiometric CH<sub>4</sub> – CO – air mixtures at T = 950 K and CO contents in the fuel of 5, 30, and 80 vol.%. P = 5 atm (<i>a</i>), P = 10 atm (<i>b</i>), P = 15 atm (<i>c</i>).<sup>46</sup>"}}]](/storage/images/resized/SXXtZKwtXte6iWcGJAjDdlddTeA38q7lk7mm2o09_xl.webp)
It follows from the above results of modelling that the generation and subsequent decrease in the concentration of reactive radicals such as OH•, H•, and О••, which determine the propagation of the process, occurs almost instantaneously at the point of ignition, whereas a significant change in the carbon monoxide concentration is extended over time and begins directly in the induction period. However, immediately after the end of the flash and a sharp decrease in the concentration of reactive radicals, the carbon monoxide concentration virtually ceases to change. This indicates that during the induction period, the formation of carbon monoxide also involves radicals and is, most likely, associated with radical decay and chain termination. The conversion of carbon monoxide takes place mainly via reaction with radicals and, hence, it stops as the concentration of the radicals decreases. As shown by experimental studies of the partial oxidation of methane, ethane, and other hydrocarbons, the major contribution to the CO oxidation and the experimentally observed yield of CO2 over long time periods is made by heterogeneous processes on the reactor surface, which leads to pronounced underestimation of the CO2 yields in calculations taking account of only gas phase mechanisms.51
When the carbon monoxide content in the mixture is low and methane is the major component of the fuel, the concentration of CO during the induction period increases, like in the ignition of methane alone, and after completion of the process, it reaches a stationary level, which depends little on the starting CO concentration. In the case of high content of carbon monoxide where it can be regarded as the major component of the fuel, the CO concentration somewhat decreases during the induction period, sharply increases to a maximum at the moment of ignition, and then decreases equally sharply to a constant value after completion of the process. A fact deserving attention is that upon variation of the CO concentration over a wide range, the maximum and final concentrations of the main reactive radicals, OH•, H•, О••, and НО2•, which carry the reaction, change only slightly, which probably attests to the predominant role of the methane oxidation mechanism in the ignition of CH4 – CO mixtures.46
A pressure rise results in a decrease in the induction period (decrease in the ignition delay time) (see Fig. 13), but all of the indicated trends are preserved. However, as pressure increases, the difference between the ignition delay times of mixtures with low and high carbon monoxide contents decreases. When P = 15 atm (Fig. 13с), the ignition delay time of a mixture with [CO] = 80 vol.% becomes greater compared to that for mixtures with low CO contents; in other words, carbon monoxide inhibits the ignition of methane.
Analysis of the oxidation kinetics of CH4 – CO mixtures in the indicated temperature and pressure ranges and component ratios 46 attests that the branched-chain methane oxidation is the key reaction mechanism for CH4 + CO mixtures in which methane predominates. Analysis of the sensitivity of particular steps to the conversion of the main reactants demonstrated that, like in almost any other methane conversion reaction, the consumption of methane mainly takes place via the reaction with hydroxyl radicals

The main channel of carbon monoxide consumption in all cases is also the reaction with hydroxyl radicals (Fig. 14)

![[{"id":"an7qSBSlXp","type":"paragraph","data":{"text":"Contributions of elementary steps to the CO balance under various conditions (for each set of conditions, six most important elementary steps are given and the rates of the most significant reactions are indicated for comparison).<sup>46</sup> M is the third body, that is, any molecule present in the gas phase and giving-off excess energy."}}]](/storage/images/resized/SdQDfXLHRucHzmUnfd7Ne6pitewSpAr454YbsiXQ_xl.webp)
However, when the carbon monoxide content is low, the major contribution to the carbon monoxide balance is made by its formation via the reaction (Fig. 14а – d ).
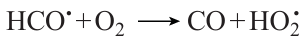
When CO concentration is high, the same reaction is the major oxygen consumption pathway.
It is also noteworthy that the reaction

plays an important role in the carbon monoxide consumption at low temperature and especially at high pressure. However, at higher temperature, the contribution of this reaction sharply decreases, although it is still noticeable as long as the pressure is high. When the CO concentration in the mixture is 95%, the process acceleration with increasing pressure at low temperature and the retardation at higher temperature, as pressure increases from 1 to 5 atm (Fig. 13с), may be due, first of all, to competition of reactions (7) and (8)
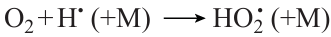
When the initial temperature is moderate and the initial pressure is high, a considerable contribution to the process is made by the reaction

A significant role in the ignition belongs to the formation of formaldehyde, which can take place not only by the reaction
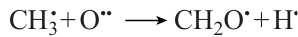
but, to a much higher extent, also by the highly exothermic reaction

which makes a considerable contribution to the heat evolution in this process.
The reactions involving formaldehyde


afford HCO· radicals, which provide for CO regeneration via reaction (6). Reaction (6) is important for several reasons. First, even at a relatively low rate, it makes a noticeable contribution to heat evolution. Second, it is this reaction that mainly provides for the increase in the CO concentration during the ignition delay period when the initial CO concentration in the mixture is not very high (see Fig. 13).
Methane is consumed, in addition to reaction (4), also in the following reaction
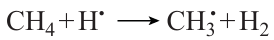
However, reaction (4), which has a lower activation energy, is still the main methane consumtion pathway. Although reaction (4) is merely a chain propagation reaction, it is accompanied by considerable heat evolution, whereas reaction (14) is slightly endothermic.
Apart from the chain termination reaction
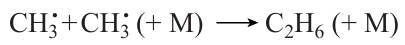
the following reaction is important:

At low temperatures, the forward reaction giving methylperoxy radicals CH3O2• predominates; this results in the formation of oxygenates or in the chain termination. However, at higher temperature, the equilibrium shifts to the left, the concentration of methylperoxy radicals sharply decreases, and their participation in the process becomes insignificant.49
As the CO proportion in the mixture increases, the contribution of the reactions associated with methane oxidation declines. Simultaneously, the role of carbon monoxide oxidation reactions (5) and (7) increases. Reaction (5) not only provides fast chain propagation, but also makes a significant contribution to the heat evolution in this process. At low temperature, as the CO concentration in the mixture increases, the reaction of formation of hydroperoxyl radicals
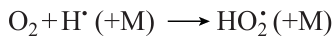
which then participate in reaction (7), becomes more important, especially at elevated pressure. This reaction plays a dual role. On the one hand, it is a major source of heat evolution in this process and, on the other hand, it is one of the main chain termination pathways. Particularly, the competition of reactions (7) and (17) may be responsible for the process acceleration with increasing pressure at low temperature and the process retardation at higher temperature as the pressure increases from 1 to 5 atm, which was observed experimentally at high CO concentrations in the mixture.
Reaction (17) with a zero activation energy at low temperature actively inhibits the ignition.51 At higher temperature, the role of reaction (7) increases. In addition to a noticeable contribution to the heat of the process, this reaction actually counterbalances the chain termination by reaction (17).
Regarding the pressure region above 5 atm, a pressure increase is accompanied by increasing role of the initiation reaction
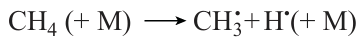
which provides for an additional, although minor, generation of H• radicals, and thus accelerates the process of ignition.
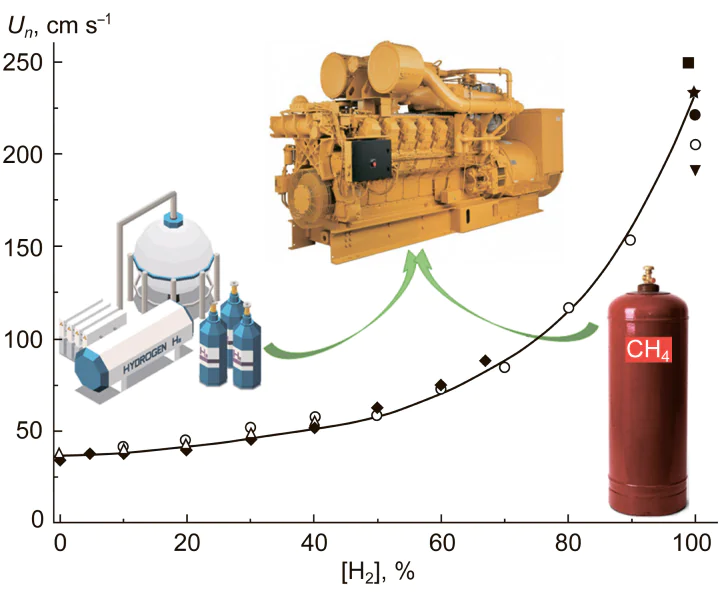
5. Ignition of methane mixtures with hydrogen
In view of the observed climate processes,3 there is wide discussion concerning the need for gradual transition to low-carbon energy sources and energy carriers.2 The most active discussion concerns the possibility of using hydrogen as carbon-free energy carrier. However, simple estimates show that this is unrealistic from an energy, resource, and economic points of view as long as there is no commercial nuclear fusion power generation.7, 53 Therefore, along with more extensive use of natural gas in energy generation and gradual replacement of coal and oil products by natural gas, the most realistic way to reduce the carbon footprint of energy generation is addition of hydrogen to natural gas (methane) and other types of fuel.
The use of methane – hydrogen mixtures with various hydrogen contents makes it possible to circumvent many intricate problems of production, storage, transportation, and distribution of hydrogen.54 – 57 These mixtures can be used, alongside with hydrogen, to feed conventional ICEs,58 – 64 in which the energy efficiency of using hydrogen is not much inferior to the efficiency of fuel cell applications of hydrogen.59 Moreover, currently ICEs are still cheaper and more reliable energy sources than fuel cells, and the use of hydrogen and hydrogen-containing mixtures in ICEs is technically more well-developed. Engine tests have shown that the use of hydrogen-enriched natural gas expands the range of lean mixtures applicable for practical use, with simultaneous decrease in the emissions of unburned hydrocarbons and carbon dioxide.65 Owing to the progress in the development of ICEs, it is expected that by 2045 their fuel efficiency would almost reach that of fuel cells.66 Therefore, the use of hydrogen and methane – hydrogen mixtures in ICEs could form a natural transition between modern ICEs powered by liquid or gas fuel and future fuel cell-based vehicles.67
The potential application of methane – hydrogen mixtures to reduce the carbon content of fuels and to expand the fuel combustion range, which would improve the economic and environmental performance of ICEs and gas turbines, require detailed analysis of the optimal conditions for their use in the existing power equipment. First of all, this concerns the detonation characteristics of methane – hydrogen mixtures and conditions of their ignition in ICEs. It is necessary to predict and control these conditions and characteristics for combustion of gas mixtures, optimize the composition of the mixtures and operating conditions of the existing power equipment using these mixtures, and to ensure their storage and transportation safety.
The main features of hydrogen and methane combustion were established long ago;68 however, there are still quite a few blanks and a large field of research that needs to be performed to provide the possibility of wide practical use of methane – hydrogen mixtures. The ignition delay time and the laminar flame velocity are among the most important parameters determining the optimal conditions and safety of using these mixtures. Although numerous publications address determination of these parameters for hydrogen or methane, there are still few studies of this type for their mixtures. Furthermore, almost all studies of the ignition delay of methane, hydrogen, or their mixtures were carried out using the shock-tubes or rapid compression machines,11, 23, 69 – 76 i.e., temperatures not below 900 – 1000 K. However, the practical use of these mixtures occurs, most often, at lower temperature. In particular, as has already been noted in Section 2, the air – fuel mixture in ICEs ignites at 500 – 900 K.10 Therefore, for optimization of the composition of methane – hydrogen fuel mixtures and their operation conditions in the engine, it is necessary to gain information on their ignition delay at temperatures below 1000 K. Investigation of the ignition of methane – hydrogen mixtures in this temperature range is complicated by the fact that exactly in this region, considerable changes in the oxidation mechanisms of both methane and hydrogen take place.49 At different (although quite similar) temperatures in this range, low-temperature oxidation mechanisms of these gases, in which peroxide compounds and radicals play an important role, is replaced by high-temperature oxidation mechanisms dominated by reactions involving Н• and О•• atoms and relatively simple OH• and СН3• radicals. These changes in the oxidation mechanisms considerably influence the hydrogen and methane ignition, which starts under ambient conditions according to the low-temperature mechanism and then switches to the high-temperature oxidation mode. In the case of methane oxidation, this gives rise to a variety of non-linear effects in this temperature range such as appearance of NTC, cool flames, inhibition of methane oxidation by oxygen, and some other.20 During the oxidation of methane – hydrogen mixtures, the mentioned changes in the oxidation mechanism of the two gases overlap with each other, which results in a complex pattern of observed phenomena, depending on the methane and hydrogen ratio in the mixture, pressure, initial temperature, and some other conditions.49
The intricate character of the effect of hydrogen on the ignition of methane was noted long ago. Gersen et al.77 measured the ignition delay for methane – hydrogen mixtures in a rapid compression machine under stoichiometric conditions for a pressure from 1.5 to 7.0 MPa, a temperature from 950 to 1060 K, and a hydrogen concentration from 0 to 100%. The results indicated that at a hydrogen concentration of < 20 vol.%, its promoting effect is insignificant, but the ignition delay time considerably decreases when the concentration of hydrogen exceeds 50 vol.%. The promoting effect of hydrogen on the ignition of methane is enhanced with increasing temperature, but decreases with increasing pressure.
It was found 73 that at T > 1000 K, the measured ignition delay time of methane – hydrogen mixtures is in good agreement with the theoretical predictions, whereas at T < 1000 K the experimental value is much smaller than the calculation result; for T ~ 800 K, the difference can be as high as three orders of magnitude.
The reflected shock wave experiments at temperature from 1000 to 2000 K and pressure from 5 to 20 atm and the kinetic modelling based on the NUI Galway mechanism 19 demonstrated that the pressure dependence of the ignition of methane – hydrogen mixtures with hydrogen concentration below 40 vol.% resembles the pressure dependence of the ignition delay of methane, particularly, the ignition delay decreases with increasing pressure.74 When the hydrogen concentration is 60 vol.%, the promoting effect of pressure on the ignition of methane–hydrogen mixtures is negligibly low. For the concentration of hydrogen above or equal to 80 vol.%, the behaviour of these mixtures rather resembles the behaviour of hydrogen: the ignition delay time demonstrates a complex pressure dependence and the effective activation energy of ignition delay follows a complex temperature dependence.
Zhang et al.75 distinguished three ignition regimes according to the content of hydrogen in the mixture: when [H2] ≤ 40 vol.%, the mechanism of methane oxidation predominates, when [H2] = 60 vol.%, combined features inherent in both methane and hydrogen oxidation mechanisms appear, while for [H2] ³ 80 vol.%, hydrogen oxidation mechanism prevails. In addition, a substantial difference was revealed between the temperature dependences of the ignition delays of methane and hydrogen at high and low temperatures. Whereas at temperatures of > 1250 K, conventional Arrhenius relation holds in both cases, at lower temperature, this dependence is more intricate for both methane and hydrogen, and the activation energy of ignition delay considerably varies (Fig. 15).
![[{"id":"bps1QUX5bk","type":"paragraph","data":{"text":"Temperature dependence of the ignition delay for methane (<i>a</i>) and hydrogen (<i>b</i>). The symbols show the experimental data, and the lines correspond to the kinetic calculations. The Figure was created using published data.<sup>75</sup>"}}]](/storage/images/resized/gAu6Uaw86v92OkOJjTq71BNI8HmpEoINvavSgqTN_xl.webp)
In view of the importance of the data about ignition conditions and combustion characteristics of methane – hydrogen mixtures in the 800 – 1000 K range, Arutyunov et al.49 determined experimentally the ignition delay times of methane – hydrogen mixtures with hydrogen contents ranging from 0 to 50 vol.% in particularly this temperature range, using the bypass installation described in detail earlier,15, 16 and performed kinetic analysis of the experimental results. The authors noted that in the high-temperature part of the studied range, the ignition delay decreases significantly with increasing hydrogen content in the mixture, that is, hydrogen promotes the ignition of methane. However, at lower temperature (T ≈ 850 K), the promoting effect is weak if at all present. An increase in pressure reduces the range of variation of the effective activation energy of ignition delay following the variation of the hydrogen concentration.
As the hydrogen concentration increases, the effective activation energy for methane – hydrogen mixtures increases, but this is accompanied by a decrease in the pre-exponential factor А. A threefold increase in the effective activation energy, from 23.4 to 73.7 kcal mol–1, takes place upon the increase in the concentration of hydrogen from 0 to 50 vol.% at T ≈ 900 K and P = 1 atm (Fig. 16). This pronounced change in Ea attests to a marked change in the ignition mechanism in this temperature range. In this respect, the effect of hydrogen on the ignition of methane substantially differs from the effect of C2 – C6 alkanes (see Section 3), for which the effective activation energy for autoignition delay was nearly invariable, being 40 ± 10 kcal mol–1 (Refs 15, 16, and 28), irrespective of the concentration of the added alkane.
![[{"id":"VOaf4Fc8U1","type":"paragraph","data":{"text":"Effective activation energy E<sub>a</sub> (<i>a</i>) and pre-exponential factor A (<i>b</i>) in the Arrhenius equation for the ignition delay of methane – hydrogen mixtures <i>vs</i>. the concentration of hydrogen at P = 1 and 3 atm.<sup>49</sup>"}}]](/storage/images/resized/TWcW5r2PHZDyJlMrdieKdL0idalkO7NWXVcKrFtt_xl.webp)
A consequence of the increase in the activation energy of ignition delay for methane – hydrogen mixtures with increasing concentration of hydrogen is increase in their sensitivity to a temperature change and, hence, decrease in the knock resistance, since not only the ignition delay time, but also the sensitivity of the fuel to characteristics such as temperature, concentration, and pressure are important. The results shown in Fig. 16 indicate that when hydrogen concentrations are below 30 vol.%, pressure has an insignificant effect on the activation energy of autoignition delay. At a higher concentration of hydrogen, the effect of H2 concentration on Ea of methane – hydrogen mixtures is less pronounced at higher pressures.
Experimental results, even those obtained in a limited range of hydrogen concentrations [H2] ≤ 50 vol.%, attest to a complex pattern of influence of hydrogen concentration on the ignition of methane – hydrogen mixtures at temperatures below 1000 K. In order to elucidate the mechanism of this influence, kinetic calculation of the ignition delay times of stoichiometric CH4 – H2 – air mixtures with different hydrogen contents was carried out.49 The kinetic modelling was based on the NUI Galway mechanism 19 as the most adequate one for the description of these processes.28 The resulting temperature dependence of the ignition delay of methane – hydrogen mixtures with various hydrogen contents at 800 – 1000 K is depicted in Fig. 17.
![[{"id":"JgjcYyosYj","type":"paragraph","data":{"text":"Calculated dependence of the ignition delay of stoichiometric CH<sub>4</sub> – H<sub>2</sub> – air mixtures on the initial temperature at H<sub>2</sub> concentrations, vol.%: 0 (<i>1</i>), 10 (<i>2</i>), 20 (<i>3</i>), 30 (<i>4</i>), 40 (<i>5</i>), 50 (<i>6</i>), 60 (<i>7</i>), 70 (<i>8</i>), 80 (<i>9</i>), 90 (<i>10</i>), 100 (<i>11</i>). P = 1 atm.<sup>49</sup>"}}]](/storage/images/resized/4YoANFXq3M0SKb7VbBWYFY2PdA9jMocKJZPTnNYm_xl.webp)
The obtained dependence is indicative of a complex pattern of influence of hydrogen on the ignition delay for methane. When the concentration of hydrogen in the mixture is up to 40 vol.%, the temperature dependence of the ignition delay has virtually an Arrhenius form (see Fig. 17, curves 1 – 5). However, at higher H2 concentrations, the dependence no longer obeys the Arrhenius law (see Fig. 17, curves 6 – 11) and has a clear-cut maximum of the effective activation energy of ignition delay at a temperature of ~900 K (Fig. 18, curves 3 – 5). Whereas the effective activation energy of pure methane (see Fig. 18, curve 1) is virtually invariable, amounting to ~30 kcal mol–1, over the whole considered temperature range, Ea of hydrogen and H2-rich mixtures increases at T ≈ 900 K approximately 3 – 4-fold compared to the Ea values at lower or higher temperatures (see Fig. 18, curves 4, 5).
![[{"id":"fprUPNM4u-","type":"paragraph","data":{"text":"Calculated dependence of the effective activation energy of ignition delay E<sub>a</sub> for stoichiometric CH<sub>4</sub> – H<sub>2</sub> – air mixtures on the initial temperature T<sub>0</sub> at H<sub>2</sub> concentration, vol.%: 0 (<i>1</i>), 40 (<i>2</i>), 70 (<i>3</i>), 90 (<i>4</i>), 100 (<i>5</i>). P = 1 atm.<sup>49</sup>"}}]](/storage/images/resized/sdTNQSYz1hvbl6PmP7znLbhQL9jMqNiTaV4roJix_xl.webp)
Note also that in the low-temperature region (T < 850 K), the effective activation energy of ignition delay is higher for hydrogen or hydrogen-containing mixtures than for methane (see Fig. 18). The causes for this are discussed below. The effective activation energy of ignition delay for mixtures containing up to 40 vol.% hydrogen monotonically decreases with temperature rise and at T > 1100 K, it becomes lower than that for methane (see Fig. 18, curve 2). In the case of methane – hydrogen mixtures rich in hydrogen or for pure hydrogen, the effective activation energy of ignition delay passes through a maximum at T ≈ 900 K, but at T > 1100 K it also becomes lower than that for methane. The higher the hydrogen content in the mixture, the lower Ea (see Fig. 18).
In this connection, the effect of hydrogen on the normal velocity of laminar flame deserves attention. This is an important parameter of combustion of methane – hydrogen mixtures, which has been addressed in many studies (e.g., Refs 78 – 88). It was shown that the addition of hydrogen to methane – air mixtures increases the normal velocity of laminar flame and extends the flame propagation limits, but rather large amount of hydrogen is required to attain a noticeable effect. The kinetic calculations carried out using the proposed mechanism 19 fully confirmed the experimental results and provided the conclusion that hydrogen present in a methane – hydrogen mixture in a concentration below 40 vol.% has a slight influence on the velocity of combustion of this mixture.89 The minor influence of such concentrations of hydrogen on the combustion velocity of methane – hydrogen mixtures is similar to the above-mentioned slight influence of such hydrogen concentrations on the ignition delay time of methane – air mixtures (see Fig. 17) and on the concentration limits of their combustion.
The sharp change in the pattern of dependence of the activation energy of ignition delay for hydrogen and hydrogen-rich mixtures (> 60 vol.%) observed at T ≈ 900 K (see Figs 17 and 18) means that a region around this temperature should be considered as a boundary between low-temperature and high-temperature regions in which the mechanism of the process crucially changes. This is also evidenced by the sharply different pattern of dependence of the activation energy of ignition delay for stoichiometric methane – hydrogen mixtures in air on the hydrogen content observed particularly for this temperature (Fig. 19). The curve for T = 900 K clearly separates two different process regimes characterized by different dependences on the H2 concentration. At low temperature (T < 900 K), the activation energy of ignition delay monotonically increases with increasing H2 concentration in the mixture, while at high temperature (T > 900 K), the activation energy passes through a feeble maximum (see Fig. 19). These temperature regions are separated by a sharply different dependence for T = 900 K in which the activation energy of ignition delay for methane – hydrogen mixtures monotonically increases with increasing concentration of hydrogen from 30.5 kcal mol–1 for methane to 117.7 kcal mol–1 for hydrogen, that is, by a factor of almost four.
![[{"id":"5Vg3oCvU9D","type":"paragraph","data":{"text":"Effective activation energy of ignition delay in air for stoichiometric methane – hydrogen mixtures <i>vs</i>. the hydrogen content at P = 1 atm. (●) experimental results (T = 900 K). Calculated data for T (K): 850 – 900 (▲), 900 (♦), and 950 – 1000 (■).<sup>49</sup>"}}]](/storage/images/resized/MN1vPXWPlS8CDhQzw4yErYBdRkvb7wWRxyn1LnTY_xl.webp)
Fig. 19 also shows the experimental results obtained for T = 900 K. Considering various factors that can distort the data,15, 16 these results are in good agreement with the results of modelling. As the concentration of hydrogen in the mixture increases from zero to 50 vol.%, the experimentally determined activation energy of ignition delay for methane – hydrogen mixture monotonically increases from 23.4 kcal mol–1 to 73.7 kcal mol–1.
A very similar change in the activation energy for hydrogen was observed in shock-tube experiments.74 At P = 5 atm and T > 1000 K, the activation energy of ignition delay for hydrogen was 39.3 kcal mol–1, whereas at lower temperature it was 126.9 kcal mol–1. At higher pressure (10 atm), a change in the activation energy for hydrogen was even greater, but it had the opposite sign relative to the temperature: 74.5 kcal mol–1 at relatively low temperature (~ 1025 K), 258 kcal mol–1 at 1108 K, and subsequent decrease to 49.2 kcal mol–1 upon further temperature rise. A similar behaviour was observed at a pressure of 20 atm.
Note that a considerable increase in the activation energy on going from the low-temperature region (T < 1000 K) to higher temperature region (T > 1100 K) was also found for methane.90 However, it cannot be ruled out that the exceptionally low ignition delay values obtained in this study at T < 1000 K, contradicting the results of modelling, are due to the inaccuracy of temperature determination in the shock wave.
Donohoe et al.,87 who studied the ignition of methane –hydrogen mixtures by the shock-wave method, also observed a sharp change in the activation energy of ignition delay for hydrogen and hydrogen-rich mixtures ([H2] = 80 vol.%), although at somewhat higher temperature (1000 – 1200 K). For mixtures with lower hydrogen and methane contents, the temperature dependence of the ignition delay time in this region was close to the Arrhenius relation. The kinetic modelling conducted by the authors adequately described the obtained results.
The effect of pressure on the ignition of methane – hydrogen mixtures follows a complex pattern, since pressure has opposite effects on the ignition of hydrogen and methane. According to the results of numerous studies obtained mainly in the shock-tubes at temperatures > 1000 K, an increase in the pressure reduces the ignition delay time of hydrocarbons, including methane. Conversely, in the temperature range from 1093 to 1170 K, the ignition delay time of hydrogen increases with increasing pressure.
The results obtained by Zhang et al.74 indicate that at T = 1093 K, the ignition delay time of hydrogen is ten times greater at a pressure of 20 atm than at 5 atm. An intricate pressure dependence was also found by Herzler and Naumann.91
The results of kinetic modelling of the ignition delay for methane – hydrogen mixtures in the considered temperature range and 1 – 15 atm pressure range are depicted in Fig. 20.49
![[{"id":"_yWKp5XBYQ","type":"paragraph","data":{"text":"Temperature dependence of the ignition delay of stoichiometric methane – hydrogen mixtures in air at different initial pressures and hydrogen contents in the mixture: [H<sub>2</sub>] = 20 vol.% (<i>a</i>), [H<sub>2</sub>] = 80 vol.% (b).<sup>49</sup>"}}]](/storage/images/resized/5l2kkidDL43AEp1gMVQaQltt3f9RMHHyxSsD265M_xl.webp)
The results show that at a low hydrogen content ([H2] = 20 vol.%, the temperature dependence of the ignition delay for stoichiometric methane – hydrogen mixtures in air corresponds to the Arrhenius law over the whole considered temperature range and at any of the considered pressure. An increase in the pressure promotes ignition at any temperature (see Fig. 20а). This supports the conclusion that the oxidation of methane – hydrogen mixtures containing a low amount of hydrogen proceeds mainly by the methane oxidation mechanism. However, the situation changes in the case of mixtures with a high hydrogen content ([H2] = 80 vol.%), which are oxidized, according to the conclusions of Zhang et al.,74, 75 by the hydrogen oxidation mechanism. The Arrhenius type of dependence is retained at high pressure (P = 15 atm), but it is distorted when P = 3 atm, and obviously no longer holds when P = 1 atm (see Fig. 20b). Furthermore, whereas in the low-temperature part of the temperature range, an increase in pressure promotes the ignition by reducing the ignition delay time, in the high-temperature part, a pressure rise, conversely, inhibits the ignition. The fundamental change in the type of pressure effect on the ignition process takes place at T ≈ 900 K. These results are quite consistent with the data of Zhang et al.,74 indicating that in the case of hydrogen, the temperature dependence of the ignition delay time approaches the Arrhenius relation as the pressure increases.
A very similar type of dependence of the ignition delay time for hydrogen and for hydrogen-rich methane–hydrogen mixtures was observed in shock tubes.91 Despite a number of differences including higher temperature, the presence of ~8 vol.% ethane in methane, which could affect the dependence of ignition delay on the conditions, and high (1 : 5) dilution of this mixture withargon, in these experiments, a clear-cut maximum of the temperature dependence of the ignition delay time of hydrogen and hydrogen-rich mixtures was also observed. When the pressure was 1 atm, the temperature at this maximum (~ 950 K) virtually coincided with the calculated value (see Fig. 20). As the pressure increased, the maximum progressively shifted to higher temperature: ~1050 K for P = 4 atm and ~ 1250 K for P = 16 atm.
The different patterns of pressure effect on the ignition delay time of methane – hydrogen mixtures at different temperatures is clearly seen in Fig. 21, which presents the calculated dependence of the ignition delay time of stoichiometric CH4 – H2 – air mixtures at P = 15 atm on the initial temperature for various concentrations of hydrogen.49 At high temperature, an increase in the hydrogen concentration promotes the ignition, whereas at low temperature it obviously inhibits the ignition, although to a small extent. These data are quite in line with experimental results, which show that at low temperature (T ≈ 850 K), hydrogen slightly promotes the ignition of methane, but at higher temperature the promoting effect of hydrogen increases.
![[{"id":"nlPNfcMQc0","type":"paragraph","data":{"text":"Calculated dependence of the ignition delay time of stoichiometric CH<sub>4</sub> – H<sub>2</sub> – air mixtures on the initial temperature at various H<sub>2</sub> concentrations. P = 15 atm.<sup>49</sup> The concentrations of hydrogen are in vol.%."}}]](/storage/images/resized/pNgyyAV7QVTF8BVzdJMPBZ0uIN6NqHxlM21WvQEK_xl.webp)
The calculated pressure dependences of the ignition delay time of methane – hydrogen mixtures with various hydrogen contents in the pressure range from 1 to 15 atm at T = 900 K obtained by Arutyunov et al.49 are shown in Fig. 22. When the hydrogen content is low, the ignition delay monotonically decreases with increasing pressure. However, in the case of pure hydrogen and hydrogen-rich mixtures at low pressure, a pressure increase leads to longer ignition delay of methane – hydrogen mixtures, giving rise to a maximum at P ≈ 3 atm. Only further pressure increase results in a monotonic decrease in the ignition delay time.
![[{"id":"4miGph5R_f","type":"paragraph","data":{"text":"Ignition delay time of stoichiometric CH<sub>4</sub> – H<sub>2</sub> – air mixtures <i>vs</i>. initial pressure at T = 900 K and at various concentrations of hydrogen.<sup>49</sup>"}}]](/storage/images/resized/tRYVydV9NrLw1BshexHhWYbcK9EWPAhyHSdNN6Cy_xl.webp)
Similar effects were found for hydrogen.74 When the temperature was above 1000 K, the increase in the hydrogen pressure from 5 to 20 atm led to noticeable increase in the ignition delay time and a change in the activation energy. For an even higher temperature (> 1100 K), apparently, because of the complete switching to the high-temperature mechanism of hydrogen oxidation, the ignition delay time and activation energy were no longer pressure-dependent.
Below we present a kinetic interpretation of the observed regularities of the ignition of methane – hydrogen mixtures in the 800 – 1000 K range.49 The fact that activation energy of ignition delay for a stoichiometric methane – air mixture is almost invariable in the considered temperature range and is substantially lower that the activation energy for the generation of active sites,
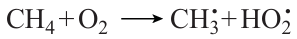
amounting to ~57 kcal mol–1 (Ref. 20), is attributable to the branched-chain mechanism of the process in this temperature range. It is noteworthy that the activation energy for the branched-chain partial oxidation of mixtures very rich in methane at similar temperatures was determined experimentally to be 46 kcal mol–1 (Ref. 20), while the activation energy found for methane-rich mixtures by Zhang et al.74 was in the range of 42.8 – 48.4 kcal mol–1, that is, it was also considerably lower than the activation energy of reaction (19).
At a temperature of < 900 K, the activation energy of ignition delay increases with increasing hydrogen concentration in the mixture (see Fig. 19), which can be formally interpreted as an inhibitory effect of hydrogen on the ignition of methane in this temperature range. The significant difference in the behaviour of mixtures with high and low hydrogen contents is apparently caused by different low-temperature (T < 900 K) mechanisms of methane and hydrogen oxidation.
When the temperature is below 900 K, the key role in the methane oxidation is played by methylperoxy CH3OO• radicals, which are formed in the reversible reaction
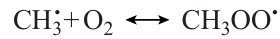
The subsequent transformations of these radicals afford methyl hydroperoxide CH3OOH, which quickly decomposes under the reaction conditions to give radicals, which results in degenerate chain branching
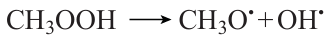
Consequently, at a temperature below 900 K, the oxidation of methane proceeds as a fast branched-chain reaction 20 with an effective activation energy markedly lower than the bond rupture energy in the active centers generation reaction (19). However, at temperatures above 900 K, the equilibrium in reaction (20) rapidly shifts to the left, the rate of formation of methylperoxy radicals and, hence, methyl hydroperoxide sharply decreases, and the reaction ceases to be branched. This leads to decreasing reaction rate, and the reaction proceeds in the NTC region. Further temperature rise induces again a branched-chain methane oxidation reaction, which now follows a different, high-temperature mechanism.20 Therefore, temperatures around 900 K form a transient region from the low-temperature to high-temperature oxidation of methane.
By coincidence, the transient region for the hydrogen oxidation mechanism is located at similar temperatures, although the causes are different. At temperatures below 900 K, the radical initiation reaction in the oxidation of hydrogen, similar to reaction (19)

gives rise to НО2• and H• radicals, which are involved in the reaction similar to reaction (20), also giving rise to the hydroperoxyl radicals НО2•. However, unlike methylperoxy radicals, НО2• radicals are low-reactive at temperatures below 900 K and mainly decay via recombination
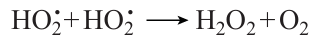
Thus, this leads to chain termination, as the hydrogen peroxide molecule, unlike methyl hydroperoxide, is relatively stable at these temperatures. Hydrogen peroxide decomposition by the reaction

similar to reaction (21), does not provide sufficiently fast chain branching. The rate of high-temperature branching in the hydrogen oxidation mechanism
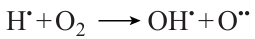
is still too low at this temperature. Therefore, the oxidation of hydrogen at low temperature proceeds as an unbranched reaction, and the addition of hydrogen to the system may even induce inhibition because of the additional consumption of the methyl radicals via the reactions
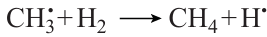
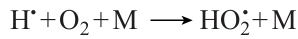
and then in reaction (23) to give hydrogen peroxide, which is relatively stable under these conditions. Apparently, this accounts for the threefold increase in the effective activation energy upon increase in the hydrogen concentration in methane – hydrogen mixtures.
Kinetic modelling gave the key pathways of reactant conversion in the chain mechanism of ignition of CH4 – H2 – air mixtures at various temperatures and hydrogen concentrations, which are depicted in Fig. 23.49
![[{"id":"yzLYXp33aW","type":"paragraph","data":{"text":"Key pathways of reactant conversion in the chain mechanism of ignition of CH<sub>4</sub> – H<sub>2</sub> – air mixtures. [H<sub>2</sub>] = 20 vol.%, T < 900 K (<i>a</i>); [H<sub>2</sub>] = 80 vol.%, T < 900 K (<i>b</i>); [H<sub>2</sub>] = 20 vol.%, T > 900 K (<i>c</i>); [H<sub>2</sub>] = 80 vol.%, T > 900 K (<i>d</i>).<sup>49</sup>"}}]](/storage/images/resized/jOGxvy4skif4WE32G0wqQQ3FW8xf6HYxZN1LUCR0_xl.webp)
When T < 900 K and the H2 concentration is low, hydrogen enhances the withdrawal of reactive radicals formed in the branched-chain oxidation of methane, which are converted to hydrogen peroxide (see Fig. 23a). On the contrary, when the H2 concentration is high, the oxidation of hydrogen is promoted by the branched-chain methane oxidation (see Fig. 23b). When T > 900 K and the H2 concentration is low, conjugate radical reactions of methane and hydrogen oxidation take place, where hydrogen promotes the oxidation of methane (see Fig. 23c). When T > 900 K and the H2 concentration is high, despite the common radical pool in the system, they are oxidized rather independently (see Fig. 23d ). This interpretation is indirectly supported by fast increase in the maximum concentration of hydrogen peroxide with increasing initial concentration of hydrogen for the oxidation of methane – hydrogen mixtures in the low-temperature (~800 K) region (Fig. 24).
![[{"id":"LvIiy3ao2P","type":"paragraph","data":{"text":"Calculated dependence of the maximum concentration of hydrogen peroxide (mole fractions) on the initial temperature at various H<sub>2</sub> concentrations (vol.%): 0 (<i>1</i>), 40 (<i>2</i>), 70 (<i>3</i>), 90 (<i>4</i>), 100 (<i>5</i>).<sup>49</sup>"}}]](/storage/images/resized/xZKI4PfNLpX1p48dLmW4xZyjXXoRTN4WiPnSDbw1_xl.webp)
As the temperature increases, the reactivity of hydroperoxyl radicals НО2• and the hydrogen peroxide decomposition rate increase. When T ≈ 900 K, the rate of hydrogen peroxide decomposition is sufficiently high, and the maximum concentration of H2O2 rapidly decreases with increasing reaction temperature (see Fig. 24). Therefore, at T > 900 K, the recombination of the peroxyl radicals НО2• no longer inhibits the reaction, which results in the considerable change in the mechanism and, correspondingly, regime of the oxidation of hydrogen and hydrogen-rich mixtures. This fundamental change of the hydrogen oxidation mechanism is called ‘H2O2 turnover’.92 In addition, an increase in the initial reaction temperature leads to fast increase in the contribution of the branching reaction (25), and the oxidation of hydrogen becomes a branched-chain reaction. When the hydrogen content in the mixture is high, this is manifested as a fast decrease in the effective activation energy of ignition delay. The above-described changes in the mechanism of hydrogen oxidation give rise to a maximum in the temperature dependence of the activation energy of ignition delay for hydrogen and hydrogen-rich mixtures (see Fig. 18).
The unique ability of methane to provide a branched-chain mechanism of oxidation at relatively low temperatures fundamentally distinguishes the low-temperature oxidation of methane not only from the oxidation of hydrogen, but also from the oxidation of its close homologues, in which the formation of alkylperoxy radical RO2•· by a reaction similar to reaction (16) in the methane oxidation
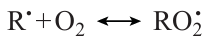
is followed by fast isomerization of the radical and the subsequent decomposition to give olefin Q= and relatively inert НО2• radical
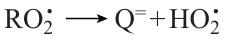
This actually leads to chain termination, which rules out the branching chain regime of the reaction. Therefore, despite a higher energy of bond cleavage and, hence, a higher activation energy of radical initiation, methane has a lower effective activation energy of ignition delay than hydrogen, ethane, propane, or even butane.15, 16, 28 However, at temperatures above 900 K, the rate of formation of methylperoxy radicals and their role in the methane oxidation are virtually eliminated, and the main features of methane and hydrogen oxidation become similar, which accounts for similar values for the activation energy of ignition delay for methane, hydrogen, and their mixtures at T > 1000 K (see Fig. 18). It is quite natural that due to the lower energy of H – H bond cleavage compared to the CH3 – H bond energy, the activation energy of ignition delay in this region is lower for hydrogen and hydrogen-rich mixtures than for methane.
As the methane and hydrogen ratio in the mixture varies, their contributions to the oxidation process change. When the initial concentration of hydrogen is low, as a result of fast branched-chain process during ignition, the concentration of methane soon decreases to zero. The concentration of hydrogen, which is an intermediate oxidation product in this case, increases to reach a maximum, which is equal to nearly one-third of the initial methane content. Then, after completion of the branched-chain process, the concentration of hydrogen rapidly decreases to some stationary value, which corresponds to the thermodynamically equilibrium composition of products (Fig. 25).
![[{"id":"Gn1CP6E9t1","type":"paragraph","data":{"text":"Kinetics of variation of the hydrogen concentration (mole fractions) at T = 900 K and initial hydrogen concentrations in the methane–hydrogen mixture, [H<sub>2</sub>] (vol.%): 10 (<i>1</i>), 20 (<i>2</i>), 30 (<i>3</i>), 50 (<i>4</i>).<sup>49</sup>"}}]](/storage/images/resized/BcBjn4PfD1DyPzGN9bubNhyQ5YCrFYb6bWkMl8Qv_xl.webp)
However, whereas the pattern of variation of methane concentration is qualitatively the same over the whole ranges of initial temperature 800 ≤ T (K) ≤ 1000 and hydrogen concentration in the fuel 0 ≤ [H2] (vol.%) ≤ 90, the behaviour of hydrogen considerably changes following the variation of its content in the mixture. Kinetic calculation of the variation of hydrogen concentration at T = 900 K and various [H2] indicates 49 (see Fig. 25) that for [H2] between 10 and 30 vol.%, a minor smooth decline before ignition is followed by a sharp increase in the hydrogen concentration immediately at the instant of ignition and then by a rapid decrease in the hydrogen concentration to some stationary value. As the percentage of H2 in the initial mixture increases, the peak concentration of hydrogen decreases, and the final stationary concentration somewhat increases.
When [H2] = 10 vol.%, the decline of hydrogen concentration before ignition is barely noticeable, but as [H2] increases, the difference between the initial hydrogen concentration and the level attained by the instant of ignition becomes more pronounced. When [H2] = 40 vol.% (not shown in Fig. 25), the peak in the curve of variation of the hydrogen concentration becomes hardly visible, while at [H2] = 50 vol.% (see Fig. 25, curve 4), it cannot be detected at all, and the pattern of variation of hydrogen concentration becomes similar to that for methane. When the initial H2 concentration is further increased, this similarity is retained. A change in the temperature does not induce a qualitative change in the picture, but the peak concentration of hydrogen increases with increasing initial temperature.
The presence of a peak of hydrogen concentration is due to the competition between its formation and consumption processes. In the absence or in the case of low initial concentration of H2 in the fuel, the formation processes obviously predominate in the early stage. As the percentage of hydrogen in the initial mixture increases and the percentage of methane, correspondingly, decreases, hydrogen oxidation prevails from the very beginning over the hydrogen formation in secondary reactions, and the peak in the kinetic curves disappears.
The ignition delay of the H2 subsystem is determined by the competition of reactions (25) and (27). Reaction (27) predominates at higher pressures, whereas reaction (25) predominates at higher temperatures due to the high activation energy. If reaction (27) is markedly faster than reaction (25), the ignition delay increases, because this reduces the chain branching rate by reaction (25). Hence, the ignition of pure hydrogen at T < 1100 K proceeds more slowly at 16 atm than at 4 or 1 atm, since reaction (27) is approximately 4 and 16 times, respectively, faster at higher pressure.91 Thus, at low temperatures, the influence of reaction (27) surpasses the effect of increasing concentration with pressure rise, while at higher temperature, the concentration effect with pressure rise prevails.
6. Effect of hydrogen and carbon monoxide mixture (syngas) composition on its ignition
Syngas is an intermediate product in processing natural gas into large-tonnage products such as ammonia, methanol, synthetic liquid hydrocarbons, as well as the most commonly available and the cheapest source of hydrogen.7, 53 Syngas is also an efficient low-carbon gas fuel, making possible a relatively «clean» use of the world’s vast coal resources for gas turbine power generation.93 – 95
Depending on its production technology and an intended use, the H2/CO ratio in syngas can vary very widely,96 so information is needed on the ignition behaviour of the H2/CO mixture over the entire range of each component concentration, from 0 to 100%. Among other reasons explaining the importance of studying the ignition dependence of H2/CO mixtures on their composition is the known evidence that these mixtures have lower ignition limits than each of the components separately.97 Moreover, in almost all syngas production processes, impurities such as water, carbon dioxide, methane and nitrogen are present in the gas and their effect on ignition is still largely unexplored.
The main combustion and ignition characteristics of syngas are determined by its hydrogen component, which is well illustrated by the dependence of the lower ignition limit of syngas on its hydrogen concentration shown in Fig. 26, where the oxidation mechanism of the latter has been the subject of extensive research for many decades.68
![[{"id":"zg5zeh83J7","type":"paragraph","data":{"text":"Lower ignition limits of syngas <i>vs</i>. hydrogen concentration in it. T = 25°C and Р = 1 atm.<sup>98</sup>"}}]](/storage/images/resized/BpQ8dPO8eQsHjVMdMFBfzikuRtQZKhveCouf1tYf_xl.webp)
Much attention was also paid to the oxidation of carbon monoxide, which is strongly influenced by the presence of hydrogen-containing impurities.40 As for syngas itself, it has also been the subject of ongoing research for almost a century and continues to be a source of challenges and questions for combustion specialists. Virtually all noteworthy results on this subject obtained before 2007 are summarized and analyzed in the very informative publication 99 and its shorter version 100. Directly in a study 99, the ignition of syngas of various compositions over a wide temperature range of 825 < T < 1400 K and a pressure of 1 < P < 45 atm was studied using reflected shock waves.
Comparison of experimental and kinetic modelling results using almost all the best known and best accepted current mechanisms, with good agreement in the high-temperature region, revealed marked discrepancies in the low-temperature and high-pressure regions. Generally, at temperatures below 1000 K, the simulation predicts a much longer ignition delay and a sharper increase as the temperature decreases than the available experimental results and their extrapolation. Analyzing the possible reasons for this discrepancy, Kalitan 99 points out the numerous sources of distortion arising when using the shock wave method at low temperature. This seems to be related to the fact that most of the results obtained so far on the ignition and combustion of syngas relate to the high-temperature and low-pressure region, usually in mixtures heavily diluted with argon.
Another source of discrepancy may be the fact that most of the kinetic mechanisms used for such analysis have been verified on the basis of maximum approximation to the experimental data of calculations based on the mechanisms of high-temperature processes. Thus, according to the assumption of Kalitan et al.,99, 100 the main reason for the discrepancy between the results of kinetic calculations and experiments may be the lack of data for testing these mechanisms in the low-temperature and high-pressure regions, as well as the inaccuracy of the rate constants of some important elementary stages.
In contrast, Dryer and Chaos 101 argued that the source of the observed discrepancies in the ignition of syngas is mainly the inadequacy of attempts to reflect the perturbations occurring during ignition on the assumption of homogeneity of the gas phase. In their opinion, these perturbations can reduce the predicted ignition delay by several orders of magnitude.
Of the more recent studies on the ignition of syngas at relatively low temperatures, the publication 102 should be mentioned, which investigated the ignition of syngas in the 15 – 50 atm pressure range and 950 – 1100 K temperature range using a rapid compression machine. It was noted that, over the entire range of parameters studied, even a slight substitution of H2 for CO, without changing the equivalence ratio, increased the ignition delay. At the same time, the inhibitory effect of CO addition increased with increasing pressure. Kinetic analysis showed that, under the conditions of these experiments, the greatest influence on the discrepancy between the experimental and calculated results was exerted by the choice of the calculated reaction rate constant (7), the optimal choice of which allows this discrepancy to be significantly reduced.
A detailed kinetic analysis of low-temperature ignition of syngas showed,103 that for CO and H2 mixtures of different compositions all current mechanisms describe the experimental results quite accurately for temperatures above 1000 K regardless of pressure. But at lower temperatures there is a discrepancy with the experimental data and none of the mechanisms were able to give a correct prediction of the ignition delay. Such a discrepancy between modelling and experiments, according to Cavaliere et al.,103 is not due to the presence of carbon monoxide, but to inaccuracies in the hydrogen oxidation mechanisms in use.
Karim et al.99 noted that the higher the hydrogen concentration in the mixture, the worse the experimental data are consistent with the modelling results. Agreement is achieved only at relatively high temperature. The fact that these discrepancies are particularly pronounced at high pressure, in the author’s opinion, indicates insufficiently reliable information on the rate constants of reactions involving HO2• and H2O2 under such conditions.
Therefore, there remains a deep discrepancy between the results of experimental determination of the ignition delay time of syngas at temperatures below 1000 K, obtained mainly by the shock wave method, and the results of kinetic modelling (Fig. 27), which may stem both from the inadequacy of this experimental method for the low temperature region and the drawbacks of the used kinetic mechanisms of hydrogen and syngas oxidation.
![[{"id":"epwFK1LezB","type":"paragraph","data":{"text":"Results of experimental temperature dependence of ignition delay times of syngas (open circles) and calculation of the same obtained using various mechanisms (lines). Syngas composition: 80 vol.% CO + 20 vol.% H<sub>2</sub>, Р = 2.6 atm (<i>a</i>),<sup>99</sup> syngas of different composition, Р = 20 atm (<i>b</i>).<sup>101</sup>"}}]](/storage/images/resized/sLWUN8DDq8lUsdeOOzttQbq6TS1JeJE3gYDyEMmE_xl.webp)
To experimentally determine the ignition delay of gas mixtures at T < 1000 K, the bypass method in a heated static reactor is commonly used.15, 16, 104 However, the presence of hydrogen in the syngas makes the mixture very flammable. Although this method was successfully used to determine the ignition delay of methane-hydrogen mixtures with hydrogen content up to 50 vol.%,49 an attempt to use it to determine the ignition delay of syngas failed. Due to the lower ignition inhibition capacity of CO compared to CH4, the ignition of even 5 vol.% H2 + 95 vol.% CO mixtures occurred at the moment of their bypass into the reactor.45 Therefore, a kinetic calculation 45 was carried out to determine the ignition delay of syngas at temperatures below 1000 K, based on the successful verification of the mechanism 19 used, by comparing it with experimental data on determining the ignition delay of H2 – O2 – CO – Ar mixtures in reflected shock waves at temperatures of 750 – 150 K and a pressure of 1 atm.105
The ignition delay was calculated for mixtures of syngas with oxygen and argon under constant volume bypass reactor conditions at an initial pressure of 1 atm and initial temperature from 800 to 1100 K.45 The process was modelled using the CHEMKIN software package included in the ANSYS software package.106 The detailed gas-phase kinetic mechanism NUIGMech1.1 (2020) was used to describe chemical transformations in the system.19
As expected, the concentration of hydrogen in the syngas has the most significant impact on ignition. The influence of CO concentration and possible impurities (CO2, CH4, etc.) is much weaker. Calculations show a sharp increase in the ignition delay when the temperature decreases below 1000 K, associated with fundamental changes in this area of the hydrogen oxidation mechanism,49 which have fallen out of sight of most of the experimental works carried out by the shock wave method, since for them this is already the maximum reachable area. Fig. 28 shows the calculation of the ignition delay of syngas for constant volume bypass reactor conditions (P = 1 atm) as a function of its composition for the higher (1000 K) and lower (800 K) temperature region.
![[{"id":"jGJOLgKbTt","type":"paragraph","data":{"text":" Ignition delay time of a stoichiometric syngas – air mixture <i>vs</i>. the composition of syngas under constant volume bypass reactor conditions at Р = 1 atm. T = 800 K (<i>a</i>), T = 1000 K (<i>b</i>).<sup>45</sup>"}}]](/storage/images/resized/aoe22dvQcFbU9PpsIZmC4MJInhcpcnryjH65AJ6Z_xl.webp)
At CO concentration in syngas almost up to 80 vol.%, this dependence is fairly weak because on ignition of syngas, hydrogen present in it obviously dominates, and CO has a significant influence only at its high concentration. At the same time, it is important that at low temperature (T = 800 K), the addition of CO initially promotes the ignition by shortening the ignition delay time. At T = 1000 K, however, due a change in the hydrogen oxidation mechanism, the addition of CO inhibits ignition from the outset.
For the same temperature values belonging to regions with significantly different mechanisms of hydrogen oxidation, the effect of pressure on the ignition delay of syngas in the range of P = 1 – 40 atm (Fig. 29) was calculated, the region being the most interesting in practical terms, including the use of syngas as a fuel for gas turbines. The pressure effect was calculated for a syngas-air stoichiometric mixture at a hydrogen concentration in the syngas of 60 mol.%.45
![[{"id":"HrCrxoe2jf","type":"paragraph","data":{"text":"Ignition delay time of syngas (60 mol.% H<sub>2</sub> + 40 mol.% CO. ϕ = 1.0; T = 800 K (<i>a</i>), T = 1000 K (<i>b</i>)) <i>vs</i>. pressure.<sup>45</sup>"}}]](/storage/images/resized/WafHZjtEi1MNEEExynKL2x7pQIdqv4Fgg8a75K1L_xl.webp)
There is also a significant difference in the behaviour of the syngas for the different temperature ranges. At T = 800 K, there is a monotonic decrease in the ignition delay with increasing pressure. In contrast, at T = 1000 K and low pressure, an increase in the ignition delay with pressure, typical of these conditions for hydrogen oxidation, is observed. Only at pressures above 10 atm its further increase reduces the ignition delay.
Despite the dominant role of hydrogen oxidation kinetics in the ignition of syngas, the role of CO in this process, especially at low temperature and high pressure, should not be underestimated. Walker et al.107 found experimentally that when small amounts of CO are added to H2 between 950 and 1100 K, as also seen in Fig. 28b, carbon monoxide inhibits the ignition, which is more noticeable at high pressure and may be related to the chain-breaking reaction
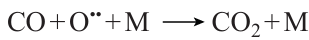
The experimentally shown promotion of hydrogen ignition by CO additives at high temperatures and low pressures seems to be due to the contribution of the chain branching reaction (31)

The potentially possible promotive contribution of reaction (7) requires further analysis. It could be significant for the region of T < 900 K, in which НО2• radicals play a prominent, if not determining, role. Their concentration increases steadily during the ignition delay period, and this is what determines the kinetics of the process. But the validity of such a statement will strongly depend on the real activation energy of this reaction, which is unlikely to be low. In any case, it was suggested that the value of the reaction (7) rate constant should be changed to more adequately reflect its increasing value at high pressure and intermediate temperatures.102
The sharp change in the ignition delay time of syngas when passing into the temperature region below 1000 K is certainly determined by the corresponding changes in the hydrogen oxidation mechanism. Even in the early studies,107, 108 a sharp change in the activation energy of hydrogen oxidation at T ≈ 850 K was observed, from which it was concluded that the change in the behaviour of the H2-air system is probably due to a change in the reaction mechanism at low temperatures and high pressures. The results described above (Section 5 and Refs 49, 108) on the ignition of hydrogen and methane-hydrogen mixtures at T < 1000 K unequivocally support this conclusion.
Thus, it can be concluded that in the 800 – 1000 K temperature range and low (a few atmospheres) pressure, the ignition kinetics of syngas at a concentration of CO < 80 vol.% is almost entirely determined by the kinetics of hydrogen ignition (see Fig. 28). In the low-temperature part of this range, the role of CO is reduced to a minor promotion, apparently due to the reaction (7), which converts inactive НО2• radicals, actually breaking the chains, into active ОН• radicals, propagating them. This effect is superior to the inhibitory effect of increasing the heat capacity of the mixture when CO is added and the higher efficiency of CO compared to H2 as the third body in the radical recombination reactions (see Fig. 28a). At temperatures near 1000 K and above, the role of НО2• radicals, and consequently, the reaction (7), becomes less significant and the inhibitory effect of CO in the reaction (28) and as a diluent becomes predominant (see Fig. 28b).
At higher pressures, the CO promotion effect due to reaction (7) may be more pronounced, although it may be partially compensated by an increased contribution of the chain-breaking reaction (30). At temperatures well above 1000 K, the role of reaction (7) is likely to be negligible due to the rapid decrease in the concentration of НО2• radicals with temperature. However, at an even higher temperature, the promotional effect of CO due to the chain-branching reaction (31) is possible.
The role of possible changes in the oxidation mechanism of CO itself when the concentration of hydrogen in the syngas changes can be excluded from consideration, since in syngas of almost any composition hydrogen and unreacted hydrocarbons are present in a concentration that is significantly higher than that necessary for rapid CO oxidation.99
Given the very high ignition propensity of hydrogen, the role of most of the remaining impurities in the syngas at temperatures below 1000 K seems to be limited to increasing the total heat capacity of the mixture and their role as a ‘third body’ in recombination and dissociation reactions. As for the effect of water vapour, no appreciable dissociation of water vapour was observed at T < 1200 K.110 Only at higher temperatures does water dissociation produce OH• radicals, which accelerate ignition. Below 1200 K, water promotes the death of Н• atoms as an effective third body in their recombination reactions, which slows down ignition. Although small additions of water vapour effectively promote CO oxidation, at [H2] = 5 vol.% and above they have little or no effect on CO oxidation.99
The possibility of promoting the syngas ignition by water vapour at low temperature and high pressure through the following reaction (32) is sometimes pointed out

where water vapour acts as the more efficient component M.111 However, based on the simulation results, the inhibitory effect due to its high heat capacity still prevails, which is most pronounced near 950 K. The effect of CO2 at T < 1000 K on the ignition of syngas seems to be quite similar to that of water vapour 111, 112 and is also reduced to an increase in the total heat capacity of the mixture and its higher, compared to hydrogen and carbon monoxide, efficiency as the third body in dissociation and recombination reactions, with an apparent predominance of inhibitory effect, although less strong than that of water vapour. The effect of dilution with nitrogen is even less pronounced due to its lower heat capacity and third-body efficiency compared to H2O and CO2. Similarly, the addition of H2O and CO2 affects the combustion of syngas.113
Of the relatively simple molecules, methane 49 and, possibly, heavier hydrocarbons have a noticeable influence on the ignition delay of hydrogen and, consequently, of syngas. Due to the high heat capacity of methane and the specifics of its low-temperature oxidation mechanism, at T < 1000 K and a methane concentration in the mixture with hydrogen of up to ca. 50 vol.%, the ignition and combustion of such a mixture proceeds by the ‘methane’ mechanism, differing only slightly from analogous parameters for methane. Only at hydrogen concentrations above 60 vol.% there is a transition to a ‘hydrogen’ oxidation mechanism. Apparently, dilution with methane will similarly affect the ignition of syngas. When the methane concentration is equal or higher than the hydrogen concentration in the syngas, its significant influence and transition to ignition of the mixture by the ‘methane’ mechanism can be expected.
It should also be noted that there are fundamental similarities in the effect of carbon monoxide additives on the ignition of methane and hydrogen.45 In both cases, the process follows the mechanism of methane or hydrogen oxidation, respectively, up to a sufficiently high (60 – 80 vol.%) concentration of carbon monoxide in the mixture, and only at a concentration of carbon monoxide above 60 – 80 vol.% its presence begins to affects markedly the ignition.
7. On the knock resistance characteristics of gas fuels and the possibility of regulating them
The widespread use of compressed natural gas (CNG), hydrogen and their mixtures (trade name Hythane), is considered as one of the most promising ways to improve fuel efficiency in transport and reduce the emission of environmentally harmful combustion products. The undoubted advantages of CNG include its availability and relatively low cost compared to other transport fuels,114 as well as its high octane number (ON), which is ~ 110.
The traditional octane number scale used to determine the engine properties of liquid fuels formally has an upper limit of 100 and is therefore not suitable for testing the antiknock quality of CNG and even liquefied petroleum gas (LPG). Accordingly, a mixture of hydrogen and methane, the so-called ‘methane scale’,115 has been adopted as a standard reference fuel for evaluating the knock resistance of gas fuels which extends the measured range beyond the traditional Motor Octane Number (MON) and Research Octane Number (RON) ranges. The measure of knock resistance of gas fuels corresponding to this scale has been called the ‘methane number’ (MN), which is defined as the volume percentage of methane in a mixture with hydrogen corresponding to the intensity of knock of the gas mixture under given engine operating conditions.
The addition of any inert components increases the heat capacity of the system and thus increases the ignition delay of gas mixtures. For example, the addition of inert gases such as N2 or CO2 in spark-ignition combustion engines markedly increases the knock resistance of gas fuels.116 Therefore, for a range beyond 100 MN, mixtures of methane and carbon dioxide are used as reference mixtures. In this case, as defined, MN is 100 plus the volume percentage of CO2 in the reference mixture of methane and carbon dioxide.115 In 1999, a consortium of European gas industry leaders decided that this method of determining the detonation resistance of gas fuels was preferred.117 Currently, there are several calculators available,118, 119 which allow calculating the methane index of a gas mixture based on its composition.115 However, an assessment of the actual knock resistant properties of gas fuels based on the methane index is not fully adequate, and in reality it is necessary to resort to engine fuel tests to determine them.
The technical procedure for determining the knock resistance of gas fuels based on the methane scale requires special facilities, of which there are few, and the procedure itself is considerably more complicated than that for liquid fuels. The possibility of adequate kinetic simulation of experiments on delayed ignition of complex gas mixtures, shown in Section 3, opens up the fundamental possibility of replacing the complex technical procedure by mathematical methods. However, kinetic modelling requires highly qualified specialists, so numerous attempts have been made to describe the ignition delay of complex gas mixtures analytically.
Spadaccini and Colket III,31 based on the experimental results obtained on the effect of ethane, n-propane and n-butane additives on methane ignition, proposed a general correlation for all types of hydrocarbon additives, accounted for as their total concentration [TC]
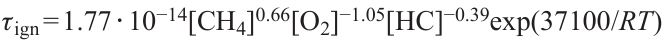
for temperatures ranging from 1300 to 2000 K, pressures ranging from 3 atm to 15 atm and an equivalence ratio ϕ = 0.43 – 1.25 with an overall pressure dependence of ~P – 0.78. A number of other correlations of a similar type were also proposed.120
The theoretical justification for the applicability of such analytical correlations can be provided by the experimentally established 16, 28 weak influence on the methane ignition delay by a particular composition of an impurity of heavier alkanes contained in the gas. On this basis, a relatively simple analytical procedure with tabulated parameters for calculating the ignition delay times of real hydrocarbon gases was proposed for a more accurate operational estimate of the antiknock rating of gas mixtures.39 Formally, it was developed for binary and ternary methane-alkane mixtures at atmospheric pressure and temperatures of 800 ≤ T (K) ≤ 1000. But since, as it has been shown experimentally and theoretically, the ignition delay actually does not depend on the specific composition of its heavier homologues present in the mixture with methane, but only on their total concentration, this method of assessing the knock resistance of gas-engine fuels can be applied to more complex mixtures, including real natural gas.
Particular attention should be paid to the assessment, on the basis of the methane scale, of the antiknock characteristics of methane-hydrogen mixtures and other hydrogen-containing gas fuels. This scale is actually based on the assumption that the motor characteristics (flame velocity, ignition delay) of methane-hydrogen mixtures depend linearly on the concentration of both components, i.e., on the additive nature of the dependence of these characteristics on the composition of such mixtures, which does not correspond to reality (see Section 5). It was shown that, at hydrogen concentrations of up to 40 vol.%, its addition slightly affects the ignition delay of methane.49, 109 Study 89 and the works cited therein found that, at hydrogen concentration in mixtures with methane of up to 40 vol.%, it also weakly affects the normal flame velocity of such mixtures. And at higher concentrations, its influence increases exponentially with hydrogen concentration (Fig. 30). Such a complex and clearly non-linear dependence of the main parameters of ignition and combustion of methane-hydrogen mixtures on the concentration of the components not only questions the validity of applying the methane scale to methane-hydrogen fuel per se, but also its adequacy in general.
![[{"id":"VyLDPK4M5B","type":"paragraph","data":{"text":"Laminar flame velocity <i>U</i>n of stoichiometric methane – hydrogen – air mixtures vs. hydrogen concentration in the mixtures at T = 293 K. Solid line correspond to calculation, symbols correspond to experimental data taken from various publications.<sup>89</sup>"}}]](/storage/images/resized/PiN4X0YcgAP7Rdup2mKW8JTOPCTq12boXv0QpGw3_xl.webp)
Despite the zero methane number attributed to hydrogen, according to literature data, it has an RON above 130, and an MON of 60 (Ref. 58), and is quite successfully used as a motor fuel in internal combustion engines (Ref. 58, 60). This indicates the inadequacy of the methane scale to characterize the knock properties, at least of the methane-hydrogen mixtures themselves. It seems that, if this criterion is acceptable, it is only for the relative comparison between mixtures of methane and its heavier homologues. However, if the mixture contains hydrogen or compounds of other classes in appreciable concentration, it is unlikely that its characteristics can be adequately assessed on the basis of the methane scale.
In terms of the results presented in Section 3 on the effect of alkane impurities on methane ignition delay, the Propane Knock Index (PKI) is a more appropriate characterisation of natural gas-based fuels. This parameter was proposed by Attar and Karim 121 and is currently used by a number of gas-piston engine manufacturers, in particular, it is calculated on a calculator.122 The PKI is a characteristic of the knock resistance of a gas mixture, defined as the equivalence percentage of propane in a mixture with methane having the same knock resistance under the same engine operating conditions as the tested mixture under test. Taking into account the almost identical effect of the impurity of all C2 – C6 alkanes on the ignition delay of methane shown in Section 3, the PKI can quite adequately reflect the knock resistance of at least methane-alkane gas fuels, i.e. real CNG, if, instead of the propane concentration, the total concentration of all C2+ alkanes contained in the mixture with methane is taken into account.
The significant decrease in the knock resistance of natural gas-based fuels in the presence of even small (~ 1 vol.%) impurities of heavier homologues of methane homologues makes the problem of their concentration reduction critically important.
Known physical methods of separation of these impurities from natural gas, such as cryogenic separation, short-cycle adsorption, membrane technologies and others require complex equipment, high energy input and, as a rule, are rarely economically viable, especially for relatively small volumes of gas fuel production.
Therefore, for this purpose, as well as for increasing the knock resistance of liquid fuels, chemical methods of increasing the knock resistance of fuels, in particular, selective oxycracking of heavy methane homologues, may be more acceptable.28, 123 This method is based on the large difference in the reactivity of methane and its homologues, which is the main reason for their different propensity to ignition. Using this difference, it is possible to selectively oxidize C2+ homologues of methane almost completely, under rather mild conditions (atmospheric pressure and T ~ 1000 – 1100 K), into CO and olefins, mainly ethylene, without affecting methane itself.123, 124
In conclusion, it is necessary to refer briefly to the safety problems of hydrogen gas fuels. Although methane-hydrogen mixtures are already used in social transport in a number of countries, data on risk and safety analyses of their use are still insufficient. Middha et al.125 conclude that the use of hythane mixtures is safer not only than pure hydrogen, which has significant risks in practice,126 but also than methane because they combine the positive properties of hydrogen (high diffusion coefficient) and methane (lower flame velocity and narrower ignition limits). The modelling results showed that, in general, the ignition propensity of the hythane mixture is slightly higher than that of methane, and for both fuels the explosion pressure is significantly lower than that of hydrogen. Similar conclusions were reached by Yang et al.127
In general, it can be concluded that methane-hydrogen mixtures with hydrogen content less than 40 vol.% are not very different from natural gas in terms of their use in various types of burners, engines and in terms of safety conditions when working with them. This makes their use in most existing types of burners and engines fundamentally possible, provided that traditional handling conditions and safety measures are taken. However, a major challenge remains the change in the strength characteristics of most structural materials when interacting with hydrogen (hydrogen embrittlement), especially at high pressure,128 which requires careful choice of the apparatus and materials to be used.
8. Conclusion
The enormous role of hydrocarbon oxidation and combustion processes in power generation and oil and gas chemistry causes a sustained interest in their study. The interest in gaseous hydrocarbons, which are becoming not only the leading type of fuel for power generation, but also chemical raw materials, is growing especially fast. The study of the ignition and combustion kinetics of liquid hydrocarbon fuels has consumed many decades and continues intensively to the present day. Despite the essentially simpler chemical structure of hydrogen, methane and gas mixtures containing them, we still cannot say that we fully understand all the subtleties and details of the kinetics of their ignition and combustion. The main reason for the complexity of these processes is their fundamental nonlinearity due to the rapidly increasing variety of intermediate products formed even in the oxidation and combustion of the simplest hydrocarbon, methane, which are actively involved in subsequent transformations.
The regularities of the influence on methane ignition of impurities of its homologues considered in the review allow us to provide a theoretical basis for the possibility and expediency of practical use of the propane knock index (PKI) and relatively simple analytical dependencies for estimating the motor characteristics of real gas fuels based on methane and natural gas. At the same time, the established peculiarities of ignition of methane-hydrogen mixtures show the inadequacy of using for this purpose the widespread methane scale, in which the knock resistance of hydrogen is taken as zero.
Insignificant influence of hydrogen on ignition and combustion of methane at its concentration in the mixture less than 40 vol.%, in principle, enables the use such mixtures for operation on the state-of-the-art gas equipment and with observance of safety measures usual for natural gas. However, the same circumstance practically eliminates the hope for the possibility of significant improvement of ecological characteristics of internal combustion engines and turbines running on gas or liquid fuels at the expense of small additions of hydrogen because of their too weak influence on ignition and combustion of the main fuel.
A unique feature of methane, which distinguishes it from other alkanes, is the possibility of low-temperature branched-chain oxidation based on the formation and decomposition of methylhydroperoxide. This property greatly complicates the dependence of ignition of methane-containing mixtures on external parameters.
No less unique is the mechanism of hydrogen ignition, in which the kinetics of hydroperoxide formation and decomposition plays a major role at moderate temperatures. Ignoring these still insufficiently taken into account features can lead to serious errors in the real assessment of the motor properties of mixtures containing these gases, impede optimization of design and operating modes of engines using them, and also lead to serious miscalculations in the assessment of the ignition ability of such mixtures, especially under non-standard conditions.
Section 6 of this study was carried out with the financial support of the Russian Science Foundation (Grant No. 22-73-00171).
9. List of abbreviations and symbols
CNG — compressed natural gas,
Ea — effective activation energy of ignition delay,
ICE — internal combustion engine,
LPG — liquefied petroleum gas,
MN — methane number,
MON — motor octane number,
Nc — number of carbon atoms,
NTC — negative temperature coefficient of the reaction rate,
NUI Galway — National University of Ireland in Galway,
ON — octane number,
P0 — initial pressure,
PKI — propane knock index,
RON — research octane number,
SBI — static bypass installation,
T0 — initial temperature,
ϕ — equivalence ratio (fuel excess ratio),
t — ignition delay time.