


Keywords
Abstract
The review covers the electrochemical reactions of oxygen reduction (ORR) and carbon dioxide reduction (CO2RR) with a focus on mechanisms, structure of intermediates, and thermodynamics of reactions. The ORR on metal,heteroatom-codoped nanocarbon substrates as catalysts is a promising source of pure electrical energy. CO2RR, being a synthetic analogue of the photosynthesis reaction, has great potential for the generation of partial or complete reduction products. Theoretical insights into the mechanism and kinetics of CO2RR are just being developed, so the systematization of advances and emerging issues is a necessary step for further progress.
The bibliography includes 242 references.
1. Introduction
The transition from fossil to renewable energy sources is one of the main challenges to increasing energy production while reducing carbon dioxide emissions, as stated in the Kyoto Protocol,1 which expired in 2020, and in the Paris Agreement that replaced it.2 Fuel cells that use the electrochemical reaction of oxygen reduction at the cathode (Oxygen Reduction Reaction, ORR) are highly efficient and environmentally friendly energy sources.3 The disadvantages of ORR are related to the fact that the formed water prevents the transport of oxygen molecules in the pores of the catalyst and to the high activation energy.4 As a result, the main obstacle to the commercialization of various fuel cells is the low rate of ORR. Various catalysts, such as platinum on activated charcoal, and various metal-nitrogen coordinated metal-organic frameworks (MOFs), etc.,5,6 are used to promote the process, with the former catalyst being used mainly until recently. Their main disadvantages are easy poisoning of platinum active sites by carbon monoxide and methanol, and also oxidation of the carbon support, high cost arising from large metal loadings due to the low reaction rate and short life of the metal support.7--10 One way to extend the life of a carbon support is to use materials with a minimum number of structural defects (graphene, nanotubes, etc.) and their high-temperature treatment.9,10 More than half a century of attempts by chemists to replace them have revealed the prospects for nanocarbon materials (NCM), doped with N, P, S, Se, Si, B heteroatoms and non-precious metals.11--19 Advances in applications of such catalysts are summurized in recent reviews.20--25
Compared to doping with other heteroatoms, Si-doped materials have received less attention, although the high (up to a certain limit, see the Sabatier principle below) oxophilicity of the silicon atom could make them promising ORR electrocatalysts. Of the experimental works, one can mention the synthesis of Si-doped single-walled nanotubes with high activity and long-term stability in ORR,26 or Si-doped graphene as a new high-performance anode material27 with unique magnetoelectronic and optical properties.28 There are few theoretical works devoted to the role of Si-doped NCMs. For carbon nanospheres and nanotubes, the high activity in ORR was confirmed by DFT calculations.26 Noteworthy are also studies related directly to ORR.29--31 Various software packages (VASP, Gaussian, ORCA) and methods (PAW-PBE, DFT) with different basis sets were used for the calculations.
The importance of the second type of reactions considered in this review, namely, electrochemical carbon dioxide reduction reactions (CO2RR) is related to the active burning of fossil fuels, which has led to a steady increase in the concentration of anthropogenic CO2 in the atmosphere. According to the Mauna Loa Observatory (USA), CO2 has increased by one-third, from 316 ppm in 1958 to 422 ppm in 2022.32 This is the maximum level of CO2 for the last 14 million years33 and it continues to grow. Carbon dioxide is the second most important greenhouse gas in the Earth's atmosphere after water vapour, whose negative effects on atmospheric34 and water35 living organisms are well known.
In contrast to O2 reduction, CO2 reduction is a complex endothermic process that can comprise thermo-, photo-, electro-, or biochemical reactions involving up to several dozen of protons and electrons (for ORR, no more than four), producing many valuable products such as carbon monoxide, methanol, methane, formic and acetic acids, ethanol, ethylene, etc.36--41
The review discusses both types of reactions, with emphasis on works published after 2015; earlier pubications are cited only when necessary and in the absence of more recent publications. Consideration of the results of synthetic works on the preparation of catalysts, or experimental works on electrochemistry of these reactions is beyond the scope of this review, and they are only mentioned when discussing studies on the mechanism of ORR or CO2RR to confirm or establish agreement between theory and experiment.
As for experimental works, the reader is referred to the reviews of the last 5 years (2019-2023) on ORR,42--44 and CO2RR,45--48 which also discuss some theoretical aspects of the mechanisms and structure of intermediates in these processes, and almost the only 2019 review focused specifically on the mechanism of electrochemical ORR on nanocarbon catalysts.49
2. Fundamental principles of electrocatalysis. Basic concepts
Electrocatalysis is characterized by the same fundamental principles as general catalysis, which allow an objective comparison of different types of catalysts for a particular process, but there are features, sometimes fundamental, that distinguish electrocatalysis from general catalysis. The general concepts include:
1) the catalyst selectivity, which determines the direction of the processes. For example, in the case of ORR, this is the ratio of the products of complete or partial reduction of oxygen to water or to hydrogen peroxide, respectively. In the case of CO2RR, this is the ratio of the C1 products, i.e. the reduction of CO2 to compounds containing one carbon atom in a varyable oxidation state (HCOOH, CO, CH3OH, CH4), and the C2+ products, i.e., compounds containing one or more C–C bonds such as CH3COOH, CH3CHO, C2H5OH, C3H7OH, C2H4.
2) activity determined by the maximum number of conversions of substrate molecules (turnover number, TON) on at a catalytic site and the turnover frequency (TOF) on this site (a number of turnovers per unit time),
3) rate-determining step (RDS) is the slowest step of the catalytic cycle determining the rate of the whole process.
Fig. 1 shows ORR free energy profile for ideal and non-ideal catalysts in the absence of an external potential and when an equilibrium potential U=1.23 V is applied to the system, corresponding to equilibrium in the system O2+4[H++e–]=2H2O. By an ideal catalyst, the one should be understood, for which, in the absence of an applied potential, the free energy decreases by 1.23 eV at each ORR step involving an electron. At the equilibrium potential, all ORR intermediates are in equilibrium with each other as well as with the staring and the reaction products. For a non-ideal catalyst, this condition is not met, and the thermodynamic profile of the reaction is characterized by the presence of unstable or, on the contrary, highly stable (potential well) intermediates. The rate-determining step is an elementary step characterized by the highest-lying transition state on the potential energy surface.
![[{"id":"hoJmBOP9eW","type":"paragraph","data":{"text":"Thermodynamic profiles of the ORR catalytic cycle on ideal (black) and non-ideal (red) catalysts"}}]](/storage/images/resized/6omVc9hbzmrGQ4E50FGuZRNgxMyWbS5WiIIpdWhj_xl.webp)
It is important to note that the catalyst nature does not affect the equilibrium thermodynamic states of the system, but only increases the rate of establishment of equilibrium between them. In terms of analysis at the molecular level, the main steps of catalysis include (i) chemisorption of the starting molecules; (ii) the sequence of elementary reactions of the catalytic cycle; and (iii) desorption of the products. The chemisorption properties and stability of adsorbates affect the kinetics and thermodynamics of the catalytic cycle, and thus the activity of the catalyst. The Sabatier principle proposed more than 100 years ago states: `The ideal catalyst should bind to the reactant with an intermediate strength that is neither too weak nor too strong'.50,51 In the first case, reactants are not adsorbed and no reaction occurs, and in the second case, the products are not desorbed, the catalyst is poisoned and there is again no reaction (Fig. 2). For a century, the Sabatier principle was considered obvious and inviolable, but recently it was shown that activation of a catalyst by electrical or light pulses increases its activity by 3-4 orders of magnitude, allowing it to overcome the Sabatier prohibition.52--54 This is achieved by switching from conventional static to dynamic catalysts. The Sabatier prohibition is related to the balance of competing reactions on the catalyst (chemisorption of reagents vs desorption of intermediates). The strategy for moving beyond these limitations is to separate and physically disconnect these processes.
![[{"id":"0Aix5kNZz4","type":"paragraph","data":{"text":"An illustration of the Sabatier principle"}}]](/storage/images/resized/kgtQVuPwfRqCQ56wXeFS7bll8C1ub5aahAqOVZnI_xl.webp)
An electrocatalyst is a heterogeneous electrode material of an electrochemical cell on which, in addition to the target reaction, electron transfer occurs, as a result of which the substrate molecules adsorbed on its surface are oxidized/reduced through the reaction on the electrode surface with charge transfer (Faradaic reaction), but the catalyst itself remains unchanged.55 As a result of the external energy supply in the form of an applied potential U, electrocatalysis is also subject to thermodynamically forbidden reactions, in particular, CO2RR,56 which is a fundamental difference between electrocatalysis and general catalysis. Also, the electrochemical constituent requires supplementing the above concepts with the following characteristics:57
4) an onset potential (volt) is the minimum required potential that must be applied to the system to initiate the reaction and the formation of products. The onset potential can be predicted theoretically as the minimum potential, the application of which to the catalytic system allows the spontaneous course of all elementary steps of the cycle involving electrons, i.e. ΔG<0 (Fig. 3). In CO2RR, the term limiting potential, UL=–eΔG, where ΔG corresponds to the limiting elementary step of the catalytic cycle involving an electron in the absence of an external potential, i.e., U=0; e is an electron charge, has become entrenched to quantify catalyst activity.
![[{"id":"ENkovEFfQJ","type":"paragraph","data":{"text":"Voltammetric characteristic curve for the catalyst"}}]](/storage/images/resized/32cEvzuCsR8MGeFYttiYtzy8wXiBK8xUOLOgldUP_xl.webp)
5) Overpotential η [V] is defined by the potential difference between the half-reaction potential (Ueq) and the potential, at which the redox process is experimentally observed (U). The value η is calculated by the formula η=U-Ueq. The overpotential is determined experimentally by measuring the potential, at which a given current density is reached (usually low). The most active ORR/CO2RR catalysts shows η values, which are close or lower than those for the platinum catalyst Pt/C. On the other hand, the overpotential can be predicted by quantum chemical calculations, provided the choice of an active site is correct, using the formula η=eUeq–ΔΔG, where ΔΔG is the free energy change for the slowest step, e is an electron charge (see Fig. 1).
6) Current density (j, A cm–2) is an amount of electricity, which can be measured on the catalyst per unit area at a given electrode potential. The value of j is calculated according to the formula j=I/S, where I is the current at a given electrode potential and S is the surface area of the catalyst. The current density j characterises the reaction rate, from the value of which the activation energy can be estimated.
7) Faradaic efficiency (FE) (%) is the ratio of the number of electrons involved in the formation of a particular product to the total number of electrons involved in the reaction at a given electrode potential. Faradaic efficiency can be calculated using the formula FEi=niZF/Q, where Z is the number of electrons involved in the formation of one molecule of product i; ni is the amount of substance obtained (mol); F is the Faraday constant, 96485 C mol–1; Q is the total charge passed through the reaction system during the electrochemical reaction (C). The FE value is widely used for CO2RR, where a wide range of different products can be formed.
8) Tafel slope (volt per decade of current), or the coefficient b in the Tafel equation η=a+blogi, is directly proportional to temperature and inversely proportional to the transfer coefficient. It is used to describe the kinetics of the process depending on the magnitude of the overpotential, the logarithm of the current density, and allows to gain a better insight into the reaction mechanism, in particular the nature of the rate-determining step.
The basis for numerous theoretical works on understanding of the mechanism and design of active sites has been the method of calculating the free energy of intermediates of electrochemical reactions, proposed in 2004 by Nуrskov et al.58 According to the proposed methodology, the free energy change ΔG of an electrochemical reaction includes six terms
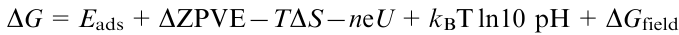
where ΔG is the free energy change (eV), Eads is the energy change by adsorption on the catalyst surface; ΔZPVE and ΔS are corrections for zero-vibration energy and entropy change in the system; T is temperature, K; n is the number of electrons involved in the reaction, e is the electron charge, U is the electrode potential, kB is the Boltzmann constant, pH is the hydrogen ion concentration and ΔGfield is the change in free energy resulting from phase transition (for simplification it is frequently taken equal to zero). The Nуrskov's equation was subsequently used as the basis for a computational hydrogen electrode (CHE) model developed by Peterson and Nуrskov,59 (see also the monograph60).
3. Electrochemical oxygen reduction reactions (ORR)
3.1. ORR nanocarbon catalysts
The advantages of nanocarbon materials as catalysts for oxygen reduction reactions (ORR) doped with metals (e.g., Co, Fe, Sn, Cu, etc.) and heteroatoms are their high performance compared to the individual components, lifetime, excellent conductivity and low cost. The catalytic activity is due to the presence of local defects induced by heteroatoms,61--74 with the combined doping with metals and different heteroatoms providing a synergistic effect.75--78 Doping facilitates the O2 adsorption on the catalyst, weakens the O–O bond thus promoting ORR.63,66 Nanocarbon support can be nitrogen- or sulfur-doped ordered carbon or graphene, carbon dots, nanofibres, nanospheres, metal-organic frameworks (MOFs).79 The use of pristine graphite is hampered by the agglomeration of layers, therefore, additives of carbon nanotubes (CNT) are used to achieve a positive effect (see Ref. 65 and references cited therein). Nitrogen-doped NCMs are effective ORR catalysts.8--12,61--68 The proximity of covalent radii of the nitrogen and carbon atoms (0.75 and 0.77 Å) ensures minimal distortion of the substrate structure. The higher electronegativity of nitrogen (χN 3.04, χC 2.55) leads to the localization of the positive charge on the adjacent carbon atoms, the appearance of oxygen chemisorption sites and facilitates the cleavage of the O–O bond.80 The nitrogen atom in NCM can exist in pyridinic, amine, pyrrolic, graphitic and N-oxide forms. Which of these is more active is a moot point; in their previous paper,81 the authors argued that the pyridinic nitrogen atom is more active, but this assumption was later rejected on the basis of experimental results and theoretical analysis.80 If an unshared electron pair of the nitrogen atom is not involved in the π-system of the carbon support, it can form strong chelate metal complexes M–Nx-C, whose activity significantly exceeds that of the metal-free complexes, although the former still lose out to the more expensive Pt/C.63,82--84 These issues are well described in recent reviews.83,85
The increased catalytic activity of such materials stems from the fact that the metal exists in the atomic state surrounded by Nx (x=1--4) of the support compared to metal-free materials. The activity of M–Nx–C catalysts in ORR depends on the nature of the metal and decreases in the series Fe≈Sn>Co>Cu>Mn>Ni,77,85,86 which makes Fe,N-doped graphene competitive against the expensive Pt/C catalyst.83
Theoretically, metal copper was expected to show higher ORR activity than other transition metals,58,87 and, given its high electrical conductivity, copper doping promotes charge transfer from the catalyst's active site to the adsorbate.88--90 Thus, Cu,N-doped NCM (Cu–Nx–C) had good voltammetric characteristics in ORR: the half-wave potential was 0.813 V, the diffusion current density was 6.0 mA cm–2, the material also proved to be tolerant to methanol poisoning and more stable in an alkaline medium compared with Pt/C.87 CuN2-doped graphene had excellent ORR activity.87,90 Even with KSCN added, which dramatically lowers the catalytic activity of Cu–Nx–C in an alkaline medium because of the rhodanide-ion coordination to the metal and the poisoning of the Cu–N sites, the catalyst showed a high current density at the half-wave potential.17
Outstanding catalytic performance for the oxygen reduction reaction (ORR) in both alkaline and acidic electrolytes, even on the addition of KSCN, and stability is demonstrated by cobalt,nitrogen-codoped highly uniform carbon nanospheres (CoN–CNS).91 In an alkaline electrolyte, they have more positive half-wave potential and higher kinetic current density than the commercial Pt/C. In an acidic electrolyte, CoN-CNS also shows good ORR activity with a high electron transfer number and its initial and half-wave potential are close to those of the platinum catalyst.
3.2. Free energy profiles of ORR
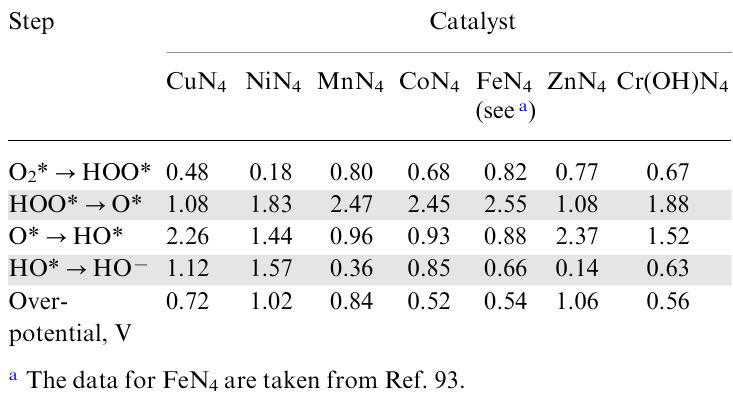
In recent works by Samara chemists,92,93 the catalytic activity of MN4-doped graphene (M=Fe, Co, Ni, Cu, Mn, Zn and Cr) in ORR including reaction thermodynamics was studied by the DFT method. Co and Cr showed the highest activity, with chromium spontaneously oxidizing to the hydroxy form.92 The results are in line with experiment, in particular at low overpotential for cobalt (~0.5 V). In an alkaline medium, all steps of the reduction of O2 to HO– proceed exergonically, as can be seen from Table 1 and Fig. 4. Fig. 5 shows the free energy profiles for all catalysts studied.
![[{"id":"yDCKBNo6_V","type":"paragraph","data":{"text":" Free energy profiles of ORR on CrN<sub>4</sub>- (purple) and Cr(OH)N<sub>4</sub>-doped graphene (green)"}}]](/storage/images/resized/wnCMHUcWabizsbY8nqvxE9gVGPuhutO3wtRyE1eE_xl.webp)
![[{"id":"3jHzS7dh3Q","type":"paragraph","data":{"text":"Free energy profiles of ORR on catalysts: CoN<sub>4</sub> (grey); CuN<sub>4</sub> (black); NiN<sub>4</sub> (blue); MnN<sub>4</sub> (red); ZnN<sub>4</sub> (dark-red); Cr(OH)N<sub>4</sub> (green). The ideal catalyst is marked dark-blue"}}]](/storage/images/resized/O6aWchODA4aQVRP781kqhMaIgZHeKsrkSU8HzUSk_xl.webp)
It was shown that FeN4-doped graphene also has a good thermodynamic activity profile in ORR.93 In the case of CoN4-doped graphene, the effect of two spin-state reactivity94 was revealed and a detailed mechanism was proposed (Fig. 6). It involves an electron transfer (2→3, 5→6, 7→8, 10→11) and association with water, in contrast to the classical mechanism comprising 1→2→5→7→ →10→12 transitions. For cobalt, the low- and high-spin states were calculated: Fig. 6 shows the lowest lying states - high-spin for 5 and 10 steps, and low-spin for all others. The inclusion of charged intermediates was found to decrease the energy of the system, facilitating the O2 adsorption. The activation barriers at the 4→5, 9→10 and 11→12 steps are due to the H–O bond cleavage and HO– desorption.
![[{"id":"GKsIr531l4","type":"paragraph","data":{"text":"Free energy profile for the proposed<sup>93</sup> ORR pathway on a CoN<sub>4</sub>-doped catalyst for 11-12 steps"}}]](/storage/images/resized/pkFWnlnnrUlKgIKcdRUq3mKaxFXGFOPc2IbWNijT_xl.webp)
Of copper's closest neighbours in the Periodic Table, silver deserves special attention as a good and relatively inexpensive ORR catalyst.85,95 Zinc per se is considered inactive, but in combination with other metals it imparts useful properties to the catalyst.83,95,96 Doping NCM with melamine and zinc nitrate revealed a crucial role of ZnNx active sites in accelerating the 4e ORR reaction on a Co-Nx–C catalyst.97 Theoretical analysis of the mechanism showed that the intermediate HO* (* denotes support, CuN2-doped graphene) at the potential U=0.8 V lies 1 eV lower in free energy relative to the unreacting system,90 so one cannot expect high catalytic activity of Cu–Nx–C in ORR. Nevertheless, the experiment indicates a satisfactory activity of Cu–Nx-C in ORR.87 The authors mistakenly thought that the free energy of the adsorption of the O2 molecule on the catalyst active site depended on the electrode potential. According to the Nуrskov's equation,58 the free energy ΔG does depend on the electrode potential U, but this is true only for the electron transfer steps. Since no electron transfer occurs during oxygen adsorption on the catalyst, the value of ΔG does not depend on the potential U. These disagreements and observed experimentally high catalytic activity of CuNx-doped graphene in ORR, as well as the lack of data on the theoretical study of the mechanism and thermodynamics of ORR on Ag–Nx–C and Zn–Nx-C complexes, prompted us to study the mechanism of ORR involving model MN4-doped single-walled nanotubes (M=Cu, Ag, Zn), which are more rigid as compared to graphene.98 This property, in combination with a sufficiently high surface area, microporous structure, excellent electrical conductivity and high chemical stability, makes carbon nanotubes unique as supports in heterogeneous catalysis.
The structure of the calculated metal-nitrogen codoped nanotubes is shown in Fig. 7. The incorporation of a metal atom into the cavity of the N4-doped nanotube markedly lowers the energy: the ΔE values calculated from the reaction equation N4-CNT2–+M2+=MN4-CNT are –30.3, –29.7 and –27.8 eV for M=Cu, Ag and Zn, respectively. The charges on the metal atoms were 0.569 (Cu), 0.654 (Ag) and 0.788 (Zn).
![[{"id":"9nAH5ARBFG","type":"paragraph","data":{"text":" Structure of metal, nitrogen-codoped nanotubes: CuN<sub>4</sub>-CNT (<i>a</i>), AgN<sub>4</sub>-CNT (<i>b</i>), ZnN<sub>4</sub>-CNT (<i>c</i>) and metal-nitrogen distances (A)"}}]](/storage/images/resized/DzLcqltdIlNI7lxwEPTLF0N396EYqjiLPrdpPUJS_xl.webp)
The charge density maps were calculated,98 which showed that on the carbon atoms labeled C(1) and C(2) at Fig. 7, which form the C2 site, the positive charge (0.113-0.144) is accumulated, which promotes molecular oxygen adsorption as the first ORR step. This suggests that the C2 sites, along with the metal atom (MN4 site), may be active in the ORR catalysis.
The structures of the resulting adsorbates are different: the O2 molecule is coordinated to the metal via one oxygen atom, and to two carbon atoms of the C2 site via both oxygen atoms, except for the catalyst CuN4-CNT, where the O2 molecule is coordinated to only one C(1) atom. Adsorption of O2 on both MN4 and C2 sites is exothermic, with the adsorption energy Eads increasing in the series Cu<Ag<Zn on the MN4 site, and in the series Zn<Cu<Ag on the C2 site.
The protonation of the O2* intermediate affords the peroxide intermediate HOO*, with a longer O–O distance in HOO* than in O2*, although smaller than in the free molecule of H2O2. The Eads of HOO* adsorbates increases in the series Zn<<Cu≈Ag on the MN4 site and in the series Zn<Ag≈Cu on the C2 site. The 4e ORR pathway suggests further protonation of HOO* adsorbate to form O* and release the H2O molecule; an alternative pathway with the O–O bond cleavage and the formation of dihydroxy intermediate 2HO* is also possible.99,100 The formation of 2HO* adsorbate is observed only on the C2 site, whereas on the MN4 site, only adsorbate H2O2* is formed. The Eads value of H2O2* on the MN4 site is –0.5 eV for M=Cu, Ag, and –0.84 eV for M=Zn. Adsorbates of atomic oxygen O* on both sites are characterised by high Eads energies, and remarkably, adsorption on the C2 site is 0.42 (Zn), 0.64 (Cu) and 1.06 eV (Ag) more favourable than on MN4. Hydroxyl HO* adsorbates have lower Eads than atomic O*, but higher than peroxide HOO*. Comparison of the calculated Eads values indicates a preference for the 4e ORR mechanism over the 2e mechanism on both catalytically active sites.98
To gain better insight into the ORR mechanism, free energy profiles in acidic and alkaline media were plotted (Figs 8-10) at different electrode potentials.98 The criterion for evaluating the potency of the catalysts considered in the profiles of Figs 8-10 is the same general appearance of the lines: in the absence of the applied potential, the free energy should decrease at each step, and in the presence of the potential, the catalytic activity is the higher the closer the intermediates lie to the zero line.
Up to potential U ≈ 0.6 V, all elementary ORR steps on the CuN4 site proceed with decreasing ΔG, i.e. the reaction is exergonic, so in the region U≤0.6 V in acidic medium ORR proceeds spontaneously. When U rises to 0.8 V, the reaction generally remains exergonic, although the second step (O2*→HOO*) becomes endergonic. The least exergonic step is the transfer of the first electron on the CuN4 site. The second electron transfer with protonation of the HOO* adsorbate can provide either H2O2* or O*. The latter is 0.43 eV more favourable (inset in Fig. 8a), indicating that the 4e mechanism is preferable. The reaction HOO*→O*+H2O proceeds with a decrease of ΔG at U<1.1 V and a slight increase of ΔG (0.16 eV) at the equilibrium potential of U=1.23 V. The transfer of the third electron (O*→HO*) is the most significant decrease over the entire potential U range. Finally, the transformation of HO* adsorbate into H2O molecule proceeds with a slight increase in ΔG. For the equilibrium potential, the maximum in the profile corresponds to O* adsorbate (0.57 eV), and the minima — to O2* and HO* adsorbates (~–0.2 eV).
![[{"id":"4BBqT_10lp","type":"paragraph","data":{"text":"Free energy profiles of ORR on the CuN<sub>4</sub> (top row) and C<sub>2</sub> (bottom row) sites at differente lectrode potentials <i>U</i> in acidic (<i>a,c</i>) and alkaline (<i>b,d</i>) media"}}]](/storage/images/resized/UlEbgQbBdBz9KayuhONxv76Q0EFKbqoNEvFKkPGd_xl.webp)
In an alkaline medium, the whole process on the CuN4 site at U<–0.2 V is exergonic (Fig. 8b), indicating good ORR characteristics. The individual steps are similar to those in the acidic medium, but the catalytic activity in the alkaline medium is significantly higher than in the acidic one. The ΔG changes, shown in Fig. 8 in the O2→O2* reaction on the CuN4 site are consistent with those known from the literature at U=0 V for CuN2-doped graphene.90 However, according to the study,90 ΔG depends on U, which is incorrect, since Eads for O2 is constant and cannot depend on U. More pertinent is the data of Li et al.,101 in which a value of ΔG ≈ 0.9 eV was obtained for CuN4-doped graphene. The ΔG value of 1.1 eV obtained by us is slightly higher, indicating a positive effect of the nanotube compared to graphene.98
The second catalytically active site is C2. The adsorption of O2 on it is only 0.1 eV less favourable than on CuN4 (see Fig. 8c). The O2* adsorbate in an acidic medium is exergonic at U≤0.94 V, although lowering the potential to equilibrium leads to an increase of ΔG by 0.29 eV. The next two steps proceed with a decrease in ΔG at all potentials (see Fig. 8c). The formation of H2O2 in ORR on the C2 site is less favourable than on CuN4, as is the 2e ORR mechanism in general. The potential window in which the 4e ORR can proceed spontaneously at the C2 site (U≤0.6 V) in an acidic medium is somewhat lower than that of the CuN4 site (U<0.75 V) and therefore, the C2 site is more catalytically active than CuN4. In an alkaline medium, all steps of the catalytic cycle at the C2 site are exergonic at U<0.12 V, whereas at the CuN4 site this potential is U<–0.2 V (see Fig. 8b,d).
Thermodynamic analysis does not provide an answer on the kinetics of the process. The calculation of transition states for such large systems, including heavy metals, usually is not performed, or is performed without frequency calculations (e.g., see Ref. 90). We carried out a complete calculation of the transition states of all elementary ORR steps on the C2 site for the most high-performance CuN4-CNT catalyst.98 It turned out that the 2e mechanism is not only thermodynamically (ΔG –0.59 vs –1.47 eV), but also kinetically unfavourable (ΔG≠ 1.90 vs 1.19 eV). However, the more important question is whether a high barrier can restrain kinetically a thermodynamically allowed reaction. The ΔG≠ barriers are in the range of 0.07-1.19 eV, except for a very high barrier for the H2O2 formation (1.9 eV), which makes the process impossible under ORR conditions. In this case, the barrier of 1.19 eV is maximal, since the solvation of eliminating water molecule should lower ΔG≠ and thus facilitate the reaction.
The adsorption activity of the AgN4 site towards molecular oxygen is 0.44 eV higher than that of copper (see Fig. 9a). At U<0.4 V, the whole catalytic cycle is spontaneous. The O2*→HOO* reaction on silver is the least exergonic step of 4e ORR in an acidic medium. The difference in ΔG between the O* and H2O2* adsorbates on silver, as on copper, is small, 0.29 eV in favour of O*, which would seem inconsistent with the formation of a minor amount of H2O2 on Ag,N-codoped graphene found experimentally.102,103 However, considering the switching of the active site during electroreduction from AgN4 to C2 for HOO* adsorbate, this difference can increase up to 1.31 eV, which is in line with the experiment.
![[{"id":"SHOQbvlJhH","type":"paragraph","data":{"text":"Free energy profiles of ORR on the AgN<sub>4</sub> (top row) and C<sub>2</sub> (bottom row) sites at different electrode potentials <i>U</i> in acidic (<i>a,c</i>) and alkaline (<i>b,d</i>) media"}}]](/storage/images/resized/pGEn7SpMqyENd2V5gfT67xCLUhqKk2qfvkVeIFFs_xl.webp)
The O2 adsorption on C2 is an endergonic step with ΔG=1.1 eV for all potentials (see Fig. 9c). However, this is an apparent problem; the real catalyst is a dynamic system in which oxygen first forms O2* adsorbate on silver with ΔG<0, and the HOO* adsorbate formed during reduction migrates to the adjacent C2 site due to the large covalent radius of the silver atom with a 0.3 eV decrease in the free energy. The proposed ORR pathway involving both AgN4-CNT sites is more advantageous than each site separately, as it avoids the formation of unfavourable O* adsorbates on AgN4 and O2* adsorbates on C2.
The Zn,N-codoped catalyst is poorly active in ORR (see Fig. 10a,b). The O2 adsorption is endergonic (ΔG=0.19 eV). This is compensated by the reduction of O2*→HOO*, which is exergonic throughout the whole potential range. But in an acidic medium there is another, the most disadvantageous step, HO*→H2O, because HO* adsorbate represents a global minimum on the reaction coordinate. Therefore, there is no potential U on the ZnN4 catalytic site, at which ORR would proceed spontaneously in an acidic medium. Only in an alkaline medium at high negative potentials (U<–1.05 V) the catalyst can show some activity (see Fig. 10b). Zinc also adversely affects the C2 site in an acidic medium, although in an alkaline medium it can exhibit weak catalytic activity (see Fig. 10b). These theoretical findings are consistent with experimental data on the catalytic activity of Co/Zn,N-doped nanotubes.98
![[{"id":"Sf54n31_7w","type":"paragraph","data":{"text":" Free energy profiles of ORR on the ZnN<sub>4</sub> (top row) and C<sub>2</sub> (bottom row) sites at different electrode potentials <i>U</i> in acidic (<i>a,c</i>) and alkaline (<i>b,d</i>) media"}}]](/storage/images/resized/KoFIlYfeTZ7fNHnh6sZdrx9kr1novH5e32SOd7T7_xl.webp)
According to the criterion proposed above for evaluating the performance of catalysts, the C2 site of the copper complex is more efficient than that of the silver complex, and in the case of the zinc complex it is the least efficient. Neither Eads nor the absolute value of free energy ΔG is a measure of catalyst efficiency. ORR intermediates are able to form hydrogen bonds with water molecules from the environment, which decreases the energy of the system, and consideration of the solvent turns out to be important in describing the ORR energy. Thus, for HO* adsorbed on a platinum support, the stabilization by water molecules was estimated to be 0.1-0.3 eV.58,104
Similar approach was used to analyze the catalytic performance of Ti,N- and Zr,N-codoped nanotubes in ORR (Fig. 11).105 Ti and Zr have outer shells of 3d24s2 and 4d25s2 configurations, in contrast to Zn, Cu, Ag discussed above, in which d shells are completely filled (d10). Ti-containing ORR catalysts are much less susceptible to poisoning with methanol,106 but their activity in ORR is quite low, such as that of TiO2.107 The mechanism of O2 reduction on the TiO2/C catalyst surface has been studied theoretically (DFT).108,109 Titanium nanocomposites,110 MOFs,111 carbide112 and nitride113 on N-doped graphene were tested as ORR catalysts. Zirconium is also promising as an ORR catalyst; for example, nanosized ZrN was recently claimed to be close in activity and stability to platinum catalyst in an alkaline medium.114 Zirconium oxynitride-doped nanotubes showed the highest activity in ORR among all oxide-type catalysts.115 TiO2- and ZrO2-doped graphene showed high activity in ORR due to the synergism between the metal and the carbon support; pathways of these reactions are considered in only two publications.116,117 DFT calculations showed that the key factor determining the catalyst activity is the interaction between the metal oxide, functional groups and graphene.118 A specific feature of titanium and zirconium is the high M-O bond energy. An important point is that, in contrast to Zn, Cu and Ag, Ti- and Zr-containing catalysts do not lose their activity on the adsorption of ORR intermediates, as in this case the C2 site is active.
![[{"id":"sQF1SzEWy1","type":"paragraph","data":{"text":"Structures of metal, nitrogen-codoped nanotubes: TiN<sub>4</sub>-CNT (<i>a</i>) and ZrN<sub>4</sub>-CNT (<i>b</i>) and metal-nitrogen distances (Å)"}}]](/storage/images/resized/X7AqSq49InlJQVXtOKEAlSmEIj0yJzWXGxfQH4O6_xl.webp)
Although the atomic radius of Ti is almost equal to those of Cu and Zn (1.40 and 1.35 Å), as well as Zr and Ag (1.55 and 1.60 Å), Ti and Zr are 1.5-3 eV weaker bound to the N4 nanotube fragment. The Egap of HOMO/LUMO in MN4-CNT is 4.07 (Ti) and 4.24 eV (Zr). Atom charge analysis in Ti(Zr)N4-CNT revealed an accumulation of the positive charge at the metal atom, its decrease at the C2 site and at the four nitrogen atoms in the metal environment. This indicates a high activity of the metal which increases when going from titanium to zirconium.105
In terms of the mechanism, ORR is a complex process that can proceed via three main pathways: (i) associative, to afford H2O2 (2e); (ii) associative, to give H2O (4e); and (iii) dissociative, to provide H2O (4e) (Fig. 12). Both the 2e- and 4e-associative mechanisms involve chemisorption of the oxygen molecule and its further protonation/electron transfer (PET) to form the peroxide HOO* adduct. The latter can be converted to H2O2 in the second PET step. An alternative 4e mechanism provides the intermediate O* adduct. In contrast, the dissociative mechanism can only produce H2O by breaking the O–O bond in the O2* adsorbate. This mechanism excludes the formation of H2O2 and is the preferred for ORR. The direction of the reaction is governed by the nature of the active sites and the activation barrier of the rate-determining step. The possibility of changing the ORR mechanism from 4e to 2e has recently been reported using a Co,N-CNT catalyst with atomically dispersed CoNx active sites.116
![[{"id":"edY_fb0gy4","type":"paragraph","data":{"text":"Two- (<i>a</i>) and four-electron (<i>b</i>) catalytic cycles of ORR in an acidic medium.<sup>105</sup> Associative and dissociative pathways are marked black and red, respectively"}}]](/storage/images/resized/aTgEAAHp1EVF3kuwFDtynDU3PmdmBcKxi416gA2B_xl.webp)
The structure of O2* adsorbates on MN4 and C2 sites of Ti(Zr)N4-CNT catalysts is similar to that of catalysts with M=Cu, Ag, Zn. The high adsorption energy Eads on MN4 site should lead to metal oxidation in the ORR, which is consistent with the high dissociation energy of the Ti(Zr)–O bond. O*O* isomeric adsorbates are ~0.3 eV more stable than O2* adsorbates, which can be considered as a rough estimate of the difference in energy between the associative and dissociative ORR mechanisms on the MN4 site in favour of the latter, which is in line with literature data.112,117,118 The further sequence of transformations looks like O*O*→HO*O*→HO*HO*→HO*→H2O*, with 2HO* dihydroxy adsorbate being the most stable [Eads=–8.50 (Ti) and –9.38 eV (Zr)], whose existence was confirmed experimentally.122 The high stability of 2HO* adsorbates on the MN4 site is probably due to the participation of all d-orbitals of the metal in the MN4(OH)2 fragment, which has an octahedral environment and a d10s2 configuration. The adsorbates on the C2 site are more than twice less stable than those on the MN4 site. Optimisation of the O*O* adsorbate on C2 results in the migration of one of the oxygen atoms to the metal atom to afford a new oxidized catalyst M(O)N4-CNT. In this regard, the possibility of ORR at the C2 site, capable of competing with the metal in M(O)N4-CNTs or M(OH)nN4-CNTs catalysts, where n=1, 2, was explored.
As for MN4-CNT (M=Cu, Ag, Zn) catalysts, free energy profiles (Fig. 13-15) at different potentials were plotted.105 These are only provided for M=Ti, since for M=Zr the free energies change in a similar way.
As can be seen from Fig. 13, the adsorption of O2 at the TiN4 site in TiN4-CNT is exergonic (ΔG=–4.40 eV). The reduction of O2* to HOO* by the associative mechanism is endergonic (ΔG=0.40 eV at U=0). The ΔG increases to 1.63 eV as the potential U increases to an equilibrium value (1.23 V). The next step, HOO*→O*+H2O, is exergonic at 2.99 eV at U=0. The resulting O* adsorbate is the thermodynamic product of ORR at the TiN4 site over the whole electrode potential range. The reduction of O* to HO* is weakly endergonic on TiN4 but exergonic on ZrN4. The last step, HO*→H2O*, is endergonic for both metals. Desorption of water from the metal (H2O*→H2O) is also endergonic (ΔG=0.82 eV). The inefficiency of the desorption process is outweighed by the much more negative values of ΔG for O2* adsorbates. This implies a predominance of oxygen chemisorption over water desorption, which allows the MN4 site to recover its activity and ensures the recycling of the process.
![[{"id":"9FCfh4b0OJ","type":"paragraph","data":{"text":" Free energy profiles of associative (<i>a</i>) and dissociative (<i>b</i>) ORR pathways on the TiN<sub>4</sub> site of TiN<sub>4</sub>-CNT catalyst at different electrode potentials <i>U</i> in an acidic medium"}}]](/storage/images/resized/bh35y2MQhq1Ob0pSr0TDvcRVbpx3gOKFWa6p6QZi_xl.webp)
While the O2*→HOO* step is endergonic, the O–O bond cleavage by the dissociative mechanism (O2*→O*O*) is exergonic (ΔG=–0.40 eV) and dominates over it. Further protonation O*O*→HO*O* lowers the free energy, so that at U>0.4 V, the HO*O* adsorbate on TiN4 is a global minimum (see Fig. 13). However, at U<0.4 V, the 2HO* adsorbate is thermodynamically more stable. On the ZrN4 site, the 2HO* adsorbate is a global minimum at U<0.95 V, but with increasing U, HO*O* becomes more stable. Further electroreduction of HO*O*, 2HO*, or O* is extremely disadvantageous due to high stability of these species, therefore, the loss of activity of this catalytic site in the ORR can be expected.
As noted above, the C2 site of the MN4-CNT catalyst is much less active than MN4, and moreover, the adsorbate can migrate from the C2 site to MN4. Nevertheless, the electroreduction of O2 on the C2 site of MN4-CNT can proceed associatively, as attempts to simulate the O–O bond cleavage in the O2* adsorbate resulted in the O* adsorbate formation on C2 in the M(O)N4-CNT catalyst for M=Ti and Zr.105 The corresponding free energy profiles are shown in Fig. 14. The adsorption of O2 on the C2 site, as well as the overall 4e ORR, is exergonic at low potentials U. For high potentials, the last ORR steps become endergonic (see Fig. 14a,b). Adsorption, like the 4e ORR in general, is exergonic at low U potentials. For high potentials, the last steps of the ORR become endergonic (see Fig. 14a,b).
![[{"id":"rF45PLVTc_","type":"paragraph","data":{"text":"Free energy profiles of the associative ORR route on the C<sub>2</sub> site of unoxidized ORR catalysts TiN<sub>4</sub>-CNT (<i>a</i>) and ZrN<sub>4</sub>-CNT (<i>b</i>) at different electrode potentials <i>U</i> in an acidic medium"}}]](/storage/images/resized/rdPleOgJrDCWHytSe5KzZt2cVam7bRec1PxFeuvs_xl.webp)
Analysis of free energy profiles of H2O2 formation by the 2e mechanism at the C2 site (see inset in Fig. 14) indicates that they are in principle possible.
However, without the applied potential, the HOO*→H2O2 process is disadvantageous compared to the O–O bond cleavage, HOO*→2HO*. Moreover, the newly formed H2O2 molecule will inevitably be trapped by the MN4 site, on which it will become 2HO* adsorbate, which is the global minimum. The formation of H2O2 on the TiO2/C cathode in ORR has also not been experimentally recorded.122
Due to the tendency of the Ti and Zr metals in MN4-CNT to irreversible oxidation, the question arises about the possibility of the secondary activity of such stable oxidized forms of the catalyst as M(O)N4 and M(OH)nN4, n=1, 2, in ORR. Analysis of the calculated charge distribution maps showed that the C2 site in the M(O)N4- and M(OH)2N4-doped CNTs exhibits charge accumulation, i.e., it can be an active site for the O2 chemisorption.49 In contrast, the C2 site in M(OH)N4-CNT has a reduced electronic density, which accumulates at the dxy orbital in the metal plane. Free energy profiles as a function of the potential U are shown in Fig. 15.
![[{"id":"1T7UxxZ3W2","type":"paragraph","data":{"text":"Free energy profiles of associative ORR route on the C<sub>2</sub> site of Ti(O)N<sub>4</sub> (<i>a</i>), Ti(OH)N<sub>4</sub> (<i>b</i>) and Ti(OH)<sub>2</sub>N<sub>4</sub> (<i>c</i>) catalysts at different electrode potentials <i>U</i> in an acidic medium"}}]](/storage/images/resized/DKaKmgjS5lvno5XUuHNIPmXPkyYC5zCoP7LW6wPl_xl.webp)
The O2 chemisorption on the C2 site decreases the free energy, which increases on going from Ti to Zr in M(O)N4-CNT and from Zr to Ti in M(OH)N4-CNT (see Fig. 14a,b). For the most stable oxidized form of the M(OH)2N4-CNT catalyst, adsorption is exergonic at 0.03 eV (Ti) and 0.36 eV (Zr), although exothermic at 1.6 eV for both metals; the difference between Eads and ΔG is due to different entropy losses.98 All steps of the electroreduction of O2* adsorbate by the associative 4e mechanism on the C2 site in the Ti(O)N4-CNT catalyst are exergonic at U<0.54 V (see Fig. 14a). At the equilibrium potential of 1.23 V, the 2HO* adsorbate is a global minimum. The ORR profile at the C2 site for Zr(O)N4-CNT is similar to that for the Ti analogue.
The free energy profiles of the C2 sites of the M(OH)N4-CNT and M(O)N4-CNT catalyst are slightly different (for more details, see Ref. 105). Finally, the C2 sites of the M(OH)2N4-CNT catalyst exhibit a good 4e profile of the O2* adsorbate reduction: for both metals all steps are exergonic at relatively low potentials. The results suggest that the primary and the most active MN4 site is oxidized to the most stable form M(OH)2N4, and then the C2 site of this oxidized catalyst is called into action. To conclude, note that the influence of acidic or alkaline media on the ORR on Ti- and Zr-doped catalysts is not significant.105
We have recently applied DFT theoretical analysis to study the catalytic activity of V,N- and Nb,N-codoped nanotubes, and in addition to thermodynamic analysis, the kinetic aspects of the problem were also studied by calculating transition states.123 Most publications on vanadium and niobium ORR catalysts are focused on their carbides and nitrides, whereas very few papers concern V(Nb)–Nx–NCM.124--126 Recent experimental progress on V- and Nb-containing nanomaterials is described.127--129 In terms of irreversible metal oxidation by chemisorption of molecular oxygen, V and Nb are similar to Ti and Zr. It is worth noting that while there is an extensive chemistry of vanadyl (VO2+) derivatives, nothing similar can be said about ’niobyl’, ’titanyl’, or ’zirconyl’ derivatives.
The structure of the M–N4–C catalysts (M=V, Nb) is shown in Fig. 16. The C(1)-C(2) distance in the C2 active site decreases when going from V to Nb from 1.414 to 1.376 Å. The energy of binding of V and Nb with the environment calculated for the reaction [N4–C]2–+M2+=M–N4–C, is 0.7 eV lower than that of Ti and Zr, respectively, and the charges on the atoms are also lower by ~0.35e, which may indicate their higher catalytic performance in ORR.
![[{"id":"ZNxe8K6xKY","type":"paragraph","data":{"text":"Structures of M,N-codoped nanotubes: V-N<sub>4</sub>-C (<i>a</i>) and Nb-N<sub>4</sub>-C (<i>b</i>) and metal-nitrogen distances (Å)"}}]](/storage/images/resized/3zcUWjXubYIBtLEfwMBtiUxpsme5mf4TeyudbFti_xl.webp)
Adsorption of the O2 molecule on metal is exothermic, Eads=–4.16 (V) and –5.08 eV (Nb), with the O–O bond elongating by 0.23-0.26 Å, and O2* adsorbates are readily converted into 2O* adsorbates with a strong decrease in both Eads values by –14.2 (V) and –15.8 eV (Nb) and free energy ΔG by –4.23 (V) and –6.05 eV (Nb). As in the case of Ti and Zr, this suggests a tendency towards irreversible metal oxidation and an inability to further promote the ORR. The Eads values for other ORR intermediates at the vanadium atom range from –0.71 (HOO*) to –10.4 eV (O*HO*), and at the niobium atom — from –1.16 (H2O*) to –12.7 eV (O*HO*).
The energy Eads of dioxygen adsorption on the C2 site to form a peroxide bridge between the C(1) and C(2) atoms is more than two times lower than that for adsorption on a metal, which means a lower activity of the C2 site in the first ORR step. However, when blocking the metal atom through the formation of stable adsorbates, the C2 site can prevail. Consideration of ORR intermediates on the C2 site in oxidized catalytic forms such as M(O)–N4–C, M(O)(O)–N4–C and M(O)(OH)–N4–C is not possible excluding the effect of the electrode potential.
The decrease in free energy upon O2 adsorption on the metal on the M–N4–C site is 2.76 (V) and 3.83 eV (Nb), which is 1.6 and 0.8 eV less than for Ti and Zr, respectively.105 The first reduction step, O2*+[H+e] →HOO*, in the associative ORR pathway (see Fig. 12, Fig. 17a,b) is endergonic, ΔG>0, over the whole electrode potential range, making this mechanism unlikely. The only possible step is the cleavage of the O–O bond in the O2* adsorbate, lowering ΔG by 1.47 (V) and 2.22 eV (Nb) and making the dissociative pathway preferable. The next step, 2O*+[H+e] →O*HO*, is exergonic at U<0.51 V on vanadium and for any potentials on niobium. Further electroreduction of O* and O*HO* intermediates is thermodynamically forbidden, ΔG>0 for any U, which means irreversible oxidation of V and Nb atoms.
![[{"id":"ILGE_gkVDO","type":"paragraph","data":{"text":"Free energy profiles of associative (<i>a,b</i>) and dissociative (<i>c,d</i>) ORR routes on vanadium (<i>a,c</i>) and niobium (<i>b,d</i>) atoms at different electrode potentials <i>U</i> in an acidic medium"}}]](/storage/images/resized/BBwgwxLv31fEbXBb3mNocM30XqJee9ODZr2GHzUX_xl.webp)
The activation barrier ΔG≠ of the O2*→2O* reaction is as low as 0.49 (V) and 0.26 eV (Nb). The same order of magnitude is also true for barriers of the reaction H*+2O*→O*HO*, ΔG≠=0.47 (V) and 0.32 eV (Nb). The second PET step, O*HO*→O*, is thermodynamically possible only on the vanadium atom. The adsorption energy Eads on the C2 site of the M-N4–C catalyst is –0.29 (V) and –0.69 eV (Nb), i.e., much lower than on the metal (Fig. 18). The O–O bond cleavage is exergonic, O2*→2O*, ΔG=–0.65 eV, but an alternative process (O2*+[H+e] →O*HO*) appears more favourable. The O*HO* reduction gives stable 2HO* and O* as global minima at U>0.76 (V) and >0.37 V (Nb), and at the equilibrium potential, the global minimum is the O*HO* adsorbate.
![[{"id":"hBc9JfAq7z","type":"paragraph","data":{"text":"Free energy profiles of dissociative ORR route on the C<sub>2</sub> site of V-N<sub>4</sub>-C (<i>a</i>) and Nb-N<sub>4</sub>-C (<i>b</i>) catalysts at different electrode potentials <i>U</i> in an acidic medium"}}]](/storage/images/resized/1xdRQ2QBkyUoqbHH2RsTfW8k8nvjKro4dNElKjUY_xl.webp)
Analysis of the free energy profiles of the C2 active site in the oxidized form of the M(O)–N4–C catalyst (omitted for brevity, see Ref. 123) indicates the reaction following the 4e dissociative pathway. According to the ωB97XD calculation, the formation of a water molecule by the equation 2HO*→O*+H2O on the C2 site of the V(O)–N4–C catalyst is endergonic at any U, since the system falls into the global minimum along the ORR coordinate at U>0.65 V. In contrast, using the PBE functional provides a virtually ideal free energy profile going downhill.123 The transition from vanadium to niobium in M(O)–N4–C adversely affects the ORR profile on the C2 site. Given that the Nb(O)–N4–C form is not only less stable than Nb(O)(OH)–N4–C at any U, but also kinetically separated from it by a high barrier, ORR at the C2 site of M(O)–N4–C is hardly possible.
The free energy profiles of the ORR reaction on M–N4–C, M(O)(O)–N4–C and M(O)(OH)–N4-C catalysts, obtained by calculating not only intermediates but also transition states (TS), are generally similar for both metals, V and Nb, so only the profile for M=V is shown in Fig. 19. From the kinetic point of view, the TS corresponding to the barrier of reaction 2O*+H*→O*HO* at the metal atom in M–N4–C lies lower compared to the barrier of formation of an unstable O2* adsorbate on the C2 site in M(O)(O)–N4–C, so the first reaction should run faster (see FFig. 19). The barrier difference, equal to 0.08 eV at U=0, raises with increasing U. The stable O*HO* adsorbate formed on the metal atom in M–N4–C, M is thermodynamically more favourable than HOO* and O*HO* intermediates formed on the C2 site in M(O)(O)–N4–C. The formation of 2HO* and O* adsorbates in the second step of PET on the metal atom of the M–N4–C catalyst is not only slower due to the high barrier ΔG≠, but for niobium, is also prohibited thermodynamically (ΔG>0). In contrast, ORR on the C2 site in M(O)(OH)–N4–C, O2*+[H+e] →O*HO*, is preferable both thermodynamically and kinetically. This confirms the assumption of the predominance of catalysis on the C2 site of the support during irreversible metal oxidation (poisoning).
![[{"id":"m0IVu8L75x","type":"paragraph","data":{"text":"Free energy profiles of ORR on V (black) and the C<sub>2</sub> site of V(O)(O)-N<sub>4</sub>-C (red) and V(O)(OH)-N<sub>4</sub>-C (dark blue) at <i>U</i>=0 in an acidic medium. Values ΔG in square brackets refer to ORR intermediates on the corresponding carbon sites. TS is transition state"}}]](/storage/images/resized/EGfcD1mv97Qx6QbRqx32umlWCCM2hh7QFRP8GKcj_xl.webp)
The most probable ORR mechanism on C2 in M(O)(OH)–N4–C involves the O2*→2O*→O*HO* reaction. Indeed, the first step of the PET, O2*+H*→HOO*, is slow for V(O)(OH)–N4–C due to ΔG≠=0.98 eV, which is higher than for O2*→2O*, and for Nb the TS cannot be localized at all. Even in the case of formation of HOO* intermediate, its barrier of the reduction to H2O2 is higher than for the HOO*→O*HO* reaction (ΔG≠=0.63 eV). As a result, H2O2 should be formed slowly on the oxidized catalyst and ORR should proceed by the dissociative 4e mechanism: 2O*→O*HO*→2HO*→O*(+H2O) →HO*→*(+H2O). The first three steps of PET are fast, and the last one, HO*+H*→*+H2O, is rate-determining, ΔG≠=1.09 (V) and 2.65 eV (Nb) on C2 in M(O)(OH)–N4–C. All PET steps are exergonic at any U for M=V, although the step of oxygen chemisorption is endergonic. For M(O)(OH)–N4–C catalysts, as U increases, not only the HO* reduction PET step but also the O* reduction becomes endergonic.
The main conclusion of this Section is that there are two active catalytic sites in MN4-doped nanotubes: a metal atom in the MN4 fragment and its neighbouring C=C bond of the support, and the activity of the latter in ORR may be higher than that of the metal, contrary to what has been assumed in the previous theoretical studies. The theoretical thermodynamic characteristics of ORR on metal,nitrogen-codoped nanotubes allow predicting the initial potential obtained experimentally from the voltammetric characteristics of real catalysts, as well as the nature of the rate-limiting step of the process.
Silicon-doped NCMs are the least studied, although their high oxophilicity, which facilitates adsorption of oxygen on the silicon atom in Si-doped substrates, makes them potentially active ORR catalysts. Similar to metal-doped nanotubes discussed above, we investigated Si-doped single-walled nanotubes SiC83 and SiC119 of different diameters (Fig. 20), the thermodynamics of 2e and 4eORR involving them, and evaluated the effect of the pyramidality η of the silicon atom, defined as η=(360– ΣSi), where ΣSi is the sum of angles at the silicon atom, on its activity in ORR.130
![[{"id":"wOqvVo82sG","type":"paragraph","data":{"text":" Structures of SiC<sub>83</sub> and SiC<sub>119</sub> nanotubes of different diameters"}}]](/storage/images/resized/8FLNbNAiOpSGxsB4IZG9M93z77hjHWNd2MBmP2jY_xl.webp)
The higher tension of SiC83 nanotube is consistent with its lower formation energy (–5.23 eV) compared to SiC119 (–9.98 eV), while the higher pyramidality of the silicon atom makes it more oxophilic. As a result, adsorption of O2 on SiC83 is more exothermic (Eads=–4.51 eV), than on SiC119 (Eads=–2.86 eV). The structure of the adducts also differ significantly: in the case of O2*–SiC119, the O and Si atoms form a ’siladioxirane’ (Fig. 21a), while O2*–SiC83 provides 1-sila-2,3-dioxetane (Fig. 21b).
![[{"id":"JCzV9--ISi","type":"paragraph","data":{"text":" Strucrures of O<sub>2</sub>* SiC<sub>119</sub> (<i>a</i>) and O<sub>2</sub>* SiC<sub>83</sub> (<i>b</i>) adsorbates"}}]](/storage/images/resized/88YgNZvYb2zvm2g7yCmC4uF5PztdWphEpTKS5Vn9_xl.webp)
Note the opposite change in pyramidality of the silicon atom in the adsorbates in Fig. 21. The value η in O2*–SiC119 increases to 74° and decreases to 43° in O2*–SiC83 due to ’pulling out’ of the Si atom from the nanotube core by two oxygen atoms in the first case and its ’pulling in’ due to the O–C bond formation in the second case.
The profiles of the ORR free energy change on both Si-doped nanotubes at different potentials are very similar, but differ for acidic and alkaline media (Fig. 22).
![[{"id":"Uyz5jv0Ukj","type":"paragraph","data":{"text":"Free energy profiles of ORR on SiC<sub>119</sub> in acidic (<i>a</i>) and alkaline (<i>b</i>) media"}}]](/storage/images/resized/5sDI5lpwqdP4U2bLqW0mBixh0NqEq4BZw0dTIaux_xl.webp)
For the 4e ORR on SiC119 in acidic medium at U=0 the value ΔG goes downhill to –1.70 eV for HO*. Increasing the potential to the equilibrium value leads to a larger decrease, to –2.94 eV, making the transfer of the fourth electron thermodynamically unfavourable. Note that the 2e ORR route is virtually impossible. The study of free energy profiles shows that in both acidic and alkaline media the 4e ORR on SiC119 reaches a minimum at all potentials, with the potential close to zero (acidic medium) or the maximum possible negative (alkaline medium) being optimal. The nanotube diameter (wall curvature) significantly affects the ORR thermodynamics: nanotubes with large diameters are preferred.
During the last decade, the interest to fullerenes C60 doped with B, N, Si, P, S grew (see Ref. 131 and references cited therein). It should be noted that when carbon atoms are replaced by a heteroatom, including silicon, fullerene retains its spherical structure.132 Advances in the synthesis of Si-doped fullerenes C59Si, C58Si2,133 and theoretical studies of ORR132,134 allowed to gain a better insight into the reaction mechanism. The importance of theoretical studies is already evidenced by the fact that fullerene per se was first predicted theoretically135,136 and only then synthesized.137 Si-Doped fullerenes C60-nSin (n=1, 2, 12) were first studied theoretically by the AM1 computational method back in 1993 (see Ref. 138) but the conclusion was uncertain: ’incorporation of silicon destabilizes the catalyst framework but not excessively’ (for a review on silafullerenes see Ref. 139).
The formation energy of the Si-doped fullerene C59Si was estimated at 5.1-5.4 eV,140--142 which fits well with our calculations (5.56 eV),131 but all these results contradict the clearly erroneous value of -3.15 eV obtained recently for C59Si.132 At first sight, this is strange, since all energy values were calculated by the same equation Eform=(ESi–NCM–ENCM)–(ESi–EC). We showed,131 that the value EC in the publication132 means not the energy of the carbon atom (as it should be and as is accepted in all other works),131,140--142 but the energy of the C atom in C60, i.e., the total energy of C60 divided by 60. This methodological error led the authors to the wrong conclusion about the low catalytic activity of Si-doped fullerene. At the B3LYP/6-311+G(d) level, the values of EC and EC60/60 differ by 8.66 eV, which when summed with –3.15 eV gives the correct value of Eform=5.51 eV, almost coinciding with the value of 5.56 eV obtained by us.
Another fundamental difference in publications131,132 concerns the structure of O2*–C59Si (incorrectly determined as O2@C59Si,132 which would imply incorporation of the O2 molecule into the fullerene cavity). The structures of the O-containing adsorbates on Si-doped fullerene are shown in Fig. 23.
![[{"id":"nkdM9IXCJK","type":"paragraph","data":{"text":" Structures of ORR intermediates on Si-fullerene C<sub>59</sub>Si: O<sub>2</sub>*-C<sub>59</sub>Si siladioxirane (<b>1a</b>), O<sub>2</sub>*-C<sub>59</sub>Si bridging (<b>1b</b>), HOO*-C<sub>59</sub>Si (<b>2</b>), O*-C<sub>59</sub>Si (<b>3</b>), HO*HO*-C<sub>59</sub>Si (<b>4</b>) and HO*-C<sub>59</sub>Si (<b>5</b>)."}}]](/storage/images/resized/YUJmnkMNPpojVc8fPOd22j24bojVPAXGsKnksMJ1_xl.webp)
Adsorbate 1b is 0.78 eV more favourable than siladioxirane 1a, although Wang et al.132 claimed otherwise. This led the authors to incorrect conclusions that Si-doped fullerene is ’the worst ORR catalyst among all other doped fullerenes’.132
The endergonic desorption of the water molecule from the silicon active site does not negatively affect ORR because the catalytic cycle is completed not by simply desorbing the H2O molecule but by the H2O*–C59Si+O2→O2*–C59Si+H2O exchange reaction. In contrast to desorption, this reaction is exothermic at 2.12 or 2.90 eV, depending on the structure of O2*–C59Si.
The free energy profiles are shown in Fig. 24. It can be seen that in an acidic medium, the adsorbate HO* 5 is the most stable. Its final protonation has a barrier from 1.49 to 2.29 eV. At equilibrium potential, the HO*O* adsorbate 2b is a global minimum; its protonation proceeds with a barrier of 0.51 eV. In an alkaline medium, without an applied potential, the adsorbates HO*O* 2b, HO*HO* 4, HO* 5 are at –4 eV (Fig. 24b) and the latter step is endergonic to more than 2 eV. As in many cases discussed above, the 2e ORR mechanism is hardly probable.131
![[{"id":"QVVZMCG2sR","type":"paragraph","data":{"text":" Free energy profiles of ORR on C<sub>59</sub>Si at different electrode potentials <i>U</i> in acidic (<i>a</i>) and alkaline (<i>b</i>) media"}}]](/storage/images/resized/ZT9yplEssHnyPQOxwILgQ8SL3XUumfNj4DUJUerq_xl.webp)
Therefore, the 4e ORR on Si-doped fullerene C59Si in an acidic medium should terminate at the step of HO* adsorbate formation at U<0.8 V, and for equilibrium U=1.23 V — at the step of HO*O* formation. The system behaves similarly in an alkaline medium. Despite the thermodynamically unfavourable last step, (5+[H++e-]→1+H2O), high exothermicity and barrierless exchange ’O2 adsorption/H2O desorption’ is the driving force of the process in the presence of fullerene C59Si, which remains one of attractive ORR catalysts.
Si-Doped graphene is a promising ORR catalyst, as the dissociation of the O2 molecule adsorbed on it is 16 times easier than on pristine graphene.143 DFT calculations showed that the kinetically preferred pathway of ORR on Si-doped graphene involves O2 dissociation and hydrogenation, HOO*→O*+H2O.30 It was concluded that Si-doped graphene is highly active in ORR, but its structure is questionable. In a study,30 it was modelled by replacing one C–C bond by a tetracoordinated silicon atom (Fig. 25a), which lowered the aromaticity of graphene. In contrast, we have incorporated a three-coordinated silicon atom into the structure with retention of aromaticity (Fig. 25b).99 Adsorption of O2 on the support (b) should be more favourable due to the instability of the three-coordinated silicon atom.
![[{"id":"AHKayg-jya","type":"paragraph","data":{"text":"Structure of model Si-doped graphene with tetra- (<i>a</i>) and tricoordinated Si (<i>b</i>)"}}]](/storage/images/resized/8k8GR22iK7npaXyVbru5MLp5kwcSNqC3wjU0YN8z_xl.webp)
The planar structure (see Fig. 25a) corresponds to a minimum on the potential energy surface but this is only a local minimum as the silicon deviation from the plane by 0.6 Å lowers the energy by 0.60 eV. The geometric parameters of the non-planar structure correspond to those previously reported.29 Adsorption of O2 on a silicon atom in both the planar and non-planar structures furnishes the same adsorbate with the silicon atom being surrounded by a nearly tetrahedral environment.
Analysis of charge density distribution maps shows that the Si atom and a-carbon atom are sites of O2 adsorption. Indeed, it was shown that O2 adsorption on silicon is barrierless (Eads=–2.40 eV); the structure was analyzed and the relative energies of all possible ORR intermediates on Si-doped graphene were estimated.99
The elongation of the O–O bond in O2* suggests the activity of the catalytic site in ORR and is observed in all O2* adsorbates as compared to the O-O bond in H2O2 (1.49 Å). Protonation of O2*–C53H18Si isomers can afford HOO*–C53H18Si and HO*O*–C53H18Si adsorbates, the latter being much more energetically favourable (~2.54 eV). It can be protonated both via free and protonated oxygen atom with the elimination of H2O or H2O2, respectively. A detailed analysis showed the preference of the 4e ORR pathway over the 2e mechanism.99
A complete analysis of the transformations on Si-doped graphene C53H18Si revealed three possible ORR pathways. The first is a common pathway through protonation of HOO* to give water and O*. The second pathway involves protonation of HOO* to HO*HO*; this is a new mechanism, it is 0.15-0.28 eV more favourable. The third one, also new, involves the incorporation of oxygen into the Si–C bond; this is the longest, but also the most advantageous (by 0.5-1.09 eV) ORR route. Free energy profiles for all three mechanisms are given in Fig. 26.
![[{"id":"CESQiRRm4Z","type":"paragraph","data":{"text":"Free energy profiles of 4e-mechanisms of ORR on Si-doped graphene in an acidic medium at <i>U</i>=1.23V: conventional route HOO*→O* (<i>a</i>); alternative route HOO*→HO*HO* (<i>b</i>); novel route via the O-O bond cleavage after O<sub>2</sub> adsorption and incorporation of the O atom into a graphene sheet (<i>c</i>)"}}]](/storage/images/resized/8A5sQlA6fpKX3sCwgKqIexrUnvEPwogW4YSzt4yR_xl.webp)
For the first and second mechanisms, the influence of the electrode potential in an acidic medium is similar: at any potential, the HO* adsorbate is the most favourable. In an alkaline medium, the following dependence is observed: the lower the potential, the smaller the energy gap between the intermediate and the product. For the third mechanism two minima, HO*O* and HO*, U-dependent in both acidic and alkaline media were revealed. It was found that by the first pathway, the reaction proceeds spontaneously in the acidic medium at U≤0.5 V, and in the alkaline medium at U<–0.33 V. For the second pathway, these potentials are U<0.57 V and U<–0.26 V, respectively, and for the third pathway, U<0.99 V and U<+0.22 V.
Therefore, metal,nitrogen-codoped and Si-doped carbon nanomaterials proved to be promising ORR catalysts.
4. Electrochemical reactions of carbon dioxide reduction (CO2RR)
The electrochemical reaction of CO2 reduction has recently attracted increasing attention.38,41 In contrast to the O2 reduction, the CO2 reduction is a complex process involving up to several dozens of protons and electrons (for ORR this number does not exceed four), depending on the reaction route (Fig. 27).37,38
![[{"id":"4H0PXD1B9a","type":"paragraph","data":{"text":" Spectrum of possible products of electrocatalytic CO<sub>2</sub> reduction up to C<sub>2</sub>"}}]](/storage/images/resized/VrYlCgTljgABjIgMb32PaLVCvyzpPNYWtAlg3lQF_xl.webp)
4.1. Direction and selectivity of CO2RR
Compared to their photochemical and biochemical counterparts, electrochemical CO2 reduction (CO2RR) features reusability of an electrolyte and renewable, environmentally friendly energy sources (water, wind, etc.), controllable electrode potential and temperature.36 The product-specific selectivity of CO2RR catalysts typically does not exceed 30% and is complicated by the competing reduction of H+ ions to H2 (hydrogen evolution reaction, HER), which occurs at an electrode potential of U≤–0.42 V vs standard hydrogen electrode. Although the electrochemical reduction of CO2 to hydrocarbons (methane, ethylene) and C2+ alcohols (ethanol, n-propanol) is thermodynamically more favourable than HER, CO2RR is slower due to a lower rate compared to HER, which stems from higher activation barrier (see below) and high overpotentials.144 Another difficulty is that to provide the formation of most products of reduction of CO2 to C2+, a potential should fall in a very narrow range between –0.4 and –0.2 V (without overpotential); otherwise the whole spectrum of products is formed, and the only way to control the selectivity is a kinetic factor.
The possibility of several processes to occur on the same catalytic site governs the potential direction of H+ and(or) CO2 reduction, and is determined by the lowest activation barrier in the reaction pathway. For example, some catalysts are able to promote only HER, others — both HER and the formation of CO and(or) other CO2RR products.
Therefore, current challenges are (i) improving the selectivity of individual reaction routes, (ii) reducing the overpotential to overcome energy barriers and inhibit HER, and (iii) achieving moderate current densities for commercial applications.46 Addressing these challenges lies at the interface between theoretical prediction of catalytic site activity depending on their environment and experimental possibilities for their targeted synthesis. Experimental advances in CO2RR are highlighted in recent reviews.37--39,46,145 This review summarizes the available sporadic theoretical concepts of CO2RR on different types of catalysts (metals, metal oxides, doped nanocarbon materials).
4.2. CO2RR catalysts
4.2.1. Metals and alloys
Metals are historically the first and still the most studied cathode materials for CO2RR. Royer146 as early as in 1870 was the first to use a zinc cathode in a solution of potassium carbonate and discovered the formation of formates. Systematic experimental studies on various metals in CO2RR were carried out by Hori et al.147 in the 1990s. All all-metal cathodes were conditionally divided into CO- (Cu, Au, Ag, Zn, Pd,148 Ru,149 Ga, Ni, Pt150) and formate-producing (Pb, Hg, In, Sn,151,152 Cd, Tl, Co), with CO generation selectivity decreasing in the series
Au>Ag>Pd>Ru>Cu>Zn>Cd>Sn>In>Pb>Tl>Hg
Note that CO is both the key CO2RR intermediate for all types of catalysts, since its adsorption energy determines the direction of further reduction, and the catalytic poison for some Ni,153,154 Fe,155 Pt,150 Ti, Ga and Co-based catalysts; in these cases, hydrogen is released as the main product (HER).38 The disadvantages of metal catalysts include high overpotential, low current densities, toxicity (Pd, Hg, Cd, Tl) and high cost.46 The catalytic activity of metals depends on particle size, morphology, structural defects and type of the crystal lattice.144,154,156,157
As noted above, the precious metals Au, Ag, Pd (see Ref. 148) and Ru (see Ref. 149) produce CO as the major reaction product (FE=91--97%) due to the low energy of CO adsorption on the metal surface, the limiting step being protonation of the *CO2-• anion radical for Au and Ag, or *COOH adsorbate in the case of Pd.144,148 Rösch and co-workers149 studied the formation of O*C*H formyl intermediate on the model Ru(0001) catalyst and showed that CO formation is not only thermodynamically, but also kinetically preferable, since desorption of CO from the catalyst surface is 0.65 eV more favourable than its protonation to give O*C*H adsorbate. Using gold–cadmium alloy Au3Cd as an example, Chorkendorff and co-workers158 theoretically showed the possibility for methanol to be formed in addition to CO production. However, this experiment showed the absence of such Au-active sites and the alloy samples showed reduced or similar activity with respect to CO formation compared to all-metal gold.
Apart from precious metals (Pt, Rh, Ir, Pd, Ag and Au), Hussain et al.150 theoretically studied Cu- and Ni-containing catalysts M(111) in CO2RR and concluded that from the thermodynamic point of view, Pt catalyst is characterized by the lowest calculated absolute value of the initial potential (as low as –0.3 V) for the formation of methanol and methane, and the onset potential decreases to –0.8 V in the series Pt>Ni>Rh>Ir>Pd≈Cu>Au>Ag, and, thus, Pt catalyst, besides high activity in ORR, should also be active in CO2RR. However, calculations of the activation energy for the protonation of a CO molecule adsorbed on Pt showed that it is quite high at 0.7 eV, which is 0.4 eV higher than that for HER, and consequently, platinum is inactive in CO2RR for kinetic reasons.
Despite the high activity of precious metals in the formation of CO from CO2, their high cost and extremely low selectivity in the formation of C1 and C2+ products explain their small applicability as CO2RR catalysts. At the same time, activity of such metals as Pb, Hg, In, Sn, Cd, Tl and Bi towards the formation of formate ion during CO2 electroreduction is explained by rather high adsorption energy of CO2-• anion radical.159 An exception is copper. In a pioneering work, Hori et al.160 used all-metal Cu as a catalyst to produce methane as the major product along with ethylene, ethanol and n-propanol, with a total current efficiency (FE) for hydrocarbons of a record 72%, while the HER product yield was as low as 21%. A detailed theoretical analysis of CO2RR on the model Cu(111) catalyst showed that the reduction of CO2 to *CO and then, through the *COH intermediate, to methane is the most probable kinetically.150 The authors note that the alternative protonation of *CO at the carbon atom to form *CHO and then formaldehyde, although thermodynamically more favourable compared to the *COH intermediate, has an activation barrier 0.15 eV higher. Recently, these results have been reexamined by another researcher group162 and compared to the results for Co, Ni, Rh, Pd, Ag, Ir, Pt, Au,161 and also to the activity of Cu(211).
Importantly, copper particles smaller than 5 nm are characterized by a sharp increase in activity in the formation of CO and H2,163 whereas hydrocarbons are formed in trace amounts.164 Apparently, ethylene is formed from two CO molecules adsorbed on adjacent Cu(111) and especially Cu(100) surface sites165,166 due to the positively charged molecular layer of water (solvated H+ or Na+), that stabilizes this intermediate as OC*C*O, otherwise its formation is endergonic, and the results of calculations in the gas phase indicate the preferential adsorption of C2O2 intermediate on the copper surface by only one carbon atom.167,168 The influence of factors such as pH, solvation, the nature of cations and anions in the electrolyte solution, in addition to the structure of the metal copper catalyst, as well as the mechanism of CO2RR from a thermodynamic viewpoint are detailed in the study.169 The unique properties of copper and copper-based catalysts were the subject of several recent reviews on bimetallic Cu–M catalysts170--172 including nanoparticles.173
4.2.2. Metal oxides
Compared to all-metal catalysts, metal oxide CO2RR catalysts have an improved performance, but low stability and electrical conductivity, which determines their limited applicability.48,174 The mechanism of action of such catalysts and the nature of their active sites are still a subject of speculation, since less oxophilic metals can be reduced under CO2RR conditions.175 Thus, some researchers argue that the activity of metal oxides stems from the formation of nanostructured metal particles on the oxide surface due to reduction,174--176 others believe that it may result from the formation of oxygen vacancies.177--180 Metal oxides can act as carriers of metal nanoclusters active in CO2RR, for which not only increased catalytic activity and selectivity of metal sites, but also changes in thermodynamic and kinetic parameters of CO2RR due to the effect of the oxide support were revealed,157,181--190 however, consideration of these issues is beyond the scope of this review.
A comparative study of the catalytic activity of Cu(111) and CuO(111) towards methanol formation revealed relatively weak adsorption of the CO2 molecule on the surface of both catalysts, Eads=–0.24 eV.191 The activation barrier for further protonation of the CO2 molecule adsorbed on Cu(111) is very high (2.32 eV), whereas in the case of CuO(111) it is 1.68 eV lower, and the resulting intermediate *COOH is in equilibrium with the starting non-reacting system (Fig. 28). The alternative protonation of *CO2 at the carbon atom in the case of Cu(111) is thermodynamically unfavourable and in the case of CuO(111) it is kinetically unfavourable. Further, the reaction on Cu(111) follows the dissociative pathway, *COOH→*CO+*OH (ΔG≠=0.61 eV), while on CuO(111), the protonation *COOH+H*→*CO+H2O* (ΔG≠=0.50 eV) is kinetically the most favourable. The CO adsorption energy on model Cu(111) and CuO(111) is –0.78 and –0.57 eV. Further reduction via the *CO→*CHO→*CH2O→ *CH3O→CH3OH route proceeds on Cu(111) with a lower activation barrier than for the first step (2.32 eV), whereas on CuO(111), the rate-determining step is *CO→*CHO (ΔG≠=1.39 eV). For both catalysts, *COH is the most stable intermediate but its participation in CO2RR was not considered by the authors. The mechanism of HCOOH and CH3OH formation was also studied by the example of Cu2O(111),192,193 but Monte-Carlo calculations showed that the catalyst was easily reduced by CO and H2 to form metal copper.193
![[{"id":"vEozgafSW7","type":"paragraph","data":{"text":"Activation energies of selected elementary CO<sub>2</sub>RR on model Cu(111) and CuO(111) catalysts.<sup>191</sup>"}}]](/storage/images/resized/b7lUTwe2pN6d69nlaGSIka88Oz2ux31l3qkiv3Nl_xl.webp)
The mechanism of electroreduction of CO2 to CH3OH on indium oxides In2O3(111) Ref. 179) and In2O3(110) (Ref. 180) differs significantly from that on copper oxides, since this reaction proceeds on oxygen vacancies and does not involve the intermediate CO formation (see Fig. 28). Carbon dioxide is better adsorbed on In2O3(110) than on In2O3(111),180 with Eads varying from –0.11 to –1.21 eV depending on the adsorbate configuration. In the first step, an unstable carbonate *OCO2 is formed, which is reduced to *CHO2 with a decrease in ΔG by ~0.5 eV (Fig. 29) relative to the non-reacting system.179 The rate-determining step is the protonation to *CH2O2 with the eventual formation of CH3OH. The electroreduction pathway CO2→*OCO2→*CHO2→...→CH3OH on In2O3(110) is retained, but the presence of water molecules on the catalyst surface reduces the activation barrier to formate-adsorbate *OCHO formation by ~30%.180
![[{"id":"m-v4rXU8UW","type":"paragraph","data":{"text":"Free energy profiles of CO<sub>2</sub>RR on indium oxide In<sub>2</sub>O<sub>3</sub>(111): CO<sub>2</sub>→CH<sub>2</sub>OOH (red); CH<sub>2</sub>OOH→CH<sub>3</sub>OH (black and deep red); CHO<sub>2</sub>→CH<sub>2</sub>OOH (green); RWGS (reverse water gas shift reaction, dark blue).<sup>179</sup>"}}]](/storage/images/resized/A8M7WX4pdWyL9hk3ipunXoBDBiZsni0yPBcpPWIg_xl.webp)
Oxides of bismuth (Bi2O3),194 tin (SnO2)195 and cobalt (Co3O4),196 like the metals themselves, are active in CO2RR at the step of formic acid formation due to the appearance of oxygen vacancies on the metal oxide surface through the reduction of surface oxygen atoms to water.194,196 Thus, a process involving adsorption of CO2 molecule and its subsequent reduction at the carbon atom on the SnO2(110) oxygen vacancy was found to be 0.46 eV more favourable than the same process on a defect-free catalyst,195 whereas CO2 protonation at the oxygen atom is extremely endergonic (>1.8 eV) and therefore unlikely. Recently, catalysts based on oxides of zinc ZnO (Ref. 197) and zirconium ZrO2,198 have been theoretically explored, but it turned out that they are only able to reduce CO2 to CO. The theoretical possibility of the formation of C1 products (HCOOH, CH2O and CH3OH) on ZnO was considered.197 Hara et al.198 found that the regeneration of oxygen vacancies is a rate-determining step (ΔE≠=2.6 eV) on ZrO2(111), which was not observed for the first row transition metal oxides. Also, it was shown that the cleavage of one of the C–O bonds in *CO2 (or *CO2H) adsorbates followed by CO (COH) migration from the ZrO2(111) surface to the Ni atom is more likely and eventually affords the methane molecule on the metal centre. Other metal oxides, for various reasons, are considered as CO2RR catalysts to a lesser extent.48
4.2.3. Nanocarbon materials
Nanocarbon materials NCMs (graphene, nanotubes, etc.) are of particular interest not only for ORR, but also for CO2RR due to their stability, high mechanical strength and good electrical conductivity.144 Pristine NCMs are inert in CO2RR because of the adsorbate instability. For example, proton transfer from the graphene surface to a CO2 molecule to give of *OCHO formate is a highly endergonic process (ΔG=2.7 eV).199
Doping with B, O, F, P, S and N heteroatoms improves the catalytic activity of NCMs.200 The nitrogen atom as a dopant of NCM is unique in that it positively affects both ORR (see above) and CO2RR. However, unlike ORR, in which carbon atoms act as active sites, pyrrolic and pyridinic nitrogen atoms are predominantly involved in the catalysis of CO2RR (Fig. 30). Relative to the above value (ΔG=2.7 eV), the formation of *OCHO adsorbate on the graphitic nitrogen atom is much less endergonic (ΔG=1.5 eV) and even lower on the pyridinic nitrogen atom (ΔG=0.7 eV). In contrast, the adsorption of *OCHO on the pyrrolic nitrogen atom is exergonic (ΔG=-0.4 eV).199 The activity of N-doped carbon nanomaterials stems not from a change in charge density as in ORR, but from the ability to effectively overlap the LUMO of the CO2 molecule localized on the carbon atom and the lone pair (LP) of the nitrogen atom (pyridinic and pyrrolic) in NCM. Obviously, localization of the LP of pyridinic nitrogen in the plane of the carbon network will result in only partial interaction with CO2RR intermediates, whereas the LP of pyrrolic nitrogen is localized in the perpendicular plane, providing a more efficient interaction. Such N-doped carbon nanomaterials promote formation of CO from CO2 due to pyridinic nitrogen atoms, while the pyrrolic nitrogen isolated from other active sites is poisoned by the *CO adsorbate (ΔG=–0.6 eV). The FECO performance of these materials is comparable or exceeds that of gold (85%), with an onset potential of as low as –0.19 V and an overpotential of ca. –0.5 V.199 In studies,201,202 similarly calculated ΔG values for adsorption of *COHO and *CO (including N-doped nanotubes) were obtained,202 and N-oxide pyridinic nitrogen as a potential active site was also considered. The performance of the latter in CO2RR was found to be lower than that of undoped graphene. The kinetic study of CO2 reduction to CO on N-doped graphene showed that the rate-determining step of the process is the reduction *OCHO→*CO+H2O.203 The activation energy ΔG≠ of this step on the pyridinic nitrogen is 0.51 eV, and on the pyrrolic one it is 0.68 eV. Activation barriers of the subsequent steps of *CO electroreduction are 0.97 and 1.92 eV, which is much higher than for the CO desorption (0.08 and 0.58 eV, respectively). In the same publication, the possibility of carbon atoms at the graphitic nitrogen atoms located at the edge of the graphene network to be involved in the reaction CO2→CO was theoretically shown (ΔG≠=0.55 eV).
![[{"id":"VdPs_cw7Ul","type":"paragraph","data":{"text":" Thermodynamic profiles of the reduction of CO<sub>2</sub> to CO on different types nitrogen of atoms incorporated into the graphene structure.<sup>199</sup>"}}]](/storage/images/resized/hTTC3hH7buN8nM0Zq92eGX7tUAjQ7Nu1V3N3oaOB_xl.webp)
Song et al.204 explored an ordered cylindrical mesoporous N-doped carbon nanomaterial and showed its 100% selectivity to the C2H5OH synthesis at -0.56 V (FE=77%). According to the authors, this is a result of grouping of active sites on the catalyst surface, which contributes to the dimerization of two closely spaced chemisorbed CO molecules.
The thermodynamic profile of the ethanol formation was examined on the example of a nanotube, in which one carbon atom was removed and the adjacent three carbon atoms were replaced with pyridinic nitrogens, and also on the example of a nanotube with two pyridinic and one pyrrolic nitrogens (Fig. 31). The calculation showed that the adsorption of two CO molecules on the pyridinic nitrogen atoms of the first structure lead to a decrease in ΔG by 1.68 eV. Subsequent closure of the C–C bond between the *CO adsorbates to form OC*C*O is a rate-determining step as ΔG increases by 0.37 eV. Further reduction of OC*C*O to C2H5OH proceeds strongly downhill in free energy (up to –4.61 eV) relative to the non-reacting system. On the contrary, reduction on the active site comprising a pyrrolic and one of the two pyridinic nitrogen atoms of the second structure provides a 0.12 eV increase in the free energy resulting from the adsorption of two CO molecules (rate-determining step), and further reduction is exergonic. Therefore, N-doped carbon nanomaterials represent a class of promising and selective CO2RR catalysts. Increasing the degree of N-doping and, consequently, the spatial proximity of the active sites determines the selectivity of the reaction to the products C1, C2 and even C3.199--206
![[{"id":"bHDCclqoIs","type":"paragraph","data":{"text":"Thermodynamic profiles of the reduction of CO<sub>2</sub> to C<sub>2</sub>H<sub>5</sub>OH on pyrrolic and pyridinic nitrogen atoms incorporated into the carbon nanotube structure.<sup>204</sup>"}}]](/storage/images/resized/2mzCNonJiTlFyzT8SMhGGPN8ea5hyt9riCs7O5Zb_xl.webp)
Metal-doped carbon nanomaterials are considered to be a metal-economic alternative to all-metal CO2RR catalysts. Such systems are characterized by the formation of nanosized metal clusters on the NCM surface, so it is difficult to differentiate active metal-carbon sites from metal sites.207,208 Another challenge is high oxophilicity of single metal atoms in the NCM structure, which can lead to irreversible oxidation of some non-precious transition metals, as was shown above for ORR, which is often neglected in calculations. Therefore, the number of theoretical works in this field is very limited. Esrafili et al.209,210 tested Pt-, Ni- and Ti-doped graphene-like structures, formed by replacing one carbon atom by a metal, as catalysts in the reduction of CO2 to HCOOH. It was shown that the rate-determining step of this process is the initial protonation of CO2 to *OCHO, and ΔG≠ goes downhill from 2.08 to 0.85 eV in the series Ni>>Pt≈Ti.
A number of metals (Ag, Au, Co, Cu, Fe, Ir, Ni, Os, Pd, Pt, Rh, Ru) incorporated into the periodic graphene model structure with one (sv-Gr) or two (dv-Gr) vacancies were studied using DFT method as single-atom CO2RR catalysts for the formation of CH3OH and CH4, and the calculated data were compared with the experimental data (Fig. 32).211 It was shown that the initial protonation of CO2 to afford *COOH or *OCHO adsorbates on Cu, Ag and Au metals is highly endergonic (ΔG>1 eV). For the Pd, Ni and Pt metals, the ΔG value is in the range of 0-0.5 eV, and for the other metals ΔG<0. The alternative adsorption of the hydrogen atom on all metals is less advantageous compared to *COOH and *OCHO, and hence HER on a given active site is unlikely. The analysis of the thermodynamic reaction profiles showed that the major product on the Ag- and Au-doped graphene is CO, with the rate-determining steps being CO2→*OCHO (ΔG=1.17 eV) and CO2→*COOH (ΔG=1.41 eV), respectively. Cu-doped graphene proved to be potentially active in the formation of CH3OH; the limiting step is similar to that for Ag, but the value of ΔG is the highest among all metals (1.48 eV). The ΔG change in the rate-determining step decreases in the series Fe (0.73)>Pd (0.62)>Ir/Rh (0.57)≈Co (0.56)>Ru/Os (0.52)>Ni (0.41)>>Pt (0.27). Based on this, the authors concluded that Pt, Ni, Os and Ru in the metal-doped graphene M-dv-Gr are the most active in CO2RR, with M=Pt and Ni producing methanol, and Ru and Os — methane. Comparison of the thermodynamic profiles of methanol formation on Pt-dv-Gr and methane formation on all-metal platinum Pt(211) showed that the profile of the former is flatter, while on Pt(211), a stable *CO adsorbate is generated due to interaction with two adjacent Pt atoms. In both cases, the rate-determining step is the *CO protonation, which on Pt(211) produces *COH (ΔG=0.75 eV), and in the case of Pt-dv-Gr, the *CHO adsorbate (ΔG=0.27 eV) is formed, which makes Pt-dv-Gr a promising catalyst for the CO2→CH3OH reaction. As noted above, the all-metal platinum cathode material is only capable of promoting H2 formation by HER.150
![[{"id":"l2tQRH4jwZ","type":"paragraph","data":{"text":"Elementary cell of a metal-doped graphene with a single (sv-Gr) (<i>a</i>) and two (dv-Gr) (<i>b</i>) vacancies. Dependence of selectivity of M-dv-Gr in CO<sub>2</sub>RR and HER based on comparison of ΔG[*COOH/*OCHO] and ΔG[*H] (<i>c</i>). Binding energy dependence on metal incorporation into sv- and dv-Gr vacancies (<i>d</i>). Thermodynamic profiles of CO<sub>2</sub>RR to CH<sub>4</sub> on Pt(211) and to CH<sub>3</sub>OH on Pt-dv-Gr catalysts (<i>e</i>). The Figure is published under the CC-BY licence.<sup>211</sup>"}}]](/storage/images/resized/74BlqCsmSIlQ17OKTXPWnJRIPyE0CpfPEHs9OcGa_xl.webp)
Stable 2D carbon allotropes, graphyne (GY) and graphdyine (GDY), containing sp- and sp2-hybridized carbon atoms (Fig. 33), are currently considered as potential catalysts for a wide range of electrocatalytic reactions212,213 due to the unique electronic structure of the materials based thereon, as well as the possibility of metal doping to build M–Csp coordination bonds. Both materials in their pristine forms are inert towards CO2RR, since the formation of staring *COOH or *CHOO adsorbates on their surface is highly endergonic (ΔG=1-2 eV).214 It is shown that replacement of some carbon atoms by nitrogen atoms in GDY can accelerate the CO or CH3OH formation depending on the doping site, with ΔG for the rate-determining step being 0.26 and 0.46 eV, respectively.214
![[{"id":"tnWpgD9jdU","type":"paragraph","data":{"text":" Structures of graphyne, GY (<i>a</i>), graphdyine, GDY (<i>b</i>) and their metal derivatives M-GY (<i>c</i>) and M-GDY (<i>d</i>)"}}]](/storage/images/resized/cDCDf5e0me9b6bZkq3tnT7p2Jw0xjg9pmJypR9mh_xl.webp)
A study of the thermodynamic profiles of CO2RR to methanol and methane on model Fex-GDY catalysts, where x=1--4, showed potentially high performance of Fe1- and Fe2-GDY in these processes.214 The rate-determining step using these catalysts at x=1--3 is the reduction of *CO to *CHO with ΔG=0.36, 0.29 and 0.57 eV, respectively, whereas with Fe4-GDY this is the reduction of *CO2 to *OCHO with ΔG=0.69 eV, although the authors note that the competitive hydrogen reduction reaction HER for this catalyst is 0.25 eV more exergonic. Recently, nickel-doped graphdyines Nix-GDY, where x=1-3, as well as heterometallic complexes MNi-GDY (Fig. 33, where M=Ti, V, Cr, Mn, Fe, Co, Cu, Zn) were studied.216 Among the homometallic catalysts, Ni2-GDY (CO2→*CO2, ΔG=0.18 eV), was potentially highly active in the methane formation, whereas for Ni1- and Ni3-GDY, ΔG of the rate-determining step CO2→*OCHO was higher, 0.43 and 0.73 eV respectively.The formation of a stable *OH adsorbate, which is an intermediate in the methanol formation, was observed for all Ni-containing catalysts (Fig. 34), with regeneration of the metal from its oxidized state determining the rate of the whole process. Thus, the reduction *OH→H2O (i.e., metal catalyst regeneration) on NiNi-GDY is slightly more endergonic (ΔG=0.28 eV), than CO2 adsorption (ΔG=0.18 eV), whereas on replacing one of the nickel atoms by Co, Cu or Fe, endergonicity increases to 0.40, 0.49 and 0.65 eV, respectively. In addition to the thermodynamic profile of methanol formation on Ni4-GDY, Yang et al.217 studied the formation of ethanol and n-propanol on the nickel atom located at the Ni4 top point. The mechanism of C2+ alcohol formation involves an exergonic step of sorption of one or two CO molecules in addition to *CH2O* (*CH2C*O) adsorbate, and the rate-determining steps are the reduction of CH3C*O to CH3CH*O (ΔG=0.40 eV) and of OC*CH2C*OH to OC*CH2CH*OH (ΔG=0.47 eV), whereas in the methane formation pathway ΔG for the step CO2→*OCHO is 0.50 eV. The obtained ΔG values allowed the authors to conclude about potential activity of Ni4-GDY in the formation of C2+ alcohols along with methane.
![[{"id":"CIehEnI1pf","type":"paragraph","data":{"text":"Free energy profiles of CO<sub>2</sub>RR to CH<sub>3</sub>OH on NiM-GDY, where M=Fe (<i>a</i>), Co (<i>b</i>), Ni (<i>c</i>) and Cu (<i>d</i>). The Figure is published under the CC-BY licence.<sup>216</sup>"}}]](/storage/images/resized/bySKv9pXibP5HZ08jLc8uepz0vx9oDre72HG4DIz_xl.webp)
The possibility of the association of several metal atoms within a single cavity to give Mx complexes with graphdyine GDY was confirmed on the example of Cux-GDY, where x=1--3, and the stability of such complexes increases with the number of metal atoms in the cavity and is maximal for x=3.218 Quantum chemical calculations of Cu3-GDY activity showed that such a catalyst is most active in HER, since the free energy change ΔG for the rate-determining step is a record low (0.02 eV), whereas for CO2RR the free energy of CO2 protonation to *OCHO is 0.7 eV higher. The formation of H2 is also faster on Cu1-GDY. In contrast, the model catalyst Cu2-GDY differs from the above by the possibility of competitive formation of stable adsorbates H* and O*CHO* (ΔG=-1.28 eV). Subsequent reduction of O*CHO* to *CO was not considered, but an alternative route of *COOH reduction to *CO and further to methane, where the rate-determining step of the latter formation, as well as for Fe2-GDY,214 is the reduction of *CO to *CHO, for which ΔG is slightly higher and equals 0.42 eV, was proposed. Due to somewhat lower copper oxophilicity, the protonation of *OH adsorbate on Cu2-GDY proceeds more readily (0.23 eV) compared to Ni2-GDY (0.28 eV,216 vide supra). Thus, the performance of M2-GDY in CO2RR to methane increases in the series Cu<Fe<Ni. Note also that the CO adsorption energy on Cu2-GDY is the lowest (ΔG=–0.2 eV) among the Mx-GDY catalysts described above, and the reduction of *CO to *CHO on this catalyst (0.42 eV) makes the formation of free CO instead of methane more likely.
Among the metal-containing graphyne catalysts M-GY in CO2RR to methane, Ti, V, Cr and Mn were explored.219 Catalysts with M=Ti, V, Mn were unsuitable because for the first two metals the reduction of *COOH to *CO within the target reaction pathway is endergonic. For M=Mn, the reduction of *CO to *CHO is endergonic (ΔG=0.78 eV), which may indicate the potential suitability of M-GY catalysts with M=Ti, V and Mn only in the CO formation. In contrast, the profile of CO2RR to CH4 on Cr-GY indicates the potential for such a process, with the reduction of *CO to *CHO (ΔG=0.04 eV) being the rate-determining step in the process from a thermodynamic viewpoint. However, the search for transition states and activation barriers suggests that the slowest step is the reduction of *CH2O* to *CH2OH (ΔG≠=0.93 eV). Furthermore, the *CH2O* adsorbate is the CO2RR bifurcation point on Cr-GY as the alternative protonation of *CH2O* to CH3O* is only 0.05 eV less favourable compared to the protonation providing *CH2OH adsorbate. Therefore, the formation of *CH2OH and CH3O* is kinetically almost equiprobable. Further reduction of CH3O* affords methane and stable *OH adsorbate. The reduction of the latter also seems to be the rate-determining step, with ΔG being equal to ~1 eV.
The combination of metal-nitrogen-codoped carbon nanomaterials M–Nx–C allows achieving activity in ORR, which is close to that of the reference platinum catalyst, while reports on their use in CO2RR are relatively recent and the research is rapidly developing.144,200 Single-atom catalysts of this type can selectively promote carbon monoxide synthesis from CO2 at moderately low electrode potentials, and at high values, methane and formic acid are predominantly produced. The influence of structural, coordination and electronic factors on the catalytic performance of M–Nx–C in CO2RR is discussed in recent reviews.220--222 In a pioneering 2015 paper by Strasser and co-workers,223 M–Nx–Gr (M=Fe, Mn) produced a mixture of CO+H2 as the main product. The catalysts selectivity (FECO) was ~80% at –0.5 V and decreased to almost zero with decreasing electrode potential. At a potential below –0.85 V, the formation of methane with a selectivity below 0.42% was recorded on the Fe–Nx–Gr catalyst. Experiment with iron-free N-doped graphene led to the conclusion about the formation of CO on the nitrogen atoms as the active sites, while the FeNx moiety was responsible for the formation of methane. Quantum chemical calculations of CO2RR on Fe–Nx–Gr, where the iron atom is coordinated to four pyridinic nitrogen atoms,* showed that the CO2 reduction on the metal site to give *COOH adsorbate is endergonic (ΔG=0.63 eV) and is a rate-determining step.224 Further reduction of *COOH produces a stable *CO adsorbate, with ΔG decreasing to –0.4 eV relative to the non-reacting system. The substitution of carbon atoms in β-position to a pair of pyridinic nitrogen atoms for graphitic nitrogen atoms decreases ΔG of the rate-determining step to 0.3 eV and lowers the adsorbate *CO stability on the metal centre. These results allow to draw a conclusion about the possibility of desorption of the CO molecule from the Fe–N4-dv-Gr metal site, which contradicts the experimental results according to which the FECO value decreases when the potential drops to –0.6 V and below. It has recently been shown that instead of *COOH, the initial CO2RR step on M–N4–Gr (M=Mn, Fe) produces more stable *OCHO adsorbate.225 Its reduction does not involve a step of *CO formation, although the predicted onset potentials are in poor agreement with those found experimentally.223
Different variations of Ni,N-doped graphenes Ni–NxCx–4–dv-Gr, Ni–NxCx–3–sv-Gr in CO2RR to CO were studied.226--228 The reduction of CO2 to *COOH on the nickel atom of Ni–N4-dv-Gr is more endergonic (ΔG ≈ 1.75 eV) compared to its Fe counterpart,224 and therefore such a catalyst should be inactive in CO2RR. The search for the optimal environment for the nickel atom indicates that the model Ni–N1C2-sv-Gr is catalytically active towards CO formation.226 For such a catalyst adsorption of *COOH is weakly exergonic at the electrode potential U=–0.12 V (corresponding to equilibrium in the CO2.CO system), whereas adsorption of hydrogen atom is endergonic (ΔG=0.52 eV), indicating high FECO selectivity and HER suppression.
Recently, CO2RR to CO on metal,nitrogen-codoped graphenes M–N4–Gr, where M=Sc–Zn, was theoretically studied at fixed electrode potentials (U=–0.3, –0.7 and –1.2 V)228 relative to a standard hydrogen electrode (SHE), and also on In–Nx–Gr, where x=3, 4, and on Sn–N4–Gr at U=0.229,230 Metals such as Sc, Ti, Co and Cu in M–N4-Gr were shown to selectively promote the target reaction in the electrode potential range of –0.7>U>–1.2 V, whereas Zn is also active at higher U values. It was noted that the formation of adsorbed anion radicals *CO2-• depending on the applied potential can play a decisive role in the activation of the CO2 molecule. The presence of structural defects in the N4 environment of the metal, in particular, vacancies due to the elimination of the nitrogen atom, increases the catalytic performance of the metal, thereby lowering the barrier of *COOH adsorbate formation.229,230
A wide range of 3d(Ti–Cu), 4d(Mo, Ru–Ag) and 5d(Os–Au) transition metals in metal,nitrogen-codoped graphene M–N2C2-dv-Gr composition were screened for catalytic activity towards the reduction of CO2 to CO, HCOOH, CH2O, CH3OH and CH4.231 Based on the analysis of the free energy changes in the first proton transfer for all transition metals, the possibility of selectively promoting CO2RR by the formation of *COOH or *OCHO adsorbate compared to H* was noted irrespective of the nature of the metal. This reaction produced the stable *OCHO adsorbate on Ti, V and Mo, ΔG is –2.2 (Ti), –1.5 eV (V and Mo), and thus these metal sites were poisoned. Similar product of the first step of reduction of CO2 (*OCHO) is formed on M–N2C2-dv-Gr, where M=Mn, Cr, Cu, Ag, Au, Pd, Ni, Pt, however ΔG in these cases is positive and is uphill in the series Cr (~0)<Mn (0.25) <<Cu (0.68)<Ag (0.78)<Au (0.91)≈Pd≈Ni (0.96)≈Pt. On the remaining metals (Os, Ru, Ir, Rh, Co, Fe) endergonic formation of *COOH adsorbate was observed, with ΔG increasing in the series Os (~0)<Ru<<Ir≈Rh<Co≈Fe. Based on their results, the authors concluded that M–N2C2-dv-Gr, where M=Cu, Co, Fe and Mn in CO2RR, is favourable. For the iron catalyst, the formation of a stable *CO adsorbate was predicted, whose reduction to *CHO is endergonic (ΔG=0.83 eV) and further affords methanol as the major product. Note that the predicted ΔG value in terms of the electrode potential (UL=–eΔG) coincides exactly with that obtained experimentally (UL=–0.85 B), but the reaction produced CH4.223 According to the authors,231 the most probable product of CO2RR on Co–N2C2-dv-Gr is formaldehyde CH2O, which formation involves the reduction of *CHO to *CH2O as a rate-determining step. Detailed thermodynamic profiles of formic acid formation from CO2 including transition states on model M–N3-sv-Gr, where M=Co, Fe and Ni, were obtained by Iranian and Thai researchers.233,234 Coadsorption of CO2 and H2 on the metal site was shown to significantly decrease the system energy (ΔG ≈ –1 eV). Further protonation can proceed via the oxygen or carbon atom of *CO2 to give *CO+*OH or *OCHO adsorbates, respectively. In the former case, the activation energy (ΔG≠) is 1.10 (Co), 1.40 (Fe) and 0.78 eV (Ni), whereas protonation at the *CO2 carbon atom proceeds much more readily, ΔG≠=0.31 (Co), 0.57 (Fe) and 0.37 eV (Ni) and indicates kinetic preference for the formation of *OCHO adsorbate. Further reduction of *OCHO on M–N3-sv-Gr is the rate-determining step; ΔG≠ is 0.51 (Co), 0.83 (Fe) and 0.67 eV (Ni).
The thermodynamic profile of HCOOH formation on Cu,N-codoped model catalyst Cu–N6–Gr was constructed.235 Molecular dynamics calculations showed that the metal atom inside a sufficiently large N6 cavity migrates in almost barrierless manner from one pair of pyridinic nitrogen atoms to another, making a full turn in ~5 ps. Chemisorption of H2 on the metal site of such a catalyst is preferable to the chemisorption of CO2, while the coadsorption of these two molecules further reduced the energy of the system. The subsequent formation of HCOOH can proceed in two routes via the *COOH and *OCHO intermediates. In the first step, dissociation of *H2 occurs so that one of the hydrogen atoms migrates from the metal to the pyridinic nitrogen with an activation barrier of ΔG≠~0.1 eV, with the process in general being weakly endergonic. Next, the hydrogen atom at the pyridinic nitrogen atom attacks the CO2 oxygen atom and simultaneously the Cu–C bond is formed to afford the *COOH adsorbate (ΔG≠=0.21 eV), whereas the attack of the hydrogen atom adsorbed on copper by the CO2 carbon atom to form *OCHO is kinetically preferable (ΔG≠=0.06 eV). Both routes produce HCOOH via low-lying transition states. In the experimental and theoretical study, Yang et al.236 showed that copper,nitrogen-codoped carbon fibres can generate methanol (FE=44% at U=–0.9 V) as the only co-product along with carbon monoxide. The mechanism of methanol formation was explored using a model single-atom Cu–N4-dv-Gr catalyst as an example. The calculation showed that the rate-determining step of the process on the Cu active site is the reduction of *CO2 to *COOH towards CO formation. Adsorption of CO on the metal site is weakly endergonic (ΔG=0.12 eV), and the rate-determining step of further reduction of *CO to methanol is protonation of *CO to *COH (ΔG=0.86 eV). Zhao et al.237 managed to prepare Cu,N-codoped porous carbon material with low metal content. The products of CO2 reduction included formic and acetic acids, methanol and ethanol (FE=2--10% for each component), as well as acetone (FE=37% at U=–0.3 V) with a maximum performance of 336 μg h–1. As a model catalyst for the reduction of CO2 to acetone, Cu–N4-dv-Gr was theoretically considered, with the nearest Cu environment representing both pyridinic and pyrrolic nitrogen atoms (Fig. 35).237 Analysis of the thermodynamic profiles involving Cu surrounded by pyridine nitrogen atoms showed highly endergonic dimerization step of two *CO molecules to give *CO*CO adsorbate (ΔG=1.67 eV) along with initial protonation of CO2 to *COOH (ΔG=1.30 eV) and therefore such active site is unable to generate C2+ products. On the contrary, at the second active site (Cu in the pyrrolic nitrogen environment), only CO2 reduction to *COOH (ΔG=1.06 eV) occurs, which is a rate-determining step. The adsorption of CO on the Cu–N4-dv-Gr active site is endergonic (ΔG=1.2 eV), while dimerization of the two adsorbed *CO molecules leads to a significant reduction of ΔG of the system, which is higher than that for the CO adsorption. Further reduction of the *CO*CO adsorbate and association with the third CO molecule to acetone is exergonic. Of special interest is the structure of *CO and the *CO*CO adsorbates where the carbon atom of the former forms a bond simultaneously with the pyrrolic nitrogen and copper, whereas in the latter both carbon atoms are attached to pyrrolic nitrogens, while the metal atom is coordinated through the C–C bond.
![[{"id":"TKrtofzOhZ","type":"paragraph","data":{"text":" Free CH<sub>3</sub>COCH<sub>3</sub> energy profiles of CO<sub>2</sub>RR to acetone on Cu-N<sub>4</sub>-dv-Gr (<i>a</i>) and structures of selected intermediate compounds (<i>b</i>).<sup>237</sup> The figure is published with the permission of OpenAccess Publishers"}}]](/storage/images/resized/VyraXbs8ufxQeUlcRvYmxDNRAgJeXGB5jO4yNAVZ_xl.webp)
The attentive reader will notice that in the periodic model of graphene with four pyrrolic nitrogen atoms shown in Fig. 35, along with the MN4 defect, half of the cyclooctatetraene fragment is present on the edges of the unit cell, creating a repeating defect in the form of a large-sized cavity in the whole network. The presence of such defects is uncharacteristic for the graphene catalysts of CO2RR and should reduce their performance through reducing the cavity formed by the N4 fragment, and thus strengthening the interaction between nitrogen and metal atoms. An alternative to this, in our opinion, not the most successful model can be the model of graphene with an active M–N4 site, which does not contain a similar cyclooctatetraene cavity (Fig. 36), which, according to preliminary data, should improve the performance of the catalyst.
![[{"id":"Ufl_rztOma","type":"paragraph","data":{"text":"Alternative structure of Cu-N<sub>4</sub>-Gr catalyst devoid of cyclooctadiene defects"}}]](/storage/images/resized/KAVnKEfXZoCSHODzmYVNo30A0RLebSb5aYHrlZlh_xl.webp)
The thermodynamic profiles of CO2RR to give C1 and C2 products on metal,nitrogen-codoped carbon catalysts containing two metal atoms M1/M2–N6-Gr (Fig. 37), where M1, M2=Cr–Zn, are highlighted in reviews.238--241 Bimetallic sites with pyridinic nitrogen atom environment exhibit moderate adsorption activity towards *COOH and *CO and excellent catalytic performance in the reduction of CO2 to CO. It is shown that the doping of M1/M2–N6–Gr with various metals provides a synergistic effect in suppressing HER, and for such metal pairs as Co/Zn, Fe/Zn, Mn/Zn and Fe/Mn, the endergonicity of the rate-determining step does not exceed 0.26 eV.239 The possibility of selective reduction of CO2 to methane was shown for Fe/Co and Co/Co metal pairs, with ΔG of the rate-determining step CO2→*COOH not exceeding 0.37 eV.240 In the study,241 the possibility of ethylene C2H4 formation on Fe/M–N6–Gr catalysts (M=Fe, Co, Ni, Cu) was shown, whereas on Cu/M-N6–Gr (M=Co, Ni, Cu), methane is the major product. As a parameter determining the activity of M1/M2–N6–Gr in CO2RR, the adsorption value of two *CO molecules was proposed, a moderate value of which provides the lowest onset potential. The carbon affinity of the adsorbates is most important for the formation of ethylene C2H4, whereas CO2RR producing methane CH4 is controlled by affinity for both carbon and oxygen. In the study,242 the CO2RR affording C1 products (CO, HCOOH, CH3OH, CH2O and CH4) on model triatomic M3–N6–Gr catalysts, where M=Fe, Co, Ni and Cu, was studied. Such catalysts, like their bimetallic counterparts, selectively promote CO2RR with respect to HER. Catalysts M3–N6–Gr, where M=Co and Cu, were the most active in the metal series in choice. Thus, for Cu3–N6–Gr, the formation of CO and HCOOH is most likely, the onset potentials are –0.67 and –0.57 V, respectively, and the reduction of *COOH is the rate-determining step. For Co3–N6–Gr, the reduction to CH3OH and CH4 (UL=-0.51 V), is most likely and the rate-determining step is the reduction of *OCHOH to *CHO.
![[{"id":"KflD4_U1Mo","type":"paragraph","data":{"text":" Structures of M<sub>1</sub>/M<sub>2</sub>-N<sub>6</sub>-Gr (<i>a</i>) and M<sub>3</sub>-N<sub>6</sub>-Gr (<i>b</i>) catalysts"}}]](/storage/images/resized/v2EnWhSQU08DUTt6ANxxUf7rPkjF3F6jtFSCblJF_xl.webp)
* Hereinafter,unless otherwise stated.
5. Conclusion
The review analyzes the results of theoretical studies in two intensively developing fields — oxygen reduction reaction (ORR) and carbon dioxide reduction reaction (CO2RR). The close attention to ORR is due to the relevance of creating efficient, environmentally friendly energy sources, while a surge of interest in CO2RR is connected with the dangerous growth of anthropogenic CO2 in the atmosphere and the possibility of producing a large number of valuable products based on this reaction. Most of the works has been published in the last five years.
Despite their different prerequisites and ultimate goals, both processes are electrocatalytic reactions using the same or similar catalysts, the same methodological approach is applied to analyze them and in both cases researchers are focused on improving activity and selectivity and replacing expensive and short-lived platinum catalysts with nanocarbon materials doped with various heteroatoms and non-precious metals. Such ORR catalysts can be based on graphene, nanotubes, carbon dots, nanofibers, nanospheres, fullerene, and other metal-organic frameworks (MOFs). Studies have been performed for various heteroatoms (B, N, Si, P, S) and a wide range of metals (Cu, Ag, Zn, Sn, Ti, Zr, V, Nb, Cr, Mn, Fe, Co, Ni) in both free and oxidized forms (TiO2, ZrO2, V=O). The nitrogen atom as a dopant is unique and very efficient in both processes, ORR and CO2RR. The possibility of efficient catalysis not only on the metal site, but also on the adjacent C2 moiety (C=C double bond) of nanocarbon substrate was shown for the first time, in particular when the stable intermediates (due to very strong adsorption) ‘poison‘ the catalyst. The claim about inefficiency of silicon doping, based on the methodological error of the authors of the 2017 publication132 is refuted, and the mechanism of ORR on Si-doped graphene, fullerene and nanotubes was analyzed. It is concluded that metal,nitrogen- and Si-doped carbon nanomaterials are highly promising as ORR catalysts. For both processes (ORR and CO2RR), the dependence on electrode potential was analyzed, as well as the dependence on the pH of the medium for ORR.
The series of metals studied in CO2RR as all-metal cathode materials is very wide and includes Cu, Ag, Au, Zn, Cd, Hg, Ga, In, Tl, Bi, Sn, Pb, Ti, Zr, V, Nb, Cr, Mn, Fe, Co, Ni, Ru, Pd, Os, Ir, Pt as well as their oxides and some alloys.
In contrast to ORR, for which the role of the catalytically active C2 site was first shown, in the CO2RR, the pyrrolic and pyridinic nitrogen atoms of the M–Nx fragment play an important role.
From the perspective of further research in this area, besides experimental studies on different ORR and CO2RR catalysts, the design (fine tuning) of active site structure to increase activity and selectivity of individual reaction routes, decrease of activation energy of rate-determining steps and thereby reduction of overpotentials and HER suppression remain relevant tasks of theoretical analysis. The solution to these problems lies at the interface between the theoretical prediction of the activity of catalytic sites depending on their environment and the experimental possibilities of their straightforward synthesis.
6. List of acronyms
2e — two-electron reduction of oxygen to H2O2;
4e — four-electron reduction of oxygen to H2O;
* — denotes the adsorption of a molecule on the catalyst surface;
ΔE — change in total energy resulting from an (elementary) reaction;
ΔE≠ — total activation energy;
ΔG — change in free energy resulting from an (elementary) reaction;
ΔG≠ — free activation energy;
ωB97XD — long-range corrected hybrid density Chai and Head-Gordon functional;
AM1 — semi-empirical quantum chemical method: Austin model 1;
B3LYP — Becke's three-parameter exchange functional in combination with the Lee--Yang--Parr correlation functional;
C1 — CO2 reduction products with one carbon atoms;
C2 — active site of an carbon catalyst in the form of two adjacent carbon atoms;
C2+ — CO2 reduction products with two or more carbon atoms;
CHE — computational hydrogen electrode model;
CNT — carbon nanotube;
CO2RR — electrochemical carbon dioxide reduction reaction;
DFT — density functional theory;
dv-Gr — graphene with a vacancy generated by the removal of two neighbouring carbon atoms;
Eads — adsorption energy;
Egap — LUMO/HOMO energy gap in the molecule;
FE — Faradaic efficiency, %;
GDY — graphdyine;
Gr — graphene;
GY — graphyne;
HER — reduction of H+ ions to H2;
HOMO — highest occupied molecular orbital;
LP — lone pair;
LUMO — lowest unoccupied molecular orbital;
M–Nx–C - metal,nitrogen-codoped carbon nanomaterial;
MOF — metal-organic framework;
NCM — carbon nanomaterial;
ORR — electrochemical oxygen reduction reaction;
PBE — Perdew--Burke---Ernzerhof functional;
PET — protonation/electron transfer [H++e–];
ppm — parts per million, 1/1000000;
Pt/C — platinum on activated charcoal;
RDS — rate-determining step;
SHE — standard hydrogen electrode;
sv-Gr — graphene with a vacancy generated by the removal of one carbon atom;
TS — transition state;
U — electrode potential;
Ueq — equilibrium electrode potential;
UL — limiting potential.