

Keywords
Abstract
The review addresses the development of the methodology of single-atom catalysts ranging from single-site to single-atom alloy systems. The preparation and characterization of single-atom catalysts and their use in a number of key catalytic reactions, including alkyne hydrogenation, are considered. The possibility of fine tuning of the surface structure of single-atom alloy catalytic systems using the adsorbate-induced segregation is analyzed for the first time.
The bibliography includes 312 references.
1. Introduction
Heterogeneous catalysts are widely used both in industry and in laboratory practice owing to their high activity, thermal stability and easy separation from the reaction mixture, in contrast to homogeneous analogues. Supported metal catalysts based on noble metals (Pd, Pt, Rh, Ir) are among the most popular heterogeneous catalysts. However, their considerable drawback is lower selectivity to target products in comparison with homogeneous transition metal catalysts. Therefore, a priority task of modern catalysis is to increase the selectivity of heterogeneous supported metal catalysts. High selectivity would enhance the economic feasibility of catalytic processes and improve their environmental friendliness, reducing the amount of waste of large-scale production facilities and mitigating the technology’s impact on the environment.1 – 9
A basic solution to the problem of increasing selectivity of heterogeneous catalysts is to develop methods for the synthesis of catalytic systems with uniform active sites. The design of heterogeneous single-atom catalyst (SAC) in which active sites consist of only one atom of an active metal (as a rule, a noble metal) attract increasing attention of researchers.10 – 13 Catalysts of this type are prepared using both traditional methods such as precipitation, co-precipitation, impregnation and adsorption of the active site precursor (e.g., metal complex) and new approaches based on stabilization of metal atoms on oxide supports by deposition of atomic metal vapour using ion implantation, laser ablation or galvanic replacement.14 However, a serious drawback of most of these methods is thermodynamic instability of the resulting structure, which hampers regeneration and reuse of the catalyst.
In this connection, of particular interest are single-atom alloy (SAA) catalysts in which the active metal atoms located on the surface of bimetallic nanoparticles are separated by atoms of an inert component, so-called host metal.15 – 17 This gives rise to a system of active sites possessing identical adsorption catalytic characteristics and high thermodynamic stability.18, 19 This approach makes it possible to achieve high selectivity of the catalytic system and enables catalyst regeneration.
According to the Chai et al.,20 in this review, isolated single-atom sites are designated as Pd1M (М is a host metal, e.g., Ag, Cu or Au).
This review gives a brief analysis of the development of single-atom active site methodology in modern catalysis, considering the synthesis and characterization of SAC and SAA catalytic systems. A separate Section is devoted to the use of SAA catalysts in key catalytic reactions such as selective hydrogenation, dehydrogenation, hydrogenolysis, cross-coupling and other processes. The last Section addresses fine tuning of single-atom sites using adsorbate-induced segregation.
2. Structure uniformity of the active sites of heterogeneous catalysts as a key condition for attaining ultrahigh selectivity
A major cause for the insufficient selectivity of heterogeneous catalysts is the non-unoform structure of active sites, which is due to a number of factors. For metal catalysts, this is different degree of nuclearity of active sites, which can include one to a few surface metal atoms. A significant role is also played by different degrees of coordinative unsaturation inherent in terrace, edge and corner metal atoms of a metal particle. As a result, catalytic transformations can occur on structurally different sites, which markedly differ in the adsorption and catalytic characteristics. Thus, the reaction follows several pathways, which give rise to by-products, thus decreasing the reaction selectivity.
A good illustration is the adsorption of carbon monoxide on the metal catalyst surface. Even in the simplest case of a flat metal surface, a СО molecule can be adsorbed in three different modes on the adsorption sites of different nuclearity: a single atom (linear on-top adsorption), two neighbouring atoms (bridge adsorption) or three atoms (threefold adsorption) (Fig. 1). The differences in the active site structure and adsorption modes give rise to considerable differences in the bond strength and energy of CO adsorption on the metal surface. The results of calculation of the adsorption energy for various forms of CO at the (111) face of the monometallic palladium cluster Pd146 indicate that, depending on the adsorption site structure and adsorption mode, the CO adsorption energy can increase by 0.57 eV (55 kJ mol–1) on going from the linear on-top adsorption on the Pd1 site to three-fold СО adsorption on the Pd3 site.21 Thus, the linearly bound carbon monoxide has a substantially lower energy and is most easily desorbed from the catalyst surface. Similar results were obtained by Mancera et al.22
![[{"id":"Lxw8X-wikh","type":"paragraph","data":{"text":"Type of CO adsorption on the palladium metal surface and corresponding adsorption energies."}}]](/storage/images/resized/M7mC96XBA9yFip8hpd5DxGp7XcVexXTuzywtwIv2_xl.webp)
While passing from the simplified model of a flat metal surface to a more realistic bulk model of a metal particle, one should also consider the possibility of CO adsorption on different terrace atoms of a metal nanoparticle and on corner or edge atoms with different degrees of coordinative unsaturation. Most often, for small monometallic particles with a face-centred cubic lattice, a cubo-octahedral shape is assumed.23, 24 It is known from the literature that ~37% of atoms on the surface of platinum nanoparticles with an average size of ~8 nm are located on (111) faces, while 28% of atoms are on (100) faces. The coordination numbers (C.N.s) of the surface atoms are 9 and 8, respectively. In addition, there are atoms located on faces with higher Miller indices and atoms with a higher degree of coordinative unsaturation located on the edges (C.N. = 7) and at the corners (C.N. = 6) of the particle.25 The ratio of faces, edges and corners depends appreciably on the particle size: as the particle size decreases, the fraction of the surface atoms with high coordination numbers decreases, whereas the number of corner and edge atoms increases.
The different degrees of coordinative unsaturation of the terrace, edge and corner metal atoms of a metal particle considerably affect their adsorption and catalytic properties. The coordination numbers of edge and corner atoms are markedly smaller than C.N.s of terrace atoms, which is reflected in the CO adsorption energy. The calculated energy of CO adsorption on coordinatively unsaturated atoms approaches the values for CO adsorbed in the three-fold sites (~ 1.47 eV) 21 and is much higher than the adsorption energy for CO linearly bound to a terrace metal atom with a high C.N. (~ 0.92 eV).
Apart from the energy heterogeneity, a considerable role belongs to the fact that sites of different nuclearity can be involved in a reaction. The existence of these energetically and geometrically heterogeneous sites on the catalyst surface accounts for the considerable differences in their adsorption and catalytic behaviour. A systematic study of acetylene hydrogenation kinetics indicated the existence of different adsorption modes of acetylene molecules (Fig. 2) and intermediates that have formed on active sites of different nuclearity on the catalyst surface.26 – 28
![[{"id":"xMlun2k1uM","type":"paragraph","data":{"text":"Modes of acetylene adsorption and reaction intermediates formed on the surface of a heterogeneous catalyst during hydrogenation of acetylene.<sup>29</sup>"}}]](/storage/images/resized/ajErrdmlgFUeWeoiz53WjTp9EfiPaQGv2hpfXqHe_xl.webp)
Acetylene (1) adsorbed initially as a π-complex is converted to the associatively chemisorbed di-σ-form (2). The subsequent addition of atomic hydrogen to the triple bond affords the vinyl intermediate (3), which is the key species in the selective hydrogenation of acetylene to ethylene.29 Meanwhile, the conceptual possibility of existence of surface-bound ethylidyne (4) and ethylidene (5) was demonstrated. These species are formed as intermediates in the hydrogenation of vinyl (3) to ethane. Also, dissociatively adsorbed acetylene (6) and vinylidene (7), which promote oligomerization and cyclotrimerization, respectively, were detected.30
Thus, the presence of structurally different sites gives rise to several reaction pathways, formation of by-products and, hence, a decrease in the catalyst selectivity.31 – 33
3. Single-atom catalysts
3.1. Concept of single-site heterogeneous catalysts in the generation of active sites with identical structure, adsorption properties and catalytic behaviour
Development of methods for the synthesis and structural studies of single-site heterogeneous catalysts is now a major avenue of research related to heterogeneous catalysis.14, 34 – 36
The term ‘single-site catalyst’ was first proposed and became widespread in the late-1970s when high activity and selectivity of zirconocene catalysts in olefin polymerization was found.37, 38 Unlike the Ziegler – Natta catalysts, zirconocene catalysts are separate metal complexes with an ordered environment. A detailed study of these catalysts revealed a relationship between the structure, the ligand environment of the metallocene and characteristics of the resulting polymer. This made it possible to control the stereo- and regioselectivity of the polymerization products.
The notion of single-site catalyst as applied to heterogeneous catalysts was introduced by Thomas and co-workers in 2005.23 According to the proposed concept, a single-site heterogeneous catalyst (SSHC) is a heterogeneous catalyst in which the active sites consist of identical numbers of atoms, have similar structure and are separated in space from one another. As a result, they have identical adsorption and catalytic properties and there is no interaction between the active sites (Fig. 3).
![[{"id":"NmxrWjlwnU","type":"paragraph","data":{"text":"Surface structure of single-site heterogeneous catalysts."}}]](/storage/images/resized/Ol7aU0EtkJc6WYpPUHnrXLFYHb0ohnLlI9XTgG0p_xl.webp)
Characteristic features of the structure and applications of SSHCs are addressed in several comprehensive reviews.14, 39, 40
Thomas et al.23 classified SSHCs in the following way:
A: catalysts in which the active sites are isolated atoms, ions, molecular complexes or small clusters of identical sizes supported on materials with a high specific surface area. The most widely used support is SiO2;
B: ship-in-bottle (host – guest) structures in which the isolated catalytic sites are located and immobilized in the pores of the support (e.g., zeolite framework). The cavities are accessible for reactants and products;
C: open-structure microporous crystalline solids (zeolites or molecular sieves) with pore diameters ranging from 3.5 to 10 Å, in which the isolated active sites are parts of the crystal structure uniformly distributed throughout the bulk.
All three types are characterized by the presence of uniform active sites of a definite structure, which can be investigated by both experimental and computational methods, in particular during a catalytic reaction.41
Single-site heterogeneous catalysts, combining the benefits of homogeneous and heterogeneous catalysts, have a number of unique properties, namely
(1) easy separation of the catalyst from the reaction products and the possibility of catalyst reuse;
(2) exceptionally high selectivity and the possibility of efficient conduction of chemo-, regio- and enantioselective reactions;
(3) owing to the identical structure of the active sites of single-site catalysts, it is possible to synthesize molecular groups that model active sites; this enables comparison of the properties of homogeneous and heterogeneous catalysts with the same active sites;
(4) the active site uniformity also makes it possible to use advanced computational approaches such as density functional theory (DFT) to carry out direct comparison with experimental results (e.g., reaction kinetics).
Type A catalysts have found fairly wide practical use.42, 43 The most numerous group consists of metal complexes supported on the surface of various materials. For example, deposition of organometallic structures based on molybdenum, tungsten and zirconium on a silica surface giving highly efficient heterogeneous catalysts for alkane metathesis and depolymerization reactions was reported in a review.44 More recent studies markedly expanded the range of applicable supports.39, 45 Thus the formation of isolated sites was detected when the active component was deposited on ceria,46, 47 zirconia,48 titania,49, 50 alumina,51 various zeolites 52 and carbon nanotubes.53
Regarding metal-supported catalysts, the number of publications devoted to the synthesis and study of the properties of catalysts representing supported metal clusters of a strictly identical size is relatively small. This is due to both the complexity of synthesis and the thermodynamic instability of small metal particles and their tendency to aggregation. The aggregation gives thermodynamically stable metal particles of larger size and is accompanied by the loss of the main characteristic of single-site catalysts, that is, active site uniformity.
3.2. Single-atom catalysts as further development of the concept of single-site heterogeneous catalysts
A milestone in the development of the SSHC concept was appearance of single-atom catalysts in which the active site consists of only one atom of an active metal, usually a noble metal (Fig. 4).33, 54, 55
![[{"id":"Iaj5-TTXcW","type":"paragraph","data":{"text":"Single-atom catalyst surface."}}]](/storage/images/resized/xQMnAPykRxjXO5rq1BWl23aurA7dRyb8DaWyjjgG_xl.webp)
Köpp and co-workers,56 whose study can be considered to be pioneering in this field, were the first to demonstrate the advantages of the atomically dispersed nickel and palladium in the Sabatier reaction. Despite the high significance of the discovery, the scientific community virtually ignored the results obtained by the authors, mainly due to the lack of obvious evidence of formation of a single-atom structure, in particular the impossibility of observing atomically dispersed sites by electron microscopy. The interest in the SAC methodology was revived in the 21st century owing to the existence of advanced instrumentation suitable for characterization of atomically dispersed metals with a very high resolution.57
The preparation of a new type of catalysts with isolated Pt atoms supported on iron oxide crystallites (Pt1/FeOx) was an important stage of development of the SAC theory and practice.54 It is in this study that the terms ‘single-atom catalysts’ and ‘single-atom catalysis’ were first proposed and the main signs of formation of isolated active sites were described, such as 100% degree of dispersion of the active metal and the identity of the catalytic performance of these sites. A study of the characteristics of the Pt1/FeOx single-atom catalyst in the CO oxidation demonstrated that the sample has an exceptionally high activity and stability and is markedly superior to the commercial Au1/Fe2O3 catalyst. Detailed investigation of the catalyst structure and theoretical calculations indicate that the efficiency of Pt1/FeOx is due to the electron density transfer from the Pt d-orbitals to the support, which is favourable not only for stabilization of atoms on the surface, but also for considerable decrease in the CO binding energy and in the activation barrier for the reaction.
Currently, SAC methodology is one of the key trends in modern catalysis, as indicated by quite a few relevant reviews published in the last 10 years.10 – 14, 34 – 36, 55, 58 – 65 Vigorous development of the SAC concept took place in the 2010s when a succession of publications appeared describing the efficient use of these catalysts in the selective hydrogenation of alkyne derivatives,66 hydroformylation of olefins,67 С – Н bond oxidation in alkanes,68 hydrosilylation of alkynes,69 cross-coupling 70 and carbonylation of terminal alkynes,71 cyclization,72 photocatalytic reactions 73 and click chemistry processes.74
The successful synthesis of single-atom catalysts was also reported by other research groups. A review by Bohme and Scwartz 75 gives analysis of the mechanism of catalytic oxidation of CO to CO2 using single Au atoms. Later, Pd1/Al2O3 catalysts with single-atom Pd active sites dispersed over the mesoporous Al2O3 surface were prepared.76 These samples showed high activity in the aerobic oxidation of allylic alcohols (TOF ~ 4000 h – 1) and exceptionally high (91 to 99%) selectivity to aldehydes, which are the target products of the reaction. The obtained Pt1/FeOx , Pd1/FeОх and Au1/FeOх catalysts for the selective oxidation of CO in the presence of H2 demonstrated higher activity/selectivity compared to commercial analogues used in these reactions.54, 77, 78 Complete conversion of carbon monoxide was detected even at temperatures below 0 °C.
Single-atom catalysts showed high activity and selectivity not only in the selective oxidation reactions, but also in the water – gas shift (WGS) reaction to obtain hydrogen. Using detailed physicochemical analysis of the structure of the Ir/FeOx catalysts with high Ir concentrations containing single atoms, subnanometre clusters and nanoparticles of iridium, the authors ascertained that the contribution of separate Ir atoms to the total activity (X ) is approximately 70%, irrespective of the amount of the active component.79 The formation of stable isolated Pt1 sites in the catalysts containing 0.5 mass % Pt on various supports was detected.80 The presence of single-atom sites ensures high activity and selectivity in the WGS reaction in the temperature range of 120 to 400 °C. Successful application of single-atom Au catalyst in the same reaction was reported.47
Wei et al.81 carried out hydrogenation of various substituted nitroarenes on the Pt1/FeOx catalyst surface. The TOF value for single-atom Pt catalyst in the hydrogenation of 3-nitrostyrene was 1500 h–1, while the selectivity (S ) to 3-aminostyrene was close to 99%, which is much higher than these values for previously known catalysts for this reaction. These high performance characteristics were attributed to the existence of positively charged platinum sites on the support surface and the absence of Pt – Pt bonds, which would promote multiatom adsorption of substrate molecules.
A recent review by Kolesnichenko et al.82 addressed the application of single-atom catalysts in the methane reactions, including dry, steam and oxidative reforming of methane, metane oxidative carbonylation and carboxylation to acetic acid, partial oxidation of methane to methanol, non-oxidative and oxidative methane condensation into ethane (ethylene), methane dehydroaromatization to aromatic hydrocarbons, etc. The authors analyzed the features of CH4 activation on the surface of single-atom catalysts and compared their performance with that of known analogous heterogeneous catalysts. In addition, prospects for the use of single-atom catalysts in methane chemistry were discussed.
Interesting features of the behaviour of a single-atom Pd/C catalyst in the Mizoroki – Heck and Suzuki – Miyaura reactions were analyzed in a publication by Ananikov’s group.83 The authors proposed an original combined approach to spatially localized characterization of the catalyst during the reaction. The use of multilevel electron microscopy, including scanning (SEM) and transmission electron microscopy (TEM) and high-resolution scanning transmission electron microscopy (HRSTEM) for the spatial positioning allowed the authors to identify metallic particles in the range from large agglomerates to single nanoparticles and atoms. The catalysts were characterized before and after cross-coupling reactions; thus, the authors were able to follow the transformation of metal particles under the reaction conditions at different scale levels and to propose the concept of 4D catalysis, which includes monitoring of the positions of catalytic sites in space (3D) on a time-bound basis (+1D). Due to the precise location of catalytic sites, the dynamic behaviour of single palladium atoms and nanoparticles in cross-coupling reactions was detected with nanometre precision. It was shown that during the reaction, some palladium atoms are leached from the catalyst surface to the solution where they exhibit very high (> 99%) catalytic activity in cross-coupling reactions compared to supported metal nanoparticles. It was found that the single-atom sites, which constitute only 1% of the total Pd amount in the catalyst, can be redeposited on the support and be again leached to the solution. Also, supported palladium nanoparticles can change their shape during the reaction and can move over the support surface, as was established by processing of the images of nanoparticle array with a neural network. On the basis of these results, the authors suggested that the high catalytic activity in cross-coupling reactions was due to leaching of single-atom catalytic sites to the solution rather than to their location on the support surface, as is usually believed.
It is noteworthy that analysis of the literature addressing single-atom catalytic systems indicates that the amount of the active metal in them is relatively low (0.1 – 0.5 mass %). First of all, this is related to the thermodynamic instability of metal atoms, which tend to aggregate. In some cases, these low concentrations can lead to insufficient catalyst activity. Nevertheless, it was shown for some reactions than the turnover frequencies (TOFs) of single-atom systems may much exceed those of conventional heterogeneous catalysts. In this case, the relatively small amount of active metal can be successfully counterbalanced by a higher catalytic activity. In addition, it should be borne in mind that a decrease in the content of the active metal increases the stability of the single-atom catalyst structure.
Since SACs combine both high activity and exceptional selectivity, development of the concept of isolated active sites would help to overcome the significant differences between heterogeneous and homogeneous catalysis.62, 67, 84, 85
3.2.1. Preparation of single-atom catalysts
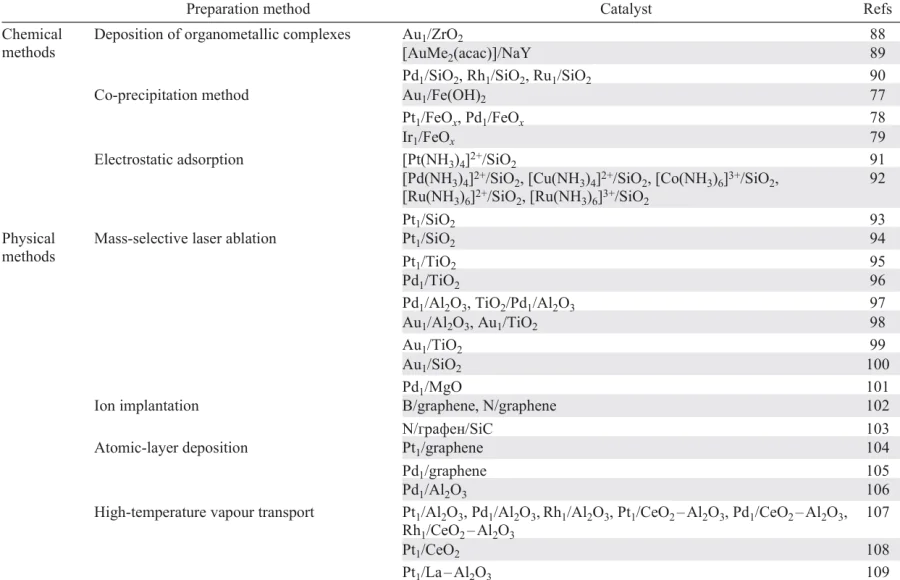
Single-atom catalysts are prepared by both conventional methods (deposition of metal complexes on mineral and polymer supports, adsorption deposition and co-precipitation) and physical methods based on stabilization of single-atom metal species on the oxide support via deposition of atomically dispersed metal vapour, ion implantation or laser ablation 14, 86 (Table 1). In addition, the preparation method of SACs based on binding the precursors by the ligand exchange mechanism proved to be highly efficient. We do not discuss this strategy here, because it has been considered in detail in a recent review.87
3.2.1.1. Chemical methods
3.2.1.1.1. Deposition of organometallic complexes
Wet chemistry methods in various combinations are actively used for the synthesis of supported metal heterogeneous catalysts.110 Most procedures for the preparation of highly dispersed metal catalysts include three successive stages: (1) deposition of atomically or molecular dispersed metal precursor on the support surface by impregnation or ion exchange, precipitation or co-precipitation; (2) drying and/or calcination; (3) activation by reduction (Fig. 5). The main benefit of these methods is that no special equipment is required to use them. Another important issue is that wet chemistry methods are actively utilized for commercial production of heterogeneous catalysts.
![[{"id":"0cQemUx_U5","type":"paragraph","data":{"text":"Scheme for catalyst preparation from organometallic complexes."}}]](/storage/images/resized/OGqB3XxfWX5RCrW2ISXDkzXgwpAXlj9JWYUmz33k_xl.webp)
Organometallic complexes are widely used as precursors, which is due to their ordered structure and the presence of active metal atoms isolated from one another.111 The main challenge of the catalyst synthesis is to bind the complexes to the support in such a way as to prevent the subsequent aggregation of metal atoms into larger cluster. In practice, strong adsorption of organometallic complexes is implemented via coordination of ligands to the surface oxide or hydroxyl groups of the substrate.23, 88, 89, 111, 112 By binding the parent organometallic complexes, it is possible to attain molecular dispersion. During subsequent treatment (calcination, reduction) of the material obtained in this way, ligands are removed and a system of isolated single atoms of the active metal is formed. The strength of adsorption of the parent metal complexes has a considerable effect on the positions of metal atoms on the support surface and the degree of isolation of atoms from one another, which in turn determines their stability to sintering.113 Tada and Muratsugu 90 reported the results of studies of heterogeneous catalysts with isolated active metal atoms (Pd, Rh, Ru) incorporated into organometallic compounds deposited on the surface of highly porous supports (e.g., SiO2). It was reported that immobilization and isolation of active sites incorporated in metal complexes are effective methods for the generation of new oxide-supported catalyst structures, which can be used in reactions such as olefin epoxidation and metathesis. In relation to Rh-containing catalysts, high stability of the resulting structures was demonstrated: leaching of the active component during the reaction was less than 0.5 mass %. In most cases, this result is related to the ligand nature and strength of interaction with the support surface groups.
3.2.1.1.2. Co-precipitation method
Co-precipitation is also widely used to prepare heterogeneous catalysts. The essence of the method is simultaneous precipitation of active metal compound and support material followed by drying and calcination. The calcination gives the catalyst active site. The calcination conditions, namely, the temperature, time, and heating rate largely determine the average size of the resulting nanoparticles and their distribution over the support surface.
By appropriate selection of the process parameters, it is possible to obtain a uniform distribution of active sites at an atomic level. However, characteristics of the final catalysts depend on a number of factors, which include the order and the rate of addition of component solutions, stirring rate, and temperature and pH of the initial solution. The co-precipitation procedure is actively used to prepare noble metal-based SACs. Qiao and Deng 77 reported the synthesis of efficient catalysts for the selective oxidation of CO in the presence of H2. The Au1/Fe(OH)x samples were obtained by co-precipitation from aqueous solutions of HAuCl4 and Fe(NO3)3 containing Na2CO3 and subsequent calcination of the precipitate in an air flow at temperatures of 200 – 400 K. The Au contents in the finished samples varied from 0.76 to 2.52 mass %. More recently, the same approach was used to obtain Pt1/FeOx (Ref. 54) and Pd1/FeОх catalysts.78 The Ir1/FeOx compositions with different metal contents that showed high efficiency in WGS reactions were also prepared by co-precipitation.79
Despite the simplicity of the co-precipitation method, it also has disadvantages such as the possibility of encapsulation of some part of active metal nanoparticles into the support material, which makes them inaccessible to the components of the reaction mixture and can decrease the catalyst activity.
3.2.1.1.3. Electrostatic adsorption
One more widely used method for the preparation of supported catalysts with isolated active sites is based on fundamental studies of the interaction of noble metal complexes with the surface of oxide supports.114 It was found that the processes at the oxide – solution interface occur as a result of gradual change in the pH of the solution, polarization of surface functional groups of oxides, and the subsequent sorption of oppositely charged ions.115, 116 The basic mechanism of the electrostatic adsorption on the silica and alumina surface was considered by Schreier 91 and later specified by Jiao and Regalbuto 92 (Fig. 6).
![[{"id":"06l5WyWihE","type":"paragraph","data":{"text":"Mechanism of electrostatic adsorption on silica and alumina surface.<sup>92</sup> Here PZC is the point of zero charge. K<sub>1</sub> , K<sub>2</sub> are the acidity constants, K<sub>ads</sub> is the adsorption constant."}}]](/storage/images/resized/a5RbOmlnqVnKemIsuWg9TjQi2tQcmvYqPXVTTS2d_xl.webp)
The oxide surface is coated by hydroxyl groups. Each oxide is characterized by a particular pH value at which the surface is neutral, i.e., has a zero charge (point of zero charge, PZC). When the support interacts with an aqueous solution of a precursor, the OH groups on the support surface are protonated (become positively charged) at pH below PZC or deprotonated (hence, become negatively charged) at pH above PZC. There are numerous examples in which oxides placed into solutions at pH below PZC adsorb anions (e.g., [PtCl6]2 – ), whereas at pH values above PZC adsorption of cations (e.g., Pt[(NH3)4]2+) takes place.114 While using this approach for catalyst synthesis, one should focus on the pH range of the support in which the electrostatic interactions are maximized. It should be borne in mind that the spatial distribution of functional groups on support surfaces is usually non-uniform and that various types of surface defects have a significant effect on the adsorption behaviour of metal complexes. In the case of impregnation of cylindrical and spherical granules of the support, pH of the solution and composition of the active metal complex may vary from the periphery to the centre of the granule and in the pores of different diameters, and this may have a significant effect on the catalyst structure. One more factor deserves attention: the composition of the complex used for adsorption may change depending on the pH.
Miller 93 reported a detailed analysis of the relationship between the conditions of adsorption and the size of the formed clusters for Pt-based catalysts.93 The author investigated the influence of the amount of active metal and calcination and reduction temperature of the Pt1/SiO2 catalysts on the degree of dispersion of the active metal. It was found that the atomic dispersion of Pt can be attained for low metal contents (< 0.1 mass %) and for catalyst reduction with hydrogen at 250 °C. A necessary condition is drying of the catalyst at 100 °C immediately before the reduction procedure. An increase in the temperature of the preliminary calcination leads to agglomeration of Pt atoms on the support surface. The size of the resulting nanoparticles can be controlled by varying the calcination temperature.
The possibility of synthesizing heterogeneous catalysts with a controlled degree of dispersion of the active metal by electrostatic adsorption was also demonstrated.92 A series of Pd-, Cu-, Co-, Ru and Ni-based catalysts were prepared using ammonia complexes of the corresponding metals adsorbed on silica as precursors. The physicochemical studies have shown that the electrostatic adsorption is a highly efficient method for the preparation of oxide-supported catalysts in a wider range of active metal concentrations in comparison with the traditional impregnation.
It is noteworthy that the main benefit of the wet chemistry methods is the possibility of synthesis of heterogeneous supported catalysts with isolated active sites in a common laboratory. However, a significant drawback of these methods is that only a small amount of active metal (~0.1 – 0.5 mass %) can be deposited due to the high probability of its surface aggregation. In this regard, a relevant trend is the development of new methods for the synthesis of catalysts with a higher content of the active component and preserved single-atom structure. This is caused by the development of the adsorbate-induced segregation technique for the control of surface structure of bimetallic catalysts (see Section 3.3.4).
3.2.1.2. Physical methods
The catalysts in which active sites are single atoms or sub-nanometre clusters supported on various materials can also be prepared using physical methods. In most cases, model systems are synthesized in this way.
One of the most interesting methods is size-selected laser ablation, which has been successfully used to obtain model catalysts based on Pt 94, 95 and Pd 96, 97 supported on MgO, TiO2 , SiO2 or other materials and Au clusters supported on Al2O3 ,98 TiO2 (Ref. 99) and SiO2.100 The efficiency of this method was demonstrated by Abbet et al.,101 who studied the dependence of the properties of metal catalysts on the size of metal clusters. The authors convincingly proved that the activity of small clusters of active metal can significantly exceed the activity of larger particles. Using size-selected laser ablation, Pdn clusters of a definite size (1 ≤ n ≤ 30) deposited on a thin MgO film (100) were synthesized. Cyclotrimerization of acetylene to benzene efficiently proceeded even on a single Pd atom at 300 K. When larger clusters were used (7 ≤ n ≤ 30), the temperature of benzene synthesis increased to 430 K.
A promising recent method is ion implantation, which is widely used for the development and functionalization of surface structures based on 2D materials such as dichalcogenides and graphene.117 The essence of this method is doping of the support material by atoms of an inert element (B, N) to generate structural defects. Using electron energy loss spectroscopy (EELS) and high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM), the conceptual applicability of this procedure to the targeted modification of the crystal lattice of graphene with nitrogen and boron atoms was demonstrated.102 Graphene doping was carried out in the concentration range from 5 to 15 vol.% at low implantation energies (50 – 200 eV). Similar results were obtained upon modification of graphene grown on SiC surface under high vacuum with nitrogen atoms.103 The configuration of nitrogen atom substitution and the highest content of the doping element (75%) at which the physical properties of the modified graphene remained unchanged were determined by scanning tunnelling microscopy (STM) and DFT.
One more technique used to prepare catalysts with isolated active sites is referred to as atomic layer deposition (ALD). The ALD technique was developed for manufacturing thin oxide films with atomic precision and first used to obtain single-atom Pt catalysts in 2013.104 Later, ALD was used to fabricate Pd1/graphene 105 and Pd1/Al2O3 (Ref. 106) single-atom catalysts. The resulting Pd1/graphene sample performed hydrogenation of butadiene with almost 100% selectivity and conversion of up to 95%. In the author’s opinion, this high selectivity is attributable to a change in the mode of butadiene adsorption and to the steric effect of the support when the reaction proceeds on an isolated palladium atom. The authors prepared Pd1/Al2O3 using an interesting method for stabilizing Pd atoms on the Al2O3 surface.106 After deposition of Pd, titanium dioxide was deposited on the Al2O3 surface regions free from Pd atoms. This resulted in Pd1 stabilization in nanocavities formed by the deposited TiO2. The titania-stabilized Pd1/Al2O3 catalyst demonstrated high activity in the methanol decomposition. Unfortunately, increase in the calcination/reduction temperature up to 200 – 300 °C induced sintering of the Pd atoms and destruction of the structure of single-atom Pd1 sites. An additional treatment of the catalyst by repeated deposition of a TiO2 layer increased its thermal stability, but decreased the activity because of blocking of some Pd1 sites.
An efficient method for the preparation of single-atom catalysts is high-temperature vapour transport. This approach can be used to deposit metals the oxides of which have a certain volatility at high temperatures, e.g., platinum.86, 107 According to theoretical calculations carried out for Pt catalysts, it was assumed that single Pt atoms can be stabilized by being deposited on CeO2 surface.108 In practice, a catalyst with single-atom Pt1/CeO2 sites was obtained by high-temperature vapour transport by mixing a Pt1/La – Al2O3 sample with a cerium oxide powder and heating of the resulting mixture at a temperature of 800 °C in an air flow.109 The authors demonstrated that Pt atoms can be deposited on a cerium oxide surface as PtO2 and can be trapped with a CeO2 surface to form stable isolated Pt1/CeO2 sites. The ability of cerium dioxide to prevent the deposited metals from sintering is attributable to high metal – support interaction energy. As a result, СеО2 is able to stabilize Pt even in the atomically dispersed state. A study of the effect of the shape of cerium oxide nanoparticles on the efficiency of platinum atom trapping demonstrated that the cube-shaped nanoparticles only retard the Pt sintering to larger agglomerates, whereas CeO2 rods stabilize Pt atoms more efficiently and sintering virtually does not take place (Fig. 7). The properties of the resulting catalysts were tested in the СО oxidation to СО2. The results of catalytic tests showed high activity for the obtained Pt1/CeO2 – Al2O3 catalyst (XCO ~ 16% at 250 °C, which is 20 times higher than the conversion attained for the traditional Pt/Al2O3 catalyst). In addition, when rod-shaped CeO2 was used, Pt atoms remained isolated from each other also after the reaction.
![[{"id":"JXYXyLqOUf","type":"paragraph","data":{"text":"Platinum nanoparticle sintering and trapping with the cerium oxide surface.<sup>109</sup>"}}]](/storage/images/resized/tqDroj4aUieg0q8ftCN6Ovsim1FllpKjFQBOpH1L_xl.webp)
The physical methods for the preparation of heterogeneous single-atom catalysts are highly efficient in the targeted synthesis of compositions with isolated active sites. However, the complexity of catalytic synthesis processes and high cost of equipment hamper the use of these methods on an industrial scale. In addition, the structure of catalysts is not sufficiently thermodynamically stable, as long-term exposure to high temperature during the catalytic reactions inevitably causes irreversible agglomeration of catalysts and disrupts the ordered structure of active sites. This leads to decreasing selectivity of the catalysts and precludes their regeneration and reuse.34, 118 It should also be taken into account that isolated metal atoms can be unstable even during their synthesis and post-synthetic treatment (e.g., reduction of the catalyst); therefore, the synthesis of heterogeneous single-atom catalysts and their subsequent operation and regeneration are nontrivial tasks.
3.3. Single-atom alloy catalysts
As has already been noted in the previous Section, despite all of the advantages of single-site and single-atom heterogeneous catalysts, their major drawback is the thermodynamic instability of the uniform active site structure. From the practical standpoint, this means that regeneration of deactivated catalysts, which is usually a combination of high-temperature redox treatments, inevitably leads to agglomeration of active sites. This results in irreversible loss of the active site uniformity, which considerably restricts the applicability of these types of catalytic systems in industrial processes. Therefore, the development of highly selective catalysts with a uniform active site structure possessing high thermodynamic stability and capable of being regenerated is a significant and relevant problem.
An obvious breakthrough in addressing this problem is the concept of SAA catalysts (Fig. 8).15, 34, 55, 119 – 121
![[{"id":"4_kW6ICLqR","type":"paragraph","data":{"text":"Surface of a single-atom alloy catalyst."}}]](/storage/images/resized/IWp0R6pyGXY0Bi59T2Of6Skagx4DIBwQ7bdbYj0P_xl.webp)
This type of catalysts refers to so-called highly dilute alloys in which atoms of the catalytically active metal (М1 = Pd, Pt, Ru) are isolated from one another by atoms of a second component (М2 = Cu, Au, Ag, Ga, In), host metal, which is inactive or has a low activity in the target process.122 This isolation results in the formation of single-atom active sites with identical adsorption and catalytic characteristics on the alloy or bimetallic nanoparticle surface and prevents the formation of multi-fold sites; as a result, the degree of uniformity of the active sites of SAA catalysts is much higher than that for conventional mono- or bimetallic analogues. Furthermore, this markedly increases the catalyst selectivity.
The key condition for the formation of a thermodynamically stable system of isolated single-atom sites is higher energy of interaction between atoms of different components (EM1 – M2) than between atoms of the same component (EМ1 – М1 and ЕМ2 – М2)
2 EM1 – M2 > EМ1 – М1 + ЕМ2 – М2
This energy benefit attained upon the formation of heteroatomic bonds provides for the thermodynamic stability of single-atom sites, which enables regeneration and reuse of SAA catalysts.
From the thermodynamic standpoint, the energy of formation of isolated single-atom sites should be correlated with the formation energy of the corresponding alloy. For the bimetallic PdAg system, which is one of the most widely used in catalysis, the energy of alloy formation was studied in detail in quite a few works using a variety of methods.123 – 125 It was found that alloy formation is characterized by a negative enthalpy and an excess entropy of mixing. The results indicated that the minimum enthalpy of mixing was inherent in the Pd0.4Ag0.6 alloy and amounted to –5.65 kJ mol–1, while for Pd0.9Ag0.1 and Pd0.1Ag0.9 , this value was –1.05 and –2.6 kJ mol–1, respectively. The authors attributed this result to the magnetic properties of the alloys: when the Pd content is below 0.4 mole fractions, the alloys are diamagnetic, while when the amount of Pd is above 0.4 mole fractions, they are paramagnetic. Similar results were reported by Myles 126 and Luef et al.,127 who demonstrated that the minimum enthalpy of mixing amounting to ΔHmix = –5.44 kJ mol–1 is characteristic of the alloy containing 35 at.% Pd. Later studies of thermodynamic properties of PdAg alloys in the temperature range of 177 – 477 °C 128 also provided the conclusion that the lowest integral enthalpy of mixing for the PdAg alloys occurs at Ag content of ~ 60 at.%, which is in line with the above cited publications.125 – 127 Density functional theory calculations led to similar results.129
Since the above data show that the heat of alloy formation is relatively small, the atomic ratio of components in an alloy or a bimetallic nanoparticle plays a significant role in the formation of a stable system of single-atom sites (in this case, Pd1) isolated from each other by atoms of the inactive metal. In some publications, a considerable excess of the host metal is indicated as a necessary condition for stabilization of isolated Pd1 active sites. It is noteworthy that the molar ratio of components needed for the formation of a stable structure of single-atom sites varies over a fairly broad range. For example, stable surface structure of isolated Pd1 sites is already formed in the Pd1Ag catalysts when the Pd : Ag ratio is 1 : 3 to 1 : 4.130 – 132 In the case of Pd1Au, this value varies from 1 : 4 to 1 : 40,121, 133 while for the Pd1Cu systems, the optimal Pd : Cu ratio is 1 : 30 to 1 : 40.134 Thus, it can be concluded that increasing content of the inactive component favours the isolation of active atoms and increases the stability of a system of single-atom active sites on the alloy surface.
In addition, it should be borne in mind that the surface structure of SAA catalysts can be strongly affected by the surface segregation of components and processes caused by the adsorption of molecules with high adsorption heat (adsorbate-induced segregation). The most stable structure is found for the SAA catalysts the surface of which is enriched in the component with a lower surface energy. This issue is considered in more detail in Section 3.3.4.
3.3.1. Methods for preparation and characterization of SAA catalysts
Most often, SAA catalysts are prepared using methods that proved to be efficient for the synthesis of bimetallic alloys.15, 17 In this review, we consider in detail three most popular approaches: galvanic replacement, sequential reduction and impregnation (Table 2).
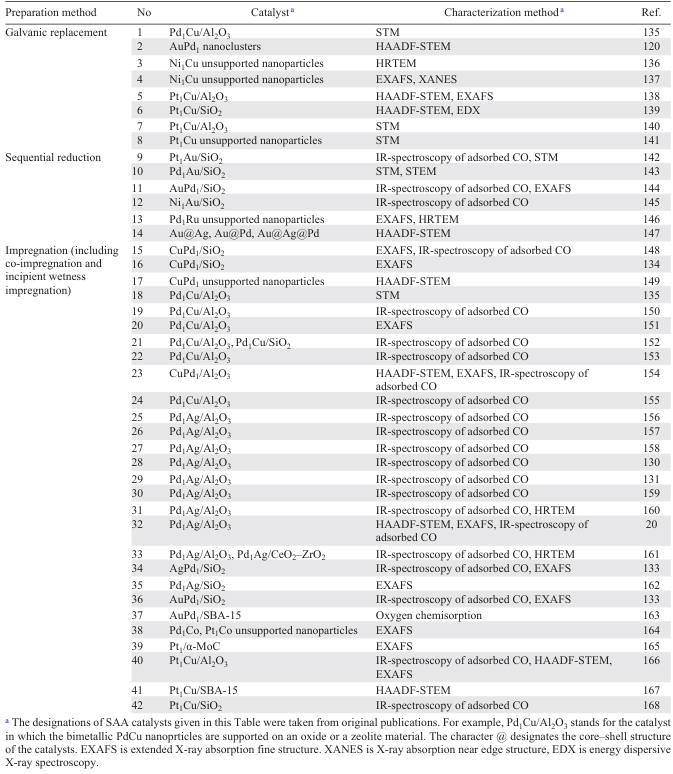
Galvanic replacement is a redox process involving oxidation of one metal with ions of another metal in solution. During the galvanic replacement, selective deposition of atom of one metal on the surface of another metal takes place if the overall reaction potential is positive. The reaction is strictly controlled by the difference between the standard reduction potentials of the two metal/metal ion pairs and is widely used if it is necessary to obtain a bimetallic alloy of a specified composition and structure (see Table 2, lines 1 – 8).
Using galvanic replacement, Zhang et al.166 synthesized the Pt1Cu SAA catalyst. For this purpose, the Cu/MgAl composition obtained by the reduction of mixed CuMgAl oxides was placed into deionized water (100 mL), and the required amount of H2PtCl4 was added dropwise to the resulting suspension. All manipulations were carried out in an ultrasonic bath in a nitrogen atmosphere for 10 min. The resulting suspension was then centrifuged, washed with distilled water and dried in a vacuum drying chamber at 60 °C (Fig. 9).
![[{"id":"tZc0onG_sJ","type":"paragraph","data":{"text":"Synthesis of the Pt<sub>1</sub>Cu SAA catalyst by galvanic replacement.<sup>166</sup>"}}]](/storage/images/resized/jZBaJpicbFaPQ8QDrsYy9zRDTijjxSkGFrgnsK3p_xl.webp)
While using the galvanic replacement method, bear in mind that a single-atom surface structure obtained by this method may be relatively unstable and may undergo considerable transformations during the subsequent redox treatment or catalyst regeneration.
Sequential reduction refers to wet chemistry techniques in which the SAA structure is formed in solution. The procedure includes two stages. First, a metallic precursor, so-called host metal, is reduced in a solution containing ethylene glycol and/or polyvinylpyrrolidone; then the precursor of the catalytically active metal is added to the solution and reduced to give the SAA structure. This procedure proved to be efficient in the synthesis of gold catalysts (see Table 2, lines 9 – 12, 14).
The impregnation of a support with a solution of the desired precursor is among the simplest methods requiring no special equipment. According to this method, a porous support is impregnated with a solution (or solutions) containing easily decomposing salts of both the active metal and the host metal followed by drying, calcination in an air flow and by reductive treatment in a flow of hydrogen or 5 – 10 vol.% H2/Ar gas mixture. The active component can be added either successively from solutions of single salts (sequential impregnation) or simultaneously from a solution containing salts of both alloy components (co-impregnation). In some cases, the calcination stage can be omitted. Also, the impregnated and dried materials are in some cases reduced directly in the catalytic reactor to prevent the material from contact with air. The incipient wetness impregnation is used to prepare catalysts with a specified metal concentration; in this method, the volume of the impregnating solution is equal to the volume of the support pores determined as the amount of a liquid that can be absorbed by a weighed portion of the support. A benefit of the impregnation method is that it is possible to prepare a large amount of a catalyst (up to several tens of grams) at once. The use of this procedure provides high-performance single-atom catalysts, Pd1M (M = Ag, Au, Cu, Co) and Pt1M (M = Cu, Co), for selective hydrogenation, dehydrogenation and reduction (see Table 2, lines 15 – 40).
The rapid development of the single-atom catalyst methodology brings about the need to characterize the catalysts. Analysis of their structure provides understanding of the nature of catalytic sites and clarifies the mechanism of their transformations during the reaction. The results obtained in this way are crucial for the understanding of reaction pathways and for rational design of catalysts with the desired activity, selectivity and stability. As the most useful methods for studying the structure of SAA catalysts, researchers consider electron microscopy, IR spectroscopy of adsorbed CO and EXAFS.
Electron microscopy is one of the simplest tools for characterization of SAA catalysts, as it may give clear and convincing evidence for the formation of isolated active metal sites on a nanoparticle surface.83 A recent study addressing the application of microscopy in analysis of single-atom catalysts even has the phrase ‘seeing is believing’ in its title.169 A microscopic image of an SAA catalyst was obtained for the first time by scanning tunnelling microscopy (STM) (Fig. 10) for a Pt1Cu composition.170
![[{"id":"Y709Bye54G","type":"paragraph","data":{"text":" STM image of Pt<sub>1</sub>Cu-SAA (<i>a</i>). The Pt atoms form small ordered regions corresponding to local structures p(2 × 2 m) (<i>b</i>) and (√3 × √3)R30° (<i>c</i>).<sup>170</sup> Published with permission from the American Chemical Society (ACS)."}}]](/storage/images/resized/PG4INSDDCexyXeHjORKF8BK8EWWevW8BPc4cd139_xl.webp)
The authors were able to demonstrate the atomic structure of metals constituting the alloy, and subsequently the STM method was widely employed to characterize systems based on Cu, Ag and Au (see Table 2, lines 1, 7 – 10, 18). For visualization of SAA catalysts with a large specific surface area, HAADF-STEM is successfully used.171 Aberration corrected transmission electron microscopy is also an efficient tool for characterization of nanomaterials of the SAA type, because this makes it possible to detect even single atoms in the catalyst.83 Several years ago, the environmental transmission electron microscope (ETEM) with atomic resolution was developed.172 This instrument can be used to investigate the structural evolution of single-atom structures during the reaction in situ; this makes ETEM a breakthrough method for real-time visualization of the dynamic state of atoms in a gas-controlled environment. It should be noted, however, that a necessary condition for obtaining high-resolution images that clearly show active metal atoms and host metal atoms is a significant difference between their atomic numbers (so-called z-difference).139, 171
IR spectroscopy using probe molecules (CO, NO, etc.) is one more tool to gain more in-depth understanding of the coordination environment of the metal atoms and determine the type of surface sites in the catalyst.173 For example, an indication of the formation of SAA structure of Pd-containing catalysts is the presence of a highly intense absorption band in the region of ~2000 – 2110 cm–1, characterizing the linear adsorption of СО molecules on the Pd1 active site, and the absence of bands in the < 1900 cm–1 region typical of multi-fold CO adsorption (Fig. 11).145, 162, 174, 175 Data on the use of IR spectroscopy of adsorbed СО to describe SAA catalysts are presented in Table 2 (lines 9, 11 – 12, 15, 19, 21 – 34, 36, 40, 42).
![[{"id":"Rz2N11j4h2","type":"paragraph","data":{"text":"Typical IR spectra recorded for monometallic Pd/Al<sub>2</sub>O<sub>3</sub> and Pd<sub>1</sub>Ag/Al<sub>2</sub>O<sub>3</sub> and Pd<sub>1</sub>Ag/SiO<sub>2</sub> SAA catalysts.<sup>131, 133</sup>"}}]](/storage/images/resized/XNKZQ9kqXpAMIp9s2pJZyyU91ut1jAd4YwspYjhi_xl.webp)
Perhaps, EXAFS is the most useful method for characterization of SAA structures. It can be used to determine structural parameters of the local environment of atoms with a selected atomic number, such as interatomic distances, coordination numbers, amplitudes of thermal vibrations, etc. Depending on the way of recording the spectra, EXAFS makes it possible to analyze the local environment of atoms located either in the sample bulk or on the surface. Owing to its versatility, the method enables investigation of catalyst structures in the gas or liquid phase, at high temperatures and pressures, and even under vacuum.176, 177 A necessary condition for the use of this characterization method is the presence of a synchrotron radiation source (see Table 2, lines 4, 5, 11, 13, 15 – 16, 20, 23, 32, 34, 36, 38 – 40).
Typical examples of application of EXAFS for characterization of SAC and SAA catalysts are depicted in Fig. 12. Using co-precipitation, Qiao et al.54 prepared Pt1/FeOx catalysts with Pt contents of 0.17 and 2.5 mass %. It was shown by HAADF-STEM that the Pt1(0.17%)/FeOx catalyst contained only isolated Pt1 atoms, whereas the Pt1(2.5%)/FeOx sample contained, apart from Pt1 , some Pt clusters with <1 nm size. A number of structural features characteristic of synthesized catalysts were established using EXAFS. The modelling of Pt1(0.17%)/FeOx revealed the presence of Pt – O (C.N. = 1.9; interatomic distance of 2.02 Å) and Pt – Fe (C.N. = 0.9, interatomic distance of 2.88 Å) coordination spheres, indicating the interaction of Pt atoms with O and Fe atoms of the support surface. The complete absence of Pt – Pt spheres attests to the formation of only single isolated Pt1 atoms. The spectrum of the Pt1/FeOx catalyst with an increased content of the platinum component (2.5 mass %) also exhibits a signal corresponding to the first coordination sphere of platinum metal with an average C.N. of ~3.8 and Pt – Pt interatomic distance of 2.53 Å. Low C.N. values and short Pt – Pt distances in the sample in comparison with the bulk platinum (C.N. = 12 and Pt – Pt distance of 2.78 Å) are indicative of the formation of small Pt clusters in the Pt1(2.5%)/FeOx samples; the cluster formation may be due to sintering of isolated metal atoms induced by the reductive treatment.
![[{"id":"TdeiUMWf6s","type":"paragraph","data":{"text":"Comparison of the EXAFS data for Pt<sub>1</sub>/FeO<sub>x</sub> SAC (<i>a</i>),<sup>54</sup> Pt<sub>1</sub>/Al<sub>2</sub>O<sub>3</sub> SAC (<i>b</i>)<sup>178</sup> and Pd<sub>1</sub>Cux/SiO<sub>2</sub> SAA (<i>c</i>)<sup>148</sup> catalysts."}}]](/storage/images/resized/TCwo7182M1cKVkhPzB24HJr2bYrH8YHIaEdBpDnO_xl.webp)
EXAFS was also used to characterize the Pt1/Al2O3 catalyst.178 According to EXAFS data, the initial Pt1/Al2O3 SAC shows a peak with a maximum at ~1.7 Å corresponding to the Pt – O distance in the first coordination sphere of PtO2. The EXAFS modelling did not reveal any Pt – Pt coordination spheres in the catalyst, which attests to the presence of single Pt1 atoms stabilized in an oxygen environment on the support surface. EXAFS modelling of Pt1/Al2O3 treated in a hydrogen-containing medium at high temperature (350 °C) predicts the appearance of Pt – Pt distances characteristic of the first coordination sphere of platinum (C.N. ~ 4.2) and confirms the formation of clusters containing up to 13 Pt atoms.
Zhang’s research group studied the structural features of a series of Pd1Cux/SiO2 SAA catalysts with different Cu : Pd ratios by EXAFS.148 Immediately before the measurements, the samples were reduced at 250 °C for 1 h. Modelling of the EXAFS spectra showed that the most intense peak in the 1.8 – 3.0 Å range inherent in the palladium metal has an intricate shape and is completely described by the model assuming the presence of the Pd metal first coordination sphere with a 2.74 Å distance and C.N. = 12. In comparison with the reference Pd sample, in the case of Pd – Cu catalysts with Pd : Cu ratios of 1 : 40 and 1 : 160, the contribution of the coordination sphere of palladium metal completely disappears, and the spectrum contains only one signal corresponding to the Pd – Cu coordination sphere (2.55 Å and C.N. = 8.7). This implies that only copper atoms are present in the local environment of palladium and points to the formation of the SAA structure. When the copper concentration increases, the major peak at ~ 2.5 Å shifts to shorter distances compared with the spectrum of the reference Pd sample (2.74 Å), as the formation of Pd – Cu bonds is accompanied by a decrease in the distance between the atoms of the first coordination sphere.
3.3.2. Specific catalytic properties of SAA catalysts in selective hydrogenation reactions
The specific properties of single-atom alloy catalysts can be analyzed from the standpoint of the commonly accepted theory of catalysis on alloys, according to which catalytic characteristics are determined by the influence of geometric and electronic factors.179 – 181 Thus, in the case of a bimetallic alloy consisting of active and inert components, the contribution of the geometric effect is determined by the change in the size of the active site (ensemble) as a result of dissolution of active metal atoms in the matrix of the host metal. From this standpoint, SAA catalysts represent a limiting case in which the active ensemble consists of only one atom surrounded by atoms of inactive (or low-activity) host metal. In addition to the geometric effect, it is also necessary to consider the electronic (ligand) effect, which is related to a change in the electronic state of the active metal atom surrounded by atoms of the inactive metal upon electron density redistribution between the alloy components.179
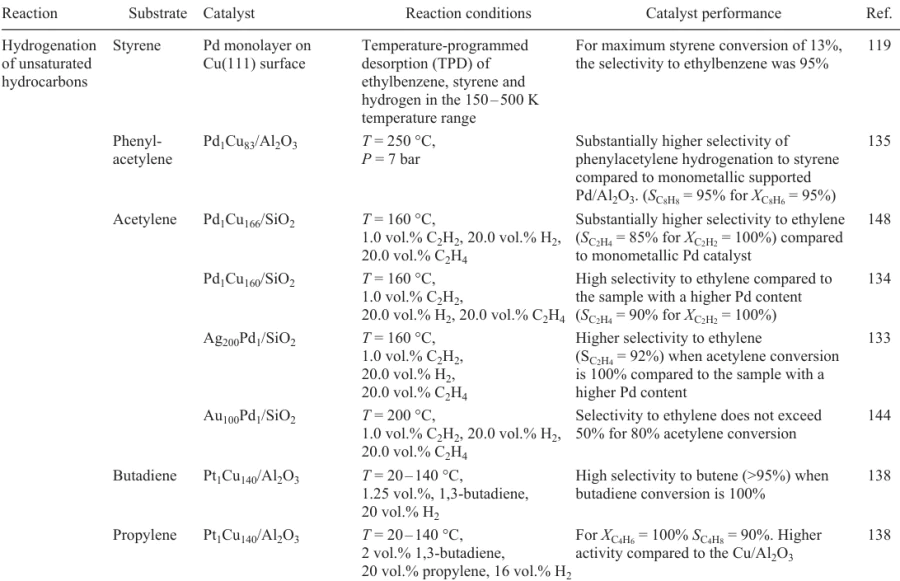
Although the concept of SAA catalysts was proposed only a few years ago, there are already publications that demonstrate how contributions of these two factors lead to unique properties of this type of catalysts. For example, quite a few publications are devoted to the use of Pd1M SAA catalysts in the selective hydrogenation of alkyne derivatives (first of all, acetylene).182, 183 For example, the use of Pd1Ag,184 – 187 Pd1Ga,185, 188, 189 Pd1Zn,185, 190, 191 Pd1Au 192, 193 and Pd1Fe 194 catalysts substantially increases the selectivity of the partial hydrogenation of alkynes to olefins (Table 3). The goal of the study was to identify specific structural and catalytic characteristics of single-atom systems and their distinctions from conventional bimetallic catalysts. In order to clarify this issue, chemists varied the ratio of metals in the catalyst over a wide range to remove the multi-fold sites consisting of several Pd atoms and generate a system of single-atom Pd1 sites on the alloy surface in which Pd atoms are isolated from one another by atoms of the inactive component. By analyzing published data, we found that Pd modification with Group 11 metal atoms plays a key role for the development of highly selective catalysts for hydrogenation of acetylene compounds. Experimental study of single-atom alloy catalysts was supplemented by computational methods. Below we consider the behaviour of the Pd1Cu, Pd1Ag and Pd1Au catalysts in the selective hydrogenation in more detail.
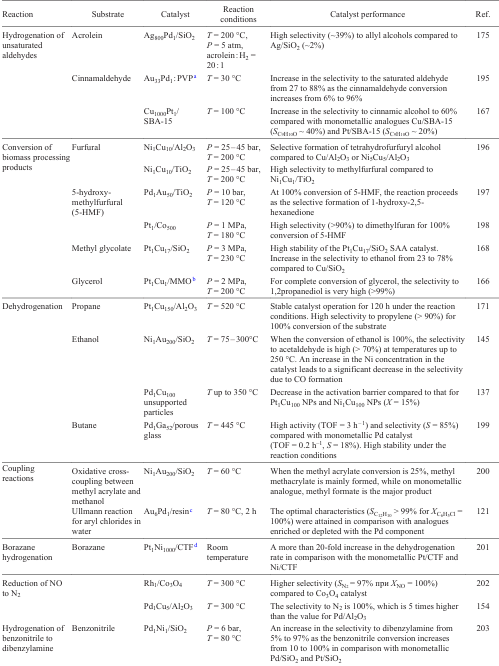

3.3.2.1. Catalytic behaviour of the Pd1Cu SAA catalysts
3.3.2.1.1. Supported Pd1Cu SAA catalysts
The applicability of catalysts based on Pd1Cu bimetallic nanoparticles for various chemical reactions was discussed in detail in a review.208 The use of Pd1Cu systems in the hydrogenation of alkynes not only increases the selectivity to the target olefin, but also markedly decreases the cost of the finished catalyst as a result of using much cheaper copper as the host metal.17, 122 As one of the first mentions of application of Pd1Cu SAA catalysts in the hydrogenation, consider the papers by Flytzani-Stephanopoulos and co-workers 119, 135 The Pd1Cu/Al2O3 catalyst was obtained by galvanic replacement of Cu atoms by Pd atoms on the surface of Cu nanoparticles and tested in the hydrogenation of styrene and phenylacetylene. The high selectivity to phenylethylene (>94%) in the presence of the Pd1Cu SAA catalyst in comparison with that on the reference monometallic Pd/Al2O3 sample was attributed to lower energy of desorption of phenylethylene in comparison with the activation barrier of its hydrogenation; this prevents complete hydrogenation of the starting alkyne to alkane.
A series of Pd1Cu/Al2O3 samples with different Cu : Pd ratios were prepared by sequential impregnation to be used in the gas-phase hydrogenation of acetylene mixed with ethylene.209, 210 According to IR spectroscopy data for adsorbed CO, in the case of high content of the copper component (Pd : Cu = 1 : 10 to 1 : 50), the catalyst contains Pd1 single-atom sites in which the Pd atoms are completely isolated by being surrounded with Cu atoms. The authors suggested that the isolated palladium atoms act as hydrogen activation sites on which H2 molecules dissociate. The alkyne hydrogenation occurs directly on the copper metal surface, which provides exceptionally high selectivity to the target alkene typical of the copper metal, while the presence of Pd1 hydrogen activation sites considerably accelerates the reaction.155, 210, 211 Similar studies were carried out for the Pd1Cu/Al2O3 catalysts with different Pd : Cu ratios in the selective hydrogenation of propyne.212 It was shown that the sample characterized by Pd : Cu = 1 : 50 provided complete conversion of the substrate with high (~86%) selectivity to propene under nearly industrial conditions. High selectivity was attained in this case without using CO as a competitive adsorbate, which is a considerable advantage over commercial PdAg catalysts for selective hydrogenation.
Cao et al.149 studied the effect of the catalyst preparation method on the catalyst selectivity in the hydrogenation of acetylene. Samples were prepared by sequential impregnation and co-impregnation or by galvanic replacement. Irrespective of the chosen procedure for the synthesis, the Pd1Cu SAA samples showed high selectivity to ethylene; however, impregnated catalysts proved to be more stable under reaction conditions. A similar result in the acetylene hydrogenation was obtained by Anderson and co-workers.212 Pd1Cu catalysts prepared by sequential impregnation and co-impregnation and by colloidal method showed the same selectivity. The content of copper was the crucial factor: the highest selectivity was found for samples with a considerable excess of copper (Cu : Pd = 50).
Fairly interesting data on the structure and catalytic performance of the SAA catalysts in acetylene hydrogenation were obtained in a series of studies by Pei et al.133, 134, 144, 148 The authors compared the structure and catalytic performance of Pd1Cu, Pd1Au and Pd1Ag SAA catalysts obtained by incipient wetness impregnation with aqueous solutions of the required salts. The Pd content was varied in the range from 523 to 2093 ppm, with the content of the inactive metal being maintained at about ~5 mass %. Despite this low percentage of Pd, the authors were able to prove the formation of a single-atom system of Pd sites on the surface of copper metal nanoparticles using a set of physicochemical methods including EXAFS, IR spectroscopy of adsorbed CO and microcalorimetric measurements of С2Н4 and Н2 adsorption energy. Subsequently, the results obtained in these studies were confirmed by DFT calculations.213 Pd1Cu/SiO2 catalysts demonstrated a high selectivity to ethylene (~85%) with complete conversion of the substrate. They were superior in the selectivity to Pd1Ag/SiO2 and Pd1Au/SiO2 catalysts. Furthermore, PdCu catalysts proved to be most stable among the whole series.
For substantiating the results, the authors proposed a reaction mechanism according to which dissociative adsorption of molecular H2 takes place on isolated Pd atoms (Fig. 13a). The resulting atomic hydrogen migrates to the neighbouring Cu atoms via the spillover mechanism. The hydrogenation of an acetylene molecule to ethylene also takes place on the Pd1 site, and the catalyst activity is determined by the distribution of atomic hydrogen over the nanoparticle surface (Fig. 13b). Owing to the low adsorption energy of π-bound ethylene on the Pd1 site, it is rapidly desorbed from the catalyst surface, which markedly increases the selectivity of the single-atom catalyst (Fig. 13c).
![[{"id":"3ahZ0ch43p","type":"paragraph","data":{"text":"Mechanism of acetylene hydrogenation on the surface of bimetallic Pd<sub>1</sub>Cu/SiO<sub>2</sub> catalyst.<sup>148</sup>"}}]](/storage/images/resized/psSz4Tabh4hfxOpwFG1WvgC8ElVbQYtfjuNMGbtn_xl.webp)
A series of studies 150 – 153 address the properties of Pd1Cu SAA catalysts in the liquid-phase hydrogenation of a number of internal and terminal alkynes [diphenylacetylene, phenylacetylene, 1-phenyl-1-propyne, 1-phenyl-1-butyne and 1-(prop-1-enyl)cyclohexanol]. The heterobimetallic Pd1Cu2(OAc)6 complex in which the Pd and Cu atoms are bound by strong acetate bridges served as the precursor of the active component. This type of structure ensures a close contact of metal atoms throughout the catalytic synthesis, thus promoting the formation of the SAA structure. The formation of Pd1Cu alloy nanoparticles was confirmed by powder X-ray diffraction (XRD) and EXAFS. Analysis of the reaction products showed a considerable increase in the selectivity to the target alkenes in the hydrogenation of any of the substrates in the presence of Pd1Cu2/Al2O3. The best result was obtained for the hydrogenation of diphenylacetylene: particularly, for 95% conversion, the selectivity to diphenylethylene was 93%, which is comparable with the selectivity of the commercial Lindlar catalyst (~ 95%).150, 151 A study of the effect of the support (SiO2 or Al2O3) showed the optimal performance for Pd1Cu/Al2O3.152 Also, the authors carried out comparative analysis of the catalysts of diphenylacetylene hydrogenation depending on the Pd : Cu ratio, which showed that the amount of the formed olefin increases with increasing Cu content in the catalyst.151, 153 For Cu : Pd ≥ 2, the selectivity was even higher than that for the commercial Lindlar catalyst.
Thus, the studies demonstrated that the formation of a system of Pd1 sites separated from one another by Cu atoms on the PdCu nanoparticle surface leads to a sharp increase in the selectivity of alkyne (acetylene, propyne) hydrogenation to olefins. This may be attributable to the decrease in the adsorption energy of the intermediate olefin, since olefins can be adsorbed on Pd1 sites only in the π-bound state. An alternative explanation is that isolated Pd1 sites perform activation (dissociation) of molecular hydrogen, while the proper hydrogenation takes place on the copper metal surface.
3.3.2.1.2. Model Pd1Cu SAA catalysts
An additional piece of evidence in favour of the alkyne hydrogenation mechanism consisting of the activation of molecular hydrogen on Pd1 sites and the subsequent hydrogenation on the Cu surface was derived from a study of model Pd1Cu catalytic systems obtained by deposition of active metal atoms on the surface of an inert component. Hydrogen activation on the surface of isolated Pd1 sites deposited over the surface of the Cu(111) single crystal was studied in detail by Sykes’s research group.214 – 216 The samples were obtained by vacuum deposition of palladium vapour and studied by STM. Theoretical calculations showed that isolated Pd atoms located on the Cu(111) surface can promote hydrogen dissociation followed by atomic hydrogen spillover on the copper surface (Fig. 14).
![[{"id":"w1l2QGv5sB","type":"paragraph","data":{"text":"Hydrogen dissociation on an isolated Pd atom supported on the Cu(111) surface.<sup>119</sup> Н<sub>ads</sub> is adsorbed hydrogen."}}]](/storage/images/resized/hWIOmEGZm5Z3X3nqdzrRW2yUzoCkZ1tih2OdcauY_xl.webp)
This is a key stage in the CO2 hydrogenation and WGS. When Au is used as the host metal, spillover does not take place, perhaps, because of the occurrence of energetically more favourable hydrogen desorption from the Au surface.
Somewhat later, it was shown that isolated Pd atoms on the Cu surface considerably reduce the energy barrier both for hydrogen adsorption on the surface and for the subsequent hydrogen desorption from Cu surface. Easy hydrogen dissociation on Pd sites and weak binding to Cu account for more selective hydrogenation of styrene and acetylene compared to that on Pd catalysts.16, 119
The data obtained for model Pd1Cu catalysts initiated the studies of single-atom Pt catalysts supported on the Cu surface.16, 141 The set of results of catalytic tests, physicochemical studies and theoretical calculations provided the conclusion that the C – H bond activation is much more efficient on Pt1Cu SAA catalysts than on the Cu metal surface. Mention should also be made of high stability of single-atom Pt1Cu catalysts, which is caused by the fact that they are resistant to coke formation, unlike Pt metal. Similar results were obtained by other rersearchers.138, 217
In conclusion, it should be noted that the use of model single-atom catalysts obtained by deposition of atomiclly dispersed metal vapour on the surface of an inert component allows detailed investigation of quite a number of fundamental characteristics of isolated active sites. The possibility of this study is associated with two factors:
(1) the use of a set of surface-sensitive methods such as photo- and Auger electron spectroscopy, ion scattering spectroscopy, low-energy electron diffraction and other methods;
(2) high degree of ordering of model systems, which makes it possible to study in detail the reaction mechanism by temperature-programmed methods and relate the obtained data directly to the active site structure at an atomic level and to the nature of intermediates, which can be identified by HRSTEM.
A drawback of this approach is that it is inapplicable for the synthesis of conventional catalysts that could be used both in laboratory and in industry. In addition, structures obtained by vacuum deposition methods are not always thermodynamically stable and can be substantially transformed at elevated temperature.16, 218
3.3.2.2. Catalytic properties of Pd1Ag SAA catalysts
An intensively studied SAA catalytic composition is Pd1Ag. This is due to several reasons.
First, PdAg catalysts are the most popular representatives of substitutional solid solutions widely employed in catalysis. They are actively used in many industrially important reactions, e.g., large-scale selective gas-phase hydrogenation of acetylene impurity in ethylene produced by pyrolysis and removal of phenylacetylene traces from styrene before polymerization.20, 133, 192, 219 – 225 In addition, selective hydrogenation of substituted alkynes is a key step of synthesis of many complex alkenes, because it can be conducted with high regio- and stereoselectivity. Palladium-based heterogeneous catalysts are used most widely in these reactions.226, 227 The PdAg catalysts used both in industry and in laboratory are bimetallic nanoparticles supported on various materials with high specific surface area, which is due to the need to use as fully as possible active metal atoms in the target process.
Second, IR spectroscopy of adsorbed CO is an exceptionally convenient and useful method for determining the structure of active sites on the surface of PdAg alloy or PdAg bimetallic nanoparticles. Using this method, the formation of Pd1 sites on the surface of PdAg nanoparticles can be detected with a high degree of certainty on the basis of disappearance of the adsorption band for СО adsorbed on several neighbouring Pd atoms. In turn, elucidation of the relationship between the active site structure and adsorption and catalytic characteristics of the surface sites makes it possible to determine the main parameters affecting the catalytic performance. For example, Somanoto and Sachtler 228 reported the results of experimental studies of the adsorption properties of Pd and PdAg alloy catalysts with different silver contents by IR spectroscopy of adsorbed СО. The monometallic sample contained all three types of adsorbed carbon monoxide molecules: linear, bridge and three-fold ones. Note that for СО adsorption on monometallic Pd catalysts, the absorption band at ~2060 cm–1 for linearly adsorbed CO is usually relatively weak, because the energy of bridge and three-fold СО absorption is much higher than that of the linear one. For PdAg catalysts with high Ag content, the intensity of absorption bands for the linearly adsorbed CO gradually increases, whereas the intensity of bands for the bridge and multi-fold adsorption sharply decreases. In addition, the energy of desorption (Edes) of CO molecules is lower for alloy catalysts (113 kJ mol–1) than for monometallic Pd samples (130 kJ mol–1). The authors attributed this decrease to the fact that geometric factors predominate over the electronic ones. Similar conclusions were drawn by Nieuwenhuys and co-workers,229, 230 who reported that in the temperature range of 250 – 340 K for monometallic sample, the desorption energy is 140 kJ mol–1, while in the case of PdAg, Edes = 112 kJ mol–1. Using temperature-programmed desorption of CO, high resolution electron energy loss spectroscopy (HREELS) and STM, Behm and co-workers 231, 232 showed that the CO adsorption is impossible on the surface Ag atoms. Also it was shown that the strength of binding of CO to Pd sites substantially decreases with increasing Ag concentration. According to STM data on Pd and Ag distribution in the surface layer, this is due to ensemble effects. When the PdAg alloy catalysts are enriched in silver, the structure of CO adsorption sites markedly alters: the Pd3 and Pd2 sites, which are preferable for the three-fold and bridge CO adsorption, become inaccessible. This is accompanied by the formation of Pd1 single-atom sites as a result of Pd isolation with Ag atoms and, hence, the CO adsorption energy decreases.
The structure of active sites of heterogeneous catalysts for partial hydrogenation largely determines the selectivity to olefin derivatives. Detailed studies carried out at the turn of the 1970s and 1980s showed that the selectivity to olefins cannot be interpreted without detailed knowledge of the reaction mechanism and the actual state of surface adsorption sites.233, 234
It should be noted that the mechanisms of selective hydrogenation of alkynes on Pd1Cu and Pd1Ag single-atom alloy catalysts are somewhat different. In the case of Pd1Cu systems, only hydrogen dissociation takes place on isolated Pd atoms located on the surface of the inert copper component, and subsequently hydrogen spillover onto copper occurs, and hydrogenation takes place on the copper surface, whereas in the case of Pd1Ag compositions, the reaction proceeds only on Pd1 sites on which both hydrogen and alkyne substrate are activated.131
The single-atom nature of the active site rules out the possibility of adsorption of the alkyne substrate and olefin product in the di-σ-bound form; hence, adsorption takes place mainly in the π-form. The adsorption energy of unsaturated compounds thus decreases, which considerably facilitates their desorption from the surface of Pd1Ag nanoparticles and increases the selectivity to the target olefin.130, 131
From this standpoint, the reaction proceeds by the classic two-step route comprehensively described in the literature 28, 30, 235 (Fig. 15).
![[{"id":"5odPA3cxlF","type":"paragraph","data":{"text":"Mechanism of selective hydrogenation of alkynes.<sup>30</sup>"}}]](/storage/images/resized/pdWc0ixq9MxPZYjJr8hwwQyKlKsP1Dy1jihJgnjG_xl.webp)
The adsorption of the alkyne molecules on the catalyst surface is followed by their conversion to the expected olefin. The product is either desorbed from the catalyst surface or is completely hydrogenated to alkane. Considering the proposed mechanism, the authors concluded that the alkene selectivity is determined by both thermodynamic and kinetic factors.
The effect of thermodynamic factor is associated with the competitive adsorption of the starting alkyne and intermediate olefin molecules. If the alkyne adsorption coefficient is much higher than the olefin adsorption coefficient (k1 : k–1 >> k3 : k–3), then the strong adsorption of alkyne leads to displacement of olefin molecules from the surface active sites of the catalyst and prevents the olefin from repeated adsorption, thus suppressing the undesirable hydrogenation to alkane. In this case, alkyne actually blocks the catalyst surface, and the excess hydrogenation of the target product does not take place as long as the alkyne is present in the reaction mixture.
The kinetic factor is significant when the adsorption coefficients of alkyne and olefin differ little, and the thermodynamic factor does not greatly influence the general course of the process. In this case, the selectivity is determined by the ratio of the rate constants of the first and second steps of the reaction (k2 and k4). High selectivity is attained if k2 >> k4. It is worth noting that, in the presence of bimetallic alloy catalysts, the reaction rate usually markedly decreases on going from the first to second hydrogenation step.236 A more detailed discussion of the influence of these factors on the selectivity of monometallic palladium and platinum catalysts in relation to acetylene hydrogenation can be found in the literature.235, 237, 238
An early attempt to investigate the performance of single-atom Pd1Ag catalysts in the selective acetylene hydrogenation was made by Pei et al.133 The authors prepared a series of Pd1Ag/SiO2 catalysts containing 2 mass % Ag and minor amounts of Pd (0.02 – 0.1 mass %) using co-impregnation of the support with a solution of palladium and silver nitrates. They found that the key step of the synthesis is the high-temperature hydrogen reduction (400 – 550 °C). Although high temperature causes sintering of the bimetallic nanoparticles and some activity loss, it also promotes surface segregation of silver and formation of Pd1 sites isolated from one another with Ag atoms.
Furthermore, high-temperature reduction enhances the electronic interaction between Ag and Pd atoms and increases the electron density on Pd atoms. The acetylene adsorption on the resulting Pd1 sites is accompanied by transfer of excess charge from the Pd atom to the π-antibonding orbital of acetylene, resulting in triple bond activation. In addition, it was shown by microcalorimetry that the ethylene adsorption energy on the bimetallic Pd1Ag catalyst is much lower than that on the monometallic analogue, which increases the catalyst selectivity by promoting desorption of the alkene.
Owing to the decrease in the adsorption energy in the acetylene hydrogenation step by 12 kJ mol–1 compared to that for the monometallic catalyst sample and to the decrease in the adsorption energy of the intermediate product, the obtained single-atom Pd1Ag catalysts have high selectivity (~92.3%) in the hydrogenation of acetylene impurity in ethylene. It was noted that a large excess of silver is a necessary condition for attaining high selectivity.239, 240
A beneficial effect of the high-temperature reductive treatment on the selectivity of the acetylene hydrogenation catalyst was also found in an earlier study for a Pd1Ag catalyst containing 0.45 mass % Pd and 0.27 mass % Ag.241 In this case, the increase in the selectivity was attributed to higher mobility of Pd and Ag atoms. This facilitates the surface segregation of Ag, enrichment of the surface in Ag and formation of single-atom Pd1 sites isolated from one another with the Ag component. The formation of Pd1 sites was proved using IR spectroscopy of adsorbed CO by monitoring the decrease in the intensity of absorption bands of the CO molecules adsorbed in the bridge and threefold mode, which was accompanied by an increase in the intensity of the bands for the linear form of CO adsorption.
Considerable effect of neighbouring Ag atoms on the adsorption energy of acetylene and ethylene molecules on Pd atoms was confirmed by theoretical calculations. Silver modification of the monometallic Pd catalyst not only modifies the active sites (geometric effect), but also increases the electron density in the palladium d-orbital because of the shift of electron density from Ag to Pd (electronic effect).219, 242 Both effects lead to decrease in the activation energy of acetylene hydrogenation from 32 – 44 to 26 – 31 kJ mol–1, acceleration of the desorption of ethylene molecules and, correspondingly, increase in the selectivity of PdAg catalysts. In addition, the authors found that the increase in the hydrogen partial pressure results in higher reaction rate, but has an adverse effect on the selectivity of monometallic Pd catalyst and Pd1Ag catalyst. Similar results were obtained in other studies devoted to selective hydrogenation of acetylene.223, 243, 244
Detailed investigation of the acetylene hydrogenation on Pd1Ag bimetallic nanoparticles identified quite a number of additional features of this reaction. Some authors noted that both dissociative adsorption of hydrogen and acetylene hydrogenation can take place on Pd1 atoms in PdAg alloy catalysts.243, 245 High reactivity of acetylene towards hydrogen and relatively slow acetylene desorption are considered as the key factors for the high selectivity of bimetallic alloy catalysts in the partial hydrogenation of acetylene.246 Khan et al.244 believe that Pd alloying with Ag sharply decreases the rate of dissociation of molecular hydrogen and the ethylene desorption energy, which also promotes its fast desorption from the surface and prevents further hydrogenation.
Despite the fact that the influence of PdAg nanoparticle structure on the activity/selectivity characteristics has been studied in sufficient detail, the catalytic performance of this system in the selective liquid-phase hydrogenation of substituted alkynes has not been adequately addressed. In this connection, worthy of note is a paper by Karakhanov et al.,247 who found high selectivity of Pd1Ag catalysts in the liquid-phase selective hydrogenation of phenylacetylene, 1-octyne and 4-octyne. Mention should also be made of a series of publications studying in detail the properties of Pd1Ag catalysts in the liquid-phase hydrogenation of diphenylacetylene,130, 131, 156 – 158, 161 phenylacetylene,156, 158 1-phenyl-1-propyne 130, 158, 159 and 1-phenyl-1-butyne.158 The catalysts were prepared by incipient wetness impregnation of the support with solutions of the required salts. Relying on the results of an integrated physicochemical study of the catalysts, the authors confirmed the formation of a single-atom structure with single Pd1 atoms isolated with silver atoms. This Pd1Ag catalyst showed a markedly higher selectivity in the hydrogenation of symmetrical and unsymmetrical internal alkynes than the monometallic Pd analogue. Regarding the process kinetics, the optimal result was attained in the hydrogenation of diphenylacetylene and 1-phenyl-1-propyne in which the rate decreased considerably for the undesirable hydrogenation of the target olefin to alkane, which greatly facilitates the kinetic control.
Study of the catalytic performance of bimetallic Pd1Ag catalyst in the hydrogenation of disubstituted alkynes with the Ag : Pd ratio being varied in the range from 0.03 to 20 revealed a number of regular patterns that relate the catalyst structure to the activity/selectivity parameters. It was shown that the selectivity to the target olefin products naturally increases with increasing amount of Ag, although this is accompanied by some decrease in the catalytic activity. In the case of samples in which the silver content is relatively low (Ag : Pd ≤ 1), high selectivity is caused by suppression of the formation of the palladium hydride PdHx phase during the formation of the PdAg alloy and by gradual decrease in the amount of the non-selective hydride hydrogen. In the case of catalysts enriched in silver (Ag : Pd ≥ 1), the Pd hydride formation is completely inhibited, as shown by Rassolov et al.,132 and the observed increase in the selectivity is, most likely, due to increasing stability of the Pd1 sites where hydrogenation is more selective. According to experiments, in the hydrogenation of diphenylacetylene and 1-phenyl-1-propyne, the influence of competitive alkyne – alkene adsorption (thermodynamic factor) on the reaction is insignificant and, furthermore, it does not depend on the Ag concentration in the catalyst. In this case, the reaction selectivity is mainly controlled by the ratio of hydrogenation rates of the alkyne and alkene compounds (kinetic factor), which increases with increasing silver content. When silver is present in a large excess (Ag : Pd > 5), the highest selectivity to the target olefin is attained (~ 99%); this is accompanied by a pronounced retardation of the complete alkene hydrogenation to alkane. The analysis of the results allowed the authors to propose the first effective kinetic model describing the liquid-phase hydrogenation of disubstituted alkynes into cis-alkenes on bimetallic SAA catalysts. According to this model, the high selectivity is provided by both suppression of alkyne hydrogenation directly into alkane and low adsorption energy of alkenes on Pd1 sites, which facilitates their desorption and prevents further hydrogenation.131, 159
Some authors also discuss the long-term stability of performance of Pd1Ag catalysts in the partial hydrogenation. According to Pachulski et al.,248 at least five ‘catalytic reaction – regeneration’ cycles can be conducted without significant loss of the selectivity of the Pd1Ag/Al2O3 sample. It is noted that the regeneration procedure should include heat treatment in a vapour – air mixture followed by reduction in a hydrogen flow at a temperature of no less than 120 °C. A similar result was obtained for the Pd1Ag/Al2O3 and Pd1Ag/CeO2 – ZrO2 catalysts in the selective hydrogenation of diphenylacetylene.161
Apart from the supported single-atom alloy catalysts, in recent years, considerable attention has been drawn by the synthesis of core – shell bimetallic PdAg catalysts. These compositions are most actively used in the electrochemical oxidation of acetic acid and methanol 249, 250 and in the production of hydrogen by decomposition of acetic acid.251 Catalysts of this type are nanostructured materials consisting of a core rich in one of the alloy components and a shell formed by several layers of atoms of the other component. Lu et al.252 reported the results of synthesis of core – shell Pd1Ag alloy catalysts with various Pd : Ag ratios. The authors found that repeated electrochemical treatment in an acid medium is accompanied by gradual dissolution of silver present on the nanoparticle surface, which affords an ultrathin Ag shell on Pd atoms. The prepared Pd1Ag@Pd/C structures showed exceptionally high activity and stability in the catalytic reduction of oxygen. Considering earlier theoretical and experimental studies, the authors found that the high activity of the catalysts is due to the synergistic effect between Pd and Ag, while the high stability of nanoparticles is attributable to the formation of the core – shell structure.
Catalysts based on the core – shell Pd1Ag nanoparticles are also effective in the liquid-phase selective hydrogenation of various alkynes, including 1-octyne, 1-phenyl-1-propyne and diphenylacetylene.253 Experiments were performed at room temperature at a hydrogen pressure of 1 bar. It was found that owing to the presence of a palladium core with a silver shell, the target olefins were formed in high yields and with selectivities of 94 – 98% almost in all cases. Owing to the unique structure of the resulting bimetallic nanoparticles, the authors were able to suppress the undesirable alkene to alkane hydrogenation (Fig. 16). In addition, it was shown that the catalyst can be easily separated from the reaction mixture and reused without significant deterioration of the catalytic performance. The effective blocking of multi-fold Pd sites by silver atoms within bimetallic Pd1Ag nanoparticles supported on TiO2 also resulted in a substantial increase in the catalyst selectivity to olefin.254
![[{"id":"lgrsY4lQyU","type":"paragraph","data":{"text":"Selective hydrogenation of alkynes catalyzed by core – shell Pd<sub>1</sub>Ag systems.<sup>253</sup>"}}]](/storage/images/resized/pQKufcDVmg8jZh6adFjiO8rfmaBEvEKa8AiuR2x9_xl.webp)
3.3.2.3. Catalytic properties of Pd1Au SAA catalysts
Analysis of the published data concerning SAA catalysts indicates that, apart from the PdCu and PdAg systems, the three catalysts that are most promising for selective hydrogenation include also Pd1Au single-atom alloys. As in the case of Pd1Ag, the presence of Au prevents hydrogenation of the target alkene and thus maintains high selectivity of the reaction. However, PdAu-based systems often show low activity at low temperatures and are rapidly deactivated under the reaction conditions.255 – 258
The studies addressing the use of Pd1Au SAA catalysts in the hydrogenation of alkynes are relatively few. Mention should be made of a series of publications by Friend and co-workers,259 – 261 who investigated the properties of dilute Pd1Au alloys in the gas-phase hydrogenation of 1-hexyne. It was shown that in the presence of the Pd1Au/SiO2 catalysts, when 1-hexyne conversion is 80%, the selectivity to 1-hexene is > 90%. It was noted that catalysts remain stable after more than 30 h of operation at 90 °C. Transmission electron microscopy examination of the catalysts did not reveal changes in the average particle size, which attests to stability to sintering. Also, the structure of the Pd1Au/SiO2 catalysts is sensitive to the preactivation conditions. For example, as the oxidative treatment temperature increases from 50 to 400 °C, the 1-hexyne conversion on Pd1Au/SiO2 increases from ~ 20 to ~ 30%, while the selectivity to 1-hexene is maintained at ~ 96%. According to EXAFS data, an increase in the activity is caused by Pd stabilization on the surface via the formation of the PdO bond.260 During the reductive treatment of Pd1Au/SiO2 with H2 , the palladium atoms are distributed throughout the nanoparticle bulk, which decreases the catalyst activity. Treatment of the catalyst with СО induces its partial reactivation even at room temperature, which is due to the adsorbate-induced Pd segregation on the surface. According to DFT calculations, high selectivity of Pd1Au/SiO2 is caused by the fact that the alkyne and alkene derivatives are bound more strongly to Pd(111) than to Pd/Au(111).259
In a previously mentioned publication,144 the catalytic testing of silica-supported Pd1Au nanoparticles in the selective acetylene hydrogenation in the presence of excess ethylene showed that a decrease in the Pd content is accompanied by a considerable increase in the selectivity to ethylene in comparison with the monometallic Pd catalyst. It was found that in the temperature range from 80 to 160 °C, the selectivity of the alloy catalyst markedly exceeds the selectivity of Pd/SiO2. The authors assumed that in the case of the Pd1Au catalyst, this is a result of the lower adsorption energy of ethylene on the nanoparticle surface. The isolation of Pd atoms with Au atoms leads to a considerable decrease in the number of multi-fold adsorption sites, which makes it possible to avoid the undesirable ethylene hydrogenation to ethane. The authors also found that a 40-fold excess of the inactive Au component is required for the efficient formation of single Pd1 active sites.
The necessary presence of an excess of gold in Pd1Au systems was also reported by Zhang et al.,121 who obtained a series of PdAu catalysts with the Au : Pd ratio ranging from 0.25 to 8. The catalysts were prepared by ion exchange and deposited on the surface of commercially available ion exchange resin. Physicochemical studies of the resulting samples by HRTEM and XRD confirmed the formation of bimetallic alloy nanoparticles. Using EXAFS and IR spectroscopy of adsorbed CO, the authors showed that an increase in the Au : Pd ratio is accompanied by a gradual change in the active site structure. As the Au : Pd ratio of ≥ 4 is reached, the Pdn adsorption sites (n > 2) virtually disappear, which attests to the predominant formation of isolated Pd1 sites. The catalytic performance of the samples was evaluated in relation to the Ullmann reaction. It was found that Au : Pd catalysts are highly efficient in the homocoupling of diverse aryl chlorides, aryl bromides and aryl iodides. As the Pd content in the alloy decreases, the turnover number (TON) sharply increases from 150 to 1150. The target products were obtained in a short time (1.5 – 6 h) and in good yields (73 – 99%). Catalyst stability studies demonstrated that this high performance is maintained over eight reaction –regeneration cycles.
3.3.3. Other reactions using SAA catalysts
Although the catalytic performance of single-atom alloy catalysts has been studied in most detail in relation to the selective hydrogenation of alkynes, SAA catalysts are also widely used in quite a few other reactions (see Table 3).
Efficient Pt1Cu and Pd1Cu single-atom catalysts were obtained for hydrogenation of butadiene, styrene and acetylene substrates and for dehydrogenation of acetic acid.119, 135, 138, 140 Apart from the selective hydrogenation, equally extensive studies of SAA catalysts were concerned with hydrogenolysis of biomass processing products.197, 198 Systems of various compositions showed excellent results in the conversion of 5-HMF to more valuable compounds such as furan derivatives and furan ring opening products, which are of considerable interest for the development of pharmaceuticals and biofuels and for the use in the polymer industry. It is noteworthy that the reaction pathway and the product yield for reactions involving 5-HMF are significantly affected by the composition of the selected catalyst. When 5-HMF is completely converted in the presence of Pd1Au SAA, the selectivity to 1-hydroxy-2,5-hexanedione exceeds 90%.197 Meanwhile, when the reaction is conducted with Pt1Co single-atom alloy catalyst, dimethylfuran is formed as the major product, with the selectivity being 90%.198
The Pt1Cu150 and Pd1Ga52 compositions have been studied in the dehydrogenation of n-propane and n-butane.171, 199 As the reference samples, monometallic Pt/Al2O3 systems and systems containing different amounts of platinum were used in the case of n-propane and the Pt/Al2O3 and Cr2O3/Al2O3 systems were used for n-butane. Single-atom alloy catalysts not only had high activity and selectivity in the considered reactions, but were also highly stable to catalytic poisons, in particular CO. In the catalytic dehydrogenation of n-propane catalyzed by the Pt1Cu150 SAA catalyst, the selectivity to propylene was ~ 90%, with the operation stability under the reaction conditions being more than 120 h.171 In the hydrogenation of n-butane, the highest selectivity to butene (> 86%) was attained for the Pd1Ga52 SAA catalyst, which provided 24 h of stable operation.199
Single-atom alloy catalysts are also used in cross-coupling reactions. In a study addressing the oxidative cross-coupling of methyl acrylate and methanol, Trimpalis et al.200 demonstrated that the use of Ni1Au200/SiO2 catalyst mainly results in the formation of methyl methacrylate, whereas the reaction catalyzed by the monometallic analogue produces methyl formate.200 Samples with different Pd contents in PdxAuy/resin were tested in the Ullmann reaction.121 The Pd/resin and Au/resin monometallic catalysts were used as reference samples. The optimal activity/selectivity balance (SC12H10 > 99% for XC6H5Cl = 100%) was found for the Pd1Au6/resin catalyst.
An interesting result was established for the Rh1Co and Pd1Cu SAA catalysts in the reduction of NO to N2. The selectivity of SAA catalysts was much higher than that of mono- or bimetallic samples with high Pd contents.154
For Pt1Ni SAA catalyst, the rate of borazane dehydrogenation was more than 20 times higher than that observed on Pt or Ni monometallic catalysts,201 and the use of Pd1Ni single-atom surface alloy in the hydrogenation of benzonitrile increased the selectivity to dibenzylamine from 5% to 97%, with the substrate conversion being 100%, in comparison with the conventional monometallic Pd/SiO2 and Pt/SiO2.203 Higher selectivity when compared with the monometallic analogue was also observed for the dechlorination of 1,2-dichloroethane catalyzed by Pd1Ag3/TiO2.204 In the glucose oxidation reaction, Pd1Au0.3 was 10 times as active as the monometallic Pd or Au catalysts and the Pd4/Au1 sample characterized by a higher Pd content.120, 205 A significant increase in the activity towards alkyne hydrosilylation was found for the Pd1Au10/SiO2 SAA catalyst in comparison with the monometallic Pd/SiO2 and Au/SiO2 catalysts and various bimetallic catalysts.207
Analysis of the published data summarized in Table 3 clearly demonstrates that the most convincing benefits of the use of single-atom alloy catalyst were obtained for structure-insensitive selective hydrogenation reactions that can proceed on a single active metal atom. A sharp increase in the amount of partial hydrogenation was independently established for this type of reactions by several research groups. The results allow considering SAA catalysts as catalytic systems that are not only of fundamental interest, but also have obvious prospects for the use in industrial processes. The range of reactions that can make use of single-atom catalysts is constantly expanding. Interesting results were obtained for hydrogenolysis, dehydrogenation, oxidation, dechlorination and some other reactions. In our opinion, these results should be treated with a certain degree of caution, although generally the trend towards the extension of the range of catalytic processes efficiently performed by single-atom alloy catalysts will undoubtedly reveal other promising areas for their use in laboratory and industrial practice.
It is noteworthy that in addition to experimental studies, quite a few recent publications describe the use of theoretical calculations for model SAA catalytic structures, including DFT calculations (Table 6). Using acetylene hydrogenation as an example, Zhang’s research group explained the higher selectivity of Pd1Cu SAA compared to Pd1Au SAA by higher electron density on the Pd atoms surrounded by Cu atoms.148 Meanwhile, Thirumalai et al.262 attributed the high ethylene selectivity of the Pd1Cu SAA catalyst to the rate of ethylene desorption from the catalyst surface. A similar conclusion was drawn by Yang and Yang,263 who attributed the selectivity of butadiene hydrogenation to butene to the desorption rate of the olefin intermediate.

Density functional theory modelling of the selective hydrogenation of acrolein to acetic acid demonstrated that acrolein is chemisorbed on the Pd1Ag SAA catalyst surface via С=С bond, which enables selective preparation of the saturated aldehyde.264 For dehydrogenation of hydrocarbons, DFT calculations revealed the causes for the absence of coke formation in the reaction catalyzed by SAA systems.141 In the analysis of methane dehydrogenation reaction, DFT was used to elucidate the possibility of activation of the СН4 molecule.265 The results may be useful for planning experiments on methane conversions reactions.
3.3.4. Fine tuning of the active site structure for SAA catalysts by adsorbate-induced segregation
A highly important parameter of bimetallic catalysts that has to be controlled regarding the catalytic performance is the composition of the surface, since particularly the surface atoms interact with the adsorbate molecules. The chemical composition and structure of active sites in bimetallic systems are initially determined by the ratio of metals during the catalyst synthesis. However, the component ratio on the surface of a bimetallic nanoparticle can differ substantially from its bulk composition.25, 268
One of the main factors inducing a pronounced change in the surface composition is the surface segregation of alloy components.269, 270 The driving force of the surface segregation is the tendency of a bimetallic system to lower its surface energy. The total energy of the system can be minimized by enriching the surface in the component that has a lower surface energy (lower sublimation energy).271 Finally, the surface composition of bimetallic nanoparticles can differ from the average bulk composition.272 For example, in the alloys of Group 8 and 11 metals, the surface is preferably filled by Group 11 atoms, which have lower sublimation energy.
The studies addressing the surface segregation are usually based on experiments (e.g., those using Auger spectroscopy) and theoretical calculations (DFT) that estimate the change in the composition of the surface layers of bimetallic nanoparticles relative to bulk composition as a result of high-temperature treatment. The investigation objects described in the literature include alloy catalysts based on AgAu,273 PtCu,273, 274 AgAl,275 NiAg,276 AgPb,277 RhPd, NiPd, RhAg (Ref. 278) and PdAu.279 For most of the alloys, it was found that at high temperatures, atoms of the component with a lower surface energy migrate to the nanoparticle surface and a near-surface layer.
For example, the Ag segregation on the surface of bimetallic Pd1Ag nanoparticles under vacuum is a known phenomenon, which is due to the substantial difference between the surface energies of two metals.280 – 284 In a fundamental publication,285 a pronounced Ag surface segregation effect was established for unsupported Pd1Ag catalysts obtained by co-precipitation. Later, using theoretical calculations for the surface structure, it was shown that at temperatures from 800 to 1000 K, the contents of Au and Ag atoms in the surface layers of bimetallic Pd1Au and Pd1Ag alloy nanoparticles are greater by more than 25% than their content in the bulk.279, 285 – 287 In the case of CuPd catalysts, this effect is somewhat less pronounced.
The situation becomes more complicated when the participants of the chemical reaction can be chemisorbed by the alloy and react with alloy components. The surface structure of a bimetallic nanoparticle can be modified upon the action of the reaction medium during the catalytic reaction.288 – 290 In this case, the component with higher gas affinity (higher adsorption energy) is exposed on the surface. This phenomenon, which was called adsorbate-induced segregation, can be used to control the surface structure, because it has a considerable effect on the surface composition and, hence, on the active site structure and catalytic properties. In addition, the nanoparticle structure can be tuned by treating the catalyst in a specially selected gas atmosphere.268, 291 – 293
Despite the active studies of the surface segregation phenomenon, the possibility of using the adsorbate-induced segregation to control the catalytic properties is virtually not discussed in the literature. Only in recent years, studies demonstrating the efficiency of this approach started to appear. For example, some publications devoted to the thermodynamic stability of bimetallic systems demonstrate that certain adsorbates (O2 , CO, C2H2) can directly influence their catalytic performance.294, 295 When carbon monoxide is hydrogenated in the presence of Pd1Cu catalysts, the surface concentration of palladium increases as a result of migration of Pd atoms to the surface induced by the adsorption of CO molecules. This results in a change in the process selectivity.295 According to IR spectroscopy data, the process is accompanied by a change in the carbon monoxide adsorption mode from linear (adsorption on one Pd atom) to bridge (adsorption on two neighbouring Pd atoms).
A study of Pt1Cu catalysts gave somewhat contradictory results about the possibility of surface segregation of the less active component of a bimetallic alloy.296 The authors found that CO desorption at elevated pressure and temperature is accompanied by enrichment of the surface layer of bimetallic nanoparticles in atoms of less active component (Cu). However, owing to the high Pt – CO bond energy, the segregation of the Cu atoms from the near-surface layer gives rise to the stable highly ordered CO/CuPt structure with isolated Pt1 active sites (Fig. 17). Apparently, the Cu segregation is due to the fact that CO adsorption on the Pt1Cu6 structure is thermodynamically more favourable than СО adsorption on a three-fold Pt site.
![[{"id":"d-ysmaj6C-","type":"paragraph","data":{"text":"Surface segregation on Pt<sub>1</sub>Cu alloys induced by CO adsorption.<sup>296</sup>"}}]](/storage/images/resized/T77pesH4itrkmfKBaojZSK2vqPbFCBxhtDYt3Qdu_xl.webp)
The possibility of controlling the performance of Pd1Cu/Al2O3 SAA catalysts for the selective acetylene hydrogenation was demonstrated by McCue and Anderson.268 Using IR spectroscopy of adsorbed CO, they found that treatment of Pd1Cu catalysts in a CO flow increases the fraction of the isolated surface Pd atoms from 4% to 5.3% and gives rise to 5.7% Pd2 dimers. This is accompanied by increasing activity of the synthesized samples, as indicated by a decrease in the temperature at which the complete conversion of the substrate is reached. The increase in the activity is accompanied by some decrease in the selectivity to ethylene, which was attributed to the formation of dimeric Pd2 sites.
The ways to control the surface structure of the bimetallic Pd1Ag catalysts have also been comprehensively investigated. Van Spronsen et al.297 carried out experiments using the model Pd1Ag system prepared by deposition of small amounts of palladium on the Ag surface. The authors found that the presence of CO or the treatment of the catalyst with oxygen leads to enrichment of the nanoparticle surface in palladium. The palladium segregation was experimentally observed at room temperature and no longer took place as the temperature subsequently decreased. This effect is attributable to the low mobility and slow diffusion of Pd atoms at low temperatures. Theoretical calculations indicate that the surface segregation of Pd is caused by the high CO to Pd binding energy. In turn, treatment with O2 at temperatures of 300 – 400 K gives rise to a surface layer of Pd oxide.
It is of interest that the surface structure formed upon the adsorbate-induced segregation remains stable even at a low CO pressure (10–6 bar).297, 298 Similar data on the effect of adsorption of various molecules (O2 , H2 , CO) on the Pd1Ag nanoparticle surface were obtained for samples synthesized by impregnation 299 and by electron-beam deposition of the metal components.300 The IR spectroscopy data for adsorbed CO also confirmed the formation of isolated active sites as a result of the surface segregation of silver atoms.
The adsorbate-induced surface segregation stimulated by adsorption of CO or O2 was successfully utilized for targeted control of the catalytic performance of Pd1Ag 301 – 304 and Pd1In catalysts 304 – 306 prepared by the traditional incipient wetness impregnation. The variation of the surface structure of bimetallic nanoparticles was monitored by IR spectroscopy of adsorbed CO and X-ray photoelectron spectroscopy (XPS), while the catalytic properties after adsorbate-induced segregation were studied in relation to the model liquid-phase hydrogenation of diphenylacetylene and gas-phase hydrogenation of acetylene impurities in ethylene as examples. This study demonstrated that treatment of Pd1Ag catalysts in a CO atmosphere at 200 °C increases the number of Pd1 sites on the surface of PdAg nanoparticles and gives rise to Pd2 structures composed of two closely located palladium atoms. The results of testing of the catalysts in the model liquid-phase hydrogenation of diphenylacetylene provided the conclusion that the treatment in a CO atmosphere increases catalytic activity, while the selectivity to stilbene (partial hydrogenation product) changes insignificantly in comparison with the untreated sample.
The treatment of catalysts based on Pd1In nanoparticles in a СО atmosphere, unlike the treatment of the Pd1Ag or Pd1Cu catalysts, has no marked effect on their surface structure or catalytic performance. Targeted modification of the surface structure of Pd1In catalysts can be attained by their oxidative treatment in O2 atmosphere. The results thus obtained indicate a significant surface modification accompanied by a change in the electronic state of supported Pd. This is caused by the fact that in the case of Pd1In systems, oxygen treatment leads to the selective oxidation of the more oxophilic In component, resulting in the formation of indium oxide. This ‘corrosive chemisorption’ markedly decreases the content of In metal in the bimetallic nanoparticles. This leads to intense formation of both dimeric Pd2 sites and multi-fold Pdn sites of greater nuclearity (n > 2). Finally, the activity may increase by a factor of 10 to 20; however, the selectivity simultaneously decreases; therefore, to attain the optimal activity/selectivity balance, fine tuning of the surface structure is required.
The processes that occur on the surface of Pd1In nanoparticles during their oxidative treatment were investigated in detail using model samples obtained by vacuum deposition of Pd and In components on the surface of highly oriented pyrolytic graphite.307, 308 The oxidation has a pronounced effect on the surface structure of Pd1In nanoparticles, since indium is selectively oxidized to InOx , due to its high oxophilicity, which gives rise to In oxide nanoparticles on the Pd1In alloy surface. It is noteworthy that this process is reversible, and the Pd1In structure can be easily restored by treating the catalyst at 500 °C in a hydrogen flow. The authors also demonstrated that cycles of the redox treatment of Pd1In nanoparticles generate a more uniform distribution of metal atoms over the bimetallic nanoparticles. It was noted that the synthesized Pd1In catalysts are stable to sintering both under ultrahigh vacuum (up to 500 °C) and under real experimental conditions (up to 100 °C; O2 pressure of 200 millibar).
Generally, the currently available data on the influence of surface segregation and adsorbate-induced effects caused by adsorption of various gases on the surface composition of bimetallic alloys provides the conclusion that this approach is promising for the control over the active site structure and catalytic performance of bimetallic catalysts.309, 310
4. Conclusion
Analysis of the published data demonstrates that the single-atom catalyst methodology is a breakthrough in modern heterogeneous catalysis. A stable structure of single-atom active sites can be generated on the surface of bimetallic nanoparticles by isolation of the active metal atoms with atoms of the metal that is inert or less active in the catalytic reaction. An important benefit of single-atom alloy catalysts is the possibility of their regeneration and reuse owing to the thermodynamic stability of the isolated active site structure on the surface of bimetallic nanoparticles. High selectivity and easy regeneration implies the possibility of extensive use of single-atom and single-atom alloy catalysts both in the laboratory practice and in industrial catalytic processes.10, 15
Nevertheless, despite the considerable progress in the synthesis and studies of single-atom catalysts, there is still a number of aspects that require further development. For example, the possibility of following the evolution of the active sites of single-atom and single-atom alloy catalysts using physical methods in operando mode has not yet been adequately investigated. The most advanced electron microscopy techniques can be used to monitor the structural changes in metal catalysts directly during the reaction; however, the resolution of the method is still insufficient to observe single atoms. This problem can be partly solved by integrated use of modern spectral methods such as EXAFS, XPS, UV-Vis and FTIR, which enables in situ or operando monitoring of the variation of the catalyst surface structure and drawing correlations with the catalytic performance.311 Determination of structural characteristics of sub-nanometre particles or isolated atoms, especially when a chemical reaction takes place, is still a challenging engineering problem. A promising approach is to use highly sensitive X-ray diffraction methods such as high-energy resolution fluorescence-detected X-ray absorption near-edge structure (HERFD-XANES); however, the application of such methods is still rare due to complexity and high cost of the equipment.169
The knowledge of transformations of the catalytic sites during a chemical reaction and causes for deactivation of single-atom catalysts is also important for the development of effective regeneration processes. It is believed that the regeneration efficiency can be increased by using the fundamental data on the modification of the catalyst active sites during the reaction. The design of a model for predicting the changes in isolated atoms taking account of the metal – support interaction and reaction conditions remains an open question.311
Since single-atom catalysts are prone to sintering, the development of research aimed at their stabilization and preventing their agglomeration is expected, e.g., the development of new methods for the synthesis of catalysts and modification of the support surface to maximize the metal – support interaction. One more promising trend is to elaborate approaches for decreasing the duration of high-temperature treatment during the synthesis of single-atom catalytic systems.312 In addition, an important objective is to obtain new highly active single-atom catalysts based on non-noble metals.169, 312
An obvious trend of development for the class of single-atom alloy catalysts is to increase the number of known metal combinations and reactions in which they can be used (in particular, selective oxidation reactions). In addition, the problem of sharp decrease in the SAA catalyst activity for highly dilute solid solutions needs to be addressed. The development and application of computational methods both for the prediction of the most active metal combinations and for the design of multicomponent systems consisting of more than two metals appears to be a promising approach. In addition, to overcome the ‘material gap’ between model systems under high vacuum and the behaviour of real systems under reaction conditions, a current trend is to carry out studies with model samples at a high pressure.15 The results provide answers to the questions about the state of the components of a single-atom alloy catalyst, their role and transformations during the reaction.
One more fairly rapidly developing trend is related to the application of the adsorbate-induced segregation to control the surface composition and structure of the bimetallic particles of single-atom alloy catalysts in order to optimize the catalytic performance. In some studies, it was established experimentally that this makes it possible to tune the selectivity and activity of catalysts for various processes. The effects attained in this way are persistent and are retained under the catalytic reaction conditions, which attests to stability of the surface structures. In this regard, it is obvious that the adsorbate-induced segregation effects need to be systematically studied in more detail by modern physicochemical methods to develop a procedure for the control of the active site structure of bimetallic catalysts at the atomic level.
The situation in the world economy, the growing demands of industry, the sanctions imposed by a number of countries and, as a consequence, the need to create new catalysts for import substitution have initiated a new stage of development and design of modern catalytic systems for various chemical processes. The increase in the catalyst activity and selectivity and more efficient use of catalysts is a key trend in the technology development for the next decade. The application of single-atom catalysts would markedly accelerate the research carried out along this line and would make it posible to meet the challenges.
We are gratefully acknowledge funding for this work from the Russian Science Foundation Grant No. 23-13-00301, https://rscf.ru/project/23-13-00301/
5. List of abbreviations and symbols
ALD — atomic layer deposition,
C.N. — coordination number,
DFT — density functional theory,
Eads — adsorption energy,
Edes — desorption energy
EDX — energy dispersive X-ray spectroscopy,
EELS — electron energy loss spectroscopy,
EXAFS — extended X-ray absorption fine structure,
FCC — face-centred cubic crystal lattice,
HAADF — high-angle annular dark field,
HAADF-STEM — high-angle annular dark field scanning transmission electron microscopy,
HREELS — high-resolution electron energy loss spectroscopy,
HRSTEM — high-resolution scanning transmission electron microscopy,
HRTEM — high-resolution transmission electron microscopy,
Kads — adsorption constant,
PZC — point of zero charge,
S — selectivity to the target product (%).
SAA — single-atom alloy,
SAC — single-atom catalysts,
SEM — scanning electron microscopy,
SSHC — single-site heterogeneous catalysts,
STEM — scanning transmission electron microscopy,
STM — scanning tunnelling microscopy,
TEM — transmission electron microscopy,
TOF — turnover frequency,
TON — turnover number,
WGS — water-gas shift,
X — substrate conversion (%),
XANES — X-ray absorption near edge structure,
XRD — powder X-ray diffraction.