




Keywords
Abstract
The experimental and theoretical chemistry of 1,2,3,4-tetrazines have been actively studied in the last 20 years. The increasing interest in this class of compounds is due to the fact that the 1,2,3,4-tetrazine ring is a promising building block for the development of energetic and physiologically active compounds. The review considers various types of 1,2,3,4-tetrazines including completely unsaturated nonannulated tetrazines and their N-oxides; nitrogen-substituted tetrazines; annulated tetrazines with a nitrogen atom common to two heterocycles; 1,2,3,4-tetrazine 1,3-dioxides fused to benzene, pyridine, 1,2,3-triazole or 1,2,5-oxadiazole ring; and also 1,2,3,4-tetrazine 1,3-dioxide fused to one more 1,2,3,4-tetrazine 1,3-dioxide ring. Methods of synthesis and reactivity of these compounds, their crystallographic features, spectral characteristics and thermal stability are described. The results of quantum chemical studies for 1,2,3,4-tetrazine derivatives are presented. The prospects of using 1,2,3,4-tetrazines as energetic compounds are discussed.
The bibliography includes 189 references.
1. Introduction
The chemistry of 1,2,3,4-tetrazines is a relatively new area of the heterocyclic chemistry, which has been successfully developing in recent decades. Biologically active representatives, in particular, a new class of nitric oxide (NO) donors, have been found among these compounds. However, the primary feature responsible for high interest in these derivatives is that the four nitrogen atoms linked to one another account for the high energy content of molecules, which is important for the development of explosives and components of solid composite propellants. The 1,2,3,4-tetrazine skeleton serves as an attractive base for the design of these molecules. In addition, 1,2,3,4-tetrazines are of considerable interest for theoretical chemistry in relation to the issue of heterocycle aromaticity.
The review considers derivatives of non-annulated 1,2,3,4-tetrazine (1)* and its annulated analogues, including completely unsaturated and partly reduced compounds. 1,2,3,4-Tetrazine N-oxides are also considered. This publication continues the previous review [1] covering the period of 1994 – 2003 and includes publications up to the beginning of 2023. New methods of synthesis, reactivity, and physicochemical characteristics of 1,2,3,4-tetrazines are discussed.

1,2,3,4-Tetrazines are the subjects of some chapters in the periodic edition Comprehensive Heterocyclic Chemistry III (2008) and Comprehensive Heterocyclic Chemistry IV (2021).2, [2] Energetic 1,2,3,4-tetrazines are considered in a special part of a monograph.[3] The synthesis of 1,2,3,4-tetrazine N-oxides is discussed in reviews,5 – 8 in a survey paper 9 devoted to fused heterocyclic energetic compounds and covering publications of 2012 – 2019, and in a review.10
* Completely unsaturated non-annulated 1,2,3,4-tetrazine 1 has not been synthesized to date.
2. Synthesis and reactivity
2.1. N-Substituted 1,2,3,4-tetrazines
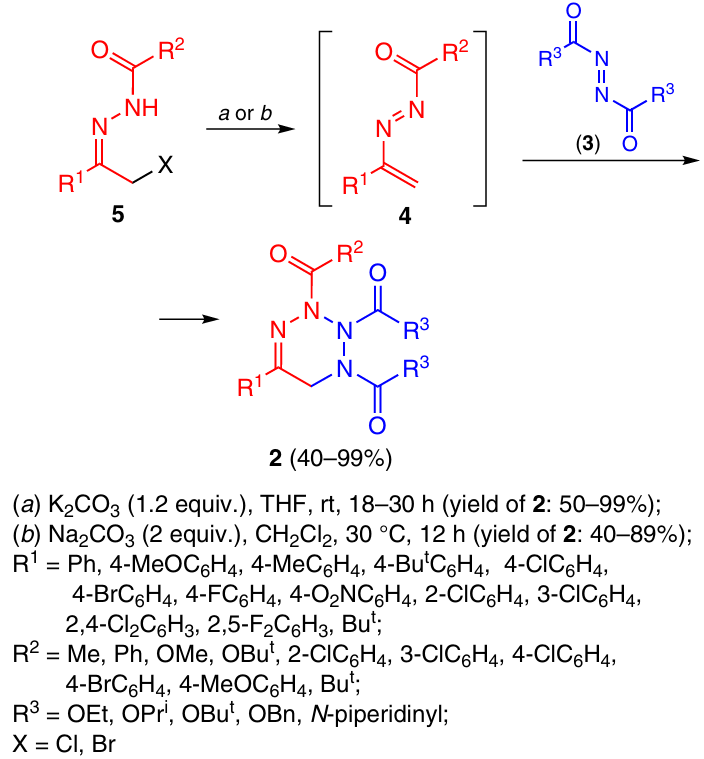
The synthesis of 1,2,3,6-tetrahydro-1,2,3,4-tetrazines 2 includes the [4+2]-cycloaddition reaction of azodicarboxylic acid derivatives (3) with 1,2-diazabuta-1,3-dienes 4, which are generated in situ from α-halohydrazones 5 under the action of bases (Scheme 1).11, 12 The reaction gives products in good yields (40 – 99%), it is easily scalable and makes it possible to vary substituents in the target tetrazines over wide limits.
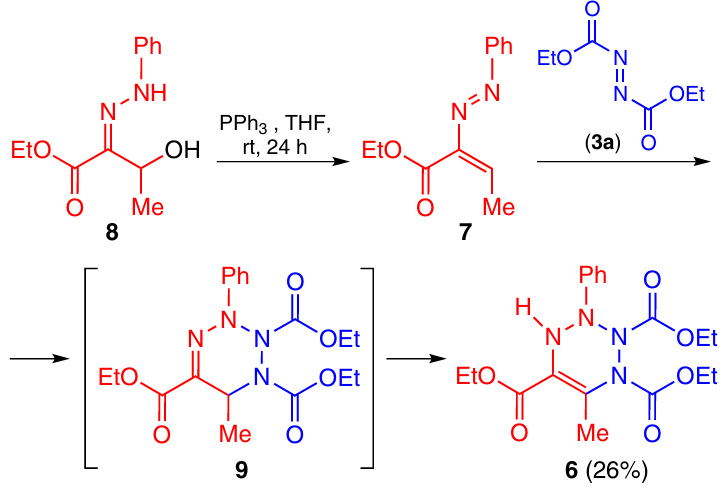
The method of synthesis of 1,2,3,4-tetrahydro-1,2,3,4-tetrazine 6 differs by the fact that 1,2-diazabuta-1,3-diene 7 is generated from hydrazone 8 on treatment with triphenylphosphine at room temperature (rt) (Scheme 2).13 The intermediate tetrazine 9 isomerizes during the reaction to give product 6.
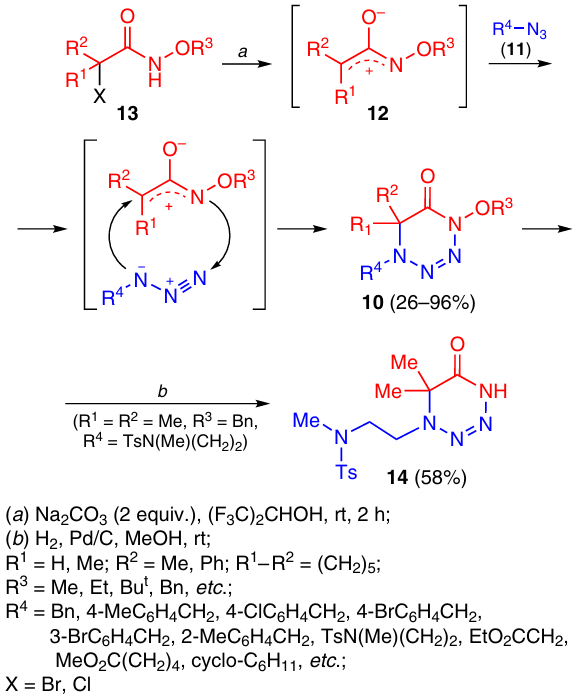
A new synthetic route to 1,2,3,4-tetrazines 10 is based on the [3+3]-cycloaddition of azides 11 to azaoxyallylic intermediates 12, which are generated in situ from the corresponding α-halohydroxamides 13 under the action of potassium carbonate in hexafluoroisopropyl alcohol (Scheme 3).14 Compounds 10 represent a new type of 1,2,3,4-tetrazines with an alkoxy substituent at the tetrazine nitrogen atom. The reaction proceeds under mild conditions and gives products containing pharmacophore groups and fragments of natural compounds such as vitamin E, cholesterol, lithocholic acid, celecoxib and zidovudine (AZT). The catalytic hydrogenation of tetrazine 10a [R1 = R2 = Me, R3 = Bn, R4 = (CH2)2N(Me)Ts, where Ts is p-toluenesulfonyl (tosyl)] is accompanied by cleavage of the N – O bond yieldng tetrazine 14.
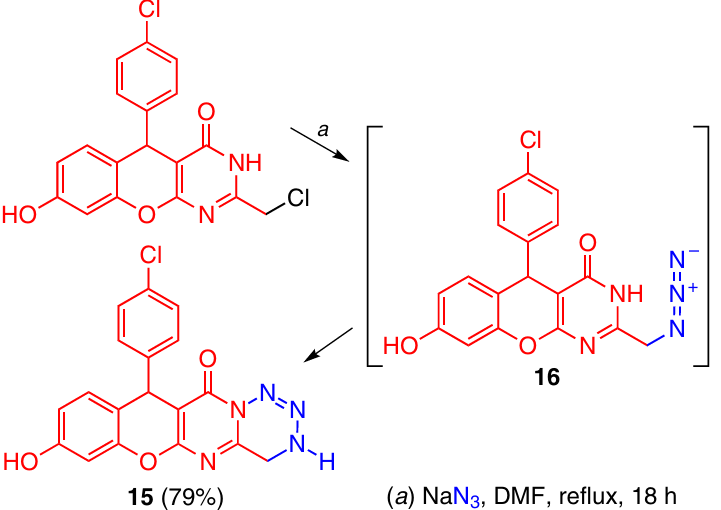
One more approach to the formation of a tetrazine ring was demonstrated in relation to the synthesis of tetrazine 15. The synthesis is based on the intramolecular reaction of the azide group with the pyrimidinone nitrogen atom in azide derivative 16. Compound 16 is prepared by the reaction of the corresponding chloro derivative with NaN3 (Scheme 4).15
A series of substituted 5-aryl-3-methylpyrazolotetrazines 18 were synthesized by cyclization of diazo compounds obtained by diazotization of arylazopyrazoles 17 with sodium nitrite in a mixture of sulfuric and acetic acids followed by treatment of the intermediate diazonium salts with sodium acetate (Scheme 5).16 Compounds 18 are fairly stable: their melting points (Tm) vary from 145 to 228 °C, depending on the nature of substituents in the benzene ring. Note that tetrazines of this type do not always exist in the cyclic form, some of them occur in the tetrazine ⇋ azo(diazo) compound equilibrium.1

Heteroannulated tetrazine perchlorates 19 and 20 were obtained by diazotization of aminopyrazole 21 and aminothiophene 22 (Scheme 6). The cyclic structure of the tetrazinium salt 19 was confirmed by X-ray diffraction analysis.17
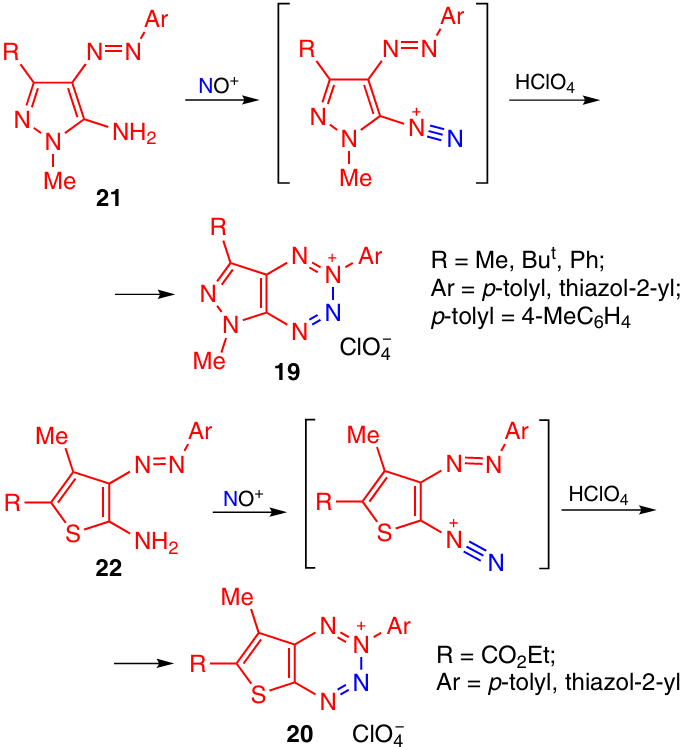
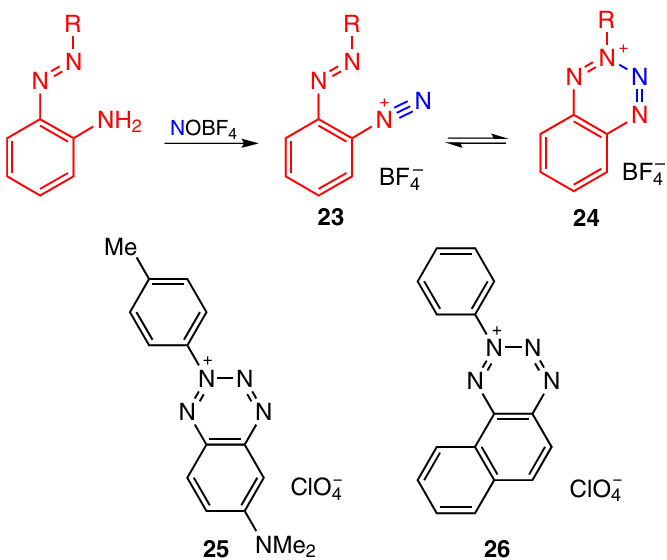
Diazonium salts 23 prepared by diazotization of the corresponding amines may exist in equilibrium with the closed-ring tetrazinium form 24 (Scheme 7). The position of equilibrium depends on the nature of the substituent R. In the case where R = Ph, the compound exists in the open form, while in the presence of electron-donating alkyl substituents, there is equilibrium in which the closed-ring form prevails (for example, for R = Me the tautomer ratio 23 : 24 is 3 : 7 according to NMR spectroscopy data).1 In 2018, it was shown that the electron-donating substituents in the benzene ring (such as a dimethylamino group) completely shift the equilibrium towards closed-ring isomer 25. Compound 26 also exists as the cyclic form.18
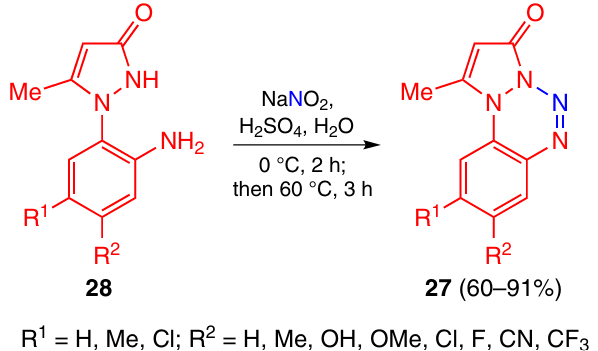
New substituted pyrazolobenzotetrazin-3-ones 27 were synthesized in good yields 19 – 21 using a previously developed procedure,22 based on the diazotization of 1-(2-aminophenyl)pyrazol-3-ones 28 (Scheme 8). Tetrazines 27 are also fairly stable (Tm = 143 – 238 °C, depending on the substituents in the benzene ring).
2.2. Annulated 1,2,3,4-tetrazines with a nitrogen atom common to two heterocycles
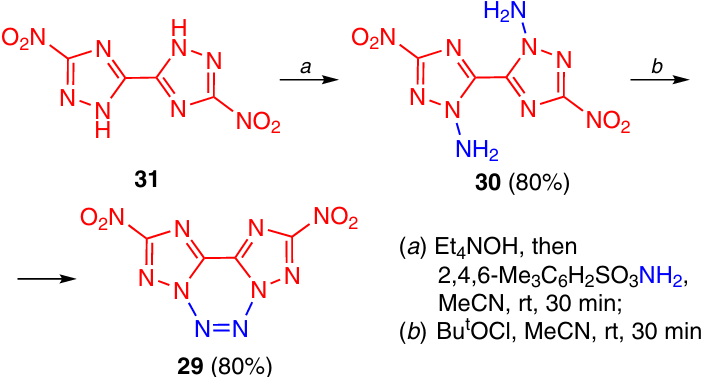
Tetrazine 29 with Tm = 138 °C (dec.) with fused to two nitrotriazole rings was formed in a high yield when diamine 30 was allowed to react with tert-butyl hypochlorite (Scheme 9).23 O-Mesitylenesulfonylhydroxylamine was used for amination of bis-triazole 31.
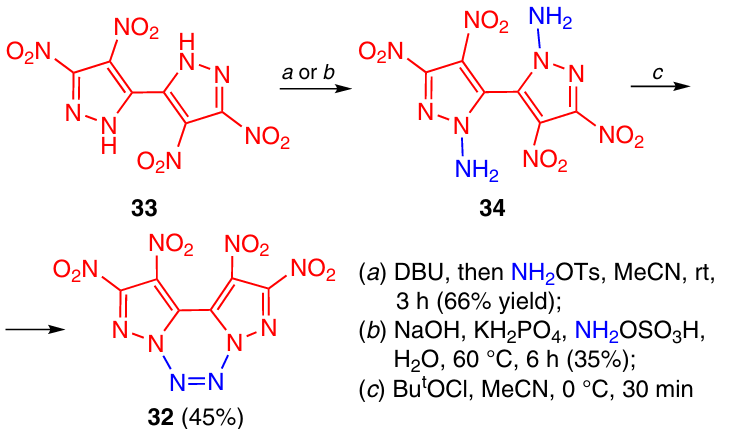
A similar synthetic route was utilized to prepare tetrazine 32 with two dinitropyrazole rings fused to the tetrazine ring (Scheme 10).24 The amination of bis-pyrazole 33 with O-tosylhydroxylamine (in the presence of 1,8-diazabicyclo-[5.4.0]undec-7-ene, DBU) or with hydroxylamine-O-sulfonic acid resulted in the formation of diamine 34, which was then oxidized into tetrazine 32 by the reaction with tert-butyl hypochlorite in MeCN. This tetrazine proved to be thermally stable: Tm = 205 °C, the onset decomposition temperature (Tonset) amounted to 233 °C.
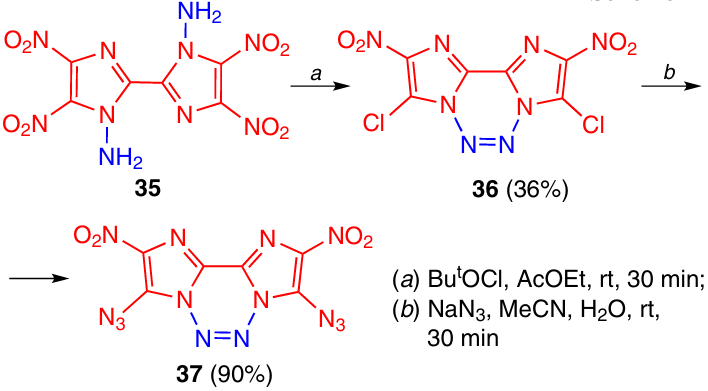
The oxidation of aminated bis-imidazole 35 with tert-butyl hypochlorite involved not only N=N bond formation, but was also accompanied by nucleophilic substitution of the nitro group in position 5 substituted with the chloride ion. This gave thermally stable dichloro derivative 36 (Tm = 215 °C) (Scheme 11).25 When this compound reacted with NaN3 in a mixture of MeCN and H2O, the chlorine atoms was easily replaced with azido groups, thus yielding tetrazine 37, which started to decompose at temperatures above >111 °C.
2.3. Completely unsaturated 1,2,3,4-tetrazines
2.3.1. Annulated 1,2,3,4-tetrazines
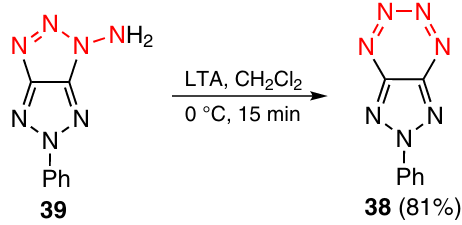
The first completely unsaturated annulated tetrazine 38 was synthesized for the first time by Ohsawa and co-workers* in 1988. The key step of the synthesis was expansion of the 1,2,3-triazole ring in compound 39 via the oxidation with lead tetraacetate (LTA) (Scheme 12).1 Tetrazine 38 proved to have low thermal stability and slowly decomposed at room temperature. Subsequently this class of compounds was not investigated.
* T.Kaihoh, T.Itoh, K.Yamaguchi, A.Ohsawa. J. Chem. Soc., Chem. Commun., 1608 (1988); https://doi.org/10.1039/C39880001608
2.3.2. Non-annulated 1,2,3,4-tetrazine 1-oxides
Benzoannulated tetrazine 1-oxides were considered in our previous review,1 and no studies devoted to compounds of this class have appeared since 2004.
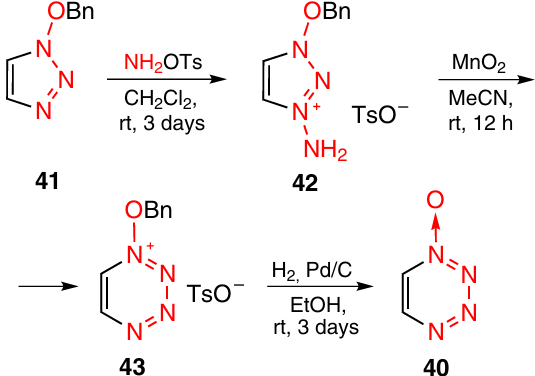
The first non-annulated 1,2,3,4-tetrazine 1-oxide 40 was obtained from 1-(benzyloxy)-1,2,3-triazole 41 (Scheme 13) in three steps.26 Initially, this triazole was aminated with O-tosyl hydroxylamine, then the resulting tosylate 42 was oxidized with MnO2 to give salt 43. The reaction mechanism is apparently similar to the mechanism of formation of tetrazine 38 (see Scheme 12) and includes a ring expansion step. The Pd/C-catalyzed reduction of salt 43 with hydrogen furnished tetrazine 1-oxide 40, which was characterized by electron impact mass spectrum and by 1H and 13C NMR spectra. The thermal stability of this tetrazine was not studied.
2.3.3. 1,2,3,4-Tetrazine 1,3-dioxides
The general approach to the formation of 1,2,3,4-tetrazine 1,3-dioxide (TDO) ring is based on the intramolecular reaction of the tert-butyl-NNO-azoxy group with oxodiazonium ion [R–N=N=O]+ followed by elimination of the tert-butyl cation (Scheme 14). The existence of the oxodiazonium ion as an intermediate species was confirmed by experiments in which this species was intramolecularly trapped by the benzene ring to give cinnoline N-oxide derivatives.27

Methods for TDO synthesis differ from one another by the way of generation of oxodiazonium ions. The first tetrazine 1,3-dioxides were prepared by the nitration method (Scheme 15), which consists in the reaction of primary nitramines, usually generated in situ, with nitrating agents (NO2BF4, N2O5).1 Recently, nitronium sulfates were used as nitrating agents.28
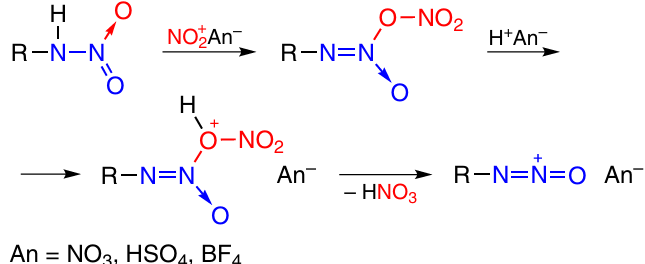
In recent years, new methods for the generation of oxodiazonium ion have been developed. The acetylation method includes the intermediate formation of O-acetylated nitramines, which dissociate in acid media, giving rise to the [R–N=N=O]+ ion (Scheme 16).29, 30 Nitramine is usually formed in situ. This method is suitable for the preparation of annulated 1,2,3,4-tetrazine 1,3-dioxides. The starting compounds used in these reactions are aromatic and heteroaromatic compounds (benzenes, 1,2,5-oxadiazoles, 1,2,3-triazoles, 1,2,3,4-tetrazine 1,3-dioxide) containing amino- and tert-butyl-NNO-azoxy groups in neighbouring positions (see Sections 2.3.3.2, 2.3.3.3).
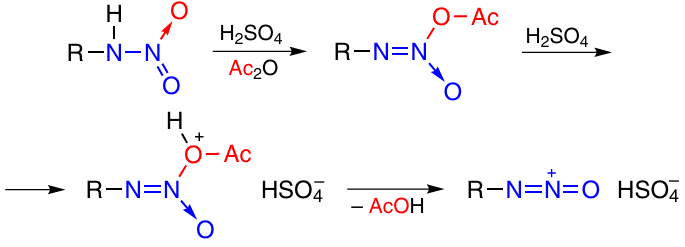
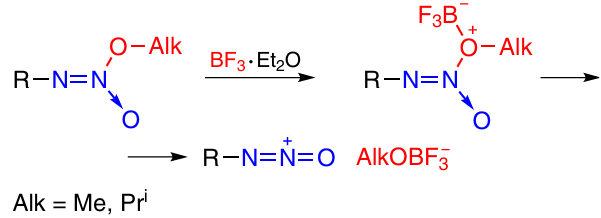
One more method for the generation of the oxodiazonium ion, O-alkylation method, includes the reaction of O-alkylated nitramines with boron trifluoride etherate or with acids (Scheme 17). This method is suitable for the synthesis of both annulated 31 and non-annulated tetrazine 1,3-dioxides (see Section 2.3.3.1.1).
2.3.3.1. Non-annulated 1,2,3,4-tetrazine 1,3-dioxides
2.3.3.1.1. Synthesis from aliphatic precursors
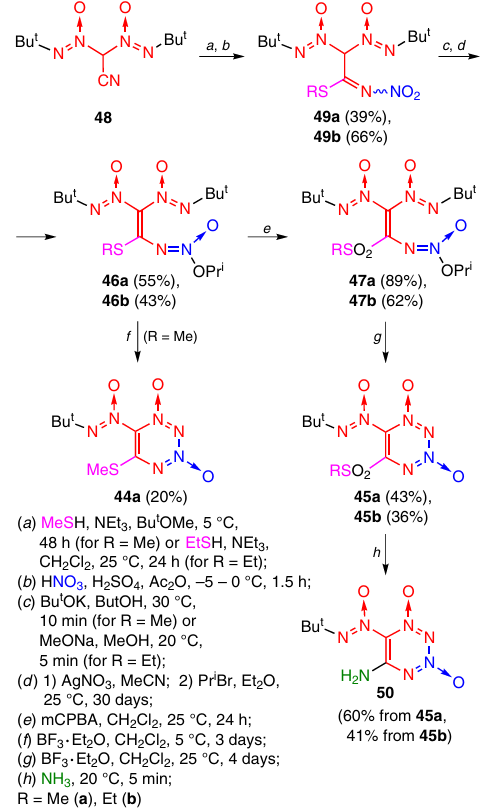
The approach to the synthesis of non-annulated TDOs from aliphatic precursors is based on the intramolecular reaction of alkenes containing a tert-butyl-NNO-azoxy group and an O-alkylated N-(nitro)amino group in neighbouring positions (Scheme 18). The cyclization is induced by boron trifluoride etherate. The plausible reaction mechanism is shown above in Scheme 17. Non-annulated TDOs 44a and 45a,b were obtained from azoxyalkenes 46a and 47a,b, respectively.32, 33 The azoxyalkenes, in turn, were prepared by reacting bis(tert-butyl-NNO-azoxy)acetonitrile (48) with alkanethiols and subsequent nitration and O-alkylation of the intermediate N-nitroimines 49a,b. The oxidation of compounds 46a,b afforded sulfones 47a,b. The best yields of TDOs were attained when sulfones with electron-withdrawing alkylsulfonyl substituents were used. The relatively low yield of product 44a (20%) can be attributed to the fact that in this case, the intermediate oxodiazonium cation is fairly unstable and easily eliminates N2O to give a vinyl cation stabilized by a methanethiol group. The alkylsulfonyl groups at the C(5) atom in TDOs 45a,b are readily substituted with ammonia to give product 50. The replacement of the methanethiol group by ammonia in compound 44a also affords TDO 50, but in a lower yield (18 – 20%).
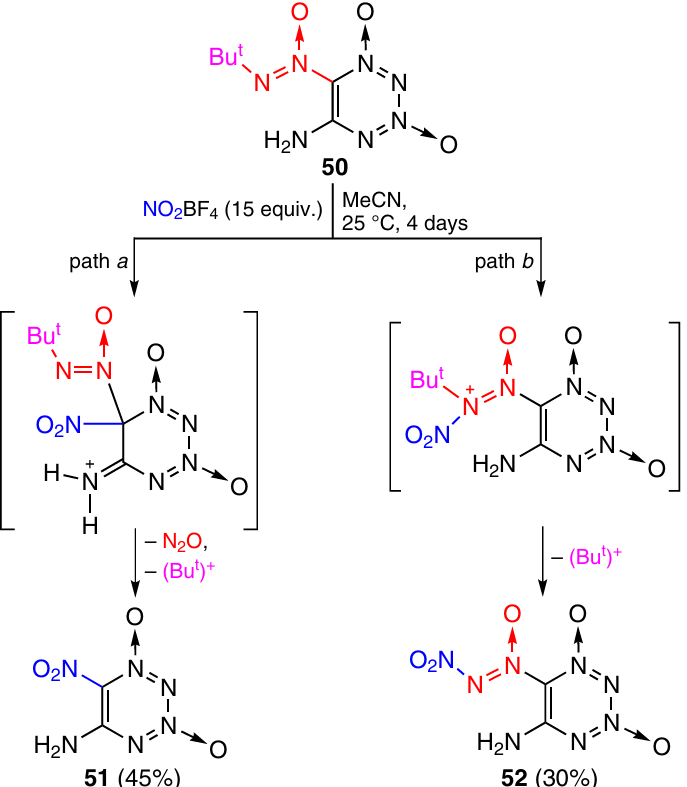
The reaction of 5-amino-6-(tert-butyl-NNO-azoxy)tetrazine 1,3-dioxide 50 with an excess of NO2BF4 in MeCN gives rise to 5-amino-6-nitro derivative 51 in 45% yield [Tm = 195 – 200 °C (dec.)]34 and TDO 52 in 30% yield [Tm = 110 – 112 °C (dec.)] (Scheme 19).35 Apparently, product 51 is formed by the electrophilic substitution mechanism of the tert-butyl-NNO-azoxy group by the nitro group (path a), while compound 52 is produced by the electrophilic substitutive N-nitration (path b).
2.3.3.1.2. Synthesis from benzotetrazine 1,3-dioxides
An alternative approach to the synthesis of non-annulated TDOs is benzene ring opening in benzo-1,2,3,4-tetrazine 1,3-dioxides (BTDOs), which can be accomplished in two ways.
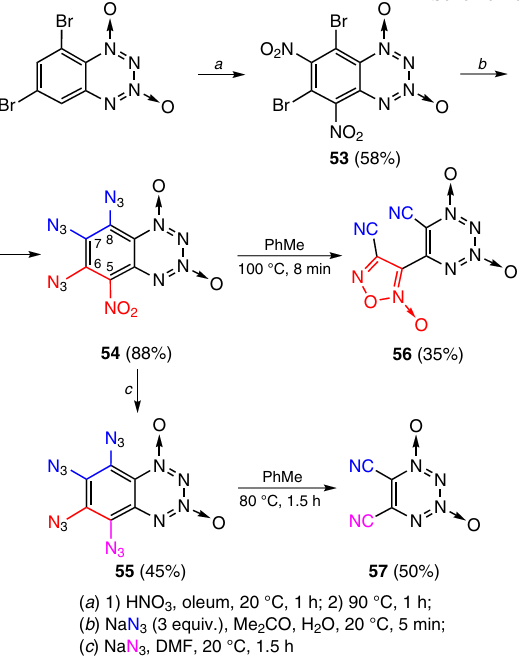
The first one is thermolysis of polyazido-substituted BTDOs. The latter were synthesized from 6,8-dibromo-5,7-dinitro-BTDO 53, which was obtained by nitration of 6,8-dibromo-BTDO (Scheme 20). The reaction of compound 53 with NaN3 led to tri- and tetra-azido-BTDOs (54 and 55) depending on the reaction conditions.36 Heating of a toluene solution of triazidonitro-BTDO 54 at 100 °C for a few minutes resulted in benzene ring opening to give non-annulated TDO 56 [Tm = 165 – 167 °C (dec.)]. 5,6-Dicyanotetrazine dioxide 57 [Tm = 110 – 130 °C (dec.)] was obtained by heating tetraazido-BTDO 55 in toluene at 80 °C for 1.5 h.
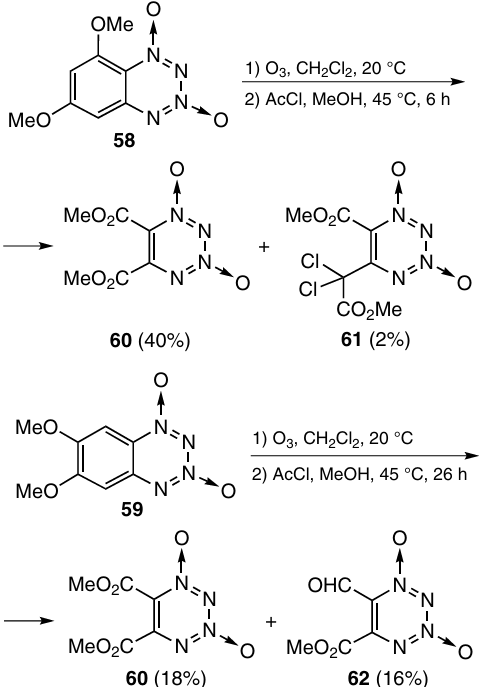
The second way for benzene ring opening is ozonolysis of BTDOs containing electron-donating substituents.37 Ozonolysis of dimethoxy derivatives 58 and 59 in CH2Cl2 with subsequent treatment of the primary ozonides with AcCl in MeOH resulted in 5,6-di(methoxycarbonyl)tetrazine dioxide 60 (Tm = 93 – 96 °C, decomposition temperature (Tdec) > 190 °C) as the major product (Scheme 21). Compounds 61 [Tm = 156 – 158 °C (dec.)] and 62 were formed in these reactions in lower amounts.
2.3.3.2. Benzoannulated 1,2,3,4-tetrazine 1,3-dioxides
2.3.3.2.1. Benzo-1,2,3,4-tetrazine 1,3-dioxides with functional groups. ‘Billiard’ reaction
Unsubstituted benzotetrazine 1,3-dioxide and some nitro- and bromo-substituted BTDOs are prepared by closure of a TDO ring in benzenes containing amino- and tert-butyl-NNO-azoxy groups in neighbouring positions (Scheme 22). The cyclization is accomplished via the nitration or acetylation method (most often, the intermediate nitramines are not isolated) (see Scheme 15 and 16) or by the O-alkylation method (see Scheme 17). The benzo derivatives thus formed can be introduced into electrophilic substitution reactions. In relation to nitration and bromination, it was shown 38 that in terms of the ease of these reactions, the positions in the benzene ring vary as follows: 5 ~ 7 > 8 > 6. The successive use of cyclization and nitration reactions made it possible to develop a one-pot method for the synthesis of 5,7-dinitro-BTDO from 2-(tert-butyl-NNO-azoxy)aniline (45% yield).39
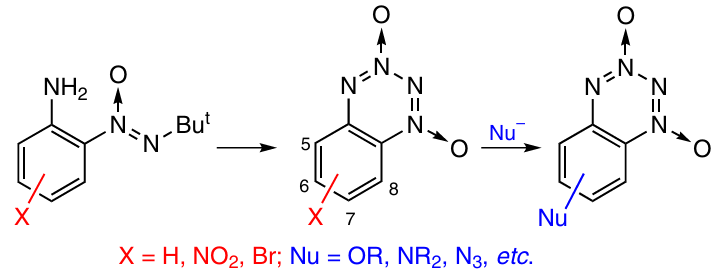
The brominated and nitro-substituted BTDOs easily undergo nucleophilic substitution reactions, in which the tetrazine ring remains intact (see Scheme 22). In relation to the reactions of mono- and di-bromo-substituted BTDOs with the methoxide anion, it was shown 40 that in terms of the ease of nucleophilic substitution, positions in the benzene ring are arranged as 6 > 8 > 7 > 5.
The unusual nucleophilic substitution with 1,2,3-triazole resulted in the discovery of a ‘billiard’ reaction for this series of compounds. In order to prepare BTDOs annulated with tetraazapentalene systems, an attempt was made to synthesize a derivative containing triazole moiety in position 5. However, the reaction of compound 63 with sodium azide did not give the expected 6-azido-5-triazolyl derivative 64, but instead furnished an isomer with the inverted positions of substituents, namely, 5-azido-6-triazolyl-BTDO 65 (Scheme 23).41
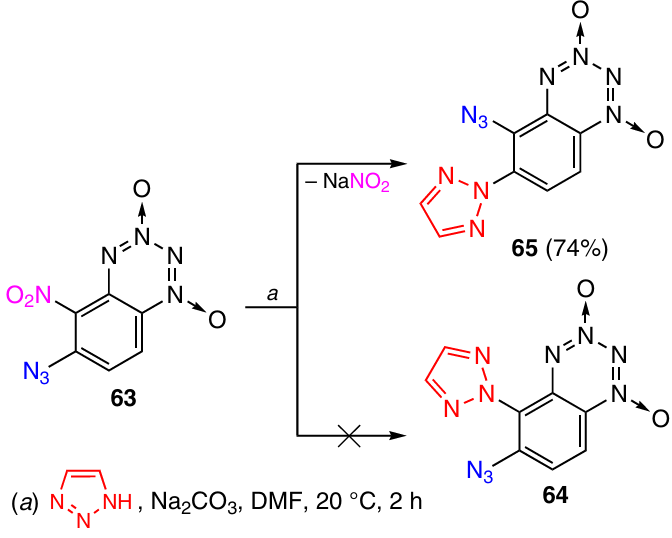
The formation of product 65 can be explained in the following way (Scheme 24). In the first step, the triazole anion attacks BTDO 63 at position 6 to give the anionic σ-complex A. This is followed by elimination of azide anion to give intermediate 66. However, the reaction does not stop at this stage: now the azide ion itself acts as a nucleophile and attacks position 5 of intermediate 66 to form anionic σ-complex B, and the subsequent elimination of the nitrite anion leads to BTDO 65.
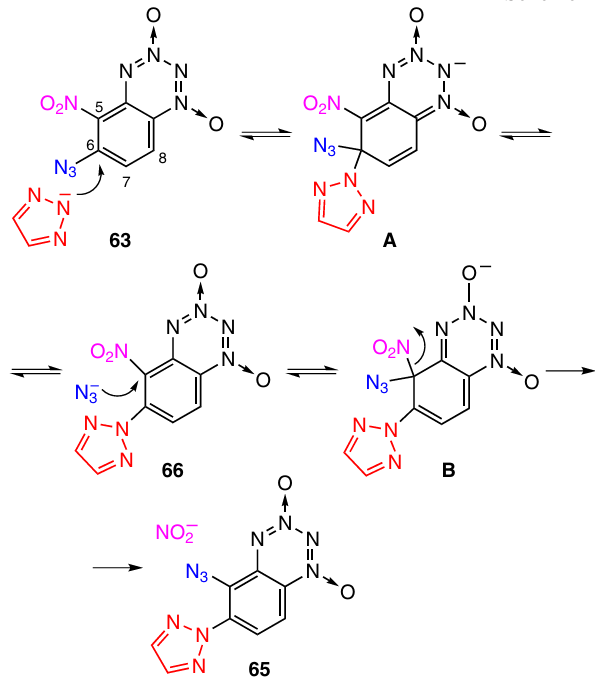
This unusual cascade of nucleophilic substitution reactions in which a nucleofuge turns into a nucleophile can be called a ‘billiard’ reaction. To our knowledge, such cascades of reactions have not been observed earlier among benzene derivatives.
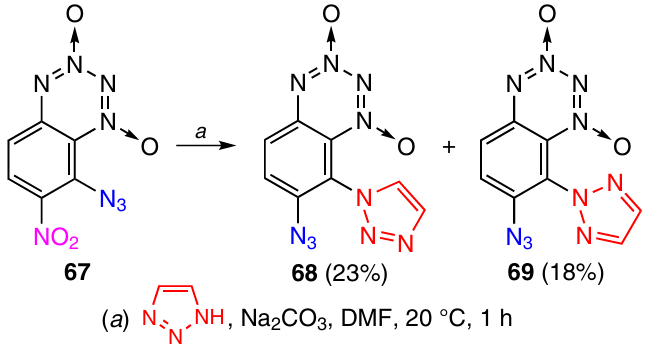
A similar ‘billiard’ process occurs when BTDO 67 reacts with 1,2,3-triazole in the presence of a base; this gives a mixture of isomers 68 and 69 (Scheme 25).41 The azido group migrates from position 8 to position 7 during the reaction.
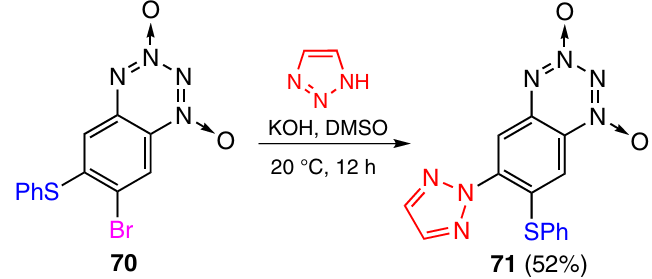
One more example of the ‘billiard’ process is the conversion of BTDO 70 to compound 71 via the reaction with 1,2,3-triazole in the presence of a base (Scheme 26).41 In this case, the benzenethiol group migrates from position 6 to position 7. Similarly to the azido group in the above examples, the benzene-thiol group acts as a nucleofuge in the first step and as a nucleophile in the second step.
2.3.3.2.2. Annulation with a second tetrazine 1,3-dioxide ring
Tetrazinobenzotetrazine 1,3,7,9-tetraoxides (TBTTOs) 72a,b were synthesized by simultaneous or successive closure of two TDO rings (Scheme 27).42 In the former case, chloroaniline 73 was converted to phenylenediamine 74 by treatment with NH3 at 250 atm. Cyclization of 74 by the nitration method under the action of N2O5 in CH2Cl2 resulted in the closure of both tetrazine rings and formation of TBTTOs 72a and 72b in a nearly 1 : 1 ratio. In the latter case, chloroaniline 73 was first cyclized on treatment with N2O5 to give BTDO 75, and then the chlorine atom was substituted with an amino group via the reaction with ammonia. Tetrazine ring closure in amino derivative 76 induced by the reaction with N2O5 in CH2Cl2 gave TBTTOs 72a and 72b in a ratio close to 1 : 2. The nitration of compound 76 with one equivalent of NO2BF4 afforded nitramine 77, which was cyclized by the phosphorylation method by treatment with P4O10 in MeCN to give unsubstituted TBTTO 72a without an impurity of nitro derivative 72b. The bromination of compound 76 yielded benzotetrazine dioxide 78, which was converted to TBTTO 79 by treatment with an excess of N2O5 in MeCN.
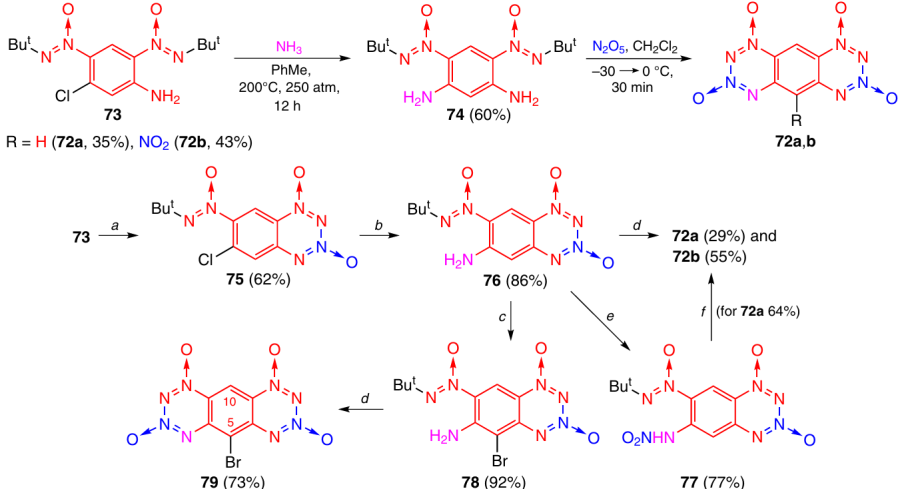
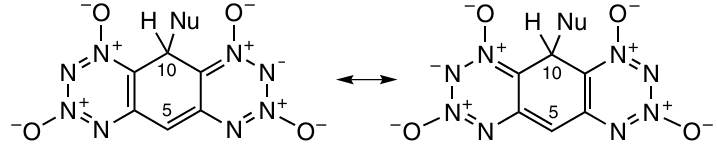
TBTTOs start to decompose without melting at 140 – 160 °C. It was found that these compounds, including the unsubstituted compound 72a, were highly sensitive to hydrolysis. This is, most likely, due to the ease of formation of the thermodynamically stable anionic σ-complex, in which the negative charge is distributed over both TDO rings, via an attack by a nucleophile on position 10.
An attempt to replace the bromine atom in 5-bromo derivative 79 with nucleophiles was unsuccessful, probably due to the fact that nucleophiles attack position 10 much more easily than position 5.
2.3.3.2.3. Annulation with tetraazapentalenes
The initial compounds used for the preparation of benzotetrazine dioxides annulated with tetraazapentalenes (T-BTDOs) represent BTDOs that contain azide and 1,2,3-triazole groups in neighbouring positions.
T-BTDO 80 with a tetraazapentalene system fused at the C(5) – C(6) bond was prepared from 5-azido-6-(1,2,3-triazol-2-yl)benzotetrazine dioxide 81 (Scheme 28).43 This precursor was obtained using two successive nucleophilic aromatic substitution reactions. First, 6-bromo-5-nitro-BTDO 82, obtained by nitration of 6-bromo-BTDO 83, was treated with 1,2,3-triazole in DMF in the presence of Na2CO3 to replace the bromine atom in position 6 with a 1,2,3-triazole moiety. Then the nitro group in position 5 was replaced with an azido group via the reaction of BTDO 84 with sodium azide in DMF.
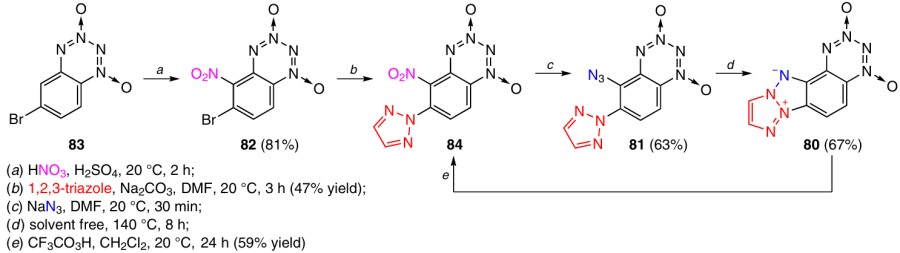
Heating of benzotetrazine dioxide 81 without a solvent at 140 °C for 8 h resulted in the formation of T-BTDO 80 in 67% yield.43 The structure of this product was confirmed by its oxidation with trifluoroperoxyacetic acid (CF3CO3H) giving 5-nitro-6-(triazol-2-yl)-BTDO 84.
According to differential thermal analysis (DTA) at a heating rate of 10 °C min–1, T-BTDO 80 decomposes without melting: the decomposition starts at 245 °C, while the onset of intense decomposition is observed at 280 °C.
A similar synthetic approach was used to obtain T-BTDOs 85 and 86 with a tetraazapentalene system fused at the C(6)–C(7) bond.44 The reaction of 6,7-dibromo-BTDO 87 with 1,2,3-triazole in the presence of a base resulted in the replacement of a bromine atom in position 6 giving rise to (triazol-2-yl)-BTDO 88 in 77% yield (Scheme 29). Triazol-1-yl-substituted isomer 89 was isolated upon this reaction in only 5% yield. Meanwhile, a good yield of this isomer was attained upon the cycloaddition of acetylene to the azido group in BTDO 90, which was obtained by reaction of compound 87 with NaN3. The bromine atom in position 7 of the triazole-substituted BTDOs 89 and 88 is readily replaced with the azido group when these compounds are allowed to react with NaN3; the reactions afford products 91 and 92, respectively. Heating of these BTDOs without a solvent at 140 °C furnishes compounds 85 and 86, respectively. The structure of T-BTDO 86 was confirmed by its oxidation to afford 7-nitro-(6-triazol-2-yl)benzotetrazine dioxide 93 (see Scheme 29).
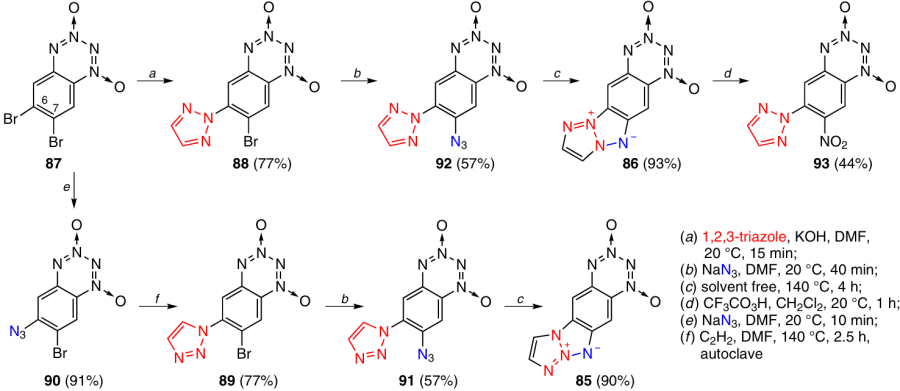
The results of DTA (heating rate of 10 °C min–1) demonstrated that T-BTDOs 85 and 86 start to decompose without melting. For compound 85, the decomposition starts at 210 °C, while the onset of intense decomposition is observed at 250 °C; for isomer 86, these values are 215 and 235 °C, respectively. These compounds are somewhat less stable than T-BTDO 80, in which annulation occurs at the C(5) – C(6) bond.
Benzotetrazine dioxides 94 and 95 with a tetraazapentalene system attached at the C(7) – C(8) bond were synthesized as described in Scheme 30.45 In the reaction of 8-bromo-7-nitro-BTDO 96 with 1,2,3-triazole in the presence of Na2CO3, bromine atom was rapidly displaced to give isomers 97 and 98 in 3 : 5 ratio. The mixture was easily separated by extraction with benzene in which the major isomer 98 is virtually insoluble. Despite the fact that position 8 in benzo-1,2,3,4-tetrazine 1,3-dioxides is activated towards nucleophilic substitution reactions to a higher extent than position 7, the triazole ring is not a good leaving group, and, hence, the nitro group at position 7 is substituted when BTDOs 97 and 98 react with NaN3; this results in the formation of products 99 and 100, respectively. Heating of these compounds without a solvent at 140 °C affords T-BTDOs 94 (95% yield) and 95 (96% yield).
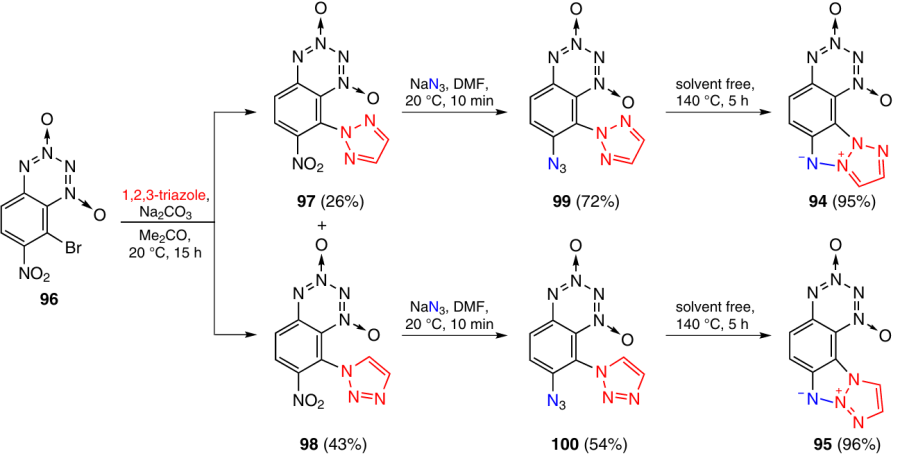
The results of DTA (heating rate of 10 °C min–1) showed that compounds 94 and 95 decompose without melting. For T-BTDO 94, the temperatures of the onset of decomposition and the onset of the intense decomposition are 220 and 245 °C, respectively. For compound 95, these values are 220 and 235 °C, respectively. In terms of stability, these derivatives resemble compounds 85 and 86 and are somewhat inferior to T-BTDO 80.
2.3.3.2.4 Annulation with a furoxan ring
The starting compounds used to prepare benzo-1,2,3,4-tetrazine 1,3-dioxides annulated with a furoxan ring (F-BTDO) are BTDOs in which the azido and nitro groups are located in the neighbouring positions. These precursors were synthesized from appropriate BTDOs via electrophilic and/or nucleophilic substitution reactions.
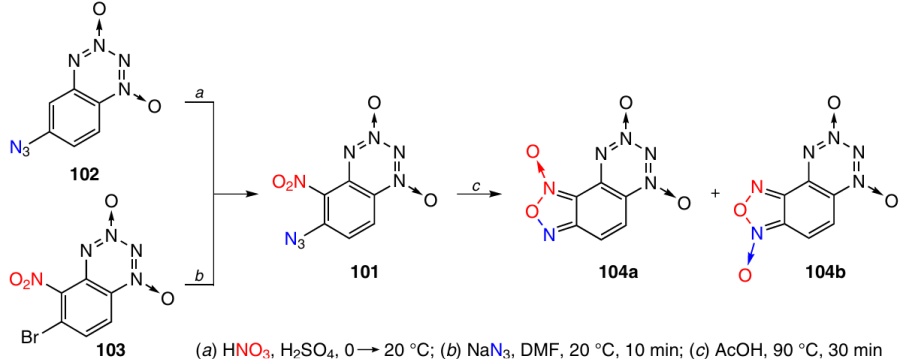
6-Azido-5-nitro-BTDO 101 was obtained in 82% yield by nitration of 6-azido derivative 102 with one equivalent of HNO3 in H2SO4 (Scheme 31). The nitration occurs almost exclusively at position 5. Compound 101 was also prepared by substitution of bromine in 6-bromo-5-nitro-BTDO 103 with an azide group on treatment with sodium azide (85% yield). The formation of the furoxan ring took place when BTDO 101 was heated without a solvent at 100 °C or in AcOH at 90 °C. The 1H and 13C NMR spectra recorded at 297 K demonstrated that F-BTDO 104 exists as a 1 : 1 equilibrium mixture of isomers 104a and 104b (Tm = 204 °C) (83% total yield).46
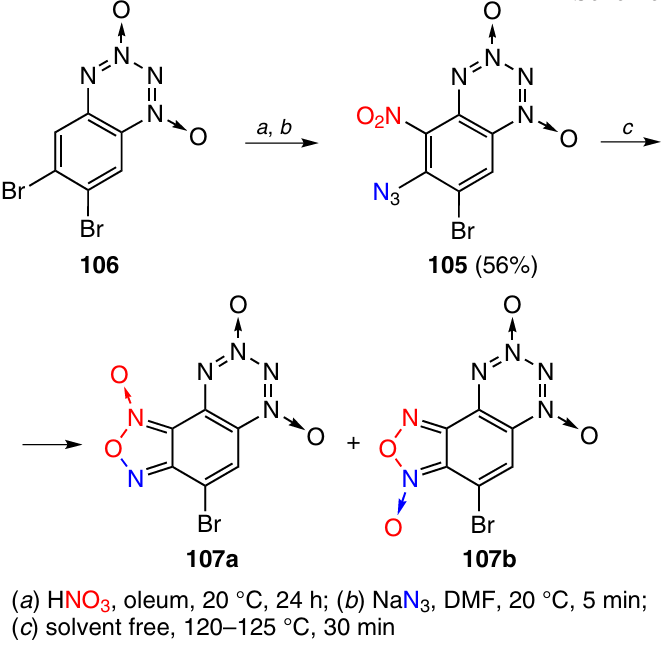
Benzotetrazine dioxide 105 was synthesized by nitration of 6,7-dibromo derivative 106 at position 5 followed by treatment with sodium azide (Scheme 32). The azido group replaced only the bromine atom at position 6. The cyclization of BTDO 105 proceeded under somewhat more rigorous conditions than in the previous case, namely, on heating without a solvent at 120 – 125 °C. The resulting bromo-substituted F-BTDO 107 (Tm = 95 – 105 °C) was also an equilibrium mixture of two isomers (66% total yield), but in this case, isomer 107a predominated (the 107a : 107b ratio was 9 : 1).
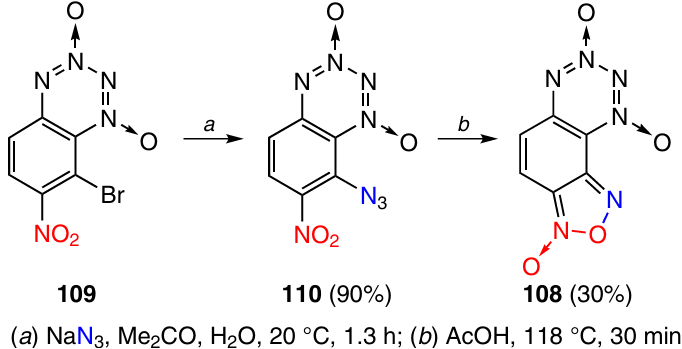
For the synthesis of F-BTDO 108 with a furoxan ring fused at the C(7) – C(8) bond, the bromine atom in 8-bromo-7-nitro-BTDO 109 was substituted with an azido group, and then 8-azido-7-nitro-BTDO 110 was kept in AcOH at 118 °C (Scheme 33). These conditions are somewhat more rigorous than those required to obtain F-BTDO 104 with fusion at the C(5) – C(6) bond. Product 108 proved to be less thermally stable than F-BTDO 104: it melted without decomposition at 74 – 76 °C and started to decompose at temperatures above 90 °C. The low thermal stability is likely to be responsible for the low yield (30%).47 The cause for the low stability of this compound is apparently in the easy interaction of the furoxan ring with the N-oxide oxygen atom of the tetrazine ring.
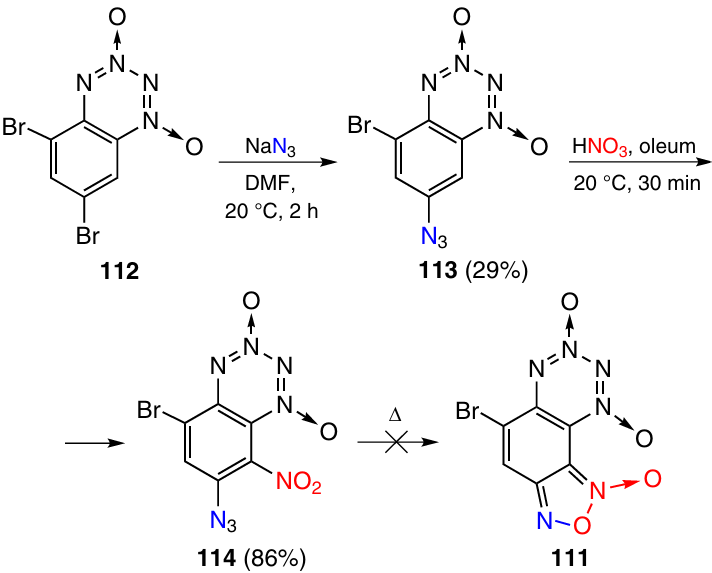
An attempt to obtain F-BTDO 111 according to Scheme 34 did not meet with success. The reaction of 5,7-dibromo derivative 112 with NaN3 gave 7-azido-5-bromo-BTDO 113 as the major product. The minor isomer, 5-azido-7-bromo-BTDO, was formed in a yield of only 12%. The nitration of compound 113 involved position 8 and gave benzotetrazine dioxide 114; heating of this product under various conditions yielded only resinification products. The cyclization is probably hampered by the steric strain in the cyclic transition state and also by low thermal stability of F-BTDOs of this type.
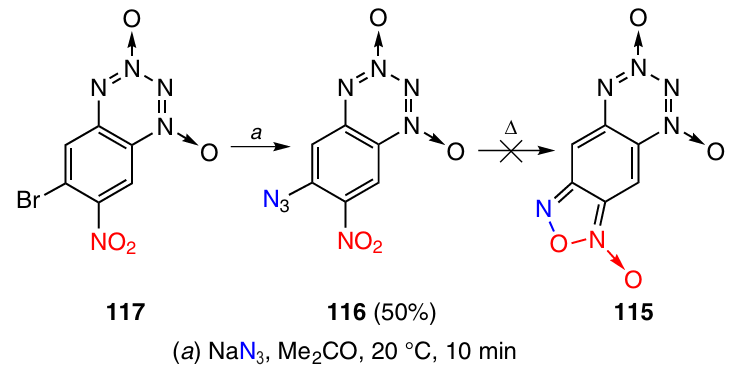
The attempted preparation of F-BTDO 115 by Scheme 35 also failed. 6-Azido-7-nitro-BTDO 116 is easily formed when the bromine atom in compound 117 is replaced with an azido group. However, heating of BTDO 116 under various conditions gave only resinification products. Probably, this is due to high reactivity of F-BTDO 115, which is susceptible to intermolecular reactions due to the quinoid structure.
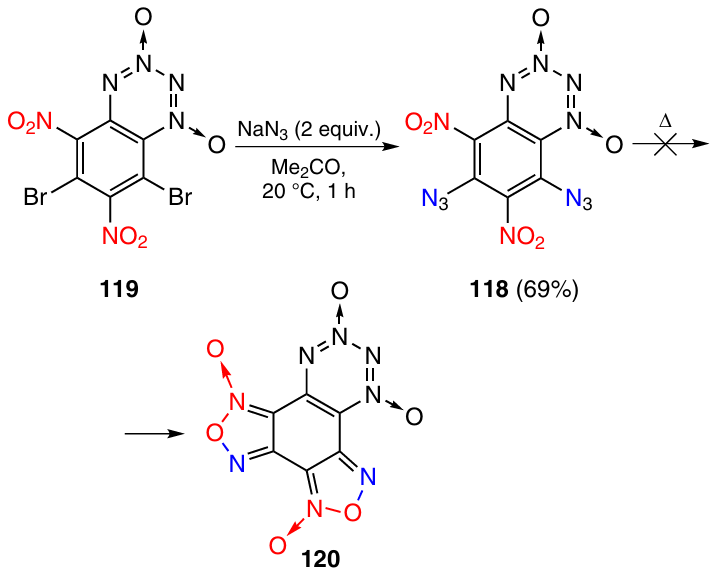
Heating of 6,8-diazido-5,7-dinitro-BTDO 118, obtained by the reaction of compound 119 with sodium azide, gave only products of resinification (Scheme 36). The target F-BTDO 120 with two furoxan rings was not formed under these conditions.
2.3.3.3. Heteroannulated 1,2,3,4-tetrazine 1,3-dioxides
2.3.3.3.1. Annulation with a furazan ring
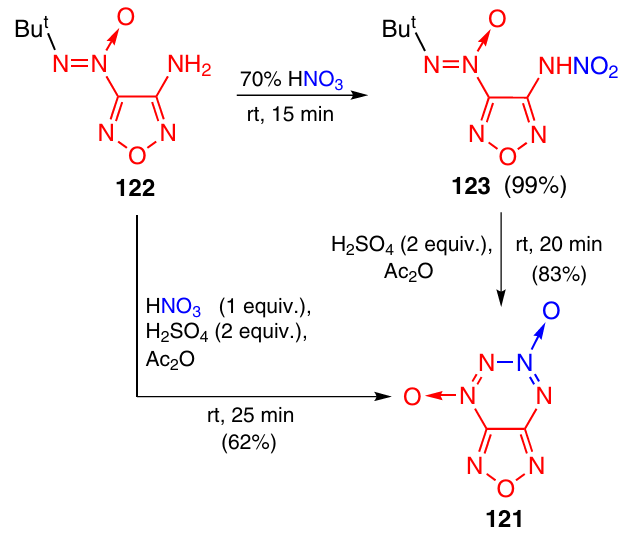
Furazanotetrazine 4,6-dioxide (FTDO) 121 was synthesized for the first time by the nitration method from aminofurazan 122 without isolation of intermediate nitramine 123.48 Later, an improved two-step method was developed; the method included the preparation of nitramine 123 by the nitration of aminofurazan 122 with 70% HNO3 and the subsequent cyclization by the acetylation method (Scheme 37).29, 30 This approach can also be used to prepare FTDO in one step, but the yield of the product is lower in this case.
At room temperature, FTDO 121 is stable to the action of water, but at 80 °C, it is completely hydrolyzed within 2 h to yield triazolofurazan 124 (Scheme 38).49 A probable mechanism of hydrolysis starts with the attack by water on the N(3) atom of the tetrazine ring.
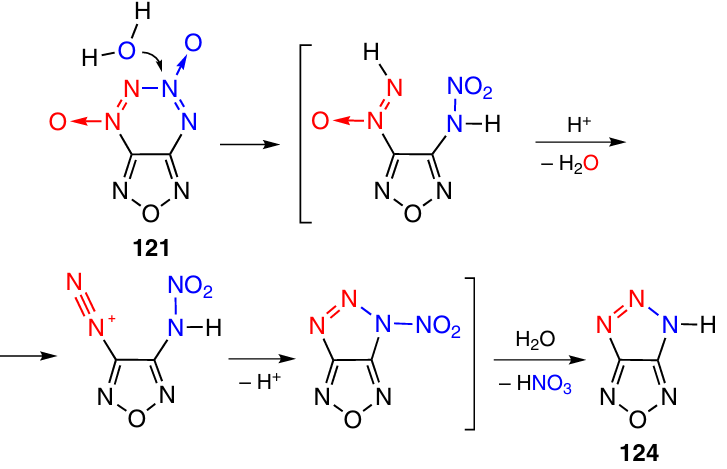
2.3.3.3.2. Annulation with a 1,2,3-triazole ring
Aminofuroxan 125 served as the starting compound for the synthesis of a number of tetrazine dioxides annulated with 1,2,3-triazole and 1,2,3-triazole 1-oxide rings. Compound 125 was synthesized from furazan 122 in three step, which included furazan ring opening to give the sodium salt of cyanoxime 126, which was then converted to amidoxime 127, and the subsequent cyclization gave furoxan 125 (Scheme 39).50 1,2,3-Triazole 1-oxides 128a – d were obtained by reacting this furoxan with aliphatic amines.51 The reduction of compounds 128a,c,d to 1,2,3-triazoles 129a,c,d followed by the reaction with the HNO3 – H2SO4 – Ac2O system afforded triazolotetrazine dioxides 130a,c,d.52 The cyclization of 1,2,3-triazole 1-oxides 128a – c induced by the same system of reagents afforded TDO 131a – c fused with triazole 1-oxides.
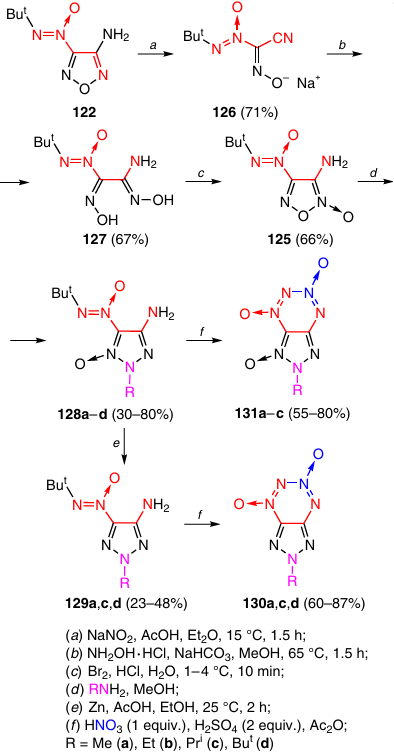
The tert-butyl group is readily removed from the nitrogen atom of triazole 128d in an acid medium to give hydroxytriazole 132 (Scheme 40).52 The treatment of this product with the HNO3 – H2SO4 – Ac2O system resulted in the formation of 1-hydroxytriazolo-TDO 133 in 62% yield. Compound 133 was also prepared in one step without isolation of intermediate triazole 132, but the product yield was somewhat lower in this case (45%).
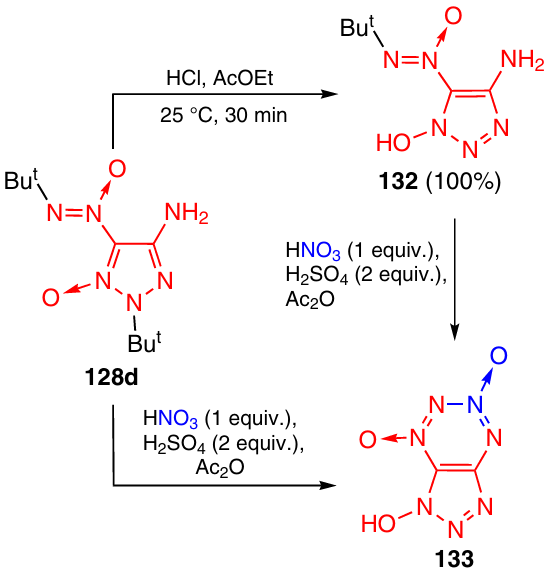
The alkylation of TDO 133 and its Ag salt 134 with various reagents (diazomethane, diazoacetone, bromoacetone, α-bromacetophenone, methyl iodide, methyl vinyl ketone or tert-butyl alcohol in acid medium) resulted in the synthesis of a number of new triazolo-1,2,3,4-tetrazine 1,3-dioxide derivatives (Scheme 41). The reaction of TDO 133 with diazo compounds and the reactions of salt 134 with halo derivatives afford, most often, O-alkylated compounds 135 – 137.53 The reaction of TDO 133 with methyl vinyl ketone and with tert-butyl alcohol in acid medium is directed towards the nitrogen atom of the triazole ring and gives N(3' )-substituted products 138 and 139, respectively.52, 53
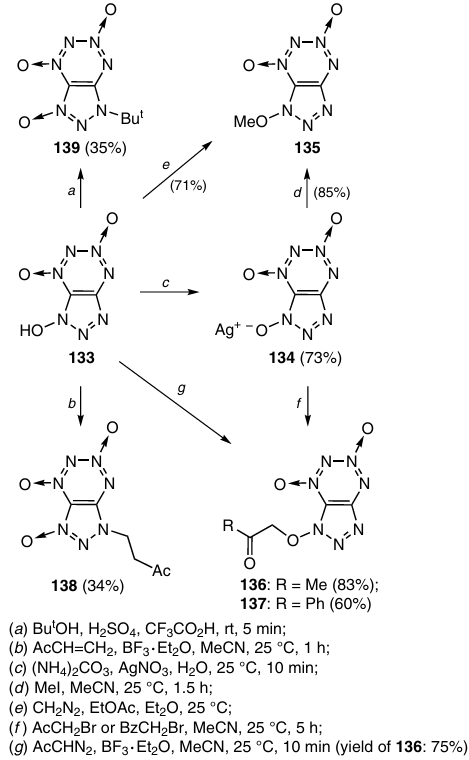
Three methods have been developed to remove the hydroxyl group from hydroxytriazolo-TDO 133 and prepare triazole derivative 140 in high yields (77 – 94%): (a) reduction of 1-hydroxytriazolo-TDO 133 by treatment with PCl3 in acetonitrile; (b) reduction of 1-methoxytriazolo-TDO 135 with Na2S2O4; and (c) reaction of 1-alkoxytriazolo-TDOs 135 – 137 with Et3N in methanol (Scheme 42). The mechanism of the latter non-classical reduction includes, most likely, elimination of the active proton to give the corresponding aldehyde, which was isolated in one case. The tetrazine ring remains intact in these reactions.54
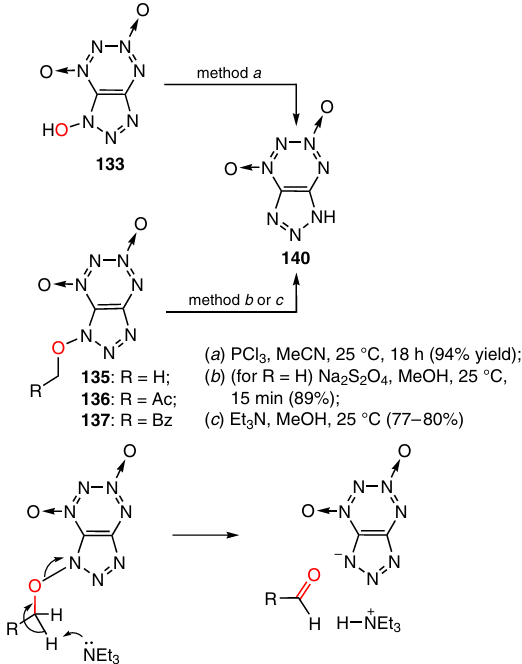
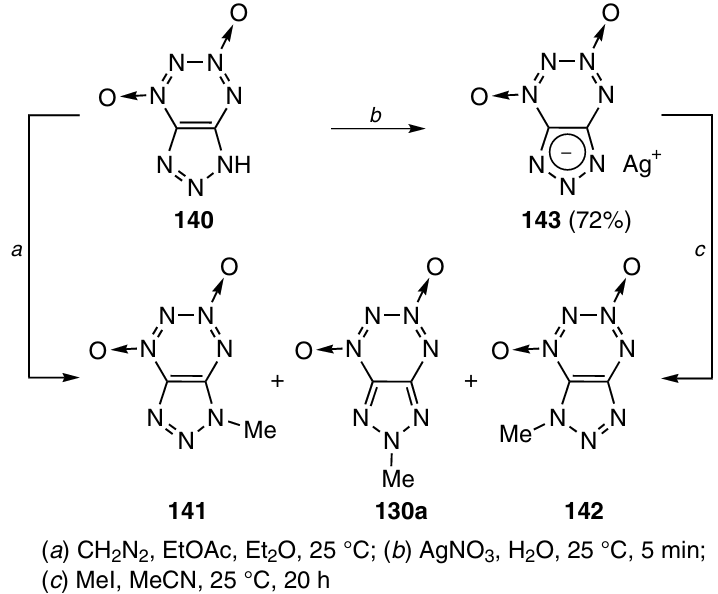
Methylation of triazolo-TDO 140 with diazomethane afforded isomers 141, 130a, and 142 in 41 : 33 : 26 ratio in a total yield of 34% (Scheme 43). Isomer 141 was formed as the major methylation product. In the case where Ag salt 143 was methylated with methyl iodide in acetonitrile, three isomers were also formed (83% total yield), but 130a was the major product (the 141, 130a and 142 isomer ratio was 30 : 56 : 14). In both reactions, the thermodynamically less stable isomer 142 was formed in the lowest amount.
The above-described approach is unsuitable for the synthesis of aryl-substituted triazolo-TDOs. For the synthesis of these compounds, an alternative route was developed, in which (tert-butyl-NNO-azoxy)acetonitrile (144) containing an active methylene unit served as the key compound (Scheme 44). This nitrile was prepared by reductive amination of cyanoxime sodium salt 126.55 1-Aryltriazoles 145a,b were obtained by the reaction of nitrile 144 with aryl azides in the presence of a base. The cyclization of these triazoles to aryl-substituted triazolo-TDO 146a,b was induced by the HNO3 – H2SO4 – Ac2O system.56

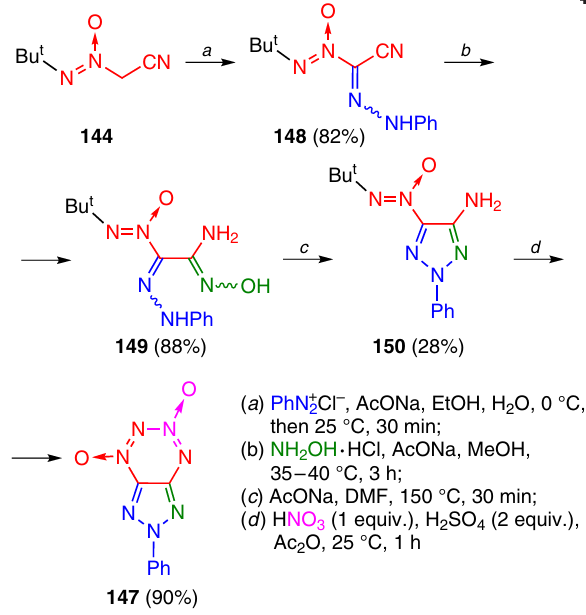
For the synthesis of 2-phenyltriazolo-TDO 147, nitrile 144 was reacted with phenyldiazonium chloride; this gave hydrazone 148, which was converted to amidoxime 149 upon the reaction with hydroxylamine (Scheme 45). On heating at 150 °C in dimethylformamide in the presence of sodium acetate, amidoxime 149 cyclized to triazole 150, which was then treated with the HNO3 – H2SO4 – Ac2O system to give 2-phenyltriazolo-TDO 147.56
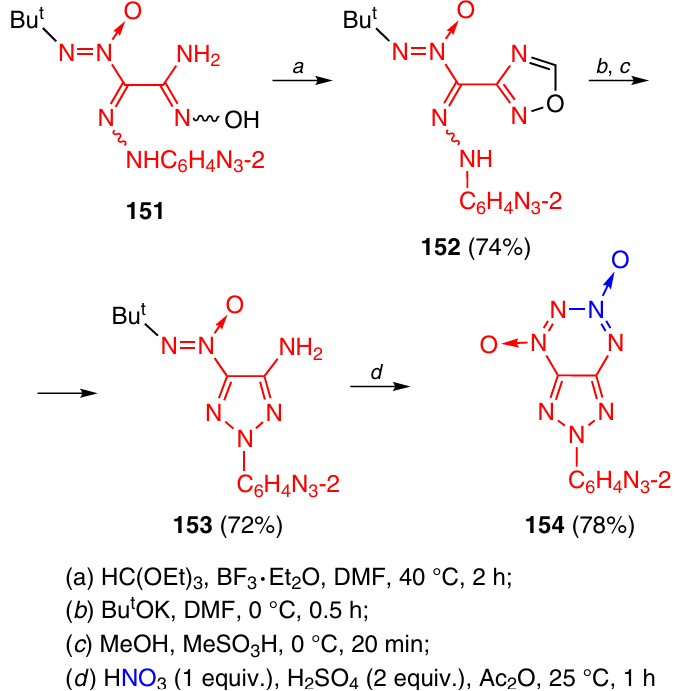
Amidoxime 151, containing a 2-azidophenyl group, was produced similarly to amidoxime 149 (see Scheme 45); however, ring closure to give 1,2,3-triazole by the heating in dimethylformamide failed. Therefore, this amidoxime was converted to oxadiazole 152 and further to triazole 153 by the reaction with a base (Boulton – Katritsky rearrangement). This was followed by elimination of the formyl group in methanol in the presence of methanesulfonic acid (Scheme 46). 2-Azidophenyltriazolo-TDO 154 was synthesized by the reaction of triazole 153 with the HNO3 – H2SO4 – Ac2O system.57
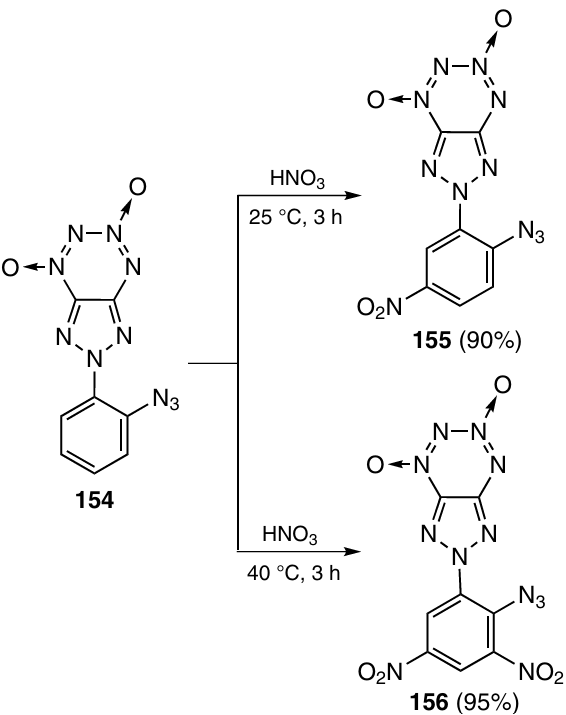
The nitration of triazolo-TDO 154 with concentrated HNO3 at room temperature furnished mononitrated product 155,57 while the reaction at 40 °C led to doubly nitrated derivative 156 (Scheme 47).58
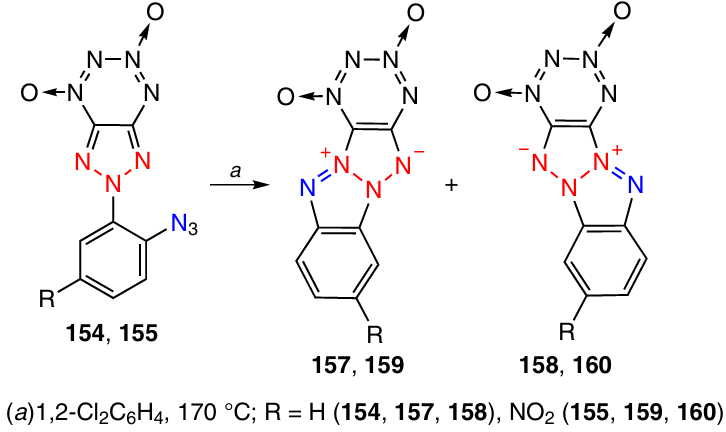
When triazolo-TDO 154 was heated in 1,2-dichlorobenzene at 170 °C for 2.5 h, ring closure took place to give isomeric tetraazapentalenes 157 (Tdec > 183 °C) and 158 in 5.3 : 1 ratio in a total yield of 75% (Scheme 48).57 The cyclization of nitro derivative 155 under the same conditions proceeded in 30 min and afforded isomers 159 (Tdec > 191 °C) and 160 in 1.1 : 1 ratio (91% total yield).
Dinitro derivative 156 cyclized on heating in toluene at 110 °C (Scheme 49).58 The azido group mainly reacted with the neighbouring nitro group, thus giving furoxan 161 (Tdec > 190 °C) in 68% yield. The isomeric tetraazapentalenes 162 and 163 were produced in 5% yield each. Markedly higher yields of tetraazapentalene 162 were attained when tetraazapentalenes 157 or 159 were nitrated with a mixture of nitric and sulfuric acids: 63 and 54%, respectively.57
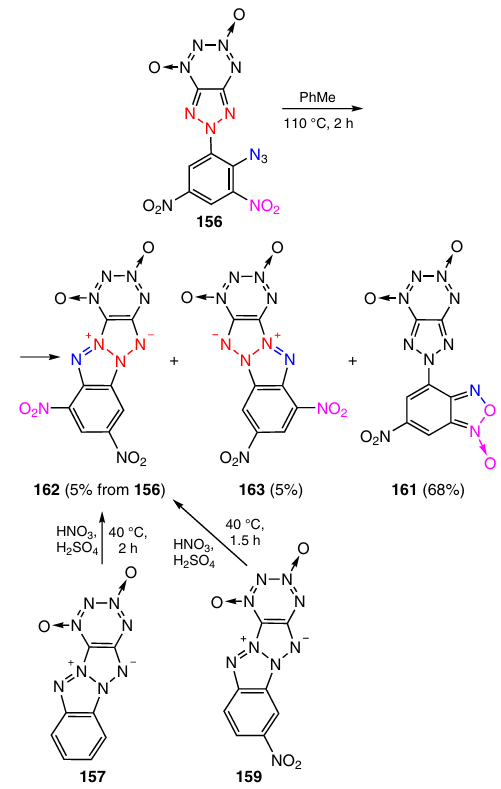
According to differential scanning calorimetry (DSC) data (10 °C min–1 heating rate), the onset decomposition temperature of tetraazapentalene 162 is 234 °C; this is much higher than that of isomer 163, which starts to decompose at 158 °C.
2.3.3.3.3. Annulation with a pyridine ring
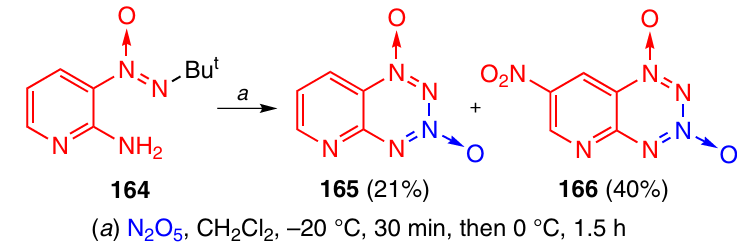
The reaction of 2-amino-3-(tert-butyl-NNO-azoxy)pyridine (164) with an excess of N2O5 in dichloromethane resulted in the formation of pyrido-TDO 165 and its nitro derivative 166 in 1 : 2 ratio (Scheme 50).59
The isomeric pyrido-TDO 167 was synthesized from pyridine 168 using nitronium tetrafluoroborate as a tetrazine ring closure reagent (Scheme 51). The use of N2O5 proved inefficient in this case.
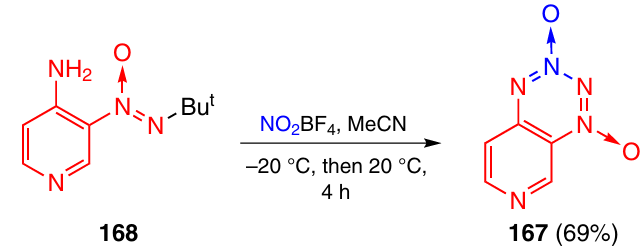
2.3.3.3.4. Annulation with a tetrazine 1,3-dioxide ring
Tetrazinotetrazine 1,3,6,8-tetraoxide (TTTO) 169 was synthesized in 22% yield from TDO 50 using the HNO3 – H2SO4 – Ac2O system of reagents (Scheme 52).33 This compound was fairly thermally stable, but it was completely hydrolyzed in 50% aqueous EtOH at 20 °C in 2 h to be converted to triazolo-TDO 140. The mechanism of hydrolysis appears to be the same as for FTDO 121 (see Scheme 38), starting with the attack by a water molecule on the N(3) atom of the tetrazine ring.
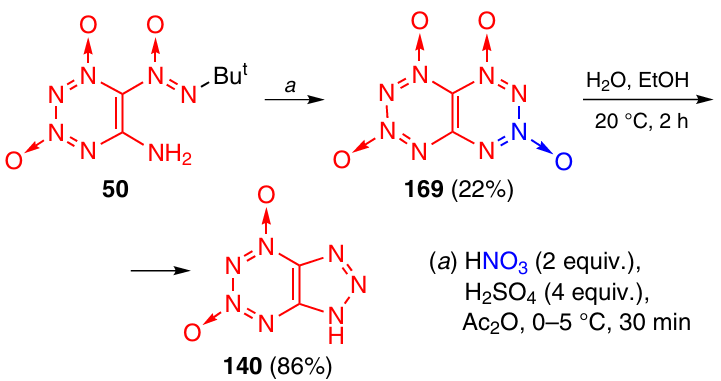
According to DSC data, for compound 169, the decomposition onset temperature amounts to 158 °C at a heating rate of 5 °C min–1 and to 165 °C at a heating rate of 10 °C min–1. The peak of the decomposition temperature is 183 °C at a heating rate of 10 °C min–1.
3. Theoretical methods for the study of 1,2,3,4-tetrazines
[]
3.1. Aromaticity
This chapter addresses the aromaticity of 1,2,3,4-tetrazines in comparison with the aromaticity of other six-membered rings.
The concept of aromaticity is widely used in organic chemistry.60, 61 By aromaticity is meant a complex multilateral phenomenon described by means of numerical ‘aromatic indices’ (related to energy, geometry, magnetic behaviour and reactivity), which determine the specific properties of a cyclic or polycyclic π-electronic system (see, for example, Ref. 62). Considerable attention is paid to the changes in aromaticity that take place when a CH group in an aromatic hydrocarbon is replaced by a heteroatom. For example, using magnetic [nucleus-independent chemical shifts, NICS(0)πzz] and energetic (extra cyclic resonance energy, ECRE) criteria, it was shown 63 that the aromaticity of azines (pyridines, diazines, triazines, tetrazines, pentazine and hexazine) remains virtually invariable. Similar conclusions were drawn by Sánchez-Sanz,64 who found that the NICS(1) and NICS(2) values for azines are close to these values for benzene. Meanwhile, the author demonstrated that NICS(0) cannot serve as a good criterion of aromaticity since at a zero distance, NICS(0) strongly depends on the number of nitrogen atoms, which is not the case for NICS(1) or NICS(2). The necessity of careful use of various types of NICS for evaluation of the azine aromaticity was also noted by Serbian scientists.65 An international research team 66 proposed an original method for evaluation of aromaticity based on a comparison of the 1H NMR chemical shifts of the methyl protons in 15,16-dimethyldihydropyrene (δ = – 4.25 ppm) and its fused derivatives. The authors proceeded from the assumption that the greater the aromaticity in the fused ring, the greater the decrease in the ring currents in dihydropyrene and, consequently, the greater the downfield shifts of methyl proton signals. The researchers stated that the aromaticity of azines can be estimated at 95 – 100% of the benzene aromaticity.
Results of a different type were obtained by Păuşescu et al.67 Relying on energetic (aromatic stabilization energy, ASE), geometric [Bird’s index (I6), harmonic oscillator model of aromaticity, HOMA] and magnetic (NICS) criteria, this research group demonstrated that all N-, P- and As-substituted benzenes are aromatic, but the aromaticity decreases in the series from N- to As-substituted compounds and with increasing number of heteroatoms in the system from one to six. It is noteworthy that the application of different criteria results in different series of aromaticity of azines. For example, according to the I6 index, the aromaticity increases in the order: 1,2,3,4-tetrazine (86) < 1,2,4,5-tetrazine (87) < pentazine (90) < 1,2,3,5-tetrazine (91) < hexazine (100). Meanwhile, HOMA predicts a different sequence: 1,2,4,5-tetrazine (0.823) < 1,2,3,5-tetrazine (0.978) < pentazine (0.993) < 1,2,3,4-tetrazine (0.995) < hexazine (0.999). It is also notable that, according to the ASE criterion, 1,2,3,4-tetrazine, pentazine and hexazine are even antiaromatic systems. Similar results were obtained by Spanish scientists,68 who used, in addition to NICS(1), the total energy density at the ring critical point [RCP(H)]. According to this criterion, aromaticity increases in the order: 1,2,4,5-tetrazine (0.0094) < 1,2,3,5-tetrazine (0.0103) < 1,2,3,4-tetrazine (0.0106) < pentazine (0.0115), which does not differ much from the HOMA series. The trend towards decreasing aromaticity in azobenzenes was noted by Pakiari and Bagheri,69 who proposed a new electric field gradient (EFG) method to estimate the aromaticity, and by Dey and co-workers,70, 71 who used the aromaticity index based on interaction coordinates (AIBIC) they developed. A decrease in the aromaticity of azabenzenes was noted by Báez-Grez et al.72 The authors emphasized that this decrease is insignificant, and a considerable degree of aromaticity is still retained for azabenzenes.
A separate task is to elucidate the relationship between the molecular aromaticity and stability, since the presence of aromaticity does not ensure by itself a high stability of the structure, which is critical, for example, for energetic materials.73 This relationship was studied in detail by Fabian and Lewars.74 One goal of the authors was ‘to locate the point or region between highly stable molecules like benzene and pyridine and the unstable hexaazabenzene’. For this purpose, they performed quantum chemical calculations of the thermodynamic and kinetic stability parameters and homodesmotic ring-opening energies of azabenzenes and estimated the aromaticity based on the geometric (Bird’s index) and magnetic (NICS) criteria. The authors stated that, although azabenzenes are aromatic, the successive replacement of the CH groups with N atoms still results in a steady decrease in the stability, which is attributable to the electrostatic repulsion of the lone pair electrons of nitrogen and/or weakening of the C – N and N – N bonds caused by n-electron donation to the antibonding σ* orbital.
Using delocalization index, Mandado et al.75 concluded that the aromaticity of tetrazines tends to increase with increasing number of N–N bonds. However, the authors also noted that the stability seems to be determined not so much by aromaticity as by other structural factors, including intramolecular electrostatic repulsion. The idea of decreasing stability as a result of electrostatic repulsion of the lone pairs of electrons was supported by Elguero et al.,68 who estimated system destabilization to be ~ 100 kJ mol–1 after the appearance of an N–N bond in the molecule. Two factors affecting the relative stability of tetrazines, namely, steric repulsion and hyperconjugation, were also considered. According to Liang et al.,76 the latter plays a crucial role.
The effect of introduction of the N→O group on the aromaticity of azines and other N-heterocycles was studied in detail by Yuan et al.77 The authors note that the appearance of the N-oxide group leads, most often, to a decrease in the aromaticity compared to the non-oxidized analogue. Similar results were obtained in a comparison of the properties of tetrazine 38 (see Scheme 12) and tetrazine dioxide 147 (see Scheme 45): aromaticity was much higher for the tetrazine ring than for the tetrazine 1,3-dioxide one (in terms of the geometric and magnetic criteria).56
3.2. Thermal stability
Politzer and Murray 78 put forward several explanations for the fact why catenation* of nitrogen atoms leads to destabilization of the molecule.
1. The energy of the N – N bond is usualy lower than the C – N bond energy; the formation of the N – N bond makes a positive contribution to the enthalpy of formation and increases the energy content of the molecule, which results in molecule destabilization.
2. The presence of two linked nitrogen atoms can facilitate the degradation of the compound because of the potential possibility of the release of a stable N2 molecule.
3. Destabilizing interaction of the molecular orbitals (the lack of stability of higher azabenzenes and the stabilizing effect of the N→O group from the standpoint of the molecular orbital theory were considered in detail by Haas and co-workers 79).
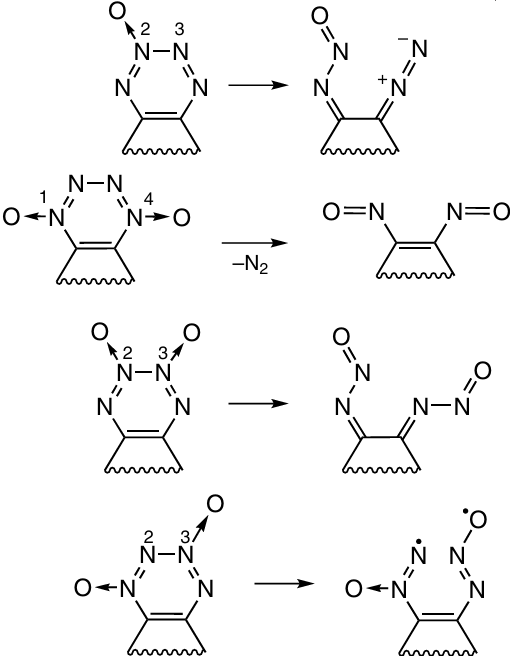
4. Repulsion between the lone pairs of electrons of the linked nitrogen atoms. In the authors’ opinion, this effect is confirmed by the highly negative electrostatic potential of azines in the region of the lone pairs of double-bonded nitrogen atoms, which is indicated by the presence of a negative charge in this space. Correspondingly, a destabilizing effect appears when these negatively charged regions overlap. One of the solutions to this problem proposed by the authors is to introduce an N→O group into the molecule.80 – 82 They assumed that this group dilutes the negative potential by spreading it over a larger area, which reduces the internal repulsion. However, note that stabilization is achieved only at a definite arrangement of N→O groups relative to each other.
The effect of the N→O group on the stability of azines and other N-heterocycles was discussed on the basis of analysis of chemical bonds, frontier orbitals and charge distribution.77, 83 The authors state that, as a rule, the introduction of this group leads to destabilization of the system, as: (i) this induces elongation and, hence, weakening of the bonds adjoining the N→O bond (this effect was also noted in another study 84); (ii) the gap between the frontier orbitals decreases, which can potentially increase the photochemical activity of the molecule; (iii) ring – chain isomerization can be facilitated, as in the case of 1,2,3,4-tetrazine 1-oxides. Presumably, the hypothetical structures depicted in Scheme 53 would also be unstable.85 For example, tetrazine 2-oxide would be unstable because it is subject to the ring–chain tautomerism to give diazonitrosoimine. Tetrazine 2,3-dioxide can readily undergo ring opening to give dinitrosoimine, while tetrazine 1,4-dioxide tends to eliminate an N2 molecule to give thermodynamically stable dinitroso compound. Nevertheless, the authors agree with the statement that the introduction of an N→O group may stabilize the molecule under certain circumstances, e.g., in 1,2,3,4-tetrazine 1,3-dioxides, in which the weakest N(2) – N(3) bond is broken with difficulty because of the formation of the energetically unfavourable azoxy radical.
The use of molecular dynamics made it possible to describe in detail the initial stage of decomposition mechanism for TTTO 169 and also reactions occurring in the condensed phase during the decomposition of this compound (see Scheme 52) under various conditions.86 According to the calculations, the activation barrier for TTTO decomposition is higher than that for the homolytic cleavage of the N – NO2 bond in RDX, HMX and hexanitrohexaazaisowurtzitane (CL-20), which suggests a high thermal stability of this compound. Degradation pathways were also discussed for superhalogens built from annulated 1,2,3,4-tetrazines.87 Pyrolysis of PTDO was studied using molecular dynamics.88 In the case of 1,2,3,4-tetranitro-1,2,3,4-tetrazinane and other cyclic polynitramines, the mechanism of pyrolysis was studied by calculation and analysis of bond dissociation energies (C – C, C – N and N – N bonds in the ring and the N – NO2 bond).89 Note that the dissociation energy of the weakest bond is often used to evaluate the thermal stability of energetic compounds.90 – 96
* Catenation is bonding of atoms of the same element into chains.
3.3. Reactivity
The cycloaddition reactions of ethylene to various azines, including 1,2,3,4-tetrazines, was addressed by several research groups.97, 98 It was noted that the reactivity of azadienes increases in the following series:
pyridine < diazines < triazines < tetrazines.
The cycloaddition was shown to be regioselective, in particular, the formation of a new C – C bond is more preferable than the formation of a C – N bond.
Alkorta and Elguero 99 investigated the annular tautomerism of various indazoles, including the 4,5,6,7-tetraaza derivative. As expected, the 1H-tautomer, i.e., indazole in which a benzene-like aromatic structure was retained in the six-membered ring, proved to be most stable in most cases.
Several studies are devoted to the complexes of azines with other molecules (H2O, CO2),100, 101 metal ions (Li+ and Mg2+)102 and supramolecules (cyclophane ExBox4+).103 It was shown that the strength of hydrogen bonds between azines and water decreases in the series
pyridine > diazines > triazines > tetrazines >pentazine > hexazine
due to the fact that the ability of the nitrogen atom to act as a hydrogen bond acceptor decreases. 1,2,3,4-Tetrazines can form both planar (more preferable) and perpendicular complexes with CO2 with a binding energy of 12.7 – 15.9 kJ mol–1. In the case of metal ions, cation – σ binding is more favourable than cation – π binding. In addition, the authors emphasize that the formation of these complexes decreases the aromaticity of the 1,2,3,4-tetrazine ring. A new approach (induced-polarization energy map) was developed for prediction of the geometry of ionic π-complexes. The potential of the method was demonstrated in relation to the complex formation of BF4– with 5,6-difluoro-1,2,3,4-tetrazine and other fluorinated azines.104 Host – guest type complexes containing 1,2,3,4-tetrazine as a component were studied 105 using an approach based on upgraded AMOEBA force field.
1,2,3,4-Tetrazines were proposed as building blocks for the construction of new systems: aromatic superhalogens.87 Leszczynski and co-workers 106 developed a method for estimating the electron affinities and the standard reduction potentials in DMF for various N-heterocycles, including 1,4-dimethyl-1,4,5,6-tetrahydro-1,2,3,4-tetrazine, and the standard oxidation potentials in MeCN. Scott et al.107 studied the adsorption of azines on carbon materials using quantum chemical methods.
3.4. Physicochemical properties
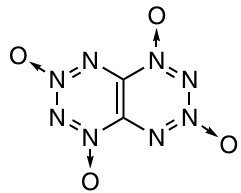
Del Bene et al.108 – 110 published a series of studies in which they found spin–spin coupling constants through one and several bonds [J(C – C), J(C – N), J(N – N) and J(N – F)] for various azines and fluorinated azines using quantum chemical calculations. Bagheri et al.111 reported the results of calculations of 1H NMR chemical shifts and spin – spin coupling constants [J(N – H), J(N – F), J(H – F)] for azine – HF complexes, where azine is pyridine, pyridazine, 1,2,3-triazine, 1,2,3,4-tetrazine, pentazine or hexazine. For iso-TTTO 170, the 13C NMR spectrum was calculated. The presence of only one signal in the spectrum was attributed to the C2h symmetry of the molecule.112
The vibrational spectra (IR, Raman) were calculated for some annulated TDOs by semiempirical and ab initio methods.112 – 115 In the case of 1,2,3,4-tetranitro-1,2,3,4-tetrazinane, theoretical IR spectra were used to predict thermodynamic properties: standard molar heat capacity, entropy and enthalpy.116 The rotation potential and the nonlinear optical properties of phenyl-substituted diazines and tetrazines were investigated by Alyar et al.117 The calculations of the photoelectron spectra and vertical ionization energies for 12 azabenzenes were also reported.118
3.5. Energetic properties
The procedure of estimation of energetic properties of a compound usually includes several stages using quantum chemical calculations.
First, the standard enthalpy of formation of a molecule in the gas phase is calculated using various empirical, semiempirical and ab initio methods.119, 120 The methods used most often include composite methods such as Gaussian-n (Gn) and complete basis set (CBS) models or density functional theory (DFT) methods using atomization and/or isodesmic reactions (see, for example, Refs 121 and 122).
The next stage is to calculate the standard enthalpy of formation of a molecule in the condensed phase; for this purpose, it is necessary to know the enthalpy of phase transition (sublimation or evaporation). It is noteworthy that, despite the availability of a variety of approximate methods (for details, see, e.g., Ref. 119), the exact calculation of phase transition enthalpies is still a challenging task. An interesting solution providing an accuracy of ± 20 kJ mol–1 based on the electrostatic potential method (Politzer scheme 123) was reported by Dorofeeva and Suntsova.124 One more approach is to use a combined method proposed by Muravyev et al.,125 which implies a high-level calculation (W2-F12 and/or W1-F12) of the enthalpy of formation of a molecule in the gas phase in combination with thermogravimetric measurement of the enthalpy of sublimation at a low external pressure (up to 0.2 Pa). Both approaches have been applied to various classes of organic compounds, including 1,2,3,4-tetrazine 1,3-dioxides.
The third stage implies the calculation of the compound density, which is performed using two main approaches: method of molecular volumes 126 and the method proposed by Ammon.127
The final stage is to predict the energetic properties, particularly, to calculate the detonation parameters using dedicated software programs, for example, BKW,128 EXPLO5 (Ref. 129), CHEETAH 130 and their later versions. Note that the validity and correctness of the data obtained with these programs is a debated issue.131 Other important characteristics of energetic materials are thermal stability (see above) and impact sensitivity. For estimation of the latter, a method has been developed under the assumption that the impact sensitivity is correlated with the free space in the crystal lattice of the compound.132 – 134
A number of energetic compounds based on annulated 1,2,3,4-tetrazine and 1,2,3,4-tetrazine 1,3-dioxide have been proposed;90, 91, 135 – 137 and the enthalpy of formation, density and detonation characteristics have been calculated for them. As substituents, the authors used commonly known explosophoric groups such as NO2, NF2, ONO2, C(NO2)3 and polynitrogen heterocycles.
Among polycyclic systems, primary attention was paid to 1,2,3,4-tetrazines and 1,2,3,4-tetrazine 1,3-dioxides fused to five-membered rings, including furazan,138 – 141 furoxan,140, 142, 143 pyrazole,144, 145 imidazole,146 oxazole,146 1,2,3- and 1,2,4-triazole,145, 147, 148 and their N-oxides. Among tetrazines fused to six-membered rings, fused dinitrobenzene,39 pyridine,149 pyrazine 93, 113, 150, 151 and piperazine 92, 93 derivatives have been studied. Benzenes fused to two 152 or three tetrazine 1,3-dioxide rings 153 have been addressed. As a rule, the calculated energetic characteristics of 1,2,3,4-tetrazines and 1,2,3,4-tetrazine 1,3-dioxides fused to five- and six-membered heterocycles are similar to or exceed those of conventional energetic compounds, which allows experts to consider such derivatives to be a promising class of explosives. In most cases, these compounds have high density (d ≥ 1.8 g cm–1), a good oxygen balance, high positive enthalpy of formation (ΔHf ≥500 kJ mol–1), detonation velocity D of ³≥9 km s–1 and acceptable impact sensitivity and thermal stability. Some representatives of this class are unique compounds, e.g., isomers of a bicyclic system containing two 1,2,3,4-tetrazine 1,3-dioxide rings, TTTO 169 (see Scheme 52) and iso-TTTO 170 (see above), which were proposed for the first time in 1999 by Churakov and co-workers.154
Later, characteristics of compounds 169 and 170 were calculaed by a few research groups, who predicted excellent energetic properties (ΔHf = 850 – 990 kJ mol–1, D ≈ 9.7 – 10.9 km s–1, heat of explosion of ~ 2000 cal g–1 and the detonation pressure of ~43 – 60 GPa) and impact sensitivity comparable with that of 1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane (HMX).86, 112, 140, 149, 153, 155 – 161 The wide scatter of the results is also due to the difficulty of calculating the crystal lattice and density for these compounds, which ranged from 1.9 to 2.45 g cm–3 depending on the basic method used.126, 127
One more outstanding compound is TDO 171, which represents a 1,3a,4,6a-tetraazapentalene system fused to two 1,2,3,4-tetrazine 1,3-dioxide rings. According to calculations, this compound has record-high density and detonation parameters (d = 2.047 g cm–3, ΔHf = 1479.9 kJ mol–1, D = 10.387 km s–1, and detonation pressure of 57.42 GPa) and low impact sensitivity together with high thermal stability.150, 153
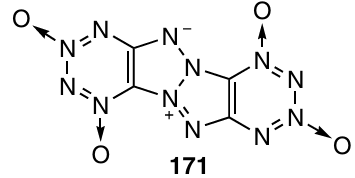
4. Experimental methods for the study of 1,2,3,4-tetrazines
4.1. X-ray diffraction studies
Over the last 15 years, X-ray diffraction data were reported for the following structural types of 1,2,3,4-tetrazines.*
Non-annulated 1,2,3,4-tetrazine 1,3-dioxides. Dimethyl-1,2,3,4-tetrazine-5,6-dicarboxylate 1,3-dioxide (60) has two crystallographically independent molecules in a unit cell. The tetrazine 1,3-dioxide ring is virtually planar and the N – N bond lengths are nearly equal, which attests to effective conjugation.37
Unlike TDO 60, methyl 5-(1,1-dichloro-2-methoxy-2-oxoethyl)-1,2,3,4-tetrazine-6-dicarboxylate 1,3-dioxide (61) contains a 1,2,3,4-tetrazine 1,3-dioxide ring in the boat conformation in which the N(3) and C(6) atoms are outside the plane of the ring, which is apparently due to the steric repulsion of bulky substituents in C(5) and C(6) positions.
6-(tert-Butyl-NNO-azoxy)-5-methylthio-1,2,3,4-tetrazine 1,3-dioxide (44a) exists as four crystallographically independent molecules in a unit cell.32
5-Amino-6-(tert-butyl-NNO-azoxy)-1,2,3,4-tetrazine 1,3-dioxide (50) has two crystallographically independent molecules in a unit cell.33
Annulated 1,2,3,4-tetrazine 1,3-dioxides. Among compounds of this structural type, the following were studied by X-ray diffraction: 2-methyl-2H-[1,2,3]triazolo[4,5-e][1,2,3,4]-tetrazine 4,6-dioxide (130a),164 1-tert-butyl-1H-[1,2,3]triazolo[4,5-e][1,2,3,4]tetrazine 3,4,6-trioxide (139),52 2-methyl-2H-[1,2,3]triazolo[4,5-e][1,2,3,4]tetrazine 1,5,7-trioxide (131a),28 1-phenyl-1H-[1,2,3]triazolo[4,5-e]-[1,2,3,4]tetrazine 4,6-dioxide (146a)56 and 2-phenyl-2H-[1,2,3]-triazolo[4,5-e]- [1,2,3,4]tetrazine 4,6-dioxide (147).56
1,2,3,4-Tetrazine 1,3-dioxides 130a, 139, 146a and 147 with a fused 1,2,3-triazole ring are nearly planar structures characterized by minor deviation of atoms from the mean plane. The oxygen atoms are also located in the plane of the heterocyclic system. The presence of two oxygen atoms in the TDO ring makes a weighty contribution to the bond length alternation, which can be seen when comparing the parameters of TDO 147 and tetrazine 38 containing no N-oxide oxygen atoms. In the crystal packing of compounds 130a and 139, apart from relatively weak and apparently non-directional O∙∙∙O, C – H∙∙∙O and C∙∙∙N contacts, there are also rather strong O∙∙∙π(N) contacts (with distances of 2.235 Å in TDO 130a and 2.884 Å in TDO 139) between the lone pair of electrons of oxygen and the π-systems of the heterocycle nitrogen atoms.
On the basis of furazanotetrazine 4,6-dioxide (121), a 1 : 1 solvate complex with benzene and a co-crystal with benzotrifuroxan (3 : 1) were obtained.165, 166
The benzotetrazine system in 5,7-dibromobenzo- (112)83, 167 and 5,7-dinitrobenzo-1,2,3,4-tetrazine 1,3-dioxides (172) is virtually planar. Compound 172 has two crystallographically independent molecules in a unit cell, which have wave-like packing.39 In the case of compound 112, the dihedral angle between the benzene and tetrazine rings is 2°. The exocyclic oxygen and bromine atoms are located virtually in the same plane as the corresponding rings. A different situation is characteristic of dinitro-BTDO 172, particularly the nitro group located in position 5 is actually coplanar to the bicyclic system. Meanwhile, the nitro group in position 7 is considerably rotated (~ 25°) relative to the plane of the fused rings due to the steric repulsion between the nitro group oxygen atoms and the tetrazine N(1) atom. A noticeable N – N bond length alternation is observed in the tetrazine ring of compounds 112 and 172.
Crystallization of 7-nitropyridino[2,3-e][1,2,3,4]tetrazine 1,3-dioxide (166) from benzene affords 1 : 1 solvate formed as a charge transfer complex, which splits off benzene in vacuum or on heating above 50 °C. The TDO 166 and benzene molecules in the crystal are parallel to one another, with the benzene ring being projected onto the pyridine ring. The molecule of 166 is almost planar; the bond lengths in the tetrazine ring differ only slightly from the corresponding bond lengths in benzotetrazine 1,3-dioxides.59
Tetrazinotetrazine 1,3,6,8-tetraoxide (169) crystallizes from benzene to give 1 : 1 solvate complex, which has a stack packing in the crystal. As in the case of compound 166, solvate of TTTO 169 and benzene is a charge transfer complex, characterized by rather strong donor – acceptor interactions. The N – N bond alternation in TTTO 169 is similar to that for benzo-TDO derivatives.33
The structure of 5,11-dehydro-5H,11H-[1,2,3,4]tetrazino[5',6':4,5][1,2,3]triazolo[2,1-a][1,2,3]benzotriazole 1,3-dioxide (157) was confirmed by powder X-ray diffraction. In the crystal, these molecules are involved in various non-specific interactions such as C–H∙∙∙O contacts. Although the molecule is planar, no π∙∙∙π stacking is present in the crystal.57
Non-annulated dihydro- and trihydro-1,2,3,4-tetrazines. The X-ray diffraction data for 4-(benzyloxy)-1-(4-bromobenzyl)-6,6-dimethyl-1,6-dihydro-1,2,3,4-tetrazin-5(4H)-one (10) were reported by Xu et al.,14 and those for diisopropyl 3-acetyl-5-phenyl-1,2,3,4-tetrazine-1,2(3H,6H)-dicarboxylate (2) were described by Zhao et al.11
Annulated 1,2,3,4-tetrazines. The structure of 2-phenyl-2H-[1,2,3]triazolo[4,5-e][1,2,3,4]tetrazine (38)168 – 170 was discussed above.
1,2,9,10-Tetranitrodipyrazolo[1,5-d:5',1'-f ][1,2,3,4]tetrazine (32) has four crystallographically independent molecules in a unit cell. The heterocyclic system is planar, and the nitro groups are rotated relative to the core because of the steric interactions with each other.24
The structure of 3,8-dichloro-2,9-dinitroimidazo[1,2-d:2',1'-f ][1,2,3,4]tetrazine (36) was determined for its complex with N,N,4-trimethylbenzenesulfamide.25
3,8-Diazido-2,9-dinitrodiimidazo[1,2-d:2',1'-f ][1,2,3,4]- tetrazine (37) has four crystallographically independent molecules in a unit cell. The heterocyclic system is planar, the nitro groups are coplanar, while the azido groups are somewhat rotated relative to the plane (by ~ 5 and 7°).25
2,9-Dinitrobis([1,2,4]triazolo)[1,5-d:5',1'-f][1,2,3,4]tetrazine (29) crystallizes as two polymorphs: α (from toluene) and β (from acetone). The heterocyclic system is planar, and the nitro groups are coplanar.23
The above data can be summarized by stating that the TDO ring is planar or virtually planar in both annulated and non-annulated compounds, with moderate alternation of the endocyclic bond lengths, which attests to efficient π-electron delocalization. Table 1 summarizes the X-ray diffraction data for the most characteristic representatives of this class (Fig. 1).
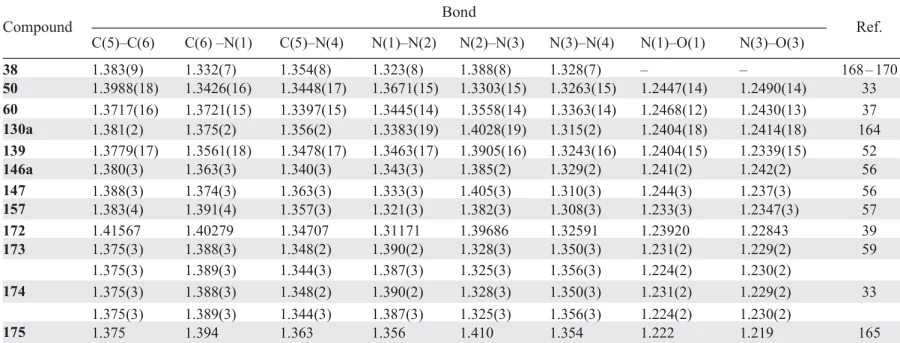
![[{"id":"GAflZ7s0qV","type":"paragraph","data":{"text":"1,2,3,4-Tetrazine derivatives studied by X-ray diffraction."}}]](/storage/images/resized/8UfzLm4LcXth1RTU3yzJTy0z3CbHNhNevYhRzww9_xl.webp)
* Earlier X-ray diffraction data for tetrazine derivatives can be found in the literature.1, 2, 162, 263
4.2. NMR, IR and UV spectroscopy and mass spectrometry
The vibrational spectra of benzotetrazine 1,3-dioxides and FTDO 121 were analyzed in detail in a previous review.1 In the period considered there, IR spectroscopy was mainly used to confirm the structures of new compounds.
Apart from the data reported previously,1, 162 UV spectra were recorded for dinitrobenzo- (Ref. 39) and tetraazapentalene-fused 59 TDOs, tricyclic compounds 43 – 45 and tetrazines 18.16
Mass spectrometry was used to characterize new compounds. The fragmentation of some tetrazine 1,3-dioxide derivatives is described in detail in the previous review.1
1H, 13C, 14N and 15N NMR spectroscopy is a routine method for determination and validation of the structures of various substituted 1,2,3,4-tetrazines. Complete signal assignment is often performed using special techniques (1H – 15N, 1H – 13C HMBC and HSQC, INEPT).
A distinctive feature of the 13C NMR spectra of 1,2,3,4-tetrazine 1,3-dioxides and 1,2,3,4-tetrazine 1-oxides is signal broadening for the carbon atom bound to the N-oxide group of the ring, caused, in particular, by the 13C and 14N spin – spin coupling. The signal of this carbon atom is observed, most often, at δ = 120 – 140 ppm. The signal of the second carbon atom of the ring appears at 145 – 165 ppm.26
The 14N NMR spectra of the indicated compounds usually exhibit two relatively narrow signals for the nitrogen atoms of the TDO N-oxide groups, which formally bear a positive charge. Previously, complete signal assignment for the nitrogen atoms of the TDO ring was performed for benzo- and furazan-fused derivatives. It was found that in the spectra of BTDO, the signal of the N(1) atom (δ = – 40 ppm, Δn1/2 = 20 Hz) is more narrow and is located at a lower field than the signal of N(3) (δ = – 48 ppm, Δn1/2 = 30 Hz). In the case of FTDO, the signal of N(3) (δ = – 44 ppm, Δn1/2 = 60 Hz) is located in the same region as that for BTDO, whereas the narrow N(1) signal (δ = –53 ppm, Δn1/2 = 10 Hz) is shifted upfield. The spectra of TDO fused to a pyridine ring 59 and non-annulated compounds are also similar to the spectrum of BTDO: the narrow signal of N(1) is located in a lower field (δ ~ – 40 ppm), while the signals of N(3) are shifted upfield to δ ~ – 60 ppm in the case of TDO 44a and 52. In the spectrum of compound 60 with two identical substituents, the signal of N(3) (δ = – 43 ppm, Δn1/2 = 70 Hz) is shifted downfield and almost coincides with the N(1) signal (δ = – 39 ppm, Δn1/2 = 50 Hz).32, 33, 37 The spectra of triazolo- (Refs 52, 54, 56) and tetraazapentalene-fused 57 TDOs are similar to the spectrum of FTDO. In the spectra of these compounds, the signal of N(1) occurs at – 50 to – 70 ppm, while the signal of N(3) is located at δ = – 37 to – 43 ppm, with the latter signal (Δn1/2 = 45 – 120 Hz) being usually twice as wide than the former one (Δn1/2 = 10 – 50 Hz). It is of interest that the spectrum of TTTO 169 (δ = – 39 [N(3,6)] ppm with Δν1/2 = 32 Hz and – 54 [N(1,8)] ppm with Δν1/2 = 13 Hz) also resembles more closely the spectrum of FTDO than the spectrum of BTDO.33
The structure of tetrazines 18 containing no N-oxide groups was also confirmed by 15N NMR spectroscopy. The signals of the N(1) atom for structures of this type are manifested at – 140 to – 150 ppm, while the signals of N(2) occur at – 20 to – 30 ppm.24, 25
4.3. Thermal stability
Thermal stability is a key characteristic of energetic materials determining the possibility of their practical use. Compounds with a decomposition temperature of not lower than 130 °C are considered to be potentially acceptable for this purpose. The structural and electronic factors that influence the thermal stability of 1,2,3,4-tetrazines and their N-oxides in comparison with other azabenzenes are discussed above from the theoretical standpoint (see Section 3.1). Below we consider the experimental data for the most prominent representatives of this class.
A comparison of the stability of pairs of compounds 38 and 147 or 112 and 176 clearly demonstrates the stabilizing effect of two N-oxide groups located in positions 1 and 3 of the tetrazine ring. Indeed, tetrazine 38 (Ref. 168) or benzotetrazine 1-oxide 176 (Ref. 1) decompose already at room temperature, unlike analogous dioxides 112 and 147.
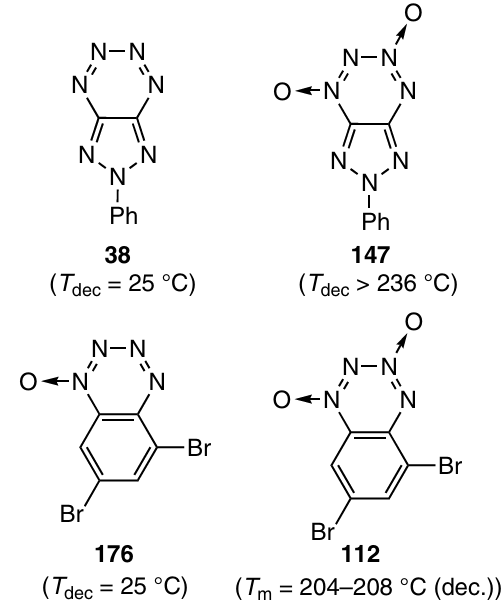
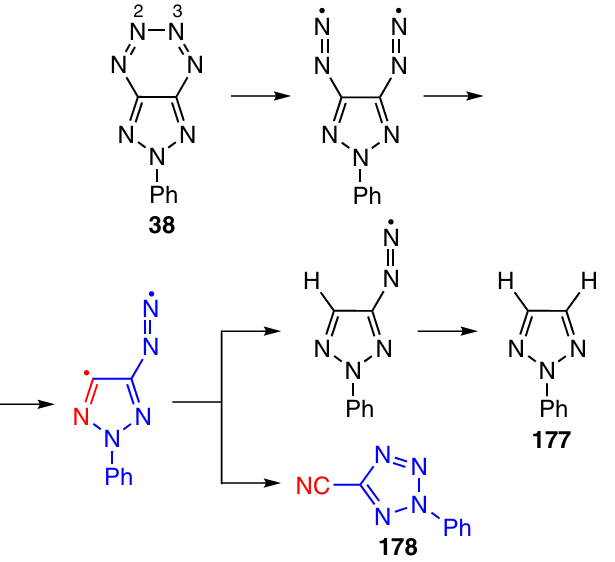
Presumably, decomposition of tetrazine 38 starts with the cleavage of the weakest N(2) – N(3) bond, which is followed by elimination of N2 to give biradical intermediate, which can either detach hydrogen atoms from the solvent and thus afford triazole 177 or rearrange to cyanotetrazole 178 (Scheme 54).
The instability of tetrazine 1-oxide 176 is associated with easy irreversible opening of the tetrazine ring, which results in the formation of o-azido nitroso compounds (Scheme 55).
Non-annulated TDOs with electron-withdrawing and/or electron-donating substituents are stable compounds with decomposition temperature above 160 °C (see Section 2.3.3.1, Scheme 18 and Scheme 19). Exceptions are dicyano derivative 57, which melts with decomposition at 110 – 130 °C 36 and 5-amino-6-(nitro-NNO-azoxy)-TDO 52 [Tm = 110 – 112 °C (dec.)].35 In the latter case, low thermal stability may be associated with degradation of nitro-NNO-azoxy group rather than the proper tetrazine ring at this temperature.
Benzotetrazine dioxides 80, 85, 94 and 95 with a fused tetraazapentalene system (see Section 2.3.3.2.3, Scheme 28, Scheme 29, Scheme 30)43 – 45 and F-BTDO 104 possess a high thermal stability with the decomposition temperature above 200 °C (see Section 2.3.3.2.4, Scheme 31, Scheme 32, Scheme 33).46 However, F-BTDO 108 (Tm = 74 – 76 °C, Tdec = 90 °C) is thermally less stable than compound 104. This is probably caused by the interaction of the furoxan ring with the N-oxide oxygen atom of tetrazine.47 The thermal stability of anthracene type TBTTO 72a,b and 79 is at a reasonable level: these compounds decompose without melting at 140 – 160 °C (see Section 2.3.3.2.2, Scheme 27).42
1,2,3-Triazolo- (140) and 1-hydroxy-1,2,3-triazolo-TDO (133) start to decompose at 150 and 182 °C, respectively.52, 54 The thermal stability of these H-forms is comparable with the thermal stability of their silver salts: Ag salt 134 starts to decompose at 197 °C, the onset of decomposition of Ag salt 143 is 182 °C.53, 54 Apparently, the temperature of ~ 150 – 200 °C characterizes the stability of the anions of these heterocyclic systems (Table 2).
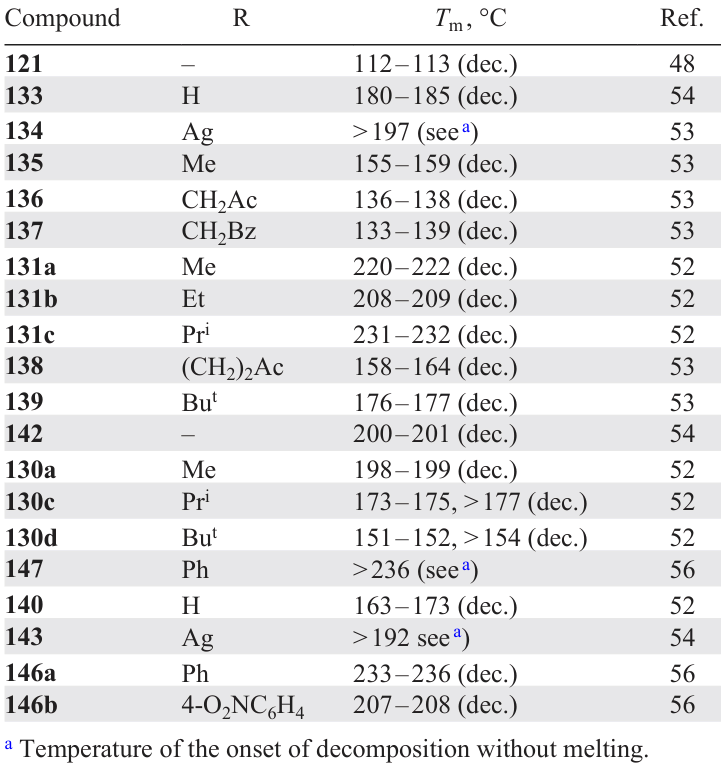
Alkyl- and aryl-substituted triazolo-TDO 130a,c,d, 142, 146a,b and 147 are ~50 °C more stable than the corresponding H-form 140 (Refs 52 – 54, 56) (see Table 2). A similar situation is observed for derivatives 131a–c. In the case of O-alkylated TDOs 135 – 137 and 3-substituted compounds 138, 139, the thermal stability is somewhat lower than that of H-form 133.53
Furazanotetrazine dioxide 121 is isoelectronic to triazolo-TDO 130a – c, 147; however, it is much less thermally stable [Tm = 113 °C (dec.)].48 Nevertheless, compound 121 is considered to be a promising component of energetic materials and rocket propellants; therefore, its thermal decomposition was studied in the solid state, in a melt and in a solution in dinonyl phthalate.171, 172 The authors obtained a stoichiometric equation for the thermal decomposition of FTDO 121 and found kinetic and thermodynamic parameters of the process. It was noted that decomposition of this compound includes two macroscopic stages, with the second stage proceeding with self-acceleration. In the initial stage, decomposition obeys the first-order kinetics.
Tetrazine dioxides 157 – 160, 162, 163 fused to a tetraazapentalene system are similar in stability to the triazole analogues and decompose without melting (see Section 2.3.3.3.2, Scheme 48 and Scheme 49).57 Dinitrobenzene derivative 162 is the most thermally stable according to DSC data (heating rate of 10 °C min–1). The temperature of the onset of decomposition of this tetraazapentalene is 234 °C. Meanwhile, its isomer 163 proved to be much less stable and started to decompose at 158 °C, which is presumably due to a change in the decomposition mechanism.
![[{"id":"W7WH4CZ4t4","type":"paragraph","data":{"text":" Structures of energetic 1,2,3,4-tetrazine 1,3-dioxides."}}]](/storage/images/resized/ANOE7Cp7g02xwBvhTWRWt90FCjPcpttKKI1JoomR_xl.webp)
Pyrido-TDOs 165 – 167 are fairly stable and melt without decomposition in the 179 – 228 °C range (see Section 2.3.3.3.3, Scheme 50 and 51).59 Compound 169 is also thermally stable, despite the hydrolytic sensitivity (see Section 2.3.3.3.4, Scheme 52).33
Among tricyclic compounds 29, 32, 36, 37, the lowest stability was found for tetrazine 29 with two fused nitrotriazole rings [Tm = 138 °C (dec.)] (see Section 2.2, Scheme 9, Scheme 10, Scheme 11).23, 173 Dinitropyrazole analogue 32 has a much higher stability (Tm = 205 °C, Tonset > 233 °C), which is attributable to conjugation of the system and the nature of the pyrazole ring.24 Chloronitroimidazole derivative 36 is also stable (Tm = 215 °C), but the replacement of the chlorine atom by an azido group induces a sharp decrease in the thermal stability of tetrazine 37, which starts to decompose even at 111 °C.25
5. Applications of 1,2,3,4-tetrazine derivatives
As indicated above, in recent years, 1,2,3,4-tetrazines have been mainly proposed as promising building blocks for the design of new energetic compounds with energy characteristics surpassing characteristics of the known compounds. Primary attention was paid to the molecules in which the 1,2,3,4-tetrazine 1,3-dioxide core was modified with various explosophoric groups or heterocycles. In most cases, these studies are theoretical; their main goal is to select the most interesting compounds out of the whole array (see Section 3.1).
Among 1,2,3,4-tetrazine 1,3-dioxides for which methods of synthesis have been developed, the greatest number of studies are devoted to FTDO 121, as this compound has been considered as a promising component of energetic materials and rocket propellants. The standard enthalpy of formation in the solid phase 174 and the enthalpy of sublimation 114 were measured for this compound. The trends of variation of the density of FTDO on long-term storage were determined by the pycnometer method, powder X-ray diffraction and IR spectroscopy.175 It was shown that the sensitivity of compound 121 to mechanical impact 176, 177 and electric pulses 178 are at the level of these characteristics for initiating explosives; this restricts the practical applicability of this compound in a pure state. Meanwhile, it was found that the co-crystal of FTDO and benzotrifuroxan (1 : 3) has a lower mechanical sensitivity.166 A number of formulations based on these compounds have been studied in detail,172, 179 – 181 with most attention being attracted by the 121 – 2,4-dinitro-2,4-diazapentane system.182 – 187 The interaction of FTDO with some organic solvents was studied.188
5,7-Dinitro-BTDO 172 was evaluated as a possible substitute for some of regularly used explosives.39 The standard enthalpy of formation of this compound in the solid phase was determined experimentally. The mechanical and electrical spark of this compound proved to be at the level of those for hexogen. Specialists placed high hopes on TTTO 169, as indicated by a large number of theoretical studies devoted to this derivative (see Section 3.1).86, 189 However, the applicability of this compound is restricted by its moderate hydrolytic stability.
Among the synthesized molecules with high nitrogen contents, compounds 29, 32 and 37 (see Scheme 9, Scheme 10, Scheme 11) with a fused tricyclic system incorporating a 1,2,3,4-tetrazine ring were considered as promising initiating and brisant explosives, combining high density, good thermal stability and calculated detonation characteristics comparable with those for CL-20.23
Previously, it was reported that benzotetrazine 1,3-dioxides are thiol-dependent NO donors exhibiting biological activity.1, 2 No recent studies on this subject are available.
6. Conclusion
Analysis of the literature devoted to 1,2,3,4-tetrazines shows that the interest in this class of compounds has persisted over the last 20 years. First of all, this is due to the fact that the 1,2,3,4-tetrazine core characterized by high energy content is a good base for the design of energetic compounds. In quite a few theoretical papers published recently, numerous energetic compounds based on this core have been proposed and their physicochemical and performance characteristics have been estimated. A large body of synthetic studies have been carried out and original approaches to the synthesis of new representatives of 1,2,3,4-tetrazines have been proposed. It has been shown that some of them can be considered as promising components of energetic materials and rocket propellants.
1,2,3,4-Tetrazines are also attractive objects for theoretical studies that discuss fundamental aspects of chemistry such as aromaticity of heterocycles and stability of long chains consisting of nitrogen atoms. Study of 1,2,3,4-tetrazines would make it possible to predict methods for increasing the stability of polynitrogen systems.
This review demonstrates that the chemistry of 1,2,3,4-tetrazines has a great potential for further research and is intended to stimulate the development of methods for the synthesis of hypothetical energetic compounds of this class, numerous representatives of which are described in theoretical studies.
The authors are grateful to A.A.Gus’kov for the help in the creation of the graphical abstract.
7. List of abbreviations and symbols
The following abbreviations and symbols are used in the review:
BTDO — benzo-1,2,3,4-tetrazine 1,3-dioxide(s),
D — detonation velocity,
DBU — 1,8-diazabicyclo[5.4.0]undec-7-ene,
DSC — differential scanning calorimetry,
DTA — differential thermal analysis,
F-BTDO — 1,2,3,4-benzotetrazine 1,3-dioxide(s) fused with a furoxan ring,
FTDO — furazanotetrazine 4,6-dioxide,
ΔHf — enthalpy of formation,
LTA — lead tetraacetate,
Nu — nucleophile,
rt — room temperature,
Tdec — decomposition onset temperature,
Tm — melting point,
Tonset — temperature of intense decomposition onset,
TBTTO — tetrazinobenzotetrazine 1,3,7,9-tetraoxide(s),
T-BTDO — 1,2,3,4-benzotetrazine 1,3-dioxide(s) fused with tetraazapentalenes,
TDO — 1,2,3,4-tetrazine 1,3-dioxide(s),
Ts — p-toluenesulfonyl (tosyl),
TTTO — tetrazinotetrazine 1,3,6,8-tetraoxide.