


Keywords
Abstract
The review is devoted to the comparative consideration of the mechanisms of transformations of isoelectronic molecules of carbon(II) oxide and molecular nitrogen in reductive conversion processes. The similarities and differences in the activation of these molecules are demonstrated. Fundamentally and commercially relevant catalytic systems are described and parallels in their operation are shown as well. Promising trends in the search for new catalytic systems and processes are noted. Related molecules with similar reductive conversion processes are indicated.
The bibliography includes 337 references.
1. Introduction
The hydrogen conversion processes of carbon monoxide 1 and molecular nitrogen 2 have been and remain an important part of the chemical industry and a subject of academic research from the first third of the 20th century to this day. Mixtures of nitrogen with hydrogen (N2/H2) and carbon monoxide with hydrogen (CO/H2) are cheap and readily available, and their conversion products are in great demand. Since the middle of the 20th century, the reduction of atmospheric nitrogen with hydrogen was the only source of primary nitrogen in industry. Syntheses based on a CO/H2 mixture (synthesis gas) allow the production of a wide range of hydrocarbon mixtures and methanol from almost any carbonaceous fuel.
The synthesis of ammonia from the elements in the classical Haber – Bosch process takes place at a temperature of ~ 500 °C, and high pressure must be used to shift the equilibrium towards ammonia formation. To move to lower pressures, reasonably priced catalysts that operate at low temperatures are being sought.
Conditions of synthesis gas conversion in the Fischer –Tropsch (FT) process are considerably milder, and the main product (higher hydrocarbons) competes with natural sources of hydrocarbons, therefore, the problem of direct conversion of synthesis gas into such valuable products as higher alcohols, alkenes and primary amines is of more interest.
Under normal conditions, CO and N2 do not interact with hydrogen; this requires extremely harsh conditions or catalytic activation. Industrial catalysts for their conversion have a complex structure and require sophisticated research methods. As a result, for a long time understanding of the mechanism of ammonia synthesis and synthesis gas conversion has been fragmentary at best. With the development of instrumental methods for catalyst characterization it became possible to study the active phase and its surface structure directly, leading to rapid progress in the study of catalytic processes,3, 4 and in the late 1970s a parallel was drawn between the Haber – Bosch and Fischer – Tropsch processes 5 as reactions involving dissociative adsorption of the starting molecules.
By the end of the 20th century, advances in computing and quantum chemistry techniques made it possible to model chemical systems directly at the molecular level, and progress in instrumentation made atomic resolution microscopy and fine methods of electronic structure characterization available. In recent years, realistic ab initio calculations of the structure of the active phase of catalytic systems and reaction pathways on model active sites have been regularly reported. This allows a step-by-step systematic comparison of the mechanisms of catalytic processes for the conversion of CO and N2 on various catalysts.
In this work we plan to consider the catalytic reductive conversion pathways of CO and N2, focusing on similarities and differences in the activation and conversion products of these isoelectronic molecules. The end goal is an overall picture into which other closely related molecules (NO, RCN, RNC, RCCR) can be incorporated. This will provide new opportunities in the search for catalysts for the hydrogen conversion of CO, N2 and related molecules, including their co-conversion with the formation of cross-coupling products (e.g., amines in the co-conversion of CO and N2).
2. Activation of CO and N2
2.1. Structure and properties of CO and N2 molecules
The CO and N2 molecules are isoelectronic (Fig. 1) and are among the most strongly bound and most inert diatomic molecules.* Unlike the chemically active triple bond of acetylene, the π-bonds in CO and N2 are in the zone of positive molecular electrostatic potential (MEP).8, 9 These bonds cannot coordinate to cations. The zones of negative MEP in these molecules are located at the lone electron pairs on the symmetry axis and are much less pronounced than, e.g., in the ammonia molecule, which forms strong complexes with many cations. As a result, both CO and especially N2 adsorb weakly to hard cations at room temperature and are easily desorbed unchanged. This made them popular probe molecules for studies of adsorption sites by IR spectroscopy.10, 11 However, due to charge separation at the subatomic scale, CO and to a lesser extent N2 can interact with the polarizable electron shells of metals at low oxidation states. Indeed, there is a diverse chemistry of compounds of d-metals in formal oxidation degrees +1 and below with a carbonyl ligand. The chemistry of similar nitrogen complexes is considerably poorer and does not include stable homoleptic** complexes.
![[{"id":"XbrlpSKTAI","type":"paragraph","data":{"text":"Schematic of the electronic structure of CO and N<sub>2</sub> molecules."}}]](/storage/images/resized/wpyevvg7DB8i2tlLWPeopIvyknZBOyLdDU4F54li_xl.webp)
To illustrate this, we have plotted MEP maps for CO, N2, C2H2 and NH3 using the P.A.M. DIRAC package 12 (Fig. 2). It can be seen that the zones of negative MEP created by lone electron pairs in CO and N2 molecules are relatively small and shallow compared to similar zones for NH3 and C2H2. At the same time, in the CO molecule, the negative MEP zone near the carbon atom is smaller in volume but deeper than that in the N2 molecule.
![[{"id":"gYvhrWl7wL","type":"paragraph","data":{"text":"Electron potential maps for CO, N<sub>2</sub>, NH<sub>3</sub>, and C<sub>2</sub>H<sub>2</sub> molecules. Crosses indicate the positions of the nuclei."}}]](/storage/images/resized/JWIowZMUK5mw8uDI8N662rbFP6QgLztSuLW3H8j9_xl.webp)
The minimum of the electrostatic potential in the lone pair of carbon is much deeper than in the lone pair of oxygen, and even in electrostatic complexes, it is the carbon atom that is being coordinated. Contrary to common belief, this does not increase the multiplicity of the C – O bond. The known small increase in the stretching frequencies of the carbon-oxygen bond observed for CO adsorbed on hard Lewis acids is caused by electrostatic interactions.13 At the same time, the interaction of CO with transition metals is accompanied by back-donation into the zone of positive electrostatic potential near the carbon atom and leads to a red shift of the stretching frequencies of the carbon – oxygen bond.
* Accurate quantum chemical modelling of bonding in these molecules requires computationally expensive methods.6 DFT methods used in quantum chemical calculations in the field of catalysis estimate bond energies with significant error.7
** A homoleptic complex is a complex in which all the ligands are identical.
2.2. Activation of CO
The direct study of CO and N2 activation on heterogeneous catalysts requires sophisticated technical equipment and is further complicated by the presence of several types of adsorption sites on most catalysts. Therefore, it is convenient to study fundamental aspects of activation on models such as coordination compounds of transition metals.
The most convenient model of CO activation is metal homocarbonyls, a long known and well studied class of compounds. Soon after the structure of metal carbonyls was reliably established, the metal-carbonyl bond was described as dative with some back-donation.14 Early quantum chemical calculations showed that the back-donation in this description was underestimated.15 Over time, the Dewar – Chatt – Duncanson (DCD) model,16 originally developed to describe complexes of alkenes with transition metals, has been adapted to describe the carbonyl ligand. In its framework, the metal-ligand bond consists of two components: direct σ-donation from the binding π-orbital of the olefin and π back-donation from an occupied d-orbital of the metal atom to an appropriate symmetry π*-orbital of the olefin (Fig. 3).17 The CO molecule usually adopts end-on coordination and the bond to the metal atom in carbonyls is usually described as a combination of σ-donation of the carbon lone pair to the metal atom and back-donation from the metal atom to the π*-orbitals of the CO molecule, with back-donation dominating.18, 19 The electron pairs of CO during the formation of carbonyl complexes push the electrons of the metal atom into the interligand space, remaining on virtually unchanged orbitals.18, 20 The results of the calculations are consistent with the experimental electron density.21
![[{"id":"H1lq8WOmp0","type":"paragraph","data":{"text":"Schematic of (<i>a</i>) donation and (<i>b</i>) back-donation of carbonyl complexes."}}]](/storage/images/resized/rRuF4fsbfvXTQGh9KmyGG0xx1uOYxestO1eS9B9z_xl.webp)
Carbonyl is a readily migrating ligand, and in some carbonyl complexes, structurally non-equivalent carbonyl ligands appear to be equivalent on the NMR time scale, even when some of them are in bridging positions.22 In solution, polynuclear/cluster carbonyls often exist as a mixture of forms with varying numbers of bridged and terminal CO ligands.23 Bonding that involves bridged CO has specific features expressed as a shift of electron density from the space between metal atoms to the carbonyl ligands.24 For example, the diamagnetic carbonyl complexes Fe2(CO)9 and Co2(CO)8 contain three and two bridging CO groups, respectively. The nature of the bonding in these carbonyls has been the subject of several studies,25 – 27 which have not revealed direct metal–metal bonding. The stretching frequency of the bridging CO ligands in these carbonyls is low (~ 1800 cm–1) and was found to be close to that in formaldehyde (1700 – 1800 cm–1). The structure can be described as a resonant hybrid, with individual structures in which one bridging CO molecule forms a two-electron three-centre bond involving a lone pair and two vacant d-orbitals of metal atoms, and the remaining carbonyl bridges form localized two-centre metal-carbon bonds.
In addition to symmetric bridges, the CO molecule can form asymmetric bridges where the CO molecule is coordinated end-on to one metal atom and π-bonded to another metal atom or even two others (FIg. 4a). The coordination of the usually electron-poor π-bond of a CO molecule is made possible by electron density transfer from the σ-coordinated metal atom due to the π-back-donation. IR bands at extremely low frequencies (~ 1300 cm–1) were reported for such ligands.28 Symmetric bridges and bridges with coordination involving π-bonds are extreme, albeit dominant cases, and a dense spectrum of intermediate structures has been found in structural databases.24, 29
![[{"id":"XZ-jeRT2IB","type":"paragraph","data":{"text":"Activation of the CO molecule in (<i>a</i>) end-on terminal and (<i>b</i>) tilt-bridging coordination by d-metal atoms."}}]](/storage/images/resized/V4GsnluJqBaV2QRuBjcJJqD4IrMjMxA68iMyf9lS_xl.webp)
In contrast to C2H2 and N2 , there are no known examples of π-type bridging coordination for CO, but an analogue with an additional hydrogen atom is known — a complex of a π-type formyl ligand with two tantalum atoms.30
The fundamental difference between the metal atoms on a surface of a metal and the metal atom in the carbonyl complex is the presence of delocalized electronic states, leading to mutual influence of the atoms. This is manifested in the mutual influence of adsorbed molecules and the dependence of adsorption properties (adsorption energy, shift of stretching frequencies, etc.) on the fraction of occupied adsorption sites.31 – 33 On isolated metal sites of metal oxides and zeolites 10 and on noble metals,34 the adsorbed CO typically shows stretching frequencies of 2000 – 2250 cm–1, i.e., the C≡O triple bond is retained. For the more active transition metals (Fe, Co), efficient back-donation from the d-level of the metal to the π* orbital of CO is possible, and for CO adsorbed at 113 K much lower frequencies 34 are observed, down to ~ 1700 cm–1. There are reports of extremely low CO stretching vibrations, as low as 1325 cm–1, attributed to adsorption in the form of an asymmetric bridge (see FIg. 4b).35 The asymmetric bridge is an intermediate in the dissociation of the CO molecule, supported by examples of such dissociation in the chemistry of cluster carbonyls.36
In carbonyls, the carbon atom is activated for attack by nucleophiles.37 Uncommon reactions with electrophiles proceed as an attack on the oxygen atom.38
When adsorbed on metal oxides, carbon monoxide can be attacked by nucleophilic surface oxygen to form carbonites (CO22– ) and/or by nucleophilic surface hydroxyl to give a carboxyl group and then a carboxylate (reactions (1), (2)):10
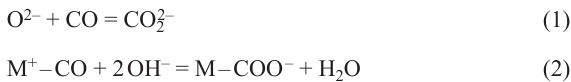
For metal oxides with variable valence, this reaction may be accompanied by carbon oxidation to form carbonates (reaction (3)):10
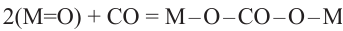
The CO molecule can also insert into metal – carbon 39 and metal – hydrogen 40 bond. These reactions can be regarded as a nucleophilic attack on the carbonyl ligand by another ligand. The acyl ligand (as well as the formyl ligand as a special case of the acyl ligand) usually occupies one coordination position in the complex and therefore, according to the Le Chatelier’s principle, excessive CO pressure is required to shift the equilibrium towards its formation.
The oxygen atom of acyl and formyl ligands is nucleophilic and susceptible to attack by electrophiles to form carbene complexes.41 In the presence of a suitably positioned hydride ligand, equilibrium reductive elimination can occur to afford an aldehyde,42 which can be hydrogenated to an alcohol. Scission the C – O bond in alcohols by hydrogenolysis is difficult because it proceeds as a nucleophilic substitution of a poor leaving group OH.
2.3. Activation of N2
The lone pairs in the nitrogen molecule are significantly less nucleophilic than the lone pair of carbon in the CO molecule. The N2 molecule is a weaker σ-donor and π-acceptor than the CO molecule and is less prone to complexation and specific adsorption.
The nitrogen molecule is isoelectronic with the CO molecule and the complexes of CO and N2 with terminal coordination therefore have a similar structure.
In end-on adsorption on sites that are strong π back-donors, the nitrogen molecule adsorbs specifically with weakening and polarization of the N≡N bond, consistent with the DCD model of donation and back-donation (Fig. 5a).44 The negatively charged uncoordinated nitrogen atom in the coordinated N2 molecule can interact with electrophiles, particularlyprotons, to form diimine derivatives and, further, hydrazine and ammonia.45 In contrast to CO, the N2 molecule is symmetric, and its binuclear complexes with double end-on bridging coordination (M – N≡N – M bridge) are known.46
![[{"id":"MuhoPqXo4z","type":"paragraph","data":{"text":"Activation of the N2 molecule: end-on terminal (<i>а</i>) and symmetrical bridging (“butterfly’-type) (<i>b</i>)."}}]](/storage/images/resized/cscTyJufPFunbg4OzHZkCzpcHtEo86gxkiAJPeZL_xl.webp)
Complexes in which the N2 molecule forms a bridge with metal atoms coordinated to one nitrogen atom are unknown. This correlates with the low stability of isodiazenes (the nitrogen analogue of carbonyl compounds), which fragment readily.47 At the same time, analogues of asymmetric bridges formed by the CO molecule are known (see FIg. 4b).45 The acetylene molecule is isoelectronic with the nitrogen molecule, and many π-complexes with d-metals are known for acetylene. However, no π-complexes have been found for nitrogen. Complexes of N2 with electropositive early transition metals are known. In these, the N2 molecule forms a bridge between two metal atoms, with both metal atoms bound to both nitrogen atoms (‘butterfly’-type bridge, see Fig. 5b). In such complexes there is a transfer of electrons from the metal atoms,48 so formally they can be regarded as complexes of the ligands N22– or N24– with double or single bond, respectively. Deeper reduction of the nitrogen molecule, e.g., for example with [Mo(R2N)3] complexes, leads to its dissociation and provides two equivalents of nitride.45, 49
2.4. Activation of CO and N2 on the surface of metals and their compounds
Metals usually have a crystal structure of one of two types: hexagonal close packing (hcp) and body-centred cubic (bcc) (Fig. 6). A hcp structure can be represented as closest-packed layers of spheres, stacked so that the spheres of the next layer fall into the hollows of the previous one (Fig. 7). In this case, once the position of one layer has been fixed and denoted by the letter A, all the other layers can have three in-plane shifts relative to the fixed layer, commonly denoted by the Latin letters A (no shift), B and C, while no two adjacent layers can have the same shift. In practice, the most common stacking patterns are ABAB, commonly referred as hcp, and the ABCABC, in which cubic symmetry emerges and which is called face-centred cubic packing ( fcc).
![[{"id":"lGgWB3_GtM","type":"paragraph","data":{"text":"Schematic of typical crystal structures of metals."}}]](/storage/images/resized/1aRdum4lvzUQn2i94xOJj2ChjbNLgX7zugzhDKOy_xl.webp)
![[{"id":"PJGtEZkJvH","type":"paragraph","data":{"text":"Comparison of <i>fcc</i> (left) and <i>hcp</i> (right) crystal lattice structures."}}]](/storage/images/resized/jawCsZxIMHpYcvr2CBiZMWSarxt3fWdAkibja5Z8_xl.webp)
Due to the undirected and unsaturable nature of the metal bond, one might expect that metals would always crystallize in a close-packed structure. In practice, this is not the case. For example, the valence electrons in alkali metals behave as an almost perfect electron gas, i.e. these simple substances are close to perfect metals. However, under normal conditions they have a body-centred cubic structure. Which structure is more favourable is determined by subtle phonon and electron effects.50 For nanoparticles, the difference in relative surface energy becomes essential.51 For example, the fcc structure is observed for cobalt nanoparticles, although bulk cobalt has the hcp structure.52
With advances in computational techniques and specialized software, the calculation of the relative stability of crystallographic metal faces in vacuo and under reaction conditions has become possible.53, 54 Various experimental studies have focused on the morphology of nanoparticles that are supported or stabilized in a solution.55 – 57 It was shown that surface formation energy is influenced by the amount and properties of the particles adsorbed onto these surfaces.
In experiments involving molecular adsorption of CO on a surface of a metal, intense bands corresponding to CO stretching vibrations are usually observed in the 1800 – 2100 cm–1 region,58 which is close to the terminal or bridging carbonyl. Indeed, the interaction of CO with the metal surface was initially described as a combination of three-atom σ- and π-interactions (Blyholder’s model 58, 59), similar to the interaction of CO with the metal atom in carbonyls. The adsorption energies of CO at terminal (on-top) and various bridge and hollow sites are found to be quite close and can vary depending on the amount of CO adsorbed.60, 61 Since back-donation to the CO molecule occurs from the d-orbitals and is crucial in the binding energy, the back-donation strength and adsorption energy correlate well with the energy of the d-level of the metal.60
On high-index* fcc/hcp surfaces, as well as on uneven low-index surfaces with ‘steps’, there are sites consisting of a pair of closely spaced atoms and a separate atom (Fig. 8). When CO molecules adsorb on them, it is coordinated by a carbon atom to one metal atom and by a triple bond to the other two metal atoms, and then dissociate.62 The formation of numerous nano-islands with a ‘step’-like borders for cobalt was directly observed by scanning tunneling microscopy (STM).63 Such ‘steps’ are thought to be low energy barrier CO dissociation sites,64 although these sites are rapidly deactivated.65 In the case of ruthenium, similar steps have been shown to be dissociation sites for molecular nitrogen.66
![[{"id":"POJqE3pUEM","type":"paragraph","data":{"text":"The (311) surface of the <i>fcc</i>-Сo structure. Rows of metal atoms offset relative to each other can be seen on which CO can be adsorbed to form an asymmetric bridging ligand."}}]](/storage/images/resized/q70D8E29dMo5nhAA0rsoUeM2CwthlsUKqgi6CvCD_xl.webp)
The α-iron crystal with a bcc structure has a complex equilibrium shape involving high-index faces,56, 67 but the proportion of (111) surfaces is relatively small. On the (110) surface of the crystal, the metal atoms form a regular triangular network that favours molecular adsorption of CO in a bridging position that hampers dissociation.68 In other cases, dissociation is much easier. On (100) surfaces, the metal atoms form a square network and on other faces a triangular network, where the metal atoms are divided into 2 – 3 families with different degrees of coordinative unsaturation. As a result, the CO molecule adsorbed on a suitable site is tilted and interaction between the CO π-bond and surface metal atoms is possible.
For a nitrogen molecule on the iron surface in a fused iron catalyst, the existence of two forms of adsorption has been established: the end-on adsorption and the ‘butterfly’-type symmetric adsorption.69 When considering the end-on adsorption, the analogy with CO and the Blyholder model is usually referred to. The second variant, by analogy with metal complexes, should be considered as a partially reduced molecule. For d-metal complexes bearing a dinitrogen ligand in the ‘butterfly’-type coordination, stabilization by coordination of the negatively charged dinitrogen ligand with alkali metal cations was shown.49 A similar role can be played by alkali metal cations used as ‘electron promoters’.
Formally, the dissociative adsorption of CO, N2 and H2 is an oxidative addition to the metal surface, since the electronegativity of the atoms forming them is higher than that of the metal atoms forming the catalyst. Such reactions are promoted by introduction of additional mobile electron density on the adsorption sites and the active phase. From a practical point of view, this can be achieved by the use of highly nucleophilic (such as calcium amide 70) or electron-donating supports (such as electrides**, 71).
In addition to studies of metallic catalysts, in recent years there has been interest in catalytic materials containing d-metals and a non-metallic element of low electronegativity, such as sulphur,72 phosphorus 73 and silicon.74 Also discussed are carbides, which can be produced under CO conversion conditions (see Section 3.2), and nitrides, which can be formed under ammonia synthesis conditions (see Section 3.1). In those compounds the d-metal atoms are electron-rich and retain the ability to bind effectively to CO, N2 and similar molecules. In the p-element-rich compounds, the average distance between metal atoms is increased compared to the pure metal. Examples of such compounds capable of reductive CO conversion are MoP- and MoS2-based systems.75 The minimum Mo – Mo distance in molybdenum metal is 0.272 nm (2.251 nm for cobalt). For molybdenum phosphide this distance is 0.321 nm. For MoS2 it is 0.316 nm and the layered structure leads to the formation of 1D edges instead of faces. These structural features prevent dissociative absorption of CO and favour the retention of the carbon-oxygen bond in the reaction products.
* Indices of crystal faces are meant. A face, indices of which do not exceed 1, is a low-index face.
** Electrides are compounds that contain a ‘free electron’ in the crystal structure, i.e. a local electron density maximum not associated with a nucleus. One of the first electrides stable at room temperature is [Ca24Al28O64]4+(e−)4 formed by reduction of the oxide precursor C12Al7. Later, the layered electride Ca2N : e– and the stable intermetallide LaCoSi became known.
3. Catalytic of reductive conversion processes of CO and N2
3.1. Conversion of N2
The industrial synthesis of ammonia from elements is based on the reaction (4).76
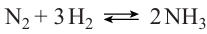
Since the ammonia synthesis results in volume reduction and is exothermic, it is promoted by high pressure and hampered by high temperature. Many variants of the industrial implementation of this process are known, differing in temperature (usually 400 – 500 °C), pressure (from 80 to 1000 atm), features of the preparation of the nitrogen-hydrogen mixture and technical implementation of the ammonia synthesis cycle.
The main catalyst used for ammonia synthesis is the so-called fused iron catalyst, based on iron and doped with non-reducible inert oxide (structural promoter) and non-reducible basic oxide (electron promoter). The industrial process using this catalyst at temperatures of 425 – 450 °C and pressures of ~ 10 – 30 MPa with recirculation of the mixture is known as the Haber – Bosch process (or Haber process). To prepare this catalyst, Fe3O4 magnetite supplemented with aluminium and potassium oxides was first melted in an arc furnace and then reduced in a reactor.77 The reduction can take hours or even days. Aluminium oxide or other difficult-to-reduce oxide increases the surface area of the catalyst and is referred to as a structural promoter.78 In the presence of a structural promoter, potassium compounds further improve catalytic activity,79 including through the additional nitrogen activation.80 An iron-based catalyst can be promoted by cobalt.81 Relatively recently, a catalyst based on wustite (Fe(1 – x)O) instead of magnetite as the iron source has been proposed showing significantly higher activity.82 Magnetite impurities reduce the activity of the wustite catalyst. The difference in activity is probably due to differences in the microstructure of the catalyst, particularly in the distribution of promoters.83 The positive effect of cobalt addition on wustite catalyst turns into a negative one on overheating.84 The iron catalyst is easily poisoned by even small amounts of oxygenate impurities in the feedstock, probably due to the formation of an oxide overlayer.85 – 87 The greatest problem is poisoning by CO, which is present as an impurity in the nitrogen-hydrogen mixture. It is removed by methanation (see below).
The composition of the modern industrial catalysts for ammonia synthesis doesn’t differ much from that originally proposed by Mittasch 88 in 1910. Despite the more than a century of history of the process, the structure of the active phase and the mechanism of action of the catalyst are still under investigation. According to X-ray powder diffractometry data, the reduction of unpromoted magnetite in a hydrogen atmosphere produces crystalline α-Fe, which remains unchanged when exposed to nitrogen. However, the α-Fe peaks observed in the diffractograms of the reduced commercial catalyst are strongly broadened.89 The broadening of the peaks can be explained by the formation of defective or nanoscale crystals. Transition electron microscopy of the catalyst shows many nanocrystalline platelets (<40 nm) of iron (sometimes called ammonia iron) forming large aggregates.90 According to X-ray diffraction (XRD) analysis data, the catalyst composition lacks Fe4N and other nitrogen-rich iron nitrides, although the formation of nitrogen-poor phases,91, 92 e.g. α'-Fe16N2 and α''-Fe16N2 cannot be ruled out. The presence of mobile nitrogen in the catalyst was confirmed experimentally: the spent ammonia synthesis catalyst releases ammonia under hydrogen flow with admixture of oxygenates due to displacement of nitrogen by oxygen.87 It was shown 91 that the major part of the surface in the activated catalyst represents the (111) crystallografic face, although the most thermodynamically stable surfaces for iron are (110) and (100), even in the presence of nitrogen and hydrogen.93 – 95 The stabilization of the (111) surface can be explained by the influence of the electron promoter, which is thought to be distributed on the iron surface in association with oxygen.80, 96 – 98
The carbon-supported ruthenium catalyst, which is extremely expensive compared to the fused iron catalyst, shows high activity at temperatures of 325 – 450 °C, allowing synthesis temperature and reactor volume to be reduced. To prepare the catalyst, the ruthenium precursor is deposited on carbon by incipient wetness impregnation, after which it is reduced and promoted by alkali metal vapour (potassium or sodium) in vacuo.99 Ammonia is not formed on the catalyst immediately, but after the induction period, which may be due to the consumption of ammonia for alkali amide formation.
An experiment with Ru(0001) surface showed that depositing a 0.01% gold monolayer reduces the probability of adsorption of a nitrogen molecule from the molecular beam by seven orders of magnitude. The gold adsorbs on the step sites on the ruthenium surface and blocks them. Therefore, the nitrogen molecule dissociates on these steps.66 This is consistent with the observed ‘size effect’ (a decrease in specific activity per active site for particularly small cobalt particles).100 Like iron, the ruthenium catalyst is sensitive to electronic factors.101
Uranium,102 rhenium,103 cobalt,104 nickel,105 rhodium, iridium and platinum,106 also showed activity in the synthesis of ammonia from the elements, although these catalysts are significantly worse in terms of efficiency/price ratio. No data on ammonia synthesis in the presence of palladium were found in the literature.
3.2. Conversion of CO
Three main processes for the reductive conversion of CO in hydrogen are used in industry: Fischer – Tropsch synthesis to produce aliphatic hydrocarbons, related methanation process and methanol synthesis. Iso-synthesis and synthesis of higher alcohols are also known.
The Fischer – Tropsch process 107 on conventional cobalt- and iron-based catalysts is described by the scheme (5)
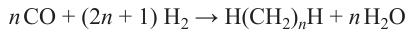
A mixture of hydrocarbons with different numbers of carbon atoms is formed. The basic kinetic model of the FT process includes the following stages: initiation of the carbon chain growth (including reduction of a CO molecule to the active fragment), chain growth (an increase in the alkyl chain length) and chain termination. The theoretical distribution of products in the Fischer – Tropsch process obtained from this model is known as the Anderson – Schultz – Flory distribution,108 and is characterized by a relatively broad peak with long shoulders.
The Anderson – Schultz – Flory distribution is obtained assuming that the chain growth rate is independent of the chain length and the chain does not branch. In practice, this is not the case. The ln W(Cn)/n plot usually shows a kink in the С8 – С12 product region, excessive formation of methane and insufficient formation of heavy products.109, 110 Also, products containing methyl groups in the side chain are formed, but no products with quaternary carbon atoms or longer side chains are observed.111
The most commonly used catalysts for the FT process contain iron 112 or cobalt 113 as the base of the active phase. Nickel is related to cobalt and is active in the FT process but inevitably shows high selectivity for methane.114 Ruthenium, which is chemically close to iron, shows extremely high activity in the synthesis of FT and provides high-molecular-weight products at low temperature,115 but is very expensive. Of the other metals active in CO conversion, copper,116 palladium 117 and platinum 118 are catalysts for hydrogenation to methanol or methane; rhodium produces a mixture of short-chain products with a high proportion of oxygenates 119 and osmium gives a mixture of C1 – C3 alkanes, predominantly methane.120
Currently, two mechanisms have been proposed for the Fischer-Tropsch synthesis: the insertion mechanism and the carbide mechanism. In the insertion mechanism, the chain growth occurs due to the CO insertion into the M – C bond. Some state-of-the-art publications shows that the insertion mechanism is relevant at least under certain conditions.121, 122 In the case of the carbide mechanism, CO dissociates on the surface generates surface C1 species which recombine and undergo hydrogenation to form a hydrocarbon chain. CO dissociation can occur by direct dissociation and hydrogen-induced dissociation, both of which can occur under certain conditions.123
In industry, the Fischer – Tropsch synthesis is usually implemented in one of two ways: high-temperature and low-temperature.124 High-temperature synthesis is carried out at 320 – 350 °C to produce mainly light olefins, gaseous under process conditions.125 Low-temperature synthesis is carried out at 200 – 250 °C to provide mainly long-chain hydrocarbons (С10 – С20 or higher), which are liquid under process conditions.126
The iron-based catalyst of the Fischer – Tropsch process is usually used unsupported.124 The oxide form can be prepared by the sol-gel method or by fusion. The catalyst is promoted 127, 128 by small additions of potassium (to lower the synthesis temperature), copper (to facilitate catalyst reduction) and aluminium or silicon (structural promoter).
The iron-based catalyst has found application in two processes: the synthesis of light terminal olefins 125, 129 at high temperature (~ 340 °C) and low pressure, and the low-temperature synthesis of waxes.126
The bcc α-Fe, Fe3O4 and several carbide phases were identified in the spent iron catalyst according to XRD data.130 Under high conversion conditions, the catalyst is poisoned by the resulting water to generate inactive oxide phase, so the process is usually run under low conversion conditions with recycling of the unreacted feedstock.125
It is currently believed that the actual active phase in the Fischer-Tropsch process on iron-based catalysts is carbides. At least five carbide phases were identified by XRD in iron catalysts spent under various conditions.131 Their activities and selectivities vary considerably. Under typical conditions, the χ-Fe5C2 and Fe7C3 phases predominate after equilibrium has been established. These phases have comparable activity but different selectivities.132 According to DFT calculations,133, 134 the surfaces of these carbides contain structural carbide carbon but allow tilted-bridging coordination of the CO molecule (see FIg. 4b). In addition, the CO molecule can interact directly with the surface carbon to form a ketenylidene moiety. Such structures are known from iron cluster chemistry and may be an intermediate in an alternative chain growth mechanism.135
For the cobalt-based catalyst of the Fischer – Tropsch process, a Co/kieselguhr/ThO2 composition in a ratio of 100/100/18 was initially proposed.136, 137 Later, radioactive thorium oxide was replaced by zirconia, titania, etc. Cobalt is used in the low-temperature Fischer – Tropsch synthesis to produce long-chain waxes,124 which can then be used directly or hydrocracked.138
It is believed that a Co2C carbide phase, active in the synthesis of light olefins and oxygenates, can be formed in a cobalt-based catalyst under certain reaction conditions and/or manganese promotion, but under typical FT synthesis conditions, the only active phase is the cobalt metal.139
The dissociation of CO on the surface of cobalt nanoparticles can be observed by instrumental methods.140 – 143 For particles in the size range of < 6 – 8 nm, the activity (the effect is more pronounced at temperatures < 200 °C) and selectivity (the effect is more pronounced at temperatures > 250 °C) are changed.144 The size effect is usually attributed to the fact that easy CO dissociation requires a step-like surface defect, which does not occur on small faces. Dissociation is hampered on defect-free low-index faces. This is supported by in operando STM data.145 Under the FT conditions, the defect-free surfaces undergo significant reconstruction.146 When a CO molecule dissociates on a step, the oxygen atom is on the terrace and the carbon atom is at the base of the step, in a position with a high coordination number. Binding to carbon stabilizes the step, resulting in formation of triangular nanoislands, the size of which depends on the chemical potential of carbon.62, 63 The 12С16О/13С18О isotope exchange data indicate that CO dissociation is rapid, reversible and doesn’t involve hydrogen.147 However, for carbon to migrate, it must bind to hydrogen, forming a mobile CHx particle.62, 63 According to STM data, the ‘assembled’ hydrocarbon chain ‘lies’ flat on the catalyst surface.148 This increases the adsorption energy of long chains, making their desorption more difficult.
Under the temperature and pressure conditions of the Fischer-Tropsch synthesis, the hexagonal phase (hcp) of cobalt is stable, but in nanoparticles, probably due to the difference in the face formation energy, the cubic phase ( fcc), which is believed to be less active, is stabilized. Although the fcc phase is related to the hcp, they still belong to different structures (see FIg. 4).
The ruthenium-based catalyst deposited on inert oxide or carbon supports does not require promoters, operates at low temperatures and is highly selective towards linear hydrocarbons.149 In industrial applications, the high activity of ruthenium does not justify its very high cost, but its activity at low temperatures makes it suitable for theoretical studies of the mechanism of the FT process.
For ruthenium-based catalysts, just as for cobalt-based catalysts, the size effect and the difference in activity between fcc and hcp particles are known,150, 151 but in contrast to cobalt, ruthenium nanoparticles usually belong to the hcp type. As with cobalt, CO dissociation is thought to occur on the step-type defects.152, 153
Methanation process is essential for the purification of the nitrogen – hydrogen mixture from residual CO in ammonia synthesis. Usually a nickel catalyst is used.154, 155 If this catalyst is not operated properly, the active phase is lost as a volatile carbonyl, limiting the applicability of the catalyst. Due to its high hydrogenation activity under all conditions, the nickel catalyst provides a significant yield of methane,156 while at high temperatures and especially in presence of excess of hydrogen, methane is the main product, which makes the catalyst unsuitable for other syntheses based on CO.
Nickel nanoparticles have an equilibrium shape that is close to spherical,57 resulting in a large number of different high-index faces with a plenty of steps. By analogy with cobalt (see above), these steps are believed to facilitate CO dissociation.157
The synthesis of methanol is described by scheme (6)158
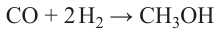
Formally, the process is the addition of two hydrogen molecules to one molecule of CO and can occur on typical hydrogenation catalysts based on Pd,117 Pt,118 Rh and Ir.159 However, in practice, CuO/ZnO catalysts are commonly used, for which nucleophilic activation of CO to form a CO2 species is relevant.160
The ZnO/Cr2O3 catalyst was used in the first industrial synthesis of methanol, launched by BASF in 1923. It operates at high temperature and pressure (300 – 400 °C, 300 atm). The hydrotalcite precursor of the catalyst is obtained by co-precipitation with a base from a metal salt solution.161 The maximum activity of the catalyst is observed for the precursor Zn4Cr2(OH)12CO3. When calcined, the latter compound transforms into a mixed oxide Zn2CrO4 of the spinel group. Under reaction conditions, zinc migrates to the surface and is partially reduced to form oxygen-deficient amorphous zinc oxide.162
The formation of surface carbonyl on zinc oxide is hampered because zinc is not capable of back-donation. IR spectroscopy studies revealed that HCOO– adsorbate is formed in the presence of CO and hydrogen,163 suggesting nucleophilic activation of CO involving surface OH– or O2– species. Oxygen vacancies on the zinc oxide surface may also be active sites.164 In a hydrogen atmosphere, zinc oxide is partially reduced on annealing, which leads to the emergence of n-type conductivity and OH groups. The appearance of conductivity indicates the reduction of the crystal due to the appearance of oxygen vacancies 165 – 168 or diffusion of hydrogen into the bulk ZnO crystal.169 – 172 This allows zinc oxide to act as a redox catalyst.
The CuO/ZnO/Al2O3 catalyst with a Cu : Zn : Al ratio of 60% : (25 – 30)% : (10 – 15)% is the basis of modern catalysts for methanol synthesis and operates under mild conditions (220 – 300 °C, 50 – 100 atm). The activity of the mixed copper–zinc catalyst significantly exceeds that of individual copper and zinc catalysts.173 In 1995, a composition doped with magnesium was proposed.174 In recent years, the effect of other promoters has been investigated.175, 176 Even minuscule amounts of hydrogen sulfide impurities in the feedstock (in parts per billion) poison the catalyst over time,177 requiring its periodic reactivation. Industrial applications did not appear until the 1960s, when methods were developed to effectively remove sulfur from the feedstock, thereby reducing the rate of catalyst poisoning.178
The CuO/ZnO/Al2O3 catalyst proposed in 1947 is prepared by co-precipitation with a base from a solution of salts.178 During the precipitate aging, amorphous particles of the freshly precipitated precursor are transformed into nano-needles of zinc-substituted malachite (Cu,Zn)2(OH)2CO3. Calcination produces spherical oxide particles, but the mesopore structure can be retained. Upon activation, most of the copper leaves the oxide phase and forms metal particles, further increasing the pore volume.179 Transmission electron microscopy (HR-TEM) analysis showed that the freshly reduced catalyst has a thin (1 – 2 nm) overlayer of graphite-like zinc oxide on Cu nanoparticles.180 In contrast to zinc, complexes of Cu and CO are known. In early studies, Cu+ ions on the surface of ZnO activating directly the CO molecule were believed to be active sites of the Cu/Zn catalyst.181, 182 However, more recently, evidence has emerged in favour of a mechanism in which methanol is derived from the intermediate CO2, with the mechanisms for the formation of methanol from CO2 and directly from CO without intermediate CO2 sharing no common carbon-containing intermediates.183 A defective zinc-doped copper oxide surface with ‘steps’ is now considered to be the active phase.184
Isosynthesis is a process of the synthesis gas conversion under harsh conditions (400 – 500 °C, 100 – 1000 atm) producing a complex mixture of products in which isobutene and other branched hydrocarbons are the main products.185 The process proceeds on oxides of Y, La, Ce, Th, Zr, Hf.186 Al2O3 shows non-negligible activity. Although the process has not found industrial application, it is still being actively researched.
Zirconia is the most studied catalyst for isosynthesis and allows high selectivity for isobutene to be achieved.187 This catalyst is prepared by precipitation of zirconium hydroxide followed by calcination. The catalyst is doped with oxides of yttrium, calcium, rare earths and zinc by impregnating zirconium hydroxide with a solution of salts prior to calcination. Isobutene is formed mainly on the surface of monoclinic ZrO2 , favoured by its high basicity, while the other phases are non-selective.187 According to other data, both acidic and basic functions are important in this process.188 Adsorption of CO and hydrogen at relatively low temperatures promotes the formation of formate and methoxy groups on the surface of zirconia.187
3.3. Methods for modifying classical catalysts
The key process-determining step of N2 activation on classical catalysts is reductive dissociative adsorption. It can be facilitated by increasing the reductive potential of the active phase and/or stabilizing the dissociation products, and hindered by ‘pumping’ the electron density out of the active phase on acidic supports.
In accordance with the Sabatier principle, the metal catalysts Mo, Fe, Ru, Os, Co and Ni showed a volcano-shaped curve for the dependence of activity on the nitrogen adsorption energy, with Fe, Ru and Os having the optimum adsorption energy.189 – 191
The reduction potential of the active phase can be improved by increasing the electron density on it. This can be achieved by direct reduction, by interaction with electron-donating species, or by using a strong reducing agent as a support.192 For example, the formation of ammonia at 200 °C and at atmospheric pressure was observed with Ru deposited on a Ca2N : e– electride,193 while on Ru/CaHF this reaction occurred at 50 °C and under atmospheric pressure.194 The use of hydride-containing supports also has a positive effect on the activity of iron- and cobalt-based catalysts.195
Current studies on synthesis gas conversion are focused on finding ways to produce oxygenates, light olefins and aromatic hydrocarbons. The synthesis of olefins requires desorption of the alkyl intermediate by the b-elimination mechanism and suppression of secondary hydrogenation activity.
To facilitate desorption, the process is carried out at high temperature (~ 400 °C).196 For cobalt-based catalysts, good results were obtained with Mn promoters,197 while for iron-based catalysts, K, Na and S additives proved to be promising.198 Of these promoters, sulfur is known to be a catalytic poison in hydrogenation reactions, while potassium and sodium, similar to the ammonia synthesis catalyst, form surface M – O – Fe groups, which increase the reduction potential of iron and prevent the readsorption of alkenes. An unexpected increase in activity of the iron-based catalyst promoted with Sn, Sb,199 Pb and Bi 200 was observed. Under the reaction conditions, those promoters appear to be present in the catalyst as molten nanoparticles in contact with the iron nanoparticles, acting as electron density donors.
The Fischer – Tropsch synthesis follows the carbide mechanism, where the first step of CO activation is dissociation. The production of higher alcohols requires conditions for the insertion of CO into the metal – carbon bond.201 One approach is to use a combination of catalysts for methanol synthesis (Cu) and Fischer – Tropsch process (Co or Fe) in a bimetallic alloy catalyst.202 Alcohol selectivity on such catalysts is usually low and the major product is hydrocarbons.
3.4. Brief overview of promising catalytic systems
At present, it can be assumed that all possible monometallic and many bimetallic catalysts for ammonia synthesis, which can be obtained by reduction of the supported precursor with hydrogen or a nitrogen – hydrogen mixture, have been tested. New highly efficient catalysts can be created on the basis of polymetallic systems or active metal compounds.
Lithium is the best known metal capable of nitrogen fixation under mild conditions. The process is not catalytic, but it inspired studies of bimetallic lithium-containing systems, among which Li – Fe catalysts,203 in which lithium is present on the surface of the catalyst in the form of a surface mixed hydride, have shown high performance.
A number of other metals, including Ti – Cr, Ta–Mo and some others, can also interact with nitrogen. Nitrides of metals with variable valence, primarily d-metals, have been considered as potential catalysts for ammonia synthesis. Currently, some of the best results have been achieved with the so-called ‘CoMo’ catalyst with an active phase of Co3Mo3N.204 A dissociative mechanism was assumed to be valid for such systems, but there are suggestions that an associative mechanism with stepwise hydrogenation of the nitrogen – nitrogen bond might take place.205
In recent years, d-metal-based hydride systems have attracted attention, such as the hydrides TiH2 and BaTiO2.5H0.5, which are comparable in activity to Cs–Ru/MgO 206 and the hydride V2H,207 which is more active than Cs–Ru/MgO. The mechanism of catalytic action of hydrides is under investigation and appears to involve nucleophilic attack of the hydride on undissociated nitrogen, making them substantially different from metal catalysts.208
LaCoSi (see Ref. 209) and LaRuSi (see Ref. 210) electrides demonstrated independent activity in ammonia synthesis. Calculations indicate the importance of anionic electrons in the activity of these materials, with lanthanum atoms being possible active sites.211
In contrast to nitrogen conversion, the Fischer – Tropsch process for the production of hydrocarbons operates under mild conditions on relatively unexpensive classical catalysts. This resulted in a lack of serious interest in finding alternative catalysts for the classical process. On the other hand, the easy availability of synthesis gas makes it an attractive feedstock, and much attention has been paid to finding processes to convert it into products with higher added value than hydrocarbons. The greatest success was achieved in the development of the synthesis of higher alcohols.
The products of synthesis gas conversion on rhodium catalysts are light hydrocarbons, mainly methane, and C2 oxygenates — acetaldehyde as the initial product and ethanol and ethyl acetate formed therefrom.212 The use of FeO as a promoter improves the selectivity of the catalyst to ethanol, but over time iron reduction and fusion with the active phase occurs shifting selectivity from ethanol to methanol and methane.213 MnO is resistant to reduction under reaction conditions and the mixed (Mn,Fe) oxide appears to be an effective promoter.214 Similarly, the use of a mixed (Co,Mn)O promoter allows ~ 70% selectivity for oxygenate formation. For CoO, in the absence of Mn stabilizing the oxide phase, the major product is methane.215 MnO free of impurities increases the performance of the catalyst, but the selectivity to oxygenates decreases. In the absence of promoters, the selectivity for the methane formation increases. Based on a combined study, it was suggested that there are two types of synthesis gas conversion sites on rhodium: active but non-selective and selective but with low activivity.212 Therefore, the role of promoters is to block the non-selective sites and activate the selective ones if possible.
Promising results were obtained with molybdenum carbide catalysts K/Mo2C and K/M/Mo2C (M = Mn – Cu). These catalysts allow the production of alcohols with selectivities > 40%, depending on the conditions and promoter.216, 217 Doping the catalyst with potassium is necessary since pure molybdenum carbide provides mainly lower hydrocarbons.218
Transition metal phosphides can also be active in synthesis gas conversion. In the electronic states of phosphides near the Fermi level, d-orbitals of the metal atoms dominate.219, 220 The insertion of the phosphorus atom increases the metal – metal distance, hampering the formation of bridged adsorbates. This makes direct dissociation of CO on phosphides difficult. In synthesis gas conversion, catalysts based on Fe and Ni (see Ref. 221), Co (see Ref. 222), and K/MoP phosphides have been tested.223 The major conversion products are methanol and higher alcohols, and the main by-product is methane. Supported phosphides are produced by the reduction of an oxide-phosphate precursor with hydrogen under harsh conditions (e.g., reduction in a hydrogen stream at T > 500 °C).224
3.5. MoS2 and systems based thereon
Layered molybdenum and tungsten dichalcogenides are promising catalysts for the conversion of CO to alcohols. These systems are used as catalysts in hydrotreatment, the largest-capacity refining processes. Hydrotreatment includes hydrogenation, hydrodesulfurization, hydrodenitrogenation, hydrodeoxygenation, and hydrodemetallation processes. In these systems, the distance between the d-element atoms is increased compared to a metal catalyst.
Molybdenum disulfide consists of hexagonal MoS6 prisms that are connected by common vertical edges to form layers with a cellular structure. The sulfide surface of the MoS2 layer, also called the basal plane, is kinetically inert, and coordinatively unsaturated (active) sites are formed on the edges (rims) of the crystallite. Interlayer bonding is maintained only by weak van der Waals forces, and the bulk disulfide readily splits into scales. The active phase of molybdenum disulfide is usually in the form of plates with an edge of 3 – 5 nm with one or a small (up to 5) number of layers.225, 226
Small (< 15 nm) freshly sulfidized monolayered molybdenum disulfide crystallites have triangular or almost triangular morphologies.227 – 229 In a reducing atmosphere, monolayer molybdenum disulfide crystallites take a distorted hexagonal shape.228, 229
The activity of molybdenum (and tungsten) disulphide in hydrotreatment is significantly improved when promoted by cobalt and nickel.230 Employees of ‘Halder Topsøe A/S’ (now ‘Topsoe’) proved the formation of a CoMoS mixed sulphide phase by Mössbauer spectroscopy.231 – 234 These conclusions were later independently confirmed by STM for (Fe/Co/Ni/Cu)MoS systems,235, 236 allowing the visualization of the mixed sulfide formation at the crystal edge of molybdenum disulfide. One of the mechanisms for deactivation of (Co/Ni)MoS catalysts is the separation of individual sulfide phases from the CoMoS/NiMoS phase.237
There are two types of CoMoS phases: CoMoS-I and CoMoS-II. The CoMoS-I phase consists of monolayer crystallites and is formed under milder conditions on oxide-supported catalysts. The more active multilayer CoMoS-II with lower dispersity is formed at high sulfidation temperatures or when heating CoMoS-I. A similar partition was later shown for NiMoS and MoS2 systems.238, 239
To describe the influence of catalyst morphology on selectivity, the ‘rim-edge’ model (FIg. 9) has been proposed.240According to this model, the upper horizontal edge of a multilayer crystallite is called the rim and the vertical face is formed by edges. The active sites on the rim are active in hydrogenation and hydrogenolysis, while the sites on the edges are active in hydrogenolysis but not in hydrogenation. By controlling the number of layers in the crystallite, the ratio of the number of sites on the rims to the number of sites on the edges can be altered, thereby controlling the selectivity of the catalyst.
![[{"id":"Ez2YFwdJfH","type":"paragraph","data":{"text":"‘Rim-edge’ model of the catalyst structure.<sup>240</sup>"}}]](/storage/images/resized/p9r1Jkm10FDvpCBowzl8yxqSV6yZ5gFvctcfWT09_xl.webp)
The number of active sites per mole of active phase depends on the catalyst morphology. As the crystallite edge length decreases, the total edge length per mole of the active phase increases quadratically, and at the same Co/Mo ratio, the proportion of unpromoted MO sites increases. Conversely, as the crystallite size increases, the total length of edges per mole of active phase decreases, and for the same Co/Mo ratio, the proportion of unpromoted Mo sites decreases. This in turn changes the activity and selectivity of the catalyst.
The properties of the active sites of molybdenum – sulfide systems were studied using the 35S radiolabel (see Ref. 241). Immediately after sulfidation, the unpromoted MoS2 catalyst contains a considerable amount of excess sulfur (‘non-stoichiometric’ sulfur), which can be easily removed by the hydrogen stream. Some of the remaining sulfur (‘mobile’ sulfur) is associated with active sites and is displaced by sulfur from thiophene fed to the catalyst under hydrotreating conditions. In the CoMoS catalyst, mobile sulfur is divided into two types, depending on the rate of exchange for sulfur from the feedstock: ‘rapid’, associated with mixed CoMo sites, and ‘slow’, associated with unpromoted Mo sites, the proportion of which depends on the composition and morphology of the catalyst.
The conversion of synthesis gas to alcohols on potassium-doped molybdenum disulfide catalysts was patented in 1984,242 and a related process for the synthesis of alcohols from a mixture of synthesis gas and alkenes was patented in 1985.243 To improve selectivity, higher pressures should be used, while excessively high temperatures (> 300 – 350 °C) and long contact times should be avoided.244, 245 Studies have shown that the properties of the support have a significant influence on the process.246 In particular, less acidic supports are preferable.247 Among the alkali metals, potassium is the optimal promoter, the other alkali metals are less effective, and lithium and barium have minimal effect.248 The addition of cobalt or nickel to a K/MoS2-supported catalyst for alcohol synthesis further shifts the selectivity towards the formation of longer-chain products.249, 250 The type-II CoMoS/NiMoS phase shows significantly higher selectivity for alcohols.251
Experimental studies of synthesis gas conversion on catalysts of different morphologies have shown that the synthesis of alcohols is more effective on CoMo sites associated with “rapid’ sulfur in the thiophene hydrotreating reaction, whereas on unpromoted Mo sites associated with ‘slow’ sulfur, the selectivity is shifted towards hydrocarbons.252 This means that the optimal molybdenum sulfide catalysts for conversion of synthesis gas to alcohols are K/CoMoS catalysts in which the proportion of promoted CoMo sites is maximaized and the proportion of unpromoted Mo sites is minimal.
Potassium is directly involved in the formation of the active phase.253 According to the DFT calculations, modification of the active sites by potassium leads to their reduction, decreases their Lewis acidity and increases their affinity for sulfur.254, 255 In the proposed mechanism for synthesis gas conversion on CoMoS catalysts, such modifications should prevent the desorption of alkanes, facilitate the desorption of alcohols and promote the insertion of CO into the metal – alkyl bond. According to calculations, later confirmed by an ethanol conversion experiment, the C – O bond scission in the chain growth cycle occurs in an aldehyde-like, not alcohol-like, intermediate.255, 256 This means that excess hydrogen pressure can inhibit the chain growth.
The synthesis of alcohols takes place at the same sites as the direct hydrodesulfurization of thiophene.252 It follows that for the synthesis of alcohols it is optimal to use catalysts in which the active phase is represented by multilayer crystallites with a large number of layers.
3.6. Enzymatic catalysis
Enzymes are natural catalysts that operate under mild conditions and have high substrate and product specificity. CO and N2 molecules are activated by enzymes containing metal atoms as a part of a metal-sulfide cluster tightly bound to the protein backbone. Selectivity is achieved by isolating the active site in the bulk of the enzyme globule and by controlled access of the starting molecules to the active site through channels in the protein structure. Nitrogenases and CO dehydrogenases act in conjunction with electron transfer agents, bringing them closer to electrochemical processes. The metal – sulfide cluster can be considered as a model of the active phase surface of a heterogeneous catalyst. As such, the metabolism of molecular CO and N2 is of interest as a source of solutions for the design of novel catalysts and electrochemical processes.
3.6.1. Enzymatic activation of CO
The biological activation of CO is carried out by three significantly different families of enzymes, but it can be said that in all three cases, the activation is of the nucleophilic type.
The family of oxygen-resistant Mo – Cu-dependent CO dehydrogenases ([MoCu] – CODH) belongs to the family of molybdenum-oxosulfide xanthine oxidases.257 [MoCu] – CODH oxidizes CO irreversibly, losing a substantial fraction of the energy stored in CO before transferring electrons to the electron transport chain of oxygen respiration. This irreversibility allows the bacteria that synthesize these enzymes to use extremely low concentrations of CO as an energy source, including free CO present in the atmosphere.258, 259 The active site in [MoCu] – CODH is the Mo – Cu oxysulfide site in which oxygen coordinated to the Mo atom acts as a proton acceptor from the water molecule and Cu atom coordinates and activates CO for nucleophilic attack (Fig. 10).260
![[{"id":"KHvHKZwskh","type":"paragraph","data":{"text":"Activation of the CO molecule at the active site of CO dehydrogenase [MoCu] – CODH.<sup>260</sup>"}}]](/storage/images/resized/cl2A8oaHRMHrUMyHzsinIhjhxV5ee3ZiwSr4r0Bd_xl.webp)
The Wood – Ljungdahl pathway (WLP) uses CO as an intermediate in the reduction of CO2 by hydrogen to acetic acid. With slight variations, the WLP is realized in a number of bacteria and archaea called acetogens. All stages of the WLP are equilibriums, allowing acetogens to use a wide range of C1 compounds, including CO, as energy and carbon sources.261 Two WLP reactions involving CO are discussed below. The family of oxygen-sensitive Fe – Ni-dependent CO dehydrogenases ([FeNi] – CODH) carry out the equilibrium oxidation of CO or, conversely, the equilibrium reduction of CO2 . When associated with a membrane complex, enzymes of this family can be involved in membrane potential generation and ATP synthesis.262, 263 Some proteins of this family, known as ‘bifunctional CODHs,’ form a stable protein complex with acetyl – CoA synthase; these complexes are discussed in detail below. There is evidence for cytoplasmic variants of [FeNi] – CODH,264 which act in tandem with cytoplasmic electron transfer agents. Despite differences in localization and role, all types of [FeNi] – CODH use the same active site and contain a fragment of the associated electron transfer chain.
Cytoplasmic [FeNi] – CODH consists of two identical protein subunits and contains five iron-sulfide clusters.265 Two [Fe4S4]-clusters called C-clusters are active sites. Two clusters are the usual [Fe4S4] clusters characteristic of electron transfer proteins and are called B-clusters. A cluster on the surface of the enzyme, also of [Fe4S4] composition, links the subunits together and is called the D-cluser. Its role is unknown, but it has been suggested that it protects the enzyme from oxidation.266
The C-cluster can be described as a cuban cluster [Fe4S4] in which Ni is located at one of the vertices and the iron atom from this position is forced to the side together with one of its sulfide ligands and fixed by coordination with amino acid residues (Fig. 11). Nickel appears to be coordinatively unsaturated and coordinates the CO molecule, which can be nucleophilically attacked by the OH group coordinated to the iron atom. A carboxyl group is formed on nickel, which readily loses a proton to form a hydrogen-bonded carboxylate. With the loss of the CO2 molecule, the active site is reduced by 2 electrons which tunnel to the B-cluster where they become available for transfer to other proteins.267, 268
![[{"id":"ahqWvNnVGt","type":"paragraph","data":{"text":"Molecules of СО and Н<sub>2</sub>О adsorbed on the active site of CO dehydroge nase [FeNi] – CODH.<sup>267, 268</sup>"}}]](/storage/images/resized/z7DZ4KhFWcQmQDJY4LIFyZanZe29D3pNErCINwqT_xl.webp)
The Fe – Ni-dependent acetyl – CoA synthase (ACS) family carries out the equilibrium synthesis of acetyl – CoA from a methyl group, a CO molecule and H – CoA. ACS can exist in a monofunctional form, but is often described in the literature as part of a complex with CODH and accessory proteins (so-called bifunctional CODH). It was shown that there is an intramolecular tunnel in ACS/CODH complexes connecting the active sites of the complex.269, 270 For the monomeric ACS of Carboxydothermus hydrogenoformans, a mechanism has been identified that controls the availability of the active site to reagents by coupling the protein configuration and the state of the active site.271 It appears that this control, together with the intramolecular channel system, prevents the loss of CO formed intermediately on CODH/ACS complexes to the atmosphere.272, 273
The active site of ACS is the so-called A-cluster, which consists of a [Fe4S4] core and two Ni atoms (Fig. 12). One nickel atom (Nip) contacts a sulfur atom of the [Fe4S4] cluster and is bonded to a proximal iron atom by a thiolate cysteine residue. The Nip atom is also bound to the second nickel atom (Nid) by two other thiolate cysteine residues and coordinates to two nitrogen atoms involved in the formation of the peptide bond of the amino acid chain of the protein.274 – 276
![[{"id":"b5xwIEAIRG","type":"paragraph","data":{"text":" Active site of [FeNi]-ACS.<sup>276</sup> Methyl group is shown in red, CO in purple and cysteine residues in blue."}}]](/storage/images/resized/7k6nmjBryHd2EMBajqbVojoJywFSyEqLfw6tK2Rc_xl.webp)
If the CO molecule is stable in the free state and can bond to the Nip atom in a barrierless mode, the methyl residue is formed in another branch of the WLP on the organic carrier. The transfer of the methyl group to the ACS requires the involvement of an intermediate carrier, which in the case of CFeSP (Corrine Fe – S protein) is methylcobalamin. The methyl group is transferred as a methyl cation. The order in which the methyl group and CO bond to the active site appears to be arbitrary. The insertion of CO into the Me – Nip bond occurs with a low barrier. Then, after coordination of CoA, reductive elimination of acetyl – CoA takes place.276, 277
3.6.2. Enzymatic activation of N2
Nitrogen is an essential component of all proteins. At the same time, biologically available nitrogen is scarce, making nitrogen-fixing organisms essential members of most modern biocenoses and autotrophic microbial communities. The industrial synthesis of ammonia is technically sophisticated and is carried out under harsh conditions. In biological systems, nitrogen fixation occurs under mild conditions, hence creating a natural desire to use it. For example, plants associated with nitrogen-fixing bacteria enrich the soil with nitrogen and can be used instead of nitrogen fertilizers.
Biological nitrogen fixation is carried out by oxygen-sensitive enzymes of the nitrogenase family. There are three families of functionally similar nitrogenases that differ in the composition of the nitrogen-fixing cofactor (Fig. 13).278 Mo-nitrogenase, discussed below, is the best studied one. Nitrogenase is a strong non-selective reductant and can reduce CO, hydrazine, cyanide, acetylene and other substrates.279
![[{"id":"MtoA7QVhXB","type":"paragraph","data":{"text":"Nitrogen-fixing cofactor of nitrogenase.<sup>278</sup> FeMo cofactor: X = S<sup>2–</sup>; M = Mo. FeV cofactor: X = CO<sub>3</sub><sup>2–</sup>, M = V. FeFe cofactor: M = Fe; X is unknown."}}]](/storage/images/resized/cTbZ7yGqlMjXgrmVz8yDle9ejKI25nYOU39MlSbk_xl.webp)
The Mo nitrogenase complex consists of two proteins, dinitrogenase, or MoFe protein, and ATP-dependent reductase, or Fe protein. Fe-protein transfers electrons from ferredoxin I to nitrogenase by consuming an ATP molecule.280, 281 MoFe-protein can be reduced by chemical reducing agents 282 or when immobilized on the electrode surface.281 In such systems, alternative substrates can be reduced but not the nitrogen molecule, i.e. Fe-protein should be necessarily involved in nitrogen reduction at nitrogenase. MoFe-protein contains four metal sulfide clusters. Two M-clusters have the composition [MoFe7S9C] and are independent active sites. Two P-clusters with the composition [Fe8S7] participate in electron transfer to the M-cluster.284 The M-cluster is buried within the enzyme and is connected to the surface by several hydrophobic and several water-filled channels.278, 285
Nitrogenase can be regarded as a nanoelectrochemical system: the Fe protein transfers electrons from ferredoxin I to the MoFe protein, i.e. it works as agenerator of electrode potential. The M-cluster of the MoFe-protein acts as a negatively charged electrode on which nitrogen molecules are reduced to ammonia with the involvement of protons from water. The activation of the nitrogen molecule occurs after the transfer of four electrons and four protons to the cluster, forming two sulfhydryl groups and two hydride ligands, and is accompanied by the release of molecular hydrogen (Fig. 14).286 Ab initio studies suggest that the electronic state of the metal–sulfide cofactor is the key factor in nitrogen activation, with the protein part playing a supporting role.288
![[{"id":"RYqaPZa3kl","type":"paragraph","data":{"text":"Schematic of the addition of nitrogen to the active site of dinitrogenase.<sup>287</sup>"}}]](/storage/images/resized/SBcKxAl5pu85kOAz2I85e2yJYeeL9fRbOedvBzv1_xl.webp)
3.7. Electrochemical reduction
An electron-rich reduction site is required to activate the nitrogen molecule under mild conditions. However, the reduction potential of the surface can be increased by applying a voltage. This means that electrochemical nitrogen reduction reaction (E-NRR) is potentially possible. In recent years, this field has attracted much attention and is covered in a number of reviews.289 – 292
In the stepwise hydrogenation of a nitrogen molecule, the most high-energy intermediate species is N2H, the formation of which by the reaction N2 + 0.5 H2 = N2H requires 253 kJ mol–1 (see Ref. 293). This intermediate can only be stabilized by complex formation. In this case, the final product of the reduction, ammonia, should be easily desorbed. Therefore, most of the binding energy should arise from strong π-bonding due to back-donation, since the strong Lewis acidity of the adsorption site will prevent desorption of ammonia.
Hydrogen reduction is the main side reaction in the electrochemical reduction of nitrogen and very often the Faraday efficiency (FE) of E-NRR is less than 10%. Electrochemical hydrogen reduction is most hindered on gold, silver and copper. The chemical inertness of gold makes it an attractive material for E-NRR. An important issue is the cost of gold, so porous composites are commonly used. As a representative result, in a publication 294 using Au – Ni composite foam, an ammonia release performance of 9.42 mg h–1 cm–2 (29.3 mg h– (mg cat.)–1) at FE of 13% in 0.1 M aqueous sodium sulfate solution was achieved. An E-NRR study on a gold thin-film electrode showed that electrolysis intermediately produces particles with single N – N bonds and NH2 groups.295 These particles are the source of the typical E-NRR by-product hydrazine.
Using a copper electrode as an example, the effect of electrolyte and potential on E-NRR performance was explored in detail.296 It was shown that the Faraday efficiency passes through a maximum with increasing voltage, and that the maximum selectivity and activity are reached at pH 13.5. When comparing the effect of the alkali metal cations, potassium proved to be the best, with a decrease in performance when switching to both lithium and cesium. This effect is probably specific to an alkaline environment, as another systematic study of the effect of electrolyte on E-NRR at a platinum-polypyrrole electrode in an acidic environment showed the order of performances of Li > Na > K; furthermore, lithium salts are generally considered to be the preferred electrolyte.297
A large number of electrode materials for E-NRR have been described in the literature.292 In particular, RhSe (yield 175 mg h–1 (mg cat.)–1, FE 13.3%) and Rh2Sb (229 mg h–1 (mg cat.)–1, FE 6.1%) can be mentioned. In general, for almost all transition metals and a number of non-transition elements, the activity of some compound in E-NRR has been established, usually at the level of tens of mg h–1 (mg cat.)–1 at FE < 5%. There is great interest in single-atom catalysts in which the catalytically active metal atom is immobilized in carbon-nitrogen,298, 299 sulfide,300 etc.301, 302 matrices. Immobilized metal complexes, such as phthalocyanines, are also considered to be single-atom catalysts.303
Another approach to optimization is to modify the solvent 304, 305 and/or use an electrode coating 306, 307 to prevent hydrogen evolution by limiting proton access to the electrode. The use of an aprotic solvent can achieve very high FE in E-NRR, but requires an external hydrogen source and an electrolyte capable of acting as a hydrogen carrier.308
A related process is the electrochemical reduction of nitrogen in the presence of CO2 to urea 309 with up to 20% FE.310 Unfortunately, this process has not received much attention.
The electrochemical reduction of CO to C2+ products occurs on copper and with less efficiency on nickel.311 CO is not reduced on the other metals. The main reaction products are methane, ethylene, C2 and C3 alcohols and aldehydes. Neither methanol nor ethane are produced in significant quantities. The reduction of CO can occur simultaneously with its formation in the electrochemical reduction of CO2.312, 313
The mechanism for electrochemical reduction of CO remains controversial. The absence of methanol and formaldehyde in the products in the presence of methane and the reduction of formaldehyde to ethanol suggest that the C – O bond cleavage occurs prior to their formation, probably in the CHO particle.314 By analogy with the known reductive coupling of CO (see Ref. 315), the key step in the formation of C2+ products in the electrochemical reduction of CO was suggested to be the reductive coupling of two adsorbed CO molecules. However, based on combined experimental studies, it has been concluded that the rate-determining step for both the formation of methane and the formation of C2 products is the reduction of CO to a C(H)O particle, and that the methane and C2+ products are probably formed at different active sites.316, 317 It has also been suggested that the formation of C2 products occurs via a ketene-like intermediate.318 This hypothesis is consistent with the formation of acetamide when ammonia is added to the electrolyte 319 and the formation of acetate in the absence of ammonia, since ketene is a very active electrophilic acetylating agent.
3.8. Alternative substrates
There are three types of readily available substrates that are isoelectronic with CO and N2: acetylenes, nitriles and isonitriles. Also, there is a related molecule, NO, which is also resistant to reduction by hydrogen at moderate temperatures in the absence of a catalyst.
There are reports on dissociation of acetylenes on metal surfaces.320, 321 For nitriles, hydrogenation reactions to amines are known, but there is no evidence in the literature for dissociative adsorption on metal catalysts.
The catalytic condensation of isonitriles to secondary amines using Raney cobalt in liquid ammonia has been reported.322 Both dialkylmonoamines and N,N'-dialkyl-α,ω-diamines are produced. Unfortunately, no further studies in this direction were found.
The reduction of nitric oxide is mainly studied in the context of combustion product purification processes (deNOx) where the target product is molecular nitrogen. NO can be reduced to ammonia on platinides.323 The hydrogenation of NO on platinum in the presence of sulfuric acid is an industrial process for the production of hydroxylammonium sulfate.324 There are reports of an electrochemical process for the reduction of NO to ammonia 325 and an industrial process for the synthesis of hydroxylamine.326, 327
A typical by-product of the catalytic reduction of NO is nitrous oxide (N2O), probably formed via the intermediate hyponitrite (–ON=NO–) and the corresponding unstable acid (H2N2O2).328 Metal catalyst-free redox condensations involving two NO molecules are also possible; in particular, the Traube reaction, the reaction of two equivalents of NO with a carbanion to give the N-nitroso-N-organohydroxylamine salt, has long been known in organic chemistry. In metal complex chemistry, the coupling of nitrosyl ligands is also reported and has been studied in the context of modelling natural enzymes.329
4. Systematization of reductive conversion of CO and N2
The Haber – Bosch and Fischer – Tropsch processes begin with the dissociation of the starting molecules to form surface particles (Fig. 15). In both cases, hydrogen is found in the products in the +1 oxidation state. It is then the reducing agent, while the CO and N2 molecules are the oxidizing agents. The dissociation products migrate over the catalyst surface.
![[{"id":"NhzTAi7Isn","type":"paragraph","data":{"text":"Schemes of reactions occurring during the fixation of N<sub>2</sub> (left) and CO (right) on the surface of a metal catalyst."}}]](/storage/images/resized/x6GTYz3QrKDBYRdpMhShBXrouux4SggyzCFioqCF_xl.webp)
First the dissociative adsorption of hydrogen takes place. The hydrogen bound to the metal atom is usually referred to as the hydride H–. However, in a number of complexes (e.g. H[Co(CO)4]) the hydride hydrogen is acidic.
Acidic protons are characterized by high mobility and indeed, adsorbed hydrogen readily migrates over the metal surface.330 It is therefore convenient to consider the adsorbed hydrogen as a hydrogen cation stabilized by the polarization of the metal’s conduction electrons.
Next, dissociative adsorption of nitrogen or CO takes place. The resulting adsorbed atoms are reduced by the mobile electrons of the catalyst and stabilized by interaction with metal atoms. The higher the coordination number of the adsorbate, the greater the effective charge it can stabilize. For this reason, the dissociation of CO and N2 occurs most readily at ‘step’-type surface defects. When such sites disappear, the mechanism of direct dissociation is not realized. The mobile cations can coordinate to the resulting negatively charged species, further stabilizing them. The catalyst surface should have a high reduction potential.
Next, recombination of CO and N2 dissociation products with surface hydrogen takes place. For carbon, condensation can occur to create the C – C bonds, whereas for nitrogen and oxygen this behaviour is not characteristic. To facilitate the recombination, it is important that the adsorbates on the catalyst surface are mobile, i.e. a high density of reduced coordinatively unsaturated metal atoms on the catalyst surface is required.
There are many similarities in the conversion of CO and N2 by a dissociative mechanism. In both cases, the activity is controlled by the reduction potential of the catalyst surface and intermediate particles can be stabilized by interaction with cations.
The Fischer – Tropsch synthesis and the Haber – Bosch process are carried out on similar catalysts (Group VIII metals), have a similar mechanism (direct dissociation of the starting molecule and subsequent assembly of the product) and, in some cases, share similar regularities. For example, the size effect for the ruthenium catalyst is observed in both processes. Other catalytic processes involving molecules of similar electronic structure can also follow the dissociative pathway. In particular, for isonitriles, a process similar to the Fischer – Tropsch synthesis is known,322 and for NO, dissociation on the surface of Pd, Pt,331 catalyzing the reduction of NO with hydrogen to ammonium, has been established.
CO and N2 can be reduced by the associative pathway involinving successive additions (Fig. 16). In the case of CO, the first two bonds are formed by the carbon atom, reflecting the asymmetry of the molecule. In the newly formed carbonyl group, the oxygen is in its characteristic covalency of 2 with two lone pairs. For nitrogen, similar compounds of the general formula R2N+=N– are extremely unstable,46 so diimine or its derivatives are formed instead. However, compounds with double and single N – N bonds are destabilized by the interaction of the nitrogen lone pairs, and it is therefore necessary to stabilize such intermediates by interaction with a suitable adsorption site. The schemes of CO and N2 activation in stepwise addition processes are given below.
![[{"id":"jaJS3mOtVj","type":"paragraph","data":{"text":"Variants of CO and N<sub>2</sub> activation in the addition mechanism."}}]](/storage/images/resized/ikTXWnSOisL8oabF6KSjJO6XtKPcIqqLOhgTrYXX_xl.webp)
Carbon monoxide is capable of reacting with strong nucleophiles, such as alkyllithium 332 or electrophiles 333 even in the absence of a catalyst. The resulting acyl intermediate reacts with the counterion to afford a carbonyl compound. The reaction is promoted by a suitable catalyst 333 (see Fig. 16a). Hydrogenation of the resulting carbonyl compound produces alcohols. Hydrogenolysis of the C – O bond initiates or continues the growth of the aliphatic chain when dissociative mechanism cannot proceed. If a hydroxide or oxide anion acts as a nucleophile, fragmentation to carbon dioxide (water shift) may occur instead of reaction with an electrophile. This is the mechanism by which CO is activated in the CODH family of enzymes.
Direct reduction of CO by potent reducing agents often leads to reductive oligomerization.334 Similar reactions are known for a number of metal complexes (see Fig. 16b).335 Electrochemical reduction of CO affords С1 – С3 products, i.e. it is logical to consider it as a related reaction.
For the nitrogen molecule, direct addition is usually thermodynamically unfavoured because of the instability of the possible products. Coordination and subsequent reduction of the nitrogen molecule creates the conditions for stepwise addition of the electrophile. In the terminal end-on coordination, the electrophile binds to an uncoordinated nitrogen atom, with a weakening of the nitrogen – nitrogen bond and a strengthening of the nitrogen – substrate bond. This can be considered as a symmetrical addition where the substrate metal atom and the electrophile are the adding groups (see Fig. 16c).The second pattern involves the addition of protons to a ‘butterfly’-type coordinated molecule (see Fig. 16d ). Hydrazine may be formed as a by-product due to desorption of the intermediate. Diimine is unstable in free form and therefore cannot be identified in the products.
The related NO molecule shows intermediate behaviour. This molecule readily forms condensation products when attacked by carbanions (the Traube reaction) or nitrous oxide when incompletely reduced,336 but can also be reduced to ammonia or hydroxylammonium salts (see above) by hydrogenation or electrochemically.
From this point of view, the known systems catalyzing the reduction of nitrogen to ammonia or amines can be divided into two large families: with dissociative and with stepwise activation. The overall process is a reduction process, but the specific mechanisms of reduction and stabilization of the intermediate steps are very different (Table 1). A similar division can be made for reductive CO conversion systems (Table 2). In contrast to N2, CO undergoes stepwise reduction in many systems, while dissociative activation outside the FT process is virtually unknown. Dissociative activation of CO occurs on the same catalysts and by the same mechanism as for N2. The CO molecule is asymmetric, but its Lewis electronic structure is symmetric. The attack on the carbon atom breaks the symmetry of the electronic structure, thus eliminating the contradiction. In contrast, both the nitrogen molecule per se and its Lewis electron structure are symmetrical, and therefore activation should be symmetrical.


5. Conclusion
The Haber – Bosch and Fischer – Tropsch processes have a rich and long history of practical application using a variety of primary feedstocks. The conditions and catalysts for these processes are well studied. The main routes of both processes start with the dissociative adsorption of the substrate (N2 and CO respectively) onto the metal surface with the cleavage of triple bonds and subsequent formation of products from the surface species. The end product of the Haber-Bosch process is ammonia. In the Fischer-Tropsch synthesis, the product is a mixture of aliphatic hydrocarbons, the composition of which depends on the catalyst in choice and the reaction conditions.
The said processes involve direct dissociation of CO and N2 to give particles with strong negative charges. They are stabilized by interaction with proximal coordination sites, e.g. on the step-type surface defects, and by interaction with positively charged species, e.g. alkali metal cations. Redox dissociation is promoted by the injection of electron density into the active phase, facilitated by nucleophilic and reducing promoters and supports.
An alternative to direct dissociation mechanism is the stepwise addition process, in which the behaviour of CO and N2 molecules is different.
In the case of CO, the molecule is asymmetric and the electron density readily shifts towards the oxygen atom. Addition takes place at the carbon atom to form a stable >C=O moiety. Such reactions are characteristic of the CO molecule coordinated to a single metal site on the carbon atom and are possible with active reagents in the absence of a catalyst due to nucleophilic attack on the carbon atom.
In the case of N2, the molecule is non-polar. The addition is usually symmetric and the products are destabilized by the repulsion of the negative charges of the lone pairs of the nitrogen atom. For the process to occur, at least one lone pair must be involved in the formation of a coordination bond and the presence of a strong reducing agent, such as an elemental alkali metal, is required.
The main challenge in industrial nitrogen conversion is the high temperature of the Haber – Bosch process. To address this issue, it is necessary to reduce the dissociation barrier of the nitrogen molecule and stabilize the resulting products. One approach is to promote the surface of the active phase with alkali and alkaline earth metal cations, which stabilize the negatively charged dissociation products of the nitrogen molecule. As part of the approach to increasing the reduction potential of the active phase, supernucleophilic and superdonor substrates for the ruthenium catalyst and strongly reducing active phases, in particular d-metal hydrides and electrides, are being investigated.
The Fischer – Tropsch synthesis operates under much milder conditions than the Haber-Bosch synthesis and produces a mixture of aliphatic hydrocarbons containing mainly medium-chain (С5 – С15) compounds. This variant of the process has low profit margins and niche applications, such as converting natural gas into more easily transportable liquid hydrocarbons. Variants of the process affording products with high added value are of particular interest. Currently, high-temperature Fischer – Tropsch synthesis is used to produce a mixture of short-chain aliphatic hydrocarbons with a high content of terminal olefins formed from the surface alkyl intermediate by the β-elimination mechanism.
In addition to dissociative activation, where CO and N2 molecules dissociate in the initial stages of the reaction mechanism, the reverse situation is also possible, where carbon–oxygen and nitrogen – nitrogen bonds are retained and other molecules add to them. Addition to N2 is usually thermodynamically unfavourable and requires the use of strong reducing agents (reduced form of the enzyme nitrogenase, electric current) and stabilization of the intermediates by interaction with the catalyst. Addition to CO is usually thermodynamically favourable. The C – O single bond is much stronger than the N – N single bond and can be retained in the final product.
A process in which the C – O bond is retained is the synthesis of methanol from synthesis gas, another industrial process with a long history. The mechanism of this process is not entirely clear and there is evidence of the intermediate CO2 formation.
A related process is the synthesis of higher alcohols using K(Co/Ni)Mo(S/P/C) catalysts. The formation of higher alcohols with low selectivity on modified Fischer – Tropsch synthesis catalysts is also known. The main side process is the formation of light alkanes and methane.
Over the last decade, much attention has been paid to electrochemical processes occurring under mild conditions that can solve the problems of storage and utilization of excess electricity without the use of high-pressure equipment. In particular, a large number of works are devoted to electrochemical conversion of CO, CO2 and N2.
In studies on the electrochemical reduction of nitrogen, the main efforts are focused on the selection of the electrode material, which determines the kinetic barriers of the competing reactions of water reduction to hydrogen and nitrogen reduction to ammonia on the electrode surface. The effect of the electrode coating material on the diffusion process of nitrogen molecules and hydrogen ions can also be used. A development of this approach is a shift to an aprotic solvent. In this case, hydrogen is delivered in molecular form to another electrode where it binds to a hydrogen carrier in the electrolyte.
In aqueous media, CO is not reduced on most metal electrodes. However, on copper, CO is reduced to a mixture of C1 – C3 products. Ethanol or acetic acid can be produced with high selectivity. The process is often carried out as an electrochemical reduction of CO2 without isolation of the intermediate CO.
6. List of abbreviations
ACS — acetyl – CoA-synthase,
bcc — body-centered cubic packing,
CoA — cofactor А,
CODH — CO dehydrogenase,
DCD — Dewar – Chatt – Duncanson model,
E-NRR — electrochemical nitrogen reduction reaction,
fcc — face-centered cubic packing,
FE — Faradaic efficience,
FТ — Fischer–Tropsch process,
hcp — hexagonal close packing,
MEP— molecular electrostatic potential,
STM — scanning tunnelling microscopy,
WLP — Wood – Ljungdahl pathway.