

Keywords
Abstract
The chemotherapy with cisplatin and its analogues widely used in medical practice is associated with undesirable side effects caused by non-selective ligand exchange and binding of the complexes to various biomolecules in the body. An alernative to classical platinum(II)-based drugs are platinum(IV) prodrugs, that is, platinum(II) complexes additionally modified with diverse biologically active axial ligands, including known pharmaceutical products. In recent years, quite a few studies devoted to the design of effective Pt(IV) prodrugs have been published, with some of the developed agents being markedly superior to clinically used cisplatin and carboplatin in therapeutic efficacy. This review summarizes the synthetic approaches to the design of Pt(IV) prodrugs and modification of the axial ligands. The second part of the review is devoted to the biological activity of Pt(IV) prodrugs reported in the period from 2018 to 2023 and comparison of various approaches to the design of effective anticancer agents based on these compounds.
The bibliography includes 239 references.
1. Introduction
Platinum(II) coordination compounds have been used in the therapy of cancer since the discovery of the cytotoxic properties of cisplatin in the mid-20th century.1 Currently, the U.S. Food and Drug Administration (FDA) has approved three Pt(II)-based drugs for clinical use, namely, cisplatin (CDDP), oxaliplatin (OLP) and carboplatin (Fig. 1). In addition, the drug nedaplatin is used in Japan for the therapy of lung and neck tumours, lobaplatin has been approved in China for the therapy of metastatic breast cancer, and heptaplatin is used in Korea to treat the gastric cancer.
![[{"id":"jG5hdR1ZTO","type":"paragraph","data":{"text":"Structural formulae of the platinum(II)-based drugs used for the therapy of cancer."}}]](/storage/images/resized/hVfOAzUwgFrdNt1dJsssCGRFikHFQHxu1rRm5yaE_xl.webp)
Platinum(II)-based drugs are square planar Pt2+ coordination compounds containing two am(m)ine ligands and two cis-arranged anionic ligands in the molecule.2 The mechanism of cytotoxic action of Pt(II) complexes has been addressed in numerous publications (see, for example, Refs 3, 4). It was proved that these drugs penetrate into cells, then the leaving ligands are exchanged for water, and the aquated Pt(II) complex binds to the N(7) atom of a purine base of DNA to give cross-links, which disrupt the cell functioning and trigger apoptosis, a process of programmed cell death. The results of studies of the last two decades also revealed alternative mechanisms of the antiproliferative action of cisplatin. In particular, binding of cisplatin to a number of proteins such as ubiquitin, G-actin and other cytoskeleton proteins causes disruption of their biological functions.5 In some studies, it is indicated that cisplatin and oxaliplatin can cause immunogenic cell death, i.e., stimulate an immune response to the appearance of malignant neoplasms.6, 7 It was also noted that cisplatin can induce apoptosis by damaging mitochondrial rather than nuclear DNA.8
Despite of being widely used in clinical practice, Pt(II)-based drugs suffer from a number of crucial drawbacks.9 A large portion of cisplatin introduced into the body (up to 90%) irreversibly binds to macromolecules in the bloodstream, and only 1% reaches the therapeutic target, that is, nuclear DNA.10 The non-specific binding is responsible for some severe side effects that accompany treatment with platinum-containing drugs such as hearing loss, nephrotoxicity and neurotoxicity.11, 12 One more important side effect is the acquired drug resistance, which decreases the efficacy of Pt(II)-based anticancer drugs due to decreasing platinum uptake by the cells or intracellular deactivation of pharmaceuticals.13, 14 Cisplatin analogues such as oxaliplatin and carboplatin have lower general toxicity; however, they are not superior to cisplatin in selectivity or antitumour actvity.6, 15
Therefore, important challenges of medicinal chemistry are to overcome the above drawbacks of the existing medications and to develop new highly efficacious drugs based on platinum. A number of approaches have been developed for addressing this task, in particular, the synthesis of cisplatin analogues with other equatorial ligands,16 non-traditional trans-platinum(II) compounds and Pt(IV) complexes.17
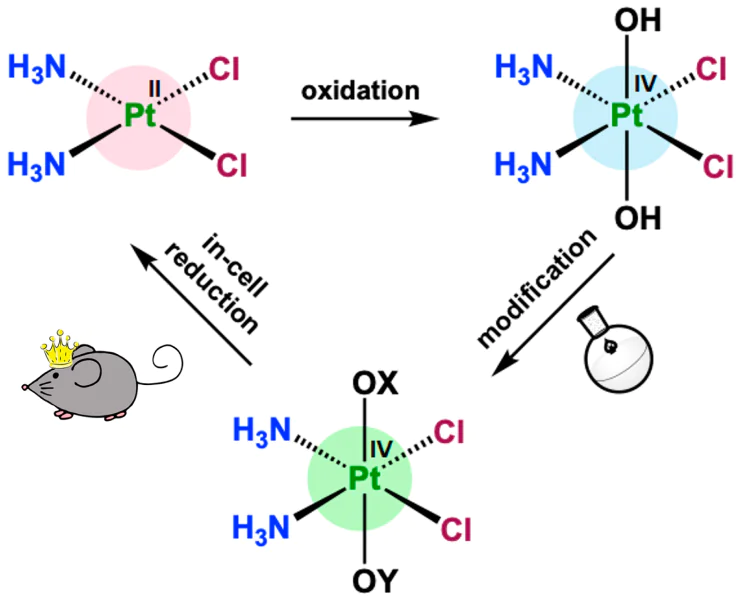
Platinum(IV)-based prodrugs are octahedral low-spin d6 coordination compounds consisting of a platinum atom, four equatorial ligands identical to those of Pt(II) complexes, and two axial ligands.18 Due to the increase in the coordination number, these compounds are less prone to ligand exchange in the bloodstream and, as a consequence, they are less likely to undergo side reactions with biological macromolecules.19 Platinum(IV) coordination compounds are unable to bind to DNA, but they can be reduced in the intracellular medium, thus releasing the cytotoxic Pt(II) complex and free ligands (Fig. 2).20
![[{"id":"ggSYUUdpAa","type":"paragraph","data":{"text":"General synthetic scheme and principle of action of Pt(IV)-based prodrugs. The grey sphere designates the biologically active axial ligand."}}]](/storage/images/resized/NxOE0H8IabQOk8ztwOU1tNu2VzQaYZtS2aGMkjWy_xl.webp)
Since the axial position of Pt(IV) complexes can be easily modified, varying axial ligands makes it possible not only to tune the physicochemical properties, but also to modify the biological activity of the products.21 – 23 Since the introduction of vector groups into the axial position of PtIV complexes is favourable for increasing the affinity of prodrugs to tumour cells,24 – 26 the introduction of a cytotoxic axial ligand may afford compounds that act on several therapeutic targets,27 – 30 while the use of compounds responsive to the external physicochemical stimuli as axial ligands may give compounds with controllable action.31 – 34
Platinum(IV)-based prodrugs have been the objects of research for more than two decades. To date, there are quite a few publications devoted to variation of the axial ligands and the starting Pt(II) complexes and to elucidation of the structure – activity relationships. A number of highly ranked reviews deal with the synthesis and biological activity of Pt(IV) prodrugs. In 2016, Johnstone et al.17 analyzed the most recent achievements in the development of new platinum-containing therapeutic agents and methods for their delivery to the tumours. In 2017 and 2019, reviews addressing the biological action of Pt(IV) prodrugs were published.35, 36 A review by Xu et al.,31 which appeared in 2021, surveys the methods of synthesis and mechanistic studies of the intracellular reduction of Pt(IV) compounds. Professor Gibson from the Hebrew University of Jerusalem presented a series of small review papers on the biological activity of Pt(IV) prodrugs. A review 37 addresses Pt(IV) compounds with a multiple biological action, while another paper 38 gives examples of increasing prodrug selectivity to cancer cells. Survey publications by Beloglazkina and co-workers are devoted by photocontrolled activation of Pt(IV) compounds 34 and combination of these compounds with non-steroidal anti-inflammatory drugs in axial positions.23
The present review integrates and systematizes the available data on the synthesis of Pt(IV) prodrugs and investigations of their physicochemical and biological properties. The first part considers synthetic approaches to the design of these drugs, with the attention being paid both to oxidation reactions in chemical media where platinum(II) compounds are converted to platinum(IV) complexes and to chemical modification of axial ligands. The second part of the review considers the biological effects of Pt(IV) prodrugs using the data of publications of the period from 2018 to 2023.
In view of the high interest in the development of new effective Pt(IV) prodrugs meant for the therapy of malignant neoplasms, the large number of publications on this subject in scientific journals, and the lack of Russian-language reviews on the synthesis and biological activity of Pt(IV) complexes, we believe that this review will be of interest to a broad range of researchers specializing in organic and medicinal chemistry.
2. Synthetic approaches to the design and modification of Pt(IV) prodrugs
Coordination compounds of platinum(IV) have been investigated for more than 40 years.39 A large body of data on the synthesis of compounds of this class has been gained to date (see, for example, reviews by Wilson and Lippard 18 and Xu et al.31). In this part of the review, we consider the key synthetic approaches used to prepare and modify Pt(IV)-based prodrugs and discuss the benefits and drawbacks of the considered methods.
The design of Pt(IV) prodrugs implies the synthesis of kinetically inert octahedral coordination compounds based on cytotoxic Pt(II) complexes. The synthetic strategy consists of the following steps:
(1) oxidation of the Pt(II) complex,
(2) replacement of the hydroxyl group at the Pt(IV) atom by various ligands,
(3) replacement of the second nucleophile at the Pt(IV) atom or further modification of the ligand introduced in the previous stage (Fig. 3).
![[{"id":"hkXr5vDg0I","type":"paragraph","data":{"text":"Approaches to the synthesis and modification of Pt(IV) based prodrugs."}}]](/storage/images/resized/pDgkl0nAi5ZRG0gsOyk2LyKFcOILkNYnH5PrrjZw_xl.webp)
Each synthetic stage is considered in detail below.
2.1. Synthesis of Pt(IV) coordination compounds by oxidation of Pt(II) compounds
The major synthetic approach used for the design of Pt(IV) complexes is the oxidation of Pt(II) complexes. The oxidants used most often for the Pt(II) atom are chlorine and hydrogen peroxide.40, 41 As a result of this reaction, two additional ligands enter the coordination sphere of the Pt(IV) ion in trans-positions. The structure of product resulting from hydrogen peroxide oxidation depends on the solvent in which the reaction is carried out.42 Thus in water, cisplatin is converted to the complex cis,cis,trans-[Pt(NH3)2(Cl)2(OH)2] (oxoplatin) Fig. 4, reaction (2)], while the reaction in acetic acid gives cis,cis,trans-[Pt(NH3)2(Cl)2(OH)(OAc)] (Ref. 44) [reaction (3)].
![[{"id":"RkP6CzzrDc","type":"paragraph","data":{"text":"Methods for the oxidation of Pt(II) complexes (the yields are indicated for L = NH<sub>3</sub>, X = Cl)."}}]](/storage/images/resized/vy3S64v0cpApqPHhFkbVmPqKnKI3djyqLJ5npf35_xl.webp)
The synthesis of asymmetric Pt(IV) complexes containing a chlorine atom and a nucleophilic oxygen atom in the axial positions via mild oxidation with N-chlorosuccinimide (NCS) [reaction (4)] was described in more recent publications.45, 46 An unusual example of oxidation of Pt(II) complex in the presence of hydrogen peroxide, acetonitrile and methanol with introduction of acetamide into the axial position has been reported [reaction (5)].47
Thus, by varying the solvent and the oxidant in the oxidation of Pt(II) complexes, it is possible to obtain Pt(IV) complexes with diverse axial ligands and with different numbers of functional groups meant for the subsequent modification. The main strategies of modification of Pt(IV) complexes used to introduce various organic groups into the metal coordination environment are considered below.
2.2. Modification of O-nucleophile at the Pt(IV) centre
In the vast majority of cases, modification of Pt(IV) complexes involves the O-nucleophile at the Pt(IV) atom (see, for example, Ref. 48); therefore, these methods are considered in a separate Section.
2.2.1. The introduction of the carboxyl group into the axial position of Pt(IV) complexes
A popular strategy for the introduction of organic groups into the axial position of the Pt(IV) atom is esterification, which gives rise of a carboxyl group and affords an ester containing a C(O)O – Pt moiety. A drawback of this approach is the necessary presence of a carboxyl group in the molecule of the introduced ligand, which restricts the range of substrates applicable as axial ligands.
2.2.1.1. Synthesis of Pt(IV) dicarboxylate complexes
Using a highly reactive acylating reagent and/or a large excess of the acylating reagent, esterification can be carried out at both available O-nucleophiles in the axial positions of platinum(IV).48 This gives symmetrical Pt(IV) dicarboxylate complexes with two identical organic ligands.
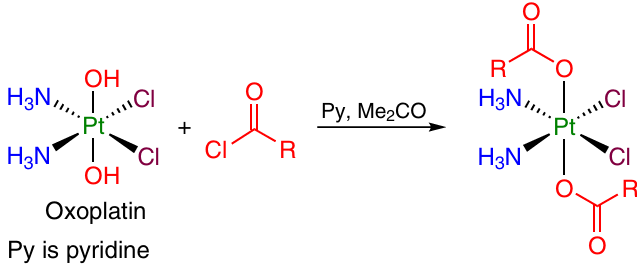
2.2.1.1.1. Synthesis from acyl chlorides
Acyl chlorides derived from the appropriate carboxylic acids can be used as the acylating reagents. Acyl chlorides react with oxoplatin to give a symmetrical Pt(IV) dicarboxylate complex (Scheme 1).
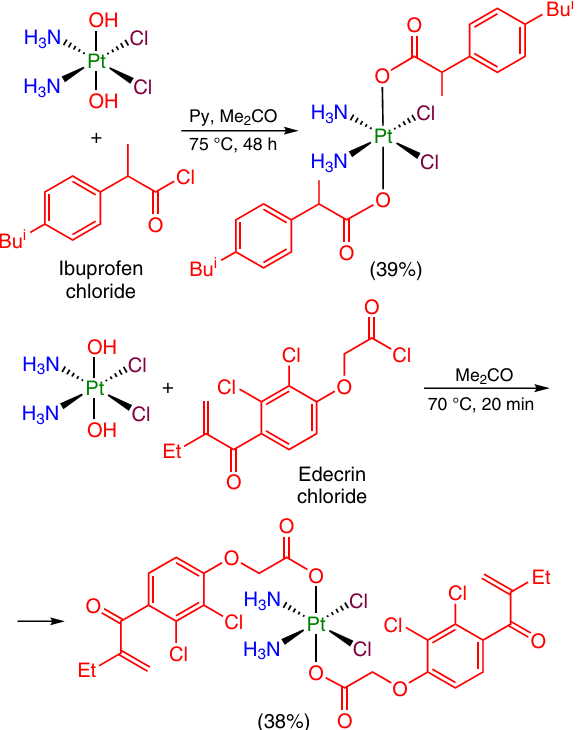
This approach was used 49 – 53 to convert oxoplatin to a variety of Pt(IV) prodrugs bearing non-steroidal anti-inflammatory drugs, which were formed in satisfactory yields (32 – 87%). In particular, cisplatin analogues containing two ibuprofen or Edecrin (ethacrynic acid) moieties are formed from oxoplatin and the corresponding acyl chlorides (Scheme 2).54
Serious drawbacks of this method are that the reactions proceed under relatively drastic conditions, which restricts the number of suitable substrates, and the lack of possibility of terminating the reaction after monocarboxylate formation.
2.2.1.1.2. Synthesis from anhydrides
In the design of Pt(IV)-based prodrugs, an important role belongs to dicarboxylic acid dianhydrides, which are less reactive acylating agents than acyl chlorides. Commercially available succinic (Scheme 3) and glutaric anhydrides are widely used for this purpose.44, 55
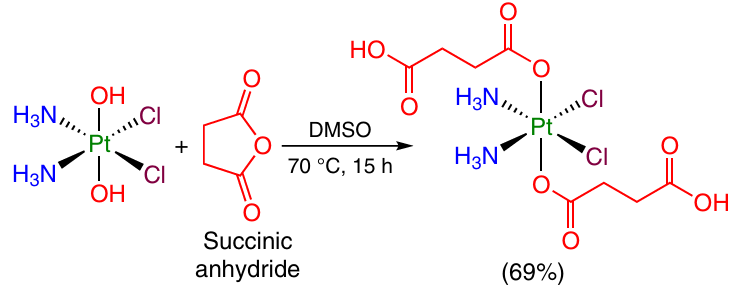
6-Azidohexanoic acid anhydride was allowed to react with oxoplatin. This gave a Pt(IV) compound with two azido groups amenable to further modification via the azide – alkyne cycloaddition reactions (Scheme 4).56
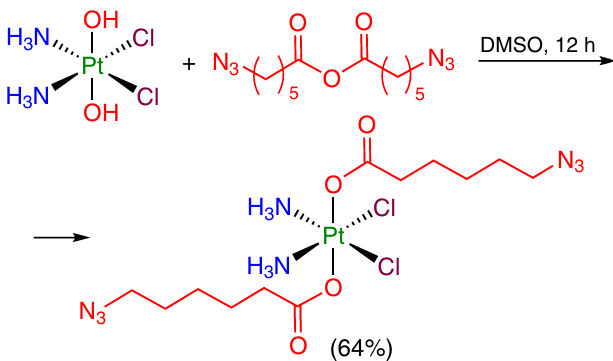
Despite the synthetic accessibility of carboxylic acid anhydrides, these derivatives are mainly used for modification of Pt(IV) monocarboxylate complexes to obtain unsymmetrical Pt(IV) dicarboxylate compounds. For this reason, they will be addressed in more detail in the following Sections of the review.
2.2.1.1.3. Synthesis from carboxylic acids
A widely used approach to the synthesis of symmetrical Pt(IV) dicarboxylate complexes implies the participation of activating reagents,57 – 60 most often, 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate 58, 59 and hexafluorophosphate 60 (TBTU and HBTU, respectively). These reagents were used to introduce diverse ligands, including conjugate of vitamin B12 with biotin (vitamin B7), the alkylating agent chlorambucil and the antimicrobial drug metronidazole, into the axial position of oxoplatin (Scheme 5).
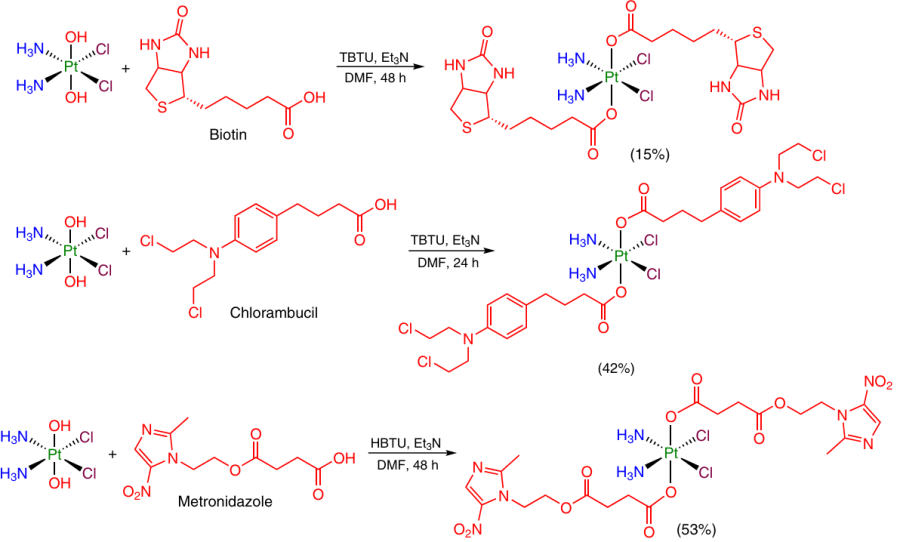
A drawback of this method is the long reaction time, which is usually 48 h, i.e., it is much longer than the time of reactions using acyl chlorides (0.5 – 2 h) or acid anhydrides (2 – 12 h).
N-Hydroxysuccinimide (NHS) esters of carboxylic acids obtained in situ are also utilized according to this approach in the presence of 1,3-dicyclohexylcarbodiimide (DCC) or 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC). For example, Pt(IV) prodrug containing two heptamethine cyanine dye moieties in the axial positions was obtained in this way (Scheme 6).57
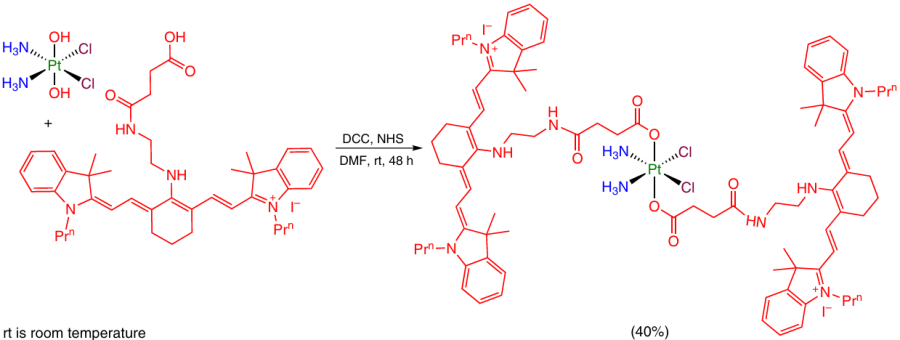
2.2.1.2. Synthesis of Pt(IV) monocarboxylate complexes
The possible control of esterification of the hydroxyl group at Pt(IV) and termination of the reaction after monocarboxylate complex has formed are of interest owing to the high biological activity of monocarboxylate derivatives.51, 61, 62 In addition, the second OH group at the Pt(IV) atom can be additionally modified using a ligand with a different type of activity, which would give a multiple-action Pt(IV) prodrug.27, 63, 64
2.2.1.2.1. Synthesis from anhydrides
Selective modification of Pt(IV) complexes at an axial OH group of oxoplatin is possible in the presence of a slight excess (1.1 – 1.5 equiv.) of the anhydride of the corresponding carboxylic acid. The anhydride can be obtained in situ in the presence of DCC as a dehydrating agent. A series of Pt(IV) coordination compounds with various benzoic acids and azidoacetic acid in the axial position were synthesized in this way (Scheme 7).61, 65
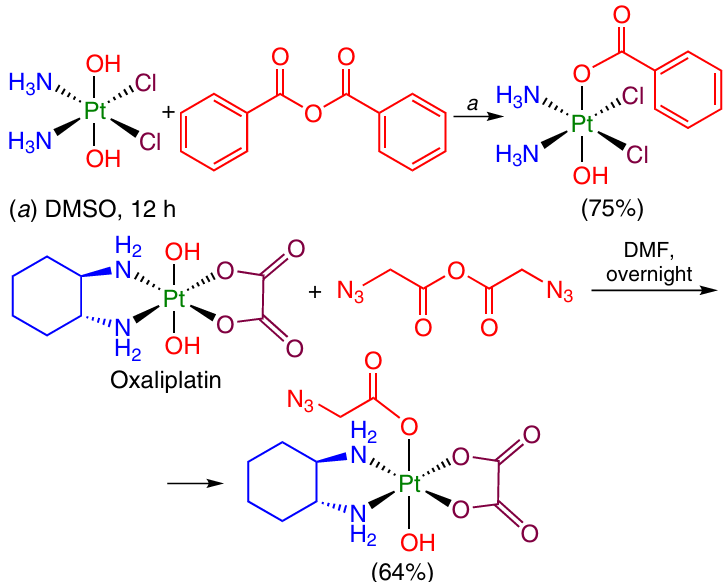
This approach was used 43, 66 to obtain Pt(IV) monocarboxylate complexes containing non-steroidal anti-inflammatory drugs (e.g., acetylsalicylic acid) (Scheme 8), alkylating agents [(4-carboxybutyl)thiophenylphosphonium bromide] anhydride and inhibitors of metabolic processes as axial ligands.
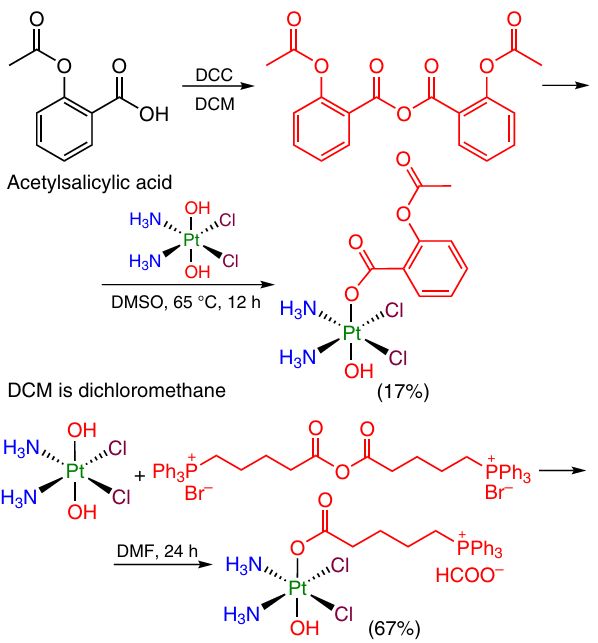
A drawback of this method is that 1 equiv. of the ligand present in the anhydride actually does not participate in the reaction and is released as a by-product.
2.2.1.2.2. Synthesis from N-hydroxysuccinimide esters
One more widely used method for the synthesis of Pt(IV) monocarboxylate prodrugs is in situ preparation of the NHS esters of carboxylic acids, which readily react with the OH group at the oxoplatin Pt(IV) atom. This method was used to obtain Pt(IV) monocarboxylate prodrugs with the ligands representing a non-steroidal anti-inflammatory drug (NSAID) (e.g., flurbiprofen) or DNA repair inhibitors (NERi) (Scheme 9).62, 67
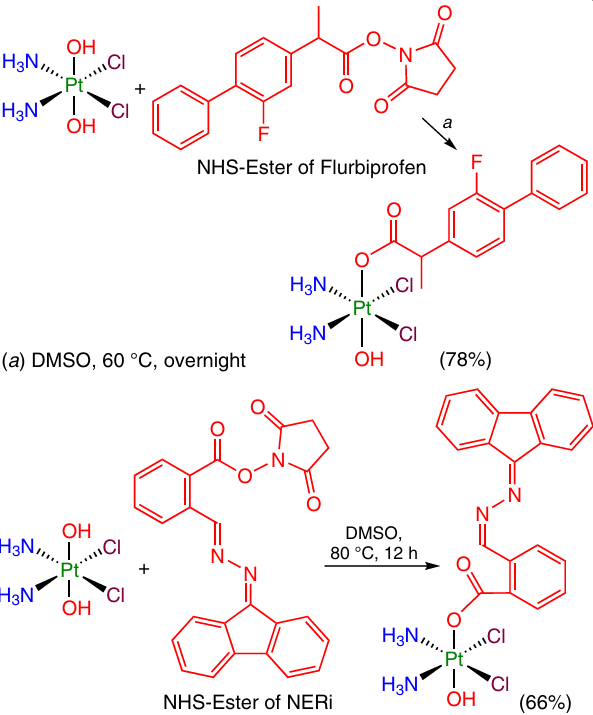
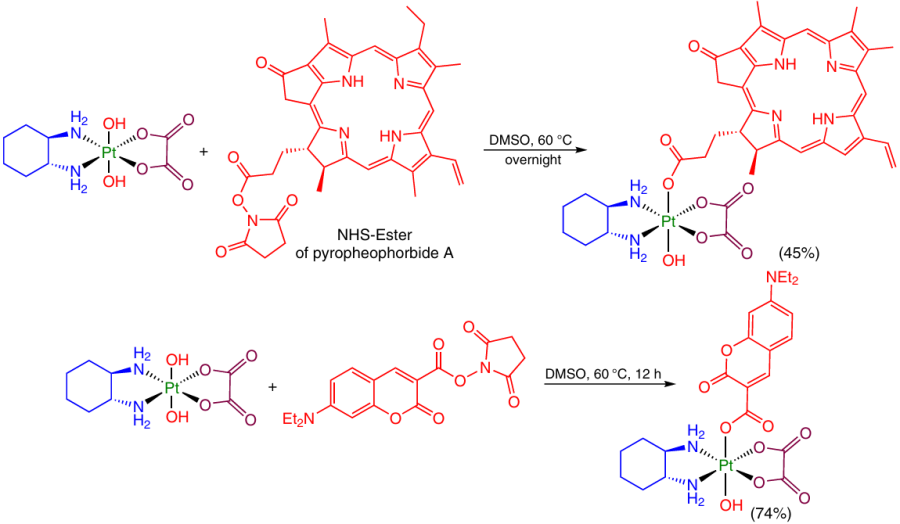
Similarly, Pt(IV) prodrugs capable of controlled activation were prepared from dihydroxy-oxaliplatin with preliminary synthesis of the NHS ester of the corresponding ligand. In this case, pyropheophorbide A and the NHS ester of 7-diethylaminocoumarincarboxylic acid served as the axial ligands (Scheme 10).32, 68
2.2.1.2.3. Synthesis from carboxylic acids
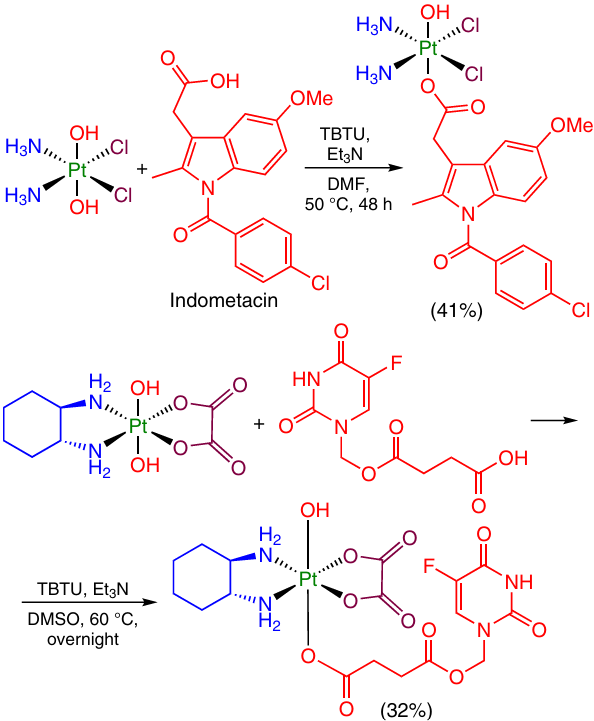
Tetramethyluronium activators of carboxyl group, such as TBTU, were proposed in a number of publications to obtain Pt(IV) monocarboxylate complexes. Platinum(IV) prodrugs containing indomethacin and 5-florouracil moieties in the axial position were prepared from oxoplatin and dihydroxy-oxaliplatin, respectively (Scheme 11).28, 46
2.2.1.3. Modification of the second OH group in Pt(IV) monocarboxylate coordination compounds
The Pt(IV) monocarboxylate complexes are modified to form unsymmetrical dicarboxylate complexes according to the scheme depicted in Fig. 5.
![[{"id":"CsegkXgNaJ","type":"paragraph","data":{"text":"General scheme for the synthesis of unsymmetrical Pt(IV) dicarboxylate prodrugs."}}]](/storage/images/resized/6Au1qi03Ighla3DG8stM5zgBFrRLcwapS7dTxIDu_xl.webp)
Modification of the second OH group is performed using methods described in the previous Sections for other classes of Pt(IV) prodrugs with addition of carboxylic acid anhydrides and tetramethyluronium activators.
2.2.1.3.1. Synthesis from anhydrides
Carboxylic acid anhydrides are the most widely used reagents for the synthesis of unsymmetrical Pt(IV) complexes. Using this approach, oxoplatin was converted to a series of Pt(IV) prodrugs containing two axial ligands with different biological action, namely, valproic acid and dichloroacetic acid residues or ibuprofen residue. The yields of products were 30 – 40% over the two steps (Scheme 12).27
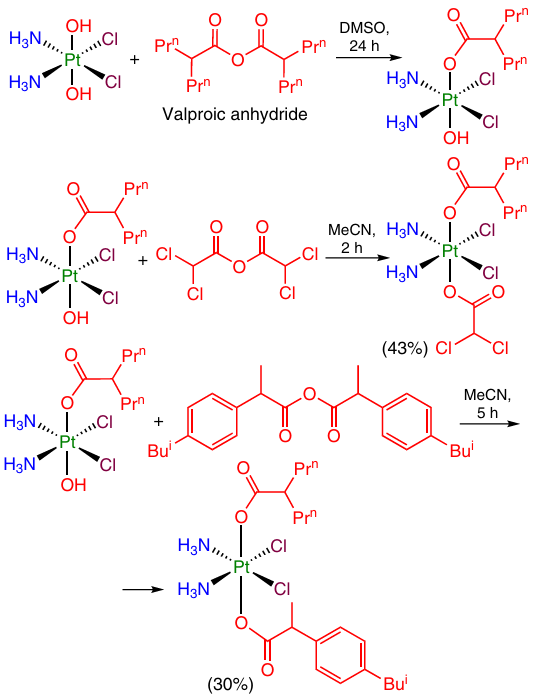
One strategy for modification of the second axial position of Pt(IV) complex is to introduce a linker moiety containing a functional group that could be further modified (see Section 2.2.3). Indeed, the above monocarboxylate complexes of oxaliplatin with pyropheophorbide A and 7-diethylaminocoumarincarboxylic acid moieties were additionally modified with succinic anhydride (Scheme 13).68, 69
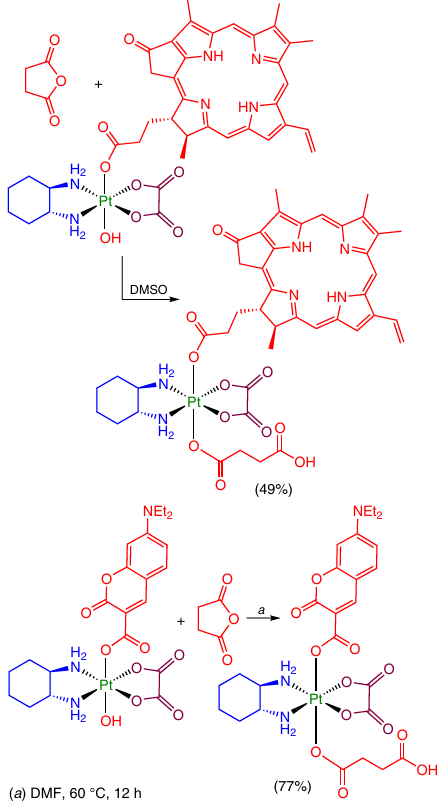
The replacement of the second OH group at the Pt(IV) atom is also meant to increase the lipophilicity of products. For example, the above-mentioned Pt(IV) monocarboxylate complexes with 5-fluorouracil and biotin were modified in the axial position with a lipophilic stearic acid moiety by treatment with the corresponding anhydride (Scheme 14).28, 70
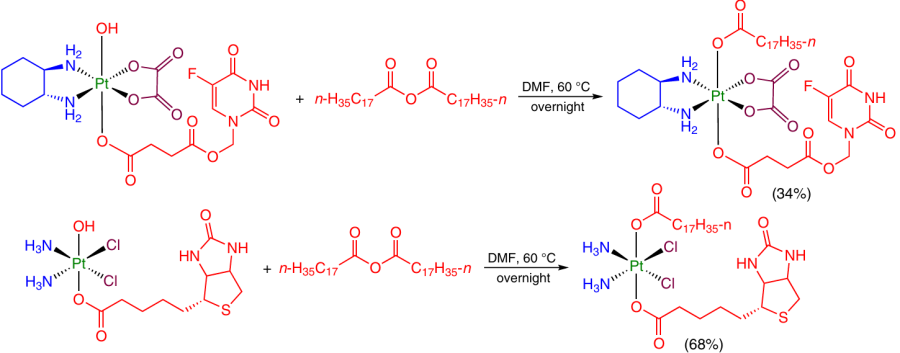
Thus, carboxylic acid anhydrides are versatile acylating agents for the synthesis of Pt(IV) prodrugs. Nevertheless, a drawback of this method is the release of one equivalent of the initial carboxylic acid during the reaction, which is undesirable if the ligand is sparingly accessible.
2.2.1.3.2. Synthesis from carboxylic acids
Unsymmetrical Pt(IV) dicarboxylate complexes can also be synthesized using reagents based on tetramethylurea, in particular TBTU. This enables more efficient use of the parent ligand than in the case of anhydrides of carboxylic acids.
For example, the activation of biotin under the action of TBTU in the presence of triethylamine was used to modify Pt(IV) monocarboxylate complex with indomethacin (Scheme 15).46
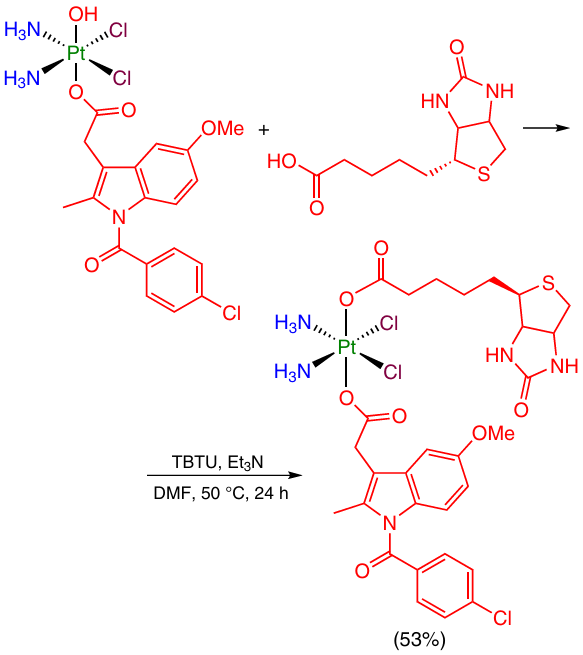
2.2.1.3.3. Synthesis from acid chlorides
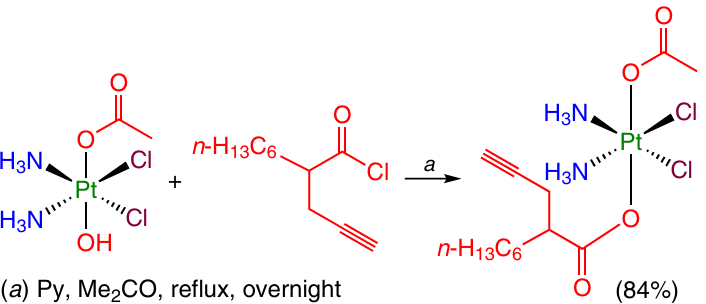
The substitution of the OH group in the Pt(IV)-based monocarboxylate prodrugs can also be carried out on treatment with acyl chlorides. For example, 2-(prop-2-yn-1-yl)octanoyl chloride was allowed to react with the complex [Pt(Cl)2(NH3)2(OH)(OAc)] to give the corresponding unsymmetrical dicarboxylate (Scheme 16).71
2.2.2. The introduction of functional groups other that carboxylate group into the axial position of Pt(IV) complexes
The Pt(IV)-based prodrugs considered above were obtained using axial ligands containing a carboxylic acid moiety. In some examples, where the biologically active ligand contained no carboxyl group, it was introduced into the axial position using a linker, most often, succinic anhydride. This approach was implemented in the synthesis of prodrugs by reactions of platinum(IV) complexes with 5-fluorouracil, metronidazole and heptamethine cyanine dye.28, 57, 60
Meanwhile, many medications used in the anticancer therapy such as gemcitabine, Taxol and estramustine contain a hydroxyl group or amino group rather than a carboxyl group. For axial ligands present in the complexes to exhibit their biological action after they have been released, they should be eliminated in an active form.72
In order to select an appropriate linker to be inserted between the Pt(IV) centre and the axial ligand, a number of synthetic approaches have been developed, which are considered below.
2.2.2.1. Carbonate-based linker
Gibson and co-workers 72 utilized the unsymmetrical carbonate RO – C(O) – OR' as a bridging moiety between the ligand and the Pt(IV) ion. The authors assumed that the carbonic acid monoester formed upon hydrolysis of the Pt(IV) complex rapidly decomposes to release an alcohol and CO2 (Fig. 6).
![[{"id":"nHsPq91wSJ","type":"paragraph","data":{"text":"General scheme of the release of the axial ligand bound to the Pt(IV) centre via a carbonate linker."}}]](/storage/images/resized/k9uko0iXpWNBMQ45VqbiLHyO0AsoLKcy9SNP1dHp_xl.webp)
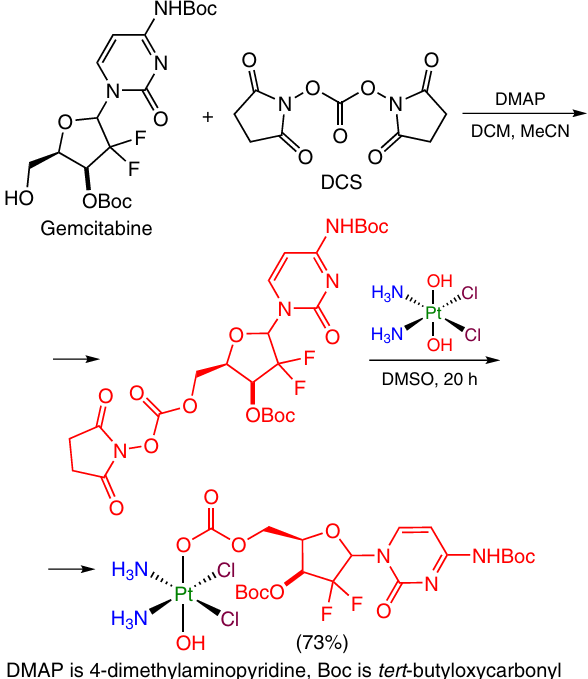
Anticancer agents targeting a site other than cisplatin does were chosen as ligands. An example is gemcitabine, which is incorporated into DNA, thus preventing its further synthesis.73 To be introduced into the axial position of oxoplatin, the OH group of gemcitabine group was activated with N,N'-disuccinimidyl carbonate (DSC), and then the product reacted with oxoplatin to give a carbamate linker between gemcitabine and Pt(IV) (Scheme 17).
A similar approach was employed to convert oxoplatin into a series of Pt(IV) monocarbonate and dicarbonate complexes in which the metal is linked to various aromatic and aliphatic hydrocarbons via a carbonate group (Scheme 18).74, 75
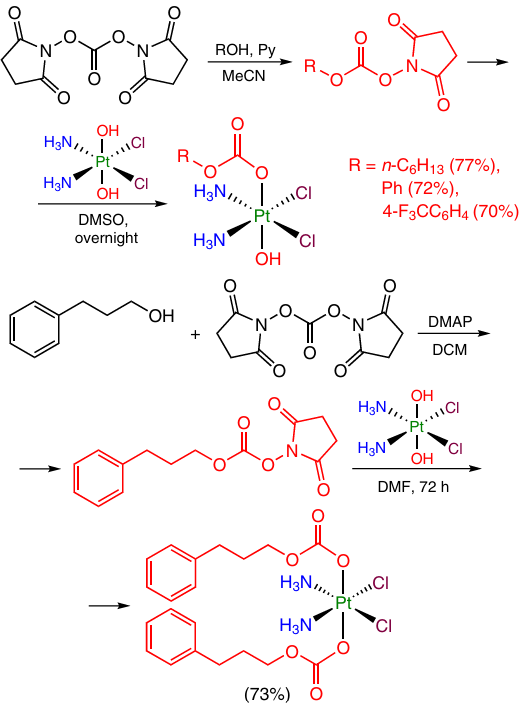
An important drawback of Pt(IV)-based prodrugs with a carbonate linker is low stability in water and fast reduction in the presence of sodium ascorbate [the reduction half-life (t1/2) is 0.5 – 3 h]74, 75
2.2.2.2. Carbamate-based linker
The carbamate moiety NH – C(O) – O is an analogue of the carbonate bridge for ligands containing an amino group in the molecule. Carbamates containing simple organic substituents (aliphatic or aromatic hydrocarbon residues) were synthesized, in some cases, using appropriate isocyanates (Scheme 19).76 – 78
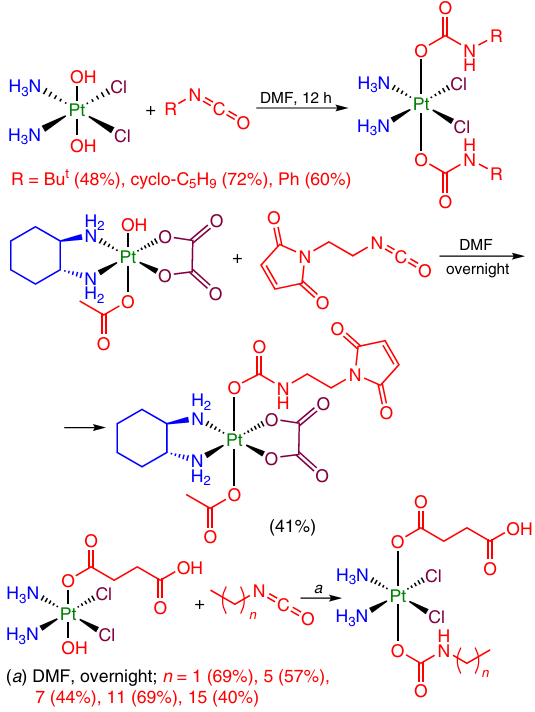
Babu et al.75 developed a method for the synthesis of Pt(IV) prodrugs with a carbamate bridge. The method includes, first, complex formation of Pt(IV) with activated carbonate as an axial ligand, which is then allowed to react with amine. This method affords the target coordination compounds in high yields over a very short reaction time (1 – 2 h). Furthermore, both aliphatic and aromatic amines are suitable for the reaction, e.g., 3-aminopropylbenzene, diethylamine and aniline (Scheme 20).
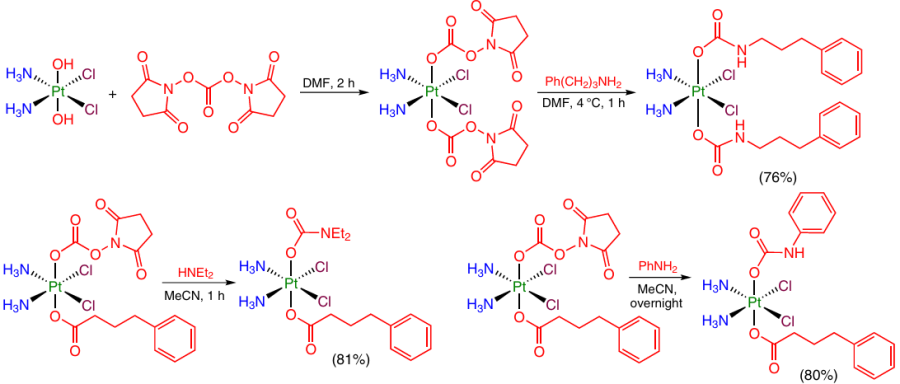
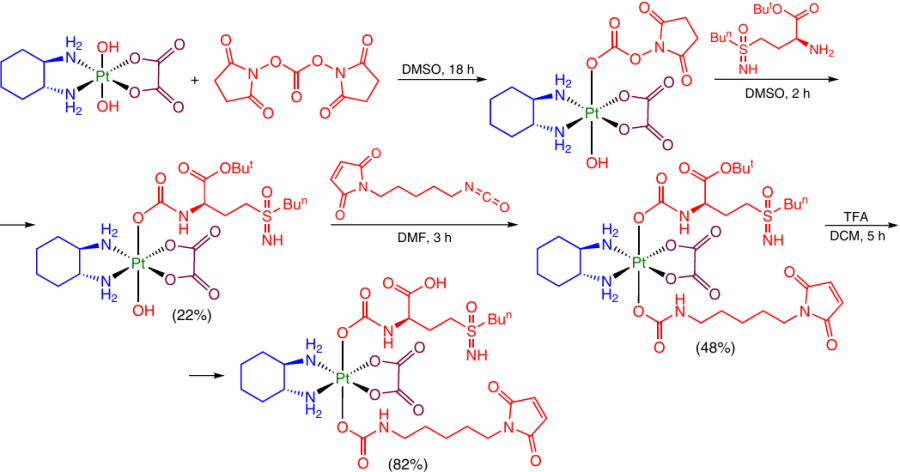
The possibility of successive modification of the OH groups at the Pt(IV) atom by introduction of two axial ligands via carbamate bridges was demonstrated in the development of a Pt(IV) prodrug capable of overcoming the oxaliplatin resistance.79 The Pt(IV) monocarbamate complex was obtained by the reaction of dihydroxy-oxaliplatin with N,N'-disuccinimidyl carbonate and an amino group-containing ligand, (2S)-tert-butyl 2-amino-4-(n-butylsulfonimidoyl)butyrate. The intermediate compound was then allowed to react with isocyanate bearing a second axial ligand (Scheme 21).
2.2.2.3 Thiocarbonate-based linker
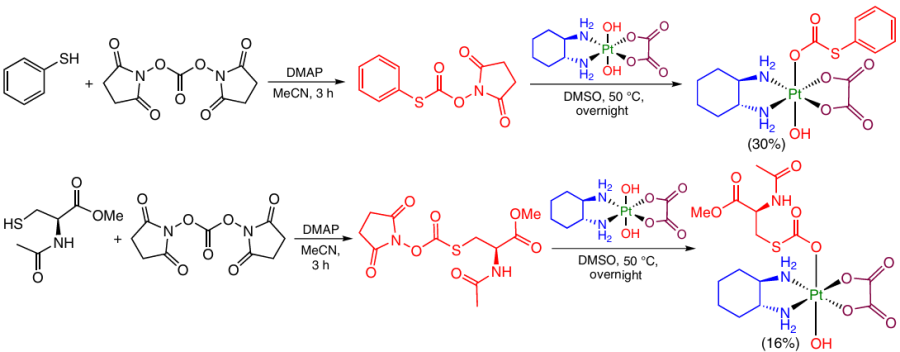
An example of unusual bridge between a ligand and the Pt(IV) centre was reported by Barth et al.,80 who demonstrated the possibility of introduction of organic groups into the axial position of coordination compounds via a thiocarbonate linker. In the first step, aromatic (thiophenol) or aliphatic thiol (methyl (R)-2-acetamido-3-sulfanylpropanoate) was reacted with DSC to give the NHS ester of thiocarbonate, which then reacted with dihydroxy-oxaliplatin (Scheme 22).
2.3. Modification of the axial ligands of Pt(IV) complexes
The methods considered above were limited to the direct acylation of the OH group at the Pt(IV) atom. The axial ligands introduced in this way become amenable for further modification. There are two main approaches proposed in the literature for introducing an additional organic moiety to the axial ligand at Pt(IV): peptide synthesis and azide – alkyne cycloaddition.
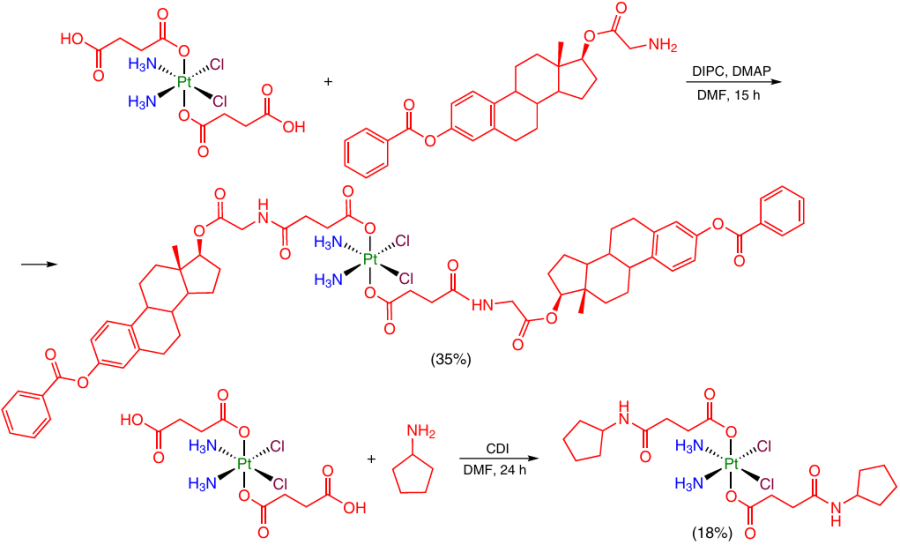
In the situations where the organic moiety has no carboxyl group, it can be introduced into the axial position of the complex by forming an amide or ester bond. For example, a bond of this type is formed when a succinic acid residue in the axial position of Pt(IV) reacts with an amino or hydroxyl group present in the organic moiety. The carboxyl group can be activated using cross-linking agents of carbodiimide synthesis (DCC or EDC) and NHS, diisopropylcarbodiimide (DIPC) or carbonyldiimidazole (CDI) (Scheme 23).28, 81
Platinum(IV) complex with 7-diethylaminocoumarin as an axial ligand was modified with a vector peptide to enhance the prodrug accumulation in the cell nuclei. The first step was the synthesis of the NHS ester of Pt(IV) complex. Then the ester was allowed to react with R8K peptide in DMSO (Scheme 24).68
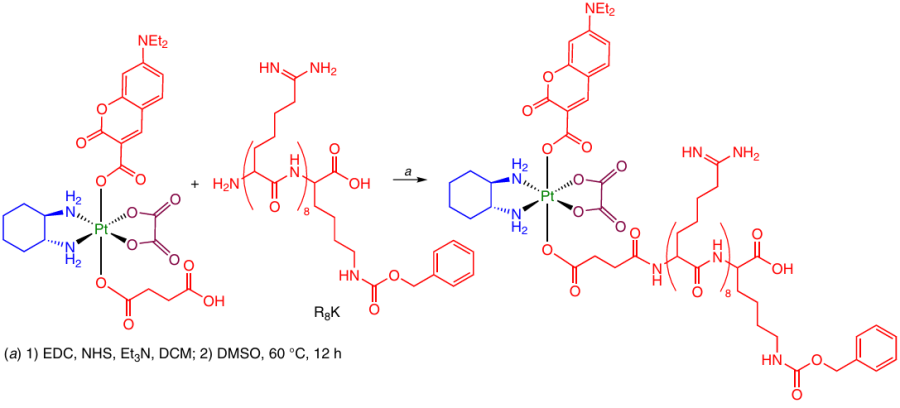
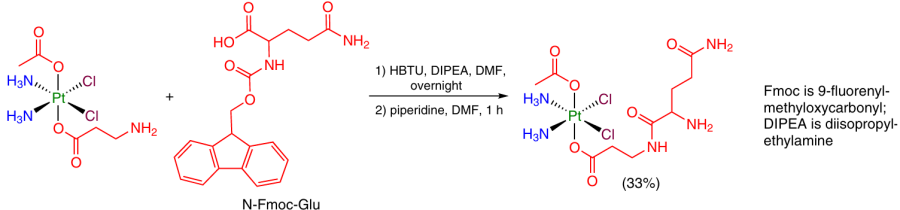
There are a few cases in which a carboxylate ligand with a protected amino group is formed in the axial position of Pt(IV) complex, while in the next step, the amino group reacts with a carboxylic acid. As an activating reagent, HBTU is used most often (Scheme 25).44
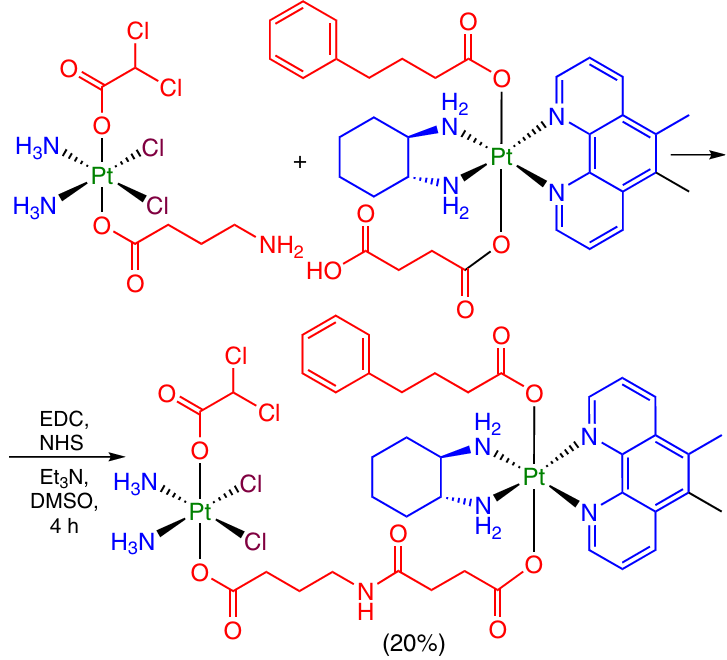
An elegant example of modification of Pt(IV) prodrugs with formation of an amide bond was reported by Petruzella et al.29 They prepared an agent with a potential quadruple action containing two Pt(IV) centres and two biologically active axial ligands, phenylbutyrate and dichloroacetate, in the molecule (Scheme 26).
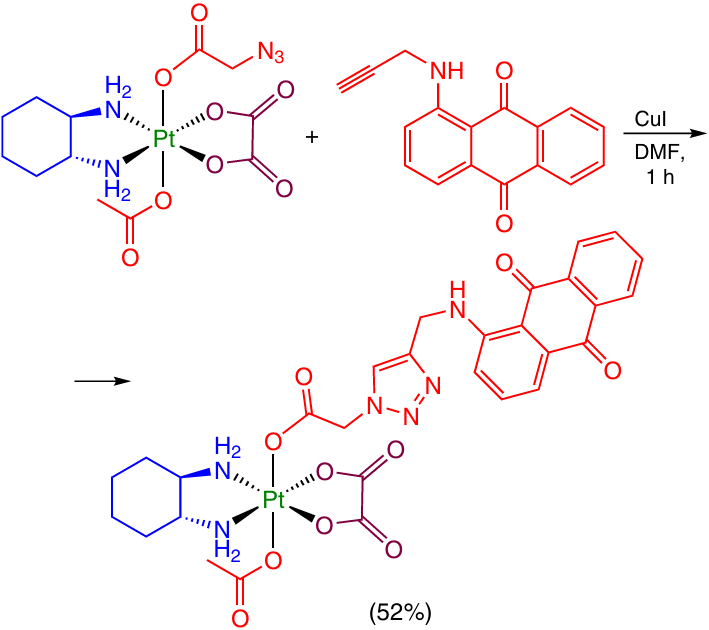
In some publications, azide – alkyne cycloaddition was used to modify an axial ligand. For instance, Hambley and co-workers 61 carried out a click-reaction between a Pt(IV) complex with an azidoacetic acid moiety and propargylated anthraquinone derivatives in the presence of a copper(I) iodide as a catalyst (Scheme 27).
A Pt(IV)-based prodrug containing an enterobactin derivative, a vector facilitating the accumulation of Pt(IV) in Escherichia coli bacteria, was synthesized using a homogeneous catalyst based on the copper(I) hexafluorophosphate acetonitrile complex in the presence of a copper-chelating ligand–tris(benzyltriazolyl)amine (TBTA).82 TBTA acted as a stabilizing ligand preventing copper(I) disproportionation and oxidation with oxygen (Scheme 28).83
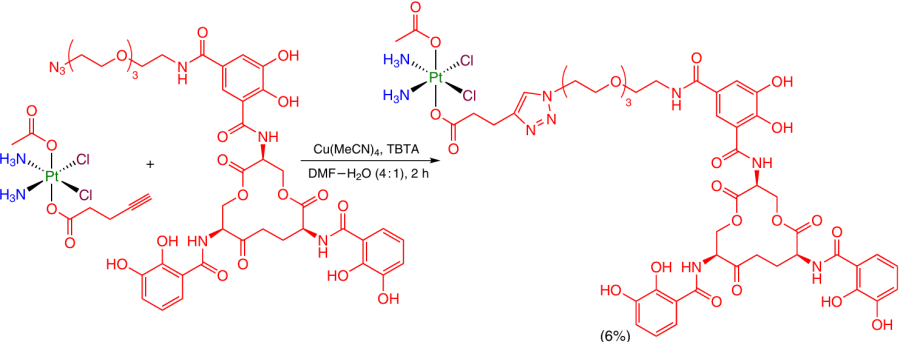
A Pt(IV) coordination compound containing two 6-azidohexanoic acid moieties was used as the substrate in the sterically promoted azide – alkyne cycloaddition reaction that did not require catalysis by copper salts.84 As a result, the axial ligand was modified with a triphenylphosphonylphosphonioalkyl moiety (Scheme 29).
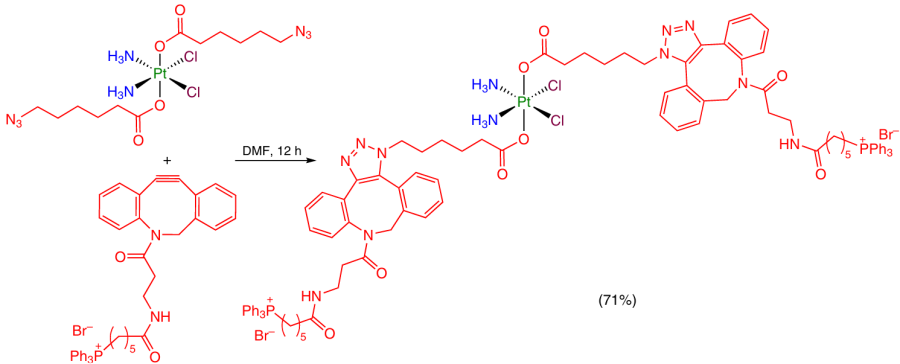
Thus, the introduction of ligands with functional groups, e.g., carboxyl, amino or azide groups, into the axial position of Pt(IV) complexes enables further modification of these compounds. This opens the way to finer tuning of the physicochemical and biological properties of Pt(IV) prodrugs.
2.4. Analysis of synthetic approaches to the preparation and modification of Pt(IV) prodrugs
Comparison of methods for the synthesis of Pt(IV) prodrugs given in this Section indicates that carboxylic acids chlorides are the optimal reagents for the preparation of symmetrical dicarboxylate complexes.49 – 54 A milder synthetic approach includes the reaction of Pt(II) compounds with carboxyl-containing organic ligands in the presence of tetramethyluronium activators.57 – 60 For the synthesis of Pt(IV) monocarboxylate derivatives and selective modification of one axial hydroxyl group in a Pt(IV) complex, it is advisable to use carboxylic acid anhydrides 43, 61, 65, 66 or NHS esters 32, 62, 67, 68 in a slight excess (1.1 – 1.3 equiv.) with respect to the initial coordination compound. The subsequent modification of the second OH group in the axial position of Pt(IV) monocarboxylate prodrugs is accomplished almost exclusively by treatment with carboxylic acid ahydrides.27, 28, 68 – 70
Apart from carboxylic acids, organic ligands containing hydroxyl or amino functional groups can be introduced into the axial position of Pt(IV) complexes using disuccinimidyl carbonate; this pathway involves the intermediate formation of carbonates or carbamates, respectively.74, 75, 79 According to an alternative approach to the synthesis of Pt(IV) prodrugs, the initial complex is allowed to react with the isocyanate of the specified ligand, which also results in the formation of the carbamate bond.76 – 78
The axial organic ligands in Pt(IV) prodrugs are mainly subjected to further functionalization by peptide synthesis methods if they contain a carboxyl or amino group.44, 55, 68, 81 One more way of modification of axial ligands is the azide – alkyne cycloaddition reaction using copper(I) catalysts or strained cyclooctynes.61, 83, 84
3. Biological activity of Pt(IV)-based prodrugs
A major benefit of the Pt(IV) prodrug design strategy is the possibility of easy variation of axial ligands. This Section of the review addresses a number of Pt(IV) complexes synthesized over the last 5 years and demonstrates the effect of axial ligands on the biological activity of the compound.
3.1. Platinum(IV) complexes with ligands possessing cytotoxic action
An efficient strategy for enhancing the antiproliferative properties of Pt(IV) prodrugs is the use of molecules possessing their own cytotoxicity as axial ligands. These agents may possess a synergistic effect, i.e., they may act more effectively than a physical mixture of the starting compounds.36, 85
3.1.1. Tubulin polymerization inhibitors
The formation of microtubules upon polymerization of α- and β-tubulin heterodimers has a crucial importance for a large number of fundamental cell functions.86 Microtubules, which play a significant role in mitosis, have been recognized as an effective target for the development of novel anticancer agents.87 Microtubule inhibitors can be classified into two types: microtubule destabilizers such as combretastatin A4 (CA4), colchicine and vinblastine and microtubule stabilizers such as taxanes.88
3.1.1.1. Combretastatin
The natural compound CA4 is a promising anticancer agent, which acts via inhibition of tubulin polymerization, thus preventing the formation of new blood vessels and destroying the vessels that are already present in the tumour.89, 90
In 2018, Li et al.91 synthesized prodrugs based on platinum(II) complexes (cisplatin and oxaliplatin) and combretastatin or its analogue AVE8063 containing an amino group instead of the hydroxyl group.
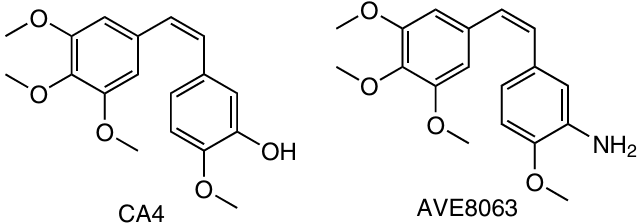
Prodrugs 1 – 4 proved to be less cytotoxic against any of the studied cell lines than the ligands CA4 and AVE8063. The activity of compounds 1 and 3 was 8 – 22 times higher than the activity of the initial cisplatin. In addition, they showed selectivity, being less toxic to normal cells.
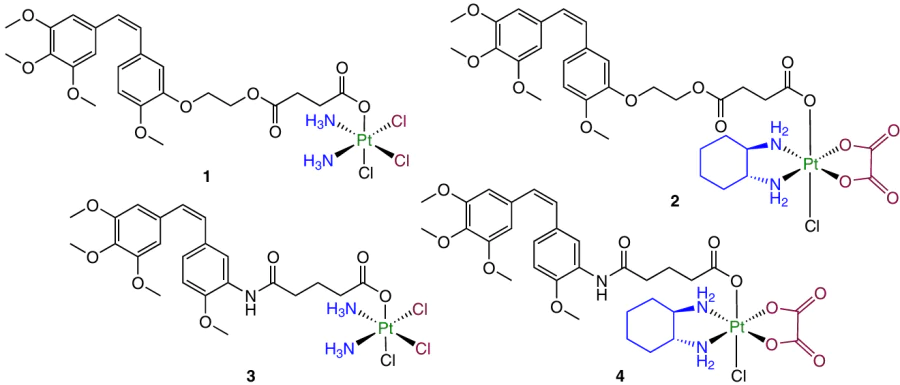
A study of the cellular uptake of prodrug 1 demonstrated cellular uptake 2.2 times as high as that for cisplatin. Complex 1 also showed activity to mitochondria and destroyed the microtubule network, which attests to the activity of CA4 moiety located in the axial position.
A study of the anticancer efficiency of prodrug 1 against SKOV-3 human ovarian adenocarcinoma xenograft model in BALB/c mice demonstrated a more pronounced tumour growth inhibition (TGI) than the parent ligand CA4, but less pronounced TGI than cisplatin. It is noteworthy that the weight loss in the group of mice administered with compound 1 was lower than that for mice treated with cisplatin.
Platinum(IV) prodrugs 5 – 7 with combretastatin in the axial position were reported in 2019 by Huang et al.92 Complex 5 demonstrated the ability to arrest the cell cycle in the G2/M phase and to inhibit the microtubule formation.
In 2021, Schmidt et al.93 described Pt(IV) prodrugs 8 – 15. These are triple-action medications containing axial combretastatin, histone acetylase (HDAC) inhibitors (phenylbutyrate and valproate), pyruvate dehydrogenase kinase (PDK) inhibitor (dichloroacetate) or octanoate, which enhanced DNA methylation.
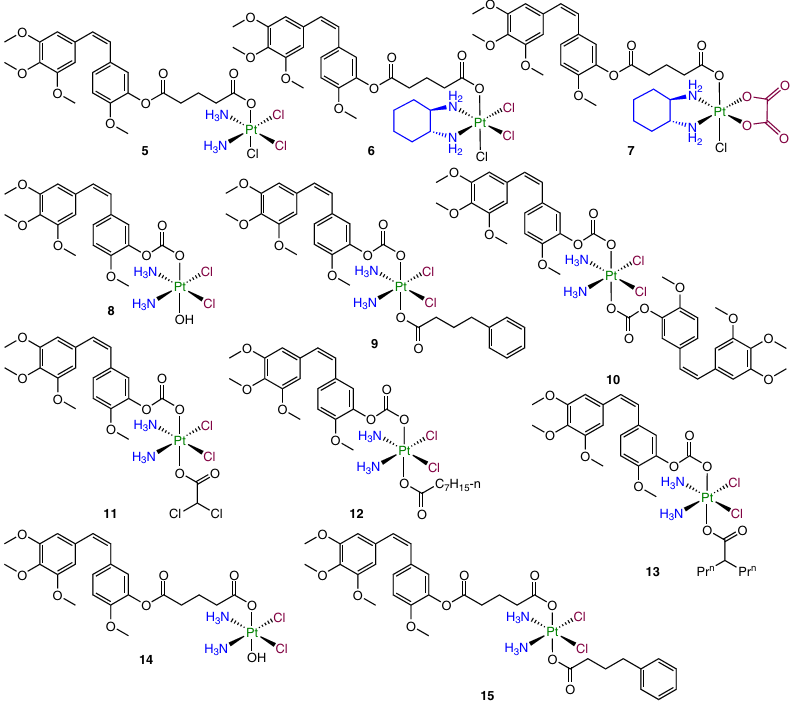
These prodrugs showed a cytotoxicity comparable with or exceeding that of combretastatin in the nanomolar concentration range (< 10 nM) and greater cellular uptake compared to cisplatin. In addition, they were found to inhibit microtubule formation. A study of the therapeutic efficacy of these compounds in vivo against the Lewis lung carcinoma model showed that CA4, which is the most potent cytostatic in vitro, exhibited the least pronounced anticancer effect in vivo. The reduction of the tumour volume induced by complex 8 was comparable to that for cisplatin (84% inhibition), while the administration of compounds 15 and 9 caused TGI of 91.5 and 92.6%, respectively.
The greatest efficacy of derivative 9 with phenylbutyrate and combretatstatin residues in the axial positions was attributed to the higher stability of triple-action prodrugs in comparison with double-action agents.
3.1.1.2. Chalcones
Chalcones, α,β-unsaturated carbonyl compounds with two aromatic cores, are classic Michael acceptors able to alkylate protein residues including thioredoxin reductases TrxRs and nuclear factors NF-κB and Nrf2.94 Chalcones and their derivatives can act as antioxidants, antibacterial and anti-inflammatory agents; they show noticeable antitumour activity, in particular through inhibition of tubulin polymerization by binding to the colchicine site.95 – 97 In view of the multiple biological activities, development of Pt(IV) prodrugs with a chalcone moiety in the axial position is of obvious interest.
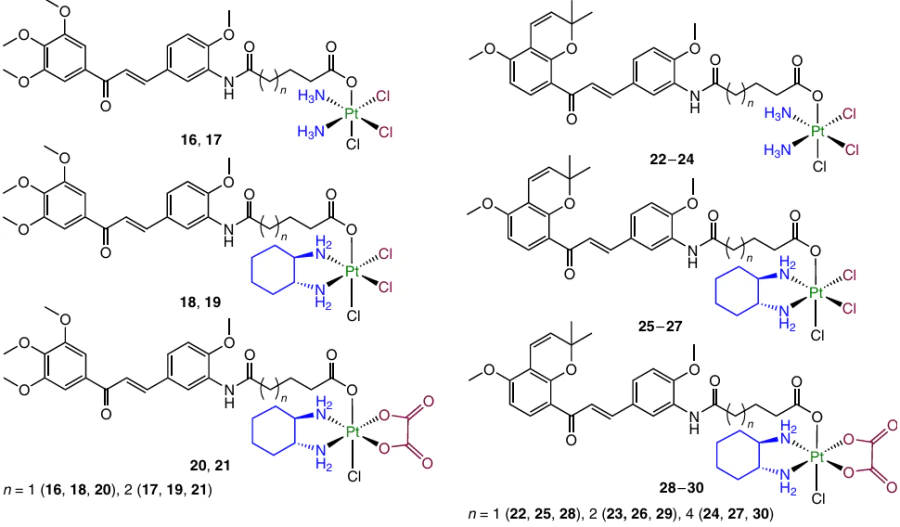
In 2018, Huang et al.98 synthesized Pt(IV) complexes 16 – 21, containing chalcone in the axial position. Prodrugs 16 – 21 exhibited cytotoxicity exceeding the cytotoxicity of the parent Pt(II) complexes against a number of cell lines, including cisplatin-resistant ones. The half-maximal inhibitory concentration (IC50) against HepG2 human hepatocellular carcinoma cells was 0.97 to 2.23 mM. Furthermore, these compounds showed an increased cellular uptake. Compounds 16 and 17 noticeably inhibited the motility of human umbilical vein endothelial cells (HUVEC), arrested the cell cycle in the G2/M phase and caused mitochondria-mediated apoptosis by regulating the expression of Bcl-2 proteins. As expected, prodrugs 16 and 17 were able to inhibit tubulin polymerization.
Millepachine is a chalcone first isolated from the Millettia pachycarpa shrub in 2013; it shows anticancer activity in vitro and in vivo and also has a potent inhibitory effect on tubulin polymerization via binding to the colchicine site.97 In 2018, Huang et al.99 developed Pt(IV) prodrugs 22 – 30 with millepachine and its homologues in the axial position.
Prodrug 22 had a higher cytotoxicity than cisplatin. The selectivity characteristics of these prodrugs were also higher than those of the parent Pt(II) agents, and the cytotoxicity decreased with increasing carbon chain length. Compound 22 showed a higher (by a factor of up to two) uptake in tumour cells than cisplatin and the ability to inhibit tubulin polymerization.
A study of the mechanism of cytotoxic action showed that prodrug 22 can induce the cell cycle arrest in the G2/M phase, change the expression of cell cycle-associated proteins and induce apoptosis via the ROS-mediated (ROS are reactive oxygen species) mitochondrial pathway. Experiments in vivo on SKOV-3 tumour xenografts exhibited efficient TGI without a clear-cut weight loss by the animals.
The inhibition of the interaction of the p53 transcription factor, regulating the cell cycle, with the MDM2 protein is an attractive therapeutic target for the development of anticancer drugs. It is known that the MDM2 protein is overexpressed in various tumours, its interaction with the p53 protein promotes uncontrolled cell proliferation, while inhibition of the p53–MDM2 interaction triggers apoptosis of tumour cells.100 In 2018, Ma et al.101 used dichloro-substituted chalcone as an inhibitor of the p53–MDM2 interaction and obtained complexes 31 and 32.
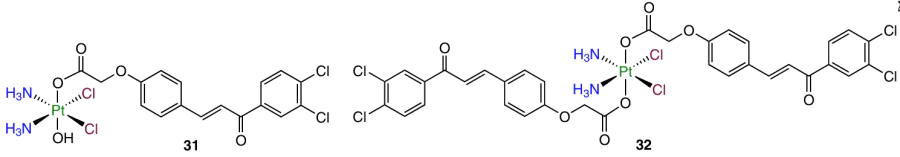
The IC50 values for monocarboxylate complex 31 were in the nanomolar concentration range, being 422 times lower than those for cisplatin (0.023 and 9.7 mM against HCT-116 colorectal carcinoma cells, respectively). Prodrug 31 also showed activity in the nanomolar range against cisplatin-resistant cell lines [0.07 and 0.14 mM against the cisplatin-resistant A2780cisR ovarian cancer cell line (A2780/CDDP) and cisplatin-resistant A549cisR lung carcinoma cell line (A549/CDDP), respectively]. Unlike cisplatin, this compound induced apoptosis and promoted a considerable growth of expression of DNA damage marker (γH2A.X). In addition, prodrug 31 was more efficiently taken up by cells (36 and 111 times higher cellular uptake than that of cisplatin in A2780 and A2780cisR cells, respectively).
The antitumour activity of complex 31 was studied in vivo against the HCT-116 tumour xenograft model in BALB/c mice. After 27 days of therapy with this agent, TGI was 80%, while for the groups of mice that were administered with cisplatin, this value was 68%.
In 2023, Cao et al.102 reported Pt(IV) prodrugs 33 – 38 with an indole analogue of chalcone in the axial position.99 These compounds showed a pronounced cytotoxicity, with complex 36 containing two methylene units in the axial ligand being the most active. Prodrug 36 showed a moderate selectivity toward normal cells (L02 human fetal hepatocytes and HUVEC) and inhibited migration of HCT-116 cells. A study of the mechanism of cytotoxic action demonstrated that prodrug 36 can destroy the Bcl-2/Bax proteins, promote the release of cytochrome C (Cyt C) and activates the cascade of caspases, thus causing mitochondria-mediated apoptosis. Using immunofluorescence assay of intracellular microtubules and molecular docking, it was ascertained that this compound can interact with the colchicine-binding site and inhibit tubulin polymerization.
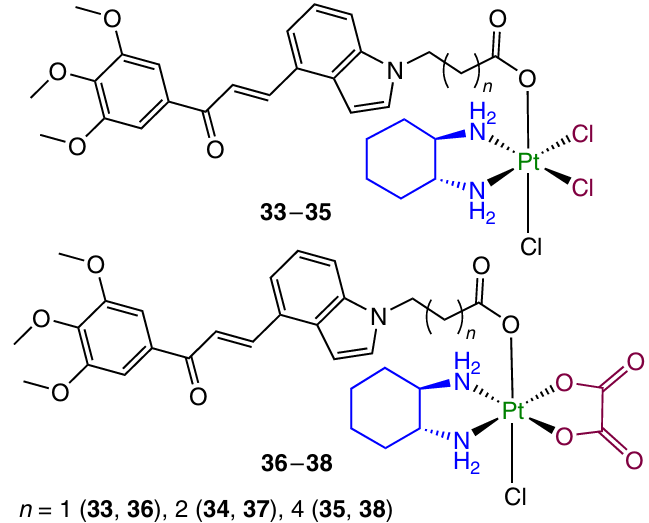
In 2023, Liu et al.103 studied analogous Pt(IV) complexes 39 – 47 containing an indolochalcone moiety in the axial position.
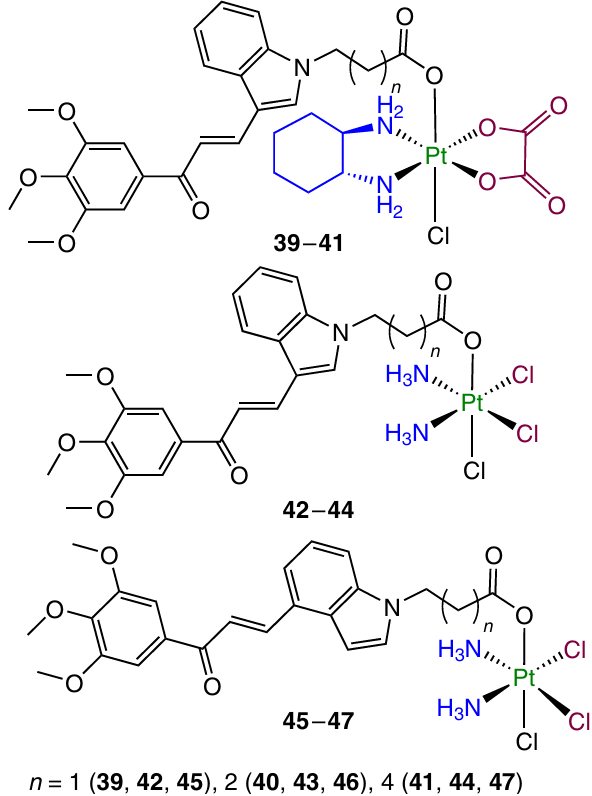
Prodrugs 39 – 47 exhibited cytotoxicity against several cell lines; their cytotoxic activity decreased with increasing number of methylene units in the chain: complexes 42 – 47 based on cisplatin proved to be more active than oxaliplatin derivatives 39 – 41. The highest cytotoxic activity was found for complex 45 (IC50 = 0.11 – 1.53 mM), in particular against cisplatin-resistant cell lines. In a study of the mechanism of cytotoxic action, it was found that this compound is efficiently taken up by the cells and triggers mitochondria-mediated apoptosis by inhibiting the activity of the Bcl-2 protein, enhancing the activity of the Bax and Cyt C proteins and activating the caspase cascade. In addition, prodrug 45 showed the ability to induce the endoplastic reticulum (ER) stress in A549/CDDP cells through the PERK/ATF4/CHOP signalling pathway.
A study of the therapeutic efficacy in vivo for A549/CDDP xenograft model in mice showed that complex 45 inhibits the tumour growth more efficiently than cisplatin (TGI of 65.9 and 25.7%, respectively) and also has a lower general toxicity.
3.1.1.3. Paclitaxel
Paclitaxel (PTX), a microtubule depolymerization inhibitor, is one of the most successful antimitotic drugs for the therapy of a broad range of solid malignant tumours.104 In addition, Pt(II) agents in combination with PTX are often clinically used for the treatment of various types of breast cancer, non-small cell lung cancer and gastric cancer.105, 106
In 2022, Zhang et al.107 described Pt(IV) prodrugs 48 – 55 containing PTX in one or two axial positions. A study of the antiproliferative activity of these complexes revealed high activity against all cancer cell lines in comparison with cisplatin; the IC50 values varied in the range of 0.13 – 5.98 mM.
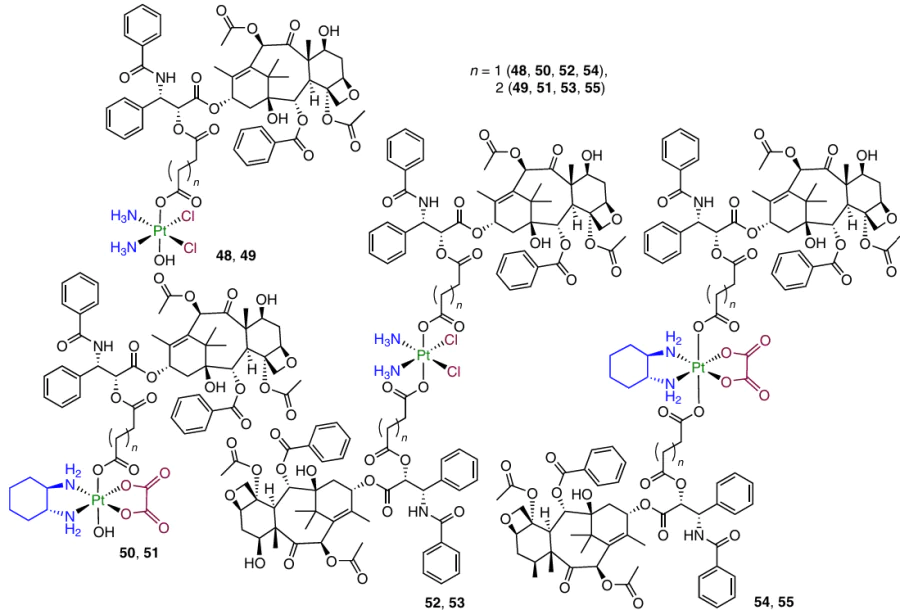
Prodrug 48 showed selectivity towards MCF-7 breast carcinoma cells: it was 344 times higher than that of cisplatin. This compound efficiently inhibited the migration of HCC1937 and MCF-7 breast carcinoma cells and was taken up by cancer cells 30 times more efficiently than the parent cisplatin. Furthermore, prodrug 48 showed the ability to induce DNA damage, arrest the cell cycle in the G2 phase and inhibit tumour metastasis. It induced mitochondria-mediated apoptosis of MCF-7 cells and increased the intracellular ROS levels. It also induced the endoplasmic reticulum stress and promoted the release of Ca2+ ions.
3.1.2. Miscellaneous cytotoxic agents
3.1.2.1. 5-Fluorouracil
5-Fluorouracil is a clinically used thymidylate synthase (TS) inhibitor. 5-Fluorouracil metabolites bind to DNA, which induces DNA damage, while TS inhibition blocks the synthesis of DNA and disrupts the repair mechanisms.108, 109
Platinum(IV) complexes 56 – 67 based on cisplatin (or oxaliplatin) and fluorouracil were synthesized and studied by Zhang et al.28
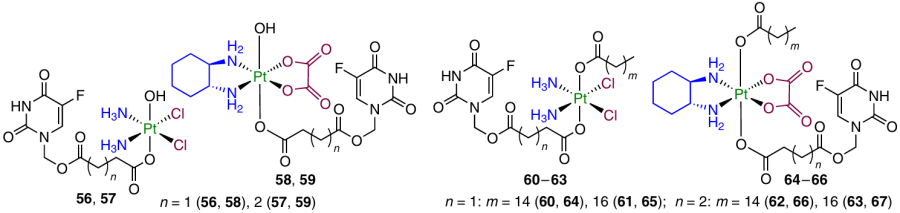
Monocarboxylate prodrug 58 proved to be less active than an equimolar mixture of oxaliplatin and fluorouracil against any of the tested cell lines. For this reason, to enhance the cytotoxic activity, the second axial position was modified with palmitate or stearate, which increased the lipophilicity of compounds. Prodrugs 60 – 67 obtained in this way proved to be 64 times more active than the corresponding Pt(II) complexes: IC50 values for HCT-116 cells were 0.13 and 8.34 mM for compound 64 and oxaliplatin, respectively. Meanwhile, when tested against normal MRC-5 lung fibroblasts, prodrug 64 was more than 2.5 times less toxic than oxaliplatin.
A study of the cellular uptake of the prodrugs in HCT-116 cells showed a high uptake for complex 64, which was 62 times as high as that of oxaliplatin. This prodrug efficiently damaged DNA and induced an increase in the expression of TS and p53 protein, markers of 5-fluorouracil activity, in HCT-116 cells. The high anticancer activity of compound 64 was also confirmed in an in vivo experiment in which the prodrug inhibited the growth of the HCT-116 tumour xenograft in NOX/SCID mice by 84.8% after 21 days of therapy. Note that for oxaliplatin and the oxaliplatin + fluorouracil combination, inhibition was 57.8 and 75.8%, respectively.
More recently, Pt(IV) prodrugs 68 – 72 bearing 5-fluorouracil and aliphatic carboxylic acids, including valproic acid as an HDAC inhibitor, as axial ligands were investigated by Ding et al.110
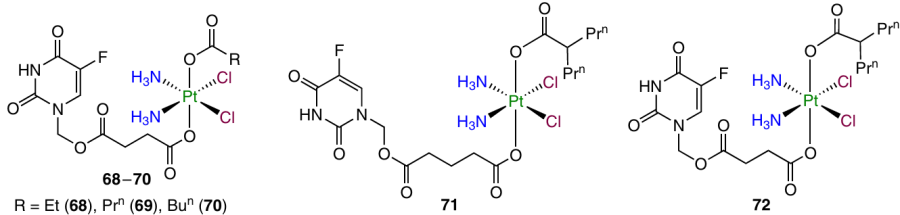
An increase in the length of the linker in the axial position entailed an increase in the cytotoxic activity of the prodrugs. The highest activity against MCF-7 cells and MDA-MB-231 triple negative breast cancer cells was inherent in complex 71, while the highest selectivity over HUVEC normal cells was identified for compound 72. The cellular uptake of 72 in HeLa cervical cancer cells proved to be three times as high as that of cisplatin, which correlates with the difference between the cytotoxic activities of these agents. In addition, prodrug 72 inhibited the HDAC expression and promoted the TS expression.
3.1.2.2. Chlorambucil
Chlorambucil is an FDA-approved anticancer drug capable of binding to the guanine or adenine N(7) atom in DNA.111, 112
In 2018, Ma et al.59 reported dual-action mono- and dicarboxylate complexes 73 and 74 based on cisplatin and chlorambucil.
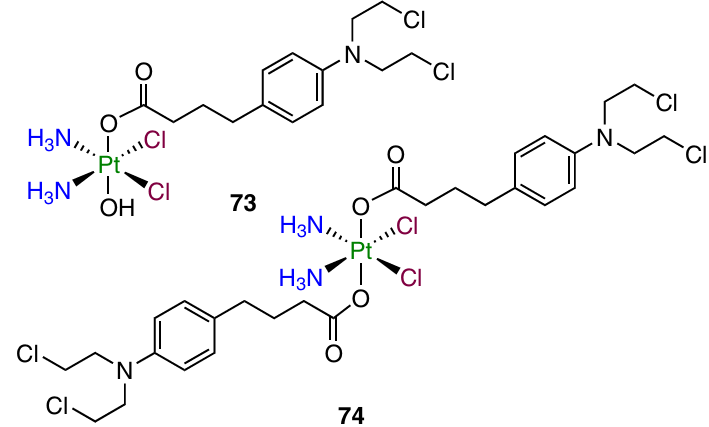
Prodrugs 73 and 74 showed activity against A549 and HeLa cells similar to that of cisplatin, with a 1.5- to 3.3-fold increase in the toxicity. The most pronounced increase in the cytotoxicity (5.5 – 6-fold) was found for MCF-7 cells. The highest efficiency relative to cisplatin was found for these compounds tested against MDA-MB-231 cell line where complex 74 was 20 times more active than cisplatin (the IC50 values were 2.5 and 51.7 mM, respectively).
The increase in the cytotoxicity of prodrug 74 was correlated with the increase in the cellular uptake. This compound induced much more pronounced DNA damage and apoptosis than cisplatin. When studied for the in vivo antitumour efficacy in BALB/c mice bearing MDA-MB-231 tumour xenograft, prodrug 74 was not superior to cisplatin; however, the therapy with this agent did not induce weight loss of the animals.
Chlorambucil was also used as an axial ligand for another class of Pt(II)-based anticancer agents, complexes 75 – 77 with equatorial ligands based on phenanthroline, which were studied by Aputen et al.113
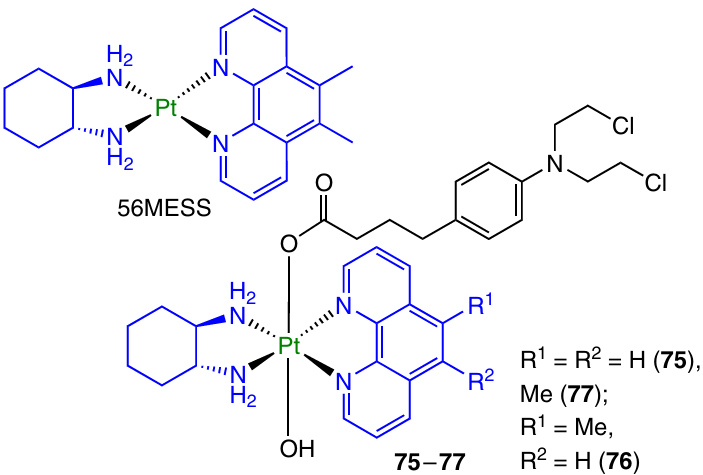
Some representatives of this class, in particular the compound designated as 56MESS, showed high antiproliferative activity against a number of cell lines, presumably due to an alternative cytotoxicity mechanism that targets the tumour by affecting mitochondria.114, 115 Prodrugs 75 – 77 showed high cytotoxicity in sub-micromolar and nanomolar concentration ranges against several cell lines. Compound 77 proved to be the most active antitumour agent: the GI50 values (concentration providing 50% cell growth inhibition) reached 2.7 nM for prostate cancer cell line (Du145) and 10 nM for cisplatin-resistant ovarian cancer cell line (A2780cisR). Despite the fact that this agent was 2800 times more toxic than cisplatin, when tested on A2780cisR cell line, it showed cytotoxicity comparable to that of the precursor Pt(II) complex 56MESS (GI50 for these cells were 10 and 13 nm, respectively). The incubation of colorectal adenocarcinoma cells (HT-29) with prodrug 77 resulted in up to three times more efficient formation of ROS than incubation with 56MESS. It is known that the high level of ROS in cells induces a significant DNA damage and activates the apoptotic cell death.116, 117
3.1.2.3. Amlexanox
Platinum(IV) mono- and dicarboxylate complexes 78 and 79 with axial position(s) occupied by the anti-asthmatic drug amlexanox possessing a pro-apoptotic effect 118 were synthesized by Guo et al.119
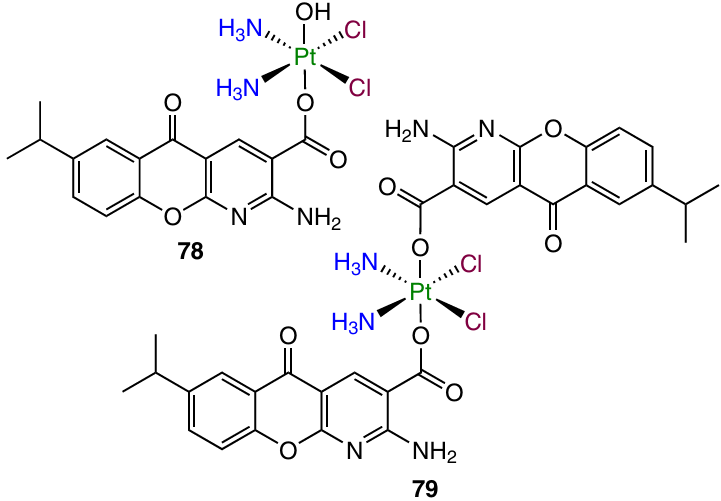
A cytotoxic activity assay demonstrated that dicarboxylate complex 79 has almost no antiproliferative properties, while monocarboxylate 78 has IC50 values in the micromolar concentration range, in particular against cisplatin-resistant Caov-3 (primary ovarian cancer) and A549/CDDP cell lines.
Detailed study of the mechanism of cytotoxic action demonstrated that prodrug 78 triggers apoptosis by a mechanism resembling that for cisplatin. In addition, this agent induces significant mitochondrial depolarization and mitochondria-mediated apoptosis of Caov-3 cells, which is due to the presence of the amlexanox moiety in the axial position. Compound 78 also induces the autophagy in Caov-3 cells.
3.1.2.4. Clioquinol
Clioquinol is an antimicrobial and antiprotozoal agent. This compound was also studied as an antitumour drug possessing anti-metastatic properties, which may markedly enhance the autophagy by inhibiting the mammalian target of rapamycin (mTOR).120
In 2023, Zhang et al.121 developed Pt(IV) prodrugs 80 – 86 with clioquinol in the axial position.
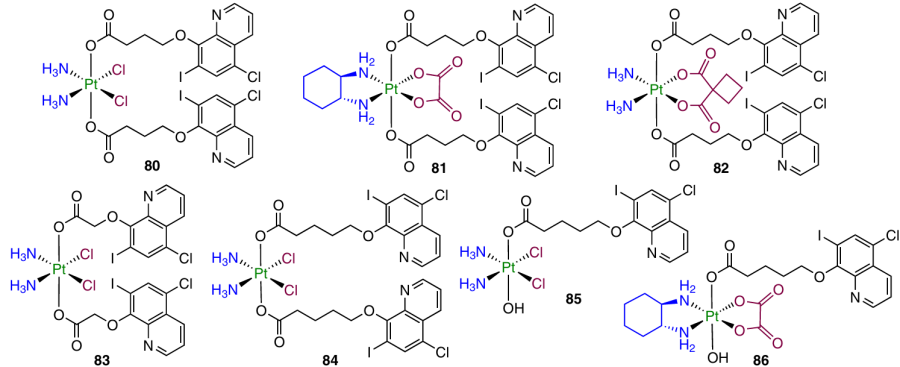
Antiproliferative activity assays demonstrated that complexes based on cisplatin have a higher activity than the prodrugs derived from oxaliplatin and carboplatin. The highest antiproliferative activity (in particular, against cisplatin-resistant cell lines) was found for complex 84 containing a valeric acid residue in the axial position (IC50 £ 0.70 mM). According to in vivo experiments on BALB/c mice bearing 4T1 mouse breast cancer, prodrug 84 was found to be less toxic than cisplatin and oxaliplatin and than monosubstituted complex 85. Higher toxicity of the last-mentioned compound is attributable to the lower stability of the agent in the bloodstream: TGI attained after the therapy with prodrug 84 was comparable with that for cisplatin. Compound 84 also showed an antimetastatic activity in in vitro and in vivo experiments. Study of the mechanism of antitumour activity for prodrug 84 identified its ability to damage DNA, increase the expression of γH2AX and p53 proteins, trigger the mitochondria-mediated apoptosis via the Bcl-2/Bax/caspase3 cascade and induce autophagy by inhibiting the PI3K/AKT/mTOR signalling pathway and activating the HIF-1α/Beclin1 pathway. Furthermore, this agent suppressed the secretion of the programmed cell death ligand (PD-L1) in tumour cells and stimulated the formation of CD4+ and CD8+ T-lymphocytes (helper and killer cells, respectively.).
3.1.2.5. Doxorubicin
In 2021, Muhammad et al.122 converted cisplatin complex 87 to prodrug 88, which contained doxorubicin and mitochondria-targeting triphenylphosphine ligand in the axial positions.
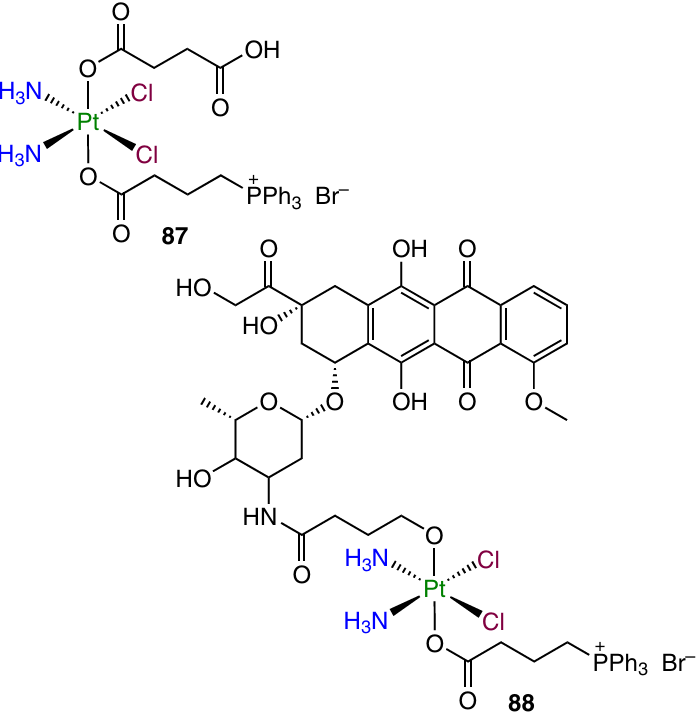
Compound 88 exhibited cytotoxicity comparable with that of doxorubicin and exceeding the toxicity of cisplatin. Prodrug 88 had a higher cellular uptake and the ability to be localized in mitochondria, arrest the cell cycle in the G2 phase and induce cell necrosis. This compound can cause mitochondrial depolarization and can form ROS inside the cells.
3.1.3. Conjugates with biologically active molecules
3.1.3.1. Phenylbutyrate and aliphatic and (aromatic) hydrocarbons
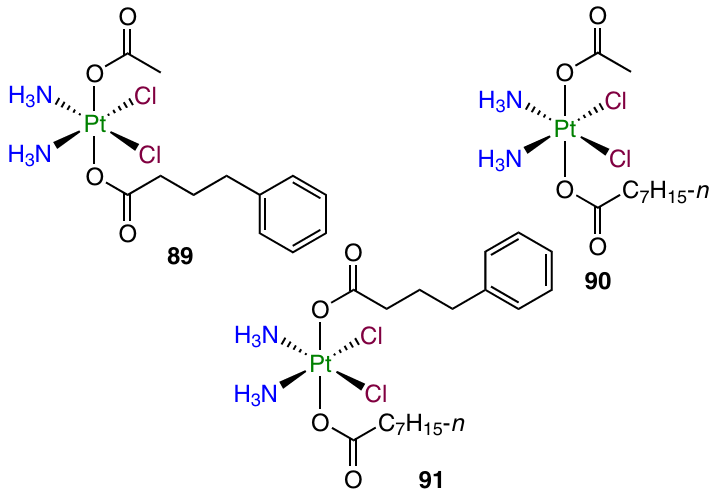
The effect of combination of various bioactive ligands on the antiproliferative activity of Pt(IV) prodrugs was studied Kostrhunova et al.123 A triple-action prodrug, platinum complex with phenylbutyrate and octanoate in the axial positions (91), was studied in comparison with cisplatin and related dual-action prodrugs 89 and 90.
Complexes 89 and 90, in which phenylbutyric or caprylic acid residue was present along with the acetate group, were 2 – 15 times more active than cisplatin, whereas prodrug 91 with three biologically active moieties had IC50 values 100 – 900 times higher than that for cisplatin. The cellular uptake level of compound 91 in MDA-MB-231 cells after a six-hour incubation was 30 and 10 times higher than those for prodrugs 89 and 90, respectively, and ~ 60 times higher than that for cisplatin, which is correlated with the lipophilicity ratio of these four compounds (log P varies in the following order: 91 >> 90 > 89 >> CDDP). Complex 91 exhibited the ability to inhibit HDAC and promoted the transmethylation of DNA, which is due to the action of axial ligands.
The physicochemical properties and bioactivity of prodrugs 92 – 102 containing carbamate moieties based on aromatic and aliphatic amines as axial ligands were investigated by Babu et al.75
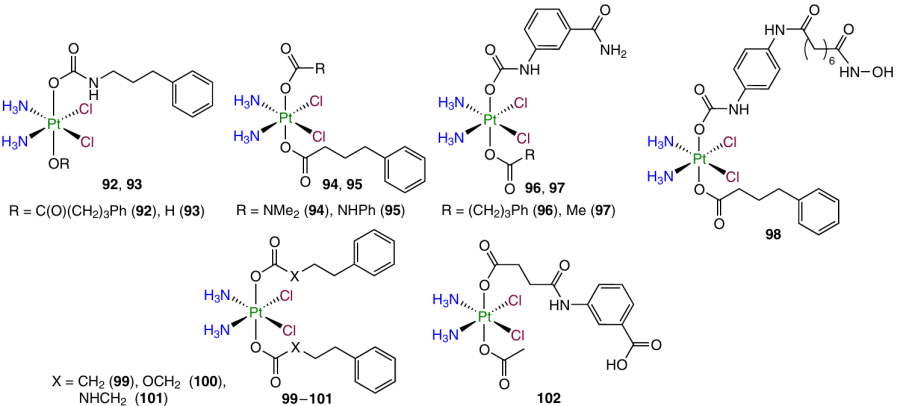
It was shown that in the presence of sodium ascorbate, the carbamate ligand of prodrugs 92 – 98 is eliminated as the carbamate anion RNHC(O)O –, which undergoes fast decarboxylation to give free amine. Succinic acid monoamide is formed as the major reduction product of complex 92. Prodrugs with a carbamate linker (92, 94, 101) showed the highest stability [half-life (t1/2) > 300 h] in the culture medium. Compound 96 with an aromatic carbamate ligand and dicarbonate complex 100 were the least stable in the aqueous medium.
The highest activity and the ability to overcome the cisplatin resistance of A2780cisR ovarian cancer cells was found for triple-action prodrug 96 containing phenylbutyrate and 3-aminobenzoate, whereas the activity of compounds 97 and 98 was similar to that of cisplatin.
3.1.3.2. 4-Halophenylacetic acids
Non-classical Pt(II) complexes with the commercial codes PHENSS and 56MESS were used by Aputen et al.124 to create Pt(IV) prodrugs 103 – 110 with 4-halophenylacetic acids.
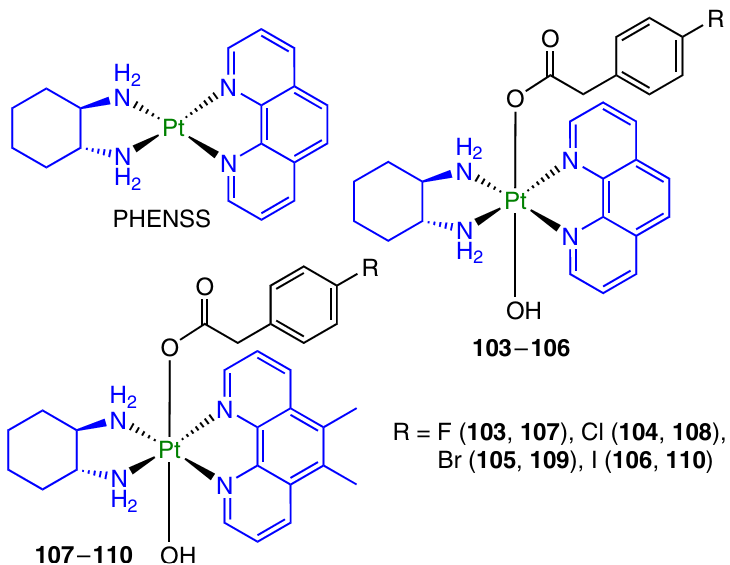
Complexes 107 – 110 showed a substantially higher cytotoxicity than the series of compounds 103 – 106, which correlates with a higher (by more than 10 times) cytotoxicity of 56MESS in comparison with PHENSS. For the most active prodrugs 107 and 109, the GI50 values indicated 1.5- to 7-fold increase in the toxicity in comparison with that of the parent Pt(II) complex and reached a value of 0.7 nM for compound 107 against Du145 cell line. Complexes 108 and 110 proved to be significantly less active than the parent complex. Prodrugs 107 and 109 were also characterized by the highest ROS level in the cells, which was three times as high as that for the parent Pt(II) complex and twice as high as that for cisplatin.
3.1.3.3. Lipoic acid
Lipoic [(R)-5-(1,2-dithiolan-3-yl)pentanoic] acid (LA) attracts attention as a compound capable of suppressing the anaerobic glycolysis of tumour cells.125 This acid is synthesized in the cellular mitochondria and has a low redox potential (E0 = –0.29 V); therefore, it inhibits the formation of ROS.126, 127 In addition, LC induces apoptosis of head and neck squamous cell cancer (FaDu) cells.128
The biological activity of kiteplatin [PtCl2(cis-1,4-diaminocyclohexane)] and its derivatives containing LA residues (111, 112) was studied by Savino et al.129
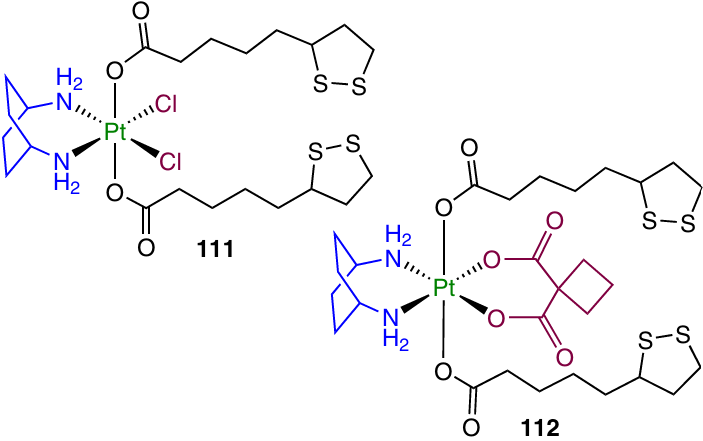
Complex 111 was active in the sub-micromolar concentration range down to IC50 = 0.1 mM against the cervical carcinoma cell line (A431). This value is 40 times lower than that for kiteplatin. In 3D spheroids of A431 cells, this prodrug was also the most active among the tested compounds (>3 times more active than kiteplatin).
Prodrugs based on cisplatin and LA (113 – 115) were reported by Liu et al.130
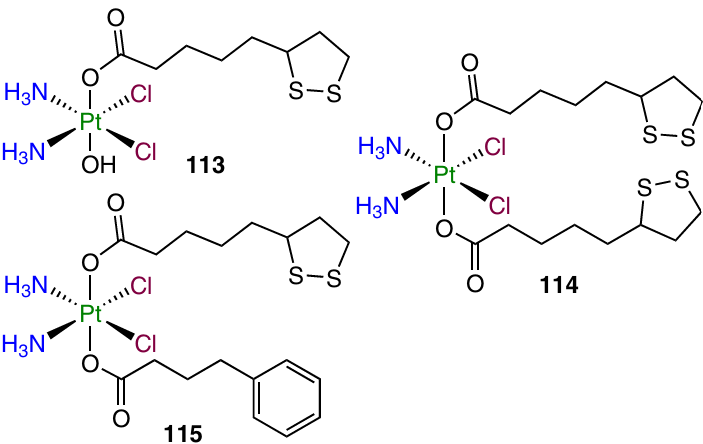
Coordination compounds 113 and 115 exhibited higher cytotoxic activity than cisplatin or an equimolar mixture of cisplatin and LA against a number of cell lines. It is worth noting that monocarboxylate complex 113 and unsymmetrical diсarboxylate 115 proved to be equally active against SW480 colorectal carcinoma cell line (IC50 = 0.74 and 0.70 mM, respectively). However, the former was 1.3 times more active against A549 cells, despite the presence of phenylbutyrate, a HDAC inhibitor, in the molecule of prodrug 115. The ability to inhibit the formation of ROS in the cell was also demonstrated for both compounds.
A series of oxaliplatin derivatives 116 – 127 containing LA and its selenium and cyclopentane analogues was studied in a more recent work by Liu et al.131
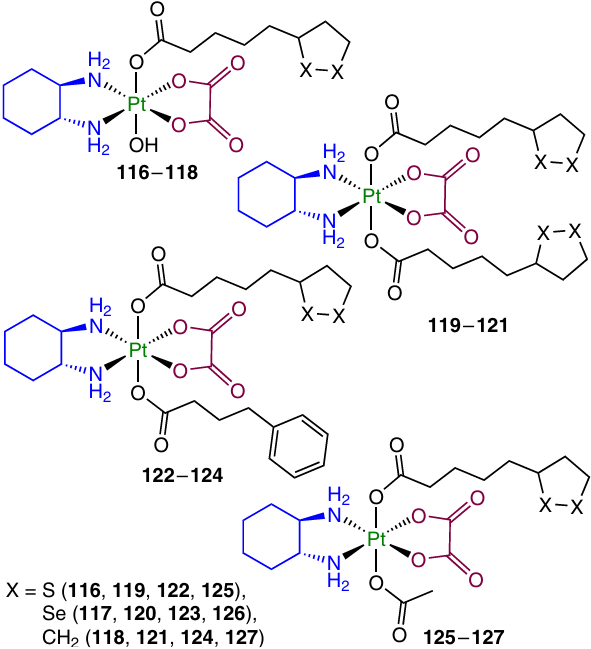
Similarly to cisplatin-based prodrugs, complex 121 proved to be stable in the presence of ascorbic acid for 72 h, unlike monocarboxylate complexes 116 – 118. The highest cytotoxic activity was inherent in prodrugs with the lipoic acid cyclopetane analogue in the axial position. The lowest IC50 values were found for prodrug 124 with phenylbutyrate: down to 18 nM against CH1/PA-1 ovarian carcinoma cells and 190 nM against SW480 colorectal carcinoma cells. The cellular uptake was examined for a series of symmetrical complexes 119 – 121; the highest platinum content was observed for compound 120, although it had the lowest lipophilicity among the three prodrugs. A possible explanation to this fact is the involvement of the active transport in the cellular uptake of the compound. The ability of complexes 116, 119 – 122 to inhibit the formation of ROS was assessed for SW480 cell line; a considerable increase in the ROS level was observed only when the concentration of the prodrugs was 50 times higher than IC50 .
3.1.3.4. Coumarin
Promising anticancer agents inhibiting tubulin polymerization and possessing activity against drug-resistant cancer cell lines were found among coumarin derivatives.132 Ma et al.133 investigated prodrugs 128 – 131 containing the residue of coumarin-3-carboxylic acid or its 6-bromo derivative.
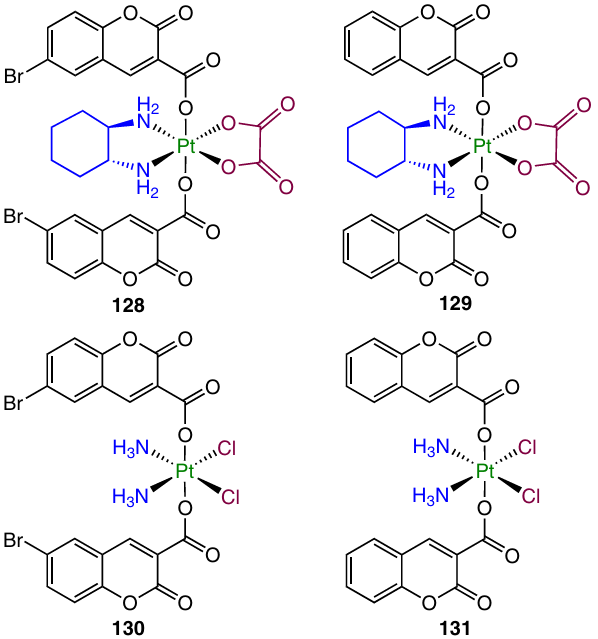
In in vitro experiments, bromine-containing complexes 128 and 130 showed the highest antiproliferative activity, and oxaliplatin derivatives 128 and 129 proved to be more active than complexes 130 and 131 based on cisplatin. Oxaliplatin-based prodrug 128 provided the possibility of overcoming cisplatin resistance for A549cisR cell line; the resistance factor (RF) was 0.81.
Cellular uptake assays for complex 128 and A549 and A549cisR cancer cell lines revealed higher uptake in A549cisR cells compared to A549 cells, while in the case of Pt(II)-based agents, the A549cisR cell line was characterized by a lower uptake than cisplatin-sensitive A549.
Evaluation of the therapeutic efficacy of complex 128 in vivo indicates that the maximum therapeutic dose (MTD) and half-lethal dose (LD50) are much higher for prodrug 128 than for cisplatin and oxaliplatin. In addition, the calculated therapeutic index (TI) (LD50/IC50 ratio) of 128 was 1.6 times higher than TI of cisplatin or oxaliplatin, indicating a reduced toxicity of complex 128 in comparison with traditional Pt(II) drugs.
3.2. Platinum(IV) prodrugs with ligands overcoming cisplatin resistance
3.2.1. Fatty acids
Resistance of tumour tissues to platinum-based drugs is a major problem of chemotherapy with cisplatin or its analogues; therefore, the search for approaches to overcome this drawback is an attractive strategy towards a better efficacy of platinum-based prodrugs.134 Complexes 132 – 134 containing a long aliphatic chain mimicking a fatty acid residue were synthesized by Jayawardhana et al.135 These agents tend to penetrate cancer cells by means of the CD36 receptor, which is overexpressed in cisplatin-resistant cell lines such as A2780cisR.
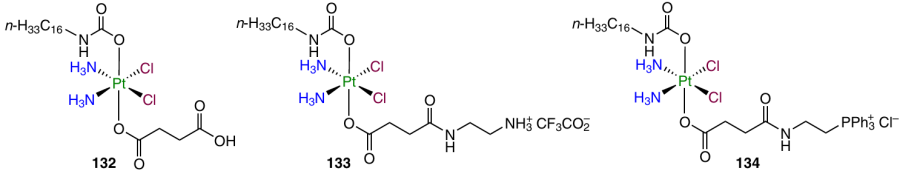
Prodrug 132 demonstrated the ability to overcome cisplatin resistance in A2780cisR cell line; RFs for the prodrug and cisplatin were 0.9 and 5.4, respectively. Compound 133 and 134 proved to be even more cytotoxic against A2780cisR cell line (IC50 = 0.24 and 0.31 mM); they were efficiently accumulated in mitochondria and decreased the mitochondrial membrane potential (MMP) of the A2780cisR cells.
3.2.2 Iron chelators
Cancer cells are more dependent on the content of iron ions than normal cells and also show increased iron uptake, accompanied by a decrease in its release.136, 137 In 2022, Pan et al.138 developed Pt(IV)-based prodrug 135 containing a clinically used chelator of iron, deferasirox (DFX), possessing a high in vitro and in vivo activity against triple-negative breast cancer.
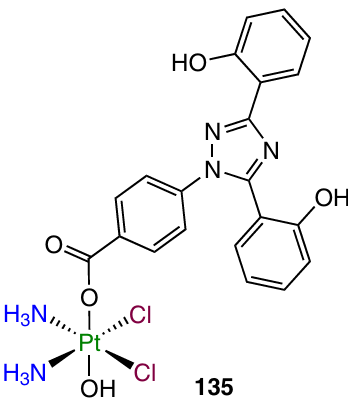
Antiproliferative activity assays showed a noticeable selectivity of compound 135 to the MDA-MB-231 cells over MCF-7 cell line or normal MCF-10A breast epithelial cells and a cytotoxic activity 43 times exceeding that of cisplatin. Studies of the cellular uptake and the ability to platinate DNA also confirmed the high activity of this complex compared to cisplatin. Despite the fact that, according to inductively coupled plasma mass spectrometry (ICP-MS) data, agent 135 did not cause a significant change in the iron content in the cell, its ability to reduce the level of the pool of chelatable iron in the cell was proved using the Phen Green dye.
The repair of DNA damages is an important mechanism for the cisplatin resistance of cancer cells. Prodrug 135 can reduce the efficiency of DNA repair in MDB-MA-231 cells and regulate the homeostasis of intracellular iron.
An in vivo study of xenograft mouse models of MDA-MB-231 tumours showed that the therapeutic efficacy of prodrug 135 was higher than that of cisplatin. At the end of the therapy, TGI was 77% for compound 135 and 41% for cisplatin; the general toxicity of the former was lower than that of cisplatin.
3.2.3. Glutathione S-transferase inhibitors
The inactivation of Pt(II) compounds with biologically active thiols such as glutathione and cysteine is considered to be one of the mechanisms of cancer cell resistance to platinum-based drugs.139 The inactivation can occur both via the passive binding of platinum complexes to glutathione and via the chemical reaction catalyzed by enzymes, in particular glutathione S-transferase (GST).140 GST inhibitors are promising axial ligands for Pt(IV) prodrugs, since agents of this type can overcome the cisplatin resistance of cancer cells.54
Oxaliplatin-based prodrugs 136 and 137, containing L-buthionine-(S,R)-sulfoximine (BSO), an irreversible GST inhibitor, in the axial position and acetate or N-(maleimidopentylcarbamate) in the second axial position, were synthesized and studied by Fronik et al.79 The maleimide moiety in the blood binds to albumin; this increases the stability of the therapeutic agent in the bloodstream and the tumour uptake of the agent.78, 141
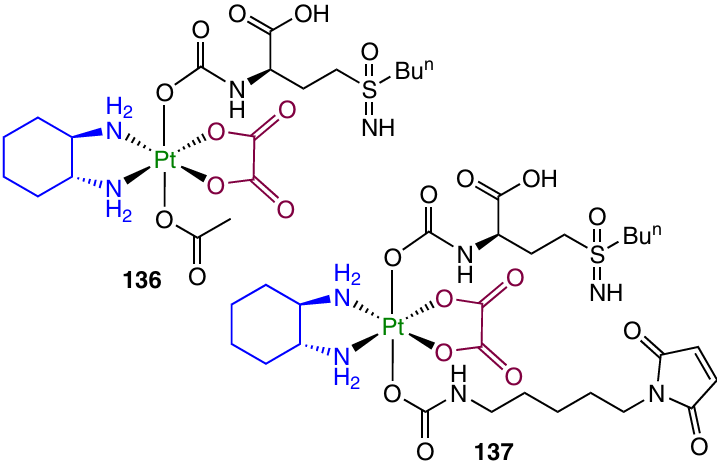
The cytotoxicity of complex 136 was assessed against HCT-116 and oxaliplatin-resistant HCT-116/oxR cell lines using oxaliplatin as the reference drug and against A2780 and A2780/cis cells in comparison with cisplatin. The cytotoxicity of prodrug 136 was found to be 10 – 50 times lower than the cytotoxicities of both Pt(II)-based drugs. However, RFs for this compound were 2.9 (HCT-116 cells) and 1.4 (A2780 cells) vs. 17.2 and 4.1 for oxaliplatin and cisplatin, respectively, for the same cell lines. Study of the cellular uptake of the agent by HCT-116 cells demonstrated that Pt(IV) prodrug 136 is accumulated in the cells five times less efficiently than oxaliplatin. It is noteworthy that the oxaliplatin uptake was two times lower in the drug-resistant HCT-116/OxR cells than in HCT-116 cells, whereas in the case of complex 136, the platinum level was identical in both cell lines.
The antitumour efficacy of prodrugs 136 and 137 was studied in vivo against CT-26 tumour in BALB/c mice. Both oxaliplatin and the test compounds showed comparable antitumour efficacy and provided a considerable (twofold) decrease in the tumour size compared to the control group by the 40th day of the therapy.
3.2.4. Inhibitors of signal transducer and activator of transcription 3
Signal transducer and activator of transcription 3 (STAT3), which regulates multiple oncogenic processes and is an important regulator of normal and cancer stem cells (CSCs), is activated in various types of cancer. This protein often serves as a therapeutic target for the development of anticancer drugs.142, 143 The cancer stem cells, which initiate the tumour formation and metastasing, are also considered to be one of the major causes for drug resistance of tumour tissues.144 Napabucasin (BBI608), acting as STAT3 inhibitor and inducing CSC death in various types of malignant growth, was approved for phase III clinical trials.145 In 2022, Wang et al.146 used a napabucasin derivative with the commercial code BBI608-OH as an axial ligand to prepare a series of prodrugs 138 – 143 based on cisplatin.
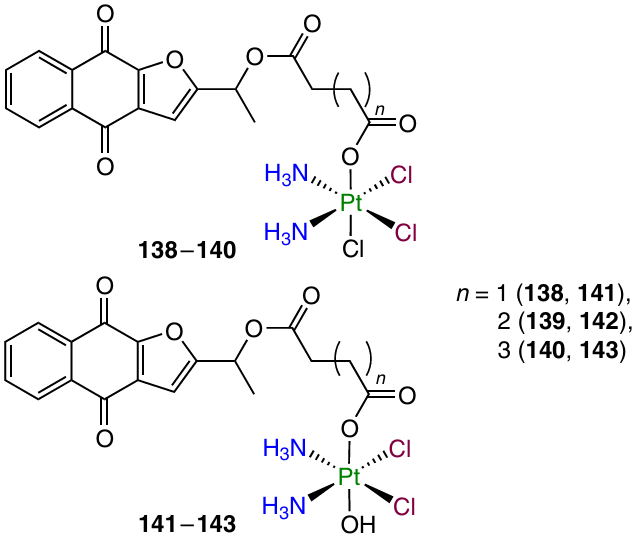
The cytotoxicity of these complexes was evaluated using a number of cell lines, including both cisplatin-sensitive and cisplatin-resistant ones (A549 and A549/CDDP). The cytotoxicity of prodrugs 138 – 140 and 141 – 143 increased with increasing length of the linker, with the highest activity being inherent in monocarboxylate complex 143 with an adipic acid linker (n = 3). Compounds 138 – 143 efficiently overcame cisplatin resistance in the A549/CDDP cells, with RF being in the 0.56 – 0.97 range.
Prodrugs 138 – 143 inhibited aldehyde dehydrogenase, the main marker of CSC; complex 143 was the most active, providing 36.31% inhibition. This compound also efficiently inhibited the CSC biomarkers, CD44 and CD133, and actively prevented the formation of A549/CDDP cell spheroids, which implies inhibition of CSC proliferation.
Complex 143 was more active in wound healing than cisplatin or napabucasin: the delay of healing was 50% relative to the control group of cells. In in vivo determination of the antitumour efficacy against A549/CDDP cells in BALB/c mice, the dose of 11.5 mg kg–1 (equivalent to 5 mg kg–1 of cisplatin in terms of Pt) induced 64.76% inhibition of the tumour growth, which was much higher than the percentage of inhibition in the group administered with cisplatin (12.77%). This indicates a high anticancer activity of complex 143 even against cisplatin-resistant tumours.
3.2.5. Poly(ADP-ribose) polymerase inhibitors
Olaparib is an anticancer drug that acts by inhibiting poly(ADP-ribose) polymerases (PARP-1, PARP-2 and PARP-3), that is, enzymes promoting the repair of DNA single-strand breaks. In the cancer therapy, olaparib is used in combination with cisplatin.147
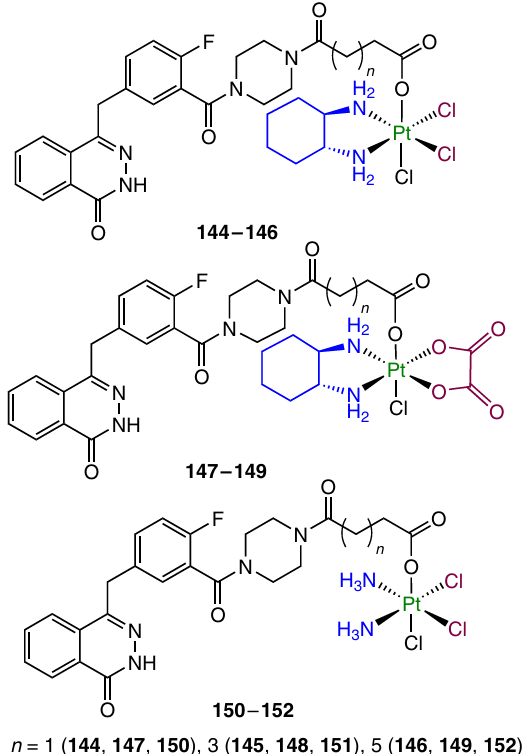
In 2023, Li et al.148 developed Pt(IV) prodrugs 144 – 152 containing an olaparib moiety in the axial position. Antiproliferative activity assays revealed good inhibitory properties of complex 151 against PARP-1 and a cytotoxic activity against MDA-MB-231 cells (IC50 = 1.13 mM) exceeding that of cisplatin, in particular against the cisplatin-resistant MDA-MB-231/CDDP cell line (IC50 = 1.72 mM). Furthermore, this compound showed high cellular uptake, the ability to inhibit DNA repair mechanisms and activate the mitochondria-mediated apoptosis. Determination of the therapeutic efficacy in vivo in MDA-MB-231/CDDP tumour xenografts in mice showed higher efficacy of prodrugs 151 compared to that of cisplatin (TGIs of 64.1 and 26.5%, respectively), along with lower general toxicity of the conjugate.
3.2.6. P-Glycoprotein inhibitors
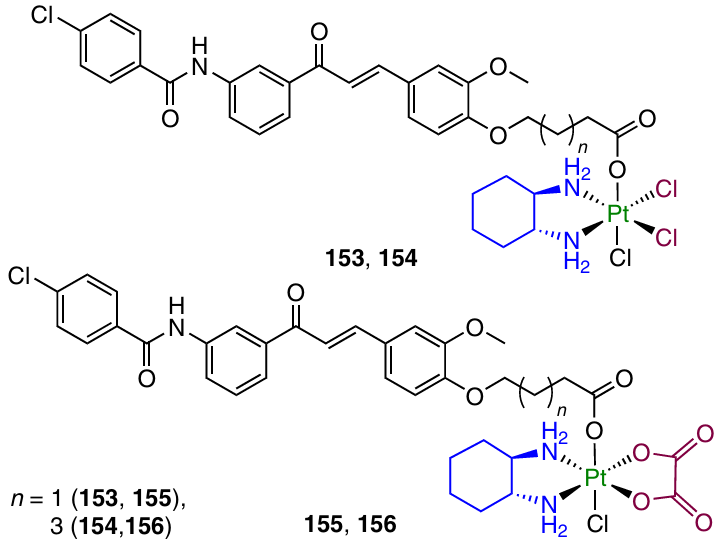
The membrane P-glycoprotein (Pgp) plays an important role in the drug pharmacokinetics. One of the mechanisms giving rise to chemotherapy resistance is the activation of Pgp, the action of which decreases the intracellular content of the drugs and, hence, reduces the therapeutic effect.149 In 2021, Cao et al.150 reported Pt(IV) prodrugs 153 – 156 with Pgp inhibitors as the axial ligands.
According to the results of antiproliferative activity assays, compound 156 was efficient against a cisplatin-resistant gastric cancer cell line (SGC-7901/CDDP; IC50 = 3.37 mM) and showed selectivity over HL-7702 normal liver cell line (the selectivity index was 6.9). Study of the mechanism of cytotoxic action demonstrated that this agent efficiently inhibits the expression of Pgp, induces the mitochondria-mediated apoptosis and arrests the cell cycle in the G2/M phase. Experiments in vivo showed efficacy of prodrug 156 for the therapy of cisplatin-resistant SGC-7901/CDDP tumour xenografts in mice, exceeding the efficacy of cisplatin or oxaliplatin (TGIs were 75.6, 25.9 and 43%, respectively).
3.3. Platinum(IV) prodrugs with non-steroidal anti-inflammatory drugs
Chronic inflammation is one of the markers of tumour tissues and a key factor in the development of the inflammatory response. A key enzyme of prostaglandin synthesis required for the development of the inflammatory response, cyclooxygenase-2 (COX-2), is overexpressed in many tumours.151, 152 Prostaglandins promote tumour cell proliferation and evasion of detection by the immune system, while downregulation of COX-2 expression has an antiproliferative effect on cancer cells.153 Therefore, NSAIDs attract attention of researchers who develop approaches to cancer therapy, in particular for the design of new platinum-based drugs.43 Combinations of cisplatin with COX inhibitors enhance the drug activity and mitigate side effects.62
3.3.1. Flurbiprofen
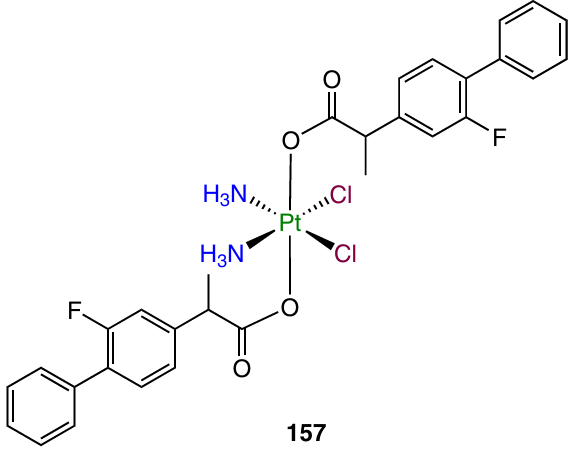
Platinum(IV) complex 157 with two flurbiprofen moieties was obtained by Tan et al.51 Prodrug 157 was found to be superior to cisplatin in cytotoxicity and to overcome the cisplatin resistance when tested on A549/CDDP cells. The RFs for A549 cell line were 0.92 and 2.7 for compound 157 and cisplatin, respectively. A study of the cellular uptake demonstrated that prodrug 157 is accumulated in the cells 20 – 50 times better than cisplatin, and the DNA platination level is 5 – 11 times higher for the prodrug than for cisplatin.
A series of Pt(IV) prodrugs 158 – 163 containing NSAIDs, naproxen, diclofenac and flurbiprofen, were prepared and investigated by Krasnovskaya and co-workers.62 The authors showed that the cytotoxicity of compounds 158 – 163 depends on the lipophilicity: indeed, monocarboxylates 158 – 160 with retention factors (log k') of 2 to 4 were toxic in the nanomolar and submicromolar ranges of IC50.
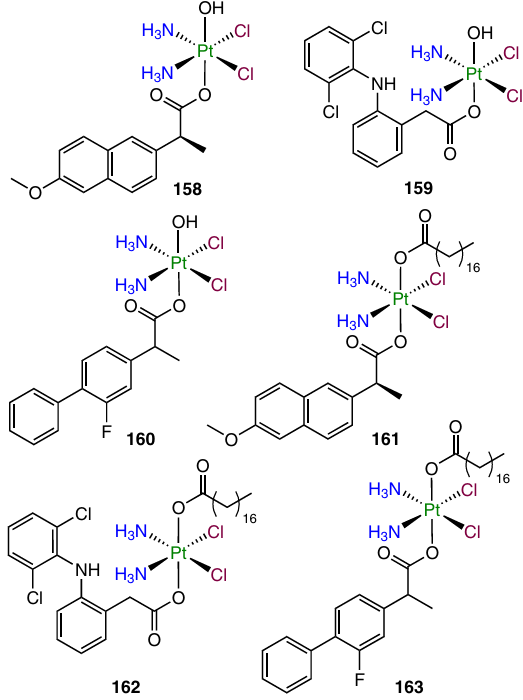
The highest activity, up to 153 times that of cisplatin, against MCF-7 cell line, was revealed for complex 160 with a flurbiprofen moiety. This compound also showed high activity against 3D cell cultures of MCF-7 cells (>30 times that of cisplatin). It was also established that prodrug 160 is an efficient agent for cisplatin delivery to the depth of MCF-7 cell spheroids. In addition, this compound efficiently delivered cisplatin deep into the EMT-6 mammary carcinoma tumour in BALB/c mice upon intravenous and intratumour injections.
3.3.2. Ketoprofen
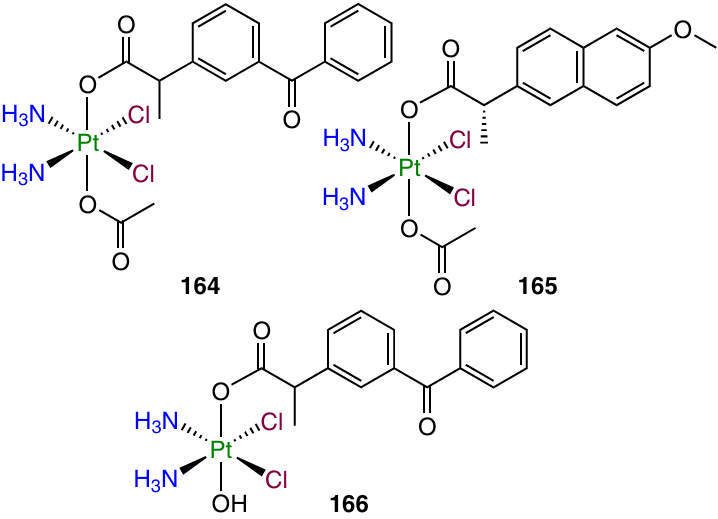
Cisplatin dicarboxylate derivatives 164 and 165 with COX inhibitors, ketoprofen and naproxen, as axials ligands were reported by Ravera et al.53
The lipophilicity of these prodrugs was investigated by high-performance liquid chromatography (HPLC) with determination of the retention factors, which directly correlated with the octanol – water partition coefficients.154 The cytotoxicity was assessed against A549, HT-29 and HCT-116 cell lines expressing COX and against MSTO-211H (mesothelioma), SW480 and A2780 cells, which do not express COX. The IC50 values for prodrugs 164 and 165 exceeded IC50 for cisplatin by a factor of 20, and no unambiguous correlation between COX expression and cytotoxicity was established. Meanwhile, the cytotoxicity was found to be directly correlated with the lipophilicity of compounds: the most active compound 165 was the most lipophilic among the series of derivatives. Study of the cellular uptake of platinum complexes in A2780 cancer cells revealed the greatest uptake for the most lipophilic prodrugs 164 and 165.
Ketoplatin 166, a monocarboxylate ketoprofen and cisplatin derivative, was investigated by Ma et al.155 Ketoplatin exhibited a cytotoxic activity 3 – 50 times exceeding that of cisplatin in a low micromolar range. Unlike cisplatin or ketoprofen, ketoplatin (166) induced a pronounced DNA damage in MDA-MB-231 cells and inhibited the cell repair and motility.
In evaluation of the antitumour efficacy in vivo, compound 166 showed retardation of the MDA-MB-231 tumour growth in BALB/c mice similar to that of cisplatin; however, unlike cisplatin, in caused no weight loss of the animals.
A series of prodrugs 167 – 175 based on cisplatin, oxaliplatin and carboplatin, containing ketoprofen and loxoprofen, were synthesized and evaluated by Li et al.156 Cisplatin-based prodrugs proved to be more cytotoxic than ketoprofen-containing compounds 167 and 166 or the loxoprofen-containing complexes 170 and 174. In addition, dicarboxylates 167 and 170 were more active than monocarboxylate analogues 166 and 174. Cisplatin derivatives 166, 167 and 170 were also able to overcome the cisplatin resistance of A549cisR cell line.
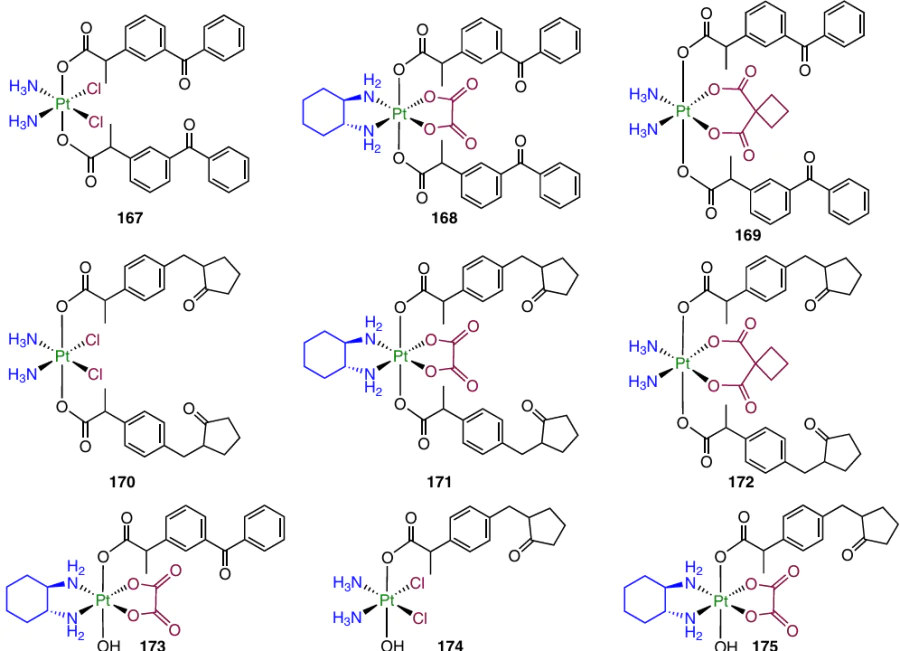
In determination of the antitumour efficacy in vivo against CT-26 colon cancer, the greatest TGI (57%) among cisplatin-based prodrugs (166, 167, 170 and 174) was found for complex 166; furthermore, this complex was less toxic than cisplatin. In addition, this compound also showed a similar TGI (54.6%) in an in vivo experiment using 4T1 tumour.
Prodrug 166 exhibited antimetastatic effect and ability to damage DNA, which was accompanied by overexpression of γ-H2AX and p53 protein (DNA damage markers) and resulted in inhibition of PD-L1 (programmed cell death ligand).
3.3.3. Naproxen
On the basis of cisplatin, oxaliplatin and carboplatin, Tolan et al.157 synthesized and studied complexes 158 (for the structure, see above) and 176 – 180 containing naproxen as an axial ligand.
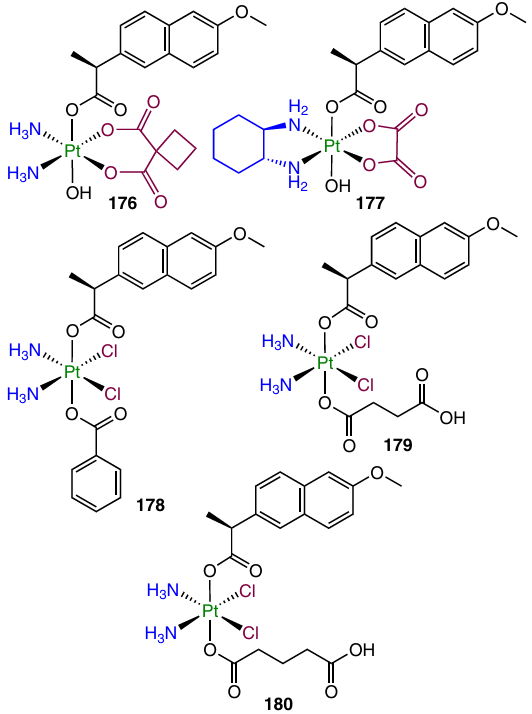
When tested against MCF-7 cells, Pt(IV) prodrugs 158 and 176 – 180 showed cytotoxicity 1.5 – 2 times as high as that of cisplatin, while in the case of MDA-MB-231 cells, they were 11 – 30 times more active than cisplatin. It is worth noting that the most lipophilic complex 178 showed the greatest cytotoxicity, as well as the ability to induce partial necrosis of MCF-7 tumour cells.
One more series of prodrugs based on cisplatin, oxaliplatin and carboplatin with naproxen 181 – 185 was reported by Chen et al.158 The cytotoxic activity of these prodrugs was evaluated against a number of cell lines including cisplatin-sensitive and cisplatin-resistant cells (A549 and A549cisR, respectively). The highest antiproliferative activity was found for monocarboxylates 181 and 183 based on cisplatin and oxaliplatin, whereas oxaliplatin dicarboxylate 184 had the greatest selectivity to tumour cells over the normal L02 cells.
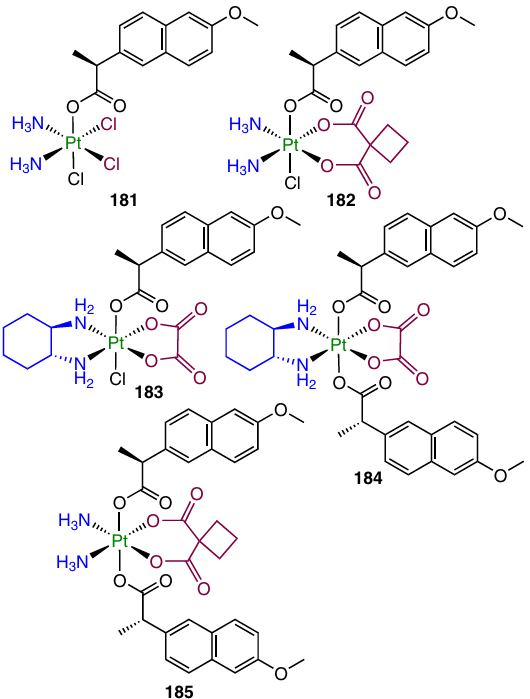
Matrix metallopeptidase 9 (MMP-9) is overexpressed in tumours, which is associated with tumour progression, metastasis and inflammation.159 Complex 183 inhibited MMP-9 expression in CT-26 tumour of BALB/c mice; its inhibitory activity exceeded that of oxaliplatin (6.8 and 8.1%, respectively). According to the study of the antitumour efficacy in vivo, this complex suppressed the growth of the CT-26 tumour to an extent comparable with those of cisplatin and oxaliplatin: the tumour volumes were 317 ± 119 mm3, 390 ± 162 mm3 and 477 ± 223 mm3, respectively.
Platinum(IV) dicarboxylate prodrugs with biotin, naproxen and stearic acid as axial ligands (158, 186 – 189) were reported by Krasnovskaya and co-workers.70
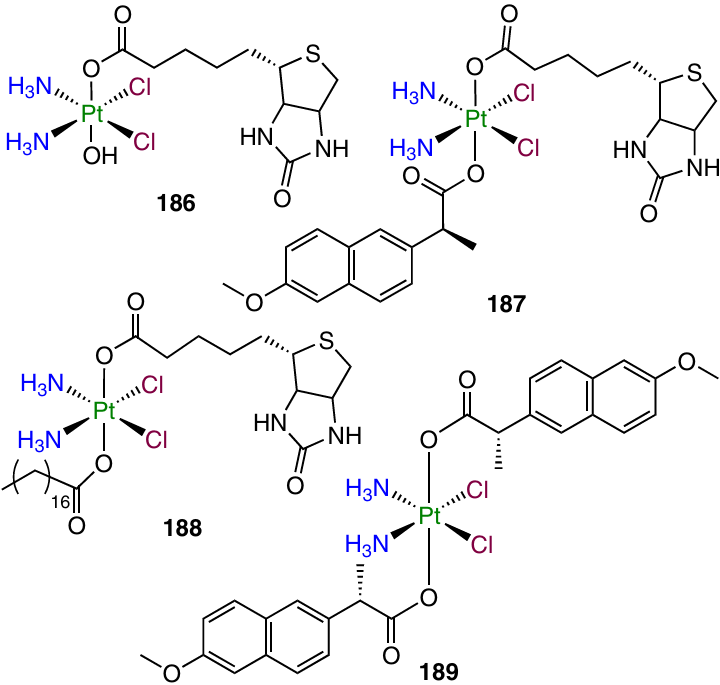
In the evaluation of the cytotoxic activity by MTT assay, naproxen-containing compound 187 showed antiproliferative activity comparable to that of cisplatin, while more lipophilic complex 188 containing stearic acid was active in the sub-micromolar and low micromolar concentration ranges (for A549 cells, IC50 was 0.87 mM). According to XANES investigation of the reduction of dicarboxylate 187 in an intracellular medium, this prodrug gradually releases the Pt(II) complex.
Cyclooxygenase-2 is not only a key enzyme in prostaglandin synthesis, but also a regulator of PD-L1 expression, which helps tumour cells to avoid detection by the immune system.125 In order to combine the cytotoxicity and the ability to activate immune response of tumour tissues in the same antitumour agent, Jin et al.58 synthesized prodrug 189, along with complex 158. When tested against MCF-7 and MDA-MB-231 cell lines and MDA-MB-435 melanoma cells, both compounds showed exceptionally high antiproliferative activity, exceeding the cisplatin activity by up to 187 times. After 24 h of incubation of MCF-7 cells with these agents, the platinum content in the cells treated with prodrugs 158 and 189 exceeded this value for cisplatin by factors of 65 and 11, respectively.
Compound 189 proved to inhibit the COX-2 and PD-L1 expression in MCF-7 tumour cells and interleukins IL-1β and IL-6 critical for the development of the inflammatory response. A study of the antitumour efficacy of prodrugs 189 in vivo in BALB/c mice bearing MDA-MB-231 tumour resulted in a substantial TGI (66 mm3 vs. 926 mm3 in the control) by the 15th day of the therapy with prodrug 189, while for cisplatin the volume of the tumour was 660 mm3.
In a XANES spectroscopy study of the intracellular reduction of prodrug 158 and 189, monocarboxylate 158 proved to have low stability, while dicarboxylate 189 was more stable, which accounts for the marked in vivo efficacy of prodrug 189.160 Evaluation of the ability of complex 189 to deliver cisplatin to tumour cells with a platinized nanoelectrode also indicated higher efficacy of this agent in comparison with cisplatin.161
3.3.4. Indomethacin and aspirin
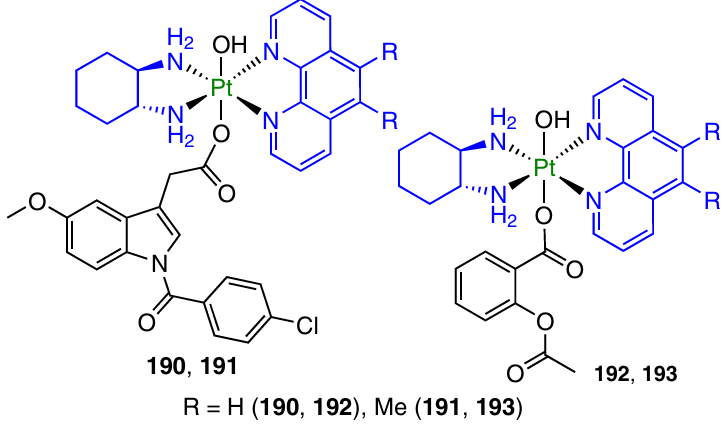
Indomethacin and aspirin derivatives 190 – 193 based on non-traditional Pt(II) complexes with commercial codes PHENSS and 56MESS were investigated by Khoury et al.162
Prodrugs 191 and 193 derived from 56MESS showed the highest antiproliferative activity: the GI50 values were, on average, 20 times lower for these compounds than for their analogues 191 and 193 based on PHENSS. Complexes 192 and 193 did not show a significant inhibitory activity against COX-2, whereas the activity of indomethacin derivatives 190 and 191 was comparable with that of free indomethacin.
3.3.5. Ibuprofen
Curci et al.50 prepared and investigated prodrug 194 based on kiteplatin containing an ibuprofen moiety in the axial position.
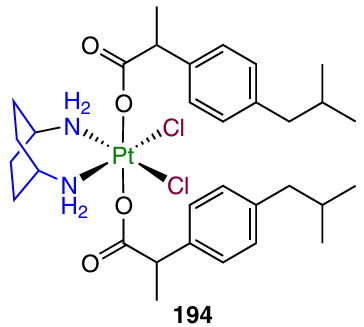
A study of the antiproliferative activity of complex 194 against HCT-115 and HCT-116 colorectal carcinoma cells resulted in sub-micromolar IC50 values, up to 42 times lower than the values for cisplatin and kiteplatin.
3.3.6. Etodolac, sulindac and carprofen
Three cisplatin complexes with NSAIDs containing etodolac, sulindac and carprofen (compounds 195 – 197, respectively) were investigated by Song et al.163
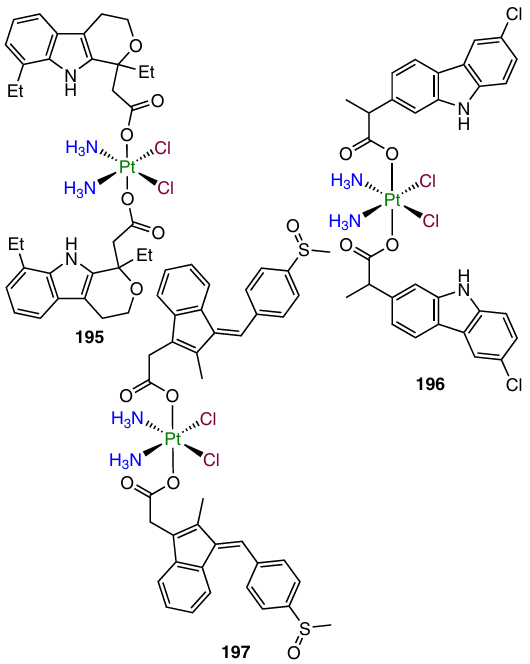
The cytotoxicity of prodrugs 195 – 197 against MCF-7, A549 and HeLa cancer cell lines was higher than that of cisplatin; meanwhile, the activity of these complexes against normal MRC-5 cell line was lower than that for cisplatin. The highest cytotoxic activity against the cancer cells was inherent in complex 195, which had an optimal lipophilicity (log P = 0 – 3).164
Lead compound 195 efficiently inhibited COX-2 and MDM-2 in MCF-7 cells; it also promoted upregulated the expression of pro-apoptotic Bax and p53 genes. In addition, this complex inhibited migration of MCF-7 cells. In experiments on determination of the antitumour efficacy in vivo, the suppression of growth of the MCF-7 tumour in BALB/c mice by complex 195 was comparable with that for cisplatin (tumour volumes were 457 and 570 mm3, respectively). However, no decrease in the animal weight was observed in the group treated with agent 195, unlike that for mice administered with cisplatin.
3.3.7. Niflumic acid
In 2023, Li et al.165 developed Pt(IV) prodrugs 198 – 201 containing niflumic acid as an axial ligand. Niflumic acid can suppress tumour metastasing by inhibiting ERK 1/2 kinases and matrix metalloproteinases.166
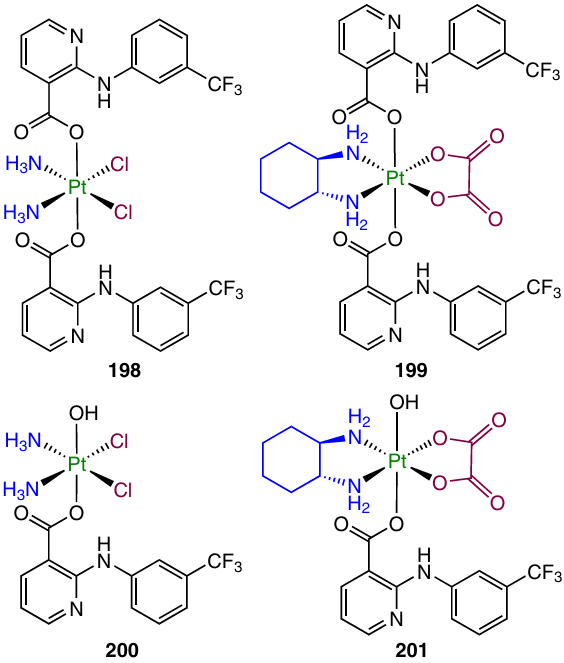
Complexes 198 and 200 had a higher cytotoxic activity than cisplatin, oxaliplatin, carboplatin or satraplatin ([PtCl2(OAc)2NH3(NH2C6H11-cyclo)], code JM216) against SKOV-3, CT26 (mouse colon cancer) and 4T1 cell lines. Prodrug 198 accumulated in the cells 4.5 times more efficiently than cisplatin. Evaluation of the in vivo therapeutic efficacy in BALB/c mice bearing 4T1 breast cancer showed similar efficacy for compound 198 and cisplatin, along with less pronounced weight loss in the former case. Prodrug 198 was found to inhibit the COX-2 and MMP-9 enzymes and also ERK 1/2 and HIF-1α. Immunohistochemical analysis showed an increase in the number of CD3+, CD4+ and CD8+ lymphocytes in tumour tissues after therapy with this agent.
3.3.8. Dichloroacetate in combination with cyclooxygenase inhibitors
A series of triple-action prodrugs 202 – 209 were investigated by Petruzella et al.27
Aspirin and ibuprofen were chosen as COX inhibitors. Dichloroacetate is an efficient PDK inhibitor and also induces cell death by damaging mitochondria.167, 168 The PDK enzyme inhibits the pyruvate dehydrogenase complex, which is significant for chain respiration.169 This complex does not function in tumour cells; therefore, the intrinsic cellular metabolism changes, and glycolysis takes place instead of glycose oxidation (Warburg effect).170 The inhibition of PDK stops this process, which results in the death of tumour cells. Phenylbutyrate and valproic acid were chosen as HDAC inhibitors.168, 171 The inhibition of HDAC causes chromatin decondensation, which makes DNA more sensitive towards platination.172
Eight prodrugs were found to be much more active than cisplatin. The average IC50 values for prodrugs 202 – 209 tested on thyroid cancer (BCPAP) and pancreatic cancer (PSN-1) cells were 51 and 71 times lower than those for cisplatin. The cytotoxicity of the compounds was also assessed against the 3D spheroids of PSN-1 pancreatic cancer cells. Complexes 205, 208 and 209 were 50 times more active than cisplatin. Study of the mechanism of cytotoxic action revealed no correlation between the inhibitory activity of axial ligands and the cytotoxicity, or between the ability of these complexes to alkylate DNA of PSN-1 cells and the cytotoxicity, which may indicate a possible synergistic effect between the ligand and the Pt(II) atom.
3.3.9. Combination of estramustine with histone acetylase and cyclooxygenase
Karmakar et al.63 developed prodrugs 210 – 214, the molecules of which contained, in addition to platinum(IV), a residue of estramustine (a steroidal anticancer drug) and various carboxylate ligands such as acetate and HDAC, PDK and COX-2 inhibitors: phenylbutyrate, dichloroacetate, valproate and o-acetylsalicylate.
According to cytotoxicity assays, prodrugs 211, 212 and 214 were 50 – 145 times more active than cisplatin, with the IC50 values against a prostate carcinoma cell line (LNCaP) being 31, 49 and 90 nM, respectively. Furthermore, all prodrugs were 13 – 50 times less active against normal MRC-5 cells. Compound 212, which was most cytotoxic against LNCaP cells, had the highest selectivity index (50) over the normal MRC-5 cell line, which was due to the effect of estramustine.
The cellular uptake of the prodrugs in LNCaP cells correlated with the cytotoxicity: compound 212 penetrated tumour cells 64 times better than cisplatin; however, the platinum level in DNA after incubation with this prodrug proved to be only 12 times higher than that for cisplatin, which indicates that other factors also make a contribution to the cytotoxicity. For the most cytotoxic prodrugs 211 and 212, no biological effect of estramustine was observed; however, an effect caused by the inhibitory activity of valproate and phenylbutyrate against HDAC was manifested.
3.4. Platinum(IV) prodrugs with immunomodulating axial ligands
Combination of immunotherapy with chemotherapy is a widely used clinical protocol for the treatment of cancer. For example, first-line therapy for patients with non-small cell lung cancer includes the combination of cisplatin and pembrolizumab.173, 174 In view of the synergism of cisplatin with immune response checkpoint inhibitors, the development of Pt(IV) prodrugs with ligands that stimulate the immune response should be of obvious interest of researchers.175
3.4.1. Inhibitors of bromodomain-containing protein 4
The programmed cell death ligand PD-L1 is a transmembrane protein interacting with the PD-1 receptor. Upon the specific binding to the PD-1 receptor on cytotoxic lymphocytes, this ligand blocks their cytotoxic activity, thus enabling the tumour to evade the immune response. The heteroannulated benzodiazepine with the code JQ1 is an inhibitor of bromodomain-containing protein 4 (BRD4), which stimulates transcription of the CD274 gene encoding the PD-L1 protein.176 Overexpression of the PD-L1 ligand may also lead to development of cisplatin resistance or radiation therapy resistance of the cell.177, 178
In 2023, Fan et al.179 synthesized Pt(IV) complexes 215 – 218 with a JQ1 moiety in the axial position. While studying the cytotoxicity, the authors demonstrated high efficacy of prodrugs 216: IC50 = 0.89 mM against a melanoma cell line (B16F10), which is 40.66 times better than that of cisplatin. This compound was found to induce cell apoptosis, decrease MMP and cause DNA damage. In addition, the parent JQ1 ligand and complex 216 considerably inhibited the expression of the BRD4 and PD-L1 proteins in cells. Experiments in vivo using mouse model of B16F10 melanoma revealed a significant therapeutic effect of prodrug 216, which exceeded the action of cisplatin or the JQ1 + cisplatin equimolar mixture and caused a less pronounced weight loss of the animals. A synergistic effect of the therapy with agent 216 and the immunotherapy with anti-PD-1 monoclonal antibodies was also detected. Immunohistochemical analysis of the tumour after treatment with compound 216 showed an increased infiltration of the tumour with the CD8+ T-cells; this provides evidence for the action of this prodrug through activation of the immune response.
3.4.2. Melatonin
Melatonin (N-acetyl-5-methoxytryptamine) is an indoleamine hormone synthesized from serotonin by the pinealocytes of the pineal gland. This hormone has an antitumour activity against breast cancer and can also affect the estrogen synthesis.180, 181 In addition, melatonin has immunomodulating properties and affects the expression of CD4+ and CD25+ lymphocytes in the tumour microenvironment.182
In 2020, Song et al.183 proposed cisplatin-based prodrugs 219 – 222 in which platinum(IV) atom is connected to one or two melatonin residues through various linkers in the axial positions.
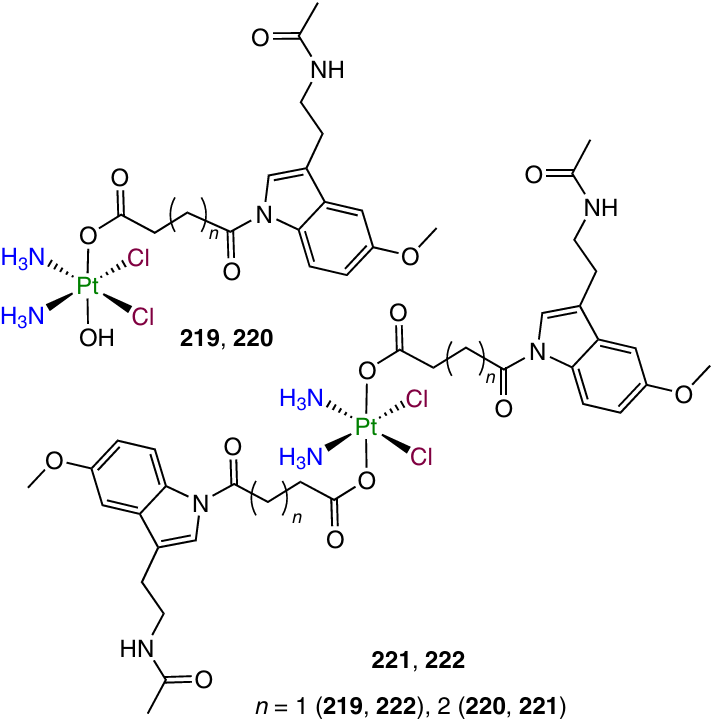
Compound 219 surpassed cisplatin in efficacy by a factor of 100 (IC50 = 0.06 mM) when tested against ER(+) MCF-7 cells; the toxicity against ER(–) MDA-MB-231 cells was only 0.36 mM. Prodrugs 219 and 220 also showed a high cellular uptake, which exceeded the uptake of cisplatin by 76 and 27 times, respectively. In addition, complex 219 stimulated the expression of the γH2AX and p53 proteins, arrested the cell cycle in the S-phase and triggered apoptosis.
Study of the therapeutic efficacy in vivo against MCF-7 tumour xenografts in BALB/c mice showed similar efficacy of prodrug 219 and cisplatin, with lower weight loss of animals in the former case. A higher platinum accumulation in spleen was found for mice treated with cisplatin. Lymphocyte proliferation in spleen was found for the groups of mice administered with melatonin and compound 219. Thus, prodrug 219 acts through immunomodulation and improves the overall survival rate of individuals with ER(+) breast cancer.
3.4.3. 1-Methyl-D-tryptophan
One of the mechanisms by which malignant cells can evade the immune response is the expression of indoleamine-2,3-dioxygenase (IDO1), which catabolizes the conversion of tryptophan to kynurenine suppressing the T-cell immunity. 1-Methyltryptophan is an indoleamine-2,3-dioxygenase inhibitor, with a higher immunogenic anticancer activity in vivo being inherent in the D-isomer.184, 185
In 2021, Fronik et al.186 investigated triple-action Pt(IV) prodrugs 223 – 226, which contained 1-methyl-D-tryptophan and succinimide in the axial positions. These compounds were compared with oxaliplatin dicarboxylates 227 – 229. The same publication describes complexes 230 – 233, which are analogues of compounds 223 – 226 containing a maleimide moiety to enhance binding of the agent to albumin.
The ability of prodrugs 223 – 229 to inhibit IDO1 was proved by analyzing cell lysates after incubation with the test compounds. Compounds 225 and 226, which are rapidly reduced, provide a more pronounced inhibition of this enzyme than complexes 223 and 224, which are reduced slowly.
Study of the therapeutic efficacy against CT26 colon cancer allografts in mice demonstrated that prodrug 230 surpassed the oxaliplatin in both the inhibition of the tumour growth and the survival rate of mice. Flow cytometry data for the immune cells isolated from the tumour tissue and from tumour-draining lymph nodes after administration of compound 231 demonstrated a significant shift in the ratio of CD4+ (immunosuppressive) and CD8+ (immunostimulatory) T cells.
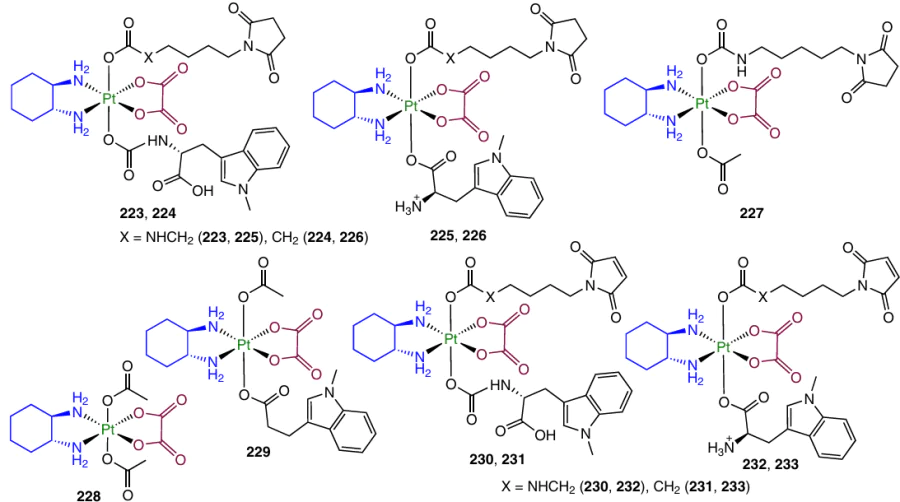
3.5. Platinum(IV) prodrugs with ligands promoting increase in the selectivity
A crucial drawback of the therapy with Pt(II)-based anticancer agents is that the lack of selectivity of cisplatin or its analogues over normal tissues, which accounts for severed side effects.187 Below we consider Pt(IV) prodrugs in which axial ligands promote higher uptake of the agents mainly in the tumours cells.
3.5.1. Carbohydrates
It was shown previously that Pt(II) conjugates with carbohydrates possess good selectivity to cells that overexpress glucose transporters (GLUT), which makes carbohydrates promising axial ligands for Pt(IV) prodrugs.188, 189 GLUT receptors are overexpressed in many tumours, including lung, breast and liver carcinomas; therefore, this group of transporter proteins are considered to be an optimal target for a Pt(IV) prodrug vector moiety.190
Wang et al.25 investigated a series of oxaliplatin-based prodrugs 234 – 239 with glucose derivatives in the axial position.
The antiproliferative activity of these compounds against HeLa, HepG2, MCF-7 and A549 cancer cell lines and cisplatin-resistant A549cisR cell line exceeded the oxaliplatin activity by 1.5 – 3 times. All prodrugs were able to overcome the cisplatin resistance of the A549cisR cell line.
The selectivity of the agents was evaluated for HepG2 cancer cells over the normal liver cells (L02). The oxaliplatin toxicity against L02 cells was somewhat higher than that against HepG2 (IC50 was 8.34 and 10.90 mM). Prodrugs 234 – 239 had a lower cytotoxicity against L02 cells, with the highest selectivity factor (24.10) being observed for complex 238.
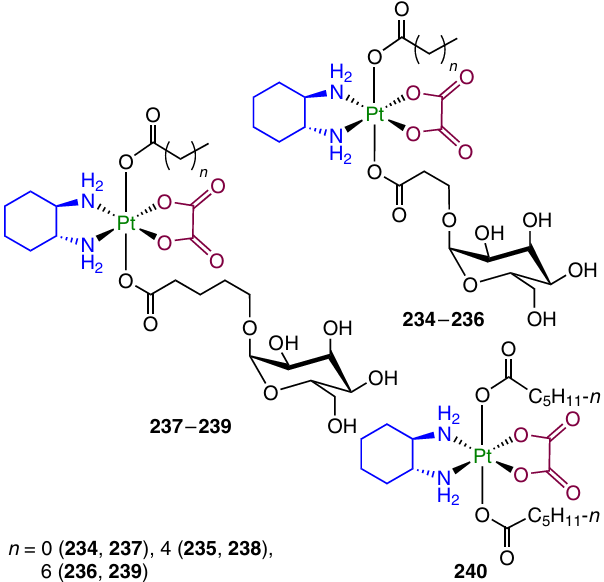
Prodrugs containing axial ligands based on carbohydrates entered MCF-7 cells 1.7 – 3 times more efficiently than model complex 240 and 10 times more efficiently than cisplatin. The cellular uptake of the prodrugs varied in the order 237 > 238 > 239, i.e., it increased with increasing carboxylic acid chain length in the axial position. The level of DNA platination with prodrugs 234 – 239, oxaliplatin and cisplatin correlated with the cellular uptake level.
3.5.2. Triphenylphosphine
Mitochondria are critical organelles of cells, as they produce most of the energy; therefore, the search for compounds affecting mitochondria is a promising task.191 Triphenylphosphonium, being a delocalized lipophilic cation, efficiently penetrates the lipid membranes and can be accumulated in mitochondrial cells with excess negative charge.192 A series of cisplatin-based prodrugs (241 – 244) and oxaliplatin-based prodrugs (245 – 248) containing an alkyltriphenylphosphonium moiety in the axial position were synthesized by Babak et al.66 In all compounds, the formate anion (HCOO – ) served as the counter-ion.
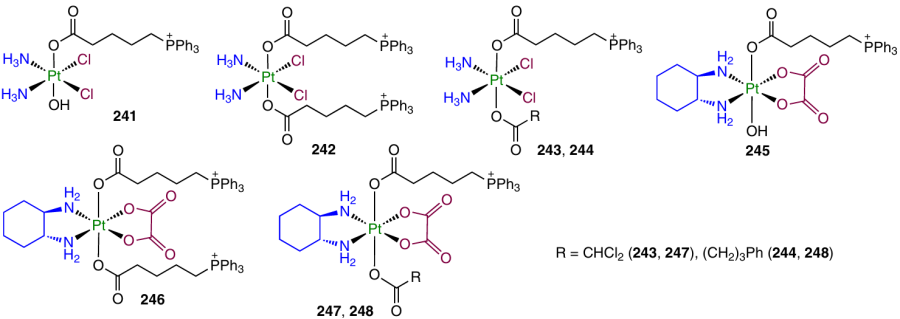
As the second axial ligand, the authors used a hydroxyl group, one more alkyltriphenylphosphonium moiety, and dichloroacetate or phenylbutyrate. The cytotoxicity of compounds 241 – 248 was assessed against A2780 cell line and MOR lung adenocarcinoma cell line and against their cisplatin-resistant analogues (A2780cisR and MORcisR). The activity of prodrugs 241, 242, 245 and 246 turned out to be lower compared to the parent Pt(II) complexes. The highest activity was inherent in complexes 244 and 248 containing phenylbutyrate as the second axial ligand; the same compounds were the least effective in overcoming the cisplatin resistance. The cellular uptake of the prodrugs correlated with the antiproliferative activity. Most of platinum in A2780 cells was accumulated in mitochondria, while the most clear-cut targeting effect was observed for compounds 243, 244 and 247, 248. Further investigation of the ability of complexes to induce mitochondrial depolarization showed the highest efficiency for prodrugs 243, 244, 247 and 248, which also markedly inhibited the mitochondrial respiratory function.
The in vivo experiments performed in BALB/c mice with CT-26 adenocarcinoma tumour model identified a high therapeutic efficacy of agent 243. Indeed, by the 32nm day of the therapy, the tumour size in the group administered with prodrug 243 was five times smaller than that in the group treated with cisplatin and eight times smaller than that in the control group. To increase the bioavailability, the liposomal form of agent 243 was obtained. After 32 days of the therapy at a dose of 1.95 mg kg–1 of platinum, complete remission of the disease was observed in the group of animals treated with the liposomal form of 243.
3.5.3. Metronidazole
Hypoxia is one of the markers of solid tumours, being important for tumour growth, angiogenesis and metastasis.193 Metronidazole is a drug widely used for the treatment of anaerobic infections; it is also able to inhibit aldehyde dehydrogenase, one of the markers of hypoxia.194 Platinum(IV) prodrug 249 containing metronidazole in the axial position was described by Krasnovskaya et al.60
For comparison, platinum(IV) complexes with 2-thioimidazol-4-one (250) and paracetamol (251) were prepared. Complex 249 was similar to cisplatin when tested for the antiproliferative activity against a monolayer of cancer cells; however, in the case of spheroids of MCF-7 cancer cells, it showed an antiproliferative activity exceeding that of cisplatin by more than 31 times.
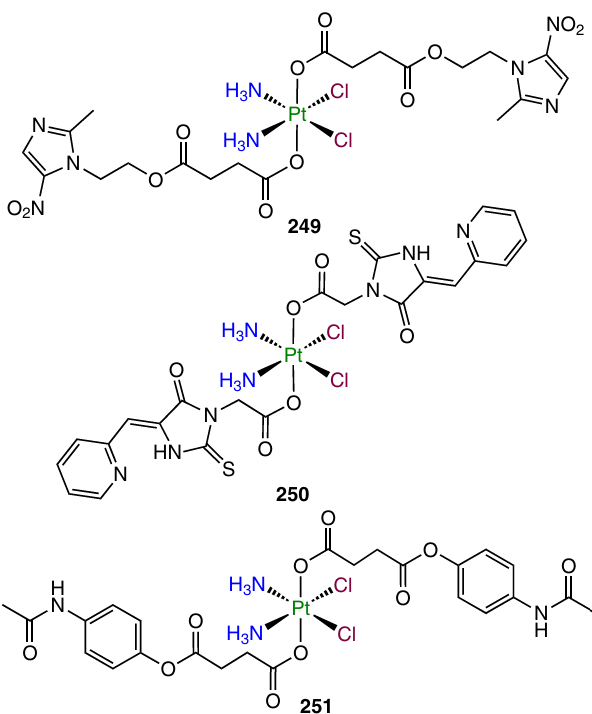
Determination of the intracellular reduction rate by XANES spectroscopy demonstrated that prodrug 249 gradually released the Pt(II) complex. After 26 h of incubation of A549 cells in the presence of complex 249, less than 60% of the starting compound was reduced. Study of the cisplatin distribution profile in MCF-7 spheroids preincubated with compound 249 using a nanoelectrode demonstrated that this complex can deliver cisplatin to the hypoxic area inside the tumour spheroid.
3.5.4. Hypoxia-sensitive agents
Carbonic anhydrases (CAIX) are transmembrane proteins that catalyze the conversion of CO2 to bicarbonate and a proton.195 These enzymes are overexpressed in many tumour tissues and acidify them.196, 197
Cao et al.195 used CAIX inhibitors in both axial positions to obtain prodrugs based on cisplatin and oxaliplatin (252 and 253).
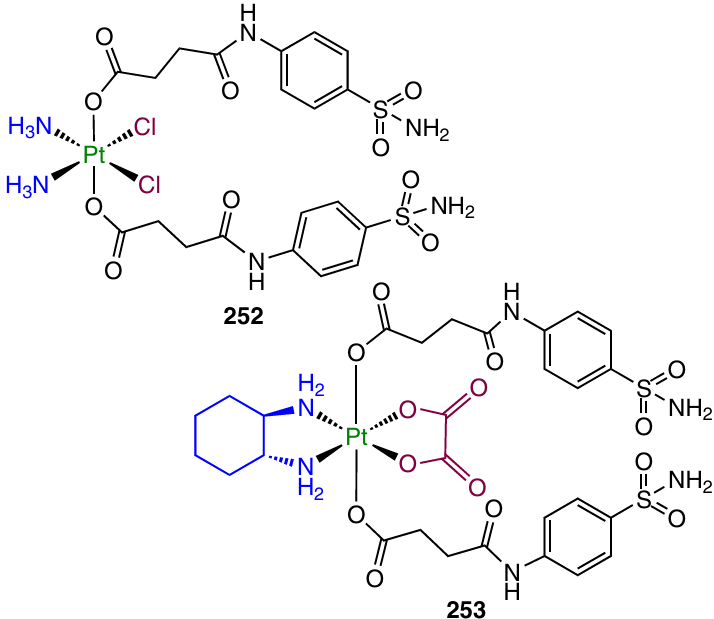
Under normoxic conditions, complexes 252 and 253 exhibited enhanced cytotoxicity against MDA-MB-231, HeLa and HepG2 malignant cells compared to normal cells–L02, HLF (human lung fibroblasts) and MCF-10A. The selectivity indices for MDA-MB-231 cells over MCF-1A cells under normoxia were 8.5 and > 7.3 for compounds 252 and 253, respectively. Under hypoxic conditions, the cytotoxicity of both prodrugs against cancer cells increased 3 – 9-fold and, hence, the selectivity index increased up to 80 and 34.5, respectively. The observed selectivity is attributable to a 10 times lower platinum accumulation in normal MCF-10A cells than in MDA-MB-231 hypoxic cancer cells.
When MDA-MB-231 cells were incubated with a CAIX inhibitor (compound encoded SLC-0111), the cellular uptake of the agents decreased; this is evidence for the contribution of active transport to the transfer of prodrugs 252 and 253 into the cells. In addition, it was shown that the oxygen content and pH of the MDA-MB-231 hypoxic cells increased after the cells had been incubated with these compounds.
The in vivo antitumour efficacy of the compounds was studied for the MDA-MB-231 tumour in BALB/c mice. After 24 days of the therapy with cisplatin, oxaliplatin and compounds 252 and 253 in 5 mg kg–1 dose, the tumour growth inhibition was 57 and 65% for prodrugs 252 and 253, respectively, and only 32 – 43% for cisplatin and oxaliplatin.
An alternative approach to the development of hypoxia-sensitive prodrugs was described by Boulet et al.198 As the axial ligands in the platinum(IV) complexes 254 and 255 obtained in this study, the authors used known fluorophores.
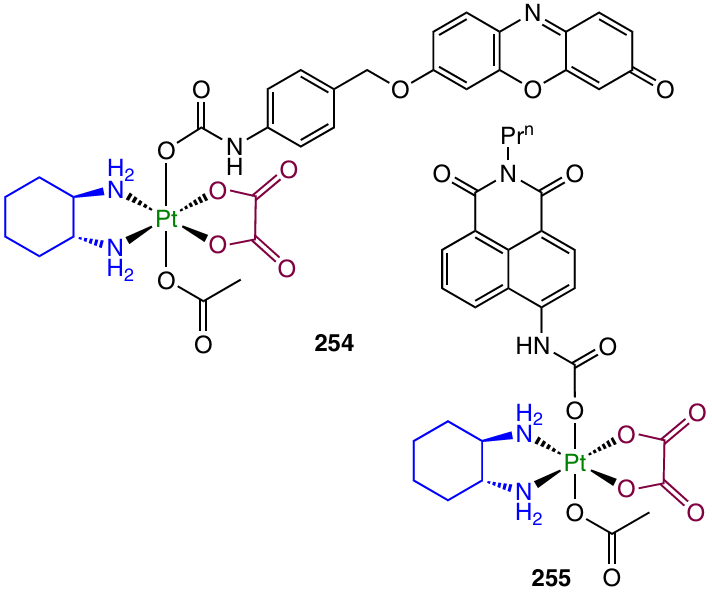
The reduction of prodrugs 254 and 255 accompanied by the release of axial ligands was monitored by dose-dependent increase in the fluorescence in the presence of sodium ascorbate. Oxygen-dependent reduction of the prodrugs and an increase in their cytotoxicity under hypoxic conditions were found. Thus, agents 254 and 255 are hypoxia markers possessing increased cytotoxicity in tumour tissues.
3.5.5. Combination of biotin with dichloroacetate
Jin et al.64 used a biotin moiety and its combination with dichloroacetate as axial ligands for cisplatin and thus obtained prodrugs 186 and 256.
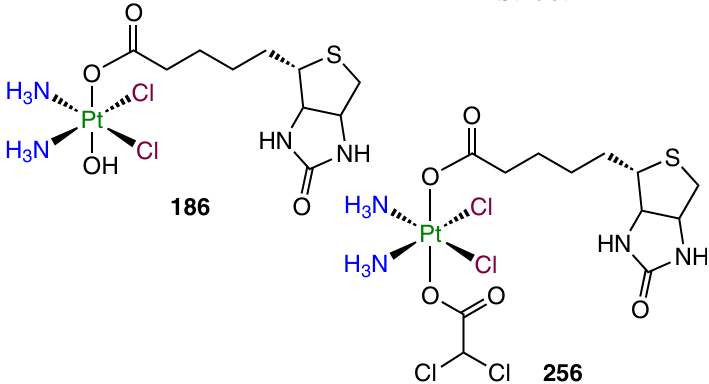
The cytotoxicity of prodrugs 186 and 256 was assessed against HeLa and HepG2 cell lines expressing biotin receptors and against HCT-116 cells in which there is no expression of biotin receptors. Compound 256 was selective to biotin-(+) cell lines (IC50 < 2 mM) but proved to be less toxic against the cells without expression of biotin receptors (IC50 > 18 mM). This complex also inhibited PDK, altered MMP of HeLa cells and induced mitochondria-mediated apoptosis, as evidenced by increased expression of cytochrome C, a marker of apoptosis in mitochondria.
3.6. Platinum(IV) prodrugs with lipid regulating agents
The change in the lipid metabolism is a distinctive feature of tumour diseases.199 Drug combinations of hypolipidemic statins and cytotoxins, such as cisplatin, have antiproliferative and proapoptotic effects.200 In 2021, Qiao et al.201 developed Pt(IV) prodrugs 257 and 258 containing bezafibrate, an FDA-approved hypolipidemic drug, in one or both axial positions.
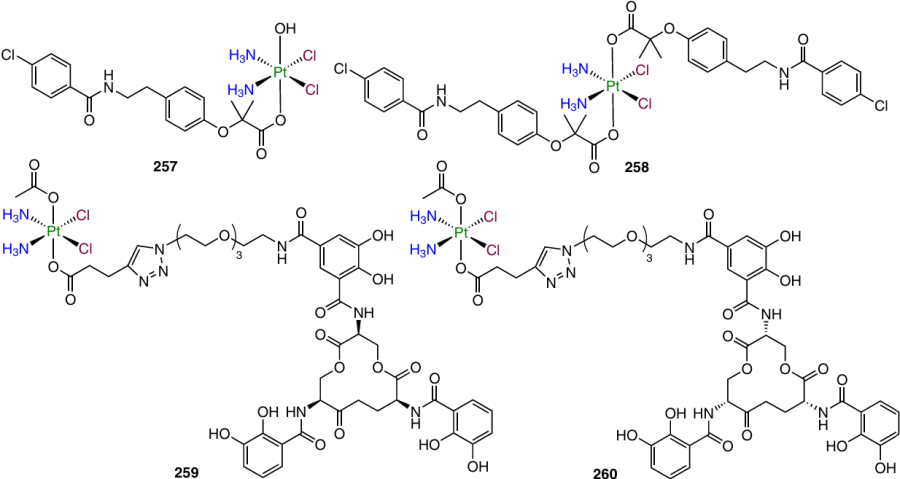
Cytotoxicity assays demonstrated that both complexes had IC50 in the nanomolar concentration range, but monosubstituted prodrug 257 was more cytotoxic: the IC50 values for compound 258 against A549 and HeLa cells were 0.15 and 0.35 μM, respectively, while those for prodrug 257 were 0.04 and 0.06 μM, respectively. It is of interest that a mixture of cisplatin and bezafibrate in a molar ratio of 1 : 1 showed a 9-fold increase in the activity against A549 cells in the absence of cytotoxicity of bezafibrate. Thus, bezafibrate promoted increase in the cytotoxicity of cisplatin even in a mixture. Complex 257 also had a 13.6 times greater cellular uptake than cisplatin, whereas in the case of compound 258, only 1.3-fold increase compared to cisplatin was observed. Study of the mechanism of cytotoxic action revealed the ability of prodrugs 257 and 258 to damage DNA, increase the intracellular ROS levels, change MMP, arrest the cell cycle in the S-phase and trigger apoptosis in А549 cells. In addition, these agents could activate 5'AMP-activated protein kinase (AMPK; 5'AMP is adenosine monophosphate), a cellular metabolic sensor.
3.7. Platinum(IV) prodrugs with antibacterial action
In 2022, Guo and Nolan 82 proposed an unusual application of Pt(IV) prodrugs 259 and 260 based on cisplatin, that is, the application as antibacterial agents. These complexes were obtained by conjugation of cisplatin with enterobactin (Ent).
Conjugate 259 showed antibacterial activity against E. coli K12 and uropathogenic isolate E. coli CFT073. Prodrug 259 also acted similarly to cisplatin, causing a filamentous morphology in E. сoli. It was shown that Ent mediated the delivery of compound 259 into the bacteria. The uptake of prodrugs 259 and 260 was ³10 times that of cisplatin (in terms of Pt). Furthermore, complex 260 had an enhanced antibacterial activity compared to L-isomer 259, probably, because the former cannot be hydrolyzed by esterases and, hence, cannot release iron. Meanwhile, human embryonic kidney cells (HEK293T) had a low uptake of this prodrug, indicating its low toxicity.
3.8. Controlled-release platinum(IV) prodrugs
3.8.1. Ligands for controlled photoactivation
One approach to the design of Pt(IV)-based prodrugs involves the use of photoactive compounds as axial ligands. Prodrugs of this type do not exhibit cytotoxic effects in the absence of radiation and are able to release a cytotoxic Pt(II) complex in a controlled manner. The use of a photodynamic therapy (PDT) agent as a photoactive ligand may produce dual-action medications, which form ROS on exposure to radiation.202, 203
Data on the photoactive Pt(IV) prodrugs obtained to date are summarized in Table 1. The Table presents the types of photoactive ligands located in the axial positions of platinum(IV), data on the increase in the cytotoxicity of complexes under irradiation and specifies the irradiation conditions used in experiments (for the chemical structures of compounds, see the relevant Sections).
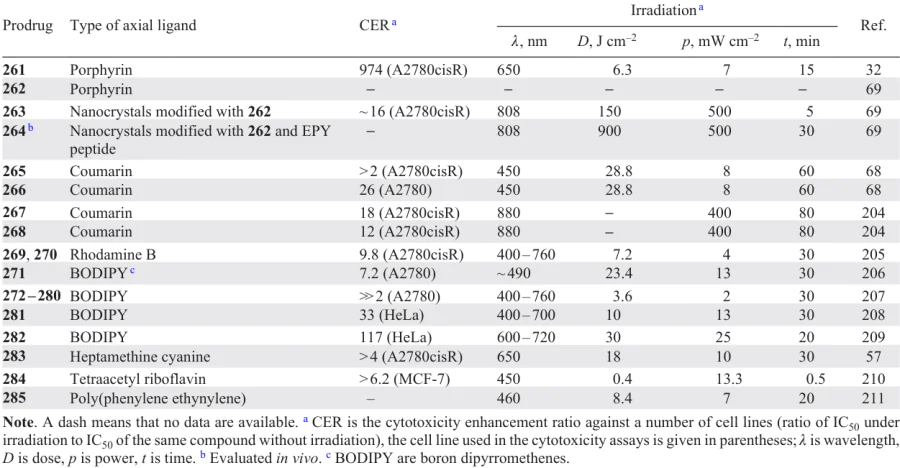
3.8.1.1. Pyropheophorbide A
One of the first Pt(IV) prodrugs that contained a photoactive axial ligand, pyropheophorbide A (PPA), (compound 261) was reported by Wang et al.32 Pyropheophorbide A absorbs light with a maximum wavelength of ~650 nm and efficiently generates singlet oxygen.212
The established mechanism of photoactivation of prodrug 261 implies the formation of PPA radical anion in the axial position of the complex upon the reaction of triplet PPA with sodium ascorbate, which is followed by a fast single-electron transfer from the ligand radical anion to the Pt(IV) centre to give oxaliplatin and free axial ligands.
Complex 261 does not possess activity in the dark; however, under exposure to red light, it is cytotoxic in the sub-micromolar concentration range. The most pronounced (1786-fold) increase in the toxicity induced by radiation was observed for MCF-7 cells. In experiments in vivo in BALB/c mice bearing 4T1 tumour, the volume of the tumour decreased by 67% on the 12th day of therapy combined with irradiation (λ = 660 nm, p = 10 mW, t = 10 min) for the mice that were treated with complex 261 compared to that for mice treated with oxaliplatin.
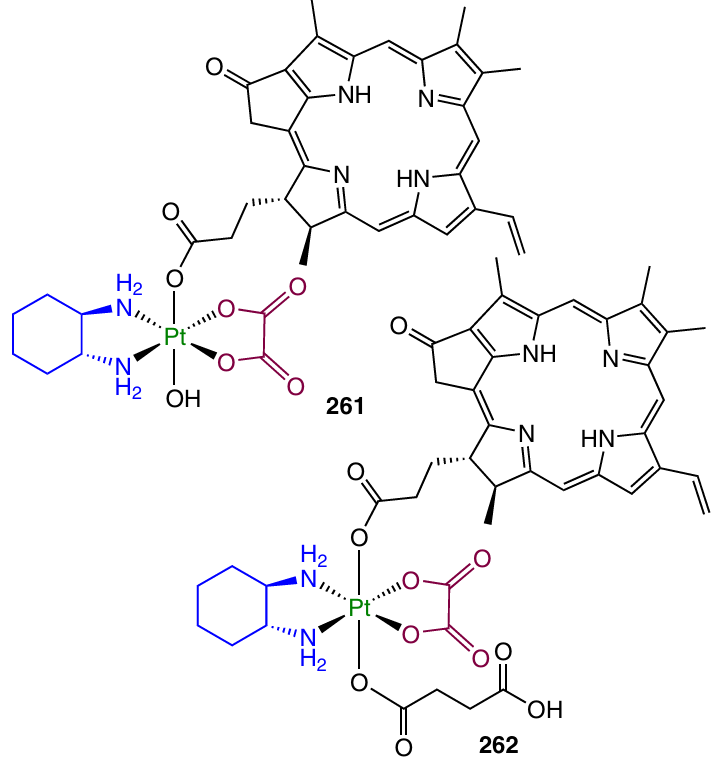
In a following study,69 the dicarboxylate analogue of 261–complex 262 with a succinic acid moiety in the axial position–was linked by covalent bond to NaYbF4 : Er@NaYF4 : Yb/Nd@NaYF4 : Ca nanoparticles to give nanocrystals 263 (Fig. 7a). In this Figure, the large grey sphere shows the nanocrystal core, complex 262 is designated by green spheres, ERY peptide is shown as orange spheres and blue zigzag lines are poly(ethylene glycol) (PEG) moieties.
![[{"id":"Lp2Wg-_N0C","type":"paragraph","data":{"text":"Schematic image of Pt(IV) prodrugs <b>263</b> (<i>a</i>) and <b>264</b> (<i>b</i>)."}}]](/storage/images/resized/HSTt63qN49KICDJr9GeEXruRFhHybT1ZsdY8i4NI_xl.webp)
On exposure to near-infrared (NIR) light, nanocrystals 263 had IC50 values in the low micromolar range, while in the absence of light, the cytotoxicity was insignificant. For increasing the selectivity to tumours, this prodrug was modified with ERY peptide specific to mouse red blood cells, which was linked to the nanocrystal by a PEG-based linker. Nanocrystals 264 obtained in this way had an exceptionally long blood circulation time (t1/2 = 907 h). In in vivo experiments, after one dose of 264 (2.5 mM of Pt per kg) and seven irradiation sessions (λ = 808 nm, p = 500 mW cm–2, t = 30 min), the average tumour volume was 109 times smaller than that in the control group, and complete remission of the disease was observed for two out of five mice.
3.8.1.2. Coumarin
7-Diethylaminocoumarin with an absorption maximum in the blue spectral range (450 nm) was chosen as an axial ligand for Pt(IV) prodrug 265.68
It was established that photoreduction of complex 265 is accompanied by oxidation of water and gives oxygen. In order to increase the ability of the complex to be accumulated in tumour cells, the second axial position of the platinum(IV) complex was modified by the R8K peptide vector, which gave prodrug 266. This product was efficiently accumulated in the nuclei of A549cisR cells; the platinum content exceeded 68%.
The phototoxicity of compound 266 was evaluated using a few cell lines, including cisplatin-resistant А2780cisR cells. The dark toxicity was similar to that observed for oxaliplatin, but under blue light irradiation, the toxicity increased 7 – 62-fold.
Deng et al.204 described carboplatin and oxaliplatin derivatives 267 and 268, which contain a coumarin-based axial ligand capable of being excited upon two-photon absorption. A xanthenone derivative was introduced into the second axial position to increase the cellular uptake of the compound. Both Pt(IV) prodrugs 267 and 268 were mainly accumulated in the endoplasmic reticulum; therefore, these compounds induced cell death by oxidation of biomolecules and formation of ROS rather than by DNA damage.
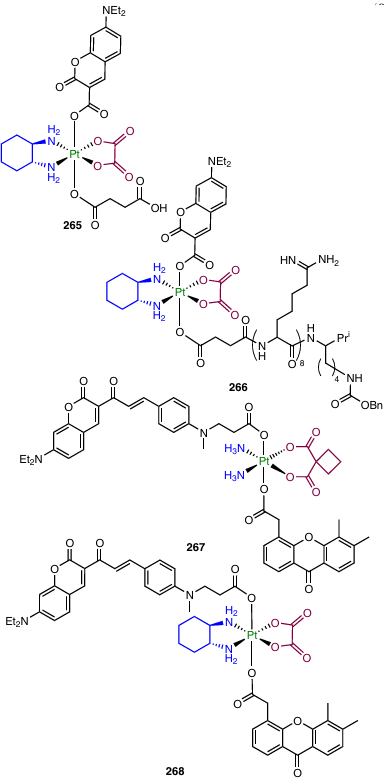
In in vitro experiments, these prodrugs were not toxic in the dark. Under laser irradiation at 880 nm (p = 0.4 W cm–2, t = 80 min), the toxicity against a number of cisplatin-sensitive and cisplatin-resistant cell lines was low: the IC50 values were in the micromolar range (2 – 5 mM). Under hypoxic conditions, the antiproliferative activity of these compounds did not decrease, indicating an oxygen-independent mechanism of phototoxicity.
Complex 267 showed a high activity in vivo against 4T1 tumour in BALB/c mice; on the 16th day of therapy, the volume of the tumour decreased by 89% compared to that in the control group. In addition, this complex suppressed metastasis of 4T1 tumour and also stimulated the immune response in the tumour microenvironment.
3.8.1.3. Rhodamine B
Rhodamine B is a widely used fluorescent dye with the absorption peak at ~570 nm. Deng et al.205 used rhodamine B as an axial ligand for Pt(IV) prodrugs 269 and 270 based on carboplatin and oxaliplatin, respectively.
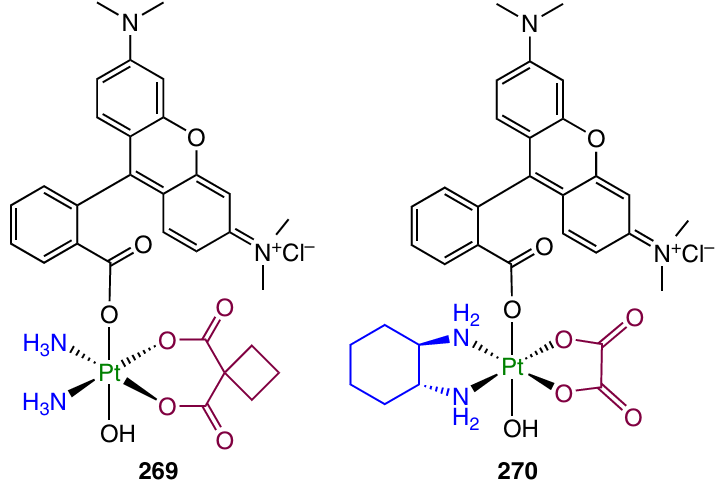
The mechanism of photoreduction under irradiation of a solution of complex 270 was studied in the presence of sodium ascorbate by detecting the ascorbate radicals by EPR; this confirmed the involvement of this reducing agent in the release of platinum(II) complex from the prodrugs. The IC50 values for a number of cell lines, including cisplatin-resistant А2780cisR and A549cisR cells, proved to be 3 – 7 times lower upon irradiation with white light (λ = 400 – 760 nm, p = 4 mW cm–2, t = 30 min) than in the absence of irradiation. Moreover, prodrugs 269 and 270 were 10 times more toxic upon irradiation than the corresponding Pt(II) compounds.
3.8.1.4. Boron dipyrromethenes
Boron dipyrromethenes (BODIPY) represent a class of organoboron fluorophores characterized by high fluorescence quantum yields, chemical stability and photostability.213 Using BODIPY, platinum(IV) complexes 271 – 282 were synthesized.
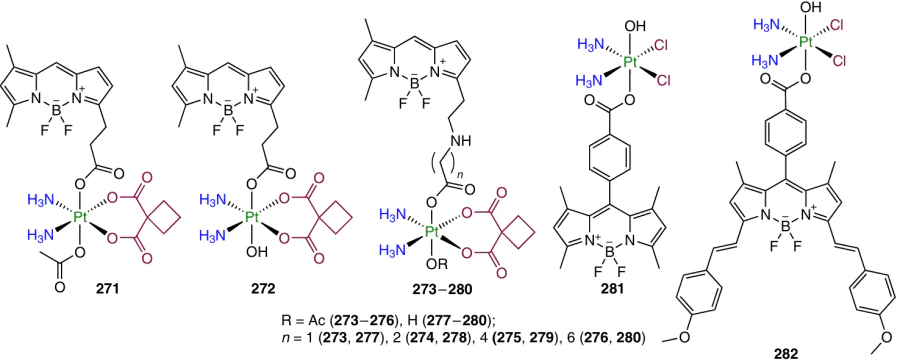
Carboplatin derivative 271 containing BODIPY in an axial position was described by Yao et al.206 Complex 267 was subjected to photoreduction by irradiation with green light either in the presence or in the absence of a reducing agent. The antiproliferative activity of prodrugs 271 were studied against several cell lines, including MDA-MB-231 and A2780. Under green light irradiation, compound 271 proved to be 2 – 11 times more cytotoxic than in the dark and 6.5 – 43 times more cytotoxic than carboplatin.
In a subsequent study, Yao et al.207 evaluated the effect of the length of the linker between the Pt(IV) centre and the photoactive ligand on the prodrug photoreduction rate. It was shown that dicarboxylates 271 and 273 – 276 are reduced on exposure to light up to 24 times faster than monocarboxylates 272 and 277 – 280. Three methylene units, as in prodrugs 273 and 277, proved to be the optimal length of the linker for the photoreduction.
Bera et al.208 used an alternative BODIPY derivative synthesized from dimethylpyrrole and p-formylbenzoic acid as an axial ligand to obtain prodrug 281. Complex 281 showed low toxicity against MCF-7, HeLa, A549 cells and lung epithelial cells (HPL1D) in the absence of radiation. However, upon irradiation with white light, the toxicity of this compound increased 10 – 25-fold and exceeded the toxicity of cisplatin by more than 10 times.
More recently, the same authors 209 obtained prodrug 282 containing a similar BODIPY derivative, which absorbed in the red spectral region. Complex 282 did not show cytotoxicity in the dark; however, red light irradiation resulted in sub-micromolar IC50 values. Prodrug 282 was also capable of inducing the formation of ROS on exposure to light and decreasing MMP upon irradiation of HeLa cells (λ = 600 – 720 nm, D = 30 J cm–2).
3.8.1.5. Cyanine dye
Cyanine dyes are widely used as PDT agents that absorb in the NIR range. Li et al.57 obtained prodrug 283 with two cyanine dye molecules in the axial positions able to absorb NIR light.
The toxicity of the complex in the dark was comparable with that of cisplatin, while red light irradiation induced a 3 – 5-fold increase in the toxicity. Prodrug 283 was equally toxic against cisplatin-sensitive and cisplatin-resistant cell lines.
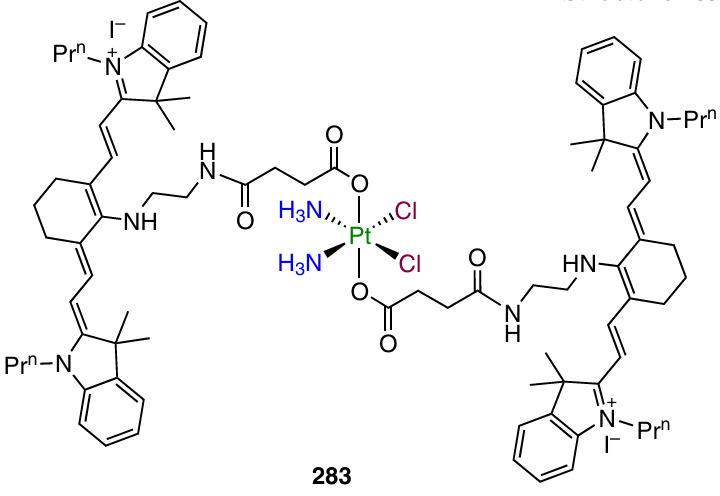
3.8.1.6. Riboflavin
Riboflavin is a group B vitamin, which plays a key role in the energy metabolism and cellular respiration. Krasnovskaya et al.210 obtained cisplatin-based prodrug 284 (riboplatin) with tetraacetylriboflavin (TARF) in the axial position.
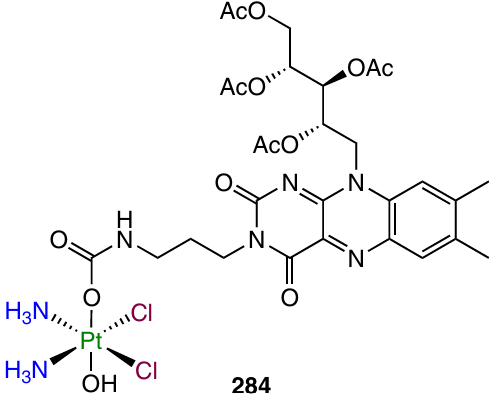
Study of physicochemical properties, the photochemical and biological activity and the photoreduction mechanism of riboplatin showed that it is a true prodrug with low toxicity in the dark releasing cisplatin upon photoexcitation with blue light.
In the absence of radiation, complex 284 turned out to be at least 3 times less toxic than cisplatin, while the efficiency attained upon blue light irradiation was more than four times higher than that of cisplatin. Moreover, riboplatin had a unique photosensitivity and was active even when the irradiation dose was 0.04 J cm–2. Compound 284 was accumulated in the cells more efficiently than cisplatin, while under the action of blue light, the cellular uptake of the agent increased 14-fold compared to that of cisplatin. A detailed study of the riboplatin photoactivity demonstrated that at an irradiation dose below 0.2 J cm–2, the cytotoxic effect is due to the release of cisplatin, whereas at irradiation doses above J cm–2, a synergistic effect of chemotherapy and photodynamic therapy was observed for this agent.
3.8.1.7. Poly(phenylene ethynylene)
As indicated above, Pt(IV) prodrugs are usually octahedral coordination compounds with a low-molecular-weight (in some cases, photoactive) ligand in the axial position. An alternative approach was demonstrated by Sun et al.,211 who used poly(phenylene ethynylene) (PPE) as the photoactive ligand. This macromolecule contains carboxyl groups in the side chain and can react with dihydroxy-oxaliplatin to give polymer 285, which has a Pt(IV) complex at the periphery.
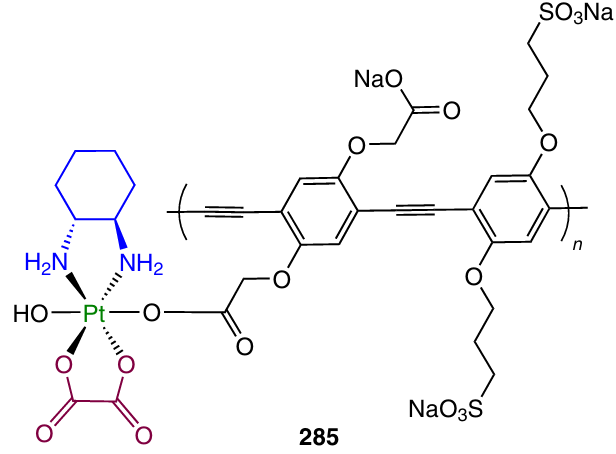
Owing to the presence of the sulfonate anion in the PPE side chain, polymeric prodrug 285 demonstrated a good solubility in water. The irradiation of complex 285 with light (λ = 400 nm, p = 5 mW cm–2, t = 120 min) resulted in the release of oxaliplatin, which was manifested in the absorption spectrum, irrespective of the presence of sodium ascorbate. The release of oxaliplatin from the prodrug under the action of blue light (λ = 400 nm, p = 5 mW cm–2, t = 30 min) was also confirmed by HPLC/MS. Prodrug 285 was reduced not only on exposure to blue light (λ = 400 nm, p = 5 mW cm–2, t = 120 min), but also due to two-photon absorption induced using 100 fs pulsed light (λ = 725 nm, p = 800 mW cm–2, t = 1 h, 50% degradation). A mechanism of photoreduction was proposed, the key step of which is the electron transfer from the polymer ligand to the Pt(IV) centre to give the PPE radical cation.
The cytotoxicity of agent 285 after irradiation with light at λ = 460 nm (p = 7 mW, t = 20 min) was comparable with the cytotoxicity of oxaliplatin; in the dark, this prodrug showed an insignificant cytotoxicity.
3.8.2. Ultrasound-induced release
Ultrasound waves represent a non-invasive and highly penetrating treatment that is widely used in neurosurgery, drug delivery and anticancer therapy.214 – 216 Liu et al.217 employed focused ultrasound (FUS) for the controlled activation of Pt(IV) prodrug 286.
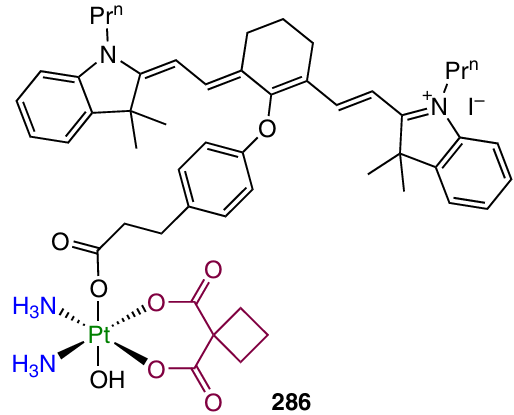
The heptamethine dye IR780 sensitive to ultrasonic treatment was used as the axial ligand.218 Platinum(IV) prodrug 286 was selectively reduced on treatment with ultrasound in the presence of sodium ascorbate, which indicates that reduction occurs via the extramolecular electron transfer. The ability to form singlet oxygen under the action of FUS (type II sonosensitization) was shown for complex 286 and the free ligand.
Evaluation of the antiproliferative activity showed that prodrug 286 was inactive without sonication, while FUS increased its cytotoxicity more than 10-fold to reach IC50 of 2 – 4 mM. Complex 286 was equally active against cisplatin-sensitive and cisplatin-resistant cell lines (А2780 and А2780cisR; or А549 and A549cisR). In 4T1 cells incubated with this compound and sonicated, markers of immunogenic cell death, including calreticulin exposure, ATP secretion and extra-nuclear HMGB1 expression, were detected. This attests to the potential ability of prodrug 286 to stimulate the anticancer immune response.
The anticancer efficacy in vivo was investigated against the 4T1 tumour in BALB/c mice. After the therapy with prodrug 286 and sonication, the tumour volume on the 18th day was 24.3% of the initial volume, and complete remission was observed for two out of five mice. In addition, markers of the antitumour immune response, CD3+ and CD4+, were detected in the tumours of mice after treatment with 286 and sonication.
Thus, the use of ultrasound is a new, fairly promising approach to the controlled activation of Pt(IV) prodrugs, which combines high penetration capacity and safety for normal organs and tissues. Prodrug 286, capable of controlled sonoactivation, showed a high antitumour efficacy in vivo, owing to the antiproliferative action caused by the release of the chemotherapeutic agent carboplatin, formation of singlet oxygen and stimulation of the antitumour immune response.
3.9. Efficacy of Pt(IV) prodrugs in vivo
A highly important characteristic of efficacy of Pt(IV) prodrugs is the tumour growth inhibition in vivo. Table 2 summarizes the results of studies of the therapeutic efficacy of Pt(IV) prodrugs described in this review.
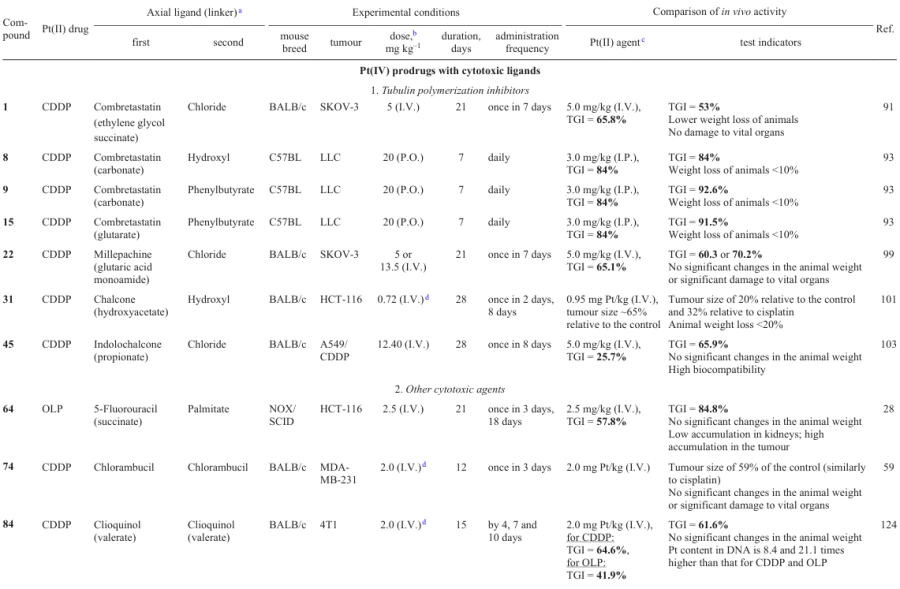
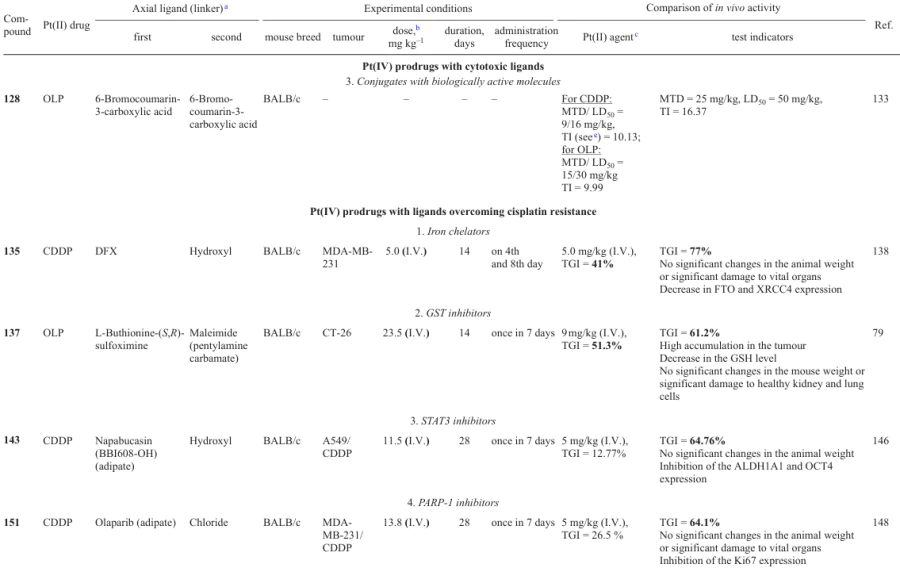
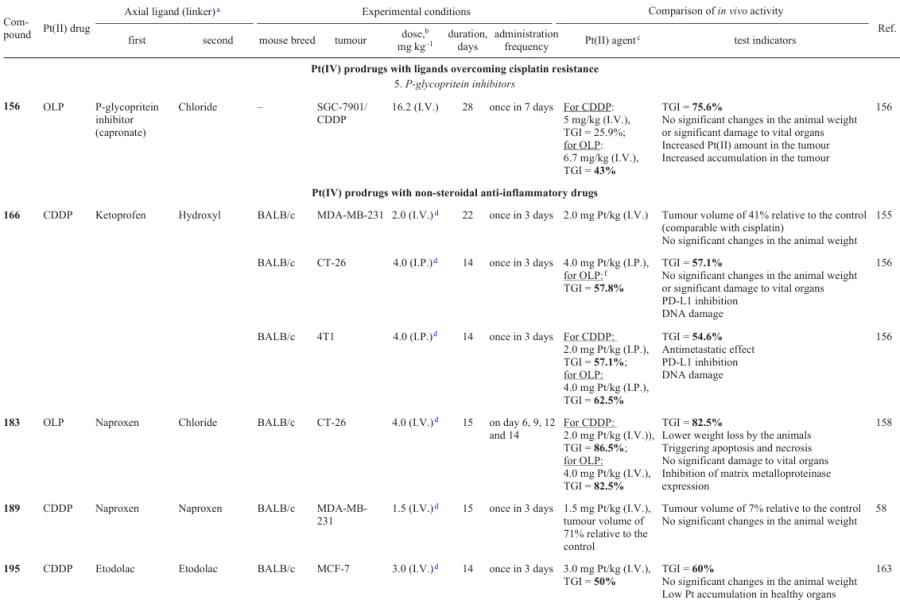
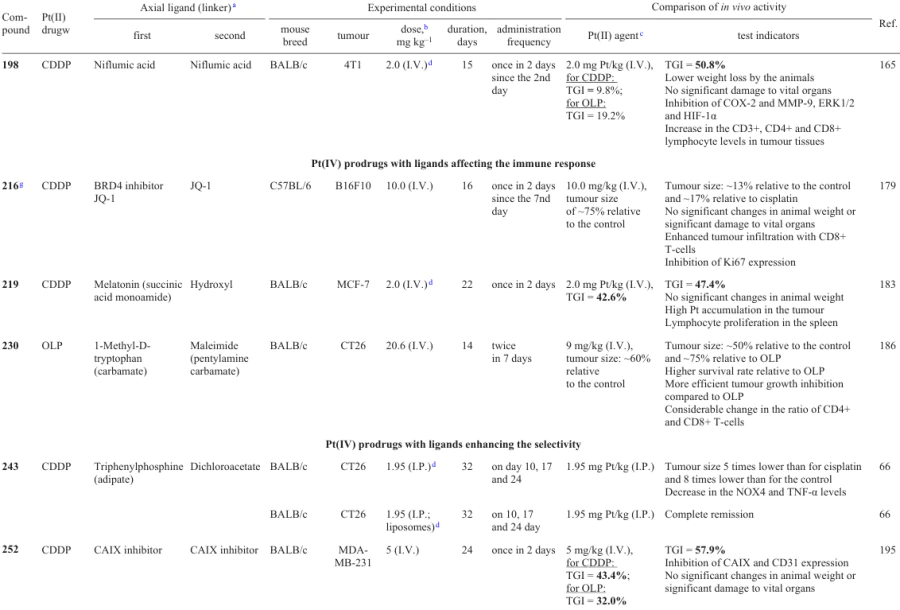
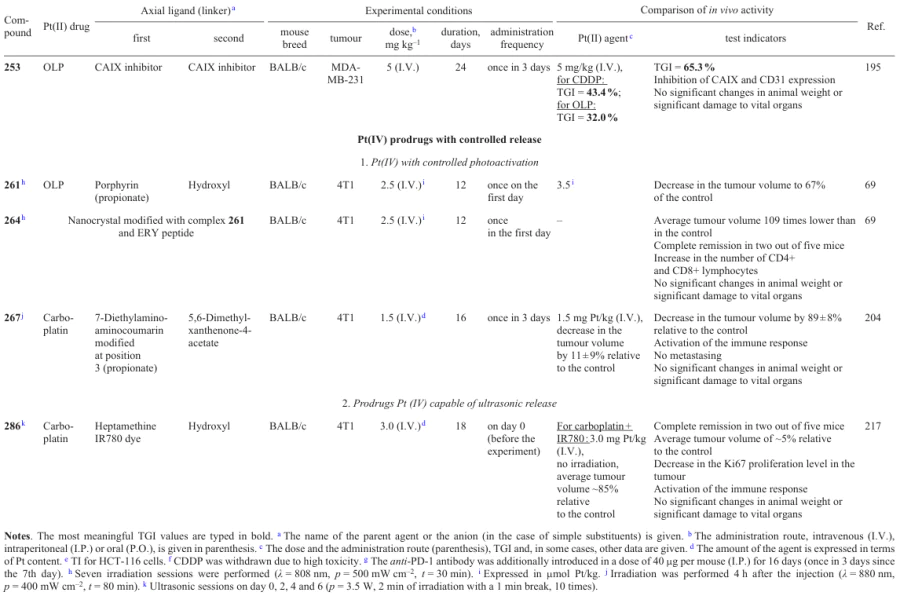
Considering these data, worthy of note is the correlation between the rate of reduction of the coordination compounds in the presence of reducing agents and their ability to inhibit the tumour growth in vivo. It can be seen that Pt(IV) monocarboxylate complexes are less stable than the dicarboxylates. The half-lives of Pt(IV) monocarboxylate prodrugs in the presence of reducing agents vary from several minutes 93 to 4 – 6 h.101, 158 Abnormally high stability (t1/2 > 24 h) is inherent in monocarboxylate oxaliplatin and carboplatin derivatives with photoactive ligands (261, 269, 270).32, 206 In in vivo assays, the unstable Pt(IV) monocarboxylates differ little in efficacy from cisplatin and show enhanced activity only against cisplatin-resistant tumours. As an exception, note the high efficacy of prodrug 31 against HCT-116 cell line.101
Meanwhile, Pt(IV) dicarboxylate prodrugs usually have half-lives of several hours 93 to > 24 h,58, 79, 163 but there are also examples of unstable dicarboxylates, which are reduced within one hour.186, 195 For Pt(IV) prodrugs 8, 15, 84 and 230, which have a relatively short (up to 12 h) half-lives in the presence of reducing agents, the efficacy in in vivo experiments was comparable to that for cisplatin. However, the stability of the agents did not directly affect their efficacy in animals. Indeed, highly stable prodrugs 189, 216 and 253 are more active in vivo than Pt(II) compounds; the ability of prodrugs 137 and 195 to inhibit the tumour growth is at the cisplatin level, despite their stability in the presence of reducing agents. This indicates that stability of Pt(IV) prodrugs in the presence of reducing agents is a necessary, but not sufficient condition for high anticancer efficacy.
The highest activity in in vivo experiments among all Pt(IV) prodrugs considered in this review was inherent in complexes 243 and 262, for which delivery vehicles ensuring longer blood circulation time have been developed.66, 69 After 32 days of the therapy of BALB/c mice bearing CT26 tumour with the liposomal form of prodrug 243 with alkyltriphenylphosphonium and dichloroacetate in the axial positions, complete remission of the disease was attained. A very high anticancer efficacy was also demonstrated for nanocrystals 264 containing a photoactive Pt(IV) prodrug 262 with pyropheophorbide A: after administration of only one dose of agent 264 and seven irradiation sessions at 808 nm (D = 900 J cm–2), remission was observed in two out of five mice on the 14th day of therapy. Thus, the efficacy of the anticancer action of Pt(IV) prodrugs is enhanced if their stability against too fast reduction and their longer blood circulation times have been attained.
3.10. Analysis of the efficacy of Pt(IV) prodrugs
Platinum(IV) complexes containing combretastatin in an axial position possess cytotoxic activity in the nanomolar concentration range.91 – 93 Prodrugs 8 and 9, in which the axial ligand is linked to the Pt(IV) centre by a cleavable carbonate linker proved to be the most active,93 while prodrugs 1 – 7 with linkers based on dicarboxylic acids had IC50 > 150 nM.91, 92 Despite the fact that prodrugs 1 – 15 were up to 6000 times more active than cisplatin in in vitro experiments, studies of the in vivo anticancer efficacy showed that the ability of 1, 8, 9 and 15 to inhibit the tumour growth is comparable to that of cisplatin.
Monocarboxylates 16 – 30 and 33 – 47 with chalcone derivatives, CА-4 analogues, in the axial positions have an antiproliferative effect in low micromolar and nanomolar ranges down to IC50 = 0.13 mM, observed for prodrug 36 against HCT-116 cells.98, 99, 102, 103 In this case, the optimal length of the linker between the ligand and the Pt(IV) centre is a short chain containing two free methylene units based on succinic anhydride. Monocarboxylate complex 31 with a p53/MDM2-inhibiting chalcone demonstrated an exceptionally high antiproliferative activity in the nanomolar concentration range: IC50 (against A2780 cells) = 10 nM.101 In in vivo experiments, the efficacy of prodrug 31 also considerably exceeded that of cisplatin. Complex 45 with indolochalcone in the axial position was active against cisplatin-resistant cells in vitro and also inhibited the growth of the А549/CDDP cisplatin-resistant tumour much better than cisplatin.
Among Pt(IV) prodrugs with known anticancer agents (paclitaxel, fluorouracil, chlorambucil and doxorubicin), clear-cut synergistic effect for the combination of two therapeutic agents in vitro and in vivo was attained only for series of compounds 60 – 67 based on oxaliplatin and fluorouracil and only after thorough tuning of the lipophilicity of the coordination compounds.28 The most active conjugate 64 exhibited a high degree of tumour growth inhibition in vivo, which much exceeded that for oxaliplatin.
Platinum(IV) prodrugs 132 – 156 were synthesized to overcome the cisplatin and oxaliplatin resistance of tumour cells. Among agents of this type, the ability to fully overcome the resistance (RF < 1) in vitro was exhibited by prodrugs 132 – 134 containing fatty acids and by complexes 138 – 143 with napabucasin in the axial position.135, 146 Nevertheless, Pt(IV) prodrugs 135, 143, 151 and 156 with the DFX iron chelator and STAT3, PARP-1 and P-glycoprotein inhibitors, respectively, meant to overcome the drug resistance showed high activity in vivo against cisplatin-resistant tumours (А549/CDDP, MDA-MB-231/CDDP and SGC-7901/CDDP).138, 146, 148, 150 This is a clear-cut evidence for efficiency of the strategy of preparing Pt(IV) prodrugs with axial ligands that inhibit the mechanism of cisplatin resistance of tumour cells.
The introduction of non-steroidal anti-inflammatory drugs into the axial positions of Pt(IV) complexes (compounds 157 – 214) markedly increases the toxicity in vitro (to reach nanomolar IC50 values). Nevertheless, in these experiments, the cytotoxicitу is affected most appreciably by the lipophilicity of Pt(IV) prodrugs.53, 62 Despite the high activity of prodrugs of this group in vitro, the ability of complexes 1166, 183, 195 and 198 to inhibit the tumour growth in vivo was comparable to that of cisplatin.155, 158, 163, 165 However, these agents showed lower toxicity against normal organs and induced a lower loss of animal weight than cisplatin. A number of Pt(IV) prodrugs 166, 189 and 198 with NSAIDs (ketoprofen, naproxen and niflumic acid, respectively) proved to stimulate the immune response by inhibiting PD-L1 and increasing the level of CD3+, CD4+ and CD8+ lymphocytes.58, 155, 165 Compound 189 also efficiently inhibited the growth of the MDA-MB-231 tumour.58
For a group of Pt(IV) prodrugs 215 – 233 with immunomodulating ligands in the axial position, in vivo experiments showed the ability to increase the amount of the immunostimulating CВ8+ Т-cells and, hence, to trigger the immune response to the malignant growth.179, 183, 186 In addition, the therapy with agents 216, 219 and 230 containing BRD4 inhibitor, melatonin and 1-methyl-D-tryphophan as axial ligands, respectively, was accompanied by a smaller decrease in the weight of animals and less pronounced damage to healthy organs than the therapy with Pt(II)-based drugs. Whereas the ability of prodrugs 219 and 230 to inhibit the tumour growth was comparable to that of cisplatin or oxaliplatin, compound 216 showed a high anticancer activity in vivo markedly exceeding the effect of cisplatin or a combination of cisplatin and the axial ligand JQ-1.179
Among the strategies towards increasing the selectivity of Pt(IV) prodrugs to tumour cells, high characteristics were found for the approach involving the introduction of glucose into the axial position: the selectivity index of complex 238 to HepG2 cells over L02 normal cells was 24.25 Prodrugs 252 and 253 with hypoxia-sensitive axial ligands, CAIX inhibitors, were also characterized by a high selectivity index for MDA-MB-231 hypoxic cells over the normoxic MCF-10A cells (80 and 34.5, respectively).195 In in vivo experiments, the activity of prodrug 253 significantly exceeded the activity of oxaliplatin.
Combination of organic fluorophores and Pt(IV) coordination compounds afforded a series of Pt(IV) prodrugs 261 – 285, which are stable and non-toxic in the absence of radiation, but in the presence of visible light, they can release cytotoxic Pt(II) coordination compounds and induce the formation of reactive oxygen species.32, 57, 68, 69, 205 – 211
The use of oxaliplatin or carboplatin as the parent Pt(II) complex led to the increase in the dark stability of the agents. The conjugation of complex 261 with the nanocrystals afforded prodrug 264, characterized by long blood circulation time; the therapy with this nanocrystal resulted in remission of the disease in two out of five mice.69
The most important results presented in this review is the successful use of two-photon absorption and sonication for the controlled release of cytotoxic agents from Pt(IV) prodrug.204, 217 The efficacy of the therapy of malignant tumours in vivo demonstrated in relation to Pt(IV) complexes 267 and 286 undoubtedly indicates the high promise of using both Pt(IV) prodrugs capable of controlled release and non-classic approaches to external stimulation.
3.11. Miscellaneous metal-containing agents in clinical practice
Along with platinum complexes, a number of coordination compounds based on other metals have been actively investigated in recent years as antitumour agents. Some compounds are already in clinical trials.
Ruthenium complexes with the codes NAMI-A and KP1019 were chosen for clinical trials for the therapy of colon cancer.219
Complex NAMI-A did not show a sufficient efficacy to continue phase II clinical trials, and compound KP1019 had a moderate efficacy in phase I trials.220, 221 Presumably, NAMI-A functions via fast ligand exchange by binding to proteins on the surface of tumour cells and preventing metastasis, while KP1019 (or its sodium salt KP1339) penetrates into the cell and binds to proteins, causing ROS formation and DNA damage. Currently, the agent BOLD100, which is a full analogue of complex KP1339, is in phase I clinical trials as a drug for the treatment of solid tumours (code NCT04421820).
Ruthenium complex TLD1433 has passed phase Ib clinical trials as a photosensitizer, a photodynamic therapy agent against the non-invasive bladder cancer (code NCT03945162).222
Two gold compounds, aurothimalate and auranofin, completed the clinical trials as protein kinase C (PKC) inhibitors, a kinase involved in cell proliferation, migration and apoptosis.
Phase I clinical trials of aurothimalate were carried out for the therapy of PKC-expressing types of cancer, in particular non-small cell lung cancer, ovarian cancer and pancreatic cancer (NCT00575393 code).223 Auranofin was subjected to phase I/II clinical trials for the treatment of chronic lymphoid leukemia, small lymphocytic and prolymphocytic lymphoma; now it is in phase II clinical trials for the therapy of ovarian cancer (9NCT03456700 code).
Titanium complexes, titanocene dichloride (CpTiCl2) and budotitanum, were the first metal coordination compounds to be subjected to clinical trials after platinum-based compounds.224, 225 In phase I trials, the maximum tolerated doses of CpTiCl2 and budotitanum were successfully determined; however, in phase II clinical trials, no noticeable beneficial effect was observed for the former compound, and difficulties in composing the formulation prevented further study of budotitanum.226 The mechanism of antiproliferative action of titanium complexes is not entirely clear; presumably, it includes the reduction of TiIV to TiII and binding to DNA.227, 228
Palladium complex with bacteriopheophorbide (TOOKADTM) was approved in Mexico for the treatment of prostate cancer and is in phase II and III clinical trials in the US (NCT04620239 code).229 This is an agent for the vascular-targeted photodynamic therapy activated by light with a wavelength of 753 nm (NIR region).230
Copper coordination compounds are also promising metal-containing agents for the therapy and diagnosis of malignant neoplasms. A combination of disulfiram with copper salts (DSF/Cu) is a Cu(II) complex of disulfiram metabolite, diethyldithiocarbamate (DDC).231
The DSF/Cu combination is in clinical trials as an anticancer agent designed to treat metastatic breast cancer (NCT03323346 code), castration resistant prostate cancer (NCT02963051), myeloma (NCT04521335), glioblastoma (NCT03363659) and sarcoma (NCT05210374). Presumably, the anticancer activity of this combination of disulfiram with copper salts is due to the ability of the coordination compound formed in situ to generate ROS owing to the high redox potential of copper and the ability to displace other ions from the enzyme binding sites and high affinity to DNA.232 In addition, a copper-containing agent for radionuclide theranostics possessing affinity to the 67Cu – CuSarTATE somatostatin receptor is currently in phase I clinical trials for the treatment of neuroblastoma (NCT04023331 code).233
Manganese, like copper, has a high redox potential, which makes manganese-based compounds promising agents for the therapy of cancer.234 Several organic manganese-containing complexes are in various plases of clinical trials.
Thus, Rucosopasem Manganese, meant for the use in stereotactic body radiotherapy (SBRT), is in phase II clinical trials for the therapy of pancreatic cancer and lung cancer (NCT04698915 code).235 This drug is a selective mimetic of superoxide dismutase, which initiates the transformation of superoxide into hydrogen peroxide and increases the efficiency and selectivity of radiation therapy.236 Avasopasem, an analogue of this agent, is also a superoxide dismutase mimetic; when tested in phase I/II clinical trials for the treatment of head and neck cancer, it increased the survival rate of SBRT patients.237
Manganese(III) porphyrins are one more promising class of anticancer agents based on manganese. The MnTnBuOE-2-PyP5+ (BMX-001 code) is also a dismutase mimetic and is in clinical trials as a radioprotector (NCT05254327 and NCT03386500).238 In addition, in vitro and in vivo assays demonstrated that Mn(III) porphyrin complexes can change the activity of the cell signalling proteins and thus influence cell proliferation and apoptosis, which makes them not only promising radioprotectors, but also potential agents for the therapy of cancer.239
Among metal-containing therapeutic agents that are currently in clinical trials, mention should be made of BOLD-100, titanocene dichloride and budotitanum. The mechanism of action of these agents is similar to that of cisplatin and includes ligand exchange, binding to proteins and DNA damage. Complexes based on manganese and copper act via redox processes accompanied by the formation of ROS.
Despite the good prospects of studies of many metal-containing anticancer agents and successful completion of early phase clinical trials, the full introduction of these drugs into clinical practice is hampered by a number of factors such as side effects and too low efficacy. This is often due to the lack of understanding of the mechanism of anticancer action and the metabolism of these compounds. Conversely, Pt(IV) prodrugs represent a unique platform for the development of highly active anticancer drugs. Their main feature is the release of Pt(II) complexes approved for use under biological conditions, the mechanism of which has been thoroughly studied over the past decades.3 – 6, 17 This is a benefit of these prodrugs over coordination compounds of other metals that undergo clinical trials.
4. Conclusion
Platinum(IV) prodrugs represent one of the most promising alternatives to Pt(II)-based anticancer agents. These coordination compounds are less prone to untargeted ligand exchange and premature binding to proteins; the axial ligand is easily modified, which allows fine tuning of the physicochemical properties and biological activity of these agents.
This review gives a systematic account of the approaches to the synthesis of Pt(IV) prodrugs from the conventional platinum(II)-based drugs and considers studies of Pt(IV) complexes with anticancer and antibacterial activity published in 2018 – 2023. There is a wide range of methods available for the synthesis of Pt(IV) prodrugs, which give coordination compounds with various axial ligands and various types of bonding between the Pt(IV) atom and the ligand and enable direct modification of axial ligands. This opens up the way to the design of Pt(IV) prodrugs optimized to overcome the specific drawbacks of the therapy with platinum(II) drugs.
Over the past 5 years, significant progress has been made in the synthesis of highly efficacious Pt(IV) prodrugs (Fig. 8). As expected, higher in vitro activity compared to that of cisplatin is inherent in the prodrugs that contain other anticancer agents such as combretastatin A4, 5-fluorouracil, estramustine and other as axial ligands. Nanomolar IC50 values were also found for prodrugs that had no cytotoxic moieties in the axial position. In particular, a considerable increase in the cytotoxicity was observed for derivatives of NSAIDs (naproxen, flurbiprofen, melatonin) and carbonic anhydrase inhibitors. In addition, high antiproliferative activity can be achieved by optimizing the lipophilicity of the agent.
![[{"id":"2JpseNWmxf","type":"paragraph","data":{"text":"Most effective Pt(IV)-based prodrugs and principles of their action."}}]](/storage/images/resized/xehVj2Ru5cDBSGHwAZinxfnd4dp5LnCQSvYLXstR_xl.webp)
The controlled activation of Pt(IV) prodrugs may solve many key problems of platinum-based therapy such as low selectivity to tumours and cisplatin resistance. Over the past five years, researchers have described prodrugs with high in vitro activity capable of controlled release under irradiation and inactive in the absence of light. Moreover, the pyropheophorbide-containing prodrug, which absorbs in the NIR region, and coumarin-containing prodrug capable of photoactivation owing to two-photon absorption exhibited a considerable antitumour efficacy in vivo.
Despite the fact that most studies published between 2018 and 2023 2 report prodrugs superior to the parent Pt(II) agents in the activity in vitro, the activity of many of these compounds in in vivo experiments was comparable to that of cisplatin. Correlation of the stability of Pt(IV) prodrugs in the presence of sodium ascorbate or glutathione with their anticancer efficacy in vivo suggests that the complexes that can be rapidly reduced under the action of electron donors decompose in the bloodstream to the parent Pt(II) complex and axial ligands and do not hit the tumour as prodrugs. Conversely, the compounds that showed a low reduction rate in the presence of sodium ascorbate were the best in inhibiting the tumour growth.
The highest in vivo activity was inherent in the Pt(IV) prodrugs used in combination with delivery vehicles that ensured longer blood circulation time, that is, liposomes or nanocrystals. A comparable efficiency was demonstrated by the approach involving controlled activation of Pt(IV) prodrugs by ultrasound. In this regard, further progress in the design of effective Pt(IV)-based antitumour agents may be related to the search and creation of optimal delivery vehicles for Pt(IV) prodrugs, which would prevent premature decomposition of the prodrugs and undesirable interactions with normal organs and tissues.
In view of the diversity of axial ligands used in Pt(IV) prodrugs investigated in recent years, it is reasonable to expect that the interest of specialists in the search for new biologically active compounds that can increase the antiproliferative activity of Pt(IV) prodrugs will continue to increase. Meanwhile, new approaches to the design of Pt(IV) prodrugs are being developed, such as controlled activation by ultrasound, and one should expect that new ways for the control of the biological action of platinum coordination compounds will appear. Although no Pt(IV) prodrugs are currently in clinical trials, we may hope that active research in this area would result in the development of highly effective anticancer agents free from the severe drawbacks of traditional platinum(II) drugs.
This review was written with the financial support of the Russian Science Foundation (Project no. 22-15-00182).
5. List of abbreviations and symbols
The following abbreviations and symbols are used in the review:
λ — wavelength,
D — radiation dose,
p — radiation power,
log k' — retention factor,
log P — lipophilicity,
t1/2 — half-life,
AMP — adenosine monophosphate,
Boc — tert-butyloxycarbonyl,
BODIPY — boron dipyrromethenes,
BRD4 — bromodomain-containing protein 4,
BSO — L-buthionine-(S,R)-sulfoximine,
CА4 — combretastatin А4,
CAIX — carbonic anhydrases,
CDDP — cisplatin,
CDI — carbonyldiimidazole,
COX-2 — cyclooxygenase-2,
CSC — cancer stem cells,
Cyt C — cytochtome C,
DCC — 1,3-dicyclohexylcarbodiimide,
DCM — dichloromethane,
DIPEA — diisopropylethylamine,
DIPC — diisopropylcarbodiimide,
DMAP — 4-dimethylaminopyridine,
DSC — N,N'-disuccinimidyl carbonate,
EDC — 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide,
Ent — enterobactin,
ER — endoplastic reticulum,
FDA — US Food and Drug Administration;
Fmoc — 9-fluorenylmethyloxycarbonyl,
FUS — focused ultrasound;
GI50 — concentration providing 50% cell growth inhibition,
GLUT — glucose transporters,
GST — glutathione-S-transferase,
HBTU — 2-(1H-Benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate,
HDAC — histone acetylase,
HPLC — high-performance liquid chromatography,
IC50 — half-maximal inhibition concentration,
ICP-MS — inductively coupled plasma mass spectrometry,
IDO1 — indolamine-2,3-dioxygenase,
LA — lipoic acid,
LD50 — half-lethal dose,
MMP — mitochondrial membrane potential,
MMP-9 — matrix metallopeptidases-9,
MTD — maximum therapeutic dose,
mTOR — mammalian target of rapamycin,
NCS — N-chlorosuccinimide,
NHS — N-hydroxysuccinimide,
NIR — near infrared (range),
NSAID — non-steroidal anti-inflammatory drug,
OLP — oxaliplatin,
PARP — poly(ADP-ribose)polymerases,
PDK — pyruvate dehydrogenase kinase,
PD-L1 — programmed cell death ligand,
PEG — polyethylene glycol,
Pgp — P-glycoprotein,
PPA — pyropheophorbide A,
PPE — poly(phenylene ethynylene),
PTD — photodynamic therapy,
PTX — paclitaxel,
Py — pyridine,
ROS — reactive oxygen species,
RF — resistance factor,
rt — room temperature,
STAT3 — signal transducer and activator of transcription 3,
TARF — tetraacetylriboflavin,
TBTU — 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate,
TBTA — tris(benzyltriazolyl)amine.
TI — therapeutic index,
TGI — tumour growth inhibition,
TS — thymidylate synthase,
Designations of cell lines:
A549 — lung carcinoma,
A549cisR (A549/CDDP) — cisplatin-resistant lung carcinoma,
A431 — cervical carcinoma,
A2780 — ovarian cancer,
A2780cisR (A2780/CDDP) — cisplatin-resistant ovarian cancer,
BCPAP — thyroid cancer,
B16F10 — melanoma,
Caov-3 — primary ovarian cancer,
CH1/PA-1 — ovarian carcinoma,
CT26 — colon cancer,
Du145 — prostate cancer,
EMT-6 — mouse mammary carcinoma,
FaDu — head and neck squamous cell cancer,
HCC1937 — breast carcinoma,
HCT-115, HCT-116 — colorectal carcinoma,
HCT-116/oxR — oxaliplatin-resistant colorectal carcinoma,
HEK293T — human embryonic kidney cells,
HeLa — cervical cancer cells,
HepG2 — human hepatocellular carcinoma,
HL-7702 — normal liver cells,
HPL1D — lung epithelial cells,
HLF — human lung fibroblasts,
HT-29 — colorectal adenocarcinoma,
HUVEC — human umbilical vein endothelial cells,
LNCaP — prostate carcinoma,
L02 — human fetal hepatocytes,
MCF-7 — breast adenocarcinoma,
MCF-10A — breast epithelial cells,
MDA-MB-231 — triple negative breast cancer,
МDА-MB-231/CDDP — cisplatin-resistant triple negative breast cancer,
MDA-MB-435 — melanoma,
MOR — lung adenocarcinoma,
MORcisR — cisplatin-resistant lung adenocarcinoma,
MRC-5 — lung fibroblasts,
MSTO-211H — mesothelioma cells,
PSN-1 — pancreatic cancer,
4T1 — mouse breast cancer,
SGC-7901 — gastric cancer,
SGC-7901/CDDP — cisplatin-resistant gastric cancer,
SKOV-3 — human ovarian adenocarcinoma,
SW480 — colorectal adenocarconoma.