


Keywords
Abstract
Development of the "methanol" economy may be a way to establish the new chemistry under decarbonization conditions. Methanol here is used as a raw material for production of a wide range of chemicals, conventionally obtained from oil. The key process for the "methanol" economy is the reduction of CO2, which, along with renewable energy, is the main carbon-containing resource in the low-carbon industry. This review summarizes recent data on the main approaches to methanol production from CO2: catalytic hydrogenation of CO2 with hydrogen on heterogeneous or homogeneous catalysts; electrochemical reduction of CO2 to methanol; and CO2 conversion using photocatalysis. The main advantages and disadvantages of each method, the mechanisms of CO2 conversion taking into account the features of each type of catalysis, and the main approaches to the efficient catalysts are discussed.
The bibliography includes 542 references.
1. Introduction
Methanol is one of the basic compounds used in the chemical industry. Its consumption exceeds 116 million t.p.a. and demand is growing at a rate of several percent per year. Methanol is used as a fuel, serves for storage and production of ‘green’ H2; it is a source of valuable products such as high-octane gasoline and methyl tert-butyl ether, dimethyl ether, dimethyl carbonate, formaldehyde, methyl and dimethyl amines and others[1-3]. Of particular note is the possibility of producing olefins, which are petrochemical feedstocks, from methanol[4-8]. These processes, implemented in China, already consume 18% of methanol.
Industrial methanol production is based on the hydrogenation of CO obtained from coal gasification or methane conversion. In the 1990s, Nobel Laureate Olah and co-workers[9-11] proposed to replace CO with CO2 and create a ‘methano’ economy. The point is that methanol can be used to produce basic unsaturated and aromatic hydrocarbons for the petrochemical industry[8, 12], thus creating a chemical industry that does not use fossil fuels (oil and gas) (Figure 1). At the time, such an idea seemed rather exotic, given the thermodynamic stability and chemical inertness of the CO2 molecule. However, in the 21st century, with the development of catalytic processes, which demonstrated the possibility of direct hydrogenation of CO2 to methanol, and combined with the urgent need to reduce the amount of CO2 in the atmosphere, the idea of creating a carbon-neutral cycle in a sustainable process of methanol production using ‘clean’ energy is of practical importance[13]. Catalysis plays a key role here, especially taking into account the volume of methanol production[14-20]. Not surprisingly, at least 300 papers are published each year on improving methanol synthesis using a variety of catalysts[21, 22].
![[{"id":"4RDlmV4rDk","type":"paragraph","data":{"text":"Ways to produce and use methanol."}}]](/storage/images/resized/9Bt6mNFG4jludKYSWljPpdGDp5f4omB4uHpjfeVO_xl.webp)
In this review, we will consider different approaches to this reaction, based on the application of different types of catalysis and energy: carrying it out both under conventional thermocatalytic conditions and under electro- and photocatalytic conditions. We will try to show the significant advances in this field, which at the same time affects both the development of catalysis and the improvement of methods and approaches, which can be applied not only to the production of methanol from CO2, but also to other, no less important products such as CO, HCOOH, CH4, HCHO and other products. Emphasis will be placed on work carried out in recent years, as the volume of material is enormous.
2. Thermocatalytic transformations
2.1. Heterogeneous catalysts for the hydrogenation of CO2 to methanol
The wide majority of industrially used catalysts for methanol synthesis from syngas contain Cu promoted by oxides, mainly ZnO, as an active component. Cu-based catalysts were first proposed by Ipatieff and Monroe[23] for hydrogenation of CO to methanol and became the basis for the development of modern industrial catalysts for its production. The complexity that occurs in this apparently very simple system from a formal chemical viewpoint, can be illustrated by the scheme of mutual transformations for syngas, CO2, H2O, methane, methanol, acetaldehyde and ethanol, given by Nørskov and co-workers[24]. Even in a simplified version, we are talking about 100 intermediates, 200 reactions and more than 2000 routes (Figure 2). This indicates the complexity to design the catalysts for the synthesis of methanol from CO2 and largely explains an enormous body of literature in this field and the problems associated with the search for efficient heterogeneous catalysts.
![[{"id":"yuD5AOhLZx","type":"paragraph","data":{"text":"Reaction pathways for the conversion of syngas to methanol and by-products (adapted from Ref. [[ type=\"anchor\" referenceId=\"6070\" ]])."}}]](/storage/images/resized/rWiIewcuoR1qVSEfnTDUtfTwybFDqlVJuiEm2wKK_xl.webp)
An ideal catalyst should provide a well-defined sequence of reactions affording methanol with selectivity close to 100%. Given that the wide majority of methanol synthesis research is carried out in the flow regime and usually under gas-phase conditions, methanol selectivity and CO2 conversion will depend not only on temperature and pressure, but also on the space velocity of the gas reaction mixture fed. The lower it is, the longer the contact time of the feed stream of reactants and product with the catalyst and the closer it is to the thermodynamically acceptable CO2 conversion. Therefore, a correct selectivity comparison should be made at the same conversion, while the comparison of catalysts should be made taking into account the difference in the feed rate. Typical gas hourly space velocities (GHSV) of CO2 and H2 mixture are between 2000 and 24000 h–1[25-27].
Other parameters mentioned in the literature include GHSV (h–1) per gram of catalyst (h–1 gcat–1; can vary from 2000 to 50000), the ratio between the flow rate expressed in moles per unit of time and the catalyst mass F/W (mol h–1 gcat–1) and the inverse ratio W/F (h gcat mol–1). The latter varies from about 0.2 to 15 in different publications. The higher the ratio, the longer is the contact time with the catalyst. It should be noted that these values depend directly on the catalyst density, which makes their comparison not always correct in terms of evaluating the activity of individual sites.
The reaction rate on a given catalyst is not only determined by these parameters, but may also depend on the CO2/H2 ratio (usually in the range of 1/3 to 1/4, although higher ratios exist) or on the presence of diluents (e.g., water vapour). Since the reaction rate for heterogeneous catalysts depends on the number of active sites per unit area, the rate may increase with increasing surface area, which must be taken into account for a correct comparison of the systems. In most cases, this factor is not taken into account due to the difficulty of estimating the number of active sites per unit area. As a rule, the catalyst activity is estimated at a given flow rate, pressure, temperature and reaction mixture composition in units such as mgMeOH h–1 gcat–1, gMeOH h–1 gcat–1, gMeOH h–1 kgcat–1, molMeOH h–1 gcat–1. In view of possible changes in activity during the reaction due to the catalyst evolution or deactivation, it is essential to compare the systems if there is no change in these parameters over time or if data on such change are available. For commercially available catalysts at 250 °C, 3 MPa and 7900 h–1, the activity ranges from ~450 to 500 g h–1 kgcat–1[18].
The conventional mechanism of hydrogenation of CO in syngas to methanol over industrial Cu/Zn catalysts involves two sequential processes: water-gas shift reaction with traces of H2O to form CO2 which binds to the ZnO surface, and hydrogenation of CO2 to formate at the ZnO/Cu interface[28]. The formation of CO2 from CO and H2O is catalyzed by Cu: CO is strongly adsorbed by Cu and reacts with H2O on its surface. The intermediate bound to the oxide phase is hydrogenated by H atoms formed by H2 activation on the Cu surface. Herein, formate is the main intermediate for the methanol formation. As shown by Rozovskii[29, 30], in the case of conventional industrial Cu/Zn catalysts deposited on Al2O3 for the production of methanol from synthesis gas (CO/H2), the presence of small amounts of CO2 is necessary for the reaction to proceed at significant rates. The formation of methanol from synthesis gas is very slow in the absence of trace amounts of CO2. Subsequent studies have confirmed that it is the carbon dioxide adsorbed on ZnO particles, formed in the reaction between CO and traces of H2O, that is the main source of methanol when synthesis gas is used as a feedstock[31-34].
Consequently, the mechanisms of methanol formation from both CO2 and CO upon reaction with H2 on Cu/Zn catalysts are similar and are called formate (Figure 3а). This mechanism is realized for most heterogeneous catalysts for the production of methanol from CO2 (see Figure 3)[26, 31, 35]. Formate obtained from CO2 in the first step is further hydrogenated by H2 activated on Cu surface to adsorbed dioxymethylene or formic acid, which further provide the products[26, 35-38].
![[{"id":"GteeIX_Z_M","type":"paragraph","data":{"text":"Scheme for the CO<sub>2</sub> hydrogenation to methanol by the formate mechanism (a); by the carboxyl mechanism with reverse water-gas shift reaction (b)."}}]](/storage/images/resized/iEJjqm7ezQEKLizABVpHTWnKZspaIagtGdkZ2dQ2_xl.webp)
At the same time, the replacement of most portion of CO by CO2 in hydrogenation leads to a number of problems. The first of them is associated with the thermodynamics of CO2 hydrogenation to methanol and with the water-gas shift reaction (reactions (1), (2)).
Under standard conditions, the formation of methanol from CO2 proceeds with a very small positive value of the standard Gibbs free energy and a decrease in volume. Low temperature and high pressure favour the equilibrium shift to methanol (Table 1). To achieve the significant rates in the gas-phase process, the reaction should be carried out at elevated temperatures. Industrial Cu/ZnO catalysts typically show high activity only at 240 – 300 °C. The decrease in the achievable equilibrium concentration with increasing temperature can be compensated by a significant increase in pressure. However, even at high pressures (5 – 15 MPa), the thermodynamically possible methanol yield is low and achieves only 14% at 2.5 MPa and 250 °C[8, 39].
![Enthalpy, entropy and Gibbs free energy for the reaction CO<sub>2</sub> + 3 H<sub>2</sub> → CH<sub>3</sub>OH + H<sub>2</sub>O[<a class="anchor" href="#reference-6087" id="mention-reference-6087">40</a>].](/storage/images/resized/n8ogxUFbF6uaHQXjImNv0gMOm9DSfeGWI3xdxBbp_xl.webp)
The hydrogenation reaction from CO2 to CO is reversible, the reverse water-gas shift reaction is endothermic, and with increasing temperature its equilibrium shifts towards the formation of CO, a by-product that is poorly hydrogenated over Cu/Zn catalysts[40-42]. While in the industrial synthesis of methanol from syngas the reaction equilibrium is shifted to CO2 due to the high CO concentration, in CO2 – H2 mixtures at temperatures and pressures employed under the gas-phase conditions of methanol synthesis this proved to be significant. With increasing temperature, this reaction becomes more pronounced and leads to a low selectivity of the hydrogenation, in which CO is formed as one of the main products along with methanol. Therefore, the ability of the systems to catalyze the water-gas shift process required for the conversion of syngas to methanol has a negative impact on the hydrogenation of pure CO2. As a result, this process is one of the most important side reactions in the hydrogenation of CO2 to methanol and its suppression is highly desirable.
Another way to improve selectivity is to create catalysts capable of hydrogenating CO per se or converting the carboxyl moiety directly to methanol. In this case, the reaction proceeds via intermediates representing the bound CO and the formyl *C(O)H moiety. For methanol to be formed, the optimum CO adsorption energy on the catalyst surface must be reached (see Scheme 1b). Low adsorption energy will lead to its formation as a by-product. High energy can favour further deoxygenation and the formation of methane and higher hydrocarbons, as with conventional Fischer – Tropsch catalysts (Co- and Fe-containing systems) and methanation catalysts (Ni-containing systems)[43]. At the optimum energy, the main reaction is methanol formation[44].
If CO hydrogenation to methanol proceeds to give no by-products, CO2 hydrogenation can only be accompanied by the release of H2O, taking place both in the target reaction and in the water-gas shift reaction. H2O contributes to catalyst deactivation by aggregating Cu nanoparticles and increasing Zn particle size. In addition, water inhibits the hydrogen activation process by adsorption on the metal surface. As shown by Christensen and co-workers[45], water is removed from syngas through the water-gas shift reaction to give H2[46]. In CO2 hydrogenation, this process can only take place when certain concentrations of CO are reached, therefore, a side reaction of CO formation is necessary to achieve optimum activity. Another side reaction involving H2O is the vapour conversion of the newly formed methanol to CO2, which reduces the process selectivity[47]. Suppression of the catalyst activity in this reaction is necessary to achieve the high selectivity.
The total amount of the available data shows that significant metal-oxide interface is a prerequisite for the formation of efficient heterogeneous catalyst for CO2 to methanol hydrogenation. Methanol formation reaction occurs close to the metal-oxide interface and its properties largely determine the characteristics of CO2 binding and activation, hydrogen activation and the hydrogenation process itself.
The primary activation of CO2 involves its binding to the catalyst surface with changes in the structure of this moiety from linear to angular[26]. CO2 often coordinates via carbon directly to a surface metal atom[48] or via carbon and oxygen atoms simultaneously[48-53]. The presence of charged species (e.g. Cu+) can also facilitate additional coordination via oxygen (see Figure 3a). In the presence of oxygen on the catalyst surface (especially in the case of oxides), the former can act as a Lewis base, converting CO2 into a CO3– species[54]. A similar interaction is possible with the catalyst surface in the presence of other groups, which are Lewis bases (e.g., nitrogen atoms, sulfur atoms, etc.).
CO2 binding via oxygen atom is facilitated by the presence of oxygen vacancies (removal of oxygen from the lattice), which are generally formed at the oxide support–metal interface, especially in a reducing atmosphere. The formation of such vacancies is accompanied by the formation of Znδ+Oх species containing atoms of incompletely oxidized Zn, or even copper oxides CuxO[55, 56]. Consequently, the formation of vacancies is followed by the formation of Mδ+ oxophilic sites at the metal/oxide interface (in particular for Cu/ZnO to generate Znδ+Ох species at the interface) and promotes the adsorption of CO2 via an oxygen atom. Calculations show that such intermediate is formed most likely by the interaction with an oxygen vacancy on the surface of Al-doped ZnO[57, 58]. Such vacancies play an important role in CO2 activation for catalysts based on In, Ti and Ce oxides. They are formed in a reducing atmosphere and their formation is promoted by metals that activate H2. CO2 binds to the catalyst surface through the oxygen atom in the presence of defects in the form of oxygen vacancies, activating this molecule and promoting its subsequent reduction (Figure 4). It is possible that any subsequent oxygen-containing intermediates formed via the oxygen atom may remain bound to the oxide surface[59, 60]. A similar pattern can be observed for sulfide vacancies in metal sulfides[61].
![[{"id":"oKsyOgKH7O","type":"paragraph","data":{"text":"Scheme for the formation of oxygen vacancies and CO<sub>2</sub> activation."}}]](/storage/images/resized/et3kZlgUi4o24K7WdHaBU9mJ8EHFzplRPKkYay7z_xl.webp)
Under reaction conditions, a dynamic equilibrium is observed between the species on the surface due to the catalyst interaction with H2 as a reducing agent and CO2 as an oxidizing agent. The catalyst composition and therefore the evolution of the interfacial boundary depends on both the process temperature and the reactant concentration. When the catalyst is activated by H2, the reduction of Zn to Zn(0) and formation of the corresponding alloy is possible, as in industrial systems. However, Cu alloy with Zn per se exists only at very low CO2 pressures. Exceptional formation of such alloy is observed at a temperature of 150 °C and CO2 pressure of 0.1 MPa. At CO2 pressures above 1 MPa and higher temperatures, the main Zn-containing phase is ZnO (formed by the reaction of Zn with CO2). As a result, upon CO2 hydrogenation, monovalent Zn atoms in the Cu alloy or on the Cu surface move to the oxide phase ZnOx, which promotes the formation of active Cu0–Znδ+ ‘interface’, and the reaction takes place at the interfaces of ZnOx – Cu species[62-64]. Muhler and co-workers[32] showed that under the reaction conditions, hydrogenation involves mainly the Cu0–Znδ+ interface[64]. Zn oxidation in the alloy and formation of such species were confirmed by in situ X-ray absorption spectroscopy (XAS)[65]. Location of the active sites for the thermocatalytic hydrogenation of CO2 at the ZnO – Cu interface was confirmed by calculations[66].
In addition to CO2 activation, the methanol synthesis catalyst should also activate H2. The latter reaction usually occurs on the metal surface (homolytic hydrogen cleavage) or at the metal-carrier interface (heterolytic cleavage) (Figure 5), which usually requires pre-activation of the catalyst in a reducing atmosphere. Successful hydrogenation requires the proximity of CO2, bound with carrier or metal-decorating oxide, to the metal surface or the coordination of carbon or oxygen atoms of the intermediate oxygen-containing species to the metal. In the Cu/Zn catalyst, low-coordinated Cu atoms, located in individual microcrystalline domains, including those on edges, steps and other defects in the catalyst, provide homolytic activation of H2.26 For Cu/Zn catalyst, the formation of such species was found to promote H2 dissociation even at room temperature[67, 68].
![[{"id":"7kPWo77S2p","type":"paragraph","data":{"text":"Examples of heterolytic and homolytic activation of hydrogen on copper oxide catalysts."}}]](/storage/images/resized/I9zTxAOX4cP2cgOm8fNPZGeqyipLYiX9kexeIojq_xl.webp)
In addition, H2 activation is promoted by strong metal–support interactions involving partial charge transfer from the metal; H2 activation here occurs near the interface and can be heterolytic[69-72]. For example, for Cu, such interactions lead to the formation of surface Cu(I) sites[73] and Cuδ+ – O – ZnO units at the interface[74], thus promoting heterolytic activation[75, 76].
It was shown that the simultaneous presence of Cu0 and Cu+ species in the catalyst is essential for hydrogen activation and subsequent hydrogenation[77]. For pure defect oxides, heterolytic activation of H2 at metal–oxygen–support bonds is possible[76, 78]. A similar situation can take place for precious metals supported on oxides capable of forming oxygen vacancies[72, 75, 76, 79, 80].
For Cu/Zn catalysts, it is assumed that after activation, a part of H2 spills over to Cu/Zn interface, where hydrogenation to formate takes place[81, 82]. Obviously, the larger the oxide–metal interface, the greater is the spillover probability and the higher is the catalyst activity[83]. It has also been shown that the high catalyst activity was achieved due to the high dispersity of Cu when its surface was decorated with Zn or ZnO nanoparticles: the smaller the latter size, the higher was the activity[35, 84].
The electronic interaction between metal and oxide components (in the industrial Cu/Zn catalyst it is Al2O3 and ZnO) largely determines the stability of the main intermediates, the dispersity of Cu on the surface and the metal – oxide interface area[85, 86]. A recent study of a catalyst obtained by supporting Cu/ZnO component on Al2O3 nanorods has confirmed that close interaction between ZnO and Cu was necessary to achieve high catalyst performance[87]. Adsorbed CO2 may interact with Al2O3 and ZnO via carbon, thus producing carbonates, or via oxygen, occupying the oxygen vacancies on the oxide surface, resulting in formate. H2 activation occurs with the participation of Cu and is promoted by the ZnOx phase present on the Cu surface. A system containing only 3% of CuZnO phase provided a 19.8% conversion of CO2 and methanol formation activity of 1.31 mmol gcat–1 h–1 at 300 °C. The shape of the Cu nanoparticles also significantly affects the catalyst activity and selectivity[88]. Cube-shaped Cu particles were ‘rounded’ and partially lost their facets (100). With this transformation, the activity increased with a slight decrease in selectivity as compared to Cu particles, which were initially spherical. The importance of the electron transfer from ZnO to Cu was confirmed[89]. Samples of the Zn1–xSixO support were obtained both with and without electron conductivity. After Cu deposition, high catalytic activity was observed only for the first sample, where partial electron transfer to the oxide oxygen at the interface was possible due to the conductivity.
The replacement of Al2O3 with Ga2O3 in the Cu/ZnO catalyst affects the formation of the partially reduced zinc phase and stabilizes the catalyst performance[90]. Due to the formation of the gallium spinel ZnGa2O4, even a small amount of Ga promotes the partial reduction of ZnO giving Cu – ZnOx particles. In addition, the formation of such spinel provides both more efficient CO2 binding and an increase in the adsorption strength of the main intermediates (HC*O, H3CO* and H2C*O), thereby improving the methanol selectivity[91]. Ga-doped Cu-containing catalysts are highly active at relatively low temperatures (210 °C)[92].
Modified Cu-containing catalysts with atomically dispersed components can be prepared by the surface modification with metal-containing individual compounds to introduce the active component[93]. Such compounds interact with hydroxyl groups of the support and form grafted groups with one or more metal atoms on the surface. Subsequent transformation of these surface species, e.g., in a reducing atmosphere, produces heterogeneous catalysts of different composition and structure. Consecutive or simultaneous complex grafting is possible, giving rise to bimetallic and polymetallic systems. This approach, described in detail in a recent review[94], has been used to prepare highly efficient catalysts with supported Cu atoms. Silica gel-supported catalysts Cu/Mn+@SiO2 (M = Ti, Zr, Hf, Nb and Ta)[95], Cu/Zn, Cu/Ga and Pd/Ga were obtained[96]. The latter were characterized by the highest performance in methanol formation as compared to other samples[96]. These Pd catalysts were prepared by grafting Pd(COD)Me(OSi(OBut)3) complex on silica gel doped with hexacoordinated Ga3+ ions, followed by the reduction and formation of Pd/Ga alloy on the surface with metal ratio close to 1 (Figure 6). As a result, the concentration of formate species on the catalyst surface and the rate of their conversion to methanol were significantly increased. This provided a selectivity of 80% and an activity that is an order of magnitude higher than that for similar Cu-based systems.
![[{"id":"VTQFhBk9mJ","type":"paragraph","data":{"text":"Synthesis of the Pd/Ga catalyst[[ type=\"anchor\" referenceId=\"6142\" ]]. The Figure is published under the Creative Common BYNCND 4.0 license."}}]](/storage/images/resized/ga8OB7DNHAB8qDn5fCIBYaBEzOp9wtWrkIfOOdDR_xl.webp)
The addition of MgO to CuO on ZnO increases the catalyst activity and methanol selectivity due to better CO2 adsorption[97]. Thus, at 20% loading of MgO, CO2 conversion reaches 8.7% with 99% methanol selectivity at 200 °C and 16% conversion with 62% selectivity at 300 °C. As a result, high activity in methanol formation with almost quantitative selectivity can be achieved. Incorporation of MgO improves the Cu dispersion, and Mg incorporation into the ZnO lattice significantly increases the CO2 adsorption. Moreover, as shown by Nørskov and co-workers[98], the presence of MgO creates the conditions for hydrogenation of the formed CO by-product at the Cu – ZnO interface.
The promotion of Cu/Zn systems by even a small amount of La (3 wt.%, catalyst 0.6 Cu/0.3 ZnO/0.03 La2O3/0.07 Al2O3) allows methanol to be obtained at 8.5 MPa of CO2 and 325 °C with a selectivity of 60%, at 300 °C with a selectivity of 65% and CO2 conversion of 20%[99]. Active catalyst for methanol production can be prepared using Cu/Fe systems, obtained by impregnating a silica support with metal salts, although even with the optimum ratio Fe/(Cu + Fe) = 0.67, the methanol selectivity at 260 °C and 3 MPa reached only 36% with CO2 conversion of ca. 12%[100].
Another way to improve the activity of Cu/Zn systems is to increase the metal dispersity using special types of support. Herrero and Ullah[101] suggested the use of structured polyhedral oligomeric silsesquioxanes, characterized by high hydrophobicity due to the presence of phenyl substituents in the polyhedron structure. Depending on the polyhedron size in the support structure at Cu and Zn deposition, catalysts with particle sizes of 7 and 15 nm can be synthesized. The first catalyst had the highest activity (the methanol yield in the batch reactor was 3.8% with CO2 conversion of 4.4% after 18 h). The use of the active phase supported on reduced graphene oxide significantly worsened the results.
Immobilization of Cu/Zn systems can also be applied to hybrid supports with high internal surface area, such as metal-organic frameworks (MOFs)[102]. In particular, a zinc- and imidazole-based framework ZIF-8 with high CO2 adsorption capacity has been used for this purpose[103]. CuO – ZnO@ZIF8 (1 : 4) catalyst showed the maximum activity (CO2 conversion 14.64% and methanol selectivity 93.41%).
Carbon-containing systems, in particular layered carbides, were used in a number of studies as supports for Cu-containing catalysts for methanol synthesis. Thus, unusual increase in the methanol selectivity due to the formation of Cu sites with specific electronic properties interacting with the support has been observed for catalyst obtained by immobilization on silica gel of the MXene phase, which is a layered carbide (Cu/Mo2CTx/SiO2, where T = –O –, –OH, –F). The support material stabilizes the catalytically active Cu – Cu+ pair via electron interactions; this material is also oxophilic, thus promoting CO2 activation. All this accelerates CO2 hydrogenation via methyl formate, favours the decrease in the water-gas shift reaction rate and enhances the methanol selectivity[104].
As mentioned in the discussion on the reaction mechanism, a significant change in the catalyst activity is possible while replacing the support for the Cu-containing catalyst with an oxide, such as Zr, Ti, Ce and In oxides, capable of forming oxygen vacancies. The presence of these vacancies provides the binding and activation of CO2, which can be further converted to methanol. Many authors have suggested that the presence of such metal oxides determines the catalyst stability and affects the reaction selectivity. By themselves, these oxides are inactive in hydrogenation catalysis due to the low H2 activation rate. Therefore, the addition of metals capable of activating H2, mainly Cu, is one of the most common methods to increase the activity of catalysts supported on the above oxide systems. At the same time, the support per se and the dopants should be ‘bad’ catalysts for the water – gas shift reaction, which is responsible for the decrease in selectivity in CO2 hydrogenation.
For example, comparing copper catalysts on CeO2, ZnO and ZrO2 supports, Pant and co-workers[105] showed that the production of methanol over the Cu – ZnO system surpasses that over the catalyst containing Cu on CeO2, but the methanol selectivity is much higher for the latter catalyst. The authors attributed the decrease in selectivity for Cu/ZnO and Cu/ZrO2 catalysts to their inability to stabilize the necessary intermediates, resulting in the reverse water-gas shift reaction between CO2 and H2 leading to decrease in selectivity. According to the available data, the latter reaction can be catalyzed by CeO2 and TiO2 and proceeds at high rates[94, 106]. At the same time, these supports are characterized by high thermal and chemical stability[107], and an increase in selectivity can be achieved by various synthesis approaches to surface modification.
It was shown that TiO2 mainly acted as ZnO, and to achieve high methanol selectivity it was necessary to create oxygen vacancies in the support with the formation of partially reduced TiO2 particles. They enhanced the binding efficiency and the CO2 reduction rate. Such vacancies can be formed either through the hydrothermal synthesis or during the reduction of pre-catalyst with H2[108, 109]. As a result, the methanol selectivity reaches 96% with almost equilibrium CO2 conversion (9.4% at 200 ºC and 3 MPa). Modification of CeO2-supported Cu-containing catalyst with magnesium slightly increased the system activity, and the highest rate was achieved at Cu and Mg loadings in the catalyst of 4 and 6%, respectively[110].
The catalysts supported on Ce or Ce – Zr oxides (Cu – Ga/CeO2 – ZrO2 and CuO – ZnO – CeO2) are characterized by relatively low selectivity values (~60% at 260 ºC and 4 MPa), regardless of whether Ga or Zn is used as a promoter component for Cu. It was shown that the optimum catalyst composition corresponded to the same molar content of Ga and Cu, while the addition of Ga changed the reaction pathway. For Cu supported on CeO2 – ZrO2, the process proceeds via –*COOH species and adsorbed CO; once Ga is added, and the reaction follows the formate mechanism typical for Cu/Zn systems[111, 112]. The presence of oxygen vacancies improves the catalyst performance in both methanol synthesis and CO formation[113].
The use of Pd can be an alternative to Cu for CeO2-supported catalysts. It was shown that at 2 wt.% Pd loading supported on CeO2 nanorods at 240 °C and 3 MPa, the CO2 conversion was 49.6%, with methanol selectivity of 69.5%[114]. Unfortunately, despite the high metal dispersion, this system retained significant activity in methanation (methane selectivity of 30.6%) due to the Pd reduction on the oxygen vacancies.
More complex systems containing bimetallic Pd and Cu particles together with mixed Zr and Ce oxides have been studied recently for methanol synthesis[115, 116]. Dendritic catalysts containing Pd/Cu particles (PdCu/Ce0.3Zr0.7O2) with spherical morphology allowed to increase the availability of both active bimetallic phase and oxygen vacancies of the support, mainly Ce3+ cations and, consequently, the oxygen vacancies providing CO2 activation. The CO2 conversion was 25.5% and the methanol yield was 6.4%. The catalyst was characterized by significantly higher stability than systems based on individual oxides or supported on mixed oxide with non-dendritic morphology. Replacing Cu with Zn maintained the catalyst activity and stability due to a similar effect of generating more oxygen vacancies. The use of Pd supported on the catalyst containing both ZnO and mixed Ce–Zr oxide (CeZrZnOx, 2 wt.% Pd) resulted in the CO2 conversion of 29.1% and methanol selectivity of 43.8%[117].
Another interesting support is nitrogen-doped carbon derived from polybenzoxazine[118]. Deposition of Cu and Ru thereon together with ZrO2 allows to reach the CO2 conversion of 37%, methanol selectivity of 75% and efficiency of 642 mg gcat–1 h–1 at 210 ºC and 1.5 MPa. Replacing ZrO2 with CeO2 improves the methanol selectivity to 92%. The catalyst prepared by simultaneous deposition of Cu and Fe nanoparticles onto a P-, N- and C-containing support allowed to reach the CO2 conversion of 12.6% and methanol selectivity of 79% at 225 °C and 2 MPa[119].
The use of ZrO2 as a support in Cu-containing catalysts leads to the formation of additional Cu – ZrO2 sites capable of binding and activating CO2 due to oxygen vacancies on the support[120]. As a result, a large number of papers have been devoted to the study of catalysts based on this support. The size of the Cu particles on ZrO2 can be controlled using porous Zr-containing structures. Thus, for Cu@ZrO2 catalyst derived from Cu@UiO67 metal-organic framework, ZrO2 framework in the resulting material provides both the formation of Cu nanoparticles and the generation of a large number of Cu+ – ZrO2 sites at the support – metal interface. This increases the activity of the catalyst by 3.5 times (3 MPa, 260 °C, methanol yield 2.28 mmol g–1 h–1) as compared to the system obtained by the conventional method[121]. Deposition of CuO nanoparticles onto Zr-containing metal-organic framework UiO-66 with additional Si-containing linkers and thus enlarged pores resulted in the catalyst providing CO2 conversion of 2.4% and methanol selectivity of 76.8% at 240 °C and 3 MPa[122].
The influence of Cu interaction with ZrO2 support is studied in detail[123]. It was shown that the methanol selectivity depended not only on the size of Cu and ZrO2 particles, but also on the degree of their interaction. The high Cu dispersity provided the significant size of Cu – ZrO2 interface, which allowed the high values of selectivity and conversion to methanol to be achieved. Similar data were obtained by Stangeland et al.[124]
The promoting effect of ZrO2 may also be due to the reduction of Zr4+ to Zr3+, which effectively binds oxygen-containing species such as CO2, CO and formate. Hydrogenation of formate to methanol effectively proceeds on Cu, whereas on the ZrO2 surface the rate of formate reduction is low[125]. At the same time, ZrO2 promotes the formation of oxygen vacancies at the ZrO2 – Cu interface and provides a high dispersion of Cu particles due to their stabilization by interaction with the support[126]. Marcos et al.[127] showed that catalysts containing only amorphous Cu particles dispersed on ZrO2, to form the large interface between Cu and ZrO2, exhibit significantly higher activity as compared to the catalyst containing crystalline Cu nanoparticles.
It is found that for Cu/Zr systems it is extremely important to have monoatomic active Cu sites bound to the ZrO2 surface. It was shown that active sites representing low-coordinated Cu atoms bound to three lattice atoms of ZrO2 in a quasiplanar structure allowed to achieve almost 100% selectivity for methanol (7% conversion at 3 MPa and low temperature 180 °C, CO2 : H2 = 1 : 3, 10 ml min–1)[128]. At the same time, Cu clusters and nanoparticles catalyzed mainly CO formation. The formation of similar single-atom sites from clusters was observed during the reaction, thus accelerating CO2 hydrogenation. H2 heterolytically dissociated at these one-atom sites involving the adjacent O atoms, allowing the formation of HCOO* species, which were further hydrogenated to methanol.
The catalyst activity can be improved by increasing the dispersity of the Cu nanoparticles. The synthesis of Cu@ZrO2 catalyst from organometallic Zr framework under low-temperature pyrolysis conditions allows the formation of highly dispersed copper with Cu0/Cu+ ratio necessary for high selectivity on the surface of hollow ZrO2 particles[129].
ZnO addition to Cu gives a ZnO – ZrO2 solid solution as a support and improves the activity of the said systems. Roger and co-workers[130] found that the Cu – ZnO – ZrO2 catalyst surpassed similar systems based on CeO2 and Al2O3 in terms of methanol selectivity, although the latter was low (33% at 280 °C and 5 MPa). The presence of ZnO in combination with ZrO2 is crucial: the performance of catalyst based on ZrO2 only is much inferior to that of CeO2-based catalyst[105].
Cu addition (<2%) to ZnZrOx leads to the formation of trimetal derivative as a promoter of methanol formation, with the rate of hydrogenation being largely determined by the H2 spillover from the Cu surface to CO2 species adsorbed on the oxide phase. Although both conversion and selectivity were low, the former was four times that obtained with the catalyst without Cu at a comparable selectivity (290 °C)[131].
The high dispersity of particles containing both Cu and Zn, with ZrO2 introduced, provides the high selectivity and activity in the hydrogenation of CO2 to methanol. These catalysts are superior in efficiency to two-component systems such as Cu-doped ZrO2, Zn-doped ZrO2 and Cu-doped ZnO catalysts[132]. It was noted that for ZrO2-supported CuO/ZnO catalyst, the highest methanol yield was observed in the presence of the tetragonal ZrO2 species[133].
The activity is also affected by the morphology of the ZnO particles. Thus, for Cu/ZnO/ZrO2 catalysts containing ZnO support in the form of ‘flowers’, plates or rods, the first type of morphology gave the best results. It was shown that this material was characterized by the highest number of oxygen vacancies and that the methanol yield increased linearly as their amount increases[134].
As for the ZrO2 content, for Cu/Zn systems, the catalyst with 10% support showed the highest performance (methanol space-time yield of 0.65 g gcat–1 h–1) at 220 °C and 3 MPa. It was found that the maximum Cu+/(Cu++Cu0) ratio was observed in this case. Cu(0) was shown to be primarily involved in H2 activation and Cu+ participated in the hydrogenation of formate to methanol[135]. The optimum ratio of ZnO and ZrO2 for Cu-containing catalyst obtained by co-precipitation was determined[136]. The catalyst for which the maximum Cu dispersion (Zn/Zr mass ratio equal to one) was achieved showed significantly lower performance than the system with 66 wt.% ZnO and 34 wt.% ZrO2, which showed the methanol selectivity of 50% with CO2 conversion of 19.6% (725 gMeOH kgcat–1 h–1 at 280 °C, 5 MPa). The authors pointed out that not only the Cu valency state and the ZnO particle size, but also the degree of ZnO – ZrO2 interaction in the support were important in designing the optimum catalyst.
Zhan et al.[137] showed that for catalysts of this composition the optimum ratio was Zr4+:(Cu2++Zn2+) = 0.5. According to the authors, in this case, a balance was achieved between CO2 binding through the interaction with oxide phase or oxygen vacancies and activation of H2 on the Cu surface. It was shown by XAS that to achieve the high selectivity (> 80% at 220 °C, 3 MPa, CO2 : H2 = 1 : 3) and productivity (> 2.5 moles per 1 kg of catalyst per hour) using catalysts prepared by the flame spray pyrolysis, it was necessary to form Cu and Zr/Zn phases separately during synthesis. The formation of atomically dispersed Zn atoms on the ZrO2 surface was crucial for the catalyst activity due to the high energy of interaction with the support[138].
Interesting results on the structure of the most active catalysts were obtained by Arandia et al[139]. The catalyst obtained by deposition of a highly dispersed ZnO phase on Cu samples containing ZrO2 or mixed ZnO – ZrO2 as support (ZnO/Cu/ZrO2 and ZnO/Cu/ZnO/ZrO2 catalysts) was significantly superior in activity to the systems in which Cu was applied after ZnO deposition. In this case, the ZnO particles on the surface of the Cu phase are much more effective in binding CO2 in the form of carbonate species and promoting their conversion to formate.
Deposition of Pd, another H2-activating metal, onto ZnZrOx with a high dispersion degree significantly increased the activity in the hydrogenation of CO2 to methanol (CO2 conversion of 12% with methanol selectivity of 80% at 320 °C and 5 MPa) by increasing the H2 dissociation rate[140]. For the physical mixture of ZnZrOx (Zn/Zr = 1/5) and Pd-coated carbon nanotubes, CO2 conversion of about 20% with over 65% methanol selectivity was achieved at 320 °C and 5 MPa. The methanol yield was 13.5% (productivity of 0.9 g g–1cat h–1), which was close to thermodynamic equilibrium. The catalyst remained stable for 600 h. Apparently, this result was achieved due to the high spillover rate of dissociated H2 from carbon nanotubes onto ZrO2[141].
It is also possible to produce ZrO2-based catalysts for the conversion of CO2 to methanol by modifying ZrO2 only with ZnO to form a solid solution. It was shown that the activity of ZnO/ZrO2 systems depended on the composition of ZrO2 crystalline phase[142]. To achieve significant methanol yields, both tetragonal and monoclinic phases must be present in the catalyst. According to the authors, this significantly changes the oxygen vacancy content. Huang and co-workers[143] showed that to achieve high selectivity for methanol, the surface of the mixed oxide should be enriched with Zn-doped ZrO2 solid solution phase. Pure ZnO, t-ZrO2, m-ZrO2 and ZnO – ZrO2 composite oxide exhibit high selectivity for CO but not for methanol. At low Zn content in the ZnxZr1–xO2 solid solution, the composition is dominated by isolated ([ZnOa]) and oligomeric ([ZnbOc]) clusters. They are located close to the oxide surface and provide the formation of Zn – O – Zr active catalytic sites on the surface. The latter activate H2 by a heterolytic pathway and significantly increase the degree of CO2 adsorption, although the activity of the system remains relatively low (8% of CO2 conversion at 350 °C and 1 MPa at a space velocity of 3600 ml gcat–1 h–1). Increase in Zn amount leads to formation of oxide particles along with clusters and subsequent downfall in activity[144].
The activity of the catalyst based on ZnO – ZrO2 solid solution can be improved by developing a mesoporous structure therein. The catalyst with such structure (surface area 109.4 m2 g–1), containing 20% ZnO – ZrO2 phase, provided the methanol formation rate of 22.1 mmol g–1 h–1 at 320 °C, 5 MPa, which was 1.35 times higher than that for the catalyst obtained by co-precipitation[145].
An original way to increase the catalyst surface area while increasing the degree of contact between the ZnO and ZrO2 phases was proposed by Zhang et al.[146] ZnO was supported on ZrO2-based nanoscale metal-organic framework. The latter comprised Zr12(μ3O)8(μ3O)8(μ3O)6 clusters linked by biphenyldicarboxylate anions. Such catalyst provided a methanol yield of 3.4 mmol g–1 h–1 at 250 °C with selectivity of more than 95%.
Surface modification with single metal-containing compounds to introduce the active component failed for Zn/Zr systems[147]; it was also used to form dispersed atomic active sites of Zn(II). However, in contrast to Cu-containing catalysts, for Zn/Zr systems, it was essential to have not individual Zn(II) sites, but highly dispersed ZnO nanoparticles in contact with ZrO2, forming active sites at the interface. The authors suggested this was due to the low activity of Zn nanoparticles and Zn(II) species in the activation of H2, in contrast to ZnO. A catalyst with the optimal composition and particle dispersion was obtained by flame-spray pyrolysis and contained 10% ZnO.
Other H2-activating components, including Au particles, can be supported on ZrO2 to improve its activity. In particular, it was shown that Au deposition on ZrO2 during the support synthesis could significantly improve the methanol selectivity and ZrO2 activity[148]. At 240 °C and 4 MPa, a catalyst containing 1% of Au provided a methanol selectivity of 70% at a CO2 conversion of 6.8% with a productivity of 59 mmol g–1 h–1. Au nanoparticles with mean diameter of 1 nm were deposited here on the cubic phase of ZrO2. The catalyst can be modified with CeO2 to enhance its stability, although the selectivity on methanol is reduced in this case[149].
ZrO2 was shown to be the optimal substrate to generate active sites for the hydrogenation of CO2 to methanol using the nickel-gallium alloy Ni5Ga3, providing optimum binding energies for intermediates and CO2[150]. For another hydrogenation component, InNi3C0.5 intermetallide, the maximum activity in the hydrogenation of CO2 to methanol was achieved due to the interaction with the monoclinic ZrO2 support[151].
Unexpected results were obtained with a catalyst containing another oxide, MnOx, deposited on ZrO2. The catalyst was characterized by low activity in the formation of methanol (4.5 mgMeOH gcat–1 h–1), but remained stable for 36 hours. The activity of the system increased 87-fold under the influence of dielectric barrier discharge (DBD) plasma (silent electric discharge), and in situ studies showed the reaction mechanism to change in this case. Plasma on the catalyst surface produced not carbonate but formate species, which were further hydrogenated to methanol due to the presence of partially reduced MnOx[152].
The activity of ZrO2 can also be increased by introducing Ga2O3 into its composition to obtain GaZrOx solid solution[153]. High dispersity of Ga2O3 and surface ratio of Ga/Zr = 0.5 makes possible the formation of a significant number of oxygen vacancies activating CO2 along with active Zr – O – Ga sites, which can be activated by H2 to form Ga – H and Zr – OH species.
Among the oxides active in CO2 to methanol hydrogenation, a special place is occupied by In2O3, for which high CO2 activation efficiency and methanol selectivity are achieved due to the easy generation of oxygen vacancies on the oxide surface with the formation of non-stoichiometric indium oxide InO2–x[154]. According to the results of theoretical modelling, the highest catalytic activity is characteristic of the defective In2O3(110) and In2O3(111) facets, and the process follows the formate mechanism[155]. The process in the reducing atmosphere is favoured by the absence of lattice oxygen on the said facets, with one or two reduced layers on the surface being optimal[156].
Various approaches and modifications have been suggested to increase the number of these vacancies, to provide the activation of H2 and reduce the water-gas shift reaction rate[157-163]. A kinetic model for CO2 conversion of methanol on In2O3 as a function of temperature, pressure and the reactant ratio has been proposed, allowing to estimate the competitive formation of CO and CH4 by-products. It was shown that high temperature favoured the reverse reaction[164].
The number of oxygen vacancies on the In2O3 surface in a reducing atmosphere can be increased by the addition of ZrO2 due to the interaction between the oxide phases[156, 165]. The most active catalysts are those in which In2O3 is combined with the monoclinic ZrO2, for which indium high dispersity is achieved in the reduction process yielding In3+ – O – Zr4+ sites together with vacancies. As a result, the reduced catalyst is not only capable of CO activation, but also of H2 dissociation to afford In – H and Zr – OH surface species[166].
Both the location and number of vacancies on the In2O3 surface are essential. For methanol to be produced, vacancies formed by the simultaneous removal of four oxygen atoms during H2 activation are important[167]. It should be noted that the hydrogenation activity of such oxide catalyst, e.g., nanostructured In2O3/ZrO2, is also affected by the calcination temperature of the support (600, 700, 800, 900, 1000 °C), which is associated with changes in its physical chemical properties, crystal structure and, thereby, its ability to adsorb and desorb CO2 and H2 under hydrogenation conditions (320 – 400 °C). This was shown to be insignificant for CO formation, but could affect methane formation[168]. Significant activity was obtained for a catalyst based on amorphous ZrO2 obtained by DBD plasma pyrolysis with 10% In2O3 supported thereon (methanol yield was 0.21 g g–1 h–1 at 5 MPa and 300 °C)[169].
A high area of the In2O3/ZrO2 interface, favouring the formation of vacancies and In3+ – O – Zr4+sites, can be achieved in the hollow mixed oxide particles obtained by pyrolysis of the metal-organic framework MIL-68@UiO-66. For this catalyst, the methanol selectivity was 84.6% with CO2 conversion of 10.4% at 290 °C, 3 MPa, and methanol yield reached 0.29 g g–1 h–1 (Ref. [170]). The data obtained indicate that the electron transfer from the ZrO2 to In2O3 surface facilitates both the dissociative adsorption of H2 and the hydrogenation of CO2 to methanol.
Deposition of In2O3 (13 wt.%) on CeO2 similarly gives the catalyst active in the hydrogenation of CO2 to methanol, but with a slightly lower methanol yield and low stability as compared to deposition on ZrO2[171]. The main reasons for the rapid deactivation are the formation of In(OH)3 on the catalyst surface and the sintering of CeO2 particles in the presence of H2O due to the high hydrophilicity of the support.
The catalyst activity can be enhanced by doping In-containing systems with metals of high hydrogenation activity, particularly platinum group metals. Particles of such metals are able to activate H2 by dissociative mechanism and significantly increase the hydrogenation rate. It has also been shown that metal doping increases the number of oxygen vacancies, which accelerate the CO2 activation. For example, small additions of Pt to In2O3 can increase methanol selectivity from 72.2 to 92.1% (220 °C) when Pt is atomically dispersed as Ptn+. The presence of Pt nanoparticles accelerates the water-gas shift reaction and decreases the selectivity[172]. The location of Ptn+ on specific crystal facets of In2O3 is important for the process selectivity and high catalyst activity. To achieve high Pt dispersity and oxygen vacancy number, the metal must be bound to the oxide (211) facet. This provides a CO2 conversion of 11.7%, a methanol selectivity of 74.8% and methanol formation activity of 0.63 mgMeOH gcat–1 h–1 at 300 °C and 5 MPa[173]. The catalyst activity can be significantly improved by introducing additional ZrO2 to obtain an In2O3 – ZrO2 solid solution as a support for Pt[174]. Density functional model calculations of CO2 hydrogenation to methanol for the Pt8/In2O3 system showed that CO2 was adsorbed at the interface between Pt and support particles and hydrogenation proceeded via adsorbed *COOH and CO species[175].
Sun et al.[176] have proposed a similar mechanism for hydrogenation via CO using deposited highly dispersed Ag. The interaction of Ag with In2O3 results in a positive charge on Ag along with oxygen vacancies on the support and promotes the formation of active sites at the Ag – In2O3 interface, allowing the formation of CO from CO2 and hydrogenation of the latter to methanol. The methanol selectivity is 58.2% at 5 MPa, 300 °C with CO conversion of 13.6% and methanol formation rate of 0.453 g g–1 h–1.
The best method to prepare the Pd – In2O3 – ZrO2 catalyst was found to be the flame-spray pyrolysis. In this case, the system 0.75% Pd – 5% In2O3 – ZrO2 provided a space-time yield of 1.3 gMeOH gcat–1 h–1 at 5 MPa and 280 °C and stable catalyst operation for 50 h[177]. Flame-spray pyrolysis allowed the synthesis of a catalyst with small Pd clusters on In2O3 monolayers dispersed on ZrO2 support. The latter was transformed into a monoclinic form under the reaction conditions. Modification of In2O3 by Pd and MnO simultaneously provided an active catalyst for the hydrogenation of CO2 to methanol with a selectivity more than 80% at temperatures lower than 280 °C for 1 wt.% Pd on MnO/In2O3[178].
Supporting highly dispersed Rh particles on In2O3 also favoured the activation of H2 with its subsequent spillover onto the oxide surface, while the formation of oxygen vacancies promoted the adsorption and activation of CO2. The conversion and methanol selectivity were 17.1% (as compared to 9.4% over pure In2O3) and 56.1% (300 °C, 5 MPa), respectively. A decrease in temperature led to increase in selectivity (up to 100%) and decrease in conversion. Nevertheless, this result was superior to those observed for other oxides[179]. Incorporation of ZrO2 into the support structure, resulting in In2O3 – ZrO2 solid solution, significantly improved the activity of Rh-supported catalyst and methanol selectivity due to the enhanced CO2 sorption ability[180].
When Re is used, the catalyst activity significantly depends on the size of the Re particles. With atomic metal dispersion (less than 1% Re on In2O3), the hydrogenation of CO2 to methanol proceeds at 5 MPa and 300 °C with high productivity (methanol formation rate is 0.54 g gcat–1 h–1) and selectivity (72.1%) due to the stabilization of oxygen vacancies and formation of hydrogen in the form of Нδ+ species on the metal surface. The presence of the latter favours the methanol production. Increasing the Re amount up to 10% results in samples containing the oxide-supported metal nanoparticles, on which negatively charged Нδ– species are generated from H2. The latter are actively involved in the deoxygenation and promote methanation[181]. The atomic dispersion of Ir on In2O3 (0.16%) also accelerates the hydrogenation to methanol by stimulating the formation of oxygen vacancies and the activation of H2 on metal atoms[182].
For unmodified In2O3, Cu deposition provides a relatively low activity system containing CuxIny alloy nanoparticles on the surface. The simultaneous use of ZrO2 and In2O3 for Cu deposition results in the stable and highly active catalyst due to the formation of mixed ZrO2/In2O3 oxide phase with higher portion of oxygen vacancies and high Cu dispersion (activity in methanol formation is 60.5 mmol gcat–1 h–1 at 270 °C, 3 MPa)[124]. For the Cu – In – Zr – O catalyst, In2O3 is responsible for CO2 adsorption, while Cu is responsible for H2 activation and hydrogenation[183].
Thus, for In-containing systems to show high activity, a high metal dispersion is important to provide the formation of active sites at the metal/partially reduced In2O3 interface. A slightly different situation was observed for the ruthenium modification. Supporting Ru on In2O3 led to methanol selectivity of 74.7% and almost two-fold increase in the yield as compared to unmodified In2O3, with complete suppression of methane formation, which is characteristic of supporting Ru on other oxides. The authors explained this effect in terms of interaction of the Ru nanoparticles with In2O3 to afford In2O3-x enriched by oxygen vacancies and to form RuOx – In2O3 – x sites active in the hydrogenation of CO2 to methanol[184].
Araújo et al.[185] compared catalysts obtained under similar conditions with different metals deposited. For In2O3-based catalysts with 0.5% metal content obtained by flame-spray pyrolysis, the methanol productivity varied in the order: Pd ≈ Pt > Rh ≈ Ru ≈ Ir > Ni ≈ Co > Ag ≈ In2O3 > Au. The catalysts with atomically dispersed metals (Pd, Pt, Rh, Ru and Ir) showed high activity in the methanol formation and reduced CO yield. Catalysts doped with metals, which formed nanoclusters (Ni and Co) or nanoparticles (Ag and Au) under the synthesis conditions, displayed low performance. At the same time, the use of non-precious metals with hydrogenation activity, such as Ni, produced active catalysts for methanol synthesis at high Ni dispersions[186]. The high interaction energy of Ni with the In2O3 crystal lattice led to the formation of a significant number of oxygen vacancies upon the H2 activation on Ni[187]. As a result, methanation did not proceed even at 10 wt.% Ni. The methanol selectivity was 64% at 225 and 275 °C and 54% at 300 °C.
As with other metals, modelling results have shown that the mechanism of hydrogenation was also changed, since methanol was formed from adsorbed CO produced in the reverse water-gas shift reaction between H2 and CO2. Calculations have shown that this pathway was the most energetically favourable for the catalyst with deposited Ni4 clusters[188]. Hensen and co-workers[189] confirmed the above mechanism by XAS and showed that, due to the strong interaction between Ni and In2O3, the properties of the resultant catalyst were similar to those of the Pd-supported catalyst. This Ni catalyst did not produce methane even at high metal contents (up to 75 wt.%); methanol formation rate was 0.25 gMeOH gcat–1 h–1 at 250 °C and 3 MPa for the catalyst containing 6% Ni.
At low Ni concentrations, a significant amount of Ni is in the atomic or clustered form, significantly lowering the activation energy of the H2 dissociative adsorption. Cannizzaro et al.[190] showed that atomically dispersed Ni produced a large amount of CO, and high methanol selectivity along with high concentration of oxygen vacancies was provided by Ni8/In2O3(111) clusters. According to the data of Frei et al.[191], some of the Ni atoms forms InNi3 phase layers on the oxide surface, which are not able to activate CO2, but are able to provide the H2 activation. As with other promoters, the activity of Ni-promoted catalysts can be significantly improved by the use of In2O3 – ZrO2 as a support and by increasing the number of oxygen vacancies[192]. The conversion at 300 °C and 5 MPa was 17.9% with methanol formation activity of 0.63 gMeOH gcat–1 h–1.
All reactions carried out over Cu/support catalysts or Zr, Ti, Ce and In oxides catalysts occur at high temperatures (200 – 250 °C, sometimes above 300 °C) and pressures (5 – 10 MPa)[28, 159, 193, 194]. The intensive development of methods to carry out reactions under milder conditions, as well as the search for new catalysts are therefore ongoing[195].
According to the results of theoretical modelling, catalysts with single Mo atoms on nitrogen-doped graphite can be highly active in the low-temperature CO2 reduction[196]. Theoretical studies suggest that ‘magic’ Cu3 and Cu7 clusters deposited on the surface of MoS2/Ag(111) support should also be highly active in the CO2 hydrogenation to methanol[197]. Hu et al.[61] showed that the use of nanolayer MoS2 with a large number of vacancies on the S atoms as a catalyst promoted the decomposition of CO2 into CO and O2 already at room temperature and allowed the temperature of methanol production to be reduced down to 180 °C (CO2 conversion was 19.5%), with methanol selectivity being very high (94.3%). Lowering the temperature helped to avoid further hydrogenation to methane. The proposed catalyst showed high stability and can operate for 3000 h without deactivation.
Xiao et al.[198] suggested a significant improvement in the CO2 reduction process. It was found that the use of pyroelectric material based on perovskite bismuth tungstate nanoflakes boosted the CO2 conversion to methanol by factors of 25 and 10 as compared to conventional Bi2WO6 and TaO2/zeolite as well as Ag/TiO2 systems, respectively[199]. The reduction can be carried out at temperatures between 15 and 70 °C with the methanol yield of 55.0 μmol g–1 after 20 cycles[198]. Another example of a low-temperature catalyst for the hydrogenation of CO2 to methanol is the Pd/Mo system obtained by the reaction of oxide precursor with NH3. This catalyst showed high stability with methanol turnover number (TON) of 0.15 h–1 at 0.9 MPa and 25 °C[200]. For the low-temperature synthesis of methanol from CO2, the approach comprising supporting Cu on rare earth (La, Ce, Y) hydrides may be promising: such catalysts allow the synthesis of methanol from syngas already at 100 – 140 °C[201].
An interesting example is the use of layered MXene (Ti3C2Tx) material decorated with bimetallic Pd50Ru50 particles in CO2 hydrogenation. Hydrogenation of CO2 with hydrogen released in situ during NaBH4 hydrolysis in ethylene glycol produces methanol with the selectivity of 78% and in 76% yield (PCO2 = 1 MPa; 150 °C, 12 h)[202]. Using ReOx/TiO2 catalyst and performing the reaction in supercritical CO2 allows the temperature to be reduced down to 200 °C with CO2 conversion of 20% and methanol selectivity >98%. High pressure and high mass transfer rates favour both catalyst activity and selectivity[203].
The so-called indirect CO2 hydrogenation on heterogeneous catalysts under liquid phase conditions in alcohols, when the reaction proceeds via corresponding carbonate or carbamate, also leads to decrease in temperature[204]. Hence, it is possible to integrate both the process of CO2 extraction by the capture solvent and the hydrogenation to methanol in this solvent[205-207]. In this case, the hydrogenation occurs on the conventional heterogeneous catalysts, mainly Cu/Zn, under liquid phase conditions using alcohols (ethanol, butanol-2, etc.) and amine additives[208-212]. The formation of methanol proceeds via formate ester of the corresponding alcohol as intermediate, making the conditions of the target product formation significantly milder[213]. Therefore, hydrogenation can be carried out under mild conditions in the presence of an amine–alcohol mixture, where the reaction involves the formation of corresponding formate on conventional Cu/Zn catalyst at 6 MPa and 170 °C. The ethanol – triethylamine and chitosan – diethylene glycol systems were found to be the most effective[212].
In conclusion, it should be noted that Cu/Zn catalysts supported on Al2O3 are being used in industry in the pilot projects for methanol production from CO2. At the same time, the catalysts and approaches proposed in many recent papers are undoubtedly of interest to industry. The resulting systems are often superior to conventional Cu/Zn catalysts, making them attractive and competitive with lower cost and increased stability.
The efficiency of thermocatalysis can be improved by reducing the process temperature and developing systems with high selectivity for the target product, methanol, at low CO selectivity. It is necessary to increase the catalyst stability towards water, which is released in significant amounts; to suppress the side reactions of reverse water-gas shift and methanol vapour-phase conversion. Particularly interesting are the results obtained with oxides capable of forming oxygen vacancies; mainly it is In2O3. Modification of such oxides with noble metals, and also Zn, Cu, etc., allows to increase the selectivity and the reaction rate. The use of special synthetic approaches, in particular oxidative pyrolysis or decomposition of metal-organic frameworks, allows to provide the maximum mutual dispersion of the components and increase the efficiency of the resulting systems. Research into the development of stable single-centre catalytic systems using different types of support is also important. Finally, the coupling of CO2 capture processes with its conversion to methanol is very promising.
2.2. Homogeneous catalysis in the hydrogenation of CO2 to methanol
The hydrogenation process starts with CO2 capture, usually using amine or alcohol in the presence of a base, followed by the cascade reaction to produce methanol. Ru complexes proved to be the most efficient catalysts for this reaction, so most authors have pre-tested different Ru complexes and selected the most active one, sometimes using several different catalysts with different metals or ligands for each stage.
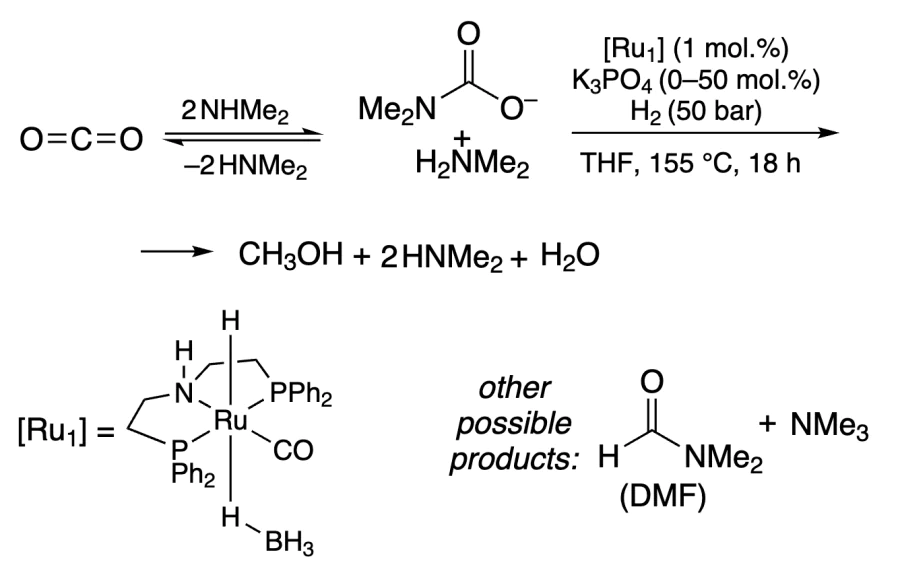
The amine variant was first carried out by Sanford and co-workers[214] using RuPNP complexes ([Ru1]) and NHMe2 as an additive (Scheme 1). It was found that dimethyl formamide and dimethylammonium formate are formed as intermediate products. At 96% conversion, the methanol yield was 27%.
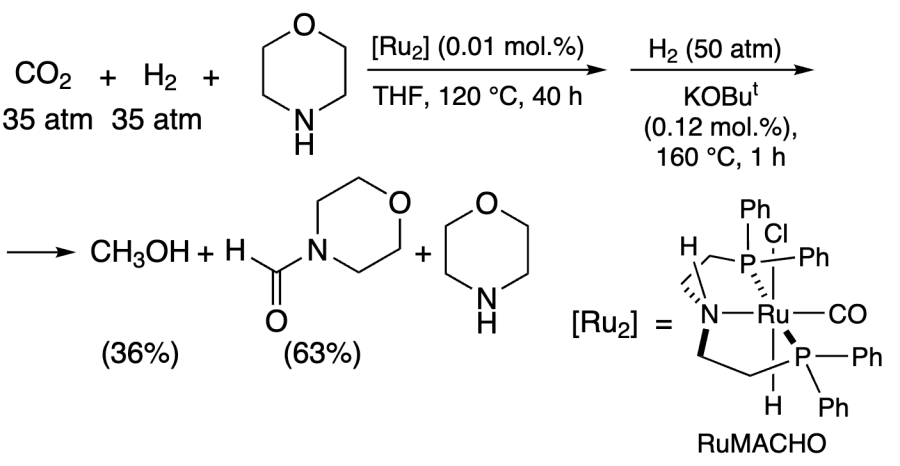
Ding and co-workers[215] improved the yield of methanol (up to 36%) and TON using morpholine and very low concentration of RuMACHO catalyst [Ru2] (Scheme 2). The reaction was shown to proceed via formylmorpholine formation. The maximum TON values (599 000) were achieved due to the high selectivity of the reaction at very low concentration of the catalyst.
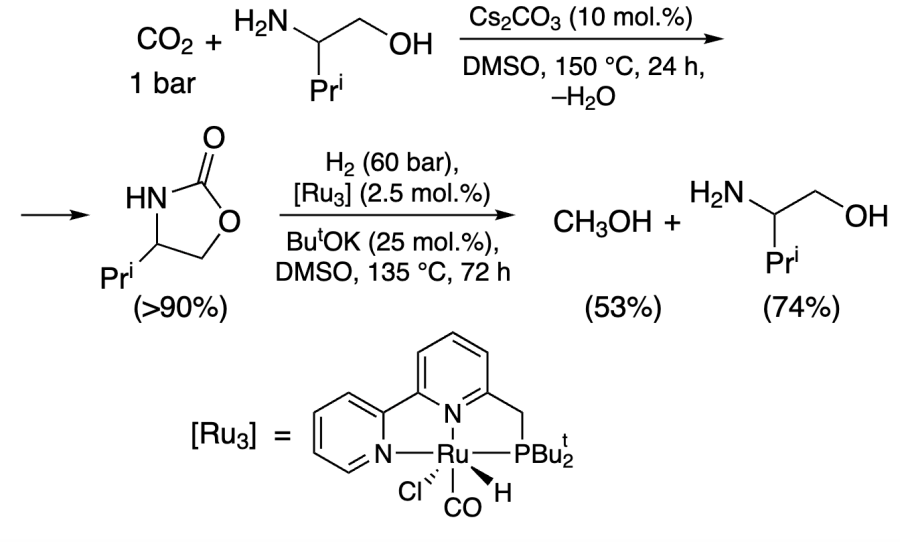
Milstein and co-workers[216] used ruthenium pincer complex RuNNP [Ru3] and valinol as an additive (Scheme 3) and carried out the reactions under atmospheric CO2 pressure, since the first step of oxazolidinone formation required only the presence of Cs2CO3 as base, not the presence of catalyst and pressure. However, Ru-catalyzed reduction of H2 proceeded at high temperature and pressure. The methanol yield was 53%[217].
Prakash and co-workers[218] obtained excellent results using various polyamines as additives (Scheme 4). The best of polyamines was pentaethylenehexamine (PEHA), which was able to capture CO2 from the atmosphere at a very low concentration (400 ppm CO2). In the presence of [Ru1] complex as catalyst and triglyme as a solvent, the yield achieved 79%. When the reaction is carried out in 2-MeTHF/H2O biphasic system, the catalyst can be recycled. After five cycles the methanol yield was 75%[219]. Immobilization of an amine on a solid support in a homogeneous CO2 hydrogenation process allowed the amine to be recycled and each step to be studied separately[220]. Under these conditions, the catalyst showed high activity and maintained it even after 10 days of operation, with TON reaching 9900, which is superior to previous results[221]. At the same time, Everett and Wass[222] carried out the hydrogenation of CO2 to methanol (1 MPa CO2 and 3 MPa H2) at 180 °C using a simpler, non-pincer Ru catalyst precursor and obtained high TON values of 8900 (per catalyst) with a catalyst turnover frequency (TOF) of 4500 h–1 (Scheme 5).
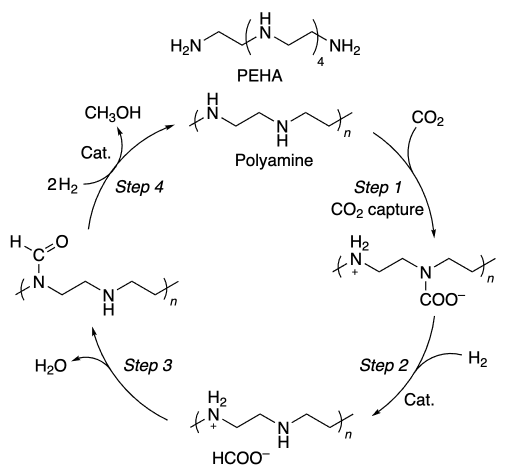
The reaction was shown to proceed via the formamide formation. The authors found that the reaction was possible only when an amine with the N – H bond was used; otherwise it stopped at the formamide formation step. Such bond may be present in the ligand of the Ru complex: in this case, metal-ligand interaction is observed.
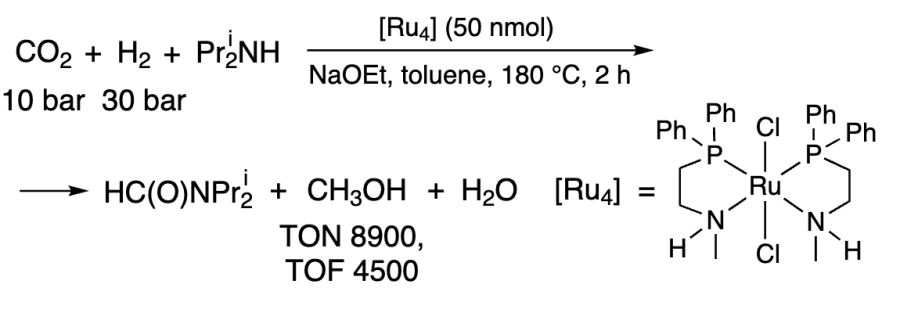
Another approach to methanol production using Ru catalysts is the use of alcohol as an additive instead of amine. This approach was pioneered by Huff and Sanford in 2011[223]. RuCl2(PN)2 can be used to produce HCOOH. A cascade process producing formic acid, its ester and then methanol as intermediates has been carried out using [Ru5], [Sc] and [Ru6] catalysts (Scheme 6).
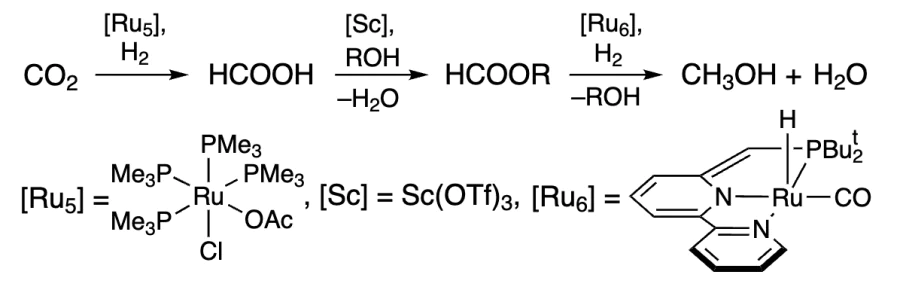
A similar approach combining different catalysts, each for a specific step to increase selectivity, was later applied by Goldberg and co-workers[224] (Scheme 7). It should be noted that the reaction was carried out in the presence of ethanol under acidic conditions at relatively low pressure and at 155 °C. Using the Ru complex [Ru7], CO2 was converted to HCOOH, the Sc catalyst gave ethyl formate, and over the Ir catalyst, CO was captured from formate and hydrogenated to methanol.
Rayder et al.[225] found that the use of multi-component catalytic system including ruthenium RuPNP [Ru8] pincer complex encapsulated in MOF (UiO-66) in the presence of alcohol led to the production of ethyl formate via the formic acid formation (Scheme 8). The hydrogenation of formate to methanol was carried out using the [Ru3] complex in MOF. Methanol was formed at 70 °C within 10 h, using ethanol regeneration. The heterogeneous catalyst was easily recycled, and TON reached 21 000 after 5 cycles.
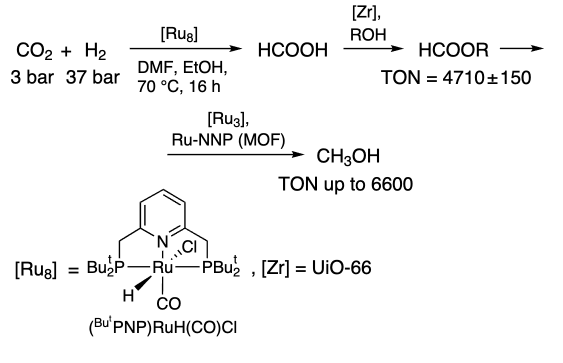
In their follow-up study[226], the authors used a catalyst encapsulated in MOF modified with different functional groups. The best results were observed for UiO-66-NH3+: the maximum TON values up to 19000 (TOF up to 9100 h–1) in 1 cycle and cumulative value of 100000 after 10 cycles were obtained. The authors showed that the acceleration was achieved at the step of CO2 to HCOOH conversion (DMF, 70 °C, RuPNP 2.2 × 10–7 mmol, RuNNP 2.2 × 10–7 mmol).
Leitner and co-workers[227] used ruthenium complexes [Ru9] and [Ru10] bearing another type of ligand, triphos (1,1,1-tris(diphenylphosphinomethyl)ethane), for the hydrogenation of CO2 to methanol in the presence of alcohol under acidic conditions (HNTf2, Tf is triflyl). The possibility of using Zn(NTf2)2 instead of acid was shown. The initial [Ru9] complex was converted to cationic [Ru10] complex capable of providing hydride transfer and protonolysis (5 MPa H2, 140 °C, TON 221). Without the addition of alcohol, methanol was formed in the 2 Me – THF/H2O biphasic system[228].
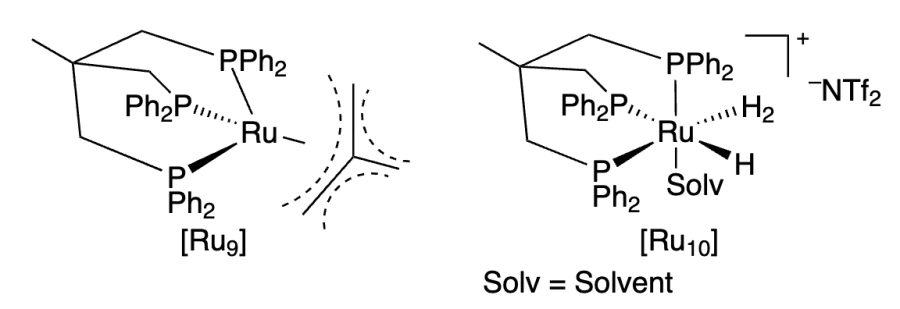
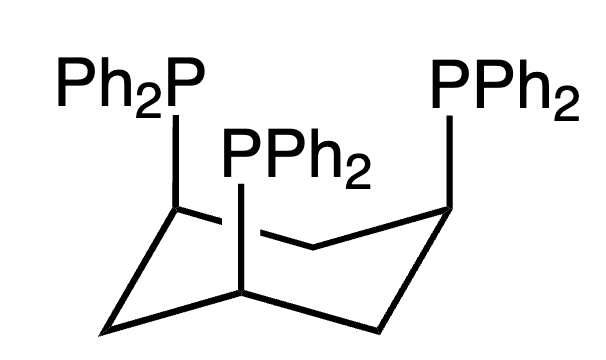
Ru catalyst proved to be more active with other tridentate ligands such as tdppcy (cis,cis-1,3,5tris(diphenylphosphino)cyclohexane). While the TON was 500 when using the complex with the triphos ligand, it was as high as 2000 in this case[229]. Alkali metal alkoxides can be added to the alcohol solution for CO2 capture instead of amines[230].
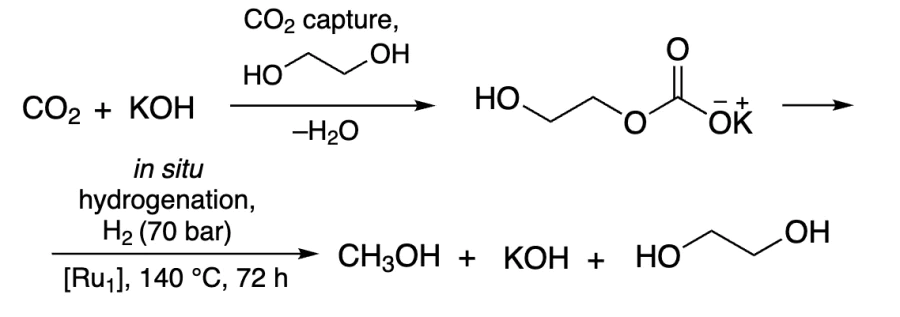
Prakash and co-workers[231] showed that when ethylene glycol and alkali were used instead of ethanol, carbonate was formed. The latter was further hydrogenated to methanol in a quantitative yield in the presence of Ru catalyst and H2, whereas ethylene glycol and alkali were released in the reaction (Scheme 9). The best results were reached by the addition of ethylene glycol salt with tertiary amine in the presence of [Ru1] catalyst (0.5 mol.%)[232]. Using a gas mixture with 10% CO2, the methanol yield was 94%.
Interesting results were obtained when studying the catalytic activity of other non-precious metals in the CO2 hydrogenation. However their efficacy, even with positive results, was still far from that obtained for Ru catalysts. Thus, Beller and co-workers[233] carried out the hydrogenation using Co(acac)3/triphos catalyst in the presence of HNTf2. TON was 50 at 100 °C, 7 MPa H2 and 2 MPa CO2, but increased up to 125 when the phenyl group in the triphos ligand was replaced by a p-tolyl group. In a follow-up study the authors showed that the Lewis acid Co(NTf2)2 could be used instead of HNTf2[234]. Prakash and co-workers[235] employed cheaper Mn and Fe instead of Ru (Scheme 10). Pincer Mn complex in the presence of morpholine or benzylamine easily produced formyl derivative of amine in the first step, but the yield of alcohol was unsatisfactory.
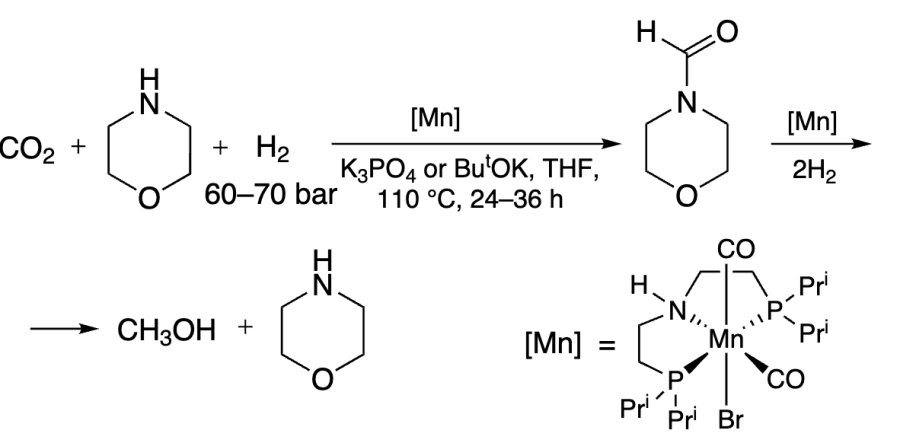
A similar reaction catalyzed by Fe complex was more successful, giving TONs as high as 1160 in the first step and 590 in the second step (Scheme 11)[236].
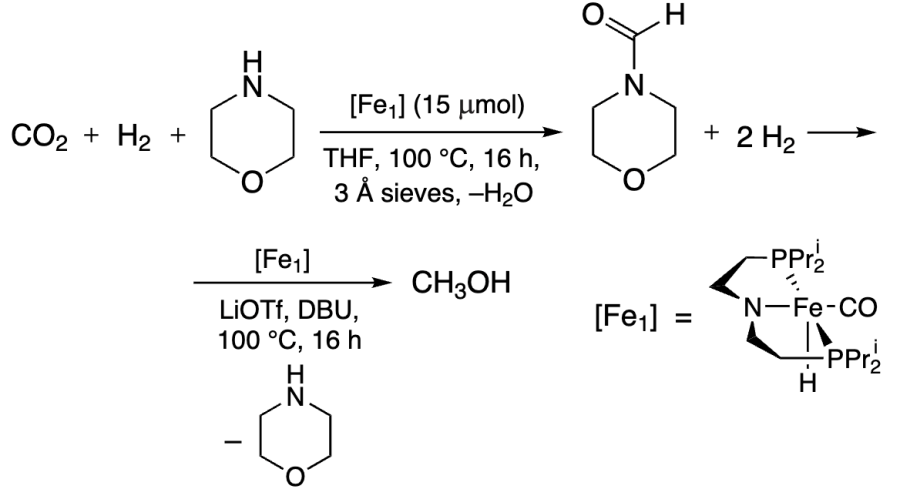
The presence of CO2 was believed to inhibit the second step, the Fe-catalyzed hydrogenation of formamide[236]. The reaction catalyzed by tripodal scorpionate Fe(II) complexes in the presence of PEHA proceeded much better (Scheme 12). The authors suggested that the pyrazole nitrogen atoms contributed to H2 activation and proton transfer. The methanol yield was 45%, and TON reached 2300[237].
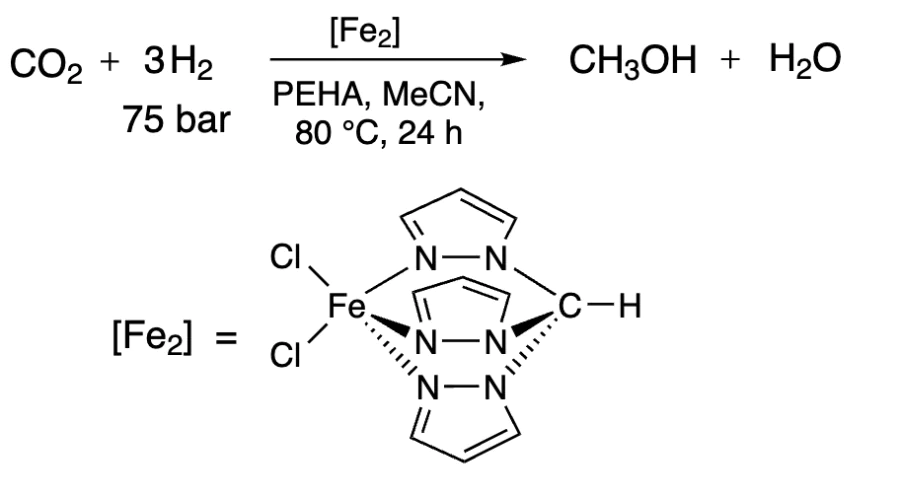
The hydrogenation of CO2 can be carried out not only with molecular H2, but also with hydride ion donors, and the latter suggest much milder reaction conditions[238]. Mn and Re as well as Co and Ni complexes performed well in the hydrogenation with silanes as hydrogen donors (hydrosilylation reaction)[239-241], whereas Pd and Ni complexes showed good results with boranes[242, 243]. However the products yields depended not only on the catalyst and reaction conditions, but also on the nature of the silane or borane. For instance, the hydrosilylation of CO2 with RSiH3 using Co pincer catalyst produced a mixture of products[239]. At the same time, the reaction of CO2 with Ph2SiH2 catalyzed by Ni pincer complex at room temperature provided a 91% yield of methanol after hydrolysis (Scheme 13)[240].
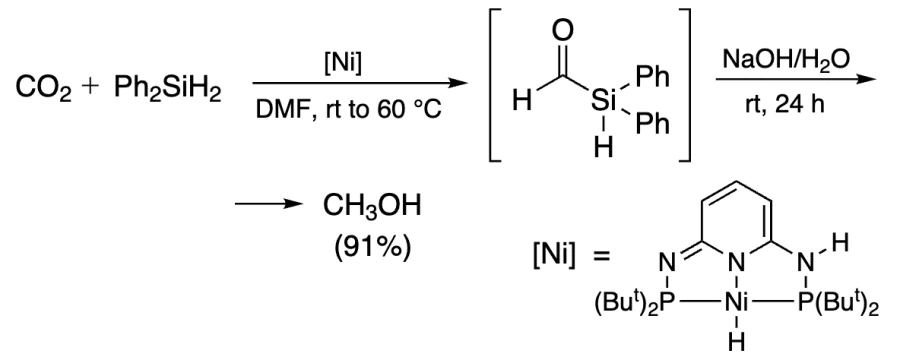
Catalysis with Re complex using Me2PhSiH afforded silyl formate, which was further hydrosilylated by PhSiH3 to methoxysilane in 53% yield (Scheme 14)[244].
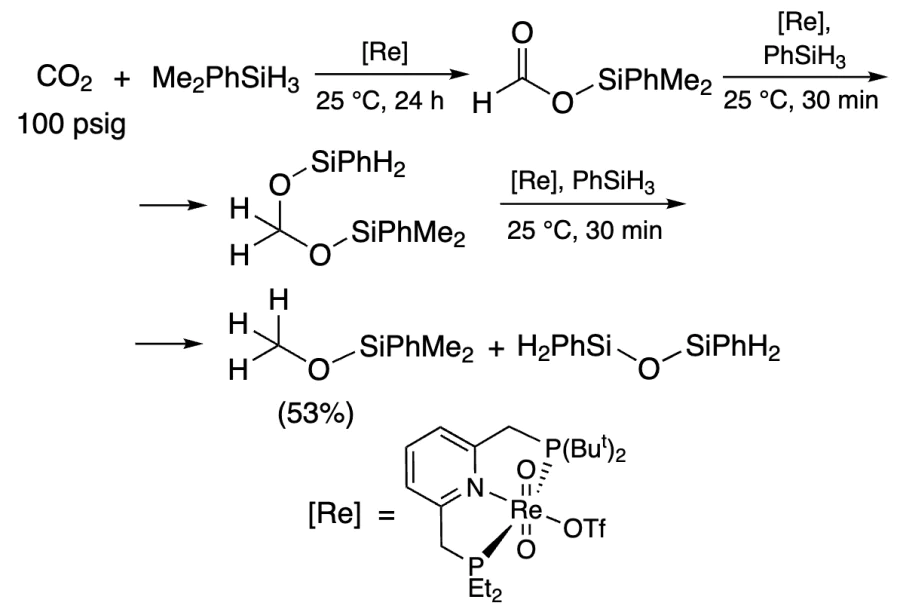
In the presence of Mn complex, methoxysilane can be obtained in 93% yield after 46 h at room temperature, but at 80 °C, the product is formed in an almost quantitative yield already within 6 h (Scheme 15)[241]. In all above cases, the active catalyst is a metal hydride. The reaction proceeds via cis-[Mn(PNPNH – Pri)(CO)2{OC(O)H}] formate, which is further reduced in three steps to methoxysilane, and then hydrolyzed to methanol (Figure 7).
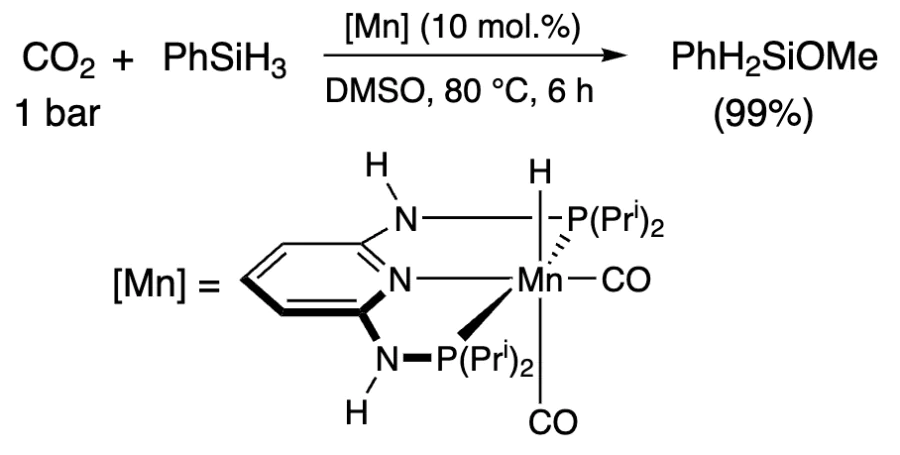
![[{"id":"ECmAkSrAAR","type":"paragraph","data":{"text":"Catalytic cycle for the reduction of CO<sub>2</sub> with phenylsilane. (Adapted from Ref. [[ type=\"anchor\" referenceId=\"6287\" ]], Copyright 2018, American Chemical Society)."}}]](/storage/images/resized/WhoxMUkG0zqFGnYZKnh1O1lb8rK3DD5sfAahQaLe_xl.webp)
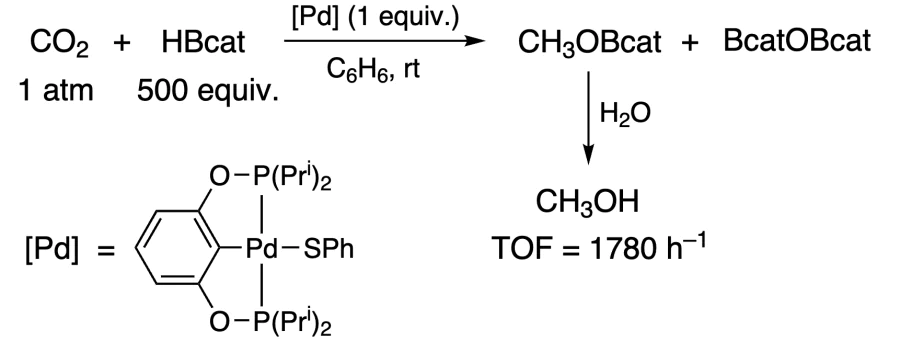
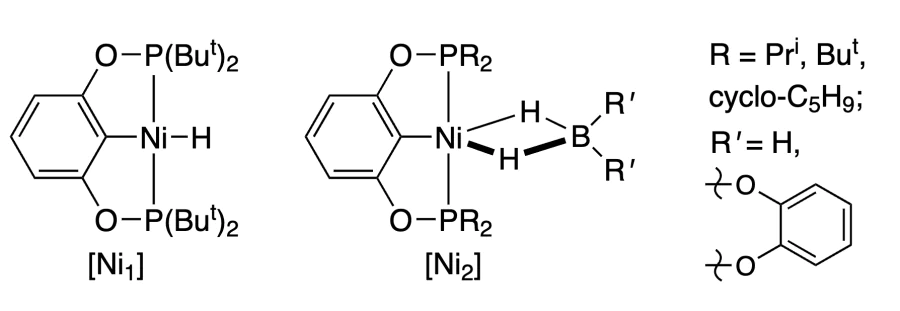
The reduction of CO2 with boranes (hydroboration of CO2 to methanol) in the presence of Pd (Ref. [242]) and Ni complexes has been described[243, 245, 246]. The reaction catalyzed by a pincer Pd complex takes place under the action of catecholborane at 0.1 MPa and room temperature (Scheme 16). Ni complex under the same conditions gives a lower TOF = 495 h–1 (Ref. [245]), but changing the substituent in phosphine to a more bulky group (complex [Ni1], R = But, cyclohexyl) increases the catalytic activity[246]. The use of the borohydride complex [Ni2] at 60 °C further improves the reaction outcome[243].
It should be noted that although reactions with silanes and boranes take place under mild conditions and give good yields, they are far from industrial use due to the high cost of such processes.
3. Electrocatalytic processes
The number of papers on electrocatalytic methanol production has increased rapidly in the last 5 years[247-262]. Such processes using ‘green’ electricity can eliminate the use of fossil fuels and reduce the carbon footprint of methanol produced[263]. While conventional methanol production from syngas emits between 0.5 and 2.6 tonnes of CO2 per tonne of methanol, depending on the feedstock (gas or coal), with no H2O emitted[264], the use of wind and solar energy in CO2 hydrogenation can reduce this to negative values. Electrocatalysis does not require the use of oxidizing or reducing reagents, making the reaction more environmentally friendly and ‘green’. The processes can be carried out at atmospheric pressure and at moderate temperatures. It is essential that such reactions can be controlled by both the electrical voltage applied and the strength of the electrical current, using different designs of electrochemical cells, the upgrade of which should significantly improve the process economy[247, 265-267]. The equipment is easy to handle and does not take up much space. Recent advances in the catalysis and technical validation of CO2 reduction, including to methanol, are described in many reviews[247-263], highlighting both advantages and disadvantages of the process. In general, the available data demonstrate the complexity of the processes involved in the multistep electrochemical CO2 reduction and challenges in achieving high methanol selectivity[268].
In the process of electrocatalysis, there is a change in the rate and selectivity of the electrochemical CO2 reduction due to the catalytic action of electrodes on the surface of which these reactions take place. The effect of the electrode material and its surface modification on the electroreduction process allows us to qualify electrode materials as electrocatalysts. It is not always possible to compare efficiencies of electrocatalysts and to evaluate their advantages over other methods due to the specificity of the indicators used and different reaction conditions. New parameters such as current and voltage are added, influencing the efficiency of electrocatalysts. It should be noted that since the process is affected by a large number of parameters, it is reasonable to compare electrochemical systems, taking into account parameters such as electrode material as catalyst, electrolyte composition, solute concentrations, membrane composition, and reaction conditions.
When comparing electrode materials as catalysts for the electrochemical reduction of CO2, several important parameters should be considered, including Faradaic efficiency, overpotential and current density. Faradaic efficiency is an analogue of selectivity for traditional catalytic processes and indicates the amount of electricity used to produce a given product, where z is the number of electrons to form the product, n is the number of moles of product obtained, F is the Faraday number (96.485 C mol–1), and Q is the total amount of charge used in the reduction process (Eq. (3)). This parameter shows how much of the electrical charge went into the target reaction to produce the product. To convert Faradaic efficiency into the traditional mole percent selectivity of chemistry, the difference in the number of electrons required for specific reactions must be taken into account. The closer is the Faradaic efficiency to 100%, the more selective the process and the more selective the catalyst[269].
The second indicator is overpotential, which defines the deviation of the electrode potential, at which the reaction proceeds, from the thermodynamic equilibrium potential. Overpotential characterizes the energy required for the process to proceed at a significant rate[270]. The higher it is, the higher is the activation energy of the process and the more energy is required for it to proceed at a higher rate. Typically, the multi-electron conversion processes of adsorbed CO2, including to methanol, require significant overpotentials, the value of which is largely determined by the properties of the catalyst[269]. Low overpotentials are typical of catalysts for the conversion of CO2 to CO or NCOOH via two-electron processes occurring along a single pathway. For multi-electron processes, the overpotential is significantly higher. Therefore, methanol is usually formed at high overpotentials and low current densities, indicating that the catalyst performance is not very high[271, 272].
In addition to the overpotential value, a third parameter, the current density at a given potential, should be taken into account when evaluating the catalytic activity. This parameter makes it possible to estimate the amount of electricity required for electroreduction per unit of active surface area of the catalyst. The overpotential value at a given current density characterizes the catalyst activity: the lower the overpotential at the same current density, the lower the activation barrier and the higher is the catalyst activity. In general, for the same catalyst, overpotential increases with increasing current density. The faster the overpotential increases, the less effective the catalyst will be. In addition, the lower the overpotential at a given current density, the more energy-efficient the process and the less energy is required to produce the same amount of methanol. Current density is an important parameter in estimating catalyst performance: the higher the current density at a given voltage, the more product is formed per unit area of the electrode.
The rate of methanol formation can therefore be defined as the number of moles of H2 formed per unit area of the electrode per unit time at a given current density and voltage. For the appropriate catalyst comparison, it is important to estimate the electrode electrochemically active area, which can differ significantly from its geometric area; these values are not always available. A catalyst with larger surface area undergoes greater concentration polarization and therefore may have higher overpotential for the formally similar electrode surface area. In general, an efficient catalyst should provide higher current density, higher Faradaic efficiency and lower overpotential, all other factors being equal. In this case, the electrochemical process requires less investment and is more economical[258].
At present, estimation of the economic efficiency of the process of CO2 electroreduction to methanol shows that its potential price is at least twice the market price, and that the process itself needs further improvement, which can be achieved by selecting efficient and long-lived catalysts and by the use of new cell designs[273]. The latter have a significant impact on the process parameters. For example, efficiency can be improved by using flow or gas diffusion cells[247, 266, 267].
To make the process of methanol production economically feasible, it is necessary to achieve a Faradaic efficiency of 70 – 80% at a current density of at least 300 mA cm–2 and overpotential of 0.5 V and below[258, 270, 274-276]. At present, the efforts of researchers are largely focused on the search for new catalytic systems to achieve these parameters. There is a number of problems which need to be solved by developing and applying new approaches to catalyst design.
As for electroreduction, electron transfer takes place to some extent over the entire surface of the electrode and the main stage is the primary adsorption of CO2 followed by its reduction and conversion to other products. Each reduction step is accompanied by electron and proton transfers (Table 2). The reduction of CO2 to methanol requires 6 electrons, which makes the process multi-step and kinetically unfavourable[277, 278].
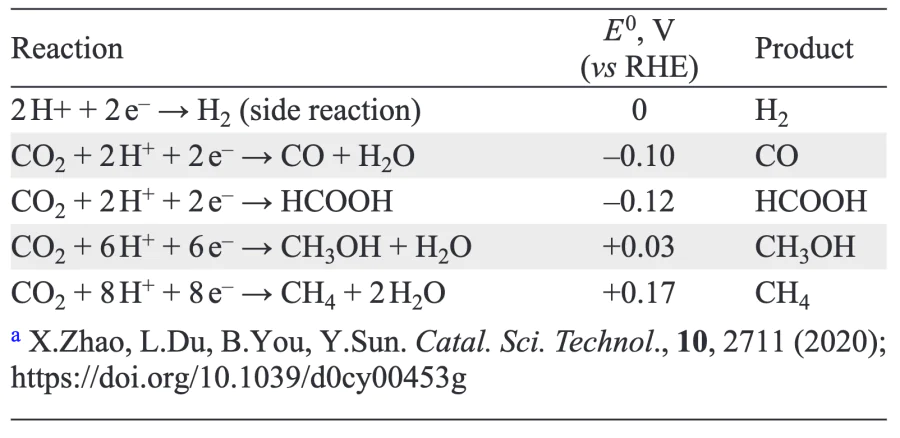
The first step involves electron transfer from the cathode surface to CO2 as it binds to the catalyst[272]. The resulting negatively charged CO2– ion is bound to the metal centre (Figure 8). This reaction is characterized by very low standard potential (–1.9 V vs normal hydrogen electrode), and a significant potential is required to hold the CO2 species on the catalyst surface. Therefore, the electrocatalyst should not only accelerate the above reaction, but also facilitate this process by reducing the standard electrode potential through the interaction with the formed species.
![[{"id":"F6m2qaqdoW","type":"paragraph","data":{"text":"Formation of formic acid and bound CO through different pathways of the CO<sub>2</sub> electroreduction."}}]](/storage/images/resized/eGH9DnDSSCVlxN7hpbrPLJ8yGxZsDdtUOLjE4RME_xl.webp)
The presence of an electric field and charged surface significantly increases the rate of electron transfer to CO2 and the reduction of the latter[279, 280]. In most cases, it is assumed that the reduction to methanol involves CO2 dissolved in H2O in the vicinity of the electrode, where CO2 participates in a series of equilibria (CO2/HCO3–, H2CO3/HCO3– and HCO3–/CO32–). As a result, the CO2 concentration depends significantly on the pH, the composition of the solution and its buffer capacity. In addition, this fact does not always allow a correct comparison of the results obtained with differences in these parameters, even under otherwise similar conditions[281-283].
The subsequent proton transfer to carbon or oxygen atom suggests two possible reaction pathways (see Figure 8). It should be noted that, unlike thermocatalysis, the formation of methanol by the formate pathway does not usually occur under electrocatalytic conditions: the formation of *C(H)O – H by hydrogenation of the HCOO* intermediate in aqueous medium is rare (Figure 8a), and this pathway mainly affords HCOOH[284, 285].
The formation of methanol from CO2 in the electrochemical reduction generally proceeds via CO adsorbed on the electrode surface (see Figs 8 b and 9) and resulting from the conversion of *C(O)OH species. After that, methanol is formed via surface-bound formaldehyde and then methoxyl moiety[269, 286-288]. Using the example of catalysts based on Cu nanoparticles, it was shown that the methanol formation does not involve formaldehyde as an intermediate, and it is most likely that the reaction proceeds via *СН2ОН[289]. As for the formation of methane, the key intermediate appears to be *CHOH. The addition of proton to the carbon atom combined with one-electron reduction gives methanol, and the elimination of OH group affords methane. Obviously, the oxophilicity of the surface (ability to bind adsorbed oxygen) is of fundamental importance. The higher the oxophilicity, the greater is the probability of oxygen being split to form methane precursors (see Figure 9)[290]. Thus, due to the high oxophilicity of the copper surface (oxygen binding energies on Cu(111) and Cu(100) facets are −4.14 and −4.71 eV, respectively)[291], the amounts of methane formed are significant[292, 293]. For metals with low oxophilicity (e.g., Au), little or no methane is formed, and for one of the most oxophilic metals (Fe), methane is the main reaction product[290].
![[{"id":"-OXLzUNbO_","type":"paragraph","data":{"text":"Mechanism of hydrogenation of the surface-bound CO to methanol."}}]](/storage/images/resized/b4K8cLcMMIxlKQYjrUCqlQpaPwEEyuGhK6CZ1C3m_xl.webp)
In all cases, the hydrogen source is formally H+, which, depending on the pH, is derived from Н3О+, H2O or other species in the aqueous solution (e.g., hydrocarbonate HCO3–, protonated buffer components such as HPO3– and HPO32–) or even from the catalyst surface. At the same time, O2 formation with H+ release occurs at the anode and the general reaction equation is as follows (reaction (4)). The oxidation of H2O to O2 requires additional energy (the difference in the standard electrode potential is slightly more than 1 V, in real systems it is much higher due to the overpotential at O2 release).
In contrast to thermal reduction to CH3OH in the gas phase, where the process selectivity is largely determined by the simultaneous interaction of the oxygen from CO2 with one of the catalyst components (usually the oxide phase) and the carbon atom with the metal at the interface, in electroreduction, the binding to the electrode surface occurs usually only via the carbon atom. As a result, dimerization of bound CO, intermediate oxygen-containing species and methylene moieties can proceed relatively easily, and catalysts allowing the production of C2 products along with C1 compounds are quite common[294-296]. Additional stabilization of intermediates by adsorbed cations can affect the catalyst selectivity[279]. For example, for CO hydrogenation to –CHO on Cu electrode, cation stabilization is provided at 0.2 eV, and for CO dimerization at 0.7 eV, making the latter reaction more favourable[297].
In addition to the formation of a large number of by-products, another competing reaction is the reduction of H2O to molecular H2. Water per se reacts as a source of protons to produce methanol. The standard electrode potential for the methanol formation is very close to that for the release of H2 during the H2O reduction, and the formation of H2 is exactly the reaction to be avoided. CO2 reduction is usually carried out under alkaline or near-neutral conditions (carbonate and bicarbonate buffer solutions)[298], which facilitates its one-electron conversion to CO2– with less overpotential and minimizes the competitive H2 release[299]. At the same time, in alkaline media, the process is complicated by the formation of bicarbonates (H2O molecule is the H2 donor; the proton abstraction from H2O gives additional OH–, and results in further CO2 binding to bicarbonate). This significantly reduces the efficiency of CO2 reduction process and leads to a number of technological issues[300]. When reducing CO2 in H2O in the neutral and acidic media, it is usually not possible to completely avoid the H2 evolution due to the low solubility of CO2 at atmospheric pressure (0.033 M). In this case, it is possible to use CO2 at elevated pressures: the solubility of this gas increases up to 0.95 M at 4 MPa, along with a slight decrease in pH[301]. It may be of particular interest to carry out the reaction at high pressures in ionic liquids, which are not only electrolytes but can also significantly affect the process efficiency[302].
As for thermocatalytic processes, the most studied catalysts for the electrochemical CO2 reduction to methanol are Cu-containing catalysts, characterized by high activity in this reaction[279, 303]. For other metals such as In, Sn, Hg, Bi, Cd, Tl and Pb, CO2 conversion to formate is more typical, whereas Au, Ag, Zn, Pd and Ga usually catalyze the reduction of CO2 to CO[271, 304]. Methanol production on heterogeneous systems based on these metals is an exception and requires special catalyst modification[251, 261]. The same is also true for metal-free catalysts, since carbonaceous systems usually catalyze the formation of CO, formate anion, C2 products such as ethanol, ethylene, etc[305].
The main challenges in carrying out the reaction on Cu-containing catalysts are associated with a variety of side processes promoted by Cu compounds, ranging from formate and CO (usually the main by-product of the electrochemical CO2 reduction) and, to a lesser extent, methane, to products containing two or more carbon atoms[306, 307]. The latter can prevail. This behaviour of Cu is due to the fact that it is this metal that preferentially binds CO rather than H atoms on its surface[252, 259]. This inhibits the evolution of H2 and free CO. The binding energy of the latter to the surface is sufficient to prevent CO elimination, but not so high as to prevent its subsequent conversion[308]. This leads not only to a two-electron reduction of CO2 to CO, but also to a multi-electron reduction of the latter to the necessary valuable products, of which methanol is often not the main one[269]. In most cases, both CO, methanol and methane, as well as C2 compounds, are formed. As a consequence, high Faradaic efficiency for methanol is not usually achieved[254].
Given the large number of reaction pathways described, even small changes in the structure, morphology and composition of the catalyst can significantly affect not only the activity but also the selectivity to the particular product. For example, using Cu(core)/CuO(shell) catalyst, the Faradaic efficiency for methanol is 2.5% and CO and HCOOH are the main products[309]. Catalysts containing Ag – Cu alloys produce methane as the main product[310-312]. Ethylene formation is characteristic of Cu-containing catalysts doped with Ni and Pd[310-312]. The formation of nanograin agglomerates with disordered ‘defective’ boundaries during the reduction has been shown to produce C2 products[313]. Dimerization is also promoted by surface hydration: the formation of hydrogen bonds between hydroxyl groups of H2O adsorbed on the electrode surface and adjacent intermediate oxygen-containing species promotes the approach of the latter[314]. Dimerization due to the approach of oxygen-containing charged species may also be facilitated by the presence of metal cations on the surface[315]. In addition, given the values of the equilibrium potentials, the reduction of H2O with release of H2 remains competitive to the formation of methanol for Cu, especially at low pH.
Moreover, Cu systems are characterized by relatively fast catalyst deactivation during the process[316, 317]. This occurs both by poisoning due to deposition of traces of other elements (Fe, Zn, Pb, Si, Pt) and by reduction of Cu oxides on the surface. Poisoning may also occur from precipitation of impurities from electrolyte or CO2 stream, impurities carried over from the anode or reference electrode, or from particles strongly adsorbed on the electrode surface due to the formation of high-molecular-weight carbonaceous products (analogues of coke)[318-320].
An increase in methanol selectivity for Cu-containing catalysts is possible by increasing the ability of the electrode surface to adsorb CO and reducing the oxophilicity, which can be achieved using systems containing a Cu foil or copper oxide particles on the Zn surface. For example, it was shown that the deposition of highly dispersed Cu2O on the electrode surface allows to achieve a Faradaic efficiency of 38% in 0.5 M KHCO3 (yield 43 μmol cm–2 h–1)[321]. Methanol formation was promoted by coordination of CO2 via oxygen on the Cu2O (100) facet, where the subsequently formed intermediate HCO* species were isolated from each other and did not dimerize. Adsorption of metallic Cu on the (111) facet occurred via the carbon atom of the CO2 molecule. The surface geometry on this facet favoured the close arrangement of intermediate species to afford C2+ products. As a result, at a potential of ~1 V, alcohol was formed predominantly on the Cu(100) surface, while ethanol and other C2 compounds were produced on the Cu(111) surface. The Faradaic efficiency for methanol decreased sharply from 70% to 10% in a few hours for the Cu – Cu2O catalyst (–1.7 V in 0.1 M KHCO3 at 5.5 mA cm–2), possibly due to the partial reduction of Cu2O on the surface in the presence of Zn[322].
When Cu-containing catalysts are modified with zinc oxide, the result also depends on the method of metal introduction. The Cu/Zn oxide catalyst obtained by co-precipitation of Cu and Zn oxides (content of the latter is 5 – 20%) was selective to ethanol (Faradaic efficiency > 22%), but for methanol, this value did not exceed 10 – 12% at the optimum potential for the reaction (0.6 V)[323]. The use of Cu1−xZnx nano-alloy particles of different compositions even led to the acetone formation, while ZnO-supported Cu nanoparticles favoured the methanol formation[324].
Modification of Cu2O film with ZnO by electrodeposition method significantly increased the Faradaic efficiency for methanol (up to 45% in 1.14 M KHCO3)[325] with 315.656 μmol cm–2 methanol obtained at a formation rate of 52.609 μmol cm–2 h–1. As a result, the proposed synthesis method improved both the rate and the selectivity as compared to the use of electrodes prepared by co-dispersing Cu2O with ZnO[326]. Modification of Zn foil with Cu nanoparticles grown on its surface upon hydrothermal reduction and morphologically representing 3D flowers, provided 48% Faradaic efficiency for methanol at a potential of 1 V and a current density of 0.05 mA cm–2. Using the copper foil support, acetate anion and formic acid were the main products. The surface structure remained almost unchanged for both catalysts[327]. ZnO allows the stabilization of Cu(I) – O – Zn species on which CO2 activation is possible, but the methanol selectivity remains insufficient and other one-carbon products predominate, since the reaction follows a formate pathway[328].
The activity of the Cu2O/ZnO system can be enhanced using soluble co-catalysts based on substituted pyridines (Py). Apparently, the interaction of CO2 with pyridine affords the corresponding carbamate (Py – CO2), which undergoes reduction to form the heterocycle-stabilized CO2– anion in the first step[329]. As a result, the overpotential is significantly reduced and the Faradaic efficiency is increased. The best results were obtained at pH = 5 with the addition of 2-methylpyridine (the maximum rate of methanol formation, r = 4.42 μmol m–2 s–1 and FE = 25.6%)[330]. Furthermore, the presence of pyridine in molar concentrations significantly increased the methanol selectivity using Cu2O-containing catalysts.
The use of silsesquioxanes (phenyl polyhedral oligomeric silsesquioxanes, PPOS) as a support for Cu/ZnO catalyst gave rise to a new material, CuZn-PPOS nanoparticles with a size of 7 – 15 nm, that improved the catalyst performance in the hydrogenation of CO2 to methanol (CO2 conversion was 4.4% and selectivity was 87.5% within 18 h, at 220 °C)[101]. Using Cu/Zn systems as an example, it was found that switching to gas diffusion electrodes increased the process efficiency and produced methanol at a rate of 50 μmol m–2 s–1 with a Faradaic efficiency of 56% (–1.38 V Ag/AgCl)[331].
Examples of improving the efficiency of the electrochemical CO2 reduction to methanol by modifying Cu-containing systems with metals other than Zn are known. Decoration of Cu2O with MoS2 particles increased current density almost twice (up to 113 mA cm–2) as compared to Cu2O and provided a Faradaic efficiency for methanol of 12.3% at –1.3 V[332]. A multicomponent catalyst consisting of Cu nanoparticles, layered graphitic carbon nitride (g-C3N4) and MoS2 was stable for 30 h of the reaction and achieved a Faradaic efficiency for methanol of 19.7% at –1 V[333].
Significantly better results were obtained introducing Ag2S into the Cu2O/Cu catalyst. This provided a Faradaic efficiency of 67.4% for methanol at a relatively high current density (122.7 mA cm–2), a potential of 1.18 V and the use of a solvent mixture consisting of 1-butylmethylimidazolium tetrafluoroborate and H2O[334]. The role of sulfur, according to the authors, was associated with changes in the catalyst electronic structure and morphology leading to an increase in the methanol selectivity, while Ag+ ions inhibited the H2 release.
The methanol selectivity can be increased using Cu alloys as catalyst components. For example, the catalyst obtained by pyrolysis of Ni/Cu metal-organic framework with benzene-1,3,5-tricarboxylic acid (carbon-supported nanoalloy Cu0.85Ni0.15) enabled the reduction of CO2 to methanol with a Faradaic efficiency of ~60% at –0.4 V and a current density of about 0.3 mA cm–2 for at least 24 h. The use of this alloy not only stabilized the intermediate anion radical •СО2– but also reduced the energy required to convert the adsorbed –ОСН3 species to methanol[335].
One of the ways to achieve the maximum selectivity in electrocatalysis is to design heterogeneous catalysts with isolated single-atom active sites, where the catalytic reaction proceeds on a single metal atom interacting only with the support. Such catalysts have demonstrated the unique specific activity and high selectivity in various processes[336, 337]. This approach can also be applied to the electroreduction of CO2[262, 338-340]. Modification by metal atoms can significantly affect the selectivity of Cu catalysts in particular[341].
Thus, the anchoring of isolated Cu atoms on nitrogen-doped carbon nanofibres by coordination with N atoms allows the production of methanol with a Faradaic efficiency of 44% at –0.91 V and maintains the catalyst stability for 50 h. The intermediate CO formed in the reduction process remains bound to single Cu atoms in this material and is further converted into methanol without the formation of C2 products due to the isolation of active sites[342]. Cu atoms supported on carbon nanofibres allowed the methanol production with a Faradaic efficiency of 44% at current density of 93 mA cm–2. The proposed electrospinning method has the potential to produce catalyst material in high yields in a continuous process[342]. Meanwhile, Cu clusters (mainly three-atom) anchored to defective graphene deposited on nanodiamond catalyzed the reduction of CO2 to methane[343]. Monoatomic catalysts obtained by anchoring Cu atoms to nitrogen-containing TiN(111) surface provided the formation of both methanol and methane[344].
The nanocatalyst, which contained atomically dispersed Sn on CuO, and on the surface of which a significant number of oxygen vacancies have been developed by H-plasma treatment, allowed a very high Faradaic efficiency (88.6%) to be obtained at a current density of 67 mA cm–2 and potential of 2.0 V (vs Ag/AgCl) (in the absence of Sn the Faradaic efficiency was 44.6%). The catalyst remained stable for at least 36 h. On the tin surface, CO2 is activated, the activation and dissociation energies of –COOH decrease. As a result, CO is formed, bound to Cu rather than to Sn, and is further hydrogenated to methanol. The optimal combination of atomically dispersed Sn atoms, a high concentration of oxygen vacancies and the presence of CuO are prerequisites to achieve such near to peak result[345].
Another example of the design of single-atom catalysts includes those prepared by etching Al atoms in the MAX phase (so called for layered hexagonal carbides and nitrides, with the general formula Мn+1AXn, where n = 1 – 4, M is a transition metal, A is a group element, X is either carbon or nitrogen) of the Ti3(Al1–xCux)C2 composition to give a bilayer carbide, which is a two-dimensional inorganic compound of the MXenes type. The final carbide contains Ti and Cu atoms, and the surface Clx groups and oxygen atoms bound to Ti atoms (Ti3C2Clx units). Cu atoms in this material are bound to the oxygen of the surface groups, isolated and carry a partial positive charge. According to the authors, the high Faradaic efficiency for methanol (59.1%) when using a carbide catalyst is achieved because the process follows the formate pathway on the isolated Cu centres with a decrease in the energy for conversion of the formate intermediate to formaldehyde[346]. Calculations for Zr-containing catalysts suggest that systems with an isolated Zr atom anchored to graphitic carbon nitride may be promising[347]. Another example of single-centre catalysts includes Ir and Rh atoms immobilized on the surface of Мо2B2. In the resulting catalyst, the platinum metal binds to the CO2 carbon and Mo binds to oxygen, providing high activity in electroreduction at low overpotentials[348]. Finally, single-centre catalysts can be prepared using supported molecular catalysts, such as supported metal complexes.
A small number of examples are known where intermetallic Cu alloys provide high selectivity and high rates of methanol formation[335]. For instance, copper modification with gallium to give the CuGa2 alloy allows the electroreduction of CO2 to methanol with high Faradaic efficiency (77.26%) and extremely low potential (–0.3 V) at current density of 21.4 mA cm–2 in 0.5 М KHCO3[349]. Increasing the potential and current density leads to a decrease in the methanol selectivity and CO formation. The authors explain the high selectivity of the process by involvement of the adsorbed formate intermediate, which is favoured by the presence of small amounts of gallium oxide. Hydrogenation proceeds due to hydrogen spillover from the Cu surface. The current density was increased to 21.4 mA cm–2 while maintaining the Faradaic efficiency for CuGa2 using a flow cell with a gas diffusion electrode[349]. Increased selectivity can also be achieved using platinum-group metal additives. When a Pd – Cu alloy aerogel was used at a current density of 31.8 mA cm–2, the Faradaic efficiency was 80%[350].
As mentioned above, one of the side reactions in the hydrogenation of CO2 to methanol is the formation of methane, which, like the formation of methanol, occurs by the reduction of the surface-bound CO. It was found[351] that this reaction can be suppressed by delocalizing the electron density of active sites containing Cu atoms. As shown by the example of copper cyanamide as a catalyst, such delocalization significantly weakens the Cu–*O – CH3 bond as compared to the O – C bond, thus improving the selectivity: the Faradaic efficiency for methanol reaches 70% at a current density of 92.3 mA cm–2 (r = 0.160 μmol m–2 s–1).
An increase in the methanol selectivity is possible passing to the systems where the reaction follows the formate pathway, and the resulting formate anion remains strongly bound to the catalyst surface and is further hydrogenated to alcohol (Figure 10а). In this case, H2 can be reduced on the surface to a bound hydride anion, which further reacts with CO2 to give formate (see Figure 10b). Thus, MoC deposited on nitrogen-doped carbon nanotubes allowed to achieve methanol selectivity of 80.4% at –1.1 V and a current density of 4 mA cm–2 at CO2 pressure of 4 MPa. According to the authors, the use of such system leads to the formation of methanol via formate anion due to the increased ability of Mo atoms in MoC to interact with oxygen atoms of CO2. At low CO2 pressures, CO becomes the main product together with H2[301].
![[{"id":"65ac5fakve","type":"paragraph","data":{"text":"Methanol formation via the formate pathway without (a) and with (b) the involvement of a hydride anion."}}]](/storage/images/resized/fDDI7WSHKYBX8coCm5EEdL4iG5Fj4RBhLsf2JnLN_xl.webp)
In addition to carbides, other catalytic systems based on metal chalcogenides for methanol reduction can be mentioned, in particular Mo–Bi bimetallic chalcogenides, which provide high Faradaic efficiency in ionic liquids[352]. The latter provide a high CO2 concentration in the electrolyte and its additional stabilization on the surface[353, 354]. The presence of Bi appears to facilitate CO2 activation[355]. The use of a composite based on multi-walled carbon nanotubes and MoBiSx significantly increased the activity and selectivity in the electroreduction of CO2 to methanol. Under optimal conditions (solvent — 40% H2O in [EMIM]BF4, potential — 0.3 V vs standard reference electrode), the Faradaic efficiency was 56.4% and the system was stable at 11.16 mA cm–2 for at least 6 h[356].
Among other chalcogenides, the highest activity and selectivity in methanol formation is expected for Mo and W tellurides[357]. High selectivity was achieved with copper selenide[358]. The Faradaic efficiency for methanol was 77.6% at a current density of 41.5 mA cm–2. It was shown that a significant increase in methanol selectivity was caused by the removal of a part of the Se atoms in nanoporous Cu2−xSe copper selenide to form a surface containing only 5% Se[359]. Finally, unusually high selectivity was observed for boron phosphide: the Faradaic efficiency for methanol was 92% at a current density of 0.2 mA cm–2[360].
Nanocomposites based on graphene modified with nitrogen atoms and ZnO – Ag particles showed high selectivity for methanol formation in hydrocarbonate buffer (Faradaic efficiency of 67.48% at –2.7 V (Ag/AgCl)). The authors constructed a special cell placed in a magnetic field and allowed UV irradiation of the electrode. The reaction rate increased 5-fold by simultaneous UV irradiation (254 nm) in a 0.07 T magnetic field[361]. It is also possible to reduce the rate of CO desorption on Zn by developing nanostructured systems such as Ag-supported Zn dendrites. The interaction with Ag changes the distances between the Zn atoms and favours the stronger binding of CO, which is hydrogenated to methanol[362].
In electrochemical reduction, as in thermocatalytic processes, systems based on In oxide are also active. To achieve high selectivity and to facilitate the formation of oxygen vacancies on the surface under electroreduction conditions, modification of the oxide surface with Ni clusters (Ni8) is necessary[190]. The use of metal compounds, which in the unmodified state usually give other products (CO, HCOOH or CH4) instead of methanol under the electrochemical reduction conditions, requires special modification that changes the electron density at the active sites. Such modification is necessary to increase the binding degree for CO and other intermediates and to reduce the adsorption degree for methoxyl species on the electrode[261, 305]. On the one hand, it is important to provide a high binding degree of the formed CO, providing its further reduction on the surface. On the other hand, the formation of methane precursors, the oxygen-free species, must be prevented by reducing the oxophilicity of the surface.
Thus, Wang et al.[363] showed that Ni-supported (CoO/CN/Ni) catalyst containing CoO deposited on a nitrogen-doped carbon layer provided a Faradaic efficiency for methanol of 70.7% at a current density of 10.6 mA cm–2. The high performance of the catalyst was explained by the electron donation from the Ni carrier to the carbon support and by the change in electronic state of CoO sites, binding CO more strongly.
High methanol selectivity is possible with the use of single-centre catalysts obtained by depositing the Co(II) tetraminophthalocyanine complex onto the electrode[364]. The authors attribute this to the formation of (4[CoII(H4L)]0) species capable of fast reaction with CO2 and subsequent 4-electron reduction to methanol. In this case, CO2 is coordinated to the Co atom. According to calculations, the transition of the phthalocyanine ligand from the dianion state to trianion radicals with the electron transfer to carbon-containing species bound to Co in the process of 4-electron reduction (for both CO2 and CO) significantly facilitates the reduction. The selectivity of such catalyst was higher than that of the system based on phthalocyanine without amino groups: the Faradaic efficiencies were 28 and 19%, respectively[365]. According to theoretical calculations, the use of coordinatively unsaturated metal-organic frameworks containing Fe(II), Co(II) and Ni(II) oxotrimers is expected to provide high activity in the methanol formation[366]. Another interesting example of the use of organo-inorganic systems for the CO2 reduction to methanol are catalysts based on organic 2D polymers of the salophen – NiN2O2 type. The introduction of quinoid motifs into the polymer structure dramatically increases the Faradaic efficiency for methanol (27%) as compared to the unmodified counterpart (8%) at –0.9 V[367].
Pd, Pt and Ru do not generally promote the formation of methanol as a major product[368]. Modification of platinum with ruthenium supported on carbon (Pt – Ru/C electrode containing 32.6 wt.% Pt, 16.9 wt.% Ru) can increase the methanol yield from 0.03 to 7.5% with a Faradaic efficiency of 75%[369]. Payra et al.[370] described an increase in methanol selectivity when using Pt – Zn alloys derived from ZIF-8 metal-organic frameworks. As a result, a high Faradaic efficiency for methanol (>80% at 0.4 mA cm–2 and potential of –0.9 V) was observed for systems containing the phase-heterogeneous PtxZn/C alloy (1 < x < 3), composed of PtZn/C and Pt3Zn/C. For Pd, an increase in the methanol yield is possible when using tin oxide as a support (Faradaic efficiency is 55%)[371]. An increase in selectivity in this case is attributed to the enhanced ability of Sn oxide to bind CO2 and to the decreased binding of the resulting CO to Pd.
For Ru-based systems, there are also few examples of methanol formation. A catalyst based on a Cu-containing metal-organic framework HKUST-1 with Ru atoms incorporated in its pristine lattice has some efficiency, with Faradaic efficiency of up to 3% (the main product being ethanol with a Faradaic efficiency of more than 45%)[372]. Deposition of RuO2 on a boron-doped diamond phase allows the methanol production at different pH values along with other products (mainly HCOOH) with Faradaic efficiency of 6 – 8%[373]. RuO2-based systems provide significant Faradaic efficiency for methanol only at very low current densities[374], with Cu modification allowing an increase of up to 41%[375]. A significantly greater effect is obtained by supporting Ru on TiO2 nanotubes: the Faradaic efficiency for methanol reaches 60.5% at –0.8 V vs the standard potential[376].
In general, despite the numerous studies, the development of catalysts for methanol synthesis by CO2 electroreduction remains an urgent problem. A large body of research in the field of electrochemical CO2 reduction has shown that the reduction to methanol is usually characterized by low and medium Faradaic efficiencies, high overpotential even at medium current densities. The process is accompanied by a number of side reactions, from the H2O-to-H2 reduction to the formation of single-carbon (CO and HCOOH) and two-carbon (ethanol, ethylene, etc.) products. The multi-electron reduction to methanol, occurring at the surface of the catalyst and electrode material, requires not only the sequential addition of 6 electrons, but also the same number of protons. The multistep process and the large number of pathways, the reduction of intermediates involved in the reaction as proton donors to H2, significantly complicate the process and make it important to develop new electrode materials providing high selectivity of the reaction. An alternative may be a combination of electrochemical reduction of CO2 to a mixture of products and their subsequent conversion to final products under homogeneous or heterogeneous catalysis conditions. This approach has been employed in the oxidative methane carbonylation via electrolysis of CO2 to CO and O2 (Ref. [377]) and certainly may be practiced with methanol. Improvements in the design of the electrolyzers and electrode cells are essential[247, 378, 265-267].
A separate problem is the possible irreversible change in the catalyst surface and its poisoning. It is necessary not only to increase the Faradaic efficiency while reducing overpotential and achieving industrially significant current densities, but also to enhance the stability of the most promising systems. Ideally, the catalyst should operate for thousands of hours without loss of activity.
4. Photocatalytic processes
Photocatalysis is a very attractive method for solving the problem of reducing CO2 into value-added products, particularly methanol. Getting closer to what nature does by developing the artificial photosynthetic processes is a goal that requires enormous effort and is still in its early stage.
Photocatalysis in the CO2 chemistry is a relatively young field with very tempting prospects, but the results achieved are far from those required for practical use. To date, conventional photoreactions are much less efficient (in terms of conversion) than thermal reactions, although they are carried out under much milder conditions. Photoreactions take place at low temperatures and pressures and yield a wide range of products (Scheme 17).
Photoreduction of CO2 to methanol was first performed by Halmann in 1978 using GaP catalyst[379]. Since then, the main effort in the field of the solar energy use for CO2 conversion has been focused on searching for a suitable semiconductor material, which has a number of requirements: it must be efficient and inexpensive, and the metal used must be non-precious and common in nature. Although many semiconductor materials capable of adsorbing sunlight are known, each has its own merits and demerits. The amount of research in this field over the last decade has been enormous, the materials obtained are being thoroughly studied, and a large number of special reviews related to CO2 photoreduction have already been devoted to the application of the most promising of them[380-390].
The process of the direct photoconversion of CO2 to methanol on semiconductors involves several steps (Figure 11). The first step is the absorption of a photon with an energy higher than the bandgap energy, with the appearance of electrons in the conduction band and holes in the valence band[391]. The wavelength of the radiation absorbed in this case is determined by the bandgap of the semiconductor. For the most common semiconductors the bandgaps are: 3.7 eV (ZnS), 3.2 eV (TiO2), 3.3 eV (ZnO), 2.7 eV (gC3N4), 2.4 eV (CdS), 2.0 eV (Cu2O)[390]. As a result, the first three semiconductors absorb light in the UV range and the last three absorb in the visible range. Changing the bandgap to utilize the widest possible spectrum of visible light is an important problem for improving the efficiency of photocatalysts for CO2 reduction.
![[{"id":"eY5hMZnF-U","type":"paragraph","data":{"text":"Process of catalytic CO<sub>2</sub> reduction to methanol, coupled with water oxidation, in the presence of co-catalysts (adapted from Ref. [[ type=\"anchor\" referenceId=\"6432\" ]], Copyright 2023, American Chemical Society)."}}]](/storage/images/resized/O8To11ES1iuREbufaJMZeDSj5whCZ9ARfL8rC9hE_xl.webp)
After photoexcitation, two processes are possible. The first of them involves the recombination of holes and electrons in bulk (bulk charge recombination) and on the surface (surface charge recombination) with the heat release. The second possible way is the charge separation[392]. At the separation process, charges migrate to the material surface (reaction sites), where reactions take place with their participation and the participation of compounds from the reaction medium. For a process to be highly effective, the migration of charge carriers to the surface (charge trapping by the surface) should dominate over recombination.
Electrons initiate the CO2 reduction through the formation of a surface-bound anion radical by a mechanism similar to that of electrocatalysis. Conversion of the anion radical gives the full range of possible products, including methanol. The position of the conduction zone is important when choosing the catalyst: for the reduction of CO2 to methanol, a potential of –0.38 V at pH = 7 should be provided, with the conduction zone being localized at more negative potentials for the reaction to proceed. For the main competitive products the potentials are higher: –0.51 V (СО), –0.61 V (НСOOН), or lower: –0.24 V (СН4) (see Figure 7).
Therefore, the position of the conductivity zone level determines the possible selectivity of the process in terms of its thermodynamics: if the conductivity zone is in a more negative region than –0.61 V, all the above products will be formed in the reaction (as for the Cu2O/g-C3N4 nanocomposite); if it is below –0.51 V (as for of TiO2 in rutile form), the primary products will be methanol and methane[380]. Furthermore, H2 may be evolved in the competing H2O oxidation reaction[393].
Formally, the reduction products are formed by the interaction of the intermediate particles with both electrons and protons. The latter are the hydrogen source (H). It is suggested that protons are produced as a result of the substrate oxidation by the interaction with holes. For the reaction to proceed, the holes must somehow obtain electrons by interacting with reducing agents or by the application of electric current. In the latter case we are talking about electrophotocatalysis, which is also a rather actively developing field[394, 395]. Under the conditions of classical photocatalysis in an aqueous medium, modelling photosynthesis, holes undergo the oxidation reaction with H2O to form O2 by the radical mechanism[393] (reactions (5), (6)). The energy of the holes in this case should be equal to the potential of O2 formation (+0.82 V).
Holes can oxidize other substrates such as H2 (then it makes sense to talk of the photochemical CO2 hydrogenation), sulfite anion, trialkylamines, triethanolamine and others. In addition, holes can interact with semiconductor per se or other compounds in its composition and thus destroy the catalyst in the process of photocorrosion[396]. Photocorrosion can also arise from the reaction with photoelectrons[397]. The photocatalyst stability is one of the main problems that can be solved, in particular, by using special protective coatings, which conduct electrons and prevent photocorrosion.
Areas of the semiconductor surface can act as catalysts for both reduction and oxidation. However, the rate of CO2 reduction by electrons and oxidation by holes on the semiconductor surface may be insufficient. The rate of reduction and oxidation can be improved by the use of appropriate co-catalysts. In this case, the system consists of material capable of absorbing a quantum of energy (preferably in the visible spectrum) and producing separated charges, and co-catalysts deposited on this material, which enable the oxidation of H2O or other substrates by interaction with holes and the synthesis of methanol by CO2 reduction with electrons (see Scheme 17). Electron and/or hole transfer to the co-catalyst promotes charge separation and prevents their recombination[398]. Metal nanoparticles, metal sulfides, phosphides and carbides, etc. can be used as such co-catalysts.
The mechanism of CO2 reduction in the presence of H2O involves the simultaneous transfer of protons generated by the oxidation of water. Such transfer can occur in a similar way to electrochemical reduction from species in solution (H2O, oxonium, bicarbonate, hydrogen phosphates) or even on the surface. The mechanism of reduction by photoexcited electrons and proton involvement is generally similar to that of electrochemical reduction. In most cases, CO and methane are identified as the main reduction products. On the catalyst surface, CO2 is converted to *COOH species affording the adsorbed CO; at low binding energy to the semiconductor and rapid desorption, CO appears to be the main reaction product. In the absence of desorption, as in the case of electrochemical reduction, CO can also be converted to *C(O)H species, which is further converted to methanol and methane. According to the current ideas about the mechanism of this reaction, for the selective formation of methanol, precisely a formyl intermediate should be formed giving rise to *СН – ОН, *СН2 – О* and СН3 – О* species, which are bound to the catalyst surface through the carbon atom, while maintaining the C – O bond, as with electrocatalysis. The cleavage of this bond promotes deoxygenation through the release of H2O and the formation of surface carbides, CH2 and CH3 species from which methane is formed[399]. As with the electrochemical process, the ability to split this bond is determined by the oxophilicity degree of the surface and the composition of the solution.
The alternative pathway of CO2 conversion in the reduction process suggests the formation of formate intermediate (HC(O)O*), bound to the catalyst surface via the oxygen atom, and can produce methanol by a mechanism characteristic of thermocatalytic processes[391]. In photocatalysis, the main competing product for this pathway is HCOOH. Methanol formation by this pathway, as in the case of electrocatalysis, is relatively rare, because it requires positively charged hydrogen to react with positively charged carbon bound to two oxygen atoms, which is unlikely if the surface of the photocatalyst is negatively charged due to the presence of electrons.
In general, it should be emphasized that achieving high selectivity is one of the key challenges in the photocatalytic synthesis of methanol, since the formation of by-products such as CO, CH4, HCOOH, ethanol and other C2 compounds is difficult to avoid, also due to the evolution of the photocatalyst surface in solution.
There is another problem that is specific to photocatalysis and affects the selectivity of the process. The separation of the oxidation and reduction processes under photochemical reaction conditions in the reaction volume is usually achieved only to a small extent, and methanol may also be available for oxidation on another species of the photocatalyst surface. The resulting methanol can then interact with the holes and be oxidized to CO. For a selective catalytic system, the rate of oxidation of methanol as it interacts with the holes, should be low, otherwise it will be converted to CO[400]. To achieve this, it is necessary to provide a spatial separation between the oxidation and reduction sites, preventing the contact between CO2 reduction products and the oxidation reaction sites.
An additional challenge in carrying out photocatalytic reactions is that these processes, like electrochemical reduction, take place in aqueous solutions, in which CO2 is poorly soluble and from which it is difficult to extract the resulting methanol. Increasing the pH of the reaction medium and using carbonates, as in electrochemical reactions, and amines, as in homogeneous catalysis, to bind CO2 can help to solve this problem.
Work in the field of photocatalysis for the conversion of CO2 to methanol aims both to achieve high selectivity and to increase the conversion rate, which is still low and energy intensive (the yield per unit of light used is no more than a few percent). At the same time, the reaction rate is significantly reduced by the recombination of holes and electrons, and the effective separation of charges, along with an increase in the degree of absorption of light radiation, is the key problem in the design of catalysts for CO2 photoreduction.
Other conditions being equal, the catalyst activity can be estimated on the basis of two parameters. The first is related to the efficiency of the absorbed radiation use. For a cost-effective process, this parameter is estimated to be around 15%[281]. The efficiency of the absorbed radiation use depends on many factors, including reactor characteristics, the power and spectral characteristics of the radiation used, catalyst properties, e.g., its porosity, and others. Reducing the degree of the bulk recombination of holes and electrons, mainly due to charge separation, should increase this index (e.g. reducing the particle size of the photocatalyst should favour the predominance of charge migration to the surface over recombination). The same objective is pursued by broadening the operating spectrum of visible light absorption for a catalyst by changing the bandgap, in particular by doping or by using the plasmon resonance effect in metal doping[401, 402]. It is also possible to increase the efficiency of radiation use applying porous or specially designed one-, two- and three-dimensional materials in which the catalyst surface area interacting with the radiation can be significantly increased (e.g., by multiple internal reflections). Such materials have a high area-to-volume ratio and allow the radiation to be used particularly efficiently[390]. In addition, increasing the specific surface area leads to an increase in the number of active sites by reducing the degree of charge recombination in bulk and increasing the probability of their capture by the surface.
Another indicator of photocatalyst activity is the methanol yield per gram of catalyst per hour. This is usually 0.02 – 0.5 mmol, whereas for industrially acceptable processes, a value of 10 mmol (or 300 mg) per gram of catalyst per hour is required. In most publications, this value is used without reference to the number of photons absorbed, due to the difficulty of accurately determining the latter. It should be borne in mind that in each specific case the characteristics of the radiation used, the CO2 concentration, the medium and the reactor peculiarities are very different, which does not allow a correct comparison of the catalyst activity values.
Many authors note that the catalyst performance can be improved with other semiconductors by optimizing the zonal structure of the material, developing defects on the surface, controlling its morphology, using both quantum dots and one-dimensional (nanowires, nanorods), two-dimensional (nanosheets) and three-dimensional materials, selecting a suitable supported co-catalyst to provide charge separation, the use of systems containing oxygen vacancies and capable of efficiently adsorbing CO2, the introduction of sensitizers to increase the light absorption efficiency, the application of special protective layers to prevent catalyst degradation, the control of pressure, temperature, pH of the reaction medium and, finally, of the reactor used.
All these factors affect the efficiency and selectivity of the process[403, 404].
In catalyst design, particular attention is paid to the formation of heterostructures of various types from several semiconductors (Figure 12), in which the electron and/or hole transfer from one semiconductor to another is possible, and which make it possible to increase the efficiency of charge separation. In a type I heterojunction, the bottom of the conduction band of the second semiconductor is lower and the top of the valence band is higher than those for the first semiconductor. As a result, electrons and holes migrate to the latter. Therefore, the transfer of electrons and holes leads to negative consequence, which is the charge accumulation on the second semiconductor and a decrease in the reducing (due to the low level of the conduction band of the second semiconductor) and oxidizing (due to the high level of it of the second semiconductor) ability.
![[{"id":"8PYgElIcRU","type":"paragraph","data":{"text":"Heterojunctions and charge transfer for heterojunctions of type I (а), type II (b) and Z-schemes, direct (c) and indirect (d) (adapted from Ref. [[ type=\"anchor\" referenceId=\"2460\" ]]. Copyright 2020, American Chemical Society)."}}]](/storage/images/resized/SMpm20pC516XZPuQLThuMr5Zp58MMn00Fj6raVcl_xl.webp)
A type II heterojunction requires the bottom of the conduction band and the top of the valence band of the second semiconductor to be lower than those of the first one. Therefore, electrons from the first semiconductor migrate to the conduction band of the second semiconductor, while holes from the valence band of the second semiconductor migrate to the valence band of the first semiconductor. This results in charge separation: electrons are accumulated on the second semiconductor, where the reduction reaction occurs, and holes are concentrated on the first semiconductor, where the oxidation reaction takes place. This significantly increases the efficiency of the use of the generated charges and can improve the catalyst activity. The main disadvantage of this approach is that the reduction and oxidation capacities are degraded due to the location of the corresponding bands.
Developing a Z-scheme heterojunction is free from the above disadvantage. The bottom of the conduction zone and the top of the valence zone of the first semiconductor are lower than those of the second. When both semiconductors are photoexcited, electrons from the first recombine with holes from the second, resulting in effective charge separation while maintaining high reducing and oxidizing capacities for holes and electrons, respectively. The electrons are concentrated on the surface of the second semiconductor, while the holes are concentrated on the surface of the first semiconductor. As a result, reduction and oxidation reactions take place on different materials. The efficiency can be further improved by using an intermediate conductor (e.g., nanocarbon systems or metal nanoparticles), providing a high rate of electron transfer from the first semiconductor to the second one (indirect Z scheme).
Semiconductors used as catalysts include base metal oxides, particularly of Zn[405], Ti[406, 407], Cu[387, 408, 409], Ce[386], sulfides and chalcogenides[410, 411], carbides[412], nitrides, a number of carbon materials[387, 413], metal-organic frameworks[414], some mesoporous materials[415, 416], and others. All these systems differ significantly in their photochemical properties, and a large number of different, often quite complex, approaches have been used to synthesize and modify each of these materials in order to reduce the degree of charge recombination, change the bandgap to increase the fraction of light absorbed, increase the rate of CO2 reduction, etc. This makes it possible to significantly change the catalyst activity and the process selectivity.
It was found that the efficiency of the ZnO-based catalyst is determined by the ZnO morphology and the method for its preparation. The composition of the products (CO, H2, CH4) depends on these factors. Carbonaceous materials such as graphene, graphene oxide, partially reduced graphene oxide can be used as the second component. The electron transfer generated by light absorption to these conductive materials significantly increases their lifetime and the efficiency of the catalyst[417].
The use of the mesoporous ZnO – ZnS heterostructure allows an almost 2-fold increase in the methanol formation rate, and its modification with 3 nm Pt nanoparticles provides an additional 20-fold increase (81.1 μmol g–1 h–1). The modification of ZnS and Pt, together with the development of the mesopore, significantly increases the probability of charge carrier transport to the catalyst surface and reduces the probability of charge recombination[418]. ZnO heterostructures containing another semiconducting chalcogenide, ZnSe, are very active in the methanol formation (1581.82 μmol g–1 h–1). The above heterostructure provides much higher light absorption efficiency due to the smaller selenide bandgap (2.5 V vs 3.5 V), while the presence of the heterojunction reduces the recombination rate and improves charge separation: the reduction takes place on the oxide particles and also involves electrons generated when the second semiconductor, ZnSe, is exposed to light. Isopropanol is oxidized by holes as an electron donor[419].
In addition, the activity of ZnO increases when using CuO-containing heterostructures[420]. Thus, the formation of a hierarchical p – n junction between CuO and ZnO nanospheres significantly increased the efficiency of electron-hole separation and, consequently, the yield of methanol formed from CO2 in an aqueous solution of DMF and triethylamine as electron donor oxidized by holes. The maximum formation rate (3855.36 μmol g–1 h–1) was achieved for the system containing 0.4 mmol CuO[421].
The use of sibunite-supported ZnO – NiO porous spheres, derived from a metal-organic framework by forming a p – n junction between oxides, also provided charge separation and a 3-fold increase in the rate compared to the use of single ZnO[422]. The presence of sibunite increases the rate of electron transfer in the heterojunction. When the agglomerated ZnO nanoparticles were modified with MoS2 nanosheets, charge separation was achieved by electron transfer to the sulfide phase, resulting in a methanol yield of 170.55 μmol g–1 h–1 in 0.5 M NaHCO3 solution under UV irradiation. In all cases, the use of porous and hollow structures provided an increase in the degree of CO2 adsorption due to the increase in surface area and the degree of light harvesting due to the refractions and reflections within the pores of the material[423].
Platinum decorated (1.5%) spinel-based ZnMn2O4 nanorods are useful photocatalysts for the CO2 conversion to methanol[424]. The catalyst produced methanol at a rate of 1906 μmol g–1 h–1 within 9 h under visible light irradiation, which is 4.25 times higher than that for the Pt-free catalyst. The doping with metal resulted in an increase in electron mobility, increased charge separation efficiency, suppressed charge recombination and reduced the bandgap. The catalyst was recycled 5 times without loss of activity[424]. Nanoparticles of non-precious metal such as Cu can also act as similar modifiers for Zn-containing spinel-type semiconductors. Thus, for the sample of ZnV2O4 modified with 10% Cu nanoparticles, the overall yield of alcohols was 6.49 μmol g–1 h–1 with a methanol yield of 3.3 μmol g–1 h–1, and its formation rate increased 1.5 times as compared to the sample without Cu. The use of triethanolamine as a reducing agent allowed to obtain methanol at a rate higher than 6 μmol g–1 h–1. The high activity was explained by a significant decrease in the hole-electron recombination rate: the electrons formed during irradiation moved from the vanadate surface to the Cu particles, where CO2 reduction took place[425].
Due to conductivity of graphene carbon materials, their use can provide efficient charge separation in the catalyst, thereby increasing the efficiency of CO2 photoreduction. Moreover, graphene structures can act as intermediates for the electron transfer from one semiconductor to another[426, 427]. The introduction of graphene oxide results in a five-fold increase in the activity of the catalyst in the methanol production reaction[428]. The use of a composite material containing ZnO nanorods and graphene allows a 2.8-fold increase in the methanol formation rate as compared to the use of nanorods alone[429]. A photocatalyst based on nitrogen-doped reduced graphene oxide decorated with ZnO nanowire arrays grown on its surface also provided a significant improvement of the rate[430].
A non-metallic semiconductor, g-С3N4, is also used to develop ZnO-based heterostructures. Due to the electron transfer from the conduction zone of photoexcited ZnO to g-С3N4, the photocatalytic system consisting of the g-С3N4/ZnO heterostructure, displays an increased activity as compared to individual materials. Here, the so-called Z-scheme is realized[431], in which electrons from ZnO recombine with holes formed during photoexcitation of g-С3N4, providing the charge separation[432]. A further increase in the catalyst efficiency is possible by constructing a hierarchical structure of ZnO microspheres and nano-sized ‘sheets’ of g-С3N4. In this case, the degree of energy absorption is significantly increased due to the scattering of the directed radiation in the pores of the material[433].
Another material commonly used in the photocatalytic CO2 reduction to methanol is TiO2. The modification of this oxide is aimed both at increasing the lifetime of the charge carriers and at changing the band gap so that the photoreactions take place in the visible rather than in the UV range. To obtain an acceptable methanol selectivity, several variants of TiO2 modification have been proposed, ranging from the surface hydrophobization and introduction of electron acceptors (metals, carbon conducting materials) to the design of heterostructures comprising this oxide. For example, fluorination of the TiO2 surface allows the methanol yield to be increased up to 247.15 μmol g–1 h–1 due to more efficient CO2 binding and electron transfer[434]. The reduction with H2 of a TiO2-based catalyst modified with Cu nanoparticles makes it possible to achieve a 17-fold increase in the methanol yield as compared to the material obtained in air[435]. According to the authors, this effect is due to the simultaneous generation of Ti3+ and oxygen vacancies and, consequently, an increase in the availability of the material surface for photoelectrons. The presence of Cu facilitates the adsorption of the resulting partially reduced intermediates.
The increase in methanol formation activity for diversely modified TiO2 is facilitated by the use of materials that provide more effective charge separation by transferring them to a different conductor. The doping of TiO2 with cobalt increases the methanol yield from 32.3 μmol gcat–1 to 730 μmol gcat–1 for 7 h (Co acts as a co-catalyst), while the introduction of graphene oxide (the conductor) improves it up to 936 μmol gcat–1[436].
Another method of modification includes the construction of layered structures from TiO2 and the formation of heteropolystructures on their basis. This can be achieved by exfoliation, a method for producing layered crystals with a small number of layers from a multilayer material using shear forces in the bulk material. The formation of the heterojunction by the interaction of exfoliated TiO2 nanosheets with CeO2 and graphene oxide promotes charge separation and provides high electron mobility in the catalyst, which contributes to a sevenfold increase in the rate of methanol formation (up to 641 μmol gcat–1 h–1) as compared to the pristine TiO2[437]. It should be noted that cerium-doped TiO2 aerogel, in which Ce is incorporated into the porous structure of the oxide matrix, showed better efficiency in CO2 reduction due to its high surface area. Ce doping led to the appearance of additional electronic levels in the oxide and allowed the use of visible light, but the main products were CO and methane[438].
Increased activity and selectivity can also be achieved by forming heterostructures with other semiconductors. For example, the material containing p-semiconductor ZnFe2O4 as a second component along with TiO2, provided the methanol production of 693.31 μmol gcat–1 h–1[439, 440]. An increase in activity in the reduction of CO2 to methanol was achieved by modifying TiO2 with the semiconductor LaYAgO4, which has a perovskite structure. Holes and electrons were separated by electron transport across the heterojunction between the catalyst materials. An additional increase in efficiency was achieved by the addition of graphene, which also facilitated electron transport between the conduction bands of the materials[441].
High performance in methanol formation was also achieved using the AgVO3/TiO2 heterostructure, where TiO2 was used in the form of nanowires: for the sample containing 25 wt.% AgVO3, the rate of methanol formation was 9561.3 μmol gcat–1 h–1. The authors assume that the high activity is conditioned by the realization of the S-scheme (analogue of the Z-scheme with the formation of a potential difference between semiconductors). As in the Z-scheme, electrons from the AgVO3 conduction band recombine with holes from the TiO2 valence band providing a high efficiency of charge separation. The charge transfer is accelerated by the formation of a potential difference between the semiconductors at the heterojunction formation. As a result, the electrons generated by high-speed irradiation migrate to the surface of TiO2 and the holes move to the surface of AgVO3[442].
Chalcogenides of various metals have also been used to produce photocatalysts, with TiO2-based heterostructures being obtained. Particular activity in methanol synthesis was observed with the systems based on TiO2 and Ag2Se[443], CuSe[444], CuCaAg2Se[445], PbSe[446], AgCuInS2[447], WSe2[448, 449].
The formation of the Mo2C/TiO2 heterostructure gave a catalyst active under visible light irradiation[450]. The electron transfer to the carbide phase after excitation reduced the probability of the charge recombination on TiO2. The efficiency of the whole process was improved by the use of an optofluidic microreactor, which, due to its special design, allowed a high efficiency of light harvesting in the flow system. The best results were obtained with the addition of 4 wt.% Mo2C: in the microfluidic reactor the methanol formation rate was ~10 μmol gcat–1 h–1. A further increase in activity can be achieved by promoting with Cu2O, another ptype semiconductor[451]. Cu2O content of 4 wt.% allowed a methanol production rate of 36.3 μmol gcat–1 h–1 under the visible light, due to more efficient charge separation and electron transfer between the components of the heterostructure.
It has been shown that materials containing only Cu2O and TiO2 are also active in the methanol production[452]. Thus, the incorporation of Cu2O nanoparticles into the structure of films based on TiO2 nanorods increases the efficiency of photon absorption by the material and provides the maximum rate of product formation (36.18 mmol gcat–1 h–1 (CH3OH) and 79.13 mmol gcat–1 h–1 (С2Н5ОН)) in a flow microreactor at the optimum Cu concentration (0.02 M)[453]. Using a similar flow system with a catalyst obtained by embedding Cu nanoparticles into TiO2 in, Albo et al.[454] achieved a CO2 reduction rate of 230.3 μmol gcat–1 h–1 under UV irradiation and a concentration of 2% wt. Cu in the material. The synthesis was carried out in the ionic liquid 1-(n-butyl)-3-methylimidazolium tetrafluoroborate, which acts not only as a template but also as a nanoparticle stabilizer. The authors showed that the methanol yield depended on many factors, including irradiation intensity, photocatalyst concentration and reactor configuration (optofluidic microreactors were used). The combined use of Cu and Ni for modification under similar conditions resulted in the mixed Ni-rich nanoparticles supported on the TiO2 surface. The resulting material was active under visible light irradiation (450 nm) and appeared more efficient than the monometallic Ni-modified catalyst in the microfluidic reactor[455].
The simultaneous introduction of Cu2O and Pd nanoparticles into TiO2 allows the efficient electron transfer to the Cu-containing particles[456]. The presence of Pd promotes the formation of oxygen vacancies on the oxide surface, which improves the ability of the material to adsorb light in the visible region and promotes charge separation. The methanol production rate in this case was 71.84 μmol gcat–1 h–1.
Ti can also be used as a doping element for other semiconductors. For example, Ti modification of WO3 nanosheets accelerates the charge transfer to adsorbed CO2 and increases the rate of CO2 conversion to methanol by 3.3 times as compared to starting WO3 (up to 16.8 μmol gcat–1 h–1), with a methanol selectivity of 88.9%[457]. Ti-containing perovskite-like oxides, in particular SrTiO3 in the form of nanofibres, also showed activity in the methanol production. By doping the above semiconductor with copper, a material with the composition SrTi1–xCuxO3 was obtained, which increased the activity of the photoreduction of CO2 to methanol by a factor of 2 as compared to the unmodified semiconductor[458]. More complex composites have also been proposed, including bimetallic Cu – Ni nanoparticles together with titanium nitride in addition to SrTiO3. This complex modification allowed to reach the 70-fold increase in the methanol production rate[459]. The authors attributed such growth to plasmon resonance for Cu – Ni nanoparticles.
Ti-containing layered systems, MXene (Ti3C2Tx), are also interesting materials for the preparation of heterostructure-based photocatalysts due to their IR absorption capability with charge carrier separation. Thus, the material with deposited CeO2 nanoparticles is active in the methanol formation in both visible and IR range, allowing the production of methanol and ethanol at rates of 102.24 and 59.21 μmol gcat–1 h–1 within 4 h, respectively[460].
In photocatalysis, as in electrocatalysis and photoelectrocatalysis, Cu2O is often used as an active component, which can be used with visible light irradiation due to the value of the bandgap energy. The application of this catalyst is the subject of a recent review[409]. The reduction of CO2 to H2O using various combinations of Cu2O with other oxides and catalysts and other Cu-containing materials under different irradiation patterns is also reviewed[408]. In most cases, the reaction produces methane and CO.
Moreover, a particular problem of Cu-containing catalysts is their stability, especially after several cycles. The above reviews include publications in which methanol was obtained via CO2 reduction. The authors consider different ways of carrying out this reaction. To increase the activity and stability of the catalysts, the same techniques are used as for other semiconductor systems: from the introduction of co-catalysts and the development of heterostructures with other semiconductors, including non-metallic ones, to the introduction of conductive carbonaceous materials. In addition, doping this catalyst with metals can generate plasmons and ‘hot’ electrons[408, 461].
To achieve high methanol selectivity, it is critical to use Cu2O with a specific morphology, since crystallites with different facet orientations differ significantly in conductivity, activity and stability[462, 463]. Spherical particles have been shown to improve the charge separation, while (111) facets promote the process towards methanol formation but have significantly lower activity[462, 464, 465]. Methanol formation is also promoted by increasing the proportion of (110) facets on which this process occurs[466]. An example of the use of co-catalysts to increase efficiency is the modification of Cu2O nanowires with Ti3C2 quantum dots[467]. During the reaction, the photoelectrons generated are transferred to the carbide particles on the surface, where the reduction of CO2 to methanol takes place. The reaction rate increases by almost an order of magnitude.
The formation of heterojunctions between Cu2O and other semiconductors requires attention to the morphology of the individual components in order to organize the interaction that provides the maximum degree of electron and hole separation. For example, the optimal results have been obtained by using p-type conducting sulfide to form a MoS2 – Cu2O heterostructure, which allows charge separation according to the Z-scheme[468]. Also the maximum activity (up to 76 μmol gcat–1 h–1) is achieved using oxide particles with a cubic morphology and a predominance of (100) facets.
A heterostructure based on Cu2O and In2O3 oxides, the former consisting of particles with a ‘flower’ morphology and the latter of spherical nanoparticles, allowed to significantly increase the rate of CO2 reduction and methanol formation via CO using Na2SO3 solution[469]. The combination of Cu2O with carbon-modified BiOI resulted in a heterostructure with a p – p junction, in which Cu2O enhanced the charge separation efficiency and carbon improved the charge transfer and light absorption. The reaction of CO2 reduction to methanol was carried out on Bi-containing material. At the optimum C/Bi ratio = 1 : 15 and 30 wt.% Cu2O, the methanol productivity reached 722.88 μmol gcat–1 h–1 within 8 h[470]. Similarly, the formation of Cu2O heterostructure with ZnV2O4 by partial reduction of Cu (10% oxide) yields 3.3 μmol gcat–1 h–1 of methanol due to faster electron transfer and higher charge separation[425]. Cu oxides can also be used at low concentrations as co-catalysts for other semiconductors such as Na2Ti6O13[471].
The performance and stability of the Cu2O-containing photocatalyst can be achieved by developing materials containing electrically conductive carbon particles. As mentioned above, these act as electron acceptors, facilitating their separation from the holes, and also act as co-catalysts. When a second semiconductor is used, such materials can act as intermediate conductors, allowing the electron transfer from one semiconductor to another (indirect Z-scheme). In general, the catalyst stability increases significantly when these are used. Thus, the combination of reduced graphene oxide with rhombic and dodecahedral forms of Cu2O (containing (110) and (111) facets, which are apparently responsible for the high rate of electron transfer to the surface) gives a high yield of methanol (355.3 μmol gcat–1, 20 h, visible light), which is much higher than the usual yield for oxides[413]. The high conductivity of graphene oxide contributed to the increased activity and charge separation. A significant increase in activity due to the efficient charge separation by the electron transfer to the carbon component was also achieved by modifying 50 nm spherical Cu2O particles with carbon nanoparticles consisting of graphene-like structures. As compared to unmodified Cu2O, the methanol yield increased 5-fold to 250 μmol gcat–1 (15 h), and the system was recycled many times without loss of activity[472]. Simultaneous coating of Cu2O nanoparticles with a carbon composite and carbon quantum dots also increased the methanol formation activity (99.6 μmol gcat–1 h–1). The carbon layer and the quantum dots increased the stability of the catalyst; the quantum dots also made it possible to change the characteristics of the radiation used in the process towards the shorter wavelengths. In addition, the resulting holes were transferred to the surface of the quantum dots, where H2O oxidation took place, while CO2 reduction proceeded on Cu2O[464].
The separation of the generated charges on oxides is achieved by introducing nitrogen-doped carbon into the catalyst structure. In this case, visible light can be used for photoexcitation. When Fe3O4 nanorods coated with such carbon were used to modify Cu2O, the methanol yield increased fourfold as compared to the Fe3O4@Cu2O system. At Cu2O content of 12%, the methanol yield was 440 μmol gcat–1[473]. Another example of improved efficiency is the preparation of a catalyst with nanoscale Cu2O (<3 nm) from layered aluminium-zinc-copper hydroxide on polymeric carbon nitride. The generated Z-scheme, where electrons from the conduction nitride band interacted with holes from Cu2O, promoted the charge separation and provided the methanol yield of 440.78 μmol gcat–1. The catalyst operated for 30 h without loss of activity[474].
In addition to the above-mentioned oxides, there are examples of the use of other combinations of semiconductors and co-catalysts to increase the efficiency of catalytic systems for the reduction of CO2 to methanol. For example, the hydrothermal synthesis of a SnS2/Bi2WO6 heterostructure with a Z-scheme heterojunction allows the methanol yield to be increased by a factor of 3.3 by increasing the width of the adsorbed light spectrum and the efficiency of the charge separation[475]. Modification of a perovskite-like material, gadolinium orthoferrite (GdFeO3), with a small amount of PdO narrows the bandgap and promotes the charge separation due to the formation of a heterojunction between oxide and orthoferrite. The catalyst containing 1.5% PdO/GdFeO3 provides 1550 μmol gcat–1 of methanol within 9 h[476]. The decoration of ZnMn2O4 nanorods with Pd leads to similar effects (narrowing the bandgap and improvement of charge separation) and allows to obtain 1.906 μmol gcat–1 of methanol within 9 h[424]. A highly active catalyst for CO2 reduction and triethylamine oxidation has been prepared by modifying CeO2 nanorods with spherical Fe nanoparticles and hexagonal Ni particles[477]. In all cases, modification significantly enhances the stability of the semiconductor materials and allows them to be reused.
Doping carbonaceous materials with nitrogen gives rise to organic semiconductors which, as can be seen from the above, can be effective components of photocatalysts for the conversion of CO2 to methanol[385, 389, 478-481]. Optimization of the g-C3N4 synthesis helps to lower the bandgap of this semiconductor to 2.4 eV, and modification with reduced graphene oxide affords a composite in the presence of which methanol is formed (114 μmol gcat–1 h–1) in CO2 photoreduction[482]. Unfortunately, a competitive reaction of H2O reduction to H2 (68 μmol gcat–1 h–1 of hydrogen) also occurs. The modification of g-C3N4 makes it possible to increase the degree of charge separation. For example, immobilization of g-C3N4 on melamine – formaldehyde – resorcinol polymer microspheres improves the activity of g-C3N4 by a factor of 10[483]. The charge transfer occurs from the C – N bond to the hydroxymethylamino group in the polymer, which catalyzes the reduction of CO2 to methanol.
Another method of modifying g-C3N4 involves the introduction of carbon dots through the specially designed linkers[484]. The holes formed in g-C3N4 are mostly transferred to specially prepared and supported on nitride quantum dots, on the surface of which H2O oxidation takes place. In this case, the electrons are accumulated in the conduction band of g-C3N4 and provide a high methanol selectivity. Wang et al.[400] showed that the predominance of graphite-like assemblies in the structure of carbon quantum dots is the prerequisite for high hole transport efficiency. Such dots are formed under microwave irradiation.
Modification of g-C3N4 can be achieved by introducing single metal atoms. The rare earth atoms act here as CO2 binding and reduction sites[485]. Thus, the introduction of Pr into g-C3N4 increases the methanol yield by 14 times. This yield is further increased by a factor of 2 by the formation of special centres (Pr1 – N4O2–) resulting from the introduction of oxygen into g-C3N4. Methanol is formed in the amount of 511.1 μmol gcat–1 h–1. A similar effect is produced by the deposition of Co atoms on the nanosheets of specially exfoliated gC3N4. The formation of single metal centers, which adsorb and activate CO2 and are able to transfer electrons from the semiconductor to the substrate, increases the rate of methanol formation by a factor of 13.4 as compared to the unmodified support, with alcohol being formed in amounts up to 941.9 μmol gcat–1 h–1 within 4 h[486]. The system remained stable for 12 cycles (48 h).
Numerous examples of the design of g-C3N4-based heterostructures for the efficient production of methanol from CO2 have been described. The process selectivity and the catalyst efficiency depend on both the particle nature and size of the second semiconductor. Thus, the use of CdSe quantum dots of different sizes affects the actual possibility of the methanol formation: at the optimal particle size (2.2 nm), the catalyst activity is 186 μmol gcat–1 h–1, which is more than 3 times that of selenide and 1.5 times that of carbide[487]. Charge separation occurs due to the transition of electrons from the conduction band of g-C3N4 to the conduction band of CdSe, where the reaction takes place. The position of the latter depends on the size of the CdSe particle. For larger sizes, the conduction band is too low and the reduction does not occur; for smaller sizes, the formation of H2 begins to prevail.
Charge separation is facilitated by the formation of the second-type heterojunction between two two-dimensional structures: Bi2MoO6 and g-C3N4. When such material is irradiated, holes are concentrated on the surface of Bi2MoO6 and electrons accumulate on the surface of g-C3N4. This enables efficient charge separation, as well as catalyst activity and stability[488]. The g-C3N4/CoS heterostructure shows a 4.5-fold increase in the methanol yield as compared to conventional g-C3N4, and the role of CoS is to activate H2O without the formation of radicals active in the OH oxidation[489].
The use of g-C3N4 and BiOBr in combination makes it possible to construct a Z-scheme heterojunction, in which electrons from the conduction band of BiOBr react with holes in the valence band of g-C3N4, providing the charge separation and improving the efficiency of CO2 reduction at the g-C3N4 surface and H2O oxidation by holes at the BiOBr surface. This mechanism improves the charge separation, reduces their recombination rate and imparts stability to the catalyst. Thus, a material based on hollow BiOBr microspheres immobilized on protonated g-C3N4 (pCN) was proposed as a catalyst for the reduction of CO2 to methanol[490]. Improved carrier separation due to heterojunction formation between the semiconductors resulted in a 2.56-fold increase in methanol yield as compared to BiOBr (1068.07 μmol gcat–1, 4 h). The use of nitrogen-enriched porous carbon nitride (10% pCN/BiOBr) increased the methanol yield by a factor of 4 under visible light irradiation[491]. Guo et al.[492] proposed a NiTiO3/g-C3N4 (NT/GCN) heterostructure characterized by the conversion of CO2 according to the Z scheme with a methanol yield 3.29 times higher than that for g-C3N4 (methanol formation rate was 13.74 μmol gcat–1 h–1). A similar type of the heterojunction is characteristic of the C3N4/In2O3 heterostructure obtained by decomposition of the metal-organic framework. Hollow hexagonal In2O3 particles interact with ultrathin layers of C3N4 to provide the active catalyst[493].
The use of another carbon material, such as carbon nanotubes (CNT), may also be promising for the methanol production by CO2 photoreduction due to their high conductivity. For example, the CNT – NiO – Fe2O3 system showed high selectivity in this reaction[494]. Carbon fibres can be used to form catalysts with plasmonic metal nanoparticles[495]. The plasmon formation and transfer of the resulting high-energy ‘hot’ electrons to the carbon support allows the efficient charge separation, especially when the carbon support is additionally modified with oxygen-containing groups. Thus, a composite of acid-treated carbon fibers decorated with Ag nanoparticles provided the methanol formation rate of 13.9 μmol gcat–1 h–1 under the visible light irradiation.
The combination of a regular porous structure with semiconducting properties is possible with the use of metal-organic frameworks, which have played a special role in the development of photocatalysis as applied to CO2 reduction[496-500]. The semiconducting or conducting properties of the frameworks allow more efficient charge separation and charge transfer to the surface, while the high surface area increases the efficiency of light absorption. The regular structure of the framework is characterized by a small diffusion distance to the surface for the formed charges and allows the separation of positive charge and electrons at the molecular level. During photoexcitation, one type of charge ends up in the nodes of the framework and other on the organic fragment. Modification of the latter can alter the catalytic properties of metal-organic frameworks, facilitating charge transfer to organic groups and affecting the bandgap width of the semiconductor[501, 502]. Moreover, such modification can increase the degree of CO2 adsorption via chemical interactions[503]. Changing the nature of the metal in the framework nodes and varying the structure of the organic fragments can also significantly influence the semiconductor properties of these materials, including the conduction band width.
Typically, frameworks with elements such as Ti, Zr, Ce, Zn in the nodes are used. These frameworks are then modified with other semiconductors to form a heterojunction. The combination of metal-organic frameworks with semiconductors, including ZnO, has proven fruitful, improving the charge transfer and hole-electron separation[504]. Thus, the simultaneous use of ZnO and the metal-organic framework UiO-66-NH2 (containing Zr6O4(OH)4) units in the nodes of the framework with terephthalic acid) increased the activity in the conversion of CO2 to methanol under visible light irradiation due to the formation of the Z-scheme with charge transfer from ZnO to the metal-organic framework. The activity of the system increased significantly (up to 34 μmol gcat–1 h–1) with the additional formation of oxygen defects on ZnO and the introduction of graphene oxide into the material[505].
Decoration of the Zn-imidazolium framework ZIF-8 with ZnO nanoparticles resulted in a heterostructure with the type II heterojunction, which significantly reduces the recombination rate of holes and electrons and provided a methanol yield of 6700 μmol gcat–1 within 1 h. Here, the large framework surface area favours a more efficient light absorption[506]. Another catalyst design using ZIF-8 suggest ZnO nanorods coated with a transparent framework layer[507]. Pt nanoparticles are deposited on the interface between the semiconductors. The electron transfer from both semiconductors to the metal nanoparticles significantly reduces the charge recombination rate and improves the catalyst performance as compared to ZnO- and Pt/ZnO-based catalysts.
The Ti-containing MIL-125 framework was modified with CuO quantum dots and further combined with g-C3N4[508]. Due to the close contact between the quantum dots and the framework, the electrons generated by irradiation on the framework and on g-C3N4 were rapidly transferred to CuO. As a result, the system showed activity in the reduction of CO2 to methanol, acetaldehyde and ethanol giving the yields of 997.2, 531.5 and 1505.7 μmol gcat–1 within 3 h respectively.
Instead of g-C3N4, another semiconductor such as LiFePO4 can be used[509]. It should be noted that the procedure for the preparation of the catalyst itself, as with other reported catalysts, is rather complex and multi-step. In the first step, it is necessary to synthesize the framework itself, modify it with CuO nanoparticles and incorporate LiFePO4 nanoparticles into the material (Figure 13). The resultant material catalyzed the formation of several products simultaneously. For the catalyst 2.5% LiFePO4/1.0% CuO@MIL-25, the formation of methanol, ethanol and acetic acid was observed in yields of 445.38, 966.36 and 844.63 μmol gcat–1 within 3 h, respectively[509].
![[{"id":"QJvUS4Hgl4","type":"paragraph","data":{"text":"Synthesis of LiFePO<sub>4</sub>/CuO@MIL125. (Adapted from Ref. [[ type=\"anchor\" referenceId=\"6552\" ]], Copyright 2022, Royal Society of Chemistry)."}}]](/storage/images/resized/OgGXpSdSHwoUe0vZQxWnfQwuN089GlDljzKLhBYP_xl.webp)
One of the challenges in the design of the catalysts based on metal-organic frameworks is associated with improving their conductivity. Modification of Ti-containing metal-organic framework NH2MIL125 with graphene oxide allows to increase the catalyst conductivity and to effectively separate holes and electrons: the former are accumulated on Ti oxoclusters, while the latter are located on organic ligands and graphene oxide[510]. The presence of amino groups increases the efficiency of CO2 binding. The use of triethanolamine as an additional reagent (oxidized in the reaction) dissolved in acetonitrile provides the methanol formation at a rate of ~5 mmol gcat–1 h–1. Mesoporous analogues of titanium-containing frameworks have been obtained as aerogels by the reaction of Ti(IV) oxoclusters with dicarboxylic acids. Due to the high porosity and availability of active sites, the resultant catalyst significantly surpassed the corresponding microporous framework obtained by the conventional method[511].
The metal-organic framework UiO66NH2, modified with Cu atoms, with [Zr6O4(OH)4] clusters in the nodes was active in the reduction of CO2 to a mixture of methanol and ethanol (5.33 and 4.22 μmol gcat–1 h–1)[512]. According to the authors, the reaction took place on Cu atoms. The above-mentioned framework can also be modified with C3N4-based quantum dots[513]. Due to the formation of the Z-scheme, the hole and electron lifetimes in this structure are significantly increased, with CO2 reduction taking place on the framework fragments and the oxidation reaction taking place on C3N4. This results in methanol formation at a rate of 386 μmol gcat–1 h–1 in DMF – H2O – TEA solution with simultaneous oxidation of triethylamine.
A metal-organic framework based on Cu and Zn, ions and 1,3,5-tricarboxylic acid fragments also showed activity in the photoreduction of CO2, although it was low (3.71 mmol gcat–1 h–1)[420]. A similar framework with Ce ions in the nodes was used to form a CeMOF/Bi2MoO6 heterostructure, which gave high CO2 conversion, albeit with low selectivity (methanol was formed at a rate of 73.48 μmol gcat–1 h–1, along with methane and formic acid). Due to the position of the conducting zones, the reduction took place on the Bi2MoO6 surface, while the H2O oxidation occurred on the framework[514].
Metal-organic frameworks can also be used as supports for metal nanoparticles, making it possible to increase the conductivity and investigate the possibility of using plasmon resonance for CO2 reduction in these systems. For example, Au particles supported on a ZIF-67 imidazolium-containing framework provided better light absorption and improved charge separation, allowing the methanol production of 2.5 mmol gcat–1 h–1 with triethanolamine oxidation[515].
In the majority of cases, the above processes require the oxidation of H2O or an organic reagent to reduce CO2 in solution under mild conditions. Another type of photocatalytic conversion is a photothermal conversion using H2 as a reducing agent. In this case, exposure to light is coupled with heating. Photothermal catalysis, combining the features of photocatalysis and thermocatalysis, can increase catalytic activity and selectivity, reduce energy costs and allow the process to be carried out under milder conditions than traditional thermal transformations (atmospheric pressure, low temperatures). Two types of materials with photocatalytic activity can be used: semiconductors and plasmonic metal nanoparticles. The latter are able to generate an intense electric field around the particles through plasmon formation under light irradiation due to the effect of surface plasmon resonance, resulting in the appearance of ‘hot’ electrons and a temperature rise at the particle surface. Recombination of charge carriers in semiconductors also leads to a temperature rise. The peculiarities of the ‘hot’ electron generation and of the photochemical thermal effect are described in detail in reviews[516-519].
The first studies on the photochemical hydrogenation of CO2 to methanol used layered hydroxides containing Cu, Zn and Ga[520]. Introduction of [Cu(OH)2]2− anions into Zn1.5Cu1.5Ga(OH)8]2+[CO3]2− · m H2O hydroxide was shown to increase the rate of methanol formation. However, the most significant results in the last decade have been related to the use of plasmonic nanoparticles for catalyst modification[384].
Thus, modification of Pd or Cu with zinc oxide allows the hydrogenation of CO2 to methanol at 220 °C and atmospheric pressure due to the generation of ‘hot’ electrons, but with low selectivity[521, 522]. It was shown that using the classical Cu/Zn catalyst (CuO/ZnO/Al2O3) at 225 °C, methanol is formed with 75% selectivity and in 30% yield when irradiated with 350 – 800 nm light. The first-order condition for maintaining the high selectivity, even at elevated temperatures, is the simultaneous photoexcitation of CuO and ZnO, which minimizes electron transfer from ZnO to Cu and produces HCOO* species. When irradiation occurs only on ZnO, the classical mechanism of photocatalysis with electron transfer to Cu and conversion of CO2 to CO is realized[523]. The addition of Al2O3 together with a slight pressure increase improves the reaction rate and the methanol yield.
It should be noted that significant improvement in productivity (up to 2200 and 8000 μmol gcat–1 h–1, respectively) is possible when the catalyst is modified by the addition of La[524] and CeO2[525]. In this case, CeO2/ZnO heterostructures are probably formed, which contributes to catalyst efficiency. Another way of catalyst modification is the introduction of ZrO2 into the former. When irradiating the Cu – ZnO – ZrO2 catalyst, the methanol selectivity increased up to 70.4%[526]. Aerosol synthesis of 6 nm ZnO – ZrO2 nanoparticles followed by Cu deposition also significantly improved the catalyst activity and selectivity[527].
The use of CaCu3Ti4O12 in photothermal catalysis under the reaction conditions allowed to develop the photoactive catalyst Cu/CaTiO3/TiO2. The catalyst was active at 0.1 MPa, and under irradiation conditions, methanol was formed (308.5 mmol gcat–1 h–1, selectivity 29.5% at 0.8 MPa)[528].
Hydroxylated indium oxides In2O3–x(OH)y are particularly promising as photothermal catalysts. Ozin and co-workers[529-531] showed that the presence of surface Lewis pairs, formed by oxygen vacancy and hydroxyl group bonded to In atom, is essential to the hydrogenation of CO2 on this catalyst. They provide heterolytic activation of H2 via its binding to In atom (In – H bond) and the hydroxyl group (Figure 14)[72, 532]. Charge separation occurs at photoexcitation: the hole transfers to the In atom bound to the hydroxyl group, and the electron transfers to the Lewis acidic In atom. This promotes the reduction process.
![[{"id":"1H5WWAwDOI","type":"paragraph","data":{"text":"CO<sub>2</sub> photoreduction over indium oxide."}}]](/storage/images/resized/EWkvfxPeJZKgFJzxy5UGzrDteB4wrkKXb1yNp4o6_xl.webp)
The methanol selectivity in CO2 photoreduction on indium oxide for such systems without the modification ranges from 7.5 to 50% with the maximum activity of 180 μmol gcat–1 h–1 and can vary significantly depending on the polymorphic form of the catalyst and development of surface defects[533, 534]. The activity can be improved by the use of doping metals[530]. For example, the introduction of 1% Bi increases the yield of methanol by a factor of 3[535]. Metal doping of oxides is also possible using ion beams[536].
The use of polymorphic forms of In2O3–x(OH)y, which combines cubic and rhombohedral phases, contributes to an increase in activity. The heterojunction thus formed provides the improved charge separation[537]. Another example of activity enhancement is the formation of heterojunctions involving TiN and TiO2, but in this case CO is the main product[538]. CO is also formed as the main product in the presence of LaInO3 promoted by Ni nanoparticles[539].
The use of 3D structured nanomaterials can also extend the practical application of photothermal catalysis in the hydrogenation of CO2 to methanol. Thus, Barrett et al.[540] used Pd3Cu nanoparticles in UiO-66 metal-organic framework at 200 °C and 1.25 MPa under low irradiation and obtained methanol at a rate of 340 μmol gcat–1 h–1. Photoradiation generates electrons from the metal-organic framework; these electrons are transferred to the adsorbed CO2* resulting in the formation of HCOO*. The interaction between CO2 and H2 is provided by the close proximity of the active sites, and the light-assisted hydrogenation takes place.
The formation of special two-dimensional structures on the support can also be promising. For example, a regular two-dimensional periodic Cu/ZnO structure was formed on Cu/Au foil[541]. The enhancement of plasmon resonance and charge separation due to the regularity allowed the rate of methanol formation under visible light irradiation to be increased by more than 180 times.
Hence, although the photoreduction of CO2 to methanol has been studied using a variety of materials and composites, the required levels of performance and stability of photocatalysts have not yet been achieved. The stability of proposed catalysts and approaches to reduce photocorrosion during CO2 reduction are still unclear. The process is complicated by the requirement to provide the effective separation of the charges formed by photoexcitation of electrons and holes, to establish the conditions for simultaneous reduction of CO2 by electrons with the participation of proton donors and oxidation of H2O to O2 by holes on the catalyst surface. The latter reaction requires special catalysts or the use of other consumable agents instead of H2O (e.g., H2). It is possible to use new alternative solvents instead of water, such as ionic liquids[542].
The design of special types of heterojunctions, the use of nanoscale particles as catalysts for reduction and oxidation, and the use of carbonaceous conductive materials make it possible to increase the catalyst productivity on methanol. The problem of developing approaches to increase selectivity for this product cannot be considered as resolved and there are still many uncertainties. In particular, there is a need to develop materials and reactors with significantly higher light absorption and also catalysts with bandgap providing maximum visible light absorption.
An interesting approach is the photoreduction of CO2 with H2 based on photothermocatalysis. Along with lowering the reaction temperature as compared to traditional thermocatalytic processes, both productivity and selectivity can be increased under thermophotocatalytic conditions. It is the activation of CO2 in the reduction reaction to methanol with the simultaneous application of photo- and thermocatalysis or photo- and electrocatalysis that may be effective in the future. An approach using multiple catalysts for sequential reduction of intermediates or by-products to methanol is also promising.
5. Conclusion
In this review we have combined two challenges facing scientists around the world:
1. How to reduce the level of CO2 emissions into the atmosphere from human activities?
2. Can we hope that the reduction of CO2 to CH3OH would produce a feedstock to replace oil?
Both problems are far from being solved. However, taking into account the efforts of researchers and the progress that characterizes the modern development of science, we can be more optimistic about the prospects of solving them.
We have already noted that the available data on the CO2 conversion to methanol using all types of catalysis are enormous and cannot be covered by a single review. Therefore, we would be grateful if the reader would appreciate our efforts and look into the directions in which research is developing. The results presented in this review show the significant progress in the development of new catalysts for the reduction of CO2 to methanol, and largely reflect the different levels of completion for the corresponding approaches.
The greatest progress from a practical point of view has been made for thermocatalytic processes: catalysts based on Cu and Zn compounds have already found industrial application in the pilot and small-scale industrial plants for the hydrogenation of CO2 to methanol using low-carbon hydrogen as a reducing agent. Electrocatalytic CO2 reduction to methanol requires the improved process selectivity and productivity for its practical implementation. Significant efforts are needed to increase the catalyst stability under electrochemical conditions, where the catalyst surface can undergo irreversible changes and poisoning. Above all, we would like to emphasize the necessity for further development of photocatalysis, which is still insufficiently successful from industrial point of view, but promising to use the limitless solar energy.
6. List of abbreviations
CNT — carbon nanotubes,
FE — Faradaic efficiency,
g-C3N4 — graphitic carbon nitride,
GHSV — gas hourly space velocity,
МАХ — layered hexagonal carbides and nitrides having the general formula Мn + 1AXn, where n = 1 – 4, M is transition metal, А is A-group element, Х is carbon or nitrogen,
MOF — metal-organic framework,
MXene — a class of two-dimensional inorganic compounds consisting of atomically thin layers of carbides, nitrides or carbonitrides of transition metals,
PEHA — pentaethylenehexamine,
PPOS — phenyl polyhedral oligomeric silsesquioxanes,
Tf — triflyl,
TOF — turnover frequency,
TON — turnover number,
RHE — reversible hydrogen electrode,
TEA — trimethylamine,
XAS — X-ray absorption spectroscopy.
References




















