




Keywords
Abstract
After the appearance of the green chemistry concept, which was introduced in the chemistry vocabulary in the early 1990s, its main statements have been continuously developed and modified. Currently, there are 10–12 cornerstones that should form the basis for an ideal chemical process. This review analyzes the accumulated experience and achievements towards the design of chemical products and processes that reduce or eliminate the use or generation of hazardous substances. The review presents the views of leading Russian scientists specializing in various fields of this subject, including homogeneous and heterogeneous catalysis, fine and basic organic synthesis, electrochemistry, polymer chemistry, chemistry based on bio-renewable feedstocks and chemistry of energetic compounds and materials. A new approach to the quantitative evaluation of the environmental friendliness of processes developed by Russian authors is described.
The bibliography includes 1761.
1. Introduction
The term ‘green chemistry’, introduced into the chemical lexicon in the early 1990s, means ‘the creation of chemical products and processes without the use or formation of harmful substances’.[1] Development of the green chemistry concept was prompted by the progressive pollution of the environment, to which the chemical industry contributes significantly. To counteract this negative trend, in 1998 Paul Anastas and John Warner formulated 12 principles of green chemistry to make chemical production less harmful to nature and humans.[2] These principles, which have remained relevant over the years,[3] include the following requirements for newly developed chemical processes and products:
— waste prevention;
— atom economy;
— less hazardous chemical processes;
— design of safer chemicals;
— safer solvents and auxiliarities;
— energy efficiency;
— renewable feedstocks;
— step-economy;
— catalysis rather than stoichiometric reactions;
— waste disposal (if any);
— real-time process monitoring;
— safer chemistry for accident (leaks, explosions, fires) prevention.
Historically, the first metrics of the environmental friendliness of chemical reactions were the atom economy (AE) criterion introduced by Barry Trost[4] in 1991, and the environmental (E) factor proposed by Roger Sheldon[5] in 1992. AE is calculated by dividing the molecular mass of the product by the sum of molecular masses of the substrates involved in the reaction, taking into account their stoichiometric coefficients, whereas the E-factor is the mass ratio of waste generated by the reaction to its product. Subsequently, these metrics were complemented by other sustainability criteria, such as the reaction mass efficiency,[6] the process mass intensity,[7] and the catalyst mass efficiency.[8] These and some other criteria make it possible to quantify various aspects of the efficiency and safety of a chemical process, and its impact on humans and the environment, and provide a theoretical basis for green chemistry in the context of sustainable development.[9-11]
To date, a number of excellent books and reviews have been published on the important role of chemical sciences in addressing the environmental issues.[12-15] However, recent review publications consider the criteria and principles of green chemistry in relation to specific, rather narrowly defined types of chemical compounds, reactions or processes.[16-20] The purpose of this review is to systematically analyze the complementarity of the green chemistry paradigm to a wide range of state-of-the-art methods of fine and industrial organic synthesis, and to assess the prospects of their application for obtaining practically relevant organic compounds and materials with minimal environmental impact. A special feature of the review is that a significant part of it is focused on green methodologies that meet several principles and criteria of green chemistry. Many of these methodologies involve the use of catalysts that reduce the activation energy of chemical reactions, allowing them to be carried out under milder conditions with less energy consumption. Organic syntheses in the presence of heterogeneous and homogeneous catalysts, including metals, their oxides and complexes, organocatalysts, photocatalysts and other types of catalysts, and their advantages and disadvantages are analyzed using a large number of examples.
The review comprises twelve chapters, and contains 1761 references to original publications, most of which have been published within the last five years. Chapter 2 considers the current metrics of green chemistry. It focuses on a recently proposed methodology for assessing the potential hazard of chemical processes to living organisms, which is based on the use of cytotoxic concentrations of all substances involved in a chemical reaction and its products as sustainability criteria. Taken together, these metrics allow a rapid and adequate assessment of the overall cytotoxicity of the reaction under consideration. Illustrative examples of bio-Profiles and bio-Strips of chemical reactions built on the basis of the obtained data are provided, and the possibilities of their application to assess the hazard of a chemical reaction for the environment and humans are demonstrated.
Chapter 3 consisting of six sections concerns the analysis of promising green methods for organic synthesis. Section 3.1 analyzes green methods of direct activation of carbon-hydrogen bonds in aromatic and non-aromatic systems, which do not require the introduction of auxiliary and protective groups and allow their direct conversion into carbon-carbon or carbon-heteroatom bonds. Such processes are extremely useful in the development of innovative technologies for the production of pharmaceuticals, plant protection chemicals and other practically relevant fine organic chemicals. Among the methods considered are green C(sp2) – H functionalization processes in cyclic systems, including catalytic processes of this type developed in recent years, and reactions of selective catalytic oxidative functionalization of aliphatic C – H groups of complex organic molecules upon treatment with hydrogen peroxide.
The paradigm of green chemistry is clearly seen in the studies aimed at developing modern variants of catalytic cross-coupling reactions of organic molecules, which is a convenient way to directly build carbon – carbon and carbon – heteroatom bonds in organic compounds. Section 3.2 shows that the Suzuki – Miyaura, Sonogashira, Heck, Chan – Evans – Lam and some other processes of this type can be carried out successfully in water (a cheap, safe and non-toxic solvent), in various water – organic mixtures, in other green solvents (ethylene glycol, glycerol, polyethylene glycols, etc.) or under neat conditions. Not only metal complexes (palladium, cobalt, iron, copper, zinc, nickel, etc.) but also their salts, oxides or nanoparticles can act as pre-catalysts, even without ligands. Such reactions can be effectively activated by microwave and ultrasound irradiation.
The asymmetric organocatalysis methodology, which is one of the most dynamically developing areas of modern organic synthesis, is also highly complementary to green chemistry. Metal-free catalysts cannot contaminate pharmacological products. They are generally stable in air and in aqueous media, which is a prerequisite for the development of green technologies suitable for the production of enantiomerically pure drugs without harmful side effects. Three promising directions of asymmetric organocatalysis that fully comply with the criteria and principles of green chemistry are discussed in Section 3.3. They include the development of highly selective, step- and atom-economic organocatalytic syntheses of biologically active compounds, organocatalysis in a continuous flow and the combined use of organo- and photocatalysts in enantioselective reactions using visible light energy.
Electric current is an extremely promising green energy source for organic synthesis. Chemical reactions, including catalytic ones, carried out under electrolytic conditions, avoid the use of traditional, often toxic, chemical oxidants and reducing agents, replacing them with electrodes that transfer or accept the electron — the most environmentally friendly reagent — from reactants. Section 3.4 summarizes the recent findings in the development of electrochemical methods for green organic synthesis. In particular, new approaches to the electrochemical synthesis of diversely functionalized complex organic molecules have been considered and the new possibilities offered by this method for modifying the reactivity of organic substrates and increasing the efficiency and selectivity of chemical transformations, including in the late synthetic stages, have been demonstrated. Much attention is paid to the use of electricity for reduction and utilization of carbon dioxide, a greenhouse gas that accumulates in the atmosphere because of the uncontrolled combustion of hydrocarbon fuels and contributes to global warming. Electroreduction of carbon dioxide converts it into single-carbon molecules necessary for chemical industry, such as carbon monoxide, formic acid, methanol, and methane, or into more complex organic compounds containing CO2 as a structural unit. The principles of electrochemical conversion of carbon dioxide into useful products, in particular, under catalytic and photoactivation conditions, and instrumentation features of such processes are considered.
A high degree of environmental friendliness is associated with chemical reactions, in which several relatively simple compounds react in series or in parallel to produce highly complex organic products in a single experimental step. Multicomponent reactions not only reduce the number of steps in chemical processes, but also significantly reduce waste related to the separation and purification of intermediate compounds. Section 3.5 presents a number of reactions of this type carried out in green solvents or reagent media using catalysts or electrocatalysis as well as ultrasonic, microwave and mechanical activation.
Heterogeneous catalysts are the most attractive for the chemical industry. Section 3.6 discusses the application of such catalysts in green redox processes of fine chemistry. In particular, the reactions of ‘hydrogen-free’ hydrogenation of multiple bonds using alcohols as hydrogen carriers, reactions of catalytic oxidation under the action of oxygen and hydroperoxides, and photocatalytic transformations of glycerol, a promising natural platform compound for organic synthesis, are highlighted. It is shown that the efficiency of mono- and bimetallic heterogeneous catalysts for such reactions can be improved by preparing them in supercritical fluid media, including carbon dioxide and alcohols.
The key to liquid-phase chemical processes is the solvent, which improve the rate and selectivity of the chemical reaction by solvating the reactants. However, once the reaction is complete the solvent should be recovered and purified, which requires additional energy and resources. In addition, many petroleum-derived solvents are toxic and pollute the atmosphere upon evaporation. It is therefore important to use alternative reaction media that are non-toxic and easily separable from reagents and products. Chapter 4 considers chemical reactions, including catalytic ones, carried out in liquid eutectic mixtures of simple and readily available non-volatile compounds, usually of natural origin, or in the liquid or supercritical carbon dioxide. Eutectic mixtures have low vapour pressures. With the proper choice of components, they are non-flammable and biodegradable. The carbon dioxide can be easily removed from the products via decompression and, being taken from the air itself, it does not contribute to global warming.
A promising trend in modern organic synthesis is the widespread use of biomass, including wood products, as a renewable raw material for the production of a variety of chemical compounds and fuels. The use of bioresources and biowaste reduces the need for petroleum products in the chemical industry and reduces greenhouse gas emissions. In addition, many natural compounds contain elements of chirality, which are essential for the development of highly effective pharmaceuticals. Chapter 5 of the review shows that natural mono- and diterpenes are excellent versatile synthons for the preparation of bioactive substances (anticancer and antibacterial drugs, antioxidants), chemical plant protection agents, chiral organocatalysts and ligands, biocompatible fluorescent markers and other useful compounds.
The application of green chemistry methods to processes providing functional organic materials, such as polymers and energetic products, is discussed in Chapter 6. Section 6.1 analyzes the features of emulsion radical polymerization in aqueous medium in the presence of surfactants as an environmentally friendly method that allows reliable control of process parameters and molecular weight characteristics of the product. The environmental impact of emulsion radical polymerization is considered and the advantages and disadvantages of new approaches proposed in recent years in this practically important field are addressed. Section 6.2 highlights the development of environmentally friendly methods for the synthesis of energetic compounds and materials. Classical nitration processes generate large amounts of waste: spent mixed acids, toxic solvents and other byproducts, the use of which is energy- and resource-consuming. The review discusses promising ‘green’ methods for carrying out these reactions without the use of sulfuric acid. In addition, green methods for producing energy-rich microsized and nanosized modifications of energetic materials in stable gases, such as carbon dioxide and low-molecular freons in liquid or supercritical state, are considered.
The implementation of the green chemistry paradigm is particularly relevant in the chemical industry, where it is necessary to account for a whole range of requirements related to economic efficiency, prevention of hazardous waste, need to increase plant productivity, minimization of fossil fuel consumption, decarbonization, changeover to renewable raw materials and a number of others. Chapter 7 of the review shows that some of these issues can be addressed on an industrial scale, based on the concept of two-phase catalysis, through the use of heterogeneous catalysts, including zeolites and metal oxides, alternative feedstock types (CO2) and energy sources (electricity or sunlight).
2. Eco-toxicological profiles of chemical reactions
Over the decades, reliable assessment of the toxic potential of chemical reactions has seemed an impossible task, mainly because of the extreme complexity of the processes involved in the interaction of chemicals with living organisms and the environment.[21-24] The increasing popularity of catalytic reactions and the development of the pharmaceutical industry in recent years has spurred the interest to possible harmful effects of the corresponding chemicals on the environment and humans.[25-28][25-28] Despite the scientific community’s desire to follow the concepts of sustainable development,[29][30] the chemical industry continues to cause significant environmental damage.
The introduction of the E factor and the concept of atom-economy helped to re-evaluate the industrial applications of chemical processes.[31-33] Metrics have been developed to calculate material balance, waste, reagent and solvent recovery potentials.[30][34] Currently, green chemistry metrics represent a specific scientific field with a significant impact on research and industrial projects.[35-37]
The methodologies that have been developed to classify chemicals according to their potential hazard to living organisms typically involve multistep procedures with testing of different biological objects and systems.[21][28][38-43] However, obtaining a reliable assessment of the potential environmental impact of a chemical process is extremely challenging. Thus, to assess the systemic effects of a chemical on an organism, many parameters should be considered, including general cytotoxicity, genotoxicity, immunotoxicity, metabolic toxicity, neurotoxicity, reproductive and embryonic toxicity, and more.[44] Therefore, none of the methodologies developed to date allow for rapid preliminary risk assessment of a wide range of chemicals and processes.
Recently, Ananikov et al.[45][46] proposed a new methodology for rapid preliminary assessment of potential hazards of chemical processes to living organisms. The idea is to use existing toxicity metrics (e.g., median lethal doses, LD50s) of substances involved in or produced by a given reaction to build diagrams (tox-Profiles) that clearly show the relative contribution of these substances to the ‘overall toxicity’ of the reaction.
The choice of toxicity metric is important. One of the most reliable metrics is considered to be the median lethal doses of substances determined in mammals such as rats and mice, since the results obtained on this basis can be extrapolated to humans with a certain degree of reliability. However, the numerical toxicity indices (LD50, IC50, EC50, etc.) currently available in the scientific literature and databases are often poorly described, and the lack of information on experimental conditions and methods of determination makes it impossible to compare indices obtained by different research groups, even in the experiments performed on the same organism.[23] In addition, chemists synthesize thousands of novel chemical compounds annually, and toxicological studies are simply failing to keep up with the synthetic ones. Ethical standards and the desire to reduce animal testing should also be kept in mind. Even for known substances, toxicity data are often not available from recognized sources such as the NLM PubChem database,[40] material safety data sheets, etc. As a result, it is currently difficult to assess the overall toxicity of even the simplest chemical reactions using the LD50 values of the compounds involved.
For this reason, we have proposed the use of cytotoxicity data to build tox-Profiles of chemical reactions, namely bio-Profiles and bio-Strips. This concept involves using the values of half-maximal cytotoxic concentrations (CC50) of all substances involved in a chemical reaction or formed during its course to estimate the ‘overall cytotoxicity’ of this reaction.[46] Cytotoxicity assays are much simpler and faster than laborious animal experiments and allow rapid cytotoxicity screening of a large number of chemicals in different cell lines. To date, this approach has been successfully applied to the analysis of such demanded chemical reactions as the Suzuki, Friedel-Crafts and Heck reactions.[46-49][46-49]
2.1. Principles of building bio-Profiles and bio-Strips of chemical reactions
The simplest way to build the bio-Profile of a reaction is to equate the area of the sections of a diagram with the mass of compounds involved or formed in the reaction, and to equate the colour of the sectors to the toxicity indices of these substances measured in a particular organism (Fig. 1a). The most toxic substance corresponds to the red colour, the least toxic substance corresponds to the green colour and all the other substances correspond to the intermediate shades of red, orange and yellow. In the given example, the starting materials SM1 and SM2 correspond to sectors of small area according to their quantities. The diagram also includes the target product P, the byproduct BP and the substance R (an auxiliary reagent). The sector with the smallest area corresponds to the catalyst CT, and the sector with the largest area corresponds to the solvent S.
![[{"id":"Xlmmhpg8eH","type":"paragraph","data":{"text":"Exemplary bio-Profiles of a chemical reaction (shown in general form above the table). In diagram <i>a</i>, the area of the sectors corresponds to the mass of substances involved in the reaction, while in diagram <i>b</i>, the area of the sectors corresponds to the ‘normalized cytotoxicity’ (NC) of the substances (the ratio of the amount of a substance to its half-maximal cytotoxic concentration (CC<sub>50</sub>)). The table shows arbitrary values used for the example. The colours of the sectors correspond to CC<sub>50</sub>s of the substances in a given cell line; the relative cytotoxicity scales are given below the diagrams. The most toxic substance is shown in red, the least toxic substance is shown in green and all the other substances are shown in the intermediate shades of red, orange and yellow. Reproduced from Ref. 46 with permission from the Royal Society of Chemistry."}}]](/storage/images/resized/YcUFOi4ZaxPcbPgyYf6hV6dk4P30v07RKThNJHzv_xl.webp)
When concentration metrics such as half-maximal cytotoxic concentrations (CC50s) are used as indicators of the substance toxicity, the relative contribution of each substance to the ‘overall toxicity’ of the reaction system can be visualized by equating the area of the sectors in the diagram to the ‘normalized cytotoxicity’ (NC) of the compounds (see Eq. (1)):
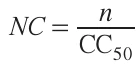
where n is the amount of a substance involved in a particular reaction (mmol), and CC50 is a half-maximal cytotoxic concentration (mmol L–1, mM) measured in a certain cell line. Accordingly, the larger the sector area, the greater the contribution of the substance to the overall cytotoxicity of the process. The colour of the sectors also corresponds to the CC50 values of the substances. Fig. 1b shows a bio-Profile of the same reaction plotted with NC of the substances. It can be seen that in this case the substances with higher cytotoxicity (i.e. lower CC50 values) have larger sectors (see, e.g., starting material SM1, catalyst CT, reagent R and product P). Conversely, the substances with lower cytotoxicity correspond to the sectors of smaller area (starting material SM2, solvent S). The byproduct BP, which has low cytotoxicity and is formed in small amounts, looks the same in both diagrams.
The bio-Strip of a chemical reaction is a compact form of the bio-Profile in which each reaction is represented by a strip consisting of sections.[31] These sections correspond to the substances involved in a particular chemical reaction and the length of the sections is equal to the ‘normalized cytotoxicity’ of these substances (see Eq. (1)). Therefore, the longer the section, the greater the contribution of the substance to the overall cytotoxicity of the reaction. The colours of the sections correspond to the CC50s of the substances in a given cell line: the substance showing the maximum cytotoxicity (i.e., having the lowest CC50) is shown in red, the substance showing the minimum cytotoxicity (i.e., having the highest CC50) is shown in green, and the remaining substances are shown in the intermediate shades of red, orange, and yellow. An example of bio-Strips for six methods of 1,1'-biphenyl synthesis is given in Fig. 2. The reactions are shown above the bio-Strips, while the common cytotoxicity scale and a list of reaction names and abbreviations are shown at the bottom of the figure. Each bio-Strip is also supplied with a bio-Factor (BF), which shows the change in the overall cytotoxicity of the reaction over time (see Eq. (2)):
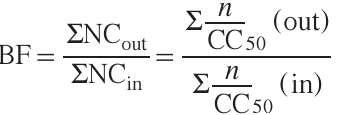
where out and in denote substances leaving the reaction (products, byproducts and reagents that can be regenerated, such as catalysts and solvents) and entering the reaction (starting materials, catalysts, solvents and other reagents), respectively. If BF >1, the overall cytotoxicity of the reaction increases over time; if BF <1, the overall cytotoxicity of the reaction decreases.
![[{"id":"jolMIavinW","type":"paragraph","data":{"text":"bio-Strips of six methods of synthesis of 1,1'-biphenyl using various catalysts (Pd(OAc)<sub>2</sub> (A), PdCl<sub>2</sub> (B) or Pd(acac)<sub>2</sub> (C)) and solvents (ethanol (A) or N-methylpyrrolidone (NMP) (B)). The reactions are shown above the bio-Strips, while the common cytotoxicity scale and explanations of the reaction names and abbreviations are given at the bottom of the Figure. The lengths of the bio-Strip sections correspond to the NC of the substances, and the colours correspond to the CC<sub>50</sub>s of these substances determined in a given cell line (here, human colorectal adenocarcinoma CaCo-2 cells). The BF values of the reactions are also given within the bio-Strips. Reproduced from Ref. 49 with permission from Elsevier."}}]](/storage/images/resized/H7hhHhWW4PmP4xL1gt7bhDn9mOrtlN7jqDUH1gT7_xl.webp)
The reactions shown in Fig. 2 differ in the starting materials, catalysts, reagents and solvents, as indicated by the first, second, third and fourth letters of the reaction names. For clarity, the same starting materials (phenylboronic acid (SM1) and iodobenzene (SM2, A), the first letter in the reaction name) and the same reagent (K2CO3 (R, A), the third letter in the reaction name) are used in all the reactions. In the case of the catalyst (CT), A = Pd(OAc)2, B = PdCl2 and C = Pd(acac)2; in the case of the solvent (S), A = ethanol and B = N-methylpyrrolidone (NMP). Looking at the bio-Strips of these methods for the synthesis of 1,1'-biphenyl, one can immediately assume that catalysts A (Pd(OAc)2) and B (PdCl2) and solvent A (ethanol) are more beneficial in terms of their contribution to the overall cytotoxicity of the reactions. This conclusion is supported by both the length of the corresponding sections of the bio-Strips and their colours.
In addition to bio-Factors, the cytotoxicity potentials of reactions are also used: (1) the initial cytotoxicity potential (CPi), or the cytotoxicity potential of the substances entering the reaction (see Eq. (3)); (2) the final cytotoxicity potential (CPf) or the cytotoxicity potential of the substances leaving the reaction (see Eq. (4)); and (3) the relative final cytotoxicity potential (CPf_rel) or the cytotoxicity potential of the substances leaving the reaction except for the target product (see eq. 5).

where out and in denote substances leaving the reaction and entering the reaction, respectively. Thus, CPi and CPf essentially quantify the hazard of a specific chemical reaction to a given cell culture, i.e. how many litres of the culture medium can be ‘poisoned’ by the substances entering or leaving the reaction. CPf_rel is a special case of CPf that does not account for the cytotoxicity of the target product, but does take into account the byproducts.
2.2. Application of bio-Strips and cytotoxicity potentials for environmental and human hazard assessment of chemical reactions
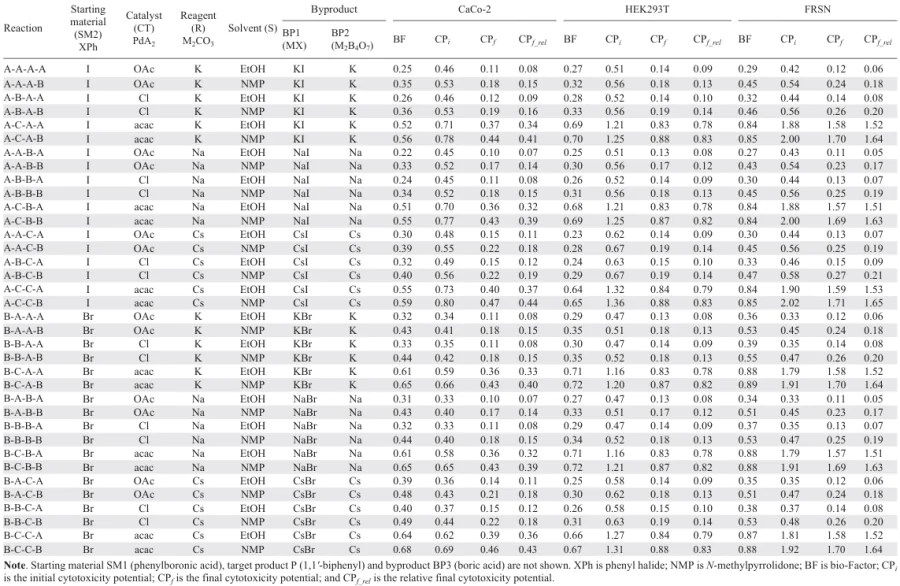
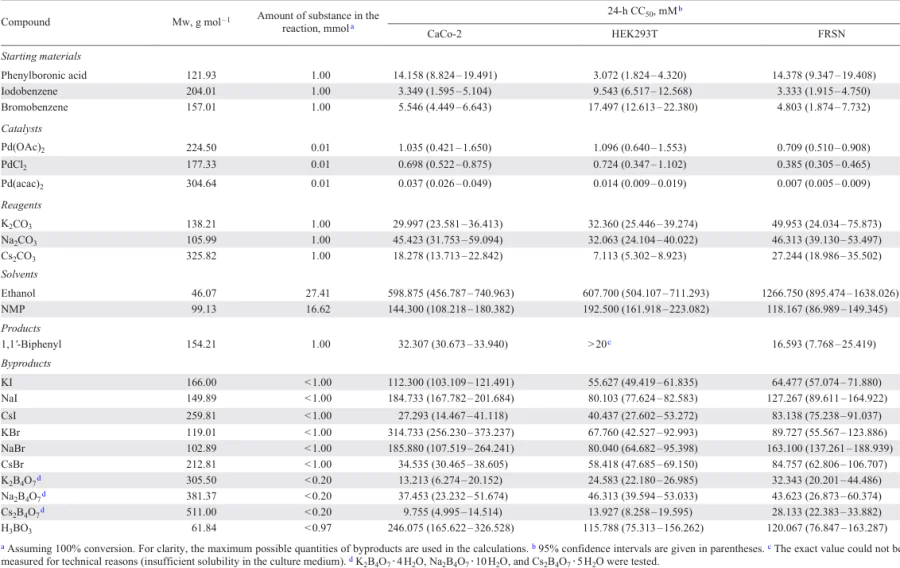
Here we analyze in detail the use of bio-Strips to identify the ‘safest’ and ‘most hazardous’ components of chemical reactions, using the synthesis of 1,1'-biphenyl as an example.[49] We consider 36 methods for the synthesis of 1,1'-biphenyl which differ in the (1) starting materials (iodobenzene, bromobenzene), (2) catalysts (Pd(OAc)2, PdCl2, Pd(acac)2), (3) reagents (K2CO3, Na2CO3, Cs2CO3), and (4) solvents (ethanol, NMP). bio-Strips for these reactions were build by using half-maximal cytotoxic concentrations after 24 hours of incubation (24-h CC50s) measured in three cell lines: CaCo-2 (human colorectal adenocarcinoma), HEK293T (human embryonic kidney) and FRSN (human foreskin mesenchymal stem cells). Summary information on these reactions, including bio-Factors and cytotoxicity potentials, is presented in Table 1; the initial data used in the calculations are given in Table 2. bio-Strips plotted against the CC50 values obtained in the CaCo-2, HEK293T, and FRSN cell lines are shown in Fig. 3, Fig. 4 and Fig. 5, respectively.
![[{"id":"PaIk5fJvOt","type":"paragraph","data":{"text":"bio-Strips of 36 methods of synthesis of 1,1'-biphenyl built by using CC<sub>50</sub> values measured in the CaCo-2 cell line. The letters in the reaction names indicate, correspondingly, the type of starting material (SM2: iodobenzene (A) or bromobenzene (B)), catalyst (CT: Pd(OAc)<sub>2</sub> (A), PdCl<sub>2</sub> (B) or Pd(acac)<sub>2</sub> (C)), reagent (R: K<sub>2</sub>CO<sub>3</sub> (A), Na<sub>2</sub>CO<sub>3</sub> (B) or Cs<sub>2</sub>CO<sub>3</sub> (C)) and solvent (S: ethanol (A) or NMP (B)). The lengths of the sections correspond to the NC values of the substances, and the colours correspond to their CC<sub>50</sub> values (see the cytotoxicity scale above the bio-Strips). The bio-Factor values are given within the bio-Strips. Reproduced from Ref. 49 with permission from Elsevier."}}]](/storage/images/resized/VTW47u27PNI2NboqOekVsbrvsCmAnOJertUA79T8_xl.webp)
![[{"id":"mVwPbPlOii","type":"paragraph","data":{"text":"bio-Strips of 36 methods of synthesis of 1,1'-biphenyl built by using CC<sub>50</sub> values measured in the HEK293T cell line. The letters in the reaction names indicate, correspondingly, the type of starting material (SM2: iodobenzene (A) or bromobenzene (B)), catalyst (CT: Pd(OAc)<sub>2</sub> (A), PdCl<sub>2</sub> (B) or Pd(acac)<sub>2</sub> (C)), reagent (R: K<sub>2</sub>CO<sub>3</sub> (A), Na<sub>2</sub>CO<sub>3</sub> (B) or Cs<sub>2</sub>CO<sub>3</sub> (C)) and solvent (S: ethanol (A) or NMP (B)). The lengths of the sections correspond to the NC values of the substances, and the colours correspond to their CC<sub>50</sub> values (see the cytotoxicity scale above the bio-Strips). The bio-Factor values are given within the bio-Strips. Reproduced from Ref. 49 with permission from Elsevier."}}]](/storage/images/resized/rLzrpbnEtckzPsBwIyo9Qqz7IM3jAYutXjZ3aI0w_xl.webp)
![[{"id":"ri6vx28F9h","type":"paragraph","data":{"text":"bio-Strips of 36 methods of synthesis of 1,1'-biphenyl built by using CC<sub>50</sub> values measured in the FRSN cell line. The letters in the reaction names indicate, in order, the type of starting material (SM2: iodobenzene (A) or bromobenzene (B)), catalyst (CT: Pd(OAc)<sub>2</sub> (A), PdCl<sub>2</sub> (B) or Pd(acac)<sub>2</sub> (C)), reagent (R: K<sub>2</sub>CO<sub>3</sub> (A), Na<sub>2</sub>CO<sub>3</sub> (B) or Cs<sub>2</sub>CO<sub>3</sub> (C)) and solvent (S: ethanol (A) or NMP (B)). The lengths of the sections correspond to the NC values of the substances, and the colours correspond to their CC<sub>50</sub> values (see the cytotoxicity scale above the bio-Strips). The bio-Factor values are given within the bio-Strips. Reproduced from Ref. 49 with permission from Elsevier."}}]](/storage/images/resized/7YHa9a6O4lPPC9JbIEARHFiiRlBbZVbqNPq31hS2_xl.webp)
Upon looking at the bio-Strips of the 1,1'-biphenyl syntheses analyzed, several immediate assumptions can be made.
1. Catalyst C (Pd(acac)2) contributes most to the overall cytotoxicity in all the cell lines tested (the effect is particularly evident in FRSN cells where the 24-h CC50 of this compound is as low as 7 mM; see Table 2 and Fig. 5).
2. Of the two solvents tested, solvent A (ethanol) was significantly less cytotoxic than solvent B (NMP).
3. Reagent C (Cs2CO3) was more cytotoxic than reagents A and B (K2CO3 and Na2CO3, respectively).
As for the starting materials, substance B (bromobenzene) contributes less to the overall cytotoxicity than substance A (iodobenzene), but this effect is weaker than that of the catalyst and solvent. However, since the phenyl halide and the reagent determine the byproducts formed, their choice requires further analysis. In the case of phenyl halide, which determines the byproduct salt, KBr has lower cytotoxicity than KI in CaCo-2 cells, whereas the cytotoxicity of these two potassium salts is comparable in HEK293T and FRSN cells. NaI and NaBr as well as CsI and CsBr show comparable cytotoxicity in all the cell lines tested. Therefore, iodobenzene and bromobenzene are similar in terms of the cytotoxicity of the bromides and iodides formed.
Potassium, sodium or cesium tetraborate is also formed as a byproduct during the reaction, depending on the reagent (K2CO3, Na2CO3 or Cs2CO3, respectively). Cs2B4O7 shows higher cytotoxicity than Na2B4O7 in the CaCo-2 and HEK293T cell lines, whereas all three tetraborates show comparable cytotoxicity in FRSN cells. This observation supports the above-discussed suggestion that K2CO3 or Na2CO3 is preferable in the reaction.
To summarize, reactions with Pd(acac)2 as a catalyst, NMP as a solvent and Cs2CO3 as a reagent seem to be more hazardous approaches in terms of the total cytotoxicity of the reactions.
Regarding the bio-Factors of the considered methods of 1,1'-biphenyl synthesis, it can be seen from Table 1 that in all cases the BF values are below 1, and consequently the overall cytotoxicity decreases during the reaction. The main reason for this decrease is the difference between the cytotoxicity of the products and that of the starting materials: the target product and byproducts, including boric acid, are mostly less cytotoxic than the phenylboronic acid and phenyl halides used as starting materials. Palladium salts, despite their high cytotoxicity, are present in both the numerator and denominator of Eq. (2) and therefore do not contribute significantly to the BF value. Nevertheless, the bio-Factors of the reactions catalyzed by Pd(acac)2 are higher than those of the reactions using Pd(OAc)2 or PdCl2 (see, e.g., reactions A-A-A-A, A-B-A-A and A-C-A-A-A or A-A-A-B, A-B-A-B and A-C-A-B in Table 1). This difference is particularly evident in the case of FRSN cells, where Pd(acac)2 shows the highest cytotoxicity. NMP as a solvent also increases BFs in some cases, but not as significantly (see, e.g., reactions A-A-A-A and A-A-A-B or A-B-A-A and A-B-A-B in Table 1).
Fig. 6 shows a comparison of CPi, CPf and CPf_rel of all the considered routes of synthesis of 1,1'-biphenyl based on the data obtained in three cell lines (see Table 1 for exact values). Such presentation of the data makes it possible to immediately identify the synthetic routes with the highest and lowest cytotoxicity potentials of the initial and final substances. Obviously, the use of Pd(acac)2 significantly increases CPi, CPf and CPf_rel in all the cases (see diagrams labeled N-C-N-N in Fig. 6, where N stands for any possible letter, i.e. A, B or C, depending on the reaction component). The contributions of NMP and Cs2CO3 appear to be much smaller or, in the latter case, negligible compared to this catalyst (see the reaction diagrams labelled N-N-N-B and N-N-C-N, respectively, in Fig. 6). Thus, the analysis of the initial and final cytotoxicity potentials of chemical reactions allows suggesting less dangerous synthetic routes from the viewpoint of the cytotoxicity of their components.
![[{"id":"du_tH7O9vO","type":"paragraph","data":{"text":"Cytotoxicity potentials of 36 routes of synthesis of 1,1'-biphenyl calculated from CC<sub>50</sub> values measured in (<i>a</i>) CaCo-2, (<i>b</i>) HEK293T and (<i>c</i>) FRSN cell lines. Reproduced from Ref. 49 with permission from Elsevier."}}]](/storage/images/resized/yZiq8v2MqN6lEFu7tKiy3JRhfdESMf4v5agqEFnp_xl.webp)
2.3. Effect of the cell culture choice on the bio-Stripes of chemical reactions
The choice of the cell line used to obtain CC50 values for the construction of bio-Profiles and bio-Strips of chemical reactions can be crucial. Three human cell lines of different origin were used in the publication[49]: CaCo-2 (colorectal adenocarcinoma cells), HEK293T (human embryonic kidney, immortalized non-cancer cells) and FRSN (foreskin mesenchymal stem cells, non-immortalized fibroblast-like cells). Due to genomic differences as well as different origins, these cells were expected to have different sensitivities to chemicals. This assumption was confirmed for at least some components of the reactions studied. A comparison of the 24-h CC50 values measured in the three cell lines is presented in the form of a heat map in Fig. 7. The colour of the cells in the Table corresponds to the given 24-h CC50 values, from the lowest (red) to the highest (green).
![[{"id":"0QM9JzvzlI","type":"paragraph","data":{"text":"Comparison of cytotoxicity of the studied chemicals in three cell lines (CaCo-2, HEK293T, and FRSN). The colour of the cells in the table corresponds to the 24-h CC<sub>50</sub> values given, from lowest (red) to highest (green). The colour legend for each cell line is shown at the bottom, with the midpoint corresponding to the 50th percentile."}}]](/storage/images/resized/q4ifhyd8GsncVS50Pue8qF9Tq6aNgIgusNpYMAyh_xl.webp)
It should be noted that FRSN cells were much more sensitive to Pd(acac)2 than CaCo-2 cells. A similar effect was observed for PdCl2, 1,1'-biphenyl, KI, NaI, KBr and H3BO3: for all these substances, the 24-h CC50 values obtained in FRSN cells were much lower than those obtained in CaCo-2 cells. The opposite effect was observed for ethanol, CsI, CsBr, K2B4O7 and Cs2B4O7 . Comparing the CaCo-2 and HEK293T cells, the latter were more sensitive to phenylboronic acid, Pd(acac)2, Cs2CO3, KI, NaI, KBr, NaBr and H3BO3, but less sensitive to iodobenzene, bromobenzene, CsBr and K2B4O7 .
Thus, no clear correlation was observed between the type of cell line and its sensitivity to the substances tested. However, it should be noted that all the cells showed the highest sensitivity to palladium salts (especially Pd(acac)2) and the lowest sensitivity to ethanol. The sensitivity to the starting materials (phenylboronic acid, iodobenzene and bromobenzene) was also quite high in all the cases. Among the tested salts, the cesium salts (both reagent and byproducts) showed the highest cytotoxicity. Based on these observations, it can be assumed that in this case the choice of cell line was moderately or even slightly reflected in the bio-Strips. Of course, it should be borne in mind that this conclusion concerns the order of cytotoxicity of the substances tested rather than the exact 24-h CC50 values and the corresponding cytotoxicity potentials. In any case, the use of a large number of cell lines of different origins allows a more reliable assessment of the potential hazard of chemical reactions to animals and humans.
The bio-Profile concept is therefore universal and can be used to visually and quantitatively assess the effect of a chemical reaction on any biological object. The choice of object or biochemical process depends entirely on the objectives of the study. For example, in the case of industrial chemical reactions, it is expedient to use the most sensitive organisms in the ecosystems that may be affected by these reactions, whereas for laboratory chemical reactions with narrower applications, it is logical to choose mammals to model the possible harmful effects of reaction components on humans. Suitable microorganisms can be used for bio-Profiles of biocatalytic processes.
Cell cultures are versatile biological systems for rapid screening for cytotoxicity of a large number of chemicals at the first stage of investigation of their toxicity potential. In particular, skin fibroblasts are often used as a simplified model in toxicology.[50][51] The low cost and relative simplicity of cytotoxicity testing make the cell cultures a suitable biological target for the construction of bio-Profiles, which can be used to pre-evaluate the harmful effects of chemical processes on living organisms and to identify the most toxic substances involved in these processes. The obtained data can subsequently form the basis for more specialized toxicological studies on higher organisms. bio-Profiles will help to select substances and processes in need of such a study.
3. Promising green strategies of organic synthesis
The key role in the design of green chemical processes belongs to the choice of the method that would provide the most efficient implementation of the process with a minimum environmental impact. According to the basic principles of green chemistry, an ideal method should produce no waste and employ safe reactants, solvents and auxiliary materials that should be mainly obtained from renewable feedstock. The method should exclude the formation of toxic products and also be resource-saving, energy-efficient and safe for humans, flora and fauna. It is clear that while developing a new synthetic approach, chemists have to sacrifice some of the above principles. Nevertheless, the set of these principles as well as the PASE (pot, atom, step economy) concept[52] must be necessarily borne in mind as important benchmarks. This chapter of the review addresses synthetic methods that largely comply with the green chemistry requirements and can be recommended for the use in research and teaching chemistry laboratories and as a possible base for the design of new, environmentally benign processes of small-scale chemical production.
3.1. Novel methods for the direct C – H functionalization in aromatic and non-aromatic systems: contribution to green chemistry
The С – Н bond is widely spread in the world of organic compounds and, therefore, it is potentially one of the most important structural groups. Quite naturally, direct C – H functionalization reactions with a variety of mechanisms have always been of interest for organic chemists. The reactions that do not require the introduction of auxiliary groups and allow direct transformation of the carbon–hydrogen bond to C – C or C – Х bond (Х = heteroatom) are especially attractive.[53] These strategies markedly reduce the number of steps in the synthesis of a target organic compound (step economy).[54] In this section, we consider the data published in recent years on the most interesting and promising methods for direct functionalization of the C(sp2) – H and C(sp3) – H bonds in structurally diverse aromatic and non-aromatic systems and analyze the benefits and drawbacks of each method.
3.1.1. Green methods for the direct C(sp2) – H functionalization in cyclic systems
First, we will analyze the green strategies for the direct C(sp2) – H functionalization of cyclic systems developed in recent years. These processes receive a great deal of attention, as evidenced by the number of publications, review articles and monographs addressing this topic.[55-65] The nature and the range of reactants suitable for the С – Н functionalization are being expanded and the reaction mechanisms are being studied. Meanwhile, many problems associated with implementation of PASE reactions,[52] in particular direct C(sp2) – H modification of organic compounds, have not yet been solved.
Herein, we consider characteristic features of non-catalytic approaches to C(sp2) – H-bond functionalization in aromatic and non-aromatic cyclic systems as well as modern protocols for conducting these reactions that include the use of metal catalysts and organocatalysts. The methods based on the catalytic activation of reactants using alternative energy sources (photoredox catalysis, electrocatalysis and mechanocatalysis) are discussed in other parts of this review.
3.1.1.1. Non-catalytic methods of the C(sp2) – H functionalization of cyclic systems
3.1.1.1.1. Electrophilic and nucleophilic C(sp2) – H functionalization
The non-catalytic C(sp2) – H functionalization methods date back to the mid-19th century and are related, first of all, to the development of the electrophilic aromatic substitution (SEAr) of hydrogen and discovery of nitration, sulfonation, acylation, azo coupling as well as the Vilsmeier, Kolbe – Schmitt, Bischler – Napieralsky reactions and many other name reactions used to introduce halogen atoms, nitro and sulfonic groups, and acyl, chlorosulfonyl, haloform, and other electrophilic residues into the aromatic ring (Scheme 1).
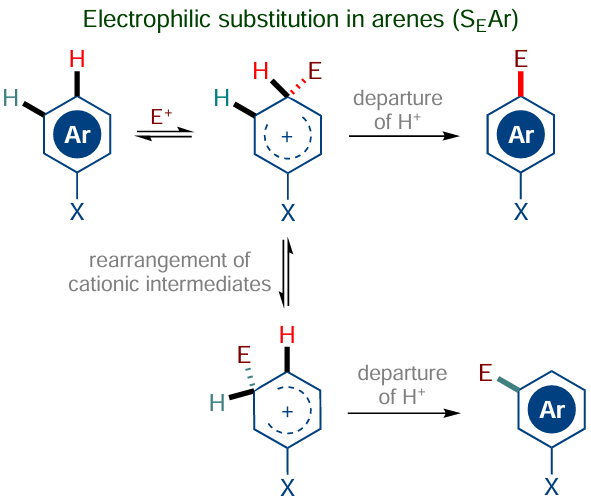
The reactions of arenes with electrophiles are a widespread and well-studied type of C(sp2) – H functionalization, regarding the reaction mechanism (SEAr) and scope of applicability. A great contribution to investigation of the SEAr mechanism and rearrangements of cationic intermediates was made by Novosibirsk chemists V.G.Shubin, V.A.Barkhash and V.D.Shteingarts under the supervision of Academician V.A.Koptyug, who were awarded the Lenin Prize in 1990 for the series of studies entitled Modern Problems of the Chemistry of Arenonium Ions, and by American chemists headed by Professor G.A.Olah, who was awarded the Nobel Prize in chemistry in 1993.
The electrophilic substitution of hydrogen in arenes appears to be quite a natural reaction, because neither the attack of the arene by an electrophilic species nor elimination of the proton from the intermediate arenonium ion is associated with a high energy barrier. A different situation arises when arenes are attacked by nucleophilic reagents to give anionic or neutral σH adducts 1 (Scheme 2), which are not prone to eliminate a proton (hydrogen atom) without assistance of an oxidant or auxiliary agent.
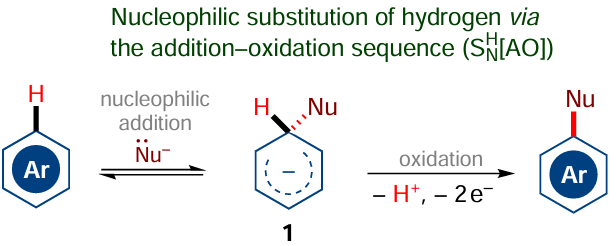
Therefore, the SEAr reactions receive much less attention in this review than nucleophilic or radical C(sp2) – H functionalization reactions, which have been developed much later. One of the first examples of nucleophilic C(sp2) – H functionalization[66-68] is the Chichibabin amination, which was first described in 1914. The reaction proceeds under fairly drastic conditions (heating with sodium amide in xylene) and produces molecular hydrogen (Scheme 3); however, the mechanism of this reaction has long been obscure.
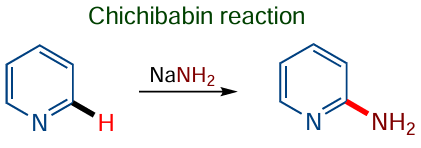
This topic was actively pursued in the 1970s, after O.N.Chupakhin initiated an extensive series of studies dealing with the nucleophilic С – Н functionalization of arenes. Together with Academician I.Ya.Postovskii, O.N.Chupakhin published the first review on the nucleophilic substitution of hydrogen in the Russian Chemical Reviews journal.[69] These reactions were designated as SNH. The Chichibabin reaction has attracted attention of many chemists both in Russia (A.F.Pozharskii,[70] A.V.Gulevskaya,[71] I.V.Borovlev,[72] etc.) and abroad (H.van der Plas,[73] M.Mąkosza[74]). The optimal conditions for the reaction were selected, more efficient oxidation systems were proposed, and views on the reaction mechanism were developed for both oxidative and eliminative types.
The mechanism of the nucleophilic aromatic substitution of hydrogen can be considered as a sort of analogue of the SEAr mechanism, but with the opposite polarity.[75] Indeed, the key intermediate of the SNH reactions is the anionic sН adduct 1 (see Scheme 2),[76] resulting from the addition of a nucleophilic species to the unsaturated ring. Rearomatization can be accomplished by oxidation of the intermediate sН adduct with the loss of a proton and two electrons (addition — oxidation, SNH[AO], mechanism).
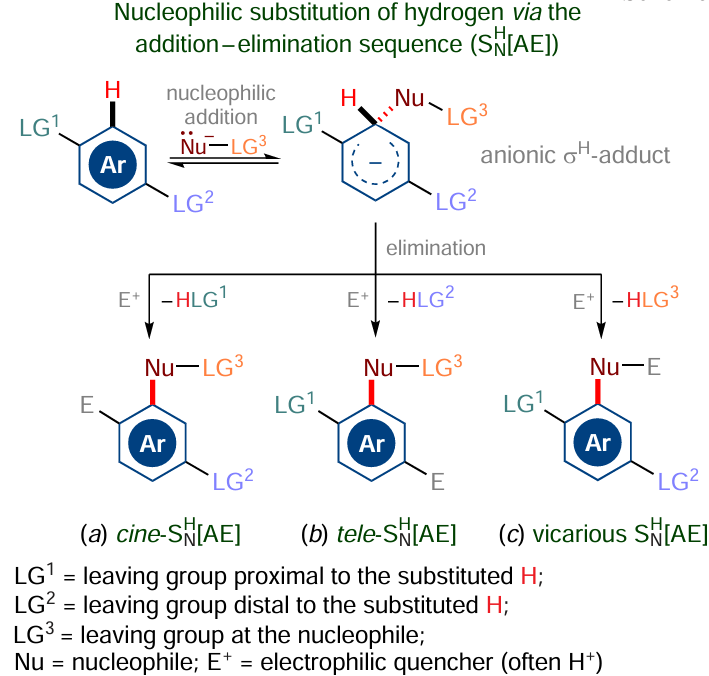
Alternatively, elimination may take place because of the presence of a vicarious leaving group in the substrate or the nucleophilic agent (addition — elimination, SNH[AE], mechanism). This accounts for the formation of unusual cine- and tele-substitution products (Scheme 4a,b) and gives rise to the reaction called vicarious nucleophilic substitution of hydrogen (Scheme 4c).[74-77]7
The range of reactions that follow the SNH mechanism is still being supplemented with new examples. For instance, in 2022, Mandler et al.[78] reported successful С – Н amination of nitro-substituted heteroarenes (pyridines, azoles and thiophenes) and nitrobenzene derivatives 2 with (hetero)aromatic amines 3 (Scheme 5).

This reaction smoothly proceeds in an open flask at room temperature in the presence of lithium bis(trimethylsilyl)amide (LiHMDS). These conditions significantly facilitate conduction of the reaction in comparison with the classic Chichibabin amination, which proceeds in liquid ammonia at low temperatures.[76] According to the results obtained by the authors, the reaction follows the SNH[AO] mechanism, with air oxygen acting as the oxidant.[78] The obvious advantages of this approach, as regards green chemistry, are the short reaction time (10 min), the use of an environmentally benign oxidant, and the possibility of carrying out the reaction in a relatively low-toxic solvent (THF) at room temperature. The drawbacks of the method include modest yields of products 4, which can be somewhat increased by using a greater excess of the starting nitroheteroarene and LiHMDS.
In the same year of 2022, Chang and co-workers demonstrated the possibility of C – H amination of pyrimidines,[79] which was distinguished by C(2)-regioselectivity. It was found that pyrimidine N-oxides 5 (Scheme 6) generated in situ by oxidation of appropriate pyrimidines react with N-nucleophile 6 in the presence of trifluoromethanesulfonic anhydride to give mainly the products of C(2)-functionalization of the pyrimidine ring in 7.
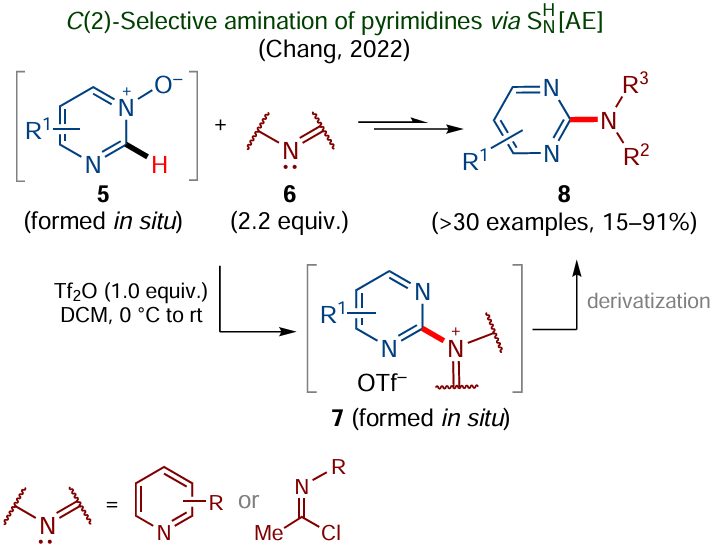
N-Nucleophiles 6 can be represented by imidoyl chlorides or pyridine derivatives containing electron-withdrawing cyano, CF3 or benzoyl substituents. However, the reaction with 4-dimethylaminopyridine (DMAP) afforded mixtures of C(2)- and C(4)-substituted products. Note that the C(2)-substituted products 7 can be subjected to subsequent one-pot derivatization to give pyrimidine amino derivatives 8.
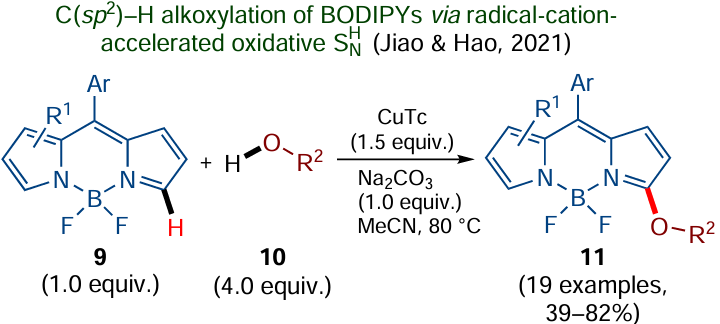
Another example of the SNH reaction unusual from the mechanistic standpoint was reported in 2021 by Jiao, Hao and co-workers[80] (Scheme 7). The authors proved the possibility of direct α-alkoxylation of BODIPY derivatives 9 via nucleophilic substitution of hydrogen; furthermore, they found that the reaction can be activated by the preliminary single-electron oxidation of BODIPY 9 to the corresponding radical cation by treatment with copper(I) thiophene-2-carboxylate (CuTc). This radical cation reacts with aliphatic alcohols 10 at one of the α-carbon atoms of the pyrromethene system. The neutral radical resulting from the addition of О-nucleophile undergoes one more single-electron oxidation, resulting in the target product 11.
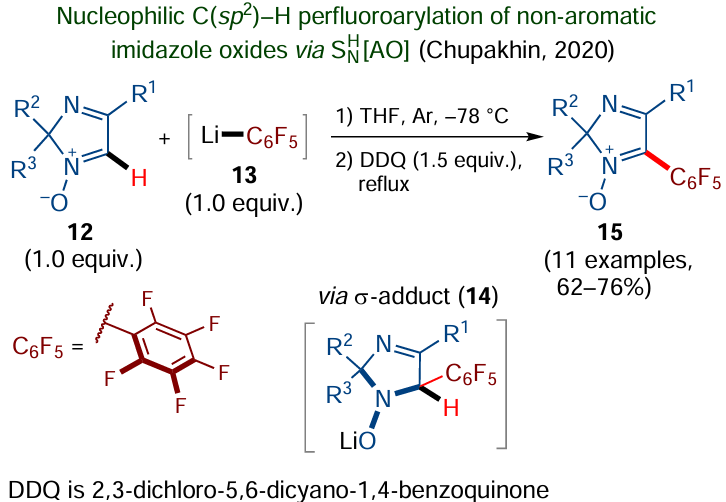
It is noteworthy that the SNH reaction concept is no longer limited to (hetero)aromatic compounds. Over the last 10 – 15 years, this strategy was extended to non-aromatic unsaturated substrates, in particular aldimine derivatives containing a C(sp2) – H bond at the azomethine moiety.[81][82] As an example, consider the reaction (Scheme 8) that is used to introduce a perfluorophenyl moiety into non-aromatic 2H-imidazole 1-oxides 12,[83] which can be regarded as cyclic nitrones. In this case, pentafluorophenyllithium 13,[84] obtained in situ by lithiation of pentafluorobenzene, was used as the C-nucleophile. The nucleophile addition to the imidazole ring gives unstable 𝜎-adduct 14, which is then transformed along both the oxidation and elimination pathways. The synthesized 2H-imidazole derivatives 15 are promising as fluorophores exhibiting the intramolecular charge transfer effect.
While characterizing the contribution of the nucleophilic C(sp2) – H functionalization to the development of the green chemistry concept, we would like to note that particularly this strategy makes it possible to avoid the introduction of auxiliary groups; if activation is still required, it can often be performed in situ, which markedly simplifies the targeted synthesis. In addition, SNH reactions can often proceed as cross-dehydrogenative coupling that increases the atom efficiency of the synthesis. Furthermore, the SNH strategy usually does not require the use of transition metal-based reagents, although in some cases, these compounds [e.g., manganese, iron(III), chromium(VI), copper and cerium salts] can be used as oxidants. Generally, an excess of oxidant or other auxiliary reagent in nucleophilic C(sp2) – H-functionalization is one of the major factors that restrict the green potential of the reaction. Fortunately, many SNH reactions can be implemented using air oxygen, an almost ideal oxidant, since it gives water as a by-product. Also, electrochemical procedures for aromatization of the relatively stable 𝜎-adducts resistant to aerobic oxidation have been actively developed in recent years.[85-87] In view of the fact that the scope of applicability of nucleophilic C(sp2) – H functionalization is constantly expanding and is no longer restricted to reactions of arenes, and also recalling the active development of the inventory of preparative methods, one can confidently predict further progress in the SNH concept and the utility of this reaction as an effective green chemistry method.
3.1.1.1.2. Radical C(sp2) – H functionalization
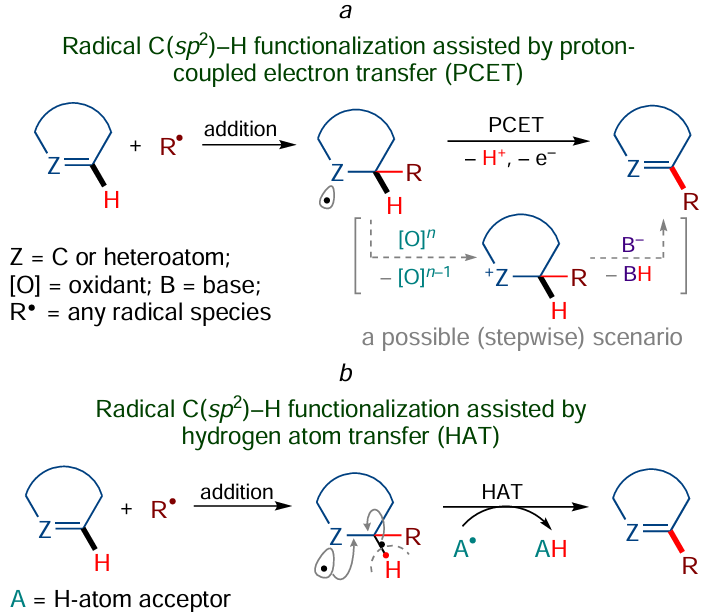
Yet another potent tool of C – H functionalization is the radical substitution reactions, which are actively used to modify not only sp2-hybridized, but also fully saturated systems. The functionalization of C(sp2) – H bonds in unsaturated systems follows two main mechanisms (Scheme 9). The first one is based on proton-coupled electron transfer (PCET),[88-90] which may occur either consecutively [single-electron transfer (SET → proton transfer (PT)] (or in the reverse order) or synchronously [concerted proton – electron transfer (CPET)] (Scheme 9а).* The latter mechanism can be regarded as a sort of the former one and implies the transfer of a hydrogen atom (HAT) as a unitary species[91-96] (Scheme 9b). Note that the greater part of PCET and HAT reactions can also occur in the presence of catalysts, in particular using photoredox catalysis.[91-94]
A useful method of radical C(sp2) – H functionalization is the Minisci reaction used to alkylate electron-deficient heteroarenes by treatment with nucleophilic carbon-centred radicals. Over more than half a century of history of this synthetic approach, various protocols have been found, some of them not requiring the use of transition metal-based catalysts.[97-99] As an example of successful use of the non-catalyzed Minisci reaction, consider direct C(sp2) – H carbamoylation of purine bases 16, particularly adenine, guanine and xanthine derivatives (Scheme 10).[100]
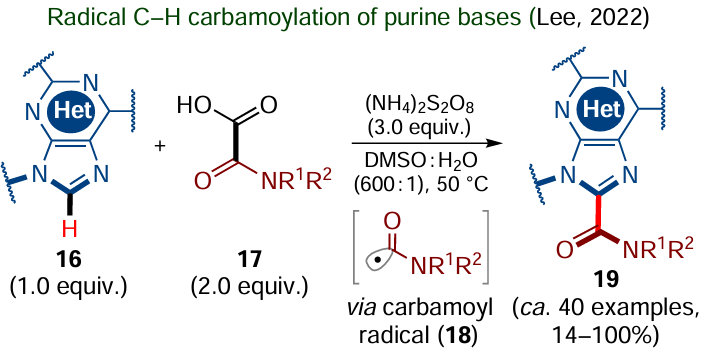
This synthetic protocol involves the generation of active carbon-centred radicals 18 by decarboxylation of N-substituted oxamic acids 17, which, furthermore, increase the electrophilicity of purine substrates 16 via their protonation. The decarboxylation is initiated by the addition of ammonium persulfate, which decomposes to give SO4–• radical anions, which in turn trigger the transfer of the unpaired electron from the oxamate molecule with subsequent elimination of CO2. The resulting carbamoyl radical 18 attacks N-protonated purine, yielding a radical cation adduct; in the presence of persulfate derivatives, the adduct is converted to the final carbamoylation product 19 via the proton and electron loss. According to the authors, this may take place by both the PCET and HAT mechanisms. The benefits of this approach include a broad substrate tolerance, good product yields and no need for transition metal-based reagents. Meanwhile, there are also drawbacks, which include the need to introduce N-protecting groups into adenines and guanines and the use of excess oxamic acids 17 and ammonium persulfate.[100]
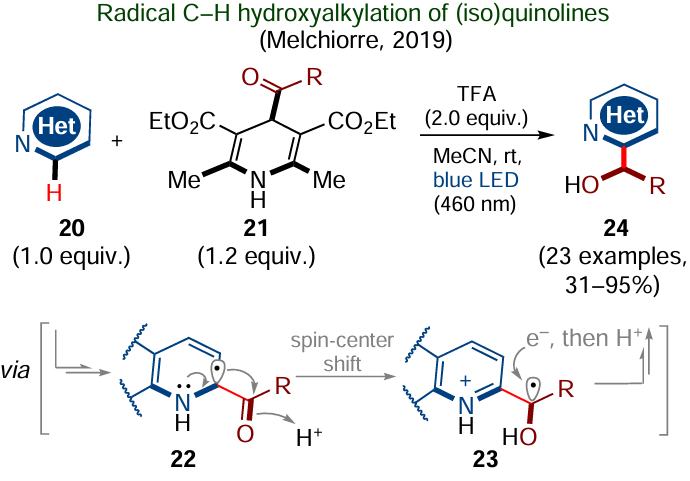
Another example reported by Melchiorre and co-workers in 2019[101] is also actually the Minisci reaction; however, the mechanism of this transformation is somewhat different from the canonical mechanism. The authors used 4-acyl-containing Hantzsch esters 21 (Scheme 11) as a source of acyl radicals. The latter were generated under the action of blue light and were trapped by N-protonated (iso)quinolines 20 as the corresponding radical cations, which were converted upon deprotonation to neutral radical intermediates 22. Then, unlike the canonical mechanism of the Minisci reaction, a shift of the spin center and protonation of the acyl oxygen atom took place. The single-electron reduction of the resulting radical intermediate 23 led to the corresponding carbanion, the protonation of which (coupled with the deprotonation of the pyridine nitrogen) gave the final α-hydroxyalkylation product 24.[101]
The most popular type of the non-catalyzed radical C – H functionalization implies the use of excess oxidant, which can be represented by peroxides, persulfates, quinones, hypervalent iodine compounds and other agents. These oxidants are rather easily available and are converted to low-toxic products. For example, (diacetoxyiodo)benzene [PIDA, PhI(OAc)2], was used by Wang and co-workers for the oxidative cross-coupling of azoles 26 with quinoxalinones 25 at room temperature (Scheme 12).[102]
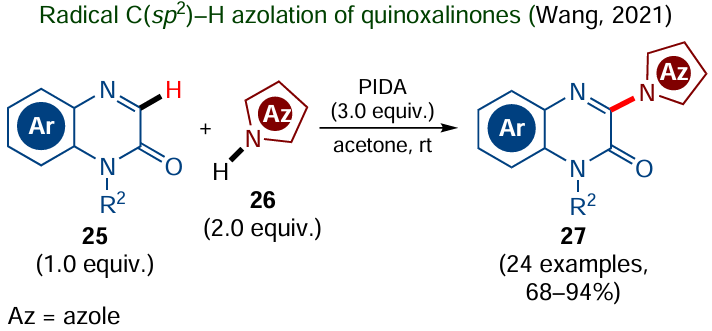
According to the mechanism proposed by the authors, PIDA is involved in the generation of the azole N-centred radical that adds to a molecule of quinoxalinone 25 to give the corresponding radical intermediate. The subsequent [1,2-H]-shift, single-electron oxidation and deprotonation afford 3-azolyl-substituted quinoxalin-2-one 27. This approach opens up the way to a fairly wide range of compounds; however, it requires the use of a twofold excess of azole 26 and a threefold excess of the oxidant.
* In earlier publications (before 2010), the term ‘proton-coupled electron transfer’ usually meant only the concerted proton and electron transfer; subsequently, this term was extended also to the consecutive SET and PT processes. In this review, the authors adhere to the current meaning of this term.
3.1.1.2. Catalytic methods of C(sp2) – H functionalization of aromatic and non-aromatic rings
As noted above, many reactions including the activation of the C(sp2) – H bond proceed in the presence of catalysts and use either air oxygen[62-64] or an electric anode[85-87] as the oxidant. According to the green chemistry principles, catalysis considerably increases the green potential of chemical reactions. However, implementation of a reaction with a catalyst does not by itself ensure an advantage over non-catalyzed reactions. The selection of reaction conditions, synthetic availability of catalysts, ligands and reagents, energy and labour costs in organization of the synthesis, as well as productivity and versatility of the process are also significant. Yet, many modern catalytic procedures comply with most, if not with all, of the above requirements, which makes them highly promising.
The history of the use of catalysis in the C(sp2) – H functionalization processes goes back more than a century and a half. One of the first examples is the catalytic benzoin condensation proposed by N.N.Zinin back in 1840 (Scheme 13). The Friedel – Crafts alkylation of arenes and other electrophilic aromatic substitution reactions are also examples of catalytic reactions that occur in the presence of Lewis acids.[103] In the 20th century, the inventory of catalytic methods was markedly extended by using transition metal catalysis. Reactions such as Cu-catalyzed Meerwein C – H arylation of alkenes (through in situ formation of aryldiazonium salts), Pd-catalyzed Mizoroki – Heck C – H arylation of alkenes (on treatment with aryl halides), and Ag-catalyzed Minisci alkylation of electron-deficient heteroarenes have become a routine practice in modern organic synthesis.[103] The discovery of the PdII-catalyzed Fujiwara–Moritani reaction in the late 1960s was an important milestone in the development of catalytic approaches to oxidative С – Н/С – Н cross-coupling of two unsaturated substrates.[104]
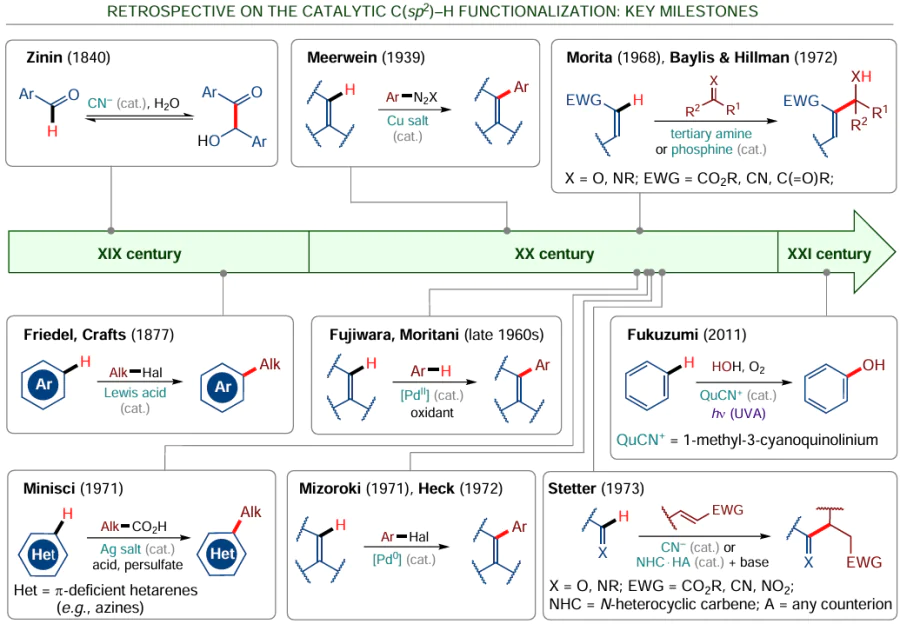
A pioneer of organocatalytic C(sp2) – H functionalization is Professor R. Breslow, whose studies of the mechanism of benzoin condensation in the presence of azolium salts have stimulated the development of numerous reactions catalyzed by N-heterocyclic carbenes (NHCs).[105][106] In the 1970s, the range of organocatalytic reactions was expanded owing to the discovery of Stetter, Morita – Baylis – Hillman and other reactions.
In the 21st century, the development of the C(sp2) – H functionalization was associated with the success in photoredox catalysis* and asymmetric organocatalysis and with the development of various hybrid procedures involving two or more interrelated catalytic cycles within one reaction. Some types of catalytic reactions involving the C(sp2) – H bond in aromatic and non-aromatic rings are considered below using selected examples.
* Scheme 13 shows a vivid example reported by Fukuzumi’s research team back in 2011,[107] which demonstrates the formation of phenol from benzene and water in a homogeneous medium, which had long been considered impossible.
3.1.1.2.1. Transition metal catalysis
Over the past few decades, the use of a variety of transition metal-based catalysts for a wide range of cross-coupling reactions has become a usual practice of modern organic synthesis. Until quite recently, these reactions required catalysts based on palladium, copper or nickel. However, currently it is known that catalytic properties are inherent in many metals. In particular, there is increasing popularity of catalysts based on iridium and ruthenium complexes, which are used in the actively developing photoredox catalysis.[108] Furthermore, in line with the trend towards green chemistry, significant efforts are being made to find approaches that utilize more readily available base-metal catalysts instead of traditional noble metal complexes.[109-111]For procedures that still involve the use of noble metal catalysts, steps are undertaken to minimize the catalyst loading without decreasing the process output and to find ways for repeated regeneration of this reaction component.
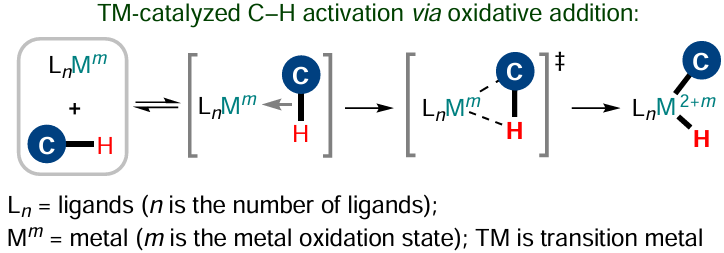
While considering the transition metal-catalyzed C(sp2) – H-functionalization reactions, it is necessary to take into account the specific features of activation of certain C – H bonds. A well-known mechanism of activation is oxidative addition according to which the metal complex initiates the C – H bond cleavage with simultaneous formation of M – C and M – H bonds, i.e., the metal complex is actually inserted into the С – Н bond (Scheme 14). This mechanism is typical of electron-rich complexes of late transition metals (Pt, Ru, Ir, Fe, Re, Os) in low oxidation states, for which the increase in the metal oxidation state or a change in the geometry of the complex caused by the potential formation of two new bonds is not a critical obstacle from the energy standpoint.[112]
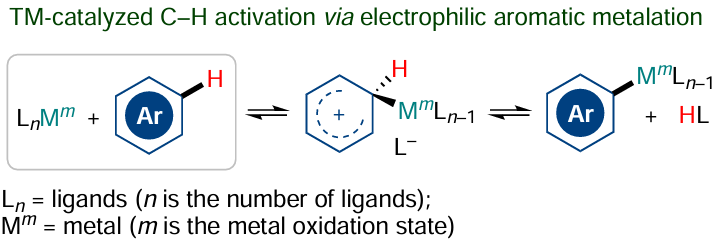
The reactions of arenes with late transition and post-transition metals possessing Lewis acidity (Pd2+, Pt2+/4+, Hg2+) follow a different metalation mechanism, which comprises interaction of the electrophilic metal centre with the π-electron cloud of the aromatic substrate, resulting in the formation of a σ-complex (Scheme 15).[112][113] The latter, in turn, is prone to deprotonation as a result of spontaneous rearomatization or under the action of a base, which may occur as one of the ligands in the metal coordination sphere.
One more C – H activation pathway involving electron-deficient metal complexes more resistant to oxidation is the σ-bond metathesis in which cleavage of the M – R and C – H bonds and formation of new M – C and R – H bonds follows a concerted mechanism without a change in the metal oxidation state (Scheme 16). This mechanism is preferable for group 3 and 4 early transition metals, lanthanides and actinides characterized by the d0 electronic configuration.[112]
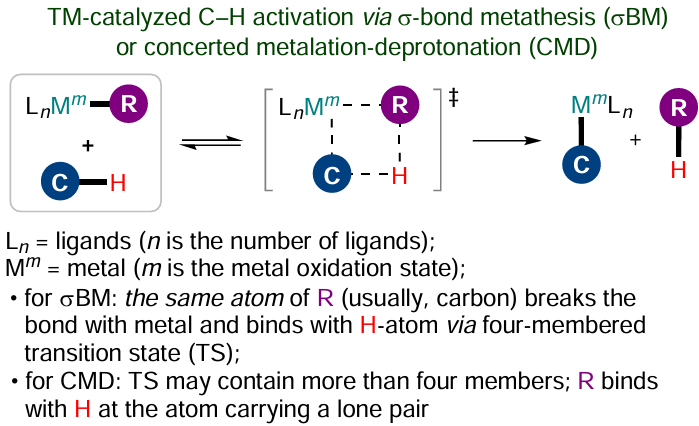
A similar transition state arises in the concerted metalation–deprotonation (CMD) reaction. Despite the fact that in the literature, CMD is sometimes considered to be the same as σ-bond metathesis,[114] most often these terms refer to different reactions. In the case of metathesis, one and the same atom (usually carbon) present in the group R (see Scheme 16) simultaneously breaks its own bond with the metal and forms a new bond with hydrogen via a four-centre transition state, thus resembling the [2σ + 2σ]-cycloaddition.[115] The transition state of the CMD process may have a larger number of members, since the concerted bond cleavage/formation at group R usually involves various atoms of this group, with the bond of R to the hydrogen atom being necessarily generated through an atom possessing a lone pair of electrons.
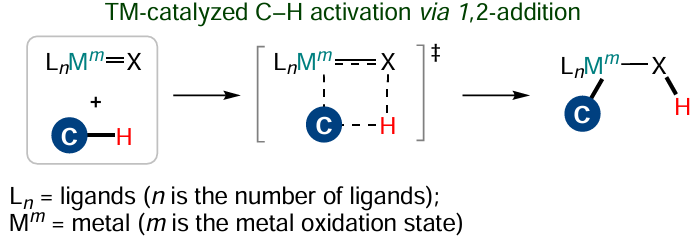
Apart from CMD, there is one more type of the C – H-bond activation that does not differ much from the metathesis. The reaction proceeds as concerted 1,2-addition of the C – H bond to the multiple bond of amido, alkoxy, alkylidene or alkylidyne complexes of early and middle transition metals (Scheme 17).[112] However, the newly formed X – H bond is not separated from the metal complex, as it utilizes π- rather than σ-electrons (or electrons of the non-bonding orbital) of the M – X bond.
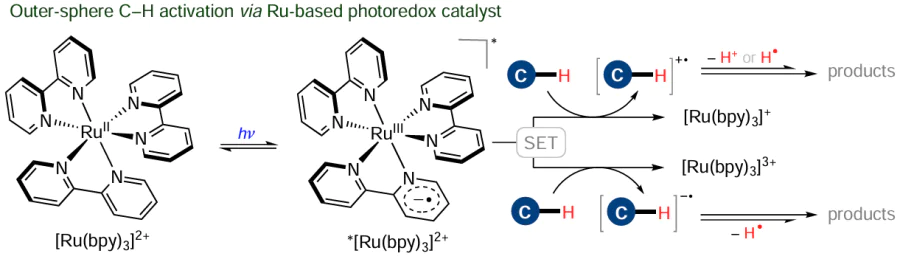
Besides the above-described inner-sphere pathways of C – H activation, there are a number of outer-sphere mechanisms in which the C – H bond interacts with the ligand environment rather than with the metal. A typical example of the outer-sphere activation is the electron transfer between the substrate and the photoexcited ruthenium polypyridyl complex in a photoredox-catalyzed process (Scheme 18).[108]
The regioselectivity of the transition metal-catalyzed C – H functionalization is influenced by directing groups (DGs) present in the substrate. These groups direct the substrate coordination to a definite site of the metal complex, which becomes preferable owing to the electronic and/or steric effects generated by these groups. The role of directing groups can be performed by either native moieties already present in the substrate molecule or substituents that are deliberately introduced for selective C – H modification and removed afterwards. The latter option is undesirable considering the green chemistry principles, because this elongates the reaction sequence; nevertheless, in some cases, this is necessary to implement the synthesis. The directing groups that can be introduced and removed without much difficulty should be used for this purpose.[116][117] Among DGs, so-called traceless directing groups that are easily eliminated from the substrate molecule upon metal-catalyzed C – H activation deserve mention.[117-119] The most promising DGs are, perhaps, transient directing groups (TDGs), which are introduced into the substrate molecule, provide the selective activation of the C(sp2) – H bond and are removed from the molecule in a single one-pot process.[120-126] Moreover, the latter option allows the use of reagents for the introduction of DGs in catalytic amounts.
As an example of catalysis involving a transient directing group, consider the Cu-catalyzed ortho-С(sp2) – H sulfonylation of benzylamines 28 with organic sulfinates 29, reported recently by Bull and co-workers[127] (Scheme 19).
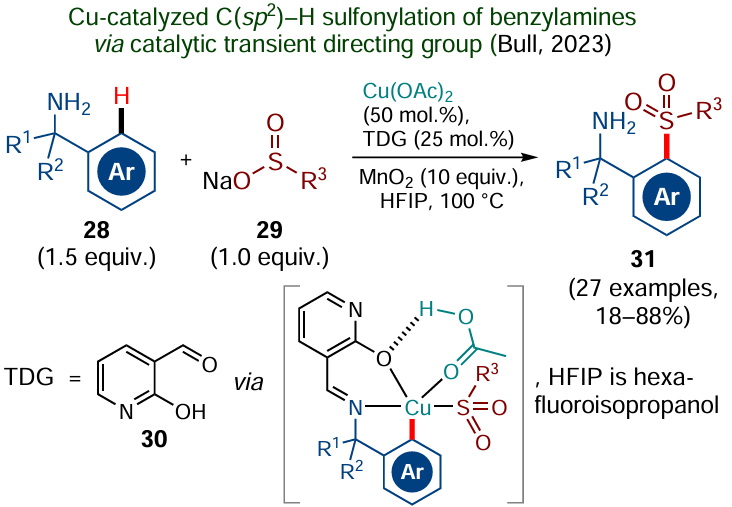
In this case, the directing group is attached in situ by the reaction of benzylamines 28 with a catalytic amount of 2-hydroxynicotinic aldehyde 30, which leads to the corresponding imines. Imine moieties are among the most popular TDGs,[122]owing to the easy introduction, pronounced directing effect, and easy removal via hydrolysis. According to DFT calculations,[127] the introduction of this directing group substantially reduced the energy barrier for concerted metalation — deprotonation, providing C – H activation of substrate 28. Using the developed approach, a series of 27 sulfonylated benzylamines 31 were synthesized. A benefit of this method is that it does not require complexes of noble metals (such as palladium), which were previously almost necessary components for this type of reactions. However, due to the need for a large amount of the copper catalyst (50 mol.%) and a large excess of the oxidant, manganese dioxide, spent for the catalyst regeneration, there is a room for improvement.
Fagnou and co-workers introduced one more ortho-directing group, N-oxide moiety, into the practice of organic synthesis.[128-131] In the context of green chemistry, a problematic feature of the N-oxide group is that it must be first introduced into the molecule (e.g., by peroxide oxidation of the aza group) and then removed by reduction, which may be incompatible with the presence of functional groups in the substrate molecule. Meanwhile, when N-oxide is a native moiety that is retained in the target product, it can be used for the subsequent derivatization. An example of such potentially useful products are cyclic nitrone derivatives, which are of interest for medicinal and analytical chemistry due to their antiradical and other useful properties. The assistance from the N-oxide moiety enabled, in particular, direct Pd-catalyzed oxidative cross-coupling of imidazole oxides 32 with π-excessive hetarenes 33 (pyrroles and thiophenes), which furnished functionally substituted nitrones 34 (Scheme 20).[132]
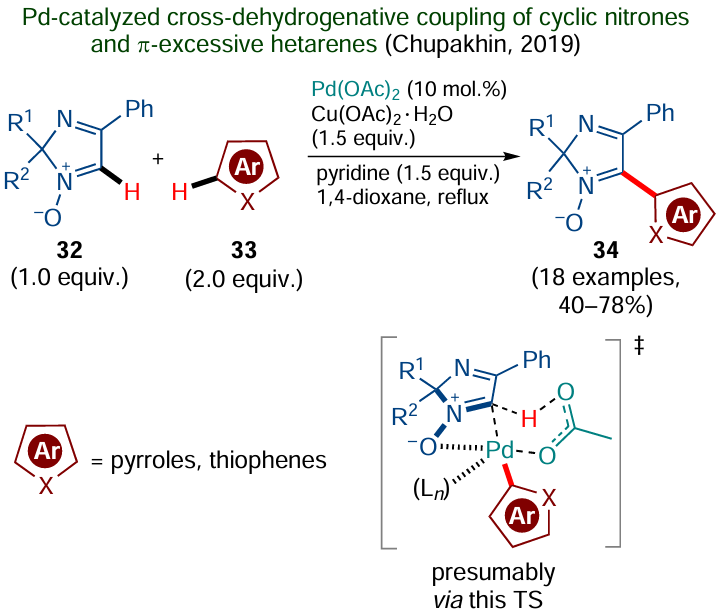
It is noteworthy that the primary C – H activation in this reaction occurs via coordination of the palladium catalyst to an aromatic heterocycle, which was confirmed by control experiments on hydrogen–deuterium exchange. The subsequent C – H activation in the aldonitrone component is likely to proceed as the concerted metalation – deprotonation, giving rise to target products 34. A benefit of this method is the cross-dehydrogenative reaction pathway. However, the use of surplus amounts of auxiliary reagents, relatively high loading of the palladium catalyst and moderate product yields leave much to be desired.
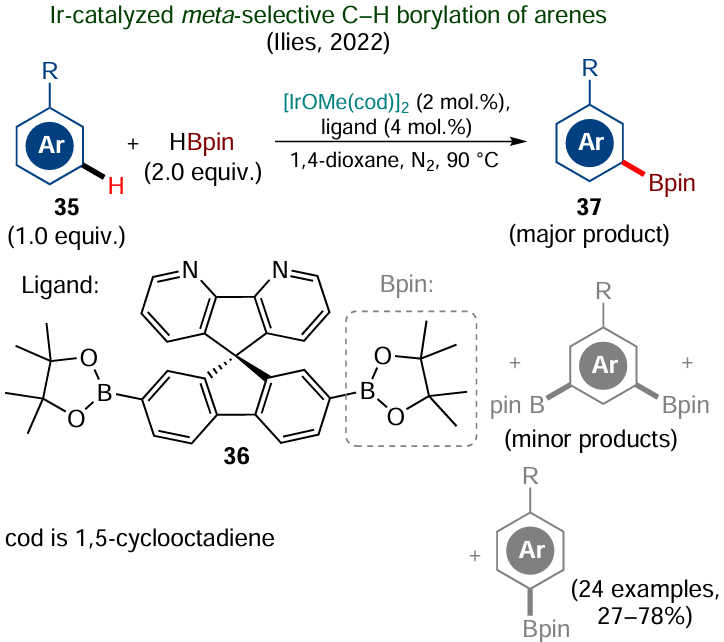
Most of the known directing groups have an ortho-coordinating effect, thus providing functionalization of the nearest C – H bond in unsaturated systems. Selective conduction of these reactions at one of remote positions may prove to be more challenging.[133][134] In principle, these transformations can be accomplished by introducing covalently linked directing groups or templates into the substrate, but this may impair the atom economy.* In this connection, the strategy of undirected C – H functionalization appears to be more interesting;[135] in this case, the reaction regioselectivity is determined by steric and/or electronic control from native substituents present in the substrate and by the proper tuning of the structure of the catalytic metal complex, the spatial geometry and electronic structure of which can have a crucial effect on the proneness of a distal C – H bond in the substrate molecule to functionalization. An example illustrating this concept is the meta-selective iridium-catalyzed C – H borylation of arenes 35, proposed by Ilies and co-workers.[136] In this case, the reaction selectivity is achieved by rational design of ligand 36, the geometry of which blocks not only the ortho- but also para-positions, while the access of the inner coordination sphere of the catalyst to the meta-C – H bonds is retained (Scheme 21). This mainly gives meta-borylation products 37. The amount of ligand 36 sufficient for the reaction to occur is only 4 mol.%, which should be regarded as an advantage of the method, even taking into account the fairly complex structure of the ligand. Unfortunately, despite the relatively high regioselectivity of the procedure, the formation of para-substituted and meta-diborylated by-products cannot be completely avoided.[136]
Generally, transition metal catalysis is still an important tool for direct C – H functionalization of unsaturated compounds, as evidenced by numerous reviews on this topic.[137-144] To increase the green potential of metal-catalyzed reactions, numerous attempts have been made to reduce the amount of chemical oxidants and other auxiliary reagents by combining transition metal catalysis with photoredox catalysis[145-154] or electrocatalysis.[149][155-164] The targeted design of ligands and templates can be used to modify С(sp2) – Н bonds both in proximal and distal groups of the substrate, while the development of transient directing groups favours a decrease in the number of chemical steps and in the amounts of reagents required for the introduction of such groups. There is no doubt that success in the field of metal catalysis will strongly facilitate further development and improvement of these methods in the near future.
* There are few examples of selective functionalization of distal C – H bonds in arenes using structurally simple directing groups in the presence of ruthenium catalysts.133 The distal orientation can be implemented due to electronic effects arising upon the arene metalation under the action of a ruthenium derivative.
3.1.1.2.2. Organocatalysis
Organocatalytic methods are finding increasing use in organic synthesis, including functionalization of С(sp2) – Н bonds. Organocatalysts often play an auxiliary role in C – H activation reactions, as they do not directly cleave the C – H bond of the substrate.[165] In this case, their possible function is to promote the activation via the formation of more reactive intermediates upon reactions with the substrate. Examples of organocatalysts are the transient ligands considered above, which are introduced into metal-catalyzed cross-coupling reactions in catalytic amounts. It can be seen that by using chiral organic molecules as ligands, one can attain enantioselectivity of coupling[126] (see chapter 3.3).
Organocatalysts (OC) can also directly activate the С(sp2) – Н bond in unsaturated molecules as a result of PCET and HAT events. A well-known class of such compounds are organic photoredox catalysts,[166][167] which are converted to relatively long-lived excited state upon irradiation, thus becoming potent donors or acceptors for the unpaired electron. This results in redox reaction with the substrate (SET, Scheme 22) or energy transfer from the catalyst to the substrate to bring the latter to the excited state, which predetermines further substrate functionalization.
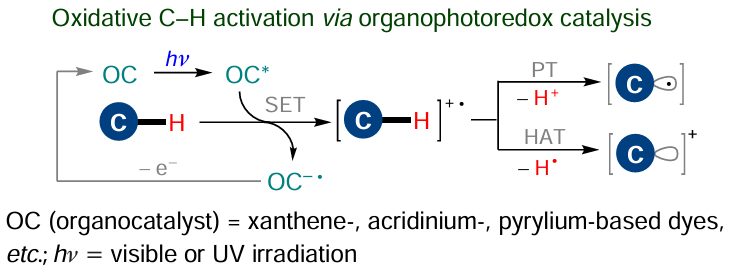
Some redox-active organocatalysts are able to initiate the C – H activation when they occur in the ground state rather than in the excited state. In particular, this refers to so-called HAT catalysts: organic radicals able to act as acceptors of a hydrogen atom or an unpaired electron (Scheme 23).168
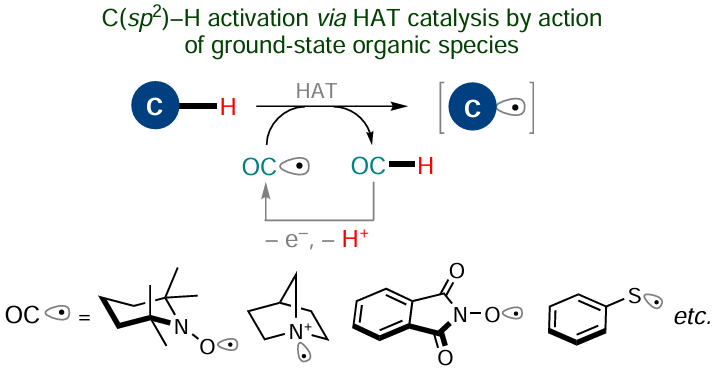
In most cases, these paramagnetic compounds (except for stable nitroxides) are formed in situ upon single-electron oxidation of the corresponding more stable forms. These reduced species are also capable of initiating reverse reactions that can take place during the process, i.e., they can be both HAT donors and single-electron reducing agents. The most frequently used organocatalytic redox pairs include 1-hydroxy-2,2,6,6- tetramethylpiperidine (TEMPOH) and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO); N-hydroxyphthalimide (NHPI) and phthalimide-N-oxyl (PINO); tertiary amines [e.g., quinuclidine or diazabicyclooctane (DABCO)] and their radical cations; thiols (e.g., thiophenol) and the corresponding thiyl radicals.[168] A similar action is characteristic of organocatalysts that are fairly strong oxidants such as quinones (for example, DDQ and chloranils), organic compounds of hypervalent iodine, and oxoammonium derivatives (for example, 2,2,6,6-tetramethyl-1-oxopiperidinium salts, which result from the oxidation of TEMPO). Note that regeneration of this type of catalysts may require superstoichiometric amounts of a terminal oxidant (or reducing agent) or the reaction with a cooperative catalytic cycle as a part of hybrid procedure. Therefore, it is necessary to compare the expenses and the general environmental footprint inherent in these organocatalytic methods with those of non-catalyzed analogues in which stoichiometric amounts of some auxiliary reagent are used.
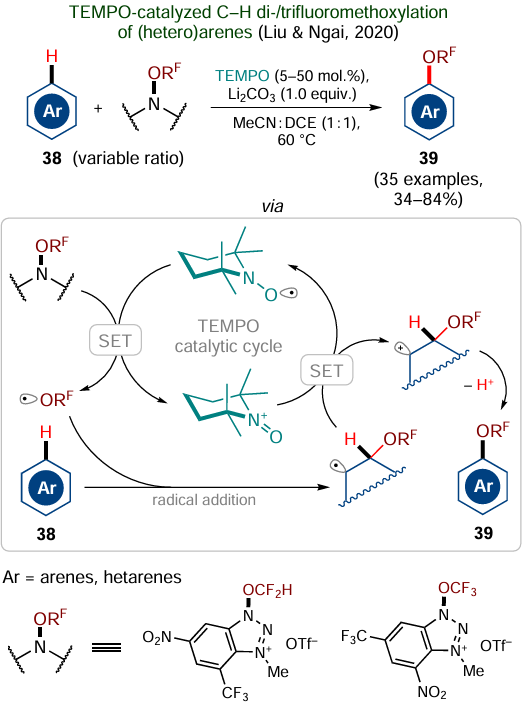
As an example, we will consider di- and trifluoromethoxylation of C(sp2) – H bonds in arenes 38 reported in 2020 by Liu, Ngai and co-workers (Scheme 24).[169] This approach is claimed by the authors as a redox-neutral method, since TEMPO used as a catalyst and its oxidized oxoammonium form generated in situ are formally capable of maintaining the catalytic cycle only through redox reactions with substrates (or intermediates); theoretically, this approach does not require additional redox agents. However, the authors noted that successful conduction of this reaction requires the use of a stoichiometric amount of lithium carbonate, which prevents the conversion of TEMPO into a catalytically inactive form via binding to an acidic by-product capable of destructive action on the catalyst. Moreover, it was shown that Li2CO3 can reduce the oxoammonium cation back to TEMPO via the SET process, thus promoting the catalyst regeneration. Since lithium carbonate is a cheap and non-toxic reagent, its use does not deteriorate the green potential of the reaction. This approach was used by the authors to prepare a series of 35 (hetero)aromatic compounds 39 in up to 84% yields.
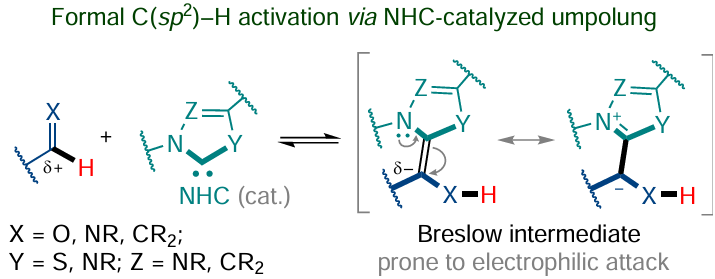
Special mention should be made of C – C coupling reactions catalyzed by N-heterocyclic carbenes, which proceed via the formation of Breslow intermediates and are based on umpolung,[105] although they cannot be classified as conventional methods for the C(sp2) – H functionalization in cyclic substrates (Scheme 25).* These reactions are characteristic of aldehydes (Stetter reaction, benzoin condensation) and their azomethine analogues.[170] Surprisingly, no examples of reactions of this type in which NHC-catalyzed umpolung takes place for cyclic aldimines are known to date.
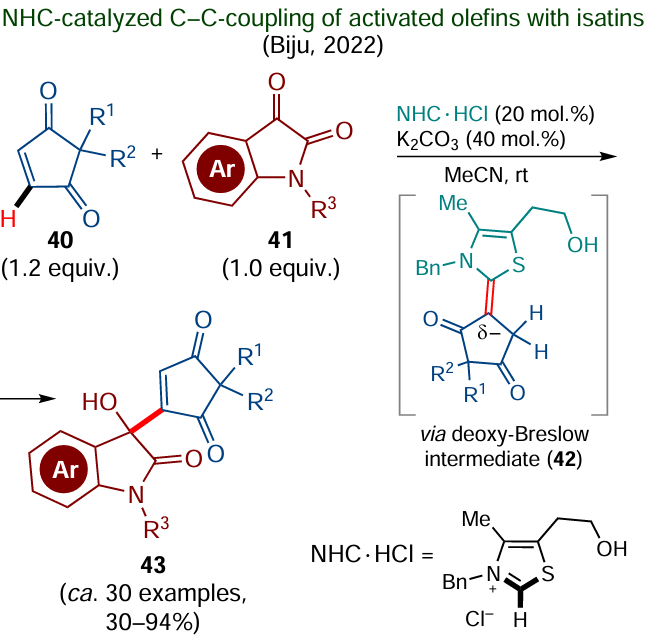
Biju and co-workers described a rare case of NHC-catalyzed functionalization of the C(sp2) – H bond occurring via an umpolung intermediate (Scheme 26).[171] In relation to cyclopent-4-ene-1,3-diones 40, the authors demonstrated the possibility of generation and isolation of pure deoxy-Breslow intermediate 42, which is formed upon the reaction of activated alkene with N-heterocyclic carbene. This intermediate can further react with the C-electrophilic centre of isatin 41 to give coupling products 43 in up to 94% yields. N-Methylmaleimide and 1,4-naphthoquinone can also be used as alkene-containing components.
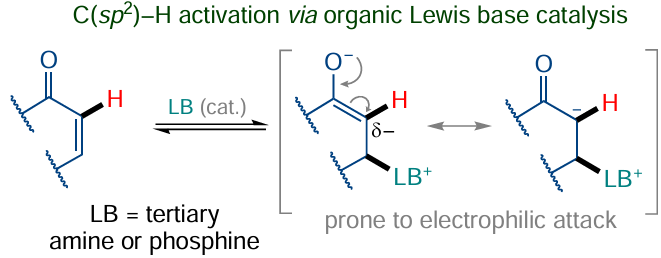
Organic bases and acids are also actively used as organocatalysts in the C(sp2) – H functionalization strategy. For example, Lewis bases catalyze the Morita–Baylis – Hillman[172] and Rauhut – Currier[173] С–С coupling reactions, thus activating the α-C(sp2) – H bonds in α,β-unsaturated carbonyl compounds (Scheme 27) and other Michael acceptors towards subsequent reactions with C-electrophilic reagents.
The basic catalysts used most often for these reactions are tertiary amines (DABCO, DBU, DMAP) or tertiary phosphines, such as tricyclohexylphosphine and tributylphosphine. The electrophiles used in the Morita–Baylis – Hillman reaction are usually aldehydes and aldimines, which are converted to (α-hydroxy)alkylation and (α-amino)alkylation products, respectively. In the Rauhut – Currier reaction, α,β-unsaturated carbonyl compounds are used as electrophiles. In the context of C(sp2) – H functionalization of cyclic compounds, the above-mentioned reactions can be applied to substrates like cyclic enones, α,β-unsaturated lactones, thiolactones,[174] lactams,[175] maleimide derivatives,[176] pyrones[177] and chromones.[178] When chiral bases are used, the reactions may proceed enantioselectively.
While discussing optically active organocatalysts, one cannot leave out the class of organic phosphorus-containing Brønsted acids such as commercially available 1,1'-bi-2-naphthol (BINOL) and 1,1'-spirobiindane-7,7'-diol (SPINOL). In recent years, these chiral phosphoric acids (CPA) have been very actively used in the synthesis of bis(hetero)aryls to initiate atroposelective oxidative cross-coupling reactions, the generalized mechanism of which is presented in Scheme 28 .[179]
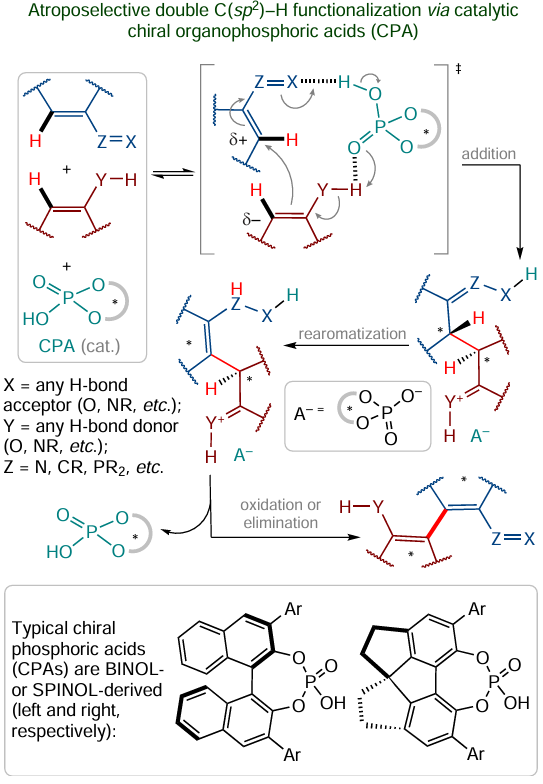
The scope of application of CPA is far from being exhausted by these reactions. Recently, the research team headed by List[180] reported that BINOL-based iminodiphosphorimidates 47, which are strong Brønsted acids, can catalyze the Friedel – Crafts reaction involving not only π-excessive (hetero)arenes, but also benzene or alkylbenzenes (Scheme 29). By the alkylation of aromatic substrates 44 with N,O-acetals 45, the authors obtained a series of (hetero)arylglycine esters 46 in up to 98% yields with high regio- and enantioselectivity.
A fairly promising type of organocatalysts for the C(sp2) – H activation/functionalization are sterically hindered organic molecules in which two functional moieties possessing Lewis acidity and basicity are separated by a carbon group preventing their interaction (so-called frustrated Lewis pairs; FLP)).[181-183] In particular, using borylation of the C(sp2) – H-bond in π-excessive heterocycles,[184] it was shown that bipolar catalysts of this type can activate the C – H bond by a mechanism similar to the concerted metalation – deprotonation (CMD) mechanism, except that in this case, the borane substituent in FLP acts as the electrophilic pseudometalating agent, while groups possessing Lewis basicity, such as amines or phosphines, are responsible for deprotonation (Scheme 30).
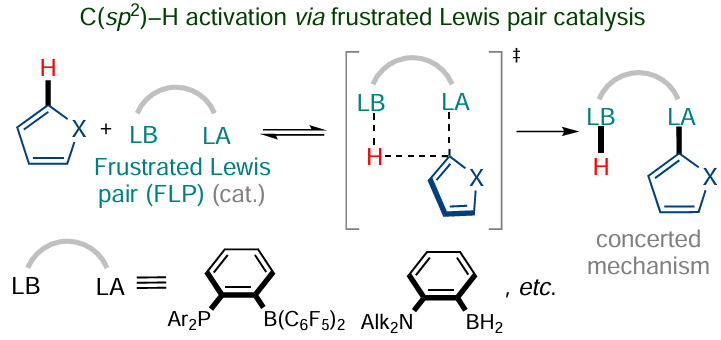
In the case of FLP in which the acidic and basic sites are located at relatively large distances from each other, the consecutive carbene-associated mechanism, which is more energetically favourable due to distance effects, can also be implemented.[185]
In general, it is obvious that direct activation of the C(sp2) – H bond in cyclic systems under the action of metal complex catalysts or organocatalysts is an exceptionally promising green strategy, allowing the introduction of various structural groups to an sp2-hybridized carbon atom in the simplest possible way with high productivity and selectivity, in particular, as late-stage functionalization (LSF).[17][57] [186][187] It is no doubt that further progress in this field can be expected in the coming years.
*Curiously, the formation of the Breslow intermediate upon the reaction of the precatalyst (azolium salt) with a carbonyl compound in the presence of base can be considered as C(sp2)–H functionalization of the precatalyst in which the carbene formation is an intermediate step.
3.1.2. Direct oxidative functionalization of aliphatic С – Н groups of complex organic molecules
Developing catalytic approaches to the selective activation of aliphatic C – H bonds in organic compounds has been listed among the ‘Holy Grails’ of synthetic chemistry.[188][189] The main application of such approaches is to provide the insertion of desired functional groups into a particular C – H bond at the ‘late’ stages of multistep synthesis of complex molecules (late-stage functionalization, LSF).[190-193] Such molecules include fine organic synthesis products in demand as agrochemicals, cosmetics, and synthetic pharmaceuticals, many of which are functionalized hydrocarbon frameworks containing several non-equivalent C(sp2) – H and C(sp3) – H groups. In the future, powerful and versatile methods for LSF of aliphatic C – H groups will facilitate the solution of challenges such as the rapid generation of vast chemical libraries of metabolites, tuning of their key pharmacological properties (therapeutic efficacy, bioavailability, pharmacokinetics, etc.) without the need to re-develop or adapt existing multistep procedures.[193][194] The processes of stereoselective oxidative C(sp3) – H functionalization are of great interest, since the absolute configuration of organic molecules is closely related to their biological activity.[195]
Recent advances in the field of selective oxifunctionalization of aliphatic C – H groups are largely due to the application of the biomimetic approach,[196] which aims at modelling the functional properties of natural oxygenase enzymes[197][198] using low-molecular-weight (synthetic) metal complexes, mainly those containing iron and manganese.[199-202] In the last two decades, non-heme (non-porphyrin) Mn and Fe complexes have been the most actively studied, offering, as compared to porphyrin complexes, much broader possibilities for structural modification and, consequently, for controlling their reactivity in oxidative catalytic processes. Mn and Fe complexes, like their metalloenzymic prototypes, catalyze direct C‒H functionalization (without forming organometallic intermediate species), which positively affects such green chemistry metrics as atom economy and E-factor.[9] When designing such systems, there is a steady trend to use cheap and environmentally friendly hydrogen peroxide as an oxidant.
This Section summarizes the state-of-the-art in the field of direct (without intermediate metal – carbon bond formation) selective oxidative functionalization of C(sp3) – H groups of organic molecules with hydrogen peroxide in the presence of non-heme (bis-amino-bis-pyridylmethyl) and similar Fe and Mn complexes. Non-heme systems generally do not require any directing or protecting groups, utilize hydrogen peroxide as an oxidant, producing water as a stoichiometric by-product, which makes them a useful tool for green chemistry.
At the current level of our understanding of the mechanisms of action of catalytic systems based on non-heme metal complex catalysts, activation of the C(sp3) – H bond (via the hydrogen atom abstraction by an active metal oxo species) occurs similarly to the C – H activation mediated by the metalloenzymes of the cytochrome P450 superfamily.[203] As a result of the H atom abstraction, a metal hydroxo species and a C-centered radical are formed (Fig. 8). There are several possibilities for further transformations of this pair in the solvent cage. If the radical is short-lived, it is likely to be captured by the hydroxometal intermediate and bind to the incipient hydroxyl group (pathway a): this option is a direct analogue of the oxygen rebound mechanism proposed by Huang and Groves.[203] Alternatively, the short-lived C-centered radical can be captured by another labile ligand X in the first coordination sphere of the metal (usually X is a residue of the carboxylic acid added to the system as a co-catalytic additive). This pathway is similar to the mechanism of catalytic action of Fe- and α-ketoglutarate-dependent halogenases.[204] The implementation of this pathway, referred to as the alternative rebound mechanism (comprising hydrogen abstraction and binding M – X ligand rather than the incipient M – OH group, pathway b),[205] makes it possible to obtain compounds with a new C – X bond.
![[{"id":"Rvpsb797QE","type":"paragraph","data":{"text":"Alternative routes for oxidative functionalization of C – H groups of organic compounds by metal-oxo species (L is chelate ligand, X is labile ligand)."}}]](/storage/images/resized/61Ff7NbALo6sAdtHZDsODXiiwoMZNTvis55acqLy_xl.webp)
In both cases, for the C – H functionalization reaction to proceed in a stereospecific manner, the encaged radical pair lifetime must be short enough such that epimerization of the radical does not take place prior to the oxygen rebound/alternative rebound step. Alternatively, the encaged radical pair can undergo formal abstraction of the second hydrogen atom[203] to give a desaturation product (pathway c).
For relatively long-lived radicals, the probability of their cage escape into the bulk solution increases; this pathway is considered the non-rebound mechanism.[206] Further transformations of the free C-radical are not necessarily controlled by the catalyst; for example, in the presence of dissolved dioxygen, organic peroxides are formed that are capable of disproportionation to alcohol and ketone (pathway d ).[207] The cage escape is often accompanied by stereoinversion, which can lead to complete racemization of the products.
In the oxidation of complex organic molecules containing multiple different C – H groups, the regioselectivity of the overall process is governed by the regioselectivity of the H atom abstraction step. In general, tertiary C – H groups are more nucleophilic than methylenic CH2 groups and therefore more susceptible to hydrogen abstraction by electrophilic metal-oxo species. This ‘innate substrate reactivity’ can be overcome by either creating steric shielding nearby the active site of the catalyst or by introducing functional groups that allow the active site to ‘recognize’ the substrate, according to the concept of biomimetic control of chemical selectivity.[196]
A significant part of recent studies is either directly devoted to, or to a greater or lesser extent concerns the issues of stereoselective oxidative functionalization of C – H groups of complex molecules.[202] This situation has developed naturally due to the fact that the main practical motivation of this work is the development of catalytic approaches to biologically active substances, primarily derivatives of natural compounds.
The catalytic activity of non-heme iron and manganese complexes in reactions of selective oxidative functionalization of C – H groups of organic compounds has been explored for more than three decades.[208][209] At the same time, the works of Chen and White,[210][211] which demonstrated the possibility of selective oxidation of aliphatic C – H groups by hydrogen peroxide in the presence of the non-heme complex (S,S)-I (Fig. 9) and revealed the main regularities responsible for the selectivity of the catalytic reaction, are considered key in this field. Thus, in the oxidation of compounds containing both methine and methylene groups, the more nucleophilic methylene groups, with lower homolytic bond dissociation energies, reacted preferentially, indicating the electrophilic nature of the catalytically active sites.[210] In such a manner, (+)-artemisinin was selectively oxidized in the presence of (S,S)-I to the C(10)-hydroxy derivative. Using the (S,S)-II catalyst bearing bulky 2,6-bis(trifluoromethyl)phenyl substituents made it possible to overcome the innate reactivity of the substrate and to direct the reaction towards predominant oxidation of the C(9) site.[212]
![[{"id":"TkQf2v5R9-","type":"paragraph","data":{"text":"Structures of complexes <b>I – III</b> and examples of their use in catalytic reactions. Hereinafter, the yields of products (%) are given. OTf is OSO<sub>2</sub>CF<sub>3</sub> ."}}]](/storage/images/resized/Uz0ycfDpH2ELsbJlMwzG3XPzmIHgq294DhL47SOX_xl.webp)
(‒)-Ambroxide lacking tertiary C‒H groups was selectively oxidized in the presence of (S,S)-I at the activated (via superconjugation with an adjacent oxygen atom) C(12) position to give the corresponding lactone, (+)-sclareolide, in high yield. (+)-Sclareolide can also be oxidized, preferably to a C(2)-keto derivative.[211] However, in complex substrates such as terpenoids and steroids, diastereotopic hydrogen atoms at the same carbon atom are characterized by different bond strengths (and different steric accessibility). For this reason, the (S,S)-I catalyst enabled the diastereoselective hydroxylation of dihydropleuromutilone 48 at the equatorial C7α position.[191] In addition to catalytic activity in selective hydroxylation and ketonization reactions, non-heme iron complexes were able to catalyze lactonization processes under certain conditions: e.g., taxane containing a carboxylic acid group was selectively converted to the corresponding γ-lactone in 49% yield (see Fig. 9).[213] Using the isotopic labelling method, it was shown that the reaction proceeds in two steps: the first step involves γ-hydroxylation, followed by intramolecular esterification (lactonization). The high selectivity for the γ-position was explained by the directing effect of the carboxylato group, through which the substrate binds to the catalytically active site. The use of iron complex in combination with strong Brønsted acid (HBF4) allowed the oxidation of substrates containing nitrogen heterocycles.[214] In particular, this approach provided the hydroxylation of the steroidal substrate 49 at the C6α position in 42% yield.[214]
It should be noted that the oxidation of methylenic groups initially produces a secondary alcohol with a more electron-rich C(OH) – H bond than in the substrate. It is therefore prone to rapid further oxidation to a ketone. At the same time, in many cases the target product is precisely the secondary alcohol. To halt the reaction at the hydroxylation stage, Costas and co-workers[215] proposed to replace acetonitrile, the conventional reaction medium, with Lewis acidic and strongly hydrogen-bond donating poly-β-fluorinated alcohols. Poly-β-fluorinated alcohols are able to deactivate the target product, the secondary alcohol, towards further oxidation by virtue of hydrogen bonding interactions.[215] Also, the use of poly-β-fluorinated alcohol solvents enables the hydroxylation of substrates that already contain hydroxyl groups. For example, using non-heme iron complex (R,R)-III, Costas and co-workers[216] prepared polyhydroxylated steroidal metabolites 50 and 51 in hexafluoroisopropanol (HFIP) without the aid of protecting groups (see Fig. 9).
The emergence of type I – III non-heme iron complexes and the discovery of their catalytic activity was an important milestone in the development of catalytic approaches to the selective oxidative C – H functionalization of organic compounds. At the same time, the inherent disadvantages of such catalysts, such as low productivity and insufficient regio- and stereoselectivity, led to their gradual replacement by catalytically active manganese complexes of similar structure, which proved to be more synthetically promising.
Bryliakov and co-workers[217] proposed the use of manganese complexes IV – VIII as catalysts for the selective aliphatic C – H bond oxidation (Fig. 10), which were able to perform up to 100 – 1000 catalytic turnovers. In this way, manganese complexes VI were used to selectively oxidize (–)-ambroxide to (+)-sclareolide[218] and tetrahydrofuran to γ-butyrolactone[219] at catalyst loadings of 0.1 – 0.2 mol.%. Such high efficiency makes manganese complexes promising catalysts not only for working with valuable complex molecules, but also for obtaining products of relatively low added value, such as the selective oxidation of benzyl alcohol to benzaldehyde.[220] As with non-heme iron complexes, catalysts based on similar manganese complexes are highly sensitive to electronic effects. In the oxidation of methylene groups, this favors the formation of ketones, since the alcohol C(OH) – H bond formed at the first oxidation step is activated by the presence of an adjacent OH group. As in the case of oxidation in the presence of iron complexes, an approach based on the use of β-polyfluoro-substituted alcohol solvents capable of deactivating the newly formed secondary alcohol due to hydrogen bonding has been used to inhibit further oxidation of alcohols to ketones.[215] Using the oxidation of cyclohexane as an example, it has been demonstrated that increasing the hydrogen bond donating ability of the solvent (in the order acetonitrile < trifluoroethanol < HFIP) improves selectivity for the secondary alcohol.
![[{"id":"qfkW36XdHQ","type":"paragraph","data":{"text":"Structures of complexes <b>IV–VIII</b> and examples of their use in catalytic reactions. OTf is OSO<sub>2</sub>CF<sub>3</sub> , 2,2-DMBA is 2,2-dimethylbutyric acid."}}]](/storage/images/resized/PTUD78FnKf2pDbZBJVI6twyR04r1aDwNIvUwHfq2_xl.webp)
To date, a significant body of information has been accumulated on the catalytic properties of non-heme manganese complexes in chemo-, regio- and stereoselective C – H oxidation reactions.[200-202][220][221] In the vast majority of cases, the selectivity of these processes is determined by a more or less successful interplay of electronic and steric properties of the substrate and catalyst. Costas’s research group followed a strategy based on a biomimetic approach,[196] which involves the specific recognition of the substrate by the active site of the supramolecular catalyst (just as it occurs in enzyme-catalyzed biochemical processes). For example, the benzocrown ether-appended catalyst (S,S)-IX allowed the site-selective oxidation of the substrate, undecylammonium tetrafluoroborate, at the C(8) and C(9) positions (Fig. 11).[222] This was achieved by hydrogen bonding between the alkylammonium group and the oxygen atoms of one of the crown ether units. This conclusion is supported by the fact that the oxidation selectivity of substrates lacking ammonium groups did not change when switching from (S,S)-V to (S,S)-IX catalyst. The authors then applied this approach to the oxifunctionalization of the aminosteroid substrate 52 at the C(16) position, with the product being obtained in the acylated form.[223]
![[{"id":"jNSQ50C08a","type":"paragraph","data":{"text":"Examples of reactions where selectivity is determined by specific substrate-catalyst interactions. TFE is 2,2,2- trifluoroethanol. "}}]](/storage/images/resized/p1QRJKm9K8mDJfBVySG9DetyA38VnsFNrGfgYC5e_xl.webp)
A slightly different version of multi-site hydrogen bonding provided efficient recognition of indane-based substrates in enantioselective hydroxylation reactions (see Fig. 11) catalyzed by the (S)-X complex, where high enantioselectivity (up to 95% ee) was achieved.[224] The hydrogen bonding interactions are thought to involve a solvent molecule CF3CH2OH. In the oxidation of substrates not capable of hydrogen bonding (devoid of a ketone group), a sharp decrease in catalytic activity was observed.
Bryliakov and co-workers[225] found that asymmetric induction in the enantioselective catalytic hydroxylation of benzylic C – H groups in the presence of complex XI stems from two processes: the enantioselective C – H hydroxylation itself and the concomitant stereoconvergent oxidative kinetic resolution of a scalemic mixture of secondary alcohols. Significantly, kinetic resolution did not occur in HFIP medium (due to almost complete suppression of ketonization of the secondary alcohol). As a result, a two-step process was developed, where in the first step, enantioselective hydroxylation was carried out in HFIP, and then the reaction mixture was diluted with an equal volume of acetonitrile. The latter ‘switched on’ the kinetic resolution, which allowed the enantiomeric excess of the secondary alcohol to be increased to 97% at the expense of reducing its yield (Fig. 12).
![[{"id":"OPFgsFmYu5","type":"paragraph","data":{"text":"Structures of complexes <b>XI</b> and their use in enantioselective benzylic hydroxylation. OTf is OSO<sub>2</sub>CF<sub>3</sub> "}}]](/storage/images/resized/7znqdFsVvuaKucy086k5pAVz2EHbjJ1n4Ui58pOL_xl.webp)
Sun and co-workers[226] developed a catalytic method for the oxidative desymmetrization of spirocyclic tetralones and indanones in the presence of the chiral manganese complex (S)-X in high yields and enantioselectivities (Fig. 13). The method also proved to be effective in the oxidative desymmetrization of spirocyclic oxindoles and dihydroquinolinones to afford the corresponding chiral ketones.[227] Moreover, after modification of the reaction conditions, enantioselective hydroxylation of spirocyclic 2,3-dihydroquinolin-4(1H)-ones was achieved to give the corresponding chiral ketoalcohols in yields up to 41% and enantioselectivities up to 99%.[224]
![[{"id":"R9Oo9tD4P4","type":"paragraph","data":{"text":"Oxidative desymetrization of spirocyclic compounds catalyzed by chiral manganese complex (S)-<b>X</b>"}}]](/storage/images/resized/6v3WuwX26cMCUyN8C7wkkwepvXDvsmFbsd5CqeeB_xl.webp)
In the context of creating libraries of biologically active compounds, substrates of natural origin that already contain one or more asymmetric centres are of considerable practical value. Therefore, the search for not only enantioselective but also diastereoselective methods of C – H functionalization is an urgent task. Bryliakov and co-workers[228] developed synthetic approaches for the regio- and stereoselective oxidative mono- and polyfunctionalization of the terpenoid substrate (–)-ambroxide (Fig. 14). Three main factors determining the regio- and stereoselectivity of the oxidation were identified including the ligand architecture nearby the active site of the catalyst, the absolute chirality of the catalyst and the nature of the solvent.
![[{"id":"9yfmiILnyA","type":"paragraph","data":{"text":"Catalytic oxidation of (–)-ambroxide in the presence of manganese complexes. CAA is chloroacetic acid, <i>N</i>-Boc-<i>D</i>-Pro is <i>N</i>-Boc-<i>D</i>-proline."}}]](/storage/images/resized/jCRsPCnIicCHWBXZqjO0cJgbCI0eLTdnbpYsXjcC_xl.webp)
The oxidation of a series of steroidal substrates has been studied and approaches to manipulating the selectivity of these processes have been proposed. For example, approaches have been developed for the regio- and stereoselective oxidation of estrone acetate and a number of its derivatives, allowing the oxidation to be directed towards either C9α-hydroxylation or C6α-hydroxylation (Fig. 15).[229] Using HFIP as solvent, high selectivity for the secondary alcohol was achieved, effectively inhibiting further ketonization. A similar approach was applied to the oxidation of a series of fully saturated steroids, derivatives of 5α- and 5β-androstane. The use of catalysts differing in steric demands and chirality provided access to regio- and stereoselective hydroxylation at the C5β, C6α and C12β positions.[230] Using the sterically hindered catalyst XIII, de Lucca and co-workers[231] carried out the hydroxylation of compound 53 at the C(2) position. Varying the temperature and oxidant excess allowed the selective production of C2-ketone or C2β-hydroxy metabolite, an intermediate for the total synthesis of ent-beyerane derivatives.
![[{"id":"wa-gL2qJ_I","type":"paragraph","data":{"text":"Catalytic oxidation of steroids in the presence of chiral manganese complexes. EHA is 2-ethylhexanoic acid."}}]](/storage/images/resized/SE5Pw6B724mkeDOfKnMLks752EtRuoxQLVj9tyDd_xl.webp)
In recent years, another promising class of selective transformations catalyzed by non-heme manganese complexes — direct C – H acyloxylation — has been identified. It was found that, along with the expected hydroxyl derivative, the oxidation often produces an ester of a carboxylic acid used as a co-catalytic additive (Fig. 16). Acyloxylation has been shown to occur by C-radical rebound to the carboxylato ligand at the metal site in the solvent cage, which competes with the rebound to the incipient OH group: this mechanism was called the ‘alternative rebound mechanism’.[205]
![[{"id":"gzmZ_5rwFW","type":"paragraph","data":{"text":" ‘Alternative rebound mechanism’ on the example of cumene oxidation."}}]](/storage/images/resized/2CmQyiSev25s6AKHjnbGeIP0wYhooxHenSlxL8Rm_xl.webp)
The selectivity for the ester is usually lower than for the hydroxy derivative. However, it increases when the acyloxylation is an intramolecular process. For example, Costas and co-workers[232] found that manganese complexes can catalyze the γ-lactonization of substituted adamantaneacetic acids with high enantioselectivity (Fig. 17). Isotopic labelling studies witnessed that the lactone formation occurs via two parallel pathways: by rebound of the OH group followed by intramolecular esterification, and by rebound to the carboxylato group. Later, this catalytic method was adapted for the diastereoselective γ-lactonization of a number of natural and synthetic α-amino acids and laid the grounds for a new synthetic approach to chiral α,α-disubstituted α-amino acids.[233] An insight into the mechanism of this reaction confirmed that the most likely reaction pathway was intramolecular hydrogen abstraction from the γ-carbon atom followed by intramolecular lactonization.[234] The high selectivity for the γ-position was explained by the directing effect of the carboxyl group through which the substrate binds to the catalytically active site. The catalyst (S,S)-XVII can activate even the strongest primary C – H bonds, thus enabling the preparation of diastereoisomeric lactones of camphanic, ketopinic and isoketopinic acids.
![[{"id":"r7vBoleKgq","type":"paragraph","data":{"text":"Stereoselective catalytic lactonization of carboxylic acids. <i>dr</i> is diastereomeric ratio."}}]](/storage/images/resized/D18st3bpZITFGqmc5U3KL47HZALdXUJbd1fTkHU3_xl.webp)
The use of the sterically hindered catalyst (S,S)-XVIII made it possible to carry out lactonization of fatty acids (Fig. 18).[235] Significantly, by varying the reaction conditions, the regioselectivity of the process can be switched between the γ- and δ-positions. In the latter case, the reaction does not appear to be directed by the carboxyl group. The formation of δ-lactone is probably due to the hydrogen abstraction from the most electron-rich C – H bond, followed by OH group rebound and intramolecular esterification catalyzed by a strong Brønsted acid added to the reaction mixture.
![[{"id":"E80Cih3ItX","type":"paragraph","data":{"text":"Regioselective lactonization of fatty acids in the presence of Mn complexes."}}]](/storage/images/resized/t7Q136fGuDBGdQNVWmMCIotc2qqAuT2Dw1IbnlVp_xl.webp)
Summarizing the above, the development of catalysts and processes for chemo-, regio- and stereoselective oxidative functionalization of C(sp3) – H groups of complex molecules is one of the priority areas of modern synthetic chemistry. The sustainability of this area is determined by the fact that this significantly reduces the step count, thus reducing the amount of byproducts, and allows avoiding the use of toxic or high-molecular-weight oxidants. Until recently, studies on biomimetic catalytic systems for the selective C – H oxidation of organic compounds were mostly fundamental. However, significant advances achieved to date open the gateway to investigations focused on practical synthetic needs, such as late-stage alteration of complex molecules of natural origin, fast generation of chemical libraries of metabolites, production of biologically active compounds and pharmaceuticals.
3.2. Green chemistry in catalytic cross-coupling reactions
The discovery of catalytic cross-coupling is a prominent achievement of organic chemistry of the 20th century. Reactions of this type are used for one-step formation of new C(sp2) – C(sp2), C(sp2) – C(sp) and C(sp2) – C(sp3) bonds, which are present in many organic compounds both occurring in nature and synthesized in laboratories.
Historically, the cross-coupling reactions were performed for the first time in Japan (Tamao, Kumada) and France (Corriu) with organolithium and organomagnesium compounds and nickel complexes as catalysts. However, the best results were obtained using a palladium complex (Murahashi). Initially, organotin (Stille, Milstein), organomercury (Beletskaya) and organozinc derivatives (Negishi) and some other compounds served as the organometallic components; however, later they were replaced with readily available, stable and relatively non-toxic (hetero)arylboronic acids (Suzuki, Miyaura). The palladium-catalyzed arylation of activated olefins (Heck, Mizoroki) can also be considered as cross-coupling. For these studies, Suzuki, Negishi and Heck were awarded the Nobel Prize in 2010. The subsequent progress in this research area resulted in the appearance of so-called carbonylative cross-coupling (Tanaka) and the discovery of a method for C(sp2) – E bond formation (E = S, Kosugi and Migita; E = P, Hirao; E = O, Beller; and E = N, Buchwald and Hartwig). The golden age of palladium catalysis was possible thanks to the development of an array of new phosphine ligands and preparation of diverse palladium complexes acting as precursors of active catalysts.
In recent years, under the influence of green chemistry concepts,[19][236] the trajectory of development of this synthetic approach has been shifting towards environmentally benign, low-waste and safe methods. In addition to widely known E-factors,[3] new criteria have been introduced for evaluation of the greenness of chemical reactions.[10][237] The need to save energy and the fact that uncontrolled use of natural fuel is no longer possible have started to be regarded as important components of green chemistry paradigm. An organic chemist who performs a catalytic reaction should evaluate the reaction utility not only in terms of TON and TOF, but also by calculating the consumed energy. There appeared the problem of production of ‘clean energy’, ‘green hydrogen’ and many other, which did not come to mind previously. Generally, the theoretical base of green chemistry continues to develop, covering an increasingly broad range of aspects and becoming a truly multidisciplinary discourse within the framework of sustainable development of society.[30][238][239]
Cross-coupling reactions are substitution reactions; therefore, as regards atom economy, they are inferior to addition reactions. Cross-coupling reactions always give salts as by-products, which most often, find no use and end as waste. Recently, toxic organometallic, first of all, organomercury, compounds were excluded from the inventory of reagents for this type of reactions. Although these reagents provide excellent results,[240][241] they are toxic and hazardous for the environment. Also, aryl halides ArX have different reactivity, cost and toxicity (in particular, considering also the salts formed in reactions) depending on the nature of X.
As regards the classic palladium catalysts, that is, complexes PdX2*L2 (L = Ph3P or other phosphines), which are reduced during the reaction to Pd(0)L2, efforts of researchers have been mainly directed towards the decrease in the palladium loading due to its high cost. The use of vanishingly low Pd concentrations of approximately 10–3 – 10–5 mass % was reported. It is clear that these low quantities of palladium were not isolated and went into waste. This brought about a new problem of palladium scattering, resulting in the inevitable decrease in the reserves of this natural element. This stimulated the development of new approaches based on catalyst immobilization on a support through linkers, use of nanocatalysts supported on soft and hard materials and replacement of noble metals such as Pd, Ru, Rh or Ir by more abundant and less expensive metals like Cu, Ni, Co and Fe.
A considerable role in the cross-coupling reactions, like in any other reactions, belongs to the solvent. Water as a cheap, nontoxic and safe solvent is the most interesting alternative to organic solvents, which have often formed the major part of waste. However, the use of water brings about other problems such as poor water solubility of organic compounds, water purification after the reaction, and the need to prevent contaminated water from getting into the environment. A closed-loop process with water recycling can be implemented in industry, whereas in laboratory, this problem is still to be addressed.
In this Section, we made an attempt to consider the above and some other issues related to cross-coupling reactions taking account of new environmental requirements.
3.2.1. Palladium-catalyzed cross-coupling in aqueous and aqueous alcohol solutions
Sheldon, who was one of the first to pay attention to the importance of waste-free methods, emphasized that transition metal catalysis by itself brings a green aspect to a chemical reaction.[242] Nevertheless, high temperatures, potentially toxic organic solvents, unsafe organometallic reagents, and the use of considerable amounts of various additives require development of alternative processes that would be markedly safer for the environment. A concise review of the methods that draw cross-coupling reactions closer to green chemistry requirements was published in 2010.[243] The advantages of conducting reactions of this type in water at room temperature without preliminary preparation of organometallic compounds is briefly considered; good prospects of C – H bond activation towards cross-coupling by simple palladium salts, thus avoiding the use of halogenating compounds, were noted.
Extensive literature is devoted to palladium-catalyzed cross-coupling reactions in water and water – organic mixtures. At the Laboratory of Organoelement Compounds of the Department of Chemistry, Moscow State University, it was shown back in the 1980s that the Heck, Suzuki and Sonogashira reactions and carbonylation reaction can be accomplished in water under fairly mild conditions by using ligand-free palladium salts [PdCl2 , Pd(OAc)2] as catalyst precursors.[244] This resulted in the discovery of palladium nanocatalysis and its wide use in organic synthesis.[245] A vivid illustration of the efficiency of this approach is successful synthesis of a wide range of substituted biaryls, which was performed using simple divalent palladium salts in water at room temperature (Scheme 31). This reaction can also be catalyzed by palladium metal (Pd black, which is actually nanopalladium, as was shown previously) also without addition of a ligand.[246]
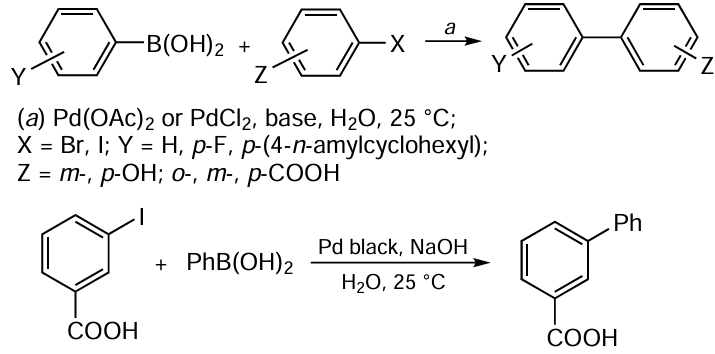
Since palladium is a precious metal and its use in catalysis, even if its amount has been effectively minimized, results in palladium scattering and non-intentional removal from natural resources, studies have been undertaken at catalyst heterogenization by immobilizing the ligand on a support,[247-250] which has blurred the distinction between homogeneous and heterogeneous catalysis. The support itself can act as a ligand that significantly affects the activity of the supported palladium nanoparticles (PdNPs). For example, ligand-free palladium supported on the copolymer of vinylimidazolone (PVI) and vinylcaprolactam (PVC) efficiently catalyzed the Suzuki –Miyaura reaction in aqueous ethanol (Scheme 32). The possibility of recycling the catalyst eight times was demonstrated in relation to the reaction of phenylboronic acid with p-bromoacetophenone.[251]
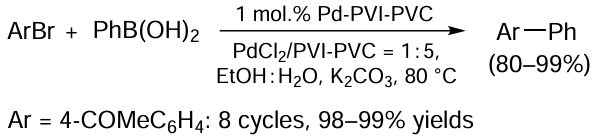
The same catalytic system can be used in one more reaction, cyanation, in which the heterogeneous catalyst can be recycled and reused 10 times (Scheme 33).[252]
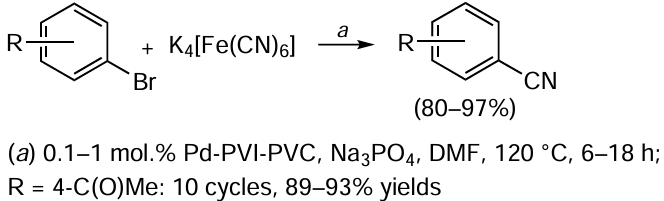
The well-known Pd/C hydrogenation catalyst is also effective in the Suzuki reaction.[253] The catalyst is easily separated from the reaction mixture by filtering, and high product yield is retained over five cycles.[254] The simplest catalysts such as Pd/C and Pd black, without the addition of ligands, enabled industrial synthesis of two medicinal agents of the Lilly pharmaceutical company (Scheme 34). The amounts of reactants were 0.5 – 1 kg, and the product yields were 67 – 93%.
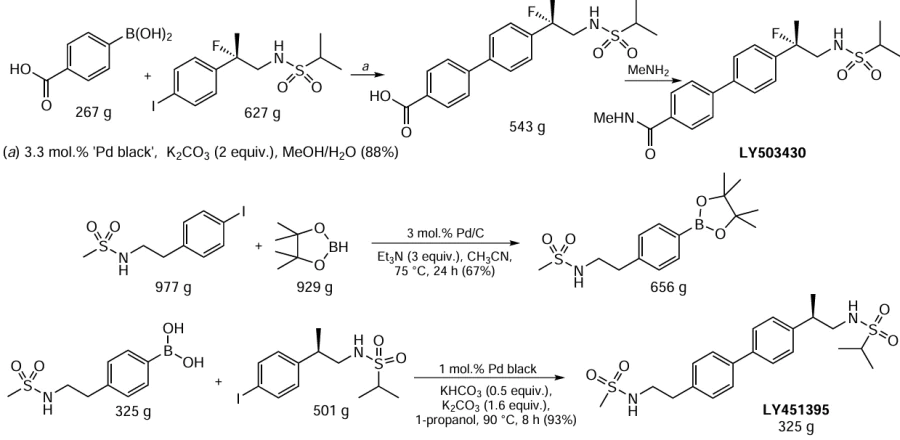
Reviews on the use of green chemistry principles in cross-coupling reactions have been regularly published in recent years; in some cases, several catalytic processes are considered in one review,[255] while other reviews address single reactions, e.g., Suzuki – Miyaura[256] or Sonogashira[257] reaction, or new protocols for known catalytic reactions using microwave or ultrasonic irradiation and mechanochemical procedures.[258] Many of these reactions are carried out in water.
The process of hydroformylation in a two-phase system in the presence of water-soluble ligands proposed by the Rhône – Poulenc company back in 1970[259] proved to be also useful for cross-coupling.[260] The suitable ligands include water-soluble phosphines,[261] hydrophilic N-heterocyclic carbenes[262][263] and other hydrophilic nitrogen-containing compounds.[264] The reactions are accelerated upon the addition of surfactants. Among recent studies, the following should be mentioned.
Palladium complexes with N-heterocyclic carbenes [NHC · H][Pd(η3-R-allyl)Cl2] in ethanol (0.3 – 0.5 mol.% Pd) were used as effective catalysts for the cross-coupling reactions involving electron-withdrawing and electron-donating aryl bromides and chlorides and heteroaromatic and sterically hindered aryl halides.[265] The phosphine ligand EvanPhos was used to synthesize biaryls by the Suzuki – Miyaura reaction in the presence of 0.5 mol.% palladium. A 2% solution of micelle-forming agent TPGS-750M (Fig. 19) in a 9 : 1 water – toluene mixture served as the reaction medium; in the case of aryl bromides, the yields of biaryls reached 97%.[266]
![[{"id":"pH9YvTJfVN","type":"paragraph","data":{"text":"Phosphine ligand EvanPhos and micelle forming agent TPGS-750M."}}]](/storage/images/resized/nhDiyIqSq6QXEdG4NZCV0nHDxdRKG75s0P3unE1m_xl.webp)
The fungicides boscalid, fluxapyroxad and bixafen were synthesized in water in the presence of ultralow loading of Pd(OAc)2 (down to 0.005 mol.%).[267] Even aryl chlorides successfully reacted under these conditions (Scheme 35).
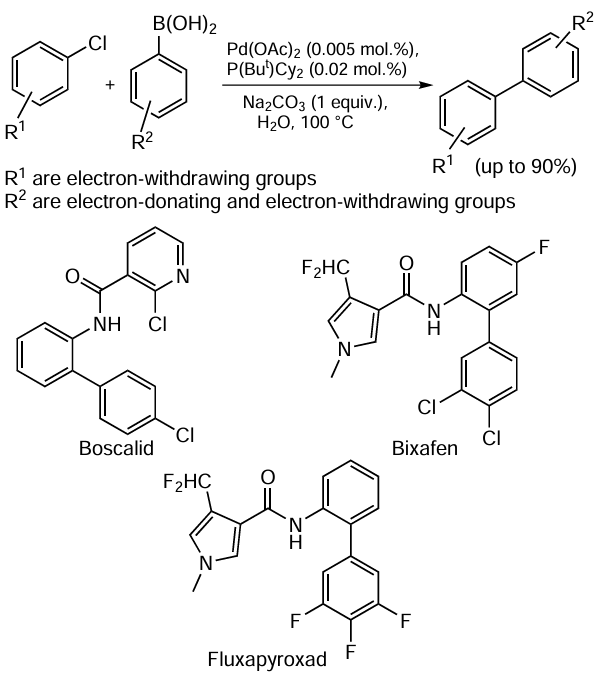
It was shown that aqueous solutions of Kolliphor EL, a well-known surfactant, form nanomicelles, which can serve as ideal nanoreactors for palladium-catalyzed cross-coupling.[268] In these systems, the Suzuki – Miyaura reaction proceeds even at room temperature.[269] The catalytic cross-coupling in aqueous micelles formed by the PiNap-750M surfactant (naphthalenediimide derivative, 2 mass %) gave rise to organic semiconductors.[270] When Pd(dtbpf)Cl2 [dtbpf = 1,1'-bis(di-tert-butylphosphino)ferrocene (2 mol.%)] was used as the catalyst, the product yields were 80 – 93% (Scheme 36).
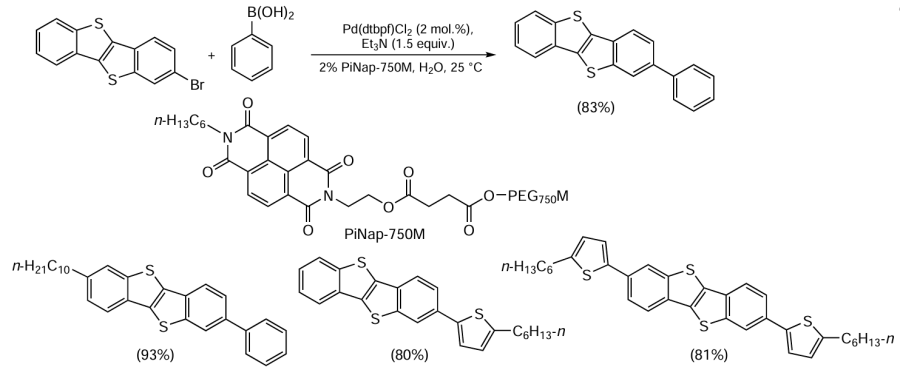
The sonidegib drug was obtained in an aqueous medium in the presence of TPGS-750M micelle-forming surfactant, with the required amount of palladium being only 0.5 mol.% (Scheme 37).[271]
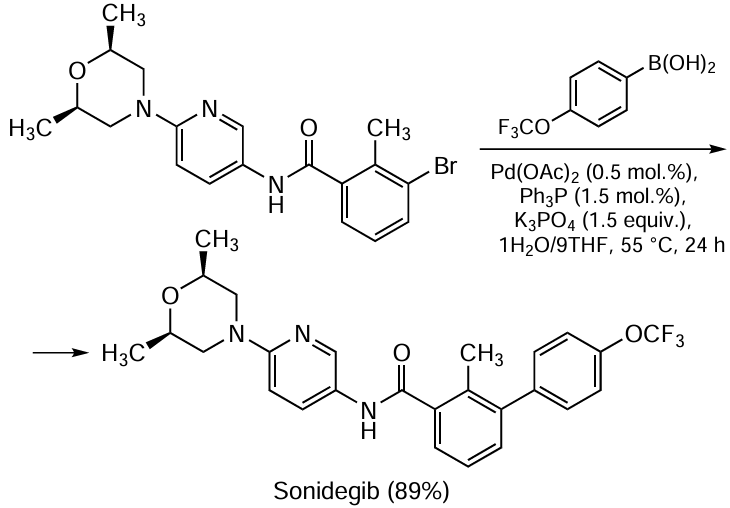
Model micellar catalysis of the reaction of p-bromoanisole in water in the presence of TPGS-1000 micelles (2 mass %) and palladium (0.5 mol%) as bacteriogenic nanoparticles from Desulfovibrio alaskensis furnished the product in a more than 99% yield.[272]Hydrogels formed from 1,3 : 2,4-dibenzylidenesorbitol modified with acyl hydrazide, agarose and 1 mol.% palladium in aqueous ethanol (1 : 3) ensured quantitative yields of biaryls in the Suzuki–Miyaura reactions and could be recycled and reused up to ten times.[273]
The water-soluble C60-TEGs/PdCl2 catalyst with an average nanoparticle size of ~60 nm obtained by supporting palladium(II) chloride on triethylene glycol (TEG)-modified fullerene (C60-TEGs) proved to be effective in the reaction of phenylboronic acid with aryl halides containing either electron-donating or electron-withdrawing substituents. The target biaryls were formed at room temperature in up to 99% yields in the presence of only 0.01 mol.% catalyst, and the catalyst could be recycled and reused up to five times without a considerable loss of the catalytic activity.[274]The use of water-soluble palladium nanoparticles stabilized by phosphinic acids in the Suzuki – Miyaura, Sonogashira and Heck reactions was reported.[275]
The application of arenes instead of aryl halides is an attractive alternative to the classical Suzuki reaction. However, the methods known to date require fairly harsh conditions and the presence of common organic solvents, which is at variance with the green chemistry principles. Recently, a mild Suzuki oxidation reaction was performed in water using hydrogen peroxide as the oxidant. The efficiency of the oxidative cross-coupling was confirmed for a broad range of substrates, including those containing various functional groups. The applicability of this green strategy for the conduction of Heck and Sonogashira reactions was also demonstrated.[276]
Nanosized palladium particles immobilized on magnetic graphene oxide modified with poly(2-acrylamido-2-methyl-1-propanesulfonic acid) (GO/Fe3O4/PAMPS/Pd) proved to act as effective nanocatalysts for the Suzuki – Miyaura reaction in aqueous ethanol, in particular for aryl chlorides.[277]
Magnetic palladium nanoparticles supported on a graphene oxide and steel composite efficiently catalyzed the Suzuki–Miyaura and Stille reactions in aqueous ethanol. Cross-coupling products were formed in up to 97% yields.[278] Magnetic iron nanoparticles containing a low (0.08 mol.%) amount of palladium combined with the S-Phos ligand (2,6-dimethoxy-2'-dicyclohexylphosphinobiphenyl) were utilized in the Suzuki–Miyaura reaction in a flow reactor. In the synthesis in a water flow containing 2 mass % TPGS-750M surfactant, the yields of products reached 95%.[279] The practical value of the method was demonstrated by the synthesis of intermediates for the preparation of drugs in amounts of up to 20 g in one experimental operating cycle (Scheme 38).[280]
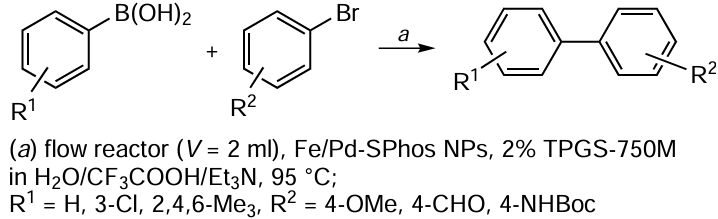
Palladium nanoparticles immobilized on titanium and zinc oxides taken in sub-stoichiometric amounts were used to catalyze the Suzuki–Miyaura reaction with the possibility of recycling. The reactions were carried out in aqueous ethanol in the presence of 0.3 mol.% palladium, with the yields of biaryls being as high as up to 96%.[281]
A number of reactions catalyzed by palladium-containing metal-organic frameworks (MOFs) have been described. Palladium attached to lanthanum MOF containing Fe3O4 nanoparticles via Schiff base was tested in the model Suzuki reaction and provided TOF of 42886 h–1, which is one of the highest values obtained for this reaction. The catalyst withstood twelve reaction–catalyst recovery cycles without a considerable decrease in the activity.[282] Metal-organic frameworks containing bis(N-heterocyclic) palladium complexes were used in the Suzuki, Heck, and Sonogashira reactions.[283] The Fe3O4@PDA – Pd@UiO-67 composite (PDA is polydopamine) proved to be an efficient catalyst for the Suzuki reaction in aqueous ethanol reusable up to eight times.[284]
The catalyst based on palladium nanoparticles (PdNPs) immobilized on reduced graphene oxide (rGO) successfully catalyzed the Suzuki–Miyaura reaction with aryl chlorides in aqueous methanol under microwave irradiation.[285] The yields of products reached 95% with the palladium loading being 0.5 mol.%. In this study, the key intermediates for the synthesis of irbesartan and fluxapyroxad drugs were obtained; however, reactions of o-substituted chloroarenes required the use of ethylene glycol or DMF as solvents (Scheme 39).
The Suzuki – Miyaura reaction involving aryl bromides in water was catalyzed by PdNPs supported on graphene acid.[286] The cross-coupling reactions were formed under these conditions in quantitative yields, and immobilized catalyst could be used five times without the loss of activity. The Pd(II) complexes (0.5 mol.%) with N-heterocyclic carbenes efficiently catalyzed the Suzuki – Miyaura reactions involving aryl bromides in water at room temperature.[287] Palladium nanoparticles (1.7 mol.%) incorporated in the pores of a covalent framework based on N,N-dimethyldodecyl terphenyldicarboxylic acid dihydrazide derivative (Pd@COF-QA) also proved to actively catalyze these reactions.[288]
Palladium(II) (1 mol.%) chelated by a porous organic polymer (POP) containing 2,2'-bipyridine moieties made possible the Suzuki reaction with aryl bromides in air in aqueous ethanol (EtOH : H2O = 3 : 2). The catalyst was used five times without decrease in the product yield.[289] Palladium-functionalized polyurethane foam also showed high catalytic activity in the Suzuki – Miyaura reaction with bromoarenes in aqueous ethanol.[290] In the presence of 1.4 mol.% palladium, the product yields reached 98%. The SBA-15 polymer modified with the N-isopropylacrylamide–methacrylic acid copolymer proved to be a convenient support for the immobilization of PdNPs.[291] The reactions were carried out in a water – ethanol mixture (4 : 1); the yields of products formed from aryl bromides and aryl iodides differed only slightly (94 – 96%).
There are quite a few recent publications describing unusual supports for immobilization of metal nanoparticles involved in catalysis. For example, PdNPs were supported on a composite in which chitosan and cellulose films were attached to corn stalk biochar. The use of this composite biomaterial in the Suzuki–Miyaura reaction in ethanol with 0.5 mol.% palladium loading gave biaryls in up to 99% yields. The catalyst could be reused many times (up to ten cycles without the loss of activity).[292]
Palladium nanoparticles immobilized on a composite comprising cellulose and volcanic pumice magnetic particles effectively catalyzed Suzuki–Miyaura reactions in DMSO, with their activity being retained even after ten regenerations.[293] A dip catalyst prepared by dispersion of PdNPs in the sugarcane bagasse proved to be effective in the cross-coupling reaction where it could be reused many times (up to 15 cycles).[294] Agar containing immobilized Pd particles (34 – 45 nm) was used to carry out the Suzuki reaction under microwave irradiation.[295]
Ultrasonic treatment is an alternative to conventional heating.[296] The efficiency of ultrasound was demonstrated in relation to Suzuki–Miyaura reactions catalyzed by Pd/C (5 mass %)[297] or Pd nanoparticles immobilized in the KIT-5 – biguanidine mesoporous structure in aqueous ethanol (EtOH : H2O = 1 : 1).[298] In the latter case, bromo- and iodobenzenes were converted to target products in up to 98% yields in the presence of only 0.25 mol.% palladium; the catalyst could be used up to six times without decrease in the performance.
Ethylene glycol, glycerol and polyethylene glycols (PEG) are of obvious interest as green solvents. Glycerol was used as a reaction medium in the Suzuki – Miyaura reaction catalyzed by palladium present in the roots of the Eichhornia crassipes plant commonly known as water hyacinth. In the presence of this plant catalyst, heteroaryl halides and heteroarylboronic acids reacted to give the corresponding bis-heterocycles in good yields.[299]
A review devoted to the use of polyols as solvents summarizes the data on the use of glycerol- and PEG-stabilized palladium nanoparticles in the Suzuki, Heck and Sonogashira reactions and the Ullmann synthesis of biaryls.[300] The reactions with medium-size (4 – 6 nm) PdNPs were noted to smoothly proceed in PEG of various molecular weights (including PEG-400) or using solid supports, for example, styrene and ethylene glycol copolymer.
The presented examples of the Suzuki–Miyaura reaction comply, to an extent, with the green chemistry requirements. Among other C – C bond formation reactions, mention should be made of the Sonogashira reaction, which can also occur in aqueous solutions. The reaction carried out in the presence of Pd(PPh3)2Cl2/PPh3/CuI (16/33/22 mol.%) in water under microwave irradiation resulted in the synthesis of polyacetylene precursors of porous organic structures (Scheme 40).[301]
The Sonogashira reaction efficiently proceeds in a flow reactor in the presence of hybrid Si-Gly-CD-PdNPs nanoparticles (the support is silica gel attached to b-cyclodextrin with a glycerol linker) in a flow of a green solvent (cyclopentyl methyl ether/water azeotrope).[302] A palladium catalyst immobilized in a styrene – divinyl benzene copolymer containing acidic cation exchange groups (sulfonic acid fragments) was successfully used in the Suzuki reaction (0.5 mol.% Pd, PriOH), Heck reaction (0.2 mol.% Pd, DMA; DMA is dimethylacetamide) and Sonogashira reaction (0.4 mol.% Pd, PriOH-H2O, 1 : 1) without adding copper as an oxidant.[303] The same behavior in the Sonogashira reaction was inherent in palladium nanoparticles supported on polyaniline (Pd@PANI), obtained by oxidative polymerization of aniline in the presence of PdCl2 , and in the mesoporous Pd-based catalyst supported on carbon nitride (g-C3N4).[304]
Among a variety of two-dimensional and three-dimensional supramolecular assemblies, Pd6L8 type structures in which Pd(II) ions are surrounded by conformationally flexible tripodal pyridine-containing tris-amide ligands proved to be fairly efficient catalysts for the Sonogashira reaction. These catalysts also do not require addition of copper or phosphine ligands to the reaction system.[305]
Magnetic nanoparticles composed of Pd(II) complexes with N-heterocyclic carbenes supported on Fe3O4 successfully catalyzed the Heck reaction in water and could be reused in five cycles.[306] Palladium-containing MOFs (PdNPs@ZIF-8,[307] Pd@ZIF-67,[308] and UiO-67 (Ref. [309])) and microporous metal-organic material based on palladium-containing nanotubes Pd@NH2-MONFs[310] were also effective in catalyzing the Heck reaction.
3.2.2. Cross-coupling reactions catalyzed by other metals
Due to high cost of palladium and the possible adverse impact of palladium-containing waste on the ecosystems, attempts have been made to replace palladium-based catalysts with catalysts containing other metals. Gold nanoparticles immobilized in the capillary pores of the Sr/Alg/CMC/GO/Au complex composite containing graphene oxide, strontium and cross-linked polymer based on the alginate (Alg) anionic heteropolysaccharide and carboxymethylcellulose (CMC) were exceptionally active in the Suzuki – Miyaura reaction. The standard reaction with aryl iodide required only 0.005 mol.% loading of the catalyst. The possibility of catalyst reuse (six cycles) with biaryl yields of up to 98% was demonstrated.[311]
The immobilization of CoCl2 on magnetic nanoparticles Fe3O4@AlO(OH) (boehmite) resulted in the formation of a catalyst active in the Suzuki – Miyaura reaction.[312] The Co(II) compounds immobilized on chitosan (CS) modified with furfural-based Schiff base was studied in the Suzuki – Miyaura, Heck, Hirao and Hiyama reactions in water (Scheme 41).[313] The CoFe2O4/asparagine nanocomposite catalyzed the synthesis of diaryl thioethers and could be recycled and reused.[314]
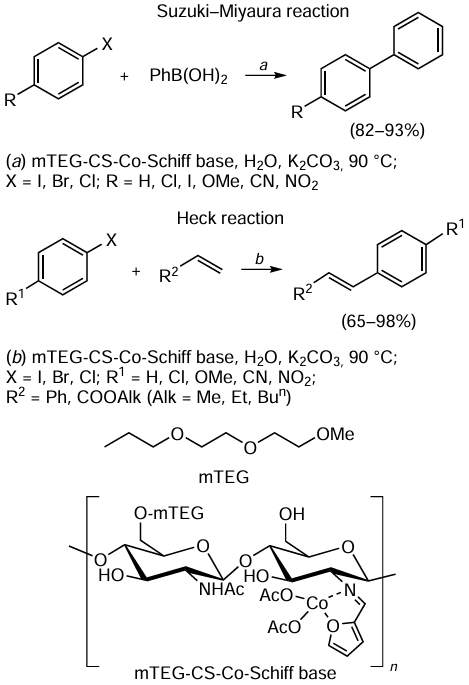
The α-Fe2O3 nanoclusters (1.8 nm) deposited on graphene oxide proved to be effective catalysts for the Suzuki – Miyaura reaction at an ultralow loading (150 ppm). At 80°C, this reaction was performed for bromobenzene derivatives containing electron-donating and electron-withdrawing groups in the para-position. The product yields were as high as 87% and the catalyst was reused four times without a decrease in the activity.[315] Sonogashira reaction conducted in the presence of iron(III) chloride complex with 1,10-phenanthroline under aerobic conditions in water was reported. Functionalized aryl iodides containing heteroaryl and sterically hindered substituents and terminal heteroarylalkynes were found to be appropriate substrates under these conditions.[316]
The palladium-free Suzuki reaction was carried out using Cu(I) complex supported on polythiophene-functionalized magnetic carbon nanotubes.[317] The ZnCl2 – [Bmim]BF4 (ionic liquid) system served simultaneously as a catalyst and as a solvent in the Suzuki – Pictet – Spengler tandem reaction, which furnished pharmacologically valuable polyfunctional 6-arylphenanthridines.[318] The zinc-catalyzed reaction of electron-rich aryl iodides with phenylacetylene resulting in the formation of the C(sp2) – C(sp) bond was also reported.[319]
In recent years, nickel-containing catalysts have found wide use for the formation of carbon–carbon and carbon–element bonds.[320][321] For example, nickel formate derived from food industry waste smoothly catalyzed the Suzuki–Miyaura reaction,[322] while the nickel composite obtained from azide-modified UiO-66 MOF, Ni(cod)2 and PPh3 proved to be an efficient reusable (up to seven cycles) catalyst for the Suzuki reaction.[323]
Nickel chloride complex with ferrocene diphosphine successfully catalyzed the Suzuki–Miyaura reaction with diverse aryl halides, including chloroarenes, in an aqueous solution of TPGS-750M. The reactions proceeded at moderate temperature (45 °C) at a nearly stoichiometric ratio of the reactants, including sterically hindered compounds (Scheme 42).[324]
The heterogeneous bimetallic Ni(0) bis(propylmalononitrile) complex (NiFe2O4@SiO2–BPMN – Ni)[325] and the nickel complex with the tetradentate N,O,O,N-salicylidenethiadiazole ligand[326] also successfully catalyzed the Suzuki–Miyaura reaction in aqueous methanol. Furthermore, the bimetallic complex was easily isolated with a magnet and could be reused seven times without noticeable loss of activity. The NiCl2(PPh3)2/CuI/PEG-400/H2O catalytic system was used to prepare various acetylenes by the Sonogashira reaction.[327] When the products were extracted with petroleum ether, this system could be reused up to six times. In the Sonogashira reaction catalyzed by nickel nanoparticles, there was no need to additionally use copper compounds, while the nanocatalyst could be utilized in five cycles.[328]
3.2.3. Catalysis by copper compounds
Obviously, reactions using copper as a catalyst form an important step towards green chemistry. The use of nitrogen- and oxygen-containing ligands, most of which are nontoxic and much more readily available than phosphine ligands, allowed the reactions to be performed under fairly mild conditions, which actually created a new Ullmann chemistry.[329][330] The mechanisms of palladium and copper catalysis have much in common, but there are also quite a few differences.[331] However, it is undeniable that copper is an excellent catalyst for the C(sp2) – heteroatom cross-coupling reaction, being especially applicable for the formation of C(sp2) – N bonds, where palladium catalysts would require the presence of expensive ligands that are often difficult to obtain. Currently, there are publications describing copper-catalyzed reactions that give C(sp2) – S(Se), C(sp2) – P and C(sp2) – O bonds using various copper compounds with oxygen- and nitrogen-containing ligands[332] or ligand-free copper.[333] The reactions are carried out in green solvents, including water,[334] ethylene glycol[335] and ethanol.[336] For immobilization of copper complexes and copper nanoparticles (CuNPs), soft and hard supports have been used.[337]
Among recent studies, the following ones may be noted. Combination of CuI with surfactants was utilized for the generation of C – S and C – N bonds.[338] A variety of diaryl sulfones were prepared by the reactions of aryl bromides and aryl iodides with benzenesulfinates in water in the presence of N-alkyl-lactosamine. The product yields reached 95% for iodides and 85% in the case of bromides. The same catalytic system proved to be suitable for the arylation of azoles such as imidazole, benzimidazole and indole, with the yields of N-aryl derivatives being as high as 80 – 90%. A polymer based on Cu(II) complex with (E)-N'-(4-(diethylamino)-2-hydroxybenzylidene)-4-methylbenzohydrazide effectively catalyzed the reaction of aryl iodides with thiophenol giving rise to the C – S bond in water at 90°C in the presence of potassium carbonate as a base (up to 98% yields).[339] The Cu(OAc)2 (10 mol.%)/Cp*Co(CO)I2 (1 mol.%) catalyst mixture was used for the amination of aryl chlorides.[340] Solvent-free and base-free reactions were carried out for aliphatic and aromatic amines and resulted in the formation of C – N cross-coupling products in 80 – 90% yields (Scheme 43). The authors believed that the cobalt complexes rather than the copper complexes were catalytically active in this case. This work is in line with other studies in which C – N bonds were generated using bimetallic cobalt and iron compounds, e.g., cobalt-containing magnetic nanoparticles[341] or nickel ferrite nanoparticles, which were found to be active in the amination reactions and in the amidation of various aryl halides.[342]
Copper(I) oxide nanoparticles can be used for the amination of aryl halides without the use of additional ligands, in some cases, in ionic liquids.[343] The use of Cu2O nanoparticles formed in situ from Cu(OAc)2 and a micelle-forming surfactant (hybrid of PEG and aminoglucose) proved to be effective for the formation of the C – S bond in water.[344] Under these conditions, aryl iodides and aryl bromides reacted with arenesulfinates to give diaryl sulfones in up to 95 – 96%.
A highly important aspect of the current stage of development of the Ullmann chemistry is the use of copper catalysts immobilized on various supports, which makes them reusable. For example, polymer-supported copper(II) complex successfully catalyzed the reaction of NH-heterocycles with aryl iodides (Scheme 44); the catalyst was recycled and reused several times without a considerable decrease in the yields of products.[345][346]
The aniline arylation with bromobenzene in the presence of copper(I) complex (0.04 mol.%) with Fe3O4-supported guanidine derivative proceeded in glycerol and gave the product in 80% yield.[347] After five catalytic cycles, the yield of diphenylamine decreased to 70% (Scheme 45).
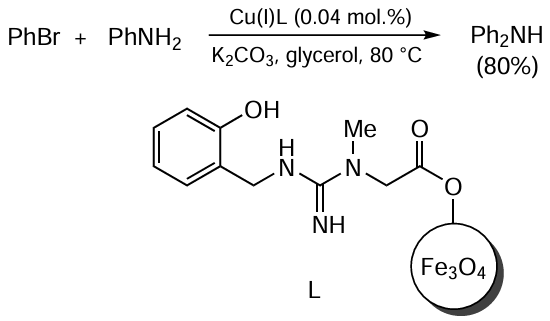
Copper(I) chloride supported on a magnetic material coated by ascorbic acid effectively catalyzed the arylation of NH-heterocycles and aromatic or aliphatic amines.[348] These reactions were carried out in water at room temperature in the presence of KOH as a base; the catalyst could be recycled and reused up to six times without a decrease in the performance. Copper(I) iodide supported on the INDION-770 cation exchange resin modified with a sulfonic acid successfully catalyzed the reactions of aryl iodides and aryl bromides with imidazole in DMSO.[349] Other polymers were also proposed for immobilization of the copper catalyst. For example, a CuI complex with Schiff base-modified chitosan proved to be an efficient catalyst for the arylation of amines, amides and azoles.[350] In the presence of this catalyst in combination with Cs2CO3, aryl iodides and aryl bromides reacted with primary and secondary amines in DMSO at 100°C to give amination products in up to 99% yields, while the catalyst retained its activity after recycling and reuse (up to five cycles).
The C – N and C – S bond formation can also be catalyzed by nickel compounds. For example, amination of aryl chlorides and aryl sulfamates takes place in the presence of the NiCl2/DME complex (DME is dimethoxyethane) in a green solvent, 2-methyl-THF.[351] The efficiency of amination increases under visible light irradiation in the presence of NaI as an activator. The catalytic reaction does not require the use of an additional photocatalyst and allows the formation of C – N bonds under mild conditions.[352] Lipshutz and co-workers[353] demonstrated that reactions resulting in the C – S bond formation can be performed using micellar catalysis with nickel compounds as catalysts. Aryl and heteroaryl halides were coupled with arene- and alkanethiols in an aqueous solution of TPGS-750M to give valuable compounds. Specifically, an intermediate for the preparation of the anticancer agent axitinib was synthesized in this way (Scheme 46).[353]
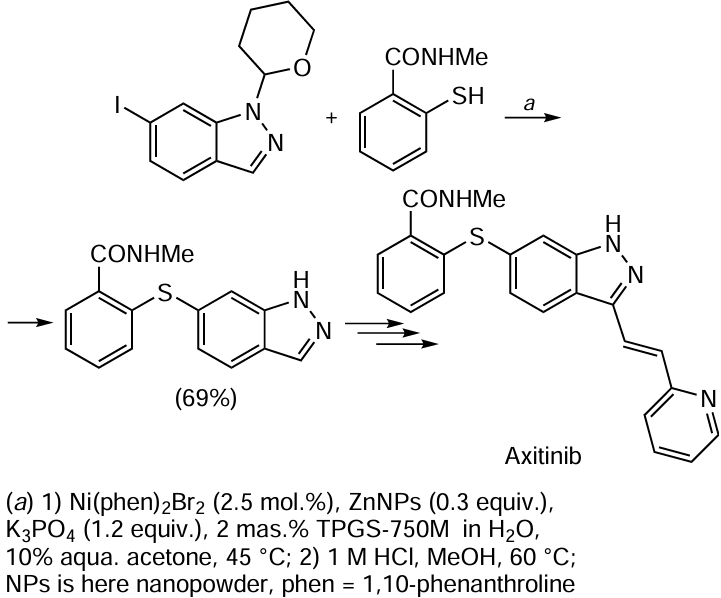
3.2.4. Chan – Lam – Evans reaction
Copper catalysts also proved to be effective in the oxidative cross-coupling of arylboronic acids with S-, N- or O-nucleophiles, discovered independently by three authors, which leads to the formation of C – N, C – S and C – O bonds.[354-356] This reaction, which involves two nucleophiles, unlike the Ullmann reaction, is widely used to synthesize a variety of alkylarylamines, alkylaryl and diaryl sulfides and aromatic ethers. We will consider some recent examples of using this strategy within the framework of green chemistry concept.
Nanomicelles formed from CuBr in the presence of a water-soluble ligand such as PEG-2000-functionalized pyridinetriazole and sodium dodecyl sulfate catalyzed the Chan–Lam reaction of diorganyl diselenides/disulfides with various aryl-, heteroaryl- and styryl-boronic acids in water at room temperature to give cross-coupling products in good yields. The aqueous phase containing the catalyst could be recycled and reused at least seven times. Transmission electron microscopy examination showed that the diameter of nanomicelles was 31.9 ± 8.7 nm in the selenide synthesis and 26.6 ± 5.0 nm in the sulfide synthesis.[357]
The Chan – Lam reactions are often performed in the presence of copper catalysts supported on various materials. For example, Cu(II) complex with a polyimide organic framework Cu@PI-COF proved to be a fairly efficient catalyst.[358] In the presence of this catalyst, the reaction in aqueous methanol (1 : 1) could be performed for aniline derivatives, aliphatic and heteroaromatic amines and azoles. The yields of products containing electron-donating or -withdrawing substituents in the aromatic rings of both reactants reached 90% (Scheme 47).
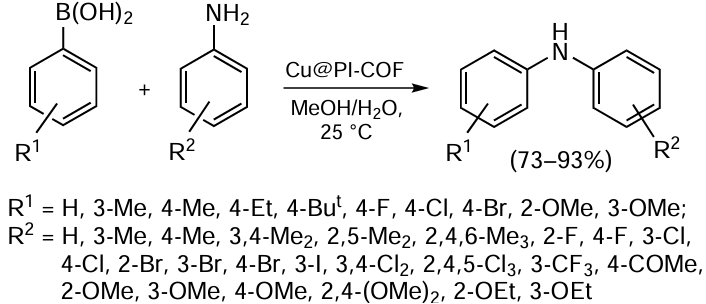
Copper nanoparticles immobilized on a modified graphene oxide (N-GO) actively catalyzed the reactions of various amines with phenylboronic acid.[359] The expected diarylamines were obtained in 76 – 98% yields, and the catalyst could be reused up to five times (Scheme 48).
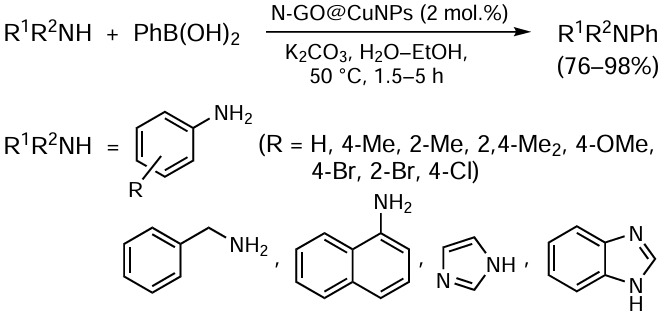
The Cu(II) complex of graphene oxide modified with n-propylsiloxane derivative containing 1-ferrocenylmethylimidazole moiety proved to be useful for the syntheses of N-arylsulfonamides.[360] A similar complex catalyzed the reactions leading to unsymmetrical diarylamines.[361] The Cu(II) complex with a Schiff base attached to graphene oxide via the (3-aminopropyl)trialkoxysilane linker was also efficient in these reactions.[362] In all cases, heterogeneous catalysts could be reused four to six times without the loss of activity.
Copper(II) oxide nanoparticles proved to be active catalysts for the cross-coupling of arylboronic acids with aniline derivatives and imidazole at room temperature; the product yields exceeded 80%.[363] When spherical CuO nanoparticles (6 nm) were used in the arylation of imidazoles, it was unnecessary to add a base to the reaction mixture.[364]
The metal-organic framework containing Cu(II) cations as the nodes and terephthalic acid (BDC, benzenedicarboxylic acid) and 4,4´-bipyridine (BPY) as the linkers provided high yields of diarylamines (up to 85%) in the reactions of aniline derivatives with phenylboronic acid in aqueous methanol and could be recycled (Scheme 49).[365]
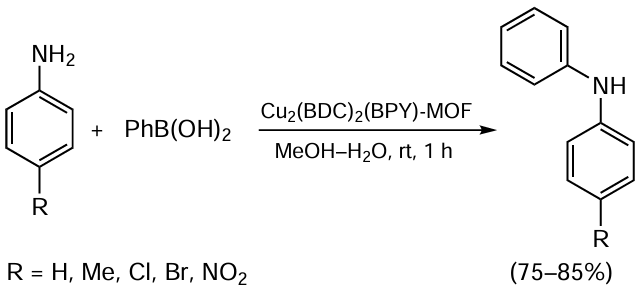
The metal-organic framework based on 4,5-bis(tetrazolyl)-1H-imidazole and Cu(NO3)2 was also effective for the arylation of amines with phenylboronic acid,[366] while a magnetic nanocatalyst based on Fe3O4 encapsulated in the Cu–apatite composite showed high activity in the Chan – Lam reaction involving imidazole and indole.[367] In both cases, the catalysts were recyclable and reusable many times.
Other methods for the synthesis of recyclable copper catalysts for the Chan – Lam reactions have also been reported, including, in particular, immobilization of copper(II) sulfate on the montmorillonite K-10[368] and immobilization of the bimetallic CuPd catalyst on the SiO2 – TiO2 support using diamine linkers.[369]
3.2.5. Cross-coupling reactions with CH-activation
An important stage in the development of cross-coupling strategy is switching to the reactions involving arenes after their CH-activation. The direct arylation of arenes and elimination of the preliminary metallation step decrease the number of steps and obviously correspond to the green chemistry requirements.[370-372] The reaction includes the metallation of the C – H bond in situ, the reaction of the metal with the partner and the reductive elimination to give the cross-coupling product. The efficiency and selectivity of the reaction are facilitated by the presence of a directing group, while Pd, Ru, Rh, Ir and, more rarely, Cu complexes serve as the catalysts.
Depending on the nature of reactants and conditions, the reaction mechanisms can markedly vary, which accounts for a great variety of reaction products. Copper-catalyzed CH arylation was described in relation to benzoxazole and pentafluorobenzene.[373] The indole arylation with diaryliodonium salt is a regiodivergent reaction yielding products at positions 2 or 3, depending on the substituents at the nitrogen atom.[374] Direct arylation of benzene with aryl halides in a photoredox reaction in the presence of iridium complex proceeds as homolytic aromatic substitution.[375] In the presence of ruthenium complexes as catalysts, the reaction can be carried out in green solvents.[376]
The CH arylation of 6-methoxybenzothiophene with p-iodomethoxybenzene catalyzed by the NiCl2(bipy) complex in a green solvent, 2-methyl-THF, was used to prepare the intermediate of the synthesis of the raloxifene drug (Scheme 50).[377] Active studies are carried out into CH alkenylation of, in particular, aromatic compounds with the acetamide directing group, which is catalyzed by Pd/C,[378] and quinoline N-oxides, which is catalyzed by iron(II) sulfate.[379] The arylation of 2-phenylpyridine (at position 2 of the benzene ring) and some other phenyl derivatives of heteroaromatic compounds was carried out in the presence of cyclometallated Ru(II) complex.[380]
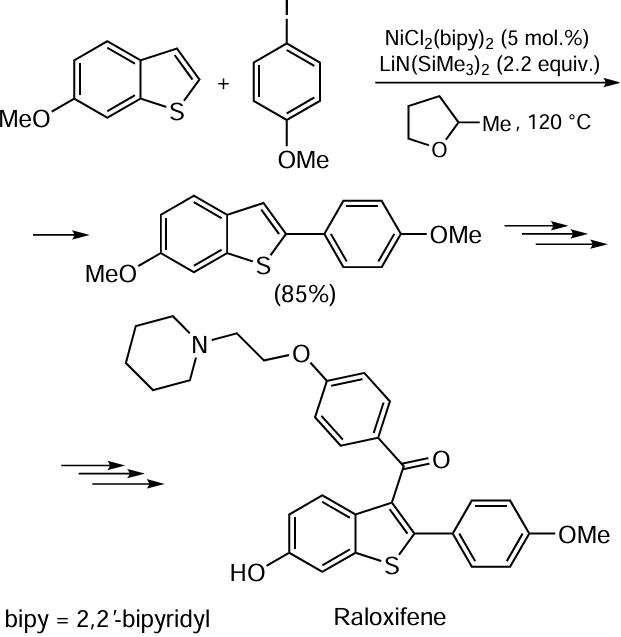
The amination reactions with CH-activation are of considerable interest. Most of reported reactions of this type are carried out with copper(II) catalysts; however, the solvents used in this case often cannot be classified as green, e.g., toluene,[381] xylene,[382] DMF[383] and nitromethane.[384] The reactions carried out in DMSO are more complementary to green chemistry principles. Roane and Daugulis[385] used DMSO as the solvent, 8-aminoquinoline as the directing group and oxygen as the oxidant for Cu-catalyzed amination of benzamides with primary and secondary amines and sulfonamide (Scheme 51).
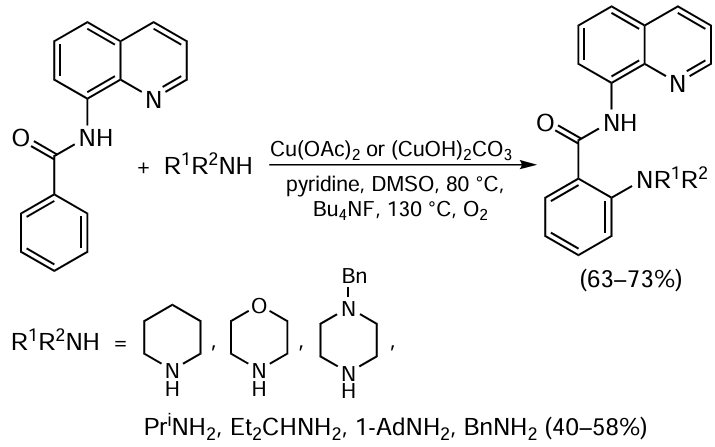
One more bidentate directing group was used by Yu and co-workers for the amidation of aryl- and heteroarylamides catalyzed by Cu(II) (Scheme 52).[386]
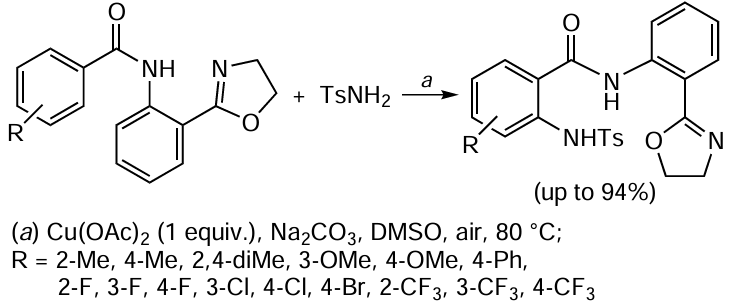
Due to the complexity of regioselective CH-amination, noble metal catalysis using metals such as Rh(III)[387][388] and Ir(III)[389][390] still plays an important role in this approach to the carbon–nitrogen bond formation, and studies along this line are in progress.
3.2.6. Replacement of common organic solvents with less toxic solvents
Many examples of cross-coupling reactions conducted in water have been given above. The introduction of other green solvents into the practice of catalytic synthesis is also being investigated. Several recent reviews address different aspects of the possible use of solvents compliant with the green chemistry principles. Thus Sherwood, Clark, et al.[391] considered various cross-coupling strategies (Suzuki, Stille, Kumada, Negishi, Hiyama, Heck, Sonogashira and Buchwald – Hartwig reactions) and analyzed the possibility of conducting these reactions in new solvents, including sulfolane, propylene carbonate, N-butylpyrrolidone, 2-methyl-THF, g-valerolactone, cyrene (dihydrolevoglucosenone). The last-mentioned solvent, cyrene, prepared from cellulose in two steps and similar to dimethylformamide in the physical properties, was shown to have no adverse impact on human health and to be efficient for the Suzuki – Miyaura reaction, where it provides high product yields and scalability.[392]
The use of green solvents in the cross-coupling reactions was considered in a review by Sydnes.[393] The author analyzed a number of solvents, including glycerol, ethyl acetate, 2-methyl-THF, g-valerolactone, glycerol carbonate, cyrene, p-cymene and limonene (Fig. 20), and concluded that most often, the use of these, mostly exotic, reaction media does not reduce the yield of target product in the catalytic reactions provided that the solvent has been appropriately selected. This conclusion echoes the results of a special study that showed that the yield of the product in the Suzuki–Miyaura reaction is generally determined by the nature of reactants but not by the solvent.[394]
![[{"id":"2BYoBqo_EG","type":"paragraph","data":{"text":"Promising green solvents."}}]](/storage/images/resized/LQTOzvrk3Co1g8l4z6vnzzjLC6OEU2ICZX4FB50X_xl.webp)
Finally, in the review by Andrade and Martins,[258] the reactants themselves, water, polyethylene glycols, ionic liquids and low-melting eutectics were considered as substitutes for the common organic solvents in the Suzuki – Miyaura reaction. Low-melting eutectics are being actively developed and start, in some cases, to replace ionic liquids. For example, the Suzuki – Miyaura reaction smoothly proceeds in these solvents at 100 °C in the presence of 0.1 – 1.0 mol.% PdCl2 . Among the considered eutectic mixtures, the best results were obtained for a mixture of choline chloride with ethylene glycol, which can be separated from the products after the reaction, together with the catalyst, and reused in the catalytic process (up to five times).[395]
Original publications often give other examples of green solvents, which are in some cases quite unusual, such as azeotropic mixture of furfuryl alcohol and water,[396] dimethyl sulfone,[319] ethyl lactate,[397] N-hydroxyethylpyrrolidone[398] and numerous technical and edible vegetable oils.[399]
According to the principles of green chemistry and taking account of the economic and environmental challenges of sustainable development, solvent-free organic reactions are preferred, because they reduce the emission of pollutants. In addition, they are characterized by lower E-factors and mass intensity (the ratio of the total mass of all materials used in the reaction to the mass of the product).[400] A number of successful examples of C – C, C – N and C – O bond formation under these conditions have been reported.[401-403] The Suzuki–Miyaura reactions can be carried out in a mixture of reagents using an inorganic base (KF – Al2O3) and mechanochemical treatment and microwave irradiation of the reaction mixture. The tolerance to atmospheric oxygen, higher yields of products and shorter reaction times make this strategy convenient and attractive. The magnetically isolated palladium catalyst was successfully used to prepare a series of biaryl compounds in the mixture of reagents, and the product yield decreases insignificantly (from 99% to 93%) by the tenth cycle.[404] Borchardt and co-workers[405] showed that the mechanochemical Suzuki polycondensation is an environmentally benign and efficient alternative to the traditional methods of poly(phenylene) preparation in solutions. By using an electromagnetic mill, Li et al.[406] carried out the Suzuki – Miyaura reaction without a solvent or a dispersant in the presence of a minor amount of palladium (0.05 mol.%).
Thus, the concept of green chemistry plays an important role in the development of new environmentally benign protocols for catalytic cross-coupling reactions. These reactions are performed, more and more often, in the medium of reactants or green solvents using low-toxic, recyclable and low-cost catalysts. There appears new equipment that makes it possible to carry out reactions in the continuous flow mode, under microwave or ultrasonic irradiation or in photochemical or electrochemical cells. Chemists have acquired a potent tool to create new types of bonds and are actively using these opportunities thus changing the adverse environmental impact of chemical synthesis and making chemistry more attractive to the society.
3.3. Asymmetric organocatalysis — the way for highly selective green chemical processes
Another essential green chemical process is the enantioselective synthesis of organic compounds under environmentally friendly conditions. The need for such processes arises from the fact that the antipodes of chiral drugs, which make up a large part of the pharmaceutical market,[407] act differently on the body’s receptors, and the presence of one of the enantiomers can lead to serious side effects.[408] Enantiomerically-enriched substances are usually synthesized from racemic or prochiral precursors in the presence of chiral catalysts, which can be natural enzymes,[409][410] metal complexes with chiral ligands[411][412] or metal-free enantiomerically pure organic molecules called organocatalysts.[413]
The most recent member of this triad is asymmetric organocatalysis, a method, the creators of which, B.Liszt and D.McMillan, were awarded the 2021 Nobel Prize in Chemistry. This methodology is complementary to green chemistry[414] and is one of the most dynamically developing areas of modern organic synthesis.[415] With a much simpler structure than the natural enzymes they mimic, organocatalysts can be obtained by convenient synthetic methods,[416] in particular from renewable raw materials.[417] Many catalytic reactions involving them are tolerant to various functional groups present in the reacting molecules and do not contaminate pharmacological products with traces of heavy metals, making them promising tools for medicinal chemistry.[418-420] At the same time, organocatalysts are generally non-toxic and resistant to atmospheric air and moisture, making it possible to carry out many organocatalytic reactions in air and even in water[421-423] and to create new sustainable chemical technologies on this basis.[424][425] In 2019, IUPAC listed organocatalysis as one of the 10 most promising chemical technologies for sustainable development of mankind.[426] The environmental potential of organocatalysis is further enhanced by modern green chemistry methodologies,[427] based on the use of solid-supported organocatalysts,[428][429] continuous flow processes,[430][431] photocatalytic[432] or electrochemical[433] reactions. Tandem organocatalytic reactions are very promising, allowing several process steps to be carried out in a one-pot fashion without the need to isolate and purify intermediate compounds.[434-436] Great hope is placed on the use of green solvents in organocatalysis such as water,[20][437] compressed carbon dioxide,[438] biobased solvents,[439] deep eutectic mixtures,[440] or on performing the reactions under neat conditions.[441-443]
Let us focus on three conceptual directions of green chemistry integrated in modern asymmetric organocatalysis, which include the development of resource-saving cascade and tandem organocatalytic methodologies, carrying out asymmetric continuous flow reactions, and the creation of combined visible-light-induced organophotocatalytic processes. Environmental aspects of asymmetric organocatalysis have already been summarized in one form or another (see, e.g., review[444]). We will only consider reactions and processes that comply several principles and criteria of green chemistry.[9] For catalysts, priority is given to the greener and more sustainable chiral amines[445] and axially chiral phosphoric acids,[446] which are structural platforms for solid-supported catalysts. In assessing solvents, we were guided by the recommendations of the European Consortium of Innovative Medicines Initiative (IMI)-CHEM21,[34] which take into account several important factors such as safe handling, potential harm to human health and potential negative impact on the environment (ozone depletion, acute ecotoxicity, potential for bioaccumulation, volatility, etc.).
3.3.1. Resource-saving cascade and tandem organocatalytic reactions
According to the green chemistry paradigm, the most efficient synthetic approach to a chemical compound of high molecular complexity should involve a minimum number of steps, carried out mainly in a single reaction vessel, without isolation and purification of intermediate compounds, thereby reducing labour costs and waste. Below are some examples of how this paradigm is realized in organocatalyst-mediated asymmetric syntheses.
Zhong and co-workers[447] proposed an original one-pot method for the enantioselective synthesis of tricyclic compounds containing fused tetrahydronaphthalene and isoxazolidine moieties from ortho-(acrylato)nitrostyrene 55, enolizable aldehydes, and N-substituted hydroxylamine (Scheme 53). The domino reaction proceeds efficiently in aqueous medium in the presence of silylated α,α-diphenylprolinol (S)-XIXa to give the products 56 in good yields and with extremely high diastereo- and enantioselectivities. The plausible mechanism of the process involves the enantioselective addition of an aldehyde activated by the catalyst (enamine activation) to the double bond of nitrostyrene 55 attached to the nitrogroup. The resulting adduct Int1 reacts with hydroxylamine to generate nitrone Int2 , which spontaneously cyclizes to the product 56. The addition of benzoic acid appears to facilitate the formation of the key enamine from the catalyst and aldehyde and its subsequent hydrolysis, thereby returning the catalyst to the catalytic cycle.
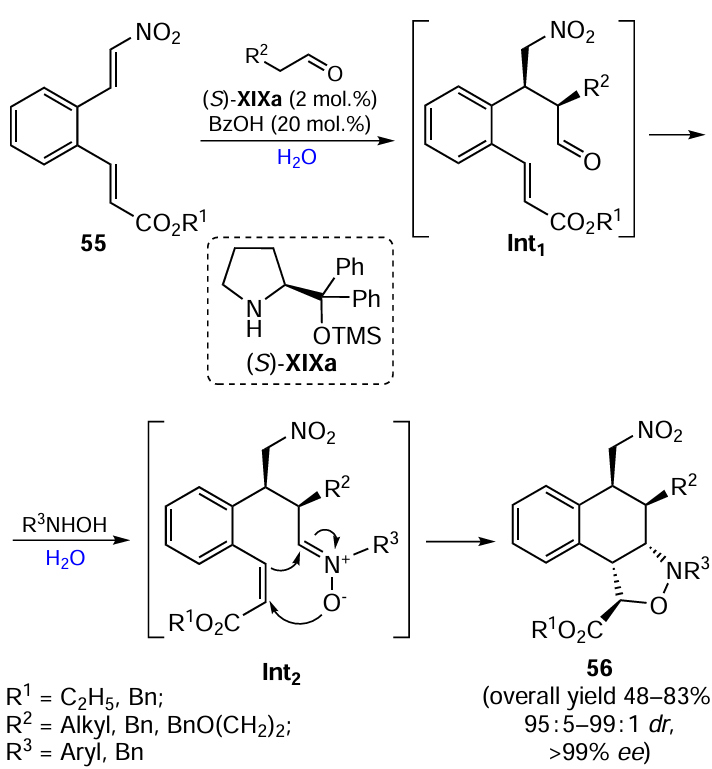
Hayashi and co-workers[448] have developed an enantioselective one-pot method for the synthesis of the bicyclic Corey lactone, a valuable intermediate in the preparation of synthetic prostaglandin hormones, which regulate important physiological processes in living organisms. The method is based on the domino reaction of ethyl 4-oxo-2-pentenoate (57) with silylated enal 58 catalyzed by (R)-XIXa in PriOH and involves two successive Michael reactions (steps 1 and 2) (Scheme 54). Functionalized cyclopentanone Int3, formed in 85% yield as a single isomer (>99% ee), was converted to the desired lactone without isolation via functional group transformation (one-pot 5-step synthesis, 50% overall yield). As a result, waste and labour costs were significantly reduced. The total reaction time for the Corey lactone was only 2.5 h, making the developed approach the most environmentally friendly and effective method for the synthesis of this useful compound. A similar methodology was employed by the same authors[449] for the enantioselective construction of a functionalized cyclopentane motif in the asymmetric syntheses of cis-hydrindane and the neuroprotective agent Clinprost.[450]
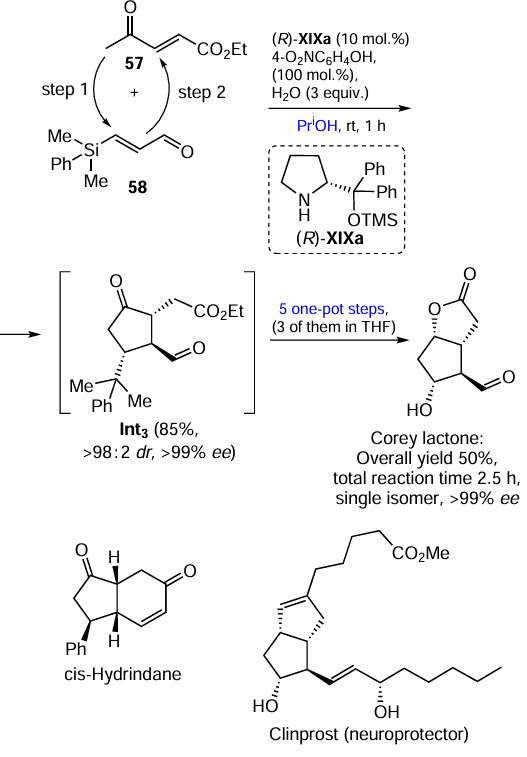
Prolinol catalysts have also been useful in the asymmetric synthesis of polysubstituted derivatives of bicyclo[4.3.0]nonane and bicyclo[4.4.0]decane, motifs found in many natural products, particularly steroids and diterpenes. Hayashi et al.[451-453] proposed efficient methods for the preparation of key bicyclic precursors to natural products via domino reactions of 1,3-diketones 59 containing a 2-positioned nitroethyl group with α,b-enals in green solvents (PriOH or THF). This procedure involves a sequence of asymmetric Michael reaction (step 1) and intramolecular aldol reaction with cyclohexane ring closure (step 2) (Scheme 55). In the presence of (S)-XIXa or (S)-XIXb catalysts, the bicyclic products 60[451][452] and 61,[453]containing five contiguous stereocentres, were formed in high yields as almost single enantiomers. The addition of a small (3 equiv.) amount of water significantly accelerated the domino reaction, apparently due to the faster hydrolysis of iminium ions formed from enal and the catalyst. Furthermore, in line with the pot-economy concept,[454] the authors greatly simplified the conversion of compound 60 to estradiol ether by reducing the number of experimental steps from 12 to 5 and increasing the overall yield of the target product from 6.8% to 15%.
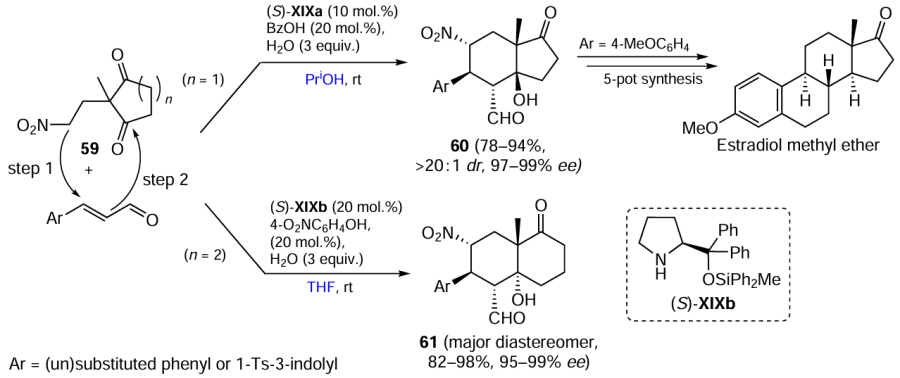
Appayee and co-workers[455] developed a highly selective one-pot cascade synthesis of cyclohexanones 62 containing three contiguous stereogenic carbon atoms from simple precursors: acetone and cinnamaldehydes (Scheme 56). This apparently simple domino reaction involves three stereoselective steps: 1,2-Mannich addition of the enamine Int4 to the iminium cation Int5 , 1,4-Michael addition of the resultant enamine Int6 to the second molecule of the iminium cation Int5 , and closure of the cyclohexane ring in the adduct Int7 via an intramolecular Michael reaction. This process features the use of two chiral aminocatalysts (S)-XIXa and (S)-XX, which are capable of forming both enamines and iminium cations with reactants. By changing the absolute configuration of just one of them, the desired isomer of product 62 can be selectively synthesized. The results obtained are well reproduced when scaling up the reaction to several grams. In addition to atom-economy, the method is attractive because it uses isopropanol, an environmentally benign and safe solvent, as the reaction medium.
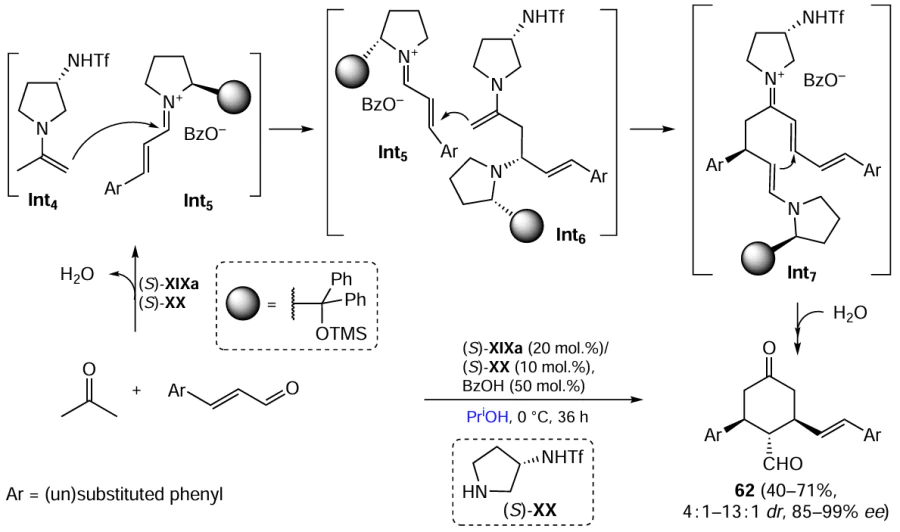
Wen and Du[456] carried out enantioselective synthesis of bispyrocyclic cyclopropanes via the reaction of 4-arylidene-2,3-dioxopyrrolidines 63 with 3-chloroxindoles 64 catalyzed by a squaramide alkaloid XXIa (Scheme 57). The domino process comprises the asymmetric Michael addition followed by intramolecular cyclization involving the catalyst-generated enolate anion and affords chiral products 65 of high molecular complexity with moderate to high yields and with excellent stereoselectivities. Screening of reaction media carried out by the authors showed that the optimal solvent is ethyl acetate, in which the reaction is easily scalable while maintaining yield and enantiomeric purity of the product.
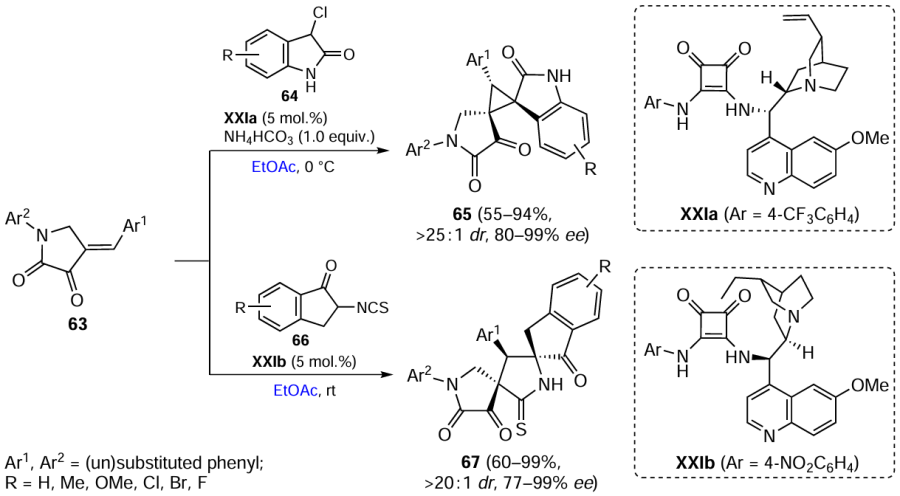
In the follow-up study, a cascade reaction of 4-arylidene-2,3-dioxopyrrolidines 63 with 2-isothiocyanato-1-indanones 66 in the presence of a pseudoenantiomeric quinidine XXIb was carried out.[457] In this case, the asymmetric Michael addition was followed by a diastereoselective cyclization via the isothiocyanate group of compound 66 to give a dispiro[pyrrolidinethione] 67 containing three contiguous stereocentres (see Scheme 57). Again, the highest stereoselectivity was achieved in ethyl acetate. The above cascade processes provide the easy synthesis of representative libraries of polynuclear heterocyclic compounds containing biogenic motifs for subsequent screening for biological activity.
For the asymmetric synthesis of spiroheterocycles, Li and co-workers[458] used another approach based on the ability of functionalized propargylic alcohols to be enantioselectively converted to axially chiral allenes when treated with chiral Brønsted acids. The authors found that 3-alkynyl-3-hydroxyisoindolinones 68 react with 3-substituted-1H-indoles 69 in EtOAc in the presence of BINOL-phosphoric acid (S)-XXIIa to give spiro products 70 in a single step in high yields and with excellent regio- and enantioselectivities (Scheme 58). The catalyst can be recovered by acidifying the reaction mixture and reused in the reaction. The reaction pathway appears to involve the catalyst-induced dehydration of isoindolinone 68 to form an adduct of propargyl-N-acylimine Int8 with the catalyst. The subsequent enantioselective 1,4-addition of indole 69 releases the catalyst to give allene Int9 , which further undergoes cyclization to afford spiro-products 70.
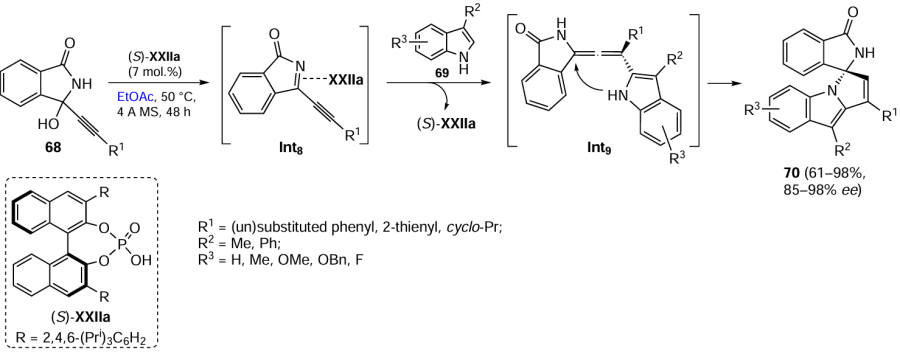
Shi and co-workers[459] have developed an interesting method for the deracemization of substituted 3,4-dihydropyrimidin-2-ones (rac-71) containing a diisopropylphosphonate moiety in position 4 of the heterocycle. The method is based on a tandem reaction involving DDQ-mediated oxidation of rac-71 to tautomeric pyrimidinones Int10 and Int11 and asymmetric hydrogenation of the C=N bond in the pyrimidine moieties of the latter compounds (Scheme 59). The best results were obtained using Hantzsch ester 72a, a biomimetic of natural reducing agents, as the hydrogen source, and a BINOL-phosphoric acid derivative (R)-XXIIa as the organocatalyst. Both steps of the one-pot redox process are carried out in the same green solvent, ethyl acetate, without isolation and purification of intermediate compounds, which greatly simplifies the experimental procedure. As a result, enantiomerically enriched products (R)-71 promising for biological studies can be obtained in a gram-scale while maintaining productivity and stereocontrol.
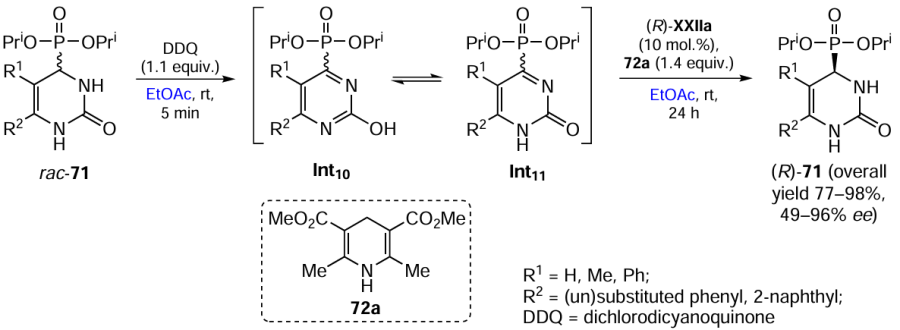
Zlotin and co-workers[460] first performed atom-economical asymmetric domino reactions in supercritical (sc) carbon dioxide, a non-toxic natural compound that is easily removed from the products and does not require disposal. In the presence of a squaramide-containing quinine XXIa, ortho-aminochalcones 73 added to nitroolefins via a cascade of two asymmetric Michael reactions (a and b) to yield functionalized tetrahydroquinolines 74 with very high diastereo- and enantioselectivities (Scheme 60). However, this was only achieved when the reaction was carried out under rather harsh conditions (20 MPa, 75 °C), providing a high medium density (~14 mol CO2/L) necessary for an efficient cycloaddition. The use of more lipophilic and better soluble in sub- and sc-CO2 bifunctional tertiary amines XXIc and XXIIIa with long-chain alkyl groups as catalysts made it possible to reduce the pressure and temperature (7.5 MPa, 35 °C) and make the process more attractive for practical implementation in academic laboratories and industry.[461] This is important because the resultant compounds are precursors to natural alkaloids and some drugs (angustureine, martinelline, etc.).
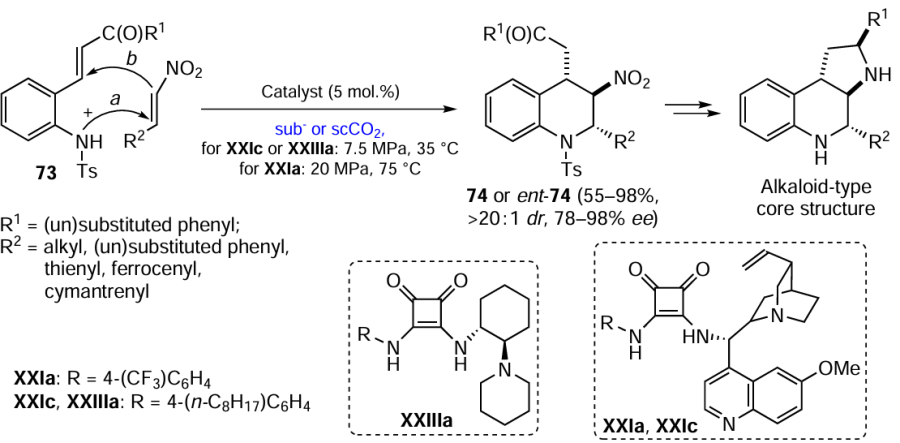
Recently, the same research group[462] found that one of the basic reactions in organic synthesis, the condensation of carbonyl compounds with arylamines, also proceeds smoothly in scCO2 , and the resulting imines can be further processed in the same reactor without prior isolation. In particular, the reaction of 4-methoxyaniline with α-ketoester 75 in scCO2 in the presence of a reducing agent (dihydrobenzothiazole 76a) and chiral BINOL-phosphoric acid (R)-XXIIa affords enantioselectively the (R)-enantiomer of the N-protected phenylglycinate 77 in a single step in 97% yield and with 96% enantiomeric purity (Scheme 61).
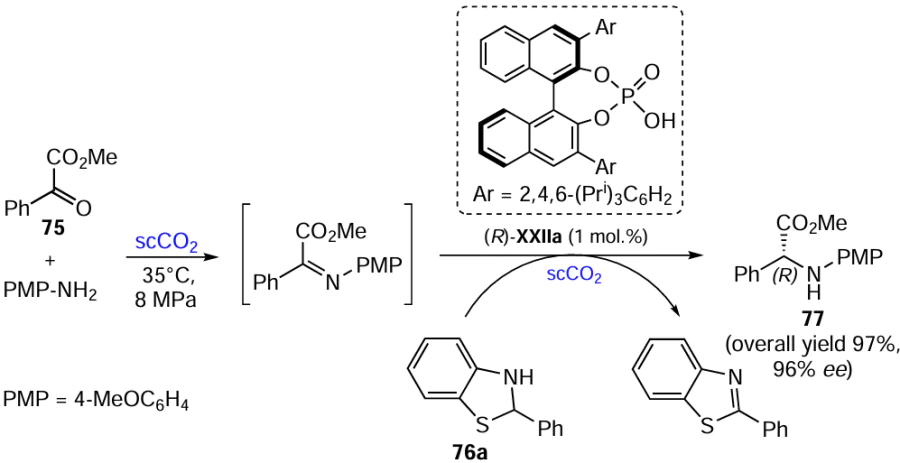
Šebesta and co-workers[463] performed enantioselective one-pot synthesis of hybrid molecules 78 comprising 2-oxoindoline and pyrazolone motifs. The tandem process involved the asymmetric Mannich addition of pyrazolones 79 to N-Boc-isatinimines 80 and diastereoselective fluorination of the resulting in situ adducts Int12 with N-fluoro-benzenesulphonimide (NFSI) (Scheme 62). In this case, the high enantioselectivity of the reaction was provided by using an alkaloid-squaramide catalyst XXId. The required catalyst loading was as low as 1 mol.%. The characteristic feature of the method is that both steps are carried out by mechanical grinding of a mixture of solid reactants wetted with a minimal amount of organic solvent (50 mL) in a ball mill.
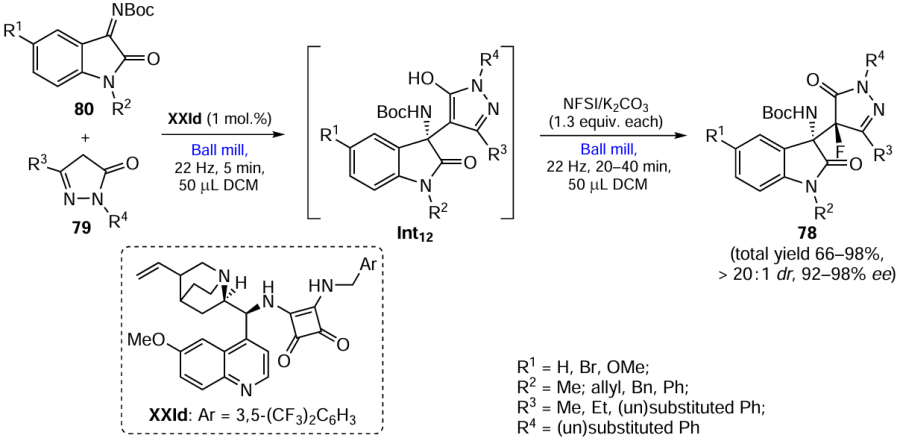
3.3.2. Asymmetric organocatalysis in a green solvent flow
Continuous-flow chemical reactions have a number of advantages over the corresponding batch processes. These include the process intensification[464] and the ability to combine multiple reactions/stages into a single process. Moreover, the accuracy of reagent dosing and control over conditions is improved, making the process safer and ensuring good reproducibility of results.[465] The use of continuous-flow processes fully comply with the principles of green chemistry,[466-469] as they significantly reduces resource, energy and labour costs, avoids isolation and purification of intermediate compounds and consequently reduces waste. These benefits are fully realized in flow systems where the working medium is green solvents that are safe for personnel and the environment.[470][471] Such systems are very promising for the selective preparation of practically relevant high-purity compounds, including drugs and their precursors.[472] Let us consider some recent examples of their application in asymmetric organocatalysis using homogeneous and polymer-supported catalysts.
Meng and co-workers[473] carried out enantioselective oxidation of β-dicarbonyl compounds 81 with cumene hydroperoxide (CHP) in a two-phase toluene/1% aqueous Cs2CO3 system in a cascade of flow microreactors using cinchonine bromide XXIVa as a chiral phase transfer catalyst (PTC) (Scheme 63). The system components were simultaneously fed into the microreactors by two pumps at a flow rate of 1.5 ml/min: one pump fed a solution of substrate, CHP and catalyst in toluene and the other pump fed an aqueous solution of Cs2CO3 . The oxidation products 82 were formed in high yields and enantiomeric enrichment of 78 – 84%. At the same time, the required residence time of the reactants in the flow microreactors (2 h) was much shorter than that for the corresponding batch reactions (24 h).
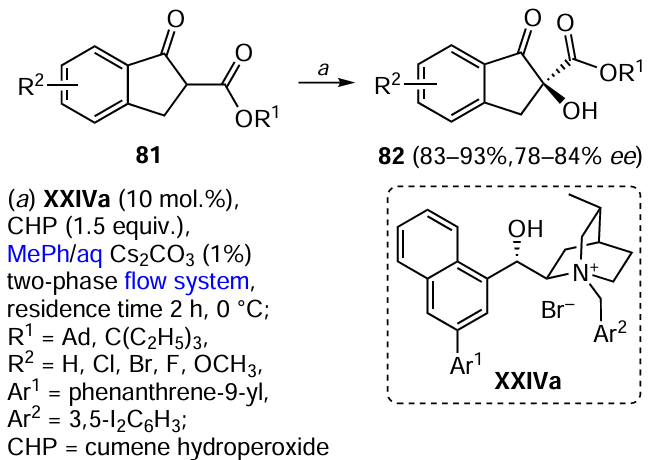
Sasai and co-workers[474] carried out an atom-economic domino reaction involving enantioselective Rauhut – Currier cycle closure in dienones 83 and [3+2] annulation of bicyclic intermediates Int13 with allene 84 (Scheme 64). The reactions were performed using chiral phosphinamide XXV in a flow system with parameters carefully optimized using artificial intelligence. Under optimal conditions (flow rate 1.7 ml min–1, temperature 80 °C), the yield of the spirocyclic product 85 (R1 = Me, R2 = Ts) was 76%, significantly higher than in a batch process (20%). Under these conditions, various dienones 83 were converted into the corresponding products 85 as single diastereomers with high enantiomeric purity in less than one minute.
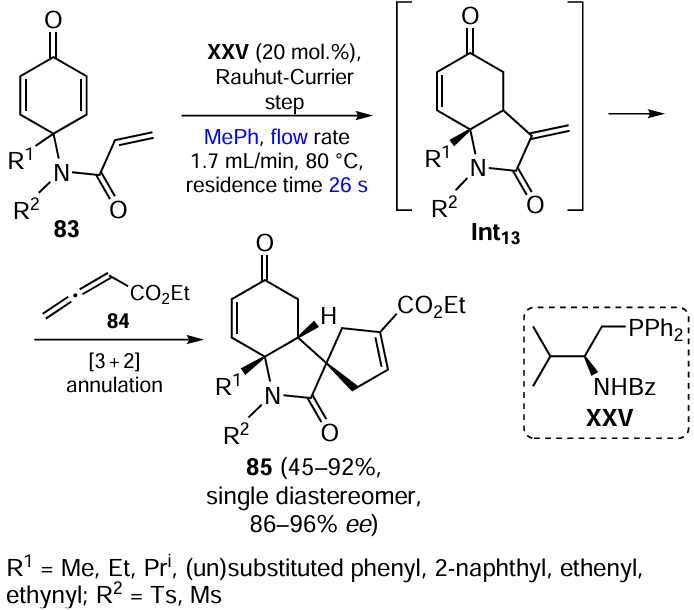
Recently, Gröger and co-workers[475] developed a tandem cascade process comprising the organocatalytic asymmetric aldol reaction and the biocatalytic reduction of the resulting aldol Int14 to diol 86 containing two stereogenic centres. The chemoenzymatic process was carried out in a continuous flow of a mixture of isopropanol and phosphate buffer (Scheme 65). The first-step catalysts were enantiomers of diphenylleucinol-substituted prolinamide XXVI,[476] and the second-step catalysts were alcohol dehydrogenases ADH030 and ADH270, providing stereoselective formation of a second stereocentre with S or R configuration, respectively. Using different combinations of organo- and biocatalyst enantiomers, the authors were able to synthesize all four possible isomers of diol 86 in optically pure form in 33 – 76% yield and with moderate to high diastereoselectivities. Importantly, the aldol Int14 did not require isolation and purification, which greatly simplified the experimental procedure. The developed process can be used as a basis for the creation of new green technologies for the enantioselective synthesis of pharmaceuticals and other useful organic compounds.
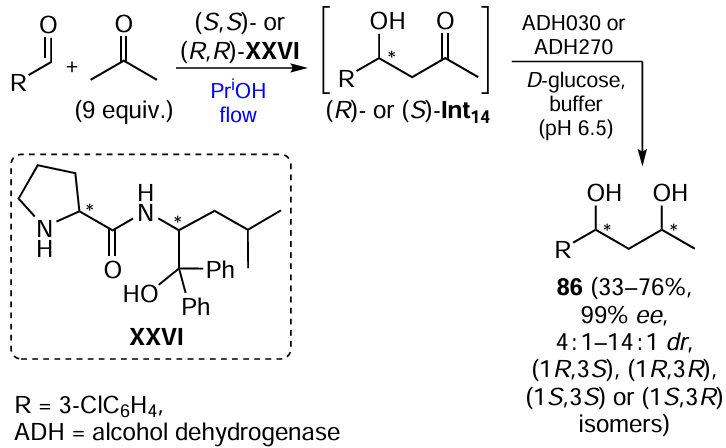
A common drawback of the above flow processes using low-molecular-weight organocatalysts is the difficulty of their recovery due to ‘washing out’ from the continuous flow unit along with the products. Nakashima and Yamamoto[8] proposed an approach to solve this problem by using the simple and accessible (S)-5-(2-pyrrolidinyl)-1H-tetrazole XXVII as a catalyst, which is poorly soluble in many organic solvents. This approach was successfully tested in the asymmetric aldol and Mannich reactions (Scheme 66). When a solution of the reactants in EtOAc or MeCN was passed at a constant rate through a column packed with catalyst XXVII, the corresponding products 87 and 88 were formed in good yields and with high stereo- and enantioselectivities. At the same time, the heterogeneous catalyst was practically insoluble in organic solution and could be used for a long time. In terms of mass efficiency (the number of grams of catalyst required to produce 1 mole of product), the catalyst XXVII is superior to many known catalytic systems.
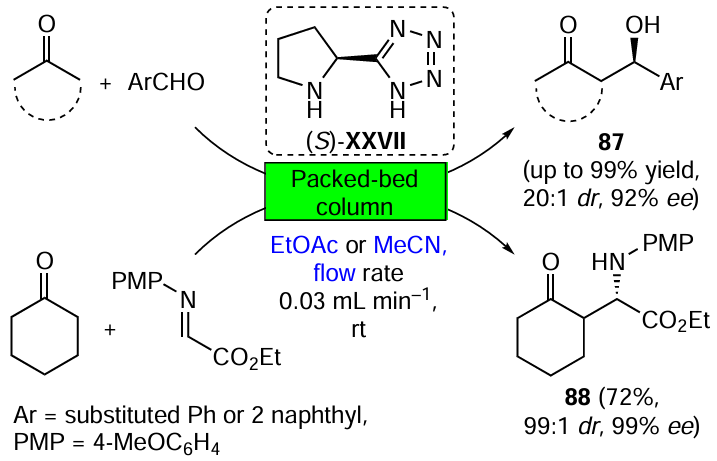
Another promising approach to immobilizing chiral organocatalysts intended for usage in flow systems is based on the creation of hybrid molecules in which catalytically active chiral units are grafted to polymers.[431] For example, Massi and co-workers[477] obtained a (S)-5-(2-pyrrolidinyl)-1H-tetrazole-containing polymer XXVII via radical copolymerization of its styryl-substituted analogue XXVII' with styrene and divinylbenzene (DVB). The resulting monolithic copolymer (S)-XXVII/PS, packed into a stainless steel column, continuously catalyzed asymmetric aldol reactions of ketones with aromatic aldehydes in a flow of aqueous ethanol for five days (Scheme 67). Enantiomerically-enriched anti-aldoles 89 were predominantly formed and the productivity of the plant was twice that of the corresponding batch reactions.
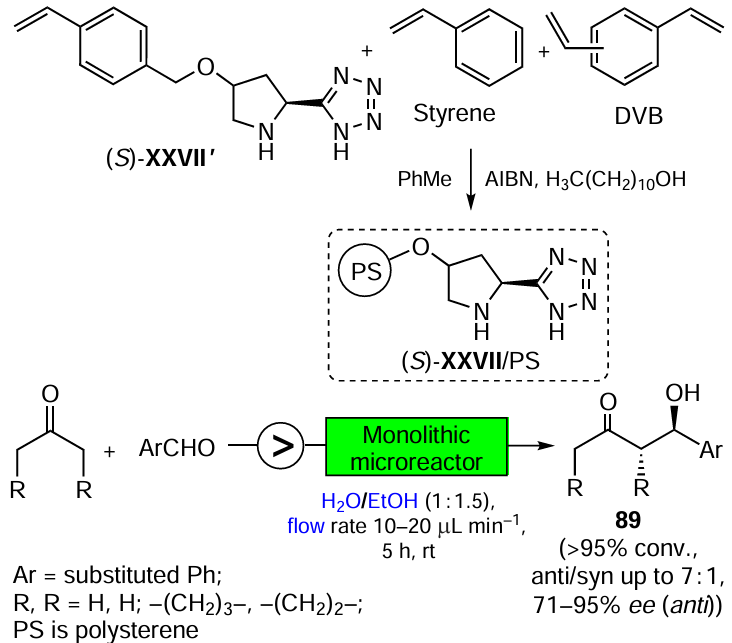
Kobayashi and co-workers[478] carried out an asymmetric aldol reaction of trifluoroacetophenone derivatives with methyl ketones under continuous flow conditions. The mixture of reactants (methyl ketone was taken up in excess) was passed through a column packed with a mixture of celite with polystyrene-supported prolinamide catalyst (S,S)-XXVI/PS (Scheme 68). A small amount of water (1 equiv.) was added to the flow system to accelerate the hydrolysis of the iminium intermediates and return the catalyst to the catalytic cycle. Under the above conditions, the reactants were completely converted to fluoromethylated carbinols 90 of high enantiomeric purity. The total catalyst operation time was 195 h. In particular, the developed continuous-flow process was used to prepare a fluorinated chiral analogue 91 of Fenpentadiol, the well-known tranquilizer and antidepressant.
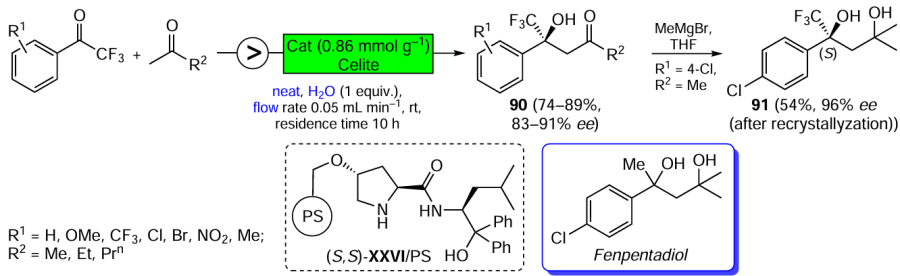
Smith and co-workers[479] performed kinetic resolution (KR) of racemic acyclic alcohols rac-92 containing a quaternary carbon atom by their enantioselective acylation with iso-butyric anhydride as the continuous flow process (Scheme 69). The catalyst was a cyclic isothiourea derivative XXVIII/PS grafted to Merrifield rubber through a spacer group. When a solution of reactants in toluene was passed through a column packed with heterogeneous catalyst (0.05 ml min–1), the S enantiomer of the alcohol was predominantly acylated to form the enantiomerically enriched ester (S)-93, while the main portion of (R)-92 remained unchanged. The resolution selectivity depended on the substituents R1 and R2 in the α-position to the hydroxyl group and ranged from 21 – 80%. The above method can be used in selective technologies for the preparation of chiral alcohols for pharmacological purposes.
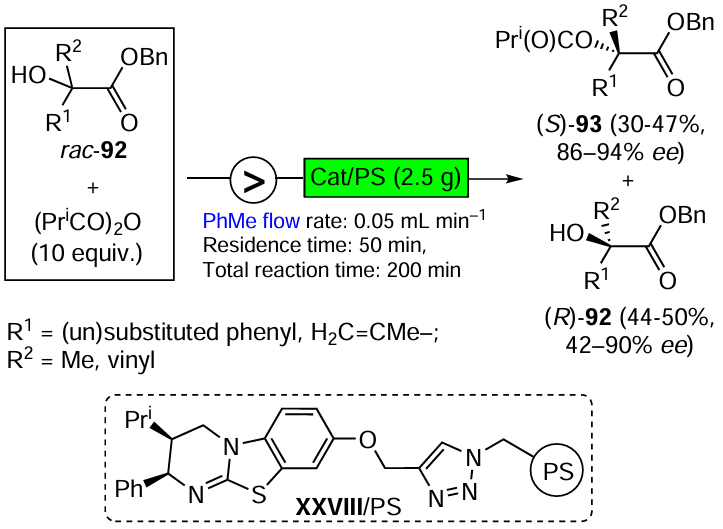
In some cases, silicon-containing polymers can be used as supports for immobilizing organocatalysts for continuous-flow enantioselective chemical reactions. For example, Massi and co-workers[480] compared the catalytic properties of chiral superbasic cyclopropenimines XXIX/Si and XXIX/PS immobilized onto silica and polystyrene, respectively. Asymmetric nucleophilic addition reactions of glycine imine derivatives 94 with acrylic acid esters to afford adducts 95 (Scheme 70) were used as a test system. The catalytic reactions were carried out in ethyl acetate in batch and continuous modes. It was found that the catalysts XXIX/Si and XXIX/PS were almost identical in terms of stereoinduction and efficiency in the batch process. However, in the solvent flow, the catalyst immobilized onto the silicon support was more stable than its polysterene counterpart, maintaining the conversion (90 – 95%) and enantioselectivity (70 – 98% ee) values over 28 h. The activity of the catalyst then decreased, but after reactivation (washing with LiOH solution in anhydrous MeOH) it continued to work efficiently for another 28 h.
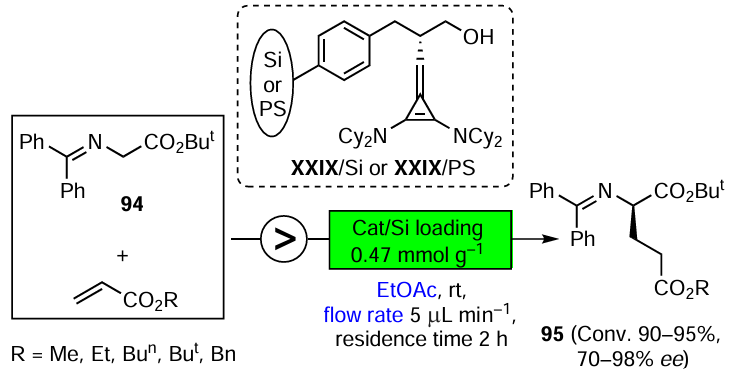
Hayashi et al.[481] carried out in a green solvent flow an asymmetric cascade reaction of a cinnamaldehyde derivative with 2-(b-nitroethyl)-cyclopentane-1,3-dione (59), a key step in the batch synthesis of estradiol esters previously described by the authors (see Refs 451,452 and Scheme 55). The catalyst was an amphiphilic α,α-diphenylprolinol ether XIXc/PEG/PS containing hydrophilic (polyethylene glycol) and hydrophobic (polystyrene) moieties (Scheme 71). A solution of reactants and benzoic acid in a specially selected solvent with an optimal combination of lipophilic and hydrophilic properties was passed through a steel reactor packed with this catalyst at a constant rate. Using the PriOH/THF/H2O solvent system, the polymer catalyst worked continuously for 60 h, maintaining the enantioselectivity of the process at 91% ee. The overall yield of product 60 was 63% with a catalyst turnover number (TON) = 81.
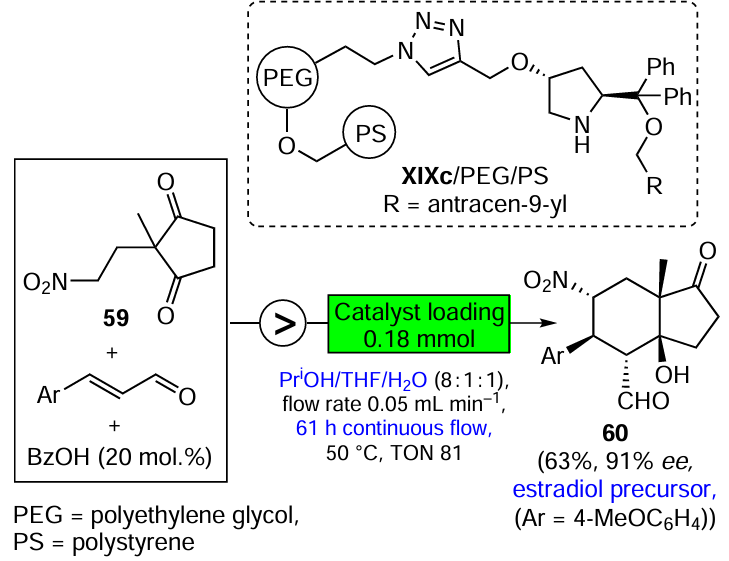
Almost at the same time, Pericàs and co-workers[482] used the catalyst XIXd/PS immobilized onto polystyrene in the continuous-flow asymmetric addition of dimethyl malonate to α,b-unsaturated aldehydes (Scheme 72). The best results were achieved in the system wherein the excess of dimethyl malonate, easily removable from the product by evaporation at reduced pressure, was used as a solvent. A series of adducts 96 with enantiomeric purity of up to 98% ee were obtained using the same portion of catalyst. At the end of the process (2.5 – 4.5 h), the catalyst was reactivated by successive washing with acetic acid and ethyl acetate and used for the reaction with another substrate. The resulting compounds included intermediates for the synthesis of the antidepressant drugs Paroxetine and Femoxetine and a peptidomimetic inhibitor. They were also diastereoselectively converted to indoloquinolizidine derivatives and δ-lactones using the Pictet – Splengler and reduction reactions, also carried out in continuous flow.
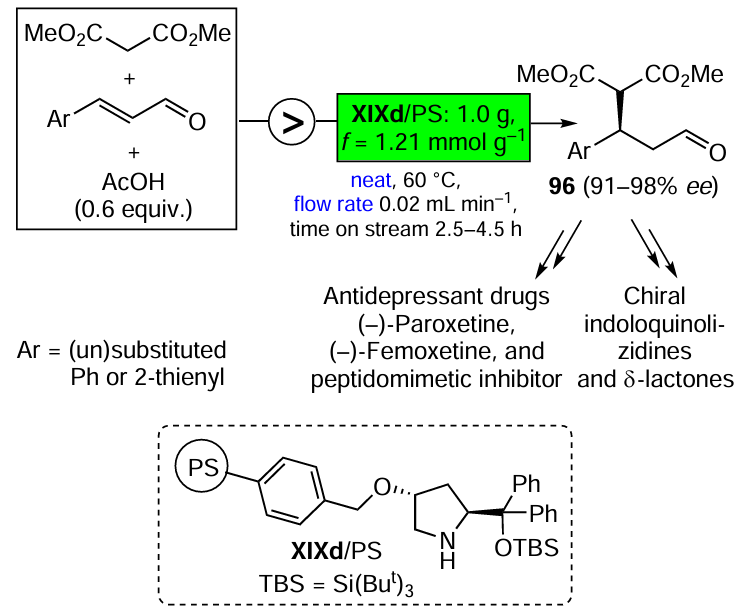
Ötvös and co-workers[483] reported an innovative method for the continuous asymmetric synthesis of rolipram, a selective cyclic phosphodiesterases type IV inhibitor. The three-step synthesis involved the enantioselective addition of nitromethane to a cinnamaldehyde derivative, oxidative esterification of the enantiomerically enriched γ-nitroaldehyde 97, and reduction of the nitro group in the γ-nitroether 98 to an amino group and concomitant lactamization (Scheme 73). To avoid side transformations of the labile aldehyde 97, the authors combined the first two steps into a single continuous ‘telescopic’ process. A solution of nitroaldehyde 97 in an excess of nitromethane, formed by passing the reactant mixture through a XIXe PS catalyst column, was mixed at the column outlet with solutions of sulfuric acid and hydrogen peroxide in methanol. The resulting mixture was subjected to oxidative esterification to γ-nitroester 98 when treated with persulfuric acid generated in situ in a coil heated to 100 °C. The reductive lactamization of ester 98 under the action of the HSiCl3 MeN(Pri)2 system was also carried out in continuous flow mode, in this case in acetonitrile. As a result, (S)-rolipram was obtained in 83% yield and with 94% enantiomeric purity.
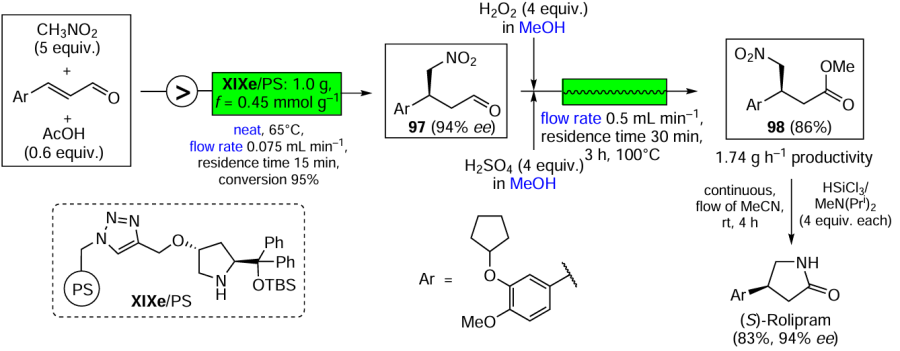
Organocatalytic flow reaction methodology has also been successfully applied to the cost-competitive asymmetric synthesis of (S)-3-aminomethyl-5-methylhexanoic acid, used for the treatment of seizures, neuropathic pain and epilepsy (trade names Lyrica, Pregabalin).[484]
3.3.3. Metal-free asymmetric photoorganocatalysis
A significant role in the development of asymmetric organocatalysis in recent years has been played by the use of new methods of reagent activation, in particular light activation.[485] Visible light, sourced from the sun, is the most environmentally friendly, safe and virtually inexhaustible source of energy for sustainable development.[486-488] However, the use of this energy for the enantioselective synthesis of organic compounds is complicated by the high reactivity of the free radicals generated via the homolytic cleavage of the covalent bond, which hampers the stereochemical control of the reaction.[486-488] The problem of selectivity has been solved by combining chiral organocatalysts with achiral photocatalysts, which can be metal (iridium, ruthenium, rhodium, etc.) complexes[134][489] or organic chromophores — derivatives of aromatic and heteroaromatic compounds, including polynuclear ones.[490] The methodology of organophotoredox catalysis is discussed in detail in the review.[491] We will give some conceptual examples of such metal-free green reactions.
Jiang and co-workers[492] described a visible light-driven enantioselective cross-coupling reaction between N-arylglycines 99 and α-branched 2-vinylpyridines or 2-vinylquinolines 100, followed by decarboxylation of the amino acid. The successful implementation of the process was ensured by the combined use of a photochrome, dicyanopyrazine (DPZ), which removes an electron from the amino acid upon light excitation, and an organocatalyst, chiral phosphoric acid XXXa, which is responsible for the enantioselective formation of the product 101 of the photochemical reaction (Scheme 74). The reaction is carried out under mild conditions in tetrahydrofuran. The practical value of the method was demonstrated by the synthesis of the antihistamine pheniramine (R)-enantiomer.
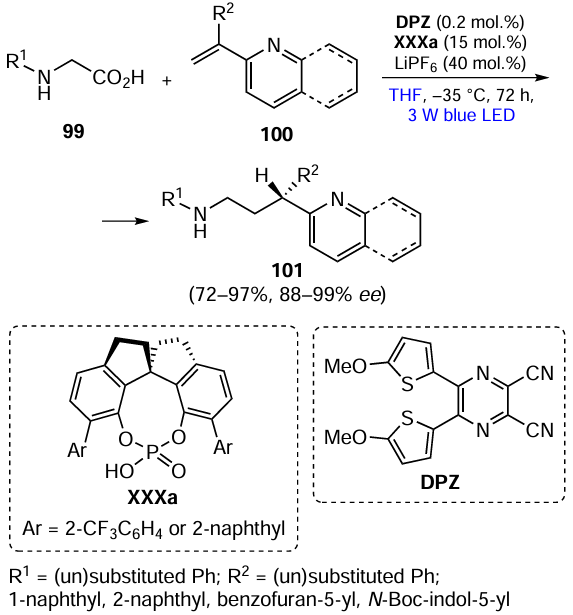
A similar organophotocatalytic system was then applied to the asymmetric [3 + 2] cycloaddition reaction of N-cyclopropylamines to N-sulfonyl-3-methylene-isoindolin-1-ones 102.[493] Under optimal conditions, the spirocyclic cycloaddition products 103 were produced in near-quantitative yields as single diastereomers with 90 – 95% ee (Scheme 75). The XXXb/DPZ catalyst combination was also useful in the reaction of N-cyclopropylamines with N-protected vinylamines 104 to give 1,2-diaminocyclopentanes 105 of high enantiomeric purity, although the diastereoselectivity of these reactions was lower for some substrates. These methods are of practical value since five-membered cycles, including those contained in spirocyclic units, are key structural motifs in many biologically active compounds.
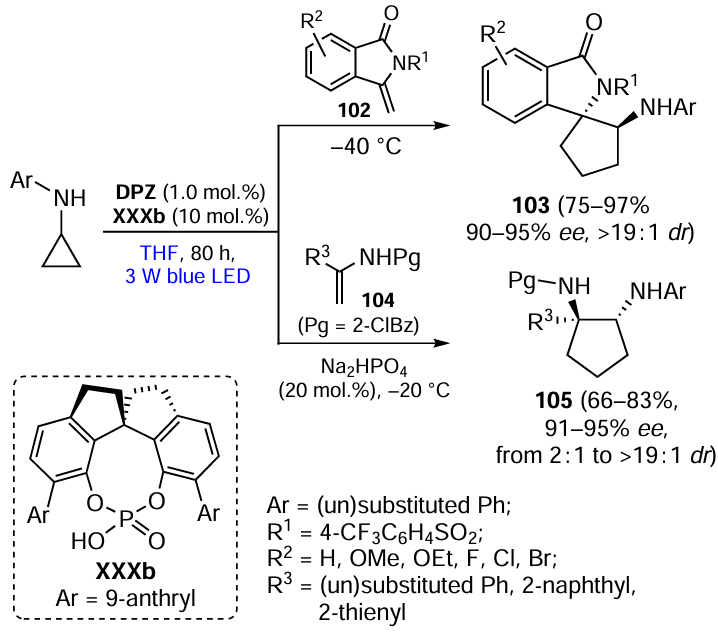
The reductive cross-coupling reactions of α-branched vinyl ketones 106 with 2-vinylazaarenes 107 (Scheme 76), where Hantzsch ethers 72b,c served as reducing agents, proved to be an excellent application of the CPA-DPZ catalytic system.[494] The above process is interesting in that two stereogenic centres at positions 1 and 4 of the linear carbon chain are formed in the products under LED irradiation as a result of enantioselective reduction/protonation cascade reactions. This gave rise to a series of enantiomerically enriched derivatives of azaheterocycles 108 containing pyridine, benzimidazole or benzothiazole units obtained in 35 – 99% yields with up to 99% ee. The efficiency of the proposed methodology of metal-free organophotoredox catalysis was also demonstrated by the authors in visible light-induced deuteration reactions of α-(chloroalkyl)azaarenes with D2O.[495]
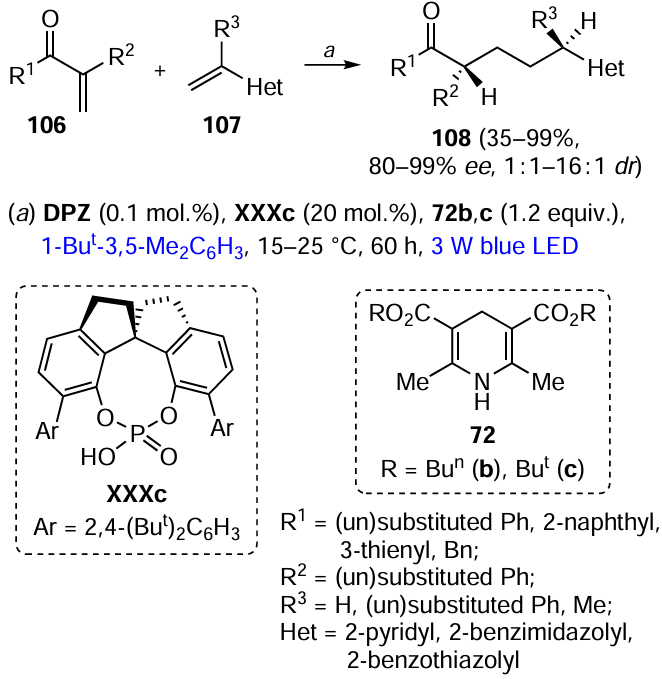
Other types of chiral organocatalysts, organic photochromes and reducing agents can also be used in visible light-induced reductive cross-coupling reactions. Recently, Jiang and co-workers[496] applied a cooperative system including the bifunctional organocatalyst XXXI based on quinine modified with a thiourea group and the polyfunctional photochrome 3DPAFIPN to the reaction of chalcones 109 with 4-cyanopyridine (Scheme 77). This reaction, accompanied by the cleavage of the cyano group from the heterocyclic substrate, produced adducts 110 in moderate yields but with high enantioselectivities (up to 92% ee). The reducing agent in this case was 2-aryldihydrobenzothiazole (76b), which was oxidized in the reaction to 2-arylbenzothiazole (76b').
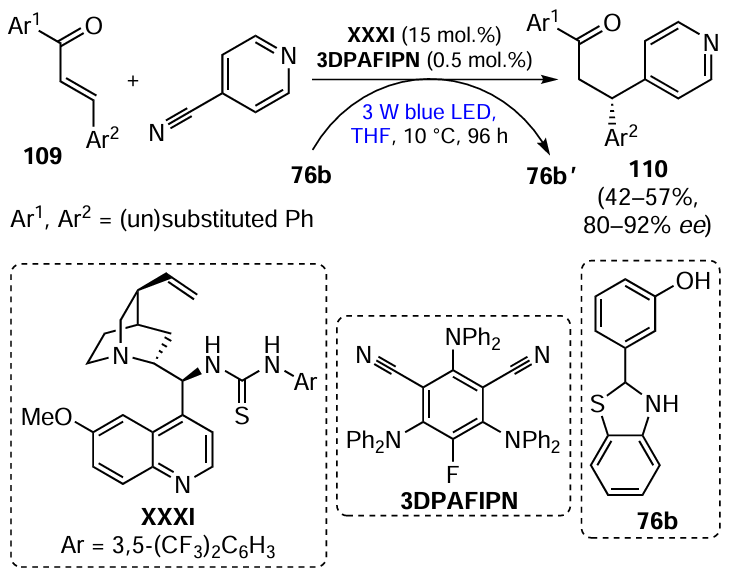
Yang and co-workers[497] described the visible-light-induced asymmetric cascade transformation of b-(ortho-aminoaryl)enones 111 to tetrahydroquinoline derivatives 112, which are widely used as biologically active substances (Scheme 78). The organocatalyst in choice was chiral BINOL-phosphoric acid XXIIb. However, in this case there was no photocatalyst in the reaction system and the light directly induced E/Z isomerization of the double bond in the starting chalcones 111, facilitating their cyclization involving amino and carbonyl moieties. The resulting quinoline derivatives Int15 were further reduced spontaneously to enantiomerically enriched (up to 99% ee) tetrahydroquinolines 112 in the presence of the XXIIb/72d system in up to 98% overall yield. The proposed method can also be used for the stereoselective preparation of cis-isomers of 2,3-disubstituted tetrahydroquinolines 112 (R3 = Me).
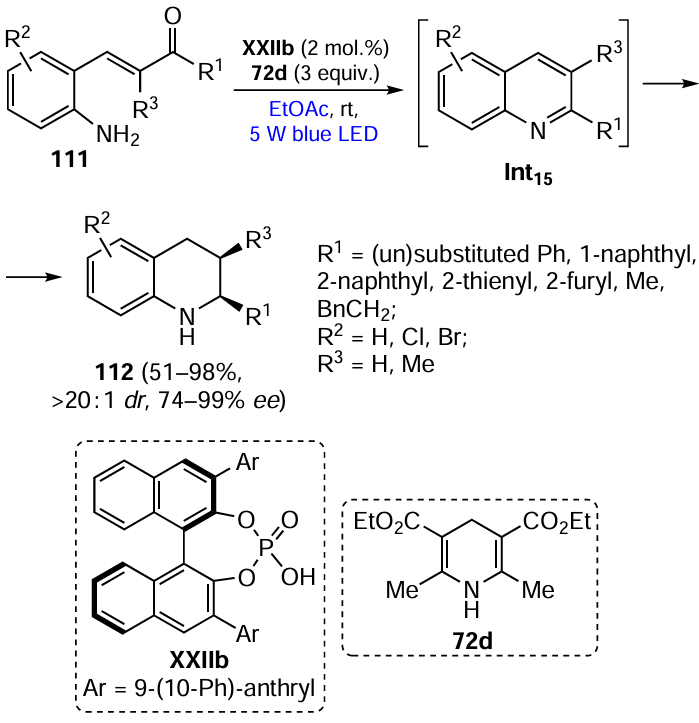
Bach and co-workers[498] proposed the use of thioxanthone XXXII, a chiral compound capable of acting as an organocatalyst by forming stereodifferentiating hydrogen bonds with the reactant and simultaneously being excited by visible light, transferring its energy to the reactant, in metal-free organophotocatalysis. This compound was used in particular for deracemization of chiral piperidin-2-one-substituted allenes 113. When irradiated with visible light at a wavelength of 420 nm in MeCN in the presence of sensitizer XXXII, racemate rac-113 was almost completely converted to the enantiomer (R)-113 (Scheme 79), while this did not occur in the absence of the catalyst. This phenomenon was explained by theoretical calculations (DFT), which showed that the distance between the carbonyl carbon atom of thioxanthone and the terminal endocyclic carbon atom of the allene unit of the substrate in complex XXXII/(S)-113 is half as much as that in complex XXXII/(R)-113. As a result, the triplet energy transfer from the light-excited catalyst to the substrate in the first complex and consequently its racemization proceeds faster than in the second complex, leading to the accumulation of the enantiomer (R)-113 in the reaction mixture. The photoinduced asymmetric conversion of achiral 3-allyl-substituted quinolones to enantiomerically enriched 3-cyclopropylquinolones in the presence of photochrome XXXII follows the similar pathway.[499]
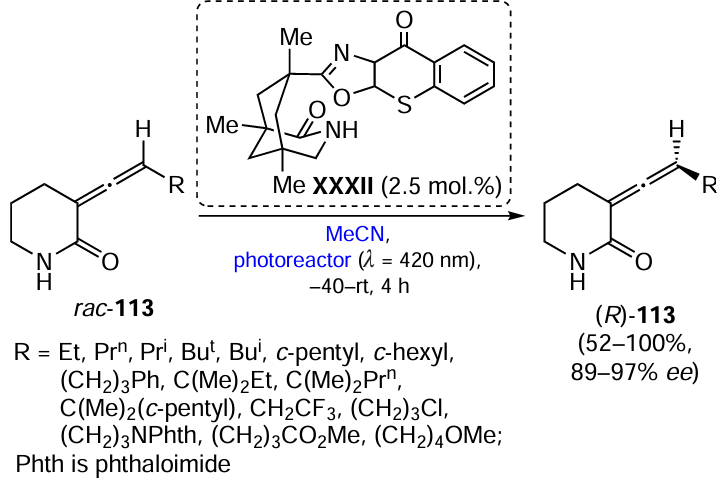
Phipps and co-workers[500] carried out the asymmetric oxidative cross-coupling of aza-heterocycles 114 (quinoline, pyridine and pyrimidine derivatives) with N-acetylated primary amines 115 (a Minisci-type reaction) under the action of light in the presence of chiral BINOL-phosphoric acids XXII. The key role in this reaction belongs to diacetyl (116), a simple, cheap and accessible compound that simultaneously acts as a photochrome, oxidant and hydrogen atom acceptor (Scheme 80). The ability of diacetyl to absorb light in the visible region of the spectrum (380 – 460 nm) eliminates the need for more complex photocatalysts. First, the light-excited molecule 116 initiates hydrogen atom transfer from N-acetylamine 115, forming the α-amino radical 115', which is necessary for the reaction to proceed, and the ketyl radical. The ketyl radical is also important: it oxidizes the primary radical cross-coupling products to α-acetylamino derivatives of aza-heterocycles 117 to give acetoin (118). A high degree of stereocontrol in the above organophotocatalytic radical process is provided by chiral phosphoric acid XXII, which forms hydrogen bonds with reactant 114 and α-aminoradical 115' in the transition state of the catalytic reaction.
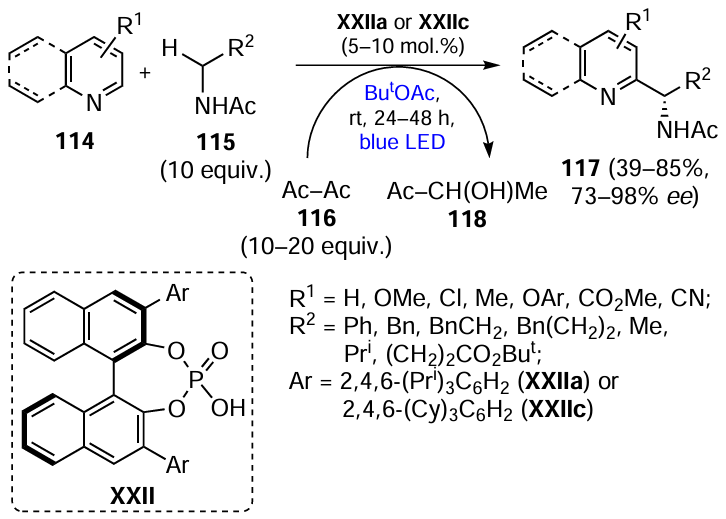
In addition to chiral Brønsted acids, chiral amines capable of forming chemically active enamines and iminium ions with carbonyl compounds can be used as organocatalysts in photocatalytic processes. Melchiorre and co-workers[501] used silylated diarylprolinol XXXIIIa in conjunction with acridinium salt XXXIV to carry out the enantioselective β-functionalization of aliphatic α,β-enals, which is not easily achievable with other photocatalytic systems. The radical sources were organosilicon compounds 119a containing a trimethylsilyl group or an aromatic ring in the α-position to the heteroatom (N or S) (Scheme 81). Light-excited photocatalyst XXXIV abstracts an electron from them, leading to the cleavage of substrate 119a into a SiR3+ cation and a C-centered radical. The stereoselective attack of this radical on the iminium cation generated upon condensation of enal with the chiral organocatalyst XXXIIIa in the activated complex Int16 and subsequent hydrolysis of the resulting adduct afford products 120, turning over the organocatalyst and the photochrome XXXIV. The best stereochemical outcome was achieved in the ‘green’ solvent acetonitrile, where branched aldehydes containing b-positioned alkyl, (hetero)aryl and protected amino groups were formed in 35 – 93% yield with up to 98% ee.
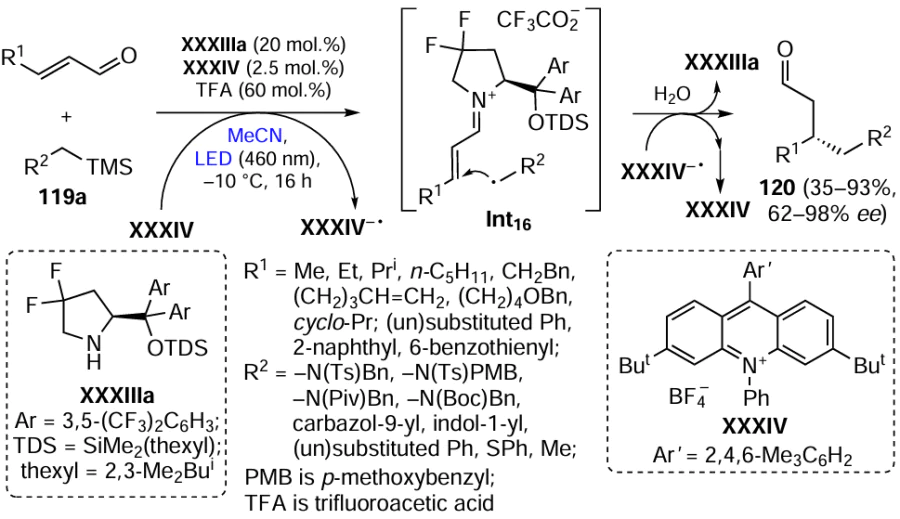
The same catalytic system has proven useful for the enantioselective preparation of 1,7-dicarbonyl compounds with a 3-positioned stereocentre.[502] These compounds occur in nature and are used as substrates for the preparation of biologically active substances. The photocatalytic process is based on the oxidative ring-opening in cyclobutanols 121 when irradiated with visible light in the presence of photochrome XXXIV. The resulting γ-keto-radicals add enantioselectively to iminium cations generated from enals and organocatalyst XXXIIIa to give 1,7-ketoaldehydes 122 in 30 – 82% yields with up to 95% ee (Scheme 82).
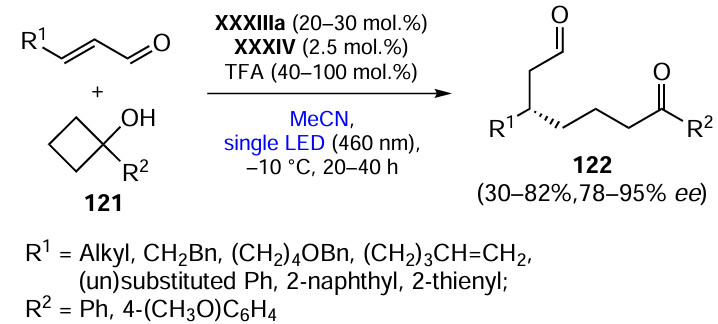
In contrast to β-alkylenals, cinnamaldehyde derivatives, which have a more extended π-electron system, undergo enantioselective photoinduced reactions catalyzed by chiral prolinol silyl ethers, even in the absence of external photochromes.[503] This is due to the ability of β-aryliminium ions to switch to an excited state when exposed to light, similar to what occurs in the organs of vision of higher organisms in nature.
Melchiorre and co-workers[504] exploited this ability to perform an asymmetric [3 + 2] annulation of cinnamaldehydes with cyclopropanols 123. The radical photochemical process involves iminium ions in the ground and excited states and allows the one-pot conversion of racemic and prochiral substrates into polysubstituted cyclopentanols 124 of high molecular complexity (Scheme 83). Apparently, the iminium ion derived from aminocatalyst XXXIIIa and enal is light-excited and abstracts an electron from cyclopropanol 123. This result in that the iminium cation is converted to the radical Int17 , and the cyclopropanol, through the opening of the cyclopropane cycle, delivers the cation radical Int18 . Subsequent recombination of the Int17 and Int18 radicals and intramolecular aldol cyclization furnishes functionalized cyclopentanol 124 and releases the aminocatalyst. Efficient control over the geometry of the three stereocentres by the catalyst XXXIIIa in the course of the organophotocatalytic process ensures high diastereo- and enantiomeric purity of the products 124.
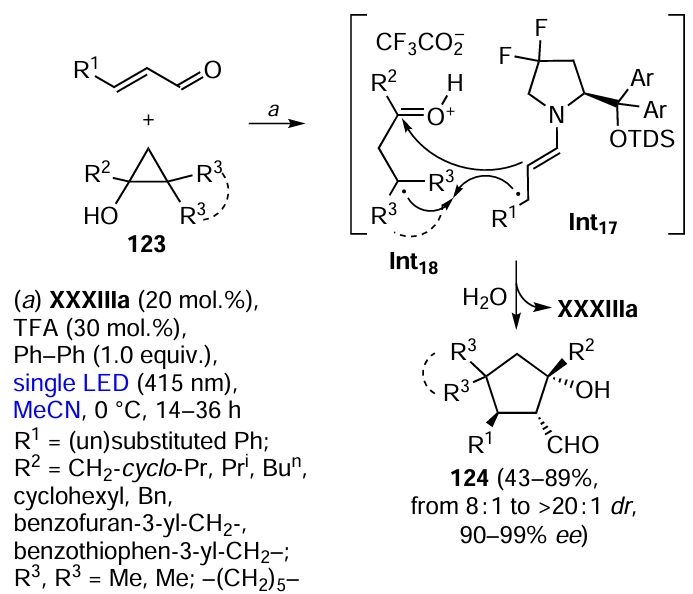
Another example is the visible light-driven enantioselective b-alkylation of cinnamaldehydes, in which 4-alkyl-substituted Hantzsch esters 72e serve as precursors to alkyl radicals.[505] In this case, diarylprolinol XXXIIIb, containing bulky perfluoroisopropyl groups in the aromatic rings, worked as aminocatalyst and the electron was transferred from the diester 72e to the photoexcited iminium cation (Scheme 84). As a result, a representative series of aldehydes 125 bearing diverse linear and α-branched substituents were obtained with enantioselectivities of up to 94% ee. In the case of α-branched R2 substituents, the diastereoselectivity of the reactions was generally not high, except for adduct of cinnamaldehyde with galactose, where the diastereomeric ratio was 9 : 1.
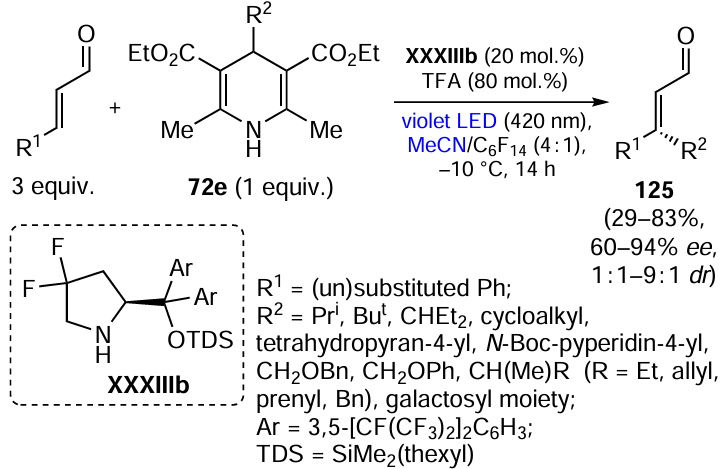
The same research group[506] proposed an original method to generate RCH2 · radicals from silylated methylamines and employ them in asymmetric addition to cyclic α,β-enones mediated by a chiral carbazole-based primary amine XXXV used as an organic photocatalyst. This catalyst was found to be able to switch to an excited state when irradiated with visible light and transfer an electron from the carbazole moiety to the double bond of the iminium cation derived from the catalyst and the cyclic α,β-enone 126 via the charge transfer π – π interaction. This transfer generates a radical at the β-position of the enone (Scheme 85). Furthermore, in the activated complex Int19 , the carbazole moiety promotes the oxidation of silane 119b to the corresponding cation radical, which is the source of the RCH2 radical. The recombination of the above radicals affords adducts 127 containing a quaternary carbon atom in β-position to the carbonyl group in good yields with up to 95% ee.
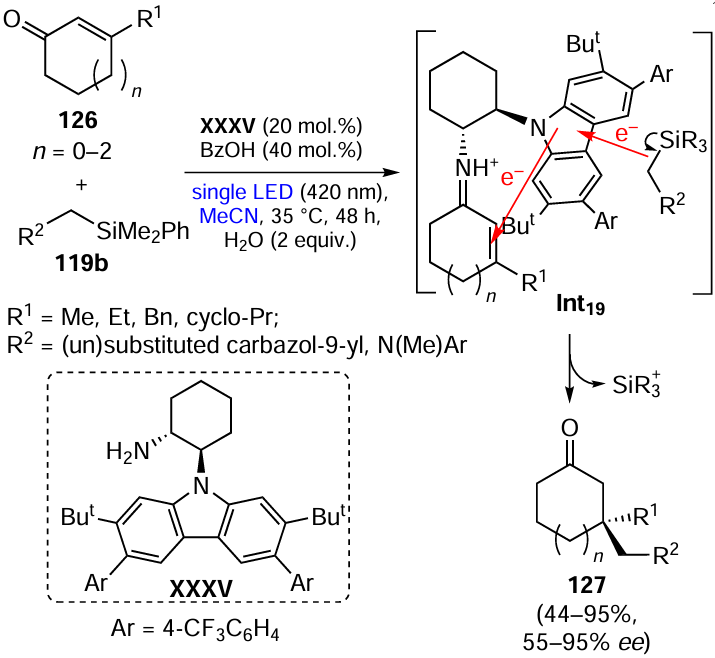
To conclude, the conceptual ideas and basic principles of green chemistry have had and continue to have a major influence on the development of asymmetric organocatalysis — an innovative field of organic chemistry. The result of their practical implementation can be the creation of highly selective, energy- and resource-saving and environmentally friendly technologies for obtaining enantiomerically pure drugs and other practically useful products of fine organic synthesis.
3.4. Electrochemistry as a powerful tool of green organic synthesis
Electric current is one of the most environmentally benign sources of energy for chemical reactions. Electrification of organic synthesis, that is, the search for alternative eco-effective approaches to the preparation of highly marginal, practically valuable products using methods of electrochemistry is growing exponentially, making up for years of quiet development.[507-525][526-530] Advantages of electrochemical methods over conventional approaches can always be demonstrated. If conventional methods are placed on one weighing pan of a balance and electrosynthesis is placed on the other pan to consider the selectivity, efficiency and economic performance of the preparation of the same product or related products, the second pan would undoubtedly outweigh (Fig. 21). The decrease in the number of steps and the atom, time and pot economy are important advantages of electrosynthesis. In addition, elimination of chemical oxidants and reductants, which are replaced by electrodes (an electron is a traceless reagent with a controlled strength), considerably simplifies the reaction system and, hence, isolation of products and eliminates the waste formation.
![[{"id":"gVXcAU-BmC","type":"paragraph","data":{"text":"Advantages of electrochemistry over conventional chemical processes."}}]](/storage/images/resized/bLBUzqZjdUgTNQT2sIVsLtDyBcxe35Zt4BvI9oq8_xl.webp)
Currently, there are more than 900 known reactions of organic compounds that take place under electrochemical conditions, out of which 7% have already been commercialized as ready industrial processes and 15% have been tested on a pilot scale.[514] However, there is some contradiction between the goals and objectives of industrial electrochemistry and fine organic electrosynthesis (Fig. 22). As a rule, large-scale electrochemical production processes aim at the synthesis of relatively structurally simple and relatively inexpensive molecules (aluminum, chlorine, adiponitrile, formic acid and so on[531]), but the scale of production and cost effectiveness depend on the cost of electricity, Faraday efficiency (current efficiency), design of the electrolytic cells, etc.
![[{"id":"_dt_jz4B27","type":"paragraph","data":{"text":"Goals of the industrial and fine organic electrosynthesis."}}]](/storage/images/resized/9sGsk09N5eCHedOkpHgRwhNrDUwK52vseAGg3OBo_xl.webp)
In turn, the goals of fine organic electrosynthesis include the search for new, more convenient and selective methods for the synthesis of costly small molecules (drugs, biologically active compounds,[507][510-522] natural compounds[523][524]), in particular from natural feedstock[525] or the products of its processing[526] (see Fig. 22). In this case, the cost of electricity is less significant, but particularly these processes take advantage of the benefits of electrochemistry, which provides control over the reactivity of polyfunctional organic substrates by implementing new, more efficient pathways of chemical bond formation and cleavage, in particular at the latest steps of the synthesis.
This Section addresses both fine organic electrosynthesis processes directed towards the preparation of useful molecules of high chemical complexity and industrial electrosynthesis processes for the production of basic organic products containing one or two carbon atoms (liquid fuels, formic acid, etc.) from carbon dioxide, a cheap and readily accessible natural raw material. The attention is focused on the latest achievements in these areas.
3.4.1. Fine organic electrosynthesis
The recent advances of electrochemical organic synthesis are largely associated with the design of new types of reactors including flow electrochemical cells, which allow scaling-up of the processes from the laboratory to industrial scale;[532-534] electrochemical cells for original alternating-current electrolysis techniques;[533] reactors combining electrolysis and light activation,[89][517] including the plasmonic excitation of the electrocatalyst.[535] New-generation electrodes with unique composition and porosity were designed; among them, mention should be made of boron-doped diamond electrodes and electrodes with immobilized catalysts.[507][508][526][532] Organic electrosynthesis makes it possible to produce a wide range of in-demand products[536] with minimized impact on the environment.[537][538] Machine learning techniques are used more and more actively to predict new electrochemical reactions.[539]
3.4.1.1. Electrochemical С – H functionalization of aromatic and heterocyclic compounds without metal catalysts
As noted above (see Sections 3.1 and 3.2), direct С – Н functionalization of molecules, without preliminary replacement of hydrogen by good leaving groups (halogen- or boron containing groups, triflate, etc.) that would increase the amount of waste, meets the green chemistry principles. As an ideal reaction, consider the dehydrating electrochemical cross-coupling of the С – H bond with various proton-containing molecules that takes place without external chemical oxidants or reducing agents (this function is performed by electrodes) and gives hydrogen or protons as by-products (Scheme 86).
Chupakhin et al.[540] proposed an atom-economic one-step synthesis of aryl-substituted aza aromatic compounds via an electrochemical oxidation reaction (Scheme 87). The С−С coupling proceeds with high yields by the SNH mechanism via the intermediate formation of s-H adducts, which have been successfully isolated and characterized by physicochemical methods, including X-ray diffraction analysis and NMR. The redox properties of these adducts determine the efficiency of the reaction, as they are oxidized before the precursors.
In recent years, considerable attention has been devoted to electrochemical С – Н phosphorylation, since the conventional methods for the preparation of organophosphorus compounds with the Р – С bonds are among the most hazardous and multistep processes.[541-545] The phosphoryl group improves the hydrophilicity and bioavailability of organic molecules that possess anticancer, antibacterial or antiviral properties, including anti-HIV activity. The choice of phosphorus precursors for these reactions is limited by the fact that many redox-active triorganyl phosphites (RO)3P and diorganylphosphine oxides R2P(O)H are not oxidized and are reduced with difficulty in the accessible potential range. Nevertheless, Xu[546] and Lei[547] and co-workers were able to prepare a broad range of aryl(heteroaryl)phosphonates, including biologically active compounds, by oxidative phosphorylation of aromatic and heteroaromatic compounds with trialkyl phosphites (Scheme 88). Some of these reactions were performed as late stages of the synthesis of biologically active products. The use of undivided and flow cells, mild oxidation conditions, and formally only one by-product, hydrogen, which is released at the cathode, indicate that these processes can be considered as complying with the green chemistry requirements.
The pathways of these reactions are of separate interest; although reaction mechanisms are proposed in many publications, convincing evidence for a particular mechanism is rarely given. Meanwhile, the knowledge of the key intermediates, their redox properties and reaction pathways is needed to expand the range of substrates, increase the yields of products and scale-up the processes. Presumably, s-H adducts, which were previously observed by Chupakhin,[540] may be intermediates for the С – Н phosphorylation;[548] other possible key species are phosphorus-centred radical cations.[549] The triorganyl phosphite radical cations and s-H adducts (dialkyl dihydroacridine-phosphonates) were isolated and characterized by physicochemical methods (EPR, X-ray diffraction, NMR spectroscopy, voltammetry, etc.) in the controlled-potential phosphorylation of acridine and its derivatives, resulting in the formation of various acridine-phosphonates (Scheme 89). The high yields in this reaction are due to the fact that s-H adducts are oxidized more readily than the precursors.[548]
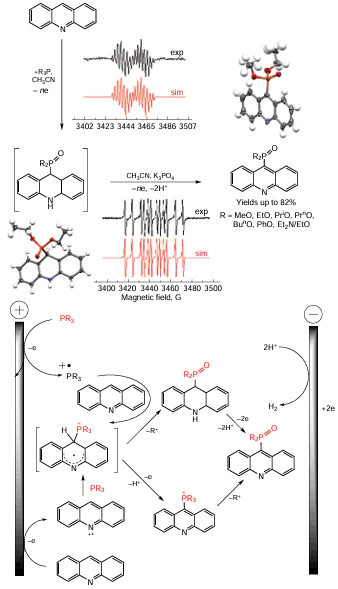
Dialkyl H-phosphonates (RO)2P(O)H and diorganylphosphine oxides R2P(O)H are not oxidized at an electrode in an accessible potential range; however, they can be involved in the С – Р bond formation with aromatic and heterocyclic compounds that can be oxidized under conditions of electrolysis. Zeng and co-workers[550] showed that co-electrolysis of quinoxalin-2(1H)-ones and xanthenes with (RO)2P(O)H or R2P(O)H results in phosphorylation of C(sp2) – H or C(sp3) – H bonds (Scheme 90). The benefits of this process include mild reaction conditions, simple design of the undivided cell, formation of hydrogen as a by-product, the absence of chemical oxidants or additives (bases and catalysts needed in the conventional methods), regioselectivity and high product yields (up to 99%).
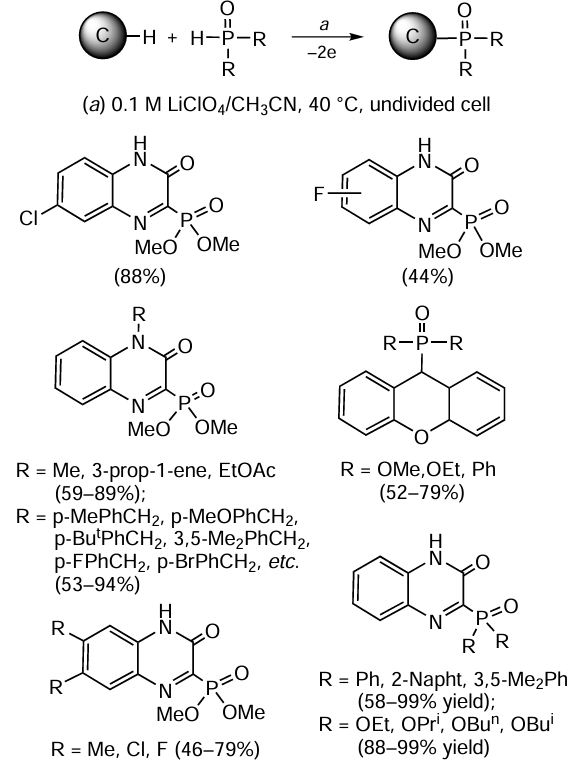
Green solvents such as water, ethanol and acetic acid are used more and more often in electrosynthesis, in particular for scaling-up the reactions. For example, a method developed for the synthesis of annulated (isoxazolidine)isoquinolin-1(2H)-ones is based on electrochemical oxidation of arylhydroxamic acid esters containing acetylene groups in aqueous EtOH (95%) (Scheme 91).[551] The reaction proceeds via the formation of amidyl radicals. The advantages of the method include a broad scope of applicability, the absence of metal catalysts or external oxidants, simple design of the undivided cell with carbon sheets as electrodes, environmental friendliness and easy scaling-up.
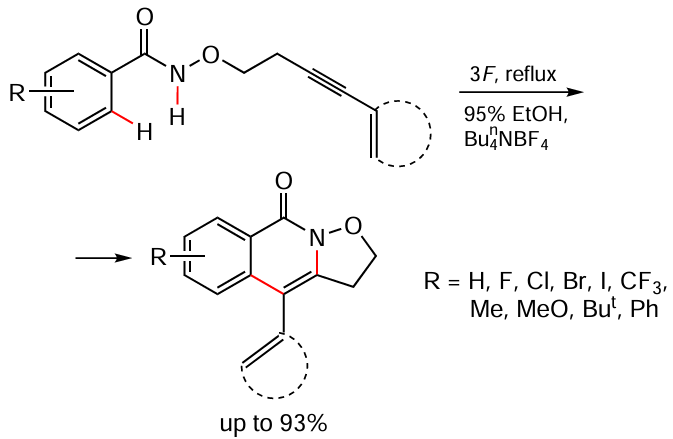
Precursors for the synthesis of many biologically active molecules contain thiocyanate groups. Recently, efficient electrochemical thiocyanation of 1,3-dicarbonyl compounds in AcOH was reported (Scheme 92).[552] The direct-current electrolysis proceeds in an undivided cell without additional halogen-containing electrolytes. Ammonium thiocyanate serves as both the source of the SCN group and the electrolyte. The reaction includes the electrochemical oxidation of the thiocyanate anion to (SCN)2 and addition of the latter to the double bond of the 1,3-dicarbonyl enol form. Some of the prepared thiocyanates exhibit a high antifungal activity.
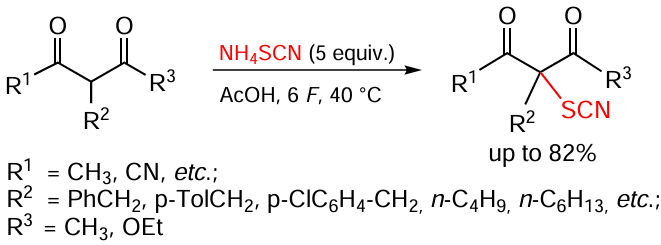
Unlike the reaction of 1,3-dicarbonyl compounds with ammonium thiocyanate, the reaction between benzylic C(sp3) – H groups and trimethylsilyl isocyanate leads to oxidation under mild electrochemical conditions to give the C – N bond.[553] The reaction can be easily scaled-up and can be used for the introduction of the isocyanate group into some pharmaceuticals and other biologically active molecules at final stages of a multistep synthesis (Scheme 93). Unfortunately, in this case, the best results were achieved when the electrochemical process was carried out in a binary solvent system containing toxic dichloroethane.
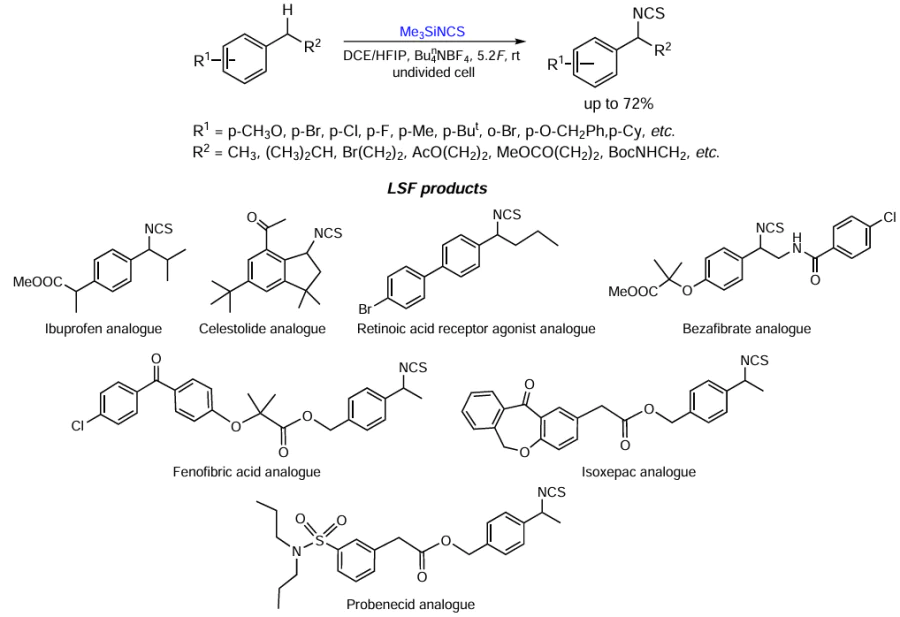
3.4.1.2. Metal-catalyzed electrochemical functionalization of С – H bonds
The С – Н substitution reactions involving metal complexes and salts functioning as homogeneous or heterogeneous catalysts (nanocatalysts) constitute a rapidly developing area of organic electrosynthesis.[543][554][555] The metal is coordinated to the substrate heteroatom and directs the substrate functionalization to the nearest С – Н bond, thus providing the selectivity. The key intermediates of these reactions are usually organometallic compounds (most often, metallacycles with metal – carbon bond), which, in some cases, can be isolated and characterized.[555-560] When this coordination is impossible, the reactions follow a radical mechanism involving phosphorus- or nitrogen-centred radicals, which are detected by EPR spectrscopy.[548][549] In this case, metals act as redox-mediators of the electrochemical reaction promoting the generation of radicals, although it should be admitted that their role is not always proved.
The oxidative phosphorylation of arenes induced by non-noble metals results in one-step synthesis of dialkyl arylphosphonates in high yields at room temperature (Scheme 94).[561] The 1% МnII/NiIIbipy bimetallic catalyst proved to be the most active and selective. This reaction was carried out for benzene and its derivatives containing either electron-withdrawing or electron-donating substituents or some coumarins. The efficiency of the process was attributed to fast catalytic cycle and regeneration of the active form of the catalyst at the electrode.
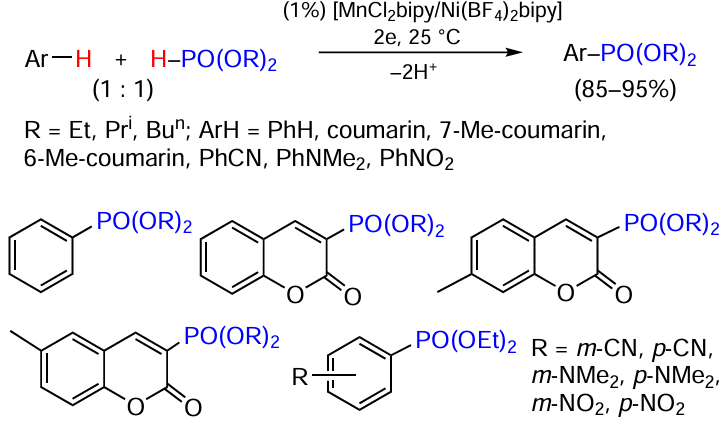
The electrochemically induced phosphorylation of azole derivatives (benzo-1,3-azoles, 3-methylindole, 4-methyl-2-acetylthiazole) with dialkyl H-phosphonates in the presence of silver salts or silver oxide (1 mol.%) proceeds at room temperature to afford dialkyl azolylphosphonates in up to 75% yields (Scheme 95).[562] According to the results of cyclic voltammetry, EPR and NMR spectroscopy, the intermediate AgP(O)(OEt)2 is oxidized before other components of the reaction mixture with elimination of a Р-radical. The EPR spectrum of this radical recorded in the presence of the PBN spin trap corresponds to published data.
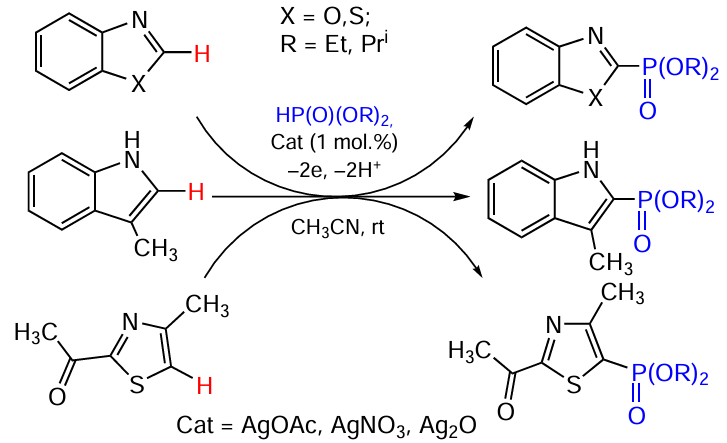
The first studies on the ligand-directed electrochemical С – H substitution involving transition metals appeared about a decade ago.[557][559][560][563][564] It was shown that coordination of the metal [Ni(II), Pd(II), etc.] to the nitrogen atom of the substrate ligand (2-phenylpyridine, benzo[h]quinoline or 1-phenyl-1Н-pyrazole) affords the expected metallacycle with the metal–carbon bond, which is electrochemically oxidized much more readily than the starting compound. The oxidation is centred at the metal and is accompanied by the transition of metal M into higher oxidation states +3 and +4.[556-560][564-567] The chemistry and electrochemistry of these metallacycles, particularly, transmetallation, reductive elimination and pathways for the control of these reactions are very interesting and deserve separate consideration, which is beyond the scope of this review. We would only like to note that in the presence of some metal salts, high regioselectivity of oxidation can be attained, for example, N-heteroarene fluoroalkylation,[557][563] phosphorylation[556-560] (Scheme 96) and amidation[556] can be carried out.
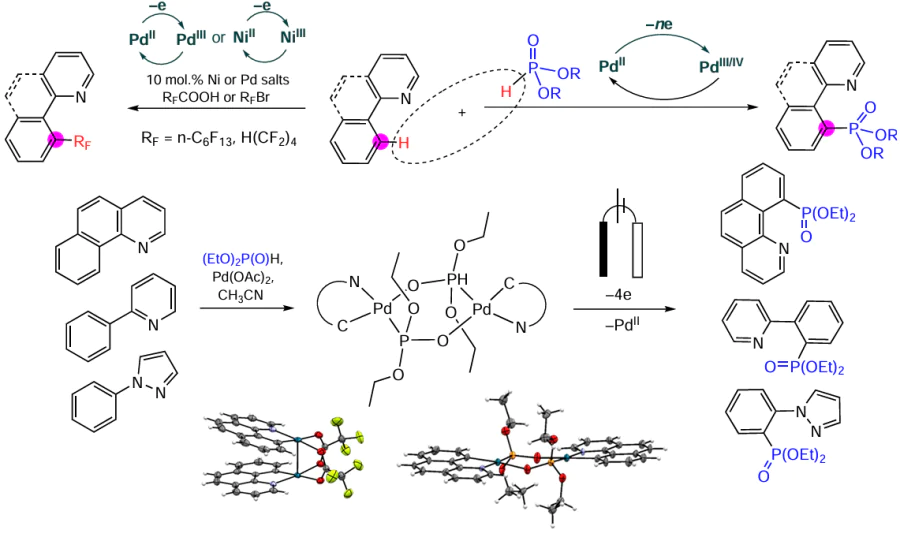
The catalyst is readily regenerated at relatively low potentials in the key steps of the reaction such as the metallacycle electrooxidation and reductive elimination of the product. There are few examples of isolated and fully characterized nickel complexes in higher oxidation states. Among them are stable Ni(III) complexes with RF substituents,[568][569] the participation of which in catalysis was confirmed by EPR data.[566] The phosphorylation of aromatic compounds in the presence of palladium salts proceeds, apparently, via Pd(II)/Pd(III)/Pd(IV) redox cycles, since the formation of the final dialkyl arylphosphonate requires two electrons per palladium(II) atom in the intermediate metallacycle.[559][563][565] The easy transition of palladium, nickel and other transition metals to higher oxidation states, needed for C – H functionalization, makes the electrochemical method preferable over conventional organic synthesis, in which only very strong oxidants, undesirable for green chemistry processes, are effective.
The possibility of controlling the selectivity of electrochemical С–Н functionalization by changing the anode potential was demonstrated in relation to the oxidative coupling of N-(quinolin-8-yl)benzamide (128) (Scheme 97).[570] By controlling the oxidation characteristics, one can regioselectively obtain various oxidative coupling products. The course of the reaction depends on the type of the reaction centre that is oxidized and the nature of the oxidant. For example, at different anode potentials, it is possible to selectively oxidize the quinolinе para-С – Н bond, amide N – H bond, or the ortho-С – Н bond in the benzene ring of benzamide 128 to give the corresponding products 129 – 132. The intermediate Co(II)[L–H]2 complex (Int20) and the C – H-activated Co(III) metallacycle (Int21) with benzamide ligands were isolated and characterized by X-ray diffraction analysis and voltammetry. The two-electron oxidation of Int20 results in the С – N coupling product 129, while the single-electron oxidation of the Co(III)-containing metallacycle Int21 furnishes the ortho-С – С coupling product 130. These electrochemical reactions proceed under mild conditions at room temperature at an anode potential of only 0.4 V without the addition of special reagents (oxidants, halides, phosphines, etc.), which are usually needed in the traditional chemical reactions. The catalyst-free electrochemical oxidation affords dimer 131 (5,5'-С – С coupling) or hydrazone 132 (N – N coupling) at potentials of 1.7 and 1.1 V, respectively.
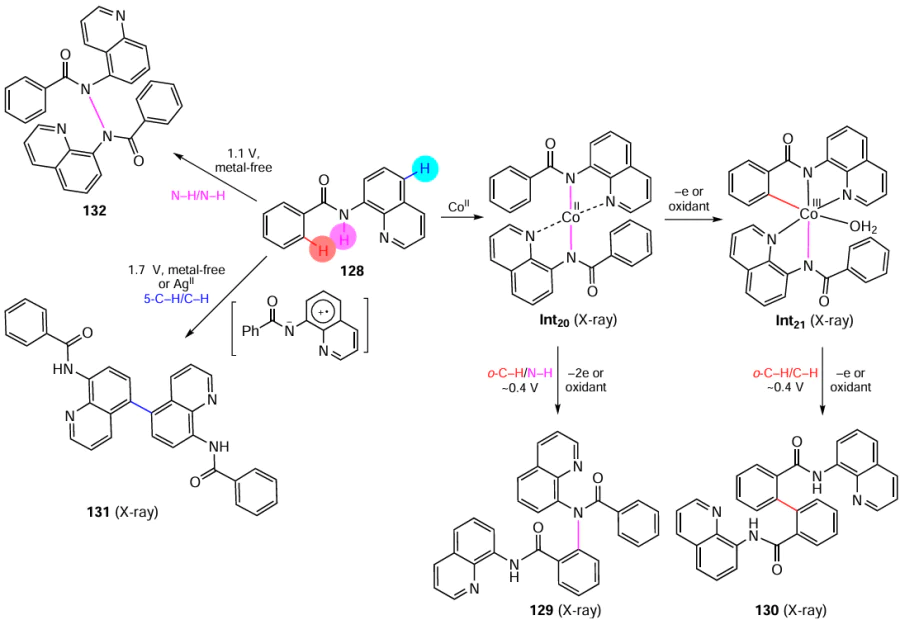
The direct electrochemical C – H/N – H cross-coupling of phthalimide with heteroaromatic compounds catalyzed by copper or silver salts results in the formation of N-substituted phthalimide derivatives (Scheme 98).[571] This reaction is characterized by good yields, mild conditions and a wide range of suitable substrates. The copper and silver catalysts give comparable results, but silver salts or oxides make it possible to monitor the reactions by EPR, because they contain no paramagnetic species, and provide higher purity of the products. The voltammetric and EPR studies of the reaction mechanism demonstrated that the free-radical mechanism can be ruled out and that the reaction proceeds via catalytic cycles involving Cu(I)/Cu(II)/Cu(III) or Ag(I)/Ag(III) redox transitions.
Electrochemical oxidative cross-coupling involving alkynes proceeds even in the presence of water. The resulting adducts can cyclize during electrolysis.[61][572-574] The reaction of disubstituted acetylenes with diphenylphosphine oxide occurs at the oxidation potential of the [Ph2P(O)Ag] intermediate to give benzo[b]phosphole oxides[572] (Scheme 99). The co-electrooxidation of propargyl aryl ethers with sulfonyl hydrazides furnishes 3-sulfonated 2Н-chromene derivatives.[574] The only by-products are hydrogen (formed at the cathode) and nitrogen (formed at the anode). Electrosynthesis of chromones from acetylenes and hydroxybenzaldehyde derivatives catalyzed by rhodium complex is favourably distinguished from the chemical methods that use oxidants by the scalability and tolerance to a variety of functional groups.[575] This strategy is applicable, in particular, to the preparation of fluorogenic peptidomimetics based on tyrosine derivatives.
Under certain conditions, nitriles (acetonitrile and benzonitrile) can serve as sources of the amide group in mild electrochemical amidation of the C(sp2) – H bonds in benzene derivatives and С(sp3) – H bonds in toluene derivatives catalyzed by Cu(II) salts (Scheme 100).[576] The reactions give N-arylamides 133 and N-benzylamides 134, respectively. This result, attained in an environmentally friendly aqueous medium, is nontrivial, since acetonitrile is widely used in organic synthesis as an inert solvent with a broad range of working potentials.
3.4.1.3. Electrochemical synthesis involving nanoheterogeneous catalysts
As noted in Chapters 3.1 and 3.2 of this review, catalytic methods based on the use of homogeneous and heterogeneous catalysts open up wide opportunities for fine organic synthesis, in particular, for reactions accompanied by C – H bond activation. In recent years, considerable efforts of researchers have been directed towards the design of nanoheterogeneous catalytic systems based on early transition metals, such as Ni, Co, Zn, Mn, Fe, Cu, and other, which are abundant in the Earth’s crust and, hence, less expensive. It is expected that these catalysts would be more active and selective than the conventional heterogeneous catalysts, while being not inferior in the durability and recyclability.[577] The properties of the nanocatalysts can be modified by varying the composition, structure, particle size and the way of immobilization.
For example, the catalytic system obtained by doping of the [(bipy)3NiII] complex into silicate nanoparticles followed by electrooxidation of the complex to [(bipy)xNiIII] found use in the oxidative fluoroalkylation of aromatic С – Н bonds (Scheme 101).[578] Coupling of 2-phenylpyridine and caffeine with perfluoroheptanoic acid in the presence of this catalyst proceeds regioselectively at room temperature up to 100% reactant conversion to give products 135 and 136 in good yields. The possibility of electrochemical regeneration makes it possible to decrease the content of the catalyst to 1% relative to substrates. The catalyst can be easily separated from the reaction mixture and reused in five cycles.

In the search for active nanocatalysts convenient for handling for the С – Н alkylation and С–Н fluoroalkylation reactions, Ag0-doped silicate nanoparticles (Ag0/+@SiO2) with a specific nanostructure in which ultrasmall silver cores are embedded into 40-nm silicate spheres were obtained.[579] The nanoarchitecture provides efficient electrochemical oxidation of Ag0@SiO2 to Ag+@SiO2 without an external oxidant. In the presence of the obtained nanocatalyst (5 mol.%), arenes are converted to the alkylation (fluoroalkylation) products in one step on treatment with carboxylic acid, while the catalyst can be regenerated by electrolysis (Scheme 102).
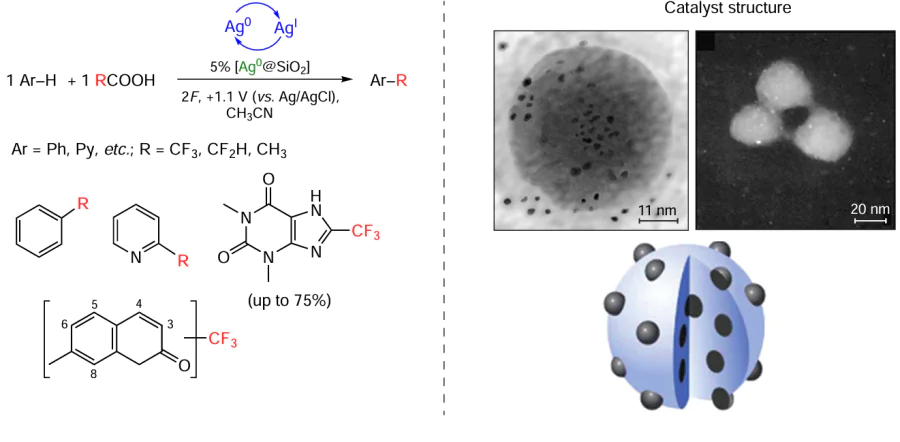
An electrochemical process underlies the environmentally benign synthesis of phosphorylated benzooxa(thia)zoles by the reaction of diethyl H-phosphonate with the corresponding azoles catalyzed by Ag0@SiO2 nanoparticles (Scheme 103).[580] The highest performance was found for the catalyst in which Ag0 nanoparticles (2 – 7 nm) were deposited on larger (approximately 140 nm) silica supports.
The oxidative C – H/N – H cross-coupling reactions can be carried out in the presence of cobalt compounds, e.g., CoIV-bipy complex obtained by electrooxidation of CoIII(bipy)3 naanoparticles enclosed in a silica matrix.[581] In the presence of this catalyst (1 mol.%), benzo-h-quinoline is coupled with tosylamine H2NTs at room temperature (Scheme 104). Optimization of the size and morphology of CoIII/IV(bipy)3-doped silicate nanoparticles gave an efficient catalyst, which could be regenerated and reused at least seven times.
Mono- and bimetallic nanocatalysts free from noble metals proved to be effective in electrochemical C – H/P – H cross-coupling reactions of substituted phosphine oxides with phenylacetylene (Scheme 105).[582] In this case, too, ions of one or two d-metals were immobilized in the SiO2 matrix. The monometallic catalysts provided the synthesis of phosphorylated acetylene 137 with retention of the triple bond, while the use of bimetallic catalysts resulted in the synthesis of the bis-phosphorylation product 138. This is the first example of electrochemical C−H/P−H coupling of acetylene and phosphine oxide using a catalyst that could be regenerated and reused in the catalytic cycle. Development of an alternative electrochemical method for the synthesis of bis-phosphine oxide 138 is practically significant, as this compound is produced by leading chemical companies (BaiFuChem, Xiamen Equation Chemical Co., Ltd and UHNShanghai) and is a precursor for pharmaceutical industry and a key component of flame retardants.
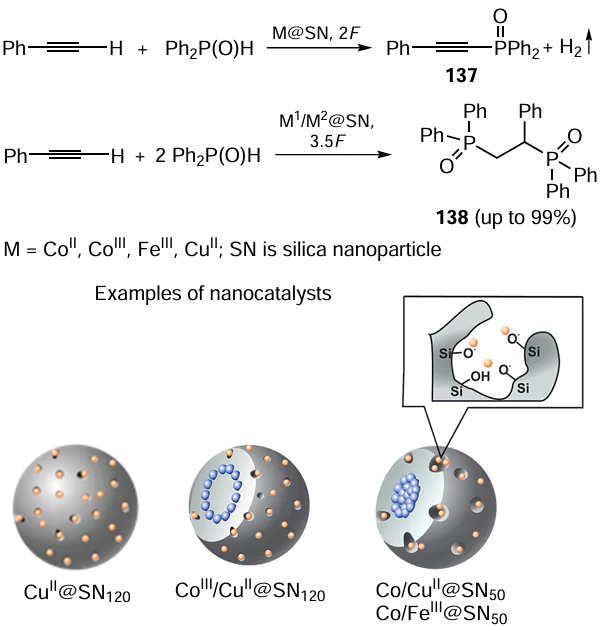
There are also published examples of successful application of electrochemical nanocatalytic systems in which the active metal in the required oxidation state is stabilized in protective matrices, e.g., in silicate or polypyrrole matrix.[583][584]
Thus, approaches based on the use of electric current as an electron acceptor and donor in fine organic synthesis have already brought impressive results and have good prospects for further development. They provide routes to diverse chemical products, including chemicals for pharmaceutical industry, by atom-economic routes, in one step, thus reducing the adverse impact of chemical processes on the environment. In addition, organic electrosynthesis can be integrated into systems with renewable energy sources, which play an increasing role in the global energy infrastructure. This makes the described processes even more environmentally attractive.
3.4.2. Electrochemical approaches in carbon dioxide reduction and utilization
An important application of electrochemical methods is the chemical conversion of carbon dioxide (СО2), a greenhouse gas released into the atmosphere in large amounts because of unbalanced combustion of hydrocarbon fuel.[585-589] Among the most significant adverse consequences of high СО2 content in the atmosphere, climate change and ocean acidification deserve attention.[590] Therefore, one of the biggest challenges today is the need to reduce CO2 emissions, while the development of new methods for CO2 conversion to fuels and other useful products is a necessary step towards solution of this global problem.[591]
Therefore, development of methods to convert carbon dioxide to organic compounds containing С – Н bonds (С1 products) and С – С bonds (С2 products) is a highly relevant task of modern chemical science. In recent years, studies aimed at the design of high-performance catalysts and relevant equipment for carbon dioxide conversion have been actively pursued.[592-599] In this regard, methods based on the use of electric current are most attractive because of low requirements to the equipment, high efficiency and environmental friendliness.[600-603] Depending on the number of electrons transferred in the electrolysis, various types of electroreduction of carbon dioxide[604-608] may furnish quite a number of practically valuable products, including carbon monoxide (CO), methane, ethylene, ethanol and other promising fuel components (Fig. 23).[609-613]
![[{"id":"eX1uaU6vHz","type":"paragraph","data":{"text":"Products of CO<sub>2</sub> electroreduction and number of electrons required for the process."}}]](/storage/images/resized/fwGBvnemlYrMuyRpsUzyWXP6qts6zFhADgXO8cSZ_xl.webp)
Recently, it was shown that carbon dioxide can be reduced to methane in the presence of unsymmetrical zirconium(IV) and hafnium(IV) pincer complexes [M(k3-NNN)Bn2] (where NNN is N-{[6-(1-H-benzimidazol-2-yl)pyridin-2-yl]methyl}-2,6-diisopropylaniline), characterized by relatively high TOF (272 h–1).[614] However, homogeneous processes of carbon dioxide reduction catalyzed by transition metal complexes require the use of large amounts of chemical reducing agents and expensive co-catalysts, which restricts their large-scale application. The conversion of carbon dioxide to industrially important compounds under electrochemical activation is free from these drawbacks and forms the basis for the design of new processes that comply with the key principles of green chemistry.
The first attempt to perform electroreduction of carbon dioxide was made by Beketov back in 1869.[615] Since then, the development of chemical methods for CO2 utilization and related issues have been a focus of interest. Indeed, significant steps towards improving the selectivity, current efficiency and service life of the electrochemical systems have been made in the last decade. However, these processes are faced with obstacles such as high reduction potentials, low reaction rates in the near-electrode space, low selectivity and low yield of the target product. Due to the high stability of the linear O=C=O molecule, for the electroreduction to occur, it should first be converted into a bent structure, which is associated with high energy consumption. Indeed, the standard potential Е0' of the single-electron step, which forms CO2–• radical anion as the primary product, is −1.90 V vs. the normal hydrogen electrode (NHE) (Eq. 6):
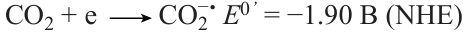
In order to decrease the energy consumption of the electrochemical process, it is necessary to use specific electrode electrocatalysts or added catalysts, the selection of which is a key task of the development of electrochemical СО2 conversion processes. The kinetics and mechanism of CO2 electroreduction and the ratio of the products depend on various factors such as electrode material, composition of the electrolyte (aqueous/non-aqueous), structure of the supporting electrolyte (especially the cation), concentration and purity of the chemicals, pH, pressure, temperature and so on.[616] This reaction is often considered as being reverse to the complete chemical oxidation of simple organic compounds (e.g., methanol) in fuel cells.[585][617-619]
The electrochemical reduction of CO2 to the most valuable С2 products can be accomplished using heterometallic systems able to catalyze not only reduction, but also С–С coupling reactions. The heterostructured Cu@Cu0.4W0.6 catalyst for the synthesis of multicarbon products (mainly ethylene, ethanol and acetic acid) from СО2 was developed relying on theoretical calculations.[620] The selectivity and activity of this catalyst in the formation of C2 products considerably depend on the mass ratio of copper and tungsten (for Cu0.4W0.6 , the current efficiency was 60.9%, and the partial current density was 121.8 mA cm–2 at –1.0 V vs. NHE). According to theoretical calculations, the Cu@Cu0.4W0.6 heterostructure inhibits the evolution of hydrogen via charge redistribution at the Cu@/Cu0.4W0.6 interface, thus promoting the formation of CO and the unsymmetrical CO – CHO coupling. High oxygen affinity of tungsten facilitates cleavage of the C – O bond in the resulting *C2H3O intermediate and promotes the formation of ethylene (Scheme 106). Raman spectroscopy experiments in situ confirmed the reaction mechanism. The cited study, together with studies addressing micro- or nanostructured and also heterostructured systems,[621-627] emphasize the importance of the synergistic effect between chemically different metals for increasing the activity and selectivity of the electrocatalytic reduction of CO2 to valuable С2 products.
The electrochemical reduction of СО2 is also considerably affected by the material of electrodes and the type of transformations that take place near the electrodes.[628][629] It was shown that in the potential range of СО2 electroreduction to СО2–•, the formed radical anion (СО2–•) may be inserted into the surface of metal (Ag, Au, Cu, Pt, Pd, Rh, Ti) and nonmetal electrodes (glassy carbon, usual and highly oriented pyrolytic graphite, graphene) used in the process. This can also occur for relatively small-size cations such as sodium or tetramethylammonium (Me4N)+, which form ion pairs with carboxylated metal species, thus being incorporated into carbon–metal matrices (Scheme 107).
The formation of a loose {Au – CO2−M+} surface layer (М = Na+, Me4N+) with a much lower electrical conductivity compared to that of the initial electrode was assumed.[630] A similar mechanism was also proposed for silver, copper and some other electrodes.[631] This method of surface carboxylation, in particular copper in non-aqueous solutions (DMF, MeCN), can efficiently protect the electrode surface from anodic corrosion.
One more actively developing area is photoelectrochemical reduction of CO2 to low-carbon products using readily available and environmentally benign solar energy. A variety of СО2 photoactivation methods have been proposed. They are based on the use of semiconductors with a specified band gap width and Fermi level energy, electron traps and conjugated heterophase photoelectrocatalytic systems; morphological surface modification; and fabrication of hybrid structures (metals, metal oxides and alloys in polymer matrices). The mechanism of СО2 photoreduction on photocatalytically active electrodes (TiO2 , p-GaP, n- and p-GaAs, p-InP, p-Si, highly doped p+/p-Si, p-SiC, zinc and cadmium sulfides and selenides) consists of several stages. Considering a semiconductor as an example, one can distinguish the light absorption stage with activation, provided that Ehn ³ Eg (Eg is the band gap). Photoexcitation gives rise to electrons in the conduction band and holes in the valence band, which are responsible for photoreduction and photooxidation. In this case, the energy of electrons should exceed the СО2 reduction potential.[632]
Most studies related to the photoelectrochemical conversion of carbon dioxide use H-type reactor with an appropriate ion exchange membrane (e.g., Nafion) (FIg. 24). Two-compartment reactors are more convenient than one-compartment reactors, since the separation of electrode compartments facilitates the separation of products.[585] Xenon arc lamps with appropriate filters are used, most often, as light sources to mimic the solar light.[591][633-636] The light should be uniformly scattered over the photoactive electrode surface.
![[{"id":"idSNTvSlGw","type":"paragraph","data":{"text":"Photolectrochemical H type reactor for the conversion of CO<sub>2</sub> to organic products."}}]](/storage/images/resized/5hsjvdOyVe7Tauh8q2ltMfK2Ed7C7ZCSQI6CU1sk_xl.webp)
Irtem et al.[637] used a filter-press type flow cell for the photoelectrochemical reduction of CO2 to formic acid. This design decreases the ohmic drop in the electrochemical cell and promotes full illumination of the photoelectrode. A 300 W arc Хе lamp with an AM-1.5G filter served as the light source. The efficiency of the reactor depended on the photon flux intensity and photocathode and photoanode areas. For the applied voltage of 1.2 V, light intensity of 200 mW cm–2 and cathode area of 2 cm2, the reaction gave formate as the major product, with the current efficiency being 53%.
Zhou and Xiang[638] analyzed the efficiency of the photoelectrochemical process for various reactor designs meant for the direct one-stage (Fig. 25a) and cascade two-stage reduction of CO2 (Fig. 25b). The efficiency of CO2 to C2H6O conversion was always higher in the two-stage reactor than in the one-stage reactor. In the first stage, two-electron reduction of CO2 to CO took place, while in the second stage, the resulting CO was converted to higher-level reduction products (ethanol or ethylene) containing С–С bonds. The current efficiency was higher by 45% in the two-stage reactor than in the one-stage reactor.
![[{"id":"I7QNy864TR","type":"paragraph","data":{"text":"Photoelectrochemical reduction of CO<sub>2</sub> in reactors of different design: (<i>a</i>) direct one-stage reduction of CO<sub>2</sub>; (<i>b</i>) cascade two-stage reduction of CO<sub>2</sub> ."}}]](/storage/images/resized/LVDYjWwFi1EMPTrOLsmAiHYwzpKs5NiCGEcniQy4_xl.webp)
The conversion of CO2 to gaseous products (CO, CH4 , C2H4) in a multilayer electrolyzer stack containing no catholyte has been reported.[639] In this case, silver nanoparticles and copper nanocubes deposited on a gas diffusion layer served as electrocatalysts. Important features of this setup include (i) low cell voltage (–3.0 V) with a high conversion efficiency (up to 40%); (ii) the absence of liquid circulation loop; (iii) the absence of problems related to the dissolution of CO2; (iv) the possibility of generating elevated pressure at the inlet of the reactor (up to 10 bar); and (v) high selectivity of the process. The major products of CO2 reduction on the copper catalyst included CO, CH4 and C2H4 , while only CO formed with the copper catalyst. The current efficiency of CO2 conversion in the multilayer electrolyzer stack reached 95%, while the current density of СО formation was 300 mA cm–2. It is expected that the use of the multilayer electrolyzer stack would markedly reduce the cost of the process and that it would be possible to adjust this design for the industrial CO2 utilization.
Thus, electrochemical conversion of carbon dioxide into practically valuable one- and two-carbon organic compounds is a relevant trend of modern green chemistry. Today, various catalytic systems (heterometallic catalysts) and devices (electrochemical cells of various types) have been developed; this made it possible to approach the highest conversion and selectivity to the desired product. Despite the difficulties caused by high thermodynamic stability of CO2 molecule,[640] in the future, the electrochemical conversion of carbon dioxide to valuable organic products may become the basis for industrial production of alternative hydrocarbon fuels, not related to non-renewable sources of energy, and thus contribute to the solution of global warming problem.
3.5. Multicomponent reactions in green chemistry
Since the appearance of the green chemistry concept, the key green chemistry statements have continuously evolved and developed. Currently, there are twelve cornerstones (green chemistry principles) that should form the basis of an ideal chemical process. One of them is the preferability of multicomponent reactions,[641-650] which markedly decrease the amount of waste that requires disposal. Since this subject area is very extensive, this section concentrates on reactions that meet simultaneously several criteria. Today, there are a number of published reviews that cover particular applications of multicomponent reactions in green chemistry, for example, solvent-free reactions,[651] reactions in water[652-657] and in low-melting eutectic mixtures,[658][659] and reactions assisted by ultrasound,[660][661] microwave irradiation[662] or mechanical activation.[663] Nevertheless, this subject area is not adequately addressed in the chemical literature.[664][665]
3.5.1. Multicomponent syntheses initiated by the Knoevenagel reaction
The Knoevenagel reaction is a potent tool actively used in many synthetic transformations, including those implemented under green conditions.[666][667] For example, this reaction served as the basis for three-component condensation of dicyanomethane (139), benzaldehydes 140 and ethyl acetoacetate (141) in water in the presence of NaOH. The target pyrans 142 were formed in nearly quantitative yields and, hence, the products could be isolated in a pure state by filtering off the precipitate formed in the reaction mixture[668] (Scheme 108). Pyrano[4,3-b]pyrans 144 were prepared by three-component condensation of malonidinitrile (139), benzaldehydes 140 and 4-hydroxy-6-methylpyran-2-one (143) induced by grinding the reactants in a mortar in the presence of ammonium acetate (10 mol.%). Some of the products showed high biological activities against MCF-7 cancer cells.[669] Malononitrile can be replaced by acetonitrtile derivatives 145, while ethyl acetoacetate can be replaced by dimedone 146; in this case, DABCO served as the catalyst for the synthesis of heterocycles 147.[670]
The mechanochemically assisted reaction of malononitrile (139), benzaldehydes 140 and hydroxynaphthalene isomer 148 or 150 afforded pyran derivatives in nearly quantitative yields. For homogenization, the reactants were mixed in a mortar, and then the mixture was kept in a furnace at 125°C. The typical yields were above 95%; therefore, products 149 and 151 were used without further purification after being just washed with water (Scheme 109).[671] The same products were formed in high yields in the presence of reusable catalyst based on ZrO2 nanoparticles. A variety of conditions were tested: in particular, the reaction was carried out without a solvent, but it was less efficient than the reaction in an aqueous medium.[672]
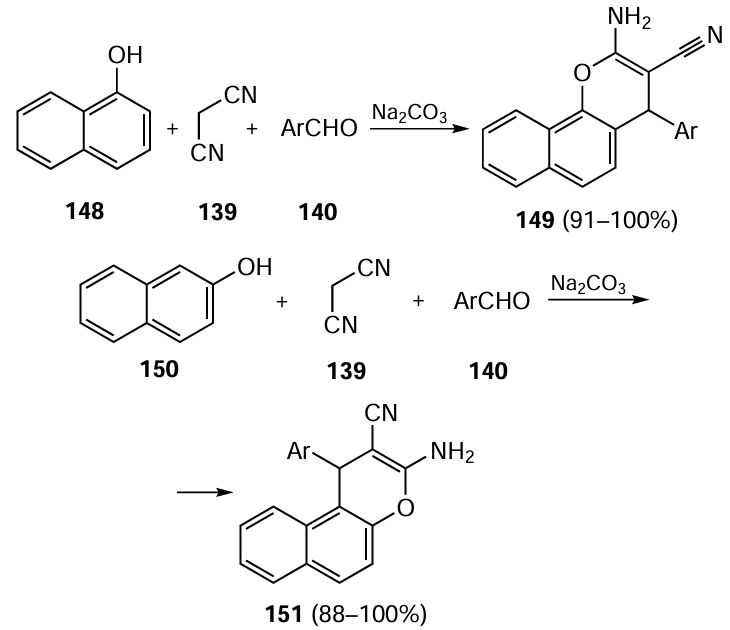
8,10-Dimethyl-12-aryl-9H-naphtho[1',2':5,6]pyrano[2,3-d]pyrimidine-9,11-diones 153 were synthesized from benzaldehydes, β-naphthol 150 and 1,3-dimethylbarbituric acid 152 in the presence of ZnO nanoparticles, which were recovered after the reaction and reused six more times without decrease in the activity (Scheme 110).[673] A similar synthesis was implemented using 1,3-cyclohexanediones 154 as methylene-active compounds. 12-Aryl-8,9,10,12-tetrahydrobenzo[a]xanthen-11-ones 155 were formed in this reaction when acids (H2SO4 or TsOH) in an aqueous medium were used as catalysts (method A) or when the reaction was microwave-assisted (method B).[674]
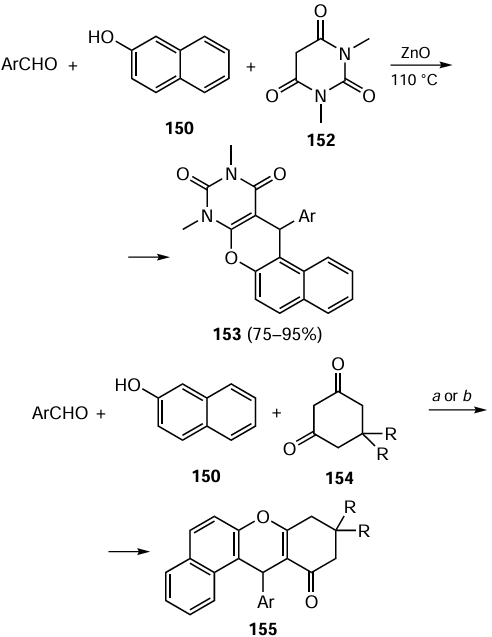
Chromene derivatives 157 can be prepared in excellent yields by the reaction of malononitrile 139 with aldehydes 140 and resorcinol 156 in the presence of a catalyst based on Fe3O4 and chitosan under ultrasonic treatment at 160 W. The magnetic catalyst can be separated from the reaction mixture without difficulty and reused four times without activity loss.[675] Using kojic acid 158, it is possible to synthesize 2-amino-4,8-dihydropyrano[3,2-b]pyran-3-carbonitriles 159 in an aqueous medium.[676] The introduction of phthalhydrazide 160 as a binucleophile into the reaction system gives rise to annulated heterocycles 161.[677] Reactions of this type were also catalyzed by citric acid[678] or silica-supported tungstic acid (STA). In the latter case, the efficiency increased under ultrasonic treatment (Scheme 111).[679]
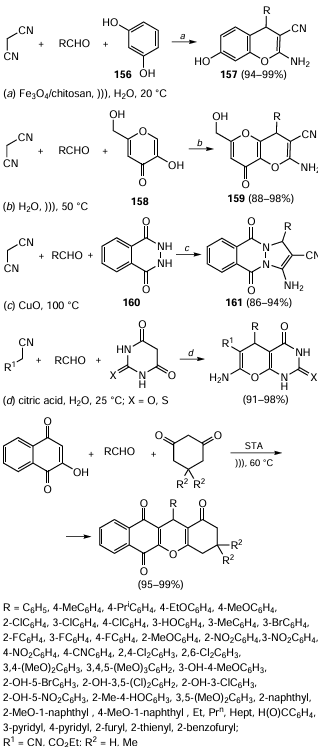
The reaction of salicylaldehyde 162 with malononitrile[680] or another methylene-active compound[681] (Scheme 112) proceeds as a tandem reaction consisting of Knoevenagel condensation followed by the attack by the phenolic oxygen atom on the nitrile group. Chromene 163 can add various nucleophiles present in the system, such as indole, 5-methylpyrazol-3-one or dimedone, to give adducts 164.[680]
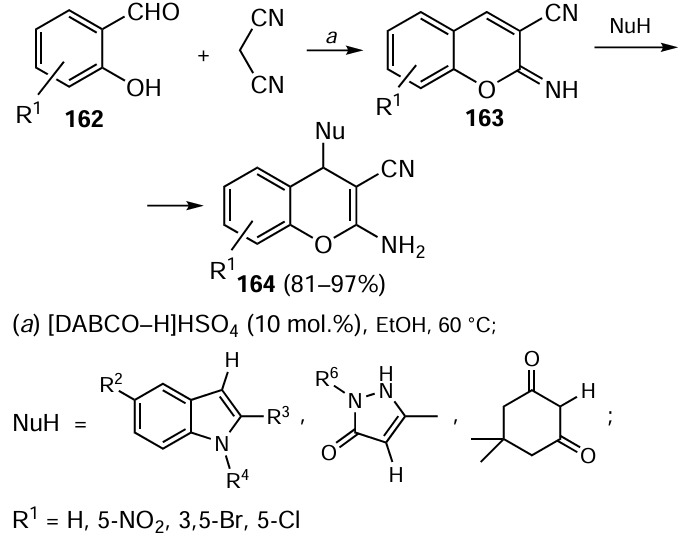
In recent years, increasing attention has been attracted by the possibility of combining conditions of a solvent-free reaction with photoactivation. Pyrano[2,3-c]pyrazoles 165 can be prepared by four-component reaction of ethyl acetoacetate with hydrazine hydrate, benzaldehyde derivative and malononitrile under neat conditions with irradiation with a 22 W compact fluorescent lamp (CFL) (Scheme 113).[682]
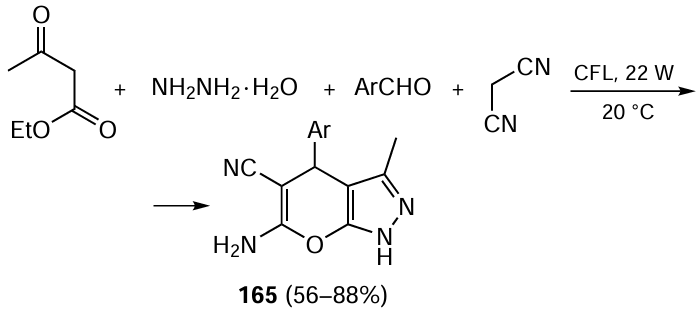
A synthesis of pyrano[3,2-c]chromen-5-ones 168 was implemented on the basis of reaction of 4-hydroxycoumarin 166 with (E)-N-methyl-1-(methylthio)-2-nitroethenamine 167 and aromatic aldehydes. The process was catalyzed by silica-supported tungstic acid, which was easily recovered and reused after the reaction (Scheme 114).[683]
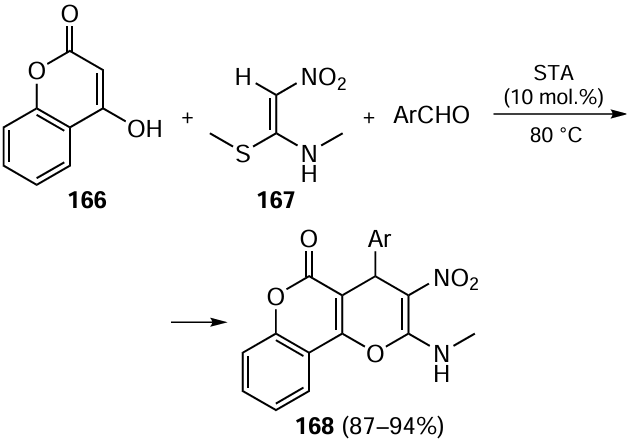
The synthesis of β-phosphonomalononitriles 170 from dialkyl H-phosphonates 169, aromatic aldehydes and malononitrile in the presence of organic bases (Scheme 115) was described.[684]
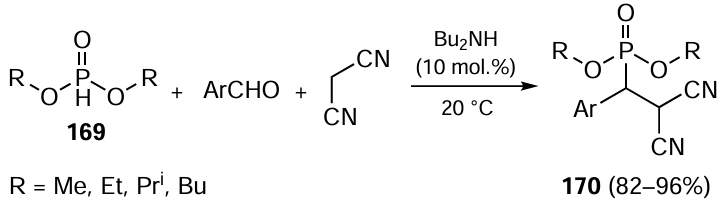
3.5.2. Multicomponent syntheses based on the Hantzsch reaction
A similar approach can be used to assemble pyridine derivatives by four-component reactions involving ammonium salts. For example, the condensation of dimedone 146 with malononitrile, aromatic aldehydes and ammonium acetate affords 1,4,6,8-tetrahydroxyquinoline derivatives 171.[685] Ultrasonic activation makes it possible to perform the reaction in water at room temperature and provides high yields of products 171 (Scheme 116). The catalysts used for reactions of this type include chitosan,[686] montmorillonite clay,[687] β-cyclodextrin monosulfonic acid,[688] aluminium-modified polyborates,[689] 1-butyl-3-sulfonic acid imidazolium chloride ([Bsim]Cl),[690] mesoporous organosilica-supported manganese,[691] glycine[692] and Fe3O4 nanoparticles.[693] The transformation can be also effected by fusing the reactants together in the absence of a solvent or a catalyst under microwave activation.[694]
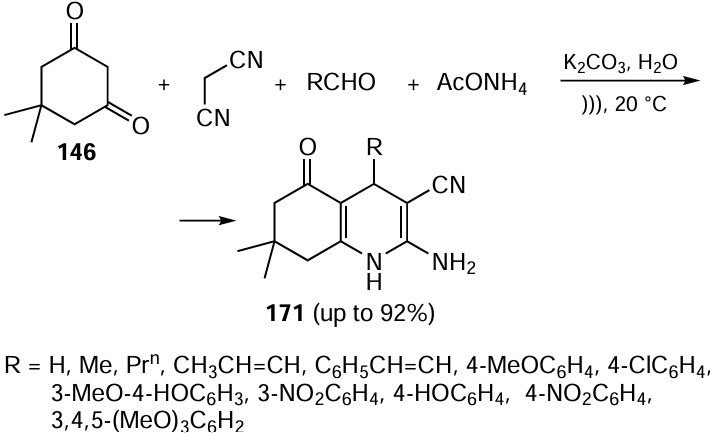
Formylferrocenes 172 also react according the same condensation pathway under microwave irradiation in aqueous medium to give ferrocenyl-containing heterocycles 173 (Scheme 117).[695]
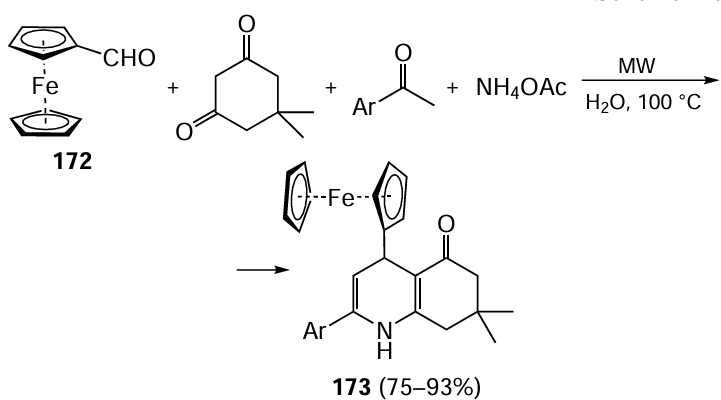
New derivatives of azapodophyllotoxin 175 were prepared by microwave-assisted multicomponent condensation of dimedone 146 with benzaldehyde derivatives and tetronic acid 174 in aqueous ammonia (Scheme 118).[696]
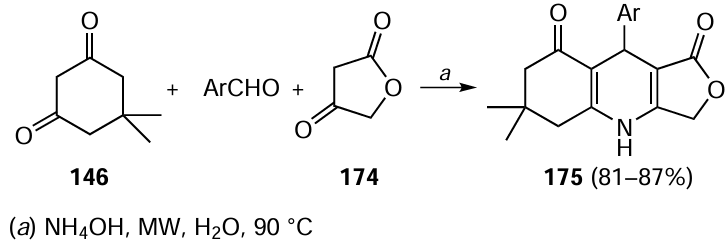
Aminoazoles can be converted to fused systems comprising pyridine and azole moieties. The microwave-assisted condensation of 2-phenyl-3-aminopyrazole 176 with benzaldehydes and barbituric acid derivatives 152 without a catalyst[697] provides a route to tricyclic derivatives 177. A similar condensation of dimedone 146 furnishes products 178.[698] The reactions with 3-aminotriazole,[699] 5-aminotetrazole 179,[700] 2-aminobenzimidazole 180[701] and aminopyrimidine 181 proceed in a similar way and afford fused systems 182 – 184 in high yields. β-Cyclodextrin (β-CD) can catalyze reactions of this type in aqueous media owing to the presence of inner cavity and numerous OH bonds (Scheme 119).[702]
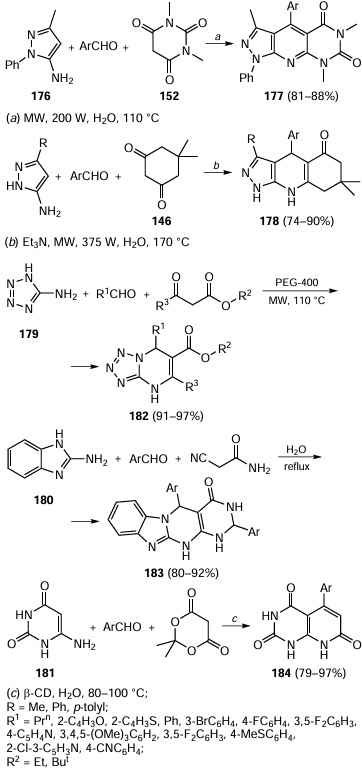
An approach to pyrido[2,3-d:6,5-d']dipyrimidines 186 is based on the reaction between two equivalents of barbituric acid 185 or its thio analogues with amines and aldehydes in an aqueous medium (Scheme 120).[703]
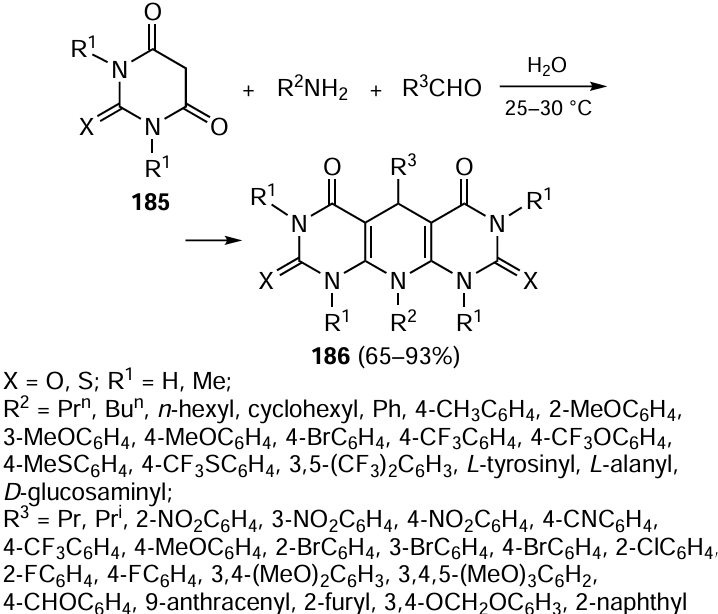
The three-component reactions of aromatic (187) or heteroaromatic (188) amines with aldehydes or cyclohexanediones efficiently proceed in ethanol[704] (Scheme 121) or in ionic liquids[705] to give annulated heterocycles 189 or 190. An ionic liquid can serve as a catalyst. For example, 1,3-disulfonic acid imidazolium hydrogen sulfate ([dsim]HSO4) is a catalyst for the synthesis of uracil derivative 190.[706] Ionic liquids based on calixarene modified with imidazolium moieties ([Cmim]HSO4) efficiently catalyze multicomponent syntheses of polyfunctional pyridine derivatives.[707]
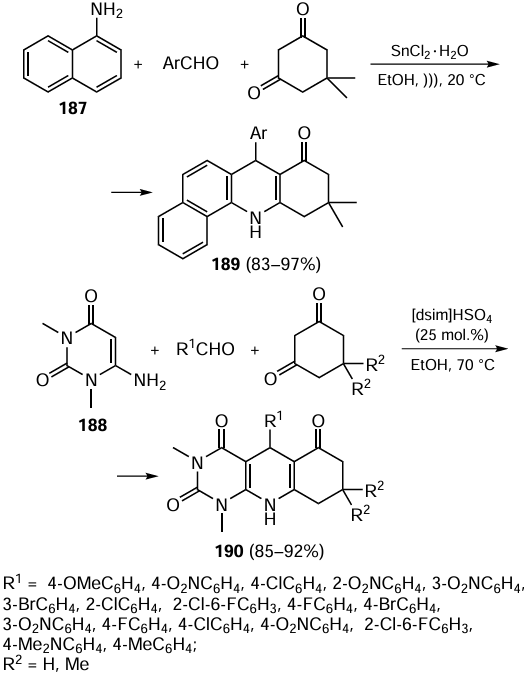
3.5.3. Multicomponent reactions of in situ-generated imines with nucleophiles
Imines are among the most popular substrates for multicomponent reactions, which is reflected in a number of reviews.[708-711] The Kabachnik – Fields reactions between amines, carbonyl compounds and dialkyl H-phosphonates 169 or trialkyl phosphites 191 catalyzed by single-walled carbon nanotubes containing iron nanoparticles result in the formation of α-amino phosphonates 192 (Scheme 122).[712] A similar transformation takes place in the presence of IRMOF-3, which is described as Zn4O(H2N-TA)3 , where TA is the 2-aminoterephthalic acid residue.[713]
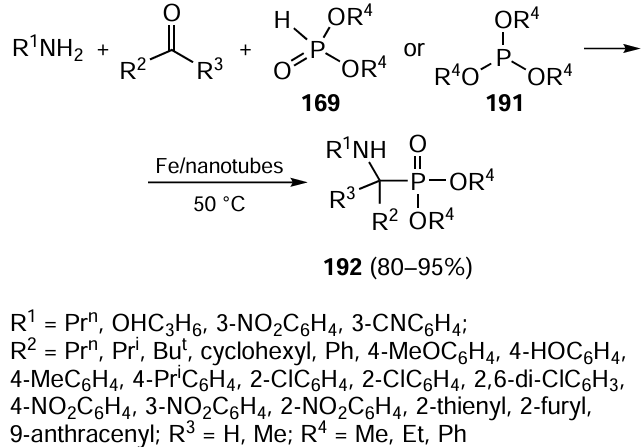
An efficient synthesis of propargylamines 195 was carried out via C – H activation of alkynes 193 in the presence of aldehydes and cyclic amines 194. The catalyst, Zn(OTf)2, was easily separated from the reaction product and could be reused.[714] 2-(Indol-3-yl)quinolines 197 were obtained by the reaction of 3-formylindoles 196 with anilines and arylacetylenes in a mill. The compounds synthesized in this way are of interest for their photophysical properties (Scheme 123).[715]
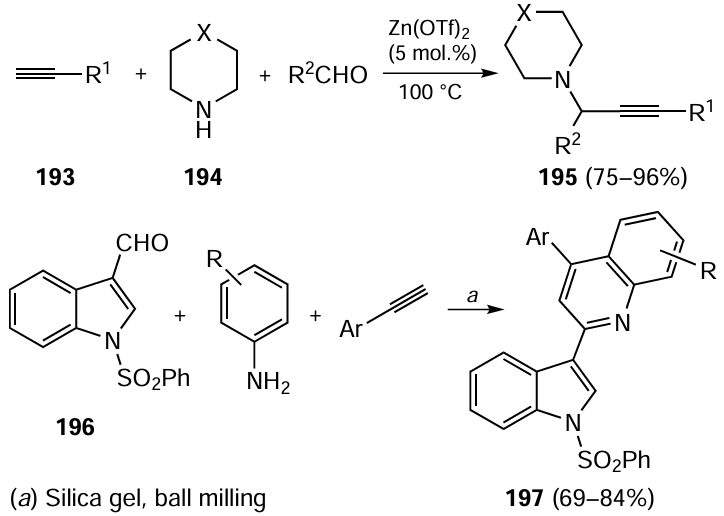
An interesting synthesis of oxazolo[5,4-b]indoles 201 is based on the multicomponent reaction involving arylglyoxals 198, enaminones 199 and amino acids 200. The reaction proceeds with high diastereoselectivity and leads to target products in up to 89% yields (Scheme 124).[716] The condensation of phenylglyoxal monohydrate with arylamines and 2-aminopyridine derivatives in the presence of iodine as a catalyst produces imidazo[1,2-a]pyridines 202. A shortcoming of the reaction is the inapplicability of aliphatic amino acids.[717]
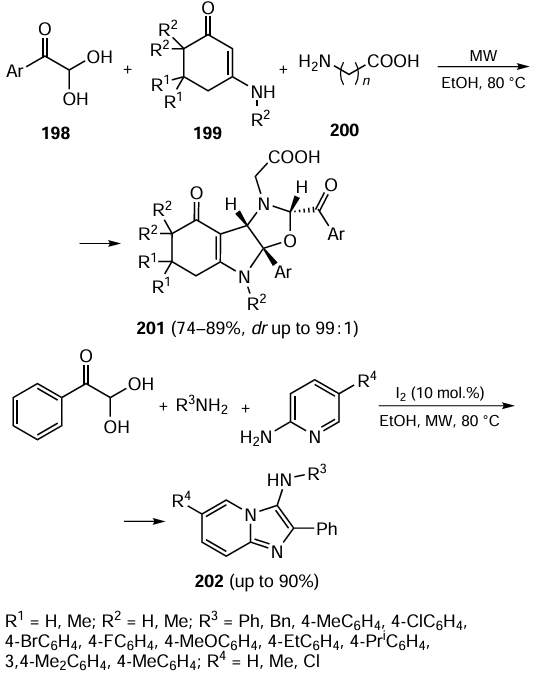
The reaction of isatins 203 with benzaldehydes and ammonium acetate catalyzed by zirconia nanoparticles furnishes the cyclic imidazo[4,5-b]indole system 204. Owing to the high biological activity of compounds 204, this strategy may find extensive use in medicinal chemistry (Scheme 125).[718]
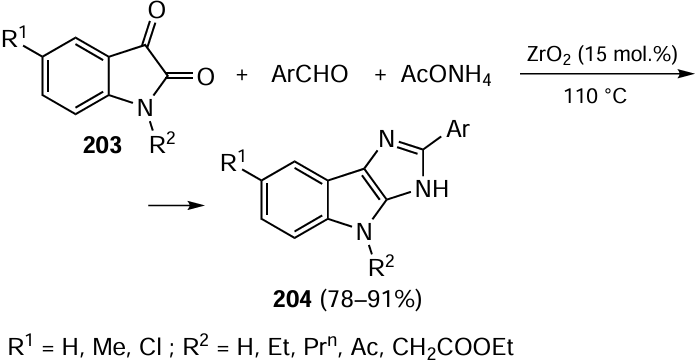
The hetero-Diels – Alder reaction of Schiff bases with imines generated in-situ from benzaldehydes and β-naphthylamine 205 opens up an access to 2,4-diaryl-1,2,3,4-tetrahydrobenzo[ f ]quinazolines 206.[719] A large synthetic potential is also inherent in the cascade reaction of 6-aminoquinoline 207 with 4-hydroxycoumarin 166 and aromatic aldehydes, which takes place in water and follows the formal [3 + 3]-annulation pathway giving polynuclear heterocycles 208 (Scheme 126).[720] Both reactions are accelerated under microwave irradiation.
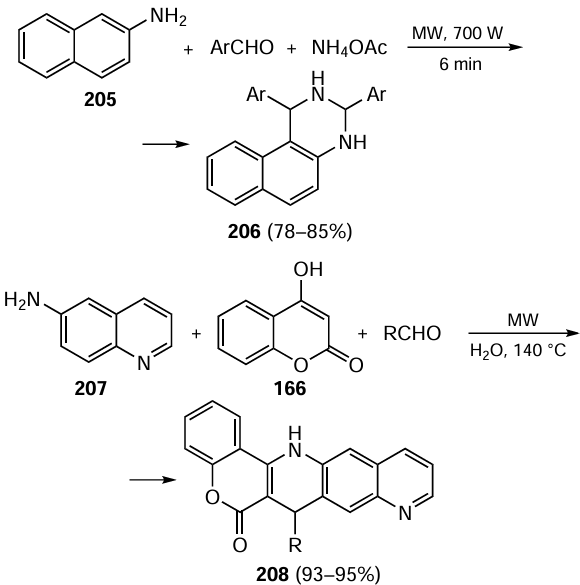
Triethylammonium hydrogen sulfate, a cheap and readily available ionic liquid, was used as a green catalyst for the condensation of 4-hydroxyquinolin-2-one 209 with formaldehyde and amines.[721] The reaction products, [1,3]oxazino[5,6-c]quinolin-5-one derivatives 210, were formed in aqueous ethanol in up to 92% yields (Scheme 127).
β-Naphthols 150 can be converted to α-amidomethyl derivatives 211 via the three-component Mannich reaction with benzaldehydes and carboxylic acid amides in the presence of maltose as an organocatalyst.[722] A similar transformation can be accomplished using β-cyclodextrin-butanesulfonic acid (β-CD-BSA) as the catalyst (Scheme 128).[723]
The three-component condensation of 4-hydroxycoumarin 166 with aldehydes and 2-aminobenzothiazole derivatives 212, resulting in the formation of thiazolo[3,2-a]chromeno[4,3-d]pyrimidin-6(7H)-ones 213, was conducted under solvent-free conditions. 1-Butyl-3-methylimidazolium tetrafluoroborate ([Bmim]BF4) used as the catalyst was easily separated from the reaction mixture and could be reused in the same reaction five times without the loss of activity (Scheme 129).[724]
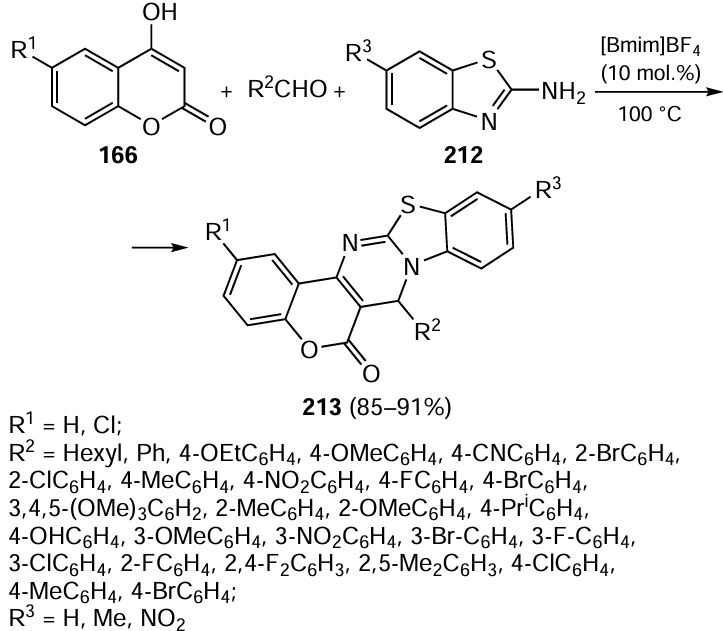
Polyphosphoric acid (PPA) is an efficient activator of multicomponent reactions. In the presence of this acid, arylhydrazines 214 reacted with 2'-aminoacetophenones 215 and alkylating agents 216 to give isocryptolepin alkaloids 217 in one synthetic step (Scheme 130).[725] The replacement of carbonyl compounds and their analogues 216 by nitroalkanes 218 increased the yield of products and allowed the reaction temperature to be reduced.[726] Compounds 217 are known for their antimalarial properties. Some of the products showed anticancer properties.[727]
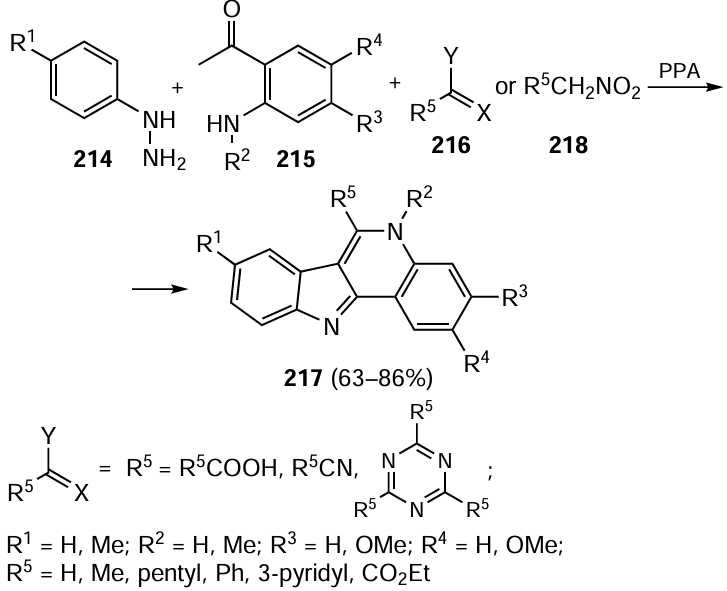
3.5.4. Reactions of α,β-unsaturated carbonyl compounds generated in situ
α,β-Unsaturated carbonyl compounds are key building blocks widely used in organic synthesis, which is reflected in numerous name reactions involving these compounds such as Baylis – Hillman[172] and Nazarov[728] reactions and Robinson annulation reaction.[729]
The combined use of the aminosugar chitosan and the [bmim]OH ionic liquid enables the three-component condensation of benzaldehydes with 1-methylpiperidin-4-one 219 and 1-(2-oxo-2-arylethyl)-1-pyridinium bromides 220, which affords furo[3,2-c]pyridine derivatives 221 (Scheme 131).[730]
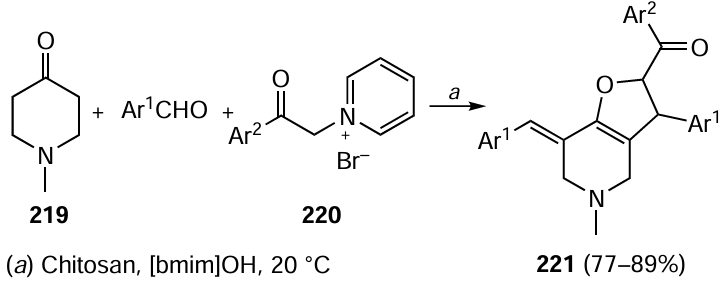
2-Acetylpyridine 222 condenses with benzaldehyde and guanidine carbonate 223 without a catalyst. The condensation products 224 are of interest as polydentate ligands (Scheme 132).[731]
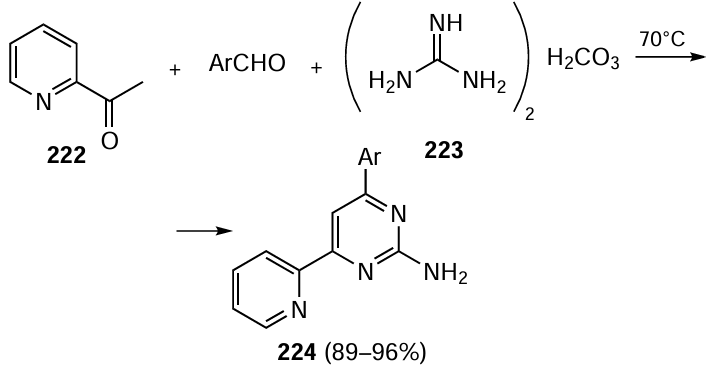
A highly efficient stereoselective synthesis of CF3-oxazinoquinolines was developed on the basis of reaction between CF3-ynones 225 and quinolines 226. Depending on the amount of ynone used, the reaction affords either compound 227 or compound 228 (Scheme 133). This reaction is 20 times faster in water than in organic solvents and gives products that do not require chromatographic purification in almost quantitative yields. This reaction can also be accomplished for other azines, e.g., pyridines 229, isoquinolines and naphthyridines. Heterocycles 230 can serve as useful building blocks for the preparation of other valuable products.[732-737]
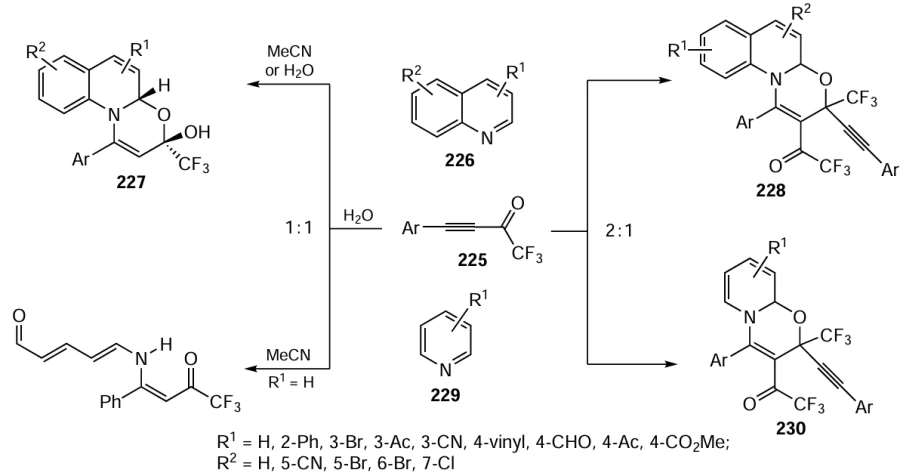
3.5.5. Multicomponent reactions based on isocyanides
The multicomponent Ugi[738] and Passerini[739] reactions have been the subjects of recent reviews and books.[740] The described use of these reactions in the synthesis of biologically active products[741] has stimulated interest of researchers.[742-744] There are examples in which reactions of this type were performed under green conditions. For example, the Ugi reaction, in which levulinic acid 231 reacts with amines and isonitriles 232, tskes place in water and gives pyrrolidin-2-one derivatives 233 in up to 95% yields (Scheme 134).[745] Subsequently, reactions of this type were carried out using choline chloride and urea eutectic mixtures[746] and in a twin-screw extruder in the absence of a solvent.[747] In the presence of Co(III) complex with a chiral ligand, the products of Ugi (234) and azido-Ugi (235) reactions were formed with high enantioselectivity.[748]
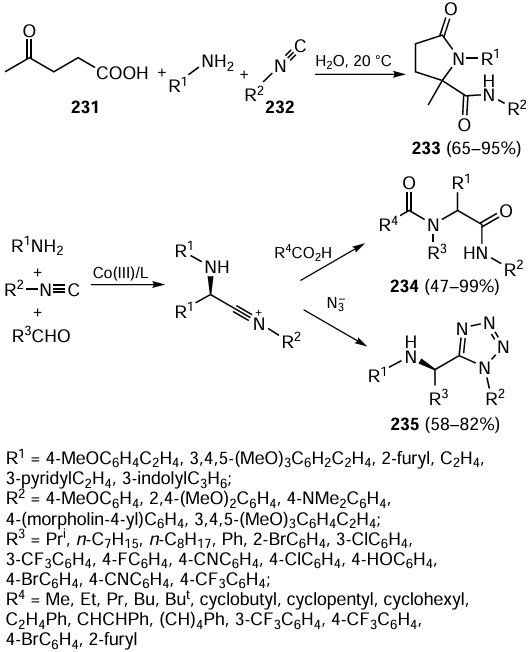
A green synthesis of amido esters 236 by the Ugi reaction in the [bmim][BF4] ionic liquid has been reported.[749][750] The three-component Passerini reaction of isatin 203, para-bromobenzoic acid and isonitriles 232 was carried out in an aqueous medium in a ball mill; isatin derivatives 237 were formed under these conditions in excellent yields.[751] This reaction can also be carried out without a solvent.[752] The Passerini reactions involving azides smoothly proceed in aqueous methanol under ultrasonic treatment to yield α-hydroxymethyltetrazole derivatives 238 (Scheme 135).[753]
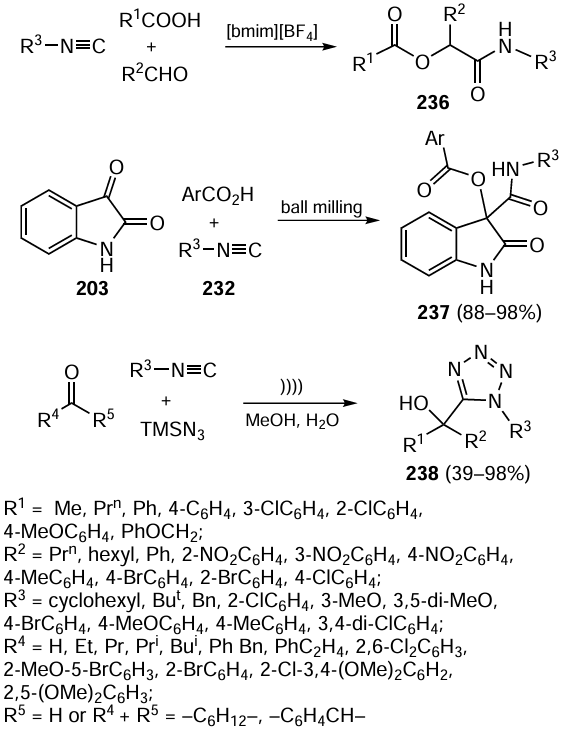
Imidazo[1,2-a]pyridines 239 can be easily obtained by three-component Groebke condensation using the Yb(OTf)3 catalyst, which can be reused after the reaction (Scheme 136).[754] A similar process providing the synthesis of benzimidazolo-imidazo[1,2-a]pyridines 239a was carried out in the presence of TsOH as the catalyst under mechanical activation[755] or in the presence of Sc(OTf)3 .[756]
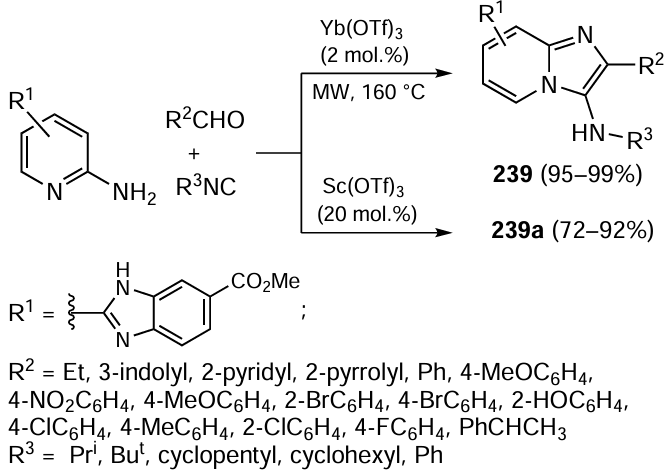
3.5.6. Multicomponent reactions of diamines with dicarbonyl compounds
Diamine reactions are widely used in the synthesis of heterocyclic compounds. Some of these reactions have been accomplished under green conditions. For example, the ultrasonically assisted condensation of o-phthalaldehyde, TMSCN and amidines in the presence of magnetic iron nanoparticles allowed the synthesis of fused heterocycles 240 in up to 92% yields (Scheme 137).[757]
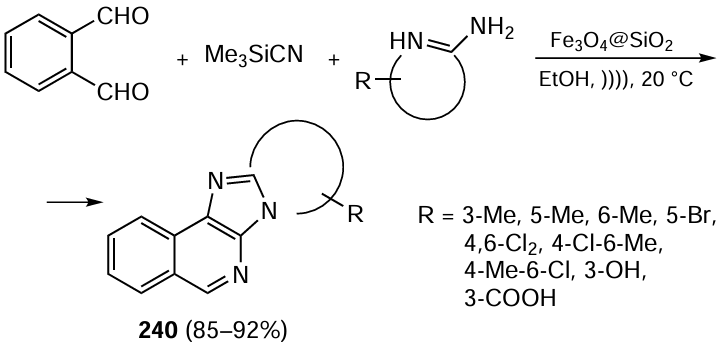
Sodium lauryl sulfate (SLS), a popular low-toxicity surfactant, proved to be an efficient catalyst for three- and four-component condensation of benzil 241 with aromatic aldehydes, ammonium acetate and aniline derivatives, or without aniline derivatives. This reaction takes place in water under heterogeneous conditions and affords imidazoles 242 in high yields (Scheme 138).[758]
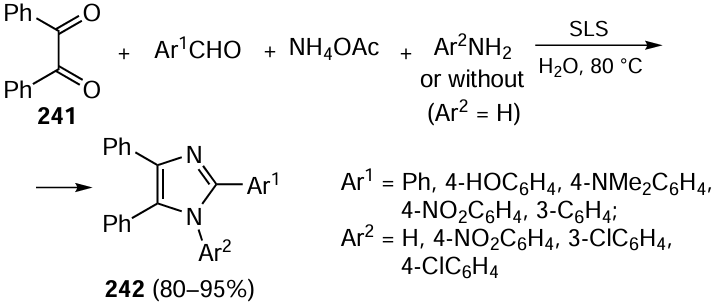
The cascade Biginelli condensation of 1,3-dicarbonyl compounds with aldehydes and urea (thiourea) 243 giving dihydropyrimidinones 244 was accomplished in the presence of 1-(3-sulfopropyl)pyridinium salt of phosphotungstic acid ([PyPrSA]3PW12O40) (Scheme 139).[759] A similar transformation is catalyzed by 1,3-bis(carboxymethyl)imidazolium chloride ([BCMIM]Cl),[760] Cu(OTf)2 (Ref. [761]) and Fe3O4 nanoparticles, which can subsequently be easily recovered from the reaction mixture with a magnet and reused.[762]
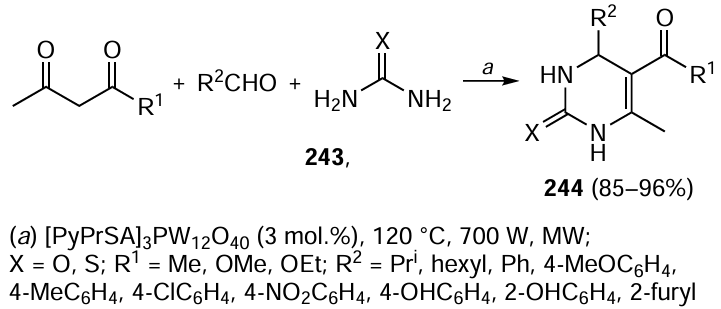
The condensation of 1,2-phenylenediamines 245 with aldehydes and tetronic acid 174 gives benzo[ f ]azulen-1-one 246. The reaction proceeds in a weakly acidic aqueous medium and, in the case of unsymmetrical diamines, it is highly regioselective (Scheme 140).[763]
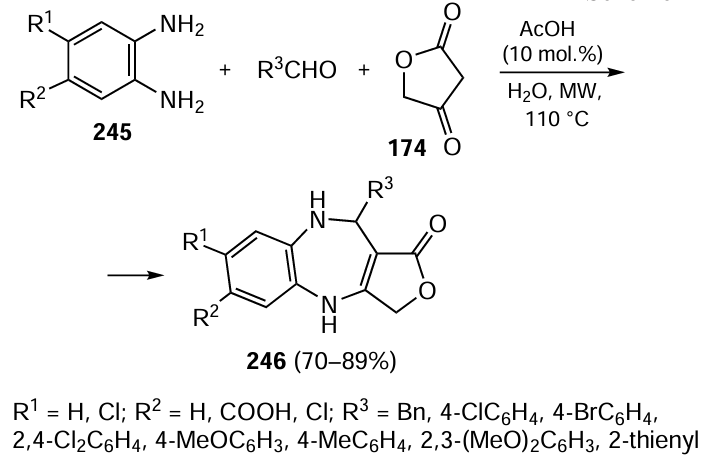
3.5.7. Spirocyclizations based on isatin and ninhydrin
Isatin is rightly considered to be a priviliged structure for organic synthesis. It can be involved in ring expansion reactions,[764][765] Friedel – Crafts alkylation,[766][767] aldol condensation,[768][769] etc. A major part of isatin chemistry are spirocyclization reactions, which are usually multicomponent.[770][771] Many of them proceed under green chemistry conditions. For example, one-pot spirocyclization involving isatin derivatives 203, tetronic acid 174, 2-hydroxy-1,4-naphthoquinone 247 and ammonium acetate to give polynuclear heterocyclic product 248 was implemented in ethanol and catalyzed by TsOH with ultrasonic assistance (Scheme 141).[772]
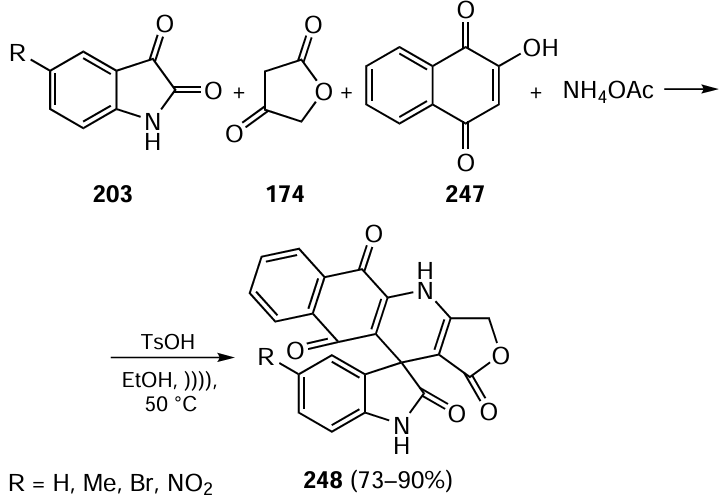
The reaction of isatins 203 with malononitrile and 1H-pyrazol-3-amines 176 in water without a catalyst furnishes spirocyclic product 249, which may then undergo ring expansion on treatment with alkali to give benzo[c]pyrazolo[2,7]naphthyridines 250, which are of great interest for medicinal chemistry[773][774] (Scheme 142). Spiro[indoline-3,4'-pyrazolo[3,4-b]pyridines] 251 were obtained via a similar transformation involving isatins 203, 3-aminopyrazoles and cyanoacetic esters. A distinctive feature of this reaction is catalysis by NaCl; however, the reaction diastereoselectivity is moderate (~3 : 1).[775] Heterocycles 253 were prepared by ultrasound-assisted four-component spirocyclization of isatins 203 with activated acetylenes 252, malononitrile and arylhydrazines catalyzed by L-proline in aqueous ethanol.[776]
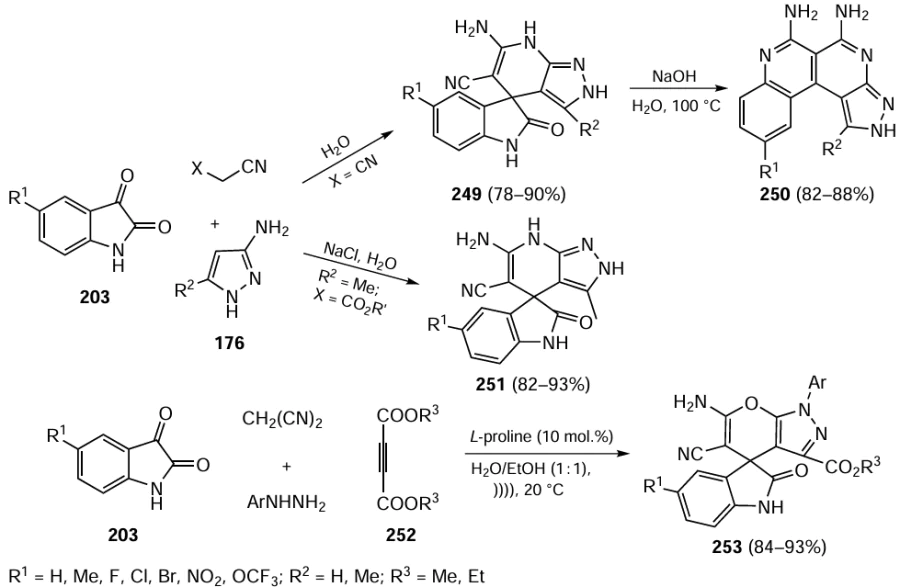
The pseudo-five-component reaction of isatins 203 with 1,1-bis(methylthio)-2-nitroethylene 254, 1,3-diketones and ammonia was utilized for the atom-economic synthesis of spiroheterocycles 255 (Scheme 143). This environmentally benign one-pot transformation catalyzed by para-toluenesulfonic acid was carried out in water.[777]
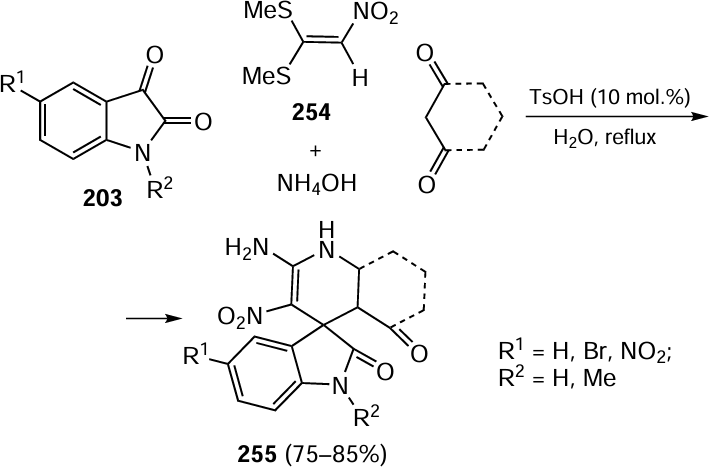
The reaction of isatins 203 with esters of acetylenedicarboxylic acid 252 and pyridine derivatives proceeds in ethanol under mild conditions as the formal [4+2]-cycloaddition to give spiro adducts 256. This approach is diastereoselective. In the case of ultrasonic assistance, the product yields reach 97% (Scheme 144).[778]
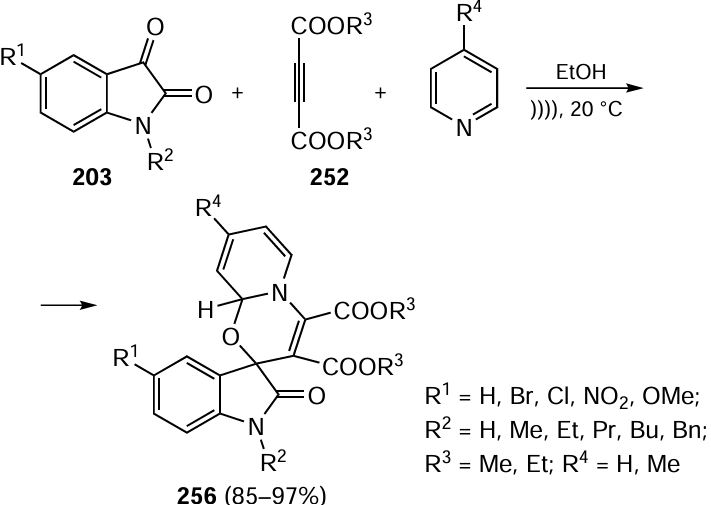
Also, the ultrasonic assistance proved to be effective in the condensation of isatins 203 with 4-hydroxycoumarin 166 and 1H-pyrazole-5-amines 176, which proceeds in water in the presence of a sub-stoichiometric amount of TsOH and results in the formation of spiro[indoline-3,4'-pyrazolo[3,4-b]pyridine]-2,6'(1'H)-diones 257 (Scheme 145).[779]
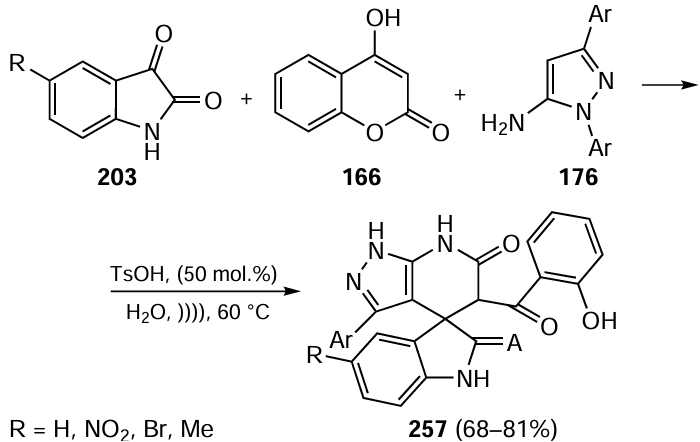
Spiro[chromene-4,3'-indolines] 258 can be prepared under very simple conditions without the use of any catalysts or promoters by hand grinding of a mixture of isatins 203, enolyzable 1,3-dicarbonyl compounds and malononitrile or ethyl cyanoacetate in a mortar (Scheme 146).[780]This process affords products in high yields via one experimental step. The three-component reaction between isatins, enaminones 259 and 1,3-diketones was performed in a similar way by mixing the reactants in a mortar and furnished indole derivatives 260 in 86 – 93% yields.[781]
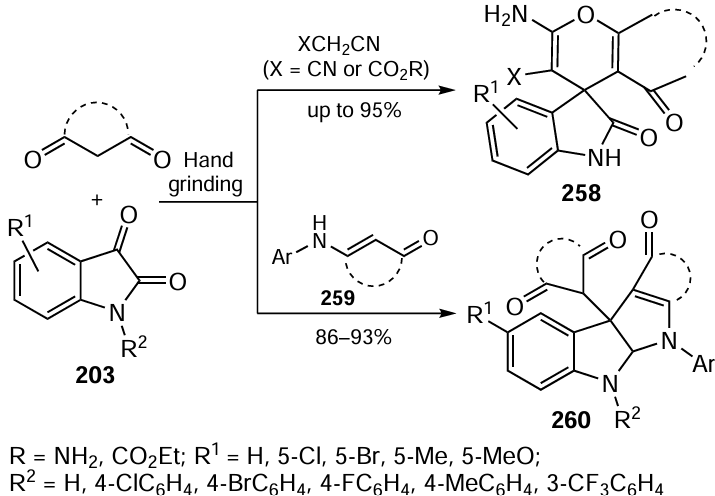
The four-component condensation of ninhydrin 261 with 1,2-phenylenediamine 245, proline and β-nitrostyrenes 263 affords valuable spirocyclic compounds 264. The reaction proceeds via the formation of azomethine ylides 262 followed by 1,3-dipolar cycloaddition, which ensures a high diastereoselectivity (Scheme 147).[782]Some of products 264 had 20 times higher inhibitory activity against acetylcholinesterase than galantamine.
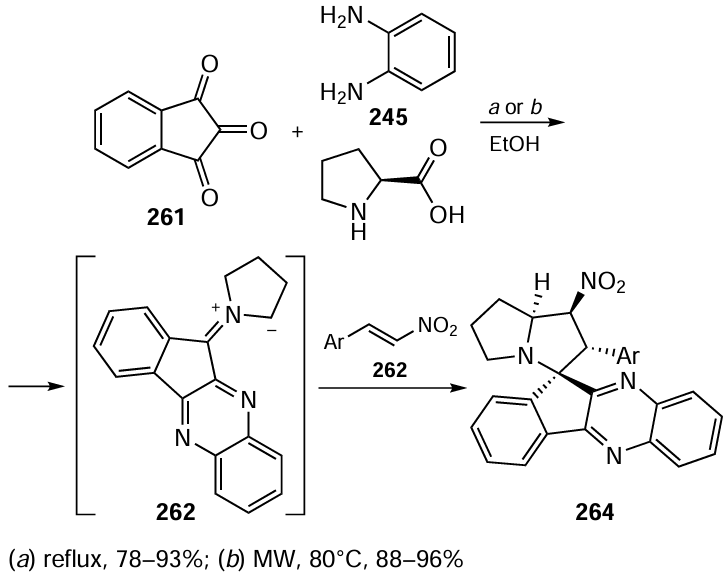
Sulfonic acid deposited on the surface of nano-sized titanium served as a green catalyst for solvent-free multicomponent reactions of isatins. This recyclable catalyst was used for the condensation of isatin derivatives 203 with mercaptoacetic acids 265 and 3-amino-5-methylisoxazole 266 to give isoxazolyl-spiro-thiazolidinones 267 (Scheme 148).[783]
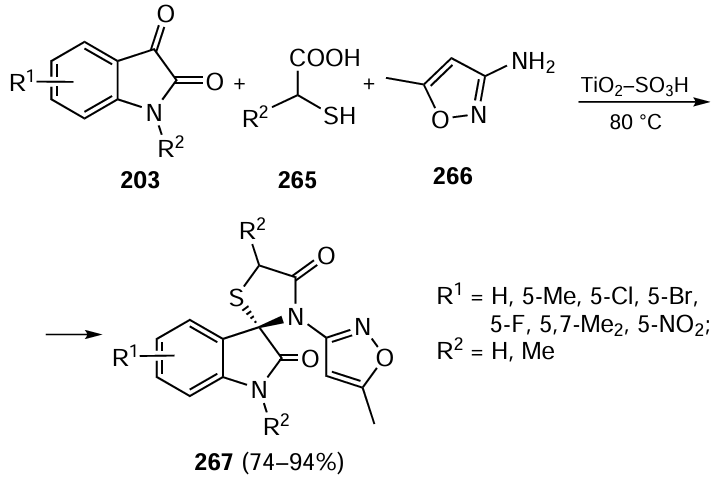
3.5.8. Electrocatalytic cascade and multicomponent reactions
The electrochemical method has occupied an important place among environmentally benign, adaptable and resource-saving methods of organic synthesis.[521][784][785] Electrocatalytic cascade[786][787] and multicomponent[788][789] reactions form a vigorously developing trend of modern organic electrosynthesis. A distinctive feature of these reactions is that the electrochemically generated species catalyze the subsequent reactions.[516]
The electrolysis in an undivided cell makes it possible to perform catalytic processes under the action of basicity gradient in the cathodic region, with the whole system being electrically neutral. The reaction is initiated by alkoxide anions electrochemically generated at the cathode, which act as nucleophiles and are regenerated during the subsequent transformations.[789] The current efficiency for the formation of target compounds markedly exceeds 100%, being as high as hundreds or thousands of percent. This is of most interest for practice as regards energy saving. Other benefits of the electrochemical generation of anions are related to the absence of chemical deprotonation agents in the reaction systems.
The first reaction of this type is the electrocatalytic transformation of 1,1,2,2-tetracyanocyclopropanes 268 into bicyclic pyrrolines 269.[790] The cyclic pyrrolines were obtained in 70 – 95% yields with 700 – 1900% current efficiencies, while the time of electrolysis amounted to 4 – 8 min (constant-current electrolysis) (Scheme 149).
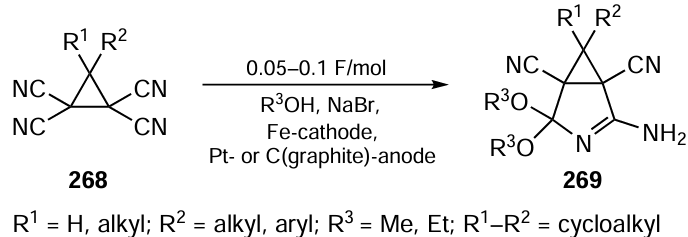
The reaction starts with the generation of the alkoxide anion at the cathode; this anion attacks the carbon atom of the CN group to give N-centred anion Int22 . The subsequent cyclization, protonation of intermediate Int23 thus formed, the addition of one more alkoxide anion and the protonation of intermediate Int24 give rise to pyrroline system 269. The alkoxide anion formed at the final step initiates the next catalytic cycle (Scheme 150).
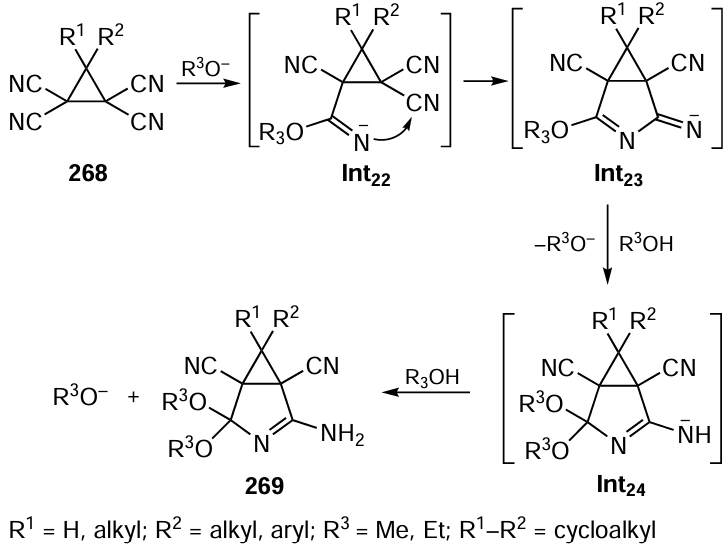
A variety of polycyclic pyrrolines 270 – 272 were obtained by this method in 75 – 90% yields with 400 – 470% current efficiency. When there was a substituent in the cyclohexane ring, the reaction was stereoselective. The yield of tricyclic products 271 were 50 – 90% yields with and the current efficiency of 260 – 460% (Scheme 151).[791] Similarly, 2,2-dicyano-1,1-dicarboxylic acids were stereoselectively converted to substituted pyrrolidones 272 in 80 – 95% yield with 400 – 480% current efficiency.[792]
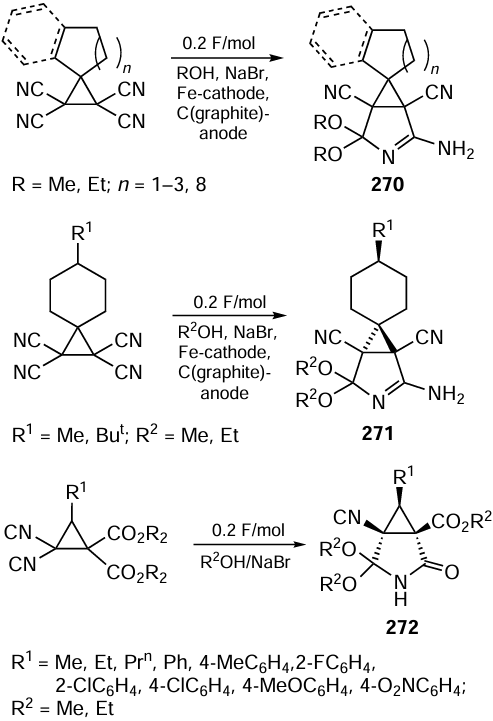
The electrochemically induced catalytic transformation of this type was successfully applied in a cascade stereoselective synthesis of pyrrolines 273 from benzaldehyde derivatives and two malononitrile molecules (Scheme 152).[793] A similar approach was used for the multicomponent electrocatalytic stereoselective synthesis of pyrrolidones 274, which contain an ester group.[794][795]
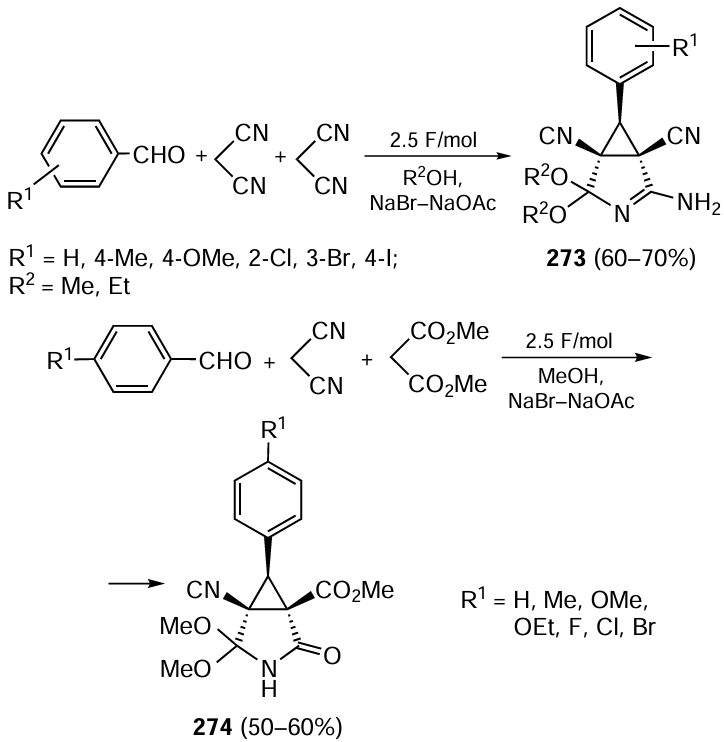
This method was useful for the electrocatalytic transformation of salicylaldehydes and malononitrile into 2-amino-4H-chromenes 275, which were obtained in 85 – 95% yields, with the current efficiency being 1700 – 1900% in 15 min of electrolysis (Scheme 153).[796] Later, a pseudo-four component process of this type was implemented to give tricyclic compounds 276 from simple molecules in one-pot reaction.[797] Various 4H-chromene systems 277 were prepared in 65 – 90% yields from salicylaldehyde derivatives and two different C – H acids, with the current efficiency of 320 – 1900%.[798]
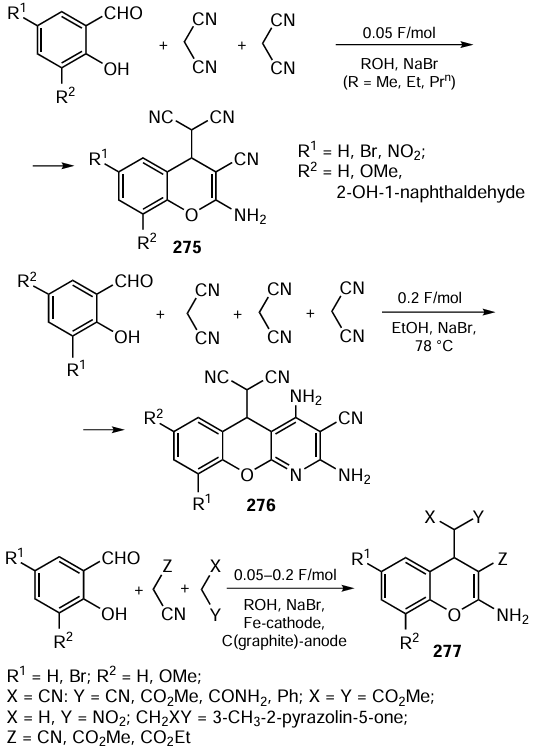
The same electrocatalytic system was successfully utilized for the reactions of isatins 203 with two C – H acids, one being malononitrile and the other being cyclic 1,3-diketone,[799] pyrazolone,[800] barbituric acid,[801] 4-hydroxyquinolin-2(1H)-one[802] or kojic acid.[803] This resulted in the synthesis of diverse spirocyclic structures 278 (Scheme 154). The reaction mechanism includes deprotonation of the alcohol at the cathode to give the alkoxide anion. The reaction between the alkoxide anion and malononitrile in the solution affords the malononitrile anion. Then a typical multicomponent reaction takes place in the solution. The addition of the malononitrile anion to isatin yields Knoevenagel adduct 279. The Michael addition of the anion of cyclic C – H acid followed by intramolecular cyclization yields spirocyclic compounds 278, with the alkoxide anion being regenerated in the latter step. This alkoxide anion starts a new cycle of the chain process by reacting with the next malononitrile molecule.
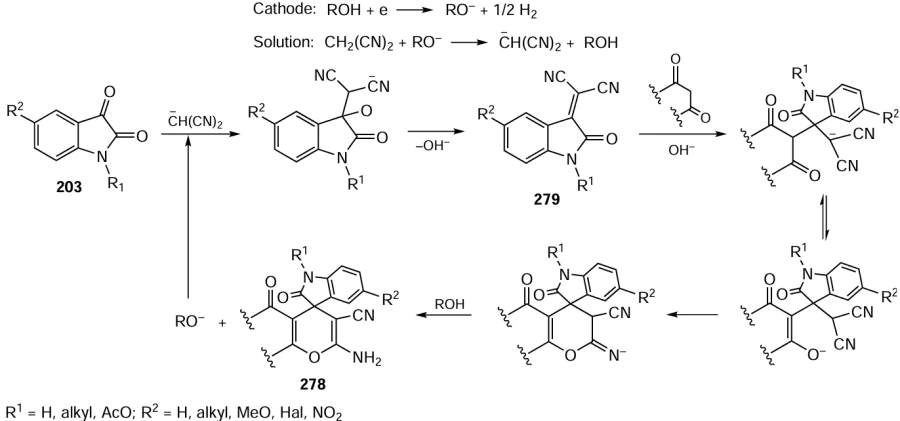
Electrochemical reactions of this type were also carried out with aromatic aldehydes, which reacted with malononitrile and cyclic 1,3-diketones 154 to give 4H-chromene-3-carbonitriles 280 (Scheme 155). The current efficiencies were 2800 – 3200%.[804] Analogous multicomponent transformations were also carried out for other C – H and N – H acids, which resulted in the synthesis of heterocycles 281 – 284.[805-808] Electrochemically induced multicomponent reactions of benzaldehydes with two, in some cases different, heterocyclic C – H acids have also been described.[809-813]
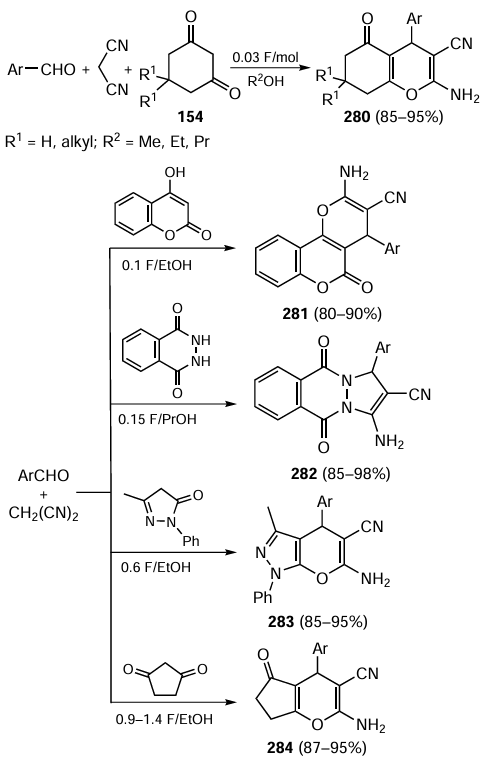
Mediated electrochemical oxidation or reduction is a trend of organic erlectrosynthesis that has been vigorously developed in recent years.[814] A considerable benefit of using mediator systems is the increase in the reaction selectivity and rate owing to a combination of chemical and electrochemical transformations.[815] The use of mediators often makes it possible to decrease the electrode potential and to conduct the process at high current densities, which decreases the energy expenditure and facilitates the control over such processes.[524] In some cases, the use of mediator systems enables targeted electrocatalytic selective transformations of organic compounds that are inactive under direct electrochemical processes.[816] In particular, this provided the development of a conceptually new approach to the cyclopropane ring construction based on co-electrolysis of CH acids and activated olefins in the presence of mediators.[816]
Halides are the best known and popular inorganic mediators.[515] The I–/I2 and Br–/Br2 pairs in which iodine and bromine electrochemically generated at the anode serve as oxidants are mediator pairs that are in high demand in organic chemistry.[817] Two catalytic cycles occur in an undivided cell with halide salts as mediators in alcohols. The alkoxide anion formed at the cathode reacts with a substrate, usually C – H acid, to form C – H acid anion A (Scheme 156). Halogens are generated at the anode and, as a result of oxidative process in solution, they react with substrate anion A to form halo derivative 285. Compound 285 is converted in solution to anion B, which initiates a series of subsequent cascade or multicomponent reactions. This approach was used to carry out linear trimerization[818] and cyclotrimerization of C – H acids to give cyclopropanes[819] in an undivided cell.
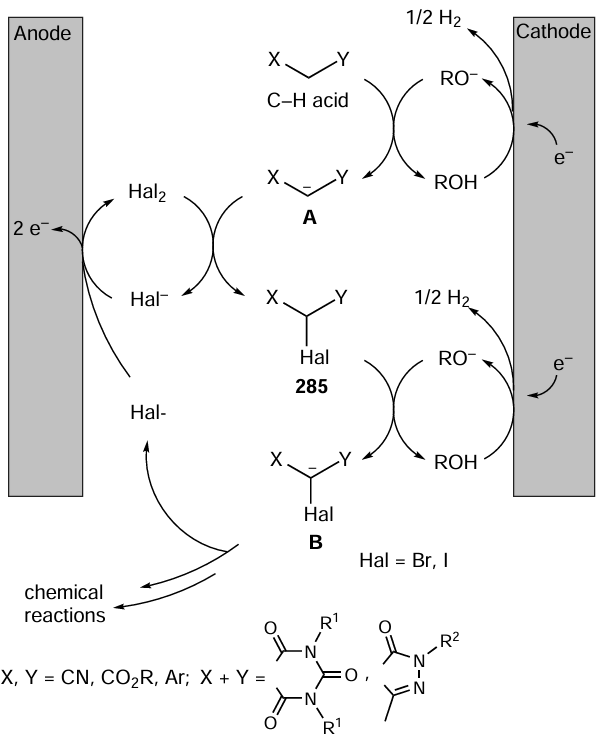
Under similar conditions, cascade electrocatalytic synthesis was performed to obtain cyclopropanes and spirocyclopropanes from activated olefins and C – H acids[794][820] and from carbonyl compounds and C – H acids.[821][822] An example is electrocatalytic stereoselective transformation of aromatic aldehydes and two 5-methyl-2-phenyl-2,4-dihydro-3H-pyrazol-3-one molecules to bis-spiropyrazolone cyclopropanes 286 (Scheme 157).[823]
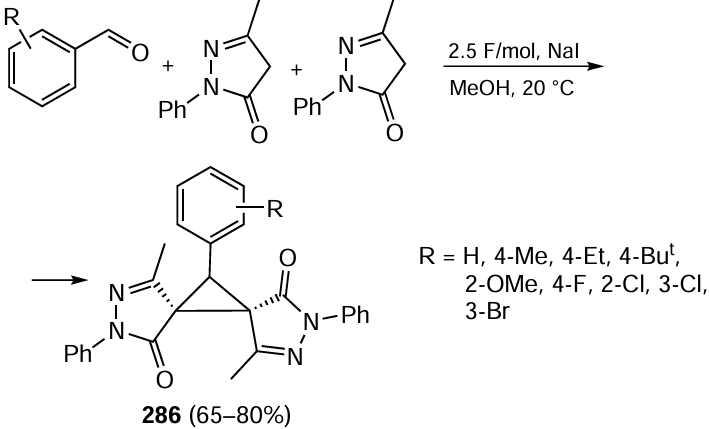
The cascade reaction of heterocyclic ketones 287 with two molecules of malononitrile via co-electrolysis in an undivided cell furnishes 6-heterospiro[2.5]-1,1,2,2-tetracarbonitriles 288 (Scheme 158).[824]
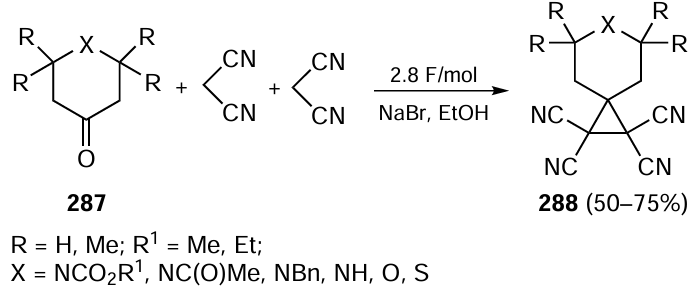
A four-component stereoselective synthesis of substituted pyrrolidones 289 from aldehydes, malononitrile, dimethyl malonate and methanol was performed under similar conditions (Scheme 159).[795]This complex electrocatalytic process includes electrochemically induced condensation of aldehyde with C – H acid to form an activated olefin, olefin cyclopropanation with participation of electrogenerated bromine,[821] and the subsequent stereoselective electrochemically induced chain cyclization of cyano-substituted cyclopropane to give bicyclic compound 289.
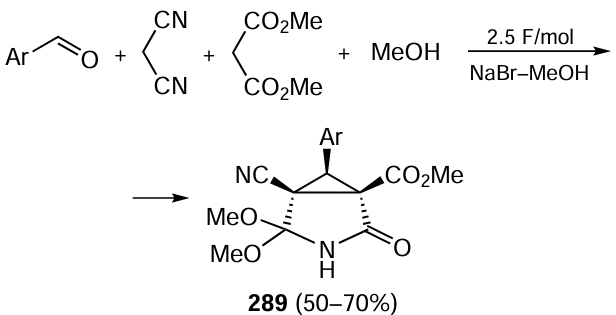
Recently, the electrocatalytic multicomponent transformation of aromatic aldehyde, dimethylbarbituric acid and 4-hydroxy-6-methyl-2H-pyran-2-one 290 was accomplished for the synthesis of spiro[furo[3,2-c]pyran-2,5'-pyrimidine]-2',4,4',6'(1'H,3'H)-tetraones 291 (Scheme 160).[825]
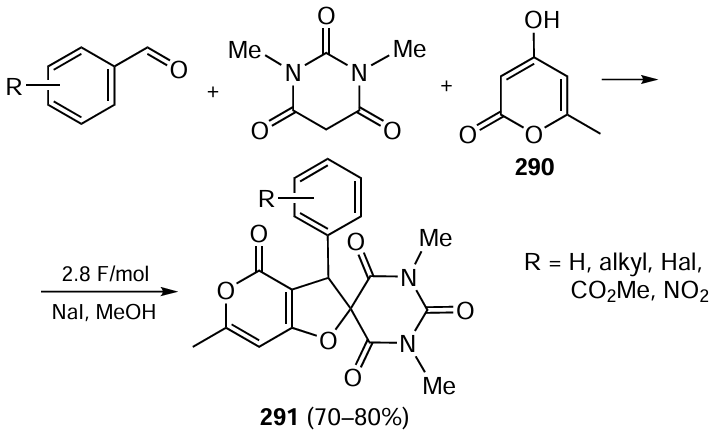
The surveyed methods and processes leave no doubt that multicomponent reactions are a powerful tool of green chemistry. The environmental friendliness of these processes is due to the fact that they markedly reduce resource, energy and labour costs and eliminate the need to isolate and purify intermediate products. In addition, this strategy provides good reproducibility of results, since all reactants are present in the reaction mixture from the very beginning and there is no need to optimize conditions of particular steps. Many reactions of this type are carried out in inexpensive green solvents, including ethanol and water, using advanced green chemistry techniques such as mechanical, ultrasonic, microwave and electrochemical assistance.
3.6. Heterogeneous catalysis of redox green chemistry reactions
The enhanced attention of the world community to human environment and environmental safety culminated in the formulation of conceptual statements of green chemistry by Paul Anatas in collaboration with John Warner in 1998;[2] later, these statements turned into universally recognized trends, known as ‘twelve principles of green chemistry’. According to the original order of these principles, the ninth one is catalysis. However, even the substantiation of the need to include this principle, given by the original authors,[13] affects the eight preceding principles, that is, waste minimization, development of new atom-economic organic reactions, implementation of advanced energy efficient chemical processes, rational use of renewable feedstock, and so on, which are impossible without extensive use of efficient catalysts. That is why, as clearly seen from the preceding sections of the review, catalysis plays an enormous role in modern organic synthesis and is one of the fundamental elements of the modern green chemistry concept.
Although new strategies for homogeneous catalysis have rapidly developed in recent years, most researchers and industrial companies recognize the priority of heterogeneously catalyzed reactions.[826][827] This section is devoted to the application of heterogeneous catalysts in fine organic synthesis.
3.6.1. Catalytic transfer hydrogenation and related reactions of organic synthesis
In recent years, there has been enhanced interest in the reduction reactions, in which one reactant serves as a source of dihydrogen (H-donor), while the catalyst provides both the release of H2 from this donor and the transfer of dihydrogen, followed by the addition of the latter to the second reactant (H-acceptor). This approach to avoiding direct participation of gaseous H2 in reactions has been referred to in the literature as the borrowing hydrogen strategy or borrowing hydrogen methodology (BHM). In some cases, these reactions are also called transfer hydrogenation (TH) or hydrogen-free hydrogenation reactions. The results of studies of some heterogeneously catalyzed TH reactions involving alcohols have been summarized in a recent review.[828] We will consider a number of additional examples of heterogeneously catalyzed TH reactions, which clearly demonstrate the advantages of green chemistry approaches over traditional methods.
It is known that transfer hydrogenation is often catalyzed or promoted by bases and alkalis. However, the use of alkaline solutions contradicts the green chemistry requirements.[829-831] A possible solution to this problem is to design solid alkaline catalysts to be used in synthetic chemistry. These catalysts[832] have already been beneficial for green TH reactions.
The heterogeneous catalysts that possess pronounced basic properties include hydroxyapatites (HAP)[833][834] and hydrotalcites (layered double hydroxides, for example, Mg – Al or Zn – Al hydrotalcites, HT).[835][836] Since hydroxyapatites are less basic than hydrotalcites, they are doped with metals to enhance their catalytic properties. For example, copper-modified hydroxyapatite (Cu0/HAP)[837] or with encapsulated g-Fe2O3 (Ref.[838]) perfectly catalyze the furfural reduction with isopropyl alcohol, in particular, in a continuous flow process (Scheme 161).
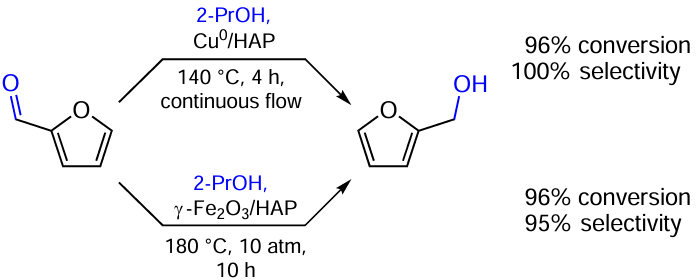
In TH reactions involving other substrates (cinnamaldehyde, 5-hydroxymethylfurfural, ethyl levulinate and acetophenone), the g-Fe2O3/HAP system also proved to be highly selective (80 – 92%) and did not lose catalytic activity after six reuses.[838]
Fatty acid esters are reduced to corresponding alcohols in the presence of metal-substituted hydroxyapatites with a somewhat lower selectivity than aldehydes or ketones. When cobalt-containing hydroxyapatite (Co0/HAP) was used as a catalyst for hydrogen-free hydrogenation of methyl stearate, the selectivity to 1-octadecanol was 68% (Scheme 162.[839] Such selectivity is high enough to use the TH strategy for production of liquid biofuel components from methyl esters of fatty acids. Other TH reactions catalyzed by hydroxyapatite modified with various metals were also reported.[840-842]
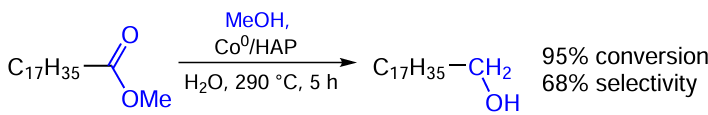
An example of using hydrotalcite is the synthesis of 1-butanol from ethanol by the Guerbet–Markovnikov reaction in a continuous flow reactor with 66% selectivity at moderate conversion (Scheme 163).[843] The synthesis consists of three consecutive steps, that is, dehydrogenation of ethanol to acetaldehyde, self-condensation of acetaldehyde to form crotonaldehyde and exhaustive hydrogenation of the latter to 1-butanol. The hydrogenation at the third step is by means of dihydrogen borrowed from ethanol during its hydrogenation. The hydrotalcite-catalyzed continuous flow process is comparable in efficiency with the corresponding homogeneous reaction carried out in an autoclave and catalyzed by ruthenium complexes under highly alkaline conditions.[844]
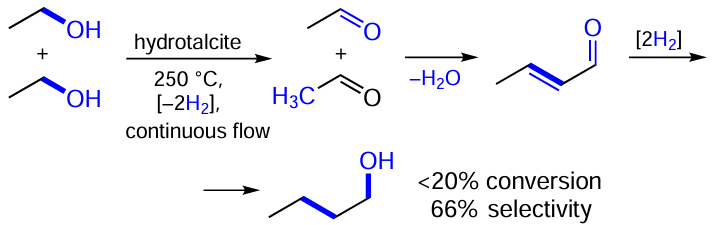
A wider range of cascade reactions can be encountered in organic synthesis based on BHM application such as the preparation of aromatic azaheterocycles from ortho-substituted nitrobenzenes catalyzed by cobalt compounds.[845] An example is the synthesis of substituted benzimidazoles from ortho-nitroanilines catalyzed by copper- and zinc-doped hydrotalcite (Cu – Zn/HT, Scheme 164).[846] In this synthesis, methanol is not only the H-donor, but also C1 source for imidazole formation.[847]
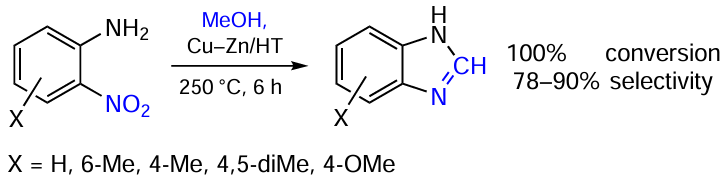
The Ru0/HT-catalyzed reduction of CO2 to formaldehyde[848] and the Fe0 – FeOx/HT-catalyzed reactions to form new C – C bonds[849] confirm the efficiency of using modified hydrotalcites for TH reactions.
One more reason for choosing hydrotalcite instead of SiO2 is its chemical stability towards methanol. It is known that silica-based materials decompose with methanol at near- or super-critical temperature to form tetramethoxysilane,[850] which catalyzes some undesirable side reactions.[851]
The partial transfer hydrogenation of crotonaldehyde to 2-buten-1-ol is quite selective when catalyzed by zirconia – silica mixed oxides. So, the chemoselectivity of this process in a continuous flow reactor in the presence of ZrO2/SiO2 catalyst (1 : 20) reaches 94% at a nearly complete conversion (Scheme 165).[852] This result contradicts the common view that the C=C bond is more reactive than the C=O bond when 2-propanol is used as the H-donor.[853]
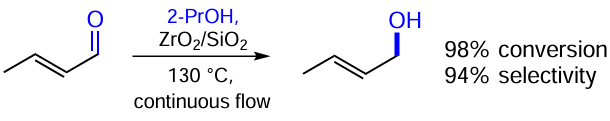
One more example of successful implementation of BHM is one-pot synthesis of N-methylanilines from nitroarenes in methanol (Scheme 166). This synthesis includes four steps: (1) dehydrogenation of methanol to formaldehyde with H2 evolution; (2) stepwise hydrogenation of the nitro group to NH2 group with borrowed H2; (3) condensation of aniline with formaldehyde to form the corresponding imine; and (4) hydrogenation of imine with borrowed H2 . In the reaction catalyzed by Rh-supported covalent organic framework heterogeneous catalyst derived from melamine and terephthalaldehyde, the product yield reached nearly 100%.[854]
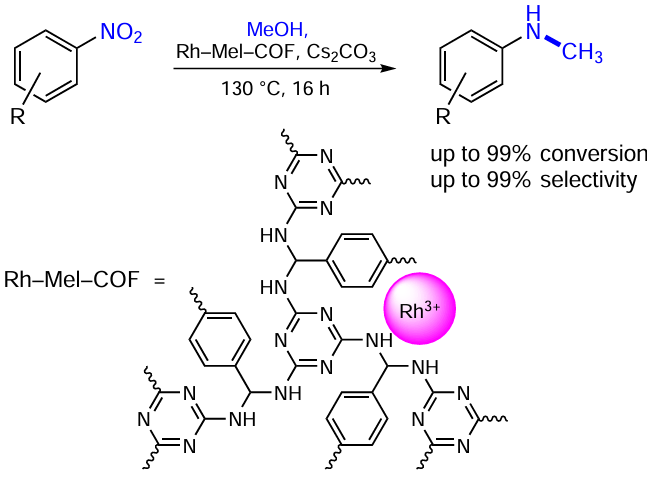
The heterogeneous catalysis for borrowing hydrogen reactions is applied not only to form new C – C and C – N bonds,[855-858] but also to generate C – S bonds. The cobalt molybdenum sulfide (Co – Mo – S) catalysts were most efficient in this case. This is surprising, since in particular Co – Mo – S materials are the main industrial catalysts for hydrodesulfurization of heavy oil feeds and refined oil products,[859] where they assist C – S and C=S bond cleavage. Nevertheless, in the presence of these catalysts, S-alkylation of arylthiols with benzyl alcohols gives arylbenzyl sulfide in a yield of 90% or more (Scheme 167).[860] When arylthiol is replaced by hydrogen sulfide, the reaction under similar conditions gives symmetrical dibenzyl sulfides via double alkylation of hydrogen sulfide with benzyl alcohol.
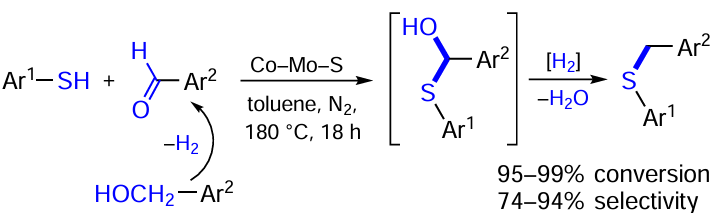
Heterogeneous TH is also useful for the asymmetric organic synthesis, in particular for the enantioselective reduction of acetophenone derivatives to benzyl alcohols (Scheme 168).[861] These reactions were carried out in a continuous flow reactor and were catalyzed by chiral iridium tetramethylcyclopentadienyl (Cp**) η5-complex immobilized on the polystyrene Wang resin.[862] The enantiomeric purity of the products was 78 – 94% ee, indicating high efficiency of the immobilized catalyst.
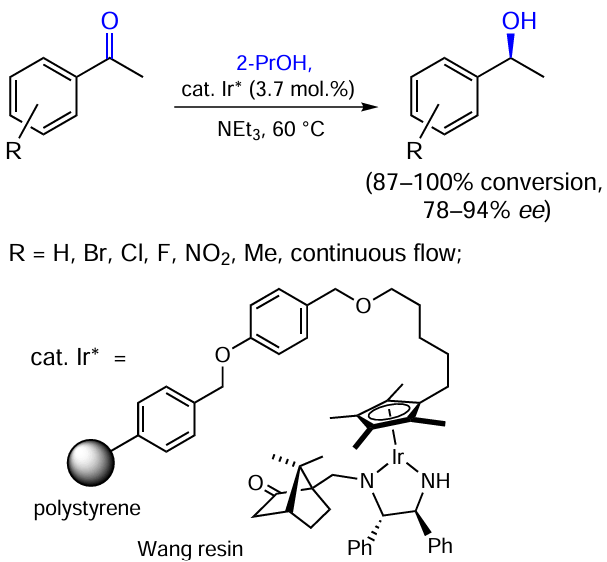
While considering the heterogeneously catalyzed TH in organic synthesis, one cannot pass over the good performance of some traditional hydrogenation catalysts in these reactions. For example the known palladium-on-carbon catalyst proved to be effective in compact portable flow microreactors, with cyclohexene being used as the H-donor.[863] Under proposed conditions, hydrogen-free hydrogenation was carried out for various functional groups (C=C, C≡C, –N3 , –NO2 , –N=N–, –CHO) in the benzene ring; the yields of the target products were 86 – 99%. There are also other examples of successful application of the Pd/C catalyst for TH reactions carried out in continuous flow reactors;[864] in the catalytic performance and compliance with green chemistry principles, these reactions were superior to conventional hydrogenation methods.
3.6.2. Green oxidants in tandem reactions
The projection of green chemistry principles onto oxidative processes in organic chemistry severely limits the choice of oxidants that meet these requirements. Most often, this includes the use of one of the three main oxidants: air (or molecular O2), H2O2 or ButOOH.[865] Co-oxidants such as TEMPO nitroxide may be added to enhance the oxidation ability.[866-868] This review addresses heterogeneous oxidation reactions involving these oxidants, first of all, reactions catalyzed by hydrotalcites.
The partial oxidation of benzyl alcohols Ar – CH2OH to aldehydes, including cascade reactions, in which the aldehydes formed in situ were involved in the subsequent transformations, often used as the test system. Green principles of the preparation of heterogeneous catalysts played an important role in these studies. Finally, a number of effective catalysts for the oxidation of benzyl alcohols have been developed, in particular γ-Al2O3@CeO2 ,[869] hydrotalcite-supported CeO2 ,[870] cerium-doped mesoporous perovskite LaMnO3 ,[871] nitrogen- and phosphorus-doped metal-free carbon spheres,[872] and other catalysts containing no noble metals. The oxidation of alcohol–amine binary systems with air oxygen catalyzed by zirconium-doped manganese oxide MnZr0.5Oy resulted in the convergent synthesis of aniline-imines 292 or benzaldimines 293 depending on the structure of the starting reactants (Scheme 169).[873] In some cases, the yields of imines were nearly quantitative, which attests to efficiency of catalysis for both steps, that is, alcohol oxidation and condensation of the amine with the aldehyde formed in situ.
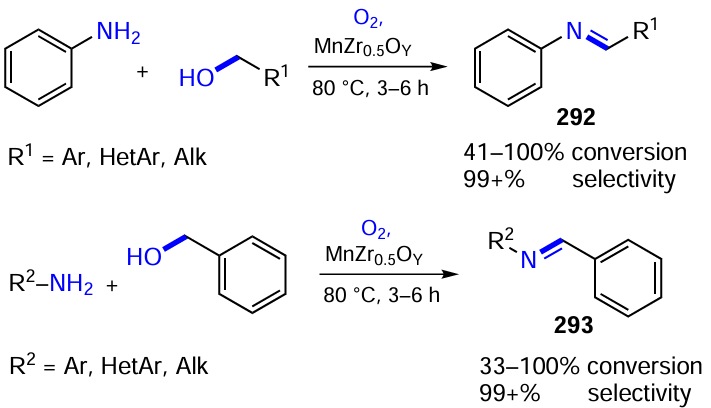
The synthesis of 2-amino-3-cyano-4H-pyran derivative 294 catalyzed by the γ-Fe2O3-immobilized N-(pyridylmethyl)imidazolium salt of tungstic acid, [(γ-Fe2O3 – Im-Py)2WO4], was also performed via a cascade process (Scheme 170).[874] In contrast to the prototype, i.e. the well-studied three-component co-condensation of aromatic aldehydes with malononitrile and dicarbonyl compounds (b-ketoesters and b-diketones), this reaction was carried out with benzyl alcohols oxidized to the corresponding aldehydes with tert-butyl hydroperoxide. This process is a green synthesis not only because of the use of a heterogeneous catalyst and a green oxidant (ButOOH), but also because of the absence of a solvent.
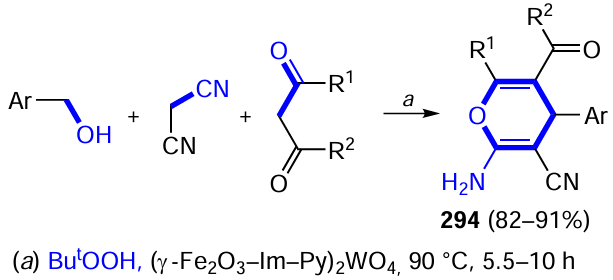
The co-condensation of benzyl alcohols with thioglycolic acid and substituted anilines also involves oxidation of the alcohol to aromatic aldehyde, in this case by molecular oxygen (Scheme 171).[875] The chemoselectivity of the reaction and high yields of 2.3-diarylthiazolidin-4-ones 295 were provided by cobalt aluminium hydrotalcite (Co – Al/HT). This result shows that hydrotalcite-based heterogeneous catalysts are effective not only in reduction, but also in oxidation reactions.
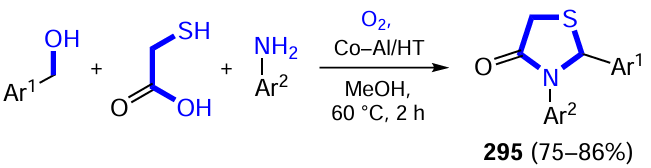
Iron(III) oxyhydroxide α-FeO(OH) supported on magnesium aluminium hydrotalcite (Fe/HT) was applied in a one-pot synthesis of 2-arylquinolines 296 from ortho-aminobenzyl alcohol and aryl alkyl ketones in the reactant medium (Scheme 172).[876] Air oxygen served as the oxidant to convert the alcohol to ortho-aminobenzaldehyde required for condensation. The yield of product 296 (R = H, Ar = Ph) was 91%. No by-products of amine oxidation[877] or oxidative transamination[878][879] were detected.
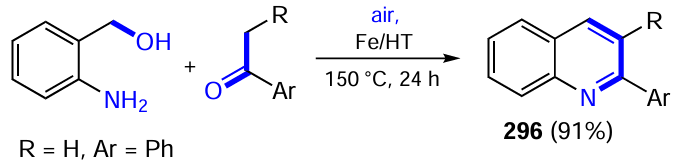
Copper–manganese bimetallic oxides supported on hydrotalcite showed good performance in the aerobic catalytic oxidation of acyloins (a-hydroxy ketones) to a-keto esters 297 (Scheme 173).[880] The proposed synthetic method, which does not include the use of any solvents or auxiliary additives, is suitable for the preparation of esters of natural sterols, e.g., cholesterol ester 298.
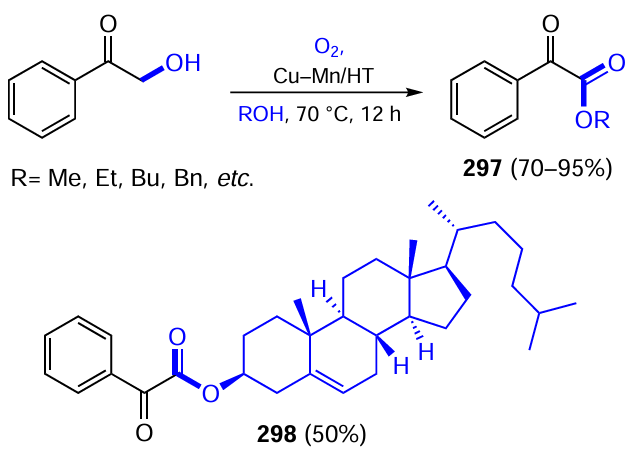
Heterogeneous catalysts can activate C(sp3) – H bonds, usually in relatively inert cycloalkanes towards selective oxidation. In particular, copper nanoparticles supported on Zn – Al hydrotalcite pretreated with a mixture of salicylic acid and NaOH (Cu/ZnAl-HT) proved to be effective. In the presence of this reusable (up to 6 cycles) catalyst, cyclohexane was oxidized with tert-butyl hydroperoxide to a mixture of cyclohexanone and cyclohexanol under relatively mild conditions (6 h, 80 °C). The cyclohexane conversion was 52% and the chemoselectivity to the major products was 97% (Scheme 174).[881] Dodecane, stable under reaction conditions, was used as the solvent. Presumably, this reaction follows a radical chain mechanism; Zn – Al hydrotalcite at least does not interfere with chain propagation.
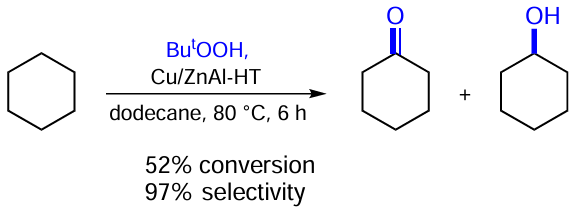
The Cu-BINAP complex supported on Mg–Al hydrotalcite ([Cu(binap)I]2/HT) showed unexpectedly a high catalytic activity in the oxidative C – S cross-coupling of substituted thiophenols with arylboronic acids, resulting in the formation of ortho-hydroxylated thioethers 299 in moderate to high yields (Scheme 175).[882] Molecular oxygen served as the oxidant in this reaction. The heterogeneous catalytic system not only exhibited high chemoselectivity toward various functional and heteroatomic substituents, but could also be re-used five times without losing activity. When potassium persulfate K2S2O8 was added to the system as a co-oxidant, meta-hydroxy- or meta-alkoxy thiophenols were converted to arylthio-1,4-benzoquinones 300 in high yields.
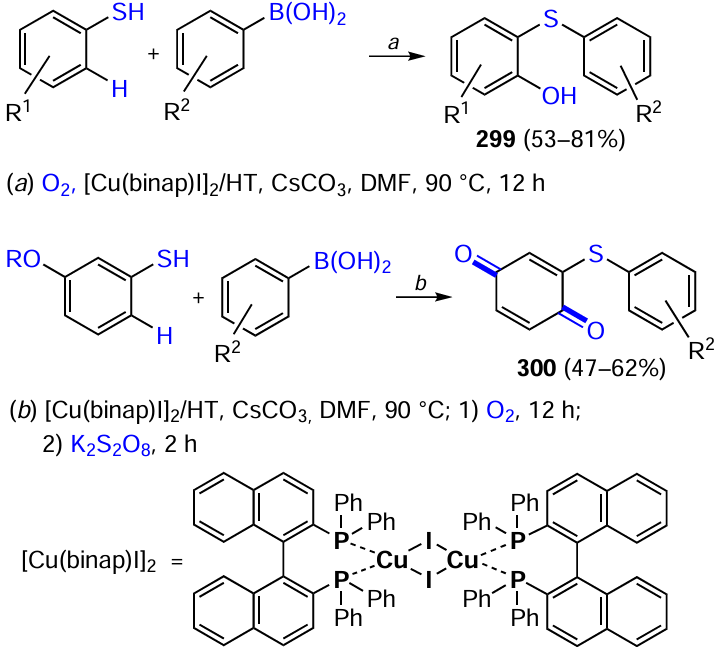
The [Cu(binap)I]2/HT complex is also useful for tandem TH reactions.[883] It is clear that these versatile heterogeneous catalysts would be more and more in-demand. Another trend in the development of green oxidation techniques may be related to photocatalytic processes, the essence of which can be conveniently considered in relation to the reactions of bioglycerol, a product of the processing of vegetable fatty oils.[884]
3.6.3. Photocatalytic transformations of glycerol over semiconductor catalysts
The discovery in 1972 of the possibility of photocatalytic water splitting to hydrogen and oxygen[885] stimulated studies of semiconductor photocatalysis (below referred to as SPC). The enhanced interest in SPC is due to its environmental friendliness: most of photocatalytic processes occur at room temperature and atmospheric pressure, with the solar light being often the source of radiation.[886]
In general, SPC is acceleration of chemical reactions under the action of light in the presence of special functional materials, photocatalysts, which interact with the reactants after absorption of light quanta and thus trigger the chemical reaction and, after that, they return to the catalytic cycle.[887] Despite the fact that SPC mainly serves for water splitting, carbon dioxide reduction or complete oxidation of hazardous organic compounds,[888][889] today it is applied more and more often to fine organic synthesis; an example is photocatalytic partial oxidation of glycerol.[890] These reactions are considered to be promising for glycerol valorization to commercially valuable products[891][892] and are more environmentally benign than other valorization methods such as steam and autothermal reforming, pyrolysis, etc.
The photocatalytic oxidative transformations of glycerol can be carried out both in aerobic (photooxidation) and anaerobic (photoreforming) atmospheres in which molecular oxygen or water, respectively, serves as the oxidant. Glycerol oxidation under anaerobic conditions results in the release of molecular hydrogen. Complete oxidation of glycerol in air and in the oxygen-free atmosphere can be described by the following reactions:[890]
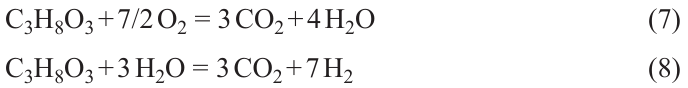
The photoreforming of one mole of glycerol can produce 7 moles of H2 (eq. 8), which makes hydrogen generation from glycerol an attractive process.[893] According to the Scopus database (Fig. 26), the number of publications on the photocatalytic hydrogen production reached a maximum in 2020 and then slightly declined. The publications on the photocatalytic oxidation of glycerol follow a similar pattern, but with a 1 – 2 year shift.
![[{"id":"dlR0fgOvM3","type":"paragraph","data":{"text":"Number of publications found by the search query [‘glycerol’ AND ‘photocatalytic’ AND (‘hydrogen production’ OR ‘oxidation’ OR ‘valorization’)]."}}]](/storage/images/resized/ej09DYbzVLjJxwIkKSEuD6B4WmRqtifhJ9n86lkx_xl.webp)
A drawback of most photocatalytic oxidation reactions of glycerol is low chemoselectivity. In the presence of the Ru/TiO2 photocatalyst under anaerobic conditions, apart from gaseous products (usually, methane, ethane, CO, CO2), the reaction gives approximately 39% of C3-products (glyceraldehyde, 1,3-dihydroxyacetone,[894][895] glyceric acid, etc.) and 61% of a mixture of C2- (glycolic aldehyde, glycolic acid, acetic acid, etc.) and C1-products (formaldehyde, formic acid).[896] Out of the listed compounds, most valuable are C3-products, which are classified, like glycerol, as platform molecules or platform chemicals. Therefore, apart from activity, visible light sensitivity and stability, an efficient photocatalyst should provide high selectivity to the C3-products of glycerol oxidation. This is attained by forming a definite phase composition, crystallinity, specific area and surface morphology for the photocatalyst as well as the ability to adsorb glycerol in molecular or ionic form.[897] The process of рhotooxidation is also affected by kinetic parameters and quantum efficiency, which depend on the substrate and photocatalyst concentrations, the acidity of the medium and so on.[898] The multivariate nature of the photooxidative transformations of glycerol markedly complicates investigation of these reactions; therefore, in most studies, only the activities of various photocatalysts are compared, whereas the influence of reaction conditions on the selectivity has not been adequately addressed.[886]
The most popular photocatalyst for glycerol valorization, as well as for other important photoinduced reactions is titanium dioxide, a relatively cheap and non-toxic material that is highly active towards oxidation. The band gap of TiO2 is 3.2 eV,[888] which makes it insensitive to visible light; however, TiO2 can generate active OH radicals upon the reaction with adsorbed water molecules.[899] The most recent published data indicate that the photocatalytic oxidation of glycerol in the presence of TiO2 leads to cleavage of the C – C bond and gives undesirable C1-products.[900][901] The activity and selectivity can be enhanced by modifying the TiO2 surface by deposition of various metals and nonmetals.[902-904]
A higher visible light sensitivity is inherent in some other photocatalysts, first of all, polymeric graphite-like carbon nitride (g-C3N4), which is more active in photoinduced processes than TiO2 and ZnO.[905][906] In addition, of considerable interest are g-C3N4/TiO2 composites with interphase heterojunctions, which increase the efficiency of photocatalysis.[907-909] In the design of these heterostructures, it is taken into account that the oxidative transformations of glycerol on the g-C3N4 surface are less prone to give C1-products and CO2 .
The rate of hydrogen formation in the photocatalytic anaerobic glycerol reforming catalyzed by g-C3N4/TiO2 composites may reach high values of 5 – 10 mmol of H2 per gram of the photocatalyst per hour,[910] which is much higher than the rates attained in the presence of single components, TiO2 or g-C3N4 . Unfortunately, the authors do not give data on the organic products formed upon the partial glycerol oxidation. Usually, high chemoselectivity to valuable chemicals is attained at lower hydrogen production rate.[911] For example, the decomposition rate of glycerol in aqueous solution and the hydrogen production rate were 2.5 times higher over the Pt/TiO2 photocatalyst than over CuOx/TiO2; however, the yield of organic C2- and C3-products was higher in the latter case. In the anaerobic reforming, the major products of glycerol oxidation over Pt/TiO2 are glycolic aldehyde (38%), lactic acid (34%) and glyceraldehyde (28%), while the reaction over CuOx/TiO2 gives, in addition to the above products (32%, 23%, and 24%, respectively), a considerable amount (21%) of ethylene glycol (Fig. 27). These examples demonstrate that in the selection of a photocatalyst for aerobic or anaerobic oxidation of glycerol, it is important to maintain a balance between the activity and selectivity in the target process, because photocatalysts that provide high conversion of glycerol tend to provide also a higher extent of oxidation.
![[{"id":"q67XRnkRjb","type":"paragraph","data":{"text":"Kinetics of hydrogen formation (<i>a</i>) and distribution of the C<sub>2</sub>- and C<sub>3</sub>-products of anaerobic glycerol reforming catalyzed by 1% CuO<sub>x</sub>/TiO<sub>2</sub> Evonik P25 and 1% Pt/TiO<sub>2</sub> (<i>b</i>).<sup>911</sup> Reproduced with permission from Elsevier."}}]](/storage/images/resized/emXHI0kaTZyy85PNgy2xXlgVRwa6kKFWb2GWhk3G_xl.webp)
The following chart was proposed to describe the photocatalytic aerobic oxidation of glycerol to organic C2- and C3-products in alkaline solutions in the presence of the Au/TiO2 Evonik P25 catalyst modified with Na2B4O7 (Scheme 176).[899] After the adsorption of borate anions B(OH)4– on the TiO2 surface, the main oxidizing species is the superoxide radical •О2– rather than the hydroxyl radical •ОН, which leads to mild oxidation of glycerol without C – C bond cleavage. For example, the selectivity of photocatalytic liquid-phase oxidation of glycerol (AM 1.5 G sunlight simulator as a source of radiation, 4 h) to C3-products at рH = 12 was 58%, with the glycerol conversion being 94%.
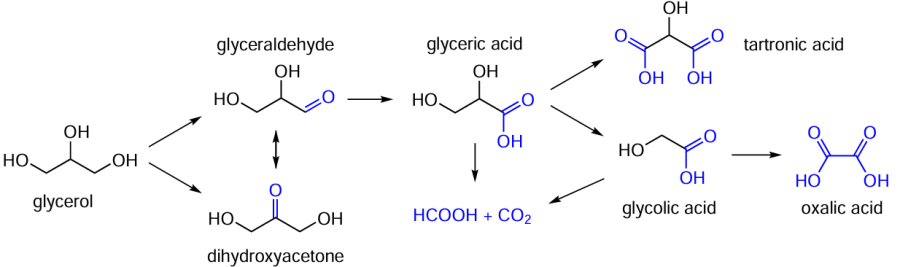
3.6.4. Green synthesis of heterogeneous catalysts using supercritical fluids
Catalyst synthesis in supercritical (sc) fluids offers significant prospects for the design of effective heterogeneous catalysts. The unusual combination of physicochemical properties of a substance in the supercritical state, or in an sc fluid as a solvent, makes it possible to obtain unique functional materials that are difficult or impossible to synthesize using media in other physical states. Heterogeneous catalysts obtained in sc fluids often acquire new catalytic properties and can be of interest in petrochemistry and organic synthesis.[828] The high diffusion rate and the absence of surface tension in sc fluids, and the enhanced solubility of sc fluids similar to that of liquids, can give rise to highly dispersed phases with enhanced structural defects and record-high catalytic activity. The green advantages of the processes using supercritical fluids, such as sc carbon dioxide, include the elimination or minimization of polluted effluents and solid waste for disposal. In addition, in sc fluids, it is possible to fabricate highly dispersed catalytic systems, including metal-containing ones, without the use of toxic metal salts (nitrates, perchlorates, etc.) as active phase precursors. In this case, the number of steps is reduced and the catalyst performance is increased.
The most popular methods for the synthesis of heterogeneous catalysts using sc fluids include supercritical antisolvent precipitation (a), solvothermal synthesis in sc alcohol (b) and deposition of the active component in an sc solvent (c) (FIg. 28).
![[{"id":"HjQ11JRRkV","type":"paragraph","data":{"text":"Methods for the synthesis of catalysts using sc fluids: (<i>a</i>) sc antisolvent precipitation; (<i>b</i>) solvothermal synthesis in sc alcohol; (<i>c</i>) sc solvent deposition of the active component."}}]](/storage/images/resized/fcUTGoNJp55Eq92Bzg8mtqPaECssj5dqPrEPeI5E_xl.webp)
The supercritical antisolvent precipitation takes place when a solution of precursors is injected into a bulk flow of an sc antisolvent, usually carbon dioxide (FIg. 28a). An amorphous vanadyl phosphate catalyst was obtained by this procedure as microspheres from an alcohol solution of VOHPO4 · 0.5 H2O.[912] In relation to the partial oxidation of n-butane to maleic anhydride in the continuous flow mode, it was shown that the catalyst activity was determined particularly by the amorphous state of vanadyl phosphate (Scheme 177).[913]
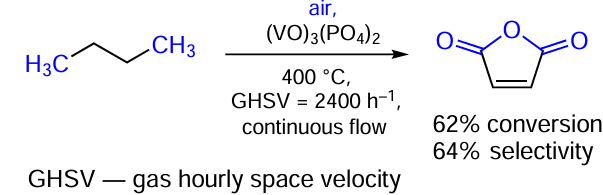
Owing to the above-indicated features,[914] the supercritical antisolvent precipitation is successfully used to obtain precursors of mixed oxide systems with a high degree of homogeneity, which may be retained even after thermolysis of these precursors during oxide phase formation. Mixed oxides are of interest by themselves as they are often used as heterogeneous catalysts or supports for active phase immobilization. For example, the precursor obtained by this method from copper and manganese acetates was used to prepare phase-homogeneous CuMn2O4 binary oxide catalyst with a hopcalite structure, which later showed a high catalytic activity in CO oxidation.[915] Highly defective copper and zinc hydroxycarbonate, with a structure of a rare georgeite mineral, was synthesized in a similar way.[916][917] This precursor was converted to a mixed oxide, which showed a very high catalytic activity in the synthesis of methanol and in the low-temperature steam reforming of CO.
Lately, the sc antisolvent precipitation has been used to obtain efficient photocatalysts. For example, TiO2-based photocatalysts prepared by this method had a surface area of 515 m2 g–1 and possessed a higher activity towards the decomposition of methyl orange and methylene blue dyes than the commercial Degussa P25 catalyst.[918] ZnO-based oxide photocatalysts doped with Eu[919] and Gd[920] showed an excellent performance in the decomposition of the eriochrome black T dye and the atrazine herbicide, respectively. The precipitation in scCO2 was successfully used to synthesize a hybrid catalyst from β-cyclodextrin and commercial TiO2 ,[921] which was active in the decomposition of β-naphthol orange dye.
The considered method can be used to prepare not only oxide systems, but also catalysts containing a metallic phase. For instance, the reduction of mixed SmCoO3 oxide, obtained by sc antisolvent precipitation, in a hydrogen flow affords a cobalt metal-containing catalyst, which is highly active in the carbon dioxide conversion of methane[922] and partial oxidation of methane[923] and is stable against coke formation.
The effectiveness of this approach to the synthesis of metallic catalysts was demonstrated in the co-precipitation of metal phase precursors and a stable oxide sol.[924][925] In particular, this method was used to synthesize metallic Ni catalysts in which the SiO2 , TiO2 , Al2O3 and ZrO2 oxides served as the stabilizing matrices.[926] The synthesized Ni/Al2O3 catalyst had a very high degree of dispersion and was fairly active in the hydrogen transfer anisole hydrogenation, which gave methoxycyclohexane as the major product (Scheme 178).[927] The ratio of nickel metal and oxide matrix phases affected the structural and catalytic properties of metal–oxide systems: the highest rate of anisole hydrogenation was observed when the nickel content was 50 mass %.
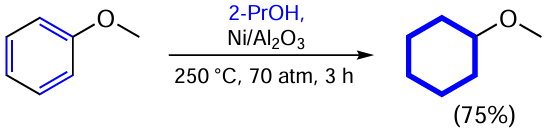
A specific feature of the obtained catalyst is the low activity in the hydrodeoxygenation of phenols and their ethers. The selectivity of heterogeneous metallic nickel catalysts was also manifested in the low selectivity of hydrogen-free hydrogenation of aldehydes, which they catalyze.[928] While planning the synthesis, one should take into account the enhanced sensitivity of these systems to the gas phase composition[929] and to some heteroatomic organic substrates.[930][931]
The proposed approach is suitable for the synthesis of not only monometallic, but also bimetallic catalysts, e.g., Ni – Cu-containing systems with the structure of the Ni1 – xCux substitutional solid solution,[932][933] which makes it possible to control the selectivity of reactions. Indeed, the addition of copper into nickel systems decreases the rate of hydrogenation of phenolic compounds in 2-propanol. The Ni – Co bimetallic systems synthesized by the supercritical antisolvent precipitation catalyze hydrogenation and hydrodeoxygenation, apparently due to the high content of structure defects.[934]
One more method for preparing metal-oxide heterogeneous systems is solvothermal synthesis (FIg. 28b) from metal-oxide precursors thermally decomposed in supercritical alcohols to form a dispersed oxide phase.[935] This method is also suitable to prepare mixed oxide systems that either contain active phase precursors or can serve as supports for the subsequent deposition of the active metal phase. For example, the synthesis of mixed Ce – Zr systems furnishes an oxide phase with the structure of a substitutional solid solution.[936] These mixed Ce – Zr oxide systems with deposited Ni – Co metal active phase showed a high catalytic performance in the carbon dioxide conversion of methane and possessed enhanced thermal and chemical stability.[937] The carbon dioxide conversion of ethanol catalyzed by metallic nickel systems supported on Ce – Zr oxides confirmed that the introduction of cobalt into this system inhibits the formation of carbon deposits.[938]
In addition to binary Ce – Zr oxide supports, the solvothermal synthesis allows the preparation of systems with more complex compositions. For example, a comparative study of the carbon dioxide conversion of methane using the Ce – Zr oxide system doped with different metals showed that titanium doping causes the formation of large amounts of carbon deposits, whereas niobium doping strongly minimizes carbonization of the catalyst.[939] Similarly, praseodymium doping sharply enhances the stability of the Ce – Zr support in the high-temperature carbon dioxide conversion of methane.[940]
This method was used to prepare metallic nickel active species on the Ce – Ti oxide support with a substitutional solid solution structure.[941] This material suppressed the detachment of nickel particles from the catalyst surface and prevented sintering of the particles. Thus, the solvothermal synthesis in a supercritical alcohol expands the possibilities of preparing mixed oxide systems and simultaneously makes it possible to dope them with various metals to improve their catalytic and service characteristics.
One more synthetic technique, the supercritical solvent deposition (FIg. 28c), is based on the ability of supercritical fluid to dissolve substrates that are convenient precursors for the active phase of the future catalyst. It is important that low viscosity and high diffusion rate in sc fluids make it possible to obtain catalytic systems with a uniform distribution of the active component throughout the support bulk.[942]
The deposition (or impregnation) in scCO2 was successfully used to obtain single-atom Co-containing heterogeneous catalysts for the selective oxidation of benzyl alcohol to benzaldehyde.[943] In the obtained heterogeneous systems, the cobalt atoms are incorporated in the structure of the nitrogen-doped carbon support. This procedure was utilized to synthesize the ZIF-8 metal-organic framework with single palladium atoms embedded into the framework,[944] which showed a high activity in the partial hydrogenation of phenylacetylene to styrene.
This method provides the synthesis of a broad range of heterogeneous catalysts and adsorbents. For example, deposition in scCO2 was used to prepare highly dispersed Pt/Al2O3 and Pt – Pd/Al2O3 catalysts for propylene oxidation[945] and a catalyst for toluene oxidation in which platinum was supported on Hβ zeolite.[946] The ionic liquid obtained from amino acid and supported on porous silica proved to be an active catalyst for the carbon dioxide cycloaddition to epoxides.[947][948] The possibility of increasing the precursor solubility in scCO2 by adding small amounts of organic co-solvents, without changing the phase state of the system, significantly expands the scope of applicability of scCO2 for the preparation of supported catalytic systems.
Supercritical CO2 can not only serve as a solvent, but also act as a mild oxidant in the catalyst preparation. A process of copper metal oxidation to give amorphous CuO oxide layer has been reported; the subsequent reduction of this material gave an amorphous metallic copper layer.[949] This catalyst showed a high chemoselectivity to C2+ products of CO2 electroreduction.
Laser ablation of palladium for obtaining metal nanoparticles and their subsequent deposition on the Al2O3 oxide support can be carried out in scCO2 .[950] The obtained catalyst showed high activity in the hydrogenation of phenylacetylene (Scheme 179). It is important that the parameters of the laser ablation of palladium and the conditions of deposition of Pd nanoparticles significantly affect the ratio of reaction products, styrene and ethylbenzene, although styrene is always the major product.
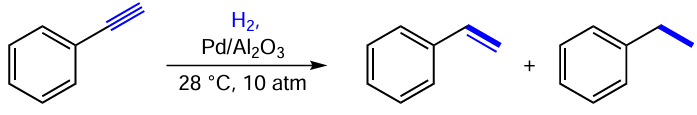
The ability of supercritical water to oxidize metals without the use of strong acids or bases can be exploited for the in situ preparation of the active heterogeneous catalysts in sc water,[951] which enhances the green nature of the synthesis. In particular, this approach was used to obtain nano-sized Fe2O3 , a catalyst for oxidation of toxic organic compounds[952] and for hydrogen generation from biomass processing products.[953] In addition, the unique properties of sc H2O expand the potential for the preparation of mixed phase-homogeneous oxide systems with a substitutional solid solution structure,[954] which can be used in organic synthesis.
* * *
Generally, the data presented in Chapter 3 of this review clearly show that the philosophy of green chemistry, which reflects society’s need to develop chemical processes that are efficient, economical, and less harmful to the environment and its inhabitants, greatly influences the development of modern methodologies for organic synthesis. The use of catalysts has become an integral part of most synthetic methods devised in recent years. The developed original methods for the catalytic activation of usually inactive C – H bonds in organic compounds make it possible to substitute a hydrogen atom in a specified position of the molecule by a carbon atom or heteroatom in a much simpler and less costly way, without resorting to auxiliary or protecting group. The design of highly efficient transition metal catalysts and nanocatalysts, especially those that can be regenerated and reused many times, served as a strong impetus for the development of a promising methodology of catalytic cross-coupling, especially in green solvents and in water. The innovative stereo- and enantioselective syntheses of organic compounds in the presence of enzyme-like metal-free chiral organic molecules (organocatalysts) have been widely spread. Multicomponent and cascade reactions, in which multistep processes are carried out one-pot without isolation and purification of intermediates, have become in high demand in organic synthesis; this significantly decreases the amount of waste, the disposal of which requires additional energy and resource expenditures. There is great potential in green chemical processes developed in recent years, including catalytic methods that use visible light or electric current as an energy source. New types of high-performance heterogeneous catalysts, which greatly facilitate the industrial implementation of the developed methods, are used more and more widely in fine organic synthesis.
4. Organic synthesis in green solvents
Organic solvents represent a major cause of the environmental pollution brought about by chemical reactions. In the syntheses of complex organic compounds, for example, the active ingredients of modern drugs, the weight of solvents is at least a half of the total weight of all materials used in the process.[7] In addition, the products are often isolated using solvent extraction, chromatography, recrystallization and other procedures that also require solvents. This problem can be solved by minimizing the amount of solvents (in the ideal case, by completely eliminating solvents from the processes) and/or by using green solvents that can be safely handled and have the least adverse effect on the personnel and the environment.[34] These requirements are met, to some extent, by water,[955] bioethanol,[956] 2Me-THF,[957] γ-valerolactone,[958] cyrene,[959] liquid polymers,[960] ionic liquids,[961] deep eutectic solvents (DESs) and low-melting mixtures (LMMs),[962] liquid or supercritical carbon dioxide (scCO2)[828] and some other. Usually, preference is given to water; however, the scope of its applicability is restricted by low water solubility of many organic compounds and by the fact that regeneration of water contaminated during the synthesis requires a lot of energy. Non-toxic organic solvents that do not pollute the atmosphere and have boiling points above 70 °C (alcohols, ketones, esters, etc.) complicate the isolation of products. This drawback is not inherent in ionic liquids; however, according to the available data, some of them may be toxic.[963]
The use of low-melting eutectic mixtures and liquid or supercritical carbon dioxide in chemical processes is fairly attractive. These compounds are stable, non-toxic and safe, and they can be easily recycled, the former owing to low saturated vapour pressure and the latter owing to high saturated vapour pressure. In addition, their properties can be modified by changing the composition of the eutectics or process conditions (pressure, temperature, etc.). In this Chapter, we consider some aspects of application of eutectic mixtures and liquid or supercritical CO2 in modern organic synthesis.
4.1. Deep eutectic solvents (DESs) in organic synthesis
Deep eutectic solvents (DESs), which are also called low transition temperature mixtures (LTTMs), are binary or ternary mixtures consisting of compounds with properties of bases and Lewis or Brønsted acids, which are liquid at ambient temperature. The mixtures contain at least one hydrogen bond donor (HBD) and at least one hydrogen atom acceptor (HBA), which provides for system structuring (Fig. 29).
![[{"id":"0mbl7kxkdT","type":"paragraph","data":{"text":"Typical components of deep eutectic solvents."}}]](/storage/images/resized/RWptuEJp6OSrWShp5pGmFHkBxthLh91iCV5gLN9k_xl.webp)
Like ionic liquids, DESs have a low vapour pressure and are not flammable. In addition, they are inexpensive, easily processable, have a low environmental footprint and can be readily synthesized.[964] The physical and chemical properties of DESs, such as polarity, hydrophobicity, viscosity and solvent miscibility, can be controlled by selecting the optimal combination of components for a particular reaction.
4.1.1. Scope and benefits of DES application in chemistry
Deep eutectic solvents, being partly composed of components of natural origin, are usually biodegradable. Considering also their low toxicity, they are excellent green solvents for analytical chemistry,[965][966] for the synthesis of functional polymer materials,[967][968] for CO2 capture and chemical fixation,[969] in particular as cyclic carbonates,[970] for biocatalytic processes,[971][972] including enantioselective processes,[973] for the synthesis of biologically active products,[974] etc. The application of DESs in organic synthesis as an environmentally benign, safe and cost-effective alternative to petroleum-derived solvents has become a popular research trend in recent years.[975-977]
The reported results[978] cast doubt on the generally accepted view that organometallic reagents such as Grignard reagents or organolithium compounds must be used in anhydrous organic solvents, under inert atmosphere, and with strict temperature control. It turned out that such reactions can be carried out in environmentally benign solvents such as water, deep eutectics, polyols derived from biomass (e.g., glycerol) and ethers [e.g., 2-MeTHF or cyclopentyl methyl ether (CPME)]. The versatility of these synthetic protocols compatible with air and moisture was demonstrated, in particular, by the nucleophilic addition of RLi/RMgX to unsaturated organic molecules, Pd-catalyzed cross-coupling reactions, etc. It is noteworthy that in some cases, the observed chemoselectivity was higher than that under conventional conditions.
The advantages of using DESs in organic and solvothermal synthesis and electrodeposition, calcination and polymerization processes were evaluated in a recent review published in Cell Reports Physical Science.[976] The evaluation was based on six criteria: (A) energy consumption, (B) requirements for equipment, (C) sustainability, (D) complexity, (E) separation and purification (F) and yield and efficiency (Fig. 30).
![[{"id":"OTLYXoShSa","type":"paragraph","data":{"text":"Evaluation of synthesis processes in DESs based on six criteria.<sup>976</sup>"}}]](/storage/images/resized/dpcHpIyPR7sCNtY4pzvaEyRjrpRTTwmegOSetUMy_xl.webp)
Analysis showed that the advantages of using DESs in organic synthesis are associated with a reduction in energy consumption energy consumption, lower requirement for equipment, compliance with the requirements for sustainable development and simpler synthetic protocols regarding the number of steps and ease of operations. However, according to evaluation made by the authors of this review, the application of DESs still offers no benefits as regards separation, purification and yields of products compared to conventional synthesis processes.
Meanwhile, owing to the wide possibilities of fine tuning of the physicochemical properties of DESs, they can be used not only as solvents, but also as acid-base catalysts. The first successful enantioselective cross-aldol reaction was carried out in 2014 via coupling of enzymatic catalysis with organocatalysis in a choline chloride (ChCl) – glycerol (Glr) eutectic mixture (1 : 2).[979] The generation of carbon–carbon bonds using enantioselective cross-aldol reaction and conjugate addition remains one of the most popular trends.[980]
Metal-catalyzed cross-coupling reactions are efficient methods for the formation of new carbon–carbon and carbon–heteroatom bonds. These reactions are also actively investigated in deep eutectic systems,[981][982] in particular using metal nanoparticles.[983]
One of the first registered DESs, a mixture of choline chloride with urea,[984] is widely used for the synthesis of heterocyclic compounds. The reactions proceed under relatively mild conditions without additional catalysts or organic solvents and give the target heterocycles in excellent yields. In addition, the reaction time is shorter and the workup of the reaction mixture is simpler than for reactions in organic solvents. Later, DESs were successfully used in the cyclocondensation reactions to prepare dihydropyrimidinones,[985] quinazolines,[986] dihydroquinazolines,[987] quinolines,[988] pyrimidopyrimidinones[989] and oxazoles.[990]
The review publications on the synthesis of heterocycles using green solvents that appeared in recent years address relatively specific aspects, e.g., the synthesis of indoles,[991][992] coumarins[993] and 5-substituted 1H-tetrazoles.[994] Some aspects of the synthesis of heterocycles in DESs via transition metal-catalyzed cyclization, cycloaddition and cyclocondensation reactions are covered in a review by Marset and Guillena.[982]
This section summarizes data on the use of DESs for the synthesis of heterocyclic compounds published over the last five years and gives examples of further modification of heterocycles in these media.
4.1.2. Multicomponent ring formation reactions in deep eutectics
Multicomponent one-pot synthesis of pyrazolopyridine derivatives was accomplished by the reaction of hydrazine with ethyl acetoacetates, ammonium acetate and aromatic/aliphatic aldehydes or ketones.[995] This condensation is efficiently catalyzed by an eutectic mixture of choline chloride (ChCl) with urea and provides the synthesis of a broad range of tetrahydrodipyrazolopyridines (301) in good to excellent yields (Scheme 180). After the simple workup of the reaction mixture, the deep eutectic mixture was reused in four cycles without a decrease in the catalytic activity. The authors compared their method with six methods described previously, including those using metal catalysis, and noted that the strategy they proposed is comparable with and even outperform the known methods.
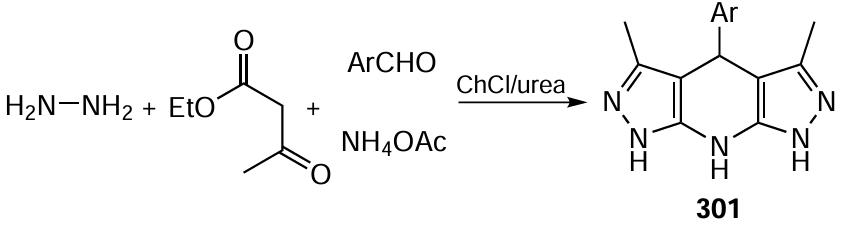
An easy one-pot synthesis of N-(hetero)aryl-2-(hetero)arylbenzimidazoles is accomplished by the condensation of aromatic/heteroaromatic aldehydes with o-nitroaniline in an SnCl2 · 2 H2O/choline chloride eutectic mixture.[996] The authors note the high rate of the reaction and the use of a simple experimental setup. The reaction involving 4-methoxy-2-nitroaniline was not regioselective and gave mixtures of 5- and 6-methoxy-substituted benzimidazoles.
The choline chloride/glycerol (ChCl/Glr) eutectic mixture containing a minor amount (0.1 mL) of water was used by Atharifar et al.[997] for the synthesis of 3,4-disubstituted isoxazol-5(4H)-ones (302). The condensation of aromatic aldehyde, hydroxylamine hydrochloride and ethyl acetoacetate proceeded over 20 min at 60 °C to give products 302 in 90 – 95% yields (Scheme 181). These reactions proceed faster in DESs than in common solvents, and the isolation of products is not difficult.
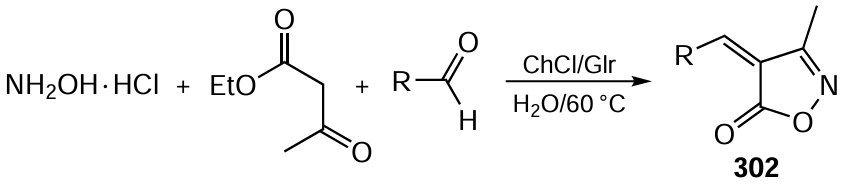
During elaboration of the green synthesis of arylacyl azides 303 from arylacyl bromides in a ChCl/Glr mixture, it was found that an increase in the reaction temperature results in isomeric 2-benzoyl-substituted imidazoles 304a and 304b being formed together with azide 303 (Scheme 182).[998] Optimization of the reaction parameters (temperature, time and amount of NaN3 used) demonstrated that imidazole isomers can be obtained in 88% total yield within 12 h at 80 °C. For comparison, pyrolysis of α-azidoketones in trichlorobenzene requires much more drastic conditions (180 – 240 °C). The authors also found that azides 303 can be converted to 2,4-diaroyl-6-arylpyrimidines (305) (45 – 88% yield) by the cyclotrimerization reaction in a ChCl/urea eutectic mixture (1 : 2) even at room temperature.
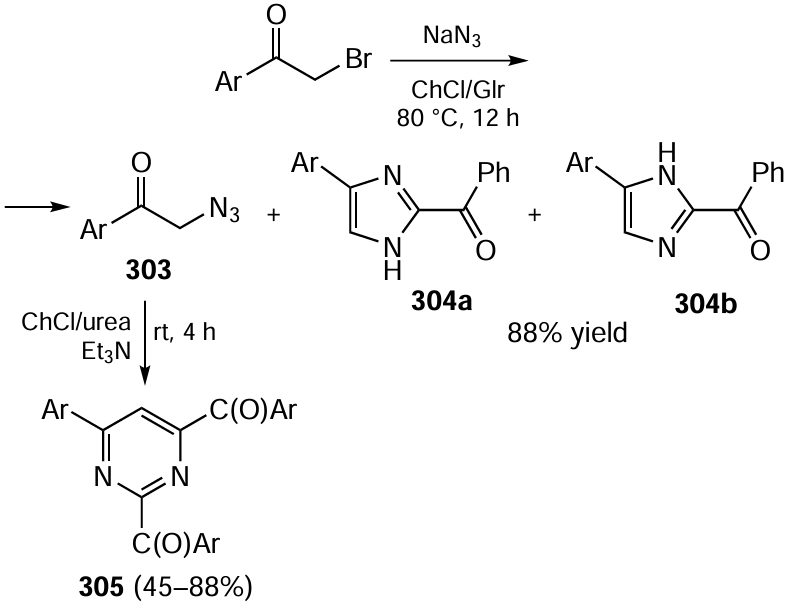
Eutectic mixtures of N-alkylated 1,4-diazabicyclo[2.2.2]octane (DABCO) salts with polyethylene glycols (PEG) of different molecular weights were successfully used for the synthesis of indoles (Fischer method) and 1H-tetrazoles (by click-chemistry methods).[999] Although the reaction in N-alkyl-DABCO with low-molecular-weight alcohols gave products in comparable yields, the use of PEG as the hydrogen bond donor in DESs provides a much safer, non-volatile medium for synthetic reactions.
The ChCl/urea DES exhibited a pronounced catalytic activity in the one-pot domino synthesis of 3-aminoimidazo-fused heterocycles 306 from aromatic or heteroaromatic aldehydes, isocyanides and 2-amino-substituted heterocycles by the Groebke – Blackburn – Bienaymé reaction (Scheme 183).[1000][1001] Appropriate substrates for this reaction are 2-aminopyrazines, 2-aminopyridines, 2-aminobenzimidazole and 2-aminobenzothiazole. Fused heterocycles 306 are formed in this reaction in excellent yields.
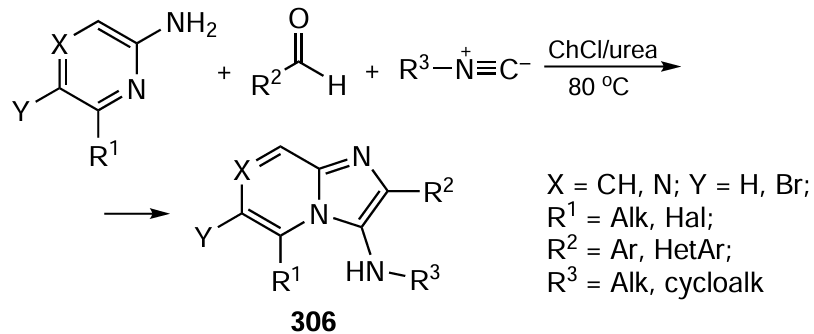
The same DES proved to be effective in the synthesis of tricyclic benzannulated seven-membered heterocycles 307 containing 1,4-benzodiazepine and 1,4-benzoxazepine moieties via one-pot three-component reaction of benzaldehydes with o-phenylenediamines or 2-aminophenols and dimedone (Scheme 184).[1002]
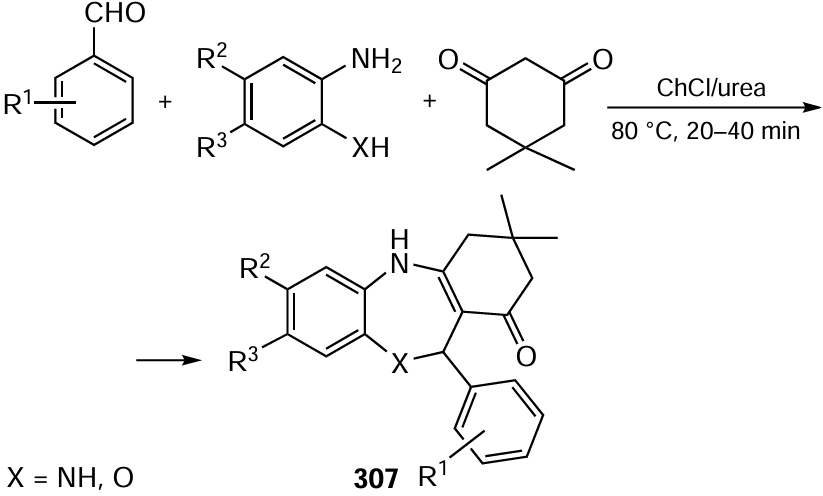
The authors noted that DES can act here as both a solvent and an organocatalyst that activates carbonyl and imine functional groups by forming hydrogen bonds with them. An advantage of DESs is their recyclability: after treatment of the reaction mixture with water and separation of crude products by extraction or filtration, DES was isolated from the aqueous phase by evaporation at 80 °C in vacuo and reused in four successive reactions without a significant loss of the catalytic activity.
Pentasubstituted pyridines 308 were synthesized in DES based on choline chloride and urea under mild conditions.[1003] The one-pot three-component reaction of structurally diverse aldehydes with aromatic thiols and malononitrile at 60 °C gave products 308 in 60 – 82% yields within 1.5 – 3 h (Scheme 185). The authors proved the reusability of DES in four cycles.
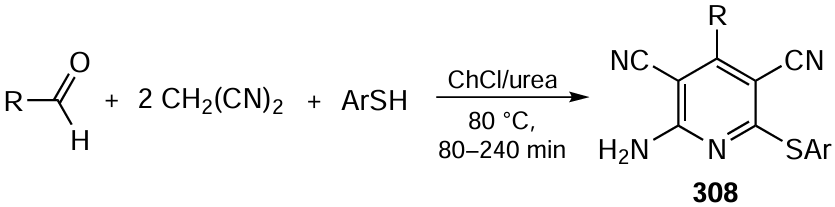
The tandem condensation of aldehydes with phenylhydrazine and ethyl acetoacetate in a choline chloride/tartaric acid (10 mol.%) eutectic mixture resulted in the formation of 4,4-(arylmethylene)-bis-(3-methyl-1-phenyl-1H-pyrazol-5-ols) (309).[1004] The ultrasonic activation of the reaction mixture intensified the reaction, which in this case proceeded at room temperature within 7 – 12 min (Scheme 186).
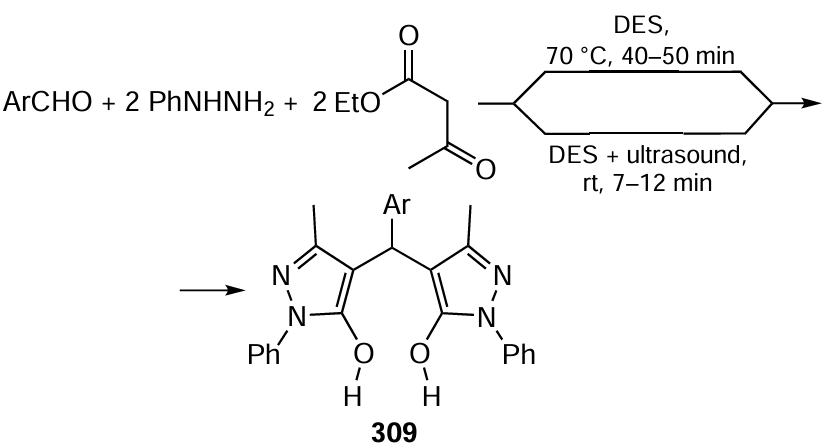
A convenient and environmentally friendly approach to the synthesis of spiro[indeno[1,2-b]quinoxalin[11,2']thiazolidin]-4´-ones (310) using sulfonated carbon as a heterogeneous acidic catalyst and a ChCl/urea eutectic mixture as a green solvent has been developed.[1005] The spiro-connected thiazolidine ring in compound 310 is formed upon a three-component reaction of indeno[1,2-b]quinoxalinone with α-mercaptocarboxylic acids and various amines (Scheme 187). The product yields are high. This method is of obvious interest since the spiro[indole-thiazolidinone] moiety is encountered in some natural bioactive molecules.
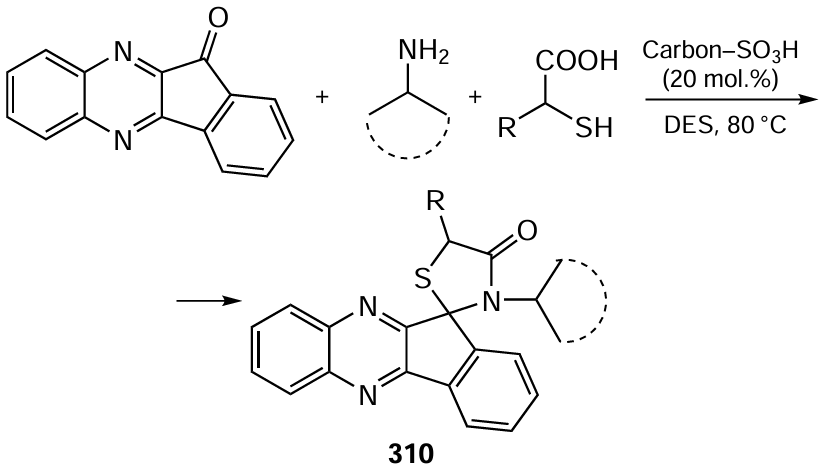
Spirooxindoles 311 and 312 were prepared by multicomponent condensation of isatins with β-ketoester, arylhydrazine and cyanoacetic acid derivative or cyclic 1,3-diketone. The reaction was carried out in water in the presence of the ChCl/urea eutectic mixture (Scheme 188).[1006]

4.1.3. Iodocyclization in deep eutectic solvents
Starting with the pioneering works of Richard C.Larock,[1007] the electrophile-promoted cyclizations, first of all, iodocyclizations of functionalized acetylenes[1008][1009] and diacetylenes[1010-1012] proved themselves as a versatile tool for the synthesis of various heterocyclic structures. One of the first examples of iodocyclization using DESs is the iodocyclization of 1-mercapto-3-yn-2-ols 313 in choline chloride-based DES reported by Mancuso et al.[1013] The reaction proceeded at room temperature under the action of I2 and resulted in the formation of 3-iodothiophenes 314 (Scheme 189). The authors were able to increase the yield of the target products (up to 80%) in comparison with the yields attained in earlier studies and to demonstrate that DES can be efficiently recycled and reused (up to six cycles). It is of interest that the starting alkynylthiols 313 can be obtained in the same eutectic mixture by direct nucleophilic addition of lithium acetylide to an appropriate α-mercaptoketone in air at room temperature.[1014]
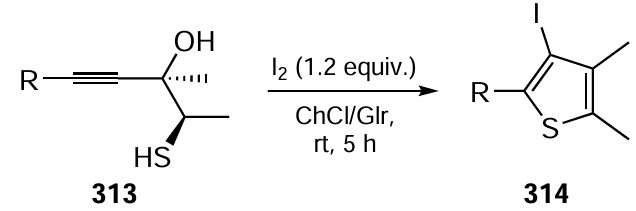
The idea of conducting iodocyclization in DESs was further developed by the same research team. It was shown that the use of DESs provides the formation of 3-iodobenzothiophenes 315 in good yields (Scheme 190).[1015] The optimal solvent for this reaction is the ChCl/urea mixture in 1 : 2 molar ratio.
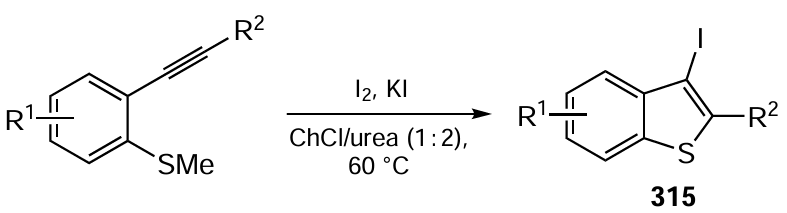
4.1.4. Synthesis and modification of heterocyclic compounds by metal-catalyzed reactions in DES
Organic-inorganic hybrid systems (MOFs) containing Zr6O4(OH)4 clusters, together with aminoterephthalic acid and urea moieties (UiO-66-Urea), proved to be effective catalysts for cascade reactions.[1016] Modification of these MOFs with choline chloride (ChCl) resulted in the in situ formation of a deep eutectics (ChCl@UiO-66-Urea) with catalytically active sites on the MOF surface. In the presence of the hybrid material obtained in this way, aromatic aldehydes reacted with malononitrile and α-naphthol or 4-hydroxycoumarin in the reactant medium to give 2-amino-4H-chromenes 316 and 317 in high yields (Scheme 191). The Zr6 nodes served as the acid sites of the catalyst, while urea moieties able to form hydrogen bonds with the reactants functioned as the basic sites. The ChCl layer on the MOF surface facilitated the contact between the heterogeneous catalyst and the reactants. The hybrid catalyst was superior in activity to other catalysts used for reactions of this type.
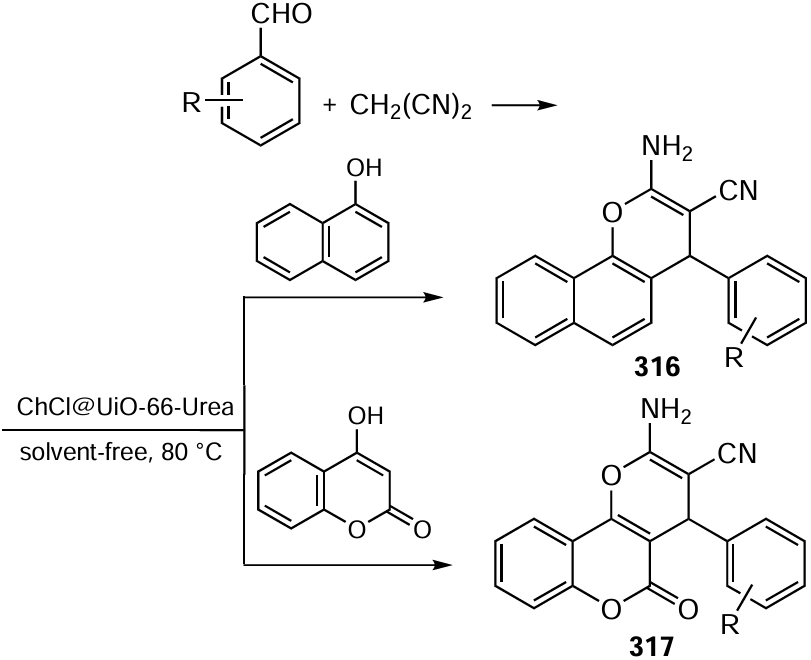
Silica-coated magnetic Fe3O4 nanoparticles [Fe3O4@ – SiO2@(CH2)3Cl] stabilized by the [urea]4/ZnCl2 deep eutectics (DES@MNP) were utilized as green catalysts for the one-pot synthesis of functionalized thieno[2,3-b]indoles 318 from cheap and readily available reactants such as sulfur, acetophenones and indoles.[1017] The reaction was carried out at 140 °C in DMF. The advantages of the developed process over the known methods include the ease of separation of DES@MNP with an external magnet and the possibility of reuse (5 cycles) without a decrease in the yield of heterocycles 318 (Scheme 192).
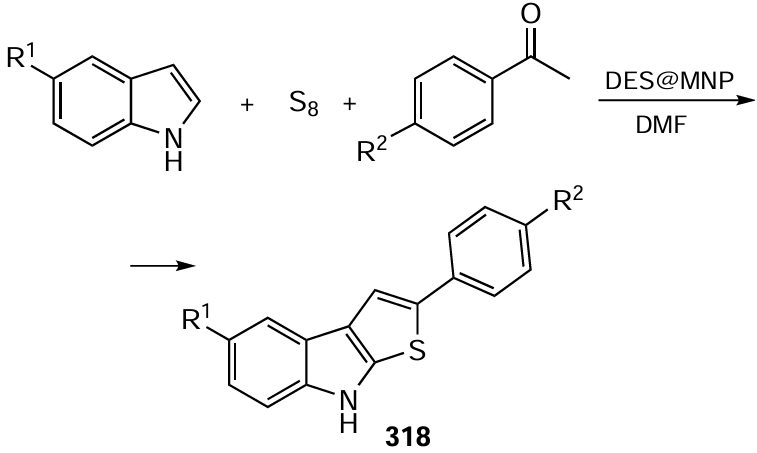
The biodegradable ChCl/Glr mixture (1 : 2) proved to be an effective reaction medium for the synthesis of arylacyl azides 303 from commercially available arylacyl halides. Using the same eutectic, azides 303 were converted to valuable symmetric 2,5-disubstituted pyrazines 319 via palladium-catalyzed reduction with hydrogen (Scheme 193).[1018]
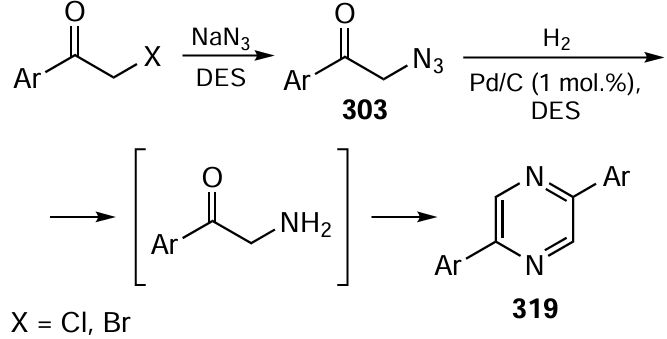
The potassium carbonate/ethylene glycol (EG) system (1 : 10) was successfully used as DES to prepare polycyclic benzo[2,3][1,4]oxazepino[7,6-b]quinolines 320 and 321 by the one-pot sequence of cyclization and Suzuki–Miyaura or Sonogashira cross-coupling (Scheme 194).[1019]
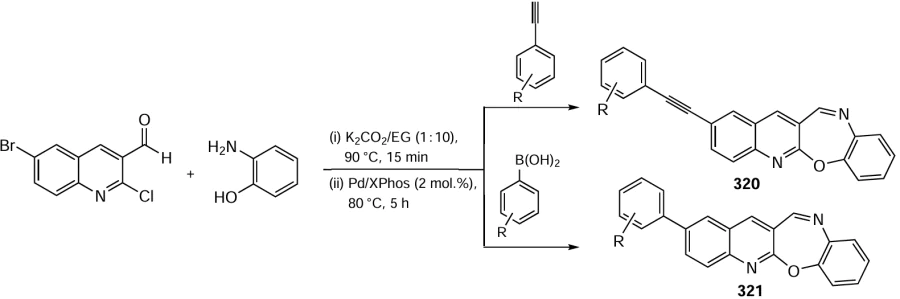
The cross-coupling of 2-bromothiophene with arylboronic acids in the presence of Ni(cod)2 and K2CO3 proceeded in the ChCl/urea eutectic mixture (1 : 2) within 5 h at 60 °C, giving rise to products 322 in high yields (Scheme 195).[1020] The high chemoselectivity of the reaction was retained even for heterocyclic boronic acids, which usually tend to undergo side protodeboronation reactions.

The tandem catalytic cross-coupling of (hetero)aryl halides with 2,3-dihydrofuran or 3,4-dihydro-2H-pyran followed by the reduction of cross-coupling products to tetrahydro derivatives 323 was performed as a one-pot process in ChCl/Glr solvent (Scheme 196).[1021] Notably, both transformations occur under aerobic conditions in the absence of additional ligands.
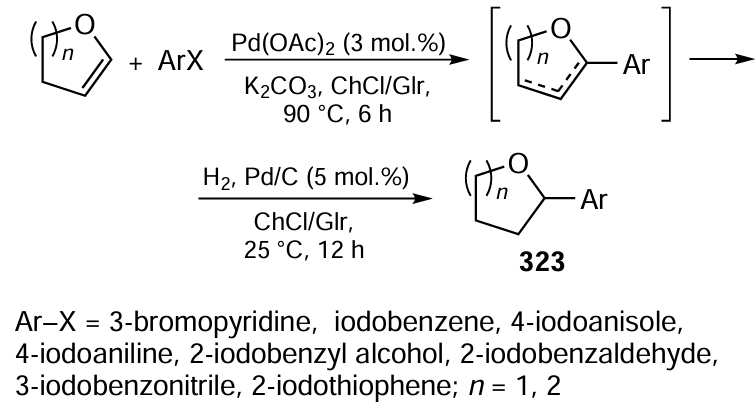
Cycloisomerization of acetylenic acids and their derivatives to heterocycles 324 catalyzed by palladium(II) oxide impregnated with magnetite can also be carried out in choline chloride-based DES (Scheme 197).[1022] Under proposed conditions, this reaction proceeds for acetylenic acids containing a terminal or an internal triple bond or alkynylsulfonylimides.

The choline chloride and glycerol eutectic mixture (1 : 2) proved to be an appropriate solvent for ligand-free Sonogashira reaction of aromatic and heteroaromatic iodides with terminal alkynes (Scheme 198). Under proposed conditions, 3-iodopyridine, 2-iodothiophene and 6-iodouracil derivatives are converted to cross-coupling products 325 in high yields when react with phenylacetylene, pent-1-yne, trimethylsilylacetylene or 1-phenylprop-2-yn-1-ol.[1023] Furthermore, DES was easily regenerated and could serve as the reaction medium in four more syntheses.
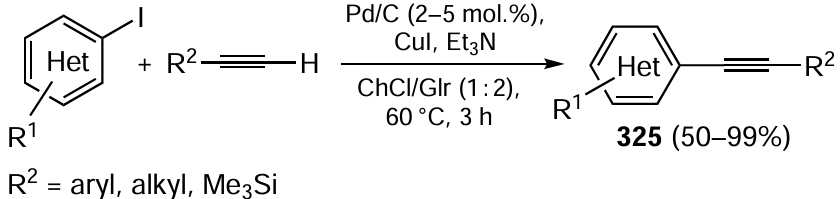
Ligand-free Suzuki – Miyara (hetero)aryl halides with water-resistant mono- and bifunctional potassium aryltrifluoroborates efficiently proceeded in the same DES in the presence of Na2CO3 in air with a minimum loading of palladium acetate (1 mol.%) (Scheme 199). The reaction afforded arylated heterocycles 326, including 3-phenylpyridine, 2- and 3-phenylthiophenes and 5-phenylindole. The catalyst, the base and DES remained intact during the workup of the reaction mixture and were successfully used in six new experiments, which reduced the reaction E-factor down to 8.74.[1024]
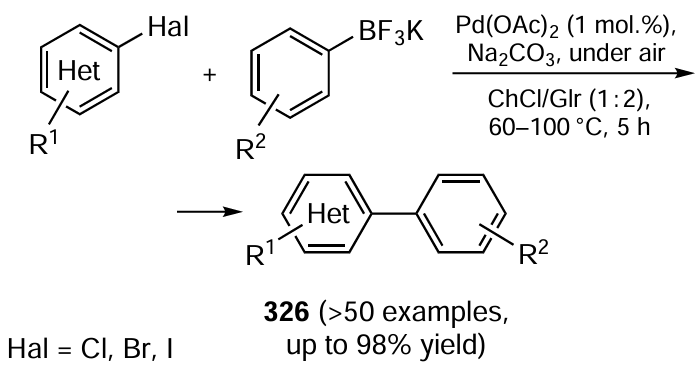
Owing to the versatility of the ChCl/Glr eutectic mixture (1 : 2), this system was also used in the Hiyama coupling of (hetero)aryl bromides with phenyltrimethoxysilane (Scheme 200). This approach to the synthesis of biaryls 327 has certain environmental and economic benefits, although organosilanes are usually less reactive than the corresponding boron derivatives. In this case, the unusual palladium catalyst was the main key to success. Apart from aryl bromides, this reaction can be performed for brominated heterocycles such as 3-bromopyridine, 3-bromofuran and 2-bromothiophene and furnishes the corresponding heterobiaryls 327 in excellent yields.[1025]
The ligand-free exhaustive cross-coupling of dihalo-substituted benzodithiophenes with various trifluoroborate salts was performed in choline chloride-containing DES (Scheme 201).[1026] These reactions, which proceed under mild conditions in air, furnished products 328 containing an extended π-electron system, which are of interest as photoactive materials. Oligomer of one of the products was obtained by the electrochemical method.
An interest green application of DESs is the recently reported N-arylation of nitrogeneous heterocycles catalyzed by magnetic nanoparticles supported on coconut shell-derived activated carbon decorated with Cu2O nanoparticles (Scheme 202).[1027]
Thus, deep eutectic mixtures are promising green solvents for a variety of useful organic reactions, including reactions leading to the formation or modification of heterocycles. Deep eutectic solvents are well combined with ultrasonic activation and application of various types of catalysts; in many cases, they can themselves act as acid – base catalysts. The catalyst–DES systems can most often be preserved during the workup of the reaction mixture and reused in subsequent experiments, thus decreasing the E-factors of the reactions.
4.2. Modern catalytic transformations in supercritical carbon dioxide
Supercritical CO2 (scCO2) is one of the most promising green solvents. Carbon dioxide is converted to the supercritical state when compressed up to a pressure above 73 atm and heated above 31 °C (Fig. 31). The specific properties in which scCO2 differs qualitatively from usual organic solvents include the sharp change in the density and viscosity, non-combustibility and the lack of toxicity. In addition, CO2 used in the synthesis does not pollute the environment and does not contribute to global warming, since it is extracted from the atmospheric air. It is noteworthy that CO2 is capable of efficient mass transfer; it is completely miscible with gaseous and liquid reagents and very easily separated from the product without drying, freeze drying or using other additional techniques.
![[{"id":"SvuOgdCL8K","type":"paragraph","data":{"text":"Schematic phase diagram of CO<sub>2</sub>."}}]](/storage/images/resized/xedtlB0G448zItWov5fOJDFann2adVLCRgG95QvA_xl.webp)
Although the first metal-catalyzed reactions in CO2 were carried out back in the early 20th century,[1028] further development of this field of chemistry was complicated by the fact that many transition metal complexes are poorly soluble in scCO2 .[1029][1030] This problem was addressed by the search for catalysts containing aliphatic[1029][1031] or perfluorinated[1032][1033] analogues of known ligands and by detailed studies of the heterogeneous catalysis in scCO2 , which has been actively developing in recent years.[1034] In this section, we consider selective organic reactions in scCO2 catalyzed by Pd, Rh, Ru and Cu compounds. Some catalytic reactions involving other metals (Pt, Ag, Au, Sn and Mo) published in 2018 – 2023 are also discussed.
4.2.1. Palladium-catalyzed reactions in supercritical CO2
Cross-coupling is the most popular type of reactions that are performed with palladium catalysts. However, only the Heck and Suzuki reactions in scCO2 have been adequately covered in the literature, and the available data allow one to draw reliable conclusions about the patterns that determine the efficiency of catalysis and factors that affect the course of these reactions in this solvent. Other Pd-catalyzed reactions in scCO2 have been less investigated.
4.2.1.1. Cross-coupling reactions
In the 1990s, it was found that the Heck reaction smoothly proceeds in scCO2 .[1035] Isomeric arylation products 329 – 331 were prepared by arylation of 2,3-dihydrofuran, ethyl 2,3-dihydropyrrolecarboxylate and cyclopentene, respectively, with iodobenzene in the presence of palladium acetate (10–1 – 10–3 mol.%) and a twofold amount of Ph3P (Scheme 203).
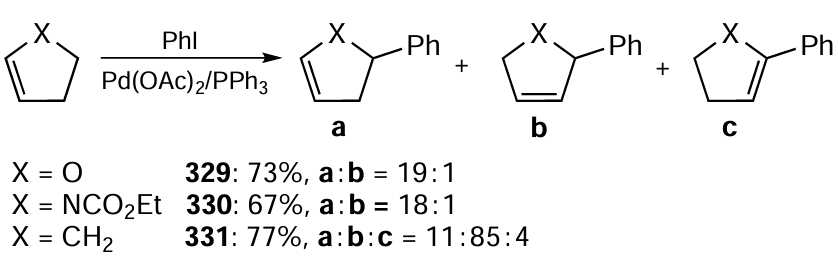
The main drawback of the synthesis was low reactant conversion, caused by low solubility of Pd(OAc)2 complexes with triphenylphosphine in the reaction medium. As expected, this stimulated the search for new catalysts. The use of fluorinated phosphines 332 and 333, obtained from chloro(diphenyl)phosphine and dichloro(phenyl)phosphine as ligands, was the first successful strategy towards the formation of palladium catalysts soluble in scCO2 (Scheme 204). A comparison of the solubility of PdCl2 and Pd(OAc)2 in the presence of these ligands and non-fluorinated phosphines [PPh3 , PCy3 , 1,1'-bis(diphenylphosphino)ferrocene] showed that only complexes Pd(332)2(OAc)2 , Pd(333)2(OAc)2 and Pd(333)2Cl2 are soluble in scCO2 .
The use of these complexes made it possible to conduct the Heck, Suzuki and Sonogashira reactions in scCO2 in moderate to high yields (Scheme 205).[1036]
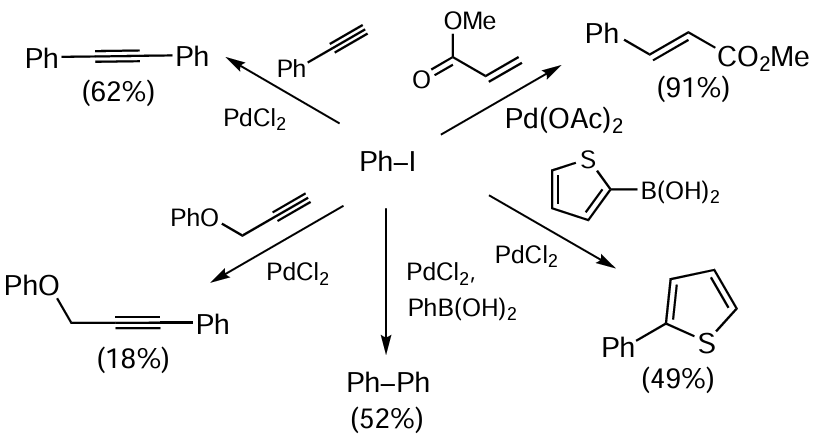
The fluorine-containing ligands P[3,5-(CF3)2C6H3]3 (334) and P(C6F5)3 (335) proved to be even more beneficial. In the presence of these ligands, not only the Heck reaction, but also the Stille reaction was carried out in scCO2 (Scheme 206).[1037]
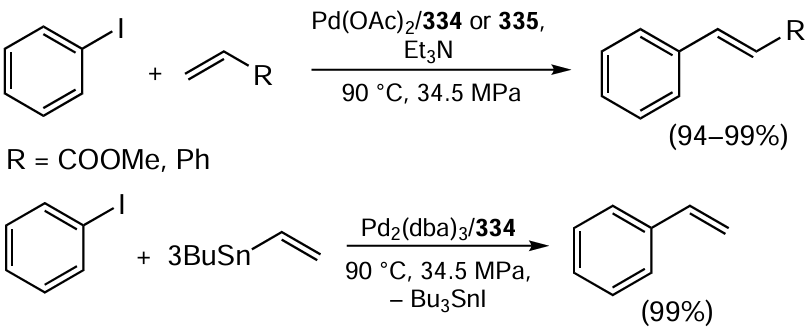
In recent years, this strategy was further developed by the synthesis of palladium complexes 336 and 337 with fluorinated nitrogen-containing ligands with a similar solubility in scCO2 (Scheme 207).[1038] Although nitrogen-containing palladium complexes are less active than phosphorus-containing complexes, the activity of the former is retained for several catalytic cycles and they are less toxic to the environment.
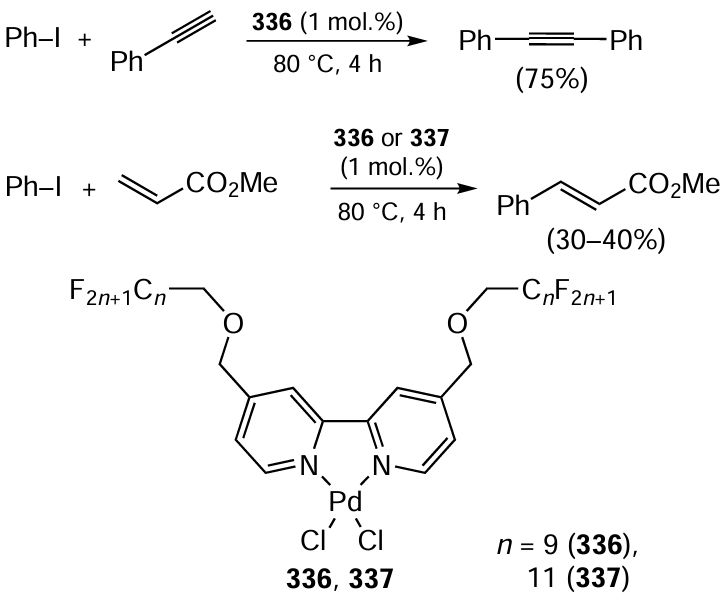
Complexes with nitrogen ligands based on ketoimines and oxazoles were proposed in 2011 for Pd-catalyzed coupling in scCO2 . These complexes, in particular complex 338, show higher activity in scCO2 than in conventional solvents and surpass in activity the commonly used phosphine ligands.[1039] In some cases, the conversion and the product yield reached 100% (Scheme 208).
Later, these reactions were carried out using combined phosphine-imine ligands.[1040] Homogeneous catalysts 339 – 341, containing perfluorinated substituents, successfully catalyze the Suzuki reactions in scCO2 (Scheme 209).
The solubility of palladium coordination compounds in scCO2 can also be increased by using palladium trifluoroacetate instead of the common acetate. For example, high yields of Suzuki reaction products were attained when Pd(OC(O)CF3)2 was used in combination with ligands 342 – 344 (Scheme 210).[1041]
The efficiency of catalysis (the yield of the target product and TOF) in scCO2 markedly decreases when going from active substrates to less active substrates. Attempts to circumvent this drawback while retaining the solubility of complexes in scCO2 led to the idea of using phosphorus-containing ligands decorated with silane or siloxane groups (Scheme 211).[1042][1043] It was found that the scCO2 solubility is correlated with the number of silicon atoms. However, the performance of silicon-containing ligands 345 and 346 was lower in scCO2 than in conventional solvents (toluene or DMF).
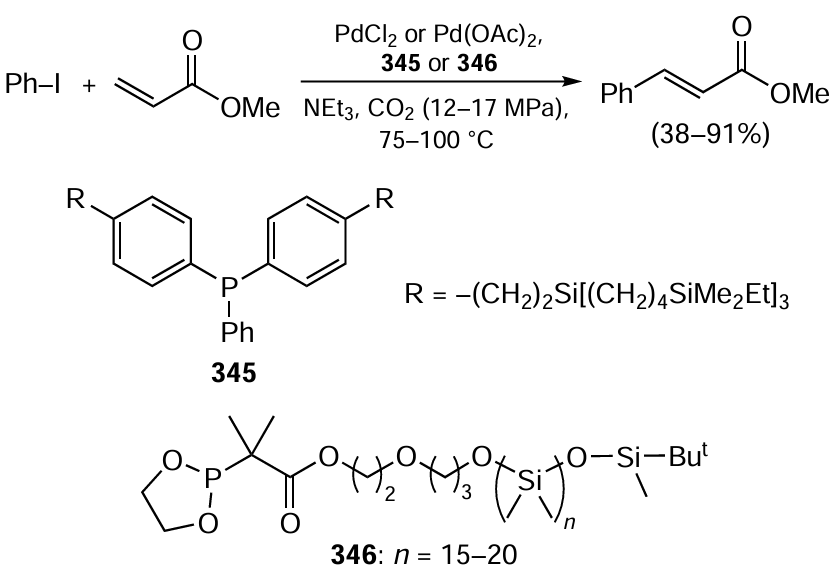
One more way to increase the solubility of palladium complexes in scCO2 is to replace triarylphosphines by trialkylphosphines, which are better soluble in scCO2 . It was shown that the system containing Pd(OAc)2 (5 mol.%) and P(But)3 (10 mol.%) can catalyze the reactions of iodobenzene with methyl acrylate and with phenylboronic acid.[1031]An additional increase in the solubility of palladium complexes can be attained by using palladium trifluoroacetate instead of palladium acetate.[1044]
Hybrid ligands containing phosphines with both aliphatic and aromatic substituents such as P(But)2(o-biphen) 347 and 2-dicyclohexylphosphino-2',4',6'-triisopropyl-1,1'-biphenyl (X-Phos) 348 were utilized for the synthesis of one more homogeneous catalyst for the Suzuki reaction[1041] and made it possible to perform the Ullmann reaction in scCO2 (Scheme 212).[1045][1046]
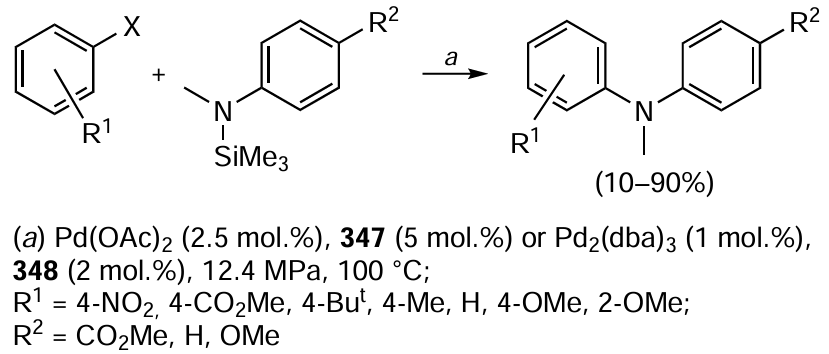
It is noteworthy that heterogeneous catalysis, which does not impose such severe restrictions on the source of palladium, is also applicable for cross-coupling in scCO2 . Even simple catalysts such as Pd/C provide high yields in the Suzuki reactions with complex substrates, e.g., iodoferrocene.[1047]
4.2.1.2. Homo-coupling reactions
The catalytic systems developed for the Heck reaction conducted in scCO2 , such as a mixture of palladium acetate or trifluoroacetate with tris-(2-furyl)phosphine, can catalyze the oxidative coupling of iodoarenes to give the corresponding biaryls in >95% yields (Scheme 213).[1048] In this case, scCO2 is not only preferable regarding green chemistry, but it is also necessary for the reaction to proceed, because the attempts to carry out analogous reactions with the same catalyst in common organic solvents did not result in noticeable amount of the target product.
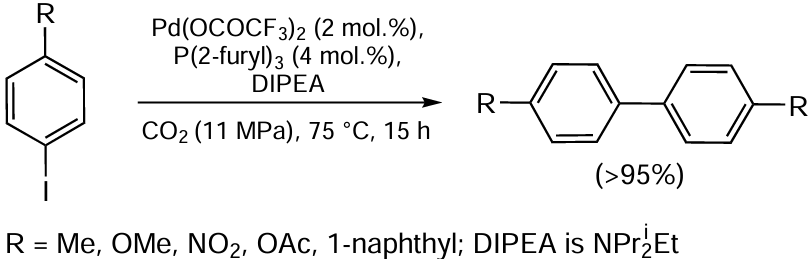
A study of the dimerization of arylboronic acids under palladium catalysis in scCO2 showed[1049] that the solubility – activity relationships similar to those described above for cross-coupling reactions also hold for homo-coupling. The yields of biphenyl with the Pd(PPh3)4 catalyst do not exceed 45%, because of its low solubility, being inferior even to the yields attained with heterogeneous catalysis in which palladium is stabilized by the PS-NMe2 and PS-NH2 polymers.
More complex heterogeneous catalytic systems such as graphite-immobilized palladium nanoparticles,[1050] proved to be effective in the homo-coupling reactions of aryl chlorides, which are less reactive substrates than aryl iodides or arylboronic acids.
4.2.1.3. Tsuji – Trost reaction
The Tsuji – Trost reaction, which involves the allylic substitution of a good leaving group by a C-nucleophile, is an important palladium-catalyzed reaction related to cross-coupling and giving rise to a C – C bond. The allylic alkylation of (E)-1,3-diphenylallyl acetate with dimethyl malonate in the presence of optically active palladium catalysts was accomplished in scCO2 (Scheme 214).[1051] The enantioselectivity of the reaction reached 90% when complex 350 was used, but replacement of the substrate with allyl acetate significantly decreased the product yield.
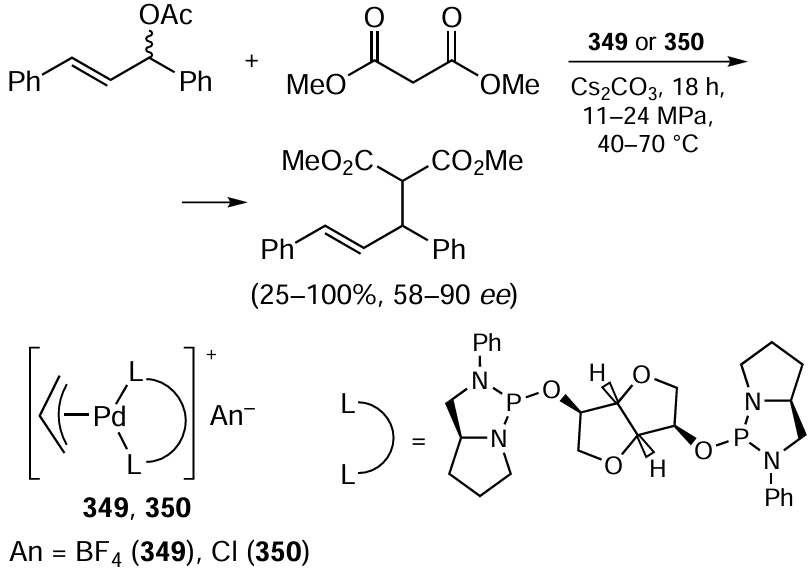
Meanwhile, when achiral substrates are used and the nucleophile is replaced with carboranemethylamine, an analogous reaction results in quantitative yields of products even in the presence of simple palladium catalysts (Scheme 215).[1052]
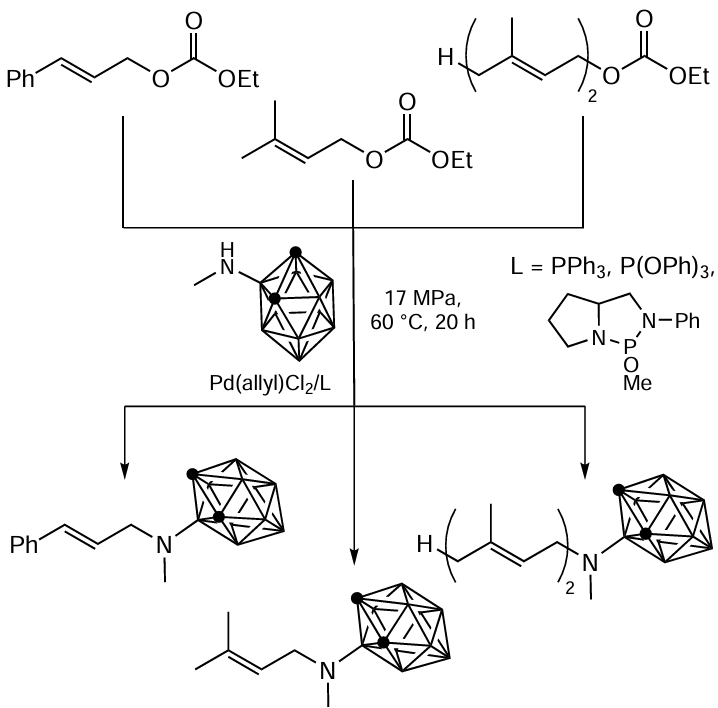
The pronounced difference between the reactivity of substrates and activity of the catalysts and very small number of examples of the Tsuji–Trost reaction in scCO2 preclude determination of the scope of applicability of the method regarding the ranges of both substrates and ligands.
4.2.1.4. Hydrogenation
Only single examples of palladium-catalyzed hydrogenolysis and hydrogenation reactions in scCO2 have been described to date. This contrast with common solvents, in which a multitude of homogeneous and heterogeneous palladium-catalyzed hydrogenation reactions are known, is attributable to the fact that in the case of hydrogenation, apart from the usual problem of low solubility of the catalyst in CO2 , there is the problem of catalyst poisoning by CO molecules that are formed from CO2 under the action of hydrogen.
Homogeneous catalysts based on palladium acetate and thiophene ligands 351 and 352 were tested in the hydrogenation of styrene, cyclohexene and 1-octene, but the yield of the target products was satisfactory only in the case of styrene (Scheme 216).[1053] The yield of the cyclohexene reduction product did not exceed 20%, while in the case of 1-octene, side isomerization reactions took place, and the yield of n-octane was also low.
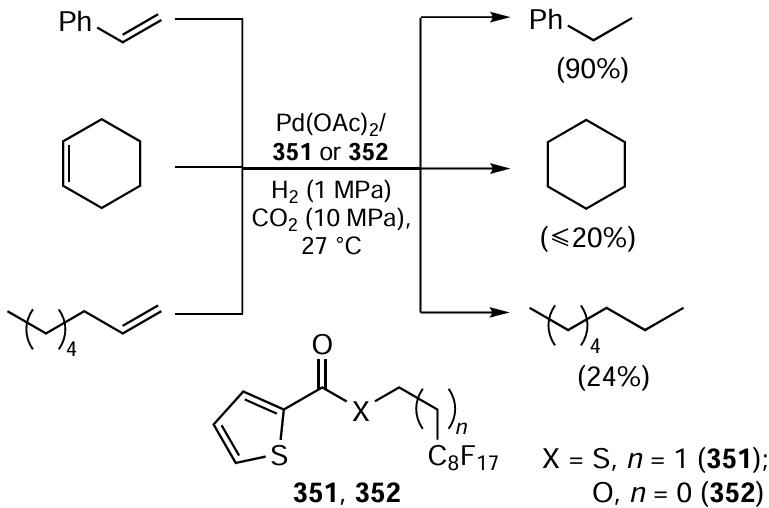
Heterogeneous catalysts were also investigated in the hydrogenation of unsaturated compounds. The natural terpenoid pulegone (353) can be selectively reduced to cis- and trans-menthone with hydrogen in scCO2 over the Pd(0.5%)/Al2O3 bimetallic catalyst (Scheme 217).[1054] The conversion reaches 99%, while the total content of menthone isomers is 98.6% as soon as 15 min after the onset of the reaction, which makes this method fairly promising. It is of interest that replacement of Pd with Ru in the bimetallic catalyst changes the course of the reaction, which then gives product 354.
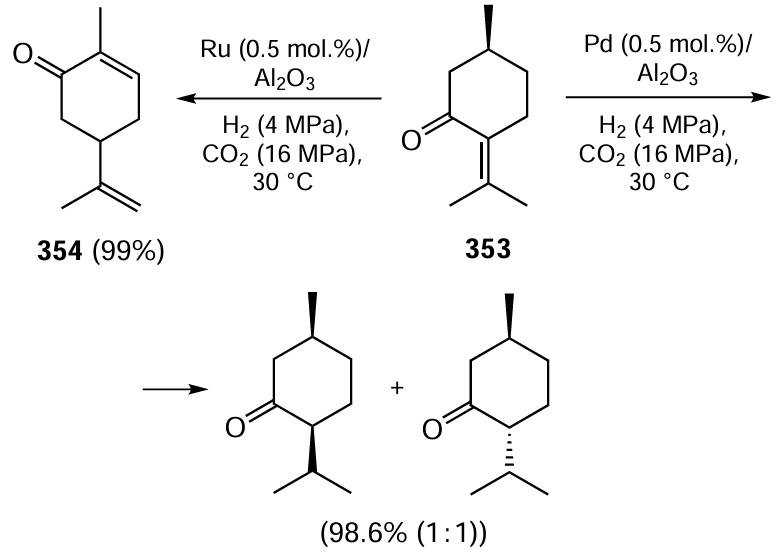
More complex catalysts based on palladium complexes immobilized on silica gel or polymer substrates were also obtained.[1053] It was shown that silica gel-immobilized palladium complex 355 with ligand 352 based on thiophenecarboxylic acid (Scheme 218) is eight times more active in the styrene hydrogenation reaction than analogous homogenous catalyst (see Scheme 216) and can be used at least in ten successive reactions without a noticeable decrease in the activity. Hydrogenation of less reactive cyclohexene proceeds over the immobilized catalyst to a conversion of 99 – 100%, but does not take place with the analogous homogeneous catalyst.
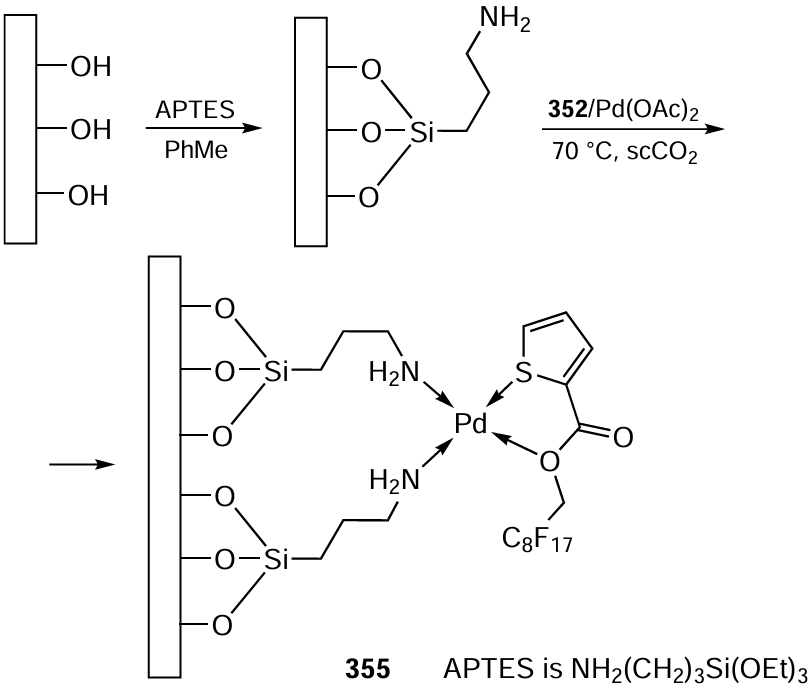
Interesting results were obtained in the hydrogenation of phenylacetylene and 2,5-dimethyl-2,4-hexadiene in the presence of supported catalysts based on cross-linked PPI and PAMAM dendrimers.[1055] The major reduction products were styrene and trans-2,5-dimethyl-3-hexene, with the selectivity to olefin reaching 73 – 98%.
The presented data indicate that all the main classes of palladium-catalyzed reactions have now been adapted to be performed in scCO2 . These reactions are efficiently catalyzed by three types of palladium catalysts: complexes with fluorinated arylphosphines and alkylphosphines and palladium trifluoroacetate. However, currently only the Heck and Suzuki reactions and the related homo-coupling of aryl halides and phenylboronic acids in scCO2 are competitive with analogous reactions in common solvents as regards the yields of the target products and the reaction rates.
4.2.2. Rhodium-catalyzed reactions in supercritical CO2
Among the diverse rhodium-catalyzed reactions, only hydrogenation, hydroformylation, hydroaminomethylation and hydroboration have been accomplished in scCO2 . Among them, selective and, in some cases, enantioselective hydrogenation reactions have been studied in sufficient detail; hydroformylation reactions have been studied markedly less comprehensively; and for other reactions only single examples are available.
4.2.2.1. Catalytic hydrogenation
Catalytic hydrogenation over rhodium catalysts in scCO2 suffers from the same drawbacks as similar reactions with palladium catalysts: catalyst poisoning by CO formed in the reaction, undesirable alkene isomerization and low solubility of the metal-containing catalysts in the reaction medium.[1033] Homogeneous catalysts 356 based on rhodium and phosphine containing SiMe2 and C8F17 groups intended for increasing the catalyst solubility in scCO2 were tested in the hydrogenation of 1-butene (Scheme 219).[1056] The complete substrate conversion to the product was attained within 100 min (353 K and 20 MPa of CO2). Unlike earlier studies on the catalytic hydrogenation in scCO2 , in this case, the authors used nanoporous silica membranes to separate the catalyst from the reaction product. In the continuous flow operation of the membrane reactor, the turnover number reached 120 000, which is the record-high value for catalytic reactions in scCO2 .
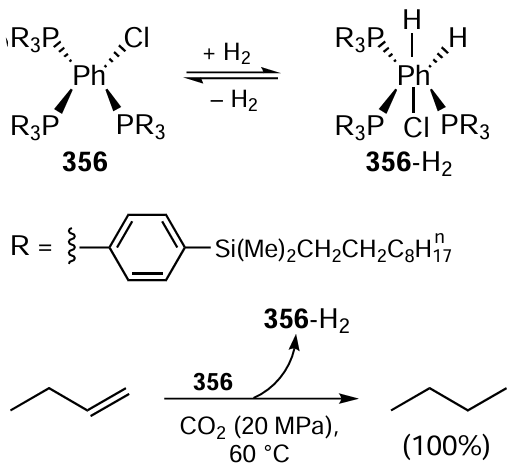
The selective hydrogenation of styrene to ethylbenzene was also performed in scCO2 using rhodium-based homogeneous catalysts.[1057][1058] A comparison of the performance of catalysts 357 – 359, differing in the number and arrangement of fluorine atoms in the ligands and counter-ions, has been reported[1057] (Scheme 220).
As for the palladium catalysts considered previously, the introduction of fluorine into arylphosphines and counter-ions increase the catalyst solubility in scCO2 and the catalyst performance. The highest yields of ethylbenzene (77%) and the highest performance (TOF = 48.4 h–1 and TON = 387.2) were attained with catalyst 358 containing fluorine in both the arylphosphine ligand and the counter-ion. The replacement of monodentate arylphosphine ligands with BINAP-containing bidentate ligands 360 increased the styrene conversion to 96% under the same conditions and provided an increase in TOF and TON to 160.7 h–1 and 482.0, respectively (Scheme 221).[1058]
The problem of low solubility in scCO2 , inherent in phosphine ligands, can be circumvented by using rhodium-based ligand-free heterogeneous catalysts. The efficiency of the reaction is boosted by high solubility of dihydrogen and high mass transfer coefficients in this solvent. The use of heterogeneous catalysis also solves the problem of catalyst regeneration after the reaction. The studies on alkene hydrogenation in scCO2 published over the last five years address particularly heterogeneous catalysts.[1055][1059][1060]
The catalysts obtained by hydrogen reduction of rhodium acetylacetonate embedded in polymer matrices in scCO2 showed a high performance in the catalytic hydrogenation of 1-octene and styrene.[1055] For example, supported catalysts containing 0.010 – 0.056 mass % rhodium provide 31 – 99% conversion for 1-octene and 37 – 100% for styrene; the use of cross-linked PPI and PAMAM dendrimers as polymer matrices almost completely suppresses the isomerization of 1-octene, one of the most side reactions observed during hydrogenation in the presence of Pd- and Rh-based homogeneous catalysts. The simplest heterogeneous catalyst Rh/C (12 mass %) catalyzes the hydrogenation of phenols and naphthalenes with preparative yields.[1059] More complex supported catalysts 361 based on rhodium nanoparticles immobilized on the surface of decyltriethoxysilane-modified SiO2 exhibit an even higher performance in the hydrogenation of substituted benzene derivatives to the corresponding cyclohexanes.[1060]The conduction of these reactions in scCO2 reduces the contribution of side reactions leading to the reduction or elimination of substituents in the benzene ring and markedly increases the conversion of the starting compounds. The heterogeneous hydrogenation reactions in scCO2 proved to be more efficient than the same reactions in heptane. For example, the reactant conversion in the hydrogenation of polyfluoroarenes is 2 – 35% in heptane (2 – 6% selectivity) and 20 – 78% in scCO2 (28 – 80% selectivity) (Scheme 222).
In particular, these reactions provided one-step high-yield synthesis of fluorinated cyclohexane carboxylates, intermediates of the synthesis of fluorine-containing polymers and liquid crystals, and other fluorine-containing cyclohexane derivatives that are difficult to prepare by alternative synthetic routes.
4.2.2.2. Catalytic hydroformylation
The catalytic hydroformylation is widely used in industry to produce aldehydes from readily available olefins on treatment with a mixture of H2 and CO. Supercritical CO2 attracted attention of researchers as a solvent for these processes back in 1991.[1061] Both H2 and CO are infinitely soluble in scCO2 . In addition, H2 and CO2 mixtures do not tend to self-ignite over a broad range of concentrations, which makes both liquid and supercritical CO2 a promising solvent not only for laboratory, but also for industrial synthesis.
To date, there are experimental data on the effect of various process parameters and rhodium catalyst structure on the hydroformylation reactions in scCO2 .[1062-1066] It was shown that the yields and activation energies of the reaction in benzene, toluene, ethanol and scCO2 are comparable. An increase in the CO partial pressure decreases the regioselectivity and the rate of hydroformylation in any of the solvents (excess CO displaces the phosphine ligands from the Rh, which results in precipitation of the catalyst and loss of activity). The main difference between the reactions in common solvents and in scCO2 is that in the latter case, the inhibitory effect of high olefin concentrations disappears. This useful feature of scCO2 is attributable to the difficulty of formation of dimeric rhodium species under supercritical conditions.[1067] Using phosphine ligands 362 and 363 (Scheme 223) containing bulky carborane C2B10H12 moieties, it is possible to retard this undesirable process and increase the efficiency and the regioselectivity of hydroformylation of styrene, 3,3-dimethylbutyl-1-ene, hept-1-ene, oct-1-ene and R-limonene.
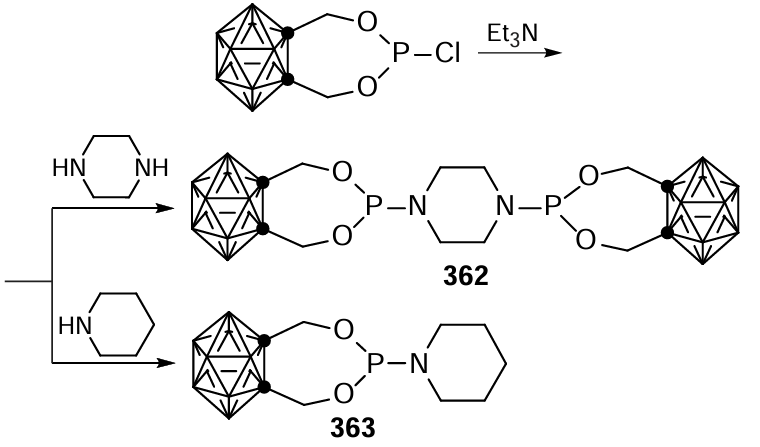
Homogeneous Rh-containing catalysts for the use in scCO2 should comply with the same requirements (low ligand polarity) as palladium complexes. The most popular ligands for Rh, like for Pd, are arylphosphines[1063] and aryl phosphites[1068] with perfluorinated substituents and simple trialkylphosphines.[1069]
The yields and selectivity of 1-hexene hydroformylation in the presence of Rh supported on phosphorylated silica gel are comparable with those for similar reactions carried out with Rh(EtPPh2)3 in toluene.[1070] It is worth noting that, like for other reactions proceeding in scCO2 , modern studies dealing with hydroformylation give preference to heterogeneous catalysts, which provide comparable yields of the target products, but are much more easily separated from the reaction mixture and can be reused many times without losing the activity.[1071][1072] Rhodium nanoparticles immobilized on polystyrene[1071] or oxidized graphene[1072] provided high yields in the hydroformylation of styrene, 4-methylstyrene, 4-bromostyrene, 4-isopropylstyrene (100% conversion, 40 – 94% selectivity to the secondary aldehyde), tert-butylethylene (100% conversion, 100% selectivity to the terminal aldehyde). The regenerated catalyst did not lose activity in the six subsequent cycles.
4.2.2.3. Catalytic hydroaminomethylation
The catalytic hydroaminomethylation opens up the access to structurally diverse amines starting from readily available olefins.[1073] Only one example of hydroaminomethylation in scCO2 has been reported in the literature.[1074] A key feature of this reaction is that CO2 serves simultaneously as a solvent and as a temporary protecting group[1075] for the NH group (Scheme 224). In organic solvents, compound 364, resulting from fast lactamization of the rhodium intermediate accompanied by reductive elimination of the catalyst, is the major hydroaminomethylation product.
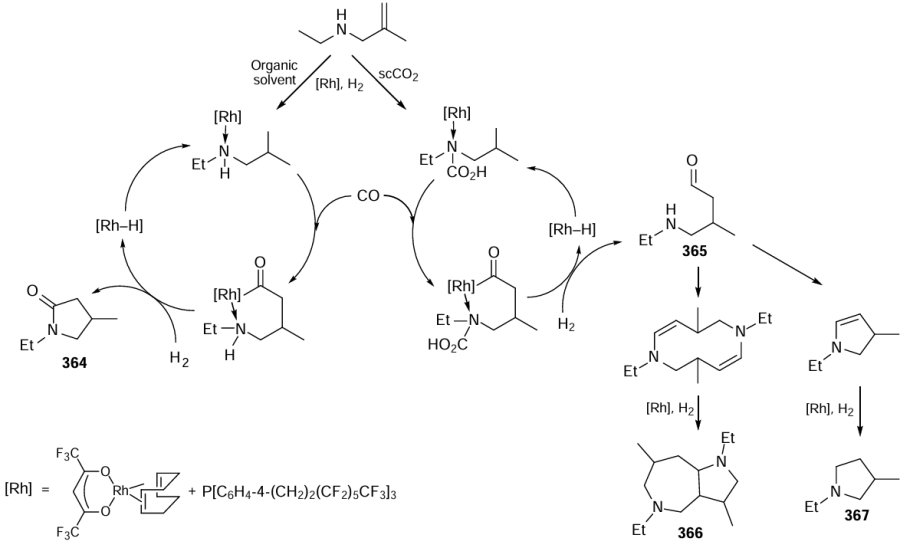
However, in scCO2 , the amino group reversibly adds a solvent molecule, being converted to carbamic acid, which markedly reduces the lactamization rate, with γ-aminoaldehyde 365 becoming the major product. Then compound 365 undergoes an intermolecular or intramolecular cyclization followed by the reduction to amines 366 and 367, which are not formed in hydroaminomethylation reactions in organic solvents. A thorough control of the reaction conditions made it possible to reach a conversion of 77% and a selectivity of 76% to the target pyrrolidine.
4.2.2.4. Catalytic hydroboration
One more vivid example in which scCO2 not only acts as a non-polar aprotic solvent, but also directly affects the course of the reaction is the catalytic hydroboration of 4-vinylanisole with pinacolborane.[1076] The transition from organic solvents to scCO2 not only preserves the high conversion (81 – 100%), but also significantly increases the selectivity (from 25 – 32% to 88 – 100%). The increase in the selectivity was attributed to the fact that CO2 molecules stabilize more efficiently the η3-benzyl-Rh intermediate (Int25), which leads to product 368, than the η1-benzyl-Rh intermediate (Int26), which leads to by-products 369 – 371 (Scheme 225).
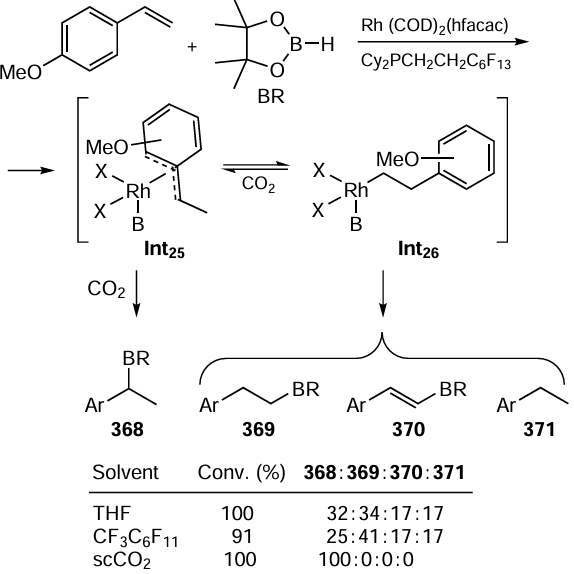
4.2.2.5. Catalytic C – H bond activation
Interesting results were attained upon catalytic C – H bond activation in the presence of the Rh2(OAc)4 complex (Scheme 226).[1077] Owing to the high solubility and stability of diazo compounds in liquid carbon dioxide, it was possible to select conditions under which selective intramolecular cyclization takes place to give β-lactam 372. For other platinum group metals, reactions of this type have not been reported.
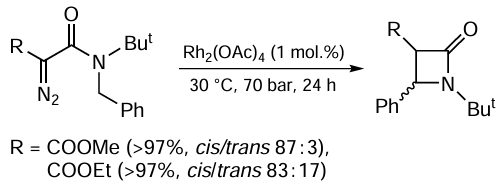
Generally, CO-tolerant rhodium catalysts, including heterogeneous and supported ones, are fairly promising for the use in the CO2 medium. In particular demand are catalytic hydrogenation reactions under these conditions that are characterized by higher yields of the target products and high TOF and TON. Furthermore, rhodium catalysts are convenient for handling and can be easily regenerated from scCO2 .
4.2.3. Ruthenium-catalyzed reactions in supercritical CO2
Coordination compounds of ruthenium are used as catalysts for olefin hydrogenation in scCO2; as a rule, their performance is comparable to that of analogous rhodium catalysts. Ruthenium complexes also catalyze hydrogenation of carbonyl compounds[1078] that cannot be reduced over rhodium catalysts. In addition, there are a few examples of application of Ru complexes as catalysts for the hydroformylation,[1079] formation of vinyl carbamates from terminal alkynes, amines and CO2 ,[1080][1081] and for olefin metathesis.[1082]
4.2.3.1. Catalytic hydrogenation
The first examples of using ruthenium coordination compounds in the catalytic hydrogenation in scCO2 , published in 1994, were related to the CO2 reduction to formic acid.[1083] Using the simplest catalysts 373 and 374 with trimethylphosphine ligands, TON = 3700 – 7200 and TOF = 1400 h–1 were attained in these reactions. Later, this procedure was adjusted to the preparation of methyl formate and dimethylformamide (Scheme 227), while the range of catalysts was supplemented by more active compounds 375 – 377, characterized by high TON up to the record-high value of 740 000.[1029][1084-1086]
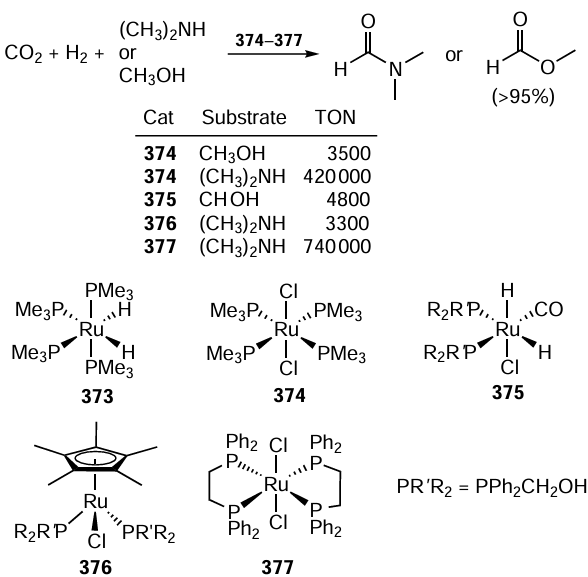
However, ruthenium-based catalysts suitable for the catalytic hydrogenation of olefins and more complex organic compounds could not be found for a long time. A considerable progress in this field was attained owing to chiral Ru complexes with binaphthyl ligands containing polyfluoroalkyl substituents.[1087] In the presence of catalysts 378 and 379, hydrogenation of dimethyl itaconate gave product 380 in 94 – 100% yield with 73 – 76% enantiomeric excess (Scheme 228).
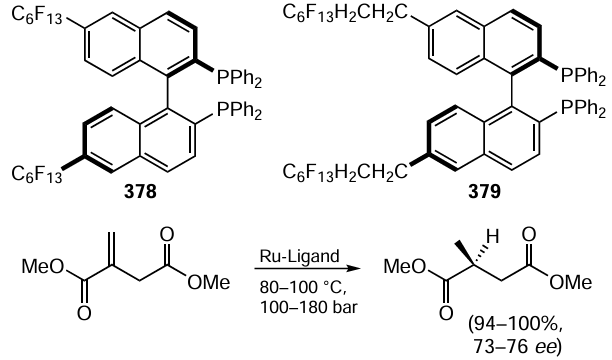
However, this reaction was more selective in methanol (95 – 96% ee). This was attributed to low polarity of CO2 , which cannot form hydrogen bonds with the substrate and/or the catalyst during the reaction. To verify this assumption, further studies are required, because the addition of methanol to scCO2 does not lead to increase in ee.
The reduction of keto esters (ethyl 4-chloro-3-oxybutyrate and dimethyl acetylsuccinate) smoothly proceeds in scCO2 containing added [Bmim]BF4 ionic liquid in the presence of analogous chiral catalysts based on [RuCl2(C6H6)]2 and (R)-BINAP at 75 °C and 11 – 15 MPa. This reaction afforded enantiomerically enriched (92 – 97% ee) β-hydroxy ester 381 and isomeric γ-lactones 382a,b in a quantitative yield (Scheme 229).[1078] The authors noted a pronounced effect of the polar co-solvent (in this case, ionic liquid) on the reaction enantioselectivity. In the absence of the ionic liquid, the enantiomeric excess in the products did not exceed 60 – 80%.
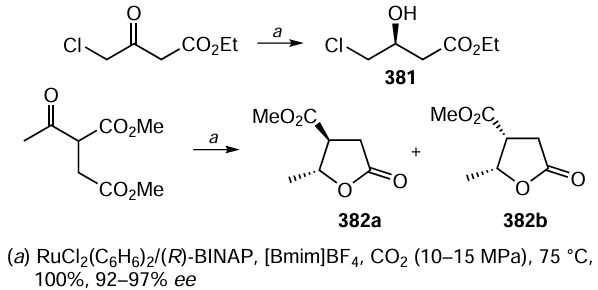
The results of studying the reduction of levulinic acid (383) to γ-valerolactone (384) indicate that, despite the low polarity of CO2 molecules, ruthenium complexes are considerably changed during the reactions (Scheme 230).[1088]
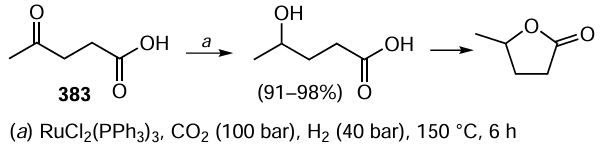
Carbon monoxide molecules, formed in a minor amount in any hydrogenation reactions in scCO2 , not only do not poison the ruthenium catalyst, but also convert the less active RuCl2(PPh3)3 complex to more active RuHCl(CO)(PPh3)3 . In the absence of CO2 , the initial complex is transformed with time to RuH2(PPh3)3 , which is inert towards hydrogenation, and this leads to a noticeable decrease in the catalytic activity. Thus the conversion of 383 in scCO2 reaches 100%, with the selectivity to γ-valerolactone (384) being 91 – 98%, whereas in methanol, the conversion does not exceed 31%, while the selectivity is 84%.
Up-to-date studies attest to the preference of heterogeneous ruthenium catalysts for hydrogenation over the homogeneous catalysts. The ruthenium additives are also useful in the heterogeneous palladium catalysis.[1089] Mono- and bimetallic catalysts have been encapsulated into polymer (polyurea) matrices. The palladium-catalyzed reduction of acetophenone to 1-phenylethanol (385) results in 70% yield and 80% selectivity. The Pd/Ru (1 : 1) bimetallic catalyst increases the conversion to 97%, but the selectivity to alcohol 385 decreases to 70%. A by-product formed in this case is 1-cyclohexylethanol, resulting from complete hydrogenation of acetophenone, which is not formed in the case of palladium catalysis (Scheme 231).
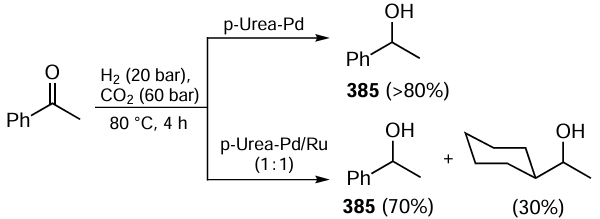
Despite the attractiveness of heterogeneous catalysis in scCO2 , associated with easy separation and higher stability of the catalyst, the effect of the support on the Ru catalytic activity has been little studied so far.[1090] It was shown that variation of the support can change the conversion in the hydrogenation of furfuryl alcohol to 1,2-pentanediol in the range of 12 – 84% under identical conditions.
4.2.3.2. Formation of vinyl carbamates
Secondary amines are spontaneously converted to carbamic acids in scCO2 , but the latter can hardly be isolated, as the equilibrium shifts towards the starting compounds upon decompression. For stabilization of carbamic acids, a strategy similar to Ru-catalyzed synthesis of dimethylformamide from dimethylamine and CO2 has been proposed.1029, 1084 – 1086 In the presence of ruthenium complexes Ru(C5H5N)4Cl2 (386) and Ru(C6H6)(PMe3)Cl2 (387), phenylacetylene adds to carbamic acid to give vinyl carbamates 388 (Scheme 232). In all cases, the reaction in scCO2 was more efficient than in saturated CO2 solutions in toluene and acetonitrile.1080
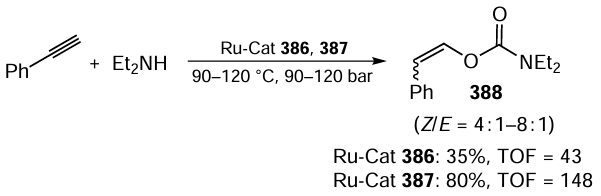
4.2.3.3. Catalytic hydroformylation and alkoxycarbonylation
Although rhodium complexes are the most popular hydroformylation catalysts both in organic solvents and in scCO2 , some examples of hydroformylation over ruthenium catalysts have also been reported.
By analogy with hydroformylation of propylene in scCO2 in the presence of cobalt carbonyls,[1061] ruthenium carbonyls have also been tested in these reactions.[1077] Ru3(CO)12 catalyzed the hydroformylation of ethylene in the temperature range of 70 – 125 °C and pressure range of 22 – 41 MPa; however, the catalyst efficiency was low: TON in scCO2 did not exceed 190 (157 in DMF). Due to low efficiency, Ru-based catalysts cannot compete with more active Rh catalysts, which have been comprehensively studied.
Ruthenium carbonyl Ru3(CO)12 can catalyze alkoxycarbonylation reactions related to hydroformylation. For example, the Ru3(CO)12/[Bmim]Cl system effectively catalyzes the formation of methyl propionate from ethylene, carbon dioxide and methanol, with the conversion reaching 100% and the selectivity to the target product being as high as 92% (Scheme 233).[1091][1092]
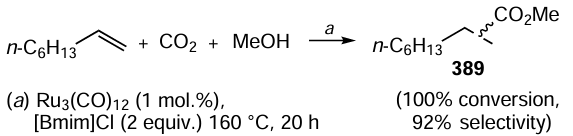
4.2.3.4. Catalytic olefin metathesis
The ruthenium Grubbs catalysts are active in the metathesis of olefins. They favourably compare with the analogues based on molybdenum and other metals in their high stability in air and tolerance to various functional groups and impurities in solvents and substrates.[1093] Despite numerous publications dealing with the use of the Grubbs catalysts and related coordination compounds in organic synthesis, there are only single examples of metathesis reactions carried out in scCO2 . It was shown that first-generation Grubbs catalysts are effective in the synthesis of macrocycles by olefin metathesis in scCO2 .[1082][1094] The course of the reaction depends on the density of the medium: when d < 0.65 g cm–3, oligomerization of olefins predominates, while in the case of d > 0.65 g cm–3, macrocyclic products are formed in up to 88% yields (Scheme 234).
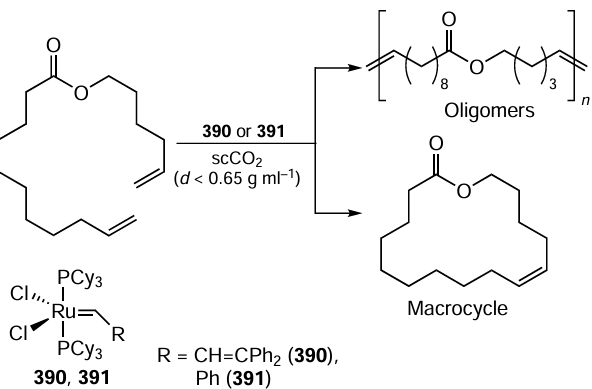
The ability of CO2 to protect amino groups of olefins by forming carbamic acid moieties with them[1075] makes it possible to increase the product yields in the metathesis involving unsaturated amines compared to analogous reactions in organic solvents. Cross-metathesis of mixtures of natural lipids isolated by supercritical fluid extraction from unicellular algae was successfully carried out in scCO2 using the first- and second-generation Hoveyda – Grubbs catalysts.[1095] The reactions were carried out at 45 °C and 30 MPa CO2 pressure in the presence of 1 mol.% of the catalyst, which gave mixtures of olefins and unsaturated carboxylic acid esters containing 5 – 11 carbon atoms in 90 – 95% yields directly from biomass.
However, today there is limited data on the dependence of the activity and scCO2 solubility of Ru complexes on the catalyst structure. The design of optimal ligands and Ru-containing catalysts with these ligands is a challenge for future research.
4.2.4. Copper-catalyzed reactions in supercritical CO2
Although copper complexes are utilized in organic reactions of various types, only three classes of Cu-catalyzed reactions in scCO2 have been systematically described, particularly, azide–alkyne cycloaddition (click-reaction), C – H activation of alkanes via carbenes and their synthetic equivalents, and oxidative coupling of alkynes. In all cases, owing to specific chemical and physicochemical properties of carbon dioxide, copper-catalyzed reactions proceed more efficiently in scCO2 than in common solvents.
4.2.4.1. Azide – alkyne cycloaddition
Copper-catalyzed azide – alkyne cycloaddition (CuAAC) is among the most valuable reactions of click-chemistry.[1096] The first examples of CuAAC reactions in scCO2 were reported in 2009,[1097] and several effective homogeneous and heterogeneous catalysts for these reactions have now been developed.
The copolymer of α-azido and unsubstituted ε-caprolactones was modified with 1,2,3-triazole moieties using the CuAAC reaction with terminal alkynes in scCO2 (Ref. [1097]) (Scheme 235). The catalyst was generated in situ from copper(I) iodide and fluorinated triamine 392 as the ligand. The product yields in this reaction reached 75 – 100% and depended little on pressure. The catalyst soluble in scCO2 was uniformly distributed throughout the polymer matrix and could be regenerated after the reaction by supercritical CO2 extraction.
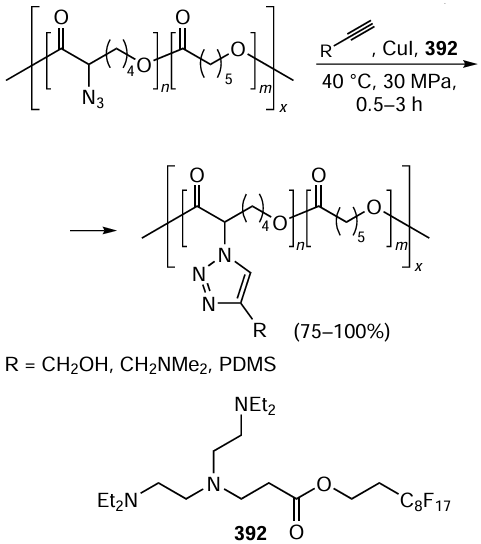
The catalytic system comprising CuBr and polymer-immobilized ligand 393 was effective in the tandem one-pot reaction including the CuAAC reaction of 2-azidoethyl 2-bromo-2-methylpropanoate 394 with ethynylpyrene and the subsequent cycloadduct-initiated polymerization of methyl methacrylate giving polymer microspheres 395 (Scheme 236).[1098] At a pressure of 30 MPa and a temperature of 60 – 75 °C, a conversion of 90 – 95% was attained within 24 – 36 h.
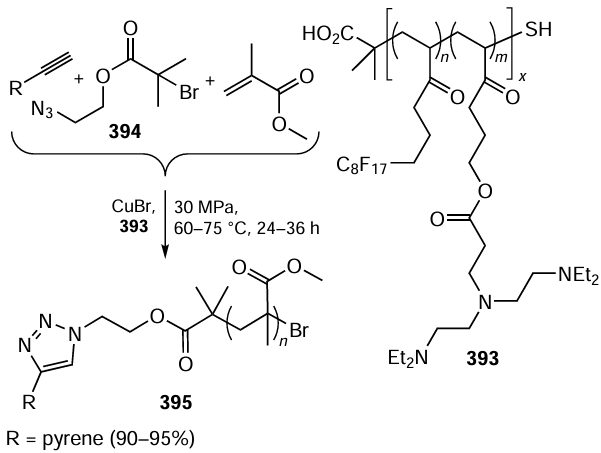
The scope of applicability of the CuAAC in scCO2 was markedly expanded after the discovery of high catalytic activity of Cu(OAc)2 · H2O in this reaction.[1099] Copper(II) salts with various anions were tested as catalysts for the reaction of phenylacetylene with benzyl azide at 36°C and 30 MPa. The halides CuCl2 and CuBr2 proved to be catalytically inactive; CuSO4 · 5 H2O and Cu(NO3)2 · 3 H2O provided 5% yield of 1,2,3-triazole, and only in the case of Cu(OAc)2 · H2O, the yield was quantitative in the presence of only 0.01 mol.% of the catalyst. The activity of the acetate did not decrease when the pressure was reduced to 8 MPa. Under optimal conditions, a pool of triazole derivatives (35 compounds) were synthesized in 90 – 99% yields (Scheme 237).
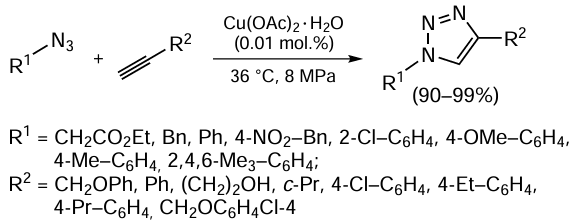
The efficiency of the Cu(OAc)2 · H2O-catalyzed CuAAC in scCO2 was demonstrated in relation to functionalization of the lactide polymer 396 containing a terminal propargyl group with 4-(azidomethyl)coumarin (397) (Scheme 238).[1100]
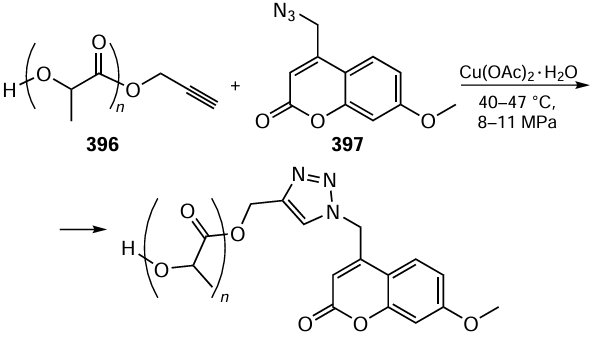
These were the first examples of successful CuAAC reactions in scCO2 in the presence of Cu(II) salts that have no catalytic activity in these reactions carried out in organic solvents. Further search for alternative catalysts for CuAAC in scCO2 resulted in the discovery of one more catalyst, copper metal, which has not been used in CuAAC reactions in organic solvents.[1101] In the presence of copper, the yield of cycloadducts in scCO2 (50 °C, 10 MPa) amounted to 90 – 99%.
Copper wire also proved to be an effective catalyst for the reaction of polylactide (396) containing a terminal propargyl group with 4-(azidomethyl)coumarin (397) under conditions similar to those shown in Scheme 238. However, a sub-stoichiometric amount of the catalyst (62 – 71 mol.%) is needed to attain a high yield of the cycloadduct (71 – 93%).[1102]
Examination of the morphology of copper wire and copper plates recovered after CuAAC in scCO2 showed that their weight has decreased during the reaction by 3 – 5%.[1103] These results are consistent with the results of another study[949] devoted to the morphology of copper nanoparticles after they have been kept in scCO2 (80 MPa). It was shown that copper nanoparticles are oxidized under these conditions and are coated with a layer of amorphous material consisting of a mixture of copper(I) and copper(II) oxides. After 12 h, this results in the formation of core–shell nanoparticles completely coated with copper oxide. Apparently, particularly the copper oxide nanoparticles formed in situ on the surface of copper wire in scCO2 act as the active catalyst species in CuAAC reactions.
4.2.4.2. C – H functionalization of alkanes
The use of diazo compounds (carbene precursors) for C – H bond functionalization in non-activated alkanes has become a potent tool of organic synthesis used to introduce the desired functional group into the carbon skeleton of the molecule in one step. Typically, alkane serves as both the substrate and the solvent, since the addition of almost any other solvent containing alternative C – H bonds complicates the reaction. The greatest challenge is the C – H activation of methane in which the C – H bond energy is the greatest among all alkanes (105 kcal mol–1).[1104] The use of scCO2 as the solvent eliminates this problem and markedly expands the range of substrates.
The possibility of functionalizing gaseous alkanes with diazo compounds in the presence of copper catalysts, in particular complex 398, was discovered in 2015 (Scheme 239).[1105] Later, liquid C5 – C6 alkanes were also included into the range of applicable substrates.[1106] The reactivity of alkanes increases in parallel with the decrease in the C – H bond energy in the series from methane to hexane, while the activity of copper- and silver-based catalysts remains approximately the same. When scCO2 is used as the reaction medium, the ratio of the isomeric products changes, apparently, due to the electron density redistribution caused by interaction of CO2 molecules with the perfluorinated substituents in the heterocyclic ligands.
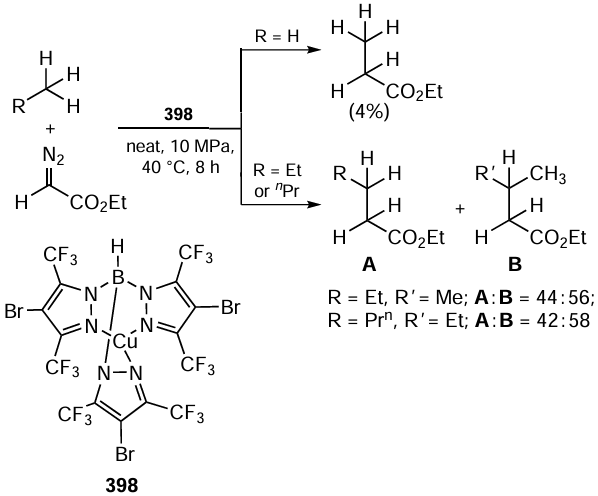
Recently, effective heterogeneous catalyst 399 for the reaction of diazoethyl acetate with n-hexane was obtained by immobilization of copper(I) coordination compound on silica.[1107] It was found that the carbene insertion follows the same patterns as in the case of homogeneous catalysis under similar conditions.
In 2021, the possibility of related amide insertion into the C – H bond in the presence of di-tert-butyl peroxide as the radical oxidant was demonstrated (Scheme 240).[1108] Catalyst 400, which is formed in situ from CuI and phenanthroline modified with the C8F17 moiety, is suitable for the synthesis of various carboxylic acid alkylamides 401. It is of interest that complex 398 (see Scheme 239), effective towards the carbene insertion into the C – H bond, has low activity in the reaction with amides.
4.2.4.3. Oxidative coupling of alkynes
The oxidative coupling of terminal alkynes in the presence of copper salts is a convenient method for the synthesis of symmetrical internal diynes, important intermediates in the preparation of heterocyclic and polyfunctional systems. The possibility of conducting this reaction in scCO2 was first demonstrated in 2006.[1109] Under optical conditions [alkyne (1 mmol), CuCl2 (2 mmol), NaOAc (2 mmol) and MeOH (1 mL); 14 MPa, 40 °C], coupling products were obtained in virtually quantitative yields (Scheme 241).
The temperature and pressure in the system have no virtually no effect on the catalyst performance. However, the addition of methanol and the types of copper(II) salt and the base were crucial: only the CuCl2 – NaOAc – MeOH catalytic system, partially soluble in scCO2 , was effective for this reaction. In the presence of other copper salts and/or other bases, the product was formed only in trace amounts. The solubility of the starting alkynes in scCO2 also plays an important role: for example, the conversion of hex-5-yn-1-ol insoluble in scCO2 is only 24% after 24 h, while the branched homologue, 2-methylbutyl-3-yn-2-ol, which is better soluble in scCO2 , quantitatively reacts over a period of 4 h.
The range of substrates was expanded by using the CuCl – DBU catalytic system (Scheme 240).[1110] The rejection of divalent copper provided high yields of the products of oxidative coupling of ferrocenylacetylene and cyclopropylacetylene, and a pressure drop to 9 MPa increased the solubility of substrates and the catalytic system in scCO2 even without methanol addition.
An interesting type of oxidative coupling is the cross-coupling of two different terminal alkynes occurring over the Cu/Pd bimetallic catalyst (Scheme 242).[1111] As in the case of homo-coupling of terminal alkynes, changes in the reaction temperature and scCO2 pressure do not influence significantly the yields and the reaction selectivity. When going from scCO2 to organic solvents, the efficiency of the reaction decreases.
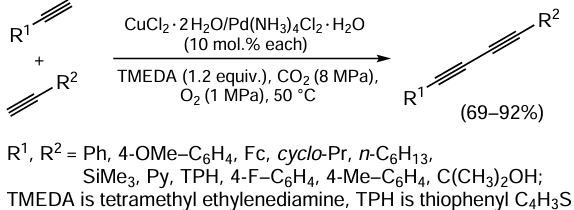
Supercritical CO2 is not a fully inert solvent in these reactions. Quite recently, it was found that in the absence of oxygen, the CuI – DBU system catalyzes direct carboxylation of terminal alkynes (Scheme 243).[1112] This reaction opens up a green synthetic route to propionic acids, important intermediates in the synthesis of various biologically active and heterocyclic compounds. Under optimized conditions it is possible to obtain the target propionic acid in a yield of up to 92%, which is comparable in efficiency with syntheses involving organometallic reagents.
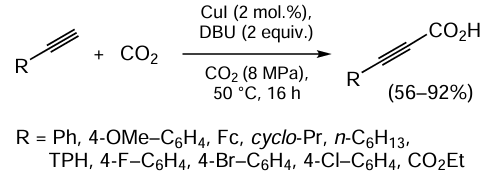
Thus, copper-catalyzed reactions in scCO2 not only represent an environmentally safe alternative to catalytic processes already known in organic solvents, but they also provide chemical transformations that cannot be conducted without scCO2 . Indeed, the CH-activation reactions of liquid alkanes in CO2 and in excess alkane are comparable in efficiency; however, the CH-activation of methane without scCO2 is impossible. Simple homogeneous (CuOAc2 · H2O) and heterogeneous (copper metal) catalysts lead to a high conversion of substrates in scCO2 , but are inactive in organic solvents. Currently, copper can be considered to be synergistic with scCO2 in which it acquires new properties not characteristic of copper under other conditions.
4.2.5. Reactions in scCO2 catalyzed by other metals
4.2.5.1. Catalytic hydrogenation (Pt)
Despite the data on low catalytic activity of supported Pt-based catalysts in hydrogenation reactions caused by fast poisoning of the catalyst surface by CO molecules,[1059] there are indications that this drawback can be eliminated by preparing catalysts of a more complex structure.[1114] A comparison of the catalyst based on graphite intercalated with platinum nanolayers with the catalyst based on platinum nanoparticles on the graphite surface in the hydrogenation of cinnamaldehyde demonstrated that free platinum nanoparticles rapidly lose the catalytic activity in scCO2 and do not provide reaction selectivity (a mixture of all possible products of multiple bond and/or carbonyl group hydrogenation is isolated). Meanwhile, the platinum nanolayers incorporated into graphite do not lose the catalytic activity and ensure the formation of cinnamic alcohol in a yield of up to 80% (323 K, 5 MPa of H2 , 10 MPa of CO2).
4.2.5.2. Cyclization of carboxylates (Ag, Sn)
It was found that allenylmethylamines 402 can cyclize to dihydro-1,3-oxazol-2-ones 403 in scCO2 in the presence of the silver complex with the N-heterocyclic carbene Ag(OAc)IPr [IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene] (Scheme 244).[1115] The CO2 molecule is incorporated into the heterocycle. The reaction proceeds both in CO2-saturated (1 MPa) organic solvents and in scCO2 (50 °C, 7.3 MPa). It is believed that this reaction is driven by the coordination of the silver ion to the allene group of the carbamic acid formed in situ, resulting in the intramolecular nucleophilic attack and cyclization. Similar products were also obtained in the presence of gold carbene complex Au(OAc)IPr. Further studies are required to elucidate the role of the metal and identify the optimal catalyst.
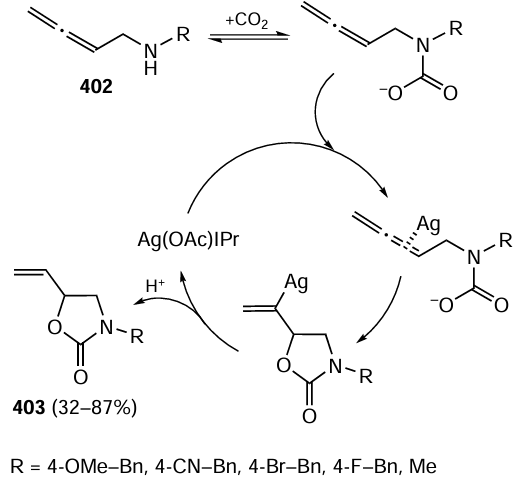
A similar heterocyclization takes place for ortho-substituted anilines in the presence of tin(II) 2-ethylhexanoate, Sn(Oct)2 , or SnO as the catalyst (Scheme 245).[1116] In this case, the reaction also starts with the formation of carbamic acid followed by the coordination of the intermediate thus formed to the metal ion. The yields of the cyclization products reached 90 – 100% provided that the reactants withstood the high-temperature reaction. The higher catalytic activity of Sn(Oct)2 compared to SnO is most likely attributable to their different solubilities in scCO2 .
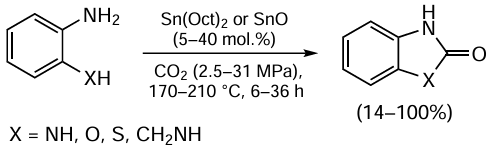
4.2.5.3. Oxidation of alkanes (Mo)
An important distinction of scCO2 from organic solvents is that the carbon atom in carbon dioxide occurs in the highest oxidation state and, hence, this solvent cannot be oxidized during a chemical reaction even by the strongest oxidants. Examples of using scCO2 as a solvent in oxidation reactions have been reported;[1117] however, most of these reactions are non-selective and give stoichiometric mixtures of products. As a rule, catalysts are not used in these reactions. An exception is the oxidation of cyclohexane to cyclohexanol with tert-butyl hydroperoxide (TBHP) in scCO2 in the presence of catalytic amounts of molybdenum coordination compounds 404 – 406 (Scheme 246).[1118]
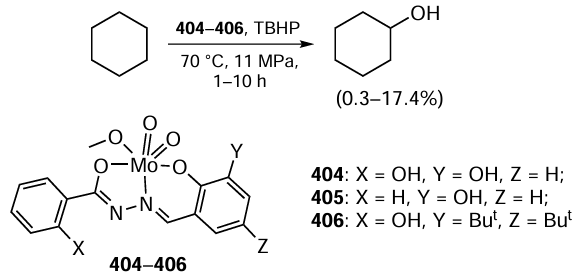
The product yields are moderate (up to 17%), and the effect of temperature and pressure on the reaction selectivity has not been investigated. The addition of the [bmim][PF6] ionic liquid as a co-solvent leads only to a slight decrease in the conversion, but does not influence the selectivity of oxidation.
4.2.5.4. Knoevenagel condensation (Zn)
The first metal-catalyzed Knoevenagel condensation in scCO2 was described in 2022.[1119] The coordination polymer based on 5-{(pyren-4-ylmethyl)amino}isophthalic acid 407, N-methylformamide and zinc nitrate was used as the catalyst. The reaction of benzonitrile with malononitrile was carried out at 40 – 60 °C and 12 MPa CO2 pressure (Scheme 247). The yield of the product in scCO2 was 62%; however, it could be increased to 100% by adding a polar co-solvent to the system (EtOH or H2O).
Generally, analysis of published data indicates that several approaches to the synthesis of catalysts for the use in scCO2 have been tested in 2010 – 2020. Apart from the synthesis of scCO2-soluble fluorinated analogues of known coordination compounds, the search for new transition metal complexes with phosphine, phosphonic and amide ligands, carbenoid-based ligands and more complex species is in progress. Using rational design of even relatively simple ligands (e.g., replacement of arylphosphines by alkylphosphines), effective catalysts soluble in scCO2 were obtained. Owing to high rates of mass transfer and diffusion in scCO2 , heterogeneous catalytic processes can also be carried out in this solvent. The advantages of using CO2 are most obvious in copper-catalyzed reactions, first of all, CuAAC reactions, which can be carried out for labile starting compounds, affording products readily separable from the catalyst traces. It is expected that more effective catalytic systems for novel or known chemical reactions that have not yet been carried out in scCO2 would be developed in the near future.
5. Organic synthesis based on the biorenewable raw materials
The use of renewable natural feedstocks is one of the basic principles of green chemistry. Plants and compounds isolated from plants can serve as the feedstock for the synthesis of many practically valuable products, being an attractive alternative to the traditional petrochemical approach. It is predicted that the bioprocessing plants would be more and more significant for the sustainable conversion of biomass to diverse chemicals and fuel, which are currently derived from crude oil. Wood, which forms the major part of the Earth’s biomass, is the most promising object for bioprocessing. In particular, conversion of wood chips to paper generates a variety of other useful products, including kraft lignin, crude tall oil and crude sulfate turpentine (CST). The production of functional molecules from renewable biofeedstocks and biowaste can significantly reduce the emission of greenhouse gases. In this chapter, the prospects that are opened by the use of natural molecules as reagents and substrates in modern organic synthesis are considered in relation to terpene compounds.
5.1. Monoterpenes and their derivatives as a promising green feedstock for organic synthesis
Crude sulfate turpentine is a mixture of bicyclic monoterpenes, including α-pinene, β-pinene and 3-carene and minor amounts of camphene and other monocyclic terpenes (e.g., limonene and terpinolene).
The amount of CST produced by palp and paper industry accounts for approximately two thirds of the world output of turpentine (~260 000 tons per year), while the rest is gum turpentine (GT) manufactured by distillation of oleoresin harvested from living trees. Currently, turpentine is used as a biofuel to generate electricity, converted into solvents (for example, α-terpineol or camphene) or fractionated into separate monoterpene components, which are then used for the production of flavoring agents (e.g., camphor and menthol), vitamins E and D and antioxidants (in particular, β-carotene). The availability of the raw material base brings to the forefront the development of efficient and scalable processes for the conversion of accessible monoterpene feedstock to biorenewable products.[1120]
An important benefit of monoterpenes containing cyclic p-menthene structural moieties as a biorenewable feedstock is the possibility of converting them to benzoid products, first of all, p-cymene, which are difficult to obtain from lignocellulosic feedstock (Scheme 248). The subsequent oxidation of the methyl and isopropyl groups of p-cymene affords ‘green’ terephthalic acid (TA), an intermediate compound for the production of polyethylene terephthalate (bio-PET). Achiral p-menthadienes, p-cymene and the products of its subsequent processing can be obtained using CST, which contains bicyclic monoterpenes differing in the enantiomeric purity and absolute configuration.[1121]
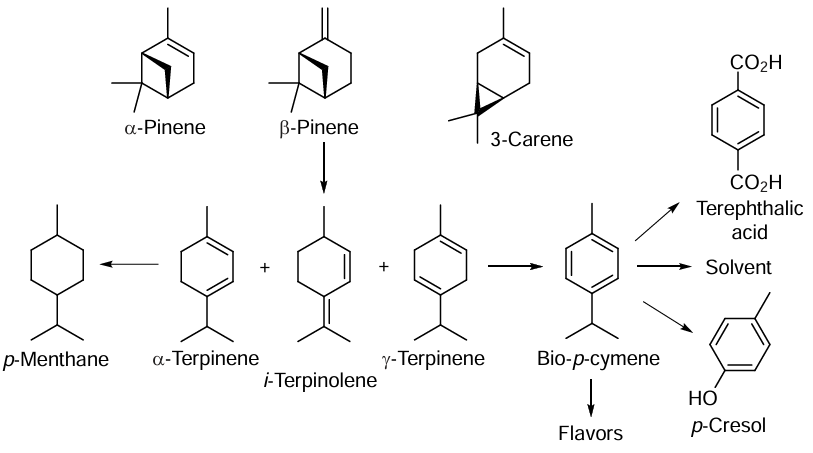
A number of catalytic reactions are available for converting monoterpenes isolated from turpentine into valuable chemicals on an industrial scale.[1122] Particularly, β-pinene and limonene derivatives meant for the use as solvents [1123-1126] as well as useful biologically active compounds such as repellents,[1127] herbicides[1128][1129] and pharmaceuticals[1130][1131] were obtained by these reactions.
Epoxidation is an important method for the functionalization of natural compounds.[1132] Subsequently, epoxides can be converted to a wide range of other polyfunctional products.[1133-1135] Terpene epoxides are regarded as chiral building blocks in the production of fragrances, vitamins, pharmaceuticals, insecticides, insect pheromones, polymer materials and other useful products. However, realization of the synthetic potential of monoterpenes and their epoxides is complicated by side reactions such as ring opening, rearrangements and hydrolysis, resulting in the formation of product mixtures that can hardly be separated. In addition, many terpenes contain di- and trisubstituted double bonds, which decreases the selectivity of epoxidation.
Therefore, special methods were developed for the synthesis of α-pinene and 3-carene epoxides, limonene bis-epoxides, and 3-caren-5-one and 3-carene-2,5-dione epoxides.[1136] The epoxidation of turpentine (technical-grade α-pinene), 3-carene and limonene was performed with 33 – 38% aqueous hydrogen peroxide in a catalytic system containing NaHCO3, MnSO4 and salicylic acid in acetonitrile at 18 – 25 °C. This procedure is suitable for selective oxidation of turpentine without isolation of single monoterpenes and for more complete oxidation of products with the same catalytic system.[1136] A useful continuous flow method for the organocatalytic epoxidation of terpenes and terpenoids makes it possible to control the accumulation of highly reactive peroxide compounds.[1137] In this case, epoxidation is carried out in a system comprising cyanamide, K2CO3 and H2O2. The residence time of the reactants in the reactor does not exceed 30 s, while the yields of α-pinene, β-pinene, 3-carene and limonene epoxides reach 83 – 92%. A by-product of this reaction is urea, which is easily separated.
Epoxidation of CST on treatment with 30% H2O2 proceeded smoothly at room temperature in the presence of inexpensive polyoxometallate catalyst in combination with the Aliquat-336 phase transfer catalyst. The resulting mixture of α-pinene oxide (408), β-pinene oxide (409) and 3-carene oxide (410) was separated by fractional distillation (Scheme 249). The polyoxometallate catalyst was regenerated and reused three times in the epoxidation of 3-carene (model reagent), with the yield of 3-carene oxide (410) remaining 64 – 75%.[1138] Epoxides 408 – 410 were hydrolyzed to the corresponding terpene anti-diols (60‒70% yield) in the presence of a heterogeneous acid catalyst, Amberlyst-15.
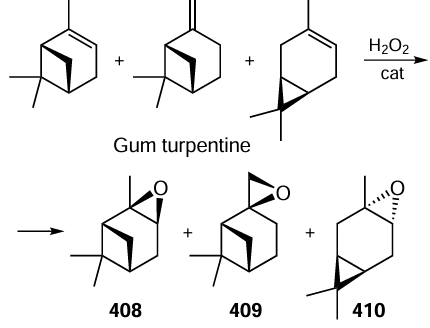
Epoxidation of monoterpenes present in untreated CST can be carried out without a solvent in the presence of the Venturello phase transfer catalyst.[1139]
A popular method for alkene functionalization is the Mukaiyama hydration in the presence of Fe-, Co- and Mn-containing catalysts in an oxygen atmosphere. However, in some cases, the stereoselectivity control remains problematic. Recently, a stereoselective method has been developed for the Markovnikov hydration of alkenes using nitroarenes as oxidants.[1140] The reaction was catalyzed by Fe(acac)3, while PhSiH3 served as the source of the hydride ion. Under proposed conditions (MeOH, 0 → 20 °C, 12 h), various monoterpenes (α-pinene, trans-pinocarveol, cis-verbenol, nopol benzoate, myrtenyl acetate, α-terpineol and terpinen-4-ol) were converted to tertiary terpene alcohols 411 – 417 in 43 – 78% yields with virtually complete stereocontrol (>20 : 1 dr) (Scheme 250). Only the product of hydration of 3-carene 418 was formed with a low selectivity (3 : 1 dr). The reaction tolerates bulky functional groups and can be used at final steps of the synthesis of natural products.
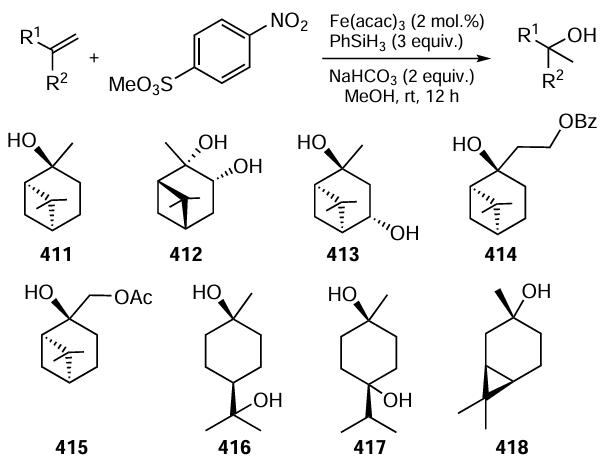
The hydroformylation (HFM) of alkenes (so-called oxo synthesis) is widely used in industry to obtain aldehydes, alcohols and acids. This reaction is usually carried out under homogeneous conditions in the presence of rhodium or cobalt catalysts. The type of solvent used in the reaction is critical for maintaining the catalyst stability. Despite the fact that biomass processing products, such as γ-valerolactone,[1141] methyltetrahydrofuran[1142] and p-cymene,[1143] and organic carbonates[1144] have been proposed in recent years as solvents for HFM reactions, the search for new alternative solvents of natural origin remains relevant.
Cheap, non-toxic and biodegradable anisole, which can be obtained from lignin, proved to be an appropriate solvent for the hydroformylation of terpenes.[1145] The substrates used in the reaction included pinane type (α-pinene, myrtenol, nopol) and para-menthane type (limonene, carveol, perillyl alcohol) terpenes, and toluene was used as a reference solvent (Table 3).
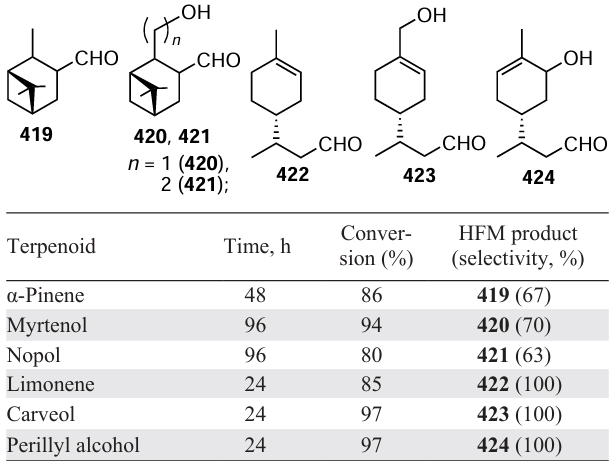
The rate of α-pinene hydroformylation to give aldehyde 419 was 30% higher in anisole than in toluene, but the selectivity remained the same (67%) because of the formation of by-products upon α-pinene isomerization to β-pinene under the reaction conditions. Myrtenol and nopol are converted to hydroxyaldehydes 420 and 421 with 70 and 63% selectivity, respectively, which is accompanied by the formation of minor amounts of intramolecular acetalization products. Limonene, carveol and perillyl alcohol containing a disubstituted double bond are converted to hydroxyaldehydes 422 – 424 much more selectively (100% selectivity), while the degrees of conversion are 85 – 97%. As a rule, the reactions are faster in anisole than in toluene, with the selectivity being retained. The resulting aldehydes have a pleasant odour and can be used in perfumery.
Primary amines are important intermediates for the preparation of valuable chemical compounds and materials. A convenient method for the synthesis of terpene amino derivatives is the reductive amination of terpene aldehydes or ketones with a mixture of hydrogen and ammonia in the presence of RuCl2(PPh3)3 (Scheme 251).[1146] This method was used, in particular, to prepare compounds 425 – 429, including steroid derivatives and pharmaceutical products. The product yields under optimal conditions were 70 – 95%.
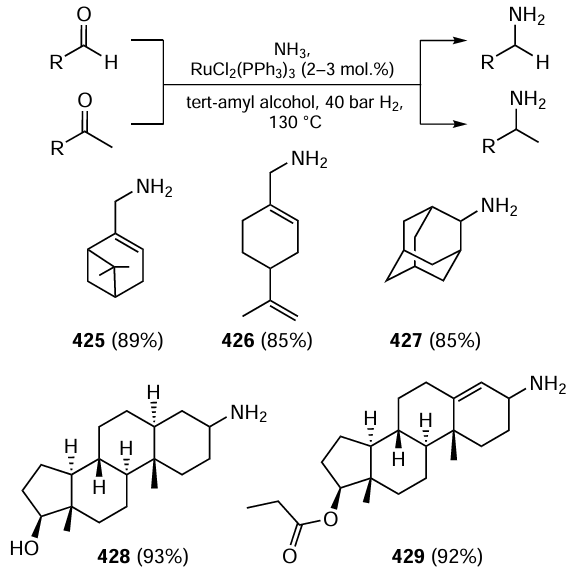
The reductive amination of carbonyl compounds by hydrogen and aqueous ammonia[1147][1148] can be carried out under microwave irradiation (MW) in the presence of magnetic nanocatalysts based on Fe, Ni and silica (Fe3O4@SiO2 – Ni), which can be regenerated and reused up to six times without a loss of activity. Apparently, the catalytic system acts as a bifunctional catalyst: SiO2 activates the substrate, while nickel nanoparticles facilitate the hydrogenation of the intermediate imine. Under optimal conditions, myrtenylamine 425 is formed with 100% selectivity. When the temperature is raised to 150 °C or the duration of MW irradiation increases, C=C bond hydrogenation takes place to give saturated amines.
Nitriles are in demand as synthons for fine organic synthesis, agrochemistry and pharmacology. As an environmentally benign method for nitrile synthesis, worthy of note is the aerobic oxidation of alcohols in the presence of ammonia (ammoxidation). The only by-product formed in this reaction is water. High activity in the ammoxidation of benzylic and allylic alcohols, in particular terpene type ones, is inherent in the Fe1 – N – C catalyst (Scheme 252), which can be easily regenerated and reused without significant losing the activity.[1149]
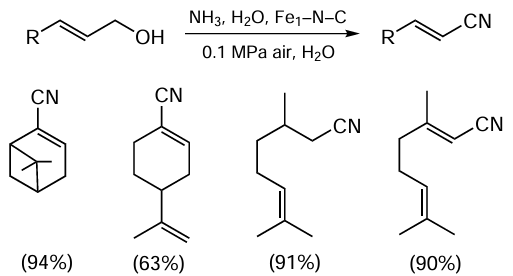
An example of a non-catalytic approach is the fast (5 – 10 min) synthesis of nitriles from the corresponding aldehydes under mild conditions using the stable and reliable H2N-DABCO as the aminating reagent (Scheme 253).[1150] Nitriles from myrtenal and citronellal were obtained in this way in 71 – 74% yields. An advantage of the developed protocol is that it does not involve the use of toxic cyanides.
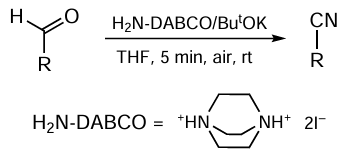
A recently reported highly efficient method for the amidation of aldehydes under visible light irradiation (450 nm, blue LED) involves the use of stable and practical reagent, N-chloro-N-sodio-carbamate (430), which is smoothly formed upon the reaction of free carbamate with trichloroisocyanuric acid (TCCA) (Scheme 254).[1151] A variety of (hetero)aromatic, aliphatic and terpene aldehydes were converted to synthetically useful N-protected amides, including myrtenal-based amide 431. The reactions proceeded under exceptionally mild conditions (30 °C, 12 h) and were highly tolerant to various functional groups. In addition, the possibility of further N-functionalization of compound 431 was demonstrated. Subsequently the authors optimized the amidation protocol by replacing trifluorotoluene with ethyl acetate, which is a green solvent.[1152]
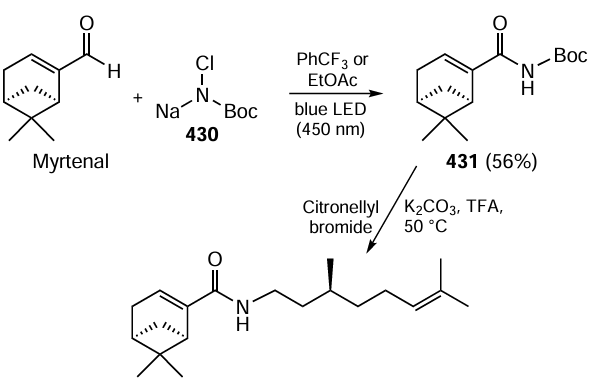
Essential oils and monoterpenoids they contain are used as repellents.[1127] However, low-molecular-weight monoterpenoids are quite rapidly evaporated, which reduces the efficiency of the oils. The repellents based on less volatile sesquiterpenoids are uneconomic due to high production costs. In order to increase the molecular weight and, hence, to decrease the volatility of available monoterpenoids, they were converted to esters of isovaleric acid 432 with the molecular weight close to that of sesquiterpenoids.[1153] Esterification was carried out by the modified Steglich procedure using the DCC – DMAP system (Scheme 255).[1154] The yields of products were 60 – 99%. Esters of α-terpineol 432a, ( – )-perillyl alcohol 432b and carveol 432c showed high short-term (2.5 h) and long-term (5 h) repellent activities. Esters of bicyclic compounds 432d – h were active in the first test and less active in the second test.
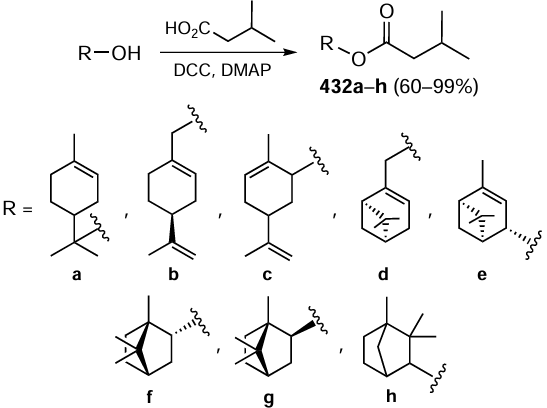
3-Caren-5-one, an important intermediate for the production of biologically active compounds, was prepared by selective allylic oxidation of 3-carene. It was converted to isomeric 3-carene-5-one oxime sulfonates 433 (Scheme 256), which were more active against the fungus P. piricola than the commercial fungicide chlorothalonil.[1155]
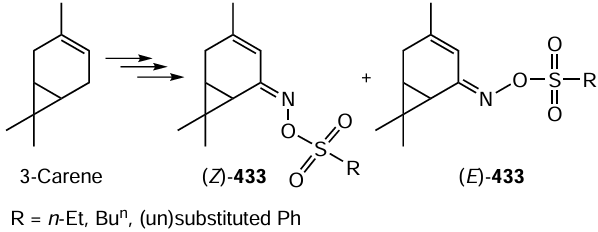
Short-chain esters of terpenes are used as flavouring agents and fragrances in the food, cosmetic and pharmaceutical industries. Therefore, interest in the development of new strategies for their industrial production has recently increased. In particular, continuous flow enzymatic (acylation with ethyl acetate in the presence of immobilized Novozym-435 enzyme) and chemical (acylation with acetic anhydride) processes for the synthesis of acetates of prenyl alcohols (geraniol, citronellol, myrtenol) combined with the purification stage were proposed.[1156-1158] The use of aqueous Na2CO3 for the extraction of residual impurities (acetic anhydride, acetic acid, etc.) furnished geranyl acetate with >94% purity.
A new catalytic system for the allylic acetoxylation of 1,1-substituted olefins, including terpenes, Pd(OAc)2 — 2-mercaptothiadiazole (434), has been developed.[1159] In the presence of this system, β-pinene was converted to myrtenyl acetate 435 in 86% yield, while limonene, containing endo- and exocyclic double bonds formed a mixture of isomeric allylic acetates 436 and 437 in a total yield of 79%. Under these conditions, α-pinene and 3-carene with a trisubstituted double bond gave allylic acetates 435 and 438, respectively, in 31 – 32% yields.
The epoxide ring in epoxyterpenes is opened under the action of nucleophiles. The involvement of amines in these reactions is of particular interest, since the resulting amino alcohols can be used as chiral ligands and auxiliary groups for the asymmetric synthesis of promising drugs.[1160] For example, (+)-3-carene was chosen as the starting substrate for the stereoselective synthesis of the diterpenoid euphorikanin A. The synthetic route included epoxidation of 3-carene, epoxide conversion to silyl enolate 439, closure of the 7-membered ring via intramolecular aldolization, and the transformation of bicyclic cycloheptenone 440 to the tetracyclic structure of euphorikanin A (Scheme 257).[1161-1163] (+)-3-Carene also served as the starting compound in the asymmetric synthesis of the diterpenoid (+)-pepluanol A.[1164] Diterpenoids of this series showed cytotoxic, antiviral and antitumour properties; they were effective for the treatment of diseases characterized by multiple drug resistance (MDR).
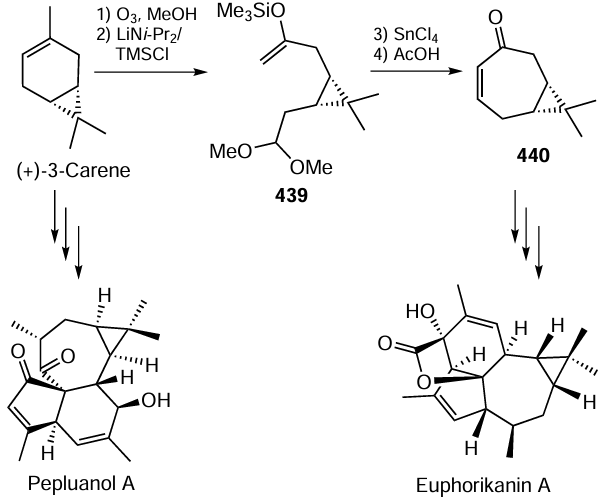
β-Amino alcohols 441 and 442, obtained from α-pinene and 3-carene, proved to be effective chiral organocatalysts for the asymmetric aldol reactions of isatin and 4,6-dibromoisatin with acetone (Scheme 258). The developed procedure allows implementing the enantioselective (up to 94% ee) synthesis of the anticancer drug (R)-convolutamydine A.[1165-1167]
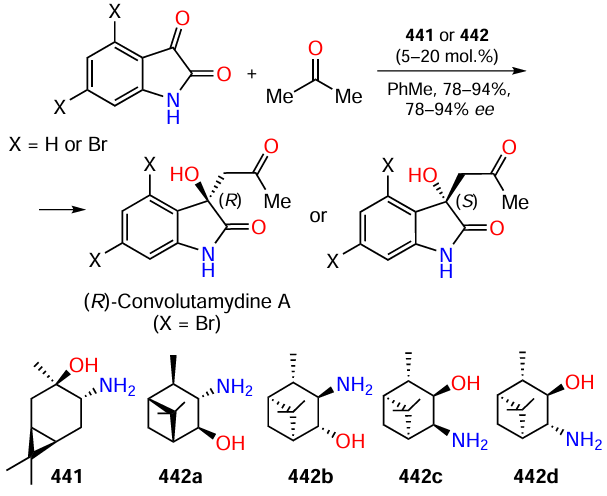
Recently,[1168] trifluoromethyl pinane (cis-verbanone, nopinone) and bornane (camphorquinone) β-amino alcohols 443‒445 were synthesized for the first time. The addition of the Ruppert – Prakash reagent to β-oxobenzyl O-oximes and the reduction of β-hydroxybenzyl O-oximes to the corresponding amino alcohols proceeds stereoselectively to give one of the diastereomers. The products may be of interest as biologically active compounds and/or their precursors and as novel chiral inducers, ligands or organocatalysts containing a trifluoromethyl group.[1169][1170]
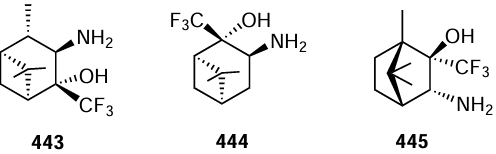
Esterification of (+)-(S)- and (–)-(R)-2α-hydroxypinan-3-ones with N-Cbz-protected α-amino acids (glycine, alanine, phenylalanine, lysine, ornithine, glutamine, histidine, etc.) followed by the deprotection and intramolecular condensation with a terpene keto group resulted in the synthesis of enantiomeric lactones 446 and ent-446. Esterification is accompanied by partial epimerization of the stereocentre of the acid; however, the cyclization is stereoselective: (1S,2S,6S)-2-hydroxypinan-3-one forms iminolactone 446 only with the (R)-amino acid ester, while (1R,2R,6R)-2-hydroxypinan-3-one gives iminolactone ent-446 only with the (S)-amino acid ester. Some of the obtained compounds inhibited proliferation of EL4, MCF7 and PC3 cancer cells.[1171]
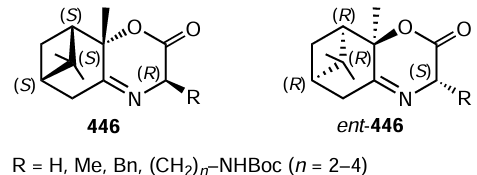
In the design of new monoterpenoid derivatives, the 1,2-diamine moiety, which is a privileged structure in medicinal chemistry, is of particular interest.[1172] A study using various in vitro models demonstrated that 2,6-diisobornyl-4-methylphenols 447a–c, containing 2α-hydroxy-3-aminopinane moieties, scavenge free radicals and have a higher antioxidant activity than the 2,6-di(tert-butyl)-4-methylphenol standard, although they are more cytotoxic.[1173-1175]
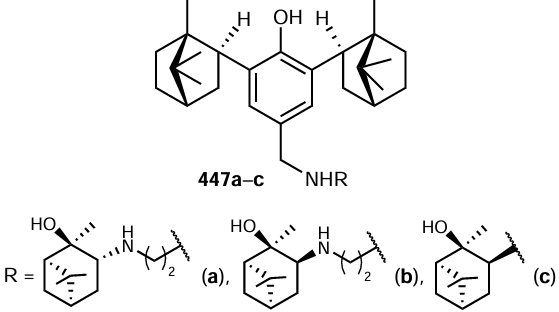
(+)-3-Carene can be converted to ε-lactams 448, precursors of polyamides, via the epoxidation step. Lactams are selectively obtained as two diastereomers, the configuration of which is determined by the stage of enzymatic or chemical oxidation (Scheme 259). Polymerization of ε-lactams 448 preserves the absolute configuration of the chiral centres and affords, depending on the monomer structure, either semicrystalline or amorphous transparent polymers, which are comparable with commercial polyamides in the thermal properties. An environmentally benign four-step one-pot synthesis of the monomer has been proposed.[1176]
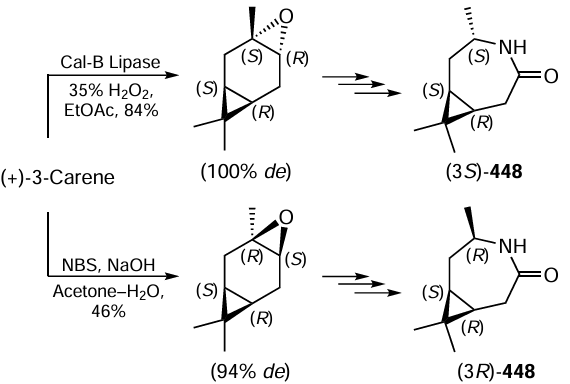
Monomeric β-lactams rac-449 and 450 were synthesized by [2+2]-cycloaddition of racemic α-pinene and (+)-3-carene to chlorosulfonyl isocyanate.[1177] Unfortunately, polyamides containing bicyclic moieties in the backbone have low thermoplasticity, which limits their applicability. One more problem is uncontrolled polymerization of highly reactive β-lactams. Nevertheless, they can be used as additives that control the glass transition temperatures of polycaprolactam and polylaurolactam. An enantiospecific synthesis of trans-β-lactams with a polyaromatic substituent at the nitrogen atom assisted by MW irradiation using (+)-3-carene-derived chiral acid has been reported.[1178]
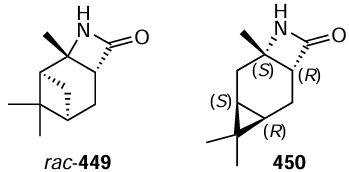
A useful method for the synthesis of practically valuable compounds from monoterpenes is ozonolysis of the double bonds present in their molecules.[1179][1180] For example, ozonolysis of (+)-3-carene and (–)-α-pinene afforded acylhydrazones 451 and 452, isoniazid derivatives containing a cyclopropane or cyclobutane moiety (Scheme 260). These compounds are promising in the design of drugs for the prevention and treatment of tuberculosis.[1181][1182]
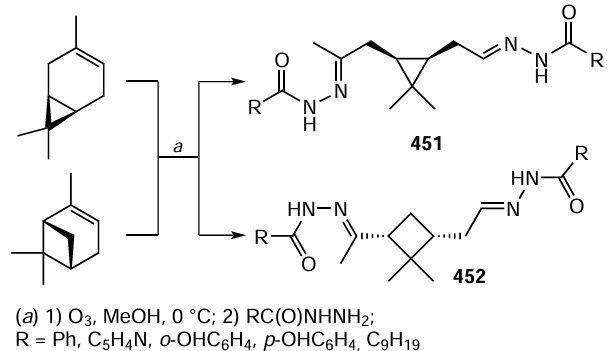
Ozonolysis of 3-carene was also used to synthesize acylthiourea derivatives 453 and N-substituted phenyl-1,2,4-triazolinethiones 454 containing a gem-dimethylcyclopropane group, which showed antifungal activity.[1183][1184]
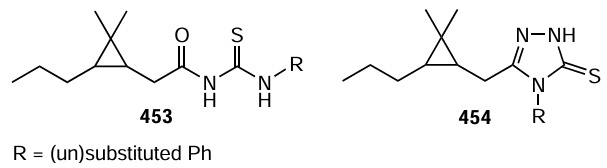
Verbenone (455), a product of allylic oxidation of α-pinene, is in fairly high demand as a fragrance and an anti-aggregation pheromone of bark beetles. In addition, it serves as the key intermediate in the synthesis of the antitumour drug taxol, potential anti-ischemic agents and anti-Alzheimer drugs. However, the known methods for the synthesis produce verbenone in not more than 50% yield.[1185][1186] An exception is the synthesis of verbenone developed in 2022, which involves the oxidation of α-pinene with the ButOOH – O2 system in ethyl acetate in the presence of Mn(OAc)3 and molecular sieves.[1187] The yield of the product was 60‒64%.
The studies aimed at the use of verbenone in organic synthesis are in progress. Recently, a number of diastereomerically pure isoxazolines 456 were obtained in up to 64% yields using 1,3-dipolar cycloaddition of (S)-455 to nitrile oxides generated in situ from the corresponding arylaldoximes and NaOCl, (Scheme 261).[1188][1189] Using in vitro cytotoxic activity assays, a product active against HT-1080 and A-549 cancer cells in the micromolar range of concentrations was identified (IC50 of 21.35 and 14.92 μΜ, respectively).[1190][1191]
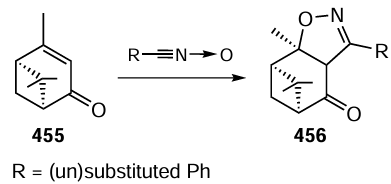
A method for the chemoselective reduction of the C=O bond in structurally diverse α,β-unsaturated ketones using a MgBu2 complex with pinacolborane, a cheap and readily available reducing agent, has been developed.[1192-1194] Cis-verbenol and cis-carveol were prepared by this method under very mild conditions (room temperature) in 70 – 94% yields and with virtually 100% diastereoselectivity.
Other promising investigation objects are myrtenol and myrtenal, bicyclic monoterpenoids present in the essential oils of many herbs, e.g., Ferula hermonis, Peruvian ginseng (Lepidium meyenii) and Chinese mint (Mentha haplocalyx)[1195] and in myrtle plants such as Campomanesia guaviroba[1196] and Myrtus communis.[1197] In the two last-mentioned plants, total content of myrtenol, myrtenol acetate and myrtenal reaches 50%. Myrtenol, myrtenal and their derivatives exhibit anticancer, antimicrobial, antimalarial, fungicidal, insecticidal, herbicidal, repellent and other useful types of biological activity. These compounds are usually prepared by oxidation of α-pinene with selenium dioxide or selenous acid. Moreover, the presence of both (+)- and (–)-α-pinene in natural sources makes both enantiomers of myrtenol and myrtenal available.
Treatment of myrtenal with the NaClO2 – H2O2 oxidative system in water gave myrtenic acid (457) (80% yield), which was converted to 2-acyl-1,2,4-triazole-3-thiones (458) and thioethers (459) containing a myrtenyl moiety (Scheme 262).[1195][1198] According to biological assays data, compounds 458a – d and 459a – c inhibit the development of fruit rot (Physalospora piricola) with a degree of inhibition of 72 – 83% (relative to the fungicide Chlorothalonil) and 90 – 98% (relative to the fungicide Azoxystrobin).
The same research team synthesized sulfamides 460[1199] containing myrtenyl and diazine moieties on the basis of myrtenylacyl chloride. Sulfamide 460a taken in 50 µg mL–1 concentration actively inhibited the development of fungi Physalospora piricola and Alternaria solani (potato blight), while compound 460b showed a high herbicidal activity (86%) against coleseed (Brassica campestris) in 100 µg mL–1 concentration.
Some organic compounds may enhance the efficacy of antimicrobial agents against planktonic and biofilm-embedded bacteria and fungi.[1200][1201] In particular, this behaviour was observed for myrtenol enantiomers.[1202] Recently, (+)-myrtenol was found to have a synergistic effect in combination with the drugs amikacin, fluconazole and benzalkonium chloride tested on clinical isolates of S. aureus and C. albicans, including MRSA and fluconazole-resistant fungi. Enantiomeric (‒)-myrtenol enhanced the inhibitory activity of amikacin and fluconazole against the S. aureus and C. albicans biofilm formation. In addition, both enantiomers significantly (16-fold) enhance the action of benzalkonium chloride against planktonic cells in an S. aureus and C. albicans mixed culture. The synergistic action of myrtenols with conventional drugs appears to be due to their interaction with the fungal cell membrane, resulting in a decrease in the membrane potential.
Nitrogen-containing derivatives of cis-myrtanic and myrtenic acids show a fungicidal activity against yeast and mycelial fungi, including phytopathogens.[1195][1203] It was assumed that the presence of several pharmacophore groups (quaternary ammonium salt and a terpenoid) in the same molecule[1204] would enhance the bactericidal properties of the prepared compounds. Myrtenic and isomeric myrtanic acids obtained from the corresponding alcohols[1205-1207] were converted to dimeric salts 461 and 462 via a sequence of reactions that included treatment of the acids with SOCl2, the reaction of acid chlorides with dimethylaminopropylamine, and quaternization of the terminal dimethylamino group with myrtenyl bromide.[1204] However, despite the presence of the quaternary ammonium group, compounds 461 and 462 have a moderate antimicrobial activity, probably due to the high lipophilicity of the molecules containing two bulky terpene moieties.
(+)-Myrtenol ether 463a and its sulfur- and nitrogen-containing analogues 463b and 463c can inhibit human platelet aggregation.[1208] According to in vitro experiments, compounds 463a – c significantly slow down ADP-, collagen-, adrenaline- and ristocetin-induced platelet aggregation and completely block the coagulation induced by arachidonic acid.[1209] The most pronounced effect was observed for sulfur-containing compound 463b, making it a promising candidate in the search for drugs to treat and prevent thrombophilia.
Terpenes are used as building blocks for the design of biocompatible fluorescent markers, which are necessary for imaging the pathogenic microorganisms and studying the mechanisms of their proliferation.[1210-1212] Luminophores based on boron dipyrromethenes (BODIPY), photostable boron coordination compounds with low cytotoxicity and high fluorescence quantum yield, are widely used for this purpose. The introduction of substituents in the meso-position of the indacene nucleus can be used to modify the spectral characteristics and the ratio of lipophilic and hydrophilic properties of the fluorophore and thus to adjust it for a particular practical task. Among the prepared biomarkers 464 and 465a,b, containing a terpene moiety,[1213][1214] the best results were found for myrtenol derivative 464.[1215]

5.2. Diterpenoids: synthesis and application prospects
Diterpene compounds are more widely spread in nature than monoterpenes; however, their contents in plants are, most often, relatively low and their isolation is, therefore, difficult and in some cases inexpedient. Abietane diterpenoids forming the basis of turpentine rosin,[1216] extraction rosin[1217][1218] and tall oil rosin[1219] are represented most widely. The rosin composition strongly depends on the method of its isolation, habitat of the source, that is, coniferous trees, and on their species,[1220-1222] but abietic 466, dehydroabietic (467), levopimaric (468) and palustric (469) acids are always the major components.[1222][1223]
Abietane derivatives are present not only in the coniferous trees, but also in flowering plants and in some fungi.[1224-1226] These terpenoids are secondary metabolites that are not essential for plant existence and do not participate in plant growth or development. They perform protective functions that allow plants to adapt to conditions of biotic and abiotic stresses arising from interaction with the environment.[1227] Owing to the broad range of their phytoprotective functions (insecticidal, bactericidal, fungicidal), these compounds can be regarded as potential drug candidates.[1228-1233]
The benefits of using these compounds as the starting reactants include their enantiomeric purity and their high content in renewable biological resources. Abietic acid and isomeric dienoic acids are unstable and prone to isomerization and dehydrogenation during chemical reactions, especially those proceeding on heating and in acidic media, being converted to more stable aromatic dehydroabietic acid derivatives. On heating with sulfur in the presence of iodine, a mixture of esters of abietic (466) and palustric (469) acids is converted to the ethyl ester of dehydroabietic acid (470) in almost quantitative yield (Scheme 263).[1234]
The oxidative transformations of abietic acid are usually non-selective and give mixtures of alcohols and hydroperoxides that are difficult to separate. A solution to this problem may be provided by introducing of a keto group in position C7 of the carbon skeleton of the molecule, for example, by treatment of methyl abietate 471 with an excess of iodine in the presence of KHCO3 (Scheme 264). The resulting ketone 472 is oxidized with air oxygen in the presence of a base to give diastereomeric hydroperoxides 473, which can be converted to more stable alcohols 474 by treatment with PPh3.[1235]
Depending on the solvent used, ozonation of methyl abietate 471 affords epoxy ketoaldehyde 475 (MeOH-Py) or epoxy ozonide 476 (CH2Cl2-Py) (Scheme 265).[1236] Ozonation of abietic acid 466 in dichloromethane furnishes ozonide 477, which showed a high cytotoxicity in vivo against human melanoma cells, but was inactive against lung adenocarcinoma and epidermal carcinoma cells. The anti-inflammatory activity of ozonide 477 is comparable with that of diclofenac.[1237]
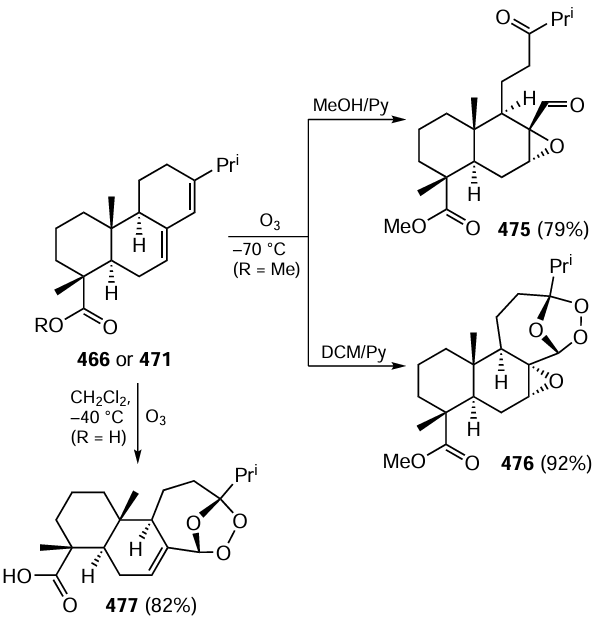
Treatment of methyl abietate 471 with the OsO4 – Me3NO oxidative system results in the selective and stereoselective oxidation of one of the double bonds thus giving vicinal diol 478 (Scheme 266).[1235] A series of transformations converts this product to enone 479, an important intermediate in the synthesis of 4-epi-parviflorons 480, which retard proliferation of tumour cells responsible for the appearance of cervical spine cancer, breast cancer and lung carcinoma.[1238]
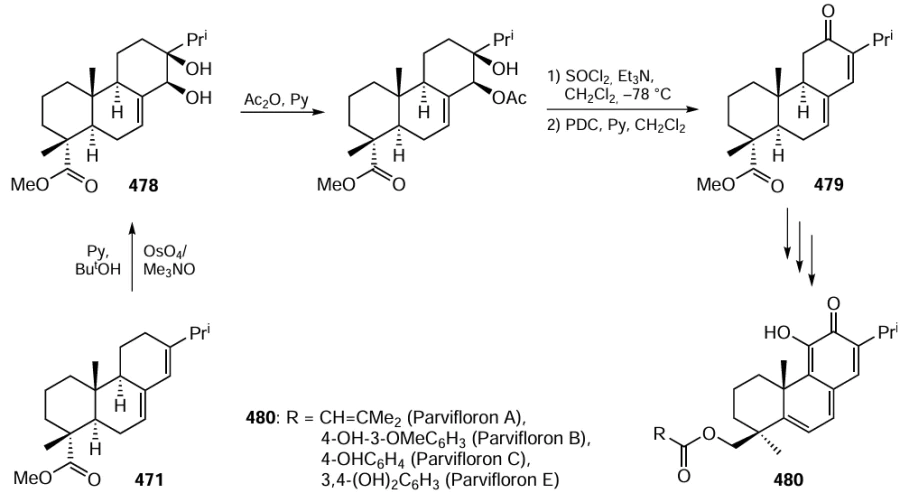
The introduction of hydroxyl groups into the aromatic ring of the dehydroabietic structure via direct oxidation has not yet been accomplished. These compounds are obtained by the Friedel – Crafts reaction with acylating reagents. Treatment of methyl dehydroabietate 481 with AcCl in the presence of AlCl3 followed by the Baeyer – Villiger oxidation of acetyl derivative 482 yielded acetate 483, which was converted to phenol 484 via alkaline hydrolysis (Scheme 267). This method is regioselective, as other positions of the aromatic ring are not acetylated under these conditions.[1239][1240]
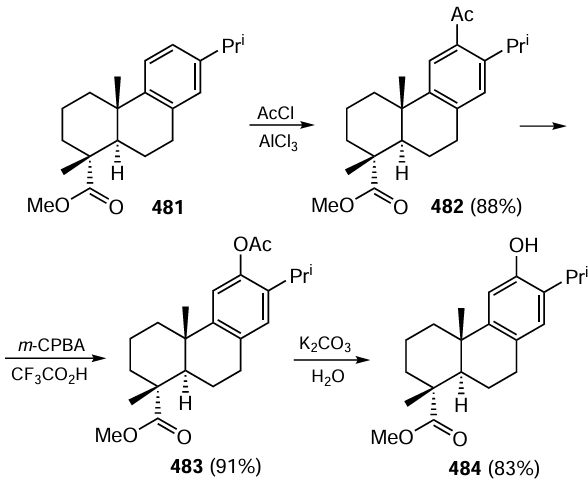
Many dehydroabietane derivatives possess various types of biological activity, including anticancer activity.[1241] Thus, taxodione and taxodone (taxodione reduction product) were found to have high activity against nasopharyngeal carcinoma.[1242] Dehydroabietic acid was inferior to reference drugs (etoposide and cisplatin) when tested against lung and colon cancer cells, but was competitive with them when tested against breast cancer cells.[1239] Some dehydroabietane oximes showed good activity against pancreatic cancer cells. It is of interest that the most active compounds of this series (compounds 485 – 487) lost the anticancer properties as the oxime group was replaced by an aromatic moiety.[1243] Taxodione, lactone 488, salvinolone and sugiol showed high antioxidant activity[1244][1245] and were effective against methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus sp. strains.[1246][1247]
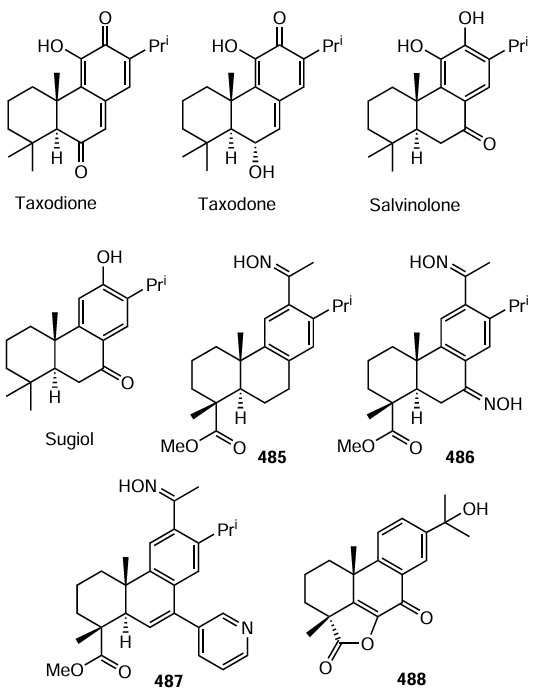
On refluxing in acetic acid, abietic acid isomerizes to levopimaric acid (468), the positions of the double bonds in which are favourable for the Diels–Alder reactions, for example, with maleic anhydride, acrylic acid, 1,4-benzo- or 1,4-naphthoquinone and so on.[1222][1248] Adducts 489 – 492, especially maleopimaric acid anhydride 489, are of interest for the synthesis of heat- and fire-resistant polymers, in particular siloxanes and polyurethanes.[1249]
Sulfur-containing abietane derivatives, which can be obtained by sulfonation of dehydroabietic acid 467, are of no less interest (Scheme 268). The sodium salt of 12-sulfodehydroabietic acid 493, known by the trade name Sodium Ecabet, is used for the therapy of gastric ulcer. It is highly active against Helicobacter pylori, causing its almost complete eradication.[1250]
A carnosic acid sulfide derivative inhibits the growth of pancreatic cancer cells and inhibits farnesyl pyrophosphate synthase, an enzyme involved in the mevalonate pathway of cholesterol synthesis. However, it also blocks signalling G-proteins responsible for the functioning of osteoclasts, the cells that cause bone demineralization.[1251]
The reactions of ethyl 12-chlorosulfodehydroabietic acid 494 with various amines (natural amino acids, their hydrazides and β-amino alcohols) yielded semisynthetic sulfonamides 495a – h (Scheme 269),[1252-1255] some of which possessed anticancer activities.[1256]
The search for diterpene compounds in natural objects is carried out very actively, but many compounds are still unexplored, as it is fairly difficult to isolate a compound in a pure state and to study its chemical and biological properties, especially if its content is relatively low. Among the diterpenoids discovered in recent years, mention should be made of cembranoids,[1257] penicichrysogenes[1258] and koninginols.[1259]
Apart from the mentioned types of biological activity, diterpenoids were found to have anti-HIV activity and activities against Coxsackie, herpes simplex, hepatitis, influenza, Zika and dengue viruses and coronavirus.[1233]
Obviously, the structural base and list of renewable natural raw materials isolated from plants will constantly expand, which will open up new opportunities for green organic synthesis and preparation of new effective pharmaceutical drugs.
6. Green chemistry synthesis of functional materials
A key objective of green chemistry is to develop new processes for the production of functional materials with desired physicochemical properties and performance characteristics under conditions that exclude or minimize the adverse effect on the environment. It should be borne in mind that for practical implementation, these processes should be not only environmentally benign, but also resource-saving and cost-effective. In this chapter, we consider some opportunities offered by the use of green chemistry techniques for the solution of this multipurpose problem in relation to the synthesis of modern polymer materials by free-radical emulsion polymerization and synthesis of high-energy compounds and materials.
6.1. Green chemistry approaches to the preparation of polymer materials
The environmental aspects of the production and application of polymers have been repeatedly discussed in the last decade by both the scientific community[1260-1262] and mass media, and through the mass media by a wider community. The high interest in this issue is mainly related to polymer waste problem and pollution of the World Ocean. Currently it is impossible to significantly reduce the production capacity or, moreover, to completely cease the production of polymers. Therefore, a systematic approach is required to address the polymer waste problem in order to select the most rational solutions. It is reasonable to start not with the polymer waste disposal, but first to analyze methods for polymer synthesis, since most of them are not perfect as regards the environmental impact.
Although advanced processes such as pseudo-living radical polymerization have been actively developed in recent years,[1263-1266] their practical significance is still low. Therefore, the environmental aspects of free-radical polymerization were analyzed in this review. This method was chosen as one of the most popular industrial methods of polymer synthesis. The attention is focused on the free-radical emulsion polymerization, which makes it possible to conduct the process in an aqueous medium and to control the molecular-weight characteristics of polymers and copolymers, which affect the biodegradability. Various aspects of the control of contaminants that appear during emulsion polymerization (EP) are also discussed.
6.1.1. History and characteristic features of emulsion polymerization
The first studies of EP were initiated by the Bayer company before World War I.[1267][1268] The monomer was emulsified in water by means of a natural polymer (starch, gelatine); therefore, according to modern views, this process is classified as suspension polymerization. In the 1920s, EP of isoprene was carried out using a surfactant and an initiator.[1269] A few sorts of synthetic rubber were synthesized in this way, but all data were kept secret by patents until the end of World War II. After the War, EP started to be used for the production of plastics, with the resulting polymers being used in latex paints, aqueous dispersions and many other products.
Emulsions are dispersions consisting of two immiscible liquid phases, which are transformed into homogeneous medium under the action of shear stress and surfactants.[1270] A typical chart of an emulsion polymerization composition is depicted in FIg. 32. The system includes monomer droplets with a size from micrometre to millimetre, monomer aggregates or nanodroplets (<1 mm), molecularly dissolved monomer, initiator molecules, primary free radicals, macromolecular free radicals (living polymers), dead polymers, monomer – polymer aggregates (surfactant-free polymer particles), free or adsorbed surfactant molecules, surfactant aggregates (micelles), monomer – polymer aggregates (monomer-swollen micelles) and monomer–polymer – surfactant aggregates (surfactant-stabilized polymer particles) immersed into the continuous phase of water molecules.[1271]
![[{"id":"fUcR7iUBuw","type":"paragraph","data":{"text":" Illustration of the emulsion polymerization mechanism. The Figure was created by the authors using published data.<sup>1271</sup>"}}]](/storage/images/resized/zQ2PK6QFhJzzCE6jC7NM1IMJ4CrZvcv7QkiDBGhv_xl.webp)
Free radical emulsion polymerization is used in industry to produce various types of polymers, including elastomers (acrylic, nitrile and polybutadiene rubber), engineering polymers (polystyrene, polyvinyl chloride, polymethyl methacrylate) and emulsions as commercial products. According to Global Market Insights, the global demand for polymer emulsions in 2022 was 30.3 billion US dollars (Fig. 33). The benefits of emulsion polymerization include easy heat removal, simple production equipment, low fire hazard of the process with water being used as the dispersion medium and the possibility of obtaining high-molecular-weight polymers (at high process rates) and highly concentrated latexes.
![[{"id":"TF49yhqUdZ","type":"paragraph","data":{"text":"Volume of the global market of synthetic latex polymers according to types, 2021 – 2032 (billion US dollars) predicted by Global Market Insights.*"}},{"id":"eL3eWCJlv2","type":"paragraph","data":{"text":"*https://www.gminsights.com/industry-analysis/polymer-emulsions-market (accessed on January 09, 2024)."}}]](/storage/images/resized/qTpSMDDrN2ELzIynsYcs6jIlSzoZ5cxTjpnyBrLQ_xl.webp)
6.1.2. Nucleation mechanisms
The mechanism of EP in the presence of surfactants has been discussed for more than half a century, and a common opinion has been reached. The Smith – Ewart – Harkins theory (1947) implied three ways of particle nucleation,[1272] which later served as the basis for three process mechanisms that include micellar nucleation, homogeneous nucleation and monomer microdroplet nucleation (Fig. 34).[1273] Decomposition of the initiator in the aqueous phase affords free radicals. According to the micellar mechanism,[1273][1274] emulsion polymerization starts with capture of free radicals by micelles, proceeds in monomer-swollen particles and ends when the monomer is exhausted. It is believed that the micellar nucleation mechanism predominates at surfactant concentrations above the critical micelle concentration. In the absence of micelles, homogeneous nucleation mechanism is likely to prevail. In the case of homogeneous nucleation,[1275-1277] monomers are dissolved in water and undergo radical polymerization to give oligomers. Oligomers coagulate giving successively seeds, nuclei and primary polymer particles. The primary particles stabilized by surfactant adsorption can grow via swelling of monomer particles or via oligomer deposition on their surface.[1278] Finally, according to the mechanism of monomer microdroplet nucleation, the microdroplets can capture oligomer radicals and be hardened to form polymer particles. Usually, this mechanism is unlikely, except for miniemulsion polymerization with hydrophobic initiators.
![[{"id":"QrsTU9ZTTy","type":"paragraph","data":{"text":"Mechanisms of particle formation in heterophase polymerization."}}]](/storage/images/resized/h4IOAQ5UMuVJG5iGO06LJ7iUH7OPDanQFRFWdWm3_xl.webp)
6.1.3. Types of emulsion polymerization
There are three types of EP differing in the particle size, aggregative stability and polymerization conditions, namely macroemulsion, miniemulsion and microemulsion polymerizations (Table 4)[1279].
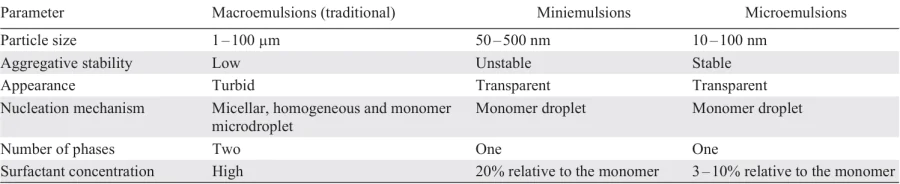
Macroemulsions must be continuously stirred to disintegrate the coalescing droplets. In the initial period, they contain micelles of the emulsifier and emulsifier-stabilized monomer droplets. Unlike macroemulsions, miniemulsions contain, apart from the surfactant, an osmotic agent preventing the Ostwald ripening of monomer droplets. A characteristic feature of miniemulsions is also the absence of free surfactant micelles in the system and, hence, predominance of the polymer chain nucleation mechanism inside droplets. Unlike microemulsions, miniemulsions are thermodynamically unstable; therefore, they are formed where strong shear stress is applied.[1280]
Drawbacks of the emulsion polymerization include the necessity to wash the polymer from the emulsifier, the monomer and the initiator and the presence of an additional stage of polymer isolation from the latex.
6.1.4. Residual monomer problem and ways to address the problem
Free-radical polymerization rarely leads to complete monomer conversion, and the residual monomer is retained in the polymer material. Although polymers have a high industrial value, the characteristic features of the polymerization mechanism have not yet been clarified in detail.[1281] The incomplete monomer conversion may be caused by the glass effect, cage effect, attachment of radicals to the surface of polymer particles and different reactivity of comonomers.[1282]
The unreacted monomers present in thermoplastics, in particular acrylonitrile and vinyl chloride are toxic.[1283] An acute problem of food industry is related to the presence of monomers in the polymer packaging materials since they can get into food products. In addition, the monomer residues influence the properties of polymers, e.g., enhance the polymer shrinkage in boiling water, change the deformation temperature and polymer colour, cause unpleasant odour, etc.[1284]
The presence of the unreacted monomer is inadmissible in the bioresorptive polymers used in surgical devices, implants, drug delivery capsules, etc.[1285] It is also highly undesirable in polymers for dentures[1286][1287] and dental inlays.[1288] The methyl methacrylate monomer present in the bone cement, which is widely used in orthopaedic surgery, exhibits cytotoxic properties[1289] and causes a change in the physicomechanical properties of the material on prolonged use.[1290] Vinyl ester and crotonic acid impurities in hair sprays and styling lotions can cause skin irritation.[1291]The presence of residual monomers is also inadmissible in vinylpyrrolidone/vinyl acetate copolymers used in pharmaceutical and cosmetic products[1292] and in ophthalmic materials where it can affect the size, transparency and biocompatibility of contact lenses.[1293][1294]
The known methods for decreasing the residual monomer content can be subdivided into two groups. The first group includes chemical methods that involve the formation of new polymer chains, for example, by adding an initiator, or formation of new compounds that can be easily removed from the system. It should be noted that these new compounds should be non-toxic. The second group comprises physical methods, that is, removal of the unreacted monomer by evacuation, heating, extraction or treatment with an ion exchange resin. The choice of the optimal method is determined by the applications and properties of a particular polymer.
6.1.4.1. Removal of the residual monomer using an initiator
A correct choice of the initiator(s) reduces the time of polymerization and the amount of the residual monomer. A frequently used type of systems contain two initiators, one operating at the initial stage of the polymerization, and the other (co-initiator with a longer half-life) starting to operate at higher temperature, thus decreasing the amount of the residual monomer.
While choosing the initiator for EP, one should take into account its solubility in both the major and dispersed phases. An initiator that is better soluble in the phase containing the greater part of the residual monomer is most appropriate. An example is the decrease in the residual monomer content in the emulsion homo- or со-polymerization of styrene by adding a system of redox initiators composed of oil-soluble (oxidant) and water-soluble components.[1295] The addition of this initiating system on reaching 97% monomer conversion decreases the residual monomer content to 1000 ppm. The major contribution to this result is made by the oil-soluble component, because the most part of the residual monomer is located in the oil phase.
The monomer conversion can be increased by increasing the process temperature and/or by using a reducing agent.[1296] The ability of the reducing component of the redox initiator, e.g., aminoiminomethanesulfinic acid and/or its salts, to exist as two tautomers facilitates the post-polymerization of the residual monomer in aqueous emulsions of the polymer.[1282]
Sulfoxylates and bisulfites are also appropriate reducing initiators of post-polymerization, while sodium or potassium persulfates, peroxides and perborates can be used as oxidizing initiators. Among combined redox initiating systems, sodium persulfate – sodium metabisulfite and hydrogen peroxide – rongalite (hydroxymethanesulfinic acid) systems are worth to mention.[1297] Organic peroxides, such as tert-butyl hydroperoxide, are more effective components of redox systems for decreasing the residual monomer content in latexes than persulfates or hydrogen peroxide.[1298]
However, an appropriate system should be chosen with caution, since the post-polymerization of latexes by using organic hydroperoxides in combination with ascorbic acid may lead to the formation of undesirable organic products.[1299] One more drawback of the residual monomer removal with an initiator is difficulty of reaching the required level of the residual monomer during scaling-up process.
6.1.4.2. Removal of the residual monomer by heating
Temperature is one of the key operating parameters in the production processes of polymer materials. The reaction and diffusion rate constants increase with temperature increasing, which leads to a decrease in the residual monomer content. However, high temperature may have an adverse effect on the polymer quality parameters such as the molecular weight distribution due to polymer chain degradation. Furthermore, too early temperature rise may lead to reaction runaway since polymerization is usually exothermic. Therefore, temperature is usually increased at the end of the main polymerization stage using the released heat, which makes the process energy efficient.[1300]
Heat treatment is used, in particular, for the manufacture of polymeric denture inlays,[1286-1290] bone cement[1301] and orthopaedic products.[1302] In the last-mentioned case, the methyl methacrylate (30 mass %) and acrylonitrile (70 mass %) copolymerization product with a conversion of ~75% is heated for 4 – 12 h at 140 – 170 °C, which decreases the amount of the residual monomer from 3000 to 40 ppm. Copolymer darkening during heat treatment does not interfere with orthopeadic applications (prostheses, soles) where a critical parameter is the minimized content of the unreacted monomer, but the colour and transparency of materials are insignificant. Heat-induced post-curing was also used to reduce the residual monomer content in poly(methyl methacrylate)-based bone cement.[1301] In this process, the degree of polymerization was limited by glass transition, and the residual monomer content thus attained (more than 20 000 ppm at 81 °C) can hardly be considered acceptable.
Conducting a thermal post-polymerization stage of the crude product (monomer conversion >95%) in a solution at elevated pressure was proposed for the production of acrylic adhesives for medicine with low contents of the residual monomer and initiator.[1303] This procedure reduced the residual content of undesirable components (monomer and initiator) in the product to 500 ppm and increased the process performance by decreasing the amount of gel deposits on the reactor wall.
6.1.4.3. Combined method for the removal of the residual monomer
If the heat treatment does not reduce the residual monomer content to the desired level, combined methods are used, in particular methods that imply the addition of a high-temperature initiator. For example, the residual monomer content was markedly reduced by refluxing a latex with continuous addition of an initiator (peroxide initiator and a reducing agent) and steam distillation.[1304] Another post-polymerization procedure consists of two stages. Initially, a redox initiator is continuously added to a crude polymer (monomer conversion ≥80%) until the residual monomer content is 1900 – 3000 ppm. The subsequent vacuum steam distillation of the remaining impurities reduces the content of unreacted monomer down to 5 – 200 ppm.[1305] It is important to mention that the two-stage procedure makes it possible to remove not only the monomer, but also the products of decomposition of the initiator and other low-molecular-weight by-products.[1306]
These methods for monomer removal are not always applicable. For example, expanded polymer beads are very sensitive to vacuum distillation and elevated temperature and uncontrollably expand under these conditions. Therefore, to decrease the residual monomer content, the monomer should be polymerized. For this purpose, granules are placed into a vessel filled with a liquid and heated in the presence of a water-soluble initiator. The liquid exerts pressure on the surface of polymer granules and, therefore, they do not expand. The water-soluble initiator absorbs monomers, which migrate from the polymer granules to the aqueous phase. Good results were obtained for polymers with glass transition temperatures below the reaction temperature. The amount of the residual monomer depends on the monomer type, but the best results (≤100 ppm) were obtained for acrylonitrile.[1307]
The content of the residual monomer can also be decreased by adding a reactive comonomer, preferably with a boiling point below 100 °C, at the post-polymerization stage. This comonomer polymerizes and thus traps the residual monomer, while excess comonomer can be easily removed from the reaction mixture. This procedure is also suitable for decreasing the residual monomer content in some copolymerization reactions.[1282]
6.1.4.4. Chemical removal of the residual monomer
A promising way to reduce the residual monomer content is to treat crude polymer with compounds that react with it to form volatile products easily removable by conventional methods. Suitable compounds are those that react with the double bond of the residual monomer, e.g., ammonia, bichromate, various sulfur-containing compounds, hydrogen, hydrogen halides, ozone, etc.[1308] Ozone, which forms non-toxic products not contaminating the polymer, when react with the monomer, is most environmentally friendly. However, highly reactive ozone can cause a decrease in the polymer molecular weight by breaking the polymer chains. In addition, it is not recommended to use ozone to remove residual monomers from unsaturated polymers.[1309]
Ozonization proved to be useful for the removal of residual monomer at the final stage of purification of poly(vinyl chloride) latexes (emulsion and microsuspension polymerization) and suspensions (suspension polymerization) in combination with usual steam distillation or inert gas distillation.[1309][1310] The proposed procedure is simple and economically sound and can be implemented using the existing production equipment. In particular, the residual monomer content in the products of polymerization of N-vinylpyrrolidone and N-vinylcaprolactam was decreased by the proposed method from 20 000 to <5 ppm.[1311] Such small residual monomer content makes the products suitable for cosmetic and pharmaceutical applications.
The residual monomer present in water-in-oil emulsions of acrylamide copolymers was removed by catalytic hydrogenation.[1312] This reaction is carried out under mild conditions (temperature of 20 – 50 °C, pressure of 0.7 – 1.2 MPa) where the polymer is not prone to degradation, and the process does not require expensive high-pressure equipment. m-Xylylenediamine proved to be an efficient reagent for scavenging acrylonitrile that has remained in ABS latexes.[1313]
A chemical method for removing the residual monomer from polystyrene type polymers includes the addition of sulfonyl hydrazide and heating of the mixture above the decomposition temperature of the adduct.[1314] Sulfonyl hydrazides have no odour and no toxicity and do not deteriorate the polymer properties.
6.1.4.5. Additional methods for removal of the residual monomer
In some cases, the amount of residual monomer can be reduced by radiation treatment, which, unlike heat treatment, makes it possible to minimize or avoid deterioration of polymer quality if the radiation dose is low. Using this approach, it was possible to improve the properties of polyvinyl chloride, polyvinylidene fluoride and polyethylene. However, radiation does not affect polystyrene and polyacrylonitrile, while irradiation of polytetrafluorethylene, rubbers and modified cellulose may induce their degradation. In any case, the effect of the radiation treatment depends on the radiation dose and irradiation time. In some cases, irradiation leads to undesirable consequences such as polymerization, cross-linking or rupture of polymer chains or formation of gases.[1315]
The residual monomer can also be removed from the system by other methods such as extraction, addition of solvents, purging with a gas or pressure decrease. In some cases, it is possible to bind residual monomers or oligomers by sorbents or ion exchange resins. The physical contact of the polymer with these materials can be attained by passing a polymer solution through a column packed with activated carbon, zeolites or acidic ion exchange resins[1316] or above a stationary adsorbent layer. It can also be efficient to mix sorbent particles (resins containing sulfonic or carboxylic functional groups) with a polymer solution and then to separate the resin with the adsorbed residual monomer by filtration.[1317] The adsorbent can be reactivated and reused.
The above methods for polymer treatment require additional costs, while degassing procedures may require additional treatment of residual water and emissions. In many cases, a combination of several methods is needed to reduce the residual monomer content to the desired level according to the application.
6.1.5. Problem of residual surfactants and ways to address the problem
Apart from the problem of residual monomers, the free-radical emulsion polymerization is faced with the problem of residual surfactants, which may also be toxic.
6.1.5.1. Traditional surfactants
Due to excess free energy, all emulsions are unstable. During EP, the monomer immiscible with the dispersion medium is dispersed,[1318] while emulsions are stabilized by adding emulsifiers. The emulsifiers promote the formation of an interfacial adsorption layer, which prevents coalescence and/or coagulation of monomer droplets or polymer. Surfactants used as emulsifiers are, most often, aliphatic molecules consisting of hydrophobic and hydrophilic parts (Fig. 35).[1319][1320] Macromolecular compounds can also act as surfactants.[1321][1322]
![[{"id":"15VEbHp23t","type":"paragraph","data":{"text":"Surfactant molecule at the interface (<i>a</i>). Surfactant stabilization of oil microdroplets in water (<i>b</i>).<sup>1322</sup>"}}]](/storage/images/resized/QWyhRBxne5byk8y7UOdoh3jRSrwiNPusP4x7V3de_xl.webp)
Surfactants are widely used as household chemicals (cleaning products and personal care products)[1323-1326] production of fabrics, varnishes, paints and paper,[1327-1329] metal processing,[1330] modern oil and mineral production processes,[1324][1331] EP processes (as stabilizers and additives)[1332] and in medicine.[1333-1335] In terms of the charge of the hydrophilic moiety, surfactants can be classified into four groups: cationic (positive charge), anionic (negative charge),[1336] nonionic (no charge) and zwitter-ionic (the charge depends on the pH of the medium) types (Table 5).[1327][1337] A considerable part of linear surfactants used in industry are alkylbenzenesulfonates, alkylethoxysulfonates, alkyl sulfates, alkylphenol ethoxylates, alkyl ethoxylates and quaternary ammonium salts.[1338][1339]
Surfactants for a particular process are usually chosen considering the hydrophilic – lipophilic balance (HLB) or the critical packing parameter (CPP). Surfactants with low HLB[1340-1342] are used to form water-in-oil emulsions, while surfactants with high HLB[1343-1347] are effective for the formation of oil-in-water emulsions (Fig. 36).[1340][1348] The CPP parameter characterizing the relationship of hydrophilic and hydrophobic parts of the surfactant molecule should also be taken into account to obtain the desired emulsion.[1349][1350]
![[{"id":"kA0QW9EDxG","type":"paragraph","data":{"text":"Applications of surfactants depending on HLB. The Figure was created by the authors using published data.<sup>1350</sup>"}}]](/storage/images/resized/S1d88kZRuAeJcGHSR1BDkYK6v331NeDTvjIz5jFi_xl.webp)
The problem is that the useful function of surfactants ends after they have been utilized in a particular process. Usually, they get into water areas together with wash water and after that, they enter soil, plants and animal and human bodies.[1351][1352] In terms of the median lethal doses (LC50), all anionic surfactants are toxic in the concentration range of 10 – 100 mg L–1, while nonionic surfactants are toxic even at an order of magnitude lower concentrations (1.0 – 10 mg L–1).[1353][1354] Compounds containing a benzene ring together with a branched aliphatic group and compounds with polyoxyethylene chains are especially hazardous.[1353]
Therefore, the replacement of currently used surfactants with natural, biodegradable, non-toxic compounds that exert no harmful effects on the environment is a major direction for the advancement of chemical engineering processes based on the use of phase transfer catalysis.[1319][1322][1355-1358] Another possible approach is the use of polyelectrolyte ‘filters’ that completely turn surfactants to stable complexes which can subsequently be regenerated.[1359][1360]
A vivid example of replacement of toxic surfactants by non-toxic ones in chemical processes is to introduce organosilicon surfactants, which have become more and more popular in recent years, in particular, for emulsion polymerization processes.
6.1.5.2. Organosilicon surfactants
Organosilicon surfactants form a huge class of organoelement compounds, which are used more and more often in various fields,[1361][1362] in particular as additives to adhesives and paints,[1363] for wood processing,[1324] as antifoaming agents in the synthesis of antibiotics,[1364][1365] in blood transfusion systems,[1366] for processing of water containing radioactive waste in nuclear technologies,[1367] as components of formation fracturing fluids in the oil production industry,[1368] and for heterogeneous polymerization in chemical industry.[1369][1370]
The amphiphilic organosilicon surfactants consist of two building blocks: a hydrophobic polydimethylsiloxane moiety bound to a polar group. They are largely superior to conventional surfactants in efficiency owing to specific structural features of the polysiloxane chain.[1371] The latter decreases the surface tension from 30 mN m–1 (characteristic of typical hydrocarbon surfactants) to 20 mN m–1 (Ref. 1358) and endows the polymer with the ability to form helical structures in which the alkyl groups are oriented outwards. Organosilicon surfactants exhibit surface activity both in aqueous and non-aqueous media[1372] and even in supercritical CO2.[1373] In this part of the review, the application of silicon-containing surfactants in the modern emulsion polymerization meant for the synthesis of polymers with a narrow particle size distribution is briefly considered in relation to styrene polymerization.
The conventional emulsion polymerization of styrene in aqueous medium occurs in surfactant-stabilized monomer droplets. The degree of dispersion of a monomer emulsion depends on the interfacial tension and the stirring rate. In addition, the degree of dispersion increases due to the transformation of heat released during polymerization into surface energy. At this stage, Oswald ripening takes place, that is, the monomer molecules diffuse from small droplets to larger ones, which further increases their size. Styrene polymerization in the presence of water-insoluble organosilicon surfactants has specific features, particularly, the surfactant layer on the surface of monomer droplets is formed via surfactant adsorption from the monomer phase. The strength of the surfactant layer formed in this way is sufficient for the onset of formation of core–shell structures even at low conversions.
Gritskova et al.[1374][1375] investigated the effect of linear (496), comb-like (497, 498) and dimeric (499 – 501) organosilicon surfactants on the polymerization kinetics, particle size and emulsion stability.
It was found[1376] that organosilicon oligomers containing reactive vinyl, vinylbenzyl and methacrylic groups can function as both surfactant and comonomer in the same reaction. In this case, stable latexes are formed at surfactant concentrations 5 – 6 times lower than in the presence of water-soluble ionogenic emulsifiers such as sodium dodecylsulfonate.
The use of functional organosilicon oligomers as surfactants in emulsion polymerization opens up great prospects for the synthesis of new latex materials for various purposes using a significantly reduced amount of the surfactant. This is especially important for the formation of homo- and copolymers used as granules or polymers obtained by bulk polymerization.[1377][1378] Considering the biological inertness of organosilicon polymers, it is possible to classify these processes as pertaining to green chemistry. The use of functionally substituted organosilicon surfactants able to be incorporated into coatings and thus completely rule out leakage of surfactants to the environment is especially promising for the production of aqueous dispersion latex compositions that form the basis for water-based paints and related products.
* * *
Thus, analysis of the main factors of polymer synthesis processes from the environmental safety point of view in relation to the emulsion polymerization provides conclusion that the major concern is associated with the residual monomer content and the surfactant used. The use of reactive organosilicon oligomers as surfactants is the major direction for advancing emulsion polymerization processes, taking into account the structural and functional variability of organosilicon oligomers. However, the residual monomer problem is more general and is much more challenging. It can be clearly seen from the foregoing data that, despite the diversity of existing approaches, none of them is universal. In this case, the use of organosilicon systems capable of being incorporated into almost any polymer backbone and containing potential traps for the residual monomer also opens up real prospects for a radical solution to the problem. This solution requires the synthesis of a wider range of organosilicon nanogels, including those containing chemically reactive groups,[1379][1380] and generation of molecularly filled polymer compositions – prototypes of new materials with high performance characteristics.[1381-1383]
6.2. Green chemistry synthesis of energetic compounds and materials
Energetic compounds and materials (ECMs) are critically important chemical products for industrial and military applications.[1384-1386] The focus of related chemistry is gradually shifting towards the search for new environmentally benign ECMs with minimized adverse impact on the environment[1387][1388] and towards development of low-waste processes for ECM production.[1389][1390] Despite the fact that it is difficult to adhere to most green chemistry principles and approaches in this specific field, some acute problems related to the use of large quantities of toxic organic solvents and mixed acids can be solved by applying new synthetic methods, new energy sources, safe and effective nitrating agents and alternative types of solvents.
6.2.1. Electrochemical methods for the synthesis of energetic compounds
Electrochemical processes, in which redox reactions take place at the electrodes without the involvement of chemical oxidants or reducing agents, are a priori considered to be environmentally friendly and are widely used in modern organic chemistry to prepare valuable products for various applications (see also Section 3.4 of this review).[511][554][1391] Electrochemistry has also been used in the synthesis of energetic compounds. For example, nitration of aromatic compounds with nitrogen dioxide (NO2/N2O4) formed in situ upon the electrochemical oxidation of a nitrite in the presence of a twofold excess of 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) in acetonitrile resulted in mononitroarenes in up to 88% yields (Scheme 270).[1392] The reaction was carried out in a glass electrolytic cell divided by a porous glass membrane with graphite electrodes. Tetrabutylammonium nitrite (NBu4NO2) served both as a safe, readily available and easily handled source of the nitro group and as the electrolyte in this process. Under proposed conditions, this nitration reaction was carried out for various substituted arenes, phenols and anilines; the scalability of the reaction was demonstrated by a 13-fold increase in the reactant amounts.
Another electrochemical process used to prepare nitro compounds is the nitration of pyrazole derivatives in the presence of Fe(III) nitrate as the nitrating agent and tetrabutylammonium salt, Bun4NBF4 or Bun4NClO4 , as the electrolyte (Scheme 271).[1393] The yields of N-nitro compounds 502 were 5 – 91%, depending on the substituent nature and position in the pyrazole ring. When the electrochemical reaction was carried out for aliphatic secondary amines, N-nitrosamines 503 were formed. The dependence of the reaction route on the substrate used was attributed by the authors to the formation of different active species in the first step: N• radical is formed in the case of pyrazole derivatives and N•+ radical cation is generated in the case of dialkylamines.
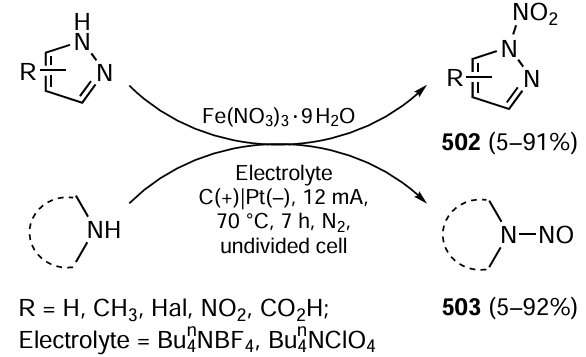
It is noteworthy that, although electrochemical nitration has not yet become widespread, examples of synthesis of energetic compounds by other types of electrochemical reactions have already been reported. For example, precursor 504 for the preparation of promising cyclobutane tetranitrate 505 (Scheme 272)[1394] and new 1,2,3-triazole 1-oxide derivatives 506 (Scheme 273)[1395] were obtained by intramolecular electrochemical cyclization reactions.
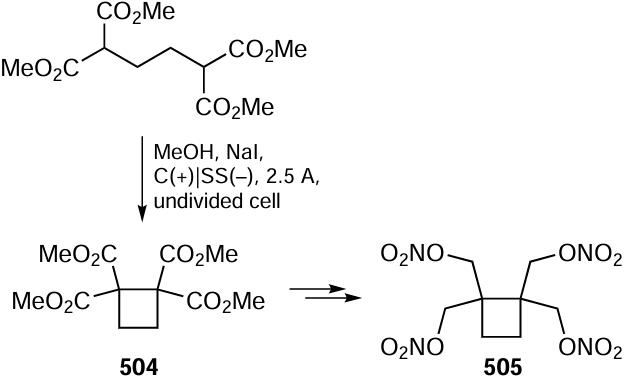
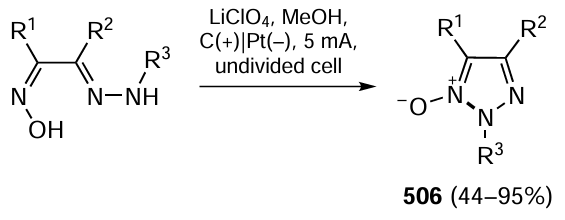
6.2.2. Microwave-assisted nitration
An advantageous approach to the development of environmentally safe processes is the use of microwave radiation, which not only provides fast heating of the reaction mixture, but can also enhance the rate and selectivity of chemical reactions.[1396][1397] For example, microwave-assisted nitration of phenol and its derivatives on treatment with Ca(II) (Ref. [1398]) or Cu(II) (Ref.[1399]) nitrates in acetic acid affords nitrophenols 507 in 60 – 85% yields within 1 min (Scheme 274). Analogous yields were attained in the microwave-assisted nitration of phenols with urea nitrate in aqueous acetonitrile (CH3CN/H2O = 95 : 5).[1400] However, in the absence of acids, the reaction time was longer (30 – 60 min).
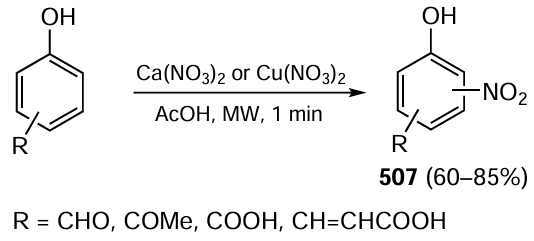
Microwave radiation also has a beneficial effect on the nitration processes involving traditional nitric acid as the nitration agent. Indeed, the time of the synthesis of promising energetic compounds, 3-nitro-1,2,4-triazol-5-one (NTO) and bis(2,2-dinitropropyl)nitramine (BDNPN), was thus decreased from 2 – 3 h to 10 – 15 min, with high yields of the products being retained (Scheme 275).[1401]
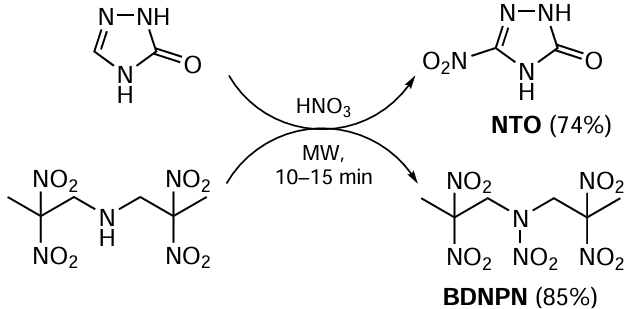
6.2.3. Nitration in green solvents
Despite environmental safety and high efficiency, the synthesis of ECMs under the action of electric current or microwave radiation still requires the use of organic solvents, for example, acetonitrile.[1392] [1400] As a consequence, transition to industrial production is faced with certain risks associated with the storage and use of potentially hazardous organic solvents on an industrial scale as well as the subsequent waste disposal. Certain aspects of the problem can be solved by using alternative types of solvents such as ionic liquids, liquid carbon dioxide or perfluorinated hydrocarbons.[1402]
6.2.3.1. Nitration in ionic liquids
Ionic liquids (ILs), that is, organic salts with melting points below 100 °C, are widely used in various fields of chemistry as environmentally benign solvents.[1403] The first mention of the use of ILs in the synthesis of energetic compounds, particularly as a medium for nitration of aromatic derivatives dates back to 2001.[1404] The reactions were carried out in ILs containing the 1-ethyl-3-methylimidazolium cation: [emim]X (X = OTf, CF3COO, NO3, AlCl4, Al2Cl7). The nitration was performed using binary mixtures NH4NO3/trifluoroacetic anhydride (TFAA), i-C5H11ONO2/BF3 · Et2O, i-C5H11ONO2/TfOH, Cu(NO3)2/TFAA and AgNO3/Tf2O. Among them, NH4NO3/TFAA in IL ([emim]CF3COO or [emim]NO3) and C5H11ONO2/BF3 · Et2O or C5H11ONO2/TfOH in [emim]OTf proved to be the most efficient systems both for the nitration and for the subsequent regeneration of ILs (Scheme 276). Under the optimal conditions, benzene and its derivatives are converted to mononitroarenes formed as mixtures of ortho- and para-isomers in a ratio ranging from 70 : 30 for anisole to 8 : 92 for fluorobenzene. Similar results were obtained in the nitration of arenes with acetyl nitrate (HNO3/Ac2O) in 1-butyl-3-methylimidazolium salts ([bmim]BF4 , [bmim]PF6) or 1-butyl-1-methylpyrrolidinium salts ([bmpyrr]OTf, [bmpyrr]NTf2).[1405]
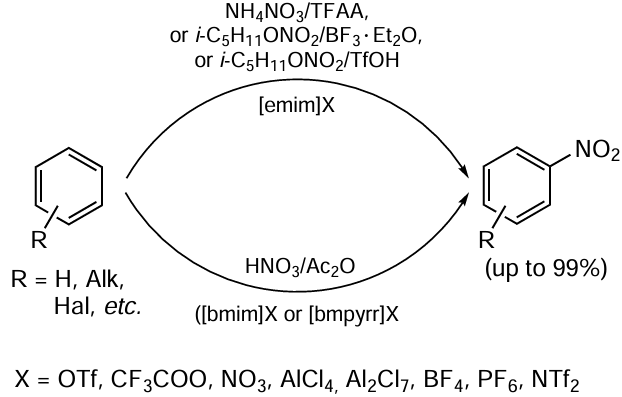
Ionic liquids proved to be effective reaction media for the synthesis of commercial explosives, such as cyclotetramethylene-tetranitramine (HMX) and hexanitrohexaazaisowurtzitane (CL-20). Nitrolysis of 3,7-dinitro-1,3,5,7-tetraazabicyclo[3,3,1]nonane (DPT) by treatment with the N2O5/HNO3/NH4NO3 system in an IL containing two SO3H-functionalized 1,3-dialkylimidazolium cations linked with a polyethylene glycol chain (PEG200-DAIL) results in the formation of HMX in 64% yield (Scheme 277).[1406] Nitrolysis of 2,4,6,8-tetraacetyl-10,12-dibenzyl-2,4,6,8,10,12-hexaazaisowurtzitane (TADB) with fuming nitric acid in 1-methylimidazolium hydrogen sulfate ([Hmim]HSO4) furnishes CL-20 in 90% yield (Scheme 278).[1407] Furthermore, the ionic liquid stable under the reaction conditions can be used in the nitrolysis at least three times with only a minor decrease in the yield of CL-20.
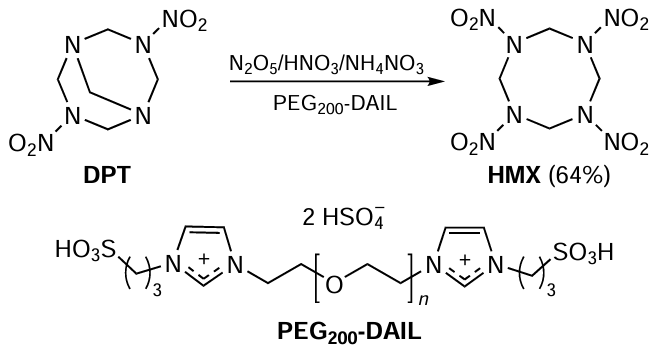
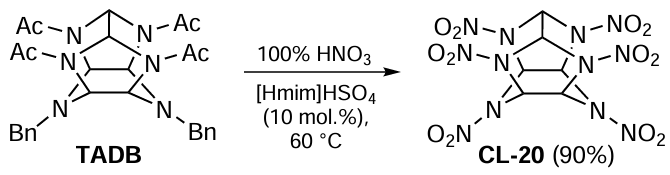
Some ILs can serve as not only effective green reaction media, but also sources of the nitro group in the nitration reactions. These ILs include, for example, 1-sulfopyridinium nitrate [Py – SO3H]NO3 in which the nitration of arenes was performed without an additional nitrating agent (Scheme 279).[1408] Apparently, NO2 gas released from [Py – SO3H]NO3 detaches a hydrogen atom from an aromatic compound, and the resulting arene radical reacts with a second NO2 radical to give the nitroarene.
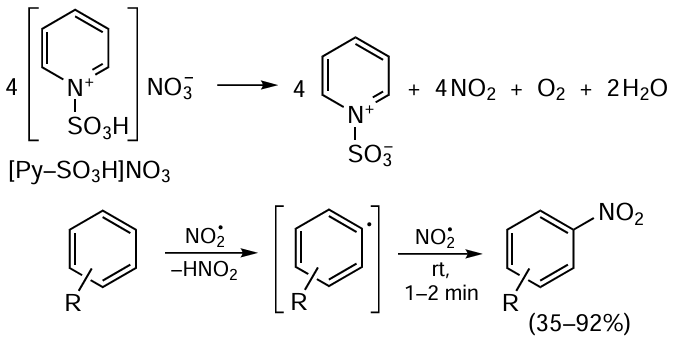
Reactions of styrene derivatives with sodium nitrite can give different products depending on the IL used. For example, the reaction in 1-butyl-3-methylimidazolium chloride ([bmim]Cl) affords β-nitroalkenes 508,[1409] while in tetrabutylammonium acetate (TBAA), benzonitriles 509 are the major reaction products (Scheme 280).
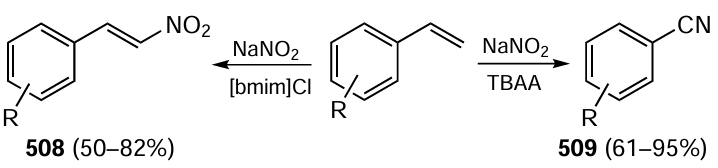
6.2.3.2. Nitration in liquid carbon dioxide
Liquid (liq. CO2) or supercritical (scCO2) carbon dioxide possesses some properties important for the chemistry of energetic compounds.[1410] It is incombustible and resistant to strong oxidants, i.e., tolerates most nitrating agents. The high diffusion coefficient and the density similar to that of liquids ensure effective mass transfer during the reaction. Finally, owing to higher heat capacity (Cр = 130.3 J mol–1 K–1 at 20 °C and 8.0 MPa[1411]) than the heat capacity of organic solvents, it can efficiently consume the heat of exothermic nitration reaction and thus minimize the explosion hazard of the process.
An early example of nitration reaction in liquid carbon dioxide is the synthesis of 3-nitroxymethyl-3-methyloxetane (NIMMO) from 3-hydroxymethyl-3-methyloxetane (Scheme 281).[1412] Dinitrogen pentoxide (N2O5) was applied as environmentally benign nitrating agent in this transformation.[1413] It was carried out by adding 3-hydroxymethyl-3-methyloxetane to a solution of N2O5 in liquid carbon dioxide at –5 °C and 6.8 MPa to afford NIMMO in 95% yield. Similar reactions using the reverse order of reactant mixing made it possible to prepare solid nitroesters such as isosorbide dinitrate (ISDN), pentaerythritol tetranitrate (PETN), D-mannitol hexanitrate (MHN) and nitrocellulose (NC) in this medium.[1414][1415]
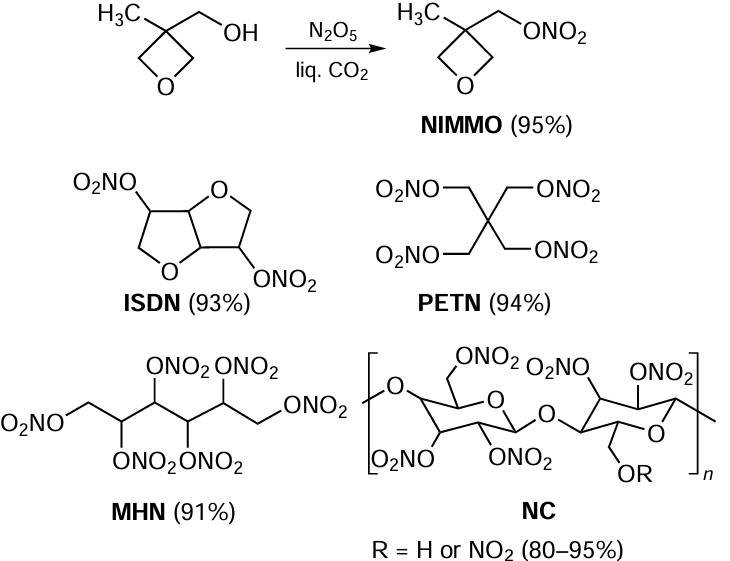
The synthesis of N-nitro compounds 510 by direct nitration of amines in liquid carbon dioxide is complicated by the formation of carbamic acid salts.[1416] However, nitrolysis of protected amines including N-silylamines 511 (Ref.[1417]) and N-acylamines 512 (Ref. [1418][1419]) effectively proceeds in liquid CO2 (Scheme 282). For example, practically valuable N-nitramines such as dinitropiperazine (513) and HMX were synthesized in this way.
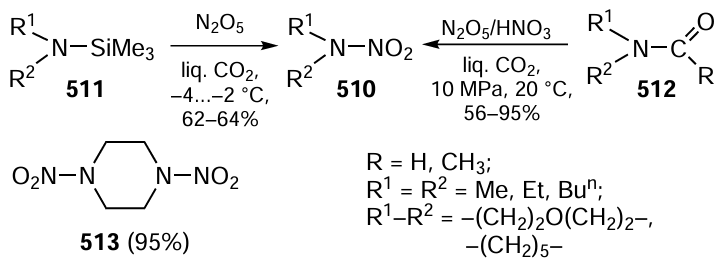
The nitration of glycoluril derivatives 514,[1420] N-alkylcarbamates 515 and carbonic, sulfuric and oxalic acid N-alkylamides 516 under the proposed conditions resulted in the formation of N-nitro derivatives 517 – 519 in high yields (Scheme 283).[1421]
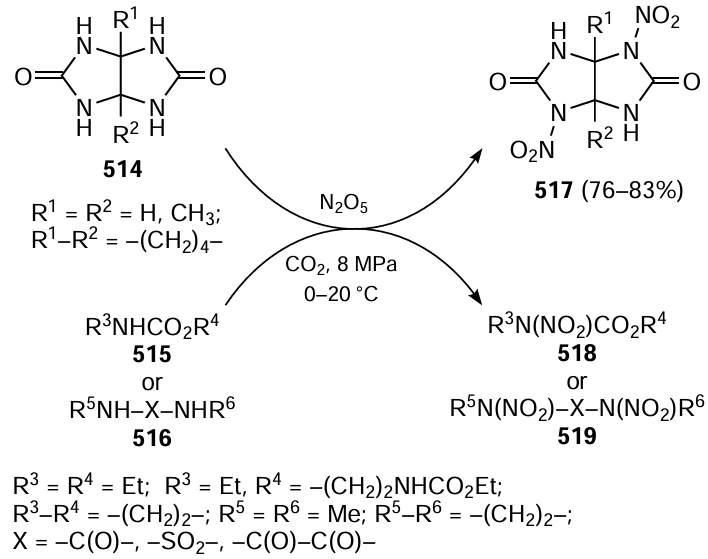
Recently, radical nitration of alkanes with NO2 under UV irradiation was carried out in liquid and supercritical carbon dioxide (Scheme 284).[1422-1424] Under these conditions, cycloalkanes (C5 , C6 and C8) were converted to nitrocycloalkanes 520 in 19 – 25% yields (95% yield in the case of adamantane[1424]). Addition of oxygen to the system (>1.6 equiv.) accelerated the reactions affording alkyl nitrates 521 as the major products in up to 65% yields.
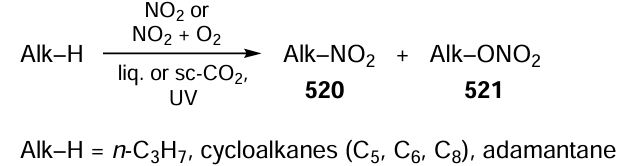
6.2.3.3. Nitration in fluorinated hydrocarbons
Another promising type of solvents used to obtain nitro compounds are polyfluorinated hydrocarbons. These solvents are close analogues of widely used chlorinated solvents, but they are inert and non-toxic. Some of them do not deplete the ozone layer and can be easily regenerated.[1425] Commercially available perfluoromethylcyclohexane, perfluorodecalin and perfluoroperhydrophenanthrene served as the reaction medium in the nitration of aromatic with nitric acid or dinitrogen pentoxide, resulting in the formation of mono- and polynitroarenes in high yields (Scheme 285).[1426][1427]
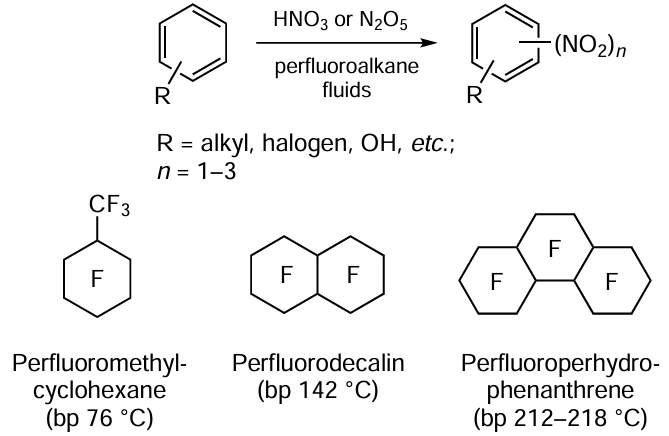
Later, 1,1,1,2-tetrafluoroethane (TFE), a component of industrial refrigeration systems, known as Freon R134a, was identified as a convenient and safe medium for nitration reactions. The chemical stability of TFE is similar to stability the above-mentioned perfluoroalkanes. In addition, owing to considerably lower boiling point (0 °C at 0.3 MPa), it is easily separable from the reaction mixture by mere decompression. Unlike reactions in CO2 , the nitration processes in TFE can be carried out at a much lower pressure (0.6 – 0.8 MPa), which does not require expensive high-pressure equipment. Furthermore, TFE can be easily re-condensed and, hence, repeatedly used in the reaction.[1428][1429]
A plethora of nitroesters,[1428] primary and functionally substituted N-nitramines[1430][1431] and nitroarenes[1432] were synthesized by the nitration of alcohols, carbamates and arenes with N2O5 in the TFE medium. Moreover, a facile continuous flow process for nitration of alcohols in a TFE flow was developed and used to prepare commercially important nitroesters, e.g., 2-ethylhexyl nitrate (EHN), nitroglycerol (GTN) and diethylene glycol dinitrate (DEGDN) (FIg. 37).[1429] The continuous process is characterized by almost two orders of magnitude higher production rate than corresponding batch process. In addition, it is safer, since the steady-state concentrations of potentially hazardous reactants and products in a flow reactor are minimal.
![[{"id":"u3-FaNoyei","type":"paragraph","data":{"text":"Continuous flow nitration of alcohols in TFE"}}]](/storage/images/resized/UK5FAIQe8EgghKUQy9WUYauPqWtvpKl8rTRQ35Km_xl.webp)
6.2.4. Production of energetic materials in liquefied gases
Green solvents, primarily liquefied gases, can be used not only to synthesize useful compounds, but also to convert them to materials such as ultra- and nano-sized powders ready to further processing.[1433] The known methods of micronization using liquefied and supercritical gases include RESS (rapid expansion of supercritical solutions), GAS (gas anti-solvent) and SAS (supercritical antisolvent) processes and their analogues (Fig. 38). The RESS process is applicable to compounds soluble in scCO2 (or in another fluid). It is based on decompression of the solution by passing it through a heated nozzle into a vessel at an atmospheric or lower pressure. As a result, the volatile fluid is rapidly evaporated, and dispersed particles of the dissolved compound are formed in an aerosol jet. If the compound is poorly soluble or insoluble in scCO2 , the GAS or SAS process is used. The scCO2 acts here as a so-called antisolvent, which induces compound crystallization from a previously obtained solution in an organic solvent or water. In these processes, either scCO2 is gradually injected into a solution of the compound to be dispersed (GAS) or both components are injected simultaneously and mixed at the inlet of the precipitation chamber (SAS).
![[{"id":"fa5gzjjTPw","type":"paragraph","data":{"text":"Micronization methods based on usage of liquefied gases."}}]](/storage/images/resized/g6xtScBISAT7UyAoJ7K5lBd0nAxVUehCMD7UbQFi_xl.webp)
1,3,5-Trinitro-1,3,5-triazacyclohexane (RDX) microparticles of the 110 – 220 nm size and a narrow size distribution were prepared via the RESS process by passing an RDX solution in scCO2 through a sapphire nozzle with the diameter of 100 – 150 mm at a pressure of 15 – 30 MPa and temperature of 343 – 348 K.[1434] Later, this process was successfully adapted to pilot production of uniform spherical RDX particles of 200 or 500 nm in size depending on the pressure in the precipitation chamber.[1435] The particles obtained in this way had a lower impact and friction sensitivity than the initial RDX sample.
Usually, ultrafine particles have a higher surface energy and tend to agglomerate thus minimizing the advantages of dispersed material. Therefore, a modified RESS process was proposed,[1436] in which an RDX solution in scCO2 was sprayed into water. The result of crystallization is affected by the acidity of the medium: high-quality RDX nanoparticles with an average particle size of 30 nm precipitate from an NH4OH buffer solution (pH = 7). More stable RDX particles suitable for safe storage, transportation or processing were obtained in the presence of water-soluble polymers–polyvinylpyrrolidone or polyethylenimine (0.005 mass %).
Despite the high efficiency of the RESS process for RDX, it has not found wide use for the micronization of other industrially important energetic materials (ECMs) that are poorly soluble in scCO2 even at fairly high temperature and pressure. In this case, better results were attained by precipitation of ECMs from organic solutions by gaseous (GAS) or supercritical (SAS) CO2 . These methods, based on the use of CO2 as an antisolvent, provide more possibilities to obtain ECM particles of desired morphology.
The efficiency of GAS and SAS processes in micronization of cyclotetramethylenetetranitramine (HMX) from solutions in various organic solvents (acetone, dimethylformamide, cyclohexanone, etc.) was investigated.[1437] The most stable and dense β-HMX particles were obtained by precipitation from acetone or γ-butyrolactone via the GAS process.[1438] Depending on the precipitation conditions, the average b-HMX particle size was 13 – 38 µm. Lower concentrations of the HMX solutions and process temperature, along with the higher CO2 flow and the stirring rates, promoted the formation of fine particles.
The optimization of SAS and GAS processes is a challenging task since the final outcome depends on numerous factors, which include concentration of the starting solution, CO2 flow rate, temperature and pressure, stirring rate and some other. Kim et al.[1439] proposed a mathematical model and determined the optimal conditions for precipitation of HMX from acetone using the GAS process.[1439] The authors evaluated the cost of CO2 and acetone regeneration and demonstrated that despite high capital cost, GAS process is profitable in the long term.
The SAS process in which scCO2 is used as the antisolvent is better suited for micronization of ECMs that do not exhibit polymorphism. RDX microparticles of 1 – 25 mm in size were obtained from a saturated solution in dimethylformamide.[1440] The finest particles were formed at a high pressure drop (11 MPa) in the nozzle.[1441]
Computational methods based on the Hansen solubility parameters were used to optimize the RDX micronization by the SAS process and to reveal the influence of process parameters on the agglomeration of the resulting particles.[1442] Relying on the computed and experimental data, the authors were able to determine the optimal conditions (65 °C, 11 MPa, solution concentration of 4.24 mass %) for the formation of ultrafine RDX particles with a low degree of agglomeration. By simulation of the ECM precipitation in scCO2 , it was also possible to elucidate the influence of equipment parameters on the particle size.[1443] For example, by analyzing the fluid movement in the precipitation chamber, the authors found a region of high-speed vortex leading to an increase in the size of RDX particles. In order to ensure a smooth straight flow, the authors changed the shape of the hanging basket for particle collection from cylindrical to conical and developed a coaxial nozzle with an internal flow mixing zone, which allowed them to obtain RDX particles with an average size of 705 nm and also RDX particles coated with fluorocarbon (F26) rubber film with an average size of 287 nm. The resulting ECM had a narrow size distribution and a lower sensitivity to mechanical stimuli.
Carbon dioxide was found to be inapplicable as the antisolvent for recrystallization of hexanitrohexaazaisowurtzitane (CL-20). In this case, both the SAS and GAS processes gave only the stable α-CL-20/0.25 CO2 solvate, irrespective of the micronization conditions.[1444] The most energetic ε-polymorph of CL-20 could be obtained when CO2 was replaced by 1,1,1,2-tetrafluoroethane (TFE). The resulting ε-CL-20 particles had an average diameter of 12 mm and a narrow size distribution. The proposed method is suitable for converting crude cage nitramine of any polymorphic composition and any quality to finely dispersed ε-CL-20. The anti-solvent (TFE) can be many times reused in a closed cycle, which makes this process green and economically sound.
The SAS process can be used for micronization of not only nitramines, but also energetic polymers. The precipitation of nitrocellulose (NC) from acetone with sc-CO2 afforded round particles with an average diameter of 190 nm (FIg. 39).[1445] The nitrocellulose composites with combustion catalysts (nano-sized Fe2O3 and/or carbon nanotubes) obtained by SAS had much better characteristics than the corresponding physical mixtures.
![[{"id":"ZQKvrvHRiM","type":"paragraph","data":{"text":"Structure and morphology of nitrocellulose: SEM images of the initial (<i>а</i>) and micronized nitrocellulose (NC) (<i>b</i>); TEM image of micronized NC (<i>c</i>); NC particle size (<i>d</i>) distribution.<sup>1445</sup>"}}]](/storage/images/resized/DdOm2VhivyMwojTw2AqGYakp4OFCBt3iPgeUAeiw_xl.webp)
* * *
Thus, the analysis has shown that green chemistry methods are becoming increasingly in demand for the production of environmentally friendly polymeric materials and for the creation of safe and low-waste methods of synthesis of energetic compounds and materials. In the former case, many environmental problems related to the presence of monomer and surfactant residues in the polymer product can be solved by broad implementation of organosilicon systems capable of minimizing the amount of harmful impurities. In the latter case, the risk of accidents and the adverse effect of harmful nitration processes on the environment can be reduced by using green energy sources (electric current, microwave radiation) and alternative reaction media (ionic liquids, liquid carbon dioxide and fluorinated hydrocarbons). In addition, apart from the obvious environmental benefits, the use of liquefied and supercritical gases makes it possible to obtain micro- and nanosized forms of energetic materials with the required particle size, morphology, and homogeneity. Further development of green chemistry approach may result in the creation of effective and safe processes for the production and processing of polymeric and energetic materials demanded by modern industry.
7. Green chemistry and industrial organic synthesis
The application of the green chemistry principles to industrial organic chemistry requires considering cost-effectiveness as an additional criterion: the green chemistry technologies can be implemented in large or medium scale industry, provided that they are economically viable. As R.Sheldon has pointed out, the term ‘green chemistry’ has no economic connotation, but primarily implies the reduction of environmental pollution through the efficient use of raw and other materials, the absence of waste of any kind, hazardous and toxic solvents and reagents. For industrial organic chemistry, the term ‘sustainable development’ is preferable: technologies and products must ensure that the needs of the world’s population are met without compromising the ability of future generations to meet their own needs. Such an approach prioritizes two issues: 1) the most efficient use of available resources with minimal depletion of them; and 2) ensuring a level of waste emissions that can be assimilated by nature without noticeable changes in it. Therefore, for industrial organic chemistry, the issues are not only the emission of hazardous waste, but also the rejection of the use of fossil fuels, decarbonization, the recycling of waste (especially polymer waste), the transition to renewable raw materials, etc.[237][1446]
At present, the very implementation of the ‘green chemistry’ paradigm is inextricably linked to economic efficiency, which is achieved, inter alia, by imposing certain restrictions.[1447] For example, in developed countries, disposal and, moreover, release of waste into the environment are punishable by heavy fines; the use of hazardous materials in production is associated with the need to comply with costly requirements for the protection and safety of employees, the cost of ensuring safe production, and so on. Compliance with other principles of green chemistry contributes significantly to economic efficiency: avoiding industrial waste minimizes the amount of raw materials used per unit of output; the same result is achieved by using by-products as raw materials for the manufacture of marketable products. The elimination or reduction of solvents and the high selectivity of the processes significantly reduce the equipment and energy costs for product separation. Reducing the number of steps in the process leads to similar results. The use of catalysts can significantly increase the productivity of an equipment unit, eliminate intermediate reagents and reduce waste and costs. As a result, it is industrial organic chemistry that has been most receptive to the implementation of green chemistry approaches in practice and the first examples of its application appeared in the 1980s and 1990s.
The move towards a decarbonization strategy, which considers CO2 as waste, and the introduction of new indicators to characterize the acceptability of the technology from a green chemistry perspective, has significantly changed the direction of research in this area. In addition to the E-factor, which suggests the additional consideration of waste in terms of CO2 equivalents,[1446] a C-factor has been proposed as the ratio of the mass of CO2 emitted during production to the mass of the product.[1448][1449] Apart from traditional research related to the development of new catalytic systems, the replacement of non-catalytic processes with catalytic ones, the use of alternative solvents, recycling and waste management, issues related to the change in raw materials for fuels and petrochemicals, the emergence of the ‘oil-to-petrochemicals’ concept, the expansion of the use of bio-feedstocks, increased attention to the use of electricity for the production of industrial organic chemistry products, the emergence of CO2 utilization technologies and the development of research into power-to-chemistry,[1450] e-fuel production, etc.[1451-1453] have come to the fore. In this chapter, the most interesting and promising works in the said directions are highlighted.
7.1. Biphasic catalysis in industrial chemistry
The usage of homogeneous metal complexes in industry is limited by the difficulty of catalyst separation and recycling.[1454][1455] In this context, the so-called biphasic catalysis has been implemented in industry in compliance with the green chemistry principles (Table 6).[1456] With this concept, the catalyst is located in one phase and the reactants and products — in another phase (Scheme 286).[1457] As a result, the liquid phase containing the catalyst can be easily separated from the reaction mass by simple decantation and reused in the process.[1458]
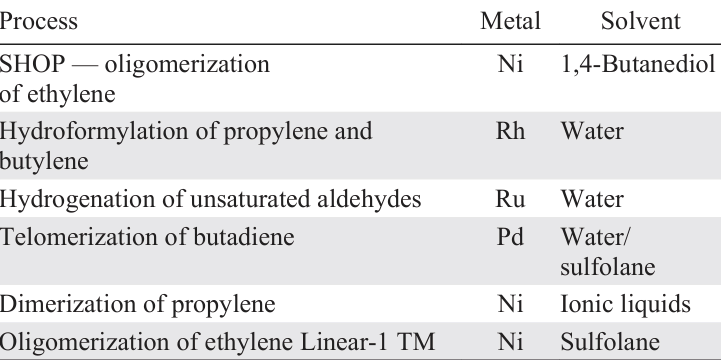
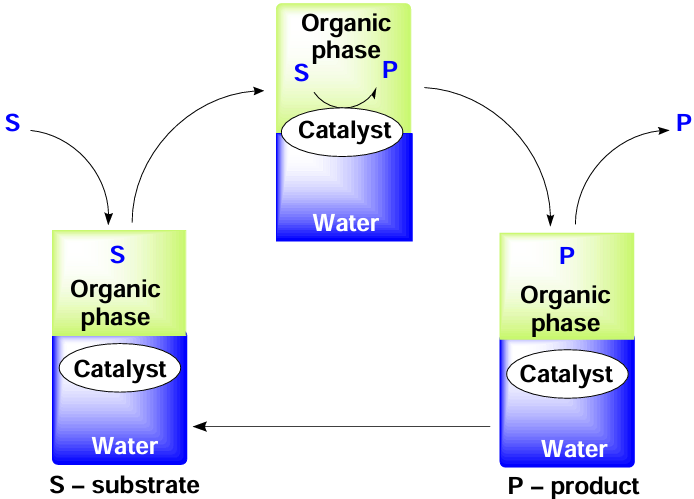
The idea of biphasic catalysis was implemented in particular in the development of the Shell Higher Olefin Process (SHOP) in the ethylene oligomerization step.[1459][1460] In this case, a nickel metal complex catalyst was dissolved in 1,4-butanediol, in which the resultant olefins were insoluble. The telomerization of butadiene to 2,7-octadiene-1-ol was achieved in a biphasic system.[1461] The reaction is carried out in an aqueous sulfolane solution at pH > 7 using a palladium complex formed in situ by the reaction of the palladium source with the phosphonium salt of sodium 2-(diphenylphosphino)benzenesulfonate carbonate or tertiary amine.[1462][1463] The resulting 2,7-octadiene-1-ol is hydrogenated to 1-octanol, the production of which by this technology exceeds 5000 tonnes per year.
The process of hydroformylation of propylene to butanols was developed, including the use of an aqueous solution of a rhodium complex with the sodium salt of tris(m-sulfophenyl)phosphine (TPPTS, Fig. 40).[1464] The success achieved motivated researchers to develop novel catalysts containing hydrophilic phosphine ligands as well as ligands based on water soluble polymers, modified cyclodextrins, calixarenes and dendrimers to be used in hydrogenation, hydroformylation, metathesis, telomerization and other processes.[1457][1458][1465-1471]
![[{"id":"pkCK8zH9YE","type":"paragraph","data":{"text":"Water-soluble phosphines used in biphasic catalysis.<sup>1458</sup>"}}]](/storage/images/resized/sd5lIxtwvINy4iBfdO76BytOpRDNrQgeodhATB8N_xl.webp)
Ionic liquids (ILs), which are salts containing bulk organic cations and organic or inorganic anions, the structure of which prevents their dense packing in the crystal and favours a lower melting point (<100 °C), can serve as alternative solvents in biphasic catalytic processes (Fig. 41).
![[{"id":"_omRGA7evy","type":"paragraph","data":{"text":"Cations and anions of ionic liquids used in catalysis."}}]](/storage/images/resized/2qYW4Hyo8b6J55ZMZAuREdtw7Xa7P8wn8eJcIjb6_xl.webp)
Many ILs are non-toxic, thermally and chemically stable and non-volatile; their properties such as polarity, acidity, nucleophilicity and melting point can be tuned by combining different cations and anions.[1457][1464][1472-1478]1472 – 1478
Ionic liquids can dissolve a variety of organic substrates, thereby increasing the rate and efficiency of reactions involving them. They are also used as components of thermomorphic multiphase systems, which allow the polar catalyst dissolved therein to be separated from non-polar reaction products by changing the temperature of the medium.[1464] [1474][1476][1478][1479]
Since the 2000s, ILs have been actively used as solvents, promoters and ligands in various catalytic processes such as hydrogenation of unsaturated compounds[1480-1483] and phenols,[1484][1485] hydroformylation,[1486-1488] hydroaminomethylation and acetalization,[1489-1492] oxidation,[1493][1494] alkylation, etc.[1493][1495] In recent years, ILs have been used not only in academic research but also in industry as solvents for extraction and purification[1496-1499] and as electrolytes in lithium-ion batteries and solar cells. The largest application of ILs is in the ISOALKY™ alkylation process,[1500] in the SHOP for the oligomerization of ethylene,[1501][1502] in the Dimersol-Difasol process for the oligomerization of propylene,[1503][1504] in the isomerization and oligomerization of γ,δ-epoxyalkenes,[1505][1506] and in hydroformylation using immobilized ILs.[1507-1509]
The ISOALKY™ technology for iso-butane alkylation of the C3 – C5 alkene fraction from catalytic cracking in the presence of ILs (Scheme 287) was developed as early as 1999.[1476][1477] A 10 barrel per day pilot plant was launched in 2005 and a 100 barrel per day demonstration plant in 2010. A full-scale plant with a capacity of 190 ktpy was launched in 2020, following optimization of process conditions and licensing.[1510]
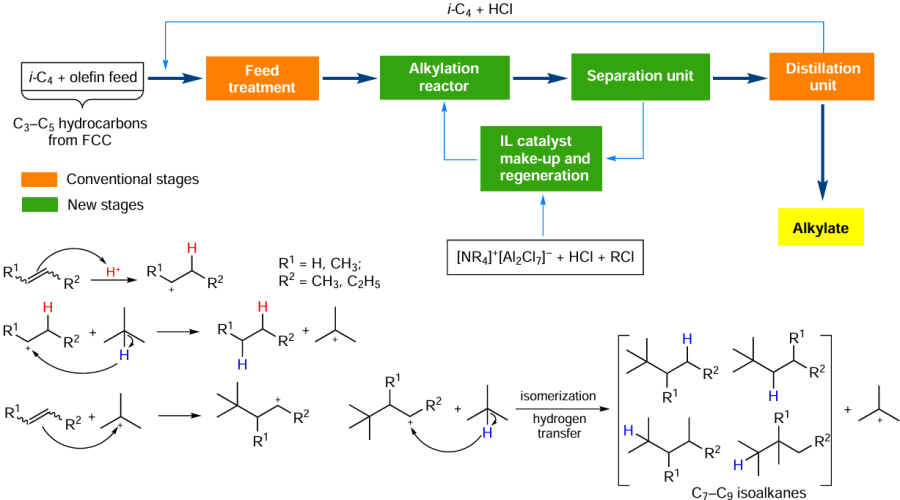
The process is carried out at 0 – 50°C in a biphasic system in the presence of a catalyst consisting of an ionic liquid ([NR4]+[Al2Cl7]–) and a co-catalyst (aqueous HCl) in a hydrocarbon medium. The interaction of [Al2Cl7]– with HCl produces a superacid which readily protonates the olefin (B), which acts as a base, thus inducing the isomerization and alkylation processes (Equation (9)). This furnishes branched alkanes C7 – C8 , the main components of gasoline for internal combustion engines.
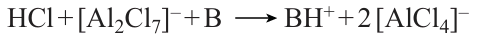
Due to the extremely high acidity of H[Al2Cl7], which directly affects the rate of the alkylation and isomerization processes, it was possible to reduce the catalyst loading to 3 – 6% by volume (cf. 50% for H2SO4 and 60 – 75% for HF) (Table 7). The research octane number (RON) of the resultant alkylate reaches 98 – 99 with an olefin conversion of 99.9%.
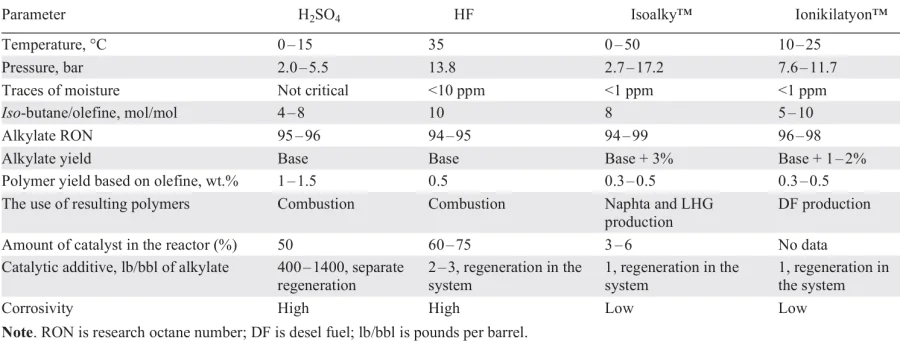
Importantly, a high concentration of iso-butane in the system hampers polymerization: the polymer yield does not exceed 0.3 – 0.5 wt.% per initial olefin loading (see Table 7). Undesirable polymers separated from the light hydrocarbon stream (propane, n-butane and alkylate) are additionally subjected to depolymerization to convert them into components of gasoline and liquefied petroleum gas (LPG), which ensures stable operation of the catalyst. The latter is easily separated from the non-polar hydrocarbon stream by coalescence under gravity. It should be noted, however, that in order to keep the catalyst in an active state and to prevent hydrolysis of the chloraluminate anions, the total water content in the system should not exceed 1 ppm. Another advantage of ISOALKY™ technology is the low corrosivity of the [NR4]+[Al2Cl7]–/HCl catalytic system compared to conventional H2SO4 or HF based systems.
An alternative to ISOALKY™ technology is Ionikilatyon™ technology, which was developed in 1994.[1476] The first pilot plant was built in 2003 and by 2020 plants with a capacity of 50 to 300 ktpy have been built to produce alkylated gasoline using this technology. The production of alkylated gasoline using Ionikilatyon™ technology includes the pretreatment of the feedstock from water, sulfur- and oxygen-containing compounds; catalytic alkylation; settling and separation; recycling of unreacted iso-butane and n-butane; purification of the resulting alkylate; and catalyst regeneration and recycling (Scheme 288).
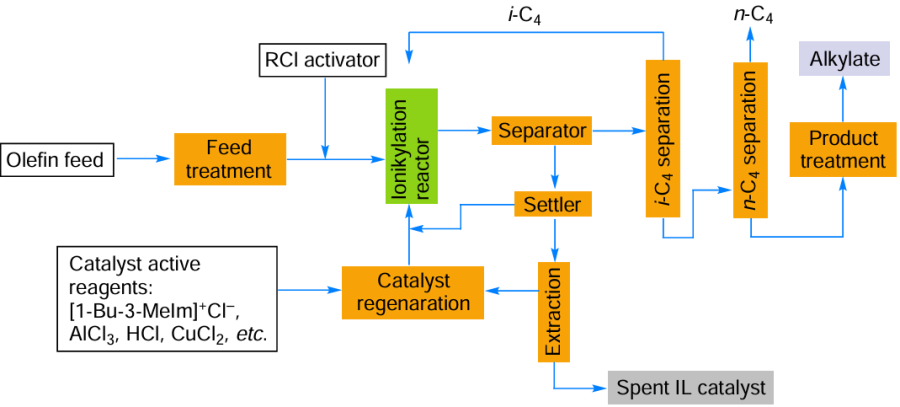
The alkylation is catalyzed by a so-called composite IL consisting of a mixture of 1-butyl-3-methylimidazolium tetrachloraluminate ([1-Bu-3-MeIm]+[AlCl4]–),[1511] HCl, Me2O, a number of aromatic compounds and CuCl2 . It is much less corrosive than conventional alkylation catalysts (H2SO4 and HF) and chloraluminate ILs. The catalyst is present in the reactor as a continuously circulating dense homogeneous phase and can be relatively easily separated from unreacted feedstock and reaction products. In this way, it is possible to obtain high-octane components of gasoline, in particular trimethylpentanes, in high yields (see Table 7) and to significantly reduce the proportion of acidic oils and nitrogen-containing compounds in the product.
A similar system has been used for the alkylation of butenes with iso-butane.[1495][1512] In this case, the catalyst represents a composite IL based on AlCl3 combined with various amides (formamide, acetamide, N-methylacetamide, urea, N-methylurea) and additives of metal chlorides (CuCl, FeCl3 , ZnCl2 , SnCl2). Such a catalyst comprises both Lewis and Brønsted acid sites. The structure of the amide, the AlCl3/amide ratio and the nature of the metal additive significantly affect the activity and selectivity of the catalyst. In the presence of urea/1.6 AlCl3/0.13 CuCl composite, the olefin conversion, C8 iso-alkane selectivity and octane number were 99.9%, 57.5% and 90.7%, respectively, under optimum conditions (15 °C, iso-butane/olefin ratio = 15 : 1 (mol/mol), reaction time 15 min). This composite was used 20 times with almost no loss of activity. The active species catalyzing the alkylation were found to be [AlCuCl5]– and [Al2CuCl8]–ions.[1513]
ILs are currently used as solvents in industrial processes for ethylene oligomerization and the dimerization or co-dimerization of C3 – C5 olefins. The catalysts of such processes are usually cationic nickel complexes (Fig. 42),[1464][1496] which work efficiently in weakly coordinating solvents.
![[{"id":"KIMF6u3jpw","type":"paragraph","data":{"text":"Cationic nickel complexes – catalysts for trimerization of ethylene and dimerization of propylene."}}]](/storage/images/resized/xdVJWRzYaZ30HBifdnPH5VAg76PoeOBSphYRPqAN_xl.webp)
Such processes (e.g., the biphasis Difasol process) in imidazolium-based ILs ([BMIm]+[AlCl4]– or [BMIm]+[PF6]–) are characterized by much higher dimerization/trimerization rate (by an order of magnitude) and better selectivity (90 – 95% of hexenes) than corresponding reactions carried out in a chlorinated solvent or under neat condition (monophasic Dimersol process).[1456][1496] The [AlCl4]– or [PF6]– anions in the IL stabilize cationic nickel complexes, while polar 1-(n-butyl)-3-methylimidazolium cations prevent the dissolution of hexenes (products of propylene dimerization or ethylene trimerization) in the IL phase, thus minimizing their superoligomerization.[1464] As a result, hexenes can be separated by a simple decantation, and the remaining catalyst/IL system can be recycled.
At present, the Difasol process using ILs has been implemented on an industrial scale. Its additional advantages include lower catalyst consumption, ability to use feedstocks with low olefin content, ability to dimerize less active C5 olefins and co-dimerize C4 and C5 olefins, and smaller reactors.
7.2. Heterogeneous catalysts in green industrial organic syntheses
A promising trend for implementing the sustainable development concept in industrial chemistry involves a shift to more stable and easily separable heterogeneous catalysts. In particular, the use of such catalysts can improve the efficiency of large-scale industrial processes for oligomerization, alkylation and oxidation of hydrocarbons and reduce the amount of waste (the use of heterogeneous catalysts in fine organic synthesis is discussed in Section 3.6).
7.2.1. Oligomerization and alkylation processes
Classic examples in this field are the use of zeolite-containing catalysts in the acid-catalyzed alkylation of olefins (e.g., alkylation of butenes with iso-butane), oligomerization of С3 – С4 hydrocarbons and the production of aromatic compounds.[1514] Oligomerization of lower olefins (propylene, butenes) to higher unsaturated hydrocarbons opens the way to motor fuels and also provides raw materials for the synthesis of various petrochemical products, including alcohols, carboxylic acids, alkylphenols and others.[1515][1516] The overall process scheme involves the formation of oligomers and their secondary reactions (cracking, isomerization, oligomerization of secondary reaction products), which can be promoted or inhibited depending on the purpose (Scheme 289).
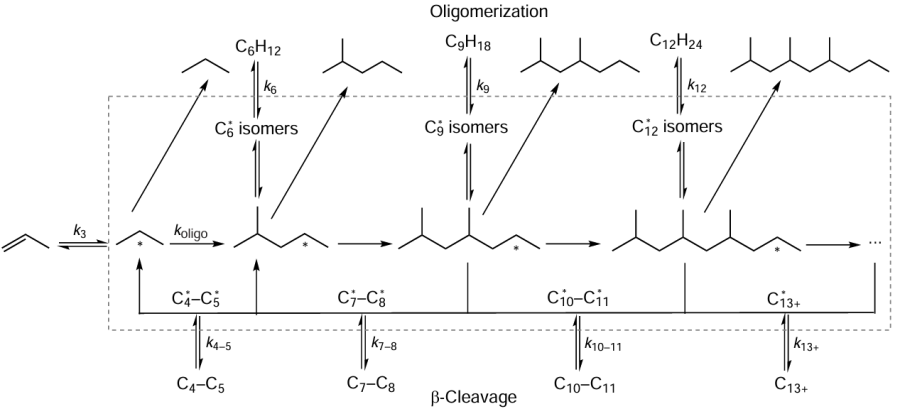
Oligomerization is one of the main processes for the production of low-carbon sustainable aviation fuels from biofuels (ethanol/butanol via ethylene/butene) or carbon dioxide (via methanol and olefins).[1517][1518] Traditional oligomerization processes use relatively unstable supported acids–mainly phosphoric acid (Ipatieff’s catalyst[1519][1520]) or Ni complexes with co-catalysts (Difasol and Dimersol processes[1521][1522]). The transition to zeolite-based catalysts significantly improves the process efficiency, and the variability of zeolite topology and acidity makes them the most suitable catalysts for the production of hydrocarbon mixtures of different compositions. As a result, industrial processes using zeolites, mainly of the MFI type, for the production of gasoline (Mobil Olefins to Gasoline and Distillate (MOGD) and PetroSA Conversion of Olefins to Distillate) appeared in the 1990s.[1515]
Intensive research in this field is focused on two directions: the search for the most efficient zeolite-based oligomerization catalysts and the design of bifunctional systems combining an acidic component (zeolite or amorphous aluminosilicate) and Ni complexes. The efforts of researchers are aimed on creating systems that maximize the yield of the target product and control secondary reactions (isomerization, cracking, hydrogen transfer processes), as well as obtaining products from ethylene and producing fuels heavier than gasoline (aviation kerosene, diesel fuel).
Zeolites are generally used for the oligomerization of propylene, butene-1 and higher olefins. The research is mainly targeted at improving the efficiency of MFI-type zeolites.[1523-1525] Crystallite size exert a significant impact on butane-1 oligomers yield and catalyst stability. Both parameters are increased with decreasing the size of the catalyst crystals.[1526] Zeolite activity is largely determined by acid sites on the outer surface and in the intercrystalline space, and partial passivation of these sites is necessary to achieve high stability.[1527] The importance of substrate and product diffusion processes has been demonstrated in the oligomerization of propylene.[1528] Selectivity and yield in the oligomerization of ethylene[1529] and butenes[1526] catalyzed by hierarchical materials based on MFI-type zeolites can be improved due to the ease of removal of coke formed in mesopores as well as on the crystallite surface.[1530][1531] Catalysts based on zeolites of other topologies, such as MEL[1531] and BEA,[1532][1533] have also been investigated for oligomerization. Oligomerization of hexene-1 proceeds readily in the presence of IM-5 zeolite of the IMF-type modified with fluorine.[1534]
An alternative to acid catalysts are bifunctional catalysts containing both an acid component and Ni compounds.[1535] Zeolites or amorphous aluminosilicates can act as the acid component and nickel phosphide can be used as an active phase.[1536] The use of such catalysts enables ethylene to be used successfully in hydrocarbon fuel production processes. Different products can be obtained depending on the support and active phase.[1537] For example, ethylene can be converted into butenes,[1538] gasolines and paraffin.[1517][1539][1540] Over nickel-containing catalysts supported on the Y- or MFI-type zeolites, ethylene can be converted into branched hydrocarbons, including components of aviation kerosene, in a single step.[1518] The availability of olefins from alternative feedstocks makes green oligomerization processes particularly attractive.
The solid acid-catalyzed alkylation of butenes with iso-butanes has been explored for many years, but an industrial process for the production of alkylated gasoline (alkylate) has emerged relatively recently.[1541-1543] In 2015, the AlkyClean process was implemented in industry. The parameters of a similar process using a CaLaHPtX catalyst were confirmed in a pilot plant. The catalyst was operated in a specially selected structured mode for 24 h with butene conversion of 97.1 wt.%, alkylate yield of 94.1 wt.% and selectivity of 76.4 wt.%.[1544][1545] The replacement of H2SO4 or HF with a solid catalyst significantly increased the environmental safety of the alkylation process. In addition, the yield of 2,2,4-trimethylpentane was higher than that in conventional homogeneous systems and comparable to that in IL-based systems.
Heterogeneous catalysts suffer from rapid deactivation due to the formation of olefin oligomerization products and the need for frequent regeneration. A sign of deactivation is an increase in the proportion of cracked products and oligomers of butene-1.[1541] Deposit formation can be slowed down by increasing the isobutane/butene ratio on the surface and in the pores of the catalyst, and by optimizing the acidity and porosity of the catalyst using its various modifications. The combination of wide cavities with medium-sized channels and the high porosity of the zeolite provided optimal diffusion of reagents.[1546] It was possible to increase the iso-butane/butene ratio within the pores of the heterogeneous catalyst by introducing CuCl into the composition of zeolite Y, which provided an increase in the process selectivity for trimethylpentanes (Scheme 290).[1547] For the BEA-type zeolite in acidic form, a similar result is achieved by significantly increasing the Si/Al ratio.
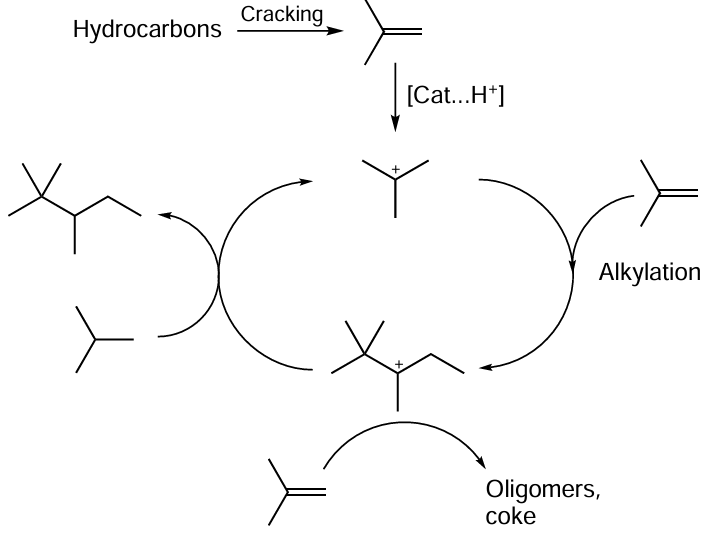
Increased selectivity for trimethylpentanes is also achieved by modifying the outer surface of the catalyst.[1548] Improving the hydrophobicity of the catalyst surface increases the iso-butane/olefin ratio and hence the stability of the catalyst.[1549] According to theoretical modelling results, zeolites such as mesoporous MOR and LTL are unlikely to provide high selectivity for alkylation, whereas BEA and Y zeolites are more promising.[1550]
The selectivity for trimethylpentanes was found to increase with an increase in the number of medium strength Brønsted active sites in the catalyst.[1551] The latter can be achieved by introducing a small loading (1%) of phosphorus in the form of hydrogen phosphate. The incorporation of lanthanum into BEA zeolite allows to tune the strength of the Brønsted sites by varying the La/Al ratio. A high La loading increases the proportion of Lewis sites, which promotes the oligomerization of butenes. A low La/Al ratio leads to a predominance of strong Brønsted sites and cracking. The optimum ratio is about 0.16.[1552]
Stability of the zeolite Y catalyst can be improved by incorporating Al from the binder component into the zeolite lattice to generate additional acid sites.[1553]Importantly, in isomerization over catalysts containing strong acid sites, the initial ratio of butene-2 to butene-1 does not affect the composition of the final alkylate.[1554] The stability of lanthanum-modified zeolite Y can be affected by modification with alkaline earth metals (e.g., calcium for alkylation reactions under slurry bed reactor conditions)[1555] or by the introduction of an acid additive (BF3).[1556] In the synthesis of substituted aromatic hydrocarbons, zeolite-based catalysts eventually replaced Lewis acid and supported acid catalysts at the beginning of the 21st century, opening the way to large-scale chemical processes for the production of xylenes, ethyl- and isopropylbenzene.[1557][1558]
Isomerization of the C8 aromatic fraction (p-xylene production)[1559][1560] and the conversion of toluene to xylene and benzene[1561] are important industrial petrochemical processes. MOR and MFI zeolites have been widely used in these processes. Though, considerable attention has also been payed to other types of zeolites.[1560][1562] The dealkylation or isomerization of ethylbenzene, which is always present in C8 hydrocarbons, can be achieved by the addition and modification of zeolite-containing catalysts.[1514][1557][1563]The use of such catalysts for the synthesis of xylenes from toluene and methanol attracts considerable attention.[1564][1565]
Zeolite-based catalysts are of major importance in the industrial synthesis of ethylbenzene and isopropylbenzene. In the last two decades, technologies using them have finally driven away processes using AlCl3 , thus avoiding harmful effluents. In the gas-phase process of ethylbenzene production, MFI zeolite catalysts[1566] can be employed, while in the liquid-phase processes other types of zeolites can be used, e.g., FAU (for catalytic distillation), BEA[1563] and MWW[1567-1570] (Table 8).
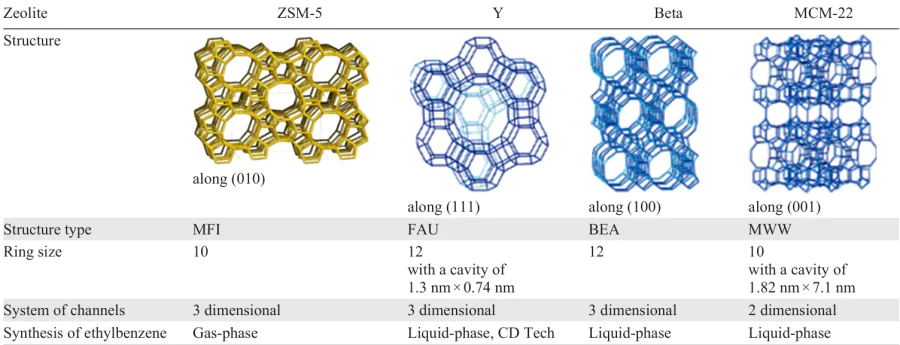
In general, when developing new catalysts, much attention is paid to maintaining yield and activity while reducing the ethylene/benzene ratio, and to increasing the selectivity for ethylbenzene by reducing the amount of xylenes (up to 10 – 3%), oligomerization products and higher alkylbenzenes.[1571] Additional heat treatment can improve the selectivity of gas-phase alkylation.[1572] The introduction of platinum makes it possible to alkylate benzene with ethanol via dehydration of the latter,[1573][1574] including over MFI zeolite nanosheets.[1575] Modification of the outer surface of the zeolite with mesoporous material helped to slow down the catalyst deactivation and increase the selectivity for p-diethylbenzene.[1576]
In the liquid-phase process at 150 – 200 ºC, catalysts based on nanoscale BEA zeolites performed best.[1577][1578] MWW-type zeolite catalysts (MCM-22, MCM-49) were preferable at lower temperatures.[1579-1581]
For isopropylbenzene, only 20% of the processes in 2020 used H3PO4 or AlCl3; in the remaining cases, the process was carried out in the presence of zeolite catalysts such as β-zeolite and zeolites of MWW or MOR types.[1582-1584] The latter is characterized by a hierarchical structure and is mainly used for transalkylation and selective production of para- and meta-diisopropylbenzenes. It is important to note, however, that in most cases diisopropylbenzenes, along with n-propylbenzene and propylene oligomers, are by-products of the alkylation of benzene with propylene.
The use of suitably modified MWW zeolite catalysts allows the process to be carried out at lower temperatures than with BEA zeolite catalyst, thus minimizing the yield of n-propylbenzene, oligomers and diisopropylbenzenes.[1585-1587] The unique properties of MWW zeolite in the alkylation of benzene with propylene largely stem from the fact that the reaction occurs on the outer surface of the crystal [001] facets.[1588] At the same time, the β-zeolite catalyst, although requiring higher temperatures, allows the use of a significantly lower propylene/benzene ratio (~2 : 1).[1589][1590] Moreover, its activity and selectivity can be tuned by changing the crystallite size and acidity.[1591][1592]
All types of catalysts require a suitable binder to achieve the required activity and selectivity.[1593-1595] The binder can be used to influence the interaction between the layers in the catalyst structure, thereby increasing its activity at low temperatures.[1596-1599]
It should be noted that apart from the study of the alkylation of benzene with ethylene and propylene, research is actively underway to develop processes for the production of higher alkylbenzenes.[1600-1602]
7.2.2. Catalytic oxidation processes using heterogeneous catalysts in industrial organic syntheses
The Green Chemistry concept has had a tremendous impact on research into the development of industrial catalytic selective oxidation processes. The transition to heterogeneous catalysts has significantly improved their environmental performance, for example, the shift from homogeneous oxidative acetoxylation of ethylene in acetic acid to ‘green’ vapour-phase acetoxylation on Au/Pd supported catalyst.[1603-1605]
It has become possible to use green oxidants (O2 , H2O2) in these processes, producing water as a by-product, as well as using nitrous oxide as an oxidant, which is converted to molecular nitrogen during the oxidation of the substrate.[1606-1609] The development of industrial catalysis allows for few-step oxidation processes, thereby reducing the overall amount of harmful emissions and waste. Finally, exothermic oxidation processes do not require the use of additional energy and are therefore characterized by a minimal carbon footprint: all other things being equal, energy production within the reactor is preferable to outerheating by fuel combustion.[1610][1611] However, the selectivity and stability issues of the employed catalysts remain unresolved in many cases, making research in this area particularly relevant.[1612-1614]
Processes such as the production of maleic anhydride by oxidation of butane with oxygen over heterogeneous vanadium-containing catalysts; the synthesis of acrylonitrile and acrylic acid by oxidation of propylene over catalysts containing mixed oxides of Mo, Bi and Sb;[1615][1616] the production of vinyl acetate from ethylene, acetic acid and O2 have been implemented in industry for a relatively long time.[1617][1618] These processes operate with relatively high selectivity and are well developed in modern petrochemistry.
In recent years, considerable attention has been focused on the catalytic oxidation of alkanes, including the methane-to-methanol oxidation and the oxidative methane-to-ethylene dimerization. The development of such processes can significantly reduce the burden on the environment compared to traditional approaches to these products.[1619-1621] In the reaction of aerobic (air oxygen) methane-to-methanol oxidation, the greatest activity has been observed for MFI, MOR and CHA zeolites[1621-1624] modified with copper compounds. The active sites in these catalysts are [Cu3(μ-O)3]2+ clusters or other polynuclear structures, making such catalysts similar to enzymes.[1625-1628] Mo and La oxide systems,[1614][1615] as well as W/Mn-containing catalysts, have proven effective in the oxidative dimerization of methane.[1629] Using oxidative methane dimerization combined with ethane cracking, Lummus Technology and Siluria have built a demonstration plant for ethylene production based on this technology.[1630]
Over the last two decades, the oxidative dehydrogenation of ethane and propane with molecular oxygen is considered as perspective source of petrochemical feedstock.[1631] The most effective catalysts here are systems based on mixed metal oxides (Mo, V, Nb, Te) containing the so-called M1 phase.[1632-1636] They allow to reduce CO2 emissions by 60% compared to the pyrolysis process.* Oxidative dehydrogenation of ethane using the ‘chemical looping’ approach, is of particular interest for chemical industry. The active catalyst phase is regenerated by the reaction with O2 .[1637-1639] The oxidative dehydrogenation of propane is complicated by the oxidation of the C – H bond in the allylic position of propylene.[1640][1641] The systems containing vanadium oxide supported on magnesium oxide and boron-containing catalysts containing the boron nitride support are active in the oxidative dehydrogenation processes.[1642]
Along with reactions using oxygen, considerable attention is being paid to processes involving milder oxidants such as sulfur or CO2 .[1643]In the latter case, it is possible to utilize greenhouse gases and to obtain carbon monoxide, which, together with olefin, is very important reagent for industrial organic synthesis.[1644] This reaction is endothermic, requiring elevated temperatures to achieve acceptable conversions. As a result, a plethora of catalysts for the synthesis of ethylene and propylene based on Ga, Cr, In oxides and supported metal nanoparticles have been proposed.[1645-1652]
Catalytic oxidation of hydrocarbons with oxygen can also deliver other petrochemical products. The most interesting results were obtained in the synthesis of acetic acid catalyzed with mixed oxides of Mo, V and Nb. For an industrial-pilot process, the Mo1V0.25Nb0.12Pd0.005Ox catalyst was proposed, which provided complete CO conversion with a selectivity of 80% at an oxygen pressure of 2 MPa.[1653][1654] The SABIC company built a plant with a capacity of 30 thousand tonnes of acetic acid per year using this technology.[1655] The efficiency of the catalyst was improved by modifying it with metals (Au, Sn, Ti, etc.). Nb can be replaced by W, Mn, Sb, Ce, Ta, etc.[1600][1656] Using Rh-ZSM-5 catalyst, this process can be carried out at low temperatures.[1657]
Improving processes for the production of acrolein, acrylic acid and acrylonitrile by oxidation of propane as a primary chemical feedstock is an important task. Such an approach can reduce emissions and waste in the production of these products. The industrial synthesis of acrylonitrile from propane was carried out using a mixed oxide catalyst containing Mo, V, Te, Sb, Nb.[1658-1662] A number of similar catalysts have been proposed for the production of acrylic acid from propane, the selectivity of which increases when using Bi3+ and K+ as promoters.[1663-1665] However, the required yield of acrylic acid (65%) has not yet been achieved.[1666] Acrolein yields were even lower.[1635] Interestingly, the direct oxidation of propane with oxygen at high temperatures using inert material (boron nitride, silicon carbide, etc.) produced propylene oxide (4%) and ethylene (5%), in addition to propylene (19%), rather than acrylic derivatives.[1667]
In addition to O2 , nitrous oxide (N2O), a by-product of the oxidation of cyclohexanone to adipic acid by HNO3 , can be an environmentally friendly oxidant. Nitrous oxide readily evolves oxygen to form nitrogen. One of the most common catalysts for oxidation processes using N2O are iron-containing zeolites. G.I.Panov[1668-1671] found that so-called alpha-sites contained therein react with N2O to form active oxygen species capable of oxidizing methane to methanol. The nature of these particles is a subject of debate and not fully understood.[1672-1674] Among the industrially relevant processes using nitrous oxide is the process of phenol production from benzene.[1675-1677] There are also reports on the use of N2O for the conversion of butenes to ketones under non-catalytic conditions[1678] and for the preparation of cyclododecanone by oxidation of 1,5,9-cyclododecatriene.[1679][1680] Nitrous oxide can also be employed as an oxidant for the dehydrogenation of propane with acceptable conversions (29 – 69%) and selectivities (45 – 69%).[1542] [1681] Finally, there is evidence for the possible use of nitrous oxide in the oxidative dimerization of methane over perovskite and samarium-based catalysts.[1609][1682-1684]
Hydrogen peroxide (H2O2) is another green oxidant that has been actively used in industry in recent years. In addition to high selectivity for oxidation products, the industrial implementation of processes using H2O2 also requires high selectivity for the peroxide per se, which is an expensive oxidant that decomposes readily even under relatively mild conditions in the presence of traces of variable valency metals. A combination of hydrogen peroxide with a heterogeneous catalyst was first used by Eni for the production of hydroquinone and pyrocatechol from phenol. The catalyst was an MFI-type zeolite with a high SiO2 content, in which Ti atoms were introduced in tetrahedral coordination (TS-1) instead of Al atoms. The process features high selectivity for dihydroxybenzene at a phenol/H2O2 ratio of ~4 : 1 and H2O2 selectivity of more than 90%.[1583][1605] The oxidation of benzene to phenol required the use of sulfolane as a solvent and the development of a process to convert the by-products (pyrocatechol and hydroquinone) to phenol by deoxygenation.[1582]
Ti-containing zeolites are among the most suitable candidates for the development of oxidation catalysts using H2O2 . In particular, they have been used in the industrial process for the production of propylene oxide[1685] and cyclohexanone oxime (caprolactam synthesis) from NH3 , H2O2 and cyclohexanone.[1605]It has long been thought that propylene oxide is best produced in the liquid phase in methanol.[1686-1689] However, despite the high yields of propylene oxide, by-products including methoxypropanol (~4%) are formed under these conditions. In water and butanol, the selectivity of the process was low,[1690] indicating the importance of solvolysis of propylene oxide during the reaction.[1691][1692] Acetonitrile was found to be a suitable green solvent for the epoxidation,[1693] and its promising application was confirmed in a pilot plant with a capacity of 2000 t h–1 and high selectivity for propylene oxide. The influence of impurities on the process[1694] was studied and the kinetic regularities of the epoxidation reaction were determined, including those in three-phase reactors.[1695]
A variant of the catalyst based on TS-1 zeolite hollow crystals with a silica-enriched outer surface was developed. This reduced the amount of methanol and increased the selectivity for propylene oxide to 96%.[1696][1697] Various methods have been developed for catalyst regeneration using solvents, oxidation with hydrogen peroxide or air.[1690][1698]
In recent years, many other zeolite-containing materials have been developed, including hierarchical ones that allow the oxidation of sterically demanding substrates.[1699] The validity of this approach has been demonstrated in the epoxidation of butenes[1700] and ethylene.[1701] Platinum metal-based catalysts have been proposed for the synthesis of H2O2 from H2 and O2 and the subsequent epoxidation of propylene. In addition to Pd, epoxide-forming Au nanoparticles immobilized onto TS-1 can be used to promote epoxide formation.[1702][1703]
The possibility of carrying out epoxidation of alkenes with H2O2 on titanium-containing catalysts in the gas phase was explored in detail.[1704-1706] It was found that this case requires a special modification of the catalyst and carrying out the process in a fluidized bed.[1707]The use of acetonitrile and hydrophilization of the zeolite surface made it possible to transfer the reaction to the liquid-phase mode inside the catalyst pores, which increased the selectivity of the process.[1708]
Another area of research in catalytic epoxidation is the use of titanium-containing zeolites, in particular TS-1, for the production of epichlorohydrin from allyl chloride.[1709-1711] Corresponding industrial process has been developed in the People’s Republic of China. It is characterized by H2O2 conversion of more than 97% and the selectivity for epichlorohydrin exceeded 96%.[1697][1712] The possibility of using methanol as a reaction medium and solvent for washing and regeneration of the catalyst has been demonstrated.[1713]
The production of cyclohexanone oxime from cyclohexanone, H2O2 and NH3 can be efficiently carried out in the presence of TS-1. This method enabled to give up polluting processes for the production of hydroxylamine,[1605][1697][1714] providing complete conversion of NH3 and cyclohexanone with 90% selectivity for the target product and for H2O2 . Further studies led to the development of a catalyst with a minimum content of TiO2 , the component responsible for the decomposition of H2O2 . The addition of polysiloxane prevented the deactivation of the catalyst due to the transfer of part of the silicon from the lattice into the solution during the interaction of the zeolite with ammonia.[1715-1718]
Other H2O2-activating catalysts include the aforementioned MFI-type zeolites containing iron and copper ions. They allow methane-to-methanol oxidation with H2O2 with rather high activity.[1670][1719-1725] In this case, it is possible to obtain H2O2 in situ,[1726] and also to use other catalysts, including those based on metal-organic frameworks.[1727]
In general, the use of alternative oxidants together with specially designed heterogeneous catalysts will remain important for the development of industrial organic synthesis over the next decades. The transition to more environmentally friendly oxidants, the reduction of the carbon footprint by switching from traditional dehydrogenation processes to oxidative ones, and the reduction of the number of steps will determine the main trends in this area of industrial organic chemistry.
* https://www.linde-engineering.com/en/processplants / petrochemical-plants/edhox-technology/index.html (last access date 16.01.2024).
8. Conclusion
Green chemistry research, which is the subject of the present review, requires fast and reliable evaluation of the environmental friendliness of the developed methods and processes. The recently proposed bio-Profiles of chemical reactions based on determination of the cytotoxicity of reactants and products using cell cultures provide a qualitative (visual) and quantitative evaluation of the effect of chemical reactions on biological subjects. Low cost of cytotoxicity tests and relatively easy handling of cell cultures compared to experiments on mammals substantially accelerates the preliminary identification of the most toxic components of a chemical process and, hence, enables timely corrections of the planned synthesis.
The analysis carried out in the review clearly indicates that the green chemistry paradigm formulated by its founders, Paul Anastas and John Warner, as twelve principles, which have already become classic, greatly influences the strategy and trends of modern organic synthesis. The most obvious consequence of this impact is the extensive use of catalysts and catalytic methods in numerous studies carried out by chemists all over the World in various areas of synthetic organic chemistry and related fields, with most of other environmental recommendations being addressed.
The use of catalysts proved to be fairly convenient for direct C – H functionalization of organic compounds, which requires no auxiliary (directing and protecting) groups and, hence, significantly decreases both the economic costs and the amount of harmful emissions into the environment; this is especially important for planning large-scale production of pharmaceutical substances and other practically significant compounds. Depending on the type of substrates, C – H functionalization reactions of arenes are efficiently catalyzed by transition metals, metal salts or complexes, organic photoactive molecules (photoredox catalysts), redox-active organocatalysts, N-heterocyclic carbenes or catalysts of other types. Although an excess of reactants, high reaction temperature, poorly accessible catalysts and ligands, and regioselectivity problems still prevent more extensive use of this promising methodology, there is every reason to expect that many of these problems will be solved in the foreseeable future.
The modern approaches to direct selective oxyfunctionalization of aliphatic С – Н groups are mainly based on using non-heme (non-porphyrin) manganese and iron complexes as catalysts and hydrogen peroxide as an oxidant. The currently available library of this type of catalysts makes it possible to convert С – Н groups in various organic substrates to alcohol, keto and ester groups with a high turnover number (TON = 100 – 1000). In the presence of chiral manganese complexes, the enantioselectivity of С – Н oxidation reactions may reach 99% ее. Currently, studies in this area are aimed at increasing the regioselectivity and adjusting the results obtained for simple model compounds to complex (including natural) molecules. In addition, it is necessary to solve the problem of using dioxygen, the most readily available and environmentally safe oxidant, in these reactions and to obtain data on the applicability of the method to replace the hydrogen atom in aliphatic С – Н groups with other heteroatoms. In the short and medium term, one should expect an increase in the number of publications, fundamental and applied studies in the field of selective С – Н functionalization at late stages of the synthesis (including electrosynthesis) of complex organic molecules.[1728]
The green chemistry concept largely determines the vector of development of the catalytic cross-coupling, a promising synthetic strategy. It turned out that many reactions of this type can be carried out in the reactant medium or in green solvents using non-toxic, recyclable and cheap catalysts. As new achievements in this field, one can mention cross-coupling reactions in a solvent flow, microwave- and ultrasound-assisted reactions and photo- and electrocatalytic reactions. The possible trends of the future development of the green catalytic cross-coupling strategies would apparently include the use of recyclable metal nanoclusters [1729][1730] and nanocages,[1731] nanoparticles encapsulated into inorganic and organic porous materials[1732] and nanocatalysts with a controlled structure and atomically precise catalysis.[1733]
Asymmetric organocatalysis, a promising trend of organic synthesis, which gained recognition of the scientific community in the 21st century, is even more complementary to green chemistry. Chiral amines and some other organocatalysts are mimetics of natural enzymes, which is the base for their high stereoinduction. However, they have a much simpler structure and are less substrate-specific than enzymes, which extends the scope of their applicability. In 2019, IUPAC included organocatalysis in the list of ten top emerging technologies in chemistry, which are able to provide the sustainable development of the humankind. The priority tasks in this field for the near future are the search for ways to increase the activity and productivity of organocatalysts up to the level comparable with that of transition metal catalysts and the design of stable heterogenized forms of organocatalysts that can operate for a long time without the loss of activity. Another line of future research is the development of scalable organocatalytic reactions in a continuous flow of a green solvent and design of hybrid processes, in which organocatalysis is combined with photocatalysis[1734] or electrolysis.[1735] The significance of artificial intelligence for the theoretical prediction of the most promising organocatalysts will apparently increase.[1736]
A promising way to switch the chemical industry to sustainable development is organic electrosynthesis–a methodology that can be easily integrated with renewable energy sources. It has been found that the efficiency of many redox CH- and NH-functionalization reactions induced by electric current increases when the reactions are carried out in the presence of catalysts, in this case, metal salts and complexes, including complexes doped with silicate particles. Some of the developed transformations were scaled-up using electrochemical microreactors. Prospects for the future development of organic electrosynthesis related to green chemistry include the search for non-toxic and safe solvents characterized by high conductivity and low environmental impact and also the development of processes in continuous flow reactors. It is expedient to carry out electrolysis in water or aqueous alcohols and to use ionic liquids. Complications are associated with the low solubility of organic compounds in water and with the fact that intermediates generated under the action of electric current are unstable in ionic liquids. A serious problem is the need to minimize the amount of supporting electrolyte used in electrosynthesis. Possible ways to solve this problem is to develop multi-site solid polymer electrolytes (polyelectrolytes)[1737] and to apply microreactors and two-phase electrolysis, which make it possible to carry out the synthesis without the addition of supporting electrolytes.[1738] It is necessary to radically improve the design of flow electrolysis cells, develop paired electrosynthesis, in which different target products are formed at both electrodes,[1739] and optimize the existing electrosynthetic processes. The solution of the problem of electrochemical reduction of carbon dioxide and increasing the selectivity of this process to valuable organic products would obviously be a breakthrough.
A persistent trend of modern organic chemistry is extensive implementation of the multicomponent reaction strategy. These processes are fairly efficient regarding green chemistry, as they substantially decrease the resource, energy and labour costs, minimize the amount of waste and exclude the steps of isolation and purification of the intermediate compounds. Most of these reactions occur in the presence of acid – base catalysts or under the action of electric current as a source of electrons. The simultaneous mixing of components increases the reproducibility of the results, as this eliminates the factors related to the rate of addition of each reactant and the subsequent distribution of the reaction mixture. It is expected that in the future, considerable attention will be paid to the development of effective catalysts for multicomponent reactions easily separated from the reaction mixture and to the conduction of reactions in green solvents or in reactants. There is no doubt that multicomponent reactions will be among the most demanded areas of organic synthesis in the future.
The synthetic methods based on the use of stable and easily recoverable heterogeneous catalysts would play an increasing role in the design of complex organic molecules for various purposes (drug substances, herbicides and pesticides, electronic and data storage materials). The catalytic systems containing hydroxyapatite- or hydrotalcite-supported metals and cobalt–molybdenum – sulfide catalysts proved to be efficient in hydrogen-free hydrogenation reactions. Metal oxide- or cerium-doped hydrotalcite and mesoporous perovskite, nitrogen- and phosphorus-doped hollow carbon spheres, and some other catalysts containing no noble metals proved to be useful in the glycerol aerobic oxidation and photocatalytic reactions. It is important that active forms of heterogeneous catalysts possessing unusual, and in some cases, unique properties can also be obtained under green chemistry conditions, in particular, in supercritical carbon dioxide (scCO2).
Carbon dioxide, a natural compound that is liquefied under pressure and easily removed from the reaction mixture after a pressure drop, proved to be useful not only for the preparation of catalysts, but also as an environmentally safe medium for various catalytic reactions. However, due to the poor solubility of most transition metal complexes in liquid and scCO2, these reactions require special ‘CO2-philic’ catalysts containing long hydrocarbon chains and/or fluorine atoms in the ligands. Palladium, rhodium and ruthenium complexes modified in this way catalyze cross-coupling, homo-coupling, allylic alkylation, hydrogenation, hydroformylation, hydroaminomethylation and other reactions in scСО2. The benefits of using carbon dioxide are especially pronounced in copper-catalyzed reactions, which proceed in this case more effectively than in conventional solvents. One more promising type of solvents for metal-catalyzed reactions are deep eutectic solvents that can be tuned to a particular reaction by merely replacing the components. It is quite likely that a wider range of catalytic processes would be implemented in these green solvents in the near future.
A basic principle of green chemistry is the preferential use of renewable natural feedstocks in chemical processes, first of all, compounds of plant origin, instead of fossil hydrocarbon feedstock. A versatile object for bioprocessing is wood, which forms the predominant part of the Earth biomass and already serves as a source for the production of more than twenty thousand names of products and items. Recycling of pulp and paper mill (PPM) waste is effective from the environmental point of view, but still unprofitable in terms of economic indicators. However, PPM by-products such as kraft lignin, crude tall oil and crude sulfate turpentine can be used for the synthesis of many practically important compounds. A number of valuable chemical products were obtained from monoterpenes isolated from turpentine. These products include organocatalysts and useful biologically active compounds: insect repellents, herbicides and pharmaceuticals. An important role in these transformations belongs to epoxidation, hydration, hydroformylation, reductive amination, allylic oxidation and other reactions; some of these reactions effectively proceed in the presence of transition metal catalysts or enzymes or upon exposure to light. Enantiomerically pure abietane-type diterpenoids isolated from rosin are quite attractive substrates for the asymmetric synthesis of biologically active products. However, because of the abundance of reaction sites, the oxidative transformations of abietinic acid derivatives are often non-selective, which necessitates the development of new synthetic methods.
An exceptionally important task is to produce functional materials with desired physicochemical and performance characteristics using green chemistry methods. Examples of such materials are polymers, being large-scale chemical products the environmental aspects of the production and application of which are under active discussion. The review analyzes the possible ways to increase the environmental safety of polymers obtained by free-radical emulsion polymerization, in particular in water. The undesirable residual monomer present in the product can be subjected to post-polymerization with an initiator, removed by heat treatment or converted to non-toxic products by treatment with appropriate reactants. Unfortunately, none of these approaches has become universal. Perhaps, a radical method for elimination of toxic ionic surfactants from the polymerization system is the replacement of these components by harmless organosilicon oligomers functioning as surfactants, which can also serve as potential traps for the residual monomer.
The green chemistry paradigm was also reflected in a specific field of chemistry pertaining to the production of energetic compounds and materials. Nitration has started to be carried out under conditions of electrolysis or microwave assistance and in ionic liquids or liquefied gases, first of all, carbon dioxide and fluorinated hydrocarbons. These alternative reaction media have made it possible to use milder reaction conditions and reduced the fire and explosion hazard of the processes, while the use of dinitrogen pentoxide as the nitration agent excluded the formation of harmful mixed acid waste, which is difficult to dispose. However, the practical implementation of these results is still hampered by the high cost of high-pressure equipment and lack of a convenient method for the synthesis of dinitrogen pentoxide suitable for industry. The use of liquefied gases may be fairly promising for the new technologies of manufacture of micro- and nano-sized forms of energetic materials (RDX, HMX, CL-20, etc.) that make it possible to control the particle morphology, size and homogeneity.
In the last two decades, the implementation of green chemistry principles in the homogeneous and heterogeneous catalysis has resulted in the appearance of new industrial processes. In particular, new processes have been designed by using zeolite-containing heterogeneous catalysts, including catalysts based on supported transition metals. Also, new catalysts for the oxidation reactions using environmentally friendly oxidants have been developed. Presumably, future studies in the field of industrial green chemistry would address the production of valuable chemicals directly from crude oil[1740-1747] and the use of electricity for synthesis of chemical products.[1748-1752] A significant aspect is the involvement of carbon dioxide in industrial processes by indirect (catalytic reduction to methanol)[1753] or direct (electrochemical and photochemical reduction) use of renewable energy.[1754-1761] The amount of research along this line gives hope for the emergence of new green chemistry production processes that utilize CO2 as a feedstock to obtain a wide range of useful organic products. Significant progress in this area is foreseen in the medium-term perspective. The desire to maximize the use of renewable and low-carbon energy would stimulate the development of new types of catalytic systems and processes, in which electricity and CO2 would serve as energy and carbon sources.
9. Acknowledgements
Chapter 2 was prepared with the financial support of the Russian Science Foundation (RSF) within the framework of the Project No. 21-13-00049 implementation (https://rscf.ru/project/21-13-00049/).
Subsection 3.1.1 was prepared with the financial support of the RSF within the framework of the Project No. 23-63-10011 implementation (https://rscf.ru/project/23-63-10011/).
Subsection 3.1.2 was prepared with the financial support of the RSF (Project No. 20-13-00032П).
Section 3.2 was prepared within the framework of the State Assignment to the Organic Chemistry Department, Faculty of Chemistry, Moscow State University (CITS Project No. AAAA-A21-121012290046-4, ‘Synthesis and Study of Physical, Chemical, and Biological Properties of Organic and Organometallic Compounds’).
Subsection 3.4.2 was prepared with the financial support of the RSF within the framework of the Project No. 23-13-00427 (https:// rscf.ru/project/23-13-00427/), in Section of heterogeneous catalysts for electrochemical СО2 reduction, and the Project No. 23-73-01215 (https://rscf.ru/project/23-73-01215/), in Section of homogeneous metal complex conversion of carbon dioxide.
Section 3.5 was prepared with the financial support of the Ministry of Science and Higher Education of the Russian Federation (FSRN-2023-0005).
Section 3.6 was prepared within the framework of the State Assignment to the Boreskov Institute of Catalysis, SB RAS; Subsection 3.6.1 was prepared with the financial support of the RSF (Project No. 21-13-00065; https://rscf.ru/ project/21-13-00065); Subsection 3.6.3 was prepared with the financial support of the RSF (Project No. 21-13-00314; https://www.rscf.ru/project/21-13-00314/).
Chapter 5 was prepared with the financial support of the Ministry of Science and Higher Education of the Russian Federation (the State task reg. No. 122040600073-3).
Subsections 6.1.1 – 6.1.3 were prepared with the financial support of the RSF (Project No. 21-73-30030).
Subsections 6.1.4 – 6.1.5 were prepared with the financial support of the Ministry of Science and Higher Education of the Russian Federation (the theme of the State task No FFSM-2021-00).
10. List of abbreviations
[BCMIM]Cl — 1,3-bis(carboxymethyl)imidazolium chloride;
[Bmim]BF4 — 1-butyl-3-methylimidazolium tetrafluoroborate;
[Bsim]Cl — 1-butyl-3-methylimidazolium chloride;
[Bsim]OH — 1-butyl-3-methylimidazolium hydroxide;
[Cmim]HSO4 — calixarene-based ionic liquids;
[dsim]HSO4 —1,3-disulfonic acid imidazolium hydrogen sulfate;
[emim] —1-ethyl-3-methylimidazolium cation;
[MIMPs]3PMo6W6O40 — 3-(1-methylimidazolium-3-yl)propane-1-sulfonate phosphomolybdenum tungsten;
[PyPS]3PW12O40 — ionic liquid based on pyrimidine and tungsto-substituted molybdophosphoric acid;
[TBA][Gly] — tetrabutylammonium glycinate;
3DPAFIPN — 2,4,6-tris(diphenylamino)-5-fluoroisophthalonitrile;
Ac — acetyl;
ADH — alcohol dehydrogenase;
AE — addition–elimination;
AES — alkyl ethoxysulfate;
Alg — alginate;
AO — addition–oxidation;
APE — alkyl phenol ethoxylate;
Ar — aromatic or heteroaromatic moiety;
AS — alkyl sulfate;
BAIL@UiO-66 — ionic liquids anchored on zirconium-based metal-organic frameworks;
BDC — benzenedicarboxylic acid;
BF — bio-factor;
BHM — borrowing hydrogen methodology;
BINAP — 2,2′-bis(diphenylphosphino) -1,1′-binaphthyl;
BINOL — 1,1′-bi-2-naphthol;
bipy — 2,2'-bipyridyl;
Boc — tert-butoxycarbonyl;
BODIPY — 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene;
BP — by-product;
σBM — σ-bond metathesis;
Bpin — boronic acid pynacol ester;
BPMN — bis(propylmalononitrile);
BPY — 4,4'-bipyridine;
CAA — chloroacetic acid;
CaCo-2 — human colorectal adenocarcinoma;
CC50 — half-maximal cytotoxic concentration;
24-h CC50 — half-maximal cytotoxic concentration after 24 h incubation;
β-CD — β-cyclodextrin;
β-CD-BSA — SO3H-β-cyclodextrin-butane sulfonic acid;
β-CD-mono — β-cyclodextrin monosulfonic acid;
CFL — compact fluorescent lamp;
ChCl — choline chloride;
CHP — cumene hydroperoxide;
CL-20 — hexanitrohexaazaisowurtzitane;
CMC — carboxymethylcellulose;
CMD — concerted metalation-deprotonation;
cod — 1,5-cyclooctadiene;
CPA — chiral phosphoric acid;
CPET — concerted proton-electron transfer;
CPf — final cytotoxicity potential;
CPf_rel — relative final cytotoxicity potential;
CPi — initial cytotoxicity potential;
CPP — critical packing parameter;
CS — chitosan;
CST — crude sulfate turpentine;
CT — catalyst;
CuAAC — copper-catalyzed azide-alkyne cycloaddition;
CuTc — copper(I) thiophene-2-carboxylate;
DABCO — 1,4-diazabicyclo[2.2.2]octane;
DBU — 1,8-diazabicyclo[5.4.0]undec-7-ene;
DCE — 1,2-dichloroethane;
DCM — dichloromethane;
DDQ — dichlorodicyanoquinone, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone;
DES — deep eutectic solvent;
DFT — density functional theory;
DG — directing group;
DMA — dimethylacetamide;
DMAP — 4-dimethylaminopyridine;
DMBA — 2,2-dimethylbutyric acid;
DME — dimethoxyethane;
DMF — dimethyl formamide;
DMSO — dimethyl sulfoxide;
DPZ — dicyanoquinopyrazine;
d.r. — diastereomeric ratio;
dtbpf — 1,1′-bis(di-tert-butylphosphino)ferrocene;
DVB — divinylbenzene;
ECM — energetic compounds and materials;
ee — enantiomeric excess;
Е-factor — the ratio of the mass of waste to the mass of desired product;
EHA — 2-ethyl hexanoic acid;
EP — emulsion polymerization;
EPR — electron paramagnetic resonance;
e.r. — enantiomeric ratio;
EWG — electron-withdrawing group;
FLP — frustrated Lewis pair;
Fmoc — fluorenyl methoxycarbonyl;
FRSN — human foreskin mesenchymal stem cells;
GAS — gas anti-solvent process;
Glr — glycerol;
GO — graphene oxide;
HAP — hydroxyapatite;
HAT — hydrogen atom transfer;
HEK293T — immortalized human embryonic kidney cells;
Het — heteroaromatic moiety;
HFM — hydroformylation;
HFIP — hexafluoroisopropanol;
HLB — hydrophilic-lipophilic balance;
HMX — cyclotetramethylenetetranitramine;
HT — hydrotalcite;
IL — ionic liquid;
IRMOF-3 Zn4O(H2N-TA)3, where TA is 2-aminoterephthalic acid residue;
LA — Lewis acid;
LABS — alkyl benzenesulfonate;
LB — Lewis base;
LD50 — median lethal dose;
LG — leaving group;
LiHMDS — lithium bis(trimethylsilyl)amide;
Ln — ligand(s);
LPG — liquefied petroleum gas;
LSF — late-stage functionalization;
MFI — zeolite morphology;
Mn@PMO-IL — manganese-containing periodic mesoporous organosilica with ionic-liquid framework;
MOF — metal-organic framework;
MW — microwave radiation;
MWW — zeolite morphology;
N-Boc-D-Pro — N-Boc-D-proline;
NC — normalized cytotoxicity (Chapter 2);
NC — nitrocellulose (Chapter 6);
NFSI — N-fluorobenzenesulfonimide;
NHC — N-heterocyclic carbene;
NHPI — N-hydroxyphthalimide;
NMP — N-methylpyrrolidone;
NP — nanoparticle;
OC — organocatalyst;
OTf — triflate (OSO2CF3);
P — product;
PAMAM — poly(amidoamine) dendrimer;
PAMPS — poly(2-acrylamido-2-methyl-1-propanesulfonic acid);
PANI — polyaniline;
PASE — Pot-Atom-Step Economy;
PCCS — modified chitosan;
PCET — proton-coupled electron transfer;
PDA — polydopamine;
PDMS — polydimethylsiloxane;
PdNPs — supported palladium nanoparticles;
PEG — polyethyleneglycol;
per-6-NH2-β-CD — peramino-β-cyclodextrin;
PIDA — (diacetoxy)iodobenzene, PhI(OAc)2;
PINO — phthalimide-N-oxyl;
PMB — p-methoxybenzyl;
PMP — polymer-monomer particle;
POP — porous organic polymer;
PPA — polyphosphoric acid;
PPI —poly(propyleneimine) dendrimer;
PS — polystyrene;
PT — proton transfer;
PTC — phase-transfer catalyst;
PVC — vinylcaprolactame;
PVI – vinylimidazolone;
R — reagent;
RDX — 1,3,5-trinitro-1,3,5-triazacyclohexane;
RESS — rapid expansion of supercritical solutions;
rGO — reduced graphene oxide;
RON — research octane number;
r.r. — regioisomeric ratio;
S — solvent;
SAS — supercritical anti-solvent process;
sc — supercritical;
SEAr — electrophilic substitution in arenes;
SET — single-electron transfer;
SHOP — Shell Higher Olefin Process;
SLS — sodium lauryl sulfate;
SM — starting material;
SN — silica nanoparticle;
SNH — nucleophilic substitution of hydrogen;
SPC — semiconductor photocatalysis;
SPINOL — 1,1'-spirobiindane-7,7'-diol;
STA — silica tungstic acid;
TBS — tris(tert-butyl)silyl;
TDG — transient directing group;
TDS — SiMe2(2,3-dimethylbutyl);
TEG — triethylene glycol;
TEMPO — (2,2,6,6-tetramethylpiperidin-1-yl)oxyl;
TEMPOH — 1-hydroxy-2,2,6,6- tetramethylpiperidine;
TFA — trifluoroacetic acid;
TFE — 1,1,1,2-tetrafluoroethane;
Thexyl — 2,3-dimethylbutyl;
TH — transfer hydrogenation;
THF — tetrahydrofuran;
TM — transition metal;
TMDPS — 4,4'-trimethylene-N,N'-sulfonic acid – dipiperidinium chloride;
TOF — turnover frequency;
TON — turnover number;
TS — transition state;
TS-1 — zeolite morphology;
UVA — near ultraviolet light.
References























![A novel and green version of the Passerini reaction in an ionic liquid ([bmim][BF4])](/storage/images/resized/qZfu8vVyaDIo81Au73p6SNE8AGGzf73yfiTC33bu_small_thumb.webp)











