



Keywords
Abstract
The strained structure of cyclopropanes serves as a kind of trigger for a variety of chemical transformations. Among others, processes involving conjugated unsaturated systems are of particular interest. The systems of unsaturated bonds are characterized by the possibility of flexibly varying their reactivity up to their full involvement in transformations. This review is the first to consider options for implementing the idea of combining the strain energy of cyclopropanes and the synthetic capacity of conjugated unsaturated systems within a single concept. A detailed analysis of processes involving activated cyclopropanes and numerous carbodiene and heterodiene systems is presented.
The bibliography includes 289 references.
1. Introduction
The idea of using saturated cyclopropanes in the synthesis of various compounds has actively attracted the attention of researchers in recent decades.[1-8] The abundance of research in this area is explained by the relative ease of obtaining and the diversity of chemistry of these compounds. The steric strain of the three-membered ring, which is 27.5 kcal mol–1 (see Ref. [9]), is the main driving force for further transformations (Figure 1). Nevertheless, sufficient kinetic inertness of the C–C bonds is maintained, which, on the one hand, prevents spontaneous violation of the ring integrity, and, on the other hand, determines the ease of its opening under mild controlled conditions. Polarization of the C–C bond due to the introduction of electrondonating and(or) -withdrawing substituents contributes to the additional activation of such a molecule. The greatest synergy is achieved for donor-acceptor cyclopropanes (DAC),[10] containing both types of substituents in adjacent positions.[11-16]
![[{"type":"paragraph","data":{"text":"Structural features of cyclopropane substrates and various aspects of their reactivity considered in this review. For the molecules in the inner circles, the angles, bond lengths and strain energies of the three-membered ring are indicated; the inner ring (a) includes the main classes of functionalized cyclopropanes; the outer zones (a, b) contain the structures of possible types of intermediates of the corresponding substrates. The following designations are used: CP is cyclopropane, M is metal, L is ligand."}}]](/storage/images/resized/nkhwH5tazxISrH7HjCNUe8gmfawDVs9iDS4xC0Dh_xl.webp)
A related strategy is the use of unsaturated carbon bonds at the three-membered ring for further metal-catalyzed activation, which is implemented for vinylcyclopropanes (VCP). In addition to substituted cyclopropanes, derivatives containing an exocyclic double bond, such as methylidenecyclopropanes (MCP) and vinylidenecyclopropanes (VDCP),[17-24] with strain energies of 40.9 and 50.9 kcal mol–1, reduced proximal bonds and increased distal bonds and valence angles C(sp3)–C(sp2)–C(sp3) of 63.9 and 62.2°, respectively (see Figure 1), are being actively studied.[25-28] In turn, the effect of the strained ring on the exocyclic C=C bond is expressed in the reduction of its length to 1.332 Å for MCP and 1.294 Å for VDCP. These structural features lead to the isolation of methylidenecyclopropane in a separate group as a special reactive motif.
All of the above cyclopropane substrates provide a large number of possibilities for opening and subsequent modification of the three-membered ring (see Figure 1).
Cyclopropanes bearing electron-donating and(or) -withdrawing substituents are susceptible to ring-opening to give 1,3-zwitterionic intermediates, and as for DAC, the possibility of further isomerization to alkenes or 1,2-zwitterionic intermediates in the presence of gallium(III) halides is also shown. For VCPs, catalysis using transition metal complexes promotes both similar ring-opening to allyl complexes and oxidative insertion of the catalytic species either in the C– C bond alone or with the involvement of the vinyl moiety. In a more general case, for derivatives bearing an unsaturated fragment on a three-membered ring, rearrangements such as vinylcyclopropane – cyclopentene or Cloke –Wilson rearrangement [29] are possible, as well as the common attack of radical particles on multiple bonds with ring-opening. The methylidenecyclopropane moiety is capable of breaking the proximal and distal C – C bonds using Lewis and Brønsted acids as catalysts or during the insertion of a transition metal complex; the acids can also react with the exomethylene bond. Finally, different species are able to attach to substituted cyclopropanes at the most activated position with the ring opening.
A large number of reactions of cyclopropanes with various unsaturated substrates have been described. Of particular interest, however, are those involving conjugated unsaturated compounds, especially when they are fully involved in the reaction. Multiple bond systems are motifs with high synthetic potential due to a number of reasons, namely, the possibility of forming carbo- and heterocycles in a wide range of molecular complexity, the implementation of the atom-economy concept, the action on two or more reaction sites in the molecule in one experimental step, as well as the potentially high diastereo- and enantioselectivity of the processes. All this explains the growing interest in these reactions over the last two decades.
![[{"type":"paragraph","data":{"text":"Schematic representation of the review structure, showing objects and processes according to sections (within a solid box) and subsections (dashed lines)."}}]](/storage/images/resized/A8ZkJvY4dipfAsIRYSBhtTzmUaTgRf1itlHbhoK2_xl.webp)
To systematize the processes under consideration, the reactions of activated cyclopropanes with conjugated systems of three main types — carbon-, nitrogen- and oxygen(sulfur)-containing — are highlighted in separate sections (Figure 2).
The reaction of the three-membered ring with the multiple bond system implies both inter- and intramolecular transformations. The main focus of the review is on those publications that consider the processes including the formation of at least one bond between the three-membered ring to be opened and the conjugated unit. In this case, at least two π-elements of the conjugated system should be involved in the formation of a new structural moiety, and priority will be given to studies in which the π-system is fully involved in the reaction. Special attention is given to the studies on the different reactivity of the same substrates under different conditions, taking into account the reaction pathways of the processes. The reactions of both cyclopropanes and methylidene- and vinylidenecyclopropanes are considered separately. The term ‘conjugated systems’ also implies double bonds of (hetero) aromatic rings. Also, cyclopropane and(or) conjugated moieties that are part of reaction intermediates are also covered in this review, since the determination of such ‘hidden’ synthons can facilitate both the analysis of known synthetic strategies and the development of new ones. A number of studies using arylcyclopropanedicarboxylates as DACs have described the formation of intermediates or the use of the pre-formed isomeric styrylmalonates. Such work is closely related to the subject of this review and is also shown schematically in Figure 2 in the form of a propene molecule.
The Büchner reaction, which occurs via the intermediate bicyclo[4.1.0]heptadiene, containing both key units in a ring, which further leads to electrocyclization to cycloheptatriene, can be regarded as one of the first examples of the reaction between cyclopropane and diene moieties.[30-33] However, this process has an independent character and will not be considered in this review.
2. Reactions of cyclopropanes with carbon-containing conjugated systems
2.1. Opening of a three-membered ring and capture of 1,3-zwitterionic intermediates with dienes and enynes
Historically, isolated examples of (4+3)-cycloaddition reactions of cyclopropanones as three-carbon synthons with cyclopentadienes, furans, pyrroles and fulvenes have been described before all others.[34, 35]
Trushkov and co-workers [36, 37] were the first to attempt to systematize such a (4+3)-cycloaddition process. As a model diene, 1,3-diphenylisobenzofuran (DPIBF) was chosen as one of the most reactive 1,3-dienes, acting predominantly as a 4π rather than a 2π component in cycloaddition reactions. As a result, a new approach to the synthesis of seven-membered rings 1 was proposed and tested on a series of donor-acceptor cyclopropanes bearing aryl, hetaryl and alkenyl substituents as donor groups. Interestingly, for some cyclopropanes, the presence of TMSOTf (TMS is trimethylsilyl, Tf is trifluoromethanesulfonyl (triflyl)) or SnCl4 and sources of external nucleophiles opens the way to the products 2 of conjugated addition, both via the direct involvement of the starting materials and via the treatment of their (4+3)-cycloaddition adducts (Scheme 1).
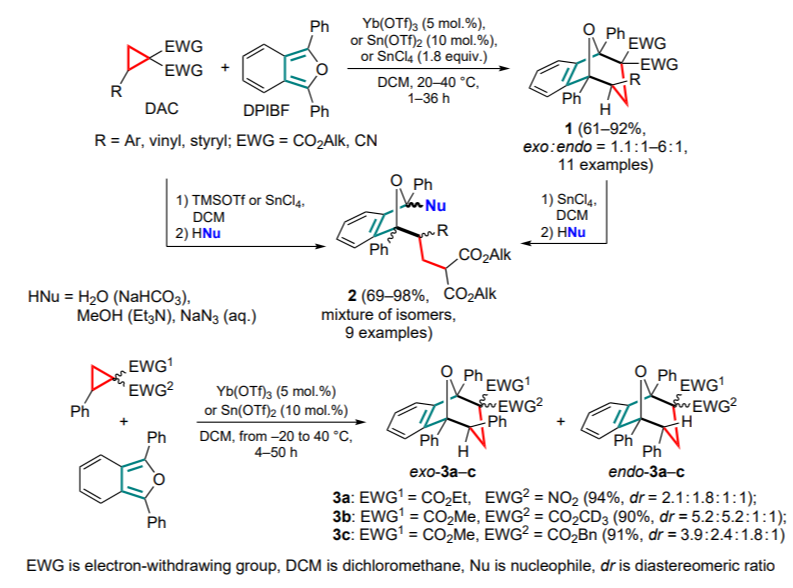
Using DAC with different acceptor groups at the C(1) atom led to a mixture of diastereomeric products 3, thus suggesting a stepwise mechanism. Later, Werz and co-workers [38] studied kinetic parameters for a number of (3+n)-addition reactions, where the said process involving DPIBF was also explored as a model (4+3)-addition reaction. For a series of substituents in the starting DAC, a rather good agreement with the parameters of the substituents in the previously studied of (3+2)-cycloaddition reactions has been found.
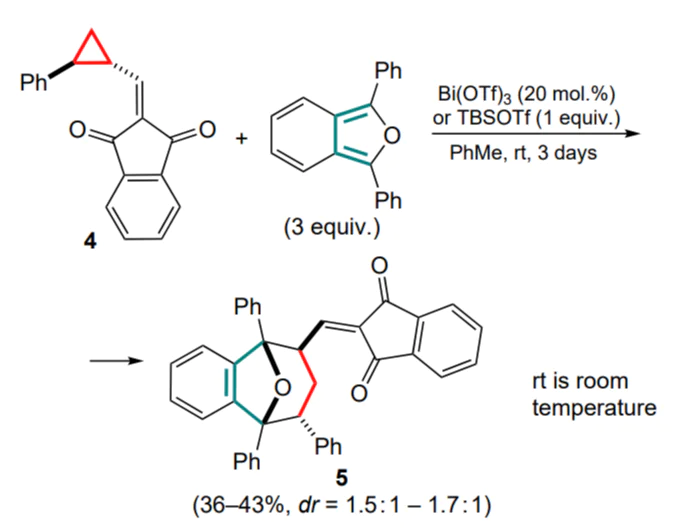
An example of a similar (4+3)-cycloaddition reaction of DPIBF with substituted cyclopropane 4 in the presence of Bi(OTf)3 is known, which gave adducts 5 as two diastereomers (Scheme 2).[39]
The first reported cycloaddition reaction involving cyclopropane and anthracene derivatives was the photochemical (4+3)-addition of trans-diarylcyclopropane to 9,10-dicyanoanthracene (Scheme 3).[40]
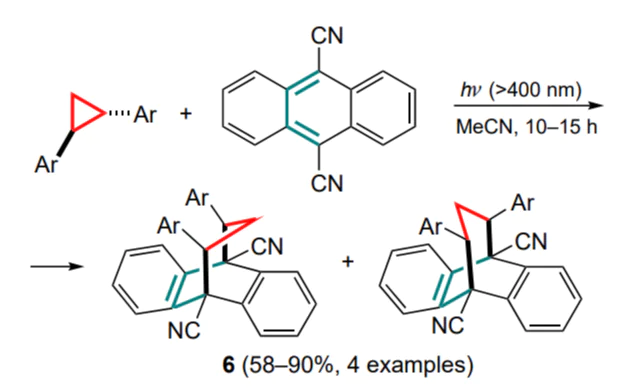
According to the mechanism proposed by the authors, the formation of the final adducts 6 (isolated as a mixture of antiand syn-isomers) proceeds via the intermediate formation of a double ion-radical pair undergoing the sequential closure of two single bonds. The photochemical addition of 2-phenylcyclopropylamines to di- and tricyanoanthracenes has been studied.[41]
This direction was further developed by Trushkov and coworkers [42] who studied the capture of 1,3-zwitterions derived from DAC in the presence of titanium(IV) chloride by anthracene and tetracene to afford compounds 7. It should be noted that in the case of donor 2-thienyl- and trimethoxyphenyl-substituted DACs, adducts 8 were also isolated as the major products, resulting from an attack of the aryl moiety on the cationic centre (Scheme 4).
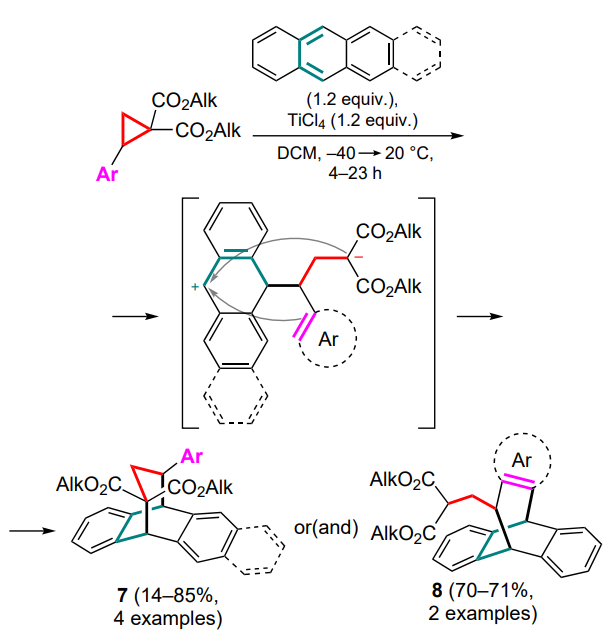
This study demonstrates for the first time that the diene substrate is completely involved in the formation of the sevenmembered ring with the incorporation of an ‘external’ aryl substituent in product 8.
The involvement of double bonds of substituted furans in the Lewis acid-promoted cyclization with DAC proceeds in a different way. Thus, 2-arylcyclopropane-1,1-diesters react with 2,5-dimethylfuran in the presence of tin(IV) chloride to afford, in the first step, the products of the conventional (3+2)-cycloaddition (9), containing a reactive vinyl ether functionality. These products can be further converted into various compounds depending on the nucleophilicity of the aryl group in the starting cyclopropane and the ratio of reagents.[43] In particular, using the same donor substrates as in the previous work, the cascade formation of products 10 was demonstrated proceeding through the nucleophilic attack of the donor (het) aryl moiety on the second furan double bond. Using two equivalents of DAC, the generated 1,3-zwitterion is attached via the double bond of adduct 9 to give the tricyclic tetraester 11 (Scheme 5).
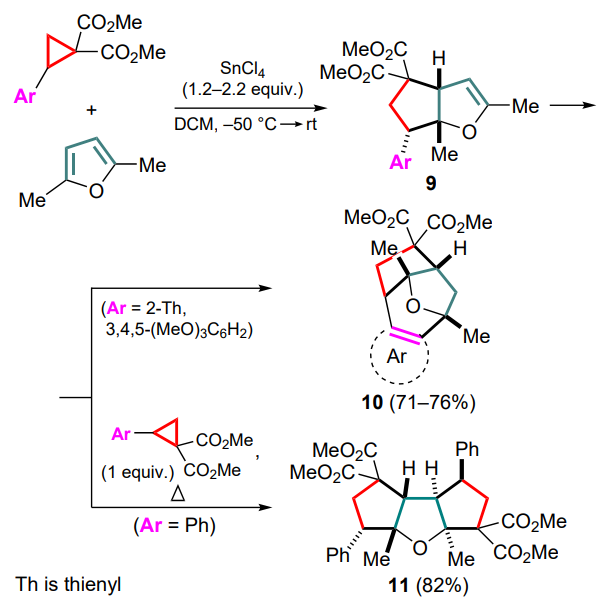
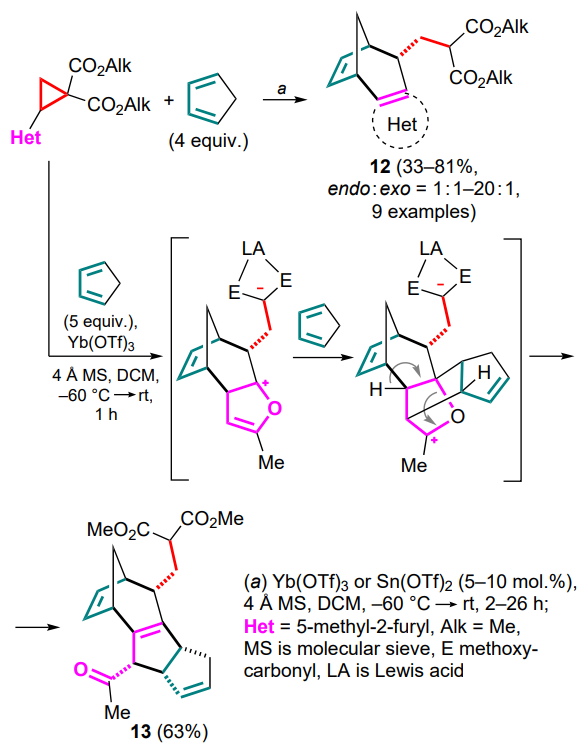
Subsequently, a more general way to create a new ring involving a donor hetaryl substituent in DAC as a nucleophile has been proposed.[44] For the first time, cyclopentadiene as a diene substrate was also involved in (4+3)-cycloaddition reactions to form DAC-based 1,3-zwitterions. This process was catalyzed by tin(II) and ytterbium(III) triflates. As a result, a series of heteroarene-annulated bicyclo[3.2.1]octa-2,6-dienes 12 were obtained. The reaction features high endo and chemoselectivity, while no reaction products were observed for malonic ester, despite its apparently higher nucleophilicity. This may be explained by a Re-face attack of cyclopentadiene on the carbocation centre of the 1,3-zwitterion, as well as a closer position of the heteroaryl moiety to the newly formed allyl carbocation. For furyl-substituted DAC, the ability to undergo a cascade sequence with an additional equivalent of cyclopentadiene to afford 5,8-methanocyclopenta[a]azulene 13 was shown (Scheme 6).
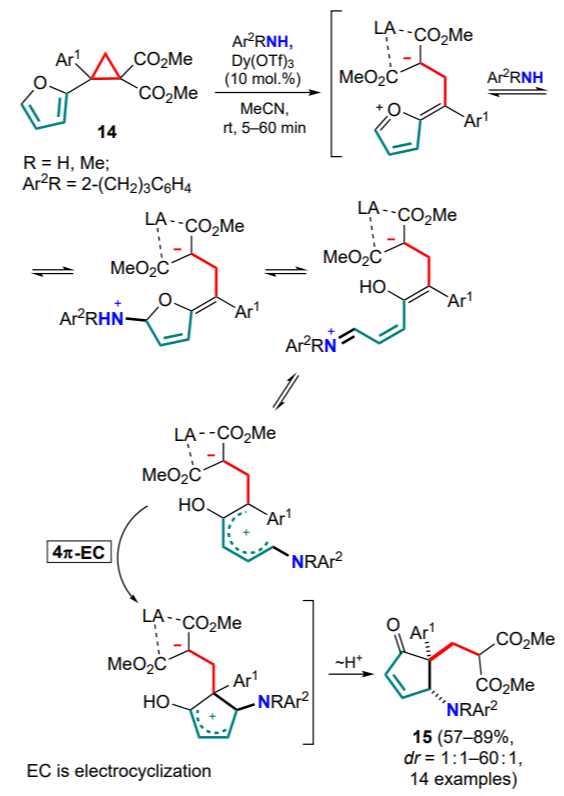
An unusual example of the intramolecular reaction between the cyclopropane moiety and the furan ring as a diene system involving an external amine has been reported.[45] Using dysprosium(III) triflate as the catalyst, tetrasubstituted DAC 14 undergoes nucleophilic attack on the furan ring, then azaPiancatelli rearrangement followed by 4π-electrocyclization to give substituted cyclopentenones 15 (Scheme 7).
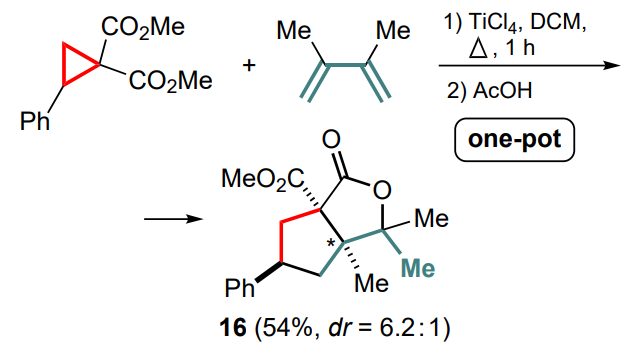
The situation has long been different for acyclic diene substrates. Initially, the full involvement of such a diene system in the reaction with cyclopropanes was quite a challenge, as it was shown that the most preferable direction is the (3+2)-cycloaddition with the participation of only one double bond.[46] However, an example of additional involvement of the second C=C bond was described in the same publication, but only for further closure of the carboxylate moiety to form a fivemembered lactone 16 (Scheme 8). Nevertheless, this transformation is also relevant to the subject of this review.
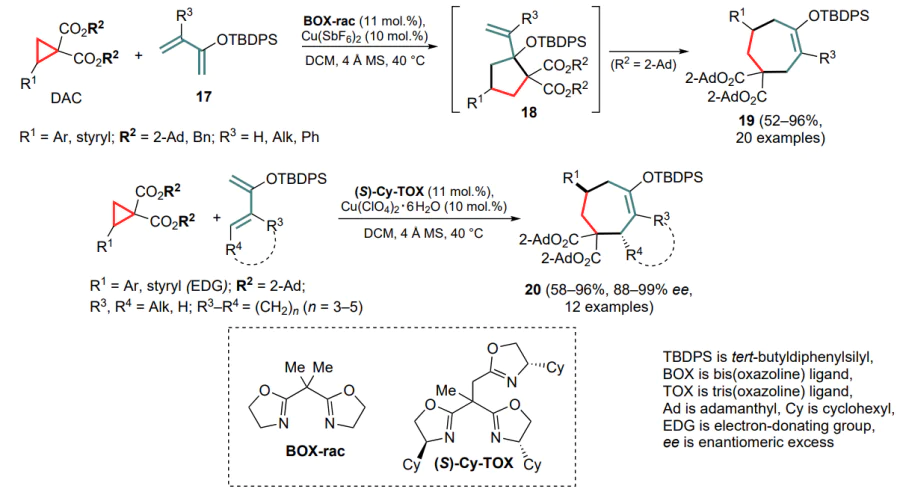
Tang and co-workers [47] were the first to achieve the full involvement of the conjugated diene system in the formal (4+3)-cycloaddition to DAC for dienylsilyl ethers 17 in the presence of copper(II) catalytic systems. The process was implemented for a wide range of substrates in both achiral and enantioselective modes using the chiral ligand Cy-TOX; however, the latter modification of the procedure was only applicable to activated cyclopropanes containing electrondonating substituents. The authors also noted the key role of sterically crowded ester functionalities in the starting cyclopropanes for the reaction to be successful. This process appears to occur mainly through the initial formation of 1,3-zwitterions as the kinetic product of (3+2)-cycloaddition on the activated double bond of the diene substrate 18 with subsequent opening of the five-membered ring and intramolecular cyclization to the thermodynamically more stable cycloheptenedicarboxylate 19 (Scheme 9). The proposed approach opens the way to the enantioselective preparation of cycloheptenes and [n.5.0]bicyclic compounds 20.
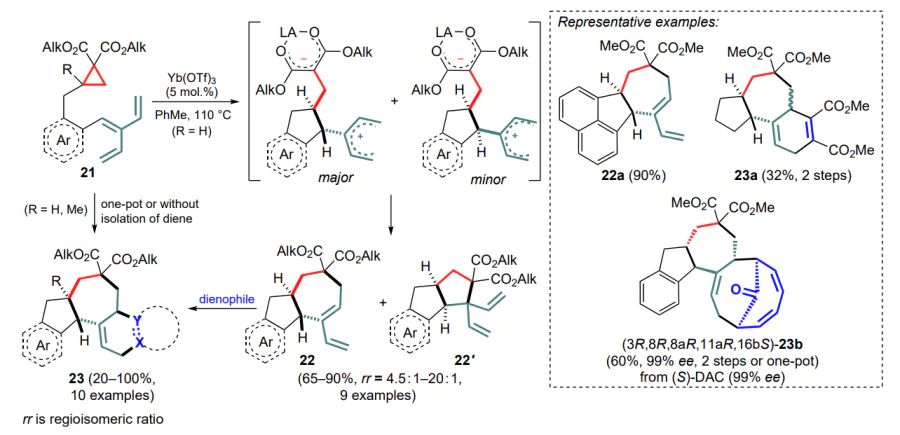
Zhang et al. [48] implemented a similar approach in an intramolecular mode. Substrates bearing cyclopropanedicarboxylate and cross-conjugated [3]-dendralene moieties were subjected to a cascade of (4+3)/IMPC (IMPC is intramolecular parallel cycloaddition) reactions catalyzed by metal triflates to yield a trans-bicyclo[5.3.0]decane skeleton 22 (Scheme 10). The reaction proceeds by sequential opening of the three-membered ring during nucleophilic attack of the internal C=C bond with possible formation of both trans- and less preferred cis-substituted cyclopentane moiety. The intermediate carbocationic centre, stabilized by two vinyl groups, can be involved in the further formation of sevenmembered (22) and five-membered (22') rings, the first pathway being preferred. The main reaction product contains a diene unit capable of (4+n)-cycloaddition reactions with dienophiles. In this way, the authors obtained a series of polycyclic structures 23 by successfully implementing the strategy of (4+3)IMPC/(4+n) reactions, in some cases optimized for one-pot use.
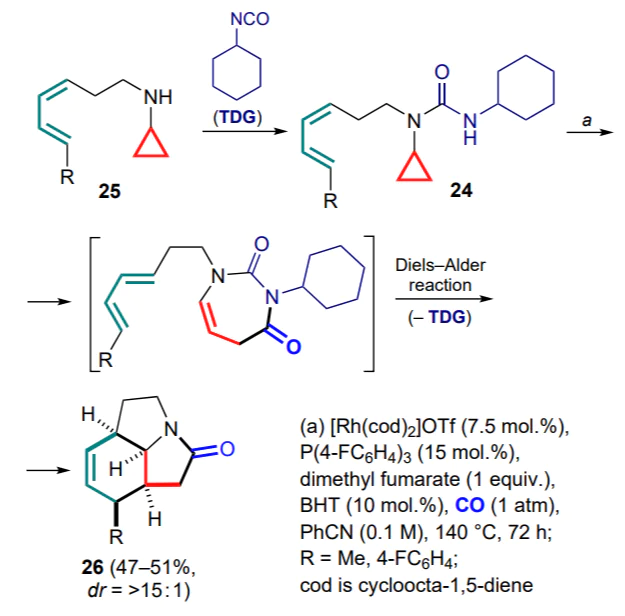
Bower and co-workers [49] described an interesting example of a tandem process involving alkadienyl(cyclopropyl)amides 24 and CO. A key feature of this approach is the use of isocyanatebased temporary directing groups (TDGs), which can be easily attached to the desired amine substrate 25 and then independently removed at the end of the process, without the need for carbonyl condensation or imine hydrolysis. In addition, these groups can participate directly in this reaction, improving its regioselectivity. In this way, the carbonylation/Diels–Alder sequence yielding diastereoselectively tricyclic amides 26 was performed (Scheme 11). In the presence of dimethyl fumarate, the reaction proceeds faster and provides higher yields of products, which the authors explained by the possible participation of this compound in the process as a labile electron-deficient ligand, accelerating the insertion and(or) reductive elimination.[50] The presence of 2,3-di-tert-butyl-4-methylphenol (BHT) helped to avoid the side reaction — radical polymerization of the diene substrate.
In parallel with the attempts to capture 1,3-zwitterions by dienes, the attention of researchers was drawn to indoles containing an unsaturated moiety conjugated to the aromatic system because of the potential use of such substrates in the synthesis of biologically active heterocyclic compounds.
Kerr and co-workers [51] pioneered in this field by proposing the cyclization of DAC with 2-alkynylindoles 27. The process involved the cyclopropane ring-opening, alkylation of the indole with the resulting 1,3-zwitterion and subsequent cyclization via the Conia-ene reaction. Zinc triflimide proved to be the most successful reagent for the simultaneous activation of both substrates. It was shown that for terminal 2-ethynylindoles 27 (R4 = H), the reaction proceeds with good yields of compounds 28 for a wide range of substituents. However, for internal 2-alkynylindoles, the presence of a substituent at the multiple bond prevents cyclization, giving only Friedel–Crafts alkylation products (29) (Scheme 12). The only exception was found for indole, which contains the ester group at the triple bond; however, as the authors noted, in this case there is a conjugated Michael addition rather than a Conia-ene cyclization. It should be noted that this work describes two examples of reactions with optically active DAC, in which the configuration of the stereocentre is retained.
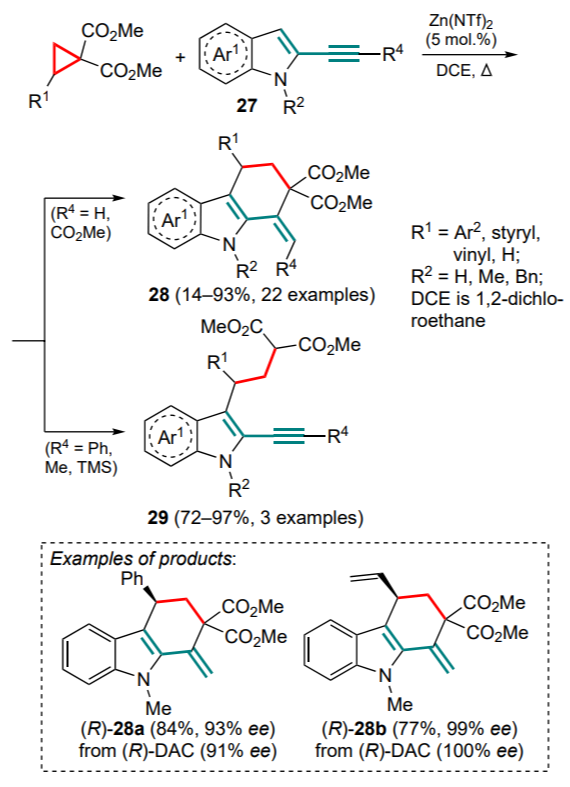
Subsequently, a similar strategy was developed, but in the enantioselective fashion.[52] This approach required the selection of special conditions for each of the stages of the ring formation. Nevertheless, the authors finally succeeded in developing a onepot method that allows to obtain, for a number of electronenriched DACs, target cyclization products 30 in high yields and with high enantioselectivities (Scheme 13). The adjustment of the stereocentre configuration takes place during the alkylation of the indole catalyzed by copper(II) triflate in the presence of the chiral ligand L1. For the second stage (cyclization), the authors proposed the use of indium(III) chloride with a small addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), which increases both the product yield and the enantioselectivity of the process.
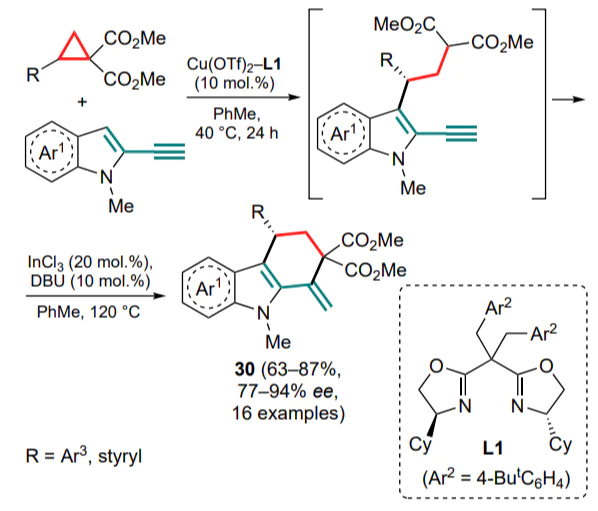
Following 2-ethynylindoles, their olefin analogues have been used in the capture of DAC-derived 1,3-zwitterions. Reactions of DACs in the presence of ytterbium(III) triflate with indoles 31 bearing a 2-positioned nitromethylidene or methylidene malonate moiety was studied.[53] In general, the yields of the malonate derivatives 32 were higher (up to 90%) than those of the nitromethyl derivatives (<=69%) (Scheme 14). For most of the tested cyclopropanes, the reaction proceeded selectively to afford a single cis-diastereomer, whereas for 2-furyl- and styrylsubstituted DACs, the diastereomer ratio decreased up to 3.5:1.
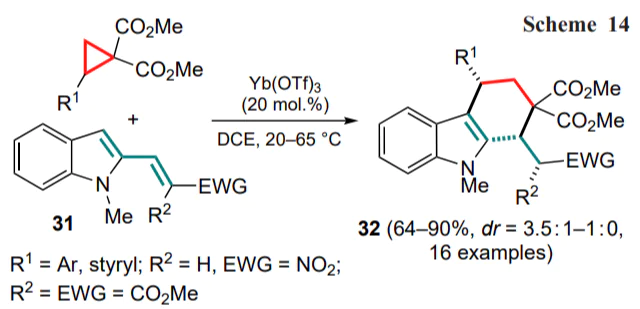
A similar concept has been implemented with β-(2-indolyl)-α,β-unsaturated ketones 33 in the presence of 2 mol.% of scandium(III) triflate.[54] In this case, as in the previous example, cis-diastereomers 34 prevailed (Scheme 15), and for optically pure DAC (R1 = Ph), the corresponding chiral product 34a was obtained with high optical purity, indicating the SN2 mechanism of Friedel–Crafts alkylation in the first stage.
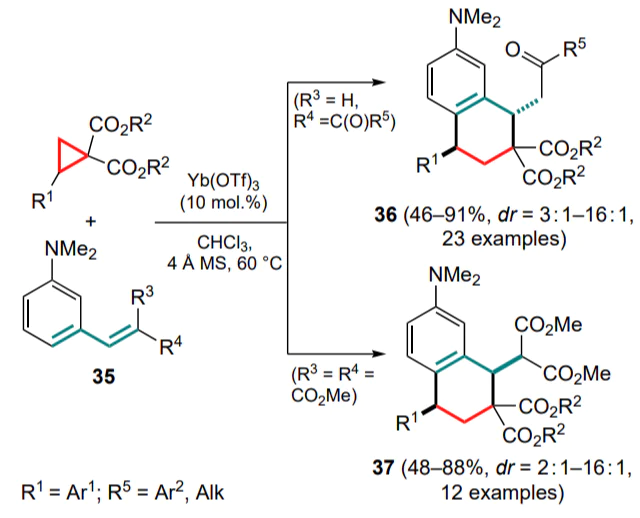
For substituted alkenylanilines, a similar strategy of the Friedel–Crafts/Michael addition reaction cascade was realized in the presence of ytterbium(III) triflate.[55, 56] The corresponding unsaturated ketones and methylidene malonates (35) were studied, the use of which delivered substituted decalines 36 and 37 with different diastereoselectivities (Scheme 16). The substrate-dependent diastereoselectivity was attributed by the authors to the steric hindrances arising between the alkene and malonate moieties prior to the Michael addition. Interestingly, depending on the substituents at the C=C bond, the most energetically favourable conformation of the intermediate switches to the opposite one, which was established by quantum chemical calculations and confirmed experimentally.
Therefore, the concept of (3+3)-cycloaddition involving donor-acceptor cyclopropanes has been demonstrated on the example of the above processes of capturing 1,3-zwitterion intermediates by unsaturated substrates containing C=C and C≡C bonds conjugated to the aromatic ring.
The abundance of biologically active compounds containing an 3,4-indole-annulated seven-membered ring motivated researchers to extend the above synthetic approach to indole substrates bearing 4-positioned C=C and C≡C bonds.
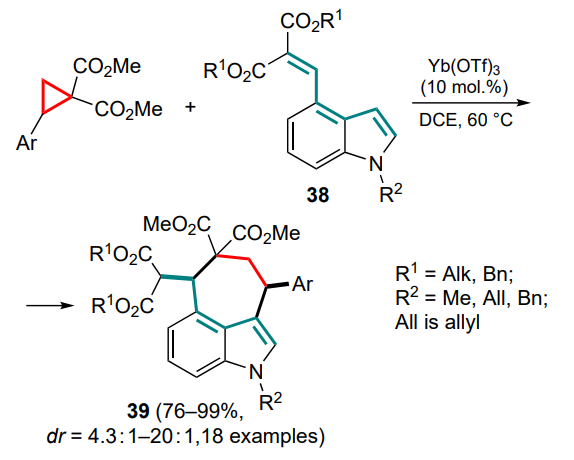
In the first work of this kind,[57] the Friedel–Crafts/Michael cascade reactions using 4-methylidene malonate derivatives of indole 38 were carried out, including a formal (4+3)-closure of the seven-membered ring. In the presence of ytterbium(III) triflate, a series of annulated indoles 39 with pronounced cisdiastereoselectivities were obtained (Scheme 17).
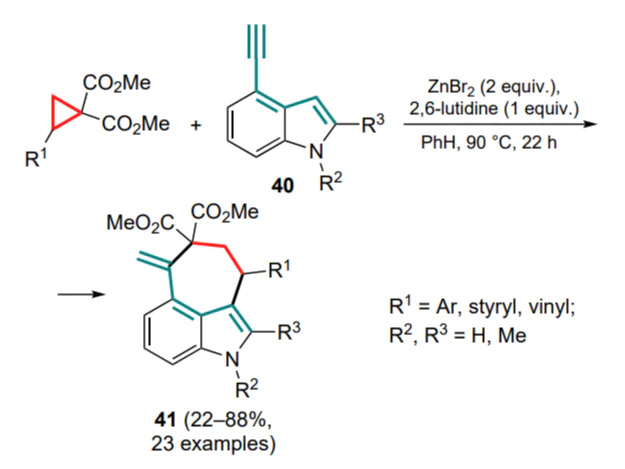
A similar Friedel–Crafts/Conia-ene cascade sequence was successfully applied to 4-ethynyl substituted indoles 40, [58] continuing the pioneering work on this subject by the Kerr’s group. [51] As in the previous case, zinc salt as a Lewis acid was the optimal choice, but in place of 5 mol.% of zinc triflimide, 2 equiv. of zinc bromide supplemented with 1 equiv. of 2,6-lutidine (Scheme 18) were used in the said transformations. It should be noted that the yields of the target products 41 (up to 88%) were higher for the N-methylindoles than for their N-unsubstituted analogues.
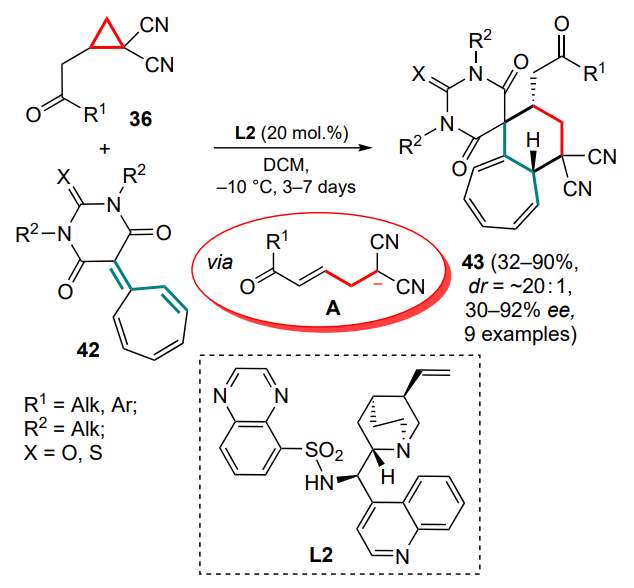
Based on the strategy of anionic activation of DAC 36 catalyzed by Brønsted bases, an enantioselective cyclization involving tropones (see Section 4.1 for details) and barbituratesubstituted heptafulvenes 42 has been proposed (Scheme 19).[59] The reaction proceeds via the intermediate formation of anion A bearing an acceptor enone moiety. For heptafulvenes 42, a quinine derivative L2 proved to be the optimal catalyst, which is indirectly consistent with the absence of additional coordination by the thiourea moiety in the case of tropones. It is noteworthy that the formation of the corresponding tricycles 43 is not accompanied by isomerization.
Modification of the cyclopropane skeleton with epoxide and alkyne moieties opened access to the diastereoselective preparation of tricyclic products 44 based on a one-pot goldcatalyzed process involving cis-2-alkyl-3-(1-alkynylcyclopropyl)oxirane 45 and substituted butadienes in the presence of water (Scheme 20).[60] Carrying out this reaction with transisomers of cyclopropanes 45 decreases its stereoselectivity or produces a complex mixture of products. The coordination of the triple bond by gold(III) chloride facilitates the subsequent 6-endo-dig electrocyclization to generate 1-oxalyl cation A, which captures the water molecule to diastereoselectively release the intermediate bicyclic alcohol 46. After filtration of the reaction mixture through silica gel and addition of the gold(I)-triphenylphosphine complex, the resulting carbocationic intermediate B is capable of diene addition.
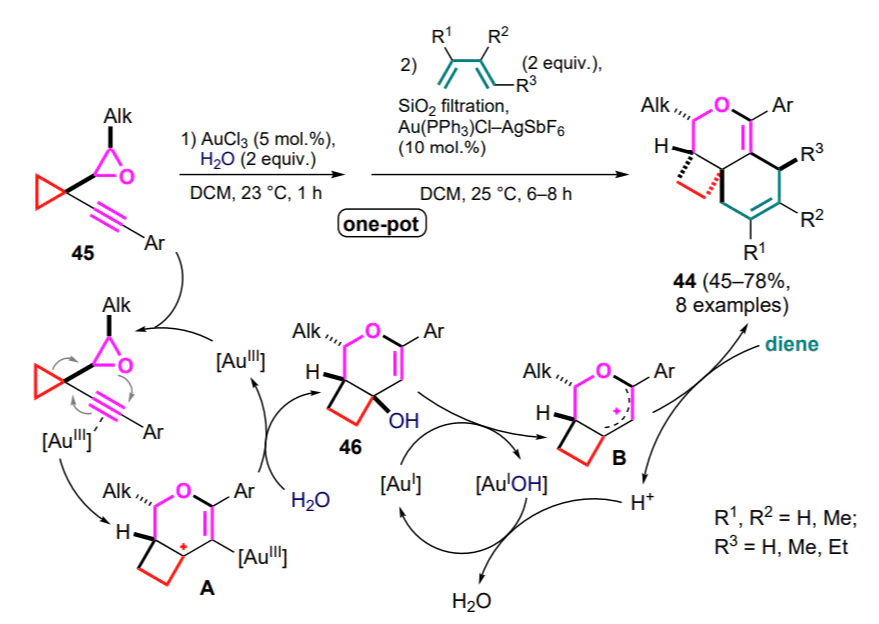
2.2. Generation of 1,2-zwitterionic Ga complexes and their reactions with dienes
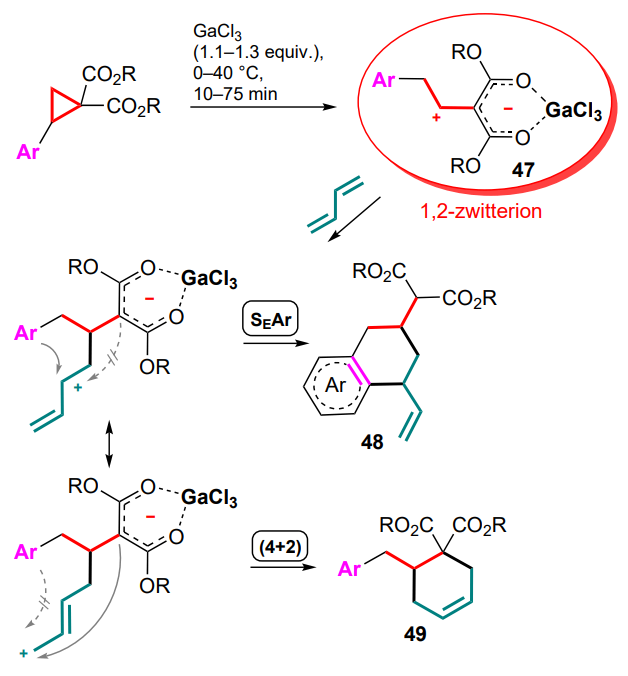
A special place in the chemistry of DACs and electronwithdrawing alkenes is occupied by reactions that occur via 1,2-zwitterion intermediates, which are usually formed in the presence of gallium(III) halides. The capture of these intermediates by various substrates has been successfully realized in numerous studies led by Yu.V.Tomilov and R.A.Novikov.[61-74] Since this review focuses on the reactions of DAC with conjugated systems, this Section presents examples of the capture of 1,2-zwitterionic intermediates by conjugated dienes.
First of all, it should be noted that the term ‘1,2-zwitterionic intermediate’ is used in this context rather conditionally, since it is obvious that in the above gallium complex 47, formed in the reaction of GaCl3 or GaBr3 with 2-arylcyclopropane-1,1-dicarboxylate or methylidene malonate, the negative charge is largely delocalized over the malonate moiety. Nevertheless, the positive charge in complex 47 is essentially localized on the a-carbon of the alkyl substituent (in contrast to the primary 1,3-zwitterion generated by the direct cleavage of the substituted s-bond in DAC), which determines the location of the electrophilic carbocationic centre and specific reactions of intermediate 47 (1,2-zwitterionic gallium complex). Theoretically, this complex can react with the conjugated double bond system in several directions, including cyclization of one or both of the diene double bonds, as well as an attack on the malonate moiety or the aromatic ring. At first sight, the formation of six-membered cyclic adducts 48 and 49 seems most likely, whereas the formation of four- or eight-membered rings is less preferable (Scheme 21).
The first attempts to carry out these reactions were performed on the example of the reaction of 2-aryl-cyclopropane-1,1-diesters as DACs with the simplest acyclic dienes, such as buta-1,3-diene and 2-methylbuta-1,3-diene, in the temperature range from –20 to 0 °C.[71] The least tendency to full involvement of the diene system in the formal (4+2)-cycloaddition was shown by unsubstituted butadiene, which gave the substituted tetralin 48a as the major product, although cyclohexene 49a was also identified in the reaction mixture. On the contrary, the DAC reactions with 2-methylbutadiene, as well as with 2,3-dimethylbuta-1,3-diene, followed mainly the (4+2) cyclization route to furnish substituted cyclohex-3-ene-1,1-dicarboxylates 49b,c (Ar = Ph) in yields of 57 and 65%, respectively (Scheme 22).
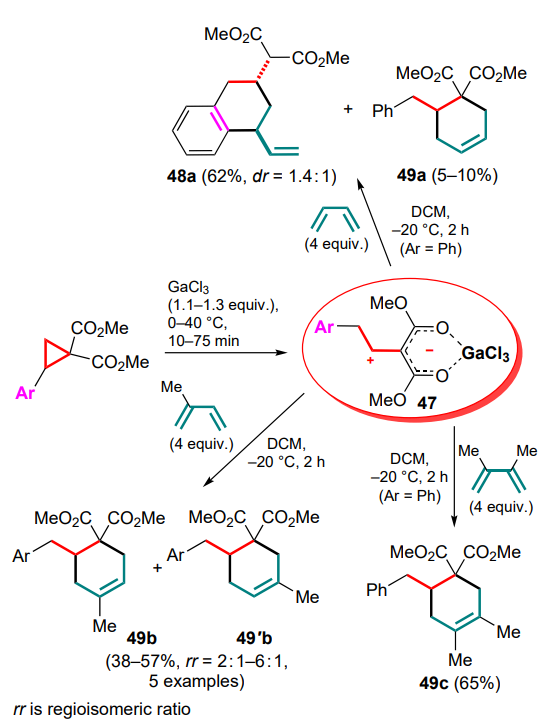
The reaction of 2-phenylcyclopropane-1,1-diester with unsymmetrical 2-methylbutadiene produced a mixture of regioisomeric methylcyclohexenes 49b and 49'b in a ratio of ~6:1, while the use of halogen-substituted arylcyclopropanes decreased this ratio to 2.3:1. However, in all cases the initial attack of the zwitterion 47 was directed towards the terminal carbon atom of the said dienes.
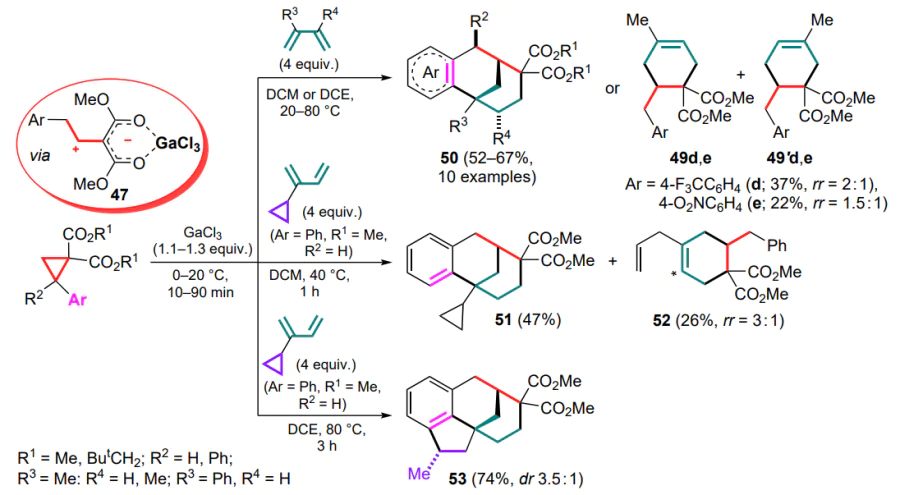
It was subsequently shown that when similar reactions are carried out at a higher temperature (40–80 °C), both reaction patterns are realized, i.e. the formation of cyclohexenedicarboxylates 49b is followed by alkylation of the aromatic ring, and benzobicyclo[3.3.1]nonanes 50 are obtained in moderate to high yields (Scheme 23).[73] For DACs bearing electron-deficient aryl substituents, e.g., 4-F3CC6H4 or 4-O2NC6H4, the reaction stops at the stage of (4+2)-cycloaddition to give the corresponding cyclohexene adducts 49d,e. The Ga complex 47a (Ar = Ph) reacts with 2-cyclopropylbuta-1,3-diene at 40 °C in much the same way as with 2-methylbutadiene to afford the corresponding 1-cyclopropylbenzobicyclo[3.1.3]nonane 51 in 47% yield. This reaction also involves a partial opening of the three-membered ring to yield regioisomeric allyl cyclohexenes 52. However, if the reaction is carried out at 80 °C from the very beginning, a triple reaction cascade delivers the tetracyclic structure 53 (see Scheme 23).
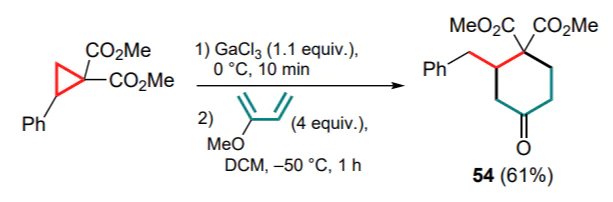
Electron-rich 2-methoxybuta-1,3-diene reacts actively with Ga complex 47a at –50 °C, but during the work-up of the reaction mixture, the resulting methoxycycloxene is readily converted to 4-oxocyclohexane-1,1-dicarboxylate 54 (Scheme 24).
Another specific direction of the GaCl3-mediated reactions of DAC with 2-methylbutadiene are transformations involving sterically demanding ester groups of cyclopropanes.[73] For example, DAC 55 bearing a 2-adamantyl or neopentyl moiety in the ester group reacts with isoprene at 40 °C in the presence of traces of water to give bicyclic lactones, 2-oxabicyclo[2.2.2]-octan-3-ones 56 (Scheme 25).
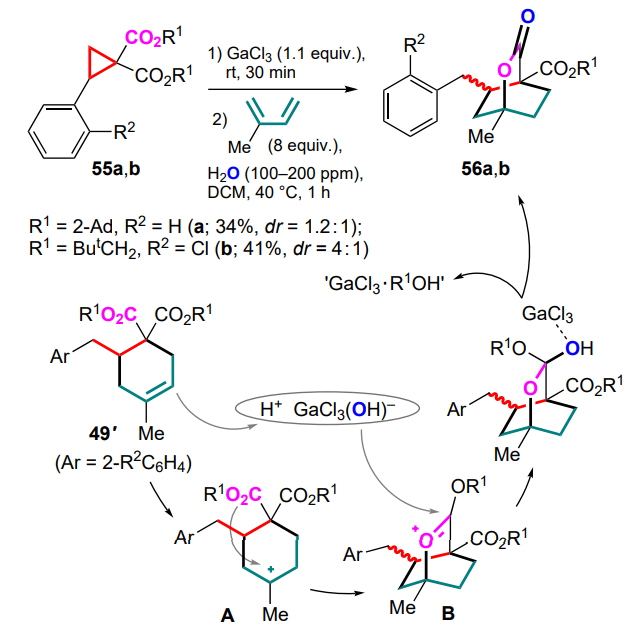
The authors believe that in the first stage of the transformation, as in previous cases, an ionic (4+2)-cycloaddition yields cyclohexenes 49, which are able to generate the carbocationic intermediate A due to the presence of the complex GaCl3 ·H2O or its dissociated form HGaCl3(OH). However, instead of an electrophilic attack on the aromatic ring, there is an attack on the oxygen atom of the ester functionality. Such a feature of reactivity is apparently due to a much weaker coordination of large substituents in ester groups with gallium cations and(or) steric blocking of the attack on the aromatic substituent. This predetermines the attack of the carbonyl oxygen on the electrophilic centre to give intermediates B. The latter are easily converted into bicyclic lactones 56 by formal addition of water and elimination of the corresponding alcohol (see Scheme 25).
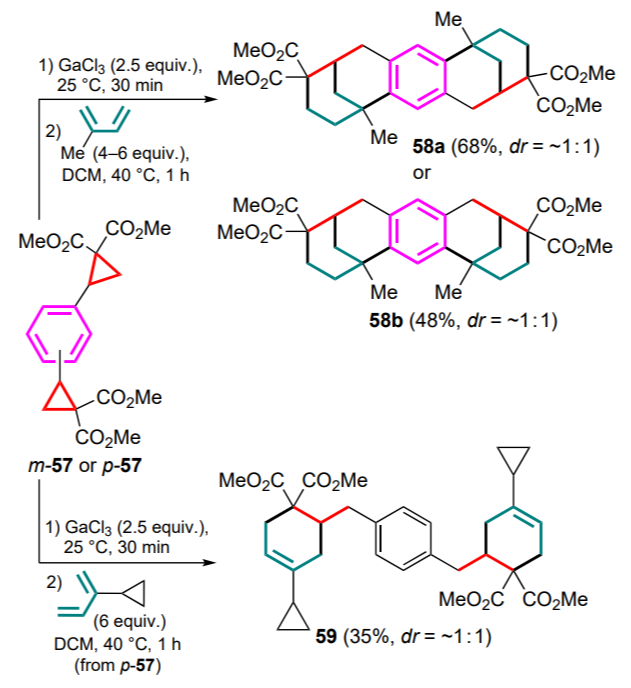
Similar reactions were carried out in the follow-up study[74] using para- and meta-substituted phenylenebis(cyclopropanedicarboxylates) 57. Starting DACs were first converted into the corresponding bis-Ga complexes under the action of anhydrous GaCl3 and then reacted with 2-methyl- and 2-cyclopropylbutadiene. The isomers 57 underwent a cascade of (4+2)-cycloaddition/double alkylation of the aryl moiety to afford the corresponding framework 58 (Scheme 26). The paraisomer selectively gave product 58a with the anti-arrangement of the bicyclononane fragments and the meta-isomer gave product 58b with the syn-orientation. Lowering the temperature to 18 °C resulted in a mixture of compounds containing primary products of the formal (4+2)-cycloaddition. It should be noted that the reaction of the bis-Ga complex derived from the p-57 substrate with 2-cyclopropylbutadiene, even at 40 °C, usually stops at the stage of the formation of cyclohexene adduct, from which the bisadduct 59 was isolated in 35% yield as a mixture of two diastereomers.
2.3. Chemical transformations of conjugated systems based on cyclopropanes with unsaturated bonds
The rhodium(I)-catalyzed ring expansion of cyclopropanes by the insertion of an unsaturated moiety can be carried out in an intramolecular fashion. The mutual arrangement of multiple bonds in the proximity of the activated σ-bonds in the threemembered ring opens the way to a variety of options to assemble medium-size rings, which has led researchers to develop convenient methods for their construction.
The first idea implemented in this direction was the strategy of generating an allene moiety based on enyne cyclopropanes 60 by taking advantage of the previously discovered[75] ability of [Rh(CO)2Cl]2 to catalyze the migration of the 1,3-acyloxy group in propargyl ethers. This approach allowed the synthesis of a series of substituted alkylidenecycloheptadienes 61 from a variety of substrates in high yields. For compounds with a substituent at the exocyclic double bond, the isomer ratio varied within quite a wide range depending on the nature of the substituent. It was also shown that the configuration of the substituents in the three-membered ring affected the regioselectivity of the single bond cleavage. Thus, trans-, gem-disubstituted and monosubstituted cyclopropanes 60 gave a single product 61, whereas cis-isomers 60' furnished a mixture of two regioisomers (61' and 61'') with low selectivity (Scheme 27).
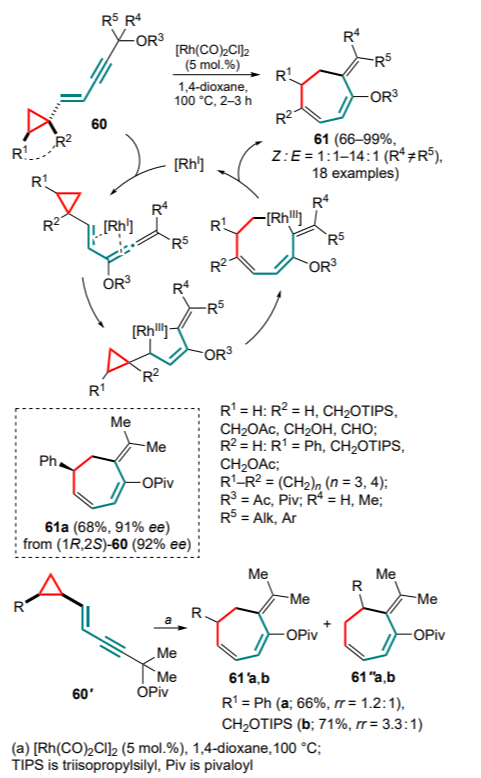
The optically active alkylidene cycloheptadiene 61a was obtained from the corresponding chiral enyne derivative of cyclopropane, with the enantiomeric excess being retained. The reaction proceeded via the formation of η4-(vinylallene)rhodium complex, oxidative cyclization to metallacyclopentene, cleavage of the three-membered ring and insertion followed by the reductive elimination of the seven-membered ring.
This direction was further developed in subsequent study with the additional insertion of the CO moiety into the ring structure. For example, a method was proposed for the synthesis of cyclooctadienones 62 based on the RhI -catalyzed reaction of buta-1,3-dienylcyclopropanes 63 with CO[76]. It was found that Z isomers of the starting dienes reacted completely when sufficient heated, whereas E isomers showed lower reactivity. At elevated temperatures, Z isomers were partially converted into the thermodynamically more stable E isomers. Despite these difficulties and the resulting incomplete conversion in most cases, it was possible to obtain a number of eight-membered carbocycles in the form of mixtures of regioisomers 62, 62' with a predominance of products with complete conjugation of double bonds. In addition, it was shown that under the reaction conditions the initially formed minor regioisomers 62' are able to isomerize into the major products (Scheme 28).
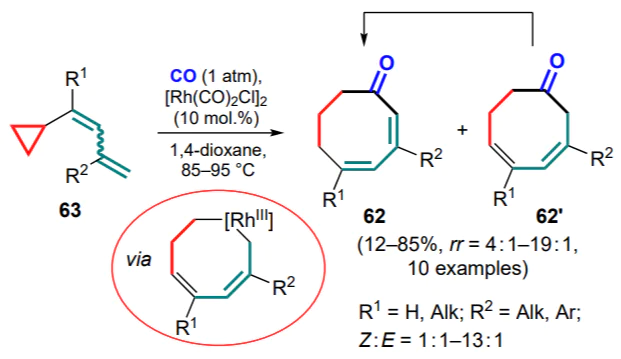
In the case of 1,1-disubstituted cyclopropanes 64, the reaction follows a different route, involving the formation of a sixmembered metallacycle, followed by the insertion of a CO molecule and the addition of an additional equivalent of the substrate, resulting in substituted cyclohexenones 65 (Scheme 29). This process is apparently due to the additional stabilization of the double bond of the metallacycle by the aryl substituent.
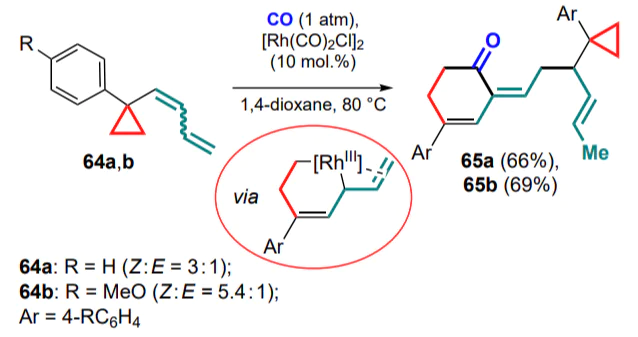
A similar approach has been implemented for cyclopropylbenzocyclobutanes 66, which act as sources of in situ generated cyclopropyl-substituted o-quinodimethanes 67 via electrocyclic thermal opening of the four-membered ring[77]. The reaction proceeds by the formation of a five-membered metallacycle and a cyclopropane ring-opening with closure of an eight-membered metallacycle, and finally insertion of CO to form benzocyclooctenones 68 (Scheme 30). A mixture of regioisomers 68' and 68'' was obtained from the methylsubstituted cyclopropane 66', with the predominance of the product formed via the cleavage of the least hindered σ-bond in the three-membered ring. A similar methodology has also been described for exocyclic 1,3-dienylcyclopropanes.[78]
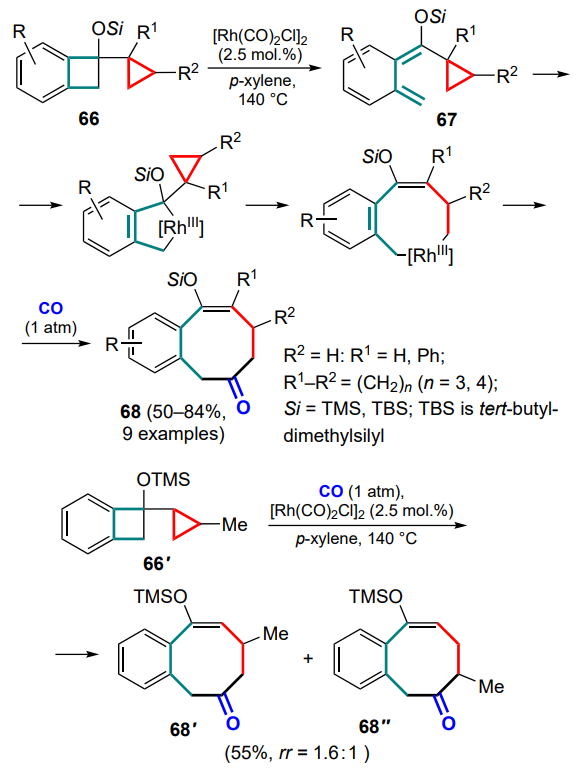
Method for the cyclization of dienylcyclopropanes 69 with release of hydrogen and formation of substituted toluenes 70 has been proposed.[79] In this case, the starting metallacycle is formed with the involvement of one of the double bonds of the diene system and the cyclopropane ring-opening. The second double bond is then inserted into the ring, followed by β-hydride elimination, insertion of the generated C=C fragment into the C(sp3)–RhIII bond, the second elimination of the hydride ion (with release of the hydrogen molecule), regeneration of RhI and isomerization to thermodynamically stable substituted toluene (Scheme 31). This method was successfully carried out on a wide range of substrates, but it was shown that the presence of the acceptor acyl substituent R2 plays a critical role, presumably in facilitating the initial elimination of the hydride ion and further aromatization.
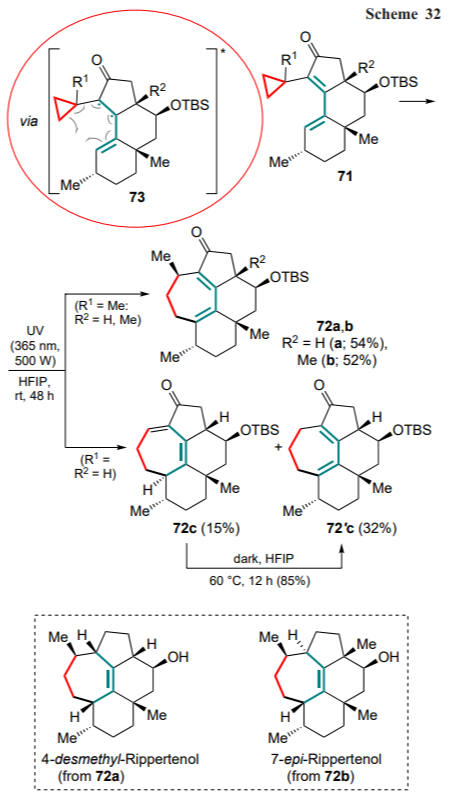
Finally, the photoinduced cyclization of the α-cyclopropyl dienones 71 was achieved for further application in the complete synthesis of 4-desmethyl-rippertenol, which had previously shown antibiotic activity against Bacillus subtilis, and 7-epi-rippertenol through the production of the key dienone intermediates 72a and 72b (Scheme 32).[80] The reaction proceeds via the formation of the triplet biradical intermediate 73, which undergoes sequential cyclization and 1,5-hydride shift. 1,1,1,3,3,3-Hexafluoropropan-2-ol (HFIP) acts both as a solvent and Brønsted acid, lowering the energy gap between the highest occupied (HOMO) and lowest free (LUMO) molecular orbitals. For α-cyclopropyl dienone 71' (R1 = R2 = H) bearing a monosubstituted three-membered ring, the formation of a mixture of isomers 72c and 72'c was observed, supporting the mechanism of the process, and the intermediate dienone 72c can be converted to dienone 72'c upon heating it in the dark.
2.4. Catalytic and radical transformations of α-cyclopropyl styrenes
One of the best-known methods for studying the mechanisms of radical reactions is the use of ‘radical clocks’, i.e. compounds with a known conversion rate. α-Cyclopropylstyrene is often used as such a compound.[81-85] Formally, cyclizations ractions involving α-cyclopropylstyrene are relevant to the subject of this review; nevertheless, these processes are often not preparative. However, a number of cases are known where α-cyclopropylstyrenes and other aryl-substituted vinylcyclopropanes are purposefully used as versatile substrates for the preparation of the corresponding dihydronaphthalenes. It should be noted that in addition to these substrates, (arylmethylidene) cyclopropanes have also been studied in similar processes (see Section 2.6). Reactions involving α-cyclopropylstyrenes do not always proceed with full involvement of the conjugated system of aryl moiety and double bond,[86] which further boosts the interest in this type of transformation.
The pioneering publication [87] on this topic appeared in 2016. The key feature of the process is the photochemical generation of acceptor radicals from the corresponding bromides using an iridium photocatalyst. The generated radical attacks vinylcyclopropane 74 to form a primary radical A, which is captured by an aryl substituent to form a more stable intermediate B, which is oxidized to the cation C as a result of the catalytic cycle completion. Another intermediate C is deprotonated with a base to afford the target dihydronaphthalene 75 (Scheme 33). In most cases, researchers focus on the introduction of acceptor radicals generated from bromodifluoroacetates or amides, but it has also been shown that the introduction of ethyl monofluoroacetate, trifluoromethyl (for example, Togni reagent II as a source), cyanomethyl and malonyl radicals is possible.
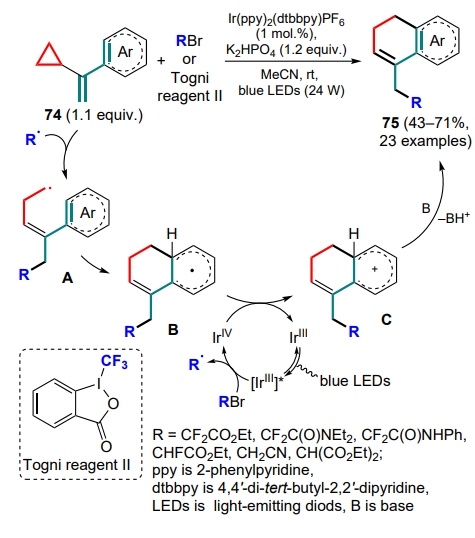
Further studies on the capture of the same substrates were continued with sulfonyl radicals.[88] As in the previous work, the authors used photochemical conditions, but the ruthenium(II) complex was used as the optimal photocatalyst. A series of target products, dihydronaphthalenes 76, were obtained in moderate to high yields (Scheme 34).
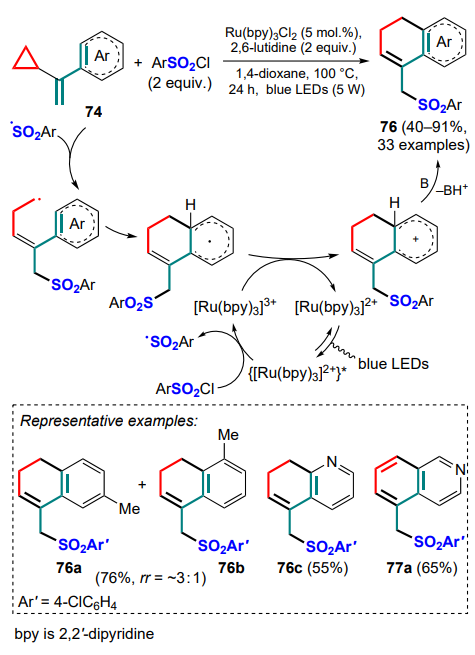
Attempts to use benzenesulfonyl chloride or vinylcyclobutane in the reaction failed. It should be noted that in the case of the m-tolyl substrate, the reaction proceeded with low regioselectivity and produced a mixture of regioisomers 76a and 76b. It is also noteworthy that the expected product 76c was derived from the 3-pyridyl derivative and the para-isomer additionally underwent ring aromatization to give isoquinoline 77a.
The same research group [89] developed a silver(I)-promoted variant of the previous reaction using sulfinates as sources of sulfonyl radicals in the presence of potassium persulfate as the oxidant. It is noteworthy that for meta-chloro-substituted a-cyclopropylstyrene 74, as for its meta-methyl-substituted analogue, the reaction showed low regioselectivity. However, the predominant product was 8-chlorodihydronaphthalene 76e rather than 6-substituted isomer 76d (Scheme 35). As in the previous study, the 4-pyridyl-substituted substrate underwent an additional aromatization to product 77b, which was not observed in the case of the ortho-substituted analogue 76f, and using benzene or trifluoromethanesulfinate in the reaction did not lead to the desired product. The general mechanism of the reaction is similar to that described above; the authors suggested two possible ways of transforming the radical intermediate into a cationic intermediate after attack on the aryl moiety involving either SO4 – • or Ag2+ as oxidizing species.
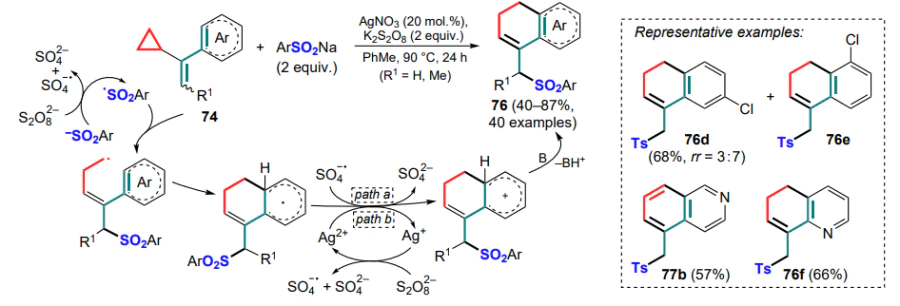
Similarly, cyclization of (1-cyclopropylvinyl)arenes 74 with ketones was carried out under conditions of α-carbonyl radical generation, giving compounds 78 (Scheme 36).[90] It should be noted that the authors failed to use acetophenone and cyclobutanone in this reaction, which may indicate that the reactivity of such ketones depends on the stability of the initially formed radical species.
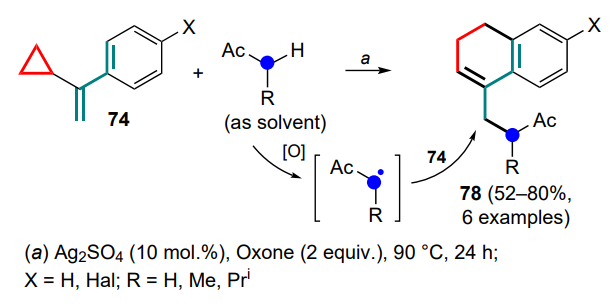
Finally, the same authors [91] proposed a similar approach for the cyclization of α-cyclopropylstyrenes 74 using cyanomethyl radicals in the presence of silver carbonate and copper(II) compounds as promoters. This transformation is particularly interesting in terms of the tandem process methodology, since in its updated modification, it can be used to form a new C – C bond and, accordingly, for the carbon chain growth, combined in one synthetic step with the ring closure. As a result, a diversity of cyanoethyl derivatives 75 were synthesised in yields up to 83% (Scheme 37). As in the previous studies, for meta-substituted substrates 74 with Ar = 3-XC6H4 (X = Me, OBn, Cl), as well as for Ar = 3,4-(MeO)2C6H3 and 2-naphthyl substituent, the formation of two regioisomers in the ratios from 7 : 3 (for 75a,b) to 17 : 3 (for 75c,d) was observed. It should be noted that under the reaction conditions, 4-pyridine-substituted vinylcyclopropane delivers the standard product 75e without further dehydrogenation, as noted earlier, but attempts to use homologues of acetonitrile or 3-thienyl-substituted vinylcyclopropane in the reaction failed.
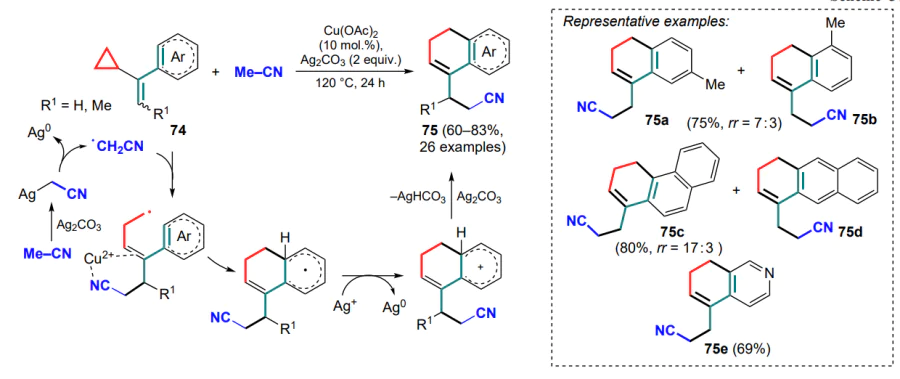
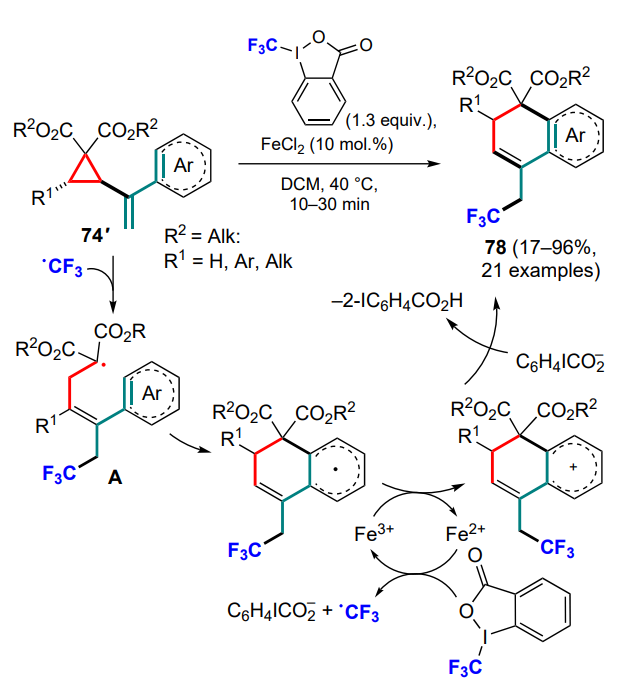
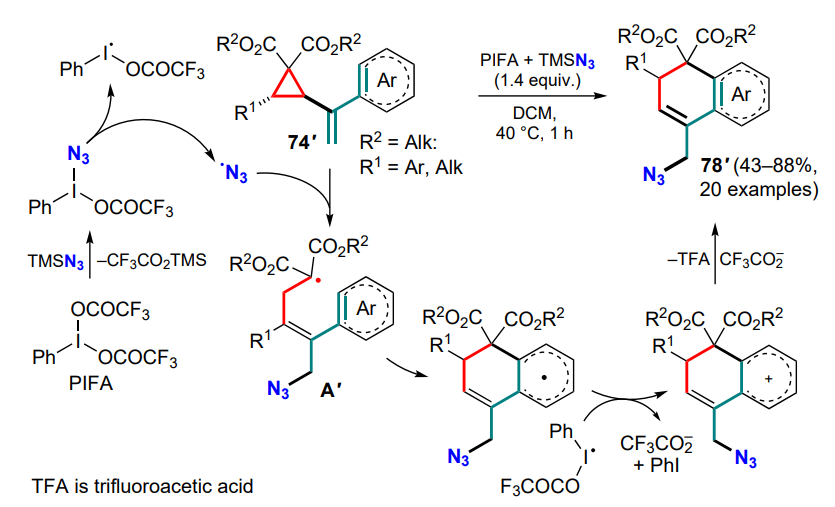
Deng and co-workers [92, 93] presented a series of works on this topic, using α-cyclopropylstyrenes 74' substituted in the cyclopropane ring and sources of trifluoromethyl and azide radicals, such as Togni reagent II or TMSN3 with bis(trifluoroacetoxy)iodobenzene (PIFA). In both cases, the three-membered ring-opening was accompanied by the generation of substituted malonyl radicals A which, after cyclization, provided the target compounds 78 and 78' (Scheme 38, Scheme 39).
2.5. Reactions of methylidenecyclopropanes with dienes and enynes
The first mention of the (4+3)-cycloaddition of methylidene cyclopropane to conjugated unsaturated substrates, given without mechanistic studies, is the Pd-catalyzed reaction with (2E,4E)-hexa-2,4-dienedioic acid dimethyl ester (79), which produced a mixture of diastereomeric cycloheptenes 80 (Scheme 40).[94]
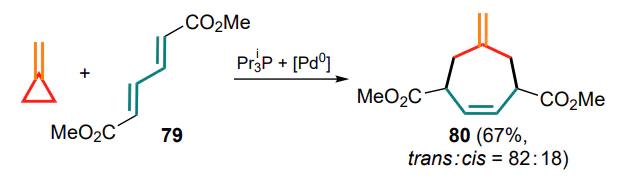
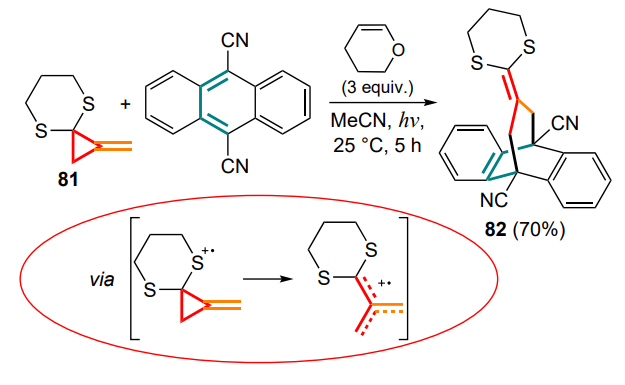
A similar reaction, described by Nakamura еt al. [95] in 1999, involved photochemical opening of the small ring and addition of the methylidenecyclopropanone thioacetal 81 to 9,10-dicyanoanthracene to yield the adduct 82. It was suggested that the process involved the formation of the corresponding cation radicals (Scheme 41).
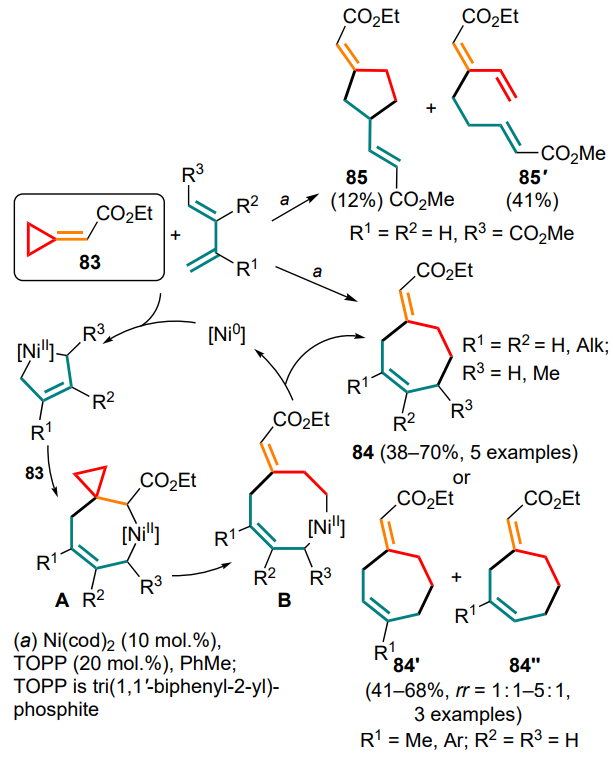
However, the first systematic studies in this direction did not appear until 2007. Saito and Takeuchi [96] proposed the use of a conjugated diene substrate as the C4-synthone. The corresponding (4+3)-cycloaddition reactions of cyclopropylidene acetate 83 to a number of dienes have been successfully carried out. Symmetrical 2,3-disubstituted substrates and penta-1,3-diene reacted to give cycloheptene 84, whereas unsymmetrically substituted dienes yielded a mixture of regioisomers 84' and 84''. In contrast to the said dienes, (E)-penta-2,4-dienoic acid methyl ester afforded a mixture of (3+2)-cycloaddition (85) and cross-coupling (85') products. The reaction onset involves the formation of a dienebased five-membered nickelacycle, into which the methylidenecyclopropane double bond is further inserted. The resulting intermediate A then undergoes cyclopropylmethyl – butenyl rearrangement to intermediate B with subsequent reductive elimination of the catalyst and formation of seven-membered adducts 84 (Scheme 42). For electron-rich trimethylsiloxy-substituted dienes, such a reaction could not be carried out.
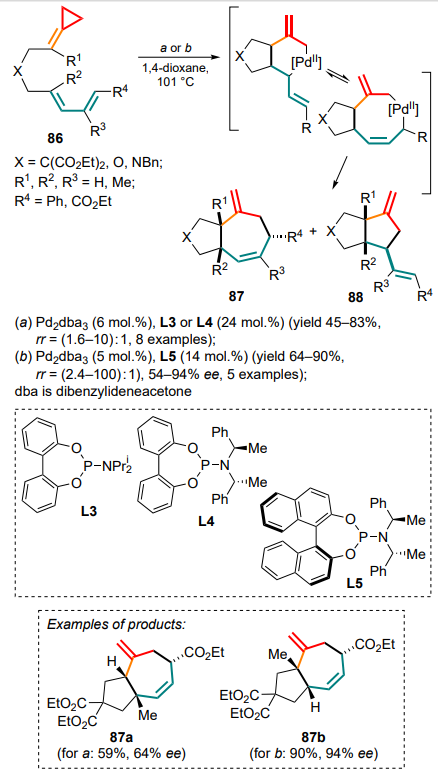
Mascareñas and co-workers [97] have proposed a slightly different principle when implementating an intramolecular variant of Pd-catalyzed cyclization involving methylidenecyclopropane and diene moieties of compound 86 in the presence of ligand L3 (conditions a). Initially, the six-membered palladacycle resulting from the opening the three-membered ring, is able to incorporate a multiple bond, thus expanding the ring to eight-membered and yielding substituted cycloheptenes 87 as the major products. However, for all the substrates studied, (3+2) cyclization by-products 88 were detected in the reaction mixture (Scheme 43). For starting compounds containing an inactivated diene system (R3 = H), the reaction produced a complex mixture of compounds. The process was made enantioselective in the presence of the chiral ligand L4 (the best ee value was 64% for the product 87a). This is the first example of an enantioselective intramolecular metal-catalyzed (4+3)-cycloaddition including the use of the cyclopropane moiety.
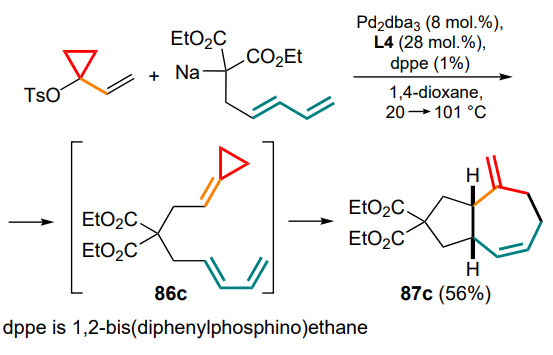
It has been shown that substrates for this reaction can be derived from the appropriate precursors under similar conditions.[97] This allowed to propose a tandem variant for the synthesis of substituted cycloheptenes 87c from 1-vinyl-1-tosyloxycyclopropane and penta-2,4-dienylmalonate via the formation of the methylidenecyclopropane intermediate 86c (Scheme 44).
Subsequently, the authors extended the initial study by optimizing the conditions for the enantioselective variant of this process (see Scheme 43, conditions b).[98] As a result, they managed to increase the ee of product 87b up to 94%. In 2023, the same research group [99] described an example of the similar transformation in the presence of a cobalt(I) catalytic system.
Saito and co-workers [100-102] combined the previous ideas and extended them to the use of the acetylene moiety as a C2-syntone for the intramolecular assembly of cyclononadienes 89, 90 involving ethyl cyclopropylidene acetate 83. The reaction has been studied for a wide range of different substrates and in some cases it was possible to obtain a single diastereomer. Notably, the use of substrate 91 containing a cyclic diene moiety in the process led to a mixture of products of cyclization (89') and further β-hydride elimination (89'') (Scheme 45). The reactions began with the formation of a π-complex between the nickel catalyst and an acetylene moiety, which is further re-coordinated to form a seven-membered nickelacycle A by incorporating the double bonds of the diene system. After the insertion of the methylidene cyclopropane fragment, the aforementioned cyclopropylmethyl–butenyl rearrangement proceeds to afford cyclononadienes 89, 90.
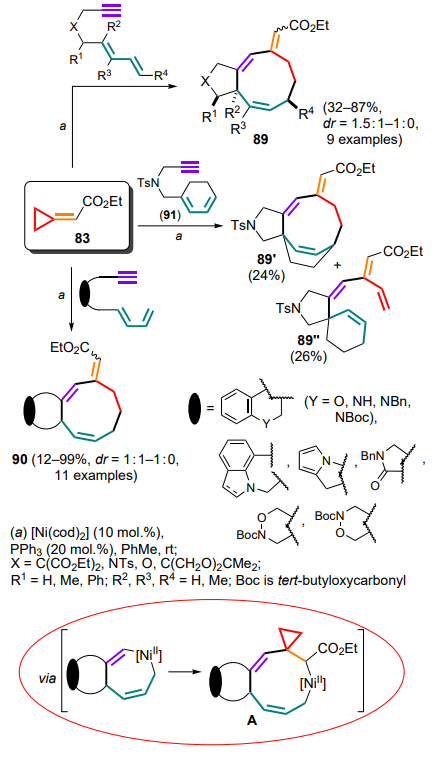
The triple bond can also act as part of the aryl ring-conjugated system 92 in a gold(I)-catalyzed intramolecular reaction involving the methylidenecyclopropane unit (Scheme 46).[103] In contrast to the 5-exo-dig or 6-endo-dig cyclizations of 1,6-enynes that are usually observed, in this case, a 6-exo-dig cyclisation occurs, which is explained by the stabilization of the cyclopropylcarbinyl intermediate A due to the reverse AuI donation. The further opening of the three-membered ring is accompanied by the Nazarov-type electrocyclization involving the aryl moiety of the starting conjugated system and the elimination of the final tetracycles 93.
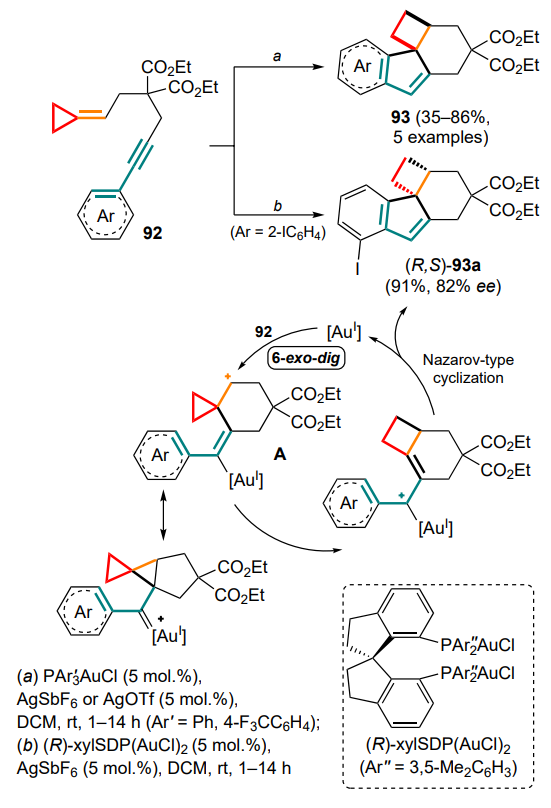
It is worth noting that the strain in the resulting alkylidene cyclobutane unit, as one of the main driving forces of this process, promotes further cyclization. The use of the chiral complex (R)-xylSDP(AuCl)2 provided an example of enantioselective modification of the tandem reaction and to obtain the chiral product (R,S)-93a from the o-iodophenylsubstituted substrate.
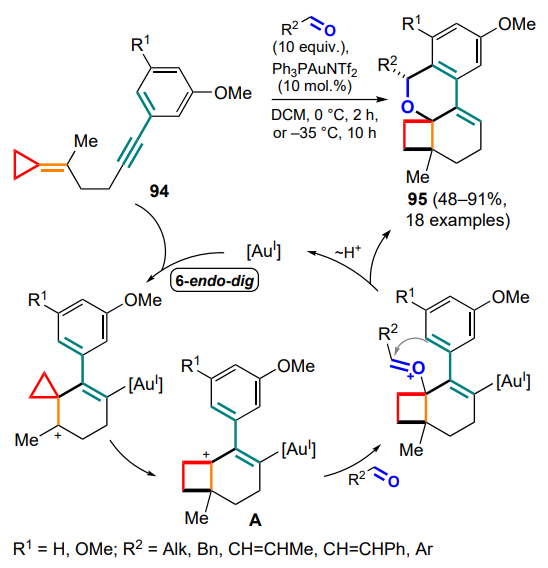
A related strategy has been proposed by Roselli and Gagné.[104] Gold(I)-mediated 6-endo-dig cyclization of methylidene cyclopropanes 94 occurs through the exomethylene bond with closure of the six-membered ring and cationic rearrangement with opening of the three-membered ring. The resulting intermediate A is capable of capturing aliphatic and aromatic aldehydes with cyclization on the adjacent electron-donor aryl moiety and additional closure of the tetrahydropyranyl ring to yield tetracyclic compounds 95 (Scheme 47).
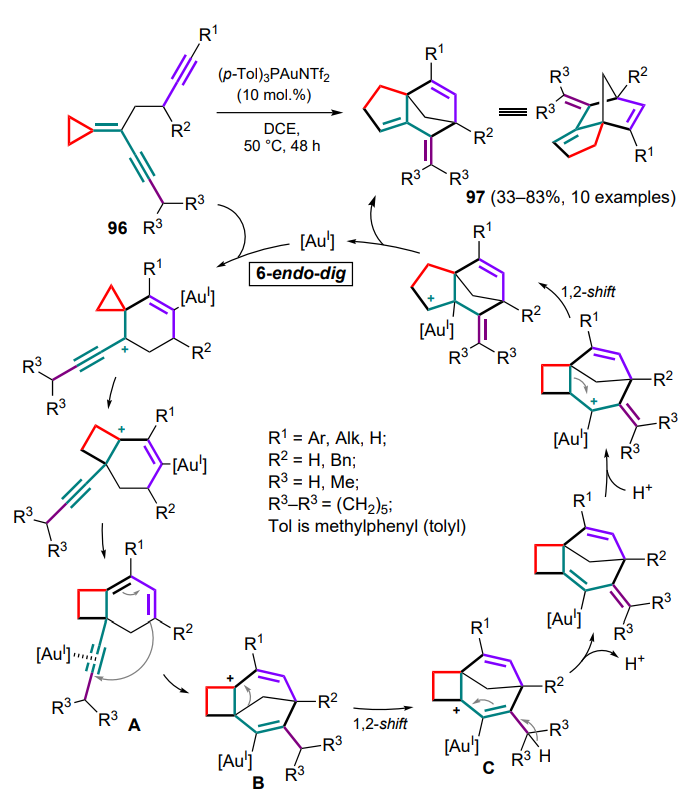
The enyne moiety as a conjugated system tailored to a threemembered ring of compound 96 can serve as a basis for cyclizations involving other multiple bonds. Thus, in the catalysis with the gold(I)-phosphine complex, a tandem process takes place involving the alkyne unit of this molecule (Scheme 48).[105] The cyclobutane intermediate A formed after coordination of the triple bond by the AuI complex contains a diene system and an alkyne moiety which undergo cyclization to generate the carbocation B. Next, there is a 1,2-alkyl shift of the methylene bridge with displacement of the carbocationic centre (intermediate C), proton migration and a final 1,2-alkyl shift to yield substituted cyclopentene-annulated methylene-norbornene structures 97.
2.6. Reactions of carbo- and heteroannulation based on (arylmethylidene)cyclopropanes
This Section includes a wide range of reactions in which the (arylmethylidene)cyclopropane moiety is involved in cyclization/annulating processes, both with a diversity of substrates and with unsaturated and heteroatomic units within the same molecule. In this context, the assignment of the material to one or the other subsection seems rather conventional.
2.6.1. Reactions of (arylmethylidene)cyclopropanes initiated by radical or electrophilic species
The first radical cyclization of (arylmethylidene)cyclopropanes 98 with diethyl malonate was described in 2004.[106] It opened an access to a series of substituted dihydronaphthalenes 99. The reaction is initiated by manganese(III) acetate generating a malonyl radical, which attacks the exomethylene bond to form a benzyl radical. The resulting intermediate undergoes further opening of the three-membered ring and subsequent oxidative cyclization involving the aryl moiety to give dihydronaphthalenes 99 (Scheme 49). It is shown that this reaction cannot be used for substrates containing an electron-withdrawing nitro group, nor for methylidenecyclopropanes containing an aryl moiety that is not conjugated to the exomethylene bond. In the latter case, instead of closing the seven-membered ring as in compound 100, there is an opening of the three-membered ring resulting in the formation of a terminal double bond. This fact once again emphasizes the special role of the conjugation of the exomethylene bond and the aryl fragment for the class of reactions in question.
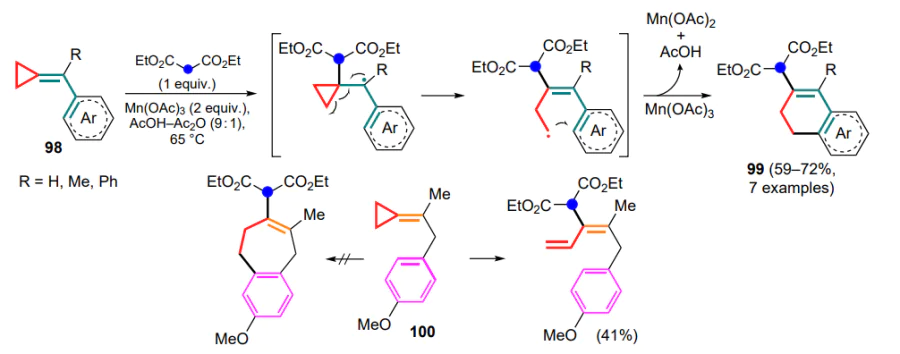
The cyclization of (diarylmethylidene)cyclopropane[b]-naphthalenes 101 with a number of nucleophiles in the presence of manganese(III) acetate as oxidant can be attributed to radical processes.[107] In contrast to the previous example, the radical centre in the (diarylmethylidene)cyclopropane molecule is initially formed by oxidation of the exomethylene bond (intermediate A), which then undergoes nucleophilic attack (B), three-membered ring-opening (C) and annulation with the adjacent aryl moiety to form tetracyclic products 102 (Scheme 50).
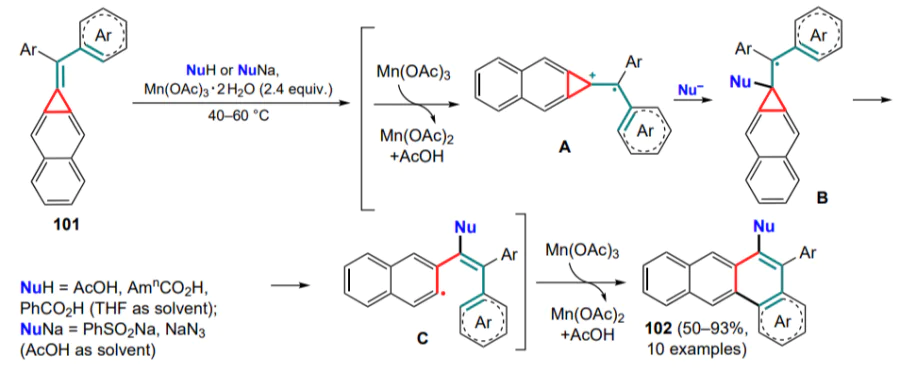
The cyclizations of unsubstituted methylidenecyclopropanes 98 using various radicals with the construction of a new C–C bond in the resulting dihydronaphthalenes 103–107 have been studied in detail (Scheme 51). Such an example was the cyclization involving trifluoromethyl radical generated from Togni reagent II.[108] A large number of substrates undergo this reaction, giving trifluoromethyl derivatives 103, and for metasubstituted (arylmethylidene)cyclopropanes, the formation of a mixture of regioisomers in the ratio of (2–3):1 was observed.
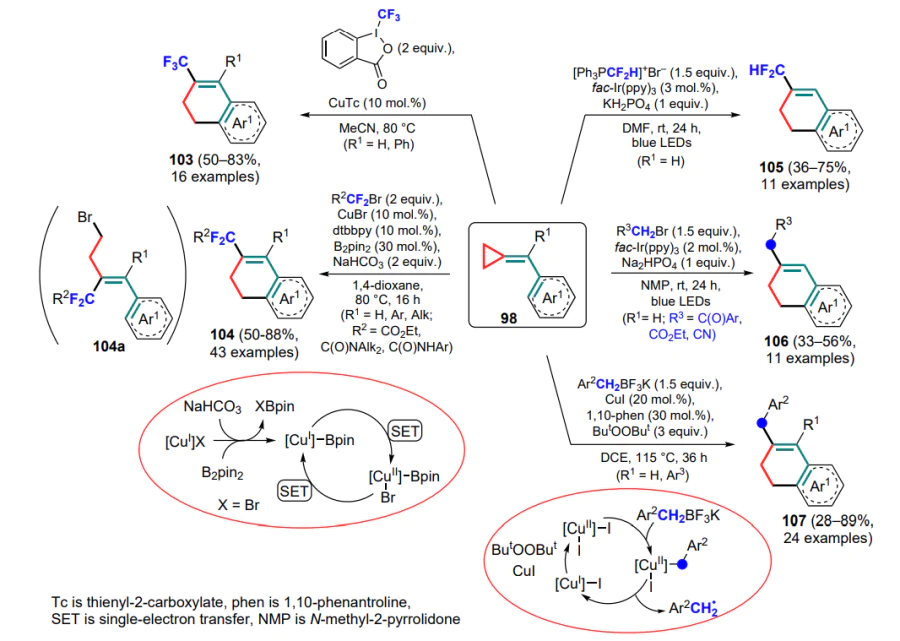
Wang and co-workers [109] continued to develop the methodology of cyclization of (arylmethylidene)cyclopropanes combined with the insertion of fluoroalkyl groups into the molecule. The authors proposed to generate difluoromethyl radicals from a series of bromodifluoroacetates in the presence of a catalytic system based on copper(I) bromide and bis(pinacolato)diboron (B2pin2). The ligand–CuI–Bpin complex formed in the presence of base acts as the main catalytic species. In some cases, in addition to the difluoroalkyl derivatives 104, e.g., the Z-alkene 104a was isolated as a by-product. It results from the ring-opening, which is unfavourable for subsequent cyclization, of an intermediate formed initially by the addition of a radical to the double bond of the substrate (see Scheme 51).
A photoredox-catalytic variant of radical cyclization was developed using difluoromethyl radicals generated from difluoromethyltriphenylphosphonium bromide and alkyl radicals formed from the corresponding bromine derivatives.[110] As a result, a series of difluoromethyl- and alkyl-substituted dihydronaphthalenes 105 and 106 were obtained in moderate yields (see Scheme 51).
Oxidative cyclization involving benzyl radicals generated from potassium benzyl trifluoroborates has also been described.[111, 112] Oxidation of the starting copper(I) complex in the presence of di-tert-butyl peroxide yields an active catalytic copper(II) species, which is further involved in the generation of benzyl radicals that undergo cyclization. The resulting radical intermediate, after rearrangement and alkylation of the aryl ring, is oxidized by peroxide to a cation with further deprotonation to the desired product, 3-benzyl-substituted tetrahydropyridine 107 (see Scheme 51).
In a number of other studies, radicals were generated by breaking the C–H or C–C bonds. For example, Liu et al. [113] proposed to use a series of ethers in the presence of tert-butyl hydroperoxide as a source of radicals to initiate the cyclization of (arylmethylidene)cyclopropanes. As a result, dihydronaphthalenes 108 were obtained using both cyclic and acyclic ethers (Scheme 52). However, despite the successful reaction with tetrahydrofuran, the use of tetrahydropyran and tetrahydrothiopyran did not yield the corresponding products due to the conversion of the starting material to benzophenone; attempts to use a number of cycloalkanes, anisole and N-methylmorpholine in this reaction also failed. This approach makes it possible to combine the roles of reagent and solvent in one compound, which is one of its main advantages.
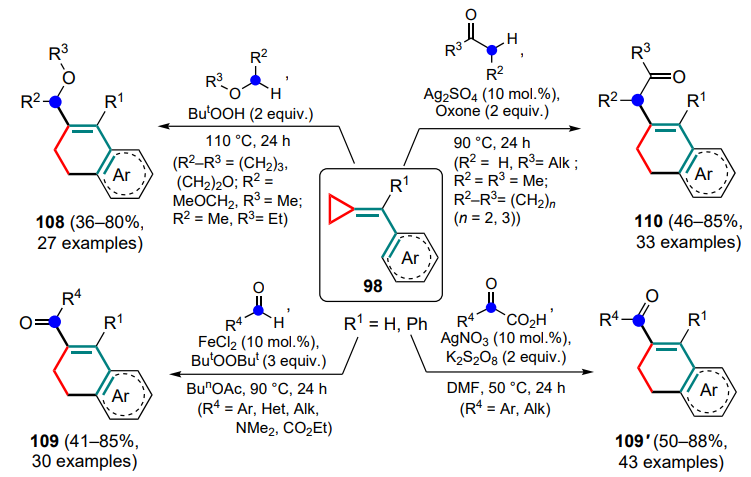
The authors also proposed a method for the cyclization of (arylmethylidene)cyclopropanes involving acyl radicals generated from the corresponding aldehydes in the presence of di-tert-butyl peroxide.[114] It was found that the presence of a Lewis acid can stabilize the newly formed radical species, with ferrous chloride showing the greatest efficiency. In addition to aryl- and alkylcarbaldehydes, DMF and ethyl glyoxylate were used to afford ketones 109, which gave the corresponding dihydronaphthalenes in moderate yields (see Scheme 52).
The use of ketone-based radicals in a similar process allowed the concept of tandem acetylation/arylation of methylidenecyclopropanes 98 to be proposed, resulting in unconjugated ketones 110. [90] Interestingly, unlike the previous reactions in this Section, it was possible to carry out this transformation with phenyl- and nitrophenyl-containing substrates. The reaction was carried out in the presence of Oxone and was promoted by silver(I) sulfate. For the initial meta-substituted (arylmethylidene) cyclopropanes, the formation of a mixture of regioisomers in a ratio from 1:1 to 7:3 was observed (see Scheme 52).
Finally, it was proposed to generate acyl radicals for the cyclization of compounds 98 to conjugated ketones 109' from α-keto acids in the presence of potassium persulfate and silver(I) nitrate.[115] It should be noted that, in addition to a diversity of substituted substrates, it was possible to use unsubstituted (phenylmethylidene)cyclopropane in the reaction, although the similar nitro derivatives turned out to be unreactive. It is shown that benzoic acid, benzaldehyde, acetophenone, benzoyl chloride and dibenzoyl gave no desired products under the reaction conditions, thus indicating the key role of radical formation by breaking the C(O) – CO2H bond under these conditions.
The possibility of cyclizing (arylmethylidene)cyclopropanes with heteroatom-centred radicals has been investigated in detail (see Scheme 52). Phosphonyl and thiyl radicals generated in the presence of azobis(isobutyronitrile) (AIBN) as a radical initiator to break P–H and S–H bonds were the first to be studied.[116, 117] The intermediate formed after cyclization participates in the radical chain by abstracting the hydrogen atom from the precursor molecule with regeneration of the attacking radical and formation of dihydronaphthalenes 111 (Scheme 53). The authors also attempted to carry out the similar reaction for selenophenol, but it was non-selective and produced a mixture of different compounds.
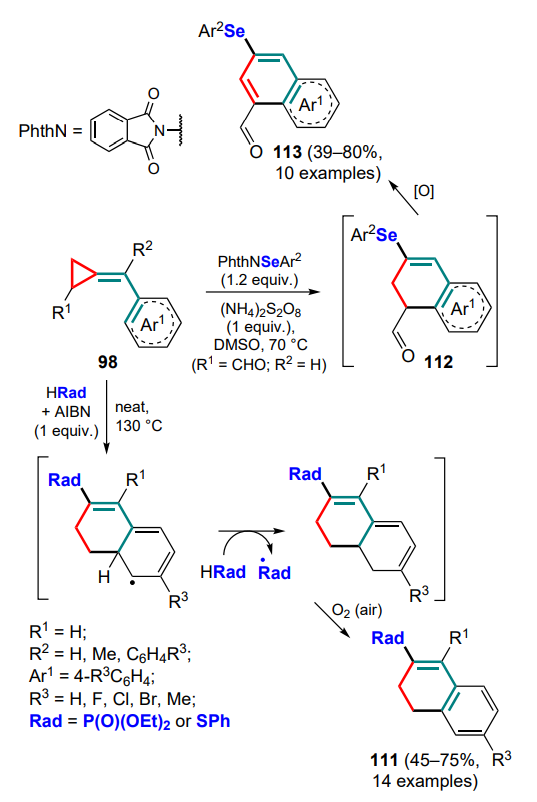
The problem of obtaining selenium derivatives was solved by the reaction of methylidenecyclopropanes 98 with N-(phenylseleno)phthalimide in the presence of ammonium persulfate as oxidant.[118] Similar to the previous transformations, dihydronaphthalenes 112 are formed first, which are then aromatized in the presence of an oxidant to seleno-substituted naphthylcarbaldehydes 113.
Shi and co-workers [119] developed a method for trifluoromethylthiolation of (arylmethylidene)cyclopropanes 98 in the presence of silver trifluoromethylthiolate and sodium persulfate as an oxidant. The resulting dihydronaphthalenes 114 can be further oxidized to the corresponding naphthalenes 115 by excess of the reagent (Scheme 54).
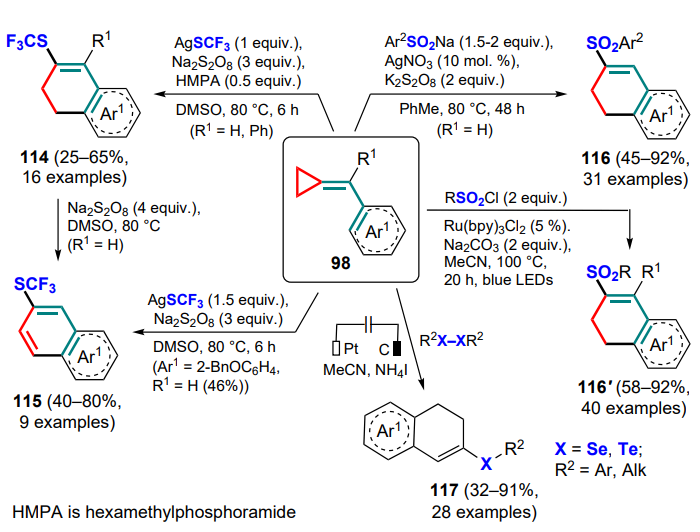
Subsequently, two methods were proposed for the synthesis of sulfonic derivatives of dihydronaphthalenes 116, 116' (see Scheme 54).[120, 121] It was shown that sulfinates can serve as sources of sulfonyl radicals in the presence of potassium persulfate as oxidant in the presence of silver(I) nitrate. For the same purpose, sulfonyl chlorides can be utilized when carrying out the reaction in the photocatalytic fashion using Ru(bpy)3Cl2 as photocatalyst. Importantly, in the second case it is possible to extend the synthetic possibilities of the reaction and carry it out for a wider range of substrates.
In 2023, a process for the electrochemical oxidative cyclization of (arylmethylidene)cyclopropanes with diselenides or ditellurides yielding 2-substituted 3,4-dihydronaphthalenes 117 was presented.[122] This method has been successfully applied to a variety of substituted diarylselenides, benzyl and 2-thienyl derivatives, and also to methoxy-, halogen- and trifluoromethyl-substituted (arylmethylidene)cyclopropanes 98 (see Scheme 54).
The unusual visible light photoredox-catalyzed generation of sulfonyl radicals from oximes of cyclobutanones 118 and potassium disulfite as the SO2 source was used to carry out the cyclization of (arylmethylidene)cyclopropanes 98 to 2-cyanoalkylsulfonic derivatives of dihydronaphthalenes 119.[123] Initially, the photoreduction and elimination of the anion stabilized by the acceptor group generate an iminyl radical A, which further undergoes ring-opening (intermediate B) and the addition of sulfur dioxide (C), after which cyclization proceeds according to the mechanism described above (Scheme 55). It is shown that other potential sources of iminyl radicals, such as derivatives of azetidinone, oxetanone or cyclopentanone, cannot be used under the conditions described, highlighting the need for the presence of a cyclobutane motif.
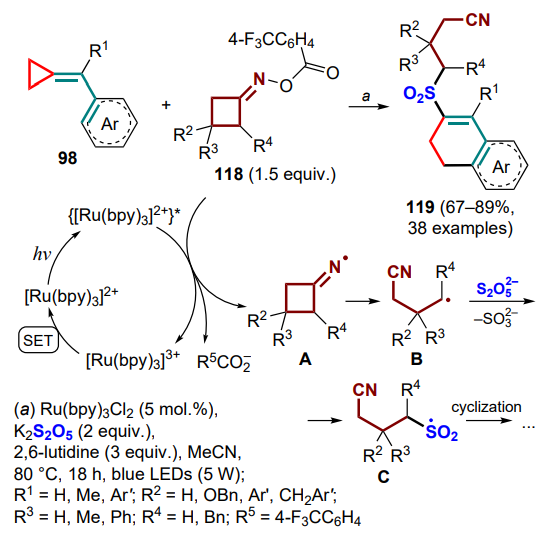
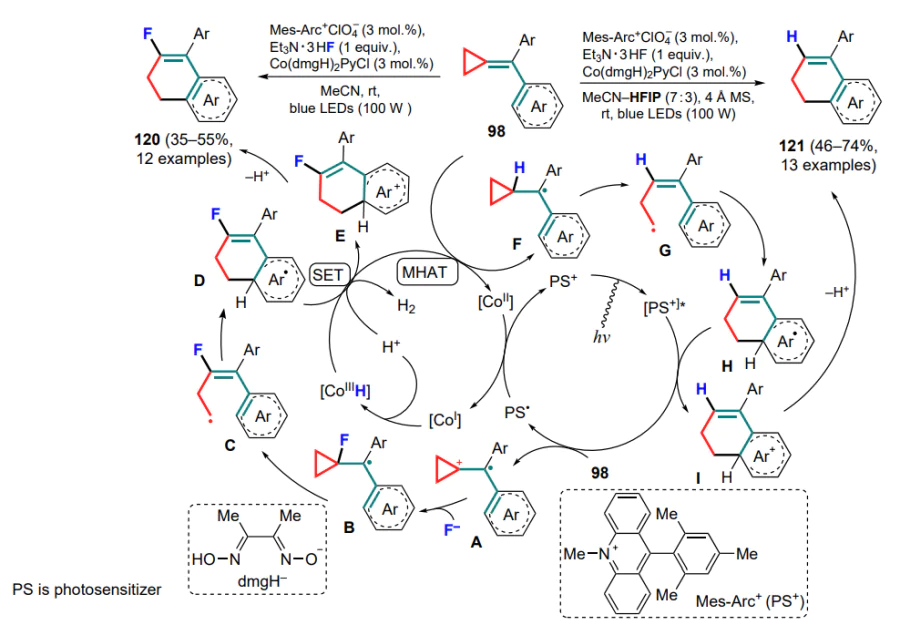
Shi and co-workers [124] pioneered in carrying out the direct oxidation of (arylmethylidene)cyclopropanes under organophotoredox catalysis conditions to produce tetrahydropyridines 120 or 121. The original idea of the study was to obtain fluorinated derivatives, which was carried out using the complex of HF with triethylamine as fluorinating agent in the presence of a proton-reducing cobalt(III) catalyst. The process starts with the photocatalytic oxidation of the exomethylene bond of the substrate to generate the cation radical intermediate A, followed by the addition of fluoride anion (intermediate B) and cyclization by the above-mentioned mechanism (C, D). The further oxidation of the radical intermediate D to the cationic (E) involves the hydride CoIII-complex, with subsequent deprotonation and release of the final product 120, as well as the formation of the CoII-complex, which is then reduced by the photocatalyst to the CoI -complex, which is capable of attaching a proton to form the CoIII-complex, thus closing the catalytic cycle (Scheme 56). The authors also found that the reaction in the presence of hexafluoroisopropyl alcohol produces tetrahydronaphthalenes which do not contain a fluorine atom. This can be explained by a fundamentally different mechanism, consisting of the initial metal-catalyzed hydrogen atom transfer (MHAT) involving the cobalt(III) hydride complex (F). After the opening of the three-membered ring in the radical G and its cyclization, the generated radical intermediate H is oxidized to cationic (I) already when treated with the photocatalyst, which undergoes further deprotonation to give the final product 121.
In a series of papers,[125-127] (arylmethylidene)cyclopropanes 98 were studied in reactions with aldehydes, aldimines and acetals in the presence of ytterbium(III) triflate and boron trifluoride–ether complex. At first, the reaction mechanism based on Lewis acid catalysis was proposed for all substrates, but in the course of further experiments, the authors gained deeper insight about the reaction pathway with the participation of aldehydes. It was concluded that in the presence of water traces, BF3 ·Et2O forms the Brønsted acid HBF3OH·Et2O, which protonates the C=C bond in the methylidenecyclopropane unit (intermediate A) and accounts for its opening after nucleophilic attack by aldehyde (B). Next, the Prins-type reaction (C) takes place between the multiple bonds of the cationic intermediate and the opening of the five-membered ring to form the stabilized carbocationic intermediate D, which undergoes Friedel–Crafts cyclization to yield indene 122 (Scheme 57).
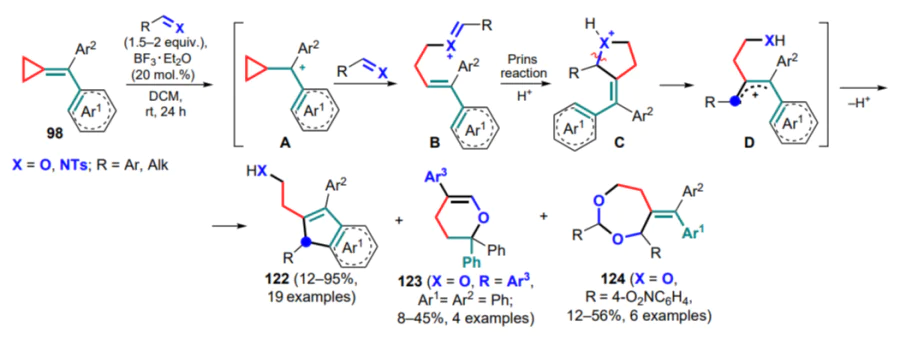
For certain substrates in the reaction mixture, other cyclization products formed without the involvement of the aryl moiety were detected (compounds 123, 124), careful analysis of which also confirmed the proposed mechanism. In the reactions of aldehyde acetals and triethyl orthoformate, the role of the Lewis acid is to form the oxonium intermediate E, which attacks the exomethylene bond of the substrate, opening the threemembered ring (intermediates F–G). This leads to a fivemembered cationic intermediate H, similar to intermediate C, which is opened via nucleophilic attack of the aryl ring (Scheme 58). In these cyclizations, the three-membered ring becomes the basis for the formation of the open-chain fragment of compounds 125–127.
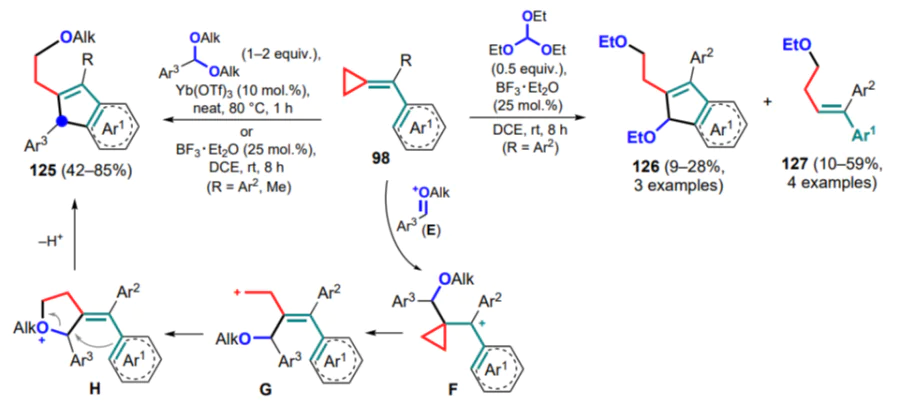
One of the first examples of reactions of (arylmethylidene) cyclopropanes with full involvement of the exomethylene bond and the aryl moiety was the dimerization of the electron-rich dimethoxy derivatives 98a,b in the presence of catalytic amounts of platinum(II) chloride in a CO atmosphere.[128] By raising the molar concentration of PtCl2 from 0.02 to 0.1, the initially complexed cyclobutene A can be dimerized to intermediate B, which is cyclized to spirane 128a (53% yield) (Scheme 59). In the case of the 3,4-dimethoxy derivative 98b, a 1:9 mixture of regioisomers 128b and 128' with the least sterically hindered isomer 128' prevailing was obtained in 87% yield.
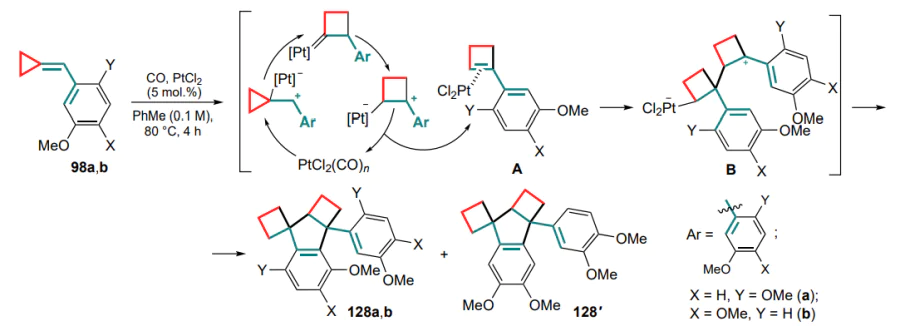
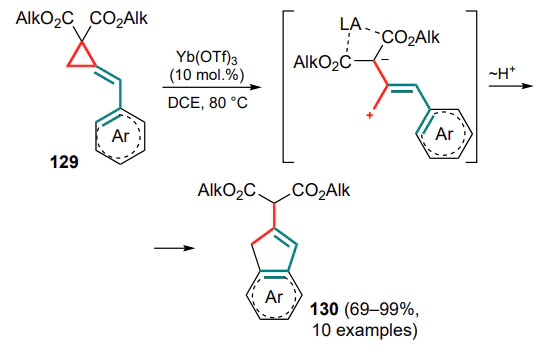
The next step in the development of the methodology for the cyclization of (arylmethylidene)cyclopropanes was an attempt to increase the polarization of the cyclopropane C–C bond by introducing electron-withdrawing ester functionalities to obtain donor-acceptor substrates 129 and to carry out the reactions in the presence of Lewis acids.[129] Thus, a method for cyclization promoted by ytterbium(III) triflate was proposed, in which the C(1)–C(3) bond of the three-membered ring is cleaved to form a carbocation, which then undergoes Friedel–Crafts cyclization to substituted indenes 130 (Scheme 60).
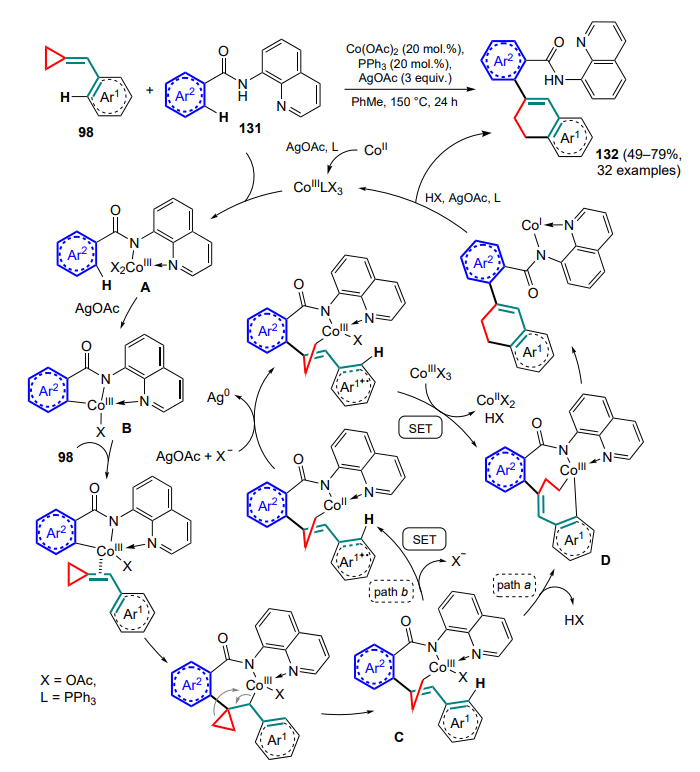
Li and Kwong[130] presented a strategy similar to radical cyclization (see Section 2.4), but implemented in a completely different way. The arylation of the C=C bond of (arylmethylidene) cyclopropanes 98 with opening of the three-membered ring and subsequent cyclization was carried out as a cobalt(II) acetatecatalyzed tandem process involving two C–H activations for each of the reactants. The arylating agents were N-(quinolin-8-yl)benzamides 131, in which the quinolinylamide moiety acted as a directing group for the cobalt(III) catalyst species (Scheme 61). In this case, complex A is formed first, which, promoted by silver acetate, abstracts an ortho-proton from the aryl substituent to form a C–Co bond. Then, intermediate B coordinates the double bond of (arylmethylidene)cyclopropane with the insertion of the C–Co bond and, after opening the three-membered ring, an eight-membered cobaltacycle C is formed. This is followed by the second C–H activation involving the aryl moiety of the MCP (see Scheme 61, pathway a). The authors also suggest, albeit with less probability, that this transformation may proceed via two successive single electron transfers (SETs) with the generation of cation-radical intermediates (see Scheme 61, pathway b). In either case, complex D undergoes a subsequent reductive elimination with closure of the six-membered ring, after which the final product 132 is formed during protonation and the catalyst species is oxidized to the cobalt(III) complex and the catalytic cycle is closed.
2.6.2. Cyclizations based on ortho-substituted (arylmethylidene)cyclopropanes
A large number of cyclizations have been described for (arylmethylidene)cyclopropanes bearing a free or substituted amino group in the ortho-position of the aryl moiety, and these processes are presented both for cyclopropanes and with the involvement of external unsaturated substrates.
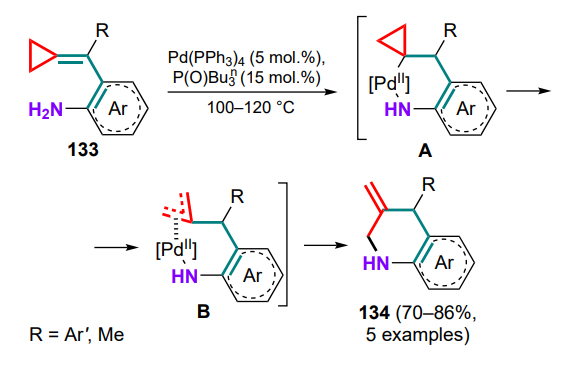
The first example of this type of Pd(PPh3)4-catalyzed intramolecular cyclization was described in 2005 for substrates 133. [131] The mechanism of the reaction implies the hydropalladation of the exomethylene bond of the substrate to yield intermediate A followed by the distal bond cleavage in the three-membered ring. This affords the π-allylpalladium complex B, which, after reductive elimination of the Pd0 catalyst, gives a six-membered nitrogen-containing ring 134 with an exocyclic C=C bond (Scheme 62). It was shown that the presence of a tri-n-butylphosphine oxide ligand significantly improves the yield of the products, whereas the reaction in a solvent leads to a decrease in yield.
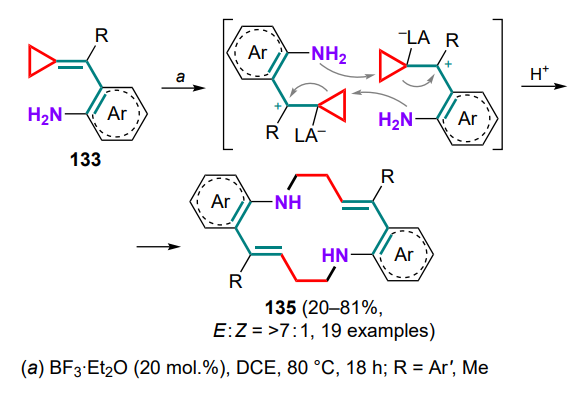
It should be noted that (o-aminoarylmethylidene)cyclopropanes 133 can react in the presence of Lewis acids, especially boron trifluoride–ether complex, as 1,7-synthones, undergoing dimerization to macrocyclic 14-membered structures 135 (Scheme 63).[132] The reaction proceeds via the zwitterionic intermediate upon Lewis acid addition to the methylene bond, followed by nucleophilic attack, opening of the three-membered ring and protonation. The predominant formation of transisomers, as well as a sharp decrease in product yield when moving from aryl substituents at the exomethylene bond to the methyl group, and the impossibility of carrying out this process for the unsubstituted substrate (R = H) were noted.
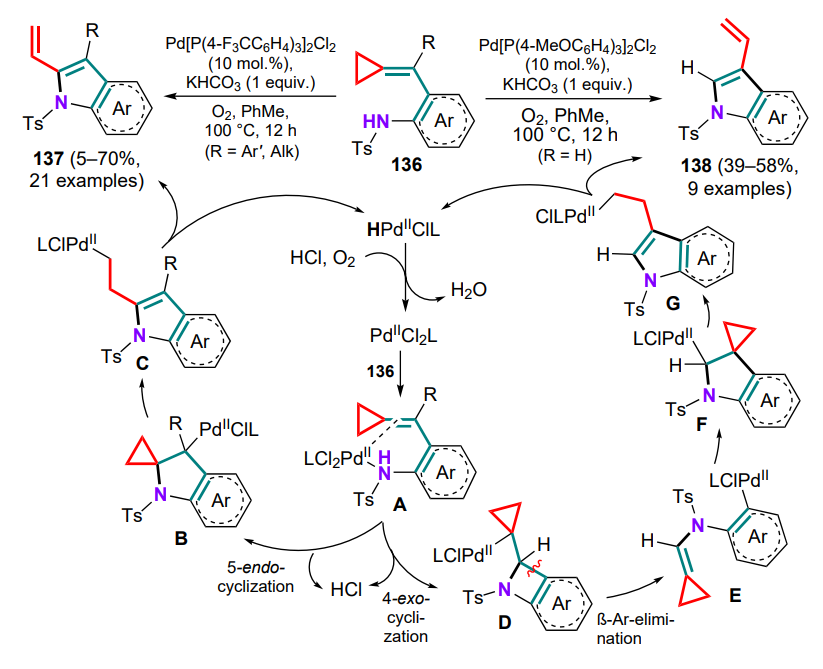
N-Tosyl-substituted (o-aminoarylmethylidene)cyclopropanes 136 can be converted into 2- or 3-vinyl-substituted indoles 137, 138 via palladium-catalyzed oxidative cyclization, with the nature of the substituent R in the exomethylene moiety having a fundamental influence on the process.[133] First, the catalyst forms a complex A with the substrate molecule through coordination via the amino group and the C=C bond, after which, depending on the presence or absence of the substituent R, either 5-endo- or 4-exo-aminopalladation occurs. In the first case, the resulting indoline intermediate B undergoes β-Celimination with cleavage of the three-membered ring proximal bond and formation of the alkylpalladium complex C, which transforms into the final 2-vinylindole 137 during the β-hydride elimination. The palladium hydride complex is then oxidized by oxygen to water and an active catalytic species, completing the catalytic cycle (Scheme 64). As for an unsubstituted methylidene bond, the tetracyclic intermediate D is formed, in which β-aryl elimination occurs to afford the arylpalladium complex E, which is inserted into a double bond (intermediate F), and similar transformations from the first cycle of the complex G gives 3-vinylindoles 138.
The cyclization of acrylamide- and vinylsulfonamidesubstituted substrates is described, where the process is initiated by a silyl radical generated from tris(trimethylsilyl)silane (TTMSS) in the presence of two photoredox catalytic systems based on cobalt(III), photosensitizers and quinuclidine as HAT catalyst (HAT is hydrogen atom transfer).[134]
In the presence of the organophotocatalyst, 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN), CoIII species are reduced to CoII, and quinuclidine is oxidized to a more electrophilic cation radical, which abstracts the hydrogen atom from TTMSS to generate the silyl radical during HAT, or the direct formation of the silyl radical takes place involving the cobalt(III) complex. The silyl radical then attacks the terminal C=C bond of the substrate (intermediate A), initiating cyclization via the exomethylene bond at a three-membered ring, intermediate B undergoes further ring-opening. The resulting intermediate radical C can be captured by the CoII particle, resulting in the photolysis of the C–CoIII bond, after which the alkyl radical attacks the C–Si bond, closing the five-membered ring and returning the silyl radical to the cycle. At the same time, the silyl radical forms a complex with the CoIII species and, upon protonation, regenerates TTMSS and CoIII, thus completing the catalytic cycle to furnish compounds 140 (Scheme 65).
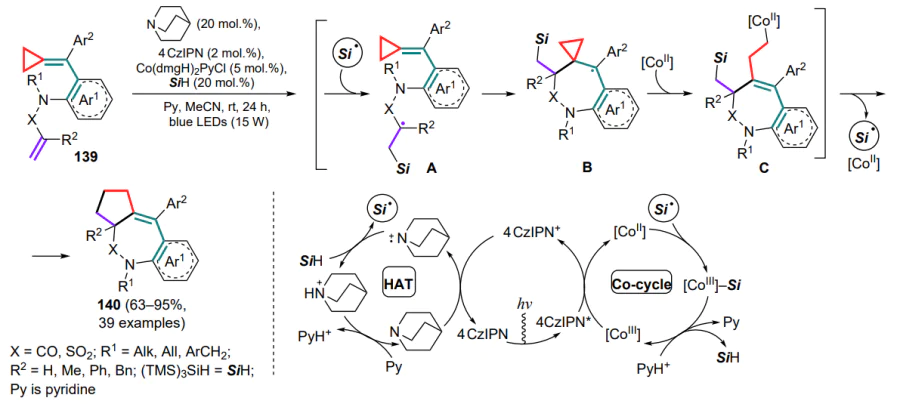
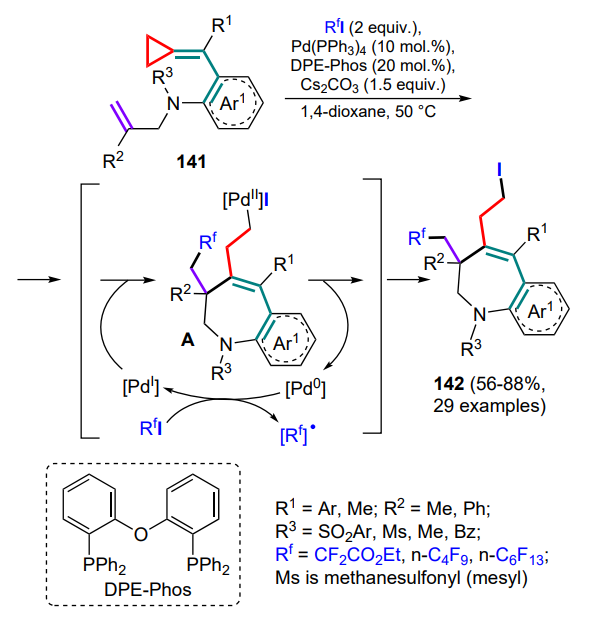
(Arylmethylidene)cyclopropanes 141, containing the C=C bond tethered to an aryl fragment through a linker, can also undergo radical cyclizations.[135] Thus, for the synthesis of 1-benzazepines 142, a combination of palladium catalysis and cyclization involving perfluorinated radicals, generated from the corresponding iodides during the oxidation of catalytic particles Pd0 to PdI , was used. The radical further initiates cyclization through a cascade of attacks first on the terminal double bond and then on the exomethylene bond, accompanied by opening of the three-membered ring and oxidative addition of the intermediate and free iodine radical to the PdI particle to afford the PdII complex A. Subsequent reductive elimination completes the catalytic cycle and yields the final product 142 (Scheme 66). The important role of substituents (R1, R2) at multiple bonds in stabilizing intermediates has been noted.
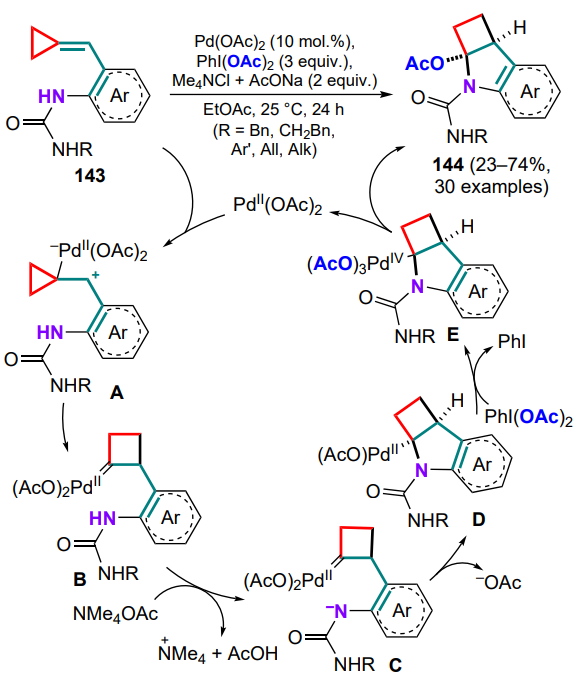
(o-Aminoarylmethylidene)cyclopropanes substituted at the amino group have found application in the development of approaches to the design of cyclobutane-based heteropolycyclic compounds. For example, the use of urea-tethered (arylmethylidene)cyclopropanes 143 proved to be a successful modification of PdII-catalyzed cyclization in the presence of an oxidant (di(acetoxy)iodobenzene) and a base (tetramethylammonium acetate).[136] The success of this oxyamination illustrates the advantage of choosing the urea moiety over other substituents at the amino group. The process runs via coordination of the Pd catalyst to the exomethylene bond with the formation of complex A and the expansion of the threemembered ring to a four-membered one in the carbene intermediate B. The urea fragment is then deprotonated upon treatment with a base (C) and the indoline ring is annulated on the carbene centre. The resulting intermediate D undergoes the oxidation of the palladium species to PdIV (E), after which the reductive elimination of the product 144 is possible and the catalytic cycle is closed (Scheme 67).
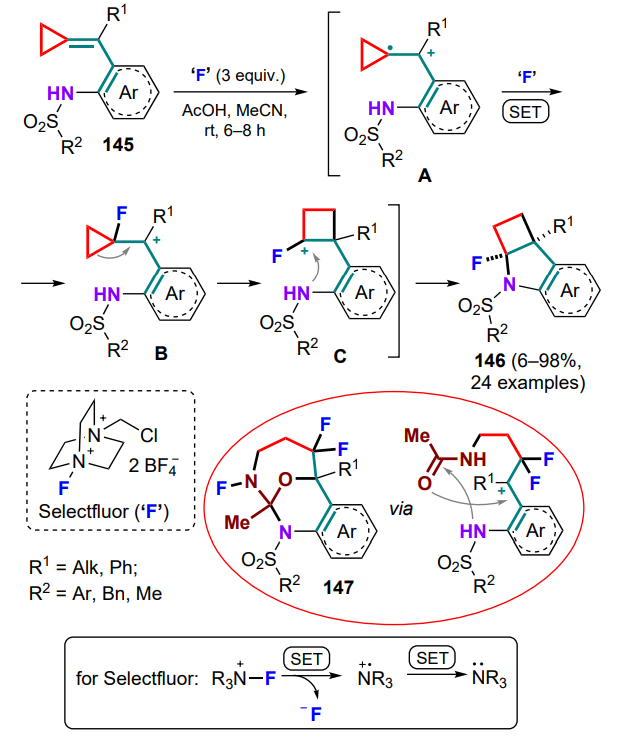
The cyclization of N-sulfo-substituted (o-aminoarylmethylidene)cyclopropanes 145 mediated with the fluorinating agent Selectfluor (1-fluoro-4-chloromethyl-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate)), which promotes the formation of fluorocyclobuta[b]indoles 146, could be counted in this category[137]. During two successive one-electron transfers with Selectfluor, the exomethylene bond (intermediate A) is fluorinated to form the carbocationic intermediate B. The latter undergoes a Wagner–Meerwein rearrangement to yield a fluorocyclobutyl carbocation C, which is further attacked by a sulfoamino group, and finally compounds 146 are obtained (Scheme 68). It should be noted that the presence of acetic acid excess improves the yield of the product. In the presence of water traces, a side process takes place producing more complex polycyclic compounds 147 (yield <10%), which involves acetonitrile used as a solvent.
Of particular interest are the reactions of (arylmethylidene) cyclopropanes with substrates containing multiple bonds. Thus, cyclization of (o-aminoarylmethylidene)cyclopropanes 133 with acetylenedicarboxylates has been developed.[138] Initially, a (2+2) cycloaddition to the exomethylene bond takes place yielding cyclobutene A, which undergoes a nucleophilic attack via the Michael reaction with an annulation of the indoline moiety. In an inert atmosphere, the transformation stops at this stage giving rise to the corresponding cyclic products 148 (Scheme 69). However, in the presence of air and in normal lighting, other rearrangement products, namely cyclopenta[b]indoles 149, prevail.
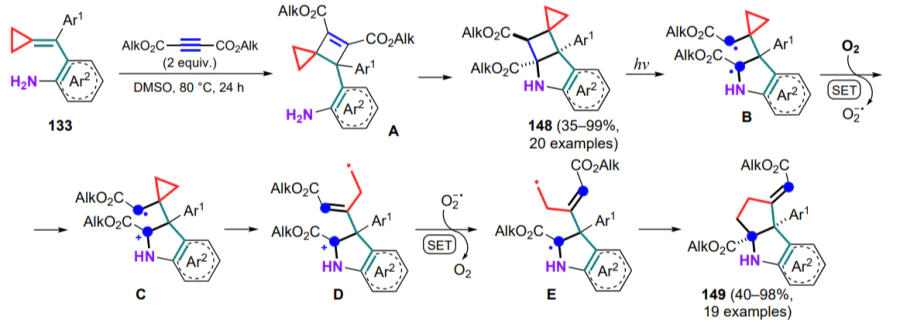
After more in-depth investigations, it was found that light induces the homolytic cleavage of the C–C bond in the fourmembered ring to generate the triplet biradical B, which is further oxidized with oxygen by the mechanism of one-electron transfer to yield the cation-radical intermediate C[139]. The latter undergoes opening of the three-membered ring and then, under the action of superoxide anion, intermediate D is reduced to biradical E, which closes the five-membered cycle 149 with an exocyclic double bond. The nature of the acceptor groups in the starting alkyne is critical, since for amide and benzoyl derivatives, as well as similar dicarboxylic acids, the reaction could not be carried out in the preparative mode.
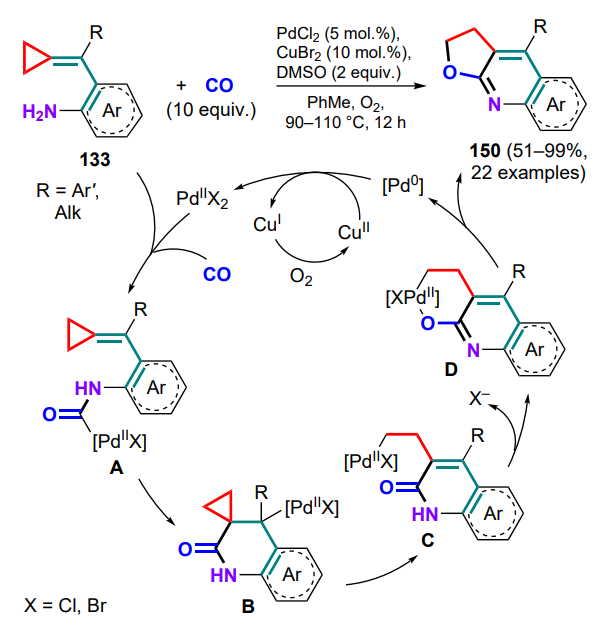
Further cyclization based on the oxidative PdII–CuII catalyzed carbonylation of (o-aminoarylmethylidene) cyclopropanes 133 afforded a series of furoquinolines 150[140]. During the intramolecular acylpalladation of the initially formed aminopalladium complex A, a six-membered ring is closed, followed by β-carbon elimination with opening of the cyclopropane ring of intermediate B and formation of the alkylpalladium complex C. Intramolecular ligand exchange involving the adjacent oxygen atom yields complex D, then, reductive elimination produces furoquinoline 150 and Pd0 complex. The latter is oxidized to the catalytically active PdII species by copper(II) bromide in the presence of oxygen (Scheme 70). The addition of dimethyl sulfoxide increases the yield of the product by reducing the catalyst losses due to the formation of palladium black.
The catalytic reaction of ortho-halo-substituted (arylmethylidene)cyclopropanes with nucleophiles may serve as another similar approach to annulated carbo- and heterocycles. It is shown that the palladium-catalyzed cyclization of (o-bromarylmethylidene)cyclopropanes 151 involving o-alkynylanilines 152 and o-alkynylphenols 153 in the presence of ligands L6, L7 can proceed via either the 6-endo or the 5-exo pathway to give 2-(2-naphthyl)anilines 154 and 5H-indeno-[1,2-c]quinolines 155 or indeno[1,2-c]chromenes 156, respectively (Scheme 71).[141, 142]
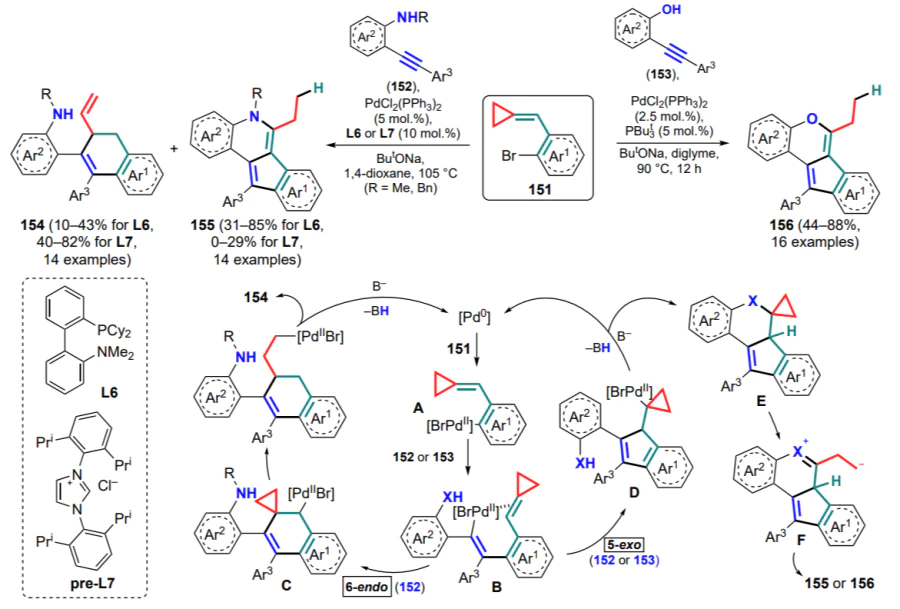
Coordination of bromopalladium(II) complex A to the triple bond of substrates 152 or 153 gives intermediate B, which is common to both reaction pathways. Furthermore, in the case of 6-endo cyclization, complex C is formed, which undergoes opening of the three-membered ring followed by the β-hydride elimination. At the same time, in the course of 5-exo cyclization, the indene ring is closed (intermediate D), after which the C –X bond (E) is formed in the coupling reaction, and then, opening the three-membered ring (F) and abstraction of the adjacent hydrogen atom afford compounds 155 or 156. It was found that in the case of anilines 152, the nature of the ligand affects the regioselectivity of the process: the phosphine ligand L6 is preferred for the formation of 5-exo-cyclization products, whereas pre-L7, the source of NHC ligand (NHC is heterocyclic carbene), favours 6-endo-cyclization. In confirmation of this observation, the choice of tri-tertbutylphosphine was optimal for the 5-exo-cyclization of phenols 153.
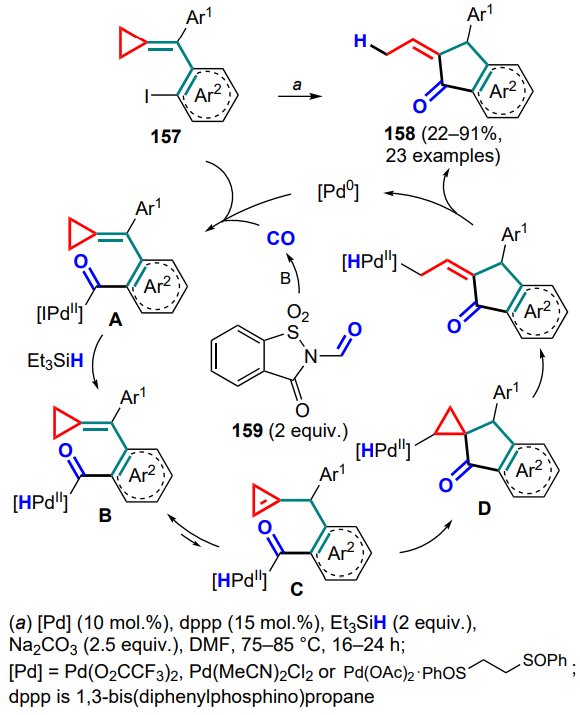
A palladium(0)-catalyzed reductive cyclization of (o-iodoarylmethylidene)cyclopropanes 157 has been developed, which is carried out in the presence of CO and triethylsilane to form substituted indanones 158[143]. A combination of N-formylsaccharine (159) and a base was used as the CO source, and the palladium catalysts were varied depending on the aryl substituents in the substrate. The reaction starts with the oxidative addition of the palladium(0) complex via the C–I bond, followed by carbonylation. In the resulting intermediate A, the iodine atom is replaced by hydrogen in the presence of triethylsilane, after which the hydride complex B is converted upon heating or when treated with a base into the cyclopropene intermediate C, which is further subjected to intramolecular reductive Heck cyclization to give the complex D. The final stage comprises the opening of the three-membered ring and the reductive elimination of the catalyst to furnish the desired indanone 158 (Scheme 72).
A similar strategy was implemented for the Pd-catalyzed dicarbonylation/amination of substrates 157 to give carbamoyl-substituted indenones 160[144]. The possibility of using diverse primary and secondary amines in the reaction has been demonstrated. The catalytic species is also a Pd0 complex, which undergoes oxidative addition of the C – I bond, followed by the insertion of a CO molecule. Next, intermediate A undergoes intramolecular acylpalladation (intermediate B) and opening of the three-membered cycle during β-C-elimination to form alkylpalladium complex C. The latter forms the final indenone 160 via the ligand exchange with an amine molecule (D) and insertion of the second CO molecule (Scheme 73).
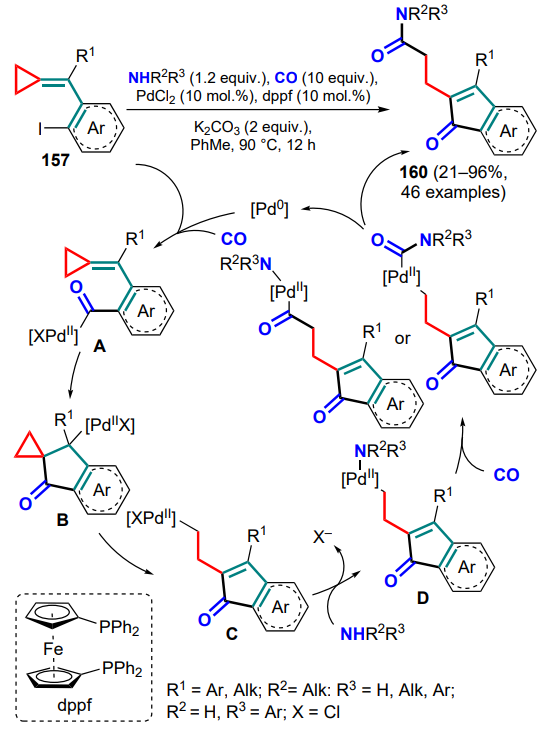
Instead of the ortho-positioned amino group in the aryl moiety of substrate 161, there is an azide functionality which serves as a source of nitrene capable of initiating cyclization.[145] As a result, a rhodium(II)-catalyzed process takes place, in which the initially formed nitrene intermediate A (or A') attacks the exomethylene bond of complex B (B') with subsequent opening of the cyclopropane ring (C or C') and closing of the four-membered ring at the nitrogen atom by a concerted 1,2-alkyl shift (Scheme 74). The resulting azetidine 162 is thermodynamically preferable over the possible cyclization product at the position 3 of the indole unit.
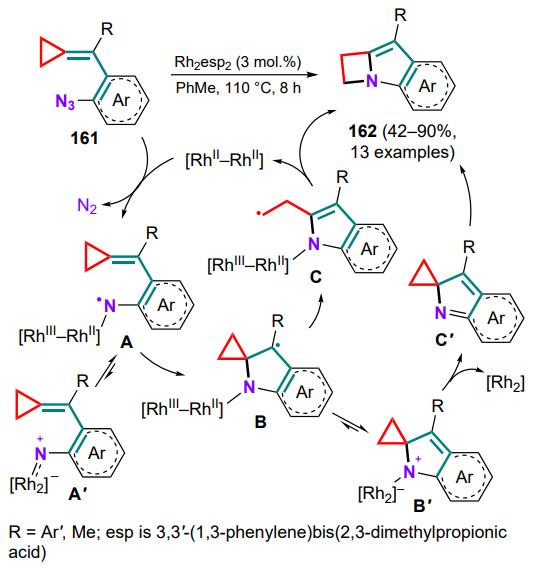
In the course of further studies, the authors also demonstrated the crucial role of rhodium catalysis in the first stage of nitrene A(A') formation. However, at later stages, its impact on the process diminishes, making the formation of azetidine more likely after removal of the catalytic species[146]. However, a competing process is the formation of a radical intermediate, which was observed in experiments with a radical trap.
The ester group in the starting (arylmethylidene)cyclopropane 163 can be converted in situ into an amide group which, after deprotonation with a strong base, acts as an initiator of anionic intramolecular cyclization to form substituted isoquinolinones 164. [147] This reaction can run directly with aromatic amines but is difficult for aliphatic derivatives, although in the latter case cyclization of amides 163' prepared in advance is possible under the same conditions (Scheme 75).
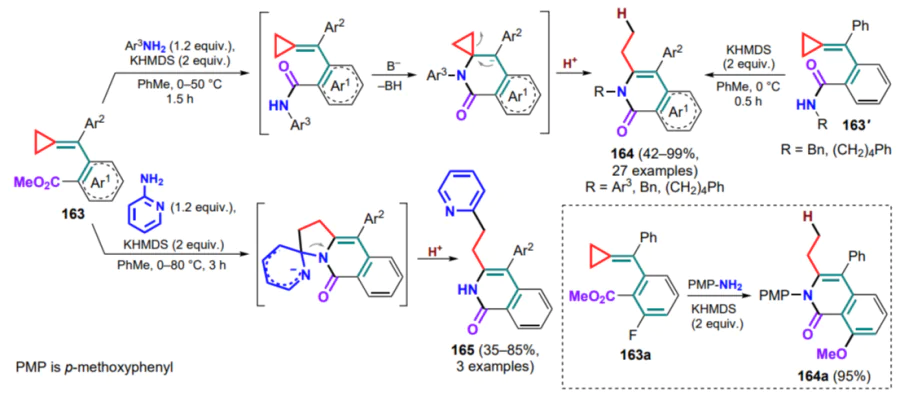
The reaction with 2-aminopyridine proceeds with migration of the pyridine ring and formation of dihydroisoquinones 165 via the intermediate formation of the Meisenheimer complex. It should also be noted that under these conditions, the orthofluoro derivative 163a undergoes an aromatic nucleophilic substitution of the fluorine atom on the in situ generated methoxy anion to give methoxyisoquinolinone 164a in almost quantitative yield (see Scheme 75).
The idea of cyclization yielding an anionic intermediate capable of further nucleophilic annulation was developed in a follow-up study.[148] The authors used Weinreb amides 166 modified to Michael acceptors 167 upon treatment with ethynyl Grignard reagents. A further acetylenide equivalent was attached to the Michael acceptor formed in situ, triggering a cascade cyclization that proceeded through attack of the double bond, opening of the three-membered ring and nucleophilic attack of the newly formed Michael acceptors. As a result, an aqueous work-up led to substituted naphthols 168 but these proved to be unstable under normal conditions. The authors then treated the reaction mixture with acetic anhydride and isolated acetyl naphthols 169 (Scheme 76).
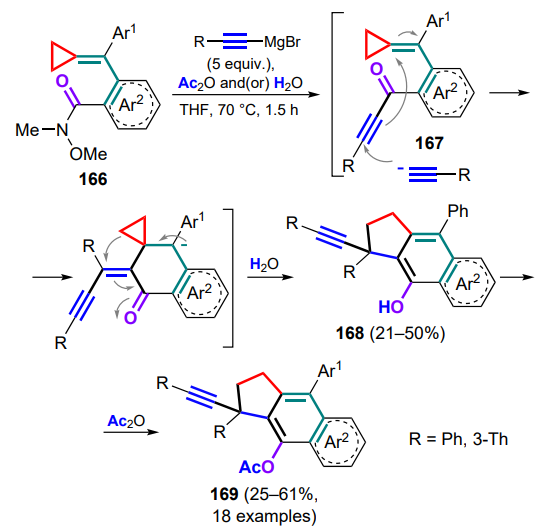
(Arylmethylidene)cyclopropanes bearing an ortho-positioned ethynyl moiety attached by a linker to the benzene ring are capable of a number of unusual intramolecular rearrangements. Such a process can be exemplified by the gold(I)-catalyzed transformation of ortho-propargyloxy-tethered (arylmethylidene)cyclopropanes 170[149]. For these substrates, a strong effect of the substituent nature on the cyclization pattern was found. Thus, compounds 170 with a terminal triple bond (R1 = H) and bulky substituents R2 (tert-butyl or tert-amyl group) in the ortho-position to the propargyloxy moiety undergo a non-standard rearrangement with migration of the methylidenecyclopropane unit (intermediates A–C) to give substituted benzopyrans 171, which is the first such gold(I)-catalyzed process. If the benzene ring contains a halogen atom, alkyl or methoxyl group instead of these substituents, the reaction proceeds under the same conditions via the threemembered ring-expansion to a four-membered one with the participation of the C=C bond (D). The subsequent attack of the carbocationic centre by an oxygen atom is accompanied by a [2,3]-sigmatropic rearrangement (E) to give 1-allenyl-2-oxabenzobicyclo[3.2.0]heptanes 172. Moreover, substitution of the triple bond with an alkyl or aromatic substituent also promotes the second-type cyclization. This was even true for the phenylethynyl substrate bearing a tert-butyl-substituted aryl moiety. Attempts at cyclization of substrates containing an ester or a TIPS functionality at the triple bond failed. The cyclization leading to the allene derivatives (R,R)-172 was additionally modified and carried out in an enantioselective mode using the chiral ligand L8 (Scheme 77).
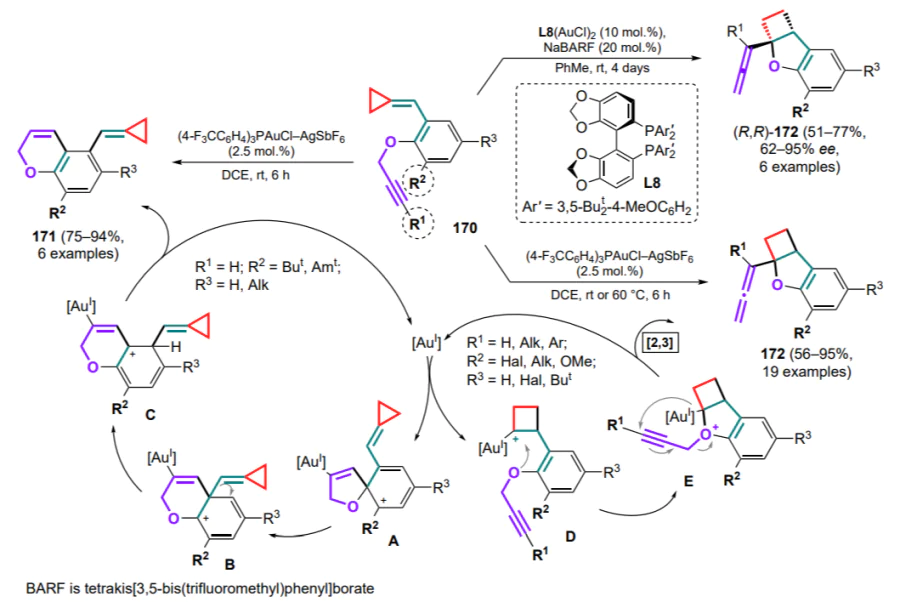
N-propargyl-N-tosylamino derivatives 173 can undergo two types of cyclization upon gold(I) or silver(I) catalysis.[150] In the first case, during AuI-catalyzed addition and cyclization via the exomethylene bond, the resulting intermediate A undergoes migration of the three-membered ring through a series of rearrangements (intermediates B, C), followed by cyclopropane ring-opening (D, E) with ring-closure to cyclobutene 174 (Scheme 78).
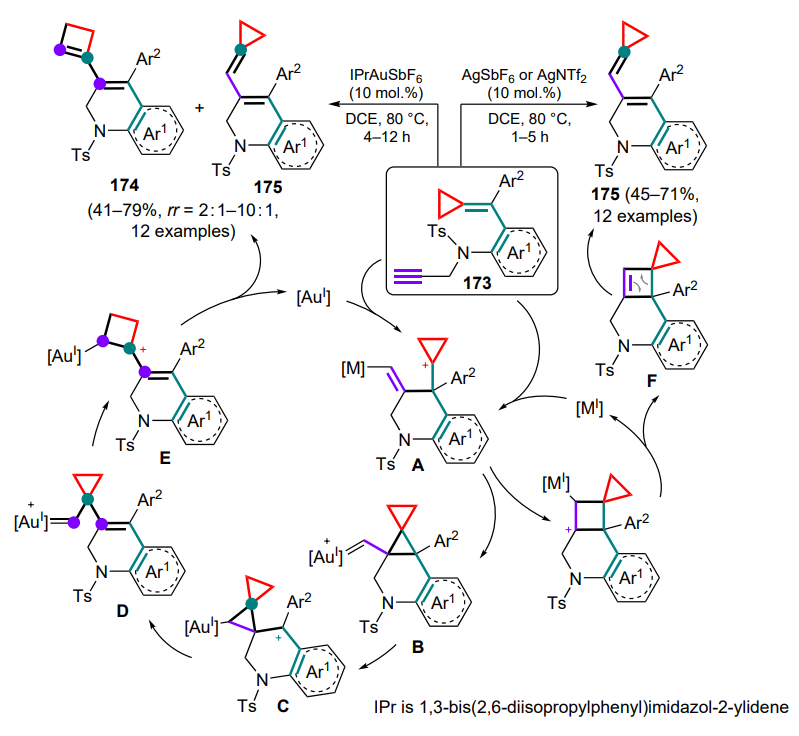
By-product 175, isolated in this reaction, could become the major product when switching to the AgI catalytic system. In this case, the initially formed carbocationic intermediate A is transformed into substituted cyclobutene F, which undergoes ring-opening to afford compound 175, a product of formal transfer of the cyclopropyl moiety.
Substrates of similar structure were used in a similar process, in which the tosylaminomethyl linker connected two conjugated systems, arylalkene and arylalkyne, each of which was involved in the transformation[151]. It was shown that cyclizations of different complexities can occur in the presence of AuI catalysts. First, the substrate undergoes 7-exo-dig cyclization with an exomethylene bond attack on the alkyne moiety activated by the catalytic complex A, then an intermediate B with a stabilized carbocationic centre is formed and then a ring expansion from a three-membered to a four-membered ring occurs. Intermediate C can react via two pathways, depending on the nature of the catalytic system. Path a involves deprotonation to form a cyclobutene moiety and the combination the conjugated systems into one in the product 177. Path b includes a Friedel–Crafts annulation involving the aryl moiety Ar2, leading to the formation of pentacyclic product 178 (Scheme 79).
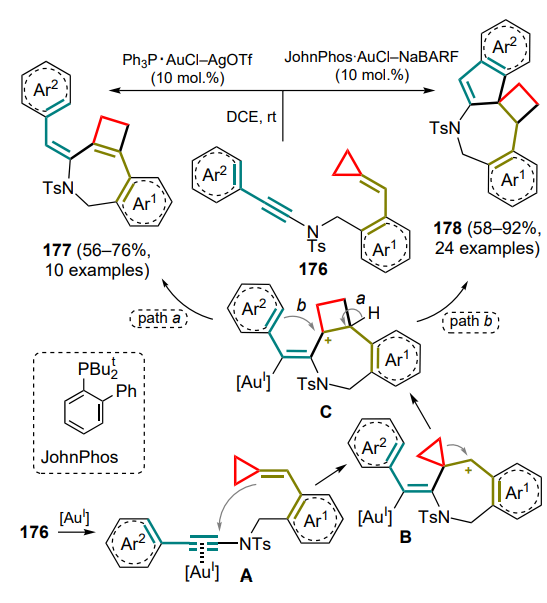
2.6.3. Cyclizations based on (о-ethynylarylmethylidene) cyclopropanes
The idea of using substituents in the aryl moiety to construct rings was continued in the cyclization of (arylmethylidene) cyclopropanes 179, which contain the ortho-positioned triple bond in the aryl substituent and thus have an elongated linear conjugated system.
The study of Zhang and co-workers[152] is considered to be a pioneering publication in this field, in which a series of cyclopenta[a]indenes 180 were obtained from the corresponding substrates through the Ni0-catalyzed process. The catalyst species is inserted into the proximal bond of the three-membered ring, followed by closure of the six-membered nickelacycle involving the triple bond. The reaction is completed by the reductive elimination and formation of the target product 180 (Scheme 80).

The next step in the study of the cyclization of (ethynylarylmethylidene)cyclopropanes 179 was the development of gold(I)- catalyzed processes[153]. It was shown that a tandem process involving additional oxidation by 3,5-dibromopyridine-N-oxide 181 and leading to naphthylacetaldehydes 182 could be implemented (Scheme 81).
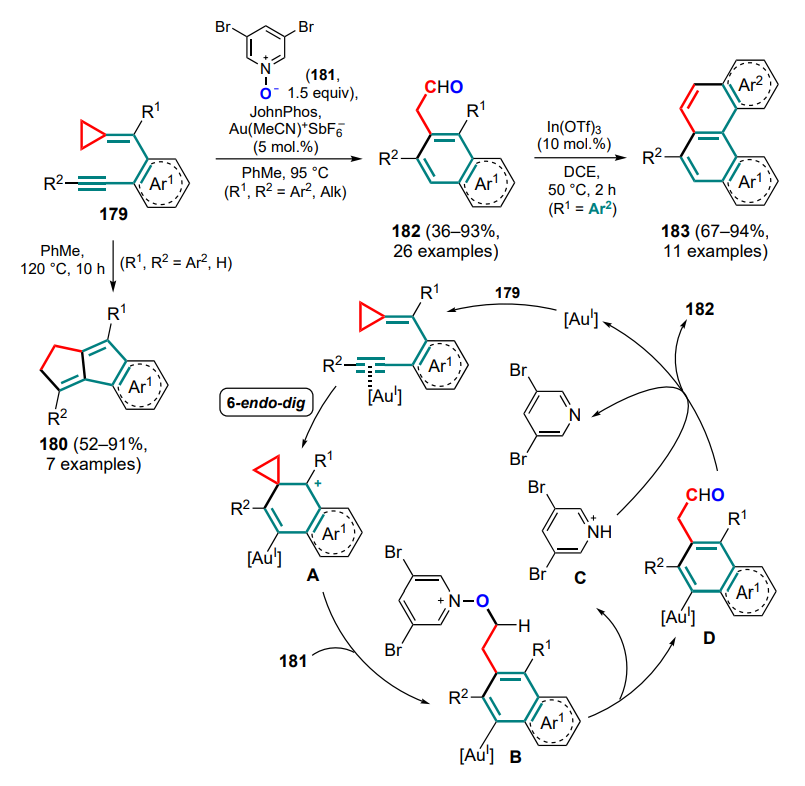
Once generated, π-complex of catalytic species of AuI and substrate undergoes a 6-endo-dig cyclization. In this case, a stabilized carbocationic intermediate A is formed, which is further attacked by an oxidant molecule with proximal opening of the three-membered ring (intermediate B) and subsequent protonation (C) and protodeauration (D) processes affording the final product 182. In addition, indium(III) triflate-catalyzed cyclization of the adjacent aryl moiety can be carried out to produce a series of polycyclic aromatic compounds 183; the two steps of this process can be combined in a one-pot procedure. It is also shown that the starting substrates are cyclized to cyclopenta[a]indenes 180 on heating in the absence of oxidant and AuI catalyst.
Another rather complicated series of transformations follows a completely different pathway for the modified substrate 179 (R1 = H, R2 = Ar2) in the absence of an oxidant and using ButBrettPhos (2-(di-tert-butylphosphino)-2',4',6'-triisopropyl3,6-dimethoxy-1,1'-biphenyl) as the ligand in the AuI catalyst[154]. In this case, 5-endo-dig cyclization occurs with further deprotonation and formation of the cyclopropane intermediate A. Repeated protonation and reactivation of the catalytic species on the exomethylene bond leads to a distal cleavage of the threemembered ring, yielding an allylic carbocation centre (B), which is cyclized on the adjacent aryl moiety (C). Subsequent deprotonation, isomerization, removal of the catalytic species and 1,3-hydride shift give rise to a series of substituted fluorenes 184 (Scheme 82).
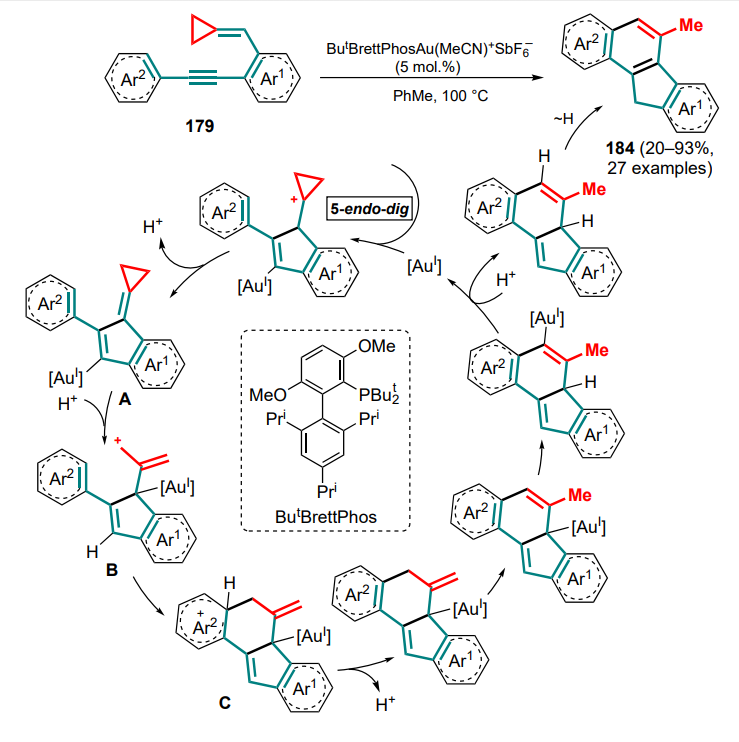
Subsequently, the acetylene moiety of the conjugated system was complicated by the introduction of an alcohol group in the propargyl position of the substrate 185, which opened access to naphtho[2,3-c]pyrans 186 (Scheme 83)[155]. It is shown that this transformation is promoted by the AuI catalyst, and it is also noted that the important role of the substituent at the exomethylene bond of the substrate determines the fundamental possibility of the reaction. The authors also suggested that steric interactions between the tert-butyl groups of the phosphine ligand and the alcoholic moiety of the substrate, which facilitate 6-endo-dig cyclization, are an important factor in the choice of transformation pathway.
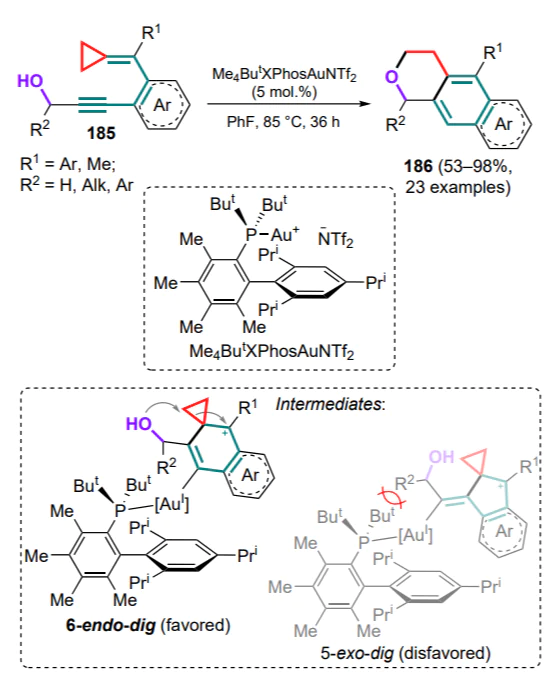
Based on similar substrates 185' containing a long alkanol chain at the triple bond of (o-ethynylarylmethylidene) cyclopropanes, Shi and co-workers[156] proposed a method for the synthesis of naphthalene-fused 8- to 11-membered macrocycles (Scheme 84). In contrast to the previous study, the reaction conditions involving the gold(I)-based catalytic system could be optimized only for substrates not substituted at the double bond. Attempted cyclizations using molecules with a short linker (n = 2) and a tosylamino group instead of a hydroxyl group, as well as compounds containing an oxopropanol group in the propargyl moiety, also failed. The mechanism initially proposed by the authors was complemented by the results of density functional theory (DFT) calculations. It was shown that the formation of the six-membered carbocationic intermediate B is preceded by a carbene intermediate A containing a strained spiro[2.2]pentane moiety. This pathway is indirectly confirmed by one of the reaction products, compound 188, derived from the octynol derivative (n = 6, Ar = 3-ClC6H4).
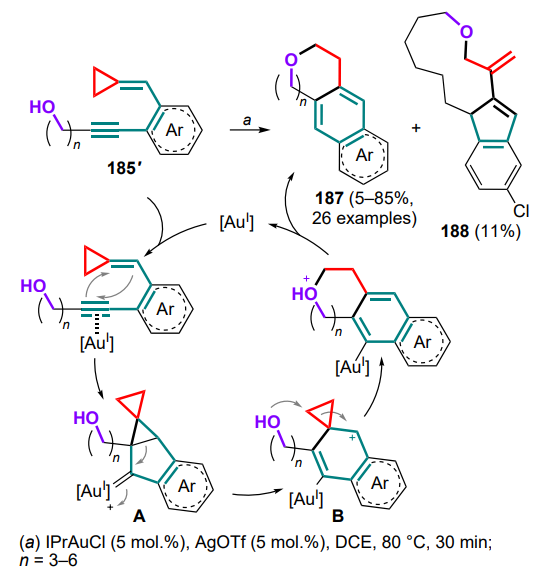
Wu and co-workers[157] studied the reaction of (o-ethynylarylmethylidene)cyclopropanes 179 with sulfonyl azides catalyzed by copper(I) iodide. The process did not stop at the standard azide-alkyne cycloaddition, but proceeded with the elimination of the nitrogen molecule and the formation of the ketenimine 189. The latter then underwent 6π-electrolysis (6πEC) and vinylcyclopropane rearrangement (VCP-RA) involving the (arylmethylidene)cyclopropane moiety (Scheme 85). As a result, a series of substituted indolines 190 have been obtained from a wide range of substrates.
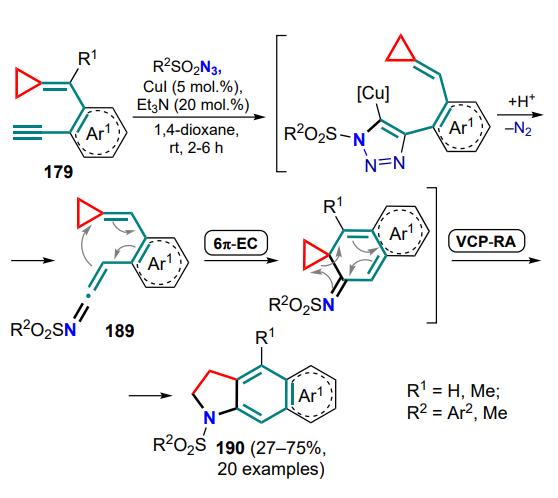
It is also worth noting the rhodium-catalyzed reactions of ortho-sulfotriazole derivatives of (arylmethylidene) cyclopropanes 191[158]. As a result of such transformations, an unusual Diels-Alder adduct, dihydrocyclobuta[a]indene 192, was obtained. This product is the result of the rhodium(0)-catalyzed formation of carbenoid A, which undergoes an intramolecular rearrangement (Scheme 86, path a). However, for a number of substrates the rearrangement can follow a different path (see Scheme 86, path b), leading to azacyclic compounds 193. It is noteworthy that for thienyl and cyclohexene derivatives, even in the presence of a catalyst, the abovementioned thermal rearrangement, giving indoles 190, remains preferable.
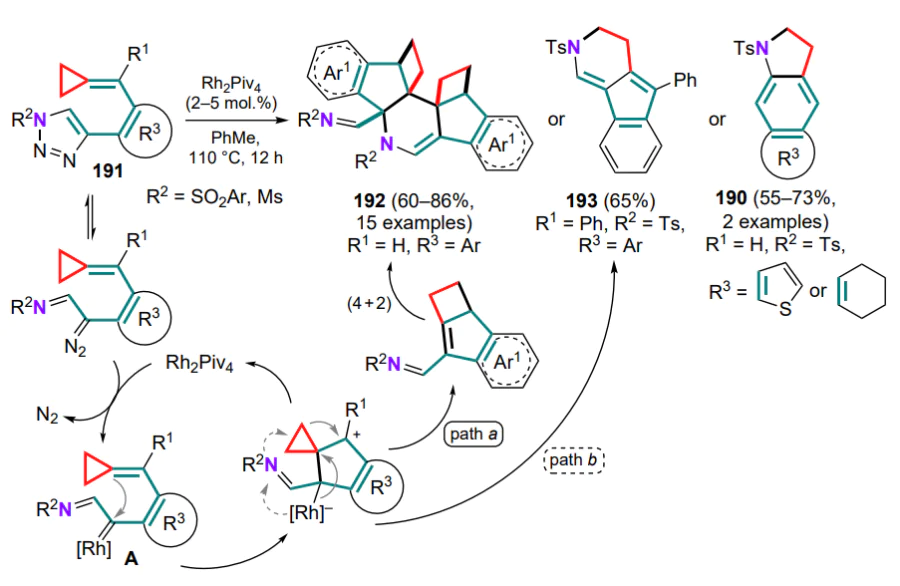
The 6π-EC+VCP-RA reaction sequence can be combined with the aza-Claisen rearrangement. Wu and co-workers[159] generated the intermediates required for these transformations in situ by a copper-catalyzed Ullmann reaction involving 1-bromoethynyl-2-(cyclopropylidenemethyl)arenes (194) and N-allyl-sulfonamides (195a). This method opens access to 9-allyl substituted 2,3-dihydro-1H-benzo[f]indoles 196 (Scheme 87).
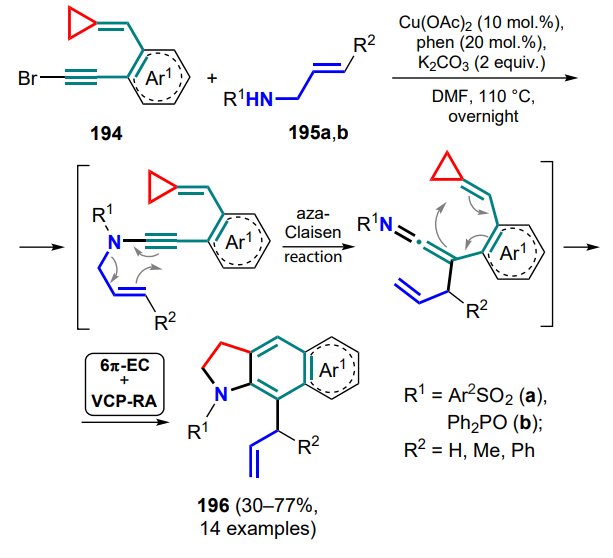
It is shown that a similar reaction can proceed with moderate product yields and with N-allylphosphonamides 195b, but not with N-allylamides and allylnosylamines.
The concept of 6π-EC+VCP-RA, which implies the possibility of the formation of a 1,2-diene intermediate from the starting (o-ethynylarylmethylidene)cyclopropane 179, has been developed in a number of papers. Thus, Shi and co-workers[160] carried out a CuI -catalyzed reaction using diazo compounds as sources of carbenes, which added with acetylenes to give allenes 197, resulting in a series of cyclopenta[b]naphthalenes 198 by the mechanism described above (Scheme 88). It is found that this process most readily proceeds for diazo compounds containing both aryl and ester substituents.
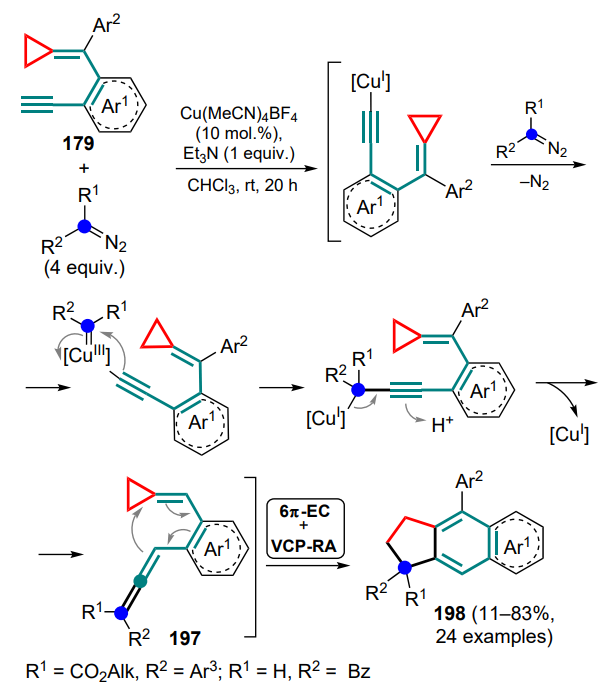
In the follow-up study[161], an allene moiety was constructed in substituted propargyl alcohols 185 treated with mesyl chloride in the presence of a base. It should be noted that all reactions were transition metal-free. Used in this process, mesyl chloride not only facilitated the cleavage of the C–O bond through the formation of a good leaving group, but also served as a source of chloride anion in the generation of chloroallene 199. The allene unit further acts as a substituent in the resulting naphthalene 200, which may be promising in terms of the subsequent functionalization of the product (Scheme 89).
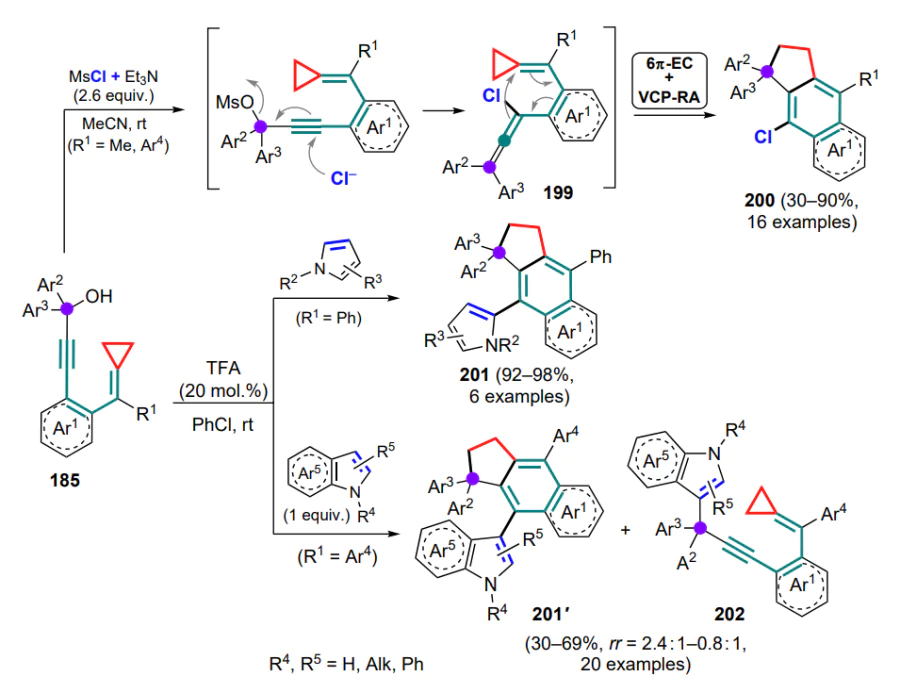
Brønsted or Lewis acid-catalyzed reactions of propargyl alcohols 185 with indole and pyrrole derivatives proceeded in a similar way[162]. The process was most effective using pyrroles, which produced substituted cyclopenta[b]naphthalenes 201 in yields up to 98%, whereas reactions with indoles often gave acyclic compounds 202 as products of conventional alkylation (see Scheme 89).
The same substrates 185 showed a different reactivity when treated with iodine[163]. In this case, the addition to the methylidenecyclopropane moiety was accompanied by intramolecular cyclization yielding the allene-substituted carbocationic intermediate A. Furthermore, the iodonium cation B was generated, which underwent an intramolecular electrophilic aromatic substitution to give the spirobiindene structure 203 (Scheme 90).
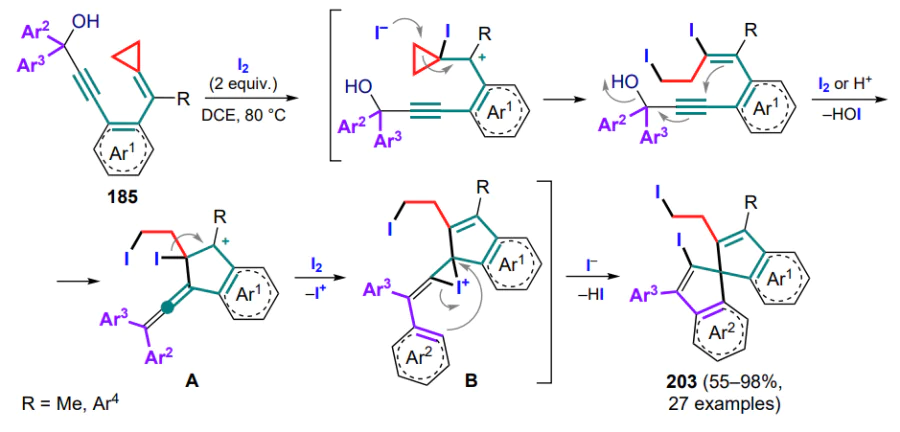
Finally, the 6π-EC+VCP-RA concept was implemented in a CuI-catalyzed three-component reaction for ortho-1,3-enynesubstituted (arylmethylidene)cyclopropane 204 using Togni reagent I and trimethylsilyl cyanide[164]. The resulting trifluoromethyl radical attacks the double bond of the enyne moiety to form a substituted allenyl radical A. This radical then reacts with the LCuIICN complex formed by the reaction between the starting CuI complex and trimethylsilyl cyanide, after which the reductive elimination of copper occurs and a cyanoallene fragment is emerged in intermediate B. The latter can undergo the 6π-EC+VCP-RA cascade reaction, furnishing the corresponding cyclopenta[b]naphthalenes 205 (Scheme 91). The isolation of a small amount of by-product 206 was explained by the complex formation of a copper with the product of 6π-electrolysis and its reaction with a ligand acting as a base.
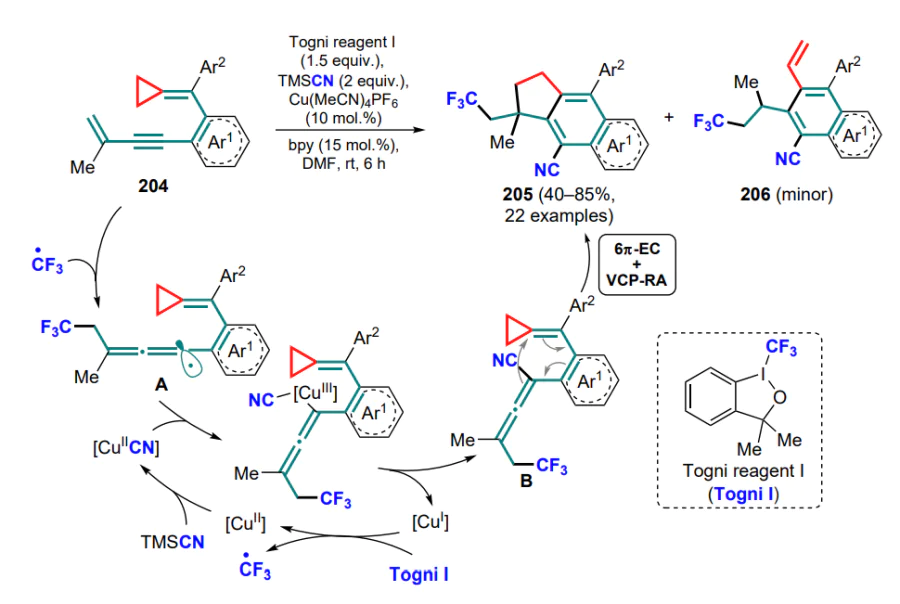
2.6.4. Cyclizations of (diarylmethylidene)cyclopropanes involving both aryl substituents
Shi and co-workers[165-172] proposed a complicated tandem process based on the cyclization of (diarylmethylidene) cyclopropanes 207. An amide or ester-spaced terminal double bond was introduced in the ortho-position of one of the aryl fragments. In this case, the radical species attacks along the less hindered C=C bond to generate a radical intermediate, which further intramolecularly attacks the sterically more loaded double bond of the three-membered ring with subsequent cyclization via the above mechanism (see Section 2.6). It may be noted that the cyclization formally involves a cyclopropanetethered cross-conjugated unit.
In the first of these studies[165], the trifluoromethyl radical generated by the Togni reagent II acted as a cyclization initiator. This gave rise to a number of polycyclic structures 208, however, no reaction was observed for N-tosyl-substituted substrates. In the case of a methoxy-substituted aryl substituent having no tethered terminal double bond, the final cyclization proceeded in an unusual manner at the ipso position, delivering a number of spiro-fused polycyclic derivatives 209 (Scheme 92).
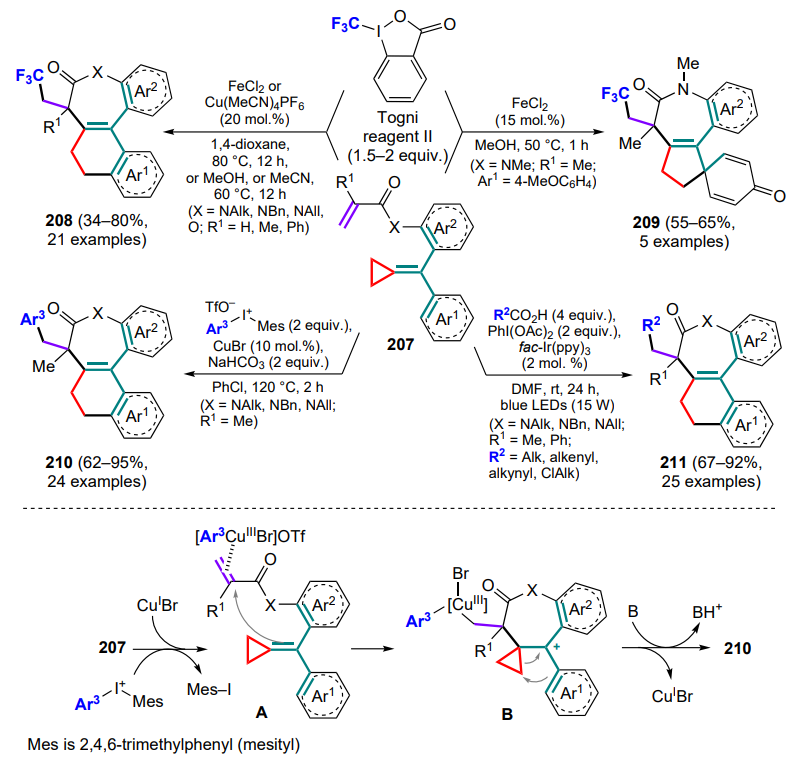
Later, the same strategy was used to promote the reaction with copper(I) bromide using aryliodonium salts, thus providing compounds 210[166]. The authors proposed that a reactive copper(III) complex is first formed with the aryl substituent Ar3 of the iodonium reagent. This complex then coordinates to the least hindered double bond (intermediate A) and initiates the closure of the seven-membered ring to give the cationic intermediate B, which undergoes further cyclization with the elimination of the catalytic particle — copper(I) bromide — during reductive elimination (see Scheme 92, transformation mechanism). As in the previous study, N-tosyl derivatives did not react under these conditions, isopropylphenyl-substituted substrate gave a complex mixture of compounds, and at R1 = Ph, only intramolecular Friedel–Crafts alkylation took place.
A photochemical variant of this cyclization involving a large number of aliphatic acids has also been proposed, which is used in the decarboxylative generation of radicals in the presence of di(acetoxy)iodobenzene and an iridium catalyst[167]. A series of substituted benzazepines 211 were obtained in high yields (see Scheme 92). Despite some good results, the authors noted that this process practically fails with ortho-substituted Ar1-group substrates and with cyclohex-3-encarboxylic acid; the use of 3,3,3-trifluoropropionic acid led to a complex mixture of products.
Tandem radical addition/arylation can also be carried out in an intramolecular mode. For this purpose, (diarylmethylidene) cyclopropanes containing ortho-positioned radical-generating groups in one of the aryl moieties have been studied. For example, the nickel(II)-catalyzed cyclization of bromoalkyl derivatives of (diarylmethylidene)cyclopropanes 212 has been proposed[168]. In the presence of zinc, the metal is reduced to Ni0 in complex species, which then undergo single electron transfer reactions, being oxidized to NiI complexes, which can then be reduced back to Ni0 either under the action of zinc or via SET. Under these conditions, a series of benzo[b]naphtho[1,2-d] azepines 213 were obtained (Scheme 93); it was found that in the presence of ortho substituents on the aryl fragment free of the bromoalkyl chain, complex mixtures of products are formed. It was also shown that a tosyl or mesyl group can be used as a protecting group for the nitrogen atom in the bromoalkyl linker, but not a nosyl group, which does not favour the reaction.
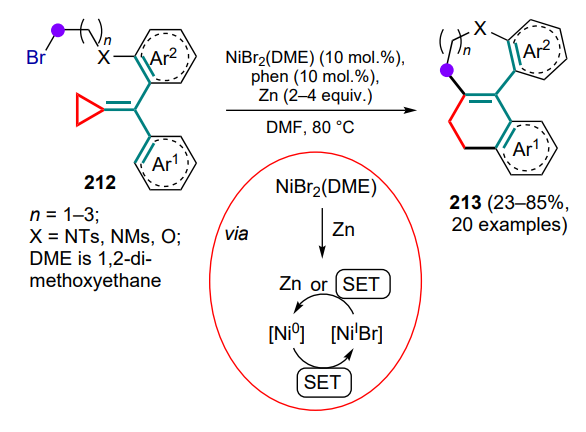
Further studies were carried out on substrates 214 with amide and ester linkers in the bromoalkyl chain. For these, a photocatalytic modification of the process has been developed[112]. The presence of Ir(ppy)3 as a photocatalyst opened access to a series of polycyclic compounds 215 containing seven-, eight- and nine-membered rings (Scheme 94).
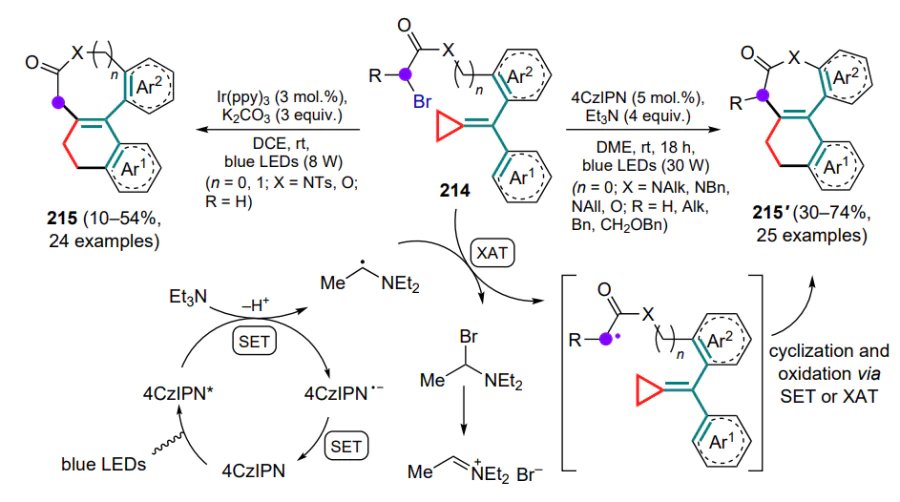
Finally, in 2022, the reaction was optimized for an organophotocatalytic version using 4CzIPN as a photocatalyst[169]. The key feature of this process is the generation of the initial radical site by halogen atom transfer (XAT) under the action of an α-aminoalkyl radical derived from triethylamine under photocatalytic conditions. Further radical intermediates undergo cyclization and oxidation (see Scheme 94) during SET or XAT to form polycyclic compounds 215' (n = 0). Naphthyl and orthosubstituted aryl moieties in the starting (diarylmethylidene) cyclopropanes hamper the conversion. The effect of the R fragment in the resulting radical site on the efficiency of the reaction has been studied on a wide range of examples and it has been shown that isopropyl and aryl groups inhibit the reaction.
For a number of aniline derivatives 216, the possibility of formation of nitrogen-centred radicals in the presence of silver(I) acetate as oxidant and subsequent cyclization by the standard mechanism was demonstrated[170]. A series of substituted [2,3-c]-dihydrocarbazoles 217 were obtained; it was shown that the corresponding carbazoles 217' and seven-membered heterocycles 217'' could be formed as by-products as a result of cyclization through the N-tosyl protecting group. Based on the process studied, a short (three-step) method for the synthesis of natural alkaloid eustifoline D was proposed with an overall yield of 66% (Scheme 95).
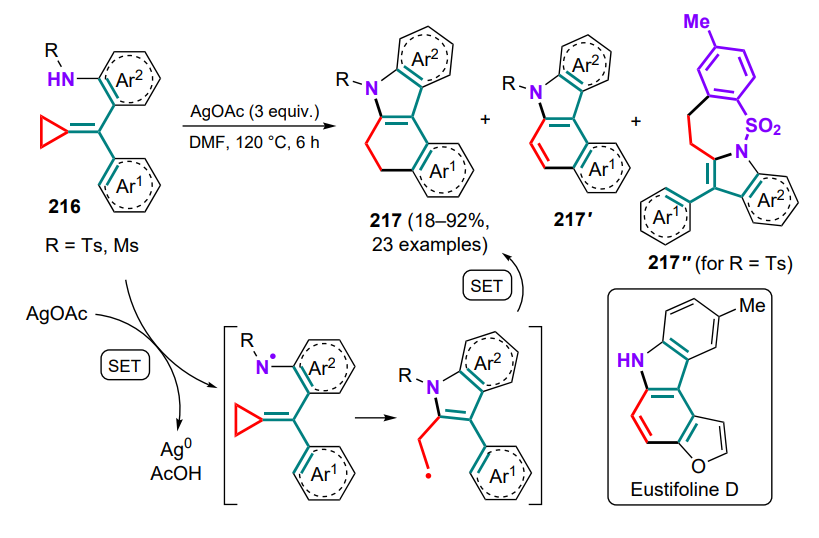
An unusual example of cyclization of (diarylmethylidene)-cyclopropanes 218 bearing an ortho-positioned o-bromaryl substituent in one of the aryl moieties is a process based on the intramolecular Heck-type reaction[171]. After oxidative insertion of a catalytic species into the C–Br bond, carbopalladation on the exomethylene bond proceeds to form the cyclopropylpalladium intermediate A. Next, a cascade of β-hydrideelimination, hydropalladation and repeated β-hydrideelimination occurs to give the cyclopropene intermediate B. The latter undergoes oxidative insertion of a catalytic species to form palladacyclobutene C, which is ring-opened to palladium carbenoid D, which undergoes cyclization on the adjacent aryl moiety (Scheme 96). Intermediate E undergoes 1,3-migration of η3-allylpalladium, intramolecular 1,4-proton shift and elimination of the catalytic Pd0 species to afford the resulting spiro-substituted fluorenes 219. It was shown that sterically crowded ortho-substituted fragments in Ar1 can hinder the desired products, as well as the 2-thiophene substituent or incompletely protected amino groups (e.g. NHTs).
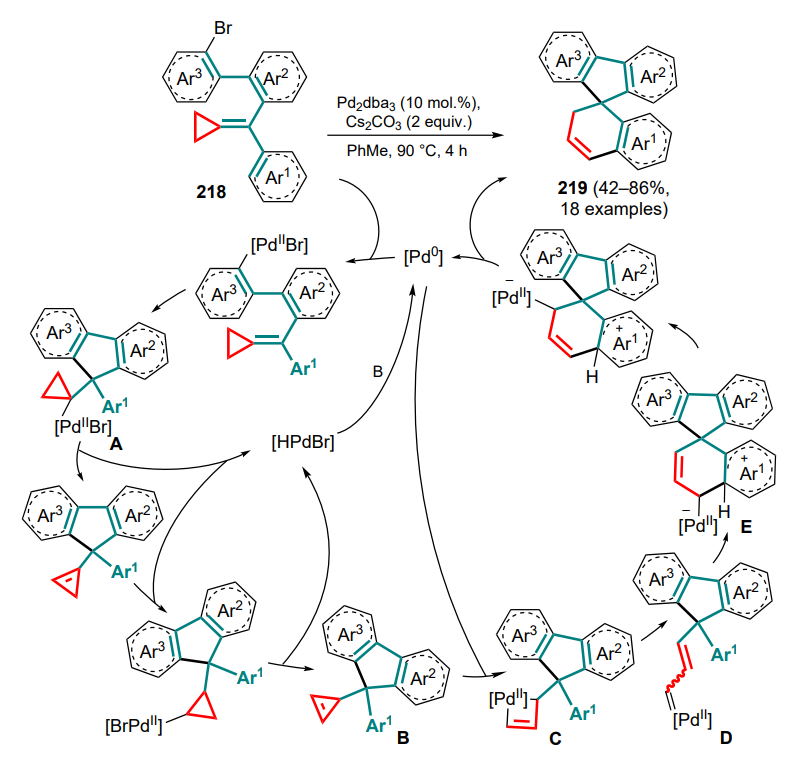
The authors later developed this approach by proposing an intermolecular variant involving the more readily available ortho-bromo-substituted (diarylmethylidene)cyclopropanes 220 and aromatic alkynes (Scheme 97)[172]. This modification allows an increase in the molecular complexity of the products compared to the previous procedure, although it has certain limitations. Thus, it has been shown that alkynes bearing electron-withdrawing and alkyl substituents, as well as terminal aryl alkynes under similar conditions, do not undergo the said reaction. It should also be noted that using prop-1-ynylbenzene led to a 6 : 1 mixture of regioisomers. The reaction mechanism is similar to the previous one, except for an additional step of alkyne insertion into the C–PdII bond (see Scheme 97).
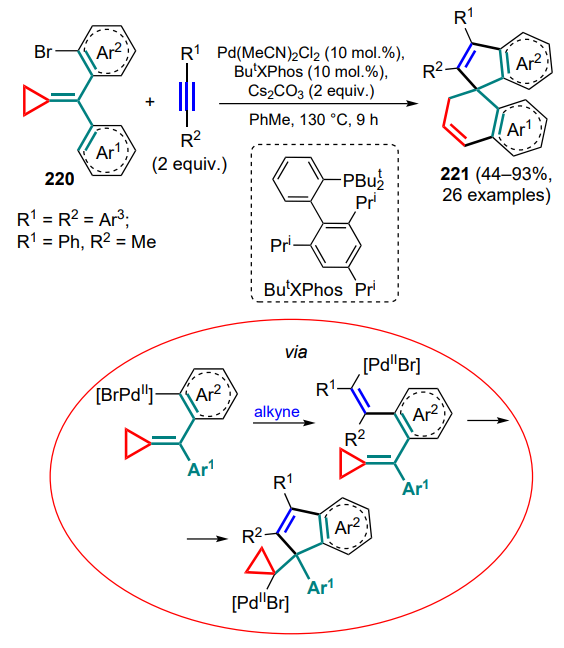
2.7. Reactions of (arylvinylidene)cyclopropanes
In 2005, Shi and co-workers[173] began a study on the rearrangements of (arylvinylidene)cyclopropanes 222 in the presence of Lewis or Brønsted acids. The authors showed that tin(II) triflate is an optimal catalyst for the preparation of the corresponding substituted naphthalenes 223 (Scheme 98). The reaction starts with the coordination of the acid catalyst species through a double bond to form a cyclopropylvinyl cation, followed by opening of the three-membered ring and subsequent intramolecular Friedel–Crafts cyclization. Catalyst release is accompanied by a series of proton shifts to yield the final product.
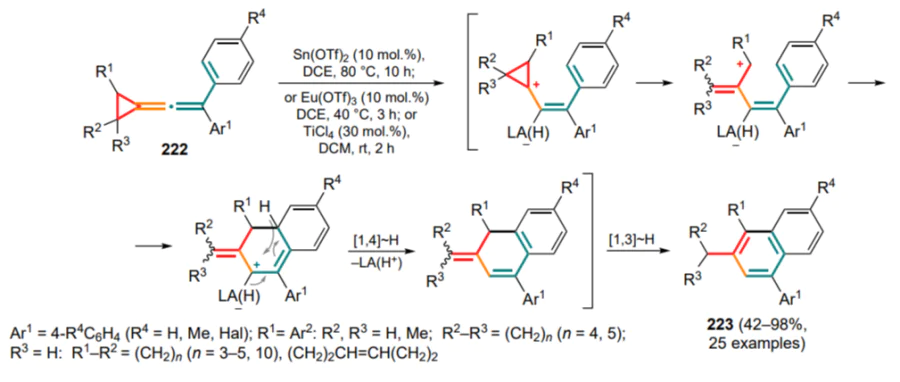
Subsequently, the authors expanded the range of substrates and also demonstrated the possibility of carrying out the reaction in the presence of europium(III) triflate as the catalyst, which allowed the temperature of the process to be reduced from 80 to 40 °C and its duration to be shortened[174].
It was later shown that the same approach could be implemented with titanium(IV) chloride to produce naphthaleneannulated medium-size carbocycles[175].
This methodology was extended to the tetramethylcyclopropyl derivative 224[176]. Using the gold(I) catalyst, the reaction provided good yields of isopropylidenedihydronaphthalenes 225 and 225' containing an exocyclic double bond (Scheme 99). The regioselectivity of the six-membered ring-closure in the presence of different aryl substituents was also explored.
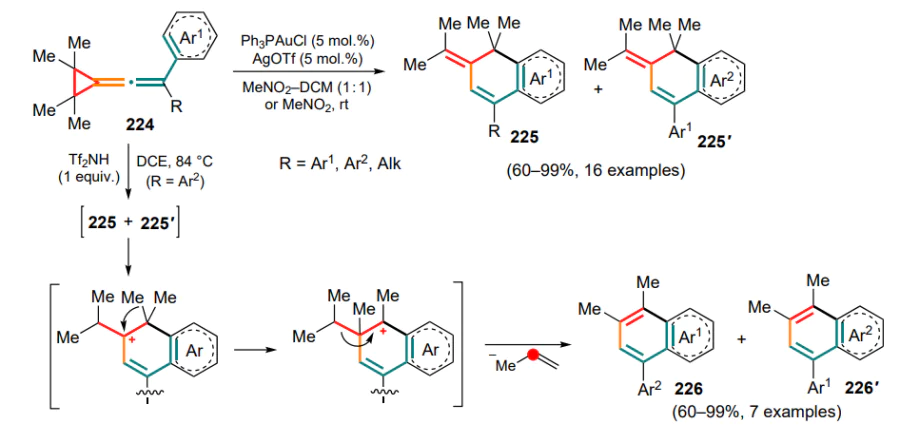
In the follow-up study[177], the authors carried out the reactions with the same substrates using different Brønsted acids. It was shown that in the presence of trifluoromethanesulfonimide intermediate, the isopropylidenedihydronaphthalenes 225 and 225' undergo protonation and 1,2-methyl shift, followed by elimination of propene to afford dimethyl-substituted naphthalenes 226 and 226' (see Scheme 99).
This idea was developed by Shi and co-workers[174, 178], who used similar substrates containing 1-alkyl-1-aryl substituted cyclopropane 227. In a similar transformation, the cationic centre is attacked by aryl moieties with additional diastereoselective closure of the five-membered ring, i.e., a cascade process delivering two C–C bonds and three stereocentres in one reaction step is realized to yield compounds 228 (Scheme 100). It is noteworthy that mixtures of cis and trans isomers of the starting (arylvinylidene)cyclopropanes can be used in the reaction without significantly affecting its diastereoselectivity. In addition, carbocycles 223, 228 were obtained in the presence of montmorillonite K-10[179].
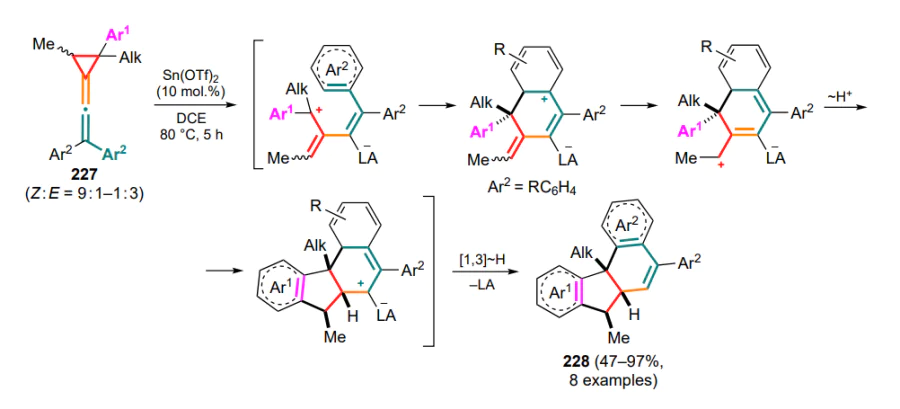
Li and Shi[180] also developed a cascade cyclization to obtain fused carbocycles 229 and 230 involving (tetramethylvinylidene) cyclopropanes 224 and substituted buta-1,2,3-trienes 231. The reaction is catalyzed by trifluoromethanesulphonimide, which protonates the initial butatriene, followed by an attack on the double bond, opening of the three-membered ring and further closure of cyclopentane (Scheme 101). The resulting cationic intermediate 232 can be attacked by adjacent aryl moieties of both the starting vinylidenecyclopropane 224 and the butatriene 231. This results in two different processes yielding two types of polycyclic esters 229 and 230 containing two different exocyclic double bonds.
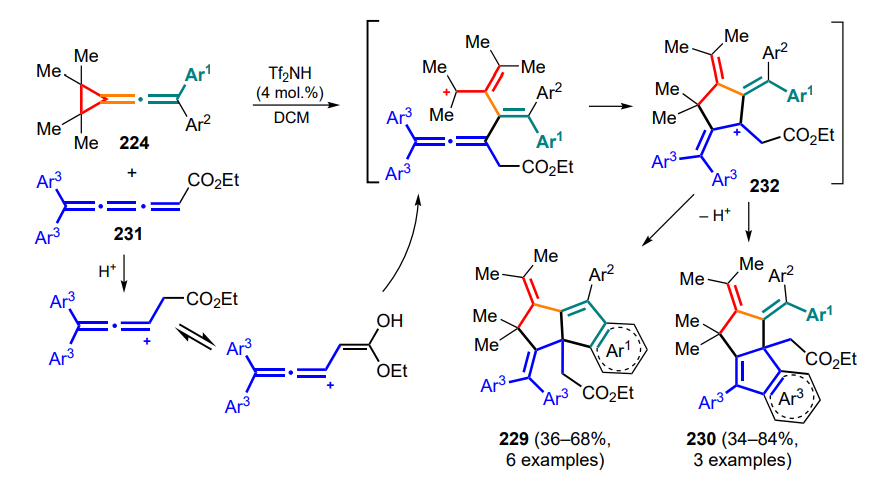
Su and Huang[181] explored the cyclization of vinylidenecyclopropanes 224 with aryl carbaldehydes in the presence of Lewis acids. The activated electrophilic complex of arylcarbaldehyde and Lewis acid A adds to the double bond of the vinylidenecyclopropane, followed by the cleavage of the distal C–C bond of the three-membered ring to generate the carbocationic intermediate B. Depending on the substituents in the three-membered ring, either nucleophilic attack of the arylcarbaldehyde moiety by an oxygen atom (intermediate C) (Scheme 102, path a) or intramolecular Friedel–Crafts alkylation of the aryl substituent (D) (path b) can proceed. In this way, a series of furans 233 and benzo[c]fluorenes 234 have been obtained from mono- and bicyclic starting vinylidenecyclopropanes, respectively.
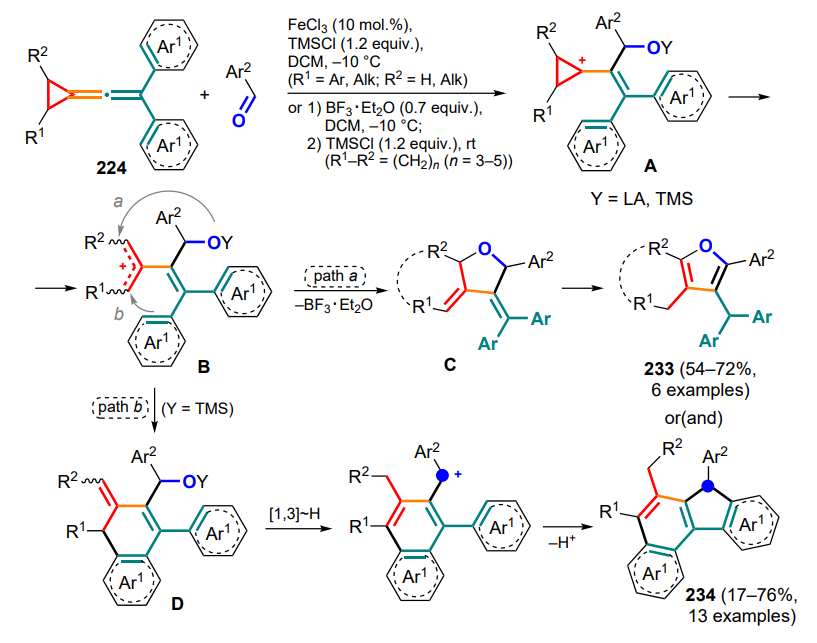
3. Reactions of cyclopropanes bearing nitrogen-containing conjugated systems
.
3.1. Reactions with 1-azadienes
As with diene substrates (see Section 2), the first methods for cyclization of cyclopropanes with full involvement of the conjugated heterodiene system comprised (4+3)-cycloaddition reactions of DAC to anthranils 235 that are aza-analogues of 1,3-diphenylisobenzofuran[182, 183]. This methodology is of particular interest for the atom-economic assembly of 1-benzazepines, a structural moiety found in a number of biologically active substances.
In a study[184], DAC underwent ring-opening under the action of scandium(III) triflate to generate 1,3-zwitterions, the carbocationic centre of which was attacked by an unshared electron pair of the anthranil nitrogen atom by the SN2 mechanism (Scheme 103). Subsequent aromatization afforded the zwitterionic intermediate 236 (shown in the Scheme for R1 = Ar, R3 = H). The authors suggest that the π – π interaction between aryl moieties promotes further selective annulation of rings to furnish endo-adducts 237. With the participation of S-enantio-enriched DAC (R1 = Ph, R2 = Me) under standard reaction conditions, compounds 237a,b were obtained with almost complete retention of the stereoconfiguration, indicating a high stereospecificity of the process.
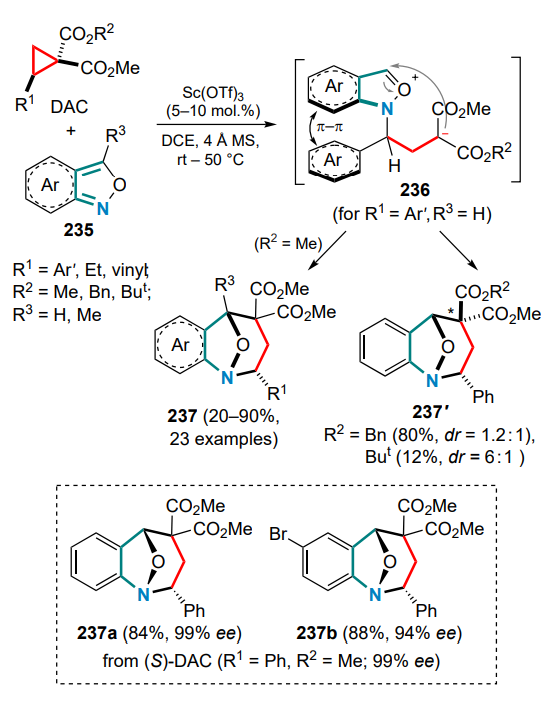
A similar protocol has been proposed using DAC 238, where the first step, similar to the previous example, gave the intermediate 236'[185]. The effect of the substituent at the C(3) atom of the starting anthranil 235 on the reaction course was also shown. In the absence of this substituent, the reaction proceeds as described above as a sequential (4+3)-cycloaddition to give the 1-benzazepine skeleton 239 (Scheme 104, path a). As in the previous case, the chiral product 239a having comparable optical purity was obtained from the enantiomerically enriched starting cyclopropane (–)-238. However, in the presence of a phenyl substituent, the intermediate 236' is fragmented to give imine A and 2-furanone B, which undergo a Povarov-type cyclization to give tetrahydroquinolines 240 (path b).
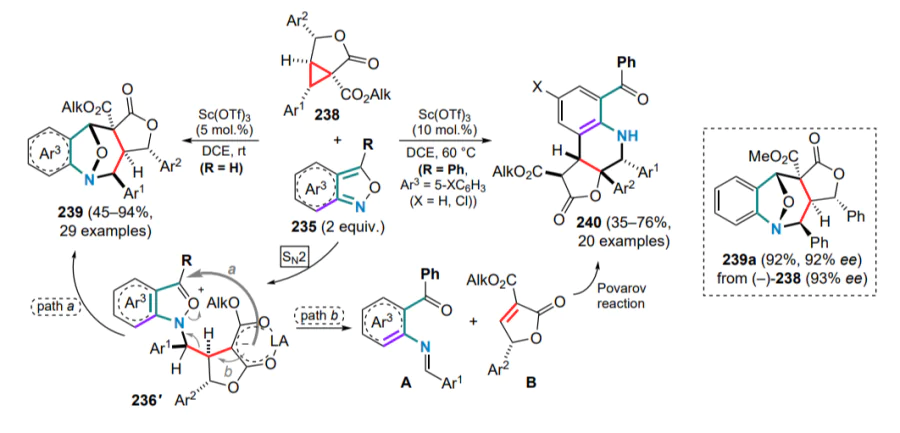
Vinylcyclopropanes are no exception, from which a coordinated open allylic zwitterion A can be generated using palladium catalysts in the presence of phosphine ligands L9 or (S)-L10 (Scheme 105)[186]. This intermediate has been successfully trapped by anthranil substrates 235a (R = H) in the presence of triethylboron, forming an activated adduct B. After addition of the palladium zwitterion A thereto, an intramolecular allylic cyclization occurs with release of vinyl-1-benzazepines 241 as well as catalytic species of Pd0 complex and triethylboron, closing the catalytic cycle. The reaction conditions were optimized for both diastereoselective and enantioselective modes; in the latter case, the chiral products 241' were isolated.

The reactions involving anthranils includes the transformation of cyclopropane intermediate A, generated from 1,5-enine 242, using gold(I) complexes as catalysts[187]. Depending on whether terminal or internal 1,5-enine 242 is used as the starting compound, two types of products can be formed — 243 or 244. In the first case (R1 = H), intermediate A can be deprotonated by the starting anthranil 235, which also acts as a base. Intermediate B undergoes ring-opening rearrangement to generate the dipole intermediate C, which adds an anthranil molecule (intermediate D) with subsequent elimination of the catalyst species. With the internal substrate 242 (R1 ≠ H), the cyclopropane intermediate A undergoes opening of the three-membered ring (E) and (4+3)-cycloaddition of an anthranil equivalent 235a with closing of the seven-membered ring and formation of the carbene intermediate F, followed by the addition of another anthranil equivalent and release of the catalytic species (Scheme 106). It was noted that the choice of the optimal anion for the catalyst changes from triflimide to hexafluoroantimonate when switching from terminal to internal alkyne.
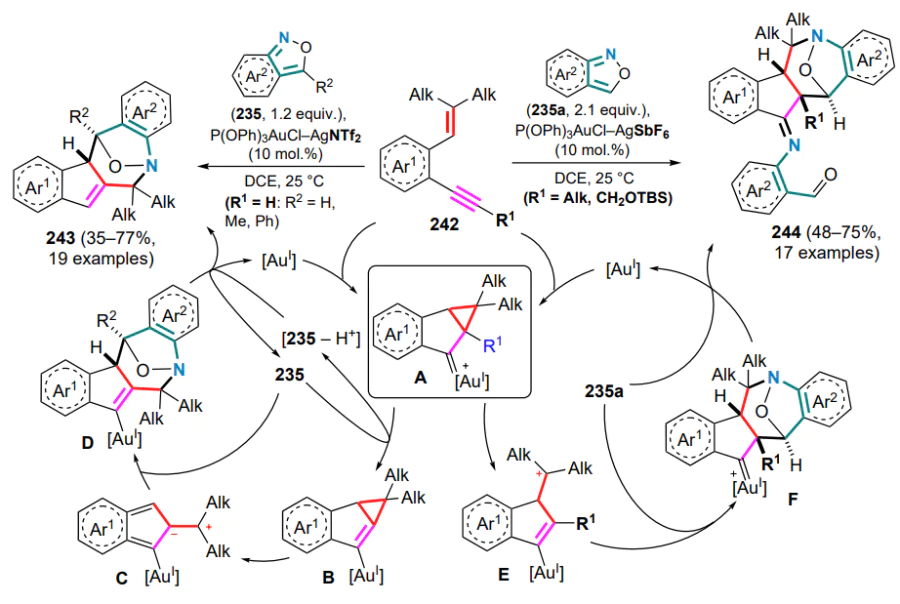
The next step in the development of this methodology was the reaction of cyclopropanes with acyclic 1-azadienes. Direct (4+3)-cycloaddition for such compounds is still unknown, but other strategies have been described that imply the full involvement of the azadiene system in the cyclization.
The first example, as with dienes (see Section 2.1 and Ref.[46]), was the stepwise insertion of the C=C and C=N bonds during the formation of a bicyclic system via a cascade of reactions: the classical (3+2)-cycloaddition of DAC 245 and azadiene 246 with the C=C bond and additional cyclization through the C=N bond involving an ester functionality[188]. Initially formed under alkaline catalysis conditions, the 1,3-zwitterionic intermediate undergoes deprotonation to give the anionic intermediate A, facilitated by the presence of an electron-withdrawing group. Next, (3+2)-cycloaddition takes place to yield the intermediate B, in which the ester function is reduced during proton migration. Cyclization of the final enamine intermediate C then gives the bicyclic product 247 (Scheme 107). A 4-nitrophenyl moiety was found to be the optimal substituent in the ester group distal from the threemembered ring. The electron-donating 4-methoxyphenyl substituent led to a sharp decrease in the yield of the target product, and with the tolyl analogue or the substrate bearing an aldehyde substituent, only side reactions took place under these conditions. At the same time, increasing the steric environment of the geminal ester groups (R = Ph) improved the diastereoselectivity of the process (dr reached 20:1). The antibacterial activity against the plant-pathogenic bacterial strains Xanthomonas oryzae pv. oryzae, X. axonopodis pv. citri и Pseudomonas syringae pv. actinidiae was studied for a series of bicyclic compounds 247.
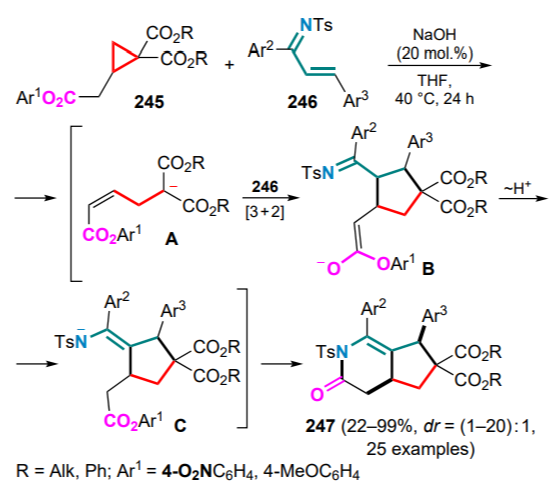
Reactions of vinylcyclopropanes with acyclic 1-azadienes — derivatives of benzofuran (248) or chalcone (249) — in the presence of a palladium catalyst can either follow the standard route of (3+2)-cycloaddition with the participation of a diphosphine ligand (dppe) to form 1-oxaspiro[4. 4]nonane 250 or, when dppe is replaced by phenanthroline, exhibit an unusual ability to react as 1,5-synthones and produce substituted ninemembered rings[189]. Of particular importance is the nature of the acceptor groups: for example, cyclopropanediesters give only (3+2)-addition products, whereas dinitrile derivatives can afford the target 6,9-dihydroazonin-5,5(4H)-dicarbonitriles 251 or 252 (Scheme 108). Switching from dppe to phenanthroline also prevents the transformation of the starting compounds into (3+2)-adducts. A slightly lower regioselectivity of the process was observed for the chalcone azadienes 249. Therefore, compared to cyclic azadienes, in these reactions not only the azadiene system but also the vinyl substituent of the cyclopropane was fully involved in the cyclization.
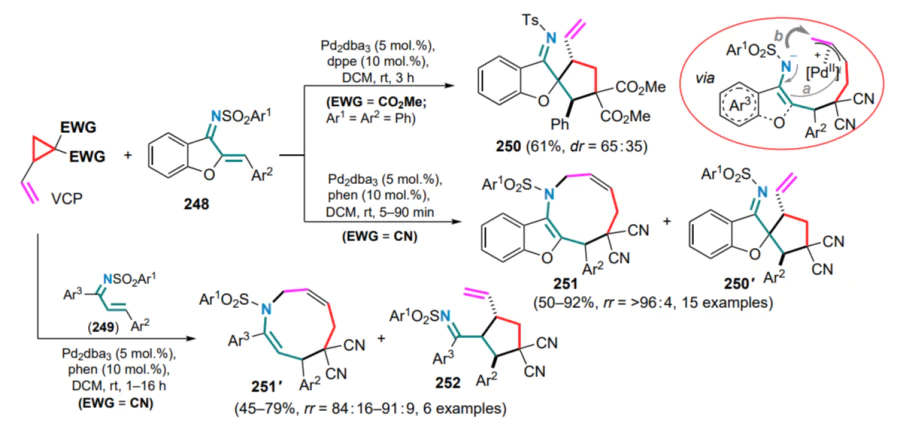
1-Azadiene can be obtained in situ from a suitable precursor substrate, such as indolyl alcohol 253, in the presence of Lewis acid (Scheme 109)[190]. This approach using DAC involves the cis-diastereoselective formation of substituted carbazoles 254, which is closely related to studies[51-53] (see Section 2.1). The nucleophilic attack of the intermediate 1,3-zwitterion A on the iminium intermediate B generates cation C, which is cyclized at the 3-position of the indole with subsequent rearrangement and deprotonation. Notably, 4-dimethylaminophenyl-substituted cyclopropane delivers carbazole 254 in the presence of scandium(III) triflate, whereas in the presence of indium(III) chloride, used under the standard conditions for this reaction, the pentacyclic product 255 is formed (see Scheme 109). The particular influence of the strong electron-donating group of the aryl substituent in DAC appears to be in the in situ formation of styryl malonate, which then undergoes the (3+2)-cycloaddition with the iminium intermediate B to give compound 255.
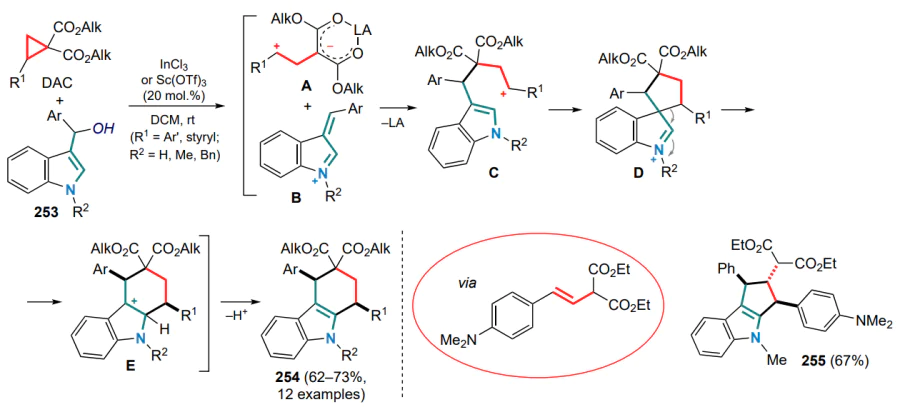
The study of the unusual reaction pathway of DAC associated with the in situ formation of isomeric styryl malonate was initiated earlier by Verma and Banerjee[191]. For a series of conformationally rigid 1-azadienes 256 and 256', it was shown that in the presence of magnesium iodide, DAC can be converted into a complex of MgI2 with styryl malonate, which undergoes a (4+2)-cycloaddition to give diastereoselectively trans,transindenotetrahydropyridines 257 and 257' (Scheme 110).
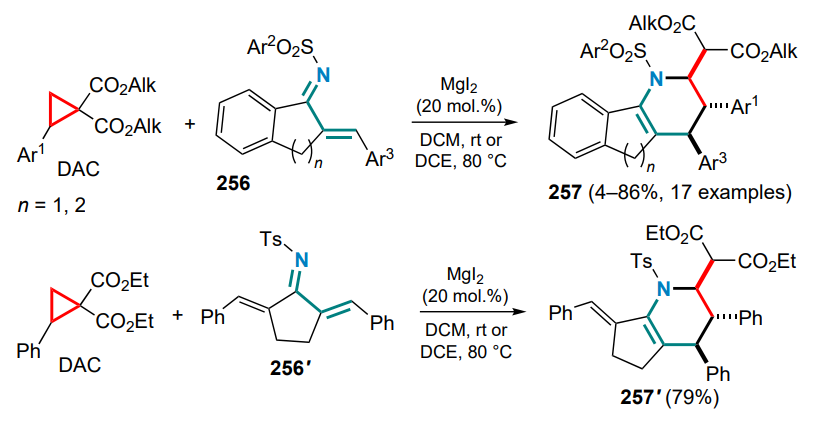
It was subsequently suggested that this approach should be extended to conformationally non-rigid 1-azadienes in order to broaden the scope of its application to the design of cyclic structures[192, 193]. By varying the reaction conditions between DAC and tosylimine of cinnamaldehyde (258a) under the action of Lewis acids, it was found that in most cases the major product is pyrrolidine 259 formed via the addition of 1,3-zwitterions to the C=N bond. Attempts to use tosylimine 258a to capture preformed 1,2-zwitterionic gallium complex 47 (see Section 2.2) also failed, apparently due to the change in stability of the complex in the presence of a stronger azadiene ligand. However, in a number of experiments, in particular when anhydrous GaCl3 was added to an equimolar mixture of DAC and azadiene 258a, the formation of products of formal capture of the intermediate styryl malonate 260a, obtained by (4+2)-cycloaddition (compound 261a) and by (2+2+2)-cycloaddition (compound 262a), together with pyrrolidine 259, which is the usual product of (3+2)-cycloaddition, was observed in low yields (Scheme 111). It should be noted that two molecules of azadiene 258a are involved in the formation of hexaza-substituted cyclohexanes 262, one of which undergoes the cyclization at the C=C bond and the other at the C=N bond. In the case of azadiene 258' bearing a phenyl-substituted C=N bond, tetrahydropyridin-2-yl malonate 261'a was isolated in 35% yield in the total absence of (3+2)- or (2+2+2)-cycloaddducts. In all cases, using different DACs, the reactions were accompanied by significant oligomerization of the substrate.
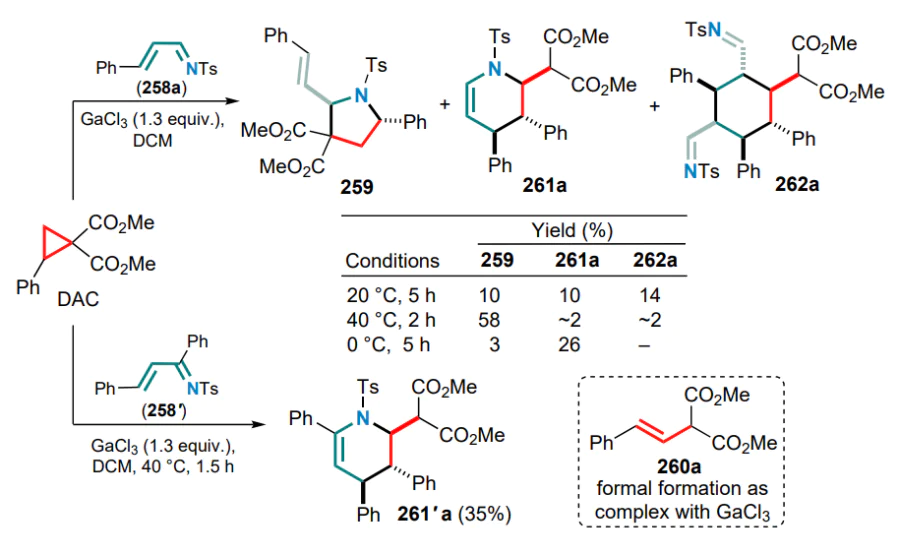
Taking into account the probability of the styryl malonate pathway, a preparative variant of the reaction of conformationally non-rigid azadienes 258, 258' with a variety of substrates 260 was proposed, which allowed to significantly improve the yield of substituted tetrahydropyridines 261 formed with high diastereoselectivities in the form of trans,trans-isomers (Scheme 112). At the same time, the use of 20 mol.% of scandium(III) triflate was most effective for the azadienes 258 with the unsubstituted C=N bond, while tin(II) triflate showed the best results for the substituted analogue 258'.
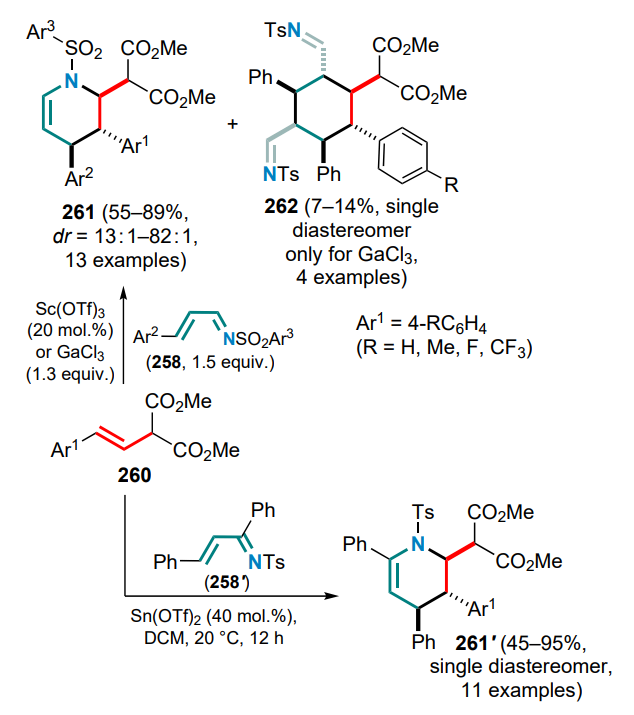
The formation of substituted tetrahydropyridin-2-yl malonates 263 also occurs via the formal (4+2)-cycloaddition of cross-conjugated azatrienes 264 to styryl malonates 260 (Scheme 113). In the case of unsymmetrically substituted azatrienes 264' (Ar3 ≠ Ar4), equimolar mixtures of regioisomers 263' and 263'' are usually obtained, but for sterically demanding substituents in the substrates at reduced temperature, the rr value can be improved up to ~4:1.
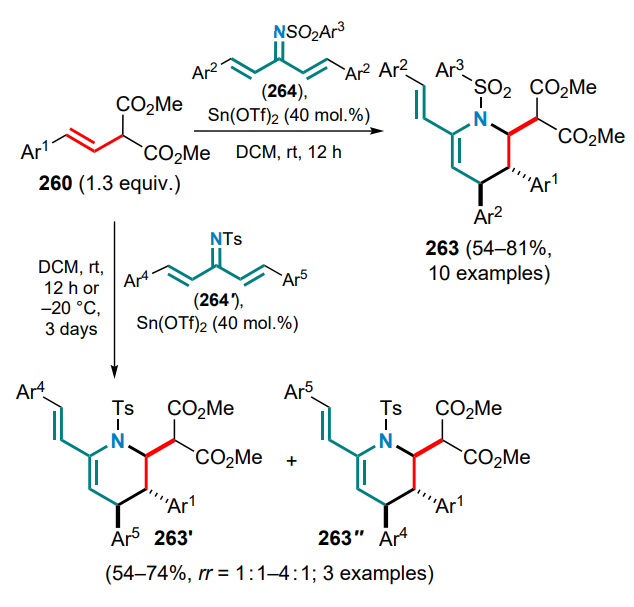
3.2. Reactions with 2-azadienes and their cationic intermediates
Reactions directly involving cyclopropanes and 2-azadienes have so far been reported in a single study[194] describing the (4+3)-cycloaddition of DAC and 3-(silyloxy)-2-azadienes 265 catalyzed by Lewis acids. Electron-rich, styryl- and aminesubstituted cyclopropanes are converted to azepanones 266, 266' (Scheme 114). A diastereoselective method using ytterbium(III) triflate and an enantioselective protocol using copper(II) triflate complex with tris(oxazoline) ligand have been developed. Two possible reaction pathways have been proposed to explain the trans-diastereoselectivity of this process. The first consists in the (3+2)-cycloaddition of the DAC complex with LA on the silylenolate C=C bond of azadiene 265 with subsequent rearrangement to intermediate A. The trans configuration of the R1 and N=CHR2 substituents helps to minimize steric interactions between the malonate moiety and the OTBS group (cf. structure). This model is in line with the findings of Tang and co-workers[47] (see Section 2). In the second case, the initial formation of an acyclic intermediate B is possible, for which the s-transoid conformation is more preferable due to the least steric repulsion between the malonate and imine moieties, and also the preferential orientation of the unshared electron pair of the nitrogen atom towards the coordinated malonate fragment (cf. structure B'). In both cases, an intermediate cyclic imine C is formed, which is desilylated to give final azepanones 266, 266'.
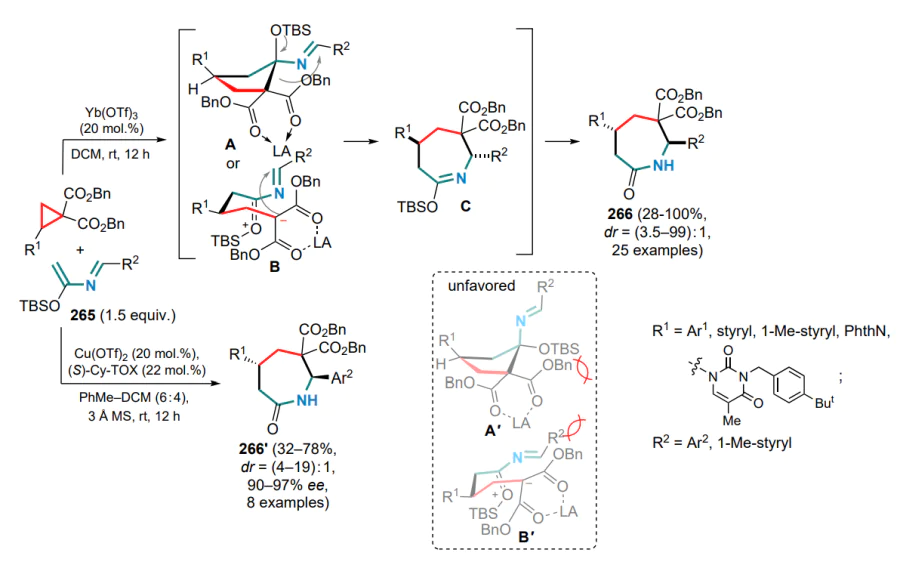
Other works referred to in this Section highlights the reaction between cyclopropane and 2-azadiene moieties as constituents of intermediates containing, in particular, a positively charged N-methylidenevinylaminium fragment (C=N+R–C=C). A natural development in this direction was the study of the reactions of cyclopropylaminium derivatives 267, characterized by the formation of the azaspiro[2.4]heptadiene intermediate 268, which combines cyclopropane and cationic 2-azadiene moieties. For example, the AuI-catalyzed reactions of 1-(alkyn1-yl)cyclopropyloxime ethers 267 with nucleophiles such as alcohols, amines and nitrones were studied[195, 196]. The proposed method provides access to substituted pyrroles 269 and pyrrolo[3,4-d][1,2]oxazepines 270 based on these substrates (Scheme 115).
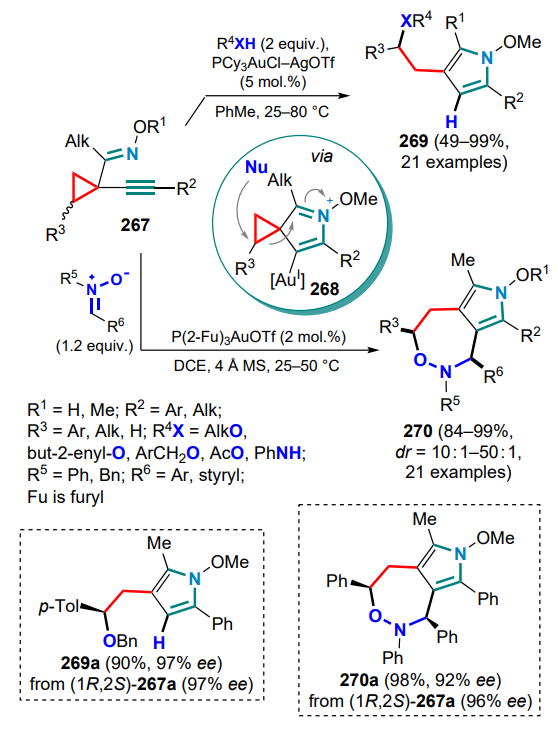
For enantiomerically pure compounds (1R,2S)-267 (96–97% ee), the products 269a, 270a with almost complete retention of the optical information could be prepared, indicating the threemembered ring opening in intermediate 268 under the action of nucleophiles by the SN2 mechanism (see Scheme 115). It was found that substrates 267' with Z-configuration of the substituents on the C=N bonds show no reaction (even on heating), apparently due to the formation of intermediate 268 being severely hampered.
A similar approach using a wide range of nucleophiles and different oximes 267 was carried out in the presence of palladium(II) salts: substituted pyrroles 271 were formed via intermediates 268[197]. It was found that for bicyclic substrates 267', in addition to seven-membered ring-annulated pyrrole 271', a minor product 272 was obtained as a result of alternative opening of the three-membered ring. For non-methylated oximes 267'', the formation of diacetyl derivatives 273 in a mixture AcOH–Ac2O was observed, whereas in the presence of methanol the same substrates gave standard products 271'' (Scheme 116).
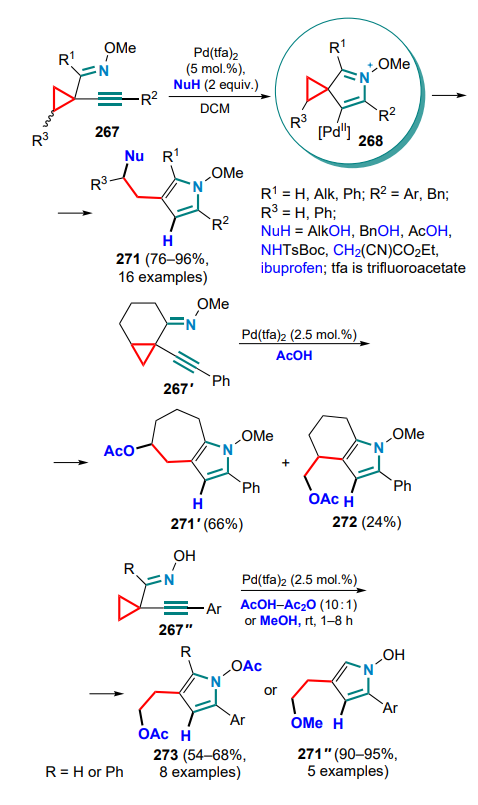
Substrates containing larger rings, 2-pyridyl or trimethylsilyl substituents on the triple bond and a number of nucleophiles (n-butylamine, ethanethiol and indole) are inactive in this reaction.
In the said processes, the oxime group can be replaced by a 2-pyridyl or 2-quinolyl moiety (274), allowing the catalysis of the gold(I) complex via a similar mechanism (via the formation of intermediate 268') to give substituted indolizines 275 (Scheme 117)[198].
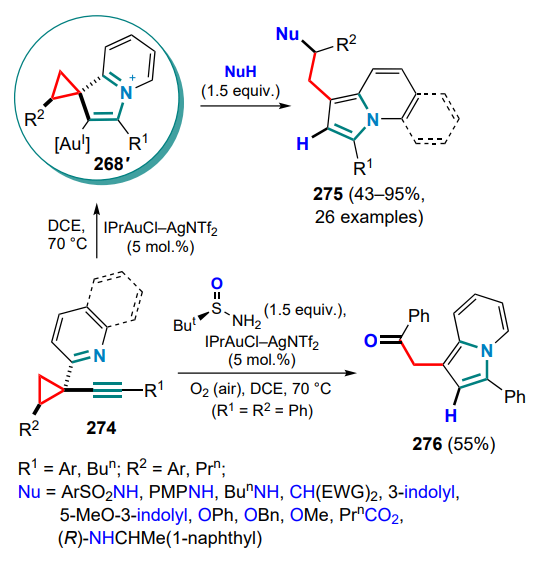
In the case of the n-propyl-tethered cyclopropane, a mixture of products is formed due to two patterns of the three-membered ring-opening by the nucleophile. The reaction with (R)-tert-butylsulfinamide in an oxygen atmosphere unexpectedly gave ketone 276, which can be explained by a nucleophilic attack of the oxygen atom on the ring with subsequent cleavage of the S–O bond.
Shi and co-workers[199] proposed a related strategy of ruthenium(0)-catalyzed cyclization of alkynylcyclopropylimines 267, affording the corresponding 3,4-cyclobutapyrroles 277. The reaction also commences with the formation of a key azaspiro[2.4]heptadiene intermediate 268, for which the resonance structure A is valid, leading to the opening of the three-membered ring and the cyclobutane intermediate B. Further elimination of the catalyst species gives the product 277 (Scheme 118).
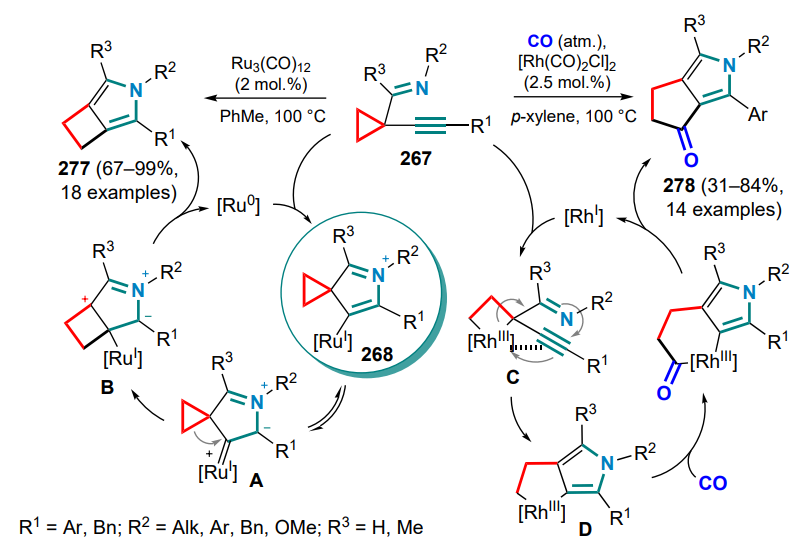
The same alkynylcyclopropylimine 267 in the RhI-catalyzed reaction with CO yields first the rhodacyclobutane intermediate C, which is rearranged to the pyrrole intermediate D as a result of an intramolecular nucleophilic attack on the activated triple bond. Subsequent insertion of a CO molecule and release of the catalytic species affords compounds 278 (see Scheme 118).
Another area of the reaction between cyclopropanes and ionic 2-azadiene intermediates was the rearrangement of O-propargylcyclopropylcarbaldoximes 279 in the presence of the RhI catalyst to afford dihydroazepine N-oxides 280[200]. Despite the fact that, according to 1H NMR spectroscopy data, the highest yield was obtained using triphenylphosphine as the ligand, the separation of the target product from the resulting triphenylphosphinoxide was difficult. As a result, the authors proposed the use of sodium diphenylphosphinobenzene-3-sulfonate (tppms) as an alternative ligand. It was shown that the reaction provides higher yields for Z isomers of the substrates than for the corresponding E isomers. The reaction starts with a nucleophilic attack of the oxime nitrogen atom on the triple bond activated by the catalyst to give vinylrhodium complex A. Once the C–O bond is cleaved and the catalytic species is eliminated, the key intermediate, N-allenylnitrone B, is formed. This step is also supported by the formation of the tetracyclic nitrone 281 as a by-product of the 4π-electrolysis. Next, N-allenylnitrone B is coordinated to the 2-azadiene moiety on the opposite side of the R2 substituent, forming the η4-complex C, which gives rise to the oxidative insertion of the azarhodacycle D with selective determination of the configuration of the exocyclic double bond. Subsequent opening of the threemembered ring (intermediate E) and reductive elimination of the catalyst species complete the formation of the target product (Scheme 119).
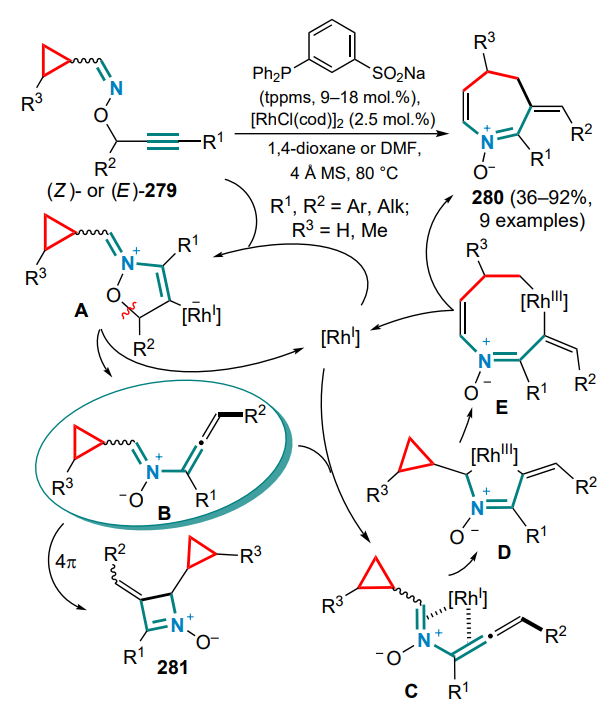
The strategy of the Cloke-Wilson rearrangement of vinylcyclopropane combined with in situ generation of iminium intermediates has been implemented. Thus, a process using cyclopropanecarbaldehyde and substituted anilines to form pyrrolo[3.2-c]quinolines 282, catalyzed by ammonium bromide or stannous triflate, was developed[201]. The initially formed imine A partially undergoes vinylcyclopropane rearrangement to form dihydropyrrole B, which undergoes the Povarov reaction with imine A. In the course of the fourcomponent condensation and taking into account the dual role of each of the reaсtants, the product 282 is obtained. o-Methyland o-methoxy-substituted anilines under these conditions gave the products of subsequent pyrrolidine ring-opening and probably of oxidative aromatization in the presence of oxygen, quinolines 283, the formation of which is probably favoured by an increase in the steric load. This is confirmed by the fact that several pyrrolo[3.2-c]quinolines 282 were converted to the corresponding quinolines 283 under oxidative conditions, and the use of endo isomers in the reaction was found to increase the yield of products compared to exo isomers. It was also found that cyclopropanecarbaldehyde reacts with a mixture of aniline and p-anisidine selectively to afford only compound 282a, with a single endo isomer being isolated (Scheme 120).
Subsequently, a similar approach with the intermediate formation of dihydrofurans from cyclopropyl ketones and with the generation of N-aryl nitrile intermediates C acting as 2-azadienes (taking into account the aryl moiety) was presented[202]. The arylation of o-formylbenzonitriles with diaryliodonium salts in the presence of a CuII catalyst delivers a cationic intermediate A, which undergoes intramolecular nucleophilic attack on the triple bond (intermediate B) and further aza-Michael rearrangement to the cationic 2-azadiene C. The latter undergoes nucleophilic attack by dihydrofuran to form the carbocationic intermediate D, which is converted to the final polycyclic compound 284 by the Friedel–Crafts reaction (Scheme 121).
N-Arylnitriles C can also be derived from the corresponding 3-hydroxy-2-phenylisoindolines 285 by dehydration with trifluoromethanesulfonic acid, which is also suitable for the in situ formation of dihydrofurans from cyclopropyl ketones[203].
Separately, mention should be made of an example of combining the rearrangement of 1-ethynyl-2-oxobicyclo[4.1.0]-heptane 286 in the presence of tungsten(0) pentacarbonyl and the subsequent capture of the resulting oxabicyclo[4.3.0]-nonatriene 287 by N,N-dibenzoylcyclohexa-3,5-dien-1,2-diimine (288) in a one-pot protocol (Scheme 122)[204]. This strategy is notable primarily for the position of the functionalized saturated site of the six-membered ring.
The rearrangement involves the intermediate formation of the tungsten-vinylidene complex A, which undergoes the ringclosure of the furan moiety (intermediate B) and the opening of the three-membered ring (C) to form a double bond remote from the original reaction site, which further undergoes (4+2)-cycloaddition with diimine 288 to give the polycyclic product 289.
3.3. Reactions of methylidenecyclopropanes with azadienes
It is shown that complex cascade syntheses of substituted 2,4-dihydro-1H-azeto[1,2-a]quinolines (benzocarbacephemes) 291 and pyrrolo[1,2-a]quinolines 292 can be carried out from methylidenecyclopropanedicarboxylic acid esters 129, 290 and N-aryl nitrones[205, 206]. The process begins with the 1,3-dipolar addition of nitrone to the exomethylene bond (intermediate A), followed by the Brandi-Guarna rearrangement, which consists of the homolytic cleavage of the N–O bond (B) and the opening of the three-membered ring to generate the biradical intermediate C. The latter attacks the aryl moiety at the nitrogen atom with closure of the eight-membered intermediate D, followed by intramolecular attack of the amino group on the carbonyl group with subsequent dehydration and proton migration to form compound 291. For the methoxy-substituted N-aryl-tethered nitrone, further spontaneous oxidation in air under reaction conditions and formation of vinylquinoline 293 as a product of opening the four-membered ring and aromatization were observed. Several similar compounds were also obtained by the subsequent oxidation of substrates 291 with 2,3-dichloro-5,6-dicyano-1,4-benzazoquinone (DDQ) (Scheme 123).
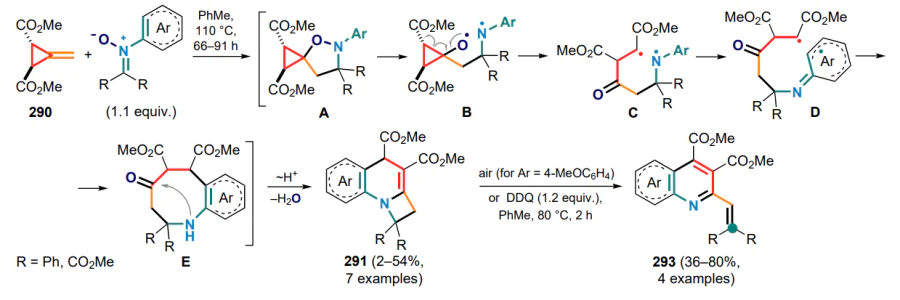
Contrary to the reaction with Feist’s ester 290, in the process with (arylmethylidene)cyclopropanedicarboxylate 129, the formation of the azetidine ring F is accompanied by its opening. This is followed by an intramolecular nucleophilic attack of the amino group on the ester moiety of the starting nitrone to give pyrrolo[1,2-a]quinolines 292 (Scheme 124).
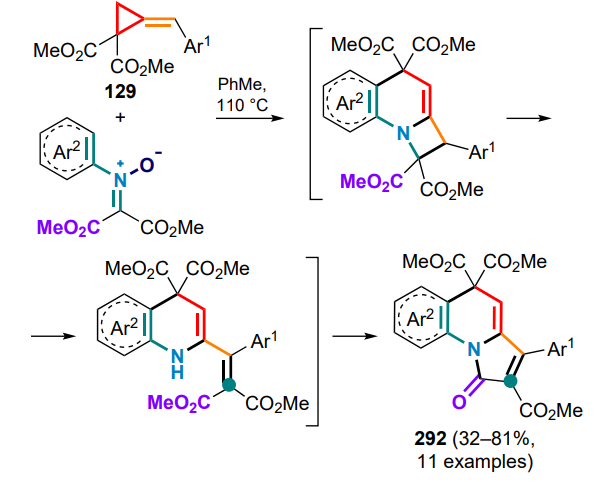
Wu and co-workers[207] developed a similar cascade strategy involving (3+2)-cycloaddition of nitrone to the C=C bond of methylidenecyclopropane 98 followed by a Brandi-Guarna rearrangement. Intermediate B then undergoes a rearrangement with subsequent intramolecular attack of the radical centre (intermediate C) on the enamine radical moiety, resulting in the closure of the eight-membered ring and the formation of benzo-7-azabicyclo[4.2.2]dec-7-en-4-ones 295 (Scheme 125).
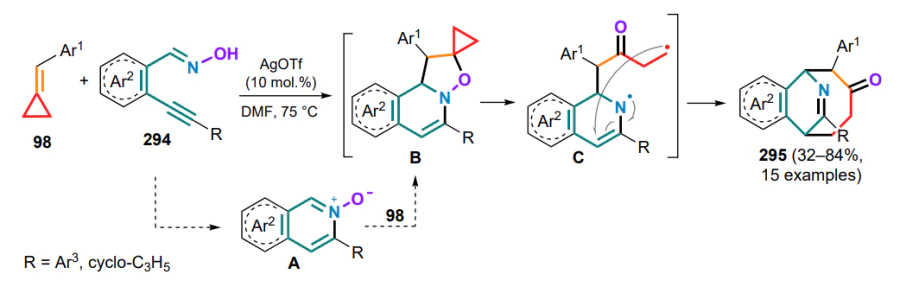
In a study of the photocatalytic cascade transformation of methylidene cyclopropanes 83' and N-vinyl-α,β-unsaturated nitrones 296 catalyzed by ytterbium(III) triflate and eosin Y, a method for the construction of nine-membered heterocycles 297 with retention of the cyclopropane moiety and insertion of an oxygen atom into the molecule was implemented[208]. The main process involves a cascade of (3+2)-cycloaddition, diastereoselective [3,3]-sigmatropic rearrangement and (4+2)-addition of singlet oxygen obtained from air via visible light-promoted eosin Y (Scheme 126). In the case of cyclopropylideneacetaldehyde (R1 = H) and nitrone 296a (R2 = R3 = Me, Ar = Ph), the possibility of an additional [1,3]-rearrangement due to the homolytic cleavage of the N–O bond yielding macrocycle 298 was demonstrated.
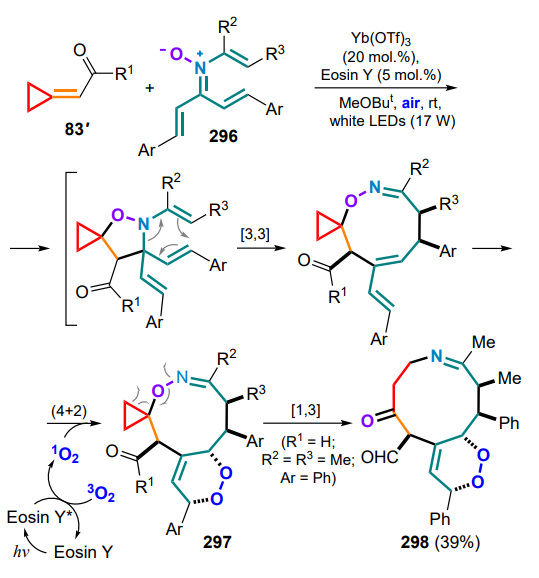
Separately, work on metal-catalyzed cascade transformations based on ortho-C–H activation of the aryl moiety, formally part of the 1-azadiene system, should be highlighted. A rhodium(III)-catalyzed cascade process combining C–H/C–C activation and annulation of (arylmethylidene)cyclopropanes 98 and N-sulfonylketimines 299 has been developed, providing access to spirocyclic benzosultams 300 with high diastereoselectivity[209]. The reaction begins with the formation of rhodacycle A, which undergoes an insertion of a molecule of (arylmethylidene) cyclopropane (intermediate B). Then, the opening of the threemembered ring (C) and the β-hydride shift afford the diene complex D. The next step can be either the 1,2-addition of the catalytic species to the least sterically hindered double bond of the diene (E), which is re-coordinated via the π-allyl complex F into the intermediate G, or the direct formation of the latter through an alternative 1,4-addition.
Finally, complex G undergoes intramolecular closure of the C–C bond by the Grignard reaction (H) to give the final product 300 and regenerate the catalytic species that closes the cycle (Scheme 127). The role of copper(II) acetate is probably to promote the C–H activation of the Ar2 moiety by the concerted metalation-deprotonation (CMD) mechanism.
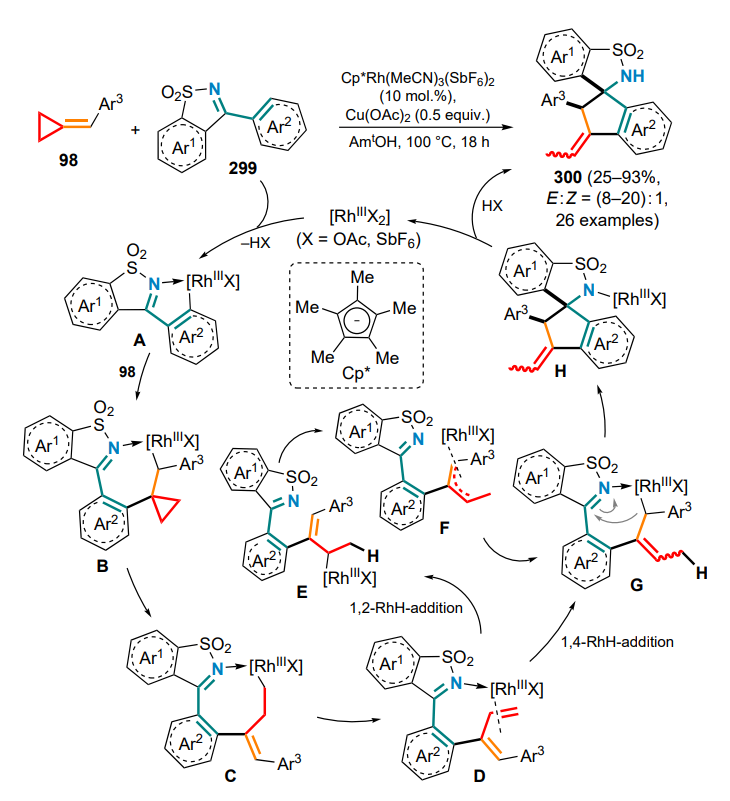
Several similar works[210, 211] have also been presented previously, which, in contrast to the previous study, described an additional bond formation between the cyclopropane and a functional group at the C=N unit. In the study[210], the reaction between (arylmethylidene)cyclopropanes 98 and aryl nitrones or azomethine imines 301a,b, as in the above example, is carried out in the presence of rhodium(III) complex and silver(I) acetate as oxidant. In the first step, the catalyst species participates in the ortho-C–H activation of the aryl moiety of the imine substrate (intermediate A), followed by the insertion of methylenecyclopropane into the C–RhIII bond (B) and the opening of the three-membered ring during β-C-elimination, to form complex C. This intermediate undergoes β-H-elimination to afford a rhodium(III) hydride complex, which is oxidized by silver(I) acetate to an active catalytic species. The diene intermediate D then undergoes intramolecular (3+2) cycloaddition with closure of the seven-membered ring and formation of the exo-products 302 or 303 (Scheme 128). A bulky substituent at the nitrogen atom of complex A apparently prevents hydrolysis and stabilizes the Z configuration necessary for the C–H activation. The impossibility for the reaction to occur under the conditions described for disubstituted methylidenecyclopropanes or substrates of type 98 containing an aliphatic substituent instead of an aryl substituent on the double C=C bond was noted.
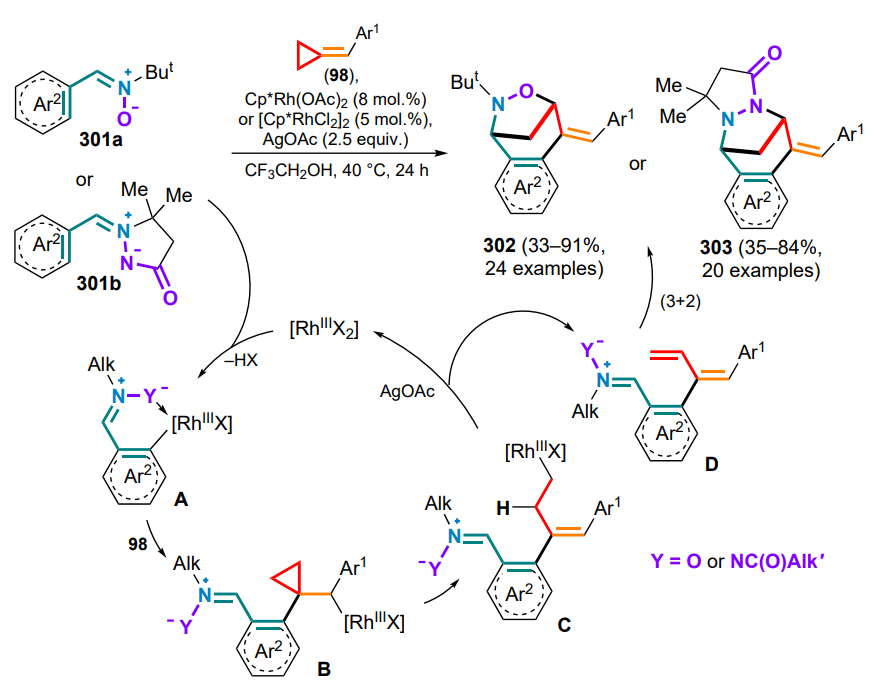
Another study[211] is devoted to the manganese(I)-catalyzed two-step one-pot cyclization of methylidenecyclopropanecarboxylates 304 with ketimines 305, which led to the diastereoselective preparation of a series of tetrahydroquinolines 306 (Scheme 129). The addition of sodium acetate to the reaction mixture improves the yields of the products, suggesting the initial formation of the Mn(CO)4OAc complex coordinated to ketimine 305, to give complex A. Next, insertion of methylidenecyclopropane 304 into the C–MnI bond (intermediate B) and the three-membered ring-opening takes place. Additional coordination of the carboxylate and double bond to the MnI atom within the new complex C promotes intramolecular nucleophilic attack on the C=N bond. As a result, the five-membered ring is closed and the intermediate D further regenerates the active complex A during protodemetallation. The resulting indene 307 undergoes the second stage of the process involving zinc(II) chloride, which consists in the intramolecular arylation of the acceptor double bond by an aniline moiety with the release of the final products 306.
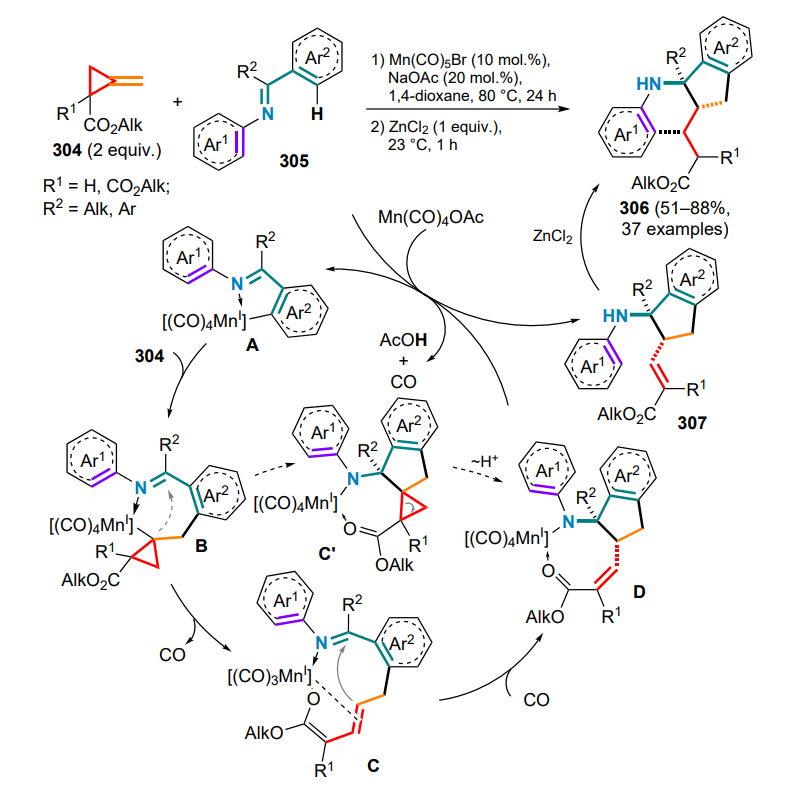
The authors also considered an alternative pathway of this reaction with a reverse sequence of nucleophilic intramolecular attack (intermediate C') and isomerization of the cyclopropane moiety to form the above complex D.
The first example of an intramolecular transformation of a cyclopropane with a nitrogen-containing unsaturated bond was the cyclization of 2-(1-cyclopropylideneethyl)pyridine (308) in the presence of catalytic amounts of tetrakis(triphenylphosphino)-palladium(0)[131]. The reaction begins with the oxidative insertion of the catalytic palladium(0) species into the distal C–C bond and protonation of the pyridine nitrogen atom in the presence of trace amounts of water. The palladacycle A further forms the π-allyl palladium complex B which, after reductive elimination, gives the intermediate C through the five-membered ringclosure. The rearrangement and deprotonation give 1,2-dimethylindolizine 309 (Scheme 130).
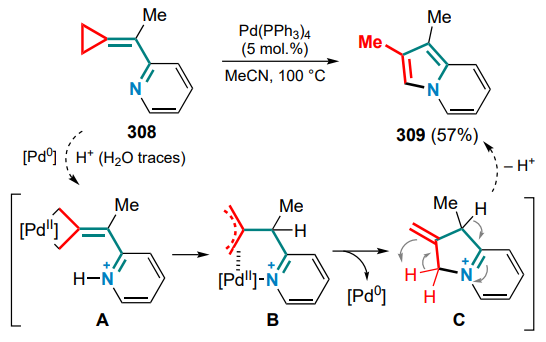
Processes involving (arylmethylidene)cyclopropanes having nitrogen-containing multiple bonds in the ortho position of the aryl moiety can be singled out in a separate group. One such process involves the in situ preparation of (o-aminoarylmethylidene)cyclopropane imines A from the precursors 133 and the appropriate aldehydes in the presence of excess magnesium sulfate, accompanied by (3+2)-cyclization with opening of the three-membered ring and formation of pyrrolo[1,2-a]indoles 310 (Scheme 131)[212].
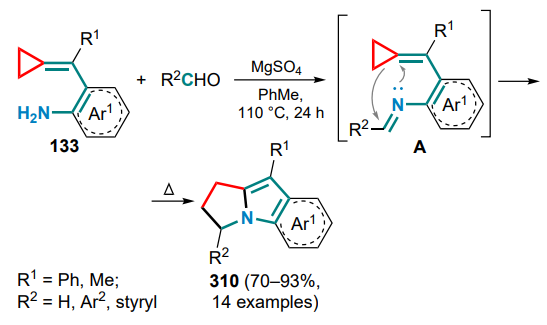
The intramolecular cyclization of (o-isocyanoarylmethylidene)cyclopropanes 311 in the presence of catalytic amounts of silver(I) carbonate can be attributed to the same series[213]. The reaction proceeds by π-activation of the C=C bond by the Ag+ cation, and the equilibrium existence of two complexes (A and B) is possible. Intramolecular nucleophilic attack of the isonitrile moiety on the activated double bond leads to the six-membered ring-closure in the intermediate C. This is accompanied by the cyclopropane ring-opening and release of the catalytic species, yielding a four-membered ring in the final 1,2-dihydrocyclobuta[b]quinones 312 (Scheme 132).
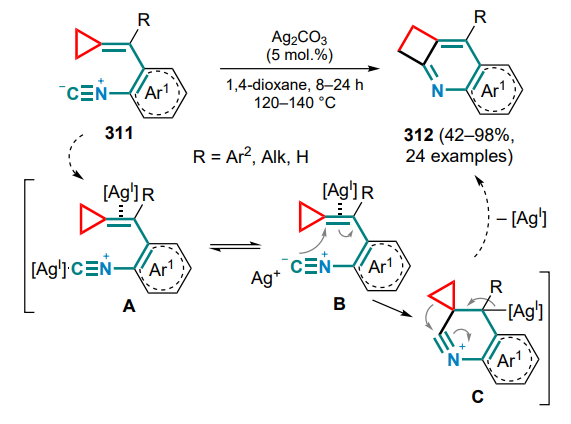
o-(N-Vinyl)aniline-tethered methylidenecyclopropanes 136 can undergo cyclization with nucleophilic reagents such as acyl halides, iodine and trimethylsilyl azide in the presence of Lewis acids to give substituted dihydroquinolines 313–315 (Scheme 133)[214, 215]. Coordination of the enamine unit by the catalyst is accompanied by the capture of an electrophilic species, e.g. proton or iodine cation, to generate the aminium intermediate A, followed by the aza-Prins cyclization and opening of the three-membered ring by nucleophilic attack.
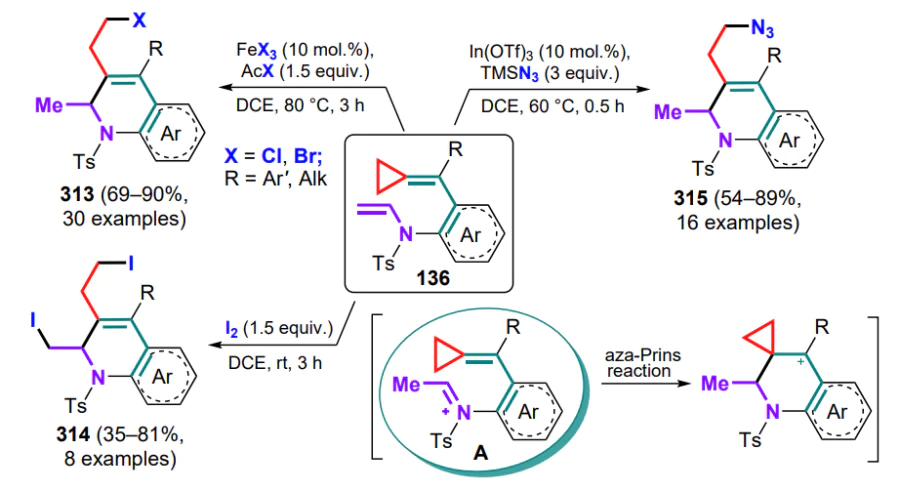
Continuing the development of the 6π-EC+VCP-RA concept (see Section 2.6.3), which involves the preparation of orthoallyl- and ortho-ketenimine-substituted (arylmethylidene) cyclopropanes via 6π-electrolysis and vinylcyclopropane rearrangement, a series of aza-modifications involving ethenimines, carbodiimides, isocyanates and isothiocyanates based on ortho azo-functionalized substrates have been proposed.
Thus, (o-aminoarylmethylidene)cyclopropanes 133 can be modified with triphosgene or carbon disulfide in the presence of a base. First, iso(thio)cyanates A are formed, which are then rearranged to quinolines 316 or 316' (Scheme 134)[216].
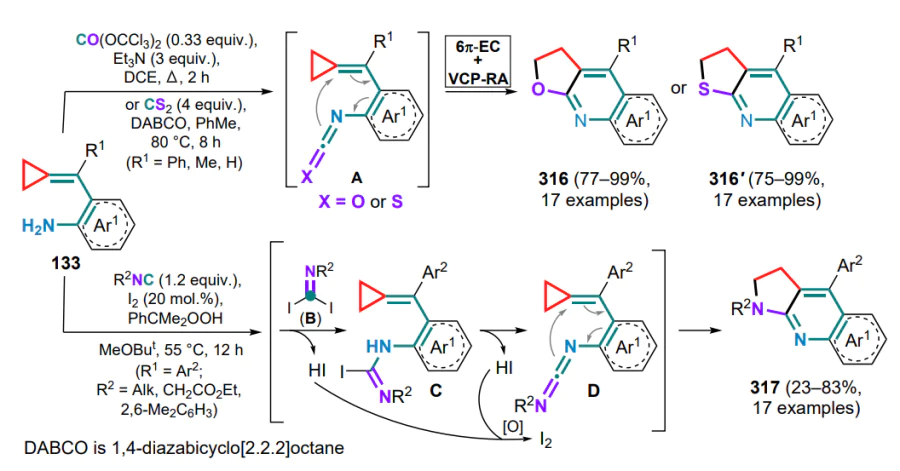
The same substrates are capable of an iodine-catalyzed reaction with isonitriles under oxidative conditions to furnish the intermediate diiodide B, which is the object of the nucleophilic attack by substrate 133, to yield the intermediate C[217]. Next, HI is eliminated, which is further oxidized to iodine by cumene peroxide, and the resulting carbodiimide D undergoes rearrangement with release of dihydropyrroloquinoline 317.
Examples of thermal cyclization of N-cyano-N-amidosubstituted (o-aminoarylmethylidene)cyclopropanes are described, in which the key carbodiimide intermediate A (see Scheme 134, where X = NR3 (R3 = Ac or C(O)CH2Cl)) is formed via rearrangement with migration of the acyl moiety to the terminal nitrogen atom. Subsequent 6π-EC+VCP-RA cyclization produces dihydropyrrololoquinolines 317[218]. The substrate with the unsubstituted acetyl group gives the product in low yield (~4%), whereas the yield for the chloroacyl analogue is 76%.
As alternative substrates, (o-azidoarylmethylidene) cyclopropanes 161 can be used as sources of nitrenes[219]. In the presence of a rhodium catalyst, they are capable of reacting with isonitriles to form the intermediate carbodiimide type A (see Scheme 134, where X = NAlk, NAr), which then undergoes similar cyclization to form dihydropyrroloquinoline type 317 in 56–80% yields.
Finally, iminophosphorane derivatives of (o-aminoarylmethylidene)cyclopropanes 318 can be subjected to the Wittig reaction with ketenes generated in situ from α-diazoketones in the thermal Wolf rearrangement, and also with isocyanates. The corresponding ethenimines A and carbodiimides B then undergo a cascade rearrangement to give cyclopenta[b]quinolines 319 or dihydropyrroloquinolines 317' (Scheme 135)[220]. Interestingly, in the presence of diisocyanates, using an additional equivalent of the starting iminophosphorane, carbodiimide (intermediate C) metathesis delivers dimeric carbodiimide D, giving rise to a series of densely functionalized compounds 320.
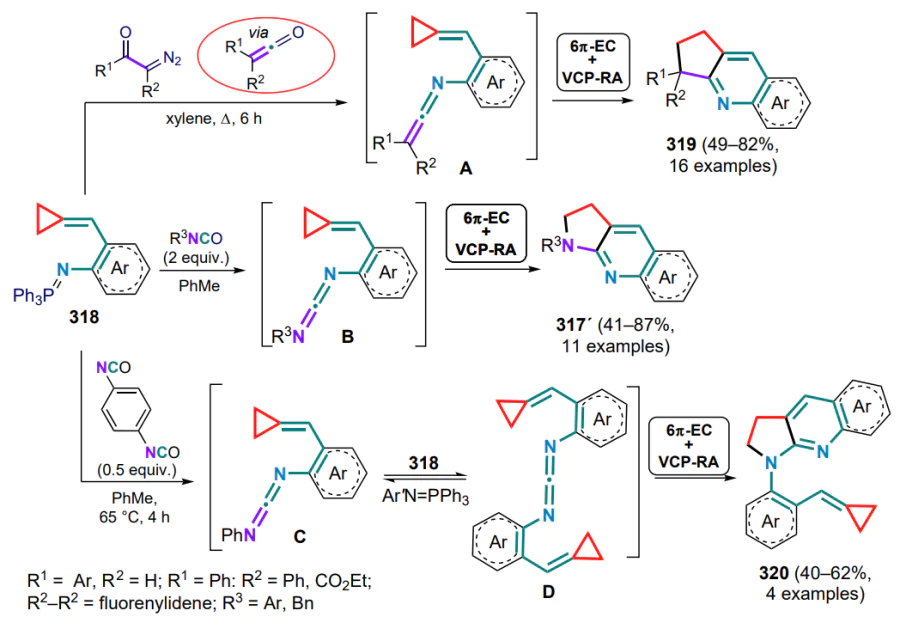
Modification of (diarylmethylidene)cyclopropanes can provide access to the intramolecular reaction between cyclopropane and aza-polyene units. For example, o-isocyanoaryl-substituted substrates 321 can undergo trifluoromethylation using the Togni reagent II under photocatalytic conditions in the presence of iridium(III) complex[221]. A cascade of radical rearrangements of the initial radical intermediate A involving the C=C bond (intermediate B) leads to the subsequent opening of the three-membered ring and annulation with the adjacent aryl moiety. The resulting intermediate C can then be oxidized with an iridium(IV) complex and deprotonated in the presence of a base to give a series of dihydrobenzo[k]phenanthridines 322 (Scheme 136). This reaction is hampered for substrates bearing heteroaryl groups and ortho-substituted Ar2 moieties due to the steric hindrance.
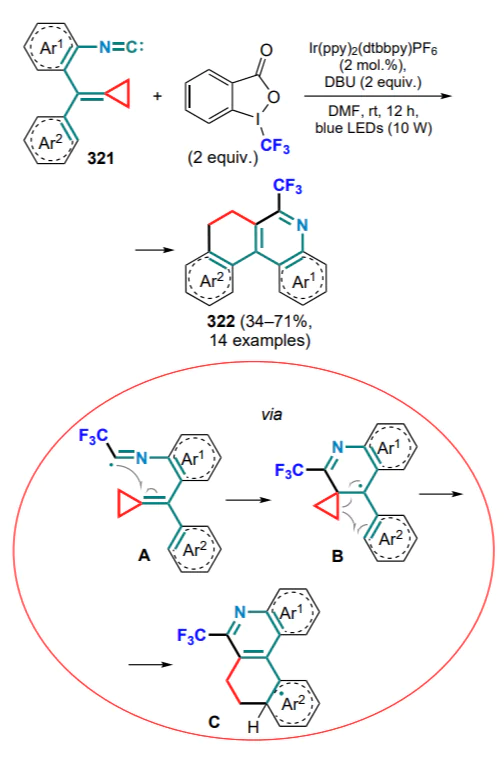
4. Reactions of oxygen- and sulfurcontaining conjugated systems
.
4.1. Reactions of cyclopropanes with oxa- and thiadienes and their cationic intermediates
One of the first examples of reactions of cyclopropanes with oxygen-containing conjugated systems was the (8+3)-cycloaddition of DAC to tropone 323 catalyzed by systems based on nickel(II) perchlorate and tin(IV) chloride as Lewis acids (Scheme 137)[222, 223]. The activated complex of DAC and Lewis acid is subjected to a nucleophilic attack by tropone to generate the tropylium zwitterion intermediate A, which is further closed into the pyran ring at the position 2 (intermediate B) or 7 (for C(2)-substituted substrates).
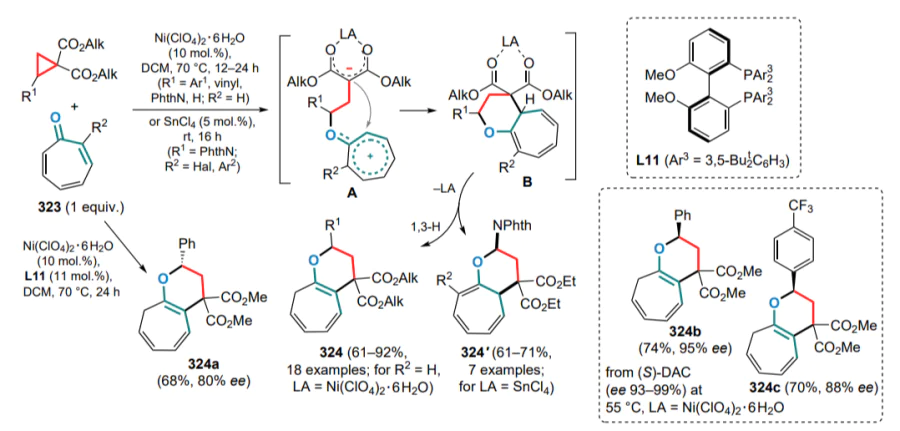
Intermediates B then undergo a 1,3-hydride shift to form oxabicycles 324, while their C(7)-substituted analogues retain the double bond arrangement in the products 324'. The NiIIcatalyzed transformation of unsubstituted tropones was realized in an enantioselective mode with the participation of the chiral ligand L11, as exemplified by the synthesis of compound 324a. Under standard conditions, the corresponding oxabicycles 324b, 324c were obtained from chiral (S)-DAC with a slight decrease in enantiomeric purity. This observation confirms the SN2 character of the attack of tropone 323 on the activated DAC bond.
Later, an enantioselective cyclization of DAC 325 with tropone 323' has been proposed using an anionic activation strategy for catalysis by Brønsted base 326 (Scheme 138)[59]. According to quantum chemical calculations, the bifunctional organocatalyst deprotonates the parent cyclopropane opening the three-membered ring and generating the Michael acceptor, which is then coordinated by the ammonium moiety to the cyano group and is bidentately coordinated by the thiourea fragment on the tropone oxygen atom through hydrogen bonds, favoured by the presence of the acceptor moiety in the amino group. In the newly formed precursor, the anionic site further nucleophilically attacks 2-positioned tropone with low enantioselectivity. The intramolecular ring-closing oxa-Michael reaction then yields the pyran ring, and the enantioselectivity of this step is high. The subsequent 1,3-hydride shift, probably resulting from a series of 1,5-sigmatropic shifts, leads to a loss of stereochemical information about the initially formed stereocentre due to the formation of a more substituted double bond in bicycles 327. The use of C(2)-substituted tropones in this reaction failed even when raising the temperature to room temperature.
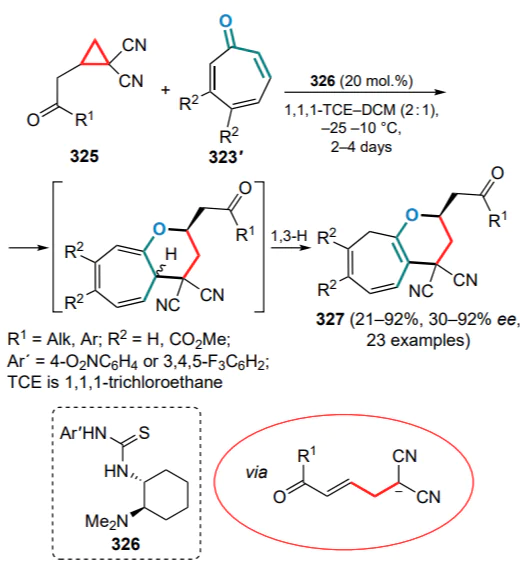
Other oxygen-containing conjugated systems that have been successfully used in reactions with DAC are benzoquinone esters 328 (Scheme 139)[224]. Methods have been developed for the synthesis of substituted chromanes 329 and benzofurans 330 based on InIII-catalyzed (3+3)- and (3+2)-cycloadditions. In the case of InCl3 catalysis, 1,3-zwitterion A is formed with further attack of the benzoquinone oxygen atom on the carbocationic centre by the SN1 mechanism and subsequent Michael cyclization. In the presence of In(OTf)3, the formation of the β-styryl malonate complex B, which undergoes further formal (3+2)-cycloaddition, becomes the most preferred.
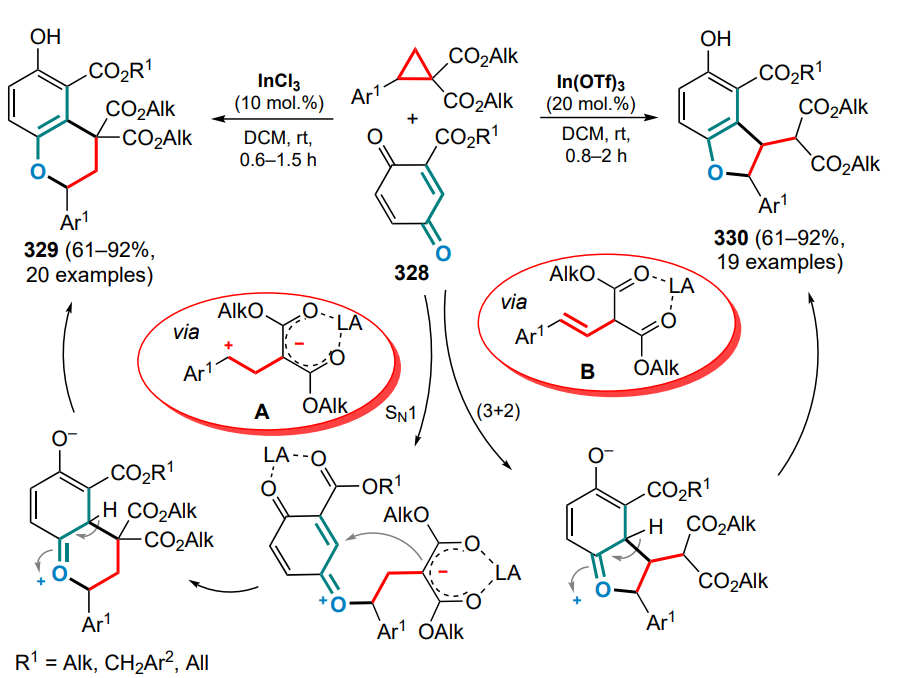
Styryl malonates 260, isomers of the corresponding DAC, have well proven themselves as preferred substrates, e.g., in reactions with aromatic aldehydes in the presence of gallium(III) chloride[63]. Although the unusual formation of indenes 331 and pentacyclic lactones 332 with high diastereoselectivity was observed in the presence of 1,2-zwitterionic intermediates generated from DAC (see Section 2.2), the use of styryl malonates 260 allowed an increase in the yields of compounds 331 and 332 due to a significant reduction in the number of side processes associated with the dimerization of the starting DAC.
The gallium trichloride-activated complex adds an equivalent of an aldehyde (intermediate A) followed by cyclization and dehydration to form indenes 331[63]. Then, depending on the conditions, an additional equivalent of the same or a different aldehyde can be readded to position 3 of the indene in a one-pot mode of the said method (Scheme 140). In this case, the cationic centre of intermediate B is attacked by an ester moiety with further deprotonation and hydration under the action of a water molecule released in the first step of the process (as demonstrated by isotopic labelling experiments). After elimination of methanol, the intermediate C undergoes Friedel–Crafts intramolecular alkylation with closure of the five-membered ring to yield product 332.
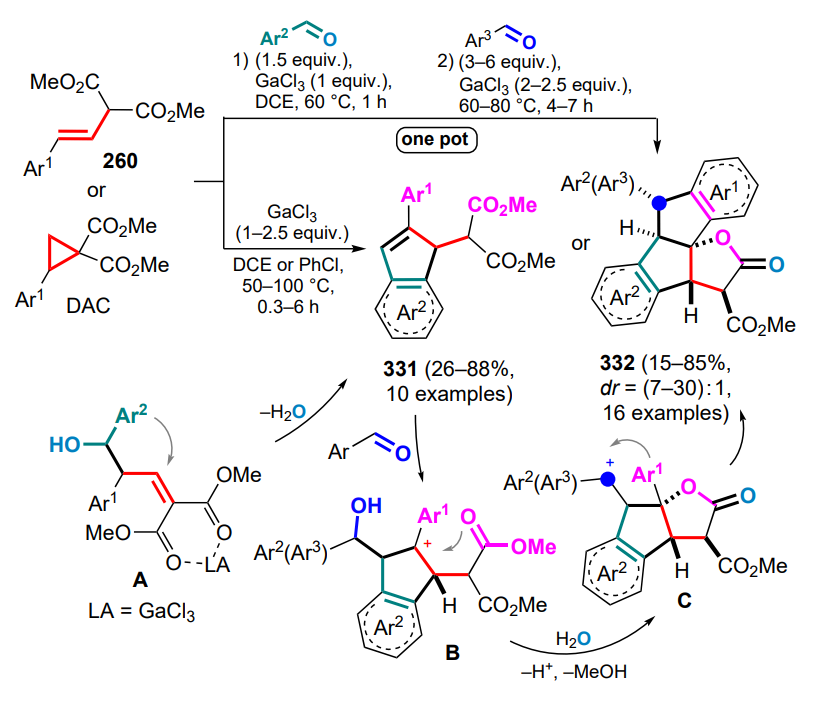
Subsequently, studies in this area were complemented by several examples of BF3 ·Et2O-catalyzed formation of cyclopentadienes 333 from styryl malonates 260 involving cinnamyl aldehydes 334 (Scheme 141)[225]. First, dihydropyranone is formed[226], which is protonated (intermediate A) and undergoes of the six-membered ring-opening followed by cyclization. Subsequent methylation in the presence of diazomethane gives the products 333.
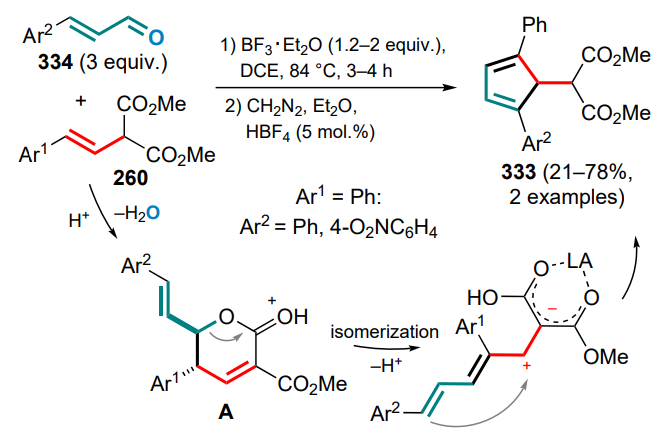
The synthetic possibilities of this approach have been extended to furan and benzofuran carbaldehydes 335 (Scheme 142)[227]. In this case, an additional hydration step of intermediate A and further hydrolysis of intermediate B furnish cyclopentenones 336, 337 as single diastereomers. The authors explain this by the preferred trans configuration of the malonate and furan moieties in the intermediate formation stage.
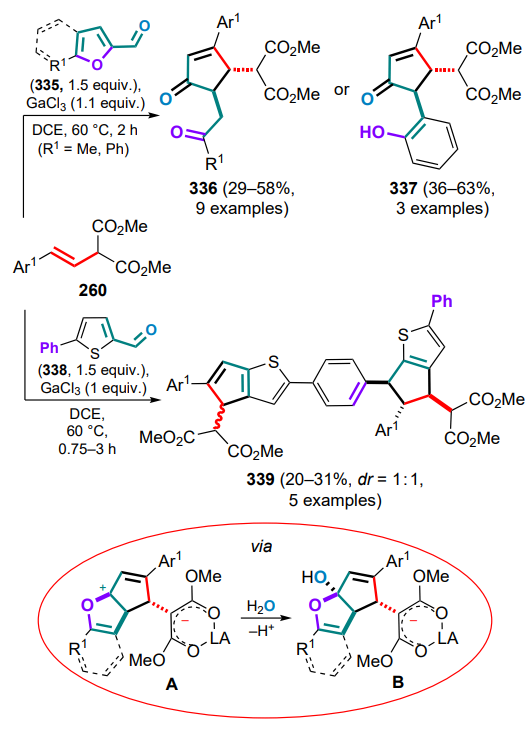
Finally, reacting styryl malonates 260 with 5-phenylthiophene-2-carbaldehyde 338 provided access to a series of polyaromatic derivatives 339, the formation of which are similar to that described above for indenes, except for an additional para substitution on the benzene ring (see Scheme 142). The UV and visible light absorption of the synthesized products was studied[228]. These examples demonstrate the promising methodology of gallium(III) chloride-promoted reaction of DAC with oxygen-containing conjugated systems involving adjacent functional groups.
Another option for cyclization of adjacent functional groups is the reaction of substituted cyclopropanecarbaldehydes 340 with salicylic aldehydes (Scheme 143)[229]. Such acceptor cyclopropanecarbaldehydes can give coumarins 341 in the presence of the carbene ligand IMes as an organocatalyst derived from the precursor IMes·HCl under basic conditions.
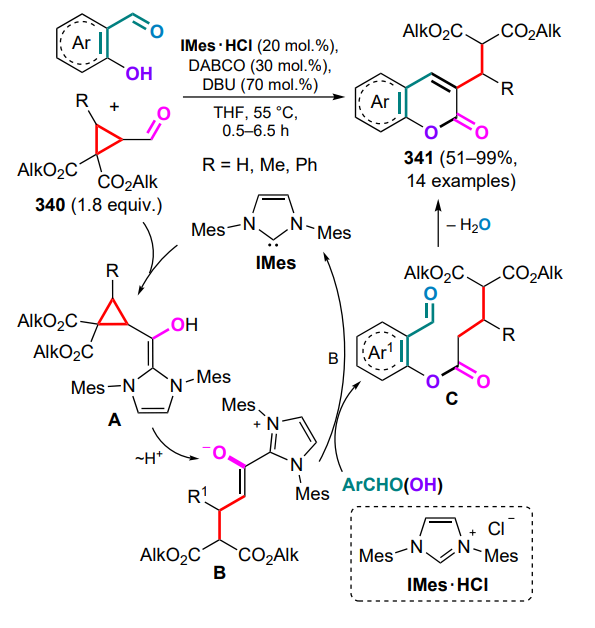
The Breslow intermediate A generated in the first step is ring-opened to give a linear intermediate B and further undergoes nucleophilic attack by a phenolic oxygen atom with closure of the C–O bond and release of the carbene catalyst. Intermediate C further undergoes intramolecular condensation with closure of the six-membered ring in the final coumarin 341.
A similar approach in the enantioselective variant was used for the synthesis of substituted δ-lactones 342 from cyclopropanecarbaldehydes 343 and chalcones in the presence of the chiral carbene organocatalyst NHC2 (Scheme 144)[230]. In this case, the Breslow intermediate is also ring-opened to a linear structure, followed by annulation and release of the carbene catalyst, affording the product 342.
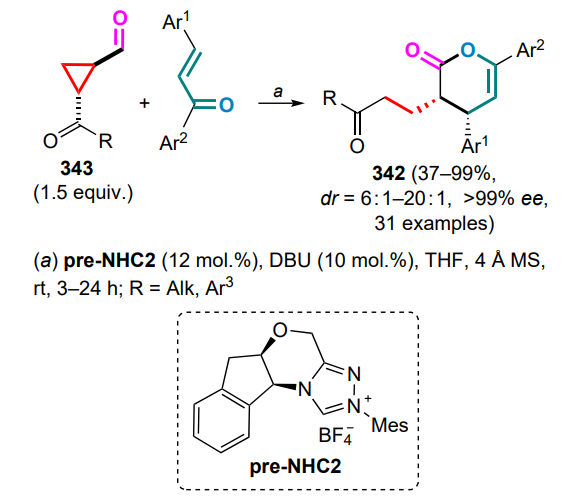
The same organocatalyst also proved successful in a similar process, namely, the reaction of cyclopropanes 340 with alkylidene oxindoles 344 or β,γ-unsaturated α-ketoesters 345 to give the corresponding δ-lactones 346 or 342' (Scheme 145)[231].
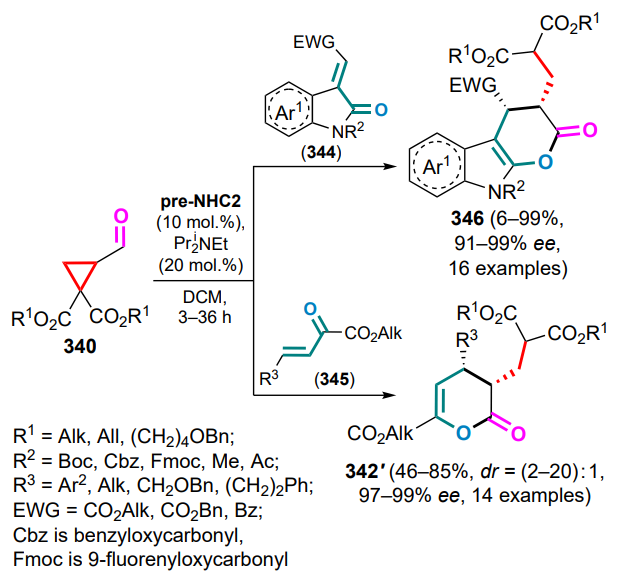
Arylcyclopropanecarbaldehydes 347 can undergo the Cloke –Wilson rearrangement in the presence of Lewis acids, which, as shown above (see Section 3.2), can be utilized in reactions with conjugated substrates. The above-mentioned benzoquinone esters 328 are no exception, and their use in this reaction leads to tetrahydrofurobenzofuran derivatives 348 as a result of a cascade of Michael addition and intramolecular nucleophilic attack of the resulting phenolic OH group (Scheme 146)[232]. The high diastereoselectivity of the process is explained by the favourable trans configuration of the intermediate and further nucleophilic attack from the least sterically hindered side.
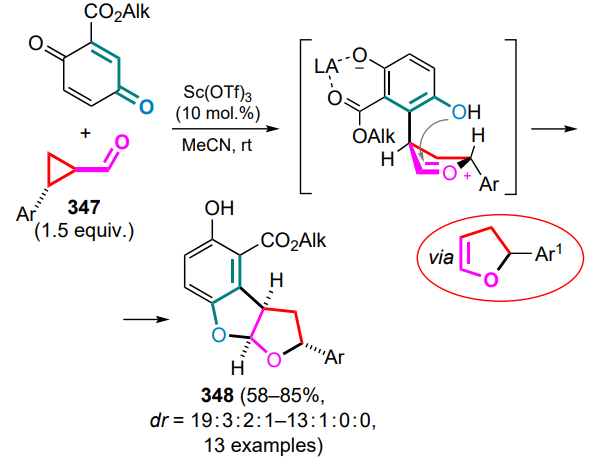
The reaction involving p-quinone methide 349 to give tetrahydrofurobenzopyranes 350 proceeded similarly (Scheme 147). In this case, the trans arrangement of the substituents in the key intermediate would contribute to an increase in the intramolecular steric loading of the molecule due to the bulk di-tert-butyl-substituted phenolic moiety, thus favouring the cis configuration of the resulting compounds. Although product 350 was isolated as a mixture of four diastereomers in ratios ranging from 9.6:3.7:1.3:1 to 4:3:1.3:1.
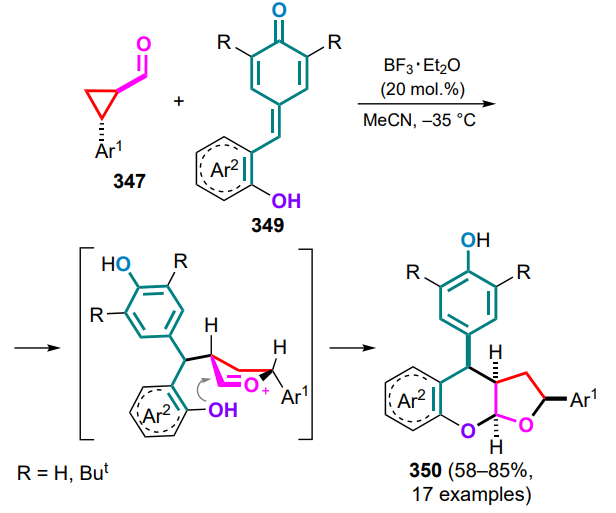
An unusual cascade reaction of DAC 351 with acryl fluorides catalyzed by NHC (IMes) and based on the Ireland–Coates–Claisen rearrangement is described, leading to diastereoselective β-lactones 352 (Scheme 148)[233]. The process begins with the formation of the acylazolium cation A, and the subsequent silyl deprotection under the action of fluoride is accompanied by a retro-aldol reaction yielding enolate B. The intermediate C formed via the nucleophilic attack undergoes a [3,3]-sigmatropic rearrangement promoted by aryl donor substituents R2 in the complex ester group of DAC. Successive aldol condensation (D) and lactonization (E) then give rise to the target products with high diastereoselectivity, with bulkier N-substituents in NHC worsening the diastereoselectivity of the process up to equimolar ratios by slowing down these steps.
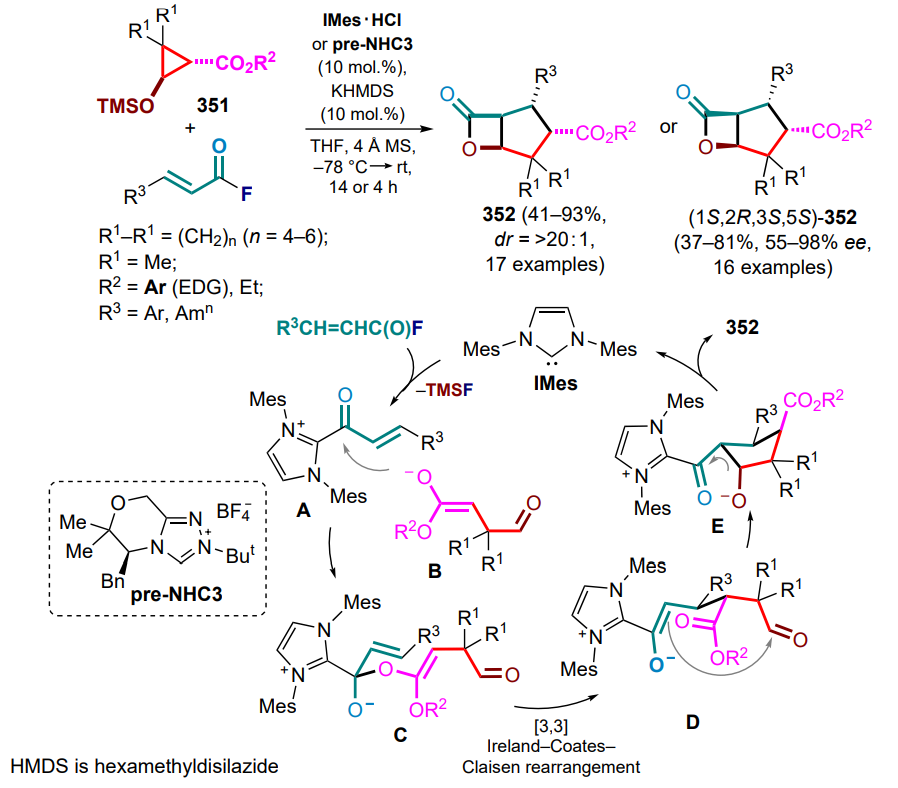
Candish et al.[234] also proposed an enantioselective modification of this reaction using a chiral triazolylidene catalyst (from pre-NHC3). The catalytic activity of N-alkyl substituted triazolylidenes was shown to be higher than that of their arylsubstituted analogues. The electron-withdrawing aryl or alkyl substituents R3 in the starting acyl fluorides decreased yields and enantioselectivities, whereas the reverse was the case for more electron-rich aryl moieties.
The gold-catalyzed 6-endo-dig cyclization of cis-1-oxiranyl1-alkynyl cyclopropanes 45, mentioned in Section 2.1, has also been carried out using enones (Scheme 149)[60]. The process involves similar steps of intermediate formation of bicyclic alcohol 46 and subsequent generation of carbocation A, which adds an enone. Intermediate B tends to add a water molecule to two successively generated oxonium moieties, giving diacetal frameworks 353 with good diastereoselectivity.
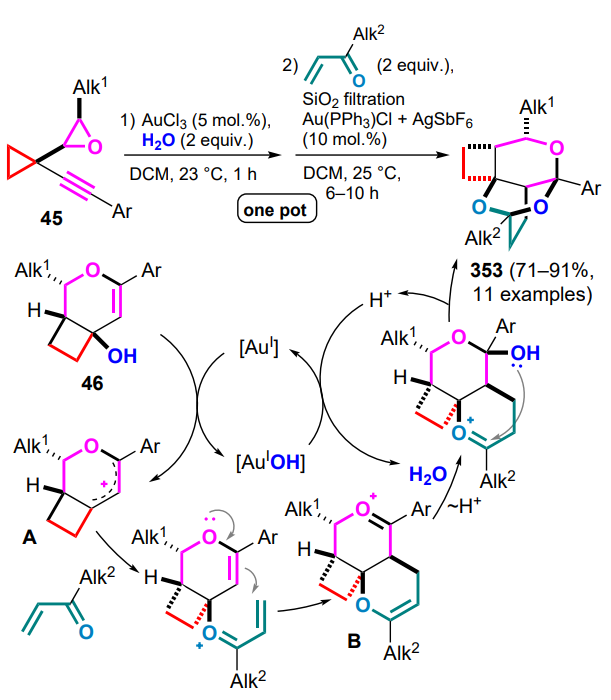
The reactions of 3-carene derivatives with electron-donating aromatic aldehydes in the presence of montmorillonite K-10 are described, and both the aldehyde group and the aryl function of the substrate are involved in the transformation[235]. High cytotoxic activity against the lymphoblastoid cell line MT-4 was found for one of the isolated products.
A method for enantioselective 1,3-dipolar (5+3)-cycloaddition using styryl-tethered cyclopropane-1,1-diesters and benzopyranones 354 capable of forming oxydopyrylium ylides A as conjugated oxygen-containing substrates in the presence of a base has been proposed (Scheme 150). Formally, this type of reaction is similar to the transformations of methylene(vinyl)-oxonium (MVO) intermediates, which will be discussed in detail below. Coordination of the chiral palladium complex to the styryl moiety in the presence of the ligand L12 promotes the 1,3-opening of the three-membered ring with its subsequent enantioselective sequential addition[236] to the ylide A, yielding 9-oxa-bicyclononanes 355.
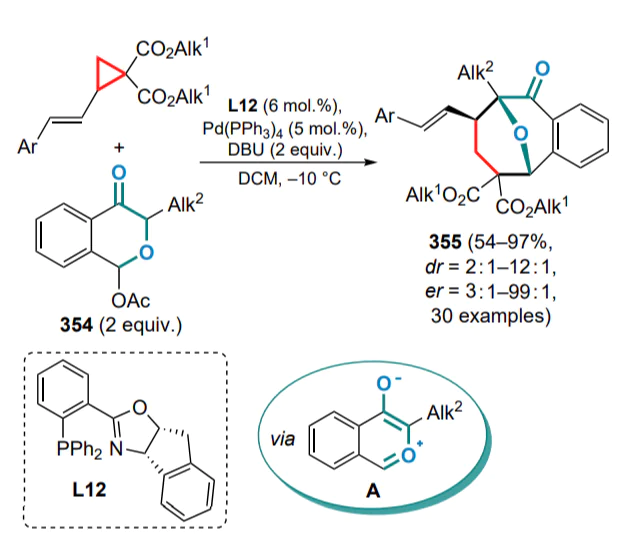
There are few examples of reactions between cyclopropanes and thiadienes. One of the first transformations of this type was the (4+3)-cycloaddition of DAC to give thiochalcones 356a, catalyzed by scandium(III) triflate (Scheme 151)[237].
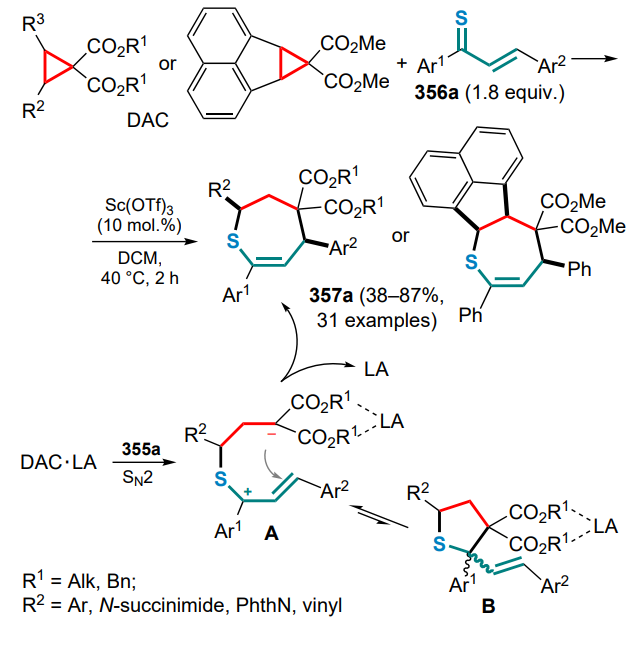
The SN2-opening of the 3-membered ring delivers the zwitterionic intermediate A, which undergoes further cyclization via the thermodynamically favourable pathway of closing the 7-membered ring to give products 357a. The (3+2)-cycloaddition product B can be obtained intermediately as a kinetic product.
The study of a similar (4+3)-cycloaddition of DAC and 3-benzylideneindolin-2-thiones 356b formed under reaction conditions from stable dimers 356'b continued the work in this direction (Scheme 152)[238]. The process was run using ytterbium(III) triflate as the catalyst. The SN2 nature of the nucleophilic monomer attack on the Lewis acid-activated complex paved the way for the synthesis of the chiral product (2S,5R)-357'b from the corresponding enantiomerically pure DAC with subsequent oxidation.
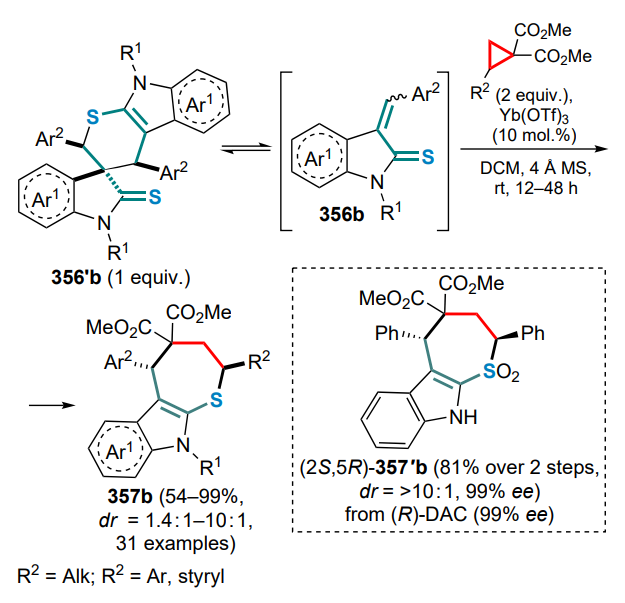
The combination of cyclopropane and oxadiene motifs in one molecule opens the way to intramolecular processes. One of the first classes of such compounds to be investigated were 1-cyclopropylprop-2-en-1-ones bearing the 1-positioned electron-withdrawing group on the three-membered ring. The initial focus was on substrates with a dimethylamino group on the oxadiene double bond as the directing leaving group.
This strategy has been implemented in a stepwise protocol using DAC 358 (Scheme 153)[239]. In the presence of ammonium acetate in acetic acid, conjugate substitution of the dimethylamine moiety by the acetate anion takes place, followed by a Cloke–Wilson rearrangement. After opening of the three-membered ring and closing of the five-membered ring, dihydrofurans 359 were formed with cis orientation of the exocyclic double bond.
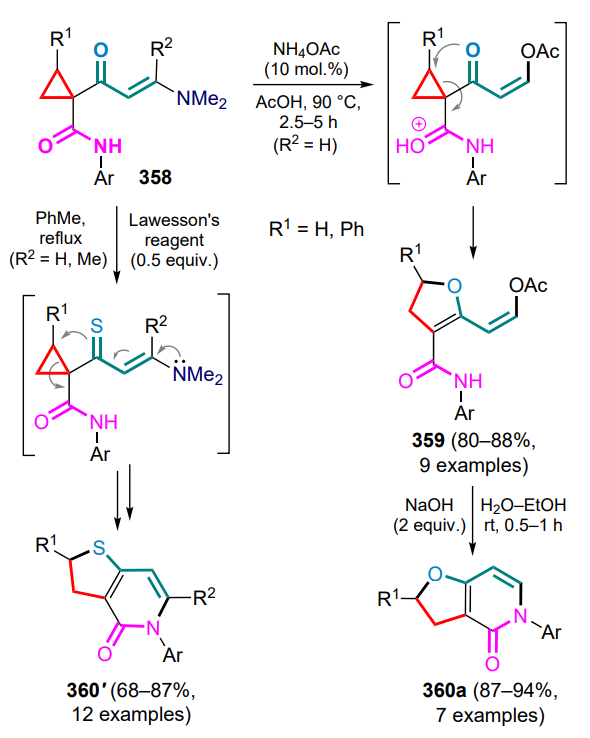
Under basic conditions, the products obtained can be easily converted into 2,3-dihydrofuro[3,2-c]pyridin-4(5H)-ones 360a. Notably, the interest of researchers in the assembly of the furo[3,2-c]pyridine core is due to the abundance of the corresponding derivatives among the alkaloids of the Rutaceae family.
Based on the same cyclopropanes 358, in the presence of Lawesson’s reagent (2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane-2,4-disulfide), thioanalogues have been obtained capable of further closing the dihydrothiophene ring by intramolecular attack of the sulfur atom on the three-membered ring activated by the donor dimethylamino group. The process is completed by aza-cyclization yielding dihydrothieno[3,2-c] pyridin-4(5H)-ones 360' (see Scheme 153)[240]. This reaction can also be considered as one of the few known examples of the reaction between cyclopropane and thiadiene moieties with the full involvement of the latter.
When treated with phosphoryl chloride or phosphorus tribromide, cyclopropanes 358 undergo cyclization with closure of only the pyridine-2(1H)-one ring to give compounds 361 (Scheme 154)[241]. The iminium intermediate A undergoes opening of the three-membered ring by the halide anion (B), followed by cyclization to pyridinone.
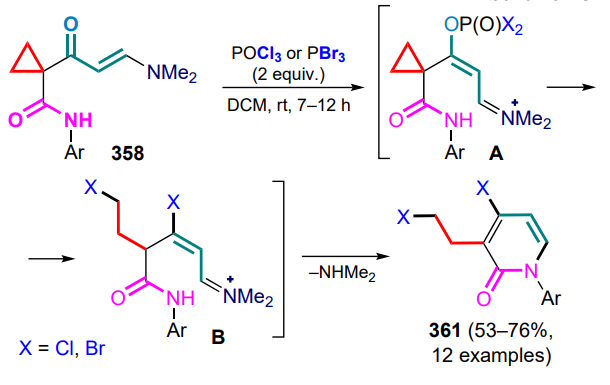
The formation of the pyridinone core can also involve an additional C1 moiety, the role of which can be taken over by the solvent (DMF) in the presence of trifluoromethanesulfonic anhydride due to the formation of highly reactive iminium triflate (Scheme 155)[242]. The latter attacks the amide nitrogen of cyclopropane 358 and the formation of iminium intermediate A is followed by two cyclizations. Finally, iminium intermediate B eliminates dimethylamine by hydrolysis to give 2,3-dihydrofuro[3,2-c]pyridin-4(5H)-ones 360b.
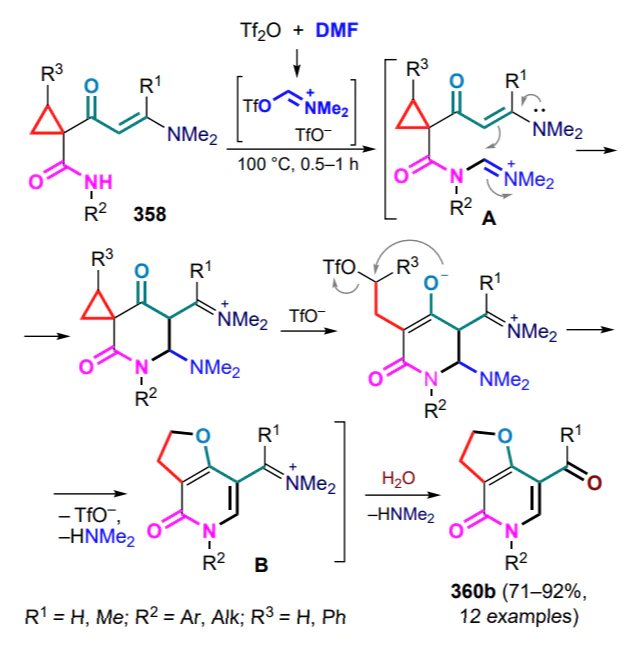
Finally, the cyclization of DAC 358' bearing the 1-positioned carbonyl group instead of an amide group can be performed. The reaction is carried out in the presence of an excess of ammonium acetate as a source of ammonia which, together with the conjugated oxadiene unit, initiates the cyclization with ringclosure of the dihydrofuran intermediate A. The latter forms a six-membered ring during the aza-cyclization and elimination of dimethylamine and, during dehydration, the intermediate B is aromatized to give 2,3-dihydrofuro[3,2-c]pyridines 362 (Scheme 156)[243].
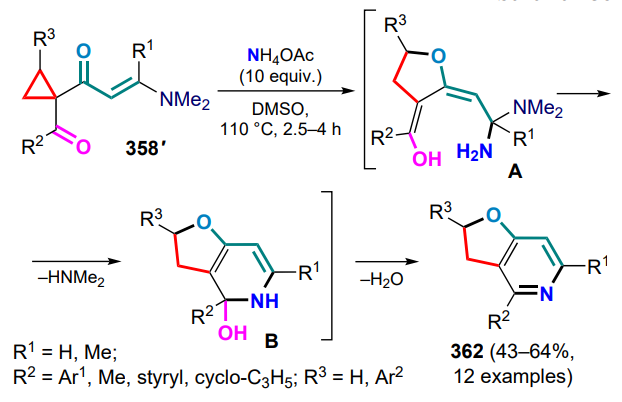
The acceptor 1-cyclopropylprop-2-en-1-ones, containing no dimethylamino group in the acrylic moiety, are also able to participate in cyclization processes with full involvement of the oxadiene unit.
For example, cyclopropanes 363 in the presence of sodium hydride in DMSO undergo anionic cascade cyclization via successive aza-Michael reactions (intermediate A) and oxoanionic opening of the three-membered ring to give an anionic intermediate, which can be represented by two resonance forms, B and C (Scheme 157)[244]. The latter (Ar1 = Ph; Ar2 = p-Tol), when treated with water, gives furo[3,2-c]-pyridinone 364 and, when reacting with aromatic aldehydes or chalcones, undergoes a C–C bond formation to afford furo[3,2-c]pyridinones 365 and 365', respectively. It should be noted that unlike aromatic aldehydes, aliphatic 3-phenylpropanal leads to a complex mixture of products.
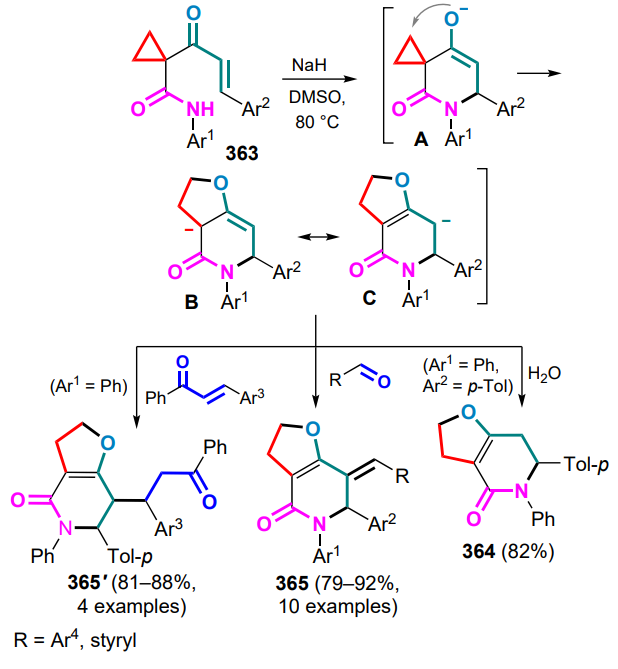
This process can be complicated and carried out in a multicomponent fashion involving 1-acetylcyclopropanecarboxamides, aldehydes and acrylonitrile (Scheme 158)[245]. Donoracceptor cyclopropanes 366 undergo aldol condensation with aldehydes to give the key oxadiene cyclopropane type 363 (see Scheme 157), which enters the aza-Michael reaction. However, intermediate A is further captured by acrylonitrile (intermediate B). A 1,3-hydride shift then occurs, after which intermediate C becomes capable of opening a three-membered ring (D), similar to the previous process. The addition of a second equivalent of acrylonitrile completes the formation of the final dinitrile 367.
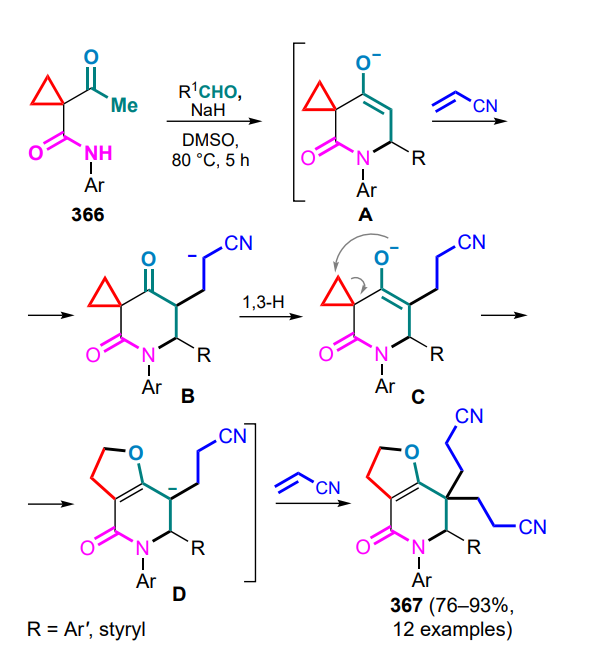
The transformations of oxadiene cyclopropanes 363 under the action of N-bromosuccinimide (NBS) and formic acid have also been studied (Scheme 159)[246]. The key bromonium intermediate A can undergo two types of intramolecular cyclization depending on the electronic nature of the substituent on the C=C bond of the oxadiene moiety. For electron-donating aryl substituents, 6-endo-tet cyclization (intermediate B) is preferred and further intramolecular nucleophilic substitution with bromide anion repulsion is possible.
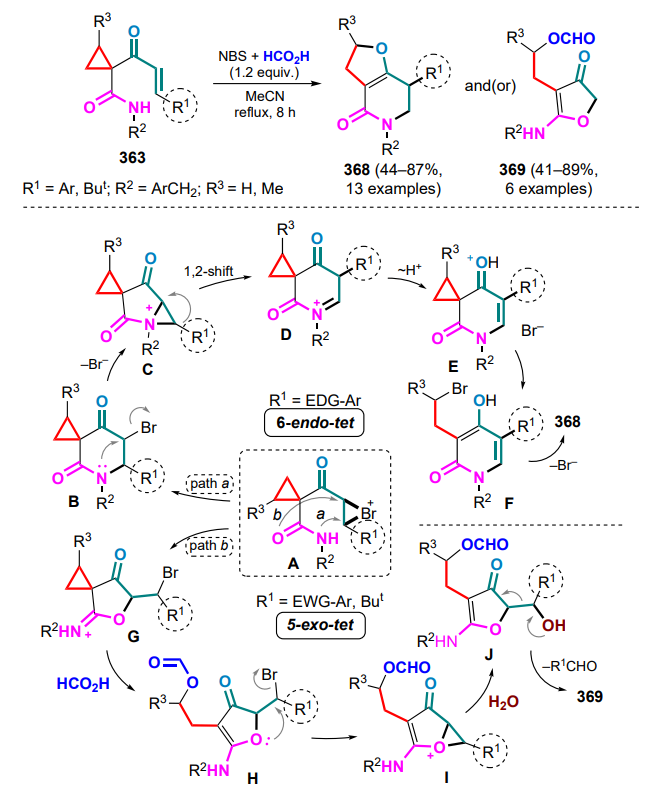
The annulated structure C relieves the ring strain due to an unusual 1,2-migration of R1 group and restoration of the sixmembered ring D. Further, the protonation of the carbonyl group by acid (intermediate E) facilitates the opening of the threemembered ring by bromide anion (F) with subsequent closure of the dihydrofuran part of the final furo[3,2-c]pyridinones 368.
In the case of electron-withdrawing and tert-butyl substituents on the double bond, the reaction route changes to 5-exo-tet cyclization promoted by the amide oxygen atom (intermediate G). The three-membered ring then opens in the presence of formic acid to give formate H, followed by the elimination of the bromide anion (I) with the anchimeric assistance of an adjacent oxygen atom, followed by the attack of a water molecule. Alcohol J releases aldehyde in the retro-aldol reaction to form the final 3(2H)-furanone 369. For phenyl and p-chlorophenyl substituents on the C=C bond, the formation of a mixture of both products is observed in moderate yields; the presence of a 2-furyl group at the same position leads to a mixture of products.
A similar cyclization can be implemented for the reaction of oxadiene derivatives of cyclopropanes 363 with primary amines in boiling ethanol (Scheme 160)[247]. As a result, 1H-pyrrolo[3,2-c]-pyridin-4(5H)-ones 370 are formed, with the highest yields observed for aryl- and benzyl-substituted amines, whereas for aliphatic amines the product yields do not exceed 34%.
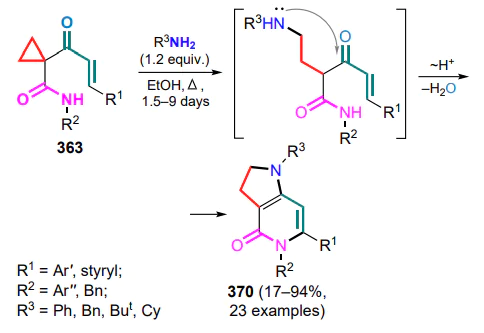
Benzoylcyclopropanes 371, due to the presence of the aryl ring, formally contain an oxadiene moiety, which allows them to be considered as one of the variants of substrates designed for the said intramolecular cyclization (Scheme 161)[248]. The ortho azide group acts as an equivalent of the amino group in the conditions of ruthenium(II) catalysis, with the possibility of further formation of imine A, which is converted to the final dihydrofuroquinolines 372.
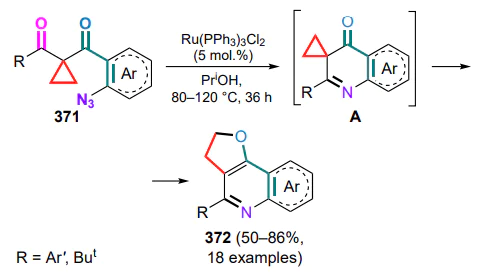
The homo-Nazarov reaction is one of the possible approaches to carry out intramolecular transformations of oxadiene cyclopropanes[249-252].
The cascade process with additional involvement of the carbonyl group based on the reaction between DAC 373 and allyl silanes in the presence of tin(IV) chloride is particularly worth mentioning in this review as one of the most suitable examples (Scheme 162)[253]. Under different conditions, both cycloalkenes 374 and hexahydrobenzofurans 375 can be obtained. In both cases, the opening of the three-membered ring in the complex of the substrate with the Lewis acid (intermediate A) is accompanied by cyclization and nucleophilic attack of the oxyallyl carbocationiс intermediate B by allyl silanes (C). Next, catalytic amounts of Lewis acid and a relatively small TMS group promotes the elimination of the silyl moiety with the Hosomi–Sakurai double bond closure (arrow a, product 374), whereas the bulky TBDPS group, less susceptible to elimination, stoichiometric amounts of Lewis acid, lower temperature and greater dilution lead to oxycyclization with the retention of the silicon atom (arrow b, product 375).
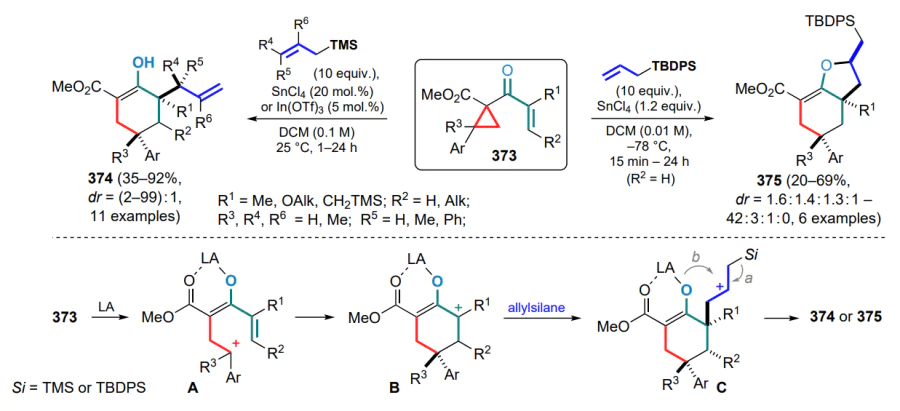
A cascade cyclization based on double catalysis by silver(I) triflate and gold(I) complex involving 1-cyclopropylprop-2-in1-ones 376, which are acetylene analogues of the abovementioned oxadiene cyclopropanes has been proposed (Scheme 163)[254]. The starting compounds 376 were in turn obtained by the rhodium(II)-catalyzed reaction of diazo ketones 377 with alkenes. Both steps can be combined in a synthetic strategy or in a one-pot protocol. The role of silver(I) triflate as a Lewis acid is to activate the carbonyl group for the ClockWilson rearrangement. Furthermore, in the presence of the gold(I) complex, the resulting dihydrofuran undergoes intramolecular nucleophilic attack of an electron-donating aryl moiety on the coordinated triple bond to yield heterocycles 378. For acetate-substituted cyclopropanes, the aromatization affords furan 379.
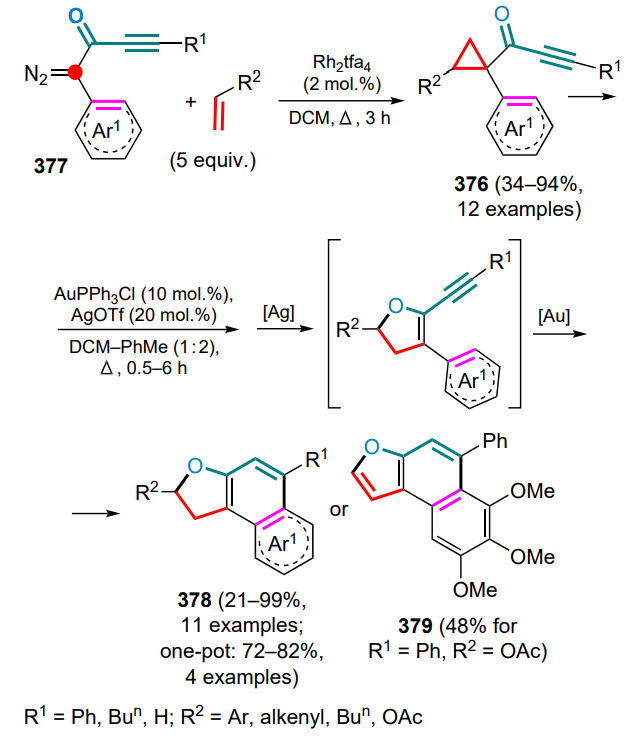
The conversion of (1-propynyloxo)cyclopropaindane 380 in the presence of the same catalysts produces pentacyclic structures 381 and 382 (Scheme 164). The formation of a mixture of such compounds is the result of competition between standard rearrangement (path a) and intramolecular alkylation of the electron-donating aryl moiety (path b).
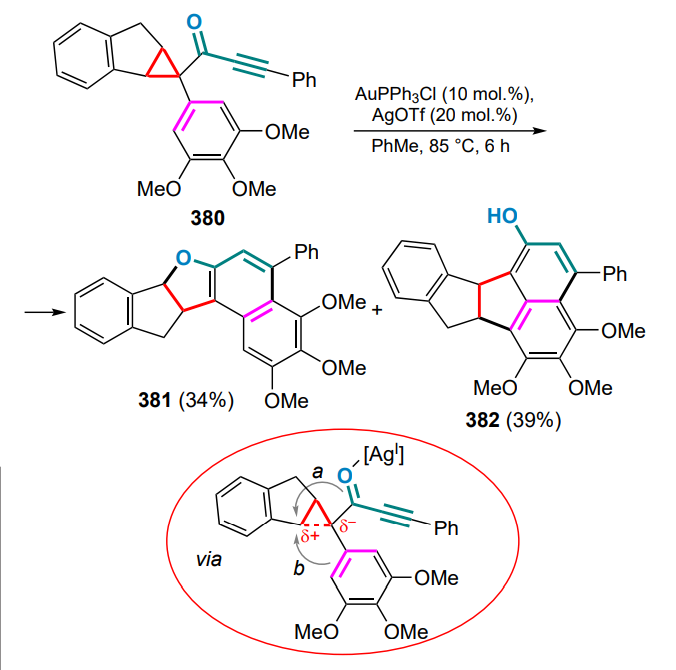
In addition to the substrates bearing a cyclopropyl-substituted carbonyl group of the oxadiene system, their analogues with a three-membered ring on the C=C bond, reacting with full involvement of the heterodienic fragment, have been described. Thus, (E)-β-chlorovinylketones 383, obtained from ethynylcyclopropane and acid chlorides in the presence of AlCl3, undergo a cascade reaction when exposed to microwave radiation (MW) to form 4-vinylfurans 384 (Scheme 165)[255]. The opening of the three-membered ring is accompanied by its expansion and ring closure to furan with release of HCl. The process is completed by isomerization of the double bond and 4π-electrocyclic ring-opening of the cyclobutene. It is noteworthy that isomeric (Z)-β-chlorovinylketones do not undergo such a reaction under similar conditions, but only hydrolyze with a low product yield.
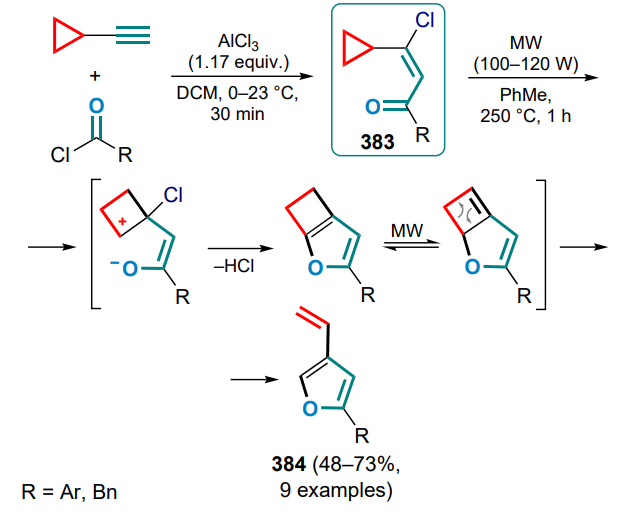
Finally, the last group of intramolecular reactions of cyclopropane and oxadiene moieties includes processes that produce conjugated oxonium intermediates, which formally can be called methylene(vinyl)oxonium derivatives. The cascade processes involving oxaspiro[2.4]heptadienium intermediates 385 as products of 5-endo-dig cyclization of starting 1-(1-alkynyl)cyclopropyl ketones 386 have been studied most carefully.
The first study[256] in this area explored the cyclization of alkynylcyclopropyl ketones 386 under the action of nucleophiles in the presence of triphenylphosphine gold(I) or silver(I) triflates (Scheme 166). Activation of the triple bond by the metal atom facilitates intramolecular attack on the heminal carbonyl group to form a key cationic intermediate 385 which is further ringopened by nucleophilic attack to give furans 386. This mechanism was further studied by DFT calculations[257]. Subsequently, a similar reaction of 1,4-oxyaminomethylation was proposed using the same substrates 386 and N,O-acetals 388, in which an intermediate iminium cation is formed to stabilize the anionic centre[258].
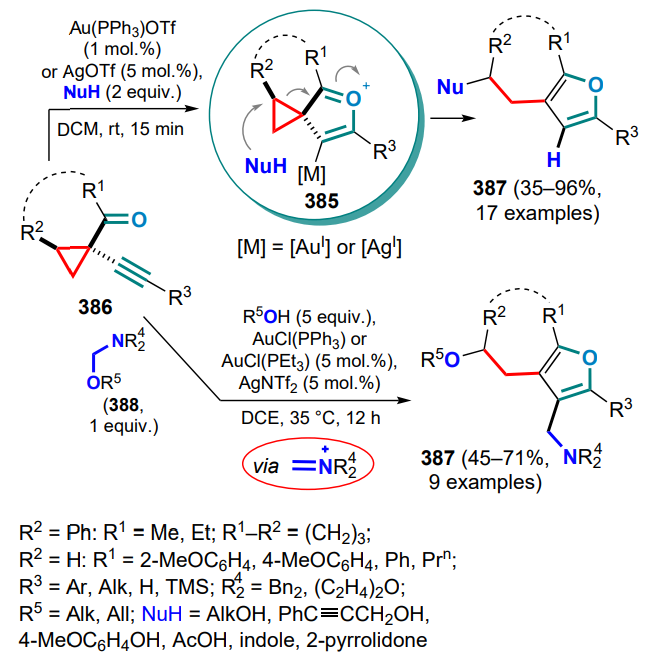
A similar reaction was reported, in which the role of activator of the triple bond was assigned to the electrophilic particles I+ or PhSe+[259]. Nucleophilic opening of the oxonium intermediate of type 385 leads to iodine- and selenium-substituted furans 387'.
Opening the three-membered ring of the oxonium intermediate can give the 1,4-dipole A with the anionic centre ‘masked’ by the metal atom, which additionally stabilizes the dipole and reduces its nucleophilicity[260]. Therefore, this can favour annulation, as has been demonstrated for MVO reactions with N-protected indoles and ketones and with 1-azadiene (Scheme 167). In this case, the gold(I) complex of carbene was preferred to catalyze the formation of annulated furans 389, 389' and 389''[261].
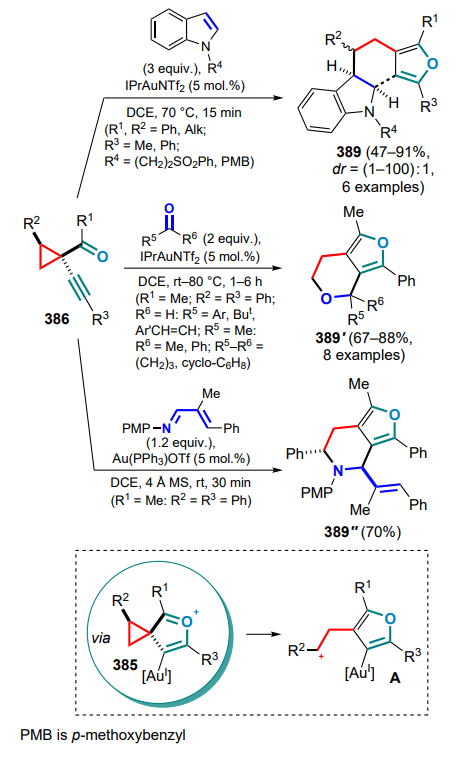
The reaction of compounds 386 with ethyl vinyl ether produces unstable bicyclo[3.2.0]heptanes 390 sensitive to silica gel (Scheme 168)[262]. Therefore, it is advisable to subject the products to further treatment, such as TsOH or other reagents, for the preparative isolation of various functionalized compounds 391. In contrast to the previous reactions, the starting oxonium intermediate 385 undergo a 1,3-dipolar cycloaddition to ethyl vinyl ether in the form of the resonance carbenoid ylide A rather than the opening of the three-membered ring. The bridging intermediate B further undergoes a 1,2-alkyl shift (intermediate C) and ring-opening to form the carbocation D. Finally, expansion of the three-membered ring and removal of the catalytic species complete the assembly of the carbon skeleton of compounds 390. The possibility of introducing hydrogen, methoxy and allyl groups into the carbocationic centre of intermediate E is also shown.
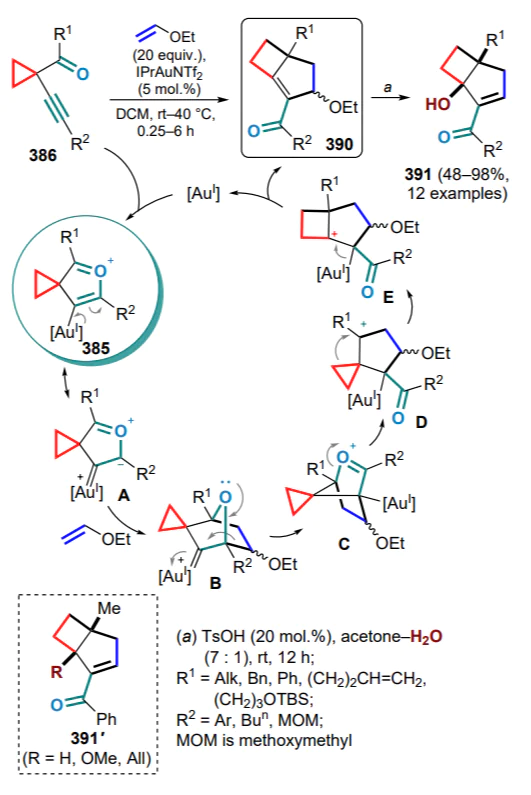
Using nitrones as 1,3-dipoles in a similar reaction, the concept of (4+3)-annulation involving oxonium intermediates 385 as an approach to the synthesis of oxazepinofurans 392 and 392’ is realized (Scheme 169)[263, 264]. As in previous cases, efficient catalysis is reached in the presence of Lewis p-acids such as copper(II) triflate or gold(I) salts, the latter being preferred. A stepwise mechanism of annulation with opening of the three-membered ring by the SN2 mechanism has been established. The high diastereoselectivity and the influence of the nitrone double bond configuration on the arrangement of substituents in the final products may indicate the predominant conformation of the intermediate A. Interestingly, in the presence of σ-Lewis acids, the chemoselectivity of the process changes: a (3+3)-annulation occurs giving no oxonium intermediate, which made it possible to propose an approach for the synthesis of tetrahydro-1,2-oxazines 393 (see Scheme 169).
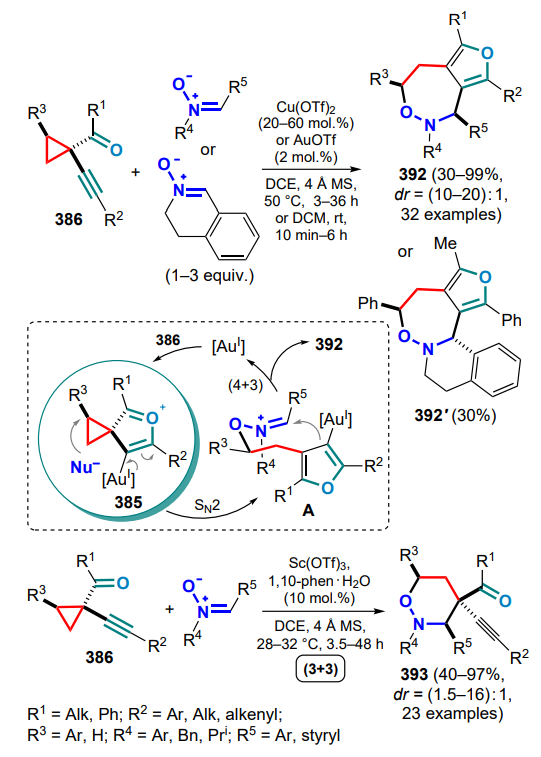
Enantioselective modification of the (4+3)-annulation using a chiral gold(I) complex led to the development of a method for the kinetic resolution of racemic alkynylcyclopropyl ketones 386 as an approach to (1R,2S)-enantiomers which are unavailable by the seemingly more obvious asymmetric cyclopropanation[265]. Enantiomerically pure alkynylcyclopropyl ketones were further converted to chiral heterocycles (1R,4R)-392 under standard conditions using the same nitrones (Scheme 170)[266].
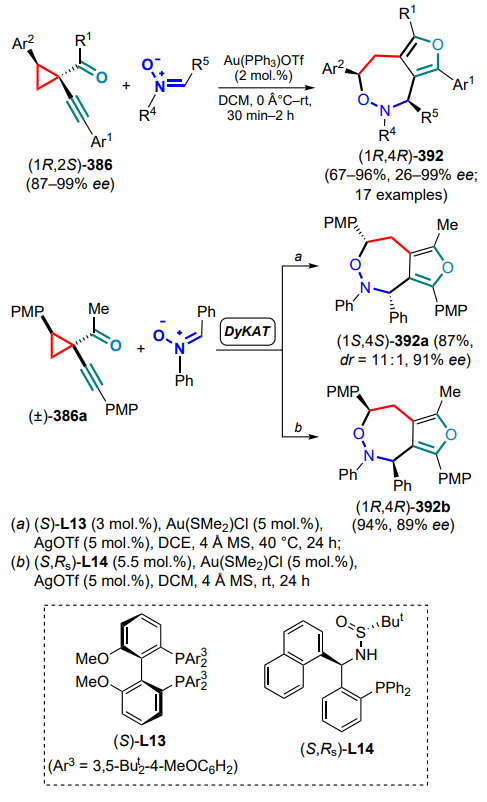
Finally, sporadic examples of the preparation of both enantiomers of hydrogenated furo-oxazepine 391a,b in a modification of the dynamic kinetic asymmetric transformation (DyKAT) of racemic alkynylcyclopropyl ketone 386a in the presence of chiral ligands L13, L14 have been described (see Scheme 170)[267, 268].
The activation of the triple bond and the σ-bond of the threemembered ring of alkynylcyclopropyl ketones 386 can be combined in a rhodium(I)-catalyzed stereoselective (4+1)-annulation in the presence of CO (Scheme 171)[269]. The resulting intermediate A is further isomerized to furan by insertion of CO followed by the reductive elimination of the catalyst and release of 5,6-dihydrocyclopenta[c]furan-4-ones 394.
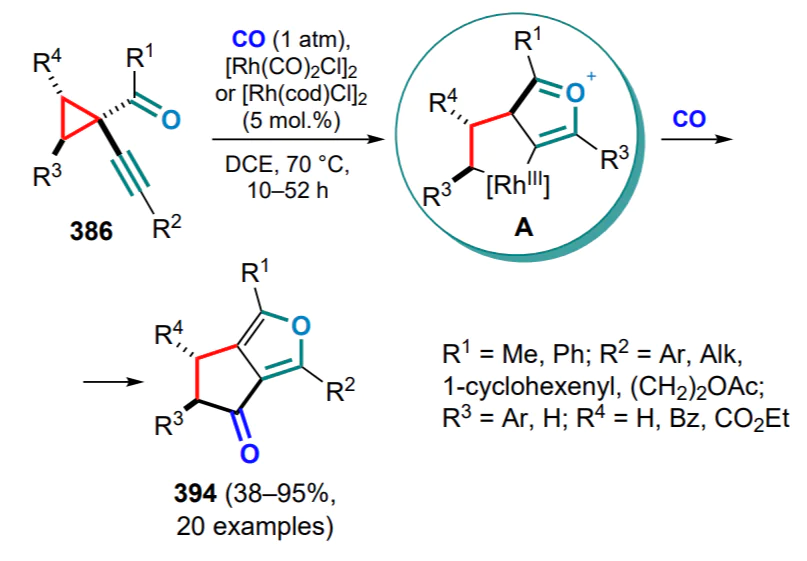
The insertion of carbonyl groups tethered to a threemembered ring (substrates 386') into the parent alkynylcyclopropyl ketones made it possible to complicate the cascade process by adding the intramolecular (4+2)-crosscycloaddition step involving 1,4-dipole A to produce furobicycles 395 (Scheme 172)[270]. This transformation proceeds in the presence of gold(I) and silver(I) salts, while nickel(II) perchlorate leads to an intramolecular (3+2)-cycloaddition (intermediate B) without furan ring closure, accompanied by the formation of alkynyl ketones 396. In addition, an azamodification of this process has been proposed involving an imine moiety obtained by condensation of PMP–NH2 with the distal aldehyde group of compound 386' (R2 = Ph, R3 = H).
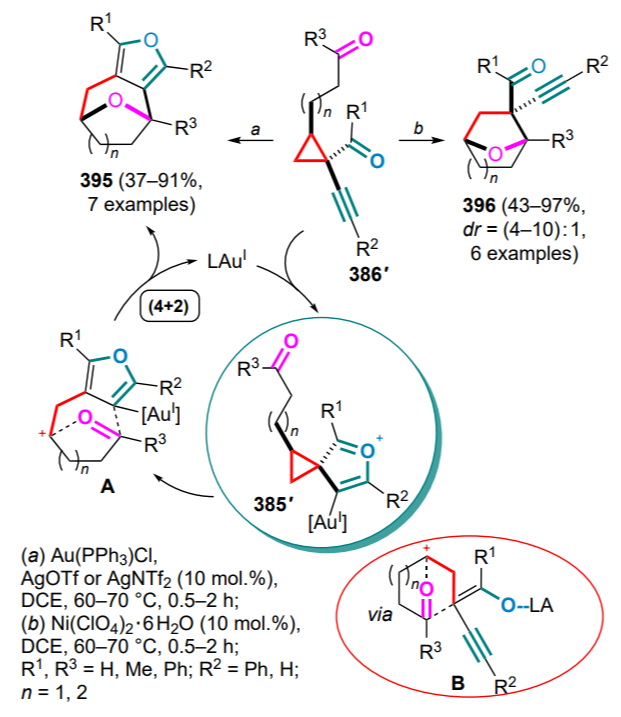
The cis arrangement of vicinal carbonyl and alkyne units in cyclopropane promotes the formation of different cyclic oxonium intermediates, namely 3-methylidene-2-oxabicyclo[3.1.0]hexenium cations A, in the gold(I)-catalyzed 5-exo-dig cyclization (Scheme 173)[271]. Such an idea has been implemented in the cascade transformation of 1-alkynyl-1-aryl2-acylcyclopropanes 386 in which the key cation A undergoes the three-membered ring opening (intermediate B) and intramolecular cyclization through the aryl moiety with release of dihydroindeno[2,1-b]pyrans 397. Replacement of the AuI catalyst by silver(I) triflate under basic conditions alters the sequence of steps, promoting the initial ring-opening (C) with subsequent cyclization and isomerization of oxepin D to arene oxide E. Further ring-opening of the oxirane with 1,2-migration of the substituent R located in the acyl moiety of the substrate, mediated by silver(I) triflate, leads to 2,4,6-substituted phenols 398a.
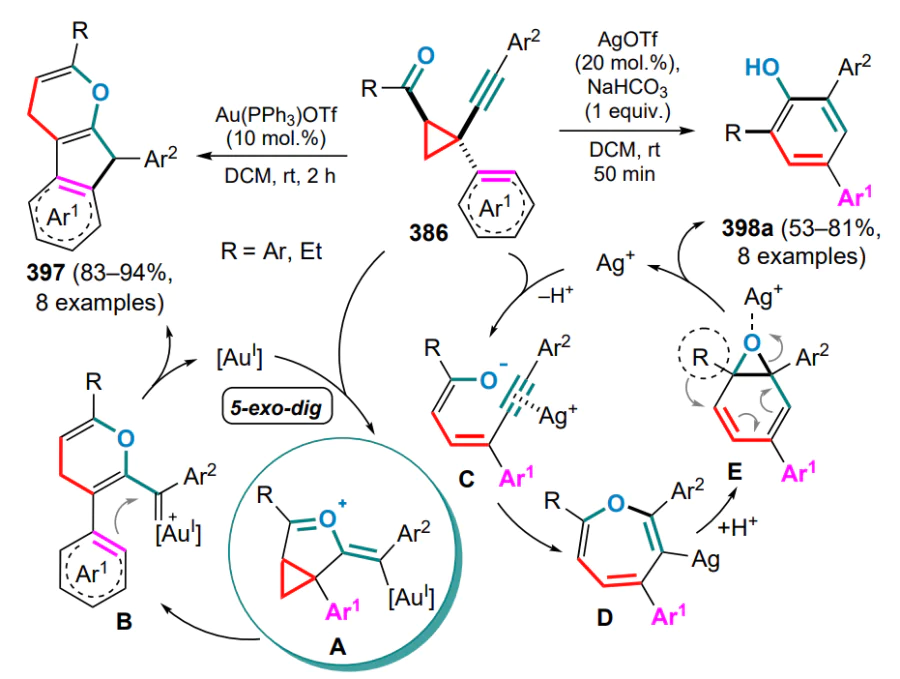
It is noteworthy that, in the presence of both catalytic systems, the 2-thiophene substrate 386b is converted only to the corresponding phenol 398b (Scheme 174)[271]. In the case of gold(I) catalysis, the authors explained this fact by the formation of the conjugated oxabicyclo[4.1.0]heptadiene intermediate A, stabilized by coordination with the metal atom, during the 6-endo-dig cyclization.
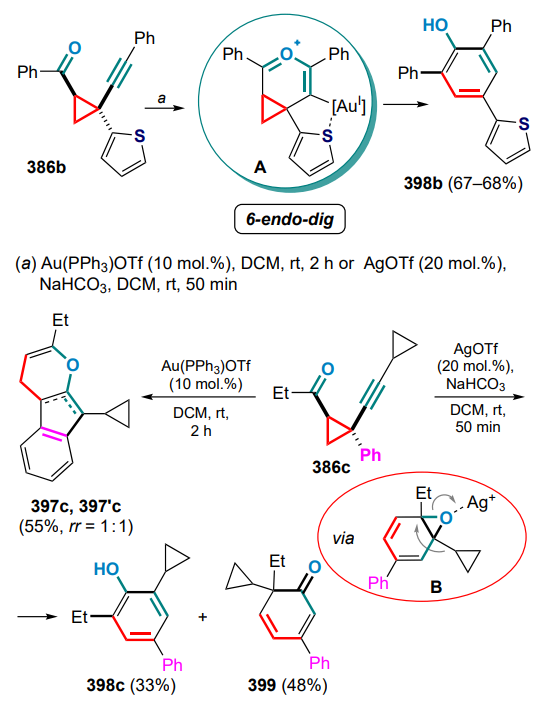
The presence of a cyclopropane moiety on the triple bond of substrate 386c also causes a deviation from the standard process: in the presence of AuI complex, a mixture of compounds 397c and 397’ with an isomeric position of the double bond is formed. AgOTf-mediated catalysis gives, in addition to the expected phenol 398c, cyclohexadienone 399, probably due to the alternative 1,2-migration of the cyclopropyl group in the intermediate B (see Scheme 174).
4.2. Reactions of methylidenecyclopropanes with oxadienes
Only a few examples of intermolecular reactions of methylidenecyclopropanes with conjugated oxadiene systems with full involvement of double bonds are described. Methylidenecyclopropanes behave as one-atom synthons designed for the assembly of dihydrofurans 400 via the (4+1)-cycloaddition using conjugated enones 401 catalyzed by nickel(0) complexes (Scheme 175)[272]. In most cases, the choice of dimethylphenylphosphine ligand is optimal in terms of product yield and diastereoselectivity of the reaction. However, only in the presence of iminophosphine L15 it was possible to carry out the reaction for the bulky tert-butyl (R2 = But ) derivative, and also to improve the yield from 57 to 98% in the case of 2-furyl substituent (R2 = 2-Fu), while the diastereomer ratio decreased from 9:1 to 2:1.
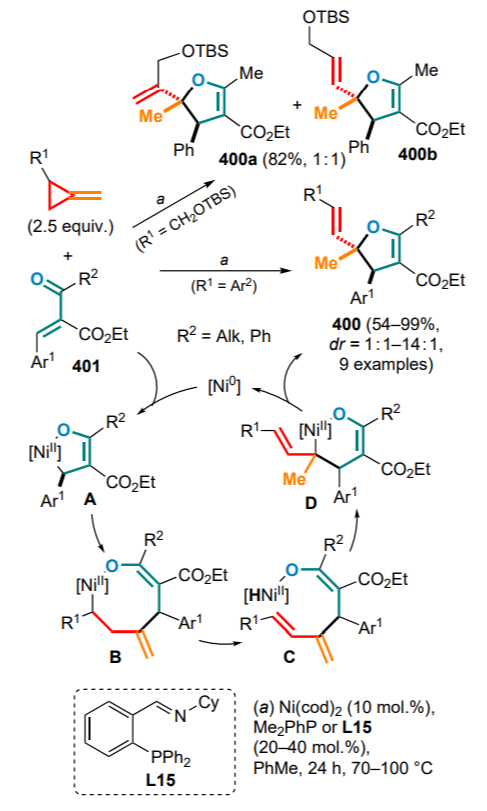
The reaction begins with the formation of oxanickelacycle A, which further inserts methylidenecyclopropane into the Ni–C bond and rearranges to oxanickelacycle B. Then, β-hydride shift delivers the complex C, which, during the subsequent intramolecular attack on the exomethylene bond, generates more stable six-membered oxanickelacycle D and, upon elimination of the catalytic species, gives the desired product. Methylidenecyclopropane bearing a CH2OTBS substituent in the three-membered ring gives in this reaction a mixture of regioisomers 400a and 400b, apparently due to two possible ways of the ring opening in the cyclopropylmethyl–butenyl rearrangement stage.
Methylidenecyclopropanes containing electron-withdrawing substituents react with cyanoenones 402 to yield dihydrofurans 403 or furans 404 when the base is added in equimolar amounts in the presence of Lewis base organocatalysts (Scheme 176)[273]. The addition of the catalyst species is accompanied by the opening of the three-membered ring in the zwitterion A, which can undergo the Michael addition in the more reactive isomeric form B to give cyanoenones 402 followed by an intramolecular closure of the five-membered ring. An equimolar amount of base promoted aromatization of dihydrofuran 403 to furan 404.
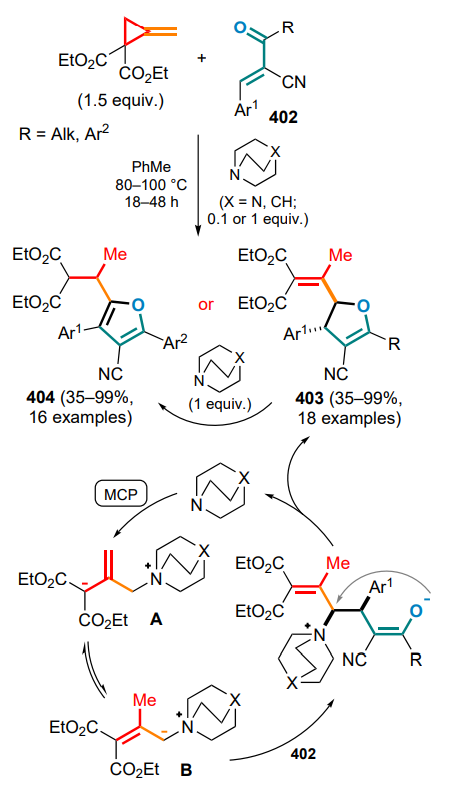
Enones 405 can undergo two types of reaction under palladium(0) catalysis, depending on the nature of the phosphine ligand (Scheme 177)[274]. The reaction of methylidenecyclopropanes with enones begins with the insertion of a catalyst species into the proximal bond of the substrate (intermediate A), followed by the opening of the four-membered ring to diene during β-hydride shift and reductive elimination (B). Further regioselective oxidative 1,4-coupling to enone yielding the key intermediate C is possible. Depending on the ligand environment, the process can follow two pathways. Thus, the sterically loaded and more electron-donating diadamantylbutylphosphine contributes to the reduction of the Lewis acidity of the catalytic species and prevents the cyclization of the intermediate, leaving only the possibility of the formation of conjugated dienes 406 via γ-deprotonation (path a). At the same time, triphenylphosphine facilitates the completion of the formal (4+2) cycloaddition with the closure of oxapalladacycle D, leading to dihydropyrans 407 (path b). Ester-free enone 405, instead of the above reactions, only undergo the methylidenecyclopropane ring-opening to diene (B), indicating the necessity of the presence of this fragment for the smooth running of the process.
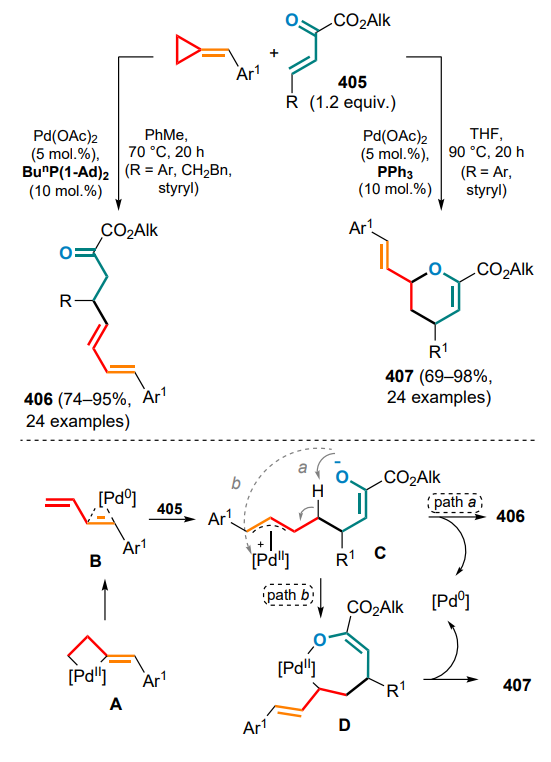
As oxadiene in the reaction with methylidenecylopropanes, o-quinone methides 408 can be used, obtained in situ from the corresponding salicylaldehydes and triethyl orthoformate in the presence of scandium(III) triflate as Lewis acid, which is also involved in other key transformations (Scheme 178)[275].
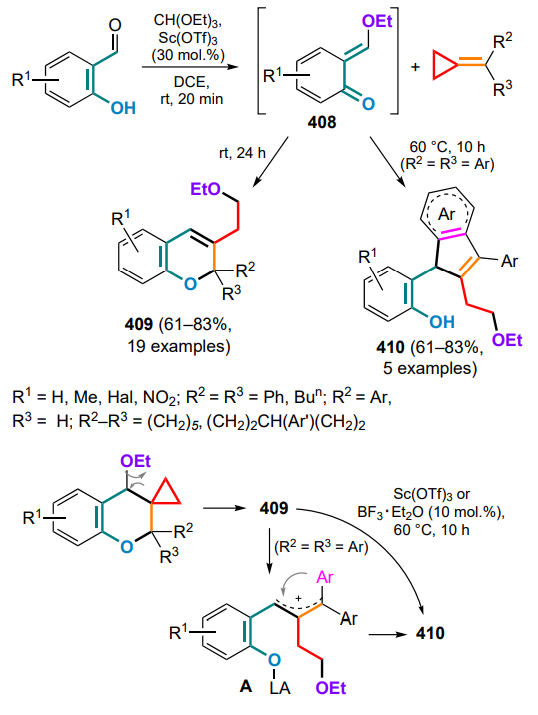
At room temperature, o-quinone methide 408 undergoes (4+2)-cycloaddition with methylidene cyclopropane, accompanied by elimination of the ethoxyl group and further opening of the three-membered ring to give 2H-chromenes 409. If the substituents on the exomethylene bond of cyclopropane are represented by aryl groups, it is possible to continue the cascade process on heating with the cleavage of the C–O bond, accompanied by the formation of the allyl carbocation A, which undergoes intramolecular Friedel–Crafts cyclization to indenes 410. This is consistent with the observed conversion of 2H-chromenes 409 to indenes 410 when heating reactants in the presence of Lewis acids.
Further work has been devoted to intramolecular reactions between methylidenecyclopropane and oxadiene moieties. An example of such a process at a sufficiently remote key reaction centres is a cascade of reactions involving cyclohexadienetethered methylidenecyclopropanes 411 (Scheme 179)[276]. In the presence of Pd0 catalyst and isopropyl alcohol, the transformation occurs to yield a mixture of tricyclic compounds 412 and 413, with the former prevailing in most cases. The catalytic species is capable of oxidative insertion into methylidenecyclopropane distal bond with subsequent transformation of palladacycle to allyl complex A, which undergoes the intramolecular Michael addition. The π-allyl complex B is then protonated under the action of isopropyl alcohol with the reduction of the enone system of intermediate C, which can further react in two ways. In the first case, a repeated Michael addition is possible (intermediate D), followed by destruction of the resulting complex by isopropyl alcohol, isolation of the product 412 and reduction of the catalytic species of complex E during the β-hydride shift (pathway a). In the second variant, the β-hydride shift occurs first and complex F is formed in which the catalytic centre is capable of double bond insertion with closure of palladacycle G and further elimination of product 412 with reduction of palladium(0) complex (pathway b). The presence of product 413 is explained by the side migration of the palladium complex in the initial intermediate A with further formation of the PdII–C(H) bond and transformation into palladacycle I, closing the catalytic cycle after reductive elimination.
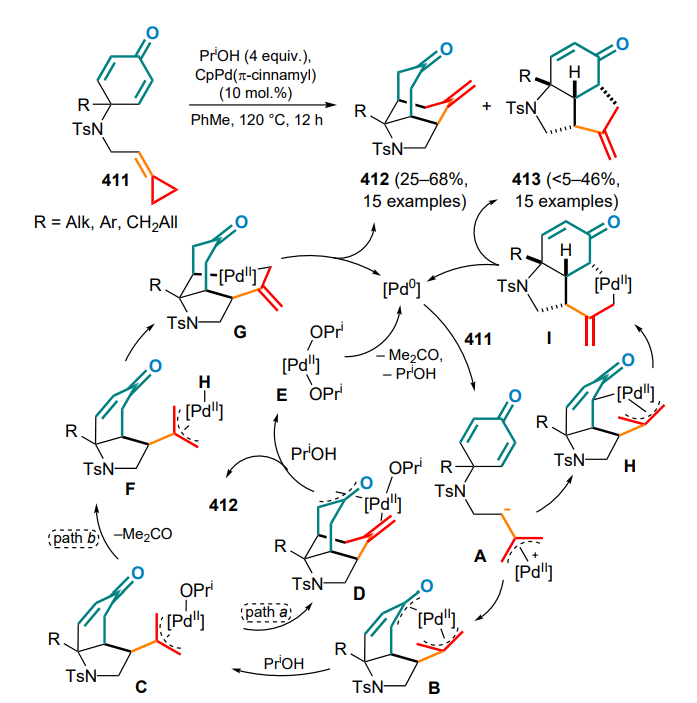
An unusual example of intramolecular cyclization is the thermal isomerization of 3-[(2-arylmethylidenecyclopropyl) methylidene]pentane-2,4-diones 414 to give spiro[2.5]octa-3,5-dienes 415[277]. In the presence of L-proline, a one-pot modification of this transformation was also carried out using substrate precursors 414, aldehydes 416 and 1,3-dicarbonyl compounds (Scheme 180). The reaction produced a mixture of diastereomers and sometimes the single trans isomer, since sterically demanding cis diastereomers were often unstable under these conditions, leading to a decrease in their content in the reaction mixture up to the complete absence. The reaction pathway consists of the rearrangement of the initial methylidene cyclopropane 414 to isomer A, which undergoes a Cope rearrangement. Prolonged heating of some condensation products of (arylmethylidene)cyclopropanecarbaldehyde 416 and cyclopenta-1,3-dione provided isolation of minor products of [3,5]-sigmatropic rearrangement (417). Substrates bearing aliphatic substituents on the C=C bond of methylidenecyclopropane did not react under these conditions.
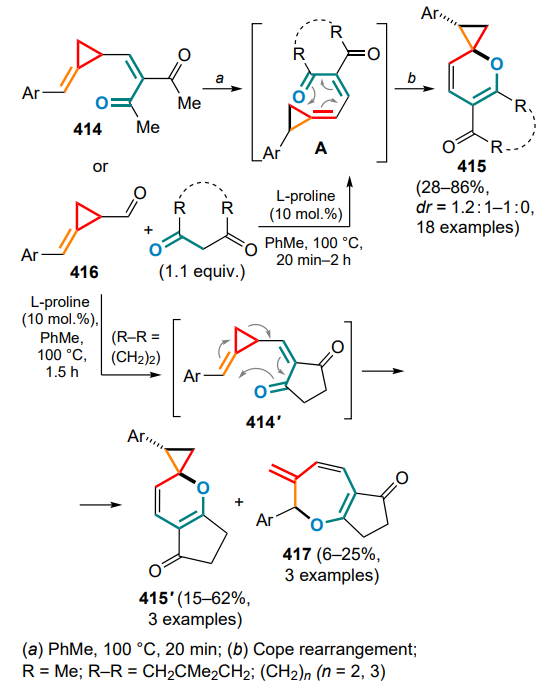
4.3. Reactions of vinylidenecyclopropanes with oxadienes
(Acylvinylidene)cyclopropanes contain closely spaced methylidenecyclopropane and oxadiene moieties. Unlike (arylvinylidene)cyclopropanes (see Section 2.7), in which the opening of the three-membered ring precedes the cyclization of the conjugated unit, in the above substrates, the activation of the exomethylene bond promotes further 5-endo-tri cyclization yielding intermediates A, similar to the oxa- and azaspiro[2.4] heptadienium intermediates 268, 385 (see Sections 3.2, 4.1).
This approach was first demonstrated for alkyl-substituted vinylidenecyclopropanes 418, which were isomerized to 2-oxabicyclo[3.2.0]heptadienes 419 in the presence of PdCl2 catalysts (Scheme 181)[278]. Aryl-substituted substrate in the presence of atmospheric oxygen gave a mixture of products 420 and 421. Having established optimized conditions, it was possible to propose an oxidative cycloisomerization for substrates bearing both types of substituents using Dess–Martin periodinane (DMP). In this case, cyclobutanones 420 were formed, which were isomerized to furanones 421 in the presence of trifluoromethanesulfonic acid.
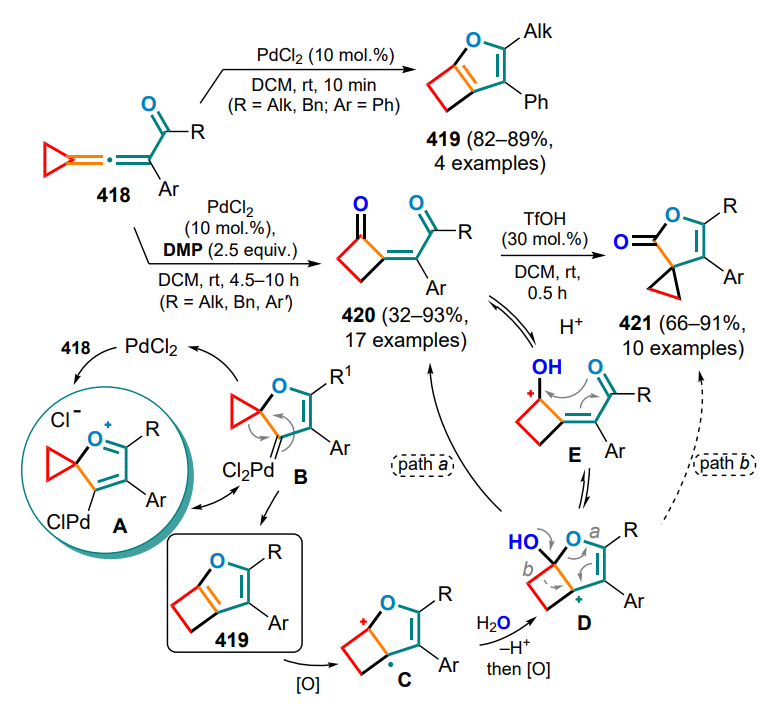
Common to these processes is an intermediate formation of 2-oxabicyclo[3.2.0]heptadienes 419 as a result of intramolecular cyclization and opening of the three-membered ring of the intermediate in oxonium (A) or carbenoid (B) forms. Next, oneelectron oxidation (intermediate C) with hydration and subsequent complete oxidation to cation D occurs. The latter tends to release a proton with the furan ring opening to cyclobutanone 420 (path a). In the presence of acid, selective protonation is possible, promoting reverse cyclization (E) and the cyclobutane ring opening to give furanones 421 (path b).
Under the action of copper(I) iodide and iodine, the intramolecular cyclization of vinylidene cyclopropanes 418 occurs via the formation of only oxaspiro[2.4]heptadienium intermediates A, followed by nucleophilic cleavage of cyclopropane and release of diiodides 422 (Scheme 182)[279]. A similar metal-free reaction was carried out using the same substrates by treatment with sodium sulfinate and acetic acid followed by addition of iodine[280]. The Michael addition of the sulfinate anion (B) is accompanied by activation of the exomethylene bond by the iodine cation with intramolecular cyclization to intermediate C, similar to copper complex A. Further attack of the iodide anion yields functionalized furans 422'.
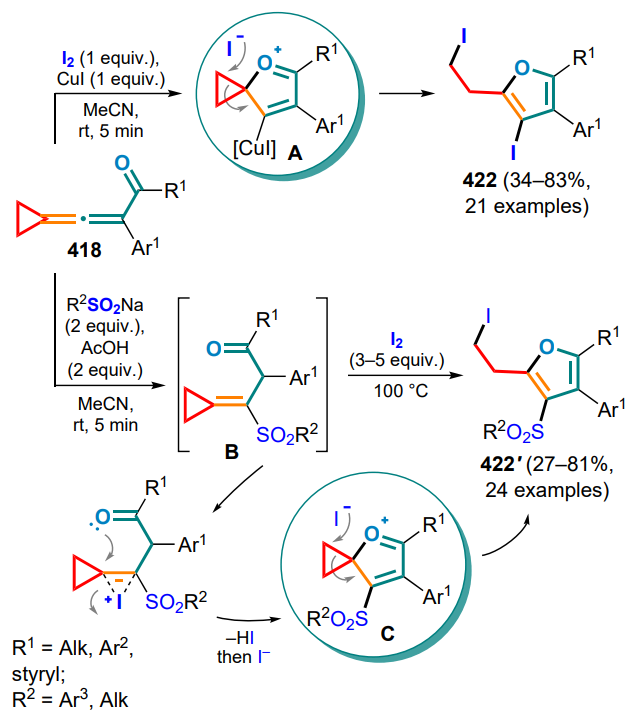
The ability of (acylvinylidene)cyclopropanes 418' to be catalytically isomerized into 2-oxabicyclo[3.2.0]heptadienes 419, containing on the one hand a cis-diene system and on the other hand, an electron-rich strain double bond as a dienophile, allows them, in the presence of copper(I) chloride, to undergo Diels-Alder dimerization (A), followed by opening of the bridging C–O bond, ring contraction and formation of polycyclic adducts 423 (Scheme 183)[281]. It is noteworthy that this transformation occurs only for substrates 418' bearing an arylsubstituted carbonyl group; with an iso-butyl substituent (418, R1 = Bui), the reaction stops at the stage of cyclobutene 419' formation. The initial dimerization product A has relatively closely spaced double bonds facing each other. This fact was subsequently exploited to photocatalytically modify the cascade process with the addition of a (2+2)-cycloaddition step in the presence of an iridium(III) catalyst and under visible light irradiation[282][282]. As a result, the corresponding cage C2-symmetric dioxabishomocubanes 424 were successfully obtained.
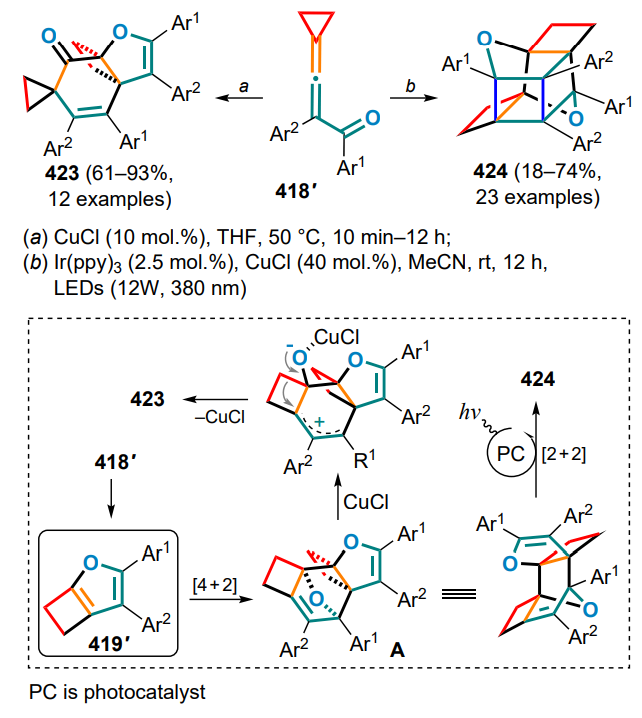
The idea of using 2-oxabicyclo[3.2.0]heptadienes 419, generated in situ from vinylidenecyclopropanes 418, in cycloaddition reactions has been further developed. A number of variations of the Diels-Alder reaction with various carbo-, oxa- and azadiene substrates, in which cyclobutenes 419 act as dienophiles, have been proposed (Scheme 184)[283, 284]. Thus, the inverse electron demand oxo-Diels–Alder reaction of 2,3-dioxopyrrolidines 425a using silver(I) hexafluoroantimonate catalyst gave pyrrolidone-fused polycycles 426a. It was found that the cycloaddition per se also takes place in the absence of AgSbF6, indicating the predominant effect of the catalyst on the formation of cyclobutenes 419 from vinylidenecyclopropanes 418. Catalytic systems based on gold(I) complexes and(or) silver(I) salts promote similar (4+2)-cycloaddition reactions with benzo[c]isoxazoles 425b, 3-acetyl-2H-chromen-2-one (425c), exocyclic azadiene 425d, reactive 1,3-diphenylisobenzofuran 425e, electron-deficient dienes 425f,g. As a result, a variety of polycyclic structures 426b–g were synthesized (see Scheme 184), demonstrating the great potential of the developed approach involving cyclopropyl, oxadiene and an additional hetero(carbo)diene moiety with the full involvement of the latter.
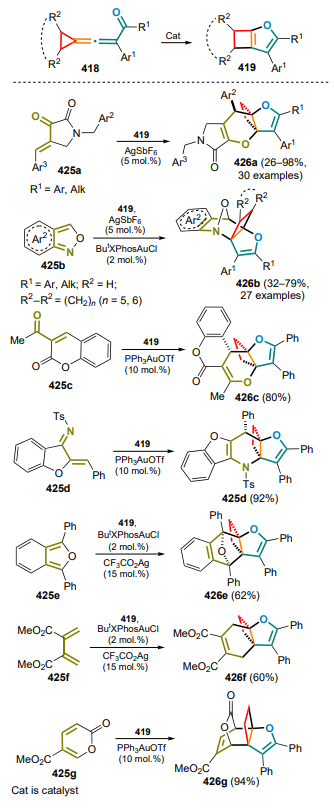
The strained double bond of 2-oxabicyclo[3.2.0]heptadienes 419 proved to be a good trapping agent in the (3+2)-cycloaddition reactions involving N-iminoquinazolinium ylides 427a, nitrone 427b and oxidopyrylium ylides 427c, generated in situ from the benzopyranone precursors in the presence of DBU. The reactions are catalyzed by gold(I) or silver compounds and proceed quite readily in oxygenated solvents or CHCl3 to yield polycyclic heterocycles 428a–c (Scheme 185).
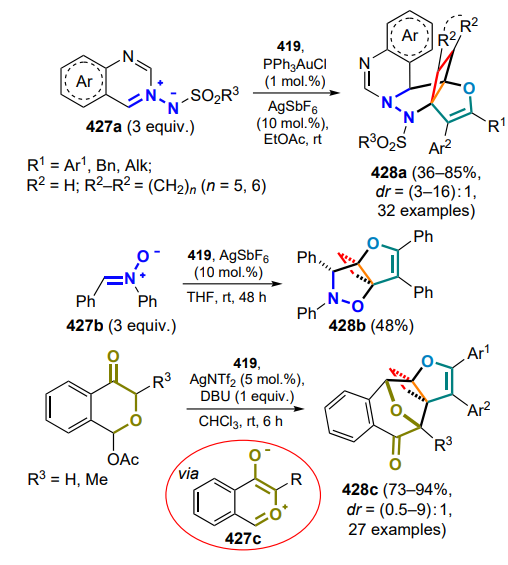
In turn, the diene nature of the cyclobutafurans 419' was revealed by their reaction with terminal alkynones 429. In this case, the reaction proceeds as a cascade process combining direct and retro-Diels–Alder reactions[285]. First, 7-oxanorbornadiene containing an annulated four-membered ring is formed, which undergoes further cleavage of two C–C bonds to form furans 430 (Scheme 186). It was found that the reaction is hampered (up to the formation of an inseparable mixture of products) by the presence of a styryl substituent on the carbonyl group of alkynone 429. Also, internal alkynones and acetylenes bearing an ester or o-nitrophenyl substituents on the triple bond are unreactive because of more preferred dimerization of the starting vinylidenecyclopropanes 418'.
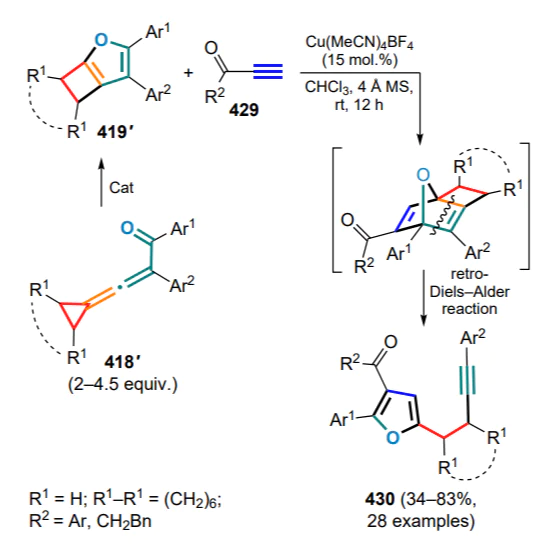
The reactivity of vinylidenecyclopropanes 418 is also altered by varying the ligands in the catalyst used. For example, the gold(I) complex of triphenylphosphine which catalyzes the (4+3)-annulation of vinylidenecyclopropanes 418 with nitrones[286], is mainly involved in the cycloaddition of the initially formed 4-oxaspiro[2.4]heptadienium intermediate A to give tetrahydrofuro[3,2-d][1,2]oxazepines 431 (Scheme 187). In the presence of the bulkier But XPhos ligand in 1,1,2,2-tetrachloroethane (1,1,2,2-TCE), further isomerization to the above-mentioned cyclobutafuran 419 is favoured. Activation of the C=C bond of this compound then occurred (B), followed by a ring contraction that actually led to the ‘transfer’ of the cyclopropane moiety to the adjacent position of the furan ring.
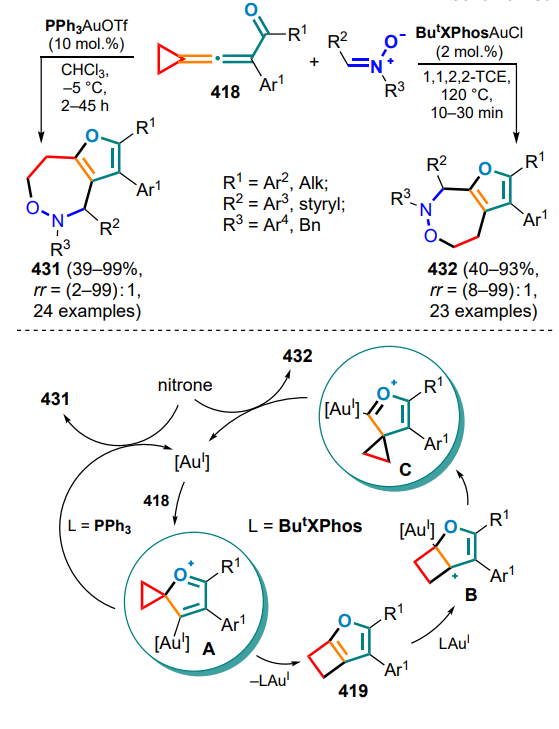
Oxaspiro[2,4]heptadienium intermediate C reacts with nitrone to give tetrahydrofuro[2,3-d][1,2]oxazepines 432 — regioisomers of bicycles 431. It should be noted that the previously discussed reaction of the same substrates (see Scheme 185, Ref. [284]) proceeds differently, without the formation of furans 431 or 432, which can be explained by the lowering of the Lewis acidity of the catalyst in the presence of oxygenated solvents.
Catalysis by gold(I) complexes bearing bulky ligands involving the formation of intermediates 419 has also been implemented in processes using a wide range of alkanols, including cholesterol, L-menthol, protected α-D-glucose, N-Boc-L-prolinol, as well as ketones and tropones (Scheme 188)[287, 288]. In the case of carbonyl compounds, these reactions proceeded as annulation yielding six- or sevenmembered oxacycles 433 or 434.
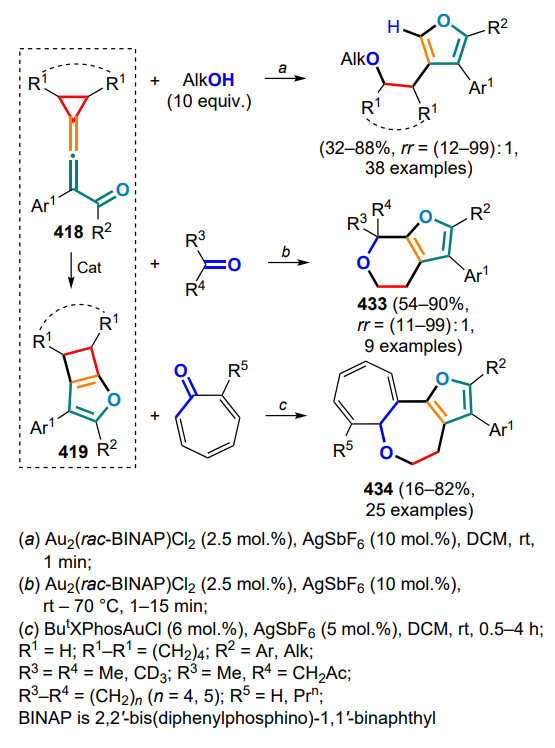
A strategy for the two-step cyclization of vinylidenecyclopropanes 418 is described in which sequential double Michael nitration with sodium nitrite in the presence of acetic acid is followed by reduction by iron in acetic or propionic acid[289]. Depending on the solvent, either dinitroketone 435 or mononitrocyclopropane 436 can be formed with high E-selectivity during nitration as a result of HNO2 elimination by sodium acetate in DMSO (Scheme 189). Further dinitro derivatives 435 are converted by selective reduction to 1,4-ketoamines A, and subsequent intramolecular cyclization and release of water and nitrous acid afford spiro(cyclopropane-2H-pyrroles) B, which undergo ring opening under the action of an acid anion to give pyrrole derivatives 437. The proposed procedure provided access to compound 438, an analogue of the hypolipidemic drug atorvastatin.
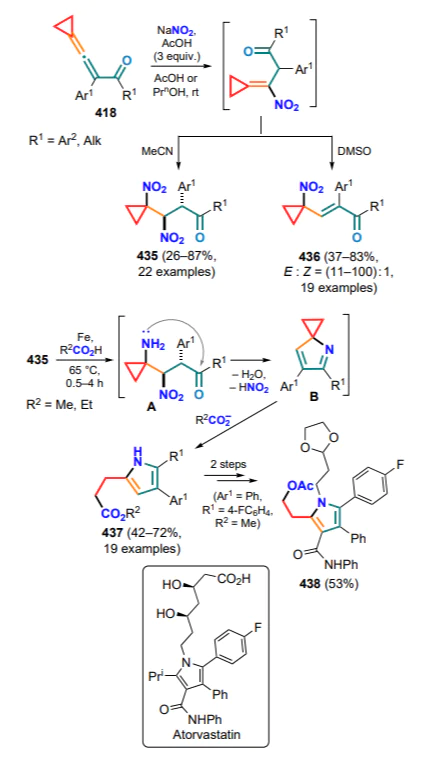
5. Conclusion
Over the last two decades, many processes involving cyclopropane derivatives and conjugated systems have been carried out using the modern arsenal of organic chemistry. A diversity of methods are described for the synthesis of relevant and complex carbo- and heterocyclic structures under thermal, electrochemical and photochemical conditions, including catalysis by Lewis and Brønsted acids, transition metal complexes and photocatalysis. A major contribution to the development of this field has been made by the use of catalysis with gold(I) complexes, which often exhibit unique reactivity. It is worth noting the plethora of cascade processes developed and the fact that the vast majority of transformations involve cyclization and(or) annulation reactions. A considerable number of publications highlight diastereo- and enantioselective methods for the preparation of compounds of different classes by this method, including those involving chiral ligands and organocatalysts. Among the most valuable are approaches to the synthesis of medium-size rings and complex frameworks. The chemistry of (arylmethylidene)cyclopropanes goes through rapid development, but there are virtually no examples of reactions of vinylidenecyclopropanes with azadienes in the literature, and only a few reactions of VCPs with thiadiene derivatives have been reported. The number of known processes involving 1,2-zwitterionic intermediates and styryl malonates as reactive structures isomeric to DAC is increasing. For some reactions, exceptional catalytic and/or substrate sensitivity was found, allowing the formation of novel products by varying the catalytic system or substituents in the starting compound. It should be noted that in published studies there are examples of obtaining potentially biologically active compounds. We believe that, despite the relatively rare use of the above methods presented in complete syntheses of complex molecules with practical applications, we should expect a further increase in the number of such papers in the future. The relevance of continued research in this scientific field is unquestionable.
6. List of abbreviations and designations
A — acceptor,
All — allyl,
AIBN — azobis(isobutyronitrile),
Am — pentyl,
B — base,
BARF — tetrakis[3,5-bis(trifluoromethyl)phenyl]borate,
BHT — 2,6-di-tert-butyl-4-methylphenol,
BINAP — 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl,
Boc — tert-butyloxycarbonyl,
BOX — bis(oxazoline) ligand,
B2pin2 — bis(pinakolato)diboron,
bpy — 2,2'-dipyridine,
ButBrettPhos — 2-(di-tert-butylphosphino)-2',4',6'-triisopropyl-3,6-dimethoxy-1,1'-biphenyl,
ButXPhos — 2-(di-tert-butylphosphino)-2',4',6'-triisopropylbiphenyl,
Cbz — benzyloxycarbonyl,
CMD — concerted metalation–deprotonation mechanism,
cod — cycloocta-1,5-diene,
CP — cyclopropane(s),
Cp — cyclopentadienyl,
Cp* — 1,2,3,4,5-petamethylcyclopentadienyl,
Cy — cyclohexyl,
4CzIPN — 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene,
D — donor,
DABCO — 1,4-diazabicyclo[2.2.2]octane,
DAC — donor–acceptor cyclopropanes,
dba — dibenzylideneacetone,
DBU — 1,8-diazabicyclo[5.4.0]undec-7-ene,
DCE — 1,2-dichloroethane,
DCM — dichloromethane,
DDQ — 2,3-dichloro-5,6-dicyano-1,4-benzoquinone,
DFT — density functional theory,
DME — 1,2-dimethoxyethane,
dmgH — dimethylglyoxime,
DMP — Dess–Martin periodinane,
DPEphos — bis[(2-diphenylphosphino)phenyl] ether,
DPIBF — 1,3-diphenylisobenzofurane,
dppe — 1,2-bis(diphenylphosphino)ethane,
dppf — 1,1'-bis(diphenylphosphino)ferrocene,
dppp — 1,3-bis(diphenylphosphino)propane,
dr — diastereomeric ratio,
dtbbpy — 4,4'-di-tert-butyl-2,2'-dipyridyl,
DyKAT — dynamic kinetic asymmetric transformation,
E — methoxycarbonyl,
EC — electrocyclization,
EDG — electron-donating group,
ee — enantiomeric excess,
esp — 3,3'-(1,3-phenylene)bis(2,2-dimethylpropanoic acid),
EWG — electron-withdrawing group,
Fmoc — 9-fluorenyloxycarbonyl,
Fu — furyl,
HAT — hydrogen atom transfer,
HFIP — 1,1,1,3,3,3-hexafluoropropan-2-ol,
HMDS — bis(trimethylsilyl)amide,
HMPA — hexamethylphosphortriamide,
HOMO — highest occupied molecular orbital,
IMes — 1,3-bis(2,4,6-trimethylphenyl)-1,3-dihydro-2Himidazol-2-yidene,
IMPC — intramolecular parallel cycloaddition,
IPr — 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene,
JohnPhos — (2-biphenyl)di-tert-butylphosphine,
L — ligand,
LA — Lewis acid,
Lawesson’s reagent — 2,4-bis(4-methoxyphenyl)-1,3,2,4-
dithiaphosphetane-2,4-disulfide,
LEDs — light emitting diodes,
LUMO — lowest unoccupied molecular orbital,
M — metal,
MCP — methylenecyclopropanes,
Mes — 2,4,6-trimethylphenyl (mesityl),
Mes-Arc+ — 9-mesityl-10-methylacridinium cation,
MHAT — metal-catalyzed hydrogen atom transfer,
MOM — methoxymethyl,
Ms — methanesulfonyl (mesyl),
MS — molecular sieves,
MVO — methylene(vinyl)oxonium,
MW — microwave irradiation,
NBS — N-bromosuccinimide,
NHC — heterocyclic carbene,
NMP — N-methyl-2-pyrrolidone,
Nu — nucleophile,
PC — photocatalyst,
phen — 1,10-phenanthroline,
PhthN — phthalimido,
PIFA — bis(trifluoroacetoxy)iodobenzene,
Piv — pivaloyl,
PMB — p-methoxybenzyl,
PMP — p-methoxyphenyl,
ppy — 2-phenylpyridine,
PS — photosensibilizer,
Py — pyridine,
rr — regioisomeric ratio,
rt — room temperature,
Selectfluor — 1-fluoro-4-chloromethyl-1,4-diazoniabicyclo[2.2.2]octane ditetrafluoroborate,
SET — single electron transfer,
TBDPS — tert-butyldiphenylsilyl,
TBS — tert-butyldimethylsilyl,
Tc — thienyl-2-carboxylate,
1,1,1-TCE — 1,1,1-trichloroethane,
1,1,2,2-TCE — 1,1,2,2-tetrachloroethane,
TDG — temporary directing group,
Tf — trifluoromethanesulfonyl (triflyl),
tfa — trifluoroacetate,
TFA — trifluoroacetic acid,
Th — thienyl,
TIPS — triisopropylsilyl,
TMS — trimethylsilyl,
Togni reagent I — 1,3-dihydro-3,3-dimethyl-1-(trifluoromethyl)-1,2-benziodoxol,
Togni reagent II — 1-(trifluoromethyl)-1λ3,2-benziodoxol3(1H)-one,
Tol — methylphenyl (tolyl),
TOPP — tri[(1,1'-biphenyl)-2-yl]phosphite,
TOX — tris(oxazoline) ligand,
tppms — sodium diphenylphosphinobenzene-3-sulfonate,
Ts — p-toluenesulfonyl (tosyl),
TTMSS — tris(trimethyl)silane,
VCP — vinylcyclopropanes,
VDCP — vinylydenecyclopropanes,
VCP-RA — vinylcyclopropane rearrangement,
XAT — halogen atom transfer.
Acknowledgement
This review was financially supported by the Russian Science Foundation (Project No. 22-13-00418).
References










