


![Solubility of initiating systems in the propylene oxide/CO2/1,4-dioxane reaction mixture at a CO2 pressure of 60 atm[61, 62].](/storage/images/resized/9m4lU6yatwVKH55nx10FyAjhOw8aGyo07ISIeqZ8_xl.webp)
![Copolymerization of cyclohexene oxide and CO2 under the action of β-diiminate complexes, ~7 atm, [epoxide]/[Zn] = 1000, 50 °C[137, 143].](/storage/images/resized/ciAtn1DcAESXN6bkZHB2aUErVKowvPBqbD8dQyQ8_xl.webp)
Keywords
Abstract
Carbon dioxide (CO2) plays a vital role in organic and polymer chemistry as a source of cheap and available raw material for the synthesis of many valuable products, including polymer materials with a specified set of properties, and as a solvent for chemical reactions. This review is devoted to the synthesis, properties and applications of polycarbonates obtained by copolymerization of CO2 with epoxides, a hot topic that has aroused great interest among the scientific community and industry representatives. The existing data on the catalytic systems used for the synthesis of polycarbonates are analyzed and summarized, depolymerization of polycarbonates, which is a key aspect in the polymer recycling, is discussed, information on the properties and applications of polycarbonates is systematized, and prospects for the development of this area of chemistry are considered.
Bibliography — 438 references.
1. Introduction
The problem of carbon dioxide utilization via transformation into valuable chemical products has attracted attention of many research groups in the last decades[1-5]. Among these products, cyclic carbonates and linear polycarbonates deserve particular attention[6, 7]. The former are actively used as solvents, reagents, plasticizers, monomers for polymer synthesis, and components of electrolytes in lithium batteries. The scope of applicability and the subsequent disposal of polycarbonates depend on not only the chemical nature of monomers, but also on the molecular weight, stereoregularity, and even the terminal groups of the polymer. Polycarbonates with the terminal hydroxyl groups are readily depolymerized under the action of metal complexes or bases or on heating to give cyclic carbonates[8]. Linear and star-shaped oligomeric polycarbonates with terminal hydroxyl groups, so-called polyols, are of interest for the production of polyurethanes[9]. High-molecular-weight aliphatic polycarbonates have relatively low glass transition temperatures (0 – 40 °C), low strength characteristics, and simultaneously good barrier properties and optical transparency, which makes them promising candidates for the fabrication of films and coatings[4]. Polycarbonates with bulky side groups and their block copolymers with cyclic anhydrides and esters are plastics and are comparable in the properties with the typical condensation polycarbonate based on 2,2-bis(4-hydroxyphenyl)propane (bisphenol A)[10]. Furthermore, polycarbonates are biocompatible and biodegradable polymers, which makes them attractive for medical and other applications (Figure 1)[11].
![[{"id":"TcPlDMCJq0","type":"paragraph","data":{"text":"Potential applications of polycarbonates."}}]](/storage/images/resized/CKKuUWi0jst1pE12Th5vtTUM7pfWZwb8IBBmGnDy_xl.webp)
Polycarbonates can be obtained by polycondensation, e.g., by the reaction of diols with carbonic acid derivatives[12-14], polymerization of cyclic carbonates[15-21] and copolymerization of epoxides with carbon dioxide[22-27]. Among condensation polycarbonates, aromatic polycarbonates, for example, those based on bisphenol A, are used most widely, owing to the optimal combination of high strength characteristics and optical transparency[28]. Polycarbonates obtained by polyaddition are still less popular because of a number of reasons: the absence of a cheap production process, insufficiently known properties and poorly developed disposal and recycling techniques. Nevertheless, studies along these lines have reached a new level in the last three to five years. This gives hope that addition polycarbonates will occupy their rightful place among other industrially produced heterochain polymers.
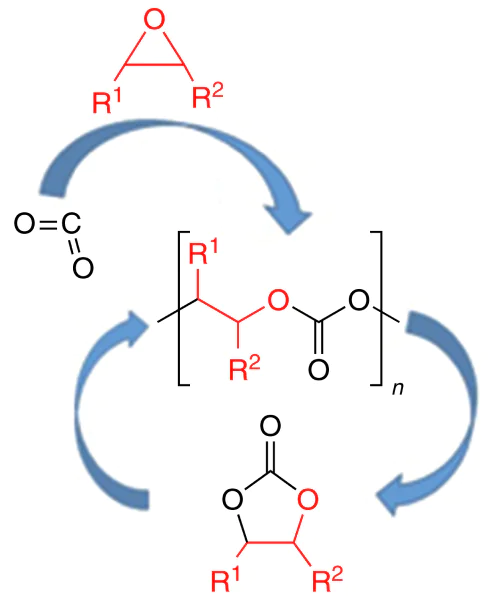
This review is devoted to the synthesis, properties and applications of polycarbonates obtained by copolymerization of CO2 with epoxides, a relevant research area that arouses great interest among the scientific community and industry representatives. Analysis of the literature shows that the formation of cyclic carbonates (pathway a) and linear polycarbonates (pathway b) in the reaction of epoxides with CO2 is induced by the same initiators (Scheme 1)[3, 5, 29-39].
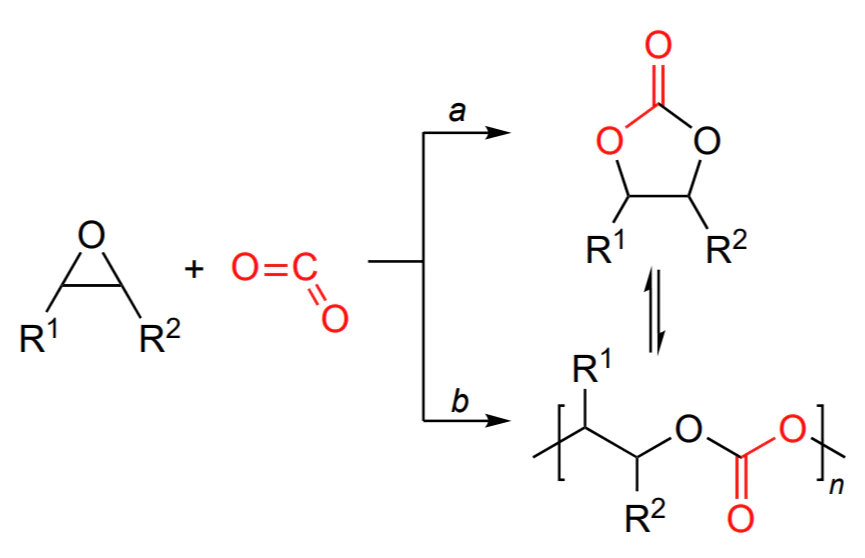
The reaction can be directed towards the formation of either cyclic or linear carbonate by changing (a) the reaction temperature, (b) the CO2 pressure, (c) the epoxide concentration, (d) the initiator structure and (e) the nature of the co-catalyst[3, 5, 40-42]. Copolymerization often follows a living mechanism (Living polymerization is the commonly accepted term in polymer chemistry, which means polymerization proceeding without chain termination and chain transfer reactions.) and is initiated by Lewis acids such as mono- and bimetallic zinc, aluminium, chromium and cobalt complexes (more rarely, compounds of other transition and rare earth metals) with phenoxide, porphyrin, salen and β-diiminate ligands and by non-metal compounds such as boranes in combination with bases and onium salts[27, 1-5]. The living polymerization makes it possible to synthesize polycarbonates with a narrow molecular weight distribution (MWD) and a molecular weight (MW) that can be controlled over broad limits (from hundreds to hundreds of thousands) and to switch from the synthesis of polycarbonates to the synthesis of polyesters by replacing CO2 by an appropriate cyclic ester, which provides the synthesis of di-, tri- and multiblock copolymers in situ[43-47].
A separate issue is the thermodynamics of the copolymerization of epoxides and CO2? This issue is usually addressed first of all in the studies of classical polymerization processes. However, in this case, it is difficult to summarize the information that is occasionally found in different sources. Most often, the authors attempt to calculate the energy states and the energies of formation of cyclic and polymeric carbonates, which depend appreciably on the natures of the catalyst, co-catalyst and epoxide. The general picture looks quite contradictory; therefore, the review barely addresses the thermodynamic aspects of epoxide and CO2 copolymerization. The main problems related to the synthesis of polycarbonates include low reaction rates, the formation of cyclic carbonates as by-products, the formation of ether linkages in the polymer apart from carbonate linkages, disruption of the chain regularity (local isomerism and stereoisomerism)[40]. These problems are solved by selecting the initiating system and conditions for polymerization.
The current studies related to addition polycarbonates can be conventionally divided into three groups. One group include studies dealing with the search for new initiators; the authors of these works have quite clear goals, which include attaining the reproducibility of synthesis; finding mild conditions to conduct copolymerization; increasing the control over MW and MWD of the polymers; minimizing the formation of by-products; attaining the control over stereochemistry; and increasing the catalyst activity and selectivity. The second group includes studies addressing the degradation of polycarbonates induced by various factors. These woks are focused on the controlled degradation of polymers to give safe or valuable products. The third group comprises studies of the mechanical and physicochemical properties of polymers, which makes it possible to predict the scope of their applicability.
This review aims not only to attract the attention of the readers to this relevant field, but also to formulate the current problems and to demonstrate various options for their solution and possible prospects of development. In our opinion, this may be of interest to specialists in polymer, organic, physical and inorganic chemistry. Below we systematically consider the listed lines of research and, in conclusion, make an attempt to formulate the prospects for the development of polymer science related to polycarbonates obtained from CO2 and epoxides.
2. Synthesis of polycarbonates
The formation of polycarbonates from epoxides and CO2 is a typical chain polymerization reaction. The initiation includes the epoxide activation via coordination on the Lewis active site. Then ring opening under the action of a nucleophile (co-catalyst and/or co-ligand) and alkoxide formation take place. This is followed by successive incorporation of carbon dioxide to give a carbonate and nucleophilic attack of the new activated epoxide molecule by the labile carbonate group. As the sequence of these reactions is repeated many times, polycarbonate is formed (Scheme 2)[48].
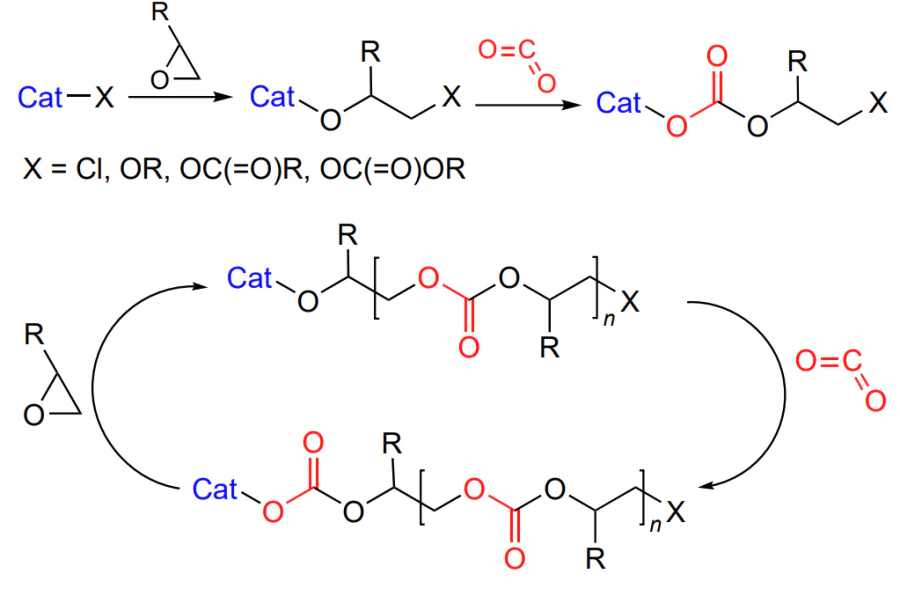
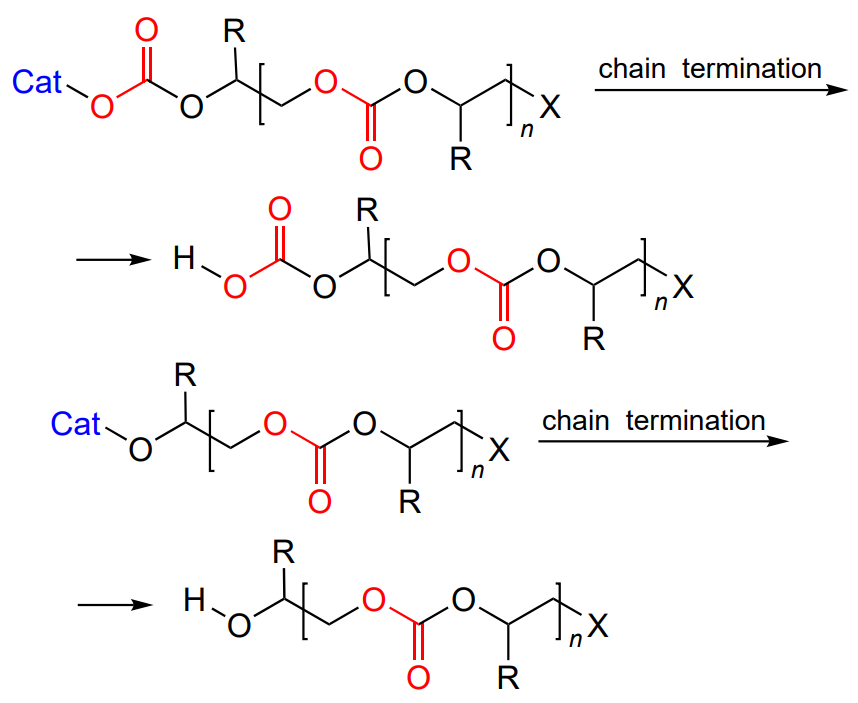
The living polymerization mechanism implies the growth of the macromolecule until no more epoxide is present. In this case, MW of the polymer is controlled by the laws known for living polymerization: it is directly proportional to the epoxide concentration and degree conversion and is inversely proportional to the initiator concentration[49]. The chain propagation is terminated by the addition of proton reagents (water or alcohols); as a result, a hydroxyl group appears at the chain end (Scheme 3).
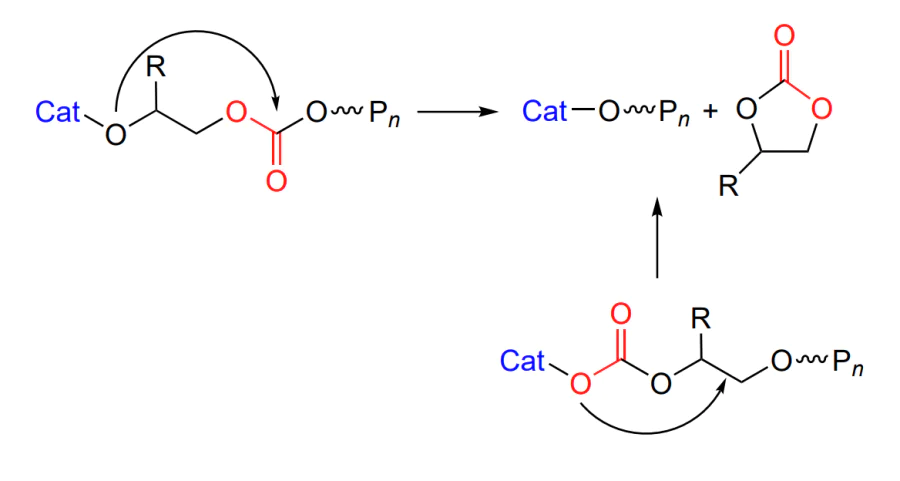
The copolymerization is complicated by several side reactions. The first one is the formation of cyclic carbonate (Scheme 4), which is related to the intramolecular cyclization (called backbiting) in which the alkoxide or carbonate oxygen atom attacks the carbon atom thus closing the ring and decreasing the macromolecular chain by one unit[50].
The second side reaction (Scheme 5) is the addition of epoxide instead of CO2 to the formed alkoxide; this gives rise to ether bonds in the macromolecule[51].
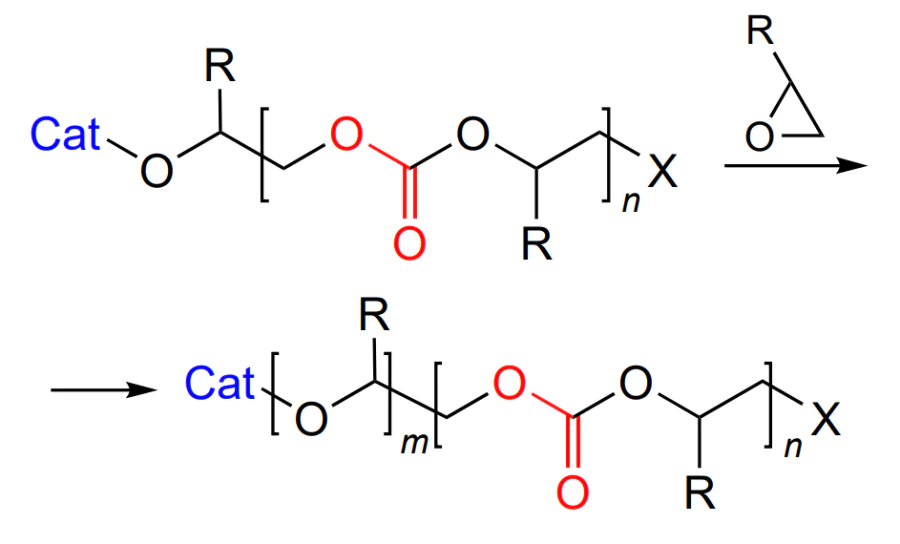
In addition, in the case of copolymerization, epoxide ring opening can take place either regioselectively or non-selectively (Scheme 6)[52].
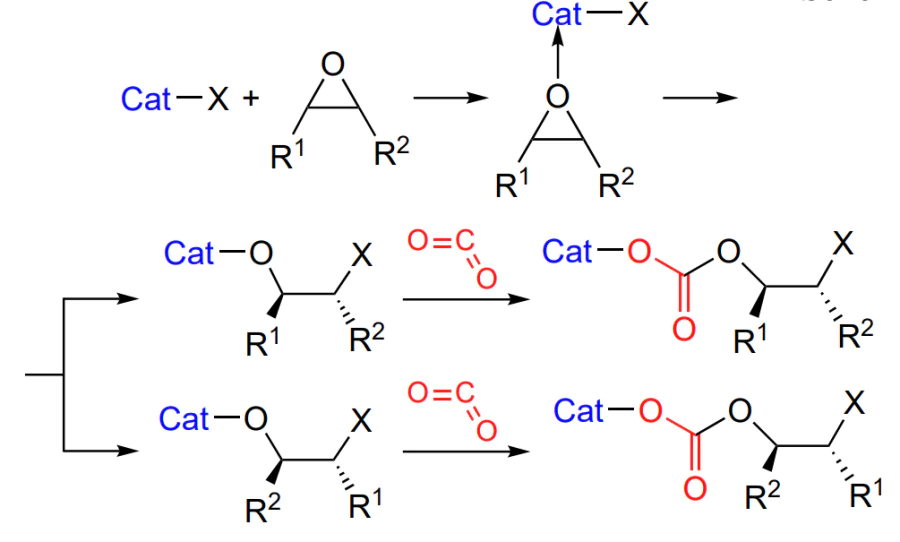
As a consequence, either regular (head-to-tail) or irregular (head-to-tail and head-to-head) polycarbonate is formed. Due to the presence of an asymmetric centre (or centres) in the starting epoxide, the formation of both stereoregular (iso- or syndiotactic) and atactic polymer is possible[53].
Finally, the general reaction pattern can be summarized as follows (Scheme 7).
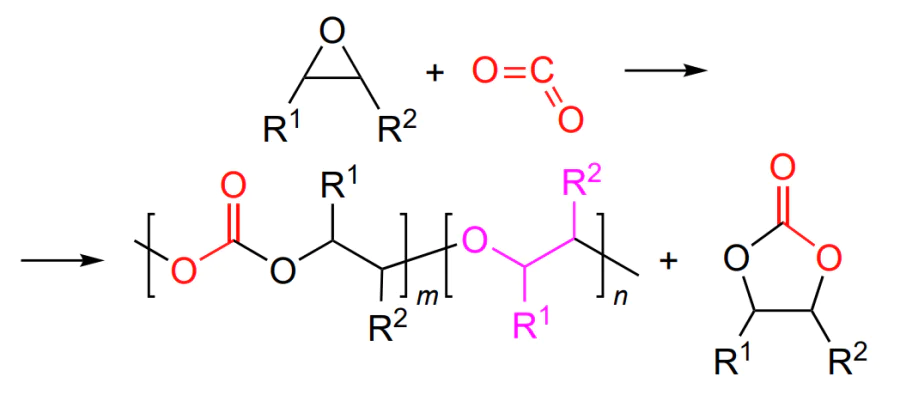
Below we address the key trends and characteristic features of CO2 and epoxide copolymerization with the most commonly used metal complexes based on porphyrins, salens, β-diiminates and phenoxyamines, which are classified according to the coordinating metal rather than the ligand. Some attention is paid to metal-free initiators (this is a new line of research in the polycarbonate synthesis that has been actively developed in recent years). In relation to various epoxide — initiator/co-catalyst system, we will demonstrate how it is possible to increase the catalyst activity and stereo- and chemoselectivity of copolymerization and will illustrate the potential of the macromolecular design, including the synthesis of polycarbonate-based block copolymers.
2.1. Organozinc catalysts
2.1.1. Zinc alkyl derivatives and phenoxides
The first advances in the polycarbonate synthesis were concerned with zinc compounds, most often, diethylzinc (catalyst), which was used together with a co-catalyst (water[54-57] or phenols[58-61]). For example, reactions of diethylzinc with phenols give alkoxide complexes, which initiate copolymerization of epoxides and CO2 at high pressures. The structure of the complexes was unknown; moreover, they were prepared immediately prior to copolymerization by adding a solution or a suspension of a mixture of reacted catalyst and co-catalyst to the epoxide, after which CO2 was fed. The absence of information on the structure of complexes and indirect data on the stereochemistry of monomer addition derived from analysis of polymer hydrolysis products brought about a variety of hypotheses for the copolymerization mechanism and active site structure and explanations for their solubility or lack of solubility in the reaction medium. It was deemed that the formation of complexes is accompanied by intra- or intermolecular association of alkoxides, which leads to their solubility or insolubility in the reaction medium. A homogeneous or heterogeneous initiating system could be obtained by varying the catalyst to co-catalyst ratio (Table 1); however, it was impossible to predict its type (homo- or heterogeneous)[61, 62]. In the case of copolymerization of propylene oxide (hereinafter, R1 = CH3, R2 = H, see Scheme 1) and CO2 induced by zinc phenoxides, cyclic carbonate was always formed together with the polymer, and the proportion of ether linkages in the polycarbonate did not exceed 6%. The diethylzinc-based initiating systems made it possible to synthesize not only poly(propylene carbonate), but also high-molecular-weight functional polycarbonates with intrinsic viscosity of 2.0 – 3.5 dL g–1, for example, from glycidol, in which the hydroxyl group was protected in advance[54-56].
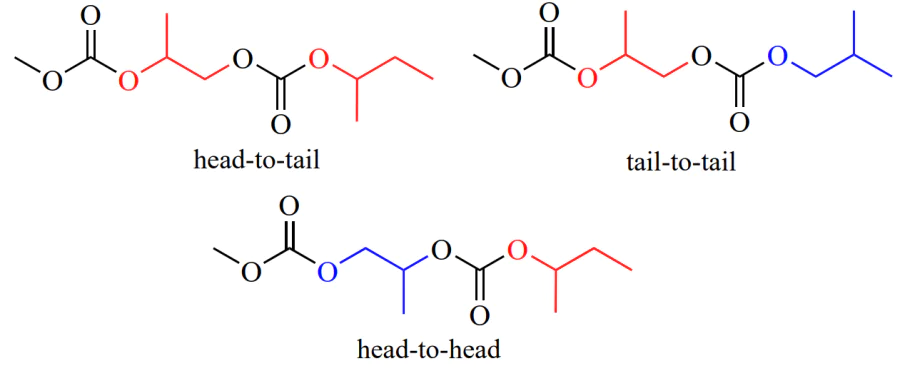
Opening of an epoxide ring containing methylene and methine groups is expected to occur via cleavage of the O – CH(R) bond. However, in the presence of diethylzinc/water or diethylzinc/aromatic alcohol or phenol system, cyclic monomers behave in different ways: in the case of R(+)-propylene oxide, the ring is opened predominantly at the CH2 – O bond[56], while ring opening of styrene oxide involves CH – O bond cleavage[57]. Moreover, it was shown[63] that copolymerization of propylene oxide with CO2 gives approximately 70% of head-to-tail units, while the other are head-to-head and tail-to-tail units (Scheme 8).
Detailed studies of the structure of zinc phenoxide complexes and their activity in the epoxide and CO2 copolymerization were started by Darensbourg and co-workers and reported in a series of publications[64-68]. The structures of phenoxide complexes were confirmed by X-ray diffraction. For example, zinc bis(2,6-diphenylphenoxide)bis(diethyl ether) was used as a mononuclear homogeneous catalyst in the CO2 co- and terpolymerization with cyclohexene oxide and propylene oxide in the absence of organic solvents[64]. Polymerization was carried out at 80 °C and at a pressure of 54.4 atm of CO2. The copolymerization with propylene oxide gives only the cyclic carbonate, whereas in the case of cyclohexene oxide, linear polycarbonate is formed. Interestingly, when two phenyl substituents in the phenoxide complex are replaced with three tert-butyl substituents, the reaction under the same conditions gives a low-molecular-weight cyclic carbonate, instead of the linear poly(cyclohexene carbonate). The cyclohexene oxide copolymerization with CO2 and terpolymerization provided that [cyclohexene oxide]/[propylene oxide] > 1 resulted in a proportion of ether linkages in the polycarbonate not exceeding 10%; however, when the percentage of propylene oxide in the initial mixture was increased, the content of ether linkages increased up to ~20%. It is significant that mixtures with a slight excess of cyclohexene oxide (10%) did not tend to form cyclic carbonate, which was found only in trace amounts. However, when the propylene oxide content in the initial mixture increased, the proportion of propylene carbonate became higher. The copolymers had Mn = (4 – 40) × 103 and broad MWD (Đ = 2.5 – 12). The turnover number (TON) proved to be higher (300 – 400) than the values attained previously for phenoxide complexes (5 – 40). The catalytic activity of the complex decreased with decreasing temperature or pressure.
A series of zinc phenoxide complexes 1a – d (Scheme 9) were synthesized and characterized by X-ray diffraction Darensbourg et al[66].
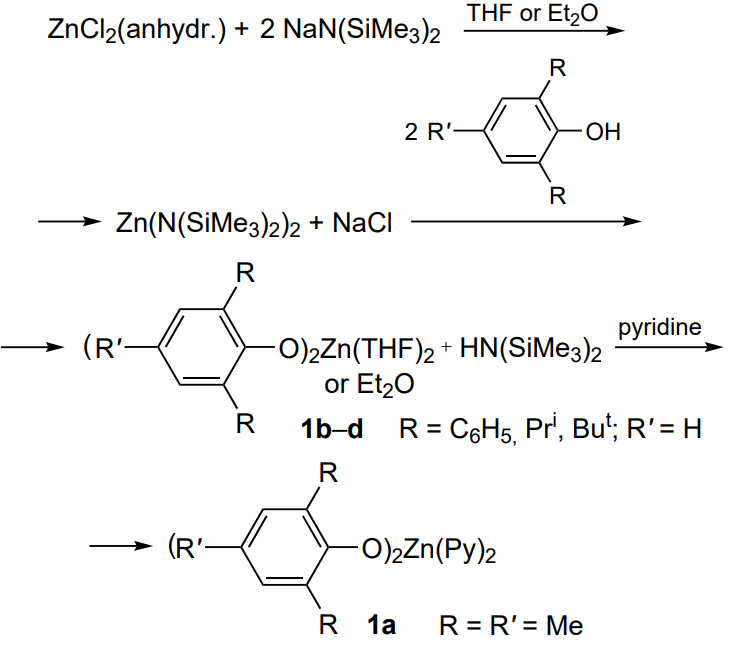
The authors determined the binding constants of ligands (THF, ether, pyridine) to the zinc atom and showed that binding is stronger for complexes with electron-donating tert-butyl substituents compared to electron-withdrawing phenyl substituents. Irrespective of the structure of the phenoxide substituents, binding decreases in the series: pyridine > THF > epoxide. Zinc bis-phenoxide derivatives proved to be active in the copolymerization of cyclohexene oxide and CO2 in the absence of an additional solvent. A study of TON as a function of the nature of substituents in the complex, polymerization temperature and time and CO2 pressure showed that TON increases in the series of substituents: isopropyl (477) < phenyl (602) < tert-butyl (677) << methyl (1441). In the complex with R = phenyl, TON increases with increasing temperature (90 for 40 °C and above 600 for 80 – 100 °C), time (from 270 in 24 h up to ~1200 in 144 h) and pressure (from ~390 at 27 atm up to ~6000 at 53 atm; further pressure rise and transition to the supercritical conditions does not lead to increase in TON of the complex). The polycarbonate formed in this reaction contains mainly carbonate (more than 90%) rather than ether linkages; an exception (53% ether linkages) is the polymer prepared in the presence of the complex with R = isopropyl (1c). The resulting poly(cyclohexene carbonate)s had predominantly syndiotactic configuration and had high MW [Mw = (45 – 170) × 103] and broad MWD (Đ = 2.5 – 4.5). Under similar conditions, propylene oxide does not undergo copolymerization with CO2, but forms only cyclic carbonate in this reaction. The terpolymerization of cyclohexene oxide and propylene oxide ([cyclohexene oxide]/[propylene oxide] > 1) with CO2 gives rise to an irregular random copolymer (atactic copolymer comprising different local isomers) with a low content of ether linkages.
Subsequently, Darensbourg et al.[67] synthesized binuclear zinc complexes 2a – c, which were characterized by X-ray diffraction analysis (Scheme 10)[67].
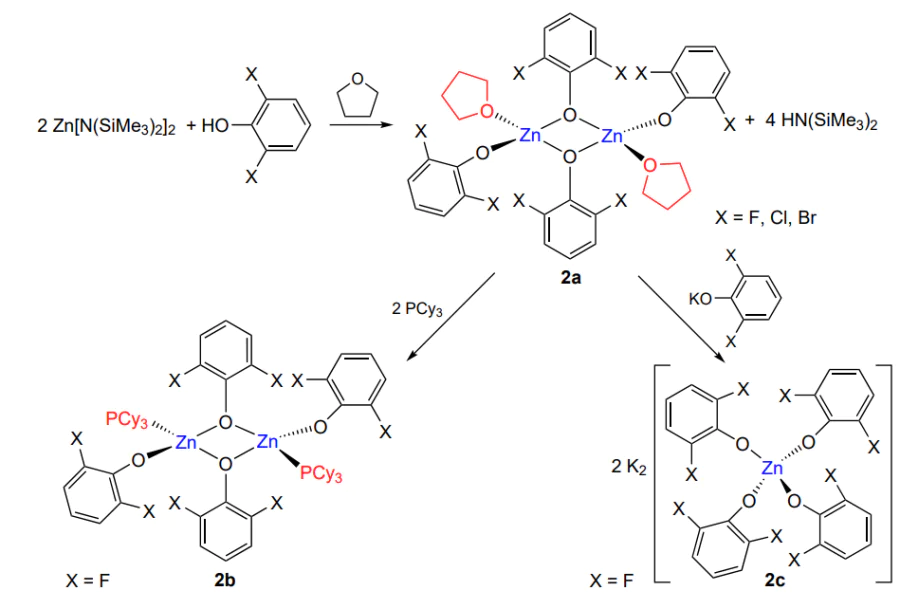
These complexes were used for copolymerization of cyclohexene oxide or propylene oxide with CO2 and for terpolymerization at high CO2 pressure (55 atm) and 80 °C. The complex based on difluorophenol with a phosphine ligand (2b) was inactive in the copolymerization, unlike the complex with the tetrahydrofuran ligand (2а), which showed TON = 790 within 48 h of the reaction and turnover frequency (TOF) of 16.5 h–1 in the cyclohexene oxide copolymerization and TON = 201 and TOF = 4.2 h–1 in terpolymerization. The copolymerization of propylene oxide and CO2 at 80 °C affords mainly the cyclic carbonate; however, a decrease in the temperature to 40 °C gives rise to polycarbonate. Unlike earlier phenoxide complexes, this complex proved to be stable to air moisture and oxygen, while retaining the activity and high selectivity; no ether linkages were detected in the cyclohexene oxide copolymer, while their concentration in the terpolymer did not exceed 3.4% (corresponds to propylene oxide). Similar properties were inherent in chlorine- and bromine-containing complexes; however, their activity was lower and decreased in the sequence F > Cl > Br.
The characteristics of poly(cyclohexene carbonate) prepared in the presence of a fluorine-containing complex were studied in more detail by Darensbourg and co-workers[68]. The slight (by 5 atm) decrease in the pressure compared to that in the previous study induced an increase in TON and TOF up to 1240 and 26 h–1. It was found that polycarbonate synthesized at 80 °C and 50 atm over a period of 48 h had not only high MW (Mw = 2.5 × 105), but also fairly broad MWD (Đ ~ 6). The broad MWD of the polymer indicated that the living polymerization mechanism was violated; this view was confirmed by the next experiment of the authors. This complex was active not only in the copolymerization of CO2 and cyclohexene oxide, but also in the ring opening homopolymerization of the latter. When the living mechanism is involved, the successive polymerization of epoxides in the absence and then in the presence of CO2 should afford a block copolymer–polyether-block-polycarbonate. When the living mechanism is violated, a mixture of products is expected to form. After this experiment, the crude product contained both ether and carbonate linkages and had two glass transition temperatures (62.3 °C for the polyether and 115.2 °C for the polycarbonate). However, after purification, the number of ether linkages in the polymer sharply decreased and the material had only one glass transition temperature. Thus, the reaction gave two homopolymers, which were separated during the purification.
Werner and co-workers[69] made an interesting attempt at using dialkyl- and diarylzinc ZnR2 (R is ethyl, n-butyl, isopropyl, cyclohexyl and phenyl) in the absence of co-catalysts or halogen-containing additives for cyclohexene oxide and CO2 copolymerization. The reaction was carried out in toluene using 0.5 mol.% ZnR2, a CO2 pressure of 20 atm and a temperature of 100 °C. Under these conditions, TON decreased in the series of substituents: isopropyl ≈ cyclohexyl (166) > n-butyl (154) > ethyl (145) > phenyl (107). A decrease in the initiator concentration or pressure resulted in decreasing TON. Apart from polycarbonate, the reaction gave cyclic carbonate, the proportion of which did not depend on the substituent R or pressure, but increased with increasing polymerization time and decreased with decreasing temperature. The chemoselectivity of all catalysts was moderate, with the content of ether linkages being in the range from 3 to 84%. The polymer had broad MWD (Đ = 2 – 10) and Mn = (4 – 80) × 103. Attempts to use other epoxides or to add co-catalysts, such as tetrabutylammonium bromide, dimethylaminopyridine, bis(triphenylphosphine) iminium chloride (PPNCl), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and triazabicyclodecene (TBD), proved to be unsuccessful: the authors observed the formation of co-oligomers of cyclohexene oxide and cyclohexene carbonate with Mn < 500.
The properties of the resulting polycarbonates depend on not only MW and MWD, but also on the chain configuration (a-, iso- or syndiotactic), which determines the physical state (amorphous for atactic polymers and partially crystalline for stereoregular ones) and, as a consequence, the mechanic properties and applications. The first attempt to accomplish asymmetric synthesis in the copolymerization of meso-epoxides (2-butene oxide, 1,2-butеne oxide and cyclohexene oxide) involved the use of a chiral catalyst based on the ZnEt2 complex with diphenylprolinol (3)[70]. Depending on the conditions, the synthesis gave either an atactic copolymer or a copolymer in which (R,R)-isotactic sequences predominated, but no MWD control was present, which is characteristic of this type of catalysts. As further development of these ideas, complex 3 was tested in the copolymerization of cyclohexene oxide and CO2[71, 72]. In the absence of additives, the complex can be used to produce polycarbonates with a broad MWD in ~60% yield, with the fraction of (R,R)-isotactic sequences being approximately 50%. The authors assumed the following mechanism of the reaction (Scheme 11).
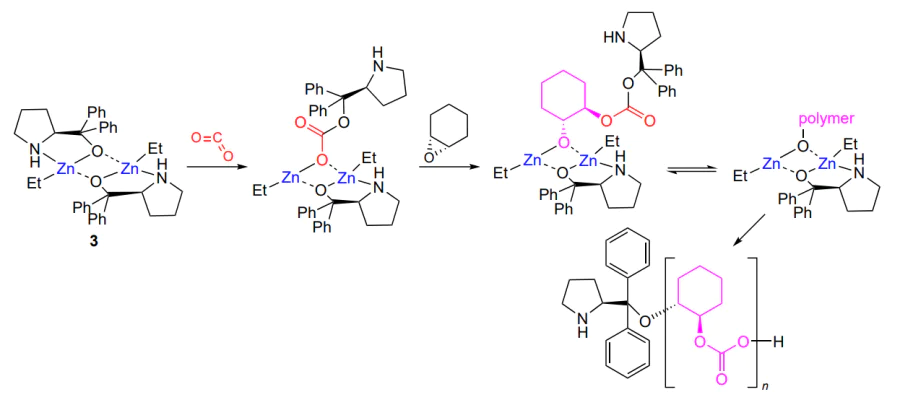
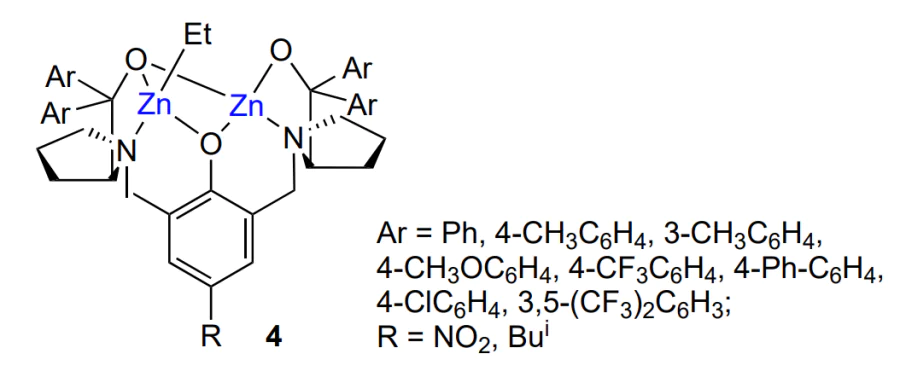
The addition of ethanol (0.2 – 0.8 equiv. relative to the catalyst) increased the polymer yield to 99%, led to narrowing of MWD to Đ = 1.2 – 1.4 and increased the proportion of isotactic sequences to 75%. Among the tested alcohols (methanol, ethanol, 2-propanol, 2-methylpropan-1-ol, benzyl alcohol and CF3CH2OH), the most pronounced effect on the polymer stereoregularity [78% of (R,R)-sequences] was found for 2-propanol and 2-methylpropan-1-ol. In turn, the variation of substituents (methyl, methoxy, CF3) and their position in the phenyl groups of the complex indicated that the complexes containing meta-monosubstituted aryl groups lose in the ability to control the selection of enantiomers during copolymerization, while those containing meta-disubstituted or para-monosubstituted aryl groups lose both the catalytic activity and enantioselectivity. Analysis of the structure of this complex and its effect on the stereocontrol of the copolymerization were reported by Nozaki and co-workers[73]. The authors synthesized the complex using (S), (R) or racemic diphenyl(pyrrolidin-2-yl) methanol. The hetero- or homochiral structure of the complex is highly important for the selection of enantiomers. Since the copolymerization mechanism implies binding of epoxide to zinc, this requires cleavage of the bridging Zn – O bond, the ease of which is determined by the structure of the complex. In addition, if the zinc dimer is not enantiomerically pure, the minor enantiomer is selectively captured by the major enantiomer to give a more stable heterochiral zinc dimer. This results in an increase in both the yield of the polymer and the proportion of isotactic units. The integration and detailed analysis of this studies were reported by Nozaki and co-workers[74]. Ding and co-workers[75] expanded the range of possible ligands and synthesized a set of compounds 4. The resulting complexes were used for copolymerization of cyclohexene oxide with CO2. Despite the lack of data on the stereochemistry, all catalysts provided polycarbonate in a high yield (>99%) and with a high content of carbonate linkages (>99%) and quite narrow MWD (Đ < 2).
2.1.2. Zinc dicarboxylates
Zinc dicarboxylates also proved to be suitable for initiating epoxide and CO2 copolymerization. They can be obtained in situ, and this approach was used in the first experiments by addition of carboxylic acids (benzoic, phthalic and other acids) to diethylzinc[76]. This provided the synthesis of high-molecular-weight products (intrinsic viscosity of ~2 dL g–1), but in low yields (15 – 20%). Subsequently, dicarboxylates for epoxide and CO2 copolymerization were synthesized separately[77-96]. These catalysts are heterogeneous and they are used for the copolymerization of epoxide with CO2 at elevated pressure and under supercritical conditions. The catalyst activity depends on the nature of the carboxylate and on the catalyst structure and can vary over wide limits (TOF varies from 3 to 630 h–1)[96]. Among known zinc dicarboxylates, of most interest is zinc glutarate, which is currently one of the few catalysts used for the industrial production of poly(propylene carbonate). However, it is noteworthy that the results obtained in the early studies on the use of zinc glutarate in the propylene oxide copolymerization with CO2 did not look very optimistic[77]. First, the concentration of zinc glutarate was high (1 g per 25 – 60 mL of epoxide), but the polymer had Mn = (5 – 30) × 103 and broad MWD (Đ = 4 – 19), i.e., it contained a large amount of the high-molecular-weight fraction. This means that only a minor part of the catalyst provided the polymer formation. In addition, the proportion of the cyclic carbonate was ~10% and virtually did not depend on the polymerization conditions (T = 40 – 85 °C and pressure of 20 – 80 atm). The content of the ether linkages in the polymer varied from 70 to 97%; this value increased with increasing pressure and increasing propylene oxide concentration with decreasing temperature. In the subsequent studies, the content of cyclic carbonate was minimized. For example, Chisholm et al.[97] carried out propylene oxide copolymerization with CO2 in the presence of zinc glutarate at 60 °C and a pressure of 50 atm. Under these conditions, the proportion of carbonate linkages in the chain was more than 95%, while the proportion of cyclic carbonate did not exceed several percent.
Due to the heterogeneous nature of the catalyst, the polycarbonate always has broad MWD. The factors that influence the polymer dispersity in MW have not been reported in the literature. Presumably, these factors include the size distribution of catalyst particles, the non-uniformity of the porosity, the degree of crystallinity, etc. The activity of the heterogeneous catalysts proved to be sensitive to quite a few factors. This is manifested, in particular, in the fact that the catalyst properties vary depending on the source and preparation method[83]. The influence of the nature of the starting compounds for the synthesis of zinc glutarate is quite significant; TON is 64 for the ZnO/glutaric acid mixture; 8.8 for the Zn(OH)2/glutaric acid mixture; 15.4 for the Zn(NO3)2 · 6 H2O/glutaric anhydride; and 2.5 for the ZnEt2/glutaric acid mixture (1 mass % catalyst, ~50 atm, 60 °C)[83]. It is noteworthy that the molecular weight of poly(propylene carbonate) decreases in the same order as TON. In addition, zinc glutarate is less active in the amorphous state than in the crystalline state[83-85, 90-93]. An increase in the proportion of the crystalline phase and in the surface area increase the catalyst activity[81-88]. In addition, the catalyst activity can be increased 1.8-fold by deposition of the catalyst onto montmorillonite[98] and increased 1.3-fold by catalyst stabilization with amphiphilic block copolymers[99, 100] or by activation (treatment with water followed by vacuum drying at 130 °C)[101]. Thus, the considerable differences between the TON and TOF values for zinc glutarate reported in the literature are due to different physical state and characteristics of the catalyst[102].
The replacement of glutarate by another carboxylate group of the same homologous series –OOC – (CH2)х – COO– has been considered as a way to increase the catalyst activity[83, 94, 95]. For example, in the case of adipate (x = 6), TOF is 580 h–1[94, 95]; for pimelate (x = 7), TOF = 530 h–1[93]; i.e., the values do not differ appreciably from the value for zinc glutarate. Similar results on the variation of the activity of zinc dicarboxylates were obtained for carboxylate activation using SO2: glutarate (630 h–1) > adipate (529 h–1) > pimelate (459 h–1) >> succinate (7 h–1)[96]. The dicarboxylate structure was determined in the first decade of the 2000s. For example, it was shown by X-ray diffraction that the glutarate substituents in zinc glutarate occurring in the bent or fully extended conformation are coordinated to one zinc atom[81, 82]. Comparison of zinc glutarate, adipate, pimelate and succinate showed that the structure of the [101] hkl-plane in the first three dicarboxylates is the same (four Zn atoms with identical spatial conformations of substituents and Zn – Zn distances of 4.6 – 4.8 Å), which is correlated with the minor difference between their activities. Meanwhile, the structure of zinc succinate is markedly different and the distances between the zinc atoms along the a, b and c axes are 4.822, 5.952 and 6.865 Å[80].
The replacement of glutaric acid by its derivatives (e.g., 2-ketoglutaric, 3,3-dimethylglutaric or diglycolic acid) leads only to a sharp decrease in the activity[98, 103, 104].
One more example is the heterogeneous fluorine-containing catalyst 5 consisting of two components, ZnO and maleic acid monoester with 3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctanol, which was proposed for the copolymerization of cyclohexene oxide in supercritical CO2[105, 106]. The authors were unable to determine the exact structure of this catalyst.
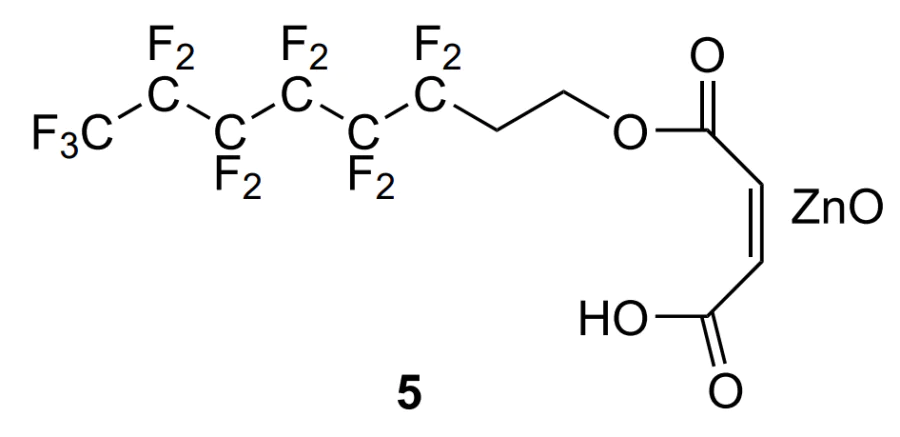
Copolymerization involving catalyst 5 was carried out in the temperature range of 60 – 172 °C and the pressure range of 40 – 340 atm. In the temperature range T = 100 – 110 °C at a cyclohexene oxide mole fraction of 15%, it is possible to obtain a polymer in ~70% yield with Mw = (5 – 18) × 104, Đ = 1.9 – 27 and TON ≈ 400, containing more than 90% carbonate linkages. Irrespective of the conditions, cyclohexene carbonate was formed as a by-product.
Experimental and theoretical structural studies made it possible to propose a bimetallic mechanism for copolymerization of propylene oxide and CO2 in the presence of heterogeneous zinc dicarboxylates (catalyst 6, Scheme 12)[80].
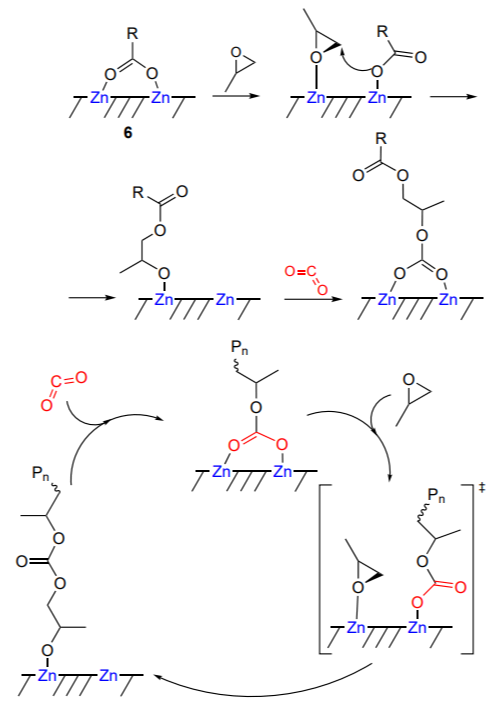
The reaction involves two Zn – Zn active centres on the zinc dicarboxylate surface. Copolymerization takes place as alternation of two reactions: the insertion of CO2 into the Zn – O bond and nucleophilic attack by the carbonate on the preactivated epoxide. The calculations show that the Zn – Zn distance is critical for the catalyst activity; for propylene oxide copolymerization with CO2, the optimal distance is in the 4.3 – 5.0 Å range. When this distance is longer, the predominant reactions are cyclic carbonate formation and epoxide homopolymerization. The greater the number of Zn – Zn pairs on the catalyst surface, the higher the catalyst activity. This necessitates preactivation of the catalyst and generation of a sufficient number of active centres on the surface.
Zinc glutarate can also be used for the terpolymerization of epoxide, CO2 or cyclic anhydride or lactone. For example, terpolymerization of propylene oxide, phthalic anhydride and CO2 (~50 atm, 75 °C) gives a low proportion of cyclic carbonate, while the ratio of carbonate and ether (propylene oxide) linkages in the terpolymer is the same as in the absence of phthalic anhydride[78]. The terpolymer has high MW and broad MWD [Mn ≈ (7 – 220) × 103, Đ = 2 – 4]; the latter value is narrower than that for poly(propylene carbonate). The terpolymerization with d-valerolactone (1 mass % catalyst, ~27 atm, 60 °C; TON = 22 – 75) results in the formation of the polycarbonate-block-polyether block copolymer with Mw in the range of (80 – 300) × 103 and Đ = 1.6 – 4.1, which has two glass transition temperatures and a melting point[83].
2.1.3. Bimetallic zinc-containing cyanide catalysts
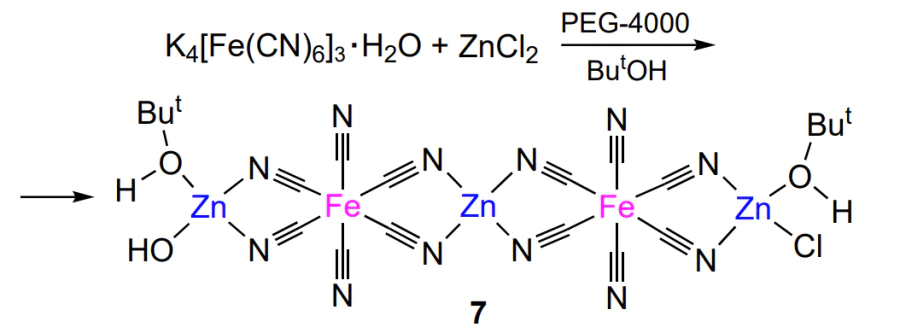
One more example of heterogeneous zinc-containing catalysts are bimetallic cyanide complexes, which attracted attention in the early 2000s and are still of interest[107-119]. These compounds described by the general formula MA,m[MB(CN)6]n · xH2O can be classified as metal-organic frameworks in which the framework comprises metal–cyanide groups, where MA = Zn2+, Fe2+, Co3+, Ni2+, Mn2+, Cr2+, Co2+, etc., MB = Fe2+, Fe3+, Co3+, Ni2+, Mn2+, Cr3+, Pd2+, etc. The use of Zn – Fe and Zn – Co cyanide catalysts in the ring opening epoxide polymerization for the synthesis of polyethers was first mentioned in the second half of the 20th century[120]. Currently, complexing agents are used for the synthesis of cyanide catalysts; this role can be played by the solvent, the molecules of which are incorporated in the catalyst coordination sphere during isolation and purification[121]. Thus, the structure of bimetallic cyanide complexes is more correctly depicted as MA,m[MB(CN)6]n · xH2O · yL (where L is the complexing agent). As an example, consider complex 7, which is formed upon the reaction of potassium ferrocyanide trihydrate and zinc chloride in the presence of PEG-4000 and tert-butyl alcohol (Scheme 13).
The catalytic activity of cyanide complexes in the synthesis of aliphatic polycarbonates is sensitive to the nature of cyanometallic precursor, complexing and co-complexing agents, metal halide, conditions of the catalyst synthesis, the degree of catalyst crystallinity, the crystal lattice type, the average particle size of the catalyst, etc[107, 111, 114, 122-127]. As an example, consider the publication by Wang and co-workers[128], which describes the Co – Zn cyanide catalyst for the copolymerization of propylene oxide and CO2 (0.01 – 0.05 mass % catalyst, 50 atm, 90 °C), characterized by TOF = 2 × 106 h–1 and high selectivity (the fraction of cyclic carbonate is ~0.7%). However, the copolymer contained not only carbonate, but also ether linkages and also had high MW (Mn = 1.3 × 105) and Đ = 2.9. The catalyst active centre is Zn2+; a change in the nature of the second metal of the complex changes the electron cloud distribution around the bridging CN ligands, which in turn, influences the Zn activity in the copolymerization. For example, the replacement of cobalt by iron or nickel was found to result in a considerable decrease in the catalyst activity[129, 130]. The mechanism of polymerization involving bimetallic cyanide complexes can be depicted as follows (Scheme 14)[131].
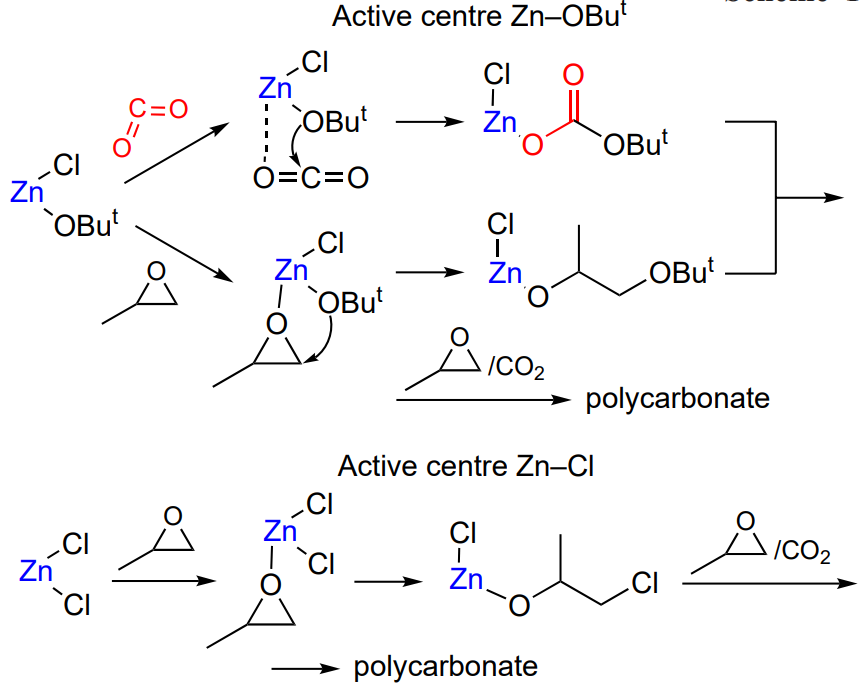
The effects of the nature of metals, co-catalysts and complexing agents and morphology of the complex on the cyanide complex activity in epoxide and CO2 copolymerization are summarized in a review by Sebastian and Srinivas[131]. It was shown that minor changes in the protocol of catalyst synthesis significantly affect the catalyst activity; therefore, fine-tuning of the catalyst activity is a priority task for many research groups. It is worth noting that many bimetallic cyanide catalysts exhibit high activities towards the synthesis of polycarbonates. It is not surprising that large commercial companies became interested in these results and patented the synthesis of bimetallic cyanide catalysts for the production of polycarbonates[132, 133]; the Co – Zn cyanide catalysts are already produced on an industrial scale.
2.1.4. Zinc complexes with β-diimine ligands
The above-described catalysts do not allow the control of MWD of the obtained copolymers. A solution to this problem was provided by the development of homogeneous single-site catalysts LnMR (Ln is organic ligand, M is the catalytically active metal atom, R is the initiating group or the growing polymer chain). Since the last decades of the 20th century, these catalysts have been used in the polymerization of olefins, cyclic oxides and esters. Examples of such catalysts are zinc complexes with β-diimine ligands (BDI)ZnX (Х is most often OAlk or OAcyl) (8a – c), which were used for copolymerization of cyclohexene oxide and CO2 (Scheme 15)[134-142].
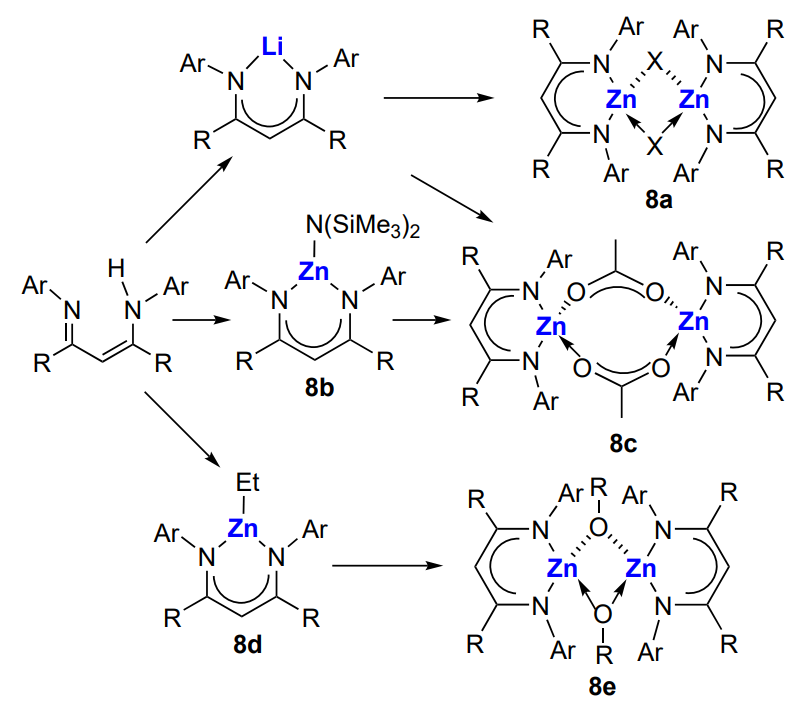
Aryl substituents (Ar) contain identical or different groups (methyl, ethyl, n-propyl, isopropyl, etc.) in the ortho-positions and mainly hydrogen in the para-position. The first use of zinc complexes (BDI)ZnX [X = OCH3, OC(=O)CH3] with isopropyl groups in the ortho-positions of Ar substituents in cyclohexene oxide and CO2 copolymerization was reported by Coates and co-workers[134]. The authors carried out the reaction at a relatively low pressure (~7 atm) and moderate temperature (20 – 80 °C) for 2 h and obtained copolymers with Mn = (2 – 3) × 104 and Đ < 1.1 with more than 95% content of carbonate linkages. The activities of both catalysts were similar [TON = 449 (OMe) and 494 (OAc); TOF = 247 (OMe) and 224 (OAc)]. According to X-ray diffraction data, the acetate complex in the solid state is a dimer in which zinc atoms are linked by acetate bridges, the substituents at the metal atom are in the tetrahedral configuration, with aryl substituents being almost perpendicular to the N – Zn – N plane of the chelate complex.
The effect of the nature of substituent in the N-aryl group and the bridging ligand on the activity of (BDI)ZnX in the copolymerization of cyclohexene oxide and CO2 ([monomer]/[Zn] = 1000, 50 °C, ~7 atm) was described by Coates and co-workers.136 It was found by variation of the group X that when X is halogen, alkyl or OH, or Х is absent, the complex cannot initiate the epoxide (cyclohexene oxide, cyclopentene oxide) copolymerization with CO2. Effective catalysis takes place when Х is an alkoxide, acyl or amine group. The structure of the aryl substituent is also significant. Generally, complexes with symmetrical structure are effective in the copolymerization of cyclohexene oxide and CO2 if they contain appropriate bulky substituents (ethyl or isopropyl group) in the ortho-positions of N-aryl moieties. Otherwise (in the presence of methyl or n-propyl group), they form inactive dimers. The turnover frequency of the catalysts is ~200 – 250 h–1, and the resulting copolymers have Mn = (2 – 3) × 104 and narrow MWD (Đ = 1.07 – 1.21). Thus, the complex can exist as the active monomer (unimer) or as the inactive dimer. The unimer ↔ dimer equilibrium (Scheme 16) for the zinc β-diiminate complexes (BDI)ZnOAc (9) and the influence of the equilibrium on the complex activity in the cyclohexene oxide copolymerization was described in more detail by Coates and co-workers[139, 143].
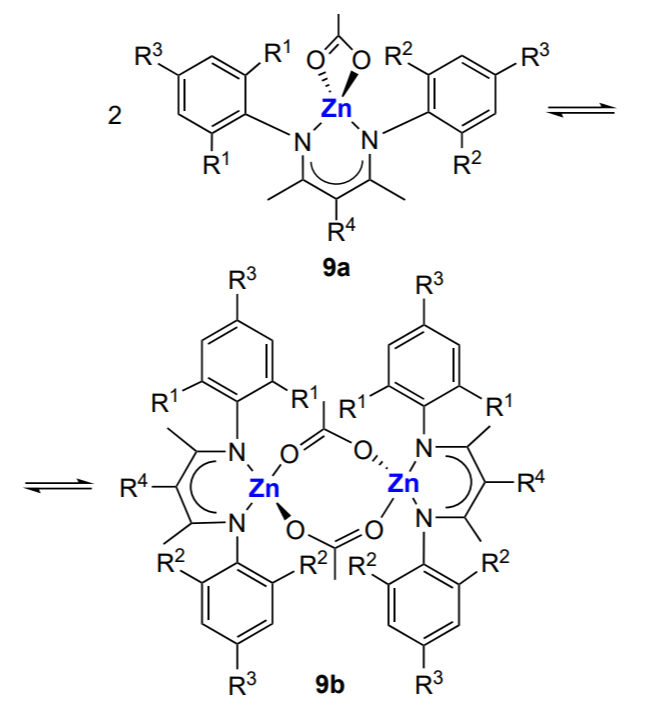
For example, the complex in which R1 = R2 = Pri, R3 = R4 = H exists mainly as the dimer in the temperature range from –80 to –20 °C; the dimer of the complex coexists with the unimer in the range from –20 to 80 °C; and the unimer predominates above 100 °C. For other (BDI)ZnOХ complexes (Х = alkyl, acyl; R1 and R2 = alkyls; R3 = Н or But; R4 = H or CN), there is the following general trend: as the steric crowding of the aryl groups increases, the equilibrium shifts from strongly bound to weakly bound dimers and finally to the dimer/unimer equilibrium.
Model experiments using equivalent amounts of the complex and the monomer [CO2 (~7 atm) or epoxide] showed that in the case of alkoxide complexes, first the activation of CO2 takes place, while cyclohexene oxide does not react with the complex. Furthermore, if Х = Me, one CO2 molecule adds to the [(BDI) ZnOMe]2 dimer, and in the case of Х = Pri, two CO2 molecules add (one molecule per Zn – OPri bond). A different situation is observed for the complex in which zinc atom is bound to the N(SiMe3)2 group: in this case, the Me3Si – N=C=O isocyanate is released and a dimeric complex in which both zinc atoms are bound to the O – SiMe3 group is formed. The complex in which the zinc atom is linked to the acetate group (mimicking the CO2 activation by the alkoxide) does not react with CO2, which is in line with the copolymerization mechanism. It was expected that this complex would easily react with cyclohexene oxide; however, an equilibrium between the reaction product and the starting reactants was established instead. The reaction could be shifted towards the product after modification of the complex (R1 = R2 = Et; R3 = Н, R4 = CN). The data on the use of these catalysts in the copolymerization are summarized in Table 2[137, 143]. It can be seen that the (BDI)ZnN(SiMe3)2 complexes 8b show high activity in the copolymerization, while combination of complexes containing ethyl and isopropyl groups in the aryl substituents actually doubles their activity. This fact suggests that a new mixed active centre is formed. It is of interest that among the (BDI)ZnOAc complexes 9a, the highest activity is inherent in the complex containing different alkyl groups (R1 = Pri, R2 = Et).
On the basis of these studies, a bimetallic mechanism (involving participation of both zinc atoms) was proposed for the first time[139] for cyclohexene oxide and CO2 copolymerization in the presence of β-diiminate complexes. Recall that a bimetallic mechanism was proposed independently also for heterogeneous catalysis[80]. Kinetic analysis is in line with the hypothesized bimetallic mechanism: copolymerization has the following orders with respect to the reactants: 1 for the epoxide (cyclohexene oxide), 0 for CO2 and ~2 for Zn. According to this mechanism, zinc alkoxide unimer А or dimer А2 add CO2 and, depending on the structure of the starting complex, they form either carbonate complexes (В or В2, which, like А and А2, exist in equilibrium with each other) or mixed alkoxide– carbonate complex АВ (Scheme 17).
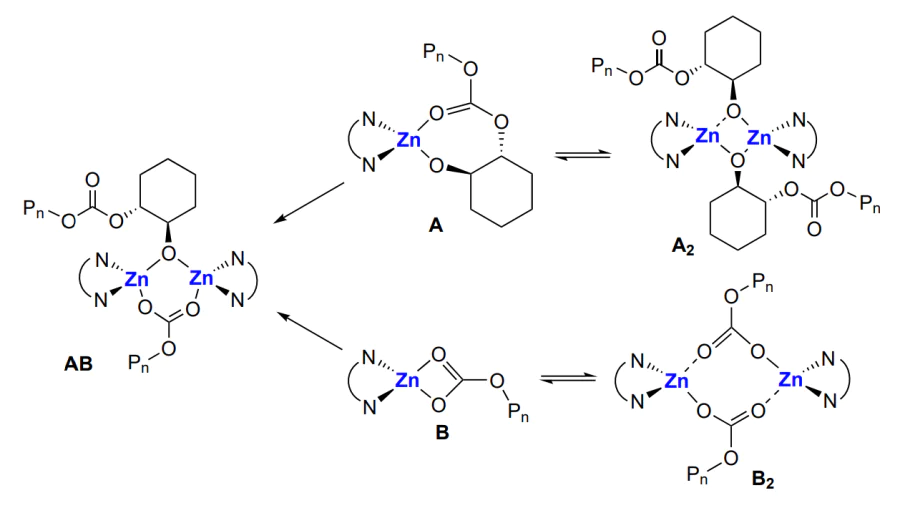
In addition, А, А2 or АВ do not react with the epoxide; in turn, В and В2 do not participate in the reaction with CO2. The latter react with epoxide and are thus converted to АВ, which then reacts with CO2 and closes the catalytic cycle of the reactions (Scheme 18).
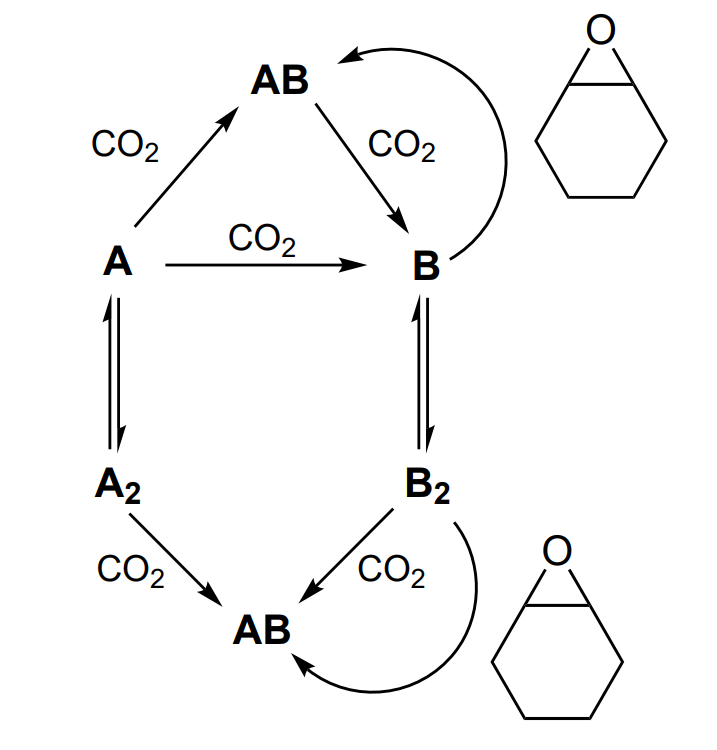
The repetition of cycles АВ → (В → В2) → АВ leads to the growth of the polycarbonate chain, the MW of which is controlled by the molar ratio of the epoxide and Zn. In this case, the bimetallic mechanism is implemented via epoxide coordination and activation on one zinc atom, while the other zinc atom provides the concerted approach and reaction of the growing polycarbonate with the epoxide ring (Scheme 19).
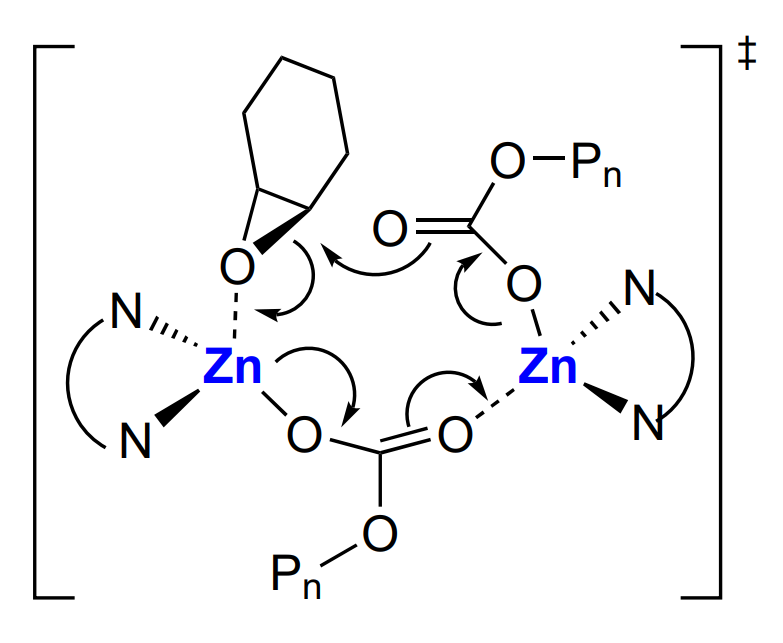
It is noteworthy that these complexes are inapplicable for copolymerization of propylene oxide with CO2 (0.1 mol.% catalyst, 10 atm, 60 – 100 °C), which is due to the intramolecular isomerization, resulting in elimination of cyclic carbonate. This reaction proceeds at a higher rate than the chain propagation reaction and inhibits the polymer formation[144].
The same complexes were used for the copolymerization of a mixture of limonene oxide diastereomers (trans : cis = 1.2 : 1) with CO2 (0.4 mol.% catalyst, 3.4 – 47 atm)[141]. When the temperature was below 25 °C or above 35 °C, the copolymerization barely took place. In the temperature range of 25 – 35 °C, virtually only the trans-isomer participated in the copolymerization, the resulting copolymer had a moderate MW (<104) and narrow MWD (Đ = 1.13 – 1.15), but the catalyst activity was low. Nevertheless, this result can be considered a success, since limonene oxide polymerizes with great difficulty. Similar results were obtained by Greiner and co-workers[145], except that they synthesized the polycarbonate with MW up to 105, while retaining narrow MWD (Đ = 1.12 – 1.19) ([monomer]/[catalyst] = 80 – 470, 90 °C, 10 atm). The (BDI)Zn – N(SiMe3)2 complex 8b proved to be suitable for the copolymerization of limonene dioxide with CO2 (25 °C, 10 – 40 atm)[146]. Epoxide is a mixture of four isomers, of which only the trans-isomer was active in the copolymerization. This resulted in moderate conversions (30 – 50%), which were attained at a relatively high catalyst concentration ([monomer]/[catalyst] = 100 – 250). The polycarbonate was characterized by unimodal, relatively narrow MWD (Đ < 1.3).
The introduction of trifluoromethyl groups (R = CF3) into (BDI)Zn – N(SiMe3)2 8b increased the activity towards the CO2 copolymerization with limonene oxide and cyclohexene oxide by a factor of 5 – 6[147]. Nevertheless, cyclohexene oxide was completely consumed in the copolymerization within less than 1 h even when the [catalyst]/[monomer] ratio was 1/2000, while limonene oxide was consumed an order of magnitude more slowly and at a higher (~6-fold) catalyst concentration[148]. This complex proved to be active in the copolymerization of propylene oxide and styrene oxide and in the terpolymerization of limonene oxide or cyclohexene oxide with propylene oxide and CO2. However, in the case of propylene oxide and styrene oxide, cyclic carbonate was formed as a by-product in a content of up to 30% or, in some cases, even higher. The same complex was used to synthesize two types of polymers: poly(3-hydroxybutyrate)-block-polycarbonate block copolymer and a random copolymer of these components in various ratios with Mn = (4.5 – 20) × 105 and dispersity Đ = 1.2 – 1.9[149, 150]. For the synthesis of block copolymers, the catalyst, epoxide (cyclohexene oxide or cyclopentene oxide) and lactone are first loaded into the reactor, lactone homopolymerization is carried out and then CO2 is injected and pressure is increased to 40 atm, which promotes growth of the polycarbonate block. In the case of limonene oxide, the reverse order is used: after loading the reactants, CO2 is introduced under pressure, copolymerization is conducted up to the desired conversion, pressure is relieved, and then the lactone block growth takes place. The random copolymerization is carried out up to complete epoxide conversion: the reactants are added and CO2 is supplied at a pressure of 3 atm (cyclohexene oxide/cyclopentene oxide) or 9 atm (limonene oxide). In each case, this gives high-molecular-weight products with moderate molecular-weight dispersity (Mn = (45 – 200) × 103 and Đ = 1.2 – 1.9).
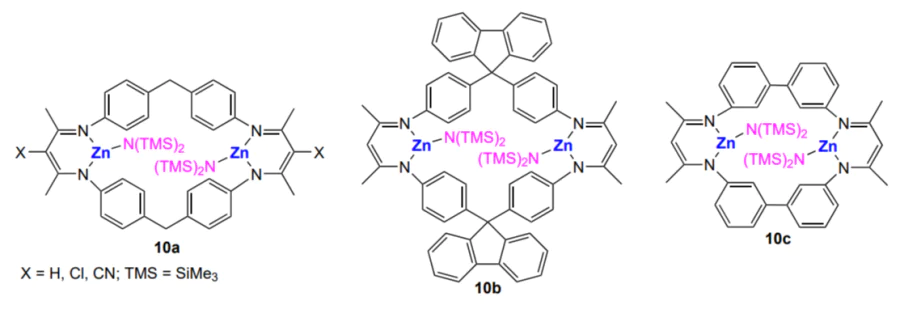
Transition to binuclear zinc β-diiminate complexes 10a – c further increased the activity of the complexes towards CO2 and cyclohexene oxide copolymerization (TOF = 1.5 × 105 h–1)[151]. The resulting polymer contains 70 – 99% carbonate linkages in the macromolecule; depending on the conditions, MW is 3 × 104 to 3 × 105 and Đ = 1.3 – 1.8.
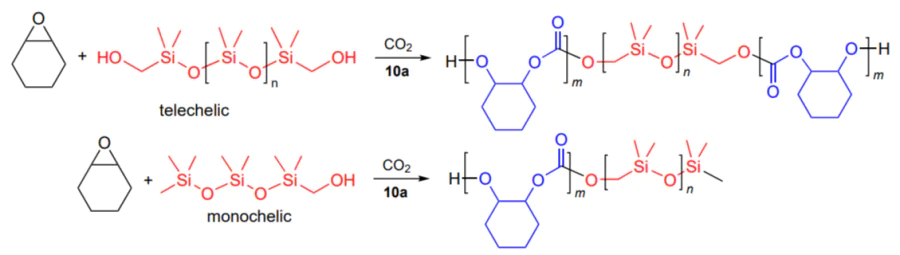
An original process for the synthesis of block copolymers by chain transfer in the copolymerization of CO2 with cyclohexene oxide catalyzed by analogous zinc β-diiminate complexes was reported by Rieger and co-workers[152]. The addition of polydimethylsiloxane with one or two terminal OH groups as a mono- or telechelic (chain transfer agent) makes it possible to obtain di- or triblock-copolymer of polysiloxane with polycarbonate (Scheme 20).
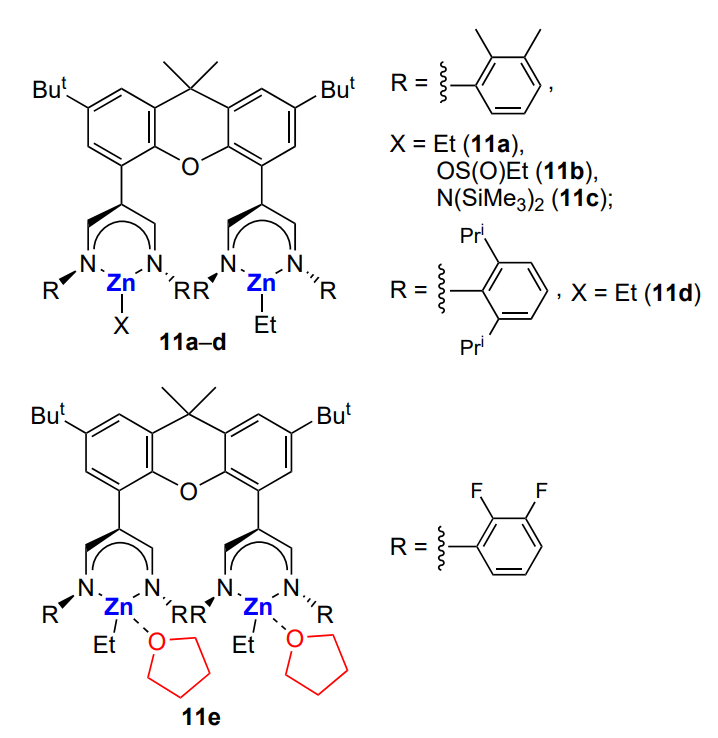
However, if complexes 11a, 11d and 11e are used instead of complex 11c (TON = 748), the catalytic activity in the cyclohexene oxide copolymerization with CO2 sharply decreases (TON = 170 – 380), while the treatment of these complexes with an excess of SO2 increases the activity (TON = 860 – 1190)[153, 154].
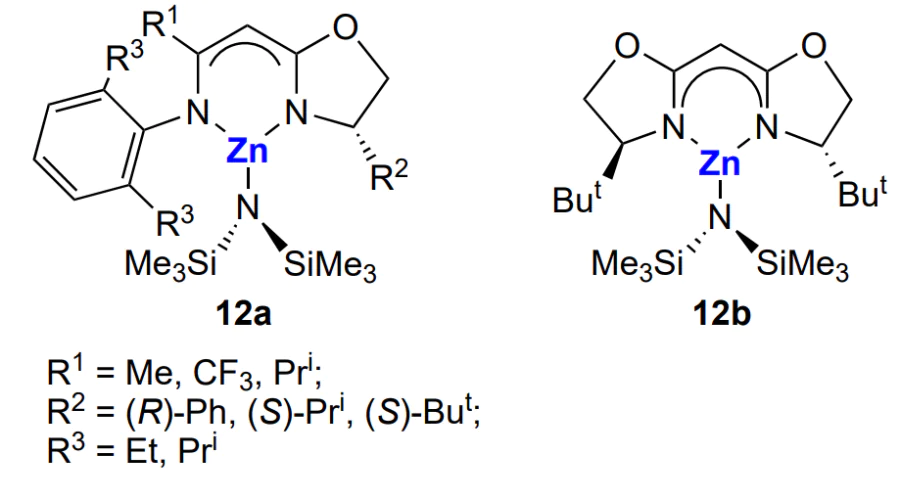
An important issue is the stereochemistry of formation of polycarbonates. It was found for cyclohexene oxide copolymerization with CO2 induced by the (BDI)ZnOX complexes that the epoxide ring opening is accompanied by stereochemical inversion and gives rise to stereocentres in the backbone of the macromolecule. In view of these results, chiral zinc-containing catalysts 12a and 12b were proposed; as expected, they could provide stereochemical control[135].
Indeed, in the copolymerization of cyclohexene oxide and CO2 catalyzed by these compounds (1 mol.%, 6.8 atm, 20 °C), the control of copolymer MWD was retained, and the reaction gave a polycarbonate containing more than 95% carbonate linkages mainly with the isotactic unit attachment (the fraction of R,R or S,S was 70 to 90%).
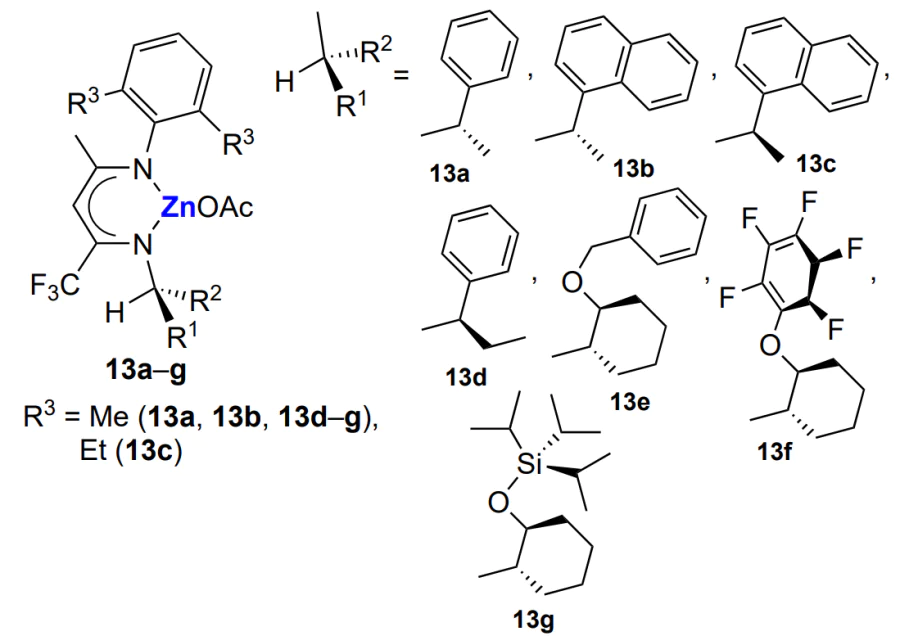
On the basis of the results of studies of epoxide copolymerization involving β-diiminate complexes[70, 71, 75, 135, 155-158], new enantioselective C1-symmetric catalysts (BDI)ZnOAc (13) were proposed for the synthesis of isotactic poly(cyclohexene carbonate) under mild conditions (22 °C, 8 atm, [monomer]/[Zn] = 400 – 4000)[159].
The choice of the R1 and R2 groups is fundamentally important, because this determines the geometry of the complex required for the coordination of cyclohexene oxide in a strictly definite way. Despite the low overall activity of catalysts (TOF = 1.2 – 190 h–1), the stereoselectivity of the formation of isotactic polycarbonate was high (above 99%). Hydrolysis of isotactic polycarbonate afforded the corresponding (R,R)-diol, while the thermal degradation of polycarbonate led to 100% yield of cyclic carbonate with retention of the (R,R)-configuration; in turn, this product can be again converted to isotactic polycarbonate on treatment with EtZnOPri. This approach illustrates wide possibilities of recycling and reuse of polycarbonates.
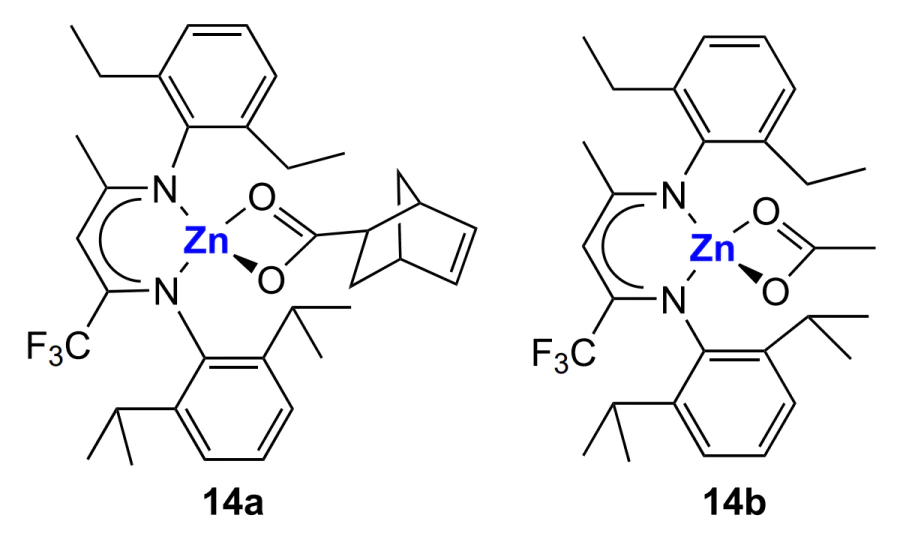
The living copolymerization mechanism involving β-diiminate complexes not only provides control over MWD and MW of polycarbonates by variation of the ratio of the monomer and the catalyst but also allows the synthesis of block copolymers. For example, using the (BDI)ZnOX complexes (14a,b), it is possible to prepare multiblock copolymers (up to six blocks inclusive) on the basis of functional derivatives of cyclohexene oxide (Scheme 21)[160, 161]. The synthesis is carried out at 50 °C, 6.8 atm and [catalyst] = 0.01 – 0.02 mol L–1. The duration of each step (> 99% conversion) under these conditions 15 – 45 min.
A combination of ring-opening copolymerization and reversible addition–fragmentation chain transfer radical polymerization (RAFT) using complex 15 was proposed by Wu and co-workers[162] (Scheme 22).
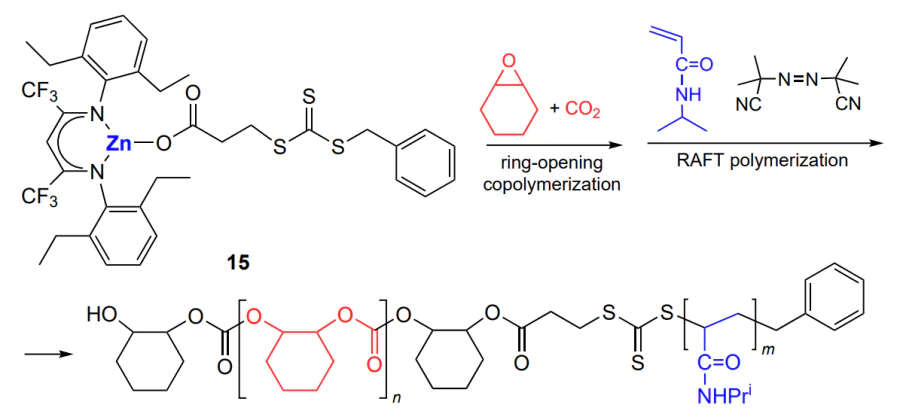
Using tandem catalyst 15, the authors successively performed copolymerization of cyclohexene oxide (or 4-vinylcyclohexene oxide) and CO2 (50 °C, 30 atm, [monomer]/[15] = 200 – 400) and polymerization of vinyl monomers, N-isopropylacrylamide and N,N-dimethylaminoethyl methacrylate, initiated by a radical initiator ([monomer]/[initiator] = 100 – 400, 65 °C) and controlled by the trithiocarbonate moiety of the complex. This gave diblock copolymers with MW = (4 – 7) × 104 and Đ = 1.1 – 1.2.
A combination of ring-opening copolymerization and group-transfer polymerization of vinyl monomer was proposed by Riger and co-workers[163]. The authors synthesized a bifunctional catalyst (binuclear complex 16, Scheme 23). Zinc complex provides the formation of the polycarbonate, while yttrium is responsible of the polymerization of 2-vinylpyridine or 2-isopropenyl-2-oxazoline.
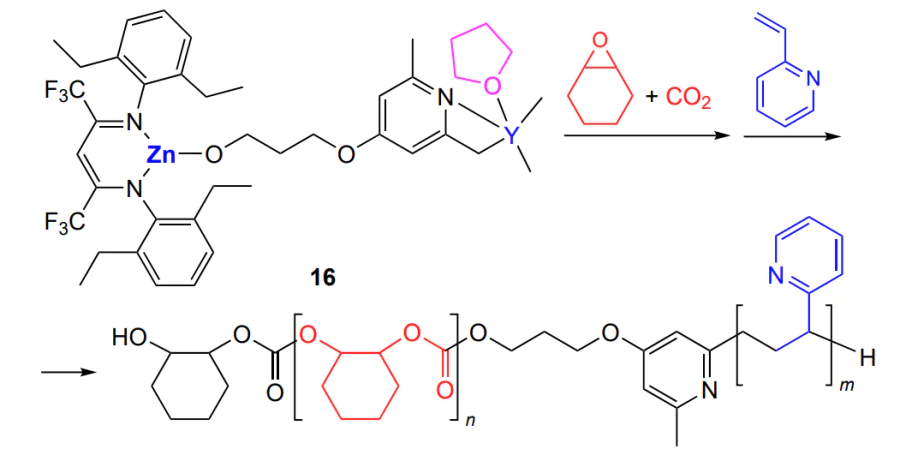
The synthesis of block copolymer can be either a stepwise (first yttrium-initiated polymerization is carried out and then epoxide and CO2 are added) or a one-pot reaction (simultaneous addition of vinylpyridine or oxazoline and cyclohexene oxide). In the latter case, group-transfer polymerization of vinylpyridine first takes place (in the absence of CO2) under the action of yttrium complex and then the poly(cyclohexene carbonate) block is built up upon CO2 supply and pressure increase to 30 atm. In both cases, polymerization takes place at 40 °C. The block lengths are controlled by the ratio of monomers and concentration of the complexes.
2.1.5. Binuclear zinc phenoxide complexes
One more example of binuclear zinc complexes exhibiting high activity in the copolymerization of epoxides and CO2 are compounds 17, which were first described by Williams and co-workers[164] (Scheme 24).
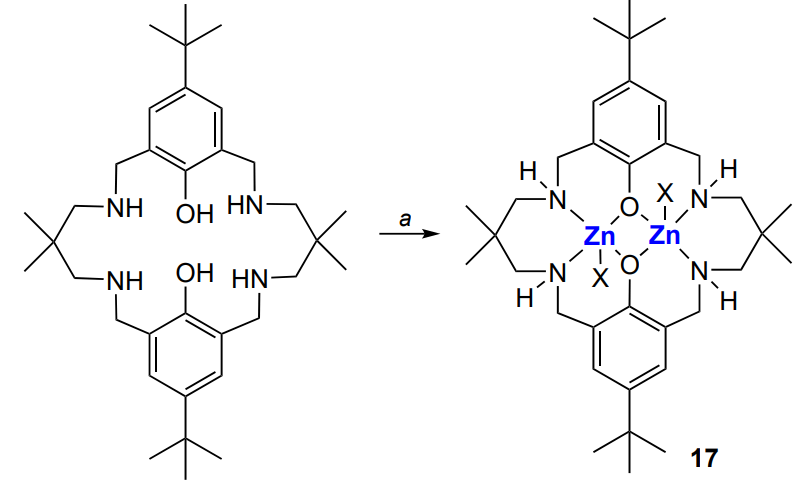
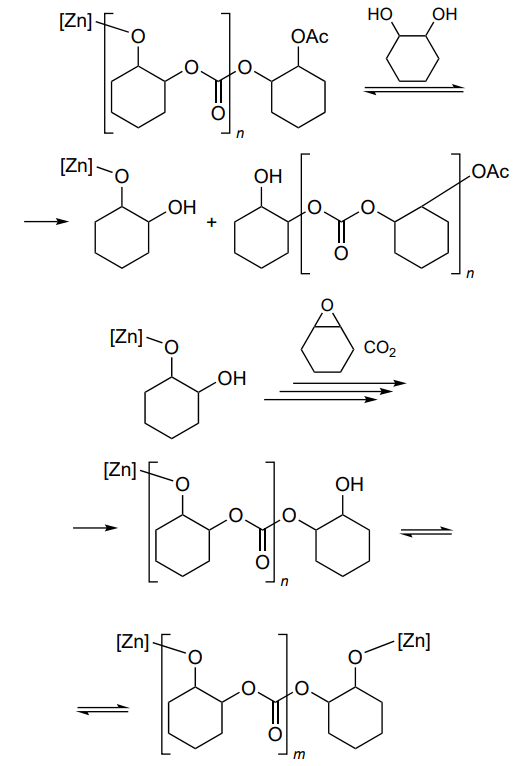
A distinctive feature of the catalysts, which were studied by X-ray diffraction[165-167], is the ability to perform copolymerization at an atmospheric pressure of CO2. The reaction was carried out at 80 – 100 °C and a [cyclohexene oxide]/[catalyst] ratio of 1000. The selectivity to polycarbonate was 94 – 96% and the proportion of ether linkages in the polymer was less than 1%. The low yield of cyclic carbonate in comparison with polycarbonate is due to the difference between the activation barriers for the formation of these products (~97 and 137 kJ mol–1)[168]. Diol was used to control MW. Finally, the resulting poly(cyclohexene carbonate) had relatively low MW and narrow bimodal MWD (Mn < 104, Đ < 1.2), which is unusual for ring-opening copolymerization. The authors found two types of macromolecules and attributed this to chain transfer reactions (Scheme 25).
The turnover frequency of the catalyst at 1 atm is relatively low (20 – 40 h–1), but increases to 140 h–1 when pressure is increased to 10 atm. The replacement of tert-butyl substituents by methyl or methoxy groups does not significantly influence the polymer characteristics or catalyst activity. The catalyst retains the activity even when the reaction system is contaminated with H2S, H2O, O2, SO2, NO or CH4 and also when Zn is replaced by Mg (in this case, the catalyst stability increases)[24].
Like β-diiminate complexes, other binuclear zinc complexes are widely used to synthesize block copolymers. For example, polylactide-block-poly(cyclohexene carbonate)-block-polylactide triblock copolymers are formed under the action of complex 18, which can be used either with[45] or without yttrium complex[47, 169]. The same complex was used to prepare polycaprolactone-block-poly(cyclohexene carbonate) block copolymers[46].
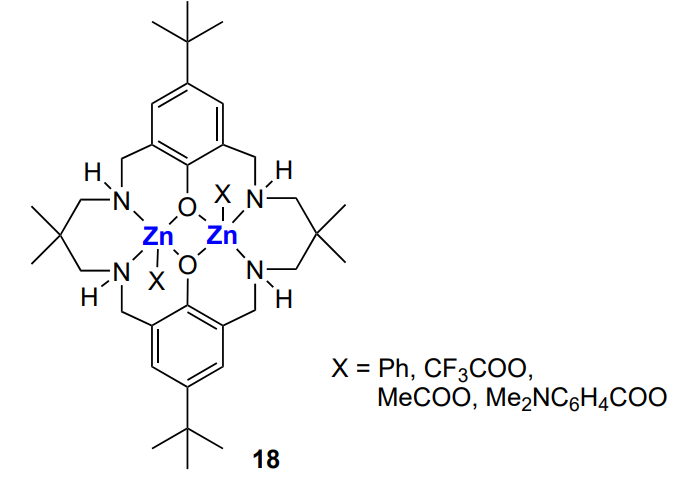
The polylactide-block-poly(cyclohexene carbonate)-block-polylactide triblock copolymer was synthesized with binuclear Mg–Zn catalyst of a similar structure with X = Ph[47]. Under the action of the binuclear zinc catalyst, АВCВА pentablock copolymer was formed in situ from a mixture of monomers (phthalic anhydride, cyclohexene oxide and ε-decalactone) and CO2 (Scheme 26)[43].
Thus, the key features and the bimetallic mechanism of polycarbonate formation in the copolymerization of epoxides and CO2 carried out with homogeneous and heterogeneous catalysts were established for the first time in relation to zinc-based complex catalysts. Polycarbonates with narrow MWD and controlled MW and optically active (stereoregular) polycarbonates were obtained. An important feature found for the copolymerization is the occurrence of two side reactions: formation of cyclic carbonate and formation of ether linkages in addition to the carbonate linkages. These reactions are most pronounced for propylene oxide and are less pronounced for cyclohexene oxide. This is why the major array of experimental data and the highest TOF values of catalysts were obtained for the copolymerization of cyclohexene oxide with CO2. Particularly the homogeneous catalysis with binuclear zinc complexes such as β-diiminate, phenoxyamine, β-diketoiminate and so on enables the synthesis of homo- and co-polycarbonates and their block-copolymers by combining various types of polymerization processes.
2.2. Organoaluminium catalysts
2.2.1. Aluminium complexes with Schiff bases
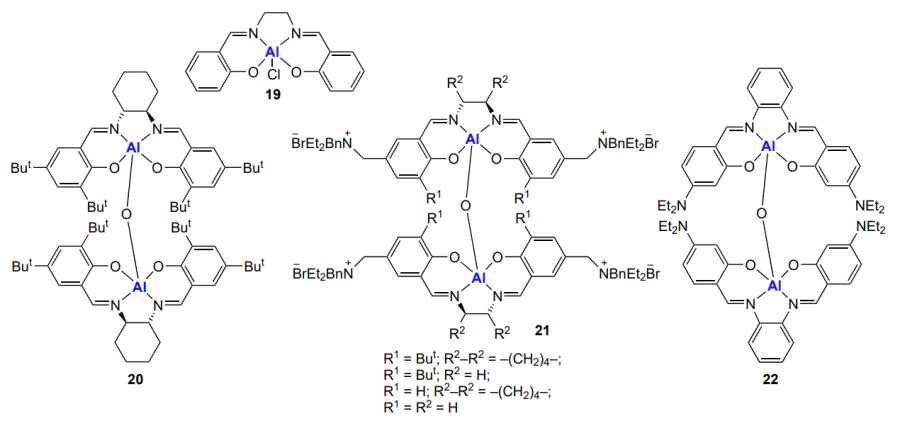
Aluminium complexes are mainly active in the cycloaddition of CO2 to epoxides[6, 25]. These complexes include, for example, aluminium compounds with salen ligands[170-176]. Salen complex 19 provides the formation of ethylene carbonate at a CO2 pressure of 160 atm and a temperature of 110 °C both by itself (TOF = 174 h–1) and with the Bu4NBr co-catalyst (Hereinafter, tetra(n-butyl)ammonium salts are used unless indicated otherwise.) (TOF = 2200 h–1)[177]. Binuclear complex 20, together with the same co-catalyst, is considerably more active, and the synthesis can be performed at 25 °C and a CO2 pressure of 1 atm. Cyclic carbonates were obtained by this method in high yields: 99% for 3-phenylpropylene carbonate (in 24 h), 88% for 1,2-hexene carbonate and 64% for 1,2-decene carbonate within 3 h[172]. A combination of both components, Lewis acid site and nucleophilic co-catalyst, in one catalyst 21 was implemented by Villuendas and co-workers[177].
An even higher activity in the cycloaddition is inherent in the binuclear salphen complexes 22, apparently, because of higher Lewis acidity of aluminium with salphen ligands[178].
However, similar aluminium salen and salphen complexes still can show activity in CO2 and epoxide copolymerization[179]. The first advances in the synthesis of polycarbonates by means of aluminium salen and salphen complexes are related to studies of Darensbourg and Billodeaux[180] and Sugimoto et al[181].
Darensbourg and Billodeaux[180] described aluminium salen complexes 23 (Scheme 27). It was found that complexes 23b, containing electron-donating tert-butyl groups in positions 3 and 5 of the phenoxide substituent, in combination with the most efficient co-catalysts (Lewis bases), phosphines (PPh3, PCy3) or bis(phenylphosphine)iminium salts (PPNX), are inactive in the copolymerization of CO2 with cyclohexene oxide (80 °C, 34 atm).
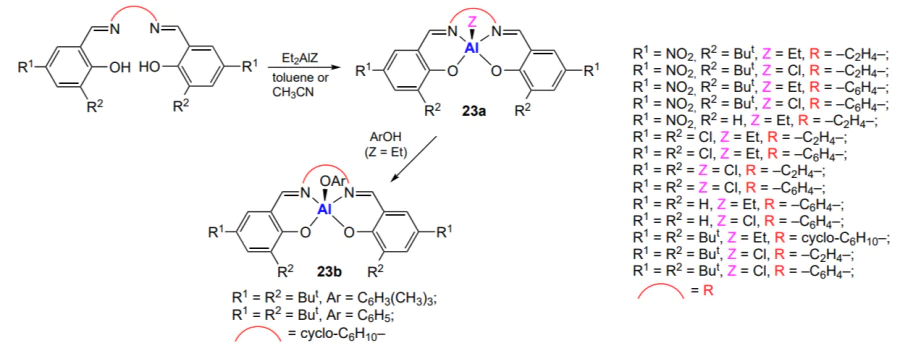
The replacement of the axial Cl or Et ligand with PhO is also inefficient. However, in the absence of substituents in positions 3 and 5 or in the presence of electron-withdrawing substituents (NO2), which increase the active site electrophilicity, poly(cyclohexene carbonate) is formed. In the absence of a co-catalyst, the proportion of cyclic carbonate is 10%, while the proportion of ether linkages in the copolymer is 26%. When the Bu4NX salts (X = Cl, N3, OAc) are used, the selectivity to polycarbonate increases to 93 – 99%, while the content of the carbonate linkages in the copolymer increases to 90 – 99%, and TOF is in the range of 7 – 35 h–1. No correlation of the salt anion or the nature of substituents and axial ligand with the selectivity and activity of the complex has been identified so far. When 1-methylimidazole, 4-(dimethylamino)-pyridine or pyridine is used as a co-catalyst, the activity of complexes decreases in the series 4-(dimethylamino)-pyridine > 1-methylimidazole > pyridine. The selectivity to polycarbonate and the content of carbonate linkages in the polymer vary in the same order.
Aluminium salphen complex 24 with tetraethylammonium acetate was used for the copolymerization of CO2 and cyclohexene oxide in CH2Cl2 at 50 atm and 80 °C[182]. In this case, polymerization follows the living mechanism; however, the polymer formed in this reaction has bimodal MWD. The selectivity of the complex to polycarbonate does not depend on the CO2 pressure (above 95%), but is sensitive to the temperature and the concentration of the ammonium salt (may decrease to 45%). The content of the ether linkages in the copolymer varies from 6 to 18% depending on the conditions.
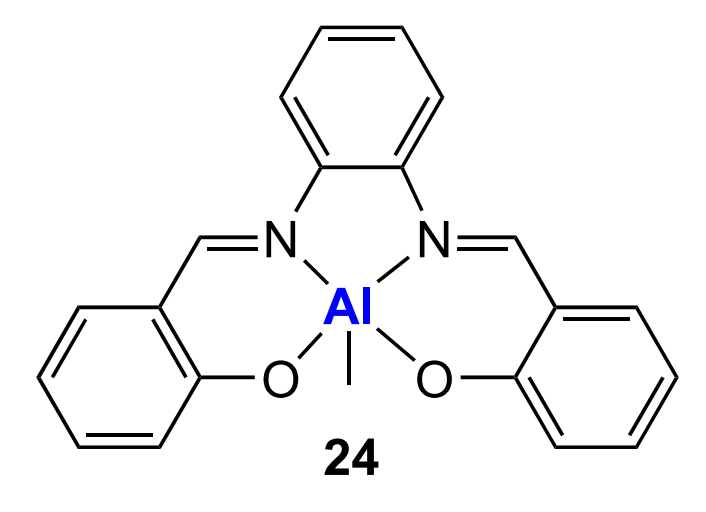
The replacement of the acetate in the ammonium salt with chloride does not have a noticeable effect on the polymer yield, the selectivity of the complex or the content of carbonate linkages in the polymer, while the replacement with bromide sharply decreases the selectivity, but does not affect the content of carbonate linkages (up to 70%). When triphenylphosphine is used, the proportion of carbonate linkages, conversely, decreases, while the selectivity remains high. The use of pyridine as a base made it possible to obtain polycarbonate with unimodal MWD and with retention of high selectivity.
Aluminium complexes with Schiff bases 25 and 26, in combination with tetraethylammonium acetate, also allow the synthesis of oligomeric poly(cyclohexene carbonate) with high selectivity and low content of ether linkages in the polymer[181].
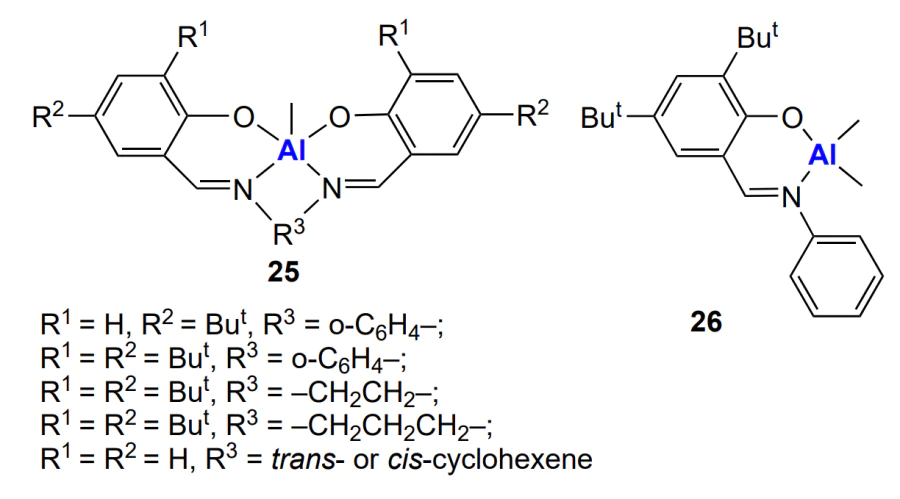
In this case, high selectivity was found only for the complexes with R1 = R2 = But, R3 = Et or R1 = R2 = But, R3 = cis-cyclohexene. Despite the living polymerization mechanism, the products had bimodal MWD. The cause for the bimodal MWD of the polymer is the formation of some binuclear complex due to the presence of impurity (H2O), which results in the formation of a high-molecular-weight polymer.
2.2.2. Aluminium phenoxide and alkoxide complexes
Aluminium phenoxide and aluminium alkoxide complexes for the copolymerization of epoxides (cyclohexene and propylene oxides) and CO2 were first described at the end of 1990s[182-184]. Commercially available Al(OiPr)3 can catalyze copolymerization of cyclohexene oxide and CO2 at high pressures (not lower than 100 atm), with the proportion of carbonate linkages in the copolymer not exceeding 20%[185]. The use of bulky substituents in the alkoxide provides high selectivity of polymer formation, but the fraction of carbonate linkages is low (7 – 20%). Under the action of alkoxides R1 – O – Al(Cl) – O – R2, where R1 and R2 are cyclohexyl and n-C8H17OCOCH=CHCO – , oligomers with broad MWD are formed (Mn = 3000 – 5000 and Đ = 3 – 4)[182]. In the case of sterically more crowded substituents (complex 27), it is possible to suppress the side termination reactions; as a result, polymerization proceeds by a living mechanism[183, 184].
Another example is the aluminium calixarene complex, (25,27-dimethoxy-para-tert-butylcalix[4]arene-26,28-dilato) aluminium chloride (28), proposed for copolymerization of propylene oxide and cyclohexene oxide with CO2[186]. However, in this case, too, oligomers (Mn < 5500) are formed, with the content of carbonate linkages not exceeding 18%, together with cyclic carbonate (4 to 14 mol.%). Similar results were obtained in the copolymerization of cyclohexene oxide and CO2 catalyzed by phenoxide complex 29[187]. Interestingly, the use of the PPNCl salt shifts the reaction towards the formation of cyclic carbonate[179]. Phenoxide complex 30 shows activity in the copolymerization of cyclohexene oxide and CO2, giving rise to a polymer in more than 65% yield and with a content of carbonate linkages of more than 50%[188].
An original aluminium aminotriphenoxide complex 31 to be used in combination with bis(triphenylphosphine)iminium chloride was proposed by Kleij and co-workers[189], who used this complex to obtain poly(limonene dicarbonate). First, copolymerization of limonene oxide was carried out to give poly(limonene carbonate), which was epoxidated at the double bond, and then the reaction with CO2 was carried out again. The monomer conversion did not exceed 50%, and the resulting polydicarbonate had relatively low MW of ~ 104 and narrow MWD (Đ = 1.19).
2.2.3. Aluminium porphyrin complexes
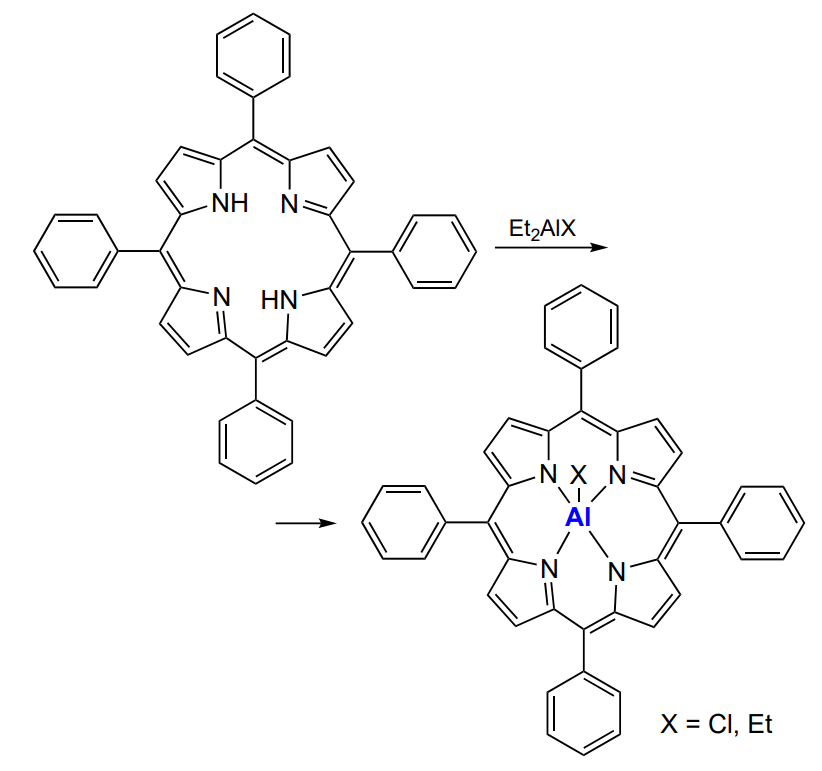
Aluminium porphyrin complexes are the most efficient organoaluminium catalysts for the synthesis of polycarbonates; they can minimize the yields of by-products–cyclic carbonate and polyether. Aluminium porphyrins are formed by the reaction of the desired porphyrin with alkylaluminium (Scheme 28).
It was found that aluminium alkylporphyrin reacts with CO2 in the presence of 1-methylimidazole under mild conditions on exposure to visible light[190]. Therefore, metal porphyrins, 1-methylimidazole and CO2 can be used for the synthesis of propionates, β-ketocarboxylic acids, alkyl methacrylates and malonic acid derivatives[191]. After treatment of aluminium complex with methanol (X = MeO), this reaction proceeds even in the dark. Carbon dioxide activated in this way readily reacts with propylene oxide to give cyclic carbonate. Cyclic carbonates are also formed under the action of aluminium porphyrin in the presence of ammonium or phosphonium salts[192]. In this case, catalyst TON amounts to tens of thousands, while the product yield varies from 30 to 100% depending on the epoxide structure.
In the absence of 1-methylimidazole, random copolymer (composed of carbonate and ether) is formed. Aluminium porphyrins containing X = Et, MeO, OH or Cl as the axial ligand provided the synthesis of propylene oxide and CO2 copolymers with narrow MWD (Đ = 1.08 – 1.15), enriched with syndiotactic sequences, under relatively mild conditions (room temperature, 1 – 8 atm)[54]. However, the introduction of 1-methylimidazole into the system after the removal of unreacted CO2 and epoxide directs the reaction towards the synthesis of cyclic carbonate. Hence, there is a possibility of fine tuning of the synthesis pathway between cyclic carbonate and copolymer.
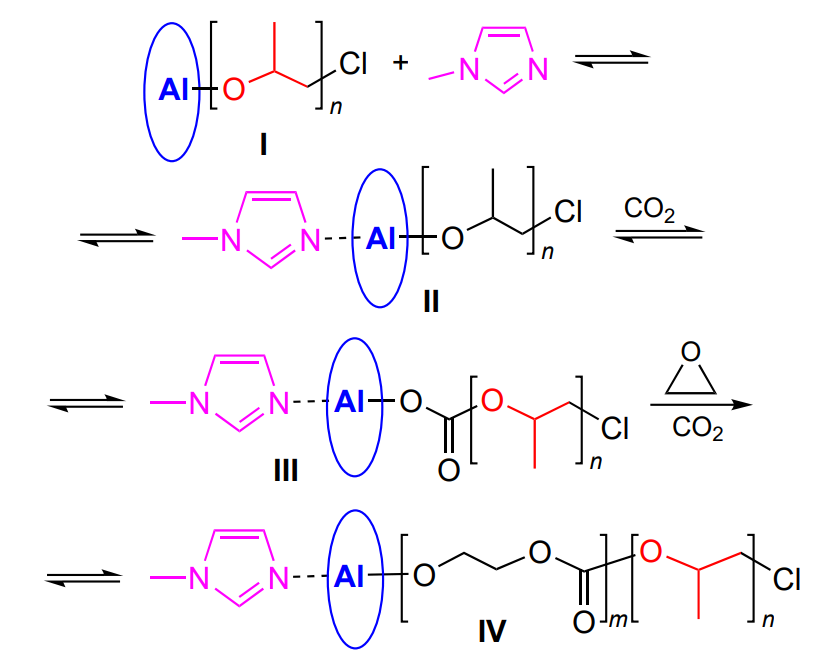
On treatment with aluminium porphyrins, oligomeric polycarbonates (Mn < 104) with narrow MWD are formed most often. For example, (5,10,15,20-tetraphenylporphyrinato) aluminium chloride in the presence of ethyltriphenylphosphine bromide provides the synthesis of poly(propylene carbonate) with Đ = 1.1 – 1.2[193]. A similar result is observed when chloride is replaced with alkoxide and the phosphonium salt is replaced with tetraethylammonium bromide. The molecular weight of poly(propylene carbonate) is controlled by the concentration ratio of the epoxide and the catalyst, which is typical of living polymerization, and copolymerization is carried out with excess CO2. If after completion of propylene oxide polymerization, CO2 and a fresh portion of propylene oxide is added, the poly(propylene oxide)-block-poly(propylene carbonate-co-propylene oxide) block copolymer is formed, in which Mn of the polypropylene oxide block varies from 1200 to 3100 and Đ = 1.07 – 1.09, while the weight of the second (growing) block ranges from 2500 to 4200 and Đ = 1.11 – 1.22. The fraction of carbonate linkages in the second block does not exceed 30%[194]. These results raised the question of how to activate CO2 towards copolymerization with epoxide under the action of aluminium porphyrin complex. A solution was proposed by Aida and Inoue[195], who successively performed the synthesisis of polypropylene oxide I under the action of tetraphenylporphyrin complex with Et2AlCl, methylimidazole complex II, complex with activated CO2 III and ethylene oxide copolymer IV with trapped CO2 (Scheme 29).
This catalytic system, tetraphenylporphyrin with diethylaluminium chloride, activated by various compounds (methylimidazole[195], ethyltriphenylphosphonium bromide[196], tetraethylammonium bromide[197]) proved to be suitable for the synthesis of alternating copolymers of CO2 with various epoxides (ethylene oxide, propylene oxide, butylene oxide (hereinafter, R1 = C2H5, R2 = H, see Scheme 1), cyclohexene oxide). In some cases, Mn of the copolymers reached ~6 × 104, while the dispersity Đ was less than 1.2. The living polymerization mechanism was confirmed by not only the linear growth of Mn with increasing monomer conversion, but also the formation of block copolymers in which the alternating propylene oxide’--- phthalic anhydride copolymer served as the initial block, and polycarbonate was the growing block[197]. It is noteworthy that combination of the aluminium porphyrin complex with quaternary ammonium or phosphonium salts allows the growth of macromolecules on both ends; in the case of synthesis of block copolymers, this results in the formation of symmetrical АВА triblock copolymer (Scheme 30).
An integrated study of the reaction of aluminium porphyrins in which X = Cl, O(CH2)9CH3, or OOC(CH2)6CH3 with propylene oxide and CO2 showed that the epoxide is incorporated into the Al – X bond in two ways, Al(porphyrin) – OCH(CH3)CH2 – X and Al(porphyrin) – OCH2CH(CH3) – X[198]. The rate of epoxide ring opening decreases in the order Cl > O(CH2)9CH3 > OOC(CH2)6CH3; however, upon the addition of a Lewis base, 4-(dimethylamino)pyridine, the order of decreasing changes to Cl > OOC(CH2)6CH3 > O(CH2)9CH3. Carbon dioxide is reversibly incorporated into the Al – OR alkoxide bond to give (porphyrin)Al – OC(=O)OR; this reaction is accelerated in the presence of a Lewis base. The coordination of 4-(dimethylamino)pyridine to aluminium porphyrin is enhanced in the order OOC(CH2)6CH3 > OOC(CH2)9CH3 >> O(CH2)9CH3. Thus, in the absence of 4-(dimethylamino) pyridine, the addition of epoxide and CO2 to alkoxide, giving rise to carbonate and ether linkages, is equally probable, while in the presence of 4-(dimethylamino)pyridine, only carbonate linkages are produced. Irrespective of the monomer structure, the polycarbonate formed in this reaction is an atactic polymer containing a higher content of syndiotactic groups.
Wang and co-workers[199] were able to attain a high activity of the metal porphyrin in propylene oxide and CO2 copolymerization and obtained a high-molecular weight polycarbonate [Mn = (1 – 6) × 104, Đ < 1.3] in a high yield and with a high selectivity. The authors introduced –(CH2)3N+Bu3X– (X = I, NO3, BF4, ClO4), F and But substituents into the para-positions of the benzene rings and varied the nature of the axial ligand (Cl, NO3, BF4, ClO4). The selectivity to polycarbonate reached 95% and decreased to 70 – 80% as the catalyst concentration decreased and the temperature increased. The turnover frequency exceeded 3000 h–1 for [propylene oxide]/[catalyst] £ 20 000, while at higher contents of the catalyst, TOF = 200 – 400 h–1. Regarding the whole set of characteristics, the best results were found for the complex with the axial NO3 ligand and (CH2)3N+Bu3NO3– and But substituents, [propylene oxide]/[catalyst] = 5000 and a temperature of 70 °C; under these conditions, the selectivity was 92%, the content of the carbonate linkages in the copolymer was 99%, Mn = 3.6 × 104, Đ < 1.08 and TOF = 450 h–1.
Nozaki and co-workers[200] used similar aluminium porphyrin chloride and bromide 32a and 32b, which showed exceptionally high activity in the copolymerization of cyclohexene oxide and CO2. A polycarbonate with Mn ≈ 2.8 × 105 was formed when the catalyst content was only 10–3 mol.%, TOF = 104 h–1.
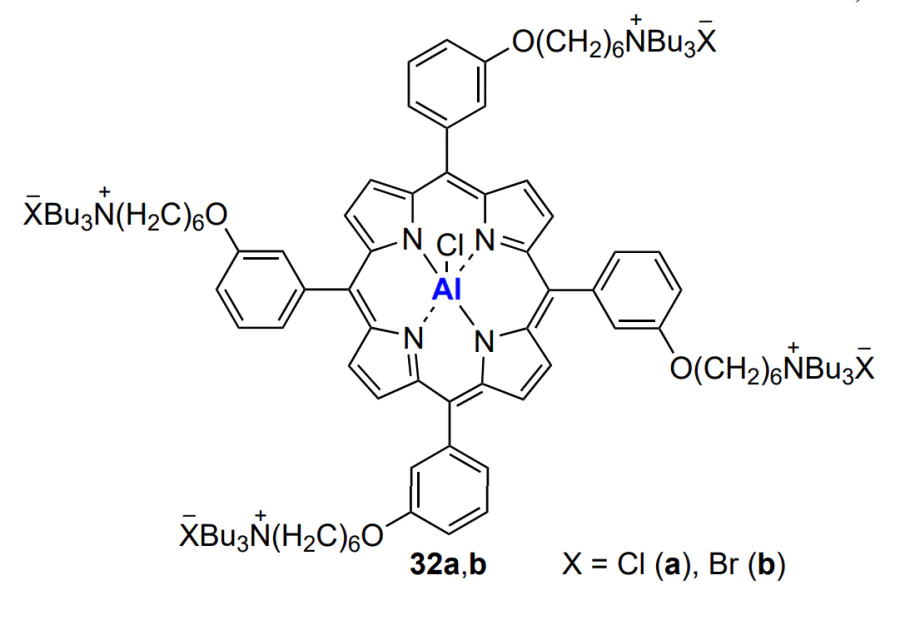
In the opinion of the authors[200], the high activity and selectivity of the catalyst are caused by the cooperative action of the metal active centre and the quaternary ammonium cation in the transition state, which both facilitate epoxide ring opening by the carbonate anion and formation of the carbonate linkage. Aluminium porphyrins activated by the bis(triphenylphosphine) iminium chloride were used for the terpolymerization of CO2, alicyclic epoxide (cyclohexene oxide) and epoxides with aliphatic substituents (propylene oxide, butylene oxide, hexene oxide, octene oxide, dodecene oxide; R1 = n-Alk, R2 = H, see Scheme 1)[201]. Despite the high content of carbonate linkages in the copolymer, the catalyst selectivity (the yield of linear polycarbonate) was less than 90%, and the polymerization products had a narrow but bimodal MWD. The last-mentioned fact indicates that there are two parallel mechanisms of the formation of macromolecules. Wang and co-workers[202] reported a systematic study of aluminium porphyrin chlorides 33 with various aromatic substituents as catalysts for copolymerization of propylene oxide and CO2.
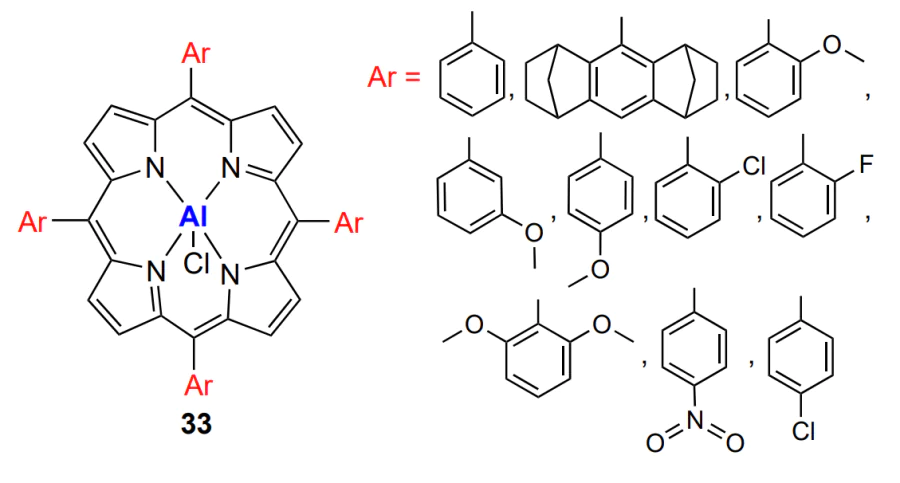
Complexes with bulky substituents provide the formation of polycarbonate with less than 1% ether linkages. The fraction of cyclic carbonate increases and TOF decreases with decreasing catalyst concentration and increasing temperature and concentration of the co-catalyst, bis(triphenylphosphine) iminium chloride. The increase in the electron-donating properties of the substituent (ortho-methoxy > meta-methoxy > para-methoxy > chloro > fluoro) enhances the catalyst selectivity and virtually does not affect the productivity. The authors[202] demonstrated that strong electron-donating and/or electron-withdrawing effect of the substituent has an adverse effect on the selectivity of the catalyst, and the best properties are inherent in the catalysts with a moderate activity of the active centre.
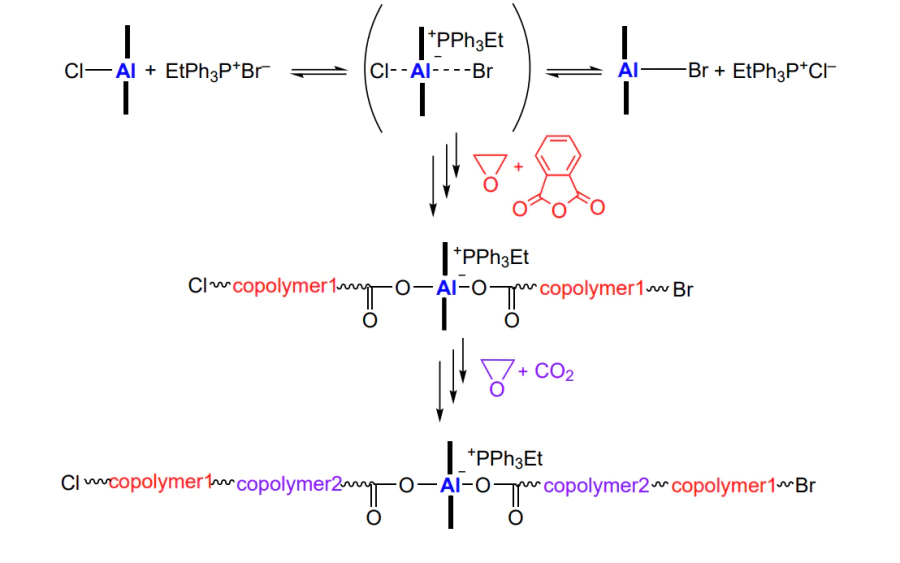
Modification of the axial ligand in the aluminium porphyrin molecule makes it possible to combine various polymerization mechanisms and, for example, to obtain block copolymers based on polycarbonate and vinyl polymer[203]. The simultaneous use of (5,10,15,20-tetraphenylporphyrinato)aluminium chloride (TPP2-Cl)AlIIICl or its derivatives in which the ortho- and/or para-positions of phenyl substituents are occupied by Cl atoms or methoxy groups, bis(triphenylphosphine)iminium chloride, and trithiocarbonate (reversible chain transfer agent, 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid, TTC-COOH) in the polymerization of vinyl monomers resulted in the synthesis of a set of hybrid block copolymers (Scheme 31). It is of interest that trithiocarbonate TTC-COOH does not react with the bis(triphenylphosphine)iminium salt, but in the aluminium complex, the axial Cl ligand is replaced with the carboxylate and, therefore, in the epoxide and CO2 copolymerization, the TTC-COO–trithiocarbonate moiety is located at the end of the growing chain.
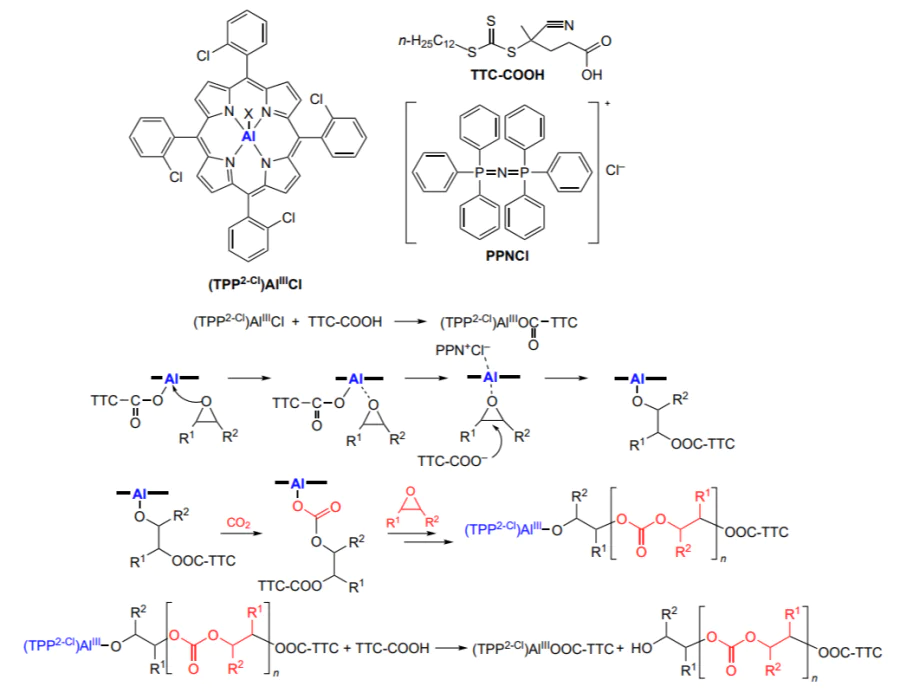
Thus, it is possible to obtain macromolecules containing a trithiocarbonate moiety and capable of providing controlled radical polymerization of vinyl monomers. This synthetic strategy was successfully implemented for the copolymerization of propylene oxide, cyclohexene oxide and hexene oxide with CO2 followed by block copolymerization of methyl methacrylate, styrene and benzyl methacrylate, yielding the corresponding block copolymer products with Mn = (12 – 21) × 103 and Đ = 1.09 – 1.14.
Catalyst 34 (Ref. [202]) provides the formation of exclusively cyclic carbonate above 75 °C and exclusively polycarbonate below 25 °C. The polymer reaction product had a narrow MWD and moderate MW (Mn ≈ (2 – 5) × 104 and Đ = 1.11 – 1.14) and contained almost no ether linkages; however, the catalyst productivity was moderate.
Wang and co-workers[204] proposed thermal and photothermal activation (laser irradiation at 635 nm) for CO2 and epoxide copolymerization induced by aluminium porphyrins and bis(triphenylphosphine)iminium chloride. The authors studied two types of monomers, alicyclic (cyclohexene oxide, 1,2-epoxy-4-vinylcyclohexane) and epoxides with aliphatic substituents (propylene oxide, butylene oxide, hexene oxide, octene oxide). Irrespective of the activation method, the resulting polycarbonates had narrow MWD and MW ≈ (10 – 25) × 103. In the case of photothermal activation of alicyclic epoxides, polycarbonates were formed in more than 99% yields and with equally high contents of carbonate linkages. It is evident that the side intramolecular rearrangement yielding cyclic carbonate did not take place in this case (Scheme 32).
A different situation is observed for epoxides with aliphatic substituents (Scheme 33). In this case, the yield of the target product, polycarbonate, depends on the type of treatment, which is either thermal or photothermal. The proportion of cyclic carbonate is 26 – 39% after photothermal activation and 8 – 11% after thermal treatment. The proportion of carbonate linkages in the copolymer does not depend on the method of activation (97 – 98% or more for propylene oxide and 99% for other epoxides). The molecular weight dispersity of the polycarbonate is greater for photothermal activation.
The twofold increase in TOF (400 – 850 h–1) and reproducibility of the synthesis upon cyclic laser on/off switching should be regarded as advantages of photothermal activation for the copolymerization of both types of epoxides over thermal treatment.
An interesting idea of using ‘oligomeric’ catalyst 35a based on oligomethacrylate with aluminium porphyrin grafted in each monomer unit (n = 4.4 – 6.8) as the side substituent was reported by Wang and co-workers[205]. Oligomeric aluminium porphyrins show a higher activity in the copolymerization of propylene oxide and CO2 (TOF = 4000 – 12 000 h–1) than usual metal porphyrin (TOF = 100 – 300 h–1). The lower the fraction of the bis(triphenylphosphine)iminium co-catalyst, the higher the activity. The reaction gives the copolymer (>99%) containing carbonate and ether linkages with Mn of up to 2 × 105 and narrow MWD (Đ < 1.3). As a continuation of this study, Wang and co-workers[206] expanded the options for both the metal porphyrin structure (35b, 35c) and the way of porphyrin attachment to the backbone.
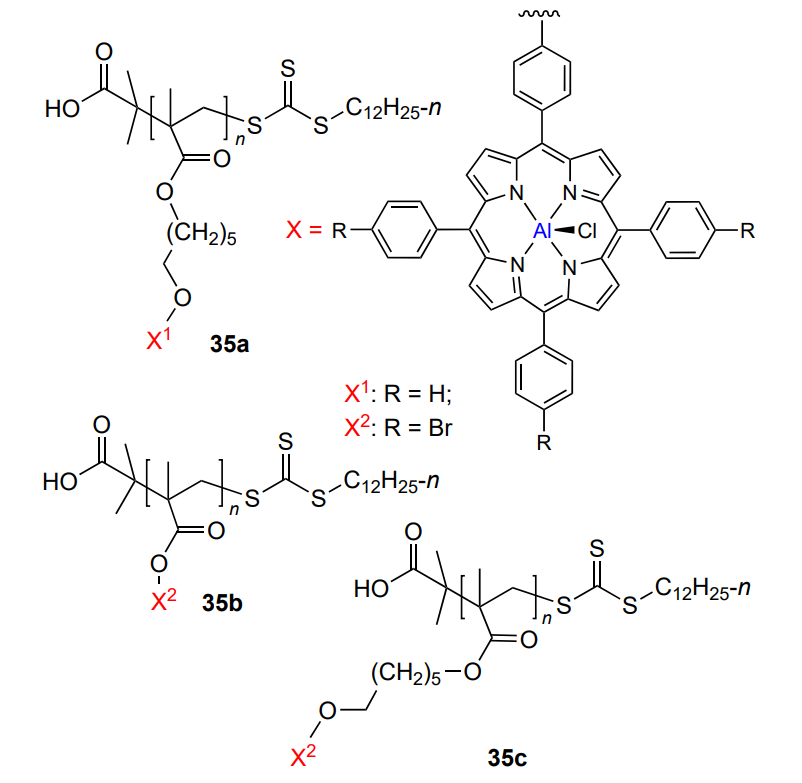
For comparison, the authors used (5,10,15,20-tetraphenyl-porphyrinato)aluminium chloride and its bromo derivative. In this case too, oligomeric porphyrin was more efficient than the monomeric one in terms of (i) catalyst turnover frequency (an order of magnitude higher), (ii) yield of cyclic carbonate (an order of magnitude lower). In the authors’ opinion, this is due to the fact that in oligomeric metal porphyrin, two aluminium atoms are involved in the monomer coordination and incorporation into the chain rather than one Al atom, as is the case for monomeric aluminium porphyrin.
Generally, aluminium porphyrin complexes proved to be quite a successful class of compounds for obtaining both oligomeric and high-molecular-weight polycarbonates with high content of carbonate linkages and for the macromolecular design involving combination of various types of polymerization processes.
2.3. Complexes of rare earth metals
Complexes based on rare earth metals were first used for catalysis of ring-opening polymerization of alkylene oxides and sulfides[207]. In parallel, publications appeared describing their use in the copolymerization of epoxides and CO2. This issue was first mentioned by Zhang and co-workers[208], who proposed mixed Y(P204)3 – AlBui 3 – glycerol complex, where Р204 means poly(2-ethylhexylphosphonate) with a degree of polymerization of 204, as the catalyst[204]. The authors tested other rare earth metals and alcohols; however, only yttrium and glycerol provided the formation of propylene oxide and CO2 copolymer in a high yield and with Mn ≈ 5 × 105, Đ ≈ 2.0. Tan and Hsu[209] used the Y(CF3COO)3 – ZnEt2 – glycerol complex. The molecular-weight-distribution of the copolymer was markedly broader (Đ = 3 – 5); however, the turnover frequency and the selectivity of the catalyst were relatively high (4000 – 6000 h–1 and 96%, respectively). The molecular weight of the copolymer increased during copolymerization and reached a limiting value of 1.2 × 105 after 24 h.
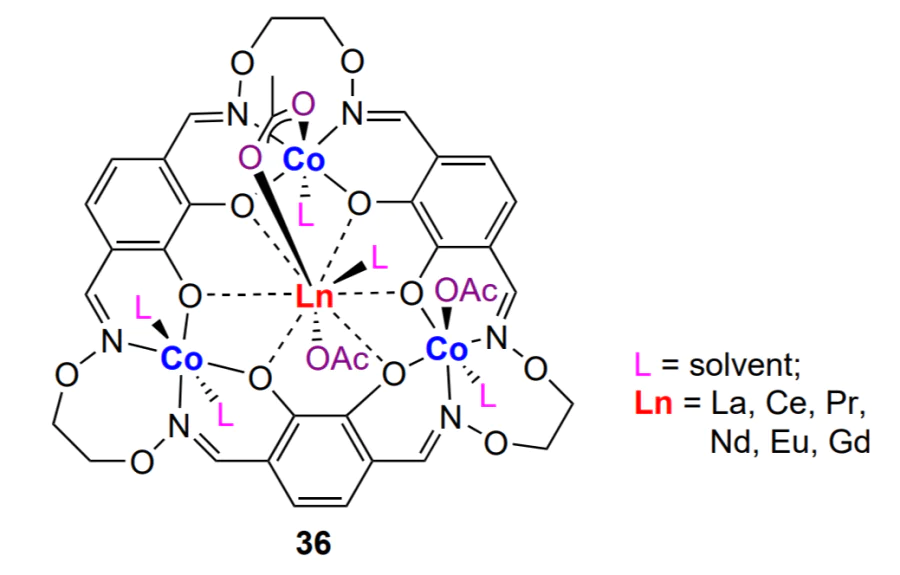
Mixed lanthanide-based complex 36 was proposed for copolymerization of cyclohexene oxide and CO2 by Nozaki and co-workers[210, 211].
Unlike common cobalt salen complexes, mixed complexes with lanthanides possess high thermal stability; hence, they can be used at elevated temperature. As a result, high catalyst activity (TON = 13000) and selectivity (up to 98%) were attained in the copolymerization at 130 °C, which gave a copolymer with a content of carbonate linkages of more than 99% and a cyclic carbonate fraction of 1 – 2%. The polycarbonate had a narrow but bimodal MWD, with average MW of the low-molecular-weight mode being half that of the high-molecular-weight mode and amounting to ~7 × 103 to ~54 × 103. On the basis of kinetic analysis, the authors drew conclusion about the cooperative action of the Co and Ln metal active centres. The copolymerization started with the epoxide coordination to the oxophilic Ln centre via exchange reaction between the ligand (solvent) and the epoxide. Then the cobalt acetate anion attacks the epoxide and forms the alkoxy complex; CO2 is coordinated to the cobalt atom of the formed alkoxy complex to give a new complex. The alkoxide group migrates from Ln to Co and adds to CO2; and after that the cycle is repeated (Scheme 34). The appearance of the second mode in the MWD curves can be caused by chain termination/transfer reactions with hydroxyl-containing compounds present in the system, including macromolecules with the terminal OH group.
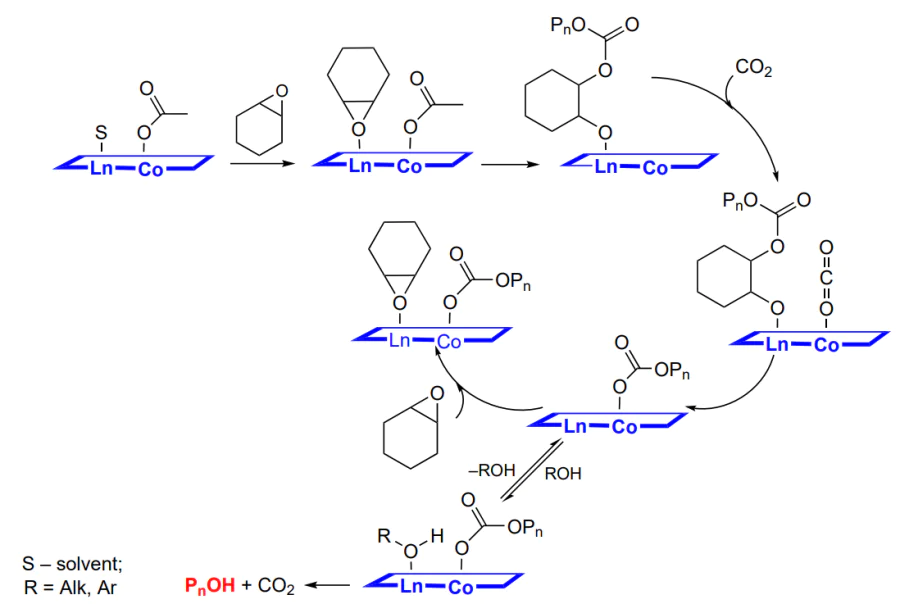
These studies were continued for cerium-containing catalyst 37 in order to optimize the copolymerization conditions and search for suitable chain transfer agents to generate necessary terminal functional groups of the polymer (Scheme 35)[211]. The authors were able to achieve a high reaction rate [TON = (5 – 10) × 103] and selectivity of the complex to obtain polycarbonates with a specified structure of terminal groups (mono- and telechelics) and a narrow unimodal MWD and MW ≈ (20 – 35) × 103.
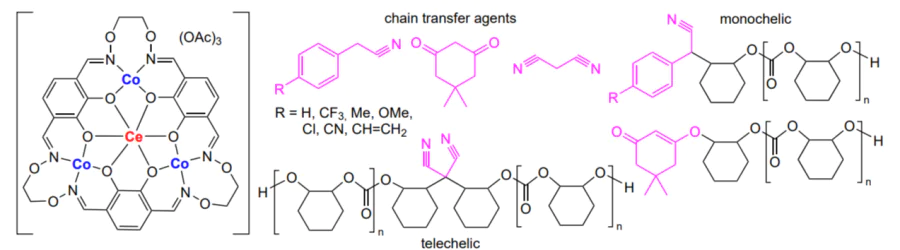
2.4. Cobalt complexes
The use of cobalt compounds in the copolymerization of epoxides and CO2 was first mentioned by Ikeda and co-workers[212]. Under the action of cobalt(II) acetate, copolymerization of propylene oxide and CO2 at 80 – 100 °C afforded a polymer insoluble in methanol, which was classified as polycarbonate. The yield of the polymer was proportional to the epoxide concentration and did not depend on the content of CO2. However, detailed studies of the influence of the structure of cobalt complexes on the stereoselectivity of epoxides and CO2 copolymerization started in the 2000s.
2.4.1. Cobalt porphyrin complexes
Cobalt porphyrins are used rather rarely in the copolymerization of epoxides and CO2[213-216]. The first application of cobalt porphyrins was reported by Sugimoto and Kuroda[214], who studied the cobalt tetraphenylporphyrin chloride–dimethylaminopyridine system[217]. The copolymerization of cyclohexene oxide with CO2 at 80 °C and 50 atm resulted in the highly selective formation of polycarbonate with a narrow bimodal MWD and more than 99% carbonate linkages. At a lower temperature (25 – 40 °C), this complex proved to be active in the synthesis of poly(propylene carbonate). Polycarbonate oligomer is formed with high selectivity even at a pressure of 1 atm. However, the overall productivity of the catalyst was moderate. A systematic study of the effect of the porphyrin structure and the co-catalyst nature on the copolymerization of propylene oxide and CO2 was carried out by Wang and co-workers[215], who varied the axial ligand (X = Cl, Br, I, OAc) and the co-catalyst (PPNCl, Bu4NCl, Bu4NBr, Bu4NI). At 25 °C and 20 atm, they were able to synthesize high-molecular-weight (Mn ≈ 48 × 103) poly(propylene carbonate) in a high yield (63%). The product contained no ether linkages and the content of cyclic carbonate was less than 1%. The copolymerization was regioselective, with the percentage of head-to-tail dyads being ~93%. However, raising the temperature even to 60 °C sharply shifted the reaction towards the formation of cyclic carbonate and decreased the selectivity, regioselectivity and MW of the polymer. A pressure increase (above 20 atm) did not affect these characteristics, while pressure drop acted in the same way as the temperature rise. Regarding the structure of the complex, the optimal results were achieved for Cl and Br as the axial ligands in combination with PPNCl as the co-catalyst. A series of metal porphyrins 38 were studied by Rieger and co-workers[213].
In the presence of PPNCl at moderate temperature (30 – 50 °C) and pressure (10 – 50 atm), these complexes showed high selectivity to poly(propylene carbonate) (the proportion of cyclic carbonate was 2 – 13%). The polymer yield followed a non-monotonic dependence on the pressure in the system and passed through a maximum at 30 atm. Generally, all porphyrin complexes have lower TOF than salen complexes. It is not surprising that the latter attract the greatest interest of researchers.
2.4.2. Cobalt salen complexes
Numerous studies devoted to cobalt salen complexes address the influence of substituents, axial ligands, (R,R) or (rac) structure of the complex and the nature and structure of the co-catalyst on the catalytic activity in the copolymerization of epoxides and CO2[41, 44, 52, 217-220]. We will represent (salen)CoX — co-catalyst complex in a simplified way as a bulky electrophile – bulky nucleophile system (Scheme 36).
In the electrophilic part of the complex, it is possible to vary the structure of substituents in the aromatic moieties (R3, R4); axial ligand Х (they are responsible for the activity of the complex, enantioselectivity and selectivity to the polymer); and chiral diamine skeleton (R1, R2, asymmetric induction and activity of the complex). In the nucleophilic part of the complex, the nature of the compound and substituents are selected in such a way as to control the nucleophilicity (activity of the complex), coordinating capacity (activity of the complex, enantioselectivity and chain microstructure) and capability for substitution (selectivity and polymer MW). A complex containing both a bulky nucleophile and a bulky electrophile significantly increases the selectivity by suppressing the intramolecular cyclization reaction giving the cyclic carbonate (Scheme 37)[221].
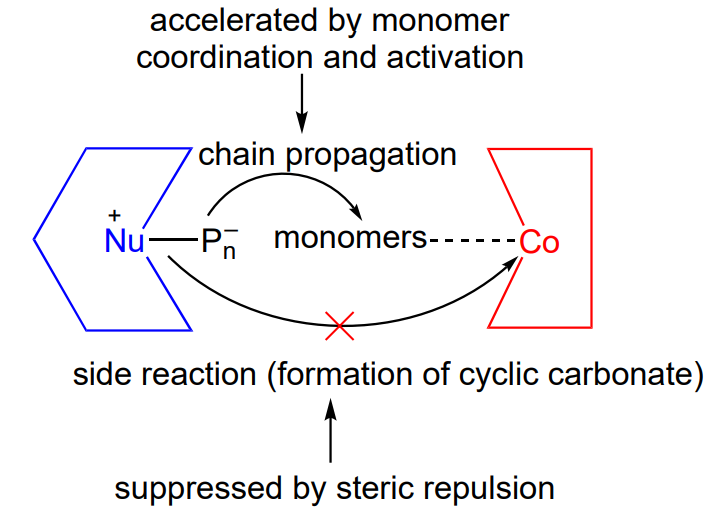
The copolymerization of propylene oxide with CO2 induced by the (salcy)Co(III)X complexes (salen complex in which R1 + R2 is cyclohexyl, see Scheme 36) is described in numerous publications[52, 142, 218, 220-226]. The development of new complexes was accompanied by studies of the kinetics of copolymerization. Regarding this issue, mention should be made of studies of Russian authors[227-231]. It was shown, for example, that under the action of complexes 39a – c at room temperature but high pressure (55 atm), polycarbonate is formed as the only reaction product (at ~75% conversion) characterized by a relatively narrow MWD (Mn ≈ 1.5 × 104 and Đ ~ 1.2 – 1.4) and containing virtually no ether linkages[222]. The turnover frequency of these complexes is noticeably lower (~50 h–1) than TOF of many zinc complexes; however, high selectivity is their advantage.
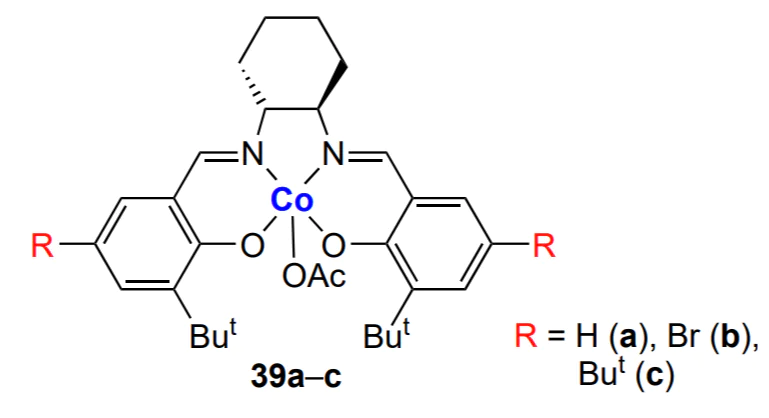
Using complexes (R,R)-(salcy)CoX and (rac)-(salcy)CoX at elevated pressure (54 atm), it is possible to obtain polycarbonates of various microstructures[223]. Isotactic polycarbonate is formed in the copolymerization of (S)-propylene oxide involving (R,R)-(salcy)CoX or (R)-propylene oxide involving (rac)-(salcy)CoX. The atactic polymer is formed from a racemic mixture of monomers under the action of (R,R)-(salcy)CoX, but when (rac)-(salcy)CoX is used, the polymer is enriched with syndiotactic triads. The heterotactic polycarbonate can be obtained by the copolymerization of (R)-propylene oxide catalyzed by (R,R)-(salcy)CoX. The catalytic activity of the complexes without a co-catalyst is moderate and is comparable with the results reported by Coates and co-workers[222]. However, the addition of a co-catalyst, bis(triphenylphosphine)iminium chloride, increases the catalytic activity of the complexes by 1 to 1.5 orders of magnitude. For example, (R,R)-(salcy)CoBr as a catalyst of (S)-propylene oxide copolymerization has TOF of 1100 h–1. The same salen complex with the axial ligand X = Cl in combination with a Lewis base (dimethylaminopyridine, pyridine, methylimidazole, triethylamine) provides 100% formation of cyclic carbonate[232], whereas linear polycarbonate is formed with high selectivity in the copolymerization of butylene oxide or cyclohexene oxide at 30 atm CO2 pressure in the presence of bis(triphenylphosphine)iminium chloride[233]. Cyclic propylene carbonate is also formed easily and selectively under the action of other salen complexes in the presence of salts as co-catalysts[234]. Thus, the reaction pathway is indeed sensitive to the structure of the complex and the co-catalyst. The use of Bu4NBr results in the predominant formation of propylene carbonate, while the replacement of Br by Cl leads to the formation of poly(propylene carbonate), and in the case of iodine, both cyclic and linear carbonates are produced[235]. An increase in the catalyst activity was inversely correlated with the formation of polycarbonate (cyclic carbonate was formed predominantly). Numerous attempts were made to increase the catalyst activity, while retaining polycarbonate as the only reaction product. In a series of studies, Nozaki and co-workers[226, 235, 236] proposed various salen complexes and a simple method for their removal and regeneration[237]. In one case, complex 40a contained two acetate axial ligands and a bulky substituent in the phenyl group; polymerization was carried out at 14 atm CO2 pressure[226]. The complex was used together with bis(triphenylphosphine)iminium chloride as the co-catalyst. In this case, it was assumed that the piperidinium moiety controls the nucleophilicity of the growing species via protonation of the anionic growing centre. As a consequence, the intramolecular cyclization reaction with the release of cyclic carbonate does not occur. The suppression of the cyclization made it possible to increase the temperature and, consequently, to increase TOF of the catalyst to ~600 h–1. Complete conversion of propylene oxide with retention of the high selectivity of the catalyst can be attained only for copolymerization in solution. As a result, MW of the polycarbonate increases to 8.4 × 104; polycarbonate has relatively narrow MWD (Đ = 1.28), while living polymerization mechanism allows the formation of block copolymer– poly(propylene carbonate)-block–poly(butylene carbonate). One more type of salen complexes 40b – e was proposed by Yamada and co-workers[235]. These complexes possess high chemoselectivity in the copolymerization of CO2 (20 atm) and propylene oxide (TOF less than 100 h–1), and some of them are chemoselective in the copolymerization of ethylene oxide, cyclohexene oxide and styrene oxide.
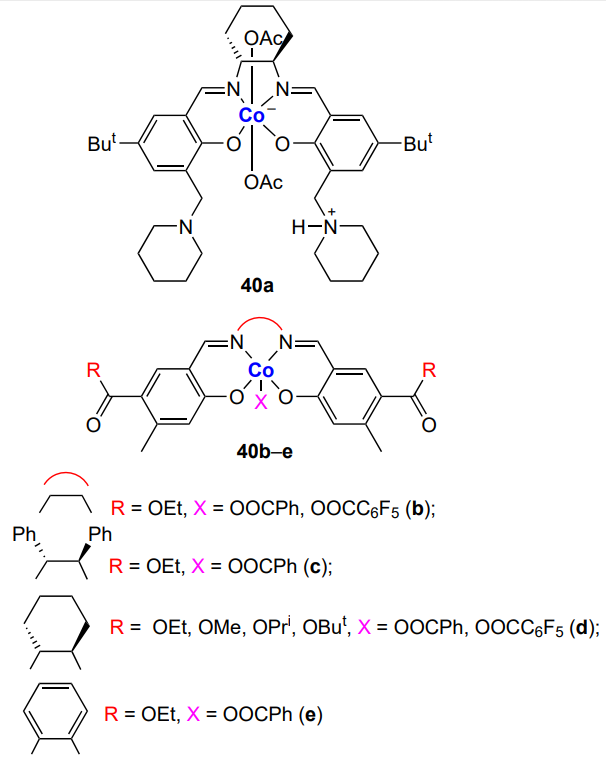
Nozaki and co-workers[236] reported salen complexes 41, which provided the formation of poly(propylene carbonate) of a specified microstructure at a CO2 pressure of 14 atm: isotactic, syndiotactic, gradient (enriched with isotactic triads) or block (iso- and syndiotactic) copolymer. These complexes contained an acetate (diacetate) or benzoate axial ligand and cyclic quaternary ammonium groups.
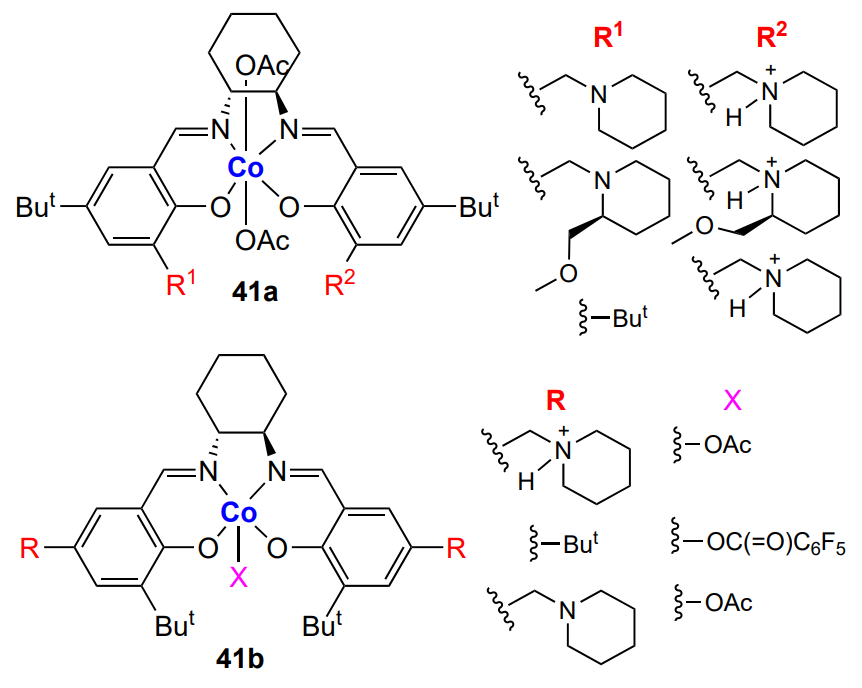
The influence of the structure of complexes 42a,b on their selectivity and activity in the copolymerization of propylene oxide with CO2 has been studied in detail[224, 232].
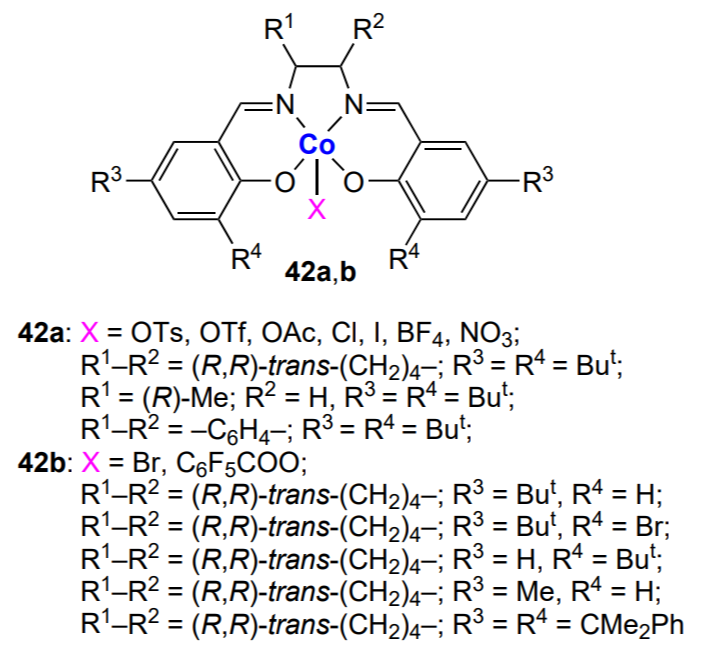
For all complexes 42a, the selectivity was above 99%, the content of carbonate linkages in the copolymer was 99%[238]. The activity of the catalyst passed through a maximum with increasing pressure (3 – 50 atm), temperature (22 – 60 °C) and co-catalyst concentration (1 – 5 equiv.). The maximum TOF was ~700 h–1 for the complex with R1 = R2 = Ph, X = NO3. Among complexes 42b, the highest activity (TOF = 720 h–1) was found for (R,R)-(salen)CoOOCC6F5 with R1 = R2 = (R,R)-trans-(CH2)4 in combination with Ph4PCl or PPNCl, or PPNOOCC6F5. The obtained polycarbonate had a regioregular structure with 98% head-to-tail connectivity[224]. Salen complexes of a similar structure [X = Cl3CCOO, (NO2)2C6H4O] with bis(triphenyl-phosphine)iminium chloride as a co-catalyst provided the synthesis of predominantly isotactic poly(cyclohexene carbonate) with narrow MWD under relatively mild conditions (6 – 25 atm CO2 pressure)[239]. As the temperature was raised from 25 to 80 °C, TOF markedly increased up to ~103 h–1, the enantioselectivity insignificantly decreased, while the selectivity almost did not change. Even higher activity of complex 43 in the copolymerization of propylene oxide and CO2 (20 atm) was attained by introducing X–Bu3N+(CH2)2Me2Si groups into the para-position of the aryl moiety[240]. This structure of the complex gave rise to Coulomb interaction between the quaternary ammonium cation attached to the ligand skeleton and the growing anionic chain, owing to which the carbonate group of the polymer chain is always located near the metal atom. Therefore, the authors expected a high catalytic activity at a low catalyst concentration and high temperature. Indeed, they obtained TOF = 3500 h–1 at 90 °C for [monomer]/[catalyst] = 25000. The introduction of four ammonium substituents into complex 44 increased TOF by a factor of ~8; TOF remained high even as the catalyst concentration decreased fourfold (CO2 pressure of 17 – 20 atm)[241]. Density functional theory calculations showed that for the (S,S)-configuration of cyclohexane in the complex, ring opening preferably takes place for (R)-rather than (S)-enantiomer of propylene oxide[242].
A more detailed study of the structure of complex 44 showed that cobalt is not bound to the nitrogen atoms and the coordination number of six is attained by means of counter-ions (Scheme 38)[243].
Bifunctional complex (45a,b), which does not require a co-catalyst, may contain both a base and an ammonium salt chemically attached to the skeleton[244, 245]. Copolymerization can be carried out (depending on the ligand structure) over a broad pressure range from 1 to 20 atm. Comparative analysis demonstrated that the catalyst — co-catalyst binary system is less selective than the bifunctional system containing the catalyst and co-catalyst in one molecule, all other conditions being the same; this is due to the difference between the activation energies of formation of cyclic and linear carbonates[158]. For complex 45а, this made it possible to attain high TOF for the catalyst (104 h–1), while maintaining high selectivity (99%), and obtain poly(propylene carbonate) with Mn = (6 – 15) × 104 and Đ = 1.09 – 1.20[244]. Both complexes were successfully used for CO2 copolymerization with epichlorohydrin[245]. High regio- and stereoselectivity in combination with narrow MWD of the copolymer were attained at 0 °C and 20 atm. As a development of this ideology, a block copolymer, poly(styrene carbonate)-block-polylactide, was obtained using complex 45b for copolymerization of styrene oxide and CO2 in combination with 1,8-diazabicyclo[5.4.0]undec-7-ene for lactide polymerization[246]. An interesting approach was the use of water to terminate copolymerization and generate the hydroxyl terminal groups required to initiate lactide polymerization.
Using unsymmetrical salen complexes 46a,b, Lu and co-workers[247] performed asymmetric copolymerization of CO2 and racemic propylene oxide at 25 °C and 10 atm with high regio- and stereoselectivity.
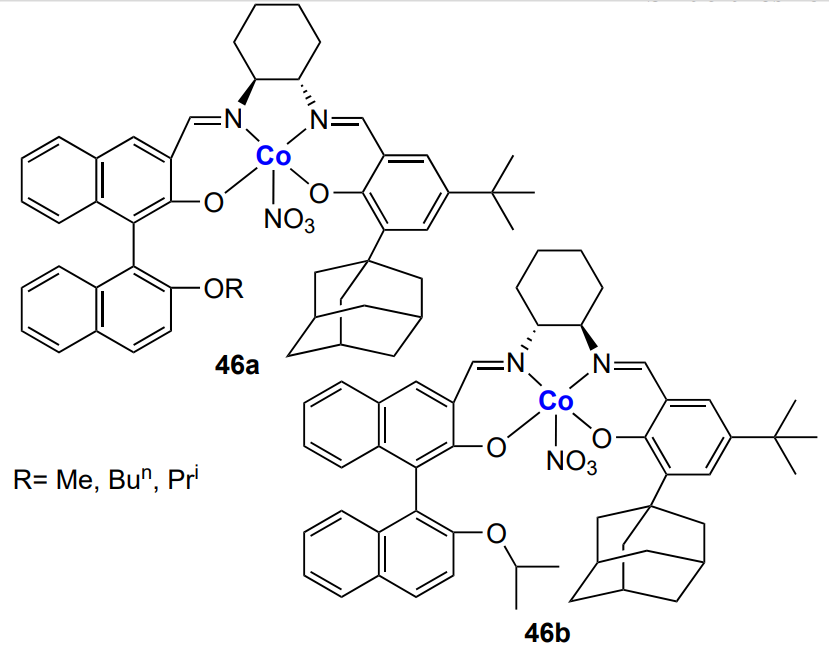
Cobalt(III) salen complexes are rarely used for cyclohexene oxide copolymerization, because they are easily deactivated via conversion to Co(II) compounds, which are inactive in the copolymerization[41]. The first synthesis of poly(cyclohexene carbonate) using a cobalt salen complex was reported by Coates and co-workers[248]. Using (rac)-(salen)CoBr (42b, R1 – R2 is cyclohexyl) without a co-catalyst at 22 °C and CO2 pressure of 54 atm, syndiotactic poly(cyclohexene carbonate) was obtained (TOF = 98 h–1). An increase in the CO2 pressure was accompanied by an increase in TOF and the proportion of syndiotactic triads in polycarbonate; the catalyst chirality (R,R-, rac-, achiral) had no influence on the polymer microstructure. The addition of PPNCl promoted an increase in TOF, but disrupted the chain microstructure. The application of salen complex 47 containing the N+Bu3X– ammonium salt as a ligand resulted in the synthesis of both poly(cyclohexene carbonate) and the cyclohexene carbonate — propylene carbonate copolymer with high selectivity and high MW (Mn ≈ 2 × 105 and Đ ≈ 1.2)[249].
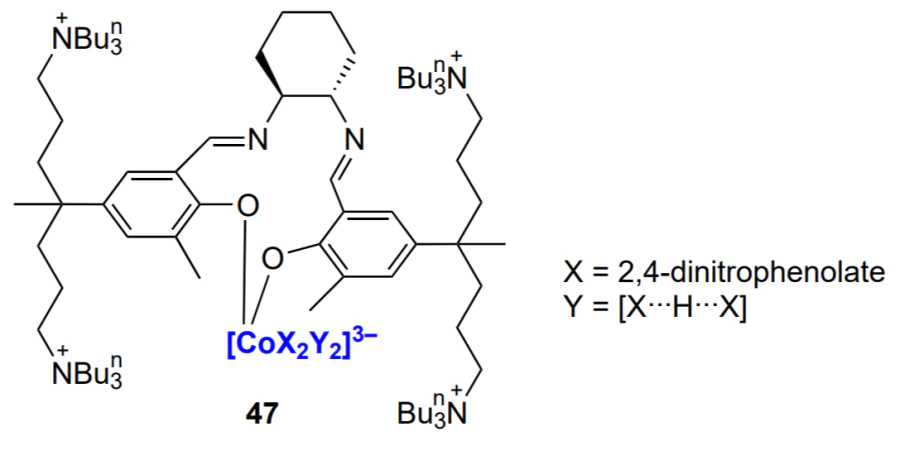
2.4.3. Bi- and trinuclear cobalt complexes
Binuclear cobalt complexes for ring-opening polymerization of epoxides were reported in a number of studies[250-252]. Complexes of this type were first proposed for copolymerization of cyclohexene oxide and CO2 by Williams and co-workers[253]. Bi- and trinuclear cobalt complexes 48a – c combined with bis(triphenylphosphine)iminium salt as a co-catalyst showed different activities in the copolymerization.
Trinuclear complex 48b proved to be too stable and had a low activity in the copolymerization, unlike binuclear complexes 48a,c. The latter had TOF = 500 h–1 in the cyclohexene oxide copolymerization at 1 atm and 80 °C and a high selectivity to the polymer. The polycarbonate had relatively low MW and narrow MWD (Mn < 104, Đ = 1. 1.3). Binuclear catalysts retain their properties at high temperature (100 °C) and a pressure of 10 atm. One more type of binuclear complexes 49 used for copolymerization of CO2 with cyclopentene oxide at 2 atm and 25 °C was described by Lu and co-workers[158, 254]. These complexes were effective in the synthesis of triblock copolymers, poly(cyclohexene carbonate)-block-poly(butylene carbonate)-block-poly(cyclohexene carbonate), and produced optically active (dextro- and levorotatory) high-molecular-weight poly(cyclopentene carbonates) with narrow MWD; however, complexes with naphthalene substituents possessed lower regio- and stereoselectivity.
Comparison of mono- (50a) and binuclear (50b,c) complexes of a similar structure proposed by Nozaki and co-workers[255] revealed similar behaviours of these complexes in the copolymerization of cyclohexene oxide with phthalic anhydride [TOF = 200 – 300 h–1, Mn = (5 – 15) × 103, Đ = 1.1 – 1.3], while in the copolymerization of cyclohexene oxide with CO2, the selectivity to the polycarbonate was higher for bimetallic complex (50b) than for monometallic complex (50a), being 97 and 88%, respectively.
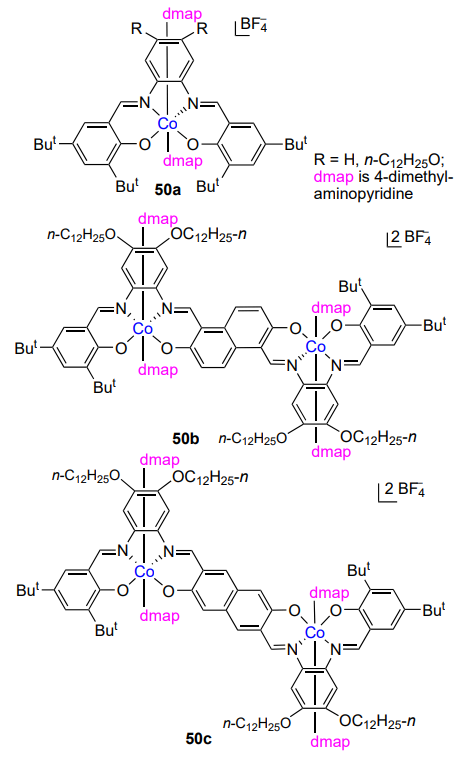
Binuclear Co salen type complexes 51 were used in the copolymerization of cyclohexene oxide with CO2 and cyclohexene oxide (or 4-vinyl-1,2-cyclohexene oxide) with phthalic anhydride[256]. Complex 51b proved to be inactive in the synthesis of polycarbonate, but effective in the synthesis of polyester (95% conversion of phthalic anhydride; more than 99% selectivity to the polyester). Complexes 51a,c were active in the copolymerization with both CO2 and phthalic anhydride, demonstrating high selectivity to polycarbonate and polyester (more than 99%). However, copolymerization with CO2 gave cyclic carbonate, the contents of which were 18 (51a) and 8% (51c). The complexes had high regio- and stereoselectivity, but low activity.
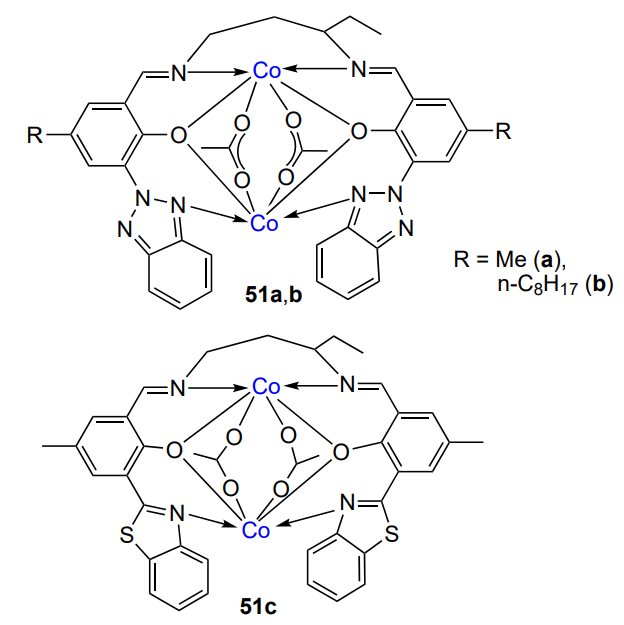
Binuclear Co/alkali metal salen types complexes 52 for CO2 and propylene oxide copolymerization were reported by Williams and co-workers[257, 258]. It is significant that in this case, there was no need to use additional co-catalysts. The CoIII/K complex showed the most satisfactory results: high conversions of monomers (alicyclic epoxides and epoxides with aliphatic substituents) and chemoselectivity, TOF = 800 h–1 for 0.025 mol.% catalyst and a broad temperature range (25 – 70 °C).
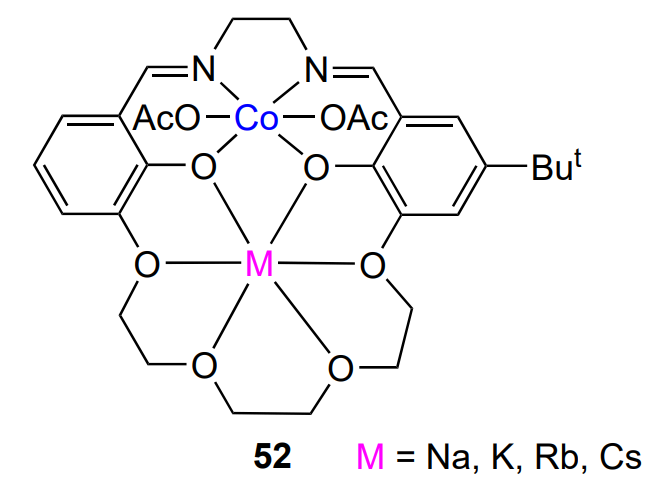
According to density functional theory calculations, CO2 is coordinated to the potassium atom, while epoxide is coordinated to cobalt. The carbonate attacks activated propylene oxide to give an intermediate and then cobalt alkoxide. The insertion of the next CO2 molecule results in the formation of cobalt carbonate. The reaction selectivity to polycarbonate depends on the equilibrium between the alkoxide and cobalt carbonate (Scheme 39), which can be controlled by changing the temperature or pressure[258]. The optimal conditions for the formation of polycarbonate are 50 – 70 °C and 20 – 30 atm.
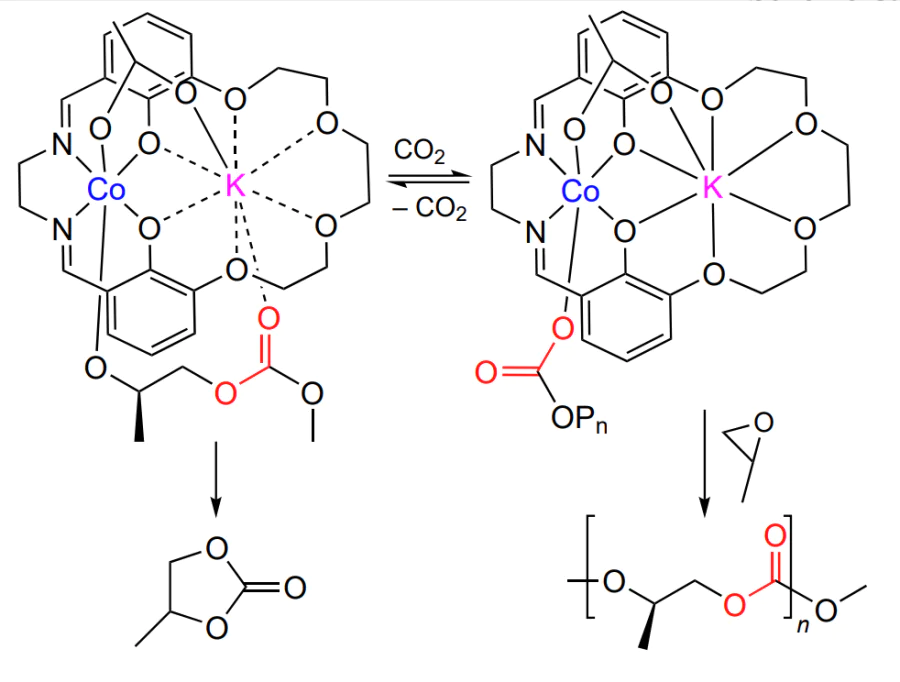
In conclusion, mention may be made of an interesting type of switchable catalysis using cobalt(II) salen complex 53[259]. This complex is active in the radical polymerization of vinyl monomers, which can follow either reversible inhibition or degenerative chain transfer pathway. Oxygen acts as an external switch by being inserted into the Co – C bond to give CoIII complex capable of initiating the epoxide copolymerization with CO2 (Scheme 40). Thus, the authors obtained triblock-copolymer – poly(vinyl acetate)-block-poly(methyl acrylate)-block-polycarbonate. The selectivity and activity of the catalyst were low in this case; however, triblock copolymers had a narrow unimodal MWD.
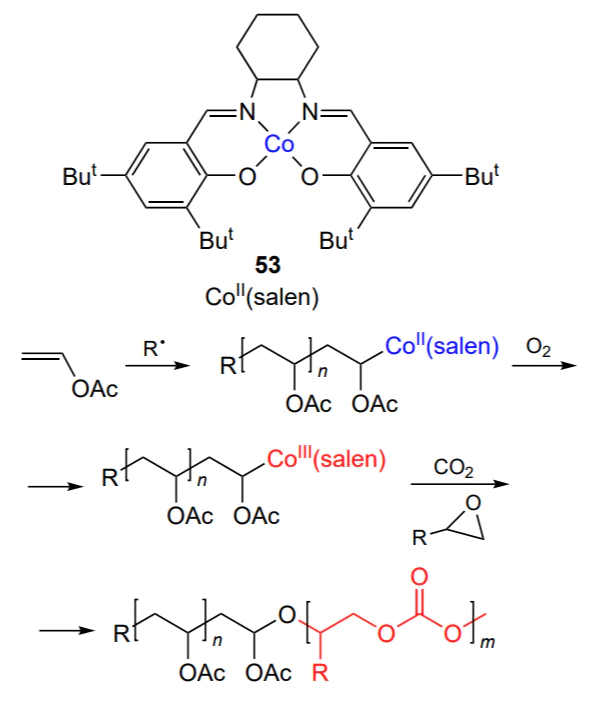
2.5. Chromium complexes
2.5.1. Chromium salen complexes
The activity and selectivity of many Cr(III) complexes in the copolymerization of CO2 and epoxides and also characteristics of polycarbonates (MW, MWD, stereoregularity) are inferior to those for analogous cobalt complexes. Moreover, the use of chromium salen complexes often results in the formation of cyclic carbonates in a high yield[260]. However, like cobalt salen complexes, they can be used for the synthesis of polycarbonates under certain conditions[261]. The first example of the preparation of poly(cyclohexene carbonate) with Mn = 8900 and Đ = 1.2 catalyzed by a salen complex, (salen)CrIIICl (54a), in combination with N-methylimidazole was described by Darensbourg et al[262]. Comparative analysis of the activation barriers for the formation of cyclic and linear carbonates in the copolymerization of cyclohexene oxide and propylene oxide with CO2 carried out in this study showed that the formation of propylene carbonate is preferable in the case of propylene oxide, while the formation of polycarbonate is preferable for cyclohexene oxide, which is actually observed in experiments. Complexes 54b and 54c proved to be ineffective in the copolymerization, and polycarbonate was formed in a low yield. The polymer contained a large percentage of ether linkages.
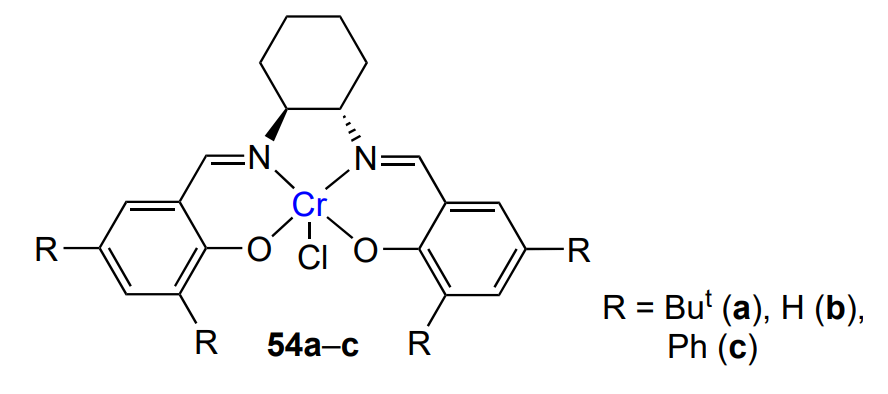
The variation of the structure of salen complex (54a, 55a–55t) and the co-catalyst made it possible to elucidate the effect of substituents, ligands, nucleophile and catalyst/co-catalyst concentration ratio on the kinetics and mechanism of epoxide and CO2 copolymerization; find conditions for the synthesis of high-molecular-weight poly(cyclohexene carbonate) with narrow MWD (Mn = 5 × 104 and Đ = 1.1) and high catalyst selectivity and activity and for the synthesis of poly(propylene carbonate) with a low proportion of the cyclic carbonate by-products; and expand the range of epoxides able to form polycarbonates[41, 50, 263-265]. Complex 54a in the presence of N-methylimidazole proved to be effective in the copolymerization of 2-(3,4-epoxycyclohexyl)-ethyltrimethoxysilane[264] and ineffective in the copolymerization of 2,3-epoxy-1,2,3,4-tetrahydronaphthalene (cyclic carbonate was formed in this case)[263]. In the presence of PPNCl, complex 54a provided the formation of poly(propylene carbonate)[265]. The highest yield of the polymer (TON > 103) in the copolymerization of cyclohexene oxide involving 2.25 equiv. of N-methylimidazole was attained by using complexes 55f,h,i,m,n,o,r,s,t[41]. High chemoselectivity (more than 99% carbonate linkages) was provided by all complexes except for 55a–c and 55p. The replacement of N-methylimidazole by PCy3 or PPNN3 led to increasing TOF for complex 55p; at 55 °C, TOF increased in the series N-methylimidazole (82 h–1) < PCy3 (346 h–1) < PPNN3 (760 h–1), and lowering the temperature to 35 °C increased TOF to 1150 h–1[50].
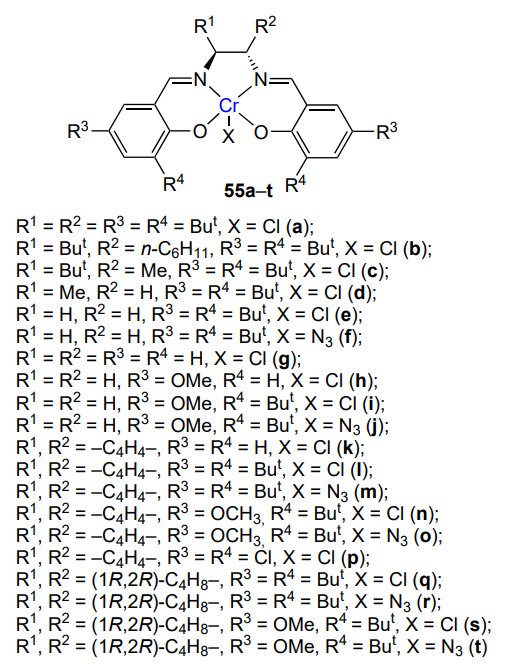
Generally, while comparing the course of development of cobalt and chromium salen complexes for the copolymerization of epoxides and CO2, one can see that different research groups chose identical or similar approaches to structural modification of complex 55 such as the introducing various substituents R3 and R4, increasing the steric restrictions by increasing the bulk of substituents R4, R1 and R2, changing the nucleophilicity of ligand Х and the co-catalyst, enhancing the nucleophilic properties of the co-catalyst, and varying the counter-ion in the case where ammonium or bis(triphenylphosphine)iminium salts are employed[44, 102, 142, 155, 219, 266-276].
Binary (catalyst – co-catalyst) and bifunctional (co-catalyst covalently bound to the catalyst) chromium salen complexes[277], binuclear chromium salen complexes[278, 279] similar to the above cobalt complexes and polymer-immobilized chromium salen complexes[280] were also proposed for the copolymerization of epoxides and CO2. A comparison of CoIII and CrIII salen complexes indicates that the latter must be used at higher temperatures because of the high activation barrier and they are less active in the copolymerization.
2.5.2. Other chromium complexes
Complexes 56a – c were tested in the copolymerization of propylene oxide and cyclohexene oxide with CO2[281].
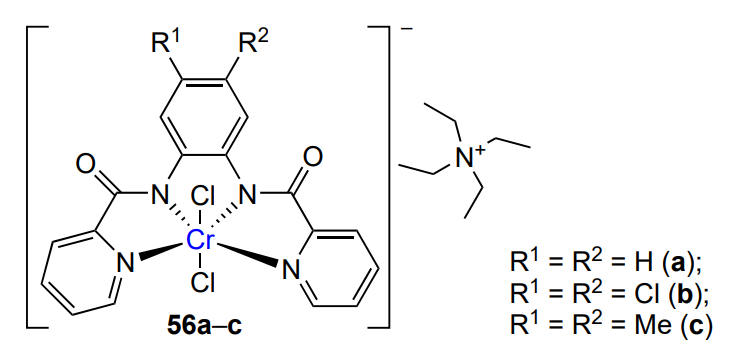
In the case of propylene oxide, only cyclic carbonate was formed, while in the case of cyclohexene oxide, the copolymer was obtained. However, despite the high chemoselectivity, the content of carbonate linkages in the copolymer did not exceed 40%.
Using complex 57, the authors compared the activity of a neutral complex (with one axial Cl ligand, 57a) and a charged complex (with two Cl ligands, 57b) in the presence of different co-catalysts (pyridine, dimethylaminopyridine and PPNCl) in the copolymerization of epoxides and CO2[282]. The reaction of styrene oxide and CO2 induced by the cationic complex 57b yielded cyclic carbonate, while in the case of cyclohexene oxide, polycarbonate was formed. In the latter case, the chemoselectivity reached 80% and the content of carbonate linkages was 92%, but the catalyst activity was moderate (TOF = 17 h–1).
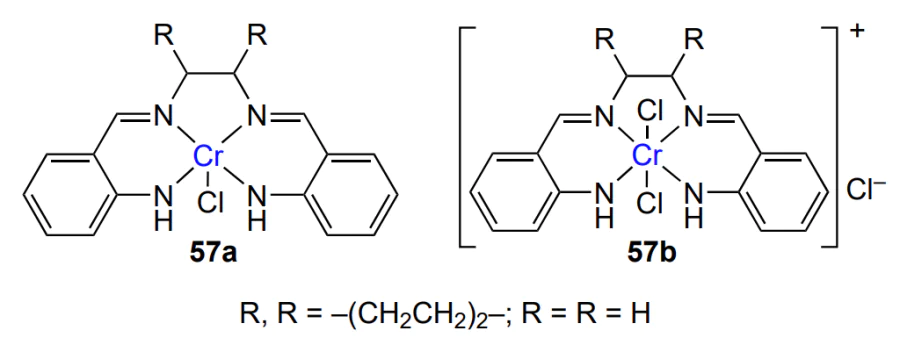
The combination of various tri- and bidentate ligands in chromium complexes 58a – f for the copolymerization of epoxide and CO2 was proposed by Masdeu-Bulto and co-workers[283]. The best results were found for complex 58d; copolymerization of cyclohexene oxide furnished polycarbonate with conversion above 90% and ~60% selectivity; the content of carbonate linkages in the copolymer was 96%; styrene oxide and propylene oxide were quantitatively converted to cyclic carbonates.
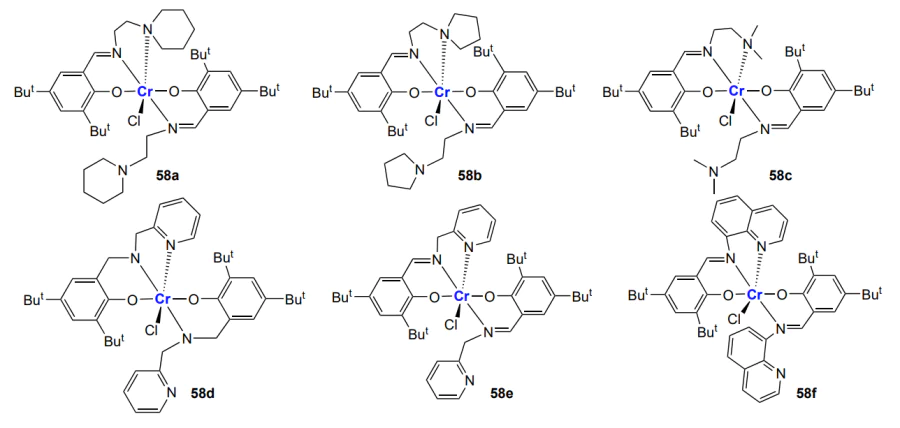
Yet another type of complexes was proposed by Duchateau and co-workers[284]; among them, complexes 59a and 59b exhibited activity in the copolymerization of cyclohexene oxide and CO2 in the presence of the PPNCl co-catalyst. In the absence of solvent, an oligomer with Mn = 3800 was selectively formed, while copolymerization in toluene resulted in higher MW values, but this was accompanied by broadening of the copolymer MWD. Despite high epoxide conversion and high content of carbonate linkages in the copolymer, the activity of the catalyst in toluene was low.
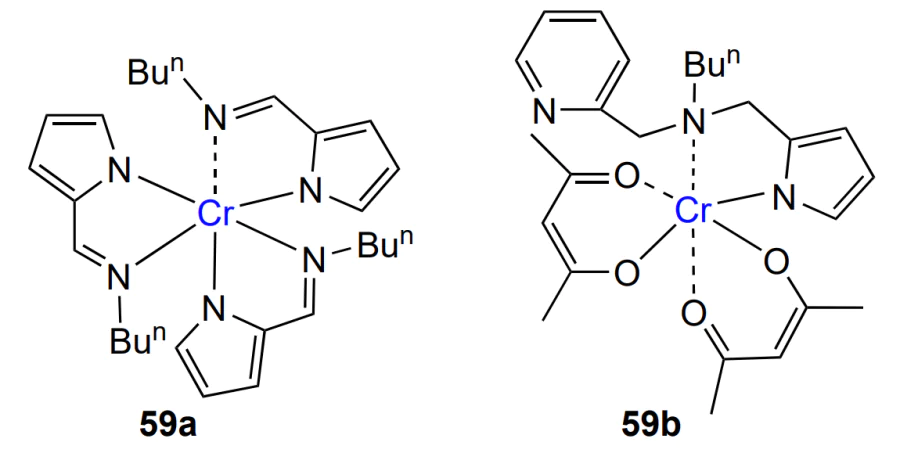
Complexes 60a and 60b proved to be active in the copolymerization of cyclohexene oxide and CO2 (Scheme 41)[285]. The monomer conversion of 76% was attained in only 3 h; however, the product was a mixture of polycarbonate and cyclic carbonate. When propylene oxide was used, only propylene carbonate was obtained, with the conversion being 99% within 4.5 h.
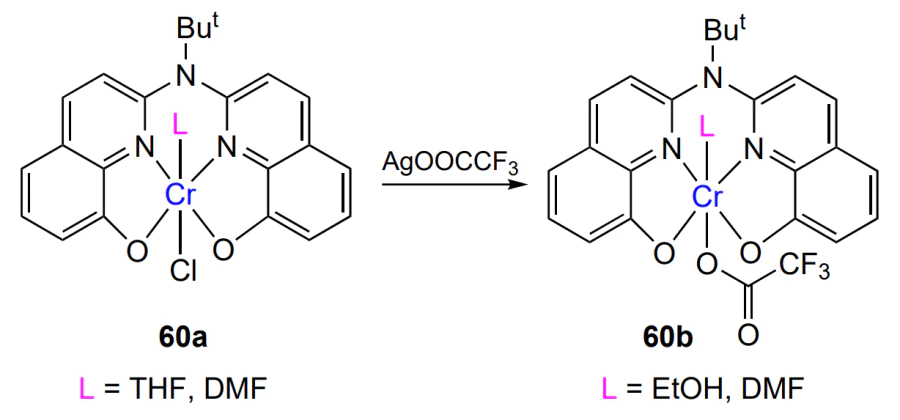
Chromium porphyrins, like cobalt porphyrins, are more suitable for the synthesis of cyclic carbonates than polycarbonates[286-289]. In some cases, the formation of oligomeric polycarbonate (Mn < 3000) is possible; this is characteristic, for example, of cyclohexene oxide[286].
Chromium chloride complexes 61a – c were tested in the epoxide and CO2 copolymerization in the presence of dimethylaminopyridine, tetrabutylammonium bromide and PPNX (X = Hal, N3) co-catalysts[290-296]. The copolymerization mechanism and the catalyst activity were sensitive to the reaction conditions, in particular, to the presence and amount of a co-catalyst. The chemoselectivity depended on the nature of the epoxide: polycarbonate was formed from cyclohexene oxide, while cyclic carbonate was obtained in the case of styrene oxide. The copolymerization of CO2 and propylene oxide at 22 – 25 °C in the presence of PPNCl gave polycarbonate as the major product, while at higher temperature and in the presence of tetrabutylammonium bromide, cyclic carbonate was mainly formed.
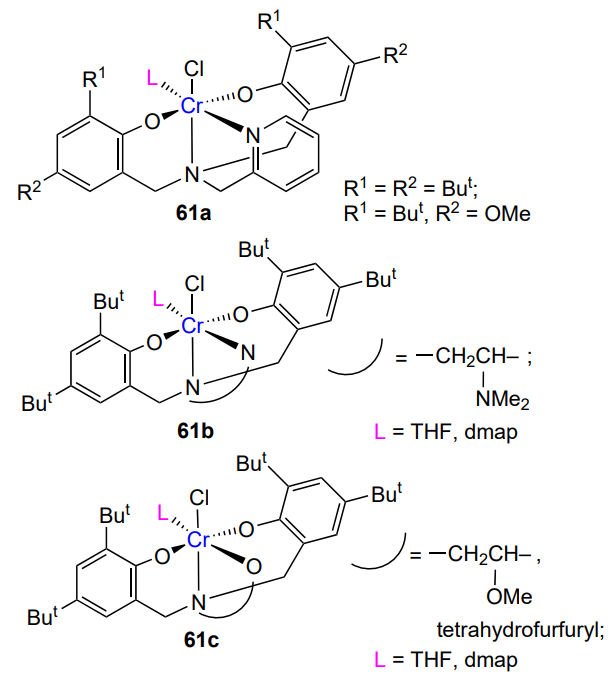
One more type of ligands, sulfur compounds, were proposed for copolymerization of 4-vinylcyclohexene oxide and CO2[297]. Complexes 62 showed a reasonably good activity (TOF = 134 h–1), a moderate selectivity (polymer proportion of 70 – 80%). The copolymers were characterized by a high content of carbonate linkages (97.5%) and relatively low MW (Mn = 1.5 × 104).
Thus, to summarize, one can conclude that among numerous chromium complexes that were synthesized and tested in the copolymerization of epoxides and CO2, there are yet no compounds that would be comparable in activity and selectivity to zinc, aluminium and even cobalt complexes.
2.6. Complexes of other transition metals
Among other transition metals the complexes of which have been tested in the copolymerization of epoxides and CO2, it is necessary to note iron and nickel, the interest in which appeared in the 2010s[44, 237, 298-305].
Binuclear iron(III) complex 63 structurally similar to cobalt complex[253] was proposed for copolymerization of cyclohexene oxide and CO2, which was carried out under mild conditions (80 °C, 1 – 10 atm, moderate catalyst concentrations)[306]. The selectivity, stereoselectivity and control of MWD increased with increasing CO2 pressure. In the presence of PPNCl, cis-cyclohexene carbonate was formed instead of the polymer. In the case of propylene oxide and styrene oxide, no polymer was produced under any conditions.
Iron tetrapyrrole macrocycles 64a,b in combination with the PPNCl co-catalyst showed a high activity (1.2 × 103 h–1) and selectivity (polymer yield > 99%) in the copolymerization of propylene oxide and CO2[237]. The polymer contained up to 17% ether linkages randomly distributed along the chain and had moderately high MW and narrow MWD [Mn = (2 – 5) × 104 and Đ = 1.1 – 1.2]. The complexes also showed high selectivity in the copolymerization of cyclohexene carbonate; however, the catalyst activity was an order of magnitude lower. The content of ether linkages in the copolymer was lower compared to the copolymerization of propylene oxide. In the copolymerization of glycidyl phenyl ether with CO2, the proportion of carbonate linkages in the polymer was very low (~10 – 20%), despite high selectivity to the formation of the polymer. This copolymer crystallized, i.e., it had a regular structure.
Iron complexes 65a,b used for the switchable synthesis of poly(cyclohexene carbonate)/cyclohexene carbonate in combination with onium salts, tert-butylammonium or bis(triphenylphosphine)iminium halide, were reported by Pescarmona and co-workers[299, 300].
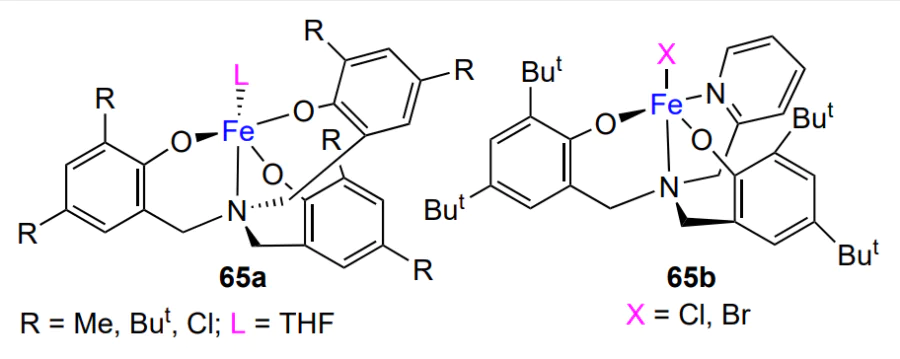
In the case of complexes 65а, when the reaction was conducted under supercritical conditions, an increase in the co-catalyst to catalyst ratio shifted the reaction towards the formation of the cis-isomer of cyclic carbonate; conversely, a decrease in the co-catalyst concentration led to the production of polycarbonate[299]. In the presence of tert-butylammonium bromide and iodide, cyclic carbonate was formed in a high yield even at a lower content of the co-catalyst. Conversely, high selectivity to the formation of polycarbonate was characteristic of tert-butylammonium and bis(triphenylphosphine)iminium chlorides. Complexes 65a with alkyl substituents R had similar activities, while R = Cl resulted in a lower solubility of the compound in supercritical CO2 and lower activity. Irrespective of substituents in the complexes and co-catalysts, the resulting polycarbonate had narrow bimodal MWD. Complexes 65b proved to be less selective in the copolymerization of cyclohexene oxide and CO2 under similar conditions[300]. They showed high selectivity in the copolymerization of 4-vinylcyclohexene oxide and CO2. In the copolymerization of styrene oxide and other epoxides with alkyl substituents, only cyclic carbonates were formed, while limonene oxide and α-pinene oxide did not react at all.
Iron complexes 66 with N,N-bis(2-pyridinecarboxamide)-1,2-benzene ligands were reported by Doring and co-workers[301]. Complexes 66 proved to be less active and selective in the copolymerization of cyclohexene oxide and CO2 than analogous cobalt or chromium complexes: monomer conversion did not exceed 55% regardless of the structure of substituents and/or ligands. Other epoxides tend to form only cyclic carbonates.
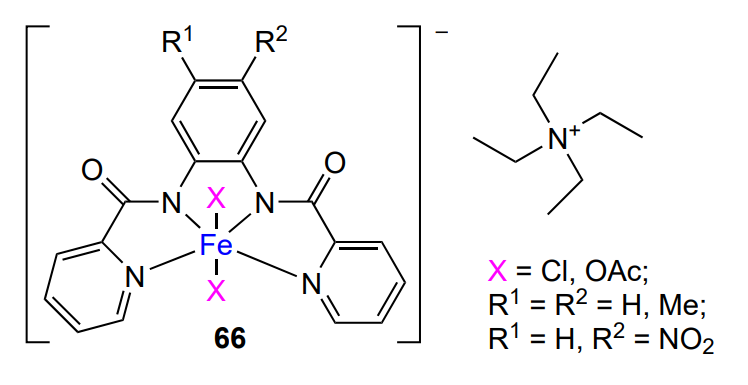
Nickel compounds attract less attention as catalysts for the copolymerization of epoxides and CO2, although they are highly effective in copolymerization. Above, we mentioned binuclear CoIII complexes (51); analogous binuclear Ni salen type complexes were also used in the copolymerization of cyclohexene oxide and CO2[256]. They were highly selective and stereoselective in the copolymerization and produced polycarbonate containing almost no ether linkages or cyclic carbonate by-product; the resulting polymer had relatively low MW and narrow MWD (Mn ~ 3 × 104 and Đ = 1.1 – 1.2)[256, 306]. Similar results were obtained upon the copolymerization of 4-vinylcyclohexene oxide with CO2 induced by the same catalysts; however, they showed low selectivity in the copolymerization of cyclopentene oxide: the proportion of cyclic carbonate amounted to 30 – 90% depending on the polymerization conditions[307]. The mentioned complexed had moderate activity; however, the recently reported binuclear nickel complexes 67 (Scheme 42) had TOF = 7.8 × 103 h–1 in the copolymerization of cyclohexene oxide with CO2 and more than 7 × 102 h–1 in the case of cyclopentene oxide[305]. The obtained polycarbonates had relatively low MW (Mn = (5 – 9) × 103) and narrow MWD (Đ = 1.1 – 1.2).
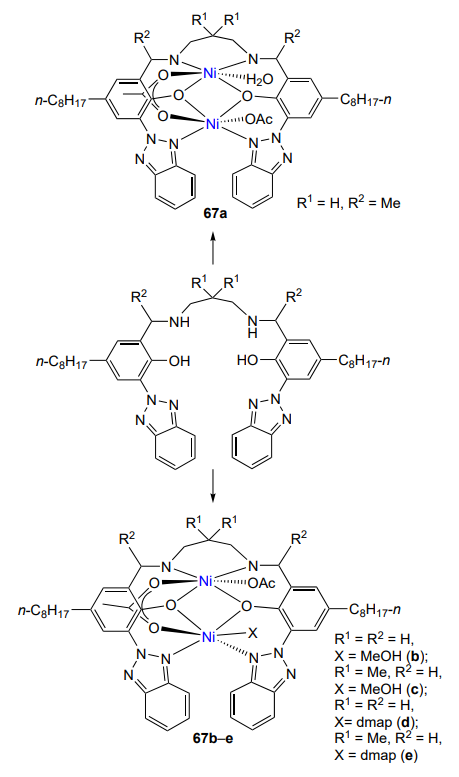
Thus, in most cases, the use of transition metal complexes in the copolymerization of epoxides and CO2 still does not provide as high activity and selectivity levels or molecular characteristics of polycarbonates as zinc or aluminium complexes.
2.7. Metal-free catalysts
In the last 3 to 5 years, the efforts of many research groups have been concentrated on the development of catalysts for copolymerization of epoxides and CO2 free from not only transition metals, but also other metals (metal-free copolymerization).
2.7.1. Catalyst – co-catalyst binary systems
The first study along this line was reported by Gnanou, Feng and co-workers[308], who induced epoxide and CO2 copolymerization using triethylborane as a Lewis acid, with onium halides and alkoxides acting as polymerization initiators (Scheme 43).
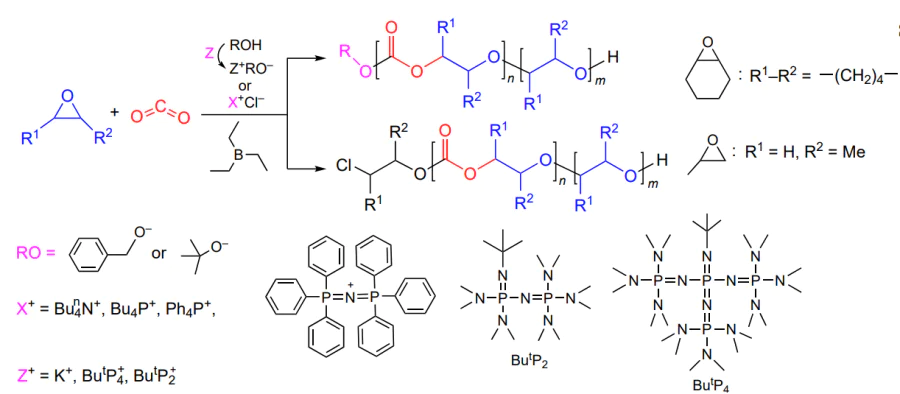
The copolymerization of propylene carbonate and CO2 was carried out at 60 °C and 10 atm CO2 pressure in THF or without a solvent. The benefits of these systems include fairly high selectivity: the polycarbonate content in the reaction product was 80 – 99%; the fraction of carbonate linkages in the copolymer was 82 – 97%; the resulting poly(propylene carbonate) had a narrow unimodal MWD and MW close to the target values (~5 × 104). A drawback was a relatively low copolymerization rate. For the polycarbonate formation, two conditions must be met. The reaction selectivity to polycarbonate requires that the rate constant for backbiting kb,12 be much lower than the propagation rate constant (kb,12 << kp,12 , kb,12 << kp,11). The chemoselectivity, implying the formation of carbonate rather than ether linkages, specifies the requirements to the ratio of cross-propagation kp,12 and epoxide homopolymerization kp,11 rate constants (kp,12 >> kp,11) (Scheme 44).
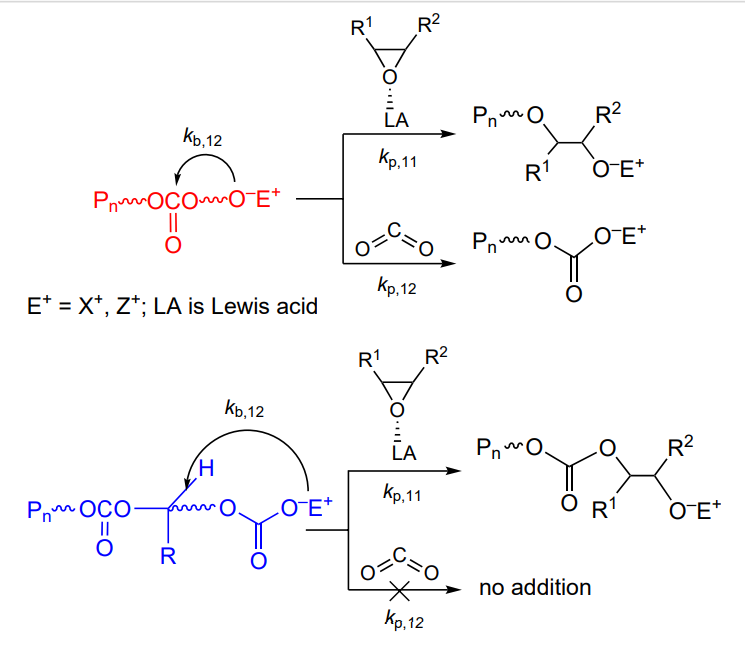
The carbonate active centre ~OC(=O)O–E+ must be able to cleave the Lewis acid-activated epoxide ring. Thus, the choice of initiator is a significant issue for implementation of this mechanism. For example, lithium tert-butoxide forms a strong O – Li bond and produces cyclic carbonate, instead of polycarbonate. Potassium tert-butoxide allows the preparation of polycarbonate as the major product (with a proportion of cyclic carbonate of 6%), with the content of ether linkages not exceeding 3%. Satisfactory results are obtained by using benzyl alcohol in combination with onium salts. The latter can be used by themselves as initiators, without additional components, for solution copolymerization in THF or for bulk copolymerization. The polycarbonate selectivity was even higher in the copolymerization of cyclohexene oxide. The molecular weight can be increased by additional purification of the reaction mixture from traces of water, because water terminates the chain propagation and is often responsible for the formation of polycarbonates with a narrow bimodal MWD[309]. The purification includes treatment of the system with tri(isobutyl)aluminium, which vigorously reacts with protic solvents: water, alcohol, etc. This approach led to outstanding results and furnished poly(cyclohexene carbonate) with Mn = 4.5 × 105 and Đ = 1.31 and poly(propylene carbonate) with Mn = 2.0 × 105 and Đ = 1.29. The selectivity of the catalyst (PPNCl in the presence of triethylboron) was high not only for cyclohexene oxide (above 99%), but also for propylene oxide (93 – 96% linear carbonate and 94 – 99% carbonate linkages in the polymer). The living nature of the polymerization provided the formation of high-molecular-weight (Mn = 1.4 × 105 and Đ = 1.25) diblock copolymer – poly(cyclohexene carbonate)-block-poly(propylene carbonate).
The first advances in this area attracted obvious attention, and dozens of publications devoted to metal-free catalysts appeared over the years of 2020 – 2023[310-328].
The tetrabutylammonium salts of carboxylic (carbonic, benzoic, succinic, m-phthalic, trimesic and pyromellitic) acids were used as initiators for propylene oxide and CO2 copolymerization in the presence of a Lewis acid – Et3B[329]. All salts exhibited high selectivity to the formation of polycarbonate (94 – 99%); the content of ether linkages in the polymer was 1 – 9% depending on the initiator, and it increased to ~50% as pressure decreased from 10 to 1 atm. The use of mono-, bi-, tri- and tetrafunctional initiator furnished not only linear, but also star-shaped (three- or four-arm) polycarbonate. In this case, the authors used a fairly high concentration of the initiator ([monomer]/[initiator] = 20 – 500), which resulted in low MW. An advantage of these initiators is that they can be recovered after the synthesis, which was demonstrated in relation to tetrabutylammonium carbonate. The recovery of the initiators is simple, but it turned out that the procedure (the sequence of addition of the reagents needed for polymer isolation and initiator recovery) is of crucial importance[330]. An improper sequence may lead to dеpolymerization of the polymer. The initiator can be immobilized (grafted) onto a substrate; polydiallyldimethylammonium chloride in which Cl–ions have been replaced by OH– ions has been used according to this approach. This initiator is water-soluble, but it is insoluble in the organic phase and can be easily separated after precipitation of the reaction mixture with water. However, a drawback of using the polyelectrolyte initiator can also be seen: the high concentration of initiating groups hampers the formation of a high-molecular-weight polymer. A series of mono-, tri- and tetrafunctional ammonium carboxylate initiators 68 were described by Hadjichristidis and co-workers[319]. They were used in the synthesis involving triethylborane to prepare linear and star-shaped poly(propylene carbonate) telechelics with terminal hydroxyl groups, and the possibility of initiator recycling was demonstrated in relation to di(tert-butylammonium) carbonate.
Tang and co-workers[313] proposed to replace the trialkylborane – onium salt combination with a trialkylborane (Et3B, Bu3B) — tertiary amine (triethylamine, tetraethylethylenediamine) pair. These initiators for the propylene oxide — CO2 copolymerization provide a high monomer conversion, low yield of cyclic carbonate (down to 1%) and the absence of ether linkages in the polymer. Their activity is comparable with that of onium salts, and control of MWD of the copolymer is also retained at a high level. The same approach was developed by Meng and co-workers[323] for copolymerization and terpolymerization of epoxides (ethylene oxide, propylene oxide, butylene oxide and cyclohexene oxide) with CO2 and cyclic anhydride. The authors varied substituents in the borane (ethyl, methoxy, methyl, phenyl) and trialkylamine (methyl, isopropyl, cyclohexyl) or diamine (tetramethylethylenediamine, triethylenediamine). Generally, amines showed high selectivity in the synthesis of polycarbonates and polyethers, even in the case of propylene oxide, but the content of the carbonate linkages in poly(propylene carbonate) was low. When amine was replaced by bis(triphenylphosphine)iminium chloride, it was more active in both co- and terpolymerization. However, when amine was replaced by phosphine (Bu3P), the initiator activity proved to be moderate, at least in the copolymerization of propylene oxide and CO2[327].
The ability of the onium salts to initiate ring opening polymerization was utilized for the switchable synthesis of block copolymers[310]. The authors used for the first time bis(triphenylphosphine)iminium chloride in the absence of other additives for the synthesis of block copolymer–poly(cyclohexene oxide-alt-phthalic anhydride)-block-poly(propylene oxide-alt-phthalic anhydride). An original feature of this approach was the use of temperature as a switch to direct the polymerization along the desired pathway. The authors demonstrated that in the polymerization of a cyclohexene oxide/phthalic anhydride/propylene carbonate mixture at 110 °C, propylene carbonate is inert and does not participate in the reaction, and the reaction of cyclohexene oxide with phthalic anhydride results in the formation of the first block. An increase in the temperature to 180 °C induces the decomposition of the cyclic carbonate and release of propylene oxide, which is involved in the copolymerization with phthalic anhydride to give the second block.
Li and co-workers[322] described initiator 69 of a more complex structure. This made it possible to carry out copolymerization of cyclohexene oxide and CO2 in the presence of triethylborane under relatively mild conditions (80 °C, 10 atm), resulting in the selective formation of polycarbonate with Mn = 2.8 × 105 containing no ether linkages. Copolymerization took place at a pressure of 1 atm, and TOF was 95 h–1. A high selectivity was found for the initiator in the copolymerization of propylene oxide and butylene oxide; however, the content of ether linkages in the copolymer amounted to 46 – 99%.
2.7.2. Bifunctional initiators
Bifunctional initiators 70 containing alkylborane and ammonium salt in one molecule were first proposed by Feng and co-workers[311].
Testing of these compounds in the copolymerization of cyclohexene oxide and CO2 showed initiator 70a to be the most active. It proved to be thermally stable and had TOF = 6500 h–1 even at 150 °C. The high selectivity to the formation of polymer and to the content of carbonate linkages and controllable MW and MWD of polycarbonate stimulated the use of this initiator for the synthesis of four-arm telechelics–oligomeric polycarbonates with terminal hydroxyl groups–meant for the production of polyurethanes. In the copolymerization of epoxides with aliphatic substituents, propylene oxide, butylene oxide and allyl glycidyl ether, the fraction of carbonate linkages was markedly lower (30 – 60%); however, the selectivity was retained at a high level (99%).
Wu and co-workers[315] proposed bifunctional initiator of type 71 containing simultaneously borane and ammonium salt. The highest activity in the cyclohexene oxide copolymerization was found for the initiator with n = 3, R = Et, X = Br (TOF = 710 h–1); it was characterized by a selectivity of more than 99%, with the resulting polycarbonate containing no ether linkages and being characterized by narrow MWD (Mn = 3 × 104 and Đ = 1.2). However, these initiators have low moisture resistance; therefore, they were replaced[328] by phosphonium initiators 72. Their activity in the cyclohexene oxide copolymerization proved to be lower, but the selectivity was high. The controlled synthesis of polycarbonate was also implemented for vinylcyclohexene oxide; however, attempts to accomplish the polymerization of monoterpenes, limonene oxide or α-pinene did not meet with success. Meanwhile, these initiators were selective in the synthesis of cyclic carbonates from epoxides with aliphatic substituents (propylene oxide, butylene oxide, styrene oxide).
Tetranuclear organoborane 73 for the copolymerization of epichlorohydrin and CO2 under relatively mild conditions (25 – 40 °C, 25 atm) was reported by Wu and co-workers[324]. The catalyst productivity was moderate; however, the final product virtually did not contain ether linkages or undesirable cyclic carbonate and had Mn = 3.6 × 104 and Đ = 1.22.
A series of bifunctional organoborane initiators 74 for the copolymerization of propylene oxide and CO2 was developed by Wu et al[318]. All initiators showed a high selectivity in the polymer synthesis, while by varying the substituent, the authors were able to obtain a polycarbonate with a controlled content of ether linkages (from 0.1 to 97%). The nature of the counter-ion (Br–, Cl–, I–) had virtually no effect on the activity of initiators in the copolymerization.
A detailed study of the effects of the cation and Lewis acid in bifunctional catalysts 75 on their activity in the copolymerization of cyclohexene oxide with CO2 or phthalic anhydride was carried out[317]. It turned out that phosphonium initiators 75b – e,g are ~3 times more active than ammonium derivatives 75a,f in the copolymerization of cyclohexene oxide and phthalic anhydride and in the homopolymerization of propylene oxide. A few tens of tertiary and quaternary bifunctional phosphonium borane initiators were synthesized by Lin and co-workers[326], who studied the kinetics of copolymerization of cyclohexene oxide and CO2. This made it possible to find conditions for the preparation of vinylcyclohexene oxide, butylene oxide and butyl glycidyl ether copolymers with high contents carbonate linkages. It is significant that tertiary phosphonium initiators also retain their activity in the presence of chain transfer agents such as diols.
Mono- and polynuclear bifunctional initiators can be used to prepare linear and branched polyester/polycarbonate block copolymers not only without isolation of intermediate compounds, but also upon simultaneous loading of the monomers into the reactor. For example, polypropylene phthalate-block-poly(propylene carbonate) (Scheme 45) was obtained in a high yield and with a high selectivity and was characterized by narrow MWD[312].
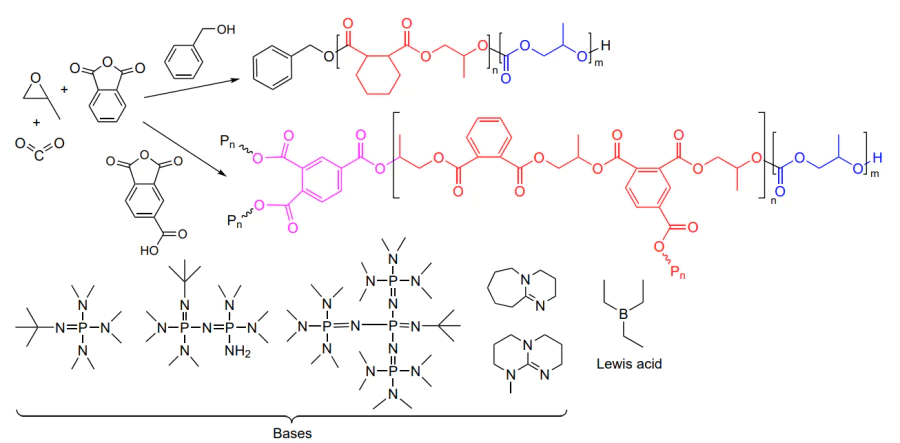
Thus, bifunctional organoboranes can be considered to be promising initiators of the copolymerization of epoxides and CO2 enabling the synthesis of both functional oligomers and high-molecular-weight polymers of linear or complex architecture.
3. Depolymerization of polycarbonates
As discussed above, copolymerization of epoxides and CO2 often leads to the isolation of a by-product, cyclic carbonate, formed upon backbiting, which is thermodynamically more favourable than the formation of linear polycarbonate[330]. This reaction is typical of both metal complex catalysis (Scheme 46) and metal-free anionic copolymerization (Scheme 47).
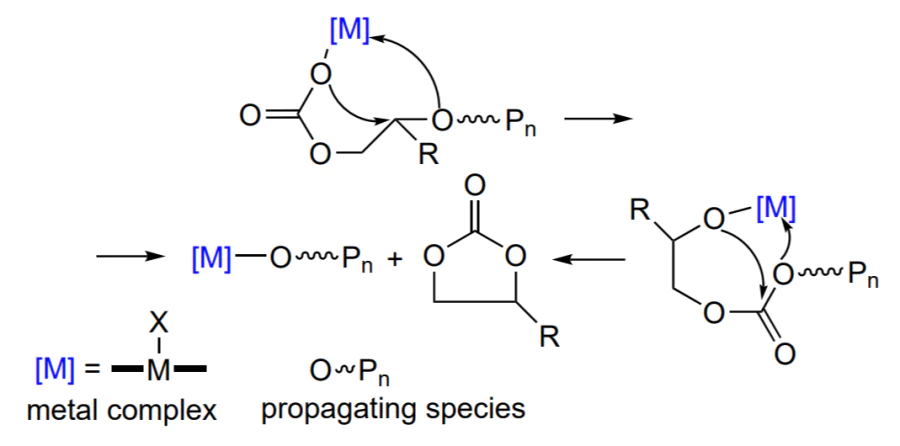
During this chain reaction, which is initiated at the end of the backbone, the length of the active centre successively decreases by one unit; hence, it is formally referred to as depolymerization.
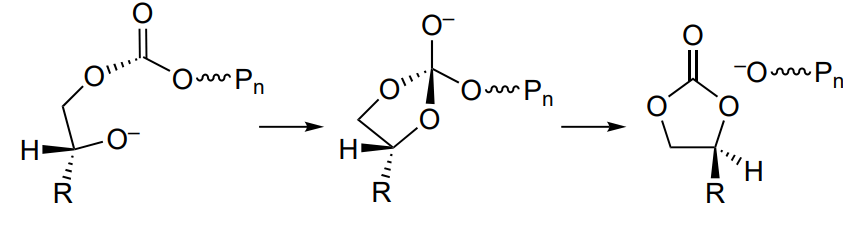
However, depolymerization gives cyclic carbonate rather than the starting monomer. Depolymerization can take place not only for the growing chain during the synthesis, but also for polycarbonate isolated from the reaction mixture[331]. In the former case, the reaction is suppressed by selection of an appropriate catalyst — co-catalyst system with the goal to achieve 100% selectivity to the target product (polycarbonate or cyclic ether). In the latter case, it can be used for recycling. In this connection, it is important to understand which factors affect, and how they affect, the rate and degree of depolymerization. Although depolymerization processes are explicitly or implicitly discussed in most studies related to the synthesis of polycarbonates, there are much less systematic works addressing depolymerization of isolated and characterized polycarbonates.
The formation of propylene carbonate on treatment of polycarbonate with diethylzinc was reported for the first time by Kuran and Górecki[331]. The authors analyzed the change in the intrinsic viscosity of polymer during the reaction, and the decrease in the viscosity suggested that the bond cleavage takes place randomly as a result of diethylzinc coordination to the carbonate group of an arbitrary polymer unit followed by rearrangement and elimination of cyclic carbonate and decrease in the macromolecule length by one unit. However, the subsequent studies did not confirm this depolymerization mechanism. Consider a series of publications[8, 41, 267, 332, 333] describing theoretical and experimental studies of depolymerization of various polycarbonates.
Slow formation of cyclohexene carbonate was detected when a solution of oligomeric poly(cyclohexene carbonate) (Mn = 3500 – 4200 and Đ = 1.2 – 1.4), synthesized under the action of (salen)CrCl and PPNCl up to 100% conversion and not isolated from the reaction mixture, was heated to 80 °C in the absence of CO2[41]. The activation energy for depolymerization was ~110 kJ mol–1. It was found that the reaction proceeds also in the absence of metal complexes; an important role in the depolymerization belongs to the co-catalyst, as was demonstrated in relation to polycarbonates synthesized from styrene oxide, epichlorohydrin and propylene oxide[51]. Tetrabutylammonium chloride, bromide and azide were used as co-catalysts. The polycarbonates isolated by a traditional procedure (precipitation in water) contained terminal hydroxyl groups. Presumably, a base deprotonates the OH group and triggers depolymerization. In the case of poly(styrene carbonate) at 70 °C, depolymerization took place within 30 min when azide was used and 12 h in the presence of bromide and did not take place for tetrabutylammonium chloride. In the last-mentioned case, a temperature rise to 90 °C did not induce depolymerization either. The hydroxyl protection via conversion into acetate retarded, but not prevented the depolymerization. The addition of the initial catalyst–chromium salen complex – retarded depolymerization due to binding of the anion to the metal complex. Similar regularities are characteristic of polycarbonates based on epichlorohydrin and propylene oxide; in the presence of metal complex, the depolymerization rate increases in the series poly(propylene carbonate) < poly(CO2-alt-epichloro-hydrin) < poly(styrene carbonate). A different behaviour is typical of poly(cyclohexene carbonate), which was stable to depolymerization even at 110 °C (20 days) in the presence of a salt; however, upon the addition of chromium complex, depolymerization proceeded slowly, but was completed within 7 days. Computer simulation of the copolymerization and depolymerization of polycarbonates clarified the obtained results and predicted the depolymerization pathways for some unexplored polycarbonates[334]. For most of polycarbonates (poly(ethylene carbonate), poly(propylene carbonate), poly(styrene carbonate), poly(cyclohexene carbonate) and poly(indene carbonate)), the heats of polymerization of the corresponding monomers are similar (~90 kJ mol–1); an outlier is poly(cyclopentene carbonate), for which this value is 1.5 times lower. Depolymerization of a free (not bound to a metal complex) carbonate anion is inhibited in the presence of bulky groups in the monomer (e.g. poly(cyclohexene carbonate)), but is accelerated when resonance stabilization is present in the transition state (e.g. poly(styrene carbonate)). It is more preferable than depolymerization of the carbonate anion bound to a metal complex. The anionic growing centre containing a carbonate group at the end exists if excess CO2 is present in the reaction mixture; in the absence of CO2, alkoxide is formed. This compound is depolymerized more easily because of the lower energy barrier (~40 kJ mol–1). The low heat of copolymerization of cyclopentene oxide and CO2 compared with other epoxides accounts for the change in the poly(cyclopentene carbonate) depolymerization pathway[335]. When depolymerization is initiated by a strong base, sodium bis(trimethylsilyl)amide, the initial monomers and some amount of cyclopentene carbonate are formed as the products. When chromium salen complex with tetrabutylammonium azide is used, the only depolymerization product is cyclopentene oxide. A similar feature is inherent in poly(limonene carbonate), which is depolymerized to give limonene oxide under the action of a strong base[336].
The depolymerization of aliphatic polycarbonates can be considered in a dual way, namely, as a desirable reaction in the case of secondary processing of polycarbonates and design of closed-loop processes or as a destructive process if polymer characteristics are deteriorared during operation. In the latter case, solution to the problem is obvious: it is necessary to protect the end groups of the polymer and prevent the formation of the anion. The protection of poly(propylene carbonate) terminal groups in order to increase the thermal stability of the polymer was first proposed by Mantell and co-workers[337]. For this purpose, poly(ethylene carbonate) and poly(propylene carbonate) containing terminal hydroxyl groups were treated with various reagents under inert atmosphere and in air. The replacement of the terminal OH group with the O – C bond (reaction of polycarbonate with methyl or phenyl isocyanate, acetyl chloride in pyridine, maleic anhydride) increased the thermal stability of polycarbonates by 20 – 40 °C; replacement of this group with O – Р bond (diphenyl chlorophosphate or chloro(diphenylphosphine) in pyridine) increased the thermal stability by 10 – 30 °C, while in the case of O – S bond (methane- or benzenesulfonyl chloride in pyridine), the thermal stability increased by 30 – 40 °C. Later, the same techniques were used by other authors[338-340].
Polycarbonates thoroughly purified from traces of the initiator (metal complex and/or base), water and a solvent are more thermally stable. The polycarbonate degradation occurs in one stage, with ~50% of the sample weight being lost at 220 °C[339, 341-343]. The activation energy of this reaction is ~100 kJ mol–1[341]. In this case, depolymerization initiated at the terminal groups of the macromolecules is accompanied by random chain cleavage (Scheme 48).
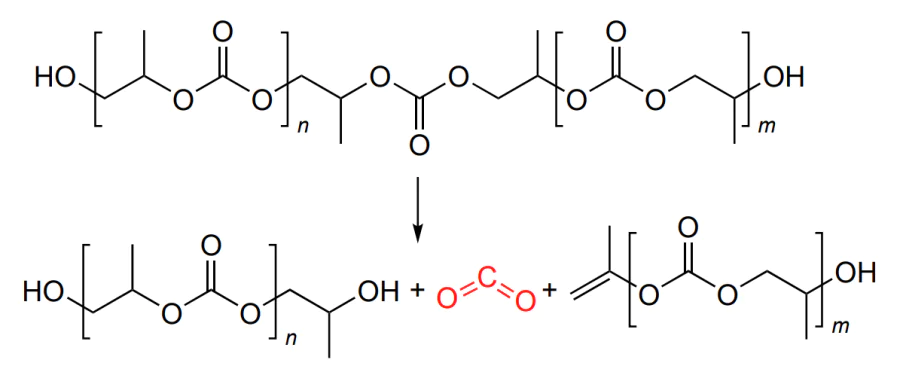
Polycarbonate testing in a weathering chamber at 60 °C at a humidity of 50% demonstrated slow degradation, as indicated by a decrease in the polymer MW and change in MWD[342]. A similar result was found upon extrusion of a melt of a high-molecular-weight poly(propylene carbonate) (Mw = 5.5 × 105)[343]. The authors observed a slow MW decrease even at 130 °C (1.5-fold in half an hour) and a very fast MW decrease at 180 °C (5–6-fold in 5 min). The activation energy for the thermal degradation of polycarbonate found in this study, 120 kJ mol–1, differs little from the values reported in other publications. Whereas depolymerization can be retarded by protecting the terminal groups, the rate of random degradation was decreased by adding citric acid, which acted as a stabilizer[339].
In some cases, an opposite effect is required, that is, to facilitate the degradation of the polycarbonates and to perform it under mild conditions. Apart from adding bases and metal complexes, recently acid and base photo- and thermo-activators of degradation, e.g., iodonium salts, tetraarylborates, etc., have been proposed for this purpose[339, 344, 345].
The structure of polycarbonates, that is, repeating carbonate linkages in the backbone, implies that they would tend to undergo biodegradation, apart from thermal degradation and depolymerization. Indeed, the conceptual possibility of biodegradation was demonstrated in relation to poly(propylene carbonate)[346]. The most complete degradation is attained by composting where polycarbonate is treated simultaneously by numerous enzymes[347]. An important stage of biodegradation is enzyme adsorption on the polycarbonate surface; therefore, the polymer hydrophilicity/hydrophobicity and physical state (amorphous/crystalline) affect the biodegradation rate[348]. Esterase and lipase enzymes play the key role in the hydrolysis of polycarbonates, but the mechanism of polycarbonate biodegradation in vivo is an open question. The issues of biodegradation and ways to control it have recently become significant due to the development of biomedical applications of some polycarbonates[349].
4. Properties and potential applications of polycarbonates
Poly(propylene carbonate) is the most studied among aliphatic polycarbonates. The interest in this compound is due, in particular, to the trend of solving environmental problems by using biodegradable plastics instead of common ones. The properties of poly(propylene carbonate) are described in numerous papers and summarized in a number of reviews[2-4, 7-9]. It is the first and so far the only one aliphatic polycarbonate that is produced on an industrial scale and the production of which has recently reached the level of tens of thousand tons per year[350]. Poly(propylene carbonate) is a relatively hydrophobic polymer readily soluble in many organic solvents (THF, benzene, 1,4-dioxane, dichloromethane, chloroform and acetone), but insoluble in water, alcohols and aliphatic hydrocarbons[7]; the contact angle is ~77°[351]. It is a dielectric possessing low nitrogen, water and oxygen permeability and high CO2 permeability. Low oxygen and water barrier performance is important for packaging materials; the film permeability can be decreased by blending, for example, with poly(butylene succinate) or low-density polyethylеne[352]. It is biocompatible[353] and can slowly degrade in vitro and in vivo[351].
The thermal properties of polymers are known to determine the temperature range for their application. Depending on the method of synthesis, poly(propylene carbonate) can have atactic or stereoregular configuration and, hence, it can be either amorphous or crystalline. Irregular polycarbonate has been studied most comprehensively; however, published data give a wide scatter of glass transition temperatures (Tc) for this polymer from 24 to 45 °C[354-357]. This is due to the fact that in the case of relatively low chemoselectivity of the synthesis, the polymer contains not only carbonate, but also ether linkages, which impart higher flexibility and decrease the glass transition temperature[358, 359]. For example, for polycarbonate containing 40 – 60% ether linkages, i.e., a copolymer of propylene oxide and propylene carbonate, the Tc value decreases to 8 °C[358]. Meanwhile, Tc depends little on the local isomerism (head-to-head or head-to-tail[22, 225, 360]) and, when MW is sufficiently high, it is rather insensitive to the terminal groups of the polymer: for polycarbonate (Mw = 7.7 × 104 and Đ = 3.7), Tc increases by 2 – 3 °C when the terminal hydroxyl groups are protected with maleic anhydride[361]. Depending on the potential application, there can be the task of either lowering or raising the glass transition temperature of poly(propylene carbonate). The former task can be efficiently solved using plasticizers that are well compatible with the polymer. For example, Tc decreases by ~20 °C in the presence of 5 mass% acetyltriethyl citrate or by 10 °C in the presence of tributoxyethyl phosphate or dibutoxyethyl adipate. One more plasticizer is propylene carbonate: Tc amounts to 7.5 °C in the presence of 15 mass% cyclic carbonate and 12.5 °C in the presence of 25 mass% carbonate[7]. In addition, pentene, hexene and octene carbonates, the homopolymers of which have Tc < 0 °C[362], incorporated in the poly(propylene carbonate) molecule, can also decrease the glass transition temperature. The latter task is solved by internal plasticizers, that is, comonomers that endow the poly(propylene carbonate) macromolecules with additional rigidity. For example, copolymerization with cyclohexene oxide, styrene oxide, phthalic anhydride, maleic anhydride and other monomers may enhance the thermal and mechanical properties of poly(propylene carbonate)[358, 363, 364]. The terpolymer of propylene oxide, cyclohexene oxide and CO2 has Tc = 70 – 80 °C[365-367]. The comonomers that enhance the rigidity may be represented by indene oxide, the polycarbonate of which has Tc = 138 °C[368], 1,4-dihydronaphthalene oxide (polycarbonate Tc is 150 °C)[158, 369], limonene oxide (polycarbonate Tc is 130 °C)[189, 370] or styrene oxide (polycarbonate Tc is 76 – 80 °C)[352]. Crystalline poly(propylene carbonate) is a typical plastic with a melting point depending on the polymer degree of purity, configurational isomerism, composition and molecular weight. For example, isotactic (R,R)- and (S,S)-polycarbonates were mixed to obtain a stereocomplex with the melting point (Tm) of 373 °C[369]. Coates and co-workers[359] synthesized isotactic poly(propylene carbonate) with Mn = 1.1 × 104 and Tm = 267 °C. Thus, in the absence of an internal or external plasticizer, poly(propylene carbonate) is a plastic the characteristics of which are determined by the molecular structure and depend on the conditions of synthesis. As noted above, poly(propylene carbonate) has a high thermal stability (180 to 220 °C)[331]. However, most of these studies dealt with the atactic polymer. Meanwhile, the degradation onset temperature of isotactic polycarbonate is 240 °C, while that of the stereogradient polymer is 273 °C[236]. Copolymerization can also increase the thermal stability of polycarbonate, as was shown in relation to multiblock copolymers with 1-hexene oxide[249, 254, 371-374] and 1-butene oxide[249] and terpolymers with cyclohexene oxide and butyrocaprolactone[371] or decalactone[375].
Poly(propylene carbonate) as a plastic cannot compete with other typical plastics in its strength chracteristics[376]. For example, poly(propylene carbonate) (Mn = 5 × 104, Đ = 1.07) with Young’s modulus of 830 MPa, elongation at break of 330% and tensile strength of 21.5 MPa has been reported[377]. A commercial sample (Mn = 2.6 × 105, Đ = 5) had a Young’s modulus of 680 MPa and tensile strength of 17 MPa. An industrial sample of China Bluechemical (Mn = 7 × 104, Đ = 3.2) with a low glass transition temperature (~20 °C) also had a low tensile strength of 4.7 MPa. Similar properties were found for poly(butylene carbonate) (Tc = 60 °C, Young’s modulus of 2.2 GPa, tensile strength of 37 MPa), while poly(ethylene carbonate) was even more elastic (Tc = 0.5 °C, Young’s modulus of 2.1 MPa and a tensile strength of 6 MPa)[352]. The set of mechanical properties of polycarbonates can be enhanced using two approaches. The first one is to create blends with inorganic fillers or with other polymers in order to enhance the strength characteristics of polycarbonate[378-386]. The second approach involves copolymerization with more rigid epoxides and/or lactones. Poly(cyclohexene carbonate) has an elastic modulus of 2.5 GPa, tensile strength of 12 MPa and a low elongation at break[362]. It resembles bisphenol A-based polycarbonate in the strength characteristics, but is more brittle[68]. The terpolymer of propylene oxide (20%), cyclohexene oxide (80%) and CO2 is similar in the properties to poly(hexene carbonate). As one more example, consider the terpolymer of limonene oxide, CO2 and decalactone, which showed an optimal combination of strength and elasticity in comparison with poly(limonene carbonate) or poly(propylene carbonate). It can form strong optically transparent films and is characterized by a wide temperature range of processing[375]. Poly(limonene carbonate) is of particular interest owing to the unique combination of optical and mechanical properties; it has a high optical transparency of 94%, Young’s modulus of 0.95 GPa, tensile strength of 55 MPa and the degradation onset temperature of 240 °C[145]. Poly(limonene carbonate) has a relatively high oxygen permeability and selectivity; in terms of its properties, it occupies an intermediate position between conventional engineering plastics and gas permeable polymers[10]. Unlike the latter, it can be processed to sheet materials, used as breathable fiberglass, cured by various methods at C=C bonds and used as a coating with good strength characteristics[387].
The applications of polycarbonates depend not only on their chemical structure, but also on their molecular weight. Oligomeric aliphatic carbonates can be considered as building blocks for the production of polyurethanes. This is due to the presence of terminal hydroxyl groups in the oligomeric telechelics[388]. A systematic study of the applicability of oligocarbonate triols for the industrial synthesis of polyurethanes was reported by Gürtler and co-workers[23]. The obtained plastic foams were not inferior in the properties to the industrial foams obtained from polyether polyols. Aqueous dispersions of these polyurethanes has higher mechanical properties and resistance to oxidation and hydrolysis than polyurethanes prepared from poly(propylene glycol) or poly(butylene glycol adipate)[389-391]. Oligocarbonates were also used for the synthesis of thermoplastic polyurethanes with good anticorrosive properties[392-394]. As a results of these studies, processes for the synthesis of oligocarbonate polyols for the production of polyurethanes were developed and implemented.
High-molecular-weight aliphatic polycarbonates can be used to form films and fibres for various purposes[395]. For example, poly(propylene carbonate) is used for the manufacture of plastic bags, shelter materials and mulch films. A biodegradable high-molecular-weight poly(propylene carbonate) film has been manufactured under the brand name PCO2® since 2011. Poly(ethylene carbonate) (QPAC®25), poly(propylene carbonate) (QPAC®40), copoly(propylene/cyclohexene carbonate) (QPAC®100), poly(cyclohexene carbonate) (QPAC®130) and poly(butylene carbonate) (QPAC®PBC) have been manufactured since 2015 as granules, solutions or films, depending on the desired use. Poly(propylene carbonate) can be used in regenerative medicine both by itself and as composites with natural polymers (starch, gelatine, chitosan, etc.) using electrospinning of the required 3D matrices (frameworks)[396-403]. The hydrophobicity of polycarbonates restricts their applications in medicine; therefore, they are often modified either during the synthesis or after the synthesis by introducing hydrophilic groups into the polymer macromolecules. These amphiphilic polycarbonates are of interest for bioimaging, medicine and development of lithium-ion batteries[8, 404, 405]. In this case, most widely used are block copolymers in which poly(propylene carbonate) serves as the hydrophobic block, while functional polycarbonate with carboxyl or hydroxyl side groups is the hydrophilic block. Block copolymers of this type are biocompatible and are able to bind conjugates, drugs and contrast agents. For example, Grinstaff and co-workers[11] described a conjugate of poly(1,2-glycerol carbonate) with the anticancer drug paclitaxel; the obtained products were tested on mice. The authors showed that the drug was localized in tumours and that the therapeutic effect was higher than that of the drug without a polymer carrier. The polycarbonate conjugate with Gd3+ can be used for tumour imaging[406]. In addition, it is easily excreted from the body due to biodegradability. The water-soluble platinum — polycarbonate conjugates can be used as carriers for drug delivery[407]. The amphiphilic polycarbonates with carboxyl groups have low Tc; they are able to provide ionic conductivity upon the addition of various organic and inorganic salts and are more stable than liquid electrolytes[408]. Therefore, they can be used for the manufacture of solid polymer electrolytes[409-413].
5. Conclusion
The interest in catalytic processes producing valuable chemical products from carbon dioxide has been growing in recent years not only in the scientific community, but also among representatives of industry[414-423]. This is promoted by the actively discussed environmental problems and restrictions imposed in various countries on the production of disposable tableware and packaging materials made of non-biodegradable polyolefins. It is not surprising that the number of scientific publications dealing with catalytic processes involving CO2 has been exponentially increasing since the late 2010s — early 2020s. A considerable part of these studies address the problem of obtaining polycarbonates by copolymerization of epoxides and CO2. The interest in these polymers is caused, first of all, by their potential biodegradability[418]. The properties of addition polycarbonates can be varied over fairly broad ranges not only via modification of the chemical structure of epoxides, but also owing to relatively simple approaches to introduction of ether or ester linkages into the polymer chain and control over the configuration of macromolecules[424-428].
The advances in the synthesis have already enabled the onset of industrial production of some polycarbonates, mainly poly(propylene carbonate), to manufacture a variety of packaging materials and to develop new polyurethane foams and adhesives. However, it can hardly be expected in the near future that polycarbonates would displace polyolefins, particularly polyethylene, from the market. The reasons are obvious. First, epoxides are more expensive than olefins. Second, the known inexpensive catalysts for the synthesis of polycarbonates have relatively low activity, while the economic feasibility of using high-performance catalysts is still low. This implies two relevant trends for the development of this area: the search for new cheap monomers or new synthetic routes to known epoxides and development of efficient, inexpensive and easily regenerated catalysts. Numerous publications devoted to the catalysis in the synthesis of polycarbonates give hope that, at least, the second task may be solved in the coming years.
The third aspect is that the knowledge of the properties of polycarbonates is still insufficient, which restricts the understanding of their potential applications. Studies along this line have been actively carried out in recent years, and one may hope that the scope of applications of polycarbonates would markedly expand[409, 404-406]. It is of obvious interest to replace condensation polycarbonates based on bisphenol А with addition polycarbonates the production of which is more environmentally friendly. The advances in the regio- and stereochemical control of the copolymerization of alicyclic epoxides and CO2 make it possible to expect an industrial production of poly(cyclohexene carbonate) and poly(limonene carbonate), which are not inferior in the mechanical and optical performance to conventional polycarbonates. Active studies aimed at the use of polycarbonates in biomedicine[4, 403] and in the solid electrolytes of Li-ion batteries[409-411] have been in progress in a few recent years.
The fourth trend is related to the life cycle and recycling of polymer materials[273, 429-438]. This requires understanding of the kinetics and mechanism of degradation of polycarbonates according to different mechanisms: biodegradation, thermal degradation, catalytic degradation and hydrolytic degradation. Development of cost-effective approaches for targeted degradation of polycarbonates to valuable low-molecular-weight substances and controlled biodegradation of polycarbonates in nature or in landfills will constitute one more step towards replacing traditional polyolefins with polycarbonates.
The area of chemistry related to the conversion of CO2 to polycarbonates is not only relevant, but also interdisciplinary and requiring efforts of specialists in polymer and organic chemistry, materials scientists, medical professionals, etc. The development of this area will not only markedly expand the fundamental knowledge in chemical sciences, but also solve many environmental problems, including environment contamination.
6. List of abbreviations and symbols
Ac — acetyl,
Ar — aryl, (BDI)ZnX — zinc complexes with β-diimine ligands,
But — tert-butyl,
Bu — n-butyl,
Bu4NX — tetra(n-butyl)ammonium salt,
Cy — cyclohexyl,
DBU — 1,8-diazabicyclo(5.4.0)undec-7-ene,
DMAP (dmap) — 4-(dimethylamino)pyridine,
Et — ethyl,
Me — methyl,
Mn — number-average molecular weight,
Mw — weight-average molecular weight,
Ph — phenyl,
Pri — isopropyl,
PPNX — bis(triphenylphosphine)iminium,
Py — pyridine,
salen — ligand formed by salicylaldehyde and ethylene diamine,
salcy — ligand formed by salicylaldehyde and 1,2-cyclohexanediamine,
TBD — triazabicyclodecene,
TMS — trimethylsilyl,
TOF — turnover frequency of the catalyst,
TON — turnover number of the catalyst,
(TPP2-Cl)AlIIICl — (5,10,15,20-tetraphenylporphyrinato)-aluminium chloride,
TTC-COOH — 4-cyano-4-[(dodecylsulfanylthiocarbonyl)-sulfanyl]pentanoic acid,
Tf — triflyl,
Ts — tosyl,
MW — molecular weight,
MWD — molecular weight distribution,
RAFT — reversible addition–fragmentation chain transfer radical polymerization,
PEG — poly(ethylene glycol),
Đ — molecular-weight dispersity of the polymer.
Acknowledgements
This work was done with the support of MSU Program of Development, Project No 23-SCH07-02.
References















