


Keywords
Abstract
Free alcohols are ubiquitous in nature, drugs, bulk chemicals, and various complex molecules. The abundantly available free aliphatic alcohols, being inexpensive, are sustainable starting materials in organic synthesis to construct value-added scaffolds. Direct C(sp3)–H functionalization of inert bonds in aliphatic alcoholic chain is challenging due to their inactive nature towards chemical reactions and also multiple similar C–H bonds are available for functionalization reaction at a time. Barton process, which is a classical method for generating alkoxyl radicals, has been widely used for the remote functionalization of alcoholic chain using the strategy of 1,5-hydrogen atom transfer (1,5-HAT). However, the requirement for the pre-activation of alcohols not only adds extra and tedious steps but also, these precursors are challenging to prepare and handle. Generating desired alkoxyl radicals from these precursors is a difficult task and also affects the atom economy of reactions. Numerous methods covering the C(sp3)–H functionalization of unprotected non-activated alcohols at diverse (α-, β-, γ-, δ-) positions to access functionalized alcohols are elaborated here. A precise and focused compilation will help to understand what has been established and how new methodologies can be developed for the future needs.
The bibliography includes 158 references.
1. Introduction
Free alcohols are abundantly available, inexpensive, and sustainable precursors for developing value-added molecules[1-3]. The existence of hydroxyl groups endow molecules with the versatile role to play a wide range of capabilities, including solvents, directing groups, nucleophiles, and also act as a proton source. Alcohols are also considered potential alkylating agents[3-5]. Consequently, alcohols are important, albeit challenging, scaffolds for various transformations in organic chemistry.
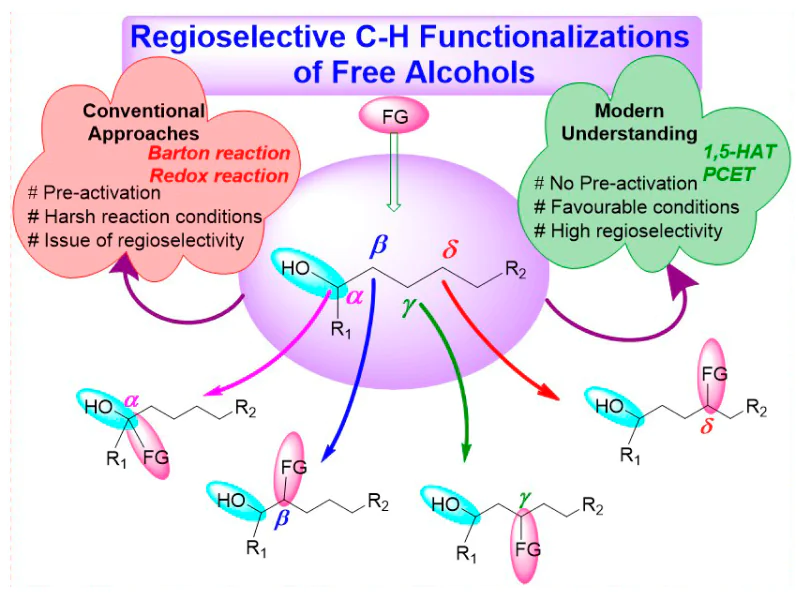
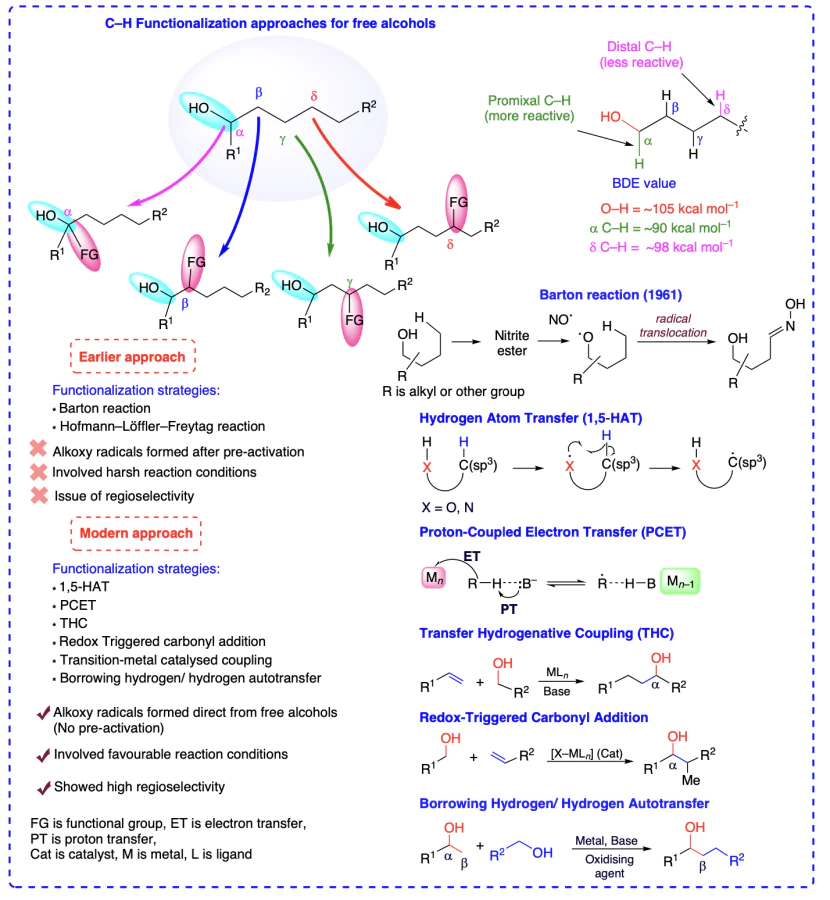
The inactive nature of alcohols towards chemical reactions renders the selective C(sp3) – H bond functionalizations of aliphatic chain a quite challenging task. Site-specific C – H functionalizations within the aliphatic chain of alcohols are also difficult attributed due to the chemical similarity of all C – H bonds. Conventional approaches for the functionalization of alcohols involve successful Barton reaction[6, 7] and Hofmann – Löffler – Freytag reaction (initially developed for the functionalization of amines)[8], although the pre-activation of alcoholic groups and tedious reaction steps need further improvement. The pre-activation of alcohols in the classical approach involved the conversion of alcohol into corresponding sulfonates or halides, using precursors, including peroxy compounds, nitrite esters, N-alkoxyphthalimides, hypohalites, N-alkoxylpyridine-2-thiones, metal alkoxides, etc., which are challenging to prepare and handle[9]. The generation of desired alkoxyl radicals, the main step towards C – H functionalizations, from these precursors requires typical conditions and affects the atom economy of the whole reaction[10]. Consequently, direct site-selective C(sp3) – H functionalizations of unprotected non-activated alcohols without pre-activation impose an attractive alternative towards scientific community, albeit a very challenging strategy owing to the high bond dissociation energy (BDE = 105 kcal mol–1) and oxidation potential (>2.0 V) of O – H bonds[11]. Additionally, the alkoxyl radicals evolved from free alcohols represents a competent methodology for position-selective (α, β-, γ-, δ-positions) C(sp3) – H bond transformation and remarkably enhances the atom economy of the process.
Modern understanding of the functionalization of alcohols excludes their pre-activation. Numerous methodologies have been developed in the last two decades on regioselective and stereoselective C – H functionalizations of alcohols, including 1,5-hydrogen atom transfer (1,5-HAT), proton-coupled electron transfer (PCET), transfer hydrogenative coupling (THC), borrowing hydrogen (BH) / hydrogen auto-transfer strategy, redox triggered carbonyl addition, transition-metal catalyzed, and metal-free functionalizations. These strategies, executed under ambient reaction conditions, are highly efficient, atom and time economic, and release very few by-products and revolutionized alcohol transformations into valuable products[3, 11-17].
Because of this, a precise and comprehensive compilation with the exploration of the reasoning behind the direct and selective C – H functionalizations in free alcohols is of utmost importance for future developments. Several reviews has been published in literature describing the distal C – H functionalization reactions and in part covers the functionalization of alcohols[18-21]. Zhang et al.[22] comprehensively reviewed seven distinct strategies for sp3 α-C – H bond activation and C – C bond formation reactions involving alcohols and ethers. Their analysis covered both methodological advancements and synthetic applications, with a particular focus on enhancing efficiency, chemo selectivity, regioselectivity, stereoselectivity, catalyst performance, substrate compatibility, and mechanistic understanding, however, limited to the a-position only. Zhang et al.[23] explored the potential of copper-mediated C(sp3) – H functionalizations via a radical relay process and covers the alcohol substrates in part. Recently, Ortiz et al.[24] published a perspective exploring the potential of enantioselective ruthenium(II) complexes in catalyzing the direct conversion of simple alcohols into structurally more intricate alcohols via a hydrogen auto-transfer mechanism. Li et al.[25] comprehensively analyzed transition-metal-mediated regiospecific functionalization reactions at distal (γ) and geminal (δ) positions of sp3-hybridized carbons including in alcohol substrates.
Current compilation incorporated and precisely highlighted the regioselective and stereoselective C(sp3) – H functionalizations of unprotected non-activated alcohols at α-, β-, γ-, and δ-positions. The prime focus of the review is to provide the readers about various hypothesis with mechanism point of view for functionalization of unprotected non-activated alcohols at various (α-, β-, γ-, and δ-) positions. Furthermore, we have presented the variegated functionalization methods within the time frame of 15 years structured as metal catalyzed and metal-free direct C(sp3) – H activation and functionalization at different positions of alcohols.
2. Common strategies for site selective alcohol functionalization
2.1. Conventional approaches vs modern understanding
The electronically and sterically stable substrates are highly challenging for selective functionalizations. To functionalize such substrates, the pre-activation strategy, which involves a lengthy, hazardous procedure that is not atom economic, is required[26, 27]. Additionally, the issues of regioselectivity and stereoselectivity arise in such molecules and methodologies. Henceforth, the scientific community realizes the dire need for some efficient and green methods to enable the direct and selective functionalizations of typical, non-activated, and remote sites of molecules[28]. Traditionally, the inception of various functionalities in alcohols at remote positions were carried out through Barton and Hofmann – Loffler – Freytag type reaction (initially developed for the functionalization of amines, similar mechanism was followed for the functionalization of alcohols). In these transformations, the free alcohol engendering alkoxyl radicals often requires hazardous precursors such as lead tetraacetate, hypervalent iodine reagents, etc. The supreme stability of O – H bond (BDE = ~105 kcal mol–1) and chemical inertness generally make the catalytic conditions for generating alkoxyl radicals challenging. In addition, the classical approaches failed to address the selectivity issues associated with the functionalization of alcohols. These processes also involved lengthy, tedious, and time-consuming procedures for functionalization.
Various methodologies have been developed in the last decade to resolve the issues of chemo-, regio-, and stereoselectivity and to devise a strategy to produce alkoxyl radicals from free alcohols under lenient reaction conditions are claimed here as modern approaches. The main focus of all these modern approaches has been to develop green and sustainable methods either without or minimal by-products (Scheme 1). As used herein, the symbol R denotes an alkyl group, that may contain any substituent at ω-position (e.g., Ar, OAlk, OAr, NAlk2, etc.).
2.2. Modern strategies for radical generation
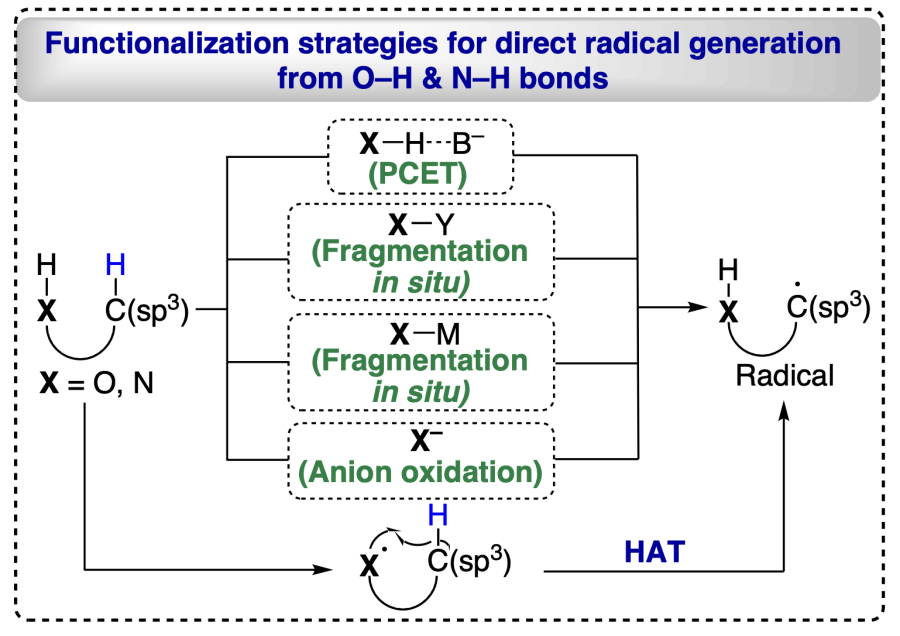
2.2.1. Proton-coupled electron transfer (PCET)
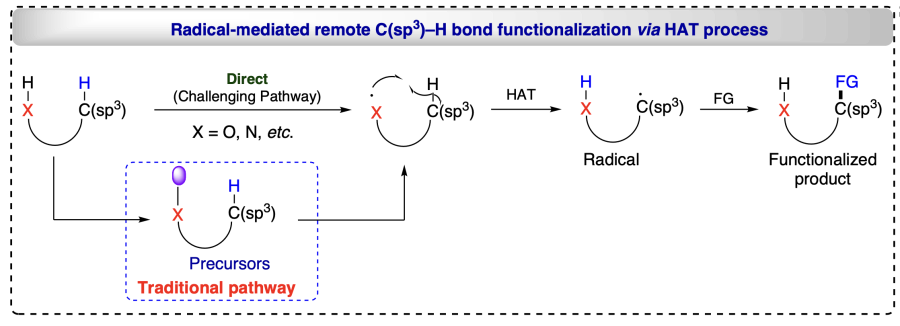
The alkoxyl radical generation has not been facilitated by alkoxides (RO–) oxidation by alkanols reacting with strong bases and oxidants, indicating that the open-shell activation model is ineffective. Thus, modern approaches involved the closed-shell activation modes such as proton-coupled electron transfer (PCET) activation, oxidation of deprotonated X– anions owning lower oxidation potential, homolysis of in situ formed O/N-halogen or O/N-metal bonds and HAT processes for position-selective transformation of C(sp3) – H bonds, for unprotected alcohols and direct engendering alkoxyl radicals (Scheme 2)[11]. Barton reaction has been pivotal in the δ-selective functionalization of alcoholic C(sp3) – H bonds using 1,5-HAT process. In the present scenario, 1,5-HAT has brought a renaissance in radical chemistry and acquired substantial attention from organic chemists. It has been found that 1,5-HAT is a thermodynamically favoured process and as the bond dissociation energy increases from 3° to 1° C(sp3) – H bonds, the stability of the resulting radical decreases, making it easier for 1,5-HAT to form 3° carbon radical than other counterparts. This compliments the increase of chemo-selectivity for the 1,5-HAT reaction. In addition, this process is highly regio-selective[29] and various research groups have utilized 1,5-HAT strategy to functionalize alcohols at α-, β-, g- and δ-positions. Mechanistically, HAT-enabled C(sp3) – H functionalizations include three steps: generation of radical center at X from where the H atom is abstracted (X = O, N, etc.) followed by regioselective HAT via exergonic intramolecular process and finally the distal functionalizations by radical trapping reagents (see Scheme 2).
2.2.2. Hydrogen atom transfer (HAT)
In conventional HAT (Scheme 3), the proton and electron embark on a simultaneous voyage from single donor to acceptor, whereas, in a multisite PCET mechanism, the electron and proton are derived from two distinct donors or end up at two distinct acceptors. Also, the PCET reaction has more scope than HAT because it has a more extensive thermodynamic range[14]. In a HAT process, the regio-selectivity depends on the polar effects, self-exchange kinetics, and reaction driving force[30].
On the other hand, in PCET there is the involvement of a separate hydrogen bond complex before the transfer of electron between the substrate and proton donor/acceptor (see Scheme 2)[31]. The PCET approach is also favoured kinetically because it involves a concerted transfer mechanism rather than stepwise pathways.
2.3. Various hypothesis for α,β,γ,δ-functionalizations of alcohols
2.3.1. Carbonyl addition via redox-triggered carbonyl addition (RTCA)
RTCA is the recently developed technique concerning direct functionalization of inactive C – H bonds of alcohols involving an exchange of hydrogen atoms between alcohol and unsaturated reactant, generating a transient electrophile-nucleophile pair and finally yielding the desired C – H functionalized alcohol product via carbonyl addition. This strategy has mainly been used for the α-functionalizations of alcohols and involves C – C bond formation[32-35].Three extensive pathways are exist for C – C coupling and alcohol oxidation to illustrate the mechanisms including i) C=C π-bond hydrometallation, ii) reductive cleavage of C – X bond, or iii) transfer hydrogenolysis of oxametallacycles. Classically, C=O addition took place via separate redox events and pre-metallated reagents. However, in this approach, the redox events are merged, and the discrete R – OH oxidation, and pre-metalated reagents [36] are not allowed (Scheme 4).
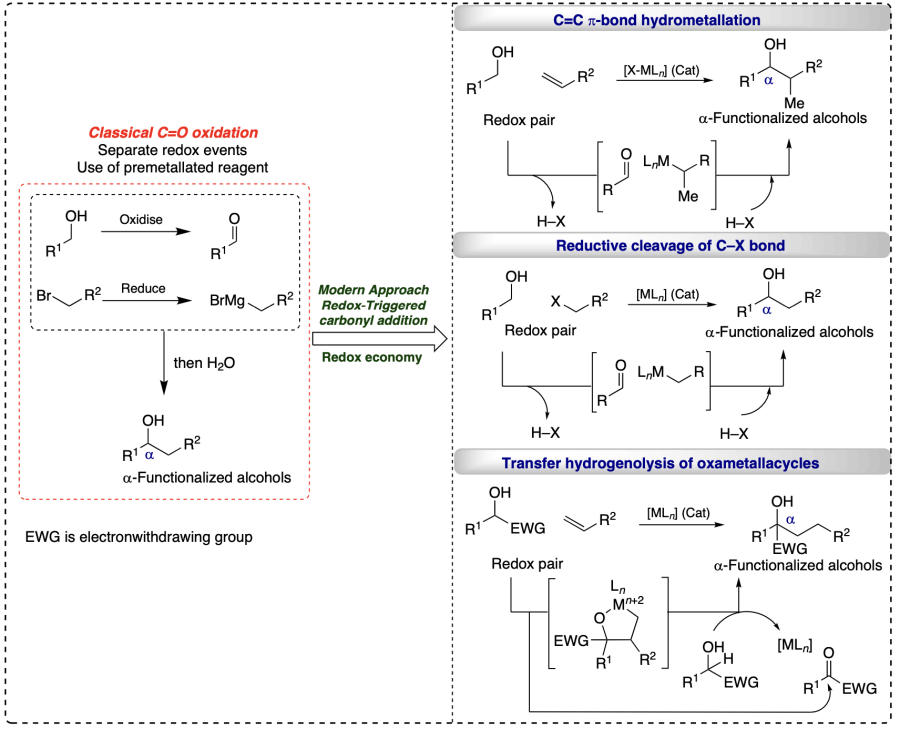
2.3.2. Carbonyl addition via transfer-hydrogenative coupling (THC)
Contrary to RTCA, the carbonyl addition using THC process involves dehydrogenation of alcohols via abstraction of α-hydrogen initially using a base and metal-ligand, resulting in the formation of an aldehyde and simultaneous metal bonding with the unsaturated reactant (say, an electrophile-nucleophile pair is formed). These both combine to form a transient state that abstracts a proton from base-H to give the desired functionalized alcohols (Scheme 5)[37-40].
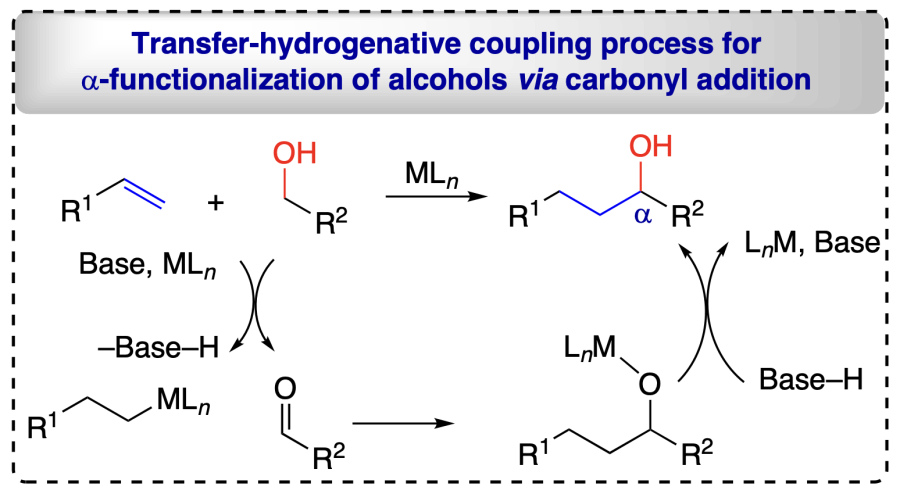
2.3.3. Borrowing hydrogen strategy
Conventionally alcohols have been oxidized to corresponding carbonyl compounds by using various oxidizing reagents. However, these methods are not efficient and sustainable due to the generation of by-products, selectivity issues, and decreased atom economy. Metal-catalyzed alcohol dehydrogenation can directly yield aldehyde and ketones without using any oxidizing agents. This approach involves transfer dehydrogenations, allowing the catalyst to abstract H-atom from alcohol, providing corresponding aldehyde or ketone. The formed aldehyde and ketone species generate an a,b-unsaturated moiety (enone) through condensation reaction. Now the temporarily taken hydrogen is back transferred to unsaturated intermediate product through hydrogenation process and the whole process is known as «borrowing hydrogen strategy» wherein, a new C – C bond is formed[41, 42]. This process utilizes alcohol as an alkylating agents to enable the functionalizations. Generation of water as only by-product is highly advantageous enabling a sustainable and green methodology for the functionalization reaction (Scheme 6).
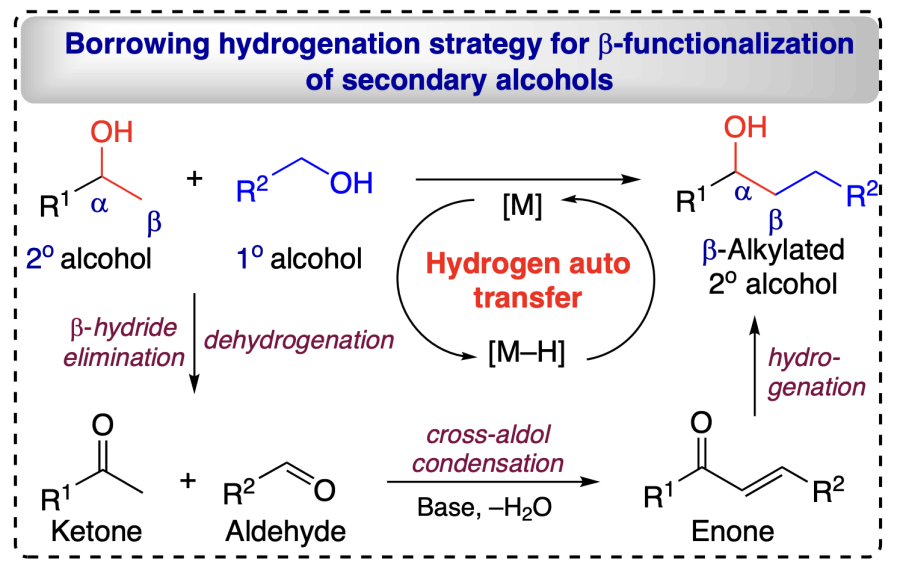
2.3.4. Hypothesis for δ-functionalizations of alcohols
Using light and oxidizing agents, metals dehydrogenate the alcohols, forming a bond with the oxygen atom. On homolysis, oxygen/alkoxyl radical is formed through a six-membered cyclic transition state, which is highly suitable for δ-functionalization reactions. For hydrogen transfer, an optimal distance of 2.5 – 2.7 Å between the carbon and the atom to which hydrogen atoms to be transferred is required[16]. As a result of 1,5-HAT a δ-carbon radical is formed, which reacts with electrophile to form the desired d-functionalized product (Scheme 7).
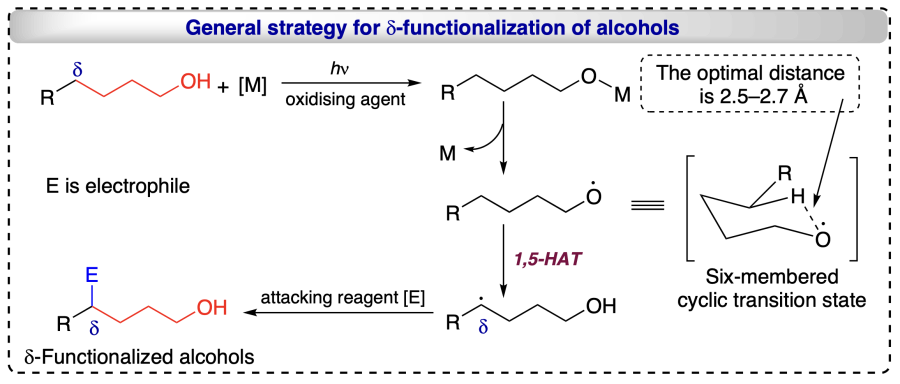
Hence, it has been observed that modern approaches realize the functionalization reactions in alcohols exclusively at the selected positions. The regioselectivities observed (at diverse (α-, β-, γ-, δ-) positions) during the functionalizations of alcohols through modern approaches such as HAT involving PCET mechanism, RTCA mechanism, etc., allow the economical, efficient and sustainable approaches. Radicals generation at the selective positions is the key for the regioselective functionalizations. Numerous methodologies has been exploited in recent time towards transformation of alcohols with high regioselectivity. Additionally, several electronically and sterically challenging substrates were also functionalized through radical approach without the preactivation of substrates and produce either green (e.g. H2O) or less hazardous by-products.
3. Direct selective functionalization of C – H bonds in non-activated alcohols
3.1. α-Selective direct functionalization for non-activated alcohols
In this section, the C – H functionalizations of non-activated alcohols at α-position under different conditions have been discussed. The schematic representations of the conditions used for the α-functionalizations, including the metal catalysts and promotors or initiators used for the transformations of alcohols have been depicted in Scheme 8. Further, this section has been classified into metal catalyzed and metal free C – H functionalization of alcohols at α-position.
3.1.1. Metal catalyzed α-selective direct functionalization for non-activated alcohols
3.1.1.1. Iridium catalyzed α-functionalization of alcohols
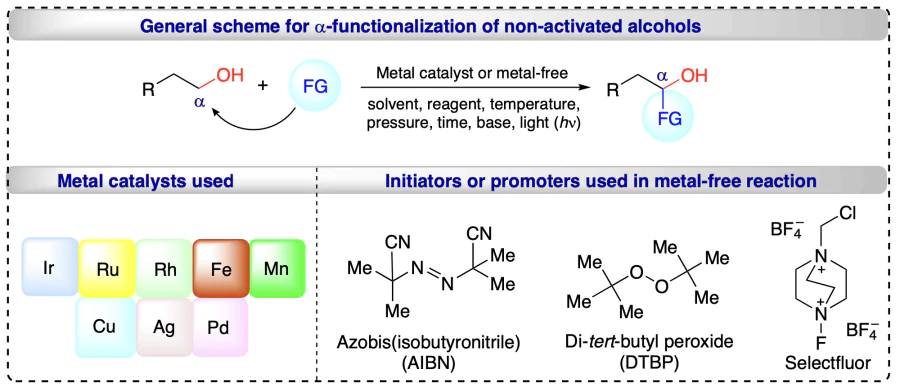
Bower et al.[37] reported the iridium-catalyzed method for the allylation at the carbonyl moiety obtained from the oxidation of alcohols and the reaction proceeding through the alcohol-allene transfer hydrogenation step. This by-product free protocol involves the reverse prenylation (3),b crotylation (6), and allylation (8) of alcohols using [Ir(cod)(BIPHEP)]BARF catalyst via hydrogen autotransfer process with good yields (Scheme 9a–c). Hydrogen auto-transfer was confirmed with the isotope labeling experiments (10) (Scheme 9d). Under standard conditions, the coupling of 1 to 9 resulted in introducing deuterium at both benzylic (>95%) and internal vinylic (85%) positions in 10.
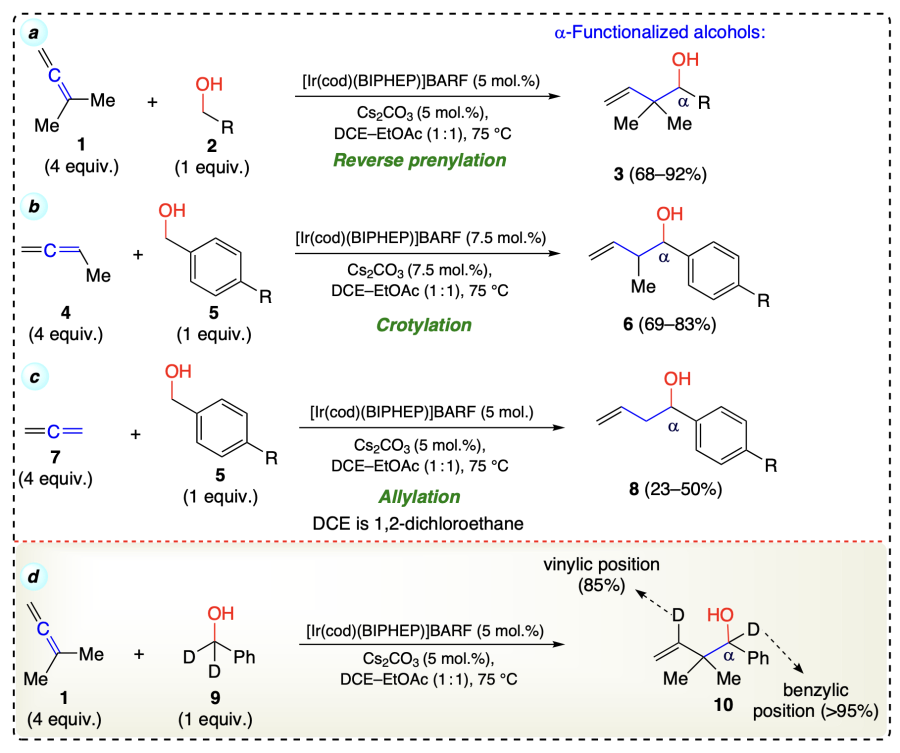
The same group[38] reported another iridium ([Ir(cod)Cl]2) catalyzed protocol involving coupling of 1,3-cyclohexadiene (11) with benzylic alcohols (12) using hydrogen auto-transfer conditions to afford the product with a single diastereomers although the quantities of regio-isomeric products were varied (Scheme 10a)[38]. Initially (without any additive, under standard conditions) there was formation of mixture of diastereomers 13 in products along with the significant amount of regioisomers 13'. The use of Bu4NI as an additive effectively minimise the formation of regioisomers 13' and most impoprtantly, it does not have large affect on the diastereoselectivity of the required products (13). Under the standard reaction conditions, deuterio-9 formed deuterio-14 incorporating 95% deuterium at benzylic position and ca. 15% in the cyclohexene ring. This incomplete deuterium incorporation might be due to fact that deuterium-hydrogen exchange with 11 (12 equiv.) occurred before C – C coupling. But on decreasing the amount of 11 in the reaction incorporation of deuterium increased to ca. 40% in the cyclohexene ring (Scheme 10b). The iridium-catalyzed dehydrogenation of alcohols (16) and hydrometallation of unsaturated reactant (15) leads to a pair of nucleophile and an electrophile. There is an involvement of a six-membered cyclic transition state that arises from iridium σ-allyl species, which further interact with aldehyde and generate the product with syn orientation. Finally, the cleavage of the alkoxide furnished the desired α-functionalized alcohols (18) with the release of a catalyst to participate in the next cycle (Scheme 10c).
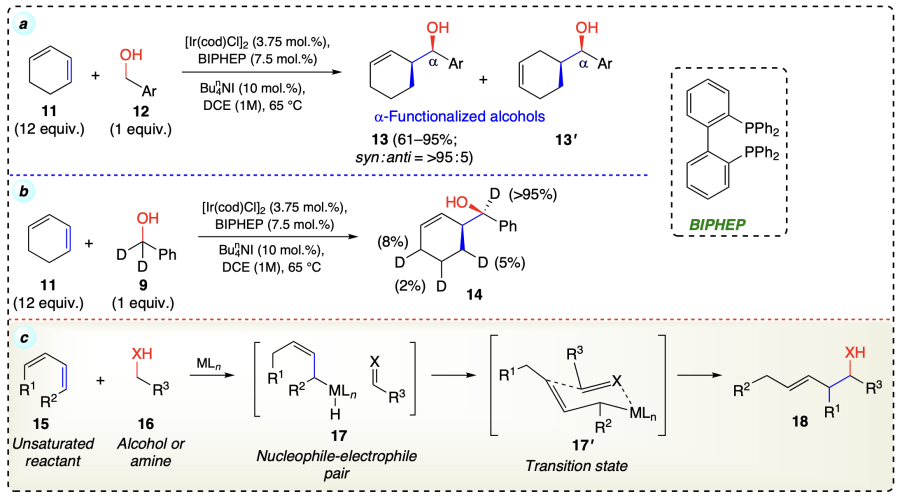
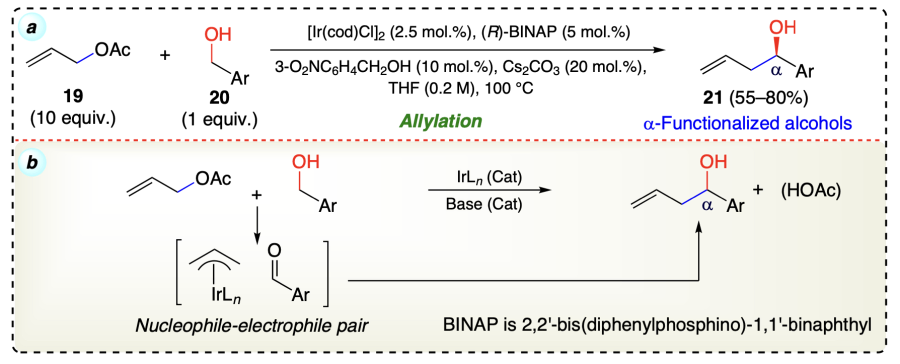
Kim et al.[43] reported the reaction of allyl acetate (19) with benzylic alcohols (20) to yield a C-allylation product (21) with exceptional enantio-selectivity using transfer hydrogenative coupling reaction by iridium catalyst (Scheme 11).
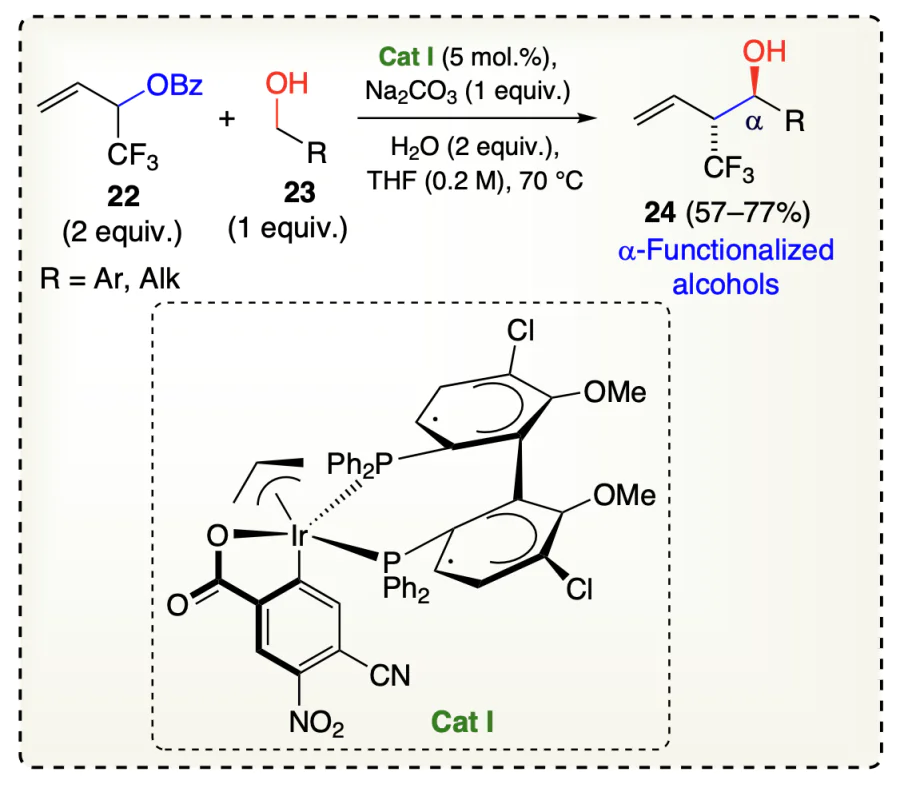
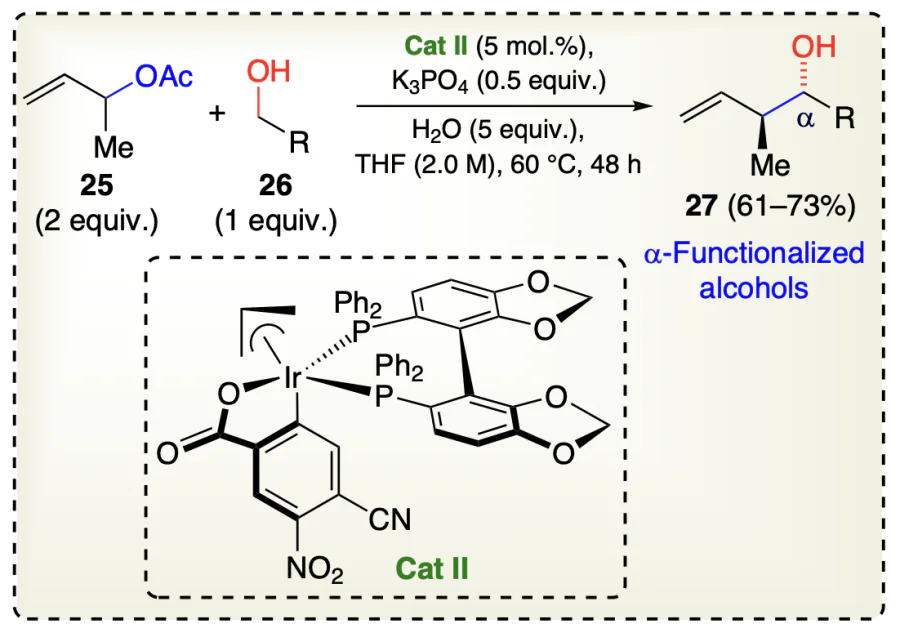
Gao et al.[44] reported a carbonyl allylation reaction for adding carbonyls to the fluorinated olefins (22) using iridium catalyst to get the α-functionalized alcohols 24. Herein, primary alcohols 23 gets dehydrogenated during the reaction sequence which triggered the reductive generation of allyliridium nucleophiles catalyzed by an iridium catalyst Cat I, thus enabling an asymmetric, anti-diastereo- and enantioselective α-trifluoro-methyl functionalized alcohols (24) (Scheme 12). Similarly, Gao et al.[45] reported a highly anti-diastereo- and enantioselective crotylation catalyzed by Ir-based catalyst Cat II in which α-methylated olefins (25) coupled with alcohols (26) for the generation of α-functionalized alcohols (27) (Scheme 13).
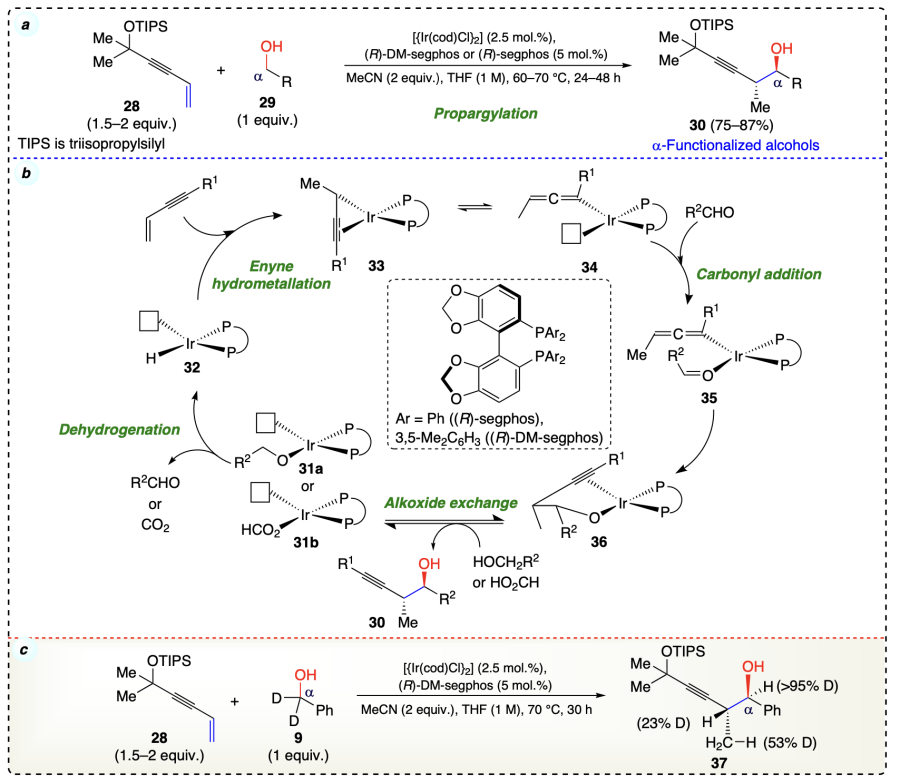
Geary et al.[32] reported a highly anti-diastereo- and enantioselective propargylation from alcohol oxidation level using iridium-phosphine complexes ((R)-segphos or (R)-DM-segphos)[46] to provide the α-functionalized alcohols (Scheme 14a). This reaction was performed without stoichiometric allenyl- or propargyl metal reagents. To start the reaction sequence, the catalyst, ligand, and alcohol substrate combined to give iridium(I) alkoxide (31a or 31b), which on dehydrogenation, afforded aldehyde and iridium(I) hydride (32). In the next step the hydrometallation of enyne molecules (second substrate) with 32 provided the σ-propargyl (33) and allenyliridium (34) species. The coordination of 34 with in situ generated aldehyde gave intermediate 35, which on carbonyl addition furnished the homopropargylic iridium(I) alkoxide (36). Finally, the desired product (30) was obtained by an alkoxide exchange with alcohol along with the regeneration of iridium(I) alkoxide (31) for further catalytic cycle (Scheme 14b). From the deuterium labeling experiment it was inferred that reversible enyne hydrometallation occurred before C – C coupling and a diastereoselective allyliridium species was responsible for carbonyl addition (Scheme 14c).
Woo et al.[33] reported a selective iridium catalyzed α-functionalization of alcohols through the introduction of propargyl group at the benzylic position of alcohols. This protocol involved silyl-substituted propargyl chlorides, which were converted to allenyliridium – aldehyde pairs through a redox-triggered reaction proceeded without any pre-metallated reagents or metallic reductants (Scheme 15a). Mechanistically, the reaction initiated with the conversion of iridium(I) halide (41a) into iridium(I) alkoxide (42a), which further on β-hydride elimination gave iridium(I) hydride (43a). Subsequently, iridium(I) hydride (44) was formed by dissociation of aldehyde. 44 on oxidative addition to second reactant propargyl halide provided propargyliridium(III) (45a) intermediate complex, that equilibrate by another intermediate allenyliridium(III) haptomer (45b). At this point an undesired C – H reductive elimination occurred and alkyne and allene were formed. These compounds together with (41b), transformed into (44) via (42b) and (43b). Alcohols (limiting reagent) promoted here reductive evolution of allenyl-iridium nucleophile from propargyl chloride and isopropanol (exogeneous reductant) compensated the removal of alcohol reactant thus, preventing aldehyde and allenyl iridium molecules from carbonyl-addition. In alternative to this pathway, propargyl (45a) and allenyliridium(III) (45b) species released HCl to generate the analogous propargyl (46a) and allenyliridium(I) (46b) complexes. 46a on association with an aldehyde, formed 47, which on carbonyl addition formed homoallylic iridium(I) alkoxide (48). Finally, intermediate 48 on ligand exchange with an alcohol gave the desired product (40) (Scheme 15b).
Han et al.[47] reported the functionalization of alcohols (50) at the a-position using trimethylsilylated olefins (49) and an iridium catalyst under transfer hydrogenation conditions (Scheme 16a). The carbonyl addition reaction occurred via a chair-like transition state that arises from the in situ generated iridium catalyst Cat III involved in the reaction originating from the (E)-σ-allyl complex. Then, the iridium complex (58), a hexacoordinate 18-electron complex, is formed by the coordination of iridium metal with a homoallylic alcohol. It does not undergo β-hydride elimination because there is no open coordination site. Rest of the mechanism was similar to mechanisms mentioned above (Scheme 16b).
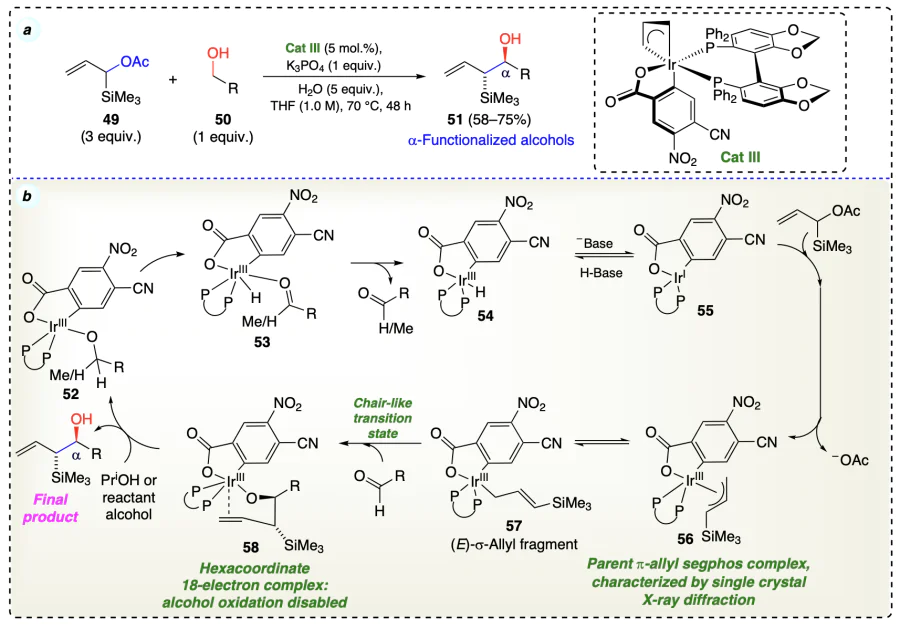
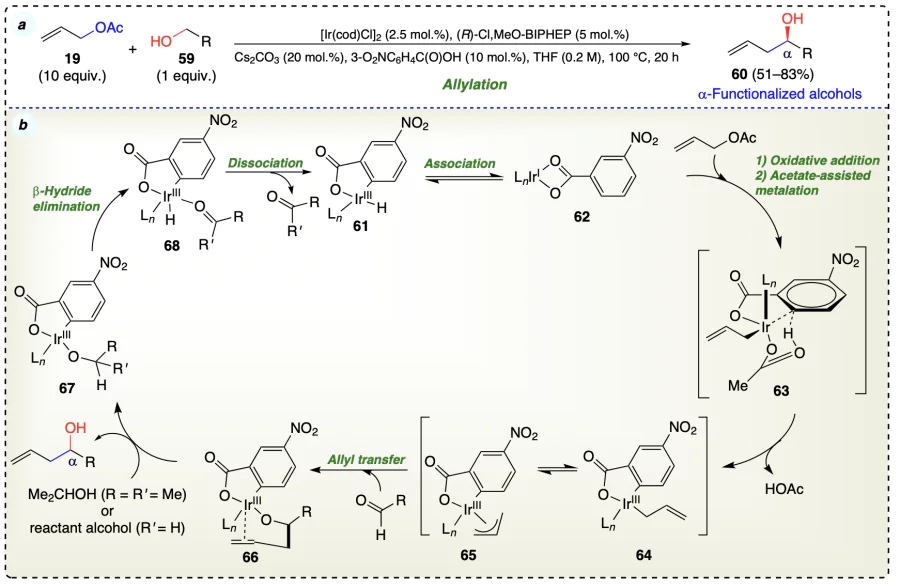
Kim et al.[48] developed an ortho-cyclometallated iridium complex catalyzed enantioselective carbonyl allylation from the alcohols using transfer hydrogenative C – C coupling reaction to afford the desired highly optically enhanced homoallyl alcohols (Scheme 17a). The reaction started with the formation of an iridium carboxylate (61) complex from the chelation of ligand (phosphine) with m-nitrobenzoic acid (used as reagent) with the iridium complex. Complex 61 equilibrate with 62, which on oxidative addition of olefin substrate (19) followed by acetate-assisted metallation via a six-centered transition state (63) provided the σ-allyl C,O-benzoate complex (64). This five-coordinate complex (64) exists in equilibrium with its corresponding π-allyl heptаmer (65). Then, the transfer of allyl species to aldehyde via chair-like transition state provide complex 66, which undergo exchange of homoallyl moiety with alcohol substrates to afford complex 67 along with the generation of desired product. Further, complex 67 underwent β-hydride elimination to form complex 68, which finally regenerated intermediate 61 for the next catalytic cycle (Scheme 17b).
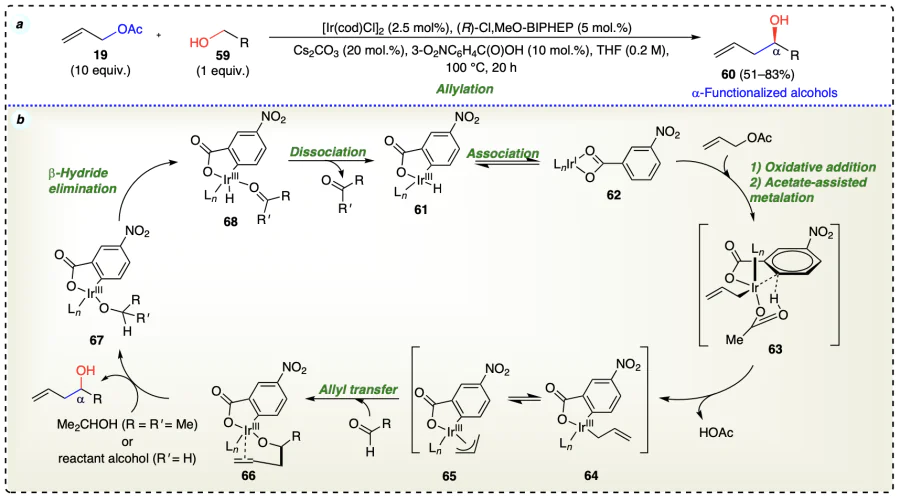
Schmitt et al.[49] displayed the iridium-catalyzed diastereo-selective allylation of ‘Roche alcohols’ (69) containing β-stereogenic center through transfer hydrogenation to provide homoallylic alcohols (70) (Scheme 18a). The reaction proceeded by cyclometalated π-allyliridium complex (71) formed with nitrobenzoic acid.
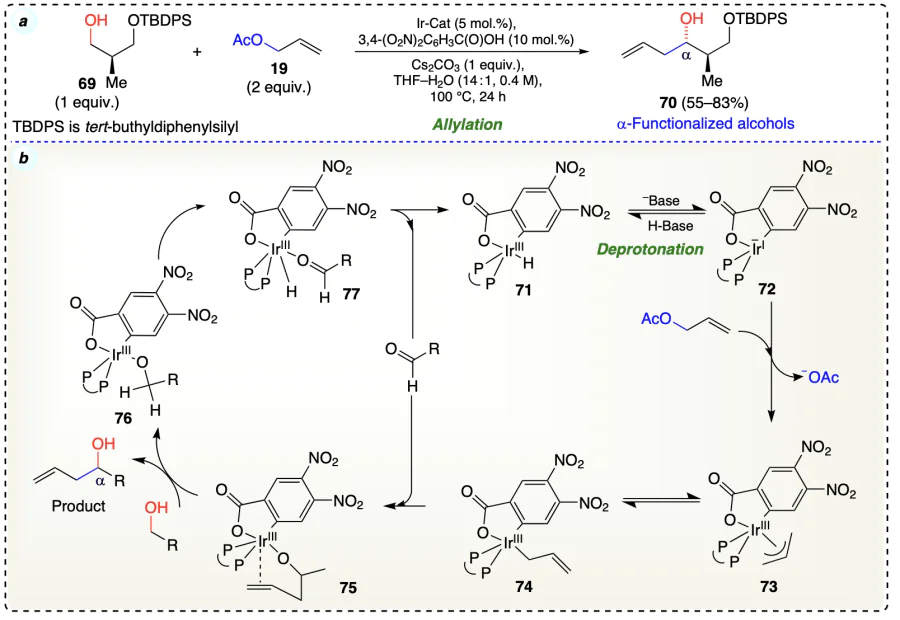
Increased Lewis acidity of iridium strengthened the sceptic interaction amongst iridium center and C – H bond of carbinol, and accelerated carbonyl addition and alkoxide exchange. Also, the removal of proton from the Ir – H species was facilitated due to strong inductive effect of 4-nitro functionality (Scheme 18b).
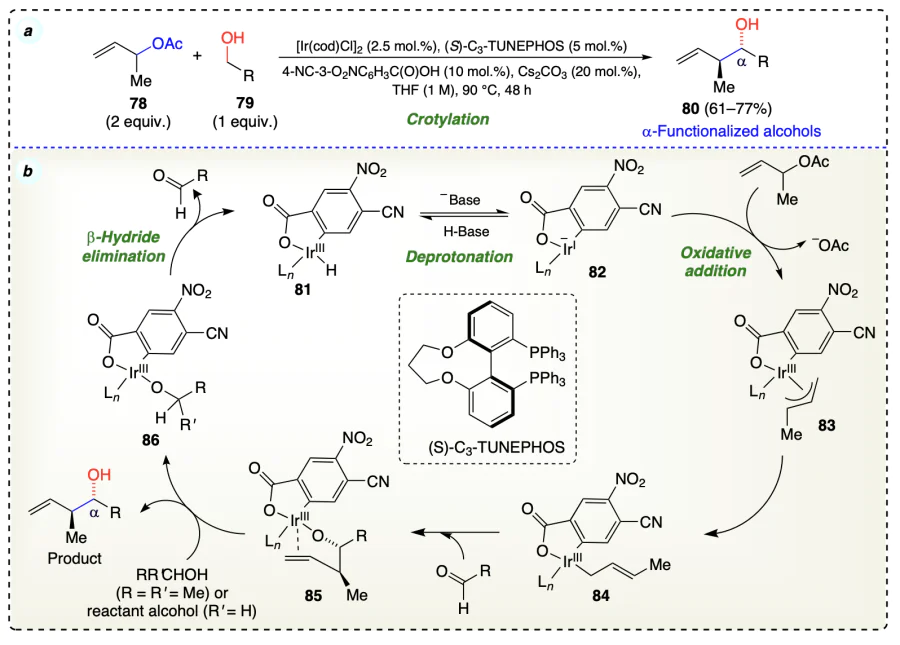
Kim et al.[50] used 4-cyano-3-nitrobenzoic acid with (S)-SEGPHOS and iridium complex for highly regio-, anti-diastereo- and enantioselective crotylation of alcohols (79) occurred via transfer hydrogenative coupling reaction sequence (Scheme 19a). Firstly, Cs2CO3 was used to deprotonate ortho-cyclometalated iridium hydride (81), forming intermediate complex 82. This intermediate then underwent oxidative addition with olefin substrate (78) to give intermediate π-crotyl complex (83). (E)-σ-crotyliridium complex (84) on addition of aldehyde via chair-like transition state generated the anti-homoallyl iridium alkoxide (85) which on alcohol exchange provides complex 86 along with the generation of the desired product. Due to the open coordination site, the β-hydride elimination from 86 regenerated complex 81 for further catalytic cycle (Scheme 19b).
Zbieg et al.[51] also used 4-methoxy-3-nitrobenzoic acid and BIPHEP [2,2'-bis(diphenylphosphino)biphenyl] for the generation of iridium based catalyst and utilised for the α-functionalization of alcohol (88) using diene substrate (87) commenced through the generation of aldehyde-allyliridium pairs via transfer hydrogenation sequence and then underwent carbon-carbon bond formation to give the desired products (89) (Scheme 20a). Herein, the butadiene hydrometallation was kinetically preferred by the s-cis conformer, which formed the anti-π-allyl and further provided the (Z)-σ-allyl stereoisomer (Scheme 20b).
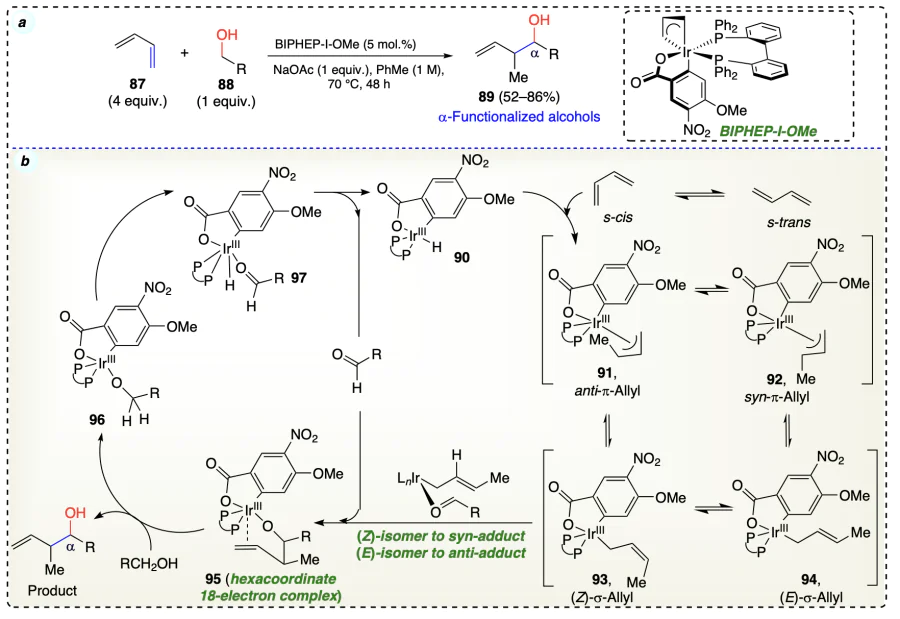
Han et al.[52] reported an iridium catalyst based method for the transfer hydrogenative reaction of allyl acetate with allylic, benzylic and aliphatic alcohols to give a-allylated and crotylated products (Scheme 21a–c). Initially, the olefinic substrate (19) coordinated to the in situ generated iridium complex (105) and formed hexa-coordinated 18-electron intermediate complex (106) which further provide the desired product via the alcohol exchange mechanism. This complex (106) has no open coordination site and did not engage in β-hydride elimination (Scheme 21d). This method was utilised for synthesizing the bryostatins[53, 54] (111) (macrolide lactone) by minimising the number of steps to half as compared to the earlier reports.
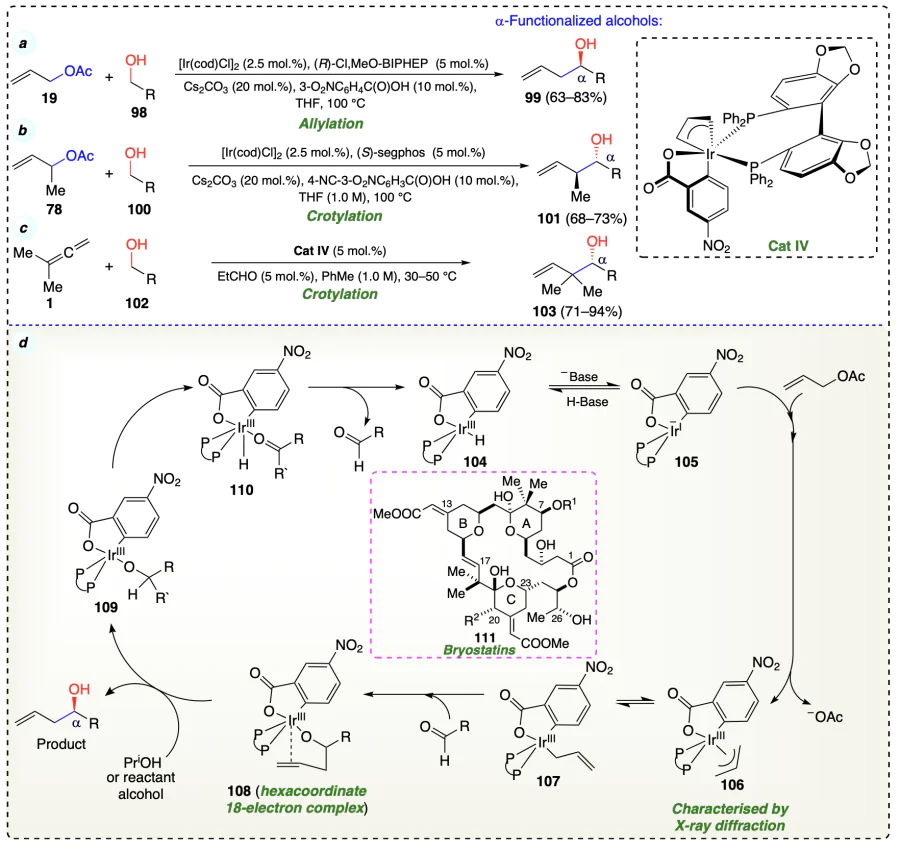
Moran et al.[55] reported first C – C coupled reaction of methanol (113) with allenes (112) to give hydrohydroxymethylation products (114) using iridium catalyst (Scheme 22a,b). The allene hydrometallation was reversible. The dehydrogenation and oxidization of methanol was the rate determining step as determined by the turnover rate of the involved catalyst. These both facts were confirmed by the deuterium labeling and competition kinetics experiments (Scheme 22c–f ).
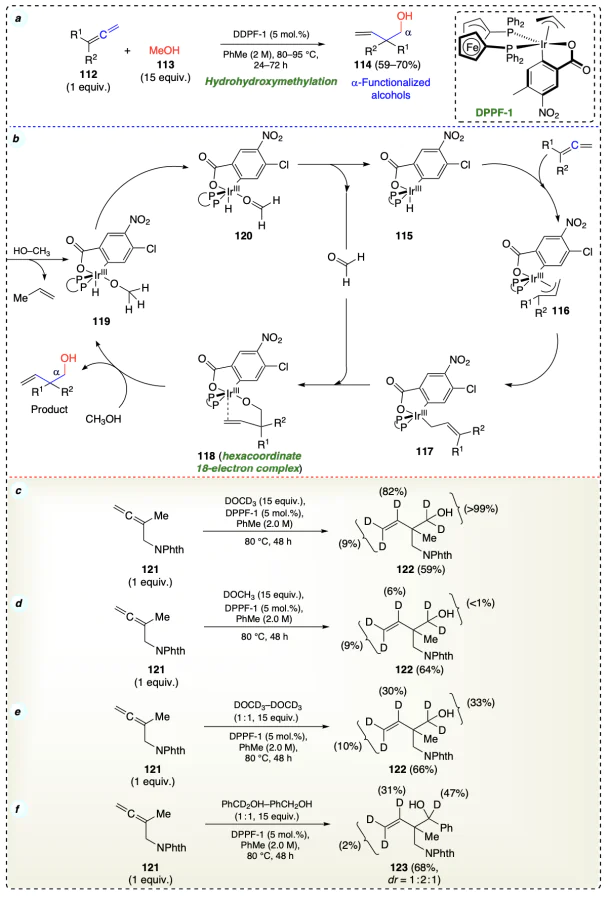
Tsutsumi et al.[56] described the allylation of alcohols (124) at the α-position using iridium [(R)-Ir – I] catalyst (Cat V) with excellent selectivities (Scheme 23a). α-Cyclopropyl allyl acetate generated the kinetically preferred (E)-σ-allyliridium haptomer 129 due to the strain in the allylic chain. Thereafter, this haptоmer was captured stereospecifically by carbonyl addition to form anti-diastereomer of α-functionalized alcohols 126 (Scheme 23b).
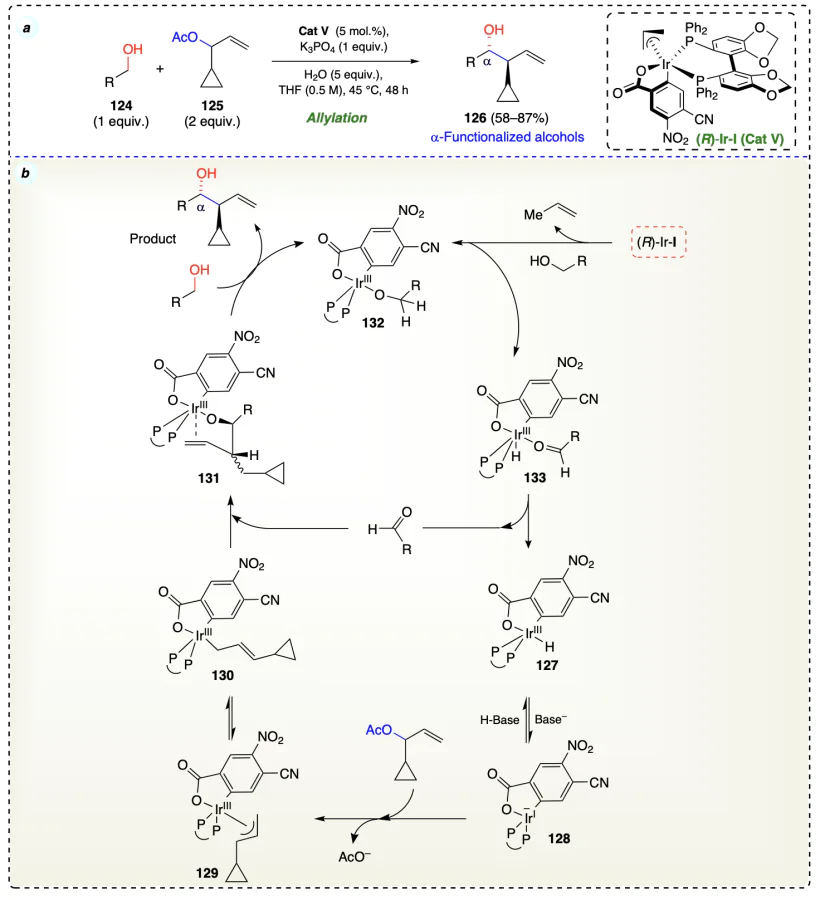
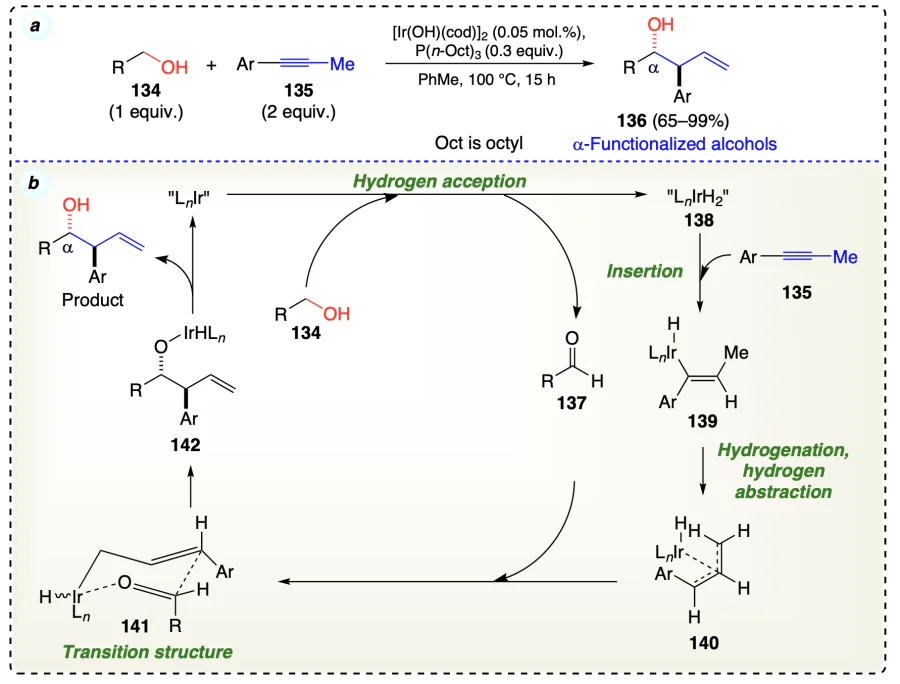
Obora et al.[57] developed an iridium-catalyzed coupling reaction between alcohols 134 and alkynes 135 for the production of secondary homoallylic products via a hydrido(π-allyl)iridium intermediate (Scheme 24a). Experiments showed that the alcohol substrate donates hydrogen to the iridium complex, forming an iridium hydride species. Initially, the iridium catalyst accepted hydrogen from alcohol (134) and gave an aldehyde (137) and an iridium-dihydride species (138). Then, the insertion of the alkyne (135) occurred by Ir – H bond of 138 and formed the hydrido(alkenyl)iridium species (139). Then 139 underwent hydrogenation by Ir – H, abstracted methyl H-atom of alkyne substrate (135), and provided hydrido(π-allyl)iridium intermediate (140), which further reacted with generated carbonyl species 137 (from alcohol dehydrogenation) and afforded the final product homoallylic alcohol 136 via a transition (six-member) state (141) (Scheme 24b).
3.1.1.2. Ruthenium catalyzed α-functionalization of alcohols
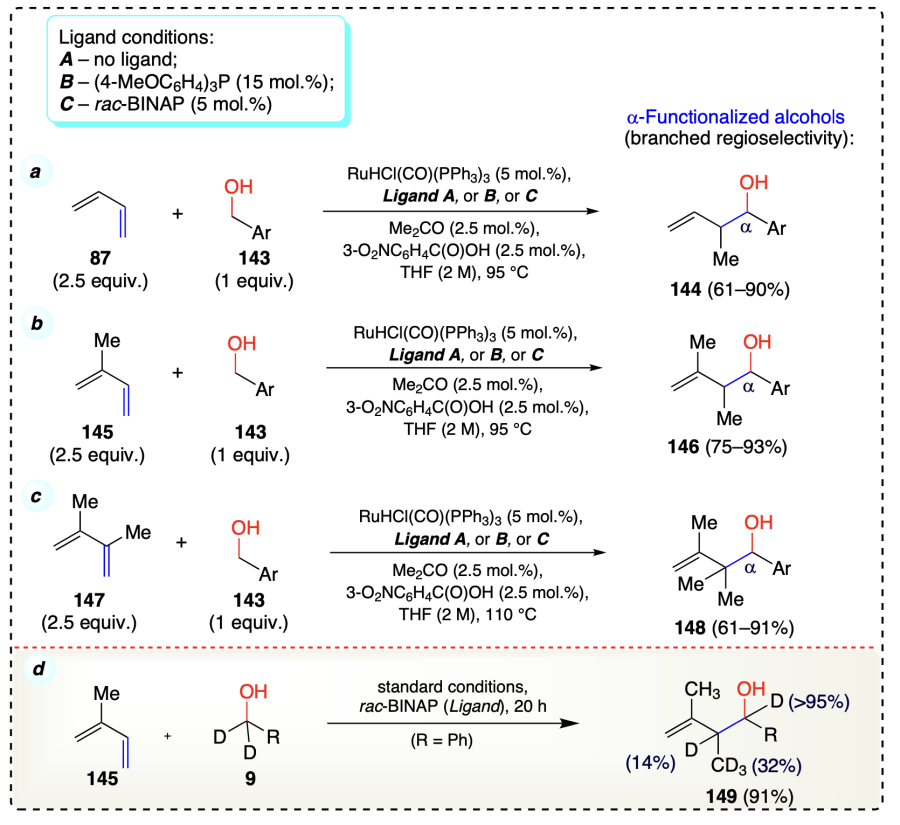
Shibahara et al.[58] reported ruthenium [RuHCl(CO)(PPh3)3] catalyzed C – C bond formation through hydrogen auto-transfer strategy by the reaction of 1,3-butadienes (87, 145, 147) with alcohols (143) to give, by-product free, carbonyl allylation products (144, 146, 148) (Scheme 25a–c). Here also the similar mechanism through the generation of a pair of nucleophile-electrophile and a six-membered cyclic transition state[38] was followed for the transfer hydrogenation reaction. Deuterium-labeling experiments confirmed the partial deuteriation of product (149) due to reversible hydrometallation of isoprene (Scheme 25d).
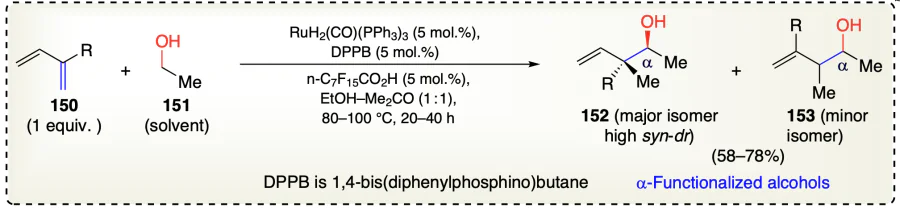
Han et al.[59] reported a reaction of alcohol (151) with 2-functionalized dienes (150) to afford neopentyl homoallylic alcohols with anti-configuration (152 and 153) under transfer hydrogenation conditions using ruthenium based catalyst (Scheme 26). This method enabled a diastereoselective synthesis under catalytic conditions without using pre-metallated nucleophiles. The coupling reaction primarily occurred at C2-position in diene enabling the generation of diastereoselective quaternary carbon centers. The same group[60, 61] earlier observed the generation of nucleophile-electrophile pairs that were engaged in C – C coupling via hydrogen exchange between alcohols and unsaturated substrates catalyzed by Ir or Ru complexes. But this study was advantageous in directly activating the alcohols such as methanol and ethanol for C – C coupling with π-unsaturated starting material.
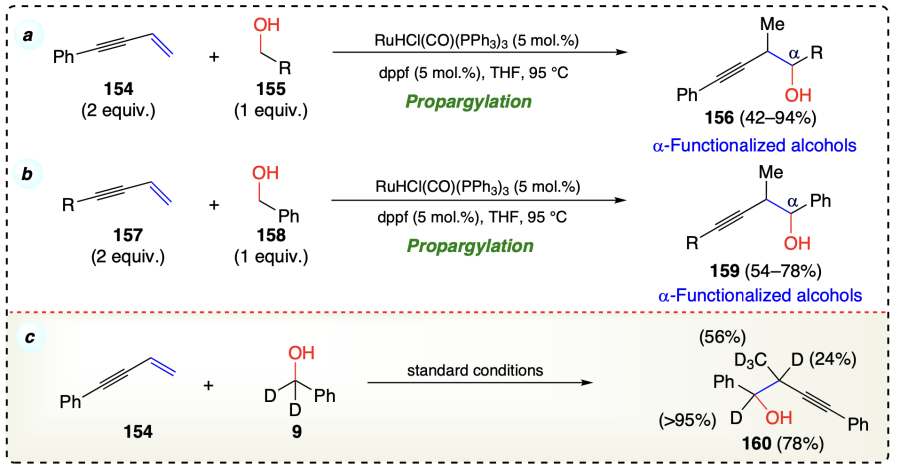
Patman et al.[39] reported a C – C bond formation via carbonyl propargylation of 1,3-enynes (154, 157) with alcohols (155, 158) to give methyl homopropargyl alcohols (156, 159) under transfer hydrogenation conditions using ruthenium based catalyst {[RuHCl(CO)(PPh3)3]}, 1,1'-bis(diphenylphosphino) ferrocene (dppf) in THF at 90 °C (Scheme 27a,b). The current process also followed the general mechanistic steps, including ruthenium hydride species generation via alcohol dehydrogenation, an allenyl metal-aldehyde/nucleophile-electrophile pair formation by enyne hydrometallation and finally, the carbonyl addition with propargylic transposition. Isotopic labeling studies under standard conditions suggested that this protocol involves reversible olefin-hydrometallation (Scheme 27c).
Patman et al.[40] reported the reaction of alkynes (154, 161, 168) with alcohols (162, 165) employing ruthenium catalyst via transfer hydrogenation conditions to achieve the vinylation through direct C – H bond functionalization of alcohols. Herein, catalytic amount of Ru(O2CCF3)2(CO)(PPh3)2 complex accomplished the reaction without any additional ligand and aiming for the altered regioselectivity of the C – C coupling reaction (Scheme 28).
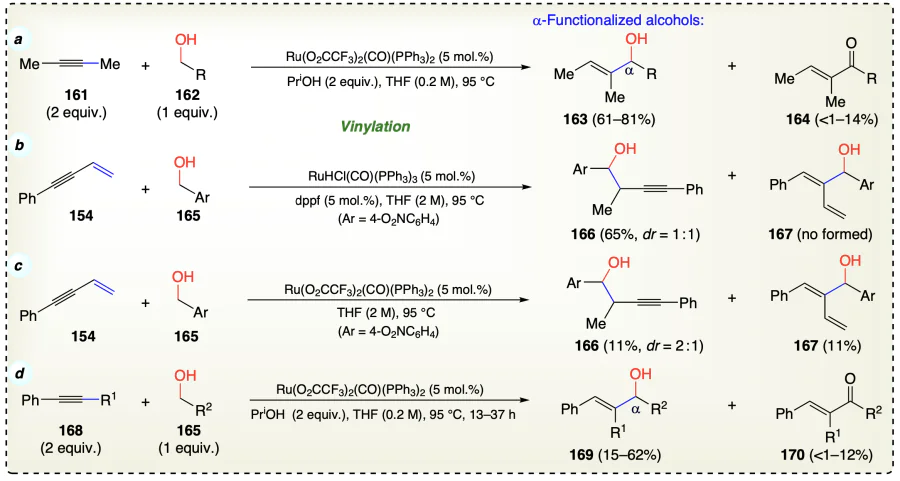
Zhang et al.[62] acomplished the C – C bond formation between tertiary (171, 174), and primary alcohols (172, 175) with ruthenium catalyst and base via activating the sp3 C – H bonds of primary alcohols to give desired α-alkylated products (173, 176) (Scheme 29a,b). The alcohol-alcohol coupling is advantageous over olefin-alcohol coupling due to the easy availability of alcohols. Firstly, Lewis acid-elevated dehydration of alcohol kinetically favored the formation of the alkene intermediate (177) followed by activation C – H bond neighbouring to the alcoholic oxygen in primary alcoholic moiety which consequently generated the radical pair 178 and then free-radical addition gave 179. Next, the ruthenium hydride ([Ru] – H) underwent reductive attack stereoselectively from the less steric hindered side in the transition state (180) and resulted in the formation of a diastereoisomer syn-173a(176a) as a significant diastereoisomer and freeing the Ru catalyst for the next cycle (Scheme 29c). It is important to note that, yield was lower when secondary alcohols were employed (42%) than tertiary alcohols because tertiary radical species is more favourable than secondary one. The deuterium labeling experiments clearly discarded the previous (I) and (II) methods for this protocol and favoured cross-coupling process (Scheme 29d).
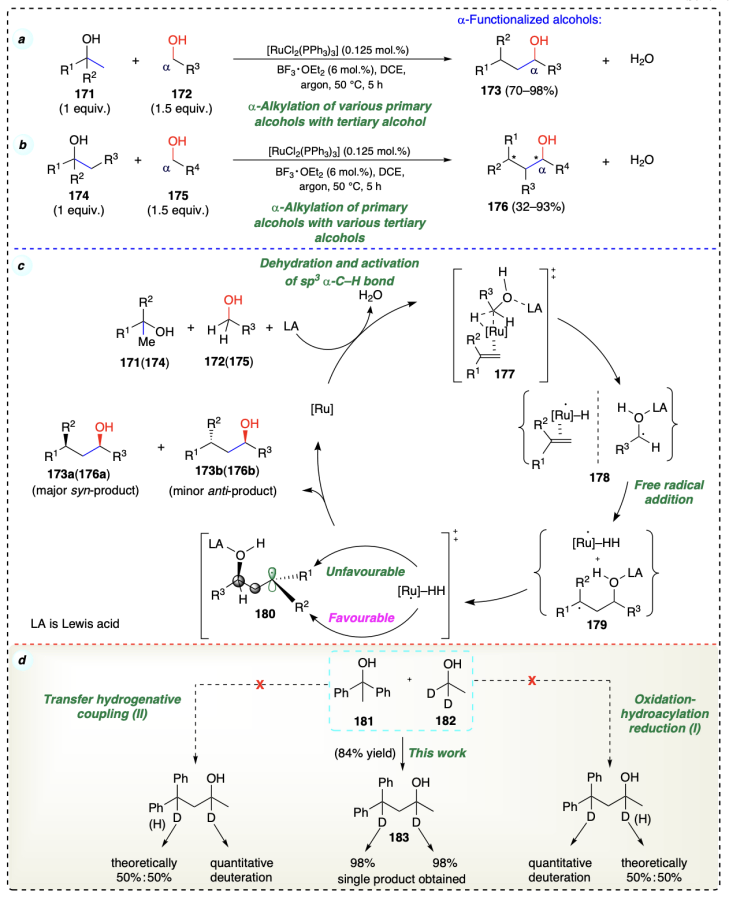
Sam et al.[34] reported a redox-triggered, Ru catalyzed highly selective carbonyl allylation of fluorinated alcohols with allenes to get α-functionalized alcohols, wherein, the alcohol dehydrogenation occurred via β-hydride elimination (Scheme 30a). Initially, there is a formation of nucleophilic allylruthenium complex by engagement of Ru – H species (187) obtained from [H(Cl)Ru(CO)(PPh3)3] and [bis(diisopropylphosphino)ferrocene] (dippf) during the process of hydrometallation of allene[63]. Next, kinetically preferred hydrometallation intermediate (188b) was observed at the π-face of allene neighboring to methyl functionality and rapid isomerization of π-allyl complex provide a π-allyl intermediate (188a), which is thermodynamically more stable complex.
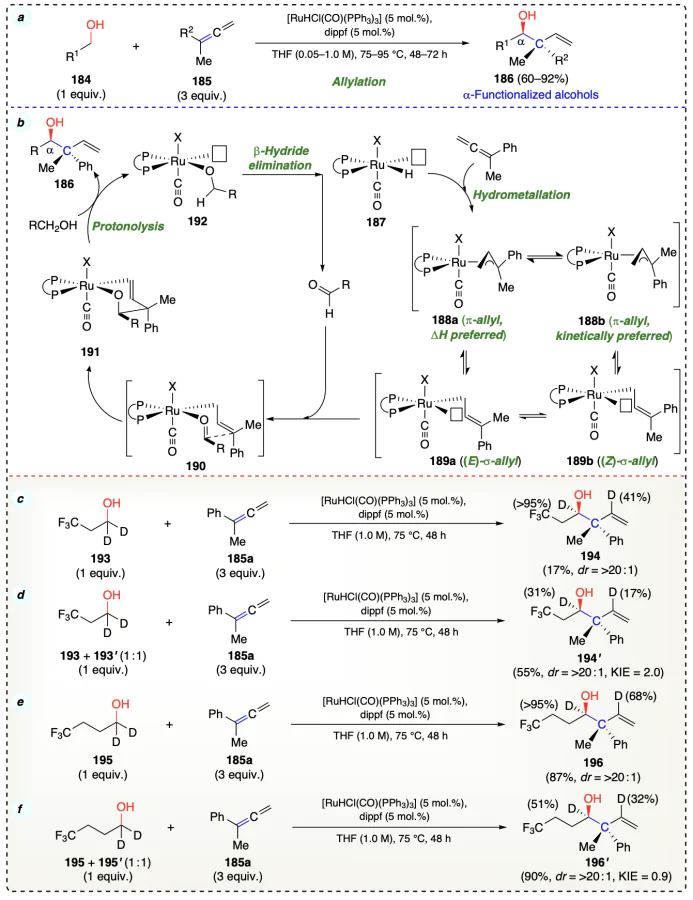
Then the (E)-σ-allylruthenium intermediate (189a), via a chair-like transition state, formed the anti-diastereomer (190) and further isomerizes to intermediate 191. Finally, the intermediate 191 on protonolysis with alcohol yielded the desired product 186. The complex 192 formed during the final step, further participate in the reaction (Scheme 30b). According to competition kinetics in redox-triggered couplings of 193 and 195 with 185a it was evident that, due to low yield, incomplete deuterium incorporation, and KIE of 2.0, for 194 β-hydride elimination was turnover limiting step. On the other hand, in 195, CF3 moiety being far from carbinol position, KIE of 0.9 was observed; thus, for 196, carbonyl addition was turnover limiting step (Scheme 30c–f ).
McInturff et al.[64] reported that the chiral anions inverted the diastereoselectivity and enantioselectivity of butadiene hydroxyalkylation for carbonyl syn-crotylation (Scheme 31c). The usage of H8-BINOL and TADDOL-acids imposed the opposite selectivities in products despite using single chiral phosphine enantiomer as ligand (Scheme 31a,b). Hydrometallation of s-cis conformer of alkene substrate favored the anti isomer, which resulted in unusual syn-diastereoselectivity. Isomerization rate is thought to be slowed down by the syn-π-crotylruthenium due to the bulky TADDOL-based phosphate ion. So, the kinetic stereoselectivity of the hydrometallation process was preserved, and the syn-diastereomer was formed by the (Z)-σ-crotylruthenium haptomer through closed Zimmerman-Traxler-type transition sate (Scheme 31d)[65].
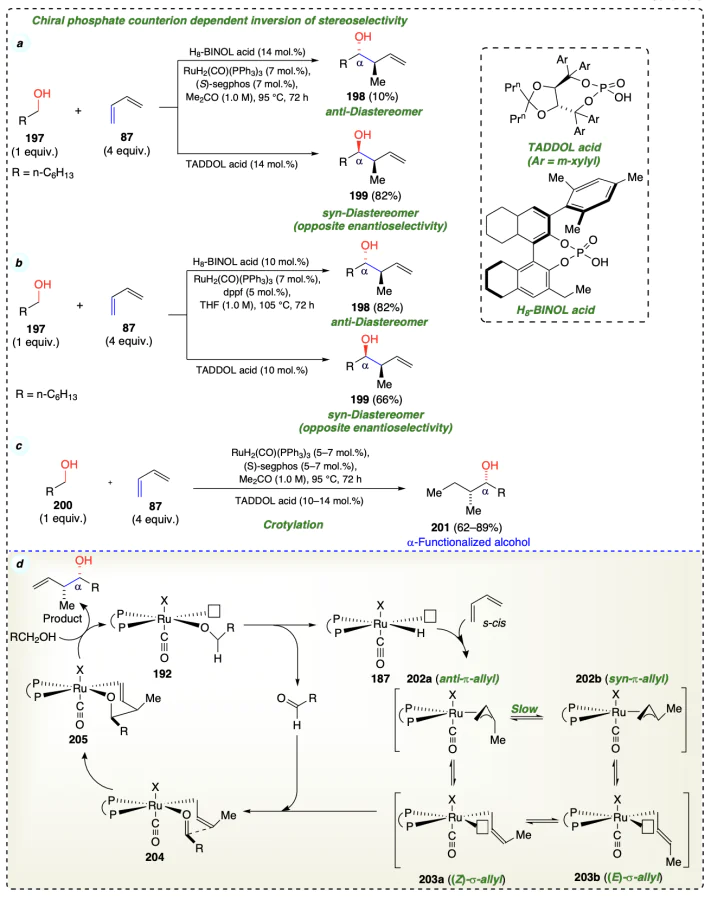
Zbieg et al.[66] reported that primary alcohols (208) and 2-silyl-butadienes (207) can be converted to crotylated carbonyl compounds with high syn-diastereo- and enantioselectivity using chirally modified ruthenium catalysts (Scheme 32a). This protocol avoids stoichiometric by-products and cryogenic conditions and was applicable towards synthesizing antisyn-stereotriads (212a,b) found in variegated natural molecules such as polyketides (Scheme 32c). Initially, the addition of a carbonyl group to a σ-bound allylruthenium complex A through a transition state produced a complex B intermediate. Then, this complex on alcohol exchange formed a pentacoordinated complex C. Finally, the vacant coordination site enabled dehydrogenation, due to which an aldehyde was formed, and Ru – H species D was regenerated (Scheme 32b).
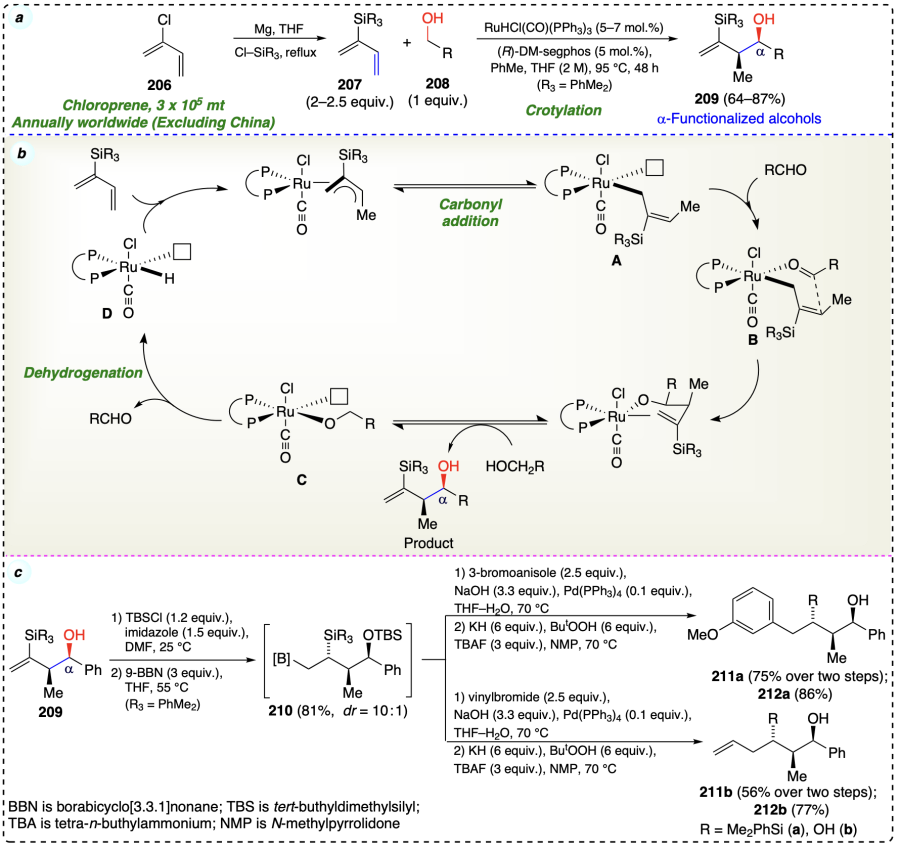
Zbieg et al.[67] also reported a ruthenium-catalyzed transfer hydrogenation reaction wherein 1,1-disubstituted allene (213) and alcohols (214) were involved in a redox-triggered formation of desired hydrohydroxyalkylation products (215) with complete branched regioselectivity (Scheme 33a). Also, excellent anti-diastereoselectivity (4 : 1 to > 20 : 1) was observed due to the Curtin – Hammett effects. The reaction did not involve any pre-metallated nucleophiles or generate stoichiometric by-products. The competitive experiments suggested that the alcohol dehydrogenation step occurred reversibly and swiftly before carbonyl addition, the rate-limiting step (Scheme 33b). The deuterium labeling suggested the β-hydride elimination as turnover-limiting step (Scheme 33c) and competition kinetics evidented that oxidation of alcohol step was not turnover-limiting (Scheme 33d).
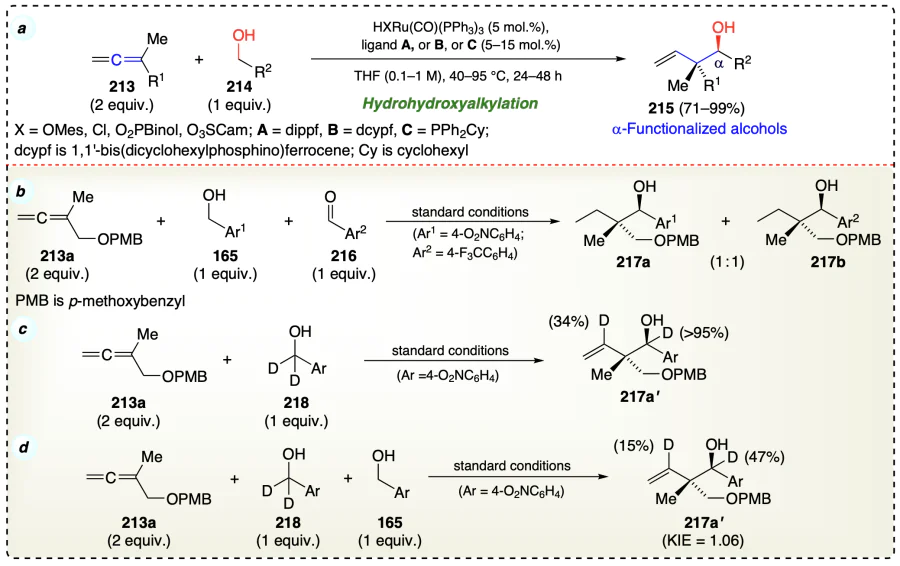
Zbieg et al.[68] also reported direct carbon-carbon bond formation between alcohol (220) and allenamide (219) to provide anti-1,2-amino alcohols (221) using ruthenium based catalyst (Scheme 34a). This method was also used for C – C coupling of 1,1-disubstituted allenes (222) to alcohols (223) to yield desired products (224) with good yields (Scheme 34b). The catalyst was generated by reacting bis(diisopropylphosphino)ferrocene with RuHCl(CO)(PPh3)3 and it conquers the limitation of earlier reported ruthenium-based catalysts[69].
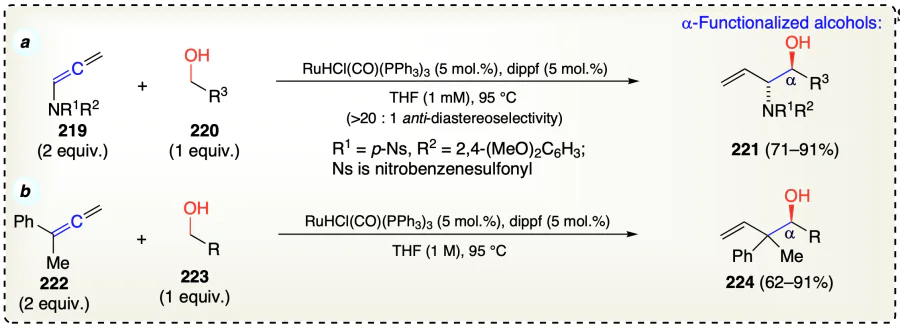
Chen et al.[70] described the prenylation and geranylation reactions of isoprene (226) and myrcene (228), respectively with 3-hydroxy-2-oxindole (225) using Ru-catalyst and under transfer hydrogenation conditions (Scheme 35a). The Ru(0) catalytic specices was synthesized by reaction Ru3(CO)12 with phosphine reagent [tricyclohexylphosphine (PCy3)]. Reaction followed the Pauson – Khand type sequence[71, 72]. The oxidative reaction of diene with carbonyl species delivered the Ru(II) oxametallacycle (230) and further isomerized σ-allyl haptomer (231) which is more stable species. Preferentially, (Z)-stereo-isomer was formed as evident from the olefin geometry in (231). Oxindole (225) on protonating 231 formed Ru(II) alkoxide (232) which generates oxo-(225) and allylruthenium(II) hydride (233) via β-hydride elimination. Then, hydride species (233), on reductive elimination, provided the final α-functionalized alcohols and zerovalent ruthenium species (Scheme 35b). Noteworthy, the reductive elimination step occurred faster than the π-facial interconversion of allyls to control the olefin geometry.
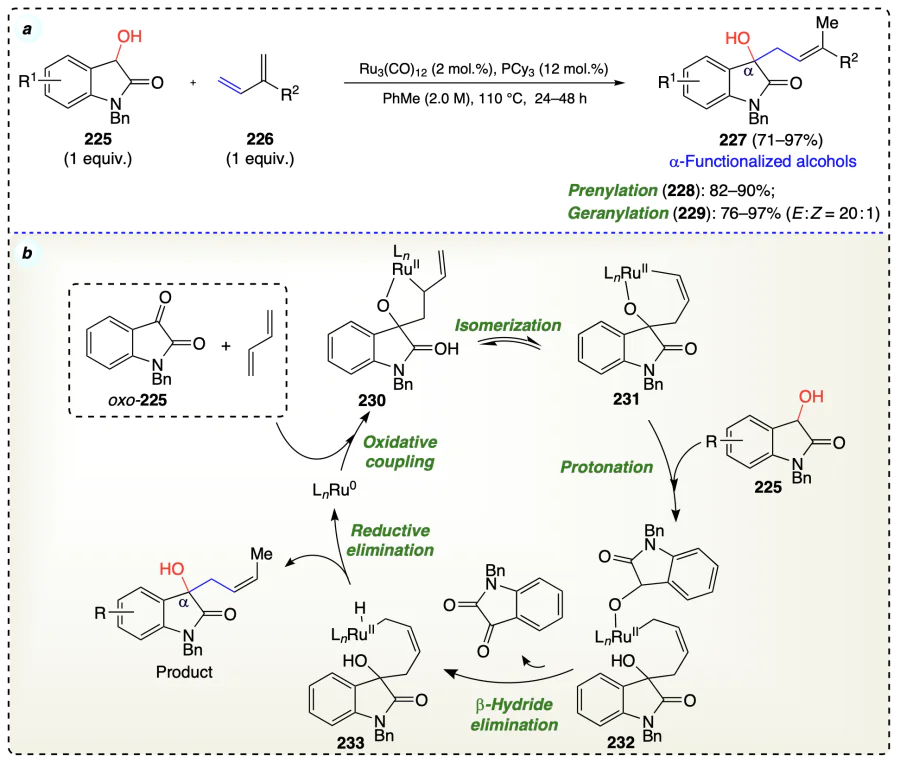
Zbieg et al.[73] described a ruthenium-catalyzed highly stereoselective carbonyl crotylation of alcohols (234) without using any pre-metallated reagents (Scheme 36a). Initially, butadiene (87) on hydrometallation delivered π-allylruthenium complex (237) followed by stereospecific carbonyl addition through σ-crotyl ruthenium haptomer via a closed transition structure. After that, two isomers (E)- and (Z)-σ-crotylruthenium isomers (238a and 238b), get partitioned, which provided the homoallylic ruthenium alkoxide (240) and resisted the dehydrogenation because of the absence of empty ruthenium coordination sites. This alkoxide exchange with alcohol reactant afforded the desired crotylation product along with the generation of a pentacoordinate ruthenium alkoxide (241). At this point, the vacant coordination sites allowed dehydrogenation to occur, forming an aldehyde and regenerating ruthenium hydride, thus completing the catalytic cycle (Scheme 36b).
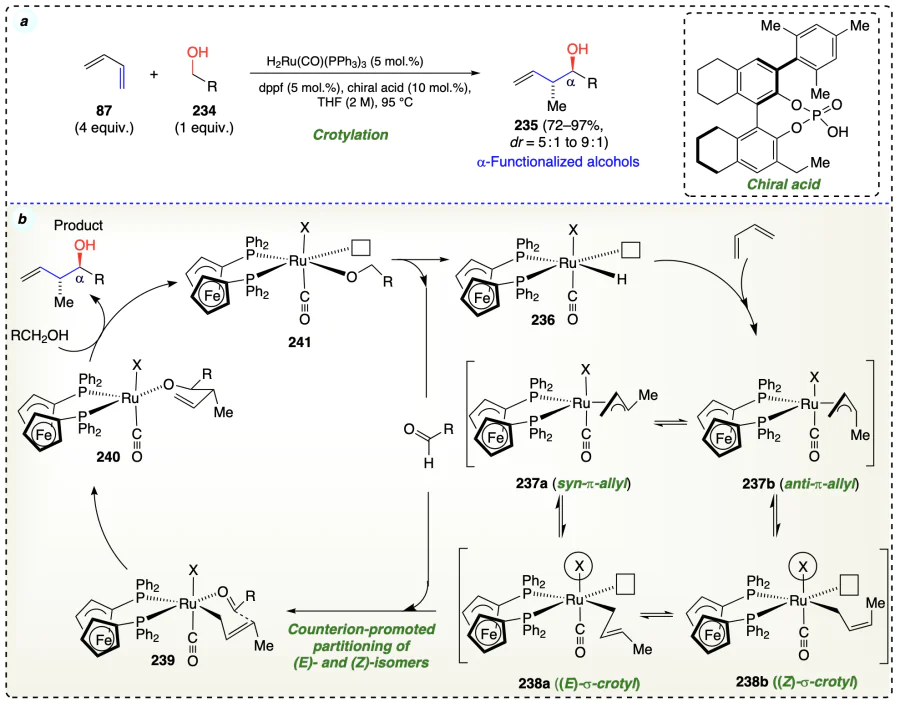
Saludares et al.[74] synthesized a ruthenium complex bound to iodide using JOSIPHOS for the reaction of primary alcohols (242) with gaseous allenes (7) to produce homoallylic alcohols (243) (Scheme 37a). The developed method was also applied to synthesize various polyketides such as spirastrellolide B and F (250) in few steps (17 vs 7 steps) than the previously reported methods (Scheme 37d). Deuterium labelling experiments showed that the breaking down an alcohol (dehydrogenation) triggers a reaction that forms a fleeting molecule (a transient allylruthenium-aldehyde pair). This short-lived molecule then participates in a subsequent reaction (carbonyl addition) (Scheme 37c). In a β-hydride elimination reaction, the pentacoordinate ruthenium(II) alkoxide complex (244) loses a hydrogen atom from the second carbon (β-position) of the alkoxide ligand. This cleavage results in the formation of an aldehyde and a ruthenium(II) hydride complex (245). Ruthenium(II) complexes, like alkoxide (244), are octahedral d6 metal ions with vacant dx2−y2 orbitals. This specific arrangement makes it very easy for these complexes to eliminate a hydrogen atom through β-hydride elimination. Allene (7) underwent hydro-ruthenation by the ruthenium (II) hydride (245) to form π-allylruthenium(II) complex (246). A special type of attachment between a ruthenium molecule and a molecule with an allyl group leads to a reaction involving a six-sided intermediate structure. This reaction results in a ruthenium(II) alkoxide molecule (247) with a homoallylic structure. Ruthenium compound (247) is replaced by an alcohol molecule and this process is sped up by TFE to complete the reaction cycle. (Scheme 37b). The isotope labelling experiment suggest that the deuterium stays on the alcohol group (carbinol position) in deuterio-248 which reveals that removing a hydrogen atom from alcohol (dehydrogenation) is difficult because alcohol (homoallylic olefin) forms a special bond with the molecule (chelation) like in complex (247). This special bond blocks the β-hydride elimination (Scheme 37c).
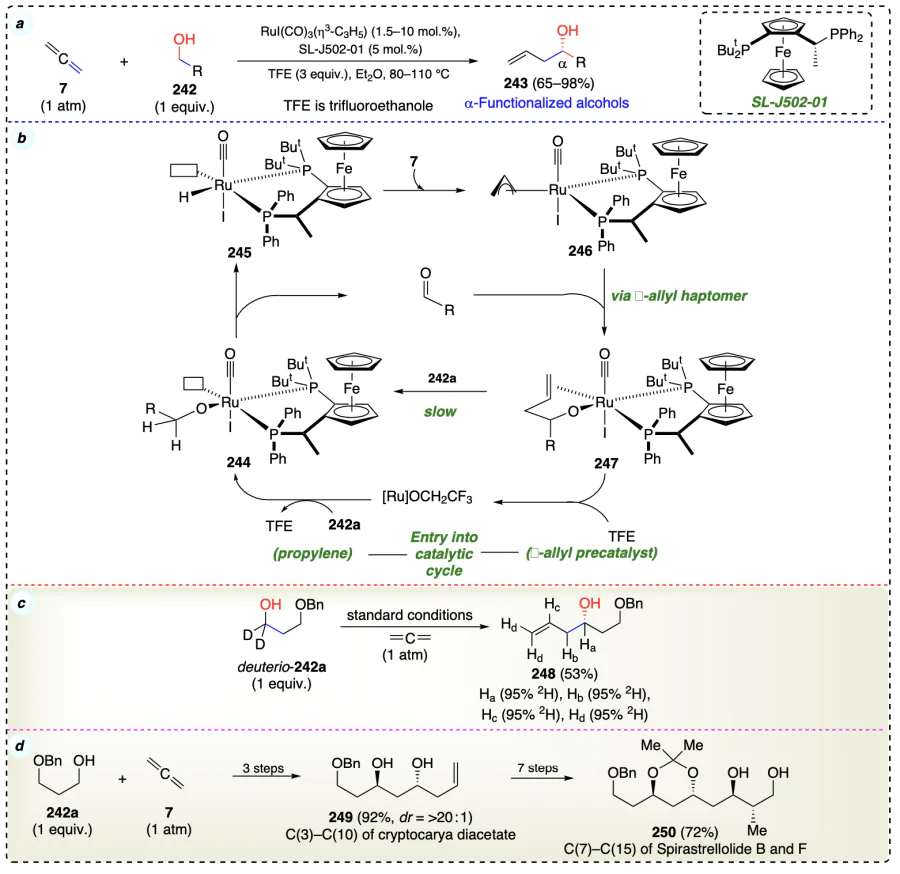
Dubey et al.[75] described a method for the carbonyl addition to combines alcohols (proelectrophiles) (252) with nonconjugated dienes (acting as allyl metal pronucleophiles) (251) using ruthenium complex in achieving high regioselectivity, anti-diastereoselectivity, and enantio-selectivity in α-functionalized products (Scheme 38a). Author introduced a new methods (demonstrated by synthesizing pironetin, 261) that simplify the process of synthesis of certain molecules (butyrate substructures) found in many natural products (type I polyketide natural products). These new methods are much faster (12 steps vs 4 steps) than earlier methods (Scheme 38d). Deuterium labeling studies revealed the incorporation of deuterium (259) at every diene-derived carbon (Scheme 38c). It support a mechanism where the addition of a metal atom (hydrometallation) to a diene (a molecule with two double bonds) is aided by a specific intermediate complex (254). This complex 254 can switch between two forms: one with a six-sided ring structure (fluxional olefin-chelated homoallylic alkylruthenium complex) and another with a five-sided ring structure (η1 form). This switching allows for the removal of a hydrogen atom (β-hydride elimination) to complete the addition process (Scheme 38b).
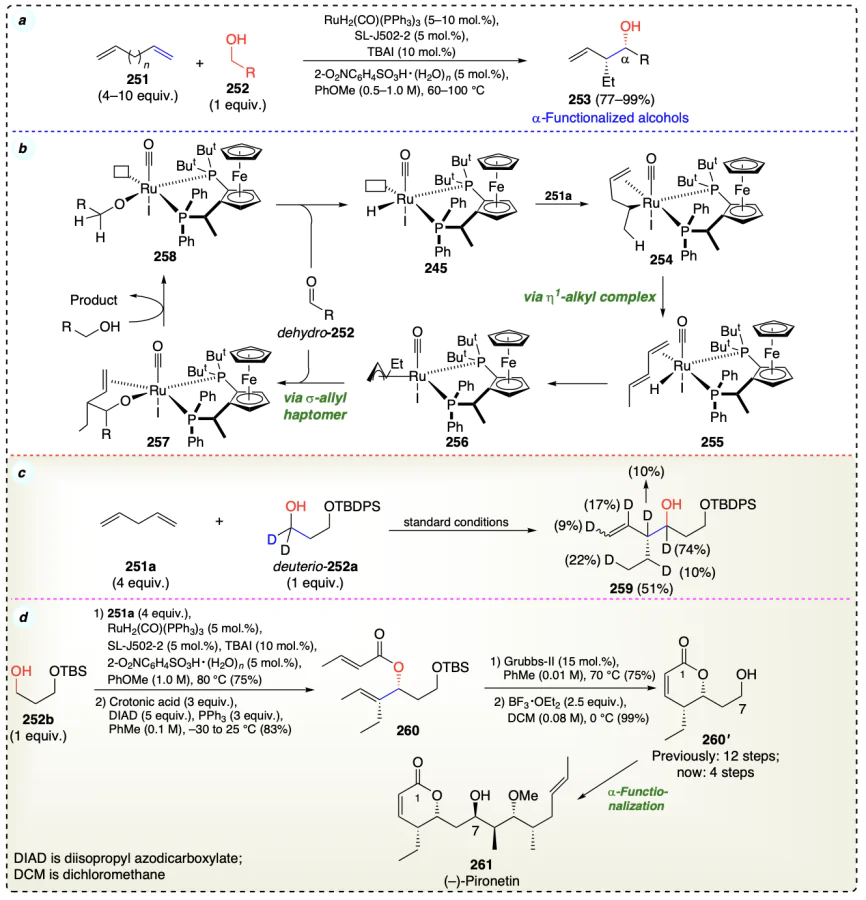
Ortiz et al.[76] reported the asymmetric ruthenium-JOSIPHOS catalyzed carbonyl vinylations accomplished via alcohol dehydrogenation. This transformation leverages a ruthenium complex bearing a JOSIPHOS ligand to facilitate the stereoselective conversion of primary alcohols (alkan-1-ols) (262) and butyne (161) to chiral allylic alcohols (263) (Scheme 39a). Author employs control experiments, specifically those utilizing deuterium labeling, to elucidate reaction mechanism (Scheme 39c). This process involves the hydrogen auto-transfer strategy to achieve high atom economy. Ruthenium-catalyzed hydrometallation of 2-butyne leads to the formation of a vinyl ruthenium complex (265). Activation of a carbonyl group via Lewis acid coordination with a ruthenium-aldehyde complex facilitates nucleophilic addition, leading to the formation of a secondary ruthenium alkoxide intermediate (266). TFE-mediated alkoxide exchange facilitates the liberation of the allylic alcohol and furnishes the primary ruthenium alkoxide intermediate (267). Subsequent β-hydride elimination liberates the aldehyde product and regenerates the ruthenium hydride catalyst (264), thereby completing the catalytic cycle (Scheme 39b).
3.1.1.3. Miscellaneous metals catalyzed α-functionalization of alcohols
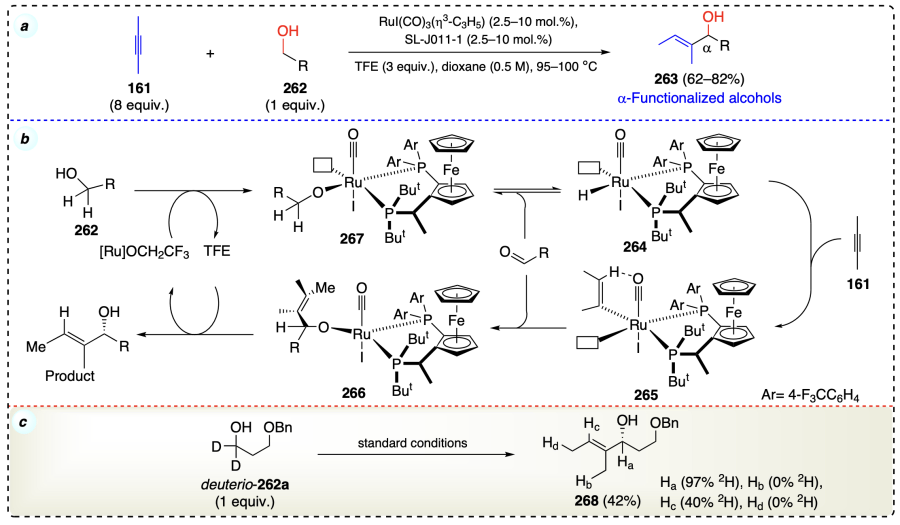
Shi et al.[77] reported a rhodium-catalyzed and Lewis acid-assisted coupling of aliphatic alcohols (270) with olefins (269) to access secondary alcohols (271) (Scheme 40a). Mechanistically, the Lewis acid coordinated with the alcoholic oxygen (272) during the reaction. Next, the coordination of alcoholic C – H bond to Wilkinson catalyst took place via oxidative addition, and subsequently, further olefins (269) coordinated with the catalyst to form a Rh(III) complex (273). After that, complex 273 generated a free radical pair (274, 275). The alcohol combined with alkene to form radicals 276 and 277; this combination was known as Kharasch radical reaction. Then, hydrogen abstraction from 276 generated the desired product and liberated catalyst 279, which will be recycled in the next cycle (Scheme 40b). PPh3 inhibited the dissociation and ligand exchange in reaction. The involvement of radical mediated mechanism was also proven by the deuterium-labeling cross-over experiments (Scheme 40c).
Zhang et al.[78] presented FeCl3-catalyzed carbon – carbon bond formation reaction between alcohols and alkenes [C(sp3) – C(sp3) bonds] (Scheme 41a). Hydroacylation was not observed during the reaction using aldehyde, PhCH2CH2CHO, and alkene as substrates under standard reaction conditions which ruled out the coupling pathway through transfer-hydrogenative coupling/oxidation or reduction. Activation of α-С(sp3) – H bond in 282 using Fe-catalyst by cleavage of the bond generated the intermediate 285. Then, a radical pair 286 was formed, which underwent free-radical addition and simultaneous dissociation to form [Fe]IV – H along with free radical (287). This metal hydride underwent hydrogen transfer in the last step to afford α-functionalized alcohol as well as regenerate the active catalytic Fe(III) species to participate further in the reaction (Scheme 41b). Furthermore, the deuterium-labeling experiment confirmed a distinctive intermolecular hydrogen transfer from alcohol to alkene (Scheme 41c). A crossover-experiment suggested that this protocol proceeded discretely intermolecularly (Scheme 41d). This iron-catalyzed reaction occurs through α-C – H bond homolysis in alcohol rather than ionic dissociation. Prevention of reactions with radical scavengers (PhSH) indicated the presence of free radical species.

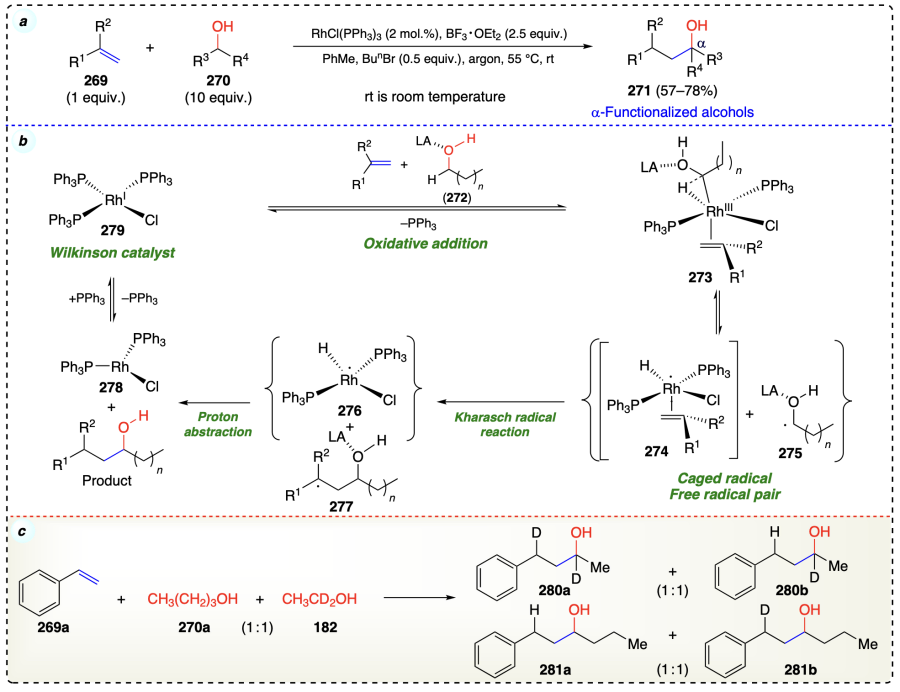
Zhang et al.[79] described a straightforward transition-metal catalyzed reaction that simultaneously functionalize vinylarenes and aliphatic alcohols. They explored a regioselective oxyalkylation of vinylarenes using the MnCl2 · 4 H2O as a catalyst in water as green solvent system (Scheme 42a). Initially, free radical initiator (TBHP) underwent homolytic cleavage in the presence of catalyst MnCl2 · 4 H2O, resulting in the generation of alkoxyl radical (296) and hydroxyl radical (297). Then, after hydrogen abstraction from 293 or TBHP, these radicals formed α-hydroxyalkyl radical (298) and free-radical (299). In the next step, 298 on addition to 292, generated the intermediate 301. Further, 297 or 299 and 301 on combination gave hydroxylated 302 or tert-butyloxylated intermediates 303. Finally, intermediates 302 or 303 gave the final product under mild conditions (Scheme 42b).
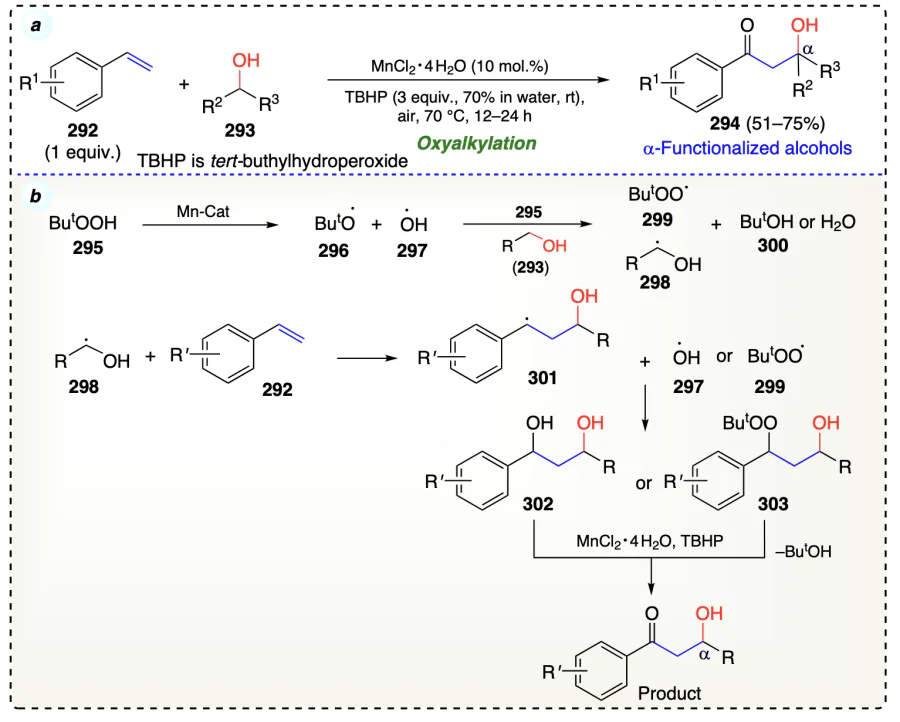
Cheng et al.[80] reported a highly effective process for the α-functionalization of alcohols with alkenes and hydroperoxides in a three-component system using zerovalent copper or cobalt as catalyst (Scheme 43a). Peroxide participates as a coupling partner. The β-peroxy alcohol (306) was further converted to give aldol product (308) (Kornblum-DelaMare rearrangement) and under reduction condition a diol (307) was formed (Scheme 43b). Copper/cobalt metal catalysts helped to generate tert-butyloxy (296) and tert-butylperoxy (299) radicals from 295. In the next step, 296 withdrew hydrogen atom from 305, forming radical 309 which gets combined with alkene substrates to provide radical intermediate (310), and further coupled with tert-butylperoxy radicals (299) to generate the α-functionalized alcohols (Scheme 43c).
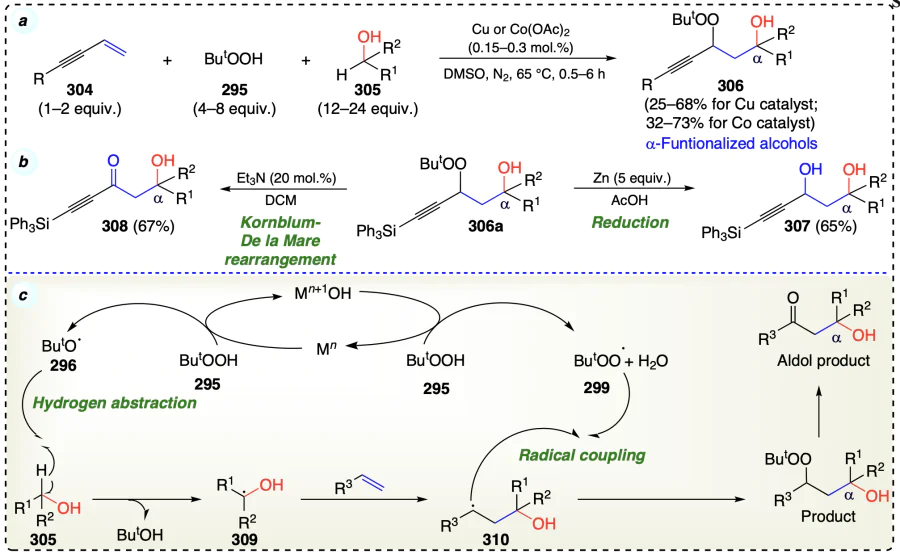
Alkoxyl radicals are important intermediate species for the δ-functionalization of alcohols, enabling the facile 1,5-HAT[16]. So, Yang et al.[81] devised a method to prevent the 1,5-HAT reaction and promote the 1,2-silyl transfer (1,2-SiT) process using a silylating agent. In this silver-catalyzed reaction, transfer of silyl group (1,2-silyl shift) was favourable over 1,5-HAT to synthesize α-functionalized alcohols (Minisci type reaction) (Scheme 44a,b). The tolerance of free alcohol was a major achievement of the process. According to the DFT studies, radical 1,2-SiT is kinetically and thermodynamically more favorable than the corresponding 1,5-HAT process. Reaction initiated with the oxidation of Ag+ to Ag2+ via oxidant K2S2O8, which underwent ligand exchange with 311 to afford an intermediate 316. Intermediate 316 further generate alkoxyl radical 317 on homolysis and Ag+ catalytic species. Thereafter, 317 underwent favoured 1,2-SiT, resulting in carbon-centered radical intermediate (318). In the next step, the intermediate 319 was generated by either an addition elimination process between 318 and 312, or trapping 318 with iminyl radical from homolysis of 312. The PhSO2 radical generated in this step was converted to PhSO3H under oxidation conditions in aqueous solution. Finally, the de-silylation of intermediate 319 afforded the final α-functionalized alcohol product under aqueous conditions (Scheme 44c).
Cui et al.[82] developed a decarboxylative olefination of alcohols with vinylic carboxylic acids, which proceeds through a radical-elimination mechanism (Scheme 45a). This reaction was further scaled up. The conditions yielding the product could be utilised for synthesis of antijuvenile hormone active precocene I and II known as chromenes (Scheme 45b). Stereospecifically E-alkenes were prepared by this protocol and also employed for the preparation of naturally occurring chromene scaffolds. Initially, homolysis of TBHP (295) occurred and formed tert-butoxy radical (296) and hydroxy radical (297). Then, 296 interacts with alcohol (322) and undergoes a hydrogen abstraction (rate-determining step) reaction to form α-carbon centered radical. This step was also confirmed by intermolecular competition kinetics experiment (KIE = 3.4) (Scheme 45d). Next, TBHP reacts with acrylic acid in the presence of copper salt to engender the Cu(II) carboxylate species. Then, α-carbon centered radical get added at olefinic α-position in species Cu(II) carboxylate species and formed an intermediate which on elimination of CO2 and Cu(I), furnished the final product. (Scheme 45c).
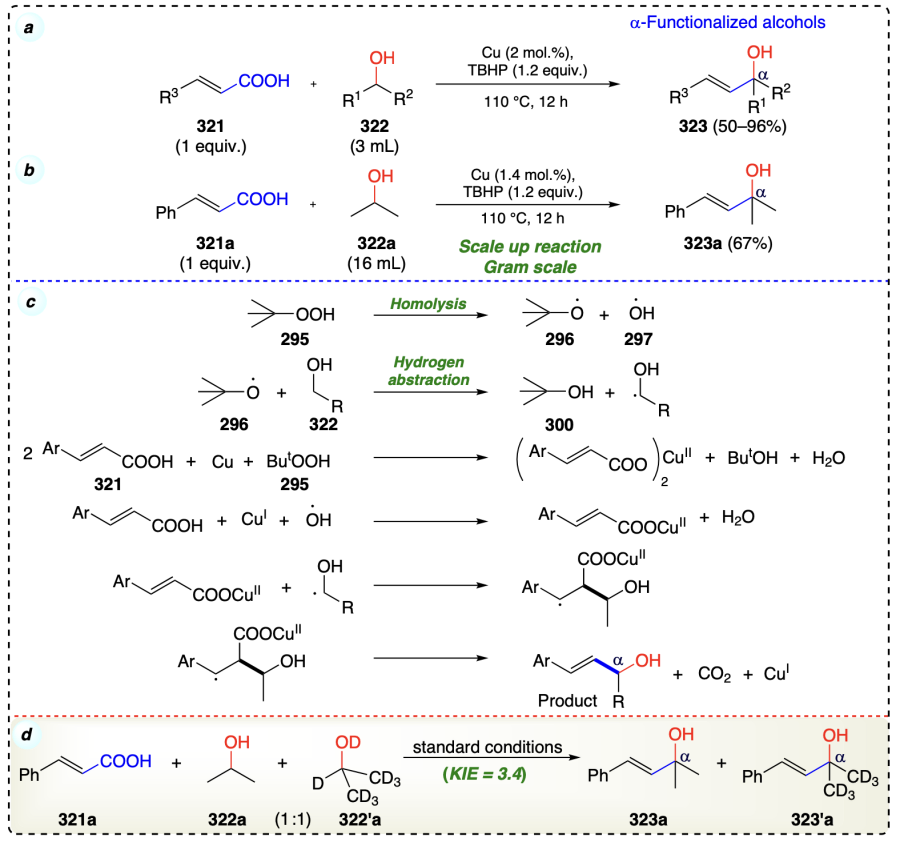
Correia et al.[83] reported a palladium-catalyzed Minisci-type reaction[84] of heteroarenes with alcohols (Scheme 46). The reaction occurred by a radical-based mechanism. At the beginning, cumyl peroxide radical was formed upon heating, to move alcoholic α-H atom, which promotes the nucleophilic radical for addition at heterocycles and re-aromatization of the heterocycles via abstraction of hydrogen. However, herein, the complete role of palladium was not investigated and postulated whether it is accountable to generate or stabilize the radical intermediate.

Zhao et al.[85] reported the phosphonation of alcohols at a-position using copper catalyst (Scheme 47a). Mechanistically, in situ oxidation of alcohol substrate occurred to provide aldehyde (326) using copper/TBHP, followed by subsequent Pudovik-type reaction[86] to synthesize α-hydroxy phosphonates (Scheme 47b).
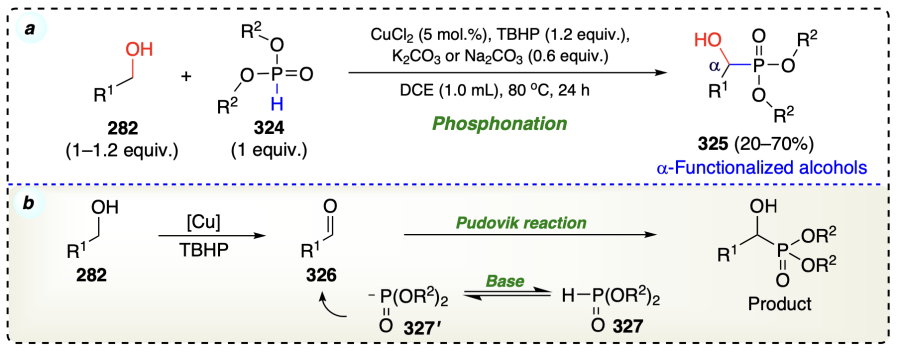
Jiang et al.[87] developed a palladium-catalyzed promoted by Lewis acid reaction between alkenes (328, 328') and alcohols (329) via selective alkene dimerization to yield γ-branched α-functionalized product (330) (Scheme 48a). The mechanism of this domino reaction[88] involved two catalytic cycles, accomplished in one operation. Catalysts enabled the dimerization of alkenes, the first step involved the activation of 328 by Lewis acid (LA) via Friedel Crafts alkylation, forming a cationic intermediate (331). Then, there was a nucleophilic attack by the Pd-catalyst giving oxidative adduct (332), which further accepted the insertion of 328', and formed a Pd(II) cation intermediate (333)[89]. Next, via intramolecular nucleophilic attack, palladacycle (334) was formed, which further underwent β-H elimination to form 335 or 340, which were in equilibrium with each other. This was followed by reductive elimination, which provide terminal alkene (335) and disubstituted alkene (336). The alkene further entered into the cycle of activation of C – H group, where it first coordinated with the Pd-catalyst to form Pd(0)-alkene complex (337). On simultaneous coordination of 329 and LA with 337, the Pd(0)-catalyst activated the nearby C – H of alcoholic oxygen in 329 to form an important transition state (338) via oxidative addition. Subsequently, insertion of alkene substrate within Pd – H bond provided complex 339 which finally provided the desired product via reductive elimination (Scheme 48b). The deuterium-labeling experiment suggested that this domino protocol is highly regioselective (Scheme 48c).
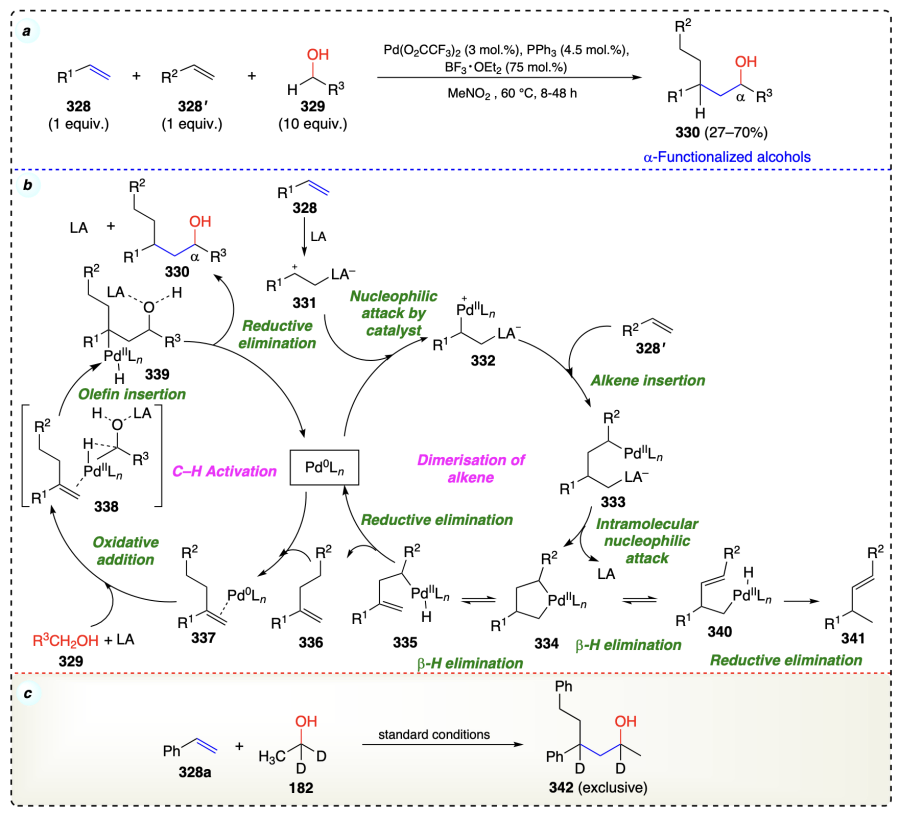
Zhu et al.[90] described a method to synthesize the complex molecules with both alcohol and amine functional groups (1,3-amino alcohols) via α-functionalization of alcohols. This method involves the reaction of alkenes (343) with anilines (345) and HFIP (344), to yield the desired α-functionalized products 346 using copper catalyst (Scheme 49a). The researchers successfully scaled up this method to produce the desired product in good yield (68%) (Scheme 49b). Mechanistically, an alkene can be transformed into a 1,3-amino alcohol through a difunctionalization reaction. During the DTBP (tert-butyl peroxide) process, heating prompts a copper(I) iodide (CuI) molecule to transfer an electron, thereby generating a tert-butoxyl radical. Subsequently, the alkoxy radical snatches a hydrogen atom from HFIP (344), creating an unstable intermediate 347. This intermediate quickly reacts with the alkene molecule (343), forming a new radical intermediate 348. The oxidation process mediated by Cu(II) converts the intermediate 348, an alkyl radical, into a carbocation (349) by abstracting an electron. This electron abstraction leads to the cleavage of a hydrogen-carbon bond in 348. In the final step, a positively charged molecule 349, reacts with an arylamine molecule to form a 1,3-aminoalcohol compound (350) (Scheme 49c).
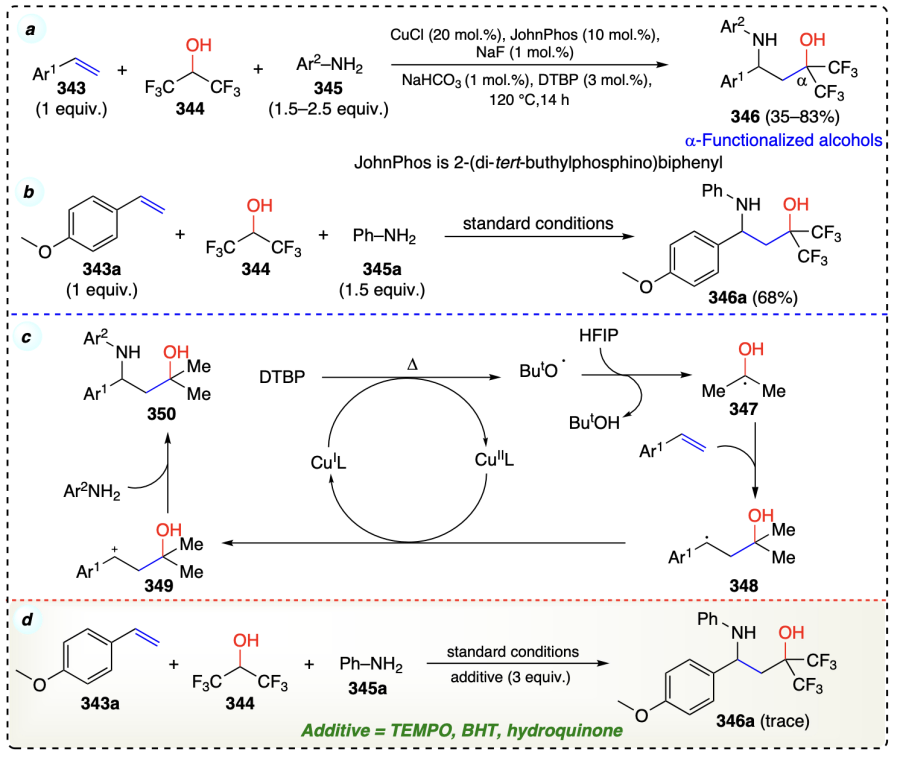
The involvement of a radical intermediate was proved by the radical inhibiting experiments using TEMPO, BHT, and hydroquinone as inhibitors (Scheme 49d).
3.1.2. Metal-free α-functionalization of alcohols
Kamitanaka et al.[91] reported α-functionalization of alcohols (352) with alkenes (351) without any metal catalyst under supercritical conditions and resulted in hydroxyalkylated products (353) in low to good yields (Scheme 50). Reactions were performed under the supercritical conditions (specific temperature and pressure for alcohols to be at supercritical state) and proceeded by the α-C – H bond fission of alcohols, which is the rate-determining step. Reaction was applicable to styrenes as well as long chain terminal alkenes.
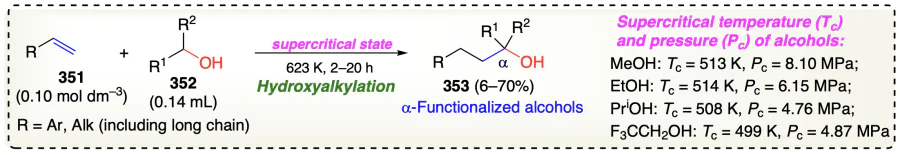
Guo et al.[92] described the metal and base-free α-C – H functionalization reaction of alcohols using diaryl(arylethynyl) phosphine oxide as alkylating agent and azobisisobutyronitrile (AIBN) as radical initiator to provide the hydroxymethyl benzo[b]phosphole oxide (356) (Scheme 51a). The C – H cleavage in alcohol could be the slowest step in the reaction, as indicated by the KIE value of 4.56 (Scheme 51c). Additionally, the inhibition of reaction using BHT or TEMPO confirms free-radical pathway for the current reaction (Scheme 51d). Initially, AIBN and TBHP generated the 2-hydroxy-2-methyl-propanenitrile (CpOH) and tert-butoxyl radicals, respectively, under thermal condition. The generated radical helps the hydrogen abstraction from 2-propanol (355a) to form α-hydoxyalkyl radical (357). Then, the inclusion of radical (357) adjacent to the P=O bond in 354 generated alkenyl radical 358.
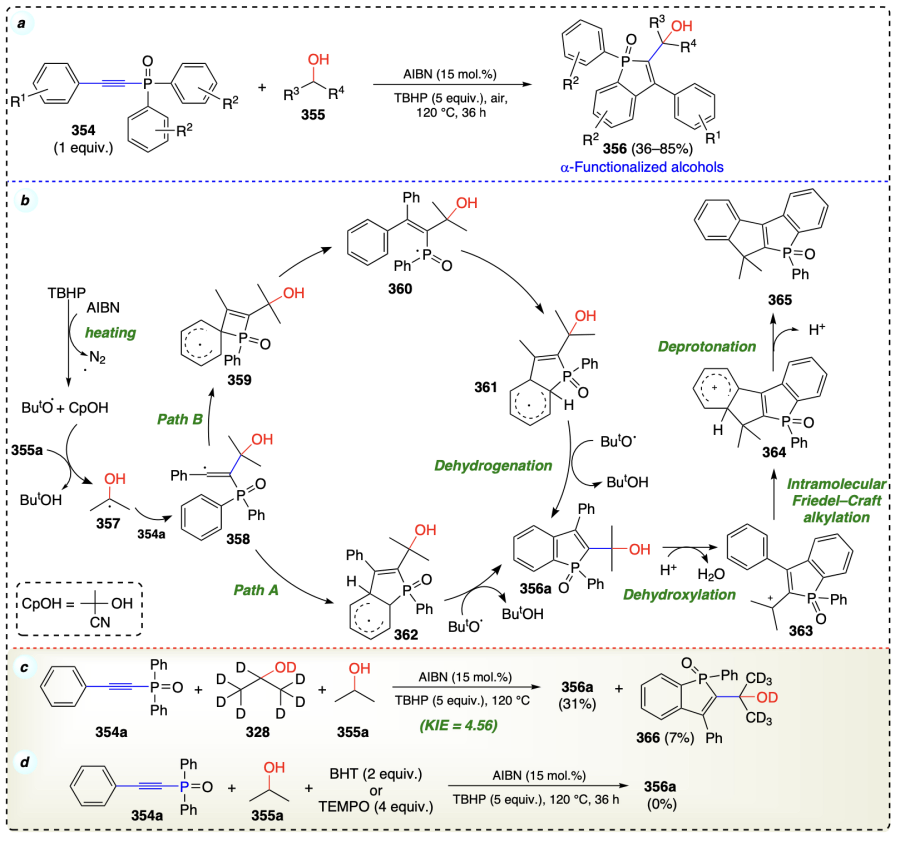
Regarding the possibilities of forming two regio-isomeric products, 358 might follow two different paths to provide the common product (356a) via dehydrogenation of intermediates 361 and 362 in path A and path B, respectively. Dehydroxylation of 356a in the presence of a Bronsted acid followed by deprotonation by TfOH may provide carbocation (363), which on intramolecular Friedel – Crafts alkylation provides 364 which finally on deprotonation provides 365 (Scheme 51b).
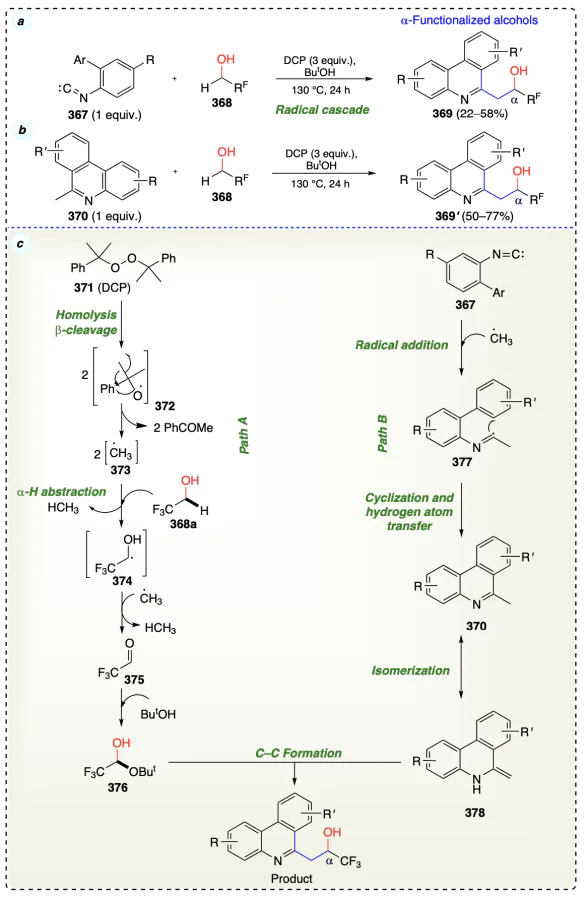
Xu et al.[93] developed an efficient metal-free protocol for α-functionalization of trifluoroethanol (TFE) (368a) and polyfluorinated alcohols (368) with isonitriles (367) using DCP (371) (Scheme 52a). The reaction proceeded through a radical-mediated addition/cyclization process, and the Friedel – Crafts pathway for the generation of C – C bond. Although the β-hydride elimination and dehydrogenation steps are reversible, the β-hydride elimination step results in 1 kcal mol–1 higher C – H bond dissociation energy in TFE than that in ethanol[94]. The reaction mechanism consisted of two different pathways (path A, and path B), which yielded the same product and thus considered a highly ordered tandem process. The first step of the first pathway involved the homolytic cleavage of DCP (371), forming a methyl radical and acetophenone as a by-product. The methyl radical abstract the C – H of TFE (368a) at a-position to give radical intermediate (374), which instantly converted into fluorinated aldehyde and finally to a hemiacetal, 1-(tert-butoxy)-2,2,2-trifluoroethan-1-ol (375) by reacting with ButOH. Conversely, in the first step of the second pathway (B), the methyl radical added to the biarylisonitrile (367) to generate a radical intermediate 377. Upon cyclization and hydrogen atom transfer, 377 radical formed 6-methyl phenanthridine (370), which isomerized into an active enamine (378). Finally, the reaction of intermediates 376 and 378 gave the targeted product 369 (Scheme 52b).
Zhuo et al.[95] reported α-functionalization of alcohols via insertion of alkenes without using any metal catalyst to access α,ω-amino alcohols (Scheme 53a). High KIE value = 5 : 1 for intermolecular competing experiment indicated the activation of C – H bond neighbouring to hydroxyl group as the rate-determining step (Scheme 53c). Scheme 53d revealed that the hydride shift occurred from the N-allylamide moiety. Homolytic cleavage of di-tert-butyl peroxide (DTBP) under heating formed tert-butoxy radical intermediate 296, which oxidized alcohol substrate (380a) and further C – H bond breaking generates α-hydroxyisopropyl radical (357). Then, on radical addition of 357 to alkene substrate 379a gave an intermediate 383. Further, the 1,3-H shift (hydride shift from N – H) in 383 formed the radical intermediate 384. In the final step, radical intermediate 384 reacted with ButOH (300), which gave the desired product, and radical 296, which further participated in the reaction cycle (Scheme 53b).
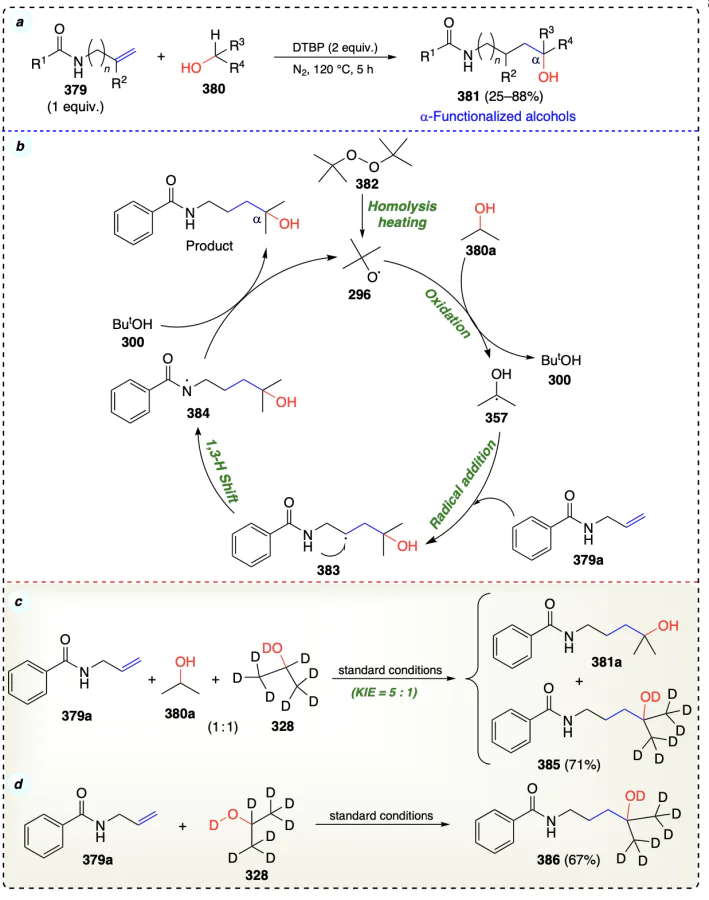
The same group[96] developed another simple metal-free method to synthesize 4-hydroxyalkyl-substituted 3,4-dihydro-isoquinolin-1(2H)-ones from alcohols and N-allylbenzamide via a radical cyclization reaction (Scheme 54a). The reaction sequence comprises the C(sp3) – H bond cleavage, oxy-alkylation of alkene functionality, and intramolecular cyclization steps. The fact that the radical scavenger TEMPO inhibited the reaction, suggests that it proceeds through a radical-mediated pathway. According to the KIE studies, the breaking of C – H bond is the rate determining step in current reaction (Scheme 54c). Reaction commenced with the generation of t-butoxy radical (296), which interacts with 2-propanol (388a) to afford 2-propanol radical intermediate (357).
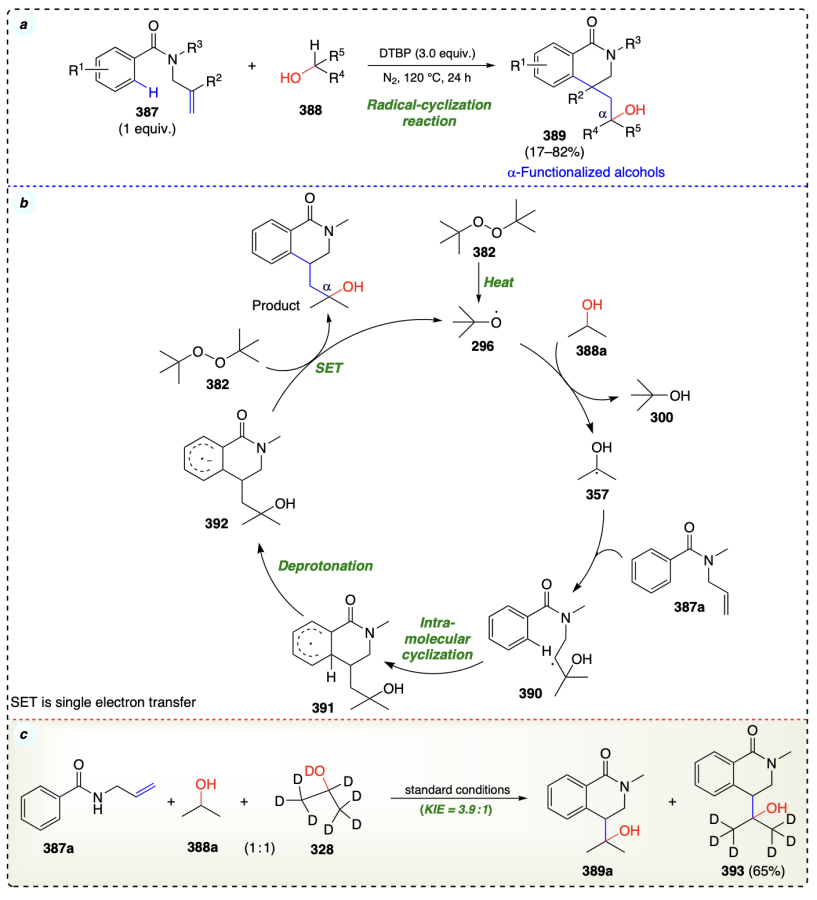
Intermediate 357 further added at the olefin moiety in second starting material (387a) and generated the carbon-centered radical intermediate (390), which underwent intramolecular cyclization to form intermediate 391. The deprotonation of intermediate 391 yielded the radical anion 392. Finally, the intermediate radical anion 392 reacted with DTBP through SET protocol to give the final product (Scheme 54b).
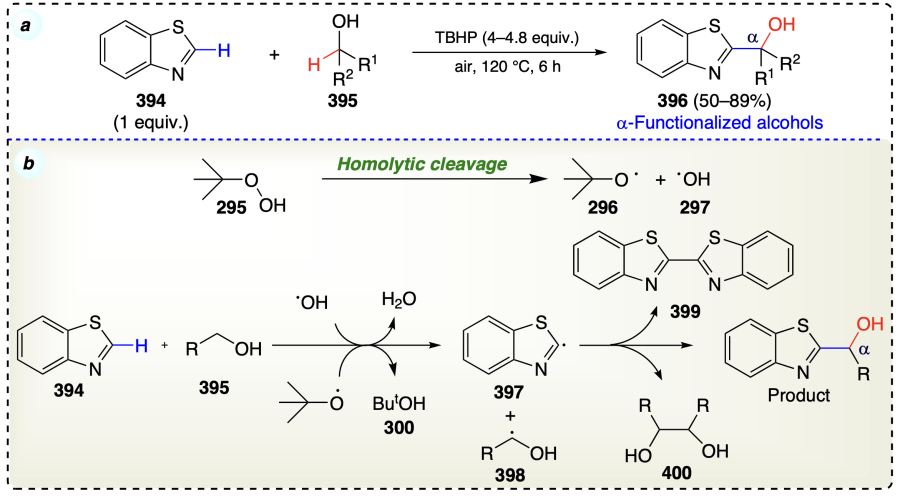
He et al.[97] displayed direct C – H alkylation of benzothiazoles using alcohols as alkylating agent and TBHP, without using any additional base or metal catalyst to get α-functionalized alcohols (Scheme 55a). The reaction mechanism involved a free-radical pathway. Initially, TBHP underwent homolytic cleavage and formed hydroxyl and an alkoxyl radical. These free radicals abstracted С(sp2) – H atom from benzazoles (394) and С(sp3) – H atom from alcohol (395). Finally, on termination of the two radicals (397 and 398) via the carbon-carbon bond formation provided the desired product (Scheme 55b).
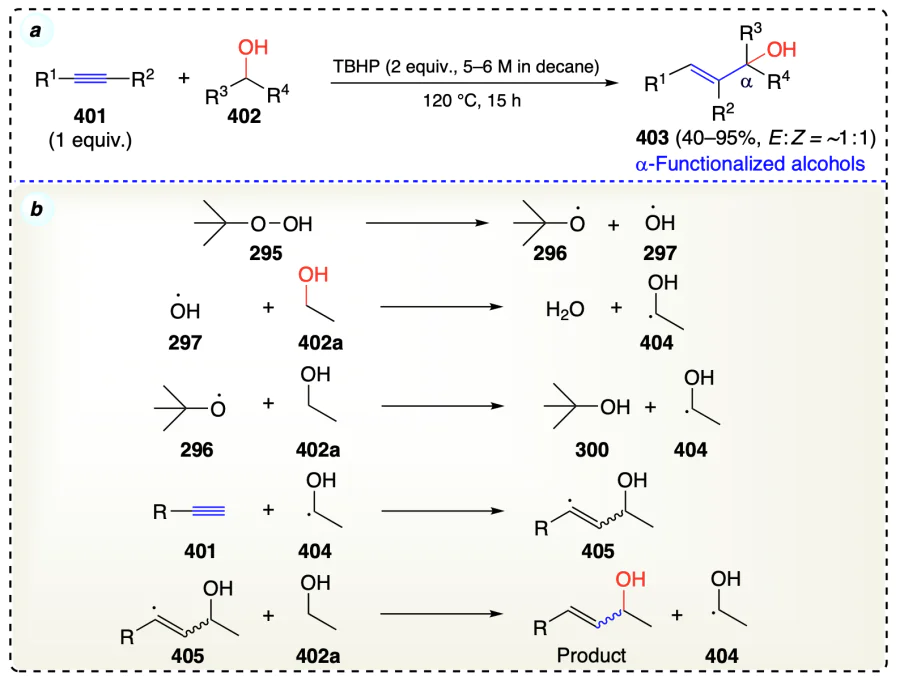
Liu et al.[98] displayed a radical-mediated (TBHP as radical initiator) and metal-free coupling of electron-rich alkynes with alcohols towards С(sp2) – С(sp3) bond formation reaction to give α-functionalized alcohols (Scheme 56a). TBHP underwent homolysis to generate alkoxyl (296) and hydroxyl radical (297). These radicals abstracted alcoholic hydrogen to form radical 404 which undergo addition with alkyne (401) to yield an alkenyl radical (405). This chain kept on growing and terminated once the alkyne was consumed entirely. Then, this alkenyl radical, on reacting with alcohol substrate, afforded the desired α-functionalized alcohols along with the generation of α-hydroxyalkyl radical (404), which further participates in the reaction cycle (Scheme 56b).
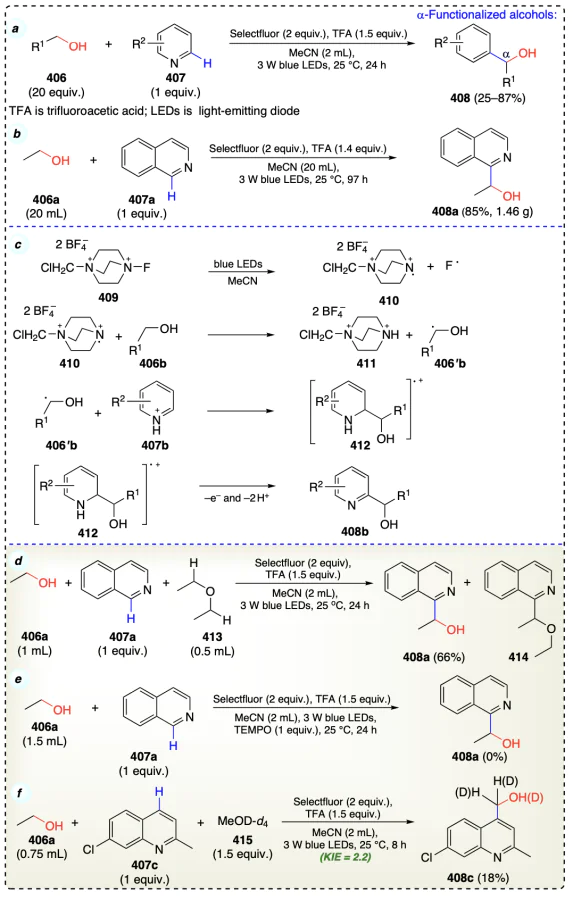
Niu et al.[99] described the photocatalytic α-functionalization reaction of alcohols with heteroarenes using selectfluor (Scheme 57a). A radical-trapping experiment (TEMPO as radical-trapping agent) suggested the radical-mediated route during arylation reaction (Scheme 57e). KIE value of 2.2 for cleaving α-С(sp3) – H confirms it as rate determining step (Scheme 57f). Mediated by visible light irradiation and a reductant, the N – F bond in selectfluor (409) was activated, which resulted in the corresponding ‘N’ radical cation (410) and ‘F’ radical. Radical 410 abstracted α-С(sp3) – H of alcohol (406b) to give hydroxyalkyl radical (406'b) and species 411. After that, the acid-protonated and electron-deficient heteroarene (407b) captured the nucleophilic radical (406'b) to generate adduct (412), and this adduct (412) provided the final α-arylated product (Scheme 57c).
3.2. β-Selective direct functionalization for non-activated alcohols
This section discusses the functionalization of non-activated β-C – H bonds in alcohols selectively to access α-functionalized alcohols. An overview has been shown in Scheme 58 for the conditions to describe the direct functionalization of non-activated alcohols at β-position. Further, this section has been classified into metal catalyzed and metal free C – H functionalization of alcohols at β-position.
3.2.1. Metal catalyzed β-selective direct functionalization for non-activated alcohols
Cheung et al.[100] introduced different ruthenium complexes for the functionalization of secondary alcohols at b-position using primary alcohols (Scheme 59a). Various ruthenium complexes supported by different ligands such as cyclopentadienyl (Cp), hydrotris(pyrazolyl)borato (Tp), and 6,6'-dichloro-2,2'-bipyridine (6,6-Cl2bipy) displayed efficient utility for the developed functionalization approach. The major catalytic pathway followed the same mechanism as mentioned earlier in literature (Scheme 59b)[101].
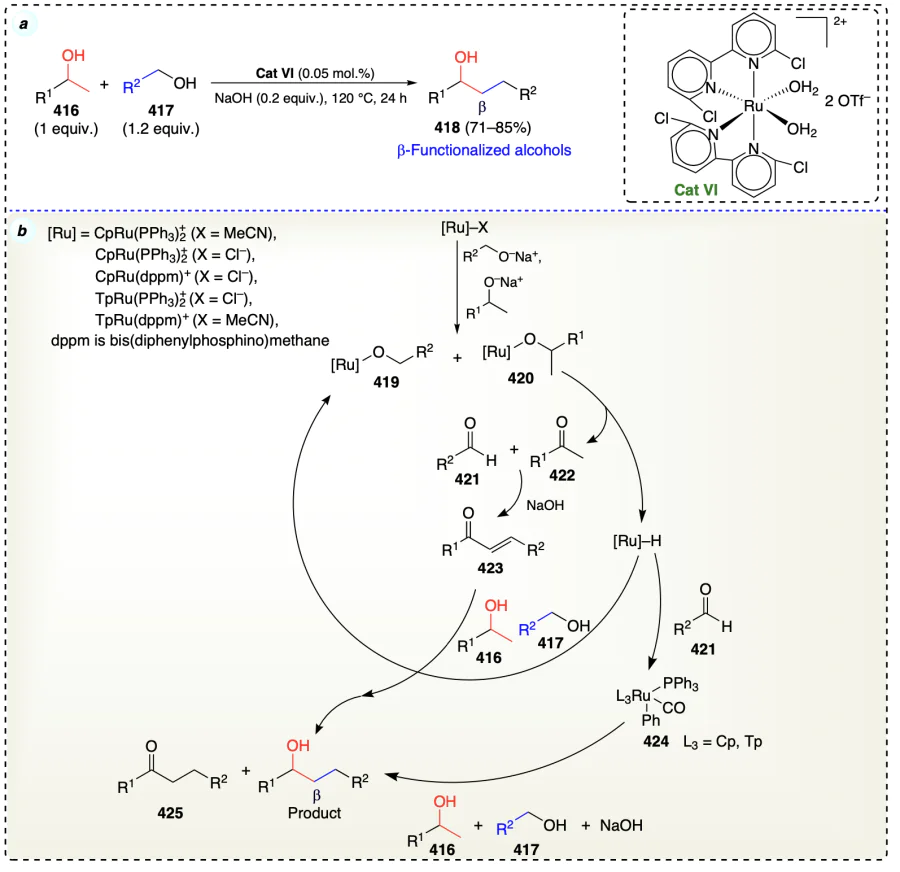
Liu et al.[102] reported a novel Mn(I) catalyst prepared from pyridyl and imidazolyl ligands without using any phosphine ligand and utilised for alkylating the secondary alcohols at b-position through a borrowing hydrogen strategy (Scheme 60a). The proposed mechanism was similar to that of borrowing hydrogen strategy (Scheme 6, section 2.3.3). Method was also applicable for the complex substrates such as cholesterol, and its derivative (429 and 431) which afforded the C-alkylated cholesterol derivatives (430, 432 and 433) on reacting with benzyl alcohols (Scheme 60b,c). In addition, the reactivities of various alcohols were tested for the current alkylation reaction and benzylic alcohols showed high reactivity comparatively to the other aliphatic primary alcohols. Furthermore, the non-cyclic alcohols displayed low reactivity comparatively to cyclic derivatives. Benzopyran derivative (435) was also prepared from simpler alcohol in one-pot reaction by employing the current methodology (Scheme 60d).
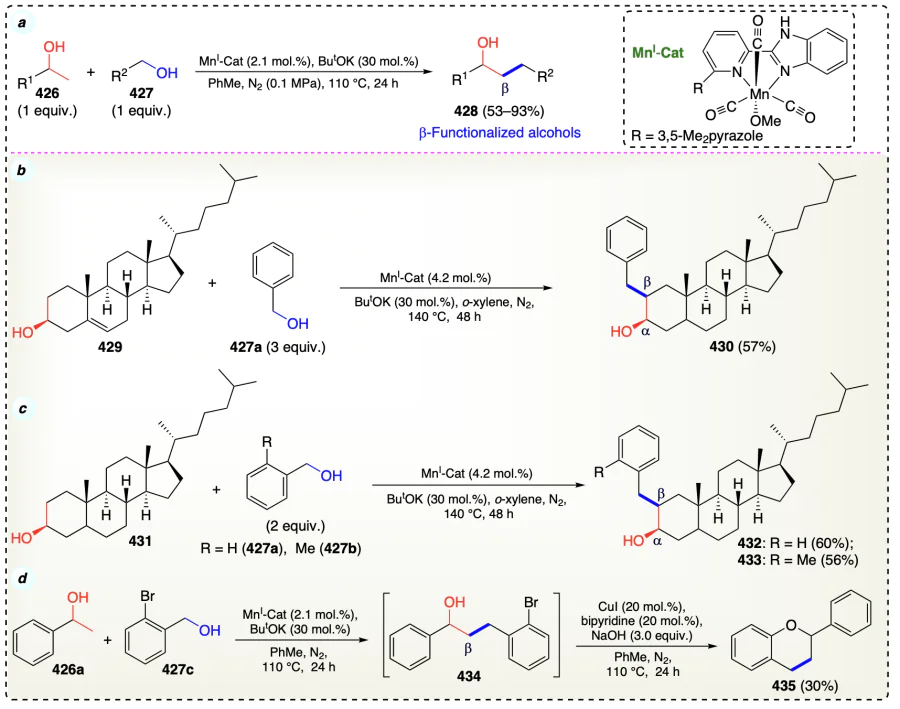
Yang et al.[103] reported a method for direct β-functionalization of alcohols (2°) using ferrocene carboxaldehyde catalyst (Scheme 61a). Initially, there is a nucleophilic attack of alkoxy anions under basic conditions on the aldehyde group of the iron catalyst (439), resulting in the abstraction of hydride by iron cation of (439) thus yielding an Fe – H entity (441) and simultaneous oxidation of 1° (437) and 2° alcohols (436) yields consequent aldehyde and ketones, respectively (redox pathway). Under basic conditions, a cross-aldol condensation reaction provides the intermediate 444 which underwent transfer hydrogenation of C=O and C=C bonds with (441), thus yielding the required final β-functionalized alcohols (Scheme 61b).
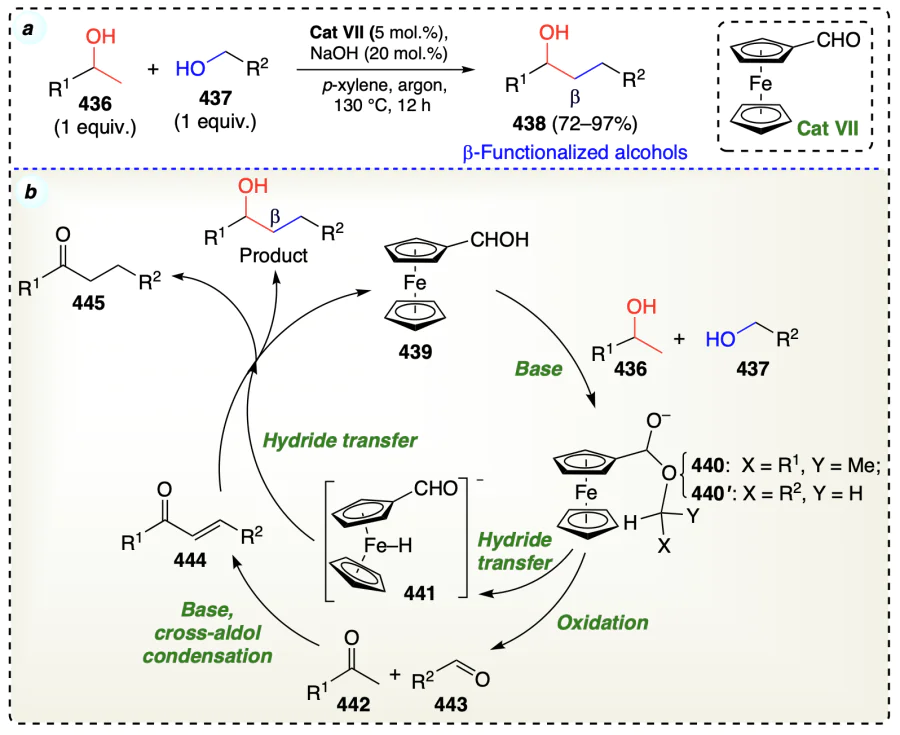
Satyanarayana et al.[104] developed a phosphorus-free β-functionalization of alcohols using tris(acetylacetonato) rhodium(III) complex as catalyst (Scheme 62a). Reaction proceeded via the redox pathway for reactants following generation of rhodium-hydrido complex. This was possible when the reaction of alcohols with KOH and Rh(acac)3 produced a rhodium(III) alkoxide compound (449). Complex 449 further underwent the β-hydride elimination and yielded ketones (450), aldehydes (451), and Rh – H (452) complex. Now 450 and 451 underwent base-mediated cross-aldol reaction to provide an α,β-unsaturated ketone (453). Ultimately, sequential transfer hydrogenation of alkene and carbonyl bonds in 453 by 452 yielded the final product. In addition to this for the current atom-economical reaction, there was no addition of hydrogen donor and acceptor in this reaction as the hydrido-rhodium species (452) was further utilised for the hydrogenation of intermediate 453 (Scheme 62b).
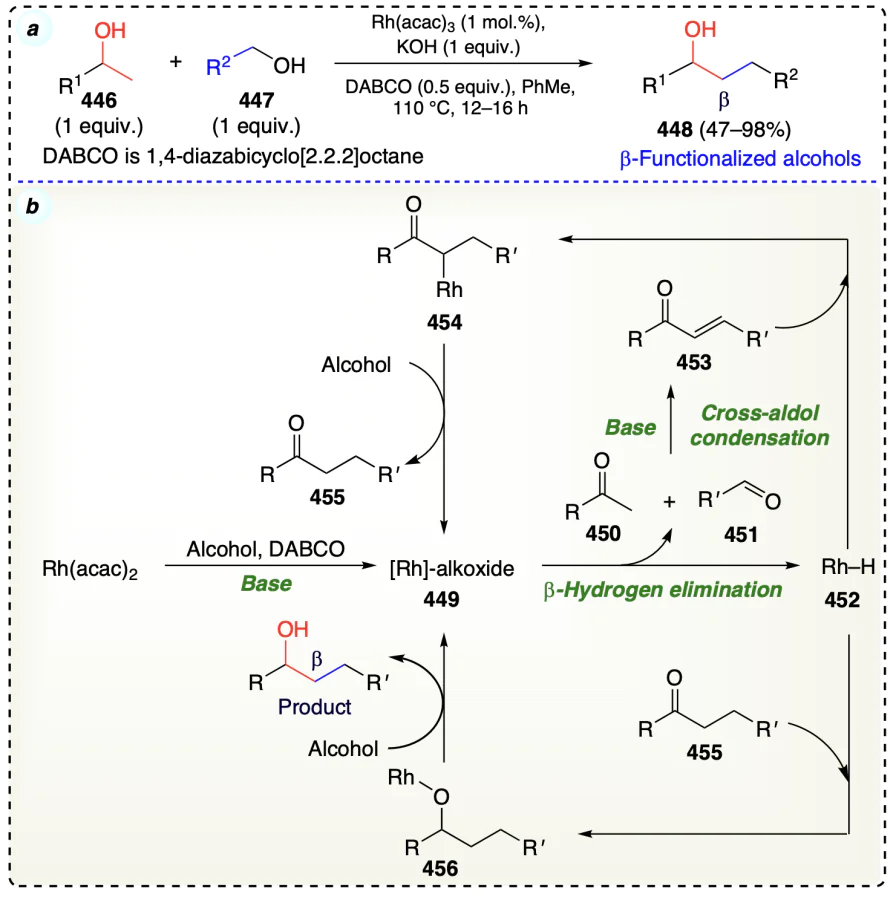
Cano et al.[105] described the iridium catalyst on magnetite for the alkylation of alcohols 457 with another primary alcohol 458 to afford β-functionalized products 459 in high yields (Scheme 63a). To probe the possible catalytic reaction pathway, initially, there is the dehydrogenation of the substrate via iridium catalytic species, resulting in the formation of aldehydes. Then, the two aldehydes (462 and 463) condensed together to form intermediate 464. This α,β-unsaturated aldehyde (464) underwent hydrogenation via Ir – H species and furnished an enolate (465) in a Michael-type fashion rather than following a general mechanism. Enolate (465) formed was protonated by water to form the aldehyde and further provide the required β-functionalized alcohol on reduction with Ir – H species (Scheme 63b). The deuterium labeling experiment provided an indirect evidence that via a Michael-type hydride addition, condensation intermediate was reduced (Scheme 63c).
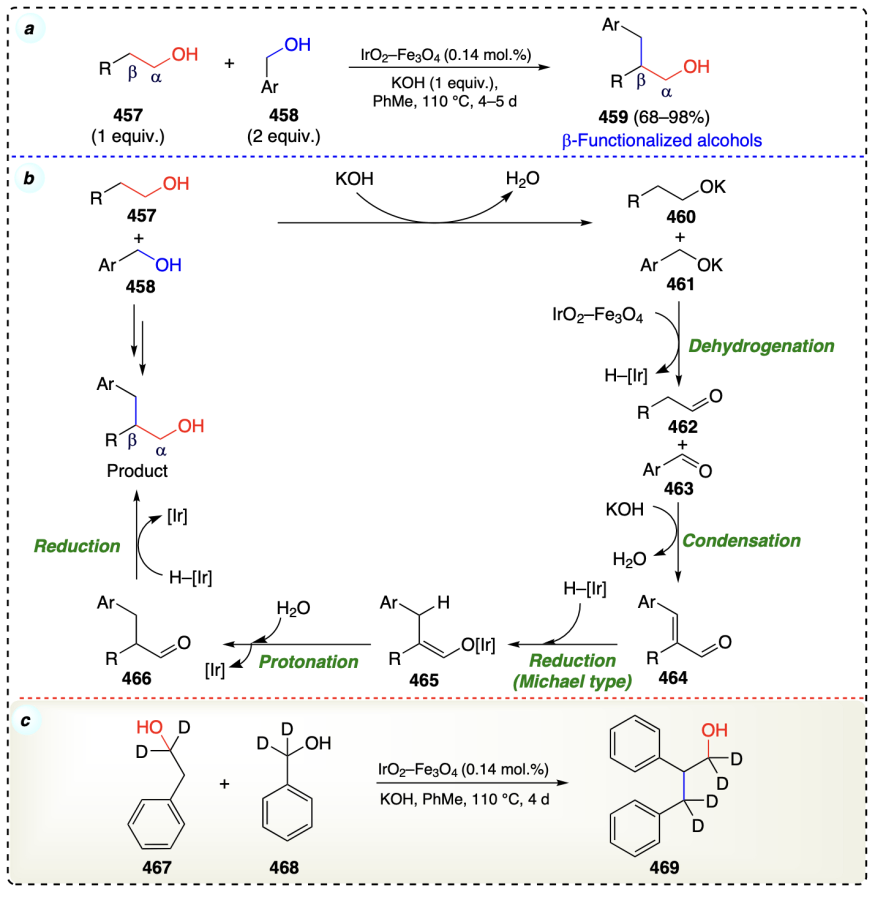
Miura et al.[106] reported a copper catalyzed protocol under anaerobic conditions to produce β-functionalized alcohols. In this protocol, two different alcohols were reacted with each other using CuI/H2/NaOH system (Scheme 64a). Mechanistically in the first step, CuBr/NaOH/(H2 or dppp) oxidise the starting secondary alcohol (470) and primary alcohol (471) to produce 473 and 474 as intermediary species. Then, 473 underwent deprotonation with M – OR (a catalytically active species) and generated an enolate (475). Next, 474 on cross-aldol condensation with enolate (475) afforded an enone species (476). Then, hydrogen transfer occurred from 471 [or 470] to 476 and 470 [or 471] to 477 to afford β-functionalized alcohols and complex 478. The complex 478 further underwent the intramolecular proton relocation and regenerated (475) for further process (Scheme 64b).
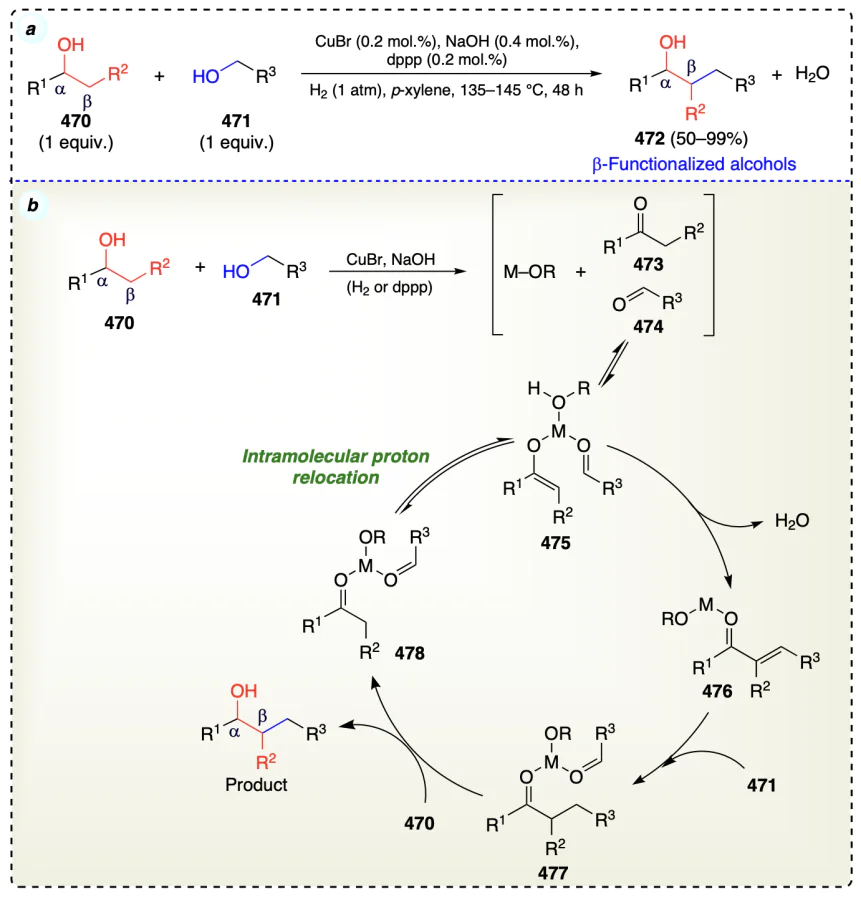
Matsu-ura et al.[107] reported an iridium catalyzed and highly efficient Guerbet reaction [108] of various primary alcohols (Scheme 65). This method provided a new way to directly produce β-alkylated primary alcohols by condensing aldehydes and hydrogenating the product. Various primary alcohols reacted via the Guerbet reaction and generated the subsequent dimeric alcohols. Guerbet reaction mainly involved three steps: (i) initial alcohol dehydrogenation to form carbonyl compound, (ii) carbonyl compounds underwent aldol condensation and crotonization, (iii) reduction of unsaturated carbonyl to provide final saturated product. This method was useful in synthesizing β-alkylated higher alcohols that might be utilised as plasticizer and detergent materials.
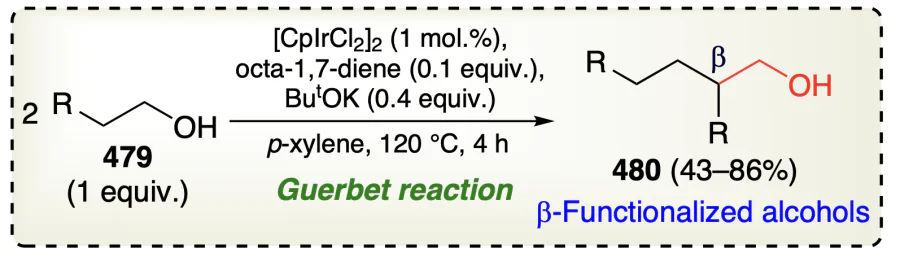
Xu et al.[109] reported an iridium based complex for the β-functionalization of alcohols (Scheme 66a). Current reaction also followed the redox pathway along with aldol condensation, dehydration, and finally the reduction of enone intermediate to produce the desired product (Scheme 66b). Authors also described that the presence of primary alcohols avoid the oxidation of secondary alcohols to ketonic form due to the occurance of β-alkylation step.
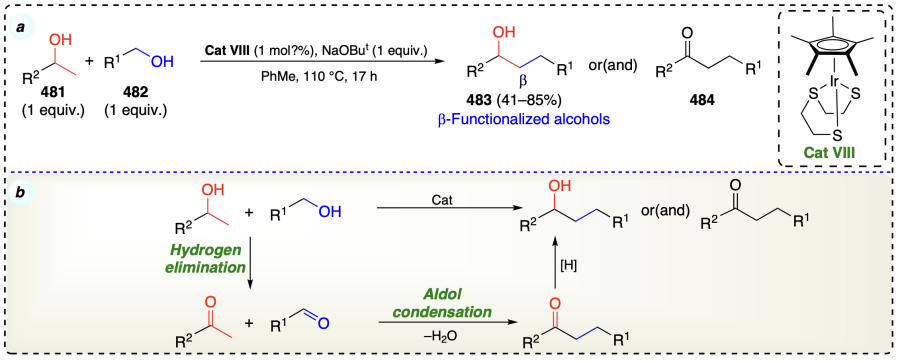
Kose et al.[110] displayed that pincer-type NHC/Pd complexes, with the help of an alkaline metal base, can effectively catalyze the alcoholic β-functionalization process under air or H2 atmosphere (Scheme 67a). Reaction also followed the sequence of redox pathway, enone formation, reduction of enone to final product through the involvement of either Pd-alkoxide or Pd – H species (Scheme 67b).
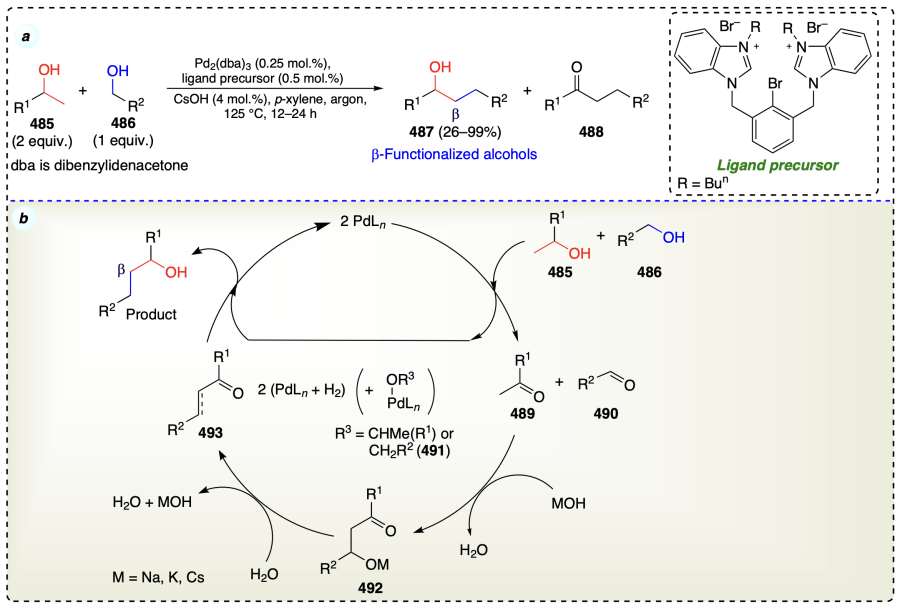
Wang et al.[111] reported the β-functionalization of alcohols catalyzed by ruthenium(III)-NNN complex through borrowing hydrogen pathway (Scheme 68a). Mechanistically, this protocol followed the mechanism similar to borrowing hydrogen strategy (Scheme 6, section 2.3.3) (Scheme 68b).
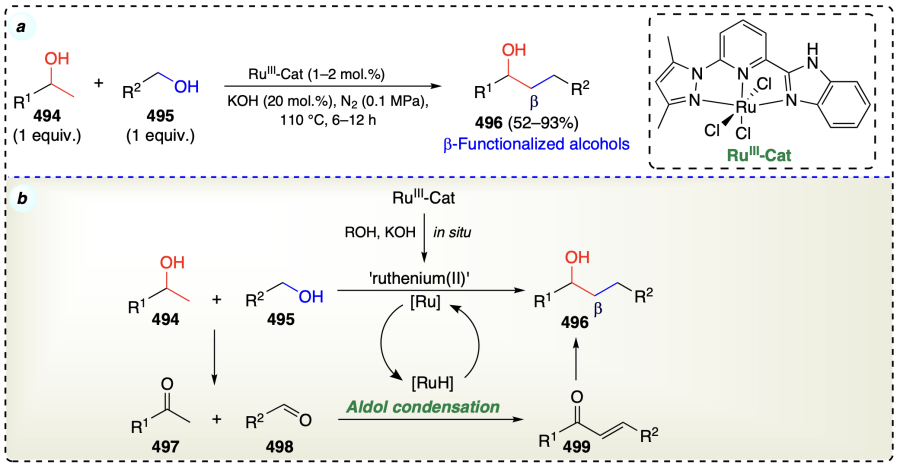
Liao et al.[112] reported an effective alcohol activation and β-functionalization in a ligand-free approach using copper catalyst (Scheme 69a). The aerobic alcohol oxidation (step 1) was fast under air and slow under nitrogen, which resulted in the oxidation of primary alcohol (500) and secondary alcohol (501) into corresponding aldehyde (503) and ketone (504), respectively. In the next step, this ketone and aldehyde underwent dehydrative aldol condensation (step 2) to give enone (505). The hydrogen transport (step 3) further included two steps (steps 3a and 3b): first hydrogen atoms from 500 and 501 were shifted to enone (505) to furnish ketone species (506), and further it was reduced to the desired product (502) along with the by-products 503 and 504. Step 3 was found to be proceeded through Meerwein – Pondorf–Verley (MPV) type six-membered transition state[12]. Hence, in the final step, 503 and 504 were recycled and condensed to give 505 again, and thus the catalytic cycle was furnished. In addition, step 3a was found to be a fast under optimised conditions, while step 3b was temperature dependent (Scheme 69b).
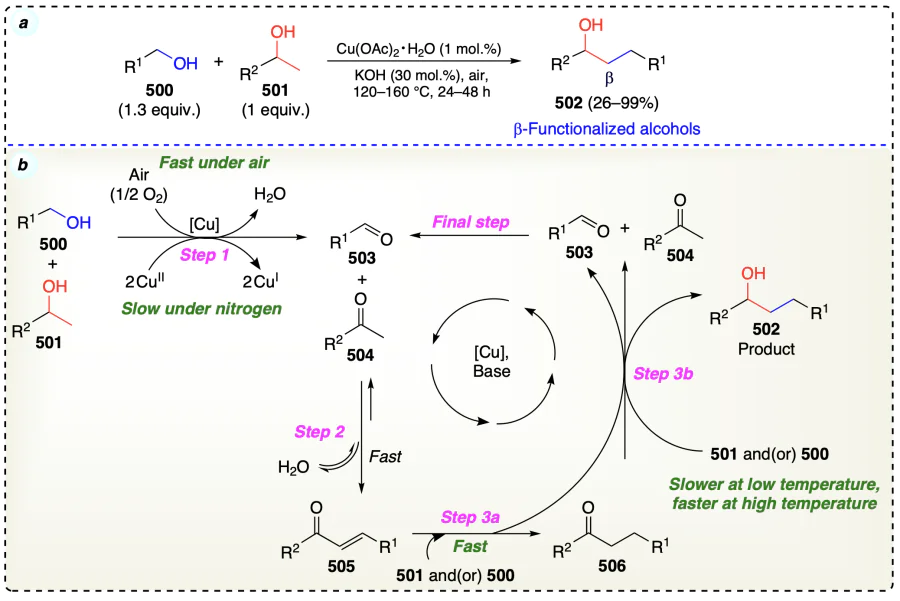
El-Sepelgy et al.[113] developed a Mn-PNN pincer complex applicable for the β-functionalization of alcohols (Scheme 70a). This Mn-catalyzed protocol followed a double hydrogen autotransfer sequence. Developed protocol followed the hydrogen autotransfer pathway as corroborated in deuterium-labeling experiments. Also, this experiment supported a monohydride involvement in pathway and indicated the role of both metal and ligand in the transfer hydrogenation process. Additionally, they investigated that dehydrogenation of alcohol was the step that took the longest and therefore a rate determining step (Scheme 70c–e). Initially, catalyst activation generate 16e– species (510) by reaction of precatalyst (Mn-1) with base. Then, 510 reacting with alcohol and formed the corresponding ketone and hydrogenated catalyst (511). Now, for the alkylation, alcohols’ (507 and 508) oxidation occurred to furnish the corresponding carbonyl compounds (512 and 513). Then, on base-catalyzed aldol condensation, 512 and 513 resulted in an enone (514) formation. Then, sequencial reduction of 514 to 515 and finally to desired β-functionalized alcohols (Scheme 70b).
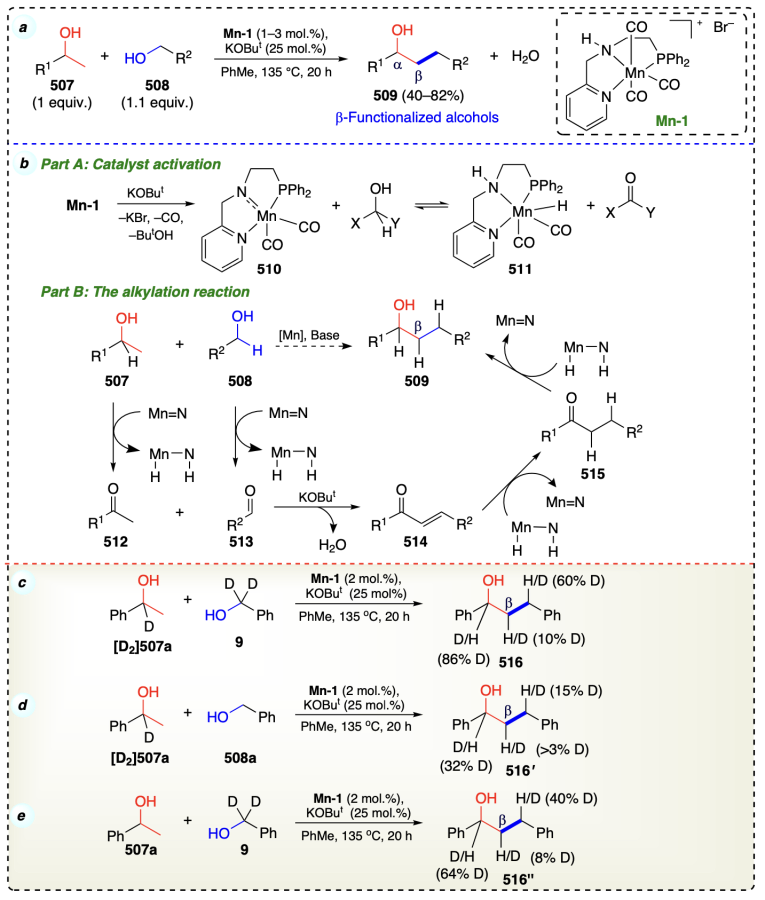
Bettoni et al.[114] reported an iron complex (diamino-cyclopentadienone iron tricarbonyl complex, Fe1) for β-functionalization of alcohols to access functionalized alcohols at β-position under mild hydrogen autotransfer[115] conditions (Scheme 71a). Initially, 520 under basic conditions formed 521. Then, 521 on decarboxylative activation formed 522. Then 522 on hydrogen release formed an unsaturated iron species (523). Thereafter, 523 coordinated with either methanol or 2-phenylethanol to form 524. Intermediate 522 was again formed when 524 underwent dehydrogenation and afforded 527.
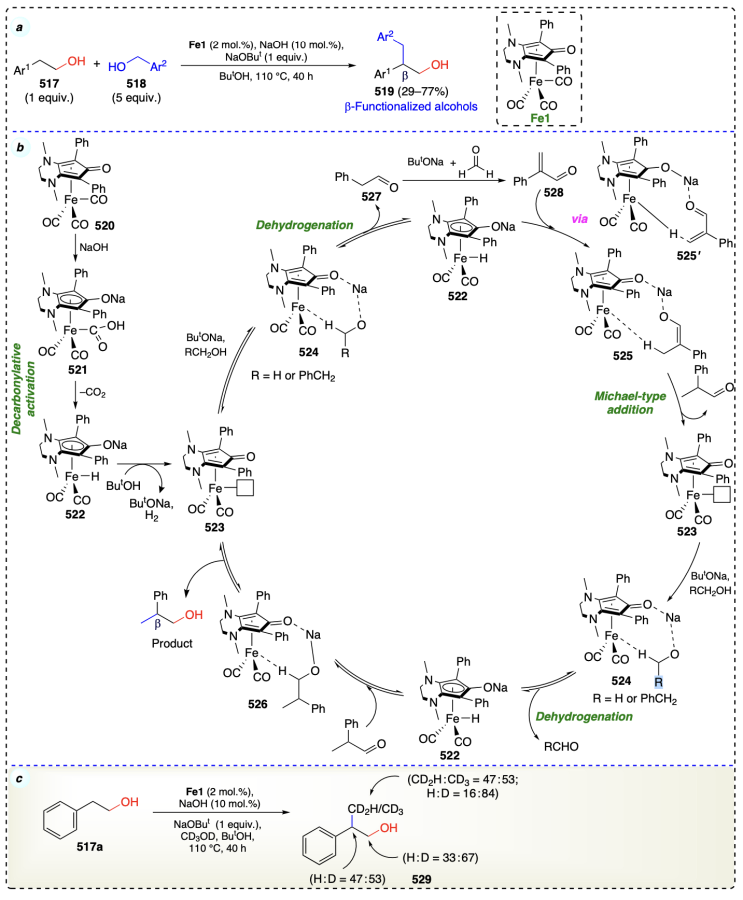
Then, 527 and enolate on condensation formed 2-phenylpropanal which via complex 525 and Michael-type addition, formed a methylated aldehyde and intermediate 523. Furthermore, an another dehydrogenation of alcohols yielded complex 522, and after reducing 2-phenylpropanal, the desired product was obtained (Scheme 71b). This mechanism was also verified by deuterium labeling experiment that both the alcohols were the source of hydride in this protocol (Scheme 71c).
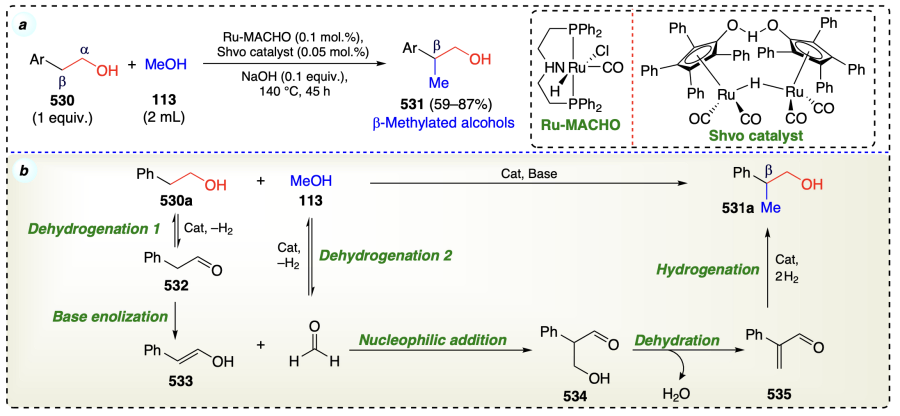
Li et al.[116] developed a protocol to nurture the challenging methylation of primary alcohols through β-functionalization approach. In the appearance of a base and collaborative mixture of two catalysts i.e., Ru-MACHO complex and Shvo’scatalyst, MeOH was used as C1 feedstock for the selective methylation of 2-arylethanols via borrowing hydrogen strategy (Scheme 72a). The process is advantageous due to escaping the carbon monooxide and hydrogen without using any high-pressure equipment. As this protocol involved ‘hydrogen autotransfer strategy’ so the proposed mechanism involved two times dehydrogenation, enolization, nucleophilic addition, dehydration and finally hydrogenation to yield the final product (Scheme 72b).
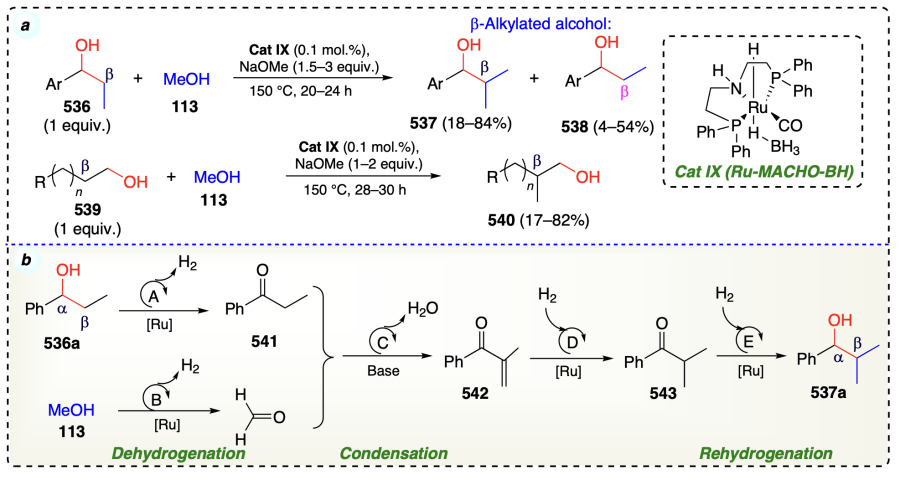
Further, Kaithal et al.[117] reported another effective protocol for β-methylation of arylic, aliphatic, and cyclic alcohols using MeOH as C1 source and organometallic ruthenium complex i.e., [RuH(CO)(BH4)(HN(C2H4PPh2)2)] (Scheme 73a). This process also involved the standard steps of borrowing hydrogen strategy and afforded the high turn over numbers for the products (Scheme 73b).
Similarly, other scientific groups, including Liu et al.[118] (Scheme 74a,b), Siddiki et al.[119] (Scheme 74c), and Polidano et al.[120] (Scheme 74d) have also described the metal catalyzed β-methylation of alcohols via borrowing hydrogen strategy.
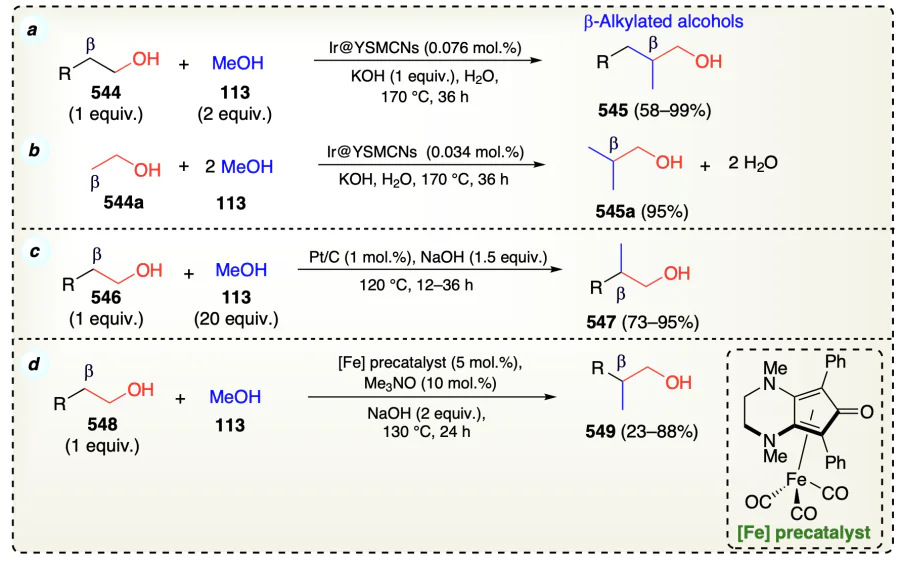
Lichosyt et al.[28] explored the palladium supported arylation methodology through β-functionalization of aliphatic alcohols using Rh complex (Scheme 75a). The results were in accordance with the intermediary of aldehydes (553 and 555) in the reaction. The mechanism involved two cycles: Ru-catalyzed activation (fast cycle) and the Pd-catalyzed functionalization wherein generation of intermediary enolate formation determined the rate of current process (Scheme 75b). The KIE study revealed that β-C – H bond breaking is the rate determining step in this protocol (Scheme 75c). In addition, the deuterium labeling experiment suggested that the oxidation of alcohol to aldehyde was faster than arylation of alcohol (Scheme 75d). Further, this protocol was applied for the synthesis of various natural product derivatives such as 552b and 552c.
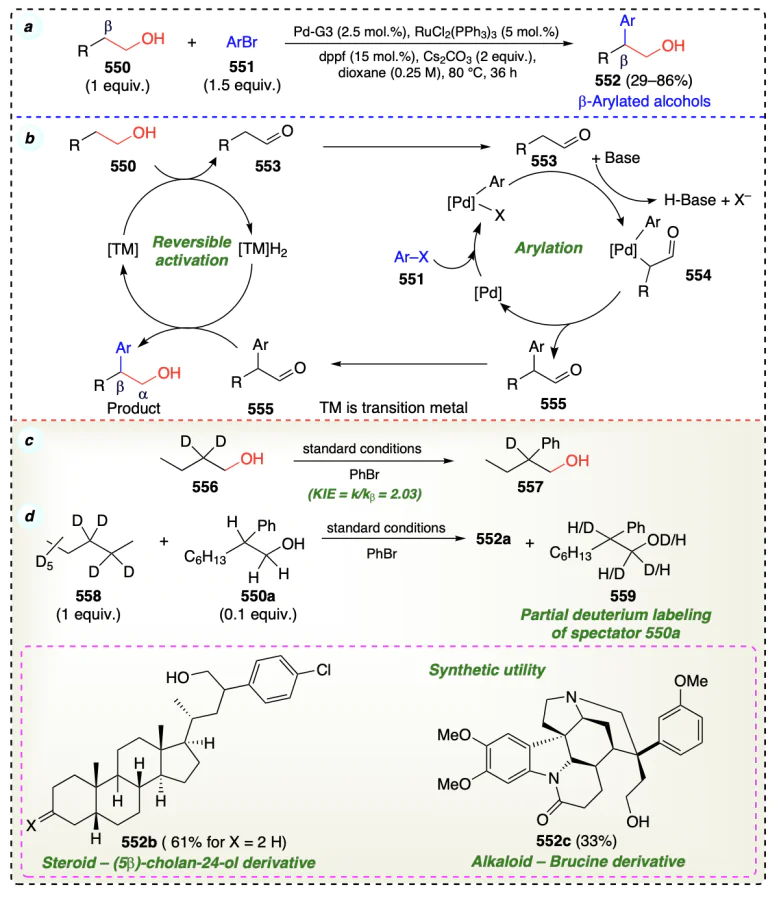
Manojveer et al.[121] reported the β-functionalization reaction to form Guerbet alcohols using ruthenium complex through borrowing hydrogenation (Scheme 76a). This method converted the 1° alcohols into corresponding β-alkylated dimeric alcohols (Guerbet reaction), and water was released as the only by-product[2, 107, 108]. This protocol was applied to synthesize rosaphen (562a) and cyclamenaldehyde (563) fragrant molecules (Scheme 76b,c). Mechanistically, this protocol follows borrowing hydrogen strategy (Scheme 6, section 2.3.3) (Scheme 76d).
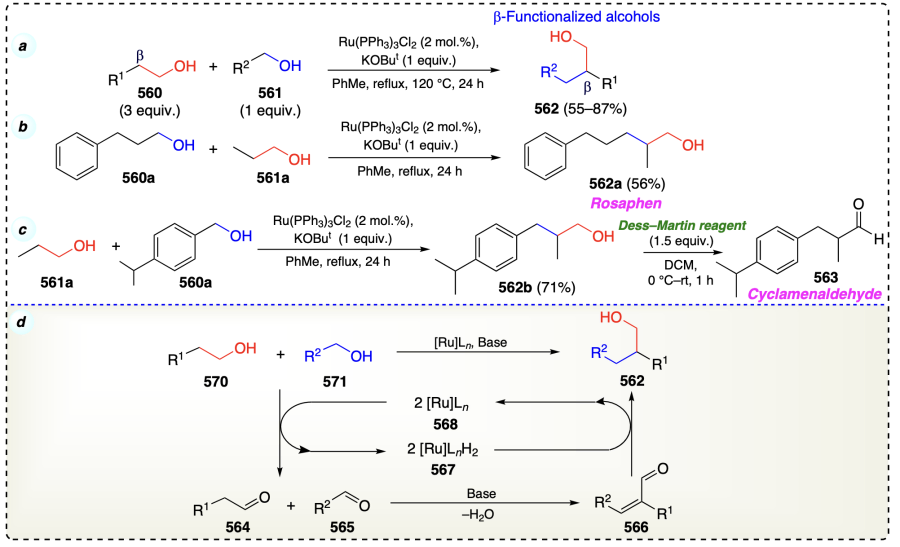
Babu et al.[122] reported nickel-catalyzed dehydrogenative C-alkylation of secondary alcohols (569) with primary alcohols (570) to prepare β-alkylated secondary alcohols (571) (Scheme 77a). The carbon-carbon bond formation proceed through borrowing hydrogen strategy (Scheme 6, section 2.3.3) (Scheme 77b). This method was further applied for synthesizing cholesterol derivative (578) (Scheme 77c). The deuterium labeling experiment suggested that alcohol underwent dehydrogenation followed by transfer hydrogenation steps (Scheme 77d). In addition, on adding radical scavengers to the reaction, product yield was not disturbed which means that this Guerbet type protocol did not follows SET mechanism (Scheme 77e).
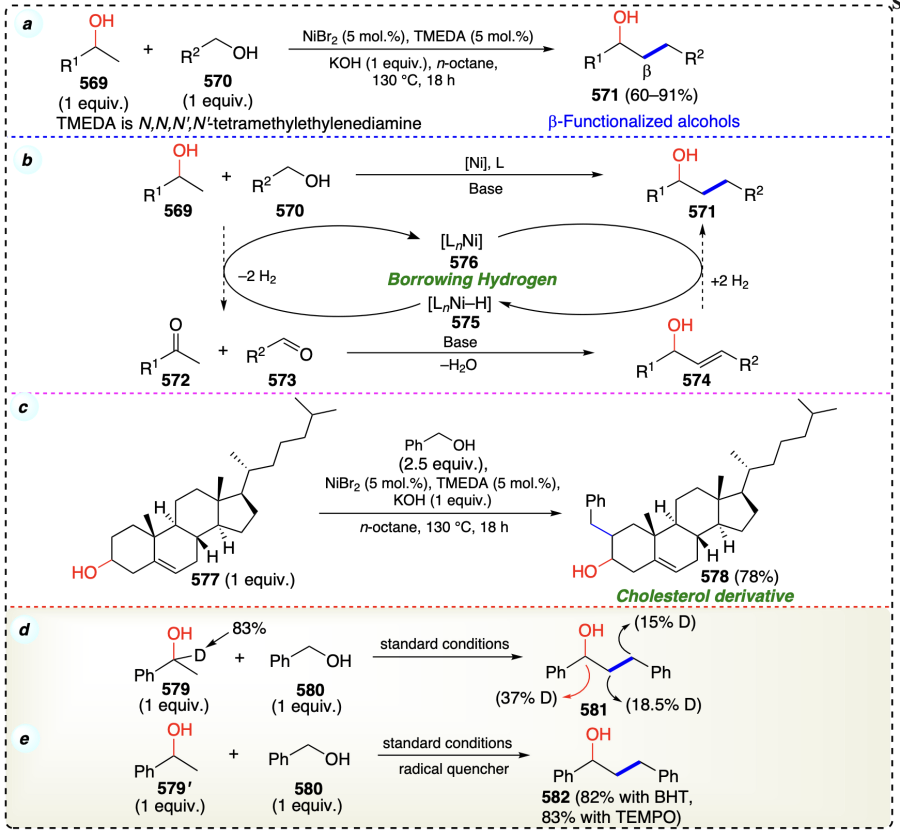
Kabadwal et al.[123] developed a nickel catalyzed coupling of secondary alcohols (583) with primary alcohols (584) to provide gem-disubstituted β-alcohols (585) through the sequence of dehydrogenation, condensation, and rehydrogenation (Scheme 78a). The catalytic cycle begins with the reaction of alcohol (583a) with Ni(acac)2. In the presence of ButONa, this reaction forms a radical intermediate 587. Studies using cyclic voltammetry (CV) and X-ray photoelectron spectroscopy (XPS) provided strong evidence supporting the conversion of Ni(acac)2 to intermediate species 587. After that, intermediate 587 is converted into intermediate 588 through the HAT step. Furthermore intermediate 588 generated the ketone (592) as intermediate along with the generation of 589. Similarly, the primary alcohol (583a) is converted into aldehyde (591) through an identical catalytic cycle (cycle II).
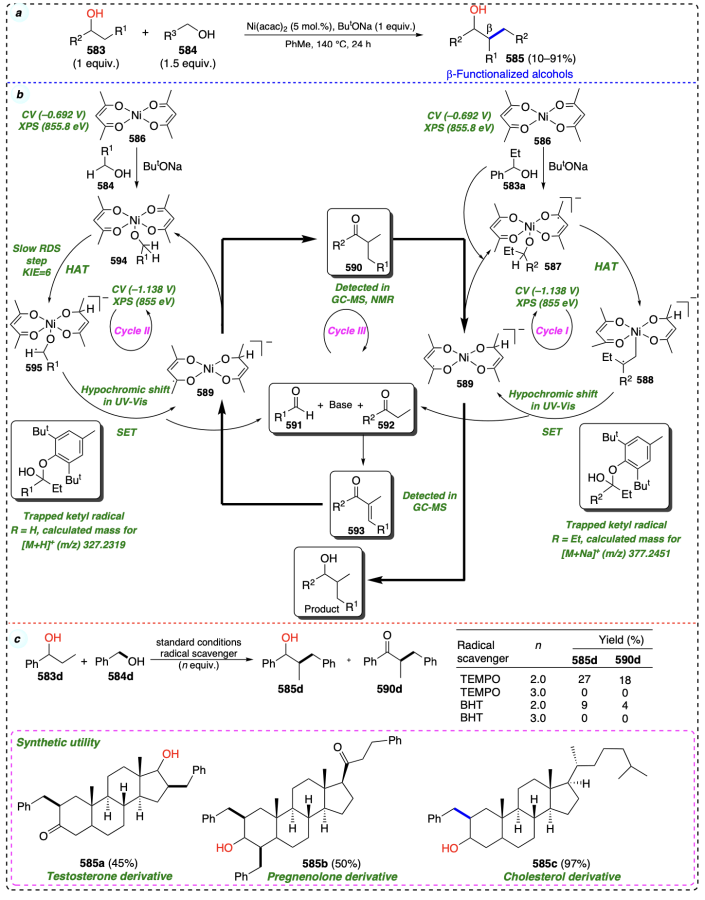
Now the base catalysed aldol reaction provide an intermediate 593, which further underwent the reduction to provide the desired product (585) (Scheme 78b). This process was efficiently applied for the synthesis of various complex molecules of importance in natural product chemistry such as 585a – c. Various mechanistic studies suggested the radical mediated mechanism for the developed protocol (Scheme 78c).
3.2.2. Metal free β-selective direct functionalization for non-activated alcohols
Nakafuku et al.[124] designed an imine (saccharin-based) radical mediated chaperone addition and removal approach for promoting functionalization of alcohols at β-position through an in situ generated hemiaminal species under photocatalytic conditions (Scheme 79a). Mechanistically, initially, alcohol substrate get inserted to an imine type catalyst (597) that formed the transient hemiaminal (600). Then, the N(sp3) atom of 600 through radical initiation generated the N-centred radical (601) with high chemoselectivity, that underwent regioselective 1,5-HAT for the generation of radical (602) at β-carbon position. Then the β-functionalized hemiaminal (602) is combined with a radical and further via intramolecular displacement it provided the aminated adduct (603), which on removal of chaperone produced β-functionalized alcohols with the regeneration of catalytic species (597) (Scheme 79b). On investigation of the various imine candidates, it was proposed that the ketone-derived imines containing electron-withdrawing groups and non-enolizable imines are favourable.
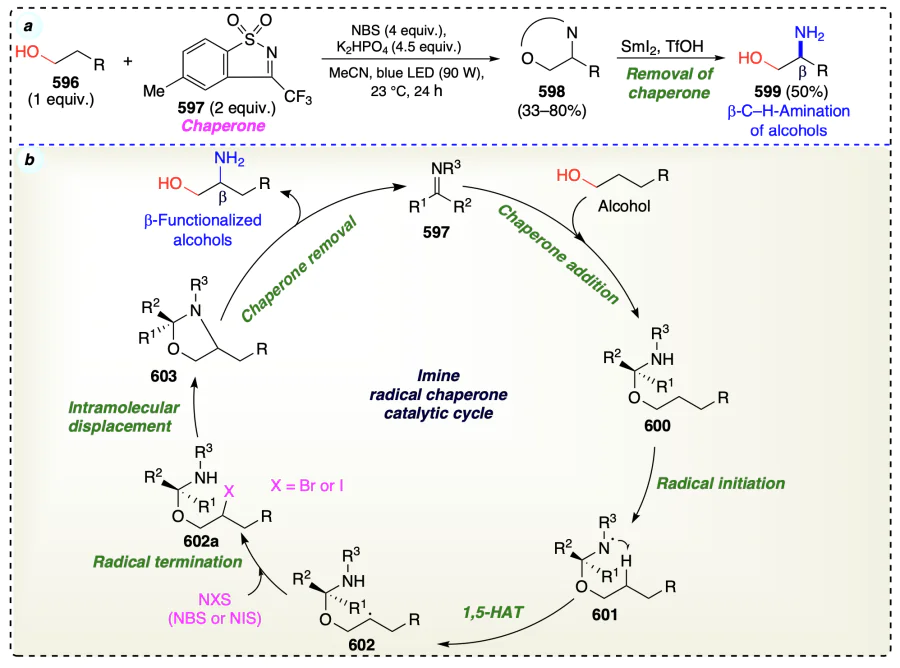
Stateman et al.[125] developed an iodine-catalyzed imidate-mediated radical relay reaction to furnish the β-C – H amination of alcohols (607, 607a) without using any transition metal catalyst (Scheme 80a,b). Here also the alcohol substrate (604) was converted into immidate and thereafter similar mechanism as of chaperone addition and removal strategies (see Scheme 79) was followed for the generation of β-aminated alcohols.
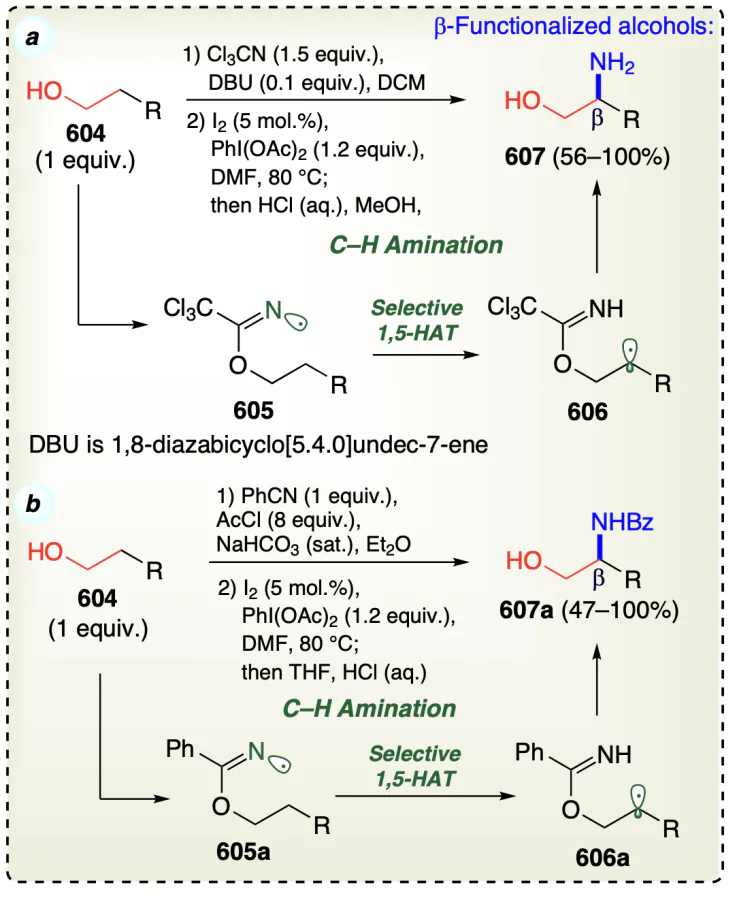
Wappes et al.[126] pioneered a radical relay chaperone approach to amination at the β-position of alcohols without using any metal catalyst and under photocatalytic conditions. 1,5-HAT and the wide functional group tolerance by radical intermediates are governing the high selectivity for current C – H amination reaction. The chaperones guided the H abstraction in current reaction by interacting with imidate radicals directly (Scheme 81a). The imidate obtained from 608a exhibited a significant primary KIE (IM) of 8.1 through an intramolecular competition between H and D and KIE of 2.1 through an intermolecular competition (Scheme 81b). This suggested that the HAT step determined the rate of reaction. Proposed iodine trap pathway was supported by the identified intermediates during reaction and explained why the subsequent nucleophilic displacement of iodide by imidate is important during the current reaction (similar to earlier mechanism, see Scheme 79).
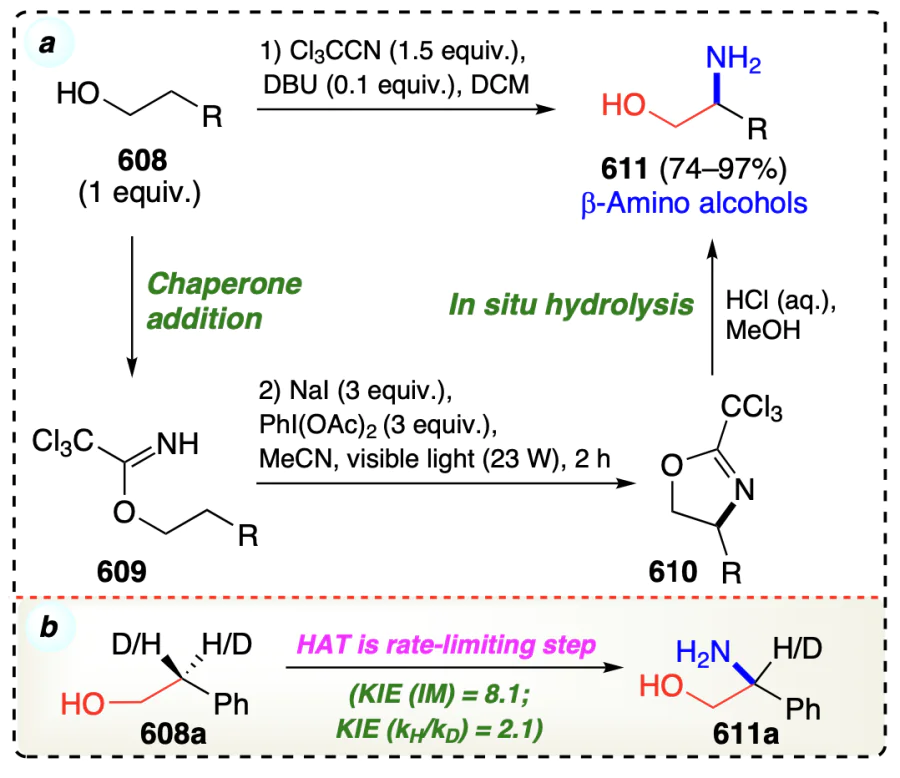
Prusinowski et al.[127] developed a method for converting alcohol to alkenes in situ via desaturation using an imidate N-radical intermediate under metal-free, photocatalytic conditions. This intermediate further underwent amino halogenation to give an iodo-oxazoline, which finally provides β-substituted amino alcohols (Scheme 82a). Acetyl iodide (AcOI) was formed first through a ligand exchange reaction between diphenyl acetate (Ph(OAc)2) and iodine (I2). Then, an imidate (613) derived from the precursor alcohol (612) underwent N-iodination to give N-iodoimidate (616). The N – I bond (weak bond) in complex 616 was photocatalytically homolyzed to give N-centered radical (617), which underwent a regioselective 1,5-HAT step and afford a nucleophilic carbon radical at b-position (618) which further provided β-alkyl iodide (619) intermediate on radical addition. Then intermediate 619 provides an alkyl triiodide nucleofuge (620) via Lewis acidic complexation of I2. In the next step, 620 underwent the elimination of HI and I2 (oxidative elimination) to generate an allyl imidate (621). Further halo-cyclization occurs either by polar amino-iodination giving (622) or by radical amino-iodination giving (623) (π addition mechanism). Thus, γ-iodo-oxazoline (614) exhibited high diastereoselectivity, suggesting a polar mechanism involving intramolecular cyclization of (622) via tethered imidate. Finally, intermediate 614 on hydrolysis gave the desired product (Scheme 82b). KIE studies substantiate that the HAT step is involved in the rate determination for intermediate C – H iodination. Also, the C – H iodination proceeded 1.6 times faster in comparison to the formation of iodo-oxazoline.
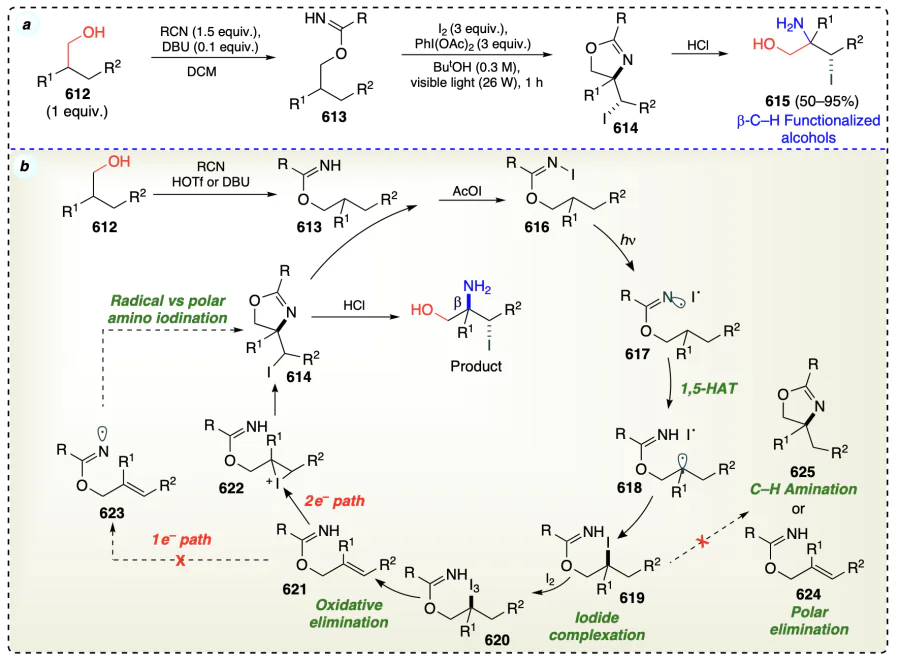
Xu et al.[128] described a transition metal-free organocatalytic protocol for β-functionalization of methyl carbinols using alcohols as alkylating agents with an aldehyde based catalyst (Scheme 83a). Firstly, the aldehyde (629) underwent Meerwein – Pondorf – Verley – Oppenauer (MPV-O) protocol with carbinol reactant (627) affording an alcohol (626) and a ketone (630) which further converted in to α,β-unsaturated ketones (631) via aldol condensation. Finally, intermediary 631 on transfer hydrogenative coupling by alcohol (626) and/or methyl carbinol (627) gave the desired product. Alkene-reduced species (632) was also generated from intermediate 631 as a by-product during the reaction (Scheme 83b).
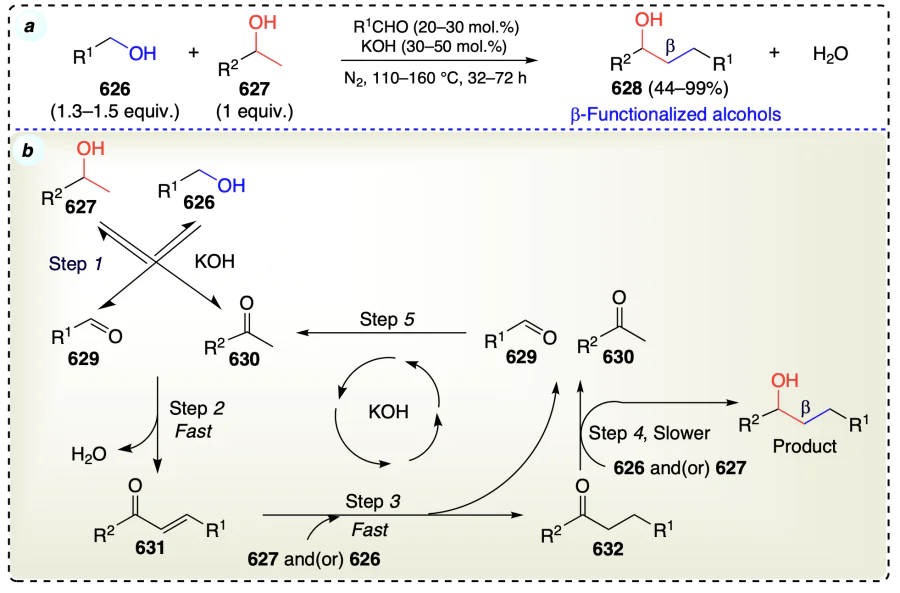
Nayak et al.[129] reported KOH-catalyzed cross-coupling of primary (634) and secondary (633) alcohols (Scheme 84a). Initially, primary alcohol underwent the base mediated deprotonation to generates a negatively charged intermediate species, benzyl alkoxide ion (636). This ion can then be further transformed into a radical anion (637), which has an unpaired electron. KOH acts as a catalyst, allowing the negatively charged phenylethanol fragment (formed by losing an electron from 1-phenylethanol) to react with acetophenone to generate intermediate 638 and water as a side product. Then 638 underwent the c-c bond formation and leading to intermediate 639. The single electron transfer from acetophenone radical anion 639 generated a potassium salt (640) and a negatively charged acetophenone molecule (radical anion). This intermediate (640) then provided a chalcone molecule (641) along with the generation of KOH. Chalcone (641) reacts with acetophenone radical anion to provide radical anion 642. This triggers a series of transfers: a base removes a hydrogen atom and an electron from benzyl alcohol, transfers the hydrogen to 642, and the electron to form an alkoxy radical anion (637). This results in the formation of β-alkylated ketone (635'). Further, 635 ́ underwent the reduction through a base-mediated transfer of hydrogen from 1-phenylethanol to provide the desired product (Scheme 84b).
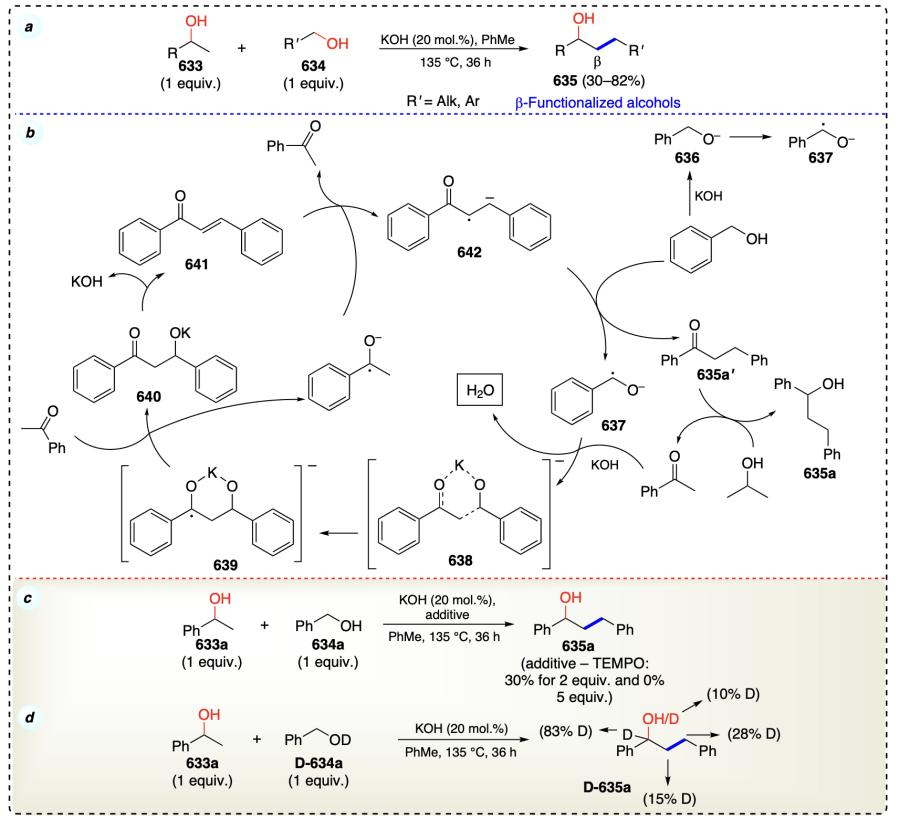
3.3. γ-Selective direct functionalization for non-activated alcohols
Herein, developments for the C – H functionalization at the remote position (γ-position) of non-activated alcohols were portrayed under different reaction conditions. An overview has been shown in Scheme 85 for the conditions used in the past by various research groups for γ-functionalization of alcohols.
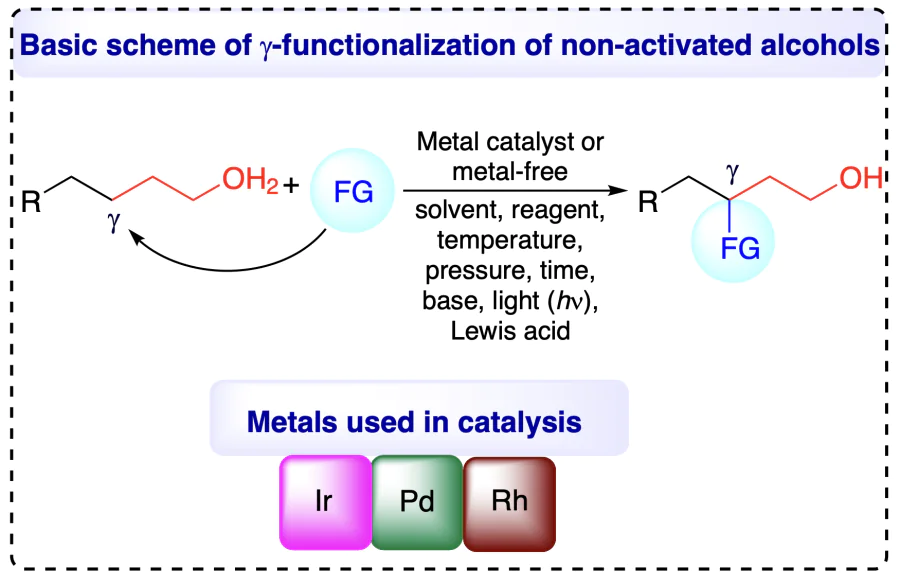
Simmons and Hartwig[130] showed an iridium catalyzed γ-functionalization of alcohols for the introduction of hydroxyl group. In this 1,4-hydroxyl-directed functionalization reaction, firstly, the silane moiety of the dihydridosilane reagent directed the functionalization by coordinating with the alcoholic oxygen (643) and forming a (hydrido) silyl ether (644). Intermediate 644 was formed by dehydrogenative coupling with 643. Secondly, the Si – H unit of 644, without removal of intermediate (644), underwent dehydrogenative silylation and thus formed oxasilolane (645). Finally, Tamao – Fleming oxidation[131-134] of 645 led to the formation of 1,3-diol (646) (Scheme 86a). This protocol was applied for the introduction of hydroxyl group at the γ-position of the alcoholic functionality bearing naturally occurring molecules. Under the standard conditions, methyloleanate (647) (a fatty acid) underwent hydroxylation at the C(23) methyl group with the help of C(3) hydroxyl functionality and led to the formation of methyl hederagenate (648) (a triterpenoid saponin). Also, methyl glycyrrhetinate (649) underwent selective oxygenation of a methyl group at C(23) to form the diol (650) (Scheme 86b).
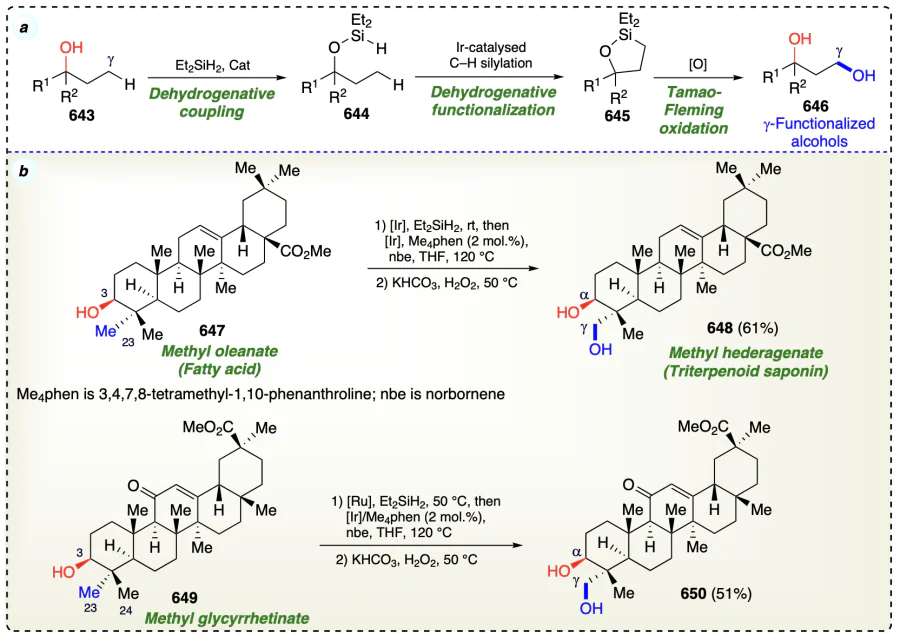
Friese et al.[135] introduced a radical translocation arylation reaction for the γ-functionalization of alcohols (Scheme 87a). This reaction involved the sulfonylation with o-iodoaryl sulfonyl chlorides and radical mediated steps for the γ-functionalizations. This cascade started with the generation of aryl radicals followed by translocation of generated radicals and sulfur to carbon center, migration of radicals, reduction, and SO2 fragmentation for the construction of all quaternary carbon centers (Scheme 87b). Arylation of the secondary C – H groups proceeded via a radical intermediate (657) and then by a chair-type cyclohexadienyl radical (658) to give 659 with R and R' substituents at equatorial positions. DFT studies further supported this mechanism.
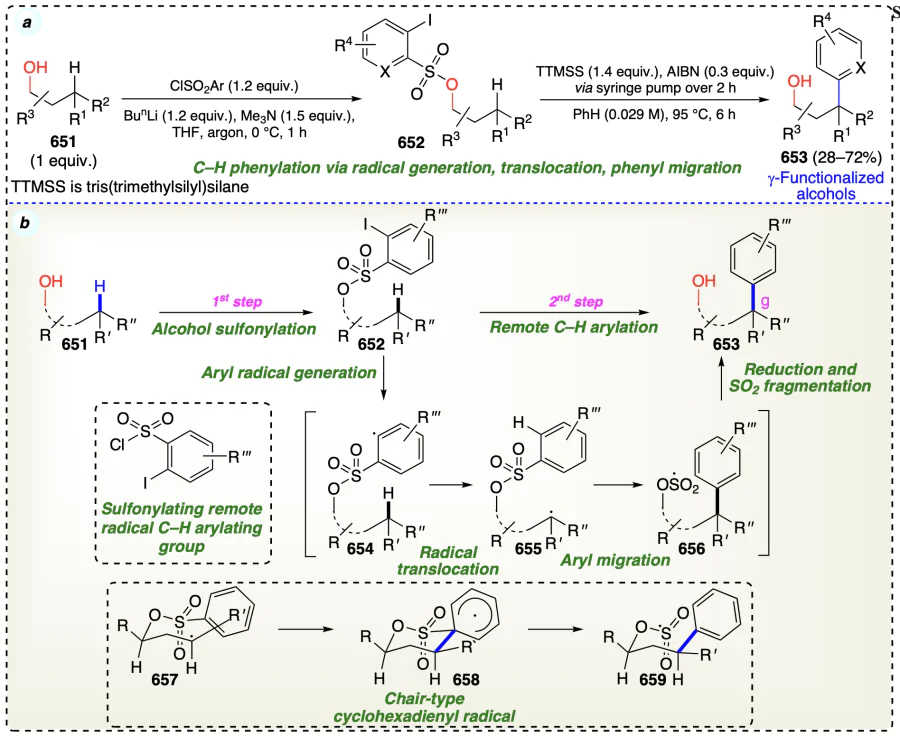
Li et al.[136] reported an iridium catalyzed intramolecular silylative γ-hydroxylation of tertiary and secondary alcohols. Reaction started with the silylation and subsequent C – Si bond oxidation resulted in the 1,3-diol as the final secondary alcohol product (Scheme 88a). This protocol had potential applications in functionalizing complex molecules such as natural products and their derivatives such as cholesterol derivative (663) (Scheme 88b). Further, KIE studies revealed that the rate limiting step was the C – H cleavage during the silylation of 2° C – H bonds (Scheme 88c).
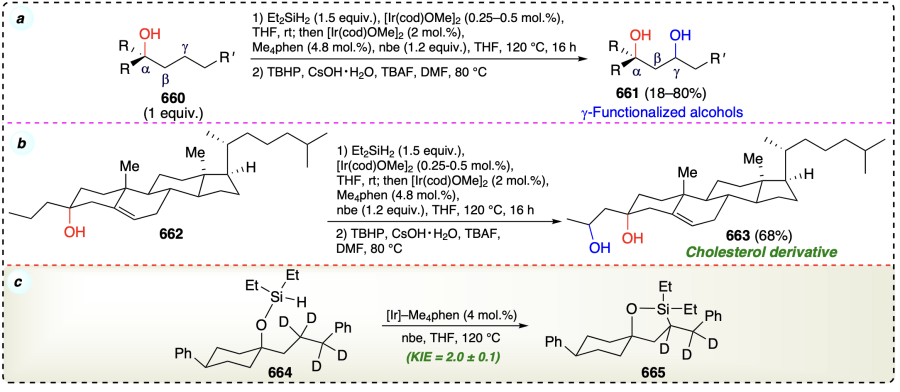
3.4. δ-Selective direct functionalization of non-nctivated alcohols
This section covers the developments in remote C – H functionalization of non-activated alcohols selectively at δ-position. An overview has been shown in Scheme 89 for the conditions used in recent times by different research groups.
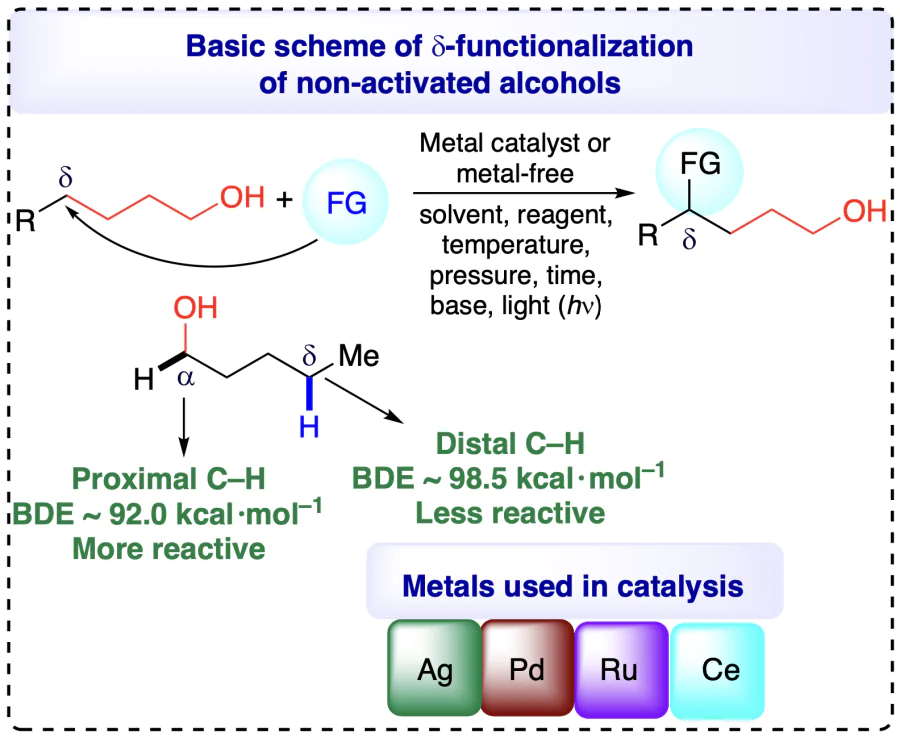
Zhu et al.[137] developed a silver-mediated method for selectively activating Csp3 – H bonds in inexpensive and readily available aliphatic alcohols. They converted the aliphatic alcohols into oximonitrile-substituted alcohols under mild conditions (Scheme 90a). The inhibition of reaction on the addition of TEMPO or BHT indicated that this transformation involved the radical-mediated pathway (Scheme 90c). Alkanols acts as chain initiator for the current radical promoted reaction and helps in β-scission, further supporting the possibility that the current silver/oxidant catalysis enables generation of oxygen centered radicals. Initially, AgNO3 (AgI) was oxidized to AgII in the presence of K2S2O8. Then, alcohol (666) coordinated with AgII and gave an intermediate (669) which underwent homolytic cleavage to give alkoxyl radical (670) and regenerate AgI. Next, 670 underwent 1,5-HAT to give radical species (671), which combined with sulphonyl reagent, yielded the radical (672). Finally, the fragmentation of radical 672 afforded the δ-functionalized alcohol as a product with the release of sulphonyl radical (673), which further transformed into benzene sulphonic acid (Scheme 90b)[138].
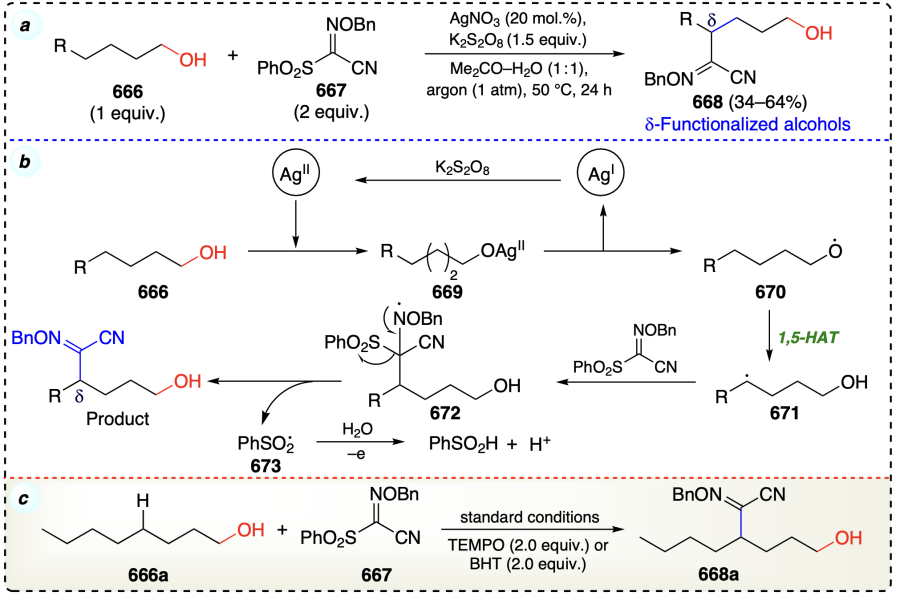
Li et al.[139] developed a protocol for the δ-functionalization of alcohols (674) to introduce heterocyclic scaffolds (675). This photoredox method is energy efficient and cost-effective, and used PFBI-OH (676) as oxidant (Scheme 91a). The protocol was efficient in synthesizing drug molecules tarocin A1 (677a) and camptothecin (677b) etc. with good yields. PFBI-OH reacts with an alcohol to form radical intermediate via single electron transfer and then followed the 1,5-HAT pathway and construction of C – C bond via Minisci reaction[140]. The isobutoxy radical undergoes β-scission and resulted in the formation of an isopropyl radical. At first, the alcohol (674) underwent alcoholysis with 676 to give 678, which on reduction via SET provide intermediates 679 and 680. The iodocenter is more electrophilic in the oxidising agent because of the neighbouring fluoro groups and facilitate alcoholysis, due to which (678) was easily reduced via SET.
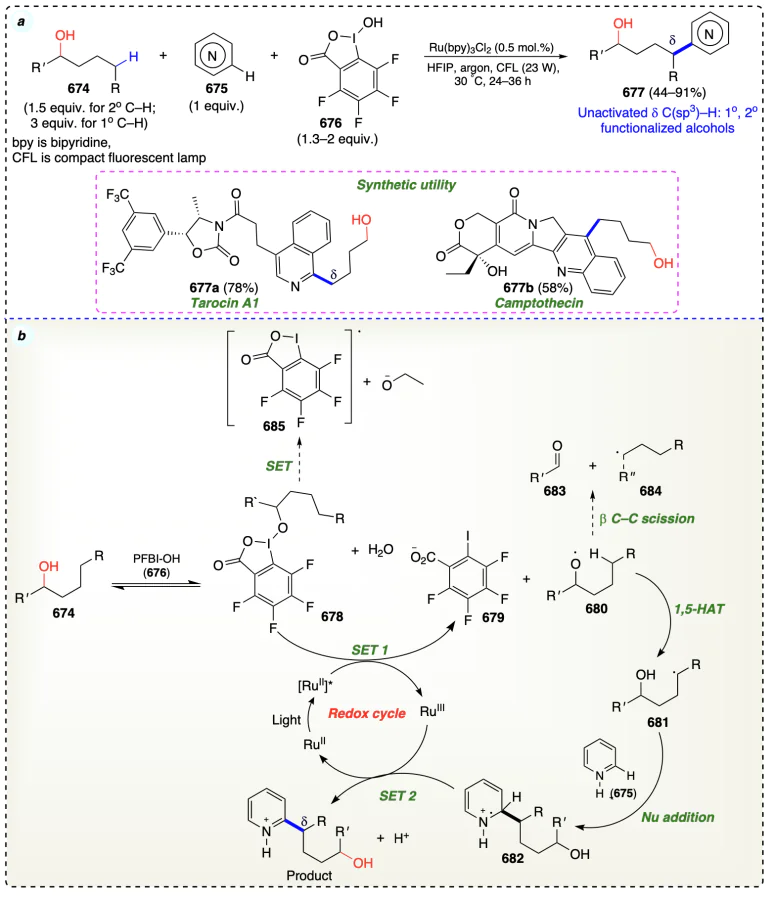
Alkoxy radical (680) on 1,5-HAT gave radical 681 combined with 675 and formed 682 intermediate. Finally, 682 underwent SET oxidation via Ru(III) and provided required δ-functionalized alcohols (677). In principle, 680 could have undergone β-scission to form 683 and radical 684. However, this path was negligible for alcohols with no β-substituents (Scheme 91b).
Barday et al.[141] developed a photocatalytic regio-selective δ-functionalization of alcohols for the introduction of thiotrifluoromethyl functionality under photocatalytic conditions. Free alcohol (686) generates an alkoxy radical under proper oxidant and visible light, which underwent 1,5-HAT and generated the δ-carbon centered radical intermediate which further reacted with SCF3 (687) to provide the desired product (688) (Scheme 92a). ArSO2SCF3 reagent have high electron-withdrawing ability and showed good Hantzsch parameters, therefore selected as a source of SCF3[142-144]. The radical scavenging experiments revealed the involvement of radical-mediated pathways during the reaction (Scheme 92c). Initially, dialkoxyiodo benzene intermediate (689) formed, which on irradiation underwent homolysis to give alkoxy radical (690). Alkoxy radical (690) on 1,5-HAT formed an alkyl radical (691), that on reacting with 687, afforded the desired product (Scheme 92b).
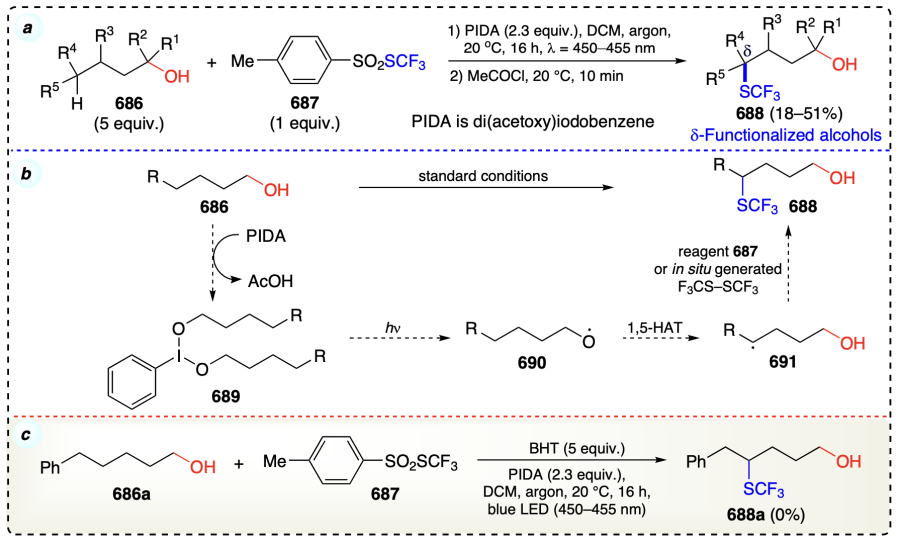
Wang et al.[145] described a silver-catalyzed methodology for selective δ-functionalization of alcohols (692) using diverse N-heterocycles (693) in water as green solvent system (Scheme 93a). With both the protonic and Lewis acids, the N-heteroarenes were efficiently activated in this Minisci-type reaction[137]. Lepidine (693a) (a natural product) was used as model substrates for reaction condition optimization. At the beginning of the optimization, they prepared radical scavenging experiment using TEMPO to confirm the involvement of radical pathway (Scheme 93c). Reaction initiated with Ag(I) oxidation to Ag(II) in the presence of S2O82–. Thereafter, Ag(II) reacted with 692 and generated Ag(II) – O species (695), which underwent homolytic cleavage and gave an alkoxyl radical (696) along with regeneratation of Ag(I). Then, 696 underwent 1,5-HAT via a six-membered ring transition state and gave δ-carbon position radical (697). Next, 697 reacted with 693 to form a radical intermediate (698) which finally oxidised to provide d-functionalized alcohols (Scheme 93b).
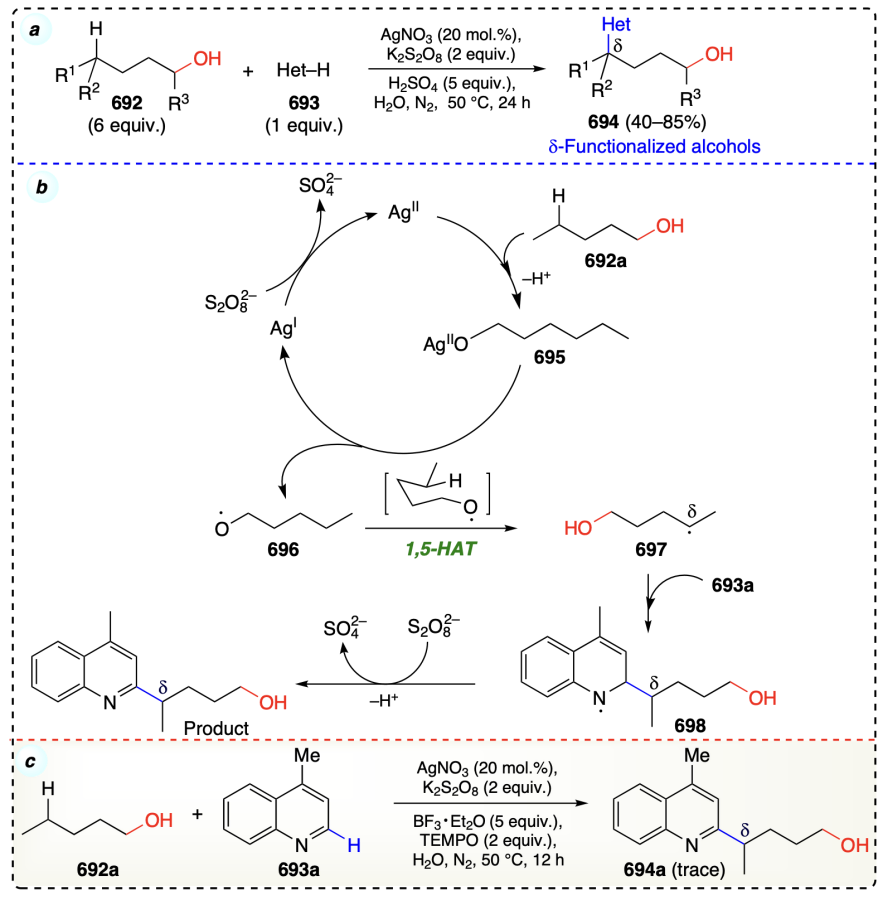
Regarding the significance and omnipresence of heteroaryl components in medicinal and biologically active molecules, intramolecular heteroarylation is less prominent than intermolecular one. Also, as per the BDE values for the C – H bonds, the hydroxyl proximal C – H groups are more reactive than the distal ones, which makes the selective functionalization at the distal site a big challenge[146-151]. Thus, Wu et al.[152] reported a regioselective intermolecular heteroarylation of alcohols via a Minisci-type reaction under photocatalytic conditions (Scheme 94a). Before this work, the primary alcohols were only used to generate alkoxy radicals. In this reaction, all types of alcohols could afford alkoxy radicals, which were responsible for triggering the heteroarylation of C(sp3) – H bonds. Mechanistically, using ether in place of hydroxyl group inhibited the reaction, thus supporting that free alcohols enabled the reaction. The mixture of alcohol (699a) and reagent (PIFA) gave a dialkoxyiodo benzene intermediate (703) which, on visible-light promoted homolytic cleavage generated the alkoxy radical (704). Radical 704 triggered the 1,5-HAT and provide alkyl radical (705). PhI and iodanyl radical were cogenerated. Further, radical 705 underwent nucleophilic addition with a quinoline salt (706) and afforded the intermediate 707, which was oxidized by PIFA or by the in situ generated iodanyl radical to give the d-functionalized alcohols (Scheme 94b).
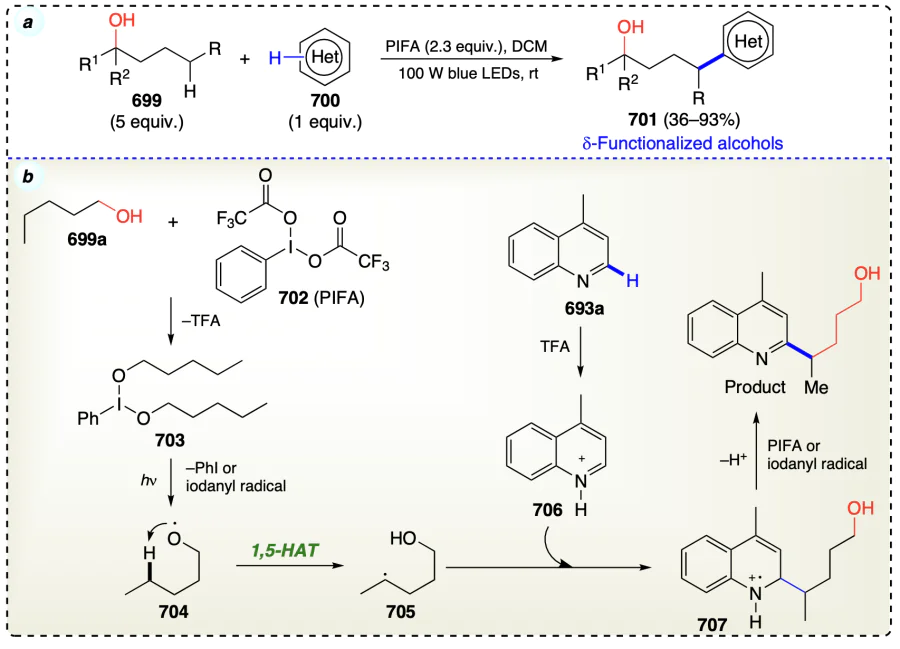
Hu et al.[153] developed a new, efficient, and easy-to-use method for selective δ-functionalization of primary alcohols with azo compounds (Scheme 95a). The photocatalytic conditions provides the green matrix to the current protocol. The authors illustrated ligand to metal charge transfer (LMCT) based catalytic process is an efficient way for the formation of alkoxy radicals. Ce(IV) complexes used here are known for the redox reactions induced by LMCT excitation, thus reducing the cerium center to Ce(III) and ligand oxidation. Alcohols easily ligate to the metal center (711) and are excited under visible-light, and afterwards the homolysis of Ce(IV) – OR generated the oxygen centered radical (712). Then the thermodynamically more favourable 1,5-HAT event activated the d-C – H bond and form a highly nucleophilic alkyl radical, which reacts with ditert-butylazodiformate to form a N-centered radical (714). Finally, the single-electron reduction of 714 provide the d-functionalized alcohols (Scheme 95b).
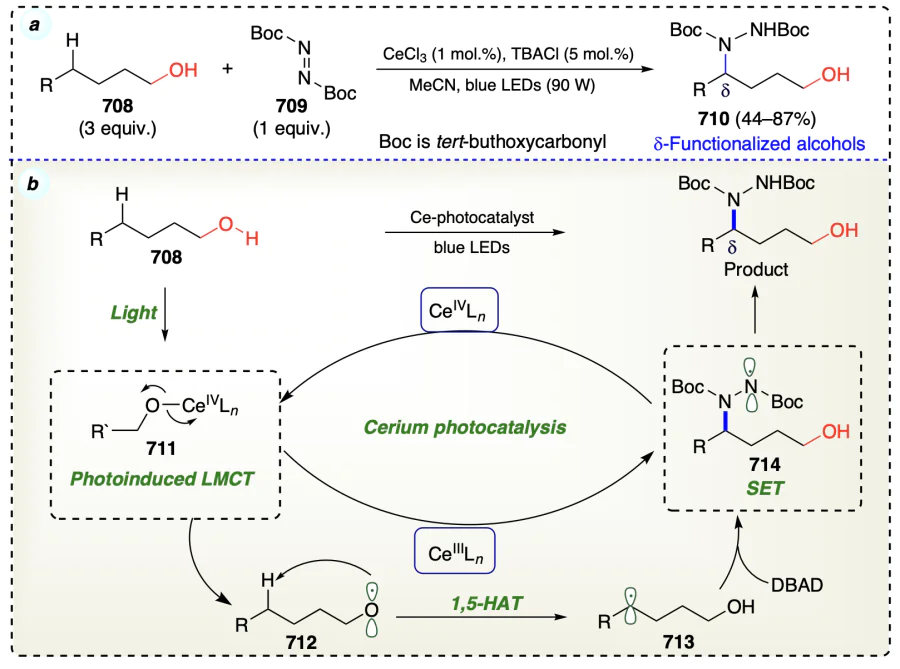
Herron et al.[154] developed a method for the δ-functionalization of alcohols to achieve the fluorination and chlorination of remote C – H bonds under moderate conditions and with high mono-selectivity. Further, this precursor also promotes the formation of remote dihalides by introducing a second halogen atom. The one-electron oxidation of directing group in the substrate 716 (which was prepared from inexpensive naturally occurring pyruvic acid, (715) initiated the decarboxylative sequence to provide the alkoxy radical (719) in addition to the generation of CO2 and acetonitrile. Radical 719 was trapped by selectfluor to provide d-mono-functionalized alcohols (724), although moderate di-halogenation also occurred to give 726 (Scheme 96).
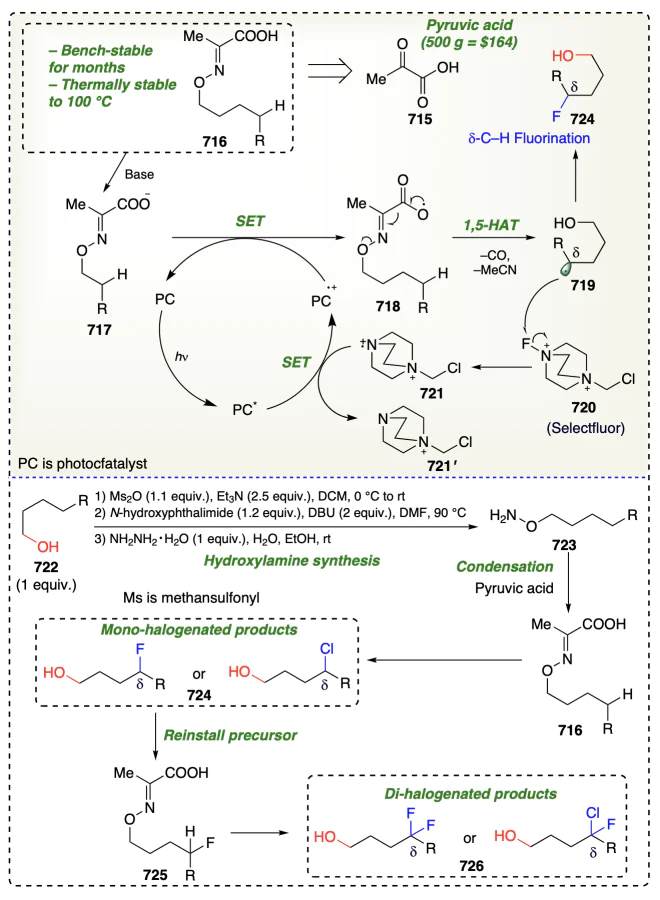
Zhou et al.[155] developed proficient δ-functionalization of free alcohols for the introduction of heterocyclic functionalities using silver based catalyst (Scheme 97a). A gram scale reaction was also performed to examine the efficiency of the method (Scheme 97b). Initially, alcohol substrate (728) coordinated with AgII species, formed by AgI oxidation using K2S2O8 giving an intermediate (730) which, on activation, provided the alkoxyl radical (731) and AgI species. Alkoxy radical (731) underwent the 1,5-HAT sequence and formed C-centered radical (733), that reacts with N-heteroaromatic salt (732) and generated the radical cation (734). The radical cation (734) on hydrogen abstraction finally afforded the desired alkylated product (Scheme 97c). The radical scavenging experiment revealed that an oxygen radical might initiate this protocol and corroborated the mechanism (Scheme 97d).
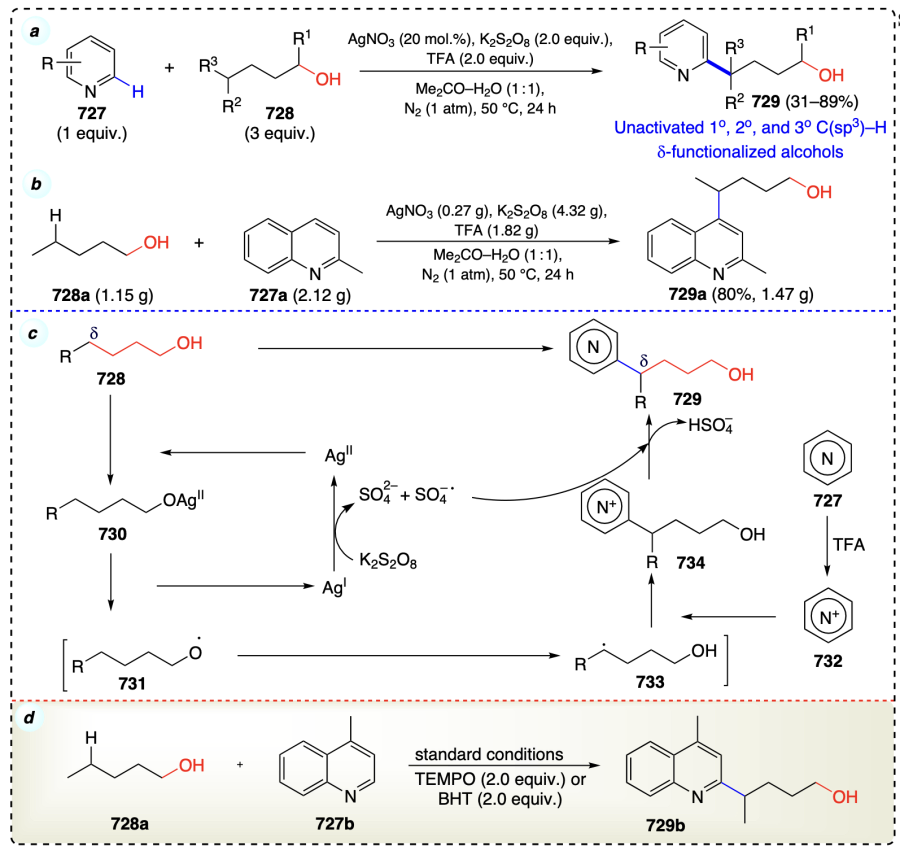
Xiong et al.[156] described a photocatalytic protocol for the amination of free aliphatic alcohols at d-position using inexpensive FeCl3 catalyst (Scheme 98a). The reaction was employed for the scaled-up reactions (gram scale) using a variety of substrates (Scheme 98b). Controlled experiments revealed the involvement of radical mediated mechanism and found that the C(sp3) – H bond cleavage is irreversible (Scheme 98d,e). Light triggers the reaction by forming an oxygen-based radical intermediate (738). The exact mechanism is unclear, but it likely involves either electron transfer from a ligand to the metal center or hydrogen abstraction by a chlorine radical. 738 underwent the 1,5-HAT to generate a carbon centered radical intermediate (739). The radical addition reaction provide an nitrogen centered radical intermediate (740). This intermediate (740) underwent the SET followed by protonation in the presence of ferrous iron (Fe2+) and hydrochloric acid (HCl) to provide the desired product (737b) along with the regeneration of iron catalyst in its trivalent form (Fe3+) (Scheme 98c).
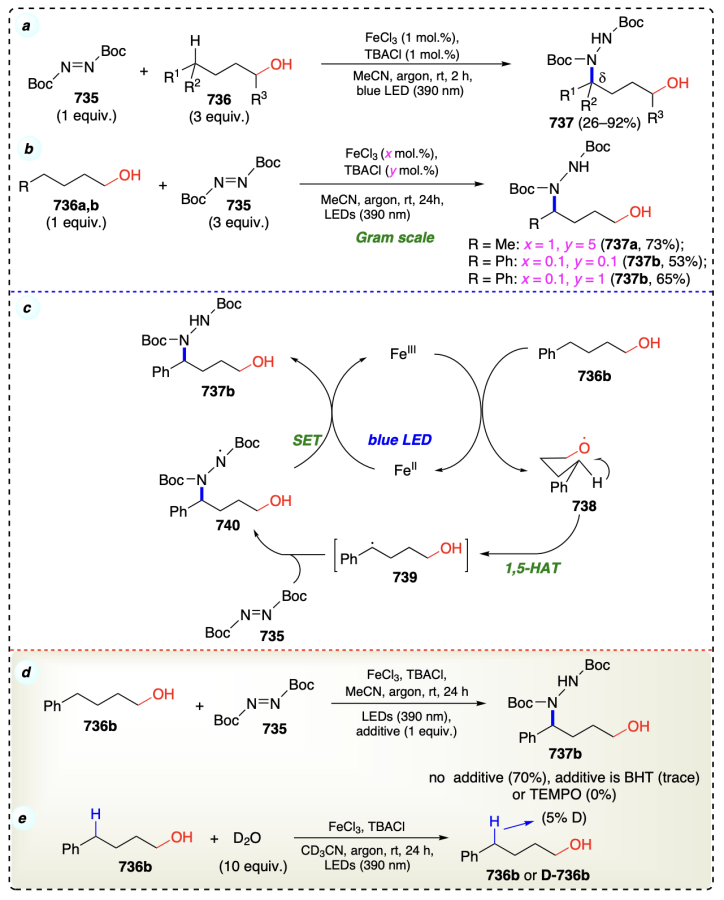
4. Conclusion and future prospects
Alcohols are captivated starting materials for the scientific community in recent years, particularly for the C – H functionalization and construction of C – C bond due to their several advantageous features such as inexpensive, easy to handle, easily storable, readily available, environment-friendly and sustainable compounds[41]. Regio- and stereo-selective direct functionalizations at diverse positions (α-, β-, γ-, and δ-positions) via various strategies have enabled access to the functional molecules for further organic transformations that are useful in chemical, biological, medicinal, pharmaceutical, and natural product fields[136, 139, 157, 158]. The utilization of alcohols as starting materials mainly for the coupling reactions provides an effective alternative for the halogenated substrates that are traditionally used for the purpose and known for the generation of toxic waste and by-products. The α-C – H bond of alcohols is more reactive than distal (β-, γ-, δ-C – H) bonds due to the lower BDE values and it makes the α-position comparatively reactive and easy for the functionalizations. Scientific community resolved the issues associated with the conventional approaches for the functionalization of alcohols through modern approaches and developments. The literature scrutiny revealed that a lot of work has been done on the functionlization of α-, and β-positions in alcohols whereas the γ-, and δ-positions still remains to be explored up to the strength. Albeit, various researchers have explored numerous transition metals based catalytic processes, however, more focus was on the utilization of Ir and Ru metal-based catalysts which are very costly, followed by Rh, Fe, Mn, Cu, Ag, Pd, etc. Expensive metal catalysts like Ru, Ir, Rh, Pd, etc. can significantly increase production costs. Hence, there is a vast scope for the development of low cost and abundant metals (Fe, Co, Ni, Cu, Zn etc.) based catalysts for the functionalization of free alcohols at diverse positions. In addition, promotor-assisted metal-free α-C – H functionalization in unactivated alcohols displayed with reagents such as AIBN, DTBP, and selectfluor that can further be applied at other positions for the development of sustainable methods. In addition, most functionalizations at δ-position in alcohols have been performed in the presence of light which itself is a highly demanding and sustainable field. Hence more efforts are required for the development of photocatalytic procedures for the functionalizations of free alcohols. The utilization of borrowing hydrogen strategies provide the highly atom economic procedures for the functionalizations of free alcohols. Researchers have extended their interest to utilize these functionalization strategies in preparing complex functional molecules and in natural product synthesis including bryostatin (111), a macrolide lactone; pironetin (261), an anticancer agent; steroidal scaffolds (429, 431, 552b, 578, 585a – c); benzopyran derivative (435), a naturally occurring class of molecules; alkaloid (552c); rosaphen (562a) and cyclamenaldehyde (563), fragrant molecules; triterpenoids (648, 650); tarocin A1 (677a), camptothecin (677b), and spirastrellolide B and F (250) natural molecules etc. During the synthesis of these important molecules, minimization of reaction steps were also observed.
The future of regioselective direct functionalization of free alcohols has immense potential. The continuous exploration of new catalyst systems with even higher selectivity and activity will further streamline complex molecule synthesis. Additionally, the development of methodologies compatible with diverse substrates and functional groups will broaden the applicability of this approach. Overall, regio-, chemo, and stereoselective direct functionalization holds the key to unlock new avenues in the realms of natural product synthesis, drug discovery, and advanced material design. The current review provides new insights for future developments in the field.
5. List of abbreviations and designations
Pc — critical pressure,
Tc — critical temperature,
acac — acetylacetonate,
AIBN — azobisisobutyronitrile,
BARF — tetrakis[3,5-bis(trifluoromethyl)phenyl]borate,
BBN — borabicyclo[3.3.1]nonane,
BDE —bond dissociation energy,
BH — borrowing hydrogen,
BHT — butylated hydroxytoluene,
BINAP — 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl,
BIPHEP — 2,2'-bis(diphenylphosphino)-1,1'-biphenyl,
Boc — tert-butyloxycarbonyl,
bpy — 2,2′-bipyridine,
Cat — catalyst,
CFL — compact fluorescent lamp,
cod — 1,5-cyclooctadiene,
Cp — cyclopentadienyl,
CpOH— 2-hydroxy-2-methyl-propanenitrile,
CV — cyclic voltammetry,
Cy — cyclohexyl,
DABCO — 1,4-diazabicyclo[2.2.2]octane,
dba — dibenzylideneacetone,
DBAD — di-tert-butyl azodiformate,
DBU — 1,8-diazabicyclo[5.4.0]undec-7-ene,
DCE — dichloroethane,
DCM — dichloromethane,
DCP — dicumylperoxide,
dcypf — 1,1'-bis(dicyclohexylphosphino)ferrocene,
DFT — density functional theory,
DIAD — diisopropyl azodicarboxylate,
dippf — [bis(diisopropylphosphino)ferrocene],
DPPB — 1,4-bis(diphenylphosphino)butane,
dppm — bis(diphenylphosphino)methane,
dppf — 1,1'-bis(diphenylphosphino)ferrocene,
dppp — 1,3-bis(diphenylphosphino)propane,
DTBP — di-tert-butyl peroxide,
E — electrophile,
ET — electron transfer,
FG — functional group,
HAT — hydrogen atom transfer,
HFIP — hexafluoroisopropanol,
KIE — kinetic isotope effect,
L — ligand,
LA — Lewis acid,
LEDs — light emitting diodes,
LMCT — ligand to metal charge transfer,
M — metall,
MPV — Meerwein – Ponndorf – Verley,
MPV-O — Meerwein – Pondorf – Verley – Oppenauer,
Ms — mass spectrometry,
nbe — norbornene,
NBS — N-bromosuccinimide,
NHC — N-heterocyclic carbene,
NIS — N-iodosuccinimide,
NMP — N-methylpyrrolidone,
NPhth — phthalamide,
Ns — nitrobenzenesulfonyl,
Oct — octyl,
PCET — proton-coupled electron transfer,
PIDA — di(acetoxy)iodobenzene,
PIFA — bis(trifluoroacetoxy)iodo benzene,
PT — proton transfer,
rt — room temperature,
RTCA — сarbonyl addition via redox-triggered carbonyl
addition,
SET — single electron transfer,
SiT — silyl transfer,
TBA — tetra-N-buthylammonium,
TBDPS — tert-buthyldiphenylsilyl,
TBHP — tert-buthylhydroperoxide,
TBS — tert-buthyldimethylsilyl,
TEMPO— 2,2,6,6-tetramethylpiperidinyloxy,
Tf — triflic,
TFA — trifluoroacetic acid,
TFE — trifluoroethanol,
THC — transfer hydrogenative coupling,
TIPS — triisopropylsilyl,
TMEDA — N,N,N',N'-tetramethylethylenediamine,
Tp — hydrotris(pyrazolyl)borato,
TTMSS — tris(trimethylsilyl)silane,
XPS — X-ray photoelectron spectroscopy.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through the Large Project number 2/454/45 and the authors acknowledge the Research Center for Advance Materials (RCAMS) at King Khalid University, Saudi Arabia for their valuable technical support.
CSIR-IHBT Communication No. 5519.
References










