



![Electronic and electrochemical properties of thiophen-containing D – A – D type systems and polymers based thereon[1184, 1193, 1195, 1196].](/storage/images/resized/cT543iO6Dzgn9TxKiLT0oGxstkgFDQTPhVI8T93B_xl.webp)
![Electronic and electrochemical properties of the D – A – D system[1184, 1193, 1195].](/storage/images/resized/t7Bt8Xic7CJXAgXpv1CZAgyZwy0P5SagqhCqZs0T_xl.webp)
![Photophysical properties of thiophen-containing conjugated systems[1184, 1193, 1196, 1197].](/storage/images/resized/ZcDcH4fL6Y0UHtbi9vG7Mpg5YhTfrPqEa3k43XAp_xl.webp)
![Ozonide cytotoxicity against cancer cells and reduction potentials (ERed)[1707, 1710].](/storage/images/resized/fLlJAcN8YAAaUAg5C5xn6kl9Mr3lGJxCbRiLPsjs_xl.webp)
![Cytotoxicity of tetraoxanes against HeLa cells[1706].](/storage/images/resized/Yu4awJKAVNvld8XCEkZma3X9mDS9njnuqK86Rais_xl.webp)
![Cytotoxic activity of colchicine analogues in vitro against tumour cell lines[2191, 2192].](/storage/images/resized/qG50G96feWzpAp9OAT2B8fOSiEwIqwE1Xy03OxeK_xl.webp)
![Ring-chain tautomerism in 1-substituted 1,2,3-triazoles[1261].](/storage/images/resized/Nt2SxdAvkHRFp4bqyGnh3WfuKuwKCmkhZylmzAEi_xl.webp)
![sPz12 molecular structure (а) and packing in the crystal (b) (CCDC 2178250)[1814].](/storage/images/resized/1WcGGDLFhi1RIRuHw3urEwxZg6ze1N4Er91gDYiq_xl.webp)



![Molecular structures of complexes of subporphyrazines sPz28b (a) and sPz29b (b) with С60 fullerene (CCDC 1571526 and 1571525)[1864, 1865].](/storage/images/resized/0qNtwvE4P2K28P8UpWnLxApU82tmGMwUu5hJZS9r_xl.webp)
Keywords
Abstract
The chemistry of heterocyclic compounds has traditionally been and remains a bright area of chemical science in Russia. This is due to the fact that many heterocycles find the widest application. These compounds are the key structural fragments of most drugs, plant protection agents. Many natural compounds are also derivatives of heterocycles. At present, more than half of the hundreds of millions of known chemical compounds are heterocycles. This collective review is devoted to the achievements of Russian chemists in this field over the last 15–20 years. The review presents the achievements of leading heterocyclists representing both RAS institutes and university science. It is worth noting the wide scope of the review, both in terms of the geography of author teams, covering the whole of our large country, and in terms of the diversity of research areas. Practically all major types of heterocycles are represented in the review. The special attention is focused on the practical applications of heterocycles in the design of new drugs and biologically active compounds, high-energy molecules, materials for organic electronics and photovoltaics, new ligands for coordination chemistry, and many other rapidly developing areas. These practical advances would not be possible without the development of new fundamental transformations in heterocyclic chemistry.
The bibliography includes 2237 references.
1. Introduction
Heterocyclic compounds are ubiquitous in nature, where they are involved in key biochemical processes and also form the basis of a significant proportion of medicinal compounds, pesticides and plant growth regulators. Traditionally, this field of chemistry has been associated with the design of energetic materials. Heterocyclic compounds are used as photoactive components in photovoltaic cells and as emitters in organic light-emitting diodes (OLEDs). Recently, heterocycles have been actively utitized as photocatalysts and photoinitiators in organic synthesis. Heterocycles are confidently taking their place in the design of polymeric materials, metal-organic frameworks and starting materials for the manufacture of sensors.
The chemistry of heterocyclic compounds has traditionally been one of the most important fields of research in Russian chemistry. A significant number of reactions bearing the names of Russian chemists are related to the synthesis and transformation of heterocycles. Examples include the reactions of Chichibabin, Yuryev, Povarov, Trofimov and others. The inexhaustible structural diversity of heterocyclic structures and their properties remains a fertile ground for exploration research and poses synthetic challenges for chemists. This review highlights the achievements of Russian chemists in this field over the last 15 – 20 years and consists of chapters by leading research groups from institutes of the Russian Academy of Sciences and Russian universities.
The generalized studies closely intertwine the methodology of organic synthesis, the search for biologically active compounds and functional materials, and the analysis of structure – property relationships. The main research focus of Russian heterocyclic chemists is the development of an effective methodology for the synthesis, functionalization and transformation of heterocyclic systems, which, inter alia, opens access to previously inaccessible structures. This area is discussed in Section 2 of this review. Section 3 analyzes the studies of the chemistry of natural heterocyclic systems and their derivatives. We believe that this review, which has gathered and analyzed a noticeable part of findings of Russian scientists in the field of heterocyclic compounds in the 21st century, will be useful to a wide range of chemists and specialists in related fields.
2. Synthesis and transformations of synthetic heterocyclic systems
2.1. SNH-Reactions in the synthesis of fused heteroaromatic systems
This Section reviews publications of the last decade on the synthesis of fused heteroaromatic systems using nucleophilic aromatic hydrogen substitution (SNH reactions) and metal-catalyzed cross-coupling reactions as complementary synthetic methodologies for the introduction of (het)aryl substituents into pyrimidines, pyrazines, furazanopyrazines and other heteroaromatic systems. The combination of these reactions provides a branched scaffold decorated with (het)aryl substituents for its subsequent transformation into heteroaromatic polycyclic systems based on π-deficient azaaromatic compounds. This opens access to fused heterocycles such as pyrimidines, quinazolines, pyrazines, quinoxalines, furazanopyrazines and indolopyrazines, as well as annulated 1,3-diazapyrenes obtained through the intramolecular SNH reactions, the Scholl condensation or photoinitiated cyclizations.
The last decade has seen a marked increase in research interest in polycyclic heteroaromatic structures, with applications as advanced fluorescent materials and chemosensors, organic light-emitting devices and semiconductors[1-7]. As an example, we can mention fused 1,3-diazapyrenes 2.1.1 – 2.1.3(Herein, the compound number, indicated by an Arabic numeral, is preceded by the number of the subsection in which its structure is given.), electrophysical properties of which allow to consider them as potential organic semiconductors[8].

To construct such polycyclic systems, mono-, bi- or tricyclic derivatives of azines, diazines or triazines are usually used as starting compounds, with structures being successively modified with aromatic or heteroaromatic substituents, thus creating a branched carbon framework for the subsequent formation of intramolecular C – C bonds between (het)aryl moieties. Polycyclic compounds 2.1.1 – 2.1.3 were obtained from 5-bromopyrimidine via the following reaction sequence: the Suzuki cross-coupling, double bromination of the aryl substituent, the double Suzuki cross-coupling and, finally, double intramolecular C – H/C – H-coupling of nucleophilic biphenyl or hetarene substituents (thiophene, furan) with the pyrimidine ring, proceeding in the presence of ferric chloride, which can be regarded as a kind of oxidative SNH processes (Scheme 1)[8].

This Section consists of two parts. The first one comprises metal-catalyzed cross-coupling reactions, oxidative and elimination SNH reactions, including C – H/C – H couplings, in which intermolecular C – C bonds are formed between hetaryl and aryl moieties, as well as their combinations, allowing the creation of a carbon framework further transforming into a polycyclic structure, are considered. In the second part, various intramolecular cyclizations are highlighted, with a focus on C – H functionalizations of (het)arenes using the methodology of nucleophilic substitution of hydrogen, the so-called SNH reactions, which are becoming increasingly widespread, as reflected in monographs[9, 10] and a series of review articles[11-21].
2.1.1. Synthesis of aryl-substituted hetarenes via the formation of intermolecular bonds
The combination of the palladium-catalyzed Suzuki – Miyaura cross-coupling of 5-bromopyrimidine with thiophene-2-boronic acid, which allows the introduction of a thiophene ring in the 5-position of the pyrimidine instead of a bromo atom, and SNH reactions in the oxidative (AO) or eliminative (AE) variants, which lead to the substitution of the hydrogen atom of the C – H bond at the 4-position by a thiophene moiety as a C-nucleophile, are very illustrative examples of the construction of C – C bonds between the pyrimidine ring and thiophene fragments (Scheme 2)[14, 22].

The combination of palladium-catalyzed cross-coupling and nucleophilic substitution of hydrogen reactions (the SNH) proved to be an efficient approach for the synthesis of a series of 4-(het)aryl-, 4,5-di(het)aryl- (Scheme 3)(The asterisk at the end of the linkage indicates the site of attachment of the moiety to the core.) (Refs. [22-24]) and 4,5,6-tri(het)aryl-substituted pyrimidines (Scheme 4)[25].


Decoration of the pyrimidine ring with two or three aryl moieties and its subsequent modification by catalytic cross-coupling reactions has brought our research group to the targeted synthesis of π-conjugated push-pull systems and fine tuning of the properties of organic dyes — potential photosensitizers for solar cells[26-28], as well as fluorophores that can be used as sensors for the detection of nitroaromatic compounds[29-31]. Push-pull fluorophores with the amide anchoring group are exemplified by compounds 2.1.4 – 2.1.6[26].

A similar tandem of reactions of nucleophilic substitution of hydrogen of the C(4) – H bond in 5-bromopyrimidine by the action of phenols and the Sonogashira cross-coupling of the resulting 4-aryl-5-bromopyrimidines with arylacetylenides, carried out by Shcherbakov et al.[32], afforded 4,5-disubstituted pyrimidines, which can further give rise to the corresponding polycyclic compounds (Scheme 5).

Another common method for the construction of polycyclic heteroaromatic compounds is the introduction of biphenyl moieties into an azaaromatic ring, which is achieved by two successive catalytic Suzuki cross-coupling reactions between 5-bromopyrimidine and appropriate arylboronic acids. Using this approach, asymmetric 1,3-diazatriphenylene cores are readily formed (Scheme 6)[33].

The regularities found in the preparation of diaryl-substituted pyrimidines also apply to the processes of introducing (het)aryl substituents into the pyrazine ring, including the formation of the biphenyl moiety. Thus, the combination of cross-coupling reactions and oxidative C – H functionalization allows to obtain (het)aryl derivatives of furazanopyrazine (Scheme 7)[34].

Similar to pyrimidines, [1,2,5]oxadiazolo- and [1,2,5]-thiadiazolo[3,4-b]pyrazines containing a biaryl moiety are formed via the Suzuki cross-coupling reaction (Scheme 8)[35, 36].

The combination of palladium(II) acetate-catalyzed Buchwald – Hartwig cross-coupling and the intramolecular nucleophilic substitution of hydrogen in the pyrazine ring proved to be the basis for the synthesis of indoloquinoxalines (Scheme 9)[37]. An alternative to the intramolecular amination is a sequence of steps in which the construction of indoloquinoxalines begins with nucleophilic C – N functionalization through the intermolecular SNH arylation of quinoxalines and formation of a new C – N bond, and ends with the Buchwald – Hartwig cross-coupling (Scheme 9)[38].

A similar process has been used for annulation of the indole fragment to furazanopyrazines (Scheme 10)[38].

The tandem SNH – SNH reactions can provide another approach to fused polycyclic systems[9, 10]. For example, the cyclization of 6,8-dimethylpyrimido[4,5-c]pyridazine-5,7-dione with diaminoalkanes proceeds in the presence of a silver permanganate–pyridine complex as the oxidant (Scheme 11)[39].

Nucleophilic substitution of hydrogen is efficiently carried out in a tandem with ipso-substitution of good leaving nucleofugal groups (SNipso), as illustrated by the annulation of the benzofuran moiety in the reaction of 5-methoxy-3-phenyl-1,2,4-triazine with resorcinol (Scheme 12)[40].

Another example of the tandem SNH – SNipso reactions affording benzo[3,2-d]pyrimidines is the oxidative combination of 5-bromopyrimidine with 4-substituted phenols, followed by the intramolecular cyclization (Scheme 13)[41].

2.1.2. Construction of polycyclic systems via the formation of intramolecular bonds
Palladium-catalyzed arylation of С(sp2) – H bonds play an important role in the construction of polycyclic heteroaromatic structures. Such reactions can be used both at the stage of introduction of biaryl moieties into a heteroaromatic ring and for the realization of intramolecular cyclizations and the formation of heterotriphenylene systems (Scheme 14)[6].

Examples of such processes include palladium acetate-catalyzed intramolecular cyclizations of 4-(3,5-dibromo-2-thienyl)-5-(het)arylpyrimidines proceeding under microwave irradiation to give fused thieno- and dithienoquinazolines in low yields, which are accompanied by the loss of two bromine atoms (Scheme 15)[42, 43].

Higher yields of dithienoquinazoline derivatives are achieved on using a different, more efficient way of forming intramolecular C – C bonds between thiophene rings through UV-catalyzed oxidative photocyclization (Scheme 16)[44].

The next approach to the intramolecular formation of 1,3-diazatriphenylenes involves oxidative C – H/C – H coupling between a biphenyl (or phenylbenzothienyl) substituent and the NH-protonated pyrimidinium ring, which is realized in the presence of FeCl3 as oxidant (Scheme 17)[33].

It should be noted that this synthetic approach has been successfully used by Kumada et al.[45] to develop emitter materials exhibiting a high-efficiency thermally activated delayed fluorescence (TADF) to be applied in organic sky-blue light-emitting diodes (Scheme 18).

Similarly, the intramolecular oxidative C – H/C – H cyclization of biphenyl- substituted furazanopyrazines gives 1,4-diazatriphenylenes fused with the 1,2,5-oxadiazole ring. This cyclization follows the ‘nucleophilic addition – oxidation’ pathway, which is typical for the SNH reactions, with the aryl substituent playing the role of nucleophile (Scheme 19)[35, 46].

An approach to 1,4-diazatriphenylenes based on the Scholl reaction has been developed[46], which allows the formation of the C – C bonds between (het)aryl substituents in disubstituted 1,2,5-oxadiazolopyrazines to be realized under oxidative conditions (see Scheme 7 for the synthesis)[34]. Such polycyclic compounds may be of interest as organic semiconductors (Scheme 20)[46, 47].

As for the mechanism of the C – C bond formation between two donor hetaryl moieties, we believe that the cation-radical reaction pathway is the most probable one (Scheme 21). In this case, one of arenes bearing electron-donating substituents is converted to a cation radical form that is activated to react with nucleophiles. However, the pathway including the formation of intermediate arenonium ions and their further involvement in the formation of the C – C bond through oxidative C – H/C – H coupling with aryl pendant substituents cannot be completely excluded.

Cation-radicals can also be generated by chemical oxidants, such as hypervalent iodine compounds PhI(OAc)2 (PIDA) and PhI(OCOCF3)2 (PIFA), or electrochemically. An example is the oxidative C – H/C – H coupling of thiophenes with phenols in hexafluoroisopropyl alcohol (HFIP), and using an anode as the oxidant (Scheme 22)[48].

Studies on the C – C bond formation between aryl rings via oxidative C – H functionalization using hypervalent iodine compounds as oxidants are highlighted in the review[49]. In particular, PIFA is a suitable oxidant for the intramolecular C – C coupling of aryl moieties in 4,5-diarylpyrimidines, which are converted into the corresponding dibenzo[f,h]quinazolines in 23 – 88% yields (Scheme 23)[50, 51].

The intramolecular cyclization of 2-phenylacetanilide into N-acetylcarbazole is an example of the intramolecular C – N bond formation mediated by the hypervalent iodine compound as oxidant (Scheme 24)[52].

In this Section we have briefly reviewed the main approaches to fused aromatic systems by completion of their carbon skeleton based on azaaromatic compounds, mainly 1,3-diazines, 1,4-diazines and their analogues. These methods include the use of palladium-catalyzed cross-coupling and photocyclization reactions of biaryl moieties and are implemented through nucleophilic C – H functionalization of hetarenes and oxidative C – H/C – H coupling reactions, including those occurring electrochemically at the anode. In selecting examples of the C – C bond formation between two sp2 carbon atoms in aromatic systems, we have given preference to those cases where one of the reactive C(sp2) sites has a pronounced electron deficiency or a cationic (cation-radical) centre is generated in situ under reaction conditions, creating the prerequisites for nucleophilic C – H functionalzation through the SNH pathway[9, 10]. Less attention has been paid to the formation of С(sp2) – С(sp2) bonds by exploiting the С – Н/С – Н couplings between aryl moieties in electron-rich aromatic systems, e.g., in the synthesis of polycyclic hydrocarbons (Scheme 25)[53, 54]. Importantly, these reactions require the presence of dichlorodicyanobenzoquinone (DDQ) as an oxidant in the reaction mixture, in an amount equivalent to the number of new C – C bonds. An excess of acid is also required, in the presence of which DDQ is able to generate cationic intermediates from arenes, which are first reduced to an anion-radical and then to the dihydrogenated form DDQ-H2[49, 53, 54]. It should be noted that the presence of DDQ is a feature not only of such cyclizations, but also of a wide range of the SNH reactions[9, 10].

In conclusion, we note that the methods discussed above, aimed at forming of new C – C bonds between (het)aryl moieties, are now being successfully applied to the design of organic semiconductors based on heteropolycyclic compounds, push-pull fluorophores as sensitizers for solar cells, emitter layers for organic light-emitting diodes, chemosensors for the determination of nitroaromatic compounds, and nonlinear optical materials[47, 55-61].
2.2. Metal-catalyzed activation of aromatic C – Н bonds in the synthesis of polyfunctional heterocycles
During the last decade, the functionalization of inactivated C – H bonds by transition metal catalysis has been intensively developed as an atom-economical and environmentally rational method to construct complex organic molecules from simple starting compounds. Numerous studies in this field have led to the discovery of a series of catalytic systems based on complex of trivalent rhodium, iridium and cobalt capable of selectively activating aromatic C – H bonds under the chelating support of certain directing groups (DG), mainly oxygen- and nitrogen-containing ones. The resulting highly reactive organometallic intermediates react readily with various electrophilic reagents to give the corresponding C – H activation products. Various unsaturated compounds such as olefins, acetylenes, aldehydes, imines, isocyanates, sulfonic acid azides, diazo compounds, etc. can act as electrophiles[62-64].
At the same time, it is well known that organofluorine compounds are now widely used in virtually all fields of science and technology, including medicine and the production of new materials. This is mainly due to the fact that the introduction of fluorine atoms or fluoroalkyl groups into an organic molecule significantly changes its physicochemical properties, often leading to an increase in its lipophilicity, thermal and proteolytic stability. In addition, fluorine atoms are able to form fairly strong hydrogen bonds with adjacent functional groups, as well as coordination bonds with metals[65].
About 10 years ago we launched a project[66] to study 2-diazo-3.3,3-trifluoropropionate in the carbenoid C – H activation of aromatic and heteroaromatic compounds catalyzed by trivalent rhodium complexes. As a result, N-arylpyrazoles[67], 6-arylpurines[68], N-pyrimidyl-substituted indoles and indolines[69], and arylhydroxamates[70] were found to be effective substrates for the simultaneous selective introduction of two pharmacophore groups (CF3 and CO2Ме) into the (het)arene molecule. The pyrazole, purine or pyrimidine rings and the hydroxamate functionality served as directing groups in this catalytic transformation (Scheme 26). The catalytically active RhIII species was easily generated from the available dimeric complex [Cp*RhCl2]2 (Cp* is pentamethylcyclopentadienyl) by ligand exchange with silver or cesium salts.

A new project aimed at developing efficient synthetic approaches to fluorinated α-amino acids containing tethered biologically important heterocyclic moieties, based on RhIII-catalyzed tandem C – H activation/annulation of (het)arenes with functionalized acetylenes, was a follow-up study in the field of aromatic C – H bond activation.
This Section presents the results of the synthesis of hetaryl-containing α-amino acids, their functional derivatives and analogues using the above catalytic strategy, as well as the use of this approach in the design of new polyheteroaromatic systems of the donor-acceptor type intended for use as potential photoactive materials.
2.2.1. Synthesis of quinolone-, isoquinolone- and isoquinolinine-containing α-amino acids
Among nitrogen-containing heterocyclic compounds, isoquinolones, their derivatives and structural analogues are recognized as one of the most important basic elements (scaffolds) in medicinal chemistry due to the abundance of their motifs in a variety of bioactive molecules and natural compounds[71-73].
At the same time, modern peptide-based drug design often focuses on the selective modification of amino acid residues with additional functional groups or known heterocyclic pharmacophores to impart the desired properties[74]. Therefore, fluorinated α-amino acids and peptides based thereon are of particular interest because of their wide application in bioorganic chemistry as selective enzyme inhibitors and biological tags, as well as in medicine for the prevention and treatment of hypertension, various types of allergies and a number of oncological diseases[75]. The development of methods for the synthesis of new members of the α-fluoromethyl-α-amino acids, including those containing tethered pharmacophore heterocyclic moieties, is attracting the attention of both fundamental and applied researchers.
O-substituted aryl hydroxamates were found to be unique substrates for tandem RhIII-catalyzed C – H activation/annulation reactions with various acetylenes, in which the hydroxamate moiety can act as both a directing group and an internal oxidant[76, 77]. Mechanistically, this redox-neutral cycloaddition proceeds via the formation of a seven-membered rhodacyclic intermediate (see below), which undergoes synchronous or stepwise C – N bond formation and N – O bond cleavage, leading mainly to NH-isoquinolones[78]. Despite the significant advances in the synthesis of functionalized isoquinolones, the development of efficient methods for their preparation using acetylene substrates remains an urgent task. It is also important to note that this metal-catalyzed C – H activation/annulation strategy has not yet been applied to the synthesis of hetaryl derivatives of amino acids.
We have established for the first time that α-propargyl-α-CF3-α-amino acids 2.2.1 are suitable acetylene components of the Cp*RhIII-catalyzed C – H activation/annulation of arylhydroxamates 2.2.2 to give isoquinolone-containing derivatives of α-amino acids 2.2.3[79]. The reaction proceeds readily in trifluoroethanol (TFE) at room temperature in the presence of 3 mol.% [Cp*RhCl2]2 complex and 2 equiv. of cesium acetate, affording the expected [4 + 2] cycloadducts 2.2.3 in good yields and with high selectivities (Scheme 27).

As α-aminophosphonic acids are important analogues of the corresponding α-amino acids with unique biological properties[80], we have shown that the conditions found can be successfully used to react compound 2.2.2 with the readily available propargyl derivative 2.2.4 to give α-CF3-α-aminophosphonates 2.2.5 (Scheme 28)[81].

The unsymmetrical internal arylacetylenes 2.2.6, which are part of the orthogonally protected α-CF3-α-amino acid, may also be involved in this catalytic transformation[82]. In this case, it was of interest to investigate the selectivity of the [4 + 2] annulation and to determine the synthetic potential of the resulting cycloadducts. The catalytic conditions found for terminal acetylenes were found equally effective for arylacetylenes 2.2.6. These reactions afford the corresponding isoquinolones 2.2.7 in high yields (Scheme 29).

The high selectivity of the [4 + 2] annulation is probably due to the insertion of the acetylene component 2.2.6 into the five-membered rhodacyclic intermediate A according to the intrinsic polarity of the triple bond (Scheme 30).

As isoquinolines are key structural elements of many biologically active compounds, including drugs[83-85], we have explored the synthetic potential of isoquinolones 2.2.7. It was found that these compounds can be readily aromatized to isoquinolines by treatment with trifluoromethanesulfonic acid anhydride in the presence of pyridine. The reactions proceed smoothly in dichloromethane at room temperature and are completed within 15 minutes to furnish the corresponding 1-OTf-substituted isoquinolines 2.2.8 in high yields (Scheme 31).

The known pseudohalogen nature of the TfO group made possible Pd-catalyzed cross-coupling reactions. It was found that isoquinoline derivatives 2.2.8 can undergo Suzuki reactions with 4-methoxyphenylboronic acid and Sonogashira reactions with phenylacetylene under standard conditions to give the corresponding combination products 2.2.9 and 2.2.10 in good to high yields. Also, the OTf group has been successfully removed in the presence of catalytic amounts of PdCl2(dppf) (dppf is diphenylphosphinoferrocene) and excess formic acid to give isoquinolines 2.2.11 unsubstituted at the 1-position in acceptable yields (Scheme 32).

While further investigating the limitations of the C – H activation/annulation strategy in the synthesis of α-amino acid derivatives containing tethered hetaryl substituents, it was unexpectedly found that changing the structure of the acetylene component can alter this catalytic process route. Thus, moving the bulky amino acid (AA) moiety [–C(CF3)(CO2Me)(NH-Pg)] one methylene group closer to the triple bond in the internal acetylene 2.2.12 (i.e. R – C≡C – AA instead of R – C≡C – CH2-AA) induces an unusual process involving a cascade of C – H activation/Lossen rearrangement of the aryl hydroxamate moiety and subsequent cyclization[86]. The reaction gives 2-quinolones rather than the expected isoquinolones 2.2.13', the traditional products of [4 + 2] cycloaddition. The highest yields of quinolones 2.2.13 were obtained in the reaction of arylhydroxamates 2.2.2 with α-(arylethynyl)-α-aminocarboxylates 2.2.12 in the presence of the same catalytic system (3 mol.% [Cp*RhCl2]2 and 2 equiv. CsOAc) in methanol at room temperature (Scheme 33).

However, if the starting hydroxamates contain electron-withdrawing (EWG) substituents such as NO2 and CF3 in the para-position of the aryl moiety, the Lossen rearrangement is completely inhibited. In this case, the reaction reverses direction to form the usual [4 + 2] annulation products, isoquinolones 2.2.14 (Scheme 34). Moreover, minor amounts of tricyclic by-products 2.2.15 were detected in the reaction mixture, resulting from the subsequent intramolecular cyclization of product 2.2.14 involving the Cbz group. Increasing the reaction temperature to 60 °C leads to the complete conversion of compounds 2.2.14 into tricycles 2.2.15.

Density functional theory (DFT) calculations revealed that the more nucleophilic aromatic carbon atom of the keto group of the para-methyl-substituted intermediate B attacks the nitrogen atom much more actively than in the case of the para-nitro derivative, leading to N – O bond cleavage to give an isocyanate moiety. At the same time, the presence of a hydrogen bond between the amide proton and the pivalinate oxygen atom (Scheme 35, see Intermediate B) is an additional factor contributing to the N – O bond cleavage. The presence of a methylene group between the triple bond and the amino acid centre changes the geometry of the molecule so that the formation of the hydrogen bond becomes unfavourable. In this case, no rearrangement products were detected[79]. A plausible reaction pathway explaining the different variations of skeletal rearrangements depending on the electronic nature of the substituents is illustrated in Scheme 35.

2.2.2. Synthesis of pirimido[1,6-a]indolone-containing α-amino acids
Our further studies have been focused on the development of synthetic approaches to fused indole derivatives such as pyrimido[1,6-a]indolones, which contain a fluorinated α-amino acid moiety. The interest in these compounds stems from the fact that this structural motif is part of many bioactive natural compounds as well as pharmaceuticals[87-89]. It is well known that chemical modification of peptides is widely used in biology and medicine, including the study of biological functions of the organism, as well as the diagnosis and treatment of various diseases. We have synthesized, for the first time, α-amino acids decorated with a pharmacophore pyrimidoindolone ring and demonstrated the fundamental possibility of introducing such motifs into peptides[90].
Using the above strategy for the model reaction of N-pivaloyloxyindol-1-carboxamide (2.2.16) with α-propargyl-α-trifluoromethyl-α-aminocarboxylate (2.2.1), the optimum conditions (1.5 mol.% [Rh], 0.5 equiv. CsOAc, MeOH, 20 °C, 2 h) were selected, which provided the maximum conversion of the starting compounds and the best yield of the heterocyclization product 2.2.17. These conditions were found to be common to a number of indole derivatives 2.2.16 resulting in a series of corresponding pyrimido[1,6-a]indolones 2.2.17 in high yields (Scheme 36).

Similarly, pyrimidoindolone-substituted α-aminophosphonates 2.2.18 have been synthesized using the propargyl derivative 2.2.4 (Scheme 37).

In order to demonstrate the fundamental possibility of using the catalytic C – H activation/annulation reaction in peptide synthesis, the propargyl-containing dipeptides 2.2.19 were prepared. The two-step synthesis involved saponification of ester 2.2.1 followed by reaction with alanine and phenylalanine esters in the presence of carboxyl group-activating reagents. The resulting dipeptides 2.2.19 were further reacted with indole derivatives 2.2.16. The same catalytic conditions were found to be suitable for the synthesis of target peptides 2.2.20 with a tethered pyrimidoindolone moiety (Scheme 38).

2.2.3. Synthesis of spirocyclic proline derivatives
Nitrogen-containing spirocyclic compounds are an important class of heterocycles with unique properties. The introduction of such motifs into an organic molecule usually significantly alters its physicochemical and biological properties by imparting high rigidity and unusual 3D geometry. In addition, the formation of spirocycles is often used as a reliable approach to more active structures in the search for potential drugs[91-93]. Heterocyclic spiro moieties, in particular azaspiro[4.4]nonane, are widely represented in natural compounds and in synthetic bioactive molecules that exhibit important pharmacological and pesticidal properties[94, 95]. Therefore, the development of efficient synthetic approaches to functionalized azaspiro[4.4]nonanes, including derivatives of α-CF3-containing spiroprolines, is a challenging task of modern organic chemistry.
As shown above, Cp*RhIII catalytic systems are good catalysts for the tandem C – H activation/annullation reaction of (het)arenes with various acetylenes under the chelating support of certain DGs. The outcome of the annulation depends on both the structural features of the heterocyclic substrate and the arrangement of functional groups in the acetylene unit. Heterocyclization can also be significantly affected by the DG structure. Thus, by replacing the acyclic hydroxamate DG, we found for the first time that the pyrrolidine ring can act as a DG in the ortho-metallation of the benzene ring in the structure of the readily available CF3 derivative of dehydroproline 2.2.21[96]. Here, the RhIII-catalyzed C – H activation of substrate 2.2.21 by symmetrical tolan derivatives 2.2.22 is accompanied by spirolization, leading to the diastereoselective formation of the corresponding spiroprolinates 2.2.23 in acceptable yields[97]. After careful screening of catalytic systems, additives, temperature regime and solvents, the optimum conditions were determined by heating 5-phenyldehydroprolinate 2.2.21 with 1.1 equiv. of alkyne 2.2.22 at 80°C in dichloroethane (DCE) in the presence of 5 mol.% [Cp*RhCl2]2, 0.3 equiv. AgOTf and 0.5 equiv. Cu(OAc)2 for 16 hours (Scheme 39).

Based on literature data[98, 99] and our findings, we have proposed a plausible mechanism for this catalytic transformation. The initially inert dimeric rhodium complex [Cp*RhCl2]2 is readily converted into the catalytically active form Cp*RhIII(OAc) by dissociation and sequential ligand exchange. Next, the C – H bond of the benzene ring is ortho-metallated to form the rhodacycle A with the chelate support of the nitrogen atom of the dehydropyrrolidine 2.2.21, after which the triple bond of the attacking tolan 2.2.22 is selectively inserted into the C – R bond to give the seven-membered intermediate B. The vinylrhodium moiety in intermediate B is then bound intramolecularly to the C=C bond of the pyrrolidine ring to generate intermediate complex C. Finally, protonolysis leads to the cleavage of the N – Rh bond to afford the product 2.2.23 and an active rhodium species intended for a new catalytic cycle (Scheme 40).

This reaction is the first example of aromatic C – H bond activation in which the dehydroproline moiety acts as a DG.
2.2.4. Synthesis of polyheteroaromatic donor-acceptor molecules
Polyheteroaromatic compounds containing π-conjugated donor-acceptor (D – A) bonds have unique electrochemical and photochemical properties that have led to their application in organic electronics and as components of luminescent materials[100, 101]. During the last decade, various electron-deficient (hetero)aromatic compounds have been widely used as acceptor building blocks in the development of advanced functional materials. Among such compounds, 2,1,3-benzothiadiazole derivatives (BTDs) are considered to be privileged ‘building blocks’ for various optoelectronic devices[102], including organic light-emitting diodes (OLEDs)[103], field-effect transistors[104], solar cells[105], and various luminescent materials[106]. Due to the unique properties of the fluorine atom, fluorinated benzothiadiazoles (FBTDs), such as 5,6-difluorobenzothiadiazole and 5-fluorobenzothiadiazole, have attracted particular attention. The introduction of a fluorine atom into the acceptor moiety generally lowers the levels of the highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals in conjugated D-A molecules, thus improving the photophysical performance of optoelectronic materials[107-109]. Based on studies of the Cp*RhIII-catalyzed C – H activation/annulation involving N-(pivaloyloxy)benzamides and functionalized acetylenes, we have proposed an efficient method for the synthesis of polyheteroaromatic π-conjugated donor-acceptor systems intended to create new photoactive molecules[110, 111]. The general concept of assembling such structures is illustrated in Scheme 41.

We have previously developed a convenient approach to BTD-containing terminal acetylenes 2.2.24, based on a sequence of two Pd-catalyzed cross-coupling reactions (Suzuki and Sonogashira reactions) from commercially available 1,7-dibromo-2,1,3-benzothiadiazole and 1,7-dibromo-5,6-difluoro-2,1,3-benzothiadiazole, which is readily formed from the corresponding difluoro-substituted o-phenylenediamine using a known procedure[112].
The hetarylacetylenes 2.2.24 proved to be suitable components for coupling with arylhydroxamates 2.2.2: under the catalytic conditions previously proposed, they provided gave good yields of a series of isoquinolones 2.2.25. The latter, when treated with trifluoromethanesulfonic anhydride, were readily converted into TfO-substituted isoquinolines 2.2.26 (Scheme 42).

Compounds 2.2.26 were also subjected to the Suzuki reaction with arylboronic acid derivatives to ‘install’ donor units traditional for optoelectronics, such as diphenylamine, carbazole and dibenzoazepine moieties, in their structure[113-115]. In all cases, the reactions proceeded smoothly under standard conditions using 5 mol.% of Pd(PPh3)2Cl2 complex to give the target D – A systems 2.2.27 in high yields (Scheme 43). The resulting products were fully characterized by modern physicochemical methods including X-ray diffraction analysis.

The study of Pd-catalyzed cross-coupling reactions revealed an interesting fact. 5,6-Difluoro-BTD-containing triflate 2.2.26 reacted with equimolar amounts of arylboronic acid to give a mixture of the expected compound 2.2.27 and the unusual C – F activation product 2.2.28 (see[116-118]) in approximately equal proportions. When the arylboronic acid loading was increased to 2.5 equiv., the reaction was completed to give only compound 2.2.28 (Scheme 44).

However, replacement of ArB(OH)2 with ArBPin allows selective formation of the cross-coupling products 2.2.27 (X = F) in good to high yields. Such products can be further used to insert another donor component (Ar2) into the D-A system via Pd-catalyzed C – F activation[119] with the appropriate arylboronic acid (Scheme 45).

The fluorine atom closer to the isoquinoline ring, which in this case acts as a DG, is exclusively substituted. The mechanism of C – F activation probably involves the initial formation of palladacycle A[120], which then undergoes ligand exchange with sodium bicarbonate to generate the intermediate complex B. The transmetallation reaction with arylboronic acid then occurs, followed by reductive elimination to afford product 2.2.28 with the release of the active species for a new catalytic cycle (Scheme 46).

The observed difference in reactivity between ArBPin and ArB(OH)2 is probably due to the fact that the rather rigid structure of the palladacycle A formed after oxidative addition prevents the more sterically demanding (compared to arylboronic acid) tetramethyldioxaborolane from being involved in the next transmetallation step of the catalytic cycle.
Also, the resulting monofluorinated structures can be further modified by nucleophilic aromatic substitution of the remaining fluorine atom[121, 122], allowing an additional donor or acceptor group, such as a carbazole ring or CN, to be introduced into the molecule (Scheme 47).

The developed strategy demonstrates for the first time the use of a combination of Rh-catalyzed C – H activation/annulation tandem reaction in the synthesis of novel benzothiadiazole-based luminophores[110, 111]. This method provides access to a diversity of photo- and electrochemically active compounds designed as donor-acceptor π-conjugated systems. The photophysical properties of such materials are currently being actively explored. Preliminary results suggest that a small variation of the substituent at certain positions of the D–A molecule allows to change the bandgap width and the luminescence properties of the compound. The direction of the intramolecular electron transfer can also be changed, indicating the great potential of this method for creating optoelectronic devices with specified properties.
In general, novel hetaryl-containing α-amino acids and their functionalized derivatives[79, 81, 82, 86, 90, 97], and also unique donor-acceptor polyheteroaromatic systems can be efficiently synthesized based on the tandem C – H activation/annulation reaction catalyzed by trivalent rhodium complexes[110, 111]. Undoubtedly, these compounds are to attract the attention of researchers working both in the search for new bioactive substances and in the chemistry of innovative materials.
2.3. Functionalization of pyrroles to design annulated N-heterocycles: how and why?
Pyrrole is a privileged aromatic heterocycle, which is a key component of physiologically important natural compounds such as chlorophyll, haemoglobin, pigments, vitamins, alkaloids. The pyrrole scaffold is found in the structure of many drugs, functional polymers (both optical and electronic), gas sensors, etc.
The pyrrole ring benefits from its high reactivity: it can be easily functionalized both at the α-position and at the nitrogen atom. In organic chemistry, functionalized pyrroles represent an important class of versatile ‘building blocks’ for the targeted design of a diversity of compounds and materials with desired properties. They are used to create new drugs, pheromones, toxins, cell division inhibitors and immunomodulators.
At the same time, it should be noted that most natural molecules containing azaheterocyclic scaffolds are represented by annulated heterocyclic systems, i.e. rigid frameworks that allow retaining the spatial mutual arrangement of atoms for the implementation of the key-lock principle, which is important in nature. Therefore, the development of modern approaches to annulated heterocycles remains an extremely relevant task on which synthetic chemists around the world are concentrating their efforts.
This Section summarizes the work carried out at the A.E.Favorsky Irkutsk Institute of Chemistry, Siberian Branch of the Russian Academy of Sciences, on methods for the synthesis of annulated pyrrole-containing heterocycles using functionalized pyrroles as ‘building blocks’.
2.3.1. Functionalization of pyrroles
Based on the reaction of ketoximes and acetylene in the KOH – DMSO superbasic system (Trofimov reaction), a synthetic approach to a wide range of α,β-substituted pyrroles has been developed[123]. It is also possible to obtain pyrroles by replacing acetylene with its synthetic analogue, 1,2-dichloroethane[124].
One of the most versatile synthetic moieties in an organic molecule is the aldehyde functionality, which can be involved in a wide range of transformations. The classic way to introduce a formyl group is the Vilsmeier – Haack reaction, which has been used for the formylation of N-vinylpyrroles 2.3.1 to give N-vinylpyrrole-2-carbaldehydes 2.3.2. This reaction can be carried out either in a DMF – POCl3 (see[125]) system or, under milder conditions, in a DMF – (COCl)2 mixture (SScheme 48)[126].

N-vinylpyrrole-2-carbaldehydes initially appeared to be promising ‘building blocks’, but it was later revealed that the vinyl moiety reacts only by a radical mechanism[127, 128], whereas the aldehyde moiety needs electrophilic activation[129-132]. This allowed us to easily direct the transformations of compounds 2.3.2 via either the aldehyde or vinyl moiety, but we were unable to get both groups to react simultaneously.
Allenic compounds are well known in organic and pharmaceutical chemistry and are characterized by reactions under the same activation conditions as the aldehyde group (and sometimes no activation is required due to their high reactivity). The method of introducing an allene moiety into the pyrrole ring using various reagents (propargyl chloride, 2,3-dichloro-1-propene or 1,2,3-trichloropropane) was described by B.A.Trofimov and co-workers[133].
The combination of above two methods described led to the development of a method for the selective preparation of a wide range of N-allylenylpyrrole-2-carbaldehydes 2.3.3 from N-allylenylpyrroles 2.3.4, which became available after the discovery of the propargylation of NH-pyrroles 2.3.5 (Scheme 49)[134].

The process was mostly selective, however, formylation of N-allylenyltetrahydroindole 2.3.4a, in addition to the target N-allylenyltetrahydroindole-2-carbaldehyde 2.3.3a, gave annulated tetrahydropyrido[1,2-a]indole 2.3.6 in a 1 : 1 ratio (Scheme 50).

Another important ‘building block’ used to construct annulated heterocycles was 2-hydroxymethylpyrrole 2.3.8 obtained through the reduction of NH-pyrrole-2-carbaldehyde 2.3.7 (Scheme 51)[135].

The reaction of pyrroles 2.3.5 with carbon disulfide and ethyliodide in the KOH – DMSO system followed by treatment of the intermediate pyrrole-2-carbodithioates 2.3.9 with methylene-active nitriles (malononitrile or cyanoacetamide) in the same system gave 2-(2-cyanoethyl)pyrroles 2.3.10 (Scheme 52)[136, 137].

Cross-coupling of pyrroles 2.3.5 with electrophilic haloacetylenes in solid oxides and metal salts gave 2-(acylethynyl)pyrroles 2.3.11 (Scheme 53). This reaction was the first example of ethynylation of pyrroles in the absence of palladium and copper, and also base and solvent[138-140].

2.3.2. Synthesis of annulated heterocyclic systems
Pyrrolopyrazines are important nitrogen-containing heterocyclic moieties in organic and pharmaceutical chemistry. Due to their unique properties and practical applications, pyrrolopyrazine derivatives have been studied for several decades.
The pyrrolopyrazine scaffold is part of a diversity of natural compounds such as longamide A and B, hanishine, stylisine D, peramine, nonazinone B, etc. Pyrrolo[1,2-a]pyrazines play an important role in pharmaceutical chemistry by exhibiting anticancer[141], antimicrobial[142], antiproliferative[143], anxiolytic[144], anti-inflammatory[145], antimalarial[146], antibacterial[147], antiparasitic[148], antiepileptic[149] activity; are inhibitors of HIF-1[150], Pl3K[151], sPLA2[152], CK2 (see[153]) and ERK2 (see[154]) kinases; act as antagonists of the selective non-competitive mGluR5 receptor[155], CRTH2 receptor[156], vasopressin 1b[157], AChE/5-HT4[158], cannabinoid receptor[159]. These compounds also have applications in materials science[160].
Substituted N-allylenylpyrrole-2-carbaldehydes 2.3.3 (R3 = H) and N-allylenyl-2-acylpyrroles 2.3.12 (R1 = Ph, R2 = H, R3 = Me, CF3) react with hydroxylamine hydrochloride in the absence of a catalyst to give pyrrolo[1,2-a]pyrazine N-oxides 2.3.13 (Scheme 54). The reaction proceeds exclusively via the E-isomeric form of an oxime, which readily undergoes cyclization due to steric factors, as has been demonstrated experimentally[161].

It is shown that pyrrolo[1,2-a]pyrazine N-oxides 2.3.13 can be effective ‘building blocks’ in the synthesis of novel heterocycles. For example, reduction of compound 2.3.13a with sodium borohydride affords pyrrolo[1,2-a]piperazine 2.3.14 (Scheme 55).

Heating pyrrolo[1,2-a]pyrazine N-oxide 2.3.13a in a KOH – DMSO superbasic system produces a mixture of two products: 1,3-dimethylpyrrolo[1,2-a]pyrazine 2.3.15 and NH-pyrrolo-2-carbaldehyde 2.3.7a in low yield (Scheme 56). This system acts as a methylating agent for the pyrazine ring and a reducing agent for the N-oxide moiety. In this case, a competing hydrolysis occurs affording the product 2.3.7a.

The reaction of N-allylenylpyrrol-2-carbaldehydes 2.3.3 with o-phenylenediamine provides two series of promising compounds such as benzimidazopyrrolopyrazines 2.3.16 and dihydrobenzimidazopyrrolopyrazines 2.3.17 (Scheme 57)[162]. This process directly depends on the amount of water in the reaction mixture. It is shown that benzimidazopyrrolopyrazines 2.3.16 (conditions a) are selectively produced when carrying out the reaction in dry methanol. Dihydrobenzimidazopyrrolopyrazines 2.3.17 are formed only as a mixture of the two products (1 : 1 ratio) when ethanol is used as solvent with added 20% of water (conditions b). These compounds are easily isolated in pure form by column chromatography.

NH-Pyrrolyl benzimidazoles 2.3.18 react with propargyl chloride in the KOH – DMSO superbasic system to give benzimidazopyrrolopyrazines 2.3.19 (Scheme 58). The first step involves substitution of the free imidazole NH moiety by the propargyl group to generate intermediate A. This is due to the fact that the NH function in imidazole is more nucleophilic than in pyrrole. Acetylene-allene isomerization leads to allene B, the sp-carbon of which is intramolecularly attacked by the pyrrole NH group via 6-exo-dig cyclization to furnish the products.

B.A.Trofimov and co-workers[163] have developed a strategy for the synthesis of pyrrolo[1,2-a]pyrazines 2.3.20 based on 2-(acylethynyl)pyrroles 2.3.11, involving the non-catalytic chemo- and regioselective nucleophilic addition of propargylamine to 2-(acylethynyl)pyrroles 2.3.11 to give N-propargylamino(pyrrolyl)enones 2.3.21 as a mixture of Z,E-isomers, and the base-catalyzed intramolecular cyclization of intermediate N-propargylamino(pyrrolyl)enones 2.3.21 into pyrrolo[1,2-a]pyrazines 2.3.20, isolated as a mixture of isomers with endo- and exocyclic double bonds (Scheme 59). The initial ratio of pyrrolo[1,2-a]pyrazine isomers appears depend on the kinetic control, since further heating of the mixture of isomers under reaction conditions leads to the predominance of the thermodynamically more stable isomer with an endocyclic double bond[163].

In a follow-up study[164], the authors developed a more efficient direct approach to pyrrolo[1,2-a]pyrazines 2.3.20. The target products can be easily obtained in high yields in a single step from 2-(acylethynyl)pyrroles 2.3.11 and propargylamine by heating the reagents in DMSO followed by the addition of equimolar amounts of Cs2CO3 to a solution of N-propargyl(pyrrolyl)aminoenone 2.3.21 (without isolation).
The pyrrolo[1,4]oxazine scaffold is part of natural compounds with a wide range of biological activities (lukianol A[165, 166], acortatarin A[167], pollenopyrroside A and B (see[168])). A pyrrolooxazine called formoxazine was isolated from the marine mudflat-derived fungus Paecilomyces formosus in 2016, but its absolute configuration could not be determined because of decomposition during the isolation. This compound showed potential as a free radical scavenger against 2,2-diphenyl-1-picrylhydrazyl (DPPH) and antibacterial activity against methicillin-resistant Staphylococcus aureus (MDRSA and MRSA strains)[169]. Pyrrolo[1,4]oxazines are inhibitors of CFTR[170-172], mTOR[173], h-ALR2[174], and also show anti-inflammatory activity[175, 176].
Propargyl chloride reacts with unsubstituted 2-hydroxymethylpyrrole 2.3.8a (R1 = R2 = H) in the KOH – DMSO superbasic system to give a mixture of three products: N-allylenylpyrrole-2-ylmethanol 2.3.22, N-allylenyl(propargyloxy)methylpyrrole 2.3.23 and pyrrolo[1,4]oxazine 2.3.24. By varying the reaction conditions (ratio of reagents, amount of alkali and degree of dilution), it was possible to achieve high selectivity to each product (Scheme 60)[177].

The introduction of a substituent capable of changing both the electron density distribution in the molecule and the mutual spatial arrangement of substituents inevitably affects the reaction pathway. Substituted 2-hydroxymethylpyrroles 2.3.8 under optimized conditions (other than those for the unsubstituted analogue, see Scheme 60) in the KOH – DMSO system undergo propargylation to give a single product, pyrrolo[1,4]oxazine 2.3.24. The reaction is regioselective and proceeds exclusively as a 6-exo-dig cyclization via the intermediate N-allylenylpyrrol-2-ylmethanol 2.3.22 formed by an attack of an oxygen atom on the sp-hybridized allene carbon atom (Scheme 61)[135].

Pyrrolizines also occupy an important place in natural and synthetic organic chemistry[178]. Many natural products, mainly alkaloids containing pyrrolizine scaffolds, have been isolated from plants, insects, animals, marine species and microbes. Pyrrolizine derivatives are promising precursors for anti-cancer drugs. The unique antitumour properties of mitomycin C have inspired chemists to synthesize various pyrrolizine systems and evaluate their potential activity against a wide range of cancer cells[179, 180]. Also, pyrrolizines have antiproliferative[181], analgesic, anti-inflammatory[182], antibacterial[183] and antiviral[184] activities. These compounds are utilized as inhibitors of aromatase[185], as well as the enzymes cyclooxygenase (COX) and 5-lipoxygenase (LOX)[186]. One of the pyrrolizines, pyrromilast, is a potent phosphodiesterase 4B inhibitor and is a promising drug for the treatment of chronic lung diseases[187]. The pyrrolizine-containing drug Lycophelone, a LOX/COX inhibitor, is used to treat osteoarthritis[188]. Moreover, functionalized pyrrolysines have proved to be promising ‘building blocks’ in the synthesis of various heterocyclic compounds[189-191].
The most popular approaches to functionalized pyrrolysines are based on multistep reactions, mainly intramolecular cyclizations of C-substituted pyrrole derivatives[192-198]. For example, 2-ethylpyrroles 2.3.10 with a polarized push-pull ethenyl unit are readily converted to methylamino-substituted iminopyrrolizines 2.3.25 when heated in aqueous or water-alcohol methylamine solution (Scheme 62)[194, 195]. It was found that the first step in this reaction (according to TLC) is the cyclization of 2-cyanoethylpyrroles 2.3.10 to 1-ethylsulfanyl-3-iminopyrrolizines 2.3.26, which further exchange its ethylsulfanyl moiety for a methylamino one.

The reaction of 2-ethylpyrroles 2.3.10 with dimethylamine and n-butylamine is similar to the formation of the corresponding 1-amino-3-iminopyrrolizines. Pyrroles 2.3.10 react more slowly and less selectively with n-butylamine in aqueous ethanol than with methylamine under similar conditions. Thus, n-butylamine reacts with 2-ethylpyrrole 2.3.10 or its cyclic isomer 1-ethylsulfanyl-3-iminopyrrolizine 2.3.26 to give a mixture of 1-n-butylaminopyrrolizine 2.3.27 and 2-(1-n-butylamino-2-cyanoethyl)pyrrole 2.3.28 in a ratio of 19 : 1 (X = CN) or 5 : 1 (X = C(O)NH2) (Scheme 63). In anhydrous ethanol this reaction is selective and only produces 1-n-butylaminopyrrolizine 2.3.27a in high yield. The bulky substituents in di(n-butyl)amine, unlike n-butylamine, hinder the reaction with compounds 2.3.10 and 2.3.26. In such a process, di(n-butyl)amine acts as the main catalyst, promoting the cyclization of ethenylpyrrole 2.3.10b to iminopyrrolizine 2.3.26b[194].

2-Ethylpyrroles 2.3.10a were found to react with aniline only on addition of an equimolar amount of triethylamine; after prolonged refluxing in ethanol (12 – 20 h), 1-anilino-3-iminopyrrolizines 2.3.29 were isolated in 50 – 80% yields (Scheme 64)[195].

The reaction of 2-ethylpyrroles 2.3.10a with substituted anilines under these conditions (in boiling ethanol) takes much longer than with aniline (63 – 65 h) and affords the corresponding 1-anilino-3-iminopyrrolizines 2.3.29 in 10 – 43% yields (see Scheme 64)[199].
1-Amino-3-iminopyrrolizines 2.3.25, 2.3.29 react with 1-chloroacetophenone in a KOH – DMSO catalytic system to give functionalized bipyrroles (Scheme 65). Using 1-methyl-3-iminopyrrolizines 2.3.25, 2,2'-bipyrroles 2.3.30 are formed, while aniline-substituted pyrrolizines are converted to 2,3’-bipyrroles 2.3.31[200].

It should be noted that 2,3-bipyrroles are obtained in low yields in the above system. However, when the catalyst in this cyclization is replaced by 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and the reaction is carried out in boiling acetonitrile, the process follows a different pathway and proceeds more readily. Thus, arylamino-substituted pyrrolizines 2.3.29 give 4-amino-1-aryl-5-benzoyl-3-cyano-2,2'-bipyrroles 2.3.30 regioselectively (up to 94% yield) instead of 2,3'-bipyrroles (Scheme 66). The reaction proceeds via the formation of intermediate ketones 2.3.32, isolated after refluxing 1-anilino-3-iminopyrrolizines 2.3.29 with 1-chloroacetophenone in the K2CO3 – acetone system[199].

The reaction of propionate 2.3.33 with diethylamine leads stereospecifically to the E-isomer of diethylamino acrylate 2.3.34, which is converted under the reaction conditions to pyrrolizin-3-one 2.3.35 (ratio 2.3.34 : 2.3.35 = 1 : 1). During chromatographic separation of this mixture on Al2O3, the adduct 2.3.34 undergoes complete cyclization (Scheme 67) and only the pyrrolizinone 2.3.35 is isolated[196].

The intramolecular cyclization of enols 2.3.36a,b, resulting from the nucleophilic addition of malononitrile to 2-(acylethynyl)pyrroles 2.3.11 in the KOH – MeCN catalytic system, produces pyrrolizines 2.3.37a,b. Here, the pyrrolizine ring is formed spontaneously during purification or drying (especially at elevated temperatures) of the adducts 2.3.36a,b. At the same time, with the stable phenyl-containing enol 2.3.36c, the synthesis of the corresponding pyrrolizine 2.3.37c is carried out in boiling ethanol in the presence of triethylamine (Scheme 68).

The cyclization to aminopyrrolizines probably occurs through the addition of the pyrrolic NH proton to the CºN bond and subsequent isomerization of the NH moiety in the iminopyrrolizine to an amino group[197]. Aminopyrrolizines are formed stereoselectively, exclusively as Z-isomers with a cis arrangement of the proton at the double bond and the presence of the ortho proton in the aryl group.
Aminoketopyrrolizines 2.3.38 have also been obtained from 2-(acylethynyl)pyrroles 2.3.11 (see[198]). The reaction sequence leading to these products involves the nucleophilic addition of benzylamine to the triple bond of 2-(acylethynyl)pyrroles 2.3.11, the subsequent reaction of the intermediate N-benzylamino(pyrrolyl)enones 2.3.39 with acylacetylenes 2.3.40 and the intramolecular cyclization of the resulting pyrrolylpentadienediones 2.3.41 (Scheme 69).

The work presented in this Section is undoubtedly just a few examples that demonstrate the potential of combining C- and N-functionalization for pyrrole to provide an extensive library of pyrrole-containing annulated heterocyclic ensembles. Sequential introduction of two functionalities or one group while retaining the NH function of the pyrrole opens access to various drug precursors, promising functional materials, optical devices, sensors and many other practically valuable compounds. At present, such unique heterocyclic systems as pyrrolopyrazines, pyrrolooxazines and pyrrolizines have been obtained, allowing us to speak with confidence of the inexhaustible potential of pyrrole ‘building blocks’ in the further expansion of classes of organic compounds.
2.4. Multicomponent reactions in the synthesis of N,O,S,Se-containing heterocyclic systems
This Section covers the key studies of the Lugansk school of synthetic chemists devoted to N,O,S,Se-containing heterocyclic systems. The attention is focused on the data on multicomponent heterocyclization reactions using methylene-active thio- and selenoamides obtained over the last 10 – 15 years. The key compounds in these multicomponent and cascade reactions are malononitrile, cyanothioacetamide[201, 202], cyanoselenoacetamide[203], malononitrile dimer[204] and 1-cyanoacetyl-3,5-dimethylpyrazole[205, 206], which are polyfunctional reagents with a great synthetic potential.
2.4.1. Synthesis of pyrrole, furan and thiophene derivatives
Malononitrile dimer 2.4.1 was used to prepare new substituted pyrrole-3-carbonitriles 2.4.2[207, 208]. The synthetic route is based on the double С-alkylation of 2.4.1 with bromoketones in alkaline medium. The mechanism of the cascade reaction involving the formation of intermediates А – Е is shown in Scheme 70.

The three-component reaction of malononitrile, phenacyl bromide and aromatic aldehydes is a convenient method for the synthesis of 2-aminofurans difficult to obtain by other methods[208]. The regiochemistry of the intramolecular cyclization is largely determined by the structure of the aromatic aldehyde and the base. For example, the use of p-bromobenzaldehyde results in the formation of furan 2.4.3; in the case of salicylaldehyde and N-methylmorpholine (NMM) being used instead of KOH, the reaction gives Schiff base 2.4.4 (Scheme 71).

The bromination of 4Н-chromene 2.4.5 is unexpectedly accompanied by ring contraction and affords substituted benzofuran 2.4.6 in a moderate yield[209]. A possible reaction mechanism involving the formation of intermediates A – C is shown in Scheme 72.

In cooperation with the colleagues from Herzen State Pedagogical University of Russia, we developed[210] an original method for the preparation of furo[3,2-c]pyrans 2.4.7 and furo[3,2-c]chromenes 2.4.8 based on the treatment of 3-bromo-3-nitroacrylates with 4-hydroxypyran-2-one under mild conditions (Scheme 73).

The reactions of cyanothioacetamide 2.4.9 with aromatic aldehydes and α-thiocyanatoacetophenone and the also the reactions of 2.4.9 with α-bromochalcones provide the synthesis of dihydrothiophenes 2.4.10. A new method for the construction of thieno[2,3-d]pyrimidine system by aminomethylation of products 2.4.10 on treatment with primary amines and excess НСНО was proposed (Scheme 74)[211].

2.4.2. Synthesis of five-membered heterocycles with two or three heteroatoms
It was found[212] that Hantzsch synthesis involving 3-amino-N-(4-methoxyphenyl)-3-thioxopropanamide and phenacyl bromide is accompanied by oxidation of the methylene group, which gives rise to α-ketoamide 2.4.11 (Scheme 75). Hantzsch thiazoles can be prepared by one-pot synthesis from thioamide 2.4.9 and 4-hydroxybenzaldehydes. The subsequent О-alkylation of intermediate compounds А affords substituted thiazoles, as shown in relation to compound 2.4.12[213].

2-(Thiazolyl)acrylonitriles 2.4.12 proved to be a molecular platform suitable for further functionalization. The bromination of these compounds proceeds selectively at thiazole position 5 to give bromides 2.4.13 (Scheme 76)[214]. This reaction does not affect the double bond, and the regiospecificity is retained even for compounds 2.4.14 containing a diene moiety[215]. 2-(Thiazolyl)acrylonitriles are readily oxidized via the Radziszewski reaction on treatment with Н2О2 in the presence of inorganic bases by a cascade mechanism, being converted to 2-(thiazolyl)oxirane-2-carboxamides 2.4.15[216]; stereochemistry of the products corresponds to the Е-configuration of the starting nitriles.

The methylene-active thiazoles undergo double С-alkylation, as shown in relation to the synthesis of compounds 2.4.16 and 2.4.17 (Scheme 77)[217].

The three-component condensation of anilines, triethyl orthoformate and cyanoselenoacetamide 2.4.18 results in the formation of a mixture of E- and Z-isomers of 3-aminoselenoacrylamides 2.4.19, which are converted to functionalized selenazoles 2.4.20 via the Hantzsch reaction (Scheme 78)[218].

2-Cyanothioacrylamides 2.4.21, obtained by condensation of carbonyl compounds with cyanothioacetamide 2.4.9, can be oxidized by treatment with a broad range of oxidants to give substituted 1,2,4-thiadiazoles 2.4.22 (Scheme 79). Effective reagents for this oxidative heterocyclization are DMSO – HCl[219], bromonitromethane[220], Et2SO – HCl[221], bromine or iodine in DMF[222] and NaNO2 in АсОН[223]. Thiadiazoles 2.4.22 are also formed upon electrochemical oxidation in an undivided cell in an aqueous solution of KBr[224]. These compounds have a pronounced antidote activity against the 2,4-D herbicide in experiments with sunflower culture[221-224]. When the oxidation is performed with the DMSO – HCl system or halogens, the product yields are nearly quantitative. It is noteworthy that treatment of cyclic analogues of thioamides 2.4.21 with oxidants, e.g., DMSO – HCl system[219] or K3[Fe(CN)6] – KOH system[225], gives disulfides 2.4.23. Selenoacrylamides 2.4.24 are oxidized in DMSO in the presence of aqueous HCl to give 1,2,4-selenadiazoles 2.4.25 in moderate yields[226].

Stepanova et al.[227] proposed an improved method for the synthesis of known 5-amino-3-cyanomethyl-1H-pyrazole-4-carbonitrile 2.4.26 by the reaction of the potassium salt of the malononitrile dimer with hydrazinium sulfate. Pyrazole 2.4.26 was successfully used to prepare pyrazolopyrimidines[228, 229] and pyrazolo-1,3,5-triazines (Scheme 80)[227].

2.4.3. Synthesis of nicotinic acid derivatives
A convenient synthetic route to nicotinonitrile derivatives is the three-component reaction of enamines, thioamide 2.4.9 and aldehydes. Nicotinonitriles 2.4.27 (Ref. [230]) and 2.4.28 (Ref. [231] and [232]) were prepared in this way (Scheme 81). The use of alkylating agents into this reaction provides the synthesis of thioethers 2.4.29 in high yields[233, 234]. Instead of cyanothioacetamide 2.4.9, malonodithioamide can be used[235]; however, the thioamide moiety is not preserved in position 3 of the product, and quinolines 2.4.30 are isolated as the final reaction products. Therefore, it is expedient to use thioamide 2.4.9 for the synthesis of quinolines 2.4.30[236]. Quinolines 2.4.30 can be introduced into the subsequent transformations without isolation, as shown in relation to the synthesis of compound 2.4.31[237].

Enamino ketones are also successfully used in multicomponent reactions. For example, compounds 2.4.32 react with СН acids and alkylating reagents in the presence of bases to furnish various isoquinoline derivatives (Scheme 82)[238].

Enamino diketones 2.4.33а react with selenoamide 2.4.18 to give diselenides, which can be converted to selenophenopyridines 2.4.34[239]. A similar reaction with Meldrum’s acid derivative 2.4.33b yields nicotinic acid derivatives[239]. However, malonothioamide derivatives behave in a different way under these conditions and form pyrimidines 2.4.35 (

Acetylenic ketones 2.4.36 (Ref. [242]) react with thioamide 2.4.9 in the presence of morpholine and KOH to give nicotinonitriles 2.4.37[243]. Obviously, the reaction involves the stage of enamino ketone formation (Scheme 84).

The Guareschi – Thorpe reaction of methylene-active thioamides with 1,3-diketones smoothly proceeds to give pyridine-2(1Н)-thiones. In the case of symmetrical 1,3-diketones, the reaction follows the expected pathway[244-246], as shown[245] in relation to the synthesis of thione 2.4.38 (Scheme 85). In the case of unsymmetrically substituted 1,3-diketones, the course of the reaction is ambiguous. Indeed, whereas the reaction of substituted β-cycloketols with thioamde 2.4.9 proceeds regioselectively to give cyclization products[247], the reactions of 2-acetylcyclopentanone and -hexanone afford mixtures of regioisomers[248]. These reactions are considered in detail in a review[249].

The three-component reaction of cyanoselenoacetamide 2.4.18 and aromatic aldehydes with cyclohexane-1,3-dione[250] or 1-cyanoacetylpyrazole[251] in the presence of a base results in the corresponding selenolates (Scheme 86).

Quinoline 2.4.39 was prepared by the reaction of thioacrylamides 2.4.21 with dimedione enamine followed by alkylation of intermediate А and [3,3]-sigmatropic rearrangement[252]. The presence of the electron-withdrawing substituent R makes it possible to obtain the Thorpe – Ziegler cyclization product, e.g., thieno[2,3-b]quinoline 2.4.40, under the same conditions (Scheme 87).

Partly hydrogenated isoquinolines are promising molecules for bioscreening[253]. Dyachenko et al.[254] described the synthesis of isoquinoline 2.4.41 by the reaction of thioamide 2.4.42 with DMF dimethyl acetal (Scheme 88).

The reaction of unsaturated dinitriles with compounds 2.4.24 or 2.4.42 follows a formal [4+2]-cycloaddition pathway and results in the formation of pyridine-3,5,5-tricarbonitriles 2.4.43а,b (Scheme 89)[255].

Nicotinonitrile derivatives 2.4.44 can be obtained by the reaction of malononitrile dimer 2.4.1 with isothiocyanates in the presence of Et3N[256]. It is noteworthy that the potassium salt of dimer 2.4.1 with phenyl isothiocyanate gives regioisomeric 2Н-thiopyran (Scheme 90).

The three-component condensation of malononitrile dimer 2.4.1, Meldrum’s acid and aromatic aldehydes gives tetrahydropyridines 2.4.45[257]. These products proved to be convenient substrates for the one-pot synthesis of substituted thieno[1,6]naphthyridines (Scheme 91).

The reaction of malonate 2.4.46 with chalcogenamides 2.4.9 or 2.4.18 proceeds as an exchange of methylene components. Dyachenko et al.[258] postulated the formation of Michael adduct А and degradation of the adduct to give acrylamides 2.4.21 or 2.4.24, which react with the starting chalcogenamides 2.4.9 or 2.4.18 to give 6-amino-3,5-dicyanopyridines 2.4.47 (Scheme 92). A similar product was also obtained by refluxing thioamide 2.4.48 with alkali in ethanol[259].

Nicotinonitriles 2.4.49a,b were synthesized by the condensation of chalcone with selenoamide 2.4.18 and alkylating agents in the presence of N-methylmorpholine (Scheme 93)[260].

Spiro-substituted nicotinonitriles 2.4.50а,b are formed upon the reaction of methylene-active thioamides with ester 2.4.51 and alkyl halides induced by sodium ethoxide (Scheme 94)[261].

The successive reactions of cyclohexanone with cyanothioacetamide 2.4.9, phenacyl bromide and 3-amino-3-thioxopropanamide results in the formation of piperidine 2.4.52[262]. Thiazole А is assumed to be an intermediate, which is confirmed by the formation of compound 2.4.53 upon the reaction of 2 equiv. of thioamide 2.4.9 with α-chloroacetanilide (Scheme 95). Structurally similar nicotinate 2.4.54 was obtained from (thiazol-2-yl)thioacetamide, diethyl malonate and formaldehyde[263].

Selenolate 2.4.55, synthesized by the reaction of diethyl ethoxymethylenemalonate with selenoamide 2.4.18, is alkylated to give selenides 2.4.56[264]. Tetrahydropyridones 2.4.57 were prepared by the condensation of aldehydes, thioamide 2.4.9, dimethyl malonate and alkylating reagents[265]. Substituted tetrahydropyridines 2.4.58 are efficiently synthesized using a Meldrum’s acid analogue as the second СН acid (Scheme 96). Some of compounds 2.4.58 were found to exhibit a hepatoprotective effect[266].

Readily available acetoacetanilides, resulting from the reactions of diketene with various anilines, smoothly react with aromatic aldehydes and cyanothioacetamide 2.4.9 to give 1,4-dihydropyridine-2-thiolates 2.4.59, which are regioselectively S-alkylated to dihydronicotinamides 2.4.60[267-272]. These compounds exhibit analgesic[267-270], anti-inflammatory[271] and antiviral properties[272]. Treatment of compounds 2.4.60 in situ with allyl bromide in the presence of alkali provides a route to nicotinamides 2.4.61 (Scheme 97)[273]. A probable reaction mechanism includes N-allylation of dihydropyridines 2.4.60 followed by [3,3]-sigmatropic aza-Claisen rearrangement[274].

2.4.4. Synthesis of thieno[2,3-b]pyridine derivatives and structural analogues
Thieno[2,3-b]pyridines[275] and their structural analogues derived from dithiolo- and isothiazolopyridine are of interest as potential biologically active compounds. Traditional methods for building the thienopyridine system imply cyclizations producing either the pyridine or thiophene ring[275]; therefore, approaches to the design of these compounds from acyclic precursors are of particular interest. For example, thienopyridines 2.4.62 were prepared by multicomponent synthesis from cyanothioacetamide 2.4.9 (Scheme 98)[276].

The four-component condensation of cyanothioacetamide (2.4.9), aldehydes, acetophenones and alkyl halides in the presence of alkali at 0 °С yields substituted thieno[2,3-b]pyridines 2.4.63[277]. On treatment of compounds 2.4.63 with excess Ac2O, double acylation product is formed, as shown in relation to the synthesis of diamide 2.4.64 (Scheme 99).

The condensation of aldehydes, thioamide 2.4.9, acetoacetanilides and alkylating agents in the presence of N-methylmorpholine at 20 °С followed by treatment with 10% aqueous KОН furnishes substituted thieno[2,3-b]pyridines 2.4.65 in good yields (Scheme 100)[278].

A multicomponent reaction involving malononitrile, hydrogen sulfide, aldehydes, alkylating agents and Et3N results in the formation of thieno[2,3-b]pyridine 2.4.66 (Scheme 101)[279]. This method is based on the in situ generation of cyanothioacetamide 2.4.9, which then participates in a cascade process.

Cyclopenta[b]thieno[3,2-е]pyridines 2.4.67 were isolated upon the multicomponent reaction of malononitrile, H2S, aldehydes, 1-(cyclopent-1-en-1-yl)pyrrolidine, alkylating agents and Et3N; thiophene ring closure was catalyzed by KОН in aqueous DMF[280]. Refluxing of intermediate thienopyridine 2.4.67 in formamide ended in the formation of tetracyclic system 2.4.68 (Scheme 102).

Pyrido[3',2':4,5]thieno[3,2-d]pyrimidines 2.4.69 and 2.4.70, which are difficult to obtain by other methods, were prepared by the multicomponent reaction of 1,3-diketones with thioamide 2.4.9 and alkyl chlorides in DMF in the presence of EtONa at 50 °С followed by refluxing with carbonyl compounds (Scheme 103)[281].

Generally, 3-aminothieno[2,3-b]pyridines proved to be convenient objects for the subsequent functionalization. The reaction of amides 2.4.71 with P4S10 in refluxing pyridine yields 1,3,2λ5-diazaphosphorines 2.4.72[282], while their reaction with ninhydrin in acid medium affords spirocyclic products 2.4.73[283]. When amides 2.4.71 react with sodium hypochlorite, unusual oxidative dimerization products 2.4.74 are formed (Scheme 104)[284, 285]. The acylation of compounds 2.4.71 with chloroacetyl chloride in refluxing toluene induces pyrimidine ring closure[286]. On treatment with phthalic anhydride in refluxing DMF, thienopyridines 2.4.71 are converted to polycyclic compounds 2.4.75[287].

3-Aminothieno[2,3-b]pyridines 2.4.76 easily react with 3,5-dimethyl-1-(cyanoacetyl)pyrazole to give the corresponding cyanoacetamides, which undergo the Camps cyclization under the action of strong bases, as shown in relation to the synthesis of dipyridothiophene 2.4.77 (Scheme 105)[288].

The reaction of dithiomalonodianilide 2.4.78 (Ref. [289]) with arylmethylenemalononitriles in the presence of morpholine furnished[290] derivatives of a rare heterocyclic system, [1,2]dithiolo[3,4-b]pyridine 2.4.79 (X = CN, Scheme 106). A similar reaction involving 3-aryl-2-cyanoacrylates[291] or 3-aryl-2-cyanoacrylamides[292-294] also gives dithiolopyridines. Some representatives of this series of compounds showed a noticeable antidote activity against the 2,4-D herbicide in experiments on sunflower seedlings[291, 294, 295].

Arylmethylidene Meldrum’s acid derivatives 2.4.80 react with dithiomalonanilide to give stable Michael adducts[296], which subsequently undergo cyclization on heating, on treatment with KOH, or on attempted alkylation to give dithiolopyridines 2.4.81 (Scheme 107)[297].

The condensation of malononitrile, aldehydes and monothiomalonamide in the presence of Et3N affords thiolates 2.4.82, which are oxidized and simultaneously hydrolyzed on treatment with DMSO and HCl to give isothiazolo[5,4-b]pyridines 2.4.83 (Scheme 108)[298].

2.4.5. Synthesis of thiazolopyridine and pyridothiazine derivatives
Dyachenko et al.[299] proposed a three-component process for the synthesis of indolyl-substituted thiazolo[3,2-a]pyridine 2.4.84 by the reaction of thioacrylamide 2.4.21 (R = indol-3-yl), ethyl 3-(morpholino)-but-2-enoate and 1,2-dibromoethane (Scheme 109).

It was found[273] that thiazolopyridine derivatives are formed in the reaction of aldehydes, thioamide 2.4.9, acetoacetanilides and chloroacetic esters, as shown for the synthesis of compound 2.4.85 (Scheme 110).

A convenient method for the construction of a partly saturated thiazolo[3,2-a]pyridine system is the condensation of ester 2.4.86, methylene-active thioamide and 1,2-dibromoethane. These products can be modified by С-alkylation, for example, for the synthesis of compounds 2.4.87 and 2.4.88 (Scheme 111)[300].

The reaction of tetrahydropyridine-2-thiolates 2.4.89 with α,α-disubstituted aliphatic aldehydes and primary amines resulted in the synthesis of thiazolo[3,2-a]pyridines 2.4.90 (Scheme 112)[301, 302].

The successive treatment of thioacrylamide 2.4.91 with diethyl malonate and 1,2-dibromoethane or 1,3-dibromopropane gives bicyclic compounds 2.4.92 (Scheme 113)[303].

Partially hydrogenated thiazolo[3,2-а]quinolines 2.4.93 were obtained by the reaction of aldehydes, thioamide 2.4.9, dimedone and 1,2-dibromoethane (Scheme 114)[304].

The reaction of isatins, malononitrile and monothiomalonamide gives spiro thiolates 2.4.94a,b, which are converted to a mixture of tautomeric alkylation products 2.4.95a,b on treatment with α-bromo ketones (Scheme 115)[305].

2.4.6. Synthesis and reactions of pyran, chromene and thiopyran derivatives
Ethyl 3-amino-3-thioxopropanoate reacts with salicylaldehyde to give coumarin-3-carboxamide 2.4.96[306]. Probably, the reaction involves the formation of intermediate A followed by hydrolysis of the thioamide group under the reaction conditions (Scheme 116).

4Н-Chromenes, which can be formed in the reaction of malononitrile, dimedone and aldehydes, can be additionally functionalized by alkylation or condensation involving the С(6) position, as shown in relation to the formation of compounds 2.4.97 and 2.4.98 (Scheme 117)[307].

The condensation of aldehydes, methylene-active nitriles, thiobarbituric acid and alkyl halides is a convenient method for the preparation of pyrano[2,3-d]pyrimidines. The introduction of additional alkylating or acylating agents makes it possible to carry out targeted functionalization of these heterocycles to give compounds 2.4.99 – 2.4.101 (Scheme 118)[308].

Derivatives of thiopyrano[2,3-b]pyran, a rare heterocyclic system, were synthesized by the reaction of malononitrile and ketosulfone 2.4.102 with carbonyl compounds (aromatic aldehydes, isatin, ninhydrin) (Scheme 119)[309].

An unexpected result was obtained upon thiophosphorylation of a pyrano[2,3-c]pyrazole derivative 2.4.103, which is formed upon a multicomponent transformation involving 3-methylpyrazolin-5-one: treatment of compound 2.4.103 with the P2S5 · 2 Py complex in anhydrous pyridine gives [1,2]oxaphosphinino[6,5-c]pyrazole in a low yield (Scheme 120)[310].

Hybrid molecules containing azine and γ-pyrone moieties were obtained by the reaction of chlorokojic acid 2.4.104 with various heterocyclic S-nucleophiles (Scheme 121)[311].

Substituted resorcinols are known as convenient reagents for the formation of functional chromene derivatives[312-314]. An example of preparation of compounds of this type is the synthesis of chromenes 2.4.105 from pyridylazoresorcinol (PAR)[314] (Scheme 122). Chromenes 2.4.105 are of interest as promising indicators and complex-forming agents.

The pseudo-five-component reaction involving malononitrile, aromatic aldehydes, cyanoacetates and acetylacetone in the presence of alkali gives 3-azabicyclo[3.3.1]nona-2,7-dienes 2.4.106[315]. It was found that the reaction proceeds via the formation of 4H-pyrans А (Scheme 123). The target compounds 2.4.106 result from the hydrolytic cleavage of 4Н-pyrans А followed by reaction with 2-cyanoacrylates formed in situ.

4-Alkyl-4Н-thiopyrans 2.4.107 were obtained by the reaction of aliphatic aldehydes with cyanothioacetamide 2.4.9 and malononitrile in the presence of morpholine in anhydrous EtOH at 20 °С (Scheme 124)[316].

Under the action of electrophilic agents and in the presence of bases, 4Н-thiopyrans 2.4.108 readily undergo cross-recyclization[317, 318]. The synthesis of pyridine 2.4.109 can be considered as an example (Scheme 125)[317].

Long-term refluxing of thiolates 2.4.89 with malononitrile dimer 2.4.1 results in the formation of fused 2Н-thiopyrans 2.4.110. On treatment with acylating agents, these products are converted to 7-thia-1,4,6,8-tetraazabenzo[de]anthracene derivatives (Scheme 126)[319].

2.4.7. Synthesis of 3,7-diazabicyclo[3.3.1]nonane, 1,3,5-triazine and 1,3,5-thia(selena)diazine derivatives
A number of methods have been described for the synthesis of pyrimido[4,3-b][1,3,5]thiadiazine[320] and -selenadiazine[321] derivatives by the Mannich reaction of compounds 2.4.9, 2.4.18, and 2.4.24 (Scheme 127). When cinnamaldehyde is used, cascade process is initiated to give tricyclic products 2.4.111[322].

Cyclic chalcogenamides – pyridin-2(1Н)-ones, -thiones and -selenones and their salts – readily undergo aminomethylation; however, the structure of the products critically depends on the reaction conditions and the substrate structure[323, 324]. For example, the Mannich reaction of tetrahydropyridine-2-thiolates 2.4.89 gives pyrido[2,1-b][1,3,5]thiadiazines 2.4.112 (Scheme 128)[272, 325-329]. These products exhibit a broad range of biological activity, including the antiviral effect against tick-borne encephalitis[272], anti-inflammatory action[328, 330, 331], analgesic effects[329] and analeptic and antidepressant effects[332-334].

However, electron-withdrawing substituents in the tetrahydropyridine ring change the aminomethylation regiochemistry, and 3,7-diazabicyclo[3.3.1]nonanes are isolated as the reaction products[335-340]. Compound 2.4.113, a derivative of malononitrile dimer 2.4.1, forms diazabicyclononenes 2.4.114 in the Mannich reaction (Scheme 129)[341].

The products of aminomethylation of 1,4-dihydropyridines 2.4.115 considerably depend on the structure of the starting substrate. In the case of thiolates and selenolates 2.4.115а,b (X = S, Se), the reaction gives tricyclic compounds 2.4.116[342-345]. Some compounds 2.4.116 in micromolar concentrations proved to be active against tick-borne encephalitis virus[345]. Azaspirane 2.4.117 is also converted to 3,5,7,11- tetraazatricyclo[7.3.1.02,7]tridec-2-ene derivatives under similar conditions[346]. In the case of compounds 2.4.115с (Х = С(CN)2), no 1,3,5-triazine ring formation takes place: the strong electron-withdrawing effect of the dicyanomethylene group decreases the nucleophilicity of the pyridine nitrogen atom; consequently, the reaction stops after the formation of diazabicyclononenes 2.4.118 (Scheme 130)[347].

1-Aminopyridines 2.4.119 are aminomethylated to be converted to bispidine derivatives fused with the 1,2,4-triazole ring (Scheme 131)[348]. The oxidized analogues, N-aminopyridines 2.4.120, behave ambiguously in the Mannich reaction to give products 2.4.121а – с depending on the conditions[348, 349].

In the general case, 2-aminopyridines 2.4.122 react with reactive primary amines and НСНО to give pyrido-[1,2-a][1,3,5]triazine derivatives 2.4.123[350-353]. In the absence of amines, the products formed in the reaction of aminopyridines 2.4.122 with НСНО undergo self-condensation to give tetrazocines 2.4.124[350]. Meanwhile, in some cases, the formation of diaminomethane 2.4.125 was noted[353]. The aminomethylaion of dicyanomethanide 2.4.126 affords[354] betaines 2.4.127 (Scheme 132). Some of these compounds exhibit a pronounced antidote action against the 2,4-D herbicide[353, 355, 356].

Derivatives of [1,2,4]dithiazolo[4,3-a][1,3,5]triazine, a rare heterocyclic system, were isolated in low yields upon the aminomethylation of xanthane hydride 2.4.128 (Scheme 133)[357].

The aminomethylation of thiolate 2.4.129 with formaldehyde and aromatic diamines gives macrocyclic structures (Scheme 134)[358].

The Mannich reaction of thiolate 2.4.130 with primary amines and НСНО yields 1,3,5-thiadiazines 2.4.131 (Scheme 135)[359].

2.4.8. Synthesis of fused polyheterocyclic systems
Treatment of isoquinoline 2.4.132 with bromide 2.4.133 triggers a cascade process giving rise to compound 2.4.134 (Scheme 136)[360, 361].

Pyrimido[5,4-b]thieno[3',2':2,3]pyrido[2,3-d]pyrimidine 2.4.135 was obtained by multicomponent condensation of benzaldehyde, thioamide 2.4.9, 4-bromophenacyl bromide and formamide (Scheme 137)[362].

Dyachenko et al.[363] studied a multicomponent condensation involving thioamide 2.4.9 and 3-(bromoacetyl)coumarin, which gave substituted chromeno[3'',4'':5',6']pyrido[2',3':4,5]thieno[3,2-e]pyridine 2.4.136 (Scheme 138).

The cascade reaction of 3-cyanopyridine-2(1Н)-thiones and polyfunctional alkylating agents is a convenient method for the construction of polyheterocyclic ensembles. For example, compounds 2.4.137 react with bromides 2.4.138 to give fused thienopyridines 2.4.139 (Ref. [364]) and react with chloromethylxanthines 2.4.140 to afford pentacyclic products 2.4.141 (Scheme 139)[365].

The azo coupling of thioamide 2.4.9 with (pyrazol-5-yl)diazonium salts gives thioamides 2.4.142, which are oxidized to thiadiazoles bearing two pyrazolo-1,2,4-triazine substituents, e.g., compound 2.4.143 (Scheme 140)[366].

The information presented in this Section demonstrates that multicomponent reactions have a great potential in applied chemistry. Studies of these reactions develop towards one-pot synthesis of target products in quantitative yields, in an atom-economic manner, with fast assembly of complex molecules and low number of by-products, as well towards time and energy saving. These features comply with most of the principles of green chemistry and also make the use of multicomponent reactions an ideal synthetic strategy, especially for synthesis directed towards chemical diversity[367]. The use of new methylene-active sulfur- and selenium-containing compounds in multicomponent condensations to obtain previously unknown biologically important S,Se-heterocyclic systems would become a promising trend of synthetic organic chemistry in the future[368, 369].
2.5. From fundamental indole chemistry to compounds with high biological activity
Indole, possessing unique properties, is a versatile pharmacophore for the design of biologically active compounds. Indole is widely used in synthetic organic chemistry, since its cyclic enamine structure provides a lot of options for changing the reaction route by introduction of electron-withdrawing substituents. This provided the basis for the discovery of a number of original cascade transformations and rearrangements and for the development of total syntheses of natural indole derivatives described in recent years. In this Section, the indole chemistry is addressed through the lens of the scientific school headed by Professor A.V.Aksenov. This part of the review presents original approaches to some compounds that are of interest for medicinal chemistry, such as 2-quinolones, iso- and neocryptolepines, β- and γ-carbolines and indolylacetohydroxamic acids, modern indole-containing scaffolds synthesized at the North Caucasus Federal University. Original rearrangements, spirocyclizations and a new type of reactions involving a conjugated nitro group similar to aza-Cope rearrangement are considered. The discussion includes a critical evaluation of the reactions we discovered, which are compared with the modern trends of indole chemistry.
Indole derivatives attract substantial research attention; synthesis and properties of compounds of this type are among the most actively developing areas of heterocyclic chemistry, owing to their wide occurrence in nature[370-372] and diverse pharmacological activities[373-375]. Currently, more than 1500 indole alkaloids of various structural complexity have been described; they were isolated from secondary plant metabolites[370, 371], marine organisms[372] and fungi[376]. Indole-based natural products, along with synthetic derivatives, attract special attention of organic chemists, medicinal chemists, biologists and pharmacists, who have been long studying their biological and chemical properties. Indole derivatives are a perfect substitute for amino acids, which is widely utilized to develop peptidomimetics[377, 378] and provides opportunities for the discovery of new drugs with various mechanisms of action[379, 380]. According to retail sales data, the list of top 200 most in-demand drugs in the USA in 2012 included seven indole-containing commercial products. Currently, there are numerous known indole derivatives that are used as drugs or are in various phases of clinical or preclinical trials.
The indole heterocyclic system, apart from being relatively stable, has an important benefit from the chemical standpoint: it is a heterocyclic analogue of enamine, which accounts for a great variety of original rearrangements and cascade reactions and makes it possible to call indole ‘the king of heterocycles’. The indole chemistry is a key area in the studies of the reactivity and biological activity of heterocycles pursued by Professor A.V.Aksenov’s scientific school in the last decade. This allows us to consider the current studies of indole derivatives through the lens of experience we gained, with our own results serving as the main story line for this Section.
The assembly of the indole nucleus is a relevant problem that is fairly widely addressed in the literature. Classification of reactions of this type is quite a laborious task, which is considered, for example, in recent reviews[381-385]. Despite the huge number of publications on the synthesis of indole derivatives, each new method increases the structural diversity of these derivatives, and the introduction of an indole nucleus into practically valuable organic molecules maintains the interest in the indole chemistry at a high level.
A traditional and reliable method for the construction of the indole nucleus is the Fischer reaction[386], which is still often used in the total syntheses of indole-containing molecular cages with different complexity. In 2023, our research group developed a new approach to 7-aryl-substituted analogues of biologically active paullone 2.5.4,с based on the reaction of 3,4-dihydro-1H-benzo[b]azrepine-2,5-diones 2.5.3 with arylhydrazines in the presence of polyphosphoric acid (PPA; the percentage of P2O5 is indicated in parentheses in the Schemes)[387]. Although reactions of this type were described previously[388, 389], the introduction of aryl substituent in position 7 and the assembly of a seven-membered ring from 4-(2-aminophenyl)-4-oxobutyronitriles 2.5.2 were carried out for the first time. This sequence of four synthetic steps provided the formation of valuable products from available 2'-aminochalcones 2.5.1 (Scheme 141).

A key approach of our research group is to change the reaction pathway depending on the conditions. This strategy has been called ‘smart reaction media’. A good illustration for this strategy is provided by 2-aryl-4-(2-aminophenyl)-4-oxobutyronitriles 2.5.2, which were synthesized for the first time by our group. It was shown that treatment of these substrates with polyphosphoric acid containing 87% P2O5 unexpectedly results in 2-(indol-2-yl)acetamides 2.5.6, which is an unusual case of umpolung (i.e., polarity inversion) of the carbonyl group and is attributable to the intermediate formation of 2-aminofurans 2.5.5 (Scheme 142)[390]. Mechanistically, this reaction resembles the transformation described in 2012 by A.V.Butin and co-workers[391].

Contrary to our expectations, no aminofuran ring closure takes place in alkaline solutions. Instead, slow formation of 2-(3-oxoindolin-2-ylidene)acetonitriles 2.5.7 is observed due to oxidation by air oxygen. After optimization of the reaction conditions, DMSO proved to be the most appropriate oxidant (Scheme 143)[392]. The procedure of the synthesis of products 2.5.7, including the addition to the cyanide anion to 2-arylideneindolin-3-ones 2.5.8 followed by oxidation with DMSO, was described previously by Velezheva et al[393].

An alternative route to products 2.5.7 was based on the reaction of ortho-nitro-substituted chalcones 2.5.10 with KCN in refluxing methanol[394]. The reaction consists of two unusual steps: nucleophilic attack of the nitro group of 2.5.11 by the enolate anion and reduction of intermediate 2.5.12 with the solvent after addition of the acid. Subsequently, it was shown that this reaction can also begin from 2 ́-nitroacetophenones 2.5.9, aldehydes and potassium cyanide. The yield of the product remains the same; hence, it can be concluded that the in situ formation of chalcones 2.5.10 is quantitative[395]. Earlier, a combination of these steps was utilized by other authors[396] (Scheme 144).

Fused indole derivatives can be prepared by a similar reaction of 6(7)-acyl-1H-perimidines 2.5.13 with nitrobenzene. The yield of 1H-1,5,7-triazacyclopenta[cd]phenalenes 2.5.14 proved to be moderate, because substrate 2.5.13 acted also as a reducing agent, which was confirmed by a control experiment with nitrosobenzene (Scheme 145)[397].

One more method providing the synthesis of heterocycles 2.5.14 consists in the reaction of nitroalkanes activated with polyphoshoric acid with 1,4,5-triaminonaphthalenes 2.5.15 or 6(7)-amino-1H-perimidines 2.5.16 (Scheme 146)[398].

A combination of acetamination of perimidines 2.5.17 with the subsequent reaction with 1,3,5-triazines affords similar products 2.5.14. In this case, nitroethane is only the source of the amino group (Scheme 147)[399].

Functionalization of indole is yet another effective strategy for the synthesis of indole-based biologically active compounds and other practically valuable products. In view of the fact that quite a number of ready building blocks containing an indole ring are currently in use, this approach is often more desirable for this class of compounds.
In continuation of the development of approaches to polynuclear derivatives containing an indole nucleus, we proposed an original method for the synthesis of a previously unknown class of heterocyclic compounds 2.5.21[400]. The method was based on the condensation of indole-4-carbaldehydes 2.5.18 with 4-oxobutyronitriles 2.5.19 and the subsequent microwave-assisted cyclization of intermediate 2.5.20. The reaction was also carried out in the intermolecular version: lactams 2.5.22 were reacted with indoles 2.5.23 to give 3-substituted 3 indoles 2.5.24 (Scheme 148)[401, 402].

In 2015, our research group synthesized 2-(indol-3-yl)acetohydroxamic acids 2.5.28, which can be considered as highly effective scaffolds due to the presence of numerous reaction centres. These compounds showed high activity in vitro against multiple drug resistant cancer cell lines[403]. Subsequently, animal tests were performed[404], and the results were patented[405-407]. The approach that we proposed include a tandem reaction of indoles 2.5.23 with β-nitrostyrenes 2.5.27 in polyphosphoric acid. These indoles (with R2 = H) can be obtained by the Fischer reaction involving phenylhydrazine 2.5.25 and acetophenones 2.5.26 (Scheme 149)[403].

The methylation of 2-(indol-3-yl)acetohydroxamic acids 2.5.28 can be carried out selectively at the hydroxyl group; however, in this case, the activity of product 2.5.29 was lower than that of substrate 2.5.28 (Scheme 150)[408].

For the subsequent investigation of the structure–biological activity relationships, we also prepared 2-(indol-3-yl)acetonitriles 2.5.30 by two different methods: reduction of the hydroxamate group[409] and a tandem transformation including the Michael reaction and reduction (Scheme 151)[410].

A similar reductive coupling of 2-substituted indoles with nitrostyrenes using polyphosphoric acid in place of formic acid gives 2-(indol-3-yl)acetamides 2.5.31 in good yields under mild conditions (Scheme 152)[411].

Convenient precursors of the above-listed compounds are 1-(indol-3-yl)-2-nitroethanes 2.5.33, which can be easily prepared by the Michael reaction involving 3-(2-nitrovinyl)indoles 2.5.32 and various methylene-active compounds. Microwave assistance (Scheme 153)[412] increases the yield of the product and decreases the reaction time and the amount of solvent in comparison with those under conventional conditions[413]. The yields can be considerably improved by conducting the reaction under microwave irradiation without a solvent in the presence of a solid calcium carbonate or sodium acetate[414].

The extension of this method to indoles 2.5.32, containing a methyl or phenyl group in position 2 demonstrated that any substituents considerably complicate the process, inducing a number of side reactions that decrease the yield of the target product. The resulting dicarbonyl compounds 2.5.33 were converted to isoxazoles or pyrazoles 2.5.34 (Scheme 154)[415].

3-(2-Nitrovinyl)indoles 2.5.32 also react with other nucleophiles, e.g., phenols 2.5.35. Owing to the presence of the ortho-hydroxyl group, a cascade process can be initiated to give benzofuran-2-ones 2.5.36 (Scheme 155)[416].

In view of the high demand for the synthesis of 3-(2-nitrovinyl)indoles 2.5.32, we developed an efficient one-step method based on the reaction of indoles 2.5.23 with available α-nitroacetophenones 2.5.37 (Scheme 156)[417].

One more type of 3-vinylindoles 2.5.40 can be prepared by condensation of indole-3-carbaldehydes 2.5.38 with methyl-substituted azines and azoles 2.5.39 under microwave irradiation without a solvent (Scheme 157)[418].

Isocryptolepines represent an important class of heterocyclic compounds exhibiting a wide range of biological activities[419-421]. Our research group proposed a convenient synthetic approach to indolo[3,2-c]quinolines 2.5.43 based on the Fischer reaction involving о-aminoacetophenones 2.5.41 and arylhydrazines 2.5.25 and the subsequent treatment with acylating agents. The anticancer activity of the new products was found to be quite high (the half-maximal inhibitory concentration IC50 was in the submicromolar range) (Scheme 158)[422].

The use of aliphatic nitro compounds as synthetic precursors of carboxylic acids produced an extensive library of isocryptolepine analogues 2.5.43, which show activity in the nanomolar range of concentrations, which is 10 000 times higher than the activity of the initial alkaloid (Scheme 159)[423].

We expected that the reaction of 2-(2-aminophenyl)indole 2.5.42 with β-nitrostyrenes 2.5.27 in the presence of polyphosphoric acid would afford paullone derivatives 2.5.4. However, in this case, the reaction was accompanied by the rearrangement with elimination of HCN and afforded indolo[3,2-c]quinolines 2.5.43, while nitroalkenes 2.5.27 functioned as synthetic analogues of carboxylic acids (Scheme 160)[424].

The synthetic precursors of о-aminoacetophenones 2.5.41 are о-nitroacetophenones 2.5.9; the latter can be used to obtain nitro derivatives 2.5.44, which can be reduced with tin metal directly in polyphosphoric acid, as was shown for the first time in our studies. However, this approach does not provide substitution at position 5, and an additional alkylation step is required to obtain products 2.5.43 (Scheme 161)[425].

The formation of the N(6) – C(4a) bond is an attractive option for the assembly of the indolo[3,2-c]quinoline ring 2.5.44; this can be done using 2-arylindole-3-carbaldehyde oximes. Although quinoline synthesis by a similar method was described earlier[426], Alonso et al.[427] reported both experimental and calculation evidence indicating that this reaction is impossible and should give 2-aryl-3-cyanoindoles instead. Nevertheless, we demonstrated that O-acetyloximes 2.5.45 are converted to desired products 2.5.43 in the presence of SnCl4, while 3-acetylindole derivatives (2.5.44, R1 = Me) undergo the Beckmann rearrangement to form acetamides 2.5.46 (Scheme 162)[428].

Isomeric neocryptolepines 2.5.48 also exhibit high biological activity[429, 430] and are found in the same natural objects as isocryptolepines[431]. We developed an approach[432] to the synthesis of these molecules based on the reductive cyclization of 3-(2-nitrophenyl)-2-quinolones 2.5.47 generated in situ from β-nitrostyrenes 2.5.27 and 2-substituted indoles 2.5.23 in the presence of polyphosphoric acid. This method can also furnish oxygen analogues of this alkaloid – compounds 2.5.49, which are scantily represented in the literature, but are known for their anti-tuberculosis effect (Scheme 163)[433].

The above ring expansion reaction resulting in 3-substituted 2-quinolones 2.5.47, difficult to obtain by other methods, was performed in our earlier study[434]. The original rearrangement of intermediate 2-(3-indolyl)acetohydroxamic acids 2.5.28 to form heterocycles of a different class, quinolones, which has no analogues in the literature, confirms the status of indole as the ‘king of heterocycles.’ The only drawback of this transformation is the non-ideal atom economy. The 2-substituent in the indole ring is eliminated in this reaction as an amide, which is possible only in the presence of at least a methyl group. Hence, the reaction does not occur for C(2)-unsubstituted indoles (Scheme 164).

The scope of applicability of this reaction was expanded to a wider range of substrates by Aksenov et al.[435], who described an alternative approach based on the reaction of 3-(2-nitrovinyl)indoles 2.5.32 with arenes, which first gives hydroxamic acids 2.5.28 and then quinolones 2.5.47 (Scheme 165).

The reacting groups can also be combined in a single substrate such as aryl 3-nitropropyl ketone 2.5.50, which is prepared by the Michael addition of acetophenones 2.5.26 to β-nitrostyrenes 2.5.27. The subsequent cascade reaction of compound 2.5.50 with arylhydrazines 2.5.25 affords 2-quinolones. The advantage of this method is that arylhydrazines can be varied, with the second substrate being the same (Scheme 166)[436].

β-Carbolines are a class of physiologically active compounds with an indole ring widely occurring in nature[437, 438]. In continuation of the studies on the synthesis of neocryptolepine derivatives[432], we made an attempt to carry out the thermal reaction of tricresyl phosphate with 2-methyl-3-(2-nitrovinyl)indole, which unexpectedly yielded β-carboline 2.5.53 in a moderate yield. Then we investigated this reaction in various solvents, among which n-butanol was the solvent of choice. This transformation is an unusual case of isomerization of a nitro compound to the aci-form accompanied by an electrocyclic reaction. The reaction involves intermediate 2.5.51, with the nitro group nitrogen being a component of the conjugation system, which was unknown previously. By using Boc-protection, the yield of product 2.5.53 can be increased 1.5-fold. The cause for this is still obscure, but apparently the protecting group precludes the undesirable alternative isomerization involving the free indole NH group rather than the 2-alkyl substituent (Scheme 167)[439].

β-Carboline N-oxide 2.5.52 is formed as an intermediate, and is then reduced with butanol during the reaction, which is confirmed by control experiments (Scheme 168)[439].

Aksenov et al.[440] carried out this reaction with new substrates and proposed several one-pot methods for the generation of 2-alkyl-3-(2-nitrovinyl)indoles. These methods are based on the reaction of indoles with N-(2-nitrovinyl)morpholines 2.5.54 and the Henry reaction involving indole-3-carbaldehydes 2.5.38 and nitroalkanes 2.5.55. Alternatively, intermediate heterotrienes 2.5.57 can be generated from spiro[indole-3,5'-isoxazoles] 2.5.56 (Scheme 169).

Previously, spiro[indole-3,5'-isoxazoles] 2.5.56 were synthesized by formal [4 + 1]-cycloaddition of β-nitrostyrenes 2.5.27 to indoles 2.5.23 in the presence of H3PO3[441]. This reaction is characterized by excellent diastereoselectivity (dr > 20 : 1) and high yields of products (Scheme 170). The resulting spirocycles showed high antitumour activity and the ability to differentiate neuroblastoma cells. Spirocyclizations based on isatins[442] and 3-arylideneoxindoles have been known previously[443]; however, structures of this type are discussed for the first time in our studies.

An alternative approach to these systems includes the addition of Grignard reagents to 3-(2-nitrovinyl)indoles 2.5.32 followed by the introduction of the resulting nitronate 2.5.58 into the Nef reaction[444]. This issue is very important, as 2-(indol-3-yl)nitroethanes 2.5.59 do not react in this way (Scheme 171).

The base-catalyzed rearrangement of spiro[indole-3,5'-isoxazoles] 2.5.56 affords 2-(3-oxoindolin-2-yl)-2-arylacetonitriles 2.5.60 with retention of the stereocentre configuration. When KOH is used, inversion of the chiral centre at the nitrile group takes place, giving a mixture of all four possible isomers in equal proportions (Scheme 172)[445].

Acid catalysis makes it possible to carry out this reaction in a cascade manner starting from the formation of indoles 2.5.23 and to introduce a substituent to the nitrogen atom (Scheme 173)[445]. Related α-ketol rearrangements of indoles are widely used by various groups of authors[446, 447] in total syntheses of natural compounds.

As mentioned above, 2-(indol-3-yl)nitroethanes 2.5.59 are completely inert under standard reaction conditions with H3PO3; however, the use of a special system for generation of nitrile oxides from nitro compounds (POCl3 – Et3N) made it possible to accomplish this transformation. The reaction proceeds in the presence of triethylamine; hence, the intermediate spiro compound 2.5.56 is rearranged into oxoindole 2.5.60 (Scheme 174)[448].

The efficient use of spiro compounds 2.5.56 was demonstrated by the synthesis of 2-(indol-3-yl)acetamides. In this reaction, the benzyl cyanide moiety migrates back to position 3, which is apparently accompanied by retention of the configuration of stereocentres. One representative of this series of compounds exhibits activity at submicromolar concentrations against the MCF-7 breast adenocarcinoma cell line (Scheme 175)[449].

2-(3-Oxoindolin-2-yl)-2-arylacetonitriles 2.5.60 possess a unique ability to eliminate a benzyl cyanide molecule to give 3H-indol-3-ones 2.5.61; this underlies the following transformations. First of all, it was shown that treatment with KOH triggers the rearrangement with migration of the aryl substituent in position 2 to give 3-hydroxyindolin-2-ones 2.5.62. N-Alkyl-substituted analogues of compounds 2.5.60 react in a different way, yielding 5-hydroxypyrrol-2-ones 2.5.63, structurally similar to the natural alkaloid aristone (Scheme 176)[450].

The related reaction of 2-(3-oxoindolin-2-yl)-2-arylacetonitriles with o-phenylenediamine 2.5.64 carried out with microwave assistance is based on the thermal elimination of benzyl cyanide[451]. The reaction gives 2-(2-aminophenyl)-3-arylquinoxalines 2.5.66 as a result of ring opening in intermediate 2.5.65 and aromatization. In the case of unsymmetrically substituted phenylenediamines, the reaction yields difficult-to-separate mixtures of isomeric products in a total yield of 70 – 96% (Scheme 177).

3-Aminoindoles 2.5.67 are fairly unstable compounds that decompose under the action of light and air oxygen. Nevertheless, chemists are concentrated on the development of approaches to these poorly accessible compounds[452, 453], because of their large practical potential[454]. An unusual reduction of 2-(3-oxoindolin-2-yl)-2-arylacetonitriles 2.5.60, furnishing products 2.5.67, was accomplished in a microwave reactor under the action of aliphatic amines or hydrazine hydrate. In order to solve the instability problem, the acetyl protecting group can be introduced into the molecule during the reaction (Scheme 178)[455].

The synthetic and reactivity aspects of indole chemistry discussed in this Section leave no doubt that this heterocyclic system has a great potential for the design of original transformations, including cascade reactions and rearrangements, which can be used in the synthesis of structures valuable for medicinal chemistry. The new indole-containing scaffolds found recently confirm the status of this heterocycle as a privileged structure for the development of effective drugs and provide opportunities for further exploratory studies in the field of new indole derivatives.
2.6. Advances in the synthesis of polynitrogen heterocyclic systems
This Section reviews the recent advances of the research group headed by Professors N.N.Makhova and L.L.Fershtat over the last 7 – 10 years in the synthesis of polynitrogen heterocyclic systems based on 1,2,5-oxadiazole, 1,2,4,5-tetrazine and tetrazole. In some cases, mechanistic features of the pathways of certain reactions of heterocyclic core assembly are presented. Particular attention will be paid to the potential for practical applications of such compounds as pharmacologically active substances (primarily as NO donors) or as promising energetic materials for multi-purpose applications.
In recent years, the development of the chemistry of 1,2,5-oxadiazole derivatives has focused on the development of synthetic approaches to various hetaryl-substituted structures containing the 1,2,5-oxadiazole (furazan) or 1,2,5-oxadiazole 2-oxide (furoxan) moiety in combination with other nitrogen heterocycles[456-462]. The relevance of this trend is mainly due to the balanced properties of such polyheterocyclic systems, which are important in the search for new pharmacologically active substances[457, 463] or components of functional materials[464-466]. Significant progress in this field was made by the implementation of the one-pot methodology, which provided the maximum efficiency of the processes.
Based on the tandem heterocyclization of furoxanylamidoximes 2.6.1 with chlorides of various aliphatic, aromatic and heterocyclic carboxylic acids, a process for the synthesis of mono- (2.6.2) and bis(1,2,4-oxadiazolyl)furoxans 2.6.3[467]. The Sc(OTf)3 catalyzed condensation of 3-methylfuroxan-4-carboxamidoxime 2.6.1a with aldehydes followed by oxidation of the intermediate 1,2,4-oxadiazolines with iodine was an alternative for the preparation of the structures 2.6.2. Interestingly, the use of 4-aminofuroxan-3-carboxamidoxime 2.6.1c in a similar reaction not only affords a 1,2,4-oxadiazole scaffold but also promotes the rearrangement of the furoxan ring into a furazan one to give carbamoylfurazans 2.6.4. This transformation is the first example of a Lewis acid-catalyzed rearrangement in the furoxan series[468]. At the same time, heterocyclization of functionalized furoxanylamidoximes 2.6.1d,e in the presence of BrCN afforded amino-1,2,4-oxadiazoles, which undergo oxidative coupling under the action of KMnO4 in hydrochloric acid to furnish energetic azo-bridged tetracyclic structures 2.6.5 (Scheme 179)[469].

Based on the reaction of the available 3-methylfuroxan-4-carboxylic acid hydrazide 2.6.6a with aliphatic, aromatic and heterocyclic carboxylic acids or their chlorides in the presence of POCl3, a method for the preparation of (1,3,4-oxadiazolyl)furoxans 2.6.7 has been developed. Nucleophilic substitution of the chlorine atom in the corresponding chloromethyl-1,3,4-oxadiazole gave new polyheterocyclic ensembles 2.6.8 incorporating furoxan and 1,3,4-oxadiazole heterocycles in various combinations[470]. Heterocyclization of hydrazides 2.6.6 under the action of bromocyan afforded amino-1,3,4-oxadiazoles 2.6.9. Chemoselective oxidation of the amino group at the furoxan ring with 85% H2O2 gave nitrofuroxan 2.6.10, in which the remaining amino group was further oxidized to the tetracyclic azo compound 2.6.11 (Scheme 180)[469].

New synthetic approaches to (1H-1,2,3-triazol-1-yl)furoxans include [3 + 2] cycloaddition of various dipolarophiles to azidofuroxans. In particular, the formation of the 1,2,3-triazole ring in the reaction between 4-azidofuroxans 2.6.12 and acetylenes occurs only upon heating in ionic liquids, and the reaction with terminal acetylenes proceeds with moderate regioselectivity to give predominantly 1,4-disubstituted 1,2,3-triazoles. However, the yields of the target products 2.6.13 were low, apparently due to the competitive decomposition of the starting azidofuroxans[471]. Interestingly, a similar 1,3-dipolar cycloaddition of 4-amino-3-azidofurazan proceeds under milder conditions and yields energetic materials based on (1,2,3-triazolyl)furazans[472].
An alternative option for the synthesis of such heterocyclic systems is the use of enolate anions of 1,3-dicarbonyl compounds as dipolarophiles (Scheme 181). In this case, the [3 + 2] cycloaddition proceeds under mild conditions of basic catalysis and the regioselectivity of the formation of (1,2,3-triazolyl)furoxans 2.6.14 is completely determined by the configuration of the intermediate enolate anion[473]. Compounds 2.6.14 can also be synthesised by Lewis acid-catalyzed Wolff cyclocondensation involving 4-aminofuroxans 2.6.15 and diazo-β-dicarbonyl compounds[474]. The construction of (4-nitro-1,2,3-triazolyl)furoxans 2.6.16 in the [3 + 2] cycloaddition reaction of 4-azidofuroxans 2.6.12 with 1-dimethylamino-2-nitroethylene catalyzed by p-toluenesulfonic acid is also characterized by high regioselectivity. It should be noted that the resulting (4-nitro-1,2,3-triazolyl)furoxans 2.6.16 are capable of releasing nitric oxide(II) in a wide range of concentrations under physiological conditions[475].

Addition of hydrazine to the nitrile group of cyanofuroxans 2.6.17a,b gives the corresponding amidrazones 2.6.18a,b, which readily undergo acid-catalyzed cyclocondensation with trimethylorthoformate to produce (1,2,4-triazolyl)furoxans 2.6.19a,b[476]. Neutralization of compounds 2.6.19 with silver nitrate produces silver salts 2.6.20a,b which, via metathesis with hydrochlorides of nitrogen-rich bases, give a series of energetic salts 2.6.21 and 2.6.22. The use of the nitrile derivative 2.6.19a in the reaction with TMSN3 (TMS is trimethylsilyl) leads to the ammonium salt 2.6.23 containing three sequentially linked azole heterocyclic rings (Scheme 182)[477]. Furoxanylamidrazones can also be utilized as substrates in the synthesis of (1,2,4-triazinyl)furoxans, which undergo a tandem hetero-Diels-Alder/retro-Diels-Alder reaction with 1-(pyrrolidino)cyclohexene and norbornadiene to give polyheterocyclic structures based on (pyridyl)furoxans[478].

The [3+2] cycloaddition of cyanofuroxans 2.6.17 to TMSN3 in the presence of NH4F is a convenient method for the synthesis of a rather large series of ammonium salts of tetrazolylfuroxans 2.6.24. This approach is based on the in situ generation of NH4N3 and is highly tolerant to the presence of various functional groups (Scheme 183)[479]. A number of nitrogen-rich energetic salts 2.6.25, 2.6.25' have been prepared by sequential metathesis of cations in compounds 2.6.24 containing explosophoric azide and azo groups. It should be noted that impact sensitivity of these structures strongly depends on the cation nature: ammonium and triaminoguanidinium salts have been found to be the most sensitive and are therefore considered to be primary explosives[480]. Neutralization of ammonium salts to free tetrazolylfuroxans is achieved by the use of hydrochloric acid. Reacting tetrazolylfuroxans 2.6.26 with 1,2-dibromoethane in the presence of DIPEA gives rise to a tandem alkylation-elimination sequence affording regioselectively 2-vinyltetrazoles 2.6.27, which can serve as precursors in the synthesis of energetic polymers (see Scheme 183)[481].

The regioselective method for the preparation of 3-nitrobifuroxans 2.6.28 is based on a one-pot reaction cascade involving initial reaction of chloroximino-1,2,5-oxadiazoles 2.6.29 with the sodium salt of dinitromethane (DNM), followed by in situ nitrosation of the resulting intermediates[482]. This approach opened access to promising non-hydrogen energetic materials with physical and chemical properties comparable or superior to the known explosives hexogen (RDX) and octogen (HMX)[483, 484]. Interestingly, this synthetic methodology can be successfully applied to the synthesis of 4-alkyl- and 4-aryl-3-nitrofuroxans using the appropriate aldoximes and N-chlorosuccinimide as chlorinating agent[485]. 3-Nitrofuroxans 2.6.28 are isomerized on heating to the thermodynamically more stable 4-nitro regioisomers 2.6.30 (Scheme 184).

A number of promising energetic materials have been obtained by tandem transformation of regioisomeric nitrobifuroxans containing a dimethylformamidine protecting group. The synthetic sequence involves deprotection of compounds 2.6.31a,b followed by one-pot oxidation of the resulting amines to the azo derivatives 2.6.32a,b or 3,4'-dinitro-3',4-bifuroxan 2.6.33a[486]. In turn, the symmetrical dinitrobifuroxan 2.6.33b was obtained by oxidation of the corresponding diamino derivative 2.6.34 (Scheme 185)[487].

A characteristic feature of 4-nitrofuroxans is their tendency to undergo substitution reactions upon treatment with various nucleophiles[475, 488, 489]. Using this approach, the first 4-hydroxyfuroxans 2.6.35 were synthesized containing an active hydrogen atom of the OH group on the C=N bond of the heterocycle, which makes possible prototropic tautomerism of the lactim-lactam type. The study of the behaviour of these compounds as ambidentate nucleophiles in the methylation reaction showed that the reaction outcome strongly depends on the nature of the methylating agent. Methylation under the action of CH2N2 proceeded with low regioselectivity affording two regioisomers — methoxyfuroxans 2.6.36 and hitherto unknown derivatives N-methyl-1,2,5-oxadiazol-4-one 2-oxides 2.6.37 in the ratio of 1.4 : 1 – 3 : 1, which provides evidence for the ambidentate nucleophilicity of hydroxyfuroxans (Scheme 186). The ratio of 2.6.36 : 2.6.37 isomers increased with increasing electron-withdrawing properties of the substituent in the aromatic ring. Methylation under the action of MeI and Me2SO4 was more regioselective compared to diazomethane. In the case of MeI, the ratio of 2.6.36 : 2.6.37 isomers was 3 : 1 – 5 : 1, and for Me2SO4 it ranged from 10 : 1 to 19 : 1. The difference in regioselectivity of methylation of 4-hydroxyfuroxans 2.6.35 under the action of MeI and Me2SO4 can be interpreted in terms of Pearson’s theory. The highest regioselectivity of O-methylation was observed with Me2SO4 (a hard alkylating reagent). At the same time, MeI as a softer alkylating agent provided a lower regioselectivity of the process.

The reduction of 4-nitrofuroxans with SnCl2 in hydrochloric acid leads to the chemoselective formation of 4-aminofuroxans, whereas the reduction of 3-nitrofuroxans under the same conditions gave exclusively 3-aminofurazans as a result of the simultaneous reduction of the nitro group and the N-oxide moiety[490]. In 2020, a convenient method was developed for the diazotization of amino-1,2,5-oxadiazoles 2.6.38 under the action of NOBF4 in trifluoroacetic acid. Some of the resulting (1,2,5-oxadiazolyl)diazonium salts 2.6.39 were isolated for the first time in free form and their azo combination with various CH-acids (dimedone, Meldrum’s acid, 2-nitropropane) gave rise to hydrazones 2.6.40 and azo compounds 2.6.41 (Scheme 187)[491]. The azo-coupling of (1,2,5-oxadiazolyl)diazonium salts 2.6.39 with electron-rich arenes gave the corresponding azo compounds capable of photoswitching under visible light irradiation. The (Z)-arylazofuroxans exhibit much higher NO donation in vitro than the corresponding E-isomers, indicating the great potential of these molecular photoswitches in photopharmacology and other biomedical applications[492]. The diazonium salts 2.6.39 were also subjected to in situ chemoselective reduction with SnCl2, and the resulting hydrazinyl-1,2,5-oxadiazoles were subjected to one-pot condensation with carbonyl compounds. This provided access to previously unknown hydrazones 2.6.42, which are considered to be isosteric analogues of therapeutic agents active against leishmaniasis, schistosomiasisand Chagas’ fever[493].

Using a synthetic protocol for the diazotization of amino-1,2,5-oxadiazoles 2.6.38, a convenient one-pot procedure has been implemented for the construction of hybrid heterocyclic systems 2.6.43 containing 1,2,5-oxadiazole and 1,2,3,4-oxatriazolium-5-olate (azasydnone) motifs. The synthetic strategy involves diazotization of the starting amino-1,2,5-oxadiazoles 2.6.38, azo coupling of the resulting diazonium salts with the potassium salt of nitroform and further double rearrangement of the in situ generated trinitromethylazo-1,2,5-oxadiazoles into azasydnones 2.6.43 (Scheme 188)[494]. Compounds 2.6.43 are promising antiaggregants with a selective mechanism of inhibition of platelet aggregation[495].

The Lewis acid-catalyzed condensation of 3-aminofuroxans 2.6.44 with trimethyl orthoformate gives iminoethers 2.6.45 which undergo intramolecular rearrangement in the presence of substoichiometric amounts of KCN to give furazanylcarbamates 2.6.46. It should be noted that a similar rearrangement of 2-aminoazine N-oxides 2.6.47 proceeds as a one-pot process using Lewis acid as a catalyst to give the corresponding urethanes 2.6.48 (Scheme 189)[496].

Another type of transformations of functionalized furazans and furoxans such as chloromethyl-1,2,5-oxadiazoles 2.6.49 gave rise to the first representatives of energetic 1,2,5-oxadiazole-based ionic liquids. Subjecting substrates 2.6.49 to the Finkelstein reaction followed by in situ alkylation of N-methylimidazole with the resulting iodomethyl derivatives leads to iodides 2.6.50, which on metathesis with silver salts containing explosophoric anions give ionic liquids 2.6.51 (Scheme 190). The energetic structures 2.6.51 are found to have hypergolic properties but are inferior to 1,1-dimethylhydrazine[497].

Hybrid heterocyclic furoxan-containing structures are attracting the attention of researchers as practically relevant scaffolds in the creation of energetic materials and pharmacologically active substances. Novel antiaggregants have been synthesized by transformations of 4-amino-3-(azidocarbonyl)furoxan 2.6.52[498]. The nucleophilic substitution reaction of the azide group in substrate 2.6.52 with 3,3-dinitroazetidine was used to obtain the hybrid compound 2.6.53, in which oxidation of the amino group gives the energetic compound 2.6.54 (Scheme 191). Condensation of dinitroazetidine with trinitroethanol (TNE) gives the corresponding trinitroethyl derivative 2.6.55, with compounds 2.6.54 and 2.6.55 having a positive oxygen balance to CO[499].

A general and regioselective method based on the nitration of oximes 2.6.59 has been developed for the synthesis of hetarylfuroxans 2.6.56 – 2.6.58, including isoxazolyl, isoxazolinyl and (1,2,4-oxadiazolyl)furoxans. Subsequent thermolysis of the resulting nitrolic acids 2.6.60 favours the formation of furoxan carbonitrile oxides 2.6.61, which undergo [3 + 2] cycloaddition with the appropriate dipolarophiles (Scheme 192)[500]. Prepared in advance 4-nitrofuroxannitrolic acid is used to obtain similar nitrofuroxans[501].

In 2023, a novel approach to assemble hybrid NO-donor heterocyclic systems comprising furoxan and sydnonimine moieties was proposed using a NOBF4-mediated tandem nitrosation-cyclization sequence of α-amino nitriles 2.6.62. This method excludes the isolation of carcinogenic N-nitrosamines and opens access to fully substituted sydnonimines 2.6.63, which gave rise to a library of heterocyclic structures 2.6.64 – 2.6.66 with the ability to release NO over a wide range of concentrations (Scheme 193)[502].

The furoxan ring can serve as a precursor to other five-membered nitrogen-containing heterocycles. In particular, monosubstituted furoxans 2.6.67 undergo cleavage under mild conditions to α-oximinoacetonitrile oxides, which are intercepted in situ by dipolarophiles to give oximes 2.6.68. The addition of a strong base (KOH) allows the one-pot preparation of functionalized furazans 2.6.69 – 2.6.71 via azole-azole rearrangement[503]. At the same time, the addition of various nucleophiles to α-oximinoacetonitrile oxides followed by oxidation of the resulting intermediates provides access to furoxans 2.6.72 and 5-amino-4-nitroisoxazoles 2.6.73 (Scheme 194)[504, 505]. A synthetic approach to 2H-1,2,3-triazoles based on the azole-azole rearrangement of (furoxanyl)hydrazones has been proposed[506].

Various polynitrogen heterocycles are extremely important, but underexplored representatives of heterocyclic compounds. However, it should be noted that it is polynitrogen heterocycles that gave rise to promising energetic materials[507, 508], as well as stable heterocyclic radicals of interest for the preparation of magnetically active devices[509].
Based on cascade transformations of the available 3-amino-6-cyano-1,2,4,5-tetrazine (2.6.74), a series of new high energy salts 2.6.75 – 2.6.77 containing tetrazinedi(N-oxide) and hydroxytetrazole moieties and nitrogen-rich cations have been synthesized (Scheme 195)[510].

Destructive nitration of cyanoacetamide with fuming nitric acid in the presence of 20% oleum gives the highly sensitive trinitroacetonitrile 2.6.78, which is not isolated in pure form but was reacted with sodium azide in the presence of acetic acid to give tetrazole 2.6.79. Compound 2.6.79 is a rather strong NH-acid and its isolation is therefore very laborious. From a preparative point of view, it is much more convenient to neutralize (trinitromethyl)tetrazole 2.6.79 with AgNO3 and then isolate the salt 2.6.80, which is poorly soluble in common organic solvents (Scheme 196). Subjecting compound 2.6.80 to the metathesis reaction with hydrochlorides of nitrogen-rich bases makes it possible to obtain a series of energetic salts 2.6.81 characterized by a record combined nitrogen and oxygen content (up to 88%), which allows them to be considered as components of energetic compositions[511].

Nucleophilic substitution of the chlorine atom in the available functionalized hydrazones 2.6.82 with an azide anion gives the corresponding azidohydrazones 2.6.83, which undergo acid-induced electrocyclization followed by one-pot deacylation. A number of previously unknown N-(heteroaryl)- and N-(aryl)-aminotetrazoles 2.6.84 have been synthesized in this way[512]. At the same time, the reaction of chlorohydrazones 2.6.82 with acetylacetone afforded diacetylpyrazoles 2.6.85 which were converted to 2H-pyrazolo[3,4-d]pyridazine-5,6-dioxides 2.6.86 through condensation with hydroxylamine followed by oxidation of the intermediate 1,4-dioximes (Scheme 197). Compounds 2.6.86 represent another subclass of exogenous heterocyclic NO donors[513].

A new method for the synthesis of 7H-tetrazolo-[5,1-b][1,3,4]thiadiazines 2.6.87 has been developed based on the condensation of α-halo ketones 2.6.88 with thiocarbohydrazide, nitrosation of the resulting hydrazinylthiadiazines and intramolecular cyclization of the nitrosation product via azido-tetrazole tautomerism (Scheme 198). It should be noted that the heterocyclic structures 2.6.87 exist in the tetrazole form both in solid and in solution, according to IR and NMR spectroscopic data[514].

Obtaining stable N-centred heterocyclic radicals is quite a relevant line in modern organic materials science. In 2023, a method for the preparation of Kuhn verdazyls 2.6.89 was optimized, consisting of the azo-coupling of aryldiazonium salts with readily available hydrazones, followed by base-induced cyclization of the in situ formed formazanes 2.6.90 with formalin (Scheme 199)[515].

A series of novel 1,4-dihydrobenzo[1,2,4][e]triazines 2.6.92 were obtained by oxidative cyclization of amidrazones 2.6.91 containing an acetyl or ester group. It was found that in this case Blatter radicals 2.6.93 are formed only in the presence of an ester substituent at the C(3) atom of the 1,2,4-triazine ring (Scheme 200). The resulting 1,4-dihydrobenzo[1,2,4][e]triazines have a high thermal stability (up to 240 – 250 °C), which opens up the possibility of using them as components of functional organic materials[516].

A promising trend in the chemistry of polynitrogen heterocyclic systems is the use of organic electrochemical methods. For example, we have implemented a new electrochemical approach to selective intramolecular N – N bond formation by oxidation of readily available oximinohydrazones 2.6.94. The developed electrochemical strategy provided access to a large series of previously hardly available 1,2,3-triazole 1-oxides 2.6.95 (Scheme 201).

Using cyclic voltammetry and quantum chemical calculations, it was elucidated that the oxidation of the N – H bond in the starting oximinohydrazones 2.6.94 with the formation of an aminyl radical is the first step in the process. It was also found that the resulting 1,2,3-triazole 1-oxides have high thermal stability and are exogenous NO donors, which allows us to consider such systems in the context of biomedical and materials science applications[517].
2.7. Synthesis of fluorine-containing heterocyclic compounds based on fluorinated alkenes and acetylenes
The strategy of synthesis of fluorine-containing compounds using fluorinated ‘building blocks’ (small molecules containing a fluorine atom or a fluoroalkyl moiety) has proved to be the most effective tool in organofluorine chemistry, as direct fluorination methods are not always sufficiently selective or require the use of expensive reagents. Fluorinated alkenes and alkynes are an excellent example of such ‘building blocks’. In this Section, methods for the preparation of fluorinated alkenes and alkynes based on the catalytic olefination and the synthesis of fluorinated heterocyclic compounds involving these reagents are discussed.
In the early 2000s, the catalytic olefination of carbonyl compounds was discovered[518, 519]. It was found that N-unsubstituted hydrazones of carbonyl compounds are converted into the corresponding alkenes under the action of polyhaloalkanes in the presence of bases (ammonia, ethylenediamine, triethylamine) and catalytic amounts of copper salts. The reaction is accompanied by the release of nitrogen and the only by-products are symmetrical azines, which are easily separated by column chromatography (Scheme 202). The reaction has great synthetic potential and allows the stereoselective synthesis of alkenes containing different halogen atoms and functional groups in yields up to 90 – 95%, with the ratio of the major product (the least sterically hindered alkene) to the minor isomer reaching 21 : 1 (see Scheme 202)[520-539]. Compared to classical methods for the olefination of carbonyl compounds, the catalytic olefination is simple, proceeds at room temperature, does not require an inert atmosphere, dry solvents or equimolar amounts of reducing metals or organoelement compounds. Procedures have also been developed for the reaction in ionic liquids[540] and in the presence of heterogeneous catalysts[541, 542]. In both cases, the process can be carried out repeatedly and the yield of target alkenes is not reduced up to 5 repeated reaction cycles.

The use of low-cost and readily available freons as olefinating reagents provides an approach to fluorinated alkenes, versatile “building blocks´ for access to more complex fluorinated compounds with important fields of application[543-545].For example, reactions with С1-freons, namely, CBr2F2 (freon 12В2)[546], CFCl3 (freon 11)[547] and CFBr3 (see[548-550]) give gem-difluorostyrenes, β-chloro- and β-bromo-β-fluorostyrenes in good yields (see Scheme 202). The use of C2-freons in the catalytic olefination opened the way to the synthesis of β-CF3- and β-CF2Cl-styrenes. Reactions with CF3CFBr2, CF3CCl3 and CF3CBr3 gave β-fluoro-[551], β-chloro- (see[552-554]) and β-bromo-β-trifluoromethylstyrenes[553, 555], and reactions with CF2ClCF2Cl afforded β-fluoro-β-chlorodifluoromethylstyrenes[551] in high yields.
Due to the rather mobile halogen atom, β-fluoro-β-bromostyrenes, as well as β-chloro- and β-bromo-β-trifluoromethylstyrenes, are convenient objects for introducing additional functional groups into the molecule via nucleophilic substitution, making these compounds promising ‘building blocks’ for obtaining a variety of fluorinated alkenes, carbo- and heterocyclic compounds[556]. For example, α-fluoro-, α-trifluoromethylacrylonitriles[557] and α-fluoro-β-arylvinylsulfones have been synthesized regio- and stereoselectively in high yields by reactions with CuCN and sodium 4-methylphenylsulfinate (Scheme 203)[558]. β-Chloro- and β-bromo-β-trifluoromethylstyrenes react with ethanethiol[559] and metal alkoxides[560, 561] to give exclusively β-regioisomers in the presence of electron-withdrawing and bulky substituents in the starting alkene. As the donor properties of the substituents increase, the proportion of α-regioisomers grows (see Scheme 203). Similarly, styrenes react with pyrrolidine (one of the most active amines) to afford β-regioisomers corresponding to CF3-enamines (see Scheme 203)[561-564]. With electron-rich styrenes, the reaction first gives an α-regioisomeric enamine, which is unstable and undergoes a reaction cascade leading to the complete loss of the fluorine atom and the formation of the corresponding guanidinium salts. However, enamines have been obtained from electron-rich styrenes by reaction with the lithium salt of pyrrolidine (an even stronger nucleophile and base). Reactions with less active secondary amines affords the corresponding guanidinium salts, and only the use of the most electron-deficient 4-nitrophenyl-substituted styrene yielded trifluoromethylenamines (see Scheme 203)[562]. Similarly, nitrophenyl-substituted styrenes react with primary amines to give β-regioisomers, which are mixtures of tautomeric trifluoromethylenamines and trifluoromethylimines. The reactions with other styrenes furnish vinylogous guanidinium salts (see Scheme 203)[565].

The reaction of the electron-deficient β-halogen-β-trifluoromethylstyrenes with N,N- and N,O-dinucleophiles first generate β-regioisomeric imines, which undergo spontaneous cyclization into fluorinated imidazolidine derivatives 2.7.1 in high yields (Scheme 204). Styrenes with strong electron-donating substituents undergo unexpected fragmentation when treated with ethylenediamine to give non-fluorinated imidazolines 2.7.2. With styrenes having no strong acceptor or donor properties, the reaction produces a mixture of both products (see Scheme 204)[566]. Fluorinated imidazolidines and oxazolidines with substituted ethylene moiety were obtained in good yields via reactions of electron-deficient styrenes with substituted ethylenediamine and ethanolamine (see Scheme 204).

Fluorinated hexahydro- (2.7.3) and/or tetrahydropyrimidines 2.7.4 were prepared by reaction with 1,3-diaminopropane (see Scheme 204)[566].
Enamines 2.7.5 are unique compounds since they contain an electron-withdrawing trifluoromethyl group and an electron-donating pyrrolidine moiety, which is a good leaving group in acidic medium. Consequently, CF3-enamines are considered as synthetic equivalents of trifluoromethylbenzyl ketones and are successfully used in the synthesis of α,β-unsaturated ketones by condensation with carbonyl compounds, as well as in the preparation of indoles and carbolines.
α-CF3-enamines 2.7.5 were found to react with substituted benzaldehydes on heating in acetic acid to give the corresponding α,β-disubstituted CF3-enones 2.7.6 in high yields. A wide range of examples showed that this reaction has great synthetic potential and provides an access to CF3-enones containing both electron-donating and -withdrawing groups in both aryl substituents. Sterically hindered substrates containing, e.g., 2,6-dichlorophenyl, 2-bromophenyl and 1-naphthyl substituents were also tolerated by this reaction. Using hetaryl-substituted aldehydes, ketones bearing heterocyclic moieties have been obtained. Of special note is also the high stereoselectivity of the reaction, which in almost all cases produce only E-isomer (Scheme 205)[567].

Due to the presence of an activated double bond, α,β-disubstituted CF3-enones 2.7.6 are good Michael acceptors and the presence of the second electrophilic centre, the trifluoroacetyl group, makes them promising objects for reactions with dinucleophiles. It was found that the use of hydrazines as free bases in boiling ethanol produces fluorinated pyrazolines 2.7.7 in good yields. Pyrazolines bearing both electron-donating and -withdrawing substituents have been synthesized by this method. The reaction proceeds stereoselectively to give diastereomers containing trans-oriented aryl substituents at the C(4) and C(5) atoms. In some cases, the reaction stops at the stage of hydroxypyrazolidine, which has been isolated and characterized. These compounds readily eliminate water when heated with TsOH in toluene to furnish pyrazolines 2.7.7 in high yields (Scheme 206)[567].

Pyrazolines 2.7.7 readily undergo aromatization under the action of DDQ. The reaction proceeds on heating in toluene and affords pyrazoles 2.7.8 in high yields (method A, Scheme 207). A convenient one-pot procedure to produce pyrazoles directly from CF3-enones 2.7.6 has been developed. The reactions of hydrazine hydrochlorides with CF3-enones produce significant amounts of pyrazole impurities in addition to pyrazoline. It turned out that crude pyrazoline with pyrazole admixture, obtained after removal of ethanol from the reaction mixture, can also be successfully oxidized with DDQ (method B). As a result, a versatile approach to fully substituted CF3-pyrazoles has been found, allowing a wide range of pyrazoles containing any combination of electron-donating and -withdrawing substituents to be obtained in high yields[567]. This approach has been successfully applied to the synthesis of a number of derivatives of the drugs Celebrex and Mavacoxib, as well as the drug with the commercial code SC-560, having an additional C(4)-substituent in the pyrazole core. The corresponding tetrasubstituted pyrazoles 2.7.8 were obtained in up to 92% yields by the one-pot method B. Such products are very attractive for further biological studies.

The presence of nitro groups in the aryl rings of α,β-disubstituted CF3-enones 2.7.6 opens up the possibility of intramolecular cyclizations after reducing the nitro group to the amine one. With the nitro-substituted α-aryl moiety, the reduction followed by cyclization gives indoles, and with the nitro-substituted β-aryl moiety, the reaction cascade affords quinolines. Both possible reaction routes were accomplished. Nitro-substituted β-aryl-tethered CF3-enones required for the synthesis of quinolines were obtained by condensation of α-CF3-enamines with substituted 2-nitrobenzaldehydes. This reaction proceeds on heating in acetic acid and provides high yields of enones diversely substituted on both aryl rings (Scheme 208)[568].

Reduction of the nitro group by heating with iron powder in acetic acid for 1 – 2 h gives an amine, which immediately undergoes intramolecular cyclization into 2-trifluoromethyl-3-arylquinolines 2.7.9 in high yields (method A, Scheme 209)[568].

The one-pot approach, in which iron powder is added to the reaction mixture obtained after heating α-CF3-enamines with substituted 2-nitrobenzaldehydes, produces the target quinolines 2.7.9 in yields up to 89% based on starting enamine (see Scheme 209, method B). An even ‘deeper’ one-pot approach uses crude enamine. In this case, the yields over three steps are in the range of 40 – 76%. An alternative route to quinolines 2.7.9 has also been accomplished, in which reduction of the nitro group is carried out prior to condensation with enamine. In this case, the aminoaldehyde can either be prepared in advance (see Scheme 209, method E) or generated in situ (see Scheme 209, method D). In both cases, the target fluorinated quinolines 2.7.9 were isolated in high yields. It is worth noting that in addition to 2-nitrobenzaldehydes, heterocyclic aldehydes containing a nitro group ortho to the aldehyde function can be used in this reaction. Fluorinated thieno[2,3]pyridines and 1,6-naphthyridines were obtained by reaction with thiophene and pyridine derivatives (see Scheme 209).
CF3-Enones 2.7.10 with nitro-substituted α-aryl moiety were prepared via condensations of various benzaldehydes with α-CF3-enamines containing the 2-positioned nitro group. Such reactions, carried out on heating in acetic acid, furnish the target compounds in good to high yields. As a result, a representative set of o-nitrophenyl-CF3-enones 2.7.10 required for the synthesis of 2-trifluoromethylindoles was obtained (Scheme 210)[569].

The reduction of the nitro group in nitro-containing enones, such as compound 2.7.10a, has been studied. A selective reaction was achieved using ammonium formate as the hydrogen source and palladium on carbon as the hydrogenation catalyst. In the first step, the nitro group is reduced to an amino group which immediately reacts with the trifluoroacetyl group via the ring-closure to give the key intermediate, indolinol 2.7.11 (Scheme 211)[569, 570].

After acid-promoted elimination of water from indolinol 2.7.11, an imine-like intermediate 2.7.12 is formed, which adds nucleophiles to give substituted 3-benzyl-2-trifluoromethylindoles. To implement this strategy, exactly 3.3 equivalents of ammonium formate should be used, which is sufficient only for the reduction of the nitro group and insufficient for the reduction of the double bond of indolinol 2.7.11[569]. With an excess of ammonium formate (5 equiv.), the double bond of intermediate 2.7.11 is hydrogenated to saturated indolinol 2.7.13, which readily eliminates water to give 3-benzyl-2-trifluoromethylindole 2.7.14a (see Scheme 211)[570].
The latter strategy is the simplest from the experimentation viewpoint. To carry out reduction and cyclization, it is sufficient to stir a mixture of CF3-enone 2.7.10 and ammonium formate (5 equiv.) in the presence of 5 mol.% Pd/C in methanol at room temperature for 1 day or for 0.5 – 1 h on heating (Scheme 212).

The addition of hydrochloric acid promotes the elimination of water from the intermediate indolinol to give the target 3-benzyl-2-trifluoromethylindoles 2.7.14 in high yields, in most cases exceeding 95% (see Scheme 212)[570].
Substituted 3-benzyl-2-trifluoromethylindoles bearing additional substituents were obtained using 3.3 equiv. of ammonium formate. An acid work-up of the reaction mixture after reduction results in the addition of a molecule of alcohol, which is the reaction medium to give methoxy-, ethoxy-, isopropoxy- and n-butoxy-substituted indoles 2.7.14 in yields up to 86% (see Scheme 213)[569].

To obtain indoles 2.7.14 by the addition of other nucleophiles, it is sufficient to change the solvent at the reduction step using non-nucleophilic THF instead of methanol, which is a nucleophile per se. It was found that the solution of indolinol A obtained by reduction of CF3-enone with ammonium formate in THF is sufficiently stable and can be stored for a long time in the refrigerator. The water can be eliminated from indolinol A using TsOH or TMSCl, depending on the nature of the nucleophile. This reaction has great synthetic potential and opens the way to indole derivatives by reactions with C-, N-, O-, S- and P-nucleophiles (Scheme 214; the reagent, yield and conditions are given under the product formula)[569].

An efficient synthetic approach to 2-trifluoromethylindoles 2.7.14 using fluorinated enamines has been developed. It was found that o-nitro-β-chloro-β-trifluoromethylstyrenes react with pyrrolidine in high yields, in some cases close to quantitative ones. Such enamines are readily converted to 2-trifluoromethylindoles by treatment with iron powder in acetic acid (the Leimgruber-Batcho indoles synthesis) (Scheme 215)[571, 572]. The first step involves the reduction of the nitro group to the amino group, after which the ring-closing intramolecular attack of the latter takes place, accompanied by the cleavage of the protonated pyrrolidine moiety. The assembly of the indole core is thus achieved via the formation of the C(2) – N bond. The reaction can also be carried out in a one-pot variant without purification of the intermediate enamine, with yields of the target indoles reaching 85%.

With nitrostyrene having an additional fluorine atom in the para position to the nitro group, there is a possibility of further modification of the structure by nucleophilic substitution. It was found that the fluorine atom undergoes substitution at room temperature in the case of pyrrolidine, whereas less active amines require heating (Scheme 216)[572]. The cyclization of enamine intermediates under indole synthesis conditions was also carried out using iron in acetic acid. As a result, 2-trifluoromethylindoles 2.7.14 bearing 5-positioned pyrrolidine, morpholine, azepine, diethylamine, methylamine and hexylamine moieties were obtained in good yields (see Scheme 216)[572].

The indole core was assembled via the sequential formation of C(2) – N and CAr – N bonds using o-bromostyrenes. In this case, the enamine formed in situ in the reaction between β-halogen-β-trifluoromethyl(o-bromostyrenes) with primary aliphatic amines (C(2) – N bond formation) undergoes intramolecular cyclization to N-substituted 2-trifluoromethylindoles 2.7.14 (CAr – N bond formation) in good to high yields (Scheme 217)[573].

CF3-Enamines were found to react with arylhydrazines in acetic acid to afford corresponding hydrazones, which were converted into 2-trifluoromethylindoles without isolation by adding 2 equiv. of stronger methanesulfonic acid to the reaction mixture (the Fischer reaction). The proposed method is universal and provides an access to 3-aryl-2-trifluoromethylindoles 2.7.14 with different combinations of electron-withdrawing and -donating substituents in the aryl and indole moieties in good to high yields (Scheme 218, Scheme 219)[574].

The use of CF3 enamines as the ‘carbonyl’ component in the Pictet – Spengler reaction was also fruitful. Thus, reacting such enamines with a series of ethyl amines containing aryl or hetaryl substituents, which are activated electrophilically under mild conditions, various fluorinated heterocycles were obtained in good yields (see Scheme 219)[574].

2-Trifluoromethylindoles are a promising class of fluorinated heterocycles in which interest has recently grown rapidly[575, 576]. It should be noted, however, that the chemical properties of compounds 2.7.14 are still underinvestigated. Reactions with certain C-electrophiles provided a number of new derivatives (Scheme 220)[572]. Unusual results were observed in the MsOH-catalyzed reactions of 2-CF3-indole 2.7.14b with aryl aldehydes in alcohols. With benzaldehydes, alkoxy derivatives were formed in good yields and the reaction with 0.5 equiv. of benzaldehyde in ethanol gave a bis(indolyl)methane derivative (see Scheme 220)[572].

Halogenation at the third position of 2-trifluoromethylindole (2.7.14b) was found to proceed readily at room temperature using conventional halogenating agents. The corresponding chloro, bromo and iodo indoles 2.7.15 are formed in 95 – 98% yields by reactions with N-chlorosuccinimide (NCS), NBS, Br2 and I2 (Scheme 221). Haloindoles 2.7.15 are promising objects for modification with nucleophiles. Reactions of these compounds with MeI, BnBr and TsCl afforded a series of N-Me-, N-Bn- and N-Ts-substituted indoles in high yields, which were further reacted with 4-methylthiophenol, copper cyanide, phenylboronic acid and phenylacetylene to furnish 3-substituted 2-trifluoromethylindoles 2.7.14 in high yields (see Scheme 221)[577].

Haloalkenes are promising ‘building blocks’ in organic synthesis and have been successfully used to obtain various alkenes, alkynes and derivatives of carboxylic acids, carbo- and heterocyclic compounds[578]. In particular, dibromoalkenes have been used to synthesize terminal acetylenes by the two-step Corey – Fuchs reaction. In this case, dibromoalkenes can be easily replaced by dichloroalkenes[579]. The inverse order of addition of hydrazone solution to carbon tetrachloride makes it possible to carry out the reaction on a scale of up to 0.2 mol. with a minimum excess of olefinating reagent, and the yields of dichloroalkenes reach 88%. The conversion of dichloroalkene to acetylene is even higher, with yields up to 97%. Taking into account the much lower cost of the olefinating agents (hydrazine hydrate and carbon tetrachloride) compared to the classic Corey – Fuchs reaction (triphenylphosphine and carbon tetrabromide), the said method is much cheaper ($ 21 vs. $ 115 per 0.2 mol.) (Scheme 222)[579]. It was also found that intermediate lithium acetylenides can successfully react with electrophiles. In particular, a series of reactions of in situ generated lithium acetylenides with ethyl trifluoroacetate were carried out. As a result, CF3-ynones 4.7.16 containing electron-donating or -withdrawing substituents as well as bulky moieties were obtained in good to high yields (see Scheme 222)[579].

CF3-ynones 2.7.16 are valuable starting materials in the synthesis of fluorinated heterocyclic compounds. The presence of a highly electrophilic trifluoroacetyl moiety and an activated triple bond opens up possibilities for modification of these compounds by reactions with nucleophiles and dinucleophiles as well as by cycloaddition reactions. Due to the strong polarization of the triple bond caused by the trifluoroacetyl group, the [3 + 2] cycloaddition of CF3-ynones to azides proceeds regioselectively to give predominantly 4-trifluoroacetyltriazoles 2.7.17, while the proportion of the minor 5-regioisomer 2.7.18 in some cases is below 1%[580]. Alkyl- and aryl-substituted azides and CF3-ynones 2.7.16 may be involved in the reaction, and a one-pot approach is used to prepare azides from alkyl halides and sodium azide in DMSO. To summarize, an efficient versatile regioselective approach to 4-trifluoroacetyltriazoles has been developed, allowing their preparation in yields up to 89% (Scheme 223)[580].

NH-unsubstituted triazoles have been synthesized by the reaction of CF3-ynones 2.7.16 with sodium azide. This reaction takes several hours and affords 4-trifluoroacetyltriazoles 2.7.19 in high yields[581]. When acetic acid is added to the reaction mixture, the direction of the process changes and affords 3-trifluoromethylisoxazole. In addition to ethanol, this reaction can be carried out in a n-heptane-water mixture. A series of 3-trifluoromethyl isoxazoles were thus synthesized in good yields. It should be noted that triazoles and isoxazoles 2.7.20 bearing electron-donating or -withdrawing substituents as well as sterically demanding moieties, have been obtained by reactions between CF3-ynones and sodium azide. Alkyl-substituted CF3-ynones were also successfully involved in such transformations (Scheme 224)[581].

The possibility of modification of 5-aryl-4-trifluoroacetyl-1,2,3-triazoles 2.7.19 at the NH moiety has been investigated. The alkylation proceeds selectively in DMF with Na2CO3 as base to give 2-isomers 2.7.21 as major products in high yields. The ratio of the minor 1-isomer 2.7.22 to the major 2-isomer does not exceed 6 : 94 in some cases and the total yield of the products reaches 93%. Reactions with tosyl and mesyl chlorides are regioselective and produce only 2-substituted triazoles 2.7.21. The aryl halides activated by electron-withdrawing groups proceed regioselectively to give 2-aryltriazoles in good to high yields. Similarly, the copper-catalyzed reaction with boronic acids leads exclusively to 2-aryltriazoles in high yields. A study of the fluorescence properties of 2-substituted 1,2,3-triazoles 2.7.21 showed that some of them exhibit the properties of UV-blue-light-emitting fluorophores with quantum yields > 60% in some cases (Scheme 225)[582].

Studying the reaction of CF3-ynones 2.7.16 with methyl thioglycolate, it was found that it proceeds readily in methanol and is promoted by sodium methoxide. In the first stage, Michael addition takes place to give thiovinyl ester, which spontaneously cyclizes into methyl 3-(trifluoromethyl)thiophene-2-carboxylates 2.7.23. The reaction tolerates both aryl- and alkyl-substituted CF3-ynones, giving the corresponding thiophenes in good to high yields. Hydrolysis of esters obtained under the action of LiOH in a THF – H2O mixture gives 3-(trifluoromethyl)thiophene-2-carboxylic acid derivatives in high yields (Scheme 226)[583].

The direction of the reaction between CF3-ynones 2.7.16 and hydrazines depends critically on the nature of the solvent. Thus, the initial Michael addition of hydrazine in DMSO gives 5-CF3-pyrazolinols, which readily eliminate water under the action of TsOH to afford 5-CF3-pyrazoles 2.7.24 in high overall yields. On the contrary, the reactions of CF3-ynones with hydrazines in hexafluoroisopropyl alcohol (HFIP) start with the attack of the hydrazine on the carbonyl group, which leads, through a series of steps, to 3-CF3-pyrazoles 2.7.8 in yields that are in some cases close to quantitative. The reaction is versatile and allows the use of CF3-ynones 2.7.16 and hydrazines with both alkyl and aryl substituents (Scheme 227)[584].

Notably, when switching from hydrazines to their hydrochlorides, the regioselectivity of this reaction in DMSO is reversed. In this case, the first step produces hydrazone 2.7.25, which is easily cyclized into 3-CF3-pyrazoles 2.7.8 in the presence of strong bases. Using this approach, a series of 3-CF3-pyrazoles containing various combinations of aryl and alkyl substituents have been synthesized in yields up to 99%. The non-steroidal anti-inflammatory drug Celebrex and the anticancer drug SC-560 were obtained in high yields using this method (see Scheme 227).
Alternative conditions for the regioselective reaction of CF3-ynones 2.7.16 with hydrazines have been developed. The salts (THD-Dipp)AuOTf (THD-Dipp is 1,3-bis(2,6-diisopropylphenyl)hexahydro-2H-1,3-diazepin-2-ylidene) and AgOTf can efficiently catalyze this reaction to afford selectively 3-CF3-pyrazoles 2.7.8 in a few minutes in yields up to 99%. The minor 5-CF3-pyrazole impurity is < 0.5% and is easily removed by column chromatography. The great synthetic potential of the above reaction was demonstrated by obtaining various pyrazoles bearing alkyl and aryl substituents as well as functional groups. The drugs Celebrex® and SC-560 have also been synthesized using this method. The mechanistical study of the reaction showed that it proceeds via a semiaminal as a key intermediate. The role of the Ag+ ion is to form a complex with semiaminal to facilitate its cyclization (Scheme 228)[585].

Hydrazones of CF3-ynones 2.7.25, readily obtained by reaction with hydrazine hydrochlorides in alcohol (yields up to 95%) undergo cyclization promoted by electrophilic reagents. These hydrazones were found to react with iodine in MeCN in the presence of NaHCO3 as the base to give iodopyrazoles in almost quantitative yields. Notably, hydrazones derived from aryl and alkyl hydrazines, as well as tosyl hydrazine, are involved in this reaction. Using NCS, Br2 and NBS as halogenating reagents, chloro- and bromopyrazoles, including 4-halogen derivatives of the non-steroidal anti-inflammatory drug Celebrex, were obtained from hydrazones 2.7.25 in high yields. The iodo derivative of this drug has been subjected to various cross-coupling reactions, successfully extending a number of Celebrex analogues (Scheme 229)[586].

The use of nitrogen-containing 1,3-dinucleophiles in the reaction with CF3-ynones 2.7.16 opened the way to the synthesis of 1,3-pyrimidines. Reactions with formamidine acetate, acetamidine acetate, benzamidine hydrochloride, guanidine carbonate, ethylisothiouronium salt hydrobromide and benzylisothiouronium salt hydrochloride in the presence of bases gave good to high yields of 6-trifluoromethyl-4-aryl(alkyl)-1,3-pyrimidines 2.7.26 and their 2-positioned methyl-, benzyl-, amino-, ethylthio- and benzylthio-substituted derivatives. This highly efficient approach provides 6-trifluoromethylated-1,3-pyrimidines in up to 97% yield (Scheme 230)[587].

The increased electrophilicity of the triple bond in CF3-ynones enables an unusual type of reaction, namely, the annulation of the 1,3-oxazine ring with azines. The first step in this process is the formation of a 1,3-dipolar complex of pyridine with acetylene 2.7.27, the behaviour of which depends on the number of CF3-ynone equivalents, the presence of water in the reaction mixture and the azine nature. For example, in the reaction between quinoline and an equimolar amount of CF3-ynone in the presence of water (or in water), the complex 2.7.27 reacts with a water molecule to give semiaminal 2.7.28, which cyclizes into 1,2-functionalized quinolines 2.7.29, which is annulated to the 1,3-oxazine ring, in yields that are in some cases close to quantitative (Scheme 231)[588, 589]. It should be noted that the reaction is stereoselective and produces a single diastereomer. Isoquinoline and 1,8-naphthyridine have also been used in this reaction.

Interestingly, with equimolar amounts of pyridine and CF3-ynone, the 3-oxazine ring is not formed. In this case, the resulting intermediate 2.7.27 undergoes the pyridine ring-opening under the action of water to give trifluoroacetylated ethenyl-5-aminopenta-2,4-dienal 2.7.30 (see Scheme 231)[590]. The reaction with 2 equiv. of CF3-ynone 2.7.16 in the absence of water runs similarly both for pyridine and its benzannulated analogues (quinoline, isoquinoline, 1,8-naphthyridine). In this case, the intermediate 2.7.27 reacts with the carbonyl group of CF3-ynone to afford betaine 2.7.31, which cyclizes into an annulated 1,3-oxazine core bearing trifluoroacetyl and alkynyl substituents. These reactions also provide products 2.7.32 and 2.7.32 in yields up to 92 – 98% but as a mixture of two diastereomers (see Scheme 231)[591, 592].
Two unusual rearrangements were found in the treatment of 1,3-oxazinoquinolines 2.7.29 with bases. The action of sodium hydroxide triggers a reaction cascade leading to 2-arylquinolines, accompanied by the loss of the fluorine atom. In the cascade initiated by morpholine, 2-aryl-3-trifluoroacetylquinolines are formed, i.e., the fluorine atom remains intact in the molecule. In both cases, the reaction proceeds in high yields and can be carried out in a one-pot variant. Derivatives of 1,8-naphthyridine have also been obtained using this method (Scheme 232)[593].

The reaction of CF3-ynones with ethylenediamine and its derivatives was found to give trifluoromethyl[1,4]diazepines in good yields. The reaction with monosubstituted N-methylethylenediamine furnishes Michael mono- and bisadducts in addition to diazepine. In the case of N,N'-dimethylethylenediamine, the Michael bisadduct is the only reaction product. The process is stereoselective and gives only E,E-isomer. Finally, fluorinated benzodiazepines were obtained in high yields by the reaction with phenylenediamine (Scheme 233)[594].

The presence of a triple bond in CF3-ynones 2.7.16 makes possible their modification by electrophilic reagents in addition to the reactions with nucleophiles. Ynones react with bromine under mild conditions, keeping the reaction mixture in dichloromethane at room temperature. The corresponding α,β-dibromo-CF3-enones were obtained in yields up to 97%. The reaction is stereoselective and gives predominantly one isomer. The reaction of the phenyl-substituted dibromoenone with hydrazine hydrate affords 4-bromo-3-trifluoromethyl-5-phenylpyrazole 2.7.24a in high yield. The process is accompanied by the formation of small amounts of 3-trifluoromethyl-5-phenylpyrazole 2.7.24b as a result of the reaction between hydrazine hydrate with CF3-ynone obtained in the halophilic reaction of dibromenone with hydrazine hydrate. The overall yield of the two products is close to quantitative. Reaction with excess of ethylenediamine is accompanied by defragmentation to give α,β-dibromostyrene and 2-trifluoromethylimidazoline (Scheme 234)[595].

Monosubstituted α-bromo-CF3-enones are accessible via the sequential bromination-dehydrobromination of CF3-enones[596]. Reactions of these compounds with a number of dinucleophiles have been studied. The chemoselectivity of the reaction of ethylenediamine derivatives with α-bromo-CF3-enones was found to be determined by the nature of the substituents in these derivatives. Thus, the reaction with ethylenediamine leads to fluorinated bicyclic aziridine-containing dihydropyrazine derivatives, 7-aryl-5-(trifluoromethyl)-1,4-diazabicyclo[4.1.0]hept-4-enes 2.7.34 (Scheme 235)[597]. The process is stereoselective and yields a single diastereomer. It is worth noting that the reaction is universal and allows the use not only of CF3-enones but also unsaturated non-fluorinated aldehydes and ketones[597]. α-Bromo-CF3-enones react with phenylenediamine to give the saturated aziridinopiperazine scaffold 2.7.35. In this case, the reaction stops at the stage of a stable semiaminal of fluorinated ketone, known for its resistance to water elimination. This reaction is also stereoselective and proceeds in high yields (see Scheme 235)[597].

An unexpected rearrangement was found when studying the reaction of α-bromo-CF3-enones with N,N'-disubstituted ethylenediamines. In this case, a heterocyclic core of piperazinone 2.7.36 was formed in high yield as a result of the trifluoromethyl group shift. The reaction tolerate a wide range of symmetrical ethylenediamines containing methyl, ethyl, benzyl, allyl, methoxyethyl groups as well as sterically hindered isopropyl and cyclohexyl groups (see Scheme 235)[598-600]. It should be noted that non-fluorinated unsaturated ketones give defragmentation products, imidazolidine derivatives[599]. The expected Michael addition product with subsequent substitution of the bromine atom was only obtained in the reaction with N,N'-dicyclopropyl ethylenediamine. Trifluoroacetylpiperazines 2.7.37 are formed as a single diastereomer in yields up to 89%, even in the case of non-fluorinated unsaturated ketones (see Scheme 235)[599].
The reactions of bromo-substituted enones with 1,2-aminoethanol and its derivatives have been studied. The outcome of the process depends critically on the nature of the dinucleophile and the reaction conditions. Thus, reaction with 2-aminoethanol or 2-aminobutan-1-ol gives trifluoromethylated morpholines annullated to the aziridine ring (4-oxa-1-azabicyclo[4.1.0]heptanes) in high yields (Scheme 236). With 2,2-dimethyl-substituted aminoethanol (2-amino-2-methylpropan-1-ol), the reaction outcome depends on the solvent and base in choice. The reaction in chloroform in the presence of triethylamine gives fluorinated morpholine derivatives, whereas the reaction in DMSO in the presence of DBU affords fluorinated 1,4-oxazepanes annulated to oxiranes (2,8-dioxa-5-azabicyclo[5.1.0]octanes). Remarkably, both transformations are stereoselective, giving predominantly a single diastereomer in high yields[601]. A similar reaction involving 2-amino-2-butylhexan-1-ol leads to fluorinated oxazepanes (see Scheme 236).

Bromoenones react with an excess of N-methyl- and N-benzylaminoethanol to give fluorinated morpholine derivatives 2.7.38 decorated with an additional amino alcohol moiety in high yields. The reaction of these substrates with 1.2 – 1.5 equiv. of N-methylaminoethanol and prolinol is accompanied by the closure of an additional oxetane ring. This gives fluorinated derivatives of the rather uncommon oxetanomorpholines 2.7.39 in yields of up to 72%[602]. The high stereoselectivity of the reaction, producing a single diastereomer, should also be emphasized (Scheme 237).

The reactions of bromoenones with amino diols proceed in a similar way to give derivatives of fluorinated morpholines. The main products are fluorinated oxetanomorpholines formed as single diastereomers (Scheme 238). These compounds were isolated in pure form but they tend to spontaneously isomerize to bis-oxazines in chloroform solution. In many cases, fluorinated morpholine derivatives bearing an additional amino alcohol moiety were identified in the reaction mixtures. With 4-fluoro- and 4-nitrophenyl-substituted bromoenones, such products have been isolated and characterized. The fragmentation of these compounds account for the appearance of fluorinated morpholines in the reaction mixture, which do not contain the aryl substituent of the starting bromoenone. These compounds were also isolated and characterized (see Scheme 238)[603].

To conclude, the use of fluorinated alkenes and acetylenes as ‘building blocks’ is a very fruitful approach to a diversity of heterocycles bearing a fluorine atom or fluorinated moiety. As the structures of the starting alkenes and acetylenes have a highly polarized multiple bond, most of their reactions with nucleophiles are chemo- and regioselective. Another important feature is the high stereoselectivity of many processes, since the trifluoromethyl group, being a strong electron-withdrawing group, is able to stabilize the semiaminal (semiacetal) moiety. In addition, this functional group is rather sterically demanding. Therefore, many transformations involving fluorinated alkenes and acetylenes are unusual and have a unique character.
2.8. Synthesis of polynuclear heterocyclic molecules based on aryl- and hetarylacetylenes
The Sonogashira reaction[604] (the coupling of terminal alkynes with aryl and vinyl halides catalyzed by palladium and copper complexes), discovered in 1975, has made a variety of acetylene derivatives available[605, 606]. Transformations involving the CºC bond are thermodynamically very favorable[607, 608]. These two circumstances have contributed to the wide application of acetylenes in organic synthesis, including the preparation of heterocyclic compounds[605, 606]. Strategies based on the electrophilic, nucleophilic and transition metal complex-catalyzed cyclizations of alkynes[609-616], as well as cycloaddition and cycloisomerization reactions have proved to be particularly fruitful in the synthesis of heterocycles[617-620]. The importance of heterocyclic compounds is well known and does not require special discussion[621]. In the reviews[609-620] one can find many examples of the use of acetylene derivatives in the synthesis of natural heterocyclic molecules and their synthetic analogues, conjugated oligomers, polymers and dendrimers based on heterocycles, as well as other heterocyclic compounds with useful optical and electronic properties. In 2022, a review[3] on heterocyclic nanographenes and other polycyclic heteroaromatic molecules was published, with cyclizations involving the CºC bond often being the key stages in their synthesis.
Here we summarize the results of studies on the reactivity of alkynyl derivatives of azines (pteridine, quinoxaline, pyrazine, pyridine), 1H-perimidin-2(3H)-one and 1,8-bis(dimethylamino)naphthalene, carried out by employees of the Southern Federal University. The review contains three main parts, in which the synthesis and transformations of the three indicated types of (het)arylacetylenes are considered sequentially.
2.8.1. Syntheses based on alkynylazines
Among the alkynylazines presented in the review, two main classes can be distinguished: structures A — monoalkynyl derivatives of pteridine, quinoxaline, pyrazine, pyridine, unsubstituted in an adjacent position or containing an ortho-substituent (Cl, CN, Ar), and B — ortho-dialkynylazines (heteroaromatic enediynes).

Alkynylazines were prepared by the Sonogashira coupling of mono- or dihaloazines with terminal alkynes. 6-Alkynyl- (2.8.2), 7-alkynyl-6-chloro- (2.8.3) and 6,7-dialkynyl-1,3-dimethyllumazines (2.8.4) were synthesized from 6-chloro- and 6,7-dichloro derivatives of 1,3-dimethylpteridine-2,4(1H,3H)-dione (1,3-dimethyllumazine, 2.8.1) using the Pd2dba3 – PPh3 – CuI – K2CO3–DMF catalytic system (dba is dibenzylideneacetone) (Scheme 239)[622-626]. In this case, the more mobile chlorine atom in position 7 was replaced by an alkynyl group at room temperature, while the coupling in position 6 occurred only when heated to 90 – 100 °C. 3-Alkynyl-6,8-dimethylpyrimido[4,5-c]pyridazine-5,7(6H,8H)-diones (2.8.5), isomeric to compounds 2.8.2, were formed under the same conditions[625, 626].

A similar method was used to synthesize compounds 2.8.6 – 2.8.13. 3-(Phenylethynyl)-2-chloroquinoxaline (2.8.6)[627], 2,3-dialkynylquinoxalines (2.8.7)[624, 627] and the dialkynyl derivative 2.8.8 (see [624]) were prepared from the corresponding dichlorides in the Pd(PPh3)2Cl2 – CuI – Et3N – DMF catalytic system at room temperature. For the synthesis of 3-alkynylquinoxaline-2-carbonitriles (2.8.11), the same catalytic system was used, but with dimethyl sulfoxide as a solvent[628]. 1,2-Bis(phenylethynyl)benzene (2.8.9) was synthesized by coupling o-diiodobenzene with phenylacetylene in accordance with a previously described procedure[629]. The synthesis of 3-alkynyl-2-chloropyrazines (2.8.12) and 3-bromo-2-(phenylethynyl)pyridine (2.8.13) was carried out at room temperature in the Pd(PPh3)2Cl2 – CuI – Pri2NH – DMSO system from 2,3-dichloropyrazine and 2,3-dibromopyridine, respectively[630, 631]. In the preparation of 1-methyl-4,5-bis(phenylethynyl)-1H-imidazole (2.8.10) from the corresponding diiodide the Pd(PPh3)4 complex was used as a catalyst[632]. In most cases, the yields of products 2.8.6 – 2.8.13 exceeded 80%.

It has been shown that the π-deficient azine ring activates the triple bond for nucleophilic attack, but does not interfere with reactions with electrophiles. The directions of nucleophilic and electrophilic attack do not coincide (Scheme 240, parts a and b). In dialkynyl derivatives of lumazine 2.8.4, due to the electronic effects of heteroatoms and substituents, one of the triple bonds is activated to nucleophilic attack, while the other becomes sensitive to electrophiles (see Scheme 240, part c). A similar electron density distribution is observed in nitrile analogues of diynes of the type 2.8.11. It has also been established that the reactions of alkynylazines are not limited to the simple addition of a nucleophile or electrophile. Transformations are usually of a tandem or cascade nature and lead to the formation of polynuclear heterocyclic molecules. In particular, taking into account the known Larock indoles synthesis[633], we attempted the oxidative alkylamination of 6-alkynyllumazines 2.8.2, which resulted in the preparation of pyrrolopteridines 2.8.14 (Scheme 241)[622, 625]. The Sonogashira coupling of ortho-aminohalides 2.8.15 with phenylacetylene led to the same compounds. Isomeric molecules 2.8.5 were similarly cyclized under oxidative alkylamination conditions. It is not entirely clear which of the particles served as an intermediate of this transformation — the SNH-amination product 2.8.16, which then underwent to 5-endo-dig-cyclization catalyzed by a silver complex, or the enamine 2.8.17, which was converted into a condensed pyrrole by an intramolecular nucleophilic attack on the electron-deficient carbon atom. As a result, heterocyclic system 2.8.18 was formed.


These enamines were stabilized by intramolecular hydrogen bonding involving a cyclic aza group. Thus, enamines 2.8.19, obtained by reacting alkyne 2.8.3a with alkylamines at room temperature, underwent heterocyclization to pyrrolopteridines 2.8.20 only when heated with potash in DMF (Scheme 242)[623, 625]. Boiling compound 2.8.3a with an excess of alkylamine led to the same pyrrolopteridines 2.8.20. Acid hydrolysis of enamines 2.8.19 and further base-catalyzed cyclization of intermediate 2.8.21 yielded furopteridine 2.8.22.

The following example demonstrates the participation of a nucleophilic group at a CºC bond in heterocyclization. Enamine 2.8.23, obtained from alkyne 2.8.3е and morpholine, was converted to pyranopteridine 2.8.24 by heating with potash in DMF (Scheme 243)[623, 625].

6-Alkynylumazines 2.8.2 were used as starting compounds in the two-step synthesis of thienopteridines 2.8.26 (Scheme 244)[622, 625]. First, dibromoalkenes 2.8.25 were obtained by reacting with bromine. The further transformation of these dibromides into thienopteridines 2.8.26 under the action of sodium thiocarbonate proceeded as a cascade process, including the nucleophilic addition of the thiocarbonate ion to the C=C bond, elimination of the CS2 molecule, intramolecular SNH-tele-substitution and, finally, dehydrobromination.

Heterofused pteridines are an important but poorly studied class of compounds (see review[634]). Representatives of this class are the molybdopterins — a group of cofactors found in most molybdenum- and tungsten-containing enzymes; the ligand in them is the pyrano[3,2-g]pteridine derivative 2.8.27. Thieno[3,2-g]pteridine 2.8.28 (urothione) is a metabolite of molybdopterin. The pteridines 2.8.14, 2.8.20, 2.8.22, 2.8.24 and 2.8.26 presented above have a similar structural basis.

The largest number of unexpected transformations was observed in the reactions of alkynylazines 2.8.3 and 2.8.6 with C-nucleophiles, the precursors of which were CH2-active compounds (Scheme 245)[627]. The interaction began with the addition of the CH-acid carbanion to the CºC bond. Next, a cascade process developed, the final result of which depended on the nature of the nucleophile used. In the case of cyanoacetic ester, the aza group took part in further cyclization, which led to the annulation of the pyridine ring to the starting heterosystem (transformation 2.8.3, 2.8.6 → 2.8.29), and compound 2.8.30 was formed as a by-product. The addition of the malonic ester residue to the CºC bond was completed by intramolecular nucleophilic substitution of the chlorine atom (2.8.3, 2.8.6 → 2.8.31). A similar process with 1,3-dimethylbarbituric acid gave the spirocyclic product 2.8.32, which was then subjected to recyclization under the action of ButOK with the elimination of the methyl isocyanate molecule (2.8.32 → 2.8.33).

Besides heterofused pteridines, o-dialkynylazines 2.8.4, 2.8.7, 2.8.8, containing a cis-3-hexene-1,5-diyne fragment, are of interest. In the 1980s, a family of enediyne antibiotics was discovered[635-637]. The mechanism of their biological action is based on a rather exotic transformation called Bergmann cyclization. When heated or irradiated, cis-3-hexene-1,5-diynes form 1,4-arynes, which in the presence of suitable hydrogen atom donors or other free radicals give the corresponding arenes. At the biological level the DNA molecule acts as a donor of hydrogen atoms, which leads to its destruction. This determines the antibacterial, antiviral and anticancer activity of enediyne antibiotics. At the beginning of our studies, some cyclizations of acyclic and aromatic enediynes were also known to occur under the action of various reagents. The corresponding data was summarized in reviews[638, 639]. Investigating the reactivity of o-dialkynylazines 2.8.4, 2.8.7, 2.8.8 and other enediynes, we discovered a number of previously unknown transformations. As already noted, the triple bonds in 6,7-dialkynyllumazines are not equivalent; in all cases, the CºC bond at position 7 is more sensitive to nucleophiles. Thus, the action of amines on compound 2.8.4a led to enamines 2.8.34, which underwent 6-exo-dig-cyclization in the presence of bases to give purple anhydrobases 2.8.35, derivatives of pyrido[3,4-g]pteridine (Scheme 246)[623, 625]. Hydrolysis of enamines 2.8.34 and further base-catalyzed cyclization of intermediate 2.8.36 made it possible to obtain the pyrano[3,4-g]pteridine derivative 2.8.37, whose heterosystem is isomeric to molybdopterin.

3-Alkynylquinoxaline-2-carbonitriles 2.8.11 showed similar reactivity (Scheme 247)[628]. On heating with alkylamines in the presence of potash in dimethylformamide, compounds 2.8.11 were converted (probably through intermediates 2.8.38) into orange cyclic amidines 2.8.39, which have fluorescent properties. Molecule 2.8.39 with an unsubstituted heteroatom in the pyridine fragment existed as tautomer 2.8.40.

The interaction of dialkynylazines 2.8.4, 2.8.7, 2.8.8 with carbanions of cyanoacetic and malonic esters began with the addition of a nucleophile to the CºC bond. The subsequent intramolecular acylation reaction involving the ester group and a cyclic heteroatom led to the annulation of two pyridine rings to the initial heterosystem and the formation of polynuclear molecules 2.8.41 (Scheme 248)[627]. When 1,3-dimethylbarbituric acid was used as a pronucleophile, a cascade process developed, including nucleophilic addition to the triple bond, 5-endo-dig-cyclization and recyclization of the barbituric acid residue with elimination of the methyl isocyanate molecule and the formation of spiro compounds 2.8.42 (Scheme 249).


Reactions of nitrile analogues of enediynes 2.8.11 with CH-acids (CH2XY) in a basic medium proceeded in different ways (Scheme 250), giving in different proportions (depending on the nature of the pronucleophile) the products of a cascade of reactions involving nucleophilic addition to the CºC-bond/6-exo-dig-cyclization with CºN-bond/HY elimination (main product 2.8.43), sequential reactions of nucleophilic addition and 6-exo-dig-cyclization (compound 2.8.44), tandem AN/SNipso-transformation (compound 2.8.45) and the process of nucleophilic addition with intramolecular acylation (compound 2.8.46)[640]. The reaction with 1,3-dimethylbarbituric acid, as in the previous cases, was accompanied by recyclization with the formation of pyrimido[4,5-a]phenazine 2.8.47.

The reaction of the ortho-dialkynyl derivatives of quinoxaline 2.8.7 and 1,3-dimethyllumazine 2.8.4 with NaN3 occurred at room temperature and led to the annulation of the triazolopyridine fragment to the starting heterosystem (Scheme 251)[624, 641]. Ortho-Dialkynyl derivatives of benzene 2.8.9, 1-methylimidazole 2.8.10, and acyclic enediynes 2.8.48 underwent a similar transformation, but at higher temperatures. The reaction appears to be a 1,3-dipolar cycloaddition of the azide ion to the CºC bond, followed by nucleophilic 6-endo-dig-cyclization of the intermediate 2.8.49 to the product 2.8.50. In the case of substrate 2.8.4, isomeric products 2.8.51 and 2.8.52 were obtained. Our studies refuted previously published data by Taiwanese chemists[642] that the interaction of acyclic enediynes 2.8.53 with sodium azide in DMF leads to the formation of benzotriazoles, isomeric to compounds 2.8.54.

Mono- and dialkynylazines also entered into 1,3-dipolar cycloaddition reactions with other types of dipoles, for example, azomethine ylides generated in situ from pyridinium and isoquinolinium salts (Scheme 252)[643]. The transformation products were indolizine derivatives or pyrrolo[2,1-a]isoquinoline 2.8.55. When o-dialkynylhetarenes were used as dipolarophiles, both triple bonds took part in the cycloaddition to give the products 2.8.56 together with compounds 2.8.55. It should be noted that indolizine is the structural basis of many natural alkaloids and bioactive molecules[644, 645]. The derivatives 2.8.55 and 2.8.56 apparently contain other pharmacophore fragments (pteridine and uracil). 1,3-Dipolar cycloaddition reactions involving alkynes and azomethine ylides are summarized in the review[646].

The reaction of alkynylquinoxalines 2.8.7, 2.8.11 with nitrile ylide generated from ethyl isocyanoacetate allowed to obtain pyrrole derivatives 2.8.57 (Scheme 253)[647]. Only ethynyl azines reacted with azomethine imines 2.8.58 under catalytic conditions. The products were 7-hetaryl-2,3-dihydro-1H,7H-pyrazolo[1,2-a]pyrazol-1-ones 2.8.59[647]. Pyrazolone is the structural basis of many widely used analgesics[648]. Tetrahydropyrazolo[1,2-a]pyrazolones have been studied as analogues of penicillin and cephalosporin antibiotics, as well as herbicides and pesticides (see references in article[647]) since the late 1980s.

Despite the π-deficient nature of the azine ring, alkynylazines are also quite reactive towards electrophiles. Under the action of halogens, interhalides and N-halosuccinimides, 2,3-dialkynylquinoxalines 2.8.7 underwent a tandem process of electrophilic addition to the CºC bond/5-exo-dig-cyclization, forming cyclopentaquinoxalines 2.8.60 or 2.8.61 (Scheme 254)[649].

The synthetic method for benzophenazines 2.8.63 proposed by us is based on the iodocyclization of 2-alkynyl-3-arylquinoxalines 2.8.63 (Scheme 255)[630]. In the case of alkyne 2.8.62 bearing a 4-methoxyphenyl substituent at the CºC bond, the cyclization leads to the formation of the spirocyclic product 2.8.64, apparently due to the electron-donating effect of the methoxy group. Phenazines are a broad class of natural and synthetic molecules with potent antimicrobial, anti-inflammatory and anticancer activity[650].

Electrophilic cyclizations of o-alkynylbiaryls 2.8.65 were key steps in the synthesis of little-studied [n]helicenes containing a heterocycle or annulated with it (Scheme 256)[631, 651-653]. Aza- and diaza[4]helicenes 2.8.66A (see[631]) and 2.8.66B (see[653]) were prepared in three steps from the 2,3-dihalogen derivatives of quinoxaline, pyrazine and pyridine by sequential Sonogashira coupling, Suzuki arylation and iodocyclization (or acid-catalyzed cycloisomerization) of 2-alkynyl-3-arylazines 2.8.65. Iodine derivatives 2.8.66 (E = I) were further subjected to Sonogashira coupling, and cycloaromatization of compounds 2.8.67 in an acidic medium made it possible to obtain double [4]helicenes 2.8.68A (see[631]) and 2.8.68B[653], as well as azine-fused [5]- and [6]helicenes 2.8.68C (see[651]) and 2.8.68D[652].

According to X-ray diffraction data, the presence of an aza group in the ‘bay region’ of [4]helicene leads to a significant flattening of the helix due to the formation of an intramolecular C – H∙∙∙N hydrogen bond (Scheme 257)[631, 653]. Heteroannelation also affects the optical properties of helicenes: the absorption and fluorescence maxima increase and the band gap decreases[651, 652]. Pyrene-containing aza- and diaza[4]helicenes 2.8.66B and 2.8.68B exhibit the highest fluorescence quantum yields, from 0.11 to 0.71[653]. In this case, their emission properties can be modulated by the pH changing. Scheme 257 shows, as an example, the values of interplanar angles between terminal rings A and D, reflecting the tendency of aza[4]helicenes to flatten, as well as the maxima of the absorption and emission bands of [5]helicenes fused with an azine core. Methods for the synthesis of [n]helicenes based on the use of acetylene derivatives as starting materials are presented in the review[654].

2.8.2. Syntheses based on 1,3-dialkyl-6,7-dialkynyl-1H-perimidin-2(3H)-ones
The synthesis of mono- and polyalkynyl derivatives of 1,3-dialkyl-1H-perimidin-2(3H)-ones 2.8.69 – 2.8.73 from the corresponding bromides was carried out in the Pd2dba3 – PPh3 – CuI – K2CO3–DMF system[655].

It has been established that 6,7-dialkynyl- (2.8.70) and 4,6,7,9-tetraalkynylperimidones (2.8.73), when heated in diglyme, undergo thermal radical cyclization with the formation of benzo[5,6]indeno[1,2,3-gh]perimidin-2-ones 2.8.74 (Scheme 258). Compounds 2.8.70 and 2.8.73 do not enter electrophilic cyclization: the action of bromine in CHCl3 leads to the addition of an electrophile to triple bonds. The review[656] deals with the chemistry of perimidines.

2.8.3. Syntheses based on alkynyl derivatives of 1,8-bis(dimethylamino)naphthalene
By coupling 1-alkynes with the 2-iodo derivative of the ‘proton sponge’ 2.8.75, 2-alkynyl-1,8-bis(dimethylamino)naphthalenes 2.8.76 were obtained. Isomeric iodide 2.8.77 was converted into 4-alkynyl derivatives 2.8.78, and 2,7-dialkynyl-1,8-bis(dimethylamino)naphthalenes 2.8.80 were synthesized from diiodide 2.8.79 (Scheme 259)[657-659].

Ethynyl derivatives 2.8.76d and 2.8.78d were used in the synthesis of 1,4-diaryl-1,3-butadiynes 2.8.81 and 2.8.82[660, 661] and a series of oligo(arylene-ethynylenes) based on the ‘proton sponge’[662, 663]. Oligo(arylene-ethynylenes) are a promising class of organic molecules with an extended conjugated π-system, that have been used to create materials for LEDs and transistors, solar cells, liquid crystal displays, chemosensors, photosensitizers for photodynamic therapy, etc[664, 665].

A change in the method of isolation of the products 2.8.76 and 2.8.80, namely replacing the extraction of the reaction mixture by evaporation in a porcelain dish, led to heterocyclizations involving a C≡C bond and a 1-NMe2 group (Scheme 260)[658, 659]. Alkynes 2.8.76a,b isomerized to benzo[g]indoles 2.8.83a,b with [1,3]-migration of the NMe group into the pyrrole ring, while under similar conditions alkyne 2.8.76c was transformed into benzo[g]indole 2.8.84 with loss of N – Me group. In the case of the dialkynyl derivative 2.8.80a, in addition to the isomerization product 2.8.85 with a migrated methyl group, the product of pyrrole ring closure and subsequent alkynylation at position 3, namely compound 2.8.86, was isolated from the reaction mixture. These processes were apparently caused by steric stress in ortho-substituted 1,8-bis(dimethylamino)naphthalenes. This was confirmed by the fact that heating 2-(phenylethynyl)-1,8-bis(dimethylamino)naphthalene (2.8.76a) with palladium and/or copper salts led to the formation of a mixture of benzo[g]indoles 2.8.83a and 2.8.87 – 2.8.89 (see Scheme 260)[658].

The proposed mechanisms for the formation of heterocyclization products are shown in Scheme 261[658]. In most cases, the reactions proceed in parallel, but under certain conditions one of these products becomes predominant or even the only one. All transformations, with the exception of 2.8.76a → 2.8.89, start with a nucleophilic attack of the dimethylamino group on the adjacent Pd2+-coordinated CºC bond (intermediate A), followed by 5-endo-dig-cyclization into the cationic intermediate B. The latter either undergoes 1,3-migration of the N-methyl group with the formation of intermediate C and then benzo[g]indoles 2.8.83, 2.8.85, or is demethylated under the action of iodide ion into product 2.8.84. Some evidence has been obtained for porcelain catalysis[658] of the 2.8.76 → 2.8.83 rearrangement. The intermediate palladium complex D is able to dimerize into compound 2.8.88 and undergo a transmetallation reaction with (phenylethynyl)copper to give product 2.8.86.

The formation of 3-aroylbenzo[g]indole 2.8.89 is the result of a Cu-catalyzed oxidative transformation (Scheme 262). Initially, oxidation/deprotonation of the NMe2 group produces iminium intermediate A, which then undergoes 5-exo-dig-cyclization, leading to closure of the pyrrole ring and the formation of cation B. The process is completed by its deprotonation and the reaction of the resulting carbene C with atmospheric oxygen.

The coupling of 2-ethynyl- (2.8.76d) and 2,7-diethynyl-1,8-bis(dimethylamino)naphthalenes (2.8.80d) with 1,8-diiodonaphthalene leads to N,N,7-trimethyl-7H-acenaphtho[1,2-b]benzo[g]indol-8-amines 2.8.91 and 2.8.92, respectively (Scheme 263)[666], in the case of the compound 2.8.76d, N,N,1,3-tetramethyl-1H-benzo[g]indol-9-amine (2.8.90). The proposed reaction mechanism involves the oxidative addition of Pd0 to the C – I bond in the primary product A of this transformation. The CºC bond of the aryl palladium complex B is activated to nucleophilic attack by the NMe2 group, which leads to closure of the pyrrole ring and the formation of palladacycle C. Reductive elimination of Pd0 and N-demethylation complete the process.

On heating with CuI in aniline, 1,4-diarylbuta-1,3-diyne 2.8.81 forms 2,2'-bibenzo[g]indole 2.8.93 in 57% yield (Scheme 264)[660]. The isomeric diyne 2.8.82 cyclizes to 2,5-diarylpyrrole 2.8.94 under the same conditions.

Thus, alkynylazines exhibit high reactivity towards nucleophiles, electrophiles and 1,3-dipoles. Transformations are usually of a tandem or cascade nature and lead to polynuclear heterocyclic molecules, including close structural analogues of natural and biologically significant pteridines, phenazines and indolizines. The synthetic potential of these reactions is far from exhausted, considering that in addition to alkynyl groups, ortho-located substituents, ring heteroatoms and side substituents at CºC bonds also participate in the transformations. The resource of the general methodology proposed by us for the synthesis of hetero[n]helicenes, promising candidates for the creation of new optical materials, has not been exhausted. Alkynyl derivatives of 1,8-bis(dimethylamino)naphthalene are accessible and convenient monomers in the synthesis of new types of oligo(arylene-ethynylenes), combining the properties of compounds with an extended π-system and strong polyfunctional neutral organic bases. The synthesis of donor-acceptor oligo(arylene-ethynylenes), containing alternating units of a ‘proton sponge’ and π-deficient heterocycles, candidates for the creation of organic conducting materials, also seems promising.
2.9. Reactions of nitrogen-containing heterocycles with electron-deficient acetylenes: recent advances
This chapter summarizes and briefly analyzes the results of recent research into the reactions of heterocycles containing C=N bonds (pyrrolines, 3H-pyrroles, pyridines, quinolines, imidazoles, benzimidazoles, etc.) with electron-deficient acetylenes (acyl and cyanopropargyl alcohols, aryl- and hetaryl(pyrrolyl)acylacetylenes, esters of acetylenic acids, etc.). Such reactions proceed via the reversible formation of 1,3(4)-dipole intermediates, which are further involved in the annelation and functionalization of heterocycles, as well as in the opening or expansion of the heterocyclic core. In some cases, an unprecedented nucleophilic substitution of hydrogen in the heterocyclic fragment is observed. The synthetic potential of such reactions is far from being exhausted, and they are attracting increasing attention of researchers due to a number of advantages. As a rule these reactions are non-catalytic, atom-efficient, energy- and resource-saving, proceed in one pot manner under mild conditions thus meeting the principles of modern ‘green’ chemistry.
2.9.1. Δ1-Pyrrolines
Δ1-Pyrrolines 2.9.1 are easily annelated under mild conditions (no catalysts) by electron-deficient propargyl alcohols 2.9.2 containing benzoyl, methoxycarbonyl, amide or cyano groups to afford functionalized hexahydropyrrolo[2,1-b]oxazoles 2.9.3 (Scheme 265)[667]. In most cases, the synthesis is stereoselective with respect to the functionalized ethenyl group located in the fused heterocyclic system.

The reaction begins with the reversible formation of a 1,3(4)-dipole by nucleophilic attack of a lone electron pair of Δ1-pyrroline 2.9.1 nitrogen at the electron-deficient triple bond of propargyl alcohol 2.9.2. Proton transfer from the hydroxyl group to the carbanionic center (at the α-position to the electron-withdrawing group) then provides a more thermodynamically stable dipole containing an oxygen-centered anionic center, which undergoes cyclization to the final product 2.9.3 (Scheme 266). This methodology provides a one-pot route to a new pharmaceutically promising family of annelated nitrogen-containing heterocycles.

The chemistry of electron-deficient pyrrolylacetylenes 2.9.4 is currently under intensive development. These compounds are readily prepared by the cross-coupling of pyrroles (indoles, tetrahydroindoles, benzo[g]indoles) with acylhaloacetylenes in the presence of solid metal oxides or salts[140]. This family of electron-deficient acetylenes is successfully involved in [3 + 2]-cycloaddition to Δ1-pyrrolines 2.9.1 (20 – 25 °С, MeCN or MeOH, 24 – 72 h). Eventually, a facile and convenient approach to the synthesis of dipyrrolo[1,2-a:1´,2´-c]imidazoles 2.9.5[668], pyrrolo[1',2':2,3]imidazo[1,5-a]indoles 2.9.6, cyclohepta[4,5]pyrrolo[1,2-c]pyrrolo[1,2-a]imidazoles 2.9.7 and benzo[g]pyrrolo[2',1':2,3]imidazo[1,5-a]indoles 2.9.8 (Scheme 267) has been developed[669].

The building of the pyrroloimidazole system onto the pyrroline ring proceeds through elementary stages similar to those shown in Scheme 266.
Photophysical studies confirm that some compounds such as benzo[g]pyrroloimidazoindoles 2.9.8 are promising building blocks for the design of thermally activated delayed fluorescence (TADF) emitters for organic light-emitting diodes (OLEDs).
An important advantage of the process is that the aqueous medium does not interfere with the annelation of Δ1-pyrrolines 2.9.1 with electron-deficient pyrrolylacetylenes 2.9.4: the yield of dipyrroloimidazole 2.9.5, synthesized from 2-methylpyrroline and benzoylpyrrolylacetylene in a MeCN – H2O mixture, is 85%. At the same time, another cascade process starts with cyanoacetylenes 2.9.9 and acylacetylenes 2.9.10: the ring opening occurs in Δ1-pyrrolines 2.9.1 to furnish ketopropylaminoacrylonitriles 2.9.11 and ketopropylaminoenones 2.9.12, mainly of the Z-configuration (Scheme 268)[670].

The vinyl carbanionic center of 1,3(4)-dipole is neutralized by a proton of water molecule to give a hemiaminal. In the latter, the opening of the C(2) – N takes place thus completing the formation of functionalized enamines 2.9.11 and 2.9.12 (Scheme 269).

2.9.2. 3Н-Pyrroles and 3Н-indoles
Dialkyl-3H-pyrroles[671, 672] 2.9.13 and 3H-indoles[673] 2.9.14, when reacted with electron-deficient propargyl alcohols 2.9.2, undergo [2+3]-annelation to regioselectively produce 2-dihydropyrrolo[2,1-b]oxazoles 2.9.15 and dihydrooxazolo[3,2-a]indoles 2.9.16, respectively (Scheme 270).

The nature of the electron-deficient group affects stereoselectivity of the process. In the case of cyanopropargyl alcohols, the product is formed exclusively as the Z-isomer, whereas the benzoyl and methoxycarbonyl derivatives give only adducts of the E-configuration.
2.9.3. Pyridines
The three-component reaction between pyridines 2.9.17, alkyl esters of acetylene carboxylic acids 2.9.18 and carbonyl compounds 2.9.19 (ethylpyruvate[674], benzofuran-2,3-diones[675], α-haloketones[676], isatines[677], benzoyl cyanide[678], tryptantrines[679]) leads to functionalized 1,3-oxazinopyridines 2.9.20 (Scheme 271).

This cascade assembly is also triggered by 1,3(4)-dipole intermediates, which are reversibly formed via the nucleophilic addition of a nitrogen atom to the triple bond. The carbanionic counterpart of the primary intermediates is selectively attacked by the carbonyl group of the third component to deliver the secondary dipole intermediate with an oxygen-centered anionic site. The latter intramolecularly attacks the positively charged α-position of the pyridine ring, thus completing the formation of the corresponding 1,3-oxazine 2.9.20 (Scheme 272).

The assembly is of general character and well tolerates quinolines[674, 678, 680],674, 678, 680 isoquinolines[679, 681], phenanthridines[682], phenanthrolines[683], imidazoles[674, 678, 684], and benzimidazoles[685].
The reaction of dimethylacetylenedicarboxylate (DMAD, 2.9.18a), 11H-inden[1,2-b]quinaxolin-11-one and pyridine (2.9.17a), quinoline 2.9.21 or isoquinoline 2.9.22 affords N-spirofuranone-2(1)-methoxyazines 2.9.23[686]. The change in the process direction is rationalized by the intramolecular addition of the oxygen anionic site of the secondary dipole intermediate to the methoxycarbonyl group with the elimination of the methoxide anion, which is then added to the carbocationic center of the azine ring (Scheme 273).

The reaction of pyridines 2.9.17 with trifluoroacetylaryl acetylenes[591] 2.9.24 or arylethoxyoxalylacetylenes[687] 2.9.25 proceeds under mild conditions as a one-pot annelation of this heterocycle with two acetylene molecules to yield diastereoselectively arylethynyl-1,3-oxazinopyridines 2.9.26 (Scheme 274).

The reaction of pyridine 2.9.17 with trifluoroacetylphenylacetylene 2.9.24 (or acylarylacetylenes 2.9.10) in water is different: an unexpectedly easy opening of the aromatic pyridine system under the action of electron-deficient acetylenes and water gives 5-[(Z)-acylethenyl]amino-2,4-pentadienals 2.9.27 (Scheme 275)[590, 688, 689]. The mechanism of the pyridine ring opening is similar to that shown in Scheme 269.

Three-component reactions between pyridines 2.9.17 (quinoline 2.9.21, isoquinoline 2.9.22 or phenanthridine 2.9.28), dialkyl esters of acetylenedicarboxylic acids 2.9.18 and malononitrile derivatives[690, 691] 2.9.29 or β-trifluoroacetyl vinyl ethers[692] 2.9.30 give the functionalized (dihydro)pyridoazines 2.9.31 and 2.9.32 (Scheme 276).

In this reaction, 1,3(4)-dipole intermediates of azines and acetylenes are intercepted by electron-deficient alkenes.
The SNH-cross-coupling of terminal acylacetylenes 2.9.10 with pyridines 2.9.17 and secondary phosphine chalcogenides 2.9.33 is carried out under mild, catalyst-free conditions (20 – 75 °C) to give 4-chalcogenophosphorylpyridines 2.9.34[693, 694]. The reaction proceeds through 2,4-migration of the chalcogen phosphoryl group in the intermediate 1-acylvinyl-2-phosphoryldihydropyridines 2.9.35 with simultaneous redox elimination of vinyl ketones and their oligomers (Scheme 277).

At 70 – 75 °C, 4-chalcogenophosphorylpyridines 2.9.36 are selectively obtained. In this original SNHAr-reaction, acylacetylenes 2.9.10 not only activate pyridine 2.9.17, but also act as oxidants, stereoselectively reducing to the corresponding olefins of the E-configuration[695, 696].
This new type of SNHAr-reaction includes the following steps (Scheme 278):
— repolarization (umpolung) of the pyridine ring 2.9.17,
— deprotonation of secondary phosphine chalcogenide 2.9.33 to form a phosphorus-centered anion and
— oxidation of the dihydrogenated intermediate 2.9.34.

Terminal[697-700] and internal[701] alkyl propiolates 2.9.18, phenylcyanoacetylene[702] 2.9.9 and bis(fluoroalkyl)phosphonates[703-705] are also involved in the three-component reaction with pyridines 2.9.17.
2.9.4. Quinolines, isoquinolines, phenanthridines and acridines
The non-catalytic solvent-free reaction of quinolines 2.9.21 with two molecules of aryltrifluoroacetylacetylenes 2.9.24 or aryloxalylacetylenes 2.9.25 leads to 3-arylethynyl-1,3-oxazinoquinolines 2.9.37 (Scheme 279)[592, 706].

The reaction mechanism is similar to that shown in Scheme 272. This one-pot protocol provides easy access to a wide range of functionalized oxazinoquinolines. The synthesis is extended to isoquinoline 2.9.22, 1,5- and 1,8-naphthyridines, and phenanthridine 2.9.28.
The reaction of quinolines 2.9.21 with oxalylarylacetylenes 2.9.25 is water tolerant. At the same time, trifluoroacetylarylacetylenes 2.9.24 trigger three-component reaction leading to the stereoselective assembly of trifluoromethylated hydroxy-1,3-oxazinoquinolines 2.9.38 (Scheme 280)[65, 588].

In this case, 1,3(4)-dipole adducts of quinolines 2.9.21 with trifluoroacetylacetylenes 2.9.24 undergo subsequent intramolecular cyclization in the presence of water (Scheme 281).

The process is diastereoselective: the cyclic products have the 3R*,4aR*-configuration. Isoquinoline 2.9.22 and 1,8-naphthyridine are also involved into this transformation. It should be noted that in an aqueous medium at room temperature the reaction is 20 times faster and gives higher yields of trifluoromethylated hydroxy-1,3-oxazinoquinolines[589] due to micellar autocatalysis.
Phenantridine 2.9.28 readily reacts (room temperature, no solvent) with oxalylacetylenes 2.9.25 and water to diastereoselectively afford (R*,R*)-2-hydroxy-4-aryl-2H,13bH-[1,3]oxazino[3,2-f ]-phenanthridines 2.9.39[707]. Acridine 2.9.40 easily undergoes simultaneous N(1)- and С(9)-functionalization under the action of the same system (aryloxalylacetylene 2.9.25 + water) to give N-alkenylacridin-9-ones 2.9.41 (Scheme 282)[708].

Quinolines 2.9.21 react with acylacetylenes 2.9.10 or phenylcyanoacetylene 2.9.9 and water in the presence of KOH to give 3-acyl- (2.9.42) or 3-cyano-2-aryl-quinolines (2.9.43) (Scheme 283)[709, 710]. This unexpected reaction looks like a formal substitution of the ethene moiety in the quinoline ring with an acyl(cyano)arylethenyl one.

It has been shown experimentally that the ring opening occurs first, accompanied by rearrangement and introduction of the electron-deficient acetylene fragment to form a dihydroquinoline intermediate. The latter contains an aldehyde function at position 4 and is further aromatized with the elimination of acetaldehyde (Scheme 284).

This unique reaction provides a novel short-cut to the functionalized quinolines.
The treatment of a mixture of quinolines 2.9.21, aryltrifluoroacetylacetylenes 2.9.24 and water with organic bases (N-methylpiperidine or morpholine) also leads to 2,3-difunctionalization products, 2-aryl-3-trifluoroacetylquinolines 2.9.44, while inorganic bases (NaOH, ButOK) catalyze the formation of 2-arylquinolines 2.9.45 (Scheme 285)[593].

2,3-Difunctionalization of quinolines 2.9.21 under the action of electron-deficient acetylenes has been further developed using acetals of cyanopropargyl alcohols 2.9.9. Finally, this reaction allows the synthesis of the corresponding 2-(1-ethoxyalkoxy)-3-cyanoquinolines 2.9.46, which upon hydrolysis (7% aqueous HCl, acetone, 20 – 25 °C) quantitatively undergo cyclization via the transformation of furo[3,4-b]iminoquinolines 2.9.47 to furo[3,4-b]quinolinones 2.9.48 (Scheme 286)[711].

Furo[3,4-b]quinolinones 2.9.48 can also be synthesized in a one-pot fashion from quinolines 2.9.21, acetals of cyanopropargyl alcohol 2.9.9 and water.
It has been shown for the first time that arylcyanoacetylenes 2.9.9 trigger the cascade assembly of dihydropyrimido[1,2-a]quinolin-3-ones 2.9.49 from quinolines 2.9.21 in the presence of KOH in an aqueous medium at room temperature (Scheme 287)[712].

The reaction proceeds in a two-phase aqueous-organic system, where hydroxide anions act both as catalysts and reactants. Here, the hydroxide anion attacks the cyanogroup in the N-cyanoethenylquinolinium cation, generating a dipole with a positive center on C(2) atom of the heterocycle, and with an anionic center on the imine nitrogen atom. The interaction of these two centers gives product 2.9.49 (Scheme 288).

This annelation method is applied to isoquinoline 2.9.22 and phenanthridine 2.9.28[713]. The reaction under question is inherently an incomplete SNHAr-process, which is terminated using the additional oxidation step (see Scheme 287).
Annelation of the 1,4-oxazepanone ring to quinoline 2.9.21 or isoquinoline 2.9.22 takes place under the action of dialkyl esters of acetylenedicarboxylic acids 2.9.18 and aromatic alcohol, functionalized hydroxybenzofuran 2.9.50 (Scheme 289) to produce polyheterocyclic systems 2.9.51[714].

When the triple C≡C bond of the second molecule of electron-deficient acetylene 2.9.18 acts as electrophile-interceptor of 1,3(4)-dipole formed from isoquinoline 2.9.22 or phenanthridine 2.9.28 and dialkyl ester of acetylenedicarboxylic acid 2.9.18, the functionalized pyridoazines 2.9.52 are produced[681, 692] (Scheme 290).

2-Methylquinoline 2.9.21b reacts with acylacetylenes 2.9.10 to give 2-{5'-(het)aryl[1,1':3',1'']terphenyl-4'-yl}quinolines 2.9.53[715]. In this case, intermediate 1,3(4)-dipoles are sequentially vinylized by two molecules of acetylene 2.9.10 with the elimination of (hetero)aromatic acid (Scheme 291).

The reaction of 2-methylquinoline 2.9.21b with phenylcyanoacetylene 2.9.9a in the presence of water (20 mol.% KOH, 0 – 25 °С) gives functionalization products on both the nitrogen atom and the methyl group (2.9.54 and 2.9.55, respectively) (Scheme 292)[716]. The process is stereoselective: the N-1-phenyl-2-cyanoethenyl substituent has the Z-configuration, and the 1,3-diene fragment at the methyl group has the E,E-configuration.

The products of stereoselective (Z)-N(1)- and (Z)-N(4)-ethenylation 2.9.56 are synthesized by the reaction of 4-aminoquinoline 2.9.21с with acylacetylenes 2.9.10 in the KOH – H2O – MeCN system (Scheme 293)[717].

1,3-Dicarbonyl compounds 2.9.57 intercept 1,3-dipole intermediates in three-component reactions with quinoline 2.9.21 or isoquinoline 2.9.22 and dialkyl esters of acetylenedicarboxylic acids 2.9.18 to give new stable 1,4-dipoles[718, 719] 2.9.58 or the products of their further transformations 2.9.59 (Scheme 294)[720]. The reaction tolerates both pyridines and 1-substituted imidazoles.

The introduction of a chlorine atom into the methylene fragment of 1,3-dicarbonyl compounds 2.9.57 changes the direction of the three-component reaction with quinoline 2.9.21a and alkyl esters of acetylenedicarboxylic acids 2.9.18 to produce pyrroloquinolines 2.9.60 (Scheme 295)[721].

The three-component reaction of isoquinoline 2.9.22, benzoylphenylacetylene 2.9.10a and nitromethane affords 2,4-diphenylpyrido[1,2-a]isoquinolinium nitrite 2.9.61 (Scheme 296)[722].

The reaction proceeds as a cascade process via intramolecular cyclization involving the CH2NO2 fragment and the carbonyl group. Further, elimination of the nitrite- anion and hydride shift occur.
In the presence of tetra-n-butylammonium fluoride (TBAF), three-component reaction of quinolines 2.9.21 or isoquinolines 2.9.22, alkyl esters of acetylenecarboxylic acids 2.9.18 and calcium carbide affords C(2)- or C(1)-ethynyl-N-alkenylquinolines or -isoquinolines 2.9.62 (Scheme 297)[723].

Such NH-reagents as amides[724] 2.9.63 or NH-azoles 2.9.64 (pyrrole, indole, imidazole)[725], in three-component reactions with quinolines 2.9.21 (isoquinolines 2.9.22, phenanthridines 2.9.28) and alkyl esters of acetylenecarboxylic acids 2.9.18 give the corresponding N-alkenyldihydroazines 2.9.65 or 2.9.66, functionalized at position 2, 1 or 6 by the carbamide fragment or heterocycle (Scheme 298).

Three-component reactions of quinolines 2.9.21 and isoquinolines 2.9.22 with electron-deficient acetylenes (acylacetylenes 2.9.10, alkylpropiolates 2.9.18 and phenylcyanoacetylene 2.9.9) and secondary phosphine chalcogenides[702, 726, 727] or bis(polyfluoroalkyl)phosphonates[728, 729] 2.9.33 yield the products of N-ethenylation and C-phosphorylation 2.9.66' or 2.9.66'' (Scheme 299).

2.9.5. Bicyclic amidines
Typical bicyclic amidines 2.9.67, the known organic superbases such as 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), are annelated with electron-deficient propargyl alcohols 2.9.2 containing cyano-, benzoyl or methoxycarbonyl groups to afford the functionalized fused heterocyclic systems, [1,3]oxazolo[3,2-a]pyrrolo[2,1-b]hexahydropyrimidines and [1,3]oxazolo[3',2':3,4]hexahydropyrimido[1,2-a]azepines 2.9.68 (Scheme 300)[730, 731].

Amidines DBN and DBU 2.9.67 react with methyl esters of acetylenic acids 2.9.18 in water to produce acetylenic amides 2.9.69 with pyrrolidone or caprolactam fragments (Scheme 301)[732].

The process involves the formation of amines by addition of water to the double bond followed by rearrangement of the adduct with opening of the six-membered ring. The formed amine is acylated with the ester group of acetylenic alcohol (see Scheme 301).
2.9.6. Imidazoles and benzimidazoles
1-Substituted imidazoles 2.9.70 undergo mild (45 – 60 °C, no catalyst, acetonitrile) stereoselective ring opening under the action of electron-deficient acetylenes 2.9.10 and water to give (Z,Z)-propenylaminoethenylformamides 2.9.71 (Scheme 302)[733].

Three-component reactions involving 1-substituted imidazoles 2.9.70, electron-deficient acetylenes 2.9.9, 2.9.10 or 2.9.18 and aldehydes 2.9.72 (20 – 25 °C, no catalyst), afford hitherto unknown vinyl ethers of 2-hydroxyalkylimidazoles 2.9.73 (see[734]) and 2.9.74[735, 736], 3-(2-imidazolyl)-3-aryl-2-acylpropanenitriles[737] and (imidazol-2-yl)methyl-1,3-propanediones[738] 2.9.75 or 1-vinylpyrrolimidazole ensembles 2.9.76[739]. The interaction occurs through the formation of 1,3(4)-dipole and carbene intermediates (Scheme 303).

1-Substituted imidazoles 2.9.70 react with phenylcyanoacetylene 2.9.9 and isocyanates[740, 741] or isothiacyanates[742] 2.9.77 at room temperature in the absence of catalyst and solvent to form (Z)-(2-cyano-1-phenylethenyl)imidazole-2-carboxamides or -carbothiamides 2.9.78 (Scheme 304). This reaction also tolerates 1-substituted benzimidazoles 2.9.79[743].

Due to the high pharmacological importance of carboxamide bearing imidazoles, new members of this series with N-cyanovinyl substituents represent promising precursors for targeted drug design.
Three-component reaction between 1-substituted imidazoles 2.9.70, phenylcyanoacetylene 2.9.9 and elemental sulfur (no solvent, 20 – 25 °C) or selenium (acetonitrile, 82 °C) leads to stereoselective formation of 3-(Z)-phenylcyanethenylimidazole-2-thiones or — selones 2.9.80[744]. The process involves the introduction of carbene intermediates into the S – S or Se – Se bonds of elemental chalcogens (Scheme 305). The dipole intermediate generated from 1-methylimidazole 2.9.70a and DMAD (2.9.18a) is directly intercepted by sulfur to generate imidazolium butenothiolates 2.9.81[745].

This reaction seems to be quite promising for the design of modern drugs.
A one-step regio- and stereoselective synthesis (acetonitrile, room temperature, 3 – 5 days) of (Z)-cyanomethylidene-1,3-oxazolobenzimidazoles 2.9.82 from benzimidazoles 2.9.79 substituted on both the imidazole and benzene ring and cyanopropargyl alcohols 2.9.2 has been developed (Scheme 306)[746].

In the presence of water (MeCN, 20 – 25 °C, 7 days or 45 – 50 °C, 6 h) this reaction leads to the opening of the imidazole fragment and cyclization of the furan fragment to give (2-{[(3E)-5-amino-furan-3(2H)-ylidene]amino}phenyl)formamides 2.9.83 (Scheme 307)[747]. This synthesis comprises multi-position cascade transformations of the intermediate hemiaminals formed from primary dipoles and water. Substituted azabenzimidazoles undergo similar transformations under the action of cyanopropargyl alcohols 2.9.2 in the presence of water[748].

The reaction of substituted benzimidazoles and azabenzimidazoles 2.9.79 with acylacetylenes 2.9.10 and water (acetonitrile, 82 °C, 46 – 120 h) gives both the products of imidazole ring opening, functionalized (Z)-aryl(pyridyl)aminovinyl ketones 2.9.84, and the products of the imidazole ring expansion, benzo(pyrido)diazocinones 2.9.85 (Scheme 308)[749, 750].

With acylpyrrolylacetylenes 2.9.4, the reaction gives only the fused eight-membered nitrogen-containing heterocycles 2.9.85. In this case, the yields of the corresponding pyrrolyl-substituted benzo- and pyridodiazocinones 2.9.85 reach 93% (Scheme 309)[751, 752]. The reaction mechanism, including the formation of 1,4-dipole and ylide intermediates, has been confirmed by quantum chemical calculations using the DFT approach.

This chapter briefly summarizes the recent works (published over the last decade) devoted to the reactions of nitrogen-containing heterocycles having at least one C=N bond with electron-deficient acetylenes. A large number of publications indicate that this research direction is developing dynamically and fruitfully. Fundamentally, such reactions are united by an initial triggering stage: the nucleophilic attack of the nitrogen lone electron pair at the antibonding π-orbital of acetylene affording 1,3(4)-dipole intermediates, which then launch various cascade processes leading (depending on the structure of the heterocycle, acetylene and third component) in one preparative step to a variety of heterocyclic systems. Finally, these reactions lead to the formation of various annulated structures, new functionalized heterocycles with rare highly reactive substituents, products of nucleophilic substitution of hydrogen in the heterocyclic fragment, as well as products of expansion or opening of the original heterocycle with destruction of the aromatic system, which is often accompanied by recyclization processes delivering new heterocyclic assemblies. The competitive advantages of these reactions are their single-stage, atom-efficient, resource- and energy-saving nature, as well as their carbon neutrality. Often such transformations occur without catalysts at room or slightly elevated temperatures. The reactions are implemented with available starting compounds, fundamental heterocycles (pyrrolines, pyridines, quinolines, isoquinolines, phenanthridines, imidazoles, benzimidazoles) and electron-deficient acetylenes (acetylene ketones, esters, nitriles), methods for the synthesis of which are now quite well developed. Various electrophilic compounds (water, PH-acids, electrophilic alkenes, aldehydes, or a second molecule of electron-deficient acetylene) are usually used as third components, easily interacting with the carbaninonic part of the dipole intermediate. The heterocyclic systems formed often contain one or more pharmacophore fragments and represent potential objects of pharmacological research in medicinal chemistry. In the coming years further in-depth development of this chemistry is expected, including in the study of the reactivity and biological activity of synthesized heterocyclic structures.
2.10. Cyclizations of functionalized acetylenes and diacetylenes for the synthesis of heterocycles
In continuation of the works and ideas of Academician Alexey Evgrafovich Favorsky and his scientific school[753], methods for the synthesis of functionalized acetylenes and diacetylenes and convenient synthetic approaches to heterocyclic systems using cyclization reactions of mono- and diacetylenes are being developed at the Saint Petersburg State University under the supervision of I.A.Balova[754]; the results of these studies are addressed in this Section.
The unique synthetic potential of the C≡C triple bond, which is able to undergo electrophilic, nucleophilic, and radical addition and cycloaddition reactions, makes functionalized acetylenes valuable building blocks for the synthesis of diverse heterocyclic structures. Among functionalized acetylenes, a special role belongs to propargylamines, which represent a versatile class of compounds widely used for the synthesis of chemically significant organic products, including a variety of nitrogen heterocycles[755, 756]. We studied the cyclocondensation of N-propargyl-о-phenylenediamine derivatives 2.10.1 with phenyl isothiocyanate, which afforded 1-(prop-2-yn-1-yl)-1,3-dihydro-2H-benzimidazole-2-thiones 2.10.2. In the case of diacetylenic analogues with conjugated triple bonds, which provides for their higher reactivity towards nucleophiles, the initially formed benzimidazolethiones undergo subsequent cyclization to give [1,3]thiazolidino[3,2-а]benzimidazoles 2.10.3 (Scheme 310)[757, 758]. The reactions of both mono- and diacetylene о-phenylenediamine derivatives with carbon disulfide in the presence of KOH give simultaneously two heterocyclic rings; however, the result depends on the nature of substituent at the triple bond. N-(Prop-2-yn-1-yl)-о-phenylenediamines with an aryl substituent and N-(penta-2,4-diyn-1-yl)-о-phenylenediamines are converted to 2-methylidene-2,3-dihydro[1,3]thiazolo[3,2-а]benzimidazoles 2.10.4 and 2.10.3, respectively; under the action of bases, these products readily isomerize to thiazolo[3,2-а]benzimidazoles 2.10.5. The cyclocondensation of N-(alk-2-yn-1-yl)-о-phenylenediamines with CS2 affords [1,3]thiazino[3,2-а]benzimidazoles 2.10.6 (see Scheme 310).

2.10.1. Richter cyclization
The Richter cyclization proceeding during diazotization of o-ethynylanilines affords cinnolinones or 4-halocinnolines, depending on the reaction conditions[759]. This cyclization of o-(buta-1,3-diynyl)arenediazonium salts formed upon diazotization of amines 2.10.7 [760] or upon acid decomposition of triazenes 2.10.8 was first proposed as a method for the synthesis of 4-bromo(chloro)-3-ethynylcinnolines 2.10.9, heterocycles containing an ethynyl substituent and a halogen atom in adjacent positions (Scheme 311)[761].

The replacement of the halogen atom in 3-ethynylcinnolines 2.10.9 by a methylamino group induces spontaneous cyclization giving pyrrolocinnolines 2.10.10 (X = NMe) (Scheme 312). Meanwhile, nucleophilic substitution of the halogen atom by the thiol group affords thienocinnolines 2.10.10 (X = S, Y = H) as a result of nucleophilic addition to the triple bond in position 3[761]. It was shown that thienocinnolines 2.10.10 (X = S, Y = I) can also be obtained from the corresponding 4-methylsulfanyl-3-ethynylcinnolines 2.10.11 by electrophile-promoted cyclization induced by iodine (see Scheme 312)[762].

It was found that the bromine atom in position 4 of the cinnoline core of 2.10.12 can be easily substituted by an azide group and that the resulting azidocinnolines 2.10.13 undergo copper-catalyzed 1,3-dipolar cycloaddition to terminal alkynes and cycloalkynes to give the corresponding 4-(1,2,3-1H-triazolyl)cinnolines 2.10.14 (Scheme 313)[763].

It was assumed that this transformation could yield a fluorogenic probe by analogy with examples described in the literature[764-766]. However, it turned out that 4-triazolylcinnolines 2.10.14 show very weak fluorescence (see Scheme 313; designated by a grey star); meanwhile, the azide group reduction yields cinnoline-4-amines 2.10.15 possessing pronounced fluorescent properties (see Scheme 313, blue star)[767]. Thus, a new type of fluorogenic and fluorochrome probes, based on the reduction of weakly fluorescent 4-azido-6-(4-cyanophenyl)cinnolines 2.10.13 to fluorescent cinnoline-4-amine 2.10.15, was found and developed (see Scheme 313).
It was also shown that the fluorescence of 6-(4-cyanophenyl)cinnoline-4-amine 2.10.15 is considerably affected by the nature of the solvent. The fluorogenic properties of the amine increase in polar solvents, with the most pronounced increase in the fluorescence being observed in water. Environment-sensitive fluorogenic properties of cinnoline-4-amine 2.10.15 in water were attributed to a combination of two types of fluorescence mechanisms: aggregation-induced emission (AIE) and intermolecular excited state proton transfer (ESPT).
The azide – amine pair was tested as a fluorogenic probe in vitro using the HepG2 cancer cell line by means of fluorescence microscopy, flow cytometry and HPLC analysis of cell lysates. The results confirmed the possibility of azide to amine conversion in the cells and the potential applicability of the detected fluorogenic and fluorochrome probe for various analytical and biological purposes[767].
The Richter cyclization involving о-ethynylarenediazonium salts formed from amines 2.10.7 was also utilized to obtain 4-oxo-1,4-dihydrocinnoline derivatives 2.10.16 as selective inhibitors of protein phosphotyrosine phosphatase 1B (PTP1B) (in cooperation with A.O.Shpakov, I.M.Sechenov Institute of Evolutionary Physiology and Biochemistry, Russian Academy of Sciences). It was shown that cinnolinones 2.10.16 can be alkylated with propargyl bromide at the N(1) nitrogen atom and then converted to triazoles 2.10.17 via Cu-catalyzed azide–alkyne cycloaddition (CuAAC)[768]. Among 4-oxo-1,4-dihydrocinnolines 2.10.16, active derivatives were identified by experiments on rats with diet-induced type 2 diabetes. These compounds appear to be promising for the development of drugs for treatment of socially important diseases such as obesity and type 2 diabetes mellitus, as well as other endocrine diseases related to insulin and leptin resistance (Scheme 314)[768-770].

In order to switch the Richter cyclization of o-arylethynylarenediazonium salts from the 6-endo-dig to 5-exo-dig pattern and to obtain 3-aryl-1H-indazoles as combretastatin A analogues, Babushkina et al.[771] studied the effect of the nature of substituents in the arylethynyl moiety and the solvent effect on the reaction regioselectivity.
It was shown that 4-bromo-3-ethynylcinnolines 2.10.9 easily undergo cross-coupling reactions. Thus, in the case of acetylenes (Sonogashira – Hagihara reaction), acyclic enedyine systems fused with cinnoline are formed[771]. This synthetic approach was used to synthesize new poly(arylene-ethynylene)s containing a cinnoline core. The three-step sequence includes the Richter cyclization in the acid-induced degradation of 2-ethynyl- and 2-(buta-1,3-diynyl)aryltriazenes 2.10.18, the subsequent introduction of trimethylsilylethynyl moieties at positions 4 and 6 to form compounds 2.10.19, and finally the sila-Sonogashira type polycondensation (Scheme 315). This gave fluorescent oligo(arylene-ethynylenes) 2.10.20 with irregular structure (head-to-head, tail-to-tail, and head-to-tail units) containing cinnoline moieties. Solutions of these compounds in THF proved to be quite sensitive to fluorescence quenching by Pd2+ ions[772].

2.10.2. Electrophile-promoted cyclization
The term ‘electrophilic cyclization’ of acetylenes was introduced by Yao and Larock[773] to describe the set of intramolecular cyclization reactions involving a C≡C triple bond and a neighbouring functional group performed in the presence of electrophilic reagents. Since the cyclization involves a nucleophilic group, it is more appropriate to call this reaction electrophile-promoted nucleophilic cyclization (EPNC)[774, 775]. The electrophile-promoted cyclization are initiated by electrophiles (most often, reagents containing an electrophilic halogen atom), which first react with the triple bond, thus activating the intramolecular addition of nucleophile, which takes place in the second step. Unlike conventional catalysis by electrophilic species (Pd2+, Ag+, Cu+, Cu2+)[754], in this case, an atom of the electrophilic reagent is retained as a substituent in the resulting heterocyclic molecule. This approach proved to be versatile, tolerating a broad range of functional groups with different relative nucleophilicities, and it was used to obtain several heterocyclic structures[612].
The electrophile-promoted cyclization for functionalized diacetylenes 2.10.21 was proposed as an approach to one-step preparation of heterocycles 2.10.22 containing an ethynyl substituent and a halogen atom. These key moieties are needed for the subsequent synthesis of unsymmetrically substituted enediyne systems 2.10.23 fused to S,N,O-heteroindenes and isocoumarin (Scheme 316)[776-778]. The benefits of this approach include ready availability of the starting substrates for cyclization and the regioselectivity of introducing different substituents at each of the triple bonds to form an unsymmetrically substituted enediyne system.

2.10.3. Copper-catalyzed azide – alkyne cycloaddition
We found that the copper-catalyzed 1,3-dipolar cycloaddition of azides to 1-iododiacetylenes 2.10.24 can be efficiently used to obtain 5-iodo-4-ethynyl-1,2,3-triazoles 2.10.25, which are versatile building blocks that have recently found a variety of applications[779]. Surprisingly, among various conditions described for CuAAC involving iodoalkynes, only the CuI(PPh3)3 — 2,6-lutidine catalytic system without a solvent proved to be applicable to iododiacetylenes, giving rise to 5-iodo-4-ethynyltriazoles in high yields under mild conditions. This reaction is highly tolerant to functional groups and in the case of alkyl azides, it proceeds with high chemo- and regioselectivity, yielding 5-iodo-4-ethynyltriazoles 2.10.25 as the only products (Scheme 317)[780].

However, in the case of aryl azides, the regioselectivity is disrupted and, together with the expected 5-iodotriazoles 2.10.26, the reaction gives unusual 4-iodotriazoles 2.10.27 (Scheme 318). The nature of the substituent in the aryl azide has a crucial influence on the regioselectivity of cycloaddition: the yields of unusual 4-iodotriazoles increase for aryl azides with electron-withdrawing substituents. It was found that the formation of 4-iodotriazoles along with the expected 5-iodotriazoles is a unique feature inherent only in the aryl azide — iododiacetylene pair of reactants. The cause for this unusual outcome of the well-studied reaction was revealed by quantum chemical calculations, which showed that the reaction follows a binuclear mechanism in which different nitrogen atoms of the azide [N(1) or N(3)] are coordinated to copper atoms of the binuclear catalyst in the transition states leading to 5-iodo- or 4-iodotriazoles, respectively[781].

The synthetic potential of 5-iodo-4-ethynyltriazoles was demonstrated by investigating two types of cross-coupling reactions (see Scheme 318). The Sonogashira – Hagihara reaction under the optimized conditions was used to prepare unsymmetrically substituted triazole-fused enediyne systems 2.10.28, which are of great interest as the starting compounds for the synthesis of natural analogues of enediyne antibiotics[780, 782]. In addition, it was found that these compounds possess valuable fluorescent properties, which can be tuned by varying the substituents [electron-withdrawing (EWG) or electron-donating (EDG) groups] in both the starting 1-iodoarylbutadiynes and acetylenes in the last step (Scheme 318, blue and orange stars). This ‘molecular meccano’ provided the synthesis of numerous fluorescent molecules 2.10.28[783]. The transformation of the second type, the Suzuki–Miyaura reaction involving 5-iodo-4-ethynyltriazoles and arylboronic acids, produced a set of 5-aryl-4-ethynyltriazoles 2.10.29, which also exhibit fluorescence and solvatochromic effect [see Scheme 318, in which the wavelengths of excitation (ex) and emission (em) maxima are separated by a slash (λex/em)][784].
A new hyperbranched polymer 2.10.30, containing s-triazine as the central core and imidazolium and triazole moieties, was obtained by CuAAC and used to stabilize palladium nanoparticles. The CuAAC click-reaction involving 2,4,6-tris(propargyloxy)-s-triazine 2.10.31 served as the cross-linking method (Scheme 319). The new heterogeneous catalyst 2.10.30 showed a high catalytic activity both in the cyanation of halides and in the reduction of aromatic nitro compounds under green conditions (in aqueous ethanol)[785].

2.10.4. Synthesis of heteroenediynes
A key line of our research is related to the preparation of heterocyclic analogues of natural enediyne antibiotics, cytostatics with strong DNA-damaging effect, due to the ability of (Z)-3-ene-1,5-diyne moiety incorporated in a strained 9- or 10-membered ring to undergo the Bergman cyclization at a human body temperature[786]. Owing to exceptionally potent antibacterial and cytotoxic properties of natural enediyne antibiotics[787-789], immediately after their discovery, studies dealing with the synthesis and investigation of their analogues and determination of the structure–activity relationships were initiated[790]. Although quite a few naturally occurring 10-membered enediyne systems and their analogues have been subjected to clinical trials, only two calechiamicin-based drugs were approved for pharmaceutical use[791], namely, gemtuzumab ozogamicin (Mylotarg®, Pfizer)[792] and inotuzumab ozogamicin (BESPONSA®, Wyeth Pharmaceuticals)[793], which are meant for the treatment of acute myeloid leukemia [CD33(+) and CD22(+), respectively].
Our studies in this area included the molecular design, synthesis and elucidation of the relationships ‘structure — reactivity in the Bergman cyclization — DNA cleavage activity — cytotoxicity’ in a series of 10-membered heterocycle-fused enediynes. Using the developed approach to the preparation of acyclic enediynes fused to heterocycles with a large set of functional groups, which could be easily varied with high regioselectivity, we tested several cyclization reactions in order to find the optimal method for enediyne ring closure.
The Nozaki – Hiyama – Kishi reaction, that is, NiII/CrII-catalyzed reaction of haloalkenes and alkynes with carbonyl compounds to give allyl and propargyl alcohols, proved to be convenient for converting cinnoline derivative 2.10.32 to cinnoline-fused 10-membered enediyne 2.10.33 (Scheme 320). It is important to note that the Bergman cyclization rate for the 10-membered cinnolino-enediyne 2.10.33 was 4 times as high as the cycloaromatization rate of its benzo analogue, which confirmed the hypothesis about the effect of the conjugated heterocycle on the Bergman cycloaromatization[794].

However, on going to benzothiophenes 2.10.34 and 2.10.35, it was found that only 11-membered ring closure takes place to give products 2.10.36a,b, whereas the attempts to obtain 10-membered enediynes failed (Scheme 321). The results of quantum chemical calculations provided the conclusion that this may be caused by increasing strain in the enediyne macrocycle annulated to a five-membered heterocycle[795].

It was found that the novel approach based on the metathesis of appropriate readily available olefins can be applied only to acyclic enediynes 2.10.37 and 2.10.38, leading to the closure of 12-membered dienediyne systems 2.10.39 and 2.10.40 fused to benzothiophene and indole[777, 796]. For smaller rings, metathesis is a thermodynamically forbidden reaction; however, this problem can be solved by complexation of one of the triple bonds of the starting enediyne with Co2(CO)8. The resulting complex 2.10.41 can be converted to the 11-membered ring 2.10.42 Scheme 322; the calculated Gibbs free energies (kcal mol–1) for the formation of E- and Z-isomers of the 12- and 11-membered rings are also indicated in the Scheme][797].

The use of the Nicholas reaction, a unique cyclization method using Cо2(CO)6 alkyne complexes, was the next stage of the study[798, 799]. According to this reaction, Co-stabilized propargyl carbocations react with various nucleophilic groups. The Nicholas cyclization was widely used for the closure of natural enediyne carbocycles in the period of rapid development of the natural enediyne chemistry by the Magnus’ research group[800]. It is noteworthy that the Nicholas reaction was not actually used in the synthesis of analogues of enediyne antibiotics; also it was not used to prepare enediyne heterocycles. We successfully utilized this cyclization, along with the previously developed approach to the synthesis of acyclic enediyne systems; this made it possible to convert substrates 2.10.43 first to complexes 2.10.44 and 2.10.45 and then to 10-membered oxa-[782, 801], carbo- (Ref. [802]) and azacyclic enediynes 2.10.46[803] fused to various heterocycles (Scheme 323; the yields of products 2.10.45 and 2.10.46 separated by a slash are indicated under the structures).

Thus, the Nicholas reaction can be considered to be a versatile and efficient method for the synthesis of strained enediyne rings, since coordination to cobalt reduces the ring strain by changing the bond angle at the triple bond from 180° in free alkyne to ~140° in Co complexes. An important issue is the applicability of this method for the synthesis of enediyne systems fused to five-membered heterocycles (benzothiophene, triazole), which cannot be obtained by other cyclization methods.
Despite the presence of a large number of publications addressing the structure — reactivity relationships in enediyne molecules and the factors that affect the activity of enediyne molecules in the Bergman cyclization[804-806], there is no general system for evaluation and comparison of the reactivity for structurally different substrates. We were the first to propose a scale for evaluation of the relative stability/activity of enediynes from the activation parameters of formation of the diradical intermediate. The calculated results obtained for a large series of such molecules were verified by experiments[802]. However, experiments with plasmids demonstrated that the DNA-damaging activity for heterocyclic enediynes may differ from the order of increasing activity of these compounds in the Bergman cyclization calculated from the activation parameters[803].
In order to develop the principles for selection of lead structures promising as DNA-damaging agents, the interactions of DNA with a series of 10-membered analogues of natural enediynes fused to various heterocycles and containing an enediyne carbo- or heterocycle were studied systematically using molecular docking techniques and metadynamics[807]. It was found that, irrespective of the nature of the annulated heterocycle, the absence of substituents in the heterocycle and the presence of substituents in the homopropargyl position of the enediyne moiety change the mechanism of compound interaction with DNA towards binding to the DNA minor groove. On the basis of these results, enediynes fused to benzothiophene were chosen as lead compounds for the subsequent modification. It was also shown that azaenediynes containing ligands to the DNA from intercalation minor groove are the most promising objects for the synthesis and experimental study of antitumour properties.
For the preparation of benzothiophene-fused azaenediynes, Nicholas aza-cyclization for compounds 2.10.47 containing three types of nucleophilic groups (amino, amido and arenesulfonamido groups) was studied. Although the secondary amino and amido nucleophilic groups possess a large potential as linkers for the attachment of clickable groups to the enediyne core, both proved to be inapplicable for the closure of 10-membered complexes 2.10.48 and azaenediynes 2.10.49, which limits the applicability of this reaction[808]. The arensulfonamide moiety is the optimal functional group for efficient synthesis of 10-membered azaenediynes via the Nicholas cyclization. In addition, this group can serve as a convenient site for the introduction of additional functional groups, such as a terminal triple bond, for further modification of cyclic enediynes by click chemistry (Scheme 324; the yields of products 2.10.48 and 2.10.49 separated by a slash are indicated under the structures)[808].

Apart from the synthesis of cyclic analogues of natural enediynes, the search for new compounds possessing anticancer activity was carried out among acyclic enediynes fused to heterocycles. Unsymmetrically substituted acyclic enediynes based on benzothiophene (2.10.50) and 1,2,3-1H-triazole (2.10.51) containing propargylamine substituents were synthesized (Scheme 325). The effects of these enediynes on the proliferation of the NCI-H460 lung carcinoma and WI-26VA4 lung fibroblast cells were evaluated in comparison with etoposide, the cytotoxic action of which is related to topoisomerase II inhibition. Unlike triazole derivatives 2.10.51, which did not show any cytotoxic activity, among enediyne benzothiophene derivatives 2.10.50, two compounds with a moderate cytotoxic activity were identified[809].

2.10.5. Heterocycloalkynes
Click reactions are a versatile tool for modifying biomolecules directly in living systems[810, 811]. The strain-promoted azide — alkyne cycloaddition (SPAAC) is a bioorthogonal transformation highly demanded for bioconjugation[812-814]. The preparation of cycloalkyne reagents that would be reactive in SPAAC, but stable to nucleophiles or electrophiles is of particular practical and theoretical interest. Two approaches for increasing the reactivity of cycloalkynes in the 1,3-dipolar cycloaddition are known from the literature[815, 816]: by increasing the ring strain of the cycloalkyne moiety upon annulation of other rings[817] and by stabilization of the transition state of SPAAC using various effects of substituents (first of all, stereoelectronic effects)[818]. We hypothesized[819] that these approaches could be combined to develop a new family of cycloalkyne reagents: heterocycloalkynes fused to heterocycles. In these molecules, concerted action of the steric and stereoelectronic effects on the reactivity and stability balance should be expected. This hypothesis was practically verified by the synthesis of a large group of heterocycloalkynes 2.10.54 with 8- to 10-membered rings fused to benzothiophene and isocoumarin, with the synthesis proceeding through the formation of intermediates 2.10.52 and 2.10.53 (Scheme 326).

It was found that particularly cyclononynes have the optimal balance between the stability and activity in SPAAC compared to their homologues: unstable cyclooctynes and cyclodecynes, which are inert in SPAAC. The unique properties of cyclononynes are due to the steric and stereoelectronic effects produced by the heterocycle and heteroatom, namely, a greater alkyne bending of staggered conformations of cyclononynes and πin*CC – σ*CX hyperconjugation and π*Het – πin*CC conjugation in the transition state of SPAAC. Successful experiments on the introduction of a fluorescence probe into azido-glycans of cells were carried out using the new SPAAC reagent IC9N-BDP-FL based on azaheterocyclononyne fused to isocoumarin (a representative of compounds 2.10.54) (Scheme 327). The synthetic approach to the design of new SPAAC reagents that we developed is general and is easily implemented; therefore, it can be used for the preparation of a variety of cyclononynes and their analogues[819].

In continuation of these studies, rational design of new oxacyclononynes IC9O-EWG (EWG = CN, CO2Me) fused to isocoumarin was successfully carried out (see Scheme 327)[820].
These compounds exhibit fluorescence, easily react with azides to form triazoles, which are also capable of fluorescence; this makes it possible to introduce fluorescent probes into biological objects without additional use of dyes.
2.11. Advances in the synthesis and study of antitumor linear hetarene-fused anthracenediones
Derivatives of anthracene-9,10-dione (anthraquinone) are of greate practical importance due to their unique spectral and redox properties, photochromism, and a wide range of biological activity. Close attention is paid to the study of the pharmacological action of anthraquinone derivatives, especially their antitumor properties. Natural, semi-synthetic and synthetic anthraquinone derivatives, e.g. doxorubicin, valrubicin and mitoxantrone etc., are used in clinical practice to treat various oncological diseases, but their use is limited by cardiotoxic and general toxic effects, as well as the emergence of multidrug resistance (MDR) of tumor cells[821]. To date many ways of a chemical modification of anthraquinone have been investigated, resulting in numerous derivatives with antitumor properties. However, annelation of heterocycles to anthraquinone is considered one of the most promising directions for the design of innovative drugs for the treatment of oncological diseases. Heterocyclic anthraquinone derivatives attract a significant interest for the development of synthetic analogues of anthracycline antibiotics. A number of hetarene-fused anthracene-9,10-diones (hetareneanthracenediones) effectively inhibit the growth of tumor cells of various histogenesis affecting new cellular targets and some representatives showed antitumor effects when studied in laboratory animals[821].
The first identified chemotype of linear hetareneanthraquinone derivatives with high antitumor activity in vitro and in vivo were 3-aminomethyl derivatives of naphthoindolediones[822]. These compounds became the ‘starting point’ for optimizing the structure and elucidation the role of individual functional groups in the activity. In particular, using the principles of structure-based analogy (scaffold-hopping) anthra[2,3-b]furan-3-carboxamides were developed — the class of compounds with a unique multitarget effect on tumor cells. Annelation of the furan core to the anthraquinone moiety was achieved by condensation of dibromoquinizarin (2.11.1) with tert-butyl acetoacetate in the presence of a base. Subsequent hydrolysis of the intermediate ester led to 4,11-dihydroxy-2-methyl-5,10-dioxoanthra[2,3-b]furan-3-carboxylic acid (2.11.2) (Scheme 328)[823].

It should be noted that methylation of the 1,4-hydroxyl groups of compound 2.11.1 helps to reduce the cyclization temperature. Condensation of 2,3-dibromo-1,4-dimethoxyanthraquinone with other β-ketoesters and β-ketonitriles opens up possibilities to vary the substituent in position 2 of anthrafurans[824].
Anthrafuran-3-carboxamides could be efficiently prepared from the corresponding acid chloride generated by the action of thionyl chloride. Treatment of cyclic diamines with acid chloride of 2.11.2 gave a series of amides, among which the (S)-3-aminopyrrolidine derivative 2.11.3 demonstrated the highest activity[823]. Antitumor effect of anthra[2,3-b]furan-3-carboxamide 2.11.3 and its ability to circumvent MDR mechanisms are attributed to a multitarget effect on tumor cells. Anthrafuran 2.11.3 binds to the DNA duplex, resulting in the blocking of topoisomerases 1 and 2. Independently, compound 2.11.3 inhibits several protein kinases and induces reactive oxygen species (ROS). The development of parenteral and oral dosage forms based on anthrafuran-3-carboxamide 2.11.3 has led to the development of a promising drug candidate with high antitumor efficacy and good tolerability, which has been studied in various laboratory animal tumor models and human breast cancer xenografts[825, 826].
The method presented above proved to be ineffective for the preparation of esters of 2-unsubstituted anthra[2,3-b]furan-3-carboxylic acid. Synthesis of this compound was of interest for the structure–activity relationship (SAR) studies in the series of its amides. In this regard, an alternative approach for the preparation of anthra[2,3-b]furan-3-carboxylic acids was developed, based on a similar method of heterocycle formation[827]. Formylation of 2-(3-haloanthraquinon-2-yl)acetic acid esters 2.11.4a,b at the activated methylene group with methyl formate in the presence of NaH resulted in key intermediates — 2-(3-haloanthraquinon-2-yl)-2-formylacetic acid esters 2.11.5a,b. Intramolecular cyclization of compounds 2.11.5a,b with K3PO4 and CuI gave the methyl ester of 2-unsubstituted 4,11-dimethoxyanthra[2,3-b]furan-3-carboxylic acid 2.11.6 in moderate yields (Scheme 329).

To further investigate the antitumor properties of hetarene-fused anthracenediones and to evaluate the influence of their heterocyclic core, bioisosteric analogues of anthra[2,3-b]furan-3-carboxamide 2.11.3, namely naphtho[2,3-f ]indole-3-carboxamides, were obtained. Oxidative nucleophilic substitution of the hydrogen atom in 2-nitroquinizarin (2.11.7) with the acetoacetic ester residue in the presence of anhydrous potassium carbonate showed the formation of tert-butyl ester 2-(1,4-dihydroxy-3-nitro-9,10-dioxo-anthracene-2-yl)-3-hydroxybut-2-enoic acid (2.11.8) in low yield[828]. However, the vicarious nucleophilic substitution of the hydrogen atom occurred when 2-nitroquinizarin (2.11.7) reacted with 2-chloroacetoacetic ester. This reaction became a favorable alternative to oxidative nucleophilic hydrogen substitution as it leads to compound 2.11.8 in significantly higher yield. Reduction of 2-(3-nitroanthraquinon-2-yl)-3-oxobutanoic acid ester 2.11.8 with hydrogen catalyzed by Pd/C in the toluene – MeOH mixture (1 : 1) was accompanied by intramolecular heterocyclization in situ at room temperature and gave tert-butyl ester of 4,11-dihydroxy-2-methyl-5,10-dioxo-1H-naphtho[2,3-f ]indole-3-carboxylic acid (2.11.9) (Scheme 330).

For the synthesis of thiophene analogues of anthra[2,3-b]furan-3-carboxamide 2.11.3, modifications of the 3-methyl group of 4,11-dibutoxyanthra[2,3-b]thiophene-5,10-dione to carboxyl were studied[829]. After several unsuccessful attempts at direct oxidation of the methyl group of thiophene 2.11.10, its multistep transformation into a carboxyl group was developed. Bromination of anthrathiophene 2.11.10 with N-bromosuccinimide (NBS) in the presence of benzoyl peroxide under light irradiation gave the 3-dibromomethyl derivative 2.11.11 (Scheme 331). Boiling of dibromide 2.11.11 with potassium acetate in acetic acid followed by acid hydrolysis of the intermediate product formed of 4,11-dibutoxy-5,10-dioxoanthra[2,3-b]thiophene-3-carbaldehyde (2.11.12). Anthrathiophene-3-carbaldehyde 2.11.12 could be effectively oxidized to the corresponding anthra[2,3-b]thiophene-3-carboxylic acid 2.11.13 by the Jones method or by boiling with Br2 in a two-phase system CCl4 – H2O.

The chemical and biological properties of anthraquinone derivatives fused to six-membered heterocycles are less studied than their five-membered analogues. Annelation of a pyridine core containing an ester group at position 3 is most often carried out by the Friedlander or Loudon-Wellings methods. To obtain derivatives of naphtho[2,3-g]quinoline-3-carboxylic acid a new scheme has been developed. The sequence of reactions includes alkylation of CH-acid esters with an anthraquinone analogue of o-nitrobenzyl bromide, reductive heterocyclization and subsequent aromatization of the heterocycle[830]. Reactions of 2-(bromomethyl)-3-nitroanthraquinone 2.11.14 with esters of β-ketoacids, cyanoacetic and malonic esters give the corresponding 3-(β-anthraquinonyl)propionic acid esters 2.11.15 in good yields (Scheme 332). Treatment of anthraquinones 2.11.15 with sodium dithionite at room temperature not only reduces the nitro group, but also leads to intramolecular cyclization resulting in dihydronaphtho[2,3-g]quinoline-6,11-dione derivatives. Aromatization of compounds 2.11.15 by heating with DDQ in toluene or by atmospheric oxygen in the presence of Pd/C in boiling o-xylene gives the derivatives of naphtho[2,3-g]quinoline-3-carboxylic acids 2.11.16 in good yields.

Traditionally, the introduction of substituents at the position 2 of quinolone has been achieved by transformation of quinolin-2-ones into 2-halogen derivatives, followed by substitution of the halogen atom with nucleophiles. The main disadvantage of this method is the relatively harsh conditions both in the halogenation stage and in the substitution of the halogen with a nucleophile. An alternative method for transforming the 2-hydroxy group of naphtho[2,3-g]quinoline 2.11.17 using peptide synthesis reagents to activate the carboxyl groups has been proposed[830].
The reaction of quinolone 2.11.17 with (benzotriazol-1-yloxy)tri(pyrrolidinyl)phosphonium hexafluorophosphate (PyBOP) yields a product that is assigned to 2-(benzotriazolyl-1-oxy)naphtho[2,3-g]quinoline 2.11.18. The reaction of 2.11.18 with piperidine in DMSO leads to 2-(piperidin-1yl)naphtho[2,3-g]quinoline 2.11.18 (Scheme 333). When 2-hydroxynaphtho- [2,3-g]quinoline 2.11.17 is treated with a mixture of piperidine and PyBOP at room temperature, 2-piperidinylnaphtho[2,3-g]quinoline 2.11.19a is immediately formed in high yield.

The ability of phosphonium derivatives of N-hydroxybenzotriazole (for example, PyBOP) to activate the 2-hydroxyl group of naphtho[2,3-g]quinoline 2.11.17 is not unique; coupling reagents based on uronium salts are also effective. Treatment of naphthoquinolone 2.11.17 with a mixture of HBTU and piperidine, as well as the reaction with PyBOP, leads to 2-(piperidin-1-yl)naphtho[2,3-g]quinoline 2.11.19a in close yields. The transformations found are applicable to the substitution of the oxo group in compounds 2.11.17 with secondary and primary amines (e.g. benzylamine) as well as to the introduction into the molecule of a residue of O- and S-nucleophiles (phenol and mercaptoethanol) — with the formation of products 2.11.19a – d. Varying the nucleophiles reveals some limitations of this method. In particular, we have not found conditions for the coupling of quinolone 2.11.17 or its activated derivative 2.11.18 with aniline, ammonium chloride or ethanol.
Mechanistic studies have shown that hetarene-fused anthraquinones inhibit the activity of oncogenic protein kinases (e.g., Aurora A and B)[823, 831, 832]. Analysis of the structure of clinically approved protein kinase inhibitors has suggested that the introduction of a cyclic amine into the heterocyclic fragment of hetarene-fused anthraquinone may lead to improved inhibition of oncogenic protein kinases. The method developed for the transformation of naphthoquinolones (see Scheme 333) has been investigated for the introduction of cyclic amines and the evaluation of the reactivity of various oxoheterocycles fused with anthraquinone[833]. Initially, transformations of several quinizarin derivatives 2.11.20 into oxoheterocyclic derivatives 2.11.21a – g were carried out (Scheme 334).

The possibility of PyBOP-mediated substitution of the oxo group in compounds 2.11.21a – g was studied using an interaction with piperidine and N-Boc-piperazine as examples. It was found that the reaction rate and the yield of amination products of the heterocyclic ring (2.11.22) depend on both the structure of the heterocycle and the nucleophile. The transformation took place under relatively mild conditions for the derivatives quinolin-2-one, quinaxalin-2-one, pyrimidin-4-one and quinaxalin-2,4-dione 2.11.21a – d (Scheme 334). In this case, the reaction with piperidine proceeded faster than with N-Boc-piperazine, probably due to its higher nucleophilicity. The introduction of a second ionizable NH group adjacent to the oxo group in the derivatives of pyrimidin-2,4-dione 2.11.21e and imidazol-2-one 2.11.21g led to a complete loss of reactivity. In the case of anthra[2,3-d][1,2,3]triazine-4,6,11(3H)-trione 2.11.21f, the reaction with N-Boc-piperazine was accompanied by the cleavage of triazinone, resulting in the formation of 3-amino-1,4-dimethoxyanthraquinone-2-carboxamide.
In studying the chemical properties of hetarene-fused anthraquinones, methods were developed for the transformation of 4,11-alkoxy groups, which are not characteristic of either aromatic compounds or most anthraquinone derivatives. When 4,11-dimethoxynaphtho[2,3-f ]indole 2.11.23a, as well as anthraquinone derivatives containing strong electron-donating groups (EDG) were heated with zinc in acetic acid, regioselective reductive elimination of the alkoxy group located in the ortho-position to EDG occurred (compound 2.11.24 in Scheme 335)[834]. Despite the fact that 4-methoxyaphthoindoledione 2.11.24 was formed in high yield, a similar modification method could not be applied to alkoxy derivatives of other hetarene-fused anthraquinones.

An original method has been developed to convert alkoxy groups into primary amines of anthrafurandiones and anthrathiophenediones. This approach is based on the oxidative dealkylation reaction of alkylamino groups. Alkoxy groups in the peri-positions of hetarene-fused anthraquinones are easily substituted by primary amines, because they are activated by an electron-acceptor effect of the quinone fragment. Thus, treatment of anthrathiophene and anthrafuran 2.11.23b,c with n-butylamine gives 4,11-dibutylamino derivatives 2.11.25a,b, respectively. It has been found that the reaction of hetarene-fused anthracenediones 2.11.25a,b with Bun4NOH in DMSO leads to the dealkylation of alkylamino groups, which gives 4,11-diaminoanthrathiophenedione and 4,11-diaminoanthrafurandione 2.11.26a,b (see Scheme 335)[835]. These and other examples clearly demonstrate that the heterocyclic ring makes a significant contribution to the chemical properties of hetarene-fused anthraquinones.
Carboxylic acids of hetarene-annelated anthraquinones 2.11.27a – i were converted into a library of amides 2.11.28aa – ii (Scheme 336) (For compounds 2.11.28 and 2.11.28', the designation is accepted in the form of two Latin letters, denoting the combination of different fragments in molecule.) using cyclic diamines in order to elucidate the effect of the heterocyclic ring on the antitumor properties of the compounds[836-840]. Depending on the position, an activation of the carboxyl group in hetarene-fused anthraquinones 2.11.27a – i for coupling with amines was carried out under different conditions: for the derivatives 2.11.27a,c,e generation of the corresponding acid chloride was the most effective, while for the remaining compounds peptide coupling reagents (for example, PyBOP) were used. Mono-N-Boc-protected 3-aminopyrrolidines, 3- or 4-aminopiperidines and other derivatives of cyclic diamines were used for amidation. Such bioisosteres of the aminosugar daunosamine, a key pharmacophore fragment of anthracycline antibiotics, are more accessible and more lipophilic, which is important for overcoming the mechanisms of an active efflux of antitumor agents from cancer cells.

Cleavage of the Boc-protecting group with methanesulfonic acid gave a series of amides 2.11.28'aa – ii (Scheme 336), isolated as methanesulfonates. Such products had a higher solubility in pharmacologically acceptable aqueous media. In studying the antitumor properties of the library of carboxamides 2.11.28aa – ii, general patterns between the structure of the compounds and their activity, solubility and lipophilicity were established. Key intracellular targets were identified (DNA duplex, topoisomerases 1, ROS generation), which act to kill tumor cells. We made the important discovery that most of the derivatives of this chemotype are able to overcome the MDR mechanisms of tumor cells associated with increased expression of the P-glycoprotein efflux transporter and deletion of the p53 tumor suppressor gene. Lead compounds (for example, 2.11.28eb, 2.11.28fb) demonstrated a dose-dependent antitumor effect in vivo in P388 murine lymphocytic leukemia models, confirming the potential of their further preclinical studies.
The valuable antitumor properties of 4,11-diamino derivatives of hetarene-fused anthraquinones and, in particular, anthra[2,3-b]furan-5,10-diones stimulated the search for new universal routes for their synthesis, allowing the diversification of substituents in the heterocyclic ring. This contributed to the development and optimization of a method for the preparation of 2-substituted 4,11-dimethoxyanthra[2,3-b]furan-5,10-diones based on a tandem of Pd-catalyzed cross-coupling/heterocyclization reactions[841]. An original method was developed to replace the halogen atom in 2,3-dibromo-1,4-dimethoxyanthraquinone (2.11.29) by treatment with the benzaldoxime anion. Accordingly, the Miller – Loudon –Schneider reaction was proposed for the synthesis of 2-bromo-3-hydroxy-1,4-dimethoxyanthraquinone (2.11.30) — the starting compound for the annelation of the furan ring (Scheme 337)[842]. Cross-coupling of anthraquinone 2.11.30 with terminal alkynes under optimized conditions: (Pd(PPh3)2Cl2 – PPh3 – CuI system as a catalyst, a mixture of dioxane and Et3N (2 : 1), 100°C) as a solvent — gave a series of 2-substituted 4,11-dimethoxyanthra[2,3-b]furan-5,10-diones 2.11.31a – g — key intermediates for subsequent modification into the 4,11-diamino derivatives of hetarene-fused anthracenediones.

Compounds with high antitumor activity and multitargeted action were also developed based on 4,11-diamino derivatives of hetarene-fused anthracenediones. For their synthesis, nucleophilic substitution of the methoxy groups in the peri-positions of anthrafurandione 2.11.23c and anthrathiophenedione 2.11.23b was proposed (Scheme 338). The use of various diaminoalkanes 2.11.32 in this reaction gave a series of heterocyclic analogues of the antitumor compound Ametanthrone (for example, compounds 2.11.33a – d). It should be noted that, in addition to the effect on duplex DNA and topoisomerases 1 and 2, this type of hetarene-fused anthraquinone derivatives modulates the activity of Sirt1 deacetylase and NADH-dependent oxidase (tNOX)[843], activating alternative pathways for inducing apoptosis of tumor cells[844, 845].

Structure modification of the terminal groups of the side chains in 4,11-diamino derivatives of hetarene-fused anthraquinones led to the development of ligands for G-quadruplex structures of nucleic acids[846-851]. G-Quadruplexes (G4) are non-canonical four-stranded nucleic acid structures that play an important role in the regulation of the function of the genome of eukaryotic cells and could be promising targets for antitumor therapy[852]. Ligands that stabilize G4 are considered as candidates for the development of new target-specific anticancer drugs. Guanidination or amidination of 4,11-bis(2-aminoethylamino)anthra[2,3-b]thiophene-5,10-dione (2.11.33d) and its furan analog 2.11.33a by treatment with pyrazole-1-carboxyminamide or iminoesters, respectively, gave the corresponding bisguanidines and bisamidines 2.11.34a – d in good yields (see Scheme 338)[845, 846, 849].
The use of bisguanidine 2.11.34d, which is able to stabilise G4 elements in the promoter region of the H-Ras oncogene, revealed an original mechanism for the regulating of the gene expression in eukaryotic cells. It was found that compound 2.11.34d affects the dynamic conversions between duplex and quadruple DNA structures in the promoter regions of genes[848]. Replacement of the guanidino groups of side chains in ligand 2.11.34d with chloroacetamidines (compound 2.11.34c) retains high affinity to G4 of the H-Ras protein, but leads to a significant increase in the intracellular accumulation of this ligand and an increase in its ability to inhibit the growth of tumor cells. An administration of bis(chloroacetamidine) 2.11.34c at a low dose (1.7 mg kg–1) leads to a significant inhibition of tumor growth and an increase in the lifespan expectancy of mice with T24 bladder cancer xenografts[850].
Subsequently, it was demonstrated that some hetarene-fused anthraquinones bearing ethylenediamine residues in the side chains also effectively stabilize G4, but (unlike guanidine derivatives) are much better at penetrating tumor cells. In particular, anthrafuran 2.11.33a stabilizes the G4 elements utr-1, utr-4 and utr-z, located in the 5'-untranslated region of the K-Ras transcript. This prevents the binding of mRNA to the ribosome and causes selective blocking of the translation of this oncogene and dose-dependent decreases p21KRAS protein levels in Panc-1 pancreatic adenocarcinoma cells[851]. Anthrafuran 2.11.33a in submicromolar concentrations activates the caspase 3, leading to cleavage of the PARP enzyme and to the induction of apoptosis in Panc-1 cells with a high expression of the K-Ras oncogene.
The ability of diamino derivatives of hetarene-fused anthraquinones to stabilize G4 nucleic acids was applied to prove the binding of small molecules to G4 in the K-Ras oncogene transcript under low-abundance cellular conditions. An approach, based on the biotin-streptavidin pull-down assay for the extraction of intracellular targets, requires the use of biotinylated ligands. PyBOP-activated conjugation of anthrafuran-2-carboxylic acid with 6-(aminohexyl)biotinoylamide 2.11.35 leads to derivative 2.11.36 (Scheme 339). Subsequent nucleophilic substitution of the 4,11-methoxy groups of compound 2.11.36 with ethylenediamine gives 4,11-di(2-aminoethylamino)anthra[2,3-b]furan-5,10-dione (2.11.37), bound to biotin through a 6-aminohexylcarboxamide linker at position 2[851]. Guanidination of the terminal amino groups of anthrafuran 2.11.37 with pyrazole-1-carboxyminamide gives 4,11-bis(guanidinoalkyl) analogue 2.11.38. Derivatives 2.11.37 and 2.11.38 provide experimental evidence for the binding of ligand 2.11.33a to the G-quadruplex of K-Ras mRNA under native conditions due to a threefold increase in the extracted K-Ras mRNA compared to the control.

For the rational design of G4 ligands based on hetarene-fused anthraquinones, a methodology for computer modeling of ligand – target interaction was developed to optimise their structure[853]. A three-stage procedure for evaluation of the interaction of 4,11-bis[(2-guanidinoethyl)amino]anthra[2,3-b]thiophene-5,10-dione (2.11.34d) with a telomeric G4 was implemented. It included sequential steps to dock the ligand to the target surface, simulate the molecular dynamics of the complex and ultimately optimise the structure. Analysis of the G4-ligand complex resulted in several promising directions for chemical modification of the initial ligand 2.11.34d. In particular, when an additional side chain with a terminal guanidino group was introduced into position 2 of the molecule, the calculated energy of complexation increased from –87.1 kcal mol–1 (for 2.11.34d) to –113.7 kcal mol–1, and the replacement of 4,11-guanidinoethyl fragments to 5-aminopentyl increased the binding energy to –136.5 kcal mol–1.
In accordance with the results of the in silico design, a series of new ligands containing terminal aminoalkyl and guanidinoalkyl groups at the positions 2, 4, and 11 of hetarene-fused anthracenediones were synthesized. The coupling of anthrafuran- and anthrathiophene-2-carboxylic acids 2.11.27b,d with mono-N-Boc-protected diaminoethane or 1,3-diaminopropane in the presence of the PyBOP and subsequent cleavage of the Boc-protecting group led to amides 2.11.39 (Scheme 340). Substitution of methoxy groups in the positions 4 and 11 of compounds 2.11.39 by treatment with diaminoethane or diaminopropane gave derivatives 2.11.40, and final guanidination of the terminal amino groups of substituents in the positions 2, 4 and 11 resulted in trisguanidines 2.11.41 with varying lengths of the linker fragment of the side chains. In order to obtain G4-ligands bearing a guanidinoalkyl moiety at the position 2 of the heterocycle and two aminoalkyl residues at the positions 4 and 11 of hetarene-fused anthracenedione, the sequence of guanidination steps and the introduction of side chains at these positions were changed in the synthesis scheme.

Experiments have revealed that G4 ligands based on anthrafuran and anthrathiophene 2.11.41, containing an additional side chain at position 2 of the heterocycle, stabilize telomeric G4 4 – 15 times more effectively than the original 2-unsubstituted bisguanidine 2.11.34d[853]. It is important to note that the proposed modification improves the selectivity of binding of ligands to G4 compared to the DNA duplex: thus, the binding constants for the lead-ligand 2.11.41 (X = O, m = n = 3) with the DNA duplex are 75 times smaller than for telomeric G4, confirming the effectiveness of the computational design of G4 ligands.
In summary, a number of effective methodologies have been developed for the annelation of a wide range of five- and six-membered heterocycles to anthracenedione, as well as new effective methods for their chemical modification. The proposed methods have significantly expanded the possibilities of function-oriented design of compounds with desired properties. Annelation of heterocycles to anthraquinone often enhances antitumor effect, helps to circumvent MDR and leads to the emergence of activity on new intracellular targets important for tumor growth (Sirt1, tNOX, protein kinases, G-quadruplexes of nucleic acids, etc.). The high antitumor activity of a number of the drug candidates developed (anthrafurans 2.11.3 and 2.11.33a, anthrathiophene 2.11.34c) confirms the advisability of further in-depth studies of this unique class of heterocyclic compounds.
2.12. Functionalized 2H-azirines as building blocks for the synthesis of heterocycles
In this Section, new convergent approaches to azirine derivatives functionalized at the C(2) atom are considered, which allow an efficient change of functional groups in the azirine ring without its decomposition. The azirines bearing one or two functional groups at the C(2) atom are characterized by fundamentally different ways of the three-membered ring opening, which provided the basis for the new methods of construction of not only pharmacologically significant nitrogen heterocycles, but also some non-nitrogen heterocyclic systems described below.
Despite its almost 100-year history, the chemistry of 2H-azirines has only recently begun to attract the attention of synthetic chemists. This is largely due to the emergence in the 2010s of new reliable and convenient methods for the synthesis of azirine derivatives, which have allowed their range to be greatly expanded. However, the main reason for the boost of research in this field is the unique reactivity of the azirine ring, which goes far beyond the simple synergism of the small ring strain and the electrophilicity of the cyclic imine. Recent advances in azirine chemistry are highlighted in a number of modern review articles[854-856], which focus on the synthesis and application of the most available hydrocarbon-substituted azirines. Such azirines can, under certain conditions, be ‘forced’ to react both with preserving the ring system and selectively cleaving of any of the three bonds, making these molecules unique building blocks for organic synthesis. Meanwhile, functionalization of a three-membered ring often changes its reactivity dramatically with respect to a number of reagents, and also allows it to be involved in those reactions that are impossible for non-functionalized azirines. This Section highlights the results of 10 years of research carried out at the Institute of Chemistry, SPbU, on the development of new synthetic approaches to functionalized azirines, the elucidation of the mechanisms of unusual reactions of such azirines and the search for their new applications in the synthesis of heterocycles.
2.12.1. Synthesis of С(2)-functionalized azirines
Among the azirine substrates most demanded for organic and medicinal chemistry are 2-carbonyl-substituted azirines A and their functionalized analogues (Scheme 341). A large number of these compounds have been synthesized in recent years with the availability of reliable and simple methods that allow rapid construction of the azirine-2-carbonyl structural unit from available isoxazoles via isoxazole-azirine isomerization[857], easy introduction of a substituent on the C(2) atom, and variation of the substituent on the carbonyl moiety. In addition, the ability to introduce the desired R2 or R3 substituent into both the precursor isoxazole B and the azirine C itself significantly expands the synthetic potential of this method, making alternative approaches to such azirines uncompetitive[854, 858].

In 2017, a new convenient method was proposed for the preparation of C(2)-halo-substituted azirine-2-carboxylic acid derivatives 2.12.2 and 2.12.3 from 4-haloisoxazoles 2.12.1 bearing an alkoxy or dialkylamino group at the C(5) atom (Scheme 342)[859]. 2-Haloazirines 2.12.2 are accessible by this method on a gram scale with 0.1 mol.% loading of dirhodium tetrapivaloate catalyst, often without chromatographic purification. The isomerization of 5-sulfanyl-substituted 4-haloisoxazoles 2.12.1 to 2-haloazirines 2.12.4 is more efficiently catalyzed by iron(II) chloride tetrahydrate. In the synthesis of derivatives of 2-bromo- and 2-iodoazirine-2-carboxylic acids 2.12.2, it is preferable to use iron(II) sulfate heptahydrate to avoid the halide exchange for chlorine in the reaction product[860].

2-Halo-2H-azirines 2.12.2 (R2 = OAlk), 2.12.3 (R2 = NAlk2) and 2.12.5 (R2 = SAr) proved to be a convenient synthetic platform for the preparation of other azirine derivatives by nucleophilic substitution of the halogen atom (Scheme 343). It is shown that the whole series of haloazirines can be derived from the appropriate bromoazirines at room temperature under rather simple conditions, using tetra-n-butylammonium halides in dichloromethane in the synthesis of fluorides 2.12.6 and chlorides 2.12.7 and using potassium iodide in acetone in the synthesis of iodides 2.12.8. Because of its simplicity and efficiency, this reaction is particularly valuable for the synthesis of hard-to-reach fluorinated azirines 2.12.6[861].

Various 2-oxygen-substituted 2H-azirine-2-carboxylic acid derivatives 2.12.9 have been synthesized in high yields by substitution of the halogen atom in 2-haloazirines 2.12.2, 2.12.3, and 2.12.5 with O-nucleophiles in the presence of triethylamine[862]. The reaction proceeds smoothly with carboxylic acids, enols and phenols having pKa values in the range 3 – 10. According to quantum chemical calculations, the substitution of the halogen atom in the starting azirine with the acyloxy group is accomplished via the cascade mechanism SN2'/SN2', in which the first conjugated substitution is the rate-limiting step. A similar approach has been used to obtain 2-azolyl-2H-azirine-2-carboxylates 2.12.10. Various NH-azoles (imidazole, 1,2,4-triazole, tetrazole), including benzannulated ones (benzimidazole, indazole, benzotriazole, etc.), have reacted to give azolylazirines 2.12.10 in good yields[863]. The reaction with substituted pyridines and azines follows the same mechanism and gives pyridinium and azinium salts 2.12.11. Tetrafluoroborate salts 2.12.11 are stable compounds, unlike halide salts[864]. In addition, in 2-halo-2H-azirines 2.12.2, 2.12.3, and 2.12.5, the halogen atom can be substituted with C-nucleophiles such as 2-pyridylstannanes. This reaction produces 2-(2-pyridyl)-2H-azirines 2.12.12 and proceeds readily with 3-, 4-, and 5-substituted 2-(tri-n-butylstannyl)pyridines and 2-(tri-n-butylstannyl)thiazole[865].
One of the strategies for the synthesis of azirine-2-carboxylic acid derivatives is the generation of unstable azirine-2-carbonyl chlorides from 5-haloisoxazoles followed by substitution of the halogen atom by N-, O-, or S-nucleophile. For example, a simple and efficient method has been developed for the synthesis of hard-to-reach azirine-2-carboxylic acids from 5-chloroisoxazoles 2.12.13 based on the isoxazole → azirine isomerization catalyzed by anhydrous iron(II) chloride (Scheme 344)[866]. This process is carried out in one synthetic step and involves the ring-opening of chloroisoxazole 2.12.13 to metalloazapolyene 2.12.14, its cyclization to azirine-2-carbonyl chloride 2.12.15 and hydrolysis of the latter. This method works well for obtaining the racemic form of natural azirinomycin. The study of the antibacterial activity of the resulting acids 2.12.16 against bacterial strains of the ESKAPE pathogens showed that 3-phenyl-2H-azirine-2-carboxylic acid was the lead compound. Acids 2.12.16 were moderately cytotoxic against APRE-19 and HEK293 cells.

The presence of a carboxyl group allows using azirine carboxylic acids 2.12.16 in the synthesis of more complex 2H-azirine derivatives (Scheme 345). Using Passerini and Ugi reactions, azirine carboxylic acids 2.12.16 have been successfully converted into azirine-containing depsipeptides 2.12.17 and dipeptides 2.12.18[867]. The photochemical reaction of azirine carboxylic acids 2.12.16 with diazo compounds 2.12.19 gives similar functionalized azirines 2.12.20, but with an ester moiety instead of an amide one. In this case, the OH insertion is selective and the azirine ring remains intact[868].

3-(Het)aryl-5-chloroisoxazoles 2.12.21, which were converted to azirine-2-carbonyl chlorides 2.12.22 using iron(II) chloride as a catalyst, were used as starting material in the synthesis of azirine-2-carboxylic acid derivatives. The intermediate 2.12.22 reacts with sodium azide to give azirine-2-carbonyl azides 2.12.23 in good yields (Scheme 346)[869]. And the in situ trapping of acid chlorides 2.12.22 with pyrazoles 2.12.24 and 1,2,3-benzotriazole furnished pyrazolylcarbonyl- (see[870]) and 1,2,3-benzotriazolylcarbonyl-substituted[871] azirines 2.12.25 and 2.12.26, respectively. The acylation of pyrazoles 2.12.24 occurs regioselectively to afford a 1,3,4-trisubstituted pyrazole ring. The benzotriazole is acylated exclusively at the 1-position.

Amides 2.12.27, esters 2.12.28 and thioesters 2.12.29 of azirine-2-carboxylic acids can be prepared in high yields at room temperature from acid chlorides 2.12.22 by reaction with N-, O-, or S-nucleophiles: primary and secondary amines, phenols, alcohols, thiophenols and aliphatic thiols (Scheme 347)[872]. To increase the yield of products it is necessary to bind the released HCl, which destroys azirine, using ortho-substituted pyridine, for example 2-picoline. It is noteworthy that the highest yields of products are obtained when using the 2-(trimethylsilyl)pyridine — ethyl chloroformate system. In the presence of a small amount of water and in the absence of other nucleophiles, the acid chlorides 2.12.22 form azirine-2-carboxylic acid anhydrides 2.12.30[872].

Diazo group-containing azirines have been synthesized from azirine-2-carbonyl chlorides (Scheme 348). This method involves the sequential treatment of acid chlorides 2.12.22 with N-isocyanotriphenyliminophosphorane (2.12.31) and water and the tosyl chloride-catalyzed dehydrochlorination of the resulting hydrazidoyl chlorides 2.12.32 with triethylamine[873]. Subsequently, the synthesis of diazoacetylazirines 2.12.33 was significantly simplified by extracting the chloride 2.12.22 from the reaction mixture with diethyl ether and treating the resulting solution with diazomethane[874]. Azirines 2.12.33 can be acylated to azirine-containing diazo ketoesters 2.12.34 in good yields[875].

The presence of an active diazo group in the azirine molecule offers wide opportunities for modification of the substituent on the C(2) ring atom while preserving the azirine system (Scheme 349)[873]. Thus, 2-(diazoacetyl)azirines 2.12.33 react with acidic reagents (HX) such as hydrogen halides, carboxylic acids and p-toluenesulfonic acid to give the corresponding 1-(2H-azirin-2-yl)-2-X-ethan-1-ones. Reaction of azirines 2.12.33 with triphenylphosphine provides quantitative yield of phosphazenes 2.12.35, which in turn undergo the aza-Wittig reaction with a preserved azirine ring and formation of azines 2.12.36. The reaction of rhodium(II) carbene complexes derived from 2-(diazoacetyl)azirines 2.12.33 using a large excess of nitrile 2.12.37 gives azirine-oxazole dyads 2.12.38. The diazo group of azirines 2.12.33 also reacts readily as a 1,3-dipole with ethyl acrylate to give pyrazolines 2.12.39 in good yields.

Another approach to azirine derivatives is based on the transformation of the bromoacetyl group at the C(2) atom of compounds 2.12.40 (Scheme 350)[873]. Phosphonium and pyridinium salts 2.12.41, 2.12.42, azidomethyl ketone 2.12.43 and also azirine-thiazole (2.12.44) and azirine-oxirane (2.12.45) dyads have been obtained in this way. In addition, the azirine system is well tolerated under the strongly basic conditions of the Wittig reaction, thus providing an access to azirines 2.12.46.

2.12.2. Synthesis of four-membered heterocycles from azirines
Important synthetic applications of 2H-azirines include the reaction of the one-atom expansion of the three-membered ring to give 2,3-dihydroazetes, which takes place under the action of α-diazocarbonyl compounds using rhodium(II) carboxylates as catalysts[876]. This reaction is currently the only method for obtaining 2,3-dihydroazetes bearing hydrocarbon substituents on the double bond in the ring. This transformation has been carried out for a wide range of 2,3-disubstituted azirines, including azirine-2-carboxylic acid derivatives. For example, azirine 2.12.47 treated with dimethyl diazomalonate in the presence of Rh2(OAc)4 forms a four-membered ring (Scheme 351)[877]. The reaction proceeds in several steps through the formation of the intermediate Rh complex 2.12.48 and the azirinium ylide 2.12.49, the opening of the latter to 2-azabuta-1,3-diene 2.12.50, the 4π-cyclization of which gives dihydroazete 2.12.51. The low yield of the final product is explained by the non-selective azirine ring opening in the ylide 2.12.49 and, as a consequence, the formation of comparable amounts of stereoisomeric azadienes 2.12.50 of which only one, the E-isomer, is able to undergo 4π-cyclization to dihydroazete.

However, it was soon possible to find an elegant solution to this challenge, which consisted of introducing a bromine atom as a directing group in the 2-position of the azirine, which provided the unambiguous stereoselective isomerization of the azirinium ylide into 2-azabutadiene[877]. The reaction of brominated azirines 2.12.52 leads exclusively to E-isomers of 2-azabutadiene, which are cyclized, albeit reversibly, to dihydroazetes 2.12.53 (Scheme 352). The hydrodehalogenation of bromodihydroazetes 2.12.53, being in equilibrium with azadienes, by the system Bun3SnH – AIBN – toluene (AIBN is azobis(isobutyronitrile)) gives rise to non-halogenated dihydroazetes 2.12.54 in good yields. It should be noted that some of the resulting dihydroazetes 2.12.54 have been found to have antitumour activity with a high level of apoptosis and low necrotic potential[877].

In contrast to the non-halogenated dihydroazetes 2.12.54, the dihydroazetes 2.12.53 are thermally unstable and reversibly open to 2-azabutadienes under the reaction conditions[878]. Nevertheless, compounds 2.12.53a,b can be produced in high yields by a procedure involving a repeated two-step process: heating of 2-azadiene – isolation of the dihydroazete by crystallization or chromatography (Scheme 353). This approach is useful when using mainly bromine, iodine or fluorine derivatives of azirine-2-carboxylic acids, as well as diazo compounds in which both substituents have a strong (–M) effect, e.g. esters of diazomalonic and dialkoxyphosphorylacetic acids. In this case, the content of the dihydroazete form in the equilibrium mixture of dihydroazete–azadiene is significant and the above method of product isolation is quite effective.

One of the challenges of using α-diazocarbonyl compounds in rhodium-catalyzed reactions with heteroatomic substrates is the deactivation of the catalyst by some N- and S-nucleophilic centres of the substrate. In particular, that is why 2-pyridyl-substituted 2,3-dihydroazetes 2.12.56 cannot be synthesized from 2-pyridylazirines 2.12.55 and diazocarbonyl compounds according to the standard procedure (Scheme 354). This problem was solved by protecting the pyridine nitrogen of pyridylazirine with trimethylsilyl group, which was removed by tetrabutylammonium fluoride in the final step of the one-pot synthesis[879]. Notably, reactions with ethyl 2-diazo-3,3,3-trifluoropropanoate proceed stereoselectively to give only the (2RS,3SR)-diastereomer, which may be due to the higher stability of this isomer and the reversibility of the 4π-cyclization of the 2-azabuta-1,3-diene intermediate 2.12.57.

When a four-membered ring is formed in the said azirine ring expansion, all three atoms of the azirine ring are involved in the product ring. An example of the inclusion of only one of the three atoms of the azirine ring, the nitrogen atom, in the four-membered product ring is the synthesis of spiro-β-lactams from azirine-2-carboxylic acid derivatives and diazo Meldrum’s acid (2.12.58) catalyzed by dirhodium tetrapivaloate (Scheme 355)[880]. This reaction is unusual in that the source of all the carbon atoms in the β-lactam ring is a diazocarbonyl compound 2.12.58, the feature of which is that it generates the rhodium carbene complex 2.12.59, which can react with azirine with comparable efficiency to give 2-azabuta-1,3-diene 2.12.60 and undergo the Wolff rearrangement to give ketene 2.12.61. Staudinger cycloaddition of the latter to the C=N bond of azadiene 2.12.60 leads to the final β-lactam 2.12.62.

A cross-version of this method using two different diazocarbonyl compounds has also been successfully carried out. Monospiro-β-lactams 2.12.64 were obtained using either a diazo diester or a diazo ketoester at the azabutadiene 2.12.63 formation step, and diazo Meldrum’s acid was introduced at the Staudinger cycloaddition step (Scheme 356)[880].

2.12.3. Synthesis of five-membered heterocycles from azirines
Methods for the preparation of various pyrrole derivatives constitute the most extensive and explored part of the reactions of 2-carbonyl-substituted azirines that furnish a five-membered heterocycle. This is mainly due to the fact that the two-atom expansion of the azirine ring to the pyrrole ring can be induced by a rather large pool of reagents, both nucleophilic and electrophilic.
The reaction of azirine-2-carboxylic acid derivatives 2.12.65 with 1,3-dicarbonyl compounds in the presence of salts and chelates of nickel(II), copper(I/II) or cobalt(II/III) provides a convenient approach to a variety of 2,4-dicarbonyl-substituted pyrroles 2.12.66 (Scheme 357, reaction 1)[873, 874, 881, 882, 869-871]. The efficiency of the catalyst depends in a rather complex manner on the nature of the C(2) substituent in azirine, and its choice for different substrates is still empirical. The construction of the pyrrole system in these reactions is achieved via the azirine ring opening at the N=C(3) bond, which proceeds sequentially via nucleophilic addition of the enolate to the multiple bond of azirine, cyclization and the three-membered ring expansion. This process can also be carried out directly from precursors of 2-alkoxy(2-amino)carbonyl-2H-azirines (5-alkoxy(5-amino)isoxazoles) using the ‘relay’ catalysis by the system FeCl2 · 4H2O – NiCl2 · 6 H2O[883]. The use of malononitrile, the reaction of which was described in 2021[884], significantly extended the number of substituents introduced into the pyrrole ring, including the preparation of pyrrole-2,4-dicarboxylic acid derivatives 2.12.67 with a 5-positioned free amino group (see Scheme 357, reaction 2).

The use of pyridinium ylides as nucleophilic partners for azirines allowed this approach to be extended to the synthesis of 3-amino-substituted pyrroles[885, 886]. The reaction of azirines with pyridinium ylides, which is generally similar to that with enolates, proved to be a very effective method for obtaining (3H-pyrrol-3-yl)pyridinium salts, which are convenient substrates for the synthesis of various 3-aminopyrroles and pyridinium ylides of a new type, (3-pyridinium)pyrrol-3-ides. For example, the reaction between pyridinium ylides and 5-methoxyisoxazoles 2.12.68, acting as precursors of azirine-2-carboxylates, produced salts 2.12.69 using relay catalysis. The latter were converted into 4-aminopyrrole-2-carboxylates 2.12.70 (Scheme 358, reaction 1) by a one-pot Zincke hydrazinolysis procedure[887-889]. In this variant, which implies the use of α-aroyl(α-acetyl)-substituted ylides, 2,3-disubstituted 2H-azirines can be used including non-functionalized analogues. However, a similar approach to aminopyrroles involving less nucleophilic ylides derived from salts 2.12.71 requires the use of only activated azirines with a strong electron-withdrawing group at the C(2) atom. In this case, both trifluoromethyl-substituted ylides 2.12.72 and aminopyrroles 2.12.73 (see Scheme 358, reaction 2) can be obtained[890]. This method opened the way to new heterocyclic betaines 2.12.75 (see Scheme 358, reaction 3) obtained from ylides, which were generated from pyridinium salts 2.12.74 (see Scheme 358, reaction 3)[891].

The developed methodology for the synthesis of 3-(N-hetaryl)pyrrole derivatives was successfully extended to imidazolium[892, 893] and triazolium[894] ylides. The water soluble α-aminopyrroles, 1-(5-alkoxycarbonyl-2-amino-4-aryl-1H-pyrrol-3-yl)pyridinium chlorides, are accessible via the reaction of 1-(cyanomethyl)pyridinium chloride with alkyl-3-aryl-2H-azirine-2-carboxylates[895].
Pyrrole-3-carboxylic acid derivatives can be synthesized via a cascade of intramolecular reactions of isoxazoles. For example, the Fe(II)-catalyzed isomerization of 5-alkoxy(amino or N,N-dialkylamino)-3-aryl(alkyl)-4-(2-R-vinyl)isoxazoles to pyrrole-3-carboxylic acid esters is mediated by 2-(2-R-vinyl)-2-(R'(O)C)-2H-azirines, which in some cases can be isolated in an individual form[896].
Another approach to the synthesis of pyrroles via azirine ring expansion is based on reactions with rhodium(II) carbene complexes generated from α-diazocarbonyl compounds. Such intermediates have electrophilic properties and, upon reaction with the azirine nitrogen atom, induce the ring opening at the N – C(2) bond, yielding pyrrole with a different position of substituents than in the above-mentioned cases. In particular, an efficient two-step procedure for the synthesis of 3-hydroxypyrrole-2-carboxylic and -2,4-dicarboxylic acid derivatives from α-diazocarbonyl compounds and 2-haloazirines has been developed (Scheme 359). This reaction first gives 2-azabuta-1,3-dienes 2.12.76, which are further cyclized without purification to pyrroles 2.12.77 under the action of tri-n-butylstannane[897, 898]. During cyclization, which was found to follow an ionic mechanism, chlorides 2.12.76 (Hal = Cl) provide the maximum yields of the product.

2-Azabuta-1,3-dienes formed in catalytic reactions of azirines with diazo compounds can be converted quite efficiently into partially hydrogenated pyrroles. In particular, azadienes obtained from azirine-2-carbonyl derivatives 2.12.78 and dimethyl diazomalonate give 1-pyrrolines 2.12.79 in good yields under the action of DBU (Scheme 360, reaction 1)[899]. This process can also be carried out in a one-pot mode from isoxazoles as a source of azirines.

6-Halo-1-oxa-4-azahexa-1,3,5-trienes 2.12.80, derived from azirines 2.12.2 and diazodicarbonyl compounds, are cyclized under the action of N- and S-nucleophiles to 3,4-epoxypyrroline derivatives 2.12.81 (see Scheme 360, reaction 2)[900]. In contrast to 1-oxa-4-azahexatrienes 2.12.80, 1-oxa-5-azahexatrienes 2.12.82, formed in the Rh2(OAc)4-catalyzed reaction of 2-formyl-substituted azirines with ethyl 2-diazo-2-cyanoacetate or 2-diazo-3,3,3-trifluoropropionate, are thermally unstable and cyclize under kinetic conditions to 2H-1,3-oxazines and at higher temperatures irreversibly isomerize to 1H-pyrrole-3(2H)-ones 2.12.83 (see Scheme 360, reaction 3)[901].
In 2022, we pioneered in carrying out an intramolecular variant of the azirine ring expansion under the action of a rhodium carbene complex. Azirinyl-substituted diazodicarbonyl compounds 2.12.34 were converted in high yields into alkyl 2-hydroxy-3-oxo-2,3-dihydro-1H-pyrrole-2-carboxylates 2.12.84 in THF in the presence of water using Rh2(OAc)4 as the catalyst (Scheme 361)[902].

Syntheses of azoles using functionalized azirines are quite rare. In 2018, an efficient method was found for the preparation of 1-amino-1H-pyrazole-4-carboxylic acid derivatives 2.12.85. It is based on the iron(II)-catalyzed rearrangement of isoxazoles 2.12.86 bearing a (2,4-nitrophenylhydrazono)methyl substituent at the C(4) atom, similar to the above-mentioned 4-vinylisoxazole-azirine-pyrrole isomerization[903]. It proceeds via the formation of the azirine intermediate 2.12.87, which can be isolated in some cases (Scheme 362, reaction 1)[903]. When reacting with O- and S-nucleophiles at room temperature, 2H-azirine-2-carbonylazides 2.12.23 undergo an unusual rearrangement to 2-(1H-tetrazol-1-yl)acetic acid derivatives 2.12.88 (see Scheme 362, reaction 2)[904]. This reaction is catalyzed by tertiary amines and hydrazoic acid.

Some of the existing approaches to N,O-azoles via azirine ring expansion involve the use of C(2)-functionalized azirines only. These include, in particular, the synthesis of isoxazoles via azirine-isoxazole isomerization, which is an effective approach to 5-alkyl(aryl)-substituted isoxazoles from 2-acylazirines. Using this method, a series of 5-cycloheptatrienyl-substituted isoxazoles 2.12.90 have been obtained from azirines 2.12.89 by catalysis with FeCl2 · 4 H2O (Scheme 363, reaction 1)[882]. 2-Acyl-2-(methoxycarbonyl)azirines 2.12.91 are quantitatively isomerized at 170°C to oxazoles 2.12.92 (see Scheme 363, reaction 2)[905].

An unusual transformation has been found for azirine tosylates 2.12.93 (see Scheme 363, reaction 3). In methanol in the presence of Et3N, such compounds undergo cleavage of the C – C bond and then cyclized to 5-methylidene-substituted oxazolines 2.12.94 (see[873]).
C(2)-functionalized azirines can also be used in the synthesis of nitrogen-free heterocycles. A process for the preparation of 5-aminobutenolides by base-catalyzed reaction of esters or amides of 2-bromo-2H-azirine-2-carboxylic acids with arylacetic acids has been developed (Scheme 364). It is noteworthy that the regioselectivity of this reaction can be controlled quite effectively by substitution of only the basic reagent, which allows butenolide-containing derivatives of both α-amino acids (2.12.95) and β-amino acids (2.12.96) to be obtained in good yields (see Scheme 364)[906].

ortho-Fused pyrroles and azoles such as indoles and azaindolizines can be obtained from azirines according to a general strategy which boils down to the preparation of a 2-(het)aryl-substituted azirine with subsequent intramolecular expansion of the three-membered ring involving two atoms of the aromatic substituent. An additional C(2)-functional substituent of the azirine is moved to the five-membered ring of the target product. For example, the FeCl2-catalyzed domino isomerization of 4-arylisoxazoles 2.12.97 to 2-arylazirines and then to indoles provides selective access to a wide range of substituted indole-3-carboxylates 2.12.98 (Scheme 365, reaction 1)[907]. Using the same scheme, thieno[3,2-b]pyrroles 2.12.99 have been successfully synthesized (see Scheme 365, reaction 2). The bicyclic framework of imidazo[1,2-a]pyridine and imidazo[1,2-b]pyridazine can be constructed in two steps by the formation of azirinylpyridinium bromides 2.12.100 followed by UV light-induced cyclization (see Scheme 365, reaction 3)[864].

The three-membered ring in esters and thioesters of 2-(2-pyridyl)azirine-2-carboxylic acids 2.12.101 can be selectively cleaved either at the N – C(2) bond using copper(II) catalysis or at the C – C bond under the action of HCl to give isomeric products of the azirine ring expansion — pyrazolo[1,5-a]pyridines 2.12.102 or imidazo[1,5-a]pyridines 2.12.103 (Scheme 366)[908]. Pyrazolopyridines 2.12.102 can be prepared from 4-bromoisoxazoles 2.12.104 by a one-pot, three-step procedure without isolation of the intermediate azirines 2.12.105.

2.12.4. Synthesis of six-membered heterocycles from azirines
One of the general approaches to six-membered nitrogen heterocycles from azirines is the rhodium-catalyzed reaction with diazo compounds via the formation of conjugated aza- or oxaza- or diazahexa-1,3,5-triene with subsequent 6π-cyclization (Scheme 367). The presence of an additional unsaturated unit (C=C, C=O, C=N bonds) either in the α-position of the diazo compound (route 1) or at the C(2) atom of the azirine substrate is a prerequisite for the formation of such a polyene (route 2).

An example of the three-atom expansion of the azirine ring following the first pathway is the synthesis of 1,4-oxazine-2,5-dicarboxylic acid ester 2.12.106 from azirine 2.12.47 (Scheme 368, reaction 1)[909]. It should be noted that such 1,4-oxazines demonstrated photochromic activity, i.e. the ability to undergo ring opening under UV light irradiation to give coloured 1,4-oxazahexatrienes which, in the absence of irradiation, are cyclized back to oxazines as the temperature rises. In the presence of an imino group in the α-position of a diazo compound, as, e.g., in indolindiazoimine 2.12.107, a similar reaction with esters and amides of azirine-2-carboxylic acids 2.12.65 affords pyrazine derivatives. This transformation was the basis for the synthesis of pyrazinoindoles 2.12.108 (see Scheme 368, reaction 2), which is accompanied by desulfonylation of the 1,6-cyclization product under reaction conditions and generally provides high yields[910]. Since azirines 2.12.65 are obtained by Rh(II)-catalyzed isomerization of isoxazole in almost quantitative yields, pyrazinoindoles 2.12.108 can be obtained directly from the isoxazole precursor without isolation of the azirine in high yields.

The transfer of the unsaturated unit from the diazo component to azirine opens the way to the synthesis of six-membered N,O- and N,N-heterocycles with heteroatoms in the 1,3-position (see Scheme 368, pathway 2). In principle, in the presence of a vinyl substituent at the C(2) atom of azirine, pyridine derivatives 2.12.109 can also be obtained but, as our studies have shown[911], the yields in such reactions are usually low because of the low stereoselectivity towards intermediate azahexatrienes (Scheme 369).

The Rh(II)-catalyzed reaction of α-diazoesters with 2-formyl- and 2-acetylazirines proved to be synthetically more relevant and may provide an access to 2,2-difunctionalized 2H-1,3-oxazines 2.12.111 (Scheme 370)[912]. Notably, the thermal stability of the resulting oxazine decreases as the electron-withdrawing ability of the R4 substituent increases, and it begins to isomerize into a pyrrolin-3-one derivative[901]. Therefore, syntheses of such oxazine derivatives should be carried out at temperatures below 40 °C. 2-Acetylazirines, unlike formyl-substituted analogues, form small amounts of thermally stable (3E)-1-oxa-5-azahexa-1,3,5-triene 2.12.112 in this reaction, which is stable to 1,6-cyclization to 1,3-oxazines.

1,3-Oxazine derivatives can also be obtained from azirine-2-carboxylic acids. For example, a series of 1,3-oxazin-6-ones 2.12.113 have been synthesized from acids 2.12.16 and diazoesters (Scheme 371)[868]. This reaction is also catalyzed by rhodium(II) carboxylates, but in most cases the more effective catalyst was bis[(trifluoromethanesulfonyl)imidate](triphenylphosphine)gold(I), which provided yields > 90%. However, for substrates such as C-unsubstituted azirine-2-carboxylic acids, higher yields of oxazinones were observed for Rh2(OAc)4-catalysis.

The reaction of α-diazoesters with azirine-2-carbaldimines 2.12.114 catalyzed by Rh2(esp)2 is a convenient approach to 1,2-dihydropyrimidines 2.12.115 unsubstituted at the 6-position (Scheme 372)[913]. The imines used in the synthesis are obtained by condensation of the appropriate aldehyde with amine and are used in the reaction without purification. The yields of dihydropyrimidines based on azirinecarbaldehyde are on average of 60 – 70% and decrease in the case of ortho-substituted anilines. Experiments and quantum chemical calculations have shown that the rhodium carbene complex formed in the reaction reacts exclusively with the azirine nitrogen atom, since the exocyclic imine nitrogen is shielded by a hydrogen atom or a substituent located at the 2-position of the azirine ring.

The high synthetic potential of azirines sometimes allows several different approaches to the derivatives of the said six-membered heterocyclic systems. In particular, an efficient method has been developed for the synthesis of methyl 6-halonicotinates 2.12.116 by domino isomerization of 4-(3-halopropargyl)-5-methoxyisoxazoles 2.12.117 using relay catalysis. The first step of the process, the conversion of isoxazole to 2-(methoxycarbonyl)-2-propargyl-2H-azirine 2.12.118, is catalyzed by Fe(NTf2)2, while the second step, the cyclization with the azirine ring expansion, is catalyzed by Ph3PAuNTf2 (Scheme 373)[914]. This methodology of pyridine ring formation has been successfully applied to the synthesis of 2,2'-bi-, 2,2':6',2''-ter- and 2,2':6',2'':6'',2'''-quaterpyridines[915, 916].

The conditions for the aerobic oxidative dimerization of azirine-2-carboxylic acid esters 2.12.119 to fully substituted pyrimidine-4,6-dicarboxylic acid esters 2.12.120 promoted with triethylamine were found (Scheme 374)[917]. The unique feature of this reaction lies in the fact that it is achieved by the formation of diethylhydroxylamine at a very low concentration and proceeds via the formation of the 2-(aminoxy)aziridine intermediate 2.12.121.

In addition to the Au(I)-catalyzed reaction of azirine-2-carboxylic acids 2.12.16 shown in Scheme 371, another approach to 1,3-oxazin-6-ones was developed using 2-acyloxyazirine-2-carboxylates 2.12.122. These azirine derivatives were the first substrates on which the free radical azirine ring expansion could be achieved. Azirines 2.12.122 reacted with tri-n-butyltin hydride and a radical initiator in boiling toluene to give mainly 5-hydroxy-1,3-oxazin-6-ones 2.12.123 in high yields (Scheme 375)[918]. A decrease in product yield was only observed in the presence of substituents with free O – H and N – H bonds, and azirines with low-volume primary alkyl R2 groups were found to be unsuitable substrates for this transformation. The reaction starts with the attack of the tri-n-butylstannyl radical on the azirine nitrogen atom to generate the aziridinyl radical 2.12.124, which is cleaved via the N – C bond to form the aminyl radical 2.12.125. The key step in this domino reaction is an irreversible O → N acyl shift, followed by a 1,3-stannyl shift, radical recombination, cyclization and hydrolysis to afford the final product. The acyloxy group in azirine acts as a ‘lock’, stabilizing the primary radical product of the three-membered ring opening and creating the conditions for the radical cascade to continue.

From the analysis of the results presented in this Section, it can be concluded that the prospects for the development of synthetic methods based on functionalized azirines depend not only on the search for new reactions, but also on an in-depth study of their mechanisms. This knowledge is necessary both for the meaningful design of experiments to optimize the synthetic procedure and to provide a basis for the search for efficient transformations involving azirine derivatives. No less important are studies aimed at developing new convergent methods for the preparation of azirines diversely substituted in the three-membered ring. In terms of demand, those approaches that allow the introduction of different substituents and functional groups, either in the azirine derivative per se or in its precursor, will come to the fore. Of particular importance are the methods for obtaining azirines with highly active and energetic substituents, which are able to ensure the realization of a wide range of orthogonal transformations while preserving the azirine system. Another important role of the functional group introduced into the ring is its action as a directing group, controlling the pathways of the azirine ring expansion into larger heterocycles. Among the variety of such transformations that will undoubtedly appear in the near future, selective methods for the synthesis of structurally isomeric heterocycles from a common azirine precursor are of particular interest in terms of the synthetic potential of available azirine substrates. Finally, new growth points for azirine chemistry can be expected to emerge in the area of radical and ion-radical reactions, which is virtually unexplored for azirines.
2.13. 1,2-Alkyl shift in the construction of heterocyclic systems
The strategy of synthesis of non-aromatic heterocyclic compounds includes a variety of approaches[919-924]. Cyclizations accompanied by sextet rearrangements, including 1,2-alkyl migrations, occupy a special place among these methods. Owing to mechanistic features of reactions of this type, this method has high stereospecificity[925]; therefore, it can be used in the syntheses based on optically active compounds for the formation of new stereocenters, including quaternary ones[926, 927].
Rearrangements involving electron-deficient intermediates are a crucial tool capable of introducing substantial structural modifications in organic substrates in one-pot manner[928]. This possibility is due to the fact that the initial formation of an electron-deficient centre may induce, in particular, multistep consecutive alkyl and hydride migrations, which may act as stages of a domino-reaction. The difficulty lies in accurately predicting the structure of the products that emerge from these rearrangements, due to their inherently unpredictable nature, making retrosynthetic analysis challenging[929]. The Nametkin rearrangement is a specific example of a 1,2-shift involving the migration of a methyl group[930]. The presence of heteroatoms in functional groups and/or π-systems that are able to interact with carbon atoms adjacent to the electron-deficient centre may cause the rearrangement to proceed owing to anchimeric assistance (stabilization of the transition state during alkyl group migration).
These cyclizations can occur through various pathways with the migration of alkyl and aryl groups (or a hydrogen atom). They may be accompanied by cyclizations involving nucleophilic centres either already present in the substrate or introduced into its molecule at one of the steps of the cascade transformation (Figure 1). It is not always possible to clearly perceive the number and nature of all elementary events leading to the final reaction products[931]. According to empirical data, such cyclizations occur predominantly with the formation of the most thermodynamically favoured 5- and 6-membered rings. However, larger rings can also be formed in certain cases, albeit less frequently[932].
![[{"id":"eYtEBNBo9f","type":"paragraph","data":{"text":"Pathways of transformation of electron-deficient intermediates."}}]](/storage/images/resized/0GCZd2CbnSrbyekxfYLZbVypCIlmAIeJT8m6MiyB_xl.webp)
The theoretical justification for the observed stereospecificity can be given in two ways. On the one hand, the process can be described as a [1,2]-sigmatropic rearrangement[933]. According to the ‘aromatic transition state’ concept, these transformations are characterized by suprafacial [1,2]-migration of the alkyl group with retention of the configuration at the migrating atom and inversion of the configuration at the atoms from which and to which the migration occurs[934]. On the other hand, stereoelectronic control of the reaction leads exclusively to the migration of groups located antiperiplanar relative to the bond orbital with the leaving group in a concerted mechanism. When considering the non-concerted mechanism for this reaction, the migration takes place for those groups that can be positioned coplanar to the vacant p-orbital of the carbocation centre[935].
Until recently, cyclizations occurring during these rearrangements have not been recognized as a tool for the preparation of heterocyclic systems. The growing amount of reported material has established this type of reaction as a developing approach to the synthesis of diverse non-aromatic oxygen- and nitrogen-containing heterocycles. Particularly intriguing are cases where the reaction centres and moieties involved in the reaction are parts of a bridged or cage structure or are directly bound to it, since such reactions often result in a change in the ring size, allowing for a more comprehensive analysis of the nature of these transformations. Exploring the potential utilization of these cyclizations on other entities becomes feasible, with the objective of synthesis of products that contain either fully or partially saturated heterocyclic moieties.
Derivatives of adamantane, homoadamantane, protoadamantane and bornane are the most popular systems in this context.To date, there are numerous known compounds that contain an adamantane moiety in the molecule and have a pronounced biological effect such as antiviral, antibacterial, anti-inflammatory, antidepressant, antiparkinsonian, hypoglycemic, etc[936-948]. In turn, rearrangements of the adamantane skeleton lead to bridged structures with a different geometry, volume, and molecular symmetry. This is significant for investigating the influence of these factors on the efficacy for analogues of existing pharmaceutical agents.
These rearrangements can cause three different outcomes as regards the topological changes of the cage (Scheme 376). The first option (reaction 1) is accompanied by an increase (decrease) in the size of one ring, while in the second case (reaction 2) a mutual change in the size of two rings occurs without a change in the topology. Migrations of substituents without skeletal rearrangements (reaction 3) can be attributed to the third case. An example of a rearrangement of the first group is the transformation of the 1-adamantylcarbinyl system A (via the formation of carbocation B) into the 3-homoadamantyl system (C)[949, 950]. The transformation of 2-adamantyl cation D into protoadamantyl cation (F) adequately illustrates the rearrangements of the second group[951-953]. Typically, the third type involves a [1,2]-migration of a methyl group, referred to as the Nametkin rearrangement[954].

2.13.1. Cyclizations accompanied by rearrangements of the first type
The synthesis of γ-sultones from adamantyl-containing alkenes and alcohols is an example of synthesis of non-aromatic heterocyclic structures. The synthesis of γ-sultones fused to a homoadamantane cage from adamantane-derived secondary alcohols was first reported by Kovalev and Shokova[955]. The authors postulate the reaction goes through sulfonation of the 1-(1-adamantyl)alkene formed in situ followed by cyclization into γ-sultones accompanied by 1,2-alkyl shift.
In the sulfonation of alkenes 2.13.1, the initial formation of β-sultones A occurs through a stereospecific concerted [2 + 2]-cycloaddition (Scheme 377)[956, 957]. Subsequently, the β-sultone undergoes ring opening, followed by a skeletal rearrangement and cyclization into γ-sultone 2.13.2. In this process, the sulfur atom acts as a linker ‘extender’, while the oxygen atom bonded to sulfur serves as a nucleophilic centre. This rearrangement is also stereospecific, resulting in the formation of a single diastereomer[958, 959].

The Ritter reaction of 1-[(1E)-3-bromoprop-1-en-1-yl]adamantane (2.13.1a, R = CH2Br) gave γ-sultones 2.13.5 – 2.13.7 with a homoadamantane moiety as a result of tandem process (sulfonation – rearrangement – cyclization), along with the expected products 2.13.3 and 2.13.4 (Scheme 378)[958].

Treatment of bromoalkenes 2.13.1a,b with a mixture of sulfuric acid and acetic anhydride in methylene chloride gave bromomethylsultones 2.13.5 and 2.13.8 and γ-acetoxymethylsultone 2.13.9 (Scheme 379), which were isolated as single diastereomers. The reaction of 1-vinyladamantane (2.13.1c) and 1-[(E)-prop-1-en-1-yl]adamantane (2.13.1c) under these conditions leads to the corresponding γ-sultones 2.13.10a,b. 1-[(Z)-3-Phenylprop-1-en-1-yl]adamantane (2.13.1d) reacts with sulfuric acid in acetonitrile to give γ-sultone 2.13.11 with syn-substituents at the asymmetric centres in the heterocyclic part of the molecule[959-961].

Adamantylated cyclopropanes can also serve as substrates for such rearrangements. For example, the reaction of 1-adamantyl-1-methylcyclopropane 2.13.12 with a mixture of nitric acid and trifluoroacetic anhydride(TFAA) gives homoadamantane-fused isoxazoline N-oxide 2.13.13 (Scheme 380)[962].

The most likely mechanistic pathway for the reaction involves the isomerization of cyclopropane to generate 2-adamantylbutene A, which is converted to carbocation B under the action of trifluoroacetyl nitrate formed in situ from nitric acid and TFAA. The subsequent skeletal rearrangement and cyclization result in the formation of the homoadamantane skeleton of 2.13.13 (Scheme 381).

The synthesis of oxygen-containing heterocyclesin which nucleophile is already present in the molecule can be exemplified by the formation of a dihydro-1,3-oxazine ring fused with homoadamantane[963], which also occurs as a tandem process. The reaction of trans-2,3-disubstituted aziridine 2.13.14a (R = Me) with TFAA in toluene leads to dihydro-1,3-oxazine 2.13.15, while monosubstituted aziridine 2.13.14b (R = H) affords a mixture of dihydro-1,3-oxazine 2.13.16 and amide 2.13.17,resulting from nucleophilic cleavage of aziridine through SN2 process (Scheme 382). This difference is probably due to the steric hindrance from the methyl group in 2,3-disubstituted aziridine 2.13.14a, which makes it difficult to cleave the aziridine ring along the bimolecular pathway.

Mechanistically, this transformation involves the initial N-acylation of the aziridine to give the electron-deficient intermediate A next to the adamantyl substituent. The subsequent rearrangement and intramolecular cyclization lead to dihydro-1,3-oxazines 2.13.15 and 2.13.16 (Scheme 383).

The reaction of aryloxymethyloxiranes 2.13.19, obtained from bromide 2.13.18,with TsOH in methylene chloride is a more rare example of cyclizations with the aryl moiety as a nucleophile. The synthesis of homoadamantane-fused [3 : 4]benzo[b]oxepin-3-ol 2.13.20 was achieved through short-term heating of the starting aryloxymethyloxiranes (Scheme 384)[964]. There are two key factors to consider in this scenario. First, a seven-membered ring is generated, and second, the involvement of an aromatic system as a nucleophile sets this example apart from related reactions.

The mechanism of this reaction includes the acid-catalyzed cleavage of oxirane accompanied by a skeletal rearrangement to a homoadamantyl cation through the participation of σ-bond electrons (Scheme 385). The subsequent intramolecular Friedel – Crafts-type alkylation results in the formation of a seven-membered ring fused with homoadamantane. As observed in other scenarios, the reaction follows a stereospecific pattern and gives rise to only one diastereoisomer characterized by a syn-orientation of substituents at the neighbouring stereocentres of oxepinol 2.13.20.

Another instance of a new C – C bond formation in a heterocyclic moiety was reported by Zonker et al[965]. This involves the rearrangement of the noradamantane cage into an adamantane one. By reacting noradamantyl-1-carbaldehyde with various amines containing an aryl group, a series of imines 2.13.21 was obtained. In the presence of an acid catalyst, these imines rearranged to yield indolines, tetrahydroquinolines and benzoazepines 2.13.22. The type of product formed was determined by the length of the linker between the amino group and the aromatic ring (Scheme 386). Interestingly, even when the aromatic substituent of the substrate contained an electron-withdrawing nitro group, a similar heterocyclic product was also obtained.

The possibility of double bond participation as nucleophilic unit in a reaction was demonstrated by the following example. Tetrahydropyridines 2.13.23 linked to an adamantyl moiety through a hydroxyethyl linker can also undergo rearrangement followed by cyclization upon treatment with trifluoromethanesulfonic acid in CH2Cl2. This tandem process leads to substituted homoadamantane[3,4]-fused 1-azabicyclo[3.3.1]non-3-enes 2.13.24 (Scheme 387)[966].

The amino alcohol 2.13.23 undergoes acid-catalyzed dehydration, resulting in the formation of electron-deficient intermediate A. This intermediate then undergoes rearrangement, accompanied by the formation of a homoadamantane skeleton, and cyclization, which occurs through electrophilic addition to the double bond. The subsequent proton abstraction from B leads to the formation of an unsaturated polycyclic ammonium cation C (Scheme 388).

Cascade transformations involving alkyl migrations for the synthesis of heterocyclic compounds are also known for non-cage carbocyclic substrates. Yu et al.[967] used a cascade reaction involving up to four chemical transformations. One of the steps in this cascade was the aza-semipinacol rearrangement. A model transformation of 3-hydroxydihydroindole 2.13.25 in trifluoroacetic acid leads to the cleavage of protective groups, rearrangement and intramolecular cyclization, ultimately yielding a propellane-like product 2.13.26 (Scheme 389).

2.13.2. Cyclizations accompanied by rearrangements of the second type
Examples of acid-catalyzed transformations of homoadamantane, adamantane and a camphor homologue illustrate the rearrangements involving changes in the sizes of two rings in the cage substrates (second type rearrangements according to Scheme 376). Among them, the most extensively studied are the transformations of homoadamantane substrates with substituents arranged in some order into a structurally similar skeleton with a different arrangement of substituents. For instance, homoadamantane hydroxyester 2.13.27 (syn- and anti-isomers) undergoes an acid-catalyzed (96% H2SO4) skeletal rearrangement resulting in the formation of lactone 2.13.28 as a single diastereomer (Scheme 390)[968].

This fact suggests that the migration of the alkyl group and the formation of the lactone ring occur as a concerted reaction. The researchers also note that adding a five-fold excess of acetonitrile as a potenial carbocation trap (competing nucleophile) did not result in the expected Ritter reaction product. According to modern theories, these rearrangements occur without breaking the bond with the leaving group and instead involve the formation of an electrophilic intermediate. For instance, when the alcohol hydroxyl group gets protonated, it creates a slight electron deficiency at the reaction centre. Based on the proposed reaction pathway and geometric considerations, the product was identified as having a syn configuration.
The migration of the alkyl group is likely to be facilitated by the nucleophilic assistance of the oxygen atoms of the ethoxycarbonyl group in intermediate A (Scheme 391). However, this assistance is only possible if the migrating group and the ethoxycarbonyl group are syn-arranged; therefore, the anti-isomer of 2.13.29 is inactive.

A related rearrangement also occurs in the 4-oxohomoadamantyl system. With some assistance, the ketone can be transformed into the geminal diol equivalent, and these transformations can be categorized as retropinacol reactions[969]. The driving force of the pinacol rearrangement is the formation of a stabilized carbonyl-onium cation intermediate, which then converts into a carbonyl compound by losing a proton. The limited occurrence of the retropinacol rearrangement can be attributed to the need for promoting factors that allow for the thermodynamic stabilization of geminal diols. Examples of such factors include the formation of five- or six-membered transition state involving neighbouring heteroatomic functional groups as well as the presence of ambiphillic reagents that lead to subsequent ring formation. The simplest example is the conversion of pinacolone (2.13.30) into tetramethylethylene sulfate (2.13.31), which happens when the substrate is treated with sulfur trioxide (Scheme 392)[970].

Similarly, the reaction of homoadamantane hydroxy(oxo)ester 2.13.32 with oleum (5% SO3) yields dioxathiolane S,S-dioxide 2.13.33, which contains a heterocyclic moiety along with the lactone ring (Scheme 393). When considering this particular case, it is worth noting that no reactions took place in less concentrated sulfuric acid or in trifluoromethanesulfonic acid. The formation of a five-membered dioxathiolane ring in the reaction appears to shift the equilibrium of the reaction as a whole, like in the case of simple pinacolone[968].

In turn, the rearrangement of α-functionally substituted homoadamantane β-ketoesters 2.13.34 (n = 1, 2) occurs both in concentrated H2SO4 and in a mixture of trifluoroacetic acid and trifluoromethanesulfonic acid (20 mol.%) (Scheme 394). Lactones 2.13.35 and 2.13.36, the structure of which was confirmed by X-ray diffraction, are obtained in moderate to high yields. In this case, the presence of heteroatomic groups in the γ- or δ-position acts as a promoting factor by providing nucleophilic assistance. An interesting observation is that in the case of γ-ketoesters 2.13.34 (n = 2), it is possible to isolate intermediate product 2.13.37, which contains only one lactone ring. However, this variant is absent for compounds 2.13.34 (n = 1)[968]. This suggests that the formation of a five-membered ring has a lower energy barrier compared to a six-membered ring.

Moreover, an instance of a related rearrangement with lactonization was also described for bicyclic bridged systems. Camphor is extensively studied as a common and easily obtainable subject in the investigation of 1,2-alkyl shifts. In particular, the product of its homologation with diazoacetic ester and subsequent alkylation (2.13.38) undergoes lactonization in sulfuric acid to form bis-lactone 2.13.39 with bicyclo[2.2.2]heptane skeleton (Scheme 395)[962].

The first step is protonation of the oxygen atom of ketone 2.13.38 by mineral acid. Similarly to the above example, the initial formation of a lactone ring involving the oxygen atom of the ethoxycarbonyl group directly bound to the cage may take place. The anionotropic 1,2-alkyl migration promoted by the nucleophilic assistance from the involved oxygen atom of the ester group occurs simultaneously. This results in the formation of the bicyclo[2.2.2]octane skeleton of intermediate C (Scheme 396).

Examples of 1,2-alkyl shifts in the bornane system, for example, in compounds 2.13.40 are also known. The reaction causes the sizes of two rings to change simultaneously, resulting in the formation of a dihydroindole unit of 2.13.41 (Scheme 397). Upon cyclization, substituted aniline 2.13.40 undergoes a rearrangement of the bornane skeleton, which can be attributed to camphene-type rearrangements[971].

2.13.3. Cyclizations accompanied by Nametkin rearrangements
The third category of rearrangements, which result in the formation of non-aromatic heterocyclic compounds, can be observed in various examples of methyl group shifts within the fenchol system. Treatment of 2-substituted 1,3-dimethoxybenzene 2.13.42 with hydrobromic acid leads to a product of demethylation followed by the Nametkin rearrangement, which involves cyclization with the formation of dihydrofuran 2.13.43 (Scheme 398)[972].

Djaidi et al.[973] reported some instances of the tandem Nametkin rearrangement followed by cyclization within the context of the Ritter reaction, utilizing substituted bicyclo[3.3.1]nonane 2.13.44 as a substrate. The oxygen atom of starting keto group appears in the structure of the oxaadamantane moiety 2.13.45 upon this transformation (Scheme 399)[973].

Thus, numerous examples of sextet rearrangements resulting in the formation of non-aromatic heterocyclic systems are available from the literature. These reactions may involve changes in the sizes of one or more rings simultaneously. The nucleophilic centre may already be present in the starting compound, or it can be formed during a multistep one-pot process. While it is challenging to predict the potential use of this method in synthesizing heterocyclic compounds, it possesses a distinct advantage over other methods due to its stereospecificity. However, high yields are not always obtained. Considering the aforementioned features of this approach, it can be concluded that it not only enables the (dia)stereoselective synthesis of various non-aromatic heterocyclic compounds but also allows for the modification of polycyclic and bridged hydrocarbon skeletons by altering the size of one or two rings or the position of substituents within the polycyclic unit.
2.14. Design of heterocyclic structures based on functionalized epoxides, dicarbonyl compounds, quinoxaline-2-ones (Mamedov rearrangement) and pyrimidin-2,4-diones
This Section summarizes the research findings in the field of chemistry of heterocyclic compounds carried out at the Arbuzov Institute of Organic and Physical Chemistry (Kazan). The Section consists of 4 parts. The first part is devoted to functionalized epoxides, their transformations into larger-sized heterocycles, including pharmaceutically significant ones. It describes rearrangements of epoxy compounds, both new ones, which are complex tandem processes, and well-known ones. Among the latter, for example, the Meinwald rearrangement (MR) is the process by which epoxides are transformed into carbonyl compounds through the migration of a hydrogen atom or substituent from one carbon atom of the oxirane ring to another. If there is a carbonyl group is in the vicinity of the epoxide ring (and we have used 3-arylglycidic acid derivatives and (2-aryloxiran-1-yl)(aryl(or methyl))ketones), MR affords 1,2- or 1,3-dicarbonyl compounds, often in situ, which opens the way for further reactions. The second part concerns the transformation of 3-halo-3-aryl(or alkyl)pyruvates, which are 1,2-dicarbonyl compounds isomeric to the corresponding glycidates, into tetrahydroindoles. In the third part, rearrangements of quinoxalinones, which can be considered as heteroprotected analogues of 3-substituted pyruvates, are presented. When reacted with dinucleophiles, such compounds are transformed into hetarylbenzimidazoles and hetarylbenzimidazolones (type I and II Mamedov rearrangements). The fourth part highlights methods for the synthesis of macroheterocycles and related open-chain structures based on pyrimidin-2,4-dione (uracil) derivatives.
2.14.1. Synthesis of functionalized epoxide and their transformations
The Darzens condensation[974], known since 1904, involving aldehydes or ketones and α-mono- or dihalocarbonyl compounds in the base medium, leads to epoxides or halodicarbonyl compounds, depending on the reactants. The great German synthetic chemist Seebach[975] suggested that ‘if carbonyl compounds have been said to be virtually the backbone of organic synthesis, the epoxides correspond to at least one of the main muscles’. We were interested in epoxides as substrates for the formation of larger heterocycles[976]. Several types of epoxy compounds were synthesized from aromatic aldehydes and mono- and dihalocarbonyl compounds under Darzens condensation conditions (Scheme 400).

Strategies for the transformation of 3-arylglycidic acid anilides 2.14.1 into larger heterocyclic systems are illustrated in Scheme 401. It is known that epoxy compounds are prone to MR under acidic conditions[977]. Depending on whether the hydrogen atom of compounds 2.14.1 migrates from position 1 to 2 or from position 2 to 1 or the aryl moiety migrates from position 2 to 1, the formation of dicarbonyl compounds A, B and C can be expected. Intermediates A, B and C may undergo intramolecular closure in a Friedel-Krafts type reaction under the reaction conditions to give 3-arylideneindolin-2-ones D, 4-arylquinolin-2-ones E and 3-arylquinolin-4-ones 2.14.2. 3-Arylquinolin-4-ones were assigned the number 2.14.2 for good reason, in contrast to the letter designations for other heterocycles, because these products were obtained in good yields in dimethyl sulfate with sulfuric acid (10 : 1 (vol.%)) on heating to 100 °C (Scheme 402)[978, 979].

3-Arylglycidic acid anilides bearing a 2-positioned nitro group in the aryl moiety (2.14.3) did not afford quinolinones 2.14.2 under the conditions outlined in Scheme 402. No individual products could be isolated from such reaction mixtures, but boiling in acetic acid with the addition of a small amount of sulfuric acid (100 : 1 (vol.)) gave almost quantitative yields of unsymmetrically substituted oxalamides 2.14.4, which are otherwise difficult to obtain[980, 981]. Not only 3-(2-nitroaryl)glycidic acid anilides 2.14.3, but also 3-(2-nitrophenyl)glycidic acid amide 2.14.5, which gave rise to oxalamide 2.14.6, have been successfully used in this reaction (Scheme 403, reaction 1)[980]. In addition, the method was extended to [2-(2-nitroaryl)oxiran-1-yl](aryl)ketones 2.14.7 and, albeit in lower yields, similar products 2.14.8 were also obtained (Scheme 403, reaction 2)[980, 982]. The mechanism of this unique, all-atom-conserving and unprecedented rearrangement was studied by DFT[983]. The use of rearrangement products 2.14.4 and 2.14.6 in the synthesis of quinazolinones is discussed below.


As early as 1895, Niementowski[984] proposed the preparation of quinazolin-4-ones using anthranilic acid and carboxylic acid amides as ‘suppliers’ of nitrogen and carbon atoms. Since then, this reaction has been modified several times to increase the diversity of quinazolines synthesized in view of their great biological significance. Having in hands previously unknown anthranilic acid derivatives 2.14.4 and 2.14.6, we converted them into the quinazolinones.
Heating compounds 2.14.4 with anilines in polyphosphoric acid at 170 °C gave 2-carboxanilido-3-arylquinazolin-4-ones 2.14.9 (Scheme 404)[981]. Only two synthetic approaches to 2-carboxanilido-3-arylquinazolin-4-ones bearing other aryl substituents have been reported. In one of them, the authors obtain quinazolinones 2.14.9 only with identical aryl moieties[985], while the other is rather laborious[986]. Compounds 2.14.9 were tested for cytotoxic activity against human tumour (M-Hela) and normal (Chang liver) cell lines. The IC50 value for compound 2.14.9a (Ar1 = 4-ClC6H4, Ar2 = 2-FC6H4) when acting on tumour cells was comparable to the activity of doxorubicin, and for compounds 2.14.9b (Ar1 = Ar2 = 4-FC6H4) and 2.14.9c (Ar1 = Ar2 = 2-FC6H4) were comparable to those of tamoxifen with no toxic effect on normal cells, unlike the above drugs.

When the anthranilic acid derivative 2.14.6 was used under similar conditions, quinazolin-4-ones 2.14.10 with a free 2-position were unexpectedly formed (Scheme 405)[987]. These compounds are precursors of the central nervous system depressants methaqualone, methylmethaqualone, ethaqualone, mebroqualone and mecloqualone, which are used medicinally as sedatives and sleeping drugs[988].

The reductive cyclization of quinazolinones 2.14.10a (Ar = 2-O2NC6H4) and 2.14.10b (Ar = 2-O2N,5-ClC6H3) afforded the fused benzimidazoquinoxaline diheterocycles 2.14.11a and 2.14.11b, 6-deoxo-6-aza analogues of the tryptanthrine alkaloid (Scheme 406)[987].

The condensation of compound 2.14.6 with 1,2-diaminobenzene (DAB) 2.14.12 led to another fused heterocyclic system, the quinoxalinoquinazolines 2.14.13 (Scheme 407)[987]. This combination of heterocycles in a fused system has only been found in one publication[989].

The presence of a carboxyl group allowed the use of compounds 2.14.4 and 2.14.6 as ligands in the synthesis of binuclear copper complexes, and the polydentate nature of these ligands ensured the construction of copper-containing one-dimensional (1D) coordination polymers based thereon[990].
Heating N-protected anilides of 2-chloro-3-arylglycidic acids 2.14.14 in TFA at 65 °C led to the precipitation of quinoline-2-ones 2.14.15 (Scheme 408)[991, 992]. Note the extreme sensitivity of the process to the conditions and nature of the substituents in the substrates 2.14.14. The conditions indicated in Scheme 408 did not provide quinolinones with a free nitrogen atom. However, such products have been isolated in lower yields (55 and 57%, 2 examples) by the in situ treatment with hydrogen chloride on unprotected 2-chloro-3-arylglycidic acid anilides[993].

Under the said conditions, only quinolinones 2.14.15 with electron-withdrawing substituents in the aryl moiety were obtained (see Scheme 408). This was due to the impossibility of isolating glycidic acid anilides 2.14.14 with other Ar substituents as they rearrange to give 3-chloro-3-arylpyruvic acid anilides 2.14.16. The latter could not be converted into quinolinones 2.14.15 under the above conditions, but in boiling TFA they were converted into 3-hydroxy-3-(chloro(aryl)methyl)indole-2-ones 2.14.17 (Scheme 409)[994].

Heating N-(4-methoxybenzyl)-3-(3-nitrophenyl)-N-phenyl-2-chloroxiran-2-carboxamide (2.14.14a) in a small amount of AcOH – H2SO4 (100 : 1 (vol.)) to 56 °C (temperature at which the substrate 2.14.14a was completely dissolved) gave dihydrocycloheptanopyrrol-2,3-dione 2.14.18, the structure of which was determined by X-ray diffraction analysis. In boiling AcOH – H2SO4 or TFA, this product was quantitatively converted to quinolinone 2.14.15a (Scheme 410)[992].

4-Methylanilides 2.14.14 were rearranged to spiro compounds 2.14.19 on heating at 45 °C in AcOH – H2SO4 – MeOH mixture (100 : 1 : 2 (vol)), and the latter furnished quinolinones 2.14.15 in boiling TFA (Scheme 411)[992]. The mechanisms of transformation of 2-chloro-3-arylpyruvic acid anilides 2.14.14 to compounds 2.14.15, 2.14.18, 2.14.19 and of 3-chloro-3-arylpyruvic acid anilides to products 2.14.16 were investigated by DFT[992]. The quinolines 2.14.15 are close structural analogues of viridicatin alkaloids. Quinoline 2.14.15b was used in a one-pot synthesis of viridicatin involving Darzens condensation and Friedel – Crafts intramolecular condensation followed by deprotection of the nitrogen atom (Scheme 412)[992]. Viridicatol was prepared from quinolinoline 2.14.15a. In this case, the sequence of transformations consisted of reduction of the nitro group by sodium dithionite, diazotization of quinolinone 2.14.20 by sodium nitrite in the presence of sulfuric acid with release of nitrogen, and deprotection of the nitrogen atom of quinolinone 2.14.21 (Scheme 413)[992].



A three-component process involving 3-arylglycidamides 2.14.22, hydrogen halides (HBr, HCl) and acetone at room temperature gives 5-(α-halobenzyl)-2,2-dimethyl-1,3-oxazolidines 2.14.23, which are converted into 5-arylideneoxazolidines 2.14.24 under the action of K2CO3 in MeOH (Scheme 414)[995-998].

Oxazolidinones 2.14.24 are hydrolyzed in acidic medium to 2-hydroxy-3-arylacrylic acid[996], which reacts in situ with 1,2-DAB to form 3-benzylquinoxalinones 2.14.25 (Scheme 415)[997].

3-(2-Nitrophenyl)pyruvic acid and its derivatives (2.14.26), obtained by hydrolysis of 2,2-dimethyl-5-(2-nitrobenzylidene)-1,3-oxazolidine 2.14.24a in concentrated hydrochloric acid or in alcohols, were used in the synthesis of indole-2-carboxylic acid, its amide and esters (2.14.27) (Scheme 416)[998].

Aryl glycidates 2.14.28 react with 1,2-DAB 2.14.12 in boiling AcOH (Scheme 417) to give, depending on the substituent in the aryl moiety, anti-dihydrodiazepinones 2.14.29 with high diastereoselectivity (the formation pathway of these compounds is coloured blue in the Scheme 417) or benzylquinoxalinones 2.14.25. The latter compounds are formed in the case of glycidates 2.14.28 containing electron-donating substituents in the aryl moiety, which is associated with their MR to give 3-arylpyruvates in the initial stage of the process[999].

Under reductive cyclization conditions mediated with sodium dithionite, epoxy compounds 2.14.7 are converted into 3-hydroxyquinolines 2.14.30 (Scheme 418)[1000-1004].

Heating 2-[(2-nitrophenyl)oxiran-1-yl](phenyl)ketone (2.14.7b) under reflux in an AcOH – H2SO4 mixture (100 : 1 (vol.)) gives 3-hydroxy-2-phenylquinoline-4-one (2.14.31a) (Scheme 419)[992] together with the product 2.14.8a (see Scheme 403, reaction 2). Other (oxiranyl)(aryl)ketones 2.14.7 rearrange under similar conditions to compounds 2.14.8 and furnish quinolinones 2.14.31 only in trace amounts.

Thus, the wide possibilities of functionalized epoxides and their transformation products in the synthesis of larger-sized heterocycles are illustrated.
2.14.2. New methods for the synthesis of tetrahydroindoles
The reaction between N-(cyclohexen-1-yl)pyrrolidine 2.14.32a and aryl chloropyruvates 2.14.33 formed the basis of the one-pot procedure developed for the synthesis of functionalized tetrahydroindoles, namely of 1-(4-chlorobutyl)-2-aryl-4,5,6,7-tetrahydroindole-3-carboxylic acid esters 2.14.34 (Scheme 420). The method comprises the following steps:
1) intramolecular nucleophilic substitution by SN2 mechanism to form an epoxide ring and elimination of Cl–,
2) pyrrolidinone ting opening in the intermediate A under the action of Cl–,
3) imine-enamine tautomerism B ⇄ C,
4) the epoxide ring opening with closure of pyrrolidinone D and
5) elimination of water.

Although aryl chloropyruvates 2.14.33[1005, 1006] used in the Stork reaction[1007] no products of the above reaction, compounds 2.14.35, were detected.
The use of N-(cyclohexen-1-yl)piperidines (2.14.36: R1 = H (a), But (b)) (Scheme 421, reaction 1)[1008] and morpholine (2.14.37) (cf. Scheme 421, reaction 2)[1009] as nucleophilic agents in reactions with various 3-halo-3-aryl(or alkyl)pyruvates leads to tetrahydroindoles with 5-halopentyl (2.14.38) and 2-(2-chloroethoxy)ethyl (2.14.39) substituents at the nitrogen atom. The reaction of ethyl 3-bromopyruvate 2.14.40 with various enamines, N-(cyclohexen-1-yl)-pyrrolidines (2.14.32: R1 = H (a), But (b)), piperidines (2.14.36a,b) and morpholine (2.14.37), gave 1-substituted ethyl tetrahydroindole-3-carboxylates 2.14.41 (see Scheme 421, reaction 3)[1010]. Reacting tetrahydroindoles 2.14.34, 2.14.38 and 2.14.41 with chloranyl in boiling xylene gave the corresponding indole derivatives 2.14.42 (Scheme 422)[1008, 1010].

Consequently, in contrast to the Stork reaction, where enamines act as effective enol synthons promoting alkylation or acylation of carbonyl compounds (aldehydes or ketones) at the α-carbon atom, in the reaction with halopyruvates, enamines give 4,5,6,7-tetrahydroindoles as a result of rearrangements.

The reactions of 3-(α-chlorobenzyl)quinoxalin-2-ones 2.14.43 with cyclohexenylpyrrolidines 2.14.32a,b afford 8,9,10,11-tetrahydroindolo[1,2-a]quinoxalin-6-ones 2.14.44 via a tandem process involving Stork alkylation of the enamine and intramolecular annulation (Scheme 423)[1011]. When the product 2.14.44a (R1 = R2 = R3 = H; Ar = Ph) reacts with chloranyl in boiling xylene, the corresponding indoloquinoxalinone are obtained.

To summarize, a new simple one-pot approach to tetrahydroindolo[1,2-a]quinoxalin-6(5H)-ones and products of their aromatization has been developed.
2.14.3. Rearrangements of quinoxalinones to hetarylbenzimidazoles and hetarylbenzimidazolones under the action of binucleophilic reagents (types I and II Mamedov rearrangement)
A new rearrangement in the 3-benzoylquinoxalinone (2.14.45a) – 1,2-diaminobenzene (2.14.12a) system has been discovered, which is induced by AcOH and proceeds with contraction of the pyrazine ring of quinoxaline to give the benzimidazole derivative 2.14.46a (Scheme 424)[1012]. The carbon numbering in the fragments coloured red show their migration in the course of the rearrangement. The pyrazine ring contraction involves the α-carbon atom of the substituent at the 3-position of the quinoxalinone system.

The rearrangement is applicable to a wide range of substrates and features high chemoselectivity. Reactions of 3-aroyl(alkanoyl)quinoxalinone derivatives 2.14.45 with 1,2-DAB 2.14.12 in acetic acid afford 2-(benzimidazol-2-yl)quinoxalines 2.14.46 diversely substituted at the 3-position of the quinoxaline (Scheme 425)[1012-1022]. It should be noted that this rearrangement tolerates the presence of a substituent on the N(1) atom of the substrate. The reaction is effective with both NH- (e.g. with compound 2.14.47) and N-alkylquinoxalinones, yielding the corresponding benzimidazole derivatives[1013-1015]. The presence of a carboxyl group in compound 2.14.46 (R1 = CO2H; R2 = R4 = H; R3 = Ph) allows the introduction of another benzimidazole moiety via the Phillips – Ladenburg reaction to give compound 2.14.48 (see Scheme 425)[1017]. In addition, the reaction also works well with compounds containing two quinoxalinone substituents, giving monopodands with terminal quinoxalinyl benzimidazole moieties[1020].

3-(Benzimidazol-2-ylcarbonyl)quinoxaline-2(1H)-one 2.14.47 reacts with 1,2-DAB in AcOH to provide high yields of 2,3-bis(benzimidazol-2-yl)quinoxalines 2.14.49 (see Scheme 425)[1023, 1024], which had an unexpected dynamic effect on the steady-state electronic absorption spectra[1025]. PEGylated (PEG is polyethylene glycol) liposomes loaded with 2-(benzimidazolyl)quinoxalines 2.14.46b – e (R1 = R4 = H; R3 = Ph: R2 = Me (b), Bun (c), n-octyl (d), dodecyl (e)) were found to be cytotoxic against the M-Hela tumour cell line (human cervical carcinoma)[1018]. The viability of 2-(benzimidazolyl)quinoxaline-loaded 2-(benzimidazolyl)quinoxaline-arginine liposomes on the M-Hela cells was 16% at a concentration of 0.15 mg ml–1. These liposomes showed lower toxicity (40%) against a normal human liver cell line (Chang liver). A mixture of regioisomeric 2-(benzimidazol-2-yl)-3-(4-fluorophenyl)-6(and 7)-(4-methylpiperazin-1-yl)quinoxalines (2.14.46f : 2.14.46ʹf = 1 : 1) showed high (IC50 = 2.8 μM) cytotoxicity against the A549 (human lung adenocarcinoma) cell line, comparable to that of the commercial anticancer drug doxorubicin (IC50 = 3.0 μM). In contrast to doxorubicin (IC50 = 1.3 μM), which was toxic to the human fibroblast cell line, this mixture of regioisomers showed only low cytotoxicity (IC50 = 34 μM) and did not cause cell hemolysis (IC50 ˃ 100 μM)[1019].
Not only 1,2-DAB and its fused derivatives, but also its aza analogues such as 5,6-diamino-2-sulfanylpyrimidin-4-ol, 2,5,6-triaminopyrimidin-4-ol, diaminomaleonitrile (as an open-chain analogue of 1,2-DAB) and arylmethanediamine, prepared in situ from various aromatic aldehydes and ammonia, react readily with 3-aroylquinoxalinones 2.14.45 to give the corresponding rearrangement products, benzimidazolylpteridines 2.14.50, 2.14.51[1026], 6-(benzimidazol-2-yl)pyrazine-2,3-dicarbonitriles 2.14.52 (see[1027]) and polysubstituted imidazolylbenzimidazoles 2.14.53[1028]. Liposomes with encapsulated (benzimidazol-2-yl)pteridin-4(1H)-one 2.14.50a (R1 = H, R3 = 2,4-Cl2C6H3) were shown to be cytotoxic towards the M-Hela cell line at the level of doxorubicin, but less toxic (37-fold) towards the normal Chang liver cell line[1029].

The putative mechanism of the above rearrangement is shown in Scheme 426. Spiro derivatives similar to structure A[1028, 1030-1033] and 2-aminoanilides of 3-arylquinoxaline-2-carboxylic acids similar to structure B[1016, 1017, 1028] have been isolated from reactions of quinoxaline-2-ones with other binucleophilic reagents. DFT Free energy calculations[1034] for the acetic acid-catalyzed rearrangement of 3-benzoylquinoxaline-2(1H)-one 2.14.45a under the action of 1,2-DAB 2.14.12a also support this mechanism.

The elucidation of the rearrangement mechanism has allowed an original suggestion to be made. The data provided indicate that 3-alkanoyl(aroyl)- (2.14.45) and hetaroylquinoxaline-2-ones (2.14.47), when reacting with 1,2-DAB 2.14.12 (see
We tested this hypothesis using several examples. We suggested that 3-(α-halobenzyl)- (2.14.43)[1030], 3-(α-aminobenzyl)- (2.14.54)[1033], 3-(2-oxo-2-arylethyl)- (2.14.55)[1031], 3-(2-aminophenyl)- (2.14.56)[1035], 3-(2-aminostyril)- (2.14.57, formed in situ from the 2-nitrophenyl analogue)[1036], 3-(2-aminobenzyl)- (2.14.58, formed in situ from the 2-nitrobenzyl analogue),1032, 1037 quinoxalinones are heteroanalogues of α-halogen ketones, α-amino ketones, β-diketones, 2-aminobenzaldehyde (formed in situ from 2-nitrobenzaldehyde), 3-(2-aminophenyl)acrylaldehyde (formed in situ from 3-(2-nitrophenyl)acrylaldehyde) and 2-(2-aminophenyl)acetaldehyde, respectively. As can be seen from generalized Scheme 427 (reactions 1 – 6), the introduction of a specific substituent at the 3-position of the quinoxalin-2-one, the use of appropriate reagents and the selection of optimal conditions opened the way to various biheterocyclic systems such as 2-(indolizin-2-yl)benzimidazoles 2.14.59[1030], 2-(pyrrole-3-yl)benzimidazoles 2.14.60[1033], 2-(pyrazol-3-yl)benzimidazoles 2.14.61[1031], 4-(benzimidazol-2-yl)quinolines 2.14.62[1035], 2-(benzimidazol-2-yl)quinolines 2.14.63 (see[1036]) and 2-(indole-2-yl)benzimidazoles 2.14.64 (see[1034, 1037]) with various substituents.

The reaction of 3-[methyl(2-phenylhydrazono)methyl]quinoxalin-2-ones 2.14.65 in PPA gives 3-(indol-2-yl)quinoxalin-2-ones 2.14.66 and 4-(benzimidazol-2-yl)-3-methylcinnolines 2.14.67 via two competing rearrangements, the [3,3]-sigmatropic Fischer rearrangement yielding the indole ring and the Mamedov rearrangement with simultaneous construction of the benzimidazole and cinnoline rings (Scheme 428)[1038]. When 3-[aryl(2-phenylhydrazono)methyl]quinoxalin-2-ones are used instead of 3-methyl analogues, the process is regioselective and affords only Mamedov rearrangement products, compounds 2.14.68 (see Scheme 428).

In all the above examples, 3-substituted quinoxalinones are rearranged when subjected to nucleophilic agents under acid catalysis to benzimidazole derivatives bearing different C(2)-hetaryl substituents. The formation of the benzimidazole system occurs by the pyrazinone ring contraction in quinoxalin-2-one and a new heterocyclic system is formed involving a nucleophilic reagent, the quinoxalinone C(3) atom and a substituent on the C(3) atom providing one[1033, 1038, 1012-1031] or several[1032, 1033, 1036, 1037] carbon atoms to construct a new heterocycle.
The use of the cyano group as a substituent at the 3-position of the quinoxalinone, due to its acceptor character and good leaving ability[1039], makes possible an attack of both amino groups of 1,2-DAB on the same carbon atom (C(3)) of the quinoxalinone 2.14.69, thus enabling the formation of a benzimidazole moiety via the intermediate spirobenzimidazole-2,2’-quinoxalinone A (Scheme 429)[1040]. In this case, the substituent at the position 3 of the quinoxalinone does not contribute any atoms to the resulting heterocyclic system. The rearrangement of the spirobenzimidazole-2,2'-quinoxalinones A gives rise to a large series of 2,2'-bis(benzimidazole) derivatives 2.14.70.

Spiro-quinoxalinones can also be obtained from other heterocyclic systems. For example, 3-chloro-2-oxoindolines 2.14.71 derived from isatinones 2.14.72 have been reacted with 1,2-DAB 2.14.12a and its derivatives, both symmetrically disubstituted (2.14.12: R2 = Me (b), Cl (c)) and asymmetrically monosubstituted (2.14.12: R2 = H, R3 = Me (e); R2 = Me, R3 = H (f); R2 = Cl, R3 = H (g))[1041, 1042]. The intermediate spiro-quinoxalinones 2.14.73 were rearranged by boiling for 6 h in AcOH to form mixtures of individual and regioisomeric 4-(benzimidazol-2-yl)quinolin-2(1H)-ones 2.14.74 resulting from tautomerism of benzimidazoles[1017, 1041] and different positions of substituents in the benzimidazole moieties (Scheme 430)[1041].

In all the above reactions, summarized in Scheme 425 and Scheme 427, the final products, 2-hetarylbenzimidazoles, are obtained via the rearrangement of the spiro-quinoxalinones (sQ1) through the intermediate 2-aminoanilides (An) of the corresponding heterocyclic acid (Scheme 431). In these cases, the spiroquinoxalinones sQ1 contain at least one mobile hydrogen atom in the spirocyclic moiety and it is this hydrogen atom, coloured green in Scheme 431, that is responsible for the rearrangement[1043-1046].

In 2012, the book ‘Organic Syntheses Based on Name Reactions’ was published by Hassner and Namboothiri[1043], in which the rearrangement of quinoxalinones according to the above schemes was named ‘MAMEDOV Heterocycle Rearrangement’.
Our further studies showed that the synthetic potential of quinoxalinones is not limited to this rearrangement alone. For example, the question arose as to whether the presence of a mobile hydrogen atom in the spiro unit was necessary, and if not, how would its absence affect the process? To answer this question, it was necessary to synthesize quinoxalinone spiro derivatives devoid of mobile hydrogen atoms. After analyzing all possible nucleophilic agents that could provide such products, we settled on enamines.
The reactions of 3-aroyl(alkanoyl)quinoxalin-2(1H)-ones 2.14.45 with commercially available enamines, namely methyl- (2.14.75a) and ethyl-3-aminocrotonates (2.14.75b) in AcOH, were found to proceed in a non-standard manner, giving a regioisomeric mixture of 1-(pyrrole-3-yl)- (2.14.76) and 1-(pyrrole-2-yl)benzimidazolones (2.14.77) with a significant predominance of the former (Scheme 432)[1047], i.e. no 2-(pyrrole-2-yl)benzimidazoles is not observed as would be the case if the above rearrangement took place (see Scheme 431).

It should be noted that the reaction of 3-benzoylquinoxalin-2(1H)-ones 2.14.45 with enamines A generated in situ from various ketones and ammonium acetate in boiling MeOH produces a single isomer, 1-(pyrrole-2-yl)benzimidazol-2-ones 2.14.78 (Scheme 433)[1048], which are minor products when commercial enamines 2.14.75 are used (see Scheme 432).

Assuming that the three-atom unit of enamine is part of amidines and iminoesters and can be easily formed by the reaction of amines and nitriles with an active methylene group, we have studied the reactions in a multicomponent system containing 3-aroylquinoxalin-2-ones, malononitrile and various nucleophilic reagents (amines and alcohols) in the presence of AcOH. In this way, we sought to determine the possibility of synthesizing some derivatives of 1-(pyrrolyl)benzimidazolones[1049].
The three-component domino reaction of 3-aroylquinoxalin-2(1H)-ones 2.14.45 with malononitrile and secondary amines or primary alcohols gives two easy-to-isolate products, 5-(benzimidazol-2-one-1-yl)pyrrol-3-carbonitriles 2.14.79 and pyrrolo[1,2-a]quinoxalin-4(5H)-one carbonitriles 2.14.80 in high total yield (Scheme 434).

As a result, we have discovered two fundamentally new transformations in the quinoxalinone series. The first is the acid-catalyzed rearrangement of quinoxalin-2-ones (Q) to 2-hetarylbenzimidazoles (BI) induced by nucleophilic agents and proceeding via o-aminoanilides (An) of the corresponding hetarylcarboxylic acids, which are formed as a result of N(1)C(2) bond cleavage in spiro derivatives of quinoxalin-2-ones (sQ1) bearing at least one mobile hydrogen atom in the spiro-forming unit (see Scheme 431) (MAMEDOV heterocycle rearrangement[1043], or type I MAMEDOV rearrangement). The second is the acid-catalyzed rearrangement of quinoxalin-2-ones (Q) to 1-hetarylbenzimidazolones (BIon), promoted by nucleophilic agents, via N-(2-isocyanatophenyl)hetaryl-2-amines (Is), formed through the C(3) – C(2) bond cleavage in spiro derivatives of quinoxalin-2-ones sQ2 which contain no mobile hydrogen atom in the spiro-forming unit (Scheme 435) (type II Mamedov rearrangement)[1044, 1046, 1050-1052].

The key step in both rearrangements is the formation of spiro-quinoxalinones, which can be synthesized based not only on quinoxalinone derivatives and dinucleophilic reagents (see Scheme 431, Scheme 435), but also on other heterocyclic systems (see Scheme 430). It should be noted that in the formation of spiro compounds from quinoxalinone derivatives, the functional group may supply one, two or more carbon atoms to the spiro unit, or none at all, but still participate in its formation, as shown in Scheme 429 on the example of 3-cyanoquinoxalin-2(1H)-ones. Alternatively, any leaving group, even a hydrogen atom, may be present in the substrate in place of the cyano group[1053], since quinoxalin-2-one is a π-deficient heteroaromatic system[1054].
Thus, among the synthetic approaches to (hetaryl)benzimidazoles, the acid-catalyzed rearrangements of 3-substituted quinoxalinones under the action of N,N- and N,C-binucleophiles, known as Mamedov rearrangements, occupy an important place and provides an approach to a diversity of 2-(hetaryl)benzimidazoles and 1-(pyrrolyl)benzimidazolones. The simple implementation and general nature of these rearrangements determine their importance in the creation of complex biheterocyclic structures — precursors of novel drugs.
2.14.4. Macroheterocycles and related open-chain structures based on pyrimidin-2,4-diones
Derivatives of nucleotide bases, including pyrimidin-2,4-dione (uracil), are important targets in the search for highly efficient biologically active compounds. Fragments of uracil have been introduced into macrocyclic structures, named pyrimidinophanes or heterocyclophanes in analogy to cyclophanes, and the structural features, self-association ability and biological properties of the resulting products have been studied. For example, pyrimidinophanes of various compositions and structures were synthesized from 1-bromoalkyl-6-methyluracils (2.14.81), 1,3-bis(α,ω-bromoalkyl)uracils (2.14.82), 1,3-bis(α,ω-ethylaminoalkyl)uracils (2.14.83) and α,ω-bis(3,6-dimethyluracilyl)alkanes (2.14.84).

In combination, these compounds were reacted with each other or with bridging reagents, α,ω-dibromoalkanes, dibromoxylylenes, formaldehyde. This furnished a large library of pyrimidinophanes containing one (macrocycles 2.14.85, 2.14.86)[1055-1059], two (regioisomeric macrocycles 2.14.87 and 2.14.87' (see[1059-1062]) and three (macrocycle 2.14.88)[1063] uracil moieties.

Dimer 2.14.84 reacts with paraformaldehyde to give pyrimidinophanes 2.14.89 with four 3,6-dimethyluracil moieties and also oligomerization products 2.14.90a,b. The dimeric moieties in macrocycle 2.14.89 are methylene-bridged through the C(5) atoms of the pyrimidine rings (Scheme 436)[1064].

Pyrimidinophanes 2.14.91 and 2.14.92, which contain not only oxopyrimidine rings but also amino- and thio-substituted pyrimidine moieties, were obtained from dibromide 2.14.82 (R1 = Me, R2 = H, n-C10H21) and the dysodium salt of N,N'-bis(4-methyl-2-sulfanylpyrimidin-4-yl)alkylenediamine (2.14.93) (Scheme 437)[1065-1067]. Pyrimidinophanes 2.14.91 and their acyclic counterparts are highly efficient ligands for silver ions. Macrocycles 2.14.91 are extremely selective and efficient in extracting silver ions even from multicomponent mixtures of metal ions, and form stable complexes therewith[1067].

Other interesting properties of such macrocycles should be noted. Pyrimidinocyclophanes 2.14.85 are able to respond to external influences, reversibly changing their conformational state in solutions as a result of protonation and deprotonation of the nitrogen atom in the spacer[1056]. This has allowed them to be used to create supramolecular ensembles, the structure of which is controlled by the pH of the medium[1058]. Pyrimidinophanes 2.14.86 inhibit acetylcholinesterase at nanomolar concentrations. In vivo experiments in rats have shown that some of these compounds effectively alleviate the symptoms of the autoimmune disease myasthenia gravis[1059]. Pyrimidinophanes 2.14.87, 2.14.87', 2.14.92 have high antibacterial activity (values of minimum inhibitory concentration against gram-positive bacteria < 1 μg ml–1)[1060, 1062, 1068] and the ability to self-organize at concentrations well below 1 mM. In aqueous solutions, such macrocycles form nanoscale aggregates with inhibitory or catalytic activity (2 – 1000-fold change) towards the hydrolysis of phosphonic acid esters[1061, 1068].
In addition to pyrimidines, other heterocyclic compounds can be incorporated into macrocycles. Thus, on based on dibromides 2.14.82, heterocyclophanes 2.14.94 – 2.14.96 containing not only uracil but also 1,3,4-thiadiazole and benzimidazole moieties have been obtained[1069].

When treated with sodium azide in DMF, dibromides 2.14.82 (R1 = Me, R2 = H) were converted to 1,3-bis(α,ω-azidoalkyl)uracil 2.14.97, from which 1,2,3-triazole-containing heterocyclophane 2.14.98 was prepared by CuAAC reaction (Scheme 438)[1070]. Condensation of diazide 2.14.97 with dec-1-yne or propargyl alcohol followed by alkylation of the 1,2,3-triazole moiety gave acyclic compounds 2.14.99 (see Scheme 438) with antimicrobial and aggregation properties[1071, 1072].

Similarly, replacement of the Br atom in the bromide 2.14.81 (R = Me) with an azido group, followed by condensation of the azide with propiolic acid hydroxy amide yielded a series of derivatives of 1,2,3-triazole-4-hydroxamic acid 2.14.100, which act as reactivators of acetylcholinesterase (AChE) poisoned with phosphorus-containing compounds. This synthetic procedure was extended to another nucleic base, adenine. The 1,2,3-triazole 2.14.101 and regioisomers of 1,2,4-triazolehydroxamic acid 2.14.102 and 2.14.103, obtained from bromide 2.14.81 (R = Me) and 9-bromalkyladenine, in which the uracil and adenines cycles are linked to the 1,2,4-triazole ring by a polymethylene bridged via N(1) or N(2) triazole atoms, were found to have the same properties[1073, 1074].

Therefore, bromalkyl and aminoalkyl uracils, as well as fused uracil derivatives provided access to a variety of structurally diverse compounds with biological activity and aggregation or complexation properties.
Summarizing the material presented in this Section, the main advantages of the research carried out by the authors can be highlighted:
— a new method for the synthesis of 3-arylquinolin-2-ones based on 3-arylglycidic acid anilides has been developed;
— a new efficient rearrangement of 3-arylglycidamides was discovered, providing a simple approach to N-(2-carboxyphenyl)oxalamides, which are excellent starting compounds for the synthesis of quinazolin-4-ones;
— the method for the preparation of a wide range of 2-aryl-3-arylglycidic acid anilides was optimized and new approaches to 3-hydroxy-4-arylquinolin-2-ones, including the natural alkaloids viridicatin and viridicatol, were proposed on their basis;
— 3-benzylquinoxalin-2-ones and indole derivatives were prepared based on 3-arylglycidamides and their heteroprotected analogues, 5-(α-halobenzyl)- and 5-arylideneoxazolidin-4-ones;
— it was found that, depending on the nature of the substituents in the aryl moiety, methyl-3-arylglycidates are converted to 3-hydroxy-4-phenyl-4,5-dihydrobenzo[1,4]diazepin-2-ones or 3-benzylquinoxalin-2-ones;
— the conversion of [2-(2-nitrophenyl)oxiran-1-yl](aryl(or methyl))ketones to 3-hydroxy-2-aryl(or methyl)quinolines has been carried out;
— a new rearrangement in the system enamine — 3-halogen-3-aryl(or alkyl)pyruvate was found, opening the way to 4,5,6,7-tetrahydroindoles, which are otherwise inaccessible;
— it was shown that N-cyclohexenylpyrrolidine can act as a source of the cyclohexane moiety when constructing tetrahydroindolo[1,2-a]quinoxalines;
— new rearrangements in the quinoxalinone series promoted by binucleophilic reagents (types I and II Mamedov rearrangement) have been discovered;
— based on pyrimidin-2,4(1H,3H)-dione (uracil) derivatives, a series of macroheterocycles and related open-chain structures with significant biological activities, promising for use in medicine, have been synthesized.
2.15. Linearly conjugated, annulated and branched thiophene-containing oligomers for organic electronics, optoelectronics and photonics
A wide range of π-conjugated organic molecules of different molecular structure and topology with interesting electronic, optical and semiconductor properties have been obtained on the basis of thiophene[1075]. The type of conjugation (linearly conjugated, annulated or their combination), the molecular topology (linear or branched) and the introduction of certain electron-withdrawing moieties or functional groups into the conjugation chain enables well-targeted control over the physicochemical properties of such materials, which are required for specific applications in organic electronics, optoelectronics and photonics. This Section highlights the main synthetic approaches and properties of linearly conjugated, annulated and branched thiophene-containing oligomers developed at the Enikolopov Institute of Synthetic Polymeric Materials of the Russian Academy of Sciences. The use of these compounds in organic field-effect, light-emitting and electrochemical transistors (OFET, OLET and OECT, respectively), gas and liquid sensors, as well as in organic and hybrid photovoltaics, bioelectronics and photonics is demonstrated.
2.15.1. Thiophene-containing linearly conjugated and annulated structures
The interest in linearly conjugated thiophene-phenylene oligomers is due to their good semiconducting and luminescent properties, which are relevant for the application of such materials in various organic electronic and photonic devices[1076]. Various cross-coupling reactions forming a new C – C bond have been used to synthesize the oligomers[1077]. This gave rise to a large library of oligomers which made it possible to identify a number of important structure-property patterns for compounds in this class. Among them, one can distinguish oligomers 2.15.1a – h containing a conjugated core of 4 aromatic units (5,5'-diphenyl-2,2'-bithiophene, PTTP) and fluorine atom or methyl, trifluoromethyl, decyl (Dec), methoxyl, tert-butyl, trimethylsilyl groups as peripheral substituents; oligomers 2.15.2a – c containing a conjugated core of 5 aromatic units (2,2'-(1,4-phenylene)bis(5-phenylthiophene), PTPTP) with decyl or trimethylsilyl substituents, and oligomers 2.15.3a – c containing a conjugated core of 5 aromatic units (PTPTP) with different numbers (from 1 to 3) of perfluorinated benzene rings (PF)[1078-1081].

Compound 2.15.1h (TMS – PTTP – TMS)[1078, 1079] was synthesized by three different routes (Scheme 439, part a). The first involved the Suzuki coupling of 5,5'-dibromo-2,2'-bithiophene with the organoboron derivative of trimethyl(phenyl)silane 2.15.4a. The second approach was based on the Kumada reaction, in which the organomagnesium derivative of trimethyl(4-bromophenyl)silane formed in situ was reacted with 5,5'-dibromo-2,2'-bithiophene in the presence of a Pd catalyst. The third method was an oxidative coupling of the lithium derivative of trimethyl[4-(2-thienyl)phenyl]silane generated in situ. All three methods provided high yields of the products.

The study of compounds 2.15.1 has shown that terminal substituents are weakly involved in the π-conjugated PTTP system, resulting in negligible changes in the absorption and emission maxima (within 0.1 eV) in the spectra of this series of oligomers. However, even a small spreading of the frontier molecular orbitals onto the substituents can almost double the photoluminescence quantum yield (PLQY) observed for But – PTTP – But and TMS – PTTP – TMS solutions (2.15.1g,h)[1080]. In addition, changing the nature of the terminal groups of the conjugated molecules that form the semiconductor active layer in organic electronic devices can change the type of the majority charge carriers. For example, organic field-effect transistors (OFETs) based on Me – PTTP – Me (2.15.1c) exhibited hole conduction (p-type), while devices based on CF3 – PTTP – CF3 (2.15.1d) exhibited electronic conduction (n-type). Meanwhile, OFETs using the TMS – PTTP – TMS oligomer (2.15.1h) showed ambipolar charge carrier transport.1081
A study of dilute solutions of TMS – PTTP – TMS and single crystals, both pure and doped with separately synthesized TMS – P4TP – TMS dopant, showed that PLQY increases from 20 to 60% when switching from solutions to single crystals[1079, 1082]. It was found that side reactions of ligand exchange, which occur in the synthesis of asymmetric conjugated oligomers by cross-coupling on Pd catalysts, can be targeted to produce molecularly self-doped oligomers in a one-pot mode (Figure 2)[1079]. The luminescence of organic semiconducting single crystals grown from such oligomers is determined by the nature of the dopant, despite its low content (< 1%). This is due to the efficient transfer of electronic excitation energy from the molecules of the target oligomer (donor) to the molecules of the dopant (acceptor) uniformly distributed in the crystal lattice of the donor, as well as due to exciton transport. The concept of molecular self-doping is quite universal and provides a simple route to highly efficient luminescent materials for organic optoelectronics[1083, 1084].
![[{"id":"XE5YHjHqI_","type":"paragraph","data":{"text":"The process of molecular self-doping in the oligomer synthesis by the Suzuki reaction (a), occurring in a one-pot mode (b), and the scheme of electron excitation energy transfer from the tar- get oligomer (donor) to the dopant oligomer (acceptor) in a single crystal (c)."}}]](/storage/images/resized/sn3M1Ohhci6H31kLEXvb1pl0CM9EWWzbUHLpLsrs_xl.webp)
The approaches to oligomers 2.15.2 containing five linearly conjugated aromatic fragments are similar to those described above. In particular, two methods have been developed for the synthesis of compound 2.15.2c (TMS – PTPTP – TMS) (see Scheme 439b)[1085], which minimize the formation of by-products with longer conjugation lengths and facilitate the purification of the oligomers. The first can be identified as a [2 + 1 + 2] coupling. In this case, the sequential treatment of trimethyl[4-(2-thienyl)phenyl]silane with n-butyllithium and 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (IPTMDOB) first gave to the organoboron derivative 2.15.4b, {4-[5-[5-(4,4,5,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-thienylphenyl(trimethyl)silane, which was subjected to the Suzuki coupling with p-dibromobenzene to give the target oligomer 2.15.2c in moderate yield[1085].
One of the disadvantages of the Suzuki reaction is the formation of a dimerized organoboron derivative by-product. In this case, oligothiophene-phenylene with four conjugated aromatic rings and a terminal trimethylsilyl group is formed. The separation of the main product from the by-product is very laborious, therefore an alternative method has been developed which can be described as a [1 + 3 + 1] coupling. 1,4-Bis(2-thienyl)benzene was first synthesized via the Kumada reaction between 2-thienylmagnesium bromide and p-dibromobenzene. Lithiation of compound 2.15.5 followed by its treatment with IPTMDOB gave the organodiboron derivative 2.15.6[1085]. The latter was subjected to the Suzuki coupling with trimethyl(4-bromophenyl)silane and after purification gave oligothiophenephenylene TMS – PTPTP – TMS in high yield.
Organic light-emitting transistors (OLETs) based on compound 2.15.2c showed ambipolar charge carrier transfer with balanced electron and hole mobility and efficient electroluminescence, while devices based on single crystals of this oligomer exhibited linearly polarized electroluminescence with a polarization degree of 0.78 ± 0.06. Single crystals of TMS – PTPTP – TMS have high emission ability despite weak H-aggregation (contrary to the common belief that J-aggregation is required for this effect). In addition, the surface molecular packing of the oligomer was shown to weaken the waveguiding effect and enhance surface emission[1086].
Macroscopic two-dimensional (2D) single crystals in the form of monolayers and multilayer films were grown from Dec – PTTP – Dec (2.15.1е) and Dec – PTPTP – Dec (2.15.2b) solutions. Polarization sensitive photoluminescence microscopy revealed the orientation of conjugated cores in such 2D crystals relative to their facets and showed that the molecules are H-aggregated. Two-dimensional organic field-effect transistor (2D-OFET) based on both oligomers showed good stability and their properties were fully consistent with the Shockley model with charge carrier mobility ( m) up to 0.2 cm2 V–1 s–1, which is one of the best values for different materials of linearly conjugated oligomers[1087]. The highest μ value was demonstrated for monolayer devices, indicating high molecular order in monolayer single crystals. Two-dimensional organic light-emitting transistors (2D-OLETs) based on oligomers 2.15.1e and 2.15.2b showed that the introduction of a 1,4-phenylene ring in the centre of the conjugated PTTP core improves the thermal stability, two-dimensional charge transfer and electroluminescence efficiency of the material. Higher thermal stability and better electrical properties of Dec – PTPTP – Dec arise from stronger intermolecular interactions in this molecule due to larger transition dipole moments, while better electroluminescence efficiency is due to the higher PLQY. The great shelf life stability of such OFETs, combined with the flexibility and optical transparency inherent in 2D materials, make these oligomers promising materials for 2D optoelectronics.
To study the influence of perfluorinated benzene rings in conjugated oligomers on their properties, compounds 2.15.3a – c containing different numbers of such units were synthesized (Scheme 440)[1088]. The reaction sequence involved a combination of organometallic synthesis under Kumada or Suzuki conditions, and also reactions of organolithium derivatives with perfluorobenzene. In the case of 2,2'-(2,3,5,6-tetrafluoro-1,4-phenylene)bis(5-phenylthiophene) 2.15.3a (PTPFTP), 2-phenylthiophene was produced on the first step in high yield by the Kumada reaction. Its lithiation with a solution of BunLi in hexane followed by treatment with perfluorobenzene afforded the target product (see Scheme 440a). 2,2'-(1,4-Phenylene)bis[5-(perfluorophenyl)thiophene] 2.15.3b (PFTPTPF) and 2,2'-(2,3,5,6-tetrafluoro-1,4-phenylene)bis[5-(perfluorophenyl)thiophene] 2.15.3c (PFTPFTPF) were synthesized by sequential lithiation of corresponding thiophene-containing precursors with BunLi and treatment with a large excess of perfluorobenzene (see Scheme 440). The low yields of the reaction products stem from the need to prepare dilithium derivatives of the thiophene-containing precursors.

Comparison of the optical properties of oligomers 2.15.3a – c in dilute toluene solutions showed that their absorption and luminescence spectra are very similar, but for PFTPTPF and PFTPFTPF a hypsochromic shift of the absorption band maxima by ~ 0.1 eV is observed. The luminescence spectra also show a hypsochromic shift that enlarges with increasing the number of perfluorinated units. PLQY values are high for all oligomers 2.15.3 and increase in the series: PTPTP (79 ± 4%) < PTPFTP (80 ± 4%) < PFTPTPF (85 ± 5%) < PFTPFTPF (90 ± 5%)[1088]. In crystal structures, fluorination leads to a switch from herringbone packing motif for PTPTP to one- or two-dimensional π-stacking for the partially or highly fluorinated derivative. In a thin-film PTPFTP-based OLET (2.15.3a), balanced ambipolar charge transport and electroluminescence were observed[1088]. Therefore, the introduction of perfluorinated benzene moiety is an effective approach to ambipolar and electroluminescent organic semiconductors for organic electronic devices. This was confirmed by the authors[1084] from NIOCh SB RAS using selectively fluorinated furan-phenylene oligomers.
One of the most promising types of molecules for applications in organic electronics are annulated structures, in particular [1]benzothieno[3,2-b][1]benzothiophene (BTBT) and its derivatives (2.15.6) due to their high charge carrier mobility[1089]. Among these, 2,7-dioctyl[1]benzothieno[3,2-b][1]benzothiophene 2.15.6b (C8-BTBT) attracts special attention[1090, 1091].

Several approaches to the synthesis of such compounds based on consecutive reactions of the Friedel-Krafts acylation and reduction of the keto group have been reported[1091-1094]. However, in the final stage, a large number of by-products can be formed (see, e.g.,[1091]), thus greatly reducing the yield of the target molecule. Therefore, a thorough research of the possible methods for reducing the keto group in the synthesis of C8-BTBT has been carried out, which has allowed developing optimal synthesis schemes, identify the advantages and disadvantages of each procedure and improve the yield of the target product[1095].
The main disadvantage of the dialkyl derivatives BTBT is the low PLQY, especially in the solid state. Therefore, 2,7-bis(4-decylphenyl)[1]benzothieno[3,2-b][1]benzothiophene 2.15.7 (DPBTBT) was synthesized with PLQY = 46 ± 4% in solution (Scheme 441)[1096]. First, a bromine atom was substituted for the n-decyl group in p-dibromobenzene, and then an organoboron derivative of p-decylbenzene 2.15.4c (DP-Bpin) was prepared and subjected to the Pd-catalyzed Suzuki coupling with 2,7-dibromo[1]benzothieno[3,2-b][1]benzothiophene 2.15.6c (Br-BTBT-Br).

Large-area 2D monocrystals consisting of one or a few molecular monolayers were prepared from DPBTBT. OFETs fabricated from such monocrystals showed high performance: their electrical properties fully matched the Shockley model with charge carrier mobility up to 1.0 cm2 V–1 s–1 for monolayer devices and 7.5 cm2 V–1 s–1 for multilayers, which is one of the highest values for organic 2D semiconductors. Two-dimensional crystals of compound 2.15.7 also showed noticeable luminescence, which helped to create the first two-dimensional OLET[1096].
The other annulated thiophene, thieno[3,2-b]thieno[2',3'':4,5]thieno[2,3-d ]thiophene 2.15.8a (tetrathienoacene, TTA), can serve as an analogue of BTBT (2.15.6a). The alkyl groups can be introduced into acenes 2.15.8 by various methods, the most effective being the Friedel – Krafts acylation. It was shown that by varying the acylation conditions it is possible to control the formation of mono- or diketones, which can be further reduced to mono- or dialkyl derivatives of TTA, respectively (Scheme 442a). OFETs based on C8-TTA-C8 (2.15.8b) have been fabricated by vacuum deposition with hole mobility of up to 0.13 cm2 V–1 s–1[1097].

In addition to the dialkyl derivatives of TTA, compounds 2.15.8d,e bearing two 4-hexylphenylene ((Hex-Ph)2-TTA) and 5-hexyl-2-thiophene ((Hex-T)2-TTA) substituents were synthesized[1098]. For this purpose, dibromide 2.15.8c (Br-TTA-Br) was subjected to the Suzuki coupling with appropriate organoboron derivatives (see Scheme 442b). The study of photophysical, electrochemical and electronic properties showed that the spectra of (Hex-T)2-TTA feature a bathochromic shift of absorption and luminescence maxima compared to those of (Hex-Ph)2-TTA. Compounds 2.15.8d,e have relatively high PLQYs in solution and a thin polycrystalline film of (Hex-Ph)2-TTA showed 6 times higher values of this parameter compared to (Hex-T)2-TTA. Ambipolar charge transport was observed in OFETs based on the oligomer (Hex-Ph)2-TTA and the maximum hole mobility reached the value of 0.6 cm2 V–1 s–1.
2.15.2. Thiophene-containing self-assembled molecules
It is well-known that charge transport in OFETs takes place in a layer of organic semiconductor only a few nanometres thick located directly on the dielectric surface[1099]. In this context, it is possible to fabricate OFETs and simple integrated circuits based on a monolayer of organic semiconductor[1100]. One of the most convenient ways to obtain monolayers is to self-assemble them from functional organic molecules, thus obtaining self-assembled monolayer organic field-effect transistors (SAMFETs)[1101, 1102].
The synthesis of a series of self-assembled semiconducting oligothiophene-containing monochlorosilanes 2.15.9a – e was first described by S.A.Ponomarenko et al[1103]. Such molecule consisted of a quater-, quinque- or sexithiophene unit responsible for the semiconducting properties, a flexible alkyl (hexyl or undecyl) spacer and a dimethylchlorosilyl anchor group, which reacted with a hydroxyl-containing support. In addition, oligomers 2.15.9a – e contained end-capping ethyl or hexyl groups to increase their solubility in organic solvents. The monolayer was formed by placing a silicon support containing surface activated silica with Si – OH groups in a dilute solution of the semiconducting oligomeric chlorosilanes in toluene. Based on a chlorosilyl derivative of quinquethiophene with an undecyl spacer and an ethyl end-capping group (2.15.9a), SAMFETs on a silicon support have been prepared with the best values of charge carrier mobility (m = 0.02 cm2 V–1 s–1) and the on-off ratio (IOFF/ION = 107) in the investigated series[1103]. Moreover, the oligomer 2.15.9a was successfully used in the first integrated circuits based on monolayer organic semiconductors[1100].

SAMFETs based on oligothiophene-substituted monochlorosilanes have found application as chemosensors. Their unique structure due to the introduction of an additional receptor layer (meso-tetraphenylporphyrin-iron(III) chloride) provided a very high sensitivity of the sensor for nitric oxide with the detection threshold < 100 ppb. Such high sensitivity can be explained by the fact that the analyte acts directly on the transistor active channel, which is a monolayer of the organic semiconductor[1104].
The production of OFETs by self-assembly of oligothiophene chlorosilanes is rather time-consuming (tens of hours), therefore methods to produce such devices in a shorter time have been explored[1105]. It was shown that using either Langmuir – Blodgett or spin coating techniques for deposition of 2.15.9a molecules to the substrate, highly ordered monolayers with vertical orientation of oligothiophene fragments were obtained, whereas the dip coating method provided only a partially covered surface. (Notably, the use of the Langmuir-Blodgett method to prepare monolayers based on non-functionalized di-alkyloligothiophenes failed). Moreover, monolayer OFETs with good semiconductor properties were obtained by the Langmuir-Blodgett technique from thiophene-containing self-assembled molecules 2.15.9a[1106]. Despite some defectiveness of the resulting monolayer, the m value reached 1.1 × 10–2 cm2 V–1 s–1, which is close to the properties of SAMFETs obtained by the self-assembly approach[1100, 1103].
Oligothiophene-containing monochlorosilanes require special storage and processing conditions due to the presence of a reactive chlorosilyl group. Since the Langmuir – Blodgett method involves contact with water and leads to hydrolysis of the chlorosilyl group yielding a disiloxane moiety, disiloxane dimers 2.15.10 – 2.15.11 have been synthesized that did not contain any reactive group and were able to self-assemble into monolayers on the water surface. Using this method, monolayers of the siloxane dimer of quaterthiophene with undecylene spacer and hexyl end-capping groups, compound 2.15.10a (D2-Und-4T-Hex), were obtained on a silicon substrate. Atomic force microscopy, optical microscopy and X-ray diffraction studies showed that such monolayers have a domain crystal structure of island character with a domain size of about 4 μm2 and a characteristic degree of filling > 90%, which allowed them to be used as an OFET channel. OFETs fabricated on D2-Und-4T-Hex monolayers showed hole mobility up to 3 × 10–3 cm2 V–1 s–1 and high long-term stability[1107, 1108].
Increasing the length of the oligothiophene fragment usually increases the charge carrier mobility. For this reason, siloxane dimers containing quinquethiophene and sexithiophene fragments have been synthesizsed[1109]. However, the solubility of such molecules is significantly reduced, making it impossible to prepare high quality monolayers suitable to OFETs.
In addition to oligothiophene-containing molecules, a siloxane dimer of oligothiophene-phenylene 2.15.10b (D2-Und-PTTP-TMS) was synthesized. The resulting Langmuir films exhibited both semiconducting and luminescent properties. It was shown for the first time that the ordered 2D packing of conjugated organic molecules in a semiconducting Langmuir monolayer does not prevent the efficient luminescence of the material[1110].
The chlorosilyl derivatives Cl-Si-Und-BTBT and Cl-Si-Und-BTBT-Hex (2.15.9f,g) and also the siloxane dimers D2-Und-BTBT and D2-Und-BTBT-Hex (2.15.11a,d), which are capable of self-assembly into monolayers on the water surface, have been described[1111]. Possible approaches to the synthesis of such oligomers were considered and an optimal route to new BTBT derivatives in high yields was selected. It involved the synthesis of monosubstituted 2-alkyl-BTBT, protection of the terminal double bond of alkenyl acid chloride with bromine, Friedel-Krafts acylation of 2-alkyl-BTBT with the resulting compound followed by the reduction of the keto group to alkane, removal of the dibromide protection with zinc and hydrosilylation. The conditions were chosen to obtain high quality monolayers by the Langmuir-Blodgett method, from which OFETs with charge carrier mobility up to 1.4 × 10–2 cm2 V–1 s–1 were obtained.
Monolayer OFETs based on compound 2.15.11d (D2-Und-BTBT-Нех) have been used to create highly sensitive gas sensors for polar gases (H2S and NH3)[1112, 1113]. On their basis, an ‘electronic nose’ with metalloporphyrins as receptor layers has been developed for the detection of toxic gases (H2S, NH3, NO2 and EtSH)[1114] and for the control of food freshness[1115]. The D2-Und-BTBT-Нех dimer has been used to create organic memristive devices that have demonstrated at least four resistive states and 25 000 cycles of sustainable operation in endurance tests with on/off current ratios (IOFF/ION) exceeding 5[1116].
Having these synthetic approaches in hand, a series of BTBT-based siloxane dimers with different aliphatic spacer lengths (C6, C7 and C11) between the semiconducting unit and the anchoring siloxane group were obtained: compounds 2.15.10c,d and 2.15.11d (D2-Hex-BTBT-Hex, D2-Hept-BTBT-Hex and D2-Und-BTBT-Hex, respectively)[1117]. The study of the influence of the aliphatic spacer length on the semiconducting properties of the dimers showed that the maximum μ value reaches 0.47 cm2 V–1 s–1 when D2-Und-BTBT-Hex is applied by Langmuir – Sheffer method as semiconducting material on polystyrene interface layer of the OFET. The device also had a low detection threshold for EtSH, used as an odourant in domestic gas supply, reaching 30 ppb in humid air. A monolayer of D2-Hept-BTBT-Hex dimer with heptyl spacer was used to fix the biotin-containing BTBT derivative in the development of a new electrolyte-gated OFET-based biosensor platform, which was successfully tested for the selective detection of influenza A virus[1118, 1119].
To explore the influence of the length of the terminal groups on the properties of siloxane dimers, a series of BTBT derivatives 2.15.11b – f containing terminal alkyl end-capping groups of different lengths (D2-Und-BTBT-Et, D2-Und-BTBT-Bu, D2-Und-BTBT-Hex, D2-Und-BTBT-Oct and D2-Und-BTBT-Trid, respectively) were synthesized[1120]. The scheme for the synthesis of such oligomers included a sequence of Friedel-Krafts acylation, keto group reduction and hydrosilylation reactions. Compounds 2.15.11 were obtained in high yields. The study of the phase behaviour showed that all these oligomers are able to form both highly ordered (smectics E or K) and disordered liquid crystalline mesophases (smectics A or C). At the same time, increasing the length of the terminal alkyl group from C2 to C8 led to higher phase transition temperatures, i.e. to improved thermal stability. The best electrical and sensory (in terms of nitrogen oxide(IV)) properties were demonstrated by OFET based on siloxane dimer 2.15.11e (D2-Und-BTBT-Oct) with octyl end-capping groups, which is probably due to the lowest surface roughness of the films formed by this compound.
2.15.3. Thiophene-containing donor-acceptor oligomers
Organic conjugated thiophene-containing oligomers of the donor-acceptor (D – A) type contain electron-withdrawing (EWG) and electron-donating (EDG) fragments in their structure, usually linked by a π-conjugated (oligo)thiophene spacer (see e)[1121, 1122]. In such systems, new molecular orbitals are formed with a significantly smaller energy difference between the HOMO and LUMO levels compared to the original orbitals. As a result, the oligomers formed have a significantly lower bandgap as well as absorption and light emission spectra significantly shifted towards the long-wavelength region compared to similar parameters for compounds of conventional structure close in molecular mass and number of conjugated units. D – A oligomers are successfully used in a wide range of organic electronic and photonic devices due to the possibility to fine-tune their properties over a fairly wide range by varying the type and number of constituent units.
Dicyanovinyl (DCV) end-capped oligomers are one of the most promising classes of D – A molecules for applications in organic optoelectronics and photonics due to the relatively strong electron-withdrawing ability of this group, availability and low cost of the starting material – malononitrile[1123]. However, the DCV group contains an active proton, which results in insufficient thermal and chemical stability of the compounds. Due to their low solubility, it is necessary to introduce solubilizing alkyl groups in different parts of the molecule, which increases the cost of the final products due to additional synthetic steps.
To improve the stability and solubility of the oligomers, we proposed to use substituted alkyl (Alk-DCV)[1124-1128], phenyl (Ph-DCV)[1129, 1130] and p-fluorophenyl dicyanovinyl (FPh-DCV) moieties instead of the standard DCV group with a free active proton[1131]. This approach provides the preparation of a family of star-shaped triphenylamine-based oligothiophenes 2.15.12a – g[1129, 1131, 1124-1126].

The process of oligomer 2.15.12b – g preparation can be divided into three main steps: synthesis of organoboron derivatives of oligothiophene ketals, preparation of bromine derivatives of donor centres and transformations giving the target D – A compounds (Scheme 443)[1126].

The versatility of this scheme is supported by its use for obtaining a large number of both star-shaped and linear D – A oligothiophenes, compounds 2.12.12 and 2.12.13, respectively. Due to a favourable combination of a complex of physicochemical properties of D – A oligomers, such a library is of great practical interest. For example, most of the compounds show effective light absorption in the visible spectral range and relatively low HOMO energies (–5.2 to –5.6 eV), which enables their application as donor materials in a mixture with a suitable acceptor in the active layer of bulk-heterojunction organic solar cells (OSCs)[1124, 1127, 1132, 1133].

It can be seen how the type of substituent in DCV in a series of four star-shaped triphenylamine-based compounds 2.15.12a,d,f,g (TPA-(2T-DCV)3, TPA-(2T-DCV-Hex)3, TPA-(2T-DCV-Ph)3 and TPA-(2T-DCV-PhF)3, respectively) affects the efficiency of OSCs with soluble fullerene-70 derivative (PC71BM) as acceptor. Switching from compound 2.15.12a bearing unsubstituted DCV group, with power conversion efficiency PCE = 2.3%, to derivatives 2.15.12d,f,g with the substituted terminal acceptor groups (Hex-DCV, Ph-DCV and FPh-DCV), all parameters of the OSCs increase significantly, reaching the efficiency of 4.36% for TPA-(2T-DCV-Ph)3, which is mainly due to differences in the morphology of the photoactive layer, the efficiency of charge transport and separation[1129, 1130].

Star-shaped compounds 2.15.12 with Alk-DCV and Ph-DCV groups can be used not only as donor materials in bulk-heterojunction OSCs, but also in single-component OSCs[1134], i.e. where the photoactive layer is not represented by a mixture of donor and acceptor materials, but by a single compound, which greatly simplifies the process of device fabrication and increases their stability. Studies of several compounds 2.15.12 with Alk-DCV and Ph-DCV terminal acceptor groups in single-component OSCs (Figure 3a) showed the promising potential of these materials as components for this type of devices[1135-1138]. The OSCs fabricated on their basis had a high open circuit voltage (up to 1.19 V), and their PCE reached 1.13%.
![[{"id":"Bv12wXYOyU","type":"paragraph","data":{"text":"Schematic diagram of the device used in single-component OSCs (the photoactive layer is designated as a «compound») and photodetectors (a); voltammetric curves of single-component OSC based on TPA-<b>(2T-DCV-Et)<sub>3</sub></b> in the light and in the dark (b), and its photosensitivity spectra (c)."}}]](/storage/images/resized/6HKA4XjU6FAZEmXmnKFiSUsys6CU3W8vbH2tolvo_xl.webp)
It is found that the photocurrent generation in such devices is higher at a negative bias, making it possible to use such single-component devices as photodetectors (see Figure 3b)[1135, 1137]. For example, the external quantum efficiency of photodetectors based on compound 2.15.12b (TPA-(2T-DCV-Et)3) increases from 23.5% to 30% at –0.5 V, and the responsivity (R) with a maximum at 560 nm increases from 0.1 A W–1 with no bias to 0.14 A W–1 at –0.5 V (see Figure 3c). The obvious advantage of such one-component photodetectors is not only the ease of fabrication and the possibility of varying the spectral range of detection, but also a relatively narrow responsivity spectrum (see Figure 3c), since there is only one material in the photoactive layer.
Given that the response speed of such photodetectors to laser pulses is in the submicrosecond range[1135, 1138], i.e. exceeds the response time of the human eye by several orders of magnitude, it opens up the prospect of creating artificial retinal photoreceptors on their basis, the so-called rods (R), responsible for twilight vision, and three types of cones, responsible for colour vision, containing short (S), medium (M) and long (L) wavelength pigments. In some eye diseases (age-related macular degeneration, retinitis pigmentosa, etc.), the nervous tissue of the retina usually remains functional and can transmit nerve impulses to the brain, which makes it possible to restore vision after replacing the damaged natural photoreceptors. The ability to fine-tune optoelectronic properties makes D – A molecules excellent candidates for artificial photoreceptors with spectral characteristics similar to those of human cones and rods (see ASC Biomater. Sci. Eng., 10, 1130 (2024); https:// doi.org/10.1021/ascbiomaterials.3c01562.)[1139]. This effect was successfully demonstrated using the example of four D–A molecules TPA-T-CO, TPA-(T-CNA-EHex)3, TPA-(2T-DCV-Hex)3, TPA-BRZ-Rh (for brevity, designated as S, R, M and L in analogy to the designation of the human retinal photoreceptors) containing carbonyl, alkyl cyanoacetate, alkyl dicyanovinyl or alkyl rhodanine electron-withdrawing moieties, respectively, for fine tuning of the optical slit (see structures S, R, M and L below)[1139].

Thin layers of such D – A oligomers were deposited from solutions by spin coating or ink jet printing (pixel diameter from 25 to 42 μm) onto glass substrates coated with ITO with a ZnO layer (Figure 4a,b), which provided an access to the simplest devices for photoresponse studies[1139]. When irradiated with low power monochromatic light, these devices, placed in a modelling biological environment, gave a response that closely corresponded to the response of a certain type of natural photoreceptor of the human retina (see Figure 4c,d ). The ability to create functional layers of such materials using inkjet printing opens up the prospect of applying multicoloured patterns (see Figure 4a), mimicking the human retina and thus making its prostheses. In the future, this will help with the treatment of one of the most common causes of visual impairment – damage to the light-sensitive cells of the retina.
![[{"id":"Q2lOnvkhL6","type":"paragraph","data":{"text":"Inkjet-printed pixel array based on <b>TPA-(2T-DCV-Hex)<sub>3</sub></b> on glass ITO/ZnO support (ITO is indium-tin oxide) (a); the cell for photocurrent and photovoltage measurements (b); normalized photocurrent spectra of the films of the D–A oligomers <b>S</b>, <b>R</b>, <b>M</b> and <b>L</b>, respectively, spin coated on glass/ITO/ZnO supports, interfaced with an electrolyte (c); and normalized absorption spectra of human retinal photoreceptors (d)[[ type=\"anchor\" referenceId=\"7710\" ]]."}}]](/storage/images/resized/zWR5QJOZsfHSoGF0UqEgJsBZvzsbXhFzkoIfVHL8_xl.webp)
Many of the D – A oligomers obtained exhibit effective fluorescence both in solution and in thin films as well as in polymer composites. The emission maximum is usually in the red and near-infrared spectral range (see g)[1140-1142], which is in demand for applications in biology and medicine. The use of the end-capping groups differing in their electron-withdrawing abilities together with Alk-DCV and Ph-DCV moieties in the D – A structures of compounds 2.15.14 based on triphenylamine (TPA) allows the width of the optical slit to be tuned in a rather simple way and their fluorescence spectra to be varied over a wide spectral range (Figure 5).

![[{"id":"xdMM-UQ67E","type":"paragraph","data":{"text":"Photographs of powders and polystyrene composites of compounds <b>2.15.14</b> (EWGs are shown) under normal and UV (365 nm) light (а) and their fluorescence spectra in a polycrystalline film (b)[[ type=\"anchor\" referenceId=\"7712\" ]]."}}]](/storage/images/resized/ijzMjEclLpPAvnVi6mB63KIxawzVIzQrYsb8wX4f_xl.webp)
Since most of the oligomers described above are p-type organic semiconductors, they can be used as hole transporting layers (HTLs) in various optoelectronic devices, such as halide perovskite solar cells (PSCs)[1143]. As an example, six D – A oligomers 2.15.15a – f, differing from each other in chemical structure (the length of oligothiophene π-spacer conjugation, the type of the donor centre, the nature of an acceptor group and the HOMO energy) were studied as undoped HTLs in PSCs[1143].

It is shown that the efficiency of most of the explored devices exceeds the characteristics of similar benchmark photovoltaic cells, but there is no clear relationship between chemical structure parameters, oligomer properties and efficiency values. This can be explained by a much greater influence of the quality of the HTL (film homogeneity, presence of holes and voids) covered above the perovskite layer on the PSC efficiency. As a result, the highest efficiencies, up to 19 and 20% respectively, were achieved in PSC based on HTLs derived from BDT-(3T-DCV-Hex)3 and TAT-(2T-CNA)3. These compounds have quite different chemical structures and HOMO levels. However, both are able to form homogeneous films over the perovskite layer, unlike the other compounds investigated, as confirmed by microscopy and spectroscopy studies with nanometre spatial resolution in the IR range. Thus, the resulting D – A oligomers can be used as HTLs in PSCs and devices based on them show PCE up to 20%, which is comparable to the record values obtained for PSCs based on organic undoped HTLs.
2.15.4. Thiophene-containing molecules with different branching centres
The first branched bithiophenesilanes with a silicon atom as a branching centre were synthesized by the reaction of a lithiated 5-hexyl-2,2'-bithiophene (Hex-2T) with (methyl)alkoxysilanes containing different number of functional groups[1144]. Depending on the number of methoxy or ethoxy groups in the starting alkoxysilane, compounds 2.15.16a – d (designated as Z1 – Z4 for brevity) were obtained, each containing from one to four Hex-2T units (linear trimethyl-5-(5'-hexyl-2,2'-bithienyl)silane was obtained using trimethylchlorosilane). Studies of the optical properties of dilute solutions of compounds 2.15.16a – d showed that the PLQY value increases from 5.8 to 20% when going from the former (Z1) to the latter (Z4)[1144]. Tetrakis(5''-hexyl-2,2':5',2''-terthien-5-yl)silane (2.15.16e, Z5) was prepared in a similar manner and first described by J.Roncali and co-workers[1145]. Molecules with longer oligothiophene units 2.15.16f,g (Z6, Z7) were obtained from tetrakis(5'-bromo-2,2'-bithien-5-yl)silane by the Suzuki coupling with corresponding bi- and terthiophene organoboron derivatives. These compounds, as well as their precursor (2.15.16e, Z5), have been used as electron-donor materials for the preparation of organic solar cells and photodetectors[1146].

The Suzuki reaction has also been used for the synthesis of compounds 2.15.17a,b, which contain terthiophene (3T, B1)[1147] and quaterthiophene (4T, B2)[1148] units in the centre of the molecule and four Hex-2T groups at the periphery. In such systems, an effective intramolecular transfer of electron excitation energy from the outer bithiophene units to the central core, which has a longer conjugation length and consequently a smaller bandgap, has been observed.
The synthesis of bithiophene silane dendrimer 2.15.18a (D1) of the first generation was described in 2005[1149]. A comparison of two approaches to organometallic synthesis via the Kumada and Suzuki reactions showed that the latter reaction was the most promising. This methodology gave rise to the preparation of dendrimers 2.15.18b – d (D2 – D4) in high yields[1150]. A detailed study of the optical properties of dendrimers 2.15.18b – d has shown that binding the bithiophene units (poor luminophores with PLQY = 1.8%) into dendritic molecules via a silicon atom improves the luminescence efficiency by up to 30% without significantly altering the spectral-luminescence properties[1151-1153].
Combining different luminophores into a dendritic molecule provides an effective intramolecular energy transfer from units with a wider bandgap (electron excitation energy donors) to units with a narrower bandgap (electron excitation energy acceptors), known as the «dendritic molecular antenna» effect[1154]. Such an effect was observed in dendrimers 2.15.18e,f (D5 (see[1147]) and D6)[1148]. However, their luminescence efficiencies were only 10 and 13% respectively for compounds 2.15.18e,f (D5 and D6) due to the low PLQY value of the 3T and 4T acceptor moieties.
The luminescence efficiency of the dendrimers could be significantly increased by introducing bis(2,2'-bithiophen-5-yl)-1,4-phenylene (PTPTP (2.15.2.a)) as acceptor moiety. In this case, the efficiency of energy transfer from the Hex-2T outer units to the central core consisting of PTPTP units was 88% and the PLQY increased to 46%[1155].
Recently, a new method based on the Stille reaction was developed for the synthesis of dendrimers containing up to 18 donor Hex-2T units and a central acceptor PTPTP unit[1156].
Challenges in the synthesis of dendrimers can be overcome by switching to branched molecules, which contain only one acceptor and several donor units linked by the silicon atoms. The latter breaks the conjugation between the units with different electronic properties but fixes them in the space at a distance of 1 – 2 nm between the centres of gravity of the chromophores in the molecule[1085, 1157-1159]. Branched thiophene-containing oligomers containing 4 or 6 donor Hex-2T units and an acceptor units include compounds 2.15.19a – f (C6H13 is n-hexyl).

There is no conjugation between the π-electronic systems of chromophores linked by a silicon atom. However, at small distances between them, Förster resonance energy transfer (FRET) can occur[1160]. Such systems are capable of capturing light quanta (photons) and non-radiatively transferring electronic excitation energy from the periphery to the core, resulting in effective absorption of light in a broad spectral range and emission in a narrow, longer wavelength region of the spectrum.
Silicon chemistry makes it easy to create branched molecular structures with different donor and acceptor units. In compounds 2.15.17 – 2.15.19, bithiophene moieties that absorb light in the UV region of the spectrum act as donor units. The choice of acceptor unit is determined by the required energy of the emitted photons, which can vary over a wide spectral range. For example, the maximum luminescence spectra of molecules 2.15.17 – 2.15.19 range from 392 to 655 nm, while their absorption maximum is in the region of 330 nm[1157]. Depending on the number of reactive groups on the silicon atoms, branched organosilicon compounds with different numbers of donor units can be obtained. Increasing the number of donor units from 4 to 6 leads to an increase in the molar extinction coefficient. The introduction of reactive alkene groups, instead of inert hexyl substituents, into bithiophene moieties of such molecules has allowed the preparation of luminescent organosilicon compositions containing up to 3% by weight of luminophores and thermostable up to 370 °C after hydrosilylation of double bonds with hydride-containing organosilicon compounds[1161].
The above organosilicon luminophores 2.15.19 have been successfully used for the preparation of plastic scintillators – materials capable of emitting visible and UV photons when exposed to ionizing radiation. Replacing classical organic luminophores with the branched molecules 2.15.19 increased the light output of the thin-layer plastic scintillators by 50%[1157]. The above-mentioned branched organosilicon compounds, with efficient intramolecular energy transfer, have been used in a new type of scintillation fibre characterized by a very short luminescence time and high light output[1162]. Studies of such scintillation fibres have shown that in the samples obtained the luminescence time is 1.2 – 1.3 ns. This is almost 2 times faster than the best standard fibre emitting in the blue region and 6 times faster than the best fibre emitting in the green region.
The ‘molecular antenna’ effect allows the creation of effective optical wavelenth shifters (WLS) for various detection systems[1163]. For example, organosilicon molecules with efficient intramolecular energy transfer have been successfully used as WLS for inorganic scintillators (undoped CsI crystals)[1164], liquid xenon scintillation detectors[1165], silicon photomultipliers[1166], and silicon indium gallium selenide (SIGS) solar cells[1167].
It is well-known that molecules with a ‘molecular antenna’ effect can also be created based on benzene-1,3,5-triyl, which acts as a branching centre[1168-1170]. In the topological concept of omniconjugation proposed by Hummelen and co-workers[1171] in 2004, substituents in the meta-positions of the benzene ring are considered as non-conjugated. However, the term ‘meta-conjugation in aromatic benzene derivatives’ can also be found in the literature[1172, 1173]. Examples of branched thiophene-containing molecules, in which benzene-1,3,5-triyl serves as the branching centre, include compounds 2.15.20a – c[1174].

The synthesis of such compounds is based on the Suzuki reaction. The absorption spectra of meta-conjugated molecules 2.15.20a – c can be represented as the sum of the absorption bands of model compounds for their peripheral (2.15.20d,e) and central (2.15.20f,g) units comprising common benzene rings which are 1,3,5-trisubstituted branching centres in meta-conjugated molecules. With increasing the number of thiophene rings in the peripheral and central moieties, the luminescence maxima in the spectra of the investigated molecules shift monotonically towards the long wavelength region, from 489 to 540 and 613 nm for compounds 2.15.20a (T-P-BTD), 2.15.20b (2T-P-BTD) and 2.15.20c (2T-P-TBTD), respectively. However, the PLQY value of such molecules depends critically on the length of linear conjugated units in their structure, decreasing from 24 to 1.3% with increasing the number of thiophene ring in the pendant arms. At the same time, the PLQY value increases to 90% in the compound with the largest number of thiophene rings in both types of fragments in 2.15.20c (2T-P-TBTD).
Quantum chemical calculations based on density functional theory made it possible to visualize the molecular orbitals of compounds 2.15.20 and to demonstrate the formation of their absorption spectra from the corresponding transitions with large values of the oscillator strength. It was shown that the binding of linearly conjugated units through the meta-positions of the benzene ring (meta-conjugation) leads to an intermediate case between fully conjugated and non-conjugated molecules due to a partial delocalization of the electron density through the 1,3,5-trisubstituted benzene branching centre[1174].

Thiophene-containing oligomers have also been synthesized using triphenylamine as the branching centre and, like their linear counterparts (2.15.22a,b), contain a thiophene-phenylene (2.15.21a,b) or benzothiadiazole (2.15.21c,d) central block[1175].
The absorption and luminescence spectra of oligomers 2.15.21a – d with triphenylamine moieties are significantly shifted towards the long wavelength region compared to the spectra of their analogues with trimethylsilyl groups 2.15.22a,b (Figure 6). At the same time, the transition from linear to branched molecules significantly increases the efficiency of light absorption in the UV and visible regions, but has little effect on the luminescence spectra.
![[{"id":"5Qug249mNm","type":"paragraph","data":{"text":"Absorption (solid) and fluorescence (dashed) spectra of luminophores based on triphenylamine and their analogues with thiophene-phenylene (<b>I, II</b> — <b>2.15.21a,b</b>; <b>I-TMS — 2.15.22a</b>) and benzothiadiazole (<b>III, IV</b> — <b>2.15.21b,d</b>; <b>III-TMS</b> — <b>2.15.22b</b>) central block and reference triphenylamine (<b>TPA</b>) in dilute THF solutions (a, b)[[ type=\"anchor\" referenceId=\"7746\" ]]."}}]](/storage/images/resized/PR333CaMWZX2Z8aYSdjE0QPM3wGQfq8roUmvv6UA_xl.webp)
The study of the photostability of oligomers 2.15.21a – d showed that it increases when passing from the linear oligomer 2.15.21a with thiophene-phenylene central block to its branched analogue 2.15.21b. However, for oligomers with a benzothiadiazole-containing central block, the stability of the branched oligomer 2.15.21d was significantly lower than that of the linear oligomer 2.15.21c. The latter showed uniquely high stability in the polymer matrix withstanding an irradiation dose of up to 120 000 MGy, which is 333 times higher than in THF. At the same time, its silicon analogue 2.15.22b withstood a dose of up to 108 000 MGy, which is >100 times higher than in THF[1175].
To conclude this Section, modern methods of organic and organometallic synthesis using organolithium, -magnesium, -boron and -tin compounds, Pd-catalyzed Suzuki, Kumada and Stille cross-coupling reactions, as well as Knoevenagel condensation, Friedel – Krafts acylation, hydrosilylation and various keto group reduction reactions, provided an access to a wide diversity of linearly conjugated and annulated oligomers of different topology (linear or branched). Such compounds have a unique set of physical and chemical properties that are in demand in various organic electronics and photonics devices. At the same time, molecules containing annulated scaffolds exhibit the highest mobility of charge carriers and therefore find application in OFETs. Thiophene-containing self-assembled molecules have been used to make SAMFETs, highly sensitive semi-selective gas sensors and ‘electronic noses’ as well as electrolyte-gated OFETs and biosensors. Thiophene-phenylene oligomers possess both high charge carrier mobility and intense luminescence in thin films, making them suitable for OLETs. Donor-acceptor thiophene-containing molecules provide an opportunity to control their absorption spectrum over a wide range and fine-tuning the frontier molecular orbitals that is why such systems have found application in OSCs and PSCs, photodetectors and photonics as luminophores emitting in the long-wavelength part of the spectrum. Branched thiophene-containing oligomers were obtained based on different branching centres such as silicon atom, 1,3,5-trisubstituted benzene and triphenylamine. Here, only silicon atoms completely break the conjugation between the linearly conjugated units bonded thereto, which opens easy access to ‘molecular antennas’ and use them as highly efficient and fast luminophores in polymer scintillators and spectroscopic components (fibres, plates and coatings) for high-energy physics.
2.16. Heterocyclic thiophen-containing conjugated donor – acceptor – donor systems: synthesis, structure – properties relationship
Over the past quarter century, oligothiophenes have attracted the attention of researchers due to their use as semiconductors for organic light-emitting diodes, field-effect transistors, electrochemical and chemical sensors, and other electronic devices. They have a number of advantages over inorganic semiconductors, including transparency, flexibility, low specific weight and high radiation resistance. Their solubility in common organic solvents facilitates their purification and processing. Such oligomers are usually accessible by methods that do not require significant energy input. Their electronic, optical and electrochemical properties can be fine-tuned by modifying their structure. One possible approach to the preparation of organic semiconductors is the synthesis of conjugated donor-acceptor-donor (D – A – D) molecules, in which bi-, ter- or quaterthiophene moieties are spaced by an electron-withdrawing heterocycle.
At the end of the XX century, Bäuerle et al.[1176, 1177], synthesized heterocyclic D – A – D oligomers consisting of 1,3,4-oxadiazole and thiophene rings in his search for new luminescent compounds. These compounds contained substituents on the terminal thiophene moieties that prevented polymerization of the oligomer. An attempt to introduce a five-membered π-acceptor heterocycle into the polythiophene chain and to study its properties was made in 2005 by the example of the electrochemical polymerization of 2,5-bis(3-methylthiophene-2-yl)-1,3,4-oxadiazole (2.16.1) (Scheme 444), but it was unsuccessful[1178]. Due to the strong electron-withdrawing character of 1,3,4-oxadiazole 2.16.1, its electrochemical oxidation started at a very high potential and afforded an unstable cation radical followed by its decomposition[1178]. To increase the stability of the cation radical, the conjugated system was extended by introducing additional thiophene rings. For this purpose, the dibromo-substituted bithiophene-1,3,4-oxadiazole 2.16.2 and (3-octylthiophene-2-yl)boronate 2.16.3 were subjected to the Suzuki coupling. However, the yield of the resulting product 2.16.4 was low.

Compound 2.16.4 was readily electrochemically polymerized to poly(dimethyldioctyl-quaterthiophene-alt-oxadiazole) (poly-2.16.4). It was the most electrochemically stable thiophene-containing polymer at the time[1178-1180]. It was reversibly oxidized and reduced, and the difference between the oxidized and reduced forms was as high as 4 V (Figure 7).

There are two general strategies for the synthesis of compounds with a D – A – D structure. One involves the formation of carbon – carbon bonds between the units of the conjugated chain by cross-coupling reactions[1178-1181]. This approach is not always efficient and is limited by the number of commercially available ‘building blocks’. The second strategy is based on the construction of the central acceptor ring from functionalized donor blocks. An approach to such thiophen-containing building blocks was proposed in 2014 at the Dostoevsky Omsk State University[1182]. This review systematizes the material on the development of this approach and its application in the preparation of conjugated heterocyclic systems of the D – A – D type, analyzes the influence of the structure of such compounds on their electronic, electrochemical and photophysical properties.
![[{"id":"b5jFokWLmr","type":"paragraph","data":{"text":"Cyclic voltammograms of р- and n-doped <b>poly-2.16.4</b> film on a Pt electrode measured at scan rates of 10, 20, 50 mV s<sup>–1</sup> (0.15 M Et<sub>3</sub>N · PF<sub>6</sub> in sulfolane)[[ type=\"anchor\" referenceId=\"7751\" ]]."}}]](/storage/images/resized/csypo5eEEsybxbx24DaIBndYPHheQOQB2MfJS9WO_xl.webp)
2.16.1. Synthesis of functionalized donor blocks
Fatty aromatic ketones 2.16.5 were used to prepare functionalized bi- and terthiophenes, which were subjected to the Vilsmeier reaction, and the resulting mixture of Z- and E-isomers of 3-aryl-3-chloroacrylaldehydes 2.16.6 was treated with mercaptoacetic acid ester in the presence of a base (Scheme 445). Yields of the products, thiophene-2-carboxylates 2.16.7, were in the range of 50 – 93%[1182, 1183]. It should be noted that both geometrical isomers of 2.16.6 were involved in this reaction and that the yields of thiophenes 2.16.7 in some cases exceeded the yield of each individual isomer. This method proved to be very versatile. It provided an access not only to the preparation of bi-, ter- (see[1182, 1183]) and quarterthiophenes[1184, 1185] but also to the construction of a more complex conjugated chain[1185, 1186], and the introduction of side functional groups[1182, 1185], long alkyl chains (including functionalized ones)[1182, 1187, 1188] or aryl substituents[1186, 1189-1191].

A similar strategy was employed in the synthesis of ethyl [2,2'-bithiophene]-5-carboxylates (Scheme 446).

In this case, ethyl 3-oxo-3-(thiophen-2-yl)propanoate (2.16.8) was reacted with POCl3 to give ethyl 3-(thiophen-2-yl)-3-chloroacrylate (2.16.9), which was treated with 2-sulfanyl acetate in the presence of sodium ethoxide. The intermediate ethyl 4-hydroxy-[2,2'-bithiophene]-5-carboxylate (2.16.10) was then alkylated with hexyl iodide to yield ester 2.16.11.
Bithiophene 2.16.12 was used to prepare diethyl esters of acids containing three thiophene rings (Scheme 447). This substrate was first acylated to ethyl 3-dodecyl-5'-dodecanoyl-[2,2'-bithiophene]-5-carboxylate (2.16.13), which conversion was accomplished in two ways. In the first case, ketones 2.16.13 were subjected to the Vilsmeier reaction and the intermediate 2.16.14 underwent the Fiesselmann reaction to give 3,3''-dodecyl-[2,2':5',2''-terthiophene]-5,5''-dicarboxylic acid (2.16.15). In the second case, ester 2.16.13 was previously hydrolyzed by alkali in alcohol and the resulting carboxylic acid was decarboxylated with formation of the compound 2.16.16. The Vilsmeier – Fiesselmann reaction cascade (via the formation of intermediate 2.16.17) furnished ethyl 3,3''-didecyl-[2,2'':5',2''-terthiophene]-5-carboxylate (2.16.18)[1191].

4-Azido- (2.16.22a – i) and 4-amino-5-arylthiophenes (2.16.23a – i) were prepared from the ketones 2.16.19 via azides 2.16.20 and 2.16.21 (Scheme 448)[1185]. Compounds 2.16.20a – i were subjected to the Vilsmeier reaction and the resulting mixtures of Z- and E-isomers of 2-azido-3-aryl-3-chloroacrylaldehydes (2.16.21a – i) were heated with methyl mercaptoacetate in the presence of cesium carbonate in dioxane or methanol.

When 1.5 equiv. of HSCH2CO2Me was used, the reaction did not proceed to completion. The use of 3 equiv. of this reagent led to the reduction of the azido group to afford amines 2.16.23a – i as the major products (61 – 78% yields). The compounds 2.16.23a – i were converted into azidothiophenes 2.16.22a – i via the corresponding diazonium salts in 49 – 78% yields. Irradiation of azides 2.16.22b – i at 356 or 254 nm afforded methyl 4H-thieno[3,2-b]indole-2-carboxylates (2.16.24b – i, 2.16.24'd,i) in 21 – 34% or 18 – 29% yields, respectively. Slightly higher yields of thienoindoles 2.16.24b – i (27 – 43% or 38 – 69%) were obtained by thermolysis of azides 2.16.22b – i in boiling chlorobenzene or in toluene in a microwave reactor (Scheme 449). An attempt to separate the thermolysis and photolysis products of compound 2.16.22a failed.

In 2021, a method for the preparation of annulated benzothiophenes based on the cyclization of 4,5-diaryl-substituted thiophenes was proposed (Scheme 450)[1186]. To select the optimal synthetic strategy, various methods of intramolecular cyclization of compounds 2.16.25 and 2.16.26 to methyl 5,6-dimethoxynaphtho[2,1-b:3,4-b']dithiophene-2-carboxylate (2.16.27a) were studied.

In the first case, the Pd-catalyzed cross-coupling took place on heating compound 2.16.25 in DMF in the presence of 10 mol.% Pd(PPh3)4 in yields of 42%. The oxidative intramolecular coupling of the substrate 2.16.26a treated by FeCl3 gave the same product 2.16.27a in higher yield (75%). However, higher yields of naphthodithiophene 2.16.27a (86 and 91%) were reached under UV light irradiation of a dilute solution of compound 2.16.25 in THF and in the case of iodine-promoted photocyclization of bithiophene 2.16.26a in THF mixed with 20 mol.% of molecular iodine. As the latter approach did not require the iodinated precursor 2.16.25 to be prepared in advance and provided high yields, it was used to obtain a series of benzo[2,1-b:3,4-b']dithiophene derivatives 2.16.27a – j (Scheme 451).

Methyl groups in compounds 2.16.26a and 2.16.27a were replaced with branched alkyl chains to improve solubility. For this purpose, compounds 2.16.26a, 2.16.27a were heated with AlI3 prepared in situ from Al and I2 and then phenols 2.16.28, 2.16.29 were alkylated with 2-ethylhexyl iodide in DMF in the presence of potassium carbonate. The reaction provided quantitative yields of intermediate phenols 2.16.28, 2.16.29 and moderate yields of compounds 2.16.30, 2.16.31 (Scheme 452)[1189].

Bithiophene 2.16.12 reacts with aryl halides (phenyl iodide, 1-bromonaphthalene, 9-bromophenanthrene, 1-bromopyrene) in DMF in the presence of Pd(PPh3)4 to afford ethyl 5'-aryl-3-decyl-[2,2'-bithiophene]-5-carboxylates (2.16.32) (Scheme 453)[1190].

Similarly, the reaction between ester 2.16.12 (and subsequently its analogue 2.16.33) and its bromination product, ethyl 5’-bromo-3-dodecyl-[2,2’-bithiophene]-5-carboxylate (2.16.34a) gave diethyl 3,3’’-didecyl-[2,2'':5',2'':5'',2'':5'',2'''-quaterthiophene]-5,5'''-dicarboxylate (2.16.35a, R = n-C10H21). A slightly higher yield (63%) of compound 2.16.35a was obtained via homocoupling of the bromine derivative 2.16.34a on heating in DMF in the presence of Pd(PPh3)4. Using the same protocol, bithiophenes 2.16.34b,c (R = n-C6H13 (b), (CH2)4NHC(O)Ph (c)) provided esters 2.16.35b,c in 49 and 25% yields, respectively. Esters 2.16.15 and 2.16.35 were hydrolyzed to the corresponding dicarboxylic acids 2.16.36, 2.16.37. It should be noted that compound 2.16.34a reacted with bithiophene to afford a mixture of quaterthiophene esters (2.16.38) and dicarboxylic acid 2.16.35a in yields of 26 and 19% correspondingly (Scheme 454)[1190, 1191].

2.16.2. Synthesis of conjugated heterocyclic donor – acceptor systems
D – A – D ensembles of five-membered heterocycles were synthesized from esters 2.16.7, 2.16.11, 2.16.18, 2.16.30, 2.16.31, 2.16.38, which were first converted to carboxylic acids and hydrazides and then used to prepare diacylhydrazines (Scheme 455)[1189, 1192-1194].

Diacylhydrazines were also obtained from hydrochloric acid hydrazine and acid halides[1192, 1193]. 1,3,4-oxadiazoles 2.16.39 – 2.16.46 and 1,3,4-thiadiazoles 2.16.47 – 2.16.49 were prepared by heating appropriate diacylhydrazines in POCl3 or toluene with Lawesson’s reagent (LR)[1189, 1192, 1193]. The reaction of the hydrazide with imidoyl chloride, formed in situ by the action of PCl5 on the anilide, gave 3,5-bis(3-dodecyl-[2,2'-bithiophene]-5-yl)-4-phenyl-4H-1,2,4-triazole (2.16.50)[1192].
Dicarboxylic acids 2.16.36, 2.16.37 and hydrazide 2.16.51 gave rise to ensembles of five-membered D1 – A – D2 – A – D1 heterocycles 2.16.52 – 2.16.55 in a similar way (Scheme 456)[1194].

Compounds containing two conjugated acceptor rings, 5,5'-bis(3-dodecyl-[2,2'-bithiophene]-5-yl)-2,2'-bi(1,3,4-oxadiazole) (2.16.57) and -bi(1,3,4-thiadiazole) (2.16.58), were prepared from hydrazide 2.16.51, which was acylated with oxalyl chloride and then cyclized under the action of POCl3 or LR[1193, 1195, 1196]. Acylation of hydrazide 2.16.51 with ethyl 2-chloro-2-oxoacetate gave diacylhydrazine 2.16.59, which was converted to ethyl 5-(3-decyl-[2,2'-bithiophene]-5-yl)-1,3,4-thiadiazole-2-carboxylate (2.16.60) by heating with LR followed by treatment with hydrazine hydrate to afford hydrazide 2.16.61. The reaction of hydrazide 2.16.61 with oxalyl chloride and subsequent Lawesson’s reagent-mediated closure of two thiadiazole rings in intermediate 2.16.62 furnished 5,5''-bis(3-dodecyl-[2,2’-bithiophene]-5-yl)-[2,2':5',2'':5'',2'''-quater(1,3,4-thiadiazole)] (2.16.63)[1196]. Using a similar reaction sequence, 5,5''-bis(3-dodecyl-[2,2'-bithiophene]-5-yl)-[2,2':5',2''-ter(1,3,4-thiadiazole)] (2.16.68) was obtained from hydrazide 2.16.61 via the formation of compounds 2.16.64 – 2.16.67 (Scheme 457)[1196].

2,6-Disubstituted benzo[1,2-d:4,3-d' ]bis(thiazoles) have also been proposed as acceptor blocks. The only known method for their preparation was based on the oxidative cyclization of N,N'-(phenylene)dithioamides. The use of strong oxidants excluded the introduction of electron-donating substituents into the molecule. It was proposed to initiate the cyclization of bis-thioamides by 440 nm light using chloranil as the oxidant[1197]. This method was used to prepare 2,7-bis(3-decyl-[2,2'-bithiophene]-5-yl)benzo[1,2-d:4,3-d' ]bis(thiazole) (2.16.72) from the corresponding bithiophenecarboxylic acids 2.16.69 and 1,4-phenylenediamine via the formation of diamides 2.16.70 and their thio analogues 2.16.71. The synthesis of compound 2.16.72 was carried out in both steady-state and flow-through photochemical reactors. The use of the flow mode allowed the product yield to be increased from 55 to 61% and the process time to be reduced from 4 – 6 h to 20 min. In order to compare the electronic and photophysical properties, the isomeric 2,6-bis(3-decyl-[2,2'-bithiophene]-5-yl)benzo[1,2-d:4,5-d']bis(thiazole) (2.16.73) was obtained from 2,5-diaminobenzene-1,4-dithiol and carboxylic acid (Scheme 458).

2.16.3. Electronic, electrochemical and photophysical properties of conjugated heterocycles. Structure – properties relationship
The position of the boundary orbitals and consequently the electron affinity (EA) and ionization potential (IP), which determine the redox, spectroscopic, electronic and photophysical properties of the conjugated molecule, depend on its structure. The tuning of these parameters is crucial for the design of any organic semiconductor. In this context, establishing the relationship between the structure of the conjugated molecule and its electronic and electrochemical properties is necessary for the synthesis of compounds with the desired properties.
Most of the above-mentioned oligomers were subjected to electrochemical oxidation and reduction. The compounds bearing unsubstituted terminal thiophene moiety were polymerized. Using cyclic voltammetry, the oxidation (Eox (onset)) and reduction (Ered (onset)) potentials were measured from the first oxidation and reduction peaks, and the IP and EA values were calculated using formulas (1) and (2)
where e is the elementary charge, and the formal potential of the ferrocene redox pair (EFc/Fc+) is 5.1 V. The energies of HOMO (–IP) and LUMO (EA) and the electrochemical bandgap were determined
Table 1, Table 2, Table 3 show the electronic and electrochemical properties of thiophene-containing oligomers, divided into two groups according to their composition: D – A – D (see Tables Table 1, Table 3) and D – A – D – A – D (see Table 2). Table 1 shows compounds which bear unsubstituted terminal thiophene rings, have different numbers of thiophene moieties and also differ in the number and nature of electron-withdrawing units. Table 3 combines D – A – D type oxadiazoles and thiadiazoles in which the electron-donating units have different structures (e.g., fused thiophenes and oligothiophenes with unsubstituted and substituted terminal rings) and contain different peripheral substituents.
![Electronic and electrochemical properties of D<sup>1</sup> – A – D<sup>2</sup> – A – D<sup>1</sup> type systems based on thiophenes and oxa(thia)diazoles[<a class="anchor" href="#reference-7765">1194</a>].](/storage/images/resized/KvMFsWUsQmggMu2iQBZktdOkiYfwk2AwnrxB0fkN_xl.webp)
The electronic and electrochemical properties of the first group of thiophene-containing oligomers are presented in Table 1 and Figure 8. Substitution of a single heteroatom in the acceptor ring of oligomers 2.16.39, 2.16.47, 2.16.50 significantly changes the electronic and electrochemical properties. In the series 4-phenyl-1,2,4-triazole 2.16.50 < 1,3,4-oxadiazole 2.16.39 < 1,3,4-thiadiazole 2.16.47, the acceptor properties of the central heterocycle increase and the LUMO level naturally decreases. In the same series, the oxidation potential increases and the band gap decreases. Thus, the replacement of the nitrogen atom by oxygen and sulfur atoms leads to a successive decrease in Egel: 3.12 (2.16.50), 3.06 (2.16.39), 2.77 (2.16.47) eV. At the same time, the HOMO level changes less significantly (see Table 1)[1184]. In contrast to compounds 2.16.50, 2.16.39, 2.16.47, the differences in the electronic and electrochemical properties of the isomeric benzobis(thiazoles) 2.16.72 and 2.16.73 are not significant[1197].
The introduction of additional electron-withdrawing rings significantly decreases the LUMO levels and slightly decreases the HOMO energy. Thus, the energy difference between the LUMO of thiadiazole 2.16.47 and quaterthiadiazole 2.16.63 is 0.77 eV and that between the HOMO levels is as low as 0.18 eV. The narrowing of the Egel energy gap is also observed in the dimerization of monomers 2.16.39, 2.16.47 (see compounds 2.16.53, 2.16.55 in Table 2)[1194] or during the expansion of the conjugation system due to the introduction of additional thiophene ring in the donor part of the molecule[1193] (2.16.41, 2.16.42, see Table 1). In this case, the boundary orbitals approach each other to a greater extent by increasing the HOMO level.
The LUMO level increases significantly during the polymerization of compounds 2.16.39, 2.16.41, 2.16.42, 2.16.47, 2.16.50. In addition, in some cases (e.g., for poly-2.16.41 and poly-2.16.42) the LUMO levels are higher than in the corresponding monomers. However, due to the fact that the increase of the LUMO level is faster, the band gap is significantly reduced (see Table 1 and Figure 8)[1193].
![[{"id":"Uv0DVY-rsz","type":"paragraph","data":{"text":"Position of the boundary orbitals in conjugated heterocyclic systems D – A – D and polymers based thereon. The energy levels of thiadiazoles are shown in grey-green, oxadiazoles in red and triazoles in blue."}}]](/storage/images/resized/WbW5l0Q2UjH7Oo3HtKoAmRjkFPLBFmiD1zufqokM_xl.webp)
Polyconjugated molecules often have poor solubility due to strong intermolecular interactions. Therefore, solubility enhancing (solubilizing) substituents, mostly long alkyl chains, are introduced into their structure.
The introduction of a substituent into the conjugated chain opens up further possibilities for modifying the redox, electronic and optical properties. The introduction of alkyl groups leads to a slight decrease in the oxidation potential, which shifts from +0.83 (2.16.39) to +0.77 (2.16.40), and from +0.63 (2.16.41) to +0.56B (2.16.44) with a single pendant alkyl substituent (see Table 1, Table 3)[1184, 1193, 1195]. The Egel decreases slightly, mainly due to an increase in the HOMO level.
The influence of alkyl groups primarily on the HOMO level was reported[1198]. The alkoxy substituent in 1,3,4-oxadiazole 2.16.43 has a similar but stronger effect due to the involvement of an oxygen electron pair into the conjugation[1184]. The effect of the aryl substituent is even more pronounced. The involvement of benzene and thiophene rings in a single conjugation system leads to a decrease in the LUMO and an increase in HOMO levels, and also to a band gap narrowing. When switching from 2,5-di([2,2'-bithiophen]-5-yl)-1,3,4-oxa- (2.16.39) and -1,3,4-thiadiazole 2.16.47 to naphthobithiophene derivatives 2.16.46 and 2.16.49, the Egel value decreases from 3.06 and 2.77 to 2.71 and 2.50 eV respectively. At the same time, the difference of Egel between aryl-substituted 2.16.45, 2.16.48 and fused structures 2.16.46, 2.16.49 is not great (see Table 1, Table 3).
Most 2,5-bis-([2,2'-bithiophene]-5-yl)-1,3,4-oxa(thia)diazoles and benzobis(thiazoles (e.g. compounds 2.16.72 and 2.16.73) are effective luminophores. They absorb light in the 360 – 420 nm range and emit light in the violet to orange part of the spectrum (430 – 594 nm), depending on the structure (see Table 4). The luminescence quantum yield of for compound 2.16.39 reaches 0.88. Replacement of the oxygen atom with sulfur leads to a decrease in the quantum yield due to the heavy atom effect[1183, 1194] and a bathochromic shift in the absorption and emission spectra. The quantum yield decreases as the conjugation chain elongates in both donor and acceptor parts of the molecule (see Table 4).
2.16.4. Application of donor – acceptor molecules
Due to their luminescent properties, functionalized thiophenes are of interest as dyes[1199, 1200]. More often, however, these compounds are used as building blocks in the synthesis of organic semiconductors. For example, dispersions of compounds 2.16.39, 2.16.50, 2.16.40, 2.16.57 derived from the appropriate thiophenecarboxylic acid derivatives, mixed with polyvinylcarbazole and 2-tert-butylphenyl-5-biphenyl-1,3,4-oxadiazole, exhibited LED behaviour (guest–host configuration), emitting blue light with a luminance of 120 cd m–2 and a luminous efficiency of 0.12 cd A–1 for non-optimized devices[1195]. Star-shaped molecules 2.16.74 – 2.16.77 derived from trimesic acid and the appropriate hydrazides, have been used as emitters in more efficient multilayer LEDs (luminance 1803 cd m–2 and luminous efficacy 3.43 cd A–1) (Figure 9)[1201].
![[{"id":"qIAWZHhM9R","type":"paragraph","data":{"text":"Structures of conjugated star-shaped donor-acceptor systems <b>2.16.74</b> – <b>2.16.77</b> and photos of their solutions in CH<sub>2</sub>Cl<sub>2</sub> under UV light irradiation (365 nm) (a), and linear D – A – D systems <b>2.16.78</b>, <b>2.16. 79</b> and a sample UV-Vis-NIR spectrum of a thin polymer film of <b>2.16.79</b> electrochemically deposited on an ITO electrode at increasing potential (p-doping) in a 0.1 M solution of TBAPF<sub>6</sub> in MeCN (b)."}}]](/storage/images/resized/k0unFBCzur6hpuJcO7119AHDn0exLSy99W04XJPo_xl.webp)
Polyconjugated D – A – D systems bearing no substituents in the terminal thiophene rings, are electrochemically oxidized to form electrochromic films on the electrode[1187, 1193, 1197]. Based on 5,8-di([2,2'-bithiophene]-5-yl)dithiazolo-[4',5':3,4;5'',4'':5,6]benzo[1,2-c][1,2,5]thiadiazoles (2.16.78) and -benzo[1,2-d][1,2,3]triazoles (2.16.79), electrochromic polymer films with high optical contrast in the IR region and a short response time were prepared. These effects make these compounds promising candidates for use in ‘smart windows’ that modulate the heat flow associated with IR radiation.
The data on conjugated thiophene-containing systems presented in this Section do not cover all the material available in the scientific literature on the preparation, properties and applications of such compounds. Only one synthetic approach to such compounds is considered here. The authors have worked closely over the years with Polish colleagues from Warsaw and the Silesian University of Technology, whose articles could not be cited in details due to space limitations.
2.17. Advances in the synthesis of heterocycles based on reactions of thioamides with azides, carbenoids and their precursors
In this Section, the special features of chemical and catalytic reactions of thioamides with azides, diazo compounds and their precursors, 1-sulfonyl-1,2,3-triazoles, are systematized for the first time. The possibility of 2-cyanothioacetamides to react with both heterocyclic azides and sulfonyl azides was demonstrated by tandem reaction of cycloaddition to 1-heteroaryl(sulfonyl)-1,2,3-triazoles and subsequent rearrangement to hetaryl(N-sulfonyl)amidines. Unlike other types of azides, sulfonyl azides can react with thioamides on heating to give heterocyclic N-sulfonyl amidines, casein kinase (CK1) inhibitors, antimicrobial and fungicidal agents. Of particular note is the ability of thioamides to react with diazo compounds and their potential precursors, 1-sulfonyl-1,2,3-triazoles, in the presence of rhodium and copper catalysts. The metal catalysis significantly increases the synthetic potential of reactions of thioamides with diazo compounds, opening the way to new heterocycles. It is shown that the key intermediates in these reactions are highly reactive and unstable carbenoids, the subsequent cyclization of which affords mesoionic thioisomünchnones and functionalized 1,2,3-triazoles.
Thioamides are an interesting and versatile class of organosulfur compounds. Cytostatic drugs containing a thioamide group, such as 6-mercaptopurine and 6-thioguanine, are well known[1202-1204]. Thioamides as amide isosters have been used in medicinal chemistry to increase the thermal and proteolytic stability and improve the pharmacokinetic properties of biologically active amide compounds[1205, 1206]. In addition, the thioamide moiety is part of natural compounds (e.g. closthioamide, methyl coenzyme reductase)[1207, 1208]. The ability to contribute to the formation of the spatial structure of proteins through non-covalent interactions makes thioamides valuable reagents for the modification of peptide chains[1209-1211].
A topical area in organic chemistry is the use of thioamides containing several reaction centers, which can also bear different functional groups, in the fine organic synthesis[1212-1214]. A variety of sulfur-, nitrogen- and oxygen-containing heterocyclic compounds, as well as transformation products of thiocarbonyl and thioamide groups such as amidines, sulfonyl amidines, enaminones, amides and nitriles, have been obtained in this way. The above-mentioned heterocyclizations of thioamides are based on cycloaddition or cyclocondensation with nitrogen and sulfur atoms and with compounds containing activated unsaturated carbon-carbon, carbon-nitrogen or carbon-oxygen bonds[201, 1215-1219].
Synthetic approaches to thioamides are well developed and are highlighted, e.g., in reviews[201, 1216, 1220]. The synthesis of heterocyclic compounds from thioamides has been further developed by reactions with hetaryl- (see[1221-1223]) and sulfonyl azides[1224, 1225], diazo compounds[1215, 1226, 1227] and 1-sulfonyl-1,2,3-triazoles[1228-1230], catalyzed by salts of transition metals and copper. As a result, new reactions have been discovered, new methods for the synthesis of heterocycles have been developed and compounds with practically useful properties have been identified. At the same time, there are no reviews focusing on the reactions of thioamides with azides and with carbenoids generated from diazo compounds by reactions with transition metals and their precursors. This Section highlights the systematization and synthesis of heterocycles obtained via reactions of thioamides with azides, carbenoids and 1-sulfonyl triazoles. Biological and optical-physical properties of compounds obtained from thioamides by catalytic reactions are also presented.
2.17.1. Reactions of thioamides with azides
The only study of the reaction between thioamides and sodium azide was performed by El-Ahl et al[1231]. The authors showed that the use of the SiCl4 – NaN3 system in the reaction of primary, secondary and cyclic thioamides 2.17.1 in boiling acetonitrile gives tetrazoles 2.17.2 in high yields (Scheme 459).

Due to their low electrophilicity, thioamides do not react with aliphatic azides. In contrast to aliphatic derivatives, aromatic azides readily react with ethoxycarbonyl thioacetamide 2.17.3 at room temperature to afford 1-aryl-1,2,3-triazoles 2.17.4 (Scheme 460)[1232].

It should be noted that in the first stage of this process, azides react with the methylene group of thioamide 2.17.3 to give triazene A (see Scheme 460). The thioamide group does not react with the azide group but with the newly formed triazene moiety to afford triazoline B. Elimination of sodium hydrosulfide completes the formation of triazole 2.17.4.
2-Cyanothioacetamides 2.17.5 react somewhat differently with aromatic azides (Scheme 461)[1221, 1222]. The presence of a cyano group in the molecule of such thioamides determines the direction of this reaction. It is the cyano group, not the thioamide group, which reacts with azides together with the methylene group of thioamide 2.17.5. The triazole ring of compounds 2.17.6 is probably assembled via the formation of triazene A. 1-Aryl-5-amino-1,2,3-triazole-4-carbothioamides 2.17.6a – g were obtained in high yields at 50 – 60 °C in aqueous alkali solution[1222] or at 0 °C in an alcohol solution of sodium ethoxide[1221].

Thioamides 2.17.5 react with 5-azidoaldehydes 2.17.7 to give quinolinotetrazoles 2.17.8 (Scheme 462; hereinafter compounds studied by X-ray diffraction analysis are denoted ‘X-ray’)[1233]. The cyclization proceeds in the absence of solvent and base at 80 °C. Apparently, the first step of the process involves the reaction of the methylene group of the thioamides 2.17.5 and the aldehyde group of the azides 2.17.7 to form azidonitriles A. Subsequent intramolecular cycloaddition of the azide group to the cyano group gives the final products 2.17.8. An alternative transformation involving the thioamide group does not occur.

This reaction tolerates both electron-withdrawing and -donating substituents in the aromatic ring of azidoaldehydes 2.17.7 but is restricted to the use of tertiary thioamides of 2-cyanoacetic acid 2.17.5. Notably, quinolinotetrazoles 2.17.8 can also be prepared in ethanol in the presence of triethylamine, but the yield of products is much lower because of tar-like product formation[1233].
The example of 5-azido-1-methyl-4-nitroimidazole (2.17.9) showed that heterocyclic azides react differently with 2-cyanothioacetamides 2.17.5 than with aromatic azides (Scheme 463)[1221]. The tandem of the 1,2,3-triazole-4-carbthioamide A formation and its subsequent Cornforth-type rearrangement[1234] to the product 2.17.10 containing two azole rings spaced by an amidine unit has been accomplished (see Scheme 463).

2-Cyanothioacetamides 2.17.5 react smoothly with azidopyrimidines 2.17.11 (Scheme 464), as does azidoimidazole 2.17.9 (see Scheme 463), in water in the presence of 1 equiv. of NaOH on cooling to give 5-cycloamino-1,2,3-thiadiazole-4-carbamidines 2.17.12 in high yields[1222].

The possibility of a one-pot synthesis of amidines 2.17.12 from available 6-chloropyrimidine 2.17.13, sodium azide and the appropriate thioamides 2.17.5 was demonstrated. Products 2.17.12 were obtained in 70 – 80% yields (see Scheme 464).
Sulphonyl azides are highly electrophilic reagents which, like heteroaromatic azides, undergo basic-catalytic reactions with thioamides. Unlike other types of azides (aryl and heterocyclic), they are able to react with the C=S group of thioamides.
Tertiary 2-cyanothioacetamides 2.17.5 react with sulfonyl azides in the presence of strong bases (EtONa, 5 equiv.) in protic solvents (methanol, ethanol, isopropyl alcohol) to give sodium salts of 5-sulfonylamido-1,2,3-triazoles 2.17.14 (Scheme 465)[1225, 1232].

The proposed reaction pathway involves the formation and cyclization of the intermediate 2-cyano-2-diazothioacetamide to 1,2,3-triazoles 2.17.14. A careful study of this reaction has shown the possibility of three directions leading to a mixture of compounds consisting of 5-amino-4-cyano-1,2,3-thiadiazoles 2.17.15, 5-sulfonylamido-1,2,3-triazole-4-carbothioacetamides 2.17.14 and 5-amino-1,2,3-thiadiazole-4-carboximidamides 2.17.16 (see Scheme 465). Optimal conditions have been found for the synthesis of individual compounds 2.17.14 – 2.17.16. Thus, thiadiazoles 2.17.15 are formed via the reaction of 2-cyanothioacetamides 2.17.5 with sulfonyl azides in pyridine at room temperature. Amidines 2.17.16 are obtained by rearrangement of salts of triazoles 2.17.14 in dilute hydrochloric acid[1225].
The reaction of thioamides with sulfonyl azides was discovered in 1984 by Zelenskaya et al.[1235] and further developed by Hatanaka and co-workers[1236, 1237]. Bakulev and co-workers[1232, 1238] first applied this transformation to the synthesis of methylene-active N-sulfonyl amidines, and then to the preparation of N-sulfonyl amidines bearing aliphatic[1224], aromatic and heterocyclic moieties[1239, 1240], including biologically active compounds[1224, 1239, 1240].
Based on this approach, a method was developed for the synthesis of analogues of modafinil[1241, 1242], a highly active dopamine transporter (DAT) inhibitor[1243], functionalized with an N-sulfonylamidine moiety. Thus, N-sulfonylamidines 2.17.18 were prepared from thioamides 2.17.17 and sulfonyl azides (Scheme 466)[1244].

The reaction can be carried out either in solvent (4‒17 h) or without solvent (1‒2 h); the yields of the products 2.17.18 are comparable, but the consumption of sulfonyl azide is reduced from 5 to 1 equiv. respectively (see Scheme 466)[1224]. Oxidation of the thioether moiety with hydrogen peroxide in glacial acetic acid gives the desired N-sulfonyl-2-(diphenylmethylsulfinyl)acetamidines 2.17.19 in moderate to high yields.
Investigation of the DAT inhibitory activity of amidines 2.17.19 showed that compared to modafinil (IC50 = 11.11 μM), compounds 2.17.19a,b are more active DAT inhibitors (IC50 = 22.07 and 26.33 μM, respectively). However, modafinil exhibits greater norepinephrine transporter inhibition activity (IC50 = 283.6 μM)[1244] than amidines 2.17.19a,b (IC50 = 87.7 and 126.6 μM, respectively).
The design of hybrid molecules containing heterocyclic and sulfonyl amidine units is highly relevant in search for new biologically active compounds, as it has been shown that the introduction of a sulfonyl amidine moiety into organic molecules can enhance the biological activity of resulting compounds or give them a different type of activity[1245-1247].
In the search for new compounds capable of inhibiting casein kinase[1248, 1249], the reaction between thioamides 2.17.20 and sulfonyl azides was used to obtain N-sulfonyl amidines 2.17.21[1236] (Scheme 467) bearing the imidazole moiety as the latter is known to impart diverse biological activities to chemical compounds[1250, 1251]. The reaction was carried out in boiling ethanol for 2‒10 h in a 1 : 1 ratio of the starting reagents. Of fifteen N-sulfonyl amidines 2.17.21 described in a study[1239], compound 2.17.21a proved to be the most potent inhibitor of casein kinase CK1ε (IC50 = 4.86 μM), but less active as compared to the PF-670462 inhibitor (IC50 = 7.7 μM) developed by Pfizer[1252].

Galieva et al.[1240] drew attention to benzothiazole and benzoxazole derivatives as objects for obtaining heterocycles bearing an N-sulfonylamidine moiety. It is known that benzothiazole derivatives have high antitumour activity[1253] and antibacterial activity against Gram-positive and Gram-negative bacteria[1254]. At the same time, benzoxazoles are structural isosters of nitrogenous bases, adenine and guanine, which allows them to interact easily with biological receptors[1255].
The optimum conditions for the reaction of benzothiazole- and benzoxazole-substituted thioamides 2.17.22 included boiling in ethanol the thioamide – azide mixture in a 2 : 1 ratio for 2‒21 h (Scheme 468)[1240].

Thioamides 2.17.24 containing trifluoromethyl (2.17.24a) and phenyl (2.17.24b) substituents on the C=S bond do not react with sulfonyl azides when heated in solvents (alcohols, DMF, 1,4-dioxane), probably because of the reduced electrophilicity of the sulfur atom[1240]. The target N-sulfonyl amidines 2.17.25 were obtained in moderate yields by carrying out the process under rather harsh conditions: heating the neat substrates with 2 equiv. of sulfonyl azide at 80 – 90 ºC (Scheme 469).

Amidines 2.17.23d,f,h,j,m,n and 2.17.25e were tested for antimicrobial activity. The dependence of optical density of solutions on the growth of bacterial and yeast cells at different concentrations of chemical compounds was determined using Gen5 software. It is shown that compounds 2.17.23d,f,h,j,m,n, 2.17.25e have antimicrobial activity against Staphylococcus aureus and the yeast fungi Candida albicans but all such derivatives are inactive against the Gram-negative bacteria Escherichia coli. The most pronounced bacteriostatic, bactericidal and fungistatic activity is exhibited by compound 2.17.23m (see Scheme 468)[1240].
To develop a synthetic approach to N-sulfonyl amidines of heteroaromatic acids, Ilkin et al.[1256] studied the reactions of thioamides 2.17.26 with alkyl- and arylsulfonyl azides (Scheme 470). It was found that thioamides 2.17.26 react with sulfonyl azides in various solvents such as n-butanol, n-propanol, toluene, ethanol, water and in the absence of solvent to form N-sulfonyl amidines 2.17.27.

A small library of N-sulfonyl amidines 2.17.27 consisting of 19 compounds was obtained under optimal conditions (solvent-free process at 88 °C and the thioamide : azide ratio = 1 : 2.5) in 67 – 92% yields[1256].
1-Aryl-1,2,3-triazole-4-carbothioamides 2.17.28 do not react with sulfonyl azides under the conditions described above for N-sulfonyl amidines (see Scheme 467 – Scheme 470). Studies of the reaction between thioamides 2.17.28 and sulfonyl azides have shown that it can be carried out in boiling n-propanol using a sevenfold excess of sulfonyl azides (Scheme 471)[1256]. The tandem process involves the rearrangement of thioamides 2.17.28 to compounds 2.17.29, which undergo iminosulfonylation to give N-sulphonyl amidines 2.17.30 in moderate to high yields.

For convenience, a one-pot method was developed for the preparation of amidines 2.17.30a,b,d from the appropriate readily available 5-amino-1-aryl-1,2,3-triazole-4-carbothioamides 2.17.28 in yields of 65, 41 and 49%, respectively (Scheme 472)[1256].

The yields of products 2.17.30 obtained by the one-pot procedure are higher than those obtained by the two-step method, and the procedure for carrying out the reaction is much simpler.
2-Aminothiazole-4-thiocarboxamide 2.17.31 reacts with sulfonyl azides to give N-sulfonyl amidines 2.17.32a,b. A similar reaction of isoxazole-containing thioamides 2.17.33 in n-propanol or n-butanol affords the products 2.17.34 in good yields (Scheme 473)[1257].

Thus, a wide range of N-sulfonyl amidines containing different heterocyclic moieties were obtained (see Scheme 467 – Scheme 473)[1239, 1240, 1256].
2.17.2. Catalytic reactions of thioamides with diazo compounds. Generation of carbenoids
Hussaini et al.[1257-1259] showed that copper- or rhodium-catalyzed reaction of thioamides 2.17.35 with diazocarbonyl compounds 2.17.36 gives enaminones 2.17.37 (Scheme 474).

In contrast to these findings, reactions of thioamides 2.17.38 with diazo compounds 2.17.39 under similar conditions yielded thioisomünchnones 2.17.40 bearing a hetaryl or aryl substituent as well as a chalcone moiety (Scheme 475)[1260].

A careful study of this reaction (variation of catalyst, solvent, temperature and reagent ratio) allowed us to find the optimum conditions for the synthesis of thioisomünchnones 2.17.40, which included the use of the catalyst Cu(OAc)2 in acetic acid (method a) or [Cu(MeCN)4]PF6 (method b) in dichloroethane at 80 – 100 °C[1260].
Preliminary studies have been carried out to find the best leaving group for thioamides 2.17.38a – f[1260]. It was found that pyrrolidine acts as such more preferably to piperidine, morpholine, aniline, dimethylamine and ammonia; the yield of thioisomünchnone 2.17.40a varies from 78 to 0% (Scheme 476).

The search for promising fluorophores for various areas of human activity, from domestic and medical to electronics and security, is currently an urgent task. Thioisomünchnones 2.17.40[1260] are yellow, orange or red crystalline substances and their solutions in organic solvents are yellow, pink or orange under visible light. The absorption spectra recorded in chloroform showed three maxima (Figure 10). The low energy absorption band at 422 – 482 nm is due to the internal charge transfer. This effect resulted from the large charge separation in the ground state of the molecules. The higher energy bands (258 – 291 nm and 309 – 334 nm) can be attributed to the π – π* transition.
![[{"id":"bp-ztQpIun","type":"paragraph","data":{"text":"Absorption (a) and fluorescence (b) spectra of thioisomünchnones <b>2.17.40</b> in chloroform (С = 5 × 10<sup>–5</sup> mol L<sup>–1</sup>) and in the solid state[[ type=\"anchor\" referenceId=\"7831\" ]]."}}]](/storage/images/resized/knq088FgIXKEwnbG6bpwgotlxX92w89mzK1Lu8vV_xl.webp)

Thioisomünchnones 2.17.40 (see Scheme 475, Figure 11) exhibit yellow, red, orange and green fluorescence in the crystalline state (see Figure 10 and Figure 11).
![[{"id":"1aJW8SQD85","type":"paragraph","data":{"text":"Photographs of thioisomünchnone <b>2.17.40c,i,k,m,n</b> (see [[ type=\"anchor\" referenceId=\"7831\" ]]) in daylight (a) and under UV irradiation (365 nm) (b)."}}]](/storage/images/resized/1CxIHGQDOAZMpbheZ4yzanZaricTWTr8NmzQKM3f_xl.webp)
The values of λex, λem, quantum yields and Stokes shifts have been measured for these derivatives. Compound 2.17.40k has the highest emission quantum yield (10%). Its analogue 2.17.40l showed red emission in solution, which was investigated in different solvents (Figure 12).
![[{"id":"Pq5HEzT7uc","type":"paragraph","data":{"text":"Absorption (С = 5 × 10<sup>–5</sup> mol L<sup>–1</sup>) (a) and emission (5 × 10<sup>–6</sup> mol L<sup>–1</sup>) (b) spectra of thioisomünchnone <b>2.17.40l</b> in various solvents at room temperature and photographs of the corresponding solutions in daylight (c) and under UV irradiation (365 nm) (d )[[ type=\"anchor\" referenceId=\"7831\" ]]. Solvent: 1 — tolulene, 2 — 1,4-dioxane, 3 — THF, 4 — chloroform, 5 — ethanol, 6 — ethyl acetate, 7 — acetone, 8 — acetonitrile, 9 — DMSO."}}]](/storage/images/resized/U4CbZzqEK89khaD3LGNt3MTcfnSCrDmvxgggzVFj_xl.webp)
Dilute solutions of thioisomünchnone 2.17.40l were shown to exhibit fluorescence in the range of 575 – 590 nm with quantum yields from 2.6 to 9.0%[1260]. The Stokes shift is significant and varies from 3022 to 3976 cm–1. The visible absorption band of chromophore 2.17.40l shows a significant solvatochromic effect due to an increase in solvent polarity from low-polar toluene (λabs = 500 nm) to high-polar DMSO (λabs = 488 nm). This suggests that thioisomünchnone 2.17.40l has a high modulus of dipole moment in the ground state. Therefore, the reported data[1260] on the photophysical properties of thioisomünchnone indicate the prospect of searching for new fluorophores in the series of mesoionic heterocycles.
Thioamides 2.17.41a – d react with the diazo compound 2.17.39a under similar conditions but in a different way – instead of thioisomünchnones, 2-triazol-4-yl-2-aminoacrylamides 2.17.42a – d are formed as E-isomers in 65 – 82% yields (Scheme 477)[1260].

Consequently, the replacement of the pyrrolidine moiety in the thioamide molecule by an NHPh group changes the reaction pathway[1260]. A careful study of the reaction of thioamides 2.17.38 and 2.17.41 with diazo compounds 2.17.39 has shown that the direction of the reaction does not depend on the conditions of the process, but is entirely determined by the structure of the thioamide used. At the same time, while the highest yields of thioisomünchnones 2.17.40 were reached in the presence of [Cu(MeCN)4]PF6 as catalyst, for enaminoamides 2.17.42, the complex (CuOTf)2 with benzene was preferred (see Scheme 475, Scheme 477).
It should be noted that replacement of the acetyl group with a cyano group in the diazo compounds 2.17.43 alters the direction of reaction with thioamides 2.17.38a, 2.17.41a,d. In this case, cyanoacrylamides 2.17.44 are selectively formed in 58 – 86% yields (Scheme 478)[1260].

The structure of cyanoacrylamides 2.17.44 was confirmed, in addition to the spectral data, by the single crystal X-ray diffraction study of compound 2.17.44a, which crystallizes as the Z-isomer[1260].
Thus, as a result of the study of the catalytic reaction of thioamides 2.17.38 with diazo compounds 2.17.39, a new synthetic approach to thioisomünchnones 2.17.40 has been developed[1260]. This method has a wide range of applications and provides thioisomünchnones 2.17.40 substituted in position 2 with aromatic and heteroaromatic substituents and containing a chalcone moiety. By varying the structure of the reagents, it is possible to change the direction of the process towards the formation of other compounds, such as heterocycle-substituted acrylamides 2.17.42 and enaminoamides 2.17.44. Optimal conditions have been found for carrying out all three transformations, giving products in high yields.
1-Sulfonyl-1,2,3-triazoles A are masked diazo compounds and exist in equilibrium with the diazoimine form B, which, using rhodium(II), nickel or silver salts as catalysts, generates a reactive carbenoid C containing electrophilic and nucleophilic centers (Figure 13)[1261].
Catalytic reactions of 1-sulfonyl-1,2,3-triazoles with C-, O- and N-nucleophiles have been reported[1262-1264]. Reactions of 1-sulfonyl-1,2,3-triazoles with S-nucleophiles such as sulfides[1265-1267] thiochromones[1230], thiiranes[1230], thionoesters[1268] and thioesters[1269] have been investigated.
Thioamides 2.17.45 – 2.17.48 were tested as substrates for reaction with 1-sulfonyl-1,2,3-triazoles (Scheme 479). A search for reactive thioamide showed that the reaction of isoxazolecarboxylic acid thioamides 2.17.45 and 2.17.46 with 1-sulfonyl-1,2,3-triazole 2.17.49 in the presence of rhodium(II) salt is accompanied by their desulfhydration to nitrile 2.17.50 (in the case of primary thioamide 2.17.45) and to amide 2.17.51 (for secondary thioamide 2.17.46). Thioamides 2.17.47 and 2.17.48 with a tertiary thioamide group remain intact under these conditions.

We hypothesized and experimentally confirmed[1270] that the creation of an additional C-nucleophilic center in thioamides 2.17.45 and 2.17.46 by introducing a double bond into the molecule would yield derivatives that would act as thiaazadienes in the RhII-catalyzed reaction with 1-sulfonyl-1,2,3-triazoles.
The scope of the reaction of thioamides of substituted acrylic acids 2.17.52a – d with 1-sulfonyl-1,2,3-triazoles 2.17.49a – d in the presence of Rh2(OPiv)4 is shown in Scheme 480. Dihydrothiophenes 2.17.53a – g were obtained in 64 – 89% yields and dr from 55 : 45 to 84 : 16. During chromatographic purification of these compounds, the sulfonyl imine group is hydrolyzed to the aldehyde group. The product 2.17.53f is formed as a mixture of (4RS,5RS)- and (4RS,5SR)-diastereomers with dr = 55 : 45 but purification by flash chromatography on SiO2 and then on neutral Al2O3 delivered the individual (4RS,5RS)-diastereomer 2.17.53f in 69% yield. Dihydrothiophene 2.17.53g was obtained after column chromatography on SiO2 as a mixture of (2RS,3SR)- and (2RS,3RS)-diastereomers with dr = 67 : 33 and the (2RS,3SR)-diastereomer was isolated in 78% yield after triturating compound 2.17.53g with diethyl ether.

Thioamides 2.17.54 with a tetrasubstituted double bond react with 1-sulfonyl-1,2,3-triazoles 2.17.49a,c to give dihydrothiophenes 2.17.55a – g containing spiro- and dispirocyclic fragments and a stereogenic center (Scheme 481). The reaction is catalyzed by Rh2(OPiv)4 and provides products in 65 – 93% yields.

To extend the scope of the reaction of thioamides of substituted acrylic acids with carbenoid precursors, a series of diazo compounds 2.17.56 have been used as the latter[1270]. A study of the reaction of thioamide 2.17.52a with diazoacetamides 2.17.39 (R1 = Ac or CH=CHAr) and 2-cyano-2-diazoacetamides 2.17.39 (R1 = CN) showed that diastereomeric dihydrothiophenes 2.17.56 are formed using Rh2(OPiv)4 (0.5 – 1 mol.%) or [Cu(MeCN)4]OTf (10 mol.%) as catalysts in benzene, chloroform or dichloroethane (Scheme 482). The Rh-complex catalyzed reaction of thioamide 2.17.52a with diazoacetamide 2.17.39a in benzene at room temperature afforded dihydrothiophene 2.17.56a/2.17.56ʹa in 97% yield with a (2RS,3SR) : (2RS,3RS) diastereomeric ratio of 57 : 43 (see Scheme 482). (2RS,3SR)- and (2RS,3RS)-isomers were separated by column chromatography and isolated in 61% and 36% yields, respectively. The diastereoselectivity of the reaction can be shifted towards dihydrothiophene (2RS,3RS)-2.17.56ʹa using [Cu(MeCN)4]OTf as the catalyst at 90 – 100 °C.

(2RS,3SR)-Dihydrothiophenes 2.17.56b,c containing a chalcone moiety were obtained in 54 and 59% yields by RhII-catalyzed reaction in benzene or chloroform at room temperature. CuI-catalyzed reaction in dichloroethane at 90 °C provided (2RS,3RS)-dihydrothiophenes 2.17.56ʹb,c in 49 and 56% yields. Dihydrothiophenes 2.17.56d – f and 2.17.56ʹd – f are formed from thioamide 2.17.52a and diazo compounds 2.17.39 in 44 – 56% ((2RS,3RS)-isomers) and 39 – 44% ((2RS,3SR)-isomers) yields in the presence of RhII catalyst (see Scheme 482).
Diazo compounds 2.17.57 react with thioamides 2.17.52 under mild conditions to furnish spiro-annulated dihydrothiophenes 2.17.58 in 10 – 60 min with low Rh2(OPiv)4 loadings in 71 – 98% yields (Scheme 483). The conditions chosen are suitable for the synthesis of both monospirodihydrothiophenes 2.17.58a – f and dispirocyclic compounds 2.17.58g – i.

The feasibility of enantioselective synthesis using chiral catalysts (Rh2((S)-PTAD)4, Rh2((S)-PTTL)4, Rh2((S)-DBPTTL)4, Rh2((S)-PTV)4, Rh2((S)-NTTL)4) is illustrated for compound 2.17.58j (see Scheme 483)[1270]. The Müller catalyst Rh2(S-NTTL)4 (see[1271]) performed best providing the highest yield of dihydrothiophene 2.17.5j (96%) and the highest enantiomeric excess (97% ee).
The data presented in this review demonstrate the use of reactions of thioamides with azides and carbenoids derived from diazo compounds or from 1-sulfonyl-1,2,3-triazoles in the synthesis of heterocyclic compounds. It has been found that the replacement of aromatic azides with heterocyclic or sulfonyl azides significantly boost the synthetic potential of such transformations. Replacing aromatic azides with heterocyclic or sulfonyl azides has been found to significantly increase the synthetic potential of such transformations.
Due to low electrophilicity, aliphatic and aromatic azides do not react with thioamides at the thiocarbonyl group. 2-Cyanothioacetamides and highly electrophilic nitrophenyl-substituted azides form 5-amino-1-aryl-1,2,3-triazoles in the presence of strong bases.
Heterocyclic azides, like sulfonyl azides, react differently with 2-cyanothioacetamides under these conditions via a tandem reaction of cycloaddition to 1-heteroaryl(sulfonyl)-1,2,3-triazoles and rearrangement to heteroaryl(N-sulfonyl)amidines. Unlike other types of azides, the reaction of sulfonyl azides with thioamides on heating produces heterocyclic N-sulfonyl amidines, among which compounds with diverse biological activities such as antimicrobial, DAT or CK1 inhibitory and fungicidal have been identified.
The review shows that thiocarbonylpyrrolidines, in contrast to thioamides bearing other amino groups, form thioisomünchnones in high yields and that replacing pyrrolidine with other amines in this reaction leads to acrylamides. In addition, the direction of the catalytic reaction of thioamides with diazo compounds is affected by the structure of the latter. Regardless of the structure of the thioamides, 2-diazo-2-cyanoacetamides form 2-cyanoacrylamides.
Thioisomünchnones were found to exhibit fluorescence both in solution and in the crystalline state. The presented data[1260] on the photophysical properties of thioisomünchnones suggest the prospect of searching for new fluorophores in the series of mesoionic heterocycles.
In contrast to 2-cyanothioacetamide, the products of its reaction with aldehydes, thiocarbamoylacrylonitriles, react readily with carbenoids generated either from 1-sulfonyl-1,2,3-trizoles or from diazo compounds. The resulting dihydrothiophenes with a high degree of diastereoselectivity may have a spirofused structure or two chiral centers.
To conclude, the availability of thioamides of different structures allows us to study the influence of their structure on their reactivity in reactions with azides, as well as with diazo compounds and with 1-sulfonyl-1,2,3-triazoles under catalytic conditions.
2.18. Phosphorus-containing heterocycles: synthesis, coordination chemistry and applications
Heterocyclic phosphorus derivatives are a class of organophosphorus compounds with a unique set of physical and chemical characteristics and practically useful properties. The relative conformational rigidity of the heterocyclic backbone of the molecule leads to a predictable arrangement of exocyclic groups. This predictability is crucial for building-up the three-dimensional architecture of pharmaceuticals for effective interaction with the target molecule, as well as for forming of the second coordination sphere of transition metal complexes in the case of coordination with the endocyclic phosphorus atom providing control over the stereo- and enantioselectivity of catalysts based on these complexes. In addition, exocyclic substituents are able to provide additional nucleophilic or electrophilic assistance for the activation of small molecules at the central ion, and to control physical characteristics of the complexes such as solubility, luminescence or magnetic properties.
In this review, we did not aim to provide comprehensive data on all known representatives of phosphorus-containing heterocycles, nor did we seek to describe all the examples of their synthesis available in the literature. Most of synthetic methods are quite trivial for organophosphorus chemistry and consist of well-known and thoroughly described stages. Multi-stage chemoselective processes, that occur under thermodynamic control, made possible by the thermodynamic stability of heterocyclic organophosphorus compounds, are much more interesting. In this regard, we focused our attention on the synthetic approaches driven by this specific character of phosphorus-containing heterocycles, allowing to synthetize the final products through a series of cascade transformations or covalent self-assembly processes.
It is worth noting that the important advantage of the methods presented below is the ability to form target phosphorus heterocycles in a single step from acyclic precursors. At the same time, despite of their attractiveness, such reactions are limited to a small number of examples. These include reactions in two- and three-component systems involving di- and trifunctional reagents, such as:
— condensation of primary phosphines or secondary bisphosphines with formaldehyde in the presence of primary amines or diamines,
— interaction of cyclic phospholide anions with organic electrophiles and polyphosphides of alkali metals with unsaturated compounds,
— cycloaddition of phosphacyclopentadienes,
— reactions of electron-rich aromatic compounds with chlorides of vinylphosphonic acids.
Such multi-stage chemoselective processes are discussed in detail below. In addition, data on the conformational behavior of the described organophosphorus cycles and macrocycles in the free state or in the coordination sphere of a transition metal are separately considered, as well as unique dynamic processes associated with the reversible transformation of the heterocyclic framework. The practical applications of these phosphorus containing heterocycles are discussed on the base of their use as antimicrobial and antitumor agents, possessing high selectivity towards tumor cells; nanosensors for determining acidity, temperature, and concentration of small biologically valuable molecules; and catalysts for hydrogen energetics. Finally, we address matters related to the improving the biocompatibility of metal complexes with P-ligands in the form of nanoparticles stabilized in a polyelectrolyte matrix, as well as to the design of materials for spintronics based on complexes of aromatic phospholide anions with d-metals.
2.18.1. Synthesis of phosphorus-containing heterocycles by cascade transformations and covalent self-assembly of acyclic precursors
Herein, we will discuss the multi-step reaction sequences, which are in general carried out without isolation of intermediate compounds and provide access to various classes of phosphorus heterocycles (benzo[e][1,2]oxaphosphinines, phosphacyclopentadienes and phosphamacrocycles).
Cyclic esters of phosphonic acids are an important class of organophosphorus compounds due to their wide range of possible applications. Being the phosphorus analogs of lactams, they are valuable precursors for the synthesis of biologically active compounds. Besides this, they can be used for the design of structurally rigid three-dimensional scaffolds possessing P=O moiety and thus capable of complexation with various metal ions[1272-1274].
Generally, cyclic phosphonates are obtained via intramolecular C – O, C – C or P – C bond formation; occasionally, various cycloadditions are also employed. Several review papers summarizing the progress in the field have appeared recently, and the interested reader is referred to them[1275-1277]. Herein, we will focus on a relatively rare approach to the synthesis of cyclic phosphonates starting from acyclic precursors based on the formation of multiple endocyclic P – O and C – C bonds in a one-pot manner.
Presumably the first example of such a reaction is the interaction of substituted salicylic aldehydes with triethyl phosphonoacetate reported in 1996, which yielded 3-phosphorylcoumarins alongside with small amounts of benzo-[e][1,2]oxaphosphinine-3-carboxylates[1278]. Subsequently, the reaction of (2-ethoxyvinyl)phosphonic dichloride 2.18.1 with various phenols has been described, yielding either cage phosphonates 2.18.2, or benzo[e][1,2]oxaphosphinines 2.18.3 (phosphacoumarins). The structure of a starting phenol has the most influence on the reaction outcome. In the case of resorcinol[1279], 2- (see[1279]) and 4-methylresorcinols, 4-haloresorinols[1280, 1281], as well as pyrogallol[1279] and hydroquinone[1282], the reaction results in the bicyclic compounds 2.18.2a – g as the sole products in 72 – 85% yield. Similarly, 1- and 2-naphthols[1283], as well as 1,8-naphthalenediol[1284] give cage phosphonates 2.18.2h – g. However, the yields of these compounds are somewhat lower, and the phosphacoumarins 2.18.3 are also present in the reaction mixtures, albeit in minor quantities.
It is interesting to compare the synthetic outcome of the reaction for the resorcinol, 2-methylresorcinol and 4,6-disubstituted resorcinols. In the same reaction conditions the first two react with the phosphonate 2.18.1 to give bicyclic products 2.18.2a,b, whereas the latter furnish phosphacoumarins 2.18.3a,b (Scheme 484)[1285]. Notably, in the case of 4,6-dimethylresorcinol the phosphonate 2.18.2 is still observed as a minor product. On the contrary, dichloro-substituted resorcinol leads only to the formation of compound 2.18.3b.

A similar trend can also be observed for naphthols. 2-naphthol reacts with compound 2.18.1 to give cage phosphonate 2.18.2h as the major product. The introduction of the bromine substituent at the 6th position switches the reaction pathway to the formation of 2.18.3d[1286]. The same result is also observed for 2,3,5-trimethyl- (see[1287]) and 3-methoxyphenol[1288], giving phosphacoumarins 2.18.3с and 2.18.3e.
Examination of these data reveals that the reaction outcome depends on the ability of a phenol to undergo the electrophilic substitution reactions, i.e., its nucleophilicity. The less nucleophilic phenols are more likely to forme phosphacoumarins 2.18.3 and wise versa — more nucleophilic phenols tend to give bicyclic phosphonates 2.18.2.
Benzophosphorinins 2.18.3 can also react with another phenol molecule, which results in 4-aryl-3,4-dihydrobenzo-[e][1,2]oxaphosphinines 2.18.4 (Scheme 485)[1287]. However, this reaction requires rather harsh conditions, namely, prolonged refluxing in toluene in the presence of trifluoroacetic acid with simultaneous passing of hydrogen chloride stream. Subsequent treatment of the compounds 2.18.4 with sulfuryl chloride in THF leads to the formation of phosphonates 2.18.5. Obviously, this reaction proceeds by the formation of corresponding acid chlorides. It also provides access to the unsymmetrical phosphonates 2.18.5 with planar chirality, albeit in the racemic form[1289-1292].

The double C=C in phosphacoumarins 2.18.3 is sufficiently reactive to undergo 1,3-dipolar cycloaddition reactions. An example is the reaction of compounds 2.18.3 with azomethyne ylides generated in situ from ninhydrine and sarcosine or L-proline[1286]. In the case of sarcosine the reaction gives spiro-derivatives of benzo[e][1,2]oxaphosphinines 2.18.6a – d. When L-proline is employed instead of sarcosine, tricyclic benzo-[e][1,2]oxaphosphinines 2.18.6e – g are formed (Scheme 486). According to NMR data, the products are formed in 60 – 90% yields. However, the isolated yields are lower, presumably due to hydrolysis during chromatographic work-up. It is noteworthy, that the presence of an electron-withdrawing substituent significantly lowers the yield of the compound 2.18.6с.

We should also note the high regio- and diastereoselectivity of the reaction, which results in a single regioisomer with dr > 95 : 5. The (SS/RR)-configuration was confirmed by X-ray analysis for the compound 2.18.6с. Interestingly, the high diastereoselectivity is also observed in the case of L-proline, in spite of the formation of three stereocentres in the course of the reaction. The (SRS/RSR)-configuration was also confirmed by X-ray analysis for the compound 2.18.6g.
The synthesis of phosphacoumarins 2.18.3 starting from terminal alkynes and 2,2,2-trichloro-benzo[d][1,3,2]-dioxaphospholes 2.18.7 should also be mentioned here (Scheme 487)[1293-1295]. Interestingly, one of the oxygen substituents in the aromatic ring undergoes ipso-substitution during the reaction. There is the also the only example of the synthesis of phosphacoumarin 2.18.3t via photocatalytic cross-coupling of (2-hydroxyphenyl)boronic acid (2.18.8) and diethyl hex-1-yn-1-ylphosphonate (2.18.9) in the presence of a palladium catalyst[1296]. In this case, phosphonate 2.18.9 acts as a synthetic equivalent of (2-ethoxyvinyl)phosphonic dichloride 2.18.1.

In summary, we can state that the reaction of electron-rich aromatic compounds, such as phenols and naphthols, with 2-ethoxyvinylphosphonates or their synthetic equivalents — ethynylphosphonates — is an effective method for the synthesis of phosphorus-containing heterocycles. The ability to carry out the reaction without isolation of intermediate products and the availability of starting compounds and catalysts make this approach attractive for the synthesis of both phosphacoumarins — analogues of natural coumarins, and bicyclic cage phosphonates. Given the ease of functionalization of phosphacoumarins, it is hoped that the chemistry of these compounds will continue to evolve.
Phospholes are five-membered 6π-systems including one or more phosphorus atoms, characterized by the lowest aromaticity in the series of N-, O- and S-heterocycles. Phospholes are able to oxidize, sulfurize and form complexes with transition metals, which fundamentally distinguishes them from classical heterocycles. Such chemical modifications allow to obtain from these substrates in one stage various organophosphorus compounds with a wide potential of practical applications. It is worth noting two areas of application for phospholes that are now being actively developed. Firstly, the possibility of coordination with transition metals determines their application as labile π-acceptor mono- and bidentate ligands for the construction of organic reactions catalysts[1297], including asymmetric one designed to obtain enantiopure substances. By varying the substituents at both the phosphorus and carbon atoms of the phosphorus cycle, the physical and chemical properties of phospholes can be easily tuned for the successful realization of catalysis[1298, 1299]. Secondly, phospholes have attracted considerable interest as ‘building blocks’ for the creation of π-conjugated materials[1300, 1301], used in molecular electronics, including light-emitting diodes, thin-film transistors, and photovoltaic cells[1302, 1303]. The processes of oxidation, sulfurization of the phosphorus atom in monophospholes or the formation of complexes with transition metals affect the shift of the absorption and emission maxima, resulting in a material with specific properties[1304]. Unlike their aromatic counterparts, such as pyrrole and thiophene, phospholes have a ‘more flexible’ electronic structure, and the slightest changes in it can cause significant changes in photophysical characteristics, indicating the great potential of these compounds for sensing applications[1305, 1306].
To date, a significant number of methods have been proposed for the synthesis of various phospholes, the main disadvantages of which are multistage, the use of toxic phosphorus(III) chloride and organometallic compounds. In this regard, the efforts of researchers are aimed at reducing the number of stages and the use of elemental (white) phosphorus (P4) in the synthesis of organophosphorus compounds. The development of white phosphorus activation methods[1307-1309] has contributed to the emergence of new approaches to the synthesis of phospholide anions[1310], the alkylation of which leads to the formation of various phospholes.
The study of the reactivity of alkali metal polyphosphides 2.18.10 towards compounds containing multiple bonds has allowed the development of a fundamentally new approach to aromatic five-membered P-heterocycles — phosphacyclopentadienide anions (phospholide anions) 2.18.11 – 2.18.14 (Scheme 488). A method for the synthesis of 2,3,4,5-tetraphenyl-1-monophosphacyclopentadienide lithium (2.18.11) based on the reaction of white phosphorus and 1,4-dilithio-but-1,3-diene has been developed[1311]. The interaction of alkali metal sodium polyphosphides (NaxPy, 2.18.10) with cyclopropenyl phosphonium salts proceeds with the transformation of the phosphorus skeleton, resulting in the formation of 3,4,5-trisubstituted sodium 1,2-diphospholides (2.18.12) containing various arene and thienyl substituents[1312]. One-pot reaction in a system containing white phosphorus, sodium and alkynes or nitriles gives exclusively 1,2,3-triphospholide anions (2.18.13)[1313] or 4-aza-1,2,3-triphospholides (2.18.14)[1314] in 61 – 72% yield. The driving force behind these processes is the aromaticity of the resulting alkali metal phospholides.

The appearance in recent years of universal and efficient methods for the synthesis of phospholide anions with various substituents has allowed a comprehensive study of the chemical behavior of 1-mono-, 1,2-di- and 1,2,3-triphospholides anions with organic and organometallic electrophiles and metal complexes. The interaction of lithium 2,3,4,5-tetraphenyl-1-monophospholide (2.18.11) with alkyl halides leads exclusively to 1-alkyl-2,3,4,4,5-tetraphenyl-1-monophospholes (2.18.15) containing substituents at the phosphorus atom, the oxidation of which gives phosphine oxides 2.18.16. The interaction of sodium 1,2-diphospholides 2.18.12a – g with primary alkyl bromides or secondary alkyl iodides leads to the formation of previously unknown 1-alkyl-3,4,5-triphenyl-1,2-diphosphacyclopenta-2,4-dienes (2.18.17) in high yield (Scheme 489)[1315, 1316].

Of particular interest is the cycloaddition of 1-alkyl-1,2-diphospholes combining simultaneously fragments of both 1H- and 2H-phospholes in one molecule. Thus, in the reaction with maleic acid derivatives, 1-alkyl-1,2-diphospholes 2.18.17 act as dienes to form the only [4+2]-cycloaddition products — anti-endo-diphosphanorbornenes 2.18.18 with yield up to 90% (Scheme 490)[1317, 1318]. At the same time, like the 2H-phospholes, the 1-alkyl-1,2-diphospholes 2.18.17 act as dienophiles and lead exclusively to 9-alkyl-3,4-dimethyl-6,7,8-triphenyl-1,9-diphosphabicyclo[4.3.0]nona-3,7-dienes in reaction with 2,3-dimethylbuta-1,3-diene (2.18.19).

Among the heterocyclic phosphorus derivatives, cyclic aminomethylphosphines attract special attention[1319-1324]. The well-known representative of this class of compounds is bicyclic triazaphosphadamantane, which has become an effective tool in the creation of a wide range of water-soluble metal complexes. These complexes form the basis of a new generation of biphasic catalysts for organic reactions and antitumor drugs[1325]. In 2006, it was discovered that coordination compounds of another type of cyclic aminomethylphosphines — eight-membered 1,5-diaza-3,7-diphosphacyclooctanes — could be considered as functional analogs of natural enzymes, specifically hydrogenases[1326-1329]. The specific catalytic activity of aminomethylphosphine complexes is attributed to the endocyclic fragment PCH2NCH2P. The two phosphorus atoms form a chelate complex with a transition metal, while the nitrogen atom, located in close proximity to the reaction center due to the cyclic structure of the ligand, provides nucleophilic assistance in the formation of the H2 molecule during proton electrochemical reduction and in the activation of the H2 molecule during electrochemical oxidation. Indeed, it has been demonstrated that nickel complexes can facilitate key catalytic reactions in hydrogen energetics at rates comparable to or even exceeding those of heterogeneous catalysts based on metallic platinum, which were previously considered unique until the appearance of the nickel complexes of aminomethylphosphine. Consequently, this opens up the possibility of replacing the expensive platinum component of catalysts, giving new impetus to the development of hydrogen energetics[1326, 1330].
The condensation of primary mono- or secondary bisphosphines with formaldehyde and primary amines is an efficient method for synthesizing cyclic aminomethylphosphines. The creation of a large library of such compounds is facilitated, on the one hand, by the availability of primary amines with a wide variety of substituents and, on the other hand, by the development of methods for obtaining the necessary phosphorus-containing substrates. Varying the substituents on the endocyclic phosphorus and nitrogen atoms allows for precise tuning of the nucleophilicity and basicity of the donor atoms, and also expands the possibilities for constructing catalysts with additional interactions in the second coordination sphere and the ability to immobilize on various substrates and in biphasic systems. Progress in the field of cyclic aminomethylphosphines over the last decade has been attributed to the development of covalent self-assembly methodology[1331] and the study of the dynamic behavior of phosphorus- and nitrogen-containing cyclophanes, corands, and cryptands, as well as the synthesis and use in Mannich condensation of new primary mono- and secondary bisphosphines with functional substituents on the phosphorus atom.
1,5-Diaza-3,7-diphosphacyclooctanes are one of the best studied classes of cyclic aminomethylphosphines. These bisphosphines can be synthesized through the stereoselective condensation of a primary phosphine, formaldehyde, and a primary amine. The reaction is typically carried out in two stages: the phosphine reacts first with the formaldehyde, followed by interaction with the amine. This results in the formation of a single isomer of 1,5-diaza-3,7-diphosphacyclooctane with a syn-configuration of the lone pairs (LP) on the phosphorus atoms. Both in the crystalline phase and in solutions, it exists in a ‘crown’ conformation, pre-organized for the formation of chelate P,P-complexes with transition metal ions[1332]. Recent progress in the chemistry of 1,5-diaza-3,7-diphosphacyclooctanes is directly related to the development of synthetic methods for primary phosphines. Chiral cyclic bisphosphines based on L-menthylphosphine (2.18.20)[1333, 1334] and their water-soluble analogs based on o-phosphinophenol (2.18.21)[1335] have been synthesized, as well as a series of polydentate cyclic compounds based on primary phosphines with additional donor fragments in the substituents at the phosphorus atoms, such as 2-pyridyl- (2.18.22a – e)[1336, 1337], 2-(2-pyridylethyl)- (2.18.23a – c)[1338-1340], and 2-(2-thienylethyl) derivatives (2.18.24a,b) (Scheme 491)[1341].

The methodology of covalent self-assembly in three-component systems consisting of primary phosphine, formaldehyde, and primary diamine with spatially separated amino groups has paved the way for the creation of a new type of cyclic aminomethylphosphines — cage tetraphosphacyclophanes 2.18.25 – 2.18.39 (Scheme 492). The driving force for the self-assembly of the macrocycles appears to be the maximum thermodynamic stability of the cyclophanes compared to other oligomeric condensation products present in the reaction mixture[1331].

In the course of studying the macrocyclization reaction, cyclophanes have been obtained in which two 1,5-diaza-3,7-diphosphacyclooctane fragments are separated by bridging groups containing two (2.18.25 – 2.18.31), three (2.18.32 – 2.18.39), and even four (2.18.40) para- or meta-phenylene fragments. In all cases, a single isomer is formed with the lone pairs (LP) of all phosphorus atoms directed towards the cavity of the macrocycle. The cavity size of the synthesized cyclophanes 2.18.38 and 2.18.39 allows the encapsulation of solvent molecules[1342]. Cyclophanes 2.18.30, 2.18.31, 2.18.36, and 2.18.37 with additional donor fragments based on 2-pyridyl and 2-(2-pyridyl)ethylphosphines have also been synthesized[1343-1345].
By condensing phenyl- or L-menthylphosphine with formaldehyde and 4,4'-bis(4-aminophenoxy)biphenyl through a process of covalent self-assembly, the first representatives of 46-membered P,N,O-containing cyclophanes 2.18.40 and 2.18.41 have been obtained (Scheme 493)[1346].

The use of secondary bisphosphines, where the phosphine groups are separated by a hydrocarbon bridge, in a three-component condensation reaction leads to the formation of the corresponding six-membered (2.18.42) and seven-membered (2.18.43) heterocycles (Scheme 494), as well as 14- (2.18.44), 16- (2.18.45 – 2.18.47), 18- (2.18.48, 2.18.49), and 20-membered (2.18.50) macrocyclic tetraphosphines (see Scheme 495), depending on the length of the bridge and the basicity of the primary amine[1347-1349].


In the case of bis(phenylphosphino)methane (n = 1), an equimolar mixture of racemic and meso-isomers of compound 2.18.42 was formed. A similar situation was observed for 1,2-bis(phenylphosphino)ethane (n = 2) when reacted with arylamines (mixtures of rac- and meso-2.18.43a – c); however, more electron-donating benzylamines produced only the meso-isomer (2.18.43d – i). By further increasing the basicity of the amine the covalent self-assembly process was realised and only 14-membered cyclic tetraphosphines were crystallized as a single isomer from the reaction mixture (rac-2.18.44)[1350-1352].
It has been demonstrated that for bis(arylphosphino)propanes, -butanes, and -pentanes, the covalent self-assembly of macrocyclic tetraphosphines becomes the main direction of the reaction. The macrocyclic phosphines are formed with high stereoselectivity: when there is an even number of methylene groups between the phosphorus atoms, a rac-isomer is formed where the lone pair (LP) of the neighboring phosphorus atoms are positioned on opposite sides of the macrocycle plane, while an odd number of methylene groups results in the formation of an RSSR-isomer. In the RSSR-isomer, the LP of one pair of phosphorus atoms connected by a hydrocarbon bridge are situated on one side of the macrocycle, and the LP of the other pair are on the opposite side. This empirical rule[1353] has been validated for numerous examples of 14- (2.18.44a – e), 16- (2.18.45a – g, 2.18.46a – j, 2.18.47)[1353-1360], 18- (2.18.48a – c, 2.18.49)[1353, 1361], and 20-membered (2.18.50) corands (see Scheme 495)[1353]. Only a few exceptions to this rule are known. For instance, there are two examples of the synthesis of RSSR-isomers of 14- and 18-membered macrocycles with bulky iso-pentyl and tert-butyl substituents at the nitrogen atoms, respectively. However, in the case of 1,6-bis(arylphosphino)hexane, a deviation from the empirical rule is observed, resulting in the formation of an RSSR-isomer 2.18.51, which is atypical for an even number of methylene groups[1362].
It is noteworthy that during the synthesis of 1,4-bis(pyridylphosphino)butane, a phospholane with a 2-pyridyl substituent at the phosphorus atom was isolated as a byproduct[1363]. This compound was used as a sterically unhindered bridging PN-ligand for the creation of bi- and tetranuclear derivatives of copper(I), silver(I), and gold(I) with unusual luminescent properties[1364-1367].
The methodology of covalent self-assembly implies the reversibility of all reaction stages and, consequently, a fairly high lability of bonds in the final reaction products. 1,5-Diaza-3,7-diphosphacyclooctanes and cyclophanes with this heterocyclic fragment did not exhibit signs of dynamic behavior. At the same time, mutual transitions of stereoisomers were observed in solutions of the seven-membered heterocycles 2.18.43d – i. For example, in the NMR spectra (1H and 31P) of the pure meso-isomer 2.18.43c, signals corresponding to the rac-isomer appeared (Scheme 496). Equilibrium in anhydrous solvents and at room temperature was established within a few days. The calculated activation energy of this transformation is less than 20 kcal mol–1, which is significantly lower than the inversion barriers of typical phosphines (30 – 35 kcal mol–1)[1348].

A similar dynamic behavior was observed for all macrocyclic tetraphosphines 2.18.45 – 2.18.51. Furthermore, the study of the solution behavior of two isomers of the 16-membered macrocycle 2.18.46h, synthesized from enantiopure primary amines and differing in the configuration of the chiral substituents at the nitrogen atoms, revealed an intermolecular exchange of endocyclic fragments. This resulted in the formation of a new isomer with different configurations of the corresponding substituents (Scheme 497). The structure of the product was confirmed by X-ray crystallography and a counter synthesis involving a racemic mixture of 1-methylbenzylamine[1355].

Direct evidence of bond lability in corands with the endocyclic fragment PCH2N was obtained from the study of the dynamic behavior of 14-membered macrocycles (2.18.44) (Scheme 498). It was shown that, reversible splitting of the macrocycle occurs in solutions, resulting in a mixture of two isomers of seven-membered heterocycles. After reaching equilibrium, the signals in the NMR spectra corresponding to the seven-membered cycles were predominant, with the macrocycle’s proportion not exceeding 5%. However, upon partial removal of the solvent from the reaction mixture, the rac-isomer of the 14-membered macrocycle 2.18.44 crystallized in an unchanged form[1350, 1352].

Consequently, macrocyclic aminomethylphosphines exist in solutions as a dynamic system of interconverting stereoisomers and smaller cycles. The rate of equilibration sharply increases in the presence of proton donors. It has been suggested that the key intermediate is an acyclic product with a planar methylenephosphonium fragment, which forms upon protonation of the endocyclic nitrogen atom (Scheme 499)[1355].

Thus, over the past decade, efficient methods have been developed to construct macrocyclic aminomethylphosphines, which represent a promising class of ligands for coordination chemistry. It has been demonstrated that compounds with a 1,5-diaza-3,7-diphosphacyclooctane backbone have sufficient stability and allow for the targeted modulation of the properties of metal complexes, imparting them with optical activity, water solubility, predictable catalytic properties, and biological activity. At the same time, phosphorus-containing corands are kinetically labile; therefore, the structure and properties of their coordination compounds with transition metals will depend on the central ion, its charge, geometry, and coordination number.
2.18.2. Coordination chemistry of phosphorus-containing heterocycles
It has been demonstrated that P,N-containing cyclophanes 2.18.31 and 2.18.30 interact with gold(I) and copper(I) derivatives to form the corresponding tetra- (2.18.52) and binuclear (2.18.53) P-complexes while retaining the 3D structure of the cyclophane (Scheme 500). In the gold complex, the linear PAuCl fragments are positioned above and below the macrocycle cavity, whereas the chelated copper ions are located inside the hydrophobic cavity. Encapsulation within the macrocycle cavity leads to the stabilization of an unusual, nearly linear geometry of the Cu2I fragment[1343].

The interaction of P2N2-macrocycles with gold(I) derivatives also preserves the overall structure and geometry of the P,N-corands in the corresponding tetranuclear complexes 2.18.54 (Scheme 501)[1356]. However, in the case of binuclear chelated copper(I) complexes, a change in the configuration of the phosphorus atoms in the macrocyclic ligand is observed. Regardless of the predominant isomer of the initial macrocycle, complexes 2.18.55 – 2.18.57 are formed with the ligand in the RRSS-stereoisomeric form, with the metal atoms positioned on opposite sides of the macrocycle[1368].

It has been shown that P4N2-corands can bind a single metal atom with all phosphorus atoms, resulting in highly stable structures due to the macrocyclic effect. The RRRR/SSSS-isomers are ideally suited for the stabilization of copper(I) ions with a tetrahedral configuration. The formation of complexes 2.18.58 with 14-membered P,N-corands preserves both the size of the initial macrocycle and the configuration of the phosphorus atoms (Scheme 502)[1368].

Metals with a square-planar geometry of the coordination center, such as nickel(II) cations, form the most stable complexes (2.18.59a,b and 2.18.60) with the RSSR-isomer of 18-membered coronands (2.18.48b,d and 2.18.49b) (Scheme 503)[1361]. It is important to note that the phosphorus atoms in the initial ligands 2.18.48b and 2.18.49b have a different, racemic, configuration. In the complexes 2.18.61a – e with octahedral iron(II) derivatives, the macrocyclic ligand 2.18.45 occupies four equatorial positions, as in square-planar complexes. In this case, the RSSR-configuration of the macrocyclic tetraphosphine is also the most favorable. In the case of the 16-membered macrocycle 2.18.45, the configuration of the endocyclic phosphorus atoms is completely preserved[1358].

Thus, the presented P,N-containing macrocycles are capable of rearranging their structure during complex formation to form the most thermodynamically stable coordination compound.
Phospholide anions 2.18.11 – 2.18.14 are also of great interest as ligands for coordination chemistry (Scheme 504). As isolobal analogs of cyclopentadenide anions, they form predominantly sandwich complexes with η5-coordination with transition metals. However, the presence of lone pairs of phosphorus atoms also provides the possibility of η1-type coordination, which allows the construction of complex supramolecular ensembles and clusters[1310, 1369]. The formation of classical sandwich complexes — 1-monophosphaferrocenes (2.18.62)[1370], 1,2-diphosphaferrocenes (2.18.63)[1371, 1372] and 1,2,3-triphosphaferrocenes (2.18.64)[1373, 1374] — has been reported by the interaction of the corresponding alkali metal phospholides with [(η5-C5H5)Fe(η6-MeC6H5)]+[PF6]–. In this case, the phospholide ligands act as a 6π-electrons donor and realize the η5-type coordination characteristic of the Cp-anion (Cp is cyclopentadienyl).

Another (μ,η1:η1 or bridged) type of coordination of the 1,2-diphospholide anion 2.18.12 with a transition metal atom has been demonstrated using manganese derivatives as an example (Scheme 505)[1375]. The study of the magnetic properties of the binuclear complexes 2.18.65a – e has allowed to fix the presence of antiferromagnetic exchange interaction between unpaired electrons located on the metal atoms, which is exclusively realized due to the bridging 1,2-diphosphacyclopentadienyl ligand. The amplitude of the exchange interaction in these systems depends both on the nature of the co-ligands at the manganese atoms (see L in Scheme 505) and on the nature of substituents in the para-position of the arene groups of 1,2-diphospholide[1376].

In addition, the binuclear Mn-complexes 2.18.65 have been studied by pulsed EPR spectroscopy. The spin echo effect has been detected for them, which makes these complexes attractive objects for applications in quantum computing and spintronics[1377].
2.18.3. Luminescent materials based on phosphorus heterocycles and their complexes
A detailed analysis of the optical and luminescence properties of 1-alkyl-2,3,4,5-tetraphenyl-1-monophospholes 2.18.15 and 1-alkyl-2,3,4,5-tetraphenyl-1-monophosphole-1-oxides 2.18.16 shows the presence of electronic absorption with maxima located in the visible (or close to visible) region of the spectrum (for absorption wavelength λabs 368 – 386 nm)[1378, 1379]. The maxima of the luminescence spectra are located in the visible region and correspond to blue-green luminescence (emission wavelength λem 475 – 511 nm). It should be noted that oxidation of the phosphorus atom, which leads to bathochromic shifts in the absorption (~ 23 nm) and luminescence (~ 34 nm) spectra, nevertheless does not affect the absolute value of the Stokes shift (103 – 125 nm), as confirmed by calculations[1380]. All characteristics of the electronic and vibrational spectra of 1-alkyl-2,3,4,5-tetraphenyl-1-monophospholеs indicate effective conjugation between the diene system of the heterocycle and the phenyl fragments at the carbon atoms, and the presence of luminescence response in the visible region of the spectrum suggests the prospects for the use of such systems in light-emitting applications of organic electronics and sensorics.
Based on the ligands 2.18.22 – 2.18.24, which have chromophoric pyridine and thienyl fragments in the substituents at the phosphorus atoms, luminescent complexes 2.18.66 – 2.18.69 with copper subgroup metals have been created. Binuclear neutral (2.18.66, 2.18.69)[1339, 1381, 1382] and charged gold complexes (2.18.67, 2.18.68)[1339, 1383] have been synthesized, in which the 1,5-diaza-3,7-diphosphacyclooctanes 2.18.22 – 2.18.23 act as bridging P,P-ligands (Scheme 506). The bridging ligand, fixed in a ‘crown’ conformation, forms a hydrophobic cavity above the plane of the heterocycle, bounded by two gold-containing fragments and two exocyclic substituents at the nitrogen atoms[1383, 1384]. Unusually high vapochromic shifts of the emission wavelength (up to 120 nm) were recorded for the gold acetylene derivatives 2.18.66a – f[1382].

In contrast to the gold(I) ion, the copper(I) ion has higher coordination numbers (3 and 4), allowing for the formation of luminescent complexes with different structures. For instance, the interaction of ligands 2.18.22 and 2.18.24 with copper(I) iodide resulted in several types of P,P-complexes, including binuclear (2.18.70, 2.18.71) and mononuclear (2.18.72, 2.18.73) complexes (Scheme 507). For the first time, competition between P,P-chelate and P,P-bridging types of copper(I) binding was demonstrated for 1,5-diaza-3,7-diphosphacyclooctanes[1385]. It is suggested that the result of the reaction is determined by the nature of the endocyclic nitrogen atom. If the nitrogen atom is conjugated with an exocyclic substituent, significant changes in the ligand’s conformation are required for the formation of a chelate P,P-complex, leading to competition between chelate and bridging binding modes. In the case of a nitrogen atom in a tetrahedral configuration, the ligand is pre-organized for chelate binding. Notably, complex 2.18.73b with intense blue luminescence was discovered, exhibiting unusual photoinduced structural transitions in the excited state[1341].

The ability of cyclic bisphosphines 2.18.23 to act as bridging ligands has been utilized for the synthesis of hexanuclear complexes with unique Cu2I3 (2.18.74a,b)[1338] and Cu2AuI3 (2.18.75)[1338] fragments (Scheme 508).

Compounds 2.18.74a,b exhibit rare and efficient dual-band luminescence, the color and intensity of which depend on a variety of external factors. This makes them promising systems for the development of OLEDs and luminescent sensors for pH and temperature.
2.18.4. Application of phosphorus heterocycles in designing catalysts for homogeneous reactions
To develop effective non-platinum catalysts for the electrochemical synthesis of hydrogen and its oxidation in fuel cells, charged nickel complexes 2.18.76 have been obtained with 1,5-diaza-3,7-phosphacyclooctanes 2.18.22b – e, which contain pyridine substituents at the phosphorus atoms (Scheme 509)[1336, 1337, 1386].

Catalysts 2.18.76 exhibit high TOF values (number of catalytic cycles per second) and relatively low overpotential. It has been demonstrated that such catalysts can be used for hydrogen oxidation in fuel cells. These model membrane electrode assemblies have shown record power density values for non-platinum hydrogen fuel cells[1387-1389].
Cycloaddition reactions in the series of 1-alkyl-1,2-diphospholеs 2.18.17 have found application in the synthesis of new chiral сaged phosphines for asymmetric catalysis[1390]. Asymmetric [4 + 2] cycloaddition reactions of both enantiopure 1-alkyl-1,2-diphospholes with a series of nonchiral dienophiles and racemic 1-alkyl-1,2-diphospholes with enanti-pure dienophiles have been investigated for the preparation of enantiopure 1,7-diphosphanorbornenes 2.18.18, 2.18.77 (Scheme 510). The interaction of chiral 1-(+)-neomentyl-3,4,5-triphenyl-1,2-diphosphole (2.18.17a) with maleic anhydride derivatives proceeds with a high diastereomeric excess (de = 88 – 91%)[1391-1393]. The [4 + 2] cycloaddition reaction of racemic 1-alkyl-3,4,5-triphenyl-1,2-diphospholes with chiral (5R)-(L-menthoxy)-2(5H)-furanone also gives a product with a high diastereomeric excess (de up to 97%)[1394]. The high diastereoselectivity of these asymmetric reactions allows the isolation of the chiral anti-,endo-diphosphanorbornenes 2.18.18a,b and 2.18.77a – c by recrystallization.

Enantiopure tricyclic 1,7-diphosphanorbornenes 2.18.18 and 2.18.77 has been tested as ligands in the Pd-catalyzed asymmetric alkylation (optical purity of the product reached 64% ee)[1395] and as catalysts in the organocatalytic [3 + 2] cycloaddition reaction of activated alkenes and allenes (68% ee)[1396]. The use of 1,7-diphosphanorbornenes 2.18.77a – c in the Pd-catalyzed asymmetric alkylation of cinnamyl acetate with ethyl-2-oxocyclohexane carboxylate provides enantioselectivity up to 52% ee[1394]. Thus, asymmetric [4 + 2] cycloaddition reactions in the series of 1-alkyl-1,2-diphospholes provide a convenient tool for the molecular design of tricyclic phosphines with a chiral phosphorus atom for asymmetric catalysis.
2.18.5. Biological activity of phosphacoumarins and phosphorus-containing macrocycles
At present, many marketed drugs have an organophosphorus moiety[1397]. So the previously mentioned paper[1386] provides data on the cytotoxicity of spiro-derivatives of benzo[e][1,2]- oxaphosphinines 2.18.6 together with their synthesis. Overall, the cytotoxicity of the studied compounds towards tumor cell lines appears to be comparable to that of 5-fluorouracil. Notably, even small changes in the aromatic moiety of the benzo[e][1,2]- oxaphosphinines, as well as introduction of the annelated ring, have significant effect on their activity. This is especially well demonstrated by comparing the IC50 values (μM) of compounds 2.18.6а and 2.18.6с,е.

The cytotoxicity of the most potent compound 2.18.10a towards HuTu 80 cell line (duodenal adenocarcinoma) is ca 3-fold higher than that of 5-fluorouracil (IC50 = 25.1 ± 1.9 μM and 65.2 ± 5.6 μM, respectively). Notably, this compound also appeared to be non-toxic to a normal cell line (IC50 > 800 μM). Thus, the selectivity index, i.e. the ratio of the IC50 towards normal cell lines to the IC50 towards cancer cells, is > 32. Such a high selectivity makes the further search for anti-cancer agents in the benzo[e][1,2]oxaphosphinines series a promising task.
The high antitumor potential of copper and gold complexes, combined with their luminescent properties, which make it possible to monitor the penetration and distribution of the drug inside the cell, highlights the importance of studying the biological activity of complexes 2.18.66 – 2.18.75. However, it was found that such complexes are unstable in biological environments and exhibit high cytotoxicity associated with this instability. To enhance biocompatibility, a stabilization method was employed by forming nanocolloidal particles with a polyelectrolyte shell using the reprecipitation method. The shells were made of polyethyleneimine (PEI)[1398], lysozyme[1399], or poly(DL-lysine)[1400].
Core-shell nanoparticles are formed by depositing PEI onto the nanoprecipitated luminescent complex 2.18.66a. This significantly increases the stability and reduces the cytotoxicity of the material. Confocal microscopy has shown that the nanoparticles penetrate cells, concentrate in lysosomal compartments, and can induce cell death through the release of cytotoxic complexes or via a photodynamic effect. The colloidal particles exhibit stable luminescence for a week, and sensory properties towards biothiols due to the complexation of the latter with Au+ ions[1398].
The methodology for obtaining nanoparticles based on complex 2.18.66a allowed for the modification of their biological activity by incorporating protein molecules such as lysozyme, pepsin, bovine serum albumin, thioredoxin, and yellow fluorescent protein. Flow cytometry and fluorescence microscopy data demonstrated the efficient internalization of PEI-2.18.66a particles in Wi-38 cell samples, leading to effective staining of all cellular organelles. The concentration-dependent cytotoxicity of PEI-2.18.66a was significantly enhanced in the presence of lysozyme[1401].
Water-soluble cationic gold complexes 2.18.67 were used to form supramolecular assemblies with anionic polynuclear rhenium clusters [{Re6S8}(OH)6]4–, [{Re6Se8}(OH)6]4–, and [{Re6S8}(CN)6]4–[1402]. As a result, colloidal systems with strong cluster-centered luminescence and significant luminescent response to pH changes in the range of 6.0 – 5.5 were obtained. Decomposition of colloids with [{Re6S8}(OH)6]4 — and [{Re6Se8}(OH)6]4– anions under acidic conditions (pH 4.5), simulating the lysosomal environment, released red-emitting rhenium cluster anions and cytotoxic cations of complex 2.18.67. Polyethyleneimine promotes the release of rhenium cluster anions from the colloids due to its counterion effect, while binding to the protein lysozyme shields the colloidal particles from protonation. The colloidal particles penetrate cells, showing a high degree of lysosomal localization in the case of [{Re6S8}(OH)6]4–, while for [{Re6Se8}(OH)6]4– under the same conditions, lysosomal membrane rupture is observed. Lysosomal localization of the colloids correlates with their cytotoxicity. The cytotoxic effect of the colloids, characterized by a significant contribution from apoptosis, differs from the action of complex 2.18.67, which induces necrotic processes[1399].
A similar electrostatically controlled self-assembly of cations 2.18.67 with molybdenum cluster anions [{Mo6I8}L6]2– (L = I, AcO) in aqueous solutions represents a straightforward method for combining therapeutic effects and cellular imaging. The incorporation of molybdenum clusters into aggregates is accompanied by an enhancement of their red luminescence. Both types of molybdenum-containing colloids undergo cellular internalization, which is enhanced when colloidal particles are embedded in a poly(DL-lysine) shell. It has been shown that nanoparticles ranging from 30 to 50 nm in size (L = I) effectively stain cell nuclei. The photodynamic therapeutic effect of the nanoparticles on M-HeLa tumor cells is due to the generation of reactive oxygen species (ROS) and correlates with their intracellular movement and aggregation[1400].
Thus, the material presented above confirms the potential of using nanoparticles based on gold complexes with heterocyclic P-ligands in the development of antitumor drugs and contrast agents for confocal microscopy[1403].
2.19. [3+2]- and [4+2] cycloaddition reactions in the synthesis of polyspirofused heterocycles
This Section highlights the results of the research of the Department of Chemistry of Lomonosov Moscow State University devoted to the synthesis and properties of 2-chalcogenimidazolidinones, namely, hydantoins and their thio and seleno analogues, containing an exocyclic carbon – carbon double bond at the 5-position of heterocycles. Such compounds, acting as dipolarophiles (dienophiles), undergo 1,3-dipolar cycloaddition with azomethine ylides, nitrile imines and nitrile oxides, as well as cyclic and acyclic dienes. As a result, spirocyclic derivatives bearing pharmacophoric pyrrolidine, imidazolidine, pyrazoline and indoline moieties are formed. The presence of the spiro unit makes it possible to fix the required steric arrangement of substituents important for interaction with biological targets in the molecule. Structures A – C represent the types of dipolarophiles (dienophiles) studied from the series of 2-chalcogenhydantoins — precursors of spirofused nitrogen-containing heterocycles.

2.19.1. Synthesis of 5-methylidene-2-chalcogenhydantoins
It is known that the ability of the C=C bonds of dipolarophiles to be involved in 1,3-dipolar cycloaddition strongly depends on the degree of substitution of the multiple bond. In general, dipolarophiles with mono- or di-substituted double bonds react with 1,3-dipoles most readily, tri-substituted ones are less active, and dipolarophiles with tetra-substituted C=C bonds in most cases either do not undergo such reactions or give target products in low yields. In this context, the best performance in reactions with 1,3-dipoles can be expected for 5-methylidene-2-chalcogenhydantoins (type A compounds) due to the spatial accessibility of the exocyclic C=C bond. Prior to our studies, sporadic examples of the preparation of 5-methylidene hydantoins and their thio analogues[1404-1406] were described, the former either having substituents on the N(1) atom[1405, 1406] or being N(3)-unsubstituted[1406]. We have succeeded in developing general synthetic approaches to 5-methylidene-2-chalcogenhydantoins, in which sulfur and selenium atoms act as chalcogen, from the available precursors, β-(morpholin-4-yl)alanine[1407] and L-serine[1408] (see below).
The starting point for developing such a procedure for the synthesis of 5-methylidene-2-chalcogenhydantoins was a study[1404], which described the preparation of 5-methylidene-3-phenylhydantoin from phenyl isocyanate and β-(morpholin-4-yl)alanine (2.19.1a). Optimization of the said procedure at each synthetic step provided an efficient approach to target 5-methylidene hydantoin 2.19.2a from available and low-cost reagents (Scheme 511)[1407].

5-Methylidene-2-thiohydantoins 2.19.3, which are not otherwise available, can also be prepared from substrate 2.19.1a in 51 – 90% yields by a similar procedure (Scheme 512)[1407].

Varying the process conditions (see Scheme 512) showed that the optimum parent amino acid is β-(morpholin-4-yl)alanine (2.19.1a) and that boiling the reaction mixture in hydrochloric acid for 1 h is sufficient for the morpholine moiety to be completely eliminated in the second step. When replacing morpholine with another secondary amine, no elimination takes place.
This synthetic approach (see Scheme 511, Scheme 512) proved inapplicable to selenium analogues of 5-methylidene hydantoin. These compounds were obtained by replacing β-(morpholin-4-yl)alanine with α-amino acid 2.19.4 bearing a C(3)-hydroxyl group, another functional group that can be eliminated[1408].
L-serine was found to react with isothiocyanates and isoselenocyanates to give the target 5-methylidene-2-thio-(2.19.3) and 5-methylidene-2-selenohydantoins 2.19.4 on subsequent treatment with aqueous hydrochloric acid (Scheme 513)[1409].

Тherefore, a general synthetic approach to 5-methylidene-2-chalcogenhydantoins has been developed based on the reaction between α-amino acids bearing an additional β-substituent (morpholine or hydroxyl) capable of elimination, and isocyanates and their thio or seleno analogues. It should be noted that thio and seleno analogues of 5-methylidene hydantoins had not been described prior to our studies.
5-Arylmethylidene-2-chalcogenhydantoins are much more available synthetically than their 5-methylidene counterparts. 5-Arylmethylidene-2-thiohydantoins 2.19.5 were obtained using the approaches shown in Scheme 514[1410]. The convenient precursors to these compounds are either 2-thiohydantoin 2.19.6 or unsymmetrical thiourea 2.19.7, derived from aniline and ethyl isothiocyanatoacetate. Condensation of these substrates with aldehydes can be carried out using both acid and base catalysis. In addition, a three-component reaction of amine, ethyl isothiocyanatoacetate and aromatic aldehyde, carried out without isolation of the urea intermediate 2.19.7, provides a similar product (see Scheme 514).

An approach using KOH in ethanol proved to be the most versatile for the synthesis of 5-arylmethylidene-2-chalcogenhydantoins, suitable for condensation with aromatic and aliphatic aldehydes for both thiohydantoins and thiourea derivatives[1408, 1409, 1411].
Methods for the preparation of 5-arylmethylidene hydantoins and 5-arylmethylidene-2-thiohydantoins have been described previously[1412], but no general approach to their selenium-containing analogues existed before we started our work. In developing this, we began by optimizing the conditions for the preparation of intermediate selenoureas 2.19.8 (Scheme 515).

The best results were obtained in the reaction of ethyl isoselenocyanatoacetate and primary amine using base catalysis (DMAP)[1410, 1411, 1413]. In this case, the products were isolated in high yields (85 – 98%). In the absence of DMAP, this reaction also proceeded, but in significantly lower (by ~5 – 10%) yield of the target product. It should be noted that using aliphatic amines, selenohydantoin 2.19.9 is produced and with substituted anilines, selenourea 2.19.8 is formed. However, on subsequent condensation with aromatic aldehydes, both selenoureas and selenohydantoins give the same product, 5-arylmethylidene-2-selenohydantoin 2.19.10 (Scheme 516).

A large series of 5-arylmethylidene-2-selenohydantoins have been synthesized with aromatic aldehydes containing substituents of different nature[1410, 1411, 1413].
5-Arylmethylidene hydantoins 2.19.11 can also be obtained via alternative synthetic routes (Scheme 517), e.g., by condensation of benzaldehydes with 3-phenylhydantoin (2.19.12) or by a two-step sequence involving methylation of 5-arylmethylidene-2-thiohydantoins 2.19.13 at the sulfur atom followed by desulfurization in acid medium. The second method proved to be much more efficient than the one-step synthesis from hydantoins[1414].

Reacting chiral amines with isothiocyanates provides an approach to optically active derivatives of 5-arylmethylidene-2-thiohydantoins. Thus, chiral α-methylbenzylamine 2.19.14 reacts with ethyl isothiocyanatoacetate to give chiral thiourea 2.19.15, which condenses with benzaldehyde and its 4-fluoro derivative without any loss of enantiomeric purity of the products 2.19.16a,b (Scheme 518)[1415].

Subsequent cycloaddition of chiral 5-arylmethylidene thiohydantoins 2.19.16 gives mixtures of diastereomers which can be separated by column chromatography or recrystallization, and subsequent debenzylation of the nitrogen atom affords enantiomerically pure N(3)-unsubstituted spiro derivatives of 2-chalcogenhydantoins (see Section 2.19.2 for details).
5-Arylmethylidene-2-thiohydantoins 2.19.17 were converted into S-arylated derivatives 2.19.18 by cross-coupling with the appropriate arylboronic acids. The optimum conditions for S-arylation of substrates are illustrated in Scheme 519. The highest yields of products 2.19.18 were obtained at room temperature in the presence of cupric acetate and 1,10-phenanthroline[1416].

A by-product of this transformation is the disulfide 2.19.19, which is formed via oxidation of the starting 2-thiohydantoin. The mechanism was studied more thoroughly using the example of 5-arylmethylidene-2-selenohydantoins (see below, Scheme 521).


A similar reaction with seleno derivatives gives 5-arylmethylidene-3-aryl-2-selenohydantoins 2.19.20 and 2.19.21 from the substrate 2.19.10a (Scheme 520)[1417].
We believe that cupric acetate first coordinates to 1,10-phenanthroline to form a dichloroethane-soluble intermediate A, which then reacts with arylboronic acid. This transmetallation process gives intermediate B. The next step involves the oxidation of CuIII to CuII via the classical Stahl (See A.E.King, T.C.Brunold, S.S.Stahl. J. Am. Chem. Soc., 131, 5044 (2009).) esterification pathway. There are two possible routes to the resulting product 2.19.21. The first involves the reaction of intermediate C with selenohydantoin 2.19.10. In this part of the catalytic cycle, there appears to be a synchronous process for the formation of the arylated product 2.19.21. Since the process does not require the presence of bases, the parent compounds 2.19.10 react in a non-deprotonated form and there is no driving force for Cu – Se bond formation. The second pathway is shown in the lower part of Scheme 522. Here, intermediate C reacts with diselenide 2.9.22, but unlike 2-thiohydantoins, diselenide 2.9.22 was not detected by liquid chromatography-mass spectrometry in the benchmark arylation reaction.

The use of isatines as the carbonyl component in condensation with 2-chalcogen hydantoins or carbethoxymethyl urea gives imidazolidine-containing dipolarophiles 2.19.23 – 2.19.25 with a tetrasubstituted C=C bond (Scheme 522). Such compounds usually undergo 1,3-dipolar cycloaddition not readily because of steric hindrances to the 1,3-dipole approach, but the presence of an acceptor isatin moiety in the molecule should on the contrary facilitates the process. We have developed a versatile method for the synthesis of 5-indolinilidene-2-chalcogenhydantoins containing exocyclic oxygen, sulfur or selenium atoms at the 2-position of the starting heterocycle, which usually provided high yields of the products, 2-thio- (see[1411, 1418]) and 2-selenohydantoins[1413, 1418].
However, the yields of isatin derivatives of hydantoin 2.19.23 by direct condensation did not exceed 40%, so an alternative synthetic approach was used starting from their sulfur-containing analogues 2.19.24 (Scheme 523)[1418].

However, despite the lower product yields, direct condensation of thiohydantoin with isatin is also used in some cases due to the availability of starting reagents and easy experimentation.
In 5-methylidenehydantoin 2.19.2a and its thio analogue 2.19.3a, additional substituents can be introduced, typically on the N(1) nitrogen or on the β-carbon atoms of the exocyclic C=C moiety. Such transformations are carried out either by alkylation, acylation or tosylation of the corresponding substrates in the presence of bases, or by their electrophilic halogenation (Scheme 524)[1419].

The alkylation of methylidene hydantoins proceeds in high yields, whereas thio analogues polymerize under these conditions. This can be explained by the generation of an ambidentate anion upon deprotonation of the N(1) atom, which can undergo alkylation at the sulfur atom, and also add to the C=C double bond of the second 5-methylidenehydantoin molecule. Bromination and iodination of 5-methylidenehydantoins also occur in high yields, whereas thio derivatives are oxidized under these conditions.
Therefore, there is a set of synthetic methods, which provide an access to hydantoins and their 2-thio and 2-seleno analogues containing an exocyclic C=C bond with different degrees of substitution at the 5-position of the heterocycle, as well as to carry out their post-modification to obtain various derivatives, e.g., with SAr and SeAr substituents in the case of thio- and selenohydantoins, with NAlk, NAc or NTs moieties, and also with halogen atoms in the case of hydantoins.
The above compounds were further subjected to 1,3-dipolar cycloaddition and Diels-Alder reactions to give spiro derivatives of hydantoins as well as 2-thio and 2-selenohydantoins of various structural types.
2.19.2. 1,3-Dipolar cycloaddition reactions of azomethine imines to 2-chalcogenhydantoins
Substituents of different electronic nature, both electron-donating and electron-withdrawing, can be introduced into the exocyclic moieties of the most thoroughly studied dipolarophiles, 5-arylmethylidene-2-chalcogenhydantoins. The reactions of 5-arylmethylidene-2-thiohydantoins 2.19.17 with azomethine imines generated in situ from isatin and sarcosine provide highly diastereoselective synthesis of a diversity of dispiro derivatives of 2 thiohydantoins.19.26 (Scheme 525)[442, 1411, 1420]. The reaction mechanism is shown in Scheme 526. In the first step, sarcosine reacts with isatin to generate an unstable spiro-oxazolone A, which then eliminates CO2 and converts to an azomethine ylide B, which further regio- and diastereoselectively adds to the double C=C bond of the dipolarophile.

A few examples of such a reaction with arylmethylidene hydantoins have been reported previously[1421], and dispiro{imidazolidinone,pyrrolidine,indolinones} shown in Scheme 525 were obtained from thiohydantoins for the first time.

In the course of optimizing the reaction conditions of arylmethylidene thiohydantoins 2.19.17 with azomethine ylides, an extremely experimentally simple and scalable (up to ~50 g of target spiro derivatives in a single batch) protocol for their synthesis has been developed. Simultaneous dissolution of all reactants followed by their gradual heating generates the reaction dipole at a lower temperature; it does not immediately undergo the target cycloaddition via the trisubstituted C=C bond and gives difficult-to-separate by-product impurities. To carry out the reaction successfully, it is advisable to add isatin to a solution of dipolarophile and sarcosine in boiling ethanol, which promotes a rapid 1,3-dipolar cycloaddition of the generated dipole to the C=C bond. In this case, the resulting dispiro-thiohydantoin 2.19.26 crystallizes from the reaction mixture and requires no further purification.
Notably, when sarcosine is replaced by amino acids with bulkier substituents (isopropyl, tert-butyl or phenyl) at the nitrogen atom, the cycloaddition to 5-arylmethylidene derivatives of hydantoins does not proceed.
The above protocol of the reaction with azomethine ylides can also be successfully used to prepare dispiro-derivatives of the 5-arylmethylidene hydantoins[1414] and their selenium-containing analogues[1413].
The introduction of spirofused moieties into the molecules of 2-chalcogenhydantoins open access to conformationally rigid scaffolds decorated with multiple pharmacophore moieties, which, with appropriate choice of exocyclic substituents, ensure strong binding to biological targets. In particular, dispiro-indolinones containing 2-chalcogenhydantoin rings have shown high activity as inhibitors of the p53 – MDM2 protein-protein interaction, which is a promising target of current antitumour drugs[442, 1411, 1422].
The cytotoxicity of the series of dispiro-indolinones 2.19.26 was evaluated on tumour cell lines of human breast adenocarcinoma (MCF7) and lung adenocarcinoma (A549) as well as on pseudo-normal cell lines of kidney (HEK293T) and lung (VA13) epithelium isolated from human embryo. The cytotoxicity of the said dispiro-indolinones was evaluated by the standard MTT assay; in addition, all compounds were tested for p53 protein activation using a transcriptional reporter. Compounds 2.19.26a – c demonstrated the best cytotoxic effect (IC50 = (1.3 – 7.5) ± 2.0 μM), but they showed a weak effect of p53 activation on the transcriptional reporter. This result may be explained by the fact that p53 protein activation is only one of several mechanisms of action of the tested compounds that affect the total cytotoxicity.

Selectivity of cytotoxic activity in various tumour cell lines has also been demonstrated for a number of dispiro-derivatives. For example, the thiohydantoin-based dispiro-derivatives 2.19.26 showed selectivity towards colon cancer cells HCT116, hydantoin derivatives were active against the cell line A549[1414]. The structure-activity relationship showed that replacement of the hydrogen atom in the 5-position of isatin with bromine significantly increases the activity and improves the selectivity of the resulting compound. The introduction of a halogen atom in the para-position of the benzene ring of the Ar2 substituent also increased the total cytotoxicity and selectivity (see Scheme 525).
For compound 2.19.26b, one of the most cytotoxic of the dispiro-indolinones[442, 1414], a number of preclinical studies were performed to determine general and acute toxicity, immunotoxicity, chronic and mutagenic toxicity, and to study experimental pharmacokinetics. The results of this work showed the efficacy and safety of dispiro-indolinone 2.19.26b, allowing us to recommend it for further clinical trials.
Dispiro-derivatives 2.19.26 (see Scheme 525) were obtained as single diastereomers but as a mixture of enantiomers, and only one of the enantiomers was found to exhibit antitumour activity (A.A.Beloglazkina. Candidate Thesis in Chemical Sciences, Lomonosov Moscow State University, Moscow, 2018.). In this context, the development of synthetic approaches to enantiomerically pure dispiro-indolinones became relevant. We proposed a methodology based on the preparation of dispiro-indolinones as single enantiomers by the introduction of 1-arylalkyl groups into the molecules of starting dipolarophiles (2-thiohydantoins) and their subsequent removal via acidolysis.
Enantiomerically pure dispiro-indolinones 2.19.27 were obtained by 1,3-dipolar cycloaddition of azomethine ylide, generated in situ from isatin and sarcosine, to 5-arylmethylidene-2-thiohydantoins 2.19.16 bearing chiral 1-arylethyl substituents at the N(3) atom. The mixture of diastereomers 2.19.28 was then separated by column chromatography or recrystallization, and the auxiliary 1-arylethyl group was removed under acidolysis conditions[1423]. The chiral 1-(2,4-dimethoxyphenyl)ethyl group (Scheme 527) was found to be the most effective, with elimination occurring rapidly and under mild conditions.

Therefore, the introduction of a chiral substituent on the N(3) atom of the thiohydantoin ring of the starting dipolarophile makes it possible to isolate individual enantiomers of dispiro-indolinones 2.19.27 and to obtain optically pure products in yields up to 40% based on the starting thiohydantoin 2.19.16.
To extend the range of resulting dispiro-imidazolidinones, we have explored two alternative synthetic approaches to the compounds 5.19.29 bearing an exocyclic arylthio moieties via reactions with azomethine ylides (Scheme 528)[1424].

Of two alternative approaches shown in Scheme 525, only the 1,3-dipolar cycloaddition of azomethine ylide to pre-S-arylated dipolarophiles (e.g. compound 2.19.18a) was found to be preparative. Arylation at the sulfur atom of dispiro-hydantoin 2.19.26d could not be achieved under any conditions, possibly due to the considerable steric hindrances posed by the rigid polycyclic framework.
5-(Indolinylidene)-2-chalcogenhydantoins 2.19.24 and 2.19.25 were successfully reacted with azomethine ylides generated from isatin and sarcosine, despite the fact that dipolarophiles with tetrasubstituted double bonds are rather inert in 1,3-dipolar cycloaddition reactions. The reaction was carried out in boiling toluene using a large excess of amino acid and paraform (8 equiv. each); under these conditions the target products 2.19.30 and 2.19.31 were formed in ~60% yield (Scheme 529)[1411, 1413]. The lowest yields were observed with substrates 2.19.24 and 2.19.25 bearing the NO2-substituted isatin moiety. When the amount of amino acid and paraform was reduced to 4 equiv., the product yield decreased by 5 – 10%, but for hard-to-reach amino acids the reaction can still be carried out under these conditions[1425].

In contrast to the dispiro-derivatives described in Section 2.19.1, amino acids bearing bulky substituents at the nitrogen atom, such as isopropyl-, tert-butyl- and 4-chlorophenylglycines (see Scheme 529, substituent R4), can be reacted with 5-indolinilidene-2-chalcogenhydantoins, in addition to sarcosine.
5-Methylidene hydantoins 2.19.2 and their thio analogues 2.19.3 having an unsubstituted terminal carbon atom of the exocyclic C=C bond readily undergo 1,3-dipolar cycloaddition with azomethine ylides derived from isatin and various α-amino acids (Scheme 530)[1426]. As a result, dispiro{indolinone,pyrrolidine,imidazolidinones} 2.19.32, 2.19.33 with a free carbon atom at the 4'-position of the pyrrolidine ring are produced in good yields.

This reaction occurs not only with sarcosine but also with more sterically demanding amino acids such as N-ethyl-, N-benzyl- and N-(4-methoxybenzyl)glycines. However, in the case of glycine derivatives with Pri-, But- and 4-chlorophenyl substituents at the nitrogen atom, no cycloaddition products were observed.
Dispiro-hydantoin 2.19.32a underwent elimination of 4-methoxybenzyl group from the nitrogen atom of the pyrrolidine ring and was converted to the spirocyclic salt 2.19.34 with the unsubstituted nitrogen atom in the central pyrrolidine ring (Scheme 531)[1426].

Compound 2.19.34 is well soluble in water, which is important for its potential biomedical applications.
2.19.3. 1,3-Dipolar addition of nitrile imines and nitrile oxides to 5-methylidene hydantoin
Due to the sterically accessible exocyclic carbon-carbon double bond, 5-methylidene hydantoins can react with nitrile oxides and nitrile imines to give 1,3-dipolar cycloaddition products with high diastereoselectivity and in high yields.
The reaction between 3-phenyl-5-methylidene hydantoin (2.19.2a) with nitrile imines affords spiro-hydantoins 2.19.35 bearing a pyrazoline moiety (Scheme 532)[1427].

For nitrile imines, examples are known where the regiochemistry of the cycloaddition changes depending on the electronic nature of the substituents at the terminal atoms of the 1,3-dipole (C-terminus and N-terminus). However, varying the substituents of nitrile imines in reactions with 5-methylidene hydantoins did not change the regioselectivity of the addition as in all cases a single regioisomeric product 2.19.35 was obtained. According to quantum chemical calculations, the transition state of TS-2 during the formation of the second possible regioisomer has a significantly higher energy (Figure 14)[1427].
![[{"id":"rg8VM85ind","type":"paragraph","data":{"text":"Energy profiles for the 1,3-dipolar cycloaddition reactions of nitrile imine with 5-methylidene-3-phenylhydantoin <b>2.19.2</b> yielding two possible regioisomeric products[[ type=\"anchor\" referenceId=\"7998\" ]]."}}]](/storage/images/resized/8OfpMCmbyXSzP4eWfhWqoEXfcGwyABZL2qrftj2u_xl.webp)
Nitrile imines and nitrile oxides are unstable 1,3-dipoles usually generated in situ in the reaction solution from stable precursors (N-hydroxymoyl and hydrazonyl halides) under the action of bases as a result of dehydrohalogenation. In the case of poorly reactive dipolarophiles, nitrile imines and nitrile oxides are prone to dimerization, which reduces the yield of the target products. To suppress the undesired dimerization of dipoles in 1,3-dipolar cycloaddition reactions involving nitrile imines and nitrile oxides, we first proposed a convenient and experimentally simple method for their generation. The reaction dipole was formed by diffusion of tertiary amine vapour into a reaction vessel containing a mixture of the dipolarophile with N-hydroxymoyl or hydrazonyl halide, which are precursors of nitrile oxide or nitrile imine, respectively.
The apparatus in which the reactant diffusion mixing process is carried out is a small open vessel placed inside a larger beaker (Figure 15). A mixture of the starting alkene and hydroxymoyl halide (or imidoyl chloride) in an organic solvent is loaded into the inner vessel. The reaction components do not have to be completely soluble in the reaction medium; the conversion was also efficient in suspension. The outer vessel contained a ~100-fold excess of tertiary amine (Et3N, or Me3N, or PriNEt2), which slowly evaporated and diffused into the solution in the inner vessel, creating a reaction dipole there. In this case, the dipole is formed slowly and gradually, “molecule-by-molecule’, which makes it possible to completely prevent its undesirable dimerization. The diffusion mixing technique is easy in synthetical implementation, does not require any special equipment and can be easily extended to other organic reactions involving dehydrohalogenation.
![[{"id":"shlRASmKqF","type":"paragraph","data":{"text":"Schematic representation of an apparatus for carrying out reactions by the ‘diffusion mixing’ technique[[ type=\"anchor\" referenceId=\"7999\" ]]."}}]](/storage/images/resized/Ct6LtUbgaFgsvYSaIdMfIG3oT3IUiyQ0i8kIdGLn_xl.webp)
The diffusion mixing method has been tested by us on a series of different activated and inactivated dipolarophiles, including 5-methylidene hydantoins[1429]. It should be noted that this method provides an access to spirocyclic hydantoins in high yields even in reactions with alkyl-substituted nitrile oxides and nitrile imines, which are most prone to dimerization. Thus, in the reaction of 5-methylidene hydantoin 2.19.2a with methyl phenyl nitrile imine under classical dropwise addition conditions[1427], the yield of the product 2.19.35a does not exceed 60%, whereas it reaches 90% using diffusion mixing[1428] (Scheme 533).

Nitrile oxides also readily undergo 1,3-dipolar cycloaddition upon diffusive mixing of the reactants to give spirocyclic products 2.19.36 (Scheme 534)[1428].

To conclude, the technique of diffusion mixing of reactants has proved to be a very effective tool for carrying out 1,3-dipolar cycloaddition of highly reactive 1,3-dipoles with less reactive dipolarophiles. The advantages of this method are the simplicity of the equipment, the possibility of varying the base (tertiary amine) and the solvent, which is usually benzene, chloroform, methanol or diethyl ether.
2.19.4. 5-Methylidene-2-chalcogenides as dienophiles in [4+2] cycloaddition
In addition to 1,3-dipolar cycloaddition reactions, 5-methylidene-2-chalcogenhydantoins can act as dienophiles in Diels – Alder reactions with cyclic and acyclic dienes such as cyclopentadiene, cyclohexa-1,4-diene, 2,3-dimethylbuta-1,3-diene and isoprene[1419]. Scheme 535 shows the products of [4 + 2] cycloaddition involving cyclopentadiene, namely, spirocyclic compounds 2.19.37 and 2.19.38.

The reactions proceed to give predominantly spiro-indolinones 2.19.37 and 2.19.38 with exo configuration of the methylene bridge in the resulting norbornene moiety relative to the C(4)=O group of the starting hydantoin. The exo : endo isomer ratio of the product is ~ 9 : 1 in all cases irrespective of the substituents in the starting dienophile.
Reactions of 5-methylidene hydantoins and their thio analogues with conjugated dienes, which are less reactive than cyclopentadiene, require Lewis acid catalysis. Thus, in the reaction of 5-methylidenehydantoin 2.19.2 with cyclohexadiene, 2,3-dimethylbutadiene and isoprene, the use of AlCl3 was optimal, whereas in the case of 5-methylidene-2-thiohydantoin 2.19.3 and the same dienes, ZnI2 performed best[1419].
[4 + 2] Cycloaddition of N(1)-unsubstituted 5-methylidene hydantoins followed by alkylation or acylation of the resulting spiro-derivatives 2.19.37 at the nitrogen atom leads to N(1)-substituted spiro-hydantoins. Under similar conditions, derivatives of 2-thiohydantoins are alkylated exclusively at the more nucleophilic sulfur atom to give 2-alkylthiospiro-imidazolidinones 2.19.39 (Scheme 536)[1419].

Furan does not react with 5-methylidene-2-chalcogenhydantoins in the absence of a catalyst, but the process takes place when promoted with AlCl3. In this case, however, the [4+2] cycloadduct was not detected and predominantly furan alkylation products 2.19.40, 2.19.41 were formed with a small admixture of Michael addition products 2.19.42, 2.19.43 (Scheme 537)[1429].

Presumably, the furan addition is preceded by a tautomerization of the starting 5-methylidene-2-chalcogenhydantoin, since no such reaction occurs with substrates substituted at the N(1) atom and not capable of tautomerization.
Thus, convenient preparative methods have been developed for the diastereoselective (and in some cases enantioselective) synthesis of spiro-derivatives of 2-chalcogenhydantoins by [3 + 2] cycloaddition to 1,3-dipoles of different nature (azomethine ylides, nitrile imines, nitrile oxides) and [4 + 2] cycloaddition reactions. Such spirocyclic compounds were tested for cytotoxicity in vitro on a number of cell lines. The lead compounds were identified and compound 2.19.26d underwent a series of preclinical trials which confirmed the potential use of this type of compound in antitumour therapy.
Further research will be focused on the extension of the developed methodology for the synthesis of spiro compounds based on 2-chalcogen hydantoins to 1,3-dipoles of other structural types (azomethine imines, nitrile ylides, nitrons, thiocarbonyl ylides). In addition, the chemo-, regio- and stereochemical patterns of reaction with different dipoles should be compared to find out the substrate limitations. The application of this synthetic approach to other dipolarophiles of similar structure, such as methylidene derivatives of rhodanines, oxazolidinones, thiazolidinones, etc., also seems promising.
2.20. Domino reactions in the synthesis of nitrogen-containing heterocycles
Nitrogen-containing heterocycles play an outstanding role in nature and human life. It is enough to recall the ‘Watson-Crick’ pairs responsible for the transmission and implementation of genetic information, the neurotransmitters serotonin and histamine, or, for example, the coenzymes NAD(P) and FAD, involved in numerous redox processes in the cell[1430]. Alkaloids, which are the secondary metabolites generally of plant origin, also belong to the nitrogen-containing heterocycles in the vast majority of cases[1431]. The title of the Hesse’s monograph[1431] ‘Alkaloids: Nature’s Curse or Blessing?’ aptly reflects the impact of these compounds on humanity. Since ancient times they have been used for medicinal purposes, as well as poisons and narcotics, and with the development of organic chemistry, azaheterocycles have become universal elements of a molecular constructor intended for the creation of synthetic physiologically active substances[1432]. Azaheterocyclic systems, which are most often found in natural products and drugs, are classified as privileged structures[1433]. In particular, indole and pyridine are top-ranking heterocycles, present in alkaloids and drugs[1431, 1432]. Berthelot’s idea[1434] that ‘chemistry creates its own subject’ developed fruitfully in heterocyclic chemistry in that synthetic chemists sometimes use heterocycles to produce... other heterocycles. Of course, such work is only worthwhile if the molecular complexity of the product increases and the starting compounds are available. The value of the method increases if two or more stages of a complex transformation, in which new chemical bonds are formed, can be carried out under the same conditions, in the same reaction vessel, and subsequent stages proceed with the participation of functional groups formed at the previous stages. Such processes are called domino reactions[1435]. The use of domino reactions in the synthesis of complex molecules allows significant savings in solvents, reagents, and energy. This is in line with the principles of green chemistry, which aims to minimize waste and conserve resources[1436].
This section discusses domino methods for the synthesis of heterocyclic systems, developed by the group of Prof. L.G. Voskressensky at the Department of Organic Chemistry of the Patrice Lumumba Peoples’ Friendship University of Russia (RUDN University) mainly over the last 10 years. Our focus was on transformations of [c]-fused tetrahydropyridines, isoquinolines and related systems, as well as 2-imidazolines under the action of electron-deficient alkynes, and reactions involving heterocycles containing an N-cyanomethyl group, including homophthalonitrile and N-propargylindole-2-carbonitriles.
The study of reactions of electron-deficient alkynes with nitrogen-containing heterocyclic systems at the Department of Organic Chemistry of RUDN University began with the work led by Prof. N.S.Prostakov[1437, 1438], who studied the interaction of pyrido[1,2-a]benzimidazole derivatives and 9,10-dihydro-1-sila-2-azaanthracene with DMAD. This topic received a powerful impetus for development at the turn of the millennium, when L.G.Voskressensky, T.N.Borisova and A.V.Varlamov[1439-1444] discovered new domino reactions of [c]-fused tetrahydropyridines (Scheme 538) with electron-deficient alkynes — DMAD, methyl propiolate (MP) and acetylacetylene (AcA). The first step in this sequence of transformations is the nucleophilic attack of the sp3-hybridized nitrogen atom at the activated triple bond with the formation of the zwitterion 2.20.1. The further fate of this intermediate depends mainly on the nature of the annulated ring, the substituent R, as well as the solvent and can lead to a whole series of different products (see Scheme 538).

In the case of an alkyl substituent R, the dihydropyridine ring can undergo Hoffmann cleavage to form vinyl derivatives 2.20.2[1439, 1442]. When the reaction is carried out in alcohols, the ring can open by nucleophilic substitution to yield alkoxy derivatives 2.20.3[1433, 1441]. One of the most interesting pathways is the intramolecular nucleophilic substitution occurring in aprotic solvents with the participation of the carbanion center of the zwitterion 2.20.1, giving azocines 2.20.4 as a result of expansion of the tetrahydropyridine ring[1442, 1443, 1445]. It is possible to assume the participation of an open zwitterion 2.20.5 in such transformations, which is clearly manifested in the formation of spiro compounds 2.20.6[1445-1447] and in the occurrence of three-component reactions involving indole, leading to triarylmethanes 2.20.7[1448, 1449]. In the case of an electron-withdrawing substituent R, the abstraction of a proton from the neighboring carbon atom of the dihydropyridine ring is possible with the formation of a nitrogen ylide 2.20.8, and subsequent Stevens rearrangement gives the vinylation product 2.20.9[1444, 1450].
Over the past ten years, the group led by Prof. L.G.Voskressensky developed preparative methods for the synthesis of various heterocyclic systems containing medium-sized rings based on this methodology. Thus, the expansion of the partially hydrogenated ring of available 1-aryl-2-ethyl-1,2,3,4-tetrahydro-β-carbolines 2.20.10 using MP and AcA granted an access to a series of 6-aryl-3-ethylazocino[5,4-b]indoles 2.20.11 (Scheme 539)[1451]. In the case of a slightly less active MP, an increase in the product yield was achieved by adding CuI or 1-methylpyrrole as catalysts.

4-Aryl-7-methyl-4,7,8,9-tetrahydrothieno[2,3-d]azocines 2.20.12 were synthesized from 4-aryl-5-methyl-4,5,6,7-tetrahydrothieno[3,2]thienopyridines 2.20.13 and various electron-deficient alkynes by gentle heating in MeCN or TFE (Scheme 540)[1452].

Hardly accessible diazonins 2.20.14 were obtained by ring expansion of 4-arylpyrrolo[1,2-a][1,4]benzodiazepines 2.20.15 in the presence of catalytic amounts of AcOH (Scheme 541)[1453]. In this case, the reaction proceeded through cyclization of the open cationic intermediate 2.20.16.

The combination of the Ugi azido reaction involving readily available 2-methyl-3,4-dihydroisoquinolinium salts 2.20.17 and subsequent tetrahydropyridine ring expansion provided a convenient synthetic route to tetrazolyl-substituted azocines 2.20.18 (Scheme 542)[1454, 1455].

Iodomethylates of 3,4-dihydroisoquinolines can be easily transformed by the addition reaction with acetylenides into 2-ethynyl-substituted 1,2,3,4-tetrahydroisoquinolines 2.20.19. The interaction of the latter with electron-deficient alkynes in trifluoroethanol has opened a new chapter in the field under consideration, connected with the preparation of an extremely difficult to access and poorly studied class of heterocyclic allenes (Scheme 543)[1456-1459].

Yields of ten-membered trienes 2.20.20 in most cases exceed 70%[1456]. The process begins with the formation of the zwitterion 2.20.21 as a result of the attack of an sp3-hybridized nitrogen atom at the MP or AcA triple bond. At the same time, the presence of an ethynyl substituent at the position 1 of isoquinoline allows a [3,3]-sigmatropic rearrangement to occur with the formation of an allene system and an enamine fragment inside the medium ring[1456]. The reaction can be carried out by heating in common aliphatic alcohols, but this leads to a decrease in product yield due to the increased reaction time[1460]. The resulting heterocyclic allenes proved to be stable during storage, making it possible to study their biological properties. Among these compounds, selective competitive inhibitors of human acetylcholinesterase with inhibition constants in the micromolar range were discovered[1458, 1459], as well as highly active inhibitors of P-glycoprotein, a transporter responsible for the transport of many cytotoxic drugs outside cancer cells[1461].
If the starting tetrahydroisoquinolines 2.20.22 contains a primary or secondary aliphatic group at the position 1 in addition to the ethynyl group, then the resulting allenes 2.20.20 (R2 = Alk) are able to isomerize in the presence of an acid into conjugated trienes with an exocyclic double bond. This made it possible to develop a method for the preparation of 8-ylidene-1,2,3,8-tetrahydrobenzazecines 2.20.23, which involved the reaction of 1,2,3,4-tetrahydroisoquinolines 2.20.22 with MP and AcA in AcOH (Scheme 544)[1461].

In the case of MP, a similar process also occurs in HFIP at 7°C, while only allenes are formed with AcA in this solvent[1461].
Heating of heterocyclic allenes 2.20.20 led to another unusual class of compounds, cyclopenta[a]indenes 2.20.24 with an aminomethylene bridge (Scheme 545)[1457, 1462]. The reaction proceeds by a radical mechanism with the formation of the cyclopropane derivative 2.20.25 as an intermediate, which can be isolated from the reaction mixture at 150 °C. However, it is completely converted to the final compound at 180 °C, thus proving to be a kinetic product.

Among the bridged cyclopenta[a]indenes 2.20.24, selective noncompetitive inhibitors of human butyrylcholinesterase with inhibition constants down to the nanomolar concentration range have been discovered[1457].
An approach towards pyrrolo[2,1-a]isoquinoline derivatives, a widespread structural fragment of biologically active natural products, is another direction developed by the group of prof. L.G.Voskressensky[1463] during the past 10 years. This method is based on the idea of annulation of the pyrrole fragment with the participation of electron-deficient alkynes and 1-aroyl-3,4-dihydro[2,1-a]isoquinolines 2.20.26 by initially attacking the triple bond with an sp2-hybridized nitrogen atom. This idea was initially implemented in a three-component version — the process took place by refluxing the starting reagents in alcohols (Scheme 546).

The first intermediate — zwitter ion 2.20.27 — attacks the carbonyl group with the anionic center, the resulting tetrahedral intermediate 2.20.28 abstracts a proton from the alcohol, and then the addition of an alkoxide ion at the double bond conjugated with the electron-withdrawing group X follows. The domino sequence is completed with the elimination of water to form pyrroloisoquinolines 2.20.29.
In aprotic solvents like ether, benzene or toluene, transformations with the participation of acetylenedicarboxylic acid esters and 1-aroyl-3,4-dihydro[2,1-a]isoquinolines 2.20.30 are realized in a different way. The tetrahedral intermediate 2.20.31 undergoes a rearrangement with the migration of the aryl group to the electron-deficient carbon atom, which leads to 10b-substituted 1,5,6,10b-tetrahydropyrrolo[2,1-a]isoquinolines 2.20.32 (Scheme 547)[1464].

The two procedures described above were applied to the synthesis of benzo[h]pyrrolo[2,1-a]isoquinoline derivatives from 1-aroyl-3,4-dihydrobenzo[h]isoquinolines. This required extending the reaction time in alcohols to 5 – 10 days and using microwave irradiation at 150 °C for reactions with acetylenedicarboxylates in toluene[1465].
Extension of this method to 4-aroyl-6,7-dihydrothieno-[3,2-c]pyridines 2.20.33 resulted in a number of surprises. Firstly, the reaction with MP in alcohols requires the use of a combined catalyst based on zinc and copper(I) salts, as well as an excess of alkyne to achieve acceptable product yields (Scheme 548)[1466].

Secondly, the interaction of thienopyridine derivatives 2.20.33 with esters of acetylenedicarboxylic acid resulted in the migration of an ester group, but not of an aryl group, as in the case of isoquinolines 2.20.30 (Scheme 549)[1466].

In addition, substitution of the solvent (toluene for acetonitrile) under microwave irradiation allowed the reaction of aromatic 1-aroylisoquinolines 2.20.34 with both acetylenedicarboxylic acid esters and MP and led to the expected 10b-arylpyrroloisoquinolones 2.20.35 (Scheme 550)[1467].

The reaction of alkynes with 2-aroylpyridines 2.20.36 proceeded similarly (Scheme 551)[1467].

An interesting development of the considered method for the synthesis of pyrrolo[2,1-a]isoquinolines in alcohols was the replacement of the latter as the third component with nitrogen-containing heterocycles of suitable acidity (NuH) and carrying out the reaction in acetonitrile. As a result, a series of 31 compounds containing a heterocyclic substituent Nu at the position 3 was obtained (Scheme 552)[1468].

The transition to C-nucleophiles in this process was very logical. Various compounds containing an active methylene group (acetylacetone, malononitrile, acetoacetic and cyanoacetic esters, dimedone, N,N-dimethylbarbituric acid, malonic ester) were used as their precursors. In most cases, the expected three-component reaction (3CR) products were obtained: 5,6-dihydropyrrolo[2,1-a]isoquinolines 2.20.37 (Scheme 553)[1469].

Using acetoacetic and cyanoacetic esters, pyrroloisoquinolone 2.20.38 was isolated in yields of 7 – 38% as a by-product of a two-component process. For acetoacetic ester, the reaction was also complicated by hydrolysis of the ester group (due to the liberated water molecule) and subsequent decarboxylation, which led to pyrroloisoquinolines 2.20.39. In the case of malonic ester, hydrolysis and decarboxylation could not be avoided in principle; mixtures of monoesters 2.20.40 and pyrroloisoquinolones 2.20.38 were isolated with 23 – 32% and 7 – 15% yields, respectively.
The introduction of 1,2-disubstituted 2-imidazolines (2.20.41) into the reaction with terminal electron-deficient alkynes proved to be effective. In this case, a pseudo-three-component reaction occurred with the formation of imidazolidines 2.20.42 — adducts of an imidazoline molecule and two alkyne molecules (Scheme 554)[1470-1472]. The process involved simply mixing the reagents in an aprotic solvent — both polar and nonpolar (e.g. in toluene, ether, dichloromethane, DMF).

As with 1-aroylisoquinolines, the sp2-hybridized nitrogen atom first attacks the triple bond to form the zwitterionic intermediate 2.20.43, which then abstracts the terminal proton from the second alkyne molecule, followed by nucleophilic addition of the resulting acetylenide ion at the position 2 of the imidazolinium ion 2.20.44 (see Scheme 554)[1470].
A wide range of imidazolines containing both aliphatic and aromatic substituents are suitable for this reaction. The presence of strong electron-withdrawing groups, such as acetyl or 2,4-dinitrophenyl, at the position 1 of the heterocycle predictably interferes with the process. Surprisingly, one can widely vary the aromatic substituents at the position 2 of imidazoline and use ortho-substituted, fused, π-rich and π-deficient heterocyclic moieties. In general, the yields of products with AcA are lower than for other alkynes due to side processes of oligomerization.
Taking into account the high activity of AcA as a Michael acceptor, it was possible to develop a 3CR method involving two different alkynes. Thus, when a mixture of MP and AcA was added to a cooled solution of imidazoline, adducts 2.20.45 containing a butenone substituent at the nitrogen atom and a propiolic ester fragment at the position 2 of imidazolidine (Scheme 555) were formed in good yields[1471].

An N-vinylpropargylamine fragment can be identified in the structure of the resulting adducts 2.20.42 and 2.20.45, allowing the [3,3]-sigmatropic rearrangement to occur—the propargyl variant of the aza-Claisen rearrangement. This process is a «trigger» for a number of domino transformations, creating a fairly wide chemical diversity based on such adducts[1470-1472].
Thermal rearrangement of adducts 2.20.42 containing an aromatic substituent at the position 3 of imidazolidine leads to pyrroles 2.20.46 with a β-(arylamino)ethyl group at the nitrogen atom under aerobic conditions (Scheme 556)[1470]. Moreover, the synthesis of adducts 2.20.42 and their transformation into pyrroles 2.20.46 can be easily carried out in a one-pot mode: an alkyne is added to a solution of imidazoline 2.20.41 in xylene and after 4 h the reaction mixture is refluxed for 30 min. Therefore, in this case the whole process represents a sequential pseudo-four-component reaction.

The simplicity of the experimental procedure and at the same time the depth of the structural changes in the substrate make it intriguing to discuss the mechanism of this domino transformation (Scheme 557)[1470]. The initiating step, as mentioned above, is the [3,3]-sigmatropic rearrangement, leading to the labile allene 2.20.47, which is converted to the non-aromatic pyrrolopyrazine 2.20.48 as a result of transannular cyclization and proton migration. This intermediate 2.20.48 can be detected by NMR spectroscopy and mass spectrometry. Then, autoxidation occurs, starting with the generation of the radical 2.20.49, followed by hydroperoxide 2.20.50 formation, the co-proportionation of which with pyrrolopyrazine 2.20.48 leads to the hemiaminal 2.20.51. The opening of the latter cyclic intermediate completes the domino sequence, being an unusual combination of pericyclic, heterolytic and radical steps.

An attempt to carry out a similar transformation with adducts containing an aliphatic substituent at the position 3 led to oligomeric products formed due to the interaction of the resulting amino group (more nucleophilic in this case) with the ester and ketone functions of the pyrrole fragment. However, this problem was easily solved by adding an acylating agent — anhydride or (thio)cyanate (Scheme 558)[1470].

The one-pot synthesis of pyrroles 2.20.52 from 2-imidazolines 2.20.41 is a sequential five-component process with 29 – 62% yields.
The rearrangement of adducts 2.20.42 in a microwave reactor in the presence of cesium carbonate as a base has allowed the synthesis of a series of pyrrolopyrazines 2.20.53 (Scheme 559)[1471]. In this case, the product of the pericyclic step, allene 2.20.47, loses a proton in the α-position to the imino group, and the anion 2.20.54 undergoes transannular cyclization, which leads to the aromatic product 2.20.53 after protonation.

A one-pot synthesis of pyrrolopyrazines 2.20.53 was successfully carried out by keeping a mixture of 2-imidazolines 2.20.41 and 2 equiv. alkyne in DMSO, followed by the addition of CsF as a base and heating under an argon atmosphere (Scheme 560)[1464].

Cross-adducts 2.20.45 were converted to the corresponding pyrrolopyrazines 2.20.54 by heating in DMSO in the presence of CsF (Scheme 561)[1471].

A completely different way of the domino transformation of adducts 2.20.42 with an aliphatic substituent in the position 3 of imidazolidine occurs in the presence of protic acids (Scheme 562)[1472]. The products in this case are pyridinium salts 2.20.55, containing an ammonium group in the side chain at the nitrogen atom.

As a rule, the reaction proceeds with almost quantitative yields on treatment with 2 equiv. of sufficiently strong acids with non-nucleophilic anions, such as TFA, HBF4, MsOH, picric acid. Tetrafluoroborates are easily isolated from the reaction mixture in crystalline form by addition of ether. The transformation does not start in the presence of simple carboxylic acids (acetic, formic) and does not reach completion when using < 2 equiv. strong acid.
It was also found that treatment of the reaction mixture with a base leads to the isolation of 1,2,3,8a-tetrahydroimidazo[1,2-a]pyridines 2.20.56 (Scheme 563)[1472].

The most convenient acidic agent in this case is TFA, and this technique makes it possible to isolate the products with a labile para-methoxybenzyl group.
The mechanism of this domino transformation can be described as follows (Scheme 564)[1472]. Protonation of the imidazolidine fragment occurs mainly at the most basic nitrogen atom in the position 3. At the same time, the migration of the proton to the second nitrogen atom triggers a [3,3]-sigmatropic rearrangement of the ion 2.20.57, and the resulting allene intermediate 2.20.58 isomerizes into the conjugated azatriene 2.20.59 through a 1,5-sigmatropic shift (or as a result of protonation–proton abstraction processes). Subsequent 6π-electrocyclization leads to imidazopyridine 2.20.56, which, however, undergoes ring opening to form the pyridinium salt 2.20.60. In this case, the released amino group binds another proton to give the dication 2.20.55.

When a base is added, recyclization occurs as a result of an attack of the amino group at the position 2 of the pyridinium salt to form imidazopyridine 2.20.56. The isomer corresponding to the cyclization at the position 6 has not been observed in this case. In general, it is possible to reversibly convert pyridinium salts 2.20.55 into imidazopyridines 2.20.56 and vice versa by adding bases or acids. At the same time, the heterocyclic system 2.20.56 is an unusual base, capable of binding two protons at once at the same nitrogen atom (the concentration of the only registered monoprotonated form, the salt 2.20.60, with the addition of < 2 equiv. acid, according to NMR spectroscopy, does not exceed 3.5% of the total amount of all forms)[1472].
The opening of the five-membered ring of imidazopyridines 2.20.56 can also be achieved by the action of acid chlorides as electrophilic reagents (Scheme 565). The resulting pyridinium salts 2.20.61 are promising antibacterial agents[1472].

The study of the adducts 2.20.62 (R1 = OMe) obtained from 6,7-dimethoxy-3,4-dihydroisoquinolines and MP, has allowed the development of a divergent approach to the synthesis of pyrrolo[2,1-b]benzazepine derivatives 2.20.63 and pyrido-[2,1-a]isoquinolines 2.20.64 (Scheme 566)[1473]. When hydrocarbon solvent is used together with the nucleophilic assistance of PPh3, the formation of pyrrolo[2,1-b]azepine system 2.20.63 occurs as a result of a [3,3]-sigmatropic rearrangement and subsequent transannular cyclization. The alternative direction is realized in a more polar solvent. It is interesting to note that adduct 2.20.62 (R1 = H), which does not contain donor substituents in the benzene ring, gives approximately the same mixture of the two products under both conditions (ratio ~ 35 : 50).

Nitriles are promising substrates for creating new domino reactions, since they combine both electrophilic and pro-nucleophilic reactivity, while the hydrogen atoms in the α-position (if present) are highly acidic. L.G.Voskressensky et al.[1474, 1475] have discovered a reaction of N-(cyanomethyl)isoquinolinium salts 2.20.65 and o-hydroxybenzaldehydes 2.20.66 in basic medium, giving chromeno[2',3':4,5]imidazo[2,1-a]isoquinolines 2.20.67 (Scheme 567). The reaction begins with Knoevenagel condensation, followed by cyclization involving phenolic hydroxyl and nitrile groups. The resulting 1-(2-iminochromen-3-yl)isoquinolinium cation 2.20.68 undergoes another nucleophilic cyclization, and the aromatization of the imidazole ring due to the transfer of a hydrogen completes the transformation sequence.

It should be noted that the use of 1-substituted N-(cyanomethyl)isoquinolinium salts 2.20.69 does not change the regioselectivity of the process: the imidazole is formed by attacking the C(1) atom of isoquinoline, but there is no aromatization of the imidazole and isoquinoline rings (Scheme 568)[1476].

The reaction has been found to be of general character, and, in addition to various isoquinolinium salts, it also works with pyridinium salts, independently discovered by the Proença[1477] group (products 2.20.70), and benzosilanopyridinium salts, which are converted into polycycles 2.20.71[1478]. Using thiosalicylic aldehydes, annulated thiochromene imidazo-[1,2-a]isoquinolines 2.20.72 can be synthesized[1479]. Due to various biological activities of azaindole and carboline derivatives, cyclization products 2.20.73[1480]. have been synthesized by the reaction of N(6)-cyanomethyl-6-azaindolium salts, and carboline derivatives 2.20.74[1481]. have also been obtained. Moderate cytotoxic activity towards tumor cells was found for many representatives of such compounds.

Reactions of 7-cyanomethyl-7-azaindolium salts 2.20.75 with o-hydroxybenzaldehydes are accompanied by hydrolysis of imine intermediates 2.20.76, and closure of the imidazole ring has been not observed (Scheme 569)[1482]. Probably, deprotonation of the N(1)-H group occurs and 7-azaindole anhydrobases are formed in this case. Thus, the loss of the positive charge on the nitrogen atom causes the reaction to lose its driving force and end up in hydrolysis. The use of N(1)-methylated 7-azaindolium salt 2.20.75a (R1 = Me) also did not allow the cyclization route.

It was possible to avoid hydrolysis and achieve the formation of the target polyheterocycles by carrying out the reaction of 1-methyl-7-(cyanomethyl)azaindolium salts 2.20.75a with o-hydroxybenzaldehydes in a microwave reactor in absolute ethanol in the presence of anhydrous K2CO3 and molecular sieves when heated to 150 °C for 7 min (Scheme 570). This approach also turned out to be effective for 4- and 5-azaindole systems: isomeric polycycles 2.20.77 and 2.20.78 were obtained in moderate and high yields, respectively.

The use of N-(cyanomethyl)thiazolium salts significantly changes the outcome of the domino reaction. The interaction of N-(cyanomethyl)thiazolium iodide 2.20.79 with o-hydroxybenzaldehyde similarly begins with Knoevenagel condensation, followed by the formation of 2-iminochromene 2.20.80 due to the intramolecular attack of the phenolic hydroxyl group at the triple C≡N bond[1483, 1484]. However, the subsequent attack of the imine at the C(2) atom of the thiazolium ion leads to ring opening and recyclization at the position 4 of chromene, forming chromenoimidazothiazine system 2.20.81 (Scheme 571). Thus, an ANRORC mechanism (nucleophilic addition — ring opening — ring closure) is implemented in this domino transformation. The reaction proceeded successfully with various thiazolium salts and o-hydroxybenzaldehydes. However, it was not possible to obtain oxazolium cyanomethyl salts, while the transformation of imidazolium salts stopped at the stage of the iminochromene intermediate with it’s hydrolysis. It is noteworthy that chromenoimidazothiazines 2.20.81 exhibit antiproliferative activity[1484].

The possibility of nucleophilic addition at the C(4) atom of 2-iminochromene has allowed us to significantly expand the boundaries of the cyanomethyl azinium salts chemistry. When carrying out the reaction of N-(cyanomethyl)pyridinium chloride (2.20.82) with o-hydroxybenzaldehyde on cooling, it is possible to stop the domino sequence at the stage of formation of this intermediate (Scheme 572)[1485]. The addition of a nucleophile to a solution of N-(2-imino-2H-chromen-3-yl)pyridinium (2.20.83) triggers further transformation, however, an oxidizing agent is required for final aromatization of the imidazole ring. Scheme 572 shows an example of the reaction of N-cyanomethylpyridinium salt with o-hydroxybenzaldehyde in the presence of nitromethane as a nucleophile and manganese(III) acetate as an oxidizing agent. The iminochromene intermediate 2.20.83 was isolated and characterized as a perchlorate, and its direct reaction with nitromethane gave the target product 2.20.84, confirming the proposed pathway.

Potassium permanganate proved to be a more versatile oxidizing agent, working with a wide range of nucleophiles (Scheme 573). The addition of π-nucleophiles to the iminopyridinium intermediate proved effective: indoles, azaindoles, and pyrrole. At the same time, phenols gave products in the form of mixtures of regioisomers and in lower yields, while pyrazole and indazole acted as N-nucleophiles. Diethyl malonate was also used as a C-nucleophile. Considering the wide variation of both o-hydroxybenzaldehydes and cyanomethyl salts, it can be stated that the sequential three-component process allows for a high degree of chemical diversity.

The possibility of isolating the iminochromene intermediate — 1-(2-imino-2H-chromen-3-yl)pyridin-1-ium perchlorate (2.20.85) — made it possible to carry out the transformation with nitromethane, occuring without an oxidizing agent, but with the elimination of a pyridine fragment (Scheme 574)[1486]. Thus, refluxing the perchlorates 2.20.85 in TFE in the presence of nitromethane and DBU led to the unusual 4-(nitromethylidene)-4H-chromene-2-amines 2.20.86. The reaction begun with 1,2-addition of nitromethane, followed by pyridine elimination and tautomerization.

The chemistry of cyanomethyl pyridinium salts has seen interesting developments in reactions with 1,3-dielectrophiles. Proença et al.[1487, 1488] have found that N-(cyanomethyl)pyridinium salts can dimerize upon prolonged reflux in acetonitrile, and the dimer 2.20.87 can undergo further reaction with acetylacetonate (R = Me) to give pyridoindolizine-10-carbonitriles 2.20.88 with substituents at the C(2) and C(4) atoms (Scheme 575). We have shown[1489] that such a transformation can occur in a one-pot manner without the need to isolate a dimer salt. We hypothesized that the use of 1,3-dielectrophiles such as malondialdehydes (R = H) in reactions with pyridinium salts would expand the range of available pyridoindolizines bearing a substituent at the C(3) atom. However, such reactions proved to be ineffective — the products were formed in low yields.

In turn, the use of synthetic precursors of malondialdehydes–vinamidinium salts 2.20.89–as nucleophiles gave the target pyrido[2,3-b]indolizine-10-carbonitriles 2.20.88 in moderate yields (Scheme 576)[1489]. It is believed that in the basic medium, the pyridinium salt dimer is able to undergo an intramolecular cyclization and is transformed into indolizine 2.20.90, which aromatizes due to the elimination of pyridine. The resulting aminoindolizine 2.20.91 condenses with the vinamidinium salt, completing the sequence. The intermediate aminoindolizine 2.20.91 can be isolated from the reaction mass, and its interaction with the vinamidinium salt 2.20.89 gives the target product 2.20.88, which confirms the proposed pathway of the process.

Other 1,3-dielectrophiles such as enaminones 2.20.92 react with substrates 2.20.82 in a similar manner (Scheme 577)[1489]. The reaction is carried out in a microwave reactor in aqueous isopropyl alcohol in the presence of sodium acetate. This method grants an access to pyridoindolizines 2.20.88 with various substituents at the C(2) atom. A study of the optical properties of such pyridoindolizines showed that they are effective fluorophores, characterized by fluorescence quantum yields of up to 81% with emission maxima in the 448 – 490 nm region.

When switching to 2-methyl-substituted salts of N-cyanomethylpyridinium 2.20.93, the type of the reaction with enaminones changes — the pseudo-three-component process becomes a two-component one (Scheme 578)[1490]. The deprotonated alkyl group intramolecularly attacks the nitrile fragment instead of dimerization, forming aminoindolizine 2.20.94. The latter condenses with enaminone, furnishing the pyridine ring. The synthesized pyridoindolizines 2.20.95 also turned out to be effective fluorophores with fluorescence quantum yields of up to 82%.

Malononitrile is widely used in multicomponent reactions as a precursor to the aminopyridine ring[1491-1493]. Much of the reactivity of malononitrile is associated with its transformation into a dimer under the process conditions. The similarity of malononitrile dimer to homophthalonitrile was exploited by the group of Prof. L.G.Voskresensky[1494] to develop a number of multicomponent reactions. Thus, heating homophthalonitrile (2.20.96) with o-hydroxybenzaldehyde in the presence of bases led to the formation of a diastereomeric mixture of chromenoisoquinolinamines 2.20.97 as a result of a pseudo-three-component reaction (Scheme 579). The use of L-proline, a cheap organocatalyst, made it possible to carry out the reaction diastereoselectively (dr = > 90 : 10). It is believed that Knoevenagel condensation followed by cyclization gives the corresponding 2-iminochromene, which can undergo another cyclization to give the iminoisoquinoline intermediate 2.20.98. Nucleophilic addition of a second equivalent of homophthalonitrile completes the transformation.

Running the process in a sequential mode allowed the introduction of different nucleophiles into reactions with chromenoisoquinolineamine (Scheme 580). Initial heating of homophthalonitrile (2.20.96) with salicylic aldehyde in a microwave reactor in the presence of ammonium acetate generated the iminoisoquinoline intermediate 2.20.98. The use of nucleophiles such as nitromethane[1495], indole, azaindole[1494] or pyrrole[1496], resulted in the corresponding products 2.20.99 – 2.20.101. Note that indole-substituted chromenoisoquinoline amines 2.20.100 have pronounced fluorescent properties and can be considered as pH sensors, since reversible quenching of fluorescence occurs upon addition of an acid[1494].

This reaction was further investigated using ammonium formate as a base (Scheme 581)[1497]. The ability of formate to be a hydrogen donor led to the reduction of the iminochromene intermediate. The corresponding chromenoisoquinolinamines 2.20.102 were obtained in good yields from various o-hydroxybenzaldehydes and homophthalonitriles. A study of the biological activity of 2.20.102 derivatives showed their high potential as antiproliferative agents. Molecular docking and in vitro experiments confirmed the hypothesis of DNA intercalation by these compounds 2.20.102[1497, 1498].

In an attempt to extend this methodology to heterocyclic dinitriles, a new process was discovered. Refluxing N-(cyanomethyl)indole-2-carbonitrile (2.20.103) with o-hydroxybenzaldehyde in alcohol in basic medium resulted in Schiff bases 2.20.104 of 1-alkoxypyrazino[1,2-a]indole-3-amines (Scheme 582)[1499]. Such reaction can begin with the addition of an alcohol molecule at the aromatic nitrile group to form imidate 2.20.105. Being a nucleophile, this imidate is able to attack the second nitrile group, and subsequent tautomerization leads to 1-alkoxypyrazino[1,2-a]indol-3-amine 2.20.106, which reacts with the aldehyde. When the reaction is carried out in the absence of aldehyde, pyrazinoindoleamines 2.20.106 can be isolated with good yields. Unfortunately, these substances are unstable in solutions and oligomerize, limiting their application and further studies. In turn, Schiff bases 2.20.104 are stable even when stored in solutions.

The formation of imidates in a basic medium was then used for the transformation of N-(propargyl)indole-2-carbonitriles (2.20.105) — in this case, an alkyne became an intramolecular trap for the nucleophilic imidate. It was shown that in the presence of bases, the N-propargyl fragment undergoes an alkyne-allene rearrangement, and the addition of an alcohol at the C≡N bond is followed by an attack of the imidate on the sp-hybridized carbon atom of the allene system (Scheme 583)[1500]. Allene 2.20.106 can be isolated from the reaction mixture, and its further heating in alcohol in the presence of a base gives the final products 2.20.107. This transformation has been carried out for a wide range of primary and secondary alcohols, and derivatives of benzyl and furfuryl alcohols have been obtained as well. When carrying out the reaction in alcohols, it is possible to use catalytic amounts of DBU (up to 5 mol.%). The ability of DNA binding was demonstrated for 1-alkoxypyrazino[1,2-a]indoles 2.20.107[1501].

Japanese scientists Yoshimatsu and co-workes[1502] reported the aza-Henry reaction involving nitriles in the presence of stoichiometric amounts and/or excess of copper iodide, DBU and cesium carbonate. The addition of nitromethane to nitrile results in the formation of nitroenamines. It turned out that N-(propargyl)indole-2-carbonitriles 2.20.105 are capable of reacting with nitromethane in the absence of copper salts, and the catalytic amounts of DBU are sufficient for almost quantitative conversion of the substrate to pyrido[1,2-a]indoles 2.20.108 (Scheme 584)[1503]. In this case, the resulting nitroenamine 2.20.109 acts as a C-nucleophile, intramolecularly attacking the sp-hybridized carbon atom.

A similar transformation was also effective in the case of malonic esters, although the use of a stoichiometric amount of DBU was required (Scheme 585)[1503]. Aromatization of the pyridine ring occurs due to the elimination of one carboxylate group. Compounds 2.20.110 are excellent fluorophores, having emission maxima in 475 – 490 nm region and fluorescence quantum yields of up to 63%.

The addition of N-nucleophiles to N-(propargyl)indole-2-carbonitriles 2.20.105 in the presence of lithium hexamethyldisilazide (LiHMDS) was nonselective–mixtures of products of alkyne hydroamination or addition to nitrile group, followed by cyclization, were formed. Lowering the temperature allowed the selective formation of compounds 2.20.111 by alkyne hydroamination, and the catalytic amounts of LiHMDS were sufficient (Scheme 586)[1504]. Anilines, (aza)indoles, pyrrole, pyrazole, and benzimidazole successfully acted as nucleophiles. The addition of methanol to the reaction mixture led to 1-methoxypyrazinoindoles 2.20.107 and completely switched the selectivity of the process. When the reaction was carried out in a step-by-step manner (first, the rapid formation of 1-methoxypyrazinoindole 2.20.107, followed by prolonged heating with a nucleophile to replace the methoxy group), yields of pyrazinoindoles 2.20.112 reached 85%, and hydroamination products were absent. The third direction of the reaction was realized through selective hydroamination at low temperature followed by intramolecular cyclization on heating. This approach was also suitable for anilines; as a result, N-arylpyrazino[1,2-a]indole-1-imines 2.20.113 were obtained. A study of the optical properties of these systems showed that, despite the low fluorescence quantum yields, the hydroamination products of 2.20.111 have very large Stokes shifts (~100 nm), being of interest for organic electronics.

Chemodivergent transformation can have the following mechanism (Scheme 587)[1504]. In all cases there is a base-catalyzed alkyne-allene rearrangement. On cooling, the anionic nucleophile adds via the allene system, giving hydroamination products 2.20.111. In the presence of methanol, the addition of the methoxide anion predominantly occurs at the nitrile group with the formation of imidate 2.20.114, which cyclizes at the allene fragment. The resulting methoxypyrazinoindole 2.20.107 undergoes nucleophilic substitution of the methoxy group under the action of an anionic nucleophile, turning into aminopyrazinoindole 2.20.112. Methoxypyrazinoindole 2.20.107, obtained by a different procedure, also gives the nucleophilic substitution product 2.20.112, confirming the proposed pathway. Cyclic imines 2.20.113 are formed as a result of isomerization of the hydroamination product 2.20.111 followed by intramolecular attack of the anilide at the nitrile group.

Thus, over the last 10 years, the group of Prof. L.G.Voskressensky has developed effective approaches for the synthesis of various heterocyclic systems, including both structures privileged for medicinal chemistry and unique, previously unknown compounds. The key to the implementation of these processes is the combination of the reactivity of the heterocyclic nucleus, the carbon-carbon and carbon-nitrogen triple bonds.
The first group of methods involves the initial attack of the sp3-hybridized nitrogen atom of a partially saturated heterocycle at the triple bond conjugated with an electron-withdrawing group (ester, ketone). Then, with the participation of the α-carbon atom of the alkyne, the heterocycle expands by 2 atoms during nucleophilic substitution or by 4 atoms as a result of a [3,3]-sigmatropic rearrangement (in the presence of an ethynyl group in a position adjacent to the nitrogen atom), giving azocines and ten-membered heterocyclic allenes. The second group of methods involves the use of partially saturated or aromatic annulated or monocyclic azines containing a carbonyl group in a position adjacent to the nitrogen atom. In this case, the attack of the sp2-hybridized nitrogen atom on the triple bond of the alkyne is accompanied by cyclization involving the α-carbon atom of the alkyne and the carbonyl group, which leads to pyrrolo[1,2-a]isoquinolines and related compounds. The third group of methods consists of divergent syntheses of various polycyclic systems based on pseudo-three-component adducts of terminal electron-deficient alkynes and heterocycles with a carbon-nitrogen double bond. The fourth group is the preparation of polycyclic systems based on the reactions of cyanomethyl derivatives of azines and azoles with salicylic aldehydes, as well as some 1,3-biselectrophiles. In these reactions, homophthalonitrile has also been used instead of cyanomethylated heterocycles. The fifth group of methods involves an isosteric transition from 1-cyanomethylindole-2-carbonitriles to their propargyl analogues. In this case, the nature of the products certainly changes, but the main thing remains the same — the ability of the substrate to undergo domino transformations.
2.21. 5-Aryloxazolidines: synthesis and chemical properties
This Section summarizes and systematizes the data on the reactions of 5-aryloxazolidines published mainly in the last ten years. During this period there has been an active development in the chemistry of these compounds, mainly associated with the discovery of a new synthetic approach, namely, the [3 + 2] cycloaddition reaction of nonstabilized azomethine ylides to aromatic aldehydes. The simplest 2,4-disubstituted oxazolidines, which are of particular interest for further modification, became available. It has been shown that the benzene ring at the 5-position of the oxazolidine significantly extends the synthetic possibilities of such molecules, complementing the reactivity of the iminium cation classical for oxazolidines by the electrophilicity of the benzylic position and the nucleophilicity of the aromatic core. Inter- and intramolecular transformations using these functional groups are considered. At the same time, since 5-aryloxazolidines contain β-phenethylamine moiety, which is one of the most common in nature and in pharmaceutical chemistry, the products derived therefrom (e.g. adrenaline) are often valuable from a practical point of view.
In general, chemical reactions of oxazolidines 2.21.1 arise from the presence of two electrophilic centres, C(2) and C(5) atoms, basic nitrogen and oxygen atoms, and a nucleophilic aromatic ring therein. Despite the simplicity of the molecular structure, the abundance of reaction centres means that all five bonds of the oxazolidine ring can be involved; cationic cyclizations with the participation of the benzene ring are also known (Scheme 588).

This review shows that substrates 2.21.1 can act as synthetic equivalents of iminium and benzylic carbocations, formaldehyde, arylethanolamine and even nonstabilized azomethine ylide.
We have attempted, for the first time, to summarize and analyze the data on the synthesis and reactivity of 5-aryloxazolidines reported mainly over the last decade. Readers may also be interested in the 2016 review[1505] on the 1,3-dipolar cycloaddition of azomethine ylides to carbonyl dipolarophiles giving rise to a variety of oxazolidines, including 5-aryloxazolidines.
2.21.1. Synthesis of 5-aryloxazolidines
In the infancy stage of 5-aryloxazolidine chemistry, the reaction between an aldehyde and an arylethanolamine was considered the most popular method for their synthesis, and unavailability of the latter determined the scope of the method in general. Since ephedrine (2.21.2), extracted from natural sources, was the most common ethanolamine, 2-substituted 3,4-dimethyl-5-aryloxazolidines 2.21.3 derived from it became the most widely used oxazolidines for several decades (Scheme 589)[1506, 1507]. The multistep synthesis of other oxazolidines made them less attractive as starting materials, severely limiting the development of the chemistry of this simple heterocyclic system.

At the same time, the retrosynthetic cleavage of the 5-aryloxazolidine molecule 2.21.1 via O – C(2) and C(4) – C(5) bonds gives the azomethine ylide A and the aromatic aldehyde 2.21.4 (see Scheme 589). Despite the high efficiency of this approach, its development was hampered by the fact that the active use of type A nonstabilized azomethine ylides in organic chemistry did not begin until the late 1980s of the previous century.
Interestingly, the first attempts to utilize azomethine ylides and intermediate 5-aryloxazolidines were known much earlier, for example in the synthesis of biogenic arylethanolamines from aromatic aldehydes. In 1943, Akabori and Momotani[1508] found that the reaction of benzaldehyde with amino acids such as sarcosine and N-methylalanine affords halostachine (2-(methylamino)-1-phenylethanol) and ephedrine, respectively, in low yields. However, the study was devoid of insights into the mechanism of this reaction.
More progress has been made by Rizzi[1509], who reported in 1970 the synthesis of phenylephrine based on the [3 + 2] cycloaddition of 3-benzyloxybenzaldehyde and unsymmetrical azomethine ylide derived therefrom. Subsequent hydrolysis of the oxazolidine mixture and reductive debenzylation afforded phenylephrine in 23% yield. The disadvantage of the above reactions and the reason for the low yields of the products is the formation of asymmetric azomethine ylides and, as a consequence, the synthesis of the target 5-aryloxazolidine together with its regioisomer. Although Rizzi[1509] discusses several variants of amino alcohol formation, it is the first time that the structures of intermediate azomethine ylides and 5-aryloxazolidines are explicitly shown.
New impetus to this area was given by Orsini et al.[1510] who correctly determined the structure of the products of the reactions of sarcosine, proline and N-benzylalanine with benzaldehydes; Grigg et al.[1511] who carried out the reaction of tetrahydroisoquinoline-1-carboxylic acid with pyridine-2-carbaldehyde; Tsuge et al.[1512] who studied the reaction of amino acids with formaldehyde and carried out the cycloaddition of ylide to 4-nitrobenzaldehyde. In addition, mention should be made of the works of Achiwa and co-workers[1513] and Padwa and Dent[1514] who synthesized one of the most successful and currently widely used (including in reactions with benzaldehydes) precursors to azomethine ylide, N-methoxymethyl-N-(trimethylsilylmethyl)benzylamine (2.21.5) (Scheme 591).

The first systematic studies on the synthesis of 5-aryloxazolidines based on the [3+2] cycloaddition of symmetrical nonstabilized azomethine ylides A were performed by Nyerges et al.[1515] (2001), who explored the reaction of sarcosine (2.21.6) and formaldehyde with benzaldehydes, and Ryan et al.[1516] (2007), who studied the reaction of the ylide precursor 2.21.5 with (hetero)aromatic aldehydes.
Nyerges et al.[1515] showed that the three-component reaction of sarcosine (2.21.6), paraformaldehyde and aromatic aldehyde 2.21.4 was highly selective, and after boiling in benzene with a Dean – Stark trap for 1 – 15 h, the corresponding 5-aryloxazolidines 2.21.1 were obtained in 58 – 98% yields (Scheme 590). Although two carbonyl components are present in the reaction medium and their ability to form azomethine ylide was previously known, azomethine ylide A is formed only from the more active formaldehyde, with the aromatic aldehyde acting as a dipolarophile. It is important to note that the authors obtained the corresponding oxazolidines as the only products. The yields of the products were high and even near-quantitative, and in the series of benzaldehydes studied, only piperonal bearing an electron-donating methylenedioxy moiety at positions 3 and 4 showed a low yield (58%). Generally, this reaction is a very efficient one-step approach to 5-aryloxazolidines 2.21.1 from available reagents.

Ryan et al.[1516] studied the reactions of the organosilicon precursor N-benzylazomethine ylide B with aromatic and heteroaromatic aldehydes 2.21.4. In contrast to the first example of such a reaction, which had already been carried out in 1987 by Padwa and Dent[1514] with benzaldehyde under the action of lithium fluoride and ultrasound, the authors chose a operationally simpler method of activation proposed by Achiwa and co-workers[1513] for cycloaddition to alkenes, namely, catalysis by trifluoroacetic acid (Scheme 591).
The reaction proceeds readily with both electron-donating and electron-withdrawing substrates. However, strongly donating or acidic starting compounds react poorly or do not form 5-aryloxazolidines. For example, 3-hydroxybenzaldehyde and pyrrole-2-carbaldehyde give no target product. Interestingly, replacement of the hydrogen atom with a methyl group at the 1-position of the pyrrole substrate did not change the reaction outcome, but decrease in the donor ability of the nitrogen atom by sulfonylation completely restored the activity of the aldehyde group in the [3 + 2] cycloaddition. The action of ylide B on 4-hydroxybenzaldehyde was accompanied by the Mannich reaction involving the benzene ring. It should be noted that this reactivity forms the base of a separate field of chemistry of nonstabilized azomethine ylides[1517, 1518].
Silane 2.21.5 is not the only precursor to N-benzylazomethine ylide B. Thus, in 2021, Kumar and Banerjee[1519] proposed an electrochemical approach for the generation of this ylide from N,N-bis(trimethylsilylmethyl)benzylamine 2.21.7 on graphite electrodes and carried out its cycloaddition to benzaldehyde and p-nitrobenzaldehyde (Scheme 592).

In 2013, Piettre and co-workers[1520] found that ylide derived from silane 2.21.5 adds smoothly to nitro- and dinitrobenzophenones 2.21.8a,b to give 5,5-diaryloxazolidines 2.21.9a,b (Scheme 593). The authors also carried out the cycloaddition of ylide to 3-formyl-1-triflylindole 2.21.10a and to indol-3-ylketoester 2.21.10b but in both cases the resulting oxazolidines 2.21.11a,b were contaminated with the dearomatization products resulting from the addition to the double bond of the pyrrole moiety, which significantly reduced the yields of oxazolidines 2.21.11.

In summary, an efficient and versatile synthetic approach to diverse 5-aryloxazolidines based on readily available aromatic substrates has emerged in the early 2000s. Cycloaddition to (hetero)aromatic aldehydes proved the easiest. The reaction of sarcosine and formaldehyde is particularly noteworthy for its efficiency, accessibility and environmental friendliness, while the acid-catalyzed desilylation of N-methoxymethyl-N-(trimethylsilylmethyl)benzylamine (2.21.5) is preferable for the addition to less active dipolarophiles.
It should be noted that the synthesis of 5-aryloxazolidines by formaldehyde-assisted cyclization of arylethanolamines has retained its relevance over the last decade. The original tandem modifications of this well-known process were the reactions of ethanolamines 2.21.12 with arylboronic acids 2.21.13 to give N-benzyloxazolidines 2.21.14[1521] or with phenylpropiolic acid (2.21.15) (Scheme 594)[1522]. The latter direction was also achieved with terminal acetylenes in a two-step process via the isolation of bis(5-phenyl-1,3-oxazolidinyl)methanes[1523]. The resulting N-benzyl- (2.21.14) and N-propargyloxazolidines (2.21.16) are valuable objects for further transformations (see Scheme 607−Scheme 609, Scheme 612, Scheme 613, Scheme 616 below).

2.21.2. Hydrolysis and related reactions
Hydrolysis of the aminoacetal methylene group in 5-aryloxazolidines is one of the most important transformations, leading to valuable amino alcohols such as adrenaline, phenylephrine and halostachine.
In 2013, we proposed a two-step synthesis of N-methyl(benzyl)arylethanolamines 2.21.17 from available aromatic aldehydes (Scheme 595)[1524]. First, the reaction of aldehyde 2.21.4, sarcosine (2.21.6) and formaldehyde gave 5-aryloxazolidine 2.21.1, which was further hydrolyzed without purification in methanol with hydrochloric acid or by treatment with hydrazine in ethyl alcohol to the final amino alcohol 2.21.17. In this way, halostachine (2.21.17a) was obtained from benzaldehyde, and adrenaline (2.21.17b) was prepared from O,O-dibenzoylprotocatechuic aldehyde. N-Benzylazomethine ylide B, formed when treating N-(methoxymethyl)-N-(trimethylsilylmethyl)benzylamine (2.21.5) with catalytic amounts of trifluoroacetic acid, was also successfully used in this reaction to give N-benzylethanolamines 2.21.17.

This one-pot method can formally be regarded as the direct nucleophilic addition of the methyl(benzyl)aminomethyl carbanion, generated from sarcosine and formaldehyde or benzylamine 2.21.5, to the carbonyl group of the starting aromatic aldehyde 2.21.4.
We have also found that in addition to aldehydes, aromatic ketones 2.21.18 can be used in the synthesis of ethanolamines (Scheme 596)[1525]. Since nonstabilized azomethine ylides are electron-donating dipoles sensitive to steric hindrance, sterically non-hindered acceptor ketones react readily with sarcosine and formaldehyde to provide expectedly high yields of the intermediate oxazolidines 2.21.19 and amino alcohols 2.21.20. At the same time, the less active benzophenone required harsher conditions: heating in DMF with the precursor 2.21.5 in the presence of LiF for five hours. Unlike oxazolidines 2.21.1, which are derived from aromatic aldehydes and hydrolyzed in methanol, the 5-substituted 5-aryloxazolidines 2.21.19 required a higher hydrolysis temperature: heating in n-butanol with hydrochloric acid at 90 °C for 1.5 h.

The hydrolysis of 5-aryloxazolidines may be accompanied by subsequent intramolecular cyclization involving the free hydroxy and amino groups. Aromatic aldehydes and ketones 2.21.21 with ester or nitrile groups in positions 1, 4 or 1, 5 relative to the carbonyl group have been found to form (aminomethyl)lactones 2.21.22 and 5-hydroxy-2-piperidones 2.21.23 (Scheme 597)[1526]. The intermediate oxazolidines 2.21.24 are accessible from low-active carbonyl compounds by boiling with N-(methoxymethyl)-N-(trimethylsilylmethyl)benzylamine (2.21.5) in DMF in the presence of LiF for 5 hours. It is noteworthy that the initially formed lactones 2.21.22 are quite stable due to conformational rigidity, and their conversion to lactams 2.21.23 requires heating in a microwave reactor at 130 °C or the action of an external nucleophile (MeOH, NaOH). Typical examples of the above products are phthalide 2.21.22a and 5-tolylpiperidone 2.21.23a.

5-Aryloxazolidines are also accessible from some sterically hindered aromatic ketones bearing an acceptor group in the α-position. For example, treatment of 3-cyanochromones 2.21.25 with an excess of sarcosine (2.21.6) and formaldehyde or precursor 2.21.5 after the formation of the primary C=C-cycloadduct 2.21.26 gives the C=O-cycloadduct 2.21.27 as a single diastereomer (Scheme 598)[1527, 1528]. The latter affords a tetracyclic amidine 2.21.28 by hydrolysis.

In 2016, Kumar and co-workers[1529] found similar transformations of substituted 3-cyanochromones and methyl esters of chromone-3-carboxylic acids, which were reacted with silane 2.21.5 to yield tetracyclic amidines and lactams. The configuration of the latter was established by X-ray diffraction analysis and was consistent with the stereochemistry of products 2.21.28 synthesized by us.
The same year, a similar reaction with chromanones 2.21.29 was reported (see Scheme 598)[1530]. Significantly, the multiple bond conjugated to the CO2Et group in compounds 2.21.29 and 2.21.30 remained unreactive towards ylide B, again highlighting the sensitivity of nonstabilized azomethine ylides to alkyl moieties in the dipolarophile. Unfortunately, the authors attributed the configuration to the spirocentre of the adduct 2.21.30 and the C(11b) atom in the product 2.21.31 having no X-ray and 2D 1H – 1H NOESY data in hands. In our opinion, this seems doubtful since in the described examples 1527 – 1529 the α-cyano group activated the carbonyl group and determined the direction of the ylide attack.
Stabilized azomethine ylides can be used in the synthesis of 5-aryloxazolidines. For example, the reaction of methyl prolinate 2.21.32 with aromatic aldehydes produces stabilized ylides C, which further undergo [3 + 2] cycloaddition with the second molecule of the aldehyde to give 2-oxapyrrolizidines 2.21.33 with high regio- and stereoselectivities[1531]. Hydrolysis of the oxazolidine ring in the latter compound affords α-(α-hydroxybenzyl)prolines 2.21.34 in 33−50% overall yields (Scheme 599).

The approach to 5-aryloxazolidines based on [3 + 2] cycloaddition is not limited to the reaction of aldehydes with azomethine ylides. An alternative method of heterocycle formation is known, which has so far only been represented by the synthesis of densely functionalized compounds. Thus, in 2018, Suga and co-workers[1532] described a method for the synthesis of substituted 5-aryloxazolidines 2.21.35 using the cycloaddition of carbonyl ylides D derived from their diazoethers 2.21.36 to imines 2.21.37 (Scheme 600). The process was catalyzed by rhodium acetate and ytterbium triflate at room temperature in dichloromethane and the resulting oxazolidines 2.21.35 were further hydrolyzed in aqueous ethanol to ethanolamines 2.21.38 in moderate to high yields using p-toluenesulfonic acid as the catalyst.

2.21.3. Reactions on the С(2) and С(5) atoms of oxazolidine
The second most common transformation of oxazolidines (after their hydrolysis to amino alcohols and carbonyl compounds) is their reaction with nucleophiles on the C(2) position. Due to the presence of an aminoacetal methylene group, oxazolidines can undergo reversible ring opening via the O−C(2) bond in the presence of Lewis or Brønsted acids to give an alcohol and an iminium cation, which can react with various nucleophiles. Among the reactions of oxazolidines at the C(2) atom, one of the simplest is the reduction to tertiary amines. This process commonly utilizes the mild reagents NaBH4 (see[1533, 1534]) and NaBH3CN (see[1535]) but more harsh reduction conditions have been described using LiAlH4[1536], AlH3 (see[1537]) hydrogen in the presence of Pd on carbon[1538], Raney nickel (Ni−Ra)[1539], and rhodium catalyst[1540].
The reduction of oxazolidines obtained by cyclization of ethanolamines with carbonyl compounds is in fact a variant of reductive amination. For example, the reduction of oxazolidines 2.21.3 derived from ephedrine 2.21.2 (or pseudoephedrine) and various aldehydes, under the action of hydrogen and Pd/C in ethanol, was used by Perron and Alexakis[1538] to give amino alcohols 2.21.39a – e and further chiral ligands based thereon (Scheme 601; the yield of the corresponding oxazolidines is given first in parentheses). As an alternative, the Hitchcock and co-workers[1541] used LiAlH4 in boiling tetrahydrofuran. It should be noted that the reduction of 2-aryl-substituted oxazolidines 2.21.3 required prolonged heating of the mixture.

The use of [3 + 2] cycloaddition of nonstabilized azomethine ylides to benzaldehydes allowed Nyerges et al.[1515] to propose an approach to 2-(dimethylamino)-1-arylethan-1-ols via reduction of 5-aryl-3-methyloxazolidines using NaBH4 in ethanol. In 2015, Ryan and co-workers[1542] explored a similar reaction involving nonstabilized N-benzylazomethine ylide, generated from the precursor 2.21.5 and a catalytic amount of trifluoroacetic acid, with phthalic anhydrides 2.21.40 (Scheme 602). Despite the fact that azomethine ylides are unreactive towards the C=O bond of esters, the carbonyl group of phthalic anhydride proved to be active enough to react with the azomethine ylide. As a result, the authors obtained a series of 3H-spiro[isobenzofuran-1,5′-oxazolidine]-3-ones 2.21.41a – e in good to high yields. The cycloadducts 2.21.41 were expectedly unstable compounds and had to be purified by chromatography on a special low-acid silica gel. Phthalides 2.21.41 were then reduced with NaBH4 to give the more stable 3-(alkylaminomethyl)-3-hydroxyisobenzofuran-1-ones 2.21.42a – e. Notably, in the absence of symmetric substituents in the aromatic ring of phthalic anhydride 2.21.40, the cycloaddition proceeded with low regioselectivity to deliver a mixture of two oxazolidines.

In addition to the reduction of ephedrine oxazolidines 2.21.3 to synthesize chiral ligands, Diéguez and co-workers[1543] carried out ring-opening of oxazolidines 2.21.43 to amino alcohols 2.21.44 with Grignard reagents (Scheme 603). The high nucleophilicity and Lewis acidity of magnesium bromides allow them to react particularly readily with 5-aryloxazolidines at the 2-position[1506, 1507].

L-Proline (2.21.45) reacts with benzaldehyde in DMSO to give stereoisomeric oxapyrrolizidines 2.21.46 in 97% overall yield, the product with the anti-position of the phenyl moieties being predominant in the mixture (Scheme 604)[1540]. The mixture of oxazolidines 2.21.46 was used to prepare α-substituted pyrrolidines 2.21.47[1544]. The initial treatment of oxapyrrolizidines 2.21.46 with Grignard reagent led to the opening of the oxazolidine ring to afford stereoisomeric amino alcohols 2.21.48 with high conversion. Subsequent oxidative cleavage of the ethanolamine moiety with Pb(OAc)4 afforded 2-alkyl- and 2-aryl-substituted pyrrolidines 2.21.47 in moderate yields of 36 – 42% based on starting proline. This one-pot procedure did not require isolation and purification of intermediates and formally represents the α-functionalization of proline 2.21.45 by replacement of the carboxyl group.

In addition to Grignard reagents, other C-nucleophiles can induce the oxazolidine ring opening in the presence of Lewis or Brønsted acids. For example, various reagents, including aqueous HCN[1545], as well as the TMSCN – BF3 · Et2O (see[1546]) and KCN – H+ systems[1547] have been used in the reaction between oxazolidines and cyanide anions. The resulting 2-[(2-hydroxyethyl)amino]acetonitriles are valuable building blocks for further transformations.
In 2016, Couty and co-workers[1548] proposed a method for the synthesis of N-arylazetidine 2.21.49 from amino alcohol 2.21.50 using oxazolidine 2.21.51 as an intermediate (Scheme 605). The target 4-methyl-1,3-diphenylazetidine-2-carbonitrile (2.21.49) was obtained by treating the intermediate amino acetonitrile 2.21.50 with mesyl chloride in the presence of triethylamine, followed by potassium tert-butoxide-promoted anionic cyclization of the appropriate chloride.

The reaction of 5-aryloxazolidines with C-nucleophiles 2.21.52 such as dialkyl malonates, ethyl benzoylacetates and deoxybenzoin has been studied in our laboratory[1549]. It was found that the reaction of these methylene-active compounds with oxazolidines 2.21.1 promoted with magnesium ethylate proceeds as a domino process (Scheme 606). Initial oxazolidine ring opening and nucleophilic attack gives the Mannich base E, which generated a terminal acceptor alkene F after elimination of the halostachine molecule. The subsequent Michael reaction with the second molecule of the methylene-active compound 2.21.52 delivers the methylene-linked 1,3-dicarbonyl compounds 2.21.53 in moderate yields. In this case, the 5-phenyloxazolidine 2.21.1 is the methylene donor and is essentially a formaldehyde analogue.

5-Aryloxazolidines show versatile reactivity due to the presence of multiple reaction centres. They are sources of iminium cations and O,N-dinucleophiles and, due to the 5-positioned aryl ring, can react to generate benzyl carbocation in the presence of Lewis or Brønsted acids. The latter direction is mainly represented by examples of Friedel – Crafts cationic intramolecular cyclizations at the benzyl position of oxazolidines (C(5) atom), and in most cases involves the initial opening of the heterocycle via the C(2) atom by a nucleophile or hydride anion[1550].
Coote et al.[1534, 1551] reported stereoselective approaches to 4-aryltetrahydroisoquinolines and tetrahydrobenz[d]azepines. The strategy involved the reduction of 5-aryloxazolidines derived from ephedrine, pseudoephedrine or halostachine and aromatic aldehydes and further intramolecular Friedel – Crafts cationic cyclization of the intermediate amino alcohol via the aromatic moiety. A 2010 publication[1552] showed that the reduction of oxazolidines 2.21.54 with LiAlH4 proceeds in high yields in the presence of donor substituents in the aromatic ring of the starting benzaldehyde (Scheme 607). Boiling of amino alcohols 2.21.55 in the presence of a 1 : 1 mixture of sulfuric acid and trifluoroacetic acid led to 4-aryl-2,3-dimethyltetrahydroisoquinolines 2.21.56 with a preferential trans position of the substituents in the case of oxazolidines 2.21.54 based on both ephedrine and pseudoephedrine. It is worth noting that the authors were able to improve the diastereoselectivity of the cyclization to 99 : 1 by using a complex of the starting amino alcohols with chromium tricarbonyl.

Extending the scope of the [3 + 2] cycloaddition of nonstabilized azomethine ylides to carbonyl compounds has led to a new synthesis of tetrahydroisoquinolines via the formation of 5-aryloxazolidines. In 2013, our group[1553] proposed a three-step procedure for the preparation of 4-aryltetrahydroisoquinolines 2.21.57 from aromatic aldehydes, sarcosine, paraformaldehyde and Grignard reagents (Scheme 608). The key step was the ring opening in 5-aryloxazolidines 2.21.1 under the action of (het)arylmagnesium bromides, furnishing N-benzyl-β-hydroxyphenylethylamines 2.21.58 in 54 – 88% yields base on the starting benzaldehydes. Treatment of the products 2.21.58 with aluminium chloride, sulfuric or polyphosphoric acid resulted in an intramolecular attack of the benzylic carbocation (former C(5) atom of oxazolidine) on the incorporated aromatic moiety to give 4-aryltetrahydroisoquinolines 2.21.57 in 32 – 65% yields (based on starting benzaldehyde). Remarkably, the cyclization in polyphosphoric acid also proceeded via the thiophene ring to afford 4-(p-bromophenyl)-6-methyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridine in 65% yield.

As can be seen from the foregoing examples, the introduction of a second aromatic moiety into the product of the 5-aryloxazolidine ring-opening under the action of a nucleophile or the reduction of 2,5-diaryloxazolidines opens up the possibility of intramolecular cyclization involving the 5-positioned carbon atom of the oxazolidine substrate. An alternative approach to tetrahydroisoquinolines was the quaternization of N-benzyl- or N-methyloxazolidines with methyl iodide or benzyl chlorides, leading to N-benzyl-N-methyl-5-aryloxazolidines 2.21.59 in high yields (Scheme 609)[1554]. Heating of these quaternary ammonium salts in 70% perchloric acid leads to ring opening at the O – C(5) bond, Friedel – Crafts alkylation at the N-benzyl moiety, elimination of the formaldehyde molecule and formation of 4-aryltetrahydroisoquinolines 2.21.57 in yields from 24 to 62% based on starting benzaldehyde.

We discovered an interesting example of the [4+2] annulation of 5-aryloxazolidines to indoles (Scheme 610)[1555]. Heating of a mixture of oxazolidine 2.21.1 and indole 2.21.60 in acetic acid follows the Mannich reaction pathway and ends in 1-aryl-2-(indole-3-ylmethylamino)ethanols 2.21.61. It is shown that heating the latter compounds in polyphosphoric acid induces an intramolecular electrophilic substitution to provide 4-aryl-1,2,3,4-tetrahydro-γ-carbolines 2.21.62 in yields ranging from 34 to 38% based on starting benzaldehyde. It is worth noting that when acetic acid is replaced by polyphosphoric acid in the reaction of 5-aryloxazolidine with indole, the process follows the route of double electrophilic substitution and immediately furnishes γ-carbolines 2.21.62.

In the above reactions (see Scheme 607−Scheme 610), the Friedel – Crafts intramolecular cyclization proceeds at the N-benzyl substituent, which becomes the aromatic ring of an isoquinoline or γ-carboline, and the 5-aryl moiety of the oxazolidine turns out to be an aryl substituent at position 4 of the target heterocycle.
In 2013, we developed a method for the recyclization of 5-aryloxazolidines 2.21.1 to 1,2,3,4-tetrahydroisoquinolin-4-ols 2.21.63 (Scheme 611)[1556]. It was shown that heating the starting compound in 6 M HCl at 60 °C leads to the oxazolidine ring opening, formation of the iminium cation G and Pictet – Spengler cyclization to the products 2.21.63 in 62−89% yields (based on the starting benzaldehyde). The presence of an electron-donating substituent in the meta-position of the aromatic ring, which promotes electrophilic substitution, was required for the reaction to proceed, whereas oxazolidines with 5-positioned p-methoxyphenyl and phenyl substituents failed to provide the desired recyclization products. It is noteworthy that, in comparison to the Pictet – Spengler cyclization with phenylethylamines, this oxazolidine recyclization is characterized by high regioselectivity and gives a single regioisomer, the product of the attack on the para position with respect to the donor group in the benzene ring.

The 1,2,3,4-tetrahydroisoquinolin-4-ols 2.21.63 thus obtained were used in reactions with electron-rich arenes in perchloric acid or in the presence of boron trifluoride etherate[1557], leading to alkylation of the arenes and the formation of 4-aryltetrahydroisoquinolines 2.21.57 (see Scheme 611). The reaction involved alkoxy-substituted arenes (good to high yields of products) as well as tetralin, benzothiophene and coumarone. In this case, the aromatic fragment of the isoquinoline was constructed from the aryl substituent of 5-aryloxazolidine.
A logical follow-up of studies on the intramolecular recyclization of 5-aryloxazolidines was the replacement of the N-methyl group in the substrate with a benzyl one. Thus, N-benzyl-5-aryloxazolidines 2.21.1, readily formed from benzaldehydes and N-(methoxymethyl)-N-(trimethylsilylmethyl)benzylamine (2.21.5), were found to be versatile intermediates in the synthesis of bridged 7,12-dihydro-6,12-methanodibenzo[c, f ]azocines 2.21.64 (Scheme 612)[1554]. In particular, heating of N-benzyl-5-aryloxazolidines 2.21.1 in 6 M HCl solution leads to recyclization to N-benzyltetrahydroisoquinolin-4-ols, whereas treatment of these oxazolidines with 70% perchloric acid reduces the donor properties of the nitrogen atom and hence its ability to form an iminium cation. This opens the oxazolidine ring at the O – C(5) bond and an intramolecular Friedel – Crafts reaction occurs at the N-benzyl moiety. Further elimination of water and Pictet – Spengler cyclization of the aromatic moiety of the 5-aryloxazolidine completes the dibenzoazocine skeleton 2.21.64. It is worth noting that in this case the final cyclization of the iminium cation H proceeded in the presence of both electron-donating substituents in the benzene ring and a halogen or hydrogen atom. However, the pronounced acceptors in the benzene ring of 5-aryloxazolidine 2.21.1 hampers both stages of cyclization.

As mentioned above, 5-aryloxazolidines react readily with strong C-nucleophiles such as Grignard reagents, methylene-active compounds, electron-deficient heterocycles (indole, furan and pyrrole) and are also capable of intramolecular attack by the benzene ring on C(2) and C(5) atoms. However, the intermolecular variant of the latter transformation remained unrealized for a long time. In 2021, we found that 5-aryloxazolidines 2.21.1 are capable of reacting with arenes in the presence of perchloric acid, leading to alkaloid analogues, alkoxysubstituted 4-aryltetrahydroisoquinolines 2.21.57 (Scheme 613)[1557]. MsOH, TfOH, BF3 · Et2O were also found to be suitable catalysts. Control experiments showed that the process follows a mechanism similar to the intramolecular recyclization of N-benzyloxazolidines. Initially, the protonation of oxazolidine and an attack on the C(5) arene atom (Friedel – Crafts alkylation of the arene by a benzyl carboxylation) take place followed by an intramolecular Pictet – Spengler reaction on one of the aromatic rings containing a meta donor group. Donor phenols and alkoxy-substituted benzenes were successfully used as arenes, less donor toluene or xylene provided lower product yields.

Notably, the absence of a meta donor group in both benzene rings hampered the Pictet – Spengler cyclization, and the reaction stopped at the monoalkylation stage to give 1-methyl-2,2-diarylethylamines 2.21.65 (Scheme 614). The latter were also obtained by preliminary demethylenation of oxazolidines 2.21.1 and subsequent reaction of arylethanolamines 2.21.17 with arenes under similar conditions.

2.21.4. Other reactions
It was found that the methyl group at the nitrogen atom of 5-aryloxazolidine[1558] 2.21.1 can be easily replaced with a cyanomethyl group: first the substrate is quaternized and then the salt 2.21.66 is heated for 15 minutes in dimethylformamide (Scheme 615). In this way, 5-phenyl-3-(cyanomethyl)oxazolidine (2.21.67) was obtained in an 48% overall yield (81% in the quaternization step and 59% after demethylation). A similar transformation was carried out for the spiro-fluorenooxazolidine 2.21.68 (yields in the first and second steps were 83 and 60%, respectively).

An unusual palladium-catalyzed reaction of N-propargyloxazolidine 2.21.69 was described in 2021 (Scheme 616)[1559]. In this case, a N – C(4) bond cleavage rare for 5-aryloxazolidines occurred through the aromatization step of the isoquinoline ring, affording 4-benzylisoquinoline 2.21.70 and 2,3-diphenyloxirane 2.21.71.

Usually difficult to classify and unexpected transformations in the 5-aryloxazolidine series are associated with the presence of leaving groups in their molecules. Thus, in 2011, Ryan and co-workers[1560] carried out the reaction of azomethine ylides derived from benzylamine 2.21.5 with isatoic anhydrides 2.21.72 (Scheme 617). Although one would expect a classical [3+2] cycloaddition at the most active carbonyl group at the 4-position, the products were not oxazolidines I but benzodiazepinones 2.21.73. The authors suggest that the intermediate spirooxazolidine I is unstable and initiates the rearrangement of the oxazine ring with carbon dioxide elimination, leading to the extension of the six-membered ring to a seven-membered diazepine ring.

The instability of 5-aryloxazolidines may be due to the presence of other leaving groups in the molecule. It has been shown that the reaction of aromatic aldehydes with 4-hydroxyproline (2.21.74) or indoline-2-carboxylic acid (2.21.75) does not stop at the stage of condensed 5-aryloxazolidines J, but continues to give N-2-hydroxyethylpyrroles 2.21.76 or indoles 2.21.77 (Scheme 618)[1561]. Only aromatic aldehydes with electron-withdrawing groups take part in this reaction.

In 2016, our research group[1562] discovered that 5-aryloxazolidines, depending on their structure (e.g. compound 2.21.1a), can undergo cycloreversion upon heating from 120 to 210 °C (Scheme 619). This ability is most characteristic of spiro-oxazolidines 2.21.78 and 2.21.79, but spiro-oxazolidine 2.21.80, which already decomposes upon heating in toluene, should be singled out. It is shown that retro-[3 + 2] cycloaddition with this compound is a convenient method for the in situ preparation of nonstabilized azomethine ylides under anhydrous conditions and can be applied to the synthesis of cycloadducts 2.21.81 from electron-poor dipolarophiles. In some cases, the use of oxazolidine 2.21.80 is the only way to carry out the reaction of ylides with a dipolarophile[1563], which does not occur under the classical reaction conditions with sarcosine and formaldehyde or N-(methoxymethyl)-N-(trimethylsilylmethyl)benzylamine (2.21.5).

The material presented above indicates that 5-aryloxazolidines have great synthetic potential and the increased interest in them over the last 10 years has not yet revealed all the possibilities of the chemistry of these compounds. Indeed, many of the known reactions have been carried out with only a few initial substrates, and the influence of particular substituents in such systems is still unclear. At the same time, it is already obvious that the oxazolidine ring, due to the presence of a 5-positioned aryl substituent, can be introduced in various reactions involving all of its constituent atoms.
The main activators of 5-aryloxazolidines are acids, and the molecules that react with them should have nucleophilic properties. This combination triggers both simple transformations, such as Mannich and Pictet – Spengler reactions or Friedel – Crafts alkylation, and complex inter- or intramolecular tandem processes leading to alkaloid-like systems fused in various ways. The stages in these transformations proceed separately, which is due to the fact that the acid catalyst activates the O – C(2) and O – C(5) bonds in 5-aryloxazolidines differently: under weakly acidic conditions, they react only as iminium cations, whereas under strongly acidic conditions they react on both positions 2 and 5 with an initial attack on the benzylic carbon atom. At the same time, a number of non-electrophilic reactions are known in the chemistry of 5-aryloxazolidines related to the low stability of the substrates due to steric stress or the presence of leaving groups. One of the most promising processes is the cycloreversion of spiroanthracenoxazolidines, which allows the in situ formation of nonstabilized azomethine ylides under anhydrous conditions.
Of special note is the presence of a pharmacophore phenethylamine moiety in 5-aryloxazolidines, as this often determines their presence in reaction products and makes 5-aryloxazolidines per se one of the most accessible building blocks for use in medicinal chemistry.
2.22. Hypervalent iodine compounds: from preparation to applications for the synthesis of heterocycles
Hypervalent iodine compounds (HICs) are widely used in modern organic chemistry as reactants and catalysts[1564-1567]. For example, HICs based on heterocyclic benziodoxole are more stable than their acyclic analogues, which opens more opportunities for the synthesis and safe use of reagents with functional ligands such as N3, CN, and CF3 groups[1568, 1569]. The higher stability of cyclic HICs is attributable to bonding between the apical and equatorial ligands at the iodine centre, which hinders pseudo-rotation and reductive elimination resulting in thermal decomposition[1570]. Currently, the chemistry of benziodoxole derivatives (Figure 16) includes a wide palette of reagents (e.g., compounds 2.22.1 – 2.22.14) that are used for the transfer of functional groups and also in annulation and heterocyclization reactions[1571-1576]. Moreover, these reagents show high reactivity both in the presence and in the absence of transition metal-based catalysts or photocatalysts. Hypervalent iodine compounds are not limited to oxygen-containing molecules. Currently, the synthesis and applications of pseudocyclic and cyclic λ3- and λ5-iodanes with a short contact between the I and N atoms in the molecule are promising lines of research, although these compounds are still poorly explored. However, it is known that this class of HICs has better physicochemical properties than oxygen-containing compounds, e.g., good solubility, higher stability and excellent oxidative activity[1577, 1578].
![[{"id":"6u_BVKNPsR","type":"paragraph","data":{"text":"Representatives of heterocyclic and pseudocyclic iodine(III or V) compounds and their analogues."}}]](/storage/images/resized/xJXSEXD6oud7mtxUWyGm6lOgZA1tcrdU9qRbP25A_xl.webp)
The chemistry of iodonium salts is an especially important area of HIC chemistry because of their unique properties and ability to function as reactants and catalysts in many reactions[1579-1582]. Arylation reactions have been studied in most detail for this class of compounds, as they possess noticeable advantages over conventional approaches using aryl halides[1583-1585].
This part of the review describes the contribution to the hypervalent iodine chemistry made by the research group headed by Professor M.S.Yusubov. Methods for the synthesis and practical applications of benziodoxoles with iodine(III or V) atoms and some representatives of nitrogen-coordinated iodanes are considered and recent data on the synthesis of heterocyclic derivatives using hypervalent iodine compounds are summarized.
2.22.1. Cyclic and pseudocyclic λ3-iodanes
In 2011, our research group[1586] reported the first synthesis and characterization of new heterocyclic compounds 2.22.15 – 2.22.17, containing trivalent iodine atoms, oxygen and boron in a five-membered ring, which were called benziodoxaboroles by analogy with benziodoxole (Scheme 620). Subsequently, studies of the chemistry of compounds 2.22.15 resulted in the synthesis of new pseudocyclic hypervalent organoiodine compounds, arylbenziodoxaborole triflates 2.22.18, under mild conditions. These compounds served as precursors for the generation of arynes 2.22.19 at room temperature under the action of water[1587]. Arynes 2.22.19 demonstrate traditionally high reactivity in certain reactions, for example, in click-reactions and Diels – Alder reactions, which afford the corresponding adducts with high selectivity and in good yields. In addition, aryne intermediates that are formed from triflates 2.22.18 selectively react with tert-butylphenol to give ortho-arylation products 2.22.20 and 2.22.21 in moderate yields (see Scheme 620) and can also form covalent bonds with graphene oxide[1588].

Benziodoxole organosulfonate derivatives are thermally stable compounds exhibiting high reactivity in oxidative transformations[1573, 1564-1568]. Benziodoxoles 2.22.6, 2.22.11 and pseudo-benziodoxoles 2.22.7, 2.22.8 and 2.22.12 are strong electrophiles and mild oxidants with respect to various unsaturated compounds[1573, 1589]. In particular, pseudocyclic triflate 2.22.7 (IBA-OTf) proposed in our study[1590] is an effective reagent for oxidative heterocyclization of compounds 2.22.22 – 2.22.24 (to give 2.22.25 and 2.22.26)[1591-1594] and oxidation of sulfides 2.22.27[1595], phenols 2.22.28 (to give 2.22.29 and 2.22.30) and unsaturated compounds 2.22.31 and 2.22.32 (to give 2.22.33 and 2.22.34)[1573], and for the reactions involving arenes 2.22.35 and alkynes 2.22.36 to prepare hypervalent iodine derivatives 2.22.37 – 2.22.40 (Scheme 621)[1595-1599], which are utilized for the transfer of functional groups[1575].

As compared with analogous hypervalent iodine compounds reported previously[1565, 1567], IBA-OTf (2.22.7) makes it possible to perform a broad range of reactions under considerably milder conditions (see Scheme 621). In addition, our research group showed that triflate 2.22.7 can be used as a recyclable reagent[1590] or even as a catalyst[1573], which complies with the green chemistry principles[1600].
Apart from IBA-OTf (2.22.7), a number of pseudocyclic tosylates IBA-OTs (2.22.8) were synthesized and their structure and reactivity were investigated. Benziodoxole tosylates 2.22.8a,b, prepared from 2-iodobenzoic acids 2.22.41, readily react with various organic substrates as iodine-centred electrophiles to give oxidation products 2.22.42 and 2.22.43 or the corresponding iodonium salts 2.22.44 (Scheme 622)[1601]. Subsequently, we have shown[1602] that IBA-OTs (2.22.8a) can be used in catalytic amounts as a mediator of heterocyclization reactions.

Yusubov et al.[1603] proposed a convenient one-pot method for the synthesis of 1-arylbenziodoxoles (ArBX) 2.22.5 from 2-iodobenzoic acids 2.22.41 on treatment with Oxone (2 KHSO5 · KHSO4 · K2SO4), an inexpensive and environmentally safe oxidant (Scheme 623). The reactivity of compounds 2.22.5 was investigated in the nucleophilic substitution reactions with N3 and F anions[1604]. It was shown that all reactions of 1-arylbenziodoxolones 2.22.5 proceed as nucleophilic substitution in which the iodonium group in the electron-deficient benzene ring of benziodoxole is the leaving group. A strong electron-withdrawing substituent, a nitro group located in the para-position to the iodine atom in benziodoxolone, sharply increases the substitution rate, whereas bulky substituents in the ortho-position of the aryl ring retard the reaction. Later, this fact was utilized to perform radiofluorination to prepare 2-[18F]-fluoro-5-nitrobenzoic acid 2.22.45 in a radiochemical yield of 39.5% and with radiochemical purity of 98.4% (see Scheme 623)[1605]. 2-[18F]-Fluoro-5-nitrobenzoic acid 2.22.45 is a promising radioligand for positron emission tomography[1580, 1606, 1607].

Hypervalent iodine organosulfonate derivatives such as Koser’s reagent and the like are in demand in organic chemistry as selective oxidants characterized by thermal stability and safety[1565, 1566]. We made use of this fact to prepare iodine(V) tosylate derivatives 2.22.46 and 2.22.47 as alternatives to known IBX (2.22.9) and DMP (2.22.10) reagents. These products are formed from IBX 2.22.9 on simple treatment with p-toluenesulfonic acid in acetic anhydride at room temperature (Scheme 624)[1608]. Tosylates 2.22.46 and 2.22.47 proved to be excellent oxidants for structurally diverse compounds, e.g., for the oxidation of alcohols 2.22.48 and 2.22.49 to aldehydes 2.22.50 and 2.22.51, which are used as key precursors in the total synthesis of polyketide antibiotics and terpenes[1608, 1609]. It should be noted that unlike IBX (2.22.9), IBX-OTs (2.22.47) is not explosive and can be stored at room temperature for several months[1608].

We isolated and characterized a thia-analogue of known IBX (2.22.9), that is, 2-iodoxybenzenesulfonic acid 2.22.11 (IBS) (see Figure 16)[1610, 1611]. Compound 2.22.11 is a more potent oxidant than IBX (2.22.9) when used in a catalytic amount[1564]. We also proposed[1611] an alternative method for the synthesis of 2-iodosylbenzenesulfonic acid 2.22.6 (IBSH), which is a promising oxidant among hypervalent iodine compounds and a precursor of functional group transfer reagents[1612].
Treatment of IBX (2.22.9) with trifluoromethanesulfonic acid resulted in the first synthesis of the most potent hypervalent iodine-based oxidant: 2-iodoxybenzoic acid ditriflate 2.22.12 (IBX · 2 HOTf)[1613]. According to X-ray diffraction data, the I – OTf bonds in ditriflate 2.22.12 are ionic, which accounts for its high reactivity in various oxidation reactions. In particular, IBX · 2 HOTf (2.22.12) can oxidize polyfluorinated primary alcohols 2.22.52, which are usually highly stable to oxidation, to give diols 2.22.53 in good yields (Scheme 625). It is important that this oxidant, which is many times more active than IBX (2.22.9), is also highly stable, non-explosive and melts without decomposition.

2.22.2. Transformations of nitrogen-coordinated cyclic and pseudocyclic iodanes
Among diverse cyclic and pseudocyclic hypervalent iodine-based reagents, iodanes with a coordination bond between I and N atoms have received a lot of attention in recent years. The high storage stability of N-coordinated iodanes in combination with high reactivity makes these reagents a convenient alternative to the well-known O-coordinated iodanes[1578, 1614]. In our study[1615], a number of N-coordinated pseudocyclic hydroxy(tosyloxy)iodosoarenes 2.22.55, 2-benzimidazolyl-substituted diaryliodonium salts 2.22.56 and their cyclic derivatives 2.22.57 were synthesized in high yields from substrates 2.22.54 (Scheme 626).

In the next stage, we studied the thermal stability and reactivity of pseudocyclic and cyclic N-coordinated λ3-iodanes[1616]. Compounds of both classes have high decomposition temperatures, which indicates that they are stable and can be safely handled. This fact correlates well with the high reactivity of these derivatives in the model oxidation of sulfides to sulfoxides in comparison with other pseudocyclic and cyclic hypervalent iodine compounds.
2-Iodobenzoic acid amides 2.22.58 were converted to bicyclic (2.22.59)[1617] and monocyclic (2.22.60)[1618] benziodazoles on treatment with m-chloroperoxybenzoic acid in acetonitrile at room temperature (Scheme 627). Benziodazoles 2.22.59 and 2.22.60 showed high efficiency in esterification of alcohols and in the synthesis of amides and could be useful in the future for the synthesis of di- and polypeptides.

Despite the high interest in the hypervalent iodine compounds containing N-centred ligands, they are still less studied than oxygen-coordinated iodanes. We hope that quite a few discoveries in this area of chemistry are still to be made.
2.22.3. Iodonium salts in the arylation of heterocyclic compounds
One more important class of HICs are iodonium salts, which are effectively used in arylation[1579, 1581] or surface modification[1582] reactions. Iodonium salts represent a more reactive alternative to conventional aryl halides, which allows conduction of arylation reactions under mild conditions and, in some cases, according to an atom-efficient protocol[1583]. Cyclic aryliodonium salts have a considerable advantage regarding the atom efficiency of reactions; therefore, they receive considerable attention. Owing to valuable synthetic properties of these compounds such as initiation of cascade reactions, they can be used for the design of complex molecules including polycyclic ones with potential application in pharmaceutics and as functional materials[1584, 1585].
In 2015, our research group proposed a convenient method for the synthesis of dibenziodolium salts, which includes the oxidative cyclization of 2-iodobiphenyl using Oxone as an inexpensive and environmentally safe oxidant in the presence of appropriate strong acids[1619]. Later, this method was used to prepare 5-iodo-1-phenylimidazole 2.22.61. Imidazole-substituted cyclic iodonium salts 2.22.62 were synthesized in this way. The cyclization was triggered by cheap and readily available Oxone – H2SO4 oxidative system (Scheme 628)[1620]. The cyclic iodonium compounds 2.22.62 obtained in this way readily underwent heterocyclization with elemental sulfur to give benzo[5,1-b]imidazothiazoles 2.22.63 in good yields.

Methods for arylation of oxadiazolones 2.22.64 and 2.22.65 (Ref. [1621]) and oxazolidinones 2.22.66 (Ref.[1622]) under the action of аcyclic symmetrical and unsymmetrical iodonium salts 2.22.67 with copper catalysis were developed (Scheme 629). This provided the synthesis of a broad range (> 75 examples) of N-aryl derivatives 2.22.68 – 2.22.70 in good or moderate yields.

An unusual approach to highly functionalized heterocycles includes the use of N-coordinated iodonium salts 2.22.71[1623]. The presence of a heterocyclic ligand was found to direct the substitution to the ortho-position of the aromatic system, which leads to the preparation of a number of valuable imidazole derivatives 2.22.72 – 2.22.81 (Scheme 630)[1623].

The interests of our research group are also related to halogen bond formation in cyclic[1624] and acyclic[1625] iodonium salts, which is important for the assembly of supramolecular systems with predictable structure and properties[1624-1632] or for the control of arylation selectivity[1633].
2.22.4. Synthesis of heterocyclic derivatives using hypervalent iodine compounds
Hypervalent iodine reagents are widely used in either catalytic or stoichiometric amounts to prepare various heterocyclic derivatives. In the formation of the target heterocycles, various hypervalent iodine species can act as substrate activating reagents or donors of heteroatoms[1591, 1634-1638].
We have developed simple and efficient methods for the synthesis of three- and five-membered heterocycles with participation of hypervalent iodine compounds. Yoshimura et al.[1639, 1640] used various olefins 2.22.82, including cyclic ones, as substrates (Scheme 631). Iminoiodane 2.22.83, stable and convenient for practical use, which was utilized to prepare aziridines 2.22.84 and 2.22.85, can also be effectively employed in the amination of silyl enolates and adamantane[1639].

Aziridines 2.22.85 were also prepared in an organocatalytic reaction meadiated by hypoiodite that is formed in situ from tert-butylammonium iodide and m-chloroperoxybenzoic acid[1640].
Subsequently, the oxidative cycloaddition of various aldoximes 2.22.86 and unsaturated compounds 2.22.87 – 2.22.89 under the action of HICs (2.22.7, 2.22.8, 2.22.90, 2.22.91) or hypoiodous acid (2.22.92) has been developed[1602, 1592-1594, 1641-1643]. These reactions carried out at room temperature for 24 h furnished various azoles: isoxazolines 2.22.93, isoxazoles 2.22.94 and 1,2,4-oxadiazoles 2.22.95 (Ref. [1592]), while in the presence of ketoenamines as dipolarophiles, 4-acyl-substituted isoxazoles 2.22.96 were isolated (Scheme 632)[1643]. In addition, this method was used to obtain a number of annulated azoles 2.22.97 – 2.22.99 (Refs[1593] and [1594]) and 2.22.100 and 2.22.101 (in an intramolecular reaction)[1602].

In these reactions, aldoximes 2.22.86 are oxidized with active hypervalent iodine species 2.22.7, 2.22.8, 2.22.90, 2.22.91 and hypoiodite 2.22.92 to highly reactive nitrile oxides, which then react with dipolarophiles 2.22.87 and 2.22.89 at room temperature to give cycloaddition products 2.22.93 – 2.22.101 in high yields. It is noteworthy that this approach is applicable for both intermolecular and intramolecular versions of cycloaddition of aldoximes and unsaturated compounds, and HICs can be formed in situ from iodides introduced into the reaction mixture in catalytic amounts.
The above material indicates that the chemistry of hypervalent iodine is quite diverse; indeed, apart from oxidation reactions, HICs are used for oxidative functionalization of various substrates and for the formation of new carbon – carbon and carbon – heteroatom bonds, in particular, for the synthesis of heterocycles. Hypervalent iodine compounds can be rightly regarded as versatile reagents with a wide scope of applicability; the chemical reactions involving these species represent a worthy alternative to conventional methods. In our opinion, these reagents will become valuable tools not only for organic synthesis, but also for related areas such as medicinal chemistry and materials science, and interest in them will ever increase. This brief review of the studies performed by our research group is aimed at attracting the attention of specialists to this rapidly developing area of organic chemistry.
2.23. Synthesis and transformations of cyclic peroxides
Traditionally, organic peroxides are considered as oxidants and sources of free radicals for radical polymerization and selective synthetic transformations, including oxidative functionalization of heterocycles[1644-1647]. The view on organic peroxides as potentially unstable compounds and strong oxidants was significantly revised in the last two decades, when numerous stable cyclic peroxides were obtained and the medicinal chemistry of organic peroxides started to be actively developed. This part of the review integrates approaches to the synthesis of cyclic peroxides based on di- and triketones and also 1,3- and 1,4-ketoesters and hydrogen peroxide. Unusual peroxide transformations with retention of the peroxide ring, important for modification and expansion of the range of available peroxide structures, are presented. Data on the biological activity of cyclic peroxides are given, indicating their high activity and selectivity to the HeLa, DU145 and HepG2 cancer cells, high antiparasitic activity against schistosomes (Schistosoma mansoni) and fungicidal activity against phytopathogenic and entomopathogenic fungi.
Cyclic organic peroxides represent an important and promising class of compounds. There are antimalarial drugs based on the natural peroxide artemisinin (structure A) and its derivatives: artesunate (structure B), artemether and dihydroartemisinin[1648-1650]. The introduction of a nitrogen atom into the artemisinin molecule resulted in the synthesis of 6-aza- and 11-aza-artemisinins (structures С and D, respectively) which have a higher antimalarial activity[1651, 1652]. The high demand for artemisinin and the complexity and high cost of artemisinin synthesis stimulate the development of methods for the preparation of synthetic cyclic peroxides. Currently, 1,2,4-trioxolanes (ozonides), 1,2-dioxolanes, 1,2-dioxanes and 1,2,4,5-tetraoxanes are most promising for applications in medicinal chemistry. In 2012, a modern and effective antimalarial drug was developed on the basis of fully synthetic ozonide–arterolane (structure E). Moreover, apart from the antimalarial activity[1648, 1649, 1653], cyclic peroxides exhibit antihelminthic[1654-1666], anticancer[1667, 1668], antituberculosis[1669-1671] and growth-regulating activities[1672-1674]. Recent studies indicate that synthetic peroxides have in vitro activity against human α-coronavirus NL63 and β-coronaviruses OC43 and SARS-CoV-2[1675, 1676].

The advances of medicinal chemistry related to compounds with a peroxide bridge stimulate the attention to the synthesis of new types of peroxides, in particular nitrogen-containing ones, since the –N – C – O – O – moiety is found in natural compounds with known biological activities. For example, fumitremorgin A (structure F) and verruculogen (structure G), alkaloids containing an eight-membered endoperoxide ring, were isolated from the fungi Aspergillus fumigatus and Penicillium verruculosum in 1971 and 1972, respectively[1677, 1678]. These alkaloids inhibit calcium-activated potassium channels of smooth muscles[1679]. Catharoseumine (structure H), a monoterpenoid indole alkaloid isolated from the plant Catharanthus roseus, shows antimalarial and anticancer activities[1680].

The most convenient and easy-to-handle precursors for the synthesis of peroxides are carbonyl compounds and hydrogen peroxide[1681-1683]. Monoketones are the most commonly used, mainly because the selective synthesis of peroxides becomes much more complicated when moving from monoketones to di- and triketones, since the reaction involves a molecule with two or three reaction centres. Multiple reaction centers increase the probability of side condensation and polymerization reactions giving peroxide or non-peroxide type products with cyclic or acyclic structure. Therefore, the preparation of peroxides from di- and tricarbonyl compounds is a challenging task. In this review, we describe the approaches to the synthesis of cyclic peroxides based on di- and triketones and also 1,3- and 1,4-ketoesters developed in the period from 2010 to 2023.
2.23.1. Cyclic peroxides from di- and triketones
The acid-catalyzed reaction of β-diketones 2.23.1 with hydrogen peroxide was used to develop methods for the synthesis of bridged 1,2,4,5-tetraoxanes 2.23.2. The effective assembly of the tetraoxane skeleton takes place in the presence of acids such as H2SO4, HClO4, HBF4, BF3 · Et2O[1684], HCl[1685] and phosphomolybdic (PMA) and phosphotungstic (PTA) acids[1686]. Since in the case of PMA and PTA, the peroxide synthesis occurs under mild conditions and the mechanism of catalysis differs from that of protic acids, by using PMA and PTA, it was possible to obtain tetraoxanes from α-unsubstituted β-diketones (Scheme 633). The peroxidation of acetylacetone under the action of acetic acid gives tetraoxane in a yield not exceeding 5%[1687].

1,2-Dioxolanes 2.23.3 – 2.23.5 and 1,2-dioxanes 2.23.6 were prepared in 15 to 62% yields by treatment of β- (2.23.1) and γ-diketones (2.23.7) with 50% aqueous H2O2 in the presence of catalytic amounts of concentrated H2SO4, I2 or SnCl2 · 2 H2O (Scheme 634)[1688].

A selective synthesis of 1,2,4-trioxolanes (ozonides) based on acid-catalyzed reaction of linear 1,5-diketones 2.23.8 with H2O2 without the need of using toxic ozone has been described[1689, 1690]. This enables the synthesis of ozonides containing the CN functional group and C=C and C≡C multiple bonds. Ozonides 2.23.9 are formed from linear 1,5-diketones 2.23.8 and H2O2 in high yields (up to 90%) in the presence of such acids as BF3 · Et2O, 98% H2SO4, TsOH or 50% aqueous HBF4 (Scheme 635). The synthesis of ozonides 2.23.9 from an alicyclic 1,5-diketone, 2,2'-methylenebis(cyclohexanone) and H2O2 was performed in the presence of BF3 · Et2O (Ref. [1691]) and concentrated HCl (Ref. [1692]) as a catalyst.

Using the 2 M HCl – 30% H2O2 system, 1,5-ketoacetal 2.23.10 was converted to ozonides 2.23.11 and 2.23.12. Ozonide 2.23.12 showed antituberculosis activity against Mycobacterium tuberculosis (H37Ra and H37Rv strains), with the minimum inhibitory concentrations (MICs) being 0.39 and 3.12 μg mL–1, respectively (Scheme 636)[1669].

The presence of a bridge in the 1,2,4-trioxolane ring makes a significant contribution to its stability; this enables the conversion of functional groups without affecting the high-energy О – О bond (Scheme 637)[1669, 1689, 1690]. The alkaline hydrolysis of ozonide 2.23.9 affords ozonide 2.23.13 containing the C(O)OH functional group, which can further be converted to amide 2.23.14. Surprisingly, the bridged ozonide ring is stable even to the action of LiAlH4. Hence, the ester group of ozonide 2.23.9 can be reduced, which gives product 2.23.15. The nucleophilic substitution reactions involving the hydroxyl group of compound 2.23.15 opened up the access to functionalized ozonides 2.23.16 – 2.23.19. Ozonides 2.23.20 and 2.23.21 were obtained in a similar way from substrates 2.23.11 and 2.23.12.

Due to the multifunctional nature of triketones, their behaviour in the reaction with hydrogen peroxide is unpredictable. This reaction can potentially give a complex mixture of monomeric, dimeric or polymeric products with either peroxide or non-peroxide structure. The complexity of peroxide synthesis from triketones is confirmed by the fact that only few examples of these transformations have been reported in the literature[1693, 1694]. A study of the peroxidation of carbonyl compounds demonstrated that the reactions of β,δ'-triketones 2.23.22 with H2O2 catalyzed by acids (H2SO4[1695], BF3 · Et2O[1696]) are accompanied by the selective assembly of tricyclic structures 2.23.23, which are peroxides containing only one O – O group in the molecule (Scheme 638). Tricyclic monoperoxide 2.23.23a exhibits a high activity against S. mansoni. The IC50 values of this compound against adult and juvenile schistosomes found in in vitro experiments were 11.7 and 14.4 μM, respectively. The worm burden in animals determined in vivo decreased by 82% upon a single oral administration[1697].

When the H2O2 (in diethyl ether) – heteropolyacid (PMA or PTA) system is used, the peroxidation of β,δ’-triketones 2.23.22 follows a different pathway. The reaction gives three types of structures: tricyclic monoperoxides 2.23.23, tetraoxanes 2.23.24 and stereoisomeric ozonides 2.23.25. An unusual fact is that one carbonyl group remains intact in the assembly of tetraoxanes and monoperoxides (Scheme 639)[1698, 1699].

In a cascade reaction, bridged ozonides 2.23.9, obtained from diketones 2.23.8 or triketones 2.23.22, were converted to α-bromo-, α-chloro- and α-iodo-δ-ketoesters or δ-diketones 2.23.26 on treatment with FeBr2, FeCl2 and FeI2 (Scheme 640)[1700].

This reaction illustrates, first, a nontrivial use of the carbonyl functional group for temporary generation of the peroxide moiety and, second, the unique properties of peroxides for formal replacement of the carbonyl group in the starting ketone by halogen atoms via the radical cleavage of the C – C bond. These results are important for the synthesis of heterocyclic compounds and useful for the understanding of iron-dependent anticancer activity of peroxides.
Although the history of peroxide chemistry has lasted for more than a century, peroxides are most often obtained under homogeneous conditions. Heterogeneous synthesis of peroxides appears to be a serious challenge for this field of chemistry, since peroxides tend to decompose on the catalyst surface[1701-1704]. A readily accessible and efficient catalyst, H3 + xPMo+612 – xMox+5O40 supported on SiO2, has been developed. In the presence of this catalyst, β- or γ-diketones can react with H2O2 and are thus converted to bridged 1,2,4,5-tetraoxanes 2.23.2 or 1,2,4-trioxolanes (ozonides) 2.23.9 in isolated yields of up to 85 and 90%, respectively (Scheme 641)[1705]. Nonpolar solvents such as toluene, benzene, diethyl ether, dichloromethane or carbon tetrachloride are suitable for this reaction, since the catalyst is insoluble in them. In the presence of H3 + xPMo+612 – xMox+5O40/SiO2, only the target cyclic peroxides are formed. It was found that the heterogeneous synthesis of bridged 1,2,4,5-tetraoxanes 2.23.2 can also be effectively performed with the Lewatit MonoPlus SP112H ion exchange resin as the catalyst[1706].

The biological activity of organic peroxides is attributed, according to the common views, to their oxidative capacity, i.e., it is believed that the higher the oxidative capacity, the greater the activity. In a series of studies[1707-1709], we demonstrated the absence of direct oxidative capacity – activity correlation for cyclic peroxides (Table 5), which attests to a different, more complex, mechanism of action. Ozonides 2.23.9a,b and 2.23.9'a,b j have a high cytotoxicity (IC50 < 1.0 μM) against HepG2 hepatic cancer cells and cause cell apoptosis. Moreover, ozonide 2.23.9a has a high selectivity index (SI, defined as the ratio of IC50 values for normal and cancer cells) equal to 28.3. Ozonide 2.23.26а has a high cytotoxicity and good selectivity to DU145 hormone-independent prostate cancer cells (IC50 = 0.4 μM, SI = 42)[1710].
The HepG2 cancer cells are highly resistant to chemotherapy due to expression of the ABCB5 membrane protein. It was found experimentally that ozonides 2.23.9a and 2.23.9b in concentrations of 1.6 and 2.4 μM can inhibit the the ABCB5 protein activity[1708]. Verapamil, which is a known ABCB5 inhibitor, was used as the reference drug and the rhodamine 123 dye served as the indicator. After HepG2 cells were treated with rhodamine, it was detected only in 20% of the cells. In the presence of Verapamil in concentration of 10 μM, the percentage of cells containing rhodamine 123 increased from 20 to 55%. The same result was also achieved with ozonides 2.23.9a and 2.23.9b, but at lower concentrations: 1.6 and 2.4 μM, respectively. The tetraoxanes 2.23.2a – e have a high cytotoxic activity against HeLa cancer cells (IC50 in the range from 0.18 to 4.27 μM), which proved to be comparable with or even higher than the activities of dihydroartemisinin and cisplatin (IC50 = 38.6 and 2.2 μM, respectively) (Table 6)[1696]. Moreover, tetraoxane 2.23.2c showed a high antischistosomal activity against S. mansoni: the IC50 values against the adult and juvenile worms in in vitro experiments were 0.3 and 0.1 μM. A single oral administration in vivo decreased the worm burden by 75%[1656, 1697, 1711].

A new application area for cyclic peroxides was discovered, in particular, they started to be used in agriculture as plant protection agents and crop protection agents against phytopathogenic fungi. Tetraoxanes have a high fungicidal activity against a large range of phytopathogenic fungal strains of various taxonomic classes[1705]. 1,2,4,5-Tetraoxanes 2.23.2e–i exhibit a more potent fungicidal action against Rhizoctonia solani (causal agent of black scurf of potatoes) than Triadimefon, with their half-maximal effective concentrations (EC50) being 4.4, 5.5, 6.5, 3.4, 4.8 and 23.2 mg L–1, respectively. 1,2,4,5-Tetraoxanes 2.23.2f and 2.23.2h are more effective against Bipolaris sorokiniana (causes root rot of wheat, barley, rye and oats) than Kresoxim-methyl and Triadimefon: EC50 = 5.0, 2.8, 8.7 and 18.2 mg L–1, respectively. Compound 2.23.2f proved to be more efficient than Triadimefon or Kresoxim-methyl against Sclerotinia sclerotiorum (causes white rot of sunflower): EC50 = 13.6, 28.2 and 18.3 mg L–1, respectively. Tetraoxanes 2.23.2d,e,i in concentration of 30 mg L–1 completely suppress the mycelial growth of Phytophthora infestans (causes potato blight)[1712].

The structures of bridged 1,2,4,5-tetraoxanes with high fungicidal activities are shown below.

Organic peroxides efficiently suppress (by up to 94 – 100%) the mycelial growth of the entomopathogenic fungus Ascosphaera apis affecting bumble-bees and bees. A mixture of diastereomeric ozonides 2.23.9b + 2.23.9'b and tetraoxane 2.23.2с are superior in activity to the commercial fungicide Triadimefon (EC50 = 4.0, 1.6 and 7.1 mg L–1 respectively). Moreover, peroxides do not reduce the ability of bumblebees to fly and do not cause their death[1713]. Thus, cyclic peroxides can be considered to be a new class of fungicides.
An atom-economic approach to the selective assembly of tricyclic mono- and diperoxides from β,γ'-triketones 2.23.27a – q and hydrogen peroxide was reported (Scheme 642)[1714]. An extensive study demonstrated that stable and structurally complex monoperoxides 2.23.28l – q or diperoxides 2.23.29a – p can be selectively obtained in high yields from simple molecules such as β,γ'-triketones and H2O2. These compounds are of interest for the design of biologically active products with antiparasitic, cytotoxic, fungicidal or antiviral properties. It was found that the size of the substituent in the α-position of β,γ'-triketones 2.23.27a – q influences the yield of diperoxides 2.23.29l – p: as the size of the substituent increased, the yield of diperoxides decreased from 87% (for 2.23.29l) to 30% (for 2.23.29n). Triperoxides 2.23.30 were not formed.

2.23.2. Cyclic amino-peroxides from di- and triketones
Alicyclic 1,5-diketones 2.23.31 can undergo three-component condensation with hydrogen peroxide and ammonia or some primary amines (aniline, benzylamine, 4-aminobenzoic acid) to give cyclic amino-peroxides 2.23.32 (Scheme 643)[1715-1717]. An important role in the assembly of these amino-peroxides belongs to the Thorpe – Ingold effect, according to which the presence of a large number of bulky substituents in the starting diketone markedly facilitates the cyclization. Therefore, 2,2’-methylenebis(cyclohexanone) and its derivatives are prone to give hydroacridines when react with ammonia or amines; hydroacridines are converted to amino-peroxides via the reaction with hydrogen peroxide.

A selective, facile and atom-economic method has been proposed for the synthesis of functionalized bridged amino-peroxides 2.23.33 and 2.23.33'. The method is based on three-component condensation of low-reactive acyclic δ-diketones 2.23.8, hydrogen peroxide and an NH-component [aqueous NH3, NH4OAc, HCO2NH4, (NH4)2CO3] without a catalyst (Scheme 644)[1718]. The isolated yields of the amino-peroxides reached 97%. However, unlike ozonides 2.23.9, amino-peroxides 2.23.33 and 2.23.33' could not be separated by column chromatography.


This approach opens access to bridged amino-peroxides with functional groups such as ester and cyano groups and multiple (double and triple) bonds. It is noteworthy that amino-peroxides can be stored at room temperature for several months without decomposition. Amino-peroxide mixtures 2.23.33a + 2.23.33'a (79 : 21 ratio) and 2.23.33b + 2.23.33´b (98 : 2 ratio) exhibit cytotoxicity and selectivity against A549 lung cancer cells and are non-toxic for normal liver cells (IC50 = 9.1 and 7.9 μM; selectivity index SI = 2.9 and 2.1, respectively)[1719].
Amino-peroxide 2.23.33 was found to be stable to the action of KOH, aqueous NH3 and NH4OAc and to be unstable to H2SO4. Amino-peroxide 2.23.33' is moderately stable to aqueous NH3 and unstable to H2SO4, KOH and NH4OAc. The stability of amino-peroxides under basic conditions opens up the possibility of their modification (Scheme 645). In particular, a mixture of amino-peroxides 2.23.33c + 2.23.33'c was converted to acid 2.23.34, which was then converted to amide 2.23.35. An important feature of these reactions is that they are the first transformations in which the amino-peroxide ring is preserved. Notably, the amino group does not react with ethyl chloroformate. This unusual behaviour for an unprotected secondary amine is due to the loss of nucleophilicity as a result of nN → σ*C−O hyperconjugation.

Despite the difficulty of peroxidation of carbonyl compounds, the three-component condensation of β,δ´-triketones 2.23.22 with hydrogen peroxide and an NH-source can be directed towards the selective assembly of amino-peroxides. This opens up the access to stable bridged tricyclic amino-peroxides 2.23.36 (Scheme 646)[1719].

Unlike usual multicomponent reactions in which the components are compatible with one another and react successively in a predictable manner, the system presented above contains competing reactants. Apart from the competition between ammonia and hydrogen peroxide for the electrophilic centre, the situation is complicated by the oxidative nature of hydrogen peroxide. In particular, H2O2 can react with ammonia and/or oxidize nitrogen-containing intermediates. Surprisingly, the pathway to these complex tricyclic products selectively blocks the possible alternative pathways leading to peroxides, hemiaminal esters, peroxy acetals and peroxy hemiaminals.
The three-component condensation of di- or tricarbonyl compounds involving ammonia and ammonium salts as N-components affords amino-peroxides (see Scheme 644 and Scheme 646). It was found that the peroxide group does not oxidize the NH group in these products, but provides an intramolecular protection of the NH group from electrophiles and oxidants via stereoelectronic nN → σ*CO interaction. As a result, no alkylation or acylation of the NH group takes place. Thus, the synthesis of N-substituted amino-peroxides proved to be a non-trivial task.
N-Substituted amino-peroxides 2.23.37 and 2.23.38 were prepared by acid-catalyzed three-component condensation of δ-diketones 2.23.8 or β,δ'-triketones 2.23.22 with H2O2 and hydrazides (Scheme 647)[1720, 1721]. This result will be useful for the synthesis of new biologically active compounds and for the general design of acid-catalyzed reactions of amines with electrophiles.

The three-component condensation – cyclization reactions between primary amines, 1,1'-peroxybis(1-hydroperoxycycloalkanes) 2.23.39 and pentane-1,5-dial induced by Sm(NO3)3 · 6 H2O resulted in the synthesis of N-substituted amino-peroxides 2.23.40 (Scheme 648). Amino-peroxide 2.23.40а shows cytotoxicity and selectivity to Jurkat, K562 and U937 lymphoid cells (IC50 = 7.47, 6.91 and 8.25 μM, respectively)[1722].

2.23.3. Cyclic peroxides from 1,3- and 1,4-ketoesters
In 1899, Baeyer and Villiger (See A.Baeyer, V.Villiger. Ber. Dtsch. Chem. Ges., 32, 3625 (1899); https://doi.ogr/10.1002/cber.189903203151.) reported a new reaction of ketones 2.23.41 with peroxy acid 2.23.42 as an oxidant to give esters 2.23.43. After more than a century, this reaction still provides a valuable interconnection between the key oxygen-containing functional groups (Scheme 649)[1723]. It is commonly recognized that the Baeyer – Villiger (BV) rearrangement occurs through the formation of the tetrahedral Criegee intermediate, resulting from the addition of peroxy acid to the carbonyl group of ketone 2.23.41. Hence, this highly energetic oxygen-rich intermediate rearranges via 1,2-alkyl shift, which is promoted, in particular, by the replacement of the weak O – O bond with more stable C – O bond[1724]. However, this intermediate has never been isolated and structurally characterized due to its instability and high reactivity.

It was found that switching from the linear Criegee intermediate to the cyclic one suppresses the 1,2-alkyl shift and results in the formation of a stable Criegee intermediate, which is meant for structural and mechanistic studies[1725, 1726]. Five- and six-membered Criegee intermediates with OOH (2.23.44, 2.23.46) and ОН (2.23.45, 2.23.47) groups were obtained for the first time. This structure stabilization effect for hydroxy-substituted acyl peroxides is attained owing to the disruption of the anti-periplanar arrangement of the С – Rm and О – О bonds (so-called ‘primary stereoelectronic effect’). The preferable conformation for the transfer of electron density from the donor (ОН group) to the acceptor (С=О group) is distorted, and the cyclic form of the Criegee intermediate becomes stable. It was also shown that the stability of cyclic peroxides further increases on going from ОН derivatives 2.23.45 and 2.23.47 to ООН products 2.23.44 and 2.23.46, due to a decrease in the donor capacity of the oxygen atom (so-called inverse α-effect)[1727, 1728].
A two-step method for the synthesis of five-membered cyclic Criegee intermediates (β-hydroxy-β-peroxylactones 2.23.45) from available 1,3-ketoesters 2.23.48 has been developed (Scheme 650)[1725]. The first step is peroxidation of 1,3-ketoesters 2.23.48 with hydrogen peroxide in the presence of a large excess of BF3 · Et2O, which affords β-hydroperoxy-β-peroxylactones 2.23.44 in 73 to 92% yields in the case of R1 = 1°Alk, 2°Alk (primary and secondary alkyl groups, respectively). The attempts to obtain the corresponding peroxide with a tertiary alkyl group as R1 did not meet with success. The second step is the selective reduction of the hydroperoxide group to ОН in the presence of a labile endocyclic acylperoxy group; this furnishes β-hydroxy-β-peroxylactones 2.23.45. The nature of the substituent R1 was found to strongly influence the possibility of formation and stability of the ring with the ОН group. For example, β-hydroxy-β-peroxylactones 2.23.45 with R1 = Me were isolated in 55 – 66% yields and could be stored at a temperature of –18 °С for several months without changes; peroxides 2.23.45 with R1 = 1°Alk were formed in 30 – 63% yields and were less stable, and peroxides 2.23.45 with R1 = 2°Alk could be isolated only as mixtures with BV rearrangement products.

It was established that β-hydroperoxy-β-peroxylactones 2.23.44 can be prepared in 30 – 96% yields by BF3-catalyzed cyclization in the presence of H2O2 starting from various acyclic β-ketoesters (2.23.48) and silyl ethers of their enols (2.23.49), alkyl ethers of their enols (2.23.50), enol acetates (2.23.51) and cyclic acetals (2.23.52) (Scheme 651)[1729]. Remarkably, irrespective of the chosen substrate, these reactions lead to the same products–β-hydroperoxy-β-peroxylactones 2.23.44. The calculated thermodynamic parameters of formation of β-hydroperoxy-β-peroxylactones 2.23.44 from enol silyl ethers 2.23.49, enol acetates 2.23.51 and cyclic acetals 2.23.52 confirm that β-peroxylactones 2.23.44 actually correspond to a deep energy minimum on the combined potential energy surface. These reactions proceed under mild conditions and open access to a wide range of β-hydroperoxy-β-peroxylactones, which are formed selectively even in those cases where alternative oxidation pathways could be expected.

The six-membered cyclic ООН derivatives of the Criegee intermediates, γ-hydroperoxy-γ-peroxylactones 2.23.46, were synthesized by peroxidation of γ-ketoesters 2.23.53 followed by cyclization in situ induced by the BF3 · Et2O – H2O2 system (Scheme 652)[1726]. The dihedral angle between the О – О and С – R1 bonds in the six-membered ring of intermediate 2.23.46 is close to 180°; hence, the BV rearrangement can take place. Weakening of the electron-donating effect of the lone pair of oxygen of the alkoxy group on the breaking С – R1 bond by the second oxygen atom provides a sufficient kinetic stabilization for the formation of γ-hydroperoxy-γ-peroxylactones 2.23.46 with the methyl substituent at C(6). Indeed, a variety of γ-hydroperoxy-γ-peroxylactones 2.23.46 with the methyl group in position 6 and a large range of substituents at С(5) were obtained in moderate and good yields. The attempts to obtain γ-hydroperoxy-γ-peroxylactones 2.23.46 with a primary alkyl or aryl group in position 6 gave the BV rearrangement products: compounds 2.23.54 and 2.23.55. The treatment of γ-hydroperoxy-γ-peroxylactone 2.23.46а with triphenylphosphine resulted in the selective reduction of the hydroperoxide group to give γ-hydroxy-γ-peroxylactone 2.23.47а (see Scheme 652). Although the last-mentioned compound is often unstable and is converted to a mixture of γ-keto acid and, possibly, γ-ketoperoxy acid, we were able to isolate and characterize product 2.23.47а. This cyclic peroxide was the first example of Criegee intermediate as a six-membered ring in which it is not protected by a pronounced change in the dihedral angle between the two breaking С – R1 and O – O bonds.

β-Cyclopropyl-β-hydroxyesters 2.23.56 react with a 30% aqueous solution of H2O2 in the presence of H2SO4 to give the corresponding β-peroxylactones 2.23.57 in 30 – 80% yields (Scheme 653)[1730]. The resulting peroxides showed a moderate activity against P. falciparum: MICs (in mg mL–1) were 10.0, > 50.0, 10.0 and 50.0 for compounds 2.23.57a–d, respectively.

An approach to a new class of organic peroxides, β-alkoxy-β-peroxylactones 2.23.58, based on the reaction of β-ketoesters 2.23.48 with Н2О2 and alcohols has been reported (Scheme 654)[1731]. Unlike other cases in which alcohols do not participate in the reactions of carbonyl compounds with hydrogen peroxide, in this case, alcohols successfully act as the second nucleophilic component for the bifunctional dicarbonyl electrophile.

According to quantum chemical calculations, the participation of alcohols in the reactions of carbonyl group with the peroxide remained unnoticed because of the absence of sufficiently deep kinetic traps on the way to more stable condensation products[1731]. Conversely, β-alkoxy-β-peroxylactones 2.23.58 are not prone to conversion to more stable β-hydroperoxy-β-peroxylactones 2.23.44, because this reaction should proceed via especially unfavourable carbonyl-substituted peroxycarbenium cation. The absence of stabilization of the cationic centre in this oxygen-free molecule is caused by the effect of an additional electron-withdrawing substituent, in this case, the carbonyl group (Scheme 655).

The reactions between the carbonyl compounds and hydrogen peroxide are convenient and facile tools for the synthesis of oxygen-free complex heterocyclic systems. These systems possess an enormous practical potential for both the design of drugs for the treatment of socially significant diseases and plant protection and for being used as building blocks in organic synthesis. Synthetic cyclic peroxides have a high cytotoxicity and selectivity when tested on HeLa, DU145 and HepG2 cancer cells, pronounced antiparasitic activity against S. mansoni, and fungicidal action against phytopathogenic and entomopathogenic fungi. The presented review demonstrates the high significance of cyclic peroxides for medicinal and organic chemistry.
2.24. Tetrapyrrole macroheterocycles for biomedical applications
The use of synthetic tetrapyrroles as analogs of natural porphyrins (chlorophylls, hemes, cobalamines, etc.) for biomedical applications is one of the most urgent directions in the development of macroheterocyclic chemistry, which corresponds to modern trends in the creation of nature-like technologies. Varying the structure of tetrapyrroles by modifying the periphery of the molecule, as well as by introducing different metal centers in the cavities of macrocycles makes it possible to obtain compounds with specified photophysical properties. Such macrocycles are intended to solve problems in biomedicine — first of all, photodynamic therapy, including its antibacterial variant, diagnostics using fluorescent methods, research of molecular mechanisms of photodynamic effect on membranes and biomolecules, as well as biophotopolymerization. The combination of various methods of photodynamic action and diagnostic techniques using tetrapyrroles is the basis for the development of agents for theranostics.
This section discusses the results obtained over the last 10 years at the Frumkin Institute of Physical Chemistry and Electrochemistry (IPCE RAS) and the Kurnakov Institute of General and Inorganic Chemistry (IGIC RAS) in the group headed by full members of RAS A.Yu.Tsivadze and Yu.G.Gorbunova[1732, 1733]. Approaches related to the preparation of (hetero)annelated and non-peripherally substituted phthalocyanines that absorb light in the near-infrared (NIR) range corresponding to the ʻtherapeutic windowʼ of tissue transparency are systematized. Methods for the synthesis of biocompatible water-soluble tetrapyrroles with cationic and anionic ionogenic groups, as well as cationic phosphorus(V) complexes with unique photophysical characteristics are considered. Heteroannelated porphyrins and ruthenium phthalocyaninates as homo- and heterogeneous catalysts and photocatalysts for the preparation of pharmaceutical precursors such as sulfoxides, cyclopropanes, secondary and tertiary amines are discussed separately. Finally, the possibilities of reversible post-synthetic modification of tetrapyrroles to control their properties and create «smart» biomedical materials are demonstrated[1734].
2.24.1. Benzannelated analogs of phthalocyanines
The search for tetrapyrrole chromophores with absorption in the NIR range is aimed at the development of photosensitizers for photodynamic therapy and bioimaging on their basis, since light in this range is characterized by the greatest penetration depth into biological tissues. The main approach to the creation of tetrapyrroles with absorption in the region of the ʻtherapeutic windowʼ is the preparation of naphthalocyanines — benzannelated phthalocyanines. However, despite the great prospects for practical application of phthalocyanines with extended π-system, a significant limitation is their tendency to aggregate due to stacking interactions, which reduces solubility and significantly affects photophysical properties.
Crown-substituted naphthalocyanines were synthesized to control aggregation, as the introduction of crown-ether moieties into tetrapyrrole molecules was previously shown to be a successful solution to their uncontrolled aggregation by cation-induced assembly of ensembles with the desired architecture[1735].
Williamson alkylation of 1,2-dibromo-5,6-dihydroxynaphthalene 2.24.1 with chloroethoxyethanol followed by Okahara ring closure reaction of the 15-crown-5-ether ring in diol 2.24.2 and Pd-catalyzed cyanation[1736] of dibromide 2.24.3 afforded 15-crown-5-naphthalonitrile 2.24.4[1737]. This compound was further used for template cyclotetramerization in the presence of magnesium or zinc acetates and DBU in boiling pentanol, leading to the first representatives of the crown-substituted naphthalocyanines M[(15C5)4Nc], where M = Mg and Zn (15C5 denotes the 15-crown-5-ether fragment, Nc denotes the naphthalocyanine) (Scheme 656(Hereinafter, unless specifically stated, alkyl substituents of normal structure were used.), path a). The nature of the base applied for this cyclotetramerization was shown to play a key role in the formation of the target naphthalocyanines. Thus, the use of lithium amylate as a base led to the cleavage of crown-ether macrocycles due to the nucleophilic attack of the amylate anion at the ipso-position C(Ar) – O in the naphthalonitrile molecule containing two acceptor CN groups[1738].

M[(15C5)4Nc] complexes are almost insoluble in chloroform, but dissolve in its mixture with methanol, thus being in the form of aggregates in the obtained solutions. Spectrophotometric study showed that the interaction of such aggregates with potassium acetate leads to cofacial supramolecular dimers {2M[(15C5)4Nc] · 4 KOAc} with an absorption maximum at 712 nm, which dissociate in the presence of [2.2.2]cryptand to form monomeric complexes exhibiting near-infrared absorption with Q-band maxima at ~ 770 nm. In contrast to photostable cofacial dimers, such monomeric forms are characterized by low photostability: the solutions containing them rapidly discolour in the light due to the generation of singlet oxygen, the formation of which was confirmed using a selective chemical trap — diphenylisobenzofuran.
To increase the solubility of naphthalocyanines and reduce their tendency to aggregation, bromination of naphthalonitrile 2.24.4 with N-bromosuccinimide followed by cross-coupling of the intermediate dibromide with hept-1-yn by Sonagashira (Scheme 656, path b) was carried out[1739]. Hydrogenation of the resulting 5,8-diheptinyl derivative 2.24.5 afforded 5,8-diheptyl-substituted 15-crown-5-naphthalonitrile 2.24.6. The template cyclotetramerization of dinitriles 2.24.5 and 2.24.6 in the presence of magnesium or zinc acetates led to the formation of the corresponding naphthalocyanines as shown by UV-Vis and MALDI-TOF mass spectra. These complexes were found in solution in monomeric forms, but they were found to be unstable and underwent rapid aerobic photodegradation. The use of copper acetate as a template resulted in non-aggregating photostable complexes, which were not capable of singlet oxygen generation due to the presence of a paramagnetic metal center. It was thus possible to study the effect of the nature of the substituents on the ability of such complexes to cation-induced aggregation using UV-Vis and ESR. The heptynyl-substituted naphthalocyanine Cu[(C7H11)8(15C5)4Nc] was shown to form a cofacial supramolecular dimer in the presence of excess potassium acetate, whereas its heptyl analog Cu[(C7H15)8(15C5)4Nc] did not aggregate under these conditions even in the presence of excess potassium acetate. In this case, dimerization was hindered by a change in the conformation of the crown ether substituents caused by the adjacent bulky heptyl groups.
In contrast to alkynyl substituted phthalocyanines, the introduction of heptynyl groups at positions 5 and 8 of the naphthalene units in naphthalocyanines did not lead to a bathochromic shift of the Q-bands. To interpret the observed spectral properties, we used a simplified approximation of time-dependent density functional theory (sTD-DFT) as a more express and accurate method for calculating the energies of electronic transitions in the spectra of tetrapyrroles compared to the full TD-DFT[1740]. The calculations showed that the UV-Vis spectra of heptynyl- and heptyl-substituted complexes do not differ substantially — both complexes are characterized by an absorption maximum at 790 nm due to the absence of orbital interaction between the π-systems of the macrocycle and acetylene groups.
To investigate how sequential benzannelation affects the optical properties of phthalocyanines, the Pd-catalyzed cyanation of diol 2.24.2 was carried out to form dinitrile 2.24.7. The latter was further introduced in a mixed template cross-condensation with 4,5-bis[2,6-di(isopropyl)phenoxy]phthalonitrile 2.24.8 in the presence of magnesium acetate and DBU (Scheme 656, path c)[1741]. Subsequent demetallation of the mixture of the resulting magnesium complexes and chromatographic separation of the reaction products allowed the isolation of the symmetric phthalocyanine and its single and double-benzannelated derivatives. The UV-Vis spectra showed that sequential benzannelation leads to a bathochromic shift of Q-bands. Thus, in the spectrum of symmetric phthalocyanine, Q-bands at 704 and 668 nm are observed, while in the single-benzannelated derivative the absorption maxima shift to 733 and 686 nm. The linear doubly-benzannelated isomer is characterized by a greater splitting of the Q-band (763 and 684 nm) compared to the angular isomer (741 and 718 nm). The observed correlations agree with the data of sTD-DFT calculations. The presence of terminal OH-groups in the molecules of asymmetric phthalocyanines allows them to be considered as promising components of hybrid materials based on nanoparticles and quantum dots[1742, 1743].
2.24.2. Heteroannelated analogs of phthalocyanines
Low oxidation potentials due to the electron-donating nature of the naphthalene groups are responsible for the reduced photostability of naphthalocyanines. In this connection, new representatives of aza-derivatives — quinoxalinoporphyrazines containing ionogenic 4-carboxyphenyl groups — were synthesized to gain access to more photostable tetrapyrroles with absorption in the NIR range[1744]. First, oxalyl dibenzoate 2.24.10 was obtained by condensation of methyl-4-formylbenzoate 2.24.9, which was reacted with 4,5-dibromo-o-phenylenediamine 2.24.11 (Scheme 657, path a). Substitution of methyl groups in the intermediate 6,7-dibromoquinoxaline 2.24.12 with amyl groups followed by Pd-catalyzed cyanation gave 6,7-dicyanoquinoxaline 2.24.13, which was further introduced into template cyclotetramerization in the presence of zinc acetate and DBU. Alkaline hydrolysis of ester groups in quinoxalinoporphyrazine Zn[(PentO2CPh)8QPz] (QPz is quinoxalinoporphyrazine) afforded a water-soluble complex in the form of its sodium salt Zn[(NaO2CPh)8QPz].

It was shown that dissolution of this salt in water leads to its aggregated form with an absorption maximum at 709 nm. At the same time, mixing a solution of this salt in DMSO with water gives a solution in which the complex is in monomeric form with a Q-band at 763 nm. An additional absorption band at 526 nm is observed in the spectrum, related to the charge transfer from the tetrapyrrole macrocycle to the peripheral aromatic substituents. It is shown that the monomeric complex Zn[(NaO2CPh)8QPz] in water-DMSO mixture is able to generate singlet oxygen with a quantum yield of 0.65, and the rate of photodegradation of this complex is 3.4 times lower than in the case of crown-substituted naphthalocyanine.
The complex Zn[(PentO2CPh)8PyzPz] (PyzPz is pyrazinoporphyrazine) was obtained by condensation of oxalyl dibenzoate 2.24.10 with diaminomaleonitrile followed by template condensation of pyrazinodinitrile 2.24.14 (Scheme 657, path b)[1745]. In contrast to QPz analogue described above, this complex was subjected to degradation by alkaline hydrolysis of the ester groups. Analysis of the electrostatic potential distribution in this molecule showed a significant susceptibility of the tetrapyrrole macrocycle to nucleophilic attack; however, this susceptibility decreased upon transfer to the free base H2[(AmO2CPh)8PyzPz]. To obtain the latter, the condensation of pyrazinodinitrile 2.24.14 in the presence of magnesium in amyl alcohol followed by demetallation under the action of trifluoroacetic acid was carried out. The carboxylate H2[(HO2CPh)8PyzPz] was reacted with zinc acetate to give the complex Zn[(HO2CPh)8PyzPz]. The studied pyrazinoporphyrazine characterized by a hypsochromic shift of the Q-band by only 10 – 15 nm compared to conventional phthalocyanines.
It is known that the introduction of alkoxyl substituents in the α-positions of the benzene rings of phthalocyanines leads to a bathochromic shift of the Q-bands by 60 – 80 nm[1740], without causing a significant decrease in photosensitivity. At the same time, the location of electron-donating alkoxyl groups next to meso-nitrogen atoms significantly increases the basicity of the latter, which makes it possible to control the properties of α-substituted phthalocyanines by reversible protonation. Thus, it has been shown that sequential protonation of octa-α-butoxyphthalocyanine and its complexes M[(α-BuO)8Pc], where M = 2 H, Mg, Zn, Ga(Cl), and In(Cl) (Pc — phthalocyanine) leads to a sequential bathochromic shift of the Q-bands by 70 – 80 nm upon introduction of each subsequent proton. In addition, a decrease in the quantum yields of fluorescence and singlet oxygen generation is observed[1746], while the ability of the complexes to act as electron acceptor during photoexcitation in the presence of electron-donating molecules increases[1747].
To extend the possibilities of controlling the properties of phthalocyanine derivatives, α-butoxysubstituted tetra-15-crown-5-oxanthrenocyanines capable of participating in the supramolecular assembly process in addition to protonation were synthesized for the first time (Scheme 658, path a)[1748]. For this purpose, the Vilsmeyer formylation of benzo-15-crown-5 2.24.15 followed by Dakin oxidation of the intermediate aldehyde to form 4'-hydroxybenzo-15-crown-5 2.24.16 were carried out. Fremy oxidation of the latter to 15-crown-5-quinone, followed by hydrogenation, led to the formation of air unstable 15-crown-5-catechol 2.24.17. The latter was introduced without isolation into the nucleophilic substitution reaction with 3,6-di-n-butoxy-4,5-dichlorophthalonitrile 2.24.18. The resulting dinitrile 2.24.19 was used to give α-substituted oxanthrenocyanines M[(α-BuO)8(15C5)4Oc], M = Mg or Zn (Oc — oxanthrenocyanine), which were in solution in the monomeric state, according to UV-Vis and NMR spectra.

It is shown that supramolecular dimerization of the complex Zn[(α-BuO)8(15C5)4Oc] in the presence of KBPh4 is accompanied by a hypsochromic shift of the Q-band from 732 to 686 nm, whereas the sequential protonation of meso-nitrogen atoms (Scheme 658, path b) leads to a bathochromic shift of this band to 798, 867, 9741, and 1028 nm, respectively. This shift is accompanied by a characteristic color change, giving this complex the name «molecular chameleon». Interestingly, both assembling and protonation are fully reversible: the addition of a base or cryptand binding potassium cations leads to the regeneration of the original form of the complex with the same spectral characteristics.
Tetra(15-crown-5)oxanthrenocyanines lacking α-BuO groups were prepared by condensation of catechol 2.24.17 with dichlorophthalonitrile 2.24.20 followed by template condensation of the resulting 15-crown-5-oxanthrenodinitrile 2.24.21 (Scheme 658, path c)[1749]. In contrast to the substituted oxanthrenocyanines, the complexes M[(15C5)4Oc], where M = Mg, Zn, showed strong aggregation in solutions. Split Q-bands in the region of 686 and 639 nm were observed in the spectra, which is atypical for phthalocyanine aggregates. The interaction with potassium acetate led to an increase in the solubility of the complexes in mixtures of chloroform and methanol due to the formation of supramolecular cofacial dimers, whose spectra also showed an atypical splitting of the Q-bands (674 and 637 nm). In contrast to the above described supramolecular dimers of naphthalocyanines[1737], the interaction of the compound {2M[(15C5)4Oc] · 4 KOAc} with [2.2.2]-cryptand did not lead to monomerization of the complex, but the formation of aggregates with a narrow particle size distribution and panchromatic absorption throughout the visible range was observed. The specific spectral behavior of the complexes was explained by exciton interactions between oxanthrenocyanine molecules in the aggregates based on magnetic circular dichroism spectroscopy data[1750].
2.24.3. Phosphorus(V) porphyrinates
Although phosphorus is not a metal, the element is able to form complexes with tetrapyrrole macrocycles, which have attracted considerable attention in terms of controlling the properties of porphyrins and phthalocyanines due to their unique photophysical characteristics[1751]. In addition, phosphorus(V) complexes with tetrapyrroles are typically cationic, since the doubly charged macrocycle and the two anionic axial ligands are unable to compensate the +5 charge on the phosphorus atom. Consequently, such complexes can be water soluble, which is important for practical biomedical applications.
5,10,15,20-tetraphenylporphyrin (H2Ph4Por), 5-(4-pyridyl)-10,15,20-triphenylporphyrin (H2PyPh3Por)[1752, 1753] and 5,10-bis(4-pyridyl)-15,20-triphenylporphyrin (H2Py2Ph2Por)[1754] were used to synthesize phosphorus porphyrinates (Scheme 659, part a). The introduction of a phosphorus(V) atom into the cavities of the first two porphyrins was made possible by the action of a mixture of PCl5 and POCl3 in pyridine. In this case, complexes with axial chlorine atoms were obtained, which could be substituted by a fragment of 3-methoxyphenol or hydroxyl groups. To synthesize a complex with a more electron-deficient ligand, H2Py2Ph2Por, containing two pyridine groups, it was necessary to use a more reactive phosphorus(V) oxobromide, which proved to be a versatile phosphorylating agent for tetrapyrroles. By interacting the intermediately formed bromide complexes with water or ethanol, complexes with axial OH- and EtO-groups were obtained.

The reaction of tetrapyridylporphyrin H2Py4Por with POBr3 followed by hydrolysis of the intermediate led to the product (Py4Por)P(OH)2+ in a low yield of 13%[1752], which is explained by a decrease in electron density in the macrocycle upon introduction of four electron-accepting pyridyl groups[1755].
Using the selective chemical trap ʻSinglet Oxygen Sensor Greenʼ (SOSG), it has been shown that the resulting complexes are characterized by moderate quantum yields of singlet oxygen 1O2 generation (ΦΔ) in both DMSO and aqueous media, determining their potential application in photodynamic therapy (Scheme 659, part b). The exception is phosphorus(V) porphyrinates containing aryloxyl axial groups — the excited states of these complexes are rapidly deactivated due to charge transfer from benzene rings to porphyrin macrocycles, so such complexes are not capable of singlet oxygen generation and do not exhibit luminescent properties[1753].
The introduction of amino groups into the axial aromatic substituents demonstrated the possibility of controlling the properties of phosphorus(V) porphyrinates to use them as molecular switches. Thus, the complex (Ph4Por)P(OC6H4NH2)2+ neither generated singlet oxygen nor showed luminesce. Protonation of the amino groups blocked the charge transfer process from the axial substituents to the porphyrin macrocycle, activating the ability of the complex to luminesce and generate of 1O2 (Scheme 660)[1756]. Unfortunately, this also led to a decrease in the photostability of the complex in aqueous solution.

A cytotoxicity study of the PyPh3PorP(OH)2+ complex against the colorectal carcinoma cell line HTC-116 demonstrated efficient accumulation of the photosensitizer in the cells, as well as significantly higher light cytotoxicity (0.32 ± 0.08 μM) compared to dark toxicity (7.2 ± 2.9 μM) when irradiated with blue light at 5 J cm–2[1757]. These characteristics are superior to the cytotoxicity of the clinically approved porphyrin photosensitizers Photofrin© and Foscan©.
Due to the luminescence of phosphorus(V) porphyrinates in aqueous solutions, such complexes can be used for luminescence diagnostics of accumulation in cells. Moreover, it is shown that the luminescence lifetimes of the compound PyPh3PorP(OH)2+, as well as the ratio of the intensities of the long- and short-wavelength fluorescence bands of this porphyrin are linearly dependent on temperature in the physiological range from 20 to 45 °C[1757]. The thermometric performance of the proposed porphyrins has been evaluated in terms of relative sensitivity (1% K–1), temperature resolution (0.1 °C) and reproducibility; the values obtained are record-breaking for porphyrin-based thermometric fluorescent sensors.
An alternative approach to the design of thermosensors based on tetrapyrrole compounds is the use of complexes of paramagnetic lanthanides with phthalocyanine ligands[1758]. Such complexes are characterized by a significant temperature dependence of lanthanide-induced signal shifts in NMR spectra[1759-1761].
A study of the possibility of using the thermometric properties of PyPh3PorP(OH)2+ to measure subcellular temperature on the CHO-K1 and HeLa cell lines showed an unexpected result: when porphyrin was accumulated in the cells, a change in its luminescence spectrum was observed, indicating dephosphorylation of the complex to form free porphyrin H2PyPh3Por, which nevertheless retained thermometric properties with a resolution of 0.1 – 0.3 °C[1762]. It was found that dephosphorylation occurs due to interaction with proteins. Thus, when the complex was incubated with bovine serum albumin (BSA) in a mixture of DMSO-d6 – D2O, the disappearance of the phosphorus atom signal at –191 m.d. of the starting complex, and the appearance of the signal at –0.8 m.d. of the free phosphate anion, were observed in the 31P NMR spectrum. Thus, it has been shown for the first time that water-soluble phosphorus(V) porphyrinate with axial hydroxyl groups can be used to deliver the hydrophobic porphyrin into the cell for photodynamic action.
A set of methods was developed to evaluate the antibacterial photodynamic activity of photosensitizers on the examples of phosphorus(V) porphyrinates with axial hydroxyl and ethoxyl groups[1754]. It included the study of adsorption of porphyrins on lipid membranes[1763-1765], molecular dynamic modeling of interaction of photosensitizers with the environment mimicking lipid membranes of bacteria, and experiments on antimicrobial photodynamic therapy (aPDT) on strains of gram-negative bacteria Escherichia coli and Acinetobacter baumannii. The summary of the results allowed the identification of the most promising photosensitizer among the synthesized compounds. The complex (Ph4Por)P(OEt)2+, characterized by the lowest values of the minimum inhibitory concentration (5.0 ± 0.4 μg mL–1 for E. coli and 4.9 ± 0.8 μg mL–1 for A. baumannii), was found to be the most promising photosensitizer among the synthesized compounds. These values were obtained by photodynamic exposure of a 405 nm blue laser with a wavelength of 5 J cm–2 to bacterial cultures in the presence of porphyrin for only one minute. The MIC values for E. coli T61 bacterial strain were two times lower than for ampicillin, and for A. baumannii NIH61 strain — comparable to those for colicin.
2.24.4. Phosphorus(V) phthalocyaninates
A major limitation to the practical use of phosphorus(V) porphyrinates as agents for photodynamic therapy is the absence of absorption of such complexes in the red and near-infrared spectral regions. In this context, phosphorus(V) phthalocyaninates characterized by the presence of intense Q-bands in their UV-Vis spectra in the range of ʻtherapeutic windowʼ have been synthesized and studied[1766-1768].
The approach used to prepare phosphorus(V) phthalocyaninates is similar to the methods described above for the synthesis of porphyrin complexes. Free phthalocyanine bases are introduced in reactions with phosphorus oxobromide, and intermediate complexes containing bromine atoms at axial positions are treated with water, alcohols, or phenol to substitute axial ligands (Scheme 661, part a, and Table 7).
![Yields and photophysical characteristics of phosphorus(V) phthalocyaninates (<a class="anchor" href="#figure-2303">Scheme 661</a>)[<a class="anchor" href="#reference-8335">1766</a>, <a class="anchor" href="#reference-8366" id="mention-reference-8366">1769</a>, <a class="anchor" href="#reference-8367" id="mention-reference-8367">1770</a>].](/storage/images/resized/bY7ryGjWtjrA5lVNYkeUOSNVvqDnC1zifJdty0Oh_xl.webp)

Switching from the unsubstituted complex PcP(OMe)2+ to β-butoxy- and 15-crown-5-derivatives [(β-BuO)8Pc]P(OMe)2+ and [(15C5)4Pc]P(OMe)2+ leads to a slight bathochromic shift of the Q-band (from 708 to ~725 nm) with preservation of the ability to luminesce and to generate singlet oxygen. At the same time, in the case of α-butoxysubstituted phosphorus(V) phthalocyaninate [(α-BuO)8Pc]P(OMe)2+ the Q-band shifts to 896 nm, thus the energy of the triplet state of such a complex turns out to be too low for 1O2 generation, moreover, such a complex has no luminescence (see Table 7)[1767] In addition, an unusual feature of phosphorus(V) phthalocyaninates compared to porphyrins is the exceptionally high quantum yield of singlet oxygen generation: ΦΔ = 0.90 in the case of the compound containing axial phenoxyl groups.
A unique reaction of reversible aromatic nucleophilic addition of hydroxide and methoxide anions has been found for the complex РсР(OMe)2+, leading to non-aromatic non-fluorescent complexes (Scheme 661, part b) characterized by a significant bathochromic shift of the Q-band (from 708 to 865 nm)[1766]. The structure of the adduct with a hydroxyl group was confirmed by X-ray diffraction. The possibility of such a reaction is determined by the electron-deficient nature of the phosphorus(V) atom, which increases the electrophilicity of carbon atoms in the α-positions of pyrroline cycles. The adducts have been aromatized in the presence of acids, regenerating the starting cationic complex.
Another type of acid-base equilibrium has been observed in the case of a complex [(β-BuO)8Pc]PO(OH) obtained from octa(β-butoxy)phthalocyanine and POBr3. It is shown that such a complex can exist in three states, including doubly protonated, partially and fully deprotonated forms (Scheme 661, part c)[1767]. Each of these forms is characterized by its UV-Vis and luminescence spectra, as well as the quantum yield of singlet oxygen generation.
The reaction of tetra(15-crown-5)phthalocyanine with POBr3 followed by addition of O-ethyl diethylene glycol affords water-soluble phosphorus(V) phthalocyaninate[1768]. The complex [(15C5)4Pc]P(Gly)2+ is also able to participate in reversible nucleophilic addition of the hydroxy group in aqueous solution. It has been shown that at pH > 9.6 this complex exists in solution exclusively as a non-aromatic adduct, while an individual cationic complex is formed in aqueous solution at pH < 4.9.
The quantum yield value of 1O2 generation for [(15C5)4Pc]P(Gly)2+ was 0.39 regardless of the nature of the solvent (water or DMSO); and this complex was not accumulated in breast adenocarcinoma cells (MCF-7) or in WI-26 fibroblasts. As a result, the complex exhibited neither dark nor light cytotoxicity at concentrations > 40 μM and ~ 20 μM against the above cell lines, which was apparently due to its too high hydrophilicity.
Due to low photocytotoxicity it was possible to use this complex for photopolymerization of biocompatible polymer — polyethylene glycol diacrylate (PEG-DA), which proceeded due to generation of 1O2 under irradiation with light of 660 nm wavelength. Carrying out photopolymerization of PEG-DA in the presence of RAW 264.7 mouse macrophages allowed us to obtain a hydrogel with living cells distributed in it. The synthesized photosensitizer is therefore promising for regenerative technologies using biocompatible polymers.
2.24.5 Cationic derivatives of aminomethylated phthalocyanines
Introduction of cationic substituents to the periphery of phthalocyanine macrocycles is considered as one of the most effective approaches to the development of photosensitizers, including those for antibacterial PDT[1771]. The reductive amination reaction of p-formylphenoxy-substituted phthalonitriles obtained by interaction of 4-nitro- (2.23.22), 3-nitro- (2.23.23) and 4,5-dichloro-substituted (2.23.20) phthalonitriles with p-formylphenol 2.24.24 (Scheme 662, step a) was chosen as a new method for introducing ammonium groups into phthalocyanine molecules[1772].

Redox amination of the resulting nitriles 2.24.25 – 2.24.27 was carried out under the action of triacetoxyborohydride in the presence of excess diethylamine in tetrahydrofuran (Scheme 662, step b). Aminomethylated phthalonitriles 2.24.28 – 2.24.30 were introduced in template cyclotetramerization reactions in the presence of zinc or magnesium acetates (Scheme 662, step c). The corresponding magnesium complexes were demetallated with trifluoroacetic acid to give free phthalocyanine bases. The amino groups were quaternized with methyliodide, thus obtaining a library of water-soluble cationic phthalocyanines with different numbers and arrangements of ionogenic groups.
The regularities of changes in the photophysical properties of such cationic complexes depending on the substituents as well as on the nature of metal centers in solutions in DMSO (Table 8) were typical for phthalocyanines. In general, the introduction of substituents in the α-positions of the macrocycles led to a bathochromic shift of the Q-band, the introduction of metal cations increased the quantum yields of 1O2 generation and decreased the quantum yields of luminescence. Aggregation was observed for a number of phthalocyanines when transferred to aqueous solutions, leading to the formation of cofacial non-luminescent H-associates incapable of singlet oxygen generation. A noticeable fraction of monomeric forms in aqueous solutions was observed only for non-peripherally substituted complexes M[(α-Ar+O)4Pc], M = Zn and Mg, the quantum yields of 1O2 for such solutions were 0.07 and 0.03, respectively.
![Photophysical characteristics of cationic phthalocyanines in DMSO (<a class="anchor" href="#figure-2304">Scheme 662</a>)[<a class="anchor" href="#reference-8339">1772</a>].](/storage/images/resized/4lgzRvGxvaLU00uWSaISO2oHRNzWuJqjwL9VzU08_xl.webp)
It is shown that these cationic phthalocyanines are converted into monomers in aqueous solutions in the presence of the nonionic surfactant Tween-80 and bovine serum albumin. The latter is especially important in the context of the fact that the vast majority of works devoted to the synthesis of phthalocyanine photosensitizers focus on the need to obtain non-aggregating photosensitizers. However, this statement is contradicted by analysis of the behavior of phthalocyanine aggregates in the biological environment[1773], where they can dissociate upon interaction with proteins and lipids to form monomeric forms capable of generating singlet oxygen. At the same time, aggregates can function on their own, leading to the generation of different reactive oxygen species from 1O2, such as radical particles[1774], which is particularly important for PDT in hypoxic environments. Finally, aggregates may exhibit alternative therapeutic and diagnostic mechanisms of action, such as photothermal and optoacoustic effects[1775].
2.24.6. Heteroannelized porphyrin derivatives
Wide possibilities of functionalization of porphyrins consist in the introduction of substituents both in meso-positions of the macrocycle and in β-positions of pyrrole fragments, including heteroannelation[1776], leading to fine-tuning of optical and electrochemical properties of the resulting derivatives. In addition, it is possible to introduce anchoring groups of different nature into porphyrin molecules in order to create hybrid and composite materials used in catalysis and photocatalysis.
To develop methods for the synthesis of porphyrins heteroannelated with imidazole and pyrazine groups, a systematic study of the reactivity of its 2,3-diamino derivative Ni[Por(NH2)2] (Scheme 663, path a) was carried out using tetra(meso-mesityl)nickel porphyrinate Ni(Mes4Por) as an example[1777]. The latter was prepared by nitration of the initial complex followed by introduction of the amino group into Ni[Por(NO2)] under the action of 4-amino-4H-1,2,2,4-triazole 2.24.31 by the Katritzky reaction. Hydrogenation of the Ni[Por(NO2)(NH2)] complex led to the target diamine, which was introduced into condensation reactions with various carbonyl compounds without further purification.

It was shown that the nature of the carbonyl compound as well as the reaction conditions determine the direction of condensation, opening the way to various porphyrin derivatives. Thus, in the reaction of diamine with 3,5-di-tert-butylquinone 2.24.32, dioxochlorine Ni(Por-dione), a convenient precursor of imidazo[4,5-b]porphyrins was formed (Scheme 663, path b). At the same time, the interaction of Ni[Por(NH2)2] with phenanthrene-9,10-dione 2.24.33 and phenanthroline-9,10-dione 2.24.34 led to heteroannelated pyrazine derivatives of Ni(PyzPor1,2) (Scheme 663, path c).
It should be noted that a more general and convenient route for the preparation of dioxochlorines is the reduction of the free bases of nitroporphyrins followed by aerobic photooxidation with an amino derivative[1778]. In addition, a similar methodology can be applied to introduce a second imidazo group into the imidazoporphyrin molecule[1779].
The direction of the reaction of Ni[Por(NH2)2] with 4-bromobenzaldehyde 2.24.35 depended on the solvent used, the presence of the catalyst (TsOH), and the stoichiometry of the reagents. Thus, in the absence of a catalyst at a 1 : 1 ratio of reagents, the above-mentioned imidazoporphyrin Ni(ImPor1) was formed with high selectivity when the process was carried out in o-dichlorobenzene at 120 °C (Scheme 663, path d). The interaction of Ni[Por(NH2)2] with 2.24.35 in the presence of TsOH in DMFA at 80 °C selectively gave pyrazinoporphyrin Ni(PyzPor3) (Scheme 663, path e).
Further development of the chemistry of diaminoporphyrins included a deeper study of the conditions for the selective preparation of imidazo- and pyrazinoporphyrins and their subsequent modification[1780]. The possibility of introducing bis(4-bromophenyl)pyrazinoporphyrin into borylation, phosphorylation, carboxylation, and Suzuki cross-coupling reactions with 4-methoxycarbonylphenylboronic acid was demonstrated, resulting in synthetic precursors of hybrid materials.
Condensation of tetra[meso-(4-butoxyphenyl)diamino]porphyrin with polyaromatic hydrocarbon-based aldehydes — 1-formylpyrene, 9-formylanthracene, and 4-(10-phenylanthracene-9-yl)benzaldehyde, yielded a series of dyads to study the nature of the electronic interaction between the porphyrin and polyaromatic moieties (Scheme 664, part a)[1781]. A Zn(PyrImPor) dyad containing a pyrenyl group exhibited unique sensory properties toward picric acid: the interaction between receptor and analyte first revealed an unusual analytical response to nitroaromatic compounds accompanied by the appearance of a new luminescence band that was attributed to exciplex emission[1782]. The porphyrin backbone of the dyad also allows the registration of intrinsic fluorescence that is weakly dependent on the presence of the analyte, providing an internal reference signal for the ratiometric detection of picric acid.

The synthesized imidazo- and pyrazinoporphyrins were used as catalysts and photocatalysts in reactions of oxidation of sulfides into sulfoxides due to the importance of this reaction for organic synthesis, including the manufacturing of pharmaceuticals[1783-1785]. Thus, manganese imidazoporphyrinates with carboxylate and phosphonate groups, Mn(CImPor) and Mn(PImPor), were immobilized on the surface of mesoporous TiO2 (Scheme 664, part b)[1783] and the obtained hybrid materials were used as heterogeneous catalysts for aerobic oxidation of thioanisole and other aromatic and aliphatic sulfides in the presence of isobutyraldehyde to obtain sulfoxides. It was shown that porphyrin with phosphonate group formed a more stable catalyst characterized by a high turnover number (TON ~ 1100) and selectivity of 98% in the oxidation reaction of thioanisole to sulfoxide, while its activity did not decrease in seven successive catalytic reactions.
The pyrazinoporphyrins H2(CPyzPor) and H2(PPyzPor) (Scheme 664, part c)[1784] showed exceptionally high activity and selectivity in the reactions of photooxidation of sulfides to sulfoxides under blue light irradiation. Even at micromolar loading of the catalyst, the conversion degree of thioanisole was 98 – 100%, TON ≈ 95 060 – 98 000, and the selectivity reached 97 – 98%. Also, a series of pyrazinoporphyrins annelated with naphthalene, phenanthrene, phenanthroline and acenaphthene groups were synthesized. It was designed to study the effect of the extension of the aromatic system on the optical and photocatalytic properties of the complexes, as well as on their photostability in sulfoxide formation reactions[1785].
Heteroannelation of porphyrins has also allowed the development of approaches to cationic complexes as components of hybrid materials (Scheme 664, part d )[1786], as well as potential agents for PDT[1787, 1788].
A study of the adsorption of β-imidazolylporphyrins M(β-Im+Por), where M = 2 H, Zn, In(Cl) (Scheme 664, part f )[1787], on bilayer lipid membranes with determination of the photodegradation rate of the potential-sensitive dye di-4-ANEPPS showed that the indium complex adsorbed better than the free base and the zinc complex. However, although the indium complex was characterized by the maximum quantum yield of 1O2 generation in solution, the degradation rate of di-4-ANEPPS and the cumulative photodynamic activity on membranes were similar for all three porphyrins. These data indicate that it is the efficiency of adsorption on the membrane that plays a determining role in the photodynamic effects. An increase in the number of cationic groups when switching to pyrazinoporphyrazines M(Pyz+2Por), where M = Ni, Zn, 2 H (see Scheme 664, part e)[1788], allowed an increase in the number of molecules adsorbed on the membrane, but the rate of destruction of the potential-sensitive dye did not increase compared to that for the complex (Cl)In(β-Im+Por).
2.24.7. Ruthenium phthalocyaninates
Among the various transformations used in the synthesis of pharmaceuticals, the carbene transfer reactions for cyclopropanation of unsaturated compounds[1789] and introduction of X – H bonds are of particular interest[1790]. Transition metal complexes, especially those of ruthenium with tetrapyrrole ligands, are being investigated as catalysts for such reactions.
To obtain ruthenium phthalocyaninates, a new reaction consisting of direct introduction of ruthenium into the cavity of previously synthesized phthalocyanines under the action of ruthenium carbonyl in boiling o-dichlorobenzene was used (Scheme 665, path a)[1791]. The main products of this reaction were ruthenium(II) phthalocyaninates with axially coordinated CO molecules. However, in a number of cases, unusual dimeric ruthenium(IV) μ-carbido-bis(phthalocyaninates), apparently formed from thermolysis products of ruthenium carbonyl, were also isolated in small yields by gel permeation chromatography.

To increase the preparative yield of these complexes, reactions of ruthenium(II) phthalocyaninates with chloroform were carried out, which in the presence of bases gave dichlorocarbene (see Scheme 665, path b)[1792]. While potassium hydroxide could be used as a base to obtain tert-butyl- and β-butoxy-substituted carbidodimers, in the case of the crown-substituted μ-carbido-dimer it was necessary to use tetramethylammonium hydroxide, which does not contain cations capable of interacting with crown-ether groups. The formation of diruthenium(IV) μ-carbido-bis(octa-mesityloxy-phthalocyaninate) was not observed in any of the reactions studied.
The carbonyl group in ruthenium monophthalocyaninates can be substituted with other axial ligands, in particular amine fragments and nitrogen-containing heterocycles. For this purpose, an approach based on oxidative decarbonylation of the complexes with trimethylamine oxide in the presence of an excess of the ligand L was developed[1793]. However, unsymmetrical complexes PcRuL(NMe3) containing coordinated trimethylamine molecules were formed as difficult-to-separate impurities to the target complexes PcRuL2. It was shown that when a mixture of such complexes is heated in boiling DCB in excess of the ligand trimethylamine is substituted for the corresponding ligands[1794]. On the example of tert-butyl-substituted ruthenium phthalocyaninates a methodology was developed for the synthesis of the complex [But4Pc]Ru(NMe3)2 (Scheme 665, path c), capable of quantitative substitution of axial aliphatic ligands for aromatic ligands — pyrazine, Pyz[1795] and 4,4'-bipyridyl[1796]. The cationic complex [But4Pc]Ru(BiPy+-Me)2 (BiPy — bipyridine) was prepared by quaternization of [But4Pc]Ru(BiPy)2 in the presence of methyliodide. The nature of the axial ligand was found to have a key influence on the ability of ruthenium phthalocyaninates to generate singlet oxygen. Thus, the highest values of ΦΔ in DMSO were obtained for complexes with carbonyl and pyrazine ligands (Scheme 665, part d ). The transition to bipyridine and its quaternized derivative led to a decrease in ΦΔ, and the lowest quantum yield was observed for the complex with coordinated trimethylamine[1796].
The catalytic properties of synthesized ruthenium phthalocyaninates were studied on the examples of cyclopropanation of styrenes and introduction of carbenes into the N – H bond to obtain secondary and tertiary amines. It was shown that ruthenium(II) monophthalocyaninate [(β-BuO)8Pc]Ru(CO)[1797] is significantly more active in both reactions compared to ruthenium(IV) μ-carbido-bis(phthalocyaninate) [(β-BuO)8PcRu]2(μ-C)[1791]. Thus, in the case of the dimer, higher temperatures were required for the interaction of styrenes and aliphatic and aromatic amines with ethyldiazoacetate (EDA) (toluene, 90 °C), whereas the monomeric complex was active at room temperature. Despite the high TON values for both types of complexes, which reached 1000 in cyclopropanation reactions, diastereoselectivity was higher when monophthalocyanate was used as a catalyst. Thus, the ratios of trans- to cis-products of the cyclopropanation of styrene and p-methoxystyrene were 4.8 : 1 and 7.3 : 1 for the monomeric catalyst and 2.3 : 1 and 3.0 : 1 for the μ-carbido dimer, respectively. In this context, it was the monomer complex that was used to systematically study the carbene transfer process in the reactions of a wide range of substituted styrenes and diazo compounds, as well as aromatic, heteroaromatic and aliphatic amines with EDA[1797]. Carbene insertion in the N – H bond for 2-aminothiazole and 2-amino-1,3,4-thiadiazole was also carried out in the presence of iron(III) tetra(15-crown-5)phthalocyaninate [(15C5)4Pc]Fe(Cl)[1798]. Catalytic reactions of these heterocyclic amines with ethyldiazoacetate were characterized by high TON values and yields of single and double insertion products.
Interesting results were obtained by studying the influence of the nature of the phthalocyanine ligand on the selectivity of carbene insertion into the N – H bond (Scheme 666). For this purpose, the catalytic activities of α- and β-butoxy-substituted ruthenium phthalocyaninates were compared (Scheme 666, path a)[1769]. It was shown that in the reactions of substituted anilines with 2 equiv. EDA, catalyzed by β-substituted complex, tertiary amines are formed, which were products of a double insertion of carbene, whereas when the α-substituted complex was used under the same conditions, only a single insertion of carbene occurred, leading to secondary amines (Scheme 666, path b).

This selectivity allowed the development of a methodology for the preparation of tertiary amines containing three different substituents. For example, the reaction of p-toluidine 2.23.36 with ethyldiazoacetate catalyzed by the α-substituted complex yielded O-ethyl-N-(p-tolyl)glycinate 2.24.37, which was isolated and reacted with diazoacetonitrile in the presence of the β-substituted complex to give tertiary amine 2.24.38 containing a cyanomethyl group (Scheme 666, path c). Similarly, secondary diamine 2.24.40 and tertiary diamine 2.24.41 were prepared from 2,6-diaminotoluene 2.24.39, where each of the amino groups was selectively functionalized with two different diazo compounds.
Finally, it was shown that ruthenium phthalocyaninate can act as a platform for the development of enantioselective cyclopropanation catalysts. The first prototypes of such catalysts were obtained from 2,6-di(hydroxymethyl)-p-cresol 2.24.41 and the naturally widespread (–)-menthol 2.24.42 as a chirality inducer (Scheme 667)[1770]. Interaction of these reagents in melt yielded bis-(–)mentyloxymethyl substituted phenol 2.24.43, which was further introduced in nucleophilic substitution reactions with 4-nitro- (2.24.22) and 3-nitrophthalonitriles (2.24.23). The corresponding (–)-mentyl substituted phthalonitriles 2.24.44 and 2.24.45 were formed as synthetic precursors of chiral tetra-α- and tetra-β-substituted ruthenium(II) phthalocyaninates [(α-ArMO)4Pc]Ru(CO) and [(β-ArMO)4Pc]Ru(CO).

X-Ray diffraction analysis of phthalonitriles 2.24.44 and 2.24.45 showed that bis-(–)-menthyl substituted aromatic groups (ArM) are arranged almost orthogonally to the phthalonitrile fragments. The results of quantum-chemical modeling indicated that the same structural feature is preserved in the obtained ruthenium(II) complexes, providing chiral environment of the catalytic centers. In the case of the [(α-ArMO)4Pc]Ru(CO) (–)-menthyl groups are located closer to the phthalocyanine macrocycle, creating prerequisites for more efficient chiral induction, and this type of substitution has been termed ʻpicket fenceʼ.
The use of [(β-ArMO)4Pc]Ru(CO) as a catalyst, in contrast to the α-substituted analog, practically did not lead to an increase in diastereo- and enantioselectivity of the cyclopropanation of styrene with ethyldiazoacetate. Optimization of the conditions for this reaction involving [(α-ArMO)4Pc]Ru(CO) showed a significant influence of the solvent on the stereoselectivity of cyclopropanation — the best results were obtained in tert-butyl acetate. For styrene, the ratio of trans- and cis-isomers was 7.3 : 1, the enantiomeric excesses of cis-(R,S)- and trans-(S,S)-isomers were 18% and 8% (for comparison — in dichloromethane the same parameters were 5.6 : 1, 10% and 4% ee, respectively).
Obviously, the results presented above are very modest compared to the extensive literature data on enantioselective cyclopropanation. However, the prototype catalysts discussed are the first examples of the use of the «picket fence» architecture for phthalocyanines. Further development of this line of research may include the creation of conformationally rigid systems, as well as catalysts with substituents that enter into stronger noncovalent interactions with substrates and orient them optimally with respect to the catalytic center.
The family of tetrapyrrole macroheterocycles is one of the most attractive platforms for the design of functional optical, catalytic and photocatalytic materials with great potential for biomedical applications, especially for photodynamic therapy and diagnostics. In this section we have presented approaches to the preparation of a number of modified porphyrins, phthalocyanines and their metal complexes, which have already been shown to be useful as photosensitizers for PDT, including antibacterial therapy, as well as in the creation of luminescent thermosensors, agents for biophotopolymerization and catalysts for the production of sulfoxides, cyclopropanes and amines. The potential of similar compounds, such as naphthalocyanines and porphyrazines, remains to be discovered, so the development of effective approaches to their synthesis is the first step towards new biomedical materials.
2.25. New heterocyclic analogues of phthalocyanines
Heterocyclic phthalocyanine (Pc) analogues containing fused aromatic heterocycles (pyridine, pyrazine, thiophene) instead of benzene rings were first described by Linstead et al.[1799] in 1937. A special chapter in the monograph The Porphyrin Handbook is devoted to methods for the synthesis and properties of porphyrazine with various annulated heterocycles[1800].
Among phthalocyanine analogues fused with six-membered heterocycles, in the last two decades, the attention has been focused on pyrazinoporphyrazines (PyzPz), which are described in detail in reviews[1801, 1802]. The chemistry of porphyrazines with 1,2,5-thiadiazole and 1,2,5-selenadiazole moieties (XN2Pz, X = S, Se), which impart more pronounced acceptor properties to the macrocycle, was initiated in the late 1990s[1803]. Currently, heterocyclic analogues of phthalocyanines containing a contracted porphyrazine macrocycle, that is, subporphyrazines (sPz) and corrolazines (Cz), are being actively studied (Scheme 668). The results of these studies, carried out mainly by Russian chemists, have not yet been integrated and are presented in this part of the review.

2.25.1. Pyrazine analogues of subphthalocyanine
Subphthalocyanine analogues sPz1 – sPz4, containing pyrazine moieties instead of benzene rings, were obtained in 2019 by template cyclotrimerization of pyrazine-2,3-dicarbonitriles in the presence of BCl3 in refluxing p-xylene or o-dichlorobenzene (Scheme 669)[1804].

Meanwhile, unsubstituted boron(III) tripyrazinosubporphyrazine (sPz1) was isolated in a yield of only 0.3%. Hexaphenyltripyrazinosubporphyrazine sPz4 was prepared from 5,6-diphenylpyrazine-2,3-dicarbonitrile in a markedly higher yield (6.2%). The benzene solvate of this compound as a single crystal was characterized by X-ray diffraction (Figure 17). It turned out that one of the benzene molecules, acting as a π-donor, forms a π-complex with pyrazine-annulated subporphyrazine as a π-acceptor.
![[{"id":"49lEknCmm4","type":"paragraph","data":{"text":"Molecular structure of the benzene solvate of sPz<b>4</b> (Cambridge Crystallographic Data Centre (CCDC), no. 1589460)."}}]](/storage/images/resized/K9HgLrpB7y3uM1ZtDQ13iW7QgLvLgVV3NNkEEfrV_xl.webp)
The cyclotrimerization of 5,6-dimethyl- and 5,6-diethylpyrazine-2,3-dicarbonitriles gave only trace amounts of hexaalkyl-substituted tripyrazinosubporphyrazines sPz2 and sPz3, which were detected by TLC, but could not be isolated or characterized. Apparently, this can be attributed to increased СН-acidity of the α-carbon atoms in the alkyl groups and to side reactions involving them. Actually, cyclotrimerization of 5-tert-butylpyrazine-2,3-dicarbonitrile in the presence of BCl3 in refluxing 1,2,4-trichlorobenzene affords tert-butyl-substituted tripyrazino-subporphyrazine sPz5 as a mixture of two regioisomers differing in symmetry (С1 and С3) in a total yield of 7% (Scheme 670)[1805].

Each of the sPz5 regioisomers was isolated in a pure state by column chromatography; note that more symmetrical С3-isomer was formed in a larger amount than the С1-isomer (1.7 : 1 ratio). Furthermore, storage of the С1-isomer was accompanied by the formation of the boron-free product sPz6, which was characterized by mass spectrometry and electron absorption spectra[1805].
Tripyrazinosubporphyrazines with fused cycloalkane moieties containing quaternary (and tertiary) carbon atoms in the α-position can be prepared in higher yields than unsubstituted subporphyrazine sPz1 or aryl and alkyl analogues sPz2 – sPz5. For example, tripyrazinosubporphyrazine sPz7 with fused camphor moieties was synthesized in 15% yield from dicarbonitrile obtained by condensation of (R)-camphorquinone and diaminomaleodinitrile (Scheme 671)[1806].

Trimerization of 5,5,8,8-tetramehyl-5,6,7,8-tetrahydroquinoxaline-2,3-dicarbonitrile containing two quaternary α-carbon atoms results in the formation of tripyrazinosubporphyrazine sPz8 in 35% yield (Scheme 672)[1807]. Upon mixed cyclotrimerization of this dinitrile with 1,1,4,4-tetramethyltetralin-6,7-dicarbonitrile, low-symmetric subporphyrazines sPz9 and sPz10, combining pyrazine and benzene moieties, were isolated in 2 – 3% yields. These subporphyrazines were used to prepare pyrazinoporphyrazines via deboration and opening of the subporphyrazine macrocycle in the presence of diiminoisoindoline in DMSO.

The halogen-substituted subphthalocyanines 2.25.1 (sPcF12) and 2.25.2 (sPcCl6) have recently been studied in detail as functional acceptor materials to be used in organic electronics (see reviews[1808, 1809]). Owing to peripheral chlorine atoms, hexachlorosubphthalocyanine sPcCl6 is one of the most promising acceptors for photovoltaics.

For increasing the acceptor properties of polychlorosubphthalocyanine, its aza analogue sPz11 was obtained; aza-substitution of carbon atoms in the benzene rings additionally enhances the electron-withdrawing properties of the peripherally chlorinated subphthalocyanine. This hexachlorotripyrazinosubporphyrazine is formed in 17% yield from commercially available 5,6-dichloropyrazine-2,3-dicarbonitrile and BCl3 in refluxing p-xylene (Scheme 673)[1810]. The structure of its benzene solvate was determined by X-ray diffraction analysis (Figure 18).

![[{"id":"q_Nzr40atv","type":"paragraph","data":{"text":"Molecular structure of sPz<b>11</b> solvate (CCDC 2003907)."}}]](/storage/images/resized/hAXOondPcqcwz2UseJIyiOlSTVXaZCYMCJOQ2K9j_xl.webp)
Subporphyrazine sPz11 was found to have the highest electron affinity among all known ѕubphthalocyanines and their analogues. According to the data of cyclic voltammetry (CV)[1810], this compound is reduced already at –0.20 V (vs. Ag/AgCl in MeCN), i.e., it is reduced much more easily than halogenated subphthalocyanines, which are normally used as acceptors (–0.43 and –0.72 V for compounds 2.25.1 and 2.25.2, respectively). Absorption and luminescence studies revealed a large extent of intersystem crossing and a longer-lived triplet state for halogenated pyrazinosubporphyrazine sPz11 compared to subphthalocyanine sPcCl6 (2.25.2)[1811].
The prospects of application in organic electronics were evaluated in a comparative study of sublimed thin films of hexachlorosubphthalocyanine 2.25.2 and its pyrazine analogue sPz11[1812]. It was shown that upon vacuum sublimation, sPz11 molecules form films with a higher degree of ordering and crystallinity than subphthalocyanine sPcCl6, which does not contain pyrazine rings. The specific conductivities of these films at 300 K are similar, but the activation energy for the photoconductivity is much higher for sPz11. It was also noted that pyrazine derivative sPz11 generates a larger number of free charge carriers than amorphous sPcCl6 upon illumination under comparable conditions. In the latter case, the efficiency of the p-CuI/n-sPcCl6 hybrid photovoltaic cell could be increased only by increasing the film crystallinity via thermal annealing[1813].
For variation of the acceptor properties of the subporphyrazine macrocycle, mixed cyclotrimerization of 5,6-dichloropyrazine-2,3-dicarbonitrile and tetrafluorophthalodinitrile was carried out, which gave unsymmetrical diaza- and tetraaza-substituted subphthalocyanines sPz12 and sPz13, combining dichloropyrazine and tetrafluorobenzene moieties in the molecule (Scheme 674)[1814]. When the ratio of the starting dinitriles was 4 : 1, the yield of unsymmetrical subporphyrazines was 4.3% (3.5% yield of sPz12 and 0.8% yield of sPz13). The use of 5,6-diphenylpyrazine-2,3-dicarbonitrile resulted in low-symmetric phenyl-substituted pyrazinosubporphyrazines sPz14 and sPz15 in much better yields: 11 and 5%, respectively[1815].

The structure of subporphyrazine sPz12 with one dichloropyrazine moiety was studied by X-ray diffraction analysis (Figure 19)[1814]. It was shown that dichloropyrazine moieties are disordered with the tetrafluorobenzene rings. This leads to alternation of subporphyrazine molecules with different spatial arrangements of the annulated moieties in the resulting stacks. Their molecular packing in the crystal is similar to that of perfluorosubphthalocyanine sPcF12, but is somewhat denser.
Quantum chemical DFT calculations demonstrated[1814] that the introduction of the dichloropyrazine moieties, instead of the tetrafluorobenzene moieties, induces a decrease in the LUMO energy (Figure 20). This leads to enhancement of the acceptor properties of the subporphyrazine macrocycle and, as shown by electrochemical CV measurements, shifts the reduction potential (E1/2Red) to less negative region [the potentials were –0.43, –0.31, –0.19 and –0.13 V vs. Ag/AgCl in CH2Cl2 for sPcF12 (2.25.1), sPz12, sPz13 and sPz11, respectively]. This effect is directly related to the introduction of the chlorine atoms into the pyrazine rings. For example, in the case of 5,6-diphenylpyrazine annulation, the LUMO energy changes only slightly, while the first reduction potential of compounds sPz14 and sPz15 (–0.42 and –0.41 V vs Ag/AgCl in CH2Cl2) remains virtually the same as in symmetrical macrocycles sPcF12 and sPz4 (–0.43 and –0.41 V, respectively).
![[{"id":"H1UWOxZLGp","type":"paragraph","data":{"text":"Effect of aza substitution and halogenation on the energy of frontier molecular π-orbitals and the reduction potentials of the subporphyrazine macrocycle."}}]](/storage/images/resized/4uJLf1PUoYnoqAmkcebm0dySpjKKmi0d8Z5ZNOXq_xl.webp)
Kovkova et al.[1814] compared the performance characteristics of subporphyrazines with fused dichloropyrazine and/or tetrafluorobenzene moieties as acceptors in prototype non-fullerene photovoltaic cells {glass/ITO/MoO3/[sPc]/acceptor/buffer/Al} containing unsubstituted subphthalocyanine as the donor. Photoconversion characteristics proved to be higher for a cell with subporphyrazine sPz12, which has one dichloropyrazine moiety, than for a cell with perfluorosubphthalocyanine sPcF12 (2.25.1) as the acceptor under the same conditions. The greatest effect was found when LiF was used as a buffer layer, and the best photoconversion characteristics were achieved in cells with bathocuproine (BCP). The increase in the device efficiency can be attributed to enhancement of the acceptor properties of the subporphyrazine macrocycle upon the introduction of a dichloropyrazine moiety and, possibly, to denser packing of the molecules. However, although the introduction of a larger number of pyrazine moieties into compounds sPz12 and sPz11 increases the electron affinity of the n-layer material compared to that of sPcF12, the material stability and ability to sublime simultaneously decrease, which prevents the formation of films of the desired composition.
Pyrazine-annulated subporphyrazines exhibit an intense absorption band in the visible range of 520 – 570 nm. The position of the maximum is considerably affected by the macrocycle periphery (the nature of the annulated moieties and substituents in them), but not by the axial ligand at the boron atom. The fluorescence spectra have relatively small Stokes shifts, while the fluorescence intensity can vary greatly depending on the properties and structure of the axial ligand. Hexaphenyl-substituted subphthalocyanines and their pyrazine analogues were used to develop fluorescence sensors for acidic pH or for alkali and alkaline earth metal ions. For this purpose, subporphyrazines sPz4 and sPz16 were allowed to react with phenols containing 4-dimethylamino or azacrown ether groups in the para-position, which furnished subporphyrazines sPz17 and sPz18 with the corresponding axial aryloxy ligands (Scheme 675)[1816, 1817].

Whereas subporphyrazines with an axial chlorine atom or phenoxy group have fluorescence quantum yields of 0.2 – 0.3, fluorescence of subporphyrazines sPz17 and sPz18 is completely quenched via photoinduced electron transfer (PET) from the nitrogen lone pair of the substituent in the para-position of the aryl substituent to the subporphyrazine π-chromophore. When the nitrogen atom of the dimethylamino group in subporphyrazines sPz17a and sPz18a is protonated, the РЕТ effect is switched off and fluorescence is enhanced to ΦF = 0.2 – 0.3. This is observed not only in organic solvents, but also in aqueous micellar solutions; therefore, it was proposed to use these subporphyrazines in biomedicine for imaging tumour tissues with increased acidity. In the case of subporphyrazines sPz17c and sPz18b – d, the PET effect is switched off when the azacrown ether coordinates an alkali or alkaline earth metal ion; this can be used for selective detection of these ions by appearance of fluorescence[1817].
2.25.2. Chalcogenadiazole analogues of phthalocyanine
One more approach to increasing the acceptor properties of phthalocyanine type macrocycles is to replace benzene rings by 1,2,5-chalcogenadiazole moieties[1801]. Until recently, among these compounds, tetrakis(1,2,5-thiadiazolo)porphyrazine [TSN2PzH2] (Ref. [1818]) (2.25.3) and its complexes with magnesium[1819], group IIIA metals[1820] and 3d metals were studied most extensively[1821, 1822].
The spectral properties and geometrical and electronic structures of TSN2Pz (2.25.3) complexes with lithium[1823], calcium[1824] and rare earth elements (YIII, LaIII, SmIII, EuIII, DyIII, ErIII and LuIII) have been studied[1825-1827]. The structural features of TSN2Pz complexes with FeII, CoII[1828], NiII (Ref. [1829]) and ZnII (Ref. [1830]) were explored using DFT and gas electron diffraction methods. The influence of the nature of chalcogen on the structure and physicochemical properties of porphyrazine derivatives was studied in relation to MgII tetra(1,2,5-chalcogenadiazolo)porphyrazine complexes[1831]. In addition to porphyrazines 2.25.3 (SN2Pz) and 2.25.4 (SeN2Pz) with 1,2,5-thia- and 1,2,5-selenadiazole moieties[1832], both symmetrical and unsymmetrical 1,2,5-telluradiazoloporphyrazines 2.25.5 – 2.25.7 (TeN2Pz) were reported for the first time[1833, 1834].
A specific feature of 1,2,5-thiadiazole-annulated porphyrazines is the existence of intermolecular interactions via the formation of S∙∙∙N bonds[1835], which determine the packing of the molecules in the crystal and endow them with functional properties of organic magnetic[1836] and semiconductor materials[1837, 1838].
Whereas 1,2,5-thiadiazole moieties fused with a macrocycle are fairly stable, 1,2,5-selena-and 1,2,5-telluradiazole rings are readily cleaved: deselenation (induced by H2S in pyridine) affords vicinal aminoporphyrazines (PzA)[1832, 1839], while detelluration (in the presence of an acid) gives diimino- or dioxoporphyrazines (Scheme 676). These heterocyclic moieties can be regarded as protective groups and as precursors for the formation of vicinal diamino-, dioxo-, and diimi-noporphyrazines, which are designated in Scheme 676 as PzA, PzE and PzF, respectively. These transformations are used to modify the porphyrazine macrocycle, e.g., for the replacement of the chalcogen atom (Se → S; Te → Se or S)[1840, 1841], or for the synthesis of pyrazinoporphyrazines (PyzPz)[1832, 1841], bis-porphyrazines with a pyrazine bridge (PyzPz2), formamidoporphyrazines (PzC)[1842] and acetamidoporphyrazines.

The oxidation of the pyrrole ring in diaminoporphyrazines PzA resulted in the formation of diaminosecoporphyrazine PzB, which undergoes condensation to give pyraziniporphyrazine PzD[1843].
It is noteworthy that attempts to perform cyclotetramerization of 1,2,5-oxadiazolo-2,3-dicarbonitrile did not give the corresponding porphyrazine due to the difficulty of closure of the 1,2,5-oxadiazolopyrrole moiety. The co-condensation of this carbonitrile with pyrazine-2,3-dicarbonitrile resulted in the formation of chlorin type porphyrazine 2.25.8 (Scheme 677), which contained an imidazolone moiety, according to mass spectrometry and IR spectroscopy data[1834].

A subporphyrazine with three 1,2,5-thiadiazole moieties (sPz19) was obtained in 9% yield by cyclotrimerization of 1,2,5-thiadiazolo-3,4-dicarbonitrile in the presence of BCl3 in refluxing p-xylene (Scheme 678)[1844]. The X-ray diffraction study revealed a dome-like structure of the macrocycle typical of subporphyrazines; upon crystallization of the product from benzene, π-complex with the solvent molecule was formed (Figure 21a)[1844].

![[{"id":"R-JyO0JHFI","type":"paragraph","data":{"text":"Molecular structure derived from X-ray diffraction data for cyclotrimerization products of 1,2,5-thiadiazolo-3,4-dicarbonitrile: sPz<b>19</b> (as a complex with benzene) (а) and <b>2.25.10</b> (b) (CCDC 188085 and 2346914, respectively)."}}]](/storage/images/resized/96MIk9j3hQNvOHPDUUtLIutGaNhJv7mJm5Eabpzx_xl.webp)
It is worth noting that, apart from the target product, two other interesting compounds, 2.25.9 and 2.25.10, were isolated from the reaction mixture and characterized. One of the products results from trimerization of 1,2,5-thiadiazolo-3,4-dicarbonitrile with formation of the central triazine ring (2.25.10) (see Scheme 678). The structure of this compound was confirmed by X-ray diffraction study (see Figure 21b). Unlike the macrocyclic boron(III) subporphyrazine complex, the triazine trimer 2.25.10 is nearly planar. This compound is considered as an n-type organic semiconductor[1845]; and owing to the combination of thiadiazole moieties with a triazine pharmacophore, it may have biological activity[1846].
The molecular weight of the other product, compound 2.25.9, found from mass spectrometry data, corresponds to the presence of four starting dinitrile moieties and one boron atom in the molecule[1847]. On the basis of UV-Vis and 11B NMR spectroscopy data and results of quantum chemical DFT calculations, a structure of porphyrazine complex containing one boron(III) atom coordinated to two neighbouring nitrogen atoms was proposed for this compound (Figure 22).
![[{"id":"hR_oFcpV56","type":"paragraph","data":{"text":"Molecular structure of product <b>2.25.9</b> according to DFT data (in two projections)."}}]](/storage/images/resized/VTXl13l55OfqBUG12Yq8p6epEPhAKRhHI9lsFpnu_xl.webp)
Subporphyrazine sPz20 with fused 1,2,5-selenadiazole moieties was prepared[1844] by template cyclotrimerization of 1,2,5-selenadiazolo-3,4-dicarbonitrile in the presence of BCl3 in refluxing p-xylene; however, the product yield was only ~ 1%, i.e., it was much lower than that in the case of the 1,2,5-thiadiazole analogue (Scheme 679).

It should be noted that replacement of benzene rings in boron(III) subphthalocyanine by electron-deficient aromatic heterocycles [pyrazine or 1,2,5-thia(selena)diazole] results in stabilization of all frontier π-МОs, which is most pronounced for 1,2,5-thiadiazole derivative (Figure 23). The least pronounced HOMO stabilization occurs for Se-containing subporphyrazine, while LUMO is less stabilized for the pyrazine-annulated compound. Hence, the spectral characteristics of subporphyrazine can be controlled by appropriate selection of the heterocycle for annulation. The position of the maximum of the long-wavelength Q-absorption band of subporphyrazines changes depending on the annulated moiety in the following order: pyrazine (530 nm) < 1,2,5-thiadiazole (538 nm) < benzene (563 nm) << 1,2,5-selenadiazole (574 nm)[1844].
![[{"id":"HrQtgLqFH7","type":"paragraph","data":{"text":"Effect of heterocyclic annulation on the energy of frontier molecular orbitals and electronic absorption spectra of subporphyrazines."}}]](/storage/images/resized/k4XohuCE1xbDbN7HJUdPuLjMfcraCkoy1MMODMJ0_xl.webp)
Cross-cyclotrimerization of 1,2,5-thiadiazole-3,4-dicarbonitrile with phthalonitrile and tetrafluorophthalonitrile (Scheme 680) gave low-symmetry subporphyrazines sPz21–sPz24, which combine one or two 1,2,5-thiadiazole moieties and benzene (or tetrafluorobenzene) rings in the molecule[1848, 1849]. Boron(III) chloride and dichlorophenylborane were used as template agents. In the latter case, the reaction gave subporphyrazines sPz23b and sPz24b with an axial phenyl group[1848]. When the molar ratio of 1,2,5-thiadiazole-3,4-dicarbonitrile to tetrafluorophthalonitrile was 2 : 1, the total yield of these low-symmetric subporphyrazines was 15%, with product sPz24b with one heterocyclic moiety being formed in a two times higher amount than the product with two heterocycles (sPz23b)[1848].

1,2,5-Selenadiazolodibenzosubporphyrazines sPz25 – sPz27 were obtained from 1,2,5-selenadiazole-3,4-dicarbonitrile and phthalonitrile or its tetrafluoro and tetrachloro derivatives (Scheme 681)[1850].

The structure of 1,2,5-selenadiazolodibenzosubporphyrazine sPz25 was established by X-ray diffraction analysis (Figure 24)[1850]. A characteristic feature of subphthalocyanine analogues with 1,2,5-chalcogenadiazole moieties that determines their molecular packing is the capability for intermolecular interactions of two types. Chalcogen bonds involving the selenium atom of one molecule and the meso-nitrogen atom of another molecule are formed between molecules of neighbouring stacks; in addition, ππ-interactions occur between the 1,2,5-selenadiazole moiety as a π-acceptor and the benzene ring as a π-donor.
![[{"id":"hCUnGiG78y","type":"paragraph","data":{"text":"Molecular structure and packing of 1,2,5-selenadiazolodibenzosubporphyrazine sPz<b>25</b> (CCDC 1971769)[[ type=\"anchor\" referenceId=\"8419\" ]]."}}]](/storage/images/resized/Du42MBaF9t11Tp86xy2UG6wXUOc5wARP1HwfOwkM_xl.webp)
The variation of the number of 1,2,5-thiadiazole moieties and introduction of fluorine atoms into benzene rings, determining the energy of frontier π-orbitals, thus make it possible to change their acceptor properties and electron affinity of the subporphyrazine macrocycle over broad limits (Figure 25). For example, the replacement of one benzene ring in subphthalocyanine by a 1,2,5-thiadiazole moiety shifts the first reduction potential to the anodic region from –1.04 to –0.74 V; replacement of two rings shifts the potential to –0.32 V, and the replacement of all three rings brings the potential to –0.10 V. Meanwhile, the exhaustive fluorination of benzene rings decreases the reduction potential by half.
![[{"id":"iHju1xS2Es","type":"paragraph","data":{"text":"MO energy diagram and reduction potentials of 1,2,5-thiadiazole-annulated subporphyrazines."}}]](/storage/images/resized/3Uul3O4soTTxiFwsO2KvapuA2D6HHRY7kqo8l3S6_xl.webp)
Owing to high electron affinity of the macrocycle in subporphyrazines with fused 1,2,5-thiadiazole moieties and high ability of these compounds to sublime, they can be used as effective acceptors in non-fullerene photovoltaic cells containing subphthalocyanine as the donor layer[1849]. It is of interest that fusion of subporphyrazine macrocycles with 1,2,5-thiadiazole moieties instead of benzene rings increases the stability of their films deposited on glass substrate compared to subphthalocyanine films[1851]. It was shown that sublimed films of subporphyrazine sPz19 containing three 1,2,5-thiadiazole moieties can act as molecular optical filters increasing the stability of perovskite solar cells against photodegradation[1852]. Considering the intrinsic photoactivity of this subporphyrazine, it is hypothesized that sPz19 may serve as a component of tandem perovskite–subphthalocyanine solar cells.
2.25.3. Thiophene analogues of phthalocyanine
Porphyrazines with 2,3-fused thiophene moieties (ThPz1) and their benzo-fused analogues (ThPz2) were prepared back in 1937 by Linstead et al.[1799] from the corresponding thiophendicarboxylic acid dinitriles. However, the interest of researchers in tetra(2,3-thieno)porphyrazines ThPz1 was moderate[1853-1855], although their 5-hexyl-substituted analogues were proposed as organic semiconductors[1856, 1857]. 2,3-Thianaphthene-annulated porphyrazines ThPz2[1858-1860] and tetrapyrazinoporphyrazines ThPz3[1861] received more attention.
Porphyrazines ThPz4 with four 3,4-annulated thiophene moieties could not be obtained, unlike their low-symmetric analogues ThPz5, which combine 3,4-thiophene and benzene rings[1862], or bridged dimers ThPz6[1863].
Subphthalocyanine analogues containing thiophene moieties instead of benzene rings were prepared from thiophene-3,4-dicarbonitrile and its 2,5-substituted derivatives[1864, 1865]. The template cyclotrimerization was carried out in o-dichlorobenzene in the presence of BCl3 at 180 °C, and the resulting boron(III) subporphyrazine was further modified by axial tert-butylphenol ligand (Scheme 682). It was found that symmetrical subporphyrazine with three 3,4-fused thiophene moieties is not formed in this case; however, cross-cyclotrimerization with phthalonitrile resulted in the formation of unsymmetrical benzosubporphyrazines sPz28 and sPz29, containing one or two 3,4-thiophene moieties. The template cyclocondensation with 2,5-unsubstituted dinitrile is accompanied by chlorination of the thiophene moiety with the released chlorine; therefore, only subporphyrazine sPz28 with one 2,5-dichlorothiophene ring was isolated in 0.5% yield[1864]. The use of 2,5-dichlorothiophene-3,4-dicarbonitrile in mixed cyclotrimerization increased the yields of mono- and bis(2,5-dichlorothiophene)-annulated subporphyrazines sPz28 and sPz29 to 8 and 4%, respectively. It is of interest that in the case of 2-nitro- and 2,5-bis(triethylsilyl)-substituted thiophene-3,4-dicarbonitriles, nitro and triethylsilyl groups were also substituted by chlorine atoms and 2,5-dichlorothiopheno-dibenzosubporphyrazine sPz28 was isolated as the final product in 1 to 3% yield[1865].

3,4-Thiophene-fused low-symmetric subporphyrazines sPz28b and sPz29b form molecular complexes with C60 and C70, the structure of which was determined by X-ray diffraction analysis (Figure 26)[1864, 1865].
Presumably[1864], subporphyrazines containing 2,5-dichlorothiophene moieties can be considered as light-harvesting materials for photovoltaics. It should be noted that the reduction potential of complexes sPz28b and sPz29b (–0.90 and –0.99 V)[1864] differs only slightly from that for subphthalocyanine, while complex formation with fullerenes, which are typical acceptors, indicates that they can be used as π-donors, unlike 1,2,5-thiadiazole derivatives, which are π-acceptors. In fact, replacement of the benzene ring in subphthalocyanine by 2,5-dichlorothiophene moieties induces only a small anodic shift of the first reduction potential (by 0.07 V for each moiety), while the effect of 1,2,5-thiadiazole rings is markedly higher (shift by 0.3 – 0.4 V) (see Figure 23).
Subphthalocyanine analogues with 2,3-fused thiophene moieties are unknown, but cyclotrimerization of benzo[b]thiophene-2,3-dicarbonitrile in the presence of BCl3 in 1,2,4-trichlorobenzene (TCB) at 220 °С affords subporphyrazine sPz30 with three 2,3-thianaphthene moieties[1866] (Scheme 683), which is a heterocyclic analogue of subnaphthalocyanine. After treatment of the reaction mixture with phenol, product sPz30 was isolated in a total yield of 8.3% as a 2 : 1 mixture of С1- and С3-regioisomers containing an axial phenoxy group. The regiosomers were separated by high-performance liquid chromatography; each of them was a mixture of enantiomers, which were separated using a column with a chiral phase.

Unsymmetrically substituted and/or annulated subporphyrazines and subphthalocyanines may be chiral. However, there are few cases of isolation of single enantiomers. The presence of a heterocyclic moiety favours the separation of chiral subporphyrazines; for example, one of the C3-enantiomers of thiazole-annulated subphthalocyanine was isolated in a pure state[1867].
2.25.4. 1,4-Diazepine analogues of phthalocyanine
Porphyrazines DzPz1 – DzPz4 containing fused quasi-aromatic 1,4-diazepine moieties (Scheme 684) were prepared by cyclotetramerization of appropriate 5,7-substituted 1,4-diazepine-2,3-dicarbonitriles[1868-1872].

A characteristic feature of these macrocycles is the ability to form complementary hydrogen bonds between the meso-nitrogen atom of one molecule and the CH-acidic methylene group of the 6H-1,4-diazepine moiety of another molecule (Figure 27)[1873-1875]. This results in the formation of strong H-dimers for porphyrazines DzPz1 and DzPz2 containing 5,7-diaryl- and 5,7-distyryl-1,4-diazepine moieties[1874, 1875] and stabilizes the sandwich lanthanide complexes DzPz4[1876-1878].
![[{"id":"N8PpT4lra4","type":"paragraph","data":{"text":"Molecular structure of Ni complex of DzPz<b>1b</b> (а) and its H-dimer with intermolecular С–N contact (b) (CCDC 2106082)[[ type=\"anchor\" referenceId=\"8443\" ]]."}}]](/storage/images/resized/0qUOgsG8EQaGdpEuUEghFSL0kf3ZXsMfAm7A4Wyx_xl.webp)
Porphyrazines DzPz5 and DzPz6 with reduced 6,7-dihydro-1Н-diazepine moieties were prepared and proved to be highly sensitive as fluorescence sensors for acidic pH[1879].
Unsymmetrical subporphyrazine sPz31 with one 1,4-diazepine moiety was synthesized (in 2% yield) by co-condensation of 5,7-diphenyl-1,4-diazepine-2,3-dicarbonitrile and tetrafluorophthalodinitrile in the presence of BCl3 in p-xylene[1880]. It was found that cyclotrimerization was accompanied by chlorination of the quasi-aromatic 1,4-diazepine ring into position 6 (Scheme 685). The annulated 1,4-diazepine heterocycle was unstable and underwent hydroprotolytic cleavage, in particular during chromatography on silica gel. As a result, it was converted to aminobenzamide derivative sPz32 (3.4% yield), which can be considered as a fluorescence sensor for acidic pH. The presence of amino group in subporphyrazine sPz32 leads to fluorescence quenching as a result of internal charge transfer (ICT), but upon protonation of the amino group in an acid medium, the ICT effect is switched off, and fluorescence is enhanced.

2.25.5. Pyrazine-annulated corrolazines
Corrolazines are contracted analogues of porphyrazines in which one of the meso-nitrogen atoms is absent (see Scheme 668). Unlike porphyrazines, corrolazines are trianionic macrocyclic ligands. Tetrapyrazinocorrolazines (TPyzCz) were first obtained in 2014 as phosphorus(V) complexes[1881] and they are still the only currently known heterocycle-annulated corrolazines.
Phosphorus(V) octaphenyltetrapyrazinocorrolazine is formed similarly to tetrabenzocorrolazines[1882, 1883] when the appropriate porphyrazine (TPyzPzH2) is refluxed in pyridine in the presence of PBr3. This compound can be isolated as the dihydroxy complex [TPyzCzP(OH)2] (Cz1a) by precipitation into water or as the dialkoxy derivatives [TPyzCzP(OAlk)2] (Cz1b,c) by precipitation into alcohol (methanol or ethanol) (Scheme 686)[1881]. The axial ligands are replaced by aryloxy groups only upon long-term treatment of Cz1a with thionyl chloride, after removal of which the intermediate dichloride complex is converted to the aryloxy complexes [TPyzCzP(OAr)2] (Cz2a–c) in the presence of an excess of the required phenol[1884]. It is noteworthy that the halide complexes [TPyzCzPHal2] (Hal = Br, Cl), formed in intermediate steps of these reactions, cannot be isolated in a pure state due to the high reactivity of the P – Hal bond.

The successive sulfochlorination and hydrolysis of macrocycle Cz1 furnishes the water-soluble form of tetrapyrazinocorrolazine Cz3 with eight 4-sulfophenyl substituents (see Scheme 686)[1885].
The annulation of the pyrazine moieties, on the one hand, and macrocycle contraction to give the corrolazine structure, on the other hand, result in enhancement of the fluorescent properties of the compound[1885]. The fluorescence of tetrapyrazinocorrolazines can be controlled by photoinduced electron transfer (PET effect) from the axial ligand to the electron-deficient macrocycle TPyzCz. For example, pH increase and deprotonation of the dihydrophosphonate moiety P(OH)2 in water-soluble corrolazine Cz3 leads to fluorescence quenching via PET from the negatively charged P(O–)2 group to the macrocyclic ligand (fluorescence quantum yield decreases almost 10-fold, from 0.18 to 0.02, as pH is changed from 4.6 to 13.0)[1885]. Thus, sulfonated tetrapyrazinocorrolazines can be considered as model compounds for the development of efficient water-soluble fluorescence sensors for pH measurement in the 4.6 – 13 range.
The photoinduced electron transfer is manifested most clearly for the lone pair of electrons of the NMe2 group in 4-dimethylaminophenoxy derivative Cz2b, which, unlike its analogue Cz2a, does not fluoresce in most organic solvents[1884]. When the NMe2 group is protonated, the РЕТ effect is switched off, and fluorescence is enhanced. This high sensitivity of fluorescence of the phosphorus(V) tetrapyrazinocorrolazine with 4-dimethylaminophenoxy groups as axial ligands can be used to detect traces of acids, which are often present in commercial haloalkanes (CH2Cl2, CHCl3).
Phosphorus(V) tetrapyrazinocorrolazines effectively generate singlet oxygen, with the 1О2 quantum yield reaching 60 – 70% even for water-soluble sulfonated derivatives[1885]. Therefore, these macrocycles can be considered as effective photosensitizers for antitumour and antibacterial photodynamic therapy. In the case of corrolazines Cz2b and Cz2c containing 4-hydroxyphenoxy and 4-dimethylaminophenoxy groups as axial ligands, the quantum yield of singlet oxygen depends on the PET effect, with the photosensitizing properties being switched on or off depending on the acidity of the medium[1884]. It can be expected that photosensitizers based on corrolazines with annulated electron-deficient pyrazine rings and axial ligands containing electron-donating NMe2 groups would be safe for normal cells (the PET effect is switched on) and phototoxic for tumour cells with increased acidity (the PET effect is switched off).
Thus, studies of the last 10 – 15 years demonstrate that combination of effects of phthalocyanine macrocycle contraction and annulation with electron-deficient pyrazine or 1,2,5-chalcogenadiazole moieties instead of the benzene rings produces new macroheterocyclic compounds containing a nonmetal as a central atom. These boron(III) subporphyrazines and phosphorus(V) corrolazines possess promising properties for practical use as acceptor materials for organic electronics and photovoltaics and as fluorescent sensors and photosensitizers for biomedical applications. Meanwhile, fused analogues of phthalocyanines with electron-rich heterocycles are still poorly studied; therefore, new studies addressing these systems are expected.
3. Chemistry of natural heterocyclic systems and their derivatives
3.1. Covalent modification of berberine for medicinal chemistry
Of all natural alkaloids (and perhaps of all natural compounds), berberine has the widest range of therapeutic and chemotherapeutic activities. The covalent modification of berberine aims not only to obtain its derivatives with more effective biological activity but also to use the berberine fragment as a ʻcargo’ to create drugs designed for targeted action on mitochondrial membranes and for coordination with DNA G-quadruplexes.
In this article we discuss methods of modifying the electrophilic berberine cation and the nucleophilic activity of its analogs obtained by the umpolung approach — dihydroberberine (the reduced form of berberine) and berberrubine.
The main directions of medical application of berberine and its derivatives can be divided into four major groups: treatment of metabolic disorders[1886-1888], cardiovascular diseases[1889-1891], neurodegenerative disorders[1892, 1893] and combating malignant tumors as a chemotherapeutic agent[1894-1896] (Figure 28). References are provided here to what we believe to be the most informative reviews for each group of diseases.
![[{"id":"gSavqG1JSg","type":"paragraph","data":{"text":"Biological activity of berberine and its derivatives."}}]](/storage/images/resized/P5559CiH0DJSdK9xBjUncAXlqHqJDcDN5UT9LuhX_xl.webp)
According to the CAS SciFinder database, berberine was observed in 11 485 publications, including 767 reviews, in the last decade (2013 – 2022). During this period, 228 publications, including 18 reviews, were devoted to methods of covalent modification of berberine. It should be emphasized that the results of berberine transformations presented in these reviews are organized not by reaction type but by their expected biological effects, such as combating type 2 diabetes or antibacterial effects. This mini-review is the first attempt to classify the most important covalent modifications of berberine based on reaction mechanisms and the nature of the underlying electrophilic-nucleophilic interactions.
The pathways for covalent modification of berberine and its derivatives correlate with the distribution of local Fukui reactivity indices ( fk)[1897]: the attack by electrophiles (E) should occur at the positions of berberine skeleton with the highest values of fk–, while the attack by nucleophiles should occur at the positions with the highest values of fk+ (Scheme 687). From our calculated distribution of Fukui indices, it follows that the π-deficient berberine (3.1.1) is prone to nucleophilic addition at the position 8 and nucleophilic substitution of the methoxy group at the C(9) atom. Most of the transformations of berberine (3.1.1) described in the literature fall into these two types of reactions. The use of the umpolung approach (polarity inversion) greatly enhances the reactivity of this molecule. For example, the reduction of compound 3.1.1 to dihydroberberine 3.1.2 opens a pathway to functional derivatives at the position 13 through interactions with electrophiles, while heating berberine in the presence of bases leads to berberrubine (3.1.3), which reacts with electrophiles at the C(12) and O(9) atoms (see Scheme 687).

The main efforts in modification traditionally focus on preserving the cationic nature of the berberine skeleton[1898]. The most common method is the nucleophilic substitution of the methoxy group at the position 9[1899], using various N- and O-nucleophiles as reagents (Scheme 688).

Interestingly, berberine (3.1.1) belongs to a rare type of organic compounds which reactivity is difficult to predict based on their structural formula. For example, the crystallization water present in its structure, which is almost impossible to remove, significantly affects the reaction mechanisms with various nucleophiles[1900]. Substitution of the 9-methoxy group with secondary aliphatic amines (such as piperidine, morpholine, or diethylamine) has not been successful, as indicated by the absence of 9-dialkylaminoderivatives of berberine in the CAS database. However, reactions with primary amines proceed quite smoothly.
Evidently, these reactions are not blocked by steric hindrance: more sterically hindered ortho-dialkylamino derivatives of dinitronaphthalene or dinitroquinoline are easily formed. As we have previously shown[1900], when berberine interacts with secondary alkylamines, a hydrolysis product, berberrubine (3.1.3), is formed. The aminated product of berberine with secondary amines, according to calculations, is thermodynamically and kinetically unstable and undergoes hydrolysis with the crystallization water under the reaction conditions forming berberrubine (see Scheme 688). According to quantum chemical calculations, berberrubine (3.1.3) can exist in two isomeric forms: the chloride of the cationic 9-hydroxy form and the hydrochloride associate of the electrically neutral 9-keto form.
The interaction products of berberine with anionic nucleophiles (electrically neutral dihydroberberines 3.1.2) are much less common in the literature than 9-substituted berberines. These compounds do not contain a quaternary (pyridinium) nitrogen atom and are therefore considered less attractive in terms of biological activity. Typically, 8-substituted dihydroberberines are considered as precursors to obtain cationic 13-alkyl-substituted berberines, in which the substituent X is eliminated from the position 8 Scheme 689).

Regarding the products of anionic nucleophile addition, only 8-allyl- and 8-acetonyldihydroberberines have been described in the literature[1899].
A large series of previously unknown C-, O-, and N-adducts of berberine at the position 8 (> 50 compounds) have been synthesized. The mechanisms of formation of 8-substituted dihydroberberines depend on the nature of the nucleophile: N-nucleophiles are first deprotonated by a base and then are added to berberine at the C(8) atom (Scheme 690)[1901].

At the same time, C-nucleophiles are not deprotonated by a base. In this case, the first stage is to add a hydroxide ion to the C(8) atom to form berberinol (3.1.4), which then deprotonates the C-nucleophile. This process is accompanied by the simultaneous elimination of water and the addition of the nucleophile (Scheme 691)[1902]. In this scenario, crystallization water acts as a bifunctional catalyst, facilitating the transfer of a proton from the nucleophile to the hydroxyl group of berberinol (3.1.4)[1902].

Systems 3.1.2 contain an enamine fragment and are therefore capable of interacting with electrophiles at the position 13, which has the highest local nucleophilicity index fk– (see Scheme 687). These systems have been introduced into reactions with various electrophiles containing electron-acceptor fragments. For the first time, methods have been developed to form carbon-carbon bonds with an sp2-hybridized center linked to the berberine skeleton. This has allowed the introduction of pharmacophoric fragments into berberine, conjugated with the π-system of the molecule (unlike the previously described 13-substituted berberines).
One of the methods of electrophilic modification involves the introduction of nitrobenzofurazan and nitrofurazan fragments, which can serve as exogenous sources of nitric oxide(II), thereby extending the spectrum of biological activity of substituted berberines (Scheme 692)[1903-1905]. Ethoxymethylidene derivatives, such as rhodamine (2-thioxothiazolidin-4-one), have been used as heterocyclic electrophiles for the modification of dihydroberberines 3.1.2. This method of obtaining berberine derivatives is considered very promising for medicinal chemistry for two reasons. First, ethoxymethylenes are easily formed from heterocycles containing an endocyclic active methylene group, which allowed the linkage of known privileged structures—rhodanine, oxindole, and barbiturates—to the berberine skeleton. Second, the target compounds proved to be quite stable, and the reactions themselves are easily scalable[1906-1908].

The molecules of 13-substituted dihydroberberines 3.1.5 and 3.1.6 contain electron-acceptor groups conjugated with the pyrrolic nitrogen atom at the position 7. This results in an intramolecular electron density transfer (calculated to be between 0.24 and 0.49 ē), which is manifested in the deep coloration of systems 3.1.5 and their good solubility in polar media. In addition, the 1H NMR spectra of these compounds show significant downfield shifts of the signals for indicator protons in the B ring and upfield shifts for the protons of the electrophilic fragment. According to the calculations, the dipole moment of systems 3.1.5 reaches 8.8 D. Electrically neutral compounds 3.1.5, 3.1.6, and their analogs can be considered as internal salts, partly retaining the cationic form of the nitrogen atom at the position 7. Such a betaine structure is responsible for many types of biological activity, while being free of the pharmacokinetic issues associated with berberine salts.

The presence of oppositely charged groups within a single molecule facilitates its coordination with G-quadruplexes of telomeric DNA fragments, which govern numerous physiological and pathological processes in the human body. The synthesis of betaine 3.1.7 based on berberine was first reported in 2021 by German researchers, who discovered its affinity for non-canonical DNA structures[1909]. The formation of non-covalent complexes with G-quadruplexes in the promoter regions of oncogenes and in the telomeric areas of chromosomes, combined with telomerase inhibition, helps to stop the uncontrolled division of cancer cells.
Another direction of umpolung modification of berberine is the involvement of berberrubine (3.1.3) in reactions with electrophiles, which is activated at the position 12 (see fk– index in Scheme 687). Indeed, berberrubine (3.1.3), unlike berberine (3.1.1), easily undergoes reactions with aryl diazonium salts at pH 7.5 – 8.0, forming 12-diazoaryl-9-hydroxyberberines 3.1.8 (Scheme 693)[1910].

According to quantum chemical calculations, in berberrubine (3.1.3), there is a low-barrier approach of the chloride anion to the hydrogen atom of the 9-OH group. This process enables the umpolung transition of berberrubine into a slightly less energetically favorable (by ~ 3 kcal mol–1) hydrochloride nucleophilic zwitterion capable of reacting with aryl diazonium salts (see Scheme 687). The barrier for Z-E isomerization of products 3.1.8 is ~ 32 kcal mol–1 according to calculated data, suggesting the use of red light or IR radiation for deep penetration into biological tissues. The potential value of compounds 3.1.8 lies in the possibility of creating DNA- or membrane-targeted derivatives of berberine with photo-controlled structures.
Derivatives of berberine are of particular interest as systems with high affinity for cell membranes and mitochondria. For instance, lipophilic berberines 3.1.9, behaving similarly to Skulachev ions, containing a long hydrocarbon fragment or a similar linker with attached ubiquinone at the position 13, have shown a high specificity for mitochondria and significantly affected the oxygen consumption by mitochondria[1911]. Such cations have been shown to have strong antibacterial activity, with the authors attributing the antibacterial effect to a possible reduction in the transmembrane potential of bacteria[1912].

All berberine derivatives mentioned in the review combine two properties: high lipophilicity and quaternary ammonium character, which means they can have high affinity for both membranes and DNA.
The main conclusion of this review is that the directions of covalent modification reactions of berberine and its derivatives can be reliably predicted using Fukui’s reactivity indices. Reactions with nucleophiles are characteristic of the original berberine, while reactions with electrophiles are characteristic of its derivatives obtained within the framework of the umpolung methodology. It should be emphasized that both the cationic derivatives of berberine and its neutral analogs possess significant therapeutic and chemotherapeutic potential, which is not yet fully understood.
3.2. Monoterpenoid heterocyclic compounds
The synthesis of novel compounds with valuable biological or physicochemical properties based on substances of plant origin is a promising area of research in organic and medicinal chemistry. Natural monoterpenes and monoterpenoids have a wide range of native biological activities and high enantiomeric purity, making them convenient substrates for the preparation of new pharmacologically active compounds. Previously, we have published reviews on the biological activity of monoterpene compounds and their derivatives[1913], in particular antiviral activity[1914], and the use of such derivatives as inhibitors of DNA repair enzyme[1915], and also reviews on the synthesis of oxygen-containing heterocyclic compounds[1916], and monoterpenoid-based five-membered heterocycles[1917]. At the same time, there is currently no studies that summarize information on the preparation of different heterocycles from monoterpene compounds.
This Section presents the results of research carried out at the N.N.Vorozhtsov Novosibirsk Institute of Organic Chemistry over the last decade in the synthesis of heterocyclic compounds, primarily biologically active compounds, based on available monoterpenes and monoterpenoids. The review is divided into two parts, discussing methods for the preparation of heterocyclic scaffolds containing at least one carbon atom of a monoterpenoid and the synthesis of heterocycles based on functional groups of monoterpenes.
An important starting material for the synthesis of oxygenated heterocyclic compounds is verbenol epoxide 3.2.1, which is obtained in two steps from the monoterpenoid verbenone 3.2.2, abundant in nature[1918]. Thus, the reaction of compound 3.2.1 and the monoterpenoid diol 3.2.3, derived therefrom under acidic conditions, with aldehydes in the presence of montmorillonite clays gives various heterocyclic compounds, including benzodioxins 3.2.4, chromenes 3.2.5 and (epoxymethano)chromenes 3.2.6 (Scheme 694)[1919-1921]. Chromenes 3.2.5 are usually formed as a mixture of diastereomers at the 4-position. The ratio of products 3.2.4 – 3.2.6 depends both on the structure of the monoterpenoid and on the nature of the acid catalyst[1922]. When 2,4,6-trimethoxybenzaldehyde was used as the carbonyl component, tricyclic epoxychromenes 3.2.7 were formed in a significant amounts (up to 25%); with other arrangements of methoxy groups in the aromatic ring such products were either obtained in minor amounts or were absent[1923].

Compounds showing high analgesic activity in in vivo experiments include benzodioxins 3.2.4 bearing 4-Cl or 4-NO2-phenyl substituents[1924], epoxychromenes 3.2.7 (see[1925]) and disubstituted chromenes 3.2.5 bearing 3-methoxy,4-hydroxy or 3-hydroxy,4-methoxyphenyl groups[1926]. All possible stereoisomers were obtained for these chromenes and the most active of them were identified[1927]. It was shown that the mutual arrangement of the hydroxy and methoxy groups in the aromatic ring has a defining impact on the mechanism of their analgesic action[1928]. The introduction of alkyl substituents in the para-position of the phenyl moiety of chromenes 3.2.5 led to the disappearance of the analgesic effect[1929].
However, chromenes 3.2.5, formed in the reaction of diol 3.2.3 with acrolein and decanal, also demonstrated high analgesic activity[1930]. Various acid-activated clays and halloysite nanotubes were used to catalyze the reaction of verbenol epoxide 3.2.2 with decanal. It turned out that the nature of the catalyst had almost no effect on the yield of chromene 3.2.5, but determined the ratio of 4S to 4R diastereomers, which varied from 1.6 to 0.8[1931]. The use of thiophene carbaldehyde and its derivatives in the reaction also gave chromenes with analgesic activity, but this effect was less pronounced than that of the analogues previously synthesized[1932]. Some (epoxy-metano)chromenes 3.2.6 were found to have moderate cytotoxic activity[1933].
When citral is used as the aldehyde component in the Prins reaction with diol 3.2.3, chromene 3.2.8 is formed, which comprises fragments of two different monoterpenes (Scheme 695)[1934]. Although citral is a mixture of E and Z isomers (geranial and neral, respectively), compound 3.2.8 has exclusively the E configuration of the substituent in the aliphatic unit, which can be explained by the previously discovered high lability of neral in the presence of K10 clay[1935]. Compound 3.2.8 showed high analgesic activity in vivo in the hot plate test[1934].

Ketones can also be reacted with monoterpenoids 3.2.3 to furnish compounds 3.2.9. The use of cyclic ketones gives spirocyclic products 3.2.10. However, this reaction requires an increased acidity of the catalyst and is accompanied by the formation of comparable amounts of dehydration products 3.2.11 and 3.2.12 in the form of a mixture of the double bond isomers (see Scheme 695)[1936]. Among these compounds, chromenols obtained by the reaction of monoterpenoids 3.2.3 with acetone and cyclohexanone showed analgesic activity. The reactions of aromatic and aliphatic ketones with diol 3.2.3 in the presence of BF3 · Et2O – H2O system gave compounds 3.2.13 containing fluorine atom instead of hydroxy group as main products, i.e. boron trifluoride etherate acts both as a catalyst and as a source of a fluorine atom[1937]. The fluorine-containing product obtained by the reaction of compound 3.2.3 with 2,4,6-trimethoxybenzaldehyde showed high activity against H1N1 influenza virus[1938].
Commercially available (–)-isopulegol (3.2.14) can also be used as a monoterpenoid component in reactions affording substituted chromenes (Scheme 696). Its reactions with ketones, as with diol 3.2.3, provided lower yields than with aldehydes, the yield of the product decreasing as the bulkiness of substituents in the ketones increased[1939]. A variety of acid-modified clays[1940-1945], mesoporous Ce-composite material[1946] and halloysite nanotubes[1947-1949] have been successfully used to promote these transformations. Interest in finding effective catalysts for these reactions was primarily due to the high biological activity found in a number of products. For example, compound 3.2.15, containing a thiophene moiety as substituent R, showed unique analgesic activity, arresting pain within one day after oral administration to mice at a dose of as low as 1 mg kg–1[1950]. Since this compound was obtained using K10 clay as a catalyst, it contains no heavy metal impurities[1951]. The products of the reaction of (–)-isopulegol 3.2.14 with substituted thiophene carbaldehydes showed moderate antiviral activity[1952], as well as the ability to inhibit fatty acid amide hydrolase (FAAH)[1953] and the tyrosyl-DNA phosphodiesterase 1 (TDP1) enzyme[1954, 1955]. The latter is a DNA repair enzyme responsible for the removal of damage caused by some anticancer drugs such as camptothecin and topotecan, so TDP1 inhibitors may improve the efficiency of anticancer therapy[1956]. The compounds 3.2.16, 3.2.17 formed by the reaction of (–)-isopulegol (3.2.14) with acetone and cyclopentanone respectively showed high activity against influenza virus[1939], and the products of reactions of monoterpenoid 3.2.14 with acrolein, butanal and 2-indanone were found to have analgesic properties[1957, 1958].

The use of the BF3 · Et2O – H2O system to catalyze the reactions of (–)-isopulegol (3.2.14) with aldehydes gave fluorinated products 3.2.18 (see[1959]) (see Scheme 696), including compounds with high activity against influenza virus[1960]. Replacement of carbonyl compounds by nitriles in the presence of sulfuric or trifluoromethanesulfonic acid as catalyst led to 1,3-oxazines 3.2.19. The product of the reaction of (–)-isopulegol (3.2.14) with benzyl cyanide was found to have significant analgesic activity[1961].
The simultaneous use of aldehydes and acetonitrile induced the Prins-Ritter tandem reaction to yield amides 3.2.20 (see Scheme 696)[1962]. Using SO3H-functionalized heterogeneous catalysts, control of the stereoselectivity of the reaction was achieved by the simple addition of water: in the absence of water, the 4S diastereomer ((4S)-3.2.20) was preferentially formed, whereas on addition of water the stereomer (4R)-3.2.20 was formed[1963]. Acetamides 3.2.21 obtained by the reaction of (–)-isopulegol with ketones (acetone, pentane-3-one, heptane-4-one and cyclopentanone) showed analgesic activity[1964].
(+)-2-Carene (3.2.22) features high activity in electrophilic addition reactions due to the conjugation of the double bond with the cyclopropane moiety (Scheme 697). Compounds 3.2.23 with an isobenzofuran scaffold were the main products in the reaction of (+)-2-carene with aldehydes in the presence of K10 clay[1965]. As minor products, 3-oxabicyclo[3.3.1]nonanes 3.2.24 were isolated, which are often identified in reactions of monoterpenes with aldehydes[1916, 1966, 1967]. Application of halloysite nanotubes as catalysts significantly improved the selectivity to products 3.2.23[1968, 1969]. The content of (+)-2-carene 3.2.22 in essential oils is usually very low, thus hampering its isolation from natural sources, but it can be obtained by isomerization of (+)-3-carene 3.2.25, one of the main components of turpentine. It has been shown that reactions with aldehydes can run with a mixture of carenes formed by heating 3-carene 3.2.25 in the presence of montmorillonite clay, instead of individual 2-carene 3.2.22, thereby increasing the yield of the target isobenzofurans[1970]. Isobenzofuran 3.2.23, obtained by the reaction of monoterpene 3.2.22 with vanillin, showed high neuroprotective activity in an animal model of Parkinson’s disease[1971], and the product 3.2.23 formed from 5-bromothiophene-2-carbaldehyde proved to be an effective inhibitor of the enzyme TDP1[1972].

Another way of activating 3-carene 3.2.25 with the shift of its double bond to the cyclopropane moiety is to subject it to the Prins reaction to give 4-hydroxymethyl-2-carene 3.2.26 (see Scheme 697). The monoterpenoid 3.2.26 reacts readily with aldehydes in the presence of montmorillonite clay[1973], to furnish, depending on the structure of the aldehyde, a variety of heterocycles such as the mixtures of isobenzofurans 3.2.27 (in reactions with aliphatic aldehydes and benzaldehyde)[1974], tetracyclic compounds 3.2.28 and 3.2.29 (with salicylic aldehyde and polymethoxy(hydroxy)benzaldehydes)[1975], and polycycle 3.2.30 (with thiophene-2-carbaldehyde)[1976]. The product 3.2.29a (R1 = OH, R2 = OMe, R3 = H) showed high cytotoxicity against the lymphoblastoid cell line MT-4, causing apoptosis of tumour cells[1975].
The assembly of a heterocyclic scaffold based on monoterpene compounds may not only involve rearrangement of the terpene core, as in the case of diol 3.2.3, isopulegol 3.2.14 and carenes, but may also involve cleavage of the C – C bond. (+)-Camphor 3.2.31 and (–)-phenchone 3.2.32 were shown to react with ortho-substituted anilines in the presence of zinc chloride in a solvent-free mode to give 2-substituted benzazoles 3.2.33 – 3.2.36 (benzoxazoles, benzothiazoles and benzimidazoles) with a significant predominance of products 3.2.33 and 3.2.35, respectively (Scheme 698)[1977]. The process proceeds as a single-step reaction. Condensation of monoterpenoids with anilines led to the cleavage of the bicyclic framework of the ketones 3.2.31 and 3.2.32 at the most substituted C – C bond: C(1) – C(2) in the case of (+)-camphor and C(2) – C(3) for (–)-phenchone. The authors proposed the plausible mechanism of these transformations.

Monoterpenoids contain a limited number of functional groups that can be involved in further chemical modifications leading to the formation of heterocyclic moieties. Typically, natural compounds contain oxygen groups such as alcohol, carbonyl and carboxyl groups. Using the natural alcohol (–)-borneol 3.2.37 as a starting material, reaction with ω-halogen-substituted aliphatic acid chlorides afforded the corresponding esters which, when alkylated with saturated nitrogenous heterocycles, gave a large series of ω-amino acid bornyl esters 3.2.38 – 3.2.40 in yields ranging from 52 to 91% (Scheme 699). Compounds 3.2.38 – 3.2.40 have linker of varying length between the nitrogen atom of the heterocyclic moiety and the natural norbornane backbone. The borneol esters of ω-amino acids showed a broad spectrum of antiviral activity. For example, esters 3.2.38 and 3.2.39 with morpholine, piperidine and N-methylpiperazine moieties were active against influenza viruses[1978]; 3-methylpiperidine and N-substituted piperazine esters 3.2.38 and 3.2.39 showed high activity as Marburg virus entry inhibitors[1979]. The vast majority of such derivatives were potent Ebola virus entry inhibitors[1980]. Compounds 3.2.38 – 3.2.40 containing morpholine, piperidine and N-alkylpiperazine rings showed high activity as inhibitors of respiratory syncytial virus (RSV) by binding to surface glycoprotein F[1981], and had antiviral activity against orthopoxviruses[1982], including smallpox virus[1983]. In addition, heterocyclic derivatives of borneol were inhibitors of SARS-CoV-2 virus entry[1984]. The antiulcerogenic activity of compounds 3.2.38 comprising various heterocycles was explored and the piperazine derivative was shown to have antiulcerogenic properties superior to those of the comparator drug Omeprazole[1985]. For compound 3.2.39 bearing a piperidine moiety, the stability in biological fluids was determined and pharmacokinetic parameters were evaluated[1986].

Acylation of (–)-borneol with 3-chloropropionic acid chloride followed by the reaction with heterocyclic N- and S-nucleophiles gave esters 3.2.41 containing aliphatic and aromatic heterocycles[1987]. The results of biological tests showed that triazole- and imidazole-substituted compounds 3.2.41 have high antiviral activity against influenza A (H1N1) virus.
Substituted 7-hydroxycoumarins 3.2.42 and 3.2.43 and also monoterpene alcohols and bromides derived therefrom gave rise to compounds 3.2.44 and 3.2.45 (Scheme 700), which showed diverse biological activities. Thus, conjugate 3.2.45 with n = 1, obtained from (+)-myrtenol, showed high inhibitory activity against the enzyme TDP1, as well as the ability to enhance the cytotoxicity of the antitumour agent camptothecin[1988], and its enantiomer showed hypoglycemic activity in an in vivo experiment[1989]. The compound 3.2.44 (R = 4-FC6H4), synthesized using geraniol, effectively inhibited the enzyme TDP1 and enhanced the antitumour effect of the anticancer drug topotecan in animals[1990]. Monoterpene-coumarin conjugates also showed antiviral activity, moderately inhibiting the reproduction of influenza virus[1991] and effectively inhibiting the reproduction of respiratory syncytial virus, which is deadly for infants and the elderly people. Among RSV replication inhibitors, the most promising appears to be the (+)-myrtenol derivative 3.2.45 (n = 2), which inhibits viral entry into the cell by binding to RSV protein F[1992, 1993] and has acceptable pharmacokinetic properties[1994]. Arylcoumarins 3.2.44 (R = Ph) derived from (–)-myrtenol and nopol demonstrated good anti-RSV activity[1995], while their analogue synthesized from (+)-myrtenol was inactive against RSV due to poorer binding to RSV protein F as determined by molecular dynamics[1996]. The 5-hydroxycoumarin derivative 3.2.46, which proved to be a highly potent inhibitor of the TDP1 enzyme in the nanomolar range, increased the cytotoxicity of topotecan against the HeLa tumour cell line but not against conventionally normal HEK 293A cells at non-toxic concentrations[1997].

Monoterpene-substituted thiazolidin-4-ones were synthesized from bromides derived from terpene alcohols[1998]. Bromides were reacted with p-hydroxybenzaldehyde, followed by multicomponent condensation with benzylamine, thioglycolic acid and terpenyl-substituted aldehyde, affording thiazolidin-4-ones in a single step. Among these derivatives, compound 3.2.47 showed the highest activity against the TDP1 repair enzyme and synergistic activity with topotecan.

A series of terpene amines were obtained from terpene alcohols using the Gabriel method. The reaction between (+)-usnic acid and amino-substituted terpenoids gave dibenzofuran-terpene conjugates, among which citronellol- and nopol-substituted compounds 3.2.48 and 3.2.49 were active as TDP1 inhibitors and showed synergistic action with topotecan (Scheme 701)[1999].

Reactions of terpene aldehydes with dimethylbispidinone 3.2.50 give diaza-adamantanes 3.2.51 with various monoterpene substituents (Scheme 702)[2000]. Compounds 3.2.51 derived from (–)-myrtenol and citronellal showed high analgesic activity. Replacement of the methyl substituents in the diazaadamantane moiety of compound 3.2.51 by ethyl substituents, as well as reduction of the keto group to a hydroxyl group, resulted in the disappearance of the analgesic effect[2001].

Another way to assemble a heterocyclic core based on the C(O)H group of a monoterpene aldehyde was proposed (Scheme 703)[2002]. 1,3-Thiazolidin-4-one 3.2.53, synthesized from campholenic aldehyde, showed pronounced antiulcerogenic and anti-inflammatory effects.

Based on (+)- and (–)-usnic acid and monoterpene aldehydes, a series of derivatives were obtained which showed the inhibitory ability towards TDP1[2003]. Among such conjugates, compound 3.2.54 performed best.

The addition of monoterpenes to heterocycles has been very productive in the design of new bioactive compounds. For example, dimethyl aza-adamanthanone was first converted to the oxime 3.2.55, which was reduced to diaza-adamantane amine 3.2.56, which reacted with monoterpenoid aldehydes to give the compounds 3.2.57. Among these products, the citronellal-containing conjugate 3.2.57a showing high activity against influenza virus should be singled out (Scheme 704)[2004]. A similar compound derived from (–)-myrtenol exhibited analgesic properties[2005].

Terpene aldehydes react with piperazine to give N-substituted piperazines 3.2.58, which were reacted with a triazole-containing terminal epoxide. This furnished a series of analogues of azole antibiotics 3.2.59 (Scheme 705), which showed excellent antifungal activity, two orders of magnitude higher than that of the comparator drug fluconazole[2006].

There are few examples of the straightforward synthesis of heterocyclic products based on monoterpene ketones. For example, a single-step diastereoselective synthesis of the spiro product 3.2.60 was achieved from (–)-phenchone 3.2.32 and anthranilamide (Scheme 706); the ZnCl2r-promoted reaction was carried out in the melt of the starting compounds[2007]. The spirocyclic compound 3.2.62 was obtained from bispidinone 3.2.50 and dihydrocarvone 3.2.61 in the presence of Amberlyst heterogeneous catalyst[2008]. A significant series of pyrazolinium salts 3.2.63 based on camphor, camphorquinone and carvone were synthesized. The study of their activity against influenza viruses showed that camphor-based salts were the most potent virus inhibitors[2009].

The imino derivatives of (+)-camphor 3.2.64 and 3.2.65 containing heterocyclic moieties acted as inhibitors of influenza viruses (Scheme 707)[2010]. Modification of the hydroxyl function of imino alcohol 3.2.66 (named camphecene) gave rise to a large library of heterocyclic derivatives 3.2.67 and 3.2.68, among which compounds bearing pyrrolidine, pyridine and pyrimidine moieties showed the most pronounced antiviral properties[2011]. Monoterpene-derived triazoles 3.2.69 and 3.2.70 were obtained via click chemistry methodology[2012, 2013].

Modification of the keto group can greatly extend the synthetic possibilities of monoterpene ketones. For example, camphor-based thiosemicarbazone 3.2.71 was successfully used in the synthesis of heterocycles 3.2.72 – 3.2.76 shown in Scheme 708. All synthesized compounds showed inhibitory activity against smallpox vaccine virus[2014].

A similar strategy was used in a study[2015]; the authors described the synthesis of thiosemicarbazones based on monoterpene aldehydes and ketones and their further reaction with bromine derivatives of usnic acid. The products of such transformations, 3.2.77 and 3.2.78 (Scheme 709), were active against TDP1 in the nanomolar concentration range and showed a synergistic effect when used together with topotecan.

The reduction of bicyclic ketone oximes can provide the corresponding primary amines. For example, derivatives 3.2.80 containing saturated N-heterocycles have been successfully synthesized from isobornylamine 3.2.79 (Scheme 710). Amides 3.2.80 differing in linker length and type of heterocyclic substituent have been studied as anti-orthopoxviral agents[1982] and as inhibitors of filovirus entry[1980]. A thiophene-substituted isobornylamine derivative 3.2.81 has shown properties as an agonist of the free fatty acid receptor 1 (FFAR1)[2016], an important therapeutic target of type 2 diabetes.

Hydrazones of natural ketones bearing an amino group suitable for further transformations are simple and accessible substrates for the preparation of conjugates of monoterpenoids and heterocycles (Scheme 711). The implementation of reactions of compounds 3.2.82 at the unsubstituted NH2 moiety significantly expanded the synthetic possibilities of the caged monoterpenoids such as camphor 3.2.31 and fenchone 3.2.32. Thus, a convenient approach to heterocycles 3.2.83 via the direct alkylation of hydrazones with dihaloalkanes has been developed; and the reaction of hydrazones with ethane-1,2-dithiol in the presence of formaldehyde and samarium nitrate gives compounds 3.2.84 containing a 1,5,3-dithioazepane moiety[2017]. Reactions of camphor hydrazone with various isothiocyanates gave N-substituted thiosemicarbazones 3.2.85, which gave rise to a large library of 2-iminothiazolidin-4-ones 3.2.86, 3.2.87 and 2,3-dihydrothiazolones 3.2.88. Among the above-described heterocyclic derivatives of monoterpenes, compounds active against smallpox virus and potent inhibitors of Marburg virus entry were identified[2018]. Pyridine-based N-acylhydrazones were explored as inhibitors of orthopox viruses[2019]. Conjugates 3.2.89 derived from camphor or fenchone hydrazones and iso-oxoindole acids were tested for inhibitory activity against orthopoxviruses and influenza viruses[2020]. Of particular interest is the use of these compounds as potent inhibitors of hantaan viruses, which cause haemorrhagic fever with renal syndrome, as drugs for the treatment of zoonotic viral diseases for which there is currently no effective chemotherapy[2021]. A stereoselective method for the synthesis of spiro derivatives 3.2.90 (see Scheme 711) based on camphor and fenchone hydrazones is also reported[2022].

Among a small number of carboxyl-containing monoterpenoids, one of the most accessible is (+)-camphoric acid (3.2.91), a dicarboxylic acid formed by the exhaustive oxidation of camphor. A series of polycyclic compounds 3.2.92 – 3.2.95, formally analogues of quinazolin alkaloids, based on (+)-camphoric acid (3.2.91) and various diamines have been synthesized (Scheme 712)[2023]. Among them, compound 3.2.93 was found to be active against a wide range of influenza viruses. The reaction of camphor anhydride, derived from camphoric acid, with various aliphatic and aromatic amides produced a large series of (+)-camphor imides, among which compound 3.2.96 showed high activity against flaviviruses such as Zika virus and yellow fever virus[2024]. Based on diamine 3.2.97, also obtained from camphoric acid, amides 3.2.98 containing N-heterocyclic moieties were synthesised[2025]. These amides were tested as inhibitors of influenza and SARS-CoV-2 viruses. It was found that N-benzylpiperazine derivatives 3.2.98 effectively inhibit the action of influenza viruses, and piperidine-substituted compounds 3.2.98 are active against SARS-CoV-2 virus.

Based on (+)-camphor-10-sulfonic acid (3.2.99), a series of sulfonamides 3.2.100 containing different nitrogen-containing heterocyclic moieties were obtained. Using the pseudovirus system as a model, these compounds were shown to be effective inhibitors of Ebola virus entry (Scheme 713)[2026]. In addition, reduction of the carbonyl group of the bicyclic ketone produces the corresponding alcohol 3.2.101, which is characterized by reduced toxicity and increased antifilovirus activity compared to the substrate[2027]. The authors of a study[2028] have scaled up the synthesis of (+)-ketopinic acid (3.2.102) based on available sulfonic acid 3.2.99 and have shown that it reacts with amidoximes in the presence of N,N'-carbonyldiimidazole to give O-acylamidoximes, which cyclize to 1,2,4-oxadiazoles 3.2.103. Heterocyclic derivatives of ketopinic acid proved to be potent inhibitors of H1N1 and H7N9 influenza viruses. (+)-Campholenic acid (3.2.104), isolated in good yield by fusion of sulfonic acid 3.2.99 with alkali, was reacted with 1,3,4-thiazol-2-amine to give compound 3.2.105, which is highly active against the DNA repair enzyme TDP1[2029].

The reaction of monoterpene carboxylic compounds with 5-adamantane-1,3,4-thiazol-2-amine afforded promising adamantane-terpene conjugates, of which compound 3.2.106 performed best as inhibitors of the DNA repair enzyme TDP1[2030].

One of the ways to modify terpenoids with an olefinic double bond is to oxidize them to epoxy derivatives. Epoxides are rarely considered as effective pharmacologically active compounds due to their high reactivity. An exception is epoxide 3.2.107, obtained by oxidation of diol 3.2.3 (Scheme 714)[2031]. Like the starting compound[2031], epoxide 3.2.107 is able to eliminate the symptoms of Parkinson’s syndrome induced by administration of the neurotoxin MPTP to mice[2031]. At the same time, it demonstrates neuroprotective and neurorestorative properties on models of Parkinson's disease with the neurotoxins rotenone[2032] and MPTP[2031]. Epoxide 3.2.108, synthesized in two steps from diol 3.2.3, gave rise to a triazolyl sulfonyl derivative 3.2.109, which displayed high antiparkinsonian activity in both in vitro and in vivo experiments[2033].

Using the available (±)-camphene 3.2.110, a bicyclic olefin with an exocyclic double bond, an efficient approach to bromoethers 3.2.111 has been proposed, which were further used as alkylating agents to give ethers 3.2.112 (Scheme 715)[2034]. Like the borneol based esters 3.2.39 – 3.2.40, compounds 3.2.112 displayed a broad spectrum of antiviral activity. They are inhibitors of H1N1 influenza virus, have activity against Ebola virus, act as virus entry inhibitors and inhibit hantaan virus entry. Propargyl ethers 3.2.113 were synthesized by the known methodology, from which triazoles 3.2.114 combining monoterpene and saturated nitrogen heterocycle moieties were obtained by click reactions[2035]. N-heterocyclic derivatives with a triazole linker showed high activity against pseudoviral particles containing Marburg and Ebola virus surface glycoproteins.

To conclude, methods for the synthesis of heterocyclic compounds containing natural monoterpenes and their derivatives as a key unit are highlighted. The high reactivity and conformational flexibility of the parent monoterpenoids allow the formation of various conjugates, including oxygen-, nitrogen- and sulfur-containing heterocycles. The presence of various, often unique, biological activities in the products obtained is also essential. The latter fact is an important stimulus for the further development of this promising field, which lies at the interface between the chemistry of natural and heterocyclic compounds and medicinal chemistry.
3.3. Recent advances in chemistry of flavones
Flavones (2-aryl-4H-chromen-4-ones or 2-phenyl-4H-1-benzopyran-4-ones) are an oxygen-containing heterocyclic system belonging to a large class of flavonoids (Figure 29). Derivatives of flavones are abundant in the plant world, many of which are secondary plant metabolites[2036], which accounts for their diverse biological activities. A number of flavones have been found to exhibit antioxidant[2037], antimicrobial[2038], antiproliferative[2039], antiviral[2040] and other activities, which explains the classification of flavones as privileged scaffolds in medicinal chemistry[2041, 2042].
![[{"id":"47qZJwgvQW","type":"paragraph","data":{"text":"Flavones of plant origin."}}]](/storage/images/resized/3ggLFyUHDZ3tIvkIq9zDJEGUGcfdrril4fQLOQd9_xl.webp)
Based on the flavone scaffold, drugs used in clinical practice have been developed such as Flaxate, Dimefline, Flacoside and Flavopiridol (Alvocidib) (Figure 30)[2043].
![[{"id":"eojC1Ia_wl","type":"paragraph","data":{"text":"Drugs based on flavones and their pharmacological action."}}]](/storage/images/resized/YodzW3evu78DQwZSmjhjwzVsb4Ke1gl9vrQtyCDc_xl.webp)
The increasing interest in this heterocyclic system is evidenced by the publication of > 500 reviews in the last 5 years. However, almost all reviews are devoted to natural flavones, methods of their isolation from plant sources and biological activity, while generalizing articles on synthetic approaches to flavone are limited to 5 publications[2044-2048], and they present virtually no methods of flavone structural modification.
The present work aims to fill this gap by providing a comprehensive discussion of the chemical and biological aspects of flavone research carried out over the last five years.
3.3.1. Methods for the synthesis of flavones
Modern methods for the construction of flavone backbone include metal-catalyzed annulation reactions. For example, Sonogashira coupling of arylalkynes 3.3.1 with aryliodides 3.3.2 (Scheme 716) (In this Section the structures are numbered according to their order in the schemes, from substrate to product.) is widely used. An approach to flavones 3.3.3 based on the Pd-catalyzed carbonylation of 2-iodophenol with terminal alkynes in the presence of piperazine as a base is described[2049]. The synthesis of flavones 3.3.4, 3.3.5 has been carried out using Et3N and Ni(OAc)2 (see[2050]) or MnCl2 catalyst[2051].

In a study[2052], Et2NH and a heterogeneous catalyst, bis(N-heterocyclic carbene)palladium(II) anchored on Merrifield’s resin were used for the preparation of flavones 3.3.6. Compounds 3.3.7, 3.3.8 were synthesized in the presence of palladium complexes ((Pd-NHC) or [Pd(Xantphos)(4-Spy)](OTf)) and metal carbonyl complexes Mo(CO)6 or Co2(CO)8 as a less toxic CO source instead of carbon monoxide (see Scheme 716)[2053, 2054].
A process has been developed for the preparation of flavones 3.3.11 by silver-catalyzed decarboxylative radical annulation of ortho-hydroxy α-keto acids 3.3.9 with arylpropiolic acids 3.3.10 (Scheme 717), which occurs via the generation of an ortho-hydroxyaryl carboxyl radical in the presence of AgI and persulfate[2055].

The synthesis of flavones 3.3.14, 3.3.16 using the Ni-catalyzed decarbonylative cycloaddition of benzofuran-2,3-diones 3.3.12 to various alkynes including terminal (3.3.13) and disubstituted (3.3.15) has been reported (Scheme 718)[2056]. Reactions with terminal alkynes require the presence of KOAc, whereas the transformations of their disubstituted counterparts occur without the base.

Phenylacetylene (3.3.17) can also be used in oxidative coupling reactions with substituted benzaldehydes 3.3.18 through activation of the C – H bond by the Rh(L-Phe)(cod) catalyst (Phe is phenylalanine) to give flavones 3.3.19 (Scheme 719)[2057].

Zhai et al.[2058] proposed a one-pot synthesis of γ-benzopyranones 3.3.22 based on the aerobic oxidation in the presence of Fe(NO3)3 · 9 H2O and (2,2,6,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) and DMAP-catalyzed cyclization of propargyl alcohols 3.3.20 (Scheme 720). The cascade cyclization of 2-(1-hydroxyprop-2-yn-2-yl)phenols 3.3.20 to flavones 3.3.23 induced by atmospheric oxygen and hydroiodic acid is also described[2059]. A cascade radical-initiated synthesis of 2-arylchromones 3.3.24 from propargylamines 3.3.21 via intramolecular 6-endo-dig annulation and hydrocarbonylation was carried out[2060]. Air was used as the oxygen source, and dimethyl-2,2'-azobis(2-methylpropionate) (AIBME) and (PhSe)2 were the initiators of the radical reaction.

The reaction between salicylic aldehydes 3.3.25 and sulfoxonium ylides 3.3.26 in the presence of RhIII catalyst furnished flavones 3.3.27, 3.3.28 (Scheme 721)[2061, 2062].

A one-pot method for the synthesis of flavones 3.3.32 based on ortho-acylation of substituted phenols 3.3.29 with cinnamoyl chlorides 3.3.30 catalyzed by the BiCl3 – RuCl3 system via cyclodehydrogenation of the intermediate o-hydroxychalcones 3.3.31 has been proposed (Scheme 722)[2063].

Pd-catalyzed oxidative intramolecular cyclization of 2-hydroxydihydrochalcones 3.3.33 yields flavones 3.3.34 (Scheme 723)[2064].

The preparation of flavone 3.3.38 by a Suzuki – Miyaura decarbonylative cross-coupling involving acid fluoride 3.3.36, generated from 2-carboxychromone 3.3.35, and tri-n-butyl(4-methoxyphenyl)stannnane (3.3.37) using Ni(cod)2 as the catalyst and the phosphine ligand PPh2Me has been reported (Scheme 724)[2065].

Historically, the most popular approach to flavones 3.3.42 has been the Claisen – Schmidt method, which is a condensation of benzaldehydes 3.3.40 with 2-hydroxyacetophenones 3.3.39 (Scheme 725). It gives α,β-unsaturated ketones (chalcones) 3.3.41, which further undergo oxidative cyclization. Flavonols 3.3.43 were obtained by treating chalcones 3.3.41 with H2O2 via the Algar – Flynn – Oyamada reaction. In the last five years, both classical and modified conditions have been used for the Claisen – Schmidt condensation (Table 9)[2066-2076]. A number of flavones thus obtained have been found to have biological properties.
![Claisen – Schmidt synthesis of flavones <b>3.3.42</b>, <b>3.3.43</b> (see <a class="anchor" href="#figure-2392">Scheme 725</a>)[<a class="anchor" href="#reference-8631">2066</a>-<a class="anchor" href="#reference-8641">2076</a>].](/storage/images/resized/F5acM4uY6kz6bDP4zDFYYeSMv3e44usXmpcM43hB_xl.webp)

3-Alkyl-substituted flavones 3.3.48, which showed the ability to inhibit SARS-CoV-2 coronavirus, were obtained by the Claisen condensation of aryl ketones 3.3.44 with benzaldehyde 3.3.45. Subsequent oxidative cyclization of the intermediate chalcones 3.3.46 was carried out in the presence of I2, and demethylation of the methoxy derivatives 3.3.47 was promoted by pyridinium chloride (Scheme 726)[2077]. Such products inhibited the RdRp strain of the said coronavirus with IC50 = 4.3 – 8.8 μM, while for quercetin this value was 6.9 μM.

The use of the heterogeneous catalyst Au@SEU-COF-1 allows the one-pot condensation of benzaldehydes 3.3.50 with 2-hydroxyacetophenones 3.3.49 to give flavones 3.3.51 (Scheme 727)[2078]. With dicarbonyl reagents, this method yields diadducts 3.3.52 – 3.3.55.

The single-step condensation of substituted 2-hydroxyacetophenones 3.3.56 with benzaldehydes 3.3.57 or benzyl alcohols 3.3.58 led to flavones 3.3.59, 3.3.60 in the presence of heterogeneous catalytic systems CuN4/CoN4@NC (@NC is nitrogen-doped carbon)[2079] and CuN4/CoN2P2@NPC (@NPC is nitrogen- and phosphorus-doped carbon) (Scheme 728)[2080].

Flavones 3.3.62 and 3-iodoflavones 3.3.63 (Scheme 729) were obtained from 2-allyloxy(hydroxy)chalcones 3.3.61 by the I2-mediated one-pot reaction in DMSO[2081].

An electrochemical oxidative cyclization process has been developed for the synthesis of flavones 3.3.65 from 2-hydroxychalcones 3.3.64 using NaI as electrolyte (Scheme 730)[2082].

The Baker-Venkataraman rearrangement is the basic transformation of 2-(aroyloxy)acetophenones to 1,3-diketones, which are cyclized to flavones under acid catalysis conditions. This reaction was first mentioned in 1933[2083], but has not lost its relevance. Using this method, a series of flavones 3.3.68 (Scheme 731) were obtained from benzoyloxyacetophenones 3.3.66 with isolation of diketones 3.3.67, among which derivatives with high acetylcholinesterase (AChE) inhibitory and antiradical activity, potential agents for the treatment of Alzheimer’s disease, were found[2084].

The reaction of 2-hydroxy-5-nitroacetophenone (3.3.69) with benzoyl chloride (3.3.70) gave a mixture of 6-nitro-substituted flavones 3.3.71 and 3.3.72 in a ratio of 15 : 1 (Scheme 732) as a result of α-acylation of an intermediate diketone[2071].

Attempts have been made to obtain flavones from 2-hydroxyacetophenones 3.3.73 and benzoyl chlorides 3.3.74 by a one-pot method without isolation of the diketones 3.3.75 (Scheme 733)[2085]. Depending on the conditions, flavones 3.3.76 or their 3-benzoyl-substituted analogues 3.3.77 were produced. Compounds with antioxidant and antitumour activity have been found in the latter series[2086].

Intramolecular cyclization of o-hydroxyphenyl-1,3-diketones 3.3.78 to flavones 3.3.79 has been shown to be mediated with trifluoromethanesulfonyl chloride[2087]; heating with zinc oxide nanoparticles in the absence of solvent gives flavones 3.3.80 (Scheme 734)[2088].

A one-pot process for the preparation of flavones 3.3.83[2089] via the Knoevenagel condensation of β-arylketosulfoxides 3.3.81 and benzaldehydes 3.3.82, intramolecular oxa-Michael cyclization and syn-elimination of sulfenic acid is described (Scheme 735). Tricine with anti-inflammatory activity in acute colitis was synthesized by this method[2090].

For the synthesis of flavones 3.3.86, salicylic aldehydes 3.3.84 and α-bromoketones 3.3.85 were used in the modified Allan-Robinson – Friedländer reaction catalyzed by the N-heterocyclic carbene (Scheme 736)[2091].

The cyclization of o-alkinoylphenols 3.3.87 to flavones 3.3.88 promoted with polar DMSO is described (Scheme 737)[2092]. A method is proposed for the synthesis of 3-hetaryl-substituted flavones 3.3.91 from ynones 3.3.89 and (iso)quinoline- or quinoxaline N-oxides 3.3.90 via a [3 + 2] cycloaddition step[2093].

The conversion of α-halo-substituted dibenzoylmethanes 3.3.92 to flavones 3.3.93 is possible via photocyclization (Scheme 738), which follows a radical pathway[2094].

3.3.2. Synthesis of fluorine-containing flavones
Fluorine-containing flavones deserve separate consideration, so they can be obtained by aromatic nucleophilic ipso-substitution of the fluorine ortho atom (SNAr). The acylation of 1,3-dicarbonyl reagents 3.3.95 with polyfluorobenzoyl chlorides 3.3.94 activated with magnesium alkoxide yields 3-alkoxycarbonyl and 3-benzoyl flavones 3.3.98 containing fluorine atoms in the aromatic ring A (Scheme 739)[2095-2097]. This process is driven by the ease of SNAr cyclization of tricarbonyl intermediates 3.3.96, 3.3.97.

A three-step procedure has been proposed for the preparation of flavones 3.3.103 containing fluorine atoms in the ring B[2098].
First, acylation of 2-hydroxyacetophenone 3.3.100 with polyfluorobenzoyl chloride 3.3.99 gives 2-(aroyloxy)acetophenones 3.3.101 (Scheme 740), which give rise to fluorine-containing 1,3-diaryl-1,3-diketones 3.3.102. Cyclization of the latter affords flavones 3.3.103.

However, for the 1,3-diketones 3.3.102 containing an ortho-fluorine atom, two competitive routes of intramolecular cyclization are realized: addition of a hydroxyl group to the carbon atom of the enolized moiety (path 1) and nucleophilic substitution of the fluorine atom (path 2) to give flavones 3.3.103 and 3.3.104 containing halogen atoms in rings A and B respectively (see Scheme 740).
For the one-pot synthesis of flavones 3.3.107 containing a fluorine atom in the pyrone ring, it was proposed to combine the fluorination of 1-(2-hydroxyphenyl)-3-phenylpropan-1,3-diones 3.3.105 using Selectfluor with H2SO4-catalyzed cyclization (Scheme 741)[2099].

The dehydrogenative oxidation of readily available flavanones 3.3.108 is still a convenient approach to flavones. The preparation of flavone 3.3.109 by Pd-catalyzed dehydrogenation at 100 °C has been reported[2100]. However, for flavanones 3.3.108 containing electron-withdrawing groups, this process can be carried out under mild conditions. For example, the treatment with iodozobenzene in DMF at room temperature produces 3-substituted flavones 3.3.110 (Scheme 742)[2101]. Using the TBAI — tert-butyl hydroperoxide (TBHP) system, flavones 3.3.111 have been obtained[2102].

To summarize, a variety of approaches to the synthesis of flavones is developed by now making these compounds available for chemical transformation and biological studies.
3.3.3. Chemical transformations of flavones
Various CH functionalization routes have been proposed for flavones with a free 3-position. For example, flavone 3.3.112 reacts with N-fluorobenzenesulfonimide (NFSI) in the presence of CuBr and a ligand, 6,6'-Me2BiPy or TEMPO, to give di(benzenesulfonyl)amide 3.3.113 (Scheme 743)[2103]. Amide 3.3.113 was also obtained by electrochemical oxidation of flavone 3.3.112 and addition of the sulfonimidyl N-radical[2104]. Reaction of flavone 3.3.112 (R1 = Ph) with diethyl phosphite using Mn(OAc)3 · 2 H2O catalyst gave phosphonate 3.3.114, reaction of the same substrate with ethyl bromodifluoroacetate in the presence of CuI and pentamethyldiethylenetriamine (PMDETA) afforded a difluoroacetate-substituted flavone 3.3.115, and direct CuI-catalyzed trifluoromethylation in the presence of 1-trifluoromethyl-1,2-benziodoxol-3-one (FB) furnished 3-trifluoromethylflavone 3.3.116 (see Scheme 743)[2062]. 3-Chloro-substituted flavones 3.3.117 and 3.3.118 are formed by treatment of flavones 3.3.112 with trimethylchlorosilane[2062] or sulfonyl chloride[2105].

The RhIII or RuII-catalyzed functionalization of the 5-position of flavones 3.3.119 by reactions with allyl alcohols[2106] and butyl acrylate[2060] yielding 5-alkyl(alkenyl)derivatives 3.3.120 – 3.3.122 has been reported (Scheme 744).

One of the most promising trends in design of novel physiologically active compounds is functionalization of hydroxy-containing flavones, since they are mostly of natural origin and have biological activity. The synthetic modification of natural flavones, diosmethine (3.3.123a) and baicalein (3.3.123b), on hydroxy groups in reactions with alkylating agents such as sulfates (3.3.124), alkyl halides (3.3.125) or carboxylic acid esters (3.3.126) is described. As a result, a series of ethers and esters 3.3.127 and 3.3.128 (Scheme 745) were obtained, which showed the inhibitory ability towards AChE and β-secretase (BACE-1)[2107].

Acylation of O-benzyl esters of luteolin 3.3.129 was used to synthesize 5-O-acylated products 3.3.130, which exhibited antitumour activity against colon (HCT116) and breast cancer (MDA-MB-231) cell lines (Scheme 746)[2108].

The reaction of 3-hydroxy-2-[4-(dimethylamino)phenyl]benzopyran-4-one (3.3.131) with alkyl halides (3.3.125) or carboxylic anhydrides (3.3.126) gave rise to a number of ether and ester derivatives 3.3.132 (see Scheme 746)[2105]. Some of them showed antifungal activity against the fungi Aspergillus flavus, Acremonium strictum, Penicillium expansum. Aminoethoxy-substituted flavones 3.3.135 were synthesized by the reaction of aminoethyl chlorides 3.3.134 with hydroxyflavones 3.3.133[2109]. Flavone 3.3.135 containing a (piperidin-1-yl)ethoxy moiety showed anti-AChE activity. 7,8-Dihydroxy-substituted flavone 3.3.136 cyclized to 8-phenyl-[1,3]dioxolo[4,5-h]chromen-6-one 3.3.137 when treated with dibromomethane[2110].
Chrysin (3.3.138) and 7-hydroxyflavone 3.3.139 react with epichlorohydrin (3.3.140) at the C(7)-hydroxy group to give epoxy derivatives 3.3.141. The latter undergo the epoxide ring opening induced by amines 3.3.142 to give 7-alkylamino-substituted flavones 3.3.143 (Scheme 747)[2111], which show the ability to inhibit type IIα topoisomerase and antiproliferative activity against DU145 prostate adenocarcinoma cells. Using 7-hydroxyflavone 3.3.139 in the Mannich reaction with primary amines 3.3.144 and formaldehyde affords oxazine-annulated flavones 3.3.145, which showed anti-tobacco mosaic virus and fungicidal activities[2112].

Modification of amino-substituted flavones can also involve the amino group. For example, 6-amino flavones 3.3.146 reacts with 2-amino-3-chloropyrimidines (3.3.147) to give flavone-pyrimidine hybrids 3.3.148 with tuberculostatic activity (Scheme 748)[2071]. In similar reactions with sulfonyl chlorides 3.3.149, flavonyl-containing sulfonylamides 3.3.150 with activity against HepG-2, A-549, Caco-2 cancer cell lines were obtained[2113].

Flavones 3.3.151 containing a bromine or iodine atom in the pyrone ring react with substituted anilines (3.3.144) or N-phenylurea (3.3.152) to give (E)-aminated aurones 3.3.153 through the aza-Michael addition, flavone ring opening and cyclization reaction cascade (Scheme 749)[2114].

The most diverse are the transformations of polyfluoroflavones induced by N-nucleophiles, since they can undergo SNAr reactions involving fluorine atoms. Examples of reactions between 3-ethoxycarbonyl polyfluoroflavones with amines are discussed in detail in a review[2115], therefore only transformations of polyfluoroflavones published over the last 5 years will be considered below.
3-Benzoylpolyfluoroflavones 3.3.154 have been shown to react with primary amines 3.3.144, including biogenic dopamine, with pyrone ring opening and deacylation to give N-substituted enamino ketones 3.3.155 (Scheme 750)[2097, 2116]. However, in the case of amino acids 3.3.156, the transformation route depends on the conditions[2097]. When the reaction is carried out under mild conditions in the presence of AcONa and DIPEA, deacylated flavones 3.3.157 are obtained whereas enamino ketones 3.3.158 are by-products. At the same time, an alternative pathway is observed in the presence of a carbonate buffer in the microwave reactor: the trifluoro-containing flavone 3.3.154 (R1 = H) gives the 7-amino-substituted products 3.3.159, while the tetrafluoroflavone 3.3.154 (R2 = F) produces a mixture of 7-amino-5,6,8-trifluoroflavones 3.3.160 and 7-amino-5-hydroxy-6,8-difluoroflavones 3.3.161. The latter are formed through the intermediate generation of the enamino ketone and its subsequent cyclization.

Under the action of γ-aminobutyric acid (GABA), the pyrone ring opening in 3-ethoxycarbonylflavones 3.3.162 takes place to give N-substituted 4-aminobutanoic acids 3.3.163. Reactions of the same substrates with β-alanine yield 3-[amino(phenyl)methylidene]-6,7,8-trifluorochromene-2,4-diones 3.3.164 as a result of chromone-coumarin rearrangement and dealkylation (Scheme 751)[2117]. Heating flavones 3.3.162 with histamine under reflux in the presence of carbonate buffer gives 3-[amino(phenyl)methylene]polyfluorochromene-2,4-diones 3.3.165. A similar reaction with dopamine in the presence of DIPEA affords the derivatives 3.3.166.

The transformations of 3-carbonyl-functionalized polyfluoroflavones 3.3.167 with cycloalkylamines 3.3.168 are characterized by the formation of 7-aminoflavones 3.3.169 as products of substitution of the fluorine atom at the activated position 7 (Scheme 752)[2097, 2117]. The reaction of 3-ethoxycarbonyltetrafluoroflavone 3.3.167 (R1 = F) with pyrrolidine and morpholine can also produce 5,7-diamino-substituted products 3.3.170[2117].

Heating polyfluoroflavones 3.3.171 with imidazole (3.3.172a) in the presence of a base gives 7-imidazolyl-substituted flavones 3.3.173. The use of carbonate buffer changes the reaction pathway since water acts as a second nucleophile. The attack of the hydroxide ion on the electrophilic centre C(2) induces the pyrone ring opening to generate tricarbonyl intermediates, which, depending on the presence of the fluorine ortho atom, undergo alternative cyclization to 5-hydroxy-7-imidazolyl-3-methoxycarbonyl-5,6-difluoroflavones 3.3.175 or 7-imidazolyl-6,8-difluorochromen-2,4-diones 3.3.174 (Scheme 753)[2118].

Flavones 3.3.176 having the para fluorine atom in the phenyl substituent were reacted with azoles 3.3.172 (imidazole (a), pyrazole (b) and triazole (c)) in the presence of a base to give azolyl derivatives 3.3.177 (Scheme 754)[2119].

In contrast, reactions of polyfluoro-substituted flavones 3.3.178 with azoles under similar conditions gave a mixture of mono- to per(azolyl)-substituted flavones (Scheme 755, conditions a)[2120]. However, for the reactions between pentafluoroflavone 3.3.178 and pyrazole (3.3.172b, PyrH), the conditions for the selective production of mono-, tri- and per(pyrazolyl)substituted flavones 3.3.179 – 3.3.181 were found (see Scheme 755, conditions b – d ). Optimized conditions were applied to reactions with triazole (3.3.172c, TrzH) and imidazole (3.3.172a, ImH), providing the synthesis of mono- and per(triazolyl)substituted flavones 3.3.182 and 3.3.183, and the monoimidazolyl-containing counterpart 3.3.184, respectively (see Scheme 755)[2121]. The use of these optimal conditions in the reactions of tetra- and trifluoroflavones allowed a more selective synthesis of both monoazolyl and tetra(pyrazolyl or triazolyl)flavones. Some derivatives were found to have fungistatic activity[2122].

Under perfluoro substitution conditions, azolyl flavones 3.3.179, 3.3.182, 3.3.184 reacted with pyrazole and triazole to give exhaustive substitution products 3.3.185 – 3.3.188 (Scheme 756), which emitted green light under UV irradiation in the solid state[2122].

Transformations of natural flavones 3.3.190, 3.3.192, tangeretin (R1 = H) and nobiletin (R1 = OMe), functionalized with reactive groups, mediated by amines 3.3.189 and 3.3.193 are described (Scheme 757). Starting from the bromine derivative 3.3.190, 5-aminoethoxy-substituted flavones 3.3.191 were obtained, and the carboxylated derivative 3.3.192 furnished amides 3.3.194. Hydrolysis of the latter gave amino acid-substituted flavones 3.3.195, which showed antiproliferative activity against the Aspc-1, SUN5, HepG-2 and HCT116 tumour cells[2123].

Condensation of carboxy-substituted flavones 3.3.196, 3.3.200 with 1-benzylpiperidin-4-amines 3.3.197 and 1-benzylpiperazinamines 3.3.198 afforded piperidine- and piperazinamides 3.3.199, 3.3.201 (Scheme 758). Some of these products displayed high antitumour activity against human hepatocellular carcinoma (HepG2)[2121].

Flavones 3.3.202 undergo condensation with 2,4-dinitrophenylhydrazine (3.3.203) as a mono-N-nucleophile through the keto group of the pyrone ring to give hydrazones 3.3.204 (Scheme 759), which showed tyrosinase inhibitory activity as well as unusual photophysical properties[2069, 2124]. The reaction with O-alkylhydroxylamine hydrochloride 3.3.205 gave O-alkyloximes 3.3.206, which showed antitumour activity against breast cancer (MDA-MB-231 cell line)[2125].

In studying the reactions of flavones 3.3.207 with hydrazine, it was found that the C(5) hydroxy group prevented this interaction[2126]. Therefore, methylation was first carried out to obtain methoxy derivatives 3.3.208, which were treated with dinucleophiles to afford pyrazoles 3.3.209 (Scheme 760). Flavones 3.3.211 reacted with substituted hydrazines 3.3.212 to give isomeric pyrazoles 3.3.213 and 3.3.214[2126]. The formation of the pyrazoles was due to the attack of the diamine on the 2-position of the flavone, leading to the opening of the heterocycle, and on the carbonyl group with subsequent cyclization. Flavones 3.3.211 react with ethylenediamine in a similar way to give 5,7-diaryl-2,3-dihydro-1,4-diazepines 3.3.215. Some pyrazole derivatives showed antifungal activity against Candida albicans (strain SC5314).

However, Solis et al.[2127] easily synthesized pyrazoles 3.3.217 (Scheme 761) from flavones 3.3.216 under similar conditions, despite the presence of a C(5) hydroxy group, and the resulting compounds showed the ability to inhibit tyrosinase. The benzamidine-mediated recyclization of flavone 3.3.218 to substituted pyrimidine 3.3.219 under basic conditions is described (see Scheme 761)[2062].

Suzuki – Miyaura cross-coupling reactions of 6- and 7-tosyl(mesyl)oxyflavones 3.3.220 with arylboronic acids 3.3.221 catalyzed by the Pd(OAc)2 – CM-Phos (CM-Phos is 1-methyl-2-[2-(dicyclohexylphosphino)phenyl]-1H-indole) and K3PO4 · H2O system led to aryl-substituted flavones 3.3.222 (Scheme 762)[2128]. Using 6(7)-bromoflavones 3.3.223 in Pd-catalyzed reactions with 1,3-azoles 3.3.224 in the presence of CuI gave 1,3-azol-2-yl-substituted flavones 3.3.225[2129].

Pd-catalyzed cross-coupling of flavones 3.3.226 containing a bromine atom in the phenyl substituent with arylboronic acids 3.3.227 furnished 2'-aryl-substituted flavones 3.3.228 (Scheme 763)[2119]. However, attempted reactions with 2-furyl- (3.3.229) and 2-thienylboronic acids (3.3.230) resulted only in the debrominated flavone 3.3.231.

6(7)-Tosyloxyflavones 3.3.232 are converted to amino derivatives 3.3.233 by the Pd-catalyzed Buchwald – Hartwig amination reactions (Scheme 764)[2128].

The Pd-catalyzed Buchwald – Hartwig cross-coupling reaction between 4'-bromo-substituted flavone 3.3.234 and diarylamines 3.3.235 afforded derivatives 3.3.236 with interesting photophysical properties (Scheme 765)[2130].

Cross-coupling of 4'-bromoflavone 3.3.237 with porphyrins 3.3.238 in the presence of the catalytic system Pd(OAc)2 and racemic 2,2'-bis(diphenylphosphino)-1,1’-binafityl (rac-BINAP) gave complexes 3.3.239, 3.3.241, the acid work-up of which afforded the conjugates 3.3.240, 3.3.242 (Scheme 766) with potential fluorescent properties and the ability to generate singlet oxygen[2131].

3-Haloflavones 3.3.243 undergo hydrodehalogenation in the presence of the CuI – K2CO3 system to give flavones 3.3.244 (Scheme 767)[2132].

Visible light-induced intermolecular [2 + 2] photocycloaddition of flavone 3.3.245 to 2,3-dihydrofuran (3.3.246) was carried out in the presence of Ir(F) sensitizer to afford stereo- and regioselectively cis-syn-cis cyclobutane 3.3.247 (Scheme 768)[2133].

The molecular oxygen-mediated oxidative reaction of luteolin and other flavones in alkaline water gives dimers and trimers of flavones, including the natural compounds dicranolomin and distichumtriluteolin (Scheme 769)[2134].

The above literature data gives an idea of the research that has been carried out in the field of flavone chemistry over the last five years. The availability of compounds in this class is ensured by the use of both classical methods, including Claisen – Schmidt condensation, oxidative dehydrogenation and Baker – Venkataraman rearrangement, and modern synthetic approaches, such as metal-catalyzed annulation reactions, for their preparation. These approaches allow to produce flavones with a diverse peripheral environment, which determines their ability to undergo chemical transformations and indicates the promising use of such compounds as polyfunctional reagents in organic synthesis. The most expected transformations of flavones are reactions of C – H functionalization and C – C coupling, O(N)-alkylation and -acylation, and reactions with mono- and dinucleophiles. It should be noted, however, that fluorine-containing flavones are prone to nucleophilic aromatic substitution and heterocycle opening reactions. The undoubted practical relevance of flavones lies not only in the development of biologically active substances with a broad spectrum of activity, but also in the development of materials with unique photophysical properties. The great applied potential of flavones should stimulate further research in this field of chemistry.
3.4. Heterocyclic colchicinoids and prodrug forms based on them
Colchicine (3.4.1), a natural alkaloid first isolated from the leaves of meadow saffron (Colchicum autumnale), has a broad range of biological activity[2135, 2136]. In modern clinical practice, this compound is used to treat acute gout, familial Mediterranean fever, Behcet’s syndrome, scleroderma, amyloidosis and chondrocalcinosis[2137-2142]. Numerous studies demonstrated the efficacy of colchicine for the treatment of allergic, chronic infectious, autoimmune, cardiovascular and neurodegenerative diseases[2143-2146]. Colchicine was the first to be described as an antimitotic agent. It inhibits polymerization of tubulin[2147, 2148], an intracellular protein present in all eukaryotic cells[2149]. Polymerization of α- and β-tubulin heterodimers is necessary for the formation of microtubules of the cytoskeleton, which are main components of the mitotic spindle of cell division. By interacting with the binding site located at the boundary between tubulin α- and β-heterodimers, colchicines form a complex with tubulin, and this complex prevents the correct assembly of microtubules and, hence, violates the formation of spindle apparatus, which arrests the cell cycle and induces apoptosis in proliferating cells[2150-2154]. Due to the non-selective action of colchicine and the presence of the target protein in all cells of the body, colchicine is toxic not only to tumour tissues, but also to normal tissues, which is responsible for severe side effects. This non-specific toxicity, particularly neuro- and cardiotoxicity, restricts the use of high doses of colchicine in clinical practice[2155, 2156].
Compound 3.4.1 is a tricyclic fused molecule composed of two seven-membered and one six-membered rings with planar and axial types of chirality[2157]. A study of the structure — activity relationships demonstrated that natural colchicine has the aR,7S-configuration, and the affinity of this stereoisomer for tubulin exceeds those of synthetic colchicines with different configurations[2158]. It was shown that the presence of methoxy groups (three in ring А and one in ring С) is necessary for binding to tubulin; note that any modification of ring А leads to almost complete loss of activity, whereas modifications of rings В and С are quite acceptable (Scheme 770). For a colchicinoid to be recognized by Р-glycoprotein, which is responsible for the development of multiple drug resistance, a nitrogen atom should be present at С(7) in ring В as either free (or substituted) amino group or an amide moiety. When there is no nitrogen atom in this position, colchicinoid can no longer function as a substrate for P-glycoprotein[2159].

There are numerous published studies aimed at decreasing the toxic side effects of colchicine, extending the range of its biological activity and preparing its structural analogues to overcome the multiple drug resistance of cancer cells to colchicine. Colchicinoids resulting from various structural modifications, in particular, of rings B and C are the subjects of review publications[2136, 2160]. A review by Ghawanmeh et al.[2161] published in 2018 addresses new colchicine-based conjugates, prodrug forms and nanoparticle systems for colchicine delivery.
This part of the review considers colchicine derivatives containing heterocyclic moieties and colchicine-based prodrug forms reported mainly between 2012 and 2023.
3.4.1. Nitrogen-containing colchicine derivatives
Structural analogues of colchicine such as allocolchicine (3.4.2)[2162], combretastatin A-4 (CA-4) (3.4.3)[2163-2165] and 4-arylcoumarins (e.g., compound 3.4.4)[2166, 2167], are ligands for the colchicine site of tubulin, possessing antiproliferative activity[2168-2171]. Biological activity assays show that two non-coplanar polymethoxy-substituted aromatic syn-rings А and С located at a definite distance from each other represent the key plarmacophore moiety of these compounds[2166].

It was shown that replacement of 3-hydroxy-4-methoxyphenyl moiety in molecules 3.4.3 and 3.4.4 with hydrophobic 1-methyl-1H-indol-5-yl moiety (compounds 3.4.5 and 3.4.6, Refs[2172] and [2173], respectively) leads to inhibition of the assembly of microtubules in vitro in the nanomolar concentration range. In 2012, the first synthetic heterocyclic allocolchicinoids 3.4.7a,b, containing a pyrrole moiety fused to ring C, were obtained[2174].

The target compounds 3.4.7a,b were synthesized starting from 3,4,5-trimethoxyphenylpropionic acid (3.4.8a) and 5-indolylboronic acid pinacol ester 3.4.9a (Scheme 771). The key steps of the reaction were the Suzuki – Miyaura cross-coupling, which gave biaryl 3.4.10a, and the subsequent intramolecular Friedel – Crafts cyclization of acyl chloride 3.4.11a, formed in situ induced by the Ghosez reagent[2175]. Modification of the С(7)-keto group in compounds 3.4.12a,b gave a number of racemic pyrrole-containing tetracyclic derivatives: alcohols 3.4.13a,b, azides 3.4.14a,b and acetamides 3.4.7a,b for antitumour activity assays. Compounds 3.4.12a,b and their hydroxy analogues 3.4.13a,b showed a high cytotoxic activity in vitro against Burkitt’s lymphoma cells (BJAB) and low non-specific toxicity. Analogous properties were found[2176] for compounds 3.4.12с and 3.4.13c obtained in 2015, which are regioisomers of molecules 3.4.12a,b and 3.4.13a,b (see Scheme 771). The derivatives containing azide (3.4.14c) and acetamide (3.4.7c) groups were the least active among the series of studied compounds.

In 2014, a method for the synthesis of colchicinoid isomers 3.4.7d,e with an inverted orientation of the N-methylindole moiety was proposed[2177]. In this case, the seven-membered ring was formed by the Pd-catalyzed intramolecular C – H arylation of intermediates 3.4.15a,b (Scheme 772). Both intermediates were prepared from commercially available acid 3.4.8a and 1-methyl-1H-indole 3.4.9c by the Weinreb ketone synthesis and Friedel – Crafts acylation, respectively. Compounds 3.4.12d,e and their modified analogues proved to be several orders of magnitude less cytotoxic than their isomers obtained previously.

Later, conceptually new approaches to the synthesis of non-racemic pyrroloallocolchicinoids from commercially available (–)-(aR,7S)-colchicine 3.4.1 were developed (Scheme 773); in this case, the configuration of the C(7) stereocentre was retained. One of these approaches is based on the synthesis of aniline 3.4.16 or its bromo derivative 3.4.17. Compound 3.4.17 was converted to pyrroloallocolchicinoids 3.4.18 in three steps, with the key steps being the tandem Sonogashira cross-coupling and the subsequent Larock 5-endo-dig-cyclization[2178, 2179]. According to another synthetic protocol, compounds 3.4.19 were obtained in three steps[2180], which included Ullmann amination and formation of the indole moiety by the Fischer synthesis. Among these pyrroloallocolchicinoids, N-methylated derivative 3.4.18b showed the highest cytotoxicity (in the nanomolar concentration range) and more pronounced apoptosis-inducing activity compared to colchicine 3.4.1 and non-methylated analogues 3.4.18a, 3.4.19a and 3.4.19'a.

Derivative 3.4.20 with expanded (eight-membered) ring В was obtained for the first time from thiocolchicine 3.4.21 in two steps using the Beckmann rearrangement (Scheme 774, reaction а)[2181].

Subsequently, Seitz and co-workers[2182] synthesized heterocyclic compounds based on colchicines containing an eight-membered ring with a nitrogen atom. The synthesis included the preparation of deacetylated colchinol О-methyl ether 3.4.22, the biomimetic transamination of which afforded ketone 3.4.23 as a racemic mixture of atropoisomers. The reaction of compound 3.4.23 with hydroxylamine hydrochloride and the subsequent Beckmann rearrangement furnished a mixture of lactams 3.4.24 and 3.4.25. The reduction of isomer 3.4.24 followed by acetylation of unstable intermediate amine 3.4.26 yielded product 3.4.27. Derivatives 3.4.24 and 3.4.27 showed a moderate cytotoxic activity against MCF-7 breast cancer cells; this may be attributable to the structural rigidity of these molecules, which does not allow them to assume the conformation necessary to bind to tubulin.
Apart from compounds 3.4.24, 3.4.25 and 3.4.27, Bergemann et al.[2182] obtained allocolchicinoids 3.4.28 – 3.4.31 with nitrogen-containing rings fused to ring B of colchicine (Scheme 775). The antiproliferative activity of compounds 3.4.29 and 3.4.30 proved to be comparable with that of colchicine 3.4.1 and (–)-N-acetylcolchinol О-methyl ether (NCME, 3.4.32) and to exceed the cytotoxicity of tetrazole derivatives 3.4.30 and 3.4.31, of which thiocolchicinoid 3.4.31 was an order of magnitude more active.

In 1998, by condensation of the colchicine tosylate derivative 3.4.33 with benzamidine or guanidine, Cavazza and Pietra[2183] synthesized colchicinoid 3.4.34 (Scheme 776). Five years later, Danieli et al.[2184] reported a two-step synthesis involving diazo intermediate 3.4.35 to give new tetracyclic colchicinoid 3.4.36, which possessed a cytotoxic activity comparable to that of paclitaxel.

Over the last two decades, a large number of methods have been developed for the preparation of arynes and their precursors, which are valuable intermediates in the synthesis of various carbo- and heterocyclic compounds[2185]. A versatile synthetic platform for the preparation of a wide range of heterocyclic colchicine derivatives was developed in 2023 by Professor A.Yu.Fedorov and co-workers[2186]. This strategy was based on the generation of aryne intermediate, which had the same chirality centre and axis as the starting colchicine 3.4.1 and can be regarded as a highly reactive chiral building block. Precursors 3.4.37а–c obtained from iodocolchinol 3.4.38 generated in situ the desired aryne, which served as the substrate for insertion and [3 + 2]- and [4 + 2]-cycloaddition reactions, giving non-racemic allocolchicinoids 3.4.39 – 3.4.42 (Scheme 777)[2187]. Biological assays using Colo357, OSA and Raji cancer cells showed that these colchicinoids have a moderate cytotoxic activity in the 4 – 20 μM range.

3.4.2. Oxygen-containing colchicine derivatives
In 2015, the first non-racemic furanoallocolchicinoids 3.4.43 were synthesized[2188] in three steps from natural colchicine 3.4.1 (Scheme 778). First, the authors prepared iodocolchinol 3.4.38, which was then subjected to Pd-catalyzed cross-coupling with alkynyl derivatives and intramolecular Larock cyclization.

Compounds 3.4.43a,b, containing a hydroxyl group in the benzyl position of the furan ring, showed cytotoxicity in the nanomolar concentration range against epithelial and lymphoid cells. Colchicinoid 3.4.43a had a pronounced inhibitory effect on tubulin polymerization. The molar ratio of the inhibitor to the protein needed to inhibit the protein activity by 50% (R) is 0.08; R = 0.3 for compound 3.4.43b, R = 0.38 for colchicine 3.4.1, and R = 0.09 for combretastatin A-4 (3.4.3). Experiments in vivo demonstrated that the most active furanoallocolchicinoid 3.4.43a effectively inhibits the growth of Wnt-1 breast tumour without causing weight loss of mice, neurological symptoms or lethal outcomes in experimental animals. The high activity of these derivatives may be attributed to their ability to covalently bind to the cysteine residue in the tubulin binding site to form compounds like 3.4.44[2189].
In 2017, Gracheva et al.[2190] reported new furanoallocolchicinoids 3.4.45 (see Scheme 778), which contain a hydroxyl moiety in the benzyl and pseudobenzyl positions. These products, which are obtained in nine steps from colchicine, showed in vitro cytotoxicity in the nanomolar concentration range, induce cell cycle arrest in the G2/M phase, and also cause hyperpolarization of mitochondrial and lysosomal membranes in cells.
While studying bifunctional derivatives 3.4.46а – с, Gracheva et al.[2191, 2192] found that the charge might have an effect on the cytotoxic activity of molecules. For example, the antiproliferative activity of compounds 3.4.46a,c with primary amino groups is lower by 1.5 orders of magnitude than that of hydroxyl-containing analogue 3.4.45b (Table 10). Acid 3.4.47a and amino acid 3.4.47b are much less active (by two or three orders of magnitude) than derivative 3.4.46b.

The presented data confirm the necessity of acylation of the C(7)-amino group and correlate with the cytotoxicity evaluation for the previously synthesized indolo- and furano-allocolchicinoids, for which the presence of free amino group decreased the activity at least 10-fold. Presumably, derivatives with functional groups that can be protonated or deprotonated under physiological conditions are less active, because they are selectively accumulated in certain cell organelles such as lysosomes and mitochondria.
A promising way of colchicine modification is the synthesis of fused heterocyclic derivatives involving a tropolone moiety. Thus the [8 + 2]-cycloaddition of diethyl malonate to colchicine in the presence of sodium affords compound 3.4.48[2193], containing a fused furan ring (Scheme 779, pathway a). The same compound was prepared by condensation of tosylate derivatives of colchicine with diethyl malonate in the presence of sodium methoxide (see Scheme 779, conditions b)[2194]. In 2021, Komogortsev et al.[2195] reported the development of the first one-pot synthesis of substituted furanocolchicinoids 3.4.49 by a multicomponent reaction of colchiceine 3.4.50, arylglyoxals 3.4.51 and Meldrum’s acid 3.4.52 (see Scheme 779).

In 2020, dihydrofuranoallocolchicinoids were described for the first time[2196] (Scheme 780). For the synthesis of these compounds, iodocolchinol 3.4.38, formed from colchicine, was alkylated with the appropriate allyl bromide. The subsequent intramolecular Heck reaction led to the target product 3.4.53 containing a 2,3-dihydrofuran ring. It was shown that colchicinoids 3.4.54 undergo acid-catalyzed isomerization to the corresponding benzofuran derivatives 3.4.55. Dihydrofuranoallocolchicinoid 3.4.54а and its fluorinated analogue 3.4.54d were cytotoxic in the picomolar concentration range against all cancer cell lines studied; therefore, these compounds can be considered to be the most cytotoxic currently known colchicinoids. It was found that the size of substituents at the exocyclic double bond in the dihydrofuran moiety is an important factor influencing the activity of colchicinoids. Colchicinoid 3.4.54a inhibits the tubulin polymerization more efficiently than colchicine 3.4.1 and combretastatin 3.4.3, while acting in sub-stoichiometric amounts. Experiments in vivo demonstrated a considerable decrease in the acute toxicity of compounds 3.4.54a,d compared to that of colchicine [the half-lethal dose (LD50) was ≥ 10 mg kg–1 for derivative 3.4.54a and 1.6 mg kg–1 for colchicine upon intravenous injection].

The benzo[b]oxepine moiety is often encountered in natural compounds such as pterulone[2197], radulanins, heliannuols, and their analogues[2198-2200]. These compounds have a wide range of biological activity. Baudoin and co-workers[2201] demonstrated that substituted dibenzo[c,e]oxepine colchicinoids can also be biologically active, which was reflected in a number of publications devoted to the synthesis of colchicine analogues with seven-membered O-containing rings.
In 2008, Professor Schmalz and co-workers[2202] reported the development of a new method for the synthesis of 6-oxaallocolchicinoids 3.4.56 using microwave-induced intramolecular Co- or Rh-catalyzed [2 + 2 + 2]-cycloaddition as the key step. Some of the obtained products had apoptosis-inducing activity against BJAB tumour cells. Subsequently, the same research group used the same reaction to prepare the first compounds 3.4.57[2203] containing a nitrogen atom in ring С. In 2012, Termath et al.[2204] described the synthesis of colchicinoid 3.4.58 by Rh-catalyzed intramolecular [5 + 2]-cycloaddition. Interestingly, whereas the cytotoxicity of colchicinoid 3.4.58 was moderate (IC50 = 1 – 10 μM against the BJAB cell line), the combination of this compound with doxorubicin significantly enhanced the cytostatic action of the latter against resistant Nalm6 cells (synergistic effect).

Wallace and co-workers[2205] synthesized a number of heterocyclic colchicine derivatives using Cu-catalyzed Ullmann cross-coupling as the key step. The most promising biological properties were found for compound 3.4.59, for which characteristics of the tubulin polymerization inhibition were comparable with those for combretastatin A-4 (3.4.3). The authors suggested that in the absence of substituents at C(1), C(5) and C(7) atoms, the molecule has a low rotation barrier along the axial axis and, hence, it can easily adopt a configuration similar to that of colchicine (3.4.1) and combretastatin (3.4.3) (aR), which is needed to bind to tubulin.
A number of diastereomeric O-containing colchicinoids 3.4.60 were obtained by Professor A.Yu.Fedorov and co-workers[2206]. In this case, the Au-catalyzed cyclization in the presence of nucleophiles was the key reaction. Compounds 3.4.60 had a moderate cytotoxicity against human tumour cell lines (HEK293, PANC-1, Colo-357 and HeLa) and murine cancer cells (Colon 26), which may be attributable to the fact that the binding geometry of these derivatives to tubulin differs from that of colchicine. Using molecular docking, it was shown that the bulky benzoxepine moiety precludes the position of the molecule characteristic of colchicine, which disrupts polar and hydrophobic interactions and may account for the decreased activity of these compounds.
3.4.3. Other heterocyclic colchicinoids
The natural alkaloid cornigerine 3.4.61, isolated from the plant Colchicum cornigerum, is an analogue of colchicine in which the vicinal oxygen atoms in positions 2 and 3 are linked by a methylene bridge. It was found that cornigerine has a biological activity similar to that of colchicine: in particular, it inhibits tubulin polymerization and cause the metaphase arrest in L1210 leukaemia cells[2207].

In 2010, Miller and co-workers[2208] reported the synthesis of heterocyclic regioisomeric colchicinoids 3.4.62a,b by the Diels – Alder reaction of compound 3.4.1 and 2-nitrosopyridines (Scheme 781). The activities of the major isomers of 3.4.62a against PC-3 and MCF-7 cell lines were similar to those of parent colchicine. The authors demonstrated that this activity is due to the fact that these compounds act as prodrugs; when injected into the body, they release colchicine as a result of the retro-Diels – Alder reaction. It was noted that the introduction of N,O-heterocycle changes the structural conformation of the molecule and thus affects the ability of colchicine to induce tubulin polymerization, which is thus almost completely inhibited.

Yang et al.[2209] obtained a series of colchicinoids in which the methoxy group of the tropolone ring was replaced with a cyclic amine such as piperidine (3.4.63), 1-methylpiperazine (3.4.64) or pyrrolidine (3.4.65). Analysis of the cytotoxicity of these compounds showed that this replacement markedly decreases the antiproliferative activity of colchicinoids to micromolar concentrations, thus confirming the significance of the methoxy group in ring C for the cytotoxic properties of colchicine and its derivatives.

3.4.4. Prodrugs based on heterocyclic colchicinoids
One of the ways to decrease the total toxicity of drugs and to improve their ADMET characteristics (ADMET is Absorption, Distribution, Metabolism, Excretion, Toxicity.) is to develop prodrug forms and to encapsulate the agents into nanosized delivery systems[2210-2212]. The development of prodrugs that can significantly expand the therapeutic potential of a drug is based on the synthesis of drug conjugates with a wide range of macromolecules[2213-2219], including antibodies, polysaccharides, lectins, serum proteins, peptides, growth factors and synthetic polymers.
The natural polysaccharide chitosan is widely used for drug encapsulation owing to a number of benefits such as biodegradability, low toxicity and large number of amino groups used to modify the nanoparticle surface[2220]. In 2016, furanoallocolchicine 3.4.43a was used to prepare[2221] chitosan conjugate 3.4.66 with a molecular weight of 40 kDa. Compounds 3.4.43a and 3.4.66 induce depolymerization of tubulin, disrupt the cell cycle and inhibit cell proliferation. Experiments in vivo using Wnt-1 breast cancer model showed that conjugate 3.4.66 inhibits the tumour growth much more efficiently than parent compound 3.4.43a; probably, this is due to better accumulation in the tumour owing to the EPR effect (EPR is enhanced permeability and retention)[2222].

This study was continued by the synthesis of fluorescent chitosan conjugate 3.4.67 with colchicinoids, fabrication of nanoparticles based on this conjugate and study of their biodistribution in the tissues and organs of mice[2223].

It was shown that increase in the diameter of nanoparticles up to 450 nm significantly affects their biodistribution, particularly, leads to drug accumulation in liver tissues. The accumulation in kidneys and lungs was low and that in spleen was higher. It was also found that conjugation of colchicinoids with chitosan decreases the gastro- and cardiotoxicity.
Liposomes are a convenient platform for the fabrication of injectable compositions for the systemic administration of hydrophobic compounds[2224]. It is known that colchicine 3.4.1, a neutral lipophilic molecule, is mainly located in the lipid bilayer; however, it tends to be displaced into the aqueous phase and to form layered aggregates[2225]. In order to retain colchicine in the lipid bilayer, it was proposed to prepare colchicine-based lipophilic prodrug forms 3.4.68 and 3.4.69[2224], which contain oleic and palmitic acid residues, respectively, and release intact colchicinoid 3.4.70 when injected into the body.
Analysis of the antiproliferative activity of these compounds against Burkitt’s lymphoma cells showed that the triazole derivative 3.4.70 has an activity similar to that of colchicine 3.4.1 (IC50 = 25 and 20 nM, respectively), while colchicinoid esters 3.4.68 and 3.4.69 (IC50 = 4 and 6 nM) are even more active than colchicine 3.4.1. This can be explained by the facilitated transport of amphiphilic compounds 3.4.68 and 3.4.69 across the plasmatic membrane followed by fast intracellular hydrolysis and transport to the tubulin molecule.

Prodrug forms 3.4.68 and 3.4.69 were encapsulated into liposomes based on natural phospholipids and were tested for the cytotoxic activity against a panel of four human cancer cell lines (HBL-100, K562, HaCaT and Jurkat). It was shown that these liposomal compositions retain the biological activity mainly against human epithelial tumour cells.
In 2015, Sitnikov et al.[2226] synthesized a series of lipophilic colchicine derivatives (Scheme 782). Colchicinoid 3.4.71 was used as the starting compound. First, it was reduced with NaBH4 to alcohol 3.4.72 and then dehalogenated; intermediate 3.4.73 was subjected to the Mitsunobu reaction to give azide 3.4.74. The subsequent dipolar [3 + 2]-cycloaddition to terminal alkynes led to 7-(triazol-1-yl)pyrroloallocolchicinoids 3.4.75a – f in 45 – 88% yields (Table 11).
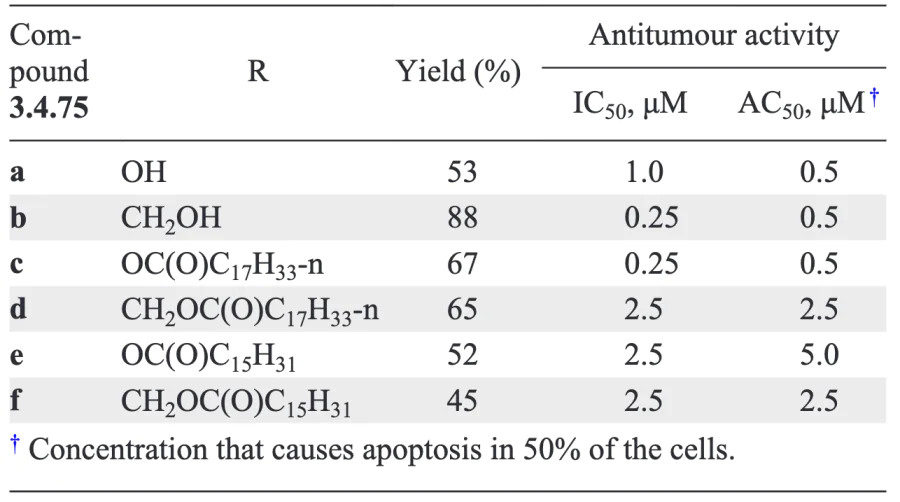

The antitumour activity of allocolchicinoids 3.4.75a – f was studied using Nalm6 human leukaemia cell line. It was shown that derivative 3.4.75с, which contains an oleic acid residue, efficiently inhibits the division of Nalm6 cells and has a high apoptosis-inducing activity (see Table 11).
Liposomes can be designed is such a way as to release the active compound at the lesion site under the action of an endogenous or exogenous trigger (change in the temperature or pH, magnetic treatment, irradiation, enzymatic cleavage, etc.). In particular, enzyme-sensitive liposomes can release the drug in pathological tissues with considerably elevated enzyme levels[2227, 2228].
In 2019, Shchegravina et al.[2229] reported the development of a new phospholipid prodrug (Scheme 783) encapsulated into enzyme-sensitive nanosized liposomes (90 – 110 nm size) based on natural phosphatidylcholine. First, colchicine (3.4.1) was converted in ten steps via intermediates 3.4.76 and 3.4.77 to active colchicinoid 3.4.78[2230]. According to the authors, under the action of increased level of phospholipase А2, which occurs in inflammation and metastasis sites[2231], hydrolysis of the phospholipid conjugate 3.4.78 results in the initial release of acid 3.4.79, which is then hydrolyzed by non-specific esterases to give colchicinoid 3.4.77 (Scheme 784). Thus, stepwise release of the active agent at the site of pathology is attained.

Analysis of the cytotoxicity of compounds 3.4.77 and 3.4.78 showed that lipid prodrug 3.4.78 is 2 – 3 orders of magnitude less active than parent colchicinoid 3.4.77, which is in good agreement with published data[2224]. Meanwhile, the cytotoxic activity of the liposomal agent (IC50 = 8 – 119 nM against PANC-1 and HaCaT cells) proved to be comparable with the activity of the starting compound. This can be attributed to fast penetration of the contents of liposomes into the cell due to facilitated endocytosis or fusion with the cell membrane, which, in turn, promotes the fast release of the active component[2232]. In order to increase the blood circulation time and storage life of liposomes that contain colchicinoid 3.4.77 and to increase the amount of loaded active component, the composition of the liposomal agent was optimized[2233]; in particular, it was proposed to incorporate cholesterol as a stabilizing agent into liposomes[2234].

The next step towards increasing the stability of conjugates and increasing the circulation time of nanoparticles was a change in their design. In 2023, a new lipophilic pH-sensitive prodrug 3.4.81 was developed on the basis of active colchicinoid 3.4.80a (Scheme 785)[2235]. Since one of the possible ways of liposome uptake by cells is endocytosis with subsequent formation of endosomes and then lysosomes with pH 5.5 – 4.5, it was reasonable to pay attention to introduction of a pH-sensitive group into the prodrugs[2236]. It is known that the tumour microenvironment is characterized as a hypoxic acidic area due to lactate overproduction by tumour cells with disrupted metabolism. Therefore, the introduction into the molecule of a group sensitive to pH decrease in the tumour microenvironment and in some cell organelles may also facilitate drug release after liposome accumulation in the tumour[2237].

Active colchicinoid 3.4.80a (see Scheme 785) was prepared from substrate 3.4.1 in eight steps in an overall yield of 28%. A diacylglycerol derivative was chosen as the lipid part of prodrug 3.4.81, while the polar tetraethylene glycol residue, which effectively interacts with the aqueous medium and mimics the phosphocholine group in natural phosphatidylcholine, allows the drug to be completely embedded into the lipid bilayer. An oxime moiety was used as the pH-sensitive group.
Analysis of the in vitro activity of conjugate 3.4.81 showed that this prodrug is three orders of magnitude less cytotoxic than the initial colchicinoid 3.4.80a. This attests to higher stability of the new conjugate 3.4.81, unlike prodrugs described previously[2229], and makes it possible to achieve a controlled release of the active drug substance. The hydrolytic stability of conjugate 3.4.81 at different pH was also studied and the pattern of variation of the rate of prodrug hydrolysis depending on the product composition was identified. It is important to note that elucidation of these trends may be useful for the development of delivery systems for antitumour agents of different classes.
The chemistry of colchicine and its derivatives has been actively developing since the second half of the 20th century. Currently, this compound is used in clinical practice as a therapeutic agent to treat gout, Mediterranean fever and some other inflammatory processes. A considerable beneficial effect of colchicine in amyloidosis, scleroderma and similar autoimmune disorders has been noted. Owing to high antimitotic activity, colchicine is of interest as an agent for the therapy of cancer; however, high systemic toxicity, low bioavailability and narrow therapeutic window limit the use of unmodified colchicine as an antitumour drug. Therefore, numerous studies are devoted to possible modifications of natural colchicine, evaluation of biological properties and analysis of the action mechanism of colchicine-based drugs. A few versatile approaches to the synthesis of numerous heterocyclic analogues of colchicine have been developed. Among these derivatives are pyrrolo- and furanoallocolchicines, which have cytotoxic activity in the nanomolar concentration range and a pronounced ability to induce apoptosis. Simultaneously with the search for new heterocyclic derivatives based on lead compounds, nanosized drug delivery systems were prepared; this made it possible to reduce the non-specific toxic effects of colchicinoids and to provide controlled release of the active agent. In general, the synthetic solutions found in this research area and the identified structure — activity and structure — property relationships are universal and can be successfully applied in various fields of organic and medicinal chemistry.
4. Conclusion
In the 21st century, chemistry of heterocyclic compounds continues to be among the most important and actively developing fields of organic chemistry. The appearance of heteroatoms in the aromatic ring multiplies the variety of possible molecules, the range of their chemical, physical and biological properties, as well as synthetic options both for ring construction and for use in further transformations.
The immensity of the subject of this review precludes the possibility of covering all of the results obtained in this field in recent years without exception, even if we limit ourselves to the works of Russian scientists only. Nevertheless, by considering studies performed by some top research groups of Russia, the review touches upon almost all the main trends in the chemistry of heterocycles; therefore, this collective work reflects quite adequately the current state of this field in the world science.
The development of synthetic strategies towards heterocyclic compounds, including heterocyclization reactions and functionalization of heterocycles already present in the molecule, is a multifaceted problem of fundamental importance for medicinal chemistry and chemistry of advanced functional materials. It is commonly known that heterocycles are frequently encountered and often play a crucial role in the molecules of pharmaceutical agents (in particular, for photodynamic therapy), agents for diagnostics and theranostics, biologically active additives, pesticides and plant growth and development regulators. In recent years, heterocyclic compounds have been actively used in advanced functional materials. Here one can list energetic compounds, materials for organic electronics (conductors, semiconductors), luminescent materials (in particular, for OLED devices), materials for sensors, photonics and photovoltaics (in particular, for organic solar cells).
The strategy of synthesis and transformations of heterocycles is not only a tool for obtaining the above practically important and vital drugs and materials, but also still pertains to fundamental science. The relevant issues are predicting the reactivity and physicochemical characteristics of heterocyclic systems and increasing their synthetic accessibility. The studies presented in this review show that even the chemistry of seemingly well-known five- and six-membered nitrogen-, oxygen- and sulfur-containing heterocyclic systems remains an inexhaustible field for discoveries. A relevant and promising trend is the search for new atom-economic approaches to self-assembly of heterocyclic systems based on multicomponent reactions that would require small numbers of steps. Novel unique heterocyclic systems are being designed, including supramolecular, oligo- and polymeric systems. In addition, Russian chemists made a significant contribution to the development of unusual heterocyclic systems such as phosphorus- and iodine-containing heterocycles, strained azirines and heterocycloalkynes and cyclic organic peroxides.
A special position belongs to the chemistry of fluorinated heterocycles, since fluorine-containing substituents have a considerable influence on the chemical and physicochemical properties of molecules. On the one hand, this accounts for extensive use of organofluorine compounds and, on the other hand, requires special synthetic approaches, which often differ from those used for other halogens. Thus, in the synthesis of fluorine-containing products of fine organic synthesis, direct fluorination of complex molecules is rarely used. As a rule, a more effective approach is the use of fluorine-containing building blocks.
One more relevant line of research of global chemical science, which was reflected in the works of Russian scientists, is the development of approaches to CH-functionalization, which are expected to open up new short and low-waste pathways to complex target structures for chemical engineering. Among the new CH-functionalization reactions implemented in the practice of organic synthesis, note nucleophilic substitution of hydrogen (SNH), redox reactions involving single-electron transfer and radical species and metal-catalyzed activation of aromatic C – H bonds.
Generally, the Russian heterocyclic chemistry appears to be highly integrated into the world science, which is manifested as the significant contribution made by Russian chemists into almost any key area of active development of this field.
5. Acknowledgements
Chapter 2.1 ʻSNH-reactions in the synthesis of fused heteroaromatic systemsʼ by the authors V.N.Charushin, E.V.Verbitskiy, O.N.Chupakhin supported by the State Task (No. 124020200072-0).
Chapter 2.2 ʻMetal-catalyzed activation of aromatic С – Н bonds in the synthesis of polyfunctional heterocyclesʼ by the authors D.V.Vorobyeva, P.S.Gribanov, S.N.Osipov supported by the RSF (17-73-30036 and 21-13-00328) and the Ministry of Science and Higher Education of the Russian Federation (Contract No. 075-00277-24-00).
Chapter 2.3 ʻFunctionalization of pyrroles to design annulated N-heterocycles: how and why?ʼ by the authors A.V.Ivanov, S.V.Martynovskaya, E.F.Sagitova supported by the State Task No. 122041100031-5.
Chapter 2.4 ʻMulticomponent reactions in the synthesis of N,O,S,Se-containing heterocyclic systemsʼ by the authors V.D.Dyachenko, I.V.Dyachenko, S.G.Krivokolylsko, V.V.Dotsenko supported by the grants VGEA-2023-0006 (No. 1023061300025-91.4.1), VGEA-2024-0004 (No. 1023082100012-41.4.1), and by the State Task FREE-2023-0002 (No. 1023030900103-8-1.4.1).
Chapter 2.5 ʻFrom fundamental indole chemistry to compounds with high biological activityʼ by the authors A.V.Aksenov, D.A.Aksenov, N.A.Aksenov supported by the Ministry of Science and Higher Education of the Russian Federation (FSRN-2023-0005).
Chapter 2.7 ʻSynthesis of fluorine-containing heterocyclic compounds based on fluorinated alkenes and acetylenesʼ by the authors V.M.Muzalevskiy, V.G.Nenajdenko supported by the grant RSF No. 23-7300014.
Chapter 2.10 ʻCyclizations of functionalized acetylenes and diacetylenes for the synthesis of heterocyclesʼ by the authors I.A.Balova, N.A.Danilkina, A.I.Govdi supported by the grant RSF No. 19-73-10077-П.
Chapter 2.12 ʻFunctionalized 2H-azirines as building blocks for the synthesis of heterocyclesʼ by the authors M.S.Novikov, N.V.Rostovskii, A.F.Khlebnikov supported by the grant RSF No. 23-13-00115.
Chapter 2.13 ʻ1,2-Alkyl shift in the construction of heterocyclic systemsʼ by the authors Yu.N.Klimochkin, M.V.Leonova, I.M.Tkachenko supported by the grant RSF No 21-73-20103 and by the Ministry of Science and Higher Education of the Russian Federation (FSSE-2023-0003).
Chapter 2.15 ʻLinearly conjugated, annulated and branched thiophene-containing oligomers for organic electronics, optoelectronics and photonicsʼ by the authors O.V.Borshchev, Yu.N.Luponosov and S.A.Ponomarenko supported by the State Task FFSM-2024-0003 (sections 2.15.1 and 2.15.4 on materials for electronics, photonics and optoelectronics) and grant RSF 19-73-30028 (sections 2.15.2 and 2.15.3 on materials for sensorics and bioelectronics).
Chapter 2.16 ʻHeterocyclic thiophen-containing conjugated donor–acceptor–donor systems: synthesis, structure–properties relationshipʼ by the authors A.S.Fisyuk, A.S.Kostyuchenko supported by the grant RSF No. 20-73-10043.
Chapter 2.17 ʻAdvances in the synthesis of heterocycles based on reactions of thioamides with azides, carbenoids and their precursorsʼ by the authors V.G.Ilkin, T.V.Beryozkina, V.A.Bakulev supported by the grant RSF No. 23-13-00248.
Chapter 2.18. ʻPhosphorus-containing heterocycles: synthesis, coordination chemistry and applicationsʼ by the authors A.S.Gazizov, A.A.Zagidullin, A.A.Karasik supported by the State Task No. 122011800132-5.
Chapter 2.20 ʻDomino reactions in the synthesis of nitrogen-containing heterocyclesʼ by the authors N.E.Golantsov, A.A.Festa, L.G.Voskresensky supported by the grant RSF No. 24-13-00132.
Chapter 2.21 ʻ5-Aryloxazolidines: synthesis and chemical propertiesʼ by the authors V.S.Moshkin, E.M.Buev, V.Ya.Sosnovskikh supported by the grant RSF No. 22-73-10008.
Chapter 2.22 ʻHypervalent iodine compounds: from preparation to applications for the synthesis of heterocyclesʼ by the authors I.A.Mironova, P.S.Postnikov, V.V.Zhdankin, M.S.Yusubov supported by the grant RSF No. 21-73-20031.
Chapter 2.23 ʻSynthesis and transformations of cyclic peroxidesʼ by the authors I.A.Yaremenko, V.A.Vil’, I.B.Krylov, A.O.Terent’ev supported by the grant RSF No. 24-43-00111.
Chapter 2.24 ʻTetrapyrrole macroheterocycles for biomedical applicationsʼ by the authors Yu.G.Gorbunova, A.G.Martynov, A.Yu.Tsivadze supported by the grant RSF 24-13-00479 (https://rscf.ru/project/24-13-00479/).
Chapter 2.25 ʻNew heterocyclic analogues of phthalocyaninesʼ by the authors P.A.Stuzhin, S.S.Ivanova supported by the grant RSF 17-13-01522п and grant RFBR 21-53-26004.
Chapter 3.2 ʻMonoterpenoid heterocyclic compoundsʼ by the authors O.I.Yarovaya, K.P.Volcho, N.F.Salakhutdinov supported by the grant RSF 23-43-10019 (https://rscf.ru/ project/23-43-10019).
Chapter 3.3 ʻRecent advances in chemistry of flavonesʼ by the authors M.A.Panova, Ya.V.Burgart, V.I.Saloutin supported by the State Task FUWM-20240009 (№ 124020200038-6).
Chapter 3.4 ʻHeterocyclic colchicinoids and prodrug forms based on themʼ by the authors A.R.Sitdikova, E.S.Shchegravina, A.Yu.Fedorov supported by the Research Laboratory of Chemistry of Natural Compounds and Their Synthetic Analogues, established under the State Task at the Research Centre ʻTechnoplatform 2035ʼ (project FSWR-2024-0002).
6. List of abbreviations
Ea — activation energy,
Egel — electrochemical band gap,
E ox (onset) — oxidation potential,
E red (onset) — reduction potential,
E1/2Red — half-wave reduction potential,
fk — Fukui reactivity index,
ΔG — Gibbs free energy difference,
ΦΔ — singlet oxygen quantum yield
ΦF — fluorescence quantum yield,
λabs — absorption maximum wavelength,
λem — emission maximum wavelength,
μ — charge carrier mobility,
3CR — three-component reaction,
А — acceptor,
AA — amino acid moiety,
AC50 — concentration that causes apoptosis in 50% of the cells,
AcA — acetylacetylene,
асас — acetylacetonate,
AChE — acetylcholinesterase,
ACHN — 1,1'- azobis(cyclohexanecarbonitrile),
Ad — adamantyl,
AIBME — dimethyl 2,2′-azobis(2-methylpropionate),
AIBN — 1,1'-azobis(isobutyronitrile),
AIE — aggregation-induced emission,
All — allyl,
Am — amyl (pentyl),
An — anion,
ANRORC — addition of nucleophile, ring opening, ring closure,
Asc — ascorbate,
BACE-1− β-secretase enzyme,
BCP — bathocuproine,
BDD — boron-doped diamond anode
BI — benzimidazol-2-yl,
BINAP — 2,2′-Bis(diphenylphosphino)-1,1′-binaphthalene,
BiPy — 4,4'-bipyridine,
bmim — 1-n-butyl-3-methylimidazolium,
Boc — tert-buthoxycarbonyl,
B2Pin2 — Bis(pinacolato)diboron,
BSA — bovine serum albumine,
BT — benzothiophene,
BTBT — [1]benzothieno[3,2-b][1]benzothiophene,
BTD — 2,1,3-benzothiadiazole,
BV — The Baeyer – Villiger rearrangement,
Cat — catalyst,
Cbz — benzyloxycarbonyl,
CDI — carbonyldiimidazole,
CI — isocoumarin,
CK1 — casein kinase,
СМ-Phos — 2-[2-(dicyclohexylphosphino)phenyl]-1-methyl-1H-indole
cod — cycloocta-1,5-diene,
COX — cyclooxygenase,
Cp* — pentamethylcyclopentadienyl
Cp — cyclopentadienyl
CuAAC — copper(I)-catalyzed alkyne–azide cycloaddition,
CVA — cyclic voltammetry,
Cy — cyclohexyl,
Cz — corrolazine,
D — donor
DAB — 1,2-diaminebenzene,
DAT — dopamine transporter,
dba — dibenzylideneacetone,
DBN — 1,5-diazabicyclo[4.3.0]non-5-ene,
DBU — 1,8-diazabicyclo[5.4.0]undec-7-ene,
DC — dipolar cycloaddition,
DCB — 1,2-dichlorobenzene,
DCC — N,N′-dicyclohexylcarbodiimide
DCE — 1,2-dichloroethane,
DCM — dichloromethane,
DCV — dicyanovinyl,
DDQ — 2,3-dichloro-5,6-dicyano-1,4-benzoquinone,
Dec — decyl,
DEGOH — diethyleneglycol,
DFT — density functional theory,
DG — directing group,
DIAD — diisopropylazodicarboxylate,
DIPEA — N,N-Diisopropylethylamine,
DMA — N,N-dimethylacetamide,
DMAD — dimethyl acetylenedicarboxylate,
DMAP — 4-dimethylaminopyridine,
DNM — dinitromethane,
DPBTBT — 2,7-bis(4-decylphenyl)[1]benzo- thieno[3,2-b][1]benzothiophene,
dppe — 1,2-bis(diphenylphosphino)ethane,
dppf — 1,1’-bis(diphenylphosphino)ferrocene,
DPPH — 2,2-diphenyl-1-picrylhydrazyl,
dr — diastereomeric ratio,
E — electrophile,
EA — electron affinity,
EC50 — half maximal effective concentration,
EDA — ethyldiazoacetate,
EDCI — 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide,
EDG — electron-donating group,
ee — enantiomeric excess,
EPNC — electrophile-promoted nucleophilic cyclization,
EPR effect — enhanced permeability and retention effect,
esp — α,α,α′,α′-tetramethyl-1,3-benzenedipropionic acid,
ESPT — excited state intermolecular proton transfer,
EWG — electron-withdrawing group,
FAAH — fatty acid amide hydrolase,
FBTD — fluorinated benzothiadiazoles,
Fc — ferrocenyl,
FFAR1– free fatty acid receptor 1,
FG — functional group,
Flav — flavone residue,
FRET — Forster resonance energy transfer,
Fu — furyl,
GABA — γ-aminobutyric acid,
HBTU — hexafluorophosphate (benzotriazol-1-yl)tetramethyluronium,
HFIP — 1,1,1,3,3,3-hexafluoro-2-propanol,
HICs — hypervalent iodine compounds,
HMDS — hexamethyldisilazane,
HMX — octogen,
HOMO — the highest occupied molecular orbital,
HTL — hole-transporting layer,
ICT — intramolecular charge transfer,
ImH — imidazole,
IP — ionization potential,
IPTMDOB — 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane,
LD50 — median-lethal dose,
LOX — 5-lipoxygenase,
LP — lone pair,
LR — Lawesson’s reagent,
LUMO — the lowest occupied molecular orbital,
mCPBA — meta-chloroperoxybenzoic acid,
MDR — multiple drug resistance,
Ment — (–)-menthyl,
Mes — 2,4,6-trimethylphenyl (mesityl),
MIC — minimum inhibitory concentration,
MP — methyl propiolate,
MR — Meinwald rearrangement
Ms — methanesulfonyl (mesyl),
MS — molecular sieves,
MW — microwave radiation,
Naph — naphthyl,
NBS — N-bromosuccinimide,
NCS — N-chlorosuccinimide,
Nc — naphthalocyanine,
NFSI — N-fluorobenzenesulfonimide,
NHC — N-heterocyclic carbene catalyst,
NMM — N-methylmorpholine,
NMP — N-methylpyrrolidone,
oct — octanoate,
OFET — organic field-effect transistors,
OLED — organic light-emitting diodes,
OLET — organic light-emitting transistors,
OSC — organic solar cells,
PAH — polyaromatic hydrocarbon,
PAR — pyridylazoresorcinol,
Pc — phthalocyanine,
PDT — photodynamic therapy,
PEG — polyethylene glycol,
PET — photoinduced electron transfer,
Pg — protective group,
Phe — phenylalanine,
phen — phenanthroline,
PIDA — (diacetoxyiodo)benzene, phenyliodine(III) diacetate,
PIFA — (bis(trifluoroacetoxy)iodo)benzene, phenyliodine(III) bis(trifluoroacetate),
Piv — pivaloyl,
PivO — pivaloyl,
PLQY — photoluminescence quantum yield,
PMA — phosphomolybdic acid,
PMB — p-methoxybenzyl,
PMDETA — pentamethyldiethylenetriamine,
PMP — p-methoxyphenyl,
Por — porphyrin,
PPA — polyphosphoric acid
ppy — 2-phenylpyridine,
PS — proton sponge [1,8-bis(dimethylamino)naphthalene],
PSC — perovskite solar cells,
PTA — phosphotungstic acid,
PTP 1В — protein phosphotyrosine phosphatase 1B,
РТТР — 5,5'-diphenyl-2,2'-bithiophene,
РТРТР — (2,2'-(1,4-phenylene) bis(5-phenylthiophene),
Py — pyridine,
py — pyridyl,
PyBOP — benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate,
Pym — pyrimidyl,
PyrH — pyrazole,
PyzPz — pyrazinoporphyrazine,
QPz — quinoxalinoporphyrazine,
RDX — hexogen,
R — inhibitor to protein molar ratio that inhibits the protein activity by 50%,
RdRp — RNA-dependent RNA polymerase,
ROS — reactive oxygen species,
RSV — respiratory syncytial virus,
rr — regioisomeric ratio,
rt — room temperature,
SAMFET — self-assembling organic field-effect transistors,
SET — single electron transfer,
SI — selectivity index,
SNAr — nucleophilic aromatic substitution,
Solv — solvent,
SPAAC — strain-promoted azide-alkyne cycloaddition,
sPz — subporphyrazine,
TADF — thermally activated delayed fluorescence,
TBA — tetra-n-butylammonium,
TBAI — tetra-n-butylammonium iodide,
TBAT — tetra-n-butylammonium difluorosilicate,
TBDMS — tert-butyldimethylsilyl,
TBDPS — tert-butyldiphenylsilyl,
TBHP — tert-butyl hydroperoxide,
TBPA — tetra-n-butylphosphonium acetate,
TCB — 1,2,4-trichlorobenzene,
Tdp1– tyrosyl-DNA phosphodiesterase 1,
TEA — triethylamine;
TEBAC — benzyltriethylammonium chloride,
TEMPO — (2,2,6,6-tetramethylpiperidin-1-yl)oxyl,
Terp — monoterpene residue,
Tf — trifluoromethanesulfonyl (triflyl),
TFA — trifluoroacetic acid,
TFAA — trifluoroacetic anhydride,
tfacac — 1,1,1-trifluoroacetylacetonate,
TFE — trifluoroethanol,
Th — thienyl,
THD-Dipp — 1,3-bis(2,6-diisopropylphenyl)hexahydro-2H-1,3-diazepin-2-ylidene,
THP — tetrahydro-2H-pyran-2-yl,
tht — tetrahydrothiophene,
Tipp — 2,4,6-triisopropylphenyl,
TMS — trimethylsilyl,
TNE — trinitroethanol,
TOF — turnover number (number of catalytic cycles per second),
p-Tol — p-tolyl,
TON — catalyst turnover number,
Togni reagent II — 1-trifluoromethyl-1,2-benziodoxol-3(1H)-one,
TrzH — triazole,
Ts — p-toluenesulfonyl (tosyl),
TS — transition state,
ТТА — tetrathienoacene,
XPhos — 2-dicyclohexylphosphino-2',4',6'-triisopropylbiphenyl.
References











![Synthesis and In Vitro Anti-Inflammatory Activity of Pyrrolo[1,2-A]pyrazines via Pd-Catalyzed Intermolecular Cyclization Reaction](/storage/images/resized/lZMRu5MhhE0HtEBGlENti9uMMEivcrzFOV6rqpGN_small_thumb.webp)
![Systematic Investigation of Fluorescence Properties of Symmetric and Asymmetric Diazolo[1,2-a:2′,1′-c]quinoxaline Derivatives](/storage/images/resized/SovkqI6gX6to8o5XBnS9RNxEOYVicn7hatkMzrvl_small_thumb.webp)
![ОТ АЦИЛЭТИНИЛПИРРОЛОВ К ПИРРОЛО[1,2-A]ПИРАЗИНАМ В ОДНУ СТАДИЮ](/storage/images/resized/QvUjpflupmitGM5fuTKfMKbjgX15XAHq011OUdeD_small_thumb.webp)





![Empirical determination of the degree of analgesic activity of some new 3-aminothieno[2,3-b]pyridines and 1,4-dihydropyridines based on a complex criterion](/storage/images/resized/ZPhzCapNX6qOhB277C8wlZiq8W4lZyaxMEZIBIYQ_small_thumb.webp)
![Study of anti-inflammatory and antinociceptive properties of new derivatives of condensed 3-aminothieno[2,3-b]pyridines and 1,4-dihydropyridines](/storage/images/resized/4KtwkDSjVAxFFa8sq4IvgEVw7HZwu0Yf4CZJ9BCm_small_thumb.webp)
![Studying the adaptogenic activity of a series of tetrahydropyrido[2,1-b] [1,3,5] thiadiazine derivatives](/storage/images/resized/F8BKAKhXkkiEcf1sXtnnZhcQVmRyVZ2KWx0FEpqr_small_thumb.webp)
![Impact of Tetrahydropyrido[2,1-b][1,3,5]thiadiazines on L-DOPA Effects in the Tail Suspension Test](/storage/images/resized/hDjFgDubMpkooGFyBh5ufdfl0YkrxcDaVm8FXGWj_small_thumb.webp)
![The Mannich-Type Synthesis of Macroheterocycles with Pyrido[2,1-b][1,3,5]thiadiazine Unit](/storage/images/resized/PmVbwfUc2aRdrnSFGtkfcabQRXGkjjb14bQC1gcl_small_thumb.webp)



![Cr(II)-promoted internal cyclization of acyclic enediynes fused to benzo[b]thiophene core: Macrocycles versus 2-methylenecycloalkan-1-ols formation](/storage/images/resized/9z7b0TQJNgUxZHYXLrTmbnjJn9y5iq56Wxtqb1Lv_small_thumb.webp)







![[3+2]-Cycloaddition of azomethine ylides to 5-methylidene-3-aryl-2-сhalcogen-imidazolones: access to dispiro indolinone-pyrrolidine-imidazolones](/storage/images/resized/BHBStD1OkQqtKFRRjS5njgNe7bXCAEgaDQVK7GTA_small_thumb.webp)














