



![Catalysis of ring-closing metathesis reactions by HG-II[39].](/storage/images/resized/Fg83i7KEXe8RAolLSaHkenGBH0jfDTIYUzzIwvh2_xl.webp)
![Comparative catalytic activity of the Hoveyda – Grubbs-type oxygen-containing complexes. Refs. [38, 46, 47, 50, 60, 40-43, 52-55]](/storage/images/resized/iF7TGNqGyY9hrEE2hzpAXYhYE0lJrf6TuZ9ZmosY_xl.webp)
![Comparative results of catalytic tests of Hoveyda – Grubbs-type sulfur-containing complexes. Refs. [81, 82, 63-65, 73-77]](/storage/images/resized/kIV2k9WbJVdIIR1hMUVwKy3KZ3DbGxiJRBlHT6vz_xl.webp)
![Comparative results of catalytic tests of Hoveyda – Grubbs-type phosphorus-containing complexes. Refs. [38, 46, 47, 109, 110]](/storage/images/resized/LtxSWhDBEzcBHUwfiikBngcyEPL5Z3EqEKntYayX_xl.webp)
![Comparative results of catalytic tests of halogen-ruthenium complexes 87 – 89 (substrate concentration is 0.2 mol L–1, CH2Cl2,argon, 25 °C)[111, 112].](/storage/images/resized/ovCsyReobuEKcX8Dd3dWLdOAxQEmhqiXQwJ8BKQF_xl.webp)
![ROMP of substrates 153 in the presence of ruthenaheterocycles 63 and 64[91].](/storage/images/resized/O7izINCqOOFc3Cr8ZxsYkiFn8PkA91zLhRQWwt4Z_xl.webp)
![Comparative results of catalytic tests of complexes 90−93 and 97b in the RCM reactions. Refs. [121, 113-117]](/storage/images/resized/TTHQ8mPExf181t7NoEkbbXm466jAyoUc59QiZlJA_xl.webp)
Keywords
Abstract
Catalytic olefin metathesis using Hoveyda-Grubbs type ruthenium complexes is a powerful tool for creating complex molecules possessing a variety of practically useful properties. This method is also applied for obtaining modern polymer materials from low-demand petroleum products. Among all ruthenium complexes containing five- or six-membered chelate rings, the commercially available HG-II catalyst is the most common. In addition, other Hoveyda-Grubbs type complexes, which include a Het→Ru donor–acceptor bond in the chelate ring, often exhibit metathesis activity equal to or superior to that of HG-II. This review considers second-generation N-heterocyclic ruthenium carbene Hoveyda-Grubbs type complexes with donor–acceptor bonds such as O→Ru, S→Ru, Se→Ru, N→Ru, P→Ru and Hal→Ru in the chelate ring. Methods of preparation, analysis of stability and catalytic activity of such complexes are compared, and examples of the application of these organometallic ruthenium derivatives in the synthesis of practically relevant products are provided. The literature from 2010 to 2023 is summarized, making this review useful for a broad audience of chemists working in heterocyclic and organometallic chemistry, as well as practitioners involved in the production of catalysts and polymers.
The bibliography includes 174 references.
1. Introduction
The catalytic metathesis of olefins is a unique reaction that occupies an important place in the arsenal of synthetic organic chemistry. The metathesis reaction was discovered in the mid-20th century during the search for new transition metal-based catalytic systems for polymer production; however, the mechanism of olefin metathesis remained elusive for almost two decades[1-4]. In 1971, Hérisson and Chauvin[5] published a work in which, based on the metathesis mechanisms proposed in the literature, they suggested their own variant, which later became classical. According to this mechanism, the key step of the reaction is the formation of metallacyclobutane, which has been proven experimentally. In addition, the hypothesis of the equilibrium nature of metathesis, which had previously been proposed on the basis of empirical data, was confirmed[6-11]. In 2005, I.Chauvin (France), R. Grubbs (USA) and R.Schrock (USA) were awarded the Nobel Prize in Chemistry with the phrase ‘For contribution to the development of the metathesis method of organic synthesis’[12].
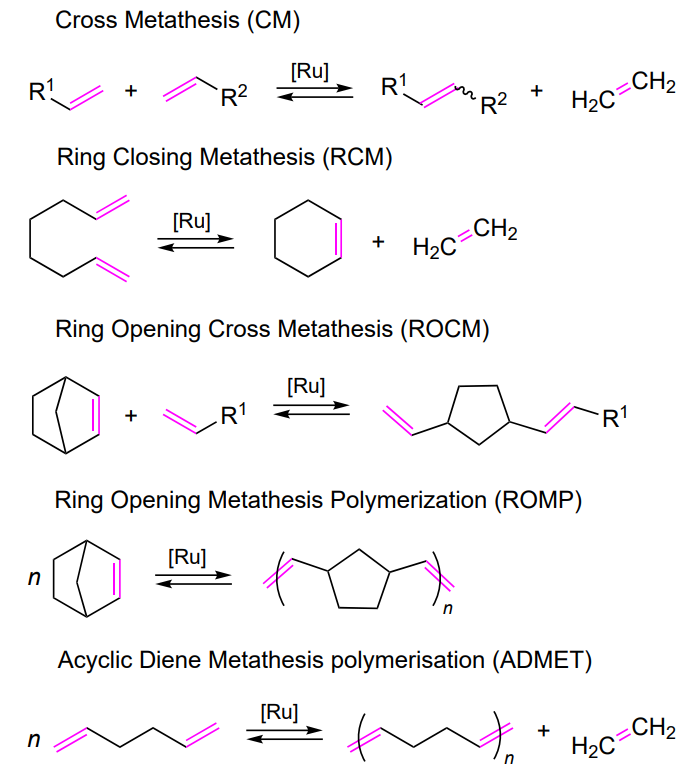
Metathesis of alkenes includes several types of transformations depending on the structure of the substrates and the resulting products. The generally accepted classification is depicted in Scheme 1[13].
Catalytic systems used for olefin metathesis can be broadly divided into two main types — those with a well-defined structure and those with an undefined structure. Systems with an undefined structure include heterogeneous catalysts such as mixtures of WCl6 or MoCl5 with co-catalysts (usually organometallic alkylating agents, e.g. Et3Al)[14]. Such systems are actively used in the large-scale synthesis of polymeric materials (polyethylene, polypropylene). Complexes with a well-defined structure include catalysts based on tungsten and molybdenum such as Schrock catalysts, and ruthenium complexes such as Grubbs and Hoveyda – Grubbs catalysts[15-17]. These complexes are used in laboratory practice for homogeneous catalysis. Figure 1 shows some commercially available ruthenium complexes for the olefin metathesis.
![[{"id":"SpLFGTZUnD","type":"paragraph","data":{"text":"Olefin metathesis catalysts with well-defined structures."}}]](/storage/images/resized/POQuBQOr64WZkDqmPtRlJRyoTK1fDWXANGAx0kQt_xl.webp)
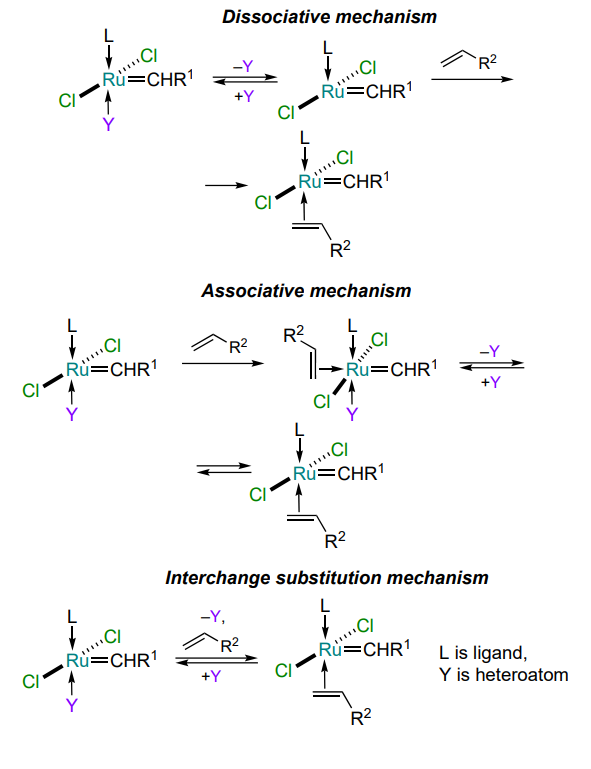
The design of olefin metathesis catalysts involves the control of their key properties by targeted modification of their structure. These properties include the rate of formation of the catalytically active 14-electron complex, its reactivity and the stability of the 16-electron structure of the precatalyst. Since most common metathesis catalysts are stable 16-electron ruthenium complexes, the mechanism of formation of catalytically active species therefrom is worth mentioning (Scheme 2). There are currently three generally accepted mechanisms for the formation of catalytically active intermediates: dissociative, associative and interchange[18, 19]. Consequently, there is a correlation between the structure of the arylidene ligand and the rate of formation of the active complex and, consequently, the rate of the metathesis reaction.
One of the most successful modifications of Grubbs complexes, apart from replacing the phosphine ligand with an N-heterocyclic carbene (NHC), has been the introduction of an additional group coordinated to the ruthenium atom in the arylidene ligand. This type of complex, known as Hoveyda – Grubbs catalysts, is attracting the attention of researchers with a wide range of possible modifications to its structure to solve specific synthetic problems[20]. Various combinations of chelating ligand and N-heterocyclic carbene have been used to obtain stable and highly reactive ruthenium metal complexes. Another advantage of such complexes is the ease of their isolation and purification compared to Grubbs complexes, as well as their tolerance to atmospheric oxygen, trace amounts of moisture and the presence of functional groups in the substrate. Reviews on various aspects of the synthesis and application of Hoveyda – Grubbs complexes are published annually (see, e.g.[21-28]), which underlines the interest of the scientific community in the topic discussed in this review.
Possible variations of second generation Hoveyda – Grubbs type complexes (HG-II) are illustrated in Figure 2. Four main directions can be distinguished: modification of the NHC ligand, modification of the benzylidene moiety, substitution of anionic ligands and variation of the donating heteroatom (Y)[28]. In this review, the last direction will be considered.
![[{"id":"mroo0Ern3p","type":"paragraph","data":{"text":"The main directions of modification of Hoveyda–Grubbstype catalysts."}}]](/storage/images/resized/vPWHwcCrkJy9mD50tGJnzSPfystu1YN8Hd3PgHOy_xl.webp)
As practice over the last thirty years has shown, the most commonly used catalysts in laboratory practices and in production are HG-I and HG-II (see Figure 1), in which the oxygen atom (Y) is coordinated to a ruthenium atom in a five-membered metallacycle containing a benzylidene unit. Chlorine atoms act as anionic ligands (X), and from the variety of N-heterocyclic carbenes, the 1,3-bis(2,4,6-trimethylphenyl) imidazolidine moiety was chosen because of its availability and the considerable steric bulk of substituents at the nitrogen atoms (see Figure 2). The reviews[28-30] provide information on the influence of the structure of NHC ligands on the properties of ruthenium complexes. The dependence of catalyst activity on the nature of the donating heteroatom Y has not been systematically analyzed. This is due both to the fact that ruthenium complexes of the Hoveyda – Grubbs type with a donor-acceptor chalcogen → Ru (except for the oxygen atom), pnictogen → Ru or halogen → Ru bond are not sufficiently covered in the literature, and to the fact that the vast majority of works dealing with them was published in the period 2005 to 2023. Nevertheless, the catalytic efficiency and stability of many of these structures are not inferior or even outperform those of commercial HG-I and HG-II catalysts (see Figure 1).
The literature review therefore focuses on the second-generation Hoveyda – Grubbs type catalysts (HG-II) with various arylidene ligands containing S, Se, N, P, Br and I atoms coordinating the ruthenium cation (Figure 3). The N-heterocyclic carbene is in all cases 1,3-dimesityl-4,5-dihydro-1H-imidazol-2-ylidene. This limitation in the choice of NHC ligand allows, in many cases, a correct comparison of the catalytic activity of ruthenaheterocycles among themselves as well as with the HG-II complex as the ‘reference’ catalyst for olefin metathesis. To facilitate comparison, the first part of the review also contains information on the preparation and properties of the main oxygen-containing complexes of the HG-II type (oxygen acts as the coordinating ruthenium heteroatom Y). Finally, modern examples of the use of organoruthenium compounds in the straightforward synthesis of organic compounds are given.
![[{"id":"B_dvZRuc8v","type":"paragraph","data":{"text":"Structures of ruthenium metallacycles considered in this review."}}]](/storage/images/resized/kiW6jDfF8ZoOPskR2XveOVOf4NQkOlVDZYtg0DqT_xl.webp)
2. Hoveyda – Grubbs type catalysts containing arylidene ligands with coordinating O, S, Se, N, P, Br, I atoms
2.1. Chalcogen-containing catalysts
2.1.1. Complexes with the O → Ru bond
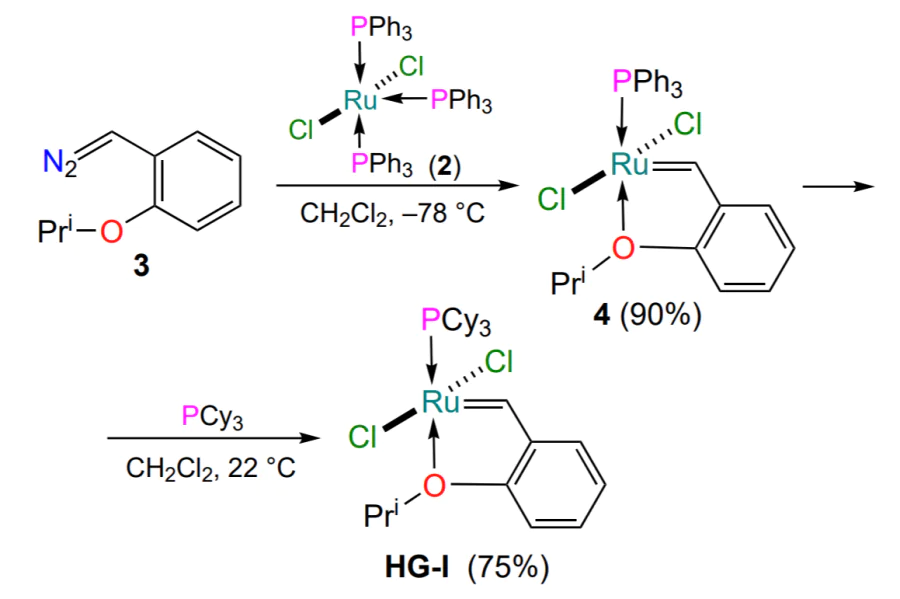
The history of arylidene ruthenium complexes starts with the work of Hoveyda published in 1997[31]. The premise for this publication was the fact that the ruthenium complex G-I was not able to catalyze the cross-metathesis reaction between 2-ethoxystyrene and diallyl ether. The authors explained this by the possible formation of a stable (passive to catalysis) 16-electron metallacycle. To confirm this hypothesis, the reaction of 2-isopropoxystyrene (1) was carried out with the first generation Grubbs catalyst (G-I), taken in an amount of 1 equiv. (Scheme 3)[31]. The Hoveyda – Grubbs catalyst HG-I was obtained as dark green crystals, and its structure was determined by X-ray diffraction (XRD). The first representative of a new type of ruthenium catalyst had a distorted pyramidal geometry of the coordination polyhedron with a transoidal arrangement of the chlorine atoms.
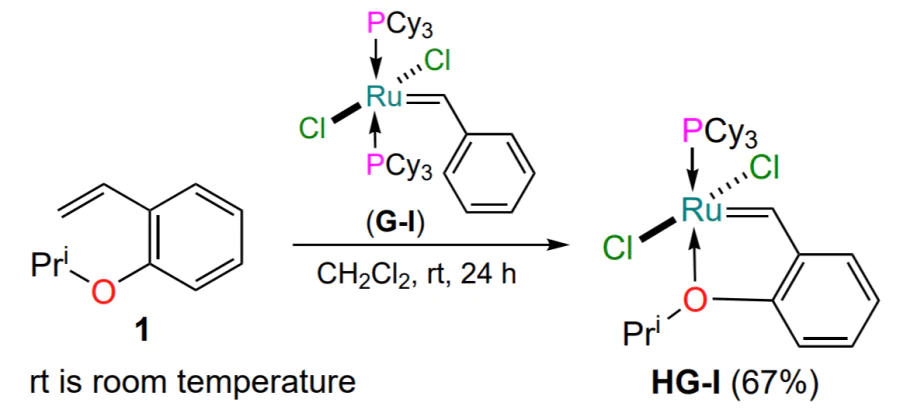
A convenient two-step procedure for the preparation of complex HG-I was developed based on Wilkinson’s complex 2 (see [32-35]) and 1-diazomethyl-2-isopropoxybenzene 3 (see [36, 37]) (Scheme 4)[36]. Complex 4 was formed in a 90% yield and its structure was confirmed by XRD. Subsequently, complex 4 was converted to complex HG-I when treated with 2 equiv. of tricyclohexylphosphine. It should be noted that the synthesis of HG-I can be carried out in a one-pot variant. In this case, the replacement of the triphenylphosphine ligand with the more electron-donating tricyclohexylphosphine is performed without isolation of the intermediate complex 4.
In 2000, two research groups[38, 39] independently published their results on the preparation of new HG-II-type complexes containing N-heterocyclic carbenes. The advantage of the phosphine-free complexes was their greater stability with respect to oxygen and moisture, as well as increased reactivity and selectivity in metathesis reactions. Blechert and co-workers[38] proposed to replace the tricyclohexylphosphine ligand in complex HG-I with 1,3-(dimesityl)imidazolidine (Scheme 5). The target HG-II was obtained in two steps in a total yield of 75%. Intermediate 5 was also isolated in pure form and characterized[38].
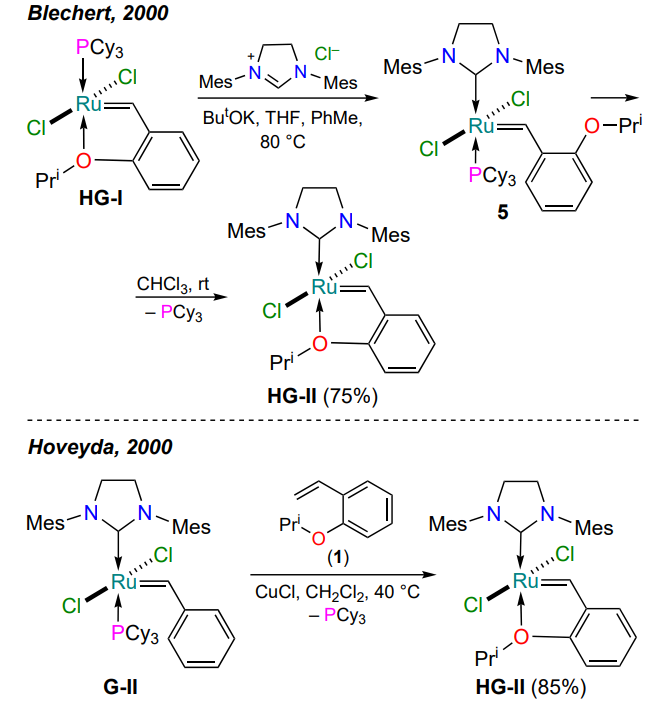
An alternative approach to the ruthenaheterocycle HG-II was proposed by Hoveyda and co-workers[39]. The second-generation Grubbs catalyst (G-II) was used as the parent ruthenium derivative. The HG-II complex was prepared from 2-isopropoxystyrene (1) in the presence of copper(I) chloride in a single step in 85% yield (see Scheme 5). Hereinafter, copper(I) salts are used to bind the tricyclohexylphosphine released during the reaction.
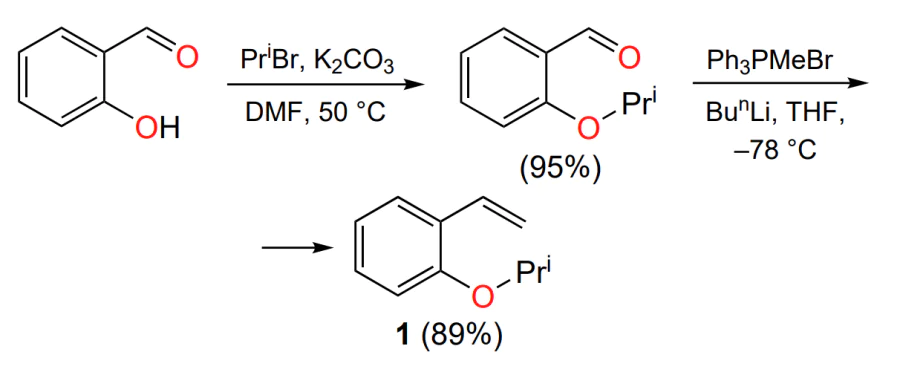
The HG-II catalyst is currently one of the most popular commercially available complexes for laboratory use. This is due to the relatively cheap production of the arylidene ligand precursor, styrene 1. The latter is prepared from salicylic aldehyde in two steps in good yields (Scheme 6)[38].
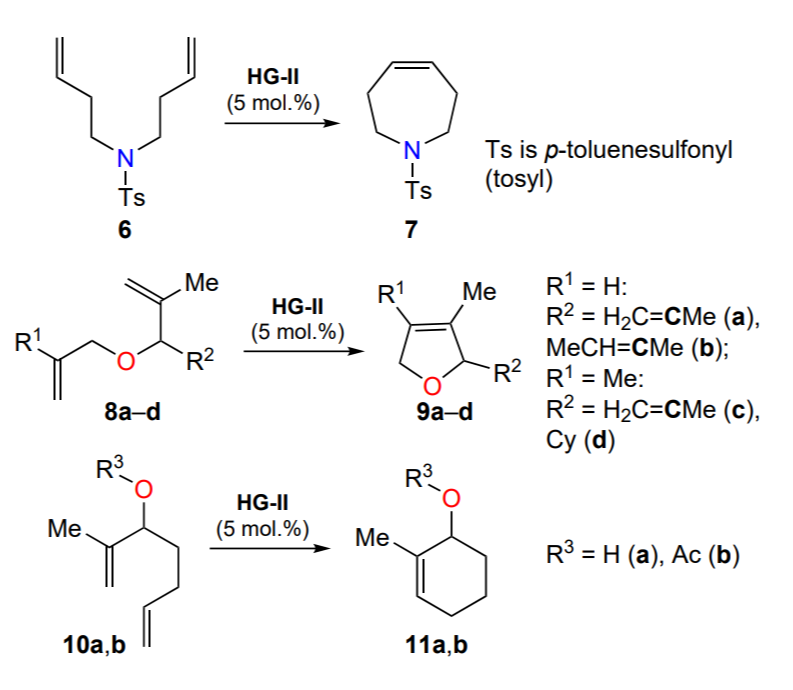
The HG-II complex showed high efficiency in ring-closure metathesis (RCM) using dienes with two terminal double bonds as substrates (Scheme 7). Most importantly, the ruthena-heterocycle HG-II was able to initiate the metathesis reaction to give tetrasubstituted cyclic alkenes. The conversion times of substrates with the most spatially accessible double bonds — N,N-di(but-3-en-1-yl)-4-methylbenzenesulfonamide (6) to 1-tosyl-2,3,6,7-tetrahydro-1H-azepine (7), dienes 8a,b,d to dihydrofurans 9a,b,d — ranges from 10 to 30 min, in the case of transformation of 3-substituted 2-methylhepta-1,6-dienes 10a,b to cyclohexenes 11a,b it reaches 1.5 – 2 h, and with tetrasubstituted alkene 9c it is up to 44 h. All processes run without heating to provide high yields of target products (Table 1). The possibility of HG-II recycling in metathesis reactions was also demonstrated[39].
The studies[38, 39] opened a new avenue in organometallic chemistry to create ruthenium derivatives with the highest catalytic activity, stability and selectivity.
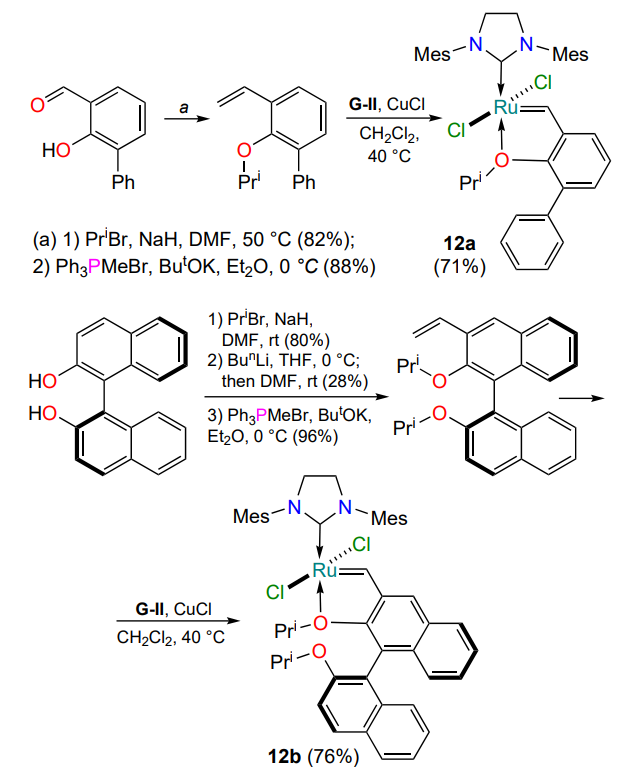
In 2003, Blechert and co-workers[40] presented the results of the synthesis of oxygen-containing complexes 12 bearing an aromatic moiety ortho to the isopropoxy substituent. The authors proved that an increase in steric hindrance favors the weakening of the O → Ru bond and facilitates its dissociation (Scheme 8). A similar dependence was found in the study of catalyst 12b based on 1,1'-bi(2-naphthol) (BINOL)[41]. Despite the high catalytic activity, complexes 12a,b had a significant disadvantage — low stability even in the simplest RCM reactions.
Further modifications of Hoveyda – Grubbs complexes HG-II involved the introduction of electron-donating and electron-withdrawing substituents in the arylidene ligand. The presence of electron-withdrawing groups in the arylidene ligand further activated the complex in olefin metathesis reactions due to a decrease in the electron density at the donating oxygen atom. Conversely, the introduction of electron-donating groups increased the stability of the chelate, hindering the dissociation of the oxygen-ruthenium coordination bond necessary for the formation of the 14-electron active complex.
The Grela’s research group[42] obtained the ruthenium complex 13a containing a nitro group in the para-position to the isopropoxyl substituent (Figure 4). This complex, which performed well in RCM reactions, gradually lost its stability in the air. Nevertheless, its high performance allowed its application in industry and laboratory studies. By analogy with complex 12a, unstable ruthenaheterocycles 13b,c with bulky R groups in the ortho-position to the oxygen atom were obtained. Interestingly, no increase in catalytic activity was observed for complexes 14 and 15 with nitro groups in the ortho- and meta-positions, respectively[42-44]. Therefore, it can be stated that the introduction of acceptor groups into the arylidene ligand promotes the activity of ruthenium chelates, which is also confirmed by computational methods[45].
![[{"id":"EzbTaZVYjd","type":"paragraph","data":{"text":" Structures of the <b>HG-II</b>-type ruthenium catalysts <b>13</b>–<b>15</b> with electron-withdrawing substituents in the benzylidene ligand."}}]](/storage/images/resized/ENLFKw5A5UkjSgECkjyoAfo7MRGKctMqiKAMTvhh_xl.webp)
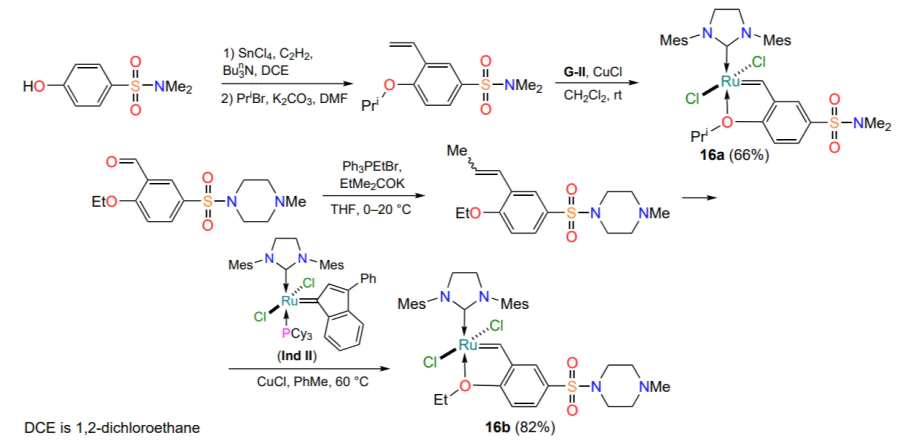
One of the most common complexes of this type is the so-called Zhan catalyst (16a), which was first described in 2007[46, 47]. It contains an additional dimethylsulfonylamide function in the arylidene ligand in addition to the isopropoxyl group, which is traditional for such complexes. Complex 16 can be recycled with almost no loss of activity. The 2021 publication[48] discloses a method for the preparation of a structural analogue of complex 16a, chelate 16b, with high potential for use in the total synthesis of bioactive molecules in ‘green’ solvents such as 2-methyl-tetrahydrofuran, anisole, ethyl acetate, dimethyl carbonate and acetone. The commercially available indenylidene complex Ind II (Hereinafter, the Roman numeral after the letter cipher of the catalyst indicates the generation by analogy with Grubbs’ complexes.) was chosen as the precursor for the preparation of ruthenaheterocycle 16b (Scheme 9)[49].
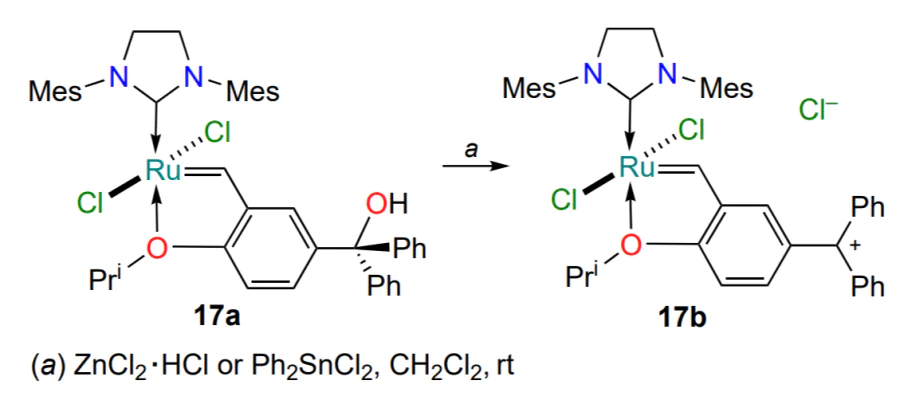
Another approach to obtaining active catalysts for olefin metathesis is the introduction of substituents containing a heteroatom with an unshared electron pair into the arylidene moiety. Such complexes are activated in the presence of acidic additives[50]. Depending on the type of substituent present in the ruthenium complex, either a strong Brønsted acid or a weak Lewis acid is required for activation. HBF4 was found to be too strong acid, leading to the degradation of complex 17a, and the optimal conditions providing high performance of complex 17b in the ring-closing metathesis reaction included the addition of 1 equiv. ZnCl2 · HCl or Ph2SnCl2 (Scheme 10).
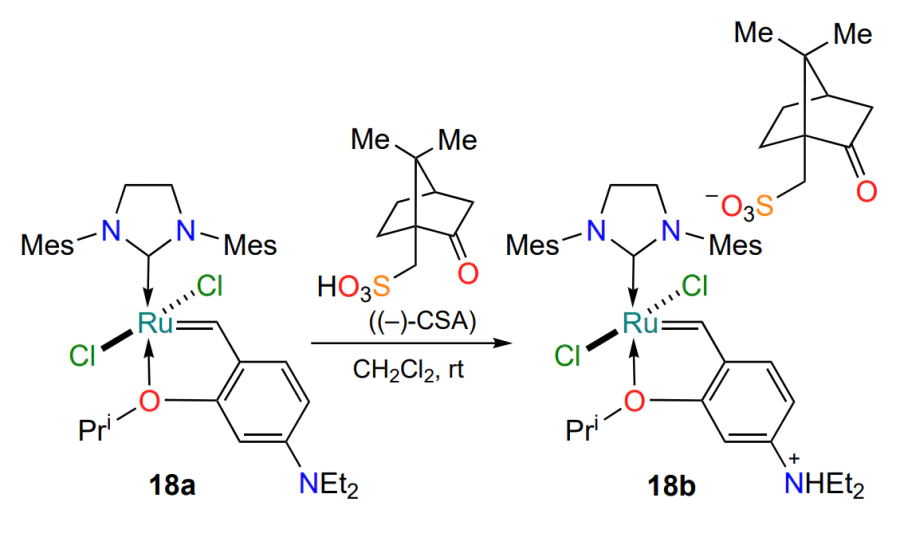
Complex 18a, described in the same study, is catalytically inert at room temperature, but when heated to 45°C, it initiates the complete conversion of terminal dienes in RCM reactions[50]. To activate it and convert it to salt 18b, (−)-camphor-10-sulfonic acid (CSA) can also be used (Scheme 11). The main advantage of the described approach is the increase in stability of complex 18a due to the introduction of an electron-donating substituent. In contrast to the above examples, where the improved stability of the complexes mostly correlates with a decrease in their catalytic activity, this is avoided in the case of catalyst 18a due to the transformation of the donor deactivating dimethylamino group into an electron-withdrawing group in acidic medium.
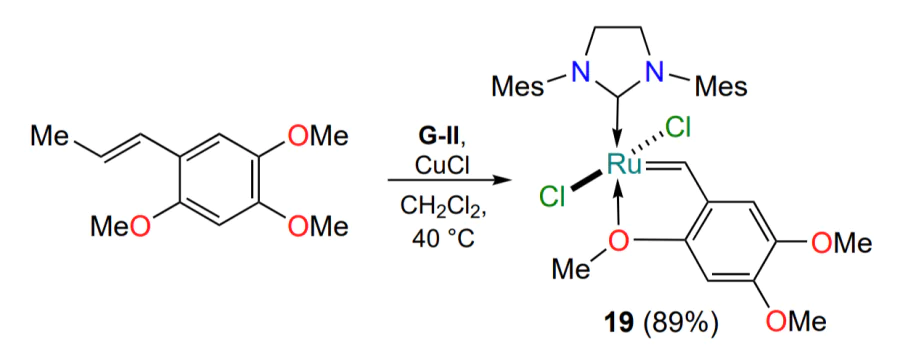
It has been shown that the presence of donor groups in the benzylidene ligand generally increases the stability of the ruthenaheterocycle[51]. In particular, catalyst 19, obtained in a single step from α-asarone (1,2,4-trimethoxy-5-propenyl-benzene), performed satisfactorily in various olefin metathesis reactions (Scheme 12). The availability of the starting alkene is also an undeniable advantage of complex 19.
The modifications of ruthenium derivatives discussed below, which are related to the second-generation Hoveyda – Grubbs catalysts, involve the replacement of alkyl substituents at the oxygen atom[52, 53]. The O-benzyl and O-phenyl substituted complexes 20 and 21 have been shown to be efficient metathesis catalysts, resistant to atmospheric oxygen and traces of moisture (Figure 5). This can be explained by the p – πconjugation between the oxygen atom and the aromatic ring, which in turn has a stabilizing effect on the transition state of the metathesis reaction. As previously shown by Grela and co-workers[42-44], the introduction of a nitro group to the phenoxyl moiety improves the catalytic activity of complex 21d in RCM reactions. Attempts were also made to synthesize catalyst 22 with a trifluoromethyl group at the oxygen atom, but it was not possible to isolate and characterize this complex[52].
![[{"id":"bManNDpce0","type":"paragraph","data":{"text":"Structures of <b>HG-II</b> type ruthenium catalysts <b>20</b>–<b>22</b> with a modified substituent at the oxygen atom."}}]](/storage/images/resized/iXS5iCPi7d5lRUEccy4pgxwjUuPQWtJ3wBmFHNRW_xl.webp)
It was hypothesized that Hoveyda – Grubbs type complexes may be stabilized by the formation of an extended system of conjugated multiple bonds in the arylidene moiety[54]. To confirm this hypothesis, a series of polynuclear heterocycles 23a–d with naphthalene and phenanthrene units were obtained (Figure 6). According to the Clar’s rule[55], phenanthrene-like aromatic systems are more stable, which probably explains the increased stability and low catalytic activity of the chelates 23b – d.
![[{"id":"TwrWuMLjLS","type":"paragraph","data":{"text":"Structures of <b>HG-II</b>-type ruthenium catalysts <b>23</b> with polynuclear arylidene ligand and their relative stabilities and activities."}}]](/storage/images/resized/guGELXYH6dhtb2jNrHuic5vqyppgS33f2uKpSWX7_xl.webp)
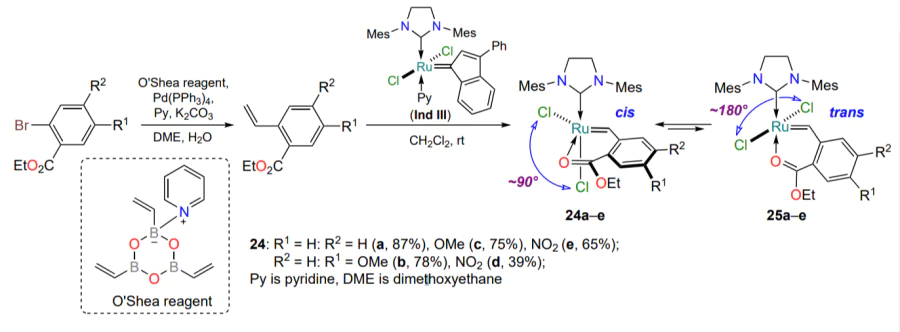
The oxygen-containing ruthenium complexes with five-membered metallacycle described above are trans isomers (the Cl – Ru – Cl angle is ca. 180°), which has been confirmed both by numerous X-ray diffraction studies and quantum mechanical calculations. Nevertheless, Slugovc and co-workers[56-58] succeeded in isolating a series of catalysts 24a – e with a cis arrangement of chloride ligands, in which the carbonyl oxygen atom of the ester group coordinates the ruthenium atom, starting from the indenylidene complex Ind III (see [59]) and the corresponding styrenes. In the same works, the influence of substituents in the arylidene ligand on the stability and reactivity of ruthenium complexes 24 was explored (Scheme 13). Experimental and computational methods showed that upon activation (e.g. heating to 80 °С) the cis complexes 24 are transformed into the corresponding trans isomers 25. The latter are effective olefin metathesis catalysts. Notably, a six-membered oxoruthenaheterocycle, a rare structural motif, is realized in complexes 24 and 25[56-58].
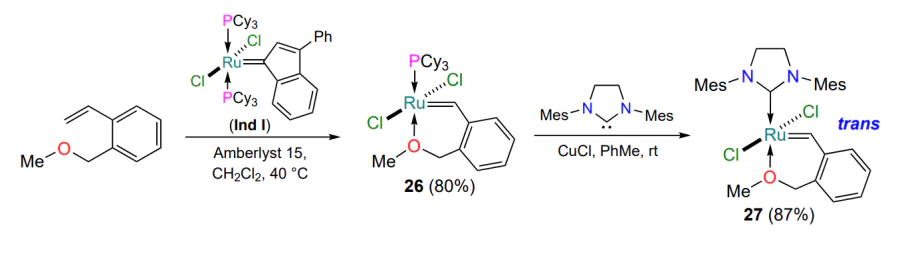
A homologue of HG-II containing a six-membered metallacycle has been described[60]. Its preparation involved a two-step procedure consisting of the isolation of an intermediate first-generation Hoveyda – Grubbs type catalyst 26 in the presence of an ion-exchange resin and its subsequent exposure to the corresponding NHC ligand (Scheme 14). The resulting complex 27 actively catalyzed RCM reactions with various substrates at both room and elevated temperatures. Attempts to introduce a substituent other than the methyl group to the donating oxygen atom in the structure of 27 were unsuccessful.
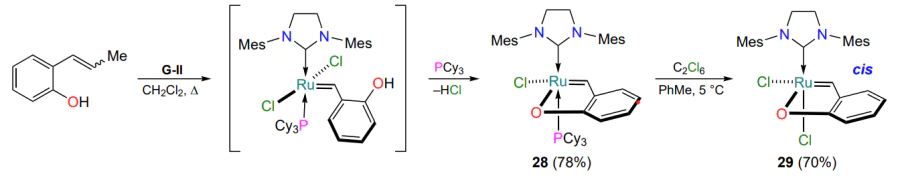
To conclude the discussion on the preparation methods and properties of the HG-II-type oxygen-containing ruthenium complexes, synthesis of ruthenacycle 28 with an anionic oxygen-containing ligand should be mentioned[61]. X-ray diffraction analysis confirmed the cis geometry of the chelate 28 (the O – Ru – Cl angle is ca. 90°) and also showed that the covalent bond formed between ruthenium and the phenolate anion changes the geometry of the coordination polyhedron (Scheme 15). The oxidative exchange of the tricyclo-hexylphosphine ligand for a halogen atom in the ruthenium(II) complex 28 was achieved using hexachloroethane (10 equiv.), while the cis configuration in the ruthenium(III) complex 29 was retained[62].
At the end of this Section, we will compare the catalytic activity of the described oxygenated ruthenium complexes in model RCM reactions. For this purpose, the two most commonly used substrates, N,N-diallyltosylamide (30) and diethyl diallylmalonate (31), have been chosen to give the products 32 and 33, respectively. Although the different of the metathesis reactions hamper the correct analysis, Table 2 provides information from which the following conclusions can be drawn.
1. Increasing the steric hindrance on the coordinating oxygen atom or in the ortho position to the alkoxy substituent of the arylidene ligand leads to an increase in the catalytic activity of the ruthenium complex.
2. The introduction of electron-withdrawing substituents to the arylidene ligand also contributes to an increase in catalytic activity with a concomitant decrease in the stability of the ruthenium complex.
3. Further stabilization (expansion) of the aromatic system of the arylidene ligand reduces the catalytic activity of the complex.
2.1.2. Complexes with the S → Ru bond
The advances made in the design of catalytic systems such as HG-II with O → Ru coordination bonds has sparked efforts to create organometallic compounds with a similar structure containing a sulfur or selenium atom in the chelate ring. The vast majority of these studies were published between 2010 and 2023. It should be noted that the replacement of the coordinating oxygen atom by a sulfur atom leads to the appearance of cis,trans isomerism of ligands in the coordination sphere of ruthenium. In addition, not only five-membered but also six-membered metallacycles are stable in the case of sulfur-containing ruthenium complexes.
In 2008, Lemcoff and co-workers[63] published their first paper on the synthesis and properties of sulfur-containing analogues of the HG-II catalyst. 2-Vinylphenylphenylsulfide 34a, required for the synthesis of complex 35a, was prepared in two steps from commercially available 2-fluorobenzaldehyde (Scheme 16). It was found that the replacement of the coordinating atom was accompanied by a change in the configuration of the ligand environment of the ruthenium atom. In all cases, complexes 35 with cis configuration (the Cl – Ru – Cl angle is ca. 90°) were identified. Complex 35a (R = Pri), isosteric to HG-II, showed high stability in solutions in the presence of air. The other metallacycles 35b – e, shown in Scheme 16, were obtained in a similar manner[64].
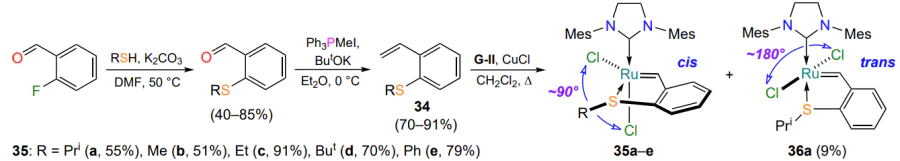
In 2010, a complex with trans-configuration 36a (the Cl – Ru – Cl angle is ca. 180°), the geometric isomer of the chelate 35a, was synthesized by joint efforts of Grela and Lemcoff groups[65] (see Scheme 16). The ruthenium derivative 36a was isolated simultaneously with complex 35a by column chromatography (35a : 36a = 86 : 14). The authors[65] did not study the catalytic properties of complex 36a. The relatively high stability of chelates 35a and 36a enabled kinetic experiments to investigate their isomerization process. It was found that the trans complex 36a is quantitatively converted into the cis chelate 35a within 50 h at room temperature in CD2Cl2. The isomerization rate showed a clear dependence on solvent polarity, with the highest conversion rate of the trans isomer 36a to the cis isomer 35a occurring in halogenated solvents like dichloromethane. The increased stability and consequently low catalytic activity of complex 35a is due to the s-donor properties of the sulfur atom, which are responsible for the formation of a stronger sulfur-ruthenium bond compared to that of catalyst 36a. In contrast, the low stability of the trans isomer 36a in solution is attributed to the trans influence of the NHC ligand. The above reasoning is supported by quantum mechanical calculations[65].
Taking into account these data, the cis ruthenium complexes 35 can be considered as products of thermodynamic control and the trans isomers 36 as products of kinetic control of the reaction[25]. The latter can be isolated under mild conditions, at lower temperature and in less polar solvents.
Subsequently, the most probable stages of the cis-trans transition between isomeric sulfur-containing ruthenium complexes were formulated based on the results of the calculations (Scheme 17)[66, 67]. Upon heating or irradiation, the sulfur-ruthenium coordination bond in the trans isomers 36 is cleaved, then the valence angle Cl – Ru – Cl is changed. The transformation is completed by the formation of a new coordination bond in the cis complexes 35. It is shown that the cis isomer 35a catalyzes ring-closure diene metathesis reactions only at either elevated temperatures or under irradiation; therefore, thermal or photoactivation is required to convert the stable and inactive cis complex 35 into the reactive trans metallacycle 36. Additional experiments were performed to demonstrate the possibility of complete deactivation of complex 35 (transition of catalytically active 14-electron species (see Scheme 17) to inactive cis complex 35) when the temperature is lowered to room temperature, after which the catalyst can be reactivated by heating without loss of activity[66]. This behavior of the catalyst has been named as ‘thermoswitchable’. The thermoswitching effect is useful for controlling polymerization processes, including 3D printing[68-71]. An alternative approach is the photoactivation of complexes 35 under UV irradiation (wavelength is ca. 365 nm)[67].
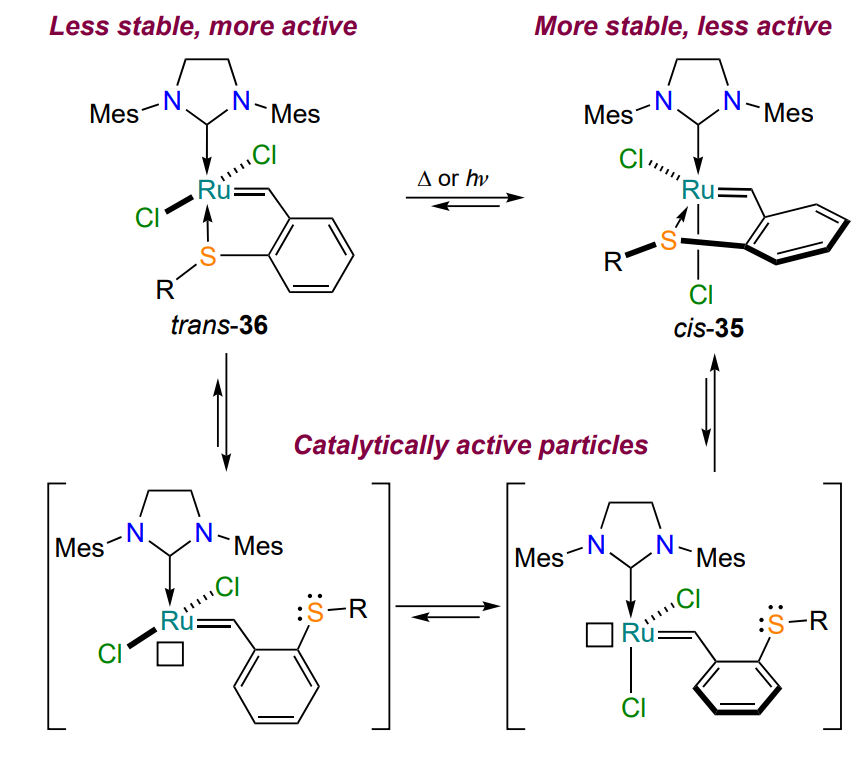
As with oxygen-containing catalysts (see Figure 5)[52, 53], the nature of the substituent at the sulfur atom affects the rate of activation of the complex: (S-phenyl)-substituted catalyst 35e performed best among such complexes, both under heating and irradiation. This is probably due to the occurrence of p – π-conjugation between the sulfur atom and the benzene ring, which stabilizes the transition states in the isomerization of chelates 35 and 36, as well as in the metathesis reaction (see Scheme 17)[72].
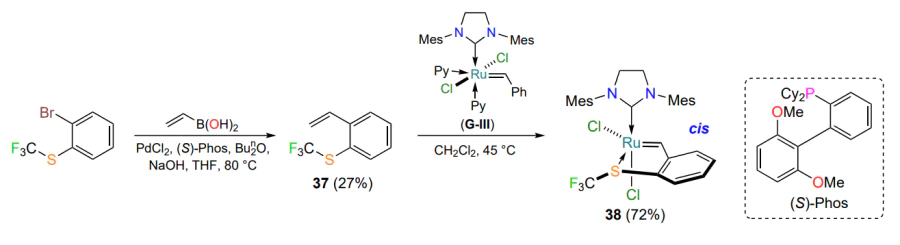
The presence of an electron-withdrawing substituent, such as a trifluoromethyl group at the sulfur atom, leads to a significant boost in the catalytic efficiency of ruthenium complexes[73, 74]. Thus, the metallacycle 38 obtained from styrene 37 and the Grubbs 3rd generation catalyst G-III (see Figure 1) was used for the RCM reaction in toluene heated to 80 °C. Complex 38 was found to be significantly superior to its S-methyl counterpart 35a in terms of efficiency (Scheme 18).
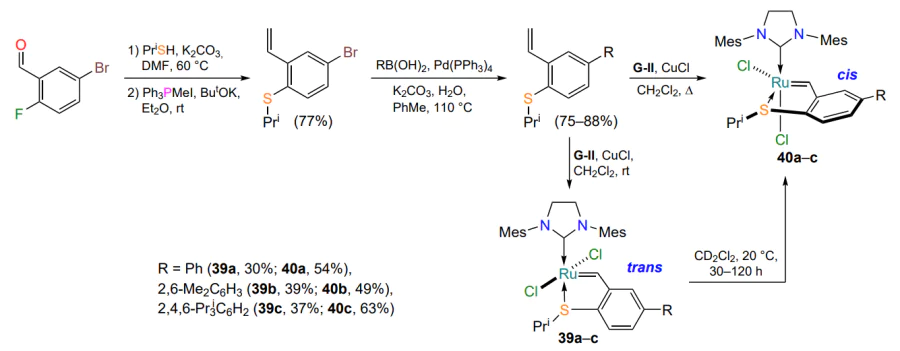
To investigate the influence of the structure of the complexes on the trans,cis isomerization, a series of ruthenaheterocycles bearing phenyl, 2,6-dimethyl and 2,4,6-triisopropylphenyl substituents in the arylidene ligand were obtained[75]. The authors developed stereoselective approaches for the isolation of individual isomers of catalysts 39 and 40 (Scheme 19). They hypothesized that the presence of bulky substituents in the arylidene ligand would increase the rate of isomerization of the trans complexes 39 to the corresponding cis isomers. However, no such dependence was found in the experiments: individual trans complexes 39 were quantitatively converted into cis isomers 40 at room temperature in polar solvents (in 30 – 120 h in dichloromethane). The authors could not explain this peculiarity in the behavior of such ruthenathioheterocycles. Trans complexes 39 showed high catalytic efficiency in the absence of thermal and photoactivation, whereas cis isomers 40 were inactive under the same conditions[75]. Grela and co-workers[76] synthesized ruthenium complexes containing a sulfoxide moiety in the coordination sphere of the ruthenium atom. The presence of this group changes the geometry of the complexes 41 and consequently increases their activity (Scheme 20). The formation of trans isomers 41 is favored, in which the ruthenium atom is coordinated to the sulfur atom of the sulfoxide group (the molecular structure of complexes 41c,d has been confirmed by XRD). Sulfoxide chelates 41 showed catalytic activity in model olefin metathesis reactions without additional thermal or photoactivation but were inferior in efficiency to commercial catalysts with an O → Ru bond, such as HG-II. It has been shown[76] that the presence of the SO group increases the activity of the complexes 41 compared to the previously described[63, 64] sulfides 35 (see Scheme 16).
The ruthenium-containing precursor Ind II gave rise to only one representative of the metallacycles 42 bearing a sulfone functionality[77]. Chelation of the ruthenium atom in sulfones 42 occurs via the oxygen lone pair of electrons to give a more labile six-membered metallacycle (see Scheme 20).
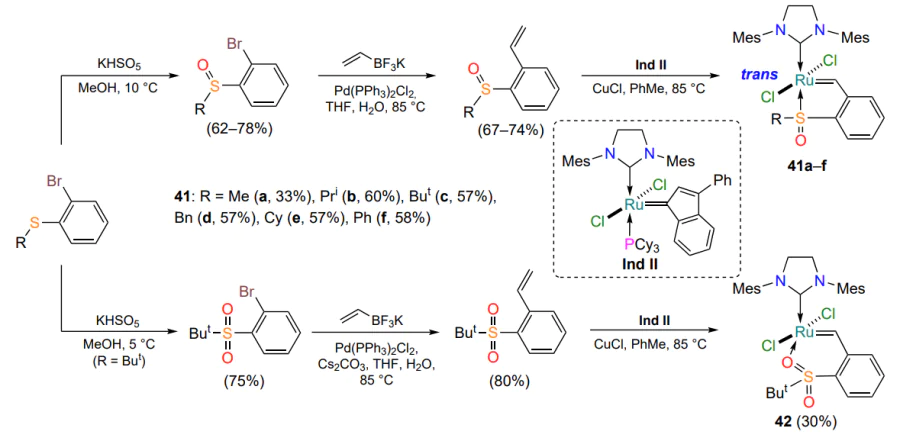
As noted above (see Figure 5), the introduction of a nitro group into the arylidene ligand of oxygen-containing ruthenium complexes improves their catalytic activity[43, 44]. However, nitroaryl-substituted sulfur-containing structures 43 and 44 were inactive in alkene metathesis reactions at room temperature (Scheme 21)[65]. Apparently, the coordination bond between ruthenium and the sulfur atom is stronger than in the case of a more electronegative oxygen atom. Similar to the sulfur-containing catalysts 35 shown in Scheme 16, thermal activation is required for chelate 44 to be used in alkene metathesis reactions.
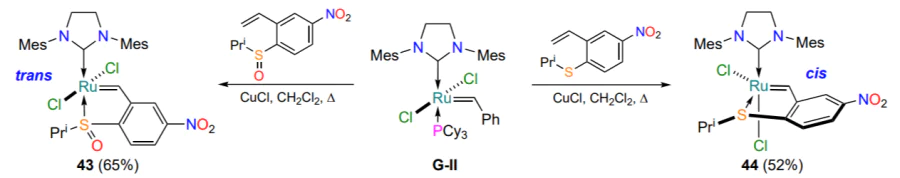
Lemcoff and co-workers[78] studied the influence of the structure of the sulfur-containing ligand on the properties and geometry of the resulting ruthenium derivatives. Catalysts 45a,b were obtained from the corresponding unsaturated thioethers (Scheme 22). In contrast to the sulfur-containing cis complexes 35[63, 64], which were catalytic inert in the absence of activation, the complexes 45 without the annulated benzene ring showed low catalytic activity already at room temperature. Thus, it can be concluded that the energy barrier for the cis-trans transition of alkylidene sulfur-containing chelates is lower compared to their arylidene counterparts. Complexes 45 have been observed to be unstable in air and in polar solvents[78].
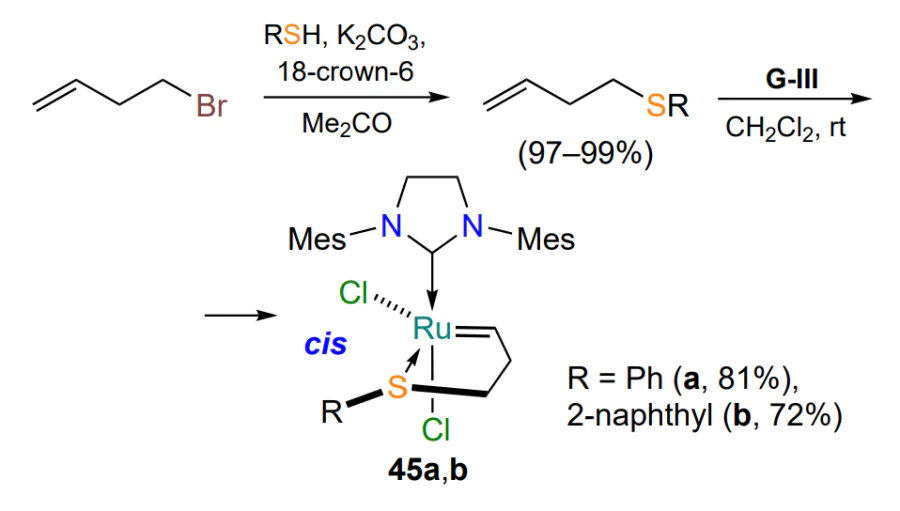
The analysis of the materials of the current subsection allows us to conclude that five-membered sulfur-containing ruthenium chelates of the HG-II-type are quite well studied. However, there are only four publications highlighting the synthesis and characterization of their six-membered homologues. In the studies of Shcheglova et al.[79, 80], based on styrenes 46a,b and Grubbs catalyst 47, a method for the construction of six-membered ruthenathioheterocycles 48a,b was proposed for the first time, but no approach to the starting styrenes 46 was provided. The structure of the complexes 48 has been confirmed by 1H and 13C NMR spectroscopy and mass spectrometry, which fail to indicate the spatial structure of the molecules. The authors note that chelates 48a,b show moderate activity in ring-closing metathesis reactions of dienes (Scheme 23)[79, 80].
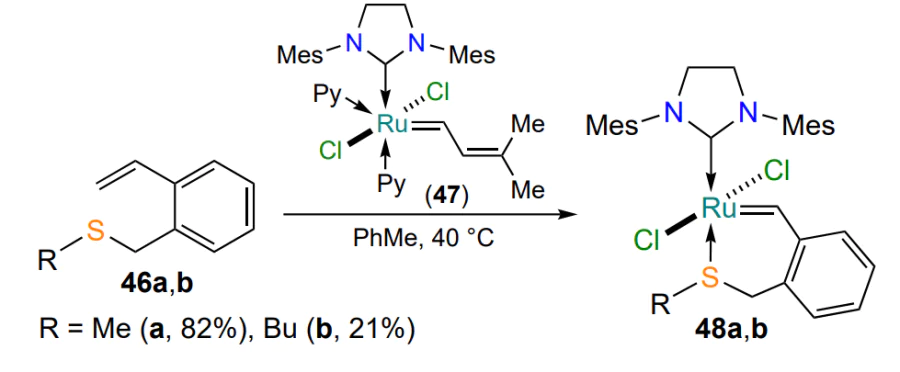
The synthesis, structure and properties of chelates 48 were described in detail in a 2023 study[81], in which six-membered metallacycles with an S → Ru bond were prepared from the precursor Ind II and ortho-substituted styrenes 46. The authors managed to selectively obtain individual trans (48) and cis isomers 49, the structure of which was determined by X-ray diffraction analysis (Scheme 24). It was shown that, like the complexes 35 containing a five-membered metallacycle (see Scheme 16), the cis catalysts 49 are only active under thermal or photoactivation, whereas the trans isomers 48b – i exhibit catalytic properties already at room temperature. The dependence of the catalytic activity of ruthenium derivatives on the steric and electronic effects of substituents at the sulfur atom has been explored in detail. Coordination compounds 48i and 49i bearing S-(2-naphthyl) substituent perform best in model reactions of olefin metathesis, while chelates 48b,c and 49b,c (R = Bun, Bui) provide the lowest conversion under the same conditions.
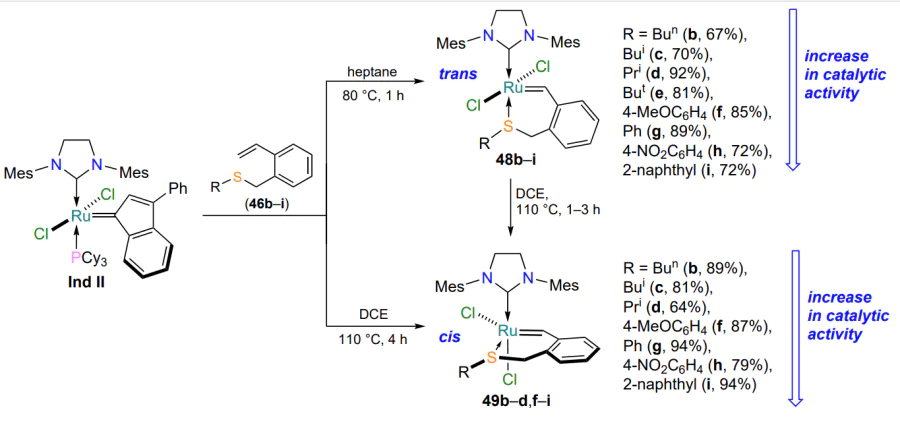
In a study[81], compound 49a (R = Me) was not synthesized because of the gaseous aggregate state and high toxicity of the starting methyl mercaptan (see Scheme 24).
In 2023, the synthesis of ruthenium dithioacetal complexes from arylidene ligands 50 was reported (Scheme 25)[82]. Complexes 51 were isolated as cis isomers. It was expected that the catalytic properties of these complexes would be similar to those of the above-described sulfur-containing chelates 35 (see Scheme 16). This assumption proved to be true only for complexes 51a,b, which were found to be inert in the absence of activation. At the same time, ruthenathioheterocycles 51c,d bearing aromatic substituents at the sulfur atoms performed good in olefin metathesis reactions at room temperature.
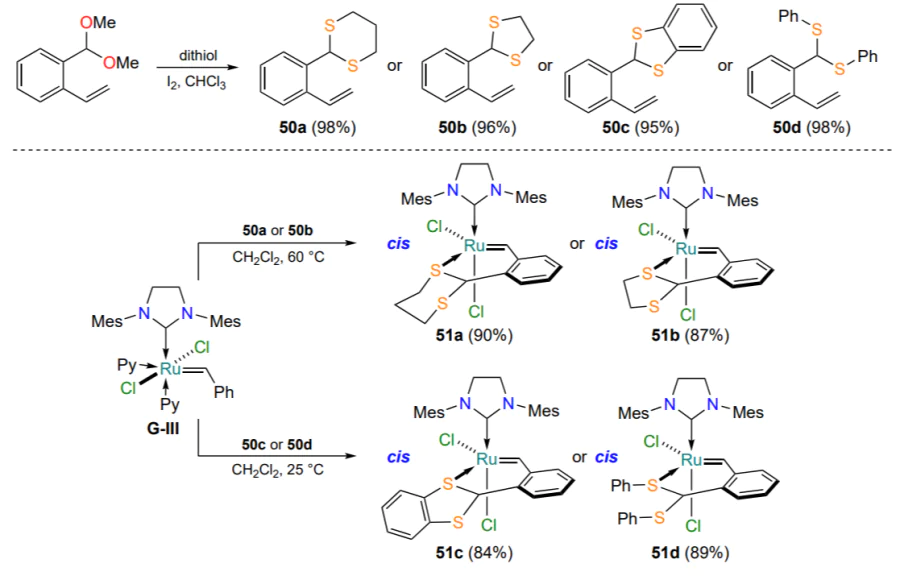
To summarize the results of this Section, it should be emphasized that sulfur-containing ruthenium complexes of the second-generation Hoveyda – Grubbs type can exist as two stable isomers which differ in the mutual orientation of the chlorine atoms in the ruthenium coordination sphere. The geometry of the complex determines its catalytic behavior: trans chelates are active at room temperature, whereas activation of cis isomers usually requires either heating or exposure to UV radiation. The possibility of thermal trans-cis isomerization of sulfur-containing chelates has been proved, with trans complexes being products of kinetic control of the reaction and cis isomers being products of thermodynamic control[83]. In terms of their catalytic activity in olefin metathesis reactions, sulfur-containing ruthenium derivatives are somewhat inferior to their oxygen-containing counterparts (Table 2 and Table 3). In general, higher molar loadings of the S → Ru catalyst are required in RCM model reactions. Complexes with electron-withdrawing substituents in the arylidene ligand, such as cis-chelate 44, show higher catalytic activity. The influence of the nature of the substituent at the sulfur atom on the catalytic activity remains unclear.
2.1.3. Complexes with the Se → Ru bond
In 2009, the group of Straub and Lemcoff[66] first characterized a selenium-containing ruthenium complex obtained from styrene 52 and the ruthenium precursor G-II (Scheme 26), similar to the previously described sulfur-containing metallacycles 35 (see Scheme 16). The synthesis of the starting seleno-substituted styrene 52 was challenging and the overall yield of this compound was 3%. The catalytic activity of the complex 53 was not investigated.
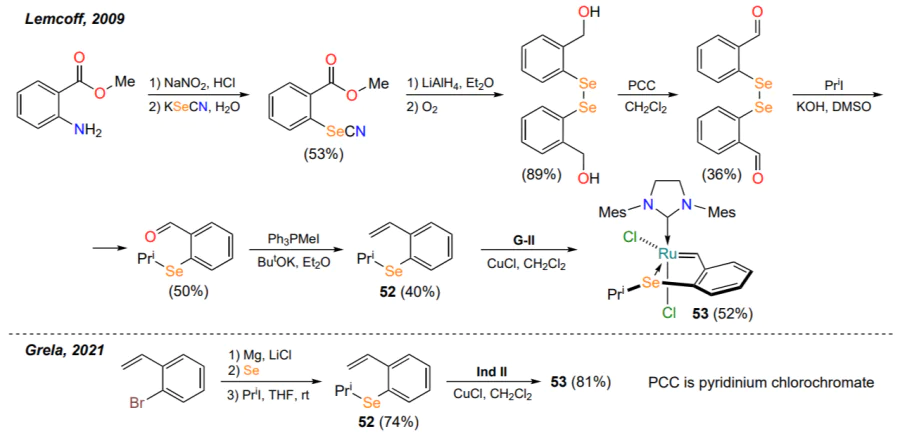
In 2021, Grela and co-workers[84] proposed a more convenient approach to the preparation of the ligand 52 and the complex 53 based thereon. The authors developed a virtually one-pot procedure for the synthesis of selenide 52 from 2-bromostyrene, minimizing the contact time with toxic organoselenium compounds.
As expected, catalyst 53 (1 mol.%) showed moderate activity in the model RCM reaction (conversion of substrate 31 to product 33, see Table 3) at a substrate concentration of 0.5 mol L–1. Under these conditions, refluxing the reaction mixture in benzene or toluene for 1 day provided pyrroline 33 in yields of 63 and 79%, respectively. Complex 53 can be photoactivated like its sulfur-containing analogue 35c (see Scheme 16 and Scheme 17), in which case the yield of product 33 is 73%. Compound 53 is stable to prolonged heating in air.
To summarize, the Hoveyda – Grubbs type complexes with Se → Ru bond are virtually unstudied. The available data suggest that selenium-containing ruthenium complexes are less active in olefin metathesis reactions than analogous sulfur and oxygen derivatives.
Ruthenium complexes with coordinating tellurium atoms of the HG-II-type have not yet been obtained, which is explained by the higher metallicity of this chalcogen in comparison with other elements of the sixteenth group. There are reasonable doubts both about the high stability of complexes with Te → Ru bonds and about the prospects of their application as catalysts for metathesis reactions. However, the development of methods for their preparation is of some fundamental interest.
2.2. Hoveyda – Grubbs catalysts with pnictogens as coordinating heteroatoms
2.2.1. Complexes with the N → Ru bond in the metallacycle
This Section summarizes the methods for the preparation of ruthenium complexes with pnictogens as coordinating heteroatoms, of which metal complexes with the N → Ru bond are the most widely reported in the literature. It should be noted that in the vast majority of cases the ruthenium and nitrogen atoms in such coordination compounds are part of a five- or six-membered metallacycle. This is explained by the availability of starting arylidene ligands that are most often derivatives of pyridine or o-vinylaniline. There are no data on the synthesis of HG-II-type ruthenaheterocycles with nitrogen as the coordinating atom, in which nitrogen and ruthenium atoms are incorporated into a seven-membered or larger ring.
The first report on the possibility of obtaining nitrogen-containing ruthenium complexes of the Hoveyda – Grubbs type seems to be a publication by Van Der Schaaf et al.[85] in 2000. Starting from 2-(but-3-enyl)pyridines 54 and a modified first-generation Grubbs catalyst 55, the authors managed to obtain ruthenium 2-pyridylallylidene complexes 56 with triisopropylphosphine ligand (Scheme 27), yields of products 56 over two steps are given).
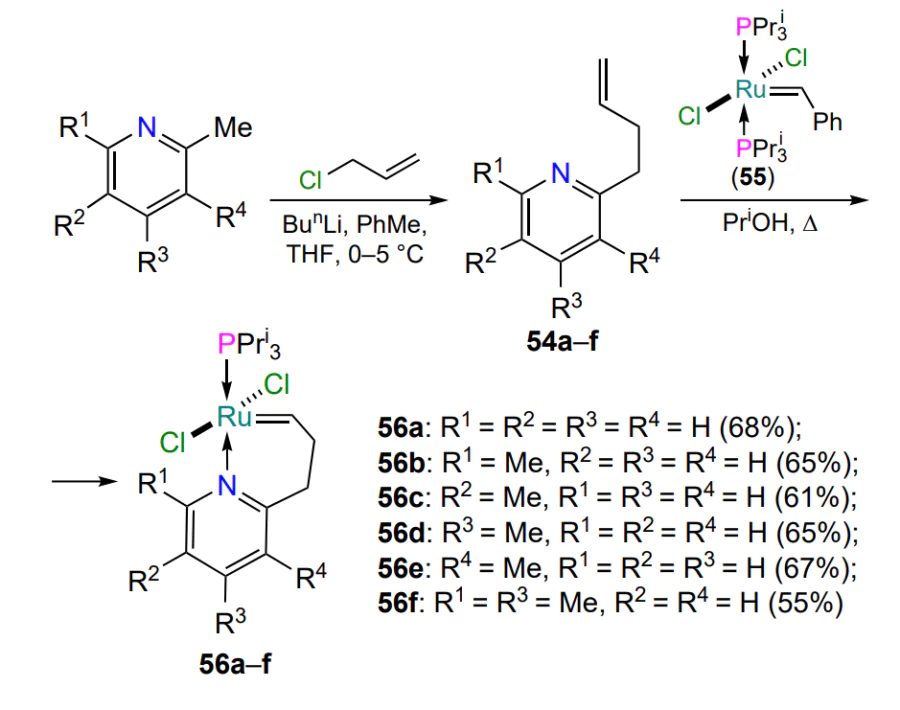
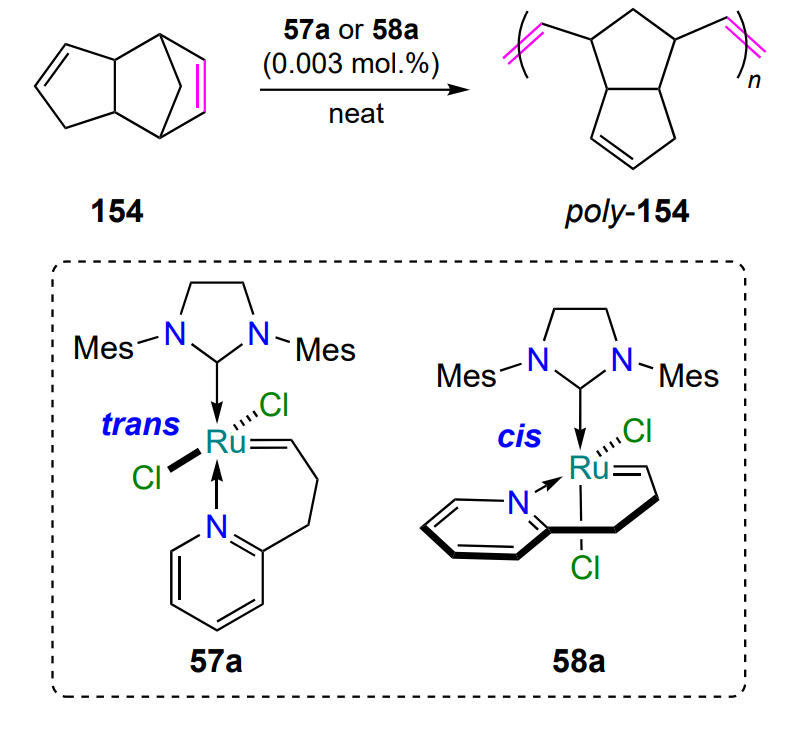
In 2004, Schrodi and co-workers[86] synthesized analogues of ruthenaheterocycles 56, ruthenium chelates 57 with NHC ligands, and explored their isomerization and catalytic activity. It was found that partial isomerization of the trans complexes 57 to the corresponding cis isomers 58 is possible upon heating to 40 °C (Scheme 28). For example, according to 1H NMR spectroscopy, when a solution of isomer 57a was kept in dichloromethane for 3 – 4 days, the ratio of 57a : 58a complexes was ~ 30 : 70. The mechanism of isomerization of 57 ⇆ 58 was clarified by density functional quantum chemical (DFT) calculations, which confirmed the experimental observations[83]. Compared to commercially available catalysts such as HG-II and G-II, complexes 57 were less efficient in the ring-closing metathesis reaction of dienes (see Table 4 below).
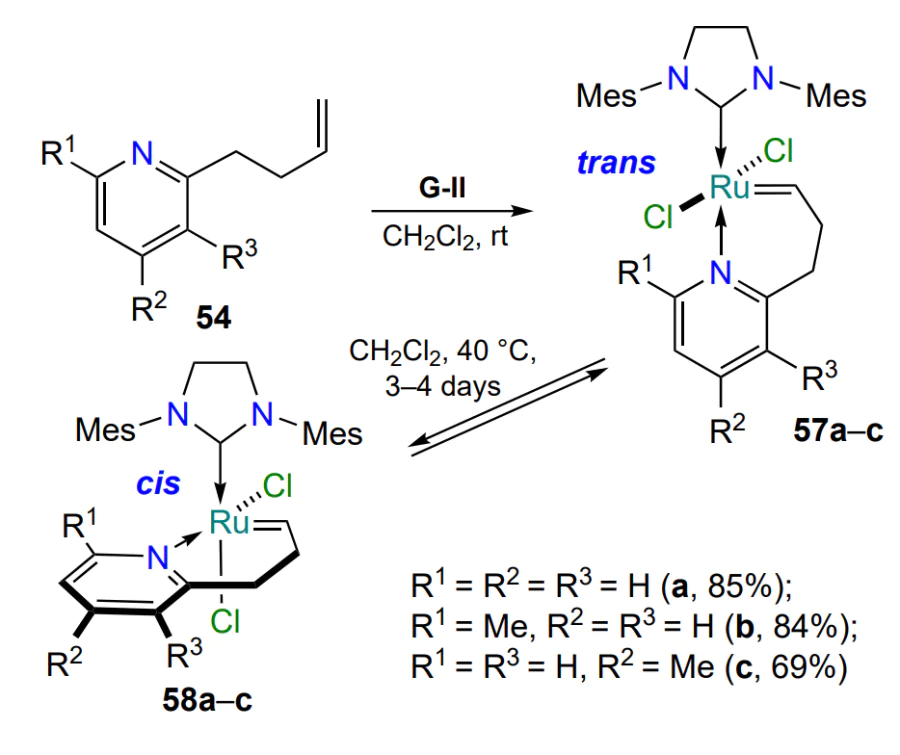
Based on arylidene ligands 59a,b, ruthenium complexes 60 with an imino group coordinating the ruthenium atom have been synthesised[87]. The authors note that the exo- or endo-cyclic position of the C=N bond in the complex significantly affects its catalytic properties (Scheme 29). Complex 60a with endo-cyclic C=N bond is stable and inactive in alkene metathesis reactions at room temperature. On the contrary, its positional isomer 60b with an exo-cyclic double bond is active under the same conditions. Later it was found that the introduction of electron-donating groups in the alkylidene ligand favorably affects the catalytic activity of ruthenaheterocycles 61[88]. The authors of the study[89] published in 2021 prepared two complexes 61h and 61i containing an intramolecular hydrogen bond N – H∙∙∙Cl, the presence of which leads to a significant decrease in catalytic activity compared to other catalysts 61.
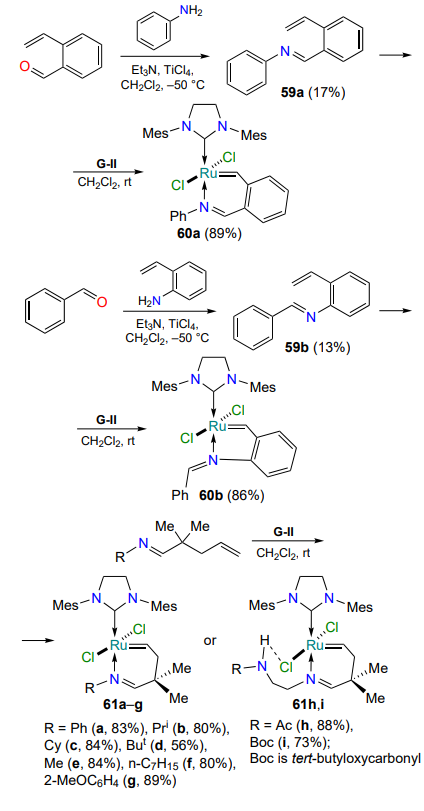
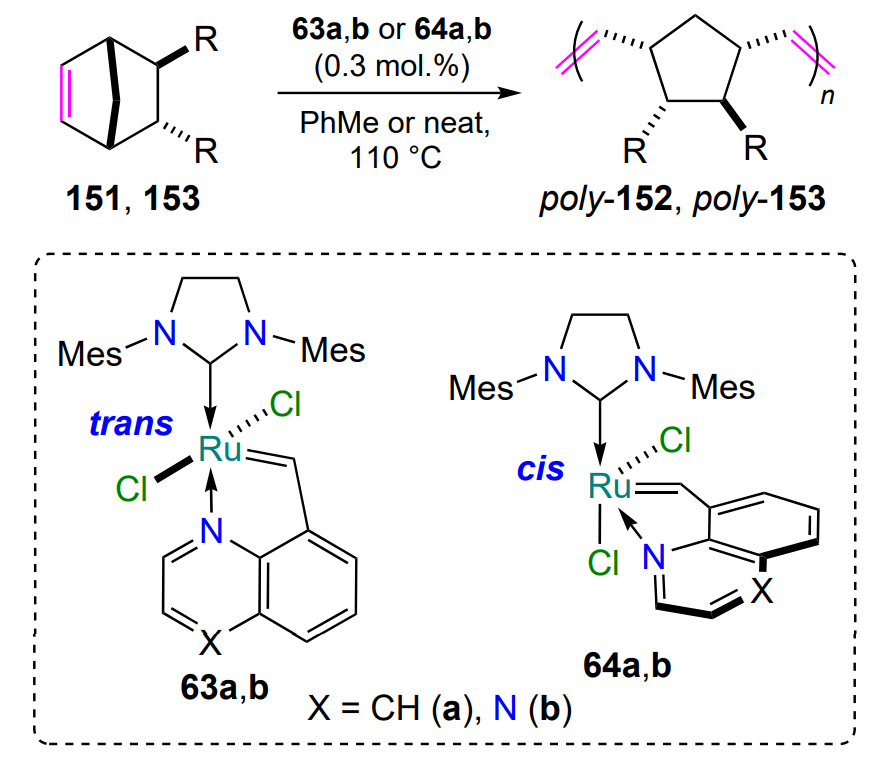
Ruthenium quinoline- and quinoxaline complexes 63a,b were prepared from styrenes 62a,b. Substrates 62a,b were obtained by the Stille reaction from 8-hydroxyquinoline or 5-bromoquinoxaline triflates, respectively[90]. Complexes 63a,b containing a five-membered metallacycle are formed as trans isomers, which upon prolonged storage in dichloromethane at room temperature transform into the more thermodynamically stable cis-forms 64a,b. Catalytic activity tests showed that the cis complexes 64 are less efficient than their trans isomers 63, with the quinoxaline-containing chelate 64b being activated more rapidly (Scheme 30). For the cis isomers, as in the case of the S-chelates 35 (see Scheme 16), a thermoswitching effect is observed[91]. In air, both cis complexes 64a,b are stable in solution for at least two weeks.
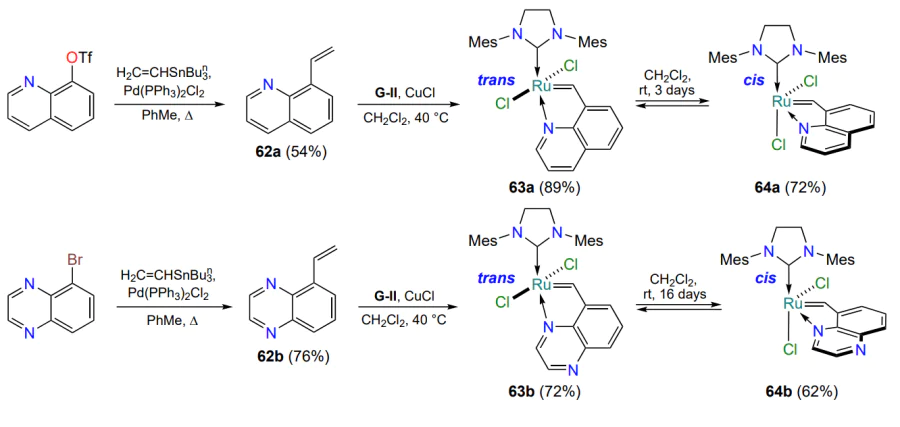
Another ruthenium complex 65a, containing a quinoxaline ligand, was unexpectedly isolated in the course of the development of the total synthesis of the drug Grazoprevir, used in the treatment of hepatitis C[92, 93]. Based on this result, a series of structural analogues of catalyst 65a, complexes 65b with five-membered (n = 0) and 65c – g with six-membered metallacycles (n = 1) was obtained (Scheme 31)[93]. The catalytic activity of these complexes varies from high to low, demonstrating once again the possibility of controlling the reactivity of ruthenium catalysts by modifying the arylidene moiety. Compound 65c (R = OPri), which contained a six-membered ruthenaheterocycle, proved to be the most active in the RCM model reaction.
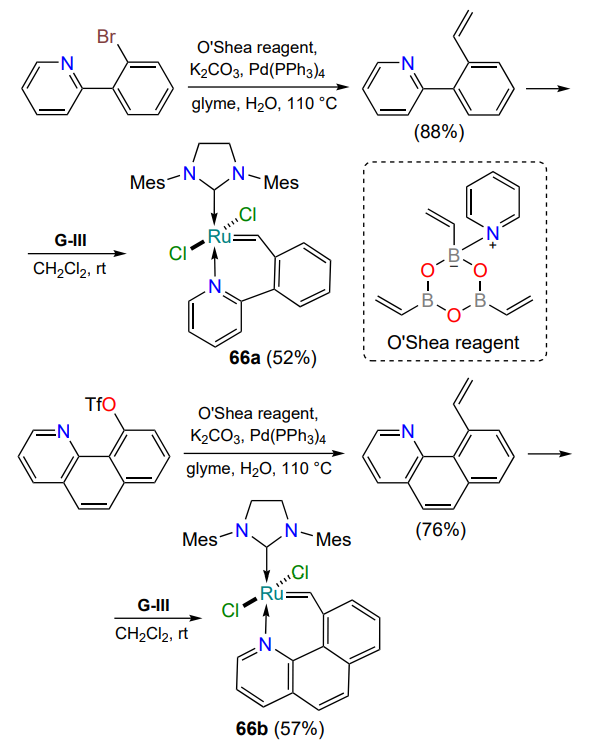
Interest in thermoswitchable complexes extends beyond the studies described above. In this paradigm, ruthenaheterocycles with bulky 2-(2-vinylphenyl)pyridine (66a) or 10-vinylbenzo[h]-quinoline (66b) moieties have been prepared[94]. Despite low substrate conversion in RCM reactions, both complexes were stable in air and did not undergo degradation in boiling toluene for 48 h (Scheme 32). Both trans complexes 66 are thermoswitchable, as demonstrated by ROMP reactions.
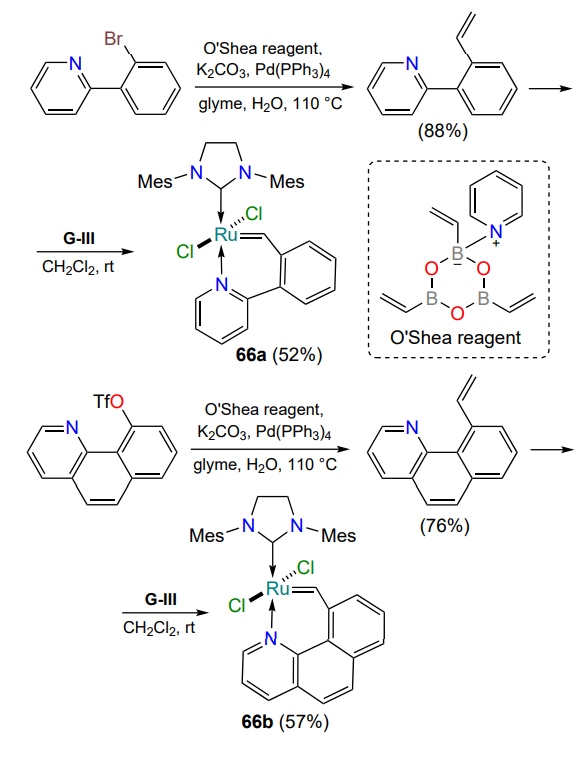
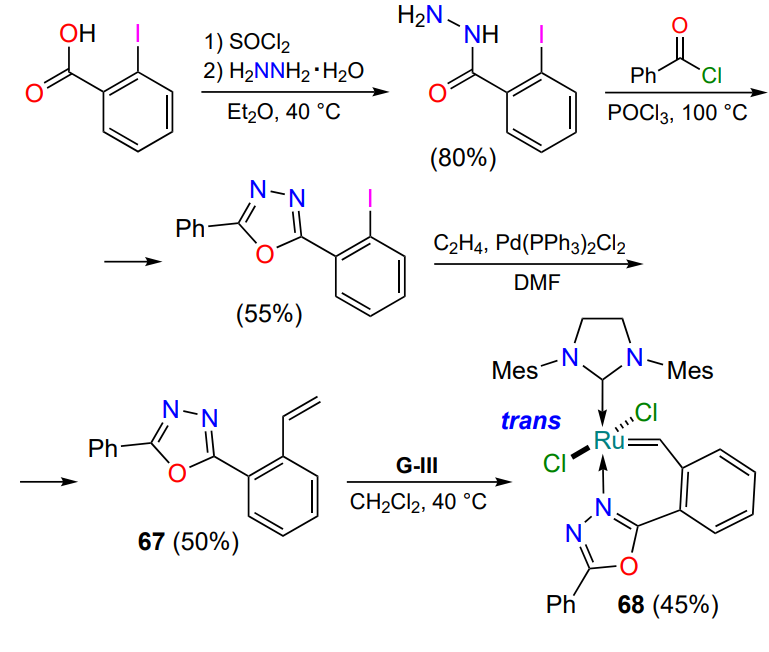
The pyridine nitrogen is not the only possible donor atom. A 2021 publication[95] discusses the properties of a complex in which the nitrogen atom of the oxadiazole ring forms a coordination bond with ruthenium. The starting styrene 67 was prepared in three steps, the key one being the cyclization of aroylhydrazine to 1,3,4-oxadiazole (Scheme 33). The trans configuration of the complex 68 was confirmed by XRD. Like most of the nitrogen-containing catalysts described in this Section, complex 68 is active in alkene metathesis reactions only at elevated temperatures.
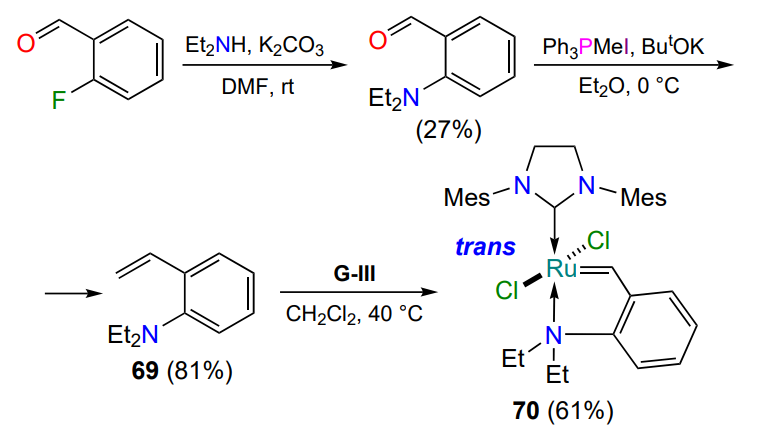
The first structural aza-analogue of the most common oxygen-containing Hoveyda – Grubbs catalyst HG-II (see Figure 1) was described in 2009[66]. Similar to the preparation of 2-vinylphenyl sulfides 34 (see Scheme 16), Lemcoff and co-workers[66] synthesized the key intermediate 69 from commercially available 2-fluorobenzaldehyde (Scheme 34). The trans geometry of the resulting chelate 70 with the N → Ru bond was established by X-ray diffraction analysis.
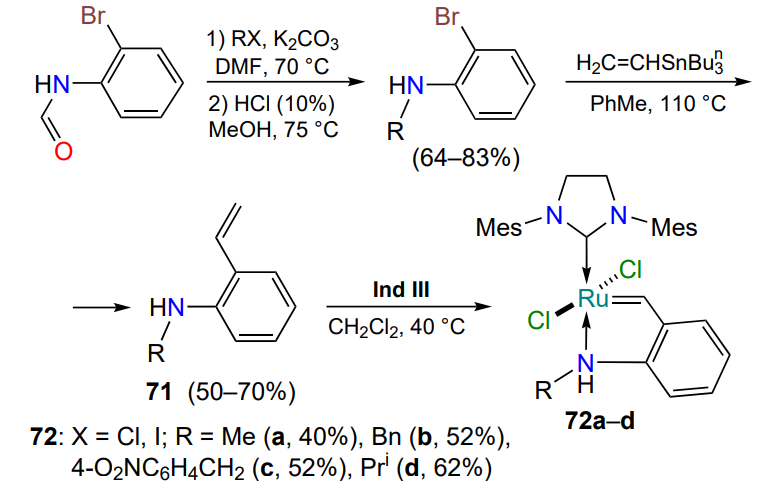
In 2012, catalysts were obtained in which the nitrogen atom of the secondary amino group of styrenes 71 coordinates the ruthenium ion[96]. The amine hydrogen atom has no effect on the process (Scheme 35). However, it was found that complexes 72 are catalytically inert in metathesis reactions without thermal activation.
Grela and co-workers[97] also explored the possibility of obtaining ruthenium complexes in which the donating heteroatom is the nitrogen atom of the primary amino group (NH2) (Scheme 36). The presence of two mobile hydrogen atoms in the molecule influences the course of complex formation. When 2-vinylaniline (73) reacts with first- (G-I) and second- (G-II) generation Grubbs catalysts, the corresponding trans-chelates 74a and 74b are formed. In the presence of excess tricyclohexylphosphine, these complexes are readily converted to ruthenaazaheterocycles 75a,b in which the nitrogen atom is covalently bonded to the metal cation[97].
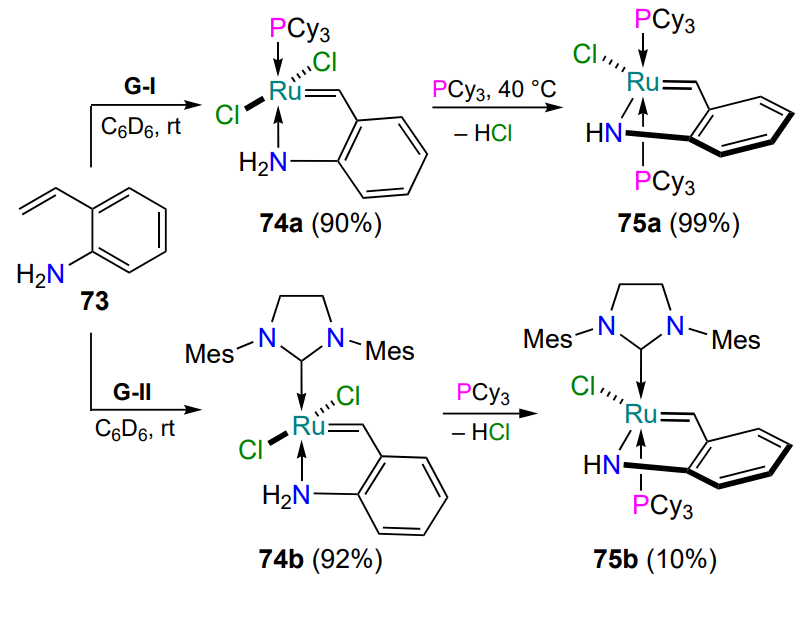
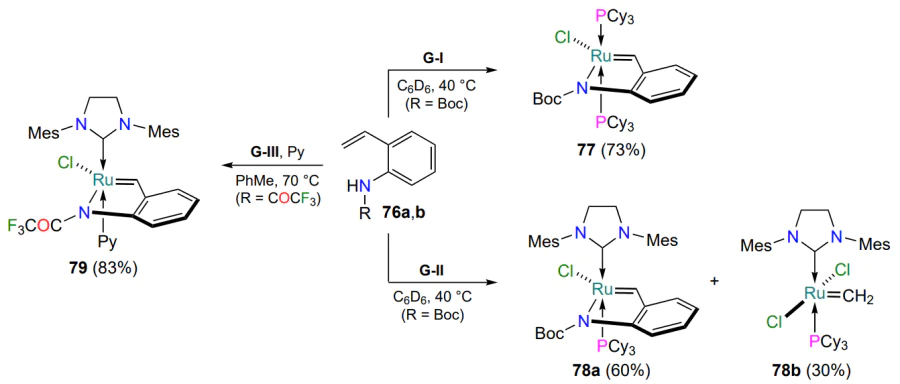
Apparently, the amide nitrogen atom is not able of forming an intramolecular coordination bond with ruthenium, but an increase in the acidity of the amide proton creates the prerequisites for the formation of the N – Ru covalent bond[97, 98]. The products 77 – 79 obtained by the reaction of styrenes 76a,b with precursors G-I, G-II and G-III are cis isomers according to the arrangement of the halogen and nitrogen atoms in the trigonal bipyramid of the ruthenium coordination sphere (Scheme 37). In the case of catalyst G-II, the trans-vinylidene derivative 78b is formed as a by-product[98]. Scheme 37 shows the yields of the products 24 h after the start of the reactions, determined by monitoring by 1H NMR spectroscopy. Catalysts 77, 78a, 79 comprising the 2-ruthenaindole ring are activated by hydrogen chloride solution in diethyl ether. Under these conditions the above complexes are not inferior to commercially available HG-II and G-II in terms of activity in alkene metathesis reactions.
A method for the preparation of structural analogues of complex 70 (see Scheme 34) containing a six-membered metallacycle with the N → Ru coordination bond has been reported[79, 99]. As with the sulfur-containing catalysts 48 (see Scheme 24), no procedure was provided for the synthesis of the starting styrenes 80 and the precursor 47 and the stereochemistry of the products 81 was not discussed (Scheme 38)[79]. Nevertheless, both the high activity of complexes 81 in the metathesis polymerization of dicyclopentadiene and their stability during storage in air were demonstrated. These observations marked the beginning of the use of ruthenaazaheterocycles 81 in industrial polymerization processes.
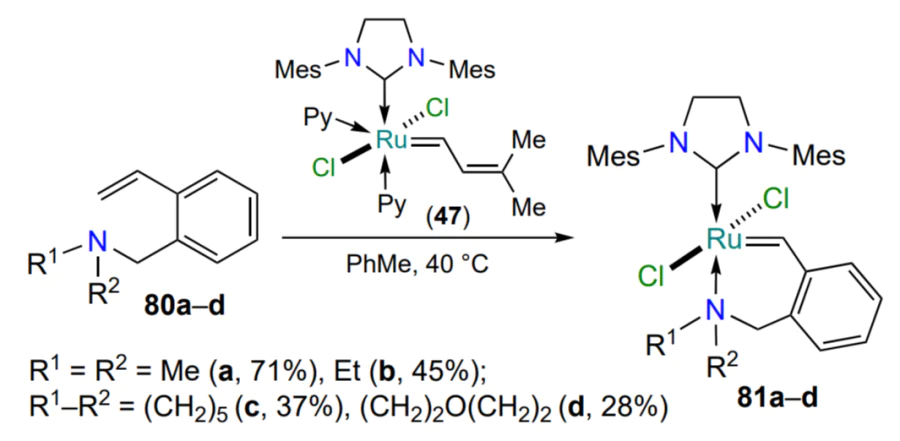
The methods of preparation and properties of nitrogen-containing ruthenium complexes with six-membered metallacycles were studied in detail in a series of works by the Zubkov’s group[100-102]. The methods of synthesis of catalysts have been optimized and the range of starting 2-vinylbenzylamines 80 has been considerably extended (Scheme 39)[100, 101]. The results of a systematic analysis of the catalytic activity of complexes 81 in metathesis reactions suggested that the presence of bulky substituents, in which the donating nitrogen atom is part of the metallacycle (81d,g – i), helps to maximize conversions of alkenes already at room temperature. At the same time, complexes 81a,b,e with N-alkyl substituents reveal their catalytic properties only in boiling chloroform. Based on styrenes 82 with additional alkyl substituents R3 in the benzylic position of the arylidene moiety, a series of metallacycles 83 have been obtained which are superior in catalytic activity to the chelates 81a – f[102]. This fact is the result of a weakening of the nitrogen-ruthenium coordination bond due to an increase in the steric crowding in the ruthenaheterocycle.
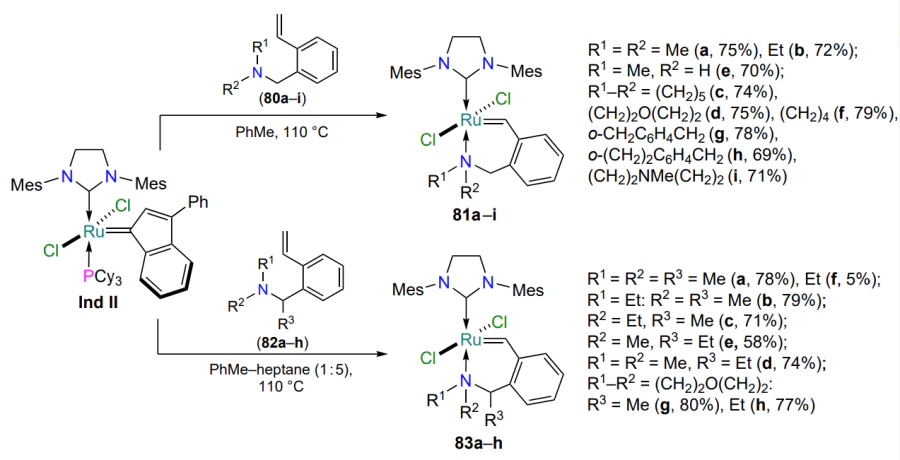
The method proposed in Scheme 39 has limitations. It has been shown that increasing the steric bulk of substituents R1, R2 and R3 in complexes 83 leads first to elongation and then to destabilization of the N → Ru coordination bond. In particular, the introduction of an isopropyl substituent at any of the modifiable positions hampered the formation of the target ruthenaheterocycle. The morpholine-containing metal complexes 83g,h performed best in RCM; chelates 83a,b showed the least activity. Under the same conditions, compounds 81d,g,h,i and 83a – h showed the best activity in the RCM benchmark reaction compared to the commercially available HG-II.
Most of the metal complexes described in this Section are air stable products that can be obtained in kilogram quantities. The presence of a nitrogen atom in the ruthenium catalyst molecule makes it possible to fine-tune its activity by selecting substituents to achieve the required rate of practically significant metathesis reactions.
In particular, Hoveyda – Grubbs type nitrogen-containing complexes 81 and 83 have proved to be effective catalysts for ring-opening metathesis polymerization. The absence or weakly expressed catalytic properties at room temperature of several such structures allows the use of ruthenaazaheterocycles in industrial polymer production; smooth polymerization occurs when reaction mixtures are heated. For example, complexes 81 find industrial applications in the production of high strength polymers from dicyclopentadiene, a toxic and teratogenic product of hydrocarbon pyrolysis[103-108]. Ring-opening metathesis polymerization is discussed in more detail in Section 4.
2.2.2. Complexes with the P → Ru bond in the metallacycle
Complexes containing a phosphorus-ruthenium coordination bond in a five- or six-membered metallacycle are known to be the most inert catalysts for the alkene metathesis reaction. For this reason, the synthesis of such structures has not received adequate attention. At the beginning of 2024, only two such compounds had been described[66, 109, 110].
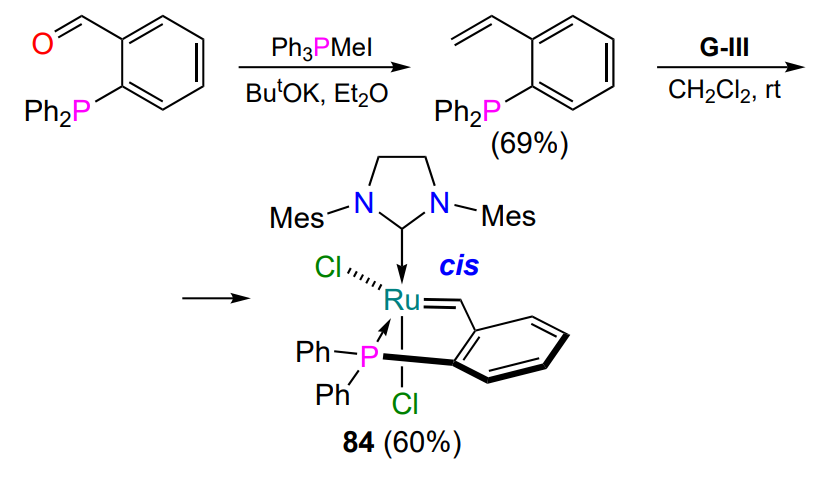
Using ortho-substituted benzaldehyde as a starting material, Lemcoff and co-workers[66] obtained chelate 84, the synthesis of which was further improved[109]. Complex 84 exists in cis-configuration and shows catalytic activity in RCM only at the boiling point of chloroform or toluene (Scheme 40).
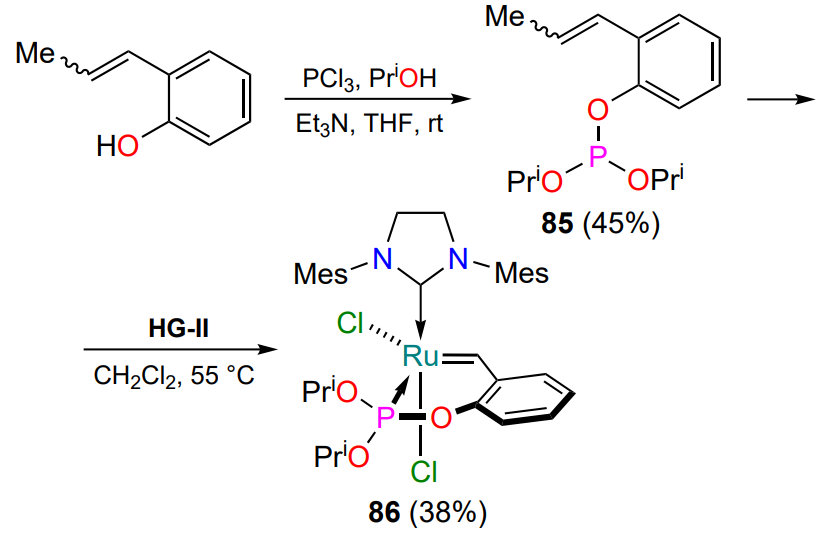
In 2018, the study on the activity of a complex with a ruthenium-coordinating phosphite phosphorus atom was reported[110]. The target ligand was synthesized from the HG-II catalyst based on phosphoric acid triester 85 (Scheme 41). Complex 86 was stable in dichloromethane and toluene solutions in the presence of air. Under thermal or photoactivation conditions, it was used to achieve moderate conversions in the RCM reaction (Table 4). At room temperature, complex 86 was inert in the same processes.
2.3. Hoveyda – Grubbs catalysts with the halogen−ruthenium coordination bond
Hoveyda – Grubbs type ruthenium complexes, in which the halogen atom coordinates ruthenium, are described in a few works[111, 112]. The authors give methods for the preparation of bromine- and iodine-containing catalysts 87 – 89, which are cis-isomers (Scheme 42). The formation of similar complexes with more electronegative chlorine or fluorine atoms is probably impossible.
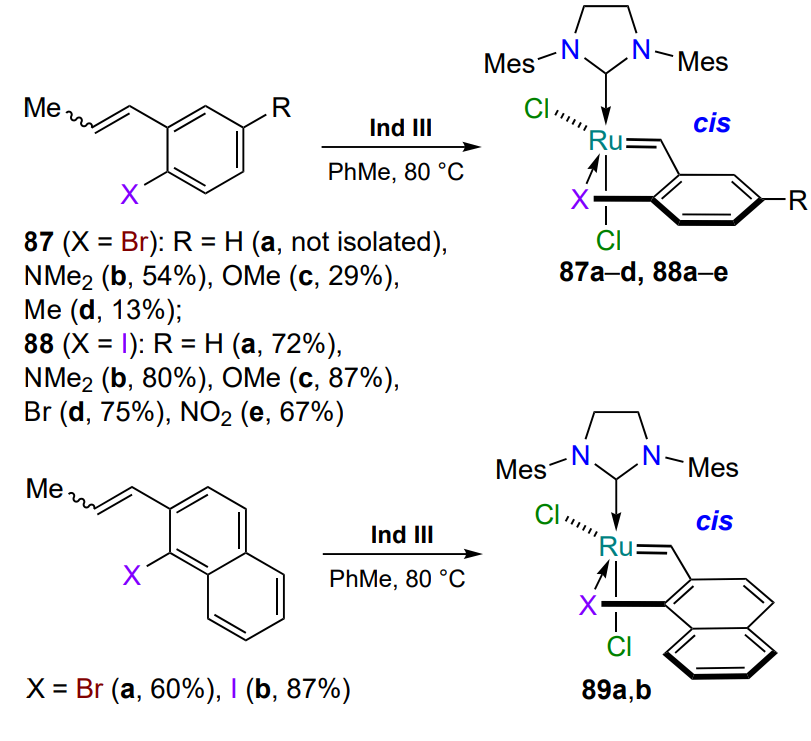
The bromide 87a (R = H) could not be isolated, but its formation was confirmed by 1H NMR spectroscopy of the reaction mixture. Electron-donating substituents R in the arylidene ligand in the para position with respect to the bromine atom significantly increase the yield of complexes 87b,c. The electron-donating naphthylidene moiety in the chelates 89 has the same influence on the yield of the products. Substituents in the para position to the more polarizable iodine atom have practically no effect on the yield of aryliodides coordinated to the ruthenium atom 88[112].
Ruthenaheterocycles 87 – 89 are stable in dichloromethane or toluene solutions in air, but show reduced catalytic activity compared to the HG-II complex in the model RCM reaction at room temperature (Table 5). The most active is the bromine derivative 87b.
2.4. Hoveyda – Grubbs type ruthenium catalysts containing tridentate ligands
The strategy of directed modification of arylidene ligands was continued with the synthesis of 18-electron complexes containing simultaneously two ruthenium coordinating heteroatoms. In this case, oxygen, sulfur and nitrogen act as donor atoms. It should be noted that the development of synthetic approaches to such metal complexes is just in its infancy, and the relationship between their catalytic activity and the nature of the heteroatoms is still unclear. There is also question about the stability of some of the ruthenium-containing bicycles.
The first studies in the field of obtaining Hoveyda – Grubbs type ruthenium complexes with tridentate ligands were aimed at constructing structures with a donating oxygen atom mimicking the well-known HG-II complex. The second coordination bond with ruthenium was expected to be achieved via the carbonyl or carboxyl oxygen (Scheme 43)[113-115]. However, it was found that the formation of 18-electron ruthenium complexes 90a – d is only observed with the coordinating oxygen atom of the carboxyl moiety. The complexes with a donating carbonyl moiety 90e – g are structural analogues of the HG-II complex, with the presence of the second heteroatom 90a – d leading to a change in geometry: the ligand environment of the central atom acquires an octahedral shape.
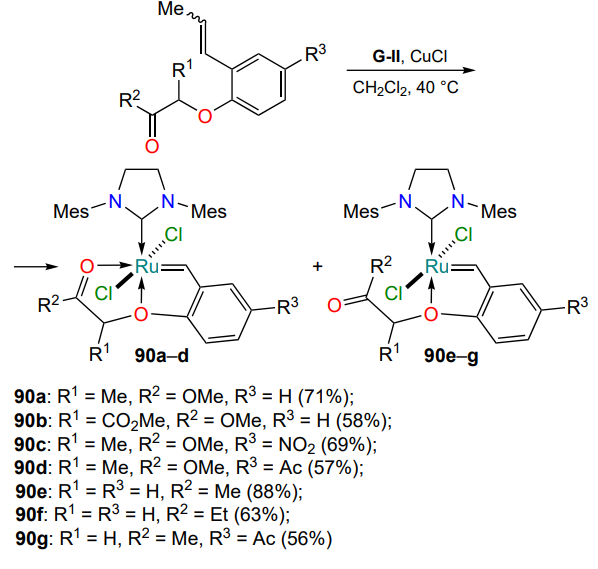
Chelates 90 were found to be efficient olefin metathesis catalysts. Their activity is superior to that of HG-II complexes catalyzing RCM reactions at 0 °C (Table 6). An interesting feature of compounds 90a – d is a pronounced E-selectivity in cross-metathesis reactions, which allows their use in the stereoselective synthesis of disubstituted alkenes.
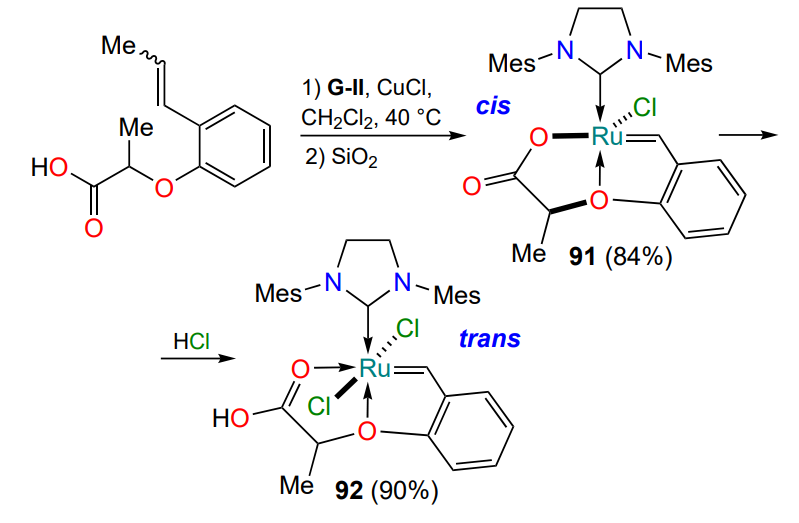
Another specific feature of the complexes 90a – d is the possibility of ligand exchange in the case of derivatives with R2 = OH (Scheme 44). In an attempt to isolate the tricycle 92 by column chromatography, its dehydrohalogenation product 91 is formed in good yield as a result of the exchange of one of the chloride anions for a carboxylate anion[116]. Complex 91 shows extremely low catalytic activity in olefin metathesis reactions, but can be activated by the addition of Brønsted acids (HCl, CF3SO3H, CF3(CF2)7CO2H) (see Table 7). Treatment with hydrogen chloride converts monochloride 91 to trans-dichloride 92.
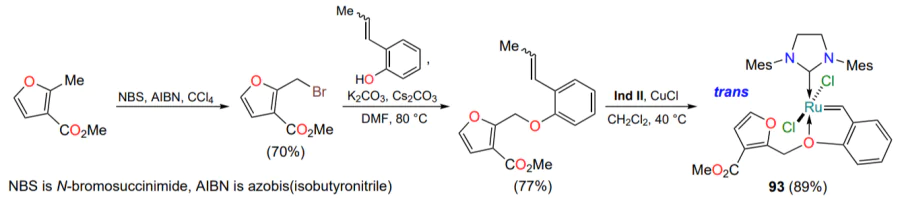
Attempts to obtain a ruthenium-containing bicyclic compound in which the second coordination of ruthenium occurs via the furan oxygen atom failed[117]. An arylidene ligand precursor (Scheme 45) was synthesized in two steps from the methyl ester of 2-methyl-3-furancarboxylic acid and reacted with the Ind II ruthenium complex. However, study of the structure of the product 93 by XRD revealed that the furan oxygen is too weak a donor. No interaction between the ruthenium atom and the oxygen atoms of the carboxylic group to form a seven-membered ruthenaheterocycle is observed. Nevertheless, the obtained complex 93 actively catalyzes the model RCM reaction at 0 °C. Its activity is shown to be superior to that of the HG-II catalyst under the selected conditions[117].
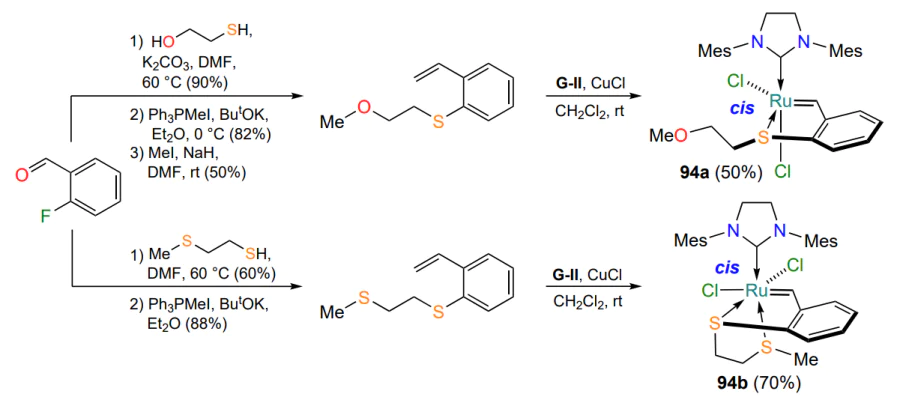
One or both donating oxygen atoms in ruthenium complexes of type 92 can be replaced by isosteric atoms, resulting in the formation of metallacycles with one or two S → Ru bonds[74]. Scheme 46 illustrates the synthesis of the corresponding sulfides and the ruthenium complexes 94 based thereon. According to the X-ray analysis data, the products 94a,b are cis-isomers according to the arrangement of the chlorine atoms. In complex 94a, the coordination bond O → Ru is not realized, whereas in its analogue 94b, the second bond S → Ru is readily formed. This is apparently due to the higher polarizability and lower electronegativity of the sulfur atom. Both ruthenium derivatives 94 (1 mol.%) proved to be inert in the RCM reaction both at room temperature and under thermal or photoactivation.
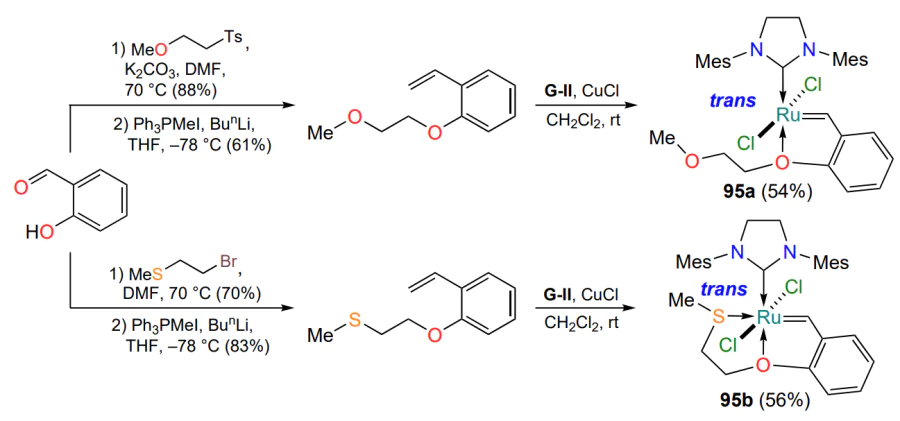
Scheme 46 is well complemented by the report of Kinugawa and Matsuo[118], published in 2023, in which the authors proposed an approach to the trans complexes 95a,b, which also contain sulfur and oxygen atoms coordinating ruthenium. The X-ray analysis showed that, as with complex 94a, the formation of the second oxygen – ruthenium coordination bond in the metallo-cycle 95a does not occur (Scheme 47). Both catalysts (95a and 95b) show high activity at room temperature in RCM reactions. The presence of the S → Ru coordination bond increases the stability of complex 95b and allows catalytic experiments in methanol (highly active commercial Hoveyda – Grubbs type catalysts are usually unstable in protic solvents)[119].
Attempts to create an additional coordination bond between the ruthenium and the oxygen (or nitrogen) atoms of the ester (or amide) group in the ruthenaheterocycles 96 were unsuccessful (Scheme 48)[120]. At the same time it was shown that dihalides 96a – c bearing a secondary amide group exhibit increased stability in benchmark metathesis reactions, probably due to the presence of an intramolecular N – H∙∙∙Cl hydrogen bond, similar to complexes 61h,i (see Scheme 29).
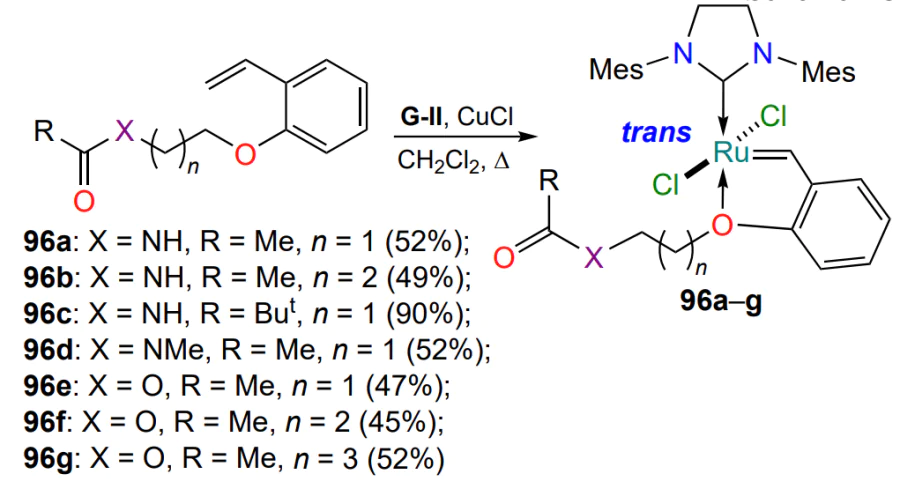
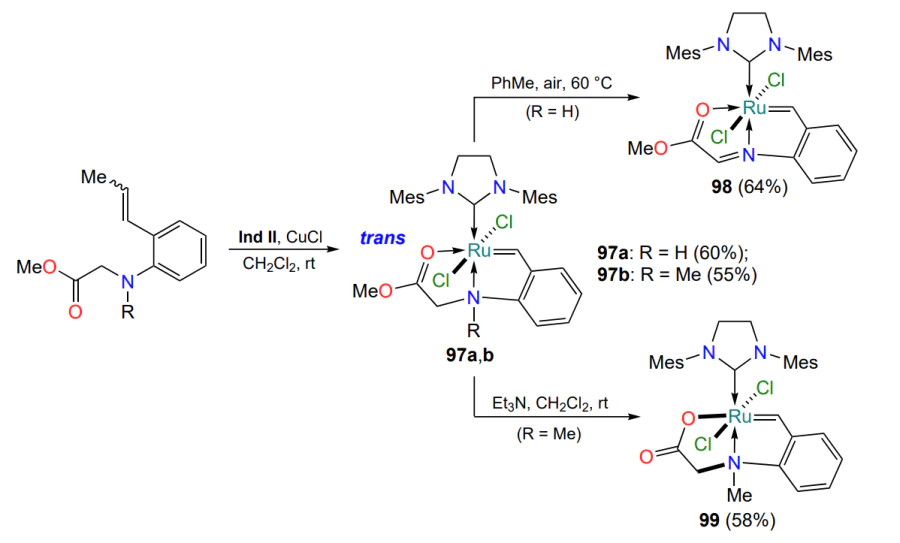
Several publications have described the synthesis and properties of the aza-analogues of complexes 90 (see Scheme 43)[121, 122]. For example, one study[121] discusses ways of modifying the esters 97a,b (Scheme 49). Depending on the reaction conditions and the substituent R, ruthenaheterocycles 97 can be converted either into imino esters 98 by oxidation of the C – N bond by atmospheric oxygen or into a 16-electron complex 99 with a covalent O – Ru bond formed as a result of methanol elimination (by analogy with complex 91, see Scheme 44). In RCM reactions, complex 97a was unstable, while chelates 97b, 98 and 99 showed catalytic activity only on heating. Compound 97b performed best, while complex 99 was the least active. Catalysts 97b, 98 and 99 have the property of thermo- and chemoswitchability; their activity directly depends on temperature and the presence of additives, while in the absence of activating factors they do not undergo degradation in solutions in polar solvents. It should be noted that the coordination of ruthenium with the nitrogen atom favors the trans position of the chlorine atoms with respect to the central metal atom. On the contrary, analyzing the structures of the ruthenium derivatives 90, 92, 94, 95 and 97−99 shown in Scheme 44, Scheme 46, Scheme 47 and Scheme 49, it can be assumed that the formation of the sulfur-ruthenium coordination bond contributes to the stabilization of the cis isomers.
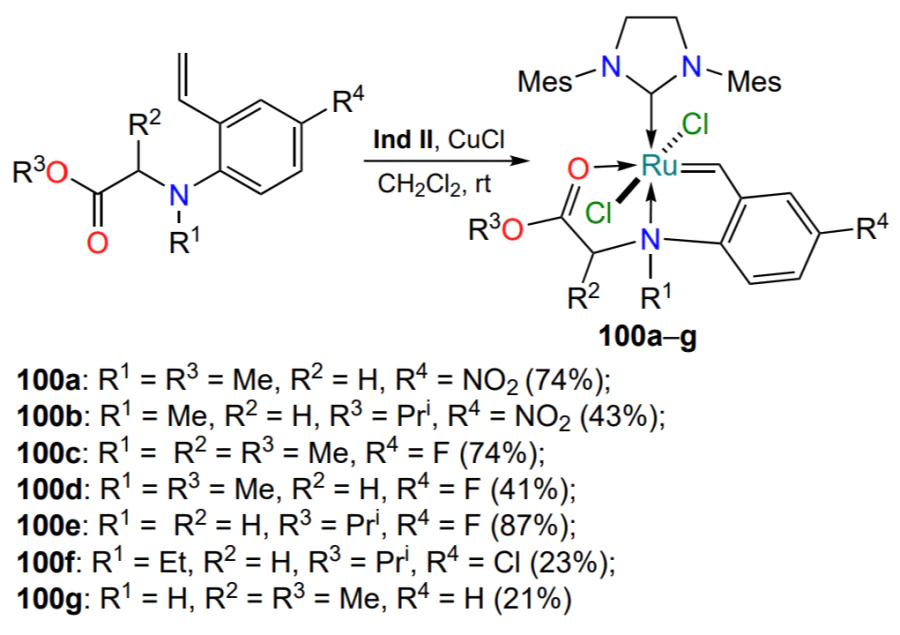
A series of ruthenium derivatives 100a – g with different substituents R2 – R4 in the benzylidene ligand were prepared using the standard approach as depicted in Scheme 46 – Scheme 49 above (Scheme 50). The study[122] does not discuss the spatial structure of the products, nor the possibility of ligand exchange, but the absence of catalytic activity in RCM reactions at room temperature was noted for all complexes 100.
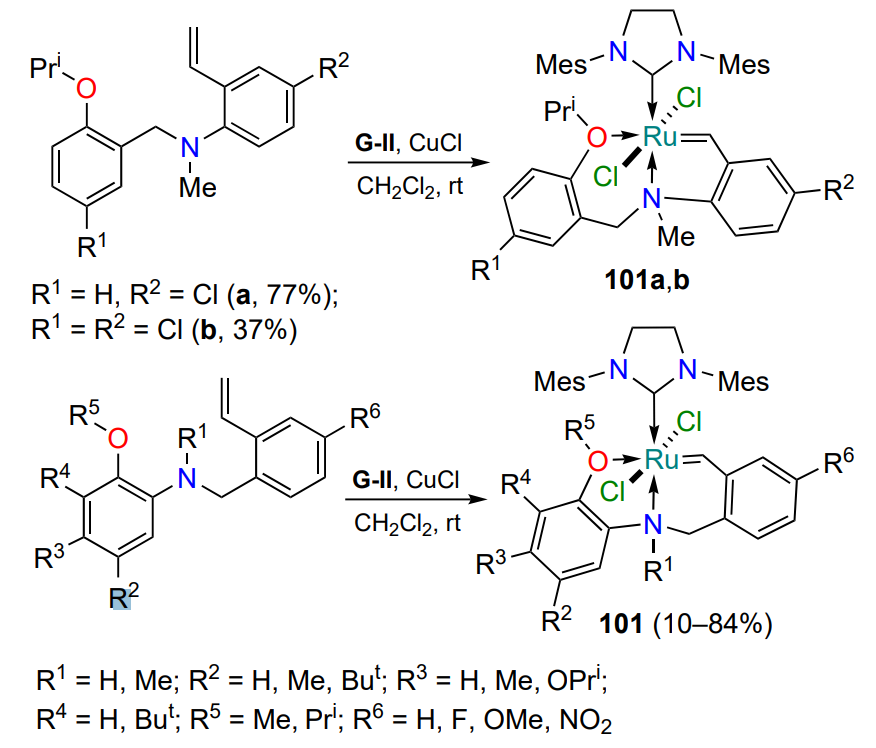
In addition to complexes with two five-membered fused rings, structures 101 with a combination of five- and six-membered fused ruthenaheterocycles are known (Scheme 51)[122]. The approaches to their preparation are similar to those described above in this Section. The catalytic activity of chelates 101 in metathesis reactions has not been explored.
The comparative catalytic activities of the metallocycles described in this Section are summarized in Table 7.
3. Metathesis in the straightforward synthesis
Over the past few decades, the olefin metathesis has become a powerful tool in the arsenal of organic chemistry methods[123-125]. Its use has simplified known and created new strategies for the synthesis of compounds with practically useful properties.
Reviews are published on this topic annually, so the first part of this Section only considers the examples that appeared in 2021 – 2023 for the preparation of complex natural molecules using the most popular commercially available oxygen-containing complex HG-II. The second part describes the use of other Hoveyda – Grubbs type catalysts with the Het → Ru bond to create scaffolds of natural compounds or potentially biologically active molecules.
3.1. Ring-closure metathesis reactions catalyzed by the HG-II complex
RCM reactions provides the one-pot synthesis of carbo- and heterocyclic units of various sizes, from five-membered to macrocycles[126, 127]. This approach is one of the most widely used in modern macrolide and cyclic alkaloid chemistry.
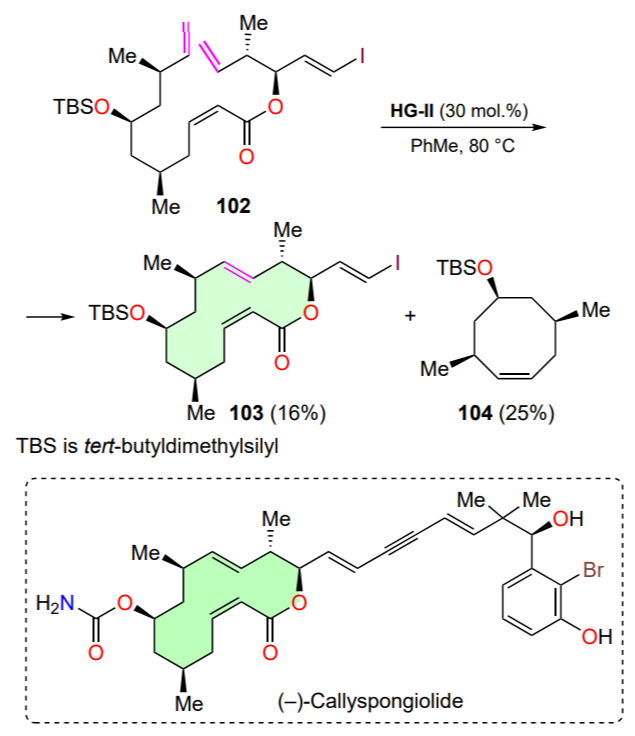
The macrolide (–)-callyspongiolide was isolated from the marine sponge Callyspongia sp. that inhabits the coast of Indonesia[128]. Structurally, (–)-callyspongiolide is a 14-membered lactone with five stereogenic centers; this compound exhibits in vitro cytotoxicity comparable to that of commercial cytostatics. The RCM reaction with tetraene 102 was used at the step of obtaining trans-lactone 103; cyclooctene 104 is also formed as a by-product (Scheme 52). The low yields of the target lactone 103 is due to steric interactions in the transition state of the intramolecular metathesis reaction[128].
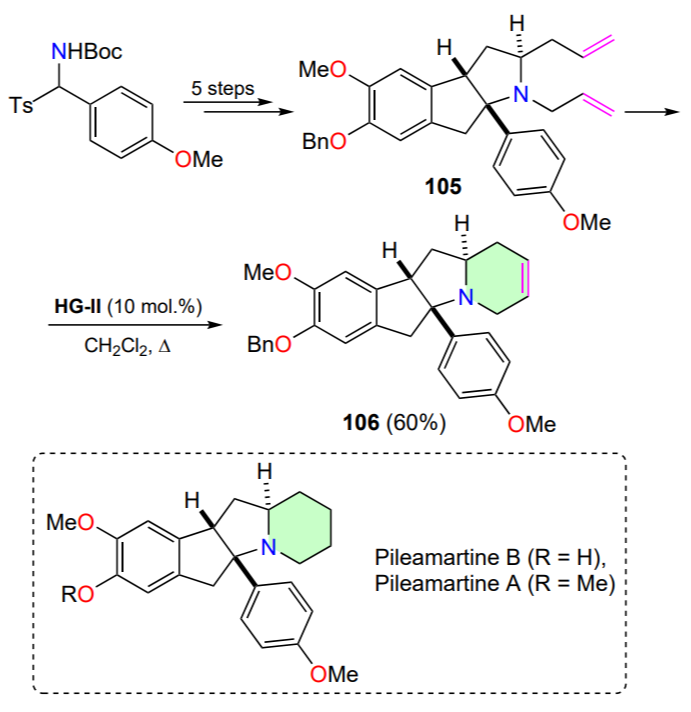
The ring-closing metathesis is a key step in the total synthesis of (±)-pileamartines A and B, a pair of tetracyclic alkaloids isolated from the leaves of the plant Pilea aff. martinii[129]. Tert-butyl[(4-methoxyphenyl)(tosyl)methyl]carbamate was used as the starting material in this method and RCM was carried out in the sixth step to cyclize diene 105 (Scheme 53). The use of the Hoveyda – Grubbs catalyst HG-II in the final step gives satisfactory yields of the target tetracycle 106.
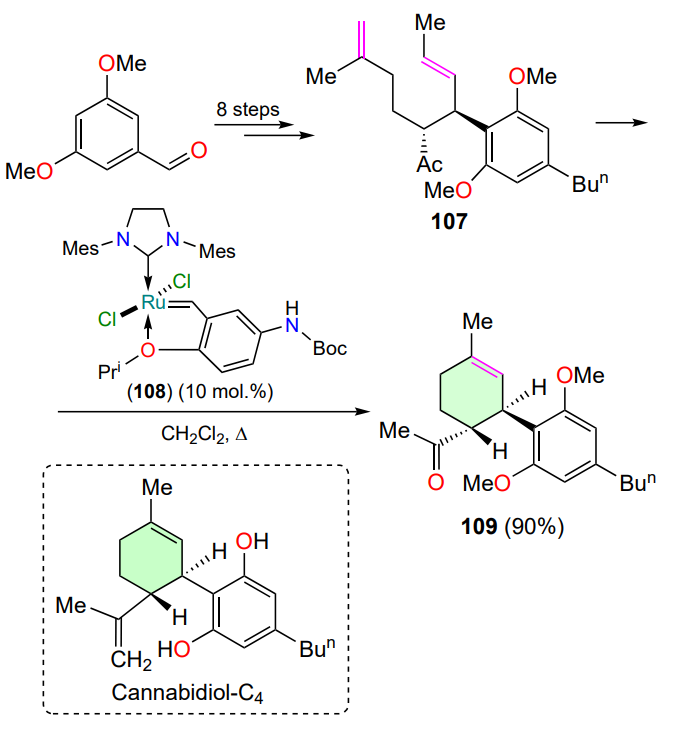
Cannabidiol-C4 (cannabidibutol, CBD-C4) was found in extracts of the Cannabis sativa[130]. Until 2022, there was no information on its isolation in individual form, so Passarella and co-workers[130] focused on the synthesis of cannabidiol-C4 and elucidation of its structure (Scheme 54). The authors developed a 12-step strategy in which one of the final steps (conversion of 107 to 109) involves an RCM reaction with substrate 107 in the presence of an analogue of HG-II, a Hoveyda – Grubbs type catalyst 108 bearing a donor substituent in the arylidene moiety[131, 132]. The product 109 was obtained in high yield.
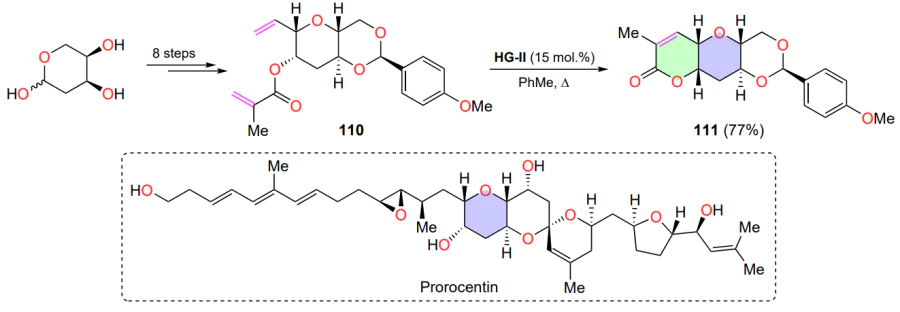
The RCM reaction found application in the total synthesis of prorocentin[133], which is of practical interest because of its potential cytotoxic and neurotoxic properties (Scheme 55). In the method proposed by Fürstner and co-workers[133], the HG-II catalyst was used for the formation of a six-membered ring in diene 110. Despite the fact that the formation of a trisubstituted double bond requires elevated temperature (boiling toluene) and a significant catalyst loadings, the yield of the product 111 was relatively high.
3.2. Cross-metathesis reactions catalyzed by the HG-I and HG-II complexes
Cross-metathesis (CM) reactions are a convenient way to functionalize terminal alkenes containing a variety of functional groups[26, 134]. Typically, a successful CM process requires the use of a significant excess of one of the interacting olefins.
The total synthesis of chabrolobenzoquinone H (a meroditerpene with pronounced cytotoxic activity) includes a cross-metathesis reaction involving allylbenzene 112 and a series of low-molecular-weight alkenes[135]. The reaction of isobutylene or methyl methacrylate with olefin 112 gives two different products, 113 and 114 (Scheme 56). The reaction with isobutylene produces the expected internal alkene 113 with complete conversion of the substrate 112. In a similar reaction with methyl methacrylate, a mixture of E- and Z-isomers of the olefin 114 was isolated as a result of migration of the terminal double bond in the starting allylbenzene 112. The authors were able to obtain compound 115, which was required for the further synthesis of chabrolobenzoquinone H, when the HG-II catalyst was replaced with a G-II complex. The reasons for this chemoselectivity were beyond the scope of the discussed study[135].
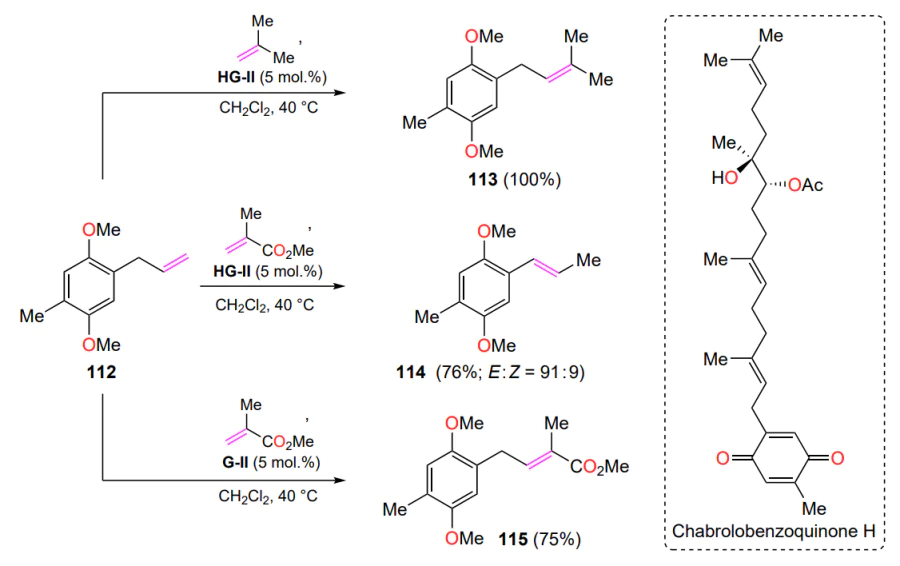
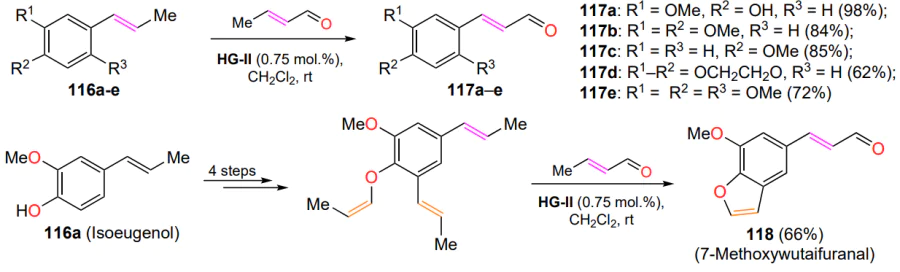
In the green chemistry paradigm, the modification of natural raw materials derived from biomass is a sought-after application of the CM reaction. One example is the conversion of isoeugenol (116a) from lignocellulose or clove oil[136]. When reacted with excess crotonic aldehyde, compound 116a and its structural analogues 116b – e are converted to cinnamaldehydes 117 in good yields (Scheme 57). As this process proved to be efficient, requiring small catalyst loadings and mild reaction conditions, a synthetic protocol was developed for the preparation of natural 7-methoxywutaifuranal (118) in a total yield of 44% over 5 steps. The transformation sequence included a tandem ring-closure metathesis reaction and cross-metathesis in the final step. Notably, benzofuran 118 was isolated from the root wood of Zanthoxylum wutaiense during the identification of plant secondary metabolites responsible for anti-tuberculosis activity[137].
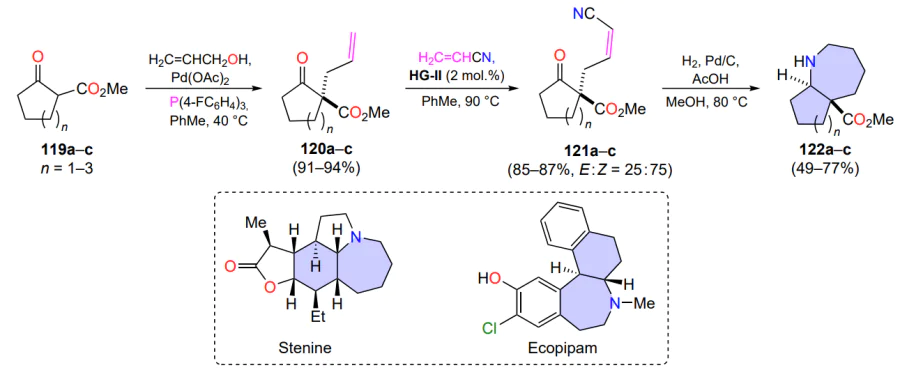
Cross-metathesis reactions are used for the synthesis of annulated azepanes, which are found in a few of biologically active structures, for example in the alkaloids stenine and ecopipam (Scheme 58). Based on cyclic ketones 119a – c, a three-step procedure has been developed[138] for the preparation of their analogues involving the CM reaction of esters 120 with an excess of acrylonitrile. Metathesis requires relatively high temperatures (PhMe, 90 °C), and no rapid thermal degradation of the HG-II catalyst was observed. Yields of intermediates 121, which were further cyclized to azaheterocycles 122, remained consistently high.
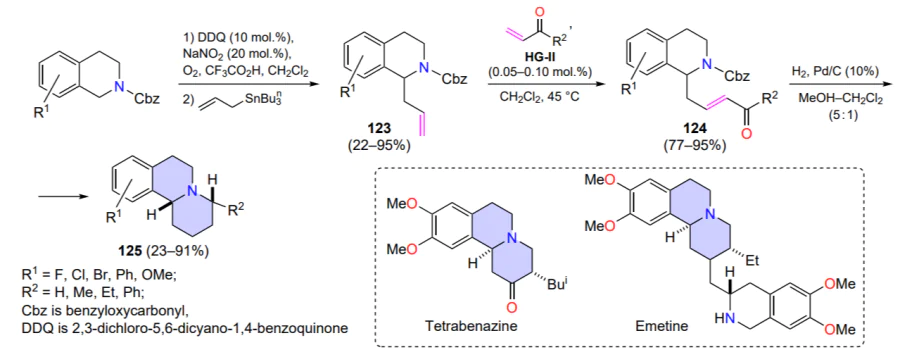
Jung et al.[139] proposed a stereoselective three-step procedure for the preparation of 4-substituted benzo[a]quinolizidines from available 1,2,3,4-tetrahydroisoquinolines (Scheme 59). The benzo[a]quinolizidine scaffold is found in alkaloids such as emetine[140] (which has antiprotozoal activity) and tetrabenazine[141] (used in the therapy of Huntington’s disease). One of the transformation steps involves the cross-metathesis reaction of allyl-substituted isoquinolines 123 and vinyl ketones, yielding the key intermediates 124. The HG-II complex (0.05 – 0.10 mol.%) afforded the cross-metathesis products 125 in good yields. However, the Z,E-selectivity of the metathesis observed in the formation of the vinyl ketones 124 was not discussed[139].
3.3. Ring-opening metathesis reactions catalyzed by the HG-II complex
Unlike the previous types of metathesis reactions discussed in this chapter, ring-opening metathesis (ROM) is relatively rarely used in fine organic synthesis. Typically, ethylene acts as the alkene to open the unsaturated ring (which requires the use of special laboratory equipment), resulting in the formation of two terminal double bonds, which are versatile functions for post-modifications.
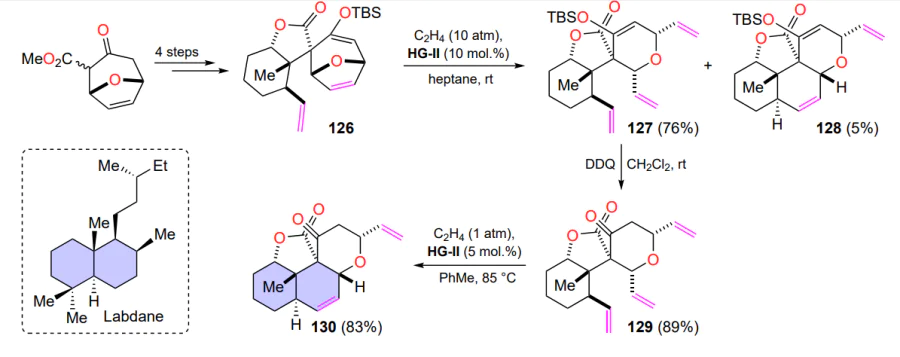
As an illustrative example of ROM, we present an original approach to the creation of a tricyclic scaffold of labdane-type diterpenes. In a study[142], the metathesis reaction occurs twice. In the first step, compound 126 reacts with ethylene in the presence of the HG-II complex to give the product of cleavage of the seven-membered ring 127 and compound 128, a by-product of the ring-opening/ring-closing metathesis (RORCM) reaction. The oxidative removal of the tert-butyldimethylsilyl protecting group from lactone 127 affords the triene 129, which is further subjected to the second metathesis reaction to furnish a six-membered ring (Scheme 60). The final product 130 contains the target diterpene scaffold.
3.4. The use of Hoveyda – Grubbs type catalysts with various donating heteroatoms in metathesis reactions
Ruthenium metallocycles, other than the oxygen-containing Grubbs (G-II) and Hoveyda – Grubbs (HG-II) complexes, are also successfully used in olefin metathesis reactions. This Section aims to analyze the prospects for the application of sulfur-ruthenium and nitrogen-ruthenium donor-acceptor complexes in laboratory practice and industry.
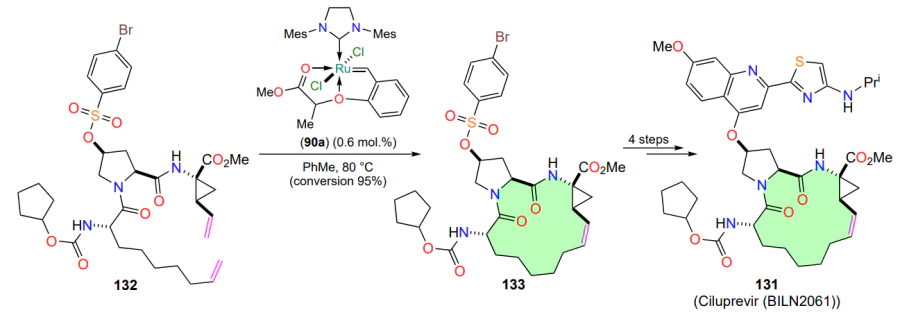
The ruthenium complex 90a (see Scheme 43) has been used in the preparation of the drug ciluprevir 131 (BILN 2061), which is used in the treatment of hepatitis C[113, 143]. The key step in the synthesis, the metathesis of diene 132 with the 15-membered ring-closure in compound 133, proceeds chemoselectively[144]. The use of the original organometallic complex 90a in the metathesis reaction was found to be more efficient than the use of the HG-I catalyst. The conversion of the starting compound 132, as determined by HPLC, was 95% at a relatively low molar catalyst loading (Scheme 61).
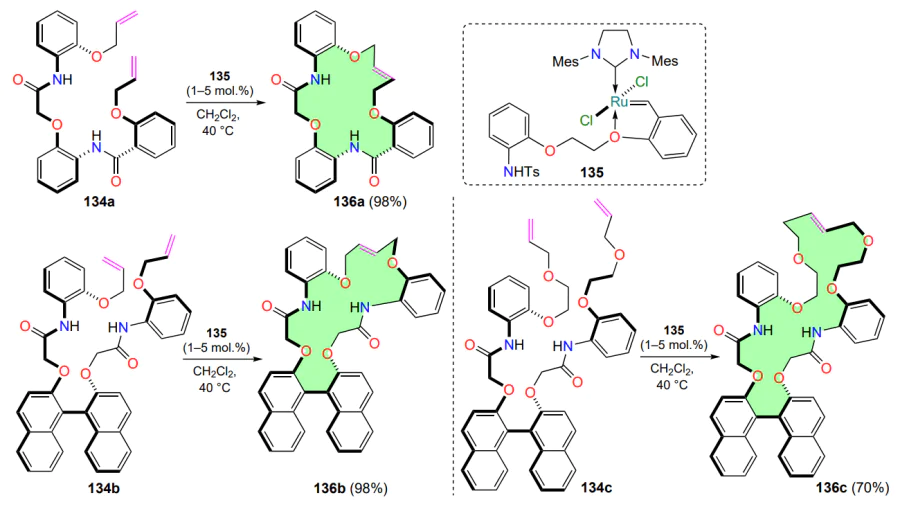
A classic example of the metathesis macrocyclization of dienes 134a – c was presented by Al-Awadi and co-workers[145]. Modified Hoveyda – Grubbs catalysts (1 – 5 mol.%), analogues of the HG-II complex, were used to carry out the transformation under relatively mild conditions (heating in dichloromethane for 1 day). It was shown that in the presence of the most active complex 135, the RCM reaction is stereoselective and produces only E-isomers 136a – c regardless of the substrate structure (Scheme 62).
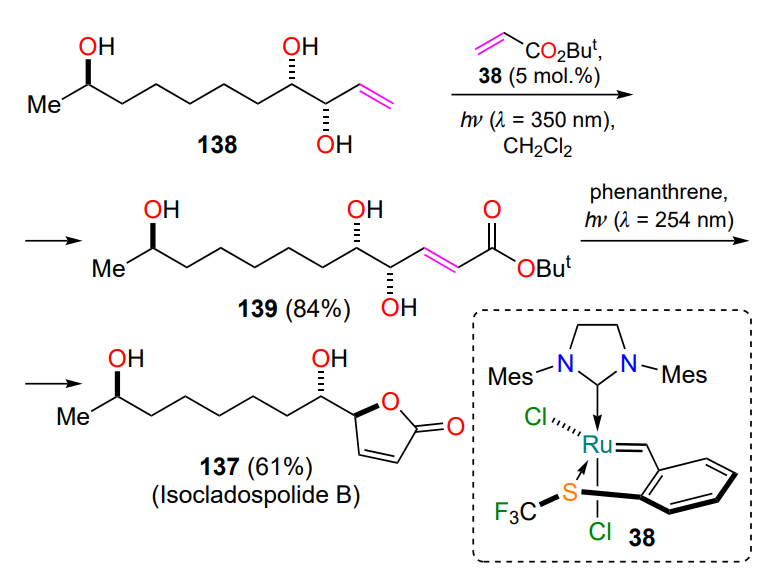
As mentioned above (see Section 2.1.2), sulfur-coordinated ruthenium derivatives can be activated by UV irradiation. In particular, catalyst 38 (see Scheme 18) was used for the synthesis of isocladospolide B (137)[68]. This representative of the class of five-membered lactones was obtained by a cross-metathesis reaction of terminal alkene 138 and tert-butyl acrylate, followed by light-induced lactonization of olefin 139 in the presence of phenanthrene (Scheme 63). The proposed method allows all stages of the transformation to be carried out under mild conditions in good yields.
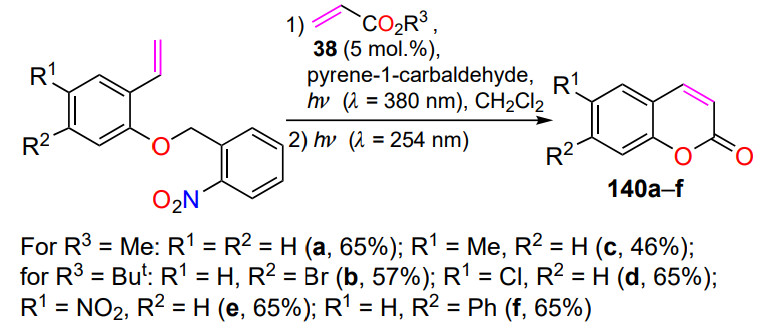
Catalyst 38 was also used in the synthesis of coumarins 140[68]. The first step involved a cross-metathesis reaction between O-benzyl-2-vinylphenols and methyl or tert-butyl acrylates under photoactivation conditions (λ = 350 nm) in the presence of pyrene-1-carbaldehyde. Next, three consecutive photochemical reactions took place under UV irradiation with sorter wavelength: cleavage of the 2-nitrobenzyl group, trans,cis isomerization of the double bond and cyclization of the intermediate to the target coumarins 140 (Scheme 64).
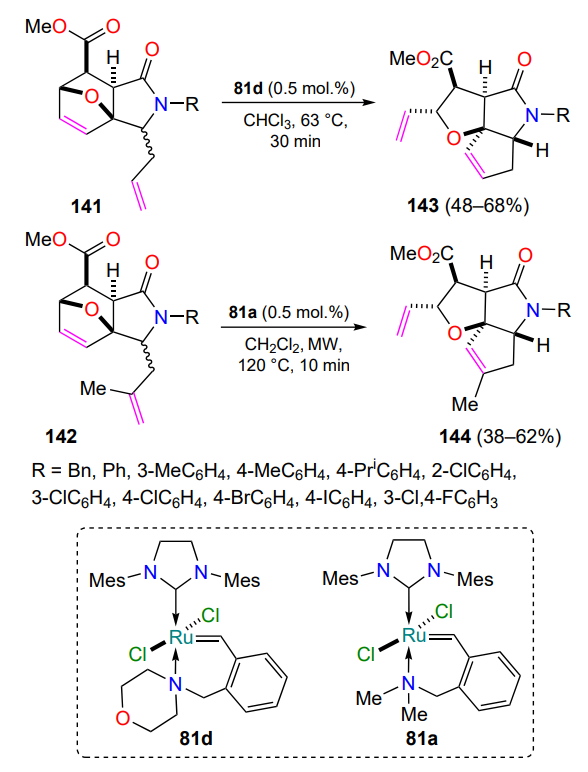
Systematic studies of furan derivatives available through the processing of renewable furanose- and pentanose-containing plant materials have provided an efficient method for the synthesis of tricyclic cyclopenta[b]furo[2,3-c]pyrroles[146]. Treatment of solutions of esters of 3-allyl- (141) and 3-metallyl-3a,6-epoxyisoindole-7-carboxylic acids (142)[82, 147, 148] with catalysts 81a,d containing a nitrogen-ruthenium coordination bond in a six-membered metallacycle afforded the target tricycles 143 and 144 respectively (Scheme 65). The ring-opening ring-closing metathesis reaction is shown to be stereospecific. Of the two possible diastereoisomers of the starting methyl 3-allyl-3a,6-epoxyisoindole-7-carboxylates 141, only the trans-isomer is able to participate in the RORCM reaction due to the mutual arrangement of the epoxide bridge and the allylic substituent. The use of two different nitrogen-containing metal complexes 81a,d is dictated by their different stability at refluxing in chloroform and under the harsh conditions of microwave activation (MW) required to create the trisubstituted multiple bond in the adducts 144 (see Scheme 65).
1,4 : 5,8-Diepoxynaphthalenes 145 – 147, readily accessible by a tandem [4 + 2]/[4 + 2] cycloaddition involving bis-dienes and active dienophiles (dialkyl acetylenedicarboxylates, hexafluorobut-2-yne, maleinimides, etc.), were used as substrates for the ring-opening cross-metathesis reaction under the action of ruthenaazaheterocycles 81a and 81d containing an N → Ru bond)[149, 150]. Depending on the process conditions, the Diels-Alder reaction can produce adducts of the ‘domino’ (145 and 146) or ‘pincer’ (147) type. The action of nitrogen-containing catalysts 81 and ethylene on the adducts 145 and 146 leads to ring-opening cross-metathesis products, compounds 148 and 149 (Scheme 66). From pincer-type adducts 147, either products of double ring-opening cross metathesis (150) or products of sequential metathesis and retro-Diels – Alder reactions (151) can be obtained under various conditions. Hydrogenated furans 148 (X = NBoc, Y = CO2Me) display pronounced antitumor activity in vitro against human prostate cancer cells[150].
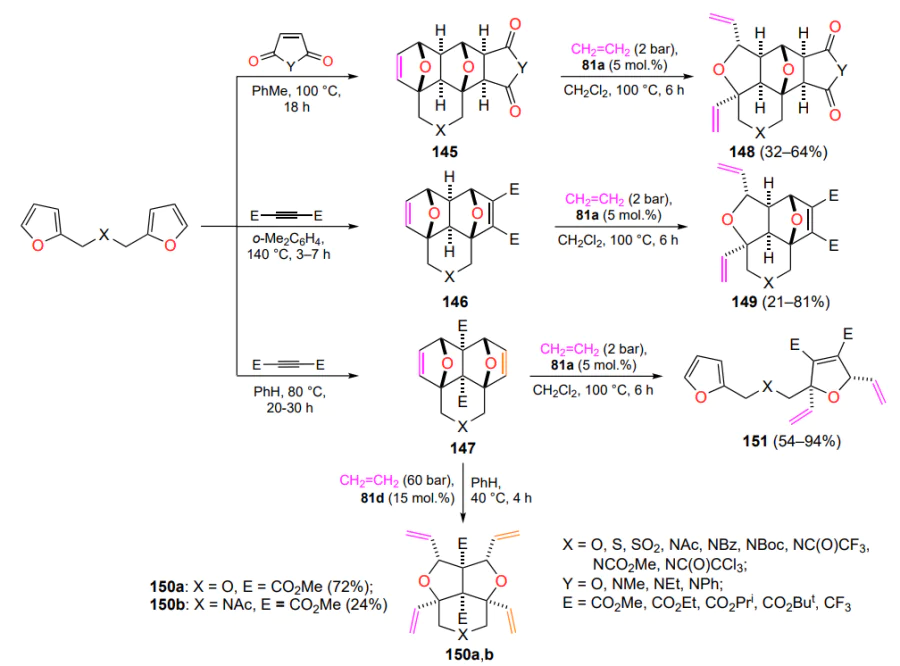
It should be noted that the starting bis-furans are products of two-step processing of molecules extracted from biomass (furfural and 5-hydroxymethylenefurfural). Consequently, Scheme 66 can be considered as a pathway for the conversion of renewable natural resources into compounds useful for organic synthesis and medicinal chemistry.
4. Metathesis polymerization of olefins in the development of novel materials
The olefin metathesis reaction is in demand not only in straightforward synthesis but also in polymer chemistry. The first successful industrial applications of this process involved the use of alkylidene complexes of transition metals (molybdenum, tungsten, ruthenium, titanium, chromium, iridium), with the structure of the catalytically active species often remaining unknown. Grubbs and Hoveyda – Grubbs catalysts are particularly effective in two types of metathesis polymerization reaction, ROMP and acyclic diene metathesis polymerization (ADMET) (Scheme 67 – Scheme 70). In the ROMP reaction, monocyclic or polycyclic olefins first undergo a ring-opening metathesis reaction and then a polymer chain is formed[151-153]. This process is atom-economical as it produces no by-products, including low-molecular-weight alkenes[154]. The ADMET reaction involves terminal dienes, resulting in the release of a low-molecular-weight alkene (e.g. ethylene)[26, 155]. With the proper selection of catalysts and monomers, it is possible to control both the structure of the polymerization product and the process rate, allowing the average molecular weight of the target polymer to be programmed.
The main substrates for ROMP are norbornenes (bicyclo[2.2.1]hept-2-enes), of which dicyclopentadiene (DCPD) is of maximum importance in industry (see Scheme 69)[156, 157]. Such bicyclic olefins are readily formed in the Diels – Alder reaction from cyclopentadiene (or furan) and various alkenes, are relatively cheap, and have high polymerization rates[158]. Most studies have used exo-isomers for polymerization, as the endo-isomers of substituted norbornenes react more slowly in metathesis processes[159]. An example of the commercial success of norbornene polymers is Norsorex®, which can be converted into Zeonex®, Arton® and Zeonor® materials with good optical properties (92% light transmission at 3 mm thickness)[160, 161].
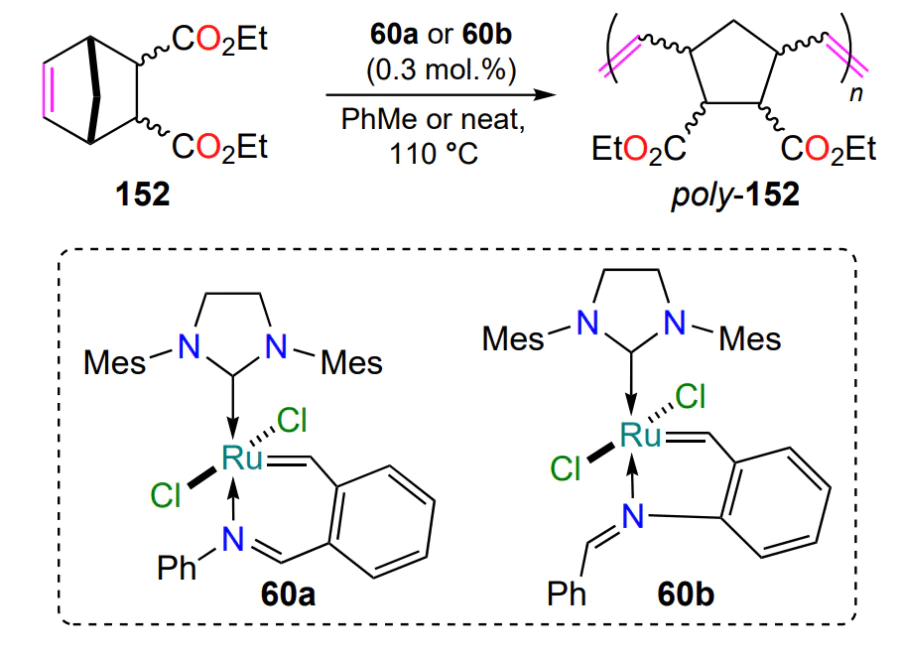
As mentioned above, nitrogen-containing ruthenium complexes (see Section 2.2.1) are often used in ring-opening metathesis polymerization due to their easily tunable catalytic parameters. For example, complexes 60a and 60b (0.3 mol.%, see Scheme 29) efficiently initiate the polymerization of norbornene dicarboxylic acid esters 152 at 110 °C either in toluene or in the absence of solvent (see Scheme 69). The time required to complete the polymerization is < 5 min[88]. The determination of weight average molecular weights (Mw) by gel permeation chromatography showed higher values for polymers obtained in the presence of catalyst 60a (16 60 000 g mol–1 in toluene and 1 540 000 g mol–1 without solvent) compared to complex 60b (1 270 000 g mol–1 in toluene and 1 340 000 g mol–1 without solvent). The polydispersity of the polymers ranged within 1.9 – 2.3.
The quinoline and quinoxaline-coordinated ruthenium complexes 63a,b and 64a,b (see Scheme 29) have also been studied in the ring-opening metathesis polymerization of substituted norbornenes 152 and 153 (see Scheme 68, Table 6)[91]. The reactions were carried out in toluene or in the absence of solvent at 110 °C, catalyst loading was 0.3 mol.% and reaction times were up to 5 h. The conversion of substrates in solutions, determined by 1H NMR spectroscopy, was slightly lower than in solvent-free reactions (see Table 6) and the isomeric ruthenium complexes trans-63 and cis-64 showed approximately the same activity at elevated temperatures.
Dicyclopentadiene 154 is a product of petroleum refining, a cheap feedstock for the ROMP reaction (see Scheme 68). DCPD-based polymeric materials (Telene®, Metton®, Pentam®, Proxima™) are widely used in engineering because of their high strength and light weight. The feature of polymerization of diene 154 is its rapidity and exothermicity. During polymerization, a reaction mixture can reach up to 200 °C, and therefore, the choice of catalyst and the rate of its initiation or thermal activation are important. Nitrogen-containing ruthenium complexes, in particular compounds 57a and 58a, were also tested in polymerization (see Scheme 28). To describe the polymerization process, the method of plotting exotherms — continuous recording of the temperature of the reaction mixture — was used. The maximum temperature and the time taken to reach it are important characteristics of the polymerization reaction. The initiation rates of the complexes 57a and 58a were different. At 30 °C in the presence of the trans isomer 57a (0.003 – 0.004 mol.%), the maximum exotherm reached 185 °C in less than 5 min, whereas in the case of the reaction with the cis isomer 58a, it was 160 °C after 25 min (see Scheme 68)[86]. These results demonstrate a common property of all Hoveyda – Grubbs type complexes: the trans isomers are more reactive in metathesis reactions than the cis form.
Other ruthenaheterocycles that are latent at room temperature, such as the sulfur-containing catalyst 35e (see Scheme 16), can also be used in ROMP reactions[156]. As noted above, such cis complexes can be activated by UV irradiation (l = 350 nm). This method of initiation has been shown to be effective in polymerization. Norbornenes 152 and 153 and DCPD 154 were used as substrates (the concentration of catalyst 35e varied in the range of 0.01 – 0.03 mol.%). The ADMET reaction is most commonly catalyzed by ruthenium complexes because of their high tolerance to a variety of functional groups in the substrates[162]. However, the process requires high temperatures, leads to isomerization of the starting dienes and, as a consequence, to the formation of atactic polymers with poor consumer properties.
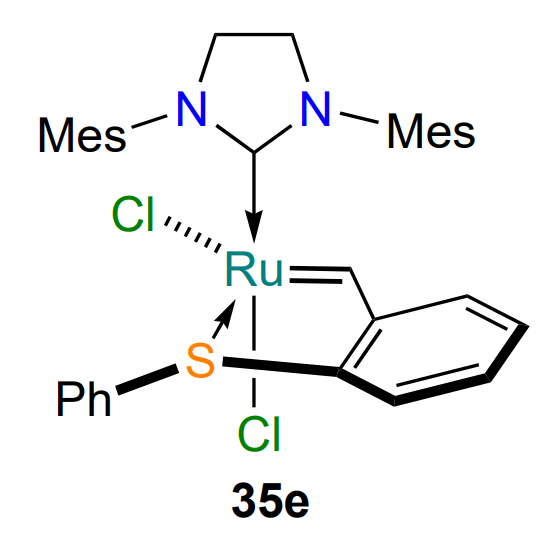
Catalyst 35e was used for the ADMET reaction with jojoba oil (Scheme 70 shows its main component 155)[163]. This produces high-molecular-weight synthetic oils 156 and hydrocarbon by-products 157. The latter can be used as fuel additives. The reaction is therefore atom-efficient, but its conditions need to be optimized. The presence of double bonds in the polymer allows it to be further functionalized to produce materials with the desired properties.
The reduced pressure in the ADMET reaction is necessary to shift the reaction equilibrium towards the products (removal of alkenes such as 157), thus eliminating side reactions such as cross-metathesis.
5. Conclusion
Of all the ruthenium complexes with five- or six-membered metallacycles containing a ruthenium–heteroatom bond, the most common is the commercially available catalyst HG-II. It is this complex that is most commonly used in organic synthesis. At the same time, other complexes such as Hoveyda – Grubbs, which contain a donor-acceptor Het → Ru bond in the metallacycle, in many cases exhibit catalytic metathesis activity that is not inferior or even superior to that of the compound HG-II. Some of these ruthenium derivatives can be prepared from less complex or cheaper starting materials, can be stored in air, are tolerant of traces of moisture and oxygen in solutions and have no patent protection (FTO, Freedom To Operate). In addition to these properties, complexes with Het → Ru bonds (especially N → Ru) offer the possibility of fine-tuning by optimizing the steric and electronic effects of substituents in the heteroatom environment, which provides the required catalyst activation rate and a given reaction rate. These factors make it possible to obtain metathesis catalysts with the required properties suitable for a wide range of olefins, including their polymerization.
From the analysis of the material presented it can be concluded that in the near future Hoveyda – Grubbs type complexes containing nitrogen and sulfur atoms coordinated to ruthenium will find (and some have already found) practical application in large scale synthesis. The catalytic activity of the nitrogen-containing ruthenium complexes 81 and 83 (see Section 2.2.1) can be varied over a wide range by altering the steric volume and electron-donating properties of the substituents at the nitrogen atom. These catalysts must have the required selectivity for a particular technological process. Sulfur-containing analogues of second-generation Hoveyda – Grubbs complexes 39, 40, 48 and 49 (see Section 2.1.2) are capable not only of thermal activation but also of reversible cis,trans- isomerization, including under the action of ultraviolet radiation. The latter property opens up the possibility of controlling alkene metathesis by light irradiation alone. This behavior of the catalysts is required for the production of polymers that are transparent in the optical and/or UV spectral range. Notably, classical HG-II catalysts are not capable of thermal and photo-switching.
Summarizing the material in this review and other works on the subject, we can formulate the following trends in the design and application of HG-II-type olefin metathesis catalysts that will be relevant at the turn of the century 2024 – 2025:
1. Development of ruthenium complexes with modified ligands for the stereoselective metathesis of alkenes. Recent studies have revealed the key structural features of Hoveyda – Grubbs catalysts that provide high Z,E-selectivity in metathesis reactions. Of special note are the synthesis of complexes with asymmetric NHC ligands[26], replacement of the NHC ligand with cycloalkylaminocarbenes (CAACs)[164] and modification of anionic ligands[165]. As our review shows, simply varying the heteroatom coordinating the ruthenium atom in HG-II-type catalysts does not control the Z,E selectivity of self and cross metathesis.
2. Metathesis involving the formation of carbon-heteroatom bonds, or hetero-metathesis. The study of the limits of applicability of Hoveyda – Grubbs type complexes in the formation of C=N (see [166, 167]) and C=O (see [168]) bonds is becoming increasingly popular. These processes are attractive not only because of the practical value of the resulting products, but also from a theoretical point of view. The vast majority of the ruthenium complexes described in this review have not been studied in these processes, although the G-II complex has already demonstrated its potential in hetero-metathesis reactions[168].
3. The use of ruthenium catalysts in industrial processes of polymerization and depolymerization, including the processing of polymers containing multiple carbon – carbon bonds[169]. In this area, it is likely to use the cheapest and most active of the complexes mentioned in the review, containing the O → Ru, N → Ru, S → Ru coordination bonds in the ruthenaheterocycle (Figure 8).
![[{"id":"yYRnX7b98_","type":"paragraph","data":{"text":"General structural formulas of catalysts used or having the potential to be used in industrial processes."}}]](/storage/images/resized/gNqJRTicoKEEmGcFCSSl8Y27U7DDOS2LkNHKZoeB_xl.webp)
4. Preparation of Hoveyda – Grubbs type complexes based on non-precious metals such as iron[26, 170], vanadium[171-173], manganese[174] and other Period IV elements of the Periodic Table. Complexes based on such metals may provide a cheaper and more environmentally friendly alternative to existing heavy metal-based catalysts. To date, it has not been possible to obtain analogues of the ruthenium complexes described above that contain a different central metal atom.
5. Modification of complexes for green chemistry processes[26], in particular the production of polymer-immobilized or water- and ionic liquid-soluble complexes. Such modifications will minimize the negative environmental impact of metathesis reactions on an industrial scale.
6. List of abbreviations
ADMET — acyclic diene metathesis polymerization,
AIBN — azobis(isobutyronitrile),
BINOL — 1,1'-bi(2-naphthol),
Boc — tert-butoxycarbonyl,
Cbz — benzyloxycarbonyl,
CM — cross metathesis,
CSA — camphor-10-sulfonic acid,
Cy — cyclohexyl,
DCE — 1,2-dichloroethane,
DCPD — dicyclopentadiene,
DDQ — 2,3-dichloro-5,6-dicyano-1,4-benzoquinone,
DFT — density functional theory,
DME — dimethoxyethane,
Mw — weight-average molecular weight,
Mes — 2,4,6-trimethylphenyl (mesytyl),
MW — microwave activation,
NBS — N-bromosuccinimide,
NHC — N-heterocyclic carbene,
PCC — pyridinium chlorochromate,
Py — pyridine,
RСМ — ring-closing metathesis,
ROCM — ring-opening cross metathesis,
ROMP — ring-opening metathesis polymerization,
RORCM — ring-opening ring-closing metathesis,
TBS — tert-butyldimethylsilyl,
Ts — p-toluenesulfonyl (tosyl),
XRD — X-ray diffraction analysis.
Acknowledgements
The review has been supported by the RUDN University Scientific Projects Grant System, project No 021409-2-000.
References










