



![Cytotoxic activity of compound 1 and cisplatin[36].](/storage/images/resized/3XLKnFYEVxFcjiX99XOj8rfWXOrTwHdroTSEicHo_xl.webp)
![Cytotoxicity and accumulation of complexes 11 – 13 in the cells[44].](/storage/images/resized/1USaVPCupiEV2xw0vh8UOi3Y0uomhzztNUL8O70c_xl.webp)
![Binding constants of compounds 32 – 37 to DNA (in 105 М–1 solutions)[51].](/storage/images/resized/8uLLirxCfKh9Gei5TNVFhpJT0AlqRdwJw2YUIz21_xl.webp)
![Cytotoxic activity of compounds 57 – 61 (IC50, μM)[68].](/storage/images/resized/bQglfIedjqawI2PABN3w2qSBqPxa75BXCh0Frpad_xl.webp)
![Cytotoxic activity of compounds 69b – 72b (IC50, μM)[71].](/storage/images/resized/iXTXZ8TkFsb6Ac83FKrXWaKw738Hs8yG5CfytDrl_xl.webp)
![Cytotoxic activity of compounds 78 – 101 (IC50, μM)[84].](/storage/images/resized/G83TKT6Dcp9mUThwPU87oH6foLLusMQrRXkYXTXa_xl.webp)
![Characteristics of binding of compounds 239 – 244 to RNA (for concentration ratio of 1 : 5)[141-143].](/storage/images/resized/QXZgUppsLit8laJlmC6mqI6pcR1TlSuqix6DarbI_xl.webp)
![Structure of the adduct of 1' with DNA[36].](/storage/images/resized/MEKOS7gg1qpTp8vfzf5ESxzLCYmoCXPYqzyYHK2S_xl.webp)
![Results of molecular docking of compound 95 into pu22[84].](/storage/images/resized/dB3ixEb75JOQG1PeHTl9FIX1tjtRm1dCBZJJ9rXP_xl.webp)
![Structure of the adduct formed by c-MYC quadruplex and compound 116[87].](/storage/images/resized/1dq3w9gVHaMWtU24J3Pmaa4znYNwsCaga37iEFEL_xl.webp)
Keywords
Abstract
Nucleic acids are important targets for many anticancer drugs. Apart from the canonical B-DNA double helix, DNA forms a number of non-canonical structures (G-quadruplexes, i-motifs, hairpins, triplexes, etc.), which play an important role in the regulation of biological processes. Binding to non-canonical DNA structures occurs mainly by π–π-stacking. Therefore, aromatic and heteroaromatic compounds such as fused polyaromatic compounds (acridines, anthraquinones, carbazoles), porphyrins, benzothiazoles, benzimidazoles, pyridines, and quinolines, as well as their complexes are used as ligands for secondary structures. These ligands should possess not only high selectivity for non-canonical structures over double-stranded DNA, but also relatively high solubility and ability to penetrate through cell membranes. This review summarizes the achievements of 2020–2024 in the synthesis and biological studies of (hetero)arenes (acridines, anthraquinones, benzazoles, xanthones, porphyrins) and coordination compounds that have exhibited anticancer activity as a result of binding to non-canonical DNA or RNA structures. Ligands of various types of non-canonical nucleic acid structures (G-quadruplexes, i-motifs, triplexes, hairpins) are considered and their cytotoxic activity and structure–property relationships are compared
The bibliography includes 166 references.
1. Introduction
Nucleic acids play a key role in the regulation of all cellular processes. DNA damage and mutations or amplification of certain DNA regions can disturb the regulation of cell cycle. In the case of changing mechanism of cell death induction, this can lead to uncontrolled cell division and development of cancer pathologies. Due to the crucial role of DNA and related RNA in carcinogenesis, these biomolecules are used as targets for many anticancer agents such as platinum compounds, anthracyclines, nitrogen mustard analogues, etc[1-4]. However, many drugs have a wide range of side effects caused by non-selective action on nuclear or mitochondrial DNA[5]. For this reason, a relevant task is the search for new agents able to selectively act on the DNA of malignant cells.
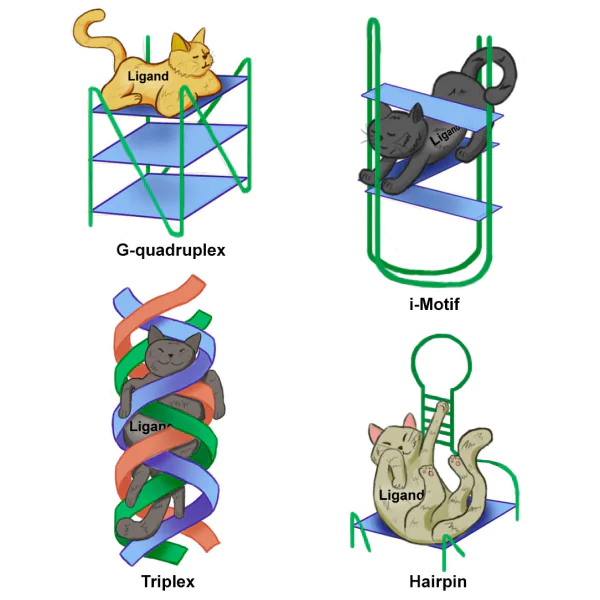
Double helix is not the only DNA structure in cells. Apart from the double-stranded B-DNA, there are so-called non-canonical DNA structures: Z-DNA, triplex DNA, hairpins, G-quadruplexes, and i-motifs (Figure 1). The sequences that can form non-canonical structures can be located in various parts of both DNA (mainly in the promoter regions of genes and in telomeres) and RNA (mainly in 5'-untranslated region). The formation of non-canonical DNA and RNA structures is attributed, on the one hand, to regulation of gene expression and protection of nucleic acid regions from damage (as, for example, hairpin RNA) and, on the other hand, to genomic instability and the development of neurological and oncological diseases[6-11]. The involvement of non-canonical structures in the regulation of gene expression, in particular oncogene expression, as well as structural differences from the В-DNA double helix make them promising targets for anticancer therapy[12-14].
![[{"id":"08dsCUUDj-","type":"paragraph","data":{"text":"Non-canonical structures of nucleic acids: G-quadruplexes (a), DNA triplexes (b), i-motifs (c), and"}},{"id":"cxHHe-48GP","type":"paragraph","data":{"text":"hairpin RNA and DNA (d)."}}]](/storage/images/resized/GtRKISb57WceziMf42cId3eY2ZfOJa5nUSlb5EDx_xl.webp)
In most cases, the strategy is to stabilize a non-canonical structure via interaction with small molecules that bind to nitrogenous bases of nucleic acids through π – π-stacking. The stabilization of the DNA secondary structure prevents the chain interaction with RNA polymerase (or telomerase in the case of telomeres), which disrupts the gene expression and/or leads to accumulation of DNA damages and, as a consequence, to cell death. Aromatic and heteroaromatic compounds of various classes have been proposed as ligands to stabilize the secondary structures of nucleic acids. In this review, we consider the achievements of the last four years in the field of synthesis of new organic and coordination compounds the antitumour activity of which is caused by binding to non-canonical DNA or RNA structures. The preceding review papers[15-20] on the subject of interest published in 2020 – 2023 focused on only one type of non-canonical nucleic acid structures. The present review addresses ligands for non-canonical structures of various types, which makes it possible to perform more comprehensive analysis of published data; to establish the relationship between the ligand structure and DNA binding affinity; and to elucidate the structural criteria responsible for the selectivity for particular DNA structures.
In view of the considerable interest in the development of new effective antitumour agents for the therapy of malignant neoplasms and the lack of reviews that summarize data on the synthesis and biological activities of ligands binding to non-canonical structures, we believe that our review may be of interest for broad circles of specialists in both organic and medicinal chemistry.
2. G-Quadruplex ligands
Among non-canonical DNA structures, G-quadruplexes (G-tetrads, G4, see Figure 1a) have been studied most comprehensively. They represent several π – π-stacked G-quartets, which are composed of four guanine bases bound by Hoogsteen hydrogen bonds. The binding of nucleic acid chains of guanosine oligo- and polynucleotides is provided by a small-sized monovalent cation, most often K+, located at the centre of the quartet. A G-quadruplex can exist as different conformers: parallel, antiparallel, and hybrid (Figure 2). The G4 conformation depends on the nucleotide sequence and the nature of the cation in the central channel of the G-quadruplex (Na+ or K+)[21]. G-quadruplex structures are involved in the regulation of key biological processes; for example, they participate in the initiation of DNA replication[22, 23], DNA transcription[24, 25], and translation[26, 27]. However, they can also prevent the replication fork progression[28] and induce genomic instability[29, 30]. In the absence of any specialized helicase, G4 formation prevents binding of DNA to proteins (DNA and RNA polymerase, transcriptional factors, telomerase), which leads to inhibition of gene expression and gives rise to single- or double-stranded DNA breaks. The presence of G-quadruplexes in mRNA inhibits the ribosome activity and, consequently, RNA transcription.
![[{"id":"whDw7IOw6I","type":"paragraph","data":{"text":"Structure of G-quartet (a) and conformations of G-quadruplex (b)."}}]](/storage/images/resized/7RISzDKQOsxKcMkEHs3drMFYhYgofEPOGJMm8L0n_xl.webp)
The participation of G-quadruplexes in the regulation of gene expression makes them an attractive target for anticancer therapy. It was shown that various small-molecule ligands can bind to G4, thus stabilizing their structure, which eventually leads to inhibition of mRNA synthesis[31] or telomerase activity[32]. The main requirement to G-quadruplex ligands is the presence of an aromatic moiety capable of π – π-stacking. In addition, the ligand binding to quadruplexes is promoted by the presence of aliphatic linkers with amine groups, which may be partially protonated and bind to the DNA phosphate groups in the acid medium of tumour cells[33]. Acridines, anthraquinones, porphyrins, benzazoles, quinolines, carbazoles, as well as coordination compounds of metals, mainly platinum and ruthenium have been proposed as G4-stabilizing compounds[14].
2.1. Coordination compounds
Coordination compounds of a number of transition metals, first of all, ruthenium, platinum, copper, and nickel were reported to bind to G-quadruplexes[34, 35]. Apart from the presence of a metal cation, which provides the cation – π-interaction with the quadruplex guanines, a general requirement to such molecules is the presence of a planar aromatic moiety capable of π – π stacking.
Zhu et al.[36] reported a mixed-ligand platinum(II) complex 1 in which the platinum ion coordinates the phenylpyridine derivative (L), heterocyclic carbene (L' ), and chloride anion. It is noteworthy that in this compound, the pyridine nitrogen of ligand L' is not coordinated to the platinum ion.
The DNA binding of complex 1 was studied in relation to Tel26, wtTel26, VEGF, c-MYC, c-KIT, and BCL G-quadruplexes and also to double-stranded DNA (dsDNA) and single-stranded DNA (ssDNA). The structure of the product of bonding of compound 1 to VEGF G-quadruplex was derived from the set of 2D NMR spectroscopy data (NOESY, COSY, and TOCSY experiments). According to the results, binding of complex 1 to DNA is accompanied by elimination of the chloride anion and coordination of the pyridine nitrogen atom of L' to the platinum ion; as a result, the emerging metal centre in 1' acquires a positive charge and a square planar geometry. Compound 1' binds to the quadruplex through the π – π stacking with G5, G9, G16, and G20 guanine moieties. In addition, a conformational change takes place in the DNA molecule: the C10 – C11 – T12 – T13 loop and the G21–T22 3'-terminal region shift in a such a way that C10 and G21 become arranged above molecule 1' (Figure 3). Thus, adaptive binding of compound 1 to the quadruplex accompanied by conformational changes in both the ligand and DNA was demonstrated.
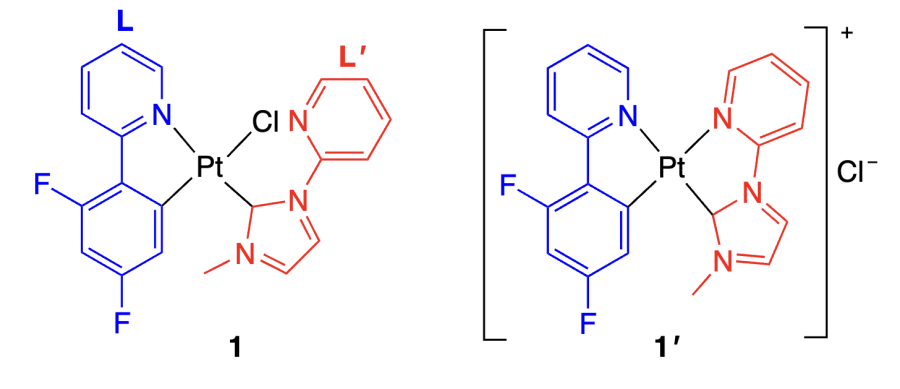
A study of the effect of compound 1 on cell viability by MTT assay [MTT is (4,5-dimethythiazol-2-yl)-2,5-diphenyltetrazolium bromide] in relation to several cell lines (HeLa, MCF-7, A549R, LO2) showed a higher cytotoxicity of 1 compared to that of cisplatin (Table 1). According to fluorescence lifetime imaging microscopy (FLIM) data with a probe specific to quadruplexes, this complex selectively binds to quadruplex DNA in living cells. Quantitative real-time polymerase chain reaction (qRT-PCR, RT-PCR, Q-PCR) and confocal microscopy with Danio rerio showed that incubation with compound 1 suppresses the expression of VEGF gene, responsible for angiogenesis, which leads to inhibition of blood vessel growth.
Farine et al.[37] prepared coordinated compounds 2 – 4, [MIIL](ClO4)2, where M = Ni2+, Cu2+, and Zn2+ and L is Schiff base. According to electronic spectroscopy of solutions of complexes 2 – 4 with calf thymus double-stranded DNA and with hTelo and c-MYC quadruplex DNA, copper complex 3 not only binds to DNA most efficiently, but is also more selective for G-quadruplex structures than complexes 2 and 4.
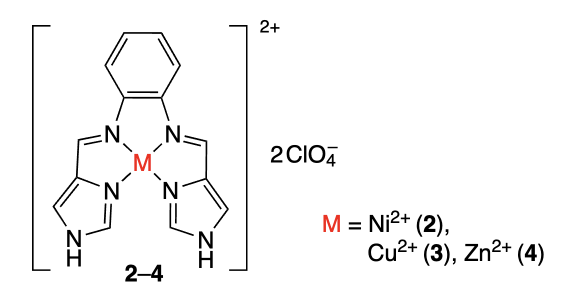
Despite the high DNA binding constant of copper complex 3 (Kb ~ 105 М–1), further Förster resonance energy transfer (FRET) measurement of the melting temperatures of the resulting complexes demonstrated that this compound stabilizes the hTelo G-quadruplex DNA by only 1.5 °C. This observation was attributed to the absence of π – π-interactions between complex 3 and the quadruplex moieties of guanine, which was confirmed by quantum chemical calculations.
McQuaid et al.[38] reported optically pure Λ-enantiomer Λ-[Ru(phen)2(qdppz)]2+ (5) {phen is phenanthroline, qdppz is naphtho[2,3-h]dipyrido[3,2-a:2',3'-c]phenazine-8,13-dione}, which was synthesized by the condensation of phenanthroline-5,6-dione with 1,2-diaminoanthraquinone (Scheme 1). Then the adduct of complex 5 with an antiparallel telomere sequence d[(G3T2A)2G3T3G3] was isolated. The structure of the adduct was established by X-ray diffraction using the single crystal grown in the presence of Ba2+ ions, which promoted crystallization. It is noteworthy that the previously reported structures of antiparallel quadruplexes and their ligand adducts were studied only by NMR spectroscopy. According to the single crystal X-ray diffraction data, the Ru(phen)2 motif is located in the DNA major groove, whereas the anthraquinone moiety resides in the cavity formed by the T10 – A12 – T11 and G1 – G9 – G13 – G21 G-quartet, being bound to all of the guanine bases in the quartet. In addition, since the quartets in the antiparallel quadruplexes are non-planar, the anthraquinone moiety is bent in the carbonyl group region at an angle of approximately 12° (Figure 4). X-Ray diffraction study also made it possible to establish the positions of three potassium ions and hydrated barium ion in the outer coordination sphere.
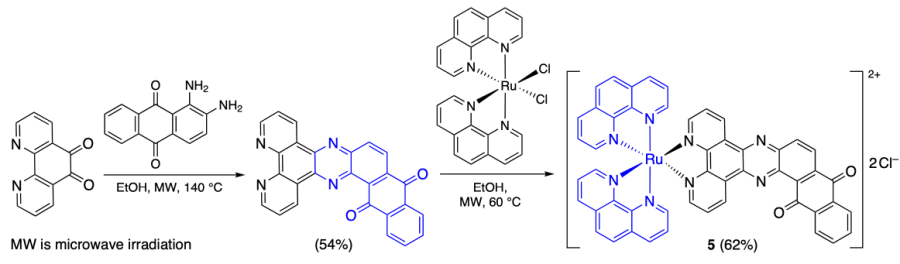
![[{"id":"LFR3QIetVB","type":"paragraph","data":{"text":"Structure of the DNA adduct of complex 5: the binding cavity of compound 5 to the triplet and G-quadruplex (а) and bending of the anthraquinone moiety (b)[[ type=\"anchor\" referenceId=\"8949\" ]]. Copyright (2022) American Chemical Society."}}]](/storage/images/resized/aMQJyg2TIrWLiXQKJdxM0Q6nIbqrqX8RBqhg6vLC_xl.webp)
The effect of complex 5 binding to the quadruplex on the replication was studied by the Klenow fragment replication assay. The assay was performed using the d(T2AG3)4 and d(G3T2A)2G3T3G3 telomere sequences. As reference samples, the authors used the Δ-isomer of complex 5 and the enantiomers of [Ru(phen)2(dppz)]2+ (6), where dppz is dipyrido-[3,2-a:2',3'-c]phenazine, which they reported earlier[39]. The most pronounced replication inhibition was found for Λ-5, and after 320 min the content of the polymerization product did not exceed 15%. Furthermore, the inhibition of replication was retained after the addition of LiCl, which is capable of disrupting the secondary structure of the quadruplex.
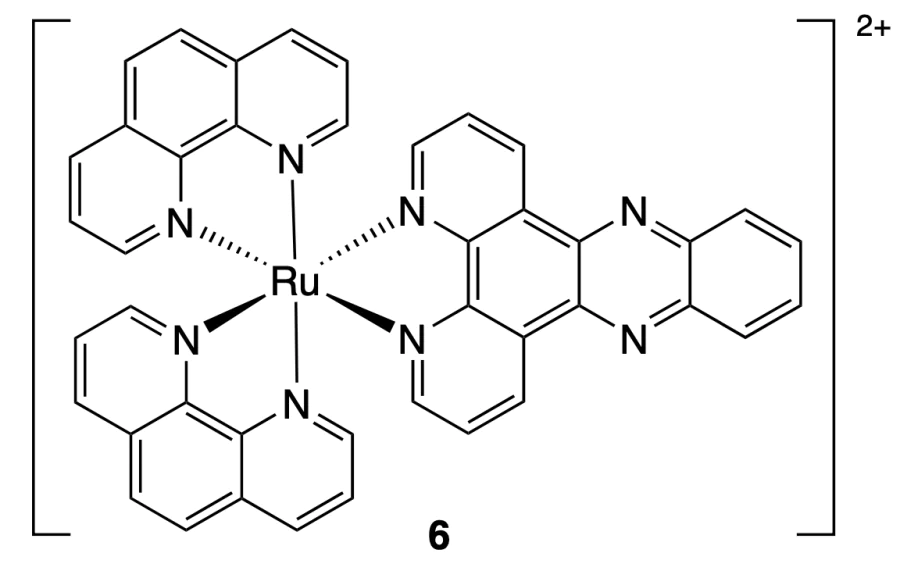
Malina et al.[40] prepared the [Ni2L3]4+ complex (7) with a cylindrical structure as a mixture of enantiomers. The resolution of the racemate by chiral chromatography resulted in the isolation of single P (right-handed helix) and M (left-handed helix) enantiomers (Figure 5)[41]. Study of the binding to c-MYC, hTelo, c-KIT1, c-KIT2 quadruplexes and double-stranded DNA by FRET and FID assay (FID is fluorescent intercalator displacement) showed a higher selectivity of compound 7 for c-MYC and hTelo quadruplexes than to the double-stranded DNA or other quadruplexes. The difference in binding was especially pronounced when the FID assay was carried out in the presence of potassium ions with a concentration of 160 mM, which corresponds to the intracellular potassium concentration. The dsDNA/G4 selectivity indexes (SI) for P- and M-enantiomers were 3.4 ± 0.3 and 5.8 ± 0.6 for hTelo and 2.7 ± 0.2 and 3.6 ± 0.5 for c-MYC. Meanwhile in the case of c-KIT1 and c-KIT2, the SI values did not exceed 2.1 ± 0.1. It is worth noting that the results for the M- and P-enantiomers of complex 7 were similar.
![[{"id":"qC0zW_UD2D","type":"paragraph","data":{"text":"Schematic view of the M- and P-enantiomers the [M<sub>2</sub>L<sub>3</sub>]<sup>4+</sup> helicates (M = Fe<sup>2+</sup>, Ni<sup>2+</sup>)[[ type=\"anchor\" referenceId=\"8952\" ]]. Copyright (2021) Frontiers."}}]](/storage/images/resized/hWqV84JsvhTbNq6ulf7wK0Pt23qVHWwglwMdk080_xl.webp)
A Taq DNA polymerase study of the effect of binding of the complex to quadruplexes on the DNA replication showed that the addition of enantiomers of 7 had no effect on the replication of c-KIT1 and c-KIT2, but inhibited the replication of c-MYC and hTelo quadruplexes, and that the inhibition by the P-isomer was more pronounced in the case of hTelo. The observed dependence is correlated with the results of FRET and FID measurements reported previously. According to МТТ assay using the HEK-293 embryonic kidney cells, compound 7 was found to have a moderate cytotoxicity: IC50 = 37 ± 4 μM. The quantitative PCR assay demonstrated that the introduction of a racemic mixture of 7 in a dose equal to three times IC50 into HEK-293 cells leads to ~ 40% inhibition of the expression of c-MYC gene, but does not affect c-KIT.
Xiong et al.[42] synthesized the optically pure Λ- and Δ-isomers of the bimetallic complex [RuIIPtIILCl]3+ (8) and monometallic ruthenium (9) and platinum (10) complexes (Scheme 2). As ligand L, all mentioned complexes contained the 1,2,4-triazine moiety with the a phenanthroline substituent.
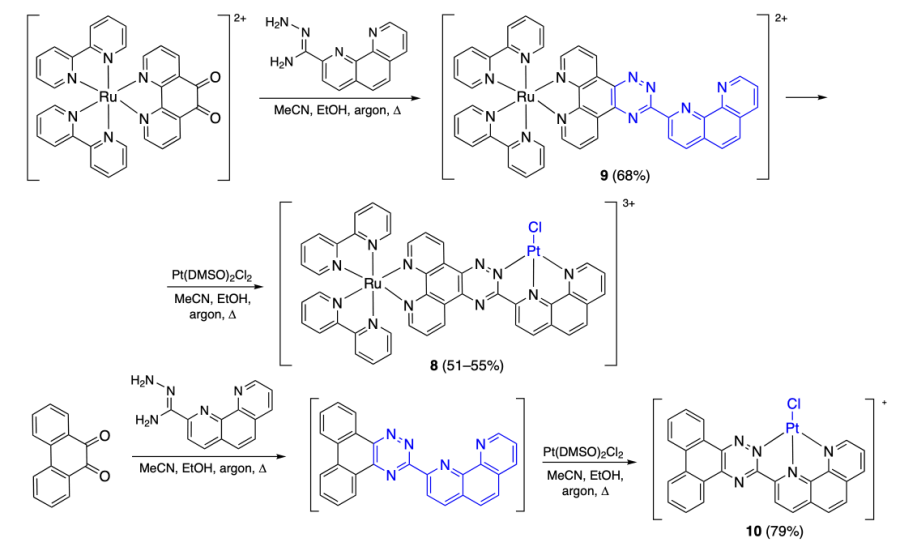
The whole set of data obtained by FRET assay and electronic spectroscopy for complexes 8 – 10 and the human telomeric quadruplex DNA (hTelo) indicated that all of the complexes can bind to DNA in a dilute solution, with the binding constant being in the range of (4.4 – 8.6) × 106 М–1. However, in a solution containing 40% polyethylene glycol (PEG200) added to simulate the intracellular environment, binding of the complexes deteriorated, but the bimetallic complex still retained a relatively high binding affinity: Kb ≈ 106 М–1 for complex 8 (in the presence of K+ ions) and 105 М–1 for complexes 9 and 10. Stabilization of the DNA quadruplex by complex 8 is also retained upon the addition of 100 equiv. of double-stranded DNA, which attests to selective binding of this complex to the quadruplex.
The difference between the binding affinity of compound 8 towards hTel quadruplex in a dilute solution and in a cell-like environment was studied by the molecular docking of compound 8 into telomeric G-quadruplex using the Surflex-Dock@SYBYL program and G4 structures taken from PDB (Protein Database) in a dilute solution (PDB ID: 1KF1) and under cell-like conditions (PDB ID: 2LD8) (Figure 6). It was found that in a dilute solution, compound 8 is bound to the top tetrad of the quadruplex via π – π- and cation – π-interactions between the platinum centre of the complex and guanine molecules. Meanwhile, under cell-like conditions, the quadruplex conformation changes. The interaction between the complex and the quadruplex weakens because of steric hindrances caused by binding of the 5'-terminal thymine and adenine moieties to the quadruplex top tetrad[43]. As a result, π – π-interaction occurs only between the guanine base and bipyridine molecules of 8, while the platinum moiety is displaced from the quadruplex plane and binds to the terminal nucleoside only by electrostatic forces.
![[{"id":"wqcVBsnLFA","type":"paragraph","data":{"text":"Results of molecular docking of complex <b>8</b> into <i>hTel</i> G-quadruplex: dilute solution (a) and cell-like conditions (b)[[ type=\"anchor\" referenceId=\"8953\" ]]."}}]](/storage/images/resized/ME3aRAuUMQ8tfvGgvgpknBlIM0rWIPR3wRvkSbaY_xl.webp)
The biological assays of compound 8 were performed using lung cancer cells (A549), cisplatin-resistant lung cancer cells (A549R), hepatic cancer cells with a low telomerase activity (SK-Hep-1), and normal lung fibroblasts (HLF). The first experiments showed low efficiency of complex 8 due to poor penetration into cell nuclei. For this reason, Xiong et al.[42] encapsulated complex 8 into a biotin-modified DNA cages. The resulting nanoparticles bearing compound 8 showed a high cytotoxicity against both A549 lung cancer cells (IC50 = 7.4 ± 0.57 μM for Δ-8 and IC50 = 8.6 ± 0.72 μM for Λ-8) and cisplatin-resistant A549R cells (IC50 = 12.7 ± 1.2 μM for Δ-8, IC50 = 19.3 ± 1.4 μM for Λ-8; for cisplatin, IC50 = 89.6 ± 8.7 μM). Compound 8 had no cytotoxicity against normal fibroblasts, which attests to selective action. Low cytotoxicity against SK-Hep-1 cells characterized by low telomerase activity, in combination with decreasing expression of the hTERT и TRF2 proteins and increasing expression of DNA damage markers, γ-H2AX and 53BP1, indicates that complex 8 can bind to the telomeric quadruplex in cells and thus inhibits the telomerase activity. The subsequent in vivo tests using mice bearing cisplatin-resistant A549R tumour demonstrated that complex 8 inhibits the tumour growth more efficiently (the decrease in the tumour volume with respect to the control was 73 and 65% for the Δ- and Λ-isomers, respectively) than cisplatin (had almost no effect on the tumour size). It is also noteworthy that the biological assays did not reveal a significant difference between the biological properties of the Δ- and Λ-isomers of compound 8.
Mei and co-workers[44] prepared a series of mixed ligand ruthenium(II) arene complexes 11 – 13 with phenanthroimidazole as a second ligand (Scheme 3). These coordination compounds differed in the position of chlorine atom in the nitrogen-containing ligand: chlorine atom in the ortho-position in complex 11 and in meta- and para-positions in compounds 12 and 13, respectively. Chlorine was chosen as a substituent relying on the results of an earlier study of the same authors[45] on the variation of substituents in analogous complexes and the assumption that a halogen bond (interaction between a halogen atom and an electron donor atom) participates in the stabilization of G-quadruplexes. The prepared coordination compounds were characterized by 1H and 13C NMR spectroscopy, elemental analysis, and electrospray ionization mass spectrometry (ESI MS) techniques. It is important to note that the reported data seem doubtful, because the m/z values of the complexes are given to the nearest integer (except for compound 13, for which this value is given to the nearest tenth), and for each complex two sets of elemental analysis data are given (with different solvation compositions of the complex).
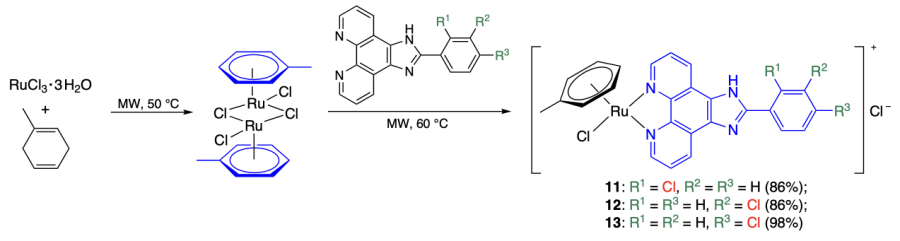
According to the electronic spectroscopy data for solutions of KRAS quadruplex DNA and double-stranded DNA in the presence of synthesized complexes, compounds 11 – 13 have a higher affinity to quadruplex DNA than to double-stranded DNA (Kb = 24.5 × 107 and 6.09 × 107 М–1 for binding of compounds 11 and 13 to KRAS G4; no other Kb values were reported). In addition, complex 12 with a chlorine atom in the meta-position has a lower quadruplex binding affinity than compounds 11 and 13. Similar dependences were observed in experiments with a complex of DNA and ethidium bromide (EB) in which the ethidium cation was displaced by ruthenium complexes (the EB displacement rates for KRAS G4 were 64.0, 26.4, and 51.3% for compounds 11 – 13, respectively; in the case of double-stranded DNA, the substitution barely took place)[44].
The mechanism of binding of the complexes to KRAS was established using molecular docking (Figure 7). It was found that complex 11 binds to guanine (G2, G3, G5) and arginine (A1) residues. Meanwhile, in the case of compounds 12 and 13, interaction with G12, G18, G19, and G20 nucleotides takes place. Despite the differences in the binding to KRAS, the calculated binding energies for complexes 11 – 13 were similar: –6.78, –7.13, and –7.23 kcal mol–1, respectively.
![[{"id":"sRlXnkh4fB","type":"paragraph","data":{"text":"Results of molecular docking of complexes <b>11</b> (а), <b>12</b> (b), and <b>13</b> (c) into KRAS G-quadruplex[[ type=\"anchor\" referenceId=\"8955\" ]]."}}]](/storage/images/resized/30C8HGZ2sHzQaLJxdB9PIoouAITG3etGcqSHHv9j_xl.webp)
Biological assays were carried out using breast cancer cells (MDA-MB-231, MCF-7), oesophageal cancer cells (EC-1), and normal mammary epithelial cells (MCF-10A). The highest cytotoxicity was found for complex 10 against MCF-7 cells (IC50 = 3.7 ± 0.2 μM); this result was comparable with the cytotoxicity of cisplatin (IC50 = 7.9 ± 0.2 μM). The cytotoxicity of complex 11 and complexes 12 and 13 against other cell lines, including normal MCF-10A cells, was low (IC50 > 100 μM). However, the results of cytotoxicity assays are poorly correlated with the data on accumulaion of the compounds in cells. For example, complex 12 was accumulated in MCF-7 cells almost two times more efficiently than complex 11, but the latter barely showed any cytotoxicity (IC50 > 100 μM), and among all tested cell lines, the highest cellular uptake of all three complexes was observed for MCF-10A normal cells (Table 2).
The subsequent biological assays using MCF-7 cells demonstrated that complex 11 can arrest the cell cycle in the G0/G1 phase. In addition, the DNA comet assay and histone γ-H2AX visualization by confocal microscopy showed that compound 11 can induce damage of DNA. Despite the reasonably good results in the DNA damage assays and cell cycle arrest, the data on the biological properties and the composition of compounds 11 – 13 are contradictory (the cytotoxicity data do not correlate with the cellular uptake data) and do not match well the electronic absorption spectra and docking results. Therefore, further investigation of the coordination compounds of this class is required.
In the next study, Mei and co-workers[46] prepared ruthenium complexes similar to those described above. In these complexes, an erianin moiety increasing the lipophilicity of the complexes was introduced into the nitrogen-containing ligand molecule; the authors expected that this would enhance the cellular uptake of the complexes. The starting complexes 14 and 15 containing bromine were used as reference compounds. The bromine atom was replaced by the erianin moiety to give compounds 16 and 17, respectively. It is noteworthy that even in this study, the results of mass spectrometry of complexes cast doubt, since the calculated and experimental m/z values differ in the first or second decimal place, which attests to a low accuracy of determination of the ion masses. For compound 14, m/z = 481.0536 ([M – ClO4–]2+ ion; the calculated value is 480.40) and 539.0770 ([M – ClO4– + NH4+]; calculated 539.055); for compound 17, m/z = 599.1655 ([M – ClO4–]2+ ion; calculated 599.1650) and 657.1889 ([M – ClO4– + NH4+], calculated 657.665).
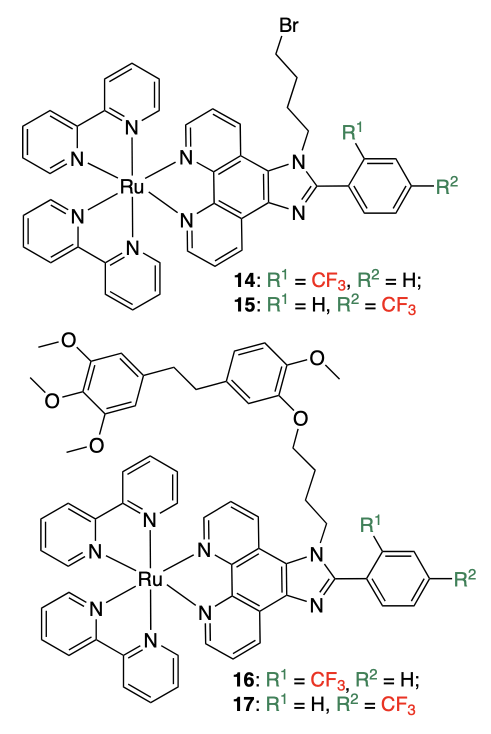
An absorption and emission spectroscopy study of these complexes in the presence of c-MYC quadruplex DNA showed that coordination compounds 16 and 17 bind to the quadruplex with high affinity, unlike complexes 14 and 15. The most pronounced quadruplex stabilization is attained for complex 17 with the CF3 group in the para-position.
In addition, the interaction of the complexes with c-MYC in the absence and in the presence of double-stranded DNA was studied by FRET. The results indicated that complexes 16 and 17 efficiently stabilize the quadruplex even in the presence of 40 equiv. of double-stranded DNA. However, this conclusion is not supported by the change in the DNA melting temperature ΔTm reported in the same paper[46].
The biological properties of complexes 14 – 17 were studied by PCR assay using Taq DNA polymerase and c-MYC quadruplex DNA as the matrix. It was shown that complexes 16 and 17, unlike complexes 14 and 15, can inhibit the synthesis of c-MYC. The results of this study generally agree with the spectral data, but unambiguous conclusions about the efficiency of binding under cell conditions and cytotoxicity may be drawn if at least a MTT assay is performed, which was not done in that study.
Kench et al.[47] obtained nickel and platinum coordination compounds consisting of two salphen moieties connected by a linker for binding to multimeric G-quadruplexes in telomeres. Monomeric complexes 18 and 19 were used as reference compounds, while polyethylene glycol chains (complexes 20 – 22) or polypeptides consisting of arginine and lysine residues (complexes 23 – 27) served as linkers (Scheme 4).
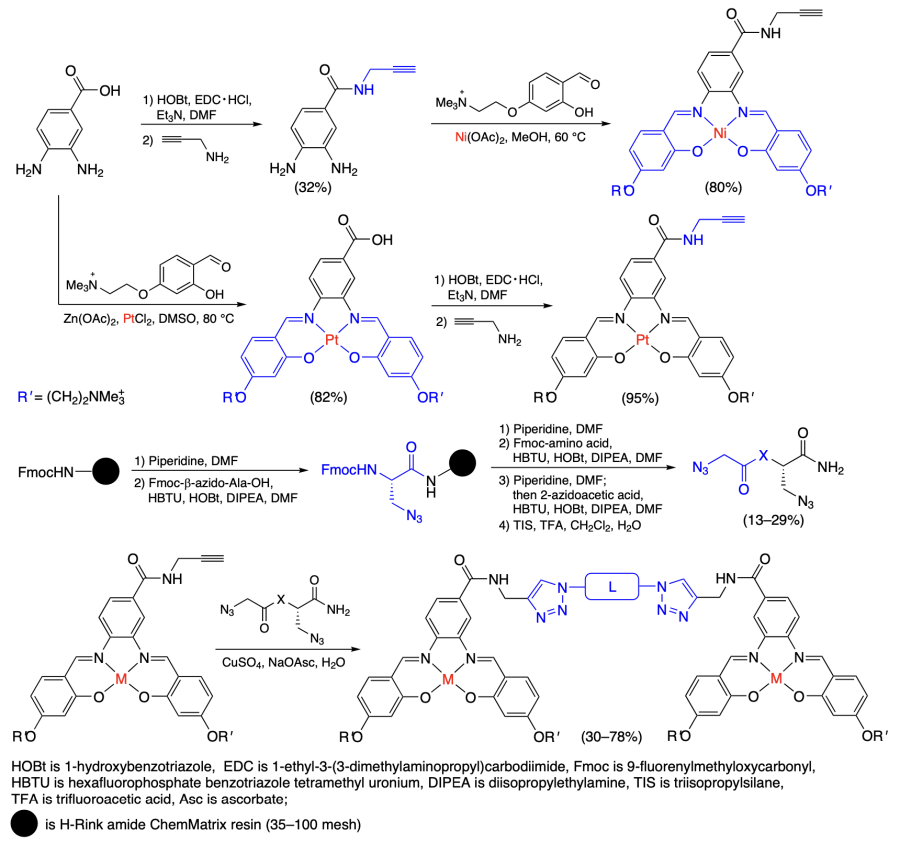
The binding assays were carried out using the hTert telomeric DNA with one quadruplex (designated by G1) and two quadruplexes with different linkers between them (G2T1, G2T6). The melting temperature measurements combined with circular dichroism (CD) spectroscopy showed that monomeric complexes 18, 19 can stabilize both monomeric and dimeric quadruplexes. The dimeric G2T1 and G2T6 quadruplexes were stabilized to a somewhat higher extent than the monomers, which was attributed[47] to the formation of sandwich adducts of DNA complexes in which the metal complex coordinates both quadruplexes. The change in the melting temperature upon the formation of adducts was somewhat smaller for complexes 20 – 27 than for 18 and 19; however, complexes 20 – 27 showed a higher selectivity for dimeric quadruplexes. The results of emission titration of platinum complex with a quadruplex DNA solution are generally in line with the results of melting experiments, with stronger binding to G4 being found for the complexes in which linkers had four and eight amino acid residues. Study of the binding to double-stranded DNA demonstrated that complexes 18 – 27 bind less efficiently to the DNA double helix; the DNA binding constants were 1 to 2 orders of magnitude lower than those for quadruplex DNA.
Despite the high binding affinity to telomeric DNA, an MTS assay [MTS is 3-(4,5-dimethylthiazol-2-yl)-5-(3-methoxycarboxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium bromide] using the U2OS osteosarcoma cell line revealed no cytotoxicity for compounds 18 – 27. The subsequent studies showed that the absence of cytotoxicity was due to poor cellular uptake of the complexes. The incubation of the cells with digitonin before addition of the complexes improved the penetrability; however, the compounds were mainly concentrated in the cytoplasm rather than in the nucleus.
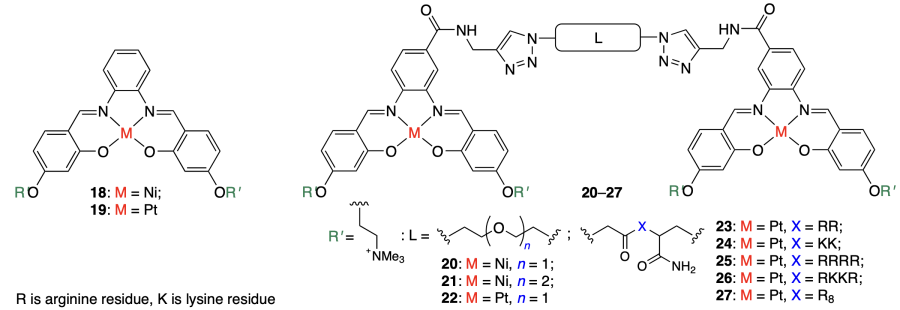
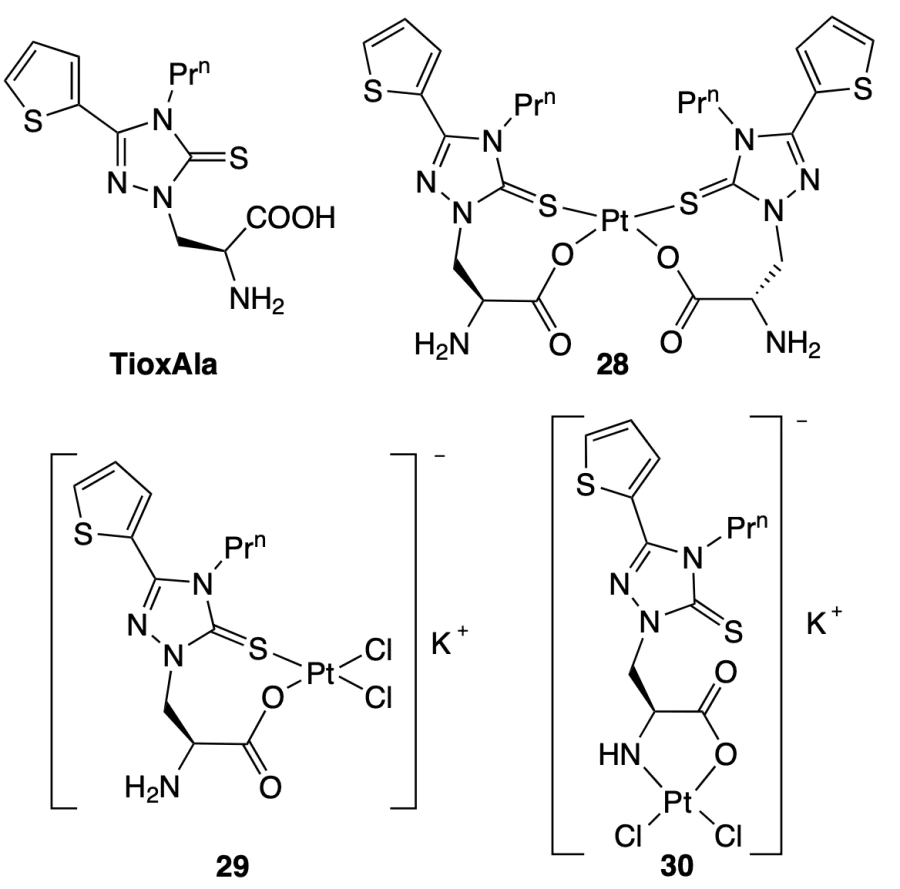
Riccardi et al.[48] and Saghyan et al.[49] prepared and studied PtII complexes, PtL2 (28) and K[PtCl2L] (29, 30), with the TioxAla amino acid. According to CD and UV spectroscopy data, the starting amino acid and complex 28 virtually do not bind to single- or double-stranded DNA or quadruplexes, whereas compounds 30 and especially 29 bind to all DNA structures, while showing no selectivity to quadruplexes.
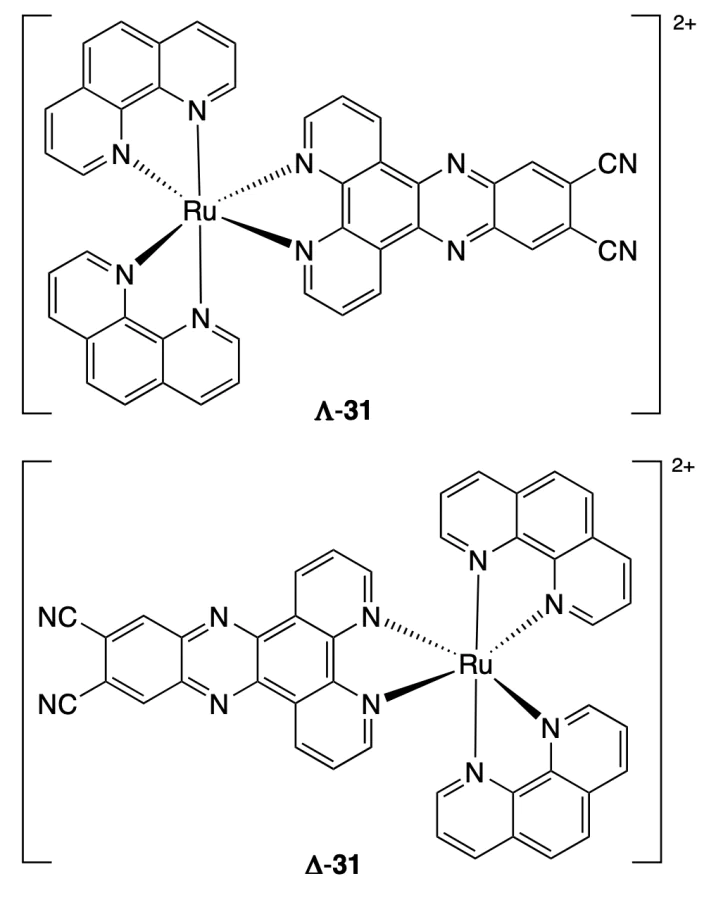
Stitch et al.[50] investigated binding of the hybrid and antiparallel hTel(K+) and hTel(Na+) quadruplexes to enantiomeric ruthenium complexes Δ-31 and Λ-31 containing phenanthroline and dipyridophenazinedicarbonitrile as ligands. According to absorption spectroscopy data, the binding constant of complex Λ-31 for the hybrid quadruplex [Kb = (1.1 ± 0.6) × 107 М–1] was an order of magnitude higher than the binding constant for the antiparallel quadruplex [Kb = (2.2 ± 0.3) × 106 М–1]. The Kb values for Δ-30 were of the order of 105 М–1 for both quadruplexes.
The further investigation of binding of the ruthenium complex to DNA by time-resolved IR spectroscopy (TRIR) and molecular docking showed that complexes Λ-31 and Δ-31 bind to hybrid hTel(K+) through π–π-stacking between the phenazine moiety and the lower tetrad of the quadruplex (G4, G10, G14, G20), while the phenanthroline moieties interact with the nitrogenous bases in the loop. The calculated Gibbs free energy of binding (ΔGbind) was lower for Λ-enantiomer (ΔGbind = –87.87 kcal mol–1) than for Δ-enantiomer (ΔGbind = –59.82 kcal mol–1), which is indicative of the higher affinity of complex Λ-31 to hTel(K+). In the case of antiparallel hTel(Na+) quadruplex, both enantiomers interact with the upper tetrad (G2, G12, G14, G21); however, compound Λ-31 binds mainly through π–π-stacking between the phenazine moiety and guanine bases, whereas complex Δ-31 interacts with the tetrad more weakly and binds mainly to thymine and adenine of the quadruplex loop. The ΔGbind values for compounds Λ-31 and Δ-31 and hTel(Na+) amount to –94.00 and –80.24 kcal mol–1, respectively. It is worth noting that the dependence of the binding energy of the enantiomers on the quadruplex structure poorly correlates with the analogous dependence of the binding constants: for complex Λ-31, ΔGbind values are comparable for both quadruplexes, while Kb values differ by an order of magnitude; in the case of complex Δ-31, ΔGbind for hTel(Na+) is lower, but Kb are comparable. Therefore, unambiguous elucidation of the relationship between the affinity to G4 and the structure of complexes requires additional experimental data on the binding of complexes to DNA (e.g., the results of fluorescence titration or melting experiments).
Reyes et al.[51] synthesized a series of platinum(II) complexes 32 – 34 with 6-arylbipyridines as ligands. To increase the solubility, the chlorine atom in complexes 32 – 34 was replaced by a mercaptoethanol moiety, which gave compounds 35 – 37 (Scheme 5).
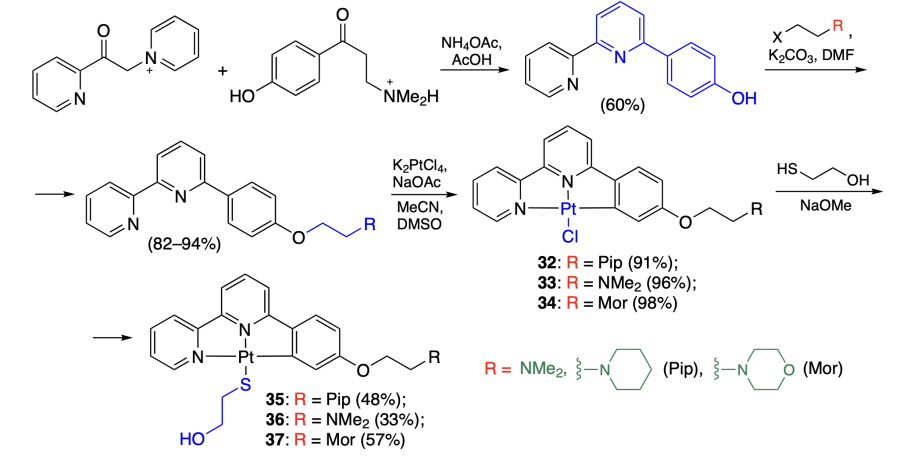
According to emission spectroscopy data for these complexes in solutions in the presence of c-MYC, c-KIT2, HTG4, hTelo(K+), and hTelo(Na+) quadruplexes and double-stranded DNA isolated from calf thymus (ct-DNA) and salmon testes (st-DNA), complexes 32 – 34 had a higher affinity to quadruplexes than to double-stranded DNA and had higher binding constants than compounds 35 – 37 (Table 3). A similar pattern was revealed in the measurements of DNA melting temperatures in the presence of platinum complexes. It is also important to note that using both methods, the highest affinity of the complexes was found towards the parallel c-MYC quadruplex, while the lowest affinity was characteristic in the case of antiparallel hTelo(Na+). The MTS assay using the U2OS cell line demonstrated that the platinum complexes exhibit virtually no cytotoxicity because of the low cellular uptake of these compounds, as was found later by confocal microscopy.51
In recent years, increasing attention of researchers has been attracted by the immunogenic cell death (ICD) process. Unlike usual apoptosis, ICD involves the development of immune response specific to tumour cells. When tumour cells that undergo immunogenic death are administered to immunocompetent mice, the tumour stops to grow in 75 – 100% of cases[52]. It was shown that ICD can be activated by many chemotherapeutic drugs such as doxorubicin[53], cisplatin[54], bortezomib[55], and other. For example, Liu et al.[56] synthesized platinum(II) complexes 38 – 40, which could potentially activate ICD. Naphthalenediimides, synthesized from naphthalene-1,4,5,8-tetracarboxylic dianhydride by a reported procedure[57], were used as ligands in these complexes (Scheme 6).
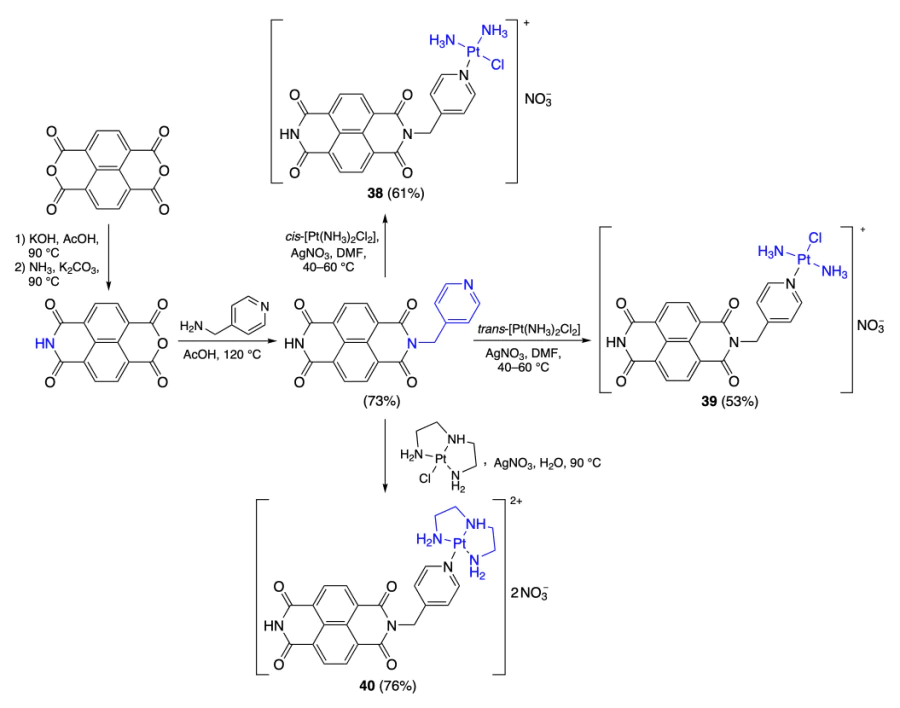
According to NMR spectroscopy data for MYT1L quadruplex in the presence of platinum complexes 38 – 40, the complexes bind to the guanine quartet through π – π interactions between the naphthalenediimide moiety and the guanine base, while in the case of compound 39, the covalent bond between the platinum ion and G6 residue in the quadruplex loop is also involved. The presence of the covalent bond accounts for the high selectivity of binding of 39 to the quadruplex and for the stability of the complex — DNA adduct.
According to МТТ assay (Table 4), compound 39 has a high toxicity against HeLa, MDA-MB-231, MCF-7, A549, and 4T1 tumour cells and also against the cisplatin-resistant A549R cells, being less toxic against normal MCF-10A cells.
![Cytotoxic activity of compounds <b>38</b> – <b>40</b> according to МТТ assay data (IC<sub>50</sub>, μM)[<a class="anchor" href="#reference-8967">56</a>].](/storage/images/resized/F4uId4lnO7o9FbT63aEI0xj9jDlAqG57zYlx7SQL_xl.webp)
Using immunofluorescence microscopy, flow cytometry, and transmission electron microscopy, it was ascertained that treatment with compounds 38 and 39 increases the levels of calreticulin (CRT) and extracellular ATP and induces the aggregation of the ends of chromatin and membrane disruption, which attests to cell death by the ICD mechanism. To study the action of the compounds in vivo, 4T1 cells were preincubated with complexes 38 – 40 or cisplatin. The mice were administered first with the dying tumour cells and, after a week, with live 4T1 cells. According to the results of 26-day monitoring of the tumour volume and the body weight of mice, no significant loss of body weight or damage of organs was observed, which indicates a low systemic toxicity of cells pretreated with these platinum complexes. In addition, the introduction of cells preincubated with compounds 38 – 40 resulted in inhibition of tumour growth by 68, 62, and 43%, respectively, while in the case of cells incubated with cisplatin, the inhibition was insignificant.
Hence, to be considered as G4 ligands, coordination compounds must contain a metal cation capable of cation – π-interactions with the guanine moieties of the quadruplex and/or a planar aromatic moiety capable of π – π-stacking. In addition, to exhibit the desired biological activity, a complex should be sufficiently lipophilic to penetrate into cells; therefore, in some cases, additional modification of the organic ligand is expedient.
Currently, the best results in the binding to G-quadruplexes and in cytotoxicity assays have been obtained for platinum and ruthenium complexes with ligands containing a bulky aromatic moiety (anthracycline, phenanthroline, or naphthalenediimide). Binding of these complexes to DNA occurs not only through π – π-stacking between DNA and the ligand, but also through binding of the metal ion to nucleotides in the case of platinum compounds. However, polyaromatic moieties of the ligands that provide binding of the complexes to DNA are also responsible for low solubility and poor cellular uptake of compounds because of π-stacking between the molecules. Therefore, additional modification of the compounds of this type is necessary, for example, by introducing alkyl substituents into the ligand molecules in order to improve their penetration through the cell membrane.
2.2. Linear polyaromatic compounds
Polyaromatic compounds such as anthraquinone and anthracene derivatives and their heterocyclic analogues (acridine, phenazine, xanthone) are well known as antitumour drugs acting via binding to DNA[58-61]. The high DNA binding affinity caused by extended π – π-system made polyaromatic compounds, first of all anthraquinone and acridine derivatives, highly popular ligands for G-quadruplexes[62-65]. In addition, mention should be made of acridine derivative, BRACO-19 {N,N'-(9-{[4-(dimethylamino)phenl]amino}acridine-3,6-diyl)-bis[3-(pyrrolidin-1-yl)propanamide]}[66] and heteroarene-fused anthraquinones[67], which have G4 binding constants of approximately 106 – 107 М–1 and binding constants to double-stranded DNA lower by two orders of magnitude. Nevertheless, despite the impressive results, acridine and anthraquinone derivatives have not yet been subjected to preclinical trials. For this reason, active studies aimed at modification of the known and synthesis of new polyaromatic compounds are currently underway.
Fukuda et al.[68] described a series of cyclic anthraquinone derivatives 41 – 44 and studied their binding to telomeric DNA (Scheme 7). These compounds were prepared from 1,5-dichloroanthraquinone by substitution of chlorine atoms by an aliphatic diamine moiety followed by acylation, including ring closure induced by dicarboxylic acid.
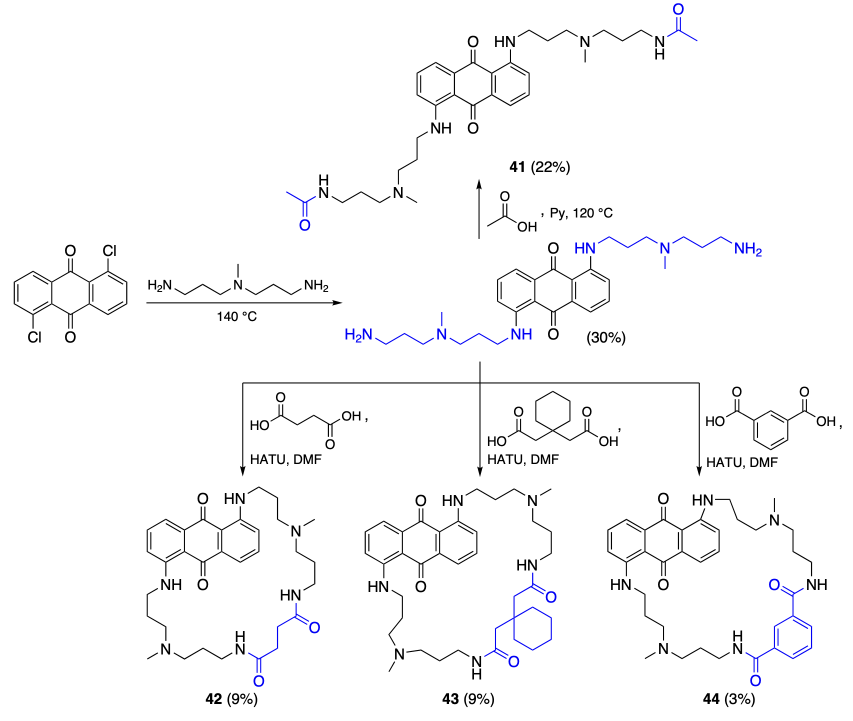
Using electronic spectroscopy and results of melting temperature measurements for telomeric G-quadruplexes, it was shown that compound 44 has the most pronounced stabilization effect (ΔTm = 8.8 °C), while having higher binding affinity to the quadruplex than to double-stranded DNA. The molecular docking of macrocycle 44 into telomeric G4 (PDB ID: 2GKU) showed that high affinity of this compound towards the quadruplex is attributable to the incorporation of the adenine moiety into the ring of the ligand and hydrogen bonding between the nitrogenous base and the phenyl group. The subsequent studies indicate that compound 44 can inhibit the growth of HeLa, Ca9-22, SAS, HSC-2, and HEK293 tumour cells, with the effect being most pronounced for the cells with high level of TERT mRNA expression and being much lower for ASF-4-4L2 and BMC normal cells (Table 5). In the tests on SAS tumour in mice, compound 44 showed inhibition of tumour growth comparable to that of cisplatin in a dose 10 times lower than the cisplatin dose.
![Cytotoxic activity of compounds <b>41</b> – <b>44</b> according to МТТ assay data (IC<sub>50</sub>, μM)[<a class="anchor" href="#reference-8978">68</a>].](/storage/images/resized/8Lczo57ffxIYI6r5G3LrtC2tOQqbdU1YNk65SqNb_xl.webp)
Hu and Lin[69] synthesized a series of dibenzophenazines 45 – 56 in order to find a compound that could actively bind to c-MYC quadruplex and also inhibit topoisomerase-1 (Topo1), which is presumably responsible for the development of resistance to G4 ligands. The choice of compounds was due to the fact that a known quinoxaline derivative QN-1 showed a high affinity to c-MYC, but not to Topo1.
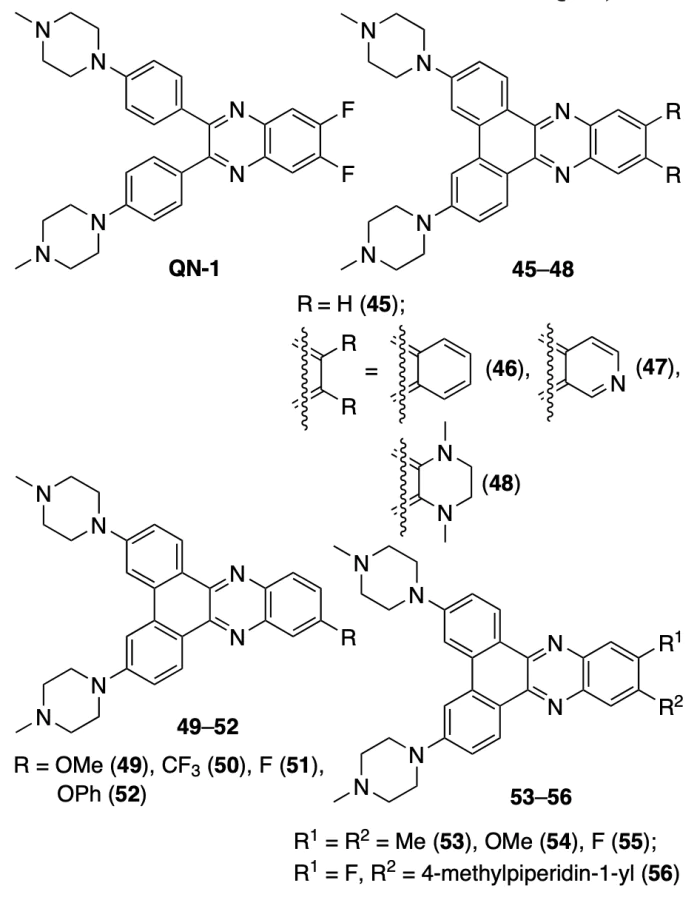
In a study of the effect of compounds 44 – 55 on the Topo1 activity towards supercoiled pHOT1 DNA, compounds 45, 47, 49, and 51 were found to be the best topoisomerase inhibitors. According to real-time PCR assay data, compounds 45, 47, and 49 also inhibited the c-MYC expression. It is noteworthy that compound 55, which differs from QN-1 only by the presence of C – C bond between the benzene rings, did not show any activity. In addition, the affinity to Topo1 and c-MYC was adversely affected by the presence of an additional ring (compounds 46, 48), bulky substituents in position 11 (compounds 52, 56), or any groups in position 12 (compounds 53 – 55). The adverse effect on going from compound QN-1 to more fused systems and to compounds with additional rings is apparently due to decrease in the solubility; also, additional and bulky substituents can prevent the intercalation of compounds into DNA. Since methoxy-substituted compounds are usually more soluble, derivative 49 was chosen as the lead compound. The absorption and fluorescence titration data indicate its higher affinity to c-MYC (the dissociation constant of the complex Kd was 0.6 μM) compared to the affinity to double-stranded DNA (Kd = 12.7 μM); melting temperature assay showed stabilization of c-MYC quadruplex by 30 °C under the action of compound 49. Using CCK8 assay, † it was shown that this compound has a high toxicity against MDA-MB-231 tumour cells (IC50 = 0.7 μM) and a lower toxicity against normal BJ fibroblasts (IC50 = 5.6 μM). The efficacy of derivative 49 against triple-negative breast cancer was confirmed by experiments on mice bearing MDA-MB-231 tumour: administration of compound 49 in doses of 2.5 and 5 mg kg–1 caused inhibition of tumour growth comparable with that of doxorubicin (2.5 mg kg–1), but, unlike doxorubicin, it did not cause loss of animal weight.
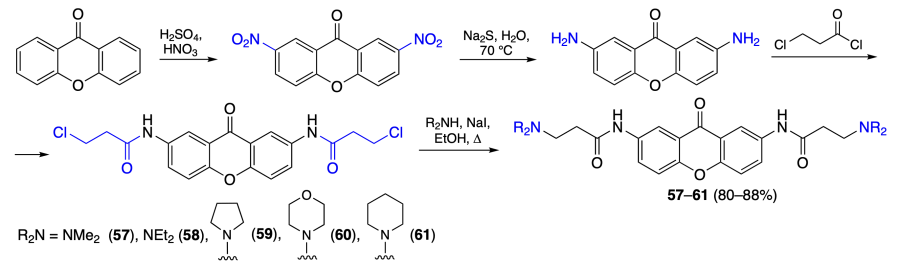
Shen et al.[70] obtained a series of xanthone derivatives 57 – 61 by acylation of 2,7-diaminoxanthone with 3-chloropropionyl chloride followed by chlorine substitution by amines (Scheme 8). Study of the cytotoxic activity of these xanthones against HeLa, MCF-7, SGC-7901, and A549 cells demonstrated that compound 57 is most efficient against any of the investigated cell lines (Table 6). Flow cytometry of HeLa cells showed an increase in the proportions of early and late apoptotic cells up to 46.86 and 25.68%, respectively.
The conformational changes in the HTG21 quadruplex in the presence of xanthone derivatives 57 – 61 were analyzed using CD spectroscopy. Upon the addition of these compounds in the presence of K+ ions, the quadruplex assumes the hybrid conformation, and the spectral peaks shift, indicating an equilibrium between a few formed adducts of different topology. The HTG21 quadruplex was stabilized by any compound of the series, with the most pronounced effect being found for xanthone 57. According to PCR assay, compound 57 can inhibit amplification of HTG21, but does not affect the HTG21mu mutant, which is unable to form quadruplexes. Fluorescence spectroscopy showed that compound 56 has a higher affinity to HTG21 than to double-stranded DNA (Kb = 6.34 × 104 and 2.43 × 103 M–1, respectively). Using the AutoDock program (Figure 8) for the molecular docking of compounds 57 – 61 into 1KF1 quadruplex and hybrid 2HY9 quadruplex, it was ascertained that in both cases, the xanthone ring is located in the quadruplex quartet plane and is bound to guanine moieties through π – π contacts. In the case of parallel quadruplex, the side chains point towards the grooves, while amino groups are located in the negatively charged regions, whereas in the hybrid G4 conformation, one side chain lies in the plane of the quartet.
![[{"id":"SpP42pov5U","type":"paragraph","data":{"text":"Results of molecular docking of compounds <b>57</b> – <b>61</b> into <i>1KF1</i> (a) and <i>2HY9</i> (b) quadruplexes[[ type=\"anchor\" referenceId=\"8980\" ]]."}}]](/storage/images/resized/GQLfqftUFBv2EvrtaDvPmx0pGDw0dcImIhQ8d2SZ_xl.webp)
Roly et al.[71] described the synthesis of new of mono- and bifunctionalized xanthones 62 – 73 containing a piperazine or morpholine moiety and studied stabilization of quadruplex DNA by these compounds.
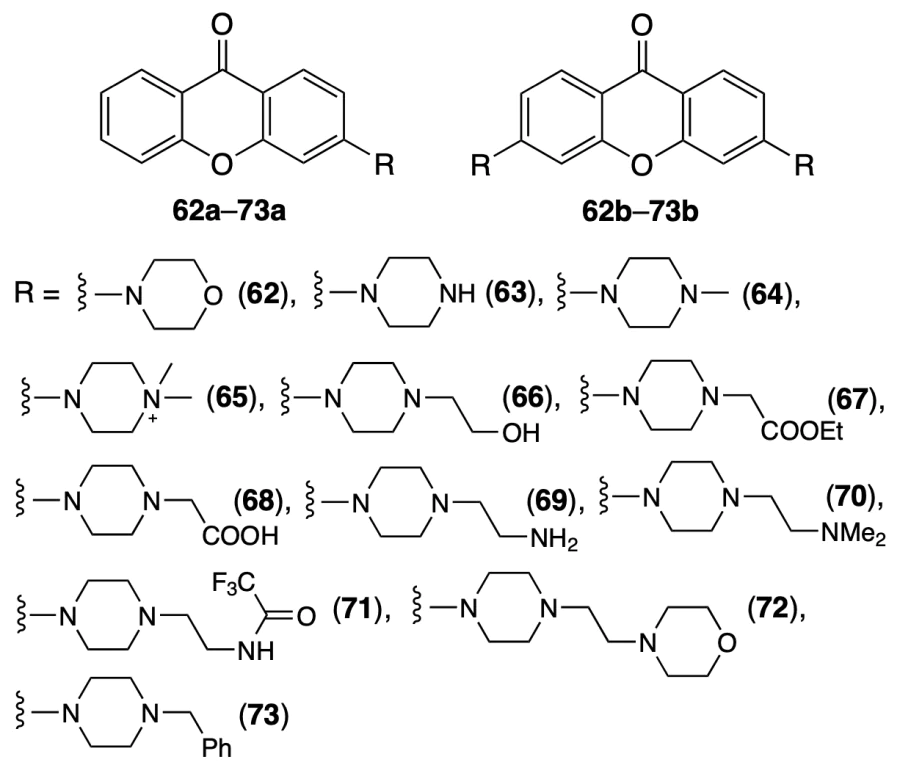
It was shown by electronic spectroscopy that all monosubstituted xanthones 62a – 73a weakly interact with G4-DNA, whereas among disubstituted analogues, low binding constants were found only for compounds 62b – 64b, in which the morpholine and piperazine rings are directly attached to the xanthone nucleus. Derivatives 69b – 72b, which showed the best results, were chosen for subsequent investigation.
DNA melting temperature measurements in the presence of the synthesized ligands demonstrated that the ΔTm values were higher in the case of c-MYC than for c-KIT or double-stranded DNA. The most pronounced effect was found for compounds 69b and 70b (for c-MYC, ΔTm = 9.4 and 11 °C, respectively; Table 7). Using fluorescence spectroscopy with ethidium bromide, it was shown that the ethidium bromide displacement rate at the same ligand concentration (20 μM) was higher for compound 70b (~ 33%) than for compound 69b (~ 26%), which attests to better binding of the former to c-MYC quadruplex DNA.
![Change in the DNA melting temperature in the presence of compounds <b>69b–72b</b> (in °C)[<a class="anchor" href="#reference-8981">71</a>].](/storage/images/resized/YZpLYg1hLJoUbJuRdSeUXUODBiceQ2wvEjoZjBiV_xl.webp)
The IC50 values for compounds 69b – 72b and three tumour cell lines and NIH3T3 normal cell line as a control were determined by MTT assay (Table 8). The highest cytotoxicity was observed for compounds 69b and 70b against MCF-7 cells (IC50 = 6 and 3.05 μM, respectively). None of the compounds in 50 μM concentration had a noticeable cytotoxic action against NIH3T3 normal cells, which attests to the selectivity of their action. According to real-time PCR data, the c-MYC transcription level in MCF-7 cells decreased in a dose-dependent manner (by 30, 50, and 55%, respectively, on treatment with 4, 6, and 8 μM of compound 70b), while transcription of the GAPDH control remains unaffected.
Dey et al.[72] investigated the effect of the length of side chains of anthraquinone derivatives 73 and 74 containing diethylamino groups on binding to G-quadruplexes in the presence of Na+ and K+ ions.
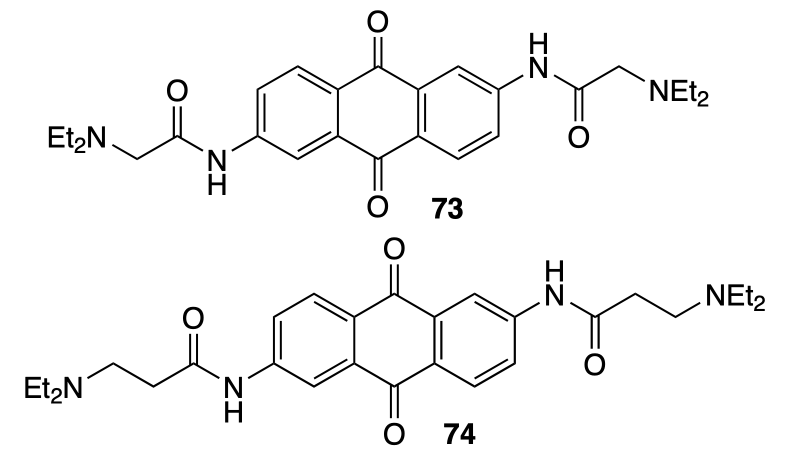
According to the surface plasmon resonance data, the best binding to the d[AGGG(TTAGGG)3] quadruplex in solution in the presence of K+ ions is observed for compound 74 (Kb = 4.8 × 106 M–1); this is an order of magnitude higher than that for compound 73 (Kb = 7.6 × 105 M–1). The set of absorption and emission spectra of ligand solutions in the presence of quadruplexes provided the conclusion that binding occurs through stacking with the G4 upper tetrad. This finding was confirmed by CD spectra. The changes in the melting temperature of HTel-22 and wHTel-26 quadruplex DNA in the presence of these ligands amounted to 10 – 13 °C.
The IC50 values for compounds 73 and 74 were determined in a 24-h assay using MCF-7 breast cancer cells and were equal to 3.4 and 1.3 μM, respectively. The flow cytometry data indicate that the proportions of late apoptotic cells 12 h after treatment were 19.94 (for 73) and 12.01% (for 74) compared to untreated cells, аnd after 24 h of incubation, the percentage of late apoptotic cells increased. In addition, a qRT-PCR assay demonstrated a decrease in the c-MYC и BCL-2 transcription in the presence of both ligands.
Andreeva et al.[73] synthesized a series of thiadiazole-, selenadiazole-, and triazole-fused anthraquinones 75 – 77. As the starting compound, the authors used 2-amino-3-nitroquinizarine, which was prepared from quinizarine by a reported[74] procedure. The introduction of the tert-butoxycarbonyl (Boc) protection followed by methylation of hydroxyl groups, reduction of the nitro group by sodium dithionite, and deprotection afforded intermediate 2,3-diamino-1,4-dimethoxyanthraquinone in an overall yield of 29%. This compound was allowed to react with thionyl chloride, selenium(IV) oxide, or sodium nitrite to give thiadiazole-, selenadiazole-, or triazole-fused anthraquinones. The final step involving nucleophilic substitution of the methoxy groups by diamine moieties yielded products 75 – 77 (Scheme 9).
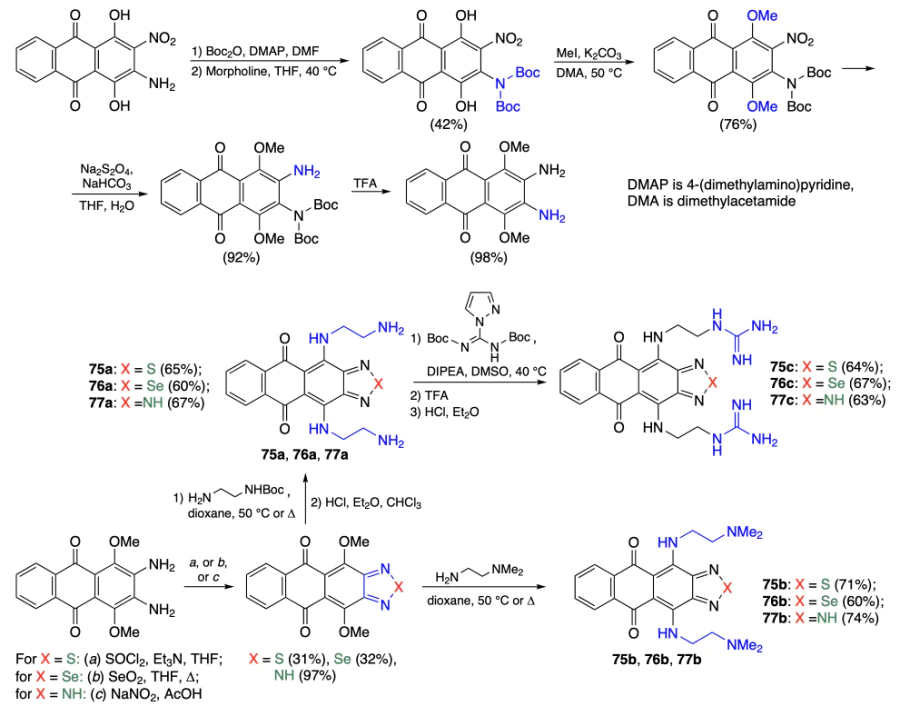
According to МТТ assay of compounds 75 – 77 with K562, HCT116, and MDA-MB-231 tumour cell lines and hFB-hTERT16 normal cell line, compounds 75а and 76а had the highest cytotoxicity against tumour cells (IC50 in the 0.6 – 0.8 and 0.8 – 4.0 μM ranges, respectively). Meanwhile, their toxicity against normal cells (IC50 = 2.4 ± 0.2 and 5.2 ± 0.4 μM) was lower than that of doxorubicin (IC50 = 0.17 ± 0.02 μM). It should be noted that triazole derivatives 77а–с and compounds 75с–77с with a peripheral guanidine moiety showed no cytotoxicity (IC50 > 50 for most cell lines).
Using FRET analysis of hybrid (22AG, BclT), antiparallel (22CTA), and parallel (c-MYC, c-KIT) quadruplexes and double-stranded DNA, it was shown that anthratriazoles virtually do not affect the DNA stability. The most pronounced stabilization of G4 at a ligand concentration of 2 μM was observed for anthrathiadiazole 75а (ΔTm = 31 – 33 °C for telomeric G4-22AG, 22CTA; ΔTm = 44 °C for BclT) and selenadiazole 76а (ΔTm = 13 – 17 °C for all G4). The selectivity of selenadiazole 76а for quadruplexes over double-stranded DNA was higher than the selectivity of thiadiazole 75а (ΔTm = 26 °C for 75а, ΔTm = 6 °C for 76а). A similar dependence was observed when dissociation constants were measured by microscale thermophoresis (MST) and fluorescence titration: compound 75а, Kd = 1.1 – 2.2 μM (for 22AG, 22CTA and c-MYC G4) and 2 – 3.7 μM (for dsDNA); compound 76а, Kd = 0.13 – 2.2 μM (for 22AG, 22CTA and c-MYC G4) and 13 ± 8 μM (for dsDNA).
A flow cytometry study of the effect of anthraselenadiazole 76а showed that the introduction of a ligand in 8 μM concentration into K562 tumour cells increases the percentage of apoptotic cells up to 46 and 67% after 24 and 48 h of incubation. Using qRT-PCR and flow cytometry assays, it was established that compound 76а inhibits expression of c-MYC gene in K562 cells after 24 h of incubation (the level of the c-MYC protein decreases by 45 – 50% and 60 – 65% with 8 and 16 μM doses, respectively). These results indicate that the cytotoxic action of compound 76a is primarily caused by interaction with the promoter of the c-MYC gene, to which it has the highest affinity, and, hence, by induction of apoptosis.
The published data described in this Section provide several conclusions. First, it is noteworthy that among the mentioned xanthone derivatives, compounds 57 and 70b with dimethylamine groups have the highest cytotoxicity and binding constants. This is attributable to the fact that the Me2N group has a moderate basicity, but it is sterically more accessible for binding to DNA phosphate groups than, for example, more basic pyrrolidine moiety. Thus, DNA binding is influenced by steric factors and by the basicity of the terminal amino group; this is confirmed by both earlier reviews on this topic[67] and publication by Dey et al.[72] In the latter publication, the most efficient binding to DNA was found for compound 74 with the longest linker and, therefore, the weakest effect of the electron-withdrawing carbonyl group on the terminal amino group. Second, among the discussed arenes, the best results were found for phenazine derivatives (in particular, compound 49 had IC50 = 0.7 μM for MDA-MB-231 cell line), containing nitrogen atoms in the ring, despite the absence of long aliphatic linkers in their molecules, as in the anthraquinone and xanthone derivatives. Since the nitrogen-containing heterocycles such as pyridine or pyrazine are less basic than aliphatic amines, the better binding of phenazines to DNA cannot be attributed to their higher basicity. Further studies are needed to disclose the causes for this relationship and to confirm the interrelation between the basicity of the terminal amino group and the type of DNA binding.
2.3. Benzazoles
Benzazoles, first of all benzimidazoles and benzothiazoles, are well-known pharmacophore groups with a broad range of biological activity[75, 76]. Among benzazole derivatives, there are known G-quadruplex ligands, including both cytotoxic[77-80] and low-toxic ones (as a rule, benzothiazole derivatives). This makes these compounds good fluorescent probes such as thiazole orange (TO)[81-83]. Most benzothiazole and benzimidazole derivatives have lower affinity to G4 than the above polyaromatic compounds (Kb ≈ 105 M–1), but nevertheless, they demonstrate a higher drug likeness because of the relatively small fused system.20
As ligands for quadruplex DNA, Wu et al.[84] obtained a series of benzazoles, including benzoxazoles 78 – 80, benzothiazoles 81 – 83, and benzoselenazoles 84 – 86, containing an ethene-linked N-substituted carbazole moiety in position 2, their azolium salts 87 – 95, and benzo analogues 96 – 98 with quaternized forms 99 – 101 (Scheme 10). In the molecules of the synthesized products, not only the heteroatoms in the benzazole ring were varied, but also the group at the carbazole nitrogen atom, while the azole nitrogen atom was quaternized.
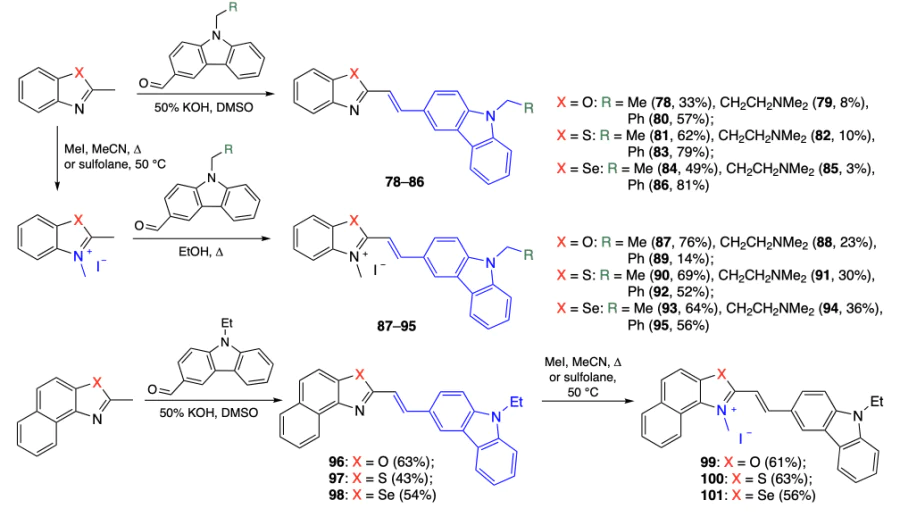
Using emission spectroscopy of the obtained compounds in the presence of pu22 (c-MYC gene quadruplex), c-KIT, htg22, and HRAS quadruplex DNA, pu22mut single-stranded DNA, and double-stranded DNA, it was shown that benzoxazoles have the lowest DNA binding affinity, while benzoselenazoles not only efficiently bind to DNA, but also possess the highest selectivity for quadruplex DNA among the tested benzazoles. In addition, it was found that methylation of the benzazole nitrogen atom usually enhances binding to pu22. An MTT assay with HepG2 hepatic cancer cells, HCT116 colorectal carcinoma cells, and NCM460 normal cells yielded a dependence similar to that established in DNA binding experiments (Table 9). Most benzoxazoles showed low cytotoxicity against all cell lines. As the lead compounds, the authors chose methylated benzoselenazoles 93 and 95, which had high cytotoxicity against HepG2 and HCT116 cancer cell lines (IC50 = 1.6 and 3.4 μM, IC50 = 3.5 and 4.1 μM for compounds 93 and 95, respectively) and low cytotoxicity against normal cells (IC50 = 29.2 and 77.6 μM for compounds 93 and 95, respectively).
The putative structure of the complex formed from compound 95 and pu22 quadruplex DNA was simulated by molecular docking (Schrödinger program) (Figure 9). It was shown that the ligand molecules bind to the upper and lower quartets of the quadruplex through π – π-interactions. Whereas the carbazole moiety was involved only in the π – π stacking, the benzoselenazole moiety was additionally bound to adenine residue, owing to the presence of the selenium atom, by a hydrogen bond with the NH2 group and a chalcogen bond with the adenine nitrogen atom. In the authors' opinion, the involvement of the selenium atom in binding to the quadruplex was responsible for the higher DNA binding affinity and higher cytotoxicity of benzoselenazoles compared to benzoxazoles and benzothiazoles.
The subsequent biological assays using RT-PCR and western blotting confirmed that compounds 93 and 95 inhibit the expression of the c-MYC gene and induce apoptosis of cancer cells. Compound 95 inhibited the tumour growth in vivo in HepG2 tumour-bearing mice at approximately the same level as doxorubicin, without exhibiting severe side effects.
Patidar et al.[85] obtained compound 102 and tested it as a fluorescent probe selective for quadruplexes. Data of emission spectroscopy indicated that fluorescence of compound 102 is enhanced in the presence of DNA, with the most pronounced effect being observed in the presence of quadruplex DNA, especially pu22.
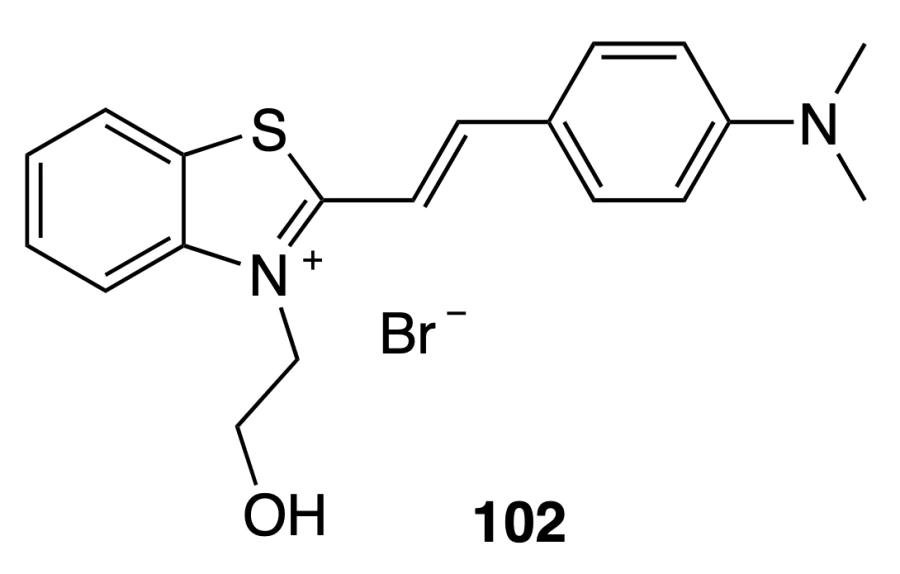
The binding constant of the ligand to the quadruplex, determined by fluorescence titration of compound 102 with a solution of pu22 DNA, was (1.12 ± 0.15) × 106 М–1; this is an average value for G-quadruplex ligands. When the complex formed by pu22 and compound 102 was titrated with solutions of other quadruplex ligands (TMPyP4, BRACO-19, PhenDC3, 360A, TO, and ThT),‡ the fluorescence intensity decreased, which was indicative of dislacement of this compound by another ligand. It should be noted that virtually complete displacement took place in all cases. A similar situation was also observed for double-stranded DNA. Hence, compound 102 can be efficiently used as a fluorescence probe to study binding of various compounds to both double-stranded DNA and quadruplex DNA structures.
Kang and Wei[86] described fluorescent probes 103 and 104 based on N-methylbenzothiazolium. The binding was studied using promoter (c-MYC, c-KIT2, BCL-2), telomeric (htg), and mitochondrial (HRCC, KSS) quadruplex DNA, double-stranded DNA (ds26), single-stranded DNA (ss17), and i-motif DNA (HTC).
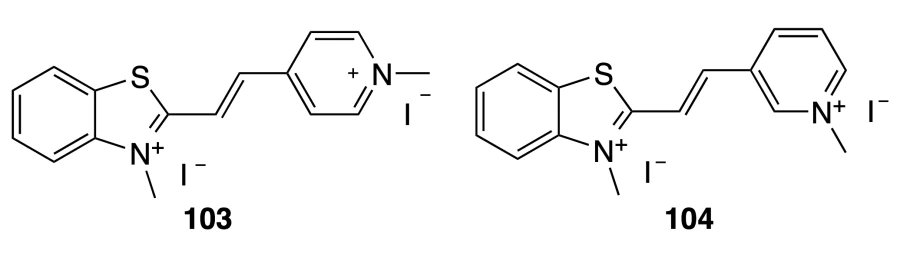
Using absorption spectroscopy of solutions of compounds 103 and 104 in the presence of DNA, it was shown that they possess high selectivity for quadruplex DNA: in the case of compound 103, Kb are in the range of (0.71 – 4.25) × 104 М–1 for G4 and (0.10 ± 0.01) × 104 М–1 for ds26; while in the case of 104, Kb = (1.63 – 4.56) × 104 М–1 for G4 and Kb = (0.83 ± 0.15) × 104 М–1 for ds26. The results of studying the fluorescence response of benzothiazoles to DNA are generally in line with the data of absorption spectroscopy; however, both compounds were found to stabilize the promoter and mitochondrial quadruplex DNA more efficiently than telomeric DNA. In addition, compound 104 with a meta-substituted pyridine moiety showed a higher fluorescence intensity after the addition of DNA. Whereas the addition of DNA to a solution of compound 104 increased the fluorescence lifetime, in the case of isomer 103, no such effect was observed.
Using HepG2 cancer cells, it was found that both compounds have a low cytotoxicity (cell survival rate exceeded 70% at up to 50 μM concentrations of compounds); this makes N-methylbenzothiazolinium salts 103 and 104 good fluorescent probes, but restricts their possible use for inhibition of tumour growth. Using confocal microscopy, it was found that compound 104, which has a high fluorescence intensity, is mainly concentrated in mitochondria and does not penetrate into cell nuclei, which probably accounts for its low cytotoxicity.
Geng et al.[87] prepared a series of benzimidazolylisoxazoles 105 – 123, which could presumably be effective against multiple myeloma by inhibiting the expression of the c-MYC gene. In order to achieve the synthesis of the target products, the authors first synthesized isoxazolecarboxylic acids by the Claisen condensation of substituted acetophenones with diethyl oxalate and subsequent reaction with hydroxylamine and alkaline hydrolysis of the ester group. This was followed by the condensation of carboxylic acids with phenylenediamine in the presence of DIPEA and [bis(dimethylamino)methylidene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) to give a benzimidazole ring, which was alkylated at the nitrogen atom (Scheme 11). Among the obtained compounds, the highest cytotoxicity against the RPMI-8226 myeloma cell line was found for compounds 112 (IC50 = 7.55 ± 0.85 μM), 113 (IC50 = 5.42 ± 1.41 μM), 116 (IC50 = 6.16 ± 0.59 μM), and 122 (IC50 = 7.50 ± 0.37 μM). However, further Q-PCR assays revealed that only compound 116 inhibits expression of the c-MYC gene, which may be attributable to better penetration of this more lipophilic dimethyl derivative through cell membranes.
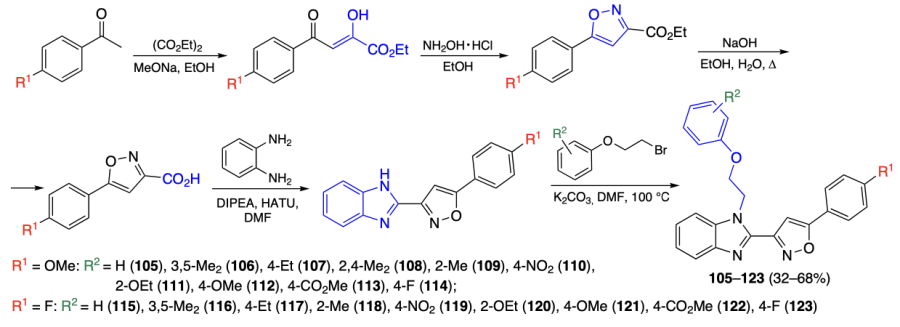
According to molecular dynamics simulation data, ligand 116 binds to the c-MYC upper quartet through π – π-stacking (Figure 10). In addition, the adduct stability is enhanced due to interaction of the benzene ring with the 5'-terminal thymine moiety.
The binding of G4-ligands to telomeric DNA may activate the immunogenic cell death. For example, Wang et al.[88] prepared a series of phenanthreneimidazoles analogous to arylimidazoles, ligands of c-MYC quadruplex, described previously by this research group[89-91]. Compounds 124 – 129 were obtained by condensation of substituted phenanthrene-9,10-quinone with heterocyclic aldehydes in the presence of ammonium acetate (Scheme 12).
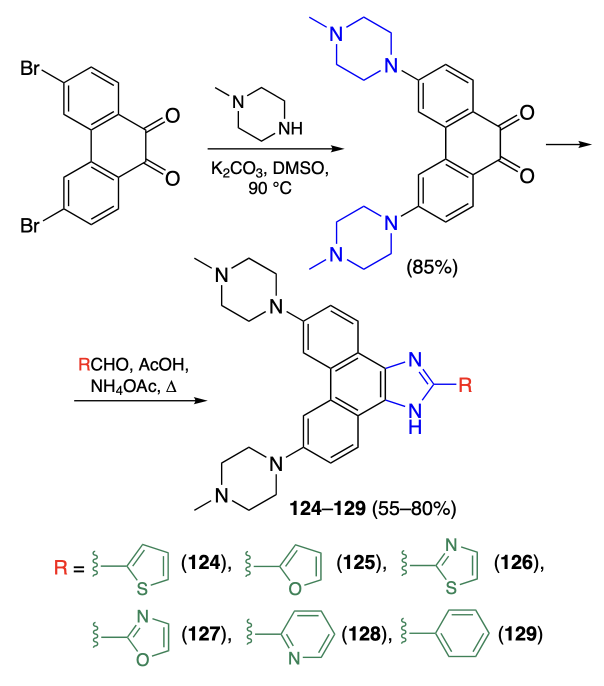
Using the data of CD spectroscopy, fluorescence titration, and melting temperature measurements, it was established that compound 125 binds most efficiently to the telomeric DNA. This compound also showed high selectivity for the telomeric quadruplex compared to other quadruplexes and double-stranded DNA. In an МТТ assay, compound 125 had the highest cytotoxicity against MDA-MB-231 and 4T1 cancer cells and simultaneously low toxicity against normal MCF-10A cells. According to immunofluorescence assay using BG4, TRF2, and γH2AX antibodies, compound 125 mainly binds to telomeres, which leads to increasing content of γH2AX, a marker of DNA damage. The subsequent studies confirmed that compound 125 activates immunogenic apoptosis in MDA-MB-231 and 4T1 cells. Experiments in vivo on mice demonstrated that the injection of 4T1 cells preincubated with compound 125 before the addition of live 4T1 cells induced a more pronounced inhibition of tumour growth than doxorubicin, without significant adverse events in animals.
As an attempt to formulate the structure – property relationship for benzazoles, it is worth comparing the results of Wu et al.[84] with the results of other studies cited in this Section. According to the available data, benzoselenazole 95 (see Scheme 9) was more efficient (IC50 = 3.5 – 4.1 μM against tumour cells and IC50 = 77.6 μM against normal cells) than benzoxazoles and benzothiazoles, owing to the presence of the chalcogen bond, as suggested by the authors[84]. Meanwhile, derivative 116 (see Scheme 11) containing an isoxazole moiety exhibited moderate cytotoxicity, and out of the series of compounds 124 – 129 (see Scheme 12), the best result was observed for furan derivative 125. Thus, the role of selenium atom and the chalcogen bond in binding to DNA is not that unambiguous, and further studies are required to clarify this issue, first of all, it is necessary to study the structure of G4 adducts with benzazoles by experimental techniques (two-dimensional NMR spectroscopy and X-ray diffraction), in addition to computer simulation. Nevertheless, it can be concluded that the use of benzazole derivatives in the form of salts enhances their binding to G4. However, when there is no relatively large lipophilic moiety in the molecule, such a ligand, despite good binding to DNA, may not penetrate into the cell nucleus and finally proves to have low toxicity (which is, apparently, the reason for low cytotoxicity of compounds 102 – 104).
2.4. Other heterocycles
Apart from the linear polyaromatic compounds (derivatives of anthracene and its heterocyclic analogues) and benzazoles, other known G4 ligands are porphyrins and complex fused heterocycles. Since the number of studies on interactions of these compounds with DNA published between 2020 and 2024 is insufficient to divide them into separate parts, they are considered together in one Section.
Ferino et al.[92] described a series of tetrapyridylporphyrins 130 – 133 with long (C12 – C18) alkyl chains and studied their binding to quadruplex structures in mRNA of RAS genes. The synthesis was carried out in two steps: first, the starting porphyrin was alkylated with 1 equiv. of the specified iodoalkane and then the remaining pyridine moieties were methylated with iodomethane (Scheme 13). Compound 134 containing a biotin moiety was also obtained from the phthalimide derivative.
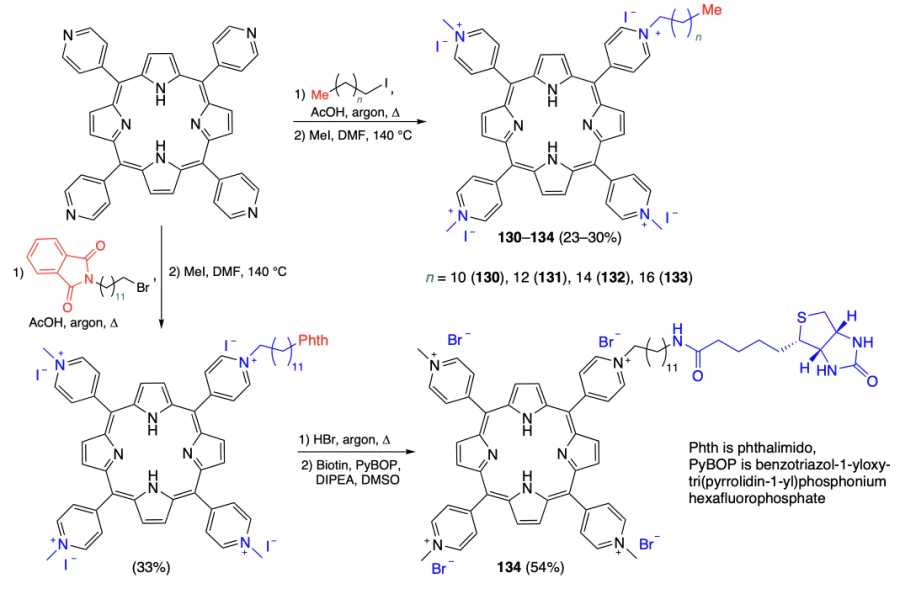
According to the absorption and emission spectroscopy data and melting experiments for quadruplex mRNA of KRAS and NRAS genes, an increase in the alkyl chain length leads to increasing RNA binding affinity of the porphyrin. In addition, the ligand to quadruplex ratio also increased with increasing alkyl chain length, which was attributed[92] to the contribution of hydrophobic interactions between the alkyl groups of compounds 130 – 134. The chain elongation is also beneficial for the penetration of porphyrins into Panc-1 cells: after 16 h of incubation, the concentration of compounds 131 and 133 was 24 and 38 times, respectively, higher than that of pristine meso-tetra(4-pyridyl)porphyrin in a similar experiment.
To prove the binding of the test compounds to G4 at a low concentration in the cell, a mixture of single-stranded and quadruplex DNA was incubated with compound 134 containing a biotin moiety in the molecule. The subsequent binding of DNA to a magnetic carrier coated by biotin-selective streptavidin attested to the DNA interaction with this ligand. Measurement of DNA concentration removed from the carrier showed that at the DNA to ligand ratio of 2 : 1 and ligand concentration of 0.2 or 0.4 μM, compound 134 was bound only to quadruplex DNA. In a similar experiment with RNA extract from Panc-1 cells, the percentage of KRAS mRNA was 25 times higher compared to the initial one. This demonstrated high selectivity of porphyrin derivatives for KRAS quadruplex mRNA.
The subsequent biological assays demonstrated that under irradiation with light, compounds 131 and 133 can generate reactive oxygen species (ROS), which leads to the destruction of RNA. The inhibition of KRAS expression was confirmed by quantitative PCR assay. In in vivo experiments with Panc-1 tumour in mice, compound 131 effectively inhibited the tumour growth under irradiation with light at 660 nm, which means that it can be promising as an agent for photodynamic therapy.
Marzano et al.[93] prepared a series of diimidazo[1,2-a:1,2-c]pyrimidines 135 – 154, similar to those reported earlier by Amato et al.[94]
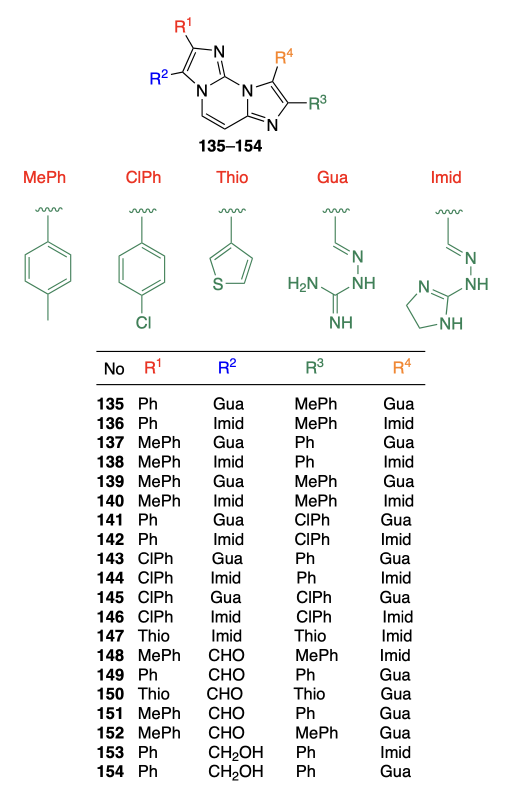
The diimidazo[1,2-a:1,2-c]pyrimidine moiety was synthesized by condensation of 2,4-diaminopyrimidine with substituted 2-bromoacetophenones. The resulting intermediates were subjected to Vilsmeier – Haack formylation, and then allowed to react with amines or reduced to the corresponding alcohols (Scheme 14).
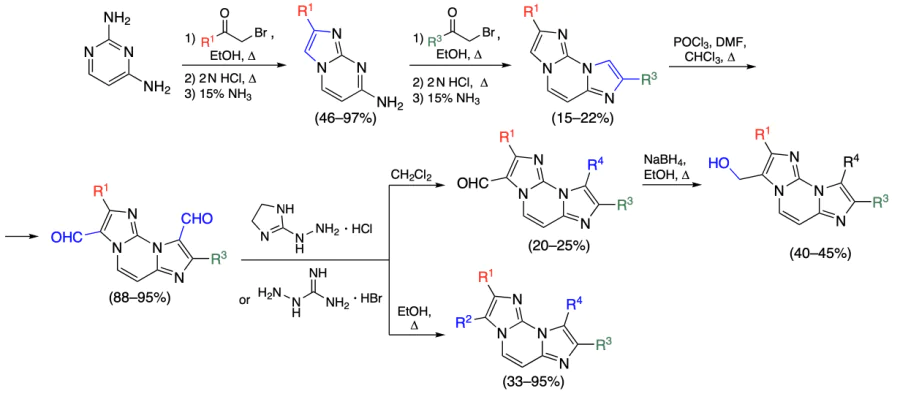
Using CD spectroscopy and melting temperature measurements for the DNA complexes of the test compounds, the authors found that the most efficient binding to quadruplexes (c-MYC, c-KIT1, c-KIT2, tel26) is inherent in compounds 135, 136, 142, 144, 149, 153, and 154. According to the data of thiazole orange displacement fluorescence spectroscopy, compounds 149, 153, and 154 did not provide 50% displacement. Meanwhile, dissociation constants of the complexes formed by ligands with quadruplexes, determined by the MST assay, occurred in the nano- or micromolar range, which attests to high affinity of the compounds to quadruplexes, but is not consistent with the results of the thiazole orange displacement experiments. The subsequent studies demonstrated that compounds 135, 136, and 142 have moderate cytotoxicity against U2SO and MNMCA1 cells; however, according to immunofluorescence assay data, compound 142, which is the most toxic (IC50 = 20.2 ± 1.0 and 23.5 ± 5.1 μM, respectively), virtually does not bind to the quadruplexes in cells. Compound 135 was found to increase the level of interferon-β (IFN-B), which may activate the immune response. Compounds 149, 153, and 154 had a higher cytotoxicity than 135, 136, and 142; in particular, compound 149 was most cytotoxic among all the test derivatives, with IC50 being 2.6 ± 0.87 and 1.9 ± 0.33 μM against U2SO and MNMCA1 cells, respectively. In addition, according to the results of confocal microscopy, compounds 149, 153, and 154 interacted with quadruplexes in the cells, despite the results of fluorescence spectroscopy on TO displacement. A distinctive feature of compounds 149, 153, and 154 compared to analogues is the presence of a non-polar phenyl group as the substituent R1 and polar CHO and HOCH2 groups as R2. Perhaps, polar R2 groups form hydrogen bonds with DNA molecule, whereas the benzene ring as the R1 substituent is involved in π – π stacking. However, this cannot be stated unambiguously without the results of NMR spectroscopy, X-ray diffraction, or computer simulation.
A promising G4 ligand is pidnarulex (CX-5461, 155), which previously proved to be able to bind to G4 and stabilize them in vitro and in vivo[95] and also exhibited cytotoxicity against DNA repair deficient cells, including BRCA1/2-dependent homologous recombination (HR) deficient cells[96]. Hilton et al.[97] reported the results of phase 1 clinical trials of pidnarulex (155). This compound has demonstrated efficacy in patients diagnosed with tumours characterized by homologous recombination deficiency (HRD), which is consistent with the results obtained in preclinical trials. Four partial responses were found in three patients with breast cancer and one with ovarian cancer, all having inherited DNA repair deficiency (2 BRCA2, 1 PALB2, 1 TP53, and BRCA2 VUS). However, not all patients with the detected BRCA mutation responded to the therapy, which was attributed, in particular, to the cross-resistance caused by previous therapy with platinum drugs or poly-ADP-ribose polymerase (PARP) inhibitors. The most common adverse events associated with the administration of compound 155 were nausea and phototoxicity, which was observed for all tested dosage levels, despite strict recommendations for UV protection. The additional clinical trials performed to determine the in vitro phototoxicity showed that the photosensitivity that occurred upon the administration of this drug was rather related to the chemical structure of the ligand and did not depend on G4 stabilization.
Li et al.[98] investigated the MTR-106 ligand (156), obtained by replacement of the homopiperazine ring in compound 155 by the octahydropyrrolo[3,4-c]pyrrole moiety. FRET melting assay demonstrated that ligand 156, like compound 155, causes an increase in the melting temperature of quadruplexes with ΔTm values being in the ranges of 4.2 – 24.8 °C (h-Telo) 1.3 – 20.8 °C (c-KIT1), and 0.8 – 13.5 °C (c-MYC), but this does not take place for double-stranded DNA, indicating higher binding affinity of 156 to quadruplex DNA than to dsDNA.
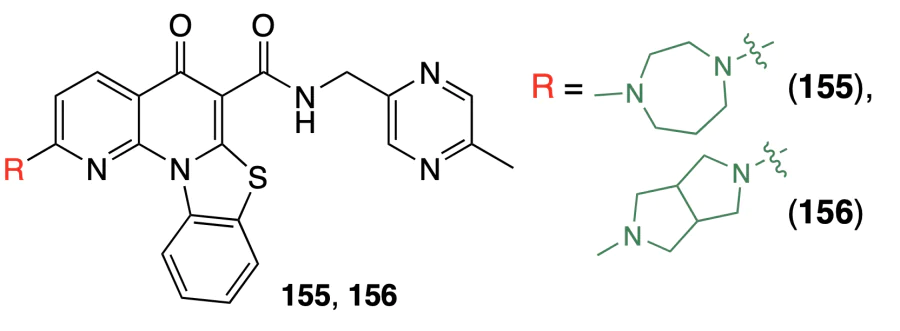
It was found that compound 156 shows an antitumour activity in vitro in cells with DNA repair deficiency, in particular with the homologous recombination deficiency, including PARP inhibitor resistant cells. In HR-deficient cancer cell lines with BRCA2, PTEN, and BRCA1 mutations, compound 156 had two times lower IC50 values (40 – 788 μM) than compound 155 (105 – 1700 μM) and had cytotoxicity 70 and 11 times higher than the PARP inhibitors olaparib and talazoparib, respectively. In addition, compounds 156 and 155 induced cell death in all olaparib- and talazoparib-resistant cell lines with significantly lower IC50.
Using flow cytometry and Caspase-Glo®3/7 assay, it was shown that treatment with ligand 156 arrests the cell cycle in the G2/M phase and induces apoptosis, which is typical of G-quadruplex stabilizers[99]. In addition, treatment with compound 156 induces accumulation of DNA double-strand breaks, resulting in increasing γH2AX levels in Capan-1 cells, as shown using western blotting, confocal microscopy, and the DNA comet method. These results indicate that compound 156 induces accumulation of DNA damages in BRCA-mutated cancer cells. Interestingly, it causes more DNA damages, as well as more pronounced cell cycle arrest in the G2/M phase and apoptosis in HR-deficient Capan-1 cells compared to 155. In in vivo experiments on MDA-MB-436 and Capan-1 tumours in mice, ligand 156 causes significant tumour growth inhibition, which additionally confirms the potential efficacy of this compound for the specific action on HR-deficient tumours.
Psaras et al.[100] performed screening of a library of fused aromatic heterocycles for stabilization of the G4 structure of KRAS gene. The results of FRET melting assay and electronic circular dichroism (ECD) indicated that compound NSC317605 (157) is the most promising ligand. In luciferase assay on Panc1 and AsPc1 cell lines, it inhibited the expression of KRAS by 65%; the IC50 values determined by MTS assay against the indicated cell lines were 55 ± 3 and 26 ± 1 μM, respectively. In order to confirm the detected activity and selectivity, compound 157 and three analogues (158 – 160) were synthesized by a different procedure. Unlike the previously reported[101] method, which implied the formation of the indole moiety by the Fischer reaction, in this protocol, the indole ring is formed upon the reaction of iodochloroquinoline with amine followed by palladium-catalyzed intramolecular Heck reaction (Scheme 15).
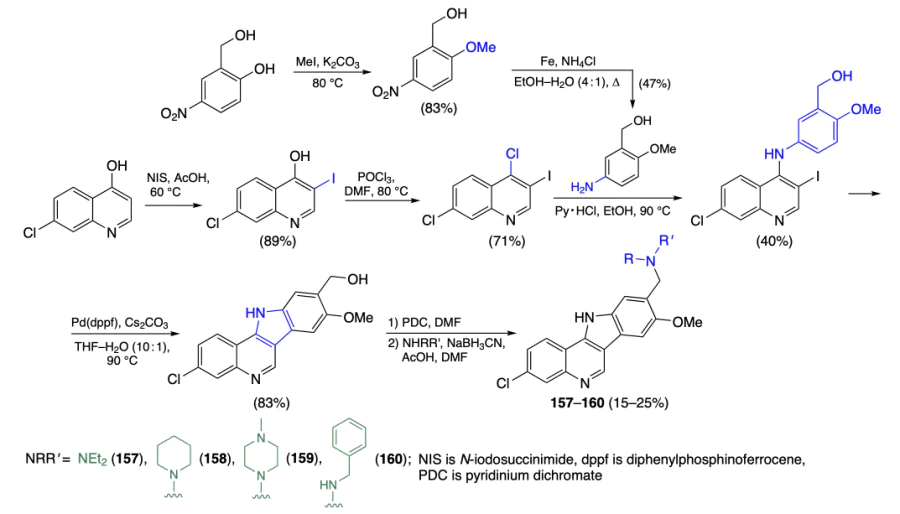
Melting temperature measurements confirmed that compounds 157 and 158 stabilized the quadruplex structure of KRAS, without affecting MYC G4. Luciferase assay of the change in the promotory activity of KRAS in HEK-293 cells demonstrated that only compound NSC317605 and product 157 (i.e., identical compounds) in 1 μM concentration decreased the activity of KRAS promoter by 34 ± 11 and 46 ± 23%, respectively. Additional experiments on determination of the cytotoxicity against KRAS-dependent AsPc1 pancreatic cancer cells and the regulatory effect on KRAS transcription confirmed these results.
Saleh et al.[102] investigated the in vitro activity of benzo-[1,2-d:4,5-d']bis(triazole)-4,8(1H,5H)-diones 161 and 162, synthesized previously and identified as G4 ligands[103]. Unlike RHPS4, a known G4 stabilizer, compounds 161 and 162 showed a lower binding affinity to telomeric G4 and Hsp90a promoter sequences in 5 μM concentration: ΔTm values were 8.4 ± 0.7 °С, 10.1 ± 0.8 °С, and 24.9 ± 3.6 °С for 161, 162, and RHPS4, respectively. The authors suggest that, apart from stabilizing G-quadruplexes, compounds 161 and 162 act on other molecular targets, since their inhibitory activity against a number of cell lines, according to the results of the MTT assay, was significantly higher than that of RHPS4.
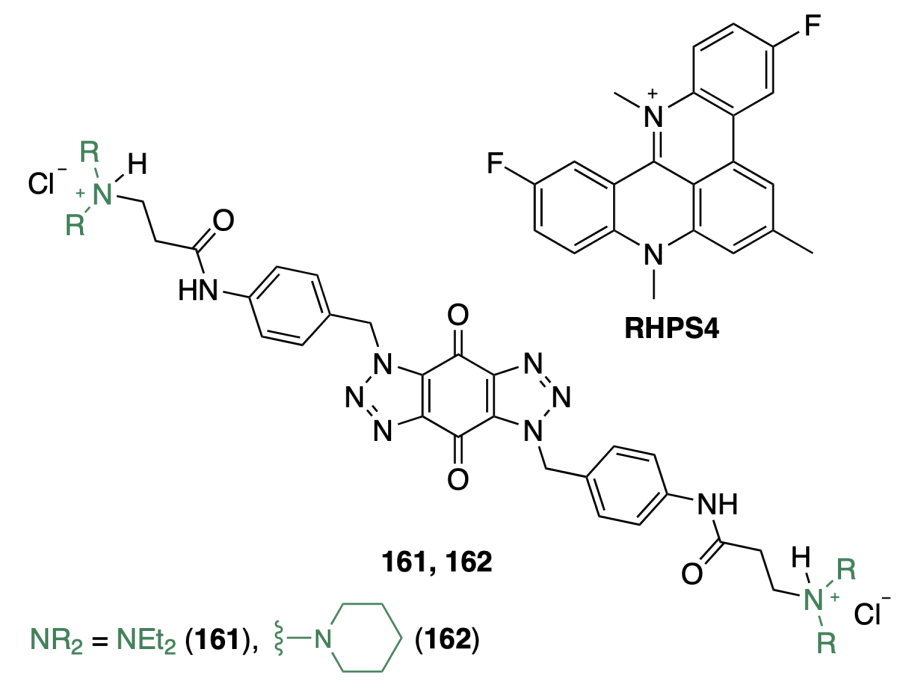
In МТТ assay, bis(triazoles) 161 and 162 retarded the growth of MDA-MB-435 cells with half-maximal growth inhibitory concentrations (GI50) of 0.226 ± 0.057 μM and 0.075 ± 0.010 μM, respectively, and showed selectivity for tumour cells over MRC-5 non-carcinogenic cell line (SI = 9.8 and 17.7, respectively). Using flow cytometry, it was found that compound 161 caused cell cycle arrest in G0/G1 and G2/M phases, while the effect of its analogue 162 did not differ significantly from the control. Cytofluorimetric analysis of MDA-MB-435 cells showed that 24 h after treatment with bis(triazoles) 161 and 162 in concentration equal to twice the GI50 value, the number of live cells significantly decreased, while the percentage of late apoptotic cells increased 10-fold compared to the control. According to western blotting results, only compound 162 (in the above concentration) significantly reduced Hsp90 levels in MDA-MB-435 melanoma cells.
Relying on the results of studies addressed in this Section, we can conclude that a promising class of G4 ligands is represented by molecules with aromatic skeleton consisting of three or four fused rings (e.g., acridine or anthraquinone derivatives). However, there is evidence indicating that an increase in the conformational mobility of the aromatic moiety promotes the DNA binding[20, 104]. Furthermore, owing to the possibility of formation of chalcogen bond, which is favourable for binding to DNA, chalcogen-containing heterocycles, first of all, sulfur- and selenium-containing benzazoles, make a good alternative to polyaromatic compounds. The use of coordination compounds generally does not provide a significant increase in the binding affinity or cytotoxicity of compounds compared to purely organic G4 ligands, while the use of complexes is more often associated with problems related to insufficient solubility and poor penetration across cell membranes. In order to enhance binding to G4, it is apparently necessary to introduce not only aliphatic linkers with amino groups (mainly dimethyl- and diethyl-substituted ones) into ligand molecules, but also hydroxyl groups (e.g., as in compounds 153 and 154). Considering the fact thay G4 ligands that have been recognized to be promising, such as BRACO-19, RHPS4, and APTO-253 {2-(5-fluoro-2-methyl-1H-indol-3-yl)-1H-imidazo[4,5-f][1,10]- phenanthroline}[105], are still not used for therapeutic purposes, the results of clinical trials of compound CX-5461 (155)[97] give some hope for biomedical application of G4 binding compounds.
3. i-Motif ligands
i-Motifs (iM) are tetrachain structures formed in cytosine-rich sequences in which semi-protonated cytosine–cytosine base pairs (C · C+) bind to each other[106, 107] (Figure 11).
![[{"id":"b_KzGsQEgx","type":"paragraph","data":{"text":"i-Motif structure."}}]](/storage/images/resized/SFVhwbiiAEqy5xWti1s0qp6SqP3mxpZJnw504RdE_xl.webp)
Since i-motifs are pH-sensitive and are formed, most often, in acidic medium, the possibility of iM formation in vivo was first doubted. However, recent studies demonstrated that i-motifs can form in biological media and at neutral pH[108]. Like G4 structures, i-motifs participate in the regulation of gene expression; however, their role is not unambiguous. For example, Kendrick et al.[109] studied triterpenoids 163 and 164 with the commercial designations IMC-48 and IMC-76, respectively. According to NMR spectroscopy data, the addition of compound 164 to the i-motif of the BCL-2 promoter leads to its destruction, while compound 163 stabilizes iM. Melting temperature measurements showed an increase in Tm of BCL-2 by 1 °С in the presence of triterpenoid 163 and a decrease by 0.5 °С in the case of compound 164. The authors suggested that destabilization of iM in the latter case is due to the interaction of triterpenoid 164 and the hairpin, which is formed after destruction of the i-motif. The driving force of binding to the hairpin is the entropy decrease upon incorporation of the lipophilic molecule of 164 into DNA and, as a consequence, a decrease in the interaction between the triterpenoid and water molecules. In the case of compound 163, the authors suggested participation of the piperidine moiety in binding to the i-motif. Using FRET method, it was found that structurally similar amides 165 – 168 with a piperidine ring but without the triterpenoid moiety decrease the fluorescence intensity of BCL-2 i-motif labelled with 6-carboxyfluorescein (FAM) and 5-carboxytetramethylrhodamine (TAMRA) by 10 – 15%, similarly to compound 164 (by 15%). A similar experiment with central and 3'- and 5'-end loop mutant i-motifs showed that no binding takes place in the case of central loop mutation. Thus, the authors proved that the piperidine moiety of compound 163 binds to the central loop of BCL-2 iM.
The effect of test agents on the BCL-2 expression was determined by qRT-PCR and western blotting using BJAB cell line in which no expression of BCL-2 gene takes place and B95.8 and GRANTA-519 cells with BCL-2 gene overexpression. It was shown that in the presence of compound 164 (in 0.25 μM concentration), the BCL-2 mRNA levels in B95.8 and GRANTA-519 decrease by 56 and 23%, respectively, with no effect in BJAB cells. Meanwhile, the addition of compound 163 in 2 μM concentration increased the BCL-2 expression by 220% in BJAB cells, but had no significant effect on B95.8 and GRANTA-519 cells. These results imply that BCL-2 iM activates the expression.
Despite the fact that the i-motif of the BCL-2 gene acts as a transcription activator, in the case of c-MYC gene, stabilization of iM leads to inhibition of the expression[110]. This observation was later confirmed by Shu et al.[111], who described a series of acridone derivatives. According to FRET and MST assay data, compounds 169 – 175 can stabilize c-MYC i-motif (ΔTm = 3.2 – 11.5 °C) and selectively bind to iM (Kd = 4.6 – 8.7 μM), unlike c-MYC quadruplex or double-stranded DNA (Kd > 50 μM). The subsequent studies of the lead compound 171 (ΔTm = 11.5 °C, Kd = 4.6 μM) using luciferase assay, qRT-PCR, and western blotting on SiHa cell line showed that the introduction of compound 171 inhibits the c-MYC gene expression (Figure 12).
![[{"id":"7SXP8bY5Wz","type":"paragraph","data":{"text":"Results of qRT-PCR study of <i>c-MYC</i> gene on SiHa cell line in the presence of compound <b>171</b> in various concentrations[[ type=\"anchor\" referenceId=\"9021\" ]]."}}]](/storage/images/resized/FMcpDY7s8M2yzLSpkqv9qMj4uVgzOzo7jcI5hM1F_xl.webp)
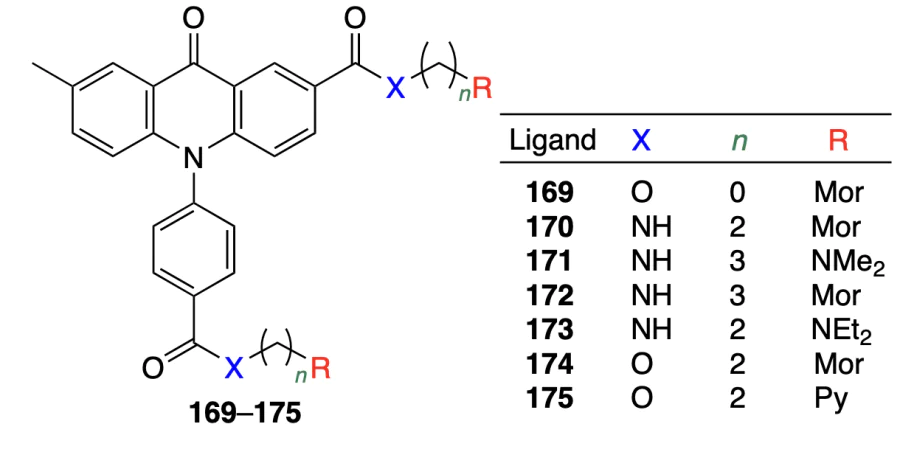
Like in the case of G4, compounds with an extended π-system, for example, polyaromatic compounds act, most often, as i-motif ligands. However, the use of versatile polyaromatic compounds complicates determination of the structural features of the ligand responsible for the selectivity only to G4 or iM. For example, Pagano et al.[112] investigated the interaction of DAP i-motif (5'-[(CCC-CCG)4CCCCC]-3´) in neutral medium with G-quadruplex ligands: berberine, BRACO-19, mitoxantrone, Phen-DC3, pyridostatin, RHPS4, and TmPyP4. DNA melting assays demonstrated that ligands destabilize iM (ΔTm varies in the range from –1.5 to –16.0 °C). According to the results of fluorescence titration of thiazole orange displacement, DC50 values (concentrations providing 50% displacement of TO) were 0.16 – 27.86 μM. These results confirm that the same compounds can act as ligands of both G4 and iM.
Previously, attempts have also been made to obtain compounds selective to a definite non-canonical structure. Debnath et al.[113] reported pyridine-2,6-dicarboxamides 176 and 177. These compounds differ in the position of the proline residue relative to the triazole substituent in the benzene ring: para-isomer 176 and meta-isomer 177. It was found that compound 176 has a higher affinity to i-motifs (Kd = 0.3 – 2.4 μM for BCL2 and c-MYC iM; Kd = 7.2 – 12.5 μM for BCL2 and c-MYC G4), while compound 177 has a higher affinity to G-quadruplexes (Kd = 5.8 – 9.5 μM for BCL2 and c-MYC iM; Kd = 1.3 – 1.9 μM for BCL2 and c-MYC G4). However, the small number of compounds and the lack of studies of the ligand – DNA complex (for example, by molecular docking or NMR spectroscopy) preclude the possibility of establishing the structure – property relationship for these products.
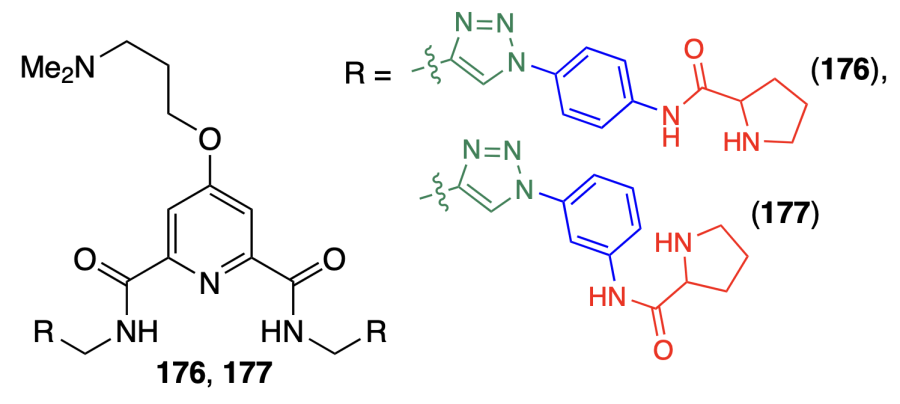
Due to the scatter and small amount of data on the biological functions of i-motifs (compared to G4) and on their potential applicability for anticancer therapy, these derivatives require further studies.
3.1. Coordination compounds
There are only a few reported examples of coordination compounds of ruthenium[114, 115], iridium[116], and terbium[117] capable of binding to i-motifs. For example, Maliszewska et al.[118] synthesized a library of platinum and gold complexes with carbene ligands. It was shown that gold(III) complex 178 is able to selectively bind to the cytosine-rich hTeloC sequence forming the i-motif. This complex does not bind to other DNA structures such as double-stranded DNA or G-quadruplexes. Hence, gold(III) complexes are potential selective ligands for i-motifs.
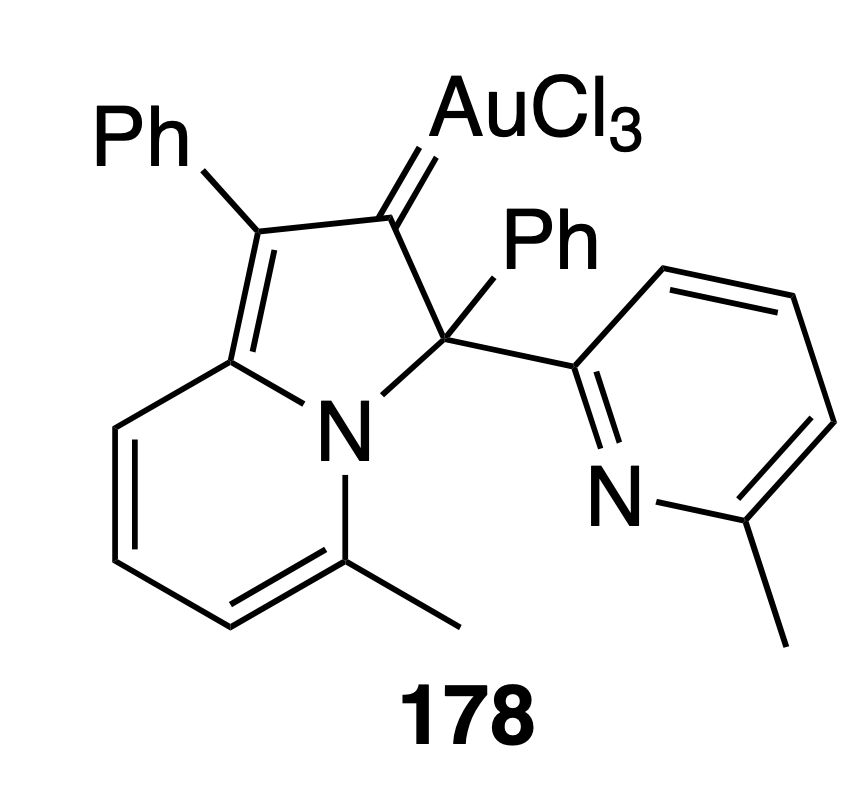
Using flavonoid 179 and thio analogue 180, Khater et al.[119] prepared ruthenium(II) complexes 181 and 182 and investigated their biological properties. It was shown that the sulfur-containing ligand and complex possess a higher antiangiogenic effect than the oxygen analogues. In addition, flavonoid coordination to the ruthenium ion reduces the cytotoxicity against MCF-7 and MDA-MB-231 cells (IC50 = 1.2 ± 0.8 μM and 43.06 ± 1.29 μM, respectively, for compound 180; IC50 > 100 μM against both cell lines for compound 182). Binding of these compounds to VEGF and c-MYC i-motifs was studied by absorption spectroscopy and melting temperature measurement, which showed minor DNA binding for both the ligands and the complexes. The authors suggested that binding occurs via electrostatic interactions with phosphate groups of DNA rather than via intercalation.
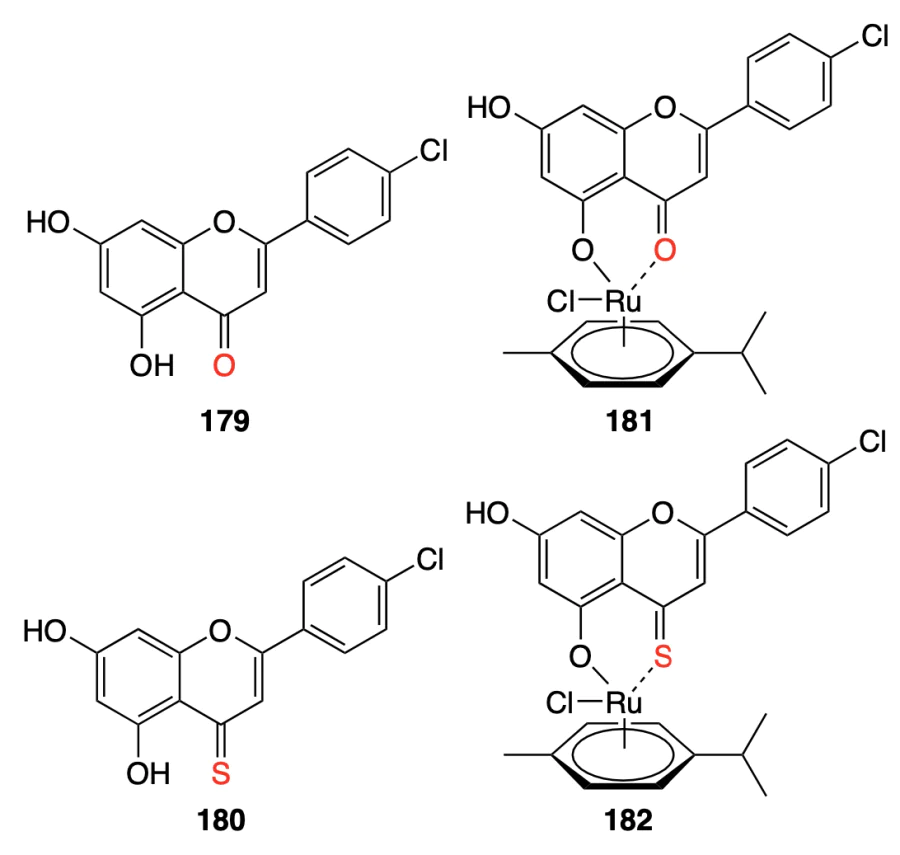
3.2. Polyaromatic compounds
It is known that polyaromatic compounds can act as G-quadruplex ligands owing to their extended aromatic system (see Section 2.2.). Although there are also examples of arenes that bind to i-motifs, for example, acridones and bis-acridines, which were reported by D.Li and co-workers[111, 120-122]. For example, in 2020, they described[120] a series of bis-acridines 183 – 206 as potential i-motif-and G-quadruplex-binding ligands (Scheme 16).
The shift of DNA melting temperatures indicates that introduction of ligands increases the thermal stability of c-MYC quadruplex (by 7.7 – 17.1 °C), while compounds 194 – 188, 203, and 204 increase also the stability of c-MYC i-motif (by 4.5 – 27.4 °C), with the melting temperature of double-stranded DNA remaining almost invariable. The results of measurement of binding constants by surface plasmon resonance are in agreement with FRET data. On the basis of the results, the authors chose derivative 187 as the lead compound, which was subjected to biological studies. According to MTT assay data, IC50 values were 0.15 – 1.22 μM for various cancer cell lines (Table 10). The inhibition of c-MYC gene expression by compound 187 was confirmed by PCR, western blotting, and luciferase assay. Experiments in vivo showed that this compound in 15 mg kg–1 concentration inhibits the growth of SiHa tumour in mice by 41.6%, virtually without affecting the animal weight.
![Cytotoxic activity of compounds <b>183</b> – <b>197</b> according to МТТ assay data (IC<sub>50</sub>, μM)[<a class="anchor" href="#reference-9030">120</a>].](/storage/images/resized/P5Urfg2xnIThoYHIRhSkR8uvr8yZB6an7NaMCIoi_xl.webp)
The interaction of bis-acridines with DNA was investigated by molecular docking of compound 187 into c-MYC G-quadruplex and telomeric i-motif (Figure 13). It was shown that in the case of quadruplex, one of the acridine rings of compound 187 is bound to the 5'-terminal quartet by π – π interactions, while the second ring is located in the DNA groove; the linker is bound by electrostatic interactions and hydrogen bonding between the hydrogen atoms of the amino groups and the DNA sugar – phosphate backbone. The complex of compound 187 with i-motif was found to involve π – π-interactions of both acridine rings with the iM cytosine moieties as well as hydrogen bonds and electrostatic interactions similar to those in the bis-acridine 187 quadruplex adduct.
![[{"id":"HFiaNJZ6SZ","type":"paragraph","data":{"text":"Results of molecular docking of compound <b>187</b> into <i>c-MYC</i> G-quadruplex (a) and telomeric i-motif (b)[[ type=\"anchor\" referenceId=\"9030\" ]]."}}]](/storage/images/resized/jTcA8ZYSXIw76QvIySGvn56kj3X9qMJEKoiCr9R8_xl.webp)
Thus, it was demonstrated that bis-acridines can stabilize both c-MYC promoter quadruplexes and i-motifs[120]. However, in the subsequent study[121], the authors screened a library of previously synthesized compounds, including bis-acridines for the possibility of binding to c-KIT promoter i-motif. The compound designated by BO5 (185) was found to bind more efficiently to the c-KITC1 and c-KITC2 i-motifs, but not to quadruplexes of the same promoter. Moreover, repeated studies by surface plasmon resonance and FRET showed that the dissociation constant of the complex of compound 185 with c-MYC i-motif is 274 μM and ΔTm is about –3 °C (the same characteristics reported in the earlier paper 120 of 2020 were 10.7 μM and 6.4 °C).
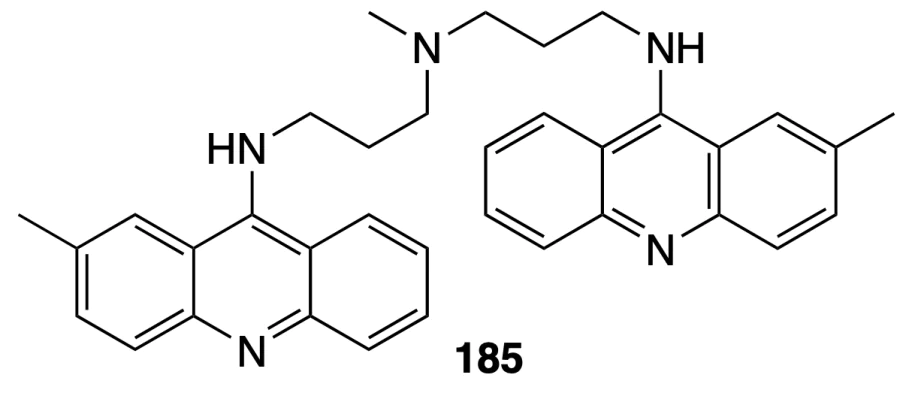
It can be seen that the results of 2023[121] are at variance with the earlier data[120]. Hence, despite the reported high efficiency of inhibiting c-MYC and c-KIT expression, this class of compounds requires further investigation.
The same research team[122] synthesized a series of acridones containing a triazole moiety in the molecule. The lead compound C4, which showed the best result in MTT assay, was found to efficiently bind to KRAS gene i-motif (Kd = 1.58 μM), but simultaneously to destabilize the DNA structure (ΔTm = –7 °C), unlike the acridines and acridones described above. Further biological studies revealed that the cytotoxic effect of C4 is associated not only with inhibition of KRAS gene expression, but also with generation of reactive oxygen species.
Tikhomirov et al.[123] modified heliomycin, a natural antibiotic, with the goal to increase its water solubility and affinity to DNA. They obtained a group of compounds 207 and 208 with an ethylene diamine moiety in different positions of the ring (Scheme 17).
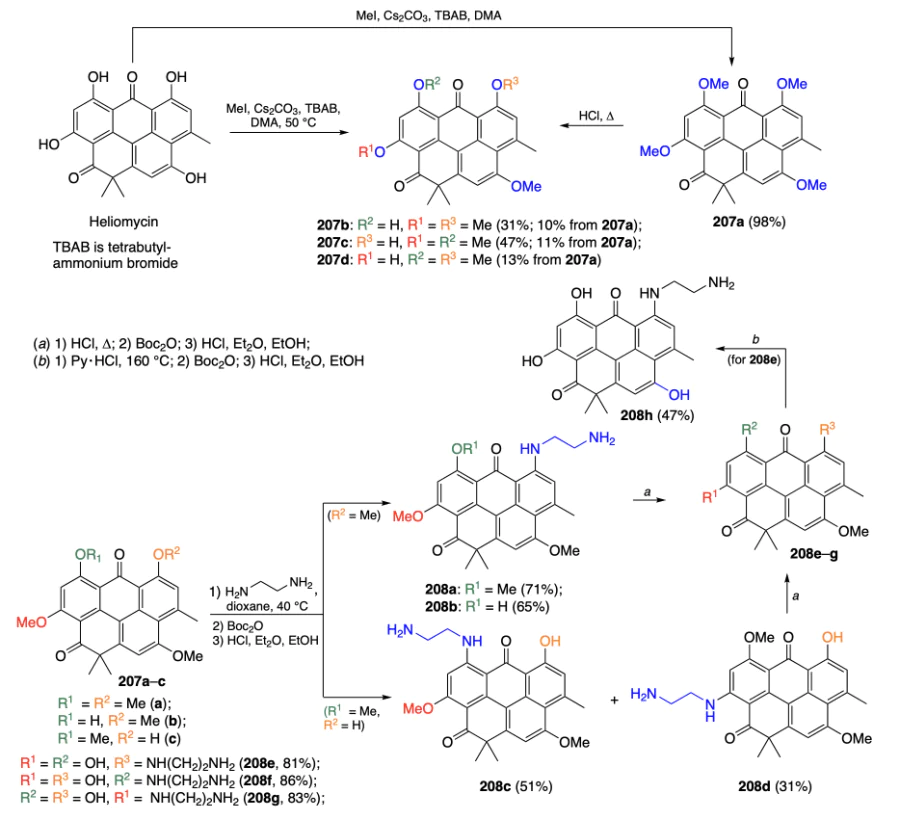
Thiazole orange displacement (FID) assay of various DNA structures (dsDNA; c-MYCC, ILPR, hTeloC, HIF1A, and DAP i-motifs; c-MYCG, hTeloG, and NASGG quadruplexes) demonstrated that the ТО displacement by the parent heliomycin did not exceed 43%, whereas in the case of compound 208е, the displacement rate was 64 – 76% for i-motifs, but markedly decreased for other structures (9.7% for dsDNA and up to 44% for G4). Measurement of the DNA melting temperature showed that compound 208е has almost no effect on double-stranded or quadruplex DNA, but stabilizes i-motifs (ΔTm = 10, 25, and 17 °C for HIF1A, c-MYCC, and ILPR, respectively). Using МТТ assay, compound 208е and its derivative 208h were found to efficiently inhibit the L1210, CEM, HeLa, and HMEC-1 cell growth (Table 11).
![Cytotoxic activity of compounds <b>208e,h</b> according to МТТ assay data (IC<sub>50</sub>, μM)[<a class="anchor" href="#reference-9033">123</a>].](/storage/images/resized/TMNf5MRa7g1sPlRkPcxsLoHwQ5Q0dPlZRXHKXOax_xl.webp)
3.3. Compounds of other classes
Zeng et al.[124] obtained a series of oleanolic acid derivatives 209 – 225 with various acyl substituents in position 2 where aliphatic, aromatic, unsaturated, and heterocyclic moieties functioned as the group R (Scheme 18). The authors hypothesized that such triterpenoid derivatives can selectively bind to the VEGF promoter i-motif. Using surface plasmon resonance and fluorescent intercalator displacement assay, it was established that compound 211 with a polyamine chain is most efficient in binding to i-motif.
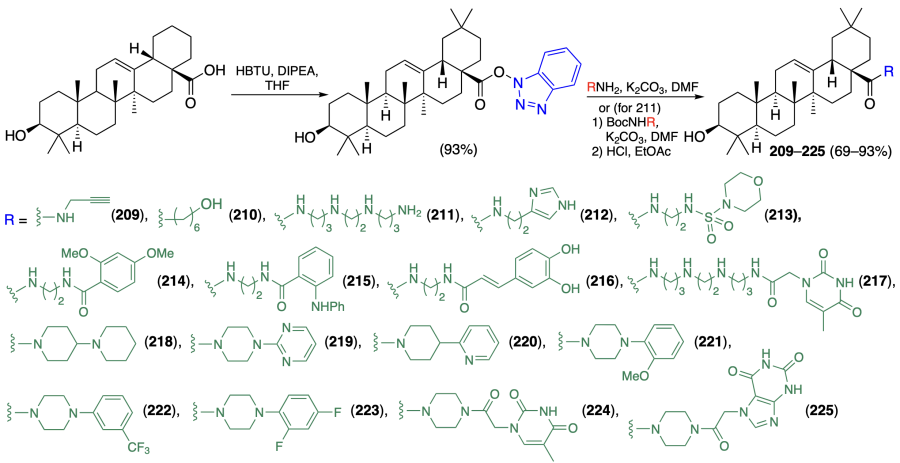
Measuring the DNA melting temperature in the presence of compound 211 indicated that the test compound increases the melting temperature of VEGF i-motif by 19.1 °C, which attests to its stabilization. In the case of VEGF G-quadruplex, double-stranded DNA, or other oncogene promoter i-motifs, no stabilization took place. This funding was confirmed by thiazole orange displacement in DNA.
The subsequent biological studies using western blotting, quantitative PCR, and luciferase assay indicated that compound 211 inhibits expression of VEGF gene. Using MTT assay and flow cytometry on MCF-7 cells, IC50 for this compound was found to be 6.5 μM; when it is present in concentration of 6 μM, the percentage of apoptotic cells increases to 39.6%.
Bag et al.[125] investigated binding of natural flavonoids, fisetin (226) and morin (227), to h-RAS i-motifs. It was shown that these compounds can selectively bind to target DNA, and the change of DNA melting temperature reaches 7 °C.
Porzio et al.[126] synthesized a series of substituted pyridines 228 – 237 with amine moieties linked through a triazole ring by a click reaction. The starting pyridine is formed upon the Hantzsch reaction from ethyl acetoacetate and benzaldehyde in the presence of ammonium carbonate followed by aromatization with sodium nitrite (Scheme 19).
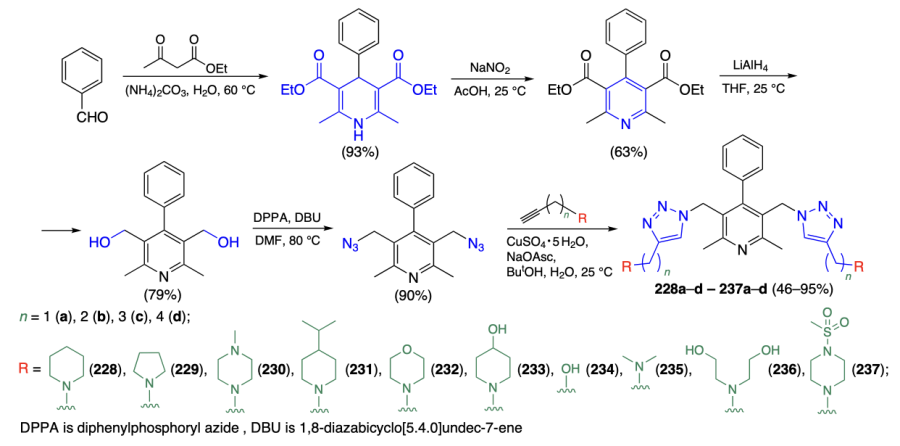
Using CD spectroscopy and DNA melting temperature measurement for non-canonical c-MYC G4, BCL-2 G4, Tel23 G4, c-MYC iM, BCL-2 iM, and hTeloC iM structures, it was found that most of compounds can bind to both quadruplexes and i-motifs. Furthermore, binding to quadruplexes results in stabilization of the secondary structure, while binding to i-motifs leads to destabilization. Compounds 228c and 231c, which showed a good affinity to quadruplexes and i-motifs, and derivatives of 233a and 236a, which had a higher selectivity for i-motifs, were chosen for further studies. Fluorescence spectroscopy studies of these compounds in the presence of c-MYC G4, c-MYC iM, and hTeloC iM demonstrated that compound 231c has a somewhat higher affinity to c-MYC quadruplex than to i-motif or telomeric i-motif; for other compounds, an opposite situation is observed (Table 12).
![DNA binding constants of 3,5-bis(triazolylmethyl)-4-phenylpyridine (10<sup>6</sup> M<sup>–1</sup>)[<a class="anchor" href="#reference-9036">126</a>].](/storage/images/resized/U1ewop08LDO4cSbgiYKTiswWyw9Xdxi6MgB0yEHi_xl.webp)
Using fluorescence microscopy of U2OS cells in the presence of these derivatives, it was found that compounds 228c and 231c induce the most pronounced decrease in the fluorescence intensity (by 70 and 60%, respectively), which attests to destabilization of i-motifs in cells.
On the basis of currently available data, it is difficult to unambiguously identify the structural characteristics of ligands necessary for effective binding to i-motifs, unlike those for G4. On the one hand, like for G-quadruplexes, polyaromatic compounds showed good results, but on the other hand, it was found[124] that non-aromatic triterpenoids can also bind to i-motifs. The information about participation of i-motifs in gene expression regulation is also ambiguous, because both DNA-stabilizing and -destabilizing compounds are present among the reported i-motif-binding ligands. In view of the fact that the use of i-motifs as objects for the targeted action of drugs still raises questions, until new data appear, these structures apparently cannot be considered as confirmed targets for anticancer therapy.
4. DNA and RNA triplex-binding ligands
DNA triplexes are formed on binding of polypurine or polypyrimidine part of the double helix with triplex-forming oligonucleotides (TFOs) through Hoogsteen bonds between nitrogenous bases. The triple helix formation may be either intramolecular or intermolecular (interaction of a DNA double helix with another DNA or RNA molecule (Figure 14)[127].
![[{"id":"xTn_A6HoYe","type":"paragraph","data":{"text":"Hydrogen bond formation in DNA triplexes (a), schematic views of intermolecular (b) and intramolecular DNA triplexes (c)."}}]](/storage/images/resized/XIIqKu1WUpagKLwhNxzj6Cbv22AUa908GQYrwfjX_xl.webp)
The polypurine and polypyrimidine sequences are mainly located in 3'-untranslated ends, promoters, and transcribed regions of certain genes[128]. The formation of DNA triplex may result in genome instability and, as a consequence, may give rise to various pathologies[129]. However, the formation of DNA or RNA triple helix, like quadruplex structures, inhibits the expression of genes, including oncogenes, which suggests the potential use of triplex-forming oligonucleotides or triplex-stabilizing molecules in anticancer therapy[130]. In most cases, oligonucleotides have been proposed for anticancer therapy[131-135], but examples of TFO conjugates with topoisomerase inhibitors or other DNA-binding compounds are also known[136, 137].
Only few examples of small molecules that can act as triplex-binding ligands are available from the literature. Lohani and Rojeswari[138] investigated the interaction of Adriamycin (doxorubicin hydrochloride) and actinomycin-D with double-stranded and triplex DNA. It was found that Adriamycin in 600 μM concentration has a three orders of magnitude higher affinity to the triplex [Kb = (8.5 ± 0.3) × 108 М–1] than to double-stranded DNA [Kb = (1.3 ± 1.0) × 105 М–1]. A review[139] presents examples of using compounds that bind to the DNA minor groove such as netropsin, DAPI, or Hoechst-33258 as triplex ligands. In particular, it was noted that various compounds have either a stabilizing or destabilizing effect on the triple helix. Strekowski's research group[140] synthesized a number of polyfused quinoline analogues, which stabilized the triplex up to ΔTm = 35.6 °C, without affecting double-stranded DNA.
The potentially higher affinity to the triple helix compared to the double helix makes the use of small molecules as DNA triplex-binding ligands a promising area of research. However, experimental data are still insufficient to unambiguously determine the structure – property relationships.
4.1. Coordination compounds
Ruthenium coordination compounds are often considered as potential anticancer drugs. In particular, studies devoted to ruthenium complexes that bind to quadruplexes have already been cited above[38, 42, 44-46]. Tan and co-workers[141-144] investigated similar complexes regarding their ability to bind to RNA triplexes. As ligands, the authors used 6-nitrodipyrido[3,2-a:2',3'-c]phenazine[141] and substituted 2-phenyl-1Н-imidazo[4,5-f ][1,10]-phenanthrolines[142-144]. Ligands 238 – 244 were obtained by condensation of 1,10-phenanthroline-5,6-dione with p-phenylenediamine or substituted benzaldehydes in the presence of ammonium acetate (Scheme 20).
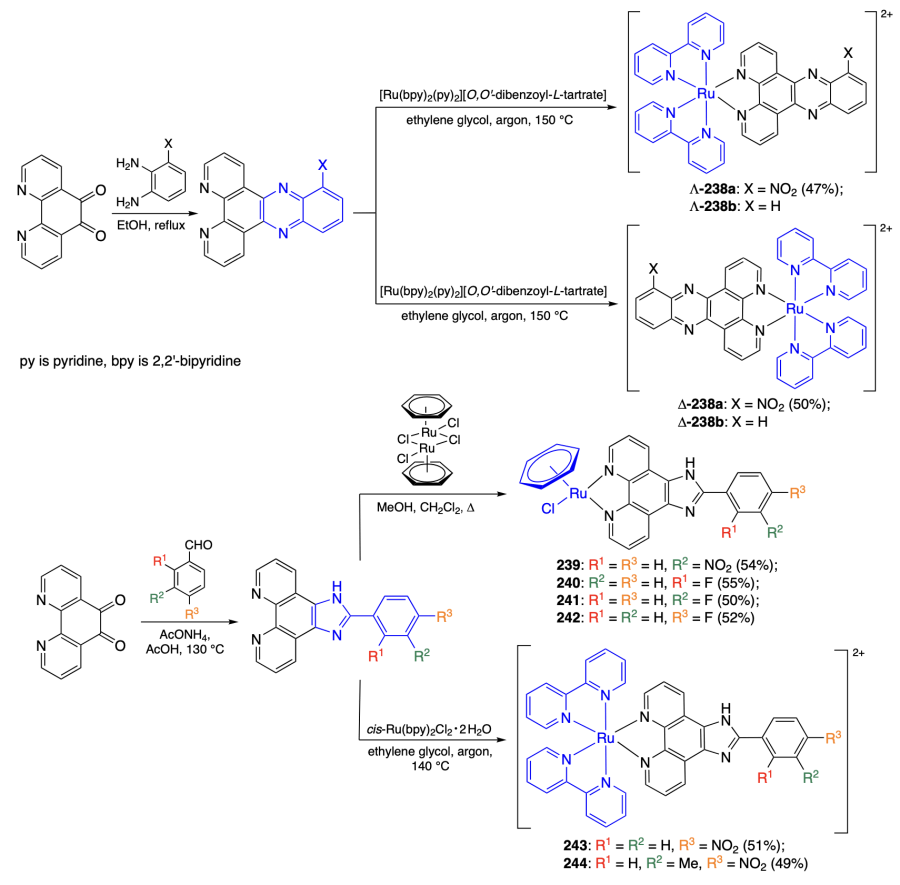
The binding constants of enantiomeric ruthenium complexes Δ-238a and Λ-238a to RNA triplex, poly(U) · poly(A) · poly(U), were determined by electronic spectroscopy at 441 nm to be (37.44 ± 9.83) × 105 М–1 and (17.78 ± 6.12) × 105 М–1, respectively. These values slightly exceed the binding constants of previously obtained complexes 238b with unsubstituted dipyrido[3,2-a:2',3'-c]phenazine [for Δ-enantiomer, Kb = (11.76 ± 3.59) × 105 М–1; for Λ-enantiomer, Kb = (6.43 ± 2.82) × 105 М–1[145]], which attests to stronger RNA binding of ruthenium complexes in the presence of a nitro group. Measurements of RNA melting temperatures showed that for complex : RNA = 0.2, the ΔTm1 values corresponding to melting of triple helix for compounds Δ-238a and Λ-238a were 9.0 and 4.5 °C, while the ΔTm2 corresponding to melting of the double helix was 7.6 and 9.0 °C. Hence, complex Δ-238a had an insignificant selectivity for RNA triplex, whereas Λ-238a was more selective for double-stranded RNA.
In the case of complexes with substituted imidazophenanthrolines, 2-fluoro- (240) and 4-fluorophenyl (242)[143]; 3-fluoro- (241) and 3-nitrophenyl (239)[142]; and 4-nitro- (243) and 3-methyl-4-nitrophenyl (244)[144] groups were chosen as substituents. A comparison of the properties of fluoro-substituted ligands indicates that the binding constants of complexes 241 and 242 with meta- and para-substituted ligands are commensurable and exceed Kb of complex 241 with the ortho-isomer (Table 13). The changes in the melting temperature of the complexes with nucleic acids were in line with Kb values; complex 241 with the meta-fluorine atom provided the most pronounced stabilization effect on the RNA triple helix. The binding constants for complexes 240 – 244 were similar and exceeded those for complex 239 containing the nitro group in the meta-position of phenylimidazo-phenathroline, which suggests destabilizing effect of the nitro group. It is also noteworthy that all complexes 239 – 244 proved to be selective for RNA triplexes, unlike the above-mentioned complexes with dipyridophenazines; compound 243 also destabilized double-stranded RNA. Despite the possible prospects of this line of research, further studies of ruthenium complexes are required, in particular, it is necessary to study binding to other nucleic acids, e.g., double-stranded DNA, and to perform biological assays in vitro and in vivo.
4.2. Quercetin derivatives
Binding of the triplex-forming oligonucleotides to the DNA double helix is highly selective, but also thermodynamically less favourable and kinetically slower than the formation of the double helix, which restricts the use of TFO in therapy. The addition of compounds that stabilize the triple helix may promote the triplex formation, which underlies the anticancer action of such drugs. Rangel et al.[146] studied a number of amino derivatives of quercetin 245 – 251 synthesized by a previously reported procedure[147] (Scheme 21).
These compounds can increase the triplex melting temperature with ΔTm up to 17.4 °C for pyrrolidine derivative 251, without affecting the melting temperature of double-stranded DNA, which attests to selective binding of quercetin derivatives to the triplex and its stabilization. This finding was later confirmed by the results of differential scanning calorimetry, CD spectroscopy, and isothermal titration calorimetry. Thus, quercetin derivatives are potential DNA triplex-binding ligands; however, drawing more unambiguous conclusions about their efficacy requires more detailed studies.
4.3. Benzazoles
Zinjic et al.[148] investigated binding of a series of cationic benzothiazole derivatives 252 – 260 to various DNA and RNA structures, including DNA : RNA hybrids and DNA triplexes. These compounds were prepared from substituted aminothiophenol and hetarylcarboxylic acids by the procedure described earlier[149-151] (Scheme 22).

According to competition dialysis assay, compounds 252, 256, and 257 had the highest affinity to double-stranded DNA : RNA hybrids and triplex DNA : АТТ (АТТ is polydA-2polydT). Melting experiments showed that compound 257 has the greatest stabilizing effect on DNA triplex, without affecting the double-stranded structure (for ligand to DNA concentration ratio of 1 : 10, ΔTm1 = 43.7 °C, ΔTm2 = 0 °C). These results were confirmed by CD spectroscopy and computer simulation.
A therapeutic effect against cancer diseases can be achieved not only by stabilizing the DNA or RNA triplex structure, but also by destroying the triplex. Rocca et al.[152] performed screening of a library of compounds to find a ligand binding to MALAT1 RNA responsible for the development of multiple myeloma and containing a stabilizing triplex moiety in the molecule. According to the results of computer simulation and melting experiments, ligand 261 was found to be the most promising among the tested compounds (Figure 15). Thus the addition of this compound to MALAT1 RNA resulted in a 2 °C decrease in the melting temperature of the RNA triplex, indicating its destabilization.
![[{"id":"qNhQq0fvwz","type":"paragraph","data":{"text":"Result of the molecular docking of compound <b>261</b> into <i>MALAT1</i> RNA.152 Copyright (2023) Wiley."}}]](/storage/images/resized/KT0MrYqh9KWRhDzyDqmwH7rCALRNqAyHc7eeTXAa_xl.webp)
Fluorescence intercalator displacement assay using thiazole orange showed that compound 261 has a moderate affinity (DC50 = 6.1 ± 0.3 μM). According to computer simulation data, pyrrolidine rings of compound 261 form hydrogen bonds with U7 and U8 uracil moieties. In addition, it is involved into electrostatic interactions with the side-chain phosphate groups, while the pyrrolepyridine system is linked to U7 and G6 bases by π – π-stacking. In experiments with myeloma cells, NCI-H929, AMO-1 (plasmacytoma), and bortezomib-resistant clone (ABZB), compound 261 showed a high cytotoxicity with IC50 of approximately 10 μM, whereas this ligand in concentrations of up to 10 μM had virtually no cytotoxicity against the normal mononuclear cells in peripheral blood.
In general, ligands binding to triplex structures of nucleic acids have been little addressed in the literature. For this reason, it is problematic to formulate the requirements to the ligand structure that would ensure their selectivity for triplexes. Since the strategy of using DNA and RNA triplexes in anticancer therapy usually implies the injection of triplex-forming oligonucleotides, this area probably does not hold much promise in the case of small organic molecules in comparison with other targets.
5. DNA and RNA hairpin ligands
Apart from the above-described non-canonical structures, DNA and RNA may have hairpin and cruciform structures (see Figure 1d ), which are formed by palindromic sequences consisting of two identical inverted repeats. The palindromic sequences can act as binding sites of nucleic acids to proteins and are frequently encountered in the genome, in particular in replication origins and gene promoters[153-157]. The formation of hairpins and cruciform structures is often attributed to the genomic instability as well as to gene amplification in tumour cells[158-162]. In view of their role in biological processes, such structures should be considered as a separate class of secondary structures of nucleic acids rather than a special case of double-stranded DNA.
A potential target for the anticancer therapy is mRNA existing as a hairpin, but able to be transformed into G-quadruplex under the action of external stimuli, e.g., in the presence of potassium cations[163, 164]. Since quadruplex structures in mRNA inhibit RNA transcription, the control over the hairpin → quadruplex equilibrium can be used to treat tumours.
Gupta et al.[165] obtained pyridine-substituted tetraphenylethylene 262 and its methylated analogue 263 (Scheme 23). Study of the HpGQ-1 system able to exist as a hairpin or quadruplex by FRET and melting temperature measurement showed that the addition of ligand 263 stabilizes the quadruplex structure of RNA. It is especially remarkable that the formation of quadruplexes in the presence of compound 263 occurs without the addition of potassium ions, which are usually required to stabilize quadruplexes. This fact was confirmed by CD spectroscopy data.
The subsequent studies were performed using luciferase assay. The HpGQ-1 sequence, the 5'-untranslated sequence of the TRF2 gene mRNA, HpGQ-2 sequence, or mutants of these sequences that cannot form quadruplexes were cloned into the 5'-untranslated region of luciferase gene. The addition of compound 244 was shown to inhibit luciferase synthesis when HpGQ-1 (luciferase activity was 27.2% of the control) or HpGQ-2 (17.5%) sequence was present in the non-coding region, but not in the case of mutant sequences. These result indicates that compound 263 inhibits translation by stabilizing quadruplex mRNA.
In most of known quadruplexes, guanine quartets are linked by relatively short loops. However, in some cases, the loop may be relatively long and contain complementary sequences, resulting in the formation of hairpins. Since the quadruplex–hairpin structures are less abundant in the genome than usual quadruplexes, compounds that could bind to both a quadruplex and a hairpin should exhibit higher selectivity than usual quadruplex-binding ligands. Yang et al.[166] screened a library of compounds in order to find a ligand selective to one of the N-MYC quadruplexes containing a hairpin. As the lead compound, the authors chose substituted 5-(pyridazin-3-yl)-1,2,4-oxadizole 264, which had a dissociation constant of 3.6 ± 1.4 μM, according to the surface plasmon resonance data.
The subsequent studies demonstrated that compound 264 does not compete in binding to N-MYC quadruplex with other ligands that bind to quadruplexes (TMPyP4, PhenDC3, PDS (3-{1-[3-(dimethylaminopropyl)propyl]-2-methyl-1H-indol-3-yl}-4-(2-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione)) or with the Hoechst 33258 dye, which binds to the DNA minor groove. According to these data and fluorescence spectroscopy with labelled DNA, compound 264 is incorporated between the quadruplex and the hairpin. The authors prepared homologue 265 with Kd = 1.5 ± 0.3 μM, which also effectively inhibited N-MYC gene expression in NBEB neuroblastoma cells.
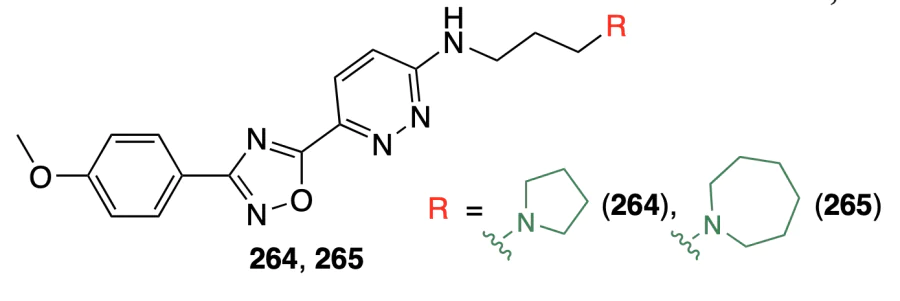
Hairpins are the least popular targets among non-canonical nucleic acid structures. However, the formation of hairpins should be taken into account while selecting a target gene, since their presence and ligand binding to them can increase the ligand selectivity.
6. Conclusion
This review covers the literature published between 2020 to 2024 devoted to the synthesis and biological properties of small molecules that bind to non-canonical DNA and RNA structures. The presented data attest to a great diversity of ligands for the possible DNA structures. These data allow researchers to select definite classes of potential ligands for non-canonical nucleic acid structures, but at the same time, complicate selection of the most promising compounds.
Among non-canonical structures, G-quadruplexes receive the most attention, as they have been most studied and, according to the currently available results, are most promising. The use of other DNA structures as targets either raises many questions about the exact mechanisms of their action (e.g., i-motifs) or is less convenient in the case of small organic molecules (e.g., DNA triplexes). However, non-quadruplex non-canonical structures should not be completely dismissed either, because they can affect the binding affinity of G4 ligands, in particular increase their selectivity for certain G-quadruplexes (e.g., hairpin structures in G4).
The papers of this period of time show that in the development of new types of ligands for non-canonical DNA and RNA structures, it is expedient to move away from the concept of a large monolithic aromatic skeleton towards more conformationally flexible molecules.20 However, according to the results of studies addressed in this review, it is obvious that the inclination towards the skeleton flexibility should not replace, but should supplement the presence of a planar polycyclic system (usually consisting of three or four rings). Thus, the conformational flexibility of the anthraquinone moiety in compound 5 was sufficient for molecule bending to be adjusted to the DNA structure,38 while CX-5461 (compound 155), which has reached clinical trials as an agent for treatment of DNA repair deficient progressive solid tumours, contains a rigid ring. Hence, it can be assumed that the most promising molecules are those similar to CX-5461, but having a fused polycyclic system with limited conformational mobility of the structure-forming moiety or with additional arylamide or stilbene groups. In the development of such ligands, it is also necessary to pay attention to other intermolecular interactions, apart from the commonly considered π – π-stacking; for example, chalcogen bonds and, as a consequence, to design the presence of sulfur and possibly selenium atoms in the G4 ligand molecules.
Finally, it may be noted that none of the compounds, ligands for non-canonical DNA and RNA structures, in particular G-quadruplexes, has been approved to date for use in clinical practice as an anticancer agent. Nevertheless, the example of CX-5461, which has reached clinical trials, gives hope for this result in the future. In conclusion, we would like to emphasize once again that success in the development of G4 ligands as anticancer drugs requires the creation of a large library of compounds and study of their interaction with various DNA structures and proteins in cells, in order to optimize the structure of the target molecule.
7. List of abbreviations and symbols
ΔGbind — Gibbs free energy of DNA binding,
360A — N,N'-bis(1-methyl-3-quinolinio)pyridine-2,6-dicarboxamide,
APTO-253 — 2-(2-methyl-5-fluoro-1H-indol-3-yl)-1H-imidazo[4,5-f][1,10]phenanthroline,
Asc — ascorbate,
bpy — 2,2ʹ-bipyridine,
BRACO-19 — N,N'-(9-{[4-(dimethylamino)phenyl]amino}acridine-3,6-diyl)bis[3-(pyrrolidin-1-yl)propanamide],
CCK8 — cell counting kit for assessment of cell proliferation and toxicity,
CD — circular dichroism,
CRT — calreticulin,
ct-DNA — calf thymus DNA,
DBU — 1,8-diazabicyclo[5.4.0]undec-7-ene,
DC50 — concentration of 50% displacement of the fluorescence agent,
DIPEA — diisopropylethylamine,
DMA — dimethylacetamide,
DMAP — 4-(dimethylamino)pyridine,
DME — 1,2-dimethoxyethane,
DPPA — diphenylphosphoryl azide,
dppf — diphenylphosphinoferrocene,
dppz — dipyrido[3,2-a:2',3'-c]phenazine,
dsDNA — double-stranded DNA,
EB — ethidium bromide,
ECD — electronic circular dichroism,
EDC — 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide,
ESI-MS — electrospray ionization mass spectrometry,
FAM — 6-carboxyfluorescein,
FID — fluorescent intercalator displacement assay,
FLIM — fluorescence lifetime imaging microscopy,
Fmoc — 9-fluorenylmethyloxycarbonyl,
FRET — Förster resonance energy transfer (FRET) melting assay,
G4 — G-quadruplex,
GI50 — half-maximal growth inhibitory concentration,
HOBt — 1-hydroxybenzotriazole,
HR — homologous recombination,
HRD — homologous recombination deficiency,
HBRU — benzotriazoletetramethyluronium hexafluorophosphate,
IC50 — half-maximal inhibitory concentration,
ICD — immunogenic cell death,
iM — i-motif,
Kb — DNA binding constant,
Kd — dissociation constant of DNA complex,
Mor — morpholin-4-yl,
MST — microscale thermophoresis,
MTS — 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium bromide,
MTT — (4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide,
NIS — N-iodosuccinimide,
PARP — poly-ADP-ribose polymerase,
PDB — protein database,
PDC — pyridinium dichlorochromate,
PEG — polyethylene glycol,
phen — phenanthroline
PhenDC3 — 3,3'-[1,10-phenanthroline-2,9-diylbis(carbonylimino)]-bis(1-methylquinolinium) triflate,
Pip — piperidin-1-yl,
PPA — polyphosphoric acid,
Py — pyridyl,
py — pyridine,
PyBOP — benzotriazolyltri(pyrrolidin-1-yl)phosphonium hexafluorophosphate,
qdppz — naphtho[2,3-h]dipyrido[3,2-a:2',3'-c]phenazine-8,13-dione,
ROS — reactive oxygen species,
RT-PCR (qRT-PCR, Q-PCR) — real-time (quantitative) polymerase chain reaction,
SI — selectivity index,
ssDNA — single-stranded DNA,
st-DNA — salmon testes DNA,
Tm — melting temperature,
TAMRA — 5-carboxytetramethylrhodamine,
TBAB — tetrabutylammonium bromide,
TFA — trifluoroacetic acid,
ThT — thioflavine Т,
TIS — triisopropylsilane,
TMPyP4 — meso-5,10,15,20-tetrakis(N-methyl-4-pyridyl)porphyrin tetratosylate,
TO — thiazole orange,
Topo1 — topoisomerase 1,
TRIR — time-resolved IR spectroscopy,
TFO — triplex-forming oligonucleotides.
References




















