



![Structures and photophysical properties of BODIPY 31 and halogen derivatives 32 – 42.[99, 100, 103, 106-113]](/storage/images/resized/JQUsbn5YWKew0eSHxDLZGBCISF8pQGTkmAZcx2Bk_xl.webp)
![Structures and photophysical properties of 1,3,5,7-tetramethyl-BODIPY 49 and 2,6-dihalogenated derivatives 50 - 52.[106, 134, 136]](/storage/images/resized/r55QvJB8AdPGhjS7vTre9jl72Gj3piTNZGvWMiVo_xl.webp)
![HOMO – LUMO energy gap as a function of slip angle (θ) in J- and H-aggregates.[243]](/storage/images/resized/QovppFg9kVMJDnaLRCmOE4Dez2nE94QIrdqXDYhv_xl.webp)
Keywords
Abstract
BODIPY chromophores (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene) are one of the most versatile fluorophores which are commonly used in a variety of scientific fields. The combination of excellent photophysical properties with the possibility of easy chemical modification of structure, chemical and photostability determines the application of BODIPY in biomedicine, photocatalysis, analytical chemistry and materials science. This review summarizes approaches to the synthesis and modification of BODIPY, discussing both classical and recently-published synthetic and postfunctionalization approaches. An influence of BODIPY structure and various postfunctionalization methods on photophysical properties of the fluorophores and their potential applications in biomedicine, materials science and biosensorics are also discussed. The bibliography includes 344 references.
1. Introduction
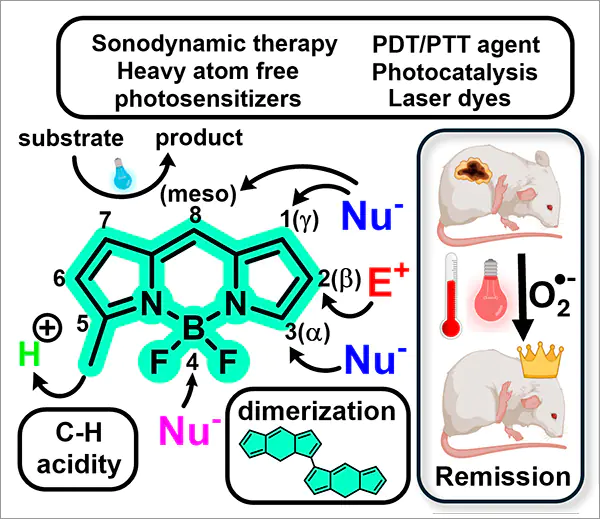
Nowadays, 4,4-difluoro-4-bora-3a,4a-diaza-s-indacenes (BODIPY chromophores, or BDP) are considered to be a universal fluorophore scaffold. BODIPY derivatives are generally characterized by high fluorescence quantum yields, facile chemical modification, valuable photophysical properties, diverse array of chemical transformations, as well as exceptional optical and thermal stability.[1] Today, BODIPY-based dyes have a wide range of applications in various fields of scientific research — from biomedicine to materials science. These compounds are used as triplet photosensitizers,[2] agents for photodynamic (PDT) and photothermal (PTT) antitumor therapy,[3] as well as photocatalysts,[4] fluorescent labels for biomolecules,[5, 6] photoremovable protecting groups [7] and light-harvesting systems.[8]
The BODIPY core is a deprotonated dipyrromethene ligand that coordinated with electron-deficient disubstituted boron atom, which is usually part of the BF2 group [9, 10] (Fig. 1). Formally, the negative charge is localized on the boron atom, while the positive charge is delocalized over the dipyrromethene system.[10] However, as the BODIPY molecule is weakly polar, the molecule is usually depicted without formal charges (see Fig. 1). Since the structure of the BODIPY core without substituents is similar to that of s-indacene, the numbering system of the BODIPY dyes is also analogous. The positions of the carbon atoms in the side rings of BODIPY are also named by analogy with pyrrole: thus, the a-position corresponds to atoms 3 and 5, the b-position — to atoms 2 and 6, and the g-position — to atoms 1 and 7. Position 8 of the BODIPY core is also called the meso-position [10] (see Fig. 1).
![[{"id":"SYOOthAur8","type":"paragraph","data":{"text":"Atom numbering in BODIPY and s-indacene"}}]](/storage/images/resized/vBa3Uk5jMAikYqSWMTVOppSrMea0IyNIi5JV1twi_xl.webp)
A unique feature of BODIPY, which favorably distinguishes them from many other chromophores, is the ability to precisely tune its photophysical properties through various postfunctionalization approaches.[10, 11] For example, replacing the boron-bound fluorine with other nucleophile can lead to an increase in fluorescence quantum yields of BODIPY, improved chemical stability, increased water solubility.[12] In addition, it also opens up possibilities for the synthesis of light-harvesting systems.[12] Introduction of chiral substituents into BODIPY results in fluorophores with circularly polarized luminescence,[13] while conjugation of photoacceptor moiety to the BODIPY core paves the way for the formation of dyads, triads and other light-harvesting systems.[14] The introduction of heavy atoms into BODIPY promotes spin-orbit interaction and stabilization of the triplet state of fluorophores, which is widely used in the design of BODIPY-based PTT agents.[15] The presence of mesomeric donors in the BODIPY structure usually causes intramolecular charge transfer (ICT) and fluorescence quenching to occur in polar solvents; this phenomena is commonly used in the development of sensors for biomolecules, metal cations and pH changes, as well as for hypoxia diagnostics.[16, 17]
The possibility of fine-tuning the physicochemical properties of BODIPY derivatives is attracting the attention of researchers, who are looking for new synthetic approaches and expanding the possibilities of the BODIPY core functionalising. A review of publications focused on these issues revealed a growing interest among specialists in both the discovery of new compounds and the modification of known synthetic methods. Thus, in 2023, new approaches were presented to the radical acylation of BODIPY,[18] the introduction of a cyano group into the core,[19] the use of photoexcitation of various derivatives to increase their reactivity,[20] and the modification of BODIPY at the boron atom with chalcogenophenes.[21] In 2024, methods for obtaining water-soluble carbohydrate derivatives,[22] BODIPY π-system expansion [23] and synthesis of fluorinated analogs were described;[24] reactions of electrophilic β-alkenylation [25] and soft electrochemical sulfonylation were studied;[26] new one-pot synthesis options for 3-substituted BODIPY were also presented.[27] Thus, the relevance of developing new approaches to the synthesis and modification of the BODIPY core is beyond doubt.
An important area of BODIPY application is the photocatalysis of various organic transformations. Thus, in 2021, BODIPY were used as photocatalysts for ion-radical addition to alkenes,[28] and in 2022, the BODIPY photocatalyzed Beckmann rearrangement was described.[29]
The enormous interest in BODIPY as highly stable fluorophores with valuable photophysical characteristics and the ability to fine-tune their photophysical properties has resulted in a large number of publications. Many works are devoted to the development of BODIPY-based agents for PTT and PDT,[30-33] for sonodynamic therapy [15] and photoacoustic imaging,[34] as well as fluorescent probes, which are used in the analysis of food objects, water, detection of biomolecules in vitro and in vivo.[35, 36]
Various areas of BODIPY application are summarized in a number of review publications. In 2014 Ni and Wu [37] published a review on the synthesis and application of NIR-absorbing BODIPY (NIR — near infrared) as fluorescent pH-sensitive probes for bioimaging. In the same year, a review was published on the structural modifications of the BODIPY core that lead to a shift of the absorption maximum in electronic spectra towards the NIR region.[38] In 2019, Chen et al.[39] generalized the data on the development of triplet photosensitizers based on BODIPY and also summarized the structural factors contributing to internal conversion. A year later Poddar and Misra [40] published a review examining optoelectronic applications of BODIPY. In 2021, a review paper dedicated to the use of BODIPY as reporters of molecular processes in a living cell was published in the Russian Chemical Reviews [41] journal. In 2023, Wang et al.[42] presented a review on BODIPY-based photosensitizers, and Zhang et al.[43] summarized the data on photoactivated BODIPY for PALM (photoactivated localization microscopy). In addition, several reviews devoted to the synthesis and chemical properties of BODIPY were published. In 2007, Loudet and Burgess [9] published a fundamental and comprehensive analysis of the preparation and postfunctionalization of BODIPY and its derivatives, including aza-BODIPY and dipyrromethenes, and a year later Ulrich et al.[1] presented a brief overview of BODIPY functionalization methods and their applications. In 2019, a detailed review dedicated to the postfunctionalization of BODIPY was published by Boens et al.[10] In the same year, Clarke and Hall [44] published a review summarizing recent advances in the synthesis of BODIPY, and Nascimento et al.[45] summarized methods for the preparation of dipyrromethanes. Of recent work, a brief review from 2023 by Yadav and Misra [46] on the synthesis, functionalization of BODIPYs and their application as PDT agents is worth mentioning.
This review summarizes recent advances in the chemistry of boron-dipyrromethenes, including synthetic tools and methods and approaches for modifying of the BODIPY structure, as well as their various applications, including as sensors, antitumor agents, and optical materials. In particular, information is presented on the synthesis of the boron-dipyrromethene core and methods for its modification, such as reactions involving the BODIPY aromatic ring and CH-acid methyl groups as well as substituents at the boron atom. In addition, the influence of chemical transformations and structural factors on the photophysical properties of BODIPY are discussed in the context of their potential practical applications. The factors contributing to the increase in the effectiveness of the BODIPY derivatives as PDT and PTT agents are analyzed, and the most successful approaches to the design of antitumor agents based on them are noted. Particular attention is paid to the use of BODIPY as fluorescent sensors, and agents for cellular organelles visualization, for detecting specific environmental conditions and specific compounds, such as biothiols, acyl chlorides, hypochlorite anion, reactive oxygen species (ROS), HgII ions. Photophysical aspects of the fluorescence response are discussed.
Given the high interest in the use of BODIPY and their derivatives in biomedicine and optoelectronics, and taking into account the large number of publications on this topic in highly rated scientific journals, we believe that this review will be of interest to a wide range of specialists in organic and medicinal chemistry. The material published in the review may be useful both in choosing a strategy for the synthesis and functionalization of the BODIPY molecules, and in designing of the BODIPY derivatives with specific photophysical properties and biological activity for biomedical applications, among others.
2. Methods of BODIPY derivatives preparation
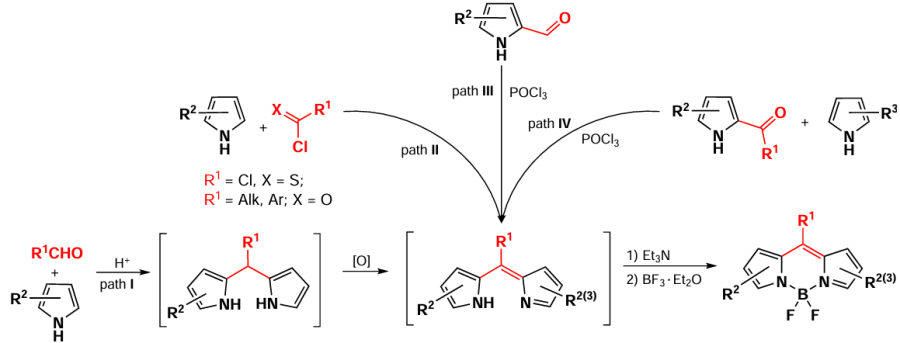
Over the last few decades, studies devoted to the design of the BODIPY fluorophores have led to the development of a large number of diverse synthetic approaches that have opened the way to the preparation of boron dipyrromethenes with different structures and different photophysical properties (Scheme 1). The methods for synthesis of the BODIPY core can be divided into two main types: preparation of dipyrromethanes with subsequent oxidation (path I) or direct synthesis of dipyrromethenes formed from pyrroles and acyl chlorides (path II), by self-condensation of acylpyrroles in the presence of POCl3 (path III) or via a reaction between acylpyrrole and pyrrole (path IV). Approaches I through IV are discussed in this section.
2.1. Synthesis from pyrroles and aldehydes
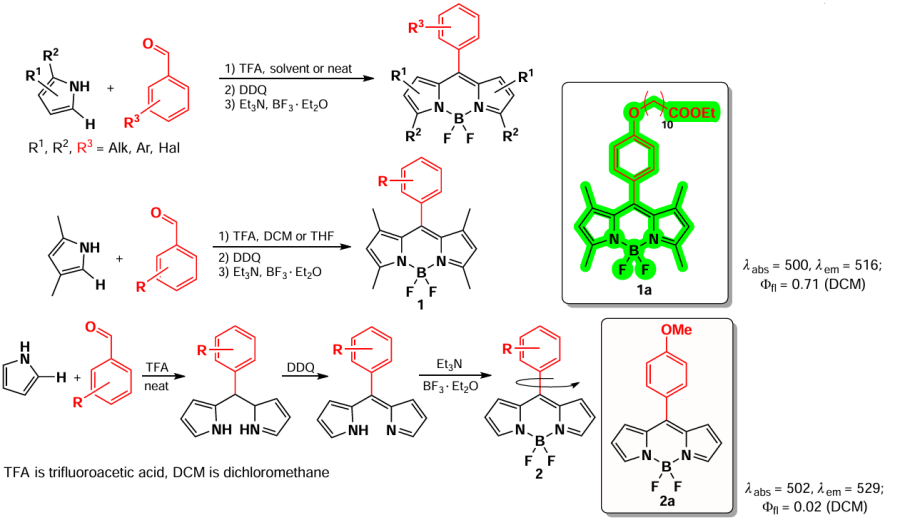
A common method for the synthesis of BODIPY is the condensation of aromatic aldehydes and pyrroles, described by Wagner and Lindsey [47] in 1996. Acid catalysis produces symmetric dipyrromethane, which is further oxidized by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and p-chloranil to form dipyrromethane. Further base action followed by complexation under the action of boron trifluoride etherate yields the target BODIPY fluorophores. α-Substituted pyrroles, such as 2,4-dimethylpyrrole, are commonly used to inhibit the polymerization side reaction.[48-50] In the case of unsubstituted pyrrole, the dipyrromethane production step is carried out directly in the pyrrole, after which the dipyrromethane is isolated and introduced into successive oxidation and complexation reactions (Scheme 2)*.[51-53] The alkyl derivatives of BODIPY, such as compound 1a, are characterized by absorption maxima in the range of 490 – 500 nm, a Stokes shift of 5 – 10 nm, and bright fluorescence. It should be noted that the BODIPY derivative 2a with free positions 1, 3, 5, and 7 exhibits almost no fluorescence due to the free rotation of the aryl substituent around the C – C bond. [54]
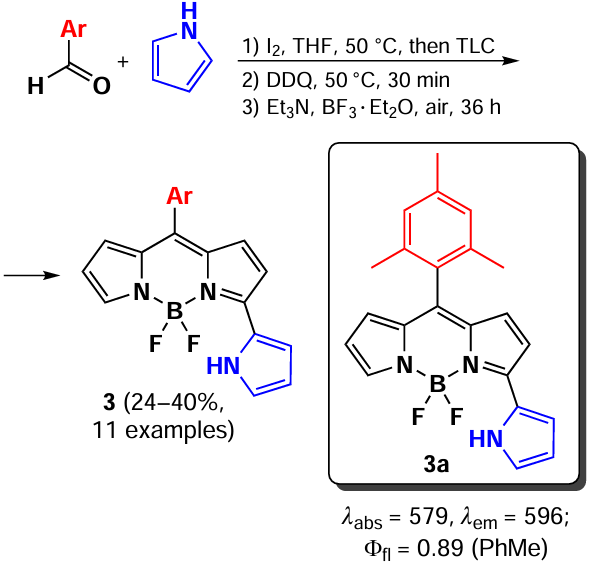
In 2024, Kang et al.[27] proposed the synthesis of meso-aryl-substituted 3-pyrrolyl-BODIPY by condensation of aromatic aldehyde with pyrrole in the presence of catalytic amounts of iodine as a weak Lewis acid [55] (Scheme 3). The key benefit of this approach is the potential for a one-pot synthesis, yielding 3-pyrrolyl-BODIPY 3 with an absorption maximum in the 580 – 600 nm spectral range.
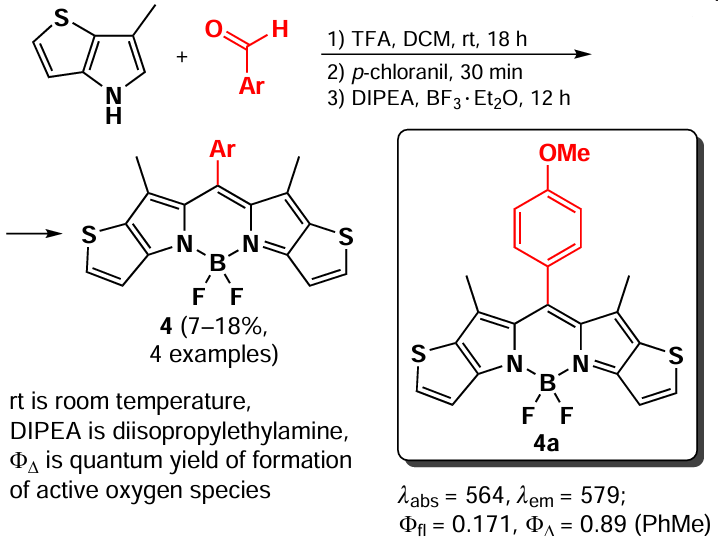
Another method of one-pot synthesis of BODIPY with a red-shifted absorption maximum is the use of condensed pyrroles as starting material. Thus, a series of PDT-active BODIPY 4 was prepared by Yoon et al.[56] from various benzaldehydes and 6-methyl-4H-thieno[3,2-b]pyrrole (Scheme 4). Due to the presence of a condensed thiophene moiety, BODIPY 4 exhibited enhanced spin-orbit coupling (SOC) in combination with a reduced energy gap between the singlet and triplet states.
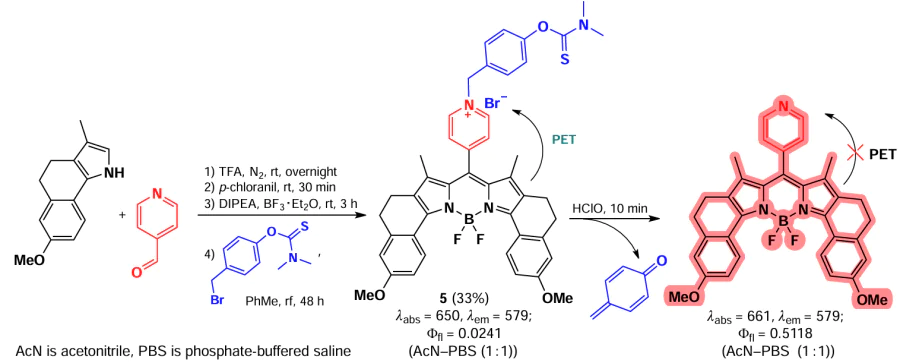
Zhao et al.[57] in 2022 synthesized a BODIPY-based fluorescent probe 5 for the detection of hypochlorous acid (Scheme 5). The fluorescence of BODIPY 5 was significantly suppressed due to photoinduced electron transfer (PET) from the electron-rich BODIPY core to the electron-deficient benzylpyridinium fragment (Ffl = 0.024). In the presence of HClO, the dimethylthiocarbamate fragment was cleaved off, resulting in an increase in fluorescence (Ffl = 0.51).
* Absorption (λabs) and emission (λem) maxima in the electronic spectra (in nm), as well as fluorescence quantum yield (Ffl) are given under the formulas of specific compounds; the solvent is indicated in brackets
2.2. Synthesis from pyrroles and acid chloranhydrides or anhydrides
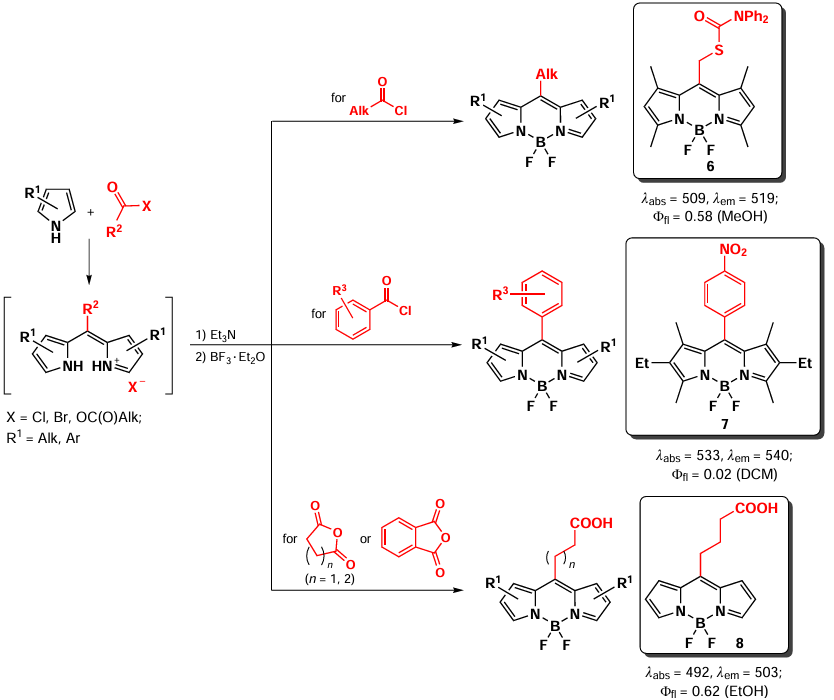
Condensation of pyrroles with carboxylic acid chloranhydrides or anhydrides is also widely used to obtain meso-substituted BODIPY 6 – 8.[58] The advantage of this approach is that both aromatic [59] and aliphatic chloranhydrides,[60] as well as chloranhydrides of bidentate carboxylic acids,[61] can be used as substrates for BODIPY synthesis. In addition, cyclic anhydrides can serve as starting compounds, yielding BODIPYs with a meso-carboxylic group [62, 63] (Scheme 6).
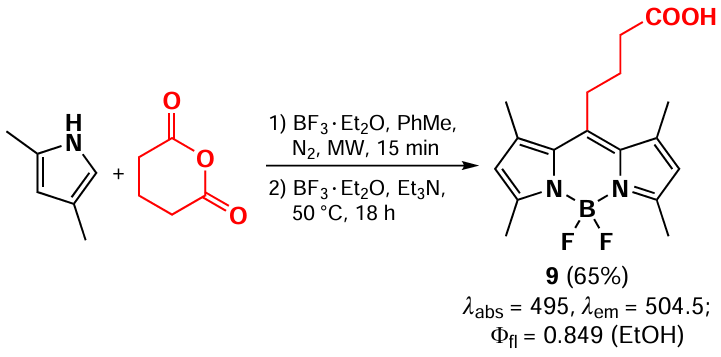
In 2020, Mhlongo et al.[64] proposed to use microwave (MW) radiation to carry out the condensation of pyrrole and glutaric anhydride, BODIPY 9 was obtained. The stage of dipyrromethene formation under these conditions took 15 min (Scheme 7).
Microwave irradiation was also used by Lama et al.[65] in 2022 for one-pot synthesis of BODIPY 10 from pyrroles and aromatic (e.g., for 10a) or aliphatic (for 10b) chlorohydrides. The reaction was carried out under gentle heating in a minimal volume of solvent, and the synthesis of BODIPY 10 in gram quantities was shown to be possible (Scheme 8).
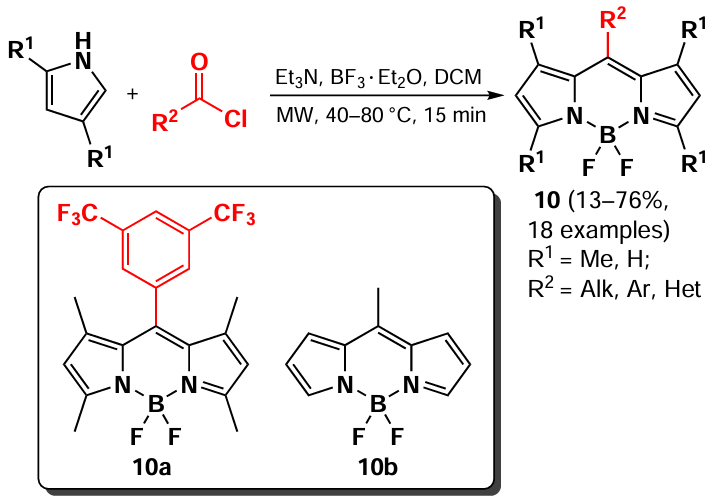
2.3. Synthesis from acylpyrroles and pyrroles
Unsymmetrical BODIPY with two different pyrrole cycles can be synthesized by condensation of α-ketopyrroles with other pyrroles. When pyrrole-2-carbaldehyde is used as a starting reagent, BODIPY derivatives without a substituent at the meso-position are formed.[66-70] This method also allows the preparation of unsymmetrical BODIPY with a carboxyl group in one of the pyrrole cycles available for further functionalization.[71, 72] When condensed pyrroles are used as substrates, the resulting BODIPYs are characterized by absorption in the NIR region and high molar absorption coefficients.[73-75] Another modification of this approach is the use of an α-iminium salt of a pyrrole, which can be obtained by the Wilsmeyer – Haack formylation, as the starting compound. It is worth noting that pyrroles [76] and their condensed analogs, particularly isoindole,[77] can be used as substrates for the formylation (Scheme 9).
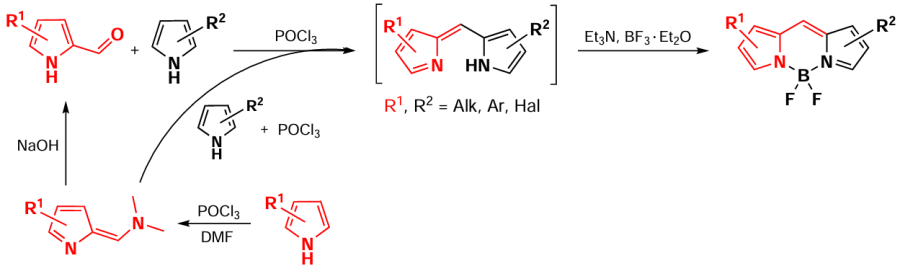
Following this approach, in 2023 Spingler et al.[78] synthesized a series of highly active PTT agents 11 based on asymmetric BODIPYs with a thiophene moiety at position 3. BODIPY 11a was highly phototoxic in the nanomolar concentration range under NIR radiation at a wavelength of 630 nm (total irradiation dose 5 J cm–2) (Scheme 10).
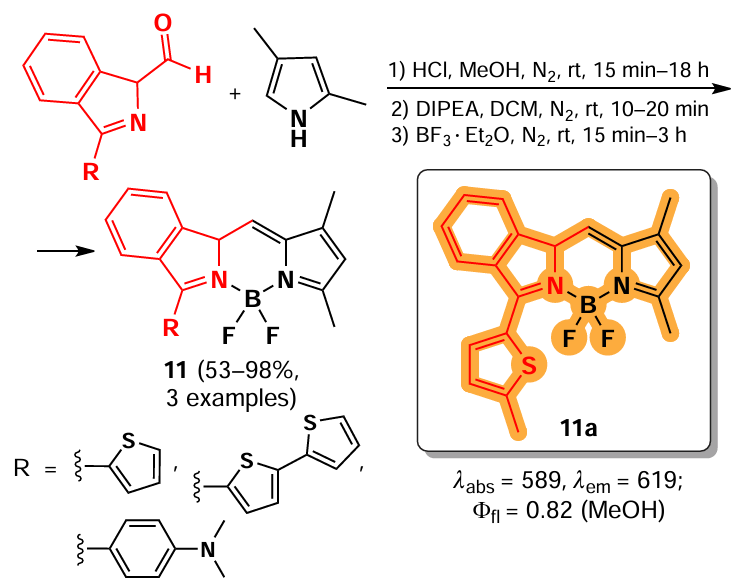
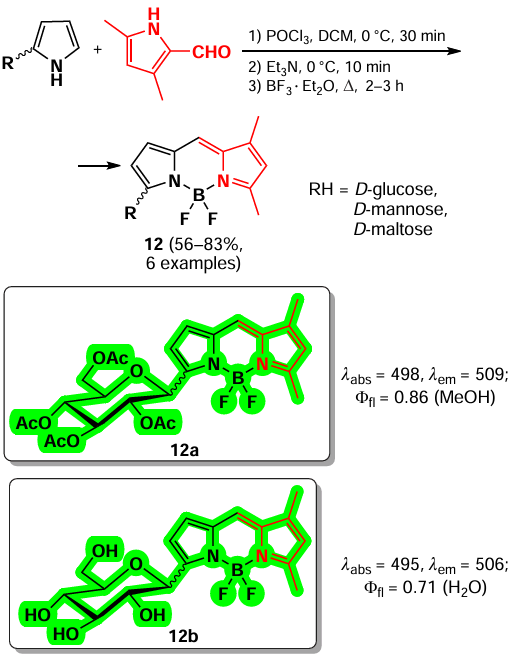
In 2024, Uriel et al.[22] have proposed an approach for the synthesis of BODIPY-based α-glycosides 12 formed by condensation of the corresponding α-glycosidopyrrole and 3,5-dimethylpyrrole-2-carbaldehyde. The BODIPY obtained, such as mannoside 12a, exhibited bright fluorescence (Φfl = 0.7 – 1) in organic solvents, while the deacylated derivative 12b showed fluorescence in aqueous media (Scheme 11).
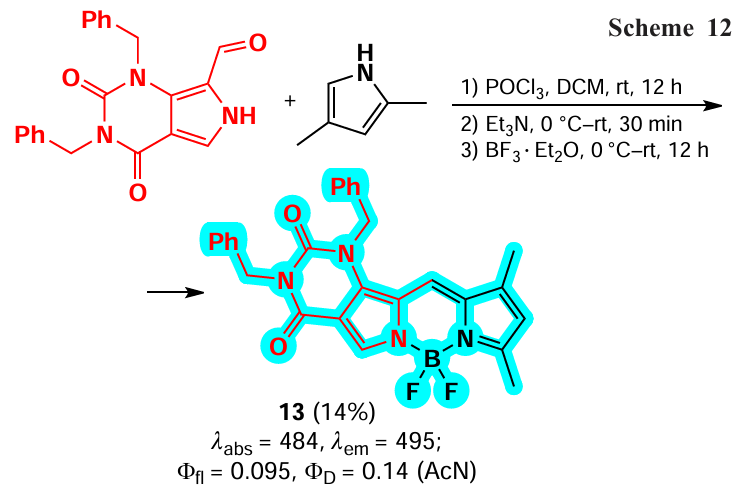
Condensed BODIPY 13 was synthesized from 2,4-dimethylpyrrole and uracil -based pyrrole-2-carbaldehyde by Nagpal et al. in 2024.[23] (Scheme 12). The BODIPY 13 showed the ability to form singlet oxygen (1O2) upon irradiation and PDT activity on rat C6 glioblastoma cells.
2.4. POCl3-catalyzed self-condensation of pyrrolcarbaldehydes
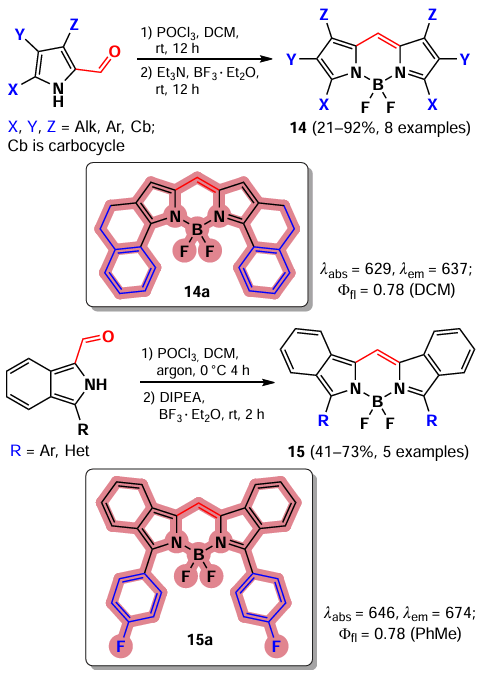
An attractive way to synthesize symmetric BODIPYs with unsubstituted meso-position is the self-condensation of pyrrolcarbaldehydes in the presence of POCl3. This approach was developed by Wu and Burgess [79] in 2008 for the synthesis of a series of BODIPY 14 (Scheme 13). The resulting fluorophores exhibited absorption in the NIR region as well as a bright fluorescence (Φfl = 0.7 – 1). BODIPY 15 were obtained in a similar way from condensed and aryl-substituted pyrroles.[70, 75]
2.5. Preparation of 8-heteroatom-substituted BODIPYs
2.5.1. 8-Sulfanyl derivatives
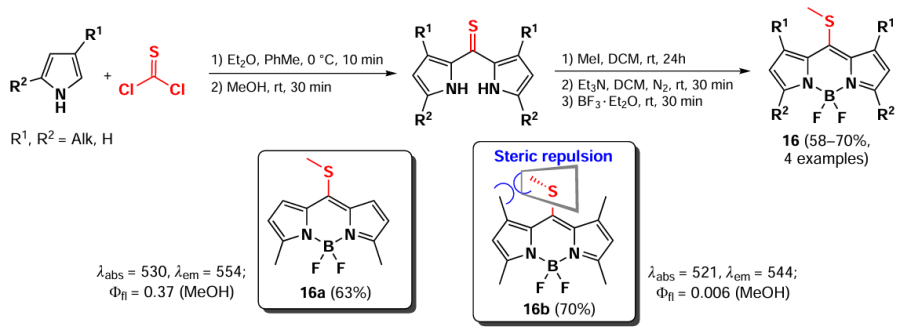
8-Methylsulfanyl-BODIPYs can be obtained by using thiophosgene as the carbonyl component in condensation with pyrroles. Such BODIPYs are utilized as substrates in nucleophilic substitution and cross-coupling reactions.[10] Three 8-methylsulfanyl BODIPYs 16, including the 3,5-dimethyl-8-methylsulfanyl-BODIPY 16a, were synthesized for the first time in 2006 by Goud et al.[80] The authors first prepared a dipyrrolylthioketone from pyrrole and thiophosgene. then methylated it at the sulfur atom followed by a complexation reaction with BF3 · Et2O (Scheme 14). In the following work,[81] a series of other 8-methylsulfanyl-BODIPYs were synthesized, including BODIPY 16b with methyl groups at positions 1, 3, 5, and 7. The emission suppression of the polymethyl derivative 16b was observed due to the removal of the 8-SMe group from electronic coupling with the BODIPY core because of steric hindrance with the methyl groups at positions 1, 7.[81] In 2022, 1,3,5,7-tetramethyl-8-methylsulfanyl-BODIPY 16b was used as a photocatalyst for Beckmann rearrangement and oxime hydrolysis.[29] The SMe-group at the meso-position promotes intersystem crossing (ISC) and the formation of a triplet excited state in which such BODIPY reacts with an electron donor to form an anion radical.
2.5.2. 8-Halogen derivatives
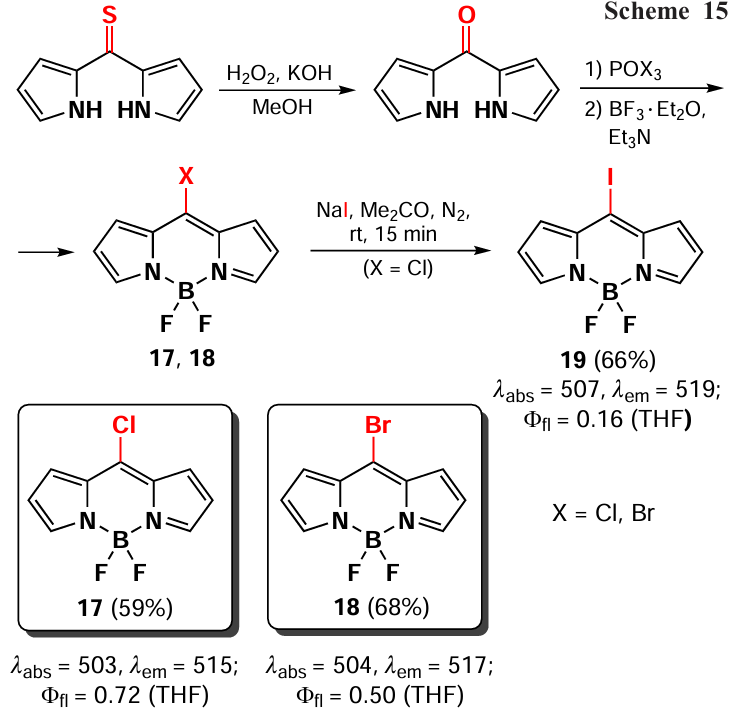
In 2012. Leen et al.[82] described the synthesis of meso-chloro- (17) and meso-bromo-BODIPY (18) by reaction of dipyrrolyl ketone with phosphorus oxochloride or oxobromide followed by treatment with base and complexation with boron trifluoride esterate. A direct synthesis of 8-iodo-BODIPY 19 using this approach is not possible, but 8-iodo-BODIPY 19 can be obtained from 8-chloro-BODIPY 17 by the Finkelstein reaction with sodium iodide. The initial dipyrrolyl ketone can be synthesized by condensation of pyrrole with thiophosgene followed by oxidation with hydrogen peroxide [83] (Scheme 15). It should be noted that the nature of the halogen atom at the meso-position has a negligible effect on the absorption and emission wavelengths of resulted BODIPYs. Nevertheless, the decrease in the fluorescence quantum yields with increasing halide ion size is in good agreement with the ‘heavy atom’ effect. The photophysical properties of halogen-containing BODIPYs are discussed in detail in Section 3.2.1.
2.6. Synthesis from pyrroles and orthoesters
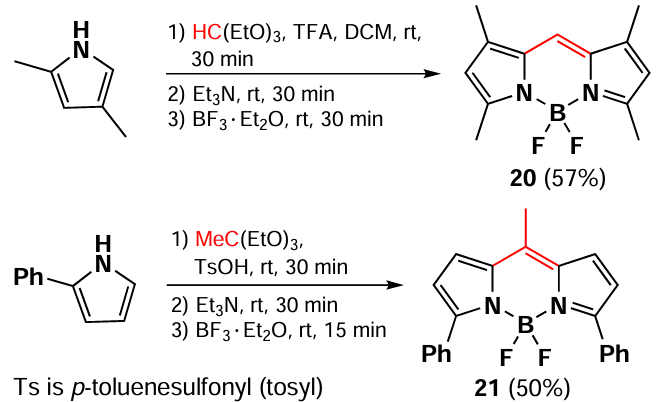
A method for the synthesis of a BODIPY with a free meso-position 20 via the condensation of 2,4-dimethylpyrrole with an orthoester was described by Sekiya et al.[84] If triethylorthoacetate is used instead of an orthoester, 8-methyl-substituted BODIPY 21 is formed from 2-phenylpyrrole (Scheme 16).[85]
2.7. Synthesis of 8-trifluoromethyl-substituted BODIPYs
BODIPYs with a trifluoromethyl group at the meso-position are of particular interest due to the fact that the electron acceptor CF3 group promotes aromatic nucleophilic substitution (SNAr) in the BODIPY core, greatly facilitating modification at positions 3 and 5.[86] When the CF3 group is present at the meso-position, the absorption spectrum of the compounds shifts into the NIR region.[34] 8-trifluoromethyl-substituted BODIPYs with methyl groups at positions 1 and 7 are of interest as PTT agents because the absence of the CF3-group rotation barrier leads to almost complete quenching of fluorescence and high photothermal efficiency.[34]
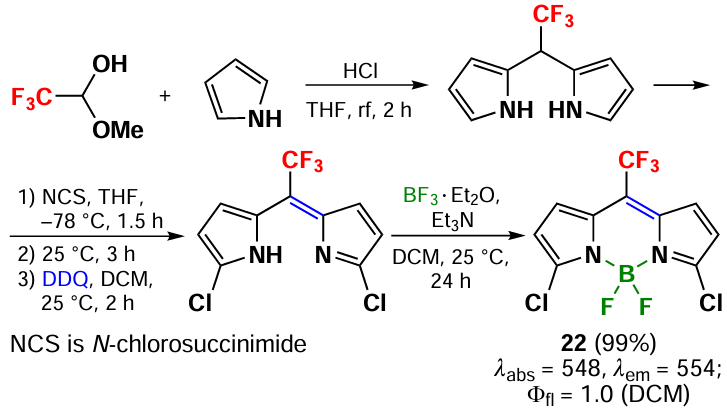
In 2008, Li et al.[86] proposed a method for the synthesis of meso-CF3-BODIPY 22 from pyrrole and trifluoromethyloxymethanol. The condensation of the reagents under acidic conditions resulted in the formation of CF3-substituted dipyrromethane, the subsequent oxidation of which and complexation with boron trifluoride etherate led to the target product (Scheme 17).
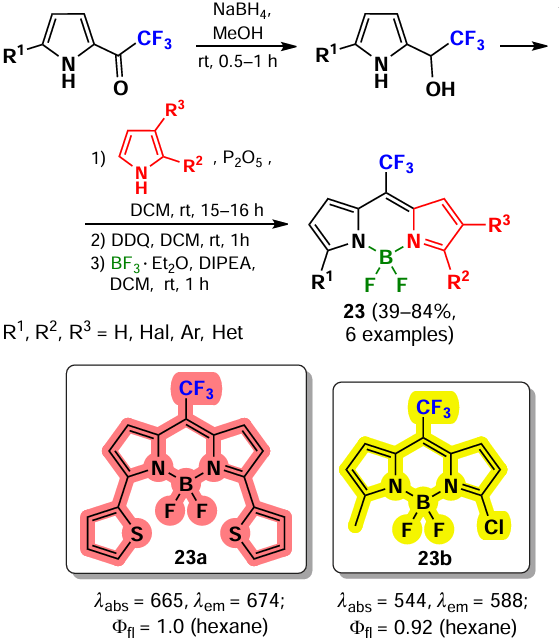
An alternative method for the synthesis of NIR-absorbing meso-CF3-BODIPY 23a and its analogs was published in 2011 by Sobenina et al.[87] The synthetic steps included the reduction of 2-trifluoroacetylpyrroles to alcohols, with further condensation reaction with pyrroles yielding dipyrromethanes, followed by oxidation and complexation (Scheme 18). Meso-CF3-BODIPYs were characterized by absorption maxima shifted to the red region of the spectrum as well as bright fluorescence (Ffl = 0.6 – 1). Using this approach, Wang et al. 2022 synthesized a series of PTT active BODIPY dimers 23b absorbing in the NIR(II)-region (see Section 3.6.10).[88]
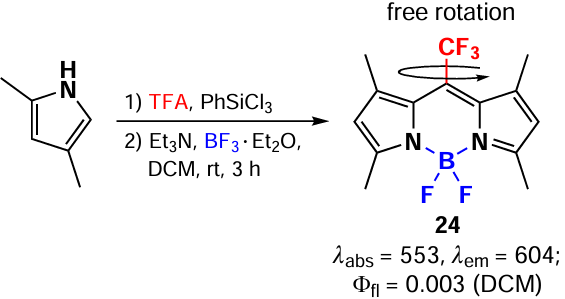
In 2014 Choi et al.[89] synthesized 1,3,5,7-tetramethyl-8-CF3-BODIPY 24 via condensation of ketopyrroles with pyrroles in the presence POCl3 (Scheme 19). The high electonegativity of the CF3 group leads to a significant decrease in the energy gap between the highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals compared to the meso-methyl-BODIPY.
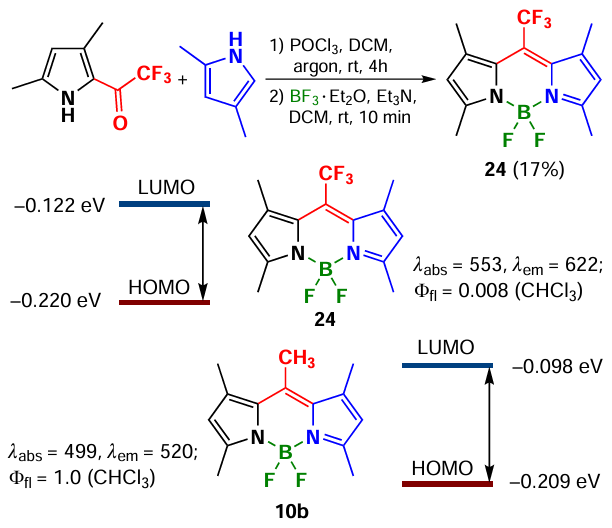
It should be noted that BODIPY 24 demonstrated an extremely low fluorescence quantum yield (0.008) due to the significant difference in the geometry of the ground (S0) and first excited (S1) singlet states. In addition, aggregation induced emission (AIEE) was observed for BODIPY 24. Thus, as the water amount in BODIPY 24 solution increased from 0 to 99%, the formation of J-aggregates occurred, accompanied by a bathochromic shift and the appearance of a new absorption band at 626 nm with a 40-fold increase in fluorescence (Φfl = 0.06). Importantly, under the same conditions, 8-methyl-BODIPY 10b exhibited H-aggregation accompanied by a hypochromic shift and significant fluorescence quenching. The main difference between BODIPYs 24 and 10b with trifluoromethyl and methyl substituents at the meso-position is the orientation of the dipoles with respect to the stacking axes. Although both of these compounds have a coplanar tilt of the dipole arrangement, the slip angle (q) is 36° for the CF3 derivative and 65° for the methyl derivative, which, according to Kasha exciton theory, corresponds to the classical examples of J-type (q < 54.7) and H-type (q > 54.7) packing.[90] The formation of J- and H-aggregates of BODIPY is also discussed in Section 3.5.1.2.
In 2017, Jiang et al.[91] proposed a one-pot method for the synthesis of 8-CF3-BODIPY 24 and 25 directly from α-unsubstituted pyrroles in the presence of 1 equiv. of trifluoroacetic acid and phenyltrichlorosilane (PhSiCl3). The key step in this approach is the reaction of TFA and PhSiCl3 with in situ chloranhydride formation (Scheme 20).

Following this approach, in 2020 Xi et al.[34] synthesized BODIPY 24 with a freely rotating meso-CF3 group in the design of a highly efficient BODIPY-based PTT agents (Scheme 21).
2.8. Preparation of boron-substituted BODIPYs
BODIPYs substituted at the boron atom are of great interest as fluorophores due to the fact that the introduction of substituents on the boron atom causes an increase in the Stokes shift and fluorescence quantum yields, increases the photostability and water solubility of the compounds, and allows the J-aggregation ability of BODIPY dyes to be controlled.[12, 92, 93] The main method of modification of boron atom in BODIPY is the nucleophilic substitution of fluoride anions. Section 3.7 is devoted to such reactions. Herein we discuss an alternative approach, the complexation of dipyrromethane with organoboron derivatives.
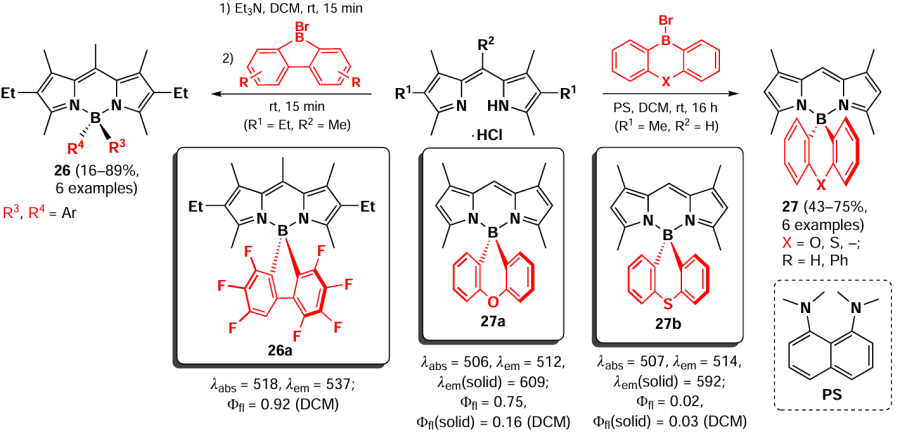
In 2009, Bonnier et al.[94] reported the synthesis of boron-substituted BODIPY 26 by complexation of dipyrromethene with perfluoroaryl borohalides such as C6F5BF2 and (C6F5)2BCl, as well as with perfluorinated 9-bromoborafluorene (Scheme 22). The resulting spiro-BODIPY 26 exhibited bright fluorescence due to the presence of a rigid chelating moiety. In 2017, Yuan et al.[95] proposed a method for the synthesis of boron-substituted BODIPY 27 by the reaction of dipyrromethene with various diaryl bromoboranes in the presence of a proton sponge (PS), 1,8-bis(dimethylamino)naphthalene. For BODIPY 27a the fluorescence quenching due to the heavy atom effect was shown, while for the oxygen-containing BODIPY 27b high fluorescence quantum yields and a large Stokes shift in the solid state were observed.
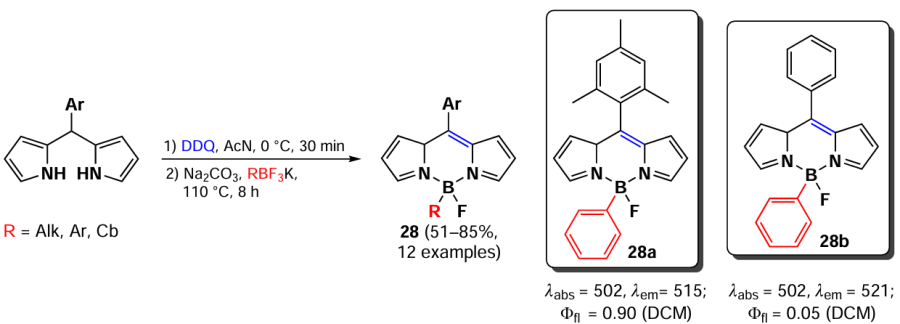
In 2019, Wang et al.[96] proposed a method for the synthesis of mono-β-aryl-substituted BODIPY 28 from organotrifluoroborates. The complexation of dipyrromethene with aryl-, vinyl-, and alkyltrifluoroborates was carried out in the presence of sodium carbonate as a base (Scheme 23). The photophysical properties of BODIPY 28 were identical to those of the BF2-analogs.
In 2016, Saikawa et al.[13] synthesized boron-dipyrromethene macrocycles in which the boron atoms are coordinated by two nitrogen atoms and two oxygen atoms (N2O2) via the reaction of macrocyclic dipyrromethene with phenylboronic acid. By varying the amounts of phenylboronic acid, a mono- (29) or binuclear macrocycle (30) can be obtained (Scheme 24; the value of the circularly polarized luminescence is also given under structure 30, see Section 3.9 for details).
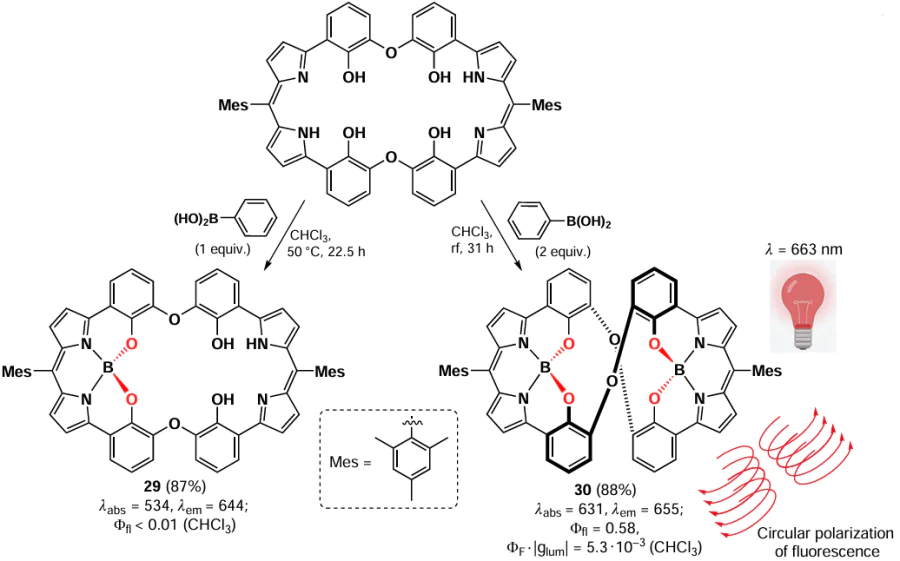
The fluorescence of mononuclear BODIPY 29 was almost completely suppressed by the PET effect, while for the binuclear complex 30 bright fluorescence (Ffl = 0.58) was observed due to its rigid structure and the binding of the oxygen lone pair. X-ray diffraction analysis revealed a twisted configuration for the binuclear complex 30, explaining the high circular polarization of the fluorescence (FF|glum| = 5.3 × 10–3) in the NIR band (λ = 663 nm). Fluorophores with high circular polarization are of interest due to the high emission resolution, which is important for bioimaging and organic electronics.[97]
2.9. Analysis of BODIPY core synthesis methods
The BODIPY core synthesis methods that were developed over the last three decades are now widely used. Given the large number of publications dedicated to these methods, it is reasonable to conclude that a specific method is optimized for solving specific tasks. Thus, the classical method of acid-catalyzed reaction of aromatic aldehydes with pyrroles [48, 49, 51, 52, 56] (see Section 2.1.) is a universal way to synthesise symmetric BODIPYs with an aromatic substituent at the meso-position. In contrast, symmetrical meso-alkyl derivatives of BODIPY are usially synthesized by reaction of acyl chlorides with pyrroles (see Section 2.2.).[60, 63, 65] A convenient method for the direct synthesis of symmetric NIR-absorbing BODIPYs is the POCl3-catalyzed self-condensation of pyrroles (see Section 2.4),[70, 79] but it is complicated by the low availability of pyrrolecarbaldehydes.
It is worth noting that the POCl3-catalyzed condensation of acylpyrroles and pyrroles is the only method for the preparation of asymmetric BODIPYs with different substituents in the pyrrole scaffold (see Section 2.3).[66, 71] The synthesis of BODIPY from two different scaffolds allows precise adjustment of the photophysical properties of the dyes and further selective functionalization of the pyrrole moieties.[22, 78]
A number of approaches could be utilized for the synthesis of BODIPY with an unsubstituted meso-position, including the above-mentioned self-condensation of acylpyrroles and condensation of pyrroles with acylpyrroles (see Sections 2.3 and 2.4).[67, 75] The use of orthoesters as reagents is also worth mentioning, as it yields meso-H-BODIPY from the available pyrroles with a high yield.[84]
A number of approaches leading to BODIPYs with specific meso-substituents should be mentioned. Thus, bis-(1H-pyrrol-2-yl)-methanethione is a key precursor for both the synthesis of 8-thioalkyl- (see Section 2.5.1) [80] and 8-halogen-BODIPY (see Section 2.5.2),[82] and to date no alternative approaches to the synthesis of meso-substituted BODIPYs have been developed. Among a number of methods leading to 8-CF3-BODIPYs, a reaction based on the in situ formation of trifluoroacetyl chloride by reaction of PhSiCl3 with TFA is worth mentioning.[91] This approach avoids the use of difficult-to-access CF3-containing precursors, such as 2-trifluoroacetylpyrroles, and uses the common substrate for BODIPY synthesis, 2,4-dimethylpyrrole. (see Section 2.7.).
The replacement of the boron trifluoride etherate with other organoboron compounds in complexation of dipyrromethenes opens the way to the direct synthesis of boron-substituted BODIPYs.[94] Such derivatives have valuable properties, such as solid state fluorescence,[95] as well as high circular polarization emission.[13]
Thus, in order to obtain a BODIPY of a given structure, one can choose the optimal method among the many described in the literature.
3. BODIPY core modification methods and their products
3.1. Substitution reactions in the BODIPY core
Analysis of the mesomeric structures of BODIPY reveals that the electron density is unevenly distributed between the atoms in the BODIPY core (Fig. 2).[9, 10] The electron density on carbon atoms in BODIPY decreases in the following series: positions 2 and 6 >> 1 and 7 > 3 and 5 >> 8. This sequence shows the change in regioselectivity in electrophilic substitution reactions and demonstrates the regioselectivity of nucleophilic substitution reactions in the reverse order. However, carbon atoms at positions 1 and 7 compared to positions 3 and 5 often exhibit less activity due to steric hindrance in the presence of a substituent at position 8.[98, 99] Therefore, according to the electron density distribution in the BODIPY core and the influence of steric hindrance, the rate of electrophilic substitution reactions decreases from carbon atoms at positions 2 and 6, to C3 and C5, and lastly to positions 1 and 7.[10]
![[{"id":"LhsHJO0rRI","type":"paragraph","data":{"text":"Regioselectivity in electrophilic substitution reactions in BODIPY"}}]](/storage/images/resized/6hYebEYnRl0xzuXZUdxh0ssPpy1Y0VLhkNLvgtan_xl.webp)
The resonance structure of the BODIPY core reveals that carbon atoms at positions 1, 3, 5, 7, and 8 are electron deficient, and thus can be involved in nucleophilic substitution reactions (Fig. 3). The rate of nucleophilic substitution reactions in BODIPY decreases from carbon atom at the meso-position, to less active C3 and C5, and to positions 1 and 7.[100] The decreased reactivity of carbon atoms at positions 1 and 7 compared to positions 3 and 5 is explained by their lower HOMO coefficients.[101]
![[{"id":"ugIn1rWgTe","type":"paragraph","data":{"text":"Regioselectivity in nucleophilic substitution reactions in BODIPY"}}]](/storage/images/resized/7Mu2eUDJ8CQmaZvUEnGMDApwvixURaIY8tMtYmbT_xl.webp)
3.2. Halogen substituted BODIPY
Synthesis of halogen-substituted BODIPYs could be conducted by several synthetic methods. The most common path involves electrophilic halogenation reactions (path I). Moreover, numerous synthetic methods based on nucleophilic substitution reactions (path II) or pre-functionalization of initial pyrroles and dipyrromethanes (path III) have been observed.
The regioselective synthesis of 3,5-dihalogen-BODIPY via electrophilic substitution reactions is not feasible. These derivatives can be obtained either through the pre-modification of the initial pyrrole or dipyrromethane or via nucleophilic substitution (Scheme 25; the amount of halogenating reagent is given in brackets).
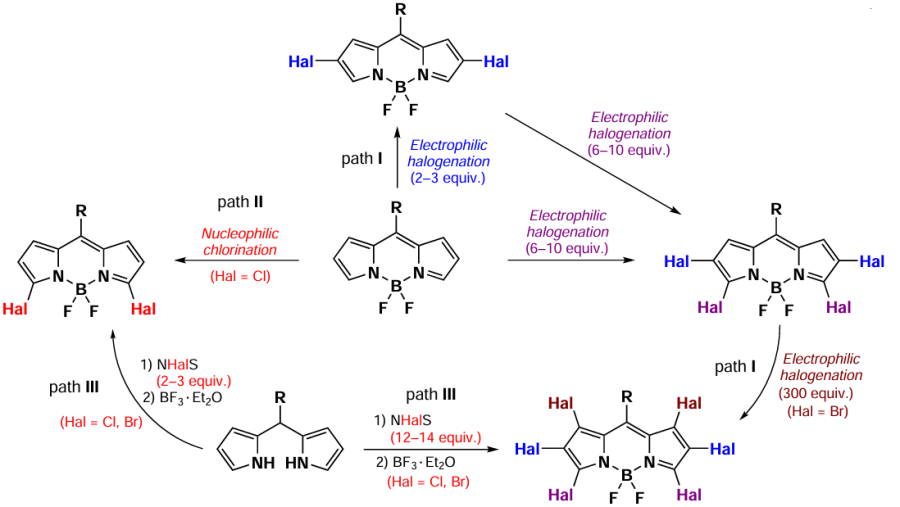
This section reviews electrophilic substitution reactions in BODIPY core, and different methods for synthesis of halogenated BODIPYs, such as nucleophilic halogenation and the pre-functionalization of the parent pyrroles and dipyrromethenes. The photophysical properties of halogen-substituted BODIPYs and their potential applications as photosensitizers in photodynamic therapy (PDT) are also discussed.
3.2.1. Photophysical properties of halogen-substituted BODIPYs
Modification of the BODIPY core with heavy halogen atoms has a great impact on their photophysical properties. Fluorescence is a spin-resolved radiation-induced deactivation of the S1 state to S0, which competes with non-radiation-induced deactivation of the S1 state to S0 (internal conversion, IC) or to T1 (ISC) (Fig. 4). The heavy atom enhances the probability of a spin-forbidden S1→T1 transition and intercombination conversion, which is known as ‘heavy atom effect’.[102, 103] The relaxation of the selection rule is due to the SOC effect, namely due to the interaction of the electron spin with the orbital magnetic moment. Thus, the modification of BODIPY with heavy atoms leads to an increase in SOC and hence ISC.[104]
![[{"id":"cSZEkaON8T","type":"paragraph","data":{"text":" Jablonski diagram: upon excitation, the molecule turns to a singlet excited state (S<sub>1</sub>) from which radiative relaxation (fluorescence), irradiation-free relaxation (IC), and spin-forbidden intercombination conversion (ISC) to the triplet excited state T<sub>1</sub> are possible"}}]](/storage/images/resized/0AqxcKXObGdpPV3d2Wwo7oIoZaeT4GPTgXJ6v7jQ_xl.webp)
The ‘heavy atom’ effect can be observed in BODIPYs directly bounded to the halogen atom, thus allowing the highest occupied molecular orbital (HOMO) and/or the lowest unoccupied molecular orbital (LUMO) of the molecule to extend to the electrons of the halogen atom. However, this effect is not observed in BODIPYs with halogen atoms in the para-position of the meso-phenyl group, and the photophysical properties of these BODIPYs are similar to those of non-halogenated BODIPYs.[105]
Therefore, the photophysical properties of non-halogenated (e.g., compound 31) and halogen-substituted BODIPYs 32 – 42 differ significantly (Fig. 5). The dihalogenation of BODIPY results in a bathochromic shift of the absorption maximum by 28 – 50 nm. For meso-aryl-BODIPY 31 this shift occurs from 500 nm to 538 – 551 nm for 2,6-dihalogen-BODIPY 32, 36 and 40. As the atomic number of the halogen increases (I > Br > Cl), both the absorption and emission maxima shift to the red region of the spectrum.[99, 106-109] The modification of the BODIPY core with four bromine atoms into the BODIPY core results in a shift of λabs of approximately 16 nm (to 554 nm) in the 2,3,5,6-tetrabromo derivative 38 in comparison to the 2,6-dibrom BODIPY 36. Moreover, further bromination of BODIPY 36 results in hexabromo-BODIPY 39, which exhibits a slight hypochromic shift due to steric hindrance to the rotation of the meso-aryl substituent.[103]
The 2,3,5,6-tetraiodo-BODIPY 42 exhibits a substantial bathochromic shift in the absorption maximum in comparison with the 2,6-diiodo BODIPY 40 (λabs = 581 and 548 nm, respectively). The 2,6-dihalogen-BODIPYs exhibit a Stokes shift of 16 – 27 nm, which is similar to that observed for unsubstituted BODIPYs. In contrast, the Stokes shift decreases to 11 nm for 1,2,3,5,6,7-hexahalogen-BODIPYs.[100, 107] Meanwhile, the halogen-free meso-aryl-BODIPY 31 is characterized by low fluorescence quantum yield due to radiation-free relaxation, which is generally < 0.1, as a result of the rotation of the meso-aryl substituent.
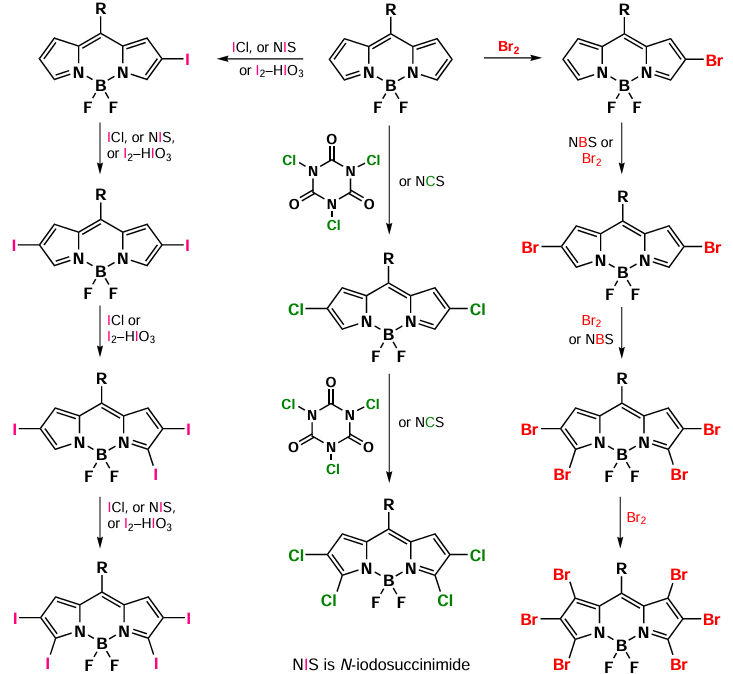
3.2.2. Electrophilic halogenation
N-Halogensuccinimides are the most common electrophilic agents for BODIPY halogenation and can be used under mild conditions (Scheme 26).[114-116] Halogenation of 1,2,3,3,5,6,7-unsubstituted BODIPY by the excess of NCS or N-bromosuccinimide (NBS) gives 2,3,5,6-tetrahalogenated derivatives.[117, 118] Similarly, polychloro-substituted BODIPYs (2,6-di- to 2,3,5,6-tetrachloro) can be synthesized by reaction with trichloroisocyanuric acid in acetic acid.[119, 120] The sequential synthesis of mono-, di-, tetra-, and hexabromo-BODIPY can be carried out with 2, 3, 6, and 300 equiv. of molecular bromine, respectively.[100, 121] The quantities of iodine chloride (5 equiv.), iodine (3.5 equiv.) and iodine trioxide (8 equiv.) or the iodine-iodine(III) oxide system in the ratio of 1 : 0.8, 2.5 : 2, 3.5 : 3 and 4.5 : 4, respectively, were employed (see Scheme 26). [99, 122-125]
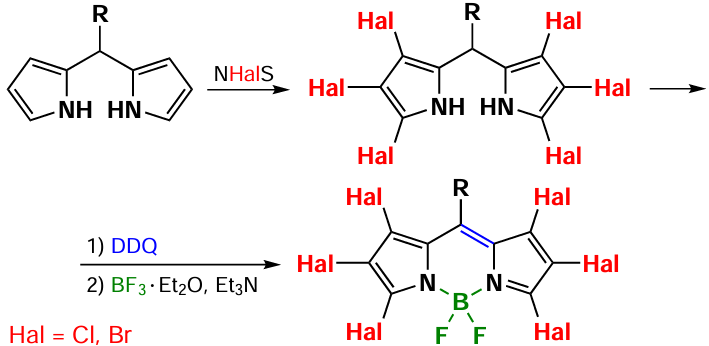
Hexachloro- and hexabromo-BODIPYs can also be synthesized by exhaustive halogenation of the initial dipyrromethane, followed by oxidation and complexation. In this case, the halogenation reaction of dipyrromethane requires the use of a modest excess of the halogenating agent, which facilitates the formation of hexachlorodipyrromethane in the presence of 14 equivalents of NCS,[107] and hexabromodipyrromethane in the presence of 10 equivalents of NBS [126] (Scheme 27).
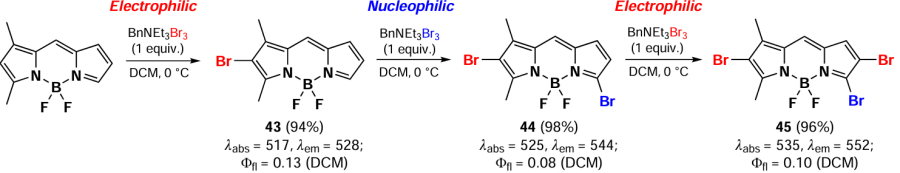
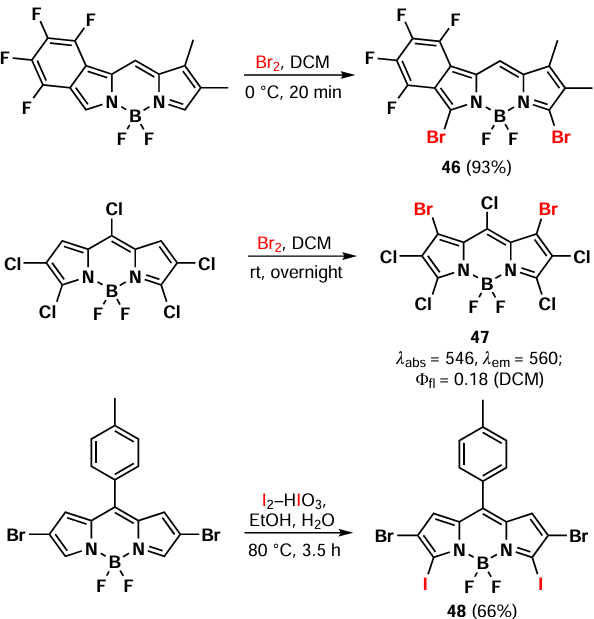
In 2012, Li et al.[127] reported an unusual regioselectivity of BODIPY bromination in the reaction of benzyltriethylammonium tribromide with 1,3-dimethyl-BODIPY. Monobromo-BODIPY 43 was synthesized by the electrophilic substitution at position 2, and following nucleophilic attack of the bromide anion at position 5 led to the formation of the 2,5-dibromo-BODIPY 44. This compound was also capable of further electrophilic bromination yielded in BODIPY 45 (Scheme 28).
The relative reactivity of the BODIPY’s carbon atoms is maintained during electrophilic substitution, provided that the C(2) and C(6) carbon atoms are not accessible to electrophilic attack. Consequently, bromination of 2,6-disubstituted BODIPY with molecular bromine occurs at positions 3 and 5, yielding BODIPY 46. Conversely, bromination of 2,3,5,6-tetrasubstituted BODIPY leads to the substitution at positions 1 and 7, yielding BODIPY 47.[74, 128] Furthermore, iodination of 2,6-dibromo-BODIPY by the I2 – HIO3 system selectively yields 3,5-diiodo-BODIPY 48 (Scheme 29). [129]
3.2.3. 2,6-Dihalogenated BODIPY as PDT agents
Increasing the population of the T1-level of BODIPY by introducing halogen atoms into the molecule is a widely used strategy in the development of drugs for photodynamic therapy (PDT) and photothermal therapy (PTT).[114, 130-132] In the triplet excited state, the photosensitiser is capable of reacting with biomolecules with radical anion formation, which subsequently reacts with water or oxygen to generate a superoxide anion or hydroxyl radical (type I PDT agent). An alternative process involves the reaction of BODIPY in the triplet excited state with oxygen, resulting in the formation of singlet oxygen (PDT agent type II). [133]
Iodinated BODIPYs are most often considered as photosensitising agents which typically exhibit high quantum yields of singlet oxygen (FD = 0.5 – 0.8). Furthermore, the photosensitising properties of such compounds are almost independent of the number of halogen atoms or their position in the BODIPY core.[99, 134] Bromo-substituted BODIPYs are also capable of acting as photosensitisers, but their singlet oxygen quantum yields are generally lower than those of their iodinated analogues, with values ranging from 0.1 to 0.6.[22, 135, 136] The fluorescence of 1,3,5,7-tetramethyl-BODIPYs is markedly diminished by the incorporation of halogen atoms, with the effect increasing in accordance with the halogen order number (see structures 49 – 52 in Fig. 6).
In 2021 Won et al.[132] synthesised a NIR-absorbing PDT agent based on 2,6-dibromo-3,5-distyryl-BODIPY 53 and an ethacrynic acid-based glutathione-S-transferase inhibitor (Fig. 7).
![[{"id":"mCrwmBrU1S","type":"paragraph","data":{"text":"Structure of a PDT agent based on 2,6-dibromo-3,5-distyryl-BODIPY <b>53</b> and schematic representation of its <i>in vivo</i> action. The figure created by the authors based on the data of the publication[[ type=\"anchor\" referenceId=\"18131\" ]]."}}]](/storage/images/resized/uTS6XpI8Y7a0istKTsZHasTb2LDKMk8T9ai7QxGd_xl.webp)
BODIPY 54 was capable of generating both singlet oxygen (type II) and NO* and ROO* (type I) species, which were detedted using of different traps. Conjugate 54 was also capable of reduction the survival of the GST-pi-expressing cell line MDA-MB-231 in the absence of light at a concentration of 5 μM, with a subsequent decrease to 30% upon irradiation with a 660 nm laser (specific power 100 mW cm–2, 10 min). In an in vivo antitumour efficacy study in mice bearing MDA-MB-231 tumour, a statistically significant reduction in tumour growth was observed after four injections of compound 54, followed by laser irradiation (λ = 660 nm, 2 W cm–2, 10 min) at week 9 of therapy.
In 2021, Tian et al.[137] obtained 2,6-diiodo-BODIPY 55 and 56 and prepared nanoparticles (NPs) based on 56 by encapsulation in poly(oligoethylene glycol methacrylate)-polyaspartic acid (POEGMA23-PAsp20), with photodynamic therapy (PDT) activity (Fig. 8). Both BODIPYs 55 and 56 exhibited absorption in the near-infrared (NIR) range, with a hypochromatic shift of the absorption and emission maxima and quenching of fluorescence due to intersystem crossing (ICT) being observed with increasing solvent polarity. Furthermore, both fluorophores demonstrated high quantum yields of singlet oxygen.
![[{"id":"4Qc292qBTz","type":"paragraph","data":{"text":"Synthesis and structure of NIR-absorbing 2,6-diiodo-BODIPY <b>56</b> with PDT activity, and a schematic representation of the<i> in vivo</i> action of NPs based on it. The figure created by the authors based on the data of the publication[[ type=\"anchor\" referenceId=\"18136\" ]]."}}]](/storage/images/resized/q4spHIZL5xxSkfVAtQUFgLGKhRj4M57gZXc80FXG_xl.webp)
The study of antiproliferative activity on 4T1 breast carcinoma and HeLa cervical cancer cell lines revealed that the half-maximal inhibition concentration (IC50) of compound 56-based NPs was 0.1 μM under 660 nm laser irradiation (10 mW cm–2, 5 min) with no dark toxicity up to 0.3 μM. In vivo therapuitic efficacy study on mice with 4T1 tumours revealed complete tumour disappearance on the 20th day of therapy after three injections of NPs and irradiation with a 660 nm laser (40 mW cm–2, 15 min).
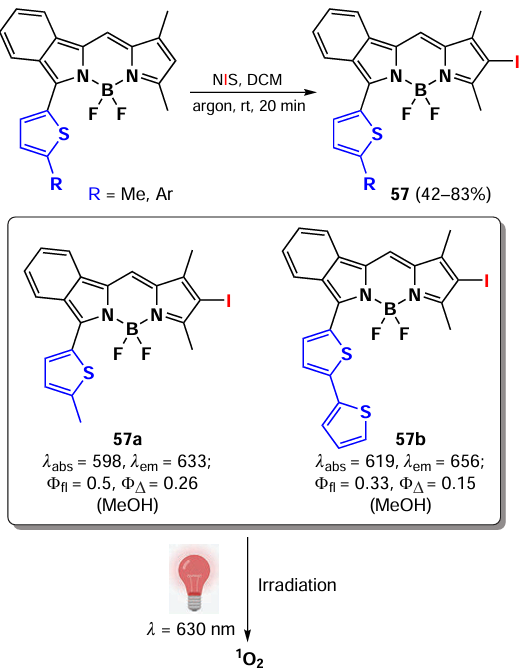
In 2023, Spingler et al.[78] synthesised a series of isoindole-based 6-iodo derivatives of BODIPY 57 (Scheme 30). BODIPYs 57a and 57b were able to form triplet states and generate singlet oxygen upon irradiation. BODIPYs 57 were non-toxic in the absence of light on HeLa cells, but when irradiated with 630 nm light (5 J cm–2), they exhibited IC50 values in the nanomolar concentration range. For instance, compound 57a demonstrated an IC50 value of 6 nm. The activity of BODIPY 57a and BODIPY 57b was not reduced under hypoxic conditions on HeLa cells, indicating an oxygen-independent mechanism of action. In particular, BODIPY 57a exhibited a hypoxia phototoxicity index exceeding 360 000. Intracellular localisation of BODIPY 57a revealed the intracellular formation of aggregates of varying sizes, which presumably exerted a photothermal effect upon irradiation.
A series of PDT agents based on cationic 2,6-dibromo-BODIPY (58a) and 2,6-diiodo-BODIPY (58b) were synthesised by Badon et al.[136] (Scheme 31). The calculation of the energy gap between the first singlet excited level (S1) and various triplet excited levels (T1 – T3) for BODIPY 58a,b demonstrated that intercombination conversion occurs via the S1 → T2 pathway.
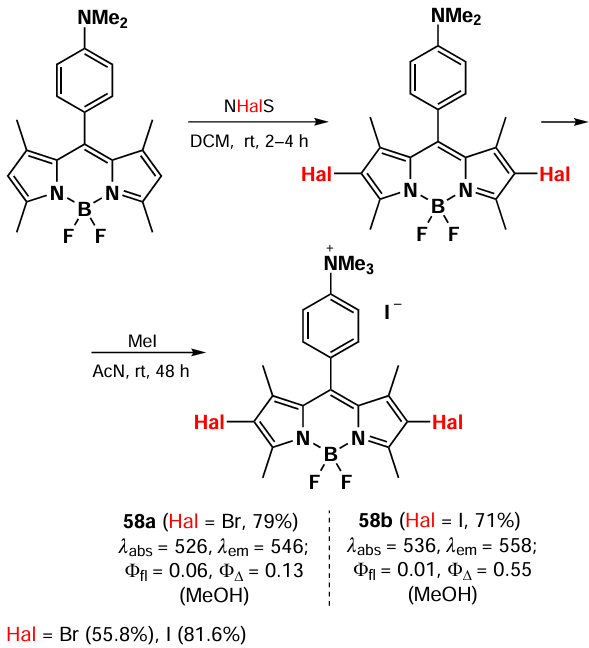
BODIPYs 58a,b were non-toxic to MCF-7 and HeLa cells in the dark, but under 520 nm laser irradiation (7 mW cm–2, 10 min) IC50 values were in the 48 – 60 nm range. Compounds 58a,b accumulated in mitochondria due to the presence of a charged quaternary ammonium group in the meso-aryl substituent.
In 2023, Jung et al.[138] reported an NIR-absorbing 2,6-dibromo-BODIPY conjugate 59 and an ROS-generating copper 59-CuII complex. This compound proved to be a promising agent for chemodynamic therapy (CDT) due to the presence of a vector fragment that targeted cancer stem cells by binding to CAIX9.[138] In the conjugate 59, the 2,6-dibromo-3,5-distyryl-BODIPY motif and the vector to CSC (cancer stem cell) acetazolamide were linked via a triazole linker (Fig. 9).
![[{"id":"97eqTwKbNE","type":"paragraph","data":{"text":"Structure of the copper complex based on 2,6-di-bromo-3,5-distyryl-BODIPY <b>59</b> with the vector to CSC acetazolamide and its<i> in vivo</i> action. The figure created by the authors based on the data of the publication[[ type=\"anchor\" referenceId=\"18137\" ]]."}}]](/storage/images/resized/J5yJti9NkRdnPgKxCAGfFhvCPEIfKjQcWPBLAzJ1_xl.webp)
The fluorescence of the 59-Cu complex was almost completely absent, and the rate of 1O2 formation was found to be 2.5 times lower under 660 nm laser irradiation (2 W cm–2, 10 min) compared to ligand 59. The 59-Cu complex demonstrated moderate activity against the MDA-MB-231 cell line in the absence of light, and high activity under 660 nm laser irradiation (100 mW cm–2, 5 min) in both normoxic and hypoxic conditions. In vivo experiments were conducted on mice bearing MDA-MB-231 tumors, which were administered three injections of the 59-Cu conjugate once a week and irradiated with a 660 nm laser (2 W cm–2, 10 min) for 12 weeks. A statistically significant reduction in tumor size was observed in the group of animals that received the complex compared to the group that received free ligand 59 and the control groups.
3.2.4. Nucleophilic halogenation of BODIPY at positions 3 and 5
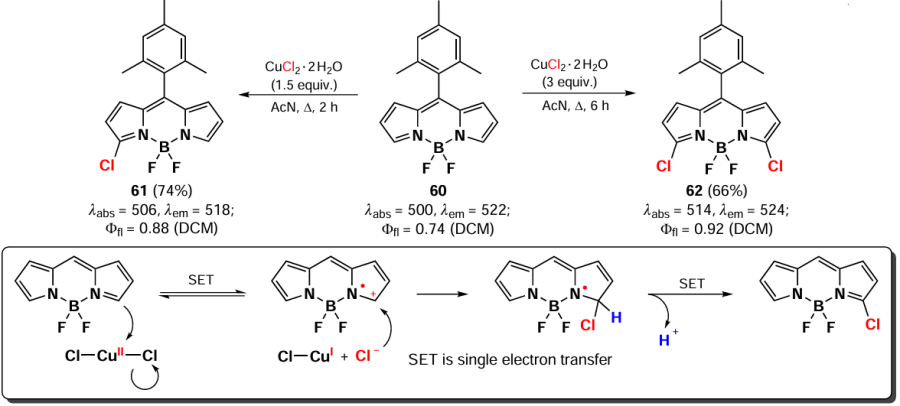
In 2015, Zhou et al.[139] described a convenient approach for chlorinating BODIPY at positions 3 and 5 by using CuCl2. This method is a one-electron oxidation of BODIPY 60 with copper(II) chloride taken in 50% excess, followed by nucleophilic addition of the chloride anion. This resulted in the formation of the monochloro-derivative 61 in 2 hours (Scheme 32). Increasing the amount of reagent to 3 equiv. and the process time to 8 hours gave 3,5-dichloro-BODIPY 62 in moderate yield.
In 2020, Frank et al.[140] developed a method for the synthesis of 3,5-dichlorosubstituted BODIPY 63 via nucleophilic substitution reaction by using the Cu(OTf)2 — TBACl system (Tf — trifluoromethanesulfonyl (triflyl), TBA — tetra-n-butylammonium). The presence of an acceptor nitro group in the meso-aryl substituent contributed to the increase in product yields up to quantitative yields (Scheme 33).
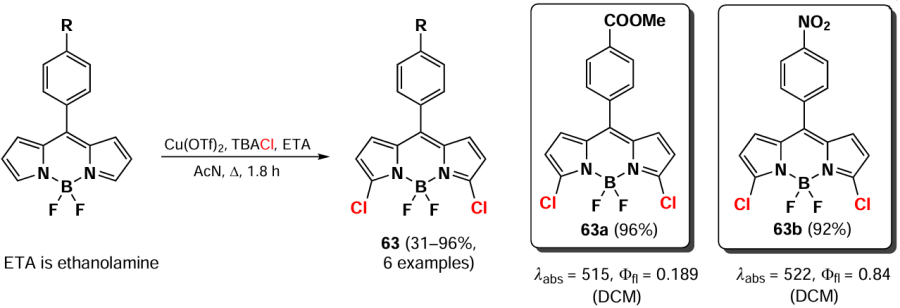
3.2.5. Synthesis of 3-mono- and 3,5-dihalogenated derivatives via pre-functionalization path
In a pre-functionalization procedure, a halogen atom can be introduced into the α-position of dipyrromethane in a regioselective manner to form α-halogen-BODIPY after oxidation and complexation reactions.[141] Using α-chlorosubstituted pyrroles 64 as starting compounds, BODIPY 65 substituted at positions 3 and 5 were obtained in yields of up to 90% (Scheme 34).[142]
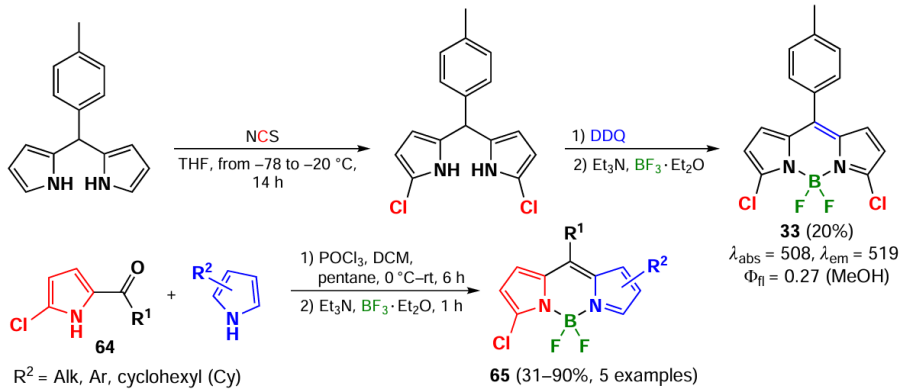
Pre-functionalisation of dipyrromomethane is also employed to obtain 3,5-dibromo-BODIPY 66. The most commonly used brominating agent for dipyrromomethane is NBS (Scheme 35).[143]
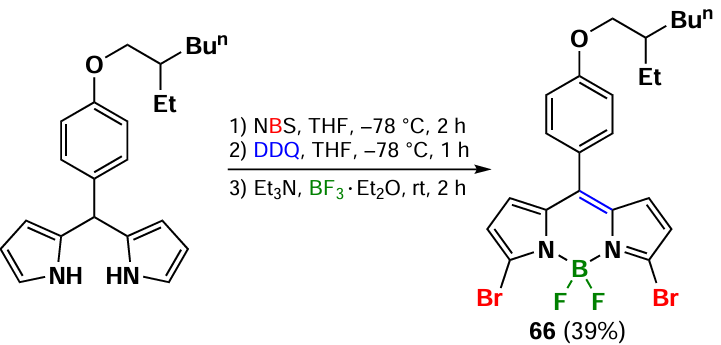
3,5-Dibromo-BODIPY can be synthesized from the corresponding bromo-substituted pyrroles. In 2014, Jiao et al.[76, 144] obtained 3,5-dibromosubstituted BODIPY 67 through the self-condensation of 5-bromo-3-methoxypyrrole-2-carbaldehyde (Scheme 36).
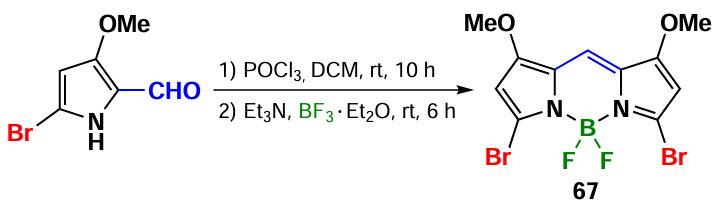
In 2021, Frank et al.[111] proposed to use the Filkinstein reaction to obtain 3,5-diiodo substituted BODIPY 68 from the corresponding dichloro- or dibromo-BODIPY. This reaction is typically employed in the synthesis of substituted BODIPYs. In the aromatic series, the Filkinstein reaction requires the use of a photo- or metal catalyst. However, due to the propensity of 3,5-dihalogenated BODIPY to undergo nucleophilic substitution, it was possible to carry out the Filkinstein reaction without a catalyst (Scheme 37).
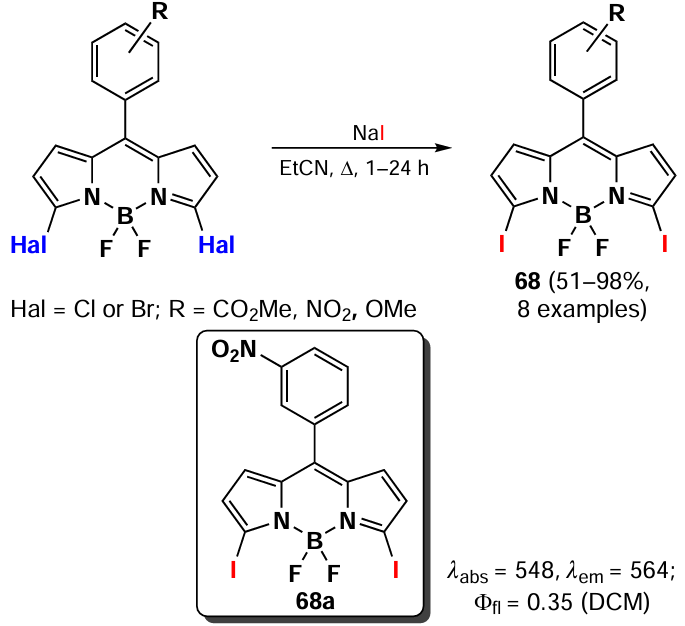
3.2.6. Fluorination of the BODIPY core
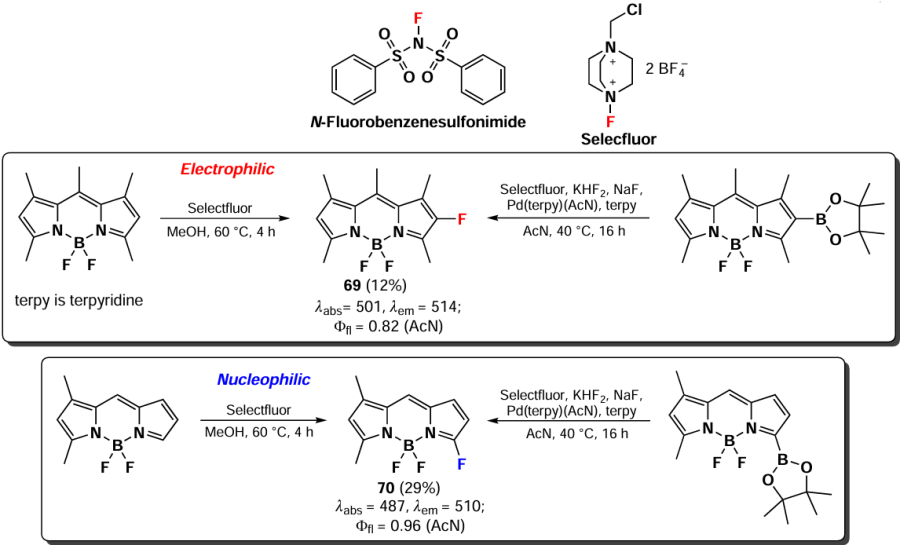
Direct electrophilic fluorination of the BODIPY core is feasible through the use of electrophilic fluorinating agents, including N-fluorobenzenesulfonimide and bis(tetrafluoroborate) 4-fluoro-1-chloromethyldiazoniabicyclo[2,2,2,2]octane (commercially named Selecfluor). Therefore, in 2016, Huynh et al.,[145] reported the fluorination of BODIPY via using the Selecfluor reagent. In the case of blocked positions 3 and 5, the electrophilic fluorination reaction proceeded at position 2 of BODIPY. Conversely, when α-unsubstituted 1,3-dimethyl-BODIPY was subjected to the same reaction, nucleophilic fluorination occurred at position 5.[139] BODIPY 69 and 70 exhibited bright fluorescence with a fluorescence lifetime of 0.8 to 1.0 nanoseconds. Given the low yields of the products in this process, an alternative indirect route for their synthesis was proposed.[146] This involved sequential reactions of halogenation, preparation of pinacol esters of boronic acid, and palladium-catalyzed fluorination, as previously described for aromatic substrates. However, the total yield of these transformations was only 1 – 2% (Scheme 38).
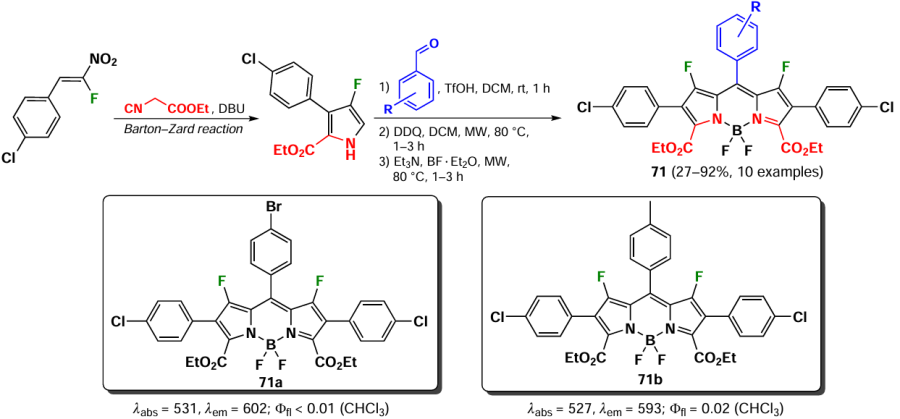
In 2024, Larkovich et al.[24] presented the synthesis of 1,7-fluorinated BODIPY 71 from fluorinated pyrroles obtained via the Barton-Zard method, which refers to the reaction of β-fluoro-β-nitrostyrenes with 2-ethylisocyanoacetate. The presence of an electron-accepting carboxyethyl group at positions 3 and 5 resulted in low fluorescence quantum yields (Ffl < 0.02) for these fluorophores. The introduction of a fluorine atom into the molecule had no significant impact on the absorption maximum in the spectrum, but led to an increase in the Stokes shift from 20 – 60 to 60 – 80 nm (Scheme 39).
3.3. Other reactions of electrophilic substitution in the BODIPY core
3.3.1. Sulfonation
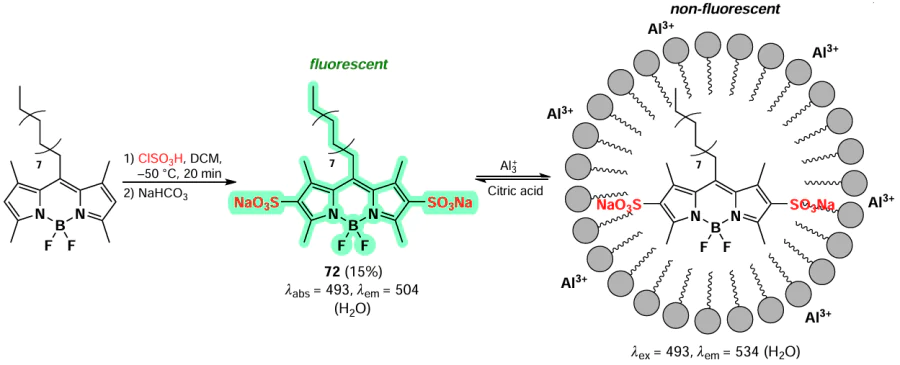
Electrophilic sulfonation reactions proceed with the formation of 2,6-substituted BODIPY under the action of such reagents as chlorosulfonic acid or SO3-pyridine complex. In 2015, an amphiphilic sensor for the detection of Al3+ cations based on BODIPY was synthesized through sulfonation of 1,3,5,7-tetramethyl-BODIPY at positions 2 and 6.[147] The presence of sulfo-groups in BODIPY 72 resulted in bright fluorescence and good solubility in water. The long aliphatic substituent at the meso-position facilitated micelle formation in aqueous solution. The fluorescence of this compound was quenched in the presence of Al3+ions due to the formation of micelles. Conversely, the addition of citric acid led to the destruction of micelles and an increase in fluorescence, with a detection limit of 5 μM (Scheme 40).
The introduction of sulfonate groups at positions 2 and 6 of benzylated 1,3,5,7-tetramethyl-8-pyrocatechin-BODIPY to form compound 73 was carried out in 2014 by Kim et al.[148] After removing the protecting groups, a water-soluble fluorescent probe for HClO detection was obtained (Scheme 41). The fluorescence of compound 74 was observed to be low due to the occurrence of resonance energy transfer (RET) from the donor fragment of pyrocatechin to the BODIPY core. In the presence of HClO or ClO–, pyrocatechin was oxidized to benzoquinone, which suppressed the RET and caused the fluorescence enhancement. BODIPY 74 demonstrated high selectivity for hypochlorite and did not exhibit fluorescence ignition in the presence of other oxidants, such as H2O2 and tert-butyl peroxide.
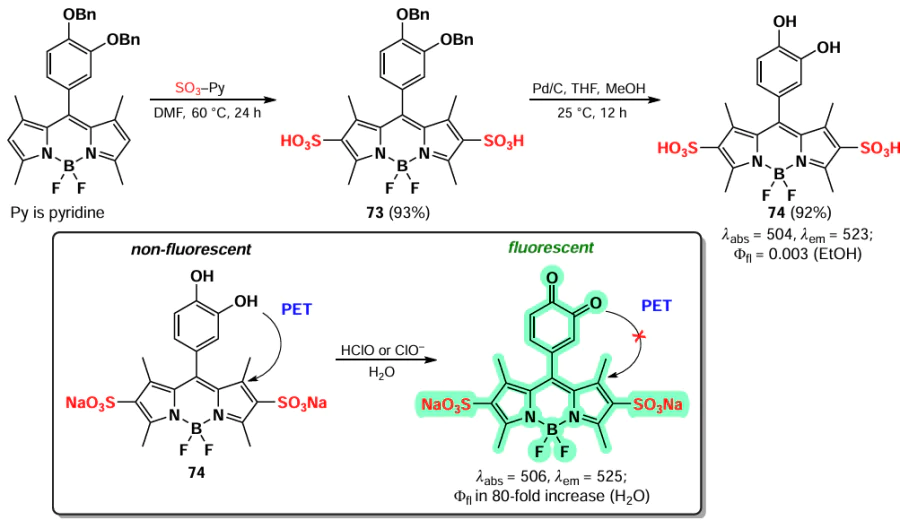
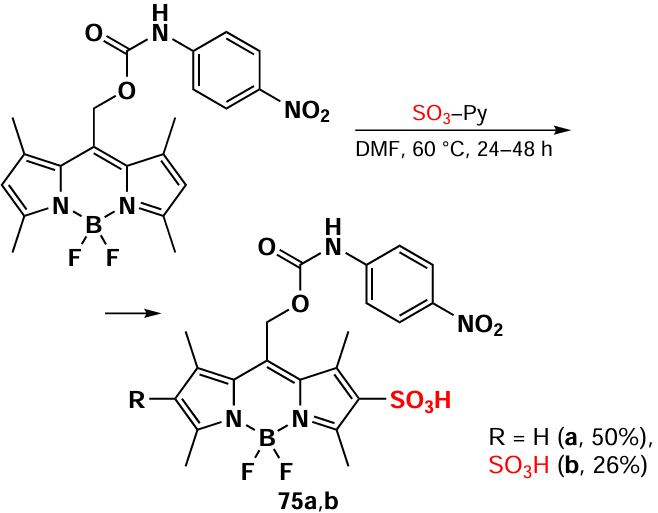
As part of the development of BODIPY-based photo-removable protecting groups, in 2020 Kand et al.[149] synthesized 2-sulfo- and 2,6-disulfo-BODIPYs 75a,b via using an SO3-pyridine complex (Scheme 42). The introduction of strong electron-acceptor sulfonate groups at positions 2 and 6 of BODIPY 75 led to a notable increase in the photolysis barrier of the C – O bond at the meso-position. This resulted in the inability of such compounds to release leaving groups upon irradiation.
3.3.2. Nitration
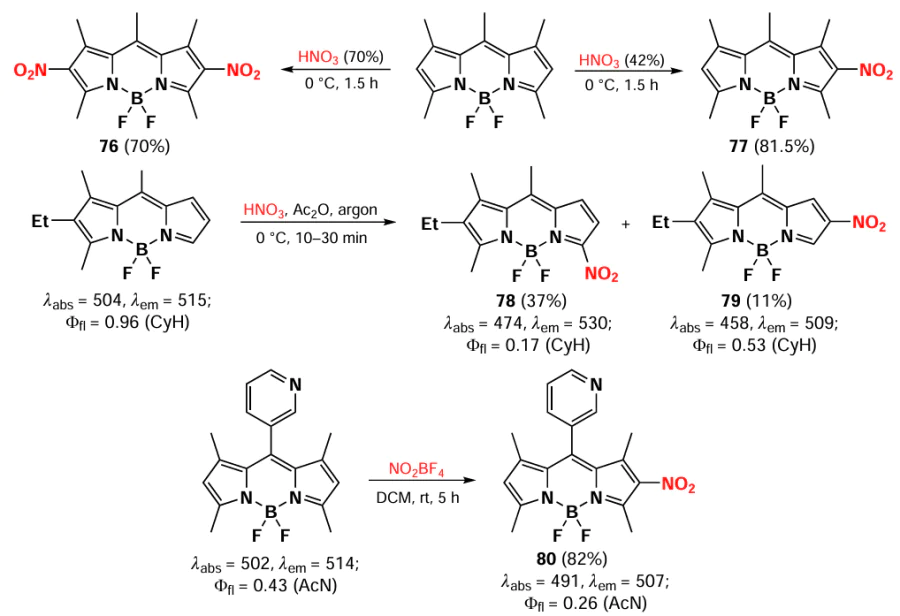
The nitration of BODIPY at positions 2 and/or 6 can be carried out with nitric acid solutions of different concentrations, resulting in the formation of mono- or disubstituted products. The use of concentrated (70%) acid leads to the formation of the disubstituted product 76, whereas the use of dilute (40%) acid gives the mono-substituted 2-nitro-BODIPY 77.[150, 151] Nitration of 1,2,3,8-tetraalkyl-BODIPY with nitric acid in the presence of acetic anhydride leads to a mixture of products 78 and 79 with a nitro group at positions 5 and 6.[152] In 2023, Ndung’U et al.[153] demonstrated that it is possible to introduce a nitro group into BODIPY at position 2, leading to the formation of product 80, using NO2BF4. Nitro-BODIPYs are characterized by a hypsochromic shift of the absorption maximum and quenching of fluorescence compared to the parent BODIPY due to ICT (Scheme 43).
3.3.3. Nitrosation
The nitrosation of fluorescent molecules represents a promising avenue in the design of sensors for the sulfide anion, which is able to reduce nitroso compounds to the corresponding amines.[154] In 2020, Gil de Melo et al.[155] presented a novel method for the nitrosation of BODIPY under mild conditions in the presence of NOBF4, leading to the formation of compounds 81a – c and 82a – e. Reactions with 1,3,5,7-tetramethyl-BODIPY yielded 2-substitution products 82a – d; nitrosation of 3-phenyl-BODIPY led only to compound 82e, an electrophilic substitution product at position 3. The regioselectivity of the nitrosation at position 2 can be explained by the preferential formation of a six-membered σ-complex intermediate stabilized by the tetrafluoroborate anion (Scheme 44).
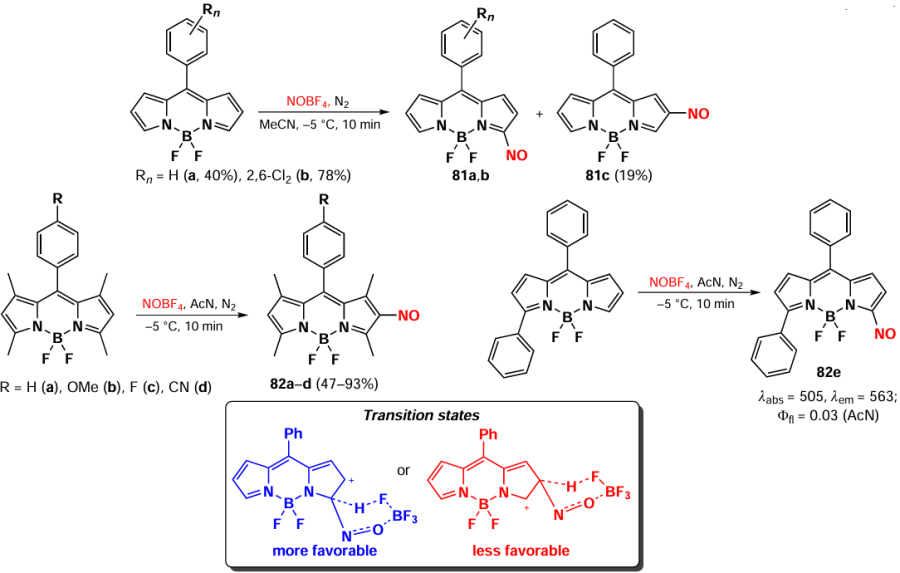
2-Nitroso-substituted BODIPYs exhibit a significant hypsochromic shift of absorption maxima, accompanied by a reduction in fluorescence intensity in polar solvents, probably due to ICT.
3.3.4. Formylation
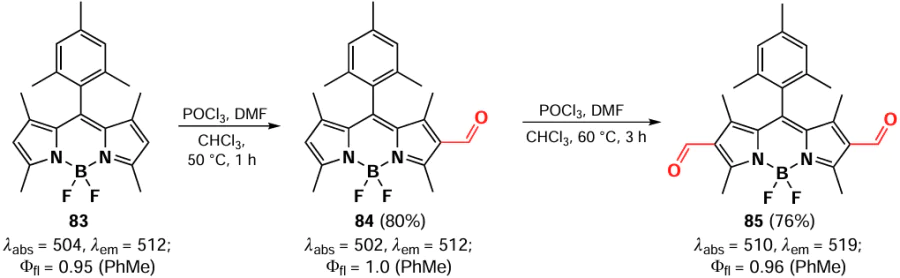
The formylation of BODIPY at positions 2 and 6 is usually conducted via the classical Wilsmeyer – Haack reaction.[105, 156, 157] The process is initiated with the formation of a monoformylation product, typically using 1,2-dichloroethane as a solvent.[158] However, the synthesis can also be conducted in DMF.[159] The photophysical properties of 2-formyl-BODIPYs are nearly identical to those of the unmodified parent BODIPYs.[105] To obtain BODIPY 83 diformylated at positions 2 and 6, the reaction is carried out in two successive steps. The introduction of a second formyl group requires an excess of electrophilic agent, long reaction times and high temperatures.[160, 161] In contrast to the formylation product of 2-formyl-BODIPY 84, 2,6-diformyl-BODIPY 85 exhibits a minor bathochromic shift in absorption (Scheme 45).[160]
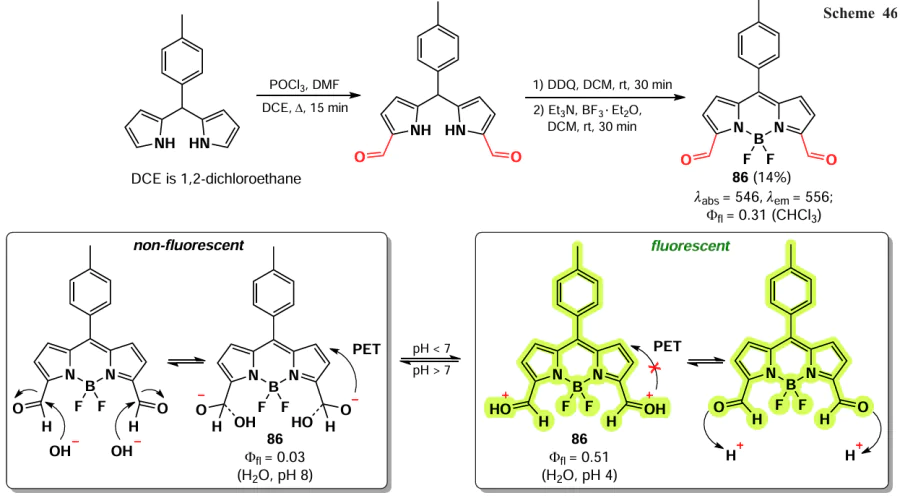
The synthesis of 3,5-diformyl-BODIPY 86 involves the pre-functionalization of the parent dipyrromethane by the Vilsmeyer reaction, followed by oxidation and complexation.[162] The resulting compound exhibits a bathochromic shift of 40 nm. In an alkaline solution, the aldehyde group of BODIPY 86 is a PET-quencher of the excited state of the nucleus, but in an acidic medium, PET is suppressed due to protonation of the aldehyde group, resulting in an up to 17-fold increase in fluorescence at pH 4 compared to the intensity at pH 7 (Scheme 46).
In 2018, Yeung et al.[163] proposed the radical reaction of electron-deficient heterocycles using trioxolane as an equivalent of formyl group (Scheme 47).
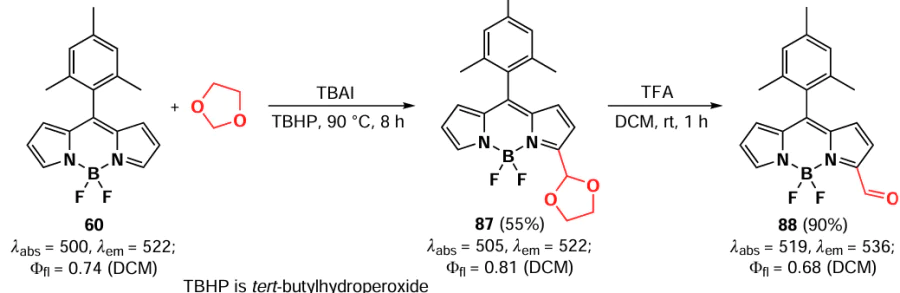
A year later, Fan et al.[164] described the formylation of BODIPY 60 at positions 3 and 5 by radical reaction with an unsubstituted 1,3-dioxolane. The transformation involved radical addition steps to generate the intermediate 87 and removal of the protecting group, resulting in the product 88 (Scheme 48). A full discussion of the radical reactions of BODIPY can be found in Section 3.6.
3.3.5. Acylation
The classical Lewis acids-catalysed Friedel – Crafts acylation is not applicable to the functionalisation of BODIPY, as the presence of protonic Brønsted acids facilitates the removal of the BF2 group.[165] Conversely, the Lewis acid AlCl3, which is commonly employed in electrophilic aromatic substitution reactions, facilitates nucleophilic substitution reactions at the boron atom (see section 3.7).[166, 167]
In 2016, Mirri et al.[168] proposed the acylation of BODIPY under the action of BF3 – Et2O as a Lewis acid. BODIPY 20 was demonstrated to undergo Friedel – Crafts acylation at position 2 in the presence of acyl chlorides. BODIPY 89 (Scheme 49) exhibited a notable decrease in fluorescence quantum yield, from 0.95 to 0.6 – 0.8.
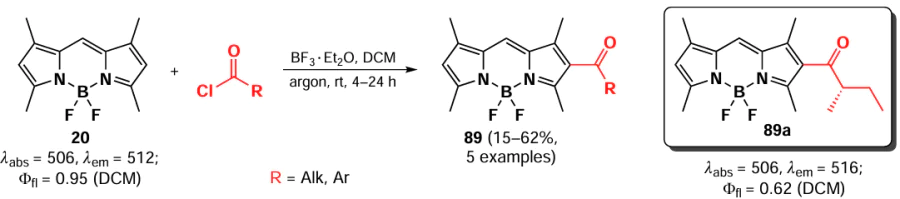
This approach was applied in 2020 by de Jong et al.[169] in the synthesis of fluorescent dyes 90 and 91, which are specific to the endoplasmic reticulum (ER). The highest co-localization ratio in the ER was observed for compound 90 (0.91), which can be attributed to the optimal ratio of lipophilic to amphiphilic parts in the molecule (Scheme 50).
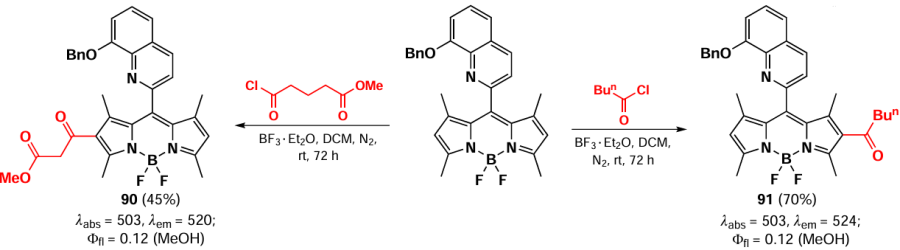
The modification of the molecule by an acyl group at positions 3 and 5 can also be accomplished by radical reaction with 1,3-dioxalanes, in a manner similar to the radical formylation described in Section 3.3.4. By varying the reaction time, mono- (92) and diacylation products (93) were obtained.[164] (Scheme 51).
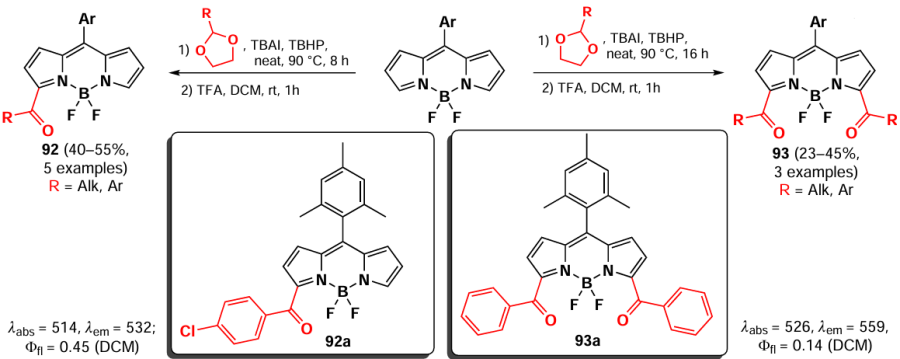
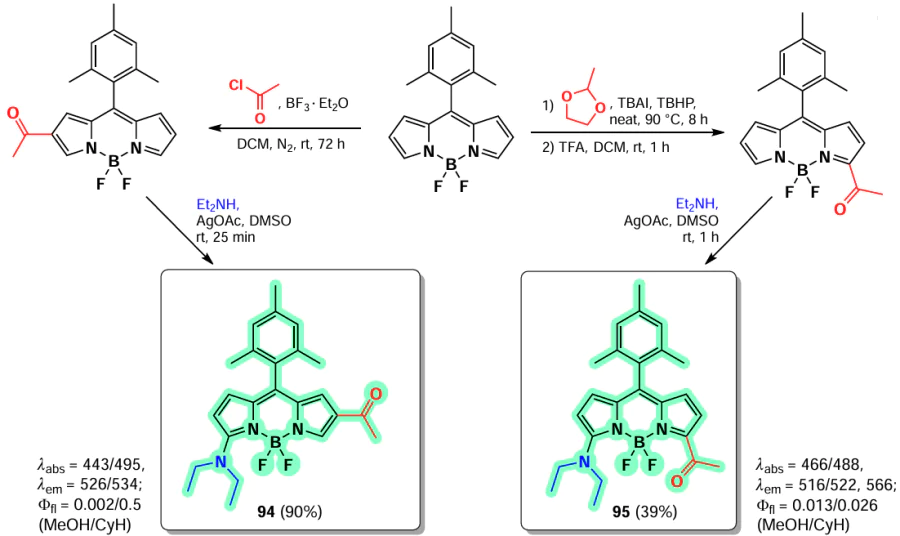
Both approaches to the acylation of BODIPY, radical and Lewis acid catalyzed, were used by Gonzalez-Vera et al.[170] in the synthesis of the fluorescent solvatochrome Prodan derivatives 94 and 95 (Scheme 52). The structure of solvatochrome dyes typically comprises an electron-donor and an electron-acceptor group. Upon photoexcitation, intermolecular charge transfer (ICT) from the donor to the acceptor occurs, resulting in an increased dipole moment in the excited state relative to the ground state. This makes the probe sensitive to environmental conditions, particularly solvent polarity. The emission in polar solvents was quenched by ICT for both BODIPY derivatives, 94 and 95. However, the former exhibited a higher degree of emission in non-polar solvents, while the latter demonstrated a comparatively reduced emission in polar solvents.
In 2023, Fan et al.[18] proposed a copper-catalysed method for the direct acylation of BODIPY at the α-position by aliphatic and aromatic aldehydes, yielding BODIPY 96 (Scheme 53).
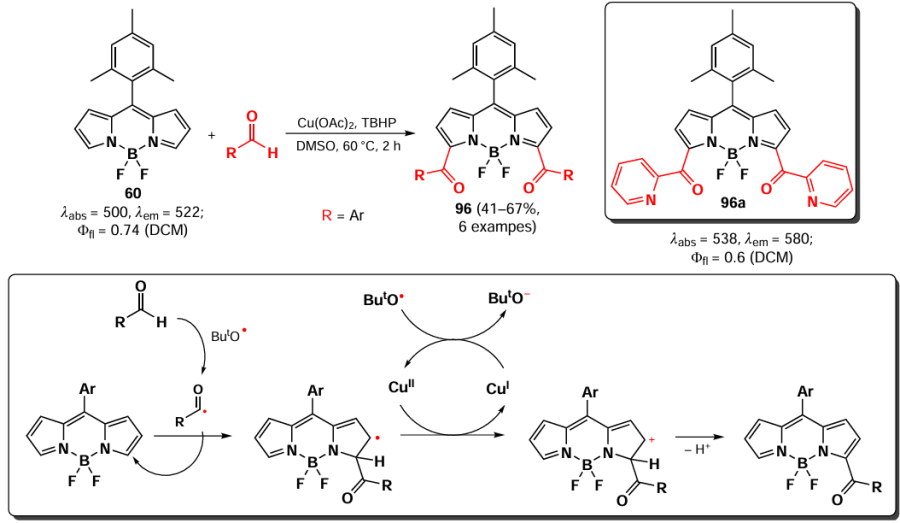
The authors demonstrated that the reaction involves radical particles. The presence of 2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) or 2,6-di-tert-butyl-4-methylphenol (BHT) was found to inhibit the reaction. The authors proposed a mechanism whereby the acyl radical attacks position 3 of BODIPY, subsequently undergoing oxidation to the cation by interaction with copper(II) ion. The resulting 3,5-diacylated BODIPY exhibited a slight bathochromic shift in the absorption maxima, accompanied by a reduction in fluorescence quantum yields.
3.3.6. Alkylation
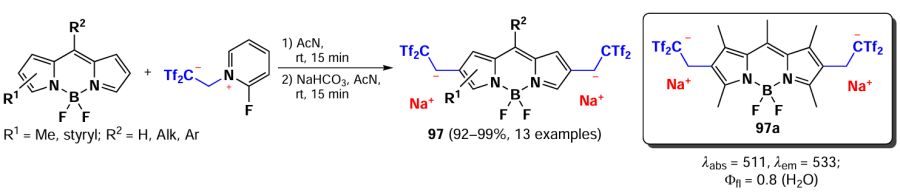
In 2021, Yanai et al.[171] synthesized carbanionic derivatives of BODIPY with high water solubility and good emission properties. The 2,6-dialkylation products 97 were obtained by alkylation with 2-fluoropyridinium ethanide to form in situ 1,1-bis[(trifluoromethyl)sulfonyl]ethene. The carbanionic BODIPYs 97 exhibited a minor bathochromic shift in their absorption and emission spectra. The relatively weak impact of the carbanionic substituent on the photophysical properties can be primarily attributed to the presence of a benzylic CH2 group between the carbanion and the BODIPY core. BODIPY 97 exhibited bright fluorescence in both organic and aqueous solutions, with BODIPY 97a demonstrating an exceptionally high solubility of > 10 000 mg L–1 (Scheme 54).
3.3.7. β-Alkenylation
In 2024, Mertinez et al.[25] reported a Lewis acid-catalized electrophilic β-alkenylation of BODIPY. The substitution occurred at positions 2 and 6, resulting in the formation of the Markovnikov adduct 98 (Scheme 55).
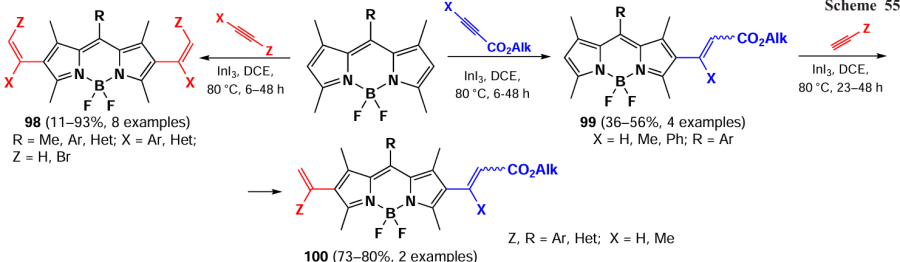
The highest yields in electrophilic substitution reactions with electron-deficient alkynes with an ester group were achieved using indium iodide as a Lewis acid, which was explained by its high oxophilicity. As a possible mechanism, the authors proposed that InI3 could coordinate with the alkyne triple bond, followed by BODIPY anti-attachment, further aromatization and protodemetallation. An intermediate alkenyl cation is also likely to form.
For electron deficient alkynes with an ester group, the authors considered σ-coordination of InI3 with the carbonyl moiety and subsequent β-addition of BODIPY to form a zwitterionic allenyl enolate. This yielded trisubstituted (Z)-alkenes 99, which could be further alkenylated to give products 100. Most of the alkenylated BODIPYs had high fluorescence quantum yields, which depended on both the nature of the alkenyl and the nature of the meso-aryl substituents.
3.3.8. Nicholas reaction
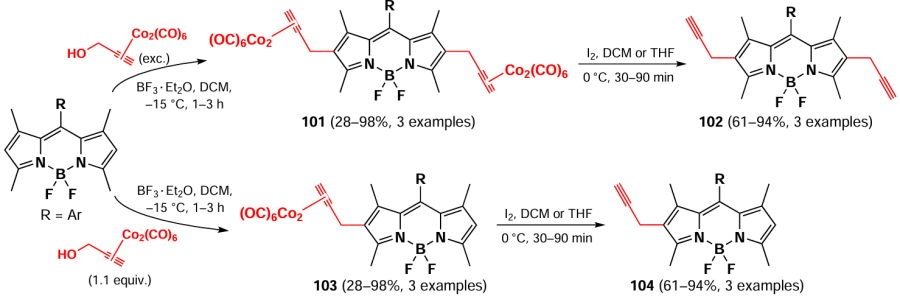
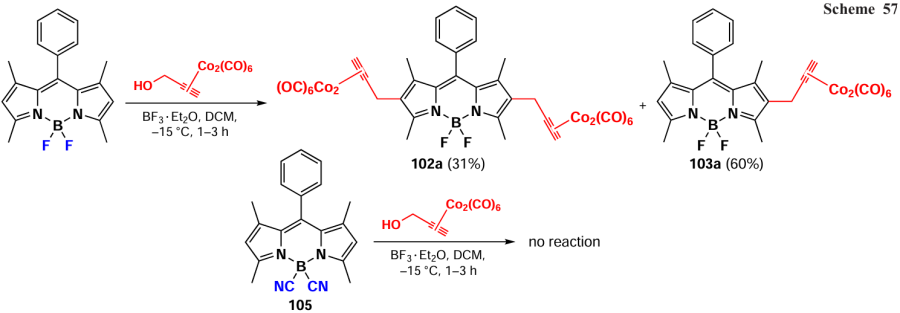
In 2021, Uriel et al.[172] showed that 1,3,5,7-tetramethyl-BODIPY is able to react with an electrophilic propargyl cation stabilized by a cobalt complex (Nicholas reaction) at positions 2 and 6. The interaction of 1,3,5,7-tetramethyl-BODIPY with different meso-substituents with the propargyl-cobalt complex in the presence of BF3 – Et2O at –15 °C led to the formation of 2,6-disubstituted BODIPYs 101. 2,6-dipropargyl BODIPY derivatives 102 were obtained by the demetallation reaction with iodine or ethylenediamine. The use of a slight excess (1.1 equiv.) of the initial cobalt complex leads to monosubstitution products in position 2 — complexes 103. BODIPYs 104 can be synthesized from corresponding complexes 103 via demetallization (Scheme 56).
In 2023, Ventura et al.[173] compared the reactivity of the B(CN)2 substituted BODIPYs with BF2-BODIPYs in the Nicholas reaction. It was shown that B(CN)2-BODIPYs barely participate in electrophilic halogenation or formylation reactions, and do not participate at all in the Nicholas reaction due to the increased electron acceptability of the CN-groups. Under Nicholas reaction conditions, the mixture of B(CN)2-BODIPY and BF2-BODIPY gave only substitution products 105a,b (Scheme 57).
3.4. Nucleophilic substitution reactions
3.4.1. Nucleophilic substitution reaction at positions 1, 3, 5 and 7
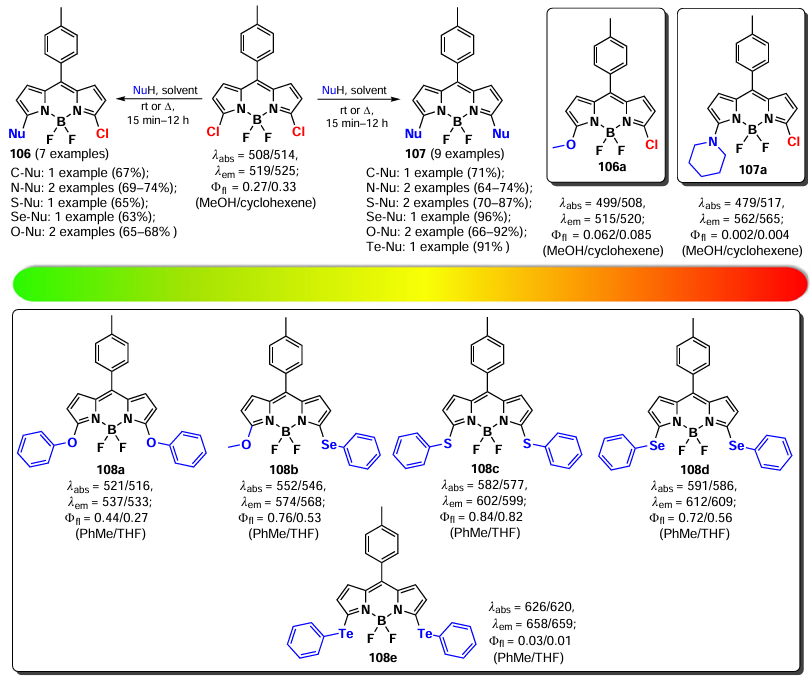
The reactivity of 3-chloro- and 3,5-dichloro-BODIPYs in nucleophilic substitution reactions with oxygen-, sulfur-, and nitrogen-based nucleophiles was initially examined by Rohand et al. in 2006.[141] Nucleophilic substitution at positions 3 and 5 resulted in a bathochromic shift of absorbtion. BODIPY 106, 107 exhibited high fluorescence quantum yields, and fluorescence quenching for compounds 106a and 107a was observed due to the ICT effect. In 2009, Vosch et al.[174] synthesized a series of 3,5-chalcogen-substituted BODIPY 108a – e from 3,5-dichloro BODIPYs. An increase in the atomic mass of chalcogen and a decrease in electronegativity from oxygen to tellurium resulted in a bathochromic shift of maxima in the absorption and emission spectra. α-Te-BODIPY 108e showed the ability to form triplet states due to the ‘heavy atom’ effect (Scheme 58).
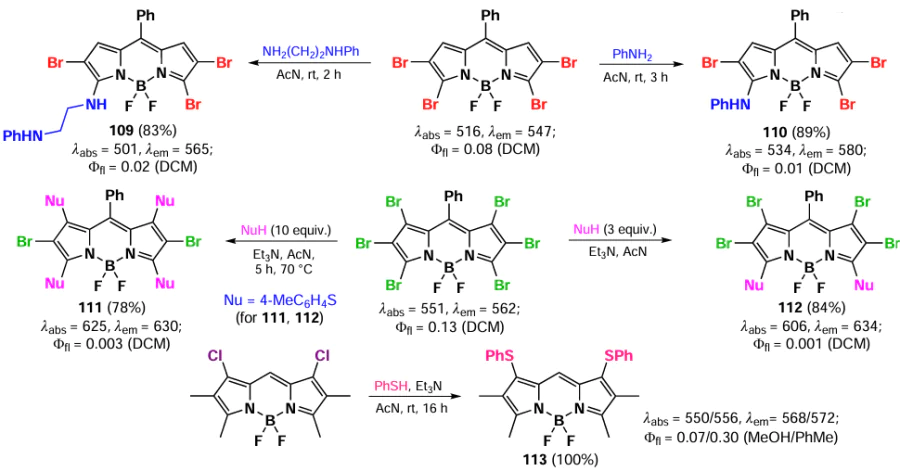
In 2011, Jiao et al.[100] investigated the reactivity of polybromo-BODIPY in reactions with O-, N-, and S-nucleophiles. The reaction of tetrabromo-BODIPY with aliphatic or aromatic amines resulted in the formation of 3-monosubstitution products 109 and 110. In the case of S-nucleophiles, the reaction gave 1,3,5-tri- (111) and 1,3,5,7-tetrasubstitution products (112). Leen et al.[101] demonstrated that nucleophilic substitution also occurs with 1,7-dihalogen-BODIPY, resulting in the formation of compound 113, which exhibits bright fluorescence, bathochromically shifted absorption and emission, and pronounced solvatochromism (Scheme 59).
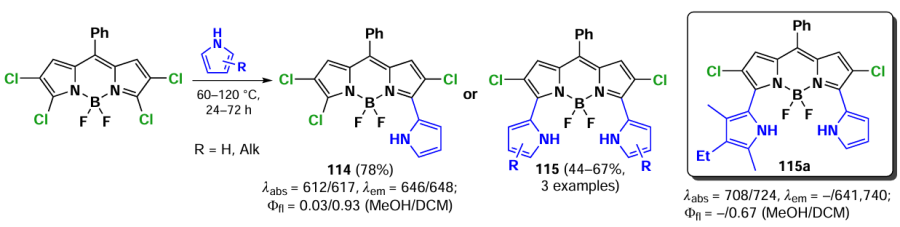
Nucleophilic substitution at the α-position of BODIPY also occurs with C-nucleophiles, foe example, pyrroles. The 3- and 3,5-dipyrrolyl-BODIPY 114 and 115 compounds display bathochromically shifted absorption and emission bands, accompanied by a notable suppression of fluorescence in polar solvents due to ICT (Scheme 60).[144]
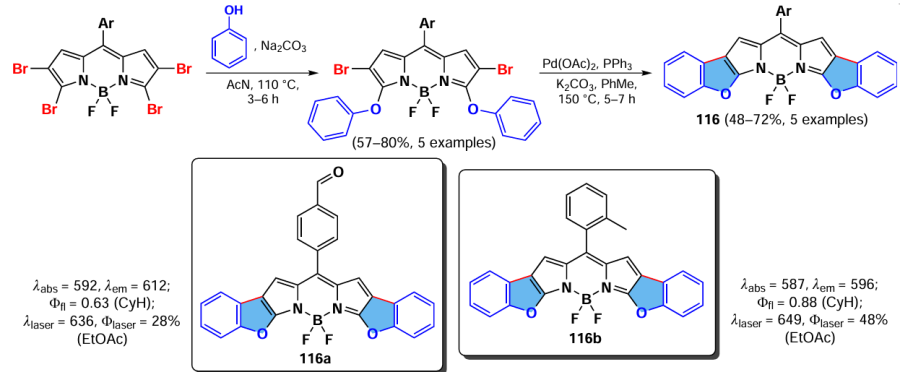
In 2019, Belmonte-Vázquez et al.[118] reported the synthesis of a condensed benzofuran-BODIPY 116 via sequential nucleophilic substitution reactions at positions 3 and 5 of BODIPY, and intramolecular Pd0-catalysed arylation. Due to high planarity, BODIPYs 116 exhibited bathochromic shifts up to 110 nm, as well as small Stokes shifts. Except for BODIPY 116a with a formyl group, compounds 116 demonstrated high laser efficiency ‡ (Φlaser > 40%) and laser emission within the λ = 642 – 647 nm range. BODIPY 116b exhibited the highest laser efficiency (Scheme 61).
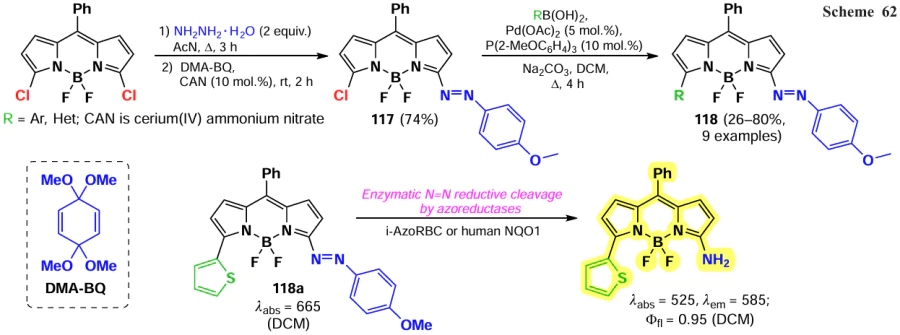
In 2022, Ribagorda et al.[17] synthesized 3-azoconjugated BODIPYs 118 that were able to act as hypoxia-sensitive fluorescent sensors. The products were obtained by nucleophilic substitution of α-halogen atoms by hydrazine, condensation with bis-(dimethylacetal) p-benzoquinone (DMA-BQ), and arylation with boronic acids in the presence of a Pd-catalyst. Under the action of bacterial (i-AzoRBC) and human (human NQO1) reductases as a result of azo-bond cleavage of probes 118, a hypsochromic shift in the absorption spectrum at 140 nm and fluorescence broadening to a value of Ffl = 0.95 were observed, including in hypoxic HeLa cells (Scheme 62).
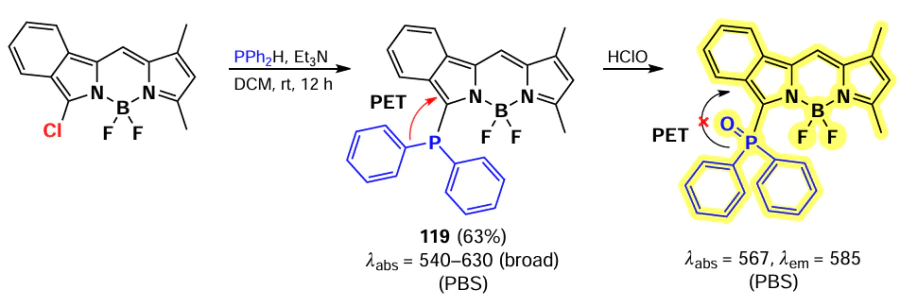
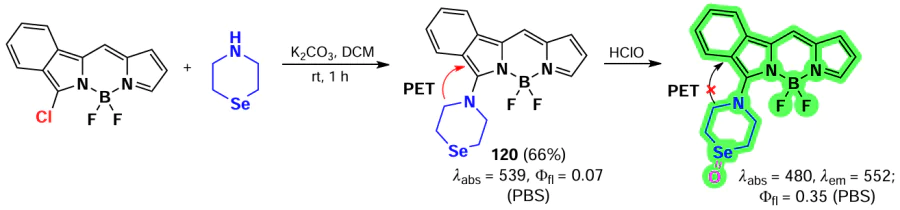
Han et al. [35, 175] developed HClO-sensitive fluorescent probes based on isoindole-BODIPYs. BODIPYs 119 and 120 were synthesized through nucleophilic substitution reactions of α-chloro-BODIPY with diphenylphosphine and selenamorpholine (Scheme 63, Scheme 64). In the presence of HClO, both probes exhibited an emission flare-up due to the oxidation of the phosphorus or selenium atom, which suppressed PET. The probes showed low detection limit (10 – 20 nm) and the ability to detect HClO in vitro and in vivo in Danio fish.
In 2023, Song et al.[176] syntesized a hydrogen peroxide/medium viscosity sensitive BODIPY 121 through a series of nucleophilic substitution reactions of a chlorine atom at the α-position and aldol condensation of the intermediate 3-formyl-BODIPY with a 2-methylindole derivative (A) (Fig. 10). In a viscous medium the rotation of the hemicyanine moiety is inhibited, followed by an increase in the emission of BODIPY 121 at λ = 600 nm. Additionally, in the presence of H2O2 an increase in both absorption (λabs = 720 nm) and emission (λem = 835 nm) were observed due to the cleavage of the α-sulfide moiety. An ability of BODIPY 121 to visualize the viscosity gradient and detect exo- or endogenous hydrogen peroxide in HepG2 cells as well as in mouse organs was confirmed.
![[{"id":"8Wru-8FDG1","type":"paragraph","data":{"text":"Synthesis scheme and principle of hydrogen peroxide and medium viscosity detection by probe <b>121</b>. The figure created by the authors based on the publication[[ type=\"anchor\" referenceId=\"18175\" ]]."}}]](/storage/images/resized/0ApNG7LZF3qNd9R3PNKQIJl4apnoSpv8oJoNtCVV_xl.webp)
3.4.1.1. Application of nucleophilic substitution reactions in BODIPY in biosensorics
Nucleophilic substitution reactions in BODIPY are commonly used in the design of fluorescent probes capable of biomolecule detection, such as biothiols cysteine (Cys), homocysteine (Hcy) and glutathione (GSH). This approach was first reported in 2012 by Yang et al.[177] 3-Chloro-BODIPY 122 was used as a selective fluorescent probe for GSH detection. Nucleophilic substitution of the α- chlorine atom in BODIPY 122 with GSH resulted in a shift of the fluorescence maximum from 556 to 588 nm. At the same time, a similar nucleophilic substitution of the α-chlorine in BODIPY 122 with Cys or Hcy resulted in an intramolecular rearrangement of α-sulfur-substituted BODIPY to α-amino-substituted BODIPY, resulting in a shift of an emission maximum to564 nm. Probe 122 demonstrated the capability of ratiometric (I588/I556) selective detection of GSH with a detection limit of 86 nm.
Further, a Cys-selective probe 123 was obtained by replacing the α-chlorine atom in probe 122 with p-nitrophenol.[178] In this case, a double nucleophilic substitution reaction took place with the product fluorescence flaring up at 564 nm due to a decrease of PET. Upon the reaction of probe 123 with GSH (Hcy), a second nucleophilic substitution reaction (α-sulfur-substituted BODIPY into α-amino-substituted BODIPY) was slower due to the less favourable transition state, resulting in a main product with emission at 588 nm. The modification of probe 122 at the second α-position with quaternised imidazole moiety yielded probe 124, capable of simultaneous detection of GSH or Cys(Hcy) (Scheme 65).[179] In the presence of GSH, the nucleophilic substitution of both thiophenol and imidazolinium resulted in a bright emission at 588 nm at an excitation wavelength (λex) of 568 nm. Nucleophilic substitution with Cys(Hcy) resulted in bright emission at 530 nm (λex = 443 nm).
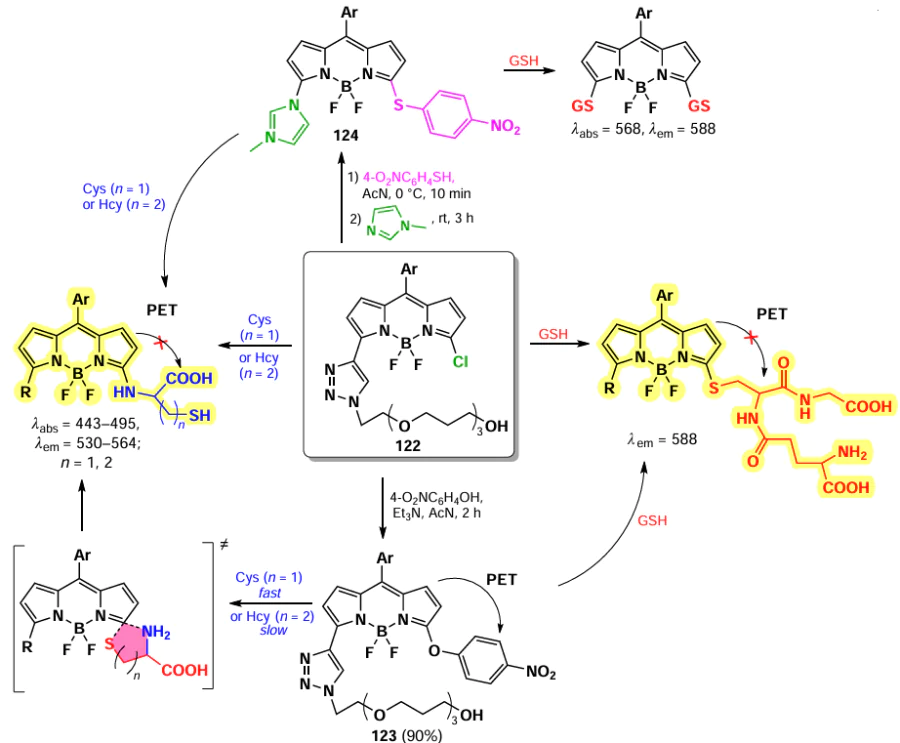
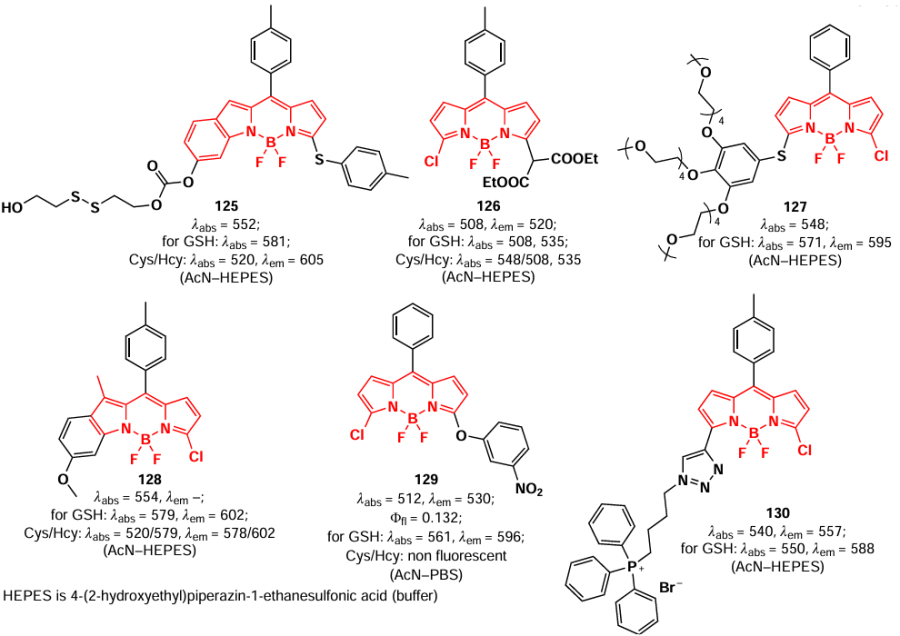
For BODIPY 125 with a disulfide moiety that cleaves in the presence of biothiols, fluorescence emission was observed at different wavelengths in the presence of GSH or Hcy/Cys.[180] Nucleophilic substitution of a similar probe 126 with Cys, Hcy and GSH gave products of different polarity, allowing the simultaneous detection of all three biothiols.[181] BODIPY 127 with hydrophilic polyethylene glycol (PEG) chains and fluorescein was capable to form non-fluorescent NPs, which undergone disaggregation and release of fluorescent BODIPY (λem = 595 nm) and dye (λem = 512 nm) upon reaction with GSH.[182] Probes 128 and 129 demonstrated the ability to detect Cys, Hcy and GSH based on differences in the fluorescence of the resulting products.[183] 3-Chloro-5-(m-nitrophenoxy)-BODIPY 129 was found to be an appropriate fluorophore for two-step ratiometric fluorescence detection of GSH across three concentration ranges, in the absence of a fluorescence response to Cys(Hcy).[184] The use of probe 130 with a vector to mitochondria and a triphenylphosphine moieties enables the detection of GSH in mitochondria.[185]
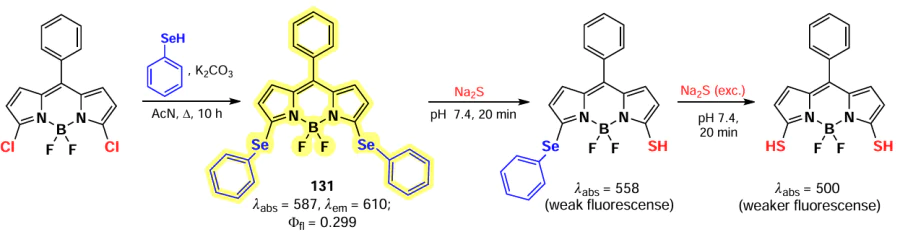
In 2017, Guo et al.[186] reported a hydrogen sulfide-sensitive fluorescent probe 131 based on 3,5- phenylselenide BODIPY. Replacement of the PhSe leaving group with a sulfonyl (SH) resulted in a 49-fold fluorescence attenuation and a hypsochromic shift of the absorption bands. The detection limit of probe 131 was 2.5 nm, and its ability to visualize GSH in living cells was also demonstrated (Scheme 66).
3.4.1.2. Oxidative nucleophilic substitution of hydrogen at 3,5-positions
In 2010, Leen et al.[187] investigated oxidative nucleophilic substitution in the BODIPY core. Positions 3 and 5 of the BODIPY molecule are electron-depleted and undergo nucleophilic attack to form a negatively charged δ(H)-complex, which is stabilized by a boron atom. Subsequent oxidation leads to the formation of substitution products. Reactions with N- and C-nucleophiles at the α-position of BODIPY can proceed in the presence of oxygen, leading to the formation of products 132 and 133 (Scheme 67).
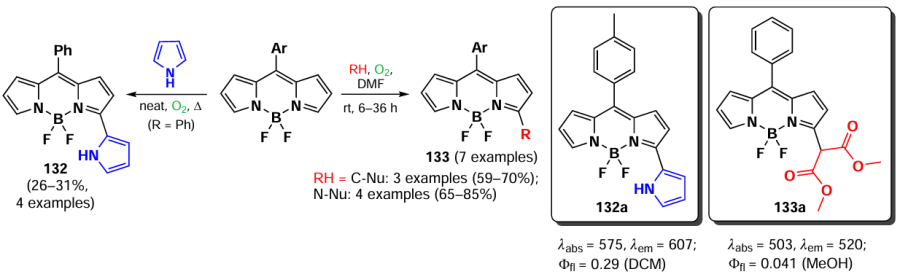
Primary and secondary aliphatic amines demonstrated the ability to act as nucleophiles in the reaction of oxidative nucleophilic substitution with BODIPY, resulting in the deactivation at the second α-position. A similar result was observed in the oxidative nucleophilic substitution reaction with pyrroles; BODIPY 134 was formed and exhibited bright fluorescence [188] (Scheme 68).
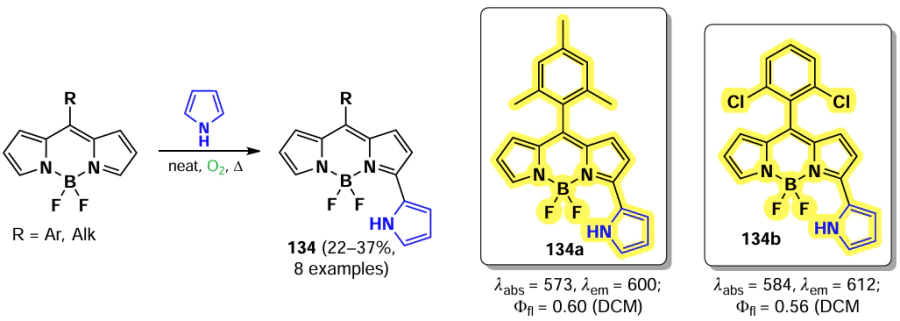
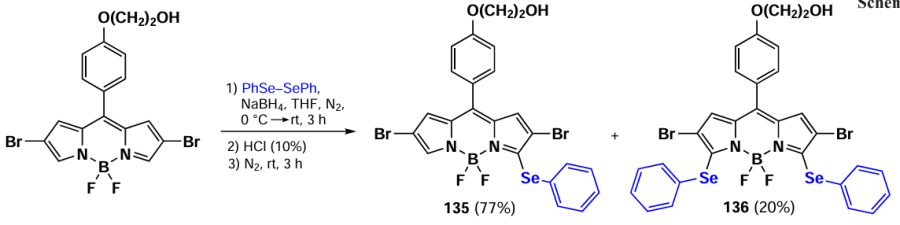
In 2021, Santos et al.[189] demonstrated the possibility of oxidative nucleophilic substitution reactions at 3,5-positions of BODIPY, even in the presence of halogen atoms at C(2) and C(6). This was achieved by reacting a 2,6-dibromo-substituted BODIPY with an in situ generated selenophenol, forming mono- and disubstituted products (135 and 136) (Scheme 69).
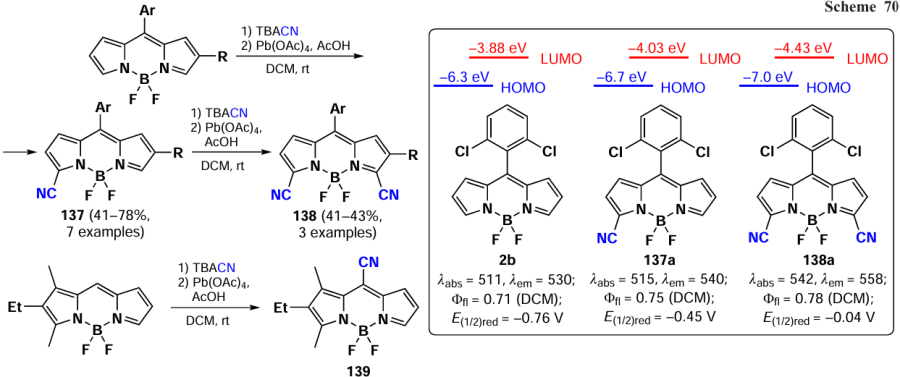
In 2023, Zuo et al.[19] proposed a new method for introducing a cyano group at 3,5-positions of BODIPY. The proposed approach involved oxidative nucleophilic substitution reactions with Pb(OAc)4 as an oxidant, resulting in the formation of BODIPYs 137 and 138 (Scheme 70). When the meso-position of BODIPY was free, the formation of 8-CN-substituted BODIPY 139 was observed. The introduction of cyano groups into the BODIPY core resulted in a bathochromic shift of the absorption and emission bands, as well as an increase in the reduction potentials (E(1/2)red). The values of E(1/2)red were –0.76, –0.45 and –0.04 V for the unsubstituted BODIPY and BODIPYs 137a and b, respectively. Also, a reduction in the LUMO energy from –3.88 eV for the unsubstituted BODIPY to –4.43 eV for the cyano derivative 138a was observed. Moreover, in the presence of triethylamine and cobaltocene, BODIPY 138a formed an anion radical, which was detected by EPR spectroscopy.
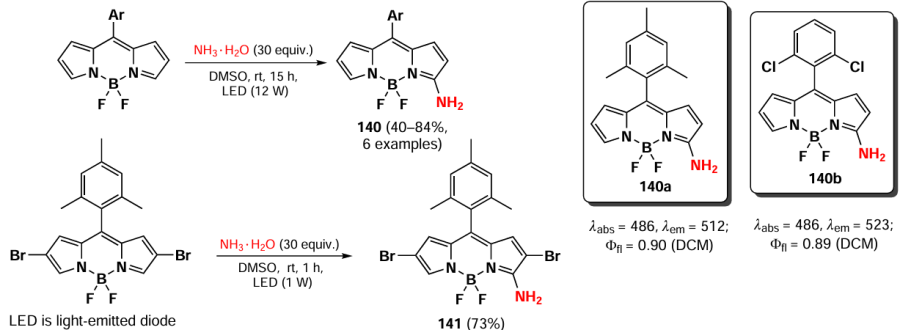
In 2023, Wang et al.[20] demonstrated that oxidative nucleophilic substitution in BODIPY with weak nucleophiles can occur under light irradiation. Light-induced oxidative nucleophilic substitution with ammonia yielded BODIPYs 140 and 141 (Scheme 71). The reaction exhibited a faster rate with the 2,6-dibromo BODIPY, suggesting that the electronegativity of BODIPY is increased in its excited state. The aminated BODIPY 140 showed bright fluorescence up to Фfl = 0.9 and a hypsochromic shift in the absorption spectra.
3.4.1.3. Vicarious nucleophilic substitution at 3,5-position of BODIPY
In 2011, Leen et al.[190] investigated vicarious nucleophilic substitution at the α-position of meso-substituted BODIPY in reactions with 2-substituted acetic acid esters. The products 142 were obtained in the presence of the bases, DBU (1,8-diazabicyclo[5.4.0]undec-7-ene), K2CO3 and ButOK (Scheme 72).
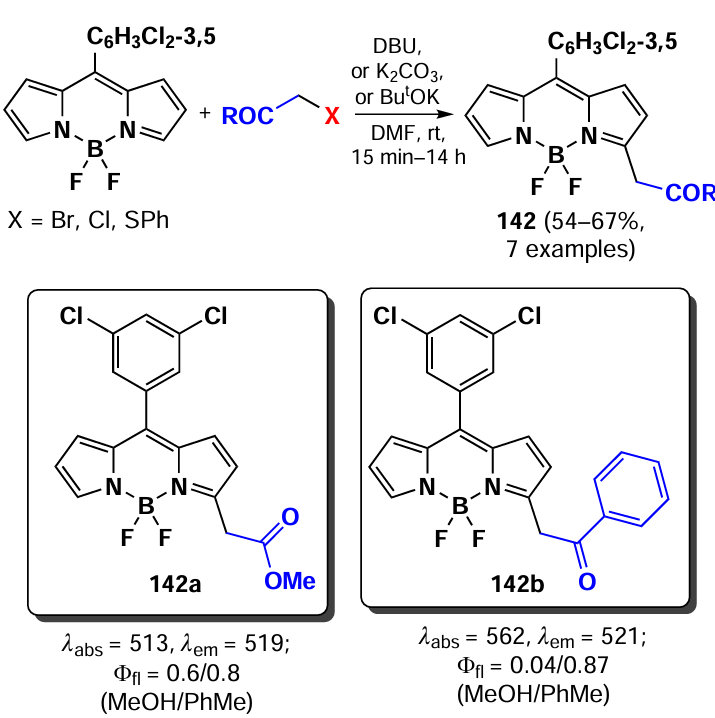
In addition, a tandem Michael addition/vicarious substitution reaction with nitroalkenes has been reported, yielding 3-styryl-substituted BODIPY 143, which was previously available for synthesis via the Knoevenagel condensation (Scheme 73).
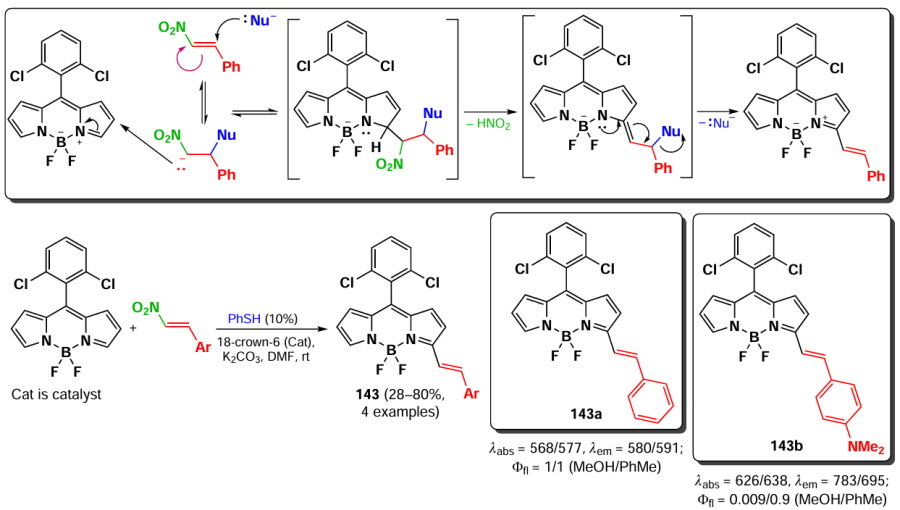
In 2015, Knight et al.[191] developed a method for amination of BODIPY via copper-catalysed vicarious nucleophilic substitution with primary and secondary amines (Scheme 74). The resulting α-amino-BODIPY 144 showed low fluorescence quantum yields due to the free rotation of meso-aryl. A reaction mechanism was proposed involving the nucleophilic attack of copper amide at position 3 of the BODIPY; followed by anion formation, protonation and HI elimination under the action of base to give the desired product.
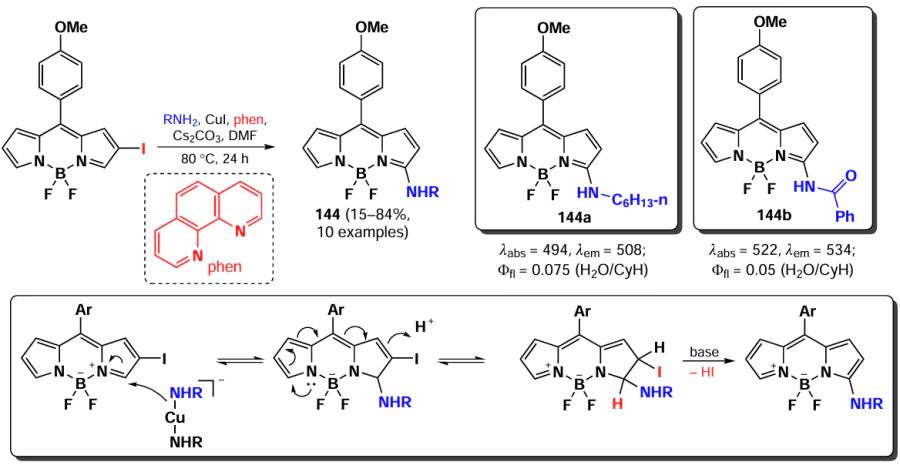
3.4.2. Nucleophilic substitution at the meso-position of BODIPY
The meso-position of BODIPY is highly sensitive to the nature of the substituent, due to the significant increase in electron density on the C(8) atom that occurs during the transition from the HOMO to the LUMO.[192, 193] Consequently, when the heteroatom is at the meso-position, the formation of a hemicyanine nucleus and a rearrangement of the electron density of BODIPY can take place.[97] In the absence of steric hindrance, the conjugation of the unshared electron pair of the nitrogen atom to BODIPY results in the formation of a new π-system, non-equivalence of pyrrole cycles, an increased LUMO energy, an increased energy gap between the HOMO and the LUMO, a hypsochromic shift, bright fluorescence in non-polar media and high laser efficiency (Fig. 11).[194]
![[{"id":"kYZObGQJaw","type":"paragraph","data":{"text":"Differences in photophysical properties and molecular orbital energies of 8-amino-BODIPY rotamers. The figure created by the authors based on the publication[[ type=\"anchor\" referenceId=\"18193\" ]]."}}]](/storage/images/resized/z6sgDlwIbm9TRAEWNbmWdi1JhAArT1dwjG1Pxr6M_xl.webp)
3.4.2.1. Nucleophilic substitution of the 8-SMe group
In 2006, Goud et al.[80] investigated the reactivity of the SMe group at the meso-position of BODIPY in nucleophilic substitution reactions. The methylsulfanyl group was readily substituted with anilines yielding 8-NHPh derivatives (145a,b), for which the hemicyanine structure was proposed based on spectral characteristics. Consequently, these derivatives did not exhibit emission properties (Scheme 75).
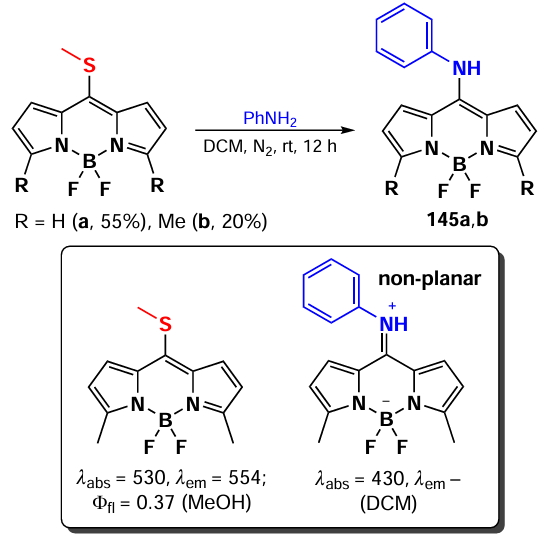
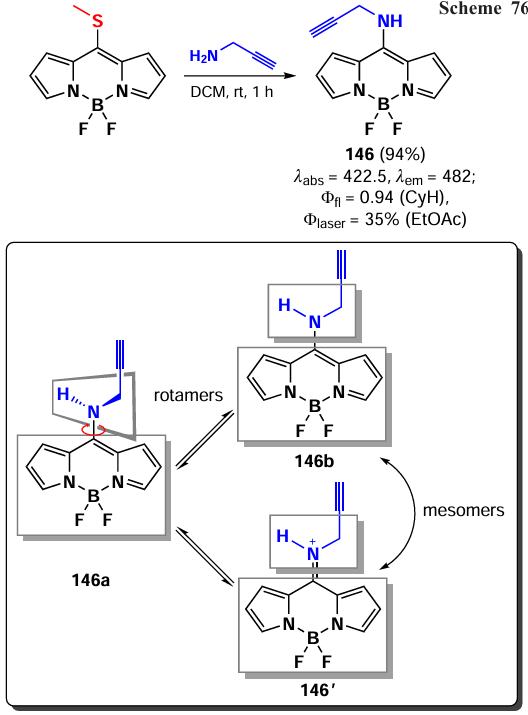
In 2020, Gomez-Duran et al.[194] synthesised 8-aminopropargyl-BODIPY 146 by a nucleophilic substitution reaction of a meso-thiomethyl group (Scheme 76). BODIPY 146 existed in two rotameric forms (146a and 146b), with the unshared electron pair of the nitrogen atom conjugated to the BODIPY in rotamer 146b. The results of quantum mechanical calculations confirmed that the HOMO is localised on the pyrrole moieties, while the LUMO is localised on the nitrogen atom, which is in good agreement with the hypsochromic shift of the absorption band and the large Stokes shift observed. The reduction in the dipole moment by 2 D in the excited state is also in accordance with the redistribution of electron density resulting from the excitation. Compound 146 demonstrated a high level of laser efficiency (35%).
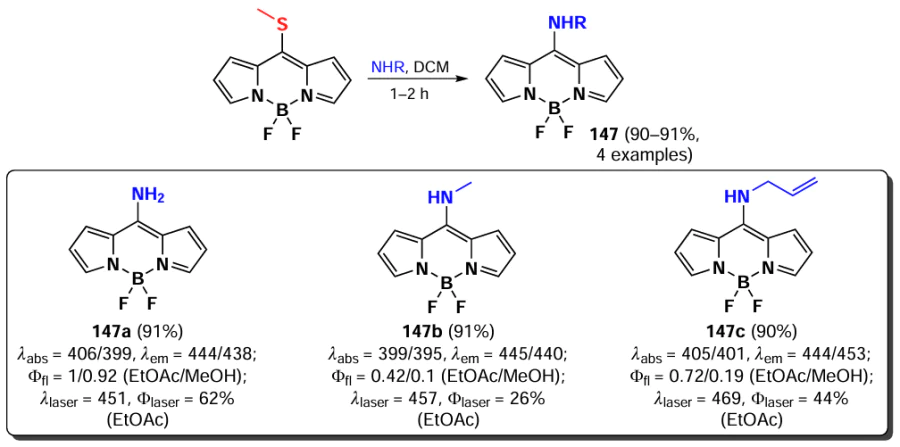
In 2011, Arbeloa et al.[195] demonstrated that an increase in the electron-donating ability of the amine in a series of 8-amino-substituted BODIPYs 147 results in a hypsochromic shift and a decrease in fluorescence quantum yields due to ICT. Some of the synthesized BODIPYs, for example, BODIPY 147a, exhibited high fluorescence quantum yields and high laser efficiency (Scheme 77).
In 2012, Peña – Cabrera et al. proved that the observed hypochromic shift and increase in Stokes shift for 8-amino-BODIPY rotamers with tert-butyl (148) and propargyl (149) were due to the destabilisation of the LUMO in the absence of meso-substituent effects on the HOMO.[196] In sterically hindered amines, the coplanar arrangement of the amine and the BODIPY core was disrupted, thereby preventing the formation of a hemicyanine. The ‘twisted’ arrangement of the amino group was also found to contribute to ICT, resulting in a lower fluorescence quantum yield. In 2013, Roacho et al.[197] demonstrated significant solvatochromism of 8-amino-BODIPYs 150a,b, and for BODIPY with the 8-NEt2 substituent 150b, a disruption of planarity of the pyrrole cycles was also observed, resulting in a complete lack of emission (Scheme 78).
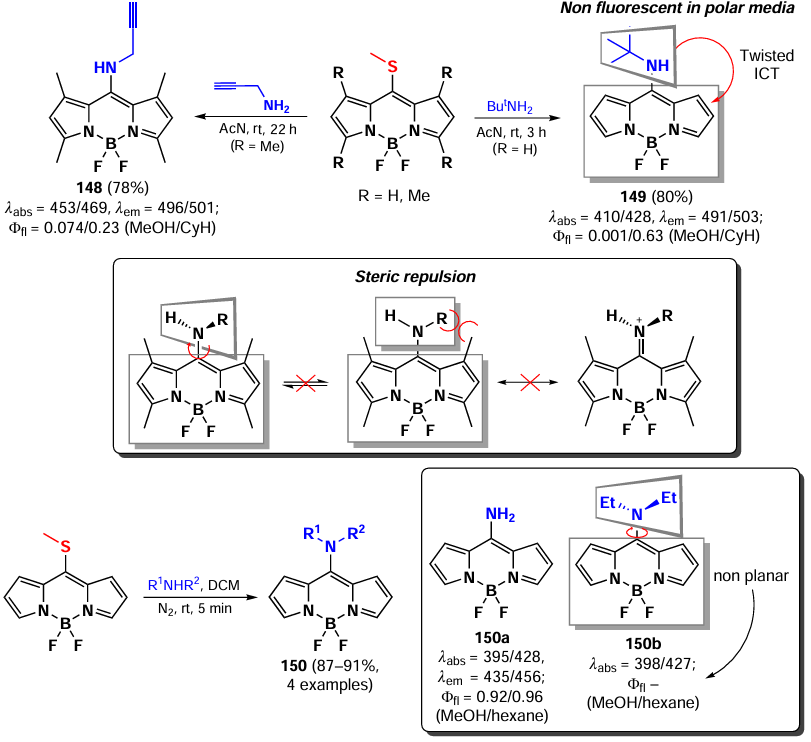
In 2013, Esnal et al.[81] reported a synthesis of BODIPYs 151 via substitution of the 8-SMe group with P,O-nucleophiles. The 8-NHMe- and 8-SMe-derivatives 151a,b exhibited a hypochromic shift, which can be explained by the conjugation of an unshared electron pair of the heteroatom with the BODIPY core. An increase in the electron-donating ability of the heteroatom results in a negative shift in the reduction potentials of the BODIPY, due to an increase in the LUMO energy, an increase in the LUMO – HOMO energy gap and a hypsochromic shift in the electronic spectra. In contrast, the 8-PPh2 substituted BODIPY 151c demonstrated a bathochromic shift in the absorption spectrum and exhibited no emission due to the free rotation of the PPh2 group. BODIPY 151 exhibited bright emission, which was enhanced by the presence of methyl groups at 3,5-positions. Additionally, high laser efficiency of BODIPYs was observed, with the exception of compound 151d. (Scheme 79).
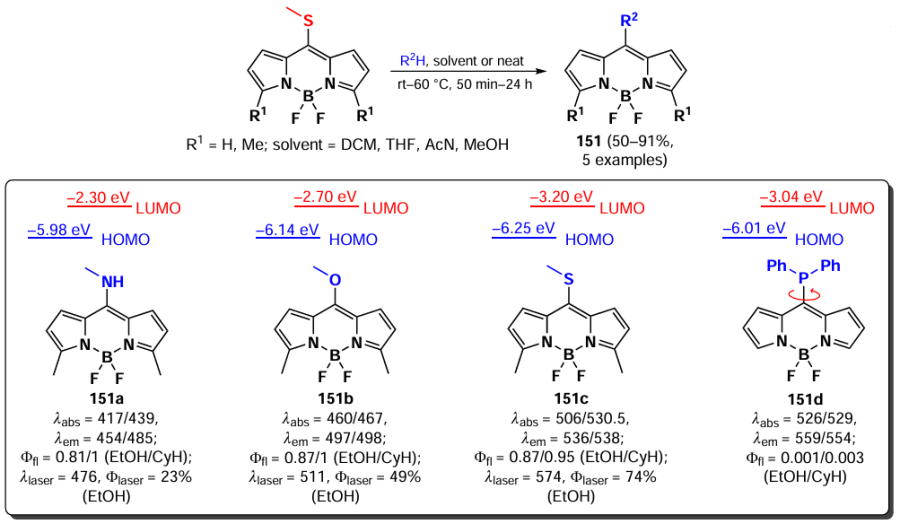
In 2015, Gutiérrez-Ramos et al.[198] reported a nucleophilic substitution of thiomethyl group under the action of sodium enolates of 1,3-dicarbonyl compounds in the presence of copper(I) thiophene-2-carboxylate (TC). The proposed mechanism involves the coordination of CuI-carboxylate on the sulfur atom, which is then attacked by soft nucleophiles (Scheme 80). BODIPYs 152a,b exhibited pH-dependent fluorescence, which was suppressed in basic medium. This suppression was attributed to the conjugation of the enol group to the BODIPY core and the enhancement of radiation-free relaxation pathways. Furthermore, these compounds exhibited high laser efficiency.
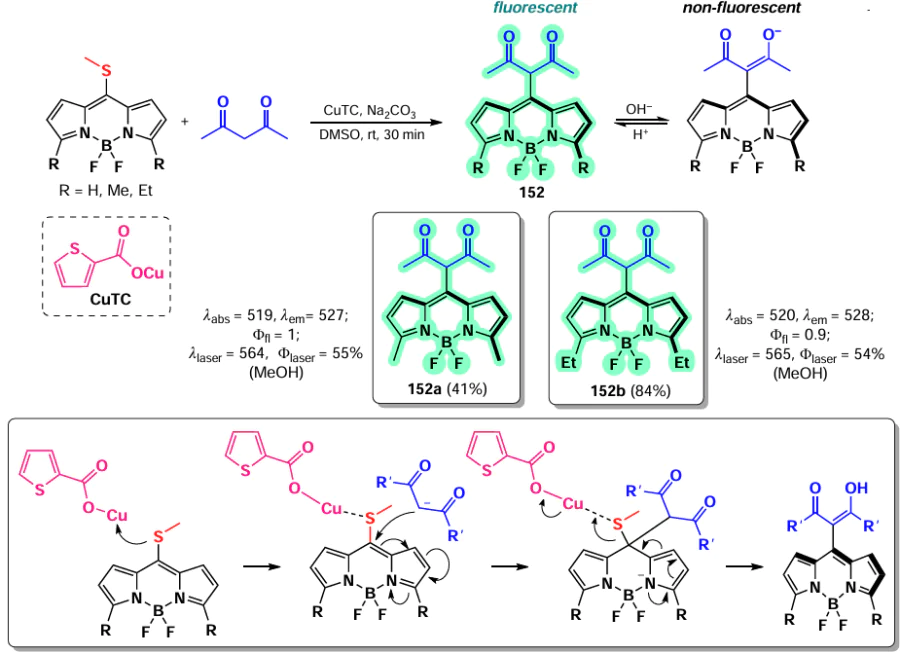
In 2015, Roacho et al.[199] reported a regioselective nucleophilic substitution of the thiophenol group at the meso-position in unsubstituted 8-SMe-BODIPY, yielding BODIPY 153. In addition, nucleophilic attack at 3,5-positions took place when THF was used as a solvent, yielding BODIPYs 154 and 155. BODIPYs 156 were obtained by nucleophilic substitution with secondary amines using dichloromethane as a solvent [200] (Scheme 81).
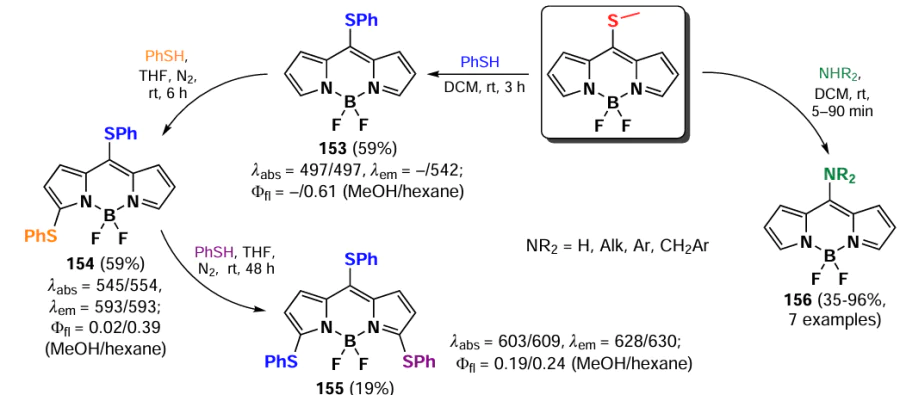
In 2021, Yagishita et al.[201] synthesised fluorescent probes for imaging aldehyde dehydrogenase 1 (ALDH1) stem cells. Meso-amino-BODIPYs 157 and 158 were obtained via a nucleophilic substitution of a thiomethyl group with benzylamine in the meso-position of BODIPY. The mechanism of ALDH1 detection was based on the permeability of the cell membrane for aldehyde-bearing BODIPYs 157 and 158, while the carboxylic acids oxidised by dehydrogenase were unable to leave the cell. Both probes demonstrated the ability to detect ALDH1 in human embryonic stem cells SBC5 (Scheme 82).
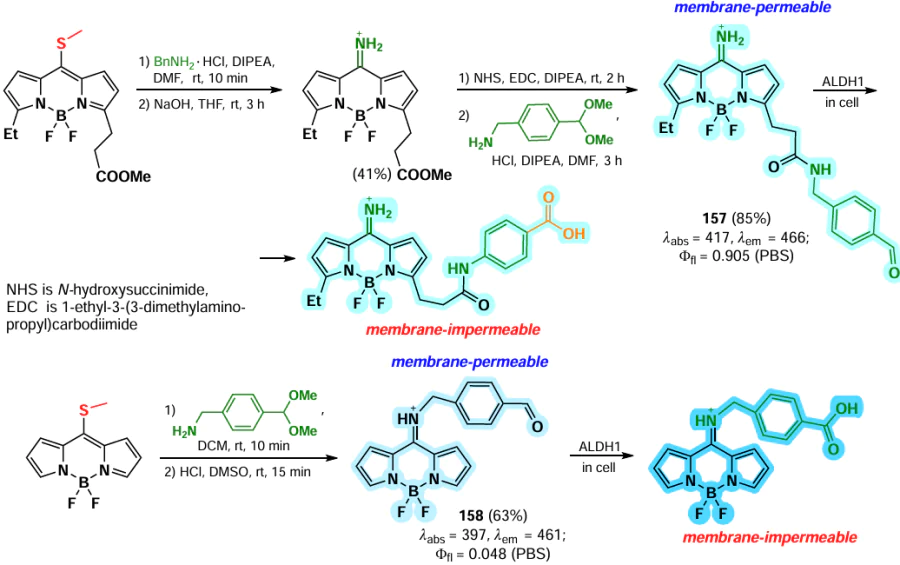
Since the presence of an amino group at the meso-position of BODIPY results in a hypsochromic shift, meso-amino BODIPYs have limited application as bioimaging probes. In 2022, Wu et al.[202] proposed a strategy to reduce the LUMO energy of meso-amino BODIPYs and to reduce the HOMO – LUMO energy gap by introducing an electron acceptor group at the meso-nitrogen atom. The resulting 8-(N-acyl)-BODIPY 159 showed a bathochromic shift in the absorption spectrum up to 770 nm and a bright emission up to Ffl = 0.99. The potential of these fluorophores for bioimaging was demonstrated in reverse-electron Diels-Alder reaction of 8-(N-acyl)-BODIPY 159a with a tetrazine cycle with trans-cyclooctene (TCO) or spiro[2.3]hex-1-ene (BCN), which was accompanied by a 128-fold increase in fluorescence. It seems reasonable to assume that fluorescence enhancement was facilitated by the presence of a flexible alkyl linker in compound 159a (Scheme 83).
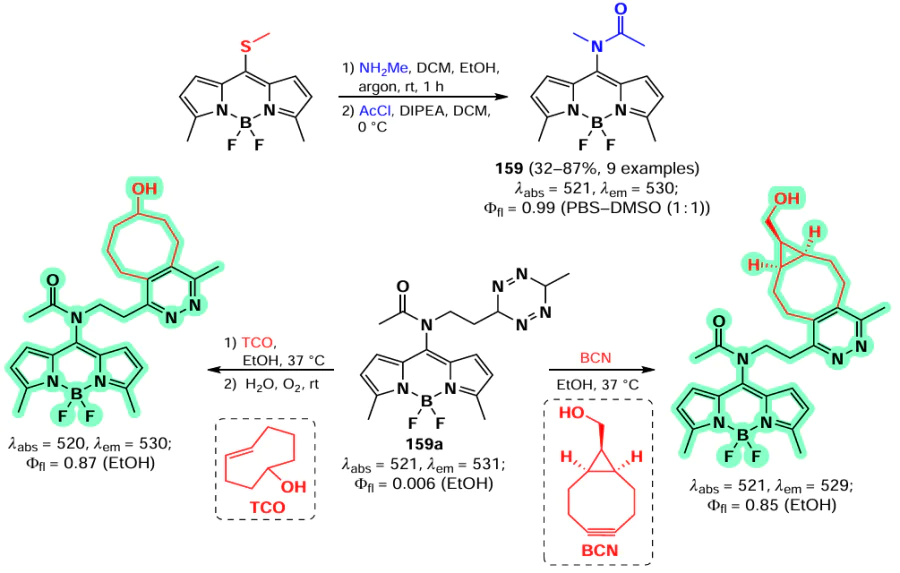
3.4.2.2. Nucleophilic substitution of meso-halogen atom
In 2012, Leen et al.[82] investigated the reactivity of 8-chloro-BODIPYs with different nucleophiles, such as anilines, aromatic and aliphatic thiols, phenols and alcohols, yielding BODIPYs 160. Meso-substituted BODIPYs 160a,c exhibited remarkably high fluorescence quantum yields, whereas no emission was detected for meso-anilino-BODIPYs 161b,d. Subsequently, Boens et al.[203] investigated the photophysical and spectral properties of BODIPYs substituted with C-, N-, O-, and S-nucleophiles. Their findings demonstrated that the introduction of a heteroatom at the meso-position of BODIPY resulted in a hypsochromic shift of the maximum in the absorption spectrum for BODIPY 161b – d. The alkynyl-BODIPY (161a) exhibited a bathochromic shift due to the conjugation between the BODIPY core and the meso-substituent (Scheme 84).
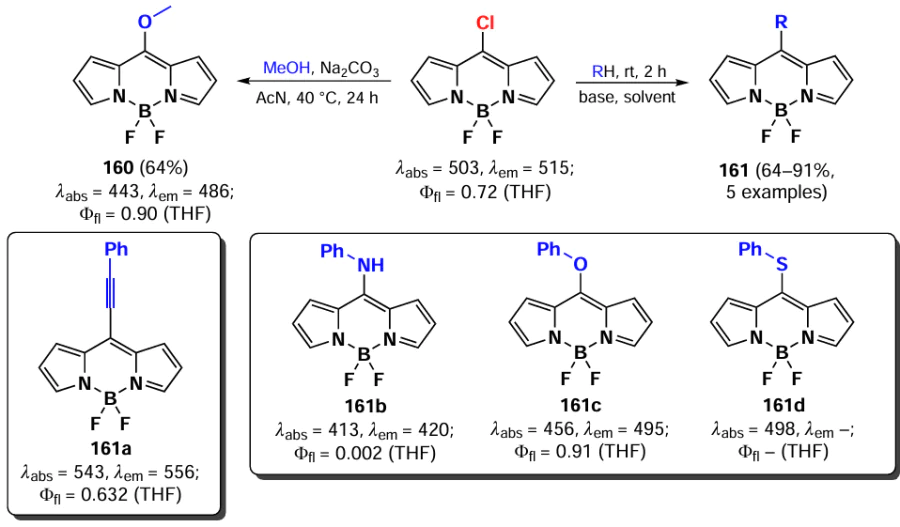
In 2014, Misra et al.[204] synthesized a series of O- and N-substituted dimers of BODIPY 162 and 163, conjugated via phenyldiol and phenylenediamine-based linkers. The resulting dimers exhibited a hypsochromic shift in their absorption spectra. Notably, the N-dimers displayed no fluorescence, while the O-dimers exhibited green fluorescence, which was attributed to the conjugation of an unshared electron pair to the BODIPY. Electrochemical studies demonstrated that the extensive delocalization of the unshared electron pair of the meso-substituent on the BODIPY nucleus facilitates the oxidation of dimers and the generation of cationic radicals (Scheme 85).
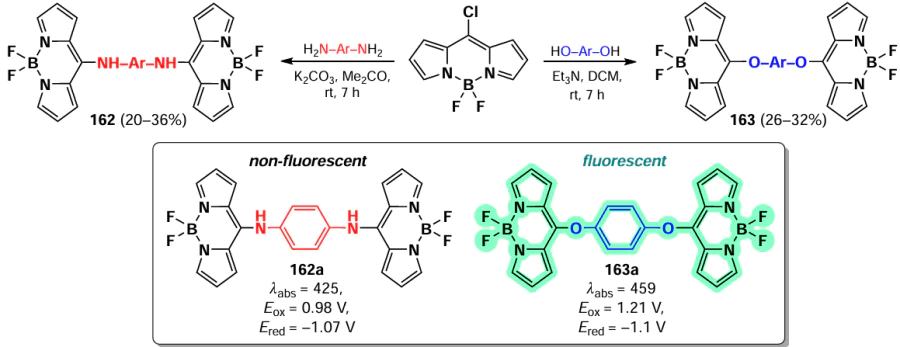
Jiang et al.[144] reported a synthesis of 8-pyrrolyl BODIPYs 164 via nucleophilic substitution of cholorine in meso-chloro-BODIPY with pyrroles. A similar BODIPY 165 was synthesized by Popov and Plenio in 2022 [205] via nucleophilic substitution of meso-chloroine in BODIPY with imidazole during the synthesis of a series of BODIPY-based photosensitisers (Scheme 86).
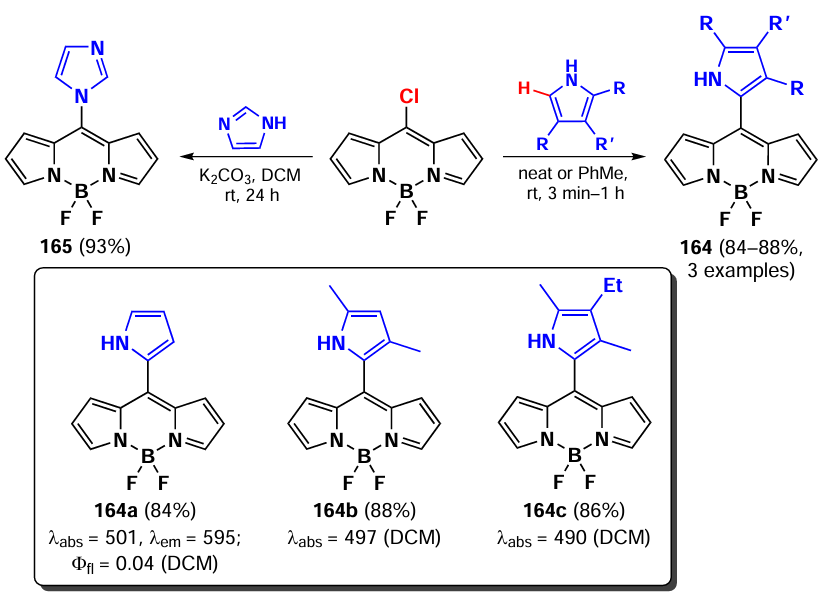
3.4.2.3. Nucleophilic substitution reactions at the meso-position of BODIPY in biosensorics
Since the meso-position of the BODIPY is sensitive to the electron-donating ability of the substituent, the ability of amino-BODIPYs to form a hemicyanine provides the potential for the use of 8-heteroatom-substituted BODIPY as fluorescent probes. Their mechanism of action is based on the disruption of the conjugation between a heteroatom and a BODIPY core, which results in a significant change in the spectral properties due to the blocking of ICT or PET processes and the rearrangement of the π-system.
3.4.2.3.1. Phosgene detection
In 2019, Wei et al.[16] synthesized a phosgene-sensitive sensor 166 based on an 8-hydrazido-BODIPY, obtained from meso-chloro-BODIPY via nucleophilic substitution. PET from the nitrogen atom to BODIPY resulted in the complete suppression of emission from the sensor. In the presence of phosgene, the oxadiazolone cycle formation took place, thereby preventing the PET process and enhancing fluorescence. BODIPY 166 exhibited a fast response time (1.5 seconds) with a low detection limit for phosgene (0.15 nanomolars), and high selectivity. In the context of the test strips, sensor 166 demonstrated the ability to detect phosgene in the gas phase at a concentration of 20 ppm (Scheme 87).
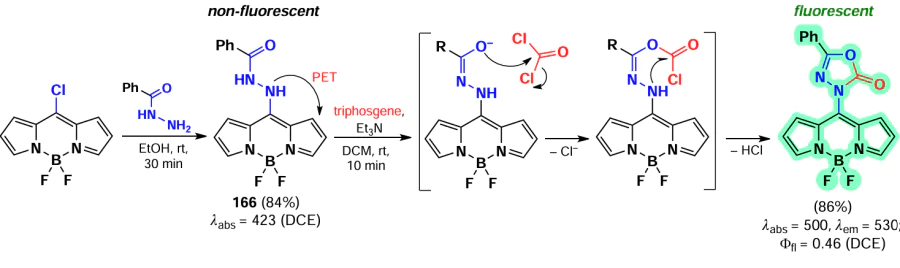
In 2022, Zeng et al.[206] presented a series of trans- and cis-8-diaminocyclohexano-BODIPY-based probes designed to detect phosgene, diethylchlorophosphate, and other acyl chlorides with a similar mechanism of toxic action. The interaction of trans-isomer 167 with phosgene and its analogues resulted in a decrease of PET, a bathochromic shift, a rapid change in fluorescence (from weak blue to bright green). BODIPY 167 showed a low limit of detection (0.52 nm). The enhanced sensitivity of the trans-isomer compared to the cis-isomer is explained by the presence of intramolecular hydrogen bonding. In the presence of acyl chlorides, the sensor showed increased blue fluorescence, with a detection limit of 0.77 nm. (Scheme 88).
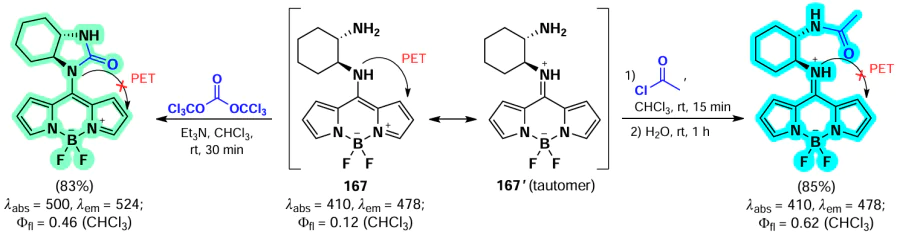
3.4.2.3.2. HClO and ClO– detection
In 2019, Xu et al.[207] synthesized 8-seleno-BODIPYs 168a – c, as an effective fluorescent probe for the detection of HClO and ClO–. The mechanism of action of these probes is based on the oxidation of non-fluorescent (via PET) selenides 168 to fluorescent selenoxides (no PET). The sensitivity of the sensors was found to be dependent on the electron-donating ability of the substituent present at the meso-position. BODIPY 168a demonstrated the highest sensitivity and fastest response, while BODIPY 168b exhibited the highest selectivity towards various ROS. Furthermore, the latter was also able to visualize exo- and endogenous HClO in living cells, with the majority of the latter localized in mitochondria. (Scheme 89).
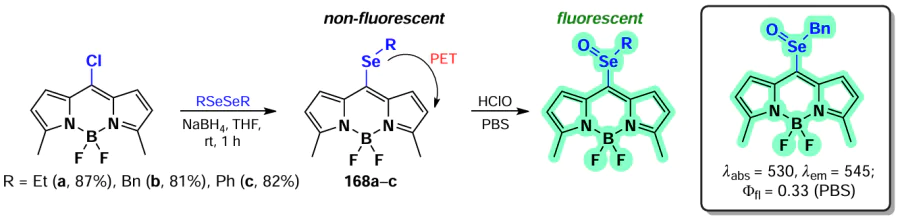
3.4.2.3.3. Biothiol detection
The detection of biothiols by fluorescent sensors is based on two distinct nucleophilic substitution reactions: the first nucleophilic attack of the sulfur atom, and subsequent an intramolecular rearrangement involving a nucleophilic attack of the nitrogen atom. BODIPY-based probes for the detection of biothiols undergoing nucleophilic substitution reactions at positions 3 and 5 are discussed in detail in section 3.4.1.1. The ability of 8-SMe-BODIPY 169 to detect Cys and Hcy by a similar mechanism has been also demonstrated.[208] Nucleophilic substitution of the methylsulfanyl group for Cys and Hcy residues, followed by intramolecular rearrangement and substitution of the sulfur atom with an amino group, resulted in a hypochromic shift not observed in the presence of other biothiols (Scheme 90).

In 2021, Ji et al.[209] synthesized a series of 8-pyridyloxy(thioxy)-BODIPY 170a – c via a nucleophilic substitution of the meso-chlorine atom, and then proceeded to quaternise the nitrogen atom in the pyridine cycle with the formation of salts 171a – c. When in cells, BODIPY 171a underwent a reaction with Cys in accordance with the aforementioned mechanism, resulting in the formation of an 8-(N-Cys) derivative with pronounced fluorescence at 620 nm (Scheme 91).
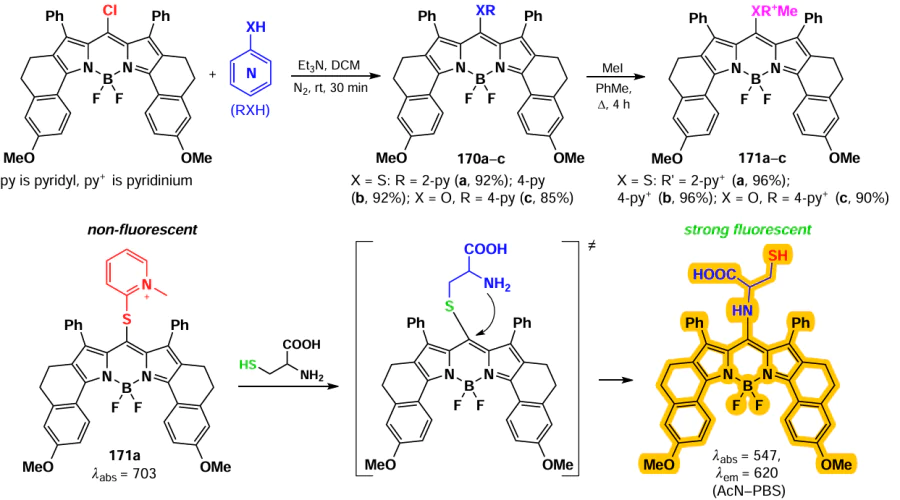
In 2023, Zhang et al.[210] reported 8-(p-methoxyphenyl)sulfanyl-BODIPY 172 as a cysteine-sensitive fluorescent probe, which was synthesized via a nucleophilic substitution reaction between meso-chloro-BODIPY and p-methoxythiophenol. The reaction of the sensor with Cys was accompanied by a change in the solution colour from blue to pink, together with a fluorescence enhancement. BODIPY 172 demonstrated a rapid reaction time (10 minutes), a high signal-to-noise ratio (3150), and an low detection limit for cysteine (11.2 nm) (Scheme 92).
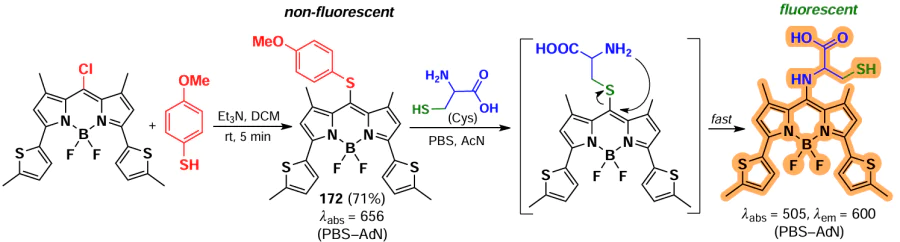
In 2024, Zeng et al.[36] developed a nitrite-anion sensor based on 8-(o-phenylenediamino)-BODIPY 173. At pH 1, the nitrite anion formed the electrophilic nitrosyl cation 174, which then reacted with the NH2-group at the meso-position of BODIPY to form a diazonium cation. Subsequently, the diazonium cation was subjected to nucleophilic attack by an amine, resulting in the formation of a triazole ring and the emission of fluorescence due to the PET disruption (Scheme 93).
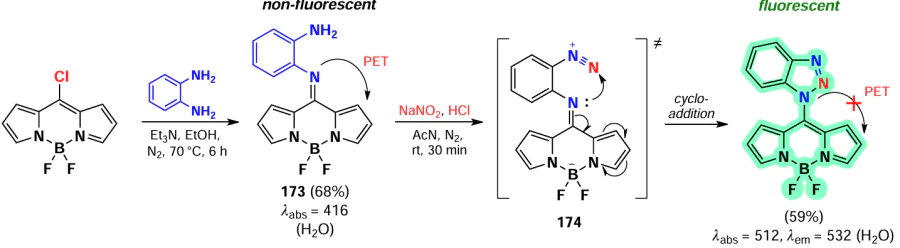
3.4.2.3.4. Hg2+ detection
In 2012, Ahn et al.[211] proposed to use the metal-catalysed nucleophilic substitution reaction of the 8-thiomethyl group of BODIPY 169 with water for the selective fluorescence detection of mercury cations in the presence of other metals. The replacement of the thiomethyl group with a hydroxyl group resulted in a hypsochromic shift of both the absorption and fluorescence maximum. Quantum mechanical calculations for BODIPY 169 and its hydrolysis product predicted a larger HOMO – LUMO energy gap for the product (3.48 eV) compared to the original sensor (3.03 eV), which is consistent with the shift of the absorption spectrum to the blue region (Scheme 94).
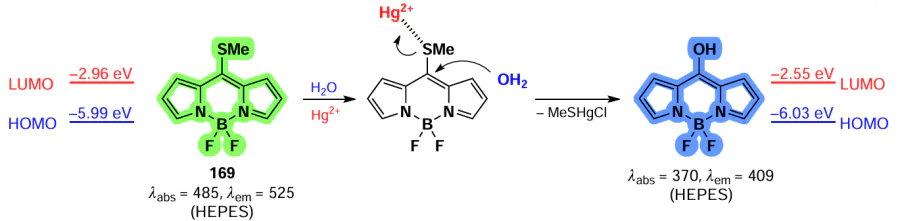
3.5. Reactions involving methyl groups
A significant number of both commercially used and described in the literature BODIPYs contain methyl groups as substituents in various positions. The presence of 1,7-methyl groups inhibits the free rotation of the meso-substituent and enhances the emission of the fluorophores.[212] In some cases, the methyl groups at C(3) and C(5) positions also contribute to the increased emission.[81]
Methyl groups at positions 3, 5, and 8 are able to form stabilized benzylic anions, cations, or radicals.[6, 213, 214] The delocalization of the positive charge at positions 1, 3, 5, 7, and 8 increases the CH-acidity of the methyl groups, opening up possibilities for modifications of the BODIPY core, leading to changes in the physicochemical, photophysical, and biological properties (Fig. 12).[10, 215] This section discusses postmodification reactions of BODIPY via modification of methyl groups, including the Knoevenagel condensation, oxidation, and electrophilic substitution reactions.
![[{"id":"cglAqg9bM4","type":"paragraph","data":{"text":"Reactions in volving the methyl groups of BODIPY discussed in Section 3."}}]](/storage/images/resized/z4QmMjqOX4Jps39iEvf7AVpRIioUXj232kP4I2RN_xl.webp)
3.5.1. Knoevenagel condensation
One of the most widely used reactions involving methyl groups in BODIPY is the Knoevenagel condensation. The methyl groups at positions 3, 5, and 8 are the most active in this condensation, while those at positions 1 and 7 are less active. This activity pattern is consistent with the calculated electron density distribution in the BODIPY core, which decreases in the order 2 and 6 > 1 and 7 > 3 and 5 > 8. The chemical shifts of the protons of the methyl groups in the nmR spectra agree with these data (Fig. 13).[216]
![[{"id":"TNSOI5S88A","type":"paragraph","data":{"text":"Chemical shifts of the protons of the methyl groups of BODIPY in <sup>1</sup>H nmR spectra"}}]](/storage/images/resized/bHdfWgIPgml44g6u5kOtdoxIUUEKoD9QQybRABCB_xl.webp)
Products of the Knoevenagel condensation are styryl-substituted BODIPYs, that exhibit a bathochromic shift in their absorption and emission spectra. Condensation of methyl groups at positions 3 and 5 results in a larger bathochromic shift compared to similar modification at positions 1 and 7.[38, 216, 217] Molecular orbital energy calculations show that the HOMO – LUMO energy gap in 3,5-distyryl-BODIPY is significantly larger than that observed for 1,7-distyryl-BODIPY.[192] The reaction conditions allow for the use of multiple aldehydes with different stereoelectronic characteristics. The introduction of donor substituents into the BODIPY molecule promotes charge transfer within the resulting mono- and distyryl derivatives, which is utilized in the design of chemosensors and molecular switches. [216]
The use of the Knoevenagel condensation to modify BODIPY and to tune the photophysical properties of the resulting products has been the subject of several reviews (see, e.g., Refs [38, 218, 219]). Styryl derivatives of BODIPY are of interest in the development of solar panels,[220] bioimaging agents [66, 221, 222] and PDT drugs.[223, 224] Due to the incredibly large variety of techniques for the synthesis of styryl-BODIPY using the Knoevenagel condensation, only the most interesting examples are discussed below.
3.5.1.1. Knoevenagel condensations at position 3
The first monostyryl BODIPY 175 was obtained by the Knoevenagel condensation by Rurack et al.[215] in 2001 (Scheme 95). The introduction of a styryl moiety with an electron-donating substituent at position 3 of the BODIPY core resulted in a bathochromic shift of the absorption and emission maxima in the electronic spectra and imparted considerable solvatochromism to the resulting fluorophore. Protonation of BODIPY 175 caused a hypsochromic shift in the absorption and emission spectra and disappearance of solvatochromism due to the disruption of charge transfer from the dimethylamine fragment to the BODIPY core. Compound 175 also showed the ability to oxidize with the formation of a cation-radical at a lower potential than BODIPY with other donor groups, indicating a high degree of charge delocalization within the molecule.
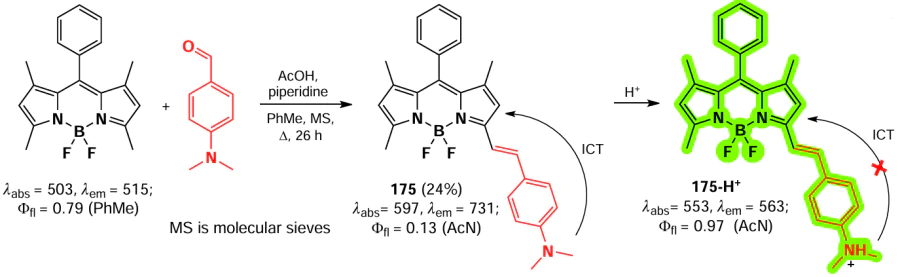
In 2021, Kaya et al.[225] synthesized styryl-BODIPY 176 as a ‘trap’ for singlet oxygen (Scheme 96). In the presence of singlet oxygen generated from the photosensitizer meso-tetraphenylporphyrin (TPP) under red light (630 nm), the opening of the furan cycle occurred, accompanied by a decrease in absorbance in the spectrum at 600 nm and the appearance of new maxima at 510 and 560 nm.
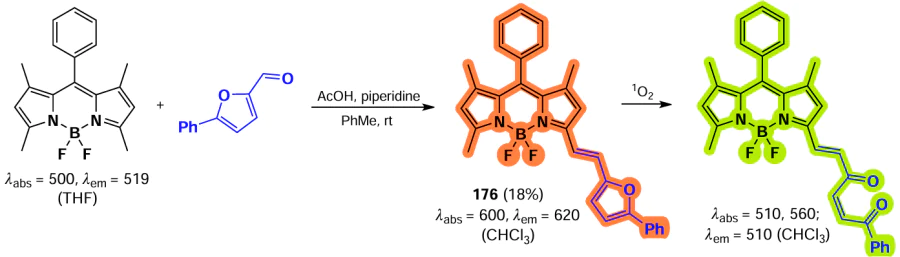
In 2020, Yang et al.[226] synthesized 2-styryl-BODIPY 177 by condensation of BODIPY with carbazole-3-carbaldehyde for use as an electron-donating material in organic photovoltaic cells (Scheme 97). The HOMO – LUMO energy gap calculated from cyclic voltammetry (CV) data for compound 177 was 1.99 eV, which is lower than that of the parent BODIPY (2.33 eV).[227] The ability to act as a solar cell component was investigated in a glass/ITO/PEDOT:PSS/active layer/PFN/Al configuration (PEDOT: PSS — poly(3,4-ethylenedioxythiophene)polystyrene sulfonate, PFN — conducting polymer), where BODIPY 177 was the electron donor in the active layer composition and the fullerene derivative PC71BM (methyl-[(6,6)-phenyl-C71]-butyrate) acted as the electron acceptor. The highest energy conversion efficiency achieved using BODIPY 177 as part of solar cells of this configuration was 6.41%.
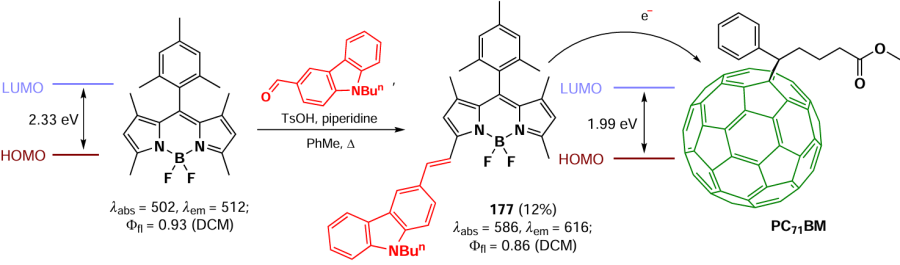
In 2012, Huang et al.[224] reported the synthesis of fullerene-conjugated distyryl-BODIPY by the Knoevenagel condensation under microwave irradiation. Eleven years later, Liu et al.[228] also used microwave irradiation to synthesize lysosome-targeted BODIPY 178 with combined activity (PDT and PTT) (Fig. 14). Compound 178 showed a fluorescence quantum yield of 0.1, indicating the predominance of nonradiative decay pathways, with a photothermal conversion efficiency (PCE) of 23%. Due to the presence of a morpholine fragment, BODIPY 178 accumulated in the lysosomes of HeLa cells and exhibited phototoxicity under 660 nm red light (1.2 W cm–2, 5 min). In vivo experiments showed that, after a single injection of this compound into mice and irradiation with a diode at λ = 660 nm (1.2 W cm–2, 1080 J cm–2, 15 min,) a significant reduction in tumor volume was observed on the 14th day of therapy.
![[{"id":"oKriZjnK7r","type":"paragraph","data":{"text":"Synthesis scheme and principle of<i> in vivo</i> action of BODIPY<b> 178</b> with a donor fragment of diethylamine with combined activity. The figure created by the authors based on the data of the publication[[ type=\"anchor\" referenceId=\"18277\" ]]."}}]](/storage/images/resized/3I4mGMjdxgj9pIfk9r2p8FGgCRK30PuUxAqqMW4w_xl.webp)
3.5.1.2. Knoevenagel condensations at positions 3 and 5
The Knoevenagel condensation of the methyl groups at C(3) and C(5) positions of BODIPY to give distyryl derivatives leads to a significant bathochromic shift of the absorption and emission maxima. Therefore, this method of BODIPY modification is widely used in the development of PDT and PTT agents, fluorescent probes, and optical materials.[219, 223, 229-237]
An expansion of the BODIPY π-system with styryl moieties at positions 3 and 5 promotes π – π-stacking and aggregation of fluorophores with the formation of J- or H-aggregates. According to the Kasha’s exciton theory, the nature of the aggregates formed depends on the slip angle between the direction of the transition dipole moment and the longitudinal axis of the centers of mass of the molecules.[90] When the slip angle exceeds 54.7°, the molecules are in a face-to-face orientation and form H-aggregates, which are characterized by a hypsochromic shift, a high degree of charge transfer, and fluorescence quenching. [238] When the slip angle is < 54.7°, J-aggregates are formed, in which the molecules are in a head-to-tail orientation. J-aggregates are characterized by a bathochromic shift of the absorption and emission maxima and an increase in fluorescence quantum yields.[239] However, J-aggregates can also suppress fluorescence and the ISC process in favor of nonradiative decay pathways,[240] or, conversely, activate ISC by reducing the energy gap between singlet and triplet states.[241] The bathochromic shift can be enhanced by a donor-acceptor-donor (D – A – D) fluorophore configuration in which the magnitude of the HOMO – LUMO gap is determined by the electronic properties of the donor and acceptor moieties,[242] thus shifting the absorption and emission bands into the NIR-II band (Fig. 15).[243]
J-Aggregation is promoted by the presence of sterically hindered groups in the structure, which strengthen the head-to-tail orientation by formation of hydrogen bonds. External and intramolecular exciton interaction, i.e. delocalization of excitation energy in the π-system of the molecule and between molecules in the aggregate, also promotes J-aggregation.[238, 244, 245] As a result of aggregation, the fluorescence of the molecule can be quenched, leaving energy dissipation via internal conversion and vibrational relaxation as the only way for the molecule to relaxa from the excited state.[246] The process of converting absorbed light into thermal energy is fundamental to PTT, whereby tumor tissues are heated by light, resulting in the induction of cell apoptosis.[247]
In 2017, Han et al.[248] presented a dyad in which a resonance energy transfer (RET) from the donor distyryl BODIPY (179a) to the acceptor (179b) was observed. The latter contains heavy iodine atoms at positions 2 and 6, promoting internal conversion and the ability of the molecule to form singlet oxygen (Fig. 16). Dyad 180 exhibited better singlet oxygen generation efficiency compared to that of diiodo-BODIPY 179b. BODIPY 180-based NPs obtained after encapsulation with folic acid and pluronic F-127 exhibited PDT activity on HeLa cell line in in vitro experiments (IC50 = 14.8 μg mL–1). In an in vivo study under the action of low-power NIR radiation (10 mW cm–2) a significant inhibition of 4T1 tumor growth was observed in comparison to the control groups.
![[{"id":"ZAa3uM7H2C","type":"paragraph","data":{"text":"Synthesis scheme of dyad <b>180</b>, its principle of action and therapeutic effect <i>in vivo</i>. The figure created by the authors based on the data of the publication[[ type=\"anchor\" referenceId=\"18297\" ]]."}}]](/storage/images/resized/hE7Dq9TgjFYRhwpZ9t7pf6wdBUg3QifSKrlDjuFg_xl.webp)
In 2021, Su et al.[240] synthesized distyryl NIR-absorbing BODIPY 181, which was capable of J-aggregation in order to design NPs with PTT activity (Fig. 17). The presence of an electronegative bromine atom at positions 2 and 6 and the electron-deficient core of BODIPY 181 promoted the formation of van der Waals Hal – p bonds and the aggregation of this BODIPY as two-dimensional sheets composed of antiparallel J-dimers (see Fig. 17).[249] Spherical NPs of 181-NP showed a PCE of 72% and high phototoxicity on 4T1 cells in vitro at a concentration of 10 μM under 785 nm laser irradiation (0.75 W cm–2, 10 min). When the therapeutic efficacy was examined in vivo, it was found that after administration of 0.9 mg kg–1 181-NP into 4T1 tumors and 785 nm laser irradiation (0.5 W cm–2, 10 min), 4 out of 6 tumors disappeared on the 21st day.
![[{"id":"ckahXcXqnx","type":"paragraph","data":{"text":"Schematic of the synthesis of <b>181-NP</b> nanoparticles with PTT activity and their therapeutic effects <i>in</i> <i>vivo</i>. The figure created by the authors based on the data of the publication[[ type=\"anchor\" referenceId=\"18289\" ]]."}}]](/storage/images/resized/s2ryZHt84tRs64WLghtQSndU36jkMF1VXRCvWv6z_xl.webp)
In 2023, Jiang et al.[32] synthesized four distyryl derivatives of BODIPY 182 with different aromatic substituents (Fig 18). The D – A – D configuration of BODIPY derivatives favored a shift of absorption to the NIR region,[250] and in aqueous media, all such compounds spontaneously formed J-aggregates. BODIPY 182-based nanoparticles proved to be effective PDT and PTT agents. The optimal ratio of ΦD and PCE values for the 182a-NP was found to be 40.76 and 54.9%, respectively. In in vitro experiments on the 4T1 cell line, 182a and 182b NPs showed the highest activity with IC50 values = 0.4 and 2.23 μg mL–1, respectively. In an in vivo study, 182a-NP and 182b-NP induced complete remission in BALB/C mice with 4T1 tumor line after irradiation with laser at λ = 808 nm (1 W cm–2, 3 min).
![[{"id":"1NkwXCPQKq","type":"paragraph","data":{"text":"Schematic of the synthesis of BODIPY <b>182</b> and the therapeutic effects of <b>182a-NP</b> nanoparticles <i>in vivo</i>. The figure created by the authors based on the data of the publication[[ type=\"anchor\" referenceId=\"18299\" ]]."}}]](/storage/images/resized/rVMnO959HtGC43PPANI3Ue5akIZ5Tv9Y31CByIcQ_xl.webp)
In 2021, Ansteatt et al.[251] synthesized a series of 3,5-diarylethynyl-BODIPY 183a – f via the Knoevenagel condensation (Scheme 98). Water-soluble BODIPYs 183g,h were obtained using a ‘click’ reaction with tetraethylene glycol azide (TEG). A significant bathochromic shift in the electronic spectra was observed for the donor distyryl-BODIPY compounds 183c – e compared to the parent BODIPYs. BODIPY 183g modified with TEG moieties exhibited moderate fluorescence intensity in the 730 – 740 nm region in polar solvents, including water, while being in micelles with Pluronic F127, Tween 20, and Triton X100.
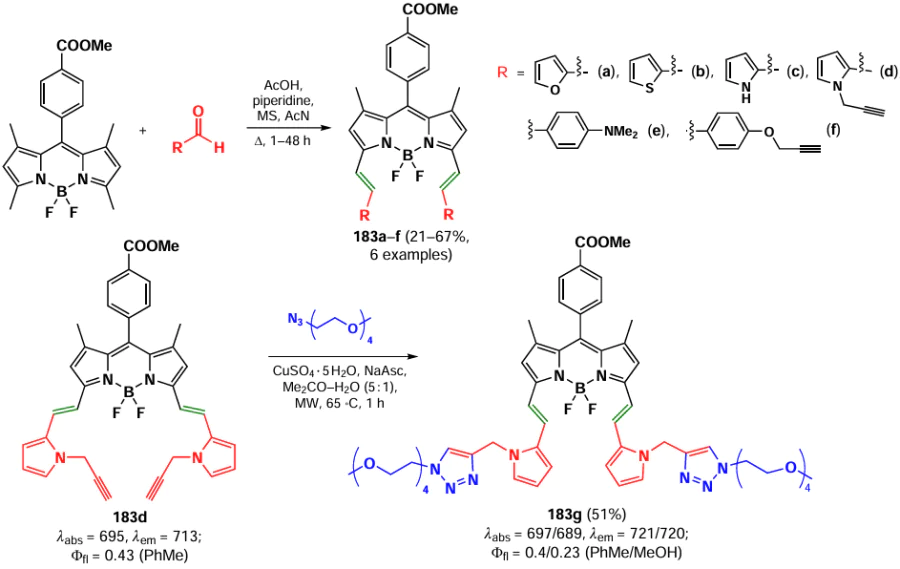
In 2020, Xi et al.[34] reported a highly efficient PTT agent based on styryl-substituted BODIPY 184 with a meso-CF3 group (Fig. 19). The PCE of 184-NP nanoparticles was 88.3%. This large PCE value was shown to be a consequence of the low transition energy between the HOMO and LUMO, which increased the fraction of nonradiative decay pathways, as well as the free rotation of the meso-CF3-group. 184-NP nanoparticles showed significant phototoxicity under low doses of red light (λ = 808 nm, 0.3 W cm–2) on HeLa, MCF-7 and 4T1 cell lines. In vivo photoacoustic (PA) imaging of 184-NP nanoparticles in BALB/C mice showed a high-contrast signal in a tumor region. The in vivo therapeutic efficacy study showed complete disappearance of 4T1 tumors after a single injection of 184-NP nanoparticles (at a dose of 300 μM in 100 μL) and light irradiation (λ = 808 nm, 0.3 W cm–2, 8 min) on the 16th day after administration.
![[{"id":"TQox4DgAep","type":"paragraph","data":{"text":"Schematic of BODIPY <b>184</b> synthesis and <i>in vivo</i> action of <b>184-NP</b> nanoparticles. The figure created by the authors based on the data of the publication[[ type=\"anchor\" referenceId=\"18034\" ]]."}}]](/storage/images/resized/1f9ibITulv36PL7j7orpTm3s5tAHqCst4FdVc5ou_xl.webp)
In 2023, Chen et al.[31] designed NIR-absorbing 3,5-diene-BODIPY with PTT activity (Fig. 20). Due to the presence of a freely rotating meso-CF3 group, the fluorescence of BODIPY 185 was negligible. Upon acidification of BODIPY 185, sequential protonation of the nme2 groups occurred, resulting in a shift of an absorption maximum from 836 to 774 and 710 nm due to suppression of ICT. However, no fluorescence enhancement was observed upon acidification, indicating that the rotation of the CF3 group is the primary nonradiative decay pathway. Despite the inability of compound 185 to generate 1O2 upon irradiation, nanoparticles based on it (185-NP) exhibited photothermal activity with a PCE of 44.4%. In an in vitro study, 185-NP nanoparticles at a concentration of 30 μM significantly reduced the survival of MGC-803 cells upon irradiation with an 808 nm laser (0.3 W cm–2, 30 min).
![[{"id":"XfO3d0Wm2y","type":"paragraph","data":{"text":"Scheme of the synthesis of BODIPY <b>185</b> and the <i>in vitro</i> action of the NPs based on it. The figure created by the authors based on the data of the publication[[ type=\"anchor\" referenceId=\"18034\" ]]. A dash indicates no emission."}}]](/storage/images/resized/mDpzwTWgsh1XoskwFtJvpn1qpV559w4qLMwwHelr_xl.webp)
In 2023, Gao et al.[30] utilized two sequential Knoevenagel condensations to synthesize BODIPY 186 with PDT and PTT activity. BODIPY 186 was designed with a lysosome-targeting morpholino moiety within its styryl substituents, and the second styryl moiety was nitrogen mustard, bis(2-chloroethyl)aniline (Fig. 21). BODIPY 186 demonstrated high PTT activity (PCE = 40%) and accumulated effectively in lysosomes. The IC50 values for the HeLa cell line irradiated with a 760 nm laser (0.5 W cm–2) were 8.75 μM under normoxic conditions and 10.5 μM under hypoxic conditions. In an in vivo study on 4T1-bearing mice, a significant reduction in tumor volume was observed on day 14 after intravenous injection of BODIPY 186 into mice, followed by irradiation with a 760 nm laser (0.8 W cm–2, 6 min).
![[{"id":"DfWglK7cFj","type":"paragraph","data":{"text":"Synthesis scheme of BODIPY <b>186</b>, its principle of action and therapeutic effect <i>in vivo</i>. The figure created by the authors based on the data of the publication[[ type=\"anchor\" referenceId=\"18030\" ]]."}}]](/storage/images/resized/XtiR1HVRCawGRZyIOJwBLrl130zjEwccBRcPU00S_xl.webp)
Methyl groups at positions 1 and 7 of meso-aryl-BODIPY exhibit low reactivity due to steric hindrance.[216] Despite this, Zhang et al.[252] demonstrated non-standard 1,7 regioselectivity in the Knoevenagel condensation. The reaction of 2,6-diformyl-1,3,5,7-tetramethyl-BODIPY with substituted benzaldehyde resulted in a disubstitution product at methyl groups at positions 1 and 7, yielding compound 187 (Scheme 99). Further reductive amination gave BODIPY 188, a pH-sensitive sensor whose fluorescence quantum yield increased more than 14-fold as the pH increased from 4.5 to 7.4. The pH-response mechanism involved the suppression of PET by the deprotonated form of the compound. In acidic conditions, probe 188 exhibited no fluorescence due to the d-PET § process from BODIPY to protonated morpholine. When evaluating the ability of sensor 188 to visualize HUVEC-C cells intracellularly, it was found that it selectively accumulates in lysosomes and produces a strong fluorescence response at intracellular alkaline pH.
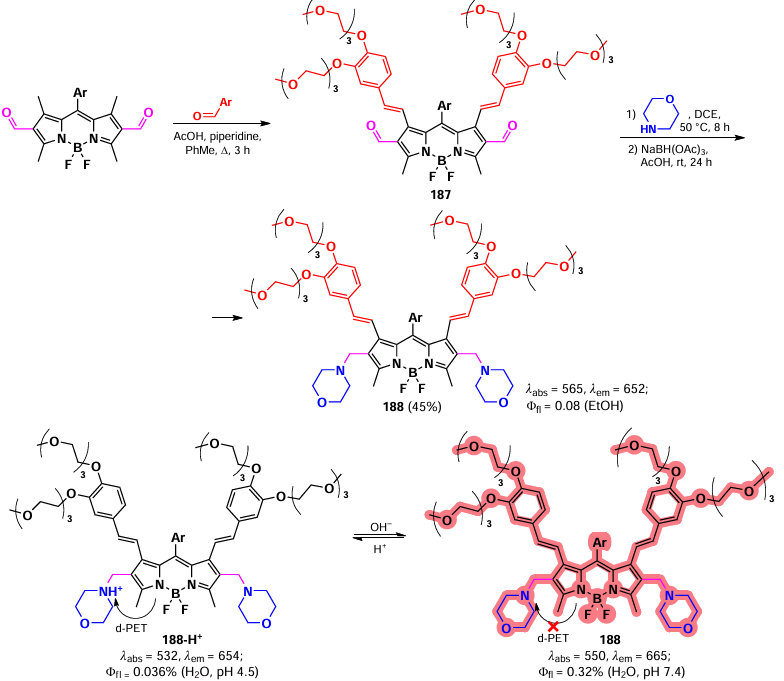
In 2023, Zhu et al.[33] reported highly fluorescent BODIPY 189 with electron-acceptor substituents for NIR-II fluorescence imaging (Scheme 100). Due to the formation of J-aggregates that were encapsulated in the NPs, the absorption and emission of 189-NP nanoparticles shifted to the NIR band in the spectrum. A high fluorescence quantum yield was also observed in the NIR-II band (Φfl = 0.005), which was attributed to the full planarity of the BODIPY core and optimal stacking for J-aggregation. In an in vivo imaging study, high-contrast real-time imaging of the mouse vascular system was obtained. Due to its ability to emit both in the visible range as monomers and in the NIR-II region in the form of J-aggregates, an anti-counterfeiting ink was developed based on BODIPY 189.
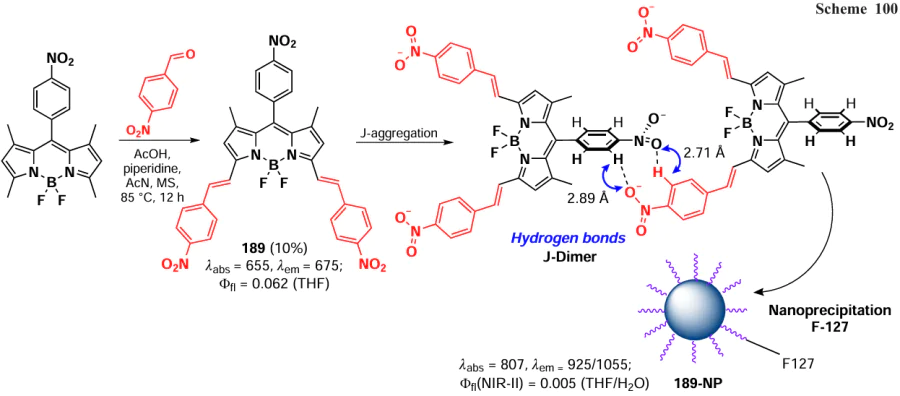
In 2024, Li et al.[15] designed 3,5-distyryl-BODIPY 190 as an agent for sonodynamic therapy (Fig. 22). Due to the «heavy atom» effect and low fluorescence intensity, compound 190 was found to be a good photosensitizer with ΦD = 0.95. It showed the ability to be sonosensitized by ultrasound (US) at 1.5 W cm–2, with singlet oxygen being the main product of this process. In the intracellular environment, BODIPY 190 could form ROS upon US exposure. The IC50 value on 4T1 cells was 3.81 μM in normoxia and 5.36 μM in hypoxia. In a study of antitumor activity in vivo, statistically significant inhibition of tumor growth was observed on the 15th day of therapy after 7 injections of BODIPY 190 every 2 days, followed by US irradiation (1.5 W cm–2, 5 min).
![[{"id":"uU-QX2NHkB","type":"paragraph","data":{"text":" Synthesis scheme of BODIPY<b> 190</b>, its principle of action and therapeutic effect<i> in vivo</i>. The figure created by the authors based on the data of the publication[[ type=\"anchor\" referenceId=\"18015\" ]]."}}]](/storage/images/resized/fLEUxtHqT6NS7E3UPiacQlnPAdSOg9MZpX8l9RxD_xl.webp)
3.5.1.3. Knoevenagel condensation at the Me-groups in positions 1, 3, 5, and 7
In 2009, Buyukcakir et al.[216] proposed a method for the synthesis of 1,3,5,7-tetrastyryl-BODIPY 191 from the tetramethyl derivative in the presence of 7 equivalents of anisaldehyde (Scheme 101). Upon the condensation at positions 1 and 7, the bathochromic shift in the spectrum was approximately 20 nm, which was much smaller than the ~ 70 nm shift upon condensation at positions 3 and 5. The fluorescence quantum yields of tri- and tetrastyryl-BODIPY remained almost unchanged when styryl substituents were introduced at positions 1 and 7, compared to 3,5-distyryl-BODIPY.
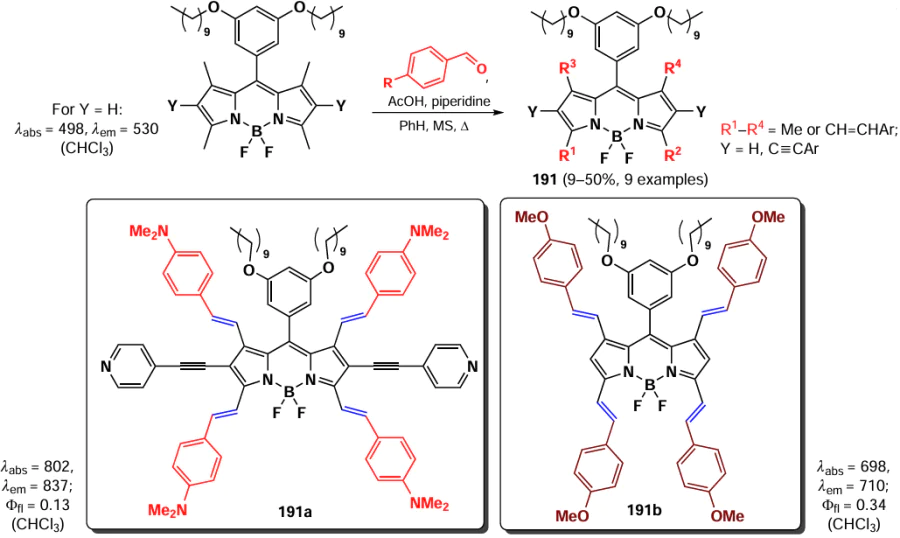
In 2014, the same research group [192] synthesized a series of 2,6-ethynylaryl-BODIPYs with two different styryl substituents at positions 3, 5 and 1, 7 (Scheme 102). The resulting BODIPYs exhibited selective response to Hg2+ cations; coordination of the Hg2+ cation to probe 193 with mercury-chelating moieties at positions 3 and 5 resulted in a shift of the absorption maximum in the spectrum from 800 to 715 nm and a fluorescence enhancement at 736 nm. Meanwhile, coordination of the Hg2+ cation to probe 192 with mercury-chelating moieties at positions 1 and 7 did not result in a shift of the absorption bands. The differences in the nature of the response of BODIPY 192 and 193 to Hg2+ were explained by the greater sensitivity of the HOMO of 3,5-distyryl-BODIPY to protonation or the binding to the cation than that of the analogous orbitals of 1,7-distyryl-BODIPY, due to the greater destabilization of the HOMO upon introduction of styryl substituents at positions 3 and 5 than at positions 1 and 7.
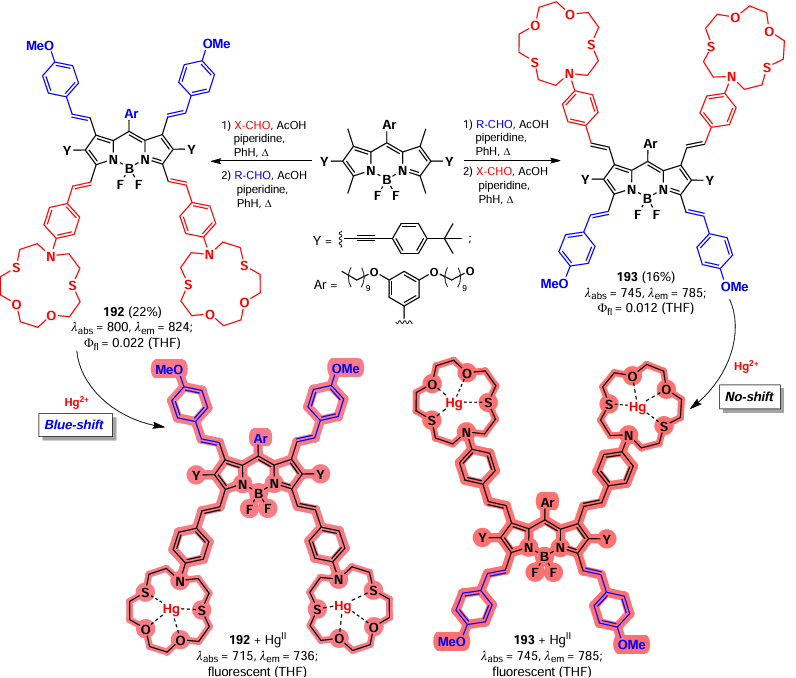
2,6-Ethynylaryl-1,3,5,7-tetrastyryl-BODIPY are also of interest as PTT agents. In 2019, Hu et al.[253] synthesized tetrastyryl-BODIPY 194 as a promising NIR-absorbing PTT agent (Fig. 23). According to density functional theory (DFT) calculations, in the antiparallel J-dimer 194 the electron density of the HOMO is completely delocalized on one of the dimer molecules and the LUMO on the other, indicating an emission-damping intermolecular charge transfer due to excitation. 194-NP nanoparticles obtained after encapsulation of 194 in DSPE-mPEG5000 showed high PCE (61%) and proved to be good PTT agents in in vivo experiments: inhibition of HeLa tumor growth was observed after irradiation of mice with λ = 808 nm laser (0.3 W cm–2, 5 min).
![[{"id":"ApCxb5O51x","type":"paragraph","data":{"text":"Synthesis scheme of BODIPY <b>194</b> and the principle of action of NPs based on it <i>in vivo</i>. The figure created by the authors based on the data of the publication[[ type=\"anchor\" referenceId=\"18302\" ]]."}}]](/storage/images/resized/QuCytlNI7u2QtYGXGvFeUA4FSnwHGqKA9k07BHqE_xl.webp)
A series of mono-, di-, and tristyryl-BODIPY 195a – c were synthesized and investigated as agents for combined PDT/PTT therapy in 2019 by Zou et al.[216] (Fig. 24). At acidic pH, the absorption of BODIPY NPs shifted to the red region of the spectrum, which was accompanied by an increase in PCE due to the suppression of intramolecular charge transfer (ICT) as a molecule relaxation pathway. In in vitro study, 195c-NP NPs showed IC50 values = 4.16 μM, the highest PCE (60.5%), and effectively inhibited HeLa tumor growth after intravenous administration and irradiation with an 808 nm laser (1 W cm–2, 8 min).
![[{"id":"Hjg8ezl_w7","type":"paragraph","data":{"text":"Schematic of the synthesis of BODIPY <b>195a</b> – <b>c</b> and the principle of action of <b>195c-NP</b> nanoparticles <i>in</i> <i>vivo</i>. The figure created by the authors based on the data of the publication[[ type=\"anchor\" referenceId=\"18266\" ]]."}}]](/storage/images/resized/eosCTXARoLBXd9lDyz9tmZ5jIWUSnQw4f2nVijvQ_xl.webp)
3.5.1.4. Knoevenagel condensation with meso-methyl group
In 2011, Chattopadhyay et al.[98] reported the synthesis of 8-styryl derivatives 196 from commercially available BODIPY PM597 (R1 = But, R2 = Me) and PM567 (R1 = Et, R2 = Me) (Scheme 103). The authors attributed the regioselectivity of the condensation to the alleviation of steric hindrance present in alkyl-BODIPY between the protons of the methyl groups at the C(1), C(7) and meso-position atoms and to the increased planarity of the molecule resulting from condensation. Nevertheless, Palao et al.[254] demonstrated that the Knoevenagel condensation proceeds regioselectively at the meso-position even in the absence of methyl groups at positions 1, 7, leading to the formation of BODIPY 197. This regioselectivity is primarily due to the preferential delocalization of the positive charge of BODIPY at the meso-position. The introduction of styryl substituents at the meso-position of BODIPY results in a small bathochromic shift (8 – 30 nm) and complete quenching of fluorescence, which is due to nonradiative relaxation caused by vibrations of the pyrrole cycles outside the plane of the BODIPY core.[255] An additional contribution to the fluorescence quenching is made by the ICT effect, which manifests itself in the case of aldehydes with both donor and acceptor substituents.
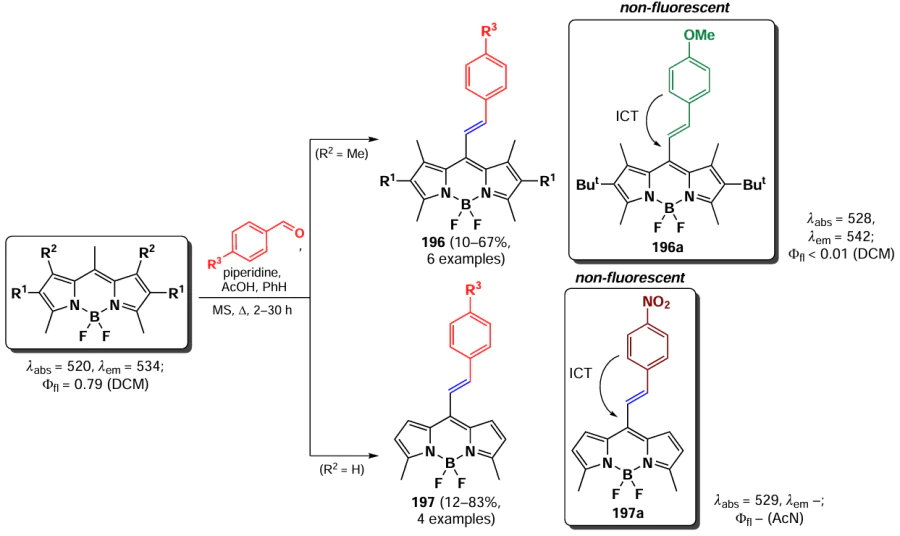
In 2012 and 2016, Yu et al.[77, 256] reported the synthesis of isoindole-based NIR-absorbing 8-styryl-substituted BODIPYs, which also exhibited almost no fluorescence (Scheme 104).
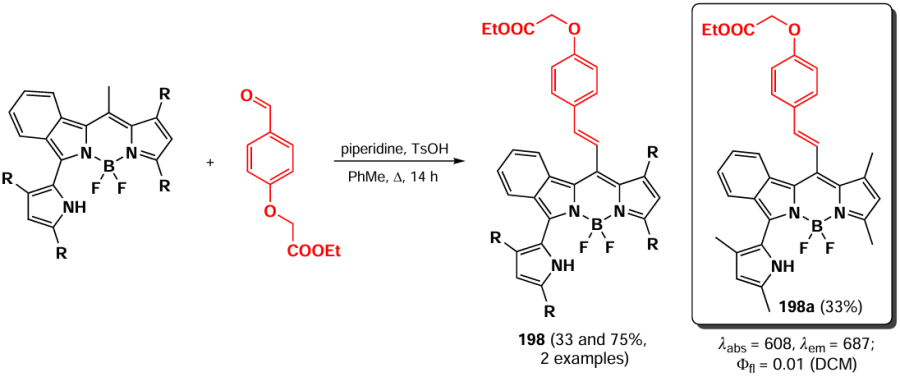
3.5.2. Electrophilic Substitution Reactions in the α-methyl groups of BODIPY
The methyl group at the α-position of BODIPY exhibits significant CH-acidity; the nucleophilic anion formed as a result of deprotonation is capable of reacting with various electrophiles. In 2009, Shandura et al.[257] demonstrated that the sequential reaction of 3,5-dimethyl-BODIPY with 1,1-di(methoxy)trimethylamine and TFA produced enamine 199a and aldehyde 199b, respectively (Scheme 105). Oxidation of the 3-methyl group caused a bathochromic shift of the absorption maximum for enamine 199a and complete quenching of the fluorescence due to the PET effect, whereas hydrolysis of enamine to aldehyde 199b restored the photophysical characteristics to those identical to the parent 3,5-dimethyl-BODIPY.
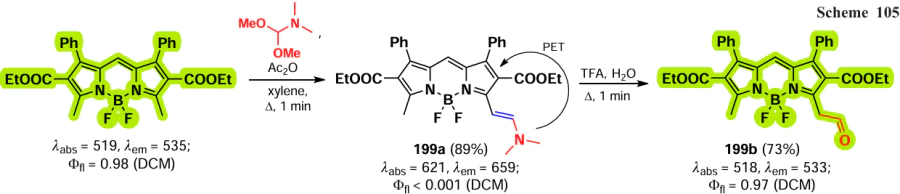
In 2012, Ulrich et al.[258] reported the electrophilic bromination of hexaalkyl-BODIPY with NBS to form unstable 3-bromomethyl- and 3,5-bis(bromomethyl) BODIPYs. These compounds readily react with various O-, N-, S-, and P-nucleophiles in nucleophilic substitution reactions yielding mono- (200) and di-substituted BODIPYs (201) (Scheme 106). The introduction of nucleophiles at positions 3 and 5 did not significantly affect the absorption and emission properties of BODIPY. However, BODIPY 201a with a tertiary amino group exhibited an extremely low fluorescence quantum yield (Φfl = 0.01) due to the PET effect.
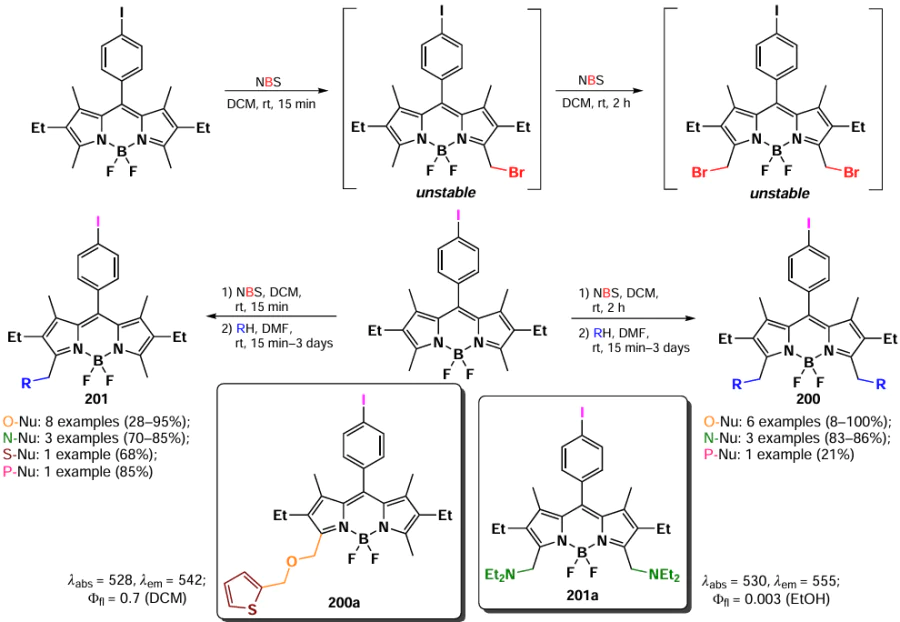
In 2020, Kand et al.[149] utilized the nucleophilic substitution reaction of the bromine atoms at positions 3 and 5 to conjugate 2-mercaptoethanesulfonic acid (MES) with the BODIPY core, yielding water-soluble photo-removable protective groups based on meso-substituted BODIPY (Scheme 107). In a study of the ability of MES-BODIPY to accumulate in cells, the mono-substituted BODIPY 202 showed a two-fold higher level of intracellular fluorescence compared to the original BODIPY. In contrast, the disubstituted BODIPY 203 was unable to penetrate HEK293T cells.
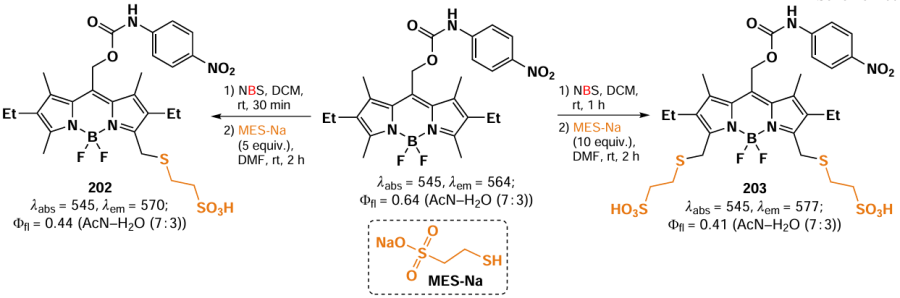
3.5.3. Oxidation of α-methyl groups of BODIPY
Due to the possibility of stabilising benzyl-type cations in the α-positions of BODIPY, peralkylated BODIPYs are susceptible to oxidation at these positions via DDQ and lead(IV) acetate.[6] In 1994, Sathyamoorthi et al.[259] synthesized 3-formyl-BODIPY 204 by oxidation of hexamethyl-BODIPY via DDQ and 3,5-bis(acetoxymethyl)-BODIPY 205 by oxidation via lead(IV) acetate (Scheme 108). Oxidation of the α-methyl group in BODIPY 204 resulted in a quenching of the fluorescence to Φfl = 0.12.
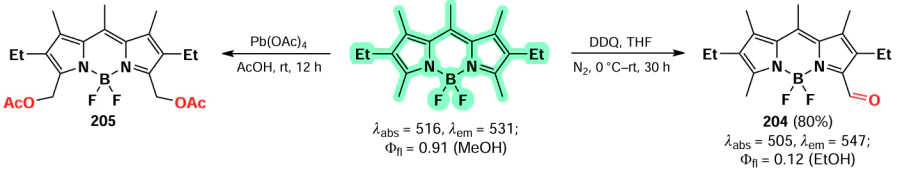
In 2016, Wu et al.[260] designed a Cys-sensitive fluorescent probe based on 1,5,7,8-tetramethyl-3-formyl-2,6-diethyl-BODIPY (204) (Scheme 109). Upon reaction with Cys a five-membered ring was closed, resulting in an increase in fluorescence and a bathochromic shift of the absorption maximum due to PET quenching. However, in the presence of other amino acids, including Hcy, no fluorescence response of BODIPY 204 was observed.
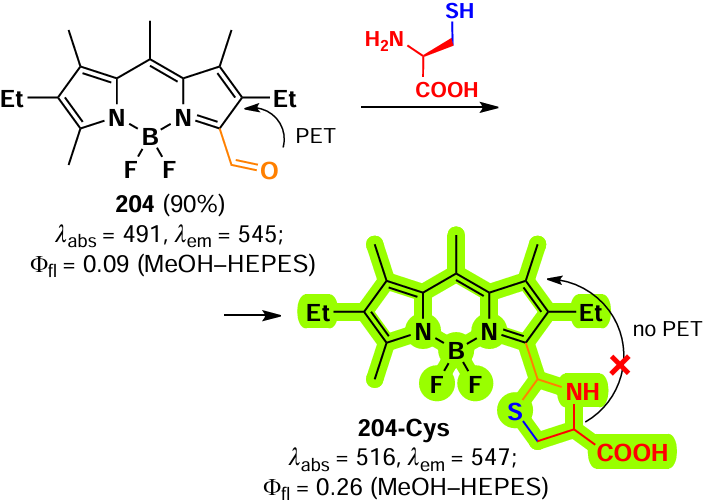
In 2022, Blázquez-Moraleja et al.[6] proposed the use of 3,5-acetoxymethyl groups as leaving groups for nucleophilic substitution reactions in the B(CN)2-BODIPY (Scheme 110). Substitution of the acetoxy group proceeded via the SN1 mechanism; acid-catalysed hydrolysis led to the formation of a stabilized carbocation, which is readily reactive with various nucleophiles. B(CN)2-BODIPY was used as a starting compound,[261, 262] stable in the presence of Lewis acids; then 3-acetoxymethyl-BODIPY was obtained through the oxidation via lead acetate, followed by the synthesis of BODIPYs 206 – 210 via nucleophilic substitution.
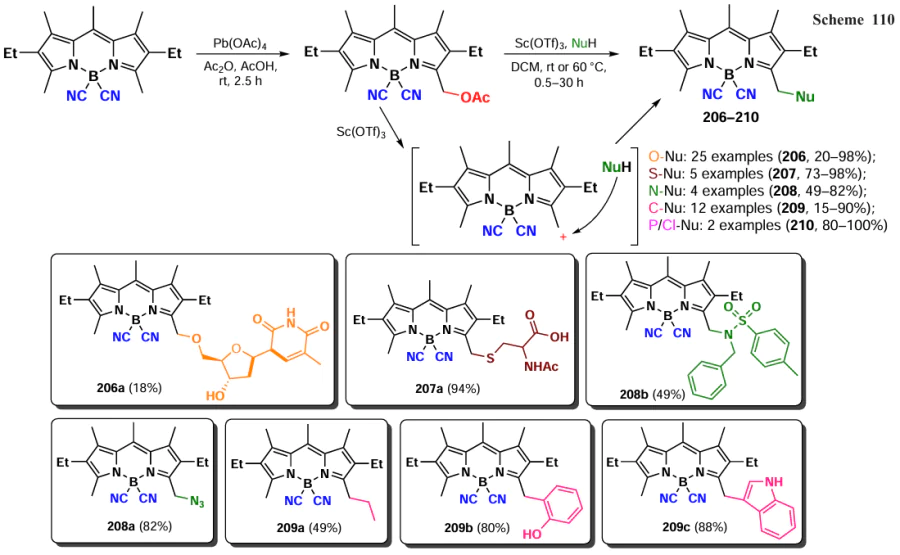
Scandium(III) triflate proved to be an optimal catalyst for nucleophilic substitution reactions. The authors investigated the reactivity of 3-methylacetoxy-BODIPY under the action of various O-, N-, S- and C-nucleophiles, yielding > 50 BODIPYs as products of nucleophilic substitution. Aliphatic alcohols (BODIPY 206a) and carboxylic acids were used as O-nucleophiles, while azide and sulfonamide were used as N-nucleophiles yielding BODIPYs 208a,b. C-nucleophiles, including the natural compounds tyrosine and resveratrol, as well as aliphatic and aromatic nucleophiles such as TMSCN (TMS — trimethylsilyl), diethylzinc (209a), indole (209b), and BODIPY at position 2, were also involved in the reaction. Sequential substitution of the two acetoxy groups in the BODIPY scaffold with different nucleophiles yielded asymmetric nucleophilic substitution products.
3.6. Radical reactions of BODIPY
In 2015, Verbelen et al.[263] demonstrated the feasibility of direct radical arylation of the BODIPY core at positions 3 and 5 using diazonium salts and ferrocene as a catalyst, yielding 3-aryl- (211) and 3,5-diaryl-BODIPY 212 (Scheme 111). Aryldiazonium salts are well-known sources of aryl radicals formed by homolytic dediazonation. The reactions occurred exclusively at the C(3) and C(5) carbon atoms of BODIPY, since the addition of the radical species at 3-position of one of the pyrrole moiety and subsequent charge delocalization over several double bonds resulted in a stabilized allyl-type radical.
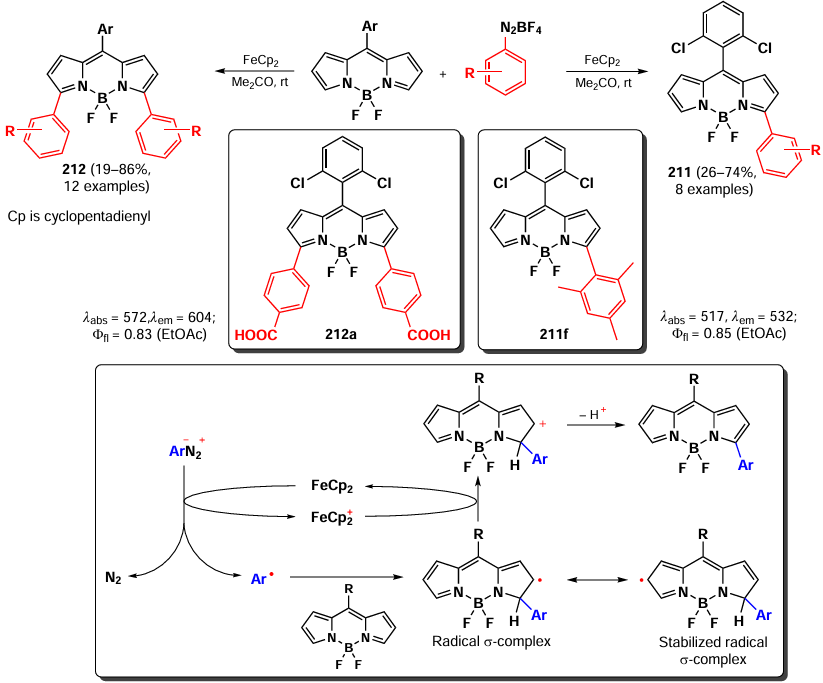
In 2015, the same research group [264] reported the radical alkylation reaction of BODIPY with potassium trifluoroborates and boronic acids, occurring at positions 3, 5, 7, using MnIII acetate as an oxidizing agent. This method allowed to obtain tri- and tetrasubstituted products, as well as unsymmetrical derivatives of BODIPY 213, by direct CH-alkylation of the BODIPY core (Scheme 112). The introduction of several sterically hindered cycloalkyl groups suppressed the formation of non-fluorescent π – π-aggregates, resulting in solid-state emissive BODIPY with quantum yields up to 0.14 for BODIPY 213b.
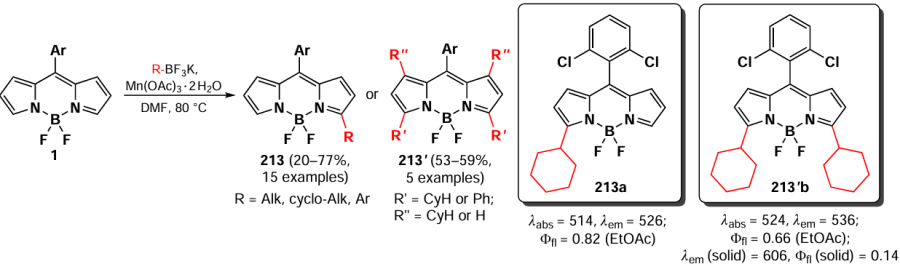
In 2016, Zhou et al.[265] proposed a method for radical arylation of BODIPY at positions 3 and 5 using diaryliodonium salts, which provided 3-aryl- (214) and 3,5-diaryl derivatives (215). The reactions were inhibited in the presence of 2,6-di-tert-butyl-4-methylphenol, thereby confirming the presence of free radicals. The proposed mechanism involved the attack of the aryl radical at the α-position to generate radical A (Scheme 113), which reacted again with the diaryliodonium salt to form intermediate B. An evaluation of the energies of the transition states after the attack of the arynium cation at positions 2 and 3 (routes a and b) was carried out; the transition state after the attack at position 3 had a lower energy (by 3.3 kcal mol–1), as a result, the product obtained after the attack at position 2, intermediate C, was not formed.
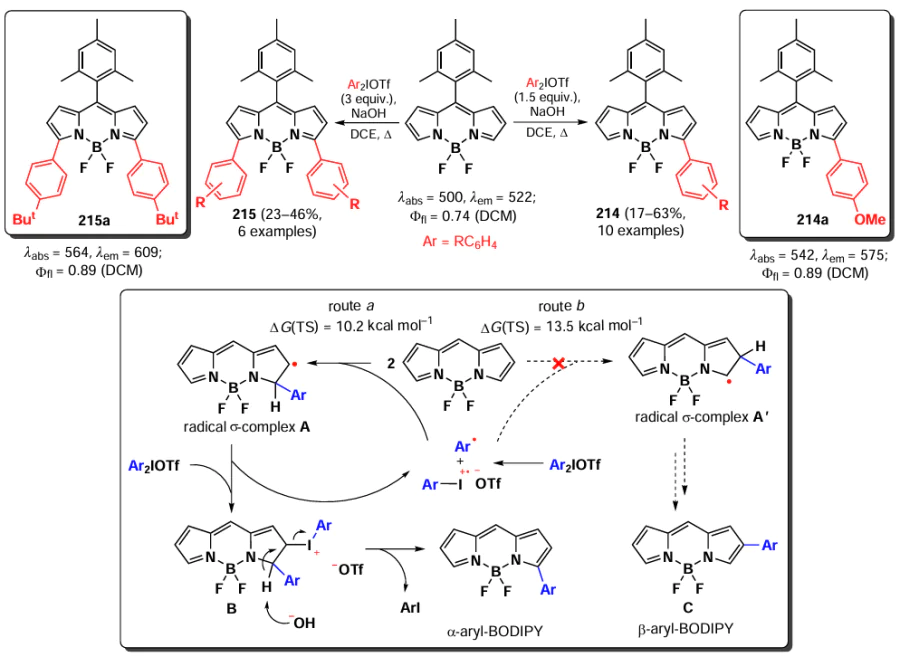
In 2017, Boens et al.[266] reported the radical alkylation of BODIPY at positions 3 and 5 with allylic alkenes and ethers in the presence of the TBAI – TBHP system. The reactions were inhibited in the presence of BHT and did not proceed at free positions 2 and 6, which excluded the electrophilic nature of the attacking particle. The mechanism proposed by the authors included the formation of an allyl radical as a key intermediate (Scheme 114). For the mono- (216) and dialkylated BODIPYs (217) obtained, an increase in fluorescence was observed compared to the parent compounds, for example, up to 0.13 and 0.64 for derivatives 216a and 217a, respectively.
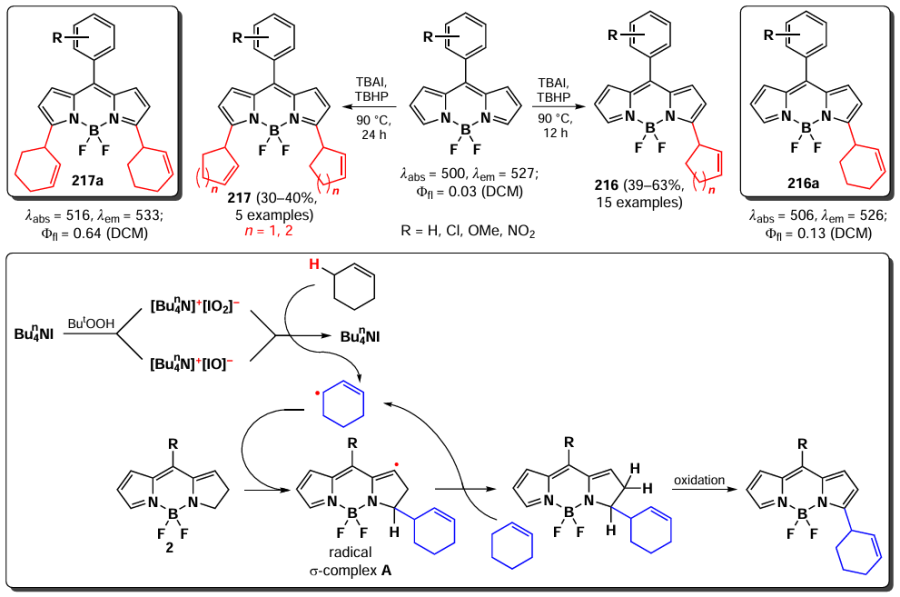
In 2018, Zhang et al.[267] proposed a method for the radical amination of BODIPY at positions 3 and 5 using silver acetate as a catalyst and oxidizer (Scheme 115). The processes were inhibited in the presence of TEMPO and BHT. The mechanism proposed by the authors involved a single electron transfer from AgI to a secondary amine with generation of an amine radical; this mechanism was confirmed by the formation of a ‘silver mirror’ during the reaction. A series of synthesized 3,5-diamino-BODIPYs 218 – 220, in particular 218a, exhibited high ER specificity together with low cytotoxicity.
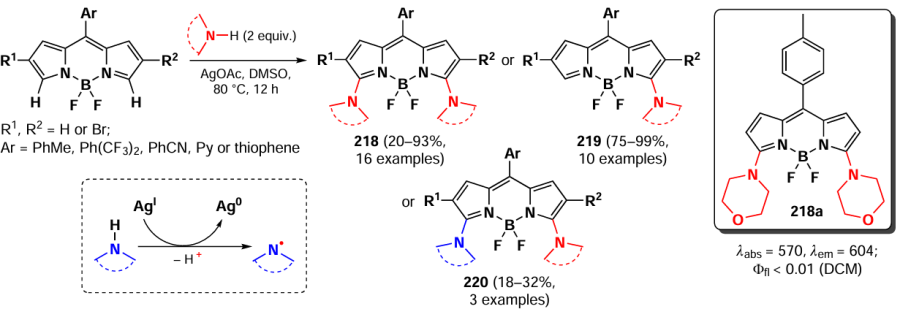
In 2019, Lv et al.[268] reported radical coupling reactions of BODIPY with thioethers in the presence of tert-butylperoxy benzoate (TBPB), which yielded 3- (221) and 3,5-dithio-BODIPY (222) (Scheme 116). These processes were also inhibited in the presence of TEMPO and BHT, confirming their radical mechanism.
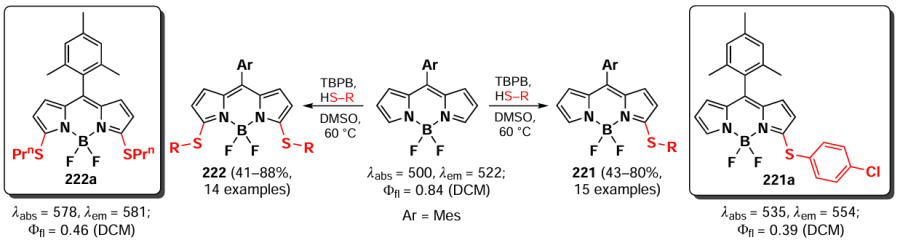
A method for the photocatalyzed coupling of BODIPY with thioethers at positions 3 and 5 was also proposed by Ma et al.[269] The authors carried out experiments using thiols with both donor and acceptor substituents, including sterically hindered ones (Scheme 117). For sulfur-containing BODIPYs 223 and 224, including BODIPYs 223a and 224a, a red-shift and fluorescence enhancement were observed upon sequential introduction of thiol substituents. The mechanism of the process involved photoexcitation of BODIPY, and then electron transfer from the thiol to the excited BODIPY, which is an oxidizing agent. The radical formed after deprotonation attacked BODIPY at the α-position.
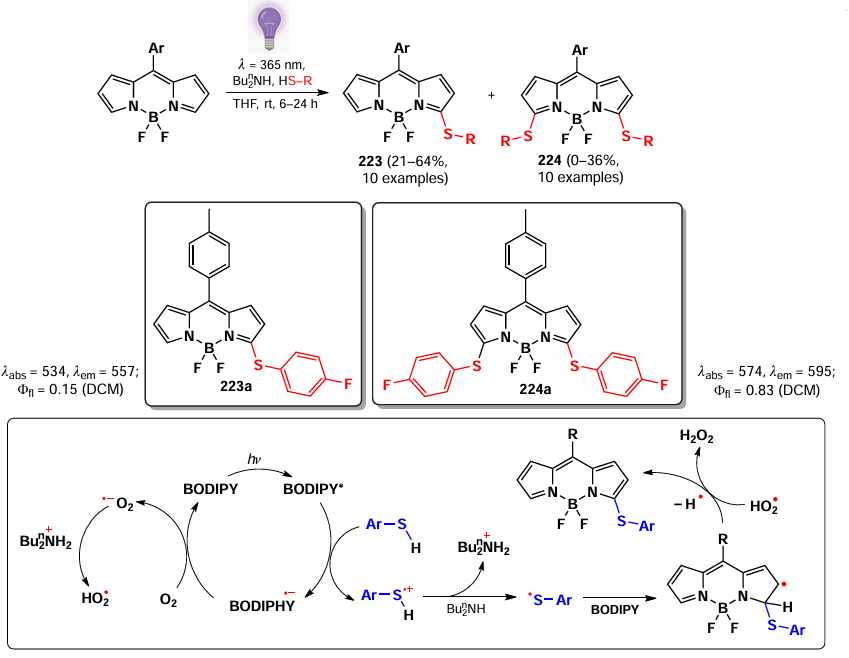
In 2024, Zhang et al.[26] developed an electrochemical approach to introduce a tosyl group at the β-position (1,3,5,7) of substituted BODIPY to obtain 2-sulfonyl derivatives 225. Electrooxidative β-sulfonylation of BODIPY with sulfonylhydrazide did not require the presence of additional oxidizing agents (Scheme 118). Compound 225a was used for fluorescent staining of the cytoplasm of human hepatocellular carcinoma cells (HepG2).

3.7. Photochemical reactions of BODIPY
[]
3.7.1. BODIPY derivatives as a source of singlet oxygen
In 2011, Jing et al.[270] proposed to use BODIPY as photocatalysts for thioanisole oxidation. BODIPYs 226 showed high photocatalytic activity, and the reaction was facilitated by the high polarity of a medium. The mechanism proposed by the authors involved the oxidation of thioanisole by singlet oxygen (Scheme 119).
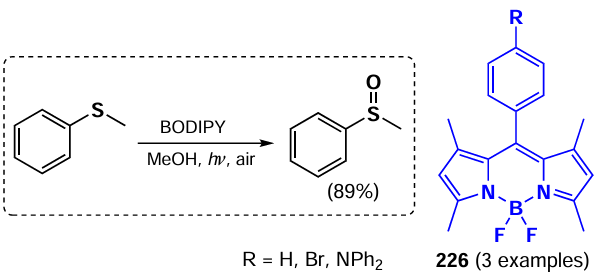
A year later, the same research group [271] used a heterogeneous catalyst, based on BODIPY, styrene and divinylbenzene, for the photooxidation of sulfides. The catalyst demonstrated high activity, but the reaction proceeded slower compared to the use of a homogeneous catalyst. In 2013, Zhang et al.[272] presented dyad PDT agents 227a – c, capable of resonant energy transfer from the non-iodinated to the iodinated moiety. Spectral studies of the transition state confirmed that the triplet state was shared between the donor and acceptor, since heavy atom-free BODIPYs were not able to populate their triplet states. The authors proposed a ‘ping-pong’ mechanism of energy transfer, namely, forward singlet energy transfer from the BODIPY-donor to the BODIPY-acceptor (singlet EnT), followed by internal conversion and backward triplet state energy transfer to the BODIPY fragment (triplet EnT). The designed dyads 227a – c acted as triplet photosensitizers, for example, for the photooxidation of 1,5-dihydronaphthalene.
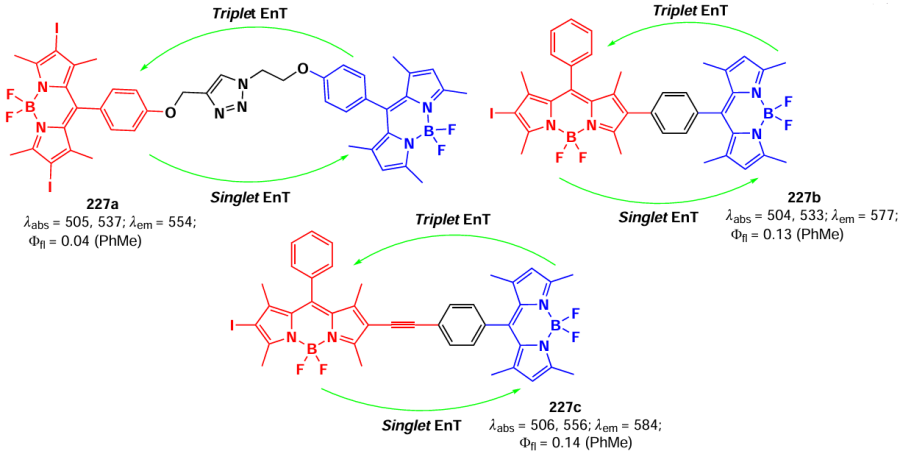
In 2013, Ma et al.[223] used iodinated BODIPY 228a for the aerobic photocatalysis of two reactions: the oxidative coupling of amines with imine formation, and the photooxidation of dihydroxynaphthalenes followed by one-pot functionalization of 1,4-naphthoquinones to produce N-aryl-2-amino-1,4-naphthoquinones (Scheme 120). The transformations occurred for amines containing protons at the α-position. A one-pot reaction of photooxidation of dihydronaphthalenes followed by 1,4-coupling of aromatic amines was also carried out. An EPR study in the presence of 2,2,6,6-tetramethylpiperidine (TEMP) and 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), which are ‘traps’ for singlet oxygen and the oxygen anion radical, revealed the participation of 1O2 in the catalytic cycle. The mechanism of oxidative coupling of amines proposed by the authors was the generation of singlet oxygen by the triplet state of BODIPY 228a, oxidation of amines to the corresponding imines, and nucleophilic attack of the free amine on the imine carbon atom (see Scheme 120).[223]
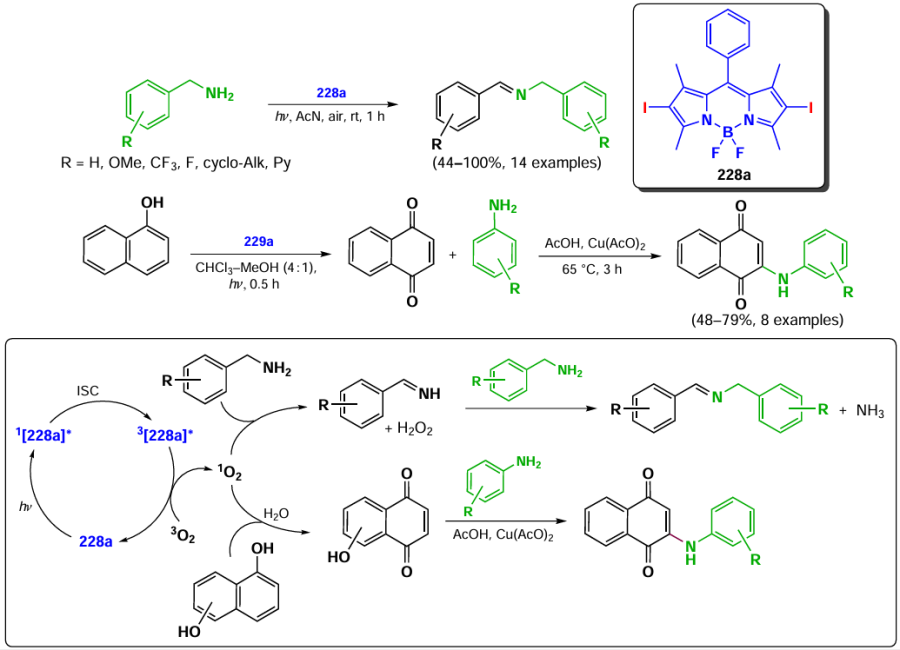
3.7.2. Photoexcited BODIPYs as oxidizing or reducing agents
In 2013, Huang and Zhao [273] investigated three redox processes photocatalyzed by BODIPY:
— the Aza-Henry reaction with tetrahydroisoquinoline and the addition of enolate anions;
— tandem oxidation/[3 + 2]-cycloaddition/oxidative aromatization reactions with tetrahydroisoquinolines and maleimides;
— C – H arylation of heteroarenes with diazonium salts.
In all cases, 2,6-diiodo-BODIPY 228a or 228b served as photocatalysts (Scheme 121, Scheme 122).
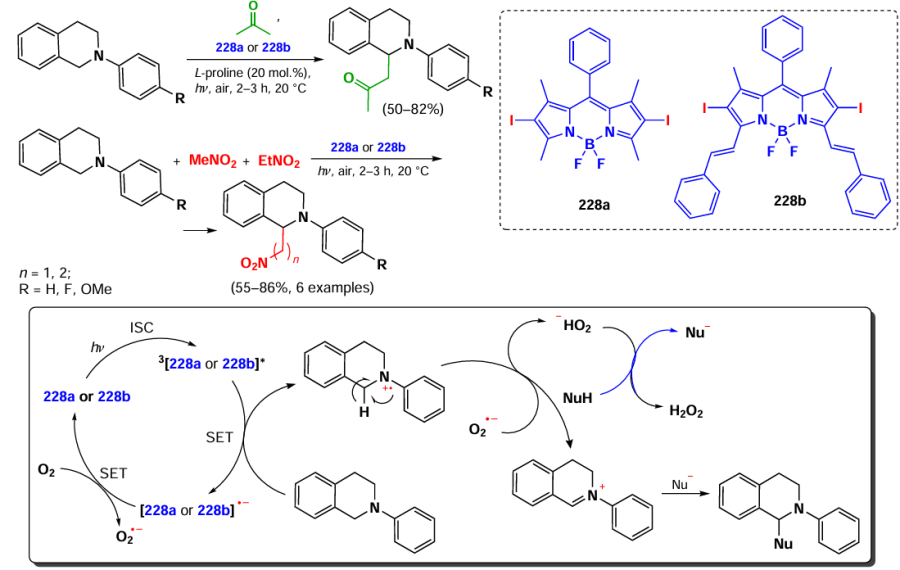
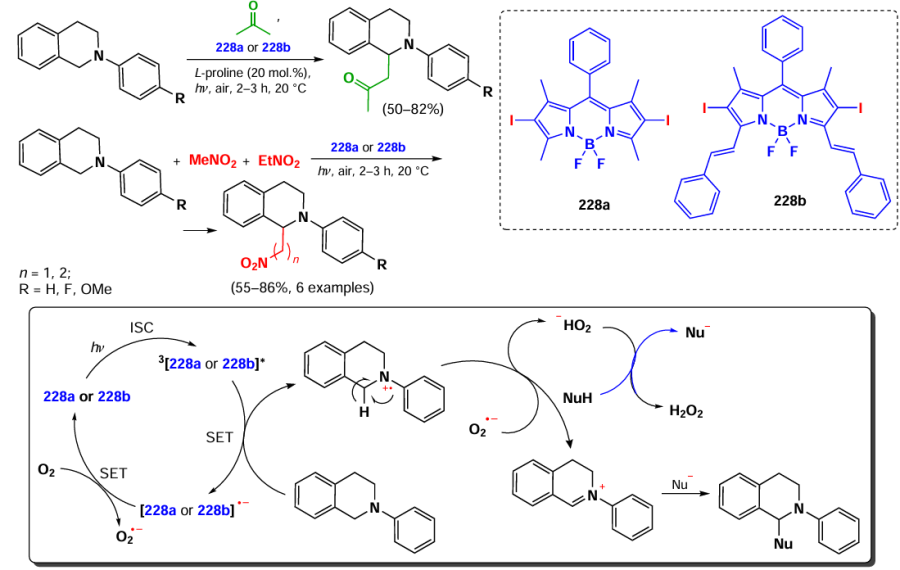
The reaction mechanism proposed by the authors involved a single-electron transfer from the amine to BODIPY 228a or 228b leading to the formation of an amine radical cation and a BODIPY anion radical, which is an oxidizing agent in its triplet excited state. Oxidation of the amine cation radical resulted in the formation of an iminium cation that reacted with a nucleophile. At the same time, the anion radical of BODIPY 228a or 228b reacted with oxygen to form a superoxide radical (O2– •), which was the oxidizing agent in the reaction with the amine cation radical.
The presence of H2O2 in the reaction mixture was confirmed by 1H nmR. When BODIPY 228b was irradiated in the presence of TEMP, the TEMP-1O2 adduct was detected, which disappeared completely on addition of the amine substrate, confirming the proposed mechanism.
A photocatalytic tandem [3 + 2]-cycloaddition/oxidation reaction with the N-substituted maleimide as a substrate was also carried out to give pyrrolo[2,1-a]isoquinolines. The photocatalytic C−H arylation of heterocycles using diazonium salts, in which BODIPY 228a or 228b served as a reducing agent, was also studied. Single-electron transfer from the photocatalyst to the diazonium salt led to the formation of an aryl radical; addition of the aryl radical to the heterocycle formed a radical intermediate. As a result of electron transfer from the radical, a carbocation was formed; and deprotonation of the carbocation led to the arylation product of the heterocycle (Scheme 123). Furthermore, the intermediate aryl radical was intercepted by TEMPO, which confirmed the mechanism proposed by the authors.
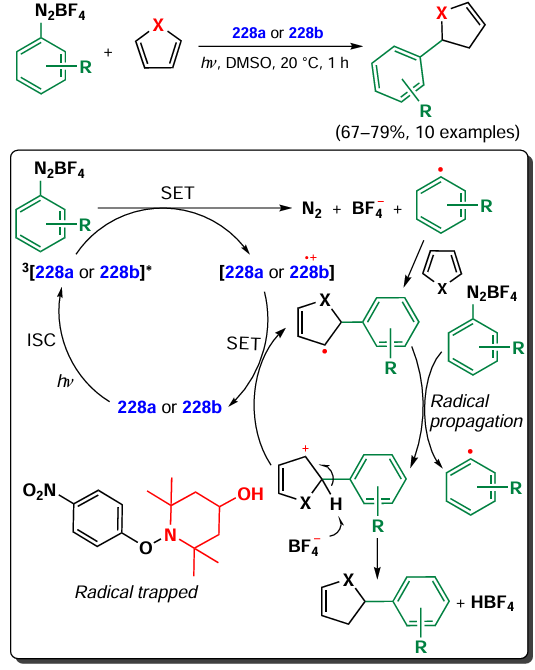
Similarly, iodinated BODIPY 229 immobilized on molecular sieves was used to obtain pyrrolo[2,1-a]isoquinoline via tandem oxidation and [3 + 2]-cycloaddition (Scheme 124).[274]
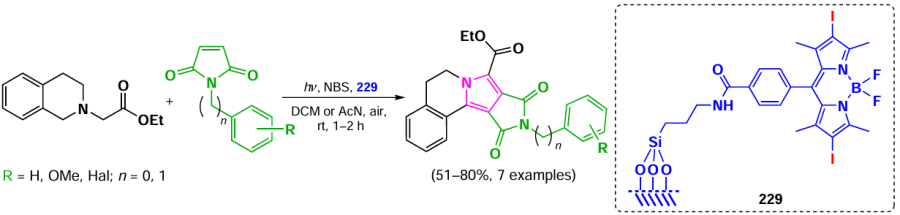
In 2014, Guo et al.[275] reported an alkynylation of tetrahydroisoquinoline in the presence of copper(I), photocatalyzed by triad 230, based on two BODIPY moieties conjugated with the 2,7-diiodo-BODIPY core via a triazole linker (Scheme 125). The triad (230) exhibited broadband absorption in the range of 400 – 750 nm and a high quantum yield of singlet oxygen, thus indicating efficient ISC. Triad 230 was found to be more efficient photocatalyst than the monochromophoric 2,6-diiodo-aza-BODIPY.
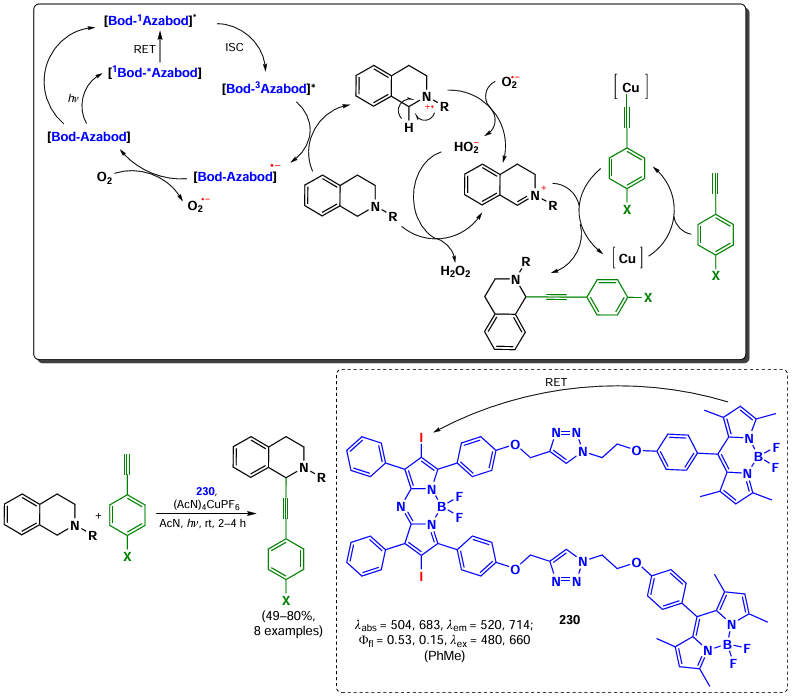
In 2015, Wu et al.[276] showed that a stable triplet state of BODIPY was not necessary for photocatalysis. Thus, BODIPY 231, irradiated with white light and free of heavy atoms, catalyzed the formation of C – C and C – P bonds in reactions with N-phenyltetrahydroisoquinoline via the formation of a reactive iminium cation. The active form of oxygen in the reactions was proven to be the superoxide anion radical (O2– •). The proposed mechanism was based on the fact that BODIPY 231 in the singlet excited state was an oxidizer and promoted the formation of an amine cation radical, the subsequent oxidation led to the formation of an iminium cation, which reacted with the nucleophile. The photocatalytic conditions were used in reactions with nitroalkanes, phosphonium esters, and also with diethyl and dimethyl malonates (Scheme 126).
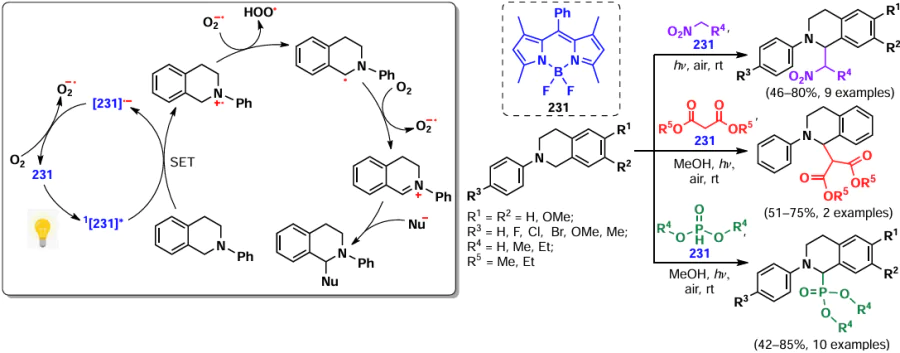
In 2019, Vairaprakash et al.[277] used the 5-hydroxymethylfurfural BODIPY 232, immobilized on a polymer resin, as a catalyst for the arylation of (het)arenes with aryldiazonium salts (Scheme 127).
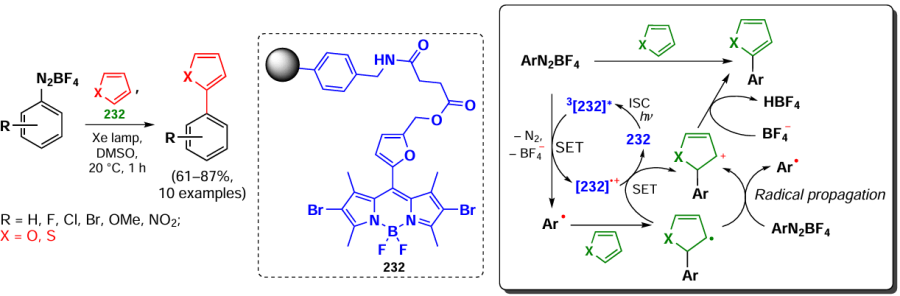
In 2022, Li et al.[29] used BODIPY for the photocatalysis of the Beckmann rearrangement. Oximes were readily oxidized to cation radicals by thiomethyl BODIPY 16b in a triplet excited state, followed by rearrangement into amides. A hydrolysis of oximes with a formation of carbonyl compounds also exhibited high yields under photocatalytic conditions (Scheme 128).
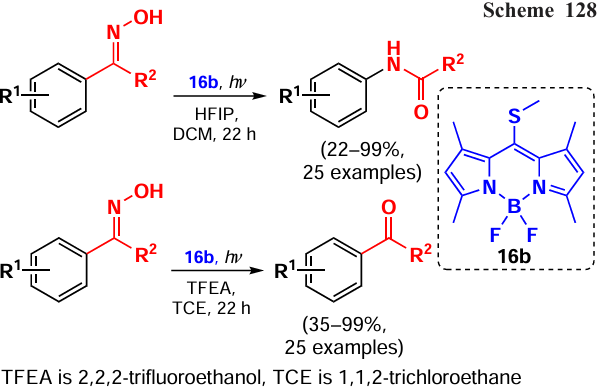
Oxygen anion radical, but not singlet oxygen, was found to be involved in the catalytic cycle, as confirmed by the use of TEMP and DMPO scavengers (Scheme 129). The proposed mechanism involved the formation of an oxime cation radical by reaction with BODIPY 16b in the triplet excited state; a reaction of the oxime cation with water led to radical formation. The Beckmann rearrangement with the formation of N-phenylacetamide took place according to path A; an alternative reaction path (B) via 1,3-transfer of a hydrogen atom ([1,3]-HAT) led to the formation of acetophenone.
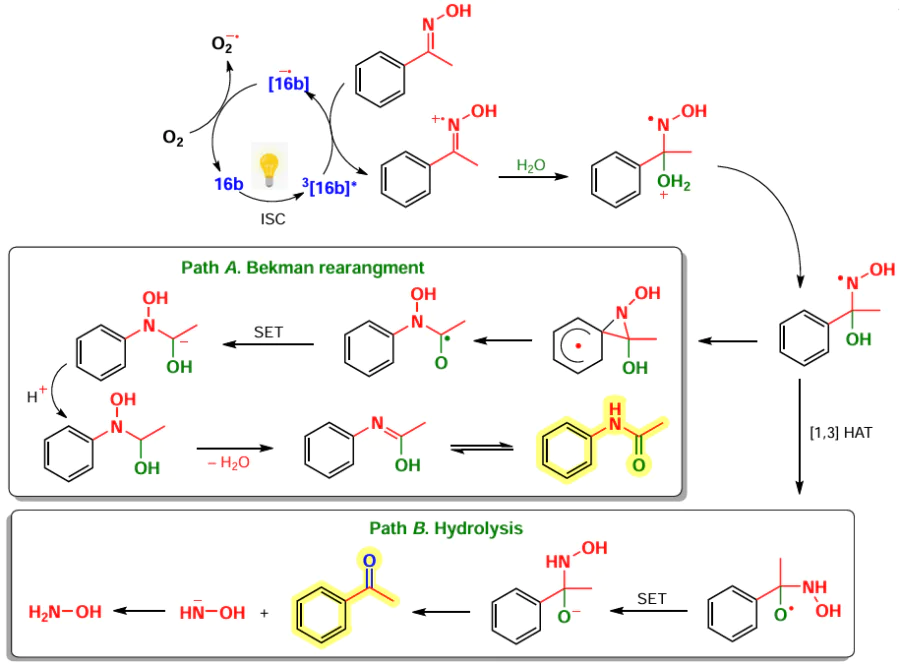
3.7.3. BODIPY catalysis of ion-radical addition to a double bond
The above-described applications of BODIPYs in photocatalytic reactions were limited to singlet oxygen generation or energy transfer. In 2017, Cozzi et al.[278] proposed an alternative application of BODIPY as a photocatalysts. Iodosubstituted BODIPY 228a was found to be an efficient photocatalyst for the radical addition with atom transfer involving bromoalkanes and alkenes. The mechanism of the process involved the formation of the BODIPY anion radical by a triplet state reaction with sodium ascorbate. The anion radical acted as a reducing agent toward haloalkane, leading to the formation of a free radical that was directly involved in the addition (Scheme 130).
In 2021, Cordero-Vargas et al.[279] investigated the photocatalyzed ion-radical addition of α-halocarbonyl compounds to vinyl ethers in the presence of 2,6-dibromo-BODIPY 233. Using this approach, lactones, protected aldehydes, tetrahydroquinolines, dihydrofurans were synthesized (Scheme 131).
According to the proposed mechanism, BODIPY 233 in the excited state was reduced by DIPEA with anion-radical formation, which upon oxidation by the α-haloketone formed radical B. The reaction of a radical B with an alkene resulted in the formation of a secondary radical C, followed by the formation of the radical oxonium cation E; the reaction of a nucleophile with E lead to the formation of the product (Scheme 132).
In 2021, Esipova et al.[28] used meso-anthracene-substituted BODIPY 234a,b dyad as a photocatalyst for the ion-radical addition of bromomalonate to alkenes. The photocatalytic mechanism was based on the excitation of the anthracene fragment (Ant) followed by energy transfer to BODIPY with the formation of a charge-separated state (CS). During spin-orbit charge-transfer intersystem crossing (SOCT-ICT), the photocatalyst transitioned to the triplet excited state, and was then reduced by sodium ascorbate with the formation of an anion radical. Thus, heavy-atom free BODIPY acquires the ability to form triplet states when it is part of a dyad. In the triplet state, BODIPY is an oxidizing agent (Scheme 133).
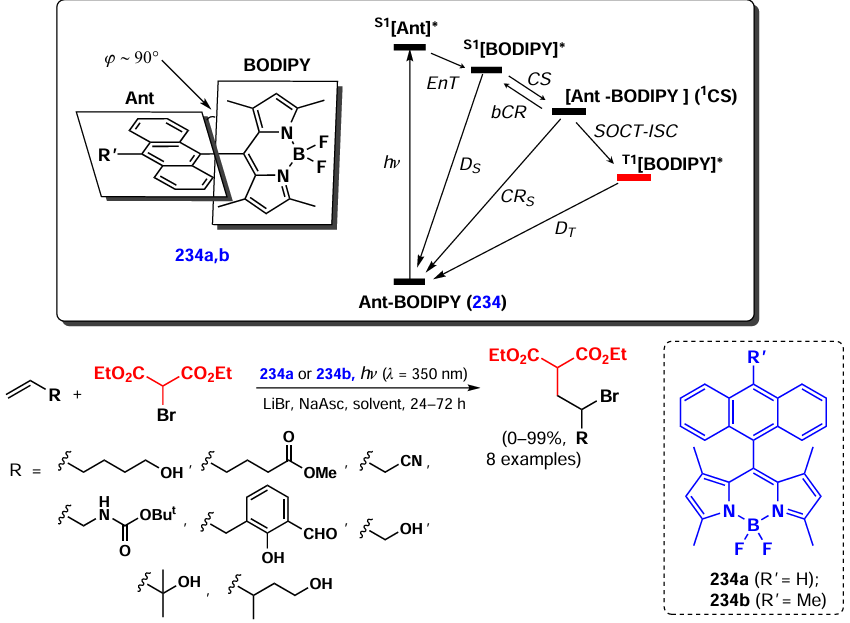
3.8. Synthesis of BODIPY dimers and oligomers
Various BODIPY dimers and oligomers have attracted the attention of researchers because the interaction between conjugated chromophores significantly affects the photophysical properties of the conjugates, thus allowing the design of materials for various practical applications (Fig. 25). Thus, α,α- and β,β-dimers based on coplanar BODIPY subunits are near-infrared dyes with extended π-conjugation. Orthogonally linked dimers (α,meso-, β,meso-, β,meso-, γ,meso- and α,γ-dimers) and oligomers exhibit exciton interactions between BODIPY subunits with effective ISC and are potential ‘heavy atom free’ photosensitizers. Exciton interactions between chromophore units lead to the removal of the degeneracy of the S1 state in α,α-dimers of BODIPY and the occupancy of their triplet state. At the same time, no exciton splitting is observed in orthogonal dimers in which two π-systems of BODIPY are located at an angle of 90°; however, such dimers are able to form stable triplet states, which allows us to consider them as promising PDT agents.
![[{"id":"vlD73bDJis","type":"paragraph","data":{"text":"BODIPY dimer types and their photochemical properties"}}]](/storage/images/resized/BU2SdOGd1DKg7x1ekKUPVlnu4edTNA0391356glb_xl.webp)
3.8.1. α,α-Dimers and -oligomers
In 2008, Bröring et al.[280] synthesized two series of BODIPY α,α-dimers with high fluorescence quantum yields and large Stokes shifts. 8-Unsubstituted α,α-dimers 235a,b were prepared from tetrapyrroles by the reaction with boron trifluoride etherate in the presence of 2,6-lutidine. 8-Aryl dimers 236a,b were synthesized by acylation of dipyrrole followed by condensation with 2,4-dimethylpyrrole (Scheme 134). X-ray crystallography data of dimer 236b indicate that the BODIPY subunits are arranged relative at an angle of 96.52° to each other. In addition, each BODIPY fragment contains fluorine atoms, which point «inward» and are 2.968 Å apart. The presence of two pairs of magnetically non-equivalent fluorine atoms observed in the 19F nmR spectrum at –140 and –147 m.d., is consistent with the proposed structure. Dimers 235 and 236 have stable fixed structure and are not prone to aggregation.
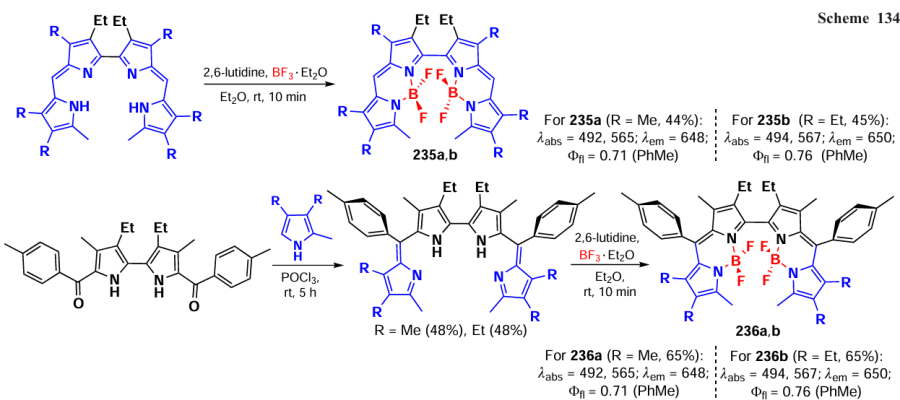
Two absorption maxima were observed in the ranges 558 – 565 nm and 489 – 494 nm, indicating exciton splitting of excited states. Hypsochromic shifts of the absorption and emission maxima and an increase in Stokes shifts compared to those for the monomers were also detected. The decrease in the fluorescence quantum yields of 235, 236 dimers to 0.67 – 0.76 when compared to those for monomers indicats an increase in the efficiency of intercombination conversion.
In 2011, Nepomnyashchii et al.[214] proposed a method for the synthesis of α,α-dimers from 3,8-unsubstituted BODIPY in oxidative conditions (Scheme 135). For dimer 237, an exciton coupling with three absorption maxima at 387, 489, and 562 nm was observed. In addition, a higher Stokes shift (88 nm) when compared to parent BODIPY (13 nm) was shown. The decrease in the fluorescence quantum yield of dimer 237 compared to parent BODIPY is due to the increased contribution of intercombination conversion.
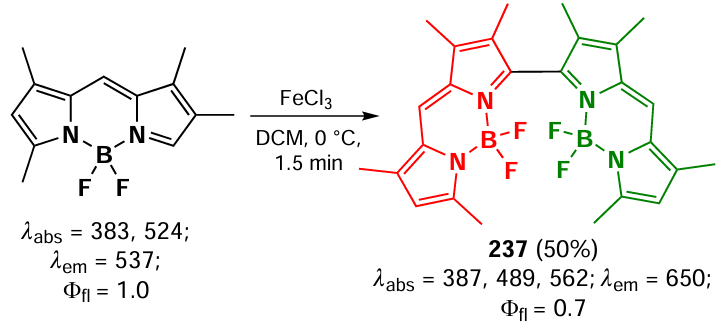
In 2014, Bruhn et al.[281] reported the synthesis of α,α-dimer of BODIPY 238 via oxidative homocombination in the presence of FeCl3, and γ,γ-dimer 239 via oxidation of the corresponding 1,2,3,5,6-pentaalkyl derivative under the action of DDQ) (Scheme 136).
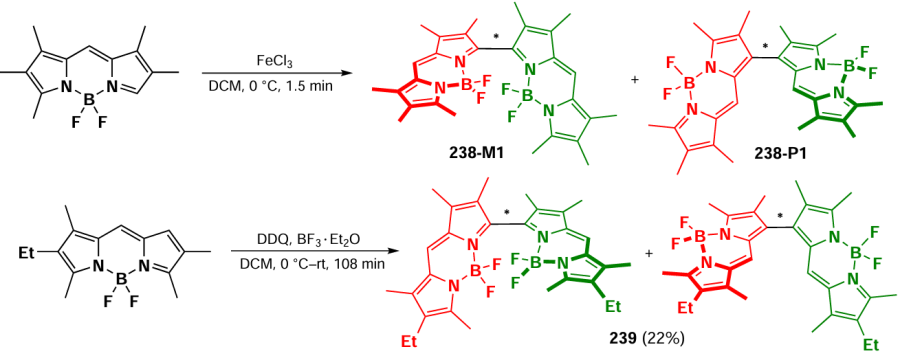
The presence of methyl substituents in the β-positions of BODIPY prevented the rotation around the C – C bond; as a result, the obtained dimers 238 and 239 were a racemic mixture of two configurationally stable aptoisomers. For α,α-dimer 238, the isomer 238-M1 with a negative compartment in the CD spectrum and 238-P1 with a positive compartment were identified by measuring circular dichroism (CD) spectra. The theoretically calculated spectrum using density functional theory (TDCAM-B3LYP) and the exciton chirality method (ECM) agreed with the experimental data. The intensity of the CD spectrum of γ,γ-dimer 239 was 5-fold lower than that of dimer 238, and the theoretical calculation (TDCAM-B3LYP) of the CD spectrum for the M-enantiomer predicted a positive couplet, which contradicted the ECM results, which expected a negative couplet for the M-enantiomer. Additional calculations and vibrational circular dichroism measurements confirmed that for γ,γ-dimers of BODIPY the exciton chirality method could not correctly predict their absolute configuration.
In 2020, Wang et al.[282] proposed a photochemical method to synthesize α,α- (240) and α,γ-dimers of BODIPY (241) under mild conditions via controlled dimerization in the presence of silver trifluoroacetate. Reduction of the oxidative potential of BODIPY in the excited state promoted the metal-induced formation of BODIPY cation radicals, leading to α,α- and α,g-dimers. The mechanism of dimer formation proposed by the authors was confirmed by a decrease in the reaction rate in the presence of TEMPO, as well as by the formation of α-adducts when the process was carried out in the presence of BHT (Scheme 137). Dimers 240 and 241 showed a significant bathochromic shift of the absorption and emission maxima. Two absorption maxima were observed in the absorption spectra of α,α-dimers 240, indicating exciton cleavage, and α,γ-dimers 241 showed the ability to generate singlet oxygen.
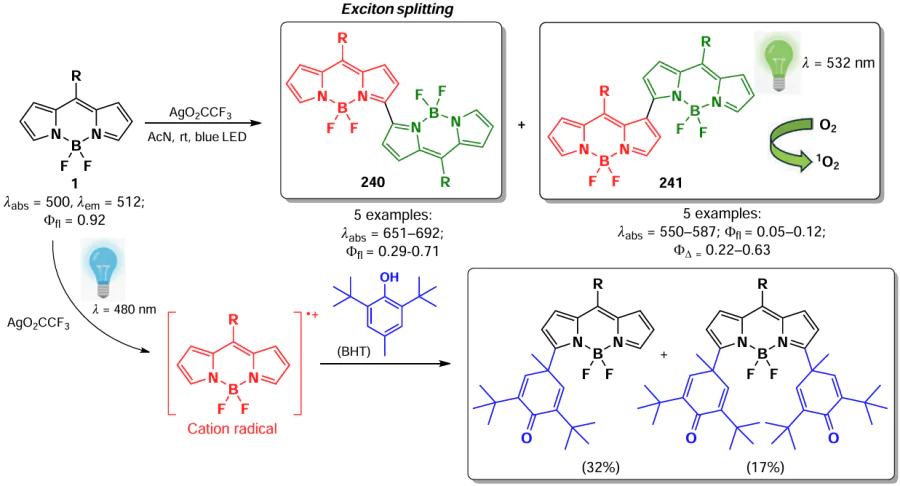
In 2021, Kang et al.[283] reported a regioselective dimerization of BODIPY yielding α,α-dimers. Theoretical calculations showed that the electron density in the cation radical of unsubstituted BODIPY is distributed between positions 2 and 3, while in the cation radical of 3-aryl-BODIPY, the electron density was at position 5 (Fig. 26). Based on this, the authors hypothesized that the oxidative homoconjugation of 3-aryl-BODIPY would proceed at position 5.
![[{"id":"w8sPDgDIfU","type":"paragraph","data":{"text":"Electron density distribution in the cation radical of unsubstituted and 3-substituted BODIPY.[[ type=\"anchor\" referenceId=\"18332\" ]] "}}]](/storage/images/resized/9bfuyRsU4yWVdmgbzRyQqgsYt7BcvimtG2hnQ8yn_xl.webp)
The regioselective oxidative dimerization of 3-substituted BODIPY under the action of iron(III) chloride proceeded at positions 5 and 5' for both 3-aryl- and 3-chloro-substituted BODIPY (Scheme 138). Two absorption maxima were observed in the absorption spectra of dimer 242, indicating exciton coupling. DFT calculations showed that dimerization leads to a decrease in the LUMO energy and an increase in the HOMO energy of the α,α-dimers compared to those for monomers, which is in a good agreement with the bathochromic shift in the absorption spectrum. By postfunctionalization, derivatives 242f,g were obtained from the dimer 242e (see Scheme 138), among which BODIPY 242g showed the ability to visualize lysosomes in living HeLa cells.
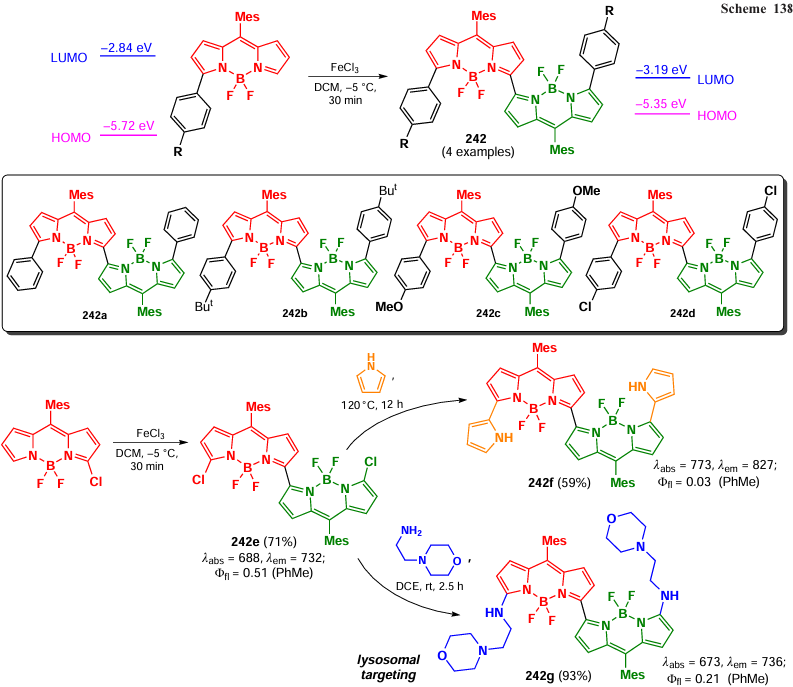
In 2022, Hao et al.[284] reported the synthesis of BODIPY 244 and 246 dimers via photochemical extrusion of sulfoxide from BODIPY precursors conjugated with sulfoxide bridges. Photoconversion was found to occur intramolecularly, without the involvement of external radical particles, and the reaction could also proceed without irradiation at high temperature. A biradical concerted process involving the triplet state was proposed as the mechanism. DFT calculations showed that the activation energy of the proposed transition state was 41.3 kcal mol–1 (Scheme 139). BODIPY 246 with a lysosome-targeting morpholino fragment showed the ability to visualize lysosomes in living cells.
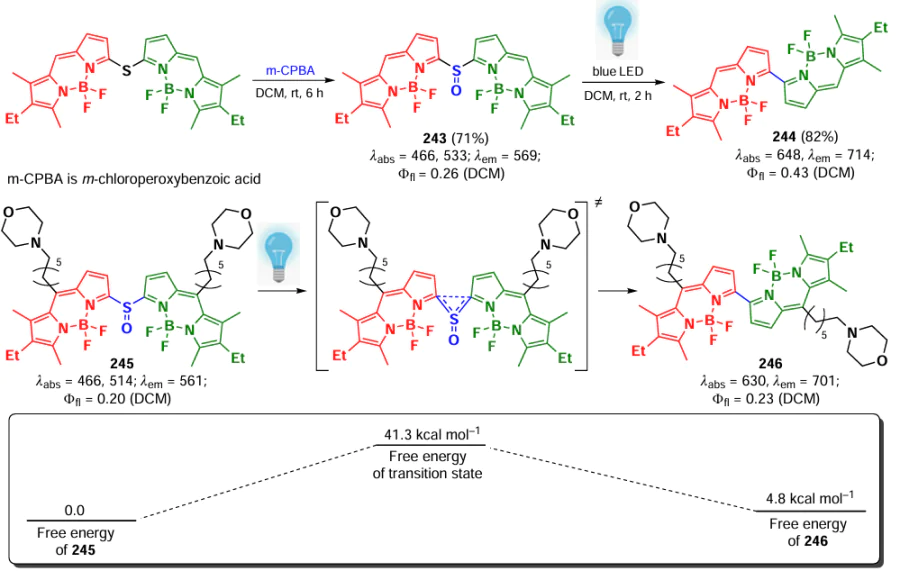
3.8.2. α,β-Dimers and -oligomers
In 2021, Teng et al.[285] described a new class of α,β-dimers of BODIPY as type I NIR-absorbing photosensitizers. Dimer 247 and trimer 248 were synthesized by the Suzuki-Miyaura reaction from 2-Bpin-BODIPY (B2pin2 is bis(pinacolato)diboron) (Scheme 140).
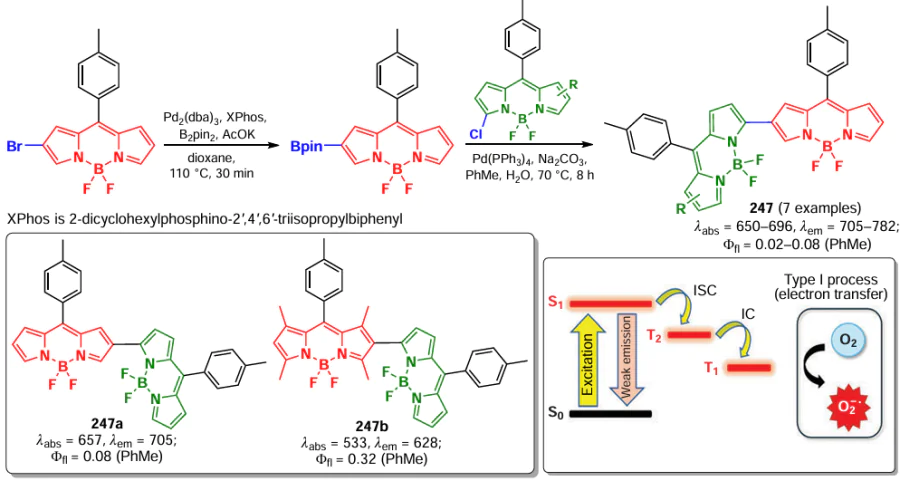
According to X-ray crystallography data, α,β-dimer 247a has almost planar structure, while in 247b two BODIPY fragments are located at an angle of 56.42° due to steric repulsion of methyl groups at positions 1, 3, 5 and 7. The planar structure ensures the proximity of the energies of the S1- and T2-states of the unsubstituted α,β-dimers of 247, which contributes to the ISC enhancement. At the same time, according to calculations, the energy of the T1-state is insufficient for the formation of singlet oxygen, so that the dimers are able to form superoxide radicals. Confirming the calculations, dimers 247 showed reduced fluorescence quantum yields compared to the monomers, as well as the ability to form superoxide radical O2–. Flash photolysis experiments showed an extremely long triplet state lifetime of 1514 μs for dimer 247a.
To shift the absorption of BODIPY to NIR region, α,β-trimer 248 was synthesized, which also formed O2– * upon irradiation and generated a long-lived (1060 μs) triplet state (Scheme 141). Trimer 248 exhibited high activity upon irradiation with λ = 730 nm (50 mW cm–2, 10 min) in normoxia and hypoxia on HepG2 cell lines with IC50 = 0.39 and 0.56 μM. In in vivo experiments, therapy of HepG2 tumors-bearing mice with trimer 248-based nanoparticles, produced using the non-ionogenic detergent pluronic 127 (F127), combined with red-light irradiation (λ = 730 nm, 120 mW cm–2, 10 min) resulted in complete disappearance of tumors on the 6th day.
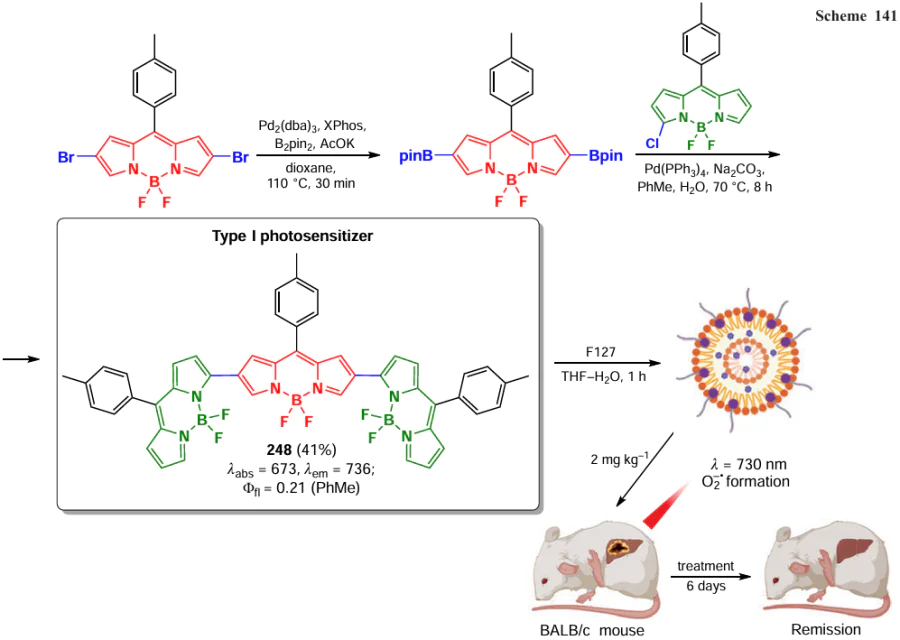
3.8.3. β,β-Dimers and -oligomers
In 2011, Hayashi et al.[286] proposed a method for the synthesis of β,β-dimers of BODIPY using the Suzuki – Miyaura reaction (Scheme 142). β,β-Dimer 249 was prepared by Pd-catalyzed borylation followed by cross-coupling. X-ray crystallography of dimer 249 and trimer 250 showed that in both compounds BODIPY subunits are coplanar due to the absence of alkyl substituents at positions 1, 3, 5, and 7.
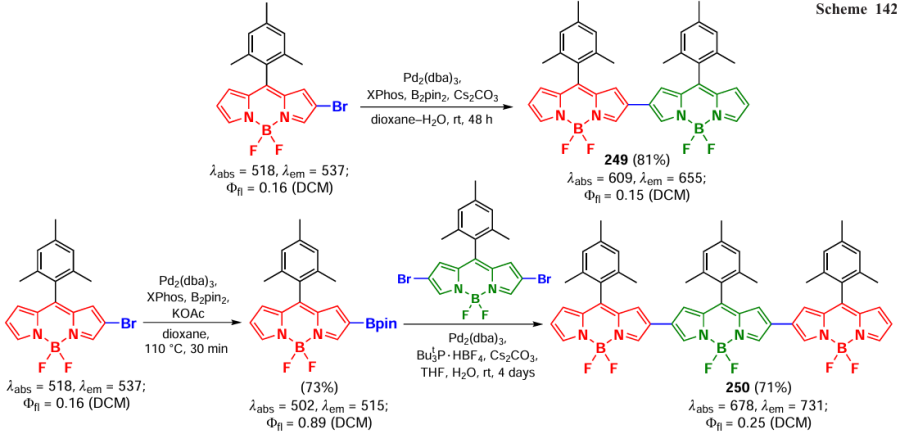
In the same year, Nepomnyashchii et al.[227] reported an oxidative coupling of BODIPY at positions 2 and 6 to synthesize the corresponding dimers and oligomers (Scheme 143). In the presence of 3.5 equiv. of anhydrous FeCl3, mixtures of dimers 251a,b (10 – 27%), trimers 252a,b (3 – 9%), and other oligomers were formed. A bathochromic shift of the absorption maxima was shown for the linear dimers 251 and trimers 252, and no exciton cleavage was observed in the electronic spectra of the β,β-dimers.
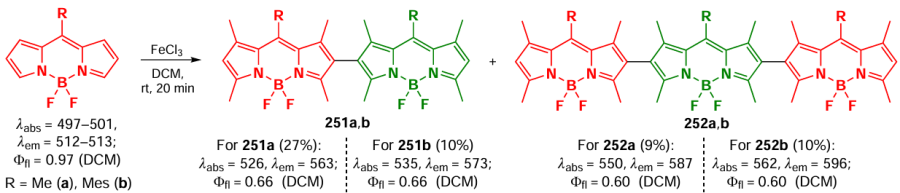
In 2011, Rihn et al.[287] synthesized symmetric β,β-dimers of BODIPY via oxidative coupling reaction with bis(trifluoroacetoxy)iodbenzene (PIFA) as an oxidant (Scheme 144). Bathochromic shifts of 40 – 70 nm as well as high fluorescence quantum yields were observed for dimer 251a and trimer 252a. Based on 2-substituted BODIPY, the corresponding substituted dimers 253 were synthesized in a similar manner. Compound 251a was modified at the boron atom by reaction with an excess of a Grignard reagent, resulting in BODIPYs 253 with ethynylpyrene moieties in its structure. When the dimer 254 was excited with 285 and 369 nm light, only one emission peak at 562 nm was observed in the emission spectra, indicating a complete exciton energy transfer from the pyrene to the BODIPY dimer.
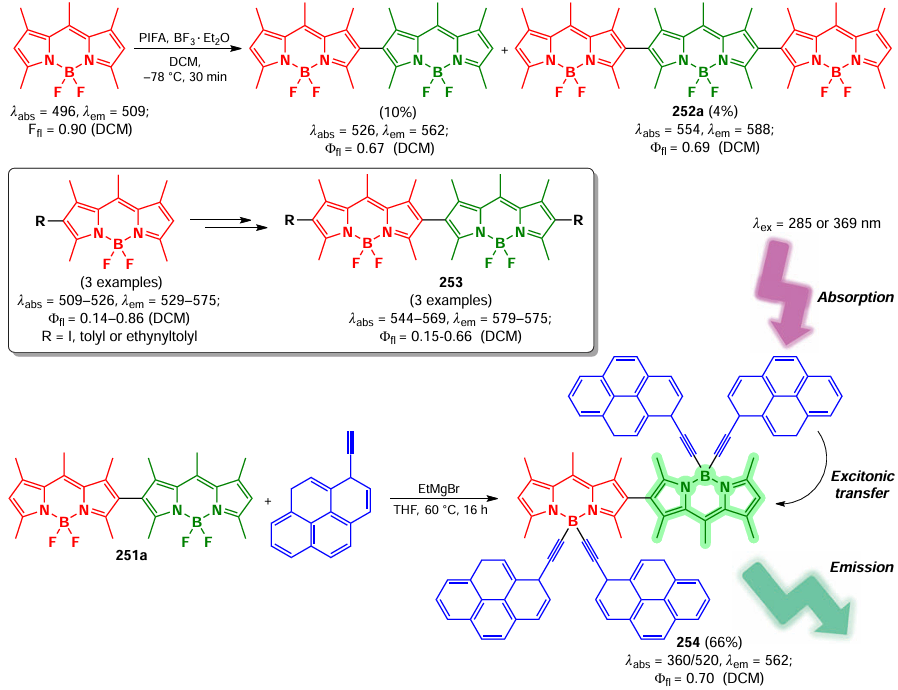
In 2018, Arroyo-Córdoba et al.[288] synthesized the asymmetric β,β-dimer of BODIPY 255 using the Suzuki reaction (Scheme 145). The obtained dimer was interesting as a platform for further functionalization to form derivatives 256 – 258 due to the high reactivity of the meso-SMe group in aromatic nucleophilic substitution reactions as well as in the Libeskind – Srogl cross-coupling reaction.
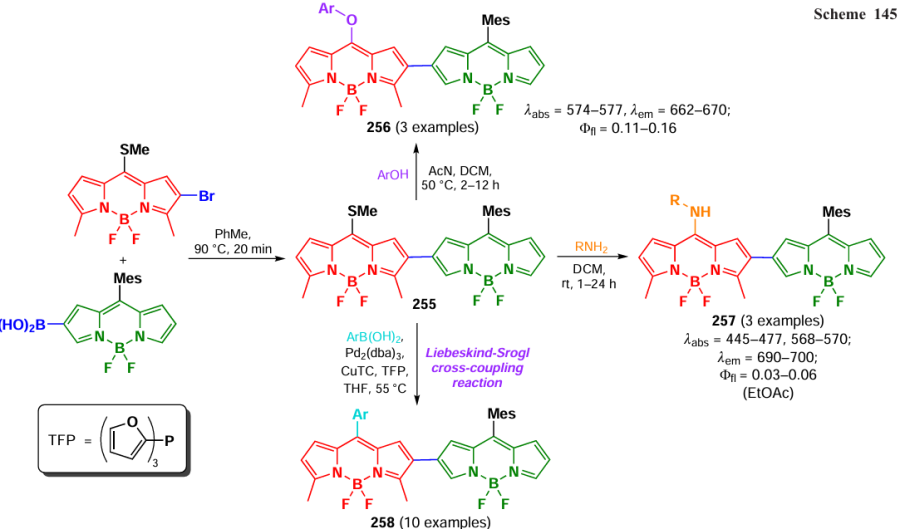
A new method for oxidative coupling of α-amino-BODIPYs was proposed by Wang et al. in 2023.[289] Under the action of m-CPBA or PIFA as an oxidizing agent, 3-amino-5-aryl-substituted BODIPY formed β,β-dimers 259. A subsequent reaction of the dimer 259a (R = 4-MeOC6H4) with PhPCl2 yielded 260 through interaction with reactive amino groups and stabilization of the coplanar orientation of the BODIPY subunits, and chlorination of this substrate upon irradiation gave the derivative 261 (Scheme 146).
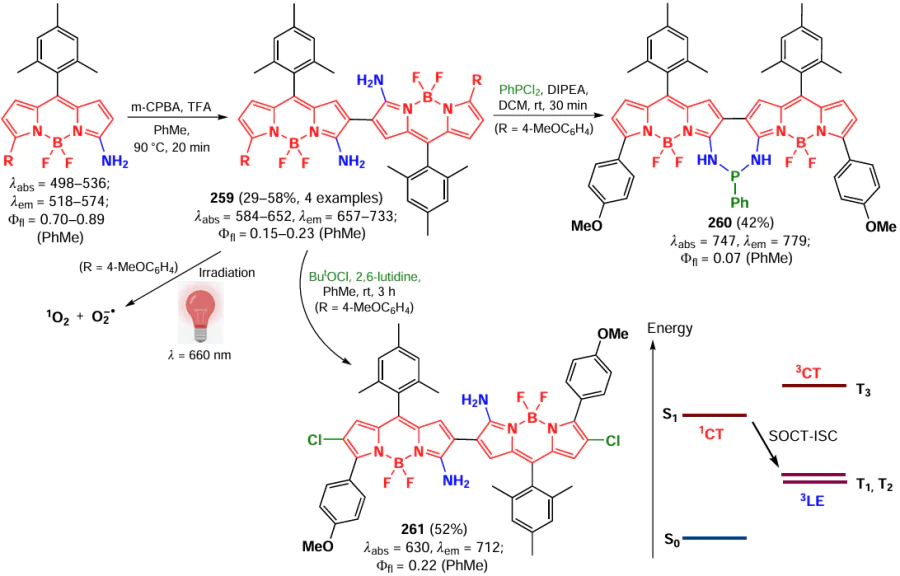
The obtained dimers showed a bathochromic shift in the absorption spectrum and a decrease in the fluorescence quantum yield compared to that of the monomers. Dimer 259a showed a solvatochromic effect as well as the ability to form singlet oxygen and superoxide radical, which is not characteristic of β,β-dimers. Quantum chemical DFT calculations showed charge transfer for the 259a dimer. Thus, distribution of the HOMO was throughout the whole molecule, whereas the LUMO was localized at the BODIPY core. The authors explain the decrease in fluorescence quantum yields and the ability of dimer 259a to form ROS as a consequence of the SOCT-ISC process.
3.8.4. β,γ-Dimers and -oligomers
In 2021, Kang et al.[290] synthesized β,γ-dimers of BODIPY 262a – d by Pd-catalyzed C – H activation; the reaction was carried out in the presence of silver trifluoroacetate as oxidant and Pd(OAc)2 as catalyst (Scheme 147). Trimer 262e, tetramer 262f, and pentamer 262g, also representing β,γ-regioisomers, were obtained with increasing the amount of oxidant.
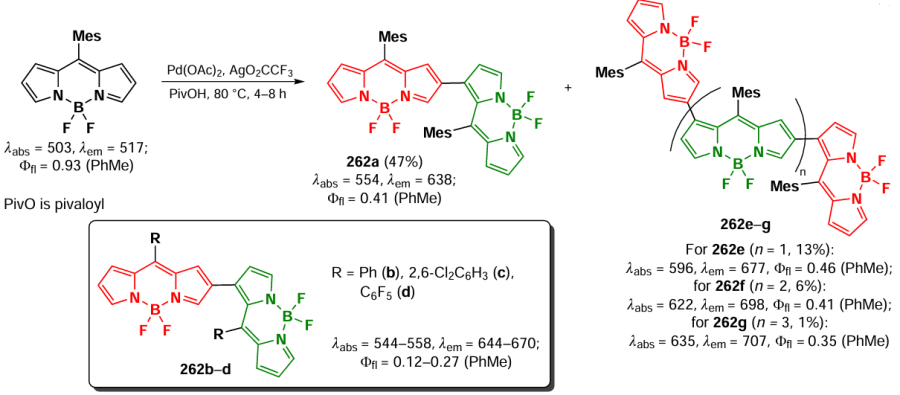
Oligomers 262a – g exhibited significant bathochromic absorption shifts in the spectra, exciton splitting, large Stokes shifts (1600 – 3230 cm–1) and moderate fluorescence quantum yields. Using DFT calculations, it was found that oligomer formation effectively stabilize the LUMO levels (–3.10, –3.22 and –3.27 eV for monomer, dimer and trimer, respectively) and destabilize the HOMO levels (–5.79, –5.73 and –5.71 eV). The calculated T1 energies for compounds 262a,e,f were 1.50, 1.47 and 1.46 eV, which is sufficient to generate singlet oxygen. To confirm the calculations, the ability to generate singlet oxygen was experimentally demonstrated for the dimers 262a – d.
3.8.5. γ,γ-Dimers and -oligomers
In 2012, Poirel et al.[291] presented a method for the synthesis of γ,γ-dimers by oxidative homocoupling reaction in the presence of PIFA. γ,γ -Dimer 263a and α,γ-dimer with a methylene bridge 263b formed via a benzyl-type cation radical were obtained. Dimers 264a – b, in which BODIPY moieties were linked through the α-positions of bithiophene, were synthesized under the same conditions (Scheme 148).
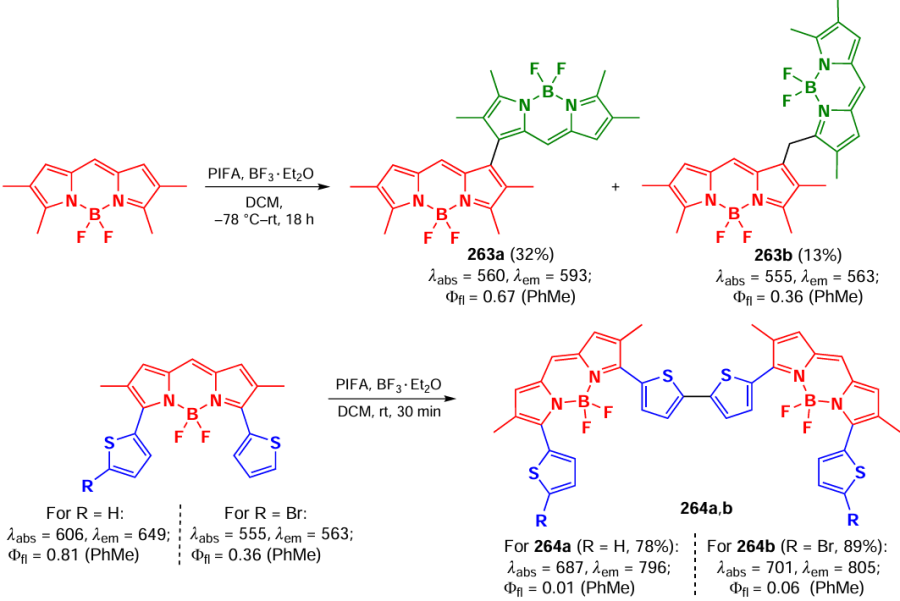
Moderate exciton coupling was observed in the absorption spectra of dimers 263a,b, and the fluorescence quantum yield and excited state lifetimes were significantly lower than those of the monomer. Dimers 264a,b also exhibited exciton coupling, low photoluminescence efficiency and reduced excited state lifetimes. The photoluminescence and fluorescence efficiencies of the dimers appeared to be solvent-dependent, thus indicating the ICT effect.
3.8.6. Meso,meso-dimers
In 2011, Cakmak et al.[158] proposed the use of BODIPY dimers as promising PDT agents. By condensation of β- or meso-formyl-substituted 1,3,5,7-tetramethyl-BODIPY with 2,4-dimethylpyrrole, β,meso- and meso,meso-dimers 265a – c were synthesized (Scheme 149).
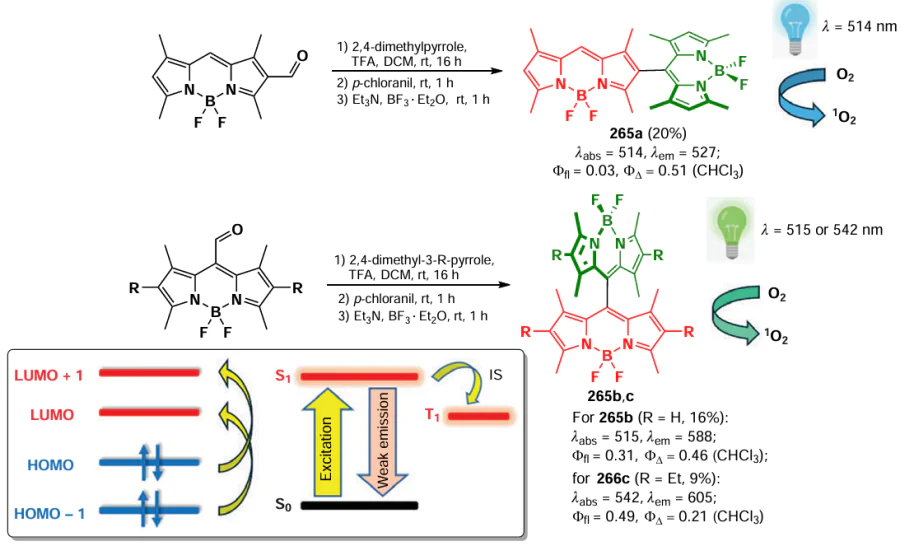
Dimers with BODIPY π-systems that are orthogonal to each other can transition to an excited state with equal probability. This doubly substituted excited state promotes the S1 → T1 transition.[292] Theoretical calculations of the molecular orbitals of the β,meso-dimer 265a showed that the transition to the S1 state represents a linear combination of the HOMO-1 to LUMO and HOMO to LUMO + 1 transitions, as well as other doubly substituted transitions, which in turn significantly hinders the S1 → S0 relaxation and promotes the transition to the T1 state. The ability to form singlet oxygen was demonstrated for the 265a – c dimer, confirming the results of theoretical calculations. The β,meso-dimer 265a showed the highest quantum yield (FD = 0.51). In an antiproliferative activity study on K562 erythroleukemia cells under irradiation with λ = 514 nm light (2.5 mW cm–2, 4 h), the half-maximal effective concentration (EC50) of dimer 265a was 50 nm.
3.8.7. Condensed dimers
In 2015, Yu et al.[293] applied an oxidative dimerization method to synthesize conjugated cyclic BODIPY dimers. Oxidation of the bridging dimer 266 with iron(III) chloride gave the condensed BODIPY dimer 267 was obtained, which was further introduced into the Knoevenagel condensation to form styryl- and distyryl-substituted BODIPY dimers 268a – d (Scheme 150).
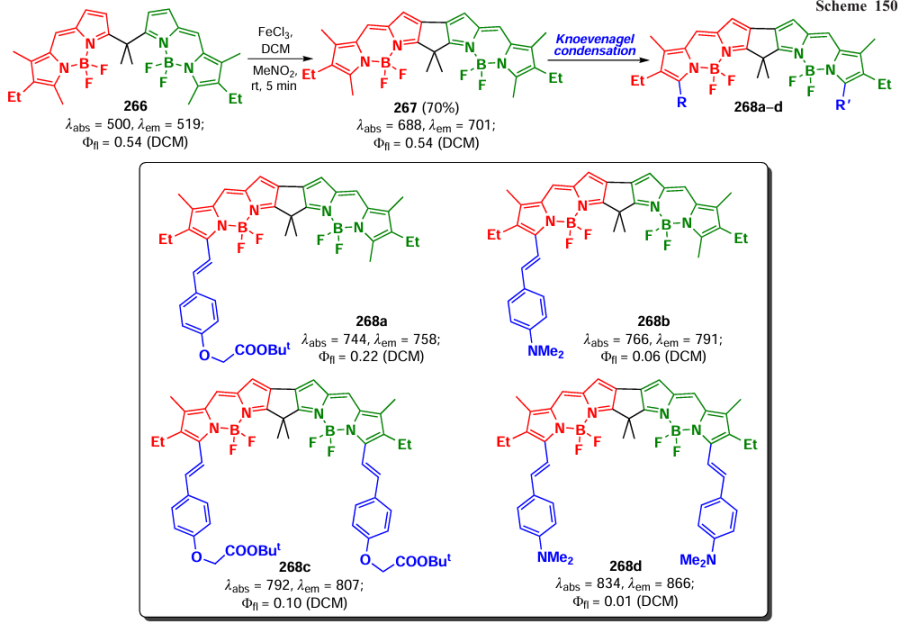
X-ray crystallography of dimer 267 showed that all aromatic rings lie in the same plane, from which only fluorine atoms and alkyl substituents deviate. A bathochromic shift was observed in the absorption spectra of dimer 267 and its styryl derivatives 268a – d compared to the bands for the original BODIPY and the unconjugated dimer 266. According to DFT calculations, the HOMO and LUMO were distributed over the two BODIPY cores; the calculated decrease in the energy gap between the HOMO and LUMO of the condensed dimer was consistent with the observed bathochromic shift.
In 2020, Wu et al.[294] described the synthesis of conformationally constrained BODIPY 269 dimers via intramolecular oxidative dimerization of unconjugated bis-BODIPYs under the action of MoCl5. The products resulting from the oxidative combination exhibited a significant bathochromic shift of the absorption maxima coupled with an increase in fluorescence quantum yields (Scheme 151). The results of DFT calculations also showed decreased LUMO energy levels and increased HOMO energy levels for bis-BODIPY and fused-BODIPY, which was in good agreement with the bathochromic shift values. In addition, dimer 269a exhibited fluorescence imaging ability in HeLa cells, in domestic cat muscle and in danio fish.
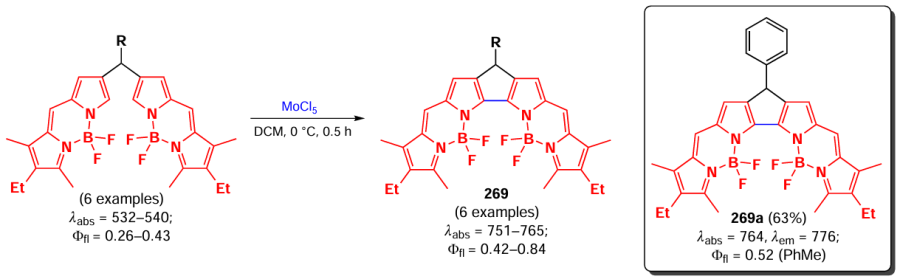
3.8.8. Dimers and oligomers with heteroatoms
In 2014, Shinokubo et al.[295] synthesized asymmetric trimers of BODIPY 270a,b by oxidation of β-amino derivatives of DDQ (Scheme 152). Such trimers exhibited significant bathochromic shifts in the absorption spectra.
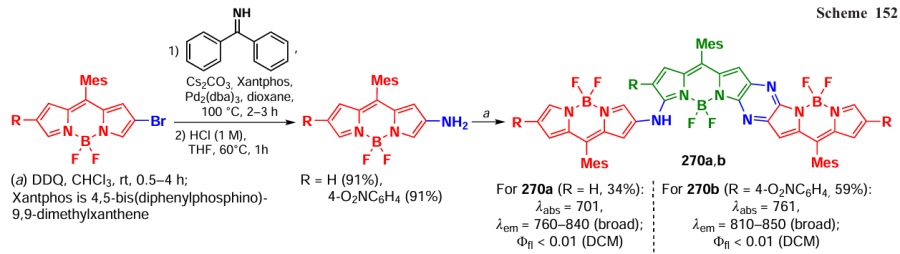
In 2021, Jiao et al.[296] reported the synthesis of dimers, trimers, and other BODIPY oligomers conjugated via a sulfide bridge using nucleophilic substitution reactions (Scheme 153). Conjugates 271 – 274 exhibited a bathochromic shift in both absorption and emission spectra. As the number of BODIPY links in oligomer increased, the fluorescence quantum yield decreased, and the ability to generate singlet oxygen increased due to the enhanced ICT effect. DFT calculations show LUMO stabilization and HOMO destabilization as the number of BODIPY links in oligomers increases.
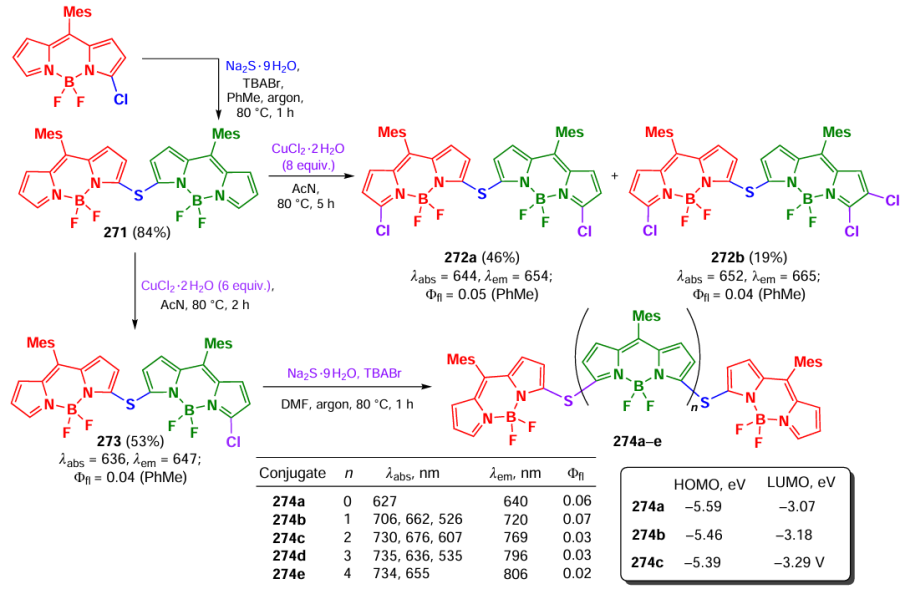
In 2021, Gong et al.[297] proposed an approach to synthesize thiophene-fused dimers of BODIPY 275. This method involves nucleophilic substitution with sodium sulfide, and subsequent oxidation. Dimers 275 were obtained from the in situ formed unsubstituted (271), mono- (273) and dichloro-substituted (272a) disulfide dimers in the presence of excess FeCl3. As the amount of FeCl3 in the reaction mixture increased, the number of condensed links in the structure of the resulting tetramers 276 – 279 also increased (Scheme 154).
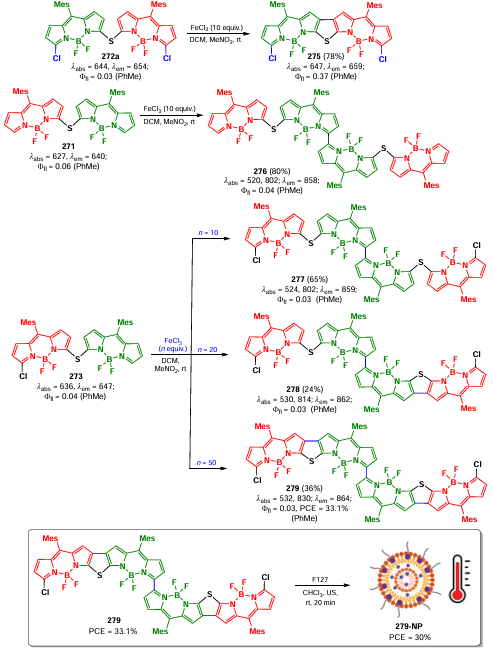
Disulfide oligomers 271, 272a, 273, 276, and 277 exhibited a significant bathochromic shift in the absorption spectrum (up to 130 nm), which increased further for the condensed dimer 275 and tetramers 278, 279 (up to 200 and 300 nm). Due to the almost complete absence of fluorescence (Ffl = 0.03), BODIPY 279 exhibited photothermal properties; the PCE was 33.1%, and 30% for the NPs based on it.
In 2024, Werz et al.[298] presented N-complexed dimers 280, trimers 281, and tetramers of BODIPY 282 obtained by the Buchwald – Hartwig amination (Scheme 155). A bathochromic absorption shift and exciton splitting were observed in the electronic spectra of compounds 280 and 281. Increasing the number of BODIPY links in the oligomer significantly decreased the fluorescence intensity, which was attributed to an increase in the contribution from radiation-free relaxation and a decrease in the S1-level transient dipole moment for higher oligomers.
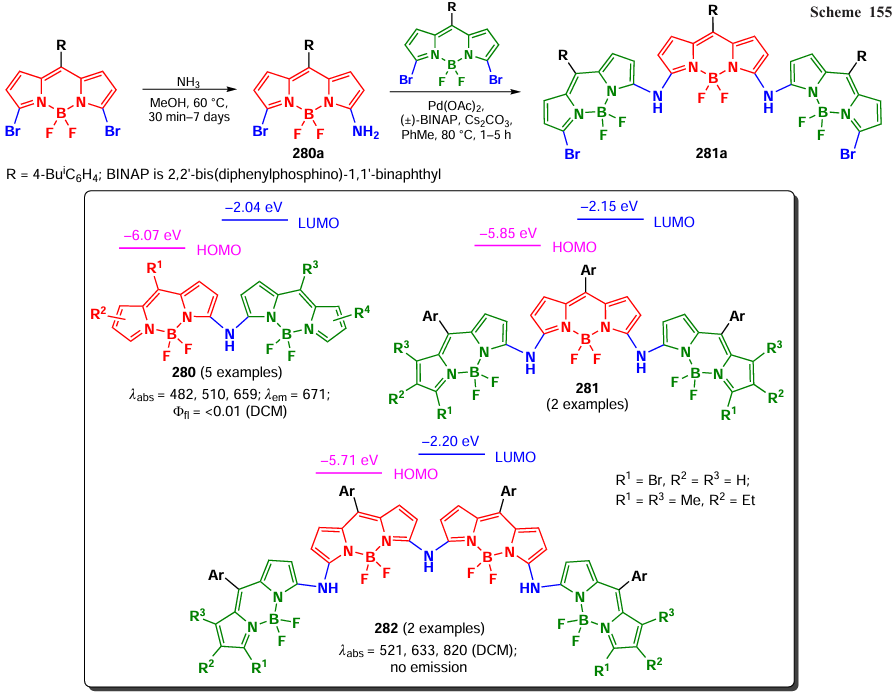
In 2024, Jiao et al.[299] described the synthesis of similar N-conjugated dimers and trimers of BODIPY via nucleophilic substitution reactions. Subsequent functionalization by the Suzuki reaction led to conjugates 284, and reductive combination involving the trimer afforded products 285 – 288 (Scheme 156). Dimers 283, 284, and 289, as well as trimers 285, 287, 288, and 290 exhibited a bathochromic shift in the absorption spectrum and showed exciton coupling. DFT calculations showed that oligomerization via N-bridged links leads to higher HOMO energies. The fluorescence quantum yields decreased as the number of BODIPY links increased. Hexamer 286-based nanoparticles showed high PTT efficiency.
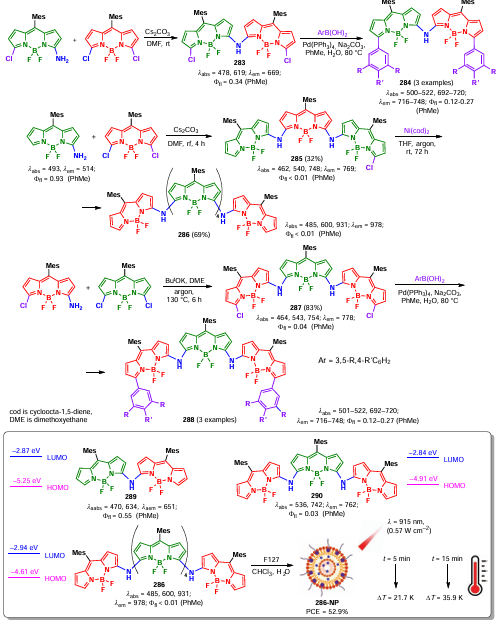
3.8.9. Dimers and oligomers with an ethylene bridge
In 2017, Patalag et al.[300] synthesized BODIPY oligomers based on BODIPY subunits linked via an ethylene bridge. Due to a high flexibility, dimers were able to adopt a coplanar conformation favoring efficient J-aggregation.[8] Dimerization of 3-methyl-substituted BODIPY was carried out in the presence of lithium diisopropylamide (LDA) followed by iodination with ICl and nucleophilic substitution of the iodine atom by Li-BODIPY. The introduction of ethylene dimer 291a with a methyl group at position 5 into the reaction under these conditions led to the formation of a mixture of tetramer 291b, hexamer 291c and octamer 291d (Scheme 157). Crystal structure of the three dimers with different alkyl substituents showed that BODIPY subunits in the dimers were coplanar, and the slip angle was 36 – 44°, which was less than the maximum possible for the formation of J-aggregates. A significant narrowing of the emission bandwidth was observed for the J-aggregates of 291a – d oligomers with increasing linker number, which makes them promising monochromatic emitters.
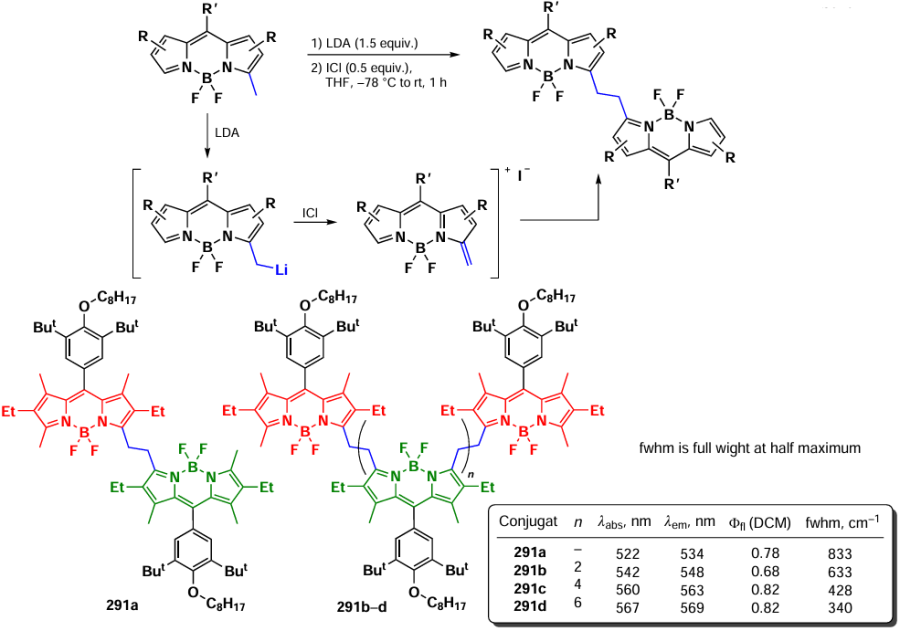
In 2021, Gong et al.[301] reported a method for the oxidative homocoupling of 3-methyl-BODIPY leading to the formation of ethylene bridge-coupled dimers and oligomers. When BODIPYs were oxidized with copper(I) iodide in the presence of Cs2CO3, four dimers 292a – d were obtained, and when the reaction time was increased to 18 h, trimers 292e,f were also obtained (Scheme 158). Investigation of the reaction mechanism using radical ‘traps’ TEMPO and N-tret-butyl-α-phenyl nitrone (PBN), as well as EPR spectroscopy, confirmed the formation of the BODIPY cation radical. This cation radical was generated under the action of a base and reacted with a second substrate molecule to form a dimer. The dimers and trimers of 292 exhibited bathochromic shifts in the absorption spectrum and high molar extinction coefficients compared to those of the original monomers.
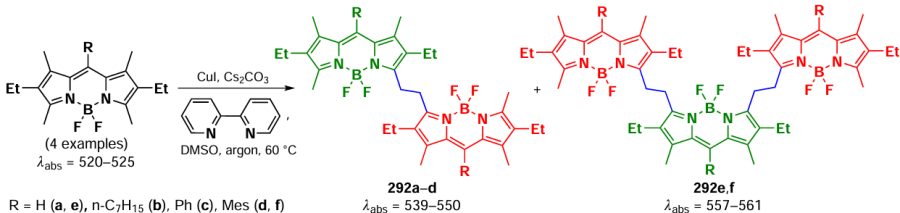
3.8.10. Double bonded dimers and oligomers
In 2020, Wu et al.[302] proposed an approach to synthesize coplanar π-conjugated dimers in which the BODIPY subunits were conjugated by a carbon-carbon double bond. Dimer 293 was synthesized by the Stille reaction from 3-chloro-BODIPY and trans-1,2-bis(tri-n-butylstannyl)ethene (ESn). Combining different α-chloro-BODIPY and stannates, trimer 294, tetramer 295a, pentamer 295b and hexamer 295c were obtained (Scheme 159).
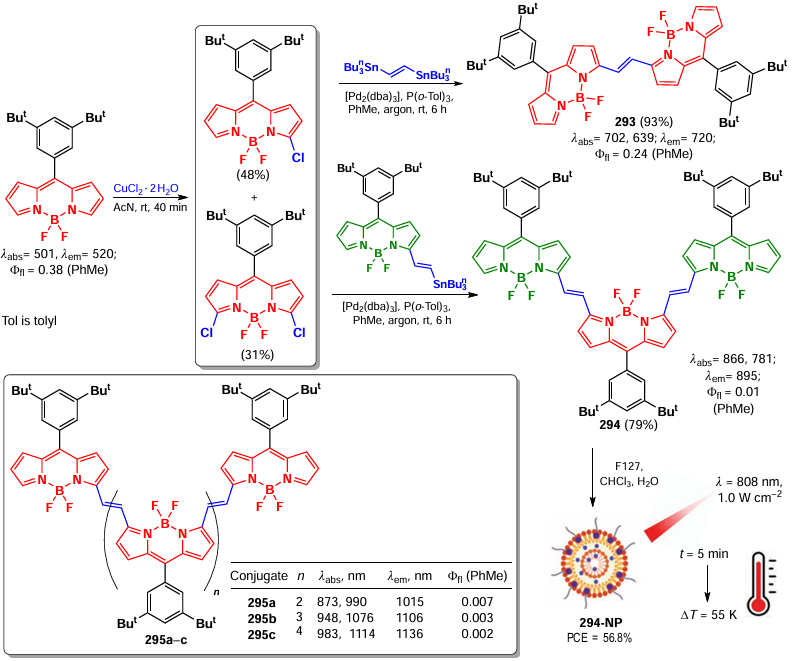
The absorption maximum in oligomers 293, 294, and 295a – c shifted significantly towards the NIR- and NIR-II-bands as the number of BODIPY links increased. The molar extinction coefficient varied nonlinearly with increasing link number, with the highest value observed for trimer 294. The low fluorescence quantum yields of oligomers 294 and 295a – c (< 1.5%) indicated the predominance of excitation-free relaxation pathways. For oligomers 294, 295a, and 295b, the photothermal conversion efficiency (PCE) was 30 – 35%, with the PCE value increasing to 56.8% after encapsulation of the 294 trimer in NPs with pluronic F127.
In 2022, Wang et al.[88] designed an ethenylene-bridged BODIPYs as NIR-II-band absorbing FTT agents. BODIPYs 296a,b (Fig. 27) were synthesized by the Stille coupling and their crystal structure was investigated. The slip angle between the monomers was 27° and they were linked by intermolecular π – π- and C – F interactions, which promoted a J-aggregation. Due to the presence of a meso-CF3- group and a tetrahydroquinoxaline donor moieties, the absorption maximum of the D – A – π-A – D dimer 296a was in the NIR-II region. In aqueous medium the absorption maximum shifted to an even longer wavelength region due to J-aggregation. 296a-NP nanoparticles encapsulated in F127 exhibited high PCE (62%) and reduced the survival of HeLa cells under laser irradiation with c λ = 1208 nm (0.8 W cm–2, 10 min) to 27% at a concentration of 30 μM. In in vivo experiments in mice with an orthotopic model of hepatocellular carcinoma, 6 h after intravenous injection of 296a-NP particles, the maximum photoacoustic signal was observed in the tumor, which was 2 – 5 times more intense than in other organs. In the in vivo therapeutic efficacy study, complete remission was achieved in mice with 4T1 tumor after therapy with 296a-NP and irradiation at λ = 1208 nm.
![[{"id":"BRnt0Bl1Pi","type":"paragraph","data":{"text":"Schematic of the synthesis of BODIPY <b>296a</b> – <b>b</b> and the principle of action of <b>296a-NP</b> nanoparticles <i>in</i> <i>vivo</i>. Figure created by the authors based on the data of the publication[[ type=\"anchor\" referenceId=\"18087\" ]]."}}]](/storage/images/resized/h1OSd3gV1BZWe2pGlKlsO1NryUBiqOwDl28G2usk_xl.webp)
3.9. Modifications at the boron atom
BODIPY fluorophores discussed above almost always contained two fluoride ligands directly bonded to the boron atom. However, nucleophilic agents can also attack the boron centre to form BR2 BODIPYs, which are the subject of this section. Although such modifications of the BODIPY core have been less thoroughly studied than the reactions of the pyrirromethene moiety, the functionalization of the boron atom is an important synthetic tool that allows significant modification of the physicochemical and photophysical properties of the products. The introduction of various nucleophilic agents onto the boron atom leads to a significant distortion of the three-dimensional structure of BODIPY and, consequently, to a change in a number of physicochemical properties, such as fluorescence quantum yield, redox potential, laser efficiency, chemical and photochemical stability, and aggregation properties.[10] In 2019, Bodio and Goze [12] published a review dedicated to the modification of BODIPY and its aza-analogues at the boron atom.
3.9.1. Lewis acid catalyzed synthesis of B(OR)2-BODIPY
In 1999, Kim et al.[303] synthesized a chiral 4,4-diaryloxy-BODIPY 297 by intramolecular esterification in the presence of a Lewis acid boron tribromide (Scheme 160).
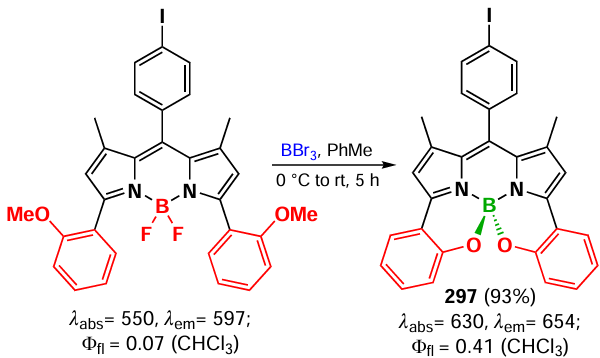
4,4-Dimethoxy-substituted BODIPY 298, synthesized according to Scheme 161, has been proposed as a fluorescent probe for the detection of NO.[304]
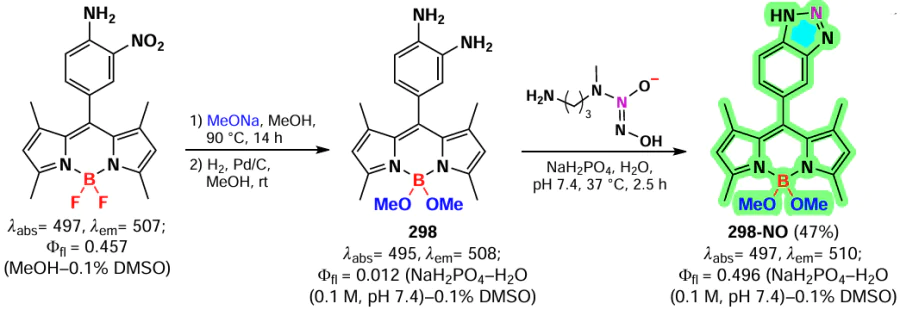
In 2007, Tahtaoui et al.[167] proposed a method for the modification of BODIPY with alcohols by the activation of the B – F bond with a Lewis acid (AlCl3), followed by nucleophilic substitution. This resulted in a large series of 4,4-dialkoxy or 4,4-diaryloxy derivatives 299 (Scheme 162). This work initiated many subsequent Lewis acid-catalyzed modifications at the boron atom.
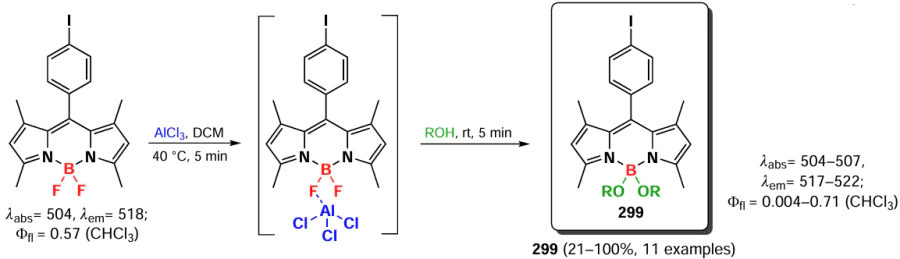
A few years later, Brizet et al.[305] reported an AlCl3-catalyzed B – O-functionalization of BODIPY to form dialkoxy and diarylalkoxy derivatives 300, which were then converted to water-soluble compounds 301a–c (Scheme 163).
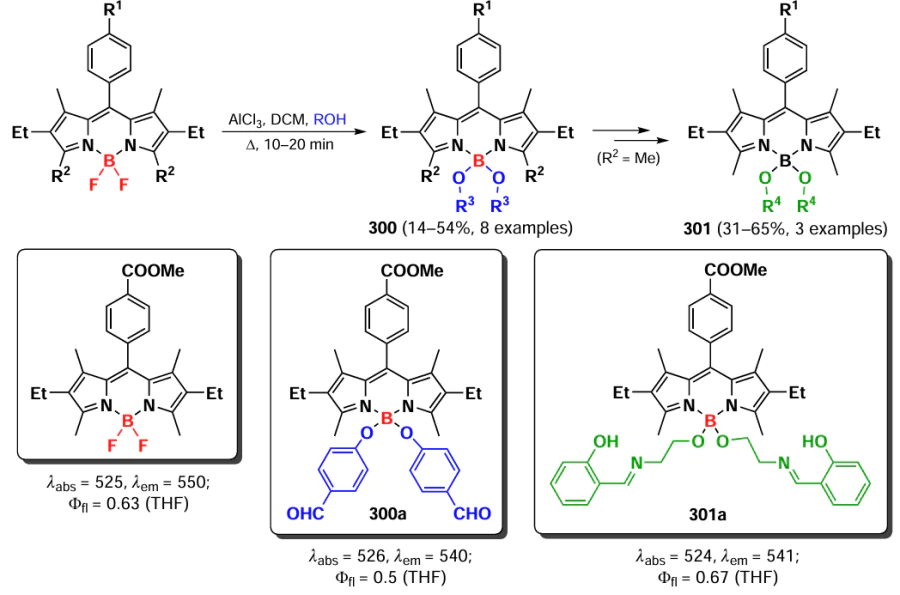
It is worth noting that boron-modified BODIPYs are of interest for the development of molecular switches. In 2010 and 2011, Wijesinghe et al.[306, 307] created a donor – BODIPY –fullerene (303a) and an antenna – BODIPY – fullerene (303b – e) triads based on derivative 302. Using femtosecond and nanosecond transition-state spectroscopy, the ability of such systems to form charge-separated particles was demonstrated: Fc+ • – BDP–[C60]– • (Fc — ferrocenyl) for the triads 303a and antenna – BDP+ • – [C60]– • for the triads 303b – e.
A similar triad donor–BODIPY–acceptor 303f based on porphyrin (Por), BODIPY and fullerene was described by D’Souza et al.[14] in 2017 (Scheme 164). Using femtosecond and nanosecond flash photolysis, ultrafast energy transfer from the singlet excited state of BODIPY to the fullerene moiety was registered, along with the formation of the triad ZnPor – BODIPY – 1[C60]* and then the triplet state ZnPor – BODIPY – 3[C60]*. Selective excitation of the porphyrin fragment in the triad led to an electron transfer to the fullerene fragment, along with the formation of ZnP+ •–BODIPY – [C60]– •.
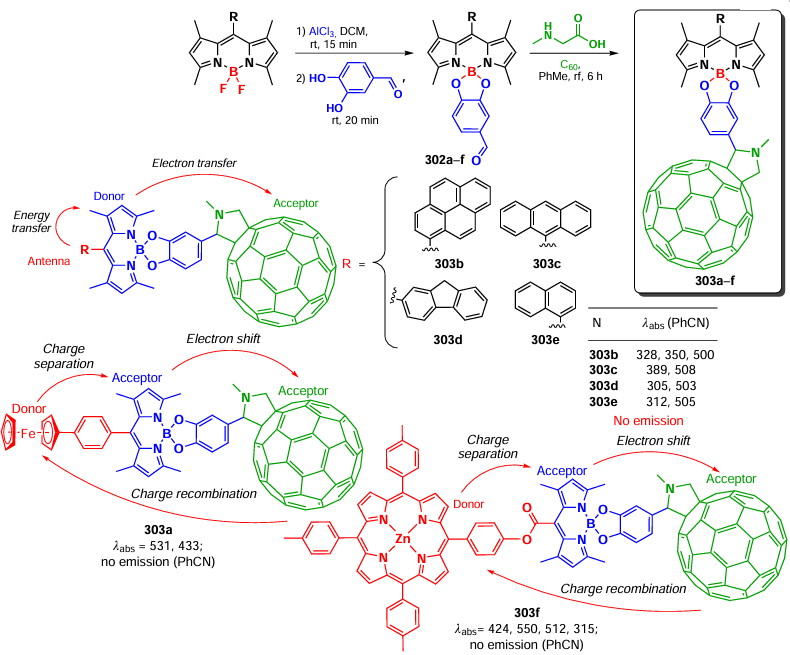
In 2012, Brizet et al.[308] presented dyads 304 and 305 consisting of BODIPY and porphyrin fragments capable of charge transfer from the former to the latter upon complexation with zinc (Scheme 165).
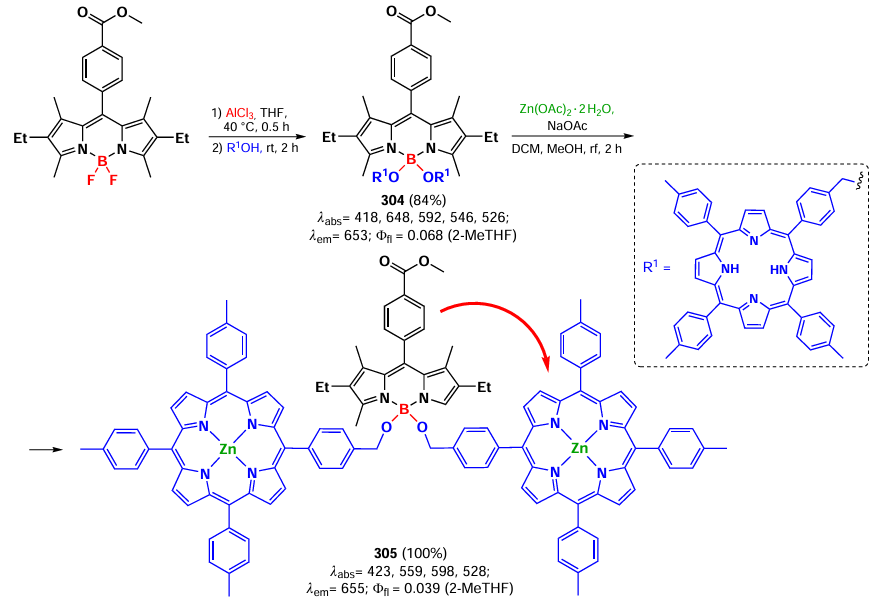
In 2014, Umeda et al.[309] proposed the use of aryloxy derivatives of BODIPYs 306 as photoremovable protecting groups. Photoactivated release of histamine from BODIPY 307 was observed under green light irradiation in HeLa cells (Scheme 166).
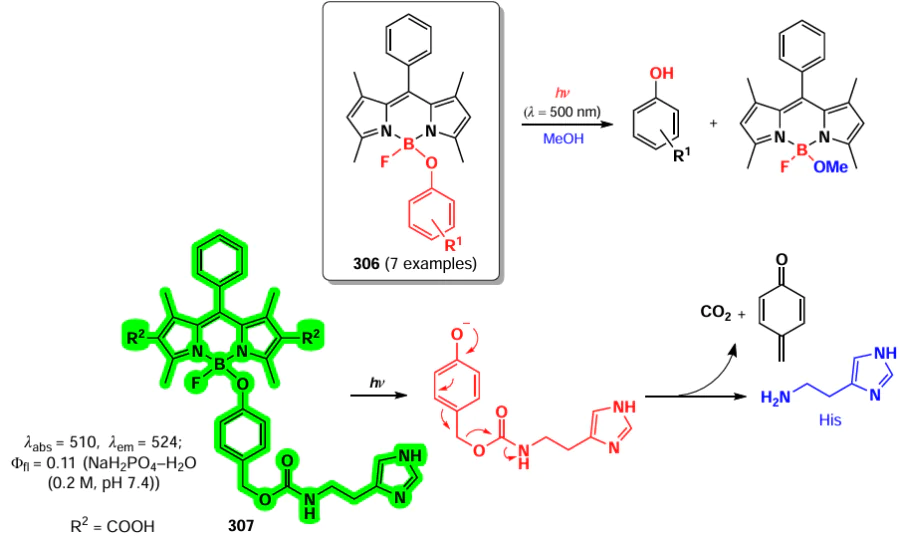
3.9.1.1. Synthesis of chiral B(OR)2-BODIPY
Circularly polarized luminescence (CPL) is a differential emission of right- and left-circularly polarized light by chiral luminescent systems. CPL is quantitatively estimated by the luminescence dissymmetry coefficient glum, whose values range from –2 to +2 (right- and left-polarized emission).[310, 311] Modification of BODIPY at the boron atom with chiral alcohols opens a way to the development of highly fluorescent CPL-active compounds.
The synthesis and chiroptical properties of chiral BODIPYs 308a,b modified at the boron atom with (R)- and (S)-1,1′-bi-2-naphthols (BINOL) were first described by Moya et al.[312] in 2014 (Scheme 167). The 1H nmR spectroscopy data indicated the diastereotopicity of the methylene group protons (ABX3 spin system), and the CD spectra showed mirror dichroic signals at ~ 525 nm. The glum values were shown to be +0.00071 and −0.00085 for compounds 308a and 308b, respectively.
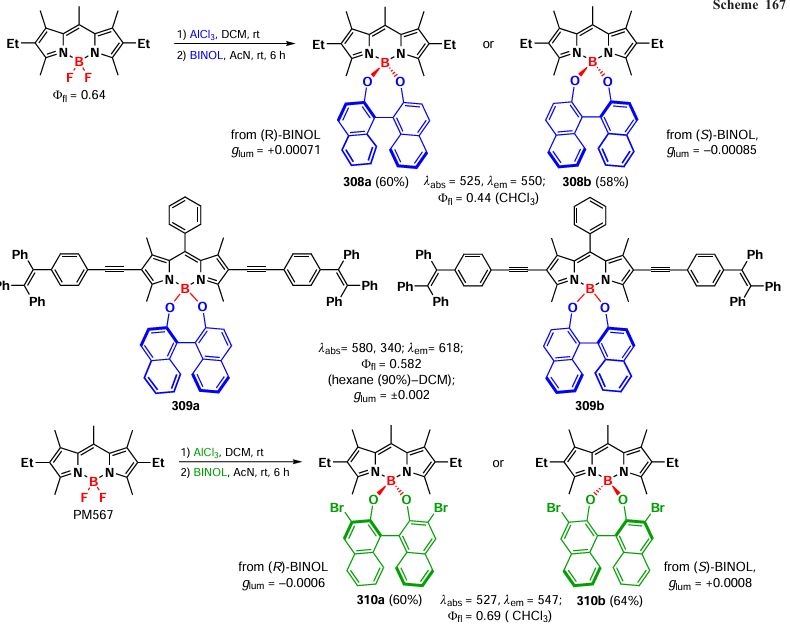
In 2015, Zhu et al.[313] presented similar BINOL-modified BODIPYs 309a,b (Scheme 167). The CD spectra of the enantiomers contained mirror signals at 630 nm and glum values were ± 0.002. In 2017, Moya et al.[314] studied the CPL for BINOL-substituted BODIPYs 310a,b, which had higher fluorescence quantum yields than the previously described compounds 308a,b and were able to produce circularly polarized laser emission.
In 2019, Moya et al.[315] demonstrated that ICT from the BINOL moiety to the BODIPY has a significant effect on the sign of CPL (Scheme 168). Thus, BODIPYs 308 and 310 show opposite glum values due to a strong ICT effect in BODIPY 308 and its absence in BODIPY 310 with BINOL containing a bromine atom substituent at the boron atom. A comparison was made of the chiroptical properties of BODIPY 311a with trifluoromethyl groups, which is incapable of ICT, and BODIPY 311b with a 2,2′-diphenyl-4-biphenanthrol (VAPOL) moiety, which is capable of ICT. The CD spectrum of compound 311a contained a negative visible band in chloroform solution, and the spectrum of the derivative 311b contained a positive visible band. At the same time, distyryl-BODIPYs 312a,b, which are not capable of ICT due to the presence of electron-donating methoxy groups, had an identical negative sign in the CD and the CPL spectra.
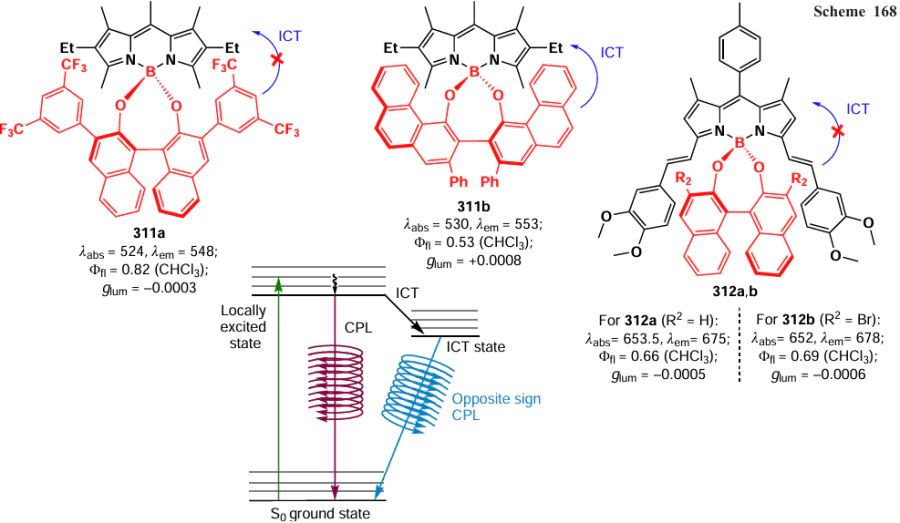
As a follow-up to this study, in 2021, Moya et al.[316] described NIR-absorbing chiral BODIPYs 313a,b with glum values +0.0016 (at λ = 780 nm) and –0.001, respectively (Scheme 169). Compound 314 showed a greater ICT effect than its analogue 313a due to the presence of a quaternized nitrogen atom. ICT included a charge transfer from the BINOL moiety to the BODIPY.
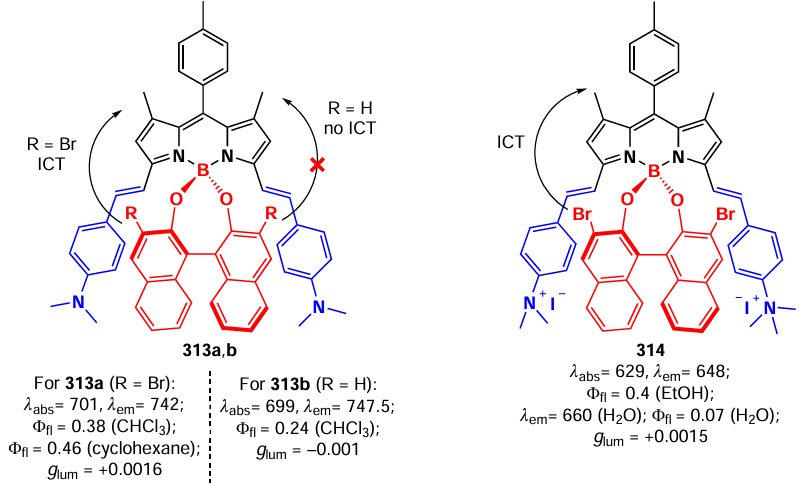
In 2016, Hall et al.[317] synthesized chiral BODIPYs 315a,b using the reaction sequence illustrated in Scheme 170. 3,5-di(2-methoxyphenyl) derivatives 316a,b were synthesized using a cross-coupling reaction, and then the methoxy moieties were demethylated with BBr3, resulting in the meso-aryl analogues 317a,b. The luminescence dissymmetry factors |glum| were shown to be 0.0033 – 0.0047 for BODIPYs 315a,b and 317a,b, and compound 315a demonstrated a record high |glum| value, along with bright fluorescence, equal to 0.0047 and 0.65, respectively.
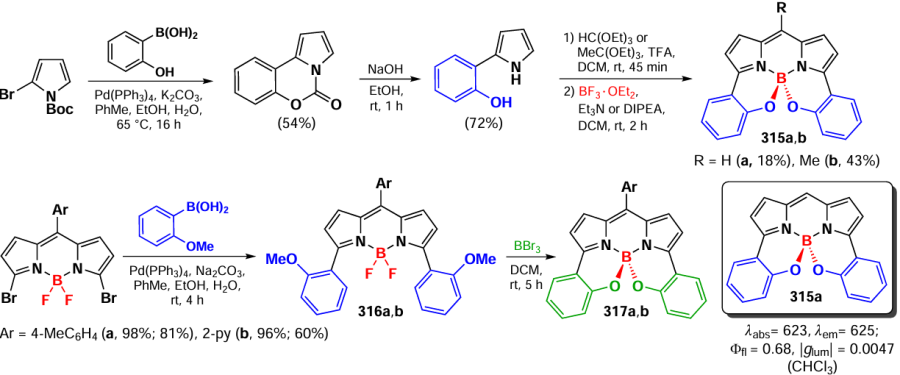
In 2017, Hall et al.[318] isolated an unexpected product 318' while preparing a similar chiral BODIPY. Presumably, this compound is formed as a result of a sequence of transformations: substitution of the BF2 group by the boron atom of (2-hydroxyphenyl)boronic acid, nucleophilic aromatic substitution (SNAr) of the bromine to the phenolic hydroxy group, Suzuki coupling with a second equivalent of (2-hydroxyphenyl)boronic acid, and chelation of the boron atom by the free phenolic hydroxy group (Scheme 171). It is worth noting that the target BODIPY 318 can be prepared by another route (see Scheme 172 below).[319]
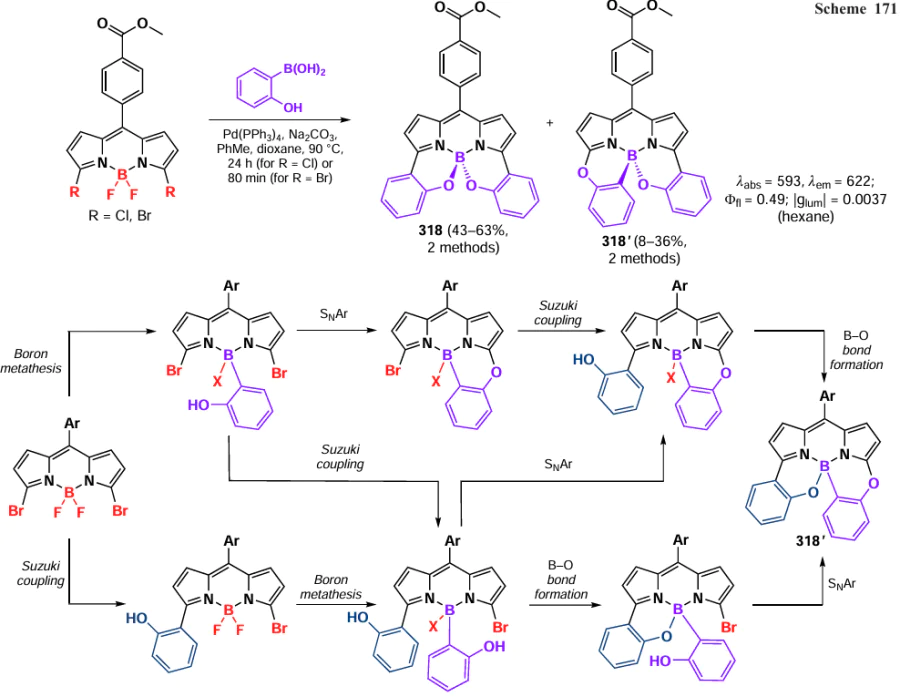
In 2023, the same research group [319] presented an optimized route to the synthesis of helical chiral N,N,O,C-BODIPY 319 from the corresponding dipyrromethenes (Scheme 172). It was also noted that Pd2+ is the key catalyst for the boron metathesis (see Scheme 171). The photophysical and chiroptical properties of this series of BODIPYs were almost identical to those of the previously described compound 318'.
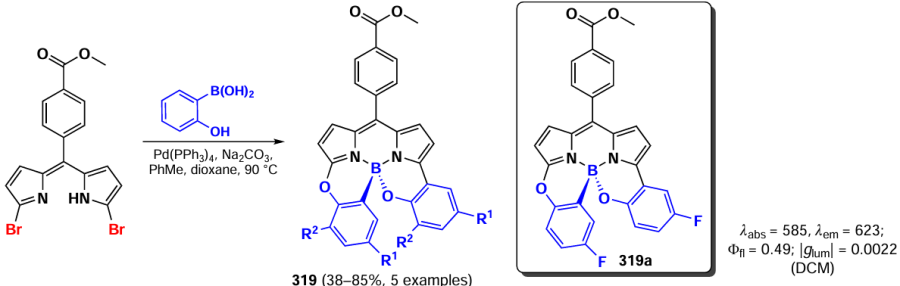
3.9.2. Synthesis of B(OR)2- and 18F-BODIPY using B(F)(OTf)-derivatives
In 2008, Hudnall and Gabbaï [320] suggested using boronium cations of BODIPY as sensors for the detection of fluoride anions (Scheme 173). The starting BODIPY (R = Me) was reacted with TMSOTf to form an unstable, moisture-sensitive salt 320a, which rapidly reacted with 4-dimethylaminopyridine (DMAP) to give a stable monosubstituted salt 321.
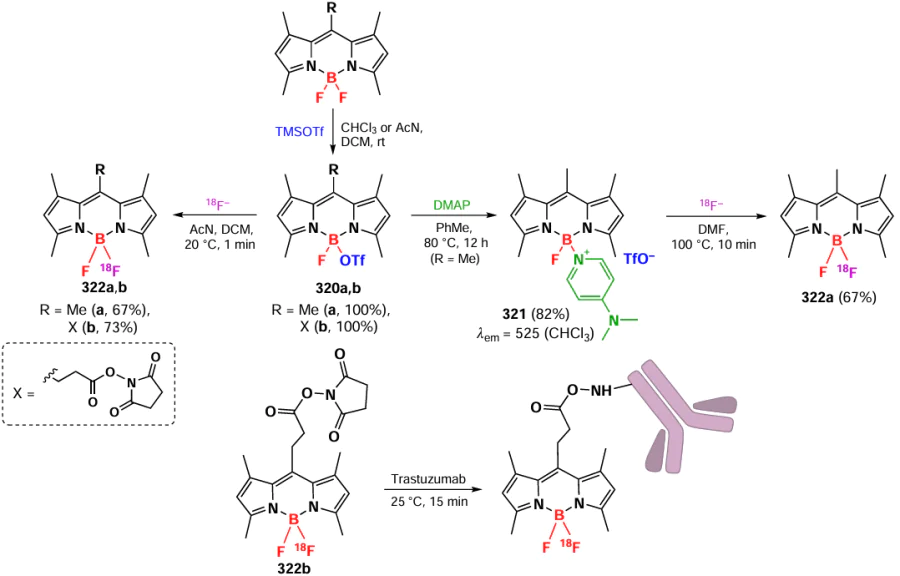
The salt 321 showed a 5-fold increase in fluorescence in the presence of fluoride anion with tetra-n-butylammonium fluoride due to the nucleophilic substitution of the DMAP moiety by the fluoride anion. However, the increase in fluorescence in the presence of chloride and bromide anions was significantly smaller (a 48% increase in fluorescence quantum yield was observed with bromide ions, and a 6% increase with chloride ions). The presence of iodide anion led to the fluorescence quenching due to the ‘heavy atom’ effect. Inspired by this result, Mazitschek et al.[321] in 2012 utilized the rapid exchange reaction of 4-dimethylaminopyridine with 18F– to obtain a labelled BODIPY 322a proposed for fluorescence imaging and PET diagnostics (see Scheme 173). The authors proposed an alternative method in which TMSOTf efficiently displaced one fluoride anion of BODIPY in quantitative yield in less than 5 min at 0°C, and the subsequent addition of 18F– instantly converted the intermediate to the 18F-labeled substrate 322a. A 18F-labeled NHS-activated ester 322 obtained by a similar procedure was used to label the antitumor drug trastuzumab, thereby demonstrating the potential of BODIPY derivatives for fluorescence imaging, PET diagnostics, and theranostics.
In 2014, Mazitschek et al.[322] applied their approach to the synthesis of water-soluble BODIPYs 323 to create highly fluorescent biological sensors (Scheme 174).
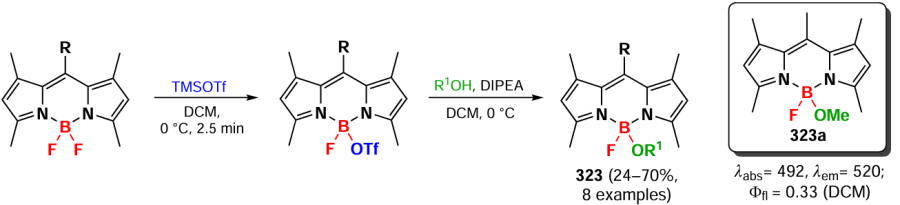
The resulting dyes were demonstrated to have the same optical properties as the starting BODIPYs, along with higher lipophilicity. Compounds 323 showed an increased cellular accumulation and cytotoxicity compared to the starting BODIPYs.
In 2015, Vicente et al.[50] used computer simulations to compare three approaches to the nucleophilic substitution at the boron atom to obtain products 324 (Scheme 175).
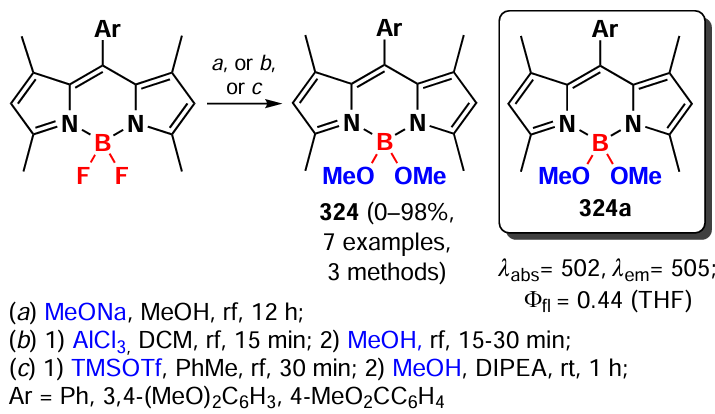
Since the substitution reactions involve the formation of the boronium cation, the Gibbs free energies of its formation from BODIPY under the conditions corresponding to each method were calculated using DFT modelling. For method a, direct dissociation of the fluoride ion from BODIPY was proposed. It occurs in methanol due to its high permittivity; the calculated energy barrier was 37 kcal mol–1. For method b, the formation of a counterion, AlCl3F–, which stabilizes the boronium cation, was proposed. In the case of method c, fluoride elimination leading to a salt of the boronium cation with the triflate anion was suggested; the potential energy barrier of 25 kcal mol–1 for this reaction can be easily overcome by boiling the reactants in toluene.
3.9.3. Synthesis of BR2-BODIPY via BCl2-derivatives
In 2012, Lundrigan et al.[323, 324] developed a method for the synthesis of 4,4-dichloro-BODIPY 325a,b under an inert atmosphere with BCl3. The corresponding dichloro-derivatives were stable under an inert atmosphere, and were found to undergo nucleophilic substitution reactions at the boron atom with both O- and C-nucleophiles under milder conditions. These conditions resulted in the formation of products 326 – 330 at a higher yield than that observed with the difluoro analogues. (Scheme 176).
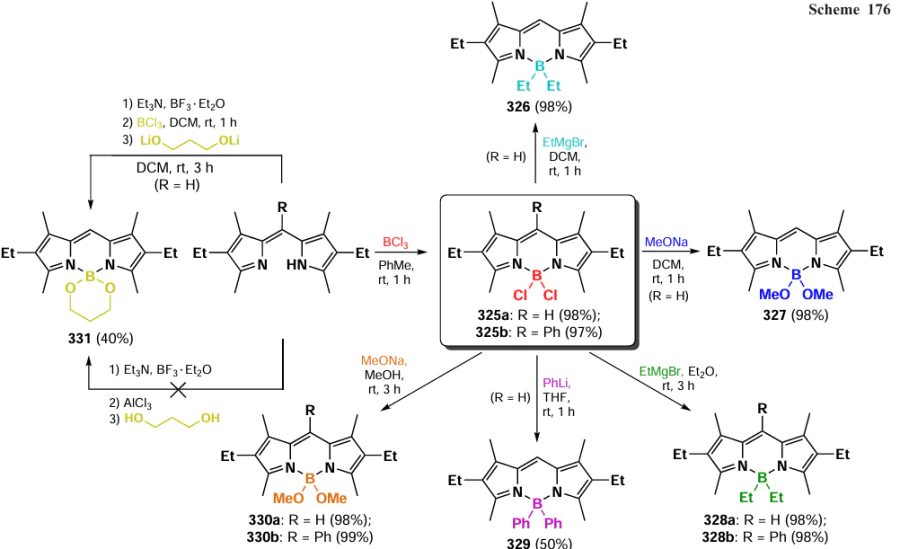
Nucleophilic attack by such a strong nucleophilic agent as a Grignard reagent occurred under the chosen conditions only at the boron atom, without affecting the substituted BODIPY core. A reaction with lithium dienolate was also carried out to obtain the O,O-ester BODIPY 331. The direct synthesis of these compounds from the difluoro derivative under the previously proposed conditions[167] using aluminium chloride catalysis did not lead to the target molecule.
In 2017, Ray et al.[325] modified the boron atom of BODIPY with N- nucleophiles using a one-pot reaction with initial treatment of the substrate with BCl3 (Scheme 177).
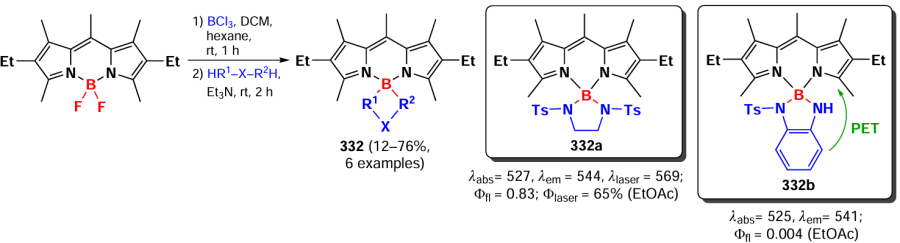
N,N-BODIPYs 332 exhibited bright fluorescence even in the solid state, as well as a noticeable laser efficiency exceeding that of the starting difluoro derivative. Compound 332b showed a weak fluorescence due to PET. Since the chelation reaction of metal cations Co2+, Fe2+, Fe3+, and Cr3+ led to the disruption of PET and to the enhancement of emission, BODIPY 332b was suggested as a fluorescent cation sensor.
In 2018, Zhang et al.[326] proposed a modification at the boron atom with N- or O-TMS nucleophiles in the presence of Lewis acids. Using such reactions, which involve the in situ formation of BCl2 derivatives, benzotriazole and trifluoroacetamide derivatives of BODIPYs 333 – 335 were obtained (Scheme 178).
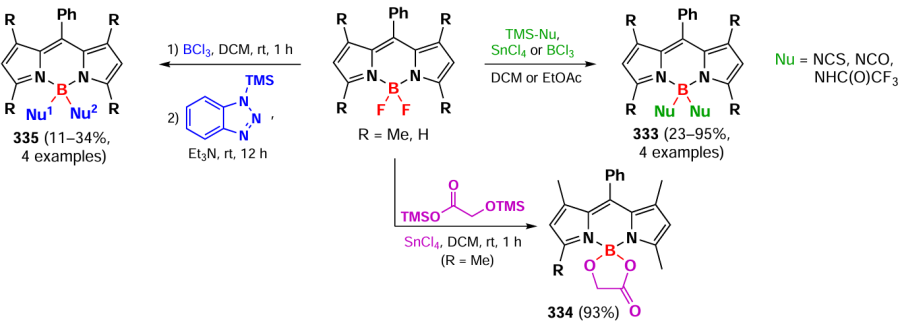
In 2021, Ray et al.[327] described N,N-substituted boron BODIPYs 336 containing an alkynyl moiety, which allowed such compounds to be introduced into a click reaction to form BODIPY derivatives of triazoles 337 (Scheme 179). The ability to accumulate in the mitochondria of PANC-1 cells was shown for compound 337a. In 2023, the same research group [328] presented unsymmetrical N,N-BODIPYs with naphthalene, camphor fragments, and morpholine fragments targeting lysosomes. The derivative 338 with an anthracene moiety exhibited FRET (fluorescence resonance energy transfer) with complete energy transfer from the anthracene moiety to the BODIPY, as indicated by absorption and fluorescence spectra.
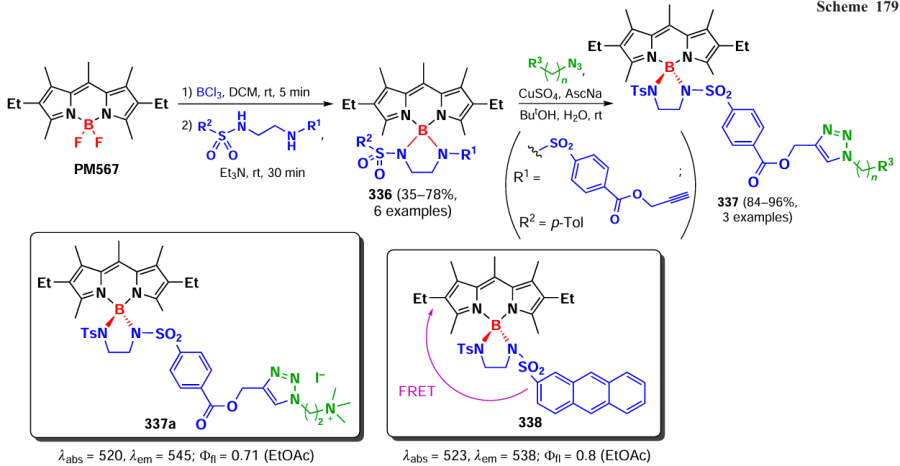
In 2019, Wang et al.[329] introduced N-protected amino acid fragments to the boron atom of BODIPY by in situ formation of BCl2-BODIPY (Scheme 180). Derivatives 339 and 340 had a slightly increased fluorescence quantum yield compared to that of the initial F,F-BODIPY due to the enhanced rigidity around the spirocyclic boron atom.
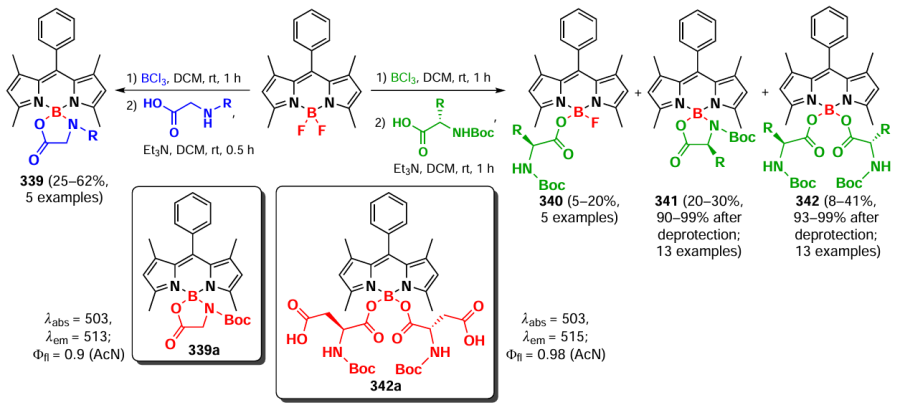
In 2021, the same research group synthesized B-spiro derivatives 341 and BODIPYs 342, containing two amino acid fragments of histidine, lysine, arginine, aspartic acid, tyrosine, serine, glutamine and methionine, as well as tripeptide (Gly)3-BODIPY (see Scheme 180). All synthesized α-amino acids with a BODIPY fragment demonstrated higher fluorescence quantum yields (Φfl ~ 0.9) than the original BF2-BODIPY (Φfl = 0.61), and N,O-bidentate spiro-Arg- and spiro-His derivatives showed toxicity on human HEP2 cells (IC50 ≈ 22 μM).[330]
In 2020, Moya et al.[331] proposed to use the in situ forming BCl2-BODIPYs to synthesise carboxylate derivatives 343. This approach made it possible to obtain the previously described BODIPYs in higher yields, to introduce different functional groups into the molecule, including the acceptor ones, and also to synthesize previously inaccessible O,O-spiro derivatives 344 (Scheme 181). Such BODIPYs demonstrated increased laser emission compared to the commercially available dye PM567.
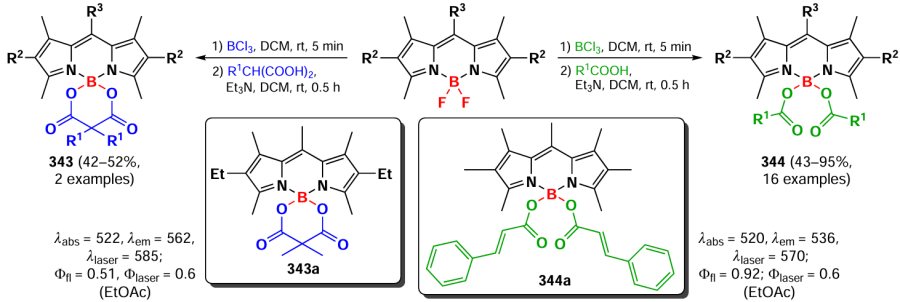
3.9.4. Modification at the boron atom of BODIPY by С-nucleophiles
Modification at the boron atom by strong C-nucleophiles, such as Grignard reagents or organolithium compounds, was first described in 2005. Ulrich et al.[332] reported reactions of BODIPY at the boron atom with 4-lithioethynyltoluene and 1-lithioethynylpyrene (Scheme 182). Products 345a,b exhibited large Stokes shifts as well as minimal fluorescence quenching upon coupling with BSA.
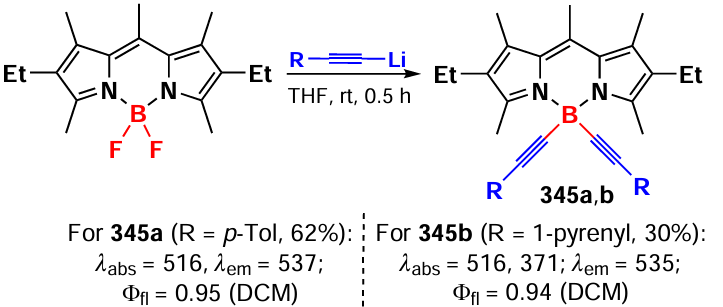
In 2006, the same research group [333] demonstrated possibility of monoarylation reactions at the boron atom using 1 equiv. of a Grignard reagent to form compounds 346, and also the possibility of substitution of both fluorine atoms using 2 equiv. of a Grignard reagent (products 347). Biaryl derivatives 348 were obtained upon modification with organolithium carbanions. In the same year, dyads 349 capable of rapid energy transfer from the pyrene and the perylene fragments to the BODIPY were presented;[334] contributions from both chromophores were observed in the absorption spectrum of the dyads, while only the BODIPY signal was recorded in the fluorescence spectrum. In 2007, Goeb and Ziessel [335] also modified diisoindolodithienylpyrromethene at the B-center with alkynyl aryl-containing chromophores using a Grignard reagent. The resulting dyad 350a exhibited a Stokes shift of 7800 cm–1 (Scheme 183).
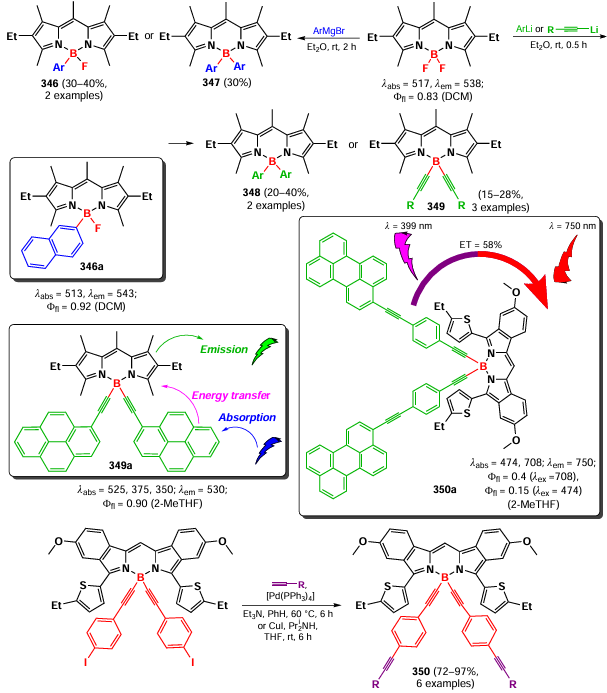
In 2008, the authors [336] proposed a method for the sequential introduction of C-nucleophiles at positions 2, 6, 8, and 4 of the BODIPY core using electrophilic halogenation reactions to form products 351 – 354. Modification at the boron atom was also carried out using a Grignard reagent (Scheme 184). It is worth noting that the selective B – C-alkylation was successfully carried out even in the presence of halogen atoms in the BODIPY core (see Scheme 1844, as well as the synthesis of compounds 355 and 356 in Scheme 185).
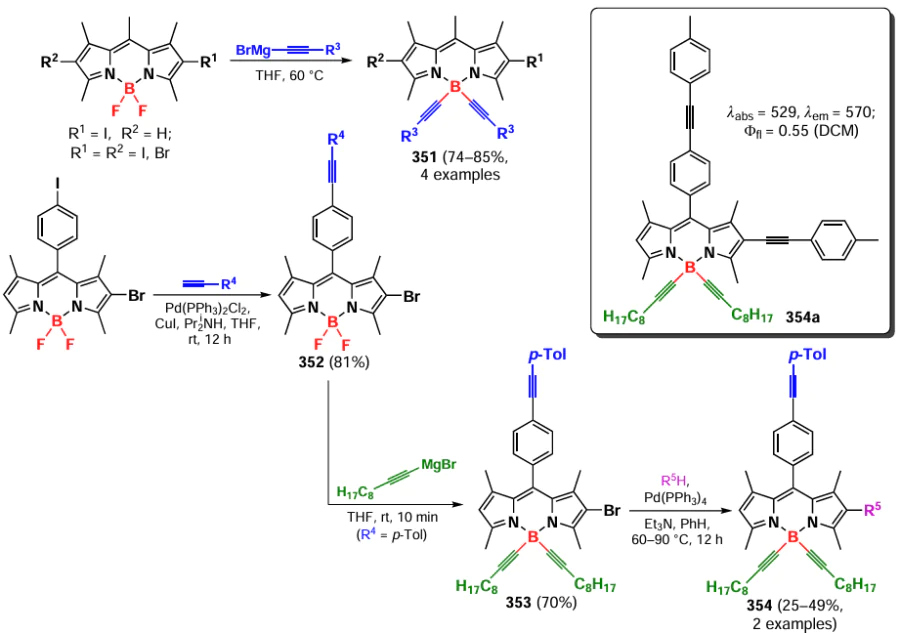
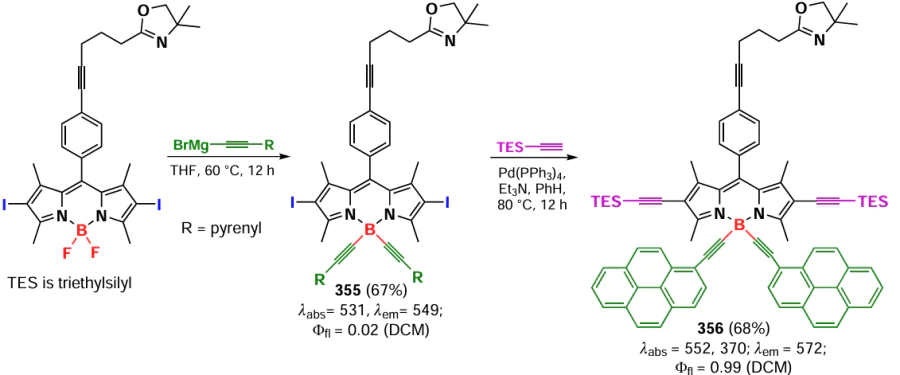
The interaction between a Grignard reagent and a boron atom of BODIPY has been successfully used by Niu et al.,[93, 337] and by Bura and Ziessel [338] to create water-soluble fluorophores 357.
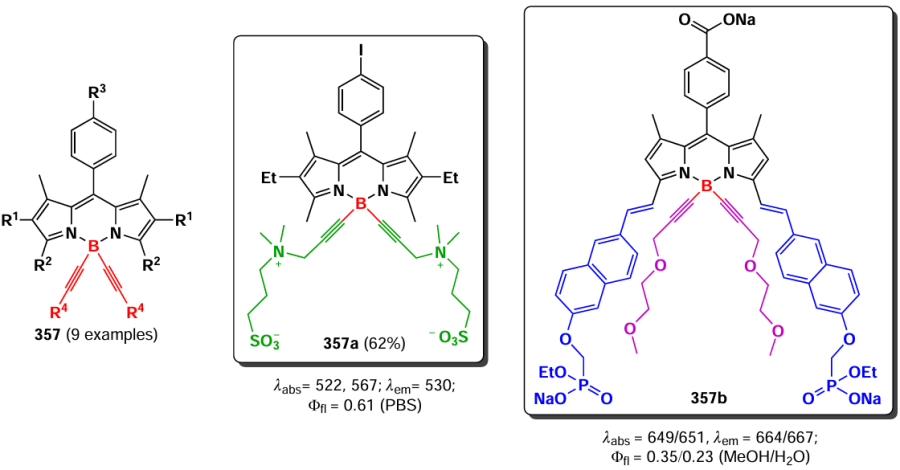
In 2010, Ziessel et al.[339] presented chiral BODIPYs 358 – 360 modified at the boron atom with naphthalene (Scheme 186). Desymmetrization of the boron atom was achieved by the oxidation of 3-methyl group; hydrogen bonding between the fluorine atom and the proton of the aldehyde group in BODIPY 359 was proven by 1H and 13C nmR spectroscopy. The CD spectra of enantiomers 360a and 360b confirmed the persistence of both enantiomers.
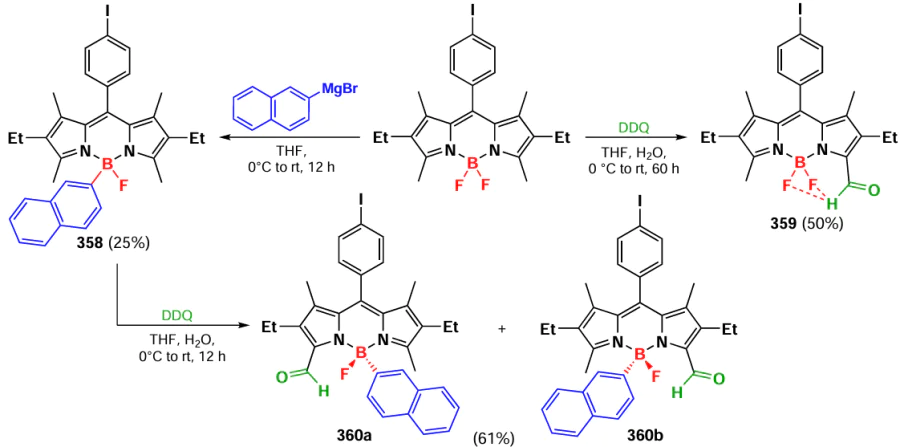
In 2008, Li et al.[86] considered the competition of reaction centres in the BODIPY core when interacting with nucleophiles. Despite the presence of electrophilic centres in unsubstituted BODIPY, which are capable of interacting with hard nucleophiles, the reaction at the B – F bond with the Grignard reagent was the fastest (product 361). Further nucleophilic substitution in a reaction with piperidine derivative proceeded at the BODIPY core, affecting positions 3 and 5 as expected (product 362) (Scheme 187).
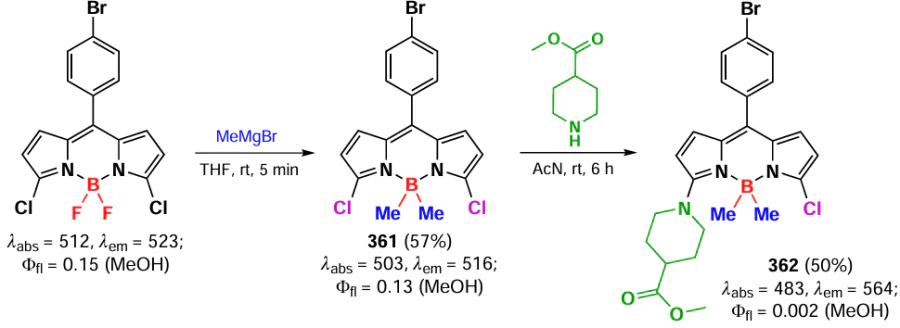
In the reaction with a softer nucleophile, cyanide anion, the result of nucleophilic substitution is determined by the Lewis acid used. In the case of tin tetrachloride, the reaction proceeds selectively at the carbon atom to form compound 363, whereas catalysis with boron trifluoride etherate promotes both types of substitution reactions, leading to tetracyanide 364 (Scheme 188).
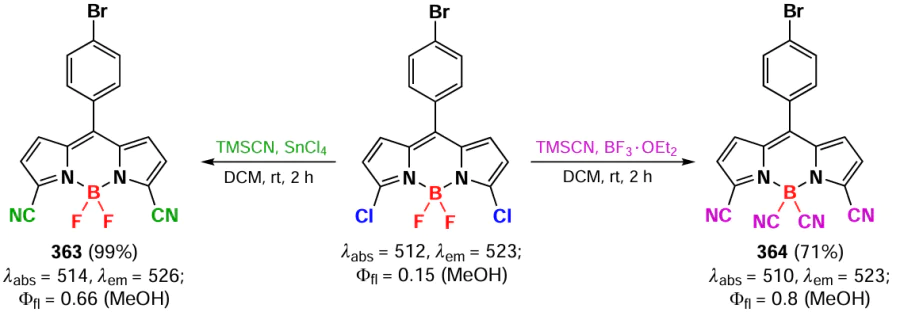
In 2014, Chattopadhyay et al.[340] suggested using the ethylation reaction at the boron atom in BODIPY as a protecting group to carry out reactions at the BODIPY core with strong nucleophiles or reducing agents capable of affecting the BF2 centre. The authors developed a convenient procedure for the substitution of fluorine atoms with ethyl groups under the use of Et2AlCl to form Et2B-BODIPY 365. Regeneration of BF2-BODIPY 366 was completed easily in the presence of BF3 · Et2O. Moreover, the preparation of a similar ethyl substrate by direct reaction with a Grignard reagent or using in situ formed BCl2-BODIPY either gave a lower yield of product 367 or required a longer time. The authors demonstrated the ability of Et2B to act as a protecting group by performing the McMurry coupling reaction, impossible for BF2-BODIPY, which resulted in the synthesis of β,β-dimer 368 with an ethynylene bridge (Scheme 189).
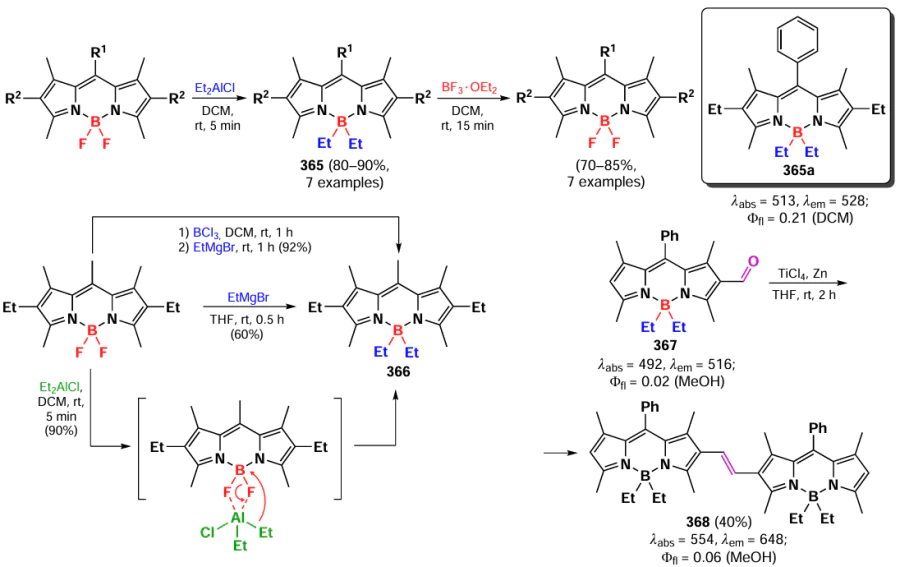
Modification at the boron atom can be carried out by using α-lithiated five-membered heterocycles. Thus, in 2023, Thompson et al.[341] presented the synthesis of BODIPYs 369a – d, modified at the boron atom with chalcogenophenes (Scheme 190). These compounds were able to generate singlet oxygen and also acted as effective PDT agents on HeLa cells with moderate toxicity in the dark and nanomolar toxicity under irradiation.
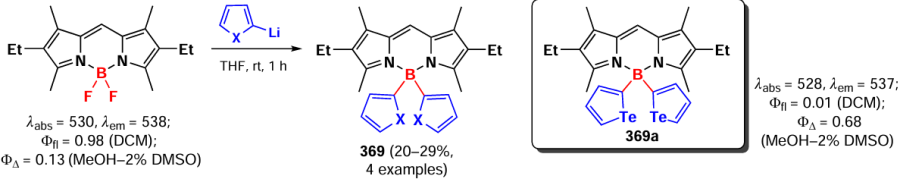
In the same year, Wang et al.[21] used AlCl3 as a catalyst to modify BODIPY with chalcogenophenes, hypothesizing that conversion of (N,N-chelate)BF2 to borenium cation [(N,N-chelate)BX]+ or [(N,N-chelate)B]2+ would allow boron metalation reactions with weak nucleophiles without affecting the pyrrole ring. Using this approach, reactions of arylstannanes with unsubstituted BODIPY in the presence of aluminium chloride were carried out to give BODIPYs 370, 371. This approach was proven to be convenient for nucleophilic substitution at the boron atom in aza-BODIPY 372 (Scheme 191).
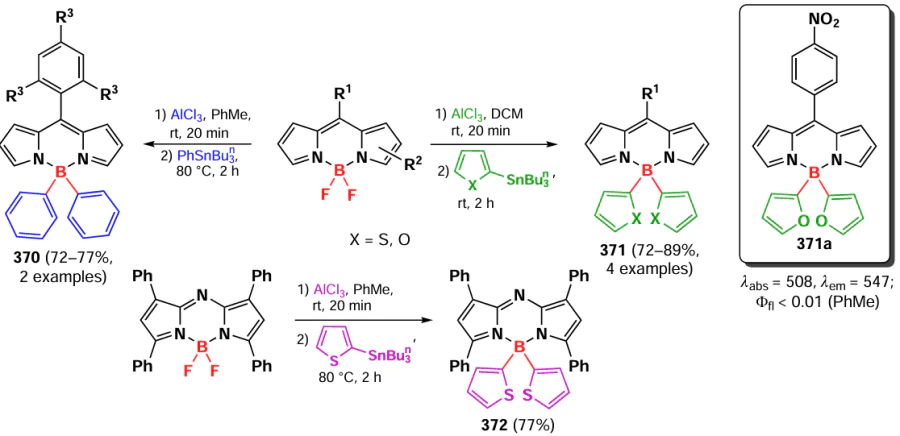
3.9.5. Synthesis of B-acyloxy BODIPY derivatives
In 2012, Zhao et al.[342] synthesized alkoxycarbonyl BODIPYs 373 and 374 containing one or two acetoxy groups at the boron atom by nucleophilic substitution reaction at the boron atom using 20 equiv. of in situ generated trimethylsilyl acetate (Scheme 192). Derivatives 373 and 374 showed significantly improved aqueous solubility compared to the original BODIPY, while maintaining its photophysical properties, such as high fluorescence quantum yield and high photostability.
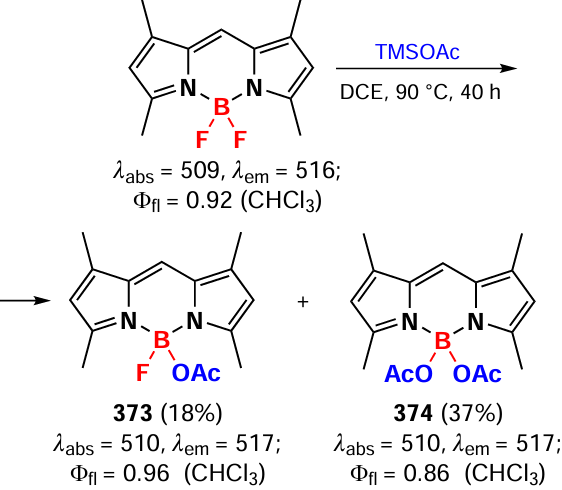
In 2013, Ortiz et al.[166] reported the synthesis of B-acyloxy-modified BODIPYs 375 – 377 by a reaction with 20 equiv TMSOAc in the presence of 3 – 4 equiv. AlCl3 as a Lewis acid (Scheme 193, Method A). Furthermore, alternative routes were developed, including a reaction with in situ generated TMSOAc or TMSOC(O)CF3 from the corresponding acid or TMSCl in the absence and presence of AlCl3 (see Scheme 193, Methods B, C). The routes B, C were more efficient compared to Method A. The BODIPY derivatives exhibited laser efficiencies exceeding those of the starting difluoro-BODIPYs, the highest efficiency was observed for compound 377a modified with trifluoroacetoxy groups.
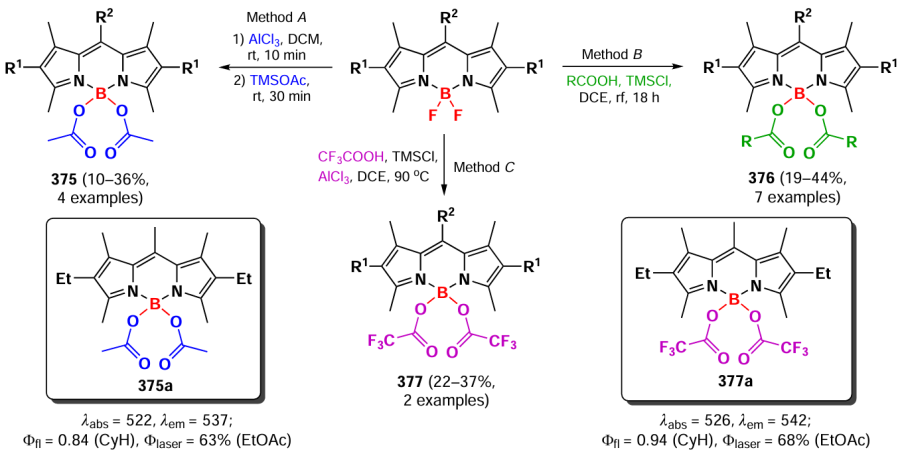
In 2016, Chiara et al.[343] suggested using microwave irradiation to synthesize boron-modified BODIPYs 378 and 379 in high yields (Scheme 194).
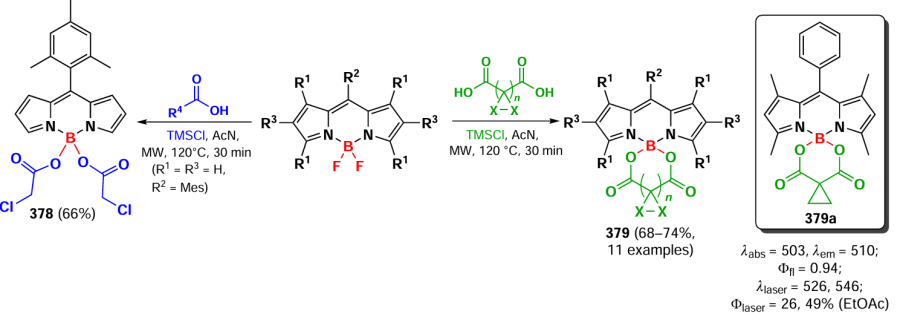
Compounds 379 exhibited high laser efficiencies and good photostability. With increasing concentration of BODIPYs in solution, a second, bathochromically shifted laser emission band appeared in their spectra at 540 – 545 nm. The dependence of laser efficiency, as well as the ratio of the bichromatic emission bands on pump energy, indicated the formation of J-aggregates in organic solvents, which was also confirmed by molecular dynamics simulations.
4. Advances in boron-dipyrromethene research
BODIPY chemistry has been actively developed over the past 30 years, and has paved the way for new fluorophores with applications as fluorescent sensors, photocatalysts, photo-removable protecting groups, anti-tumor agents, laser dyes and light harvesting systems.
The main synthetic approaches to obtain the boron-dipyrromethene core were developed 10 – 30 years ago.[47, 58, 79, 80] Currently, the known methods are being improved and new, more advanced methods are being developed. Thus, it is worth noting the direct synthesis of α-pyrrolyl-BODIPY 3 with absorption at ~ 600 nm [27] and the use of microwave radiation in the preparation of BODIPY from acid chlorohydrides, which significantly reduced the reaction time.[65] The expansion of the range of starting substrates for BODIPY synthesis has resulted in the synthesis of new compound libraries. In particular, water-soluble α-glycosido-BODIPY 12 with bright fluorescence in aqueous media are formed from α-glycosidopyrroles.[22] The use of organoboron compounds as an alternative to boron trifluoride esterate allowed the direct synthesis of 4-substituted BODIPY 26 – 28.[95, 96]
An important advantage of 2,6-dihalogen-BODIPY was their ability to induce ISC and exhibit photosensitizing properties. 2,6-Dibromo-BODIPY 53 and 54, which belong to both the type I and type II PDT agents, demonstrated high antiproliferative activity in vivo.[132] The 2,6-dibromo-BODIPY complex with copper 59-Cu formed ROS not only as a photosensitizer but also via a copper-mediated mechanism. As a result, it exhibited cytotoxic activity under hypoxic conditions in vitro, and also significantly reduced tumor growth in vivo.[138] Monohalogenated BODIPYs can also act as PTT-agents. For example, 6-iodo-BODIPY 57a was active in the nanomolar concentration range under low-power light due to the formation of PTT-active aggregates in the intracellular medium.[78]
Many new methods of modifying BODIPY via electrophilic substitution reactions have been developed in recent years. Thus, the interaction of BODIPY with 2-fluoropyridinium ethanide leads to the formation of a stable carbanion 97 with good water solubility.[171] Indium-catalyzed β-alkenylation of BODIPY allows the synthesis of 2,6-alkenyl-BODIPY 98 – 100, [25] and 2- and 2,6-dipropargyl-BODIPY have been obtained by the Nicholas reaction involving BODIPY and the propargyl alcohol Co-complex 101 – 104.[172]
New synthetic strategies for the modification of BODIPY, such as oxidative nucleophilic substitution, developed in the last few years have opened a direct route to the formation of α-seleno-BODIPYs 135, 136 and α-cyano-BODIPY from unsubstituted analogs.[19, 189] Photochemical synthesis of α-amino-BODIPY by reaction with aqueous ammonia proves the possibility of BODIPY modification by increasing its the reactivity in the excited state.[20]
The susceptibility of BODIPY to nucleophilic substitution reactions together with its sensitivity to the donor-acceptor properties of the substituent is widely used in the design of fluorescent probes for the detection of various organic and biological analytes. The fluorescent probes presented in this review act either through the nucleophilic substitution reaction of BODIPY with the analyte, which leads to a change in the photophysical or chemical properties of the probe due to differences in the electronic properties of the leaving group and the nucleophile, or by reducing PET-induced fluorescence quenching due to the reaction of the PET-donating scaffold with the analyte and the quenching of BODIPY fluorescence. Thus, probes 122, 130, and 169 are able to selectively detect cysteine, homocysteine, or glutathione due to the different emission maxima of the nucleophilic substitution products of these biothiols.[177, 185, 207] Probes 123 – 125, 128 and 129 have suppressed emission in the initial state, their reactions with different biothiols result in products with emission at different wavelengths, allowing selective detection of biothiols ratiometrically.[183, 178-180] The mechanism of action of probe 126 is based on the emission properties not being affected after reaction with biothiols, but the resulting products differ from each other in polarity.[181]
Reduction of PET-induced fluorescence quenching as a result of specific reaction with the analyte is a preparatively convenient way to detect acids and other electrophiles due to the significant difference in fluorescence of the starting fluorophores and products. This principle was utilized in the development of sensors 119 and 120 sensitive to HClO, as well as sensors 167 and 173 for the detection of acyl chlorides and nitrite anions, respectively.[16, 35, 36, 175, 205] It should be noted that the analytical signal may be caused not only by the flaring of fluorescence due to the removal of the PET effect, but also by the quenching of emission due to ICT. This is the operating principle of the 131 sulfide-anion sensor.[186]
The CH-acidity of BODIPY methyl groups is widely used in a variety of synthetic approaches to tune the photophysical properties of fluorophores. In particular, the π-system expansion via the Knoevenagel condensation is a common approach to create NIR-absorbing BODIPY fluorophores. The suppression of BODIPY emission due to J-aggregation contributes to nonradiative decay, thus J-aggregates based on distyryl-BODIPY are promising PTT agents that have repeatedly proven their therapeutic efficacy in vivo. Thus, NIR-absorbing self-aggregating BODIPY 182 in nanoparticles have been developed, acting as combined PDT and PTT agents, which exhibit remarkable antitumor efficacy in vivo.[32] Styryl derivatives of 8-CF3-BODIPY 184 and 185 are of great interest as PTT agents due to the free rotation of the CF3 group, for which the record high PCE of 88.3% and the possibility of photoacoustic imaging of tumor tissues in nanoparticle form have been shown.[31, 34] Distyryl-BODIPY 190 can exhibit the properties of sonodynamic therapy agents and effectively inhibit tumor growth in vivo under ultrasound irradiation.[15] The ability of BODIPY 189, containing electron-withdrawing substituents, to form J-aggregates allowed the development of a bioimaging agent with fluorescence in the NIR-II band, as demonstrated by real-time imaging of the mouse vascular system.[33]
The α-methyl groups of BODIPY can also be modified by various nucleophiles. For example, the oxidation of 4,4-dicyano-BODIPY by Pb(OAc)4 followed by reaction with nucleophiles, described in 2022, allowed the synthesis of a library of more than 50 compounds and the introduction of a variety of O-, N-, and C-nucleophiles at the α-position of BODIPY via a methylene linker. This transformation opens up the possibility of easily modifying BODIPY by creating a new C – C bond that does not require metallocatalysis.[6]
Approaches to the modification of BODIPY by radical mechanism with the creation of C – C, C – S, and C – N bonds are being actively developed due to the ease of radical processes, the availability of reagents, and the possibility of introducing sterically loaded functional groups that are difficult to introduce by alternative nucleophilic substitution or cross-coupling reactions.[264] For example, the introduction of thiol groups into the α-position of BODIPY can be easily accomplished by the action of radical initiators such as tert-butylhydroperoxide [268] or under irradiation.[269] The use of radical reactions makes it possible to introduce sterically loaded functional groups into the BODIPY core. Thus, BODIPY 213'b with cyclohexyl fragments at positions 3 and 5 was obtained, which exhibits solid-phase fluorescence.[264] An alternative synthetic route to create the C – C bond is the use of metallocatalysis or organometallic compounds, which require the introduction of protecting groups at the boron atom, which is prone to reactions with harsh C-nucleophiles (see Section 3.9.4).
Due to their excellent photochemical properties, BODIPYs have also been the subject of interest as photocatalysts. 2,6-Dihalogen-BODIPYs and BODIPYs devoid of heavy atoms are capable of functioning as photocatalysts in the singlet or triplet excited states, acting as both oxidants [275] and reducing agents.[273] BODIPYs have the ability to function as a source of singlet oxygen in oxidation reactions and as an electron donor or carrier, thereby facilitating the formation of С – С, С – N, С – Р bonds.[272-275]
BODIPY derivatives prone to ISC, such as 8-SMe-BODIPY 16, are employed as photocatalysts for the Beckmann rearrangement, facilitating the conversion of oximes to amides.[29] In recent years, an approach to the use of BODIPY as catalysts for ion-radical addition to multiple bonds has also been developed. Furthermore, 2,6-dihalogen-BODIPY 233 has been identified as a suitable photocatalyst for the ion-radical addition of α-halogenecarbonyl compounds to vinyl ethers, leading to the synthesis of several classes of organic compounds, including lactones, protected aldehydes, and others.[279] In regard to dyad 234a – b, the potential for photocatalysis in the radical addition of malonic esters to the carbon-carbon double bond was demonstrated.[28]
The formation of dimers and oligomers from monomeric BODIPY is a topic of significant interest, given the distinctive photophysical properties exhibited by these higher-order structures, which are not observed in individual chromophores. The position at which dimerisation occurs has a significant impact on the properties of the resulting oligomers. To illustrate, α,α-dimers 235 – 239, which exhibit a low degree of conjugation between BODIPY links, are characterized by exciton splitting of absorption maxima and high fluorescence quantum yields.[213, 279, 280] The α,α- and γ,γ-dimers 238 and 239 possess helical chirality and are a mixture of optically active aptoisomers. Planar α,α-dimers 240 – 246 exhibit a large bathochromic shift in the electronic spectrum with no exciton splitting.[282-284]
It has been demonstrated that dimers at the α,β-, α,γ-, β,γ- and meso,meso-positions, which do not contain heavy atoms in the structure, are capable of acting as PDT agent. The orthogonal α,γ-, β,γ-, β,meso- and meso,meso-dimers (e.g. compounds 241, 262, 265) are predominantly type II PDT agents, producing singlet oxygen upon irradiation. In contrast, the β,meso-dimer 265a has antiproliferative activity with EC50 values in the nanomolar range.[158, 281, 288] In contrast, α,β-dimers 247 are exclusively type I PDT agents and therefore exhibit high antiproliferative activity upon irradiation under both normoxic and hypoxic conditions.[285] The condensed oligomers 279 and 286, which exhibit absorption in the near-infrared region, are regarded as prospective FTT agents. The emission of these compounds is suppressed due to the diminished energy gap between the HOMO and LUMO.[295, 297] BODIPY oligomers 294 and 296a, in which the BODIPY subunits are linked via an alkenyl linker, also display high FTT activity.[88, 300] These oligomers are distinguished by a high PCE value (56 – 62%) and the additional property of emission in the NIR-II band.
The variation of substituents at the boron atom enables the enhancement of fluorescence quantum yields, the improvement of photostability, and the development of water-soluble BODIPY derivatives. BODIPYs with substituents at the boron atom have been identified as a promising class of light-harvesting systems. The incorporation of chiral substituents, such as BINOL, at the boron atom in BODIPY results in the formation of fluorophores exhibiting circularly polarised luminescence.[310, 311] The BODIPY 314 synthesised by Moya et al.[316] represents the inaugural instance of a fluorophore exhibiting NIR-band CPL in an aqueous milieu. The ICT effect control permits the alteration of the sign of the circular polarisation.[315] Another example of light-gathering systems based on the change of substituents at the boron atom are BODIPY dyads and triads, which are created by the introduction of a fragment of ethynylene pyrylene, resulting in an energy transfer from the pyrene to the BODIPY core.[334] Moreover, in these light-gathering Stokes systems, the shift reaches values as low as 200 nm.[336] The functionalisation of BODIPYs can be achieved through the modification of substituents at the boron atom, while maintaining the photophysical properties of the molecule. This allows the generation of probes with bright fluorescence.[328] Boron-substituted BODIPYs are also promising agents for PDT, as exemplified by compound 379a, modified at the boron atom by tellurophene and exhibiting nanomolar toxicity due to the ‘heavy atom’ effect.[341] The introduction of such chalcogenophenes is feasible not only with the corresponding organolithium derivatives, but also with arylstannanes in the presence of AlCl3.[21]
This section presents an analysis of the most significant advances in the design and property studies of the various BODIPY derivatives considered in this review.
5. Conclusion
Boron-dipyrromethene dye-based fluorophores (BODIPY) are one of the most popular and widely used classes of fluorescent organic compounds. Over the past few decades, research into the development of BODIPY with tailored photochemical properties has led to many applications for such derivatives, such as bioimaging agents, fluorescent probes, antitumor agents, laser dyes, components of organic photovoltaic cells, and photocatalysts. Such a diversity of practical applications is due to the photophysical properties of BODIPY, as well as the ability to fine-tune the photophysical and photochemical properties of these fluorophores to suit the required tasks through simple structural modifications accessible by classical methods of organic synthesis. This paper systematizes information on the synthesis, modification and application of boron-dipyrromethene dyes, with an emphasis on studies published from 2017 to 2024.
Approaches to the creation of BODIPY fluorophores with specified photophysical properties and high biological activity are currently being actively developed. At the same time, significant progress has been made over the past 5 years in developing new methods for modifying the structure of BODIPYs and tuning their photophysical properties. A number of new synthetic and post-modification methods allow the target BODIPYs to be obtained under milder conditions, in higher yields, or in fewer steps. In particular, a significant reduction in process time can be achieved by using microwave radiation. Approaches to the creation of new methods for modifying BODIPY by electrophilic and nucleophilic substitution reactions, as well as by means of radical reactions, are also being actively developed. A number of methods, such as radical С – H-activation and oxidative nucleophilic substitution of the hydrogen atom, allow the introduction of target groups into the BODIPY core directly via the С – Н bond and under mild conditions. Thus, further research into the chemical transformation of BODIPY can be aimed both at creating more efficient methods for the synthesis of already known BODIPY derivatives and at developing new ways to optimize the structure of boron-dipyrromethene dyes. The methods for modifying BODIPY developed in recent years can be further applied to the creation of new fluorescent probes, therapeutic and diagnostic agents, as well as laser dyes.
The use of BODIPY as imaging and detection tools has become attractive due to the high extinction coefficients and large fluorescence quantum yields of boron-dipyrromethene complexes. Over the past 5 years, methods for detecting a large number of objects, such as metal ions, hypochlorite and biothiols, have been improved. Fluorescent probes created in recent years make it possible to selectively determine target compounds in the nanomolar concentration range, including in living systems. Taking into account the wide variety of chemical properties of BODIPY derivatives, it is reasonable to expect that new probes based on them will be developed with sensitivity to metal ions, biomolecules and other objects. In addition, the creation of fluorescent probes with NIR emission for effective use in living systems, remains an urgent task. BODIPY fluorophores with emission in the NIR-II region have shown great promise for in vivo imaging, so it is reasonable to expect that the creation of new active fluorescent probes with NIR-II emission based on them.
PDT- and PTT-agents containing the BODIPY core are particularly attractive because they combine high antitumor activity under the influence of radiation with low toxicity in the absence of light. Due to the poor solubility of BODIPY in water and the shift of absorption and emission ranges to the NIR region, promising PDT- and PTT-agents are used in the form of J-aggregates, which form water-soluble nanoparticles in combination with polymers. The bathochromic shift resulting from J-aggregation enables NIR-II emission of BODIPY-based medications, opening up the possibility of their use as theranostic medications. In addition, recent advances in the development of PTT-active BODIPYs exhibiting activity in the nanomolar range of IC50 values demonstrate the still incompletely explored potential of BODIPYs as antitumor agents and prove the feasibility of searching for new structural modifications to enhance their effectiveness.
6. List of abbreviations
Ered — reduction potential,
Eox — oxidation potential,
glum — luminescence asymmetry factor,
|glum| — luminescence dissymmetry factor,
λabs — absorption wavelength,
λem — emission wavelength,
λex — excitation wavelength,
λlaser — lasing wavelength,
Ffl — quantum yield of fluorescence,
FD — quantum yield of reactive oxygen species formation,
Flaser — quantum yield of laser irradiation,
A — acceptor,
AcN — acetonitrile,
AIEE — aggregation-induced emission,
ALDH1 — aldehyde dehydrogenase 1,
Ant — anthracenyl,
Asc — ascorbate,
AT — atom transfer,
BCN — spiro[2.3]hex-1-ene,
BHT — 2,6-di-tert-butyl-4-methylphenol,
BINAP — bis(diphenylphosphino)-1,1′-binaftyl,
BINOL — 1,1′-bi-2-naphthol (1,1′-bi-2-naphthol),
B2pin2 — bis(pinacolato)diboron,
Boc — tert-butoxycarbonyl,
BODIPY (BDP) — 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene,
Brettphos — 2-dicyclohexylphosphino-3,6-dimethoxy-2′,4′,6′-triisopropyl-1,1′-biphenyl,
CAN — cerium(IV)-ammonium nitrate,
Cat — catalyst,
Cb — alicyclic carbocycle,
CD — circular dichroism,
CDT — chemodynamic therapy,
cod — cycloocta-1,5-diene,
Cp — cyclopentadienyl,
CPL — circularly polarized luminescence,
CS — charge separated state,
CVA — cyclic voltammetry,
Cy — cyclohexyl
Cys — cysteine,
D — donor,
dba — dibenzylideneacetone,
DBU — 1,8-diazabicyclo[5.4.0]undec-7-ene,
DCE — 1,2-dichloroethane,
DCM — dichloromethane,
DDQ — 2,3-dichloro-5,6-dicyano-1,4-benzoquinone,
DFT — density functional theory,
DIPEA — diisopropyl ethylamine,
DMAP — 4-(dimethylamino)pyridine,
DME — dimethoxyethane,
DMPO — 5,5-dimethyl-1-pyrroline-N-oxide,
dr — diastereomer ratio,
DSPE-PEG — 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)],
EC50 — half-maximal effective concentration,
ECM — exciton chirality method,
EDC — 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide,
EnT — energy transfer,
ER — endoplasmic reticulum,
ESn — trans-1,2-bis(tributylstannyl)ethene,
ETA — ethanolamine,
EWG — electron-withdrawing group,
Fc — ferrocenyl,
FRET — fluorescence resonance energy transfer,
F127 — pluronic F127,
GSH — glutathione,
HAT — hydrogen atom transfer,
Hcy — homocysteine,
HEPES — 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (buffer),
HFIP — hexafluoropropan-2-ol,
HOMO — highest occupied molecular orbital,
m-CPBA — m-chloroperoxybenzoic acid,
IC — internal conversion,
IC50 — half-maximal inhibition concentration,
ICT — intramolecular charge transfer,
ISC — intersystem crossing,
LDA — lithium diisopropylamide,
LED — light emitting diode,
LUMO — lowest unoccupied molecular orbital,
MES — 2-mercaptoethanesulfonate sodium,
Mes — 2,4,6-trimethylphenyl (mesityl),
MS — molecular sieves,
MW — microwave radiation,
NBS — N-bromosuccinimide,
NCS — N-chlorosuccinimide,
NHS — N-hydroxysuccinimide,
NIR — near infrared (spectrum),
NIS — N-iodosuccinimide,
NP — nanoparticles,
PA — photoacoustic,
PALM — photo-activated localization microscopy,
PBN — N-tret-butylphenylphenylnitron,
PBS — phosphate-salt buffer,
PCE — photothermal conversion efficiency,
PEDOT:PSS — poly(3,4-ethylenedioxythiophene)polystyrene sulfonate,
PDT — photodynamic therapy,
PEG — polyethylene glycol,
PEG-PCL — aminopoly(ethylene glycol)-b-poly(ε-caprolactone),
PET-photoinduced electron transfer,
phen — phenanthroline,
PIFA — bis(trifluoroacetoxy)iodbenzene,
PivO — pivaloyl,
por — porphyrin,
PS — proton sponge, 1,8-bis(dimethylamino)naphthalene),
PTT — photothermal therapy,
Py — pyridine,
py — pyridyl,
RET — resonance energy transfer,
ROS — reactive oxygen species,
rt — room temperature,
Selectfluor — bis(tetrafluoroborate)4-fluoro-1-chloromethyl diazonium-bicyclo[2.2.2.2]octane,
SET — single electron transfer,
SNAr — nucleophilic aromatic substitution,
SN1 — monomolecular nucleophilic substitution,
SOC — spin-orbit coupling,
SOCT-ISC — spin-orbit charge-transfer intersystem crossing,
TBA — tetra-n-butylammonium,
TBHP — tret-butyl-hydroperoxide,
TBPB — tret-butyl-peroxybenzoate,
TCBQ (p-chloranil) — tetrachloro-1,4-benzoquinone,
TC — thiophene-2-carboxylate,
TCE — 1,1,2-trichloroethane,
TCO — trans-cyclooctene,
TEBAC — benzyltriethylammonium chloride,
TEG — tetraethylene glycol,
TEMP — 2,2,6,6-tetramethylpiperidine,
TEMPO — 2,2,6,6,6-tetramethylpiperidine-N-oxyl,
terpy — terpyridine,
TES — triethylsilyl,
TFA — trifluoroacetic acid,
Tf — trifluoromethanesulfonyl (triflyl),
TFEA — 2,2,2,2-trifluethoethanol,
TFP — tri(2-furyl)phosphine,
TMS — trimethylsilyl,
Tol — tolyl,
TPP — meso-tetraphenylporphyrin,
Ts — p-toluenesulfonyl (tosyl),
TTEG — tetraethylene glycol,
US — ultrasound,
VAPOL — 2,2′-diphenyl-4-biphenanthrol,
Xantphos — 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene,
X-Phos — 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl.
References
















