



Keywords
Abstract
Discovered in 2008 by T.Ogoshi and co-workers, pillararenes have already attracted much attention due to their highly organized rigid structure and electron-rich cavity capable of accommodating various cationic and neutral guests. Pillararenes are successfully used in supramolecular tectons, liquid crystals, porous materials, organic–inorganic hybrid systems, drug delivery systems and, most importantly, in various metal-supported or metal-free catalytic systems. The current review highlights the most representative examples of the use of pillararenes for the catalytic applications. In particular, the metal-free pillararene-based catalytic systems for the Knoevenagel condensation reactions as well as supramolecular artificial enzymes and phase transfer catalysts will be analyzed. Among the metal-based catalysts the pillararene-supported catalysts for C–C-coupling reactions, including asymmetric or cycloaddition reactions, as well as pillararene-supported nanoparticles or polyoxometalates will be analyzed. Special attention will be paid to pillararene-mediated photocatalytic reactions.The bibliography includes 151 references.
1. Introduction
The term ‘pillararene’ (PA) was first introduced in 2008 by Ogoshi et al.,[1] who described this new representative of cyclophanes, namely, pillar[5]arene (P5A). Pillar[6]arenes (P6As) were first obtained in 2009.[2] Since then, various synthetic strategies towards P5-6As [3-6] and larger (up to 15 units) PAs[7] have been reported. At present, PAs are widely used in many research areas, such as porous materials,[8-10] materials for gas storage/separation,[11-15] organic–inorganic functional materials,[16-18] liquid crystals,[19][20] molecular devices and machines,[21] fluorescent and AIE-materials (AIE-aggregation induced emission),[22][23] materials for the DNA fragment extraction,[24] nanotheranostics[25-28] and drug delivery systems.[29-34]
Similar to calixarenes and other supramolecular systems,[35][36] PAs are considered as convenient platforms for catalysts or catalyst carriers.[32][37-42] The main advantage of PAs (most commonly P5As and P6As) is their larger symmetric and highly rigid cavity of up to 6.8 Å in diameter (for P6A[43]) bearing electron-rich oxygen-containing moieties from the both sides. Due to π-cation or π-dipole interactions, this electron-rich cavity can readily accommodate small neutral or positively charged molecules/species of various sizes and geometries, such as metal cations, ammonium cations, alkyl nitriles, etc. In some cases, pillararenes can coordinate to metal cations, as catalytic species, via the outer π-surface of aromatic rings so that metal ions remain outside the cavities of the pillararenes.[44][45] In addition, the oxygen moieties can be selectively modified in order to either provide an extended cavity to accommodate the guest/catalytic species or to create additional or the only receptor arms/chelating units capable of interacting with guest molecules and/or catalytic species through non-covalent interactions, such as ionic bonds, hydrogen bonds, Van-der-Waals forces, etc. The proposed coordination modes for PA (P5A is shown) for the metal-based catalytic species are shown in Fig. 1. In addition to metal-based catalysts, PAs can be used for the metal-free catalysis via the involvement of PA cavity/rim and/or side-arms. In addition, the PA cavity may provide a suitable environment for the proper orientation of the reacting species or for better stabilization of the transition state compared to an external environment.
![[{"id":"BiXIpE0fo8","type":"paragraph","data":{"text":"Proposed <b>P5A</b> coordination modes for the metal-based catalytic species"}}]](/storage/images/resized/bQVj1c6agIAETjcMp1jcmaOzckPQZGLyHfF4KTqi_xl.webp)
To date, several reviews on catalytic applications of PAs have been published.[32][37-39][42] Among these reviews, only in some cases have the authors attempted to analyze the role of PAs in the catalytic process [42] or to classify the PA-based catalysts according to reaction types.[39] This review highlights the most recent information on the application of PA-based catalytic systems, and in the framework of this work we have arranged all the reported PA-based catalytic systems according to a catalyst nature/catalysis type and, within these rows, according to a reaction type.
2. Most common synthetic approaches to pillararenes
Methods for the synthesis of pillar[n]arenes have been well analyzed in several review articles as well as monographs and book chapters.[36] [46-50] In a typical case, pillar[5]arenes (Scheme 1) can be obtained by cyclooligomerization reaction between 1,4-dialkoxybenzenes and paraformaldehyde with acid catalyst at room temperature. This process is catalyzed by various Lewis acids, such as boron trifluoride,[1] [51][52] ferric chloride,[53-55] as well as trifluoroacetic acid,[56] trifluoromethane- (Ref. [57]) or p-toluenesulfonic acid [58][59] and even sulfuric acid.[5] It is worth mentioning that in a cyclooligomerization reaction, a single aromatic monomer could be used to afford homopillar[5]arenes or two different aromatic monomers could be involved to produce co-pillararenes.[60-64]
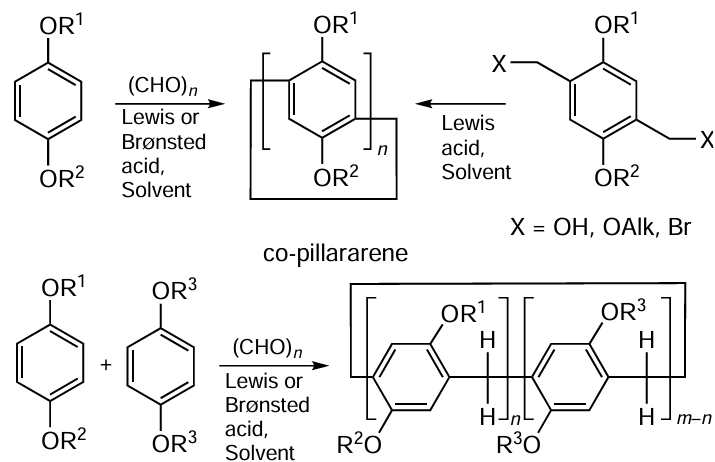
Nierengarten and co-workers[65] reported another two approaches to pillar[5]arenes, namely, the p-toluenesulfonic acid-catalyzed condensation of 1,4-dialkoxy-2,5-bis(alkoxymethyl)benzene or cyclooligomerization of 2,5-dialkoxybenzyl alcohols or 2,5-dialkoxybenzyl bromides promoted by an appropriate Lewis acid (see Scheme 1).
As for selective synthesis of pillar[6]arenes, these macrocycles can be obtained as the major product of the Friedel – Crafts cyclooligomerization by using monomers bearing bulky alkoxy groups, by switching the catalyst for a Lewis acid[53] or using a bulky chlorinated solvent. For example, Ogoshi and coworkers[66] reported the synthesis of a pillar[6]arene containing 1,4-bis(methylcyclohexyl ether)phenylene units in 87% yield using chlorocyclohexane as the solvent. The authors suggest that the bulky chlorinated solvent acts as a template for the formation of the larger pillar[n]arene.
Finally, we have reported a convenient and chemoselective approach to pillar[6]arenes via the solvent-free reaction between 1,4-dialkoxybenzenes and paraformaldehyde by grinding the reactants in the presence of a catalytic amount of H2SO4.5
It is worth mentioning that higher pillar[n]arene homologues, pillar[6 – 15]arenes, have been synthesized through the ring expansion of pillar[5]arene.[67]
3. Metal-free pillararene-based catalytic systems for Knoevenagel condensation reactions
Metal-free size-selective catalysis is of key importance in the green synthesis and in the conversion of crude oil or biomass. Similar to calixarenes, PAs can be readily involved in the size-selective catalytic systems. Examples of metal-free PA-based catalytic systems are presented below.
The formation of the C – C bond plays a fundamental role in organic chemistry, as it aims to build molecular scaffolds of different geometries. New catalytic systems for this type of reaction are therefore in great demand.
Ma and co-workers [45] synthesized P5A1-integrated 3D framework porous polymers P5A1PP1-3 (Scheme 2, Fig. 2). The reaction of tetrakis(4-aminophenyl)methane (TAPM), tris(4-aminophenyl)amine (TAPA), or tetrakis(4-aminophenyl)-1,4-benzenediamine (TABD) with benzaldehyde-difunctionalized pillar[5]arene P5A1 in dimethyl sulfoxide at 80°C for 24 h afforded three highly thermal and chemically stable 3D network structures P5A1PP1-3. According to the authors, the presence of porous pillar[5]arene core provides high size-selectivity, making P5A1PP1-3 suitable for catalytic applications.

![[{"id":"NxgUXhnD3f","type":"paragraph","data":{"text":"3D network structure of <b>P5A1PP1</b> and illustration of its size selective catalysis"}}]](/storage/images/resized/ngHsRQ9zRT54cG0fxUJgEfMsEri69lPqYdixQdDU_xl.webp)
The permanent porosity of P5A1PP1-3 was evaluated by nitrogen adsorption–desorption isotherms at 77 K, and all the isotherms show a type II characteristic according to the IUPAC classification. This indicates the presence of micropores and mesopores in the polymers. In addition, the Brunauer – Emmett –Teller (BET) surface areas were found to be 114 m2 g–1 (P5A1PP1), 103 m2 g–1 (P5A1PP2) and 86 m2 g–1 (P5A1PP3), respectively. Based on these results, polymers P5A1PP1 and P5A1PP2 derived from TAPM and TAPA respectively, exhibited the highest surface areas. And the largest pore size, based on the NLDFT analysis, was found for the TABD-based polymer P5A1PP3, while the smallest was observed for the polymer P5A1PP1.
Based on the ability of pillararenes to encapsulate various electron-deficient guests, the next step was to perform Rhodamine B (RhB) adsorption experiments. In these experiments, P5A1PP1 showed an adsorption capacity of up to 36.9 mg g–1, a similar value of 37.1 mg g–1 was found for P5A1PP3, while a much lower value of 29.0 mg g–1 was observed for P5A1PP2.
In the final step, the catalytic activity of the resulting polymers was explored using the Knoevenagel condensation reaction as an example, with the polymer P5A1PP1 showing the highest catalytic activity together with a good stability. The involvement of the pillararene core of P5A1PP1 in the catalytic process was confirmed. The reaction between 4-ethylbenzaldehyde and malononitrile was almost instantaneous because, according to the authors, both substrates can move freely through the P5A cavity, while as the size of the substrates increased, the reaction rate decreased in the following order: 4-isopropylbenzaldehyde > 4-tert-butylbenzaldehyde > 3,5-di-tert-butylbenzaldehyde. This confirms the important role of the P5A cavity size in PA[5]-integrated porous polymers, which exhibited apparent size-selective catalytic activity.
Du et al.[68] developed the synthesis of the P5A-based [1]rotaxane P5A2 via aminolysis of the P5A-based monoester with octamethylene diamine and direct capping of the amino-terminated pseudo[1]rotaxane by the SN2 reaction with 1-(bromomethyl)-3,5-dimethoxybenzene (Scheme 3). The structure of the [1]rotaxane, namely, the orientation of the amine chain inside the P5A cavity, was confirmed by 1H and 13C NMR, 2D HSQC and COSY NMR as well as by theoretical calculations at the B3LYP/6-31G(D) level using the PCM model matched well with 2D NOESY. The obtained P5A2 catalyzed the Knoevenagel condensation reaction between acetone and malononitrile and, according to the authors, the reaction proceeded smoothly and followed second order kinetics in the presence of P5A2. Despite the fact that tertiary amines can catalyze the Knoevenagel condensation reaction, the reported case can be considered as proof of concept for the use of P5A2 as catalyst for the size/geometry-selective reactions, including those for biomolecules.

Gong and co-workers [69] described the Tröger’s base (TB)-derived porous organic polymer P5A3, which was constructed using an octamethyloxy-pillar[5]arene-based platform (Fig. 3). The porosities of P5A3 and the model polymer P1 were evaluated by a nitrogen adsorption isotherm measured at 77 K. The authors argue that the adsorption isotherm of both P5A3 and the model polymer P1 exhibited a pattern closely related to a Type IV isotherm (based on the IUPAC classification). Based on the BET isotherms, the surface area of P1 was found to be 66 m2 g–1, while the surface area of P5A3 was found to be 26 m2 g–1, which, according to the authors, is due to the extra space occupied by the pillar[5]arene. The mesopore nature of P5A3 and P1 was confirmed based on the pore size distribution plot, which showed a mean pore width of 2.15 nm (P5A3) and 16.5 nm (P1). And the pore volume was determined to be of 0.040 cm3 g–1 (P5A3) and 0.085 cm3 g–1 (P1), which, according to the authors, is consistent with pillar[5]arene occupying a specific space within the material. The presence of both the TB moiety and the P5A platform allows above polymer to be used as a catalyst for the Knoevenagel condensation reaction between aromatic aldehydes and malonic acid. The authors observed a certain substituent effect in the aldehydes. For example, aldehydes with electron-withdrawing groups provided higher yields than their counterparts with electron-donating groups. No reaction was observed with 9-anthraldehyde, which may be due to the steric effect and may indicate the size-selective catalytic behaviour of P5A3.
![[{"id":"TZuBcXpqpC","type":"paragraph","data":{"text":" Pillar[5]arene-based polymer <b>P5A3</b> as a metal-free heterogenous catalyst for the Knovenagel condensation and CO<sub>2</sub> conversion"}}]](/storage/images/resized/wtCn1cMtej7ponMoa2xAiG8XBxEFmZpzbc8chsrd_xl.webp)
The authors hypothesized that, due to the presence of multiple receptor sites, the P5A3 cavity effectively retains the malonic acid molecule after it is moved into the cavity, reducing molecular movement and creating the so-called confinement effect, leading to substrate activation.
Pillar[n]arenes[13] [70-72] and Tröger’s base-derived materials[73] have been widely reported as materials for CO2 capture/transformation. Therefore, P5A3 was further explored as a ‘host’/catalyst for the CO2 utilization via its reaction with oxiranes to give 1,3-dioxolan-2-ones.
4. Pillararene-based supramolecular artificial enzymes
In living organisms, enzymes are responsible for a diversity of (bio)chemical transformations. These nanometre-sized three-dimensional structures are formed by the folding and self-assembly of polymeric chain-like components through supramolecular interactions. The most common biological reaction catalyzed by a large number of cationic type enzymes is phosphoryl group transfer. In this case, the negatively charged phosphate anions are targeted by positively charged moieties of such enzymes.
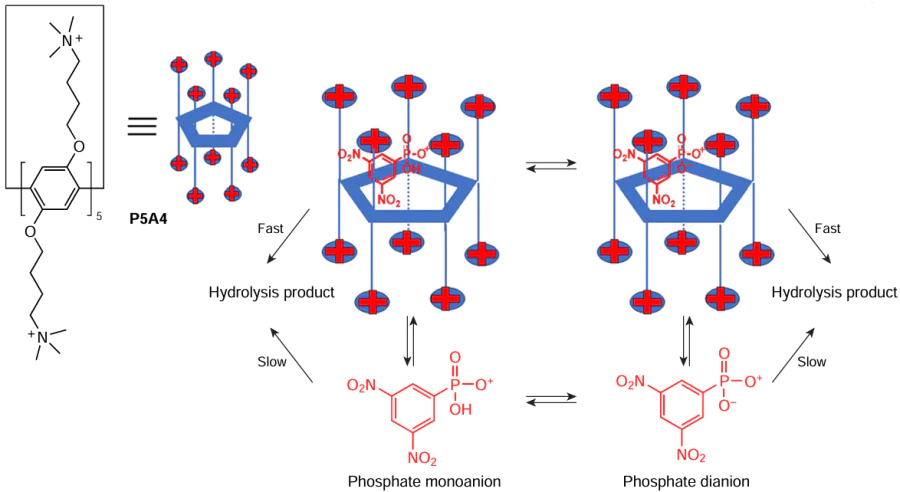
With this concept in mind, Nome and co-workers[74] reported the pillar[5]arene P5A4 decasubstituted with alkylammonium moieties as a biomimetic model of phosphatase (an enzyme that uses water to cleave a phosphoric acid monoester into a phosphate ion) for the model reaction of hydrolysis of the phosphate monoester, 2,4-dinitrophenylphosphate (DNPP) (Scheme 4). According to the authors, P5A4 is capable of encapsulating DNPP in both the monoanion and, with stronger binding, the dianion forms. Based on 1H NMR experiments and molecular dynamics calculations, the catalytic effect should be attributed to the formation of a 1 : 1 ‘host – guest’ complex between P5A4 and DNPP. The hydrolysis of DNPP molecule is possible both in bulk water and inside the P5A4 cavity. As a result, in the presence of P5A4, the DNPP hydrolysis constant increased by a factor of 4 and 10 for the monoanion and dianion, respectively. The main factors favouring the catalytic hydrolysis of the phosphate monoester inside the pillar[5]arene cavity are: (a) electrostatic stabilization (fundamental for the incorporation of the dianion into the cavity of P5A4) at the expense of reducing the negative electron density on the PO3– oxygens; (b) steric effects of the wall of close NMe3+ groups surrounding the PA cavity, which interfere with the stabilizing H-bonding to water and consequently destabilize the monoester dianion accelerating the reaction. Analysis of the kinetic data gives the binding constant for the association of the dianion with P5A4, K = (5150 ± 660) M–1, and the rate constant for the hydrolysis of DNPP inside the cavity of P5A4, k = (7.80 ± 0.22) × 10–5 s–1. And, according to the authors, this value is higher than that in water, k = 8.27 × 10–6 s–1. This confirms the apparent catalytic effect of about one order of magnitude provided by the involvement of the P5A4 cavity.
In the follow-up study,[75] pillar[5]arenes bearing ten imidazole moieties (to obtain simultaneously a cationic and a nucleophilic receptor), namely, P5A5, were reported as another example of highly reactive and selective supramolecular artificial enzymes (Scheme 5) for the hydrolysis of the symmetrical phosphate diester bearing two 2,4-dinitrophenyl moieties (BDNPP) or asymmetrical ones bearing the 2,4-dinitrophenyl moiety and either ethyl (EtDNPP) or phenyl moiety (PDNPP).
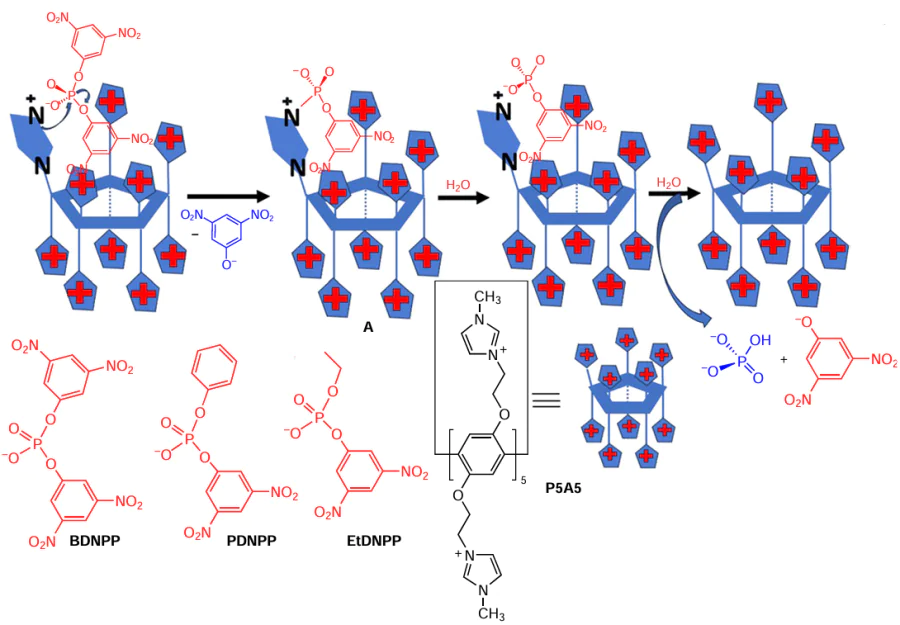
Due to both the presence of a pillararene cavity, capable of encapsulating 2,4-dinitrophenyl moieties (BDNPP, EtDNPP and PDNPP), and anchored imidazoles in a pillar[5]arene matrix, P5A5 strongly accelerated the phosphate diester hydrolysis by a factor of 104. According to the authors, in the case of the P5A5-assisted hydrolysis of BDNPP, two consecutive processes took place with the release of both 2,4-dinitrophenolate leaving groups (see Scheme 5). Thus, in the first step, a nucleophilic attack of the imidazole moiety of the P5A5 on BDNPP took place with the release of 2,4-dinitrophenolate anion. The in situ formed phosphoroamidate intermediate A decomposes relatively rapidly, allowing catalyst regeneration, as confirmed by 31P NMR. In the final step, hydrolysis of the 2,4-dinitrophenyl phosphate monoester occurred with the release of free P5A5. For the reaction of 5 mM P5A5 with BDNPP, the first-order rate constant at the plateau is 1.9 × 10−3 s−1, which is a value that is 270-fold greater than the rate constant calculated for the reaction of the substrate with 5 mM of free neutral imidazole (7.1 × 10−6 s−1), using the reported second-order rate constant of 1.42 × 10−3 M−1 s−1. It should be noted that, although EtDNPP, PDNPP and BDNPP contain the same leaving group, 3,5-dinitrophenolate, changes in the substrate structure promote large differences in reactivity, with an increase in rate constants of 1 : 7 : 520 for EtDNPP:PDNPP:BDNPP, respectively. Thus, P5A5 reacted 520-fold faster with BDNPP than with EtDNPP, whereas only a 5-fold increase effect was observed in bulk water, confirming an apparent supramolecular effect. In summary, P5A5 acts as a supramolecular artificial enzyme.
5. Pillararenes for phase-transfer catalysis
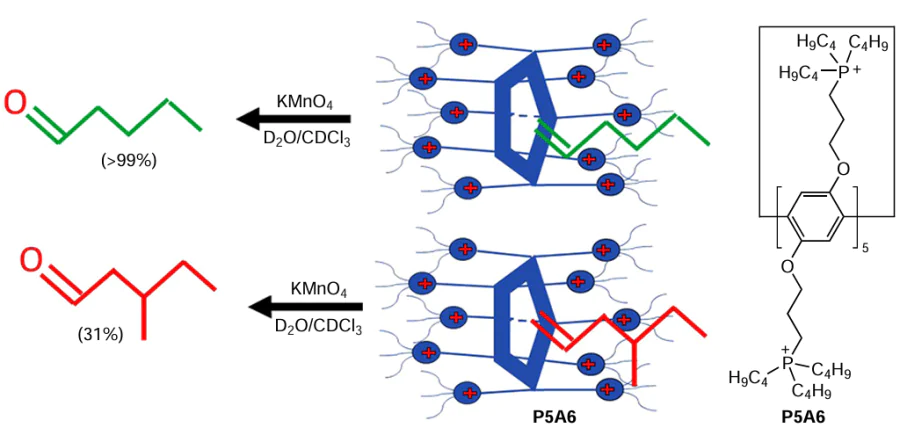
Phase transfer catalysis is a powerful tool in the synthetic organic chemistry.[76] Similar to other macrocyclic platforms,[77][78] PA-based materials can also be used as phase transfer materials. For example, Ogoshi et al.[79] developed the P5A6 phase transfer catalytic system based on amphiphilic pillar[5]arene bearing 10 tetraalkyl phosphonium bromide moieties (Scheme 6). P5A6 efficiently accommodated various guest molecules, such as 1-hexene, in its cavity, which was confirmed by the results of the 1H NMR studies.[79] In the presence of KMnO4 as an oxidant, P5A6 demonstrated very high selectivity for the oxidation of linear 1-hexene over the more sterically constrained 4-methylhex-1-ene to afford the corresponding aldehyde in up to 99% yield vs. 31%, respectively. According to the authors and based on the literature data, the driving force for complex formation is the CH-π interactions. Using the Job plot analysis, the authors estimated the stoichiometry of the complex as 1 : 1, and the association constant K for the binding of 1-hexene in the cavity of P5A6 was calculated as (65 ± 16) M–1 using non-linear curve fitting analysis. The involvement of the pillar[5]arene cavity in the catalysis process was confirmed by the decrease in the degree of conversion of 1-hexene to the aldehyde in the presence of competitive guest molecules, such as 1,4-dicyanobutane (the association constant of with P5A6 in the case of the 1 : 1 complex was as high as K = (1.62 ± 0.026) × 103 M–1 and a decrease in the degree of conversion in the presence of CH2Cl2 (the structure of P5A6 · CH2Cl2 complex was confirmed by single crystal X-ray diffraction analysis).
In 2018, the syntheses of pillararenes P5A7 – 10 substituted with methyl, sodium methyl carboxylate or PEG-tethered 1,2,3-triazole groups on both sides of the macrocyclic core were reported.[80]

The presence of amphiphilic groups gives P5A8 the ability to act as a phase transfer catalyst. Surface tension measurements at the air/water interface revealed a slight decrease in the surface tension with the addition of P5As, while no clear micelle formation was observed. In the presence of carboxylate-substituted P5A8 (P5A – (Me)10 – x – (CH2COONa)x, where x is equal to 3.7 and 8.6 respectively), the authors observed a decrease in the surface tension from 63 to 37 mN m−1 using catalytic concentrations of P5As, while at higher carboxylate substitution (x = 10), a decrease in the surface tension from 72 mN m−1 for pure water to 59 mN m−1 at 0.032 mol L−1 of P5A was observed. To elucidate the limits of applicability of the P5A8-based catalyst, its activity in rhodium-catalyzed hydroformylation of 1-decene and 1-hexadecene was studied. Rh catalyst was formed in situ by the reaction of Rh(CO)2(acac) with triphenylphosphine-3,3',3''-trisulfonic acid trisodium salt (TPPTS) as a ligand. Increasing the number of sodium carboxylates (x) up to ~ 8 increased the conversion of 1-decene up to 85% (72% for 1-hexadecene), whereas introducing more carboxylate residues reduced the conversion due to a possible decrease in the catalytic ability of the P5A substrate. In the model experiments for the hydroformylation of 1-decene in the presence of other phase transfer agents, e.g., modified cyclodextrin (RAME-β-CD), P5A8 showed similar catalytic activity with higher regioselectivity (up to 4.1 vs. 1.8 (for RAME-β-CD)). However, the hydroformylation chemoselectivity was lower (78% vs. 98%). These facts suggest an interaction between the substrate and the low phosphine-coordinated Rh-catalyst within the RAME-β-CD cavity. In the case of P5A8, the phosphine ligand does not interact with the cavity of this macrocycle, the equilibria are not altered and mainly linear aldehydes are formed. However, in the case of 1-decene, the ‘protective’ effect of the P5A cavity against side reactions was lower compared to cyclodextrin, most probably, due to less efficient incorporation of 1-decene. It should be noted that for the PEG-appended pillararenes P5A9 and P5A10 a higher or comparable chemoselectivity of the hydroformylation of 1-decene was observed (77% (P5A9) and 92% (P5A10)), while the regioselectivity was lower than that for P5A8 (2.4 (P5A9) and 2.6 (P5A10)). In addition, according to the authors, P5A8 was reusable over five consecutive catalytic runs.
In summary, P5A-based systems, such as P5A8 – P5A10, can be used as supports in the Rh-catalyzed hydroformylation of 1-decene and 1-hexadecene, and the regioselectivity of these reactions is higher than that in the reactions carried out under homogeneous conditions, suggesting the involvement of the P5A-core in the process. However, the stereoselectivity of such conversions is still modest compared to those mediated by cyclodextrin (92% vs. 98%), which is possibly due to the lower ability of P5A8 – P5A10 to prevent the isomerization of the substrates, caused by the only partial encapsulation of these alkenes by the P5A cavity.
6. Pillararene-supported metal catalysts
The main challenge for the metal-based catalysts is to provide their high regioselectivity, chemoselectivity, as well as reusability and high TON (turnover number) values. The introduction of macrocyclic carriers, such as pillararene, allows a more precise control of the catalytic nature for particular application. Some of the most representative examples of PA-supported metal-based catalysts are highlighted below.
6.1. Pillararene-supported metal-based catalysts for C – C-coupling reactions
New metal-based catalysts for such reactions, especially with high TON values [81] are in high demand. Some examples of the use of pillararenes in the construction of such catalysts are discussed below.
6.1.1. Pillararene-based N-heterocyclic carbene Pd(II) complexes formed in situ
Imidazolium salts are typical precursors for N-heterocyclic carbenes (NHC), which are widely used as ligands for transition metal complexes for the catalytic applications. Despite many examples of calixarene-supported NHC-Pd complexes for the catalytic applications including cross-coupling reactions,[82-87] only very few of them have been reported for other macrocycles,[88] including pillarerenes.
For example, Wang and coworkers [89] successfully developed pillararene P5A11 (*10 PF6–) as an NHC ligand for Pd-catalyzed Suzuki-Miyaura cross-coupling reactions. According to the authors, the well-organized hydrophobic P5A cavity can easily host aryl substrates. These data agree well with the above-mentioned results on an artificial P5A11-based enzyme (see Scheme 5),[75] which readily accommodated aromatic substrates. Using P5A11 (*10 PF6–) as a carbene ligand for Suzuki – Miyaura cross-coupling reactions in model experiments, the 90 – 99% yields were obtained at reaction temperatures ranging from room temperature to 65°C. Much lower yields were observed for non-pillararene-based carbene ligands, confirming the involvement of P5A11 (*10 PF6–) in the catalytic cycle through both the coordination of the Pd catalyst by carbene moieties and encapsulation of bromoarenes by the pillar[5]arene cavity.
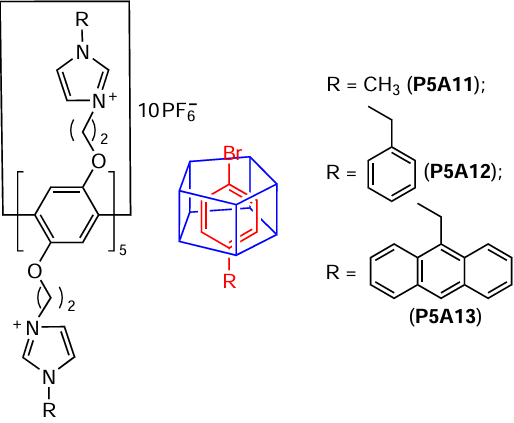
The similar catalytic system P5A12-13, bearing phenyl and bulky 9-anthranyl substituents in the imidazole core, has been successfully developed by the same research group[90] for the Heck reactions between styrene and aryl halides, and in the case of iodoarene up to 99% yield was achieved. The authors found that the resulting Pd complexes showed different reactivities depending on the type of the aryl moiety in the NHC ligand. For example, 1.3 mol.% P5A12 provided higher yield than 2.0 mol.% P5A13, most likely, due to the greater steric hindrance in the 1-(9-anthracenylmethyl)imidazole NHC ligand P5A13. Again, the coordination of the Pd catalyst by carbene moieties and encapsulation of haloarenes by the pillararene can be suggested.
6.1.2. Pillararene-rotaxane-based Pd(II) complexes
Rotaxanes are highly promising and attractive carriers for the supramolecular catalysts.[91] And PA-based rotaxanes have already found wide applications in materials chemistry.[92-95]
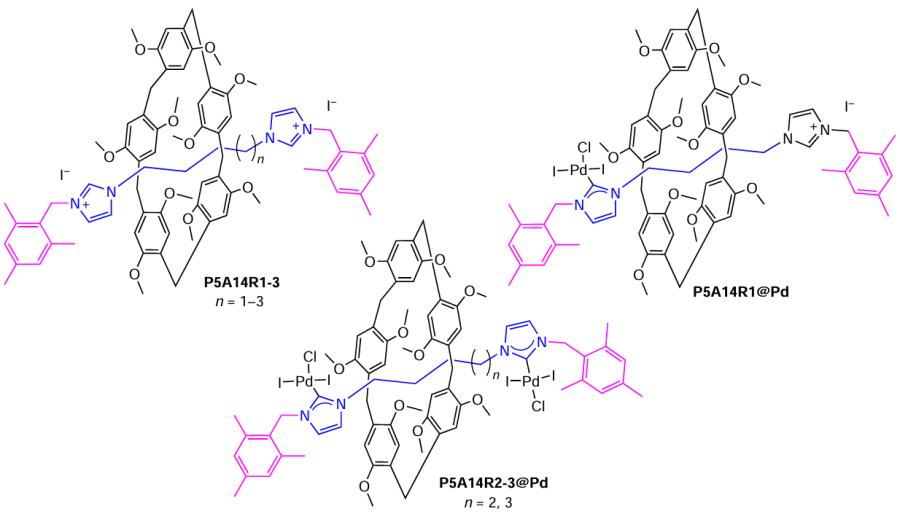
In 2019, a series of [2]rotaxanes P5A14R1-3 composed of a decamethylpillar[5]arene P5A13 and guest molecules based on bis-imidazolium cations (rods) were reported as precursors of N-heterocyclic carbenes.[96] It was found that NHC moieties at the end of the rod in P5A14R1-3 effectively coordinated Pd(II) cations when reacted with Pd(CH3CN)2Cl2 in pyridine to give complexes P5A14R1@Pd or P5A14R2-3@(Pd)2 depending on the length of the linkers. The authors suggest that, the introduction of metal complexes to the ‘rod’ restricts the shuttling motion of the pillar[5]arene along the entire length of the [2]rotaxane, providing a simple method of influencing shuttling motion. Unfortunately, the catalytic activity of the resulting Pd(II) complexes was not investigated.
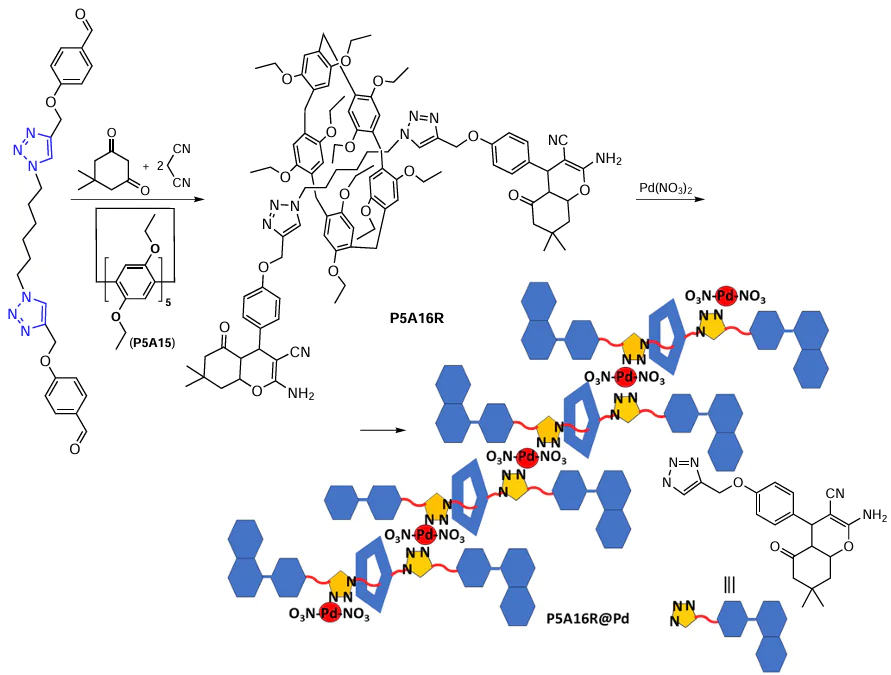
In 2020, the P5A-based [2]rotaxane P5A16R based on the decaethylsubstituted pillar[5]arene P5A15 was reported as a platform for the preparation of a Pd-coordinated polymeric catalyst for the Suzuki – Miyaura cross-coupling reaction (Scheme 7).[97] The formation of the [2]rotaxane was confirmed by a number of physical methods, including 2D NOESY NMR spectroscopy. The main idea of the study was to use two 1,2,3-triazole units on the central axle of P5A16R as N,N-anchoring sites for the coordination with Pd(II) to obtain the coordination polymer, namely, poly(P5A16R@Pd), which was formed by hinges of small particles with a diameter of 50 nm (according to the results of transmission electron microscopy (TEM)). Energy dispersive X-ray spectroscopy (EDX), X-ray powder diffraction (XRD) analysis and scanning electron microscopy (SEM) mapping confirmed the incorporation of Pd into the polymeric materials. The resulting polymer showed good catalytic activity and stability even in the reaction with chloroarenes, and the target 4-phenylpyridine was obtained in up to 95% yield. Furthermore, in the model experiment, poly(P5A16R@Pd) exhibited good reusability and can be used up to 3 times without losing the degree of conversion. For the comparison, Pd(NO3)2 and the G@Pd complex were used as catalysts and both showed very poor catalytic activity and recyclability compared to poly(P5A16R@Pd). According to the authors, the higher catalytic activity and recyclability of poly(P5A16R@Pd) is due to the conversion of Pd(II) ions into Pd(0) nanoparticles (NPs) and their incorporation into the polymeric materials after the first catalytic cycle. Therefore, P5A16R@Pd can be considered as a promising catalyst for cross-coupling reactions. However, the P5A moiety has no influence on the reaction selectivity, and even in the case of sterically hindered aromatic substrates, such as 3,5-di-C(CH3)3C6H3Br, the reaction yields were still high (up to 92%).
6.1.3. Pillar[5]quinone-based Pd(II) complex
Among the so-called unorthodox ligands for complex formation with Pd, quinone-based ones are very underrepresented.[98]
In 2019, a pillar[5]quinone (P5Q)-based porous polymer was reported,[99] which was prepared by crosslinking P5A by p-xylene dichloride via the Friedel – Crafts reaction followed by oxidation (Scheme 8). The oxidation afforded poly-P5Q. The introduction of the 3D P5A moiety increased the polymer porosity, and based on the nitrogen adsorption-desorption isotherms, the surface area of P5A-based polymer was 400 m2 g–1. Surprisingly, the surface area of the P5Q-based polymer decreased to 272 m2 g–1, which is possibly due to a decrease in the size of the groups attached to the pillararene core (namely, the =O group vs. the OCH3 group). In the final step, poly-P5Q was further used to prepare the Pd catalyst with high loading ratio (up to 12.0 wt.%). According to the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and SEM data, the Pd catalyst was well dispersed on the solid support in the form of Pd(II)-species. In addition, after the Pd loading, the surface area decreased further to 202 m2 g–1 due to the introduction of the Pd catalyst and partial occupation of the porous structure. The efficacy of the Pd-loaded P5Q-based polymer was investigated in model Suzuki-coupling reactions between bromoarenes and phenylboronic acid, and compared with reactions catalyzed by Pd/C. In these reactions, in the presence of Pd-loaded poly-P5Q, the target bisarenes were obtained in up to 99% yields with TOF values ranging from 9700 to 29 700 compared to 10 to 96% yields for the Pd/C catalyst with TOF values ranging from 20 to 980. It is worth noting that, Pd-loaded poly-P5Q showed good thermostability and recyclability (up to six cycles) with just a little decrease in reactions yields. However, the introduced P5A moiety has no or little impact on the Pd-loaded poly-P5Q catalyst performance and selectivity, and in the case of 2-bromo- or 3-bromo-1,1'-biphenyl-based substrates no decrease in the reaction yields was observed. A little influence of the electronic nature of the substrate on the reaction yield was observed with 2-bromopyridine, with the yield of the target 2-phenylpyridine equal to 69%, although this value does not exceed the yield obtained using the Pd/C catalyst (31%).

6.2. Pillararene-supported asymmetric catalysts
Asymmetric catalysts are important both for mimicking natural enzymatic processes and for target drug design. Pillararenes, as planar chiral macrocyclic arenes, are of interest for the creation of catalysts for the asymmetric synthesis.[100] To date, only two examples of the use of pillararenes for asymmetric addition/C – C coupling reactions have been reported.
6.2.1. Ti(IV)-based pillararene-supported chiral catalyst
Huang and co-workers [101] obtained pillar[5]arene-based polymer poly(P5A17) starting from per-(2-bromoethyl)-pillar[5]arene P5A17 and the (R,R)-tetraaryl-1,3-dioxolane-4,5-dimethanol derivative (TADDOL-OH). The resulting chiral 3D polymer network was further converted to the O-titanate derivative poly(P5A17@Ti(OPri)2) (Scheme 9) by the reaction of poly(P5A17) with Ti(OPri)4. The morphology of both P5A-based polymers was confirmed by solid-state 13C NMR, ICP-MS and energy-dispersive X-ray spectroscopy (EDX). According to the authors, the structure of both materials allows them to swell in organic solvents, which allows organic substrates to pass through the chiral environments of active sites, promoting the catalytic ability of both polymers in the asymmetric catalysis. For example, poly(P5A17@Ti(OPri)2) was used as an efficient asymmetric catalyst in the addition reaction of Zn(Et)2 to aromatic aldehydes to afford 1-aryl-propan-1-ols in up to 92% yield with up to 96% ee. In the recyclability tests, a slight decrease in both the reaction yield (from 88% to 80%) and ee (from 94% to 85%) was observed after six runs. The presence of both the pillararene moiety and the O-titanate containing the chiral core in the catalyst structure seems to be crucial. According to the authors, in the case of bulky aldehydes (4-ButO – Ph, 1-naphthyl and 1-pyrenyl), the decrease in both reaction yield (83 – 85%) and ee (82 – 86%) was observed.

6.2.2. Pd-based pillararene-supported chiral catalyst
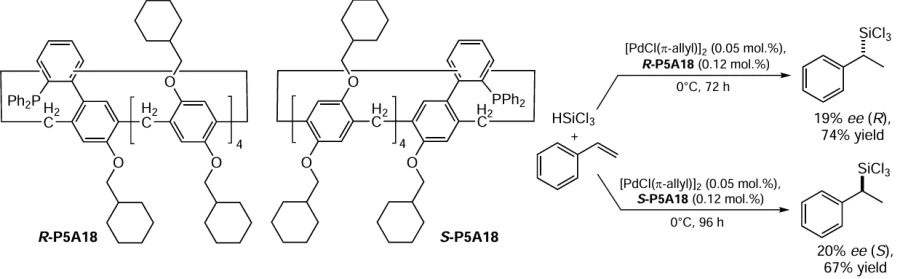
The Ogoshi’s group[102] reported a P5A-based chiral phosphorus ligand P5A18 with cyclohexylmethoxy side chains and a triphenylphosphine substituent in the aromatic moiety of the P5A core (Scheme 10). The catalytic potential of these chiral ligands was investigated in the Pd-catalyzed hydrosilylation reaction. Both R-P5A18 and S-P5A18 showed catalytic activity to afford chiral α-trichlorosilylethylbenzene. According to the authors, the low enantioselectivity of the reaction, such as 19% ee (R-enantiomer, 74% yield) and 20% ee (S-enantiomer, 67% yield) suggests that the planar chirality was transferred from the pillar[5]arene to the freely rotating biphenyl moiety. To the best of our knowledge, this is the first example of using a planar chiral P5A as a ligand for the Pd-catalyzed asymmetric reaction.
6.3. Pillararene-supported Co(III) salens for cycloaddition reactions
Salen metal complexes are promising catalysts for cycloaddition reactions, involving oxygen [103] and, more importantly, CO2,[104][105] opening up new prospects for the conversion of (greenhouse) gases into valuable industrial products.
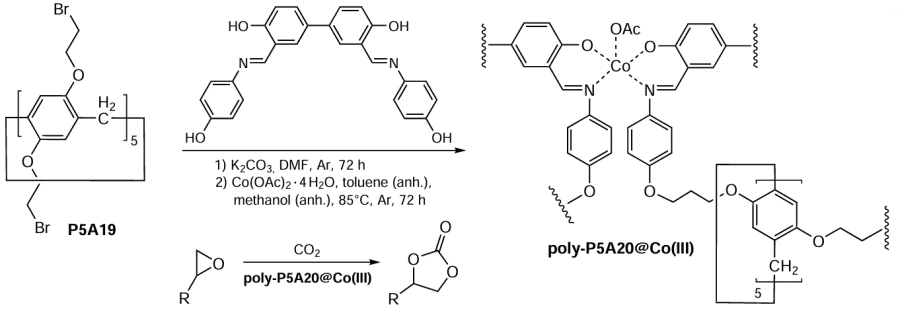
In 2003, a novel pillar[5]arene-based Co(III)-salen-loaded organic polymer was reported as a heterogeneous catalyst for the preparation of cyclic carbamates via the epoxide/CO2 cycloaddition reaction (Scheme 11).[106] The catalyst was obtained via condensation of deca-O-bromoethane pillar[5]arene P5A19 with a Schiff base to afford poly(P5A20), which was then reacted with Co(OAc)2 to give the target poly(P5A20@Co(III)) bearing salen – Co(III) units. Under mild conditions (30°C, 1 atm CO2), poly(P5A20@Co(III)) shows general applicability to afford the desired cyclic carbonates in up to >99% yield, which is much higher than under catalyst-free conditions (47 – 82%). The influence of the pillararene cavity on the efficiency and selectivity of the catalytic process was observed. For example, a decrease in the reaction yield was observed in the case of cyclohexane-fused epoxide and 2-(phenoxymethyl)oxirane a decrease of the reaction yield was observed. The results obtained demonstrate the excellent catalytic performance of poly(P5A20@Co(III)) for the conversion of CO2 .
7. Pillararene-supported nanoparticles
Metal/metal oxide nanoparticles have already demonstrated their efficiency in various reactions as greener catalyst for organic synthesis.[107-110] However, there are some challenges in using NPs, such as the need for precise size and shape control, which affects their potential application, as well as ecological aspects related to the toxicity of NPs and their leaching.[111] Some examples of the use of PA-based NPs are highlighted below.
7.1. Reduction reactions
7.1.1. Pillararene-supported Au(0) nanoparticles
Imidazoles are known to form stable assemblies with Au species of various nature.[112][113] Huang and co-workers [18] synthesized imidazolium-appended pillararene P5A21 (*10Br–) as a stabilizer for the preparation of Au NPs to prepare pillar[5]arene-protected Au NPs (Fig. 4). The formation of P5A21-stabilized Au NPs was confirmed by the presence of the characteristic optical signal of gold colloids: a surface-plasmon resonance in the visible region in the range of 560 to 512 nm range upon increasing the concentration of P5A21, such as 560 nm (2 mM), 545 nm (4 mM), 534 nm (10 mM), 519 nm (50 mM) and 512 nm (200 mM) along with an increase in the intensity of the Au NPs absorption peak. According to the authors, such a noticeable shift could be due to the decreasing size of the newly formed Au NPs, and the decrease in the intensity of the Au NPs’ absorption peak with decreasing concentration of P5A21 is due to the formation of larger-sized Au NPs.
![[{"id":"JUemevFEqF","type":"paragraph","data":{"text":"Scheme for the stabilization of the Au NPs with pillararene <b>P5A21</b> and its linear analogue and their appearance of the Au solutions.[[ type=\"anchor\" referenceId=\"14805\" ]]"}}]](/storage/images/resized/uoYddhVvHRbrDFPhUPy4LzEwRvejTvwcnsLLTILr_xl.webp)
According to the TEM results, at a concentration of P5A21 of 200 mmol L–1, spherical Au NPs with a diameter of 1.88 ± 0.58 nm were observed, and at a very low concentration (10−6 mol L–1), some polydisperse Au NPs with an average particle size < 6 nm were detected. The morphology of the Au NPs could be changed into microtubes either by keeping them at pH 7 for 4 weeks or by adding water to the THF solution and mild sonication for 5 min.
Finally, the P5A21-stabilized Au NPs were used as recyclable catalysts for the reduction of isomeric nitrophenols with NaBH4 in solution, and the corresponding anilines were obtained in 30 min in 95% – 99% yield. According to the authors, the main role of the P5A21-stabilized Au NPs is in the efficient electron transfer from the BH4– anion to the nitro compounds absorbed on the NP surface, mediated by the large Fermi level shift of the nanoparticles, and the catalytic reduction takes place on the surface of the Au NPs. In a series of model experiments with Au NPs with sizes ranging from 3.09 ± 0.79 nm, 3.86 ± 0.91 nm and 5.95 ± 1.64 nm, the rate constants were estimated to be 5.73 ± 10–3 s–1, 2.72 ± 10–3 s–1 and 8.17 ± 10–4 s–1, respectively, indicating that the smaller Au NPs perform better in catalyzing the reaction.
Liao et al.[114] synthesized a thermoresponsive trithiocarbonate-tethered P5A22-containing polymer based on poly(N-isopropylacrylamide) (PNIPAM) via reversible addition-fragmentation chain transfer (RAFT) polymerization. The presence of trithiocarbonate groups allows this polymer to stabilize gold nanoparticles by chemisorption (Scheme 12). In aqueous solution, in the absence of Au NPs, the formation of P5A22 micelles with a diameter of 21 nm was observed, whereas in the presence of Au NPs, P5A22-PNIPAM@Au aggregates with a diameter of 58 nm were formed. In addition, the presence of terminal P5A22 moieties in the PNIPAM polymer chains allows the resulting material to encapsulate electron-deficient guests. For example, in the presence of n-octylpyrazinium hexafluorophosphate, linked nanoparticles with a diameter of ~106 nm were obtained. In fact, the host-guest interaction made the polymers more hydrophobic facilitating the aggregation of the individual particles. The authors argue that the use of the n-octylpyrazinium guest lowers the cloud point due to aggregation between the alkyl chains of n-octylpyrazinium and PNIPAM.

The complexation event was detected by the UV-VIS absorption spectra, and the maximum absorption of P5A22-PNIPAM-coated AuNPs was red-shifted from 523 to 529 nm. Upon the addition of guest molecule, the absorption was red-shifted to 541 nm indicating the formation of Au aggregates via host – guest complexation. The authors believe that P5A22-PNIPAM-coated AuNPs can be used as sensors for detection by surface-enhanced Raman spectroscopy.
The shift of the maximum absorption was explained by the fact that PNIPAM is a thermoresponsive polymer and dissolves in water below its lower critical solution temperature (LCST), and aggregates at higher temperatures. P5A22-PNIPAM@Au NPs demonstrated good temperature responsiveness. With increasing temperature, the absorbance increased dramatically with a red shift of the surface plasmon resonance (SPR) peak caused by the contraction of the PNIPAM chains leading to an increase in light scattering and aggregation of Au NPs. According to the TEM data, aggregated Au NPs with an average diameter of ≈ 1074 nm (based on dynamic light scattering ((DLS)) were observed at a temperature of 40°C. As the temperature decreased, the hypsochromic shift of the absorbance took place with a concomitant blue shift of the SPR peak, and the polymer chains became soluble in water.
Finally, the resulting material was effectively used as a recyclable thermoresponsive catalyst for the NaBH4-mediated reduction of p-nitrophenol (< 3% yield loss after 5 catalytic cycles). The catalyst was recoverable by heating above the cloud point of the composite (³ 40°C).
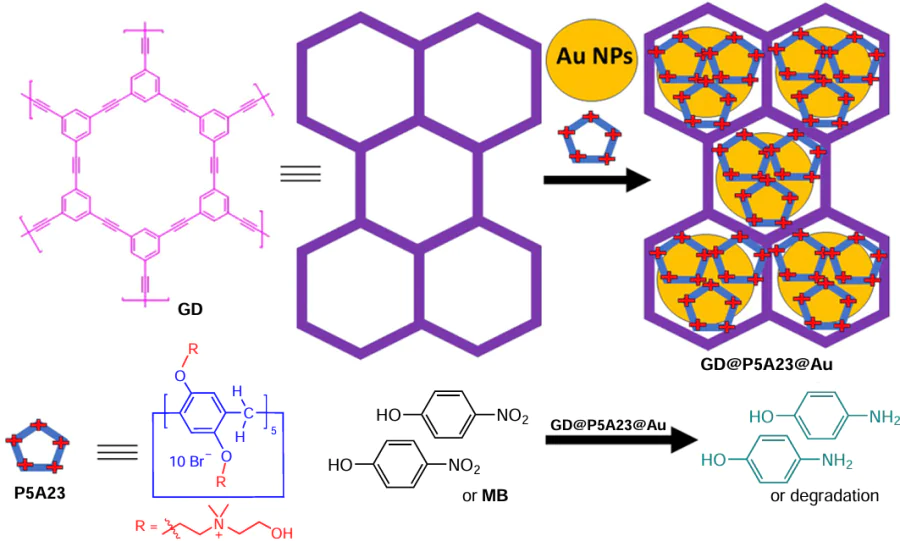
In 2019, Tan et al.[115] reported a 2D heterogeneous hybrid nanomaterial GD@P5A23@Au loaded with Au NPs dispersed in the 2D porous structure of graphdiyne (GD). These 2 – 3 nm Au NPs were prepared in situ from GD and P5A23 (Scheme 13). The resulting material showed enhanced catalytic efficiency for the reduction of 4-nitrophenol and the degradation of methylene blue (MB) in the presence of NaBH4 . According to the authors, this reaction featured a shorter reaction time and a higher degree of the reduction compared to that using the commercial Pd/C catalyst. It should be noted that the rate constant (k) of the reduction of 4-nitrophenol and for the degradation of MB was 11.3 times higher than that of the Pd/C-catalyzed reaction (k = 0.042 min–1). The MB degradation rate constant increased by a factor of 15.77 times. The main advantage of the above-mentioned P5A-based materials is the small size distribution of the catalytically active AuNPs within the material. In addition, due to the insolubility of the 2D catalyst, high stability and recyclability were observed without changing the size and catalytic behaviour of the Au NPs after five cycles. Therefore, these materials are good supports for the metallic NPs.
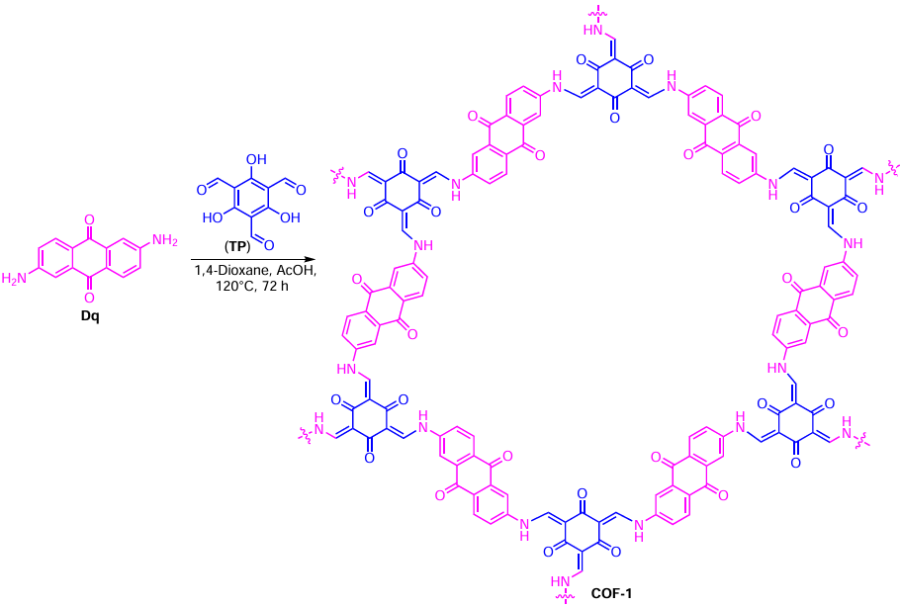
In a follow-up study,[116] the 2D heterogeneous catalyst based on P6A1-stabilized Au NPs within a covalent organic framework (COF) as a solid support was prepared (Scheme 14, Scheme 15). This supramolecular assembly was obtained by the reaction between cationic pillar[6]arene P6A1 and Au NPs within supramolecular organic polymeric framework COF-1 (see Scheme 15). The porosity of P6A1-Au-COF-1 was estimated by N2 adsorption experiments using the BET method. The N2 adsorption isotherms of P6A1-Au-COF-1 and COF-1 are type I reversible isotherms with the surface areas of 328 and 695 m2 g−1, respectively. The reduction in the specific surface area of P6A1-Au-COF-1 is evident and is caused by the loading of Au NPs. Furthermore, the pore size distribution plot of COF-1 and P6A1-Au-COF-1 shows two peaks at 8.2 and 10.5 Å. The reduction in pore size is attributed to the blocking of the cavity of COF-1 by Au NPs. The authors believe that these data can further confirm that Au NPs are successfully anchored on the surface of COF-1.
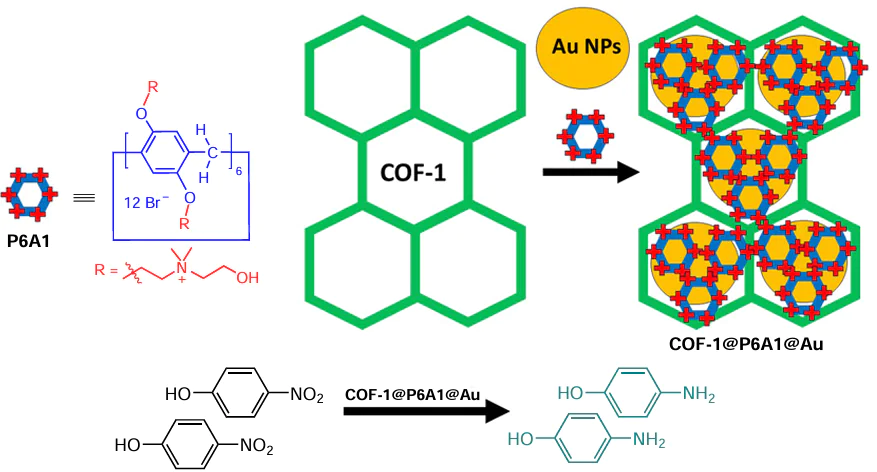
Compared to the commercially available Pd/C catalyst, P6A1-Au-COF-1 demonstrated superior catalytic performance for the NaBH4-mediated reduction of o-, m- or p-nitrophenols with rate constants of 9.05, 5.11 and 33-fold higher, respectively. This confirms the influence of the P6A cavity on the selectivity of the process. Such reduction efficiency was provided by the high porosity and crystallinity of the catalyst, which allows accumulation of the nitrophenol isomers at the surface of the COF-1, and interaction of the electron-rich Au NPs with the electron-deficient nitrophenol isomers.
7.1.2. Pillararene-supported Rh(0) nanoparticles
Li et al.[117] reported the use of P5A-functionalized Rh nanoparticles (P5A24@Rh NPs) as the catalyst for the reduction of toxic nitrophenols and azo dyes with NaBH4 (Scheme 16). These nanoparticles were obtained by the reduction of the carboxylated P5A sodium salt and RhCl3 with NaBH4 and were characterized by TEM, XRD, X-ray photoelectron spectroscopy (XPS), IR spectroscopy, and UV-VIS spectroscopy. The average diameter of P5A24@Rh NPs was ~ 2.9 nm. In the NaBH4-mediated reduction of isomeric nitrophenols, as well as methyl orange and Congo red, the above-mentioned P5A-supported Rh NPs exhibited good catalytic activity, which was comparable or higher than that of the previously reported metal NPs-based catalysts. It should be noted that the P5A moiety has some influence on the catalytic activity of P5A24@Rh NPs, and in the case of m-nitrophenol, a decrease in the degree of conversion can be observed. In addition to the excellent catalytic activity, P5A24@Rh NPs have superior photothermal ablation capability towards Staphylococcus aureus under 808 nm laser irradiation.

7.1.3. Pillararene-supported Pd(0) nanoparticles
Wang et al.[118] reported the preparation of Pd(0) NPs using terpyridine-substituted pillar[5]arene P5A25 as a ligand (Scheme 17). The complex P5A25@Pd(II) was obtained by the reaction of P5A25 with PdCl2 . Its reduction with NaBH4 in solution afforded the desired P5A25-supported Pd(0) NPs. According to the TEM- and DLS data, the average particle size of the resulting NPs was 2.25 nm with narrow size distribution. In addition, the binding energies of 338.13 and 343.43 eV, which were attributed to Pd(0) 3d5/2 and 3d3/2 , respectively, obtained from the X-ray photoelectron spectroscopy (XPS) data, were consistent with those reported for the Pd(0) NPs. According to the HAADF-STEM imaging and EDX mapping analysis, C, N, O, and Pd atoms were uniformly distributed among the above-mentioned NPs. Finally, the PXRD pattern showed that 2θ values of the materials of 2θ ≈ 40°, 47°, 68°, and 82°, which could be assigned to the (111), (200), (220), and (311) lattice planes, respectively, are consistent with those of the Pd(0) NPs. In the final step, the P5A-supported Pd(0) NPs were tested as reusable catalysts for the reduction of nitrophenols to anilines in aqueous solution in up to 97.5% yield. Such an excellent catalytic activity (the reaction took several minutes) was attributed to the large size of pillar[5]arene, which inhibits the aggregation of PA-supported Pd(0) NPs, thereby providing the availability of the reaction sites to ambient air, and sufficient electron-transfer channels for the reaction.

7.2. Oxidation reactions
In the presence of different metal ions (Ag+, Bi3+, Pb2+, Pt4+ or Hg2+) or their combinations, Au NPs exhibit peroxidase-, oxidase-, or catalase-like activities.[119] Metal cations are deposited on the surface of Au NPs, and the catalytic activity of these cation-deposited Au NPs results from the different oxidation states of the surface metal atoms/ions. This feature of Au NPs is important for the construction of enzyme-like oxidants for the selective oxidation of biomolecules and other substrates. In continuation of such research, Park et al.[120] developed pillar[5]arene-coated Au NPs P5A26@Au by a one-pot hydrothermal method starting from HAuCl4 (Scheme 18). Based on the TEM data, the resulting P5A26@Au NPs were about 17 nm in diameter, and the hydrodynamic diameter of ~ 24 nm was estimated by the DLS. The above-mentioned P5A26@Au NPs were stable at different pH values, which was attributed to the stabilization of Au NPs by polycarboxylate moieties of multiple P5A26 ligands. The P5A26@Au NPs were then aged with AgNO3 , whereby the Ag+ ions were deposited on the AuNP surfaces through aurophillic interactions.
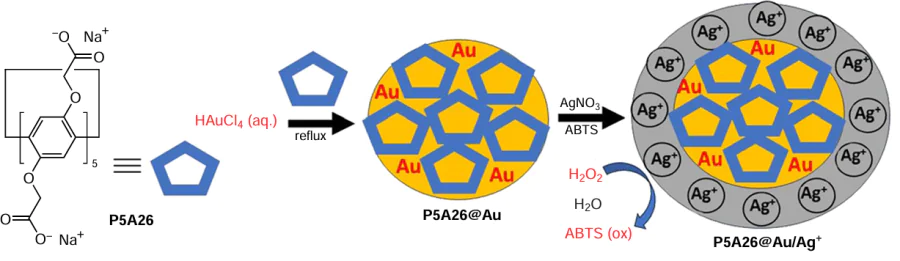
As a final step, to study the peroxidase-like activity of the P5A26@Au/Ag+ catalyst, the reaction of 2,2'-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS) with H2O2 used as an electron acceptor was investigated. The catalyst demonstrated high oxidative activity in this reaction. P5A26@Au/Ag+ NPs exhibited peroxidase-like activity by catalyzing a one-electron oxidation of ABTS to form the ABTS radical.
8. Pillararene-supported polyoxometalates
Polyoxometalates (POMs) are molecular metal oxide clusters with a 1D, 2D and 3D structure and have already found wide applications in homo- and heterogeneous catalysis. However, they have serious drawbacks mainly related to the use of POMs in solutions, such as low selectivity, low stability, tendency to aggregation and/or precipitation, etc. Therefore, to extend the scope of applications of POMs, different strategies have been developed for the deposition of POMs on different substrates.[121]
8.1. Pillararene-supported polyoxometalates for oxidation/degradation reactions
Chen et al.[122] fabricated 2D hybrid nanosheets by the self-assembly of imidazolium-appended pillar[6]arenes P6A2 and H3[PW12O40] used in a 1 : 1 ratio at room temperature in water (Scheme 19). The formation of the P6A2*POM ionic assembly was confirmed by a Tyndall effect. According to the authors, the calculated molar ratio of POM to P6A2 was about 3.91 (1 : 3.91), indicating the complete incorporation of POM into the obtained hybrid material. The zeta-potential of the hybrid material was determined to be 10.5 mV, indicating that the outer surface of the assembly is covered by the anionic POMs, and thus, cationic sites of P6A2 can interact with this surface through electrostatic interactions. Based on a non-localized DFT (NLDFT) method in an N2 sorption experiment at 77 K, the porous structure of the hybrid material was confirmed with two main peaks at around 2 and 2.5 nm.
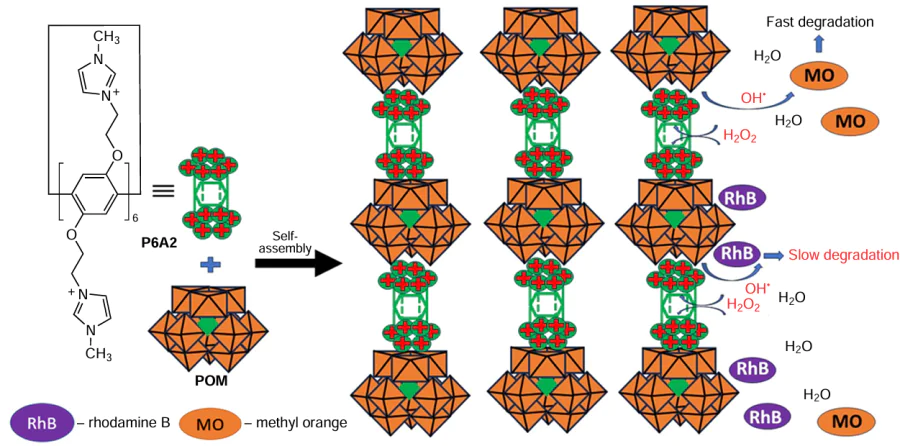
One of the most common applications of POMs is the photodegradation of organic pollutants in water.[123] To confirm the catalytic performance of the P6A2:POM nanosheets, the degradation of typical organic pollutants such as the anionic dye methyl orange (MO) and the cationic dye rhodamine B (RhB) was studied in the presence of H2O2. Complete degradation of MO was achieved in 60 min. According to the authors, in the P6A2@POM system, polyanionic POMs are isolated and surrounded by the macrocycles P6A2 via hydrogen bonding and electrostatic interactions. The main role of P6A2 is to provide a transport of H2O2 molecules through the cavity towards the POM catalytic sites to form radicals WV – O – O• for the subsequent generation of •OH radicals (in the presence of water) and their return to the bulk solution through the P6A2cavity. Dyes underwent degradation more efficiently, than when using the A1@POM@H2O2 system. In addition to MO, the authors also observed the degradation of RhB, which reached 91.32% after 180 min. The faster degradation of the anionic dye MO compared to the cationic dye RhB suggests that the degradation of the dyes takes place in the bulk solution rather than on the surface of the hybrid material. And in the case of cationic RhB it is readily absorbed onto the negatively charged surface of the P6A2@POM cluster, preventing H2O2 from approaching the surface of the material to activate them. As a result, the lower concentration of active H2O2 molecules resulted in slower degradation of RhB.
Zheng et al.[124] reported similar approach for the construction of the catalytic system based on cationic pillar[5]arene P5A27 and anionic Anderson polyoxometalates Na3[CrMo6O18(OH)3] (hereinafter designated as CrMo6) (Scheme 20).
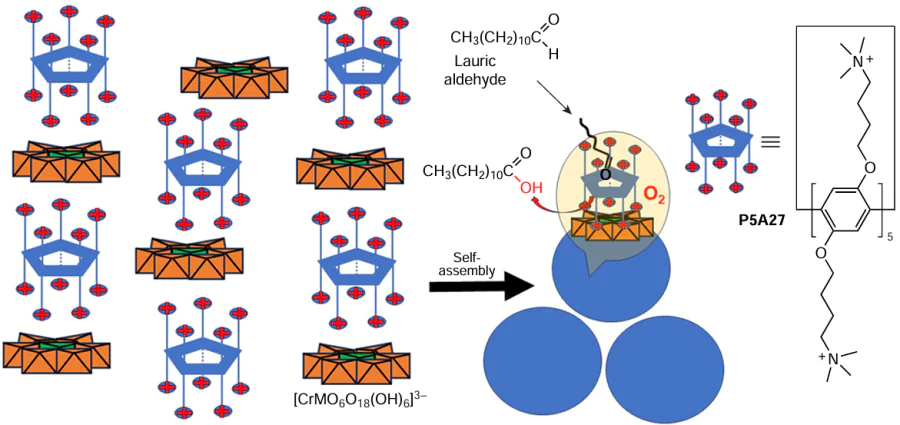
Based on the DLS data, changing the charge ratios of the ionic complexes (P5A27@CrMo6) leads to a change in the diameters of the nanospheres. In addition, based on the TEM and SEM data, the zeta-potential of the aggregates was determined to be +30 mV, indicating that the outer surface of these aggregates is, probably, covered by the cationic P5A27. According to the elemental analysis data, the composition of these nanospheres was CrMo6@P5A27 = 3.3 : 1, i.e. each CrMo6 anion is surrounded by the cationic P5A and can be represented as P5A27@(CrMo6)3.3. Based on the DFT calculations, the diameter of P5A27 (10.0 Å) is larger than that of CrMo6 (8.7 Å), and the surface occupancy of CrMo6 was greater than 2/3. As a result, a classical reverse bilayer cannot be formed, suggesting a dense packing of rigid balls, which is consistent with the detection of solid aggregates by SEM and TEM techniques.
In the next step, the P5A27@CrMo6 catalyst was used for the oxidation of aldehydes in the presence of oxygen. Thus, aldehydes bearing electron-donating groups afforded the corresponding acids in up to 99% yield, while heterocyclic aldehydes or aldehydes bearing electron-withdrawing groups gave the corresponding acids in lower yields. Sinergy between P5A27 and POM plays an important role. In the control experiments, the reaction of POMs with Na3[CrMo6O18(OH)3] afforded benzoic acid in 90% yield in 4 h. In the presence of only P5A27, the reaction took 6 h to give the same product in 90% yield, while in the auto-oxidation process, benzoic acid was obtained in 8 h in 90% yield. The involvement of P5A27 was confirmed by the example of oxidation of linear aliphatic aldehydes, such as lauric aldehyde. In this case, high oxidation yields (91%) were observed, which can be attributed to the strong host-guest interaction between the cationic pillar[5]arenes and lauric aldehyde or lauric acid. Such interactions could result in the transfer of water-insoluble lauric aldehyde into the positively charged P5A27 due to the non-covalent interactions, making this aliphatic aldehyde more susceptible to oxidation.
In addition, the recyclability and stability of the nanosphere catalysts were investigated, and it was found that the reaction yield was greater than 80% after six cycles.
9. Pillararene-сatalyzed photoreactions
Photoredox catalysts are used to convert light into chemical energy,[125] making them the more sustainable alternative to traditional catalytic systems. Ambient light is the most environmentally friendly energy source compared to high temperatures or hazardous chemical reagents, and the sustainability of photocatalyzed reactions is based on their low E-factors.
9.1. Photodegradation of organic contaminants
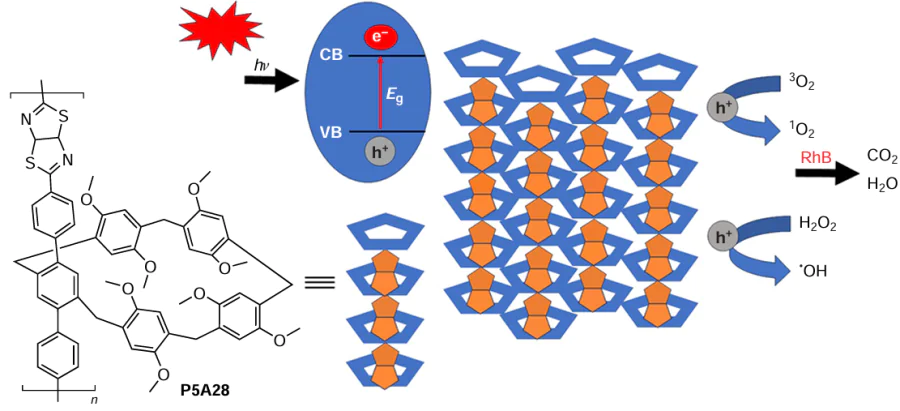
Wang and co-workers [126] reported the synthesis of polymer P5A28, in which P5A moieties were connected by thiazolo[5,4-d]thiazole bridges (Scheme 21). In addition, the terphenylene TP1-based polymer was prepared and used for model experiments. The porosity of P5A28 was measured by N2 adsorption – desorption at 77 K, as well as CO2 adsorption –desorption at 273 K. The adsorption isotherm of TP1 exhibited type II isotherm with a BET surface area of 41.1124 m2 g–1, and the gas sorption experiments revealed P5A28 to be nonporous. The authors believe that it can be due to the interpenetration of P5A, which occupies the entire polymer space, between different polymer chains. In addition, the morphologies of P5A28 and TP1 were recorded by SEM, and micrometer-scale blocky structure of P5A28 with a convex surface was observed.


According to the authors, the presence of both P5A cavities and the heterocyclic moiety provide better photophysical properties of P5A28 than TP1 – P5A28, such as bathochromic shift of the absorption maximum to 700 nm in the UV spectrum and longer lifetime at 580 nm. The possible reason is a more extended electron delocalization within the P5A core of P5A28 compared to TP1. Moreover, the P5A28 exhibited a narrower band gap energy of ~ 1.85 eV compared to that of TP1 of ~ 2.05 eV, indicating the stronger reduction/oxidation abilities of photogenerated electrons (e–) and holes (h+) to improve the photocatalytic activity of TP1.
Being a promising photocatalyst, P5A28 was further studied for the degradation of organic contaminants under visible light irradiation (300 W Xe lamp, 420 nm). In the RhB dye photodegradation model experiments, P5A28 (0.4 mg mL−1) demonstrated lower activity (4 h for the complete photodegradation of RhB) than TP1 (0.1 mg mL−1) (complete photodegradation of RhB in 40 min).
Based on the data from electrochemical impedance spectroscopy (EIS) and electrochemical Mott – Schottky plots, it was found that P5A28 has a lower resistance than TP1, suggesting that the introduction of pillararenes with electron-rich cavities provides the high carrier mobility due to the improvement of interfacial charge transport capability and photocatalytic properties. According to the data obtained, both polymers are n-type semiconductors, as their Mott – Schottky plots exhibited positive slopes. The calculated conductivity band (CB) potential (0.63 V vs. saturated calomel electrode (SCE)) of TP1 is more negative than the reduction potential from •O2 to •O2– (0.55 V vs. SCE), and TP1 was therefore feasible for the production of superoxide species and enhancement the photodegradation of dyes. The more positive valence band (VB) position of P5A28 makes it suitable for the oxidation of H2O to generate reactive oxidation species (ROS) as confirmed by the higher VB potential (1.73 V vs. SCE) than the oxidation potential of H2O to H2O2 (1.54 V vs. SCE). The •OH radical can be produced from H2O2 as a secondary ROS. According to the authors, h+ is primarily responsible for the photodegradation, suggesting that the predominant redox reactions are different from those of TP1, where •O2– and e– play key roles in the photodegradation process (Scheme 22). Therefore, P5A28 has a stronger oxidation capacity, indicating the importance of the P5A cavities. Finally, the recyclability of both polymers was explored, and after three cycles both showed excellent recyclability and stability under prolonged visible light irradiation.
9.2. Dehalogenation reactions
Dehalogenation reactions play an important role in the development of efficient approaches to the management of industrial and household plastic waste.[127] Dehalogenation under photocatalytic conditions appears to be the most promising in terms of its efficiency and sustainability.[128]
In 2023, Wang et al.[129] synthesized a dimeric meso-functionalized ethoxy pillar[5]arene P5A29, by combining two pillar[5]arenes (Scheme 23). In this structure, two properly connected P5A moieties form a tetraphenylethylene (TPE) moiety, a common AIEgene, in the central part of bis-P5A. The structure of P5A29 was confirmed by a single-crystal XRD analysis. According to the X-ray data, this TPE-based bis-P5A adopted a symmetrical rigid conformation with all ten diethoxyphenylene subunits perpendicular to the molecular plane of a twin pillar[5]arene host. Thus, the TPE moiety provides a link between two pillar[5]arenes with an average valence angle along the C=C axis of the bond between the cavities of about 112°. According to the authors, the introduction of the TPE moiety into P5A29 restricts the intramolecular rotation (RIR) of the benzene rings in TPE, leading to AIE in the low-aggregation state, observed as strong blue emission in CHCl3 solutions.

P5As and P6As are known for their ability to encapsulate readily aliphatic guests bearing electron-withdrawing moieties, such as cyano, tetramethylammonium, pyridinium, etc., to form host-guest complexes as well as supramolecular assemblies. Therefore, to study the host-guest properties, the above-mentioned dimeric P5A29 were exposed to dinitrile-based guests bearing linkers of various length, such as n-butyl (G1), n-octyl (G2), and 1,4-bis(pentyloxy)phenyl (G3). According to the authors, the morphological control and fluorescence properties of the resulting host – guest assemblies depend on the guest type, and the type of host – guest complex formed depended on the nature of guest molecules. Thus, short guest G1 is completely embedded in the cavities of the P5A to form a 1 : 1 inclusion complex, the structure of which was confirmed by the 1H NMR titration studies and a single-crystal X-ray diffraction analysis. In the case of longer guest G2, a linear supramolecular polymer was formed, the structure of which was confirmed by 1H NMR DOSY experiments and a single-crystal X-ray diffraction analysis. Finally, the guest G3 leads to the formation of a supramolecular layered polymer, in which each cavity of P5A29 encapsulated the alkyl cyanide unit of one side of G3, and the resultant inclusion complex was self-assembled via the π – π stacking between the central benzene rings of G3 (see Scheme 23).
According to the TEM data, the formation of a densely-packed lamellar structure was observed for P5A29@G3. Powder X-ray diffraction (PXRD) patterns indicated good crystallinity. Comparison of the PXRD patterns for P5A29@G3 and P5A29 revealed a significant difference, which the authors attributed to the formation of the aggregates in the former case.
In addition, P5A29@G3 showed excellent AIE properties, and upon adsorption of Eosin Y (EsY) as an acceptor by the layered structure, the energy transfer (via the Föster energy transfer (FRET) mechanism) between the P5A29@G3 as a donor to the EsY acceptor was confirmed by UV-VIS and fluorescence spectroscopy. In the last step, the P5A29@G3@EsY FRET pair was used in the photocatalytic degradation of bromoacetophenones. In the presence of 0.5 mol.% of P5A29@G3@EsY complex in aqueous solution, a-bromoacetophenone gave a dehalogenated product, acetophenone, in good yield under white light (20 W) irradiation (as a solar light simulator) for 2 hours.
Another example of the P5A catalysts for the photodegradation of bromoalkanes are the phenazine-appended pillar[5]arenes P5A30 – 32 developed by Schmidt and Esser[130] as model photocatalysts.
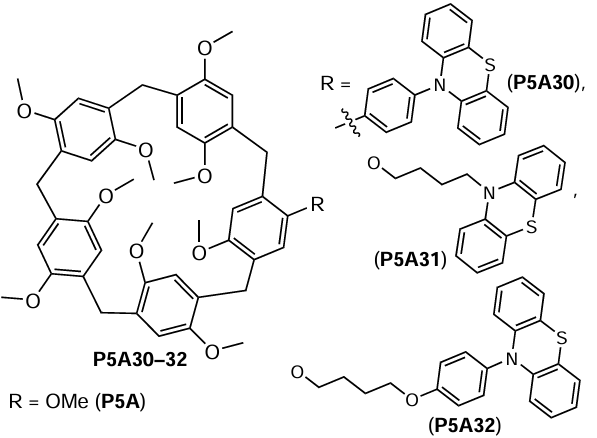
Compounds P5A30 – 32 were synthesized by mono-demethylation of permethylated P5A followed by the covalent introduction of the phenazine moiety via linkers of various lengths (Scheme 24). The authors then used all three P5A30 – 32 catalysts in the model reaction of the reductive dehalogenation of ethyl 4-bromobutanoate and 5-bromopentanenitrile upon irradiation with a 365 nm light source.

It was found that the presence of both P5A cavity and phenothiazine-based antennas efficiently promoted the reductive dehalogenation by keeping the substrate locked in a close proximity to the photocatalyst. Thus, the association constant for the complexation of 5-bromopentanenitrile by P5A was as high as 1.7 × 104 M–1, which was 30 times higher than that for the debrominated product, pentanenitrile (5.7 × 102 M–1).[131] The authors suggested that the dehalogenation product is readily released from the P5A after the photocatalyzed dehalogenation regenerating the molecules of the P5A30 – 32 catalyst. In addition, the DFT calculations using the TURBOMOLE v7.5.0 program package showed that the structure of the complex of P5A30 with 5-bromopentanenitrile has the perfect fit of guest molecule into the P5A cavity with a close proximity of the bromide atom to the photocatalyst. Finally, the authors demonstrated the applicability of P5A30 – 32 in the reductive dehalogenation of ethyl 4-bromobutanoate and 5-bromopentanenitrile upon photoexcitation at 365 nm, and up to 61 – 63% yields of dehalogenated alkanes were obtained. The encapsulation of both bromoalkanes by P5A cavity of P5A30 – 32 was suggested based on higher Stern – Volmer quenching constants compared to N-phenylphenazine.
To confirm the involvement of the P5A cavity in the process, the reactions were carried out with sterically hindered substrates that cannot fit into the cavity of P5A30, P5A32, such as 4-bromo-2,2-diphenylbutanenitrile, (bromomethyl)cyclohexane, 4-bromoanisole and 4-bromobenzonitrile. In all the cases, yields similar to those of the cavity-free N-phenylphenazine were observed, indicating the importance of the P5A cavity for the photocatalyzed debromination reaction.
In 2019, a two-step FRET photocatalytic system based on a non-covalent supramolecular assembly based on the water-soluble pillar[5]arene P5A33 and a bola-type tetraphenylethylene-functionalized dialkyl ammonium derivative (TPEDA) was reported (Scheme 25).[132] In the presence of TPEDA, a significant opalescence of the aqueous solution of P5A33 was observed with an obvious Tyndall effect, suggesting the formation of large amounts of TPEDA @P5A33 supramolecular nanoparticles.
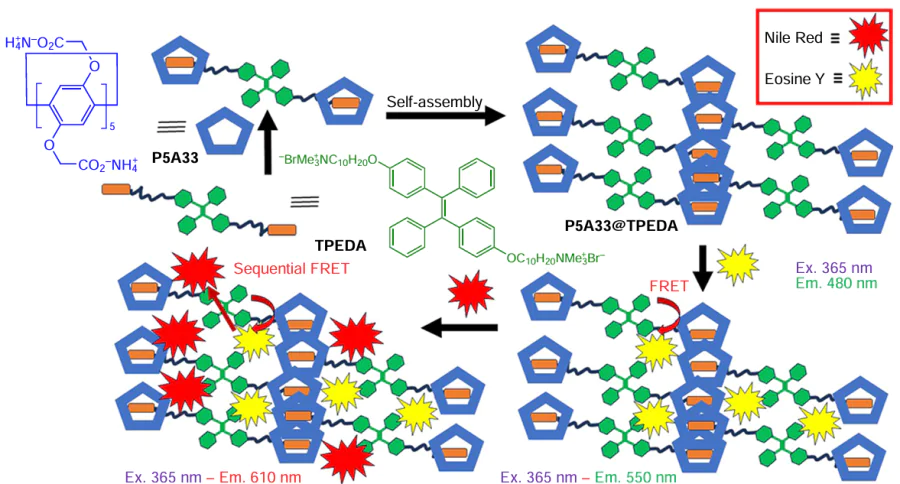
The TPEDA @P5A33 association constant was determined to be (1.69 ± 0.27) × 105 M−1. The TPEDA/P5A molar ratio during aggregation was 5 : 1 at the critical aggregation concentration (CAC) of TPEDA and P5A33 assembly of 0.014 mM. The size and morphology of TPEDA@P5A33 NPs were investigated by DLS, and these NPs showed a narrow size distribution with a mean diameter of 180 nm. In addition, TEM images revealed a dark spherical structure with a diameter similar to that calculated from the DLS results, suggesting the formation of a multilayer vesicular structure. In addition, due to the high positive zeta-potential (19.84 mV), the electrostatic repulsive force around the surfaces of the NPs can prevent their agglomeration and improve their stability in aqueous solution. In TPEDA@P5A33, both components are stacked via π – π bonds and this assembly proved to be an ideal donor for the construction of an artificial light-harvesting system in aqueous medium.
Further, the fluorescent dye EsY was introduced into this system. The authors found that EsY can be easily entrapped into the hydrophobic layer of TPEDA@P5A33 vesicles via non-covalent interactions, leading to significant shortage of the distance between the donor and acceptor, which ensures the efficient FRET effect. To study this effect, the fluorescence behaviour of the EsY-loaded vesicles was examined, and it was found that with increasing concentration of EsY, the fluorescence intensity of TPEDA (donor) at 480 nm decreased, while the fluorescence intensity of the EsY (acceptor) at 550 nm increased (λex = 365 nm). In addition, the light blue fluorescence of TPEDA@P5A33 changed to a bright yellow – green (TPEDA@P5A33@EsY) due to an efficient energy transfer. The light-harvesting properties of the TPEDA@P5A33@EsY assembly were confirmed by the fluorescence decay experiments: for TPEDA@P5A33, the decay curve was fitted as a double exponential decay with fluorescence lifetimes of τ1 = 1.79 ns and τ2 = 5.40 ns, while for the TPEDA@P5A33@EsY assembly, the fluorescence lifetimes decreased to τ1 = 1.15 ns and τ2 = 3.64 ns, indicating that the TPEDA@P5A33@EsY system functions as an artificial light-harvesting system with energy transfer from the TPEDA@P5A33 assembly to the acceptor EsY. Finally, the two-step energy transfer TPEDA@P5A33@EsY@NiR light-harvesting system was prepared by adding Nile Red (NiR) dye as the second acceptor. After the addition of NiR, the decrease in the emission intensity of the TPEDA@P5A33@EsY system at 550 nm attributed to EsY was observed along with the appearance of the new emission band of NiR at 610 nm. This resulted in a change in fluorescence colour from bright yellow-green to off-white due to the formation of the TPEDA@P5A33@EsY@NiR assembly. Along with the changes in the emission intensity and colour, a change in the fluorescence lifetime to τ1 = 1.05 ns and τ2 = 2.68 ns was observed. According to the authors, this phenomenon indicates a sequential energy transfer process from TPEDA@P5A33@EsY to NiR mediated by EsY. The fluorescence quantum yield of TPEDA@P5A33@EsY@NiR was found to be 5.01%, and the energy-transfer efficiency was calculated to be 56.28% with the molar ratio of TPEDA (donor)/EsY (acceptor I)/NiR (acceptor II) = 200 : 1 : 1. The most common practical application of these TPEDA@P5A33@EsY@NiR FRET systems is both in function as nanoreactors to provide a favourable space for the photocatalytic reactions in aqueous environment and as systems for an efficient transfer of UV light to the longer-wave emission.
Finally, the said FRET-system was used as a photocatalyst for the dehalogenation of α-bromoacetophenone in aqueous solution, and an excellent yield of 96% was observed compared to the control group using EsY or NiR alone.
Using the same P5A host, P5A33, Hu and co-workers [133] developed two other water-soluble FRET systems for the photocatalyzed dehalogenation of α-bromoacetophenone in aqueous medium (Scheme 26). Two supramolecular assemblies were constructed by reacting the water-soluble pillararene P5A33 as the host and a bola-type salicylaldehyde-azine derivative (HNPD) as the guest. As a common AIE fluorophore, HNPD achieves high emission in the aggregates and serves as an excellent energy donor for the light-harvesting processes in an aqueous phase.[134-139] The introduction of water-soluble P5A moieties into the supramolecular assembly lowers the CAC of HNPD thereby enhancing the AIE effect. Thus, in an aqueous solution of HNPD containing P5A33, the P5A33@HNPD inclusion complex was formed, which further self-assembled into globular nanoparticles exhibiting green fluorescence under 365 nm excitation in a quantum yield of 2.94%. The resulting P5A33@HNPD aggregates exhibited a narrow size range and an average hydrodynamic diameter of 176 nm (based on DLS). According to the TEM data, a core-shell globular morphology was observed with diameters ranging from 120 nm to 170 nm. Finally, the zeta-potential measurements showed that P5A33@HNPD NPs had a relatively high negative values (–23.41 mV), indicating that the repulsive interaction of their shells can prevent their agglomeration and further enhance the solubility of P5A33@HNPD. EsY and NiR as energy acceptors were successfully entrapped into the hydrophobic interior of P5A33@HNPD (as a donor) to form P5A33@HNPD@EsY and P5A33@HNPD@NiR nanoparticles, respectively. Again, similar to the above TPEDA@P5A33, the overlap of fluorescence emission of the P5A33@HNPD NPs with the UV absorption region of EsY and NiR was observed, resulting in an efficient FRET process to EsY and NiR with a high energy transfer efficiency (80.5% and 60.2%, respectively). According to the authors, the FRET process can be observed by the fluorescence change from green to yellow and red, respectively. In addition, these assemblies showed an excellent antenna effect of 32.5 for the P5A33@HNPD@EsY assembly and 30.1 for the P5A33@HNPD@NiR assembly with a high energy donor/acceptor ratio ([HNPD]/[EsY] = 250 : 1 and [HNPD]/[NiR] = 200 : 1). Due to the AIE enhanced FRET efficiency, the fluorescence self-quenching could be avoided, while the harvested solar energy could be efficiently converted.
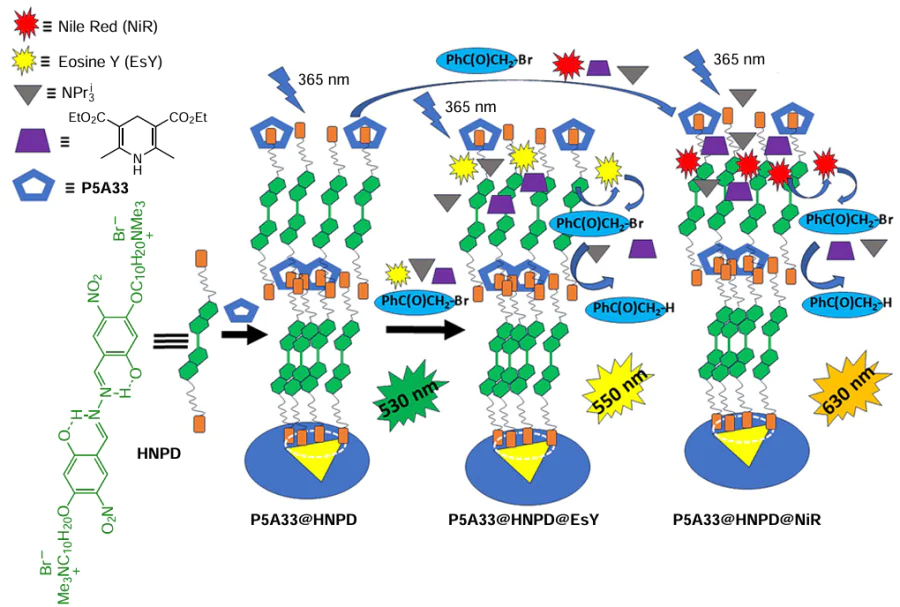
The EsY and NiR dyes are widely used in the photoredox catalysis due to their ability to excite free radicals in certain wavelength ranges. The redox potential (E 0) values for EsY and NiR calculated from the cyclic voltammetry data are –0.75 and –0.9 V, suggesting that EsY and NiR are suitable for the photocatalytic dehalogenation reaction of, e.g., α-bromoacetophenone (E 0 = –0.49 V).[140] When excited with the Xe lamp, both free EsY and NiR promoted the photocatalytic dehalogenation of α-bromoacetophenone, however, in very low yields (7% for EsY and 9% for NiR).[140] This may be due to the limited absorption of EsY and NiR in the UV region. Compared to free EsY and NiR, P5A33@HNPD@EsY and P5A33@HNPD@NiR FRET assemblies exhibited an improved absorbance of solar light in the UV-vis region, and, thus, an improved efficiency in the photocatalytic dehalogenation of α-bromoacetophenone. As a result, under UV excitation (365 nm), due to the high efficiency of the FRET process, both P5A33@HNPD@EsY (with an emission peak at 550 nm) and P5A33@HNPD@NiR (with an emission peak at 630 nm) showed an increase in the yield of α-bromoacetophenone dehalogenation product up to 55% (for the P5A33@HNPD@EsY system) and 65% (for the P5A33@HNPD@NiR system) in shorter reaction times.
9.3. Photooxidation reactions
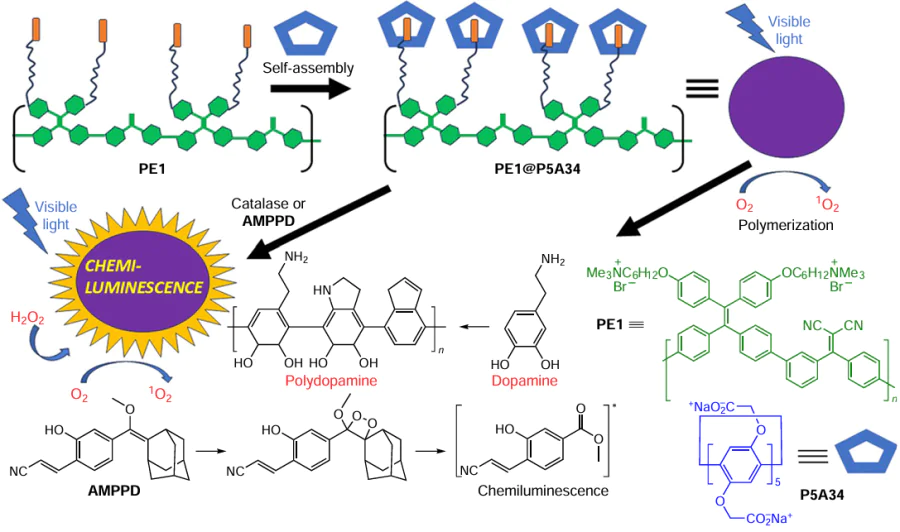
In a follow-up study, Wand and co-workers[141] reported AIEgen-appended P5A34-based singlet oxygen (1O2) generation system for the photooxidation of dopamine to form polydopamine (Scheme 27). In the first step, a typical donor-acceptor structure P5A34 was prepared containing a polymeric AIE photosensitizer based on tetraphenylethylene (TPE1), which was responsible for the efficient generation of 1O2 . C The optimum molar ratio for the TPE1@P5A34 was found to be 7 : 1 at the inflection point, and CAC of PE1@ P5A34 was determined to be 1.8 × 10−6 mol L–1. The formation of stable amphiphilic host-guest complex via encapsulation of tetraalkylammonium groups of the pendant alkyl moieties of TPE1 into the cavity of P5A34 is followed by the self-assembly of supramolecular nanoaggregates in aqueous solution. The nanoaggregates formation was confirmed by DLS (average diameter of 185 nm) and TEM (shrunk nanoparticles with diameters around 150 nm) measurements. The results showed a negative zeta-potential (‒38.5 mV) for the resulting nanoparticles, indicating that the electrostatic repulsive force is able to prevent nanoparticle agglomeration and improve their stability. Along with significant 1O2 photogeneration, the obtained PE1@ P5A34 NPs exhibited enhanced fluorescence. The 1O2 quantum yield of the PE1@ P5A34 NPs was calculated to be 0.89 (using Rose Bengal as a reference). In addition, by encapsulation of catalase enzyme, the PE1@P5A34 supramolecular system showed an increased 1O2 generation efficiency in response to H2O2 . Encapsulation of an energy donor such as adamantane derivative (AMPPD) afforded a supramolecular chemiluminescent nanosystem. Upon UV irradiation of the resulting system, high efficiency chemiluminescence/fluorescence including post-luminescence was observed. This is probably due to the FRET process between encapsulated AMPPD (donor) and PE1 (acceptor). A narrow size distribution with a mean diameter of 241 nm was shown for PE1@P5A34@AMPPD nanoaggregates. TEM data confirmed the formation of spherical nanoparticles with a diameter of ~200 nm. Due to the developed surface area of PE1@P5A34 nanoparticles, their ability to encapsulate guest molecules and their excellent 1O2 generation capability, PE1@P5A34 NPs were also successfully used as nanoreactors for the photoactivatable polymerization of dopamine to polydopamine. The authors argue that the above-mentioned smart 1O2 generation system may be considered as a new insight into fabrication of a novel photothermal/photodynamic system.
Yang and co-authors[142] developed a highly efficient two-step FRET system based on pillar[5]arene-decorated metallocycle (Scheme 28). This FRET-system was constructed via a supramolecular approach using an anthracene-containing building block (L1) as the first fluorescence donor, coumarin – platinum – iodine complex (L2) as a bridge and the BODIPY-substituted pillar[5]arene P5A35 (L3). The self-assembly of the building blocks L1 and L2 led to the formation of the coordination Pt metallocycle (M1) and was driven by FRET (from anthracene (donor) to coumarine (acceptor)); the rate constant for the self-assembly was determined to be 4.2 × 10−4 s−1. Under UV excitation at a wavelength in the anthracene absorption region, the M1 metallacycle showed strong emission at 465 nm. In the next step, the host – guest complex between the coumarin moiety of L2 and P5A35 was formed to afford the metallocycle-containing M1@P5A35 assembly. As a result, two effective FRET pairs were formed, such as anthracene (L1) — coumarin (L2) and coumarin (L2) — BODIPY (from L3). The efficiency of both FRET-processes was confirmed by fluorescence studies. According to the data obtained, the gradual addition of L3 (0 – 3.0 equiv.) to the M1 solution shows a decrease in fluorescence with an emission peak at 465 nm with a simultaneous increase in fluorescence with an emission peak at 552 nm, which is attributed to FRET. Based on the UV/VIS spectroscopy data, the host – guest binding constant between the long-chain alkyl bromide L2 and the P5A moiety of P5A35 (L3) was measured to be 98.46 M−1. Based on the ratio between the fluorescence intensities of the donor (coumarin) in the absence and presence of the acceptor (P5A-appended BODIPY), the FET from coumarin to BODIPY was determined to be 79.6%.
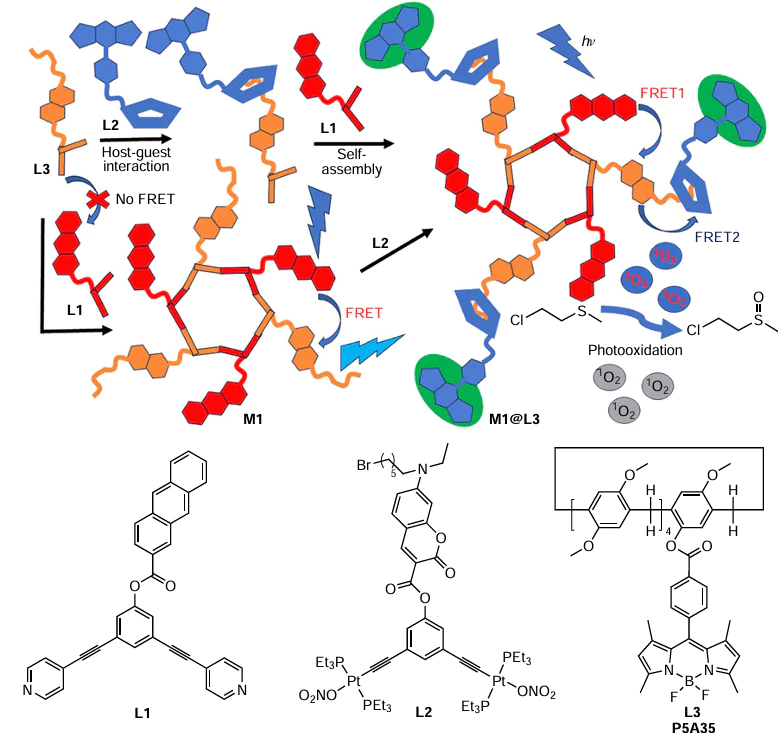
In addition to stepwise assembling of M1@P5A35, a concerted approach was realized by simply mixing three different building blocks L1, L2, and L3 in a 1 : 1 : 1 ratio to afford the target assembly.
The M1@P5A35 system was found to be the most effective for the photogeneration of singlet oxygen. Based on the ability of singlet oxygen to oxidize sulfides to sulfoxides, the photooxidation activity of M1@P5A35 together with other FRET systems was tested in a model reaction of oxidation of 2-chloroethyl ethyl sulfide (CEES, a chemical warfare agent simulant of mustard gas) to non-toxic 2-chloroethyl ethyl sulfoxide (CEESO). And after 20 h of photoexcitation (Xe lamp, 340 nm) in the presence of M1@P5A35 (1 mol.% based on CEES), the reaction provided 42.1% conversion of CEES to CEESO (for other FRET systems it was < 40%) with a turnover frequency (TOF) up to 1.4 times higher than those for other FRET systems. Such an improved photooxidation activity of M1@P5A35 was attributed to its enhanced photosensitization productivity due to very efficient FRET process. The possible role of P5A in this case is likely to provide better communication between the FRET components in the two-step FRET processes from anthracene to coumarin and then to BODIPY.
Qiang et al.[143] developed an approach to the P5A-based conjugated macrocyclic polymer network P5A37, based on the Sonogashira cross-coupling reaction of thetriflate-substituted P5A, namely, P5A36, and tris(penthynylphenyl)trazine TR1 (Scheme 29). The model polymer PTR1 and the P5A-containing polymers P5A38 were also obtained as possible carriers for the photocatalytic oxidation of sulfides to sulfoxides. According to the SEM data, the morphology of the resulting polymer depend on the nature of both components. Thus, in case of P5A37 and PTR1, the polymer particles were independent on each other, whereas in the case of P5A38, the interconnected particles were found. The BET surface areas were calculated from the N2 adsorption/desorption isotherm at 77 K. The P5A-containing polymers P5A37 and P5A38 exhibited a type II isothermal curve with the BET areas of only 23.67 and 8.56 m2 g–1, respectively. Based on the data obtained, the low BET areas of P5A37 and P5A38 were speculated to be related to the non-porous structure of pillar[5]arene. The pore volume of PTR1 was found to be 0.12 cm3 g–1, while for the P5A37 and P5A38 these values were as low as 0.007 and 0.0012 cm3 g–1, respectively, which again is due to the presence of macrocyclic cavities in P5A-based polymers.
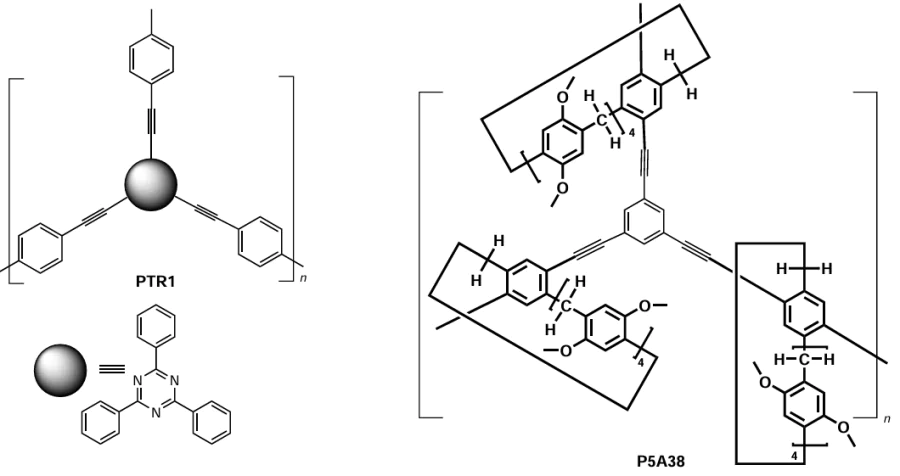
Based on the UV-vis diffuse reflectance spectroscopy (DRS) data, the authors considered the possibility of using the resulting polymers as photocatalysts under visible light irradiation in such an important reaction as the oxidation of sulfides, e.g., containing a linear alkyl nitrile moiety (compound S-1) or aryl-substituted alkyl group (compound S-2). When irradiated with a blue LED lamp (420 – 460 nm, 200 W) in open vessels, a smooth oxidation of sulfides to the corresponding sulfoxides took place (Fig. 5). The ability of the P5A cavity to accommodate linear sulfides such as S1 but not the diphenyl substituted sulfide S-2, was confirmed by model experiments. Thus, when P5A-free PTR1 was used as the catalyst, both sulfides (S-1 and S-2) showed similar conversion yields after 30 h of irradiation (92% for SO-1 and 93% for SO-2), whereas when the P5A37 catalyst was used, the conversion yields of S-1 and S-2 differed significantly: the conversion of S-1 to SO-1 was 80%, and that of S-2 to SO-2 was only 10%. Furthermore, the use of a competitive guest such as butyronitrile, in the model experiments caused a dramatic inhibition of the formation of the sulfoxides. This confirms the involvement of the P5A cavity in the photooxidation process. The model experiments confirmed that polymers can generate 1O2 under visible light irradiation and OH from the solvent H2O or dissolved O2 , and that these reactive oxygen species attack the S group in the oxidation reaction of sulfides.
![[{"id":"Vfzg-_Zjb7","type":"paragraph","data":{"text":"Formation of <b>P5A37</b> and the illustration of its use for the singlet oxygen generation in the chemoselective oxidation of sulfides"}}]](/storage/images/resized/sPve2P73odw7WrUWyDK6l1Kw8bEKoWcEdOAMUvmF_xl.webp)
9.4. Photopolymerization reactions
Interconversions of pillar[6]arenes to pillar[5]arenes or higher PAs and/or undesired linear polymers have been reported.37 Recently, Chang and co-workers[144] reported the use of per-O-ethylated pillararene P6A3 as an initiator for free-radical photopolymerization (Scheme 30). Under excitation with 295 nm light (corresponding to the absorption maximum), the absorption of P6A3 at 295 nm decreased, and a new absorption peak appeared at 350 nm. Under prolonged excitation, a decrease in absorption peak at 295 nm was observed together with a decrease of a newly generated absorption peak at 350 nm. Based on this observation, the authors concluded that P6A3 undergoes intense photodegradation under UV-light irradiation to form unstable intermediate species (Ph6(OEt)12 biradical) (see Scheme 30). To confirm the ability of the unstable intermediate species to initiate photopolymerization, different monomers such as PEG200DA, as well as other acrylic monomers, such as monofunctional (HEA), bifunctional (TPGDA) and trifunctional (TMPTA) monomers were used to give the corresponding copolymers P2 – 5 (see Scheme 30). In all the cases the photopolymerization was successful, however, for the monofunctional (HEA) and bifunctional (TPGDA) monomers, the final conversion rates were much higher than that for the trifunctional monomer (TMPTA), which was explained by the low viscosity of HEA and TPGDA. The presence of the Ph6(OEt)12 unit in the newly formed polymers was confirmed by the presence of a strong absorption of benzene ring at 295 nm in the UV absorption spectra. A cross-linked polymer was also obtained by mixing HEA and PEGDA (10 mol.% based on HEA).
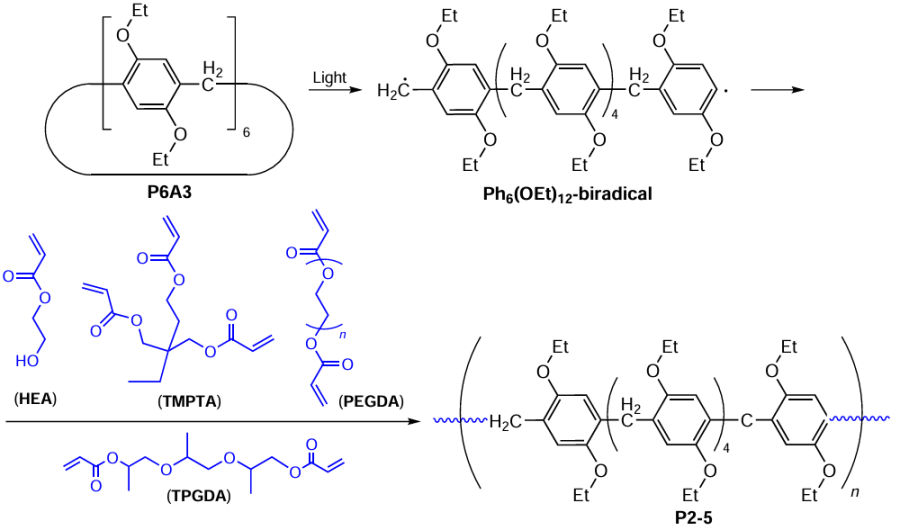
It should be noted that compared to commercial photoinitiators, such as the type I initiator benzil dimethyl ketal (BDK) and the type II initiator benzophenone (BP), P6A3 showed significant photopolymerization initiating activity and had a much lower migration rate and cytotoxicity. This report was a very first example of the use of macrocyclic molecules such as P6A3, as a photoinitiator for the reactions following the macrocycle fracture mechanism.
10. Miscellaneous reactions
Yu et al.[145] reported the preparation of MoS2/reduced graphene oxide (MoS2/rGO-NP) electrocatalytic system for hydrogen evolution. The synthesis was based on the reaction between P5A11, graphene oxide (GO), Na2MoO4 and L-cysteine. Due to the presence of five positively charged moieties on each side, P5A11 was able to interact with the negatively charged surface of GO. This helps to mitigate the charge incompatibility between MoO42− and GO, which favours better hybridization. The introduction of P5A11 rendered the hybrid material a graphene-like morphology; the MoS2 sheets with cracked fringes and increased interlayer spacing are well dispersed over rGO, providing more exposed active edge sites for the hydrogen evolution reaction (HER). As an electrocatalyst for HER, MoS2/rGO-P5A11 exhibits much higher current density with lower Tafel slope of 44.5 mV dec–1 and greatly improved kinetics than model materials, such as MoS2 and MoS2/rGO. The authors believe that such an outstanding electrocatalytic performance of MoS2/rGO-P5A11 makes it promising electrocatalyst for cost-effective hydrogen production.
Wu et al.[146] obtained an organic-inorganic hybrid material based on decamethylsubstituted pillar[5]arene P5A13 and titanium dioxide (TiO2) by a convenient sol – gel method starting from tetrabutyl titanate. The resulting porous organic-inorganic hybrid material was loaded with Pt nanoparticles to form a supramolecular organic-inorganic assembly, which had a good stability and an enhanced photocatalytic efficiency for hydrogen production under visible light irradiation (λ > 420 nm). The optimized catalyst P5A13@TiO2@Pt (5.2 wt.%) exhibited a photocatalytic hydrogen production rate of 1736 mmol g−1 h−1. In addition, this catalyst had good long-term durability after 10 cycles of 50 h testing. These results suggest that pillararene/TiO2 functional materials are promising hybrid photocatalysts for the visible light-driven hydrogen production.
Hydrazones are important building blocks for synthetic organic chemistry and for bioconjugation strategies.[147][148] The hydrolysis of hydrazone-based bonds is used in medical biotechnology to couple drugs to targeted antibodies, e.g., antibodies against a certain type of cancer cells.[149] Therefore, approaches for the selective hydrolysis of hydrazones are in high demand. For instance, Kosiorek et al.[150] developed an approach for the selective hydrolysis of arylhydrazones using pillararene carboxylic acids P5ACO2H and P6ACO2H. Similarly to the above-described synthesis of artificial enzyme, the design of P5ACO2H and P6ACO2H was aimed on stabilization of the substrates, namely, arylhydrazones in the PA cavity and their selective and effective hydrolysis. It was assumed that the stabilization of substrates due to the electrostatic interactions of the carboxylic groups with proximal C=N moieties would facilitates the hydrolysis to aldehydes. The authors suggested that based on the dimensions of P5ACO2H and P6ACO2H (Fig. 6) and arylhydrazones (Table 1), the hydrolysis of AH3 should be most efficient with P5ACO2H, whereas the selectivity and efficiency should be lower in the case of P6ACO2H due to the larger cavity size.
![[{"id":"RIlQ4BzGCT","type":"paragraph","data":{"text":"Pillararene-based carboxylates for the hydrolysis of arylhydrazones"}}]](/storage/images/resized/wISPkOiSv3a7PPjfZJ3OzWZYt3nU04Nl6KGzMU7H_xl.webp)
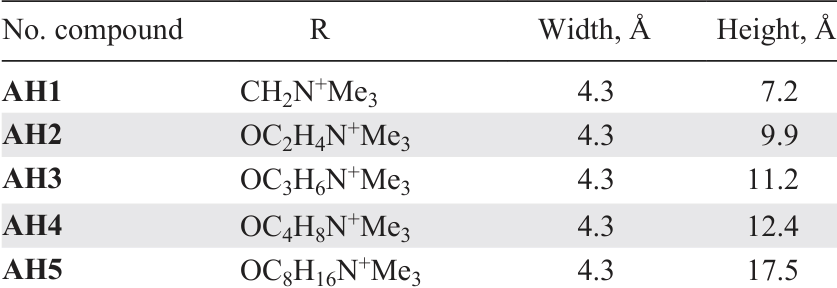
The experimental data obtained for P5ACO2H were consistent with the suggestion that the highest reaction rate was observed with AH3. Surprisingly, in the case of P6ACO2H, the highest rate was observed with AH2, while similar reactivities were observed for the substrates bearing the shortest (AH1), medium (AH4) and the longest (AH5) substituents. According to the authors, the presence of a large and hydrophobic macrocyclic cavity (width ≈ 7.5 Å) in P6ACO2H provided more degrees of freedom for the substrates AH1-5 to pass through this cavity, and the distance between the both rims and the hydrazone group became shorter, making the substrates more reactive. In P5ACO2H, the cavity is narrower (width ≈ 5.0 Å), and due to the presence of the bulky quaternary ammonia moiety (size ≈ 4.3 Å), substrates AH1-5 remain outside, and the Coulombic stabilization is weaker in polar media, causing the ammonia group to remain solvated, and the entropy of the hydrolysis process to decrease. In other words, the ability of the arylhydrazone substrates to pass through the macrocyclic cavity directly affects the reactivity of the hydrazones. For example, in the case of P5ACO2H the transport of hydrazones through the cavity is hampered, which made them to be positioned more firmly, and thus large differences in the reactivities of AH1-5 were observed. In the case of P6ACO2H, the different conformations of the substrates are possible and the substrates themselves pass through the cavity unhindered.
11. Conclusion
Pillararenes can be considered as promising catalysts and/or catalyst carriers for the most important/common organic reactions due to the high stability and rigid shape of these macrocycles, as well as the ability for the functionalization of both ends of the PA core to achieve/modulate the target properties. These properties include solubility in aqueous or organic media, ability to accommodate various guest molecules via encapsulation within the PA cavity or via coordination at the periphery of the PA core, and many others. However, there are several issues that need to be addressed. Firstly, a better understanding is needed to evaluate the synergy between the size of the PA cavity and its ability to accommodate/stabilize the reaction synthons, intermediates or transition states. A deeper insight into the influence of the nature of functionalities on the PA core on its catalytic activity in the certain types of reactions (hydrolysis, reduction, oxidation, dehalogenation, elimination, etc.) is necessary. Better control is needed to achieve size-, chemo-, regio- or, most importantly, stereoselectivity for PA-catalyzed reactions, for example by appropriate selection of size of the PA cavity or the targeted functionalization of its periphery. To date, only a few examples[79][80][89][90][102][143][150] have evaluated the effect of PA cavity size on selectivity and/or yield and reaction rate. Finally, it is necessary to explore the catalytic ability of PAs towards other types of reactions, e.g., fundamental types of reactions such as the C – C bond-forming reactions (aldol condensation, Witting reaction, Michael reaction, etc.), heteroatom bond-forming reaction (hetero-Michael addition, the Shiff base formation, etc.) as well as Pd-catalyzed processes and, as an example of modern synthetic approaches, photocatalyzed[151] reactions. Furthermore, the stability of PAs under certain conditions still remains an issue, which may hamper an application of PA-based catalysts in the presence of strong acids or under highly acidic conditions.
Acknowledgement. This research was financially supported by the Ministry of Science and Higher Education of the Russian Federation (State Assignment for the IOS UB RAS, No. Gos. Reg. 124020100137-7), the Council for Grants of the President of the Russian Federation, Project No. NSh-1223.2022.1.3 (Chapter 10) and the Russian Science Foundation, Grant No. 23-23-00538 (Chapter 6).
12. List of abbreviations
ABTS — 2,2'-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt,
AIE — aggregation induced emission,
BET — Brunauer–Emmett–Teller method,
BODIPY — 4,4-difluoro-5H-4λ5-dipyrrolo[1,2-c:2',1'-f]-[1,3,2]-diazaborinin-4-ylium-5-ylide,
CAC — critical aggregation concentration,
CB — conductivity band,
CEES — 2-chloroethyl ethyl sulfide,
CEESO — 2-chloroethyl ethyl sulfoxide,
DFT — density functional theory,
DLS — digital light scattering,
DNPP — 2,4-dinitrophenylphosphate,
DRS — diffuse reflectance spectroscopy,
EDX — energy-dispersive X-ray spectroscopy,
EIS — electrochemical impedance spectroscopy,
EsY — Eosin Y,
FRET — Föster resonance energy transfer,
GO — graphene oxide,
HAADF-STEM — high angle annular dark field scanning transmission electron microscopy,
HER — hydrogen evolution reaction,
MB — methylene blue,
MO — methyl orange,
NiR — Nile Red,
NHC — N-heterocyclic carbene,
NLDFT — non-localized DFT,
NPs — nanoparticles,
PA — pillararene,
PNIPAM — poly(N-isopropylacrylamide),
POM — polyoxometalate,
PXRD — powder X-ray diffraction,
RAFT — reversible-addition-fragmentation chain-transfer polymerization,
rGO — reduced graphene oxide,
RhB — Rhodamine B,
RIR — restricted intramolecular rotation,
ROS — reactive oxygen species,
SCE — saturated calomel electrode,
SEM — scanning electron microscopy,
TB — Tröger’s base,
TEM — transition electron microscopy,
TOF — turnover frequency,
TON — turnover number,
TPE — tetraphenylethylene,
VB — valence band,
XPS — X-ray photoelectron spectroscopy,
XRD — X-ray diffraction.
References
![para-Bridged symmetrical pillar[5]arenes: their Lewis acid catalyzed synthesis and host-guest property.](/storage/images/resized/iLiQsFqFaSEx6chlGQ5fbAwF6VYU3WWa08hkss0g_small_thumb.webp)


![Recent advances in pillar[n]arenes: synthesis and applications based on host–guest interactions](/storage/images/resized/leiAYcRDGTSl5B1eCnwpSGqmDEUEfDPPoYisFGhT_small_thumb.webp)


![Supramolecular Vesicles Based on Amphiphilic Pillar[n]arenes for Smart Nano-Drug Delivery](/storage/images/resized/5YZtvLvkPZuc2JHOaZsjCvGSHFCuC3drUwN3YAc5_small_thumb.webp)

![Copillar[5]arene Chemistry: Synthesis and Applications](/storage/images/resized/xqixcltwJYe6H8Uco2JbAFfIOzt7UNKH0OcPOPzO_small_thumb.webp)
![Synthesis and host-guest properties of pillar[6]arenes](/storage/images/resized/voXLqlsvTwv5p3iMQ8Dhs95nqB4AXOG7Taj7G4ra_small_thumb.webp)
![Synthesis of 1,4-Bis(n-propoxy)pillar[7]arene and Its Host-guest Chemistry](/storage/images/resized/6EJSsoiR4fNMNMPFBpMh5yKuYKcPXJDQKvKVeEMy_small_thumb.webp)

![Pillar[5]arene Based [1]rotaxane Systems With Redox-Responsive Host-Guest Property: Design, Synthesis and the Key Role of Chain Length](/storage/images/resized/4QWA67eqfcfyOiA8Wk7YnqroHFqQbTsmDJUYTCTg_small_thumb.webp)
![Covalent organic frameworks bearing pillar[6]arene-reduced Au nanoparticles for the catalytic reduction of nitroaromatics](/storage/images/resized/LsKy6OnmmmRGcAU6CZgWQvNiP1polbaSLNrN7zqj_small_thumb.webp)