


Keywords
Abstract
The urgency of improving the efficiency of known methods and developing new ones for the synthesis of unsaturated silanes is caused by the need to obtain useful compounds, including biologically active ones, on their basis. This review considers methods for the preparation of unsaturated silanes, their transformations and prospects for their use as precursors in organic and organoelement synthesis. A large number of substituted organosilicon products are given, including those capable of further functionalization and the formation of heterocycles, including silaheterocycles. The bibliography includes 205 references.
1. Introduction
Unsaturated organosilanes are convenient and important reagents in both organic and organoelement chemistry. The substitution of hydrogen or carbon atoms in an organic molecule with silicon activates the substrate for many transformations due to steric and electronic effects created by Si-containing substituents when introduced into the molecule.
The use of silanes as reagents allows the formation of organic frameworks with the introduction of an organosilyl group into the molecule, which can significantly alter the reactivity of the molecule, as well as acting as a protecting or leaving group (in desilylation reactions) and being bioisosteric to various functional groups.
Si-functionalized organic molecules are easily analysed by NMR spectroscopy, which greatly facilitates the determination of the structure of the resulting products. The unsaturated silanes are compounds with one or more vinyl, allyl or alkynyl groups at the silicon atom. This review focuses on the properties of the first two classes of compounds. Commercially available allylsilanes are allyl(trimethyl)silane, allyl(triethyl)silane, allyl(triisopropyl)silane, allyloxy(trimethyl)silane, diallyl(dimethyl)silane. Vinylsilanes are represented by a wider range — vinyl(trimethyl)silane, vinyl(triethyl)silane, vinyl(triisopropyl)silane, vinyl(dimethyl)phenylsilane, vinyl(methyl)diphenylsilane, vinyl(triphenyl)silane, vinyl(triethoxy)silane and 1,3-divinyl-1,1,3,3-tetramethyldisiloxane.
Due to the electronegativities of the silicon atoms (1.9) and carbon atoms (2.5), the Si – C bond is polarised towards the carbon atom, which gives carbon a certain nucleophilic character. The energy of the Si – C bond is ~ 76 kcal mol–1, which is significantly lower than the energy of the C – C bond (83 kcal mol–1).[1] The Si – C bond is quite stable to homolytic cleavage, whereas the C – H bonds in organosilicon compounds are more reactive to free radicals than the Si – C bonds. According to the Pauling electronegativity scale, C – Si bonds are ~ 12% ionic and therefore more susceptible to reactions involving the ionic mechanism.[1]
The β-effect of the silicon atom, which consists in stabilising the carbocation formed in electrophilic addition reactions and which largely determines the regioselectivity of the reaction depending on the electrophile attached, is most important for understanding the reactivity of vinyl silanes. The resulting conjugation significantly weakens the Si – C bond, which favours the desilylation processes that often accompany reactions of unsaturated silanes and to which a separate chapter of this review is devoted. Vinylsilanes readily react with a wide range of electrophiles to form both addition and desilylation products (substitution by addition/elimination).
The development of methods for the synthesis of unsaturated silanes is also inextricably linked to the development and understanding of the capabilities of catalytic systems.[2-9] For example, Pd- and Ni-catalysed cross-coupling of silicon-containing electrophiles (silyl halides, silyl triflates) with C-nucleophiles has recently been actively developed. The silyl – Heck, silyl – Negishi and silyl – Kumada – Koryu reactions allow the preparation of saturated and unsaturated organosilanes under mild conditions.[10]
Despite the fact that hydrosilylation offers almost unlimited possibilities for obtaining silicon-containing products due to the wide range of substrates and transformations that can be used, and has been extensively studied, a large number of papers on this topic have appeared in the literature in recent years.[11-15] It is the most popular and widely used route to organosilicon compounds, both in laboratory syntheses and in industrial processes.[8] The method is used not only for the preparation of functionalized silanes [16-18] and polymeric compounds,[19] but also for the production of composite materials and surface functionalization. Hydrosilylation also plays an important role in organic synthesis, in particular as a selective method for the reduction of various functional groups (carbonyl, nitrile, imine).[20]
In recent years, several reviews have been published on the influence of silicon-containing substituents on reactivity. Reviews [21, 22] have considered the importance of the β-effect of the silicon atom in unsaturated silanes in reactions involving the 1,2-silyl shift. Acyl and amidoylsilanes undergo photochemically attack or thermally initiated Brook 1,2-rearrangement to form basic and nucleophilic intermediate siloxycarbenes, which exhibit unique reactivity, including C – H bond insertion, the ability to react to form 1,2-dicarbonyl compounds and 1,4-coupling reactions, and cross-coupling reactions in the presence of transition metals, opening the way to hard-to-reach silanes.[23-26] Recently, the reactions of β-silyl acrylates (R3SiCH=CHCOOR1) [27] and β-silyl methylene malonates (R3SiCH=C(COOR1)2),[28] which are efficient reagents in the synthesis of natural products and bioactive molecules and in hydrodimerisation reactions, organocatalytic asymmetric Michael addition reactions, inter- and intramolecular Diels – Alder reactions, 1,3-dipolar cycloaddition reactions and many others have been reviewed in detail. Recent advances in the use of various silanes and siloxanes in the organic synthesis and preparation of functional polymers and materials are discussed in monographs [29, 30] and reviews.[31-35]
The chemistry of silanes has a long history, despite the fact that organosilicon compounds are unknown in nature; however, the methods of C – Si bond formation, the study of its reactivity and its effects on functional groups continue to be actively developed.The motivation for writing this review was the fact that unsaturated organosilicon compounds have been undeservedly neglected in reviews of the last 30 years and the activity of organosilicon chemistry research slightly decreased in the early 21st century. It is only in the last decade that an increasing interest in the chemistry of unsaturated organosilicon compounds has been observed, and one of the aims of this review is to further stimulate research in this area.
2. Synthesis of unsaturated silanes
2.1 Hydrosilylation of alkynes
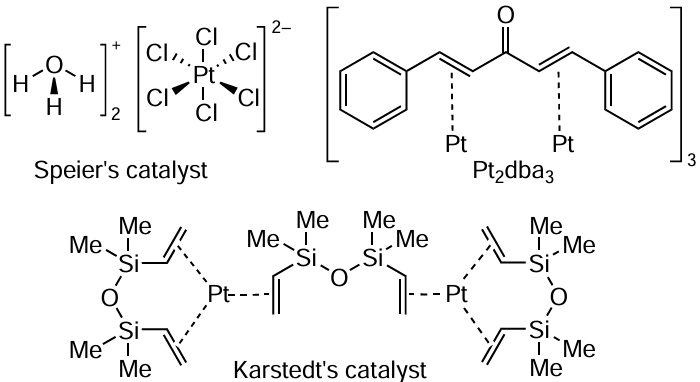
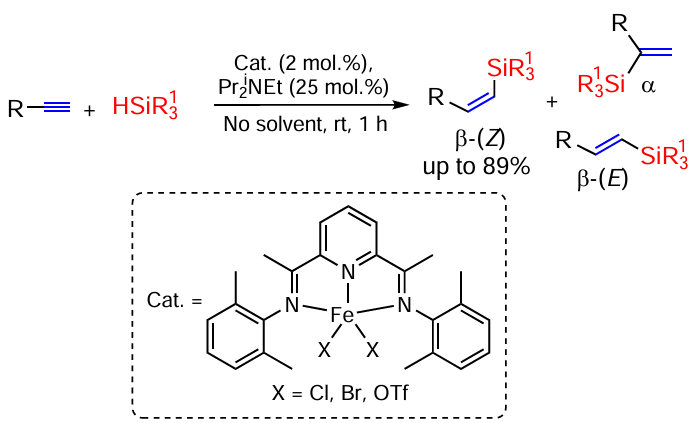
Hydrosilylation of C=C and C≡C bonds is one of the most important reactions for the synthesis of organosilicon compounds. These reactions began to be actively developed with the introduction of the Speier catalyst (H2PtCl6) [36] and the Karstedt catalyst (Pt(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane) in the middle of the 20th century.[37] Later, the use of the Pt(0)tris(dibenzylideneacetone) complex (Pt2dba3) was considered for the hydrosilylation of alkenes and alkynes (Scheme 1).[38]
The use of new heterogeneous catalysts in hydrosilylation reactions has recently been described, mainly alkenes and, to a lesser extent, alkynes and allenes.[39] Reactions of these substrates leading to unsaturated silanes and related to the 2017 – 2024 work are the subject of this section.
A model scheme for the hydrosilylation reaction of terminal alkynes is shown in Scheme 2.
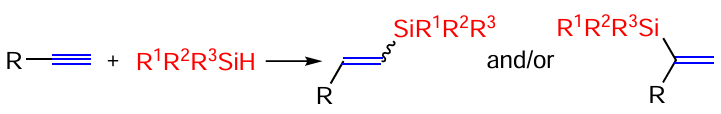
The reaction can proceed as α- or β-silylation, and in the latter case both Z and E isomers of vinylsilanes can be obtained. Thus, the reaction of acetylenes (R1 = Alk, Ar) with phenylsilanes PhR2SiH2 (R2 = H, Me, Ph) in the presence of Co(acac)2 complexes with different ligands proceeds as β-silylation with an average yield of 82% and selective (up to 99 : 1) formation of E-isomers of the products.[40] The formation of β-silylated adducts is also observed in the reaction of terminal (hetero)aromatic and aliphatic alkynes with the phenylsilane PhSiH3 in dimethoxyethane solution at 60°C in the presence of the diphosphine copper complex Cu(MeCN)4PF6 · Xantphos (the structure of the ligand is shown in Scheme 3), which also produces E-isomers.[41] E-isomers of β-silylstyrenes ArCH=CHSiR3 are also formed when styrenes react with silanes R3SiH in THF at 70°C in the presence of a diphosphine complex of manganese tricarbonyl,[42] but in this case the reaction proceeds as a hydrosilylation/dehydrogenation (see Scheme 3).
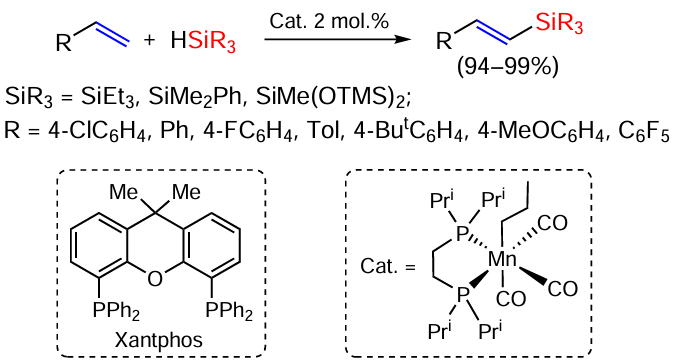
This approach (hydrosilylation/dehydrogenation) makes it possible to obtain vinylsilanes not from alkynes, but from alkenes, including functionalized ones, e.g., from enamides.[43] The reaction with various silanes proceeds in the presence of tert-butyl peroxybenzoate, does not require catalysis by metal complexes and shows high stereoselectivity, up to stereospecific formation of E-isomers.[44]
Reversal of stereoselectivity (formation of Z-isomers of vinyl silanes) occurs when terminal alkynes react with (Me3SiO)2SiHMe in the presence of the rhodium complex RhHCl(CO)(H2IMes)(PCy3),[45] with Et3SiH in the presence of monothiolate-bridged binuclear rhodium complexes, or with (Me3Si)3SiH and Eosin Y as a photocatalyst (Scheme 4).[46]
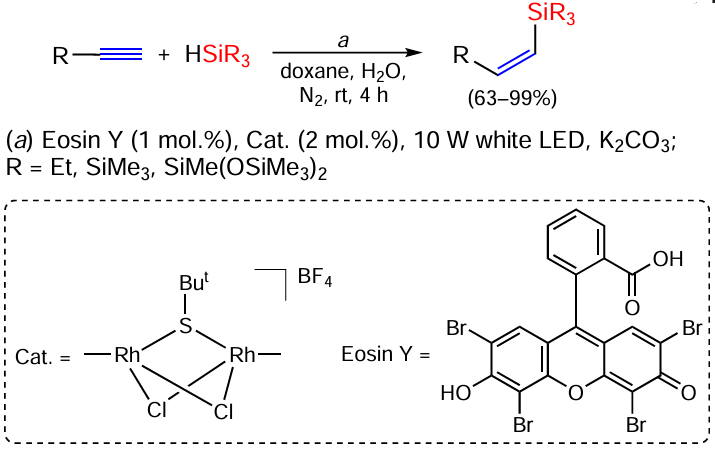
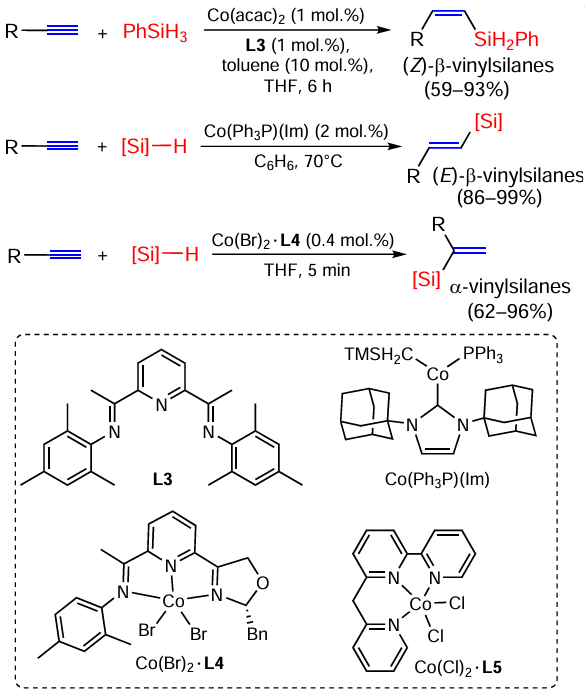
The hydrosilylation of terminal alkynes in the reaction with Ph2SiH2 on cobalt phosphiniminopyridine complex (catalyst A) is highly (Z)-β-selective.[47, 48] However, the reverse regioselectivity (formation of α-substituted vinylsilanes) is observed on the cobalt catalyst B. Thus, alkyl- and arylacetylenes react with primary, secondary and tertiary silanes in the presence of Co(II) complexes to give α-vinylsilanes in high yields under mild conditions (40°C) (Scheme 5).[49]
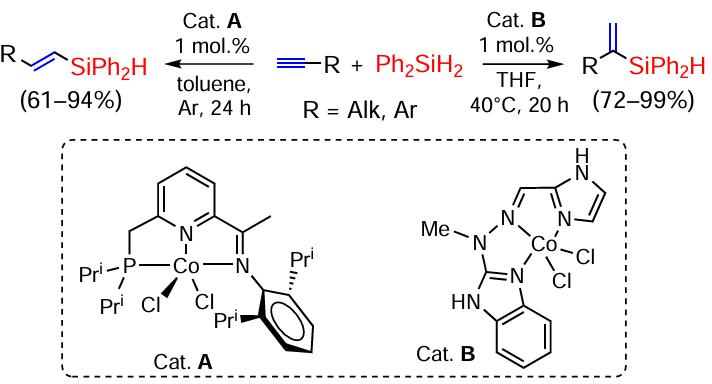
As noted by Deng et al.,[50] the [49] is a rare example of α-silylation of terminal alkynes with different silanes, and for tertiary silanes this catalyst often fails. Zhang et al.[51] proposed the use of the binuclear cobalt carbonyl complex [(IPr)2Co2(CO)6] (IPr = 1,3-di(2,6-diisopropylphenyl)imidazol-2-ylidene) for such silanes and showed it to be effective for alkyl and arylacetylenes and for a wide range of tertiary silanes.
The same regioselectivity is observed on other cobalt catalysts for a wide range of arylacetylenes in reactions with PhSiH3 .[51, 52] However, on Co(acac)2 doped polymers with different ligands, terminal aryl(hetaryl)acetylenes are silylated regio- and stereoselectively under the action of PhSiH3 , giving E-isomers of the adducts in yields of 42 – 83%.[53]
The platinum complex with heterocyclic carbene shows high activity as a catalyst for the hydrosilylation of terminal alkynes with a wide range of secondary silanes. Varying the ratio of reagents allows mono- and disubstituted products to be obtained (Scheme 6).[54]
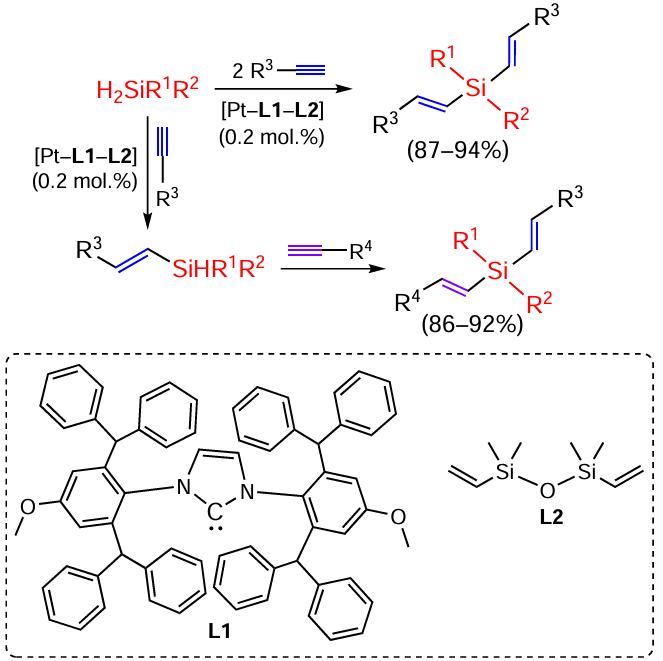
For a large number of terminal acetylenes, hydrosilylation on Rh/NHC/COD complexes yields mainly E-isomers of β-silylation products, the content of α-silylation products does not exceed a few percent.[55]
Mono- and disilylation of terminal acetylenes proceeds smoothly and with high α-selectivity on binuclear cobalt carbonyl complexes with bulk NHC ligands (Scheme 7).[56, 57]
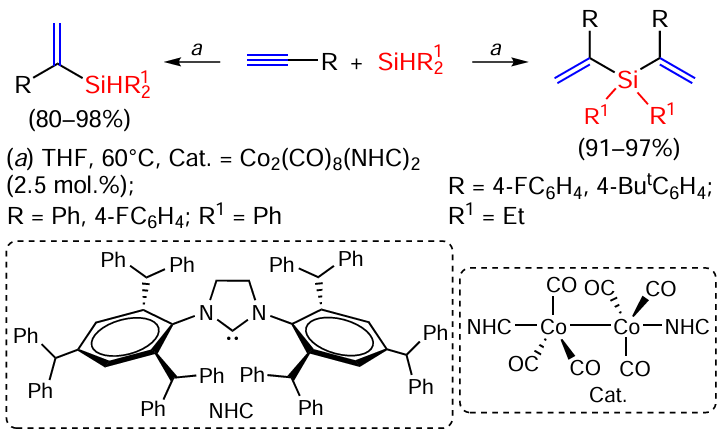
Interesting results were obtained by studying the hydrosilylation reaction of 1-octyne C6H13C≡CH, tolan PhC≡CPh and 1-phenyl-1-propyne PhC≡CMe with silanes HSiMe2Ph and HSiEt3 on rhodium catalysts on graphene support.[58] In the reaction of 1-octyne and tolan with HSiMe2Ph, the yield of the cis-isomer of the β-silylation product reaches 99%, while the contents of the trans-isomer and the α-silylation product vary between 0 – 12 and 0 – 6%, respectively. In contrast, in the reaction of 1-octyne with HSiEt3 , the content of the latter two products can reach 27 and 20%, respectively. The reaction of HSiMe2Ph with tolan proceeds almost exclusively as a cis-addition, but with 1-phenyl-1-propyne the Z : E-ratio of the vinylsilane isomers varies from 20 : 80 to 54 : 46. (E)-β-Selectivity of acetylenes hydrosilylation on different cobalt complexes varies from low (3%) to high (65%).[40, 47, 53, 59, 60] As for hydrosilylation on iron complexes, already in 2012 the FeH(CO)(NO)(PPH3)2 catalyst was proposed for the hydrosilylation of internal alkynes and it was shown that the E/Z-selectivity of the reaction depends on the silane structure.[61] Terminal alkynes were (Z)-β-selectively hydrosilylated on an iron complex with bis(imino)pyridine (Scheme 8),[62] but the use of a xanthene diphosphine iron complex (the complex was prepared from FeCl2 and Xantphos, the structure of the ligand is shown in Scheme 3) allowed the selective preparation of (E)-β-isomers in moderate to high yields (44 – 89%).[63]
All of the above possible directions for the reaction of terminal alkynes with silanes have been investigated for a wide range of substrates in reactions with HSiMe2Ph, HSiMePh2 and HSiEt3 on a rhodium complex containing methylimidazole and carboxylate moieties (see Scheme 8).[64]
Typically, the predominant direction, up to > 99%, is the formation of the β-(Z) product. However, the result depends on the silane structure: for example, in the reaction of Et3SiH with AnC≡CN the β-(E) product content reaches 81%, whereas in the case of tBuC≡CN the β-(E) and α-product contents are 33 and 21%, respectively, and 33% of the hydrosilylation/dehydrogenation product is formed, although in other cases its content is not higher than 3 – 4%.[64] A similarly high β-(Z)-selectivity of hydrosilylation has been shown by the same authors for catalysis with triazole rhodium catalysts.[65] At the same time, referring to previous work from 1995 to 2015, the authors note that in most cases hydrosilylation reactions are characterised by β-(E)-selectivity and that obtaining β-(Z)- or α-isomeric vinylsilanes is a challenging task.
A vivid example of the critical dependence of the direction of hydrosilylation on the nature of the ligand in the complex with the same metal (Co) is the [66] on the hydrosilylation of terminal alkynes with phenylsilane and comparison of the results with the data of other authors [67-70] (Scheme 9).
The hydrosilylation reaction of terminal alkynes RC≡CH with the silane Ph2SiH2 on tridentate cobalt pyridine complexes (see Scheme 9, Co(Cl)2 .L3) proceeds as α-silylation and with diphenylacetylene the E-isomer of the adduct PhCH=C(Ph)SiHPh2 is formed.[71]
A method for the hydrosilylation of 2-alkynes over a cobalt catalyst with π-bond migration has been proposed, leading to α-vinylsilanes in moderate to near quantitative yields (Scheme 10).[72]
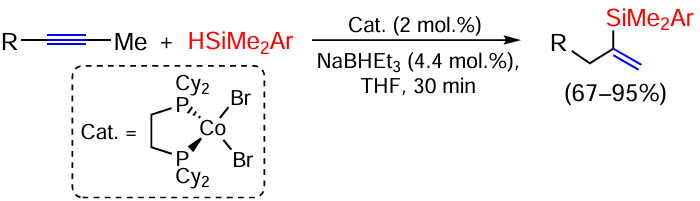
Hydrosilylation of unsymmetrical aryl-substituted alkynes ArC≡CR1 (R1 = TMS, Bu) by silanes HSi(OEt)3 and HSiPh3 on a low-valent Co(I) complex (HCo(PMe3)4) proceeds selectively to the α-position to the aryl group to form vinylsilanes R23SiC(Ar)=CHR1.[73]
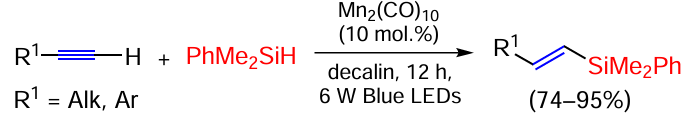
Photoactivation of the Si – H bond in silanes in the presence of manganese carbonyl Mn2(CO)5 catalyst provides high anti-Markovnikov regioselectivity and stereoselectivity, leading to Z-isomers of vinylsilanes in high yields (Scheme 11).[74]
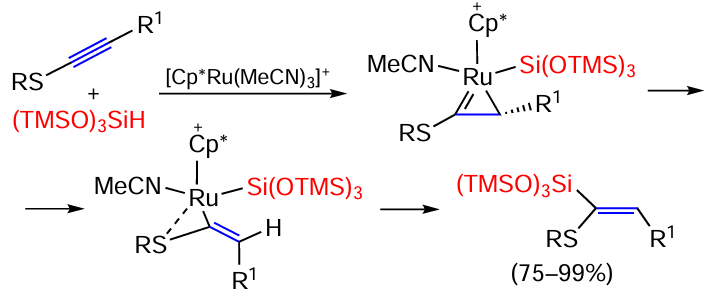
RS – C≡C – R1 thioacetylenes are hydrosilylated in up to quantitative yields using tris(trimethylsiloxy)silane (Me3SiO)3SiH over a ruthenium catalyst so that the silyl substituent is inserted in the α-position to the sulfur atom (Scheme 12).[75]
A unique reversal of regioselectivity was observed for the hydrosilylation of ArC≡CH alkynes by RSiH3 silanes depending on the aryl groups in the 2,9-positions of FeCl2 complexes with 1,10-phenanthroline (Scheme 13).[76]
Despite the successes in the field of acetylene hydrosilylation on non-noble metals (Fe, Co), noble metals (Ru, Rh, Pd, Pt) continue to attract the attention of chemists due to their high efficiency and often minimal loading, which largely compensates for their high cost. Thus, the study of the regio- and stereoselectivity of the hydrosilylation of functionally substituted terminal alkynes and disubstituted acetylenes, including those containing Me3Si group at the triple bond, on platinum nanoparticles under the action of silanes containing chlorine atoms, alkyl and alkoxy groups has shown that the reaction proceeds with preferential or exclusive formation of β-(E)-silylation products with yields close to quantitative. The content of α-(E)-silylation products increases to 16% in the reaction of Et3SiH with some arylacetylenes and reaches 70% in the reaction of Et3SiH with propiolic acid.[77] The reaction of R3SiH silanes with terminal alkynes in the presence of 3-iminoisoindolin-1-one platinum complexes leads to α- and β-(E)-silylation products. Yields depend on the structure of the reagents and vary from moderate to quantitative, with regioselectivity varying from 81 : 19 to 5 : 95.[78] The yield reaches 94% for disubstituted acetylenes and the ratio of α-(E)- to β-(E)-products also varies from 81 : 19 to 45 : 55. The catalyst is effective at concentrations up to 0.01 mol.%, below which the yields drop sharply.[78]
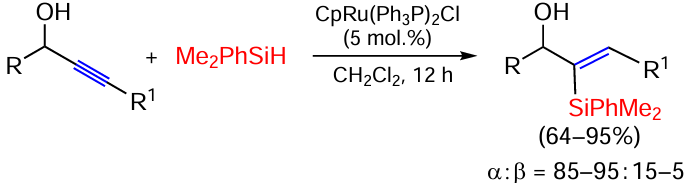
Styrenes react with silanes on the rhodium catalyst [Rh(COD)Cl]2PPh3 via the silylation/dehydrogenation pathway, producing (E)-isomers of vinylsilanes Ar – CH=CH – SiR3 in yields up to 95% and with high selectivity.[79] The same authors showed that the reaction on a binuclear rhodium complex with 1,3-bis(diphenylphosphino)propane proceeds similarly, with the content of saturated hydrosilylation products varying from 0 to 7%.[80] Previously, they also found that propargyl alcohols react with Me2PhSiH on the ruthenium diphosphine catalyst CpRu(Ph3P)2Cl at room temperature with high yields and regioselectivity (Scheme 14).[81] The regioselectivity is due to the polarisation of the triple bond to the OH-containing substituent and the attack of the silicon atom on the acetylated carbon atom with higher electron density.
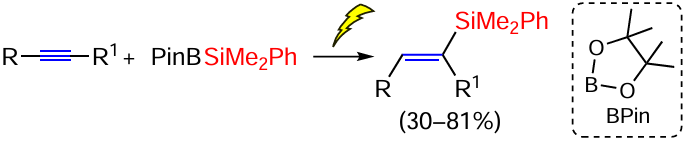
An interesting example of hydrosilylation of disubstituted acetylenes under the action of the boron-silicon reagent PinB – SiMe2Ph (Pin = pinacolin, Suginome reagent) by electrochemical activation with selective formation of E-isomers of vinylsilanes was found (Scheme 15).[82]
The synthesis of vinylsilanes by electrochemical hydrosilylation/decarboxylation of acrylic acids has also been described.[83] The reaction proceeds under mild conditions, in contrast to the previously proposed method for the preparation of the same silanes in the traditional oxidative system tBuOOH/CuCl under heating (Scheme 16).[84] In both cases, the reactions are stereoselective.
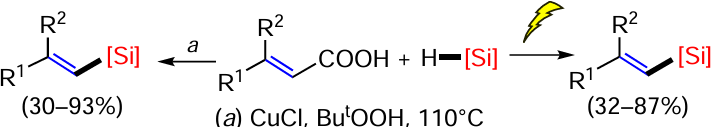
Not only hydrosilylation, but also the reaction with disilanes can lead to vinylsilanes by cleavage of the Si – Si bond and addition of two silyl groups to the C≡C bond, and the addition can proceed both intermolecularly and intramolecularly (see references in [85]). Naka and Kobayashi [85] carried out an unprecedented trans-disilylation of alkynones under mild conditions in the presence of norbornadiene (nbd) rhodium tetrafluoroborate complex. The reaction is stereoselective and gives good yields (Scheme 17).
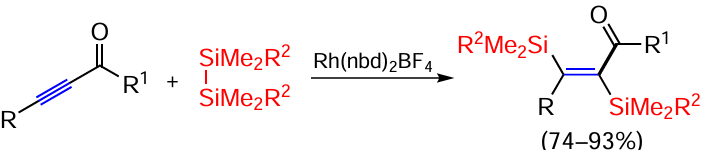
The reaction of o-bromostyrenes with hexamethyldisilane Me3SiSiMe3 catalysed by the palladium diphosphine complex Pd(OPiv)2Xantphos leads to the substitution of both the bromine atom and the vinyl hydrogen atom and opens the way for further functionalization of the resulting products (Scheme 18).[86]
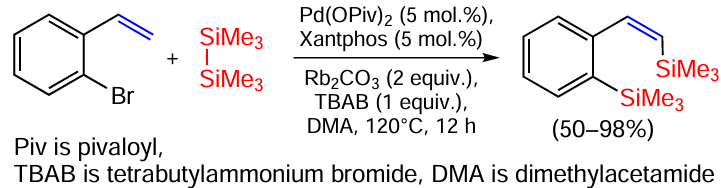
Acetylene carbonates react with disilanes catalysed by gold nanoparticles on zirconium dioxide with decarboxylation and dephenylation leading to allenylsilanes in yields up to 93% (Scheme 19).[87] In the same work, alkyl, allyl and benzylsilanes were obtained in high yields.
The cis-addition of silyl and alkyl groups to the C≡C triple bond in the three-component reaction of diarylacetylenes, silyliodides R3SiI and EtZnI (or Et2Zn) catalysed by the palladium complex (PPh3)2PdCl2 proceeds with near quantitative yields. Сatalysis by the complex (DrewPhos)2PdI2 shows the same stereoselectivity. However, replacing even one aryl substituent in the ligand with a tert-butyl group (complex (JessePhos)2PdI2) leads to a complete reversal of the stereoselectivity (Scheme 20).[88] This reversal is due to the significantly higher sterical hindrance of the DrewPhos catalyst than JessePhos.
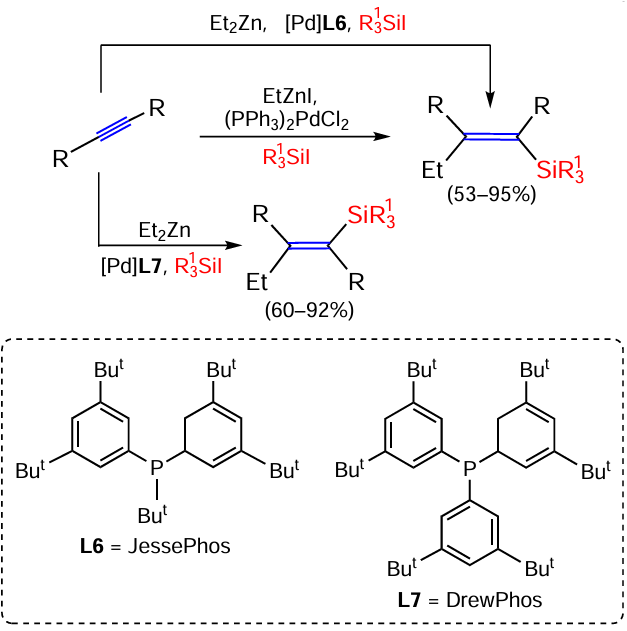
Note that it has previously been shown by Watson et al. that the (JessePhos)2PdI2 complex effectively catalyses the Heck silyl-type reaction of terminal alkenes with Me3SiI, leading to double bond migration and the formation of allylsilanes Me3SiCH2CH=CHR,[89] as well as unsaturated organosilicon heterocycles such as 1,1-disubstituted 2-methylidene-1-silolane, -silinane and -silepane.[90]
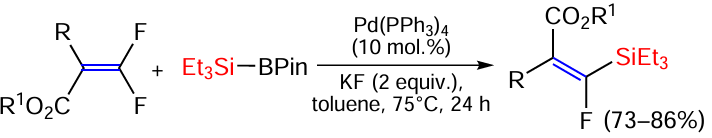
The above boron-silicon reagent PinB – SiEt3 participates in a Pd(PPh3)4 catalysed defluorosilylation reaction with β,β-difluoroacrylates to give β-fluoro-β-silylacrylates with excellent stereoselectivity (> 99 : 1) (Scheme 21).[91]
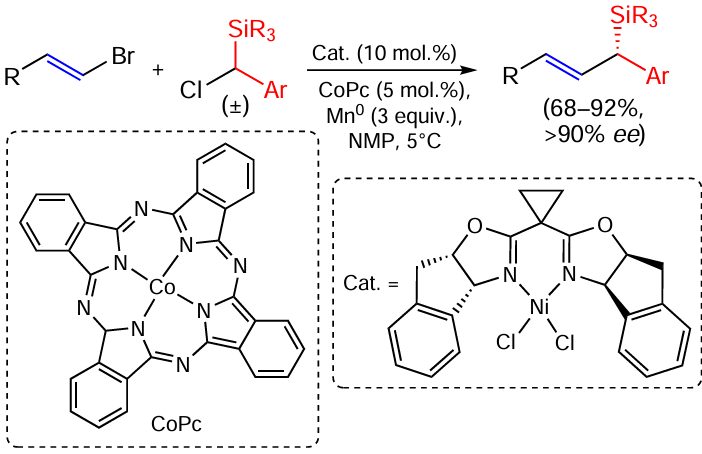
Coupling reaction of vinyl bromides with α-chloro-substituted organosilanes catalysed by an optically active nickel oxazoline complex (Scheme 22) has given optically active allylsilanes with a silicon atom at the asymmetric centre.[92] The reaction proceeds under mild conditions and allows the introduction of various functional groups. The resulting chiral silanes undergo various transformations, including intramolecular Hosomi –Sakurai reactions, with high asymmetric induction.
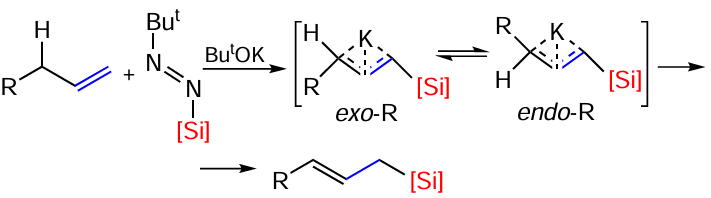
Recently, a method has been described for the silylation of allylic C(sp3) – H bonds under mild conditions without transition metal catalysis, allowing the preparation of allylsilanes from terminal olefins. The study of the reaction mechanism revealed the key role of interconversions of exo- and endo-isomers of allylpotassium (Scheme 23).[93]
Although the Pd-catalysed coupling of vinyl halides with silyl derivatives has been known for at least 40 years, non-noble metal complexes (Cu, Ni) have only recently been used in these reactions.[94-96] Of note is the copper triflate-catalysed silylation of vinyliodonium salts of silyl-substituted zinc chloride to form vinylsilanes in high yields. The reaction is insensitive to oxygen and humidity and allows the introduction of various functional groups into the molecule (Scheme 24).[97]
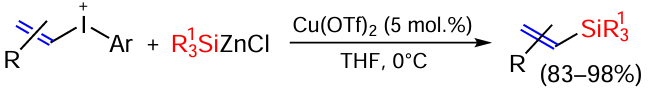
2.2 Hydrosilylation of dienes, allenes and enynes
The reaction of 2,3-allenols and disilanes in the presence of the catalyst Pd2dba3 and the base Cs2CO3 gives 2-silylated 1,3-butadiene as a result of dehydration of the primary adduct formed. In the absence of Cs2CO3 , disilylation products are formed at the internal allene double bond, which upon further base treatment eliminate the silanol to give the final product, suggesting the mechanism shown in Scheme 25.[98]
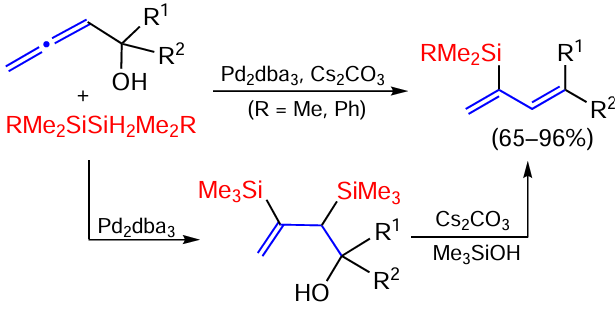
The same authors showed that the silylation of the allenols shown in Scheme 25 under the action of PhMe2SiZnCl or Ph2MeSiZnCl catalysed by iPrCuCl proceeds in the same way.[99]
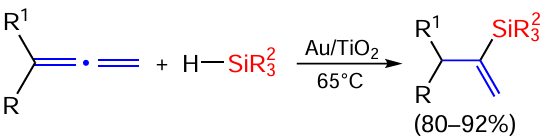
The ligand-free hydrosilylation reaction of terminal allenes with R3SiH silanes catalysed by gold nanoparticles on titanium dioxide proceeds predominantly or exclusively via the internal double bond in 80 – 92% yield (Scheme 26).[100]
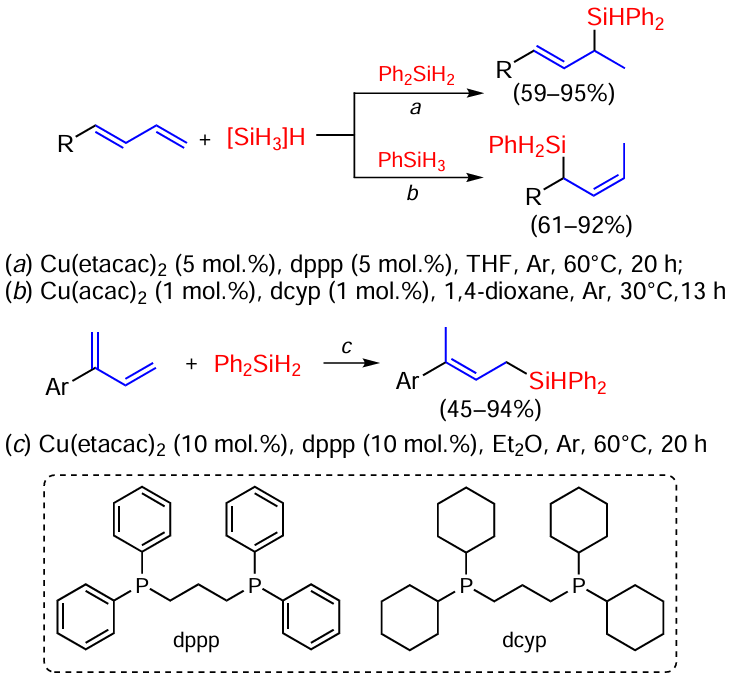
The reaction of s-trans- and s-cis-1,3-butadiene with the diphenylsilane Ph2SiH2 catalysed by copper acetylacetate (Scheme 27) gave structurally diverse allylsilanes.[101]
Enynes with an electron acceptor group at the triple bond, RCH=CH – C≡C-EWG, enter the silylation reaction with PinB – SiMe2Ph when catalysed by copper complexes.[102] Catalysis by the complex with substituted 2,2'-bipyridine gives racemic mixtures, whereas in the case of the optically active complex with a 2-pyridinoxazoline-type ligand, enantiomerically enriched 1,3-bis-silylpropenes were obtained with enantioselectivities up to 94% (Scheme 28).
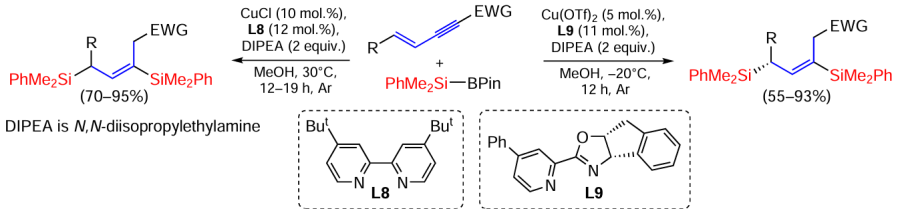
The reaction of 1,3-enynes with PhR3SiH2 silanes catalysed by cobalt complexes showed high regio- and (E)-stereoselectivity (Scheme 29).[103]
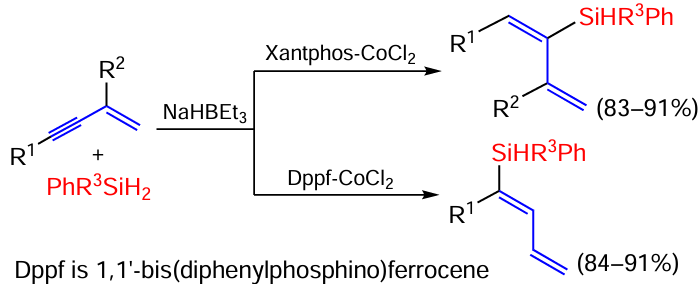
Enynes isomeric to those shown in Scheme 29 are silylated with phenylsilane when catalysed by an iron-containing catalyst containing the FeN3 fragment in a similar manner by cis-addition via a triple bond (Scheme 30).[104]
In summary, the hydrosilylation of carbon-carbon multiple bonds is one of the most important reactions in organosilicon chemistry. Starting more than half a century ago, these processes have evolved with the development of new catalysts and the expansion of the range of substrates and silanes. The regio- and stereoselectivity (α- and β-silylation, formation of Z- and E-isomers) is controlled both by substituents at the multiple bonds, which determine their polarisation, and by steric requirements, which determine the accessibility of a given carbon atom. The structure of the catalyst can also have a critical influence, apparently also as a result of the different ease of silylation on one or another carbon atom in the complex of the substrate with the catalyst. Such a variety of reaction courses allows the researcher to purposefully synthesize unsaturated organosilicon compounds of predetermined structure by varying the catalyst, substituents in the substrate and/or reagent, and reaction conditions, using both mono- and disilylation processes. Varying the above factors, as well as photo- and electrochemical activation of the Si – H bond, is also an effective tool for increasing the yield of target products. Finally, the use of metal complex catalysts with optically active ligands opens the way to optically active organosilicon compounds.
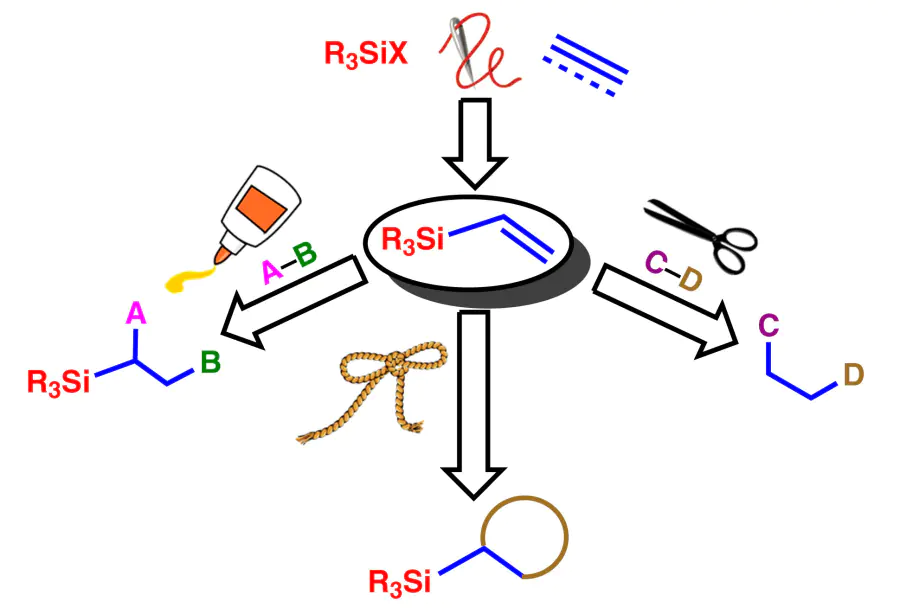
3. Reactions of unsaturated silanes with the silyl group retained at the carbon atom
3.1. Hydrogenation, reduction, hydrosilylation and hydroalkylation of vinylsilanes
Efficient reactions of asymmetric hydrogenation of vinylsilanes in the presence of Ir-N,P complexes are known. The reaction gives chiral organosilanes with high enantioselectivity (65 – 91%) (Scheme 31).[105]
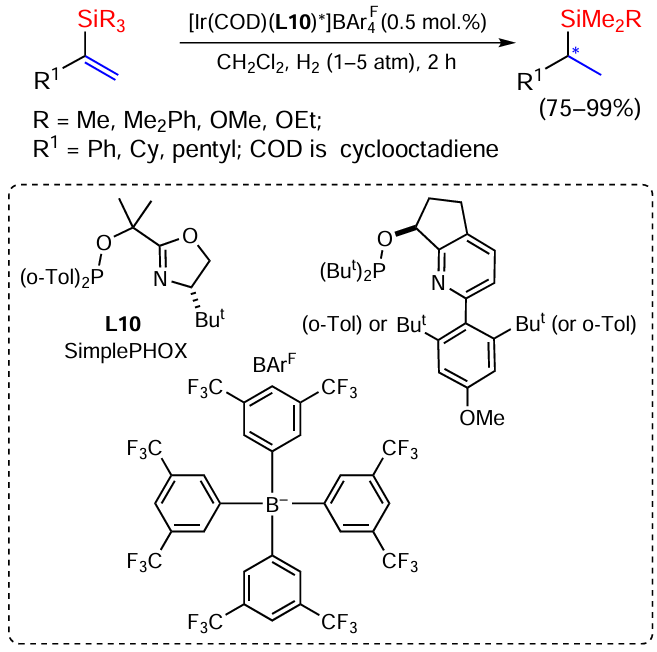
α-Trimethylsilylstyrene as substrate allows complete conversion with enantioselectivities ranging from 13 to 88%. The reaction with the corresponding cyclohexyl-substituted vinyl silane for 7 h gave even better results. Pyridine phosphinite ligands and oxazoline-based ligands (SimplePHOX) proved to be the most effective. The presence of a more sterically hindered dimethylphenylsilyl substituent in the substrate leads to a dramatic decrease in reactivity compared to the SiMe3 analogue. To accelerate the reaction, hydrogen was supplied at pressures up to 5 atm. Under these conditions, both SimplePHOX and pyridine phosphinite gave the product with complete conversion and enantioselectivity of 81% and 91% respectively.[105]
Recently, the asymmetric hydrogenation of N-sulfonylarylsilylimines was achieved for the first time. The reaction was catalysed by a palladium complex of the P-stereogenic diphosphine ligand QuinoxP*, yielding chiral α-aminosilanes, analogues of natural α-amino acids, in high yields and with excellent enantioselectivity, since some of the silyl groups are bioisosteres of carboxyl groups (Scheme 32).[106]
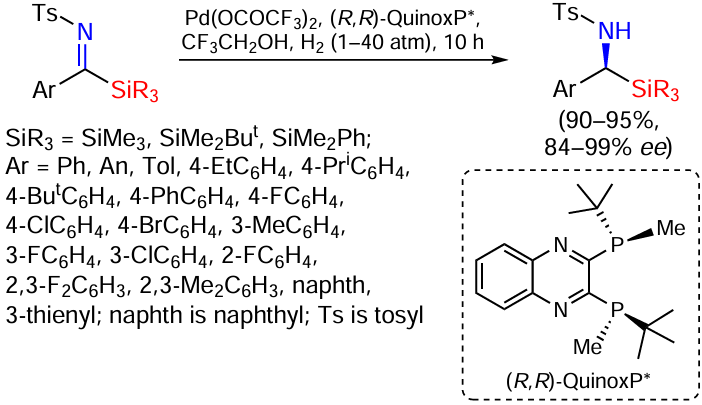
The tert-butyl dimethylsilyl substrate gives the product with the highest enantioselectivity of 99%.[106] The sulfonyl group also significantly affects the reaction efficiency. High yields and enantioselectivities were observed for substrates containing phenylsulfonyl or 4-methoxyphenylsulfonyl groups, whereas no hydrogenation product was formed for the substrate containing 4-(trifluoromethyl)phenylsulfonyl group. Reducing the hydrogen pressure from 40 to 15.5 and even 1 atm allows the reaction to be completed in less than 10 h with a slight decrease in enantioselectivity from 99.6% (40 atm) to 99.4% (15 atm), 98.5% (1 atm) (data given for the model derivative with Ar = Ph). At a hydrogen pressure of 40 atm, a wide range of arylsilylimines containing both electron-donating and electron-withdrawing substituents at the 4-position of the aryl groups were studied, yielding the corresponding amines with excellent ee (e.g. 99% for products with Ar = 4-MeC6H4 , 4-EtC6H4 , 4-MeOC6H4) and somewhat lower for electron-withdrawing substituents (98% in the case of Ar = 4-FC6H4 , 4-ClC6H4 , 4-BrC6H4). The transfer of the substituent from the p- to the m-position of the aryl groups had practically no effect on the enantioselectivity. Substrates with naphthyl and 3-thienyl groups also gave products with enantioselectivities of 99% and 98% and yields of 95% and 91% respectively. However, benzylsilylimines did not give any hydrogenation products under these conditions. The corresponding silylamines with the preserved chiral central configuration were obtained by removing the sulfonyl protection.[106]
In the presence of CoBr2 , alkoxy(vinyl)silanes also undergo hydrosilylation by various hydrosilanes with anti-Markovnikov regioselectivity.[107] Similarly, in the presence of platinum, vinylsilanes, vinylsiloxanes and vinyl(ethoxy)silanes undergo hydrosilylation with methyl(diethoxy)silane to form anti-Markovnikov products in high yields of 93 – 97%.[108] The hydroboration of vinylsilanes also proceeds with anti-Markovnikov regioselectivity in the presence of triazine complexes of Co(III) and Cs2CO3 (Scheme 33).[109]
β-Phosphorus-substituted silanes are formed in hydrophosphorylation reactions of, for example, vinyl(triethyl)silane or divinyl(methyl)phenylsilane with bis(trimethylsiloxy)phosphine, and also against the Markovnikov rule.[110]
Hydrogermanylation reactions of allylsilanes are also known.[111] Some aspects of hydrosilylation reactions have been discussed in recent reviews.[2, 112, 11-13]
Nickel-based C – H silylation catalysts are almost unknown in catalysis; there are only two examples. The first, described in 1992, requires the presence of reagents with a strained Si – Si bond.[113] A second example of a CH-silylation reaction has recently been described. The reaction of C6F5H and H2C=CHSiMe3 in the presence of [Pri2Im]Ni(η2-H2C=CHSiMe3)2 gave exclusively the C – H silylation product C6F5SiMe3 and ethylene. Catalytic silylation of the C – H bond can also be carried out with fluorinated aromatic substrates containing two fluorine atoms in the o-position or with 1,2,3,4-tetrafluorobenzene. Less fluorinated substrates react more slowly (Scheme 34).[114] Vinylsilanes also undergo perfluoroiodalkylation under free radical conditions.[115]
In the presence of Cu(I) salts, a catalysed conjugated addition of Grignard reagents may occur followed by diastereoselective protonation to form (E)-α-trialkylsilyl-β-alkyl(aryl)-α,β-unsaturated esters with diastereoselectivity (dr) reaching more than 20 : 1 with an excess in favour of the anti-diastereomer (Scheme 35).[116]
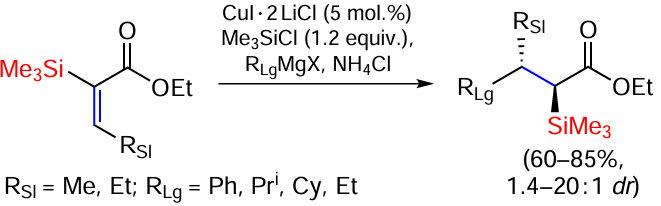
The addition of PhMgBr to the initial ester in the presence of CuI – 2LiCl (5 mol.%) and Me3SiCl (1.2 equiv.) at –10°C initially gave a saturated ester as a result of protonation of the putative intermediate silyl ketene acetal in aqueous NH4Cl solution with dr = 5 : 1 in favour of the anti-stereoisomer. The reaction with iPrMgCl (2.5 equiv.) proceeds better (dr = 15 : 1 also in favour of the anti-stereoisomer). The cyclohexyl Grignard reagent gave 85% yield and a high dr value (up to 20 : 1). The addition of EtMgBr at –40°C gave the product in 75% yield, but dr decreased significantly to 1.4 : 1.[116]
Reactions of cyclopropenylsilanes with Grignard reagent in the presence of CuI are also known. This reaction gives access to a wide range of different substituted cyclopropylsilanes and is highly diastereoselective. The reaction has several limitations, mainly due to the difficulty of introducing sterically hindered substituents at the geminal position. Nevertheless, several representatives of hexasubstituted cyclopropanes have been obtained (Scheme 36).
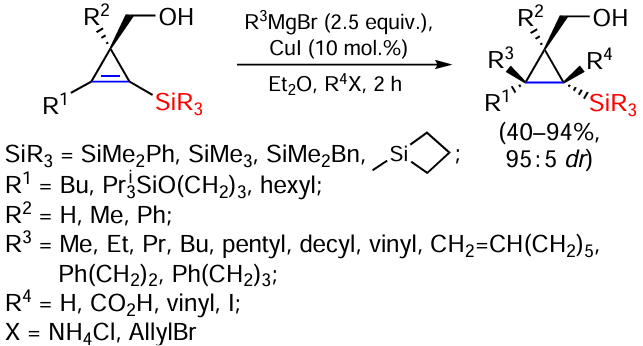
The reaction proceeds in Et2O at room temperature. The addition of both primary and allyl Grignard reagents to the cyclopropenyl ring gives equally good results, but Grignard reagents with a secondary carbon atom do not react. The substituents at the silicon atom can also be varied in this reaction (PhMe2Si, Me3Si, PhCH2Me2Si, Et3Si, Me2HSi) and the products have been formed in comparable yields. The substrate with a bulky group ButMe2Si in the cyclopropenyl ring did not give the target products. Hexa-substituted silacyclopropanes were obtained, including the use of allyl bromide as R4X. In all cases the α-silylated cyclopropyl substituted products obtained showed good configurational stability and no epimerisation was observed.
Recently, a one-pot synthesis of 3-silyl-3-sulfolenes from propargylsilanes was carried out. The reaction was carried out in excess of SO2 and in the presence of 10 mol.% TsOH at room temperature for 16 h, with yields of cyclic products reaching 84%.[117] For product A, reductive desulfurization to silylolefins was studied. Using lithium in liquid ammonia as the reducing agent, the C – S bond in the sulfolenes was broken with complete conversion of the initial substrate (Scheme 37).
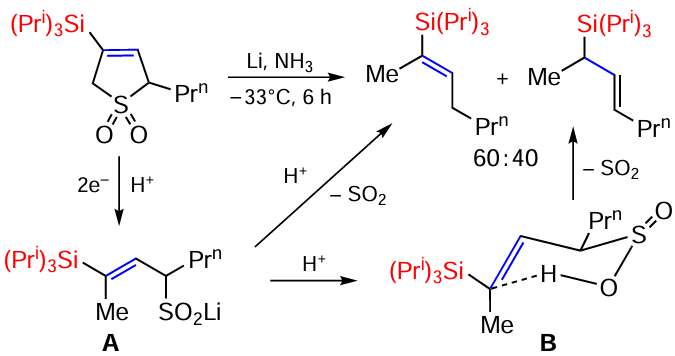
The corresponding vinylsilane was formed in this reaction together with its allylic isomer in a ratio of 60 : 40. The first product was formed by direct elimination of SO2 from intermediate A. The formation of the allylic product can be explained by retro-ene elimination of SO2 from intermediate B, formed by protonation of intermediate A. Changing the reaction conditions (temperature, addition of THF as co-solvent, additional base, different proton source or substitution of reducing agent) did not change the course of the reaction and the ratio of products was also maintained.[117]
The reactions of alkylazaarenes and vinylsilanes occur in the presence of strong Brønsted bases (Scheme 38).[118]
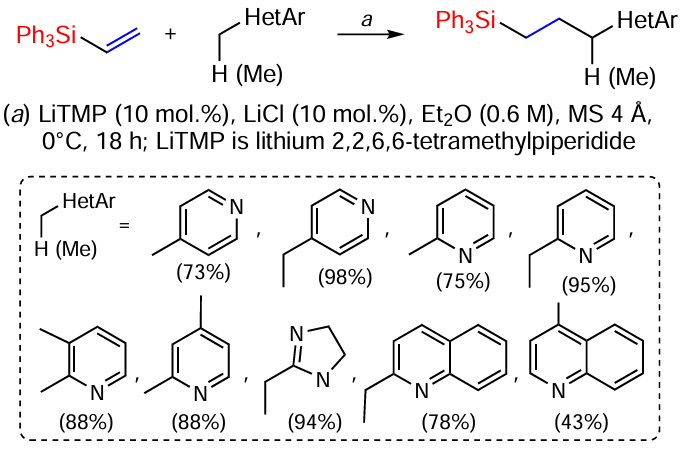
For example, the reactions of alkylpyridines and their analogues with vinylsilanes occurred in moderate to high yields in the presence of catalytic amounts of LiTMP, LiCl and MS 4 Å. 4-Methylpyridine as substrate gave slightly lower product yields than 4-ethylpyridine. 2-Alkylpyridines, such as 2-ethylpyridine, also gave products in 75 – 95% yield. 2,3-Dimethylpyridine gave the product only at the 2-methyl group in high yield (88%). The selectivity can be explained by the much higher acidity of the proton of the 2-methyl group than of the 3-methyl group, as well as by the different degree of stabilisation of the formed anion due to charge delocalisation after deprotonation. In addition, for 2,4-dimethylpyridine the reaction proceeded with high regioselectivity exclusively at the 4-position. This may also be due to the greater acidity of the hydrogen atoms of the 4-methyl group compared to the 2-methyl group. The reaction of vinyl(dimethyl)phenylsilane proceeded in good yields, but only on heating due to its reduced reactivity.[118]
N-Isopropylmethyl-3-pyridine and N-isopropylmethyl-3-thiophene can be efficiently alkylated with vinyl(trimethyl)silane in the presence of Zr(NMe2)4 . The reaction requires the presence of 40 mol.% of the catalyst (Scheme 39).[119]
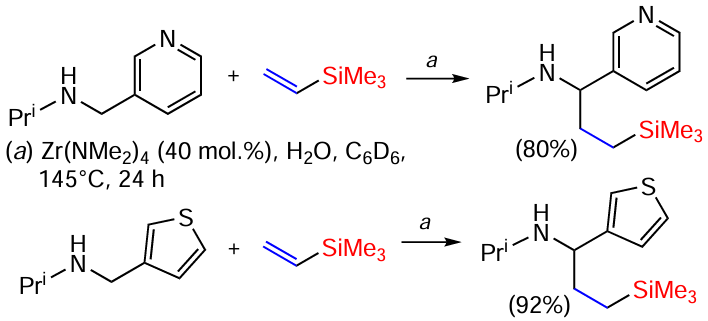
In the presence of titanium mono(aminopyridinate) as a catalyst, vinyl silanes undergo hydroaminoalkylation; in the most cases, the reaction proceeds with the formation of branched products (Scheme 40).[120]
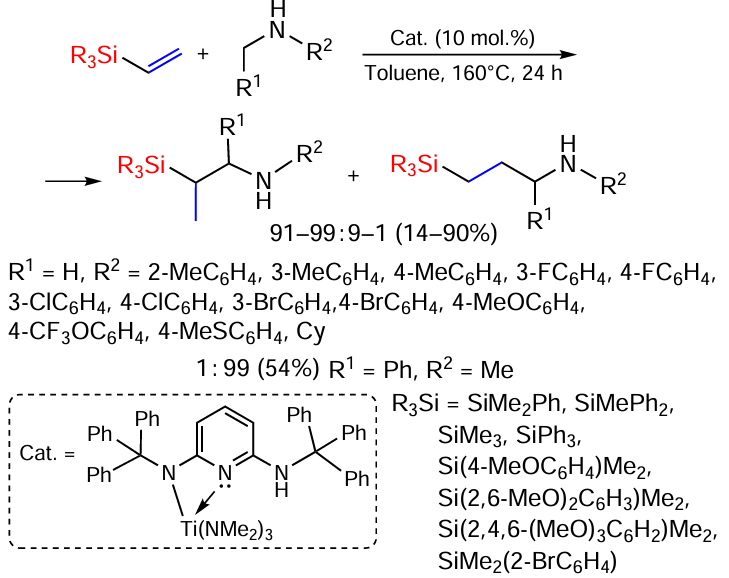
A number of vinyl silanes have been studied as substrates using N-methylaniline as an example. The reaction of dimethyl(phenyl)silane, which is the only example of the formation of a linear hydroaminoalkylation product (it proceeds in the presence of a formamidinate titanium complex), is remarkable. Vinyl(triphenyl)silane, as one of the most sterically hindered substrates, gave mostly a branched product in 75% yield. Among the methoxyphenyl-substituted vinylsilanes, the p-monoisomer gave the best result, the product being formed in 83% yield. While (2,4,6-trimethoxyphenyl)dimethylvinylsilane decomposed under the reaction conditions, (2,6-dimethoxyphenyl)dimethylvinylsilane gave a mixture of products in 68% total yield. The reactions of a wide range of secondary amines with vinyl(dimethyl)phenylsilane have also been studied. The reaction is strongly dependent on the steric volume of the amine. Sterically less hindered N-methyltoluidines gave hydroaminoalkylation products in much better yields than N-methyl-o-toluidine. It should also be noted that o-halogen(F,Cl,Br)-N-methylanilines did not react under the above conditions. The study of reactions with p- and m-substituted N-methylanilines showed that in addition to substrates with alkyl substituents in these positions, F, Cl, Br, OMe and SMe-substituted substrates also react. In all these cases the hydroaminoalkylation proceeded with excellent regioselectivity in favour of the branched isomer and in good to excellent yields (63 – 90%).[120]
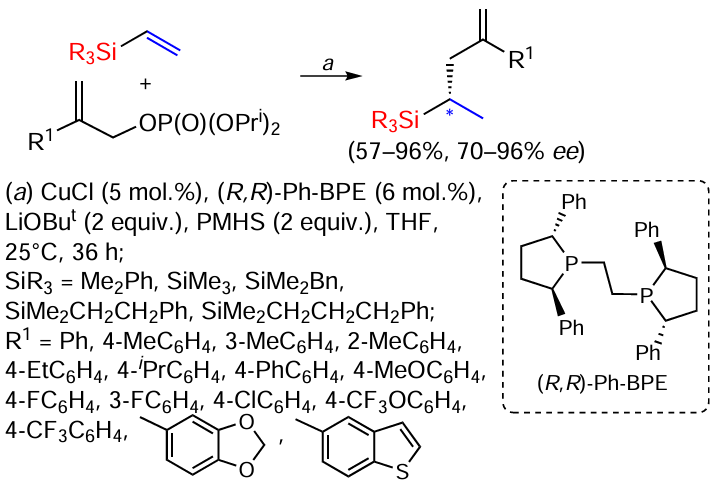
The method of asymmetric hydroallylation of vinylsilanes with allyl phosphates under mild conditions is well known. The process demonstrates high chemo- and enantioselectivity, and no noble metals are used as catalysts (Scheme 41).[121]
The catalytic C-alkylation reaction of carboxylic derivatives, such as amides, esters, and sulfonamides, with vinylsilanes under mild conditions in the presence of a strong Brønsted base, potassium hexamethyldisiloxanolate (KHMDS), and 18-crown-6-ether is known (Scheme 42).[122]
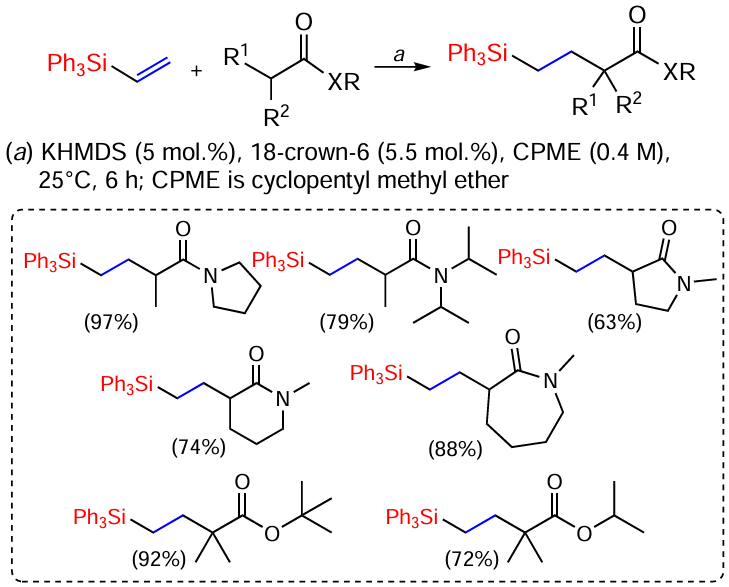
The reactions proceed smoothly for the substituted (tertiary) amides and esters, leading to the formation of the target products in high yields at room temperature. N-Methyllactams also react as tertiary sulfonamides (not shown in the scheme) in good to high yields.[122]
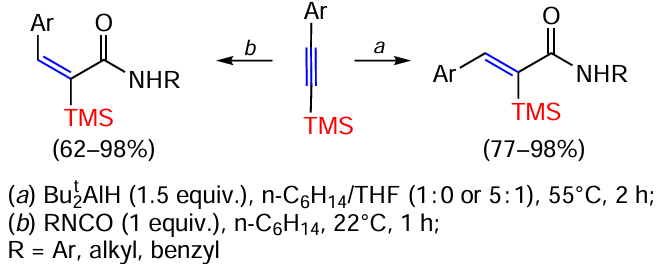
α-Silylated α,β-unsaturated amides were prepared in high yields by hydroamination of TMS-substituted aromatic alkynes with isocyanates under mild conditions under the action of diisobutylaluminium hydride. The reaction gave diastereomerically enriched products, the ratio of which depended on the solvent (Scheme 43).[123]
Regiodivergent hydroalkylation of vinylsilanes is known, in which the reaction course depends on the auxiliary ligand used on a nickel catalyst (Scheme 44).[124]
Various primary alkyliodides react to give branched hydroalkylation products. When the catalyst is replaced, the same alkyliodides give linear products, but in lower yields.[124] In the presence of Rh(II) and 8-aminoquinoline, the selective o-C – H alkylation of arylsulfonamides with vinylsilanes proceeds to give branched products. Benzenesulfonamide and p-substituted arylsulfonamides selectively give the corresponding monoalkylated branched products without side adducts on activated C – H bonds (Scheme 45).[125]
m-Substituted aryl sulfonamides gave the corresponding products in good yields (70 – 80%) and good selectivity (75 – 90%). O-methyl-substituted sulfonamides gave lower yields (41 – 46%) and a selectivity of 88 : 12 (with respect to the linear product). In the case of p-substituted aryl sulfonamides, the corresponding branched alkylation products were formed with a selectivity of 90 : 10 and the best yields (80%). The introduction of different functional groups, such as OMe, alkyl, F, Cl, NHCOCH3 , CF3 , and benzyl chloride, also allowed to obtain stable products. It should be noted that no dialkylation products were detected in the reaction. The use of vinyl(trimethyl)silane, 1,1,1,3,5,5,5-heptamethyltrisiloxane, vinyl(dimethyl)phenylsilane and vinyl(methyl)diethoxysilane as alkylating agents did not affect the efficiency of the reaction and comparable results were obtained.[125] Similar reactions have been studied for carboxamides.[126]
A method for the synthesis of α-stereogenic allylsilanes in the presence of CuI and Pd using vinylsilanes and styrenes has recently been developed. The reaction is enantioselective and gives a variety of allylsilanes in good to excellent yields (Scheme 46).[127]
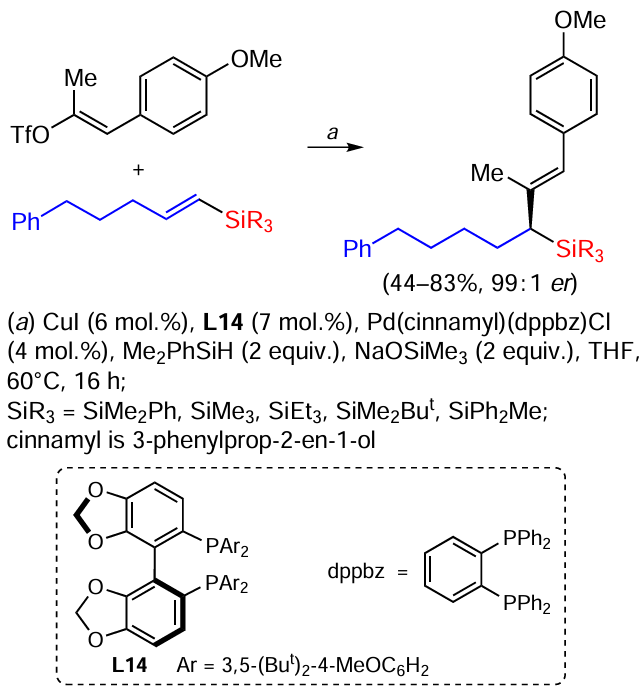
Allylsilanes containing SiMe2Ph and SiMe3 groups, which are the most commonly used in allylsilane chemistry, can be obtained by this reaction. As the size of the substituent on the silicon atom increases, the yield decreases in the SiMe3 , SiMe2Ph, SiMe2But series. This is due to the slower copper metalation of the olefin fragment or trans-metalation as a result of steric hindrance in vinyl silanes. It is also possible to obtain allylsilanes with acyclic and cyclic substituents with yields up to 73% and high enantiopurity (99 : 1).[127]
The synthesis of anti homoallyl alcohols from 1,3-dienylsilanes and aldehydes proceeds in the presence of copper (Scheme 47).[128]
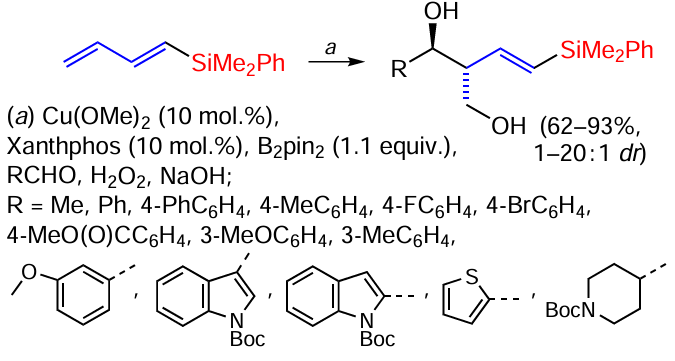
In general, the reaction proceeds well with a wide variety of aldehydes. For example, aromatic aldehydes with a substituent in the p-position, regardless of their electronic properties, reacted to give products in yields of 79 – 92% and anti/syn = 10 – 20 : 1. Reactions with m- or o-substituted aromatic aldehydes gave alcohols in yields of 62 – 88% and diastereoselectivity of 12 – 20 : 1. With heteroaromatic aldehydes, diols were formed in yields of 70 – 88% and diastereoselectivity of 9 – 18 : 1, as with aliphatic aldehydes (61 – 75% at anti/syn ratio of 10 – 20 : 1). Dienylsilanes with silyl groups of different sizes were investigated to see if the size of the 1,3-diene silyl group affects the stereochemistry of the reaction. The reaction of the less sterically hindered trimethylsilyl 1,3-diene with benzaldehyde gave a diol product in 68% yield and an anti/syn selectivity of 10 : 1. The diphenylmethylsilyl-substituted 1,3-diene also reacted to give the product in 86% yield with an anti/syn selectivity of 12 : 1. The reaction of benzaldehyde with the sterically more hindered SiPh2But-substituted diene gave a 2 : 1 (anti/syn) mixture of stereoisomers in 91% yield. The formation of a product with the Z-configuration of the double bond was not observed in any of the transformations investigated under these conditions. Overall, the results suggest that the size of the silyl group of the 1,3-diene influences the anti/syn selectivity, especially in the case of the bulky SiPh2But-substituted diene.[128] A similar reaction in the presence of acyl fluorides led to the synthesis of (Z)- or (E)-β,γ-unsaturated ketones.[129] Silane reactions catalysed by copper complexes have been discussed in detail in a 2019 review.[4]
The transfer of the trimethylsilyl group from vinylsilane to the terminal carbon atom in (E)-1,3-dienes catalysed by the [RuHCl(CO)(PCy3)2] complex led stereoselectively to (E,E)-dienylsilanes, the proportion of which relative to the E,Z and Z,Z-isomers reached 68 – 100% (Scheme 48).[130]
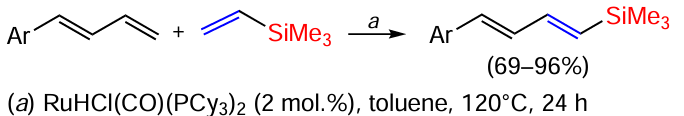
3.2. Vinylation and allylation reactions with unsaturated silanes
The reaction of a wide variety of different aldehydes with trimethyl[(Z)-(2-phenylsulfanyl)-1-(trifluoromethyl)vinyl]silane in the presence of tetrabutylammonium fluoride (TBAF) under mild conditions has recently been described (Scheme 49).[131]
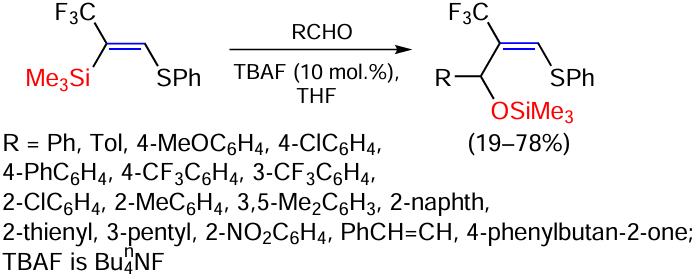
The synthesised 2-(trifluoromethyl)allyl OTMS esters are useful building blocks for the preparation of more complex CF3-containing molecules. The reaction is applicable to a wide range of aldehydes and is characterised by yields of 40 – 75%. Aldehydes containing methyl, methoxy, chloro, phenyl and trifluoromethyl groups at various positions at the aromatic ring are effective substrates for this reaction. 2-Methylbenzaldehyde gave a low yield (19%), apparently due to steric hindrance of the aromatic ring. 2-Naphthaldehyde, 2-thiophenecarbaldehyde and 2-ethylbutanal gave the corresponding adducts in 57 – 64% yield.[131]
The use of a combination of catalysts (so-called cooperative catalysis) allows direct α-allylation of linear prochiral esters in the presence of vinylsilanes. The silicon-containing substituent allows control of the regio- and enantioselectivity of C – C bond formation in products containing pentafluorophenyl ester and vinylsilyl moieties (Scheme 50).[132]
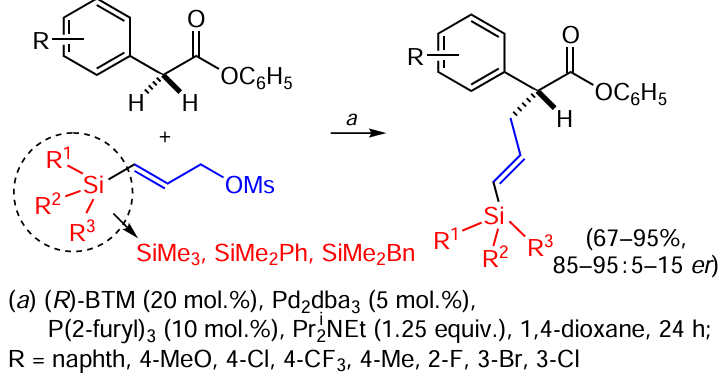
An auxiliary ligand on the palladium atom is used to prevent the formation of by-products. Nucleophiles with electron-donor groups in their structure gave the best results, while the electron-acceptor group CF3 gave the lowest enantioselectivity in 60 – 84% yield, but this was found to be due to racemisation of the products over time due to the increased acidity of the stereogenic proton in the reaction product.[132]
The copper-catalysed addition of allenylsilanes to ketones to form enantio-enriched tertiary homoallyl alcohols is well known. In the presence of PhSiH3 and a phosphine-ferrocene ligand, branched products are formed with high diastereoselectivity and enantioselectivity (Scheme 51).[133]
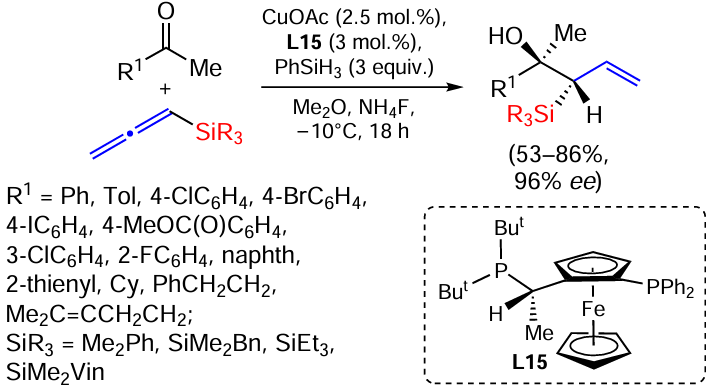
A variety of (hetero)arylmethyl ketones are suitable substrates for the synthesis of chiral allylsilanes in 53 – 96% yields. Aliphatic ketones gave products in lower yields, 77 – 85%; enantioenriched ketones were also investigated. Allenylsilanes gave products in good yields (44 – 82%) and excellent enantioselectivity (85 – 90%) regardless of the amount of silyl group. 1,1-Disubstituted allenes did not give branched products.[133]
Palladium-catalysed C – H vinylation of aromatic hydrocarbons has recently been shown to be an efficient, reliable and practical route to functionalized arylated vinylsilanes (Scheme 52).[134]
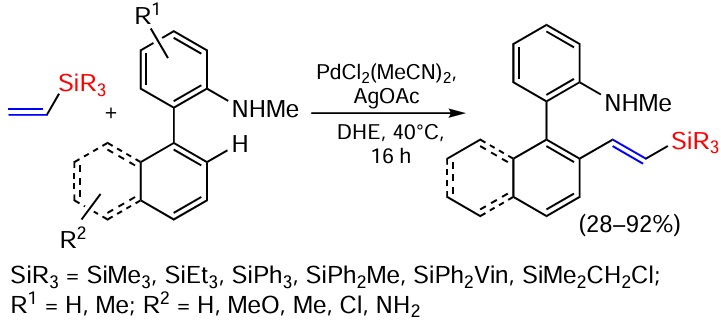
Under the conditions considered, vinylsilanes with different substituents at the silicon atom, including those with a chloromethyl group that tends to react with the amino group, behave in principle in the same way. The general nature of the vinylation reaction has been demonstrated for the substituted 2-aminobiaryl derivatives. Methyl and phenyl groups as substituents in the aromatic ring have a negligible effect on the course of the reaction. The presence of a chlorine atom increases the yield to 87% compared to the electron-donating methoxy group in the same position (60%).[134] Some other realisations of this reaction for hydroxylimino derivatives instead of arylamines are given in [135].
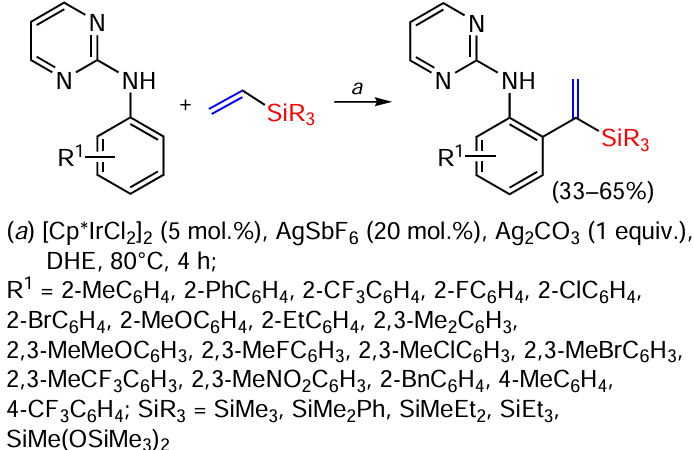
The highly selective C – H vinylation of aniline derivatives with vinylsilanes takes place in the presence of Ir(III). In this case, the main condition for the reaction is the presence of a pyrimidine substituent in the amino group of aniline as a directing group. The reaction is the first example of selective C – H vinylation of aniline derivatives by vinylsilanes, proceeding at the α-carbon atom of the C=C bond of the silane. The reaction is applicable to a wide range of anilines functionalized with groups such as OMe, NO2 , CF3 , Br, Cl, F (Scheme 53).[136]
Benzylation reactions of vinylsilanes in the presence of nickel salts are known.[137] Vinylsilanes with a bromomethyl group in position 3 relative to the silyl group and unstabilised benzylzinc derivatives have been used as reagents. The regioconvergence of this radical reaction is regulated by the presence of the silyl substituent,[138, 139] and high enantioselectivity is achieved by the use of the chiral ligand PyBOX. The reaction is a rare example of enantioconvergent allylbenzylation in the presence of hard benzyl nucleophiles (Scheme 54).[137]
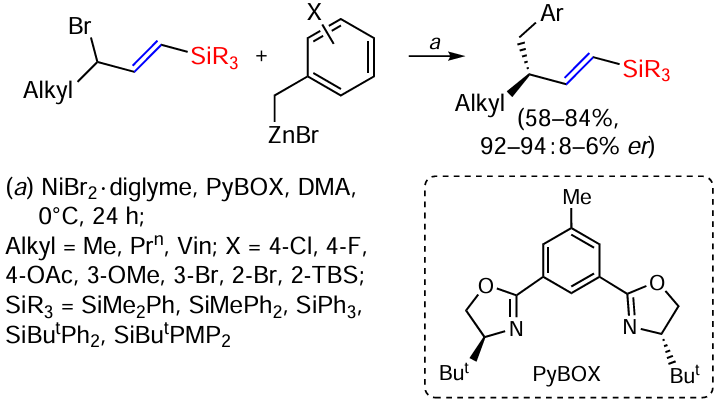
Compounds with not only methyl but also n-propyl and vinyl groups at the bromallyl carbon atom have been investigated as substrates. However, substrates with substituents other than methyl give lower enantioselectivity. Benzyl zinc derivatives bearing both electron donating and electron-withdrawing substituents show high enantioselectivity. Replacement of the ButPh2Si group with the less sterically hindered Me2PhSi and MePh2Si groups also increases the enantioselectivity of the reaction.[137] Vinylation and allylation as well as hydroalkylation in the presence of unsaturated silanes and other Zn derivatives and aryliodides using NiBr2 as catalyst have been described in recent studies.[124, 138, 140]
Polyfunctional allylsilanes are good substrates for the synthesis of alkaloid analogues. In the presence of TBAF, intramolecular allylation occurs via the Sakurai reaction, which is the cyclization of geminal bis(silyl)enamide with indoline (Scheme 55).[141]
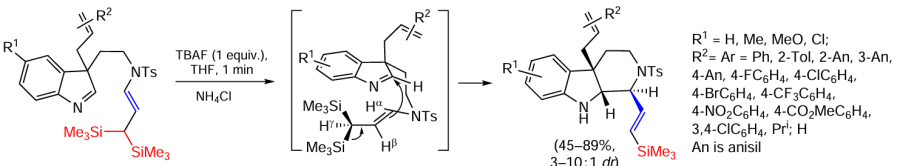
The key role in cyclisation is played by the steric effects of the substrate and the possibility of realising the effect of hyperconjugation in the geminal bis(silyl) product, leading to the formation of a C=C bond in the final hexahydropyrido[3,4-b]indole with a vinylsilyl moiety.[141] The reaction is carried out in THF at room temperature for 1 min and the reaction must be quenched by the addition of a saturated aqueous solution of NH4Cl, otherwise the final product decomposes. The stereochemistry of the cyclisation is explained by the reaction of addition antiperiplanar to the C3-allyl group in the indole fragment. In order to minimise the steric repulsion between the geminal bis(silyl) fragment and the indole ring, the former is turned away from the indole, resulting in a cis,trans-position of the hydrogen atoms and the allylic substituent in the piperidine ring, as shown in Scheme 55. In addition, the protons Hβ and Hγ are in a trans position to each other to avoid additional strain in the cycle, with elimination of one of the silyl groups resulting in the formation of an E-vinylsilyl moiety. The resulting products are precursors to the basic structure of akuammiline-type alkaloids.[141]
Cascade cyclisation/alkenylation of alkynyloximes with various vinylsilanes to form (β-isoxazolyl)vinylsilanes occurs in the presence of palladium diacetate (Scheme 56).[142]
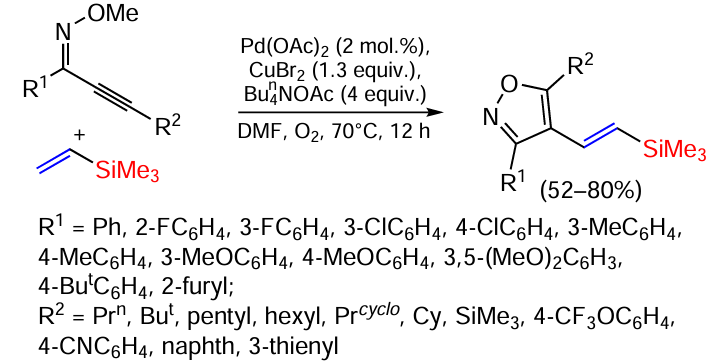
Both electron-donating (Me, OMe, But) and electron-withdrawing (F, Cl, CF3 and CO2Me) substituents in the phenyl ring of O-methyloximes react well with vinyl(trimethyl)silane in yields of 58 – 75%. Notably, the presence of substituents in the p-, m- and o-positions does not affect the reaction efficiency. A substrate with a 2-furyl substituent gave the product in 66% yield. Substrates with electron acceptor groups such as trifluoromethyloxy and CN also gave 67% and 70% yield respectively.[142]
3.3. Cyclization reactions of vinyl and allylsilanes
Allylsilanes are among the most widely used organosilicon compounds in organic chemistry. They are stable in air, relatively moisture resistant, low toxic and easily functionalized. Recently, stereoselective synthesis of optically active cyclopropylsilanes has been achieved using various allylsilanes and functionalized diazoesters (Scheme 57).[143]
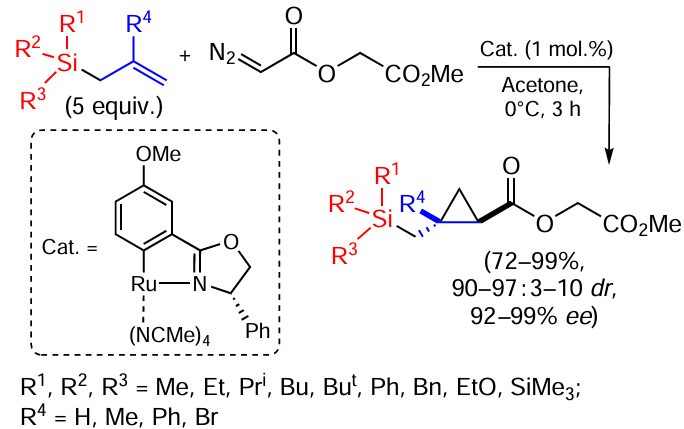
Allylsilanes give the corresponding cyclopropanes in good yields, with high enantio- and diastereoselectivity. 2-Bromallyl(trimethyl)silane, dimethyl(2-methylallyl)phenylsilane and allyl(trimethyl)-2-phenylsilane gave products in 83 – 98% yield and 97 – 99% enantioselectivity, but the diastereoselectivities for the latter two substrates were 68 : 32 (cis) and 56 : 44 (cis).[143] The vinylsilanes RMe2SiVin reacted with methyl(diazoacetoxy)acetate in the presence of a 10-fold excess of silane and 3 mol.% catalyst (under optimised conditions) to give products with lower yields (35 – 61%) but excellent enantio- and diastereoselectivities (> 99 : 1 dr, 98% ee). The cyclopropylsilanes obtained are building blocks for the synthesis of biologically active compounds (Im-7, Imatinib-7) and anti-HIV drugs.[143]
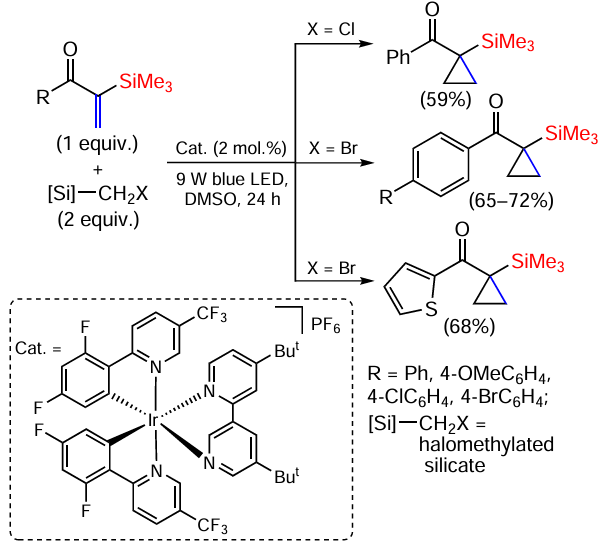
The stabilising effect of introducing a silyl group is also used in photooxidation-reduction catalytic reactions. Under these conditions, the cyclopropanation of vinylsilanes with (halomethyl)silicates as sources of methylene groups proceeds by the Giese reaction under mild conditions, in a neutral environment and without additives. For example, even vinylsilanes containing electron acceptor substituents gave 1-acyl-substituted silylcyclopropanes from 1-arylvinyl ketones in good yields under these conditions (Scheme 58).[144]
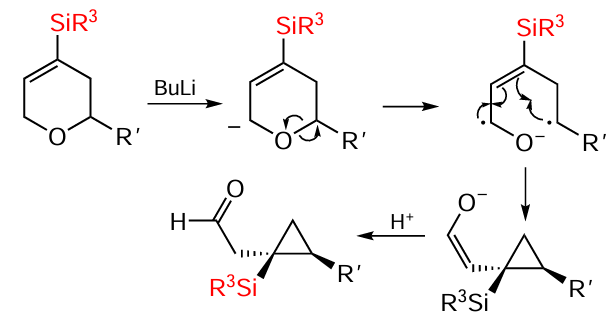
The synthesis of silylcyclopropylacetaldehydes with different silyl groups by Wittig [1,4]-rearrangement of 4-silyl-5,6-dihydropyrans is well known. High selectivity of the reaction has been achieved using substrates with electron-neutral or electron-donor substituents that undergo a 1,4-shift (Scheme 59).[145]
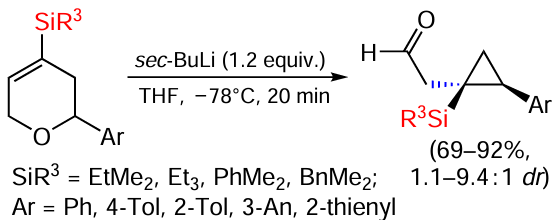
In general, the diastereoselectivity of the [1,4]-rearrangement is such that the bulkier groups (silyl and aryl/alkyl) are in cis-position to each other. The sterically less bulky EtMe2S group afforded silylcyclopropylacetaldehyde in 85% yield with a diastereoselectivity of 3.3 : 1, whereas the bulkier Et3Si group afforded a silylcyclopropane derivative in slightly lower yield (70%) but with a higher diastereoselectivity (11 : 1). Substrates containing electron-donating groups, such as the 4-methyl group in the phenyl ring, gave silylcyclopropanes only in the presence of PhMe2Si and BnMe2Si groups, and the reaction proceeded in good yields but with low diastereoselectivity. The presence of an o-methyl group in the aryl ring gave silylcyclopropane in 91% yield and a diastereoselectivity of 8.3 : 1. The 2-thienyl substrate gave silylcyclopropylacetaldehydes in 69% and 71% yield respectively.[145] However, in contrast to all previous examples, the main diastereomer in this case had a trans-position of the substituents. The result can be explained by the fact that 2.2 equiv. BuLi was used in the reaction, since the 2-thiophenyl group simultaneously undergoes competitive deprotonation at position 5.[145] The reaction proceeds in a stepwise manner via Wittig [1,4]-rearrangement and is accompanied by homolytic C – O bond cleavage and intramolecular recombination of the radical and anion radical. The selectivity of the rearrangement is determined by the ability of the silyl group to stabilise the allyl radical and direct the recombination process towards the Si-containing carbon atom (Scheme 60).[145]
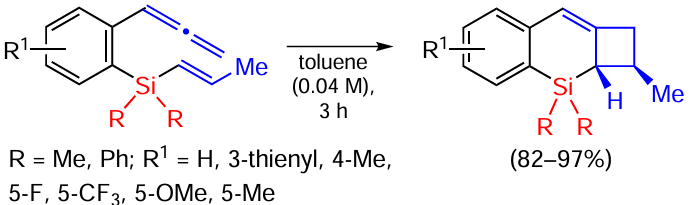
Vinyl(dimethyl)phenylsilanes containing an allenyl group in position 2 of the phenyl ring underwent the [2 + 2]-cycloaddition reaction at the distal (remote) C=C bond of the allene fragment on boiling in toluene without catalyst in up to 90% yield. The addition of catalysts or the use of blue LED irradiation did not lead to higher yields (Scheme 61).[146]
In selecting the catalyst and searching for the best variant, it was found that the complex of hexafluoroarsenide JohnPhos and gold gives the product in up to 87% yield, but in this case the reaction proceeds via the proximal (near) C=C bond of the allene fragment (Scheme 62).[146]
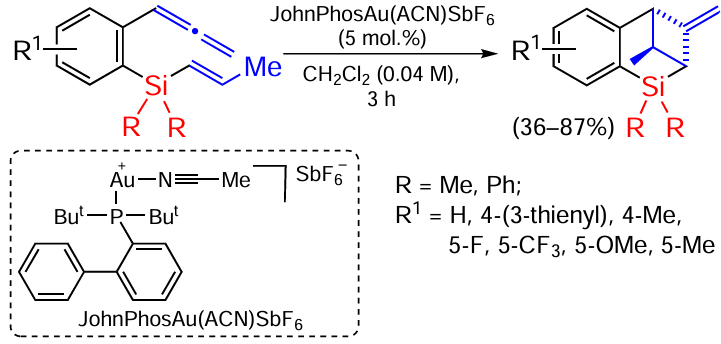
Studies have shown that the formation of products containing a cyclobutyl fragment is not significantly affected by changing the substituent in the phenyl ring. On the other hand, the yield of the proximal double bond reaction product is strongly dependent on the substituent at the phenyl ring of the substrate. The thienyl group significantly reduces the yield, whereas the methyl group gives an excellent yield. Substituents in the 5-position also have a significant effect on the yield of the product: electron-withdrowing groups decrease the yield, electron donor groups increase the yield.[146]
The cyclobutyl product was formed in the absence of a catalyst under thermal conditions, with the distal allene double bond and the vinylsilane double bond orthogonal to each other, favouring thermal [2 + 2]-cycloaddition (Scheme 63).[146]
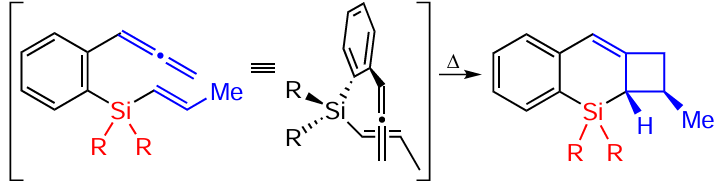
The second product was formed in several steps. First, the catalyst coordinated to and then activated the proximal double bond of allene (intermediate A). Next, the internal carbon of the vinyl silyl group nucleophilically attacked the central carbon of allene to form intermediate B and a β-silyl cation. The latter attacked the anionic centre at the benzyl position to form the final product (Scheme 64).[146]
The asymmetric hydrogenation of alkenes is one of the most convenient approaches to the synthesis of optically active compounds, as it is an atom-economical and relatively simple method and is widely used in the pharmaceutical, agrochemical and fine organic synthesis industries. The main limitation is the use of noble metal-based catalysts.[147] Nevertheless, a method for the highly efficient enantioselective hydrogenation of 1,1-disubstituted vinylsilanes has recently been realised (Scheme 65).[147]
The asymmetric hydrogenation of o-methyl-substituted chiral benzylsilanes has been further subjected to intramolecular silylation/dehydrogenation using ruthenium-based catalysts to form optically active silanes without racemisation of the stereogenic centre at the α-position with respect to the tetracoordinated Si atom.[147] Similarly, dihydrobenzosilanes can be synthesised from the intramolecular cyclisation of 2-hydrosilyl substituted styrenes in the presence of Rh(III).[148]
Hydrosilanes containing 1,1-dichloroalkenyl moieties undergo reductive intramolecular cyclisation. The reaction is catalysed by nickel complexes (Scheme 66).[149]
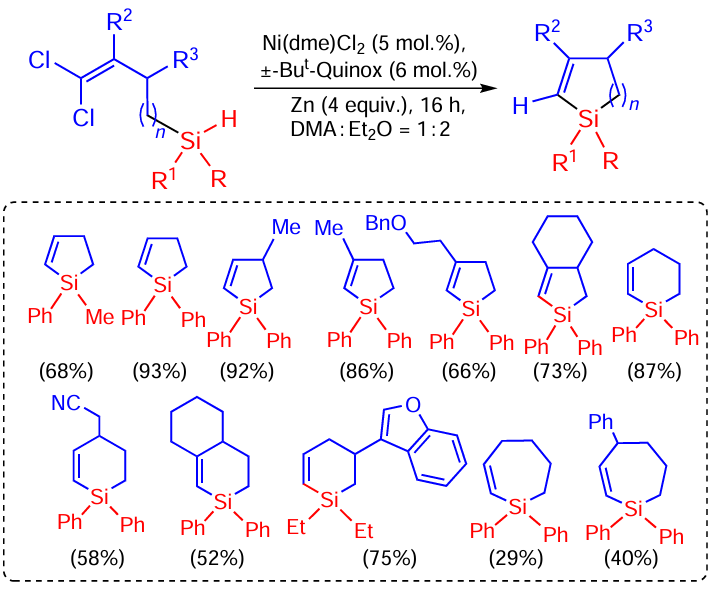
As can be seen from Scheme 66, the products formed in this reaction are unsaturated five- or six-membered organosilicon cycles formed in good yields. Two examples of seven-membered ring formation reactions have also been demonstrated, albeit in lower yields (29 and 40% respectively).[149]
Silacyclobutanes are synthetic equivalents of vinylsilanes. Recently[150] the first example of the ring expansion reaction in (2-alkenyl)aryl-substituted silacyclobutanes has been demonstrated. The reaction is catalysed by a Ni(0)/PR3 system with a chiral phosphine ligand. The approach is an efficient method for the synthesis of benzosilanes with an asymmetric silicon atom containing a wide range of functional groups both in the phenyl ring and at the double bond (Scheme 67).[150]
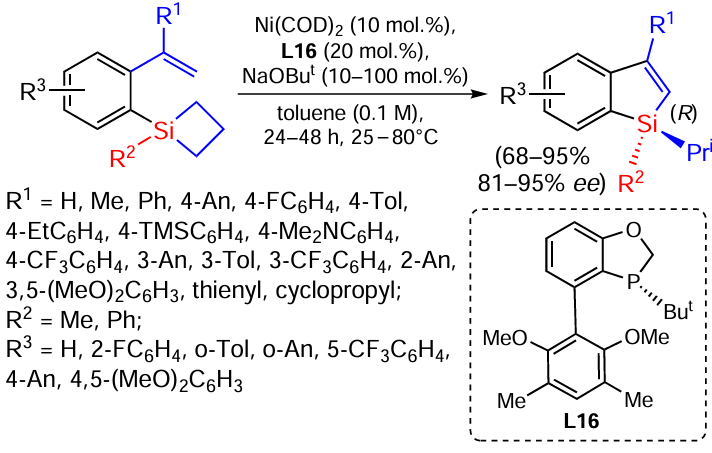
Two examples of bis-silicon derivatives have also been prepared using this approach. The reactions proceed with excellent selectivity and good yields (Scheme 68).[150]
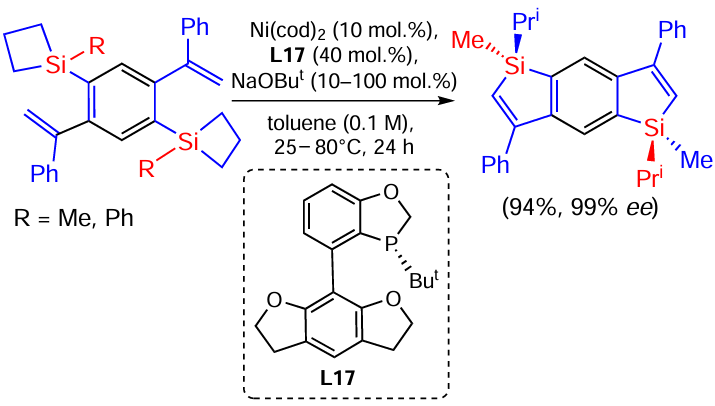
The latter compounds exhibit fluorescence, Cotton effect and CPL (circular polarised luminescence) activity, i.e. they have high potential for the design of new chiral optical materials based on enantio-enriched chiral benzosilanes.
The reaction can follow two pathways. Pathway a involves the coordination of Ni(0) with an alkenyl fragment and the formation of complex A or its pentacoordinated form B. Intermediate B undergoes cyclisation to Ni-silacycloheptene C, followed by cleavage of the hydride ion to form alkyl nickel hydride D. This is followed by reductive elimination to form the final product and regeneration of the Ni(0) catalyst. Pathway b involves the formation of intermediate C by oxidative addition of the C – Si bond of silacyclobutane to the catalyst and subsequent intramolecular addition of the alkenyl moiety through the formation of intermediate E (Scheme 69).[150]
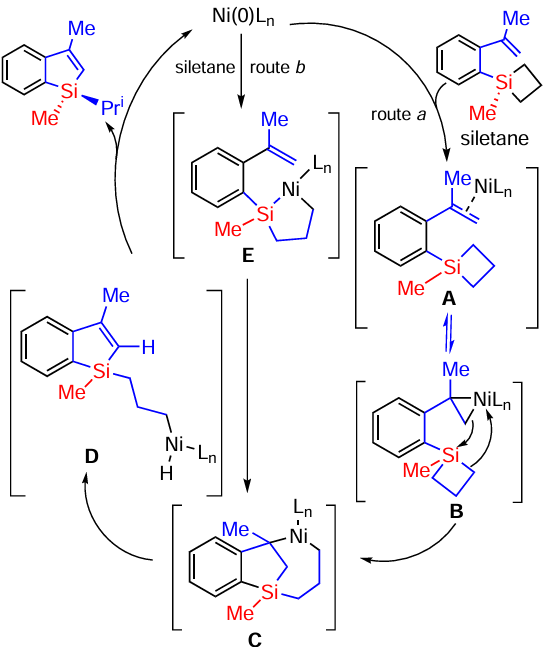
Benzosyloles have been successfully synthesised by the cyclisation of styrenes with a dimethylvinylsilyl(dimethylprophenylsilyl) group at position 2 (Scheme 70).[151]
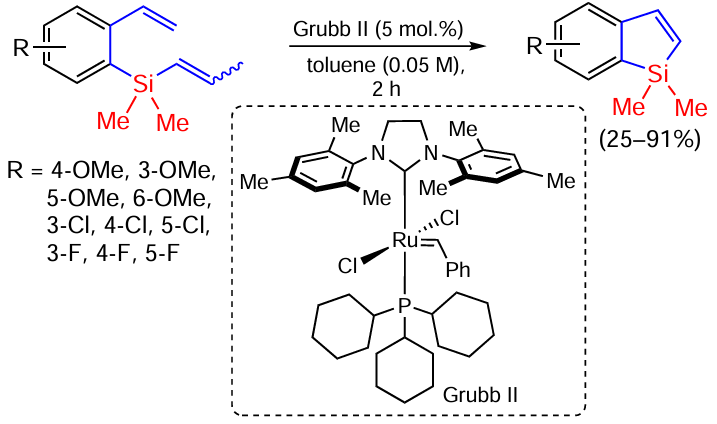
The best result was obtained by dissolving the substrate and Grubbs II catalyst (5 mol.%) in toluene (0.05 M) followed by boiling. The yield of the 4-MeO derivative was 91%. The lowest yield (25 – 35%) was obtained for 3-substituted styrenes, apparently due to steric effects. The introduction of electron-donating substituents accelerated the reaction.[151]
The photocatalysed cycloaromatisation of o-alkynyl(aryl)vinylsilanes and arylsulfonyl azides to form naphthyl-condensed benzosilanes with a wide range of functional groups in their composition is well known. The reaction is a unique combination of sequential S – N/C – S bond cleavage and α-silyl radical rearrangement (Smiles rearrangement) (Scheme 71).[152]
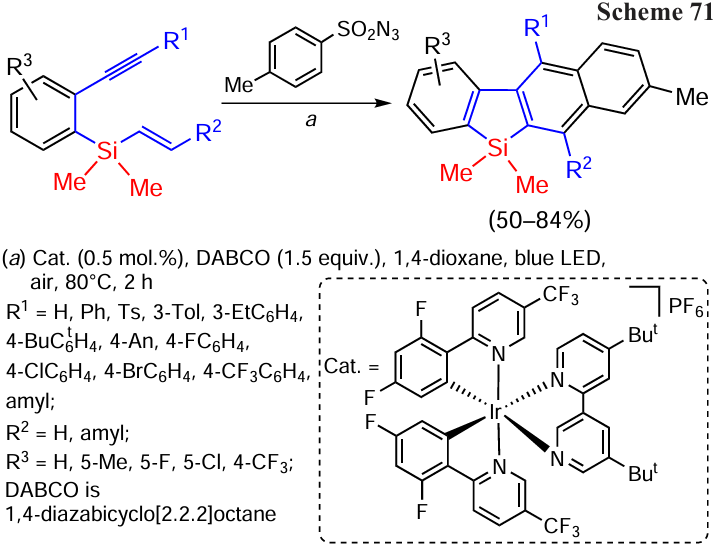
The obtained π-highly conjugated benzosiloles are promising targets for the preparation and investigation of new luminescent materials.
The reaction of various o-alkynyl(aryl)vinylsilanes with p-tosylazide has been studied in detail. Silaenynes having electron-donor substituents (Me, Et, But, MeO), halides (F, Cl, Br), strong electron-acceptor substituents (CF3) and even a vinylsilyl substituent at the p-position of the alkynylphenyl group enter the carbocyclisation and aromatisation reaction giving 4-(4-aryl)-2,3-benzosilafluorenes (50 – 71%). Similarly, m-tolyl-substituted alkynyl(aryl)vinylsilane has given the product in 63% yield.[152]
The reaction proceeds by the addition of a sulfonyl radical to a double bond in the substrate, leading to the formation of α-silyl C-radical A (its formation energy is 5.6 kcal mol–1 according to DFT calculations), which undergoes further radical cyclisation by interaction with an alkynyl fragment to form vinyl radical B (–11.6 kcal mol–1). Further intramolecular cyclisation of intermediate B proceeds by attack on the benzene ring followed by rearrangement to the spirocyclic intermediate C (–12.7 kcal mol–1) and desulfonylation to form β-silyl radical D (–29.9 kcal mol–1). The reaction is completed by cascade radical cyclisation to intermediate E (–41.4 kcal mol–1) and aromatisation to the final reaction product (Scheme 72).[152]
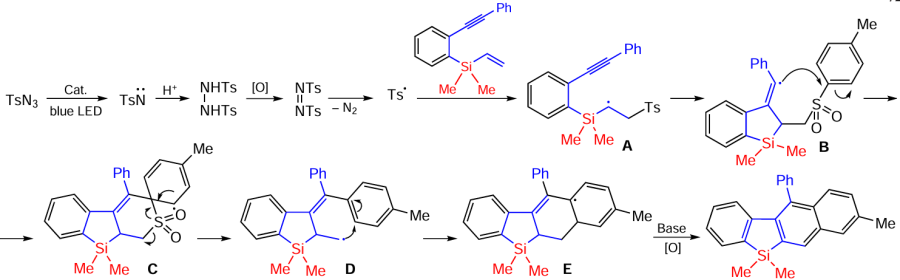
The synthesis of thiosulfone-bifunctionalized benzosilacycles by photocatalytic cyclization of silaenynes in the presence of thiosulfonates has recently been described. The reaction is characterised by highly selective radical coupling to form S – C(sp2) and S – C(sp3) bonds. Thiosulfonates with electron-donating substituents and halogen atoms in the p-position of the phenyl ring at R2 are most effective in this reaction (Scheme 73).[153]
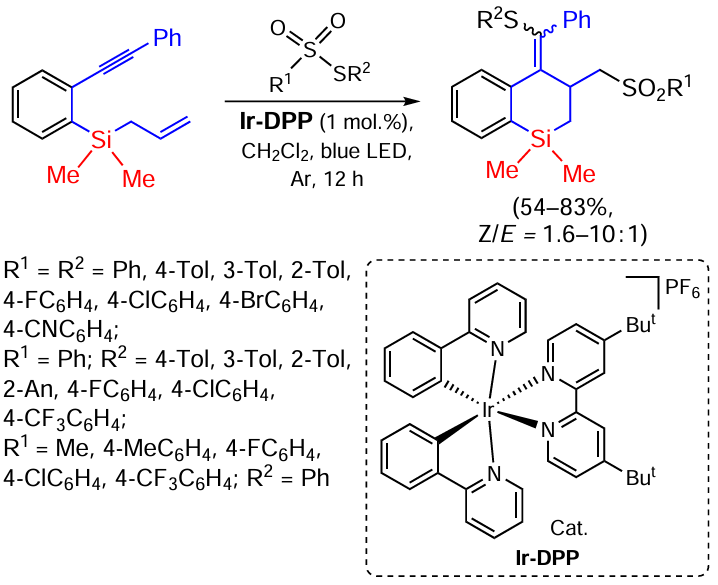
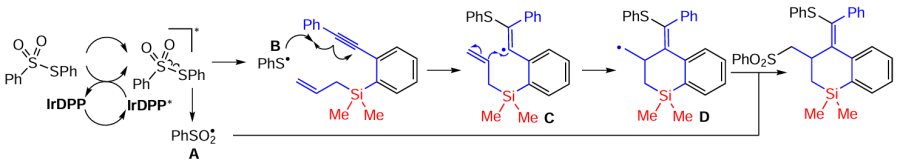
The reaction proceeds in the presence of Ir-DPP and involves the initial formation of the thiosulfonate transition state excited by direct blue light irradiation [PhSO2SPh]*, which undergoes further homolysis to form the phenylsulfonyl radical A and the phenylthio-radical B. The radical addition of radical B to the alkynyl moiety of silaenyne to form the vinyl radical C is followed by radical cyclisation to form the alkyl radical D. Recombination of the alkyl radical D and the phenylsulfonyl radical A completes the process leading to the formation of thio and sulfonyl-bifunctionalized benzosilinanes (Scheme 74).[153]
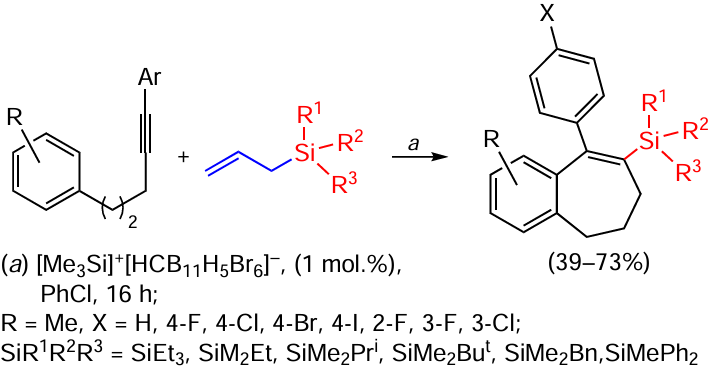
The carboxylation of alkynes with cyclization proceeds in the presence of silylium ions. The process is initiated by electrophilic activation of the triple bond by silylium ions and the catalytic cycle is then maintained by protodesilylation of stoichiometrically added allylsilane reagent. The reaction exhibits highly selective formation of benzocycloheptenes with a fully substituted vinylsilyl moiety (Scheme 75).[154]
The development of new methods for the synthesis of spirooxindoles containing a spirocyclic fragment at the C3 position of the oxindole nucleus is of great importance in the chemistry of natural alkaloids, medicinal chemistry and the preparation of drugs with a broad spectrum of activity (antiviral, anticancer and antimalarial agents). Spirocycles and, in particular, spirooxindoles exhibit stereospecific biological activity, indicating the importance of ensuring a high degree of diastereo- and enantioselectivity in the development of synthetic methods for the preparation of such scaffolds. For such compounds, the selective assembly of a spiro-quaternary carbon atom heavily loaded with functional groups, including for their further transformation, is a challenging task. Nevertheless, on the basis of unsaturated silanes, namely when using allenylsilanes in the reaction of alkylidene oxindoles in the presence of Sc(OTf)3 , enantioselective [3+2]-cyclization to form silicon-containing cyclopentene spirooxindoles is achieved (Scheme 76).[155]
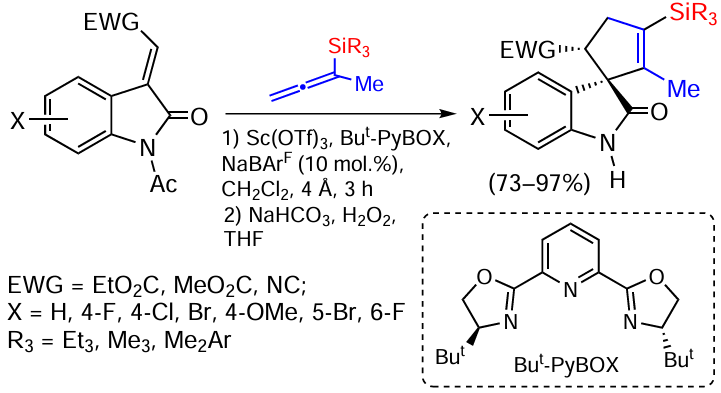
The low nucleophilicity of the substrates requires the use of a chiral complex Sc(OTf)3 for substrate cyclization, which is diastereoselective (dr > 94 : 6) and enantioselective (ee > 80%).[155] The authors have not given a detailed mechanism, but it is obvious that in the active cationic complex with Sc(OTf)3 the SiR3 group migrates to the central carbon atom of the allenyl fragment.
A similar reaction also occurs with allylsilanes. In the same work, the competitive kinetics of the spirocyclization of allenyl and allylsilanes was studied, monitoring the ratio of the products by 19F NMR spectroscopy (for fluorinated alkylidene oxindole) and by 1H NMR spectroscopy (Scheme 77).[155]
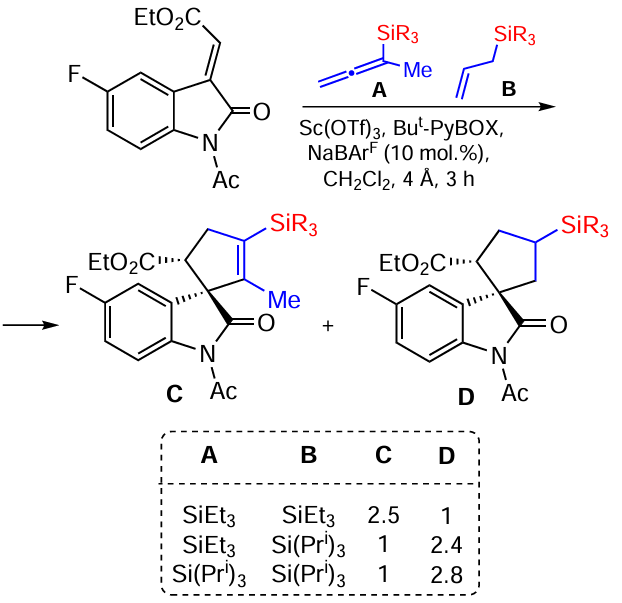
The reaction of allenyl(triethyl)silane and allyl(triethyl)silane with alkylidene oxindole under the conditions considered indicates the increased nucleophilicity of allenylsilane, which favours the formation of spirocyclopentene in a ratio of 2.5 : 1 relative to spirocyclopentane, and only these two cyclisation products are formed. When changing to allyl(triisopropyl)silane in a competitive reaction with allenyl(triethyl)silane, the process proceeds with reverse chemoselectivity, with the formation of mainly spirocyclopentane in a ratio of 2.4 : 1 to spirocyclopentene. At the same time, the reaction with allyl(triisopropyl)silane and allenyl(triisopropyl)silane shows an increased nucleophilicity of the allylsilane, for which the products are formed in a ratio of 2.8 : 1 (see table in Scheme 77). The dependence obtained suggests that allylsilane with a less bulky SiEt3 group is more nucleophilic. At the same time, judging from the comparative kinetic data shown in Scheme 74, allylsilane with a bulky SiPri3 group in its structure is more nucleophilic. This means that cyclization of allylsilane occurs via the formation of an α,β-stabilized intermediate carbocation (formed by a 1,2-silyl shift), which is preferred for silyl groups with bulky substituents. With allenylsilane, cyclization can proceed via concerted cycloaddition, which is again more efficient for the less bulky silyl group. Thus, both the nature of the π-system of the unsaturated silane and the size of the substituent at the silicon atom influence the relative rate and yield of the products in this case.[155]
As mentioned above, indoles are structural elements of many pharmaceuticals, natural compounds, agricultural preparations and functionalized polymers. Vinylsilanes are convenient building blocks in the synthesis of indoline derivatives. The reaction proceeds in the presence of transition metal catalysts, such as Rh(III), and aniline derivatives containing a 2-pyrimidine group at the amino group (Scheme 78).[156]
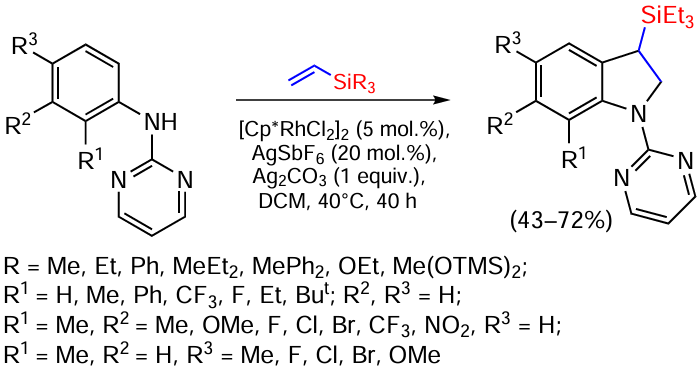
The reaction proceeds in the presence of pentamethylcyclopentadienylrhodium dichloride dimer [Cp*RhCl2], AgSbF6 as additive and Ag2CO3 as oxidant in dichloromethane under a nitrogen atmosphere. The efficient reaction requires a substituent on the nitrogen atom of aniline to coordinate the formation of a complex with Rh(III). The pyrimidine substituted at the C2 atom of the cycle proved to be the best. Substituted pyrimidines, pyridine and acetamide gave much lower yields of products. Anilines with both electron-donor and electron-acceptor groups were used as substrates. Even the presence of a large tert-butyl group in the o-position of aniline does not critically affect the yield of the product (62%).[156] Vinylsilanes containing alkyl, phenyl and alkoxy groups at the silicon atom allow the cyclization of anilines in good yields. In this case, the reaction of vinyl(triethoxy)silane gives a mixture of indoline and linear alkene, the product of the vinylation of aniline with the terminal carbon atom of the vinylsilane double bond in the o-position to the amino group. Other trialkoxy(vinyl)silanes, such as vinyl(trimethoxy-, triethoxy- and triisopropoxy)silanes, do not give reaction products. The reaction proceeds by activation of the C – H bond via o-metalation, with experiments on the o-deutero derivative showing that the C – H bond cleavage is reversible (kinetic isotope effect value was 0.97). The addition of radical inhibitors does not affect the reaction, whereas competitive reactions with substituted anilines indicate that the electron-donating groups in the cycle accelerate the reaction so that the resulting complex with rhodium is electrophilic in nature and is converted to an intermediate of o-rodiylation, which is further cyclised to the final product. It has been shown that C – H bond cleavage is not a rate-determining step and that six-membered cationic Rh(III) complexes are the key catalytic intermediates (confirmed by FAB-MS and NMR).[156, 157]
The synthesis of 2,4-diaryltetrahydropyrans by a multicomponent reaction involving the formation of a quaternary stereoactive centre has recently been published. The process is accompanied by a 1,2-shift of the phenyl group from the Si atom to the C atom (Scheme 79).[158]
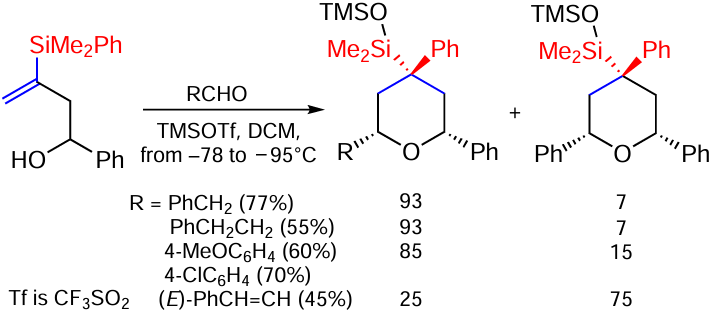
The reaction is applicable to a wide range of different alcohols and aldehydes. In addition, when benzyl alcohols are used, the reaction involves a Cope rearrangement of the oxonium type which competes with direct cyclisation, the course of which depends on the nature of the aldehyde used and the presence or absence of its excess. A two or three-fold excess of arylaldehydes or enals gives almost exclusively (55 – 60%, 80 : 20) Prince-type cyclisation products.[158]
The proposed mechanism of cyclisation and by-product formation is shown in Scheme 80.[158]
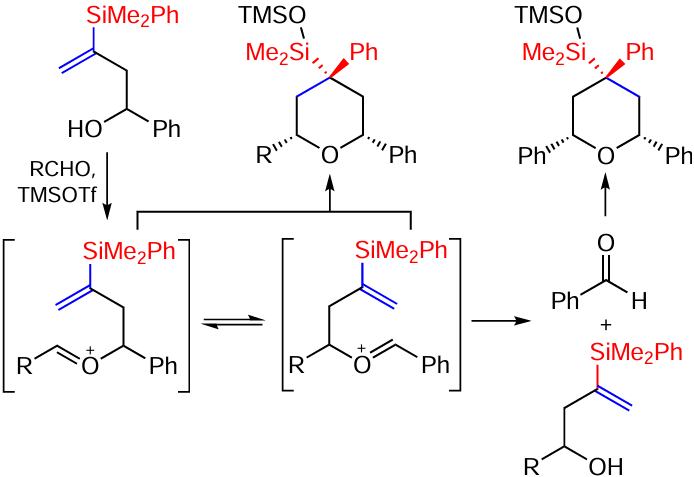
Vinylsilanes containing a phenyl group at the silicon atom undergo cyclization under the action of LiAlH4 with the double bond intact, forming five-membered cyclic vinylsilanes (Scheme 81).[159]
The product skeleton can be considered as a dihydrofuran into which a silicon atom is introduced. The compounds may be of interest for the synthesis of new silicon-containing bioactive small molecules.[30, 159]
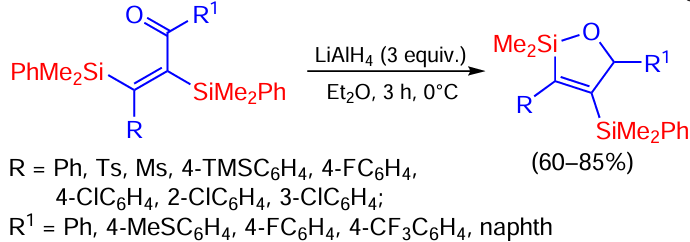
A vinyl(trimethyl)silane containing an ester group in position 2 is oxidized under the action of molecular iodine to give a silalactone containing a vinyl group at the silicon atom. The product can be further reduced by the action of LiAlH4 to a silafuran similar to that shown in the previous scheme (Scheme 82).[160]
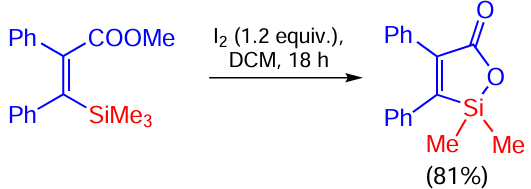
Vinylsilane containing a bromine atom in the 2-position and a benzyl group in the α-position is cyclized to 1-acetyl-2-tri(isopropyl)silylindene in good yield under the action of palladium acetate in the presence of XPhos, the double bond not being involved in the reaction (Scheme 83).[161]
A method is also known for the repeated trans-silylation of vinylboronates with vinylsilanes in the presence of Ru(CO)Cl(H)(PCy3)2 in polyethylene glycols to form 1-boryl-1-silylethenes (73 – 96%, 90 : 10).[162]
3.4. Reactions of N,S-nucleophile addition to vinyl and allylsilanes
The reaction of 2-mercapto-1,3-benzoxazole and 2-mercapto-1,3-benzothiazole with vinyl(triethoxy)silane was studied. At high temperatures in argon atmosphere, the reaction proceeds as an anti-Markovnikov addition in the absence of solvent and catalyst (Scheme 84).[163]
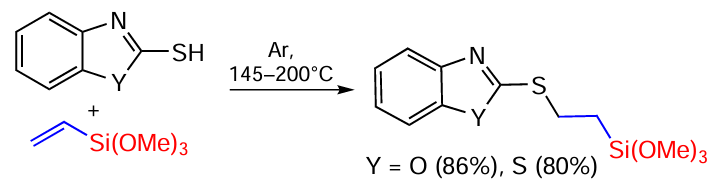
The addition of 2-mercaptobenzazoles to vinylsilane proceeds regioselectively against the Markovnikov rule at the terminal carbon atom. The presence of electron acceptor substituents at the silicon atom in vinylsilane (R = OMe) leads to additional polarisation of the C=C bond compared to vinyl(trimethyl)silane, increasing its reactivity. The reaction is carried out at reflux for 34 h in the absence of solvents and catalysts and proceeds by the radical mechanism in the presence of a small excess of vinyl(trimethoxy)silane, which acts as a radical acceptor. During the reaction, the temperature of the reaction mass was gradually raised to 145°C in order to achieve thiol-ene addition. It should be noted that the reaction of 2-mercaptobenzoxazole with vinyl(trimethoxy)silane in a sealed ampoule without access to atmospheric oxygen is faster (in 1 h) and gives the target product in 86% yield.[163]
The addition of 3-mercaptopropionic, thioglycolic or thioacetic acid to 1,3-divinyltetramethyldisiloxane, 1,3,5-trivinyl-1,3,5-trimethylcyclotrisiloxane and 1,3,5,7-tetravinyl-1,3,5,7-tetramethylcyclotetrasiloxane upon thermal or photochemical activation proceeds with similar regioselectivity and is accompanied by polymerization for the latter two substrates.[164]
The intermolecular hydroamination of a number of commercially available vinylsilanes by various amines in the presence of yttrium complexes has been studied. The reaction is highly selective for benzyl and aliphatic primary and secondary amines (Scheme 85).[165]
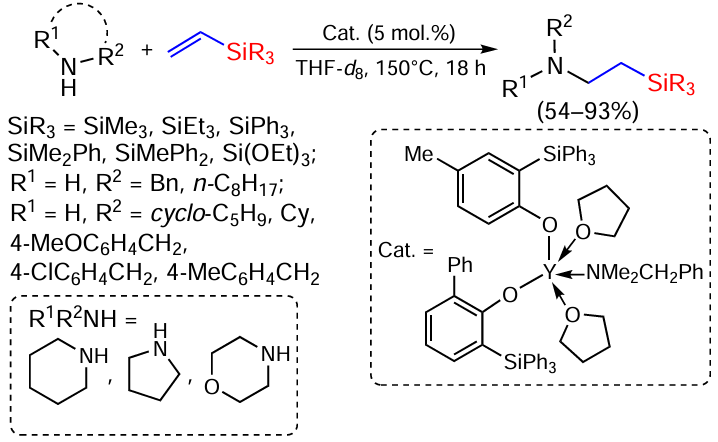
The introduction of trialkyl(vinyl)silanes into the reaction leads to a complete conversion of octylamine, in contrast to the sterically more hindered benzylamine, for which the conversion reaches only 18 – 26%. Vinyl(dimethyl)phenylsilane gives high yields, as does vinyl(methyl)diphenylsilane, vinyl(triphenyl)silane, but also only with n-octylamine. Benzylamine is also poorly reactive, apparently due to increased steric requirements.[165] The use of vinyl(triethoxy)silane leads to deactivation of the catalyst, presumably due to the strong coordination of the silane to the metal centre acting as a Lewis acid in the catalyst. Various primary and secondary, including cyclic, aliphatic amines also react, with the reaction being completed within 2 h in the case of pyrrolidine. p-Substituted benzylamines give products in 80 – 93% yield. However, the sterically more demanding N-methylbenzylamine gives the product in 21% yield regardless of the reaction time.[165]
Recently, the amination of alkenylsilanes with an allyl group in position 3 of the carbon chain in the presence of phosphineselenide as catalyst and PhI(OAc)2 as oxidant has been reported (Scheme 86).[166]
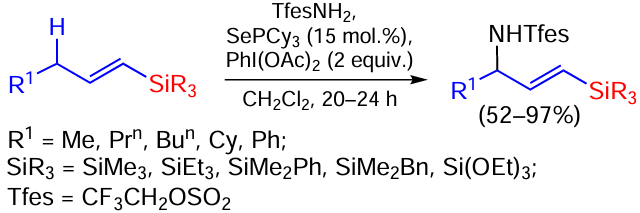
A notable feature of this reaction is that it proceeds without competitive reaction at C=C or C – Si bonds. Electron-deficient nitrogen sources, in this case sulfonamides, are good aminating agents for various carbofunctional groups. Various terminal vinylsilanes containing alkyl and aryl substituents at the silicon atom are involved in the reaction under consideration. Silanes with an internal double bond are also excellent reactants. In this case, the amination proceeds through a fragment distal to the silyl group. Amination of benzyl substrates and substrates containing a tertiary CH reaction centre is also efficient under these conditions. The resulting compounds can be converted to the corresponding silyl(allyl)amines by removing the sulfonamide protection.[166]
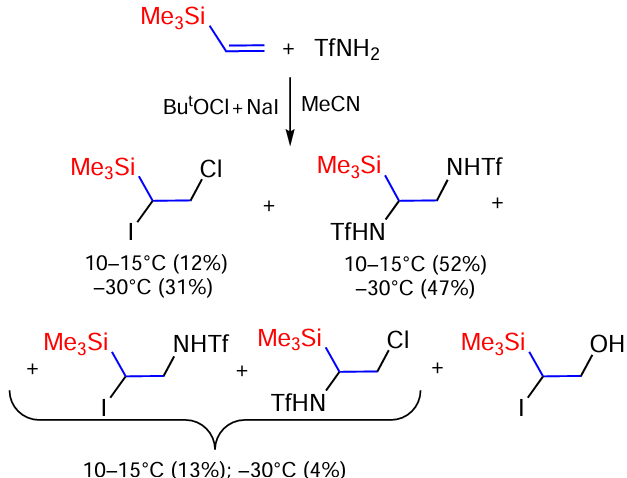
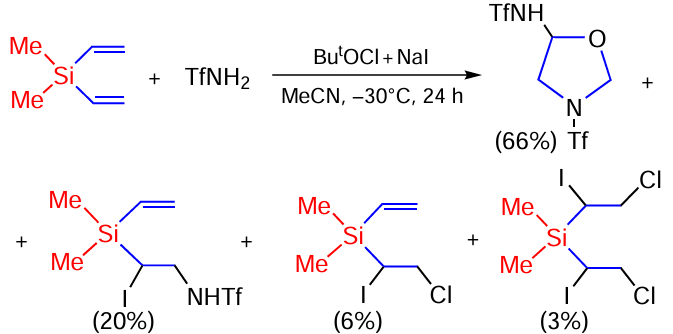
The oxidative addition of sulfonamides to vinyl(trimethyl)silane and divinyl(dimethyl)silane has been studied.[167, 168] Vinyl(trimethyl)silane reacts with triflamide at 10 – 15°C or –30°C to form five oxidative addition products (Scheme 87).
Arensulfonamides react differently with vinyl(trimethyl)silane over the temperature range of –30 to +45°C, leading to the formation of the halogenation product as the minor (6 – 30%) and aziridines as the major ones, in total yields of up to 91% (Scheme 88).[167, 168]
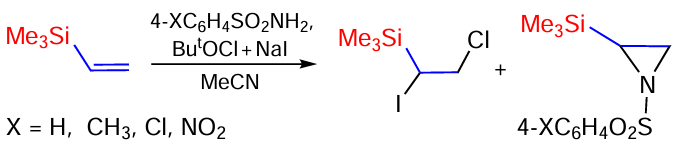
The reaction of divinyl(dimethyl)silane with sulfonamides was studied under the same conditions. The reaction with triflamide proceeds in high total yield. The main product is oxazolidine; in addition, an iodotriflamidation product is formed and small amounts of mono- and bis(iodochlorination) products have been isolated (Scheme 89).[167, 168]
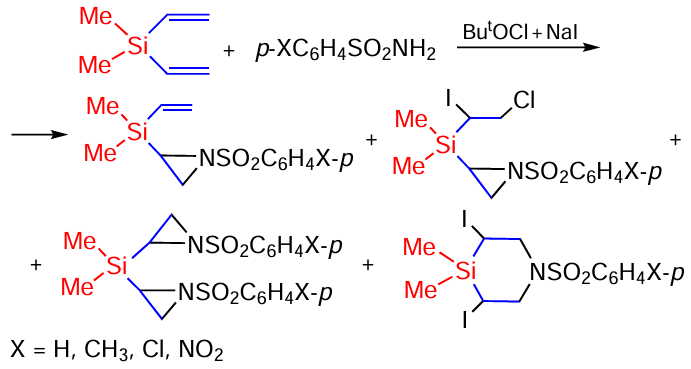
The reaction of divinyl(dimethyl)silane with arenesulfonamides produces aziridination products, 1,4-azasilinanes. The only products identical to those formed in the reaction with triflamide are those shown in Scheme 90.[167, 168]
The reactions of various sulfonamides with vinyl(trimethyl)silane in the presence of N-bromosuccinimide (NBS) have been studied. The reactions studied are regioselective and lead to bromosulfonamidation products capable of undergoing further transformations. Thus, the reaction of vinyl(trimethyl)silane with TsNH2 in CH2Cl2 in the presence of NBS was studied. At a sulfonamide : NBS ratio of 1 : 1, the yield of the target product was 88%. 1,2-Dibromoethyl(trimethyl)silane (7%) was present as an impurity. Based on the selection made from the results of the reaction with TsNH2 , the reaction of vinyl(trimethyl)silane with various sulfonamides was carried out in equimolar ratios (Scheme 91).[167, 168]
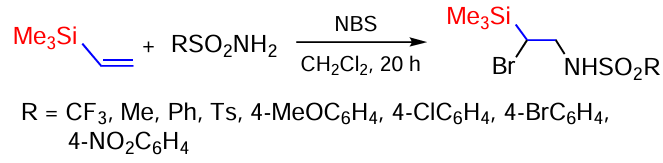
Studies have shown that, with the exception of the reaction with methanesulfonamide (48%), the yields of β-silylated sulfonamides are sufficiently high (72 – 88%) and the reaction can be used as a preparative method for their synthesis.[167, 168]
When vinylsilanes react with arenesulfonamides in the presence of NBS in acetonitrile, the corresponding acetamidines are also formed and one of the bonds of the divinylsilanes is halogenated (Scheme 92).[169]
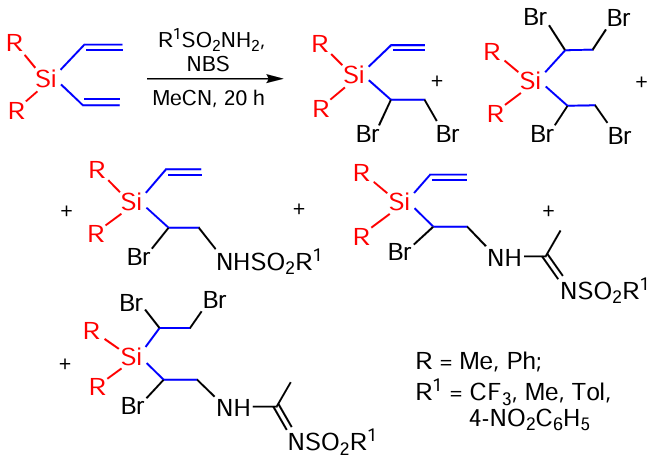
Some products are further cyclized to form otherwise hardly accessible heterocycles (Scheme 93).[169]
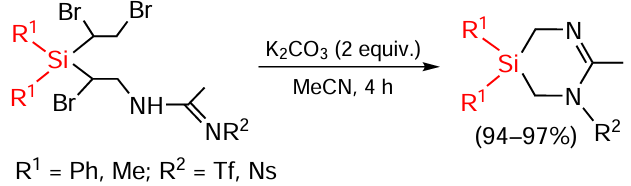
Corresponding amidines have also been obtained by reactions of sulfonamides and carboxamides with vinyl, trivinyl and tetravinyl silanes.[170-172]
The reaction of triflamide with 1,1,3,3-tetramethyl-1,3-divinyldisiloxane is an unusual example of oxidative sulfonamidation and leads to the formation of two heterocyclic products, N-((6-iodo-2,2,7,7-tetramethyl-4-(triflyl)-1,4,2,7-oxaazadisylepane-3-yl)methyl)triflamide and 2,2,4,4-tetramethyl-6,8-bis(triflyl)-3-oxa-6,8-diaza-2,4-disilabicyclo[3.2.2]nonane (Scheme 94).[173]
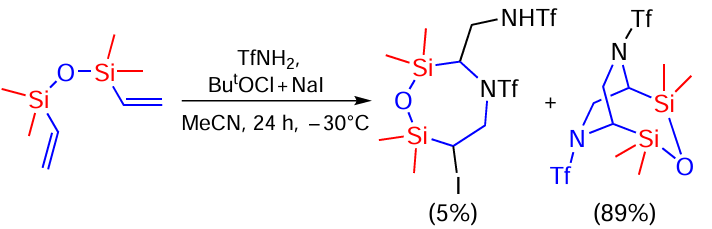
The structure of the compounds suggests that they can be formed sequentially. In this case, the first step may be a sequential attack of the substrate by the TfNHI intermediate formed in the BuOCl/NaI system with the formation of the α-iodo-β-amination product on both double bonds, which undergoes cyclisation to the final product under the action of TfNHI (Scheme 95).[173]
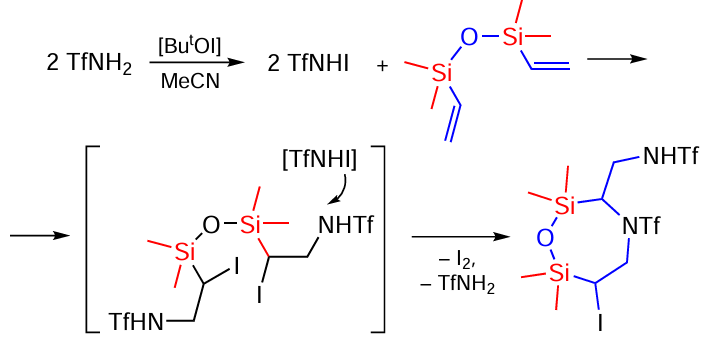
In the second step, a bicyclic compound is formed (Scheme 96) under the action of TfNHI.
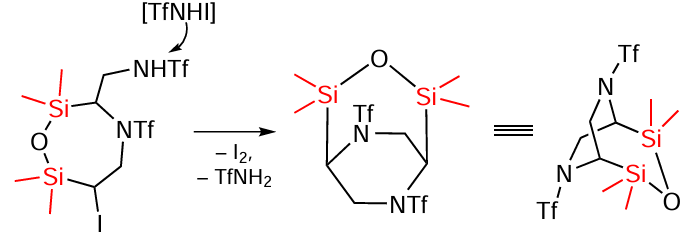
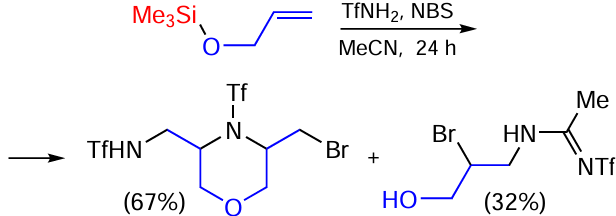
The reaction of triflamide with allyloxy(trimethyl)silane gives N-((5-(bromomethyl)-4-(triflyl)morpholin-3-yl)methyl)triflamide and N-(2-bromo-3-hydroxypropyl)-N'-(triflyl)acetamidine in a ratio of approximately 2 : 1 (Scheme 97).[173]
In the presence of Cp*Co(III), regioselective [4 + 2]-cyclization of N-chloramides and vinylsilanes proceeds to form 4-silylated isoquinolones (Scheme 98).[174]
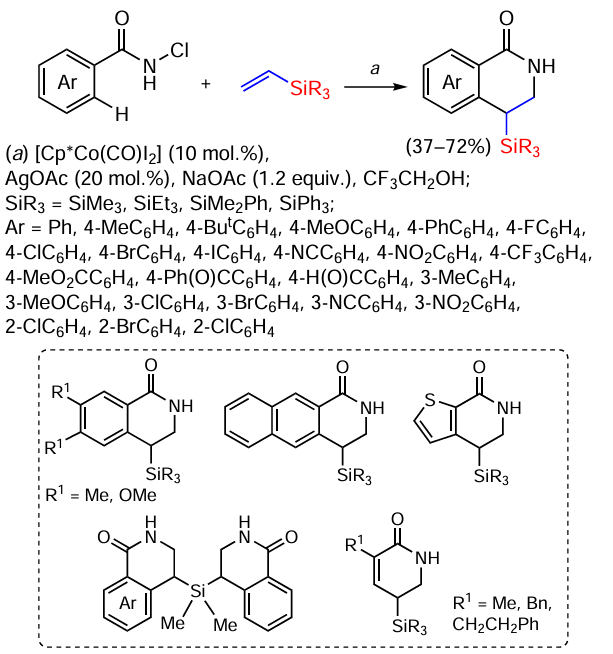
The reaction of N-chlorobenzamides with vinyl(trimethyl)silane, vinyl(triethyl)silane or vinyl(dimethyl)phenylsilane gives products in 61%, 46% and 62% yield respectively. In addition, vinylsilanes with two vinyl groups, such as divinyl(dimethyl)silane, give the corresponding monoannelated (61%) and bis-isoquinolones. Vinyl(triphenyl)silane does not react under these conditions.
N-Chlorobenzamides with electron donating groups such as Me, OMe, But and Ph in the p-position give cyclic products in 50 – 70% yields. Benzamides with halogen atoms (F, Cl, Br and I) in the p-position also react to give products that can be further functionalized. The reaction also proceeds with substrates containing electron-acceptor groups CN, NO2 and CF3 to give the corresponding 4-silylated isoquinolones in 69, 42 and 66% yields. In addition, N-chlorobenzamides with electrophilic functional groups such as ester, ketone or aldehyde functional groups also participate in the reaction (yields 61 – 62%). The reaction proceeds worse with o- and m-substituted substrates. In the case of the N-chloramide derivative of 2-naphthalene, regioselective activation of the C – H bond was observed with formation of the cyclisation product in position 3 of the substrate in 36% yield. N-chloroacrylamide derivatives also reacted to form products in low to moderate yields (32 – 62%). The lower yields can be explained by the low reactivity of the olefin bond for CH activation under the conditions considered.[174]
The reaction of pyridine-2-sulfenyl- and pyridine-2-selenenyl halides with vinyl(trimethyl)silane leads to the formation of new annelated compounds, 2-functionally substituted 2,3-dihydro[1,3]thiazolo- and 2,3-dihydro[1,3]selenazolo[3,2-a]pyridines (Scheme 99).[175]
4. Reactions of vinyl- and allylsilanes with desilylation
Recently, the first example of a carbonyl-ene cyclisation-type reaction of 2-substituted vinylsilanes with α-ketoesters has been found. 1,2-Disubstituted vinylsilanes, acting as nucleophiles, react intramolecularly with the keto group of the 2-oxoacetyl moiety of the substrate to form indenols with a quaternary carbon atom (Scheme 100).[176]
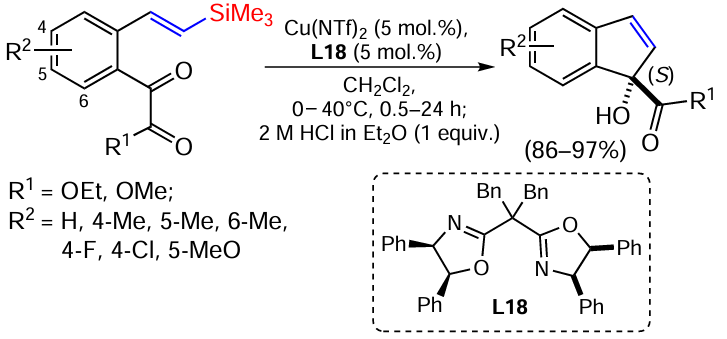
The complex of N,N-bis(triflyl)imidate of copper(I), CuN(Tf)2 , and a chiral bis(oxazoline) ligand containing benzyl groups at the central carbon atom gave the best yield. The reaction was enantioselective (94 – 97% ee), in high yield and under mild conditions.
Substrates with a methyl group at the C4, C5 or C6 position react at room temperature and give the best yields. Substrates with electron-withdrawing groups (F, Cl) at the C4 position are less reactive, so the mixture must be heated to 40°C to complete the reaction. An electron-donor group (MeO) at the C5 position increases the reactivity of the substrate, giving the product in 91% yield in 30 min at room temperature, but the enantioselectivity decreases to 71% ee. Cooling to 0°C increases the yield to 97% and the enantioselectivity to 77% ee.
(E)-2-Oxo-2-(2-(2-(trimethylsilyl)vinyl)phenyl)acetate shows high reactivity in cyclisation with vinylsilanes because the intermediate carbocation is stabilised on the one hand as a β-silyl cation, possibly due to anchimeric assistance, and on the other hand as a benzylic cation (Scheme 101).[176]
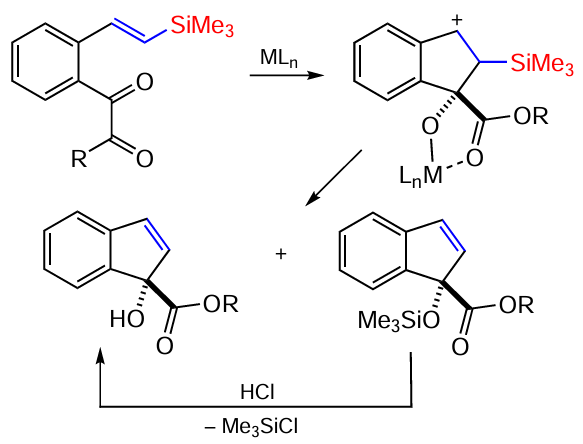
At a catalyst concentration of 10 mol.%, the intermediate Me3SiO-substituted product is formed in up to 72% yield, but when the concentration is reduced to 5 mol.%, the yield drops to 12%.[176] Washing with HCl solution allows complete conversion of the trimethylsilyl ester to the target (S)-1-hydroxy-1H-inden-1-carboxylate.
Homoallyl alcohols containing a triethylsilyl group at the double bond react with aldehydes to give 2,6-disubstituted 3,6-dihydro-2H-pyrans with (R,S)-configuration of the chiral carbon atoms in good yields (Scheme 102).[177]
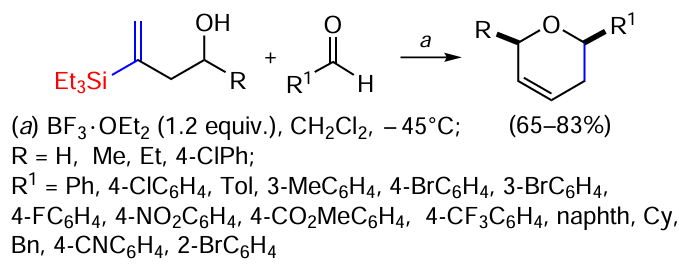
The reaction is applicable to a fairly wide range of aldehydes, particularly substituted benzaldehydes. 2-Naphthaldehyde is also active in this reaction, giving regio- and stereoselectively the product in 65% yield. Reaction with aliphatic aldehydes (cyclohexyl- and benzyl-substituted) gives mixtures of regioisomers with different double bond positions in 2 : 1 ratio. Depending on the substrate, the reaction proceeds as a concerted or stepwise oxonium-ene condensation with cyclization. For benzaldehyde derivatives, concerted condensation leads to the formation of a single regioisomer (Scheme 103).[177]
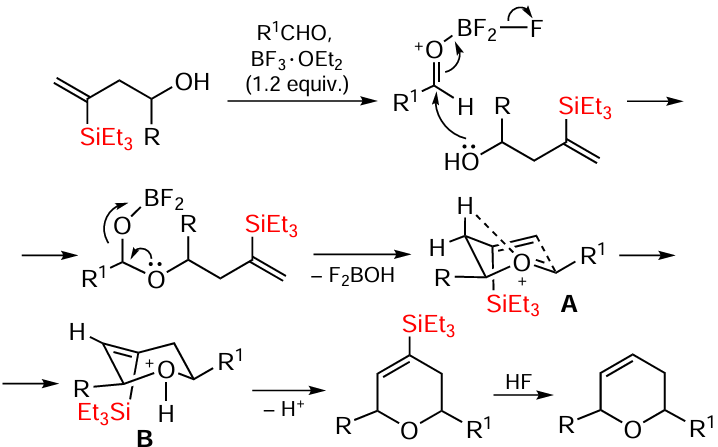
A Lewis acid activates the carbonyl group for nucleophilic attack by the hydroxyl to form the oxocarbenium ion A, which undergoes a 1,3-prototropic rearrangement to the oxonium intermediate B. Its deprotonation and desilylation under the action of in situ generated HF leads to the formation of the target dihydropyrans. The formation of a mixture of regioisomers can be explained by a Claisen rearrangement in intermediate A. The reaction is diastereoselective and applicable to the synthesis of biologically active dihydropyrans.[177] Not only homoallyl alcohols but also their silyl esters in the presence of BiBr3 can be used in the reaction, in which case only one regioisomer of the double bond position is formed.[178]
Similarly, vinylsilanols, which have a trimethylsilyl group at the terminal position of the double bond and are homoallyl alcohols, react with 2,3-dihydrofuran or 3,4-dihydro-2H-pyran to form 5,6-dihydro-2H-pyrans or 4-chlorotetrahydropyrans.[179] It is worth noting that in the main diastereomeric products, the substituents in the α-position to the oxygen atom are in the equatorial positions of the cycle, as is the chlorine atom at the C4 atom (Scheme 104).
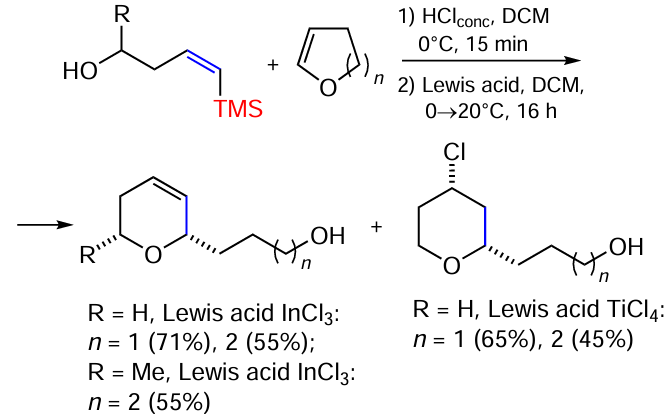
The reaction is regio- and stereoselective. When TiCl4 is chosen as the Lewis acid, the reaction leads exclusively to the formation of the corresponding 4-chlorotetrahydropyrans, presumably via protodesilylation following cyclisation. In contrast, the corresponding 5,6-dihydro-2H-pyrans are formed in good yields, including the enantiomerically pure 6-methyl-substituted product when InCl3 is used.[179]
An aza-silyl variant of the Prins reaction of vinylsilanes containing a homoallylamine moiety with aldehydes in the presence of Lewis acids is also known, giving tetrahydropyridines as products (Scheme 105).[180]
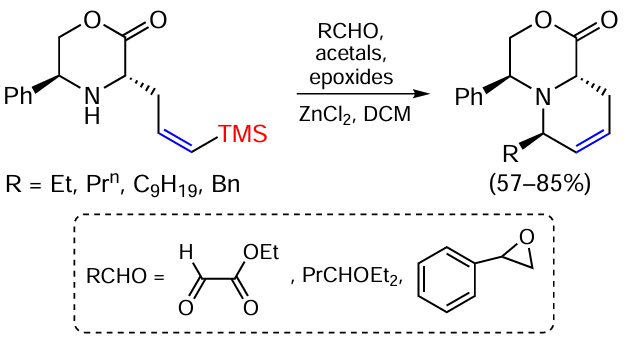
The reaction proceeds via an iminium intermediate and a secondary carbocation stabilised by a silyl group in the β-position. In the presence of ZnCl2 , the reaction is accompanied by desilylation to give bicyclic compounds in high yields, including as individual diastereomers and enantiomers, as shown in Scheme 105. The reaction also proceeds when acetals or epoxides are used as substrates, although in slightly lower yields.[180]
Allylic alcohols chemo-, diastereo- and enantioselectively undergo an allyl-allyl coupling reaction with α-silyl substituted allyl boronates. The reaction is catalysed by an iridium complex with a chiral ligand (Scheme 106).[181]
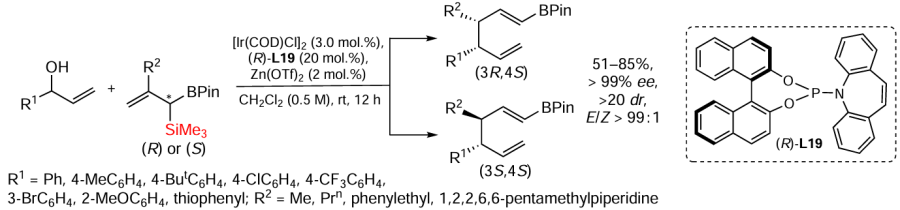
As shown in Scheme 106, α-silyl-substituted allylboronates react with allyl electrophiles to give a variety of enantioenriched (E)-1-boryl-substituted 1,5-dienes in good yields, with desilylation far prevailing over deborylation. This reaction allows the synthesis of the corresponding (3R,4S)- or (3S,4S)-diastereomers by simply changing the chirality of the starting α-silyl-substituted crotyl boronate ester. The use of secondary allyl alcohols with electroneutral, donor and acceptor substituents at the p-position of the benzene ring has hardly any effect on the efficiency of allyl-allyl bond formation (yields up to 61%). Secondary allylic alcohols containing substituents in the m- or o-position give slightly higher yields (~ 71%). The method allows the formation of vicinal stereocentres in 1,5-dienes using chiral reagents and a catalyst, and the synthesis of all four possible stereoisomers of (E)-1-boryl-substituted 1,5-dienes with high chemo-, diastereo- and enantioselectivity. DFT calculations showed that the selectivity of the process is influenced by several factors, including steric, β-effect of the silicon atom and high desilylation rate.[181]
The dehydration/functionalization reaction of allylsilanes with concomitant desilylation to form 1,5-dienes is known (Scheme 107).[182]
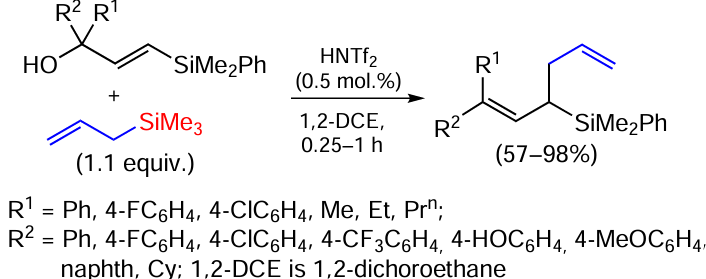
Triflimide as a strong Brønsted acid was used as a catalyst in the reaction. Allyl(trimethyl)silane gave the corresponding products in good to quantitative yields. Homoallyl alcohols containing aryl and alkyl groups were used as substrates. The reaction proceeds with complete control of regioselectivity.[182]
An Rh-catalysed acylation reaction of vinylsilanes has recently been described. The reaction demonstrates the feasibility of a wide range of vinylsilanes (Scheme 108).[183]
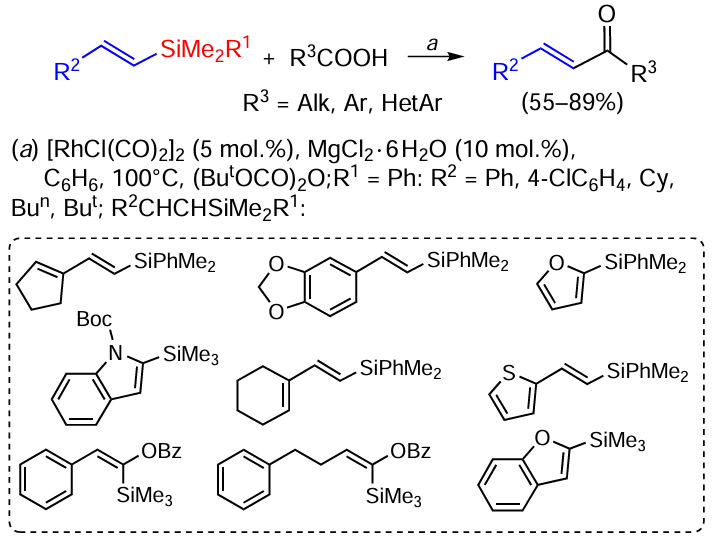
Homologues of benzoic acid, 2-naphthylacetic acid and other aromatic carboxylic acids, acrylic acid, alkyl carboxylic acids, including those with cyclic, secondary and tertiary linear substituents, give good yields. Carboxylic acids with basic heterocyclic substituents of particular interest in medicinal chemistry (2-tetrahydrofurancarboxylic acid, 2-thiophenecarboxylic acid, 2-furancarboxylic acid, benzofuran-2-carboxylic acid, and 1-methyl-1H-indole-2-carboxylic acid and indomethacincarboxylic acid) also react well under these conditions, including in the synthesis of biologically active compounds. The reaction is an efficient method for the preparation of α,β-unsaturated ketones.[183] Similar reactions in the presence of acid anhydrides are described in a recent review.[184]
Enones can also be prepared from vinylsilyl alcohols by the reaction with TBAF in the presence of KOH and subsequent oxidation of the hydroxyl group to a carbonyl group (Scheme 109).[185]
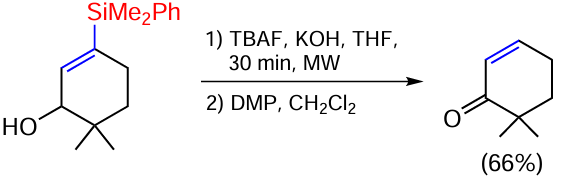
The synthesis is a protodesilylation of the substrate, which consists in the substitution of the phenyl group at the silicon atom with a hydroxyl group to form a silanol (recorded by NMR), which under the action of fluoride ion (from TBAF) is desilylated to form an alcohol. The resulting allyl alcohol is oxidised by DMP to give the corresponding enone.[185] TBAF is an effective desilylating agent, including in the reaction with vinylsilanes having two silyl groups in the trans position at the double bond (Scheme 110).[159]
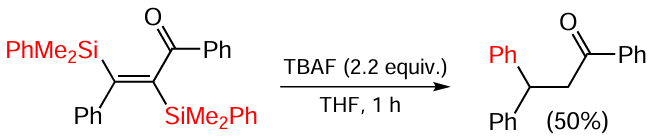
Scheme 110 shows that the ketone formed is the result not only of double desilylation of the substrate, but also of phenyl migration from a silicon atom to a neighbouring carbon atom.[159]
The silyl groups at the double bond can be easily differentiated chemically by the action of bases. The group with Ar = o-C6H4C(O)N(Pri)2 of the two silyl groups in the Z-isomer of α,β-disilylated styrene (Scheme 111) appears to be more reactive than SiMe3 and is selectively cleaved by TBAF to give E-β-(trimethylsilyl)styrene in 97% yield.[186]
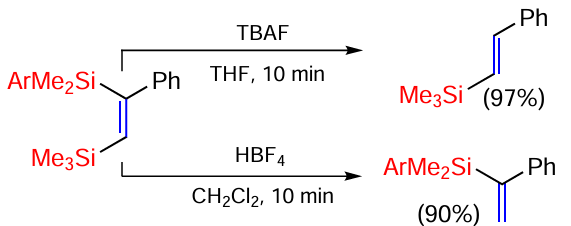
On the other hand, the acid-catalysed desilylation reaction leads exclusively to the cleavage of the SiMe3 group to give α-silylated styrene in 90% yield. Silaflavone can be obtained by a similar reaction in 72% yield as a result of the desilylation of vinyldisilane under the action of BF3 (Scheme 112).[186]
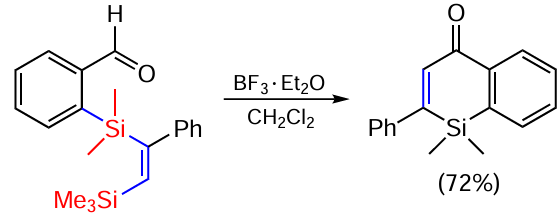
The reaction proceeds as a desilylation, cyclisation and oxidation to form a flavone sila-analogue in 72% yield.[186] Under the action of BF3 – Et2O, the desilylation of tert-butyl(vinyl)dimethylsilanes also proceeds readily, with retention of configuration of the double bond.[187]
The silyl group undergoes electrophilic substitution by electrophilic iodine sources such as N-iodosuccinimide (NIS). Substrates with an aryl group at the allyl position undergo desilylation with simultaneous intramolecular cyclisation to form indenes (Scheme 113).[161]
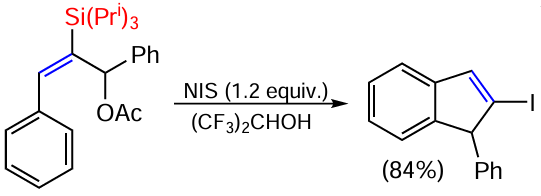
Vinyliodide is formed in 84% yield when substrates with a lower ionisation potential of the allylic moiety are used due to the electron-donating effect of the trialkylsilyl group and hexafluoroisopropanol is replaced by 2,2,2-trifluoroethanol (Scheme 114).[161]
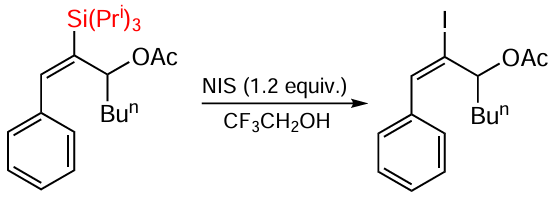
In similar desilylation reactions, Ag2CO3 or AgOAc additives are sometimes used to bind free I2 and HI formed from NIS in order to avoid the formation of by-products and thus increase the yield of the target products.[187, 188]
Iododesilylation is also observed when vinylsilanes with fully substituted double bond are treated with a small excess of ICl, and the product is formed in near quantitative yield, with an E/Z isomer ratio of 95 : 5 (Scheme 115).[189]
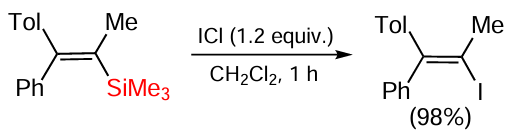
Olefination of α-trialkylsilyl-β-alkyl-α,β-unsaturated esters with aliphatic aldehydes by Peterson desilylation is known to yield isomeric substituted conjugated diene products with high selectivity (Scheme 116).[190]
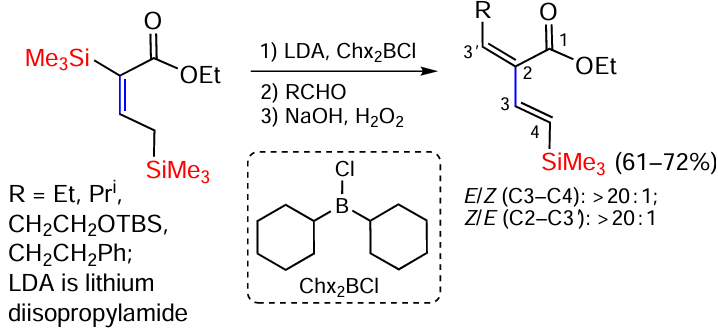
The proposed mechanism involves four consecutive stages (Scheme 117).[190]
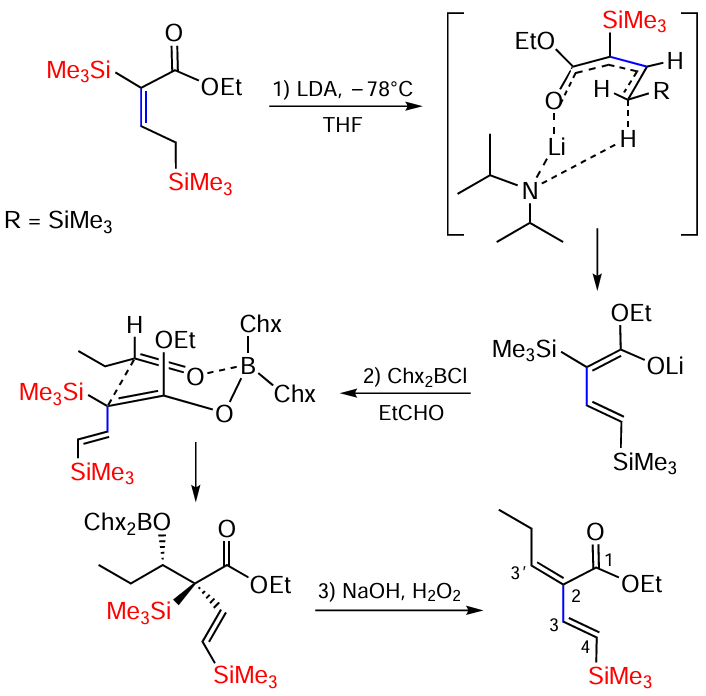
In the first stage, due to the CIPE effect (complex induced proximity effect), an eight-centred cyclic transition state is formed under the action of LDA, which is deprotonated with high stereoselectivity in the configuration of both double bonds (E/Z, Z/E > 20 : 1). In the second stage, a lithium enolate is formed and then trans-metalation proceeds under the action of excess Chx2BCl (dicyclohexylchloroborane) to form a boron dienolate intermediate, preserving the stereoconfiguration of the C1=C2 and C3=C4 double bonds. The dienolate then undergoes aldol condensation with propanal according to the Zimmermann-Trexler model, which assumes that the stereocentre configuration of the initial enolate is maintained in the reaction product during aldol condensation. Desilylation also occurs at the same stage. The final step is the oxidation of the intermediate borinate under basic conditions to the conjugated diene.[190]
Similar α-trimethylsilyl-β-alkyl(aryl)-α,β-unsaturated esters undergo desilylation under the action of N-halogen succinimides NXS (X = Cl, Br) in DMF to form β-substituted α-halogenated α,β-unsaturated esters in moderate to high yields with dr values > 20 : 1 (Scheme 118).[191]
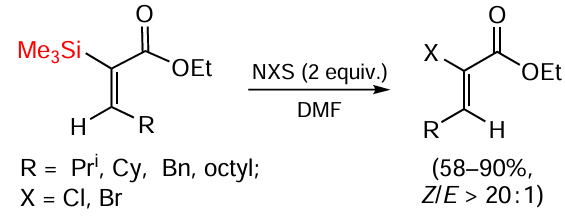
The reaction involves inverting the stereochemistry in the reaction products relative to the substrates (Scheme 119).[191]
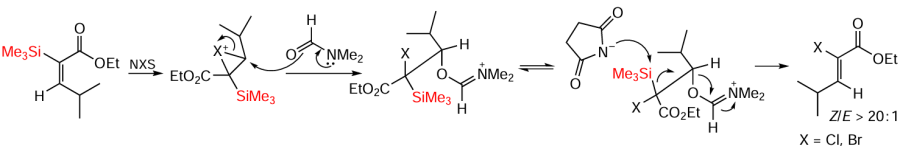
The halogenonium cation formed in the reaction with NXS undergoes regioselective ring-opening with DMF via the SN2 mechanism. Monitoring of the reaction revealed almost exclusive formation of Z-alkenes as reaction products (Z/E ratio > 20 : 1), so Probasco and Jennings suggested[191] that only a rotamer with an anti-periplanar arrangement of the eliminating groups, which is more favourable than syn-periplanar from a stereoelectronic point of view, enters the desilylation reaction by the E2 mechanism upon action of the succinimide anion. The simultaneous elimination of the trimethylsilyl group and the DMF residue leads to the stereoselective formation of reaction products with a reversal of the configuration of the substituents at the double bond.[191]
Desilylation also occurs during the synthesis of 1,4-dienes in the presence of gold-based catalysts via the reaction of vinyl diazo compounds and alkenylsilanes. In most cases, the reaction is regio- and stereoselective, with the silyl group acting as a regio- and stereocontrolling group (Scheme 120).[192]
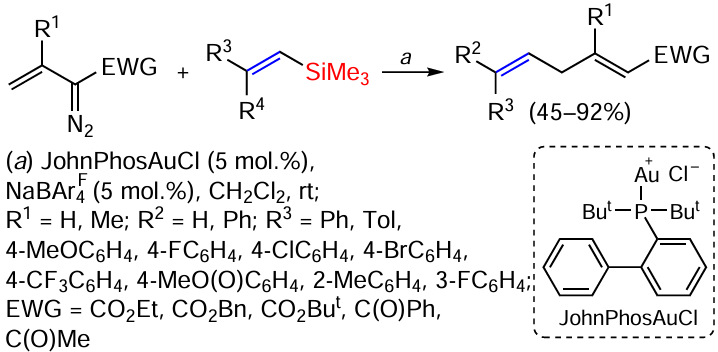
Effective reaction requires the presence of an aryl group in the β-position of the vinylsilane relative to the silicon atom. For example, vinyl(trimethyl)silane and β-furyl- or β-alkyl-substituted vinylsilanes do not react.[192] The reaction starts with the decomposition of the diazo derivative to form the Au-carbenium cation A, in which the terminal olefin carbon atom is attacked by the α-carbon atom of the vinylsilane (relative to the TMS group) to form the benzyl-type carbocation B, which is further stabilised by conjugation to the TMS group in the β-position. The latter undergoes intramolecular 1,4-migration of the TMS group to form the intermediate C, which gives the target diene in the presence of trace amounts of water in the reaction medium (Scheme 121).[192]
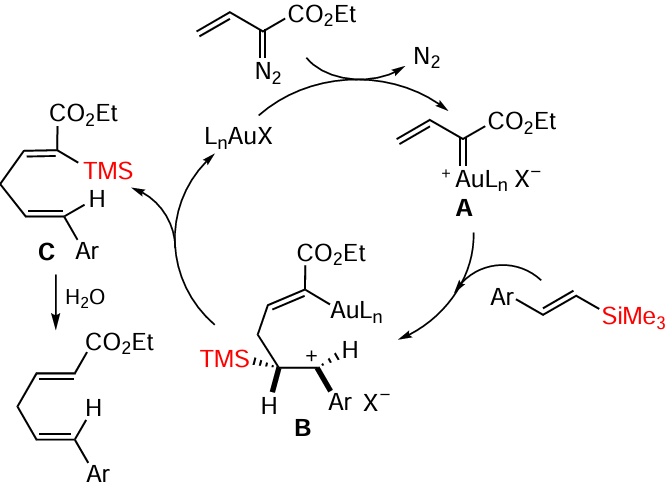
Desilylation of fluorinated vinylsilanes is one of effective methods of introducing fluoroalkenyl moieties into the aryl ring. For example, in the presence of CuOBut and 1,10-phenanthroline, a cross-coupling reaction of dimethyl(phenyl)trifluorovinylsilane with iodobenzene to give α,β,β-trifluorostyrene occurs in good yields (Scheme 122).[96]
The palladium-catalysed cross-coupling of acid- and base-stable organosilanes with aryl and alkenyl halides is known (Scheme 123).[193]
The reaction proceeds in the presence of CuI · P(OEt)3 and Bu4NF – (tBuOH)4 and is accompanied by desilylation and formation of di- and trisubstituted alkenes with complete retention of configuration. The method is versatile for the synthesis of highly substituted alkenes. Benzyldimethylsilyl substituted alkenylsilanes are quite stable towards acids and bases, but are easily converted to pentacoordinated silicate anions upon treatment with TBAF. Alkenyl derivatives of copper(I) are then formed in the presence of Cu(I), which react with organohalides to form coupling products (Scheme 124).[193]
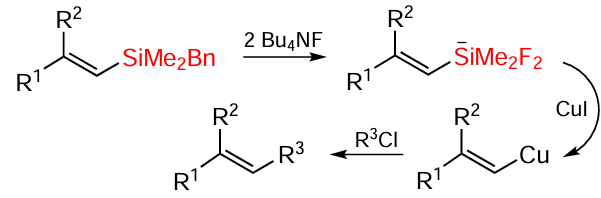
Similarly, in the presence of CuI and tetrabutylammonium difluorotriphenylsilicate (TBAT) as a fluoride ion source, vinyl(triethoxy)silanes and allyl bromides are desilylated to form 1,4-dienes.[194] Vinyl(triethoxy)silanes also undergo cross-coupling reactions with alkynyl bromides in the presence of [Cu(MeCN)4]PF6 and tetrabutylammonium bifluoride (TBAHF2) accompanied by desilylation.[195]
Recently, a method for the N-vinylation of amides and azoles by vinyl(trimethoxy)silane has been developed. The reaction is catalysed by CuF and DMAP, proceeds at room temperature in good yields and allows the synthesis of a wide variety of N-vinyl monomers (Scheme 125).[196]
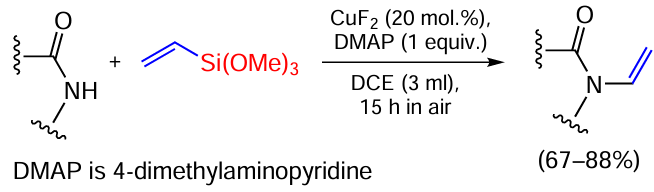
The reaction yields vinyl derivatives of β-lactams, imides, amides, oxazolidinones, succinimide, pyridinone, phthalimide, isobutyramide, oxazolidin-2-one, 5-methylpyridin-2(1H)-one, quinoline-2(1H)-one, isoquinoline-1(2H)-one, acridin-9(10H)-one, quinoxaline, 5,5-dimethylhydantoin, pyrazinamide, nicotinamide, benzamides.[196]
The reaction of diallyl(dimethyl)silane with triflamide under oxidative conditions leads to desilylation. The products were obtained in yields of 61 and 28%, respectively (Scheme 126).[197]
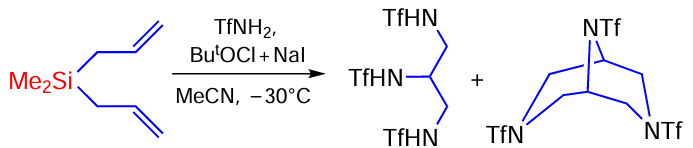
Among the desilylation reactions, the Hiyama – Denmark reaction [198] (a modification of the Hiyama reaction), which is a palladium-catalysed cross-coupling of deprotonated silanols with vinyl and aryl halides, is of great importance.[199] However, this reaction requires the use of vinylsilanols or silanolates, which are very difficult to work with because they readily form siloxanes in the presence of air moisture, or silicon-activated vinylsilanes, the range of which is very limited. The Hiyama-Denmark reaction sometimes does not require a fluoride anion as an activator,[199] so it can be used with substrates carrying additional silyl protecting groups. The process is feasible in large tonnage reactors.[198]
A variant of such a reaction using cyclic vinyl silanes is known (Scheme 127).[200]
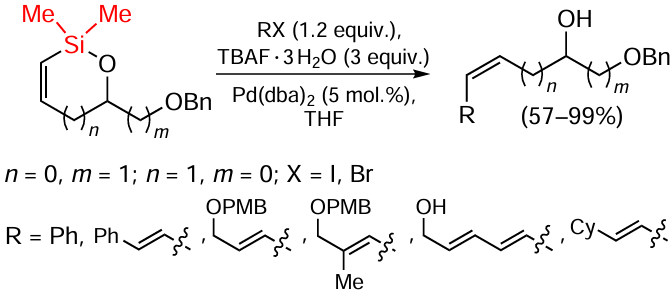
Scheme 127 shows the use of the reaction for the synthesis of polyenes. Pd(dba)2 is used as catalyst and TBAF as activator. The reaction is carried out in THF at room temperature. Cross-coupling in the presence of iodobenzene leads to both 5-membered 1,2-oxasylol and its 6-membered analogue 1,2-oxasiline in 98 – 99% yield. It should be noted that replacing the methyl groups at the silicon atom with ethyl groups significantly slows down the reaction (from 15 – 120 min to 24 h).[200]
Recently, a variant of this reaction has been developed involving inactivated (no oxygen atom at the silicon) tetrasubstituted vinylsilanes, important tetracoordinated silicon compounds, and aryl halides. The source of the vinyl group was dimethyl(5-methylfuryl)vinylsilanes (Scheme 128).[201]
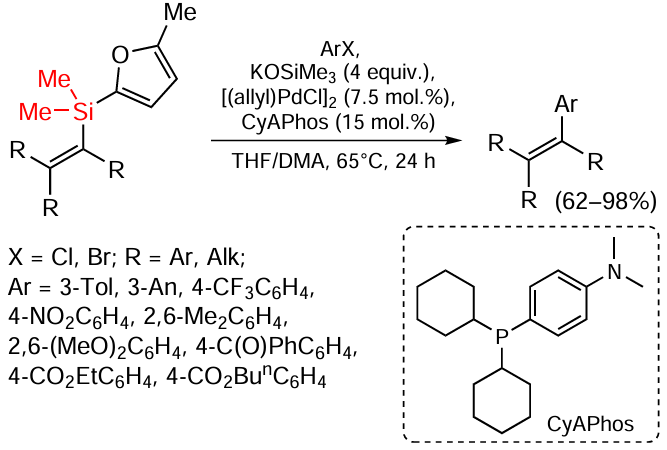
Cross-coupling proceeds well with aryl chlorides and aryl bromides, the reactions proceed to form tetrasubstituted alkenes with high stereospecificity. The range of substituents in the vinylsilane molecule is very wide. In addition, a mild base (KOSiMe3) and common solvents (THF/DMA) are used in the reaction. Importantly, no toxic additives such as 18-crown-6-ester are required to activate the reaction. The conditions are also suitable for 1,1,2-trisubstituted vinylsilanes as substrates.[201]
Vinylsilanes can be precursors of nucleosides with a stereogenic centre. The cyclisation reaction proceeds through a step of radical transfer of the vinyl group, which is efficient under conditions of photooxidation-reduction catalysis (most efficient in the presence of hexafluorophosphate 4,4´-bis(1,1-dimethylethyl)-2,2'-bipyridin-N1,N1']bis[2-(2-pyridinyl-N)phenyl-C]iridium(III) (Ir(dtbbpy)(ppy)2PF6), activated by irradiation of the reaction mixture with a blue light-emitting diode). The transfer of the vinyl group from the silicon to the radical centre can proceed from either the re- or si-side of the radical plane,[202] and the authors have shown that the corresponding transition states differ by 3.2 kcal mol–1 in favour of the experimentally observed kinetically controlled product shown in Scheme 129.
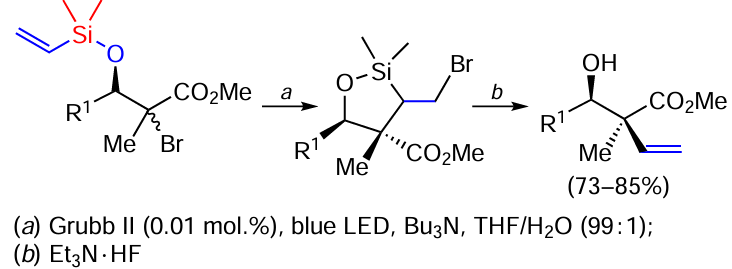
The final product is then subjected to lactonisation to synthesise new nucleoside analogs that exhibit biological activity against gastric cancer cells.[202] The final desilylation step can be carried out in the presence of HCl in methanol.[203] Some examples of the use of unsaturated silanes in asymmetric reactions catalysed by visible light are described in a recent review.[204] Examples of desilylation reactions of other silanes are given in [23, 27, 28, 35].
5. Conclusion
This review shows that recently there has been a steadily growing interest in new methods of preparation, chemical transformations and applications of unsaturated silanes both in organic and organoelement synthesis and as building blocks for the design of new drugs and agricultural products, luminescent, optically active and other functionalized compounds. The most important reaction for the synthesis of unsaturated silanes was and still is the catalytic hydrosilylation of acetylenes, which began with the use of platinum catalysts by Speier (1957) and Karstedt (1973). The renaissance in this field has been caused both by the involvement in hydrosilylation reactions of various highly unsaturated substrates — allenes, dienes, and enynes — and by the use of various metal complex catalysts based on manganese, iron, cobalt, nickel, ruthenium, rhodium, palladium, iridium, and platinum. The hydrosilylation of alkynes can proceed as α- or β-silylation, with different degrees of regioselectivity and, in the latter case, stereoselectivity, giving Z- or E-isomers of vinylsilanes and offering structural diversity and the possibility of targeted synthesis of products by choice of conditions and catalysts. The hydrosilylation of conjugated dienes proceeds with different regioselectivity depending on the reagent and the structure of the diene, and can give both 1,2- and 1,4-addition products.
Alternative routes for the synthesis of unsaturated silanes include the reaction of disilanes with alkynes, dienes or enynes, yielding both mono- and disilylated products, and the coupling of vinyl halides with organosilicon derivatives.
The variety of vinyl silane transformations includes asymmetric hydrogenation with high enantioselectivity, addition reactions with regioselectivity according to or against the Markovnikov rule. Vinyl-, allyl- and allenylsilanes undergo vinylation reactions with aldehydes, ketones and arenes to form carbo- and heterocycles. A wide range of hydroamination and oxidative amination reactions of vinylsilanes have allowed to obtain various functional derivatives with organosilicon substituents capable of further functionalization and formation of heterocycles, including silaheterocycles and heterocycles with exocyclic silicon-containing substituent. Cyclisation reactions can be followed by desilylation to give carbocycles, O- or N,O-heterocycles. The yields of the resulting products are usually quite high.
Further development of the chemistry of unsaturated organosilicon compounds can be predicted in the direction of the synthesis of new drugs with different pharmacological activities on the basis of functionalization reactions with suitable nucleophilic reagents, as well as the development of synthesis methods and the use of unsaturated silanes as building blocks in organometallic and organic chemistry.
6. List of abbreviations
An — anisyl,
BTM — 2-phenyl-2,3-dihydroimidazo[2,1-b][1,3]benzothiazole,
Cinnamyl — 3-phenylprop-2-en-1-yl,
Co(acac)3 — cobalt acetylacetonate,
COD — cyclooctadiene,
COE — cyclooctene,
Cp — cyclopentadienyl,
CPME — cyclopentyl methyl ether,
Cy — cyclohexyl,
DABCO — 1,4-diazabicyclo[2.2.2]octane,
Dba — dibenzylideneacetone,
1,2-DCE — 1,2-dichloroethane,
Dcyp — 1,3-bis(dicyclohexylphosphino)propane,
DIPEA — N,N-diisopropylethylamine,
DMA — dimethylacetamide,
DMAP — 4-bimethylminopyridine,
Dppf — 1,1'-bis(diphenylphosphino)ferrocene,
Dppp — 1,3-bis(diphenylphosphino)propane,
DrewPhos — tris(3,5-di-tert-butylphenyl)phosphine,
JessePhos — tert-butyldi(3,5-di-tert-butylphenyl)phosphine),
LDA — lithium diisopropylamide,
LiTMP — lithium 2,2,6,6-tetramethylpiperidide,
1,10-Phen — 1,10-phenanthroline,
Nbd — norbornadiene,
NHC — N-heterocyclic carbene,
NIS — N-iodosuccinimide,
NMP — N-methyl-2-pyrrolidone,
Ns — p-nitrophenylsulfonyl, 4-NO2C6H4SO2,
Pin — pinacol,
Piv — pivaloyl,
Phth — phthaloyl,
PMP — 1,2,2,6,6-pentamethylpiperidine,
TBAB — tetra-n-butylammonium bromide,
TBAF — tetra-n-butylammonium fluoride,
Tf — CF3SO2, triflyl,
Tfes — trifluoroethylsulfamate, CF3CH2OSO2NH2,
Ts — tosyl,
Xantphos — 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene-9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene-(9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphine).
References










