



![Rearrangement of O-allyl- and N-allyl(propargyl)anthraquinones 19 – 22 in ionic liquids (see Scheme 8) [38, 39]](/storage/images/resized/gPSO4xDOziZFh9IAmayanfVv98Il0a0PTx06Nd4f_xl.webp)
![Cross-coupling of iodoanthraquinones with alkenes (see Scheme 28) [60, 65, 66]](/storage/images/resized/ASw53nnYT4KDjHPI0kKMBLPU53X7SqKoy1VMs8oP_xl.webp)
![Cyanation of haloanthraquinones (see Scheme 46) [92-94]](/storage/images/resized/0vWv72x5X8TqnRIT3TUE6fGgKvNIddzQEs9kzpPu_xl.webp)
![Chlorination of anthraquinones (see Scheme 50) [103-109]](/storage/images/resized/8YzJhoVi7UM3abnUzU33J5ggE6K4j1PNVQ6NrMcq_xl.webp)
![Bromination of anthraquinones with molecular bromine (see Scheme 51) [94, 110-114]](/storage/images/resized/ggGluqLRsu96w8FBHH98oq7X2I7AqG50rgDn2kbV_xl.webp)
![Bromination of anthraquinones with N-bromosuccinimide (see Scheme 52) [89, 104, 105, 115-117]](/storage/images/resized/OX4Ixxkaw0aX7OWtdosV3d0EL9a9TJpQyRxfwwiX_xl.webp)
![Bromination of anthraquinones with bromide ion (see Scheme 53) [104, 118, 119]](/storage/images/resized/5XJxwlqCLG09MoVmCYix2R7X03sYz0hv2rplTyIc_xl.webp)
![Synthesis of brominated anthraquinones via diazo compounds (see Scheme 54) [120-123]](/storage/images/resized/nsSoHjauBsZD2wAGkHHXeKTiU6s0D8Qp2FVY904s_xl.webp)
![Iodination of anthraquinones with molecular iodine (see Scheme 55) [60, 103, 104, 124-127]](/storage/images/resized/dQErVcWTZZIMJclmNIUeGdGjtF5zXUaz6YGmDMjE_xl.webp)
![Synthesis of iodinated anthrquinones via diazo compounds (see Scheme 56) [130-136]](/storage/images/resized/v1i5lBezPMGjre8LijQfNoH6LAQ3xLfk17cv6M45_xl.webp)
![Iodination of anthraquinone-2-carboxylic acid derivatives (see Scheme 60) [138, 139]](/storage/images/resized/SQo3OqtxyF8lUVrkKHJvgGYIkG7AlipJtaBgK9b9_xl.webp)
![Synthesis of hydroxyanthraquinones by halogen atom substitution (see Scheme 61) [140-142]](/storage/images/resized/Erb3hpKwKUvwJDrbwVRl42jpsqAAuXEXohevo3jo_xl.webp)
![Alkoxylation of anthraquinones (see Scheme 70) [147-150]](/storage/images/resized/dukKHjpAbmW6oc7F9SqDFHABSfG9nv94FP3KqfwS_xl.webp)
![Direct sulfonation of anthraquinones (see Scheme 73) [151, 152]](/storage/images/resized/MPUcgqm6XLoBmsa1Pq7Dx5FNUnMDECEBiuTGkvsB_xl.webp)
![Formation of the C—S bond via diazo compounds (see Scheme 75) [154, 155]](/storage/images/resized/VJZVsp1rYhBwsyV4HJVVyuqbB0VWiYuVjavCEqc2_xl.webp)
![Substitution of the halogen atom in anthraquinones by a sulfanyl group (see Scheme 76) [156-158]](/storage/images/resized/xLTmqCan7GsGUeI30rAInRMBAj4cXZOyZOKuRCUY_xl.webp)
![Selenation of anthraquinones (see Scheme 103) [212]](/storage/images/resized/rLdKPx0BI5OwqLGYjJE0LQ98feXRVAHvrGoa9LFE_xl.webp)
Keywords
Abstract
Anthraquinone derivatives (anthracene-9,10-dione) have found applications in medicine, chemical technology, and analytical chemistry, and are also promising for the creation of new materials. The development of methodologies for introducing functional groups into anthraquinones is fundamental for the targeted synthesis of new compounds. The structural features of anthraquinones, particularly the presence of electron-withdrawing carbonyl groups, significantly affect their chemical properties, making the modification of such derivatives often a non-trivial task. This review examines and analyzes methods for forming the most important bonds (C–C, C–N, C–O, C–S, C–Hal, C–Se, C–B, C–P) with anthraquinone, as presented in the scientific literature since 2000. The review of methods for modifying anthraquinone derivatives aims to systematize and evaluate the synthetic possibilities of constructing these bonds. The analysis of both new and classical methods for transforming this molecular scaffold will help to expand its structural diversity, identify new areas for studying its chemical properties, and facilitate the task of targeted synthesis of anthraquinone derivatives. The bibliography includes 224 references.
1. Introduction
Anthracene-9,10-dione (anthraquinone) derivatives are of great practical importance due to their unique spectral and redox properties,[1] photochromism [2] and diverse biological activities.[3, 4] These valuable properties, together with scientific interest in the fundamentals of anthraquinone reactivity, have attracted the attention of synthetic chemists to this scaffold.
Much of the research into the synthesis and study of the chemical properties of anthraquinones has been devoted to the development of industrial dyes, pigments and luminophores.[5] The focused study of these compounds has led to the development of disperse, cationic, vat and other types of dyes (Fig. 1). This historically first area of application, systematically developed from the laboratory production of anthraquinone in 1869 until the 1980s, contributed to the development of the methodology for the synthesis of anthraquinone derivatives. However, many of the approaches developed to modify the structure of anthracene-9,10-dione have been based on processes used in the industrial production of dyes.
![[{"id":"tAZh1VcHTB","type":"paragraph","data":{"text":"Anthraquinones and their applications"}}]](/storage/images/resized/opeBV5sFa9QMQEpdqWLFOfB0AP8MU1lAk5TIcYPh_xl.webp)
The second stage in the development of the chemistry of anthraquinones derivatives, in which particular attention was paid to their antitumour properties, is associated with the study of biologically active compounds.[6] The natural anthracycline antibiotic doxorubicin (see Fig. 1), which contains an anthraquinone motif, has been used in the cancer therapy since the 1970s. Since the discovery of the first anthracyclines, about a thousand of their derivatives and analogues have been produced, and several dozen anthraquinone-based drug candidates are in clinical trials as antitumour agents. The development and introduction into medical practice of semisynthetic anthracyclines (epirubicin, idarubicin, etc.) and synthetic anthraquinone derivatives (e.g. mitoxantrone, see Fig. 1) testify to the importance of this structural motif for the development of new chemotherapeutic agents.[3, 7, 8] Another example of an anthraquinone-based drug is diacerein (see Fig. 1), which is used for the treatment of primary and secondary osteoarthritis due to its anti-inflammatory effect.[9]
Other promising practical applications of anthraquinone derivatives that have been actively developed in recent years include the creation of new materials for optoelectronics,[10] 3D photoprinting,[11] redox flow batteries [12] and chemosensors for biopolymers,[13] and use as initiators for photopolymerization.[14] Numerous studies have been devoted to the potential applications of anthraquinones as organic polymers [15] and analytical reagents,[16] etc. The above-mentioned areas of practical applications of compounds of this class underline the usefulness of systematizing the developed approaches and reactions used to modify the anthraquinone structure.
The formation of new С – С, C – N, С–О, С – Hal, С – S, С – Se, C – B, C – P bonds, directly or through the transformation of functional groups, is the basis of organic synthesis, which plays a key role in the construction of complex molecules with desired properties.[17-19] Obviously, for the efficient formation of new bonds, it is necessary to consider the structural features and reactivity of the starting compound. X-ray diffraction studies and quantum chemical calculations showed that anthracene-9,10-dione is a planar tricyclic system of two isolated benzene rings linked by CO groups and does not contain the α,β-unsaturated ketone moiety typical of other quinones.[20] The presence of carbonyl groups reduces the electron density in the benzene nuclei, significantly hampering the reactions characteristic of aromatic compounds. The rigid geometry of the molecule, which precludes rotation of the aromatic ring relative to the carbonyl groups, also plays an important role in the reactivity of anthracene-9,10-dione. This increases the steric influence of the carbonyl groups in the peri-positions of the benzene rings and leads either to steric hindrance in the substitution reactions or, conversely, to facilitation of the process through additional coordination. For example, the non-catalytic sulfonation of anthracene-9,10-dione to position 1 is difficult due to the bulky attacking species and is directed exclusively to the β-position, whereas nitration, which is less sensitive to the steric factor, goes to the α-position due to the coordination of the attacking species to the carbonyl oxygen atom.[20]
Analysis of the possibilities of forming the above bonds in the anthracene-9,10-dione series are important for several reasons:
1) the introduction of substituents and functional groups into the anthraquinone core can affect its applied properties,
2) transformations of substituents and functional groups open the way to obtain derivatives inaccessible otherwise,
3) the comparison of different methods allows their evaluation and the selection of an effective approach to the development of multistep schemes for the synthesis of anthraquinone derivatives.
One of the most important works in the field of chemistry of anthraquinones is the monograph by M.V.Gorelik,[20] published in 1983, which presents data on the structure, methods of preparation and reactivity of anthraquinones. The closest in structure and importance is the book ‘Anthracycline Chemistry and Biology I. Biological Occurrence and Biosynthesis, Synthesis and Chemistry’,[21] edited by Krohn in 2008, which summarizes methods of anthraquinone functionalization, but the main part of this monograph is devoted to methods of anthraquinone core assembly. Since then, several reviews have appeared focusing on particular aspects of the chemical properties, transformations or structure of anthraquinone derivatives. For example, reviews have been published on the synthesis of anthraquinone-based electroactive polymers,[15] the total synthesis of natural anthraquinones,[22] the preparation methods and applied aspects of acetylene derivatives of quinones, including anthraquinones,[23] the synthetic potential of anthraquinone-containing diazonium salts,[24] and the reductive Claisen rearrangement of a number of compounds in this class.[25] However, no systematic review of anthraquinone functionalization has been published for more than 20 years.
Obviously, since the monographs of M.V.Gorelik [20] and Krohn,[21] a sufficient number of new methods for molecule functionalization have been successfully adapted for modification of the anthraquinone nucleus. In this context, it seems appropriate to review and systematize recent advances in the methodology of functionalization of anthracene-9,10-diones, as well as to recall the main traditional methods for their modification that are currently actively implemented. This review presents methods for anthraquinone carbon–carbon bond formation involving sp 3-, sp2- and sp-hybridized carbon atoms via reaction with C-nucleophiles, Mannich and Marschalk reactions, metal-catalyzed cross-coupling, radical processes, etc. Various methods for the introduction of halogen atoms as well as sulfur-, oxygen-, nitrogen-, boron-, selenium- and phosphorus-containing functional groups based on substitution reactions or on metal-catalyzed approaches are considered, in which direct CH functionalization reactions play a special role. The review considers the transformations that have been reported mainly in publications over the last 25 years.
2. Reactions with the formation of the carbon – carbon bond
The formation of a carbon backbone of a given structure is a key challenge for organic synthesis. A considerable pool of reactions has been developed for this purpose at different times: some of them are universal synthetic methods, while others are specific and applicable to a narrow range of substrates. Many anthraquinone derivatives with useful properties contain functionalized substituents, which has led to an increasing practical interest in the development of methods for their preparation.
Despite the presence of two benzene rings in the structure, the chemical properties of anthraquinones differ from those of the aromatic compounds. Most anthraquinones, due to the deactivating effect of the carbonyl groups, do not participate in the reactions of the C – C bond formation, such as Friedel – Krafts alkylation and acylation, halomethylation, Vilsmeier formylation, Mannich aminomethylation, etc., which are traditional for arenes. In some cases, for activated compounds containing electron-donating groups (mainly hydroxy and amino groups), electrophilic substitution reactions (SEAr) with C-electrophiles can be carried out. Another partial solution to this problem was found in the 70s and 80s of the 20th century: for amido-, hydroxy- and aminomethylation reactions, the starting anthraquinones are first reduced to their leuco form (hydroquinones), then the SEAr reaction is carried out and the intermediates are reoxidized to anthraquinone (the so-called Marshalk method [20]). Some examples of the application of this methodology are given in the monograph by M.V.Gorelik,[20] but certain reactions remain relevant, as will be demonstrated in this review. Although the strong electron-withdrawing influence of the quinoid moiety on the anthracenedione aromatic system limits the SEAr reactions for anthraquinones, this effect opens up the possibility of realizing processes of a different type. In some cases, anthraquinones are capable of direct nucleophilic substitution reactions (SNAr) under mild conditions, which are difficult for benzene analogues without metal complex catalysis.
Obviously, solving the problems of modern function-oriented synthesis of anthraquinone derivatives with valuable properties requires the development of universal approaches to the formation of C – C bonds in such compounds. In this Section, the advances made in the development of new methods for the introduction of carbon-containing substituents leading to the formation of a bond between sp3-, sp2- and sp-hybridized carbon atoms and the anthraquinone nucleus are discussed sequentially, and the most vivid examples of the optimization of previously proposed methods of chemical modification of this type are presented.
2.1. Bond with the sp3-hybridized carbon atom
Nucleophilic substitution reactions of halogens, nitro groups and even hydrogen in the series of anthraquinone derivatives have been adapted for the introduction of carbon-containing moieties. The most interesting of these is a two-step procedure based on the condensation of 2,3-dihaloquinizarins 1 and 2 with malonic or acetoacetic ester derivatives and the subsequent cleavage of intermediate anthrafurans.[26, 27] Nucleophilic substitution of halogen in compounds 1, 2 by C-nucleophiles followed by base-catalyzed cyclization gives the angular anthra[1,2-b]furans 3a – c (Scheme 1), which, unlike their linear counterparts, are poorly stable in acidic medium.
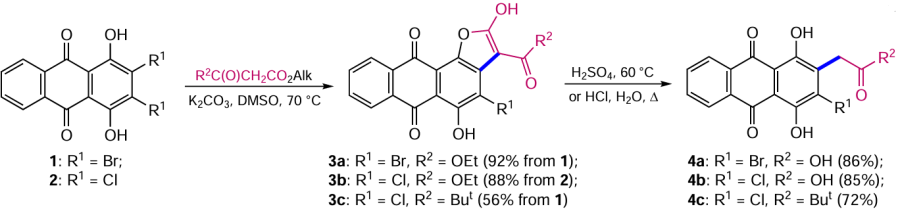
Heating of 4-haloanthra[1,2-b]furan-3-carboxylic acid esters 3a,b or anthra[1,2-b]furan 3c with strong mineral acid resulted in cleavage of the heterocycle to give compounds 4a – c (see Scheme 1).[26, 27] Replacement of the acyl or alkoxycarbonyl moiety in the reacting C – H-acids with other electron-withdrawing substituents, such as a nitro or cyano group, is expected to extend the synthetic possibilities of this approach.
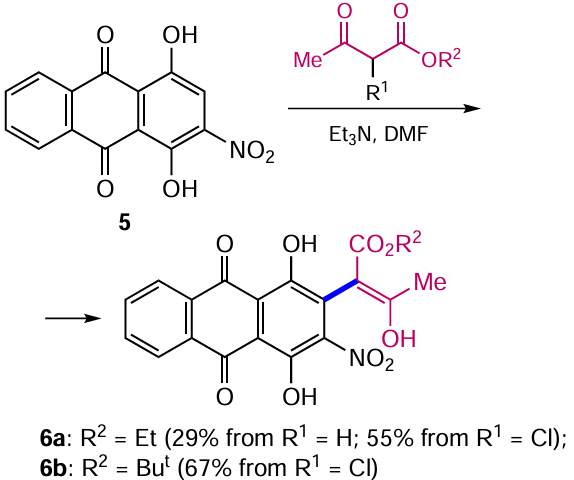
Quinizarine derivatives undergo direct oxidative nucleophilic substitution of hydrogen (ONSH) in the β-position by C-nucleophiles. As an example, the reaction of 1,4-dihydroxy-2-nitroanthracene-9,10-dione (5) with acetoacetic ester under mild conditions afforded the product 6a, albeit in low yield (Scheme 2).[28] Replacing the acetoacetic ester with its 2-chloro derivative improves the efficiency of the method by implementing a different reaction mechanism, vicarious nucleophilic substitution (VSN), instead of ONSH. The change in the mechanism is due to the difference in the efficiency of the leaving group cleavage: in the case of the unsubstituted acetoacetic ester, the intermediate adduct contains a ‘bad’ leaving group, hydride ion (H–), the cleavage of which involves oxidation. In contrast, in 2-chloroacetoacetic ester, the chloride ion (Cl–) eliminates much more readily.[28] Interestingly, the use of the 2-bromo analogue of acetoacetic acid ester does not increase the yield of anthraquinone 6a, whereas the substituent in the ester group affects the yield of the substitution product: the use of the tert-butyl 2-chloroacetoacetate in the reaction promotes an increase in the yield of the corresponding ester 6b.[29]
The Mannich reaction is one of the methods of the C – C bond formation widely used in organic synthesis, e.g. of biologically active compounds.[30] However, in the series of anthracene-9,10-dione derivatives, this reaction can only be achieved for anthraquinones containing strong activating substituents. Thus, in the search for new antitumour compounds based on the natural anthraquinone emodin (7), a series of derivatives 8a-f bearing an aminoalkyl substituent at the 2-position were synthesized under Mannich reaction conditions in 56 – 83% yields (Scheme 3).[31] This technique proved effective with aromatic aldehydes containing both electron-donating and electron-withdrawing substituents, but no clear dependence of product yield on substituent structure was observed. Aminomethylation of emodin (7) with formaldehyde in the presence of aniline unexpectedly gave anthra[2,3-e][1,3]- oxazine-6,11-dione 9.[31]
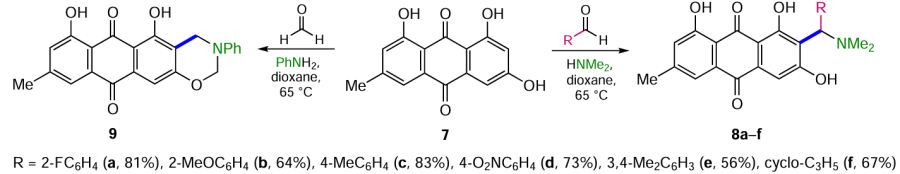
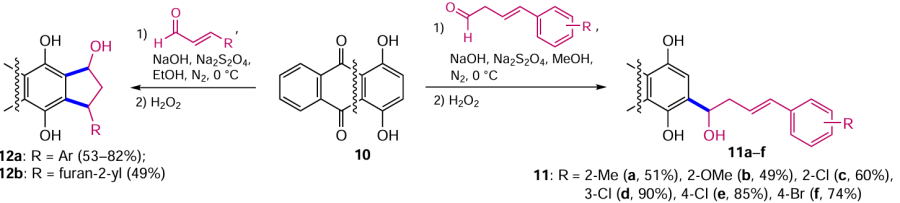
As mentioned above, to increase the efficiency of hydroxymethylation and alkylation of the β-position of anthraquinones by the Marschalk method, the substrate containing hydroxy or amino groups is converted to the leuco form (dihydroanthraquinone) under the action of reducing agents.[20] Thus, by reacting quinizarine (10) with various aldehydes in the presence of Na2S2O4 and NaOH, followed by oxidation of the leuco-semiproducts with hydrogen peroxide, it was possible to introduce the substituted shikonin moiety into anthraquinones 11a – f (Scheme 4).[32] Despite the considerable remoteness of the substituents in the aldehyde benzene ring from the reaction centre attacked by anthraquinone, ortho-substituted aldehydes give products 11a – c in lower yields. The use of α,β-unsaturated aromatic aldehydes under similar conditions leads to the annulation of cyclopentane to anthraquinone (compounds 12a,b, see Scheme 4),[33] which proceeds via an additional Michael reaction. The structure and position of the substituents in the aromatic ring of an aldehyde have little influence on the yield of the products; however, in the absence of an aromatic moiety in the aldehyde, as in the case of acrolein or croton aldehyde, no cyclization products are formed.
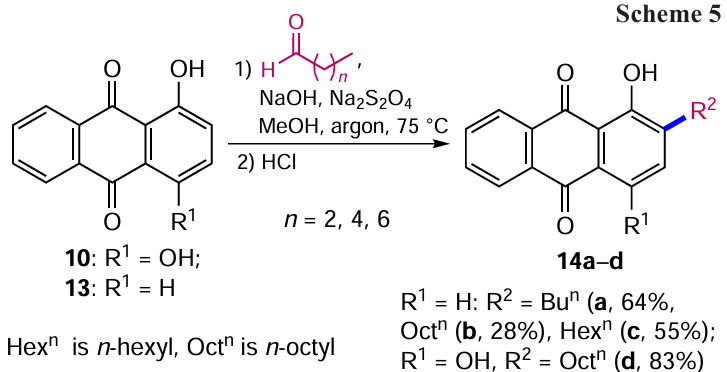
Increasing the reaction temperature and the amount of reducing agent in some cases allows the reduction of hydroxyalkyl moieties to alkyl ones.[20] Using this method, 2-alkylanthraquinones 14a – d were obtained from 1-hydroxyanthraquinones 10, 13 (Scheme 5). The introduction of an additional hydroxyl group into anthraquinone, as expected, leads to an increase in the yield of the alkylation product of quinizarine (14d).[34]
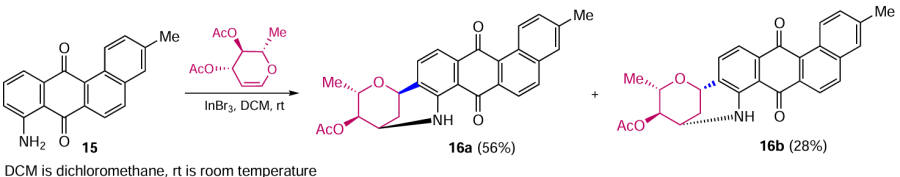
Examples of electrophilic alkylation, even for activated anthraquinone derivatives, are extremely rare and such reactions are usually sporadic. For example, the Lewis acid-catalyzed N,C-glycosylation of 8-amino-3-methyltetraphen-7,12-dione (15) was used as one of the key steps in the total synthesis of the antitumour angucycline marmycin A.[35, 36] Catalysis with indium bromide allowed the 2,3-dihydropyran moiety to be annulated to aminoanthraquinone 15 at room temperature, yielding two diastereomers 16a,b (Scheme 6).[35] Maugel and Snider [36] carried out the same reaction using In(OTf)3 as a catalyst, which proved to be less sensitive to moisture, and the yields of the isomeric products 16a and 16b decreased to 27 and 14% respectively.
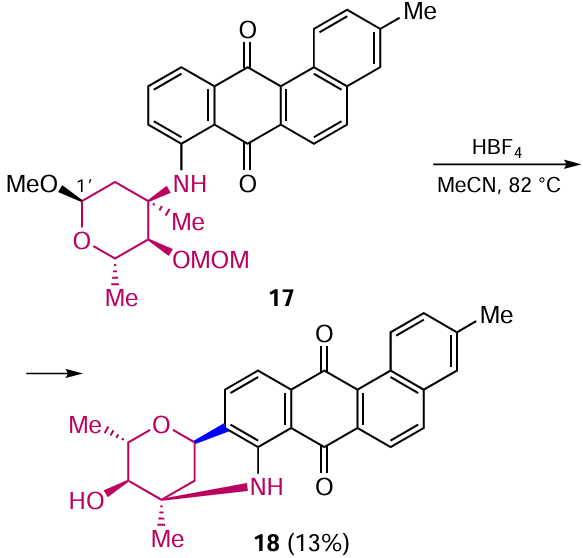
Subsequently, Cañeque et al.[37] described another approach to the synthesis of marmycin A, the most complicated step of which was the acid-catalyzed C-glycosylation of an anthracene-9,10-dione derivative with an aminosugar residue. Notably, the aminosugar moiety was introduced into the anthraquinone core by substitution of the trifluoromethanesulfonic group by the Ullmann reaction.[37] The Friedel – Crafts intramolecular cyclization of derivative 17 proceeds with the formation of the C – C bond between the C(1') carbon atom of the aminosugar and the β-position of the anthraquinone with cleavage of the methoxymethyl (MOM) protecting group (Scheme 7).
The Claisen rearrangement, which is a [3,3]-sigmatropic shift in ortho-allyl ethers of phenols, can be applied to the C-functionalization of anthraquinones. For this series of compounds, the above method is most effective after prior reduction of the parent quinone to the leuco form with reagents such as zinc in acetic acid, sodium dithionite or glucose.[20, 25]
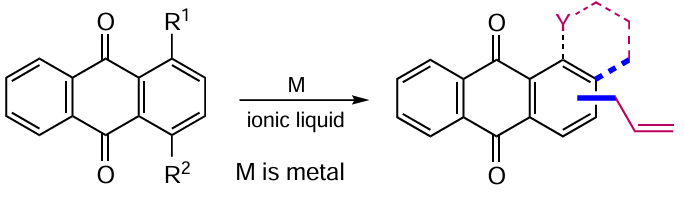
Nadali et al.[38] studied the reductive rearrangement of mono- and di-(allyloxy)anthraquinone derivatives 19 and 20 mediated with iron powder in a mixture of environmentally safe ionic liquids (Scheme 8, Table 1). Transformation of these anthraquinone ethers results in the formation of a new C – C bond to give rearrangement products 23 and 24. Varying the molar ratio of reducing agent to substrate or the reaction time for derivatives with multiple allyl groups at the α-position of an anthraquinone allows control of the preparation of both β-mono- and β,β-diallylanthraquinones. In 2023, Albooyeh and Aghapour [39] demonstrated the possibility of zinc-promoted intramolecular rearrangement of 1-(N-allylamino)anthraquinones 21 in 1-hexyl-3-methylimidazolium tetrafluoroborate to 1-amino-2-(prop-2-enyl)anthraquinone (25) (see Table 1). Replacement of the allyl moiety with a propargyl moiety (compound 22) resulted in an intramolecular cyclization product 26.
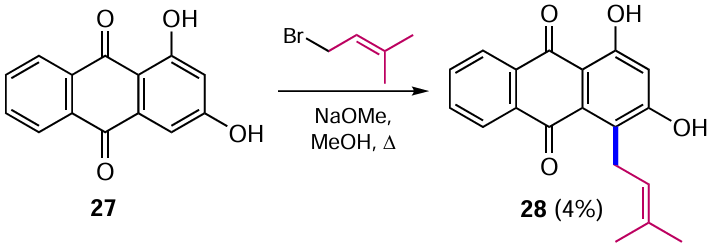
The Claisen rearrangement can also proceed without reduction of the quinone moiety, but with much lower efficiency. Alkylation of 1,3-dihydroxyanthraquinone (27) with prenyl bromide in boiling methanol gave 2,4-dihydroxy-1-(3-methylbut-2-en-1-yl)anthracen-9,10-dione (28) as the only isolated product (Scheme 9), which appears to result from a number of rearrangements.[40] Unfortunately, the authors do not discuss either the reaction mechanism or the unusual regioselectivity of the process at the α-position, although it is usually accompanied by the formation of β-allyl derivatives of anthraquinone. It should be noted that product 28 selectively inhibited the proliferation of human hepatocellular carcinoma HepG2 cells, in contrast to other tumour lines.
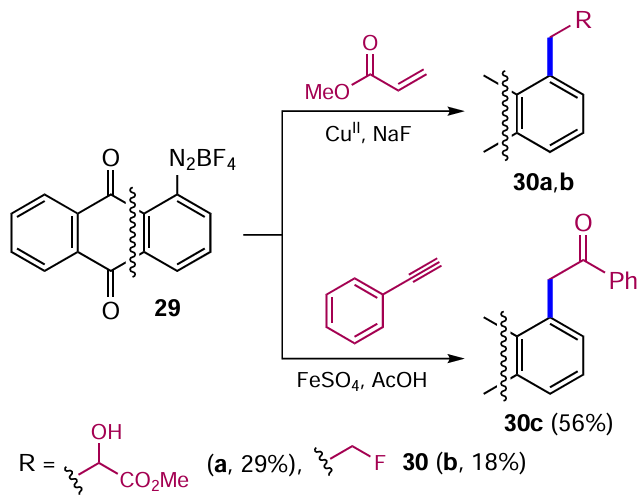
In addition to nucleophilic and electrophilic substitution reactions, anthraquinone derivatives can be modified by radical processes. For example, diazonium salts of anthraquinones are used as intermediates to introduce carbon-containing groups into aromatic nuclei. One of the products of arylation of unsaturated compounds with diazonium salts (Meerwein reaction) are alkylarenes.[41] This method of modifying anthraquinones was discovered a long time ago, but some modern work has demonstrated its new possibilities. For example, the reaction of anthraquinone-1-diazonium tetrafluoroborate (29) with activated alkenes, e.g. methyl methacrylate, produces not only vinylation products (see Section 2.2 below), but also functionalized alkyl derivatives 30a,b (Scheme 10).[42] The reaction of compound 29 with phenylacetylene in the presence of ferrous sulfate in acetic acid under optimized conditions gave the ketone 30c in moderate yield (see Scheme 10).[43] Stasevich et al.[24] published a review devoted to the synthetic potential of diazonium salts of the anthraquinone series in which they considered methods of functionalization of anthraquinones via transformation of the diazonium group.
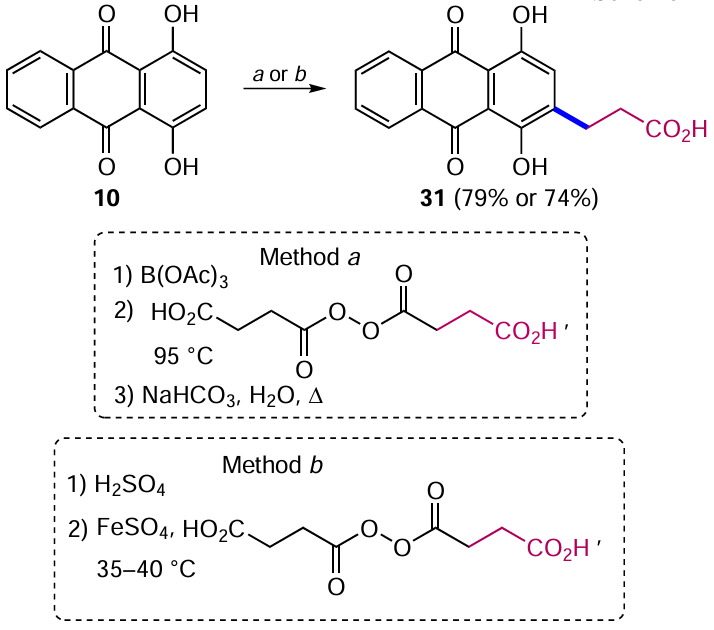
Arinich and Gorelik [44] developed a method for introducing alkyl substituents in the β-position of quinizarine based on radical carboxyethylation. To enhance reactivity, the starting 1,4-dihydroxyanthraquinone (10) was converted to a boron complex or protonated form, thereby activating the anthraquinone nucleus to react with weakly nucleophilic alkyl radicals (Scheme 11). Subsequent addition of succinic acid peroxide under strong (95 °C, method a) or weak (35 – 40 °C, method b) heating and hydrolysis of the intermediate boron complex in the former case afforded 2-(2-carboxyethyl)-1,4-dihydroxyanthraquinone (31) in similar yields.
A methodology for the radical cyclization of aromatic 1,3-dicarbonyl adducts obtained by a four-component Ugi reaction was developed to prepare a library of polysubstituted isoindolinones. Microwave irradiation (MW) of an anthraquinone derivative 32 in the presence of a pair of the radical initiator (oxidant) [Bun4N]2S2O8 and (2,2,6,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) leads to the angular naphtho[2,3-e]isoindole 33 as a single regioisomer (Scheme 12, reaction pathway is shown in Scheme 13).[45]
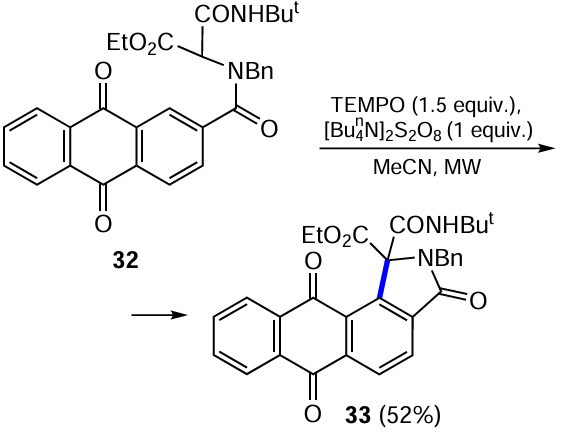
The authors note that tetra-n-butylammonium persulfate decomposes under the action of microwave irradiation to two equivalent radicals •SO4−(Bun4N+), one of which acts as a radical initiator in the cyclization and the formation of intermediate radical I via the single electron transfer (SET). Another radical oxidizes TEMPO to species A, which, after intramolecular radical 5-endo-trig-cyclization of intermediate compound I, oxidizes intermediate aromatic radical II.[45] The final step involves an acid-base rearomatization of cation III to product 33.
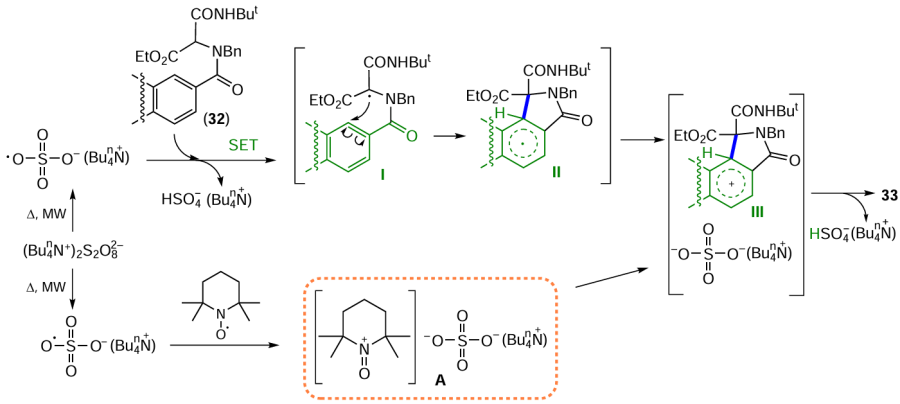
Among anthraquinones, as well as other aromatic compounds, cross-coupling reactions catalyzed by transition metal complexes are one of the most important approaches to the formation of new C – C bonds.[46] Optimization of the Suzuki – Miyaura reaction conditions allowed cross-coupling of boronic acid derivatives with halogenated anthraquinones.[47] For example, the methyl group was introduced into the molecule by cross-coupling of 7-bromo-1-hydroxyanthraquinone (34) with methylboronic acid in the total synthesis of barleriaquinone-I (1-hydroxy-7-methylanthracene-9,10-dione, 35) (Scheme 14).[47]
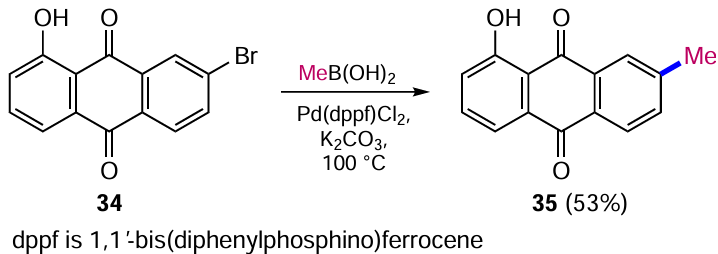
Another example of the application of the Suzuki reaction to form the C – C(sp3) bond is the step used in the synthesis of an intermediate component of an anthraquinone-based polymer intended for the development of modern batteries. Kawai et al.[48] carried out a cross-coupling reaction in which two anthraquinone nuclei were ‘cross-linked’ via a bicycloheptene moiety. The reaction of 2-iodoanthracene-9,10-dione (36) and (anthraquinone-2)-boronic acid ester 37 with bicyclo[2.2.1]hepta-2,5-diene in the presence of the Pd(OAc)2 – PPh3 system gives the derivative 38, which a monomer for further polymerization (Scheme 15).
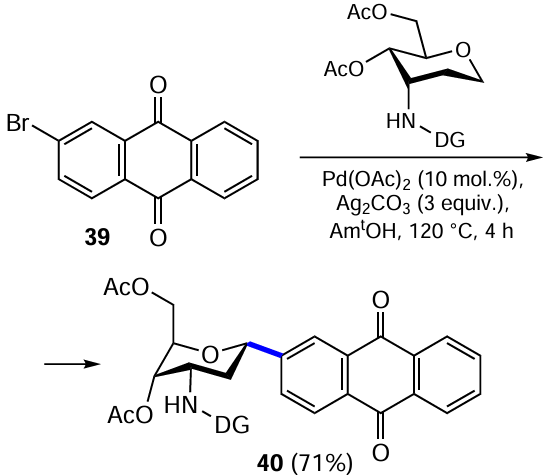
A method for the diastereoselective Pd-catalyzed C(sp 3) – H bond activation involving the anomeric carbon atom of an aminosugar has been developed for the synthesis of C-(het)arylglycosides with preferential formation of the α-anomer.[49] This method is suitable for a wide range of substrates diversely substituted in the aromatic nucleus. The formation of the product 40 with an α-configuration of the anomeric centre is exemplified by the reaction of 2-bromoanthraquinone (39) with an aminosugar derivative (Scheme 16). Interestingly, this high selectivity was achieved due to the presence of a picolinic acid residue on the amino group of the monosaccharide, which acted as a directing group (DG) in the implementation of the bidentate coordination mechanism.
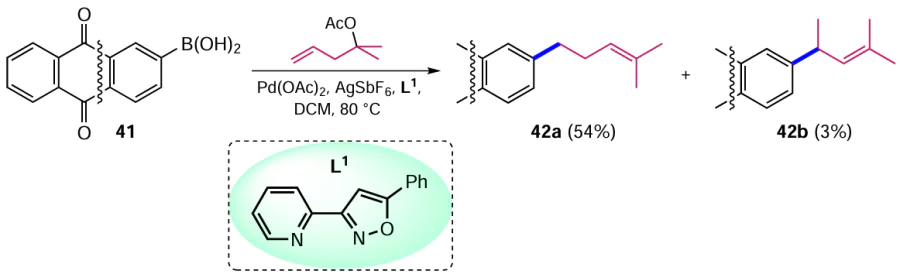
Muto et al.[50] proposed a method for the Pd-catalyzed arylation of alkenes, which, in contrast to the classical Mizoroki – Heck reaction, proceeds as a redox process with β-acetoxy elimination. A key feature of the reaction is [Pd] – H dissociative ‘chain walking’, where no alkene is formed until β-elimination of the acetoxy group occurs.[51] Treatment of (anthraquinone-2-yl)boronic acid (41) with (2-methylpent-4-en-2-yl)acetate catalyzed by the Pd(OAc)2–AgSbF6 system in the presence of 3-(2-pyridyl)-5-phenylisoxazole (L1) produced a mixture of 2-(4-methylpent-3-enyl)anthraquinone (42a) and its isomer 42b in a ratio of 16 : 1 (Scheme 17).[50] It has been shown that 2-(4-methylpent-3-enyl)anthraquinone (42a), which is hard-to-reach otherwise, has antifungal activity.
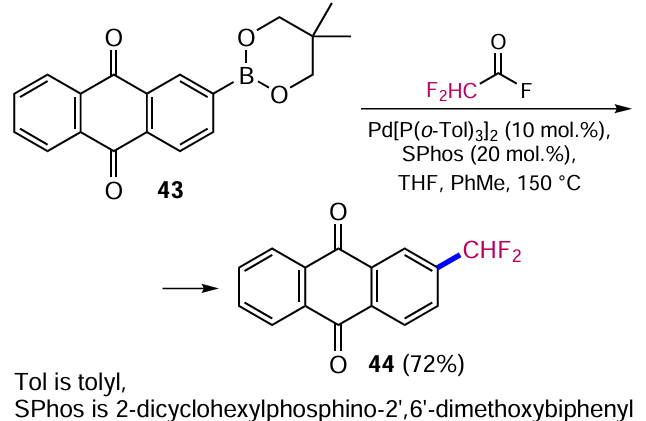
Lalloo et al.[52] have developed a unique approach to form a bond between aryl and fluoroalkyl functional groups, based on the reaction of difluoroacetic acid fluoride with arylboronic acid esters. As an example, the Pd-catalyzed reaction of anthraquinone-2-yl-boronic acid ester 43 with 2,2-difluoroacetyl fluoride leads to 2-(difluoromethyl)anthracene-9,10-dione (44) in good yield (Scheme 18).
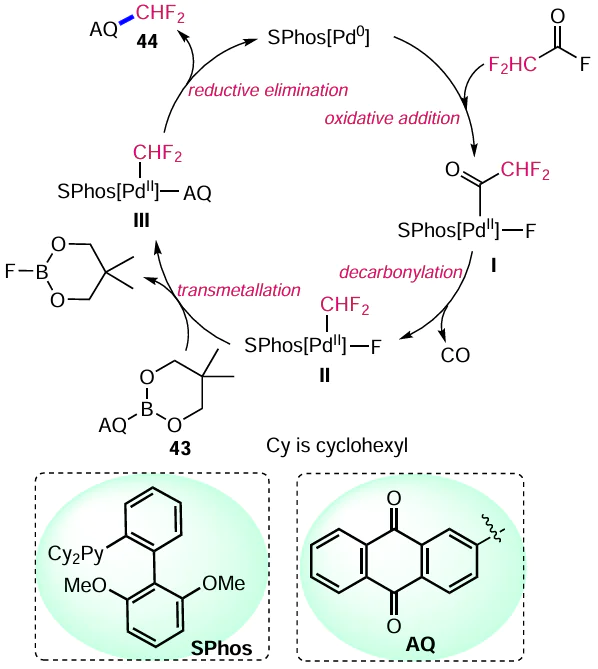
The authors proposed a catalytic cycle for this Pd-catalyzed decarbonylative fluoroalkylation of arenes (Scheme 19).[52] In the first step, the electrophile CF2C(O)F is oxidatively inserted into the in situ activated Pd0 catalyst to give a PdII complex I with an incorporated fluorocarbonyl moiety. This is followed by decarbonylation to form Pd complex II, which undergoes transmetallation: an anthraquinone moiety (AQ) replaces the fluorine atom. The final step is completed by the reductive elimination of the catalyst and the construction of an arylfluoroalkyl bond.
Matsumura et al.[53] synthesized tetraalkylanthracenes (pentacenes) via the Ru-catalyzed C – H alkylation of anthraquinone derivatives with various alkenes. Ruthenium-catalyzed alkylation of unsubstituted anthraquinone (45) proceeds regioselectively to α-positions relative to carbonyl groups and furnishes predominantly linear α-tetraalkylanthraquinones 46a – d (Scheme 20).
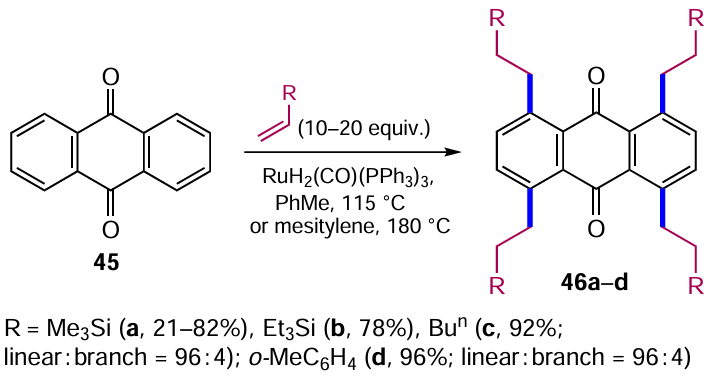
Temperature significantly affects the outcome of the above cross-coupling; heating anthraquinone (45) with vinyltrimethylsilane at 115 °C in toluene gives the product 46a in low yield (21%), whereas raising the temperature to 180 °C in mesitylene (1,3,5-trimethylbenzene) increases the yield of this compound to 82% (see Scheme 20). Interestingly, using alkyl- and aryl-substituted ethylenes (R = Bun and о-МеС6Н4), alkylation products 46c,d of both linear (through the β-position of the alkene) and branched structure were obtained in a ratio of 96 : 4.
2.2. Bond with the sp2-hybridized carbon atom
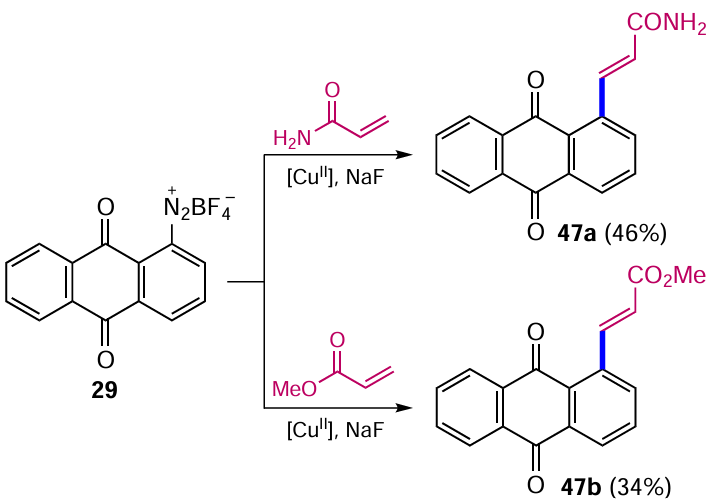
The formation of bonds between anthraquinone and substituents with the sp2-hybridized carbon atom is in most cases based on radical reactions or processes catalyzed by transition metal complexes. The previous Section (see Scheme 10) gave an example of the formation of a С–С(sp3) bond with an anthraquinone core by the Meerwein reaction, but this method is traditionally used for the arylation of unsaturated compounds. The reaction of anthraquinon-1-yl diazonium tetrafluoroborate (29) with acrylamide gives 3-(anthraquinone-1-yl)acrylamide (47a), and treatment of this salt with methyl acrylate gives methyl 3-(anthraquinon-1-yl)acrylate 47b (Scheme 21) together with the alkylation products 30a,b (see Scheme 10).[42] The authors do not specify which copper salt is suitable for catalyzing such cross-coupling.
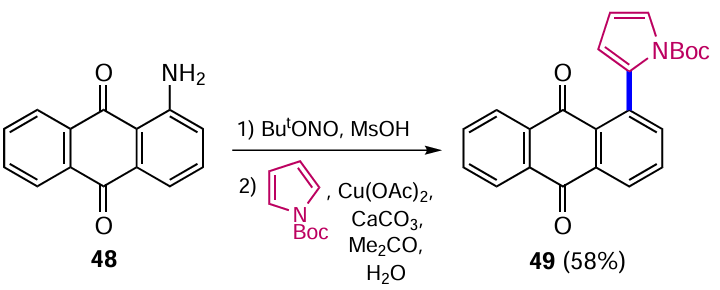
Honraedt et al.[54] used the Meerwein reaction for the arylation of pyrroles, with the arendiazonium salts generated in situ from the appropriate arylamines. Using an anthraquinone derivative as an example, it was shown that the sequential treatment of 1-aminoanthracene-9,10-dione (48) with tert-butyl nitrite and methanesulfonic acid (MsOH), followed by the addition of copper acetate, N-Boc-protected (Boc is tert-butoxycarbonyl) pyrrole and calcium carbonate, led to 1-(pyrrole-2-yl)anthraquinone 49 (Scheme 22).
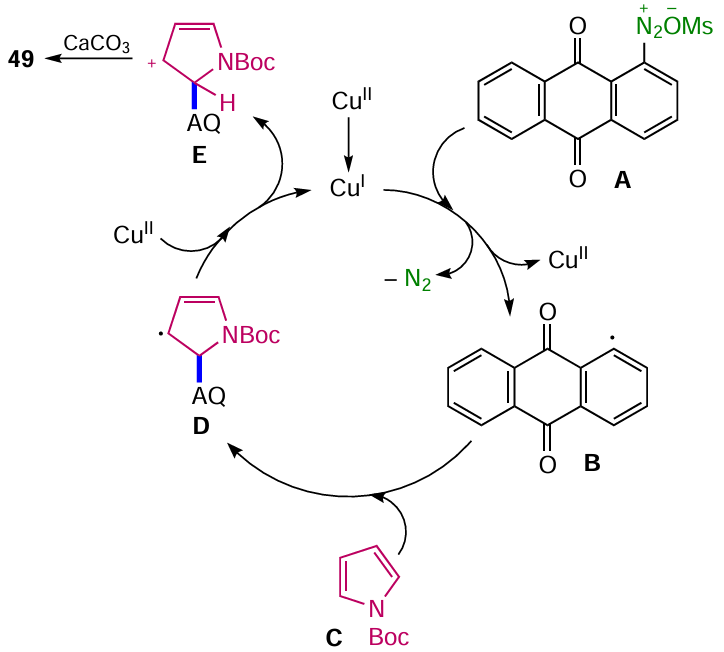
The arylation pathway involves CuI-catalyzed homolytic cleavage of a nitrogen molecule from anthraquinone diazonium A to generate an aryl radical B, which attacks pyrrole C (Scheme 23). The intermediate radical D is further oxidized by CuII to the corresponding cation E, which upon deprotonation affords the target product 49. The high regioselectivity of the arylation at position 2 of the pyrrole core is due to the higher stability of the radical D compared to the isomeric intermediate when attacked at position 3 of the pyrrole. The authors noted the dual role of calcium carbonate in the arylation: first, it acts as a buffer to prevent the formation of the azo compound, and second, it deprotonates the intermediate carbocation E.
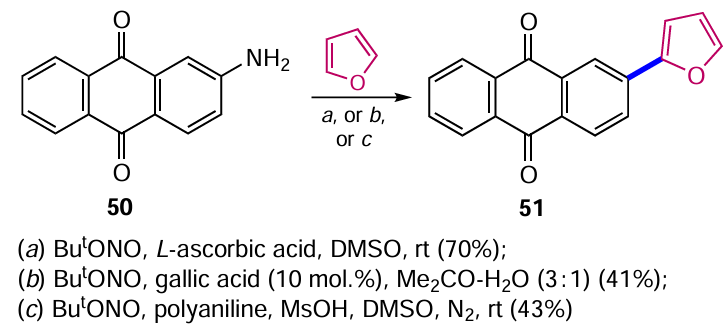
Modifying the conditions for the arylation of (het)arenes helped to eliminate the need for metal-containing catalysts. Quite high efficiencies were achieved when L-ascorbic acid was used as an initiator for intermolecular C – H arylation, for example when furan was treated with 2-aminoanthraquinone (50) (Scheme 24).[55] Alternative conditions for the arylation of furan with compound 50 catalyzed by gallic acid [56] or by a reduced form of polyaniline in the presence of MsOH [57] are also presented, but the yield of 2-(furan-2-yl)anthraquinone 51 was significantly lower in these cases.
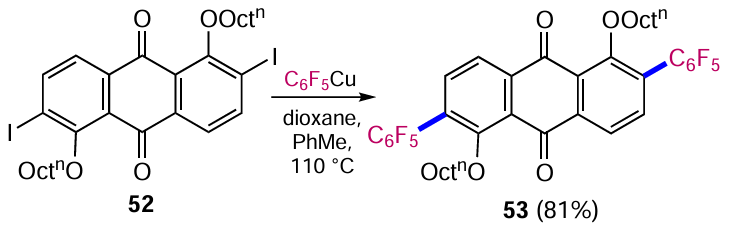
The use of organometallic compounds as reagents for the construction of carbon-carbon bonds is an essential tool in organic synthesis.[58, 59] At the same time, the application of this method to the modification of quinones is limited by side reactions of addition to carbonyl groups, which in some cases can be minimized. The use of moderately reactive organocopper compounds, such as pentafluorophenylcopper, allowed the coupling with 2,6-diiodoanthraquinone 52 to give the 2,6-diphenylanthraquinone derivative 53 in high yield (Scheme 25).[60]
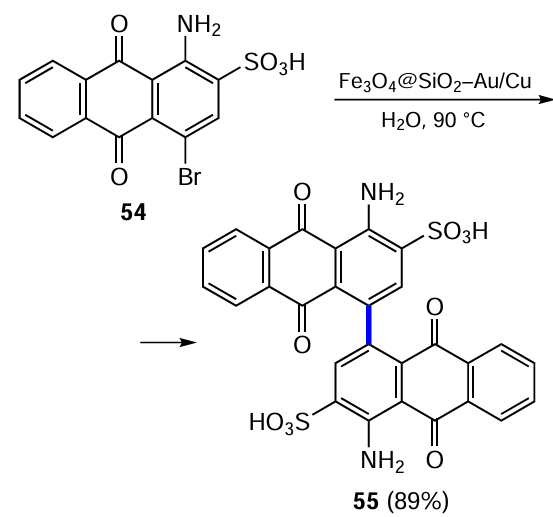
The Ullmann reaction is considered to be one of the pioneering methods for copper-catalyzed cross-coupling of two aryl groups.[61] However, factors such as high temperature and long process time, as well as the need for stoichiometric amounts of copper, have stimulated the search for more efficient conditions for its performance. Gao et al.[62] developed a new catalytic system based on Fe3O4@SiO2 – Au/Cu bimetallic nanoparticles for the synthesis of compound 55, a key intermediate of the high-quality organic pigment PR 177. 1-Amino-4-bromo-9,10-dioxoanthracene-2-sulfonic acid (54) gave the dimerization product 4,4'-diamino-1,1'-dianthraquinon-3,3'-disulfonic acid 55 under the action of catalytic nanoparticles (Scheme 26). The advantage of this catalytic system is the possibility of easy separation of the magnetic nanoparticles from the product and their subsequent regeneration.
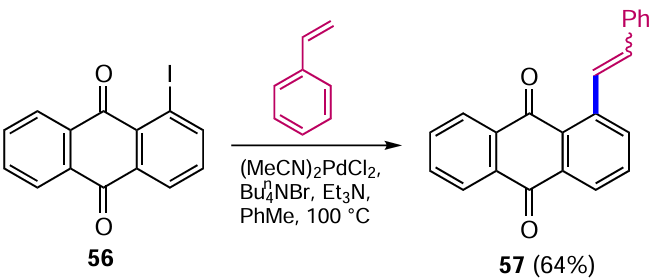
The Heck reaction is also widely used in organic synthesis for the construction of new C – C bonds.[63] The versatility of this method of introducing an alkene unit into the anthraquinone nucleus has been confirmed in a significant number of studies. An example is the cross-coupling of 1-iodoanthraquinone (56) with styrene in the presence of (MeCN)2PdCl2 catalyst leading to the product 57 (Scheme 27).[64]
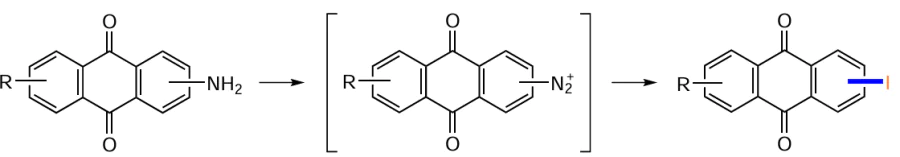
In the development of new materials for optoelectronic devices, a series of cross-coupling reactions of diiodoanthraquinones at the β-position have been carried out. The Pd(OAc)2-catalyzed Heck reaction of anthraquinone 52 with methyl acrylate gave compound 58 — the product of substitution of two iodine atoms (Scheme 28, Table 2).[60] In another study,[65] the introduction of a 4-substituted styryl moiety into an anthraquinone core was explored to prepare analogues of the dye Disperse Violet 17 based on 1-amino-2-bromoanthraquinone 59. Optimal conditions include the use of Pd(OAc)2 and the ligand P(o-Tol)3 , and the yields of the products 60a – e were almost independent on the structure of the substituent in the styrene.
Investigations into environmentally acceptable variants of the Mizoroki – Heck reaction led to the development of conditions based on the use of the biodegradable solvent Cyrene (dihydrolevoglucosenone or 6,8-dioxabicyclo[3.2.1]octanone) and the more available catalyst Pd/C.[66] It has been experimentally confirmed that under the proposed conditions 2-iodoanthraquinone (36) is converted to ethyl 3-(anthracene-2-yl)acrylate 61 in 50% yield (see Table 2). Notably, Cyrene can be recovered and reused up to 4 times, reducing the amount of waste generated and the overall cost of the technology.
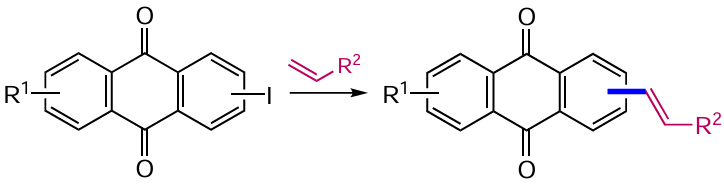
The Stille reaction, based on the Pd-catalyzed cross-coupling of organotin derivatives with haloanthraquinones, was an equally efficient approach to the vinylation of anthracene-9,10-diones. Li et al.[67] carried out the reaction of 1,5-dichloroanthraquinone (62) with vinyl(tri-n-butyl)tin in the presence of Pd2(dba)3 (dba is dibenzylideneacetone), PBut3 and cesium fluoride to afford 1,6-divinylanthraquinone 63 (Scheme 29).
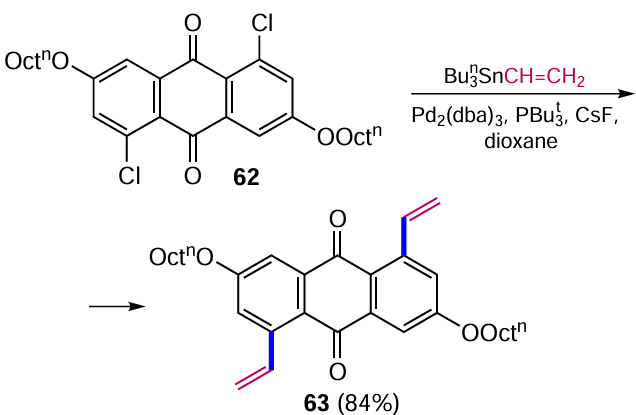
Vinylation of 2-iodoanthraquinone (36) with vinyl(tri-tert-butyl)tin at the β-position catalyzed by Pd(PPh3)4 was carried out by a similar method to give 2-vinylanthraquinone (64) (Scheme 30).[68]
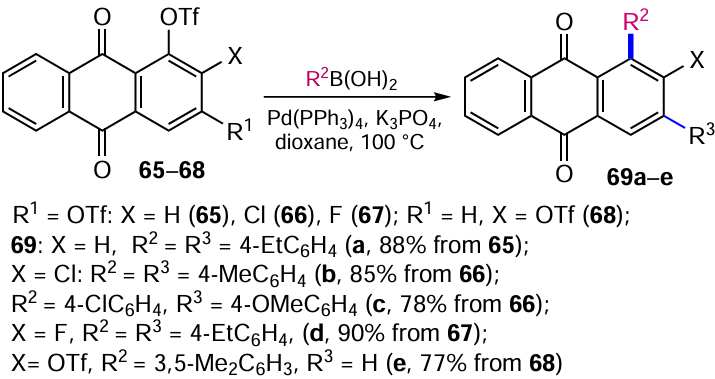
The Suzuki-Miyaura reaction is widely used for the arylation of anthraquinones. The Pd(PPh3)4-catalyzed reaction of (trifluoromethanesulfonyloxy)anthraquinones 65 – 68 with arylboronic acids affords arylanthraquinones 69a – e in high yields (Scheme 31).[69, 70] Notable, anthraquinones 65, 66 and 68 bearing two TfO groups undergo regioselective arylation initially at the α-position, whereas in the case of the 2-fluoro analogue 67 such selectivity is not observed. The use of 1 equiv. arylboronic acid in the reaction with anthraquinone 68 affords 1-(3,5-dimethylphenyl)anthraquinone 69e, the product of substitution of only α-OTf group.[70] Although position 3 is the least hindered, the chelating effect of the carbonyl group required for Pd(0) coordination and the greater electron deficiency of position 1 compared to position 3 were the determining factors for the initial formation of the α-mono derivatives.[69, 70] This was supported by the fact that the sequential cross-coupling of 4-chloro- and 4-methoxyphenylboronic acids with 1,3-di(trifluoromethanesulfonyloxy)-2-chloroanthraquinone (66) gave compound 69c (see Scheme 31).[69] This method is also applicable to the coupling of iodoanthraquinones with other (het)arylboronic acids.[71]
The Hiyama reaction [72] involves the reaction of aryl, alkenyl and aryl halides with organosilicon compounds as a possible alternative to toxic organotin reagents.[67, 68] This method has been successfully adapted to produce a copolymer based on tetrathiafulvalene and anthraquinone that can be used in solar cells.[73] Specifically, the Pd-catalyzed coupling of anthraquinone 36 with 1,3,5,7-tetravinyl-1,3,5,7-tetramethylcyclotetrasiloxane gives 2-vinylanthraquinone 64 in high yield (Scheme 32).
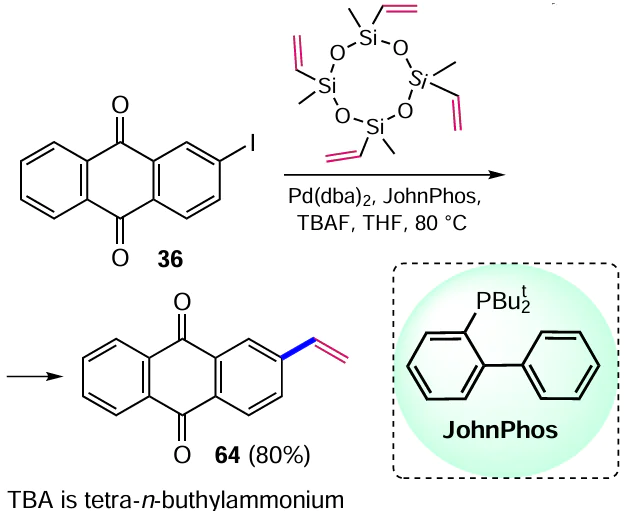
To obtain various aryldiazoacetates, a Pd-catalyzed cross-coupling reaction of aryliodides with diazoacetoacetic ester was developed,[74] which allowed the formation of a C – C bond in the β-position of anthracene-9,10-dione. 2-Iodoanthraquinone (36) reacts with diazoacetoacetic ester in the presence of Pd(PPh3)4 to give ethyl 2-(anthracene-2-yl)-2-diazoacetate (70) in moderate yield (Scheme 33).
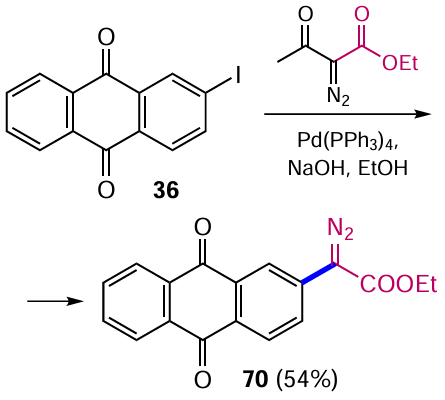
The reaction can follow two pathways (Scheme 34).[74] In the first the intermediate adduct A is formed by oxidative addition of iodide 36 to the palladium catalyst. Further coordination of intermediate A with the diazo ester generates species B, which undergoes nucleophilic attack by the hydroxide ion to give intermediate C (path a). Alternatively, the acyldiazoacetate can first be activated by nucleophilic attack of the hydroxide ion to form intermediate E, which after coordination converts complex A into intermediate C (path b). Interestingly, adduct B does not eliminate N2 , despite the stability of this molecule.
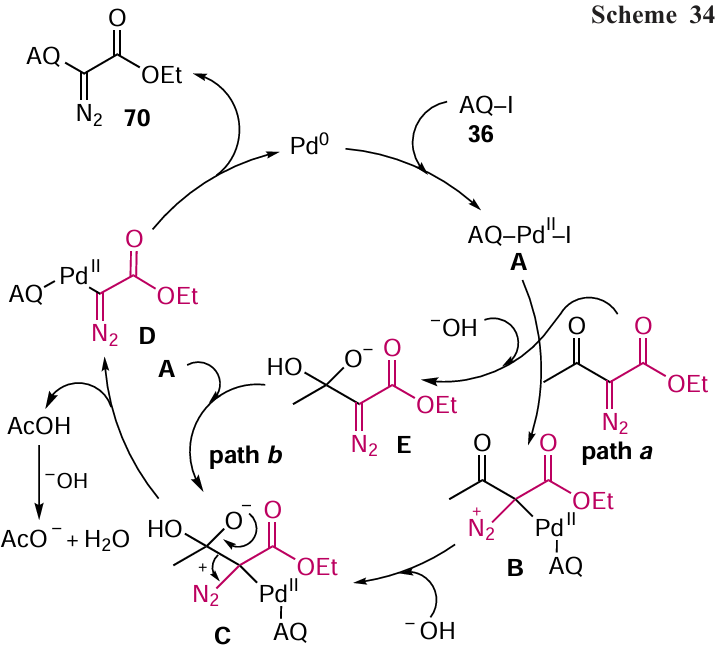
When developing organic semiconductors based on anthracene-9,10-dione, the arylation of unsubstituted anthraquinone (45), based on the C – H activation using Ru catalyst, has been used.[75] For the C – C bond formation in the reaction of anthracene-9,10-dione (45) with arylboronic esters, Kitazawa et al.[75] used a RuH2(CO)(PPh3)3 catalyst, which directs the reaction selectively to the α-position of the anthraquinone core by coordinating ruthenium to the carbonyl groups of anthraquinone in pinacolin (3,3-dimethyl-butan-2-one) medium. Anthraquinone (45) reacted with substituted phenylboronic acid esters to afford 1,4,5,8-tetraarylanthraquinones 71a – e (Scheme 35). The use of unsubstituted phenylboronic acid esters significantly reduced the yield of the arylation product 71a. The authors note that the reaction is accompanied by the formation of by-products, α,α-di- and triaryl-substituted anthraquinones (yields 3 – 5%).
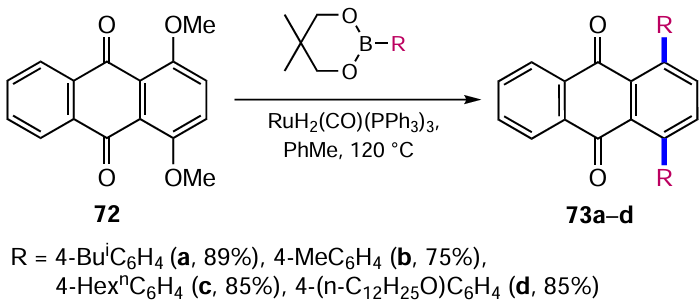
In order to obtain anthracene-9,10-dione-based polycyclic compounds with valuable properties for semiconductors, Suzuki et al.[76] modified the ruthenium-catalyzed cross-coupling described above and used a non-standard substrate, 1,4-dimethoxyanthraquinone (72), as the starting compound. Taking into account the different reactivity of C−H and C−O bonds in the α-positions of the anthraquinone nucleus, the arylation proceeded with the substitution of mainly alkoxy groups of anthraquinone 72 leaving the remaining α-hydrogen atoms intact to give anthraquinones 73a – d (Scheme 36). This difference helps to control the Ru-catalyzed cross-coupling and to introduce successively different aromatic substituents into the anthraquinone core.[77]
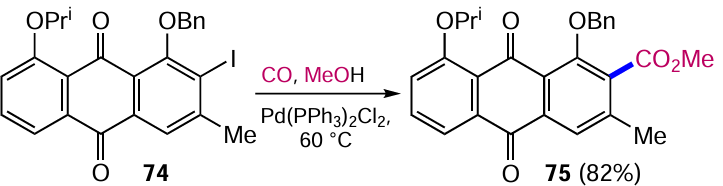
The ester functionality can be introduced into aromatic compounds via Pd-catalyzed methoxycarbonylation reactions of aryl halides or boronic acid esters.[78] Anthraquinone-based substrates can also be used in such cross-coupling reactions, as exemplified by the synthesis of the natural antitumour compound (±)-BE-26554A.[79] In one of the stages of its preparation, we carried out the carbonylation of 2-iodoanthraquinone 74 with carbon monoxide catalyzed by Pd(PPh3)2Cl2 to obtain ester 75 (Scheme 37).
Under similar conditions, methyl anthraquinone-2-carboxylate (76) was obtained from (anthraquinone-2-yl)boronic acid ester 43 by the action of carbon monoxide and the Pd(OAc)2 – PPh3 catalytic system in the presence of 1,4-benzoquinone (BQ), which is required for the oxidation of Pd0 to PdII (Scheme 38).[80]

Friis et al.[81] carried out catalytic conversion of the halogen atom of aryl and heteroaryl bromides to amides of various structures. Aminocarbonylation, demonstrated by the reaction of 2-bromanthracene-9,10-dione (39) with carbon monoxide and morpholine catalyzed by a palladium complex with the ligand XantPhos (4,5-bis(diphenylphosphino)-9,9-dimethylxanthene), gives anthraquinone-2-carboxamide 77 (Scheme 39). The XantPhos ligand in the precatalyst composition is superior to other ligands (e.g. PBut3 , dppf, 1,3-bis(dicyclohexylphosphino)propane (dccp), 2-dicyclohexylphosphino-2'-(dimethylamino)biphenyl (DavePhos), etc.), providing complete conversion of the starting aryl halide and high product yields under moderate heating.
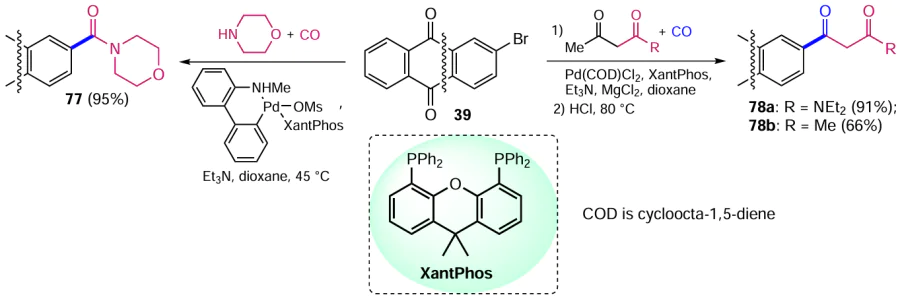
Another group of researchers [82, 83] developed a method for palladium-catalyzed carbonylation/deacetylation of aryl halides by reacting them with 1,3-dicarbonyl compounds and carbon monoxide. Treatment of 2-bromoanthraquinone (39) with carbon monoxide and acetylacetone or N,N-diethylacetoacetamide in the presence of a complex catalyst system gives the products 78a,b, the yield of the amide 78a being higher than that of the derivative 78b (see Scheme 39).
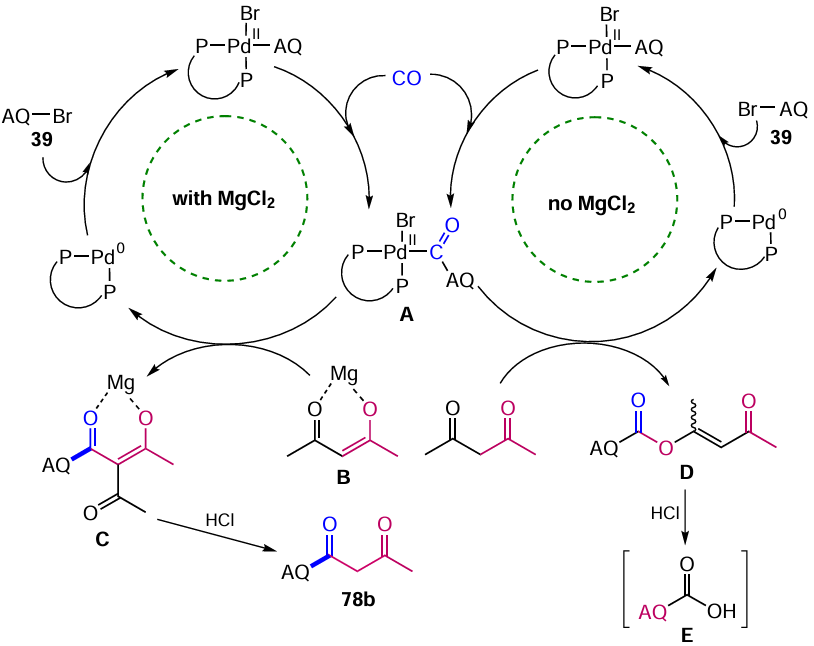
The putative mechanisms of the catalytic carbonylation cycles are summarized in Scheme 40.[83] Initially, in both cases, an oxidative addition of ArBr to Pd0 takes place, followed by the CO insertion and the formation of complex A. The magnesium chloride-activated complex B attacks intermediate A via nucleophilic substitution or ligand exchange to give compound C by reductive elimination. The triketone C is deacetylated by heating in aqueous HCl to give the 1,3-dicarbonyl compound 78b. In the absence of MgCl2 , the competing attack of complex A occurs on the more reactive oxygen atom to yield the enol ester D, which is converted to the acid E when treated with hydrochloric acid.
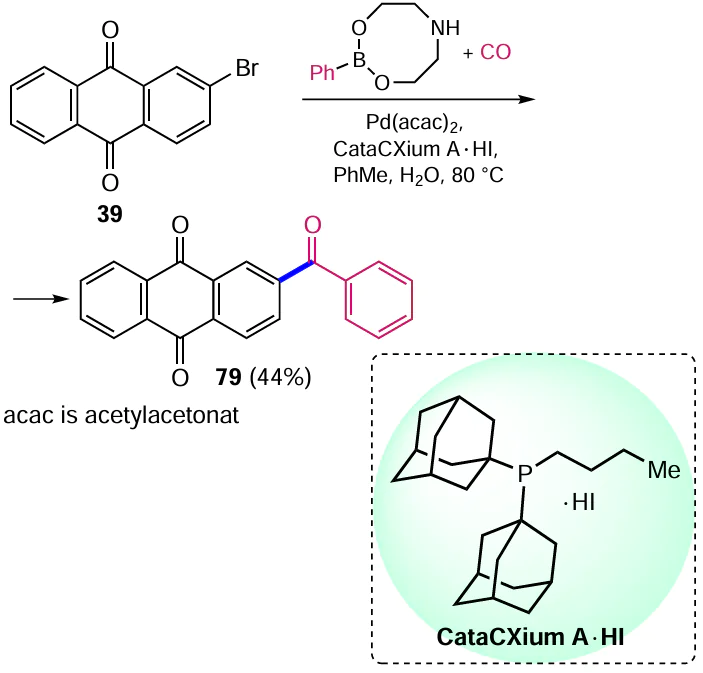
For the synthesis of unsymmetrical aryl ketones, Bjerglund et al.[84] carried out tandem carbonylation/cross-coupling of Suzuki – Miyaura (het)aryl bromides with phenylboronic acid derivatives in the presence of carbon monoxide. The reaction was adapted for the electron-deficient 2-bromoanthraquinone (39), which afforded 2-benzoylanthracene-9,10-dione (79) in good yield via Pd-catalyzed reaction with CO and 2-phenyl-1,3,6,2-dioxaazaborocane (Scheme 41). The use of di(1-adamantyl)-n-butylphosphine hydroiodide (CataCXium A*HI reagent) as a ligand provided high substrate conversion and good selectivity in the synthesis of diarylketone 79.
2.3. Bond with the sp-hybridized carbon atom
The formation of new C – C bonds with an sp-hybridized carbon atom is necessary for the preparation of alkynyl and cyano derivatives of anthraquinone. It should be noted that recent advances in the synthesis and application of acetylenic derivatives of quinones and, in particular, anthraquinones are discussed in detail in the review by Vasilevskii and Stepanov,[23] published in 2022. Therefore, only a few of the most important methods for the preparation of alkynyl anthraquinones will be discussed in this Section.
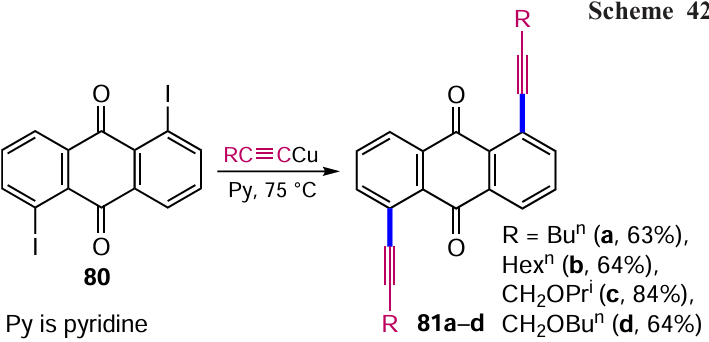
One of synthetic approaches to alkynyl-containing aromatic compounds is the Castro – Stephens reaction. At present, the validity of this method has diminished considerably due to the development of a more efficient and experimentally convenient Sonogashira cross-coupling. Nevertheless, Baranov et al.[85] used the reaction of various copper acetylenides with 1,5-diiodanthracene-9,10-dione (80) in pyridine to give 1,5-dialkynylanthracene-9,10-diones 81a – d (Scheme 42).
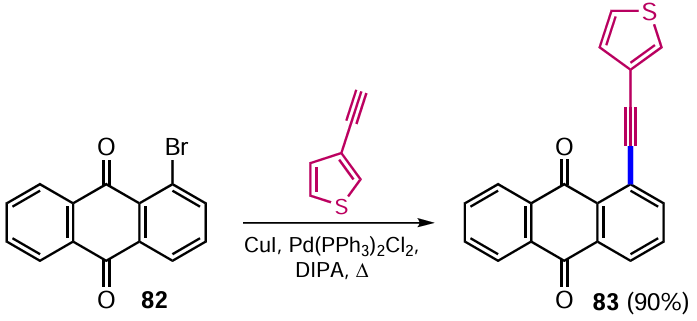
To date, Sonagashira cross-coupling is recognized as the most efficient way to form a C – C bond with an sp-hybridized carbon atom, allowing the introduction of various alkynyl groups into the molecule and providing high yields of the products.[86] This reaction was a key step in the preparation of thiophene-containing anthraquinones, which were explored as potential organic molecules for applications in molecular electronics. 1-Bromoanthraquinone (82) reacts with 3-ethynylthiophene in the presence of Pd(PPh3)2Cl2 , CuI and diisopropylamine (DIPA) to afford 1-(thien-3-yl-ethynyl)anthracene-9,10-dione (83) (Scheme 43).[87]
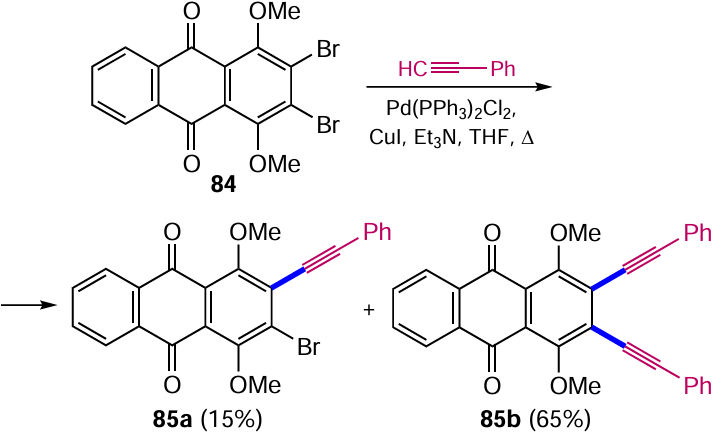
Sonogashira cross-coupling also proved to be an efficient method for the preparation of β-alkynylanthraquinones. Condensation of 2,3-dibromo-1,4-dimethoxyanthraquinone (84) [88] with equimolar amounts of phenylacetylene catalyzed by the Pd(PPh3)2Cl2 – CuI system gives predominantly the product 85b with two halogen atoms substituted (Scheme 44). The increased reactivity of 2-bromo-1,4-dimethoxy-3-(phenylethynyl)anthraquinone (85a) compared to the starting dibromoanthraquinone 84 was observed, which may be due to better coordination of the catalyst by the introduced alkynyl moiety.[88] A similar approach was applied to the synthesis of ligands based on 3,5-alkynylanthracene-9,10-diones that stabilize G-quadruplexes of nucleic acids.[89]
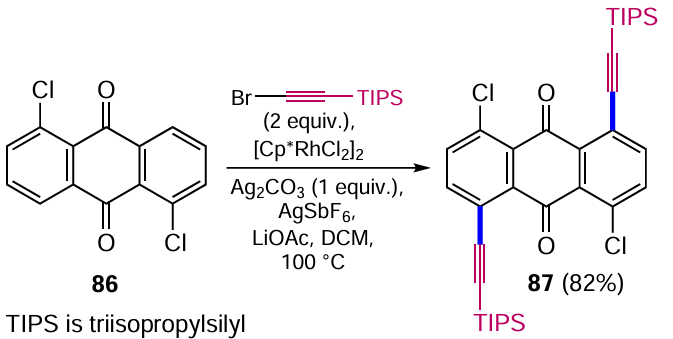
Tan et al.[90] developed a procedure for the Ru-catalyzed alkynylation of C – H bonds by bromalkynes, which is an inverse Sonogashira reaction. The main advantage of the new approach over the classical Sonogashira cross-coupling is that it does not require the prior introduction of leaving groups (Hal, OTf, etc.) into the anthraquinone core and is characterized by a high regioselectivity of the alkynylation of the α-position of the anthraquinone. Cross-coupling of 1,5-dichloranthracene-9,10-dione (86) with (bromoethynyl)triisopropylsilane promoted by complex ([Cp*RhCl2]2) (Cp* is pentamethylcyclopentadienyl) leads to the double alkynylation product 87 (Scheme 45) without affecting the chlorine atoms. The reaction mechanism involves ortho-CH activation via electrophilic aromatic substitution of hydrogen by a ruthenium complex, followed by insertion of the bromalkyne and elimination of the bromide ion. The anthraquinone carbonyl groups act as directing groups, directing the process towards the formation of the α-alkynylation product.
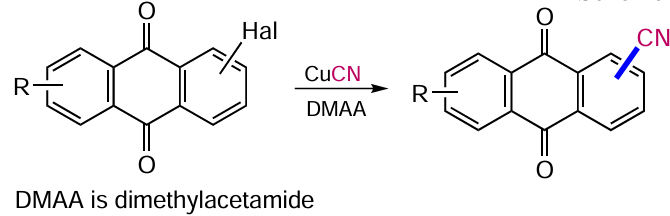
The Rosenmund--von Braun reaction is one of the most convenient ways to introduce a cyano group into aromatic compounds, which can be further modified to obtain other functional derivatives. This approach is based on the substitution of halogen atoms by copper(I) cyanide in aromatic compounds,[91] including anthraquinones. For example, treatment 1,8-dichloranthracene-9,10-dione (88) with CuCN gave 1,8-dicyanoanthraquinone (89), which is a valuable intermediate that can be further converted to acid derivatives (Scheme 46, Table 3).[92, 93]
The brominated anthraquinones undergo cyanation under similar conditions, but at a much lower temperature.[94] In the presence of two halogen atoms in the α- and β-positions of an anthraquinone (compound 90), the α-bromine atom is predominantly substituted (product 91), even with an excess of cyanating reagent (see Table 3). A similar compound 92 containing the β-alkyl group gives the nitrile 93 in quantitative yield.
Increasing the temperature in the Rosenmund – von Braun reaction also allows the substitution of the β-halogen in compound 84 to give product 94 (Scheme 47).[95] Although this cyanation method does not require additional catalysts, its disadvantages, such as the use of toxic cyanides and the generation of equimolar amounts of copper-containing waste, have encouraged the development of other approaches to the synthesis of nitriles.
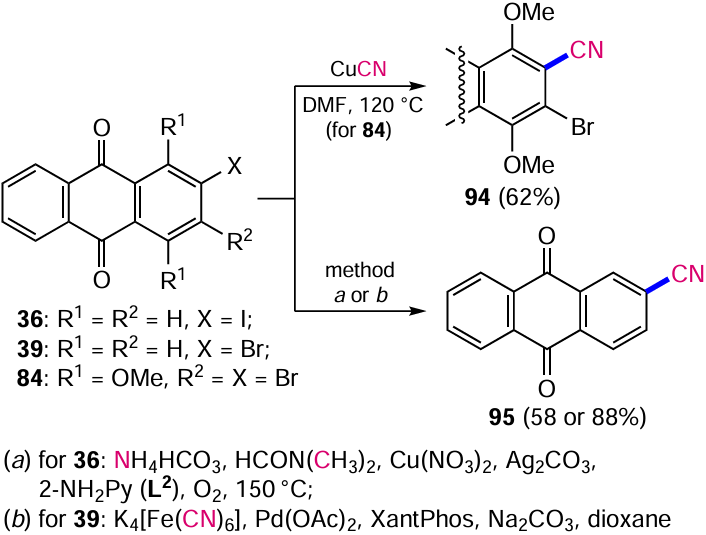
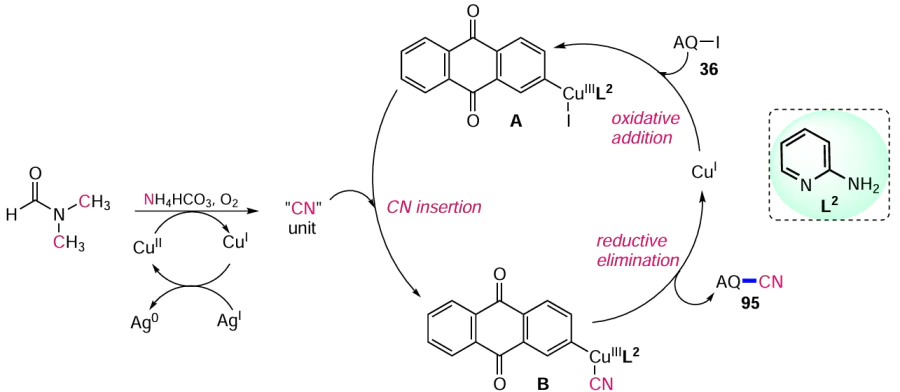
Pawar and Chang [96] developed an alternative Cu-catalyzed reaction to safely introduce the nitrile group into the aromatic nucleus. In contrast to the classical Rosenmund – von Braun reaction, in which the cyanide ion acts as a nucleophile, the new method involves the in situ generation of the cyanide ion. Ammonium bicarbonate is used as the nitrogen source and DMF performed best as the carbon source. Cyanation is catalyzed by a mixture of Cu(NO2)2 and Ag2CO3 in an O2 atmosphere. The method has also been tested on 2-iodoanthraquinone (36), from which 2-cyanoanthracene-9,10-dione (95) is formed (see Scheme 47, method a). The putative reaction mechanism is shown inScheme 48.
While searching for other approaches to introduce a cyano group into the aromatic nucleus, a unique methodology was developed for the mechanochemical solid-phase Pd-catalyzed cyanation of aryl bromides under the action of K4[Fe(CN)6]. The reaction was carried out in an electromagnetic mill using ferromagnetic rods, and to increase the number of collisions of the components of the reaction system, a small amount of dioxane was added for better grinding (so-called liquid-assisted grinding). Optimized cyanation conditions for substrates with electron-withdrawing substituents, including 2-bromoanthraquinone (39), include the Pd(OAc)2 – XantPhos system (see Scheme 47, method b).[97]
The reaction mechanism (method a, Scheme 47) shown in Scheme 48 involves the Cu-catalyzed formation of cyanide from DMF and ammonium bicarbonate, with reduction of copper(II) to copper(I).[96] The resulting monovalent copper salt undergoes oxidative addition via the halogen–anthraquinone bond, forming a copper-based complex A with the 2-aminopyridine ligand L2. The resulting cyanide species is inserted into the complex via the Cu – I bond and leads to the adduct B, which undergoes reductive elimination to give 2-cyanoanthraquinone (95). In particular, the silver(I) salt is required for the reoxidation of CuI to CuII.
3. Reactions with the formation of the carbon – halogen bond
Halogenated anthraquinones are among the most important intermediates used for the subsequent functionalization of the anthraquinone nucleus. Halogen atoms represent a universal leaving group capable of substitution directly under the action of nucleophiles or in various catalytic reactions, providing access to the formation of virtually any C – heteroatom bond. The role of the halogen atom in anthraquinones is highlighted by a significant number of examples of its transformation into various functional groups to form carbon-, oxygen-, nitrogen-, sulfur-, selenium- and phosphorus-containing anthraquinones. In addition to the synthetic significance of halogen derivatives, this class of compounds is characterized by valuable biological (e.g. topopyrones A and B, chloroemodines), spectral (dyes, including dispersed polyester blue) and other applied properties.[4, 5]
Traditionally, chloro, bromo and iodo derivatives of anthraquinone are prepared by electrophilic halogenation of the anthraquinone nucleus under various conditions. The bromination and iodination reactions are best for anthraquinones bearing strong electron-donating substituents (OH, NH2). Halogen atoms can be introduced during the anthraquinone nucleus construction, as well as by addition reactions, nucleophilic substitution of another halogen or diazonium group with a halide ion.[20] The latter method is often used as an approach to fluoro and iodo anthraquinones. The modern methods of anthraquinone core halogenation are based on palladium-catalyzed processes, including metathesis reactions.
Despite progress in the development of halogenation methods, direct fluorination reactions of anthraquinone derivatives are virtually undescribed. The synthesis of fluorinated anthraquinones involves the substitution of a fluorine atom by functional groups or the direct assembly of the anthraquinone core from fluorinated intermediates (e.g. Friedel – Crafts acylation of hydroquinone with fluorinated phthalic anhydride).[98]
Among the approaches based on the transformation of functional groups, the Shiemann fluorination is still relevant for the synthesis of fluorinated anthraquinones. For example, diazotization of 2-aminoanthracene-9,10-dione (50), followed by treatment with tetrafluoroboric acid and thermal decomposition of the resulting diazonium tetrafluoroborate gives 2-fluoranthraquinone (96) in moderate yield (Scheme 49, method a).[99] Using slightly different steps, 2-fluoranthraquinone (96) was synthesized from 2-aminoanthraquinone (50) in a higher yield of 69%.[100] Importantly, 1-fluoroanthraquinone was also obtained from 1-aminoanthraquinone in the same way.[100]
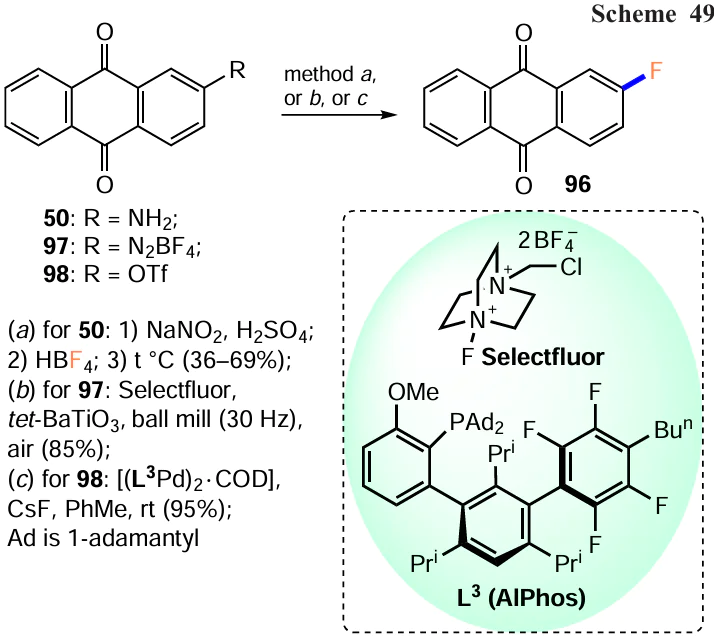
Recently, Wang et al.[101] proposed a unique method to obtain aryl fluorides from an aryldiazonium salt and Selectfluor reagent (1-fluoro-4-chloromethyl-1,4-diazabicyclo[2.2.2.2]octane bis(tetrafluoroborate)) using piezoelectric material in a ball mill. Mixing anthraquinone-1-diazonium tetrafluoroborate (97) in a ball mill with tetragonal barium titanate and Selectfluor reagent gives 2-fluoranthraquinone (96) (see Scheme 49, method b). The reaction follows a radical pathway with Selectfluor acting as a fluorine source and reducing agent.
Sather et al.[102] described an efficient Pd-catalyzed method for the fluorination of activated (het)aryltrifluoromethane sulfonates or aryl halides. To achieve this, they obtained a biarylmonophosphine ligand, di-1-adamantyl(4''-butyl-2'',3'',5'',6''-tetrafluoro-2',4',6'-triisopropyl-2-methoxy-m-terphenyl)phosphine (AlPhos, L3) (see Scheme 49, method c), which allows the introduction of a fluorine atom into the aromatic nucleus at room temperature. The Pd0-catalyzed reaction of (anthraquinone-2-yl)trifluoromethanesulfonate (98) with cesium fluoride gave 2-fluoranthraquinone (96) in almost quantitative yield. It should be noted that the direct nucleophilic substitution of halogens or other leaving groups by fluoride ion occurs at temperatures > 100 °C, and in some cases at 300 °С,[20] making the catalytic method more preferable for the modification of polyfunctional anthraquinones.
A large number of synthetic methods are known for the construction of the C – Cl bond in anthraquinone derivatives, mainly due to the possibility of direct chlorination. Anthraquinones can undergo electrophilic chlorination reactions with substitution of hydrogen, nitro or sulfo groups using various reagents, allowing the introduction of a chlorine atom in the α- and β-positions.[20] For example, chlorination of emodin (7) with N-chlorosuccinimide (NCS) in the presence of a catalyst (e.g. ZrCl4) gives 2,4-dichloroemodin (99) in 77% yield (Scheme 50, Table 4),[103] but the feasibility of such catalysis is not entirely clear, since in the absence of zirconium chloride, when NCS was used in THF at 60 °С, the yield of product 99 was quantitative.[104] At the same time, Koerner et al.[105] showed that the addition of ZrCl4 during the chlorination of emodin (7) with N-chlorosuccinimide is able to control the regioselectivity and give a 2-chloro derivative of emodin in 76% yield. Interestingly, treatment of emodin (7) with sulfuryl chloride gives mainly 4-chloroemodin 100 (see Table 4).[105] The reason for the high regioselectivity of the chlorination of this compound with SO2Cl2 remains unclear.
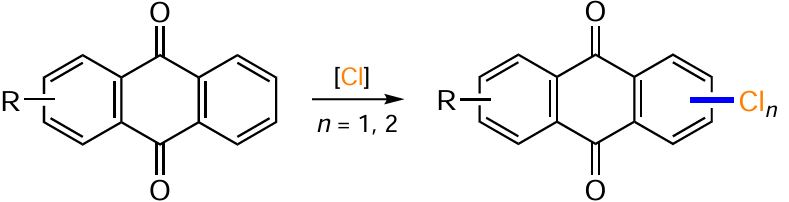
Other chlorinating agents can be used to chlorinate emodin (7): system HCl – H2O2 in trifluoroethanol, HCl – MnO2 in acetic acid, SO2Cl2 , HClO3 and others. For example, different ratios of HCl and hydrogen peroxide give a mixture of mono-, di- and trichloro derivatives, but the use of 5 equiv. HCl and 10 equiv. H2O2 gives predominantly 2,4-dichloroemodine (99) (see Table 4).[104] Chlorination of 2-methylquinizarin (101) using the HCl – MnO2 system furnished the β-chloro derivative 102.[106, 107]
Heating 5-nitro-9,10-dioxoanthracene-1-potassium sulfonate (103) to reflux in hydrochloric acid with HClO3 leads to the formation of 5-nitro-1-chloroanthraquinone (104) as a result of the ipso-substitution of the sulfonic group by a chlorine atom (see Table 4).[108]
The introduction of a chlorine atom, as well as other halogens, into anthraquinones, can be carried out by the Sandmeyer reaction by treating the diazonium salt with copper(I) chloride. For example, 2-iodo-1-chloranthraquinone (106) is obtained from 1-amino-2-iodoanthraquinone (105) by treatment with the NaNO2 – H2SO4 system followed by substitution of the diazonium group under the action of copper(I) chloride (see Table 4).[109]
Direct electrophilic halogenation with molecular bromine is the most common approach to bromine derivatives of anthracene-9,10-dione. The regioselectivity and the number of bromine atoms introduced can vary considerably depending on the reaction conditions and the structure of the substrate. In the presence of electron-donating substituents on one of the benzene rings of anthraquinone, bromination occurs exclusively at the activated ring. Thus, 1,2-diaminoanthraquinone (107) reacts with Br2 in acetic acid to give 1,2-diamino-3-bromoanthraquinone (108),[110] and the bromination of 1-hydroxy- (13) and 1-aminoanthraquinones (48) affords 2,4-dibromo derivatives 109a,b (Scheme 51, Table 5).[94, 111, 112]
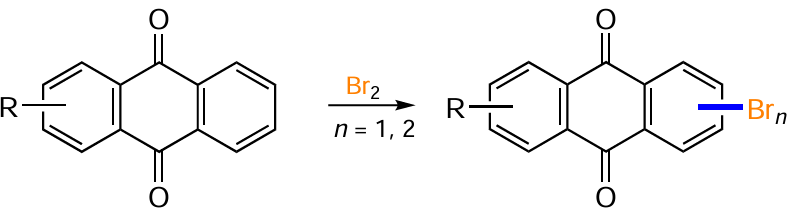
Blocking of position 2 by substituents (e.g. in compounds 110) leads to bromination in the para position to the amino or hydroxy group to give products 111 (seeTable 5).[94] Malik et al.[111] synthesized 1-amino-4-bromoanthraquinones 113 containing both electron-donating and electron-withdrawing substituents in position 2 of the anthraquinone nucleus by brominating compounds 112 using molecular bromine in DMF. The reaction of the sodium salt of 1-aminoanthraquinone-2-sulfonic acid with Br2 at room temperature gives a 4-bromo derivative, but on mild heating the reaction is accompanied by ipso-substitution of the sulfonic group with bromine and leads to 1-amino-2,4-dibromanthraquinone.[111] The bromination of 1,8-dialkoxyanthraquinone 114 in the presence of AcONa in chloroform preferentially produces the 4,5-dibromo derivative 115,[113] while the ortho positions to the alkoxy groups are not affected.
Phadtare and Shankarling [114] carried out the electrophilic bromination of 1-aminoanthraquinones 116 with molecular bromine in a deep eutectic solvent (a mixture of choline chloride and urea) to afford brominated anthraquinones 117 (see Table 5). The use of this environmentally friendly solvent not only shortens the reaction time but also improves the product yield compared to that in chloroform and methanol, as exemplified with bromination of 1-aminoanthraquinone.
N-Bromosuccinimide (NBS) is also widely used as a reagent to introduce a bromine atom into the anthraquinone nucleus containing electron-donating substituents. In most cases, hydrogen substitution under the action of NBS is achieved in a non-catalytic mode, although some additives can improve the regioselectivity of the reaction.
The bromination of 2-amino- and 2-hydroxyanthraquinones proceeds predominantly to positions 1 and 3 of the anthraquinone nucleus. However, due to the electron-withdrawing effect of carbonyl groups, the α-bromine atoms are labile and can be selectively removed, allowing only the product of the b-substitution to be obtained. Reaction of 2-aminoanthraquinone 50 with NBS in DMF and subsequent reductive dehalogenation of the intermediate 2-amino-1,3-dibromoanthraquinone gives 2-amino-3-bromanthraquinone (118) (Scheme 52, Table 66).[115]
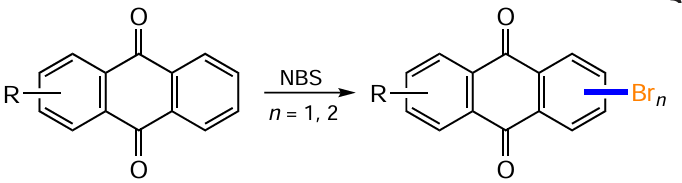
The use of the NBS-DIPA system in DCM has some value for performing b-halogenation of 1-hydroxyanthraquinones: bromination of compound 119 at room temperature produces mainly 2-bromo derivative 120.[116] Under similar conditions, it is possible to introduce two bromine atoms in the ortho positions of 1,8-dihydroxyanthraquinone (121), while the low yield of the main product 122 is due to insufficient selectivity of the bromination of the parent compound.[89] The authors do not report any by-products formed in this process. Other di- and trihydroxyanthraquinones, such as compounds 123 and 7, also react readily with NBS to give the corresponding (di)bromanthraquinones 124 and 125, depending on the substrate structure (see Table 6).[104, 105, 117]
It should be noted that the treatment of emodine (7) with HBr (4 equiv.) and H2O2 (5 equiv.) in trifluoroethanol also allows the efficient preparation of 2,4-dibromoemodine (125) (Scheme 53, Table 7).[104] Halogenation in this case can be stopped at the stage of formation of 2-bromo-1,3,8-trihydroxy-6-methylanthraquinone (126) by reducing the amount of hydrogen bromide and H2O2 , but it should be noted that the process is accompanied by an incomplete conversion of the starting emodin (7).[104]
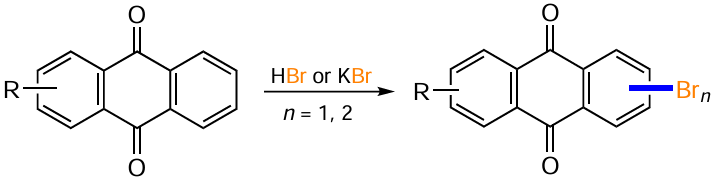
A similar method has been used to introduce a bromine atom into aminoanthraquinones 127 (see Table 7). In particular, Patil et al.[118] brominated such derivatives using the HBr – H2O2 or HBr – mCPBA system to obtain 2,4-dibromanthraquinones 128. In this case, 1-(N-alkylamino)anthraquinones gave products monobrominated at the 4-position, probably due to a steric factor. The use of mCPBA as oxidant significantly accelerated the formation of bromoanthraquinones.
Alternatively, anthraquinone bromination can be carried out under mild conditions using KBr in the presence of 1,9-bis(peroxy)nonanedioic acid.[119] The efficiency of this halogenation method was demonstrated on anthraquinones 129 containing electron-donating substituents, which gave bromine-substituted anthraquinones 130 in this reaction (see Table 7).
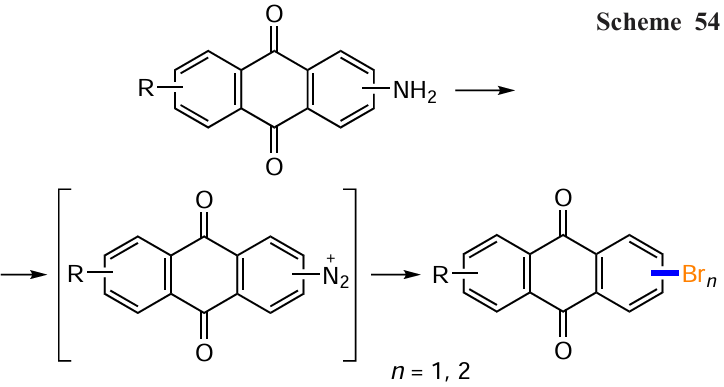
Bromo derivatives can be obtained from aminoanthraquinones by sequential diazotization and Sandmeyer reactions. Typically, α- or β-aminoanthraquinones, such as compounds 131 or 50, are first diazotized by treatment with tert-butyl nitrite followed by CuBr2 in acetonitrile to give products 132 and 39, respectively (Scheme 54, Table 8).[120-122] The classical conditions (NaNO2 , HBr and CuBr) have been used to convert the polyfunctional 2-amino-3-methyl-1,4-dimethoxyanthraquinone (133) to the 2-bromo derivative 134.[123]
A number of methods based on electrophilic substitution reactions are effective for introducing the iodine atom into the anthraquinone nucleus. For anthraquinones activated by electron-donating substituents, iodination is usually achieved by the action of molecular iodine in the presence of various oxidants. For example, treatment of 3-amino-1,8-dimethoxyanthracene-9,10-dione (135) with a mixture of I2 and Ag2SO4 in ethylene glycol allows the iodine atom to be selectively introduced into the α-position of the most activated ring, giving 3-amino-4-iodo-1,8-dimethoxyanthracene-9,10-dione (136) (Scheme 55, Table 9).[124]
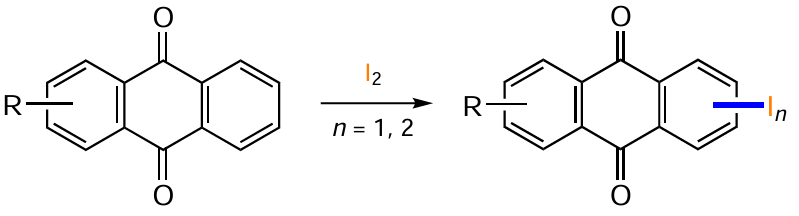
The iodination of 1,5-dibutoxyanthraquinone (137) in the presence of [bis(trifluoroacetoxy)iodo]benzene (PIFA) proceeds regioselectively in the para position relative to the alkoxy groups to furnish the product 138 (see Table 9).[125] The presence of the I2–oxidant system also allows the introduction of an iodine atom in the b-position of anthraquinone. Interestingly, when the oxidant in the I2 – PIFA system is replaced by I2 – HIO3 when using 1,5-dialkoxyanthraquinone 139, electrophilic substitution at positions 2 and 6 occurs to provide high yields of compound 52 (see Table 9).[60] Other examples using the I2 – HIO3 system, in dioxane [126] or in a CCl4 – AcOH mixture [60] confirm the efficiency of this method (see Table 9, synthesis of the product 140 and the conversion of substrate 141 to compound 142).
The iodination of anthraquinone can be achieved by using molecular iodine in the presence of bases.[103, 104, 127] The reaction of emodin (7) with a small excess of I2 (2 – 4 equiv.) and NaHCO3 in aqueous THF gives the mono-iodination product 143 in 78% and 100% yields respectively, while a significant excess of iodine under the same conditions leads to the 2,4-diiodo derivative.[104] It should be noted that the iodination of emodin (7) to the 2-iodo derivative 143 is also efficiently carried out in the presence of I2 – Н2О2 or N-iodosuccinimide (NIS).[104] The transformation of 2,6-dihydroxyanthraquinone (144) to 2,6-dihydroxy-3,7-diiodoanthraquinone (145) (see Table 9) demonstrates the possibility of using pyridine as a base.[127]
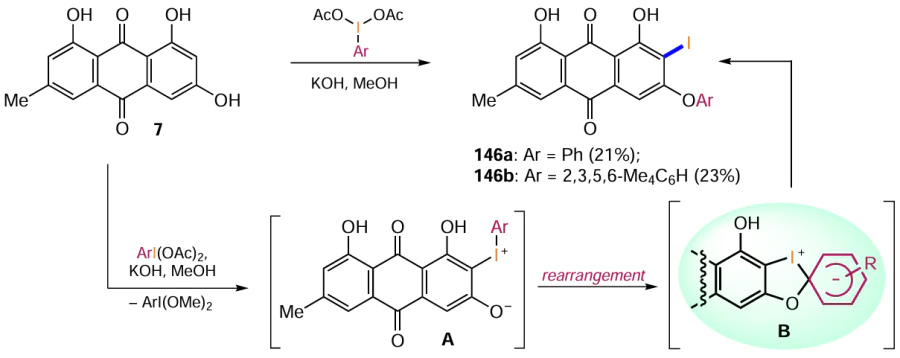
Approaches to the synthesis of iodine derivatives of anthraquinone can be further extended by the use of various iodine-based reagents. For example, Daub et al.[128] obtained the derivatives 146a,b through the oxidation of emodin (7) with (diacetoxyiodo)arenes in methanol in the presence of potassium hydroxide (Scheme 56). A mechanistic study into the reaction showed that the iodination proceeds via the formation of an intermediate iodide A, which is rearranged first to the spirocyclic Meisenheimer complex B and then to the iodoanthraquinones 146a,b. The authors note that (diacetoxyiodo)arenes ArI(OAc)2 are converted to (dimethoxyiodo)arenes ArI(OMe)2 under strongly alkaline conditions (e.g. KOH in MeOH).
Tetramethylammonium dichloroiodate is an unusual reagent for the iodination of electron-deficient aromatic nuclei.[129] During the selection of conditions for introducing the iodine atom into unsubstituted anthraquinone (45), it was found that only the use of Me4N+ICl2– leads to 2-iodoanthraquinone (36), albeit in low yield (Scheme 57, conditions a). It is worth noting that the addition of silver sulfate in sulfuric acid helps to increase the electrophilicity of the iodinating reagent, which seems to be accompanied by an increase in the yield of the target product 36 to 73% (see Scheme 57, conditions b).
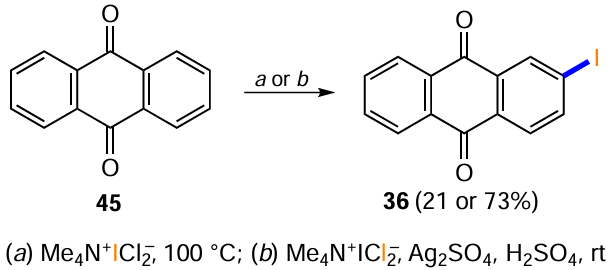
The substitution of an amino group with an iodine atom via a diazo intermediate, as in the case of other halogens, is often used for the synthesis of iodine-containing anthraquinones. Krasnokutskaya et al.[130] developed a one-step procedure for the conversion of anilines into the corresponding iodoaromatic derivatives; in one example, the treatment of 1-aminoanthraquinone (48) with p-toluenesulfonic acid, NaNO2 and KI in acetonitrile gave 1-iodoanthraquinone (56) (Scheme 58, Table 10). An alternative modification of the method of diazotization of 1-aminoanthraquinone (48) using a polymer suppored nitrite reagent has been proposed, but it provides a much lower yield of product 56 (see Table 10).[131] Bringmann et al.[132] successfully diazotized electron-deficient 1-aminoanthraquinone-2-carboxylic acid (147) and then substituted the diazo group with iodide under classical conditions to give 1-iodoanthraquinone-2-carboxylic acid (148).
The applicability of this approach to the preparation of β-iodoanthraquinones has been demonstrated on a number of substrates (see Table 10). The substitution of the amino group of 2-aminoanthraquinone 135 for the iodine atom occurs with varying efficiency.[133-135] For example, it can be achieved in a single step using isopentyl nitrite and diiodomethane, but with lower product yields.[136] In the case of 2-aminoanthraquinone (50), the maximum yield of product 36 was obtained by sequential treatment with NaNO2 in hydrochloric acid and KI.[135]
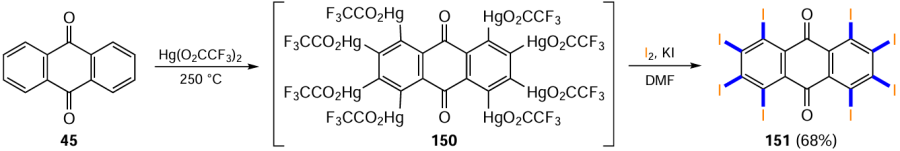
Jiang and Jin [137] have developed a method for the synthesis of octaiodoanthraquinone 151 based on the polymercurated anthraquinone 150 by mercury-iodine exchange under the action of a mixture of iodine and potassium iodide (Scheme 59). The authors note that due to the electron-withdrawing effect of carbonyl groups in the anthraquinone nucleus, as well as the low reactivity of most iodinating reagents (such as ICl, HIO4 , I2 – H2O2 system), polyiodoanthracene-9,10-diones are virtually inaccessible by direct electrophilic substitution. In particular, the polysubstituted anthraquinone 150 was synthesized by keeping anthraquinone (45) with mercury(II) trifluoroacetate at high temperature (see Scheme 59). The authors also obtained intermediate 150 by an alternative route via Hg-catalyzed oxidation of anthracene to anthraquinone and subsequent mercuration, similar to derivative 45. The yield of intermediate 150 is not reported in the original paper.
Lee and Morandi [138] proposed a variant of the metathesis of aroyl and aryl halides in which the interconversion of functional groups takes place mediated with a phosphonium ligand, which is part of the catalyst. The efficiency of this method was also demonstrated on anthraquinone derivatives 152 – 154. The reaction of anthraquinone-2-carboxylic acid chloride (152) or its 1,8-diacetoxy derivative 153 with iodobenzene in the presence of the palladium complex Pd2(dba)3 and the XantPhos ligand afforded 2-iodo-(36) or 1,8-diacetoxy-3-iodoanthraquinone 155 (Scheme 60, Table 11).
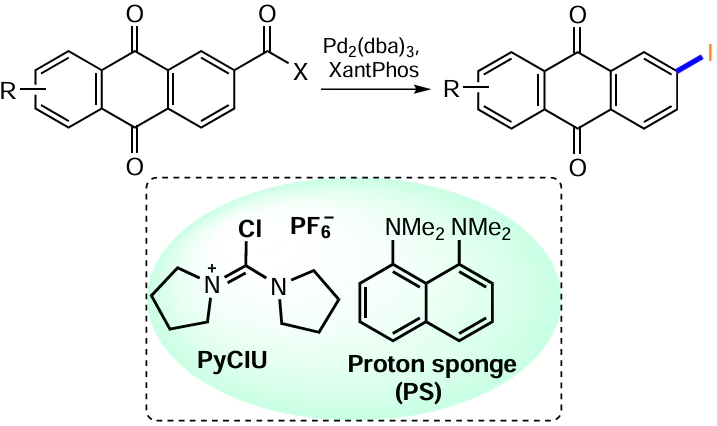
The said method for the preparation of aryliodides has been modified by using iodobutane as an iodine source, with aryl and vinyl carboxylic acids as substrates.[139] Thus, the treatment of acid 154 with 1-[(pyrrolidin-1-yl)chloromethylidene]pyrrolidinium hexafluorophosphate (PyCIU) generates in situ the corresponding acid chloride, which reacts with iodobutane under catalysis with Pd2(dba)3 and XantPhos in the presence of the proton sponge (PS) to give iodoanthraquinone 155 (see Table 11).
4. Reactions with the formation of the carbon – oxygen bond
The introduction of oxygen-containing functions (hydroxy, alkoxy, acetoxy groups, etc.) is a widely demanded trend in chemical modification of anthraquinone derivatives, which serves mainly to obtain biologically active compounds, dyes and analytical reagents. As with other classes of aromatic compounds, the formation of the C – O bond in anthraquinones is in most cases achieved by the transformation of some functional substituents into a target alkoxy or hydroxy group. Obviously, the possibility of introducing a hydroxy group in a given position on an anthraquinone is limited by the availability of suitable substrates.
Substitution of halogen atoms in the aromatic nucleus with a hydroxy group is a classical synthetic approach to phenols. The halogen atoms in the α-position to the carbonyl groups of anthraquinone can be directly substituted under the action of alkali, but for the β-functionalization such a method has not yet been described.
Reaction of aryl halides with 2-butyn-1-ol and potassium tert-butoxide under microwave irradiation gives propargyl ethers which are further isomerized in situ to allenyl ethers and then hydrolyzed to phenols.[140] This method was tested on the preparation of 1,4-dihydroxyanthraquinone (10) from difluoroanthraquinone 156 (Scheme 61, Table 12).
Sodium trimethylsilanoate (NaOTMS) is an effective reagent for the introduction of a hydroxy group via SNAr ipso-substitution of the fluorine atom for activated arenes.[141] Derivatives of fluoranthracene-9,10-dione or fluorobenz[g]isoquinoline-5,10-dione 157 (presented in general form in Table 12) were converted to 1-hydroxy-containing products 158 when treated with NaOTMS in THF at different temperatures followed by hydrolysis. It is worth noting the high chemoselectivity of the process: boiling 4-fluoro-1-chloroanthraquinone with excess NaOTMS led to isolation of the product of substitution of only the fluorine atom (see Table 12).
Škalamera et al.[142] synthesized β-hydroxyanthraquinone 160 by heating 2,6-dibromoanthraquinone (159) with CsF followed by treatment with aqueous potassium carbonate solution. The intermediate 2-bromo-6-fluoroanthraquinone probably undergoes hydroxylation with substitution of the fluorine atom due to its greater mobility compared to the bromine atom in aromatic nucleophilic substitution reactions. The low yield of anthraquinone 160 may be related to the formation of the intermediate difluoroanthraquinone during the bromine atom substitution with fluoride anion, but the authors do not discuss the by-products.
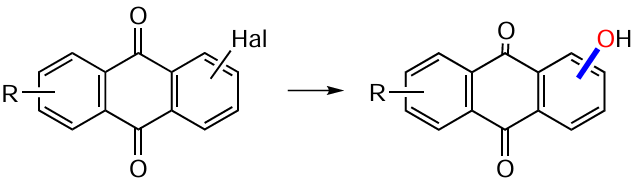
An efficient way to prepare β-hydroxyanthraquinones has been the nucleophilic substitution of various leaving groups (LG) in compounds 161 (e.g. halogen atom, nitro and sulfonic groups) by the action of benzaldoxime anion (Miller-Loudon – Snyder reaction, Scheme 62).[95] It is noted that electron-donating substituents R, located in the ortho-position to the leaving group, significantly reduce the tendency of compounds 161 to nucleophilic substitution, whereas electron-withdrawing groups, on the contrary, activate it and easily yield β-hydroxyanthraquinones 162. The presence of β-positioned ionizable functional groups (R = NH2 , OH, NHAc) completely deprives the molecule of the ability to substitute the leaving group.
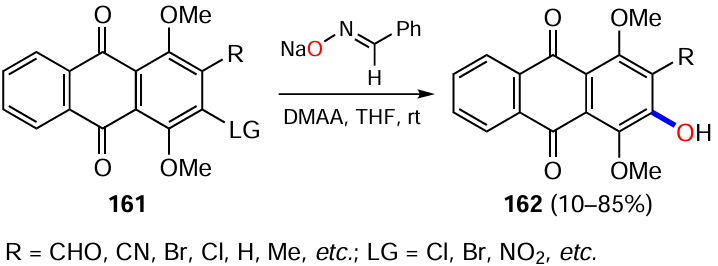
Hu et al.[143] managed to introduce hydroxy groups into the aromatic ring using oxime N-methylpyrrol-2-carbaldehyde and acetohydroxamic acid as hydroxylating agents in a basic medium, with the dimethylsulfide group in the arene being the leaving group. By this method, the derivative 163 was converted into 2-hydroxyanthraquinone (164), the yields of which were close for both reagents (Scheme 63, conditions a and b).
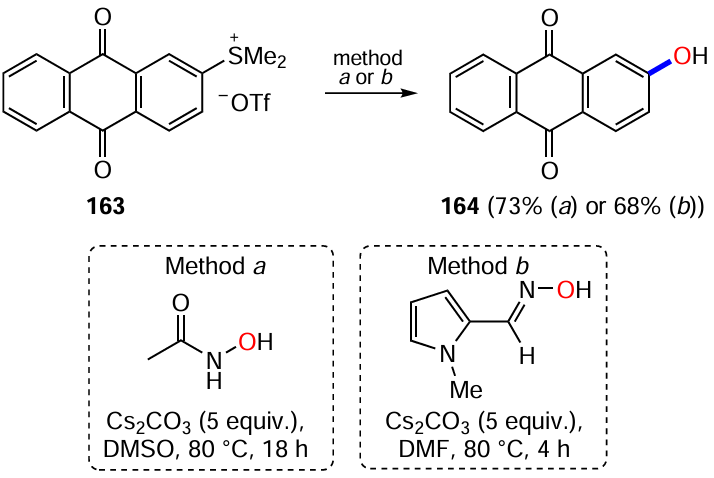
Diazotization of aminoanthraquinones followed by substitution of the diazo group with O-nucleophiles is used for the synthesis of both α- and β-hydroxyanthraquinones.[142, 144, 145] Varying the conditions of diazotization of 1-aminoanthraquinone (48) and the introduction of a hydroxyl group allows the preparation of 1-hydroxyanthraquinone (13) with different efficiencies (Scheme 64).[144, 145]
The scope of this method was also demonstrated for 2-aminoanthraquinone (50), where treatment with NaNO2 followed by refluxing in water gave 2-hydroxyanthraquinone (164) in high yield (Scheme 65).[142]
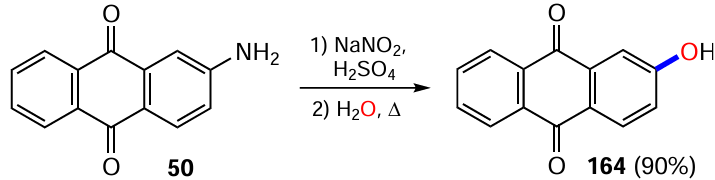
Arylboronic acid esters, which often act as masked analogues of hydroxy derivatives, can be derived from the appropriate aryl halides. Following this methodology, Yamamoto et al.[80] obtained β-hydroxyanthraquinone (164) by oxidative cleavage of the ester functionality in 2-(5,5-dimethyl-1,3,2-dioxaborinan-2-yl)anthracene-9,10-dione (43) to the phenol one (Scheme 66). Such esters are accessible via the metal-catalyzed borylation of halogen- or TfO-substituted anthraquinones with diboronic acid derivatives such as bis(pinacolato)diboron (B2Pin2) or by the assembly of an anthraquinone core from fragments containing a boronic acid ether residue (see Section 7.1 below for details).
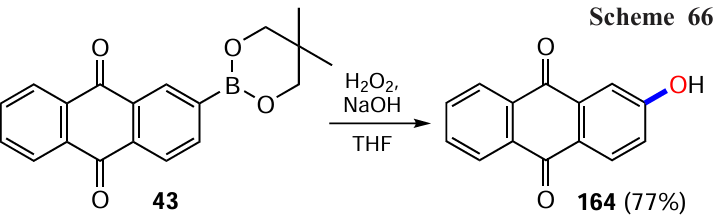
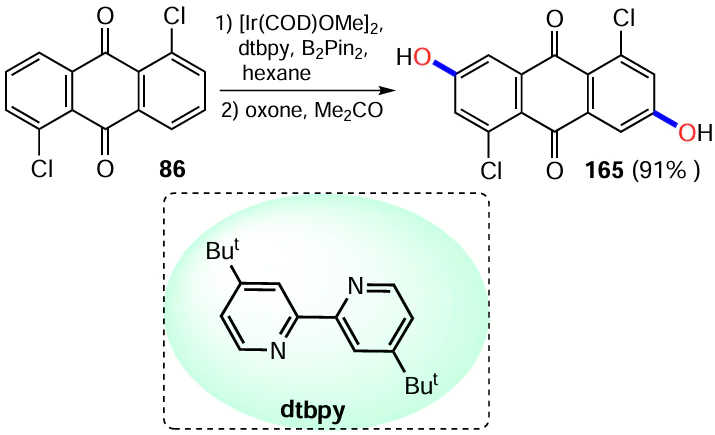
Li et al.[67] used oxone to oxidize the boronic ether to the hydroxyl group in the anthraquinone derivative, allowing the synthesis of dihydroxyanthraquinone 165 in high yield (Scheme 67). Interestingly, the intermediate 1,5-bis(boronato)anthraquinone was obtained without isolation and purification from 1,5-dichloranthraquinone (86) by direct C – H activation of the β-position of the anthraquinone nucleus with an iridium complex containing 4,4'-di-tert-butyl-2,2-dipyridyl (dtbpy) as the ligand.
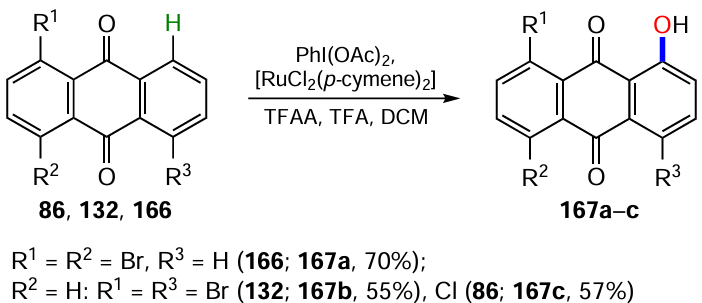
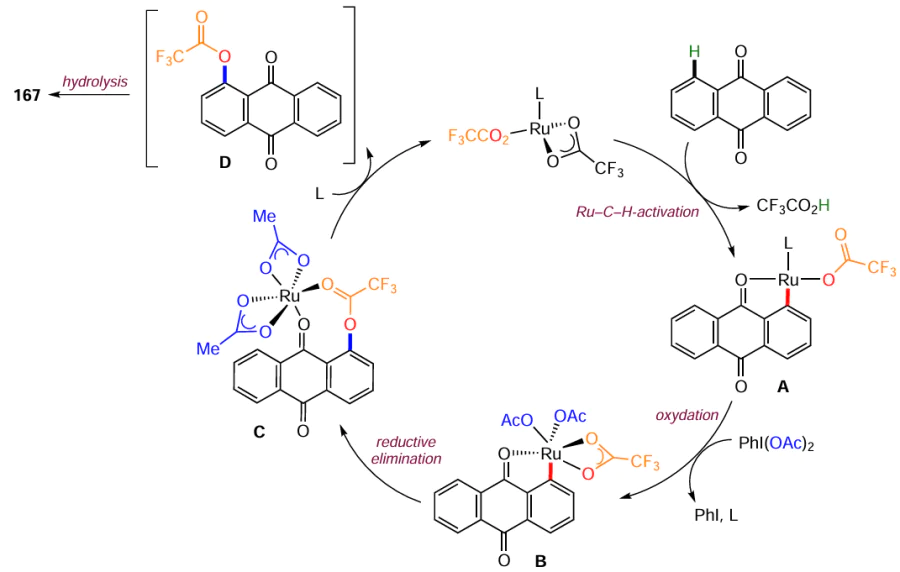
The direct C – H hydroxylation of aromatic compounds has undeniable advantages over classical functional group conversion reactions because it reduces the number of synthesis steps, allows modification in the final steps, has high atomic efficiency and complies with the principles of green chemistry. However, the methods of C – H hydroxylation of anthraquinone, as well as other aromatic compounds, have been investigated to a much lesser extent compared to the methodology of nucleophilic substitution. Dias et al.[146] developed a method for the regioselective introduction of a hydroxy group at the α-position of naphtho- and anthraquinones. Using 1,4-dibromo- (166), 1,5-dibromo- (132) and 1,5-dichloroanthraquinones (86) as examples, it has been shown that the reaction proceeds with the preferential formation of 1-hydroxyanthraquinones 167a – c (Scheme 68), without affecting the halogen atoms. The hydroxylation proceeds under the action of an oxidant, (diacetoxyiodo)benzene (PIDA), in a mixture of trifluoroacetic anhydride (TFAA), trifluoroacetic acid (TFA) and DCM, and a catalyst based on a ruthenium(II) complex, which gives the α-hydroxylation product with high regioselectivity due to coordination with the carbonyl group.
The putative mechanism for the model substrate is shown in Scheme 69 and involves the classic steps of metal-catalyzed cross-coupling.[146] The PhI(OAc)2 reagent acts as an oxidant of ruthenium(II) in complex A to ruthenium(IV), which affords complex B. Intermediate B then undergoes reductive elimination, forming a C – O bond in the anthraquinone to give intermediate C, from which the Ru complex and 1-trifluoroacetoxyanthraquinone D are released. Hydrolysis of intermediate D gives 1-hydroxyanthraquinone. The solvent mixture, TFAA and TFA, was found to be critical for the most efficient chelation of the C – O bonds in the aromatic core.
The alkoxylation of anthraquinone is usually carried out using methods developed for the introduction of hydroxy groups. The substitution of functional groups such as sulfonic-, nitro-, halogen- and even hydrogen atoms for alkoxy- and aryloxy groups under various conditions has been performed and studied for a long time.[20] Nevertheless, this method is still relevant for the synthesis of anthraquinones with valuable properties. In the search for photocontrolled ionophores, Mart’yanov et al.[147] obtained α-phenoxyanthraquinone derivatives 168, 169 from dichloroanthraquinones 86 and 88 (Scheme 70, Table 13) by substitution of chlorine atoms with phenoxide ions in the presence of copper powder. However, the results do not allow us to draw a definite conclusion on the necessity of using catalysis: the yield of the reaction product is comparable to or does not exceed that of a similar non-catalytic substitution.
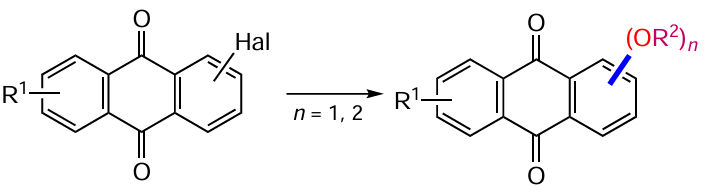
The substitution in 1,8-dichloroanthracene-9,10-dione (88) could be carried out in the absence of catalysis even with weak nucleophiles such as 3- and 4-hydroxybenzoic acids to afford the phenoxy-anthraquinone derivatives 170a,b.[148] The non-catalytic reaction of 1,2,3,4-tetrachloroanthraquinone (171) with sodium phenoxide at room temperature gave the monosubstitution product 172 in good yield.[149] Kim and Chae [150] showed that the substitution of the β-halogen atom of anthraquinone 173 with an O-nucleophile (see Table 13) proceeds quite efficiently without additional catalysis even in the presence of electron-donating groups that deactivate the molecule for aromatic substitution, and the products 174 are formed in moderate to high yields.
An intramolecular Pd-catalyzed Buchwald – Hartwig O-arylation has been implemented for the synthesis of 2-substituted 2,3-dihydroanthra[2,3-b]furan-5,10-diones 176. The potential of this method has been illustrated by the example of several anthraquinone derivatives 175: their treatment with Pd(OAc)2 in the presence of a phosphorus-containing ligand is accompanied by the substitution of a bromine atom with the formation of the C – O bond and a five-membered heterocycle in the product 176 (Scheme 71).[123]
The electron acceptor properties and the structure of the anthraquinone core determine in some cases the direct substitution of hydrogen by various nucleophiles, including alkoxide ions. In the synthesis of marmycin A, it was found that treatment of the anthraquinone derivative 177 with sodium hydride at low temperature leads to intramolecular hydrogen substitution by an alkoxy nucleophile to afford the cyclization product 178 (Scheme 72).[37]
5. Reactions with the formation of the carbon – sulfur bond
As with most aromatic compounds, the sulfonation of anthraquinones is one of the most common ways of constructing the C – S bond. The introduction of a sulfonic group into an anthraquinone nucleus is carried out by different methods depending on the structure of the starting compound. Sulfonation of 1-aminoanthraquinone (48) with chlorosulfonic acid affords the sodium salt of 1-aminoanthraquinone-2-sulfonic acid (179) (Scheme 73, Table 14).[151] The reaction of 1,4-diaminoanthraquinone (180) under similar conditions gives the product 181, but in much lower yield.[151] In the presence of an electron-withdrawing substituent in the anthraquinone core, the introduction of the sulfonic group can be achieved by replacing the halogen atom with sodium dithionite, as shown for the conversion of compound 182 to the anthraquinone-2,4-disulfonic acid salt 183.[151]
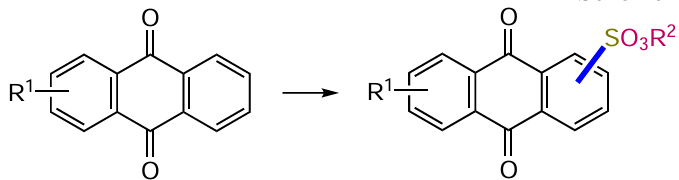
Direct electrophilic sulfonation of anthraquinone-2-carboxylic acid (184) with fuming sulfuric acid under microwave irradiation proceeds in the unsubstituted ring, yielding a mixture of 2,6- and 2,7-disulfoanthraquinone-2-carboxylic acids 185a and 185b in a 1 : 1 ratio in high yield (see Table 14).[152]
As mentioned above, the transformation of the diazonium group plays an important role in the functionalization of the anthraquinone core, as confirmed by many examples, including the formation of C – S bonds.[153] Diazotization of β-aminoanthraquinone (50) with sodium nitrite and subsequent chlorosulfonylation of the diazonium salt with SO2 and CuCl2 afforded anthracenedione-2-sulfonyl chloride 186 with high efficiency (Scheme 74).
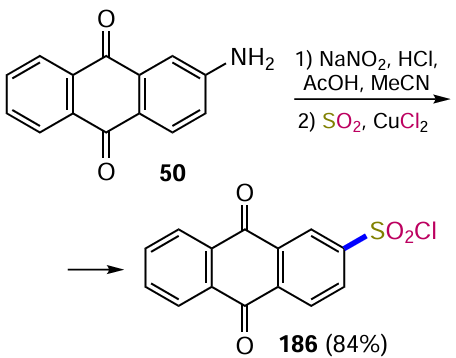
Zvarych et al.[154] proposed a method for the synthesis of anthraquinone mono- and dithiocarbamates based on the reaction of diazonium salts with in situ generated amides of dithiocarbaminic acid. The diazonium salt derived from aminoanthracene-9,10-diones was treated with dithiocarbamide in aqueous medium without the addition of a catalyst. This method gave rise to a library of α- and β-dithiocarbamate-substituted anthraquinones (e.g. compounds 187 and 188). Similarly, treatment of the diazonium salt with potassium rhodanide followed by reduction afforded 1-sulfanylanthracene-9,10-dione 189 (Scheme 75, Table 15).[155]
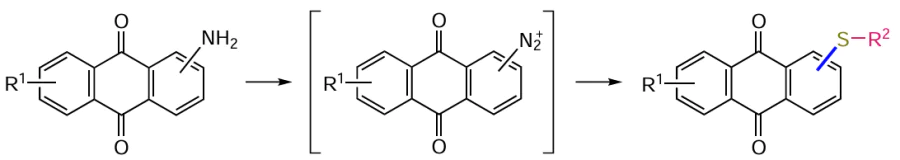
The electron-withdrawing quinone moiety in anthracene-9,10-diones activates halogen atoms and affects some other functional groups, making them prone to substitution by strong nucleophiles. To study the properties of analogues of the anticancer drug mitoxantrone, Huang et al.[156] synthesized a series of symmetrical 1,5-dithioanthraquinones 190 by refluxing 1,5-dichloranthraquinone (86) with thiols in the presence of sodium methoxide (Scheme 76, Table 16). Liu et al.[157] carried out SNAr substitution of the halogen atom in arenes containing electron-withdrawing groups by thiophenols, noting the efficiency of N,N-dimethylacetamide as solvent and K2CO3 as base. For example, the anthraquinonyl thioether 191 was obtained from 2-bromoanthraquinone (39) in quantitative yield. It is known that 1,4-dihydroxyanthracene-9,10-diones exist in equilibrium with the tautomeric form of 1,4-diquinone, thereby activating halogens in such compounds for nucleophilic substitution. Treatment of 2-bromoquinizarine (192) with sodium sulfide and subsequent alkylation of the intermediates gives β-sulfanyl-substituted anthraquinones 193a,b (see Table 16).[158]
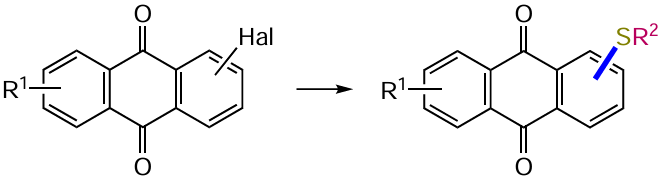
Copper-catalyzed thiolation of aryliodides have been carried out to obtain sulfur-containing naphtho- and anthraquinones.[159] The best results were obtained using copper(I) thiophene-2-carboxylate (Tc) as a catalyst. The example in Scheme 77 shows the conditions for the formation of the sulfanyl derivatives 194a and 194b from 1-iodoanthraquinone (56) and silver benzylthiolate or trifluoromethanethiolate. The authors note that CuTc provides internal coordination of the sulfur atom to the metal, and thiophene-2-carboxylate, as part of the catalyst, is able to further stabilize the structure of complex A (Scheme 78).
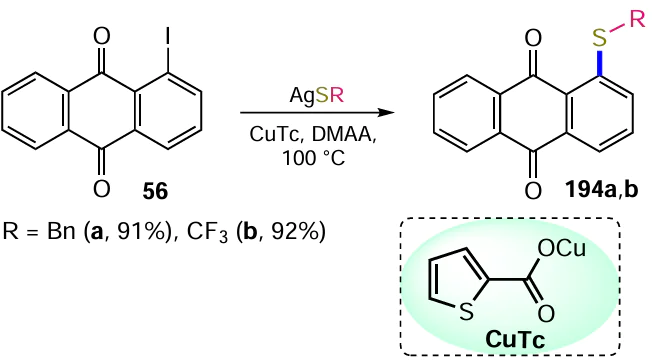
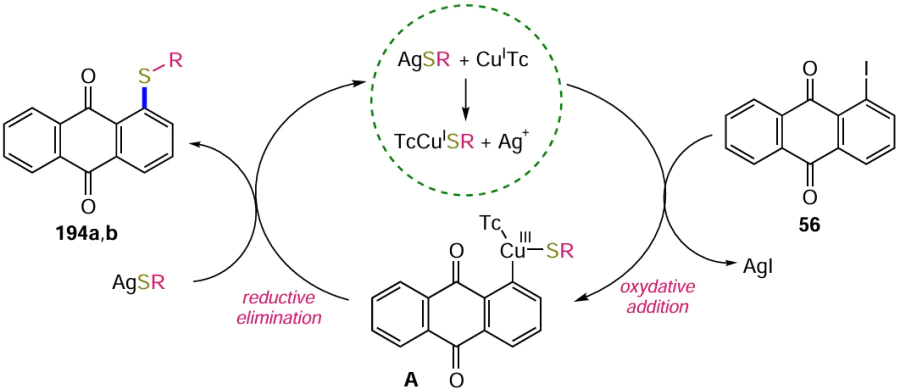
The reaction pathway involves the initial formation of a complex between the organosulfur anion and copper(I) thiophene-2-carboxylate (see Scheme 78), leading to the formation of the TcCuISR active species. It is then oxidatively bound to the C – I bond. The resulting trivalent copper-based complex A undergoes rapid reductive elimination to give thiolation products 194a or 194b.
Qiao et al.[160] developed a unique procedure for Pd-catalyzed cross-coupling of aryl and alkyl halides or triflates using convenient and environmentally friendly sodium thiosulfate as the sulfur source to yield aryl thioethers. The results of this study are remarkable in that the C−S bond is formed without the use of thiols or thiophenols. The versatility of the method is demonstrated by the reaction of 2-(trifluoromethanesulfonyloxy)anthraquinone (98) with Na2S2O3 and 1-chloro-2-ethoxyethane using PdCl2(dppf) catalysis to give the thioanthracene-9,10-dione 195 (Scheme 79).
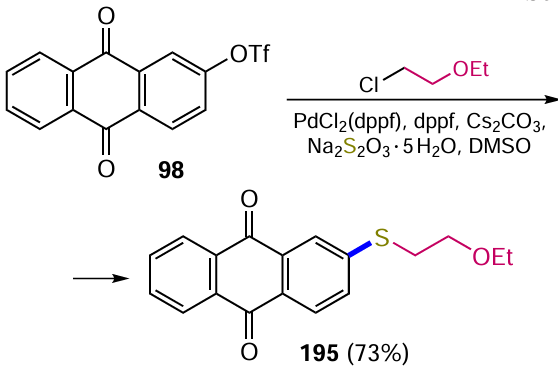
The cross-coupling reaction begins with the oxidative addition of substrate 98 to Pd0 to afford intermediate A (Scheme 80).[160] The alkylation of the sulfur atom is thought to occur even before the CArS bond is formed by the reaction of 1-chloro-2-ethoxyethane with thiosulfate to give sodium S-alkylthiosulfate B. Subsequent substitution of the OTf moiety with an S-containing ligand yields species C, which is further converted to complex D via the SO3 elimination. In this process, the electron-withdrawing effect of SO3 weakens the coordinating properties of the sulfur atom towards palladium. The reductive cleavage of PdII is accelerated by the sulfur oxide released from intermediate C. In the final step, complex D is converted to thioether 195 with regeneration of Pd0.
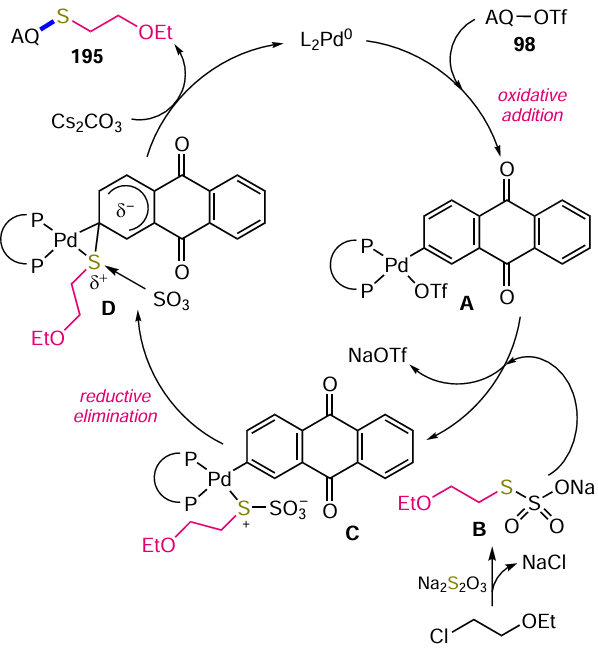
The substitution of the unactivated halogen atom in the β-position of anthraquinone by a nucleophilic reagent can be boosted by catalysis with copper salts. The addition of copper iodide allows the substitution of the bromine atom in 2-bromo-6,7-dihydroxyanthraquinone (196) with aromatic thiophenols to give thioanthraquinones 197a,b in moderate yields (Scheme 81).[161]
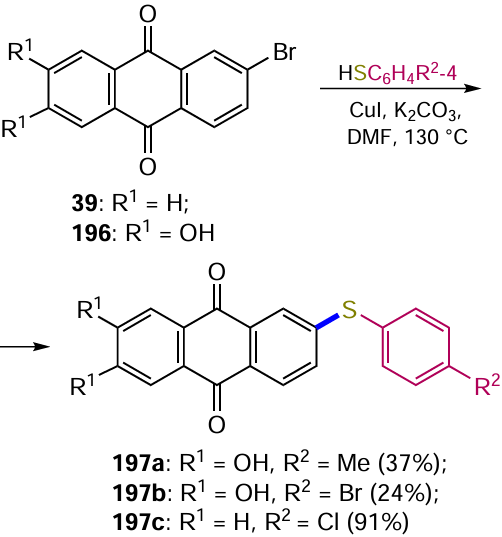
In the absence of deactivating groups in the anthraquinone, thiolation reactions are much easier. The reaction of 2-bromoanthraquinone (39) with 4-chlorothiophenol delivers 2-(4-chlorophenylthio)anthracene-9,10-dione (197c) in high yield.[162] Interestingly, the need for copper catalysis is not proved by the authors, as a number of publications [156-158] reported high yields of products without the addition of any catalyst (see, e.g., the synthesis of compounds 190, 191, 193a,b in Scheme 76).
It was possible to introduce the sulfide group into the aromatic core by Pd-catalyzed thiomethylation of various aryl chlorides with a mixture of potassium dimethyl carbonate and potassium thioacetate.[163] It was observed that thiolation with methanethiol is much more difficult than using a masked thiolating agent (KSAc) followed by alkylation of the sulfanyl group in the arenethiol. The use of dimethyl carbonate as a methylation reagent allows the negative effect of sulfur on the catalyst to be reduced. The efficiency of this method is demonstrated on the example of aryl chlorides containing both electron-donating and electron-withdrawing substituents. In particular, 2-chloroanthraquinone (198) is converted to 2-(methylthio)anthracene-9,10-dione (199) in the presence of Pd(OAc)2 catalysis in almost quantitative yield (Scheme 82).
The annulation of heterocycles to anthraquinone also activates halogen atoms for nucleophilic substitution. In two independent studies,[110, 164] substitution of chlorine and bromine atoms in 4-haloantra[1,2-c][1,2,5]thiadiazole-6,11-diones 200a – c (Scheme 83) with various S-nucleophiles was carried out to obtain antitumour compounds, in particular topoisomerase 1 inhibitors.
The possibility of substitution of both halogen and hydrogen atoms in position 4 of azoloanthraquinones by nucleophiles was first demonstrated by M.V.Gorelik,[20] but the current research has considerably extended the potential of this method. It was shown that the thiol structure mainly determines the reactivity and yield of the products 201 (see Scheme 83). For the oxidative nucleophilic substitution of the hydrogen in position 4 of anthra[1,2-c][1,2,5]-thiadiazole-6,11-dione (200c) by various thiols, iron(III) chloride addition was used.[164]
6. Reactions with the formation of the carbon – nitrogen bond
Anthraquinone derivatives bearing nitrogen-containing substituents are of particular applied value, especially for the development of antitumour drugs and dyes. The most common method of N-functionalization of the anthraquinone core is nitration.[20] Depending on the conditions and structure of the starting compound, electrophilic substitution affords mono-, di- and polynitro-substituted anthraquinones.[20] In some cases, due to the availability of some amino anthraquinones, it is possible to oxidize the amino group to the nitro group with trifluoroacetic anhydride in 30% H2O2 .[165]
Nitroanthraquinones can be converted into the corresponding amino derivatives when treated with reducing agents (Fe – AcOH, Zn – HCl, Na2S2O4 – NaHCO3 , H2 – Pd/С, etc.).[166] These processes were well studied in the 60 – 80s of the XX century and systematized in detail in the monograph by M.V.Gorelik.[20] A review of current literature shows that no significant discoveries have been made in the methods of nitration of anthraquinones and reduction of nitro derivatives; therefore, these reactions are not considered in this review.
Another common way to introduce an amino group into the anthraquinone core is by aromatic nucleophilic substitution of functional groups, namely nitro, halogen, alkoxy-, sulfonic-, hydroxy groups and even hydrogen atoms. Reactions of this type are fundamental and are often used for anthraquinone modification. The reactivity of the leaving groups in the α- and β-positions of anthracene-9,10-dione depends on many factors, including the reaction conditions, the presence and location of other substituents in the anthraquinone, and the nucleophilicity of the reagent. Although these processes were also discovered in the last century, new aspects of the reactivity of anthraquinones and the possibility of their N-functionalization are still relevant.
Among the halogens in the anthraquinone nucleus, the fluorine atom is the most mobile in nucleophilic non-catalytic substitution reactions.[20] In the synthesis of cyclodextrin-modified anthraquinones, the selective substitution of a fluorine atom in 1,4-difluoroanthraquinone (156) with propargylamine was carried out (Scheme 84, Table 17).[167] Lehmler et al.[168] synthesized mono- and dialkylaminoanthraquinones 202 by prolonged heating of 1,4-difluoroanthraquinone (156) with alkyl and fluoroalkyl amines in DMSO, varying the reaction time (see Table 17). The low yield of products 202 may be due to the introduction of electron-withdrawing substituents into the nucleophile molecule (e.g. for fluoroalkylamines), which affect their reactivity and lead to a decrease in the selectivity of the process, as diaminoalkyl anthraquinones are additionally formed. It should be noted that the chlorine atom in anthraquinones is also easily substituted by piperidine in toluene at 80°С.[169]
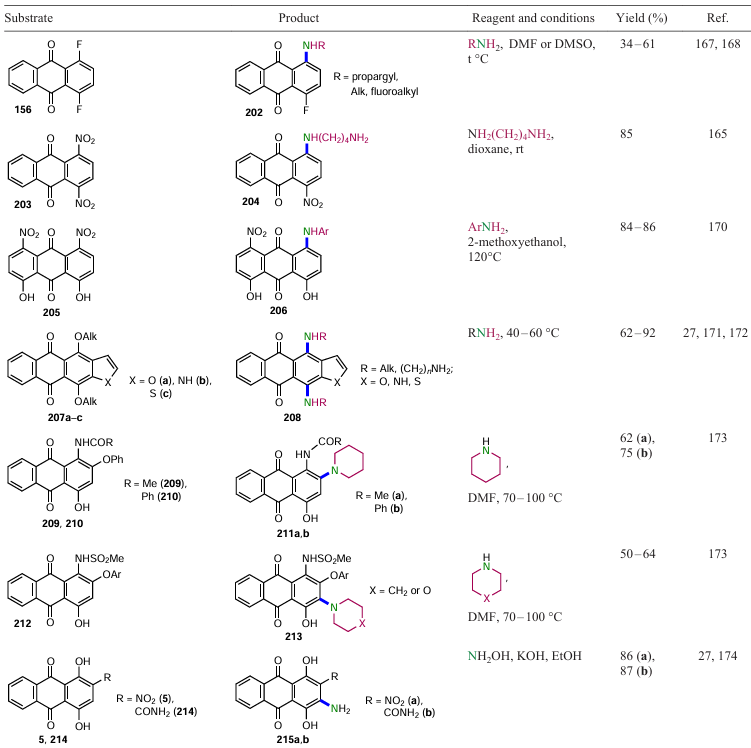
1,4-Dinitroanthraquinone (203) reacts with 1,4-diaminobutane at room temperature to give nitroanthraquinone 204,[165] while under more harsh conditions (DMSO, 100 °C) the second nitro group is also substituted.[165] Interestingly, the nitro group in the α-position of anthraquinone can be substituted, even in the presence of deactivating substituents, with arylamines containing electron-donating groups. Treatment of 1,8-dihydroxy-4,5-dinitroanthracene-9,10-dione (205) with an excess of aniline derivative on heating in methyl cellosolve gives mono-N-aryl-substituted anthraquinones 206, but the prolonged reaction time does not lead to substitution of the second nitro group (see Table 17).[170]
The annulation of heterocycles with anthraquinone activates substituents in the peri-positions to such an extent that nucleophilic substitution of even bad leaving groups such as alkoxy groups is possible under mild conditions. For example, when 4,11-dimethoxyanthra[2,3-b]furan-5,10-dione (207a) is reacted with diaminoalkanes at 50 °С in THF, the corresponding 4,11-diamino derivatives 208 are formed (see Table 17).[171] Other heterocycles, such as the thiophene- and pyrrole-fused anthraquinones 207b,c, feature similar reactivity and product yields.[172] This method has proved versatile for introducing aminoalkyl moieties into the peri-positions of heteroarene anthraquinones obtained in the development of new anticancer agents.[27]
Nucleophilic substitution can also be achieved for the leaving groups at the β-position of the anthraquinone. The amination of 2-aryloxy-1-acylamino-4-hydroxyanthraquinones 209 or their benzoyl analogues 210 with piperidine in DMF was found to give substitution products of the aryloxy groups 211a,b.[173] It is noteworthy that the replacement of the acylamino group in position 1 of compound 209 by a methanesulfonylamino group (derivative 212) under the same conditions leads exclusively to hydrogen substitution products 213 (see Table 17). The authors explain this fact by the steric hindrance to nucleophilic attack at position 2 for the derivatives of 1-methanesulfonylaminoanthraquinone.
Direct nucleophilic substitution of hydrogen is an attractive method from a practical point of view because it does not require the introduction of additional functional groups into the molecule. In the case of anthraquinone derivatives, this reaction has been used mainly for the synthesis of hydroxy and amino derivatives. For example, the reaction of 2-nitroquinizarin (5) and quinizarine-2-carboxylic acid amide (214) with hydroxylamine to afford the amino derivatives of anthraquinone 215a,b can be mentioned (see Table 17).[27, 174] It has been observed that the introduction of electron-withdrawing groups significantly activates the anthraquinone nucleus for vicarious nucleophilic substitution of hydrogen upon hydroxylamine action.[27]
The search for efficient methods for the arylamination of anthraquinones led to the development of inexpensive and environmentally friendly catalysts.[175] Thus, choline hydroxide promoted nucleophilic aromatic substitution of halogen in the α-position relative to carbonyl groups was carried out on derivatives 216. This reaction with substituted anilines was the basis for the synthesis of a series of N-arylanthraquinones 217 (Scheme 85). In the presence of other bases such as NaOH, KOH, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), Py, K2CO3 , etc., the products of substitution of the bromine atom by aniline are formed in much lower yields. With anthraquinone bearing two bromine atoms in the α- and β-positions, the reaction proceeds with high α-regioselectivity.
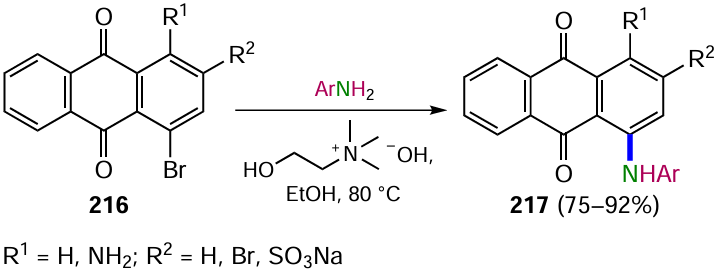
Increased efficiency of hydrogen substitution in aromatic compounds, including anthraquinones, can be achieved by using oxidative nucleophilic substitution. Teich et al.[176] obtained 4-amino derivatives of emodin 218b,c, 219a,b by selecting a hypervalent iodine compound (PIDA) as the oxidant (Scheme 86, Table 18). In this study, the reaction of emodin (7) in boiling piperidine leads to 4-amino- and 4-[N-(2-hydroxy-ethyl)amino]emodin as the main products. The authors also report that various cyclic derivatives of the 3-hydroxyl group of emodin (7) are formed at room temperature, which is apparently due to the low yields of products 218b and 219a,b.[176] Notably, Chalothorn et al.[103] carried out amination of emodin (7) with ethylenediamine and ethylamine under similar conditions, and product 218b was isolated in higher yields.
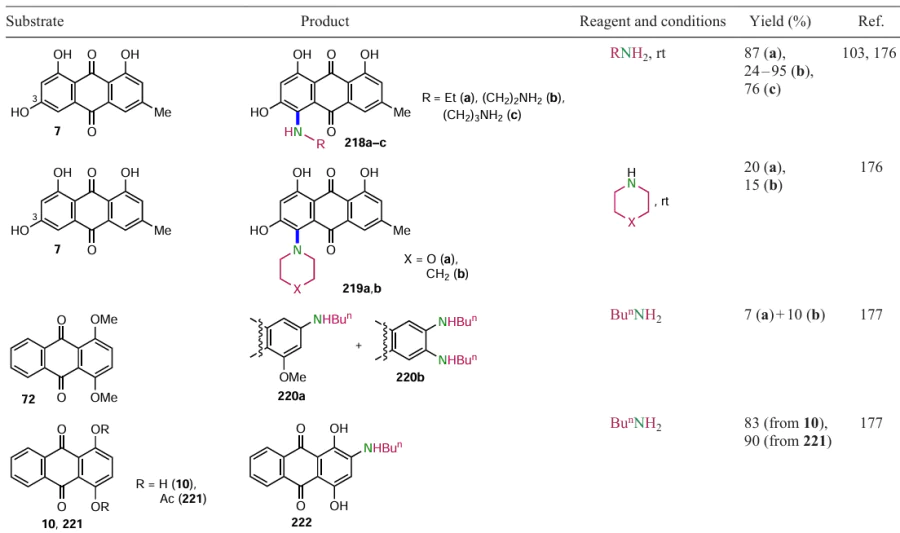
The study of the reaction of butylamine with 1,4-dihydroxy- (10), 1,4-diacetoxy- (221) and 1,4-dimethoxyanthraquinones (72) showed that the presence of PhI(OAc)2 as catalyst improves the yield of amination products by about 10 – 20%.[177] However, the reaction of 1,4-dimethoxyanthraquinone (72) with n-butylamine in the presence of PhI(OAc)2 is characterized by low substrate conversion, accompanied by demethoxylation, and results in a mixture of products 220a,b in low yield. Amination of 1,4-diacetoxyanthraquinone (221) is followed by deacetylation and gives quinizarine 222 (see Table 18).
Teich et al.[176] suggest that the initial step in amination is the addition of (diacetoxyiodo)benzene to the 3-hydroxy group of emodin (7), followed by attack of the amine at the α-position, which causes dearomatization (Scheme 87). After elimination of iodobenzene and acetic acid, complex B is rearomatized to the 4-amino derivative of emodin 219. The mechanism of reaction of quinizarin (10) with amines is similar to that of emodin (7) and is presented in another study,[177] which also investigated the dimethoxylation pathway of 1,4-dimethoxyanthraquinone (72) in reaction with n-butylamine in the presence of PIDA.
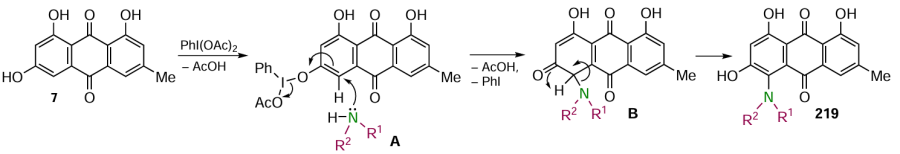
A series of diaminoanthraquinone derivatives 223a – c were synthesized by Pd-catalyzed amination of chloroanthraquinones (Buchwald – Hartwig reaction) in the works of academician I.P.Beletskaya and co-workers.[178] Cross-coupling of 1,8-dichloranthraquinone (88) with aromatic and aliphatic amines under Pd(dba)2 catalysis afforded 1,8-diaminoanthraquinones 223a – c (Scheme 88). BINAP was found to be the most effective ligand for the synthesis of most of these derivatives.[179] Although the authors were able to use sterically hindered amines such as morpholine and piperidine in the reaction, it was not possible to substitute the chlorine atom with acyclic secondary amines (N-methylaniline, diethylamine).[178] It is worth noting that Pd-catalyzed amination can be used to successfully perform deamination of not only 1,8- but also 1,5-dichloroanthraquinone, and the presence of chiral ligands leads to enantiomerically enriched planar chiral macrocycles.
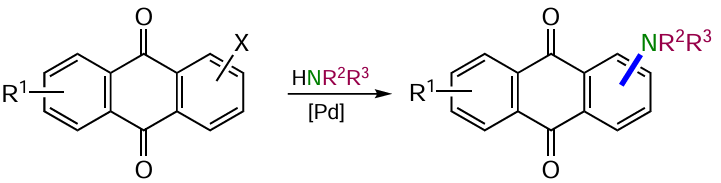
The catalytic system Pd2(dba)3 – BINAP – Cs2CO3 in toluene has been used for the arylation of 3-aminocarbazole derivatives with chloroanthraquinones to give aminoanthraquinones 225 in 42 – 80% yield. An example, the reaction of 1,4-dichloroanthraquinone (224) with 3-amino-N-2-hexylcarbazole, is given in Table 19.[180] Despite the obvious steric hindrance at the α-position of anthraquinone, another group of researchers [181] adapted this method for the use of amines with bulky substituents. For example, 1,5-dichloroanthraquinone (86) reacts with 3,6-dimethoxy-9H-carbazole in the presence of Pd2(dba)3 and the dppf ligand to give the product 226 of arylation at the nitrogen atom of the carbazole heterocycle.[181]
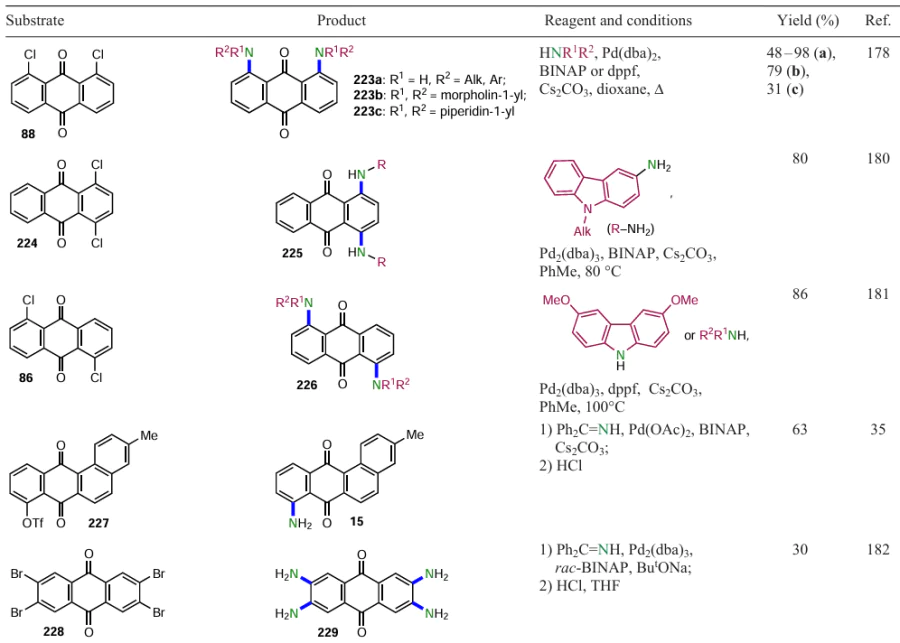
The use of imines for Pd-catalyzed cross-coupling allowed the introduction of an unsubstituted amino group it the α- and β-positions of the anthraquinone core. The reaction of 3-methyl-7,12-dioxo-8-trifluoromethanesulfonyloxytetraphene (227) with benzophenone imine using Pd(OAc)2 catalysis and BINAP ligand and subsequent acid hydrolysis gave 8-amino-3-methylbenz[a]anthraquinone (15) (see Table 19).[35] Xia et al.[182] synthesized 2,3,6,7-tetraaminoanthraquinone (229) in 30% yield via a two-step procedure using the complex catalytic system Pd2(dba)3 – rac-BINAP – ButONa.3
A nickel-catalyzed amination of phenols using 2,4,6-trichloro-1,3,5-triazine (TCT) has been developed to introduce an amino group into the aromatic core.[183] The said reagent activates the C – O bond by forming an intermediate ether containing a triazine moiety which is attacked by the amine in the presence of a catalytic nickel complex. The method was adapted for the preparation of 2-aminoanthraquinone (50) from 2-hydroxyanthraquinone (164), using ammonium carbonate as the source of the amino group (Scheme 89).
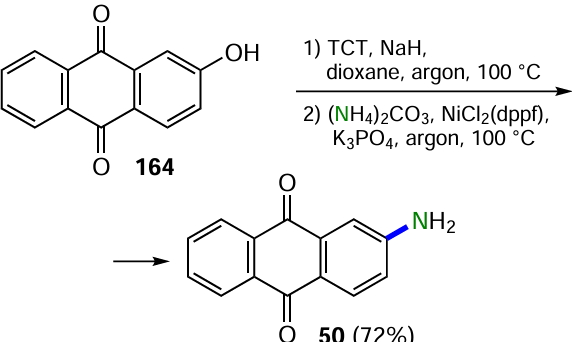
Harmata and Hong [184] proposed a method of Pd-catalyzed N-arylation of (R)-sulfoximine with chloroarenes for the synthesis of sulfonimidoyl arenes. The reaction of 1,5-dichloroanthraquinone (86) with (R)-sulfoxime catalyzed by the Pd(OAc)2–rac-BINAP – Cs2CO3 system under microwave irradiation afforded compound 230, which is the product of the substitution of a chlorine atom (Scheme 90). The detailed mechanism of the reaction is not provided in the study.
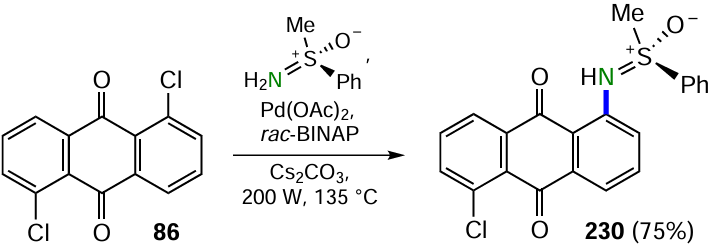
Along with the modification of the α-position, the Buchwald – Hartwig cross-coupling is also used to introduce aryl-, diaryl- and heterocyclic amines into the β-position of anthraquinone. Thus, the reaction between 2,6-dibromoanthraquinone (159) with sterically hindered diarylamines or hetarylamines is efficiently realized in the presence of the catalytic system Pd2(dba)3 – SPhos and ButONa as base (Scheme 91).[185] The use of a palladium N-heterocyclic carbene complex (PEPPSI-IPr) as a catalyst gives anthraquinone derivatives of similar structure in similar yields.[186] The resulting compounds 231a-f, bearing diaryl and heterocyclic amines as electron-donating groups and a π-deficient anthracene-9,10-dione nucleus as acceptor, are potentially useful as materials for optoelectronic devices.[185]
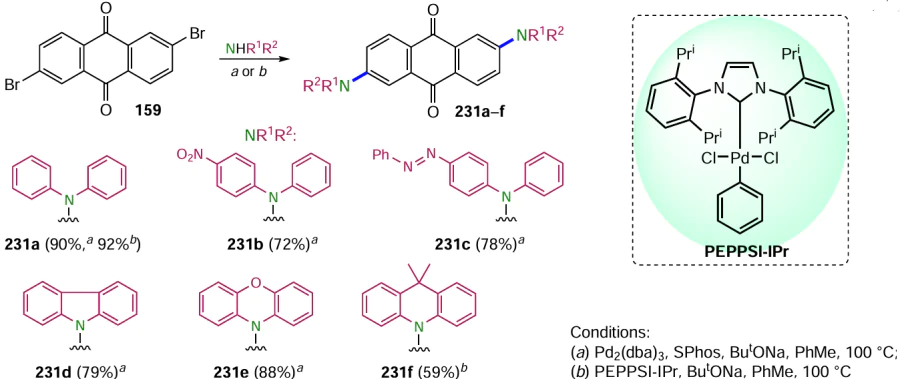
The Ullmann reaction is an effective method for the arylation of phenols, anilines, thiophenols, etc.[187] However, in the case of anthraquinones, the classical conditions for this transformation require the use of activated amines, harsh conditions and prolonged heating, which significantly reduces the yield of products. In this context, many studies have attempted to optimize the conditions for the preparation of amino derivatives of anthraquinone by this method. Some modifications of the Ullmann reaction have formed the basis for the synthesis of a large library of 4-N-substituted 1-amino-2-sulfoanthraquinones, which are of interest for the development of new dyes,[188, 189] purinergic P2 receptor antagonists and ectonucleotidase inhibitors.[190, 191]
For example, Glänzel et al.[151] treated the sodium salt of 1-amino-4-bromoanthracene-2-sulfonic acid 182 with anilines in the presence of the reducing agent Na2SO3 and obtained derivatives 232 (Scheme 92, Table 20). Later, Weyler et al.[192] used CuSO4 as catalyst, but the reaction time was 2 days. Although the yield of the target products is in a wide range and depends on many factors, a number of studies have shown that the use of copper powder and the addition of sodium phosphate buffer under microwave irradiation has a positive effect on product yield and reaction rate, and also allows the introduction of various aniline moieties into the molecule.[189-194]
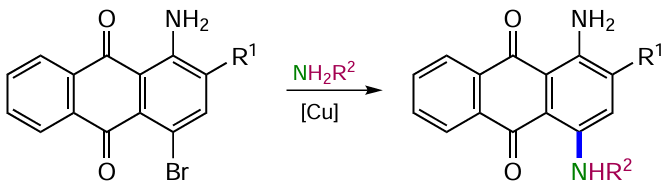
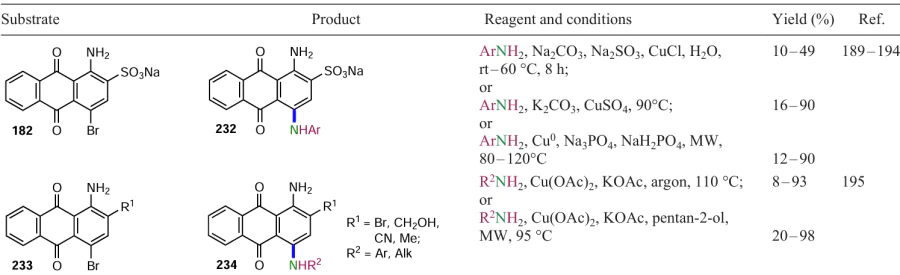
Malik et al.[195] found that for 1-aminoanthraquinone derivatives 233 (see Table 20) bearing substituents such as Br atom or CH2OH, CN and Me groups in position 2, the Ullmann reaction proceeds efficiently at the 4-bromine atom. The process is carried out in a liquid amine medium catalyzed by Cu(OAc)2 in the presence of KOAc in an inert atmosphere to obtain 4-aminoanthraquinones 234. The amination of anthraquinones with anilines containing electron-withdrawing substituents in the aromatic core was performed in the presence of Cu(II) salt under microwave irradiation.[195]
Catalytic substitution of chlorine atoms in 1,8-dichloroanthracene-9,10-dione (88) by p-toluenesulfonamide followed by treatment with H2SO4 gave 1,8-diaminoanthraquinone (235) (Scheme 93, conditions a).[196] In other work,[197] the anthraquinone core served as a ligand for a ruthenium-based complex catalyst. One of the steps in the synthesis of such a complex was the preparation of 1,8-diamino-anthraquinone (235) by Cu-catalyzed substitution of chlorine atoms in 1,8-dichloro-anthraquinone (88) with phthalimide and further acid hydrolysis of the intermediate 1,8-(diphthalimido)anthraquinone (see Scheme 93, conditions b).
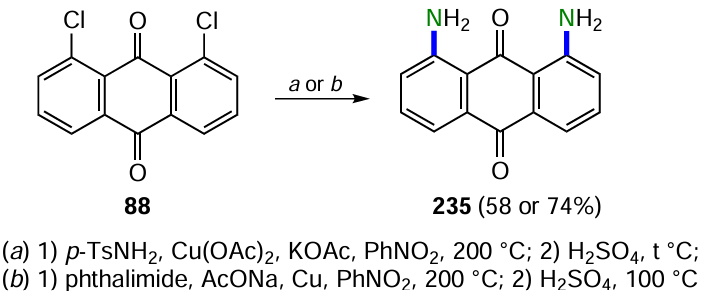
As noted above, the annulation of heterocycles to anthraquinone largely activates nucleophilic substitution reactions, including for direct substitution of hydrogen.[20] The amination of anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (200c) with aromatic and aliphatic amines is promoted by CuOAc under microwave irradiation to give anthrathiadiazoles 236 (Scheme 94), which have been investigated as potential topoisomerase 1 inhibitors.[110]
Despite the obvious advantages of direct substitution of hydrogen, in some cases the a chlorine atom was preliminary introduced in anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (200c) and subsequently substituted with an N-nucleophile.[198] Unfortunately, the authors did not correlate the yields of the products with the method of synthesis, so it is not possible to compare the efficiency of substitution of hydrogen and chlorine atom, which only indirectly indicates the limitations of the SNAr – H reaction.
Khoumeri et al.[107] developed an efficient method for the intramolecular cyclization of anthraquinone benzenesulfonamides 237 to 2-substituted 1-(benzenesulfonyl)-1H-naphtho[2,3-f ]indoline-5,10-diones 238 in high yields (Scheme 95). Optimization of the conditions showed that the best yield of the model compound (Ar = Ph, 84%) was reached after 1.5 h with CuI catalysis in the presence of tetrakis(N,N-dimethylamino)ethylene (TDAE) ligand and K2CO3 base under microwave irradiation. The reaction carried out under heating gives the compound 238 (Ar = Ph) in 73% yield within 5 h.
An important method for the synthesis of primary amines, including aromatic amines, is the Curtius rearrangement of carboxylic acid azides.[199] Some modifications have been used to introduce an amino group in the β-position of the anthraquinone core.[136] Under classical conditions, the Curtius rearrangement of azides proceeds via the formation of isocyanates, which on hydrolysis give the corresponding amines.[136] Treatment of anthraquinone-2-carboxylic acid 239 with diphenylphosphoryl azide (DPPA) led to isolation of an intermediate azide, which, on refluxing and hydrolysis, gave β-aminoanthraquinone 135 (Scheme 96). Dende et al.[47] obtained similar β-aminoanthraquinone 241 from acid 240 without isolation of the azide and hydrolysis step, demonstrating the benefits of a practical one-pot reaction over classical conditions.
One of the most actively studied methods for the N-functionalization of aromatic hydrocarbons is the metal complex-catalyzed C – H amination. Shin et al.[200] developed a method for ruthenium-catalyzed α-C – H amination of aryl ketones with sulfonyl azides, the regioselectivity of which is ensured by coordination of the ruthenium complex to the carbonyl oxygen. Using anthraquinone (45) as an example, it was shown that its reaction with p-toluenesulfonylazide in the presence of the [Ru(p-cymene)Cl2]2 – AgSbF6 – Cu(OAc)2 catalytic system gives rise to 1-tosylaminoanthraquinone (242) (Scheme 97).
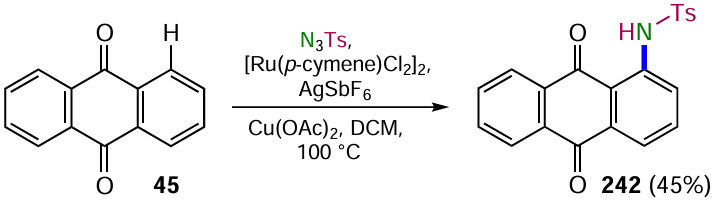
An increase in the efficiency of the direct metal-catalyzed C – H-amidation of anthraquinones 45, 72, 86, 88 and 243 was achieved by choosing the proper catalytic system. Camelio et al.[201] succeeded to obtain high yields of the product 244c and almost complete conversion of the unsubstituted anthraquinone (45) and to reduce the yield of disulfonylamide by using a complex catalytic system based on an iridium complex under optimized conditions. The reaction of the electron-deficient 1,5- and 1,8-dichloroanthraquinones 86 and 88 with p-dodecylphenylsulfonylazide afforded the products 244a,b in low yields (Scheme 98). However, the methoxyanthraquinone derivatives gave α-mono- and disulfonylamides 244e,g more readily. The authors note that hydroxy and amino anthraquinones are inert to this type of sulfamidation.
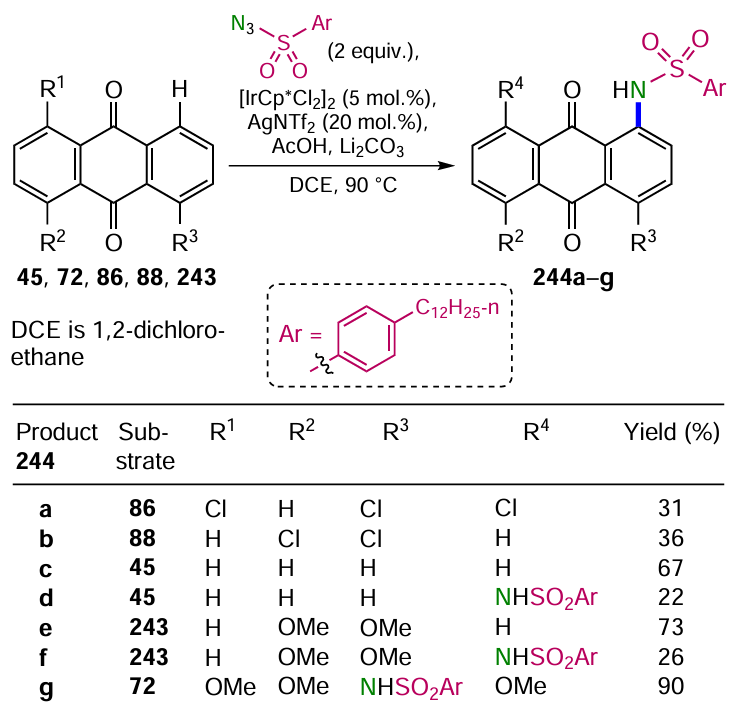
7. Reactions with the formation of bonds with other elements
7.1. Carbon – boron bond
Fragments of boronic acid or its esters that are part of organic compounds are important functional groups of interest because they solve many synthetic challenges.[202] A wide range of transformations of organoboron compounds allows the introduction of substituents and functional groups such as halogen atoms, hydroxy and alkoxy groups, nitrogen-containing fragments, etc. into various molecules.[202] One of the most important applications of boronic acid esters is in metal-catalyzed cross-coupling reactions to form C – C bonds.[202]
The most common method of introducing a boronic acid ester residue into the anthraquinone core is the Pd-catalyzed cross-coupling of haloanthraquinones with diboronic acid esters (Miyaura reaction).[50, 201] Treatment of 1,5-dichloroanthraquinone (86) with bis(pinacolato)diboron in the presence of the XPhos-Pd-G2 catalytic system in dioxane gives ester 245 in 62% yield.[201] Heating the 1,5-dibromo analogue 132 to reflux in the presence of Pd(dppf)Cl2 catalyst helps to isolate the cross-coupling product 245 in higher yield (Scheme 99, Table 21),[203] confirming the differences in reactivity of halogen atoms. The higher mobility of the bromine atom and the higher yield of products 247 in the C – B bond formation reaction are exemplified by the reaction of 2,6-dibromo- (159) and 2,6-dichloroanthraquinones (246) with B2Pin2 (see Table 21).[201, 204]
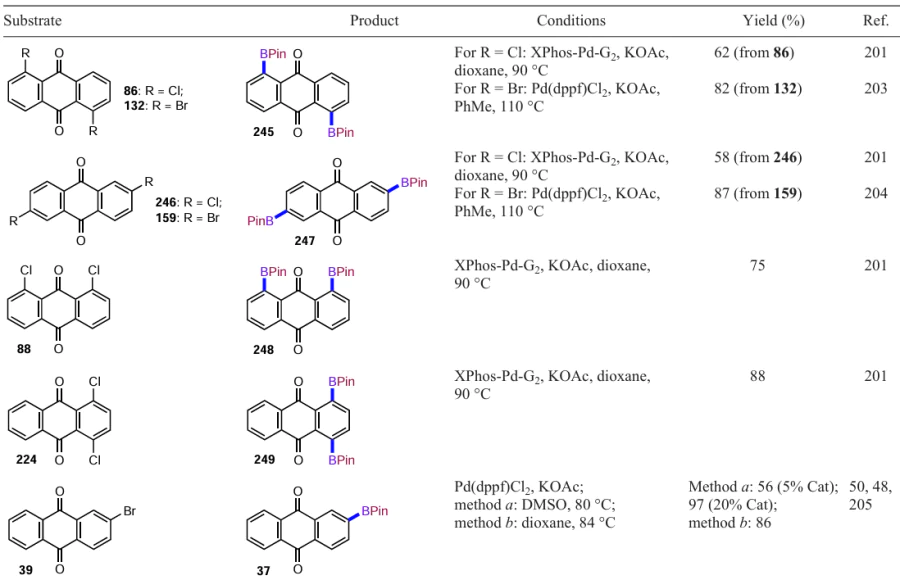
The example of 1,5- and 2,6-dihaloanthraquinone pairs also shows that the position of the leaving group has little impact on the efficiency of the cross-coupling with the diboronic acid ester. A similar procedure was used to prepare 1,8- and 1,4-dimethylanthraquinones 248 and 249 from dichloroanthraquinones 88 and 224, respectively.[201]
The synthesis of 2-(1,3,2-dioxaborolan-2-yl)anthracene-9,10-dione (37) from 2-bromoanthraquinone (39) and B2Pin2 with widely varying yields has been reported in several publication (see Table 21). The use of 5 mol.% Pd(dppf)Cl2 catalyst in DMSO gives the borylation product in 56% yield,[50] which increases to quantitative yield when the catalyst loading is 20 mol.%.[48] When the solvent is changed to dioxane and 10 mol.% catalyst is used, the yield of product 37 is 86%.[205]
Camelio et al.[201] examines the possibility of direct borylation of the C – H bond of the anthraquinone nucleus. The pinacolate ester residue of boronic acid was introduced into anthraquinone (45) using catalysis with an iridium complex. The choice of conditions showed that the use of the [Ir(COD)Cl]2 – dtbpy catalytic system in heptane allowed the introduction of the BPin unit into anthraquinone (45), but this produced a mixture of isomeric esters of 2,6- and 2,7-diboronic acids 247 and 250 (Scheme 100).[201] Li et al.[67] carried out a similar cross-coupling of 1,5-dichloroanthraquinone (86) with bis(pinacolato)diboron using the complex [Ir(OMe)COD]2 as the catalyst in cyclohexane. The borylation proceeded selectively to the 3,7-position of the substrate without affecting the chlorine atoms, but the resulting boronic acid ester was used for further transformations without isolation.
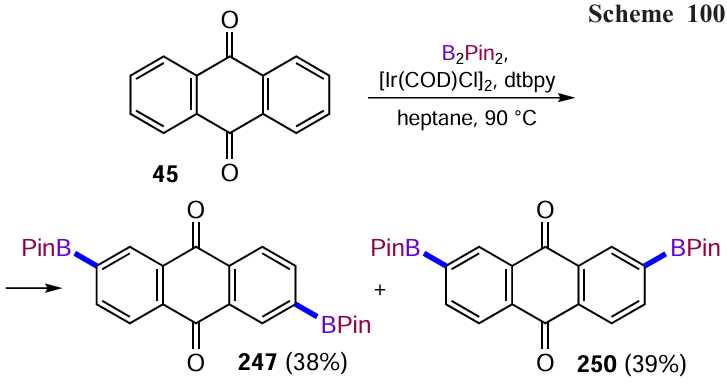
7.2. Carbon – selenium bond
Natural and synthetic organoselenium compounds have various types of biological activities, including antitumour, antioxidant, anti-inflammatory, etc.[206] Selenium-containing quinones have also attracted interest due to their involvement in various redox processes, which is reflected in their biological properties. For example, selenoquinones have been found to downregulate the levels of oncoproteins Bcl-2 and Ki-67, and activate caspase-8 expression in HepG2 hepatocellular carcinoma cells.[207] Selenoquinone derivatives have shown high cytotoxicity against various tumour cell lines, with half-maximal inhibitory concentrations (IC50) below 1 mM.[208] In 2024, the search for new anti-tumour compounds identified a class of anthraquinones fused to selenadiazole, which proved promising in terms of anti-proliferative activity and high selectivity for binding to G-quadruplex structures of nucleic acids.[209]
A simple and efficient way to form a C – Se bond with the anthraquinone core is the copper-catalyzed selenation of the appropriate haloanthraquinones. For example, Nakanishi et al.[210] obtained a series of 1-(arylselenyl)anthraquinones 252a – g by the reaction of 1-chloroanthraquinone (251) with diaryldiselenides (Scheme 101). It was found that the presence of electron-withdrawing substituents in the aromatic nucleus of diaryldiselenide leads to a slight decrease in the yield of the products.
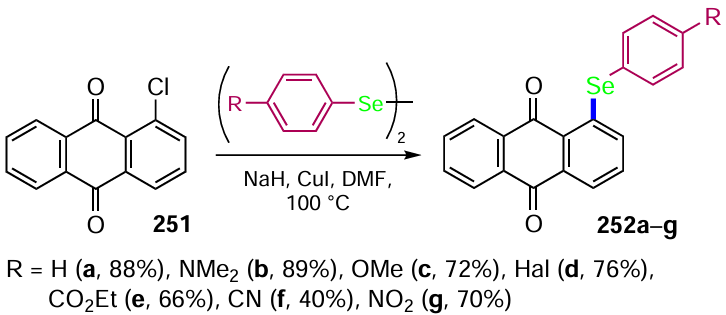
In another study,[211] selenolate was generated from diaryldiselenide by treatment with sodium borohydride and further reaction with 1,8-dichloroanthraquinone (88) in the presence of CuI, resulting in the formation of derivatives 253a,b (Scheme 102).
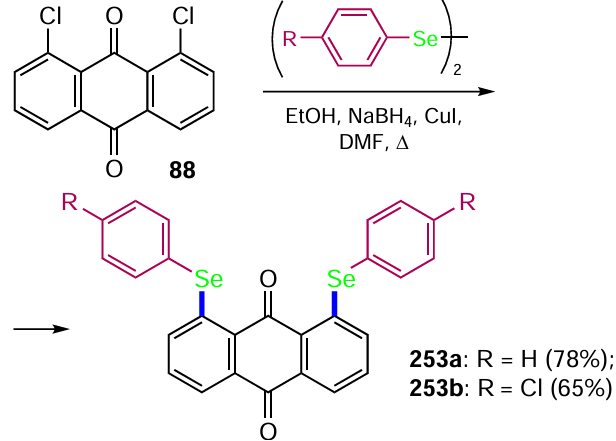
Jardim et al.[212] developed three efficient strategies for the synthesis of selenium-containing naphthoquinones and anthraquinones. The first method is based on the in situ generation of Santi’s reagent from arylselenyl chloride and zinc and reacting it with 1-iodoanthraquinone (56) in the presence of thiophene-2-carboxylate to give 1-(arylselenyl)anthracene-9,10-diones 252a,c,d and 254a (Scheme 103, Table 22).
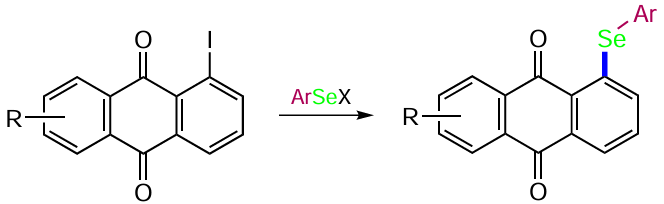
An alternative approach to selenation of anthraquinones with high efficiency has been realized using ArSeAg with the addition of the copper complex Cu(PPh3)3Br (see Table 22). This approach was tested on substituted anthraquinones and for anthraquinone containing two halogen atoms, in the α- and β-positions, the reaction proceeds predominantly to the 1-position to provide a high yield of the product 259. Selenium-containing anthraquinones were also obtained in the presence of silver selenide (ArSeAg) and CuFe2O4CNT catalyst (nanotube-supported copper ferrite) (see Table 22).[212] The use of such a nanocatalyst, as opposed to the Cu(PPh3)3Br complex, significantly reduced the catalyst consumption (up to 0.3 mol.%) and to obtain the desired compounds 262, 252a, 263a,b in moderate yields.
Photocatalytic radical di- and trifluoromethylselenation by introducing a selenium-containing functional group into the aromatic core turned out to be promising.[213, 214] The applicability of this method has been demonstrated on a wide range of substrates including electron-deficient anthraquinone. Treatment of anthraquinone-1-yl-diazonium tetrafluoroborate (28) with a selenium reagent (F3CSeTs-p) under irradiation in the presence of the photocatalyst eosin Y gives the product 264 in good yield (Scheme 104).[213]
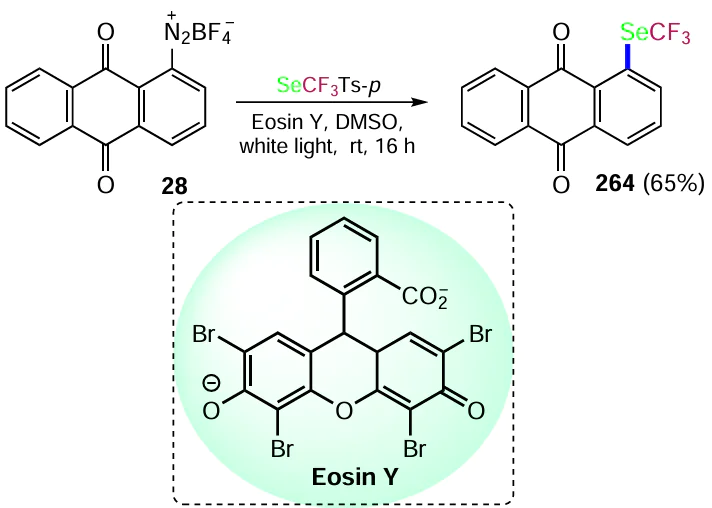
Later, Lu et al.[214] modified this reaction by replacing eosin with the photocatalyst Rose Bengal, which has a similar structure. Various anilines were used as substrates, which were treated with p-toluenesulfonic acid and tert-butyl nitrite to generate in situ a diazonium salt. Visible light irradiation of 1-amino- (48) and 2-aminoanthraquinones (50) in the presence of Se reagent and photocatalyst allows their conversion with close efficiency to [(difluoromethyl)selanyl]anthraquinones 265 and 266 (Scheme 105).
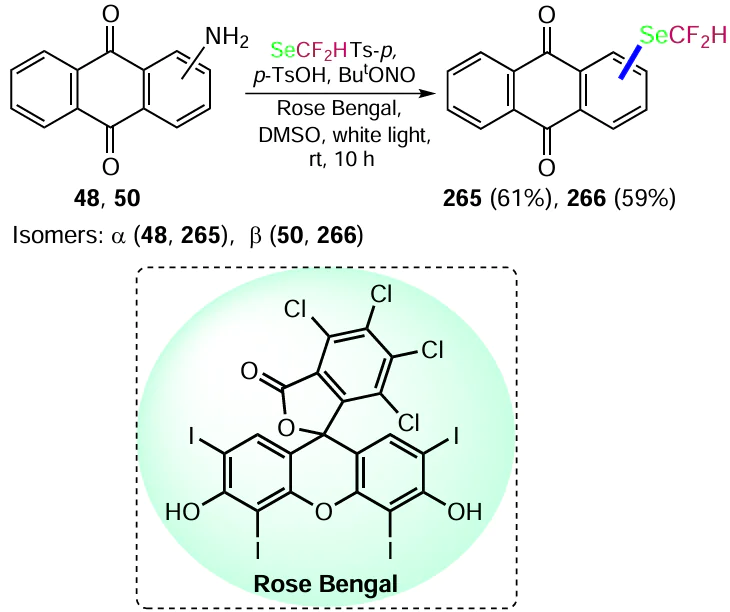
7.3. Carbon – phosphorus bond
Organophosphorus compounds are widely used in various fields of science and technology, such as catalysis, optoelectronics, pharmaceuticals, agrochemistry, etc.[215] However, studies of phosphorus-containing anthraquinones have received much less attention than other functional derivatives of this class. Methods of the C – P bond formation in the anthraquinone series are presented in only a few patents and publication from the 60s and 80s of the last century. Among these, isolated examples of the reaction of α- and β-haloanthraquinones with the following reagents are described:
— triphenylphosphine [216] and tributylphosphine,[217] to give the corresponding quaternary phosphonium salts;
— triethylphosphite to give diethyl(anthraquinonyl) phosphonate;[218, 219]
— phosphorus trichloride to give anthraquinone-1-phosphoric acid.[220]
However, the significant application potential of organophosphorus compounds and anthraquinones separately suggests that the role of phosphorus-containing anthraquinone derivatives is underestimated and that it would be useful to develop this area of research.
In fact, the only work published over the last 40 years describing the C – P bond formation in anthraquinones is the study by Wang et al.[221] in which a mild enantioselective Ni-catalyzed cross-coupling method with the C(sp2) – P bond formation was developed and used to obtain P-stereogenic phosphine oxides. The reaction of racemic secondary phosphine oxides with alkenyl or aryl bromides in the presence of a catalyst and a chiral ligand furnishes such products in high yields and with high enantioselectivity. For example, 2-bromanthraquinone (39) reacted with tert-butyl(phenyl)phosphine oxide using Ni(COD)2 as the catalyst and the (S,SP)-Ph-Phosferrox (L4) to give (S)-2-[tert-butyl(phenyl)phosphoryl]anthracene-9,10-dione (267) (Scheme 106).
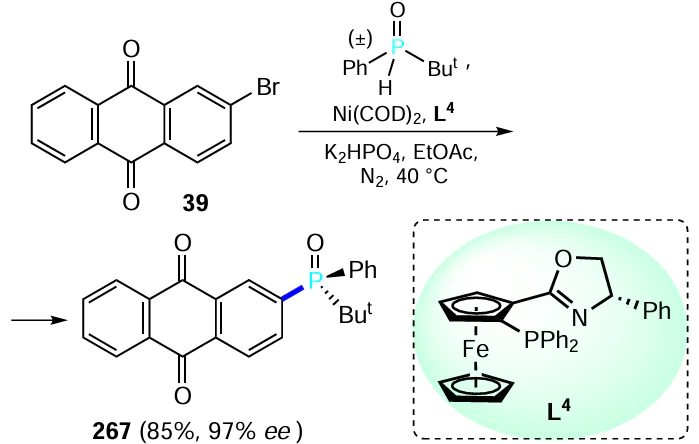
Methods of bond formation between anthraquinone and other heteroatoms, not mentioned in this review, are practically not found in the literature. It should be noted that the methods for the C – Si bond formation by silylation of anthraquinones are unknown, but such compounds can be obtained directly during the assembly of the nucleus from silicon-containing building blocks.[222] At the same time, organosilicon derivatives of anthraquinone can be promising materials for use as organic semiconductors, photocatalysts, photovoltaic elements and transistors.[223] The significant applied interest of these compounds can provide impetus for the development of methods for C – Si functionalization of anthraquinones as well as for the preparation of previously unknown organoelement derivatives of anthraquinone.
8. Conclusion
Since the publication of M.V.Gorelik’s monograph ‘Chemistry of anthraquinones and their derivatives’,[20] which is still one of the most important works in the domestic and foreign scientific literature on the structure and chemical properties of anthraquinones, the range of methods for their modification has expanded considerably. Modern trends in organic synthesis have extended to compounds of this class. Modern trends in organic synthesis have extended to compounds of this class. Metal-catalyzed cross-coupling and radical reactions have become important methods of functionalizing the anthraquinone core. Cross-coupling has made it possible to efficiently form C – C and C – heteroatom bonds, giving access to a wide range of anthraquinone derivatives. The use of these approaches has in some cases even provided access to selective and efficient activation (functionalization) of C – H bonds without the need to introduce additional leaving groups. It seems that in the near future these two methodologies will form the basis of directed atom-efficient modification of anthraquinone derivatives to obtain compounds with a given structure.
As has been shown in this review, the structural features of anthraquinones determine the chemical properties of this class, hampering many processes that occur readily with conventional aromatic compounds. Despite the existing limitations, examples of intramolecular variants of Friedel – Crafts cyclization have been demonstrated in the anthraquinone series, and the scope of Mannich reaction, Claisen rearrangement, amidomethylation and hydroxymethylation reactions, etc. have been considerably expanded. At the same time, the electron-deficient nature of anthraquinone allows the direct introduction of residues of C, N and S nucleophiles via oxidative substitution of hydrogen or vicarious nucleophilic substitution. The development of these methods of anthraquinone modification is also of considerable value due to the availability of reagents and the absence of complex metal complex catalysts.
The development and study of the peculiarities of cross-coupling reactions in a series of anthraquinone derivatives have allowed the efficient formation of C – C(sp3), C – C(sp2) and C – C(sp) bonds, opening the way to a significant complication of the molecular periphery. It is important to note the high chemoselectivity of the Suzuki reaction, related to the different reactivity of the leaving groups in the α- and β-positions of the anthraquinone, which makes it possible to effectively control this process and introduce different functional groups into the compounds. The use of Ru- and Rh-based catalysts provided an access to α-substituted alkyl, aryl and alkynyl anthraquinones with high regio- and chemoselectivity. A separate contribution to the modernization of chemistry of anthraquinone is the development of environmentally friendly cyanation methods, which are an excellent alternative to classical approaches to cyanoanthraquinones.
The importance of the methods for the preparation of halogenated anthraquinones cannot be overestimated, as they serve as a tool for the synthesis of not only important building blocks, but also compounds with practically useful properties that determine the applications of anthraquinones. The classical reactions of electrophilic halogenation of anthraquinones and substitution of a diazo group by a halogen atom still play a dominant role. However, the emergence of new methods for the conversion of other functional groups to halogens, based on metathesis reactions and metal-catalyzed substitution of the OTf group, emphasized the importance of efficient synthesis of such substrates. Although modern approaches to the C – Hal bond formation have only been described for iodo and fluoro derivatives of anthraquinone, their optimization allows the introduction of other halogens into the molecule and expands their possibilities for the modification of anthraquinones, especially those of natural origin.
The formation of C – O, C – N and C – S bonds with an anthraquinone core occurs mainly by substitution of halogen atoms or suitable functional groups under various conditions. Metal complex catalysis plays an important role in such transformations: for example, the Pd-catalyzed cross-coupling of arylamines with haloanthraquinones allows the introduction of bulky substituents even in the sterically hindered α-position of an anthraquinone. Methods for direct C – H functionalization of anthracenedione derivatives in the presence or absence of oxidants or catalysts are also being actively developed. However, for example, Ru- or Ir-catalyzed methods for introducing such groups are limited to the preparation of α-derivatives and sometimes cannot be carried out for hydroxy- and aminoanthraquinones.
The formation of С–B, C – Se, С – P bonds with anthraquinones is also mainly based on transformation or substitution of functional groups. The borylation of anthraquinones in the α- and β-positions is achieved by Pd-catalyzed substitution of the halogen atom. An important achievement in the preparation of boron-containing anthraquinones was the Ir-catalyzed method of the C – B bond formation at the β-position of the anthraquinone core. This approach is particularly significant as other options for functionalization of the anthraquinone core based on activation of the C – H bond are mainly used for the preparation of α-derivatives of anthraquinone. The chemistry of selenium-containing anthraquinones has also been actively developed in the last decade. For example, efficient reactions with arylselenides for selective seleno-functionalization of the α-position of the anthraquinone nucleus have been described. The methods of photocatalytic selenation have been successfully applied to obtain both α- and β-difluoromethylselenoanthraquinones. Other organoelement derivatives of anthraquinone have not been studied at present. For example, there is only one publication from the last few decades that deals with the formation of the C – P bond, using the example of the stereoselective introduction of a i-butyl(phenyl)phosphoryl group into anthraquinone.
Obviously, a significant part of the new methods of anthraquinone core functionalization are of an individual nature and are presented by one or a few examples, which does not allow to assess unambiguously their efficiency with respect to a wide range of available anthraquinone derivatives. Nevertheless, a review of such methods is of great value to synthetic chemists solving applied problems to obtain compounds of a given structure. The demonstration of the potential of modern synthetic strategies also contributes to the interest in focused research and identification of the limits of their application to open the way to polyfunctional anthraquinone derivatives, such as (hetarene)anthraquinones with antitumour activity.[224]
In conclusion, the methodology of anthraquinone functionalization has been actively developed in the last decades by creating new approaches to the C – heteroatom bond formation. However, the straightforward synthesis of anthraquinone derivatives of a given structure is still a non-trivial challenge in many cases, highlighting the importance of further searching for efficient methods of modification of anthraquinones and other related polyannulated quinones.
This review was partially supported by the Russian Science Foundation (Project No. 24-73-10199, https://rscf.ru/project/24-73-10199/
9. List of abbreviations
Following abbreviations and designations are used in the review:
acac — acetylacetonate;
Ad — 1-adamantyl;
AlPhos — di(1-adamantyl)(4''-butyl-2'',3'',5'',6''-tetrafluoro-2',4',6'-triisopropyl-2-methoxy-meta-terphenyl)phosphine;
Am — amyl (penthyl);
AQ — anthraquinone residue;
BINAP — 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl;
Boc — tert-buthoxycarbonyl;
B2pin2 — bis(pinacolato)diboron;
BQ — 1,4-benzoquinone;
Bzmim — 1-benzyl-3-methylimidazolium;
CataCXIum A*HI — di(1-adamantyl)-n-butylphosphine hydroiodide;
COgen — 9-methyl-9H-fluorene-9-carbonyl chloride;
COD — cycloocta-1,5-diene;
Cp* — pentamethylcyclopentadienyl;
Cy — cyclohexyl;
DavePhos — 2-dicyclohexylphosphino-2'-(dimethylamino)biphenyl;
dba — dibehzylideneacetone;
DBU — 1,8-diazabicyclo[5.4.0]undec-7-ene;
dccp — 1,3-bis(dicyclohexylphosphino)-propane;
DCE — 1,2-dichloroethane;
DCM — dichloromethane;
DG — directing group;
DIPA — diisopropylamine;
DIPEA — diisopropylethylamine;
DPPA — diphenylphosphoryl azide;
dppf — 1,1'-bis(diphenylphosphino)ferrocene;
dtbpy — 4,4'-di-tert-butyl-2,2'-dipyridyl;
Hexn — n-hexyl;
hmim — 1-hexyl-3-methylimidazolium;
IC50 — half-maximal inhibitory concentration;
L — ligand;
LG — leaving group;
M — metal;
mCPBA — m-chloroperoxybenzoic acid;
Mes — mesityl;
MOM — methoxymethyl;
Ms — methanesulfonyl (mesyl))
MW — microwave irradiation;
NBS — N-bromosuccinimide;
NCS — N-chlorosuccinimide;
NIS — N-iodosuccinimide;
Octn — n-octyl;
ONSH — oxidative nucleophilic substitution of hydrogen;
PEPPSI-IPr — dichloro[1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene)](3-chloropyridyl)palladium(II);
PIDA — (diacetoxyiodo)benzene;
PIFA — ([bis(trifluoroacetoxy)iodo]benzene;
PS — proton sponge;
Py — pyridine;
PyCIU — 1-[chloro(pyrrolidine-1-yl)methylidene]pyrrolidineium hexafluorophosphate;
rt ‒ room temperature;
Selectfluor — (1-chloromethyl-4-fluoro-1,4-diazoniobicyclo[2.2.2]octane bis(tetrafluoroborate));
SET — single electron transfer;
SPhos — 2-dicyclohexylphosphino-2',6'-dimethoxybiphenyl;
TBA — tetra-n-butylammonium;
Tc — (thiophene-2-carboxylate;
TCT — 2,4,6-trichloro-1,3,5-triazine;
TDAE — tetrakis(dimethylamino)ethylene;
TEMPO — (2,2,6,6-tetramethylpiperidin-1-yl)oxyl;
Tf — trifluoromethanesulfonyl;
TFA — trifluoroacetic acid;
TFAA — trifluoroacetic anhydride;
TFE — 2,2,2-trifluoroethanol;
TIPS — triisopropylsilyl;
TMS — trimethylsilyl;
Tol — tolyl;
Ts — toluenesulfonyl (tosyl);
VSN — vicarious nucleophilic substitution;
XantPhos — 4,5-(bis(diphenylphosphino)-9,9-dimethylxanthene;
XPhos-Pd-G2 — chloro(2-dicyclohexylphosphino-2',4',6'-triisopropyl-1,1'-biphenyl)[2-(2'-amino-1,1'-biphenyl)]palladium(II).
References












![Efficient synthesis of 3,6,13,16-tetrasubstituted-tetrabenzo[a,d,j,m]coronenes by selective C–H/C–O arylations of anthraquinone derivatives](/storage/images/resized/ex6KJoZujZOZFZh7jGfeHauiftuB3CI7iwJVFRDg_small_thumb.webp)






![Structure-based strategies for synthesis, lead optimization and biological evaluation of N-substituted anthra[1,2-c][1,2,5]thiadiazole-6,11-dione derivatives as potential multi-target anticancer agents](/storage/images/resized/9z7b0TQJNgUxZHYXLrTmbnjJn9y5iq56Wxtqb1Lv_small_thumb.webp)
