



Keywords
Abstract
This review is devoted to small molecules (compounds with molecular weights of up to 800–900 Da) that show activity against viruses causing hemorrhagic fevers that occur in Russia. The review presents compounds of various chemical classes that have shown antiviral effect against the causative agents of three acute infectious diseases, Omsk hemorrhagic fever, Crimean-Congo hemorrhagic fever, and hemorrhagic fever with renal syndrome, or alleviate symptoms of these diseases. Despite the severity and high prevalence of hemorrhagic fevers, it should be noted that there are no effective medications for the prevention and treatment of hemorrhagic fevers in Russia and that the search for new antiviral agents is highly relevant. The synthetic nucleoside ribavirin is commonly used for the treatment, but it is not sufficiently effective. The structures and activity parameters (half maximal inhibitory concentration) of the indicated compounds are given and putative mechanisms of their action are discussed. The review summarizes the data published over the whole period of studies of these diseases.The bibliography includes 160 references.
1. Introduction
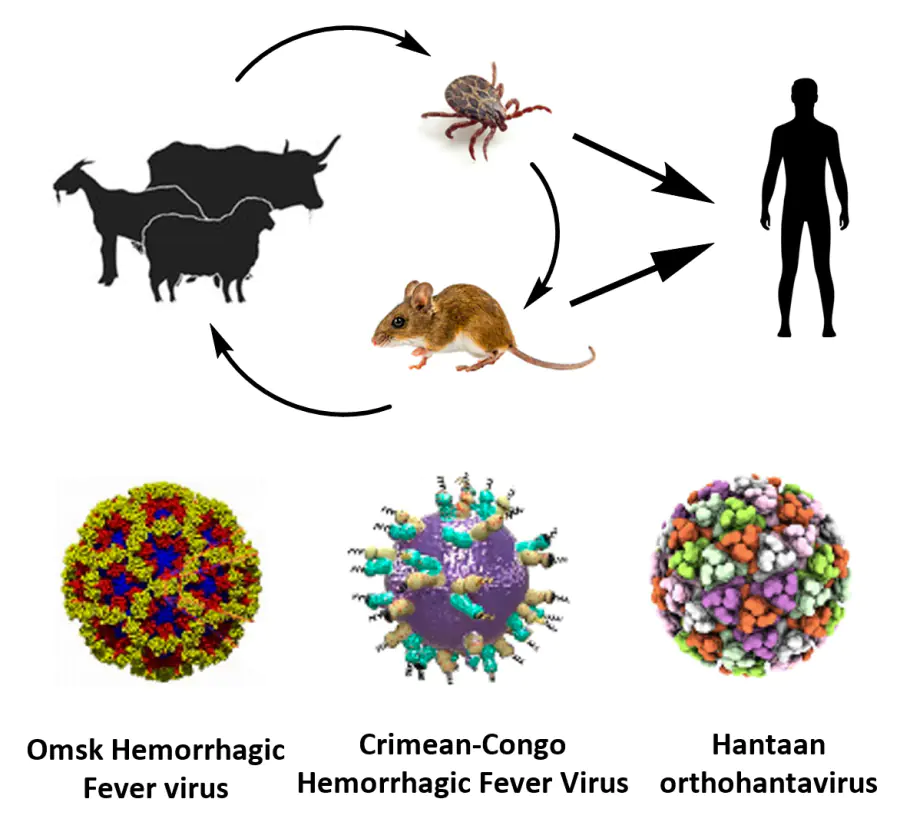
Hemorrhagic fever is an acute viral disease characterized by elevated body temperature and, as a rule, vascular damage and bleeding. Publications devoted to these diseases appeared in scientific journals back in the 19th century. An example is the report[1] about the yellow fever outbreak in Memphis, USA, in 1873. Currently, the name ‘hemorrhagic fevers’ combines quite a few known infections. In terms of epidemiology, these diseases are subdivided into four groups. Hemorrhagic fevers may be transmitted by mosquitoes (dengue fever, Rift Valley fever, yellow fever), ticks [Omsk hemorrhagic fever, Crimean-Congo hemorrhagic fever (CCHF), Kyasanur forest disease], rodents [hemorrhagic fever with renal syndrome (HFRS), hantavirus pulmonary syndrome, Lassa fever, and Argentine and Bolivian hemorrhagic fevers], and by bats (Marburg and Ebola hemorrhagic fevers).[2][3] Hemorrhagic fevers occur more frequently in tropical and subtropical climates, but they can easily spread to other geographical belts because of international tourism and globalizaion.[4] Some types of hemorrhagic fevers (Marburg, Ebola, Lassa, and several others) are highly severe diseases for humans and are fatal in the majority of cases (> 80%).[5] This high case fatality imposes limitations on the conduction of effective clinical trials, especially randomized controlled trials, since some patients will receive only placebo.[5][6]
The causative agents of hemorrhagic fevers are viruses. The major part of these viruses (representatives of Flaviviridae, Nairoviridae, and Phenuiviridae families) belong to the ecological group of arboviruses, that is, viruses transmitted to humans from blood-sucking arthropods. Apart from arboviruses, some hemorrhagic fevers can be caused by representatives of RNA-containing Arenaviridae viruses, arenaviruses, e.g., human pathogenic Lassa, Junin, and Machupo viruses. Among the causative agents of this disease, representatives of Filoviridae viruses (Marburg and Ebola fevers) and Hantaviridae orthohantaviruses (HFRS) deserve mention.[2-4]
Despite different etiologies and epidemiologic features, a common clinical syndrome can be noted for all viral hemorrhagic fevers. It has an incubation period of 3 to 8 days, gradual or abrupt development of the disease symptoms and signs of damage to vital organs lasting for approximately 3 days with possible short periods of relapse, and, finally, a sudden rapid deterioration of the condition on the 3rd or 4th day. These diseases are characterized by bleedings and complications; however, only some types of hemorrhagic fevers (Crimean-Congo, Lassa, Ebola, Marburg) were noted to be transmitted from human to human, especially in hospitals.[7]
Despite the threat that the disease may become an epidemic, neither acceptable vaccines nor effective small-molecule antiviral drugs have yet been developed for some types of hemorrhagic fevers. One of the important causes for this situation is too small number of biological laboratories with an appropriate safety level in the areas where epidemic outbreaks of hemorrhagic fevers occur most frequently.[8] A significant factor is the limited area of occurrence of a particular type of fever, the wrong opinion that the foci of the disease have almost vanished, and considering hemorrhagic fevers to be neglected diseases. For example, difficulties in treatment and vaccination of the population resulted in the Ebola epidemic in West Africa in 2014, although more than a dozen vaccines and antiviral drugs to prevent and treat the disease had been developed by that time.[9]
In this review, we consider in detail three types of hemorrhagic fevers that are most characteristics of Russia: Omsk and Crimean-Congo hemorrhagic fevers and the hemorrhagic fever with renal syndrome. Brief information about the diseases and the viruses that cause them is given. The importance of development of effective drugs for the prevention and treatment of hemorrhagic fevers in Russia is demonstrated, considering the set of data on these diseases available from the literature. For all three diseases, no optimal antiviral drugs have been found as yet, and the number of studies dealing with the search for these drugs is scarce. There is only one published review[10] addressing the potential small-molecule inhibitors of viruses that cause HFRS; however, the review mainly gives information on a narrow range of compounds meant, first of all, for infectious disease specialists. Therefore, we summarized for the first time the published data of small molecules that exhibit activity against viruses causing the indicated types of hemorrhagic fevers and/or alleviate their symptoms. The structures of these compounds are quite diverse and include nitrogenous heterocycles, aliphatic cyclic molecules with hydroxyl and amino groups, and natural compounds and their analogues. The putative mechanisms of action of the antiviral compounds are also addressed provided that they are available from the literature. We hope that this review would be useful for both synthetic and medicinal chemists as well as virologists, infectious disease specialists, epidemiologists, and other health care professionals.
2. Omsk hemorrhagic fever
Omsk hemorrhagic fever is a typical representative of recurrent viral infections. The investigation of this disease was motivated by large epidemic outbreaks in the mid-1940s in several forest steppe areas of the Omsk region. More than 600 people fell ill, and the patient’s tests and symptoms were indicative of a previously unknown disease. It was suggested (and later confirmed) that the ornate cow tick (Dermacentor reticulatus) and the ornate sheep tick (D. marginatus) may be involved in the infection transmission from the causative agent to humans. In 1947, viruses were isolated from the patients and from the ticks and proved to be identical.[11] Later, cases of humans being infected through skin lesions on contact with, or bites from, infected muskrats have been reported; this is considered to be a more common route of transmission of the disease.[12] It should be noted that no cases of human-to-human transmission have been noted. As a result, the nosological independence of the disease was proven and it was called ‘Omsk hemorrhagic fever’. Currently, the Omsk hemorrhagic fever virus (OHFV) has been assigned to the Flavivirus genus of the Flaviviridae family. A specific feature of this type of fever is the rather narrow nosoarea, practically not going beyond the forest steppe regions of Western Siberia. Cases of the disease have been registered in Russia only in the Omsk, Novosibirsk, Tyumen, and Kurgan Regions.[11] However, upon thorough investigation of the spread of the disease, the Omsk hemorrhagic fever was also found in Kazakhstan, in the Almaty area, at a distance of up to 1500 km from the Russian focus, both in patients and in natural sites.[13]
When the disease occurs in humans, the virus affects the endothelium of the capillaries, adrenal glands, and autonomic nervous system. The incubation period is typically 3 to 7 days, but it can also be shorter or last for up to 10 days. The sudden onset of the disease is characterized by fever with increased body temperature (39 – 40°С), which lasts for 5 – 12 days. After the first wave of fever, a second wave occurs a few days later in about 40% of cases, and it is often more severe than the first one. The mortality of this disease varies in the range from 0.4 to 3.0%. Patients die either after the rapid onset of hemorrhagic signs or, at a later stage, as a result of septic complications.[14] The Omsk hemorrhagic fever is diagnosed by virological and serological methods and treated using the pathogenetic and symptomatic therapy, for example, antibiotics against the secondary infection. Despite the high protective effect against the virus, the formalin-inactivated vaccine produced from the tissue of white mice infected with OHFV has not been clinically used for many years due to severe side effects;2 but the tick-borne encephalitis vaccine can be used to prevent this disease.[14]
The Omsk hemorrhagic fever is caused by a filterable RNA virus that is unstable to various physical and chemical impacts. A study of the primary structure of fragments of the NS5 and Е genes showed that this virus is a separate species among flaviviruses,[15] but in the antigenic properties, it resembles the tick-borne encephalitis virus.[16] It was found that the 5’-untranslated region of OHFV markedly differs from the 5’-terminal sequence of tick-borne encephalitis viruses, which determines the replication ability of this virus.[17] The virus has a spherical shape and a diameter of ~ 40 nm and a bilayer envelope composed of lipids and proteins. The OHFV genome comprises 10 787 bases with an open reading frame of 10 242 nucleotides encoding 3414 amino acids. This frame encodes a large polyprotein that is cleaved by cellular and viral proteases during and after translation into three structural (C, prM, E) and seven non-structural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) proteins. The viral non-structural proteins NS2B, NS3, and NS5 are involved in the viral replication mechanism in the host cell.[14][18] Formerly, it was believed that there are two serological types of OHFV that differ in the level of homology of genes that encode the surface proteins and viral polymerase. One type combines the strains from eastern areas, while the other one includes strains from the western areas.[11] However, sequence analysis of OHFV strains isolated at different times in three regions of Western Siberia showed that the genetic diversity of the virus is represented by three rather than two subtypes.[19]
Muskrats and white mice are most susceptible to OHFV.[11] The results of infection with OHFV in a study using young white BALB/c mice, published for the first time, demonstrated that, unlike encephalitis flaviviruses, this virus causes fever without overt signs of encephalitis or meningitis, despite the presence of virus in the cerebellum in the late stages of the disease.[20] This indicates that OHFV is unique among other flaviviruses. Tigabu et al.[21] investigated some clinical characteristics of the diseases caused by the tick-borne encephalitis virus and OHFV after the peripheral inoculation in two different lines of adult mice. It was found that BALB/c mice are more severely affected by the OHFV infection, while the encephalitis virus causes a more severe disease in black C57BL/6 mice. These results confirm that differences in disease severity are caused by the body response and prove the utility of the mouse model for the development of vaccines or therapeutic agents. Furthermore, it was shown for OHFV that the BALB/c and C57BL/6 mice can efficiently reproduce the infection pathology in human patients with mild meningoencephalitis characterized by minor brain damage and significant cerebellar damage.[22]
3. Small-molecule inhibitors of the Omsk hemorrhagic fever virus
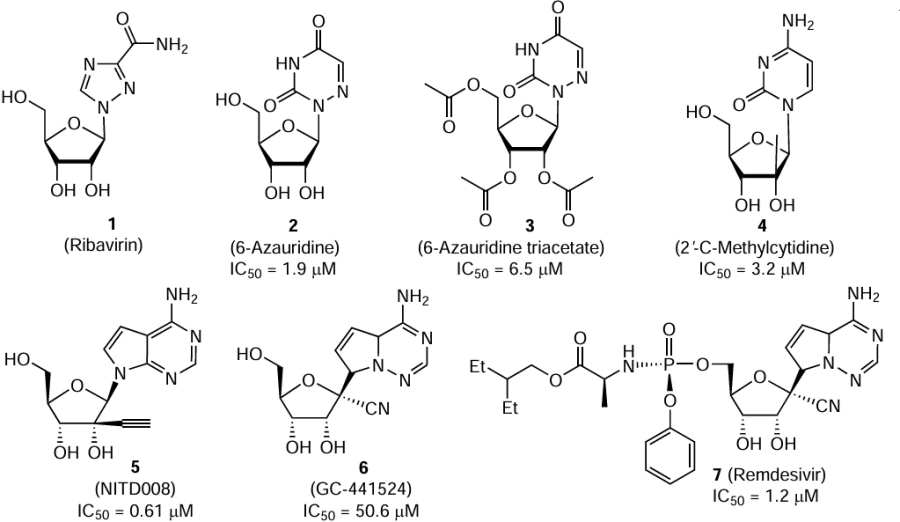
In 1970, ribavirin, or virazole (1), a nucleoside containing a 1,2,4-triazole moiety, was synthesized from natural D-ribose and proved to be active against some DNA or RNA viruses in experiments with tissue cultures.[23] The compound was effective against viral infections with different routes of inoculation in mice and rabbits. Despite the detected teratogenic effect of ribavirin on hamster and rat embryos [24] and the fact that it caused anemia when taken in high doses (³30 mg per kg of body weight per day), this drug was used to treat viral hemorrhagic fevers.[25][26] An important aspect of ribavirin activity may be related to its ability to act on the virus by several mechanisms simultaneously. Being a purine analogue, ribavirin can potentially affect the virus life cycle at any of its stages, in particular by inhibiting the protein translation, preventing the RNA synthesis by binding to the active site, ambiguous incorporation into RNA causing increased mutation and formation of non-viable genomes, or enhancement of the antiviral immune response. A few direct and indirect evidences for the mechanisms of action of this drug have been proposed, but there is still no explanation for the action on DNA viruses.[27] Currently, ribavirin is used as an antiviral drug to treat severe infections.
Ribavirin was tested against the Omsk hemorrhagic fever using cell lines and infected animals.[28] High concentrations of the drug moderately inhibited the viral reproduction in cell culture (up to 99.8% at 250 μg/mL concentration), while upon a single intramuscular administration in 200 μg dose, it showed a moderate efficiency against an experimental fever model (the survival rate of white mice was 55%). The fever in chinchilla rabbits is usually mild, and treatment with ribavirin reduces the duration of the disease by 2 days. The ribavirin toxicity was evaluated for chinchilla rabbits and ordinary white mice weighing 6 – 8 g: the maximum tolerated doses were 700 and 171 mg/kg, respectively.[29]
Compounds that were previously found to be active against representatives of the Flaviviridae family (e.g., against the hepatitis C virus) were tested on A549 human lung carcinoma cells against some tick-borne flaviviruses, in particular OHFV (Bogoluvovska strain).[30] 6-Azauridine (2), its triacetate (3), and 2’-C-methylcytidine (4) inhibited OHFV replication with IC50 from 1.9 to 6.5 μM,* determined by analysis of cytopathic effect. Since the antiviral activity of compound 2 decreased upon the addition of cytidine, but not guanosine, the authors concluded that the target activity was due to inhibition of pyrimidine nucleotide biosynthesis. Additional experiments showed that the antiviral action of compound 4 is caused by binding of the catalytic site of the virus polymerase NS5. In the opinion of the authors, these compounds may serve as the starting points for further search for effective drugs.
It was shown that compound 5 (designated by NITD008), adenosine nucleoside analogue, inhibits the replication of mosquito-borne flaviviruses (West Nile, yellow fever, and dengue viruses.).[31] The presence of an ethynyl group in the molecule is important for the activity, because the replacement of the ethynyl group by a methyl group resulted in an approximately 20-fold decrease in the activity against the dengue virus. The action of compound 5 on tick-borne flaviviruses, in particular OHFV, was tested using A549 cells.[32] The IC50 values for the inhibition of viral replication were determined by three different methods and, in the case of OHFV, they were in the 0.14 – 3.04 μM range. A value of 0.61 μM (with SI above 164) was found by analysis of cytopathic effect. It is noteworthy that inhibition of viral transcription and replication through the action on virus polymerase by nucleoside analogues is a promising strategy for the development of antiviral drugs. However, investigation of the antiviral activity of NITD008 was discontinued due to the detected toxicity of this compound.[33]
1'-Cyano-substituted nucleoside analogue 6 (GS-441524) was developed as an agent against RNA viruses capable of causing a pandemic.[34] The action mechanism of this compound implies conversion to the triphosphate metabolite capable of downregulating the activity of viral RNA polymerases. The cyano group located in position 1' of compound 6 provides the efficacy and selectivity of action against viral RNA polymerases. However, because of the slow kinetics of phosphorylation, modification of the monophosphate moiety was required to generate a high intracellular concentration of this compound. The resulting monophosphate 7 (GS-5734) was later called remdesivir. This compound showed a high antiviral activity and acceptable safety, in particular for primates infected with the Ebola virus.[35] Currently, it is used as a broad-spectrum antiviral drug. The activity of compounds 6 and 7 against various viruses, including flaviviruses, in particular OHFV (Bogoluvovska strain), was studied using A549 cells.[36] The IC50 values for compounds 6 and 7 determined by analysis of the cytopathic effect were 50.6 and 1.2 μM, respectively. Remdesivir showed a noticeable activity (IC50≤ 4.2 μM) against the tested flaviviruses, but was inactive against the Crimean-Congo hemorrhagic fever virus.
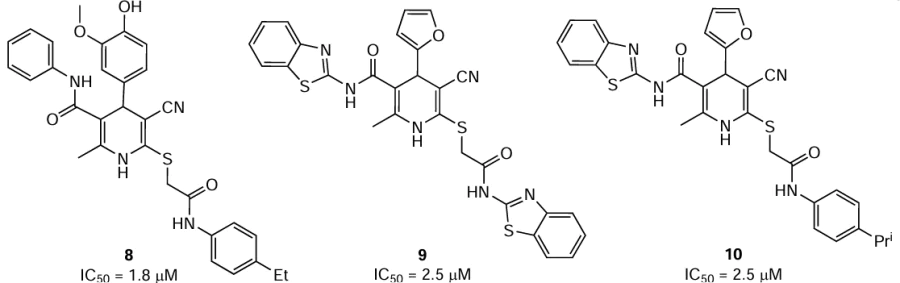
Homology models for the glycoprotein E of tick-borne flaviviruses were built.[37] In order to identify small molecules capable of inhibiting the reproduction of these flaviviruses via the interaction with the glycoprotein E in the hydrophobic pocket, molecular docking of a library of 5886 heterocyclic compounds was carried out. Eighty nine compounds, substituted 1,4-dihydropyridines and pyrido[2,1-b][1,3,5]thiadiazines, were selected for the experimental tests in vitro. In the plaque reduction assay using porcine embryo kidney cell line, 17 heterocycles noticeably inhibited (IC50 ≤ 7.2 μM) the reproduction of at least one of three flaviviruses: two viruses that cause encephalitis and OHFV (Nikitin strain). Only substituted 1,4-dihydropyridines inhibited the reproduction of OHFV; however, these derivatives were inactive against two other flaviviruses. Compounds 8, 9, and 10 showed the most pronounced effect against OHFV with IC50 of 1.8 (SI = 17.2), 2.5 (SI = 20.8), and 2.5 μM (SI = 11.6), respectively. It is noteworthy that compound 8, which was most active against this virus, was the only derivative containing a hydroxyl group. In the authors’ opinion,[37] the structure — activity relationship for these compounds has not yet been elucidated, because this is complicated by different ways of binding to proteins. It can only be noted that the presence of hydrogen bond acceptors in the substituents of 1,4-dihydropyridine is probably important for the effect of the compound on the virus. Generally, using virtual screening and molecular design, compounds with pronounced inhibitory activity against tick-borne flaviviruses and particularly against OHFV were found for the first time. However, additional studies are required to both determine the antiviral activity and mechanism of inhibition of viral reproduction and optimize the compound structures.
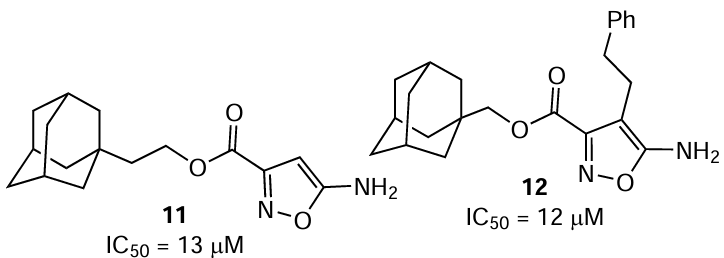
After several promising compounds that exhibited activity against the tick-borne encephalitis virus have been found, Vasilenko et al.[38] set themselves the task to obtain a new series of compounds based on a cage that was not evaluated against the tick-borne flaviviruses before and to study their activity against two viruses that cause encephalitis and OHFV (Nikitin strain). Isoxazole derivatives were chosen owing to their compact structure, relatively simple synthesis (3 steps), and the possibility of diverse functionalization. Among synthesized isoxazoles, there were compounds containing various lipophilic groups, substituents in the 1-adamantyl moiety and in the isoxazole ring, and various linkers. The plaque reduction assay using porcine embryo kidney cells performed for the series of isoxazoles showed that, unlike compounds 8 – 10, in this case, a compound that affected the OHFV reproduction also inhibited the reproduction of the other two flaviviruses. Compounds 11 and 12 had the highest activity against OHFV with IC50 values of 13 (SI = 32) and 12 μM (SI = 34.7), respectively. For compound 12, it was noted that the introduction of a lipophilic β-phenylethyl group in position 4 of the isoxazole ring markedly reduces the toxicity. The presence of this bulky substituent in isoxazole changes the mode of binding to protein E, and the oxygen atom is no longer hydrogen-bonded to the amide group of glutamine, but forms a hydrogen bond with the phenolic hydroxyl group of tyrosine. In addition, the adamantyl substituent is prone to hydrophobic interactions, but its modification has virtually no effect on the activity. Generally, heterocycles 11 and 12 can be considered to be promising models for further structure optimization to increase the antiviral activity.
* In this review, the antiviral activity of compounds is characterized by the half maximal inhibitory concentration (IC50) expressed in μmol/mL (μM) and the selectivity index (SI), equal to the ratio of the compound concentration that causes the death of half of normal cells to IC50 .
4. Crimean-Congo hemorrhagic fever
The Crimean-Congo hemorrhagic fever (CCHF) is a serious human arboviral disease accompanied by fever, hemorrhagic syndrome, and severe intoxication. The first documented outbreaks of CCHF (~ 200 cases) were reported since the summer of 1944 in a Crimea steppe region. In 1969, it was found that the viral strains that caused the Crimean hemorrhagic fever and isolated by M.P.Chumakov, a Soviet virologist, were antigenically and biologically related to the Congo fever virus isolated in 1956 and later.[39][40] The CCHF virus refers to the Orthonairovirus genus of the Nairoviridae family. It is transmitted by ticks and has been isolated from various types and subtypes of ticks, most often Hyalomma, which has one to three hosts. Domestic and wild animals are included in the viral cycle. The infection transmission is possible only through direct contact with a sick person.[41]The CCHF virus has the widest geographic distribution among the viruses that cause this type of tick-borne diseases and holds the second position among arboviruses after the dengue virus.[42] This infection is found in various countries in Africa, Europe, Asia, in the Balkans and in the Middle East, and the number of CCHF cases is as high as 15 000 per year. In Russia, the disease is registered in the southern regions of the European part and peaks in the summer months (June and July). The people that are most at risk of CCHF infection include agricultural workers and military personnel.[2]
The virus that causes CCHF infiltrates mainly the reticuloendothelial cells. The endothelial damage occurs due to both direct action of the virus and viral replication in the cells and as a result of an immune-mediated response.[41] The clinical course of the disease includes the following periods: incubation, pre-hemorrhagic and hemorrhagic stages, recovery, and long-term effects.[42] In some cases, the damage of central nervous system occurs, which is usually associated with a poor prognosis.[2] As a rule, the symptoms last for 10 – 12 days, but in the case of severe course of the disease, a person can recover only after 2.5 months.[41]The case fatality rates vary from 5 to 50% and even up to 80%, with patients dying, most often, due to shock, blood loss, or intercurrent infections.[43] The disease is treated using pathogenetic and symptomatic therapy; ribavirin is used in severe cases, but its efficacy against CCHF has not yet been convincingly demonstrated in clinical trials.[40] Despite the undertaken efforts, there is still no safe and effective vaccine against CCHF.[44] Thus, wide geographic distribution, possible epidemic outbreaks, high fatality rate, and limited therapy make this disease an important public health problem.
The CCHF virus is a single-stranded RNA virus. It has a single-stranded negative-sense genome, which must first synthesize a complementary positive-sense antigen, subsequently used to create a genomic RNA strand. The CCHF virion has a spherical shape and a diameter of ~ 90 – 100 nm. The lipid envelope of the virus includes glycoproteins Gn and Gc, which are responsible for the virion binding to cellular receptors.[43] The genome of the CCHF virus consists of small (S), medium (M), and large (L) RNA segments, which encode nucleoprotein (N), surface glycoproteins (Gn and Gc), and RNA-dependent RNA polymerase, respectively, to initiate transcription and genome replication in the host cell. The segment L consists of > 12 000 nucleotides.[45][46] The 3'-, 5'-terminal sequences of nucleotides of viral RNA are conserved, like those of other nairoviruses.[47] Terminal base pairing provides functional regions for the interaction with viral RNA-dependent RNA polymerase. Thus, the CCHF virus has a more complex genome structure than most of the viruses that cause human diseases and produces a positive-sense viral RNA using the negative-sense viral RNA to initiate the production and replication of viral proteins.[43] [46] The CCHF virus itself is fairly unstable, but in the absence of environmental impacts, it can retain viability for up to two years. Embryonic kidney cells are most sensitive to the virus. Although the CCHF virus is not adapted to a human body, high virulence of the virus is apparently due to the presence of interferon antagonists, inhibiting the interferonogenesis, which results in a severe course of the infection.[41]
The Hazara virus, a representative of the Orthonairovirus genus of the Nairoviridae family, was first isolated in 1954 from ticks collected in Pakistan. It is non-pathogenic to humans and, hence, it can be handled in laboratories with a low biosafety level, but it is lethal to newborn mice and causes cross-protection against CCHF infection in adult mice. The Hazara virus is an alternative model for studying the CCHF virus due to their serological and phylogenetic similarity.[48] Thus, it was shown that the infection of mice with the Hazara virus can serve as a useful surrogate model to test compounds for the activity against the CCHF virus.[49] Evaluation of potential therapeutic agents against CCHF was previously hampered by the absence of animal models that reproduce the distinctive features of this disease in human patients. In many cases, after the animals were infected, the viruses entered the bloodstream and spread throughout the body, but no clinical signs of the disease appeared.[50] However, in recent years, several promising animal models, including murine models, have been described, and in vivo studies in non-human primates have been conducted.[51] Using STAT129 mice, the main signs of fatal CCHF in humans were demonstrated. This model proved to be useful for testing therapeutic strategies and for exploring ways to mitigate the effects of the virus.[52] Using IFNAR–/– mice lacking type I interferon receptor, it is possible to reproduce the features of the disease in humans, including liver damage; this model can be used to evaluate the efficacy of drugs against the CCHF virus.[53] The STAT2–/– hamster model of CCHF infection provides additional information about the clinical picture of the disease and viral pathogenesis and can be used to test potential drugs and vaccines.[54] It has been shown that macaques infected with the virus exhibited all signs of human CCHF with remarkably similar pathway of virus spread, organ pathology, and disease development.[55]
5. Small-molecule inhibitors of the Crimean-Congo hemorrhagic fever virus
Currently, there are no recommendations on the treatment of CCHF depending on the severity of the disease. During epidemic outbreaks, only ribavirin (1) is actually used in practice, as it has the strongest evidence base for application efficacy. However, a number of clinical trials indicated that the intake of ribavirin has no noticeable effect on the treatment[56] and does not decrease the case fatality,[57] while a review[58] states that the available data on the use of this drug to treat CCHF are contradictory and that it is impossible to draw conclusions about the drug efficacy. Meanwhile, the intake of ribavirin for 10 days starting with a single dose of 30 mg/kg and ending with a dose of 7.5 mg/kg reduced the case fatality from 58 to 12%.[59] According to systematic reviews,[60][61] ribavirin should be regarded as an antiviral drug used for patients with CCHF, especially in the early stages of the disease.
Additional intake of corticosteroids, together with ribavirin, can efficiently alleviate the course of the disease, especially in the hemorrhagic phase.[61] The use of glucocorticosteroids, which have an anti-inflammatory effect and reduce the production of leukotrienes and prostaglandins, helps to reduce the acute inflammatory response of the body to the infection. For example, the intake of methylprednisolone (13), a synthetic glucocorticosteroid, in a high dose (10 mg/kg every day for 3 days and then 5 mg/kg) resulted in increasing leukocyte and platelet counts and decreased the degree of hemorrhage in patients with severe CCHF, which is a good result.[62] High doses of methylprednisolone (5 – 30 mg/kg per day for up to 20 days) were also efficacious for CCHF patients who had the reactive hemophagocytic lymphohistiocytosis in the hemorrhagic phase of the disease, because of dysregulation of the immune response.[63] It was found[64] that the use of ribavirin decreases the mortality in CCHF patients, especially in the case of moderate course of the disease. Meanwhile, the use of another synthetic glucocorticosteroid, dexamethasone (14), in a single dose of 15 – 20 mg also decreased the mortality, but mainly among patients with severe course of the disease.

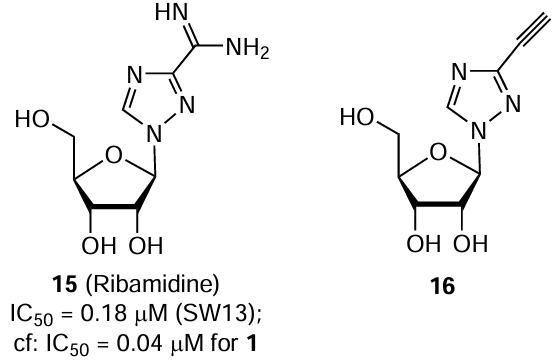
In a search for antiviral agents, ribavirin and four its structural analogues, in particular compound 2, were tested for the activity against four CCHF viral strains differing in the geographic origin. Studies were performed on infected SW13 (human adrenal gland carcinoma) cells using analysis of the cytopathic effect, which was determined by neutral red uptake assay.[65] Apart from ribavirin, only ribamidine (15), which is the ribavirin prodrug, showed antiviral action, but it was 4.5 – 8 times lower than that of ribavirin. The other three compounds, including 6-azauridine (2), did not show a noticeable activity against the used strains of CCHF virus. No significant differences in the drug sensitivity was found among the strains of this virus; for the strain Hy-13, which was isolated from a tick from China, the IC50 values of compounds 1 and 15 were 0.04 (SI > 1875) and 0.18 mM (SI > 417), respectively.
A series of 3-substituted 1-β-D-ribofuranosyl-1,2,4-triazoles were investigated to find a compound that would possess a unique antiviral ability, like ribavirin.[66] 1-β-D-Ribofuranosyl-3-ethynyl-1,2,4-triazole (16), synthesized in two steps from commercially available methyl 1-(2,3,5-tri-O-acetyl-β-D-ribofuranosyl)-1H-1,2,4-triazole-3-carboxylate, showed a considerable activity against two orthohantaviruses. According to focus forming unit assay results, compound 16 in a concentration of 30 μM also inhibited the CCHF virus (strain IbAr 10200) in Vero E6 cells by approximately 75%.
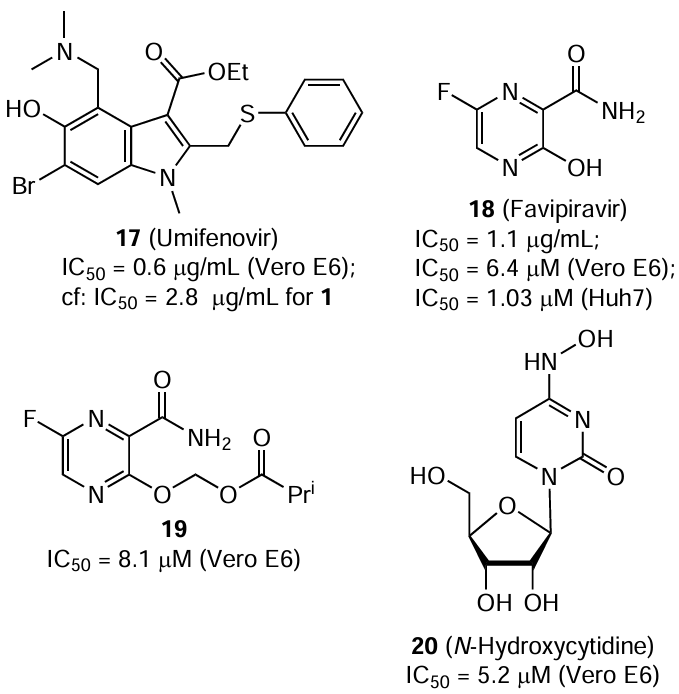
Oestereich et al.[53] tested umifenovir (Arbidol) (17) and favipiravir (T-705) (18), apart from ribavirin, in in vitro and in vivo models in order to identify effective CCHF inhibitors. The former drug is used to treat respiratory infections, while the latter previously showed inhibitory activity against a broad range of viruses, including those causing hemorrhagic fevers.[67] Compounds 1, 17, and 18 inhibited the replication of the CCHF virus (strain Afg09-2990) in Vero E6 cells, with IC50 values ranging from 0.6 to 2.8 μg/mL and SI ³ 11.5. The in vivo assays were carried out with IFNAR–/– mice, in which the course of the CCHF disease resembles that in humans. The intraperitoneal administration of umifenovir in a daily dose of 150 mg/kg induced no effect compared to the placebo and did not increase the survival rate of animals. Meanwhile, when ribavirin was administered in a daily dose of 100 mg/kg, some of the mice survived, but more than a half of mice died on the 6th day of post-infection. All animals who received favipiravir 1 h after being infected and every day in doses of 15, 30, and 300 mg/kg or 2 days after being infected and every day in a dose of 300 mg/kg survived; they had no signs of disease and did not show the presence of virus in blood and organs. Favipiravir had a higher efficiency in vivo than ribavirin, which is usually applied to treat CCHF. Co-administration of compounds 1 and 18 in vivo gave rise to a synergistic effect and resulted in the survival of all mice with the daily favipiravir dose being 7.5 mg/kg.[53]
As a possible mechanism of favipiravir action against the CCHF virus, Oestereich et al.[53] proposed incorporation of its ribofuranosyl 5'-triphosphate derivative into an arising RNA and inhibition of further chain elongation or induction of lethal mutagenesis. It was also shown that the 5'-monophosphate favipiravir derivative inhibits the inosine 5'-monophosphate dehydrogenase enzyme in the host cell much less efficiently than ribavirin derivatives. For this reason, this enzyme is not the main target for compound 18, unlike ribavirin. A key role in the favipiravir conversion to 5'-monophosphate derivative belongs to a purine metabolic enzyme–hypoxanthine guanine phosphoribosyltransferase. The activation of metabolism and the presence of antiviral activity requires the presence of a hydroxyl group in position 3 of 2-pyrazinamide rather than fluorine atom in position 6, which would form hydrogen bonds with amino acid residue.[68] In relation to influenza virus, it was ascertained that compounds 1 and 18 act as purine pseudo-bases, with the latter compound being an active competitive inhibitor of viral polymerase. The synthesis of viral RNA in infected cells is completely inhibited by both compounds at concentrations of ³50 μM, while at lower concentrations of these compounds, non-infectious species are formed and random point mutations are accumulated in the viral genome. The mutagenic effect of favipiravir (18) is twice as high as that of ribavirin (1).[69] Favipiravir was developed in Japan to treat influenza, while in Russia, it is used for the therapy of COVID-19.
Although the treatment of CCHF with ribavirin (100 mg/kg per day intraperitoneally) was successful at early stages of the disease, ultimately it did not prevent the terminal disease stage in IFNAR–/– mice infected with this virus. Conversely, favipiravir (300 mg/kg) showed a therapeutic effect in this mouse model even if the treatment was started at later stage of the clinical progression of CCHF. Some animals had a relapse of the disease after the favipiravir intake was discontinued. Hence, favipiravir demonstrated a noticeable activity against two strains of the CCHF virus.[70] The efficiency of favipiravir in a 300 mg/kg dose per day for the treatment of CCHF was studied using macaques infected with the strain Hoti.[71] It was found that the drug is well tolerated and does not cause adverse effects; it also inhibits the spread of the virus and reduces its content in the blood, tissues, and organs. Thus, the antiviral action of favipiravir is beyond doubt and it needs to be subjected to clinical trials in CCHF patients.
Wang et al.[72] evaluated the efficiency of several nucleoside analogues including favipiravir (18) and remdesivir (7), against the CCHF virus (strain YL16070). Remdesivir did not give any effect, whereas compound 18, derivative 19 (obtained by the reaction of favipiravir with bromomethyl isobutyrate with the goal to increase the bioavailability and safety), and N-hydroxycytidine (20) inhibited the infection in vitro in Vero E6 cells. Using immunofluorescence assay, the IC50 values for compounds 18, 19, and 20 were found to be 6.4 (SI = 151), 8.1 (SI = 12.6), and 5.2 μM (SI = 46.7), respectively. Mention should be made of higher (approximately 10-fold) cytotoxicity of ester 19 compared to this value of hydroxy derivative 18. The antiviral effect in vivo was demonstrated in IFNAR–/– mice infected with the CCHF virus. Instead of compound 20, its 5'-O-isobutyric ester was used in these experiments, but administration of this prodrug even in a daily dose of 1000 mg/kg did not protect the animals from death. Favipiravir and its derivative 19 in daily doses of 75, 150, and 300 mg/kg protected all mice from the lethal outcome even if the first administration was 24 h after the mice were infected; however, in the case of compound 19 administered 48 h after infection, only five out of seven mice survived. In the opinion of the authors,[72] compound 19 can be considered to be one of the most promising candidates for the treatment of CCHF.
For identification of new antiviral agents, Welch et al.[73] created a new recombinant CCHF virus, the inhibition of which can be quantitatively determined by measuring the decrease in the fluorescence in infected cells treated with test compounds.[73] The screening was performed with Huh7 human hepatocarcinoma cells using ribavirin (1) as the control. The authors analyzed 38 nucleoside analogues as compounds able to inhibit viral replication and also mycophenolic acid (21) and its prodrug, mycophenolate mofetil (22), which, like ribavirin, inhibit inosine 5'-monophosphate dehydrogenase. Only four compounds out of 40 candidates were able to markedly inhibit the replication of the CCHF virus. Unlike ribavirin, IC50 of which in Huh7 and Vero E6 cells were similar [12.5 (SI > 12) and 11.5 μM], in the case of favipiravir (18), IC50 values found for the above cells differed more than sixfold: 1.03 (SI > 50) and 6.4 μM, respectively. Compounds 21 and 22 provided a higher inhibition of the virus (IC50 ≤ 390 nM) than ribavirin or favipiravir, but they had moderate selectivity characteristics (SI ≤ 18). Among the test compounds, the highest efficiency was inherent in 2'-deoxy-2'-fluorocytidine (23) with IC50 = 61 nM (SI > 820), which did not show cytotoxicity at the highest tested concentration (50 μM). This compound was also found to strongly inhibit replication of the virus (IC50 = 95 nM) in SW13 human adrenal gland carcinoma cells. A combination of compounds 18 and 23 in 2 μM and 210 nM concentrations, respectively, also showed a 51% synergistic effect. However, no synergism was observed when compound 23 was combined with ribavirin.
The baculovirus expression system is based on infecting insect cells with special baculoviruses with an embedded gene of the desired protein. These viruses are pathogenic only to insects and are safe for warm-blooded animals. After introduction of the infection, the infected cells express a foreign, or recombinant, protein. The expression of recombinant full-length L-protein of the CCHF virus was used as a tool for the development of novel antiviral agents.[74] This approach can be used to screen large libraries of small molecules, because investigation of RNA-dependent RNA polymerase does not require high biosafety level.
The RNA synthesis was inhibited using ribavirin and favipiravir triphosphates. It was found that these compounds inhibit the viral polymerase activity and act like adenosine 5'-triphosphate or guanosine 5'-triphosphate, but incorporation of the last-mentioned two compounds into the growing RNA strand occurs at a higher rate. The inhibitory effects of nucleotide analogues, 2'-deoxy-2'-fluorocytidine 5'-triphosphate (24) and 2'-deoxy-2'-aminocytidine 5'-triphosphate (25), were also studied; they enhanced inhibition of viral polymerase because of higher incorporation rates and proved to be better substrates for this enzyme than triphosphates of compounds 1 and 18. In this study, compound 25 was recommended as a novel inhibitor of the CCHF virus polymerase.[74]

Nitric oxide is a mediator with a diverse biological action and inhibitory properties against various pathogens. The inhibitory properties of exogenous NO against the CCHF virus (strain IbAr10200) were investigated.[75] Using Vero E6 cells, it was found that S-nitroso-N-acetylpenicillamine (26), a known NO donor, has a clear-cut activity against this virus. Compound 26 in 400 μM concentration decreased the number of virions by 99% when the cells were treated a few hours before being infected and by ~ 82% upon treatment 1 h after initiation of the infection. Meanwhile, 3-morpholinosydnonimine hydrochloride, which is a peroxynitrite (ONOO–) donor, did not show a noticeable effect against the CCHF virus: the inhibition of viral replication by this compound in 400 μM concentration did not exceed 40%. This rules out the assumption that the pronounced activity of compound 26 against the CCHF virus is due to the formation of minor amounts of peroxynitrite via the reaction of NO with O2–. In addition, it was noted that S-nitroso-N-acetylpenicillamine specifically inhibits the N and Gn proteins of this virus and does not affect the production of proteins by the host cell.
Clathrin-dependent endocytosis is known to be a route of entry of the CCHF virus into cells.[76] Potential activities against the CCHF virus were evaluated for several compounds capable of inhibiting this route.[77] A noticeable inhibitory activity against the virus was found for the antimalarial drug chloroquine (27) and the neuroleptic chlorpromazine (28), approved by the Food and Drug Administration (FDA); these compounds showed activity against a few other types of viruses. The inhibition of the CCHF virus was demonstrated on Vero E6 and Huh7 cells infected with two viral strains (IbAr10200 and ArD39554) using neutralization and plaque reduction assays. For compounds 27 and 28, IC50 were 39.4 (SI = 26.9) and 10.6 μM (SI = 2.8) for infected Vero E6 cell and 43.4 (SI = 21.3) and 4.3 μM (SI = 7.1) for infected Huh7 cells, respectively. Experiments with various times of addition of test compounds relative to initiation of the infection showed that these compounds directly affect both the degree of infection and the spread of CCHF virus, and they still remained effective even when added 24 h after the cells were infected. Combinations of ribavirin with compound 27 or 28 demonstrated a synergistic effect against the CCHF virus. The rather low selectivity index of chlorpromazine necessitates its further chemical modification, while chloroquine has a good potential for the treatment of this disease. The synergistic effect of combining compound 27 with ribavirin makes it possible to reduce the required dose of chloroquine and thus mitigate its side effects. For example, the intake of chloroquine, which is a weak base, causes dysfunction of some enzymes, including acid hydrolases, which may affect the pH-dependent entry of the virus.[77]

Zivcec et al.[78] developed a system mimicking the life cycle of the CCHF virus and forming analogous virus-like particles in the cell culture that do not cause infection and, hence, do not require the use of special laboratory. These virus-like particles simulated four pathogenic strains of the CCHF virus, and the inhibitory effects of ribavirin and chloroquine against these strains were assessed in SW13 cells. In the case of compounds 1 and 27, the average IC50 values amounted to 47.5 (SI = 6.5) and 23.2 μM (SI = 4.1), respectively. The IC50 value for chloroquine was quite consistent with the value found in another study;[77] however, the selectivity was much lower.
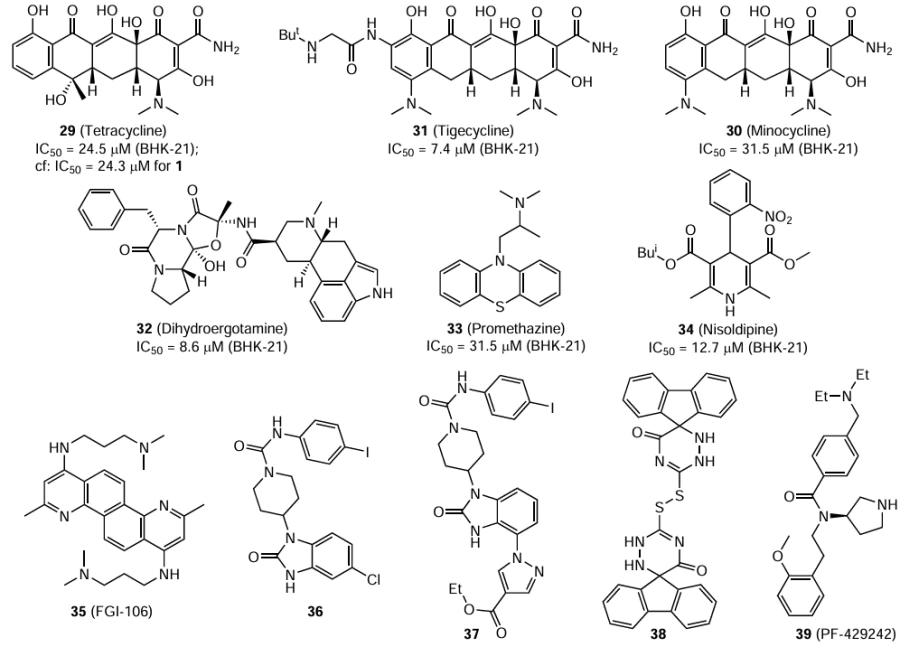
A minigenome system consisting of two orthonairoviruses and the CCHF and Hazara viruses was developed to study the viral replication by exogenous introduction of viral proteins and genomic RNA elements.[79] These minigenome systems make it possible to reproduce a viral replication machinery in vitro and to perform screening or investigate the action mechanism of small-molecule inhibitors.[80] This surrogate model was used to test a library of FDA-approved compounds.[79] The replication of the minigenome was assessed by analysis of luciferase activity. The luciferase expression found for a solution of the test compound in DMSO was compared with that for pure solvent. Screening of this library revealed 14 candidates that markedly decreased the luciferase activity. Considering the side effects, availability, and cytotoxicity, seven compounds were selected and studied using BHK-21 cells (kidneys of newborn Syrian hamsters) in different concentrations. Tetracycline (29), minocycline hydrochloride (30), tigecycline (31), dihydroergotamine mesylate (32), promethazine hydrochloride (33), and nisoldipine (34) had an activity against the CCHF virus (IC50 ≤ 31.5 μM) comparable with the ribavirin activity (IC50 = 24.3 μM). Compounds 32 and 34 were less active against the Hazara virus (IC50 ³ 23.8 μM) than tigecycline (31), which had a considerable effect on this virus (IC50 = 6.8 μM). The mechanism of inhibition was studied for the most active compound 31; it was found that tigecycline disrupts the interaction between the viral protein N and viral RNA. The authors suggested that the combined use of tigecycline and a nucleotide analogue active against the CCHF virus may be effective for the control of this disease. Generally, Hirano et al.[79] demonstrated the principle of testing the antiviral activity using animal models and development of a drug for the treatment of CCHF based on the most active derivatives.
Sharifi et al.[81] carried out a virtual computer screening to find inhibitors of protein N of the CCHF virus among FDA-approved drugs. As a result, they selected two tetracycline antibiotics, doxycycline and minocycline. The computation results suggest that both compounds are potential inhibitors of the nucleoprotein of the indicated virus. Although these drugs are used to treat bacterial infections, their use as antiviral agents, for example against dengue virus, cannot be ruled out either.[82]
Small molecule 35 (FGI-106), which has already demonstrated an antiviral effect against various RNA viruses in Vero E6 cells, considerably affected the viability of some Bunyavirales viruses, including the CCHF virus (strain IbAr 10200).[83] When present in 1 μM concentration, compound 35 decreased the activity of the CCHF virus approximately 16-fold. The pharmacokinetic profile of FGI-106 identified a substantial effects of the agent on the critical organs of mice (lungs, liver, kidneys, and spleen). The agent acts as an inhibitor that blocks the entry of the virus into the host cells.
In order to identify compounds possessing activity against RNA viruses, Tampere et al.[84] carried out phenotypic analysis of the small-molecule library available at their disposal. Due to the complexity of screening of antiviral agents under high biosafety level conditions, they used SW13 cells infected with Hazara virus. Out of the available library, 14 compounds in 10 μM concentration provided 90% inhibition of the primary viral infection, with the cell survival rates exceeding 80%. Screening identified substituted benzimidazolones 36 and 37 providing 90 and 95% inhibition of the virus titer, respectively. After Vero E6 cells were infected, treatment with compound 36 in 10 μM concentration reduced the titer of the CCHF virus (strain IbAr 10200) by 70%. Compound 37 showed a higher efficacy against the Hazara virus than analogue 36 in a dose-dependent assay and was selected for further investigation. It should also be noted that replacement of the benzimidazolone moiety in compound 36 with unsubstituted indole moiety resulted in the loss of antiviral activity.
Using a computational approach based on determination of ligand binding coordinates and correlation analysis, Kocabaş and Ergin [85] identified the pocket that serves as an inhibition site for the CCHF virus protease, which is a part of L segment. Quantum chemical calculations of the binding energy for the selected 300 possible protease inhibitors revealed compound 38, which did not show cytotoxicity in the assays in various cell lines. It is important to note that the authors [85] did not test the compounds for the antiviral properties; however, this approach may be useful for the search for drugs applicable for the therapy of CCHF. Finally, mention should be made of aminopyrrolidine 39 (PF-429242), which acts as an effective inhibitor of the SKI-1 protease, essential for the function of the Gn protein in the CCHF virus.[86] This compound is stable, has low toxicity, and possesses pharmacokinetic properties that make it suitable as a promising candidate for the treatment of the disease in question.[87]
* Here and below, the IC50 values are followed by the names of cell lines given in parentheses; in some cases, the same value for another drug is also given for comparison (cf).
6. Hemorrhagic fever with renal syndrome
Hemorrhagic fever with renal syndrome is an acute zoonotic viral infection characterized by high temperature and general intoxication of the body accompanied by vascular damage, hemorrhagic syndrome, and kidney disorder in the form of acute nephritis and development of acute renal failure.[88] The disease has been encountered in Asia for hundreds of years, but attracted attention only in the 1950s, during the Korean War, when 3 000 army men fell ill.[89] In 1978, a group of scientists succeeded in isolating the causative agent of HFRS, that is, the Hantaan virus, from the lung tissue of mice.[90] Subsequently, other HFRS pathogens belonging to the Hantaviridae family of the Orthohantavirus genus, namely, Puumala, Dobrava – Belgrade, Seoul viruses, were registered.[91] The human infection with this type of hemorrhagic fever is directly or indirectly associated with mouse-like rodents. Humans get infected through a contact with animals or through objects contaminated with animal excreta. The virus enters the human body through mucous membranes or damaged skin, but most often it enters the respiratory tract with airborne dust. People inhale the virus together with dust; therefore, the disease usually affects able-bodied men who perform agricultural or construction work. The virus is not transmitted from human to human.[2] The hemorrhagic fever with renal syndrome is a severe problem for people in Asia. Thus, every year, up to 50 thousand cases of infection with this virus are registered in China, and up to several thousand cases occur in Russia and Korea. Cases of infection have been reported in the Balkans, the Scandinavian Peninsula, Western Europe, Malaysia, and Japan.[91] In Russia, HFRS ranks first among zoonotic infections, with the greater part of affected persons living in the European part and only a small part being in the Far East. The Volga Federal District accounts for the largest number of HFRS infections in Russia (~ 85% of their total number).[88]
During the incubation period, the virus replicates and accumulates in the vascular epithelial cells; this stage can last from 4 to 45 days, but usually it lasts from 7 to 14 days.[92] The disease may be of various degrees of severity and may be accompanied by hemorrhagic, renal, and neuropsychiatric syndromes and disorders.[2] The combined treatment of HFRS includes the prevention and elimination of complications, administration of sedatives and analgesics, and maintenance of fluid balance in the patient body. Among etiotropic drugs, ribavirin is used, favipiravir may also be taken, and in severe cases, glucocorticosteroids are prescribed.[2] The mortality rate of this disease does not exceed 3%, but in some cases, it may reach 12%. The mortality of HFRS varies depending on the type of the virus: high mortality rates are inherent in the Hantaan and Dobrava–Belgrade viruses, a medium mortality is characteristic of the Seoul virus, and in the case of Puumala virus, the mortality rate is low.[93] The surviving patients acquire life-long immunity to the disease. Vaccines against the corresponding strains of the virus are available in China, Korea, and Japan. In Russia, the prevention of HFRS remains an open issue, because the developed Russian vaccine combining the European and Asian strains has not been registered.[88]
According to molecular analysis, the Hantaan virus genome consists of three negative-sense single-stranded RNAs that share the common 3'-terminal sequence of three genome segments.[94] The small (S), medium (M), and large (L) segments encode the nucleoprotein (N), envelope glycoproteins (Gn and Gc), and L protein or viral RNA-dependent RNA polymerase, respectively. The total size of RNA genome is 11845 nucleotides. Despite their different geographic locations and disease consequences, orthohantaviruses are highly homologous in their nucleic acid sequences and exhibit similar stages of life cycles. The virions of orthohantaviruses are usually spherical, have diameters from 80 to 120 nm, and are coated by a lipid shell. A single open reading frame functions in each of the genomic segments of orthohantaviruses. All three RNA segments have the same characteristic sequence AUCAUCAUCUG at the 3'-ends.[95][96] It was found that pathogenic orthohantaviruses enter cells via β3 integrins, which are representatives of transmembrane heterodimeric cellular receptors.[97]
A lethal infection with a non-mouse adapted strain of Hantaan virus was investigated on a newborn mice model.[98] Having studied the histological and virological features of the infection, the authors formulated the immunopathologic basis of the disease caused by Hantaan virus, which subsequently could be extended to humans. This experimental animal model was used to study the efficacy of ribavirin for the treatment of HFRS.[99] The results of another study[100] indicate that the virulence of the virus depends on the type of infected animals (newborn mice or rats) and on the particular strain. In a study of the Dobrava virus, high levels of viral replication and elevated levels of nitric oxide production were found in most lethally infected mice.[93] Sanada et al.[101] investigated infection of Syrian hamsters with the Puumala virus; this infection was found to be very similar to the orthohantavirus infection in natural rodents. These results suggest that a Syrian hamster may be a suitable animal model in the search for drugs to treat HFRS. The first described model of orthohantavirus infection in non-human primates showed that the cynomolgus macaque is also suitable as a model to study the pathogenesis of infection caused by the Puumala virus and to create new diagnostic methods, immunization strategies, and treatments.[102] Studies of the effects of vaccines in rhesus macaques made an important contribution to the development of a DNA vaccine to protect humans against HFRS.[103]
Laboratory models of hantavirus infection are known only for certain types of hantaviruses, which greatly holds up the development of chemotherapeutic agents effective against these viruses. The difficulties in the culturing of these viruses are related to the fact that under usual culturing conditions, hantaviruses have no visible cytopathic effect on a cell culture and are accumulated in small amounts even after long-term incubation. The applicability of МТТ assay for determination of the replication of the hantavirus strain Hantaan 76-118 was demonstrated,[104] and pseudovirus systems containing glycoproteins of the viruses inducing HFRS on the surface were developed.[105][106]
7. Small-molecule inhibitors of viruses of the hemorrhagic fever with renal syndrome
Ribavirin is the only antiviral drug currently used to treat HFRS in Asia, although it has serious side effects. By acting on inosine-5’-monophosphate dehydrogenase, this drug causes a pronounced decrease in the cellular level of guanosine 5’-triphosphate, which inhibits the synthesis of viral RNA.[107] In addition, ribavirin induces mutation of the Hantaan virus genome, leading to errors in viral replication and, hence, the formation of non-infectious viral particles.[108] A prospective randomized clinical trial was conducted in China, which implied a placebo control and double-blind intravenous injection of ribavirin (in the initial dose of 33 mg/kg; then 4 days in the 16 mg/kg dose; and the next 3 days in the 8 mg/kg dose) to parallel groups including 242 patients definitely diagnosed with HFRS.[109] The mortality among patients who took ribavirin was much lower (3.2%) than in the placebo group (9.5%). Ribavirin therapy also reduced the risk of disease progression to the oliguric phase and decreased hemorrhage events. Anemia completely reversible after the end of the therapy was the only side effect observed. In Russia (Vladivostok), patients with severe HFRS caused by Hantaan and Seoul viruses were first intravenously administered with ribavirin in 16 mg/kg dose four times a day for three days and then orally administered with ribavirin in a daily dose of 1000 mg for five days.[110] As a result of treatment, the body temperature became normal in a shorter period, the abdominal and lower back pain disappeared, the oliguric phase was shorter, and only a mild hemorrhagic syndrome was observed. The patients with mild to moderate forms of HFRS caused by Seoul virus did not require antiviral therapy. However, the results of a study [111] involving patients in Korea only suggest that intravenous administration of ribavirin reduces the frequency of oliguria and the severity of kidney diseases and require additional placebo control. In addition, a randomized open trial, conducted in the European part of Russia, on the efficacy and safety of intravenous administration of ribavirin in the treatment of HFRS caused by Puumala virus did not demonstrate sufficient efficacy and safety of this drug.[112]
Orthohantaviruses predominantly infect endothelial cells and induce a host immune response to the viral infection that includes a pro-inflammatory cytokine response, the inhibition of which may be useful for fighting the virus.[113] The first attempt to use glucocorticosteroids for this purpose dates back to the HFRS epidemic in the Korean War. Сortisone (40) was administered to patients either intramuscularly or orally in a dose of 100 to 300 mg per day.[114] The patients who took cortisone had the same stages of the disease and the same mortality, but the toxic febrile state was markedly alleviated and the mortality from the infectious toxic shock decreased; however, full clinical trials of the effects of shock in HFRS have not been conducted. However, in China, it is recommended to use glucocorticosteroids for patients in shock or in the presence of signs of severe disease as injections of hydrocortisone (41) in a dose of 100 mg intravenously once or twice a day for 3 to 7 days.[92] Apart from hydrocortisone, it is possible to use methylprednisolone (13) or dexamethasone (14). In Russia, glucocorticosteroids are also recommended for severe forms of HFRS, e.g., prednisolone (42) in a dose of 0.5 – 1 mg per kg of body weight.[2] When the infusion therapy is ineffective, the vasoactive drug norepinephrine (43) should be used for patients with HFRS-induced shock; the initial dose should be 8 to 12 μg/min and then the dose should decrease. The patients with oliguria should be treated with diuretics. The drug of first choice is, in this case, furosemide (44), which should be administered in a single dose of up to 200 mg and a daily dose of up to 800 mg.[92]
No data on clinical trials of favipiravir (18) for the treatment of HFRS is available from the literature. Favipiravir and ribavirin showed a moderate inhibition of the Dobrava virus (strain Sotkamo), with the IC50 values in Vero E6 cells being 93 (SI = 52) and 72 μM (SI = 17).[115] These agents inhibited the infection caused by Hantaan virus (strain 76-118) in Vero E6 cells in a dose-dependent manner, showing very low IC50 of 3.89 (SI = 4300) and 2.65 μM (SI = 490 000), according to the data from highly sensitive quantitative real-time polymerase chain reaction.[107] In the case of combined therapy, the efficacy of ribavirin increases when favipiravir is added. The highest antiviral activity corresponding to > 99% inhibition was attained for ribavirin and favipiravir concentrations of 18.8 and 12.0 μM, respectively. It was assumed [107] that by decreasing the guanosine 5'-triphosphate level, ribavirin enhances the effect of the nucleoside analogue, favipiravir, which is then incorporated into RNA chains and causes viral mutagenesis. Ye et al.[116] found a moderate inhibition of replication of the Hantaan virus (strain 76-118) in Vero E6 cells by favipiravir with IC50 = 150.8 μM (SI > 6.6).
The same publication [116] describes the effect of another influenza medication, baloxavir acid (45), on the Hantaan virus replication. Compound 45 is an active form resulting from hydrolysis of baloxavir marboxil. Compound 45 provided a noticeable inhibition only in 6.25 μM concentration (IC50 = 27.2 μM; SI > 3.7). The results of docking suggest that compound 45 binds to the endonuclease domain of Hantaan virus L protein and inhibits the action of the virus.
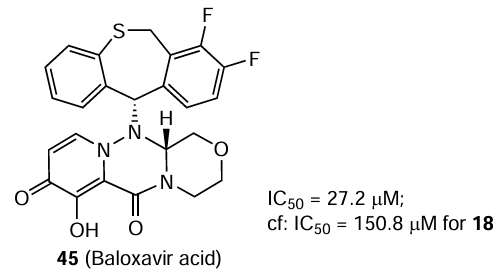
Nucleotide analogue 16 showed a pronounced antiviral activity against the Andes and Hantaan orthohantaviruses (strain 76-118);[66] in the latter case, the 30 μM concentration of the compound provided up to 94.3% inhibition in Vero E6 cells according to the focus forming unit assay results. Mycophenolic acid (21) in 6.25 μM concentration and ribavirin in 60 μM concentration provided 94 and 99% inhibition of the Hantaan virus, respectively. The IC50 values for compound 16 determined by enzyme immunoassay and focus forming unit assay were 27 (SI > 33) and 10 μM (SI > 88), respectively. Meanwhile, IC50 values for compound 21 and ribavirin were 67 and ³ 50 μM, respectively, i.e., compound 16 was more active against the Hantaan virus than ribavirin. The percentage of surviving newborn mice in the control group and in the groups receiving ribavirin for 14 days in a dose of 50 mg/kg or compound 16 in doses of 12.5 and 25 mg/kg were 10, 35, 25, and 26.3%, respectively. The antiviral activity of compound 16 is, most likely, due to the decrease in the guanosine 5'-triphosphate levels in cells as a result of inhibition of inosine 5'-monophosphate dehydrogenase; however, direct interaction of this compound with the viral polymerase is also probable. Study of the activity of ribavirin and compound 21, inosine 5'-monophosphate dehydrogenase inhibitors, against the Hantaan virus may indicate that the antiviral activity of ribavirin is primarily due to the interaction of ribavirin triphosphate with the viral RNA-dependent RNA polymerase rather than to the inhibition of inosine 5'-monophosphate dehydrogenase.[117]
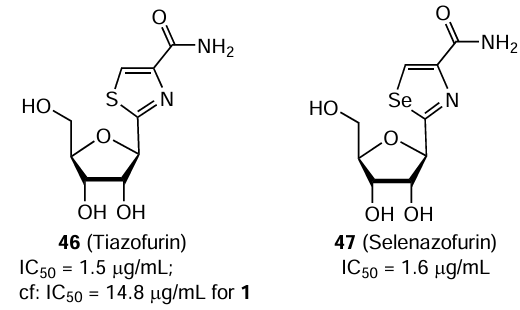
The antiviral activity of three related nucleoside carboxamides, ribavirin (1), tiazofurin (46), and selenazofurin (47), was studied in vitro against some DNA and RNA viruses, in particular the Hantaan virus (strain HBL7990).[118] The IC50 values measured by the plaque reduction assay in Vero E6 cells were 14.8 (SI ³ 68), 1.5 (SI ³ 666), and 1.6 μg/mL (SI ³ 625) for compounds 1, 46, and 47, respectively. All three compounds had a similar antiviral spectrum, but the overall antiviral activity was markedly higher in the case of selenazofurin. The compounds showed no toxicity in various cells. Combinations of two different carboxamides did not produce a synergistic effect against the Hantaan virus, but demonstrated additive effects.[119]
N1-(3-Fluorophenyl)hypoxanthine (48) and N1-(3-fluorophenyl)inosine (49) were synthesized from inosine and tested for the activity against the Andes and Hantaan orthohantaviruses (strain 76-118).[120] The IC50 values determined by the plaque reduction assay in Vero E6 cells were 234 (SI > 1.9) and 94 μM (SI > 3) for compounds 48 and 49, respectively. Analysis of the metabolism of compound 49 revealed low conversion to 5'-triphosphate in the cells and no inhibitory effect on inosine 5'-monophosphate dehydrogenase. This compound did not reduce the levels of viral RNA and did not increase the frequency of RNA mutations. Possibly, the antiviral activity is due only to the interaction of compound 49 or its metabolites with viral or host proteins that affect the level of virus release.
Out of the tested compounds, in addition to ribavirin, acyclovir (50), a guanosine analogue, used in the clinical practice as an antiviral drug also inhibited the replication of the Hantaan virus (strain 76-118) in infected Vero E6 cells.[121] The minimum effective concentration of acyclovir determined by immunofluorescence assay was 156 μg/mL, and it was nontoxic in doses of up to 625 μg/mL.

Triazavirin (51), 7-methylthio-3-nitro-1,4-dihydro[1,2,4]triazolo[5,1-с][1,2,4]triazin-4-one sodium salt dihydrate (a synthetic analogue of guanine), which is an antiviral drug developed in Russia, may become a prospective agent for the treatment of HFRS caused by the Hantaan virus.[122] This drug in a concentration of 100 mg/mL reduced the reproduction of Hantaan virus in SPEV by a factor of 100. The prolongation of the life span of newborn white mice infected with this virus (by more than 3 days) and a significant (tenfold) decrease in the level of virus accumulation in the brain of animals reflect the useful effect of triazavirin (50 mg/kg for 5 days) on the course of experimental infection. The substitution of nitro group via reactions with sulfur-containing amino acids and peptides may occur in vivo and is believed to contribute to the mechanism of antiviral action of triazavirin.[123]
The activity of umifenovir (17) against the Hantaan virus (strain 76-118) was studied in vitro and in vivo.[124] The plaque formation assay in Vero E6 cells demonstrated that this compound has a considerable inhibitory activity against the Hantaan virus, when added before and after the viral infection, with IC50 of 0.9 (SI = 17.4) and 1.2 μg/mL (SI = 13.0), respectively. In in vivo experiments, newborn BALB/c white mice infected with Hantaan virus, were orally administered with umifenovir 24 h before the infection for 10 days in a dose of 5 – 20 mg/kg per day, which is lower than the median lethal dose (78.4 mg/kg). All mice that received placebo died, whereas administration of the drug increased both the animal survival rate (60% for a dose of 20 mg/kg) and the mean time to death, and also decreased the histopathological changes, viral load, and expression of the viral antigen. A study of the mechanism of action showed that umifenovir can alleviate the pathological changes and acute inflammation caused by this virus by inhibiting the production of inducible nitric oxide synthase.[125] The authors concluded that umifenovir modulates the inflammatory response of cytokines or chemokines in the Hantaan virus infection.
Testing of Amixin, the active ingredient of which is tilorone dihydrochloride (52), oral inducer of endogenous interferon, in two-week-old mice infected with the Hantaan virus (strain 76-118) showed that this compound is effective in the case of both oral and subcutaneous administration.[126] For oral administration, the highest efficacy was observed when the drug was taken 96 h before introduction of the infection in 10 mg/kg dose, with the protection from death being 61%. In the case of subcutaneous administration, the dose was 1 mg/kg and 65% protection from death was provided. It was found that compound 52 effectively inhibited the reproduction of the virus in brain tissue.
Chloroquine (27) was found to have a pronounced antiviral activity against several species of orthohantaviruses including viruses distributed throughout Eurasia and America.[127] The IC50 values determined in Vero E6 cells by the quantitative real-time polymerase chain reaction were 10.5 μM (SI = 24.8) for the Hantaan virus (strain 76-118) and 9.5 μM (SI = 27.3) for the Dobrava–Belgrade virus (strain SK/Aa). In in vivo studies, pregnant C57Bl/6 mice were daily subcutaneously injected with chloroquine 2 days before the birth of the offspring, with the assumption that it would be excreted from the female body with her milk. Within 24 h after the birth, the mice were infected with the Hantaan virus. The percentages of surviving mice for chloroquine doses of 10, 5, and 1 mg/kg were 72.7, 47.6, and 4.2%, respectively; higher doses resulted in the death of offspring. When newborn mice were treated with the drug after introduction of the infection, chloroquine was less effective: only 16.7% of animals survived. Generally, chloroquine, which increases pH of intracellular membrane organelles, inhibits the release of the virus, and has an immunomodulatory action, can be considered to be an effective drug against Hantaan virus infection.
Compound 35 (FGI-106) exhibited a noticeable activity against the Andes and Hantaan orthohantaviruses (strain 76-118) in Vero E6 cells.[83] When used in 1 μM concentration, it decreased the virus titer approximately 10-fold, while ribavirin was inactive in this concentration.
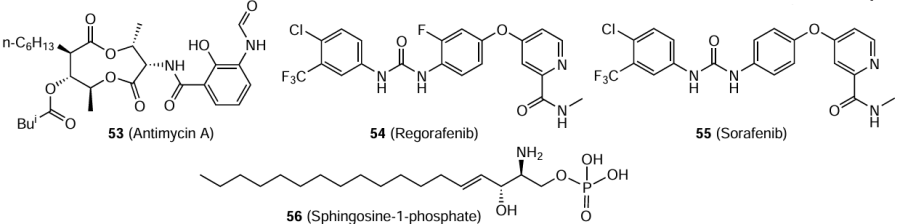
Using high-throughput flow cytometry, the Prestwick Chemical Library of small molecules was screened for inhibitors of orthohantavirus infection.[128] Only three compounds in 10 μM concentration markedly (by more than 60%) inhibited the Hantaan virus-induced infection in Vero E6 cells. Among them, the lowest cytotoxicity and the highest activity (more than 80% inhibition) was found for antimycin A (53). This compound directly binds to viral particles.
Among the FDA-approved drugs, urea-based multi-kinase inhibitors regorafenib (54) and sorafenib (55) showed excellent antiviral activity against a number of viruses and low cytotoxicity.[129] In concentration of 3 μM, both compounds were active against the Hantaan virus (strain 76-118) in HEL 92.1.7 cells where they reduced the viral load and expression of the viral antigen by ~ 85%. The antiviral activity of these drugs is apparently due to blocking of cell signalling pathways that are involved in viral replication. It was found that a signalling sphingolipid, sphingosine 1-phosphate (56), blocks the permeability of HUVEC endothelial cells for orthohantaviruses, in particular the Hantaan virus (strain 76-118); therefore, it is a potential therapeutic agent for treating diseases caused by these viruses.[130]
Natural compounds and their synthetic analogues are widely used as antiviral agents with a broad spectrum of activity.[131, 132] Studies of inhibition of the Hantaan virus (strain 76-118) in vitro and in vivo carried out by Chinese scientists revealed various natural compounds that have low toxicity to cells, directly inhibit the virus, and retard the progression of HFRS. The following compounds were investigated: gypensapogenins A (57) and B (58), triterpenoid saponins Gynostemma pentaphyllum,[133, 134] aphanamixoid A (59), limonoid with a new carbon skeleton from Aphanamixis polystachya,[135] eryngiolide A (60), cytotoxic macrocyclic diterpenoid with an unusual skeleton from the edible mushroom Pleurotus eryngii,[136] chukrasones A (61) and B (62), limonoids with a new carbon skeleton from Chukrasia tabularis,[137, 138] myriberine A (63), alkaloid with a pentacyclic skeleton from Myrioneuron faberi,[139] incaviranone A (64), structural hybrid with an unusual carbon skeleton from the plant Incarvillea delavayi,[140] sarcabosides A (65) and B (66), compounds with a new carbon skeleton from Sarcandra glabra,[141, 142] racemosin A (67), new bis-indole alkaloid from green algae Caulerpa racemosa,[143] lycojaponicumins A (68), B (69), and C (70), alkaloids with an unusual skeleton from Lycopodium japonicum,[144-146] kadcoccitone A (71), tetracyclic triterpenoid from Kadsura coccinea,[147] zoarenone (72), structurally related metabolite of an alkaloid from Zoanthus sp. polyp,[148] linderolide H (73), sesquiterpenoid lactone from roots of Lindera strychnifolia,[149] orientin (74), flavone and luteolin C-glucoside from Cecropia pachystachya.[150] These compounds showed considerable protective effects in animals (mice, hamsters, gophers, and shrews) infected with the virus, alleviated the course of the disease, and reduced the mortality of animals. Thus, these compounds can be considered as effective and safe drugs for the treatment of HFRS. However, it should be noted that their biological properties are described only in Chinese patents and have not been published in peer-reviewed scientific journals.
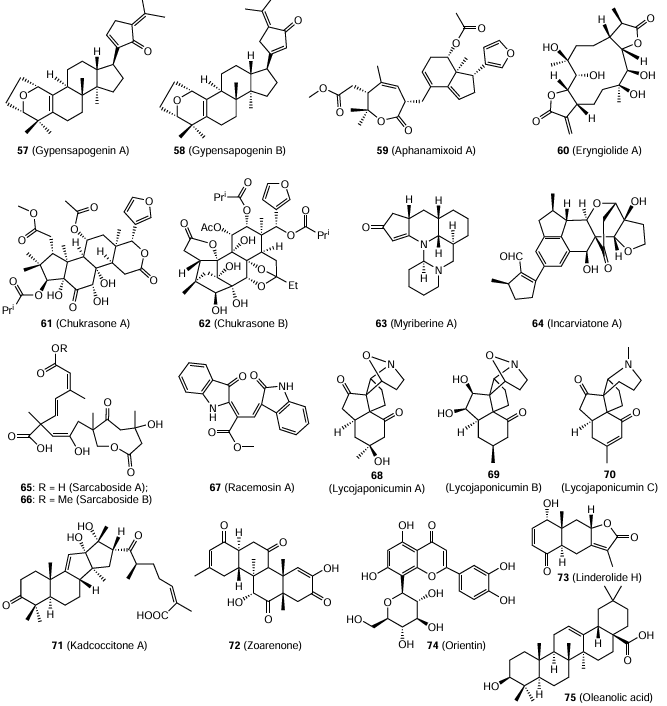
Antiviral activity was found not only for pure compounds isolated from plant sources, but also for fractionated extracts. For example, active compounds were extracted with hot water and alcohol from the Chinese herb, Alternanthera philoxcroides. The product was fractionated by treatment with diethyl ether, ethyl acetate, and alcohols.[151] Four extracts showed antiviral activity against three Hantaan virus strains (HV114, HV435, and A9) in Vero E6 cells. The highest efficiency was found for the initial extract; the IC50 values for three strains determined by plaque reduction assay were ~ 155 μg/mL. A similar activity with IC50 ≈ 205 μg/mL (SI ³ 2.5) was found for oleanolic acid (75) isolated from the first fraction.
Romurtide (76), a synthetic lipophilic derivative of muramyl dipeptide, is the minimum structural unit of bacterial cell wall components necessary for stimulation of biological action.[152] The effect of romurtide on the resistance to viral infection in newborn mice was studied;[153] 24 h after birth, the mice were infected with a lethal dose of the Hantaan virus (strain 76-118). If before infection, the mice were administered with 50 or 100 μg of romurtide, the survival rates (50 or 67%) were higher than those in the control group (17%). The effect was noticeable even when romurtide was injected 7 days after infection. The viral titers isolated from animal organs 12 days after infection were about 30 times lower in mice treated with this drug. Compound 76 also increased the peripheral leukocyte and splenocyte counts. The results indicate that this agent enhances the resistance of newborn mice to the Hantaan virus in a systemic infection model by promoting hematopoiesis and immunity.
Larson et al.[154] described a cyclic peptide consisting of nine amino acids and inhibiting the entry of the Hantaan virus into cells. Considering the similarity with this peptide, peptidomimetics were chosen and studied for the antiviral activity.[155] First, biological screening of such molecules tested in 1 μM concentrations revealed effective inhibitors of pathogenic orthohantaviruses in the micromolar range, which were approximately 1000 times more active than the pristine cyclic peptide. Finally, three peptidomimetics that showed dose-dependent inhibition in Vero E6 cells were selected; among them, compounds 77 and 78 provided the most pronounced inhibition of the infection caused by the Hantaan virus (strain 76-118). The mechanism of action of these molecules is based on the involvement of the β3 integrin receptor. Comparison of their activities indicates that the presence of an aliphatic ring containing a nitrogen atom reduces the activity, while the activity is increased by chlorine atoms in the ortho- and para-positions of the benzene ring or two fused rings that mimic amino acid residues in the interaction with the orthohantavirus entry receptor.
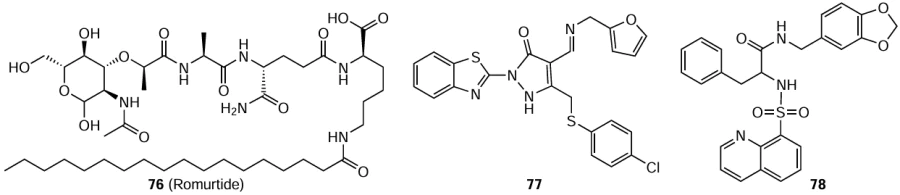
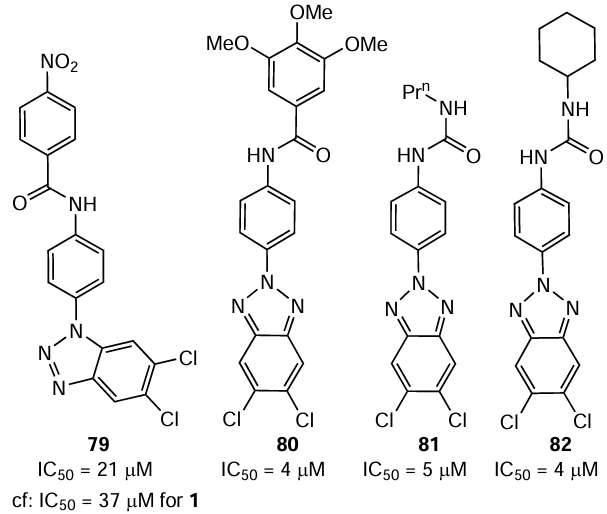
Sanna et al.[156] tested various substituted 5,6-dichloro-1(2)-phenyl-1(2)H-benzo[d][1,2,3]triazoles with the goal to find new inhibitors of RNA viruses. The antiviral activity of these compounds against the Hantaan virus (strain 76-118) in Vero E6 cells was determined using chemiluminescence analysis. Only one 1-phenylbenzotriazole derivative, compound 79, was active against this virus (IC50 = 21 μM), while three 2-phenylbenzotriazoles 80 – 82 proved to be rather potent inhibitors of the Hantaan virus with IC50 in the 4 – 5 μM range (SI > 6), i.e., the activity was markedly higher than those of ribavirin (IC50 = 37 μM) and favipiravir (IC50 = 151 μM). Derivatives 80 – 82 in 20 μM concentration substantially decreased the viral titer compared to the control experiment. The production of the virus decreased 1200-fold when the cells were treated with compound 81 and 260- and 140-fold on treatment with compounds 82 and 80, respectively. Meanwhile, ribavirin in a concentration of 50 μM provided only a 60-fold decrease compared to the control. Complete loss of antiviral activity resulted from the replacement of two chlorine atoms in the 2-phenylbenzotriazole derivative by hydrogen atoms or methyl groups or replacement of the propyl group in compound 81 by ethyl or butyl group. Generally, the detected considerable antiviral activity and low cytotoxicity of phenylbenzotriazoles suggest that compounds of this class could be potential candidates for further search for effective agents to treat HFRS.
Yarovaya et al.[157] synthesized N-acyl derivatives of camphor, fenchone, and norcamphor hydrazones and presented them as a new class of inhibitors of the Hantaan virus (strain 76-118). The paper describes the method of synthesis and detailed investigation of the antiviral activities of 3a,6-epoxyisoindole derivatives in which four most important positions were varied, namely, the C(4)=C(5) double bond, the terpene moiety, and substituents at the N(2) nitrogen atom and at the C(6) bridgehead carbon atom, as shown in Fig. 1.
![[{"id":"z-bjQ6WdEP","type":"paragraph","data":{"text":" Study of the antiviral activity of 3a,6-epoxyisoindole derivatives.[[ type=\"anchor\" referenceId=\"8586\" ]] Copyright (2021) Elsevier"}}]](/storage/images/resized/yYIvZDbOu5Rp8v3yh2Uz1rVe8G3ifnA6ANVYZNWs_xl.webp)
A cytopathic model was developed for testing, and a pseudovirus system was used to study the mechanism of antiviral activity. Triazavirin (51) and ribavirin (1) drugs were chosen for comparison. The former was active against the Hantaan virus, showing IC50 = 14 μM (SI = 26), while the latter proved to be inactive and toxic in experiment using Madin – Darby canine kidney cells (MDCK). Among the test compounds, note structurally similar mixtures of diastereomers 83 and 84. Camphor derivative 83 showed a noticeable antiviral activity with IC50 = 14 μM (SI = 10), and fenchone derivative 84 showed both a considerable activity with IC50 = 7.6 μM (SI = 140) and a markedly lower toxicity than compound 83. Presumably, an isoindole moiety bound to the bridgehead carbon atom is needed for the presence of the target activity, because compounds unsubstituted in position 6 of isoindole are inactive against the Hantaan virus. In addition, the antiviral effect is promoted by the presence of a double bond in the isoindole moiety and the absence of substituents in the para-position of the benzene ring and the linker between the benzene ring and the isoindole nitrogen atom. The binding of these compounds to the nucleoprotein of the Hantaan virus was evaluated using molecular docking. According to calculations, all derivatives can interact with the nucleoprotein at the site of its binding to viral RNA and form intermolecular bonds with amino acid residues. However, the compounds that were more active against the Hantaan virus showed better affinity to the binding site.
Using commercially available (±)-camphene as the starting compound, Sokolova et al.[158] synthesized a series of ethers containing various saturated N-heterocyclic moieties. Among this series, compounds 85 and 86 were found to have a pronounced activity against pseudovirus particles containing Hantaan virus glycoproteins on the surface (strain 76-118). It is noteworthy that compound 85 has a broad spectrum of antiviral activity: it is an effective inhibitor of H1N1 influenza virus and Ebola virus.
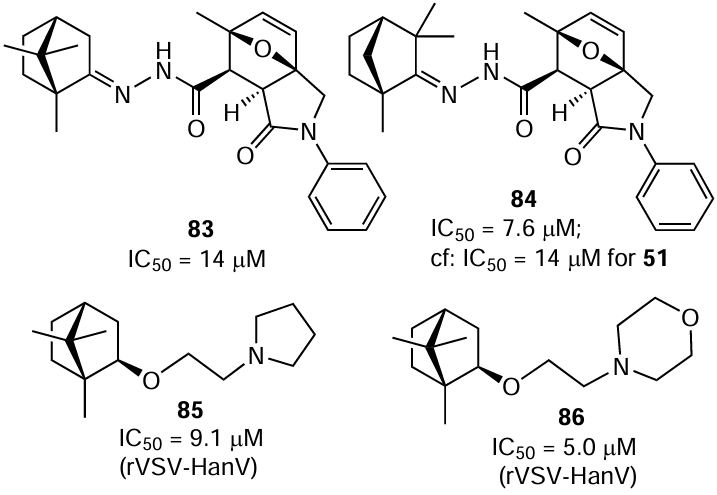
In order to find and identify new inhibitors of viruses causing hemorrhagic fevers, it is important to investigate both active and inactive compounds. However, researchers very rarely publish negative results, as they believe that compounds with low activity may be perceived as irrelevant or insignificant for the development of science. However, even few published results concerning inactive compounds may provide valuable information for the use of computer-aided drug design.[159] Osolodkin et al.[160] carried out phenotypic screening of a series of spiro-annulated oxepanes and azepenes against a broad spectrum of viruses, including Puumala virus causing HFRS. It was shown that these compounds inhibit the reproduction of DNA virus (C5 adenovirus) and are inactive against RNA viruses.
8. Conclusion
The present review is devoted to analysis of published data on small-molecule compounds that show activity against viruses causing hemorrhagic fevers encountered in Russia (Omsk hemorrhagic fever, Crimean-Congo hemorrhagic fever, and hemorrhagic fever with renal syndrome). The data available from the literature provide the conclusion that only a few compounds of this type are currently known, and the search for new drugs for the prevention and treatment of these diseases is quite a relevant task. For all three types of hemorrhagic fever, no medications with effective antiviral action have been developed so far. Most studies dealing with the development of these medications are limited to nucleoside analogues or known drugs that have proved themselves as antiviral agents. Due to the great pathogenicity of these viruses, it is difficult to arrange a large-scale search for active agents among numerous structurally diverse compounds. Today, such studies are very rare and, therefore, they are highly valuable. Of particular note is the detected considerable activity against the Hantaan virus in 2-arylbenzotriazoles 80 – 82 and in fenchone derivative 84.
The key goal of chemists engaged in the modification of natural compounds is the understanding of the structure–activity relationships, the principles of which are demonstrated in this review using particular examples. The relationship between the chemical structure and activity can be unambiguously established only using specific studies including a broad variation of the structures of compounds. The characteristics of the antiviral activity (IC50 values) reported in the literature can be correctly compared only within a single study. Even for one and the same compound, these values can vary over broad limits depending on the method of determination and the cell line used. Further accumulation of experimental data should help to solve this problem. The modern animal models, including those using non-human primates, provide prospects for this understanding. It is noteworthy that there is insufficient number of publications addressing the mechanisms of action of small molecules active against viruses that cause hemorrhagic fevers. In conclusion, it should be emphasized that pronounced antiviral effects, including the effect against HFRS-causing viruses, have been found in various natural compounds. It can be assumed that particularly these compounds would be used as scaffolds in the design of new promising drugs for the treatment of hemorrhagic fevers encountered in Russia.
9. List of acronyms
The following acronyms are used:
CCHF — Crimean-Congo hemorrhagic fever;
FDA — Food and Drug Administration;
HFRS — hemorrhagic fever with renal syndrome;
IC50 — half maximal inhibitory concentration;
OHFV — Omsk hemorrhagic fever virus;
SI — selectivity index.
References























