



Keywords
Abstract
Ligand design is a critical element in homogeneous catalysis. Over the past two decades, a variety of selenium-substituted compounds have been used as ligand scaffolds in the development of transition metal catalytic systems. Air and moisture stability, good solubility in various solvents, stability towards oxidation and excellent electron-donating properties of the selenium atom are some of the key features of organoselenium compounds that make them suitable candidates for application as ligands. The metal complexes of selenium ligands are widely known for their ability to catalyze various organic reactions, including but not limited to the Sonogashira coupling reaction, Heck coupling reaction, Suzuki–Miyaura coupling reaction, imidazole arylation, transfer hydrogenation reaction, oxidation of alcohols, and ortho-arylation of phenols. In some cases, exceptionally high catalytic efficiencies have been observed. In addition, catalysis often allows for a wide range of substrate in numerous scenarios, as it is tolerant of different functional groups. This review highlights the main synthetic methods used in the recent development of organoselenium compounds as ligands for the synthesis of transition metal complexes. It also considers their use in various preparative organic transformations.The bibliography includes 133 references.
1. Introduction
Although organoselenium compounds have been known for over 150 years, they have been largely overlooked for a long time due to their unpleasant odour and unstable properties. The toxic properties of organoselenium reagents were discovered in the 1930’s, but the early 1970’s saw a sharp boost in the chemistry of organoselenium compounds, spurred on by the recognition of their utility in organic synthesis and chemical biology.[1-7] Since then, a considerable pool of organoselenium compounds has been synthesized for various applications in research fields.[8-12] A variety of organoselenium reagents have been introduced into synthetic practice, leading to the description of chemo-, regio-, stereoselective, and stereospecific reactions [13-16] in various synthetic transformations, including selenenylations, selenocyclizations, selenoxide eliminations and 2,3-sigmatropic rearrangements.[17-20] Numerous publications, including books,[21-23] book chapters [24-30] and review articles [12, 31-38] have appeared recently, providing comprehensive coverage of various facets of organoselenium chemistry. To date, organoselenium reagents have received particular attention, mainly due to their utilization as ligands in metal complexes for catalytic processes.
The proper selection of the organic ligand is a key step in the design and synthesis of transition metal complexes with the desired properties necessary for the application in catalytic processes. The choice of ligand influences the selectivity of the reaction in the case when more than one product is formed, the solubility of the metal complex as well as the catalyst lifetime. Phosphine ligands are among the most widely used organic compounds to modulate the chemical reactivity of palladium and other transition metal-based catalysts. However, their use is subject to the several limitations, such as toxic environments, drastic reaction conditions and high levels of air and moisture instability. All of these circumstances have limited their use in large-scale synthesis and have prompted the search for other non-phosphine-based ligands among the various classes of compounds as viable alternatives. Various organoselenium compounds have been used as basic components in the preparation of numerous transition metal complexes.[39-41] These complexes are catalysts for various chemical reactions. Such ligands and their complexes have many characteristics that make them suitable candidates for application in the field of catalysis. Air and moisture stability,[42, 43] good solubility in various solvents, oxidation stability and excellent electron-donating properties of the selenium atom are among the most important ones.[40, 44] On the other hand, the availability of commercial precursors for the synthesis of the desired organoselenium ligands, the mild reaction conditions, and the possibility to modify their structural backbones by standard chemical transformation make them a convenient and suitable choice in the field of ligand development.[45, 46] Different methods have been used to obtain organoselenium ligands. One method is nucleophilic substitution, where selenolates react with organic halides.[47-50] Another approach involves addition-elimination reactions to afford imine-substituted ligands, which are further reduced to give secondary amines.[51, 52] To date, the most exploited classes of organoselenium ligands in the field of organocatalysis are organic selenides, selenocarbonyls, imine-substituted organoselenium compounds, N-heterocyclic carbenes (NHC) and pincer compounds, which have more than one donor atom in a molecular backbone. The fact that organoselenium ligands have proved to be convenient building blocks for the preparation of different types of metal complexes (Pt, Pd, Ru, etc.) can be attributed to the excellent polarizability of the selenium atom as a cause of strong bonding and interactions with metal atoms.[53-55] In addition, selenium possesses unique chemical properties, such as their ability to act either as a non-metallic element or as a metalloid under certain circumstances and the tendency to form intramolecular Se∙∙∙H interactions.[56, 57]
The first examples of catalytic systems involving organoselenium compounds used in different coupling reactions were related to the development of homogeneous catalytic systems used in different coupling reactions with high efficiency.[40] The major drawbacks of using homogeneous catalysts are high cost, waste by-product and difficulty in separating the product from the catalyst. These disadvantages have led to the development of other organoselenium-based catalytic systems, mainly heterogeneous and nanocatalytic systems. Today, heterogeneous catalytic systems are a well-recognized concept due to their recyclability and easy separation of the products. The use of organoselenium compounds in heterogeneous catalysis is based on immobilization of preformed ligands or complexes on solid supports (polymers, carbon, silica, metal oxides). Given the high toxicity and cost of palladium, the development of methods for its recovery is becoming one of the main challenges in the field of catalysis. On the other hand, in nanocatalytic systems, organoselenium compounds have been used as stabilizers to allow uniform dispersion of nanoparticles.[58-63]
The purpose of this review is to present the recent syntheses of organoselenium compounds as ligands for the synthesis of transition metal complexes. Their application in various preparative organic transformations is also included with particular emphasis on cross-coupling synthetic strategies for the formation of C – C and C – O bonds. Advances in the field of selenium-ligated metal catalysis for the formation of C – C and C – O bonds over the past last 10 years is highlighted.
2. C – C Coupling reactions
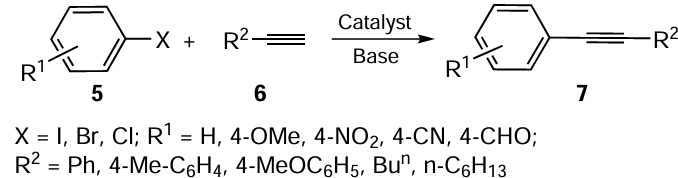
The construction of a carbon–carbon bond is an extremely important task in organic chemistry, considering that the design and synthesis of many molecular structures require the formation of C – C bonds (preferably biaryls), which are considered as privileged scaffolds in the development of pharmaceuticals, agrochemicals, materials, and various other chemical products.[64] Transition metal-catalyzed C – C coupling reactions developed by Suzuki,[65] Heck [66] and Sonogashira [67] have become the gold standard for the efficient and straightforward construction of carbon-carbon bonds.
2.1. Sonogashira reaction
The formation of the C – C bond in Sonogashira coupling involves the reaction between a terminal alkyne and an aryl/alkenyl halide in the presence of a palladium catalyst and a copper-based co-catalyst using an amine as the base. These reactions have generally been carried out under anhydrous and anaerobic conditions. The careful selection of organoselenium compounds as building blocks of the catalytic systems used in Sonogashira coupling overcomes some of the limitations of this reaction protocol. The advantages are often reflected in the mild reaction conditions, the acceptable scope of the reaction, no need for a co-catalyst and, in some cases, the possibility of the catalyst reuse.
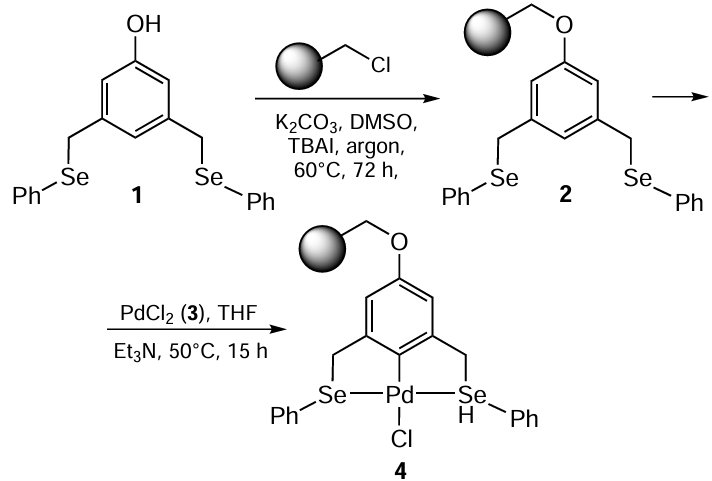
Mohammadi and Movassagh [68] prepared palladium complex 4 from the polystyrene-supported SeCSe pincer ligand 2. Ligand 2 was obtained by a four-step procedure, in which the OH moiety was introduced in the molecule prior to binding to the solid support. The functionalized polymer beads were prepared by stirring 3,5-bis((phenylselanyl)methyl)phenol 1 with chloromethylated polystyrene resin in the presence of K2CO3 and tetra-n-butylammonium iodide (TBAI) at 60°C under an inert atmosphere (Scheme 1). The resulting catalyst 4 was further used in the Sonogashira coupling reaction. Various aryl halides 5 (1 equiv.) react with aliphatic and aromatic terminal alkynes 6 (2 equiv.) on using 2 equiv. of tetra n-butylammonium fluoride (TBAF) as a base and 0.3 mol.% of catalyst 4 in 1 mL of NMP at 70°C (Scheme 2). Although similar catalytic system had previously been used in various cross-coupling reactions, this was the first example of a copper-free Sonogashira reaction. The method was particularly effective for aryl iodides. Eighteen compounds bearing both electron-rich and electron-poor substituents gave the corresponding products in high yields (up to 95%) within 0.25 – 3 h of the reaction time. 1,2-Disubstituted acetylenes 7 were also obtained in moderate to high yields from various chlorides and bromides with low catalyst loading (0.3 mol.%). The use of β-bromostyrene in this transformation provided a stereocontrolled route for the 1,3-enynes with excellent E/Z ratios.
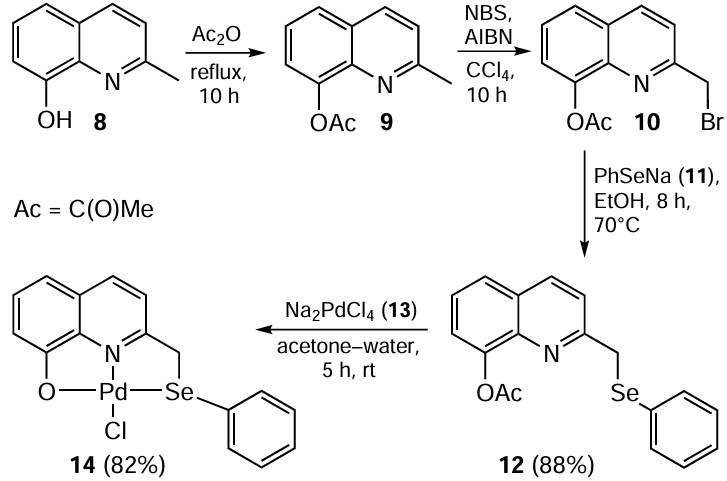
Based on the unsymmetrical quinoline-based pincer (O−,N,Se) ligand, Kumar et al.[69] prepared the palladium complex 14 used in Sonogashira coupling, which was both amine- and copper-free. The idea behind the research was to combine different hard and soft donor sites in a ligand molecule, which could potentially affect the hemilabile structure of the ligand and facilitate its oxidative addition to the metal centre. The key starting material for the preparation of ligand 12 was 2-(bromomethyl)quinolin-8-yl acetate 10, obtained by acetylation of 2-(hydroxymethyl)-quinolin-8-ol 8 followed by treatment with N-bromosuccinimide (NBS) in the presence of azobis(isobutyronitrile) AIBN in CCl4 (Scheme 3). Ligand 12 was obtained in 88% yield by the reaction of compound 10 with in situ generated PhSeNa (11). A pale-yellow palladium complex 14 was then prepared by stirring a solution of ligand 12 in acetone and Na2PdCl4 (13) in water. The successful coupling of an aryl halide with a terminal alkene in the presence of catalyst 14 gave disubstituted acetylene in up to 90% yield under the conditions involving 0.5 – 1.0 mol.% catalytic loading, using K2CO3 as base and DMF as solvent at 90°C. The yields of the coupled products were slightly lower in the case of aryl chlorides compared to those obtained with aryl bromides or iodides. A total of 11 reactions were successfully completed in 6 – 15 h with aryl halides substituted with different electron-donating and electron-withdrawing moieties. The reaction was also carried out with palladium complex of quinone-based sulfur-containing ligand. It was reported that catalytic activity of selenium-containing palladium complexes was slightly better than that of the sulfur-based ligands.
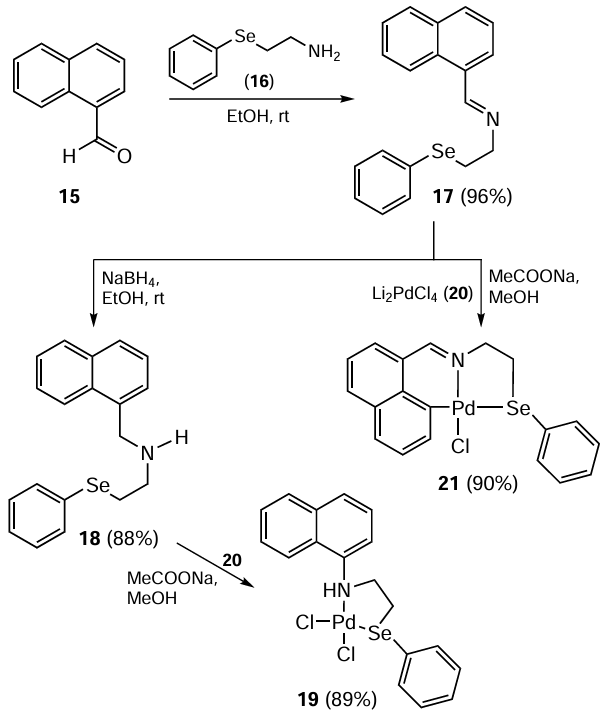
Bhaskar et al.[70] have reported an interesting research work on the application of palladium complexes based on organoselenium ligands in both Suzuki – Miyaura (SMC) and Sonogashira coupling reactions of aryl halides in the absence of Cu and amine. For this purpose, the air- and moisture-insensitive Schiff base 17 prepared from 1-naphthaldehyde 15 and 2-(phenylthio/seleno)ethylamine 16 as well as its reduced form 18 were explored as ligands. The reaction of the ligands 17, 18 with NaOAc and Li2PdCl4 (20) at room temperature yields an unsymmetric (C‾, N, Se) pincer 21 and bidentate (N, Se) 19 complexes (Scheme 4). For the activated substrates, such as p-bromobenzonitrile, p-bromoacetophenone, p-bromobenzaldehyde and p-bromonitrobenzene, the catalytic loading of 0.05 mol.% was efficient for the synthesis of the coupled products 7 (see Scheme 2) in 1 hour and in up to 99% yield. For the deactivated substrates, the yields were significantly lower with the possibility of its improvement after prolonging the reaction time and using a catalytic loading of 0.1 mol.%. The formation of palladium-containing nanoparticles (NPs) of ~ 2 – 7 nm size was observed during these coupling reactions. This work is the first example to describe the in situ formation of palladium-containing nanoparticles during a coupling reaction and their role in catalysis. The nanoparticles were able to independently catalyze the coupling reaction.
In 2017, Dubey et al.[71] reported the synthesis of trinuclear Pd(II) complex 27 by reacting 3-methyl-1-(2-(selanyl)ethyl)-1H-benzo[d ]imidazol-3-ium iodide 25 with Ag2O followed by treatment with Pd(MeCN)2Cl2 (26) in a 3 : 2 metal-to-ligand ratio, i.e. transmetallation (Scheme 5).
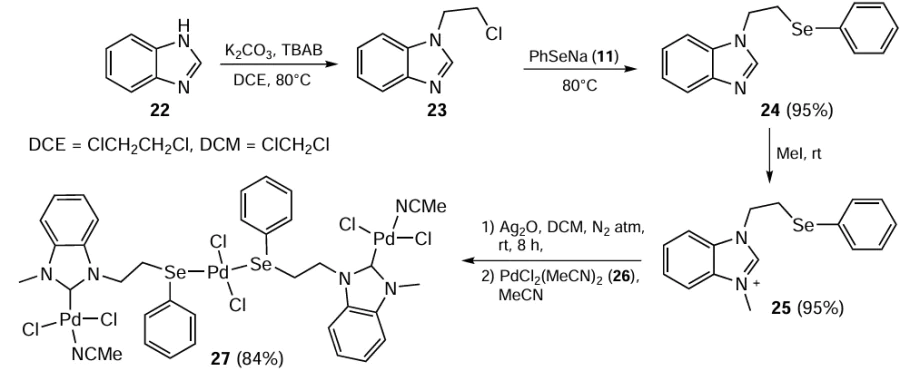
The ligand 25, which serves as a precursor to the selenium-substituted NHC, was synthesized by first reacting benzimidazole 22 with 1,2-dichloroethane, and then treating the resulting chloroethyl derivative 23 with PhSeNa (11) and methyl iodide. Palladium complex 27 was used as a catalyst for the interconversion of nitriles and amides as well as for carrying out the amine-free Sonogashira coupling reaction promoted by CuI. Although the sulfur ligand-based complex has proved to be a more effective catalyst, the use of the organoselenium-ligated complex showed that the developed methodology has a high level of tolerance to C – C coupling. The reaction worked well with an optimum loading of 1.0 mol.% of organoselenium-based complex in the presence of K2CO3 and CuI (5 mol.%) in DMSO under a N2 atmosphere at 110°C. The reaction was completed within 8 h giving up to 82% yield of the product. The aryl bromides and iodides reacted readily, especially in the case of the activated examples, but the yields of the coupled product obtained using aryl chlorides were satisfactory.
Due to the colour change of the Sonogashira coupling reaction mixture to black, the formation of colloidal Pd(0) in the reaction was proposed. Mercury and triphenylphosphine poisoning tests were performed to know whether the discrete Pd(0) or Pd(0) cluster is the catalytic species in the Sonogashira coupling reaction. The negative results of the test indicated that Pd(0) was the actual catalyst. The ESI-MS peak at m/z = 581.24 for complex I was concluded to be the most plausible discrete Pd(0) species driving the Sonogashira coupling and the peak in the spectrum of II to be its transmetallation product. Although complex 27 does not activate aryl chloride efficiently, it is still much better than the ferrocene-based Pd complex and the imidazole-based Pd complexes of N-heterocyclic carbenes (NHCs).
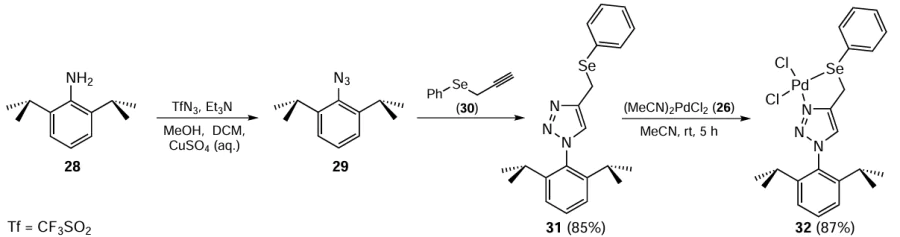
In 2016, Singh and co-workers [72] reported the synthesis of Pd complex 32 and further tested its catalytic potential in the SMC and Sonogashira coupling reactions. Ligand 31 was obtained by reaction of 2-azido-1,3-diisopropylbenzene 29 with propargyl phenyl selenide 30. Subsequent treatment of ligand 31 with Pd(MeCN)2Cl2 (26) at room temperature for 5 h afforded the Pd(II) complex 32 in 87% yield (Scheme 6). Complex 32 was found to be effective in promoting couplings for a wide range of aryl bromides, even for deactivated ones. In the case of Sonogashira coupling, the product was obtained in up to 99% yield when the catalyst 32 was used in the range of 0.001 to 2.0 mol.% and CuI was used as a co-catalyst (2 – 4 mol.%) with K2CO3 as the base. For deactivated aryl bromides, such as p-bromoanisole and p-bromotoluene, the product was obtained in much lower yields. Similar sulfur-ligated palladium complexes were also synthesized. The comparative study showed that such complexes performed better than catalyst 32 in both the SMC and Sonogashira coupling reactions.
Bhanage and co-workers [73] carried out the copper- and phosphine-free Sonogashira reaction catalyzed by palladium(II) complexes such as [PdCl(SCH2CH2NMe2)]3, [PdCl(SCH2CH2CH2NMe2)]2, [PdCl(SeCH2CH2NMe2)]3, [PdCl(SeCH2CH2CH2NMe2)]2, and PdCl(SeCH2CH2NMe2)(PPh3)], which gave coupling products in moderate to good yields under aerobic conditions. The investigation showed that increasing the catalyst concentration from 2.0 to 3.0 mol.% resulted in an increase in the expected product yield. However, further increases in catalyst concentration did not significantly affect the product yield. 1,4-Dioxane was the solvent of choice for the reaction and of the various bases tested, Et3N gave product in high yield.
2.2. Heck reaction
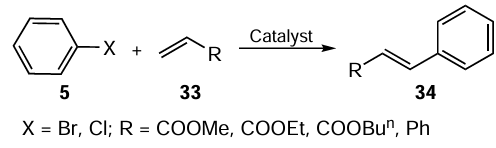
Organoselenium compounds have also found application in one of the most important coupling reactions for the C – C bond formation, the Heck – Mizoroki reaction, as an excellent alternative to the homogeneous catalytic system based on air- and moisture-sensitive phosphine ligands (Scheme 7). As previously discussed, the recently developed methodologies had implied either the design and synthesis of heterogeneous catalysts immobilized on different solid supports, or the selenated-NHC pincer complex as a superior option.
Rangraz et al.[74] have developed an organoselenium-based palladium complex that is stable in both air and moisture. This complex is immobilized on silica-coated Fe3O4 magnetic nanoparticles (MNPs) 36, as shown in Scheme 8. Notably, this study represents the pioneering achievement of successfully grafting the selenotetrazole ligand onto the surface of MNPs. The synthesis of catalyst 43 involves several steps. First, Fe3O4 MNPs 35 are prepared using a chemical co-precipitation technique. These nanoparticles are then coated with a thin layer of silica via the Stöber method to prevent their aggregation, resulting in silica-coated magnetic nanoparticles 36. Subsequently, functionalization of the silica-coated MNPs is carried out using (3-aminopropyl)triethoxysilane (APTES) to yield Fe3O4@SiO2-APTES 37. At the same time, the selenotetrazole ligand 41 is synthesized by converting p-aminobenzoic acid 38 into a diazonium salt, which is then reacted with KSeCN to produce phenylselenocyanate 39. Finally, tetrazole 40 was obtained by reacting phenylselenocyanate 39 with sodium azide. Finally, the nanoparticles 41 containing Se and N as chelating groups on their surface are metallated with Pd(OAc)2 42 in ethanol. The desired Pd complex 43 supported on the nanoparticles designated as Fe3O4@SiO2 – Se-T/Pd(II) was further used as a catalyst in the Heck– Mizoroki cross-coupling reaction.
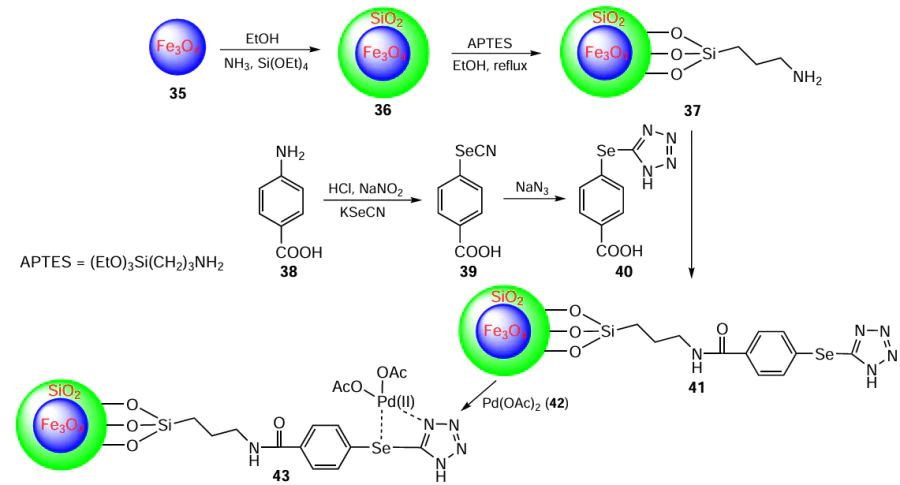
The plausible reaction mechanism involves four steps. In the first step, Pd(II) is reduced to its active form Pd(0) to initiate the reaction (Scheme 9). In the next step, the Pd(0) species undergoes oxidative addition with the aryl halide 5, where the palladium atom is inserted into the carbon-halogen bond. This step produces an intermediate species 44, which is a σ-arylpalladium complex. The palladium(II) atom of intermediate 44 reacts with acrylate 33 to form a π-complex. The acrylate is then inserted into the palladium–carbon bond via a syn-addition to give a new complex 45. The next step is a β-hydride elimination, which involves the abstraction of a hydrogen atom from the β-position of complex 45. Finally, the resultant complex decomposes, releasing the desired coupling product 34. The catalytic cycle is completed by regeneration of the active Pd(0) species by base-assisted reductive elimination of the palladium(II) compound.

A number of different products 34 (12 examples) were obtained by C – C bond formation by reaction of aryl halides 5 with acrylates 33 in the presence of catalyst 43 within 6 h achieving yields up to 98%. The advantage of the developed methodology is reflected in the reusability of the heterogeneous palladium complex without significant loss of activity or its degradation. It was found that aryl chlorides and bromides, as examples of inexpensive substrates, also react smoothly with terminal alkynes, and 1,3-enynes could be prepared in a stereoselective manner.
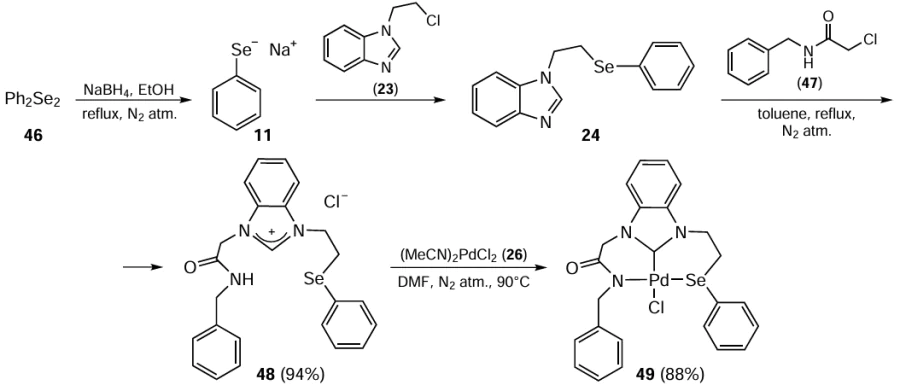
Excellent catalytic activity in the Mizoroki – Heck reaction of aromatic bromides/chlorides was also demonstrated by the palladium complex 49.[75] This was the first example of the selenium-containing NHC pincer ligand used for screening the coordination behaviour and catalytic activity. The synthesis of this ligand starts with the reduction of diphenyl diselenide 46 with sodium borohydride followed by the reaction with 2-(1-chloroethyl)-1H-benzo[d ]imidazole 23 to give 1-(2-(phenylselanyl)ethyl)-1H-benzimidazole 24 (Scheme 10). Further reaction of N-benzyl-2-chloroacetamide 47 with compound 24 afforded the selenium-NHC pincer ligand 48 in excellent yield. Finally, the reaction of the NHC ligand 48 with PdCl2 26 afforded complex 49. The results of the Mizoroki – Heck coupling reactions using the Pd complex 49 were quite encouraging. The developed procedure uses a low concentration of catalyst Pd(II)-NHC 49 (0.2 mol.%), water as solvent in the presence of K3PO4 and TBAB at 110°C. The catalyst was found to be potentially active for both the electron-donating and electron-withdrawing substituents, giving very high yields of coupling products within 12 h. Up to 94% yield was obtained when aryl/heteroaryl bromides 5 were reacted with alkenes 33. It was observed that the use of aryl chlorides 5 reduced the product yield to 70% (see Scheme 10). It has been suggested that the palladium-selenium NPs (Se : Pd ratio is 2 : 7) formed during the reaction act as a catalytic palladium species.
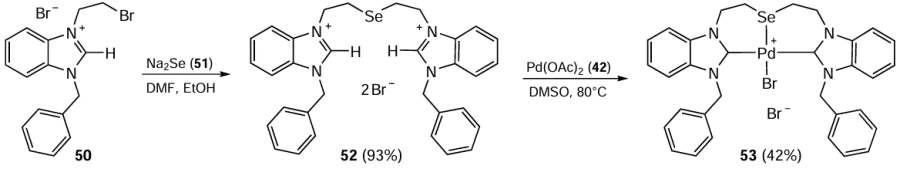
Rishu et al.[76] presented the first example of a carbene ligand with selenoether bridge (L) 52 and used it to prepare the pincer ionic complex [PdBr(L-κ3CSeC)]Br 53. 1-Benzyl-3-bromoethylbenzimidazolium bromide 50, the building block of the ligand 52, was obtained by reaction of 1-benzyl-1H-benzimidazole with 1,2-dibromoethane. Further nucleophilic substitution in compound 50 with the selenide ion afforded selenoether-bridged bis-benzimidazolium dibromide 52. The reaction of ligand 52 with an equimolar amount of Pd(OAc)2 42 at 80°C in DMSO afforded a pincer complex [PdBr(L-κ3CSeC)]Br 53, in which the backbone Se atom was coordinated to Pd(II) (Scheme 11). The idea behind the research was to investigate the effect of the soft donor Se atom in the ligand molecule on the C – C bond formation in the mono-Heck reaction of aryl bromides 5 with methyl acrylate 33. The reaction worked well in the presence of 0.7 mol.% of Pd complex 53 and Na2CO3 as base in DMA at 140°C. In addition to the smooth formation of mono-coupled products in moderate yields, the bis-arylation products were observed in some cases. The developed procedure did not require the use of excess substrate or additive, and the catalytic loading was lower compared to some previously published methods.
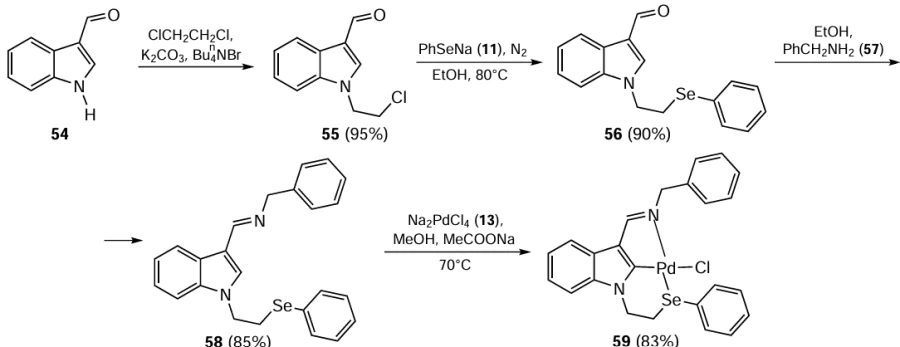
Singh et al.[77] pioneered the synthesis of asymmetric (N,C,Se)-type pincer ligand precursors based on an indole core.[77] The synthetic procedure involves the preparation of 1-(2-chloroethyl)-1H-indole-3-carbaldehyde 55 by reaction of indole-3-carboxaldehyde 54 with 1,2-dichloroethane (Scheme 12). To obtain good results, K2CO3 and Bu4NBr were essential for the reaction. Subsequently, the nucleophile PhSe− 11, generated in situ by reducing diphenyl diselenide 46 with NaBH4 , was reacted with compound 55 to give the selenium-substituted indole carboxaldehyde 56. The resulting aldehyde 56 was condensed with benzylamine 57 to afford (N,C,Se)-type pincer ligand precursors 58 in 85% yield. The Pd complex 59 was then obtained by reaction of ligand 58 with Na2PdCl4 13 in methanol in the presence of sodium acetate. A similar Pd complex with an indole-based sulfur-containing ligand was also prepared. Both complexes with sulfur and selenium ligands showed good results when used for Heck coupling. The optimized reaction conditions include 0.1 mol.% of catalyst in the presence of K2CO3 base and N,N-dimethylacetamide (DMA) as solvent to give 95% of the product.
2.3. Suzuki – Miyaura reaction
The Suzuki – Miyaura coupling reaction is one of the key C – C bond formation reactions in synthetic organic chemistry. It involves the coupling of nucleophilic and electrophilic partners in the presence of a transition metal catalyst, in particular the palladium catalyst. Typically, boronic acids, esters and trifluoroborate salts have been used as nucleophilic coupling partners while aryl/vinyl halides and triflates have been used as electrophilic coupling partners in these reactions. A general scheme for the Suzuki – Miyaura coupling reaction is shown in Scheme 13. Various selenium-ligated transition metal complexes have been successfully used to catalyze the Suzuki – Miyaura coupling reaction in good yields. Interesting features of these complexes are their catalytic activity in very low loadings, and their reusability gives additional value to these complexes. These reactions showed good tolerance to different electron-donating and withdrawing functionalities in the aromatic rings.
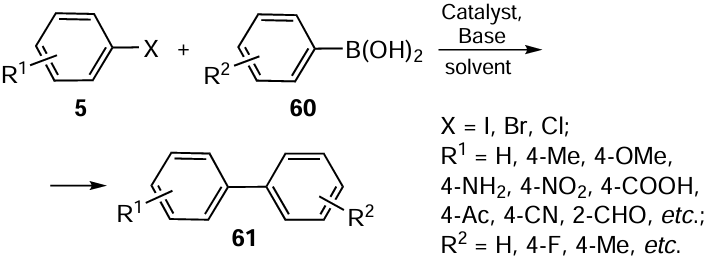
In recent years, intensive scientific efforts have been made to develop more environmentally friendly methods. The catalyst 49 obtained by Sharma et al.[75] for use in the Mizoroki – Heck reaction was also employed in the Suzuki – Miyaura cross-coupling in water selected as the best alternative to other organic solvents. In contrast to aryl chlorides 5 and phenylboronic acid 60, which didn’t undergo coupling, switching to the aryl bromides provided up to 94% yields of coupled products. The process required only 0.01 mol.% of catalyst and was completed in less than 2 h under mild conditions including K2CO3 as base and water as solvent at 5°C (see Scheme 13).
Rangraz et al.[78] have prepared the palladium(II) complex 65 with Fe3O4-supported organoselenium ligand modified by SiO2/azidopropyltrimethoxy silane (Scheme 14). The synthesis of Pd(II) complex 65 starts with the preparation of magnetic Fe3O4 NPs 35 and their subsequent coating with silica to give NPs 36, which is carried out as shown in Scheme 8. These silica-coated nanoparticles are then subjected to post-grafting to introduce 3-azidopropyltrimethoxy silane to afford compound 62. A click reaction between the azide-functionalized magnetic nanoparticles and 2-chloroacetonitrile was used to construct the tetrazole ring in compound 63. The introduction of the selenoether is achieved by the reaction of the PhSe‒ 11 anion with compound 63 to give the ligand 64. Finally, the magnetically recoverable nanoparticles containing chelating selenium and nitrogen atoms on their surface react with palladium acetate 42 in ethanol at 80°C. This process affords the desired Pd complex 65 attached to the nanoparticles as shown in Scheme 14. This is the first example of magnetic solid-supported metal complexes based on organoselenium ligands. These complexes have been used in SMC reactions to facilitate the synthesis of various biaryls 61 from aryl halides 5 and arylboronic acids 60 (see Scheme 13). The high tolerance to the wide range of the functionalities has been observed under very mild reaction conditions. A total of 13 biaryl products 61 were obtained in a yield of up to 95% when aryl halides 5 substituted with various electron-withdrawing and electron-donating groups were subjected to the coupling reaction in the presence of palladium complex catalyst 65 using K2CO3 as a base and the mixture of ethanol and water as the solvent at 60°C within 2 hours. In addition, the immobilization of the ligand has provided good recyclability and easy recovery and the catalyst could be used continuously for seven runs with negligible loss of activity.
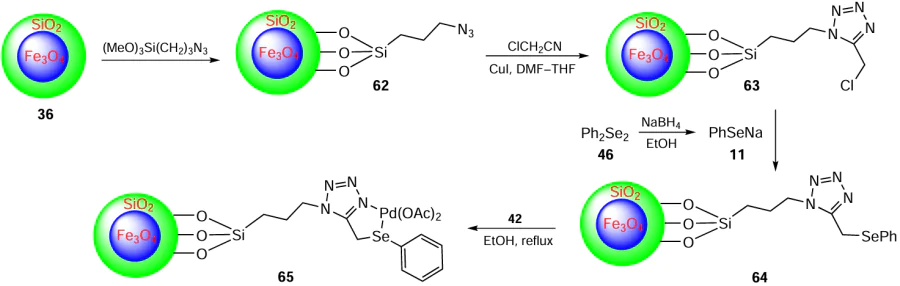
The proposed coupling reaction pathway involves the in situ formation of Pd(0) by reduction of Pd(II) in the presence of phenylboronic acid and a base. The oxidative addition of aryl halide to Pd(0) then generates a σ-arylpalladium intermediate 66, which is the rate-determining step involving oxidation of Pd(0) to Pd(II). Next, the phenyl group from phenylboronic acid replaces the halide via transmetallation to give the Pd(II) diaryl complex 67. Finally, reductive elimination yields the coupled product 61 together with regeneration of Pd(0) and completion of the catalytic cycle (Scheme 15).
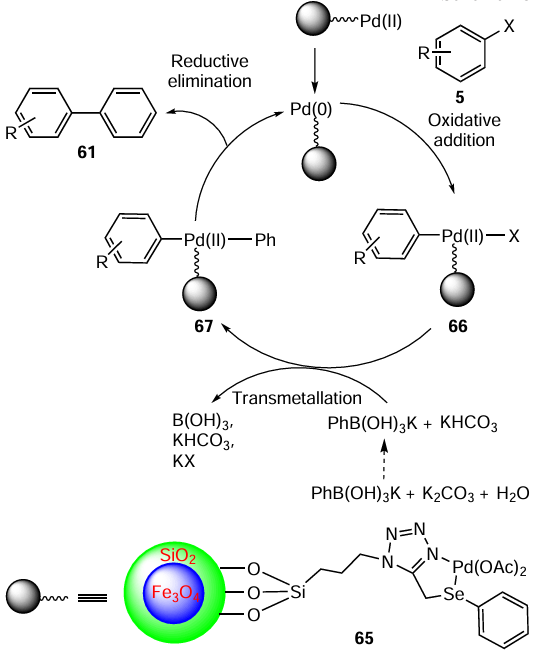
Sharma et al.[79] used selenium and sulfur-substituted secondary amines 71 as stabilizers for the palladium nanoparticles 72 that were used as catalysts in the Suziki – Miyaura coupling reaction. The organoselenium-based ligand 71 was obtained by the reduction of the corresponding imines 70 with NaBH4 (Scheme 16). The study showed that Pd NPs 72 stabilized by organosulfur-based ligands (Pd : L ratio 1 : 1) performed better than their organoselenium analogues except in the case of the reaction with 4-bromobenzaldehyde where the catalytic performances of all tested systems were excellent with 99% yield on using 0.1 mol.% catalysts in aqueous DMF. This was explained by the greater electron-donating ability of the sulfur atom in contrast to Se and the combination of sulfur and nitrogen donor sites to achieve NP stabilization.
The same research group [80] has presented efficient magnetically retrievable Fe3O4@SiO2@ – SePh@Pd(0) nanoparticles 74 as a catalyst for the C – C/C – O coupling reaction in water. For the preparation of the NPs, silica-coated Fe3O4 36 was used, which was treated with PhSeCl to afford compound 73 and its further reaction with PdCl2 3 gave Fe3O4@SiO2@ – SePh@Pd(0) NPs 74 (Scheme 17). The developed protocol has the advantages for the Suzuki – Miyaura coupling such as an easy separation of the catalyst and the simple experimental procedure. The C – C coupling reaction ran excellently within 2 h with an optimum loading of 0.01 – 1.0 (in mol.% of Pd) of 74 in the presence of water as solvent and K2CO3 as base at 80°C. Regarding the influence of the substituents in the benzene ring of ArX, it was found that higher yields were obtained with aryl halides bearing electron-withdrawing groups, especially those in the para position.
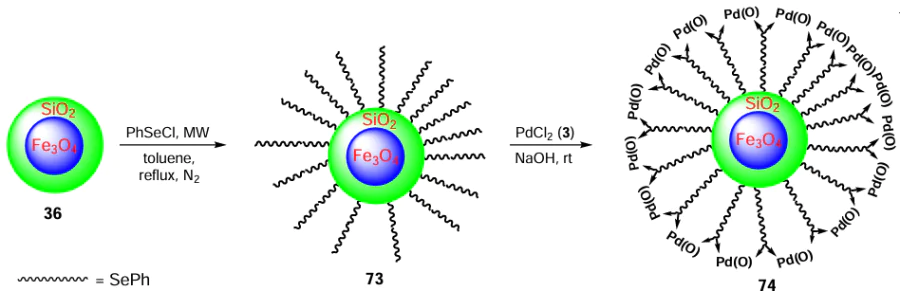
In 2019, Oswal et al.[81] synthesized organoselenium/sulfur ligand containing an anthracene core and an amine moiety and used it to stabilize the ultra-small palladium nanoparticles 78 (size 1 – 6 nm). The ligand 77 was prepared by reacting anthracene-9-carbaldehyde 75 with 2-(phenylselanyl)ethan-1-amine 16 to give imine 76, which was reduced with NaBH4 to give the desired product 77. The Pd(0) NPs 78 were further obtained by the reaction of the ligand 77 with Na2PdCl4 13 (Scheme 18). The resulting NPs have shown good catalytic activity in the Suzuki – Miyaura coupling of phenyl boronic acid 60 with various aryl halides 5 along with the potential for recyclability. From this study, it is evident that the NPs stabilized by the secondary amine-containing sulfur ligand are more catalytically efficient as compared to its selenium analogue. The high catalytic activity is attributed to the relatively small particle size and high dispersion homogeneity. This study has also revealed that the catalytic activity was strongly influenced by the ligand/metal ratio of the NPs. Among the selenium-based species, the best catalytic activity was observed a the ratio of 1 : 4 (ligand: Pd), which was due to the good dispersion of very small NPs.
N,N-Diphenylacetamide-based selenoether ligand ((Ph2NCOCH2)2Se) 81 was used to prepare air and moisture-stable complex [((Ph2NCOCH2)2Se)2PdCl2] 82 by reaction with Na2PdCl4 13. Further, the complex 82 was subjected to thermolysis with tri-n-octylphosphine (TOP) to give Pd17Se15 nanoparticles 83 as shown in Scheme 19.[82] Both the nanoparticles 83 and the complex 82 were tested for their activity in C – C/C – O coupling reactions. The complex 82 was shown to be more catalytically active than the nanoparticles derived therefrom. For optimal results, 0.0001 – 0.01 mol.% of Pd catalyst 82 was required along with the use of K2CO3 in aqueous DMF at 100°C. The good results were reflected in high yields (up to 96%) and short reaction times (< 3 h), which were achieved with aryl bromides bearing electron-withdrawing groups. When aryl chlorides were used, very poor or no conversion was observed in some cases. The recyclability of complex 82 was also investigated, which showed that the catalyst could be reused for two cycles with the yield decreasing in the third and fourth cycles. Palladium–sulfur complexes were also synthesized by a similar procedure and showed comparable efficiencies to complex 78 in catalyzing the Suzuki – Miyaura reaction.

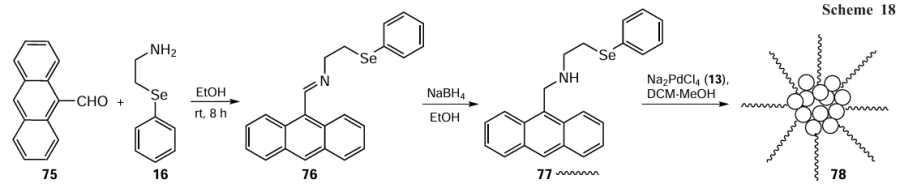
The first example of ferrocene-based organoselenium Schiff base used in the synthesis of cyclopalladated complex 86 was reported by Sharma et al.[83] in 2017. Ligand 85 was obtained by the reaction of ferrocene-carboxaldehyde 84 with 2-(phenylselanyl)ethylamine 16. Furthermore, the reaction of ligand 85 with Na2PdCl4 13 in the presence of sodium acetate gave cyclopalladated complexes 86 (Scheme 20). Good aryl bromide conversions were observed, with a reaction time of 6 h, catalyst’s loading of 0.01 mol.% and turnover number (TON) values up to 9300 (TOF = 3100 h–1). The catalytic activity of complex 86 was reported to be higher than that of the complex with the corresponding thiol-based ligand.
Gonzalez et al.[84] obtained novel ferrocenylated chalcogen (Se and Te)-containing imidazolium salts. These imidazolium salts were used as starting materials for the synthesis of ferrocenyl-NHC selenones, ionic palladium(II) complexes and silver NHC complexes, in which the imidazolium ligand acted as the cation. The silver carbene transfer reaction with the bidentate (Se, CNHC) coordination of the ligand was used to synthesize the Pd NHC(Se, CNHC) complex from the appropriate imidazolium salts.
The Pd(II) complex 32 of the organoselenium ligand 31 (see Scheme 6) synthesized by Singh and co-workers [71] was also investigated in the SMC reactions. The 0.01 mol.% of Pd(II) complex 32 efficiently catalyzed the Suzuki – Miyaura coupling reaction in water. The product yield was 99% after 12 h. It was noted that the reaction was faster when using Pd(II) complex with organosulfur ligand compared to the organoselenium ligand.
Singh and co-workers [85] reported the synthesis of palladium(II) complexes of pyrazolated selenoethers. The reaction of PhSeNa 11 with 4-bromo-1-(2-chloroethyl)-1H-pyrazole 87 gave selenoether ligands 88 and 90 (Scheme 21). These ligands were then used to synthesize [PdLCl2] 89 and [PdL2Cl]BF4 91 complexes by the reaction with bis(acetonitrile)palladium(II) dichloride 26 in acetonitrile at 70°C. The catalytic performances of the resulting complexes, which have the advantage of stability under normal environmental conditions, were investigated in the Suzuki – Miyaura coupling reaction. Complexes 89 and 91 showed significant catalytic activity towards various aryl bromides, including electron-rich ones. The reduced amount of catalyst (∼ 0.01 mol.%) proved to be effective, in several cases achieving significant conversions within a reaction time of 2 h. The highest yield (96%) was obtained with p-bromobenzonitrile and p-nitrobromobenzene, particularly with complex 91 as the catalyst. It was observed that this complex underwent in situ formation of Pd4Se and PdSe nanoparticles. However, the isolated nanoparticles did not appear to be very active compared to the NPs generated in situ and it was observed that a higher amount of isolated NPs was required for comparable conversion rates compared to complexes 89 and 91. It was therefore concluded that the nanoparticles formed by complexes 89 and 91 are the actual catalysts and that switching from organoselenium to the sulphur-containing ligand does not make much difference.
Jain and co-workers [86] reported the Suzuki coupling in the presence of [PdCl(SeCH2CH2NMe2)]3 , [PdCl(SeCH2CH2CH2NMe2)]2 and [PdCl(SeCH2CH2NMe2)(PPh3)] complexes. The best results were obtained with the [PdCl(SeCH2CH2NMe2)(PPh3)] complex. The catalyst was used with different aryl iodides and aryl bromides. The coupling of aryl iodides showed excellent performance, yielding biaryl products in high yields within 6 h. The comparable yields were obtained with other catalysts. The coupling of electron-deficient aryl bromides was found to give the corresponding biaryls in high yields (92%) compared to electron-rich aryl bromides. However, the yield was slightly improved by increasing the reaction time.
Singh and co-workers [87] described the synthesis of ligand 93 and its Pd(II) complex 94. The reaction started with the reaction of sodium benzeneselenolate 11 with (2-chloroethyl)(phenyl)sulfane 92 to give phenyl(2-(phenylselanyl)ethyl)sulfane ligand 93 (Scheme 22). Ligand 93 was further reacted with Na2PdCl4 13 to form a Pd(II) complex 94 which was tested as a catalyst in similar Suzuki–Miyaura coupling reactions. In the presence of the catalyst and K2CO3 base at 100°C, the reaction was completed within 1 h to provide a good yield of the product. Catalyst loading between 0.02 – 1.0 mol.% was reported to be sufficient for good conversion. Analogues of Pd(II) complex 94 containing sulfur- and tellurium-containing ligands instead of organoselenium one were also prepared and tested as catalysts in the same coupling reaction. Similar to the selenium-containing Pd(II) complexes, the sulfur-based counterparts showed good catalytic activity while the complex with an organotellurium ligand was not as efficient. The catalytic activity is attributed to the in situ generated palladium NPs. However, in the case of the tellurium ligand, the large inactive Pd NPs aggregates thus reducing the catalytic performance.
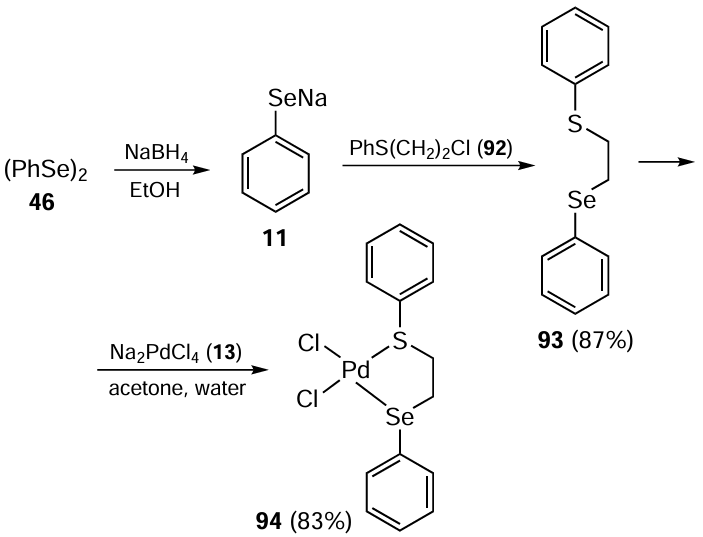
The research group of Singh [88] has also described the square planar palladium(II)-selenated Schiff base complexes 97 (Scheme 23). These complexes were derived from a ligand 96, which in turn was obtained by the reaction between 2-(phenylselanyl)propylamine 68 and various benzoate esters of 2,4-dihydroxybenzaldehyde 95 such as 4-methoxy, 4-decyloxy, 2,3,4-trisdecyloxy, and 4-octadecyloxy. The subsequent complex formation involved the reaction of Na2[PdCl4] 13 with ligands 96. The synthesis of organoselenium-stabilized Pd(0) nanoparticles is also reported. These complexes, loaded at 0.5 mol.%, demonstrated high efficacy in catalyzing similar coupling reactions under mild conditions to afford biaryl products in up to 95% yield. Notably, the complex with R1 = OC18H37 and R2 = H performed best among others with different ligand substitutions. The catalytic pathway involved the formation of organoselenium-stabilized Pd(0) nanoparticles. The observations revealed the unprecedented influence of the length of the alkyl chain in the complex molecule on the composition and dispersion of these particles thereby affecting the catalytic performance.
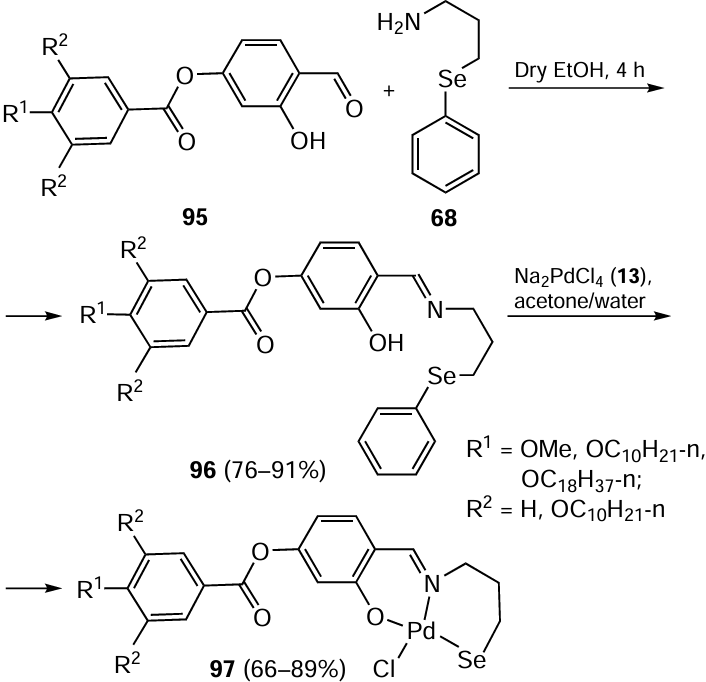
In a separate study, Kumar et al.[51] detailed the synthesis of potentially hexadentate [O−,N,E:E,N,O−] chalcogenated bisimine ligands (E = Se, S, Te) based on the reaction between 4,6-diacetylresorcinol 98 and 2-(phenylselanyl)ethylamine 16. On reaction with Na2[PdCl4] 13, the ligand 99 underwent partial hydrolysis, which is probably metal-promoted. This process gave the complexes 100 and 101 (Scheme 24). Both complexes were found to be stable to heat and air. has been shown to be an efficient catalyst in Suzuki – Miyaura coupling reactions, giving yields of up to 100%. This coupling reaction showed exceptional performance when carried out at 100 – 110°C in DMF for 24 h, using K2CO3 as the base, with a minimum catalyst loading of 0.1 mol.%, without any additional promoters. The sulfur-ligated palladium complexes were found to be as catalytically active as complex 100 in the Suzuki – Miyaura cross-coupling reaction while the Pd complex with tellurium-containing Schiff base ligand was the least active. The Pd(II) complex 100 derived from the selenated ligand 99 also showed activity in catalyzing the coupling of 2-chlorobenzaldehyde with 3-chlorotoluene.

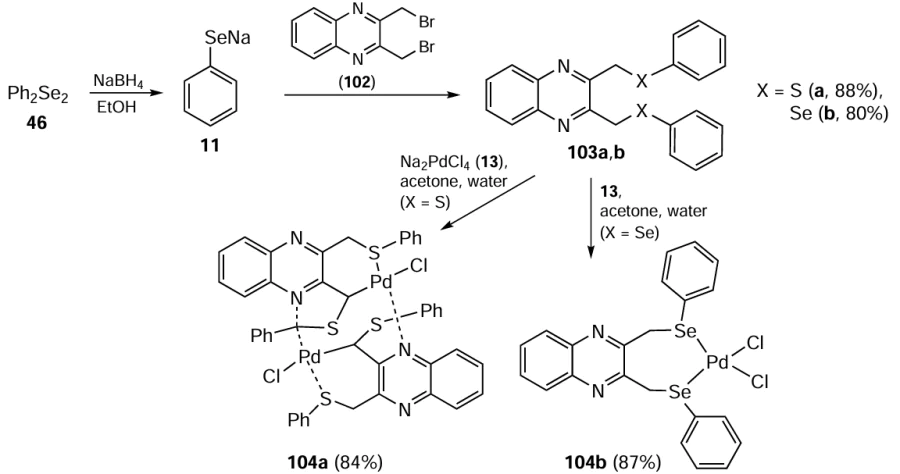
Saleem et al.[89] described the synthesis of palladium complexes of 2,3-bis[(phenylchalcogeno)methyl]quinoxaline ligands for use as catalysts in the Suzuki – Miyaura reaction. The process involved the reaction of 2,3-bis(bromomethyl)quinoxaline 102 with PhS(Se)Na 11 to afford ligands 103a,b (Scheme 25). These react with Na2PdCl4 13 to give complexes 104a,b. Interestingly, activation of the benzyl C(sp3) – H bond of the ligand 103a in the absence of an external base led to palladation of the ligand to give palladacycle 104a. The catalytic activity of complex 104a was investigated in Suzuki – Miyaura coupling reactions of various aryl bromides including the deactivated ones. Comparative analysis of 2,3-bis[(phenylthio)methyl]quinoxaline-Pd complex 104a with its Se-analogue 104b revealed that 104a exhibits superior catalytic activity. The coupling reaction performed well to give the coupling product in up to 93% yield in 3 h at 80°C using 1.0 mol.% catalytic loading of complex 104b. The catalytic process appeared to occur via in situ generated nanoparticles, which were characterized by a size of less than 2 nm consisted of palladium and either sulfur or selenium. These nanoparticles were shielded by ligands and exhibited catalytic activity even after isolation. Results from a two-phase test indicated a hybrid catalytic mechanism partly homogeneous and partly heterogeneous suggesting a cocktail-type catalytic effect.
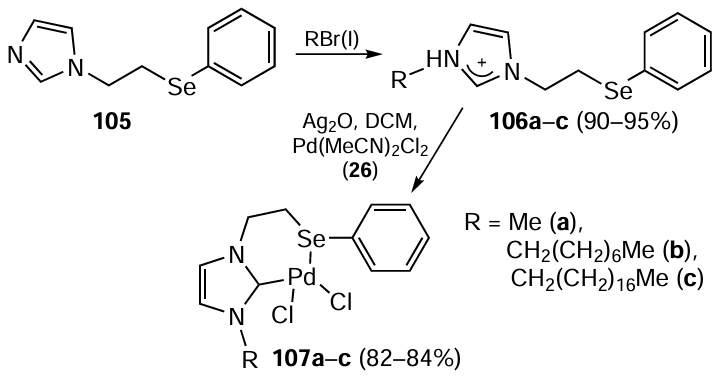
Sharma et al.[90] reported the synthesis of three types of N-alkyl-N'(2-ethyl-1-selenophenyl)imidazolium salts 106a – c differing in the alkyl chain length in the ligand. 1-(2-Phenylselanylethyl)-1H-imidazole 105 was used as a precursor for the ligands and their NHC complexes 107a – c. For the synthesis of Pd complexes 107 from imidazolium salts 106, a route involving the silver carbene transfer reaction was used (Scheme 26). The resulting complexes 107a – c were thermally stable, moisture- and air-resistant and also showed significant catalytic activity in Suzuki – Miyaura coupling reactions by achieving up to 96% yields in 2 hours at 80°C. In these reactions, NPs with a size range of 2−5 nm (accounting for approximately 80−85% of particles) were initially observed when the reaction mixture reached 80°C. These nanoparticles appear to play a pivotal role in the catalytic process potentially acting as Pd(0) dispensers containing both Pd and Se. The ratio of Pd to Se in these NPs was approximately 3 : 2, 4 : 5, and 1 : 1 for those formed from compounds 107a – c, respectively. Notably, the catalytic efficiency of complex 107c, which has the longest alkyl chain among the three complexes and contains the (Se, CNHC) ligand, exceeds that of complexes 107a,b with shorter alkyl chain ligands. The length of the alkyl chain in the complex probably influences the catalytic activity by regulating the dispersion of the NPs, which are spontaneously formed during the catalytic process.
2.4. Allylic alkylation
Allylic alkylation is one of the key reactions for introducing the alkyl functionality in the allylic position in organic synthesis. Various transition metals, in particular, palladium, have been successfully used as catalysts to carry out these reactions under mild reaction conditions.
The palladium-catalyzed allylic substitution is a unique tool for the formation of C – C and C – heteroatom bonds. Various organoselenium-ligated Pd complexes have been used as catalysts in the allylic alkylation reactions.[91-93] In addition, the chiral selenium-ligated palladium catalysts have contributed significantly to the development of various enantioselective allylic alkylations.[94]
In 2019, Diéguez and co-workers [95] synthesized a library of carbohydrate-derived thioether- and selenoether-phosphite ligands 108. The catalytic performance of these ligands was evaluated in a Pd-catalyzed asymmetric allylic substitution reaction (Scheme 27), where the catalyst was formed in situ from π-allylpalladium chloride dimer ([PdCl(η3-C3H5)2]) and an appropriate ligand. The reactions provided full conversions with high selectivities of up to 99% ee for hindered substrates and 91% ee for unhindered systems. Ligands with a chiral center in the alkyl backbone adjacent to the phosphite group and also an enantiopure biaryl phosphite group provided the high enantiomeric excess. Selenium-containing ligands were as effective as their sulfur-containing counterparts.
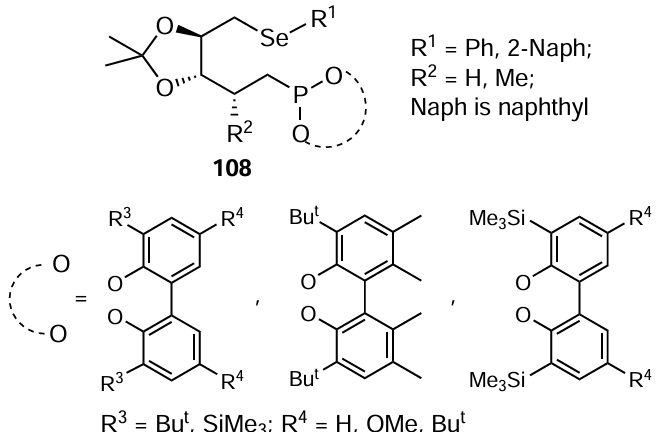
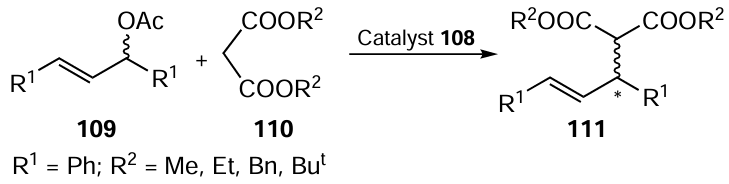
In 2022, You and co-workers [96] synthesized organoselenium-based ligands from a chiral aminoselenide 112, which reacted with chiral BINOL in the presence of DIPEA as base and PCl3 in DCM to give P,Se-ligands 113 and 114 in 88% and 83% yields, respectively (Scheme 28). Both ligands were used in the Pd-catalyzed asymmetric allylic alkylation reaction of CH2(CO2Me)2 with rac-(E)-1,3-diphenylallyl acetate in acetonitrile at 40°C to provide a good product yield with 75% ee and 87% ee, respectively. The reason for the enantioselectivity of both ligands is the matching and mismatching effect of point chirality and axial chirality. Based on the above results, compound 114 was selected as a promising ligand for optimising the reaction conditions. Using different solvents and bases it was found that the best results were obtained by reacting 5.0 mol.% of [Pd(C3H5Cl)]2 and 10 mol.% of ligand 114 in the presence of 3 equiv. of Cs2CO3 in acetonitrile. The (S)-product was obtained as the main product in the above reaction. Using the ligand 114 in reactions with nucleophiles other than malonate such as acetylacetone, malononitrile, bis(phenylsulfonyl)methane and indole, the products 111 were successively prepared in 77 – 92% yield with up to 90% enantiomeric excess (see Scheme 27). N-nucleophiles such as benzylamine were also found to be compatible with the given reaction conditions. rac-(E)-1,3-Diaryl-2-propenyl acetate with electron-donating or electron-withdrawing substituents at different positions on the phenyl ring is also suitable for this reaction. Sulfur-containing ligands have also been prepared and were found to perform slightly better in the asymmetric allylic alkylation reaction.

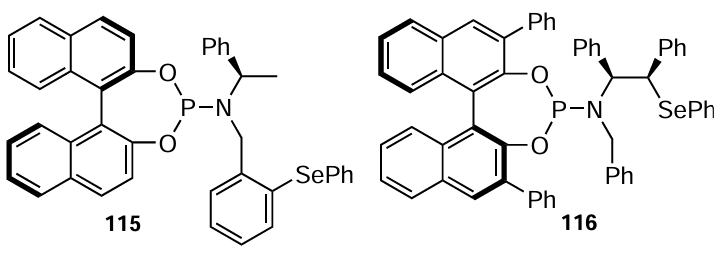
You and coworkers [97, 98] have also synthesized structurally modified selenide-phosphoramidite ligands 115 and 116 which were tested in the Pd-catalyzed asymmetric allylic alkylation reaction. Both ligands were obtained according to the above-described procedure.[96] Ligand 115 afforded allylic alkylation in 85% yield with 72% ee. Its sulfur-containing analogue required a shorter reaction time and gave a slightly better yield. The longer reaction time for the selenium-based ligand suggests that the selenium atom in 115 can suppress the activity of the metal complex. Ligand 116 has also been tested in the Pd-catalyzed asymmetric C-3 allylic alkylation of functionalized indoles, giving excellent yields with up to 99% enantiomeric excess.
2.5. Miscellaneous C – C coupling reactions
In 2016, Dresch et al.[99] obtained a number of nickel(II) complexes [NiBr2(N^Se)2] 120 bearing bidentate N^Se ligands as catalysts for ethylene oligomerization. The synthesis of bidentate arylselenyl – pyrazolyl ligands 118 involved the reaction of chloroalkyl pyrazoles 117 with the appropriate nucleophilic organoselenium compounds in a mixture of THF and ethanol (Scheme 29). Further, the reaction of 2.0 equivalents of ligands 118 with NiBr2(DME) 119 (DME is 1,2-dimethoxyethane) afforded the corresponding [NiBr2(N^Se)2] complexes 120. X-ray diffraction analysis of Ni complex 120 (n = 1, R1 = Me, R2 = Cl) revealed an octahedral geometry of the nickel atom with a selenium–nickel bond. After activation with methylaluminoxane (MAO), all nickel complexes showed moderate to good activity in ethylene oligomerization with turnover frequencies (TOF) ranging from 6.2 – 23.0 × 103 (mol ethylene) (mol Ni)–1 h–1, giving predominantly α-C4 as the main product.
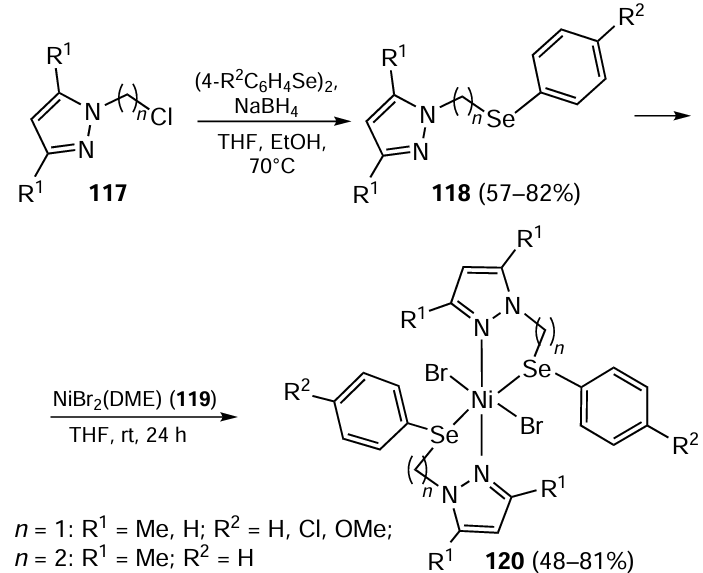
The catalytic activity and selectivity towards 1-butene were influenced by the ligand environment, in particular the substituents on the arylselanyl and pyrazolyl moieties and also by the reaction conditions. Under optimized reaction conditions, including 400 equivalents of MAO at 40°C for 20 min in toluene, the Ni complex (n = 1; R1, R2 = Me, H)/MAO catalytic system exhibited TOF values of £ 109.2 × 103 (mol ethylene) (mol Ni)–1 h–1 with high selectivity towards α-C4.
In 2018, the same research group [100] detailed the synthesis of nickel(II) bromide complexes 122. These complexes were synthesized in high yields using selenium-based tridentate ligands 121 in reaction with NiBr2(DME) 119 (Scheme 30). The catalytic activity of the complexes 122 in ethylene oligomerization was evaluated using MAO (20 wt.% TMA; TMA is trimethylaluminium) as a co-catalyst. When activated with MAO, the complexes 122 showed moderate to good activities in ethylene oligomerization (TOF = 4.3 – 25.7 × 103 (mol ethylene) (mol Ni)–1 h–1) with 1-butene being the predominant product (89.2 – 94.3 wt.%). The catalytic performance was influenced by the nature of the substituents on the ligand, in particular, on the pyrazolyl moiety. Under optimized conditions (toluene, 500 equiv. of MAO, 30°C, 20 min), the Ni complex 122 (R1 = Me; R2 = Cl)/MAO catalytic system showed improved TOF values, namely, 47.2 × 103 (mol ethylene) (mol Ni)–1 h–1, favourable for α-C4 (83.8 wt.%).
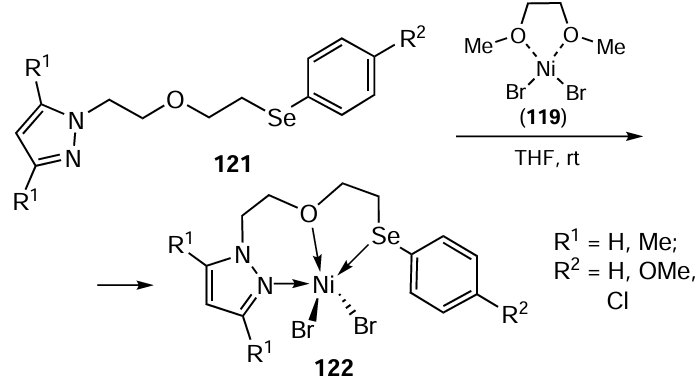
In 2022, Kumari et al.[101] presented a novel approach for the preparation of selenium-ligated ruthenium complexes involving a selenium-guided ortho-vinylation of benzyl selenide using a selenated NHC – half-pincer Ru(II) complex as a catalyst. The synthesis of the organoselenium ligand Ru(II) of complex 127 commenced with the reaction of benzimidazole 22 with dichloroethane to give 1-(2-chloroethyl)-1H-benzimidazole 23. The subsequent reaction with diphenyl diselenide 46 afforded 1-(2-(phenylselanyl)ethyl)-1H-benzimidazole 20, which was reacted with 2-bromomethylpyridine 124 to give the desired ligand 125 (Scheme 31). The final step involved the silver carbene transfer reaction, in which the ligand 125 was reacted with Ag2O in CH2Cl2 under an inert atmosphere at room temperature in the dark. Subsequently, [η6-(C6H6)RuCl(μ-Cl)]2 126 suspended in methanol was added to the reaction mixture to give the ruthenium(II) complex 127.
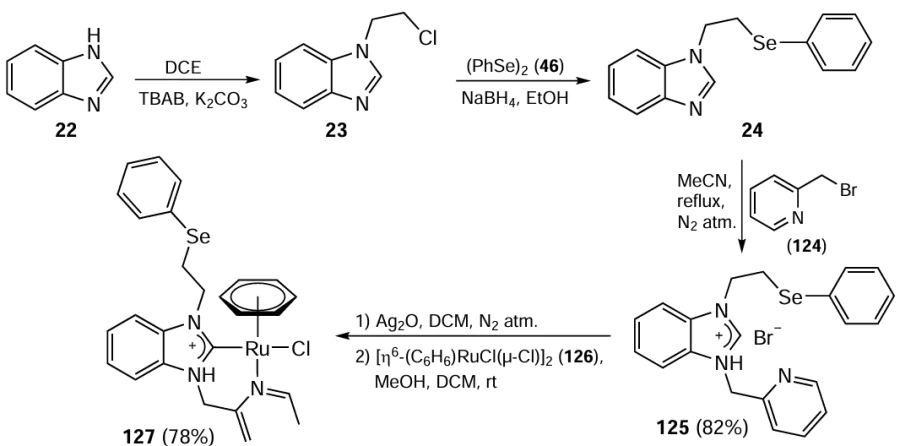
The catalytic activity of the resulting Ru(II) complex 127 was then tested in the ortho-vinylation reaction. This innovative reaction methodology is based on the C – H activation by selenium acting as a directing group. Moderate yields of products 129, 130 were obtained using 4.0 mol.% of the Ru(II) complex 127 and 0.05 mol.% Cu(OAc)2 as an oxidant at 120°C (Scheme 32). It should be noted that the reaction tolerates different types of vinyls 33 and various substituted benzyl selenides 128. This method is an effective synthetic approach to the synthesis of organoselenium compounds using selenium as the directing group.
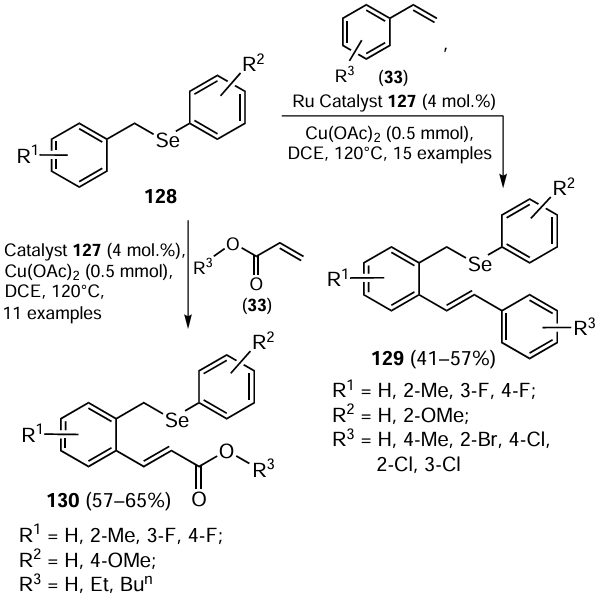
In 2016, Sharma et al.[102] obtained a potentially chelating bidentate (N,Se)-benzotriazole-based organoselenium ligand 132 by reacting 1-(chloromethyl)benzotriazole 131 with the in situ generated PhSeNa 11 (Scheme 33). Further, the ligand 132 was treated with silver nitrate in MeCN at 70°C to give the polymeric silver(I) complex 133. The complex was reported to be light-, air- and moisture-insensitive.
The Ag complex 133 was found to be an efficient catalyst for the A3-coupling of aldehydes 134, amines 135 and phenylacetylene 6 to afford propargylamines 136 in yields up to 94% at 60°C (Scheme 34).[102] The A3-coupling reaction ran efficiently in the presence of 2.0 mol.% of the polymeric Ag complex 133 under aerobic conditions at 60°C within 3 – 8 h. The proposed reaction mechanism suggests that [AgL(amine)]+ is the actual catalyst. The sulfur-containing analogue of complex 133 showed similar catalytic activity.
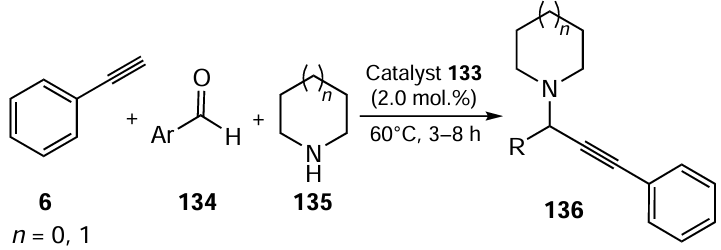
In 2019, Elhampour and co-workers [103] obtained a copper(I) complex with organoselenium ligand supported on Fe3O4 nanoparticles modified with SiO2/aminopropyltriethoxysilane. The resulting magnetic nanocatalyst was tested for catalytic activity in the A3 coupling reaction (see Scheme 34). The product yield was good to excellent with various substituted aldehydes, secondary amines, and terminal alkynes. The catalyst was easily recoverable and could be reused for up to five cycles without any significant loss of its activity.
The same year, Jain and co-workers [104] obtained the copper(I) complex 139 of the 4,5-bis((phenylseleno)methyl)acridine ligand 138 by the synthetic route shown in Scheme 35. The characteristic feature of this complex is its impressive thermal stability and resistance to moisture and air. The ligand in complex 139 coordinates with copper in a pincer-like fashion to form two six-membered chelate rings. The coordination arrangement results in a distorted tetrahedral geometry around the copper atom due to the donor atoms.
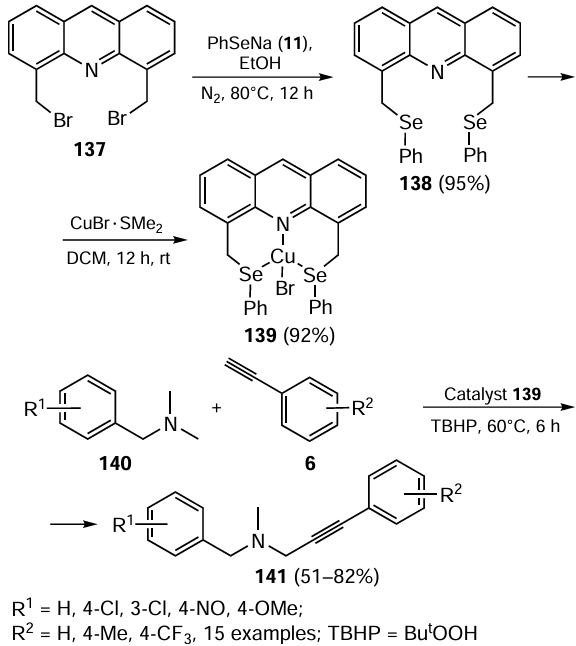
The complex 139 proved to be a highly efficient catalyst for two specific reactions. The first reaction was the cross dehydrogenative coupling of unactivated tertiary amines 140 with unactivated terminal alkynes 6 (see Scheme 35). The C – C coupling reaction was carried out under oxidative solvent-free conditions in the presence of tert-butyl hydroperoxide (TBHP) at 60°C with a catalyst loading of the 1.0 mol.% to provide moderate to good yields of the product. It was found that substrates with electron-donating groups on the aromatic ring of amines or alkynes gave higher yields of the corresponding propargylamines 141 than substrates bearing electron-withdrawing groups. However, the chloro derivatives of N,N-dimethylbenzylamine had no noticeable effect on the reaction. In general, catalyst 139 showed slightly better catalytic activity compared to its sulfur-ligated counterpart.
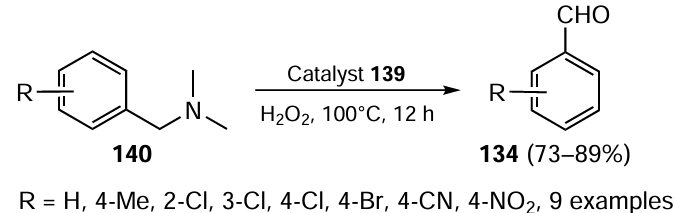
Another reaction discussed in the context of the use of complex 139 is the selective method for cleaving the C – N bond in N,N-dimethylbenzylamines 140 to give functionalized aldehydes 134 (Scheme 36). The high yield of the products 134 was achieved using the catalyst loading of 1.0 mol.%, 2 equiv. of H2O2 at 100°C under an oxygen atmosphere. The reaction was completed in 12 h and showed exceptional selectivity towards aldehydes with no cases of over-oxidation to acids. It was also concluded that the substrates containing electron-donating groups and halogens gave higher yields than those with electron-withdrawing groups such as CN and NO2 .
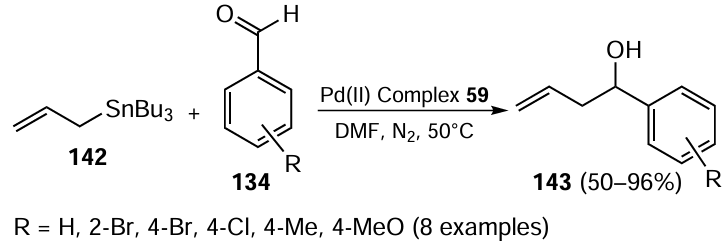
Palladium(II) complex 59 (see Scheme 12) was also used as a catalyst for the allylation of aryl and heteroaryl aldehydes 134. The optimized reaction conditions involved 1.0 mol.% of the complex catalyst in DMF (Scheme 37). The reaction was completed within 16 h at 50°C. The yield of alcohol 143 was found to be depended on the electron-withdrawing or electron-donating groups in an ortho or para position to the CHO functionality. The catalytic activity of the complex was higher for the substituents bearing electron-withdrawing groups.
3. C – O coupling reactions
C – O coupling reactions (Scheme 38) are a useful tool for the preparation of o-arylated products with applications in agrochemistry, biology, materials science and pharmaceuticals.[105, 106] Moreover, many of the compounds obtained exhibit significant antifungal, antibacterial and herbicidal activities.[107] The advantages of using organoselenium ligands in C – O coupling reactions include shorter reaction times and lower temperatures required to achieve high conversions of starting materials to coupled products.
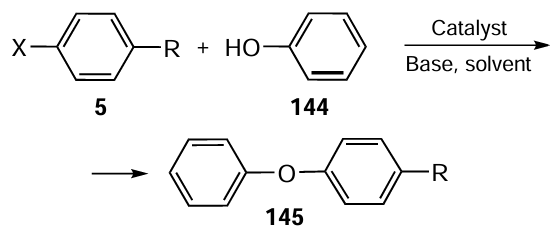
Arora et al.[108] have developed a Pd(II) complex 148 using an N-pyrene organoselenium-based compound 147 as the ligand (Scheme 39). The synthesis of the ligand 147 was carried out by reacting 1-pyrenecarboxaldehyde 146 with 2-(phenylselanyl)ethylamine 16 in ethanol. Compound 147 combined an imine functionality and the selenium donor atom in a single molecule. Further reaction of the ligand 147 with bis(acetonitrile)palladium(II) dichloride 26 as a source of palladium afforded the molecular palladium complex 148. The Pd atom has an almost square planar geometry in the complexes with a (Se,N)-bidentate coordination of the ligands. Screening of the catalytic activity in the o-arylation of phenols showed an excellent efficiency of the complex 148, even much better than that of a variety of known palladium complexes and of analogous organosulfur-based complexes used in the same study (see Scheme 38). The product yield reached 99% using K2CO3 as the base and DMSO as the solvent at 110°C.
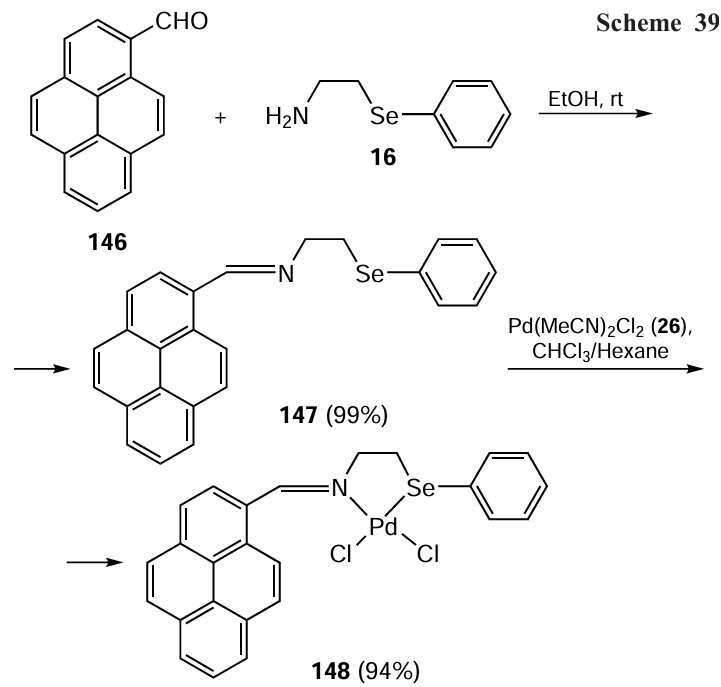
The proposed mechanism consists of three steps; the first is the oxidative addition of ArX to LnPd(0) to form the intermediate 149; the second involves the transmetallation of the metal phenolate to give 150 and the last step is the reductive elimination of Ar – O – Ar to generate LnPd(0) (Scheme 40). The possible explanation for the good catalytic activity is that the organic moiety present in the catalyst protects and stabilizes the Pd(0) species formed during the reaction.
The previously described magnetically retrievable nanoparticles 74 (see Scheme 17) used in Suzuki – Miyaura coupling were also tested in the ortho-arylation of phenols chosen as an example of C – O coupling reactions.[79] Using 0.1 mol.% catalyst and NaOH, together with water as solvent, the maximum yield of 95% was obtained. The reaction of various aryl chlorides, iodides and bromides 5 with phenols 144, even in the case of less activated compounds, afforded a wide range of coupled products in good yields. The said nanoparticles showed high catalytic performance even with heteroaryl halides, indicating their versatility and potential usefulness. The substitution pattern was found to have a significant effect on the product yield. The ortho substituents have a negative effect on the yield of the reaction, while the para substituents have a less detrimental effect or may even increase the yield.
The catalytic activity of selenoether-based complexes 86 and nanoparticles reported by Sharma et al.[83] (see Scheme 20) used in the Suzuki – Miyaura coupling reaction, was also investigated for the formation of C‒O coupled products from the aryl halides 5 and phenol 144 (see Scheme 38). The reaction worked well using 0.5 mol.% catalyst 86 in the presence of Cs2CO3 in DMSO at 110°C. The similar sulfur-based complex was found to be more effective than its organoselenium counterpart. The influence of the electronic effects of the substituents of the aryl halide was quite similar to that observed in the Suzuki – Miyaura reaction and the C‒O coupling product 145 was obtained in up to 95% yield within a reaction time of 3 h.
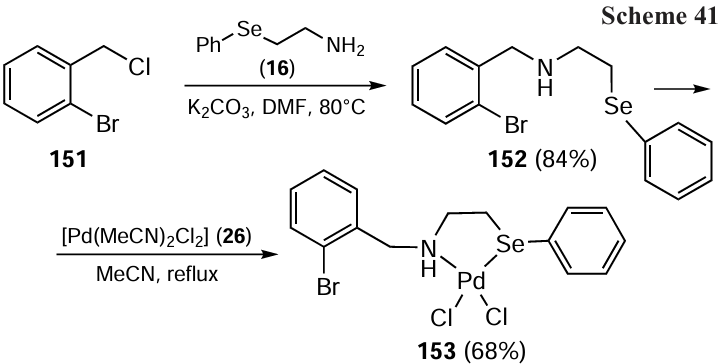
In 2020, Bhatt et al.[109] developed the synthesis of palladium complex 153 with a (N,Se)-bidentate ligand (Scheme 41). The new ligand, N-(2-bromobenzyl)-2-(phenylselanyl)ethanamine 152 was obtained reacting 1-bromo-2-(chloromethyl)benzene 151 with 2-(phenylselanyl)ethanamine 16 in the presence of K2CO3 and DMF at 80°C within 5 hours. The Pd complex 153 was then obtained by refluxing the ligand 152 with [PdCl2(MeCN)2] 26 in acetonitrile. The resulting complex showed a distorted square planar geometry around the palladium centre. It was thermally and air stable and promoted the arylation of imidazole and phenol. For the ortho-arylation of phenol, a catalytic loading of 2.0 mol.% was found to be efficient at 110°C and gave impressive results (71 – 92%) while the coupling reaction showed tolerance to a wide range of functional groups. A total of nine substrates were used in these reactions. Additionally, a similar sulfur-ligated complex was synthesized. On comparing the catalytic efficiency of sulfur and selenium-coordinated ligands, it was evident that the selenium-containing complex outperformed its sulfur-containing analogue in catalytic reactions.
4. C‒S/Se coupling reactions
Lang and co-workers [49] described a series of bis(2-pyridyl)diselenoethers 155 and used them as ligands to obtain Cu complexes with copper iodide. The ligands 155 were synthesized by reacting bis(2-pyridyl)diselenide 154 with NaBH4 with further addition of the corresponding dibromoalkane (Scheme 42). These ligands were reacted with metal ions of different hardness (Cu+, Ag+, Cu2+, and Co2+) to furnish six new complexes. For example, the ligand 155 (n = 2) reacts with CuI to give [Cu4I4{(2-PySe)2CH2}2] 156.
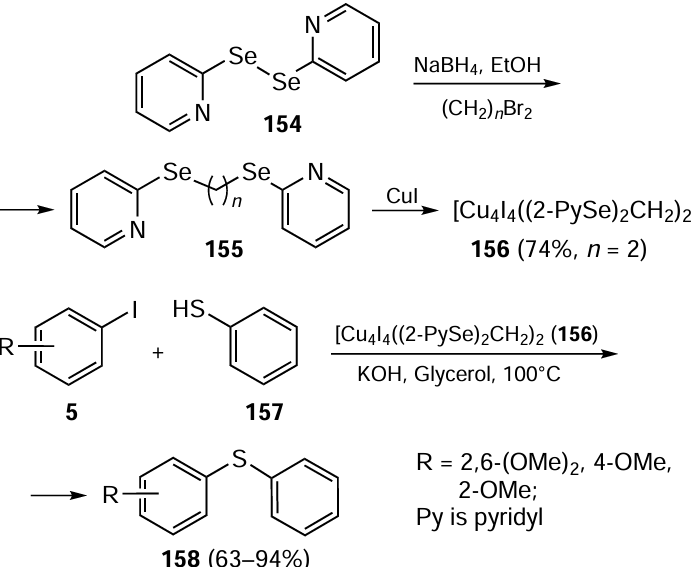
To evaluate catalytic activity of this new class of compounds, the complex [Cu4I4{(2-PySe)2CH2}2] 156 was tested in the C – S coupling reactions. The reaction of 1.0 equiv. of aryl iodide 5 with 1.5 equiv. of thiophenol 157 in the presence of KOH and Cu(I)-complex catalyst 156 at 100°C for 24 h afforded the C – S coupling product 158 (see Scheme 42). The coupling reaction proceeded smoothly with 10 mol.% of catalyst 156 to give the coupling products 158 in high yields (up to 94%). The disadvantage of the process is that the catalyst is inactivated after a single use.
In 2019, Coelho et al.[110] described copper complexes bearing ligand 160 as useful catalysts for carbon–sulfur and carbon–selenium coupling reactions. The ligand 160 was synthesized in good to high yields by the nucleophilic attack of PhSeNa (11), generated in situ by the reaction of diselenide 46 with sodium borohydride, with the chlorinated compound 159, derived from pyrazole in two chemical steps (Scheme 43).
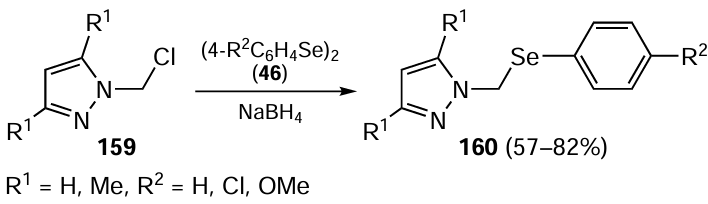
Ligands 160 were investigated for their effectiveness in C – Se and C – S bond formation, particularly in the synthesis of chalcogenoacetylene 162 in the presence of a copper catalyst (CuI) and showed good performance (Scheme 44).
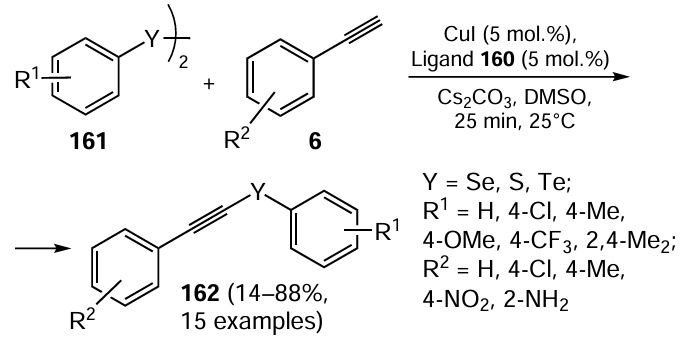
Chalcogenoacetylenes 162 were obtained by the reaction of the ligand 160 with CuI. The best results (yield 82%) were achieved with the ligand 160 (Y = Se, R1 = Me, R2 = H) under mild and aerobic conditions. Aromatic and aliphatic diselenides 161, bearing various electron-donating and electron-withdrawing groups, reacted with substituted phenylacetylenes 6 to afford the product 162 in moderate to good yields. A putative mechanism for the selenoacetylene synthesis was proposed which was supported by the 77Se NMR study indicating that compounds 160 functions as a hemilabile ligands. The efficiency of these catalysts in C – S cross-coupling was evaluated by reacting aryl halides with thiols to afford products in good to excellent yields.
5. C‒N bond formation reactions
The formation of the carbon – nitrogen bond is a chemical process that is essential for the construction of various organic molecules, pharmaceuticals, materials, and agrochemicals. This can be achieved by a variety of different methods. In this Section, we will discuss organoselenium ligand-metal complexes that are involved in the formation of the C – N bond.
In 2018, Srinivas and Prabusankar [111] described the synthesis of mononuclear copper(I) complexes 164, 165. The complexes were obtained by the reaction of copper(I) halides with 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-selone 163 in methanol in high yields (Scheme 45). The authors also succeeded in isolating cationic copper(I) complexes 167 – 169 in very good yields. The mononuclear copper(I) complex 166a reacted with compound 163 to give complexes 167, 168, while the Cu(I) complex 166b was treated with [Cu(MeCN)4]PF6 to give complex 169.
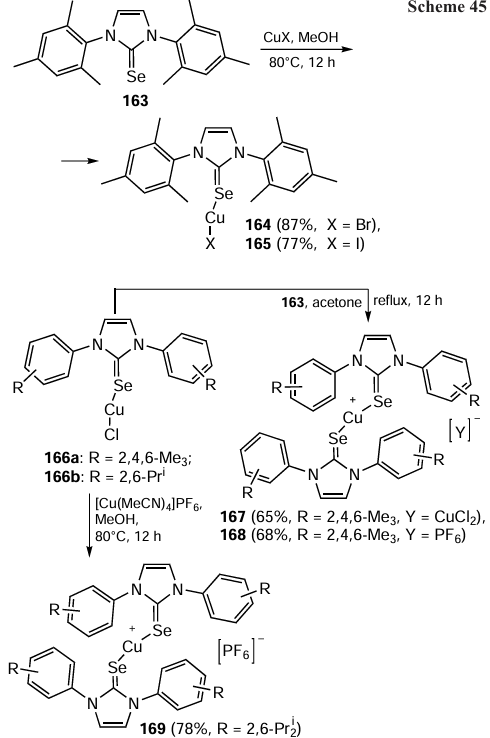
The above-mentioned NHC=Se-containing copper(I) complexes were used in a click reaction.[111] The catalytic solvent-free reactions were carried out under ambient conditions at room temperature as depicted in Scheme 46. It should be noted that the catalysts 164 and 165 provided excellent conversion rates (70 – 92%) within only 1 h, while the linear copper(I) chalcogenones 167 – 169 gave moderate yields (68 – 76%) of the triazole 171. Coordination polymers were also synthesized and were found to be as effective as the linear chalcogenones in this catalytic process. The copper(I) catalyst is expected to coordinate with both the terminal alkyne and the azide to form the intermediate 172, which then eliminates the triazole 173 to release the catalyst for the further reaction (Scheme 47).
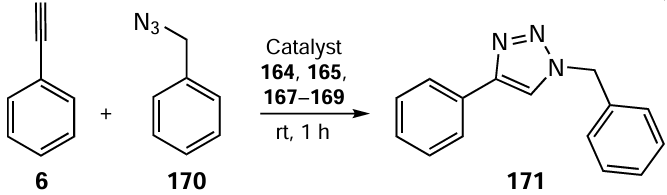

Dubey et al.[112] reported the synthesis of Ru(II) complexes of organoselenium ligands for the N-alkylation of amines. The Schiff base 76 was synthesized by condensation of anthracene-9-carbaldehyde 75 with 2-(phenylselanyl)ethylamine 16 (see Scheme 18). Complexes 174 and 176 were obtained by the cleavage of the chloro bridges in [(η6-C6H6)RuCl-(μ-Cl)]2 126 and [(η5-Cp*)RhCl(μ-Cl)]2 175, respectively (Scheme 48). Next, the reaction with ligand 76 proceeded at ambient temperature and was assisted by anion exchange using NH4PF6 . Complex 177 was obtained by the reaction of complex 175 and ligand 76 in the presence of MeCOONa at 50°C. All the resulting complexes were insensitive to air and moisture.
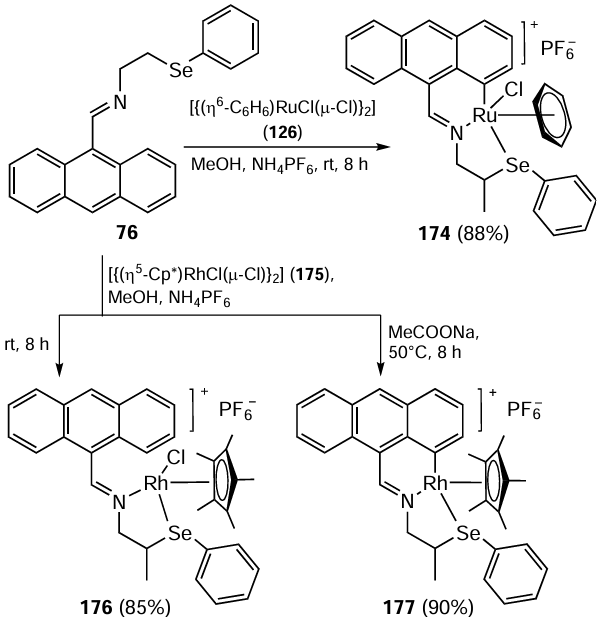
In the study,[112] the possibility of N-alkylation of aniline 178 and its derivatives with benzyl alcohols 179 using complexes 174, 176 and 177 as catalysts was investigated. Benzyl alcohols 179 with electron-donating and electron-withdrawing substituents at the para-position react with anilines 178 to give N-alkylated anilines 180 in good yields (Scheme 49). Among the above complexes, the catalytic activity of 176 was found to be superior to that of 174, whereas the complex 177 was the least active.
6. Arylation of imidazoles
Direct arylation of the imidazole nucleus represents valuable synthetic approach to a wide variety of compounds that have found applications as pharmaceuticals, polymers or functional materials.[113] The use of palladium complexes as catalysts in these transformations favours arylation at C-2 and C-5 positions of the imidazole ring rather than at the C-4 position (Scheme 50). In addition to the non-selectivity of arylation at C-2 and C-5 positions, there are also some other limitations to overcome, such as high catalyst loadings, long reaction times, the need for an inert atmosphere and a relatively narrow range of substrates.
In recent years, N-heterocyclic carbenes have been developed as a unique class of ligands for the preparation of metal complexes as catalytic systems with excellent stability/reactivity ratios. In continuation of this trend, some of the research groups have studied the effect of the ligands combining NHC and an organoselenium moiety on improving the performance of metal complexes.
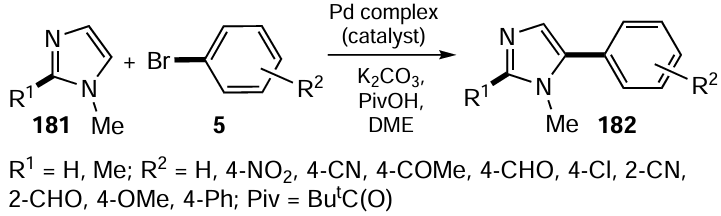
An interesting work, describing the efficient use of novel selenium-containing imidazolium bromide as a Se,CNHC,Se type pincer ligand for the synthesis of palladium complexes 185, appeared in 2020.[114] The ligand, 3-(2-(phenylselanyl)ethyl)-1-(2-(phenylthio)ethyl)-1H-imidazolium bromide 184, was synthesized by the reaction of 1-(2-(phenylselanyl)ethyl)-1H-imidazole 105, with ((2-bromoethyl)thio)benzene 183 in MeCN at 120°C under a nitrogen atmosphere (Scheme 51). After this, the Pd(II) complex 185 was prepared from imidazolium bromide 184 and Pd(MeCN)2Cl2 26 in the presence of silver oxide. The bromide ion was then changed to BF4- by the reaction with silver tetrafluoroborate. The resulting complex 185 was tested in the C – H bond arylation of imidazoles 181 (see Scheme 50). A wide range of cross-coupled products 182 were obtained from structurally different aryl bromides 5 in excellent yields (up to 95%) and with good regioselectivity for C-5 arylation using only 0.5 mol.% catalyst together with K2CO3 base and pivalic acid (PivOH) as an additive at 100°C. The introduction of electron-donating groups in the aryl bromide, as well as variation of the ortho – para positions of the substituents, didn’t affect the reaction outcome. The homogeneous nature of the catalysis was confirmed by mercury and triphenylphosphine poisoning tests. It was also shown that the catalyst 185 could be recycled several times without losing its catalytic potential. It was observed that the catalytic performance of the Pd complex bearing an organoselenium ligand was much better than that of the sulfur-containing analogues.
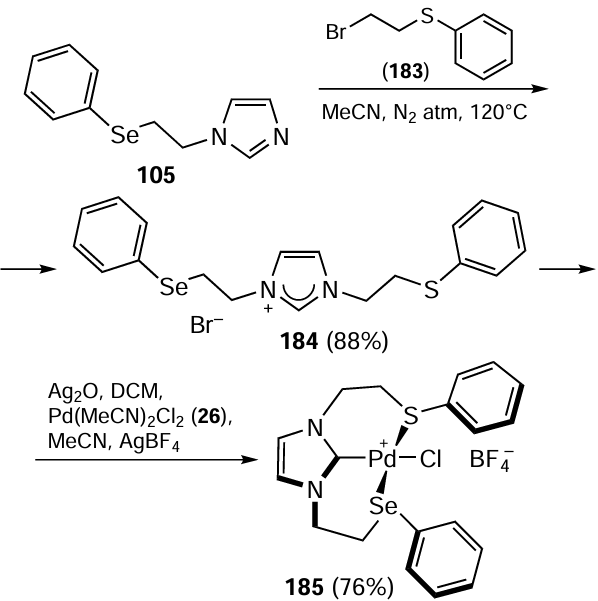
Kumar et al.[115] have presented the synthesis of the macrocyclic palladium(II) complex 191 based on the air-stable bidentate organoselenium ligand 190 (Scheme 52). The ligand was prepared from 2,2-(octane-1,8-diylbis(oxy)dibenzaldehyde 188 which in turn was obtained by reacting 1,8-dibromooctane 187 with an excess of salicylaldehyde 186 in DMF in the presence of K2CO3 . Compound 188 was isolated in moderate yield and was further reduced with NaBH4 followed by chlorination with thionyl chloride to give 1,8-bis(2-(chloromethyl)phenoxy)octane 189 in 91% yield. Compound 189 was further reacted with the sodium salt of diphenyl diselenide 11 to furnish the organoselenium ligand 190. The nineteen-membered cyclic compound 191 containing a macrocyclic palladium(II) complex was obtained in 54% yield by reacting the selenium ligand 190 with Pd(MeCN)2Cl2 26. The resulting complex 191 was tested as a catalyst for the regioselective arylation of imidazoles (see Scheme 50). The advantages of this method are the high yields of the products obtained (73 – 95%) and C-5 regioselectivity. The catalytic arylation reaction is completed within 10 h and is a homogeneous in nature. A wide range of substituted aryl halides 5 react with imidazoles 181 using only 1.5 mol.% of complex 191 as catalyst in the presence of K2CO3 and PivOH as base and additive, respectively at 100°C to give aryl imidazoles 182.
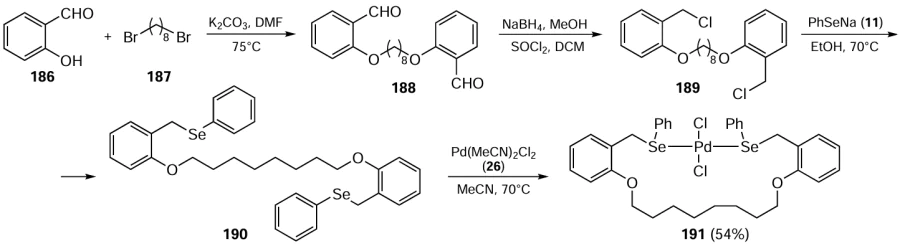
Bhaskar et al.[116] described the precursors of chalcogenated acetamide-functionalized 1H-benzimidazolium salts 195 (Scheme 53). The reaction of benzimidazole 22 with 2-bromo-N-(2-phenylselanyl)ethyl)acetamide 193 gave compound 194 which was treated with methyl or benzyl bromides(iodides) to furnish ligands 195. Further, the reaction of ligand 195 with bis(acetonitrile)palladium(II) dichloride 26 and silver oxide afforded Pd complexes 196. These complexes were reported to be almost square planar with the NHC rings almost perpendicular to the palladium coordination plane. The complexes were insensitive to both air and moisture. Complexes 196 with methyl and benzyl substituents performed well in the regioselective arylation of imidazoles at the C-5 position in the presence of air. Optimum conversions were obtained with a 0.5 – 1 mol.% catalyst in the presence of PivOH additive and K2CO3 at 110°C. A wide range of aryl chlorides and aryl bromides could be used with these catalysts. It should be noted that the benzyl-substituted complexes turned out to be more effective than their methyl-substituted analogues. Also, the catalysts with organoselenium ligands proved to be more active than their sulfur-containing counterparts. All complexes 196 showed recyclability for up to six cycles in the regioselective arylation of imidazole with the minimal loss of efficiency.
7. Reduction reactions
Transfer hydrogenation (TH) is a process in which hydrogen is transferred from one molecule to another without the direct use of gaseous hydrogen. This method is commonly used in organic synthesis and catalysis for the reduction of various compounds. Metal complexes have been reported to be efficient catalysts that facilitate the transfer of hydrogen from the donor to the substrate.
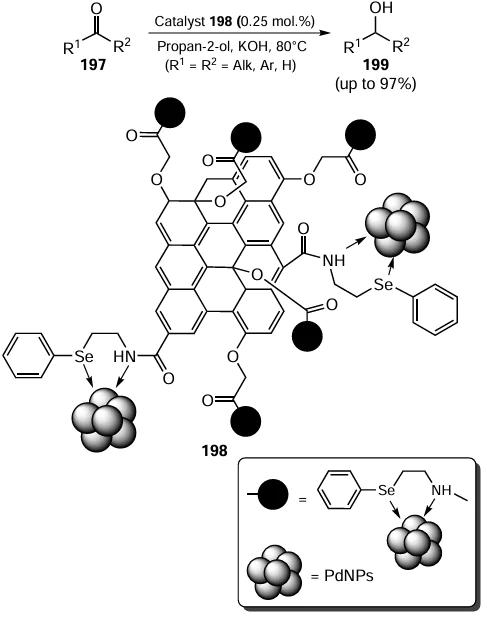
Complexes with organoselenium ligands were found to be highly active catalysts in the reduction reactions. In 2017, an interesting work [117] was published describing the functionalization of the graphene oxide (GO) surface with 2-(phenylselanyl)ethylamine 16 as a chelating ligand. Furthermore, the reaction of the resulting ligand with the solution of sodium tetrachloropalladate(II) 13 in the presence of NaOH affords GO – Se anchored with Pd(0) NPs 198 (Scheme 54).[53, 117] The efficiency of the catalytic system 198 was confirmed by the conversion of aldehydes(ketones) 197 to the corresponding alcohols 199 in good, in some cases quantitative yields. The conversion of aldehydes was slightly higher than that of ketones.
In 2014, Prakash et al.[50] pioneered the synthesis of half-sandwich complexes of rhodium(III) and iridium(III) bearing (Se,Se) ligands. The ligand 1,2-bis(phenylselenomethyl)benzene 201 was synthesized by reaction of PhSeNa 11 with 1,2-bis(bromomethyl)benzene 200 under nitrogen atmosphere (Scheme 55). The iridium(III) complex 203 and rhodium(III) complex 204 were then prepared by the reaction of 1,2-bis(phenylselenomethyl)benzene 201 with [(η5-Cp*)IrCl(μ-Cl)]2 202 and [(η5-Cp*)RhCl(μ-Cl)]2 175, respectively, in the presence of NH4PF6 at ambient temperature. The air- and moisture-insensitive half-sandwich complexes were obtained in good yields.
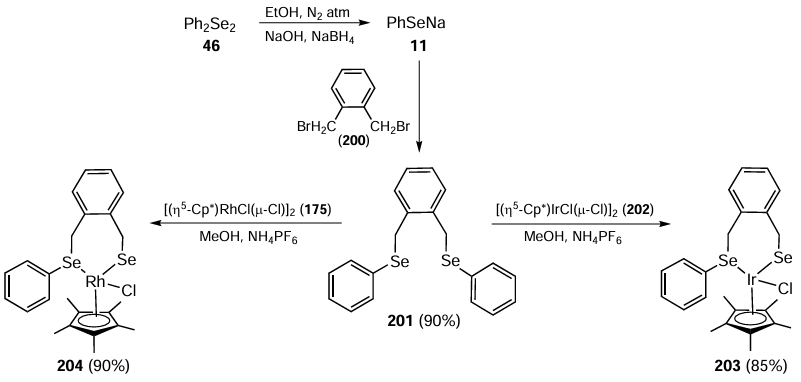
Complexes 203 and 204 were studied in the transfer hydrogenation of carbonyl compounds 197 using glycerol 205 as solvent and hydrogen donor (Scheme 56). The reaction was carried out at 120°C to give alcohols 199 in good to excellent yields. Dihydroxyacetone 206 was obtained as a by-product of the dehydrogenation of glycerol. When comparing the catalytic efficiencies of the (Se,Se)- and (S,S)-ligands with other co-ligands held constant, the Se ligand showed higher activity due to its stronger electron-donating tendency towards the metal centre. In general, the Rh complex 204 performed slightly better than its Ir counterpart 203.[50]
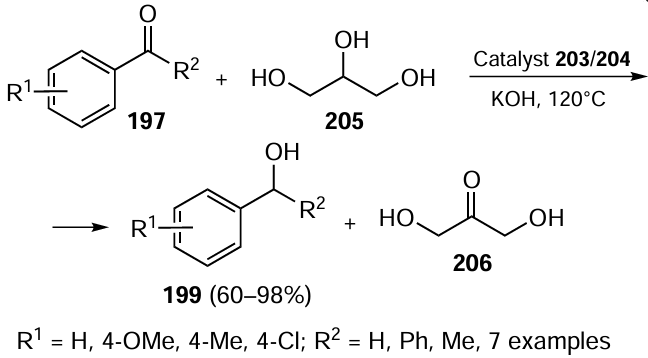
Dubey et al.[112] reported the base-free catalytic TH of the carbonyl group in compounds 197 using complexes 176 and 177 (see Scheme 48) as catalysts with a maximum loading of 0.3 mol.% in the presence of 2-propanol as solvent and a hydrogen source under ambient conditions (Scheme 57). It was found that benzaldehydes with electron-withdrawing groups provided high conversions even with 0.2 mol.% of the catalyst whereas the substrates with electron-donating groups required 0.3 mol.% catalyst loading for the same conversion. Compared to aldehydes, complexes 176 and 177 were less efficient in the reduction of ketones to the corresponding alcohols 199 but by increasing the catalyst loading to 0.5 mol.%, the products were obtained in good yields. The reduction of aromatic ketones bearing electron-donating substituents provides access to the secondary alcohols in up to 83% yield.
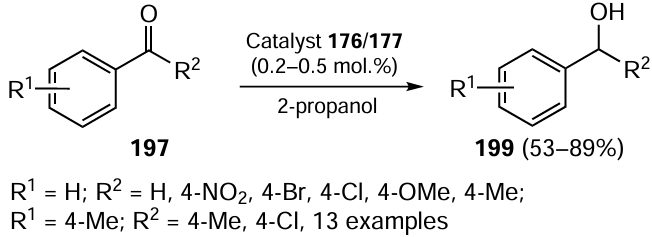
In 2020, Singh and co-workers [118] reported the synthesis of Ru(II) complexes 211 and 212. The ligand (E)-N-(2-(phenylselanyl)ethyl)-1-(pyridin-2-yl)methanimine 208 was obtained by the reaction of 2-(phenylselanyl)ethan-1-amine 16 with pyridine 2-carboxaldehyde 207 (Scheme 58). Further, the ligand 208 was reduced with NaBH4 to 2-(phenylselanyl)-N-(pyridin-2-ylmethyl)ethan-1-amine 209. The reaction of [Ru(PPh3)3Cl2] 210 with ligands 208 and 209 gave Ru(II) complexes 211 and 212, respectively. Both resulting complexes had distorted octahedral geometry around Ru. Further investigations delved into the application of these complexes to the transfer hydrogenation of aldehydes and ketones. It was found that using propan-2-ol as the hydrogen atom source along with 0.1 mol.% of complexes 211 and 212 as catalysts, the reaction is completed in 15 min at 80°C to give the products in excellent yields. Similar complexes with a sulfur-containing ligand have been reported to be equally potent for this reaction.
Singh and co-workers [119] have carried out extensive research on ruthenium(II) complexes of 1,2,3-triazole-based organoselenium ligands 213 and 214. Half-sandwich complexes [(η6-benzene)RuLCl]PF6 215 and 216 were prepared by reacting ligands 213 and 214 respectively with [{(η6-C6H6)RuCl(μ-Cl)}2] 126 followed by treatment with NH4PF6 (Scheme 59). Complex 215 with ligand 213 has the N(3) bonded to the ruthenium atom, whereas in ligand 214, the coordination to ruthenium involves the N(2) atom. The resulting complexes were also tested as catalysts in the catalytic reduction of ketones. Complexes 215 and 216 were used in 0.01 – 1 mol.% catalyst loading. High conversions of ketones were observed using 2-propanol as the hydrogen donor and KOH as the base at 80°C. The catalytic efficiency of the organosulfur Ru complexes was also investigated and was found to be comparable to that of the selenium-ligated complexes. Complex 216 involving N(2) of the 1,2,3-triazole ring and Ru for the complex formation was reported to be a superior catalyst among all the synthesized complexes for the catalytic reduction of ketones as it requires low catalyst loading. The reaction proceeds via the formation of a metal hydride intermediate.
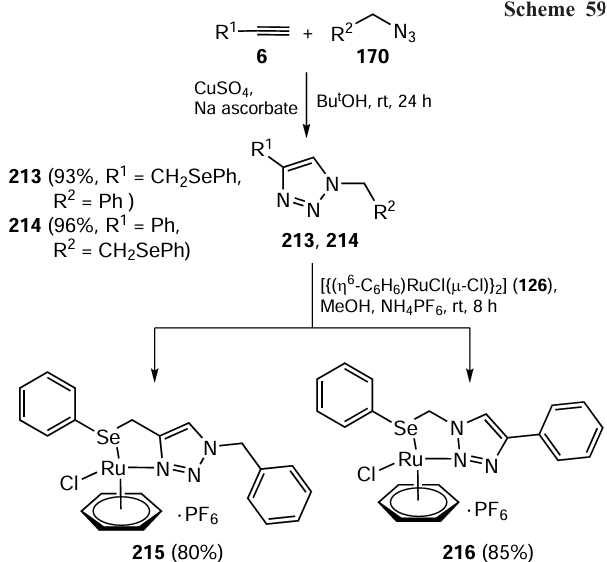
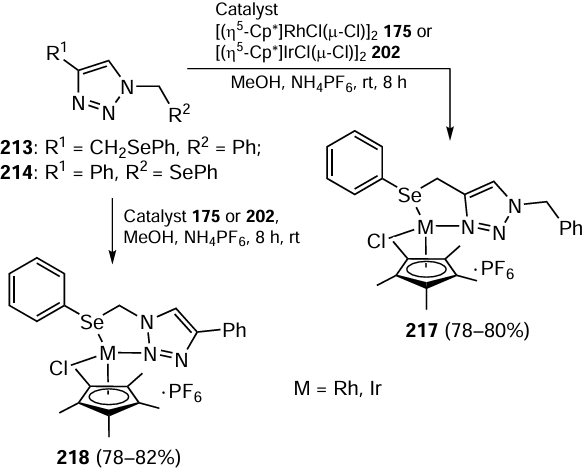
Later, similar type Rh and Ir complexes were synthesized by Saleem et al.[120] Complexes 217 and 218 were obtained by reactions of organoselenium-tethered triazoles 213 and 214 with [(η5-Cp*)RhCl(μ-Cl)]2 175 and [(η5-Cp*)IrCl(μ-Cl)]2 202, respectively under the aforementioned reaction conditions (Scheme 60). Moreover, less studied half-sandwich type rhodium and iridium complexes were explored for their potential use as catalysts for the reduction of carbonyl compounds. The conversion was quite good in short reaction time using 0.01 – 0.001 mol.% of Rh and Ir complexes 217 and 218 with 2-propanol as the hydrogen donor at the moderate temperature (80°C) in the presence of KOH. The rhodium complexes showed greater efficiency in catalytic processes compared to their iridium counterparts. Additionally, similar to complex 216, it was found that complexes 218 with N(2) of the triazole ring coordinated to the metal atom exhibit the superior catalytic activity compared to complexes 217 in which the N(3) atom was involved in the ligation. Complexes with selenium-containing ligand were more reactive than their sulfur-containing analogues.
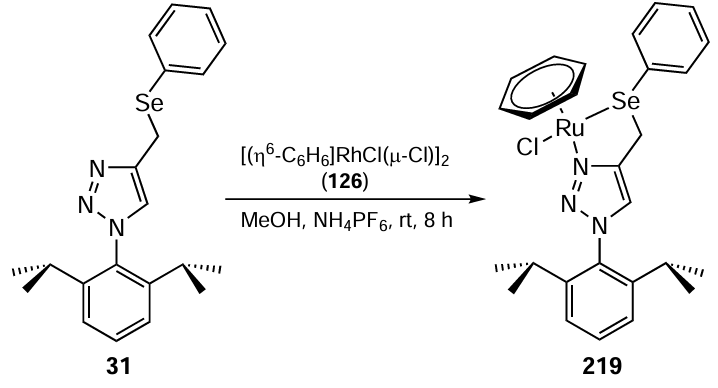
In 2016, the same research group [72] prepared the complex [(η6-C6H6)Ru(L)Cl]PF6 219 using the triazole-based organoselenium ligand 31 (Scheme 61). The Ru complex 219 had a pseudo-octahedral arrangement of donor atoms similar to a ‘piano-stool’ configuration. The catalytic activity of this complex was investigated in the transfer hydrogenation of aldehydes and ketones. It was found that the optimum catalyst loading for this reaction was in the range of 0.1 to 0.4 mol.%. The hydrogenation reactions were carried out using water as the solvent and glycerol as an environmentally friendly hydrogen source. The catalytic efficiency of this complex was found to be comparable to that of other catalysts reported for these hydrogenations using glycerol or propan-2-ol as the hydrogen sources.
In a follow-up study, Saleem et al.[121] synthesized another complex bearing 1,2,3-triazole-based organoselenium ligand, [(η6-C6H6)RuClL]PF6 223, as an efficient catalyst for the reduction of carbonyl compounds. The complex 223 (Е = Е1 = Se) was obtained by the reaction of 1,4-bis(phenylselenomethyl)-1,2,3-triazole 222 with ruthenium compound 126 and NH4PF6 (Scheme 62). It was effective in the reduction of carbonyl compounds using glycerol and propane 2-ol as the hydrogen source with the product yield ranging from 95% to 100%. The reduction was carried out with 0.01 mol.% of complex 223 for 0.5 h.
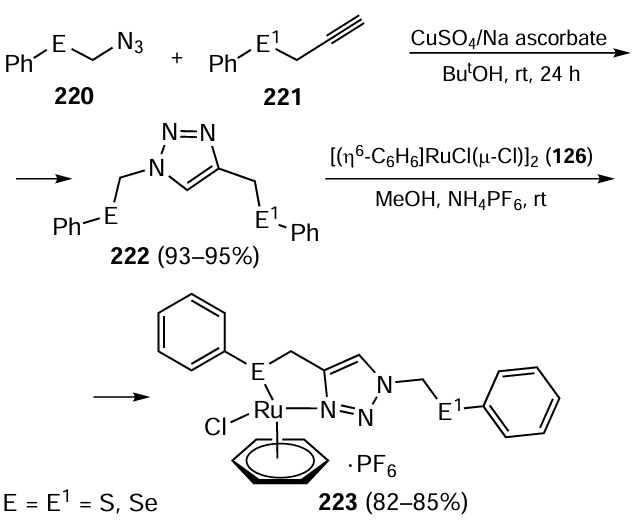
In 2023, Kumar et al.[122] reported a detailed synthesis of a 17-membered cyclic ligand 225 obtained by the reaction between 1,8-bis(2-(chloromethyl)phenoxy)octane 224 and Se powder (79). Further treatment of the macrocyclic ligand 225 with bis(acetonitrile)palladium(II) dichloride 26 gave trans-palladium(II) dichloride complex 226 (Scheme 63). The metal centre in this complex has a distorted square planar geometry. Notably, both the new ligand and complex showed resistance to moisture and air while remaining stable at ambient temperature.
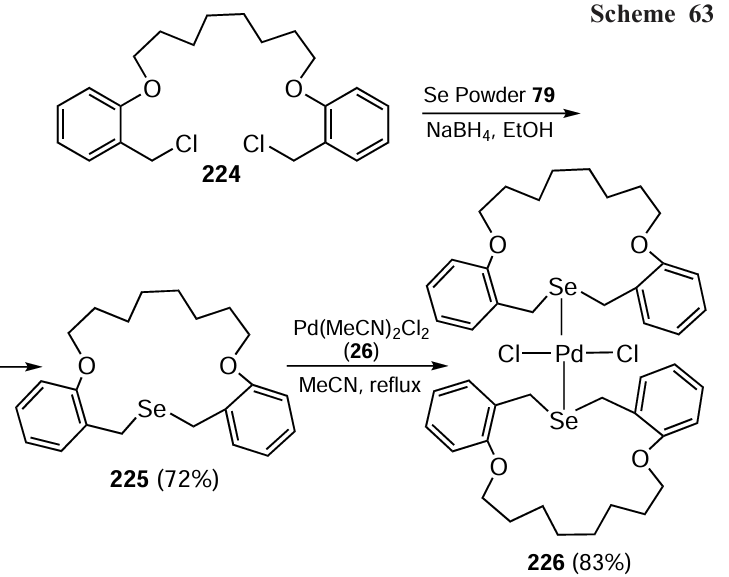
Palladium complex 226 was used as a catalyst for the dehydroxymethylation of dihydroxy compounds 227 containing long alkyl chains (Scheme 64). Typically, two single catalysts are tested in the dehydroxymethylation, one for the alcohol oxidation and another for the aldehyde decarbonylation. However, in this case, the catalyst 226 showed dual action in the dehydroxymethylation giving up to 91% efficiency at only 5.0 mol.% catalytic loading. This catalyst is suitable for a wide range of substrates and shows good tolerance to different functional groups. Interestingly, the same dihydroxy compounds with long alkyl chains were found to undergo macrolactonization when exposed to a ruthenium catalyst.
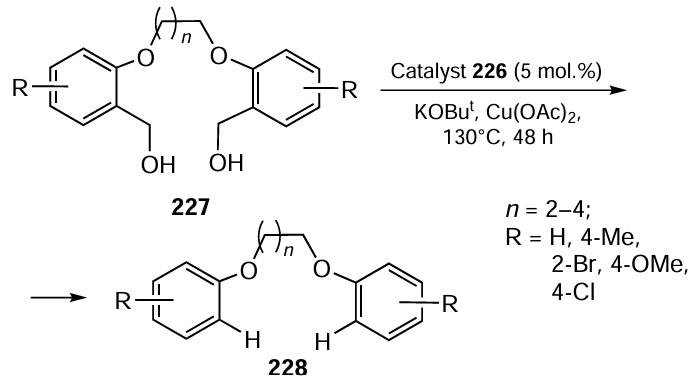
Arora et al.[123] described the preparation of the selenoether ligand 231 (L) by the reduction of [pyren-1-yl-CH=N – (CH2)2 – SePh] with NaBH4 followed by its transformation into organoselenium compound-stabilized copper NPs 232, which exhibit long-term stability (Scheme 65). These stabilized NPs 232 have been non-covalently immobilized onto a chitosan-layered magnetic support 230 to give Fe3O4@CS@L-stabilized-CuNPs 233.
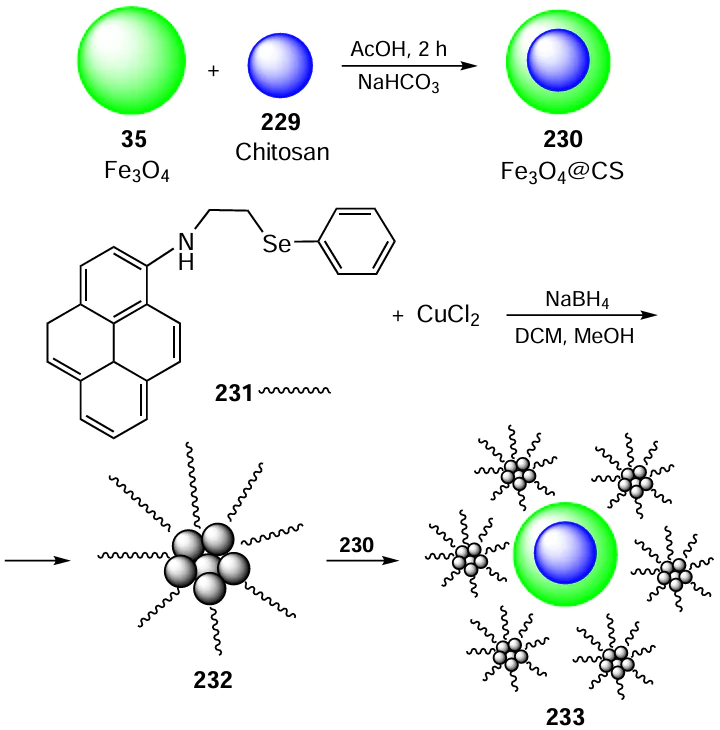
The resulting system Cu-NPs 233 was found to be a highly effective and reusable catalyst for low cost and environmentally friendly reduction reactions. Catalyst 233 performed well with various nitroarenes 234 in water at room temperature by achieving conversions to aminoarenes 178 of up to ³99% within remarkably short reaction times (Scheme 66). In addition, the catalyst 233 offers other advantages such as its magnetic separability and recyclability. Importantly, the catalyst demonstrates robustness by preventing the leaching of copper species and the ligand into the solution during the reduction reactions.
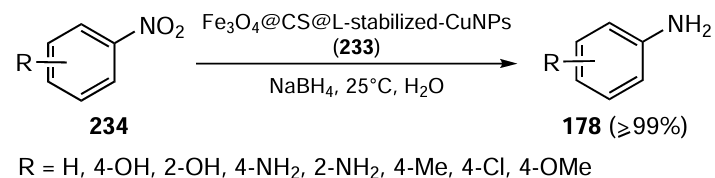
8. Oxidation reactions
The oxidation of alcohols to the corresponding carbonyl compounds is an important chemical transformation that is often promoted by transition metal-based catalysts. Complexes based on ruthenium, rhodium, and iridium, which belong to the class of transition metals, exhibit significant catalytic performance when paired with click-generated organoselenium ligands. Key features of these catalytic processes include: (i) exceptional stability of the catalyst in the presence of air and moisture which is advantageous for practical applications; (ii) high TON values, which indicate their efficiency in catalyzing multiple reactions with a single catalyst molecule; (iii) shortened reaction times, which is beneficial for increasing productivity and throughput in chemical processes and (iv) adaptability in the selection of oxidants for the reaction. Depending on the nature of the oxidant, these reactions can be categorized into two different types: the Oppenauer-type oxidation and classical oxidation facilitated by N-methylmorpholine N-oxide (NMO) as the oxidant.
The catalytically active complexes show high TON values in NMO-based oxidation reactions. The product yields obtained from the NMO-based oxidation are also significantly higher than those observed in the Oppenauer-type oxidation. However, a disadvantage of the NMO-based oxidation is that the reactions are typically carried out in dichloromethane.
Acetone has traditionally been used as a solvent in the Oppenauer-type oxidation process. As well as acting as a solvent, acetone also acts as an oxidant, which is a distinct advantage as it simplifies the reaction set-up. In addition, acetone is non-toxic, which is an advantage from a safety point of view. However, despite these advantages, the product yields obtained from the Oppenauer-type oxidation may not be as high as those obtained from the NMO-based oxidation. In both types of oxidation reactions, potassium carbonate and/or tert-butanol are commonly used as bases. These bases help to improve the yield of the desired products.
Ruthenium(II) complexes 211, 212, 215 and 216 (see Scheme 58, Scheme 59) were found to be quite efficient in the oxidation of primary and secondary alcohols to carbonyl compounds in the absence of base. The products were obtained in good to excellent yields with a catalytic loading of 0.01 – 1.0 mol.% in the presence of NMO. Catalysts 211 and 212 required 5 h to complete the reaction, whereas catalysts 215 and 216 showed complete conversion in 3 h. N-Methylmorpholine and water were the by-products of these reactions.[120, 121]
In NMO-based oxidation, rhodium and iridium complexes together with the base such as K2CO3 give very high product yields (Scheme 67). The Rh- and Ir-complexes 217 and 218 (see Scheme 60) were also used as catalysts with a catalyst loading of 0.1 – 0.01 mol.% for the oxidation of alcohols in the presence of NMO and by the Oppenauer-type oxidation reaction using acetone as both oxidant and solvent. It was found that the Oppenauer-type oxidation proceeded slowly compared to the NMO-based oxidation. The oxidation using NMO was completed within 3 hours whereas the Oppenauer-type oxidation required 6 h for a similar conversion. Nevertheless, the Oppenauer-type oxidation is significantly more cost-effective than the NMO-based oxidation and the isolation of the product is easier compared to the latter reaction.
9. Amide functionalization
Joshi and co-workers [124] reported a half-sandwich complex [(η6-C6H6)RuCl(Se)]PF6 236 as a highly effective catalyst for the conversion of aldehydes to amides. The organoselenium ligand 88 was synthesized from the chlorinated pyrazole 87 and was further treated with the rhodium compound 235 in ethanol to afford the novel Ru(II) complex 236 in 72% yield (Scheme 68). Similar complexes bearing sulfur- and tellurium-containing ligands were also obtained. Single crystal X-ray diffraction study has revealed a pseudo-octahedral half-sandwich piano-stool geometry around the Ru centre in the complex 236.
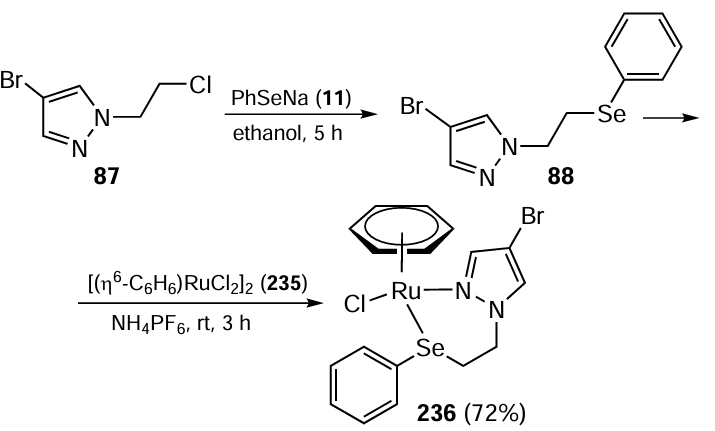
Complex 236 was found to be thermally stable and insensitive to moisture and air. The remarkable efficiency in the conversion of aldehydes 134 to amides 237 was observed when the reaction was carried out with a catalyst loading of 0.1 mol.% and a temperature of 100°C under aerobic conditions (Scheme 69).
Furthermore, this catalytic process requires no hazardous additives and produces amides in good to excellent yields without any by-products. It was found that the complex bearing an organoselenium ligand performed better than those with sulfur- and tellurium-containing analogues and the reason for the good efficiency was the stronger S-donor coordination properties of the selenium atom.
The same authors [61] pioneered the synthesis of Fe3O4@SiO2@SePh@Ru(OH)x nanoparticles 239. These NPs were prepared by coating nanostructured magnetic oxide Fe3O4 with silica and then reacting it with phenylselenyl chloride and RuCl3 · x H2O 238 under a N2 atmosphere in an aqueous medium (Scheme 70). The weight percentage of Ru in the catalyst was reported to be 5.48%. The resulting NPs 239 effectively promoted the one-pot conversion of aldehydes, nitriles, and benzylamine to primary amides. Notably, this conversion process takes place in water without the need for organic solvents. The synthesis of amides from aldehydes was successfully carried out by reacting aldehyde, NH2OH · HCl and NPs 239 in water at ∼ 120°C under aerobic conditions. Thus, the NPs 239 exhibit sustainable and efficient chemical transformations, providing a greener alternative to traditional synthetic methods.
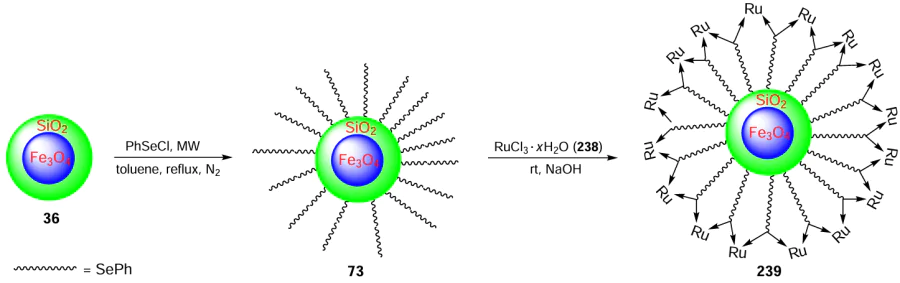
The hydration of nitriles was carried out in water. The product 237 was obtained in 92% yield by heating the nitrile 240 with Fe3O4@SiO2@SePh@Ru(OH)x 239 at 120°C under aerobic conditions (Scheme 71). Once the reaction is complete, the catalyst could be easily separated using an external magnet. The NPs 239 could be reused for the same transformation up to seven cycles without any loss in their catalytic activity. The reaction product was obtained in up to 86% yield when the same reaction was performed at the gram scale.

10. Olefin metathesis
Grela and co-workers [125] found that replacing the sulfur atom in the Hoveyda – Grubbs type structure with selenium imparts interesting light activation properties to the resulting catalyst. The cis-dichlorido seleno-chelated Hoveyda – Grubbs type complex 243 was synthesized by a simple and highly efficient three-step procedure. The selenium-containing styrene derivative 242 was obtained in high yield from 2-bromobenzaldehyde 241. In addition, the Hoveyda-type complex 243 was successfully obtained in 81% yield by the reaction of ruthenium indenylidene complex (Ind – II) with the selenium-containing styrene derivative 242 in the presence of anhydrous CuCl at 80°C (Scheme 72).
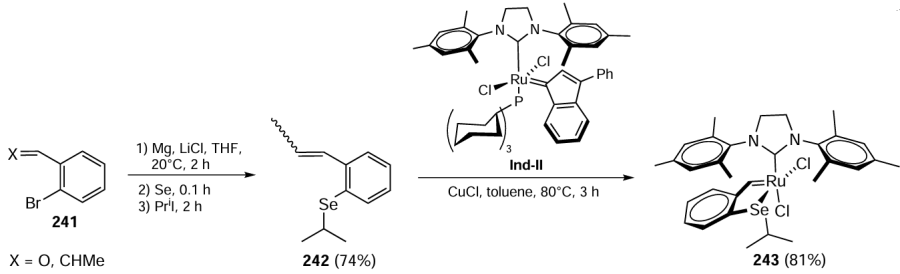
The catalytic properties of Ru complex 243 have been thoroughly investigated. The versatility of this complex was demonstrated by both thermal and light activation. The seleno-chelated ruthenium catalyst showed thermally switchable behaviour in the ring-closing metathesis (RCM) of diethyl diallyl malonate 244. The reaction proceeds on heating compound 244 with the complex 243 at 80°C (Scheme 73). Cooling the reaction mixture to room temperature terminates the reaction, which can be restarted by heating to 80°C. Complex 243 showed no signs of decomposition even after several cycles of heating and cooling. Upon irradiation of the reaction mixture of diethyl diallyl malonate and Ru complex in benzene at 365 nm, the reaction proceeds rapidly but stops in the absence of light. The latency of this complex was also confirmed in the ring-opening metathesis polymerization (ROMP) of cyclooctene 246 and the self-cross metathesis (self-CM) of methyl oleate. In all these reactions, Ru complex 243 exhibited high activity and efficiently catalyzed the polymerization reactions of norbornene and cyclooctene with a catalyst loading as low as 10 ppm. In addition, the self-CM of methyl oleate showed excellent selectivity of more than 99% towards the C=C bond shift.
11. Hydrolysis of phosphate diesters
Recently, Peralta and co-workers [126, 127] synthesized four different organoselenium-copper(II) mononuclear complexes 250. All ligands were obtained according to the known procedure (Scheme 74).[128]
Complexes 250 were obtained as green solids by the reaction of ligand 249 with CuCl2 in ethanol at room temperature within 1 h in 80 – 92% yields. Subsequently, the mononuclear copper(II) complexes 250 were used as the catalysts for the cleavage of phosphate diesters. Bis(2,4-(dinitrophenyl)phosphate) (2,4-BDNPP) was chosen as the model substrate. The hydrolysis of the active substrate was evaluated by the increase of its absorbance at 400 nm over time due to the formation of 2,4-dinitrophenolate (2,4-DNP). The best results were obtained at the optimized pH of 6.5. In a slightly acidic medium the water molecules lose a proton to form [CuL(OH2)(OH–)]. This intermediate is catalytically active and respresents a hydroxo nucleophile that facilitates the cleavage of the P – O bond. The turnover rate of the reaction for the substituted complex was reported to increase in the order p-H >p-Me > p-Cl > p – OMe.
The mechanism for the above reaction was proposed based on the results obtained and the literature data.[129] Intermediate 251 represents the proposed structure in the solid state, whereas in aqueous solution it is converted to form 252 due to the exchange of the weaker chloro ligand with water (Scheme 75). Intermediate 253 is the active species in the catalytic reaction with a hydroxy group acting as a strong nucleophile. Intermediate 254 illustrates the substrate entry and subsequent nucleophilic attack. The 2,4-DNPP group is then released and the catalytic cycle starts again with species 255.
12. Conclusion
This review covers the synthesis of a variety of organoselenium ligands and their application as key synthetic intermediates for the formation of various transition metal complexes. The resulting selenium-ligated transition metal complexes have been investigated for their catalytic potential in various catalytic reactions and have shown high catalytic activities in synthetically important organic transformations including carbon-carbon and carbon-heteroatom bond formation reactions. The reusability and low catalytic loadings of the selenium-ligated transition metal-based catalysts make these catalytic processes more relevant to the organic synthesis. Various sulfur-ligated metal complexes have also been prepared by several research groups as sulfur has a better ability to coordinate with metals compared to the selenium atom, which has been studied as a catalyst in similar catalytic reactions. The catalytic potential of the selenium-ligated complexes has been compared with that of the sulfur-ligated metal complexes and in some reactions the sulfur-ligated complexes outperformed their selenium-containing counterparts, while in a few reactions the reverse was true. The use of selenium-ligated transition metal complexes has advantages over traditional metal catalysis in terms of higher catalytic activity, better control of selectivity and the ability to operate at moderate temperatures. The main disadvantage of these complexes is the challenge of storage due to their stability in air and wet conditions. Our review article will provide an ultimate platform for the organic chemists who are working in the developments of organic synthesis by involving catalysis to develop new synthetic approaches to synthetic and medicinally important compounds including natural products. The review will also inspire the inorganic chemists to design new transition metal complexes with organoselenium and other ligands and evaluate their catalytic potential in organic synthesis.
Acknowledgement
R.M. acknowledges the support from the Department of Science and Technology, New Delhi, India, for WOS-A project SR/WOS-A/CS-107/2018. M.D.K. thanks the Ministry of Science, Technological Development and Innovation of the Republic of Serbia (Agreements No. 451-03-66/2024-03/200378, 451-03-66/2024-03/200122 and 451-03-65/2024-03/200122). F.V.S. is thankful to VIT Chennai for providing infrastructure for preparing the review article and CSIR, New Delhi for providing the research grant [Grant No. 02/(0330)/17-EMR].
13. List of abbreviations
AIBN — 2,2'-azobis(isobutyronitrile),
APTES — (3-aminopropyl)triethoxysilane,
DCE — 1,2-dichloroethane,
DMA — N,N-dimethylacetamide,
DME — 1,2-dimethoxyethane,
GO — graphene oxide,
MAO — methylaluminoxane,
MNP — magnetic nanoparticle,
NBS — N-bromosuccinimide,
NHC — N-heterocyclic carbene,
NMO — N -methylmorpholine N-oxide,
NMP — N-methyl-2-pyrrolidone,
NP — nanoparticle,
PivOH — pivalic acid,
ROMP — ring-opening metathesis polymerization,
SMC — Suzuki – Miyaura coupling,
TBAF — tetra n-butylammonium fluoride,
TBAI — tetra n-butylammonium iodide,
TBHP — tert-butyl hydroperoxide,
ТМА — trimethylaluminium,
TOF — turnover frequency,
TH — transfer hydrogenation,
TON — turnover number,
TOP — tri-n-octylphosphine.
References









