


Keywords
Abstract
This review highlights the dynamics of the development and synthetic application of the recently discovered reaction of acetylenic carbanions, generated in superbase media, with the C=N bond of different classes of substrates. A fundamental feature of this reaction is its synthetic divergence, i.e. its ability to proceed in different directions, which manifests itself in novel transformations to selectively deliver structurally different synthetically important products (depending on the structure of the acetylenes and substrates with C=N bond). The review also discusses cascade processes, in which the key intermediates containing the C=N bond add acetylenes thereby participating in the self-organization of diverse and potentially useful compounds. The competitive advantage of the reaction and its daughter branches is environmental safety (neutrality), based on the fundamental chemical nature of these reactions as addition processes that occur without the release of by-products.The bibliography includes 133 references.
1. Introduction
Nowadays, the application of acetylene, its substituted analogues and functional derivatives as building blocks for the design of complex molecular architectures is one of the focal points of organic, medicinal chemistry and materials science, as evidenced by the ever-growing number of original research papers and reviews.[1-13] Such situation is predetermined by the industrial availability of acetylene,[14, 15] its prospects to be renewable raw material (production from charcoal and lignocelluloses),[16-18] as well as high and multifaceted reactivity of the triple bond. The competitive advantage of acetylenes as an efficient and environmentally benign source of basic organic chemicals is that their reactions are mainly the addition processes, which exclude the release of any hazardous by-products, i.e., such reactions are safe (neutral) for nature and humans. In addition, most reactions involving acetylenes are exothermic, i.e., energy-saving. Another unique feature of acetylenes is the dichotomy of their chemical properties: they can react simultaneously (or sequentially) as nucleophiles and electrophiles that enables the self-organization of several acetylene molecules into complex ordered structures incorporating other small molecules.
The role of acetylenes in the organic synthesis is difficult to overestimate. The addition of acetylenic carbanions to the C=O bond alone, which usually occurs in the presence of bases (Favorsky reaction),[19, 20] is a special direction in industrial and fine organic synthesis. A breakthrough in the development of this reaction occurred, when superbase media began to be systematically used to promote it.[21-23] In the presence of superbases, aldehydes and ketones add acetylene much more actively than under traditional conditions: various acetylenic alcohols can be synthesized in high yields without pressure, cooling and large amounts of solvents (mandatory conditions of the classical ethynylation).
At the same time, despite the known examples of acetylene addition to imines promoted by salts and complexes of transition metals,[24-26] the addition of acetylenic carbanions, generated from terminal acetylenes in strongly base media (superbase media), to the C=N bond remained for a long time a Terra incognita of organic chemistry. At the first glance, a hurdle is that the attack of the weakly electrophilic C=N bond by the acetylenic carbanion is kinetically unfavorable. It has since been shown that this hurdle can be overcome by carrying out the reaction in superbase media, of which the simple, cheap and easy to use are systems such as KOBut/DMSO (pKa ~ 30 – 35).[27, 28] Indeed, in such a medium, the addition of acetylenes to the C=N bond is not only possible, but easily realizable.
2. Reaction of acetylenic carbanions with N-arylimines
It was found that aryl- and hetarylacetylenes react with N-aryl(hetaryl)ketimines in the presence of the KOBut/DMSO system (40 °C, 10 min), providing a one-pot synthesis of propargylamines (Scheme 1).[29] The reaction involves the addition of acetylenic carbanions to the C=N bond. This is a first example of the nitrogen analog of the Favorsky reaction.
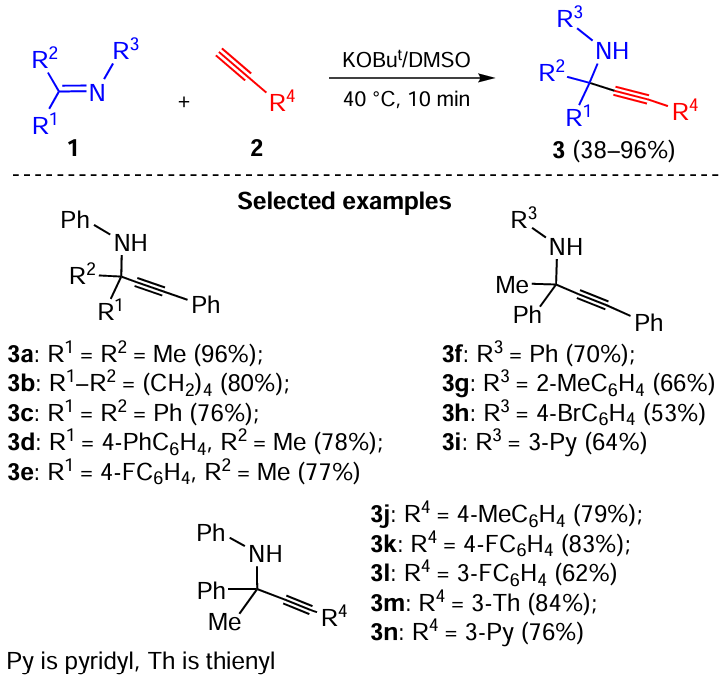
The reaction is efficient for N-aryl(hetaryl) ketimines 1 and a large series of aryl- and hetarylacetylenes 2 giving 38 – 96% yields of internal propargylamines. Evidently, this reaction is made possible, because in the superbase media, the concentration of acetylenic carbanions is increased with simultaneous enhancment of their nucleophilicity (due to desolvation). Also, electrophilicity of the C=N bond is strengthened due to the coordination of the potassium cation with the electron lone pair of the nitrogen atom (electrophilic assistance). Acetylene is also activated by coordination with the potassium cation (Tedeschi complexes) [30] and by complexation with dimethylsulfoxide (crystalline complexes of acetylene with DMSO are known).[31]
Generally, for the whole reaction, the integral substituent effect is consistent with the mechanism of the process, which represents a nucleophilic addition to the polarized C=N bond (Scheme 2).
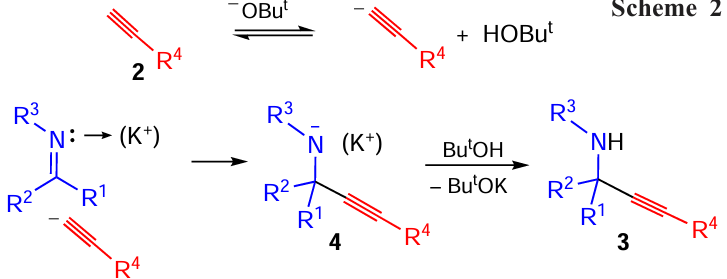
Indeed, the formation of the tetrasubstituted carbon center (intermediate 4), resulting from the initial nucleophilic attack of the acetylenic carbanion at C(sp2) atom, should be sterically hindered, while the acceptor substituents, both in ketimine and acetylene, should facilitate the process, in particular by increasing the concentration of the acetylide anions and by distributing the negative charge appearing on the nitrogen atom in intermediate 4 over the adjacent aromatic (heteroaromatic) system (aza-analogue of benzyl anions).
The reaction is thus general for various acetylenes and ketimines and opens a straightforward access to a large number of propargylamines, which find application as privileged molecules [32-35] due to their synthetic [36, 37] and pharmaceutical [38-42] importance. The first syntheses of these compounds by catalyst-free (K.Mannich) [43] or copper-catalyzed (W.Reppe) [44, 45] aminoalkylation of acetylenes were complemented in 2001 by the iridium-catalyzed three-component reaction between primary amines, aldehydes and acetylenes.[46, 47] This approach (A3 coupling) is currently being implemented in the presence of various transition metal catalysts.[48-52] The key step of A3 coupling is shown [29, 53, 54] to be the nucleophilic substitution of the hydroxyl group in intermediate α-hydroxyalkyl amines by π/σ-metal acetylene complexes. At the same time, the similar reactions with ketones (especially with aromatic ketones), amines and acetylenes (KA2 coupling) and thus the synthesis of the corresponding propargylamines with a quaternary carbon atom, remain undeveloped due to the lower reactivity of ketones in this process.[48, 53, 55-59] The above reaction between C=N bond and acetylenic carbanions now fills this gap.
The parent and most fundamental member of acetylenes, unsubstituted acetylene, has been out of the reaction scope for some time. Meanwhile acetylene now occupies a specific place among alkynes due to its double C – H acidic functionalities, higher and multifaceted reactivity, and much grater availability (industrial production). In this respect, the superbase-promoted reaction of acetylene gas with C=N bond to give terminal propargylamines is of paramount importance, particularly, because the latter are much more reactive [60-63] compared to their internal congeners.
It turns out that ethynyde-anions, generated from acetylene gas, add to the C=N bond of ketimines even more easily than acetylenic carbanions generated from aromatic and heteroaromatic alkynes: the reaction proceeds in the KOBut/DMSO system at room temperature under slightly excess acetylene pressure (~ 2 atm), providing terminal propargylamines 5 in up to 94% yield (Scheme 3).[64]
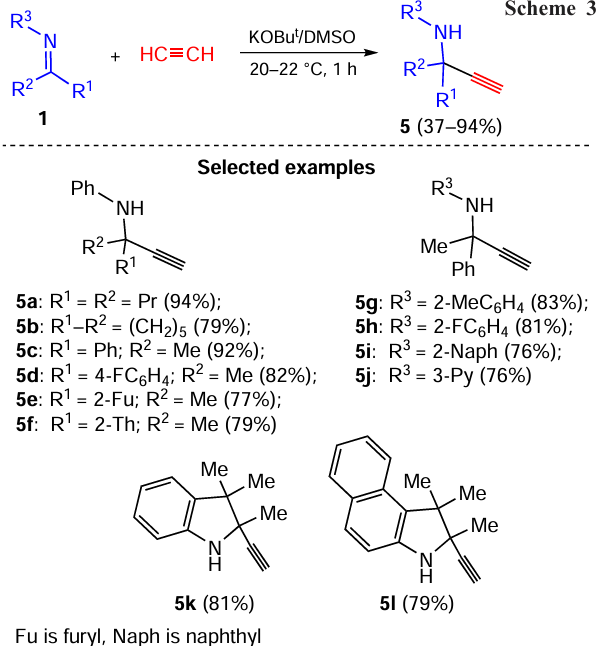
The synthesis covers various ketimines having aromatic and heteroaromatic substituents both in ketone and amine counterparts. Under atmospheric pressure, the ethynylation of ketimines with acetylene gas proceeds with almost the same efficiency. A facile ethynylation of indoles 1k,l shows that the terminal acetylenic substituent can be directly introduced into nitrogen heterocycles.
The reaction is remarkably chemoselective: the expected products of the double ethynylation, 1,4-diaminobutynes, are not observed. This is due to a lower CH acidity and steric screening of the acetylene substituent at the quaternary carbon atom. It is noteworthy that the oxygen analogue of this synthesis (Favorsky reaction) [19] is commonly accompanied by the formation of acetylenic diols along with major products (acetylenic alcohols).
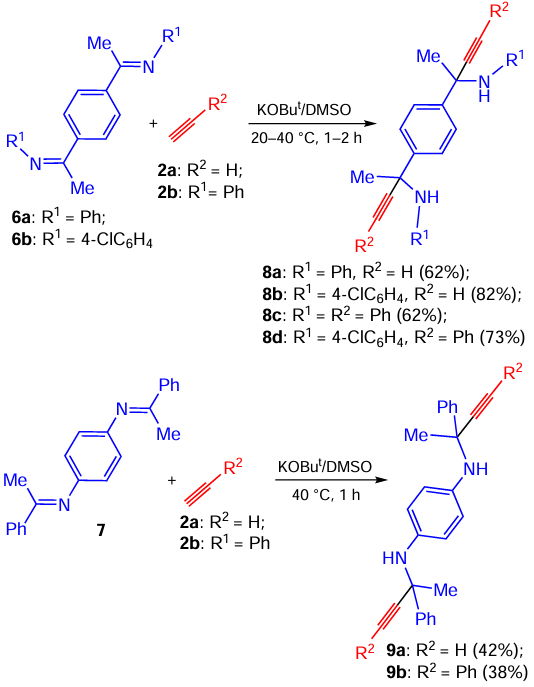
As a further development of the ethynylation of C=N bond-containing compounds, the KOBut/DMSO-promoted addition of acetylenes to structurally analogous C- and N-linked 1,4-bis(imino)benzenes 6, 7 was studied.[65] The reaction proceeded under mild conditions (room temperature or slight heating) to produce double ethynylation products 8, 9 (Scheme 4), attractive precursors for the preparation of novel functional materials.[66, 67]
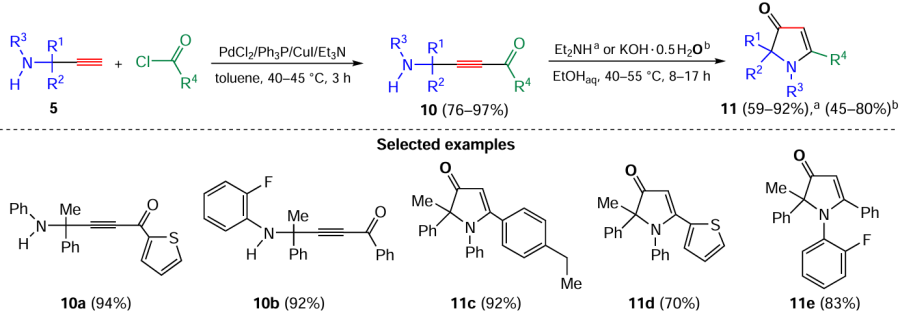
The above new families of propargylamines, particularly those with terminal acetylenic moiety, are now becoming rewarding objects for organic synthesis. So, Pd/Cu-catalyzed cross-coupling of propargylamines 5 with aromatic acyl chlorides providing α-(het)arylaminoacetylenic ketones 10 was realized.[68] After the treatment with bases (KOH or Et2NH), these ketones undergo intramolecular cyclization to pharmaceutically promising [69-72] 1,2,5-tri(het)aryl-1,2-dihydro-3H-pyrrol-3-ones 11 (Scheme 5).[68] The substrate scope of this atom-economy approach allowed the simultaneous introduction of various aromatic, condensed aromatic and heteroaromatic substituents in 1, 2 and 5 positions of the pyrrolone scaffold.
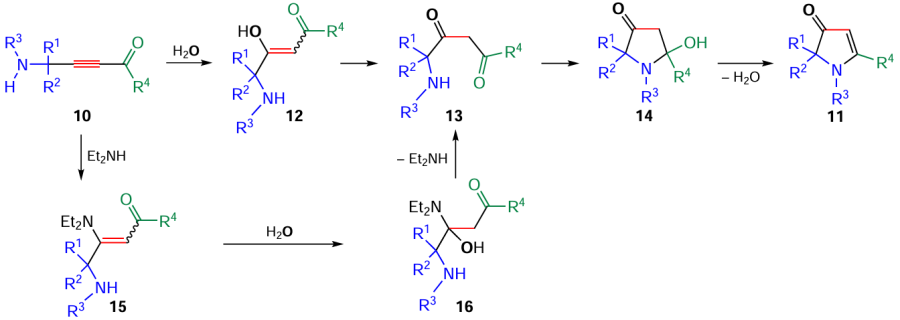
The key step of the cyclization (Scheme 6) is the addition of a water molecule (in case of KOH · 0.5 H2O catalysis) giving enols 12, which rearrange to 1,3-diketones 13. The latter undergo the ring closure by the attack of NH moiety at the carbonyl group to afford intermediates 14, which, after elimination of a water molecule, produce pyrrolones 11. In the case of Et2NH catalysis, the first step of the cyclization is the addition of Et2NH to the triple bond to form intermediate enamines 15, which add a water molecule giving hemiaminals 16. The latter, after release of Et2NH, lead to the same 1,3-diketones 13, which are transformed to pyrrolones 11.
The synthesis of 3H-pyrrolones 11 can be implemented in a one-pot version by consecutive treatment of propargylamines 5 with acyl chlorides and bases.[68]
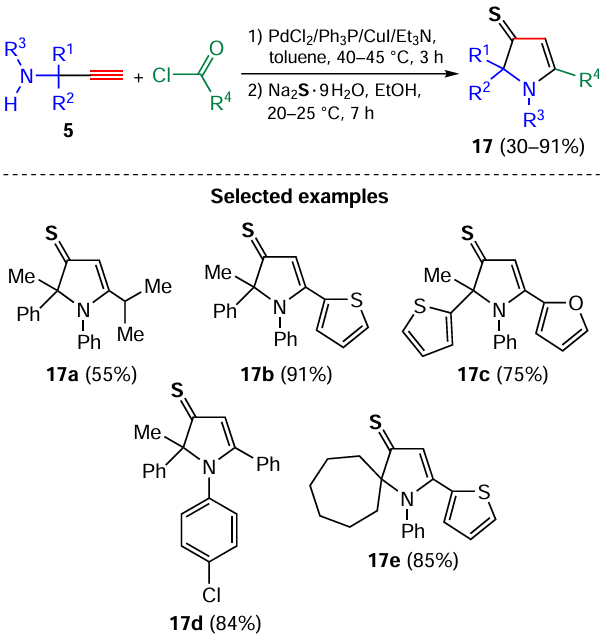
The efficient one-pot synthesis of 1,2,5-trisubstituted-1,2-dihydro-3H-pyrrole-3-thiones 17, essentially new heterocyclic systems, by the successive treatment of propargylamines 5 with acyl chlorides and sodium sulfide has been developed (Scheme 7).[73] The synthesis comprises the addition of hydrosulfide-anions to the aminoacetylenic ketones 10 formed, followed by dehydrative cyclization of the prototropically rearranged adducts.[73]
A fundamental feature of the reaction of acetylenic carbanions with the C=N bond is its substrate-driven divergence: each new class of the starting C=N bond-containing compounds gives structurally different products. For example, aldimines 18 react with acetylenes 2 in the superbase triad KOBut/DMSO/THF under mild conditions (14 °C) to afford 1-azadienes 19 of the E configuration relative to the C=C bond (Scheme 8).[74] The reaction has been successfully extended to aryl(hetaryl)acetylenes and aldimines derived from aryl(hetaryl)substituted aldehydes and aryl(hetaryl)amines, providing a straightforward atom- and energy-economic access to 1-azadienes, versatile building blocks in organic chemistry[75-77] and privileged intermediates in the total syntheses of natural molecules.[78-81]
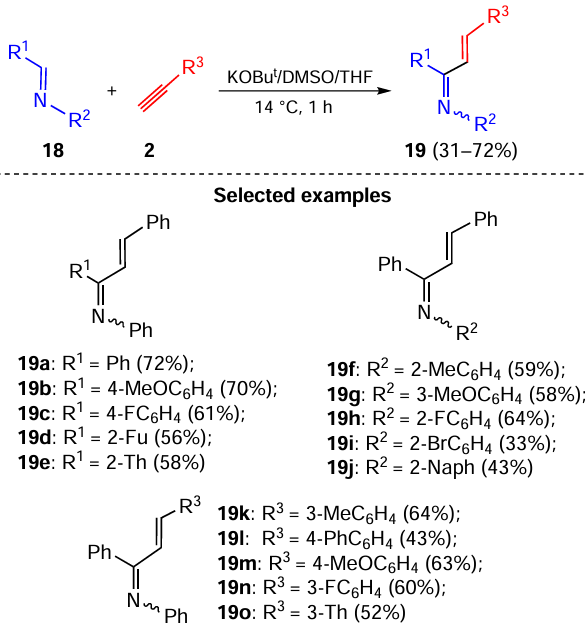
Apparently, the reaction starts with the addition of acetylenic carbanions to the C=N bond of aldimines 18 (Scheme 9). The propargylamines 20 thus formed undergo further prototropic isomerization to 1-azadienes 19 via allenic intermediates 21. The fast double prototropic isomerization at low temperature (14 °C) has been proven experimentally: the specially synthesized intermediate propargylamine 20a [82] is immediately and quantitatively transformed to 1-azadiene 19a under the reaction conditions (see Scheme 9).
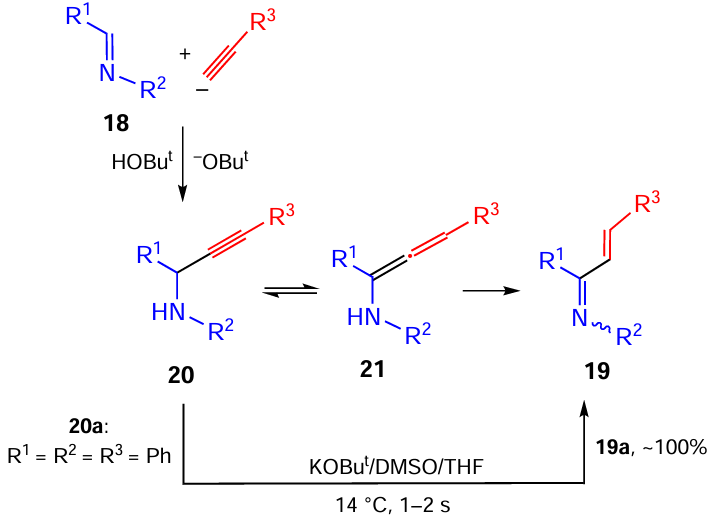
The result of the reaction of 2-iminopyridines 22 (another group of C=N bond-containing substrates) with aryl(hetaryl)acetylenes 2 turned out to be unexpected: in the NaOBut/DMSO system at room temperature, the formation of benzyl imidazo[1,2-a]pyridines 23 was observed (Scheme 10).[83]
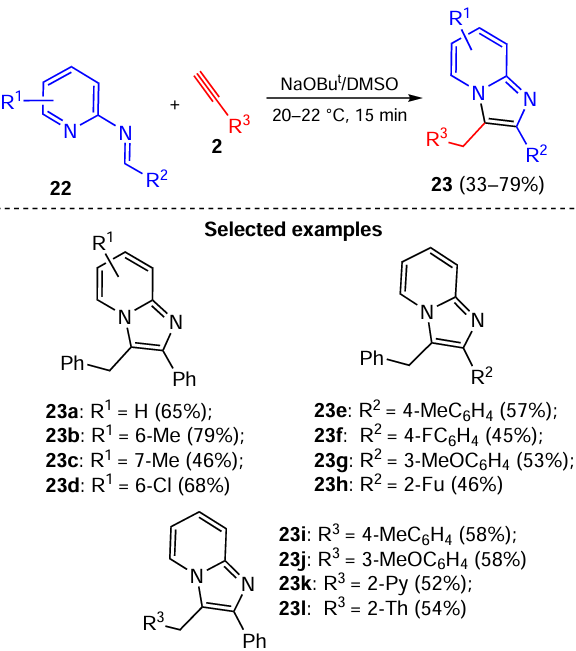
Even more unexpected was the result of the interaction of acetylenes 2 with a 2-fold excess of 2-iminopyridines 22: under similar conditions, stilbene imidazo[1,2-a]pyridines 24 were stereoselectively assembled from two molecules of 2-iminopyridine 22 and one molecule of acetylene 2 (Scheme 11).[83]
These results can be understood as the addition of acetylenic carbanions to the C=N bond of imines 22 and further evolution of the intermediate propargyl-1,3-diaza-1,3,5-trienyl anions 25 involving their intramolecular cyclization to anions 26, which are intercepted by a proton of the medium to give benzyl imidazopyridines 23 or by a second molecule of imine 22 followed by elimination of 2-aminopyridine from adducts 27 to give stilbene imidazopyridines 24 (Scheme 12).[83]
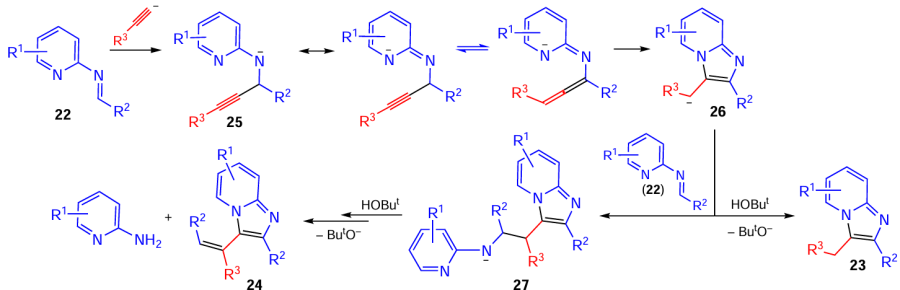
This cascade process (see Scheme 12) is characterized by the following features: 1) addition of acetylenic carbanions to the C=N bond with the long-range distribution of the negative charge appearing at the imine nitrogen atom over the whole pyridine ring of anionic intermediates 25; 2) easy (room temperature) and fast (15 min) intramolecular nucleophilic addition of the pyridine nitrogen atom to the propargyl (allenyl)moiety of anions 25; 3) extremely facile addition of benzylic carbanions 26 to the C=N bond of imines 22 followed by stereoselective elimination of the 2-aminopyridine molecule. Generally, this reaction provides a simple route to products incorporating the stilbene,[84-86] pyridine,[87-89] and imidazole [90-92] structural units, which are the focus of current medicinal research and materials science. Indeed, these compounds became the objects of photochemical researches. It was found that stilbene imidazo[1,2-a]pyridines 24 undergo selective oxidative cyclization upon UV irradiation (λ = 365 nm) to 5,6-diarylnaphtho[1',2':4,5]imidazo[1,2-a]pyridines 28, which fluoresce intensely in the blue region (λem = 417–441 nm) with quantum yields of 0.20 – 0.60 (Scheme 13).[93]
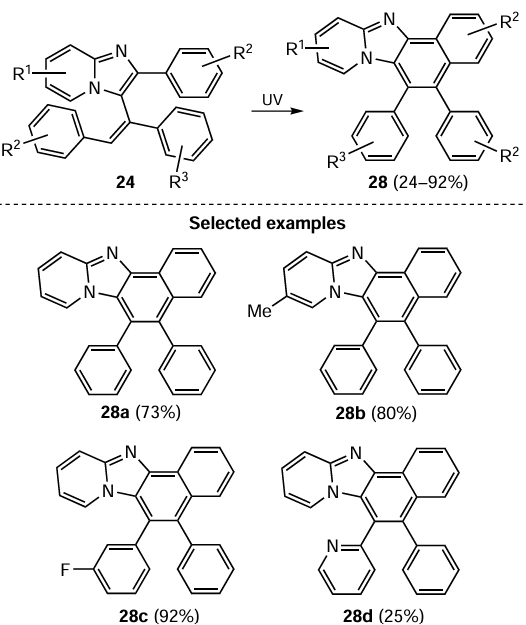
As a logical development of the acetylenic carbanion/C=N bond interaction, it was shown that aromatic aldazines 29, the substrates containing two conjugated C=N bonds (2,3-diaza-1,3-dienes), readily react with arylacetylenes 2 in the NaOBut/EtOH/DMSO superbase triad at room temperature to give 4-arylmethyl-3,5-diaryl-1H-pyrazoles 30 (Scheme 14).[94]
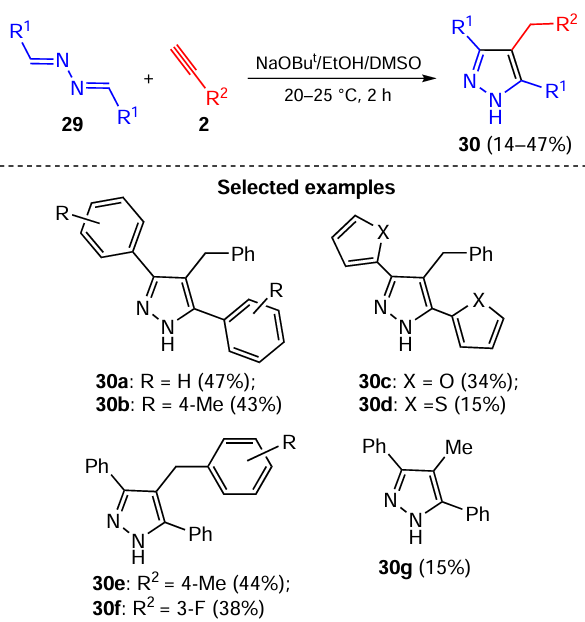
The reaction was rationalized [94] as proceeding via the diazaallyl anions 31, the adducts of acetylenic carbanions to the C=N bond, which further undergo the proton transfer processes and intramolecular cyclization to pyrazoles 30 (Scheme 15).
3. Reaction of acetylenic carbanions with N-benzylimines
The reaction of N-benzylketimines 32 with acetylene gas in the KOBut/DMSO system proceeded in a quite unexpected direction (another appearance of the synthetic divergence): instead of the formation of the corresponding propargylamines, the stereoselective assembly of 2-azadienes 33 was observed (Scheme 16).[95]
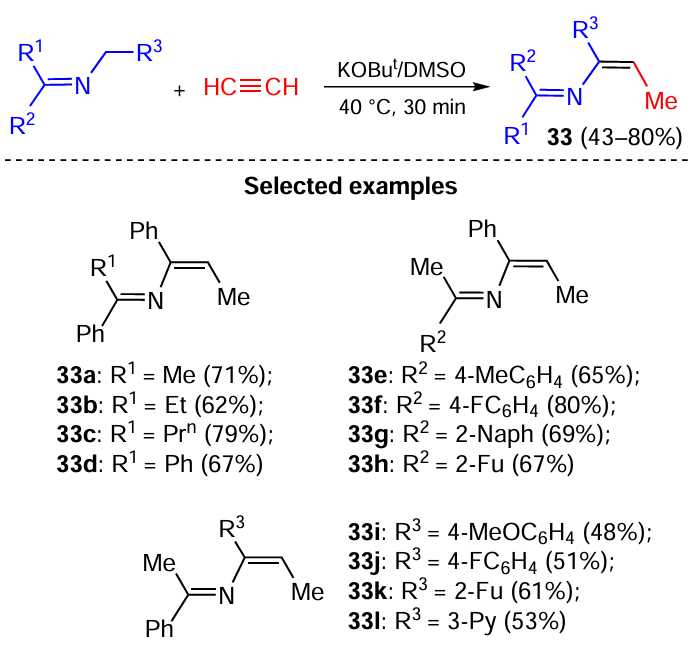
This is due to the higher acidity of the N-benzylketimines (pKa ~ 24) [96, 97] compared to acetylene (pKa 29.7).[98] Consequently, the key step of this reaction is the addition of the deprotonated N-benzylketimines (azaallyl anions 34) to acetylenic triple bond (Scheme 17). The electron density delocalization in anions 34 is extended over the adjacent aromatic (heteroaromatic) substituent that additionally stabilizes these anionic species and increases their concentration in the reaction mixture. The stereoselectivity of the reaction is likely originated from the chelation of the potassium cation with both sites of the azaallyl anions 34. After the insertion of the acetylene molecule between the carbanionic center and the potassium cation and neutralization of the vinyl carbanions 35, the intermediate nonconjugated azadienes 36 are formed. The latter are prototropically isomerized, still being retained in the coordination sphere of the potassium cation that ensures the Z orientation of the forming Me group to give conjugated 2-aza-1,3-dienes 33.
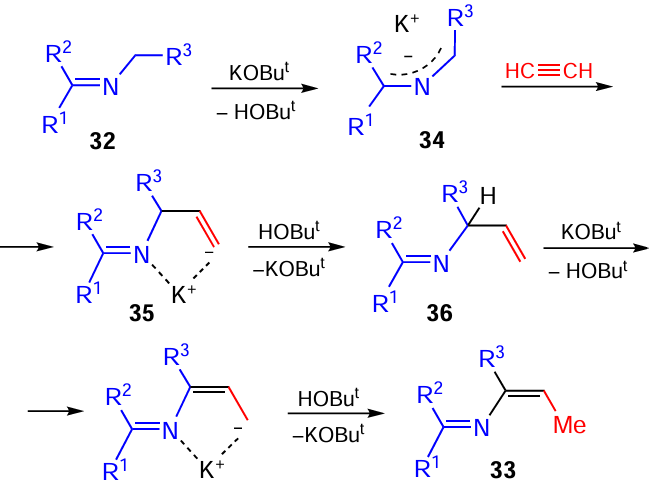
Another direction of the acetylene/C=N bond interaction in strongly base media is the formal [3 + 2] cycloaddition of N-benzylketimines 32 to aryl(hetaryl)acetylenes 2 to afford polyarylated 1-pyrrolines 37, which are in situ oxidized to 2,3,5-tri(het)aryl-2H-pyrroles 38 (Scheme 18),[99] promising objects for applications in heterocyclic synthesis.[100, 101] The intermediate 1-pyrrolines 37, valuable templates for new drugs [102] and their precursors,[103-105] can be isolated separately in up to 91% yield.
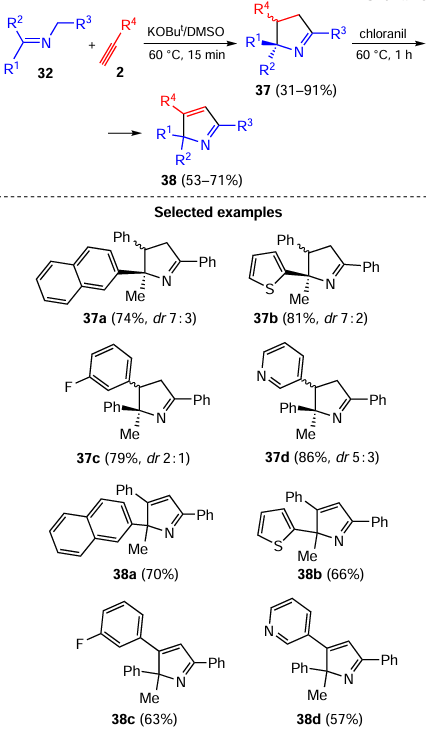
The assembly of pyrrolines 37 includes the addition of deprotonated N-benzylketimines (azaallyl anions 34) to the triple bond of acetylenes 2 followed by the prototropic isomerization of carbanions 39 to the conjugated intermediates 40. The latter attack the C=N bond to close the pyrroline ring (Scheme 19).
When N-benzylaldimines 41 were used instead of the N-benzylketimines 32 for the reaction with acetylenes 2, the process proceeded again differently, giving 2,3,5-triaryl-1-pyrrolines 42 as two tautomers with 1,2- and 1,5-location of the double bond in a ratio of ~ 4 : 1 (Scheme 20).[106] A distinctive feature of this further branch of the acetylene/C=N bond interaction is its diastereoselectivity: both pyrroline tautomers are formed as trans-diastereomer. The oxidation of the tautomeric pyrrolines 42 (without their isolation from the reaction mixture) gave inaccessible [107-109] polyarylated 1H-pyrroles 43 (see Scheme 20).
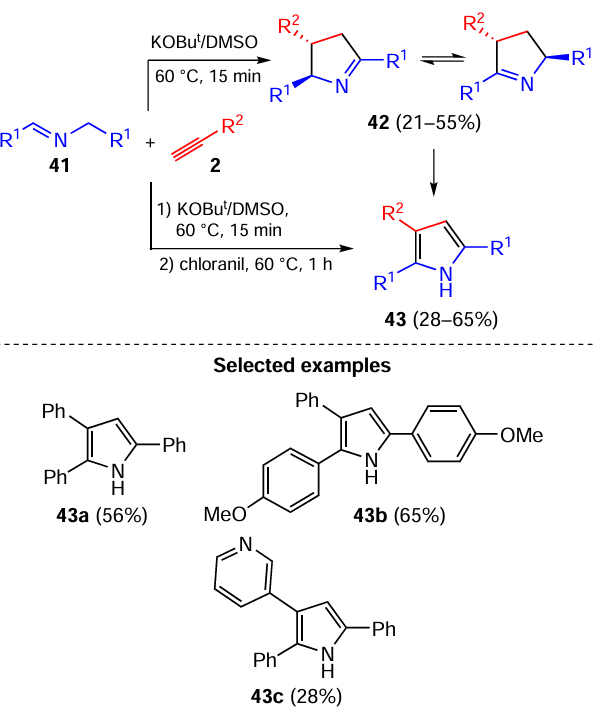
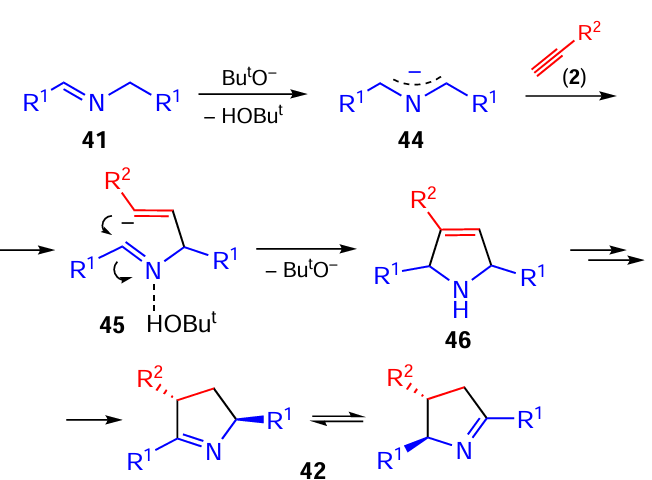
The reaction was assumed [106] to involve the addition of azaallyl anions 44 to the acetylenic triple bond (Scheme 21). Emerging carbanions 45 attack the C=N bond closing the pyrroline cycle, probably with the electrophilic assistance of a proton from the medium. Intermediate pyrrolines 46 are rearranged by a series of proton transfers to give the final tautomeric pyrrolines 42. The concerted nature of the cyclization stage has been supported by quantum-chemical calculations (B2PLYPD2/6-311+G**//B3LYP/6-31+G*).[110]
4. Addition of acetylenic carbanions to the C=N bond as a key stage in the self-organizations of complex molecules
In the context of the acetylene/C=N bond interaction in superbase media, a phenomenon of self-organization of complex molecular structures from several molecules of acetylene and amines has been observed. These processes are initiated and controlled by the dichotomy of acetylene reactivity, i.e. its ability to act alternatively both as an electrophile and a nucleophile.[111] The key intermediates of this reaction cascade contain the C=N bond, which adds acetylenic carbanions thereby triggering the self-organization. Typical examples of the above self-assemblies are analyzed below.
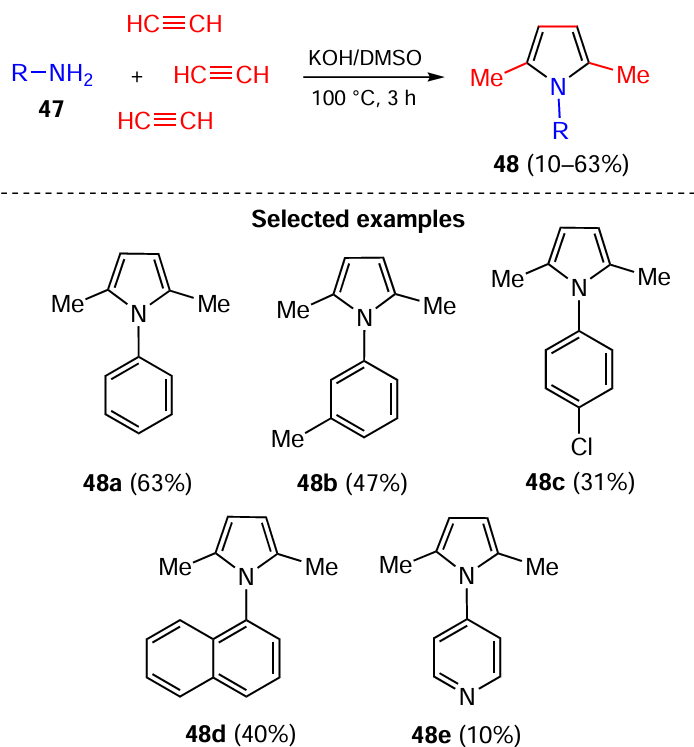
Arylamines 47 react with excess acetylene gas in the KOH/DMSO system to form 1-aryl-2,5-dimethylpyrroles 48, which are built up from three molecules of acetylene and one molecule of amine 47 (Scheme 22).[112] Various substituted anilines, condensed aromatic and heteroaromatic amines tolerate the reaction.
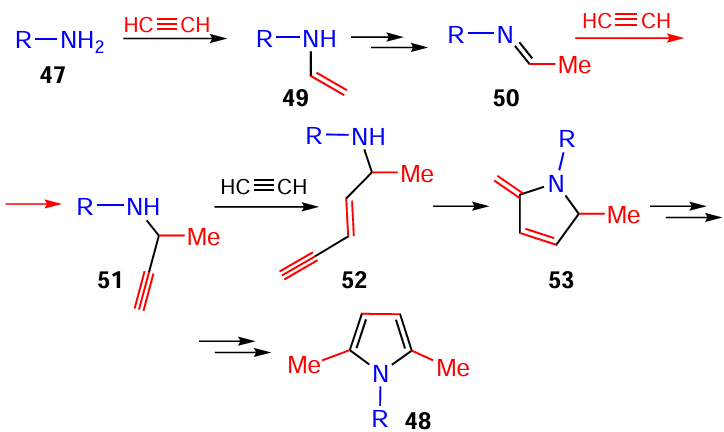
The reaction is initiated by the nucleophilic attack of arylamines 47 at the acetylenic triple bond to give N-vinylamines 49, which, after the prototropic isomerization, are transformed to imines 50 (Scheme 23). The latter accept the second acetylene molecule (in carbanionic form) to generate intermediate propargyl amines 51. The third acetylene molecule (in carbanionic form) is then inserted into the triple bond of propargyl amines 51, and the intramolecular cyclization of intermediates 52, after aromatization of dihydropyrroles 53, gives the final pyrroles 48. This mechanism has been supported by DFT calculations (B2PLYP(D3)/6-311+G**//B3LYP/6-31+G*) of activation barriers and thermodynamics of all steps.[113]
An even more sophisticated reaction cascade led to the self-organization of 1-aryl-3-ethyl-4-vinylpyrroles 54 from arylamines 47 and four molecules of acetylene, when the KOH/DMSO system was replaced by a more basic KOBut/DMSO system (Scheme 24).[114]
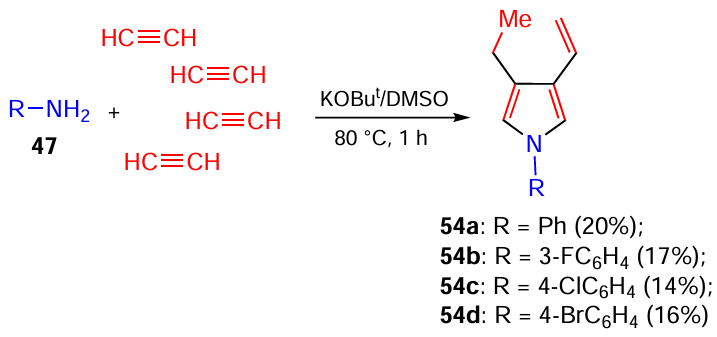
The few known syntheses of 3,4-disubstituted pyrroles are either multistep,[115, 116] provide poor yields and restricted coverage,[117] or require costly chemicals [118] and catalysts.[119-121] Meanwhile, 3,4-disubstituted pyrroles with both a-positions unsubstituted are important in the synthesis of porphyrins [122] and other bioactive compounds.[115, 116]
Interesting example of the self-organization phenomenon is the assembly of 1-acetyl-1,3-bis(haloarylamines) 56 from two molecules of o-halo arylamines 55, three molecules of acetylene gas, and one molecule of water (Scheme 25).[123] Notably, 1,3-bisamines are important intermediates for the synthesis of natural alkaloid manzacidin A,[124, 125] and HIV-1 protease inhibitors A-74704.[126, 127]
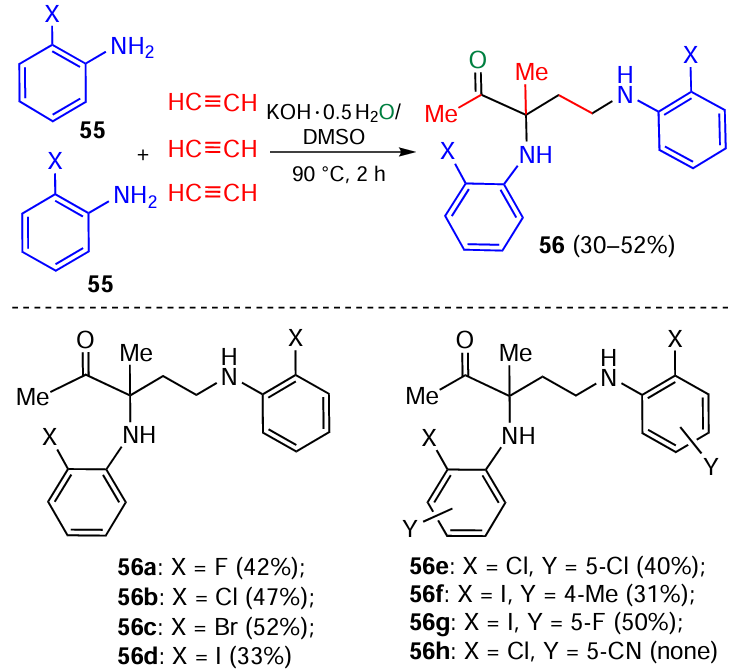
The construction of 1-acetyl-1,3-bis(haloarylamines) 56 (Scheme 26) starts from the nucleophilic addition of amines 55 to the first molecule of acetylene to give N-vinyl amines 57, which are prototropically isomerized to imines 58. The latter add the second acetylene molecule (in carbanionic form) to the C=N bond, forming propargylamines 59, which are transformed to azadienes 60. The acetylene carbanion generated from the third acetylene molecule adds to the C=N bond of these intermediates to produce acetylenic amines 61, which are attacked by the second molecule of amine 55, and adducts 62 undergo hydration across the triple bond.
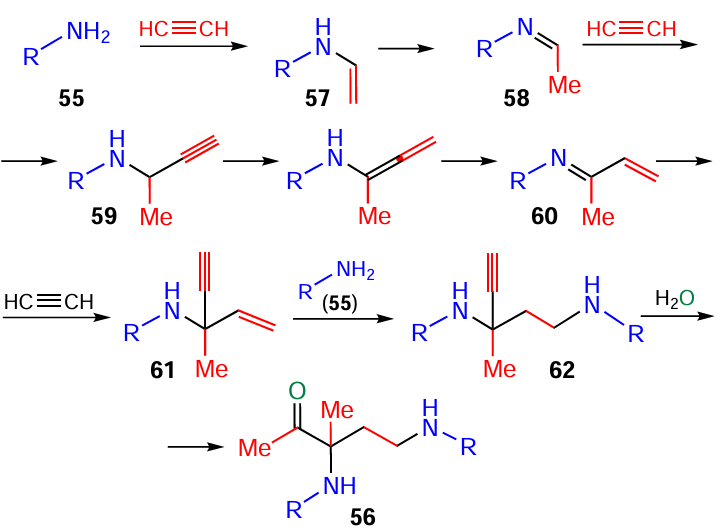
Quantum-chemical analysis (B2PLYP(D3)/6-311+G**//B3LYP/6-31+G*+PCM (B3LYP/6-31+G*)) [128] does confirm that bisamines 56 are assembled via the sequential alternating addition of amine anions to acetylene and acetylenic carbanions to the C=N bond of the resulting intermediates. The driving force of this cascade process is that each subsequent stage proceeds with better thermodynamics (lower energy) than the previous one. According to the quantum-chemical calculations,[128] the reaction is catalyzed by a solvate complex of potassium hydroxide with five molecules of dimethylsulfoxide (KOH · 5 DMSO, Scheme 27), in which the hydroxide ion is significantly separated from the potassium cation (by ~ 0.5 Å vs the normal K – OH bond length) that imparts it superbasicity.[129, 130]
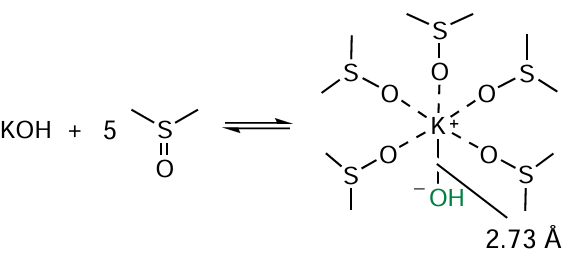
According to the DFT analysis,[128] the mechanistic feature of the assembly of 1-acetyl-1,3-bis(haloarylamines) 56 is the shuttle-like proton transfers from neutral molecules (to transform them to anions) to hydroxide ion and back from the water molecule thus formed to new anionic intermediates produced by accepting acetylene or arylamine molecules (Scheme 28). The hydroxide ion in the complex KOH · 5 DMSO plays the role of a ‘hydrogen hub-dispatcher’ ensuring proton migration (Н+ + НО– → Н2О) throughout the cascade process.
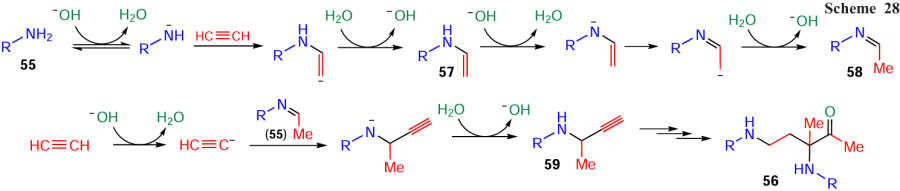
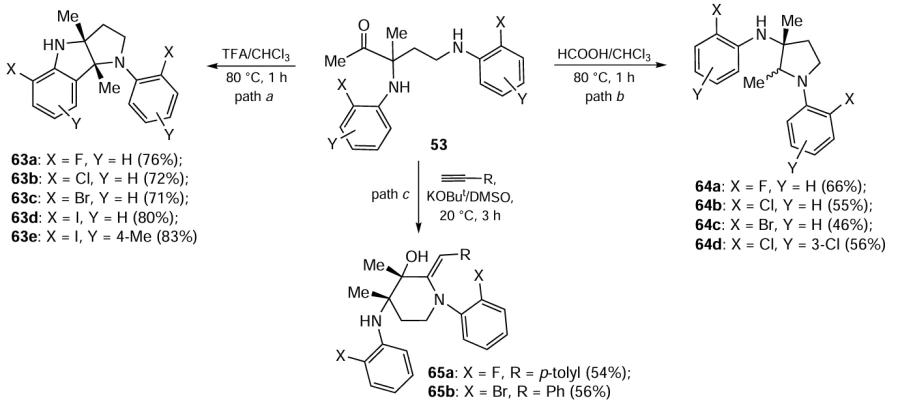
Apart from its obvious theoretical interest (see Scheme 25), this reaction is of a considerable synthetic value since it opens up simple routes to molecular complexity and diversity.[123] In particular, bisamines 56, upon treatment with trifluoroacetic acid, undergo double intramolecular cyclization to diastereomerically pure hexahydropyrrolo[3,2-b]indoles 63 (Scheme 29, path a). In the presence of formic acid, bisamines 56 have been subjected to reductive cyclization to 3-aminopyrrolidines 64 (Scheme 29, path b). Bisamines 56 also react readily with arylacetylenes 2 under the action of superbase KOBut/DMSO at room temperature to give benzylidene piperidinols 65 stereoselectively (Scheme 29, path c).
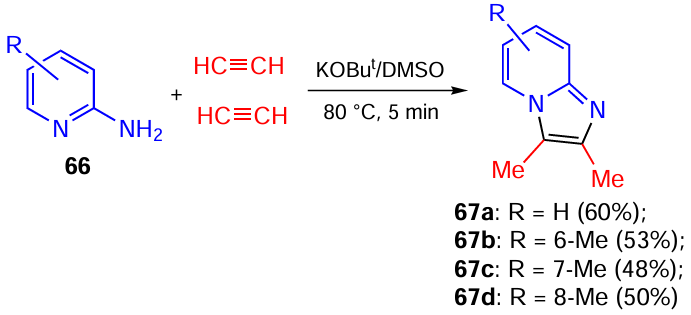
Another case of multi-molecular self-organization phenomenon, in which the addition of acetylenic carbanions to the C=N bond plays a leading role, is the reaction of substituted 2-aminopyridines 66 with acetylene gas. In the KOBut/DMSO system upon heating (80 °C) for a short time (5 min), the above reactants afforded imidazo[1,2-a]pyridines 67 (Scheme 30).[131] The structure of these products indicates that they are assembled from one molecule of 2-aminopyridine 66 and two molecules of acetylene.
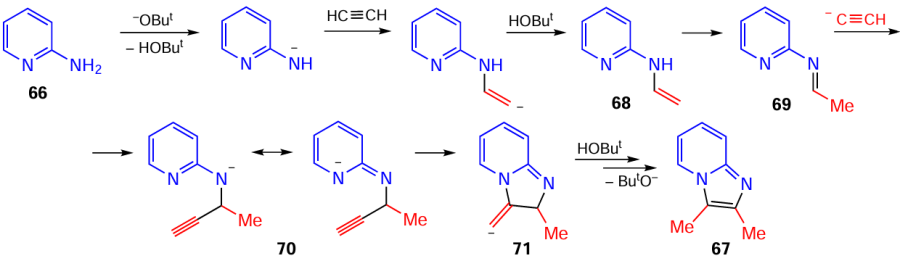
The assembly of imidazo[1,2-a]pyridines 67 includes the addition of deprotonated 2-aminopyridines 66 to acetylene (Scheme 31). The intermediate N-vinyl aminopyridines 68 isomerize to imines 69, which add acetylenic carbanions across the C=N bond to form the nitrogen-centered anions 70, the negative charge of which is transferred to the pyridine ring. The pyridine nitrogen atom closes the imidazole cycle by the addition to the triple bond and gives imidazopyridines 67 after protonation of anionic adducts 71 and prototropic rearrangement. Thus, in this cascade reaction, the addition of acetylenic carbanions to the C=N bond formed in situ is a driver of this assembly.
It was found that 2-aminobenzo[d]imidazole 72a reacted with acetylene gas under similar conditions (KOBut/DMSO, 80 °C, 20 min) to give benzо[d]imidazo[1,2-a]imidazole 73a and benzo[4,5]imidazo[1,2-a]pyrimidine 74a formed via the self-organization of one molecule of 2-aminobenzo[d]imidazole 72a with two or three molecules of acetylene (Scheme 32).[132] This result suggests that the process involves all three nitrogen atoms of the starting 2-aminobenzo[d]imidazole 72a. Similar products were obtained by the reaction of N-methyl-2-aminobenzo[d]imidazole 72b with acetylene under the same conditions (see Scheme 32).
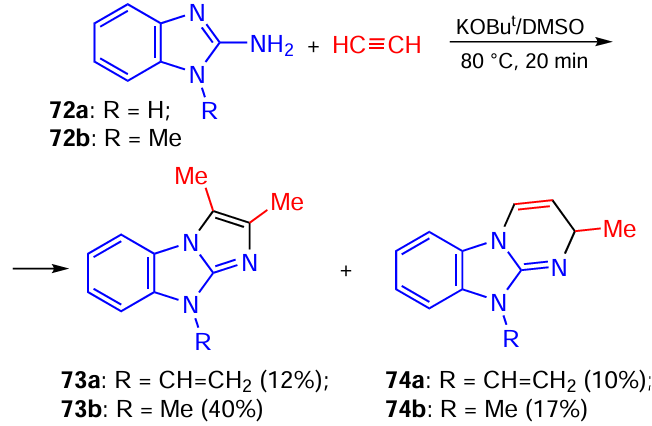
The self-assembly of heterocycles 73 and 74 (Scheme 33) probably begins with the nucleophilic addition of 2-aminobenzo[d]imidazoles 72 to acetylene. The resulting N-vinyl adducts 75 isomerize into imines 76, which are attacked by the second acetylene molecule (in carbanionic form) to generate nitrogen-centered anions 77, in which the negative charge can be transferred to the imidazole ring. The intramolecular addition of anions 78 to the α- or β-carbon atom of the triple bond results (after protonation) in the formation of imidazo[1,2-a]imidazoles 73 or imidazo[1,2-a]pyrimidines 74, respectively. In the case of 2-aminobenzo[d]imidazole 72a, the resulting products are further vinylated at the NH group by the third acetylene molecule.

As can be seen from Scheme 33, the process also involves the sequential addition of nitrogen-centered anions to the triple bond of acetylene and the addition of acetylenic carbanions to the C=N bond formed in situ.
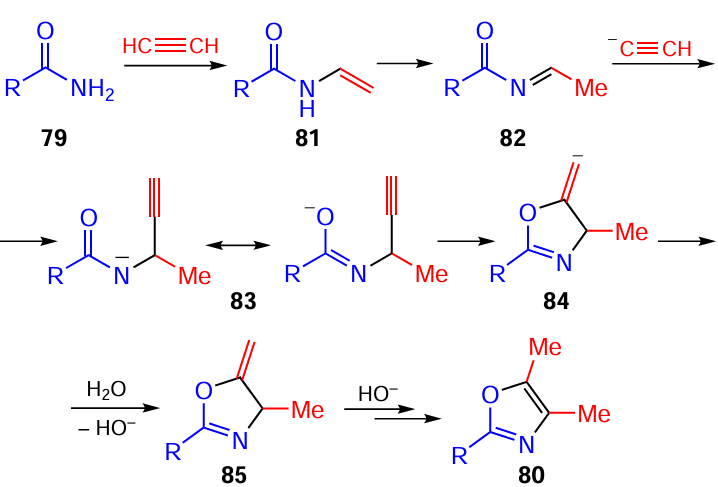
It was found that a molecule of primary amides 79 and two molecules of acetylene gas in the KOH/MeOH/DMSO superbase triad underwent a complex cascade transformations to give 2-substituted 3,4-dimethyloxazoles 80 (Scheme 34).[133]
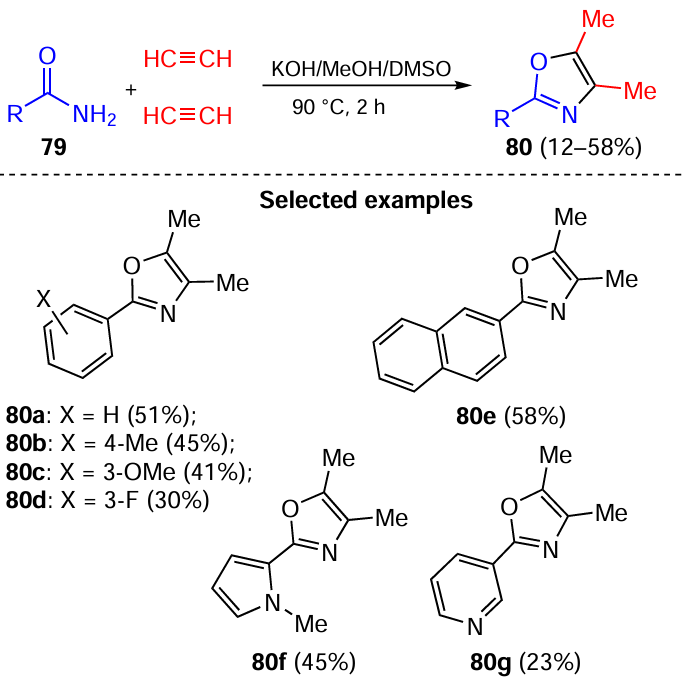
Apparently, the cascade sequence of oxazoles 80 assembly is triggered by the vinylation of amides 79 with acetylene (Scheme 35) followed by prototropic isomerization of vinyl amides 81 to imines 82, which are attacked by acetylenic carbanions at the C=N bond to form propargyl amide anions 83. These anionic intermediates in their oxygen-centered form are cyclized via the intramolecular vinylation involving the triple bond. Protonation of anionic adducts 84 and prototropic rearrangement of methylene-dihydrooxazoles 85 provides oxazoles 80.
5. Conclusions
As can be seen from the above, the reaction of acetylenic carbanions, generated in superbase media, with C=N bond, has now received a rapid development due to its obvious synthetic advantages such as mild conditions, simplest starting reagents (acetylenes, imines, amines), one-pot implementation, energy-saving, and environmental safety. In addition, the reaction occurs in the presence of only biogenic ions (K+, Na+, HO–, Н+) in a non-toxic, easily recoverable solvent (dimethylsulfoxide), which allows large-scale syntheses to be implemented in a closed cycle. The characteristic feature of the reaction is its synthetic divergence, which is expressed in its metamorphosis into essentially new processes by changing the structure of the starting C=N bond containing compounds. Within a short period of time (6 years), this new fundamental reaction became the generator of a number of earlier unknown daughter reactions with high synthetic potential. The diversity of transformations of acetylenic carbanions with the C=N bond opens a straightforward access to novel groups of terminal and internal propargylamines, bis(propargyl)benzenes, 1-azadienes, substituted pyrazoles, benzyl- and styryl imidazo[1,2-a]pyridines. Unexpected reactions of acetylenes with N-benzylimines in superbase media have provided one-pot selective syntheses of 2-azadienes, 1-pyrrolines, 1Н- and 2Н-pyrroles. No less importantly, the addition of acetylenic carbanions to the C=N bond appeared to be the key step of the self-organization of complex molecules involving acetylene and amines, another vast unexplored area of organic synthesis.
Further development of the reaction of acetylene carbanions with the C=N bond offers exciting possibilities for targeted structural diversification of synthesized products with a view to application in other segments of organic synthesis, including carbon dioxide fixation (assembly of methyleneoxazolidinones from propargylamines and СО2), reactions with carbonyl compounds (synthesis of methyleneoxazolidines), isothiocyanates (formation of thiazole derivatives), and other electrophiles for the preparation of new functional heterocyclic systems, potential drugs and their precursors, as well as starting compounds for the assembly of complex molecular architectures.
6. List of abbreviations
dr — diastereomeric ratio,
Fu — furyl,
Naph — naphthyl,
Py — pyridyl,
Th — thienyl.
References












