


![Selected bond lengths, and Hirshfeld bond orders and atomic charges of the archetypal HCs.[38, 40, 103]](/storage/images/resized/Y66whISNhZu4g8BiCFjJSKFiWLBEq5oldYHe4hdT_xl.webp)
![Bond lengths (XRD, DFT) and Hirshfeld bond orders (DFT) for HC 17 and HR 2.[38, 40]](/storage/images/resized/usjlMI0FZ0lJVZZu1KmrzGzCv88Q0KmkfMOJFYzc_xl.webp)
![Selected UV-VIS data of Herz cations, radical cations and dications.[7, 28, 29, 100, 101, 106, 117, 234, 237, 238]](/storage/images/resized/dHRttQjC1ftyTUBdba0zwVESLqyUA5KQtFJUxRGP_xl.webp)
![Selected 15N, 77Se, and 125Te NMR data of HCs.[25, 33, 38, 40, 100, 101]](/storage/images/resized/bSrS90vBXfs1OQnB8u2XoFmI2FYznEMSa5orXEbc_xl.webp)
![Selected experimental a and g values of HRs A5 (Figure 1) and their R4 – R7 tetrafluoro derivatives.[18, 38, 40, 41, 185, 25-27]](/storage/images/resized/h5F6Fg526UPH9ujEbR8cbqYYxkJQ5cwGk61EvU6f_xl.webp)
![Selected bond distancesa in σ-dimers [147, 150, 160, 176, 233, 235, 250, 254-257] of HRs at normal pressure.b](/storage/images/resized/3CPz0OZhFs8uVFrJ8U32HJDEUivvtfECemHNB7tJ_xl.webp)
![Selected UV-VIS data of HRs.[41, 170, 260]](/storage/images/resized/av4tohg2zBLxsS5Np34EJGSH4Fp4jPq99hbaQiau_xl.webp)
![Electrical conductivity of solids formed by HRs, σ-dimers, bipolar ions, mixed-valence and RC salts [31, 32, 39, 42, 110, 127, 155, 157, 192, 233, 240, 241, 244, 247, 250, 275, 279, 280, 34-37, 143-148, 150-152, 159-161].](/storage/images/resized/mSb5WG2JryEq0cFOS2z6QIudi8u1N5nnC2OfNzoc_xl.webp)
![Magnetism of solids formed by HRs and RCs.[37, 127, 145, 148, 152, 159, 239, 240, 243, 247, 248, 275, 279, 286, 281-283]](/storage/images/resized/flzwajPXqf5ifcTrEriwYFHXwjSpbn91ZfzPBIvT_xl.webp)
![The π*-SOMO and solution EPR spectrum of HR 2 (top) and RC 14 (bottom). Experimental hfc constants a (mT): 2: 0.82 (aN), and 0.37, 0.24, 0.10, 0.08 (aH);[38] 14: 0.16 (aN, dithiazole), 0.08 (aN, phenazine), 0.01 (aH, phenazine).[117]](/storage/images/resized/N89V8UUJ85KaNnhjxAO6y1lPhFFpQVXRiDShf54N_xl.webp)

Keywords
Abstract
This review discusses the achievements of Herz chemistry over its first century, together with challenges and prospects. The discussion focuses on the synthesis, structure, and reactivity of various closed- and open-shell chalcogen-nitrogen π-heterocyclic species. The latter are derivatives of the (het)areno-fused 1,2,3-dichalcogenazole ring system with S, Se, and less often Te, chalcogens in various spin and charge states encompassing cations, radicals, bipolar ions, and quinoid antiaromatics/diradicaloids. They are important for fundamental chemistry and materials science, specifically for the design and synthesis of metal-free conductive, magnetic, and optoelectronic materials. The potential for further extension of Herz chemistry to other non-transition elements is also considered. A comparative analysis of Herz species and their 1,3,2-isomers (Wolmershäuser species) is provided. The bibliography includes 344 references.
1. Introduction
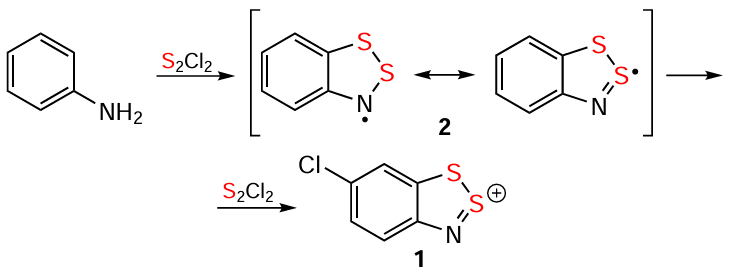
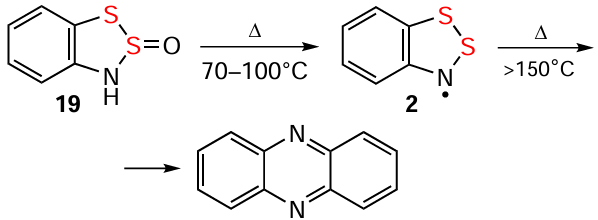
The chemistry and applications of 1,2,3-benzodithiazoles were initiated a century ago by Richard Herz (1867 – 1936)[1] with the cyclization of ArNH2 into 1,2,3-benzodithiazoliums (Herz cations, HCs), e.g., 1 (Scheme 1), by the action of S2Cl2 and their isolation in the form of chloride salts (Herz salts, HSs). The cyclization revealed the general nature and wide industrial applicability for the production of various synthetic dyes, and was named the Herz reaction.[2-13] Its limited extension to HetNH2 is the hetero-Herz reaction.[13-18] Both reactions belong to various chemical transformations involving organic amines and sulfur halides.[12, 19-23] Other milestones in the field include the EPR detection of persistent 1,2,3-benzodithiazolyls (Herz radicals, HRs), e.g., 2 (see Scheme 1) as the key intermediates of the Herz reaction by Roland Maier (1927 – 2013) [24] et al.;[18, 25-27] extension to Se derivatives by Lev S. Éfros et al.;[28-30] isolation of HRs in the form of thermally-stable solids with conductive and/or magnetic properties by Richard T. Oakley et al.;[31-43] generation of otherwise inaccessible HRs, e.g., polyfluorinated, by thermolysis or UV photolysis of polysulfur-nitrogen rings; and direct preparation of analytically pure HSs by reaction of 1,2,3,4-benzodithiadiazines with SCl2.[38, 44, 45] Important bioactivity of Herz species relates mostly to monocyclics.[46, 47]
Formally, HCs and HRs are π-heterocyclic derivatives of NS+ and NS•, respectively, which have been observed in interstellar space, and NS• has also been found in the coma of comet Heil – Bopp;[48-55] EN+ and EN• (E = Se, Te) have been studied to a lesser extent.[50, 51, 56] Stable acyclic derivatives of NS• are thioaminyls.[57, 58]
Herz chemistry is highly relevant and rapidly developing as an integral part of polysulfur-nitrogen chemistry,[59, 60] which is currently evolving into polychalcogen-nitrogen chemistry,[61-64] which is itself part of main group chemistry (i.e., chemistry of the elements from the groups 1, 2, 13 – 18). The family of Herz species includes variously substituted/annulated 1,2,3-benzodichalcogenazoles and encompasses cations, radicals, radical ions, and neutral molecules (Fig. 1). The latter can be classified as antiaromatic quinoids (Herz quinoids, HQ), bipolar ions, and/or singlet diradicals/diradicaloids, and they have attracted significant interest in current main group chemistry.[2, 13, 43, 65-73] For species with two radical centres, it is reasonable to distinguish between diradicals and disradicals; the former reveal significant (J ≠ 0) and the latter insignificant exchange interaction (J ~ 0) between the paramagnetic centers.[72] Of particular importance are real and/or putative high-spin (S ≥ 1) Herz di- and polyradicals [74, 75] — by themselves and as parts of a large family of persistent and stable metal-free high-spin species.[73, 76] Overall, Herz species are intriguing due to their multifaceted functionalities in chemistry. Particularly, they exhibit ring-modification, -contraction, and -opening reactions, and are involved in the synthesis of various compounds that are otherwise difficult to access.[77-80] Additionally, they are important for materials science as conductive, magnetic, and optoelectronic materials.[74, 75, 81-85] Acyclic aminodisulfide analogues of Herz species are also of great current interest in chemistry, materials science, and life sciences.[86]
![[{"id":"A357MIO8p-","type":"paragraph","data":{"text":"Representative <b>A1-A16</b> archetypical (E = S, Se; E = S, Se, Te) and specified <b>3</b> – <b>15</b> Herz scaffolds. For simplicity, from now on, π-delocalized HRs are shown as N-centred to avoid resonance superpositions."}}]](/storage/images/resized/4qwEioWEkyEmVXXSnv23ZzxHrE4cX6YYtI1qRmib_xl.webp)
Herz chemistry and its applications have been discussed in numerous review articles and book chapters, primarily focusing on S derivatives, monoradicals, and functional materials.[2, 3, 5, 6, 43, 47, 69-71, 81-84, 87-89] In addition to updating these topics, this review expands to include heavier-chalcogen species, high-spin radicals, radical ions, and neutral quinoids (singlet diradicals/bipolar ions). The overall goal of this review is to examine Herz chemistry and its applications within the broader framework of main group chemistry. Consequently, this review is more conceptual than descriptive. For this reason, and for clarity, in Chapter 2 (Synthesis and reactivity), reaction byproducts and substituents in Herz species, including annulated derivatives, are unspecified/omitted where not significant; i.e., the main emphasis is made on the transformations of scaffolds. Similarly, HS anions are typically [Cl]– unless otherwise indicated; π-delocalized HRs are depicted as N-centred (cf. Fig. 1) to avoid resonance superpositions. However, the substituents and anions are specified in Chapters 3 (Molecular and electronic structure), 4 (Crystal structure), and 5 (Materials science). Units/dimensionalities of physical quantities and abbreviations are specified in Abbreviations and units Section.
The synthesis and reactivity of Herz species are considered together because of the interconvertiblity of the compounds; less studied are 3H-1,2,3-benzodithiazole 2-oxides (Herz bases, HBs), which contain the S(IV) asymmetric atomic centers and can introduce chirality into reaction products. In terms of reactivity, the main focus is on heterocyclic transformations; the carbocyclic reactivity of Herz species is typically associated with nucleophilic or radical substitution. HCs and HRs are systematically compared for the first time with isomeric 1,3,2-benzodithiazoliums and -dithiazolyls (Wolmershäuser cations and radicals; WCs and WRs, respectively).
Monocyclic Herz species are not considered in this review, with the exception of their involvement in the synthesis of HQs. The best known of these is AC,[90] which has a different chemistry to HCs.[47, 74, 91-94] Monocyclic radicals, unlike HRs, demonstrate a tendency to C-centred recombination and are stable only when the corresponding reaction centres are sterically hindered;[87, 95-98] bicyclic radical cation representing tetrathiadiazafulvalene scaffold [98] is stable enough to be isolated in the form of salts.[94]
2. Synthesis and reactivity
2.1. Cations and radical cations
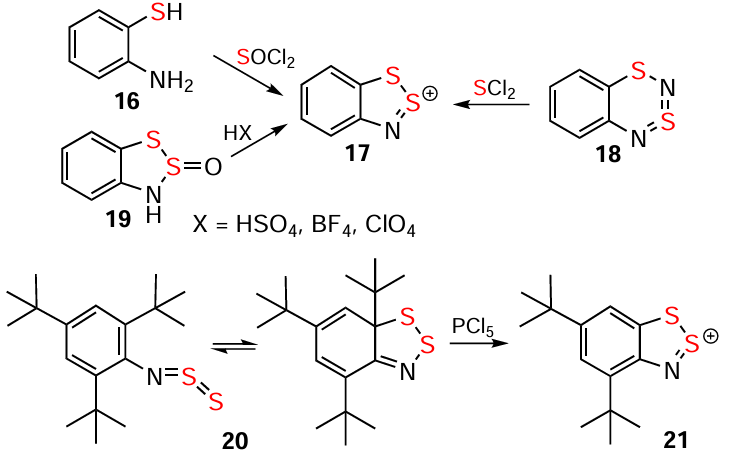
In addition to the Herz and hetero-Herz reactions,[89, 94, 99, 3-9, 12-18] four other preparative approaches include the reaction of 2-aminothiophenols or their N – R derivatives with S2Cl2 or SOCl2;[3, 4, 13, 33, 38, 40, 41, 89, 100] the reaction of 1,3,2,4-benzodithiadiazines with SCl2;[13, 44, 101-103] the treatment of HBs with strong acids;[7, 13, 89, 104-106] and the reaction of 7aH-1,2,3-benzodithiazoles with PCl5 [89, 107, 108] (Scheme 2). These approaches utilize ring-closure, ring-contraction, and ring-modification reactions (Scheme 1 and Scheme 2); and the approach based on aminothiols can be considered as retrosynthetic one. The Herz reaction involves chlorination of the carbocycle at position 4 by substitution of an H atom (see Scheme 1) or an electron-acceptor group but not an electron-donor group. Non-S2Cl2 approaches (Scheme 2) typically avoid chlorination and the loss of the initial substituents.
Preparative approaches to Se containing HCs also employ the same three reactions, with ring-modification occurring through chalcogen exchange (Scheme 3).[28, 29, 33, 38, 40, 43, 70, 103, 109, 110] The only reported Te containing HC has been synthesized by a ring-closure reaction (see Scheme 3),[40] while other attempted approaches yielded Te-free HCs;[100] this underscores the unique behavior of Te among the chalcogens.[111]

In HSs obtained by the Herz/hetero-Herz reaction, the anion is [Cl]–, and they are poorly soluble in nonpolar organic solvents except But derivatives.[100, 103, 107] The poor solubility is likely caused by chalcogen bonding (ChB) [112, 113] between HCs and [Cl]–,[114] a phenomenon known for halide salts of WCs.[115, 116] Replacing [Cl]– with low-coordinating anions, such as [MCl4]– (M = Al, Ga, Fe), [SbCl6]–, [CF3SO3]–, [ClO4]–, etc., improves solubility and enhances the general utility of HSs in various preparations and applications.[7, 32, 38, 39, 41, 71, 114, 117]
2.1.1. Herz and hetero-Herz reactions
The Herz reaction is primarily limited to S chemistry and is used for single and double ring-closures. Attempts to extend the reaction to heavier chalcogens using EnCl2 (E = Se, Te; n = 1, 2) [118-124] have not been reported. The reaction has a wide scope, encompassing numerous carbocycle- and N-functionalized (–NH – OH, –NH – NH2 ,–N=O, –N=S=O, =N – OH) substrates, including nonbenzoid ones. The substitution/annulation patterns of HCs exhibit considerable diversity, which is significant for both fundamental chemistry and applications. Reaction conditions are crucial, including solvent dependence.[9, 10, 13, 20, 89, 100, 103, 114, 117, 2-6, 125-128] In the presence of organic bases, e.g., DABCO or Et3N, the reaction route is changed to Ar – N=S=N – Ar and/or A – N=S=S instead of HCs.[129, 130] ArNH2 with an ortho Me groups give 2,1-benzoisothiazoles [131] (cf. similar cyclization of ortho-methylated [ArNSN]– anions);[132] and those with ortho NH2 groups, 2,1,3-benzothiadiazoles.[133, 134]
In the Herz reaction, the reagent S2Cl2 , typically used in excess with respect to ArNH2 , has both oxidizing and chlorinating properties.[11, 18, 21, 22, 135, 136] The oxidizing ability is essential for the transformation of the intermediate HRs into the final HCs (Scheme 4).[23, 25] Elemental chlorine generated during the reaction also participates in both oxidation and carbocycle chlorination.[9] The latter is a hallmark of the Herz reaction. Generally, chlorination occurs at the para-position to the N-function if the position is unsubstituted (see Scheme 4) or carries an electron-acceptor group. An electron-donor group at this position remains intact, and chlorination occurrs at another position, such as the ortho position in the case of Ar = 4-AlkOC6H4.[137] Bulky substituents or annulation prevent chlorination (cf. HCs 30 – 32; Scheme 5).[11, 103] However, full understanding is complicated by byproducts including chlorinated/nonchlorinated and cyclic/acyclic.[23, 136, 137] Particularly, in the reaction ArNH2 + S2Cl2 (Ar = 4-MeOC6H4) at 70°C the major non-chlorinated HS was formed along with minor ortho chlorinated one; whereas at 50°C ortho chlorinated ArNH2 was observed as the major product, together with minor target HS.[137]
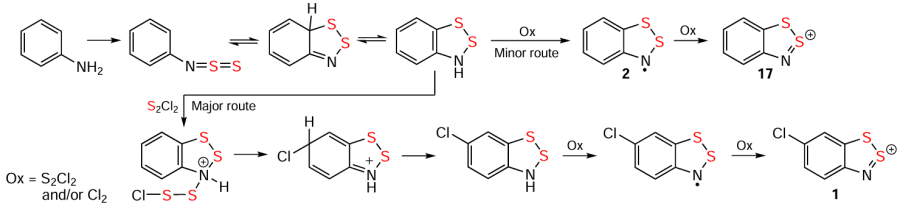
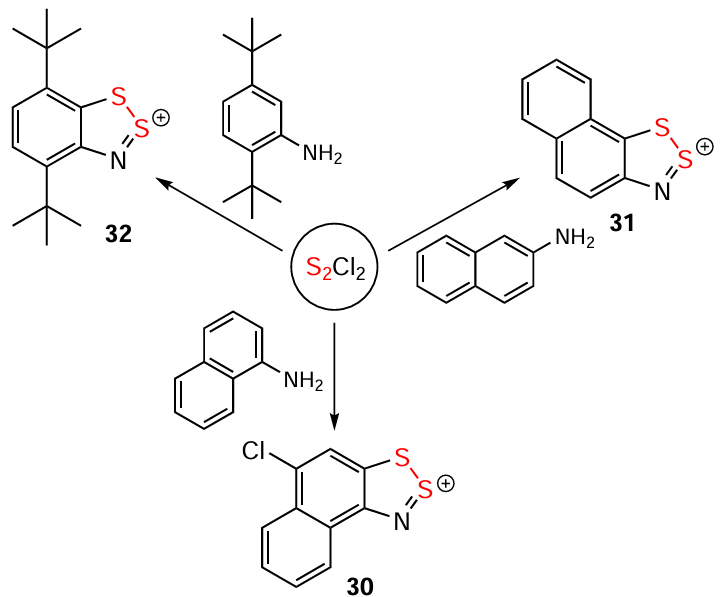
The ring-closure in the Herz reaction is regioselective, e.g. cyclization of 2-acetamidonaphthalene proceeds at position 1.[99] The reaction of naphthalene derivative 33 with S2Cl2 leading to HC 34 in MeCN [114] and 3H-1,2,3-dithiazole 35 in AcOH (alternative product 36 is not observed) gives an example of solvent dependence (Scheme 6).[138] The ring-closure reaction can also involve leaving groups other than H atoms. Particularly, the transformation 37 → 38 and synthesis of 39 demonstrate the utilization of a Br atom and carboxyl group, respectively (Scheme 6 and Scheme 7).[139, 140] For benzo-fused heterocycles, a bifurcation between Herz and hetero-Herz pathways occurs depending on the position of the NH2 group.[18, 130, 13-15] Overall, the approach encompasses a wide range of heterocyclic substrates, including thiophenes, pyrazoles, isothiazoles (but not isoxazoles),[17, 18] and benzothiadiazoles [125, 141] (cf. HCs 39 – 44) (see Scheme 6 and Scheme 7).
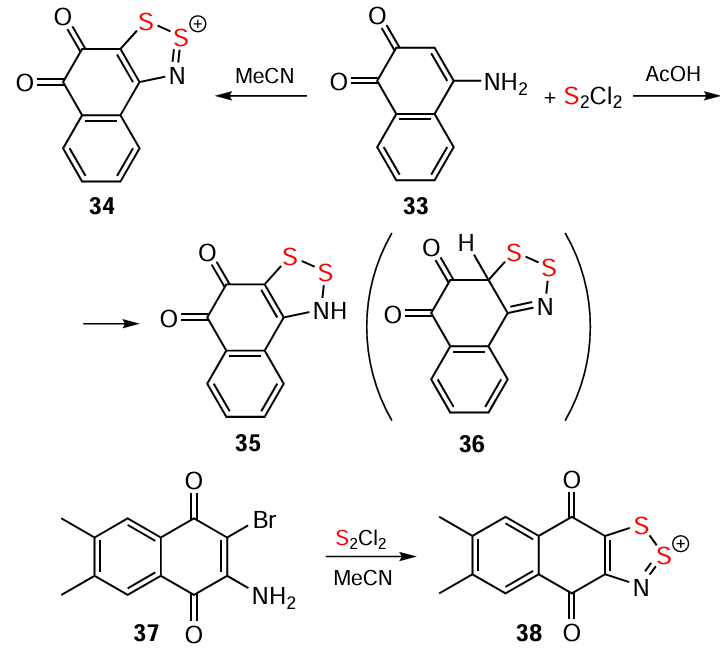
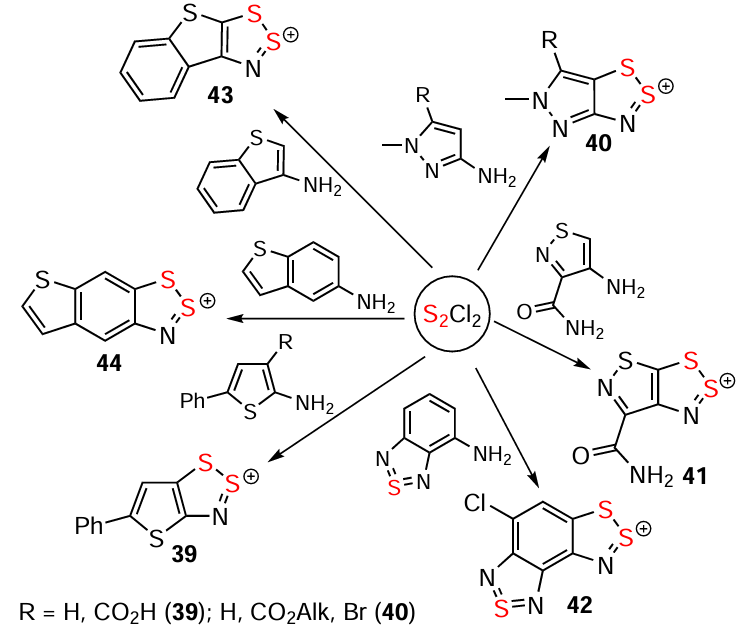
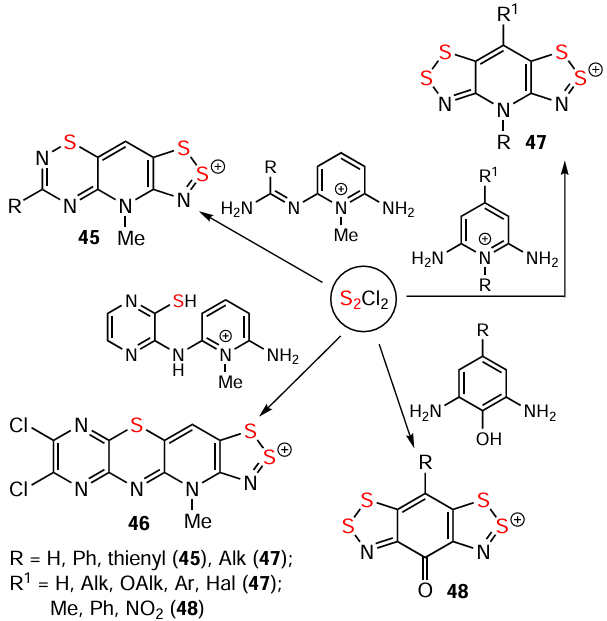
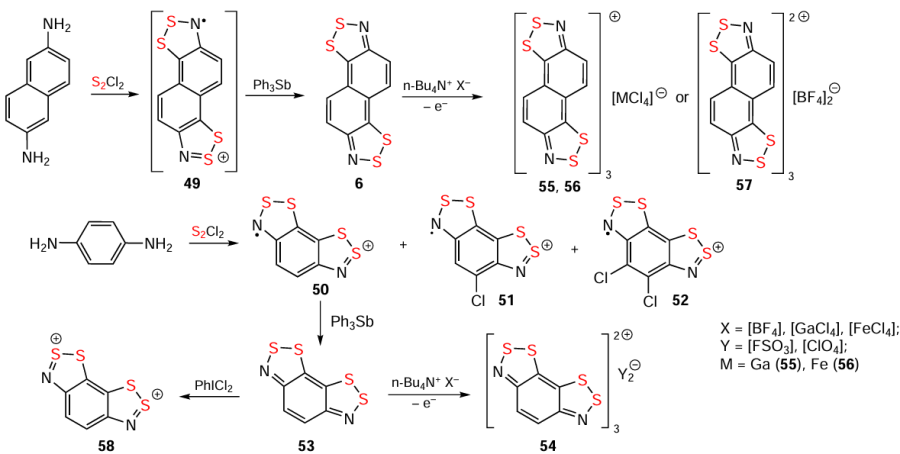
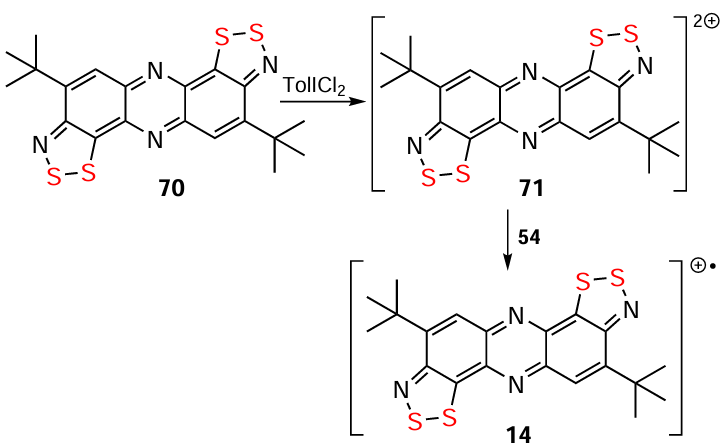
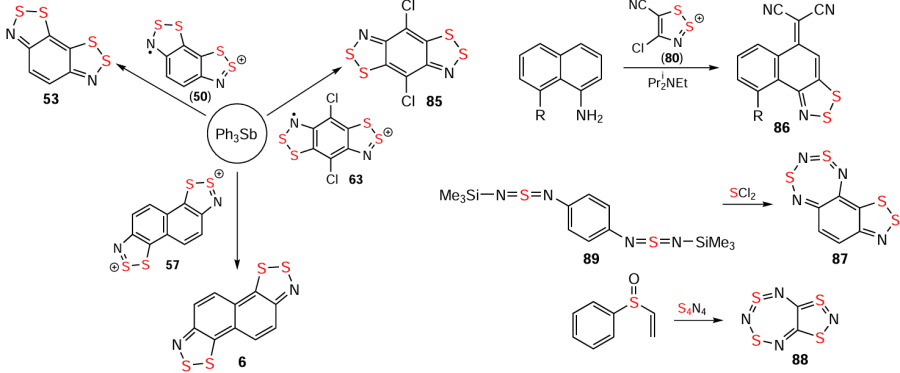
The hetero-Herz reaction, accompanied by additional cyclization processes, and the double (hetero)-Herz reaction, lead to the formation of polycyclic HCs 45 – 48 (Scheme 8) [16, 35, 127, 142-154] and RCs 49 and 50, together with chlorinated derivatives 51 and 52 of the latter (Scheme 9). For isolation and identification, 49 and 50 were chemically reduced into corresponding closed-shell HQs 6 and 53. Subsequent chemical and electrochemical oxidation of these HQs gave the chloride salt of dication 54, and/or mixed-valence HSs 55 – 57 (see Scheme 9).[31, 32, 155-157]
The Herz and hetero-Herz reactions appear to involve multiple stages, and the overall situation regarding regioselectivity of ring-closure, carbocyclic substitution, and solvent dependence is rather intricate, necessitating further research, mainly through quantum chemical modeling. The inherent chlorination is not necessarily a drawback, as halogenated Herz species hold a particular interest (see below).
2.1.2. Other ring-closures
Alternative ring-closure methods to the Herz/hetero-Herz reaction encompass S, Se, and Te containing substrates and are facilitated by SOCl2, SnCl2 (n = 1, 2) and PCl5 reagents. The reaction of 2-aminothiols with SOCl2 yields nonchlorinated HCs, whereas S2Cl2 or S2Cl2/Cl2 afford chlorinated products. The alternatives include –N=PR3 derivatives instead of –NH2 ones (Scheme 2 and Scheme 10; cf. Scheme 9).[2, 13, 32, 39, 40, 117, 158-162]
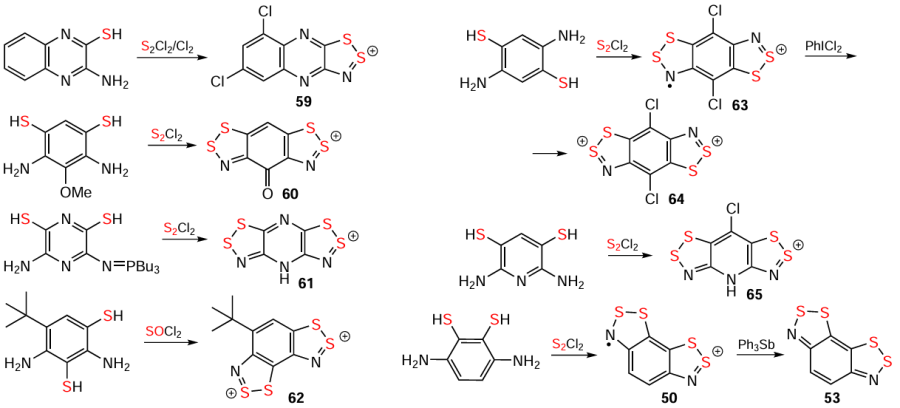
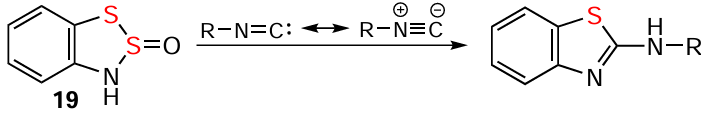
The reaction of compound 20 (existing as an equilibrium between dithiazole and N-thiosulfinyl,[138, 163] a peculiarity of the N=S=S but not of the N=S=O group [130]) with PCl5 , which closes the ring of the N-thiosulfinyl and oxidatively modifies the dithiazole, provides HC 21 (see Scheme 2). Extending this singular result to other Ar – N=S=S [2, 23, 107, 108, 164] (putative intermediates of the Herz reaction, Scheme 4) remains challenging.
Reactions of 2-aminochalcogenols with EOCl2 (E = S, Se), SeCl4 or H2SeO3, and 2-aminothiol with TeCl4 , produce Se HCs [33, 38, 40, 70] and Te HC,[40] respectively; whereas attempts with 2-aminotellurol derivative yielded only Te-free HCs with Te containing anions [TeCl6]2–, [Te3Cl14]2–, or [Te4Cl18]2 – [100] (see Scheme 3).
2.1.3. Ring-modifications
Ring-modification approaches encompass dehydration of S containing HBs under the catalytic action of strong protic acids, yielding analytically pure HSs, e.g., that of 17, and chalcogen-exchange in HBs, directly or through HCs, under the influence of H2SeO3 or SeO2 , producing Se HCs, e.g., 23 [6, 29, 70, 104, 109, 146] (see Scheme 3).
2.1.4. Ring-contractions
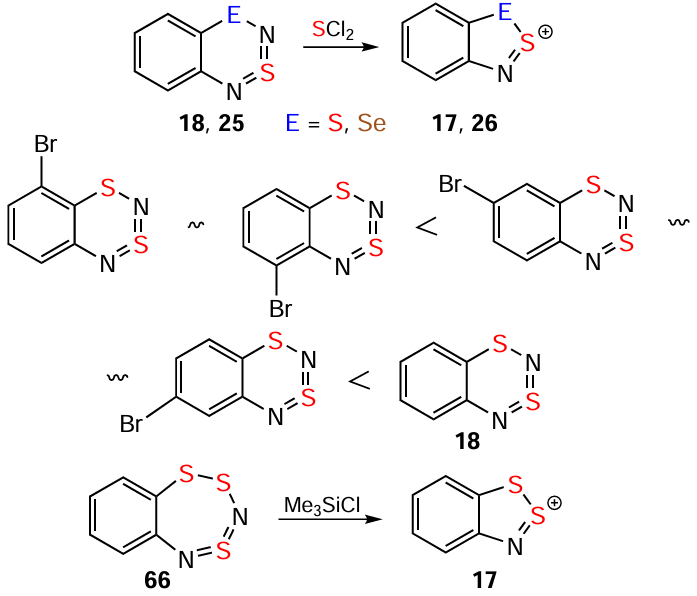
Oxidative hetero-ring contractions of benzo-fused dithia-/selenathiadiazines 18/25 with SCl2 provide pure HSs without carbocycle chlorination (except for the 6-Me derivative of 18). The reaction rate for 18 and its derivatives significantly depends on the nature and positions of the substituents in the carbocycle. Halogens Cl, Br, and I slow down the reaction, especially if they are adjacent to the heterocycle (for relative reaction rates of isomeric monobromo derivatives of 18, see Scheme 11).[44, 101-103] Ring contraction of 18 and its derivatives into HCs with S electrophiles C6F5SCl and [NS2]+ yields numerous byproducts.[82] Trithiadiazepine 66 is converted to HC 17 under the influence of Me3SiCl (see Scheme 11); similar transformation of its 1,3,5,2,4-isomer into the corresponding WC occurs notably faster.[165-167]
2.1.5. Miscellaneous
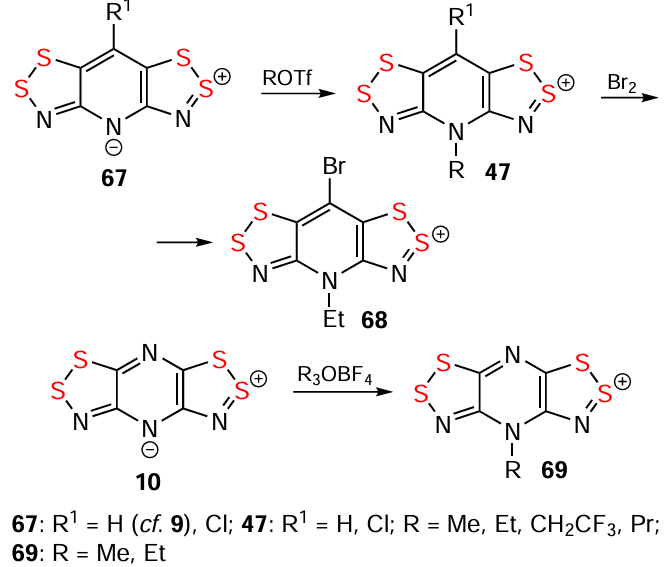
Alkylation of bipolar ions 10 and 67 with R3OBF4 or ROTf, respectively, generates HCs,[147, 149] which can then be brominated (compounds 47, 68, 69; Scheme 12).[146] Chemical oxidation of HQ 70 yields dication 71, which comproportionates with the starting HQ to produce RC 14 (Scheme 13).[117]
2.1.6. Reactivity
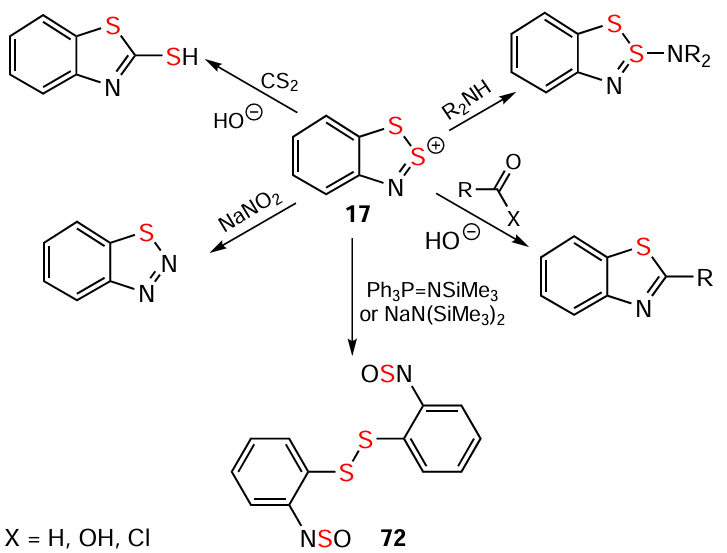
HCs exhibit thermal stability. The most important chemical properties of HCs are the reduction to HRs (below) and hydrolysis to HBs. Under mild hydrolytic conditions, S-HCs convert into S-HBs; whereas Se-HCs do not readily form stable Se-HBs. Strong protic acids reverse the reaction, converting HBs back into HCs (see Scheme 2). Their reactive site is mainly position 2, and numerous reactions transform HCs into a wide range of heterocyclic [2, 6, 10, 45, 140, 168] or acyclic [169] compounds (Scheme 14; for the sake of simplicity, the archetypal HC 17 is shown; the 17 → 72 transformation requires the air’s O2 to participate [81, 170]).
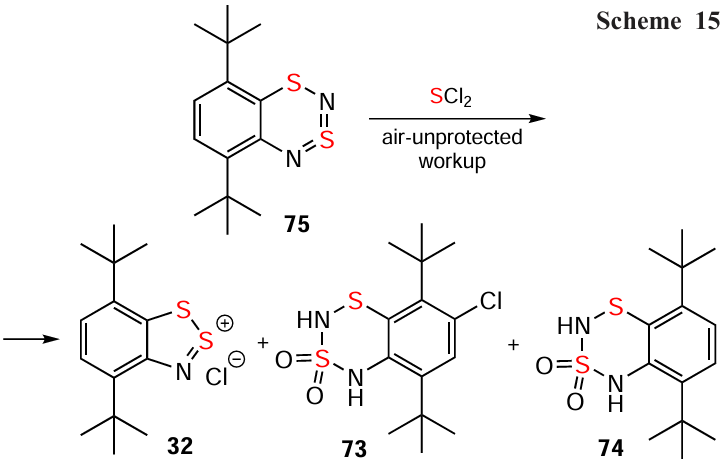
The HC 28 forms the Lewis-acid complex 28 GaCl3.[40] The complex of HC 32 with S,S-dioxides 73/74 (Scheme 15) is formed by the reaction of dithiadiazine 75 with SCl2 under air during the work up process. A putative precursor of 73/74 is RC [75]•+ (cf. [171]), formed by the oxidation of 75 with SCl2, followed by chlorination and exposure to atmospheric O2/H2O during the work up.[171]
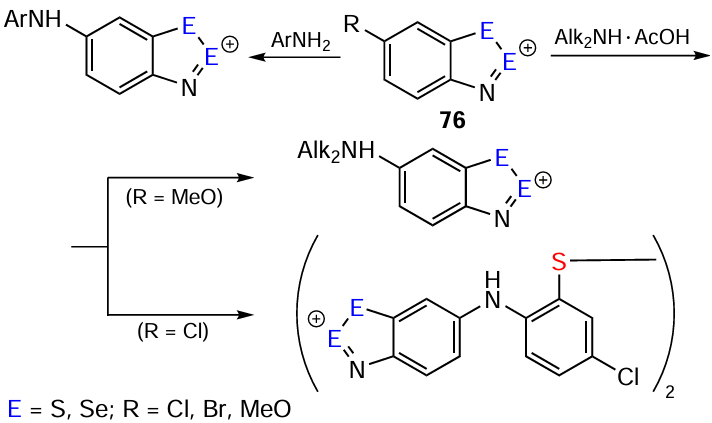
The carbocyclic reactivity of HCs is associated with nucleophilic substitution of Cl, Br, and MeO under the action of organic amines; the substitution rate depends on both the substituent and the chalcogen, with a particularly pronounced dependence on the chalcogen in position 2 mentioned above (Scheme 16).[2, 6, 106, 172-175]
2.2. Radicals
2.2.1. Reduction of cations
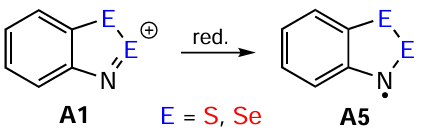
Numerous S- and Se-HRs have been obtained via reversible (electro) chemical reduction of their corresponding HCs (Scheme 17), which is the main approach to these Herz species. The chemical reduction agents used are usually Zn, Zn/Cu, Ph3Sb, [I]–, FeCp2 , FeCp*2, and tetrakis(dimethylamino)ethylene (TDAE); and the solvents are nonpolar hydrocarbons, e.g. heptane, cyclohexane, or benzene. The electrochemical potentials at a stationary Pt electrode are quite low, varying from –0.26 to 0.18 V vs. SCE; typical solvents are DME or MeCN. For the archetypal HC 17, replacing the S atom at position 2 with a Se atom (HC 23) causes an anodic shift of ~ 0.1 V, while there is no effect at position 1 (HC 26); replacing two S atoms (HC 24) shows a combined effect with the same ~ 0.1 V shift. The reduction is visually monitored by the change in color of the reaction mixtures (compared to HCs, HRs are deep-colored from red to green; see below) and the disappearing of the poorly soluble starting HSs [16, 18, 35, 40, 81, 114, 127, 144, 145, 154, 162, 31-33, 150-152, 176-179]. Reduction of Te HC 28 results in unidentified diamagnetic products.[40] Elemental halogens Cl2 and Br2 convert HRs back to HCs.[18]
2.2.2. Ring modifications
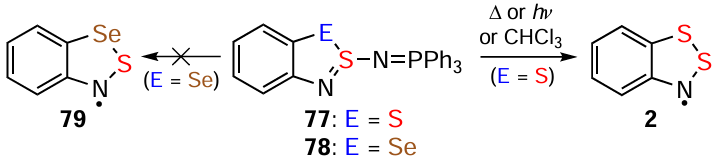
HRs can be obtained by thermolysis of various precursors, e.g., HBs (cf. 19 → 2).[25, 180] In the hydrocarbon and fluorocarbon series, compounds 77 transform into HRs upon thermolysis or simply upon dissolution in CHCl3 . However, for their Se congener 78, the corresponding HR 79 was not observed under the same conditions (Scheme 18).[181-183]
2.2.3. Ring contractions
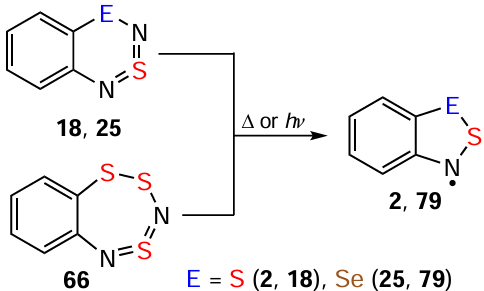
Many HRs, including those inaccessible by other methods such as polyfluorinated ones, can be generated from dichalcogenadiazines 18 and 25 in dilute organic solutions by mild thermolysis or UV photolysis. This method also extends to trithiadiazepines 66 (Scheme 19). This approach is suitable for both EPR and mechanistic studies.[44, 81, 103, 170, 181, 182, 184-189]
2.2.4. Ring closures
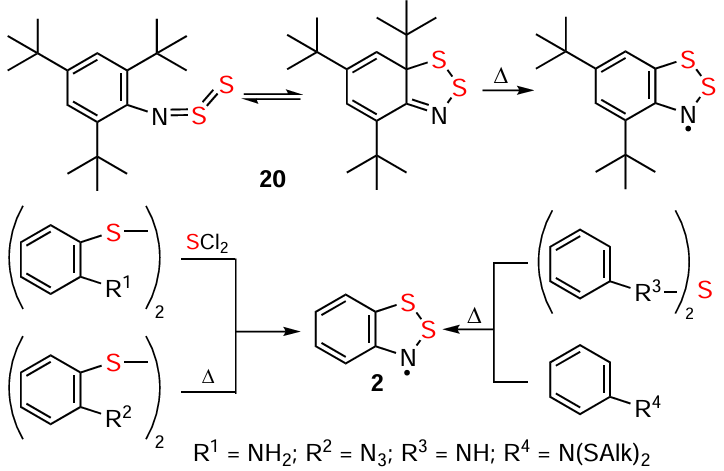
N-thiosulfinyl 20 upon heating under anaerobic conditions eliminates But • and transforms to EPR-detected HR corresponding to HC 21; thermolysis in air provides the corresponding HB and amine, and p-toluenesulfonic acid catalyst increases their yields. In addition, the ring closures towards HRs are EPR-observed for some other reactions of various acyclic sulfur-nitrogen derivatives (Scheme 20).[25, 89, 180, 190, 191]
2.2.5. Reactivity
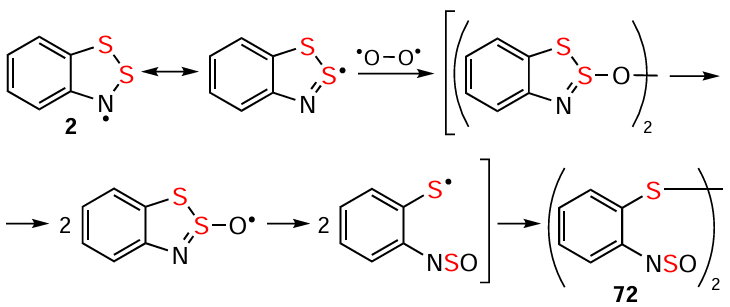
HRs are thermally stable, with benzo-annelated derivatives up to ~ 120 – 150°C. Above, various HQs and azaarenes are formed, e.g., phenazine and derivatives, most likely by self-condensation of HRs via π dimers and some other transformations (see below).[2, 12, 25, 33, 44, 81, 89, 117, 182, 183, 191] HRs are unstable in protic solvents, as well as in those containing acidic admixtures. In particular, they decompose rapidly in CHCl3 with the formation of an unidentified salt-like precipitate,[23] most likely caused by traces of HCl. Protected from H2O/O2 and UV light, HRs remain persistent in aprotic organic solutions at ambient temperature, and many HRs are isolated in a pure form (below). Reaction with O2 involves recombination followed by heteroring-opening to form O=S=N-functionalized diaryl disulfides 72 (Scheme 21, the archetypal HR 2 is shown for simplicity; cf. Scheme 14).[81, 170] The expected products of reacting with H2O under unaerobic conditions are 1,2-aminothiols, i.e., 16 and its derivatives. However, the corresponding protocols appear never to have been published, and in practice these compounds were used without isolation.
HRs exhibit electrochemical redox activity and are reversibly oxidised to HCs.[23] The oxidation potential depends on the chalcogen atom in position 2 (S or Se) and the working electrode material (Hg or Pt). Electrochemical reduction is irreversible, e.g., for 2 and 79.[30, 40] 1,2-Naphthoquinone-based HR has stable cationic (i.e., HC) and anionic (i.e., naphthoxide) states.[114]
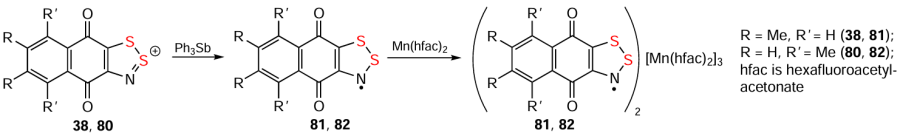
Furthermore, HRs are practically unexplored, but highly promising paramagnetic ligands for metal coordination compounds. In particular, HCs 38 and 80 were reduced to HRs 81 and 82, which with Mn(hfac)2 formed complexes [HR]2[Mn(hfac)2]3 (Scheme 22) exhibiting an S = 13/2 ground state. In the complexes one Mn atom is bonded to two O atoms and each of two other Mn atoms is bonded to the O and N atoms.[139]
2.3. Quinoids, bipolar ions and radical anions
HQs (A13, A14, 6 – 10, 83 – 86; Fig. 1 and Scheme 23) can be considered as 4n π-electron antiaromatics, singlet diradicals or bipolar ions;[31, 32, 39, 192, 193, 65-68] as well as 4n + 2 π-electron aromatics (87, cf. Wolmeshäuser-type quinoid 88; Scheme 23).[166, 194, 195] They are typical for polysulfur-nitrogen chemistry in general.[59, 60] In the hydrocarbon series, HQs are obtained by various methods, including carbocyclic nucleophilic substitution in HCs;[125, 142, 196] reduction of RCs/dications;[31, 32, 39] and miscellaneous reactions.[166, 194, 195, 197] Compared to the isostructural phenazulene and azulene,[198] the quinoids 87 and 88 prepared from bis(sulfur diimide) 89 and SCl2 , and phenyl vinyl sulfoxide and (SN)4 , respectively (see Scheme 23), have reversed molecular polarization: the seven-membered ring bears a negative charge; and the five-membered ring a positive one.
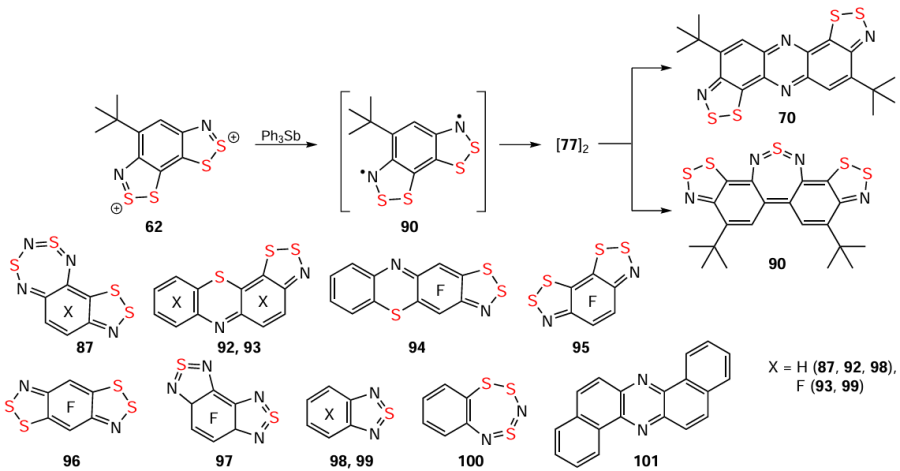
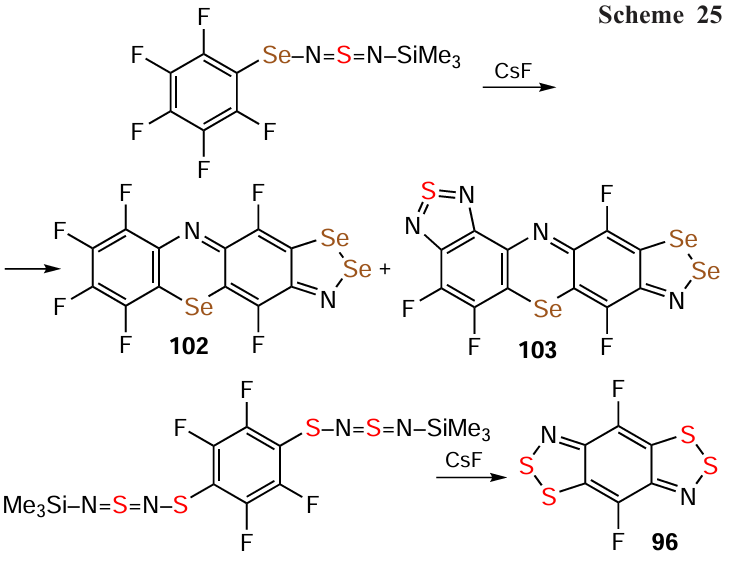
Importantly, HQs are also obtained by the self-condensation of HRs in concentrated solutions, as exemplified by the formation of 70 and 90 from diradical 91 via isomeric π dimers [91]2.[117] The similar structures of the hydro-/fluorocarbon scaffolds suggest that the self-condensation reactions leading to HQs 87 and 92 – 96 involve non-electrophilic/nucleophilic annulation processes, but rather radical ones. The isolation of other heterocycles, such as 66 and 97 – 101, further reinforces the complexity of the involved transformations (Scheme 24).[2, 12, 25, 33, 44, 81, 117, 182, 183] In addition to HQs 92 – 97 from the self-condensation of HRs, fluorinated HQs 96, 102, and 103 are obtained from transformations of 1,3-C6F4(–S – N=S=N – SiMe3)2 and C6F5 – Se – N=S=N – SiMe3 under the influence of CsF (Scheme 25), together with other chalcogen-nitrogen π-heterocycles (cf. transformation 89 → 87;[194] Scheme 23); and 93 is also found among the products of the reaction between tetrafluorinated dithiadiazine 18 and Ph3As.[137]
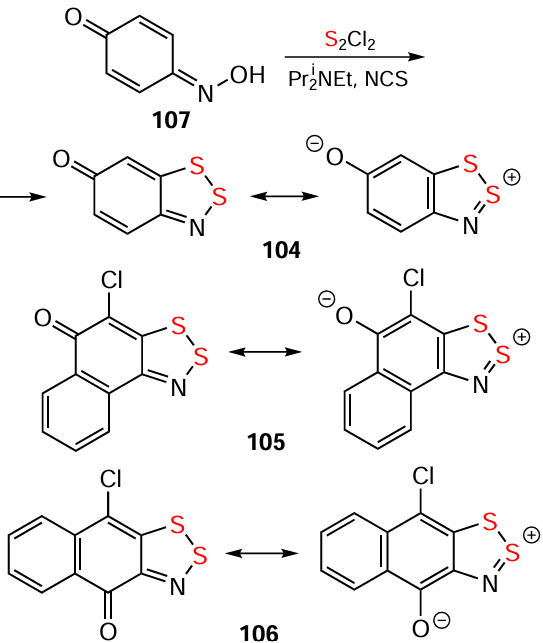
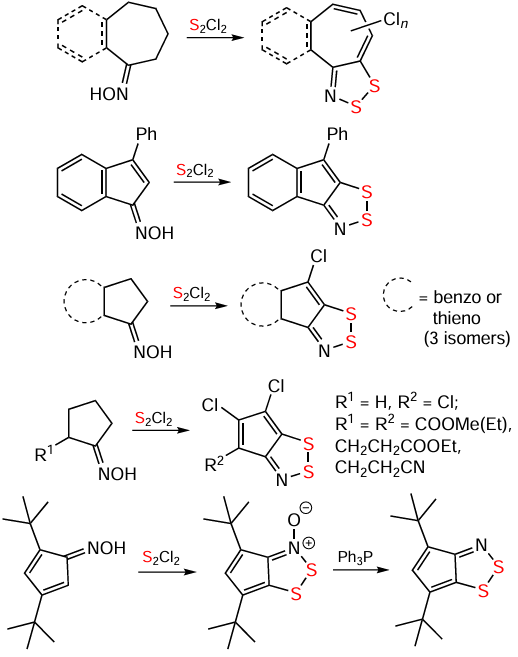
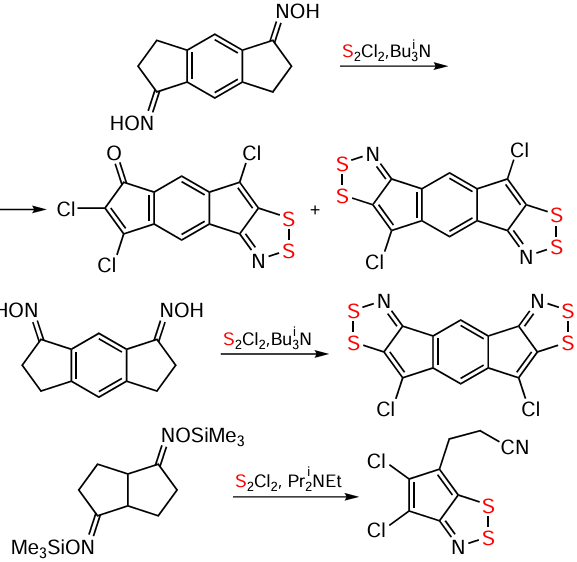
Oximes allow extension of the Herz reaction to nonaromatic starting materials. In particular, hydrocarbon HQs and related S,O-bipolar ions have been synthesized from oximes in both aromatic (compounds 104 – 107; Scheme 26) and nonaromatic (Scheme 27) series. In some cases with the nonaromatics, the presence of strong bases, such as Bui3N or Pri2NEt (Hünig’s base), was requireded (Scheme 28). In some other cases, the assistance of NCS or Ph3P was required (see Scheme 27). The reactions are often accompanied by carbocycle chlorination.[20, 60, 128, 199-203]
HQs 108 – 110 have also been obtained from neutral derivatives of AC and related compounds by miscellaneous reactions (Scheme 29).[80, 204]
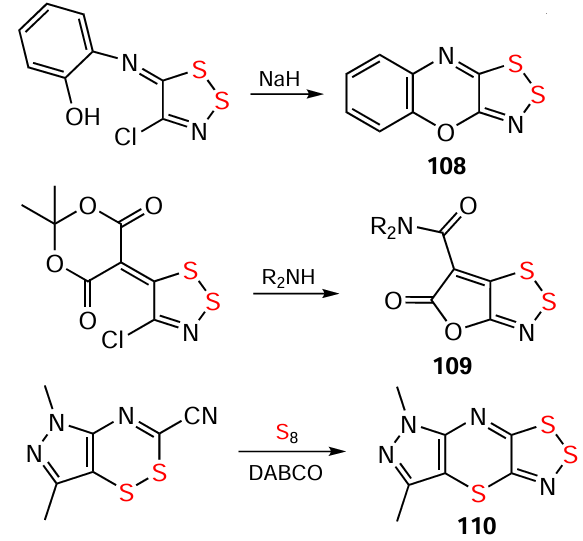
HQs exhibit selective S → Se chalcogen exchange in position 2. For example, S-HQs 111 transform into Se-HQs 112 under the influence of SeO2 (Scheme 30). The exchange is thought to be thermodynamically-driven, as the SO2 byproduct is more preferable than the starting SeO2 .[109, 115, 205] In particular, the incorporation of Se into heterocycles is a hot topic in current chemistry and biomedicine.[206]
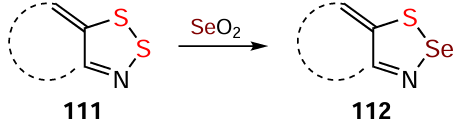
The redox behavior of HQs depends strongly on their structure. HQ 70 produces persistent RA 15 (see Fig. 1) under electrochemical reduction (Scheme 31) in addition to electrochemical oxidation to RC 14 (Scheme 13). HQ 84 forms persistent RA 113 on both chemical and electrochemical reduction; and its archetype 114 is isolated in the form of thermally stable salts. At the same time, HQs 105 and 115 do not afford the expected RAs 116 and 117 (see Scheme 31).[117, 141, 193, 199, 205] According to DFT calculations, the SOMO of RA of 118 (the nonhalogenated archetype of 85 and 98) is strongly antibonding, which leads to cleavage of one on the S – N bonds with the formation of the acyclic structure of the S(–)∙∙∙N(•) type [39] (see below).
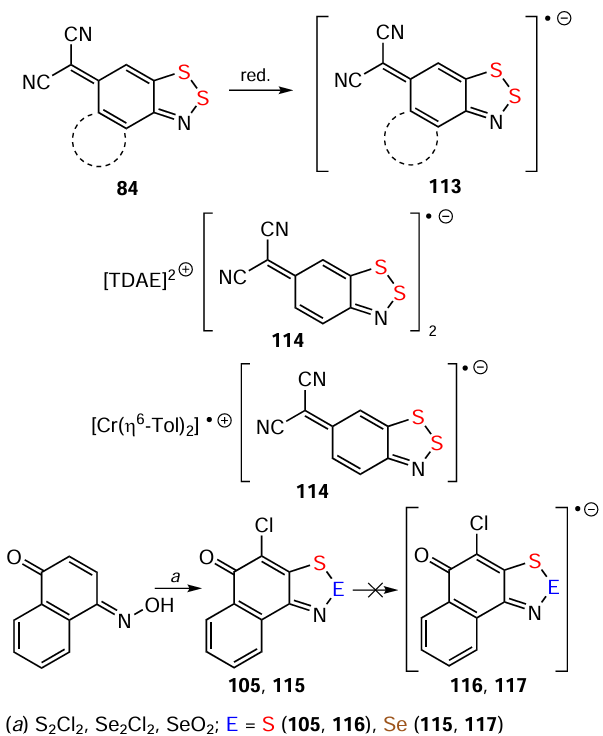
The S,O- and S,N-bipolar ions 7 – 10 are synthesized by deprotonation of HCs 119 – 121 with 1,8-di(dimethylamino)naphthaline (aka the proton sponge) which has a strong tendency to attract protons; the ion 122 is obtained from HC 123 through unstable HRs 124 and 125 (cf. 8, 47, 48, and 67 for 9, 119 – 121, and 123). Likewise, HR 126 transforms into bipolar ion 7 (Scheme 32); however, Se analogues of 124 and 126 do not undergo this transformation.[143, 149, 154, 162, 176, 207, 208]
2.4. Bases
Mild thermolysis of HBs leads to EPR-detected HRs, which under harsher condidions yield phenazines (Scheme 33; here and below, the archetypes are shown for simplicity).[25, 180, 191] The mechanism of the first transformation is unclear, whereas for the second, π dimerization of HRs is proposed, followed by isomerization of the dimers and elimination of sulfur.[89]
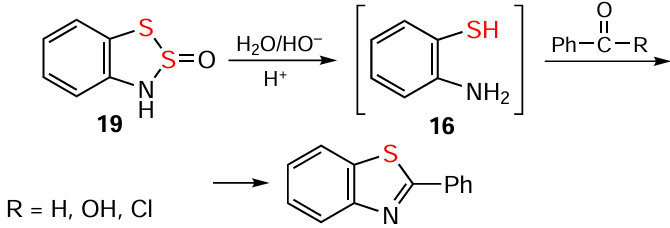
As mentioned above, strong protic acids convert HBs to HCs (see Scheme 2). Mild alkaline hydrolysis of HBs leads, after neutralization, to the formation of 1,2-aminothiols, i.e., 16 and its derivatives, which are normally used without isolation for the synthesis of dyes (i.e., thioindigo) and some other products, e.g., benzothiazoles (Scheme 34; cf. Scheme 14).[6, 7, 10, 140, 2-4, 104-106] The interaction of HBs with organic isocyanides also yields benzothiazoles (Scheme 35).[8]
2.5. Main-group expansion
Both fundamental and practical considerations motivate the further extension of Herz chemistry to include the lightest chalcogen oxygen (another unconventional element in this group) and the heavier pnictogens phosphorus and arsenic in the heterocyclic compounds, as well as the polyhalogenation of carbocyclic systems. Today, only a few polyhalogenated (F, Cl) HQs and bipolar ions are known in this context (Schemes 23 – 25).
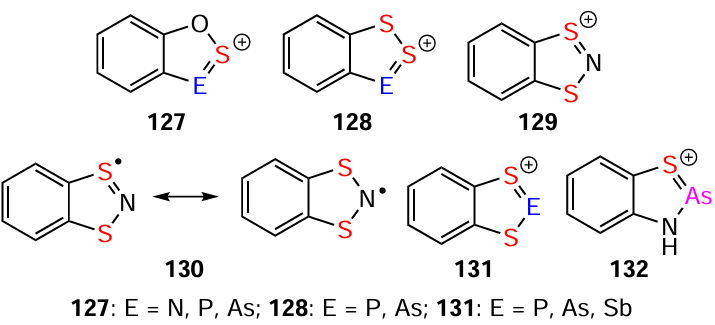
DFT calculations suggest that HCs 127, 128 and their Se analogues are quite feasible, as are the corresponding HRs.[137] In particular, the related WC and WR 129 and 130 are known,[88] and the former have P, As, and Sb congeners 131 and 132 (Scheme 36).[209-216] These findings suggest that S-oxides 133 [217] (for monocyclic sulfamidites, see [47]) could potentially be transformed into HCs 134 by strong protic acids. Furthermore, hydrocarbon [218, 219] and fluorocarbon derivatives of the Ar – O – N=S=O motif could be converted into O containing Herz species 135 – 138 (Scheme 37) using well-established Herz chemistry methods.[44, 115, 165, 166, 182, 185, 220-222] These reactions could potentially be used to synthesize novel heterocyclic and carbocyclic compounds with diverse properties.
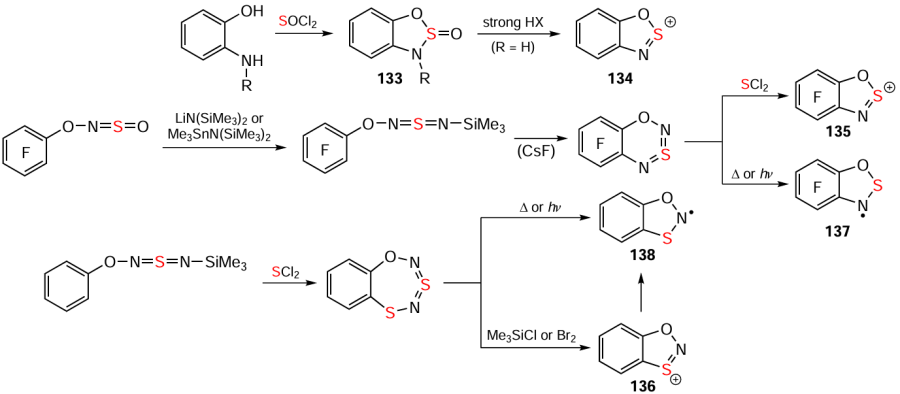
In the carbocyclic context, where polyfluorinated HCs are unknown and EPR-observed HRs have not been isolated [44, 81, 188, 189] (in contrast to WCs/WRs),[81, 137, 221, 223, 224] DFT calculations suggest that the archetypal HCs are HRs are thermodynamically more favored than the corresponding WC and WR by ~ 11 and ~ 14 kcal mol–1, respectively. Tetrafluorination changes this thermodynamic balance only minimally.[223] Monofluorinated ArNH2 undergo the Herz reaction, but the resulting HSs are transformed into 1,2,3-benzothiadiazoles without isolation.[10] Given the significant impact of (poly) halogenation of arenes, including those fused with π-heterocycles, on electronic structure, crystal packing, and electrical/magnetic properties,[224-231] (poly)halogenated HCs, HRs and HQs warrant further investigation.
3. Molecular and electronic structure
Chemical bonds are not directly observable and can only be conceptualized through theoretical descriptors.[232] For Herz chemistry, the various descriptors used throughout its history do not show consistent relationships even for the most relevant compound libraries, posing a significant challenge for further theoretical investigations. Over the years, numerous HCs, HRs, HQs, radical and bipolar ions, dications have been structurally defined by XRD, whereas HBs have only recently been characterized by this method. By EPR, HRs were initially assigned to 1,2-thiazetyls,[180, 191] which was corrected with 33S isotopomers.[18, 26, 27]
3.1. Cations and dications
According to XRD and quantum chemical calculations (Fig. 2 and Fig. 3; Table 1 and Table 2), HCs, RCs, and dications exhibit planar molecular scaffolds and delocalized π-systems [45, 100, 103, 107, 114, 117, 127, 149, 206, 31-33, 38-40, 233-235] typical of unsaturated chalcogen-nitrogen heterocycles.[61, 236] Structurally, HCs are rigid, and variations in the counterion only lead to minor alterations in their solid-state geometries. Gas-phase DFT calculations accurately reproduce the experimental geometries of HCs 17, 23, 24, 26, and 28; ~ 60% of HC charge is localized on the heterocycles. Typical Hirshfeld atomic q values are ~ 0.23, 0.34 and –0.05 for S1, S2 and N3, respectively. The nucleus independent chemical shift (NICS) values of ~ –6 and –15 for carbo- and heterocycles, respectively, suggest the aromaticity of HCs. At the same time, bonds C4–C5 and C6–C7 are shorter, and their bond orders are higher than other C–C bonds, implying a certain ortho-quinoid character of HCs. Replacing of S with Se significantly modifies the atomic charges and bond lengths/orders only at the involved and neighboring sites.[38, 40, 103, 173]
![[{"id":"gfsuj6TBxt","type":"paragraph","data":{"text":"XRD-defined HCs and dications, and CCDC deposition numbers (here and below: multiple numbers for a given Herz species indicate different polymorphs or solvates, or temperatures of XRD measurements; and for charged species, also different counterions). Anions are [Cl]<sup>–</sup> for <b>21, 26, 32</b> (complex with <b>73/74</b>), <b>34</b>; [TfO]<sup>–</sup> for <b>28, 34, 62, 71, 141 – 144, 147</b>; [BF4]<sup>–</sup> for <b>17, 26</b>; [AlCl<sub>4</sub>]<sup>–</sup> for <b>64, 152</b>; [GaCl<sub>4</sub>]<sup>–</sup> for <b>17, 23, 24, 28, 34</b>; <b>62, 71</b>; [FeCl<sub>4</sub>]<sup>–</sup> for <b>34</b>; [SbF<sub>6</sub>]<sup>–</sup> for <b>65, 139, 140, 148</b>; [SbCl<sub>6</sub>]<sup>–</sup> for <b>21, 145, 149, 150</b>; [SbCl<sub>6</sub>]<sup>–</sup>/[Cl]<sup>–</sup> for <b>17, 26</b>; [TeCl<sub>6</sub>]<sup>2–</sup> for <b>150</b>; [Te<sub>2</sub>Cl<sub>10</sub>]<sup>2–</sup> for <b>28</b>; [Te<sub>3</sub>Cl<sub>14</sub>]<sup>2–</sup> and [Te<sub>4</sub>Cl<sub>18</sub>]<sup>2–</sup> for <b>21</b>; [C<sub>4</sub>F<sub>9</sub>SO<sub>3</sub>]<sup>–</sup> for<b> 146</b>."}}]](/storage/images/resized/IvtiVm4Ru73eOAAfZpfavqnIFhEluE16hkiGhfBo_xl.webp)
![[{"id":"1KtWyHYksH","type":"paragraph","data":{"text":"XRD molecular structures of selected HCs; color code: C grey, H light grey, N blue, S yellow, Se light orange, Te dark orange."}}]](/storage/images/resized/S40AN2rYTV3a6oWfyTzn0sjIONYU9EizKUs5jzQM_xl.webp)
The linear dications 64 and 152 also feature elongation of the heterocyclic C–C bonds prior to equalisation of all others, in contrast to the angular dication 62. The XRD structure of the linear/angular dication 71, known for three HCs with two counterions and two solvates, suggests that 1,2,3-dithiazoliums are structurally more rigid than benzene or pyrazine rings.[39, 117]
Absorbance in UV-VIS and chemical shifts in heavier-nuclei NMR spectra of HCs are solvent and counter-ion dependent. The UV-VIS spectra of perchlorates in CF3CO2H reveal the λmax bands in the range ~410 – 560. Substitution of S by Se at position 2 results in a bathochromic shift Δλmax ~ 20 – 25 [7, 172, 174] (Table 3). The spectra of the dications are quite similar to those of HCs, except for the UV-VIS spectrum of 62, which has a λmax of ~ 400.[117] The δ77Se of Se-HCs falls within the downfield half of the window reported for Se – N compounds; and the δ77Se for position 1 is upfield compared to the δ77Se for position 2 (~ 1310 – 1420 and ~ 1470 – 1600, respectively). The DFT-calculated δ77Se and δ15N values are in reasonable agreement with the experiment, although δ77Se is slightly underestimated; and δ15N is overestimated [38, 234] (Table 4).
3.2. Radicals and radical ions
XRD, EPR, and DFT studies (Fig. 4 and Fig. 5; Table 5 and Table 6) have confirmed that HRs and RCs have planar molecular geometries and delocalized π-systems. The (het)arene-, e.g., pyridine-, bridged HRs are resonance-stabilized in that spin delocalization is enhanced by a resonance interaction between open- and closed-shell 1,2,3-dithiazole rings. The HC → HR transformation lengthens E – N bonds (E = S, Se), decreasing their bond orders, and markedly, but not completely, aligns C–C bonds [16, 41, 42, 89, 110, 117, 126, 127, 139, 160, 176, 177, 192, 207, 233, 31-38, 142-153, 155-157, 239-250] (see Table 5). According to the Bader analysis (QTAIM), this transformation significantly decreases the delocalization indices of the S – E bonds (E = S, N, C), exhibiting significant differences between HCs and HRs, with higher values for HCs; and the lowest calculated delocalization index of 0.96 corresponds to the internal S∙∙∙S bond in σ-dimer 153 (cf. with 1.35 for Me – S – S – Me).[251] This transformation also reduces the q values for all atoms, in particular by ~ 0.2 – 0.3 for the N and E atoms (E = S, Se) and ~ 0.1 for the C6 atom. For the N atoms, q is negative, whereas for the chalcogen atoms it is positive; for the latter, q is larger at position 2 than at position 1. The overall charges of the carbocycles and heterocycles of HRs are negligible.[38]
![[{"id":"FSai7xFzQl","type":"paragraph","data":{"text":"XRD-defined HRs, σ-dimers, and RCs, and their CCDC deposition numbers. Anions are [Cl]<sup>–</sup> for <b>49</b>; [ClO<sub>4</sub>]<sup>–</sup> for <b>50, 211</b> (for [<b>211</b>]<sub>3</sub>[ClO<sub>4</sub>]<sub>2</sub>); [BF<sub>4</sub>]<sup>–</sup> for <b>49</b>; [GaCl<sub>4</sub>]<sup>–</sup> for <b>14, 49, 63</b>; [GaBr<sub>4</sub>]<sup>–</sup> for <b>63</b>; [FSO<sub>3</sub>]<sup>–</sup> for <b>50</b>; [TCNQ]<sup>• –</sup> for <b>49</b>. L = hfac."}}]](/storage/images/resized/tjfILmVxeE2SRKyWCaKVJgidAaT9swO38Q7XOf1R_xl.webp)
![[{"id":"npQ0xzW5ng","type":"paragraph","data":{"text":"XRD-defined HRs, σ-dimers, and RCs, and their CCDC deposition numbers. Anions are [Cl]<sup>–</sup> for <b>49</b>; [ClO<sub>4</sub>]<sup>–</sup> for <b>50, 211</b> (for [<b>211</b>]<sub>3</sub>[ClO<sub>4</sub>]<sub>2</sub>); [BF<sub>4</sub>]<sup>–</sup> for <b>49</b>; [GaCl<sub>4</sub>]<sup>–</sup> for <b>14, 49, 63</b>; [GaBr<sub>4</sub>]<sup>–</sup> for <b>63</b>; [FSO<sub>3</sub>]<sup>–</sup> for <b>50</b>; [TCNQ]<sup>• –</sup> for <b>49</b>. L = hfac.[[ type=\"anchor\" referenceId=\"9456\" ]]"}}]](/storage/images/resized/L7wYLTxb3KlMFibcuXdzq7MCr1zKdKOpQ71Klf0K_xl.webp)
![[{"id":"70gGDJafq1","type":"paragraph","data":{"text":"XRD molecular structures of selected HRs, σ-dimers, and RCs; color code: C grey, H light grey, Cl green, F light green, N blue, S yellow, Se light orange."}}]](/storage/images/resized/Qcog0SVyoX6AIx27o1dTfVjpZpVo2FCKyX5QLlm4_xl.webp)
![Selected bond lengths, Hirshfeld bond orders and atomic charges, atomic spin densities and hfc constants (<i>a</i>) of HRs <b>A5</b> (Figure 1) from B1B95/cc-pvqz calculations.[<a class="anchor" href="#reference-9242">38</a>]](/storage/images/resized/A3DCj8Op7QKXR47TcL76eYV7uYb6xlWVtKsJBbnd_xl.webp)
Resonance-stabilized HRs A6 and A7 (E = S, Se) are dimorphic, existing in two different molecular and crystal paramagnetic/diamagnetic modifications consisting of individual molecules and, in the space group P21/c, e.g., the same as for (SN)x,[253] unusual centrosymmetric σ-dimers, respectively.[147, 150, 160, 176, 241, 245] The peculiarity of the dimers 153 and 206 – 209 is the hypervalent four-centre six-electron E – E∙∙∙E–E (E = S, Se) bonds. A characteristic of these nearly linear bonds is that the terminal E–E distances, corresponding to the sum of covalent radii, are shorter than the internal E∙∙∙E distance which is intermediate between the sums of covalent and van der Waals (VdW) radii, whereas the C – E distance is intermediate between those for the C – E single and C=E double bonds (Table 7; to emphasize this, the pseudo-quinoidal C=E designation is used for the σ-dimers in Fig. 4 and Table 7). At normal pressure and temperatures below 400 K, dimers 204 do not undergo thermal dissociation 204 → 2(174); when exposed to external pressure at ambient temperature, they remain intact up to 8 GPa. The dimers 205, however, undergo the 205 → 2(175) topochemical dissociation at 0.8 GPa. Dissociation of 204/205 can be induced by visible light of 650 nm, and the photoinduced HR pairs persist up to 150/242 K for HRs 174/175, respectively, before reverting to the dimer states. Arrhenius activation energies for the HR ↔ σ-dimer interconversion are 8.3 and 19.6 kcal mol–1 for 2(174) ↔ 204 / 2(175) ↔ 205, respectively. DFT and CASSCF calculations suggest that the ground-state interconversion is symmetry-forbidden, requiring a configurational change (σ2+)2 ↔ (π)+(π)–, but is allowed photochemically.[150, 176, 247] With pyrazine linkage, σ-dimers 153 and 210 exhibit bimodal association characterized by the presence of S – S and C – C bonds, respectively; 210 is kinetically, and 153 thermodynamically, favored. Replacing the N – Et group of 153/210 with N – Me gives non-dimerized 183.[160, 161]
The antibonding π*-SOMO (Fig. 6) shows isolobal behavior for S and Se incarnations of HRs. The spin densities are mainly concentrated on the chalcogen-nitrogen fragments, especially at the N atoms. In 2 and its analogues, only ~ 23% of the spin density is transferred to the carbocycle, presumably encompassing atoms C4, C6 and C7a, similar to the spin density distribution observed in benzyl radicals. The substitution of S by Se introduces minor perturbations in the electronic structures, including spin density distributions and a magnitudes, primarily affecting neighbouring sites. Due to the strong spin-orbit coupling (SOC, which scales with the atomic number Z as Z4) [258, 259] at Se atoms and the large anisotropy of g tensors, the resulting EPR spectra in solution are significantly affected, sometimes becoming unresolved. In contrast to MP2 calculations, DFT simulations using B1B95 and B3LYP functionals (but not PBE) and 6-31G and cc-pvtz basis sets provide reasonable approximations of experimental a constants magnitudes (see Table 6).[33, 38, 45, 81, 88, 125, 182, 185, 189, 235, 242, 40-42]
The UV-VIS spectra of isostructural HRs/HCs exhibit remarkable shape similarity; the HCs → HRs transformation primarily induces bathochromic shifts in λmax (Table 3 and Table 8) and modulates the fine structures of bands.[18, 23, 25, 41, 170, 179] In particular, the spectra of HRs 2 and 156 show three solvent-dependent absorption bands that increase in intensity from long to short wavelengths; and the longest wavelength band of 2, a fine structure with four maxima spaced at distances of ~ 400 cm–1.[179]
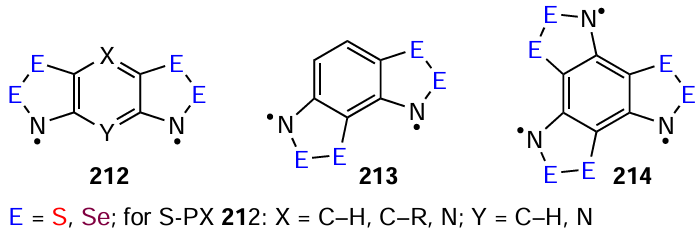
High-spin HRs have only been investigated theoretically, where the quantitative estimation and interpretation of spin-energy gaps of polyradicals remains challenging.[261] For proposed diradicals 212 and 213 (Scheme 38), CASSCF and DFT simulations indicate that the ring closure mode and the nature and positions of chalcogen atoms influence the sign/magnitude of the singlet-triplet splitting ΔEST. The ground state of the all-S linear radical 212 is a singlet. Replacing S with Se decreases the absolute value of ΔEST and results in a triplet ground state for the all-Se derivative.[81, 88] Electron-donating substituents in the central ring stabilize the paramagnetic state of all-S 212 and its aza-congeners; while electron-withdrawing substituents stabilize the diamagnetic bipolar state.[177, 262] For angular diradicals 213, irrespective of S and Se combinations, the ground state is a triplet with a much larger ΔEST compared to linear 212. For C3-symmetrical angular triradicals 214, CASSCF / DFT calculations propose a quartet ground state. Replacing S with Se has a negligible impact on the doublet-quartet splitting, with two doublet states nearly degenerate.[81, 88, 262, 263]
Similar to HCs/HRs, RCs 14, 49, 50, 63, and 211 [31, 32, 117, 192, 239, 155-157] and RAs 15, 113, and 114 [141, 196, 199] exhibit π-delocalization (Figs 6 – 8). In 113 and 114 the spin density and negative charge are distributed throughout the molecule; but the density has a higher concentration on the heterocyclic moieties. Persistent RAs 113 and 114 are chromophores with λmax values of ~ 660 (for neutral precursors M, λmax values are ~ 575); and 15 (Fig. 7) is a NIR chromophore with λmax ~1000.[117] In the transformation from M to RA, DFT calculations indicate the lengthening of the heterocyclic S – E (E = C, N, S) and N–C bonds, accompanied by a shortening of the C–C bond, reflecting the antibonding character of the π*-SOMO (see Fig. 7; cf. HCs/HRs). For 114, the archetype of RAs 113, magnetic properties of the radical-ion salt [Cr(η6-Tol)2][114] suggest its solid-state π dimerization.[141, 196, 199] Compared to 15 and 113, RAs 116 and 117 (see Scheme 31) are highly unstable. DFT calculations on the related RA 215 and its Se congener suggest a σ*-SOMO with an abnormally elongated S∙∙∙E (E = S, Se) distance (Fig. 8), i.e., weakening or breakage of the corresponding bond [205] (cf. RA 118).[39]
![[{"id":"fknVkbNJOF","type":"paragraph","data":{"text":"Molecular structure (<i>a</i>), the π*-SOMO (<i>b</i>), and solution EPR spectrum (<i>c</i>) of RA <b>15</b>; hfc constant a<sub>N</sub> (mT) 0.48 (dithiazole).[[ type=\"anchor\" referenceId=\"9321\" ]]"}}]](/storage/images/resized/RKNjUMZuVzkhUI3Gj5pnwN6M7fIR2YtVsdKyYqpI_xl.webp)
![[{"id":"7LPuKi2o3N","type":"paragraph","data":{"text":"Top: The π*-SOMO and solution EPR spectrum of <b>114</b>; hfc constants (mT): a<sub>N</sub> ~ 0.82 (dithiazole) and ~0.04 (CN), a<sub>H</sub> ~ 0.25, 0.10 and 0.01.196 Bottom: the σ*-SOMO and DFT-optimized structure of <b>215</b> with an unusually long S∙∙∙S distance (Å).[[ type=\"anchor\" referenceId=\"9409\" ]] Color code: C grey, H light grey, Cl green, N blue, O red, S yellow."}}]](/storage/images/resized/vHqiwZDPqFkEC7loWbOKfvbrLhTDYPKoZpolgqLE_xl.webp)
3.3. Quinoids and bipolar ions
The stereoelectronic properties of HQs are varied. Structurally characterized HQs (Fig. 9 and Fig. 10) show localized double and single bonds. Some HQs are formally antiaromatic, which can also be assigned to singlet diradicals in the diamagnetic ground state, while others are formally aromatic, e.g., 87 and 216. For HQ 118, DFT calculations suggest that the excited triplet state is 0.74 eV higher in energy than the singlet ground state; conversely, for its 1,3,2-isomer, the triplet ground state is 0.48 eV lower in energy than the singlet state. The bonding situation in many HQs can be represented by superposition of nonpolar and bipolar structures, i.e., of S,O/S,N-bipolar ions, with the charge differentiation arising from the separation of the π-system into two distinct subunits (Fig. 9).[31, 32, 39, 85, 114, 117, 128, 141, 149, 162, 192, 193, 199, 200, 264, 65-67]
![[{"id":"qYZZBa91ey","type":"paragraph","data":{"text":"Selected XRD-defined HQs and bipolar ions, and their CCDC deposition numbers. For naphthoxide <b>221</b>, cation is [Li]<sup>+</sup> in ([Li][<b>221</b>] · H<sub>2</sub>O)<sub>4</sub>."}}]](/storage/images/resized/r0D3GVWmPOXhouVWyy5H2YDmDrftGxuNbFZtKMdn_xl.webp)
![[{"id":"eQRJ0yDmVN","type":"paragraph","data":{"text":"XRD molecular structures of selected HQs and bipolar ions; color code: C grey, H light grey, Cl green, F light green, N blue, O red, S yellow, Se light orange."}}]](/storage/images/resized/bvth9FvEncAyz7lfpU2Y5GZQenLC9kV2UU4PtieU_xl.webp)
HQs exhibit chromophoric properties with λmax ~ 460 – 660.[31, 32, 39, 183, 194, 200] In particular, 6-6-5 tricyclic angular and linear isomeric HQs 105 and 106 reveal λmax ~ 460 and ~ 600, respectively;[203] whereas 5-6-6-6-5 and 5-6-7-6-5 pentacyclic HQs 70 and 90 serve as NIR dyes with λmax ~ 690 – 760 and 745 – 795, respectively.[117] For HQ 6, the low-lying excited states (in particular, a thermally excited triplet state was detected by EPR, ΔEST = 0.05 eV) are attributed to its singlet diradical character.[193] For benzoquinone-bridged E,N/E,O bipolar ions A15 (E = S, 7; E = Se, 234) λmax ~ 730.[143, 208]
3.4. Bases
The HBs are structurally defined by XRD only in the form of a dichlorinated compound 236 and 88 : 12 cocrystal 237 of mono- and dichlorinated derivatives;[137] the structures exhibit a shortened N – H∙∙∙O contact ~ 2.1 Å (sum of VdW radii, ~ 2.7 Å),[255] i.e. a hydrogen bond, together with planarity of the C – N(–H) – S fragment. According to DFT, this fragment is nonplanar for a free molecule, i.e. the N atom is chiral, as well as the neighboring S atom (Fig. 11).[137] The presence of two chiral centres agrees with the observation of diastereomers in solution 1H NMR spectra of HBs.[104]
![[{"id":"oezGm4Abic","type":"paragraph","data":{"text":"DFT molecular structure of the archetypal HB <b>19</b> and XRD molecular structures of chlorinated HBs <b>236</b> and <b>237</b> (88 : 12 cocrystal of mono and dichlorinated derivatives).[[ type=\"anchor\" referenceId=\"9341\" ]] Color code: C grey, H light grey, Cl green, N blue, O red, S olive / yellow."}}]](/storage/images/resized/lRb4dDlGO1lutgeQk4eixArQuvGDWD4TwPzrphhv_xl.webp)
4. Crystal structure
4.1. Salts
In HSs from the Herz reaction, the anions are typically [Cl]–. Normally, such HSs are poorly soluble in organic solvents due to strong anion-cation electrostatic interactions and do not form single crystals suitable for XRD; acetic and sulfuric acids are more suitable. Metathesis with various low-coordinating anions easily provides HSs soluble in organics. Crystalline HSs [17][BF4], [23][BF4], [24][BF4], [28][GaCl4] and [151][GaCl4] exhibit centrosymmetric dimers characterized by shortened E2∙∙∙N3 intermolecular contacts of ~ 3.24, ~ 2.87 and ~ 2.53 Å for E = S, Se and Te, respectively.[33, 38, 40] The sum of VdW radii of the E and N atoms is ~ 3.35, ~ 3.45 and ~ 3.60 Å for E = S, Se and Te, respectively.[255, 265] These shortened contacts are typical of chalcogen-nitrogen compounds, particularly those containing Se and Te, and are indicative of SBIs (i.e., interactions resulting in interatomic contacts that are longer than the sum of the corresponding covalent radii but shorter than the sum of the corresponding VdW radii), namely, ChB driven by σ-holes [266] and enhanced by increasing atomic number/polarizability [267, 268] of chalcogen. The holes are regions of low electron density/positive electrostatic potential spatially located on the outer-side extensions of the σ-bonds.[112, 266, 269-271] In contrast, no shortened intermolecular E∙∙∙N contacts are observed in [24][GaCl4] and [26][Cl].[38, 103] However, it should be emphasized that for the most studied SBI, i.e., the halogen bond (XB), the interaction distances exceed the sum of the VdW radii up to ~ 20%.[272]
Owing to ion-ion interactions, most chloride HSs are nonvolatile and exhibit poor solubility in organic solvents, e.g., [26][Cl].[103] In contrast, chlorides of isomeric WCs, e.g., 129 and its substituted derivatives, exhibit good solubility, and salt [129][Cl] sublimes in vacuo.[221] Overall, HCs have a lower propensity to form neutral ion pairs than WCs; however, XRD studies have identified pairs or dimers for chlorides of But-substituted HCs, which also exhibit high solubility in CHCl3 and CH2Cl2 .[100, 103] Crystals of [21]2[Te4Cl18] exhibit discrete neutral units composed of the anion and two cations and good solubility in CHCl3 and MeCN. In contrast, crystals of poorly soluble [21]2[Te3Cl14] contain an infinite number of double chains of the anions bound by numerous interactions with four chains of the cations.[103]
As mentioned for HCs, the S/Se replacement causes only minor changes in the molecular and electron structures, often manifested in the isomorphism of the S and Se incarnations of HSs. Thus, [HC][SbCl6] (HC = 21, 145, 149, 150), [HC]3[SbCl6]2[Cl], and [HC][BF4] (HC = 17, 26) are isomorphous; and [HC][SbCl6] (HC = 149, 150) form mixed crystals.[38, 100, 107]
4.2. Radicals and radical ions
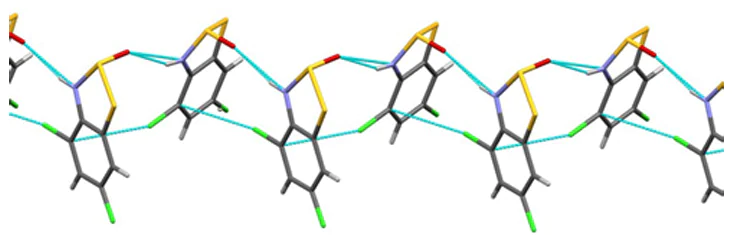
In the crystalline state, HRs (see Fig. 1) form 0D dimers, 1D stacks, planar 1D ribbons, or 2D layers, whose aggregation results in various 1D-3D architectures. Cofacial dimers typical of chalcogen-nitrogen heterocyclic π-radicals with long E∙∙∙E intermolecular distances (S∙∙∙S, ~ 3.1 – 3.3 Å) are observed only for the naphthalene/naphthoquinone derivatives 156/82 (one of two polymorphs). The dimers of another polymorph of 82 are cofacial only for the heterocyclic moieties; similar dimers are observed for 154, whereas HR 38, an isomer of 82, does not form dimers. HRs 155 and 157 form head-to-tail coplanar dimers. Dimerization by the formation of the C – C σ-bond is known only for 210. Planar σ-dimers 153, 204 – 209 are packed into stacks with a strong offset. Stacks of HRs can be equidistant or alternating, perfectly coplanar or not, and with varying degrees of slippage and inclination. Interplanar distances are normally within ~ 3.3 – 3.5 Å typical of π-stacking interaction.
The SBIs, manifested by shortened intermolecular contacts, are most typical for the E atoms in position 2. The E2∙∙∙E2' contact is often accompanied by the N∙∙∙E1' contact forming a cycle composed of N atom and three E atoms. Overall, cyclic supramolecular synthons based on the E∙∙∙E and E∙∙∙N contacts are widespread in crystalline HRs and have also been observed in HCs. They form both planar and non-planar ribbons and layers.
Dimorphic resonance-stabilized HRs A6 and A7 (E = S, Se) exist in paramagnetic and diamagnetic modifications composed of individual HRs and their σ-dimers, respectively.[150, 176, 245] Among paramagnetic modification, the most interesting is the group of isomorphous tetragonal pyridine-bridged HRs. For 175, 176, 181, 182, 189, 190, 194 – 197, and 201 – 203, neighboring infinite stacks of HRs are connected by shortened intermolecular E∙∙∙E and E∙∙∙N contacts forming columns composed of four parallel stacks. In the stacks, HRs are strongly shifted so that the magnetically interacting SOMOs of the neighbours are nearly orthogonal, allowing ferromagnetic (FM) ordering (see below) in accordance with the Kahn orbital model (KOM), which requires for the FM state a strict orthogonality of magnetically-coupled SOMOs that excludes their spatial overlap; and for the antiferromagnetic (AF) state a non-orthogonality that allows a small overlap.[273, 274] The diamagnetic modification is exemplified by the centrosymmetric σ-dimers 153 and 206 – 209 crystallizing isomorphously in the monoclinic space group P21/c,[147, 160, 233, 235] e.g., the same as for (SN)x.[253] The space group allows approximately coplanar HR ribbons, bridged by four-center [E∙∙∙N]2 (E = S, Se) SBIs, i.e. ChBs, which are suitable for least-motion topochemical conversion of the E–E bonds of HRs into the internal E∙∙∙E bonds of the dimers (for the formation of σ-dimers by pyridine-bridged HRs, see Scheme 39).[147] The size of the side groups is important for the conversion, e.g., exchanging N – Et of 153 for N – Me of 183 suppresses dimerization of the latter.[160, 161] Other space groups observed for related HRs exclude the [E∙∙∙N]2 SBIs and thus the formation of σ-dimers.[147]
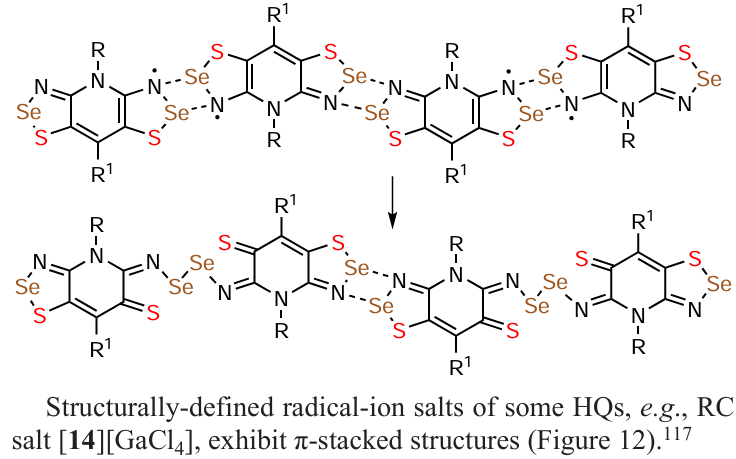
Structurally-defined radical-ion salts of some HQs, e.g., RC salt [14][GaCl4], exhibit π-stacked structures (Fig. 12).[117]
![[{"id":"MxKdtGA_ZB","type":"paragraph","data":{"text":"Fragments of XRD crystal structure of RC salt [<b>14</b>][GaCl<sub>4</sub>] (cation omitted) featuring infinite uniform π-stacks with interplanar separation of ~ 3.4 Å. Color code: C grey, H light-grey, N blue, S yellow."}}]](/storage/images/resized/jHZ9NYvqaSfUCbAxjDSkPRck8tsRdK59FOXQotAd_xl.webp)
4.3. Quinoids and bipolar ions
In the crystals of HQs and related bipolar ions (Fig. 9), the most common structural motif is π-stacking with typical interplanar separation of ~ 3.4 – 3.6 Å. Bipolar ions display head-to-tail and head-to-head stacking with offset, allowing electrostatic interaction of their unlikely charged parts. Shortened intermolecular E∙∙∙E and N∙∙∙E contacts in crystalline HQs and bipolar ions form planar 1D ribbons and 2D layers.
Compound 220 forms non-planar infinite chains with shortened S∙∙∙O contacts. In crystalline 122, each molecule is connected by S∙∙∙O contacts with four others giving rise to 3D structure. Among other oxo compounds, 7 forms 2D layers, whereas 105, 222, 223, and 234 form π-stacks. Nearly planar dimers of 222 are formed by the H∙∙∙O hydrogen bonds between OH and C=O groups.
Mixed-valence salts formed by RCs 49, 50, 63, 211 with corresponding HQs 6, 53, 85, 231 show stacks with alternating distances and slippage. RC salt [50][ClO4] has no stacks, whereas RC salts [6][Cl] and [14][GaCl4] form infinite equidistant stacks. Crystals of [85][TCNQ] exhibit ...74 ... 74 ... TCNQ ... TCNQ ... infinite stacks and nearly planar layers, in which each RC and RA contact with four counterions.
4.4. Bases
5. Materials science
Herz species are real or potential building blocks of conductive, magnetic and optoelectronic materials. The electrical conductivity of solid HRs is facilitated by a low Coulombic barrier to CT between their π*-SOMOs. For resonance-stabilized HRs, the nature of the spacer and chalcogen atoms, as well as the positions of the atoms, are influential. For some, band-gap closure with formation of the metallic state is observed under external pressure [31, 32, 39, 42, 110, 114, 126, 127, 192, 193, 207, 233, 262, 273, 34-37, 143-153, 157-162, 239-250, 275-286]. From a magnetic point of view, Se and yet unknown Te derivatives are particularly exciting, since the spin density on the heavier atoms with stronger SOC [258, 259] allows for increased isotropic and mediated anisotropic exchange effects, magnetic hysteresis and anisotropy, and is involved in singlet fission (SF; i.e., the transformation of a singlet excitation into a singlet spin-correlated triplet pair state, potentially enabling the thermodynamic Shockley – Queisser limit of 33% for the solar cell efficiency to be overcome),[287-289] access to triplet excited states,[290] and triplet-triplete interconvention,[291] which are of interest for materials science. Isotropic and anisotropic FM exchange can also be enhanced indirectly by the incorporation of heavier atoms into nonspin-bearing sites, where they can contribute to multiorbital/multicenter SOC.[285] Such heavier atoms may not only belong to the chalcogen family but also to the halogen family, which is capable of participating in XB,[292, 293] i.e., another SBI relevant to σ-hole driven interaction [266]. The latter is especially important; in particular, in crystalline [Mn(hfac)2]3[81]2 (see Scheme 22) the S∙∙∙O ChBs form Heisenberg spin chains, allowing long-range magnetic ordering.[242] In some cases, external pressure increases the tempetature of bulk magnetic ordering, i.e., the Curie/Nèel temperature (TC/TN).
Overall, the S/Se replacement strengthens solid-state SBIs, first of all ChB, towards increasing electrical conductivity and magnetic exchange interactions and magnetic anisotropy.[273, 285, 286] Many Se-HRs (see Fig. 3) are electrical conductors and low-temperature magnetics, i.e., FMs/AFs, in the latter case including also systems spin-canted by Dzyaloshinskii-Moriya mechanism (DMM),[294] i.e., weak FMs; however, heavier-halogen containing HRs (see Fig. 3) are less studied. The conductive/magnetic properties of some HRs-based materials are driven not only by external pressure, but also by heat and/or light, making them smart materials.[174, 245, 277]
Of particular interest are HQs capable of forming stable radical-ion salts, e.g., RC salts [63][GaCl4],[157] [14][GaCl4],[117] and [63][GaBr4],[239] and RA salts,[117, 141, 196] e.g., [Cat][114],[196] promising as both conductors and magnetics. Potentially, even more important is the singlet diradical or quinoidal/Baird (anti)aromatic character of HQs (in Baird theory, electronically-excited states of ground-state antiaromatics are aromatic;[295-297] in particular, this allows photoswitching of local aromaticity/antiaromaticity [298]). Together with multicenter SOC in heavier-atom derivatives, chromophoric/fluorophoric, redox, and semiconductive properties, these make HQs promising materials for small-molecule optoelectronics [299-302] including unconventional SF materials.[288, 303] However, HQs are less studied in these contexts.
The polymorphism, typical of Herz species, obviously affects their conductive, magnetic and optical properties.
5.1. Conductors
The open- and closed-shell Herz species, e.g., HRs and their dimers, bipolar ions, and mixed-valence salts can be paramagnetic/Mott insulators (Ea >> 0, σ ≤ 10–7), semiconductors (Ea > 0, σ ≥ 10–6) or/and metals (Ea ~ 0, σ ≥ 103; where Ea is the activation energy of the thermal electrical conductivity expressed in eV, and σ is the room-temperature electrical conductivity expressed in S cm–1). Besides the external pressure and SBIs, the electrical conductivity of HRs strongly depends on a complex interplay of many factors, including the nature/molecular positions of chalcogens, the nature/bulkiness of the substituents, the structure and energy of the π*-SOMOs forming the solid-state electron-energy bands. For oxobenzo-bridged HRs relevant to A4 HCs, the π*-SOMOs can be constructed from group-orbitals of the heterocyles, C=O and other carbocyclic substituents. In this sense, their conductivity is driven by multiorbital effects via corresponding electron-energy bands. Exteremely important is the SBIs-related dimensionality of the crystal packing where uniform 1D π-stacks or 2D π-sheets are the most favorable.[143, 144, 148, 155, 207, 279, 280] The A15 benzoquinone-bridged diradicals/bipolar ions (E = S, 7; E = Se, 234) exhibit different crystal packing as Cmc21 for 7, and P21/c and R3c for two polymorphs of 234, together with C2/c for 234 · 2 DMI solvate. Normal-pressure (~ 100 kPa) σ is ~10–3 for 7 and R3c-234, ~ 10–5 for P21/c-234, and < 10–7 for 234 · 2 DMI. High-pressure σ is 1 for P21/c-234 (8 GPa), > 10 for 7 (8 GPa), and ~ 102 for 7 (12 GPa) and R3c-234 (6 GPa), with metallization (Ea ~ 0) in the last three cases [143, 208] (Table 9).
The σ-dimers 206 – 209 of pyridine-bridged HRs 169 – 171 and 174 are small bandgap semiconductors with normal-pressure σ ~ 10–6; Ea ~ 0.32 and 0.36 for 206 and 209, respectively. At an external pressure of 4 – 5 GPa, σ ~ 10–2, 10–1, 1 and 10 for 209, 207, 208, and 206, respectively. For 206 at ~ 5 – 9 GPa, Ea ~ 0 indicates metallization. A weakly metallic state arises from intra/intermolecular changes including a collapse of the HOMO – LUMO gap (and hence a near coalescence of the valence and conduction bands, as suggested by solid-state calculations) and increased SBIs, causing band edge broadening and overlap. Near 5 GPa, intramolecular changes include an electronic configuration switch from a hypervalent S – Se – Se – S σ-bond to a π-bonded arrangement via buckling of 206 molecules accompanied by a construction in the S – Se – Se – S interatomic distances. The intermolecular changes are a concertina-like compression of the π-stacks. Overall, the σ-dimer → π-HR switch transforms a semiconductor to a metal.[147, 233, 250]
For the σ-dimer/monomer dimorphic pyridine-bridged 205/175, σ ~ 10–7/10–4 at 0.5/5 GPa.[150] For the pyridine/pyrazine-bridged 193/183, normal-pressure σ ~ 10–3; at 8 GPa it increases to 10/1 remaining activated. The benzoxo-bridged solvate 167 · MeCN exhibits σ ~ 10/5 – 8 GPa.[147, 148, 161]
The record normal-pressure σ is ~ 0.5 for the mixed-valence salt [63][GaCl4]; whereas ~ 102 for E,N/E,O bipolar ions 7 (E = S) and 234 (E = Se) (i.e., A15) [143, 208] is the record higher-pressure σ (Fig. 14). All these values are in the upper part of the σ range of conventional semiconductors ~ 10–6 – 103, and 102 is not far below the Mott – Ioffe-Regel (MIR) limit of metallic electrical conductivity.[302] For HR 160, a 2D Fermi-liquid (i.e., quantum liquid composed of interacting fermions featuring delocalized states and exhibiting no phase transition) metallic state is observed at 6 GPa.[279] The best thiazyl conductor, polymeric sulfur nitride (SN)x, formally composed of NS• radicals,[50, 51] is a genuine metal, exhibiting σ ~ 103 (~ 105 at ~4 K) along the macromolecules and becoming a superconductor at ~0.3 K (0.54 K at 0.9 GPa).[253, 289, 304, 305] The metallic state of (SN)x is caused by the peculiarities of its crystal packing in the space group P21/c leading to the degeneration of the frontier MOs of macromolecules with the formation of half-filled 1D electron-energy band responsible for conductivity. Numerous SBIs (S∙∙∙S, 3.47 – 3.70; S∙∙∙N, 3.26 – 3.38 Å) transform the Peierls-unstable 1D band into a stable anisotropic 3D one. This situation is unique and the metallic state of (SN)x is a whim of nature that apparently cannot be used to design other molecular metals; in any case, all known oligomeric analogues of (SN)x with terminal and/or chain-incorporated arene groups are insulators.[306-309]
![[{"id":"hzTEYAvn76","type":"paragraph","data":{"text":"Herz conductors with record σ values (see Table 9)."}}]](/storage/images/resized/GrmPNhJtvAGTecUAR4xc9ZkQWUCHdTTbFQ5viFGR_xl.webp)
The RC salt [14][GaCl4], which has not been studied in this context, has uniform π-stacks of the RCs with shortened interplanar spacing in the crystalline state (see Fig. 12) [117] and is a potential conductor.
The mixed-valence RC salts [63]2+[BF4]–2 and [63]+[MCl4]– (M = Ga, Fe) demonstrate nonlinear electrical transport in the charge-ordered insulating state.[31] For M = Ga the room-temperature low-field negative magnetoresistance has been observed,[157] which is rare, e.g., for (SN)x it has only been detected below ~4 K.[253]
The semiconductive bipolar ion 7, which exhibits high stability and low solubility in both neutral and charged states associated with strong S-based solid-state SBIs, together with efficient charge delocalzation, is a promising electrode material for lithium-ion batteries. Its Li cells have been successfully cycled at a high rate for 400 cycles with 94% capacity retention, and such Herz species are promising for further design and synthesis of new fast-charging organic electrode materials.[154]
The individual WRs are insulators, but their CT complexes with TCNQ or I2 are semiconductors with record σ ~ 35.[223]
5.2. Magnetics
Conductive HRs are are often also bulk FMs/AFs [110, 127, 148, 155, 161, 248, 273, 281, 284, 286, 310, 311] (Table 10). The most interesting is the A6 group (see Fig. 1, E = S, Se) of isomorphous tetragonal pyridine-bridged/resonance-stabilized HRs 175, 176, 181, 182, 189, 190, 194 – 197 and 201 – 203 (see Fig. 4), which in the crystalline state exhibit infinite π-stacks connected by shortened E∙∙∙E and N∙∙∙E contacts. In the stacks, the magnetically interacting SOMOs of the neighboring HRs are nearly orthogonal, allowing FM ordering according to KOM.[273, 274] All FM-coupled HRs 190, 194, 197, and 201 – 203 contain the Se atom in position 2 and belong to this structural type. Notably, their all-S tetragonal congeners do not exhibit magnetic ordering (cf. AF coupled polymorphs of 172 revealing different exchange networks;[243] for 175, spin crossover [312] between paramagnetic species and their diamagnetic σ-dimers is hysteretic) [150] (some WRs exhibit room-temperature magnetic bistability with hysteric loops of ~ 10 – 90 degrees).[223, 224, 313, 314] The only tetragonal 2,1,3-thiaselenazolyl derivative 189 is canted-AF below 14 K. The magnitudes of TC and coercive fields are higher for all-Se-HRs 201 – 203 than for their S,Se congeners 190, 194, and 197.
DFT, MP2 and CASSCF calculations on π-dimers of HR 169 revealed multicentred bonding similar to that in π-dimers of the phenalenyl radical. The dispersion interaction is the largest attractive term of the bonding,[315, 316] and the bonding strength controls the macroscopic magnetic behavior of crystalline 169 (metamagnetism below 5 K) [35, 151, 275] in such a way that increasing the interdimer distance from ~ 3.2 to 3.9 Å leads to an AF → FM crossover.[276] Overall, the current awareness of the essential significance of literally omnipresent dispersion interactions in chemistry and materials science should be emphasized.[305]
The σ-dimers 204 – 209 of pyridine-bridged HRs 169 – 171, 174 and 175 are diamagnetic; 205 exhibits a sharp thermal hysteretic (T↑ = 380, T↓ = 375 K) 205 ↔ 2(175) interconversion into the paramagnetic state.[147, 150] The dimers 204/205 dissociating into Curie – Weiss paramagnets 174/175 under low-temperature irradiation with visible light at λ = 650 nm can be considered as photomagnetic materials. The photoinduced HR pairs persist up to 150/242 K for HRs 174/175, respectively, before reverting to the dimer states. The Arrhenius activation energies for the HR ↔ σ-dimer interconversion are 8.3 and 19.6 kcal mol–1 for 2(174) ↔ 204/2(175) ↔ 205, respectively. DFT and CASSCF calculations suggest that the ground-state interconversion is symmetry-forbidden, and the photochemical one is symmetry-allowed. Variable-temperature high fields/frequencies EPR of 205 below ~380 K has detected ~ 2% of radical defects, which cannot be assigned to residual 175.[150, 176, 245, 277]
Overall, the FM ordering of HRs is very sensitive to small changes in molecular and crystal structure caused by chemical modifications of their scaffolds or/and external pressure.[273] The record TC of 24 and 27.5 are revealed at ~ 2 GPa by pyridine-bridged all-Se-HRs 202 and 203 (normal-pressure TC 17.5 and 10.5, respectively; Fig. 15), whereas 200 displayed strong AF coupling at lower temperatures.[146, 148, 241, 247, 248, 284] Among the S/Se pyridine-bridged HRs, 197 showed TC ~ 1 [155] (see Fig. 15), while 193 showed strong AF coupling.[148] The pyrazine-bridged all-S HR 183 revealed strong AF coupling,[161] while other tested all-S HRs remained paramagnetic.[110] The benzoxo-bridged all-S HR 161 is ordered as a spin-canted AF at TN = 8 at 2 K; for such AFs, field-induced spin-flop transitions to FMs are known.[127] Among other thiazyl magnetics, 4-R-1,2,3,5-dithiadiazolyl (R = 4-NCC6F4), which is spin-canted AF, has TN = 36 K at normal pressure and 70 K at 1.6 GPa.[190, 310, 311]
![[{"id":"6TYSBYgAXc","type":"paragraph","data":{"text":" Herz ferromagnets with record T<sub>C</sub> values (see Table 10)."}}]](/storage/images/resized/kU8NCGlJS1F1UbbYWMGuFktOalaBqAdX0EfEKAMp_xl.webp)
The RC salt [14][GaCl4] exhibiting uniform π-stacks of the RCs with shortened interplanar spacing (see Fig. 9) [117] is potentially magnetic. The RC salt [63][GaBr4], having in crystal a kagome-coupled chain structure, exhibits spin frustration.[239]
5.3. Optoelectronics
As mentioned above, some HQs are formal antiaromatics/singlet diradicals featuring low-lying excited states, which may be triplet or singlet;[85, 295-298] it is important since most of the functional properties of HQs are π-electronic properties. For small-molecule optoelectronics towards light-emitting diodes, solar cells, and, especially, field-effect transistors, antiaromatics (or, more broadely, π-delocalized scaffolds incorporating 4n π-electron subunits)/singlet diradicals are advantageous due to lower first ionization energies and higher first electron affinities (in MO terms, smaller HOMO – LUMO energy gaps and corresponding excitation energies) favorable for charge and energy transport.[99, 317-322] Similar compounds are considered an emerging class of organic semiconductors.[66, 322-325] With the exception of the HQ-based mixed-valence RC salt [63][GaCl4], the HQs do not belong to the best-conductive Herz species, such as benzoxo-, benzoquinone-, and pyridine-bridged 1,2,3-dichalcogenazole scaffolds. However, HQs are less sensitive to the atmosphere than HRs and exhibit chromophoric/fluorophoric and redox properties. The chromophoric properties of HQs are typically manifested in λmax range of ~ 460 – 660,[31, 32, 39, 128, 183, 194, 200] and for 70 and 90 of ~ 690 – 760 and 745 – 795 (i.e., in NIR area), respectively; and 70 has five long-lived and differently colored redox-states.[117] As HQs possess chromophoric/fluorophoric, redox, semiconductive and potentially, multicenter-SOC properties, they are promising materials for small-molecule optoelectronics [299-302] including SF materials.[288, 303] More broadly, for heavier-atom HQs any heavy-atom effects [326] typical of chromophores/fluorophores can be expected. However, and in contrast to hetareno/areno-fused 1,2,5-chalcogenadiazoles (chalcogen = S, Se), which have found exciting applications in small-molecule [327, 328] and polymer [329] organic optoelectronics, HQs have not yet been studied adequately in this regard. DFT calculations on putative analogues of A13 (E = S, cf. non-fluorinated analogue of 96) with consequently increasing number of annulated benzene rings bridging terminal 1,2,3-dithiazoles suggest a relationship between the diradical character and the longitudinal second hyperpolarizability (significantly exceeding that of the relevant polyacenes) of these HQs, which can be used in the design of optoelectronic materials.[85] Their naphtho congener 6 [31] experimentally exhibits nonlinear optical properties with a large third-order nonlinear susceptibility of 10–11 Fr, which can be associated with a number of degenerate electronic states caused by its singlet diradical nature.[193]
6. Conclusion
Contemporary Herz chemistry is a rapidly expanding subfield of future polychalcogen-nitrogen chemistry, encompassing fundamental research and practical applications. Among HRs, resonance-stabilized bridged scaffolds, including σ-dimers, are the most remarkable for materials science, and they and their modifications deserve further attention. Heavier-chalcogen/polyhalogenated Herz species, particularly polyradicals, are the most captivating. A combination of heavier atoms contributing to the overall SOC with multiorbital approach [330] seems especially promising for magnetics. For small-molecule optoelectronics, the antiaromatic HQs typical of Herz chemistry, stabilized by the first-order heteroatom perturbations of π-systems, seem encouraging. Furthermore, even if in the beginning Herz chemistry was focused on HCs/HSs and HBs, and then increasingly on HRs, nowadays, i.e., in its second century, HQs seem the most exciting for fundamental studies and applications. Practically forgotten HBs, still having no Se analogues, are of special interest due to chirality, e.g., as ligands for metal coordination compounds; notably, isomeric 1,3,2-benzodithiazole S-oxides are optically active too.[331] Also, HBs are promising starting materials for HQs, e.g., through thermal transformations. RAs of polyconjugated compounds are potential materials with unusual conductive, magnetic and optical properties,[332] and those of Herz species should be studied intensively. The chemistry and applications of NS• [50, 51] and its heavier analogues, conventional sources of unpaired electrons for HRs, are also worthy of attention. They are unstable in condensed phase but seemingly can be stabilized as ligands at metal centers, or/and adsorbates in porous frameworks. Notably, NS•, in addition to (SN)x, is the archetype of two other (SN)n (n = 2, 4), binary sulfur nitrides exhibiting non-trivial structure, bonding, and physical and chemical properties.[61, 118, 305, 333] In particular, (SN)2 is proposed for fingerprint and inkjet-trace imaging;[334] while (SN)4 is a strong brisant explosive comparable to pentaerythritol tetranitrate.[335]
While the preparative potential of Herz reactions, particularly hetero-Herz reactions, remains undiminished, the development of novel synthetic approaches and further extension of Herz chemistry to the main-group elements are warranted. The reaction mechanisms remain challenging and demand cutting-edge quantum chemical modeling for a comprehensive understanding.
Finally, an important emerging field beyond the scope of this review is the biological applications of Herz chemistry, which at present mainly involce monocyclic 1,2,3-dithiazoles but show potential for extension to fused derivatives. In any case, and despite synthetic challenges, the 1,2,3-dithiazole scaffold, as well as the 1,3,2-dithiazole one, is promising in the context of various biomedical applications.[46, 47, 336, 337]
7. Terminology, abbreviations and units
The radical-related terms, such as stable radical, persistent radical, radical ion, radical center, etc, are used as specified by the IUPAC [338] and accepted in common practice.[339]
AC — Appel cation (4,5-dichloro-1,2,3-dithiazolium);
AF — antiferromagnets(ic);
CCDC — Cambridge Crystallographic Data Centre;
ChB — chalcogen bonding;
CT — charge transfer;
DABCO — 1,4-diazabicyclo[2.2.2]octane;
DMI — 1,3-dimetyl-2-imidazolidinone;
DMM — Dzyaloshinskii – Moriya mechanism (of spin canting);
E — chalcogen atom (S, Se, Te);
Ea — activation energy (of thermal electrical conductivity, eV);
FeCp2/FeCp*2 — ferrocene/decamethylferrocene;
FM — ferromagnet(ic);
HB — Herz base (3H-1,2,3-benzodithiazole 2-oxide);
HC — Herz cation (1,2,3-benzodithiazolium);
hfac — hexafluoroacetylacetonate;
hfc — hyperfine coupling;
HQ — Herz quinoid;
HR — Herz radical (1,2,3-benzodithiazolyl);
HS — Herz salt;
KOM — Kahn orbital model;
MIR — Mott – Ioffe – Regel limit (of metallic electrical conductivity);
NCS — N-chlorosuccinimide;
NICS — nucleus independent chemical shift;
NIR — near infrared (area);
OTf — triflate ([CF3SO3]–);
RA — radical anion;
RC — radical cation;
SBI — secondary bonding interaction;
SCE — saturated calomel electrode;
SF — singlet fission;
SOC — spin-orbit coupling;
SOMO — single-occupied molecular orbital;
TDAE — tetrakis(dimethylamino)ethylene;
TC/TN — Curie/Néel temperature (K);
VdW — van der Waals (radius);
WC — Wolmershäuser cation (1,3,2-benzodithiazolium);
WR — Wolmershäuser radical (1,3,2-benzodithiazolyl);
XB — halogen bonding;
XRD — X-ray diffraction;
a — hfc constant (mT),
δ — NMR chemical shift (ppm);
J — exchange coupling constant (cm–1);
λmax — UV-VIS longest wavelength (nm);
q — electrical charge (e);
σ — room-temperature electrical conductivity (S cm–1).
Other abbreviations are standard/common.
Conflicts of interest
There are no conflicts to declare. Alphabetically arranged authors contributed equally.
Acknowledgments
The authors are grateful to all their coauthors cited below for their contributions in Herz chemistry, and Dr. Ekaterina A. Radiush also for the graphical Abstract. The paper has been jointly supported by the Russian Science Foundation and Government of the Novosibirsk Region (project no. 24-23-20101).
References

![Benzo[1,2,3]dithiazole Compounds: A History of Synthesis and Their Renewed Applicability in Materials and Synthetic Chemistry, Originating from the Herz Reaction](/storage/images/resized/MjH1ITP7lMYGxeqUZfkt2BnVLgjkk413jwBV97XX_small_thumb.webp)


![Synthesis of 2-mercapto-substituted thieno[2,3-d]thiazoles and ben zothieno[3,2-d] thiazoles](/storage/images/resized/voXLqlsvTwv5p3iMQ8Dhs95nqB4AXOG7Taj7G4ra_small_thumb.webp)











