


![Comparative table of different techniques for saturation of heterocycles[172, 173]](/storage/images/resized/ctgAXnybxRUOSTvpSnaVp7HGUK0XeFrzIFevsYLP_xl.webp)

Keywords
Abstract
The review summarizes the known data on various methods for saturation and dearomatization of heterocyclic moieties, forming diverse aromatic compounds. Since the classical hydrogenation of substrates with molecular hydrogen has a number of fundamental drawbacks that hinder both its use at the laboratory level and its widespread implementation in industrial production, the main attention is paid to non-classical methods described in the literature over the last 20–25years. The methods of ammonia-free Birch reduction, transfer hydrogenation, reduction by Hantzsch esters and some other reactions involving heterocyclic compounds are summarized and systematized. It is shown that such methods of reduction or dearomatization of heterocycles can in some cases be more efficient than classical procedures. The bibliography includes 173 references.
1. Introduction
Straightforward approaches to saturated rings in hetarene molecules (Fig. 1) provide a simple and atom-economic access to the corresponding heterocycles, which are structural moieties of a variety of biologically active and natural compounds.[1] In terms of ease of synthesis, saturation (including enantioselective) is an attractive route because it minimizes the manipulation with functional groups to obtain the target heterocyclic compounds, including those in enantiomerically pure forms. Compared to other well-studied substrates (e.g., ketones, imines, olefins, etc.), the preparation of saturated heterocycles from the corresponding hetarenes is more challenging. The main problems of such transformations are the high stability of the heteroaromatic compounds and the negative effect of heteroatoms [2] contained in both substrates and reaction products on the active sites of the catalyst (see Fig. 1).
![[{"id":"x74C1VyYdo","type":"paragraph","data":{"text":"Possible saturation pathways for model annulated (<i>a</i>) and substituted (<i>b</i>) heteroaromatic compounds. Y = NH or NAlk, O, S; R, R’ are hydrocarbon moieties of different nature, n is the number of carbon atoms."}}]](/storage/images/resized/S161ZsZTkab1CRHmIZCFQiJCEdzIdYsKvKyL75GJ_xl.webp)
As hetarenes represent a large family of compounds with different structural characteristics, their reactivity in saturation processes varies greatly depending on the nature of the heterocycle. Saturation of six-membered monocyclic heteroaromatic compounds such as pyridines, pyrimidines and pyrazines is hampered because of their high aromaticity.[3] Five-membered heteroaromatic compounds containing nitrogen, oxygen or sulfur atoms are also chemically active substrates due to their ability to deactivate metal-containing catalytic sites and participate in side reactions.[4-6] For example, in most cases indole or pyrrole derivatives can react via the N – H bond, so for the hydrogenation process to be efficient, the nitrogen atom is often protected by an electron-withdrawing group such as acetyl (Ac), p-toluenesulphonyl (Tosyl, Ts) or tert-butoxycarbonyl (Boc). These protecting groups act as catalytic coordinating groups and also prevent deactivation of the catalyst by the basic nitrogen atom. The absence of auxiliary coordination between the heteroaromatic substrate and the catalyst can result in multiple low-energy directions of the reaction between the substrate and the metal centre, with an overall low (enantio)selectivity.[7]
In bicyclic heteroaromatic compounds, the heteroatom-containing ring is usually hydrogenated to a carbocycle, with the more stable ring often remaining unsaturated during the hydrogenation.[8] It should be noted that this is true when applied to the classical methodology of catalytic hydrogenation, i.e., carrying out the process in the presence of molecular hydrogen and heterogeneous or homogeneous catalysts. To date, the most successful examples in this field relate to annulated aromatic and monocyclic heteroaromatic compounds containing nitrogen and/or oxygen atoms, but single reactions of S-heterocycles have also been reported.
The stability of heteroaromatic compounds requires the application of harsh hydrogenation conditions (H2 pressure up to 150 bar, temperature up to 250°C) to break the aromaticity of the starting molecules.[3, 9-16] For example, in the case of nitrogen-containing heterocycles, most of the known methods have been developed for condensed ring substrates such as quinolines,[17-35] quinoxalines[36-44] and similar polycyclic molecules, which are more active due to reduced aromatic stabilization. Pyridines, the most aromatic compounds and therefore the most stable substrates are not often reduced under catalytic conditions.[20, 45-49] High pressure requires the use of special equipment to carry out the reaction, and high temperature can result in low enantioselectivity of the target hydrogenation products.[50-56] In addition, such conditions are not suitable for a number of functional groups [14, 57-59] and there is a high probability of heterocycle ring-opening.[60-65] These facts are the main reason for the development of new catalysts active in the hydrogenation of heteroaromatic compounds. Many examples of the successful application of enantiomerically pure transition metal complexes in the asymmetric hydrogenation of heteroaromatic compounds are based on the ability of the substrate to form a chelate-like intermediate with a metal-containing centre, which must necessarily contain the hydrogenated double bond and a donor atom of the substrate.[66] The development of the ‘ligand tuning’ approach has enabled the design of efficient catalytic systems for some heteroaromatic compounds.[67-69]
An elegant strategy called ‘relay catalysis’ has been proposed.[70] It involves the initial partial reduction of the starting heteroaromatic compound to a prochiral heterocyclic structure using an achiral catalytic system. The subsequent reduction of the intermediate heterocycle to the final enantiopure derivative is carried out in the presence of a second catalytic system, which is also present in the reaction mixture and is responsible for the enantioselectivity of the process. In addition, a novel method for a cascade enantioselective hydrogenation promoted by a metal complex catalyst and an organocatalyst has been reported.[71] This cascade process mimics the cofactor function of the nicotinamide adenine dinucleotide (NADH) cycle using NADH mimetics and molecular hydrogen, which are readily recycled.
A method of substrate activation is also known in which the substrate is converted to a related heterocyclic system that is more susceptible to hydrogenation reactions, including asymmetric ones. One embodiment of this approach is to facilitate hydrogenation through the formation of positively charged hetarene derivatives. In this way, catalyst deactivation resulting from the binding of the substrate to the metal centre can be avoided. It should be noted that in this strategy, the coordinating ability of the ligating groups of the substrate and/or product with respect to the catalyst is neutralized.[8] The ligating auxiliary group can be introduced into the hetarene by various chemical reactions. Coordination of such a group with a metal centre promotes the formation of a chelate-like intermediate and facilitates hydrogenation. This method of substrate activation involves the formation of a model heterocyclic compound with broken aromaticity via an acid- or base-induced double bond migration process.[8]
This review highlights the literature published over the last 20 – 25 years on new trends in the catalytic hydrogenation and reduction or dearomatization of N-, O- and S-hetarenes. It should be noted that informative reviews [14, 72-74] published between 2014 and 2020 focuse on homogeneous catalysts for the hydrogenation of such compounds. However, with minor exceptions, all the information presented in these reviews relates to the ‘classical’ method of hydrogenation, i.e. involving the use of molecular hydrogen and a catalyst. In addition, a 2024 review [75] analyzes known methods for the hydrogenation of indoles to 4,5,6,7-tetrahydroindoles. We considered mainly transfer hydrogenation reactions, as well as a number of other reduction protocols, e.g. under the action of alkali metals in alcohol,[76] in the presence of Hantzsch esters,[77-81] silanes [82, 83] and others,[84] which are not often found in the literature. As mentioned above, the classical method of hydrogenation requires complex equipment for working with gaseous hydrogen at relatively high pressures and temperatures, strict safety precautions due to the explosive nature of this reagent, and catalysts that quickly become ineffective.
By publishing this review, the authors wish to draw attention to methods of hydrogenation and/or dearomatization of hetarenes which, in some cases and under certain conditions, have proved to be no less effective than the generally accepted protocols. The following reactions are classified according to the type of saturation of the heterocycle and the nature of the heteroatom present in the substrate.
2. Transfer hydrogenation
Transfer hydrogenation reactions are an attractive alternative to conventional hydrogenation methodology. This method does not require complex experimental design as the hydrogen donor in this case is a molecule of an organic compound such as a solvent (called a liquid hydrogen carrier). The latter is converted by hydrogen transfer to another stable compound that ideally does not react with either the substrate or the reaction products. Transfer hydrogenation usually takes place at moderate temperature and pressure in the presence of organic or organometallic catalysts, many of which are chiral, allowing efficient asymmetric synthesis.[85, 86]
2.1. Nitrogen-containing heterocycles
Fragments of N-heterocycles with varying degrees of saturation are ubiquitous in natural products and their synthetic analogues, pharmaceuticals and agrochemicals (neonicotinoids), and are also present in many products of low-tonnage chemistry.[68] It is therefore not surprising that hydrogenation of these molecules has been studied most intensively compared to other hetarenes. It should be noted that the work on the hydrogenation of N-heterocycles mainly provides information on the adsorption of six-membered cycles (e.g., pyridine) on various metal surfaces. Although five-membered cyclic systems are intermediates in a number of catalytically relevant processes, the nature of the interaction of such compounds with the catalyst surface has not been well studied. This is due both to the lower stability of azoles compared to their six-membered analogues, which makes the former less attractive for modelling studies that involve complicated surface chemistry during and/or after adsorption,[5] and to the relative conformational rigidity of the five-membered ring.[6] This is probably the reason why the selection of catalytic systems for the hydrogenation of N-heterocycles is still empirical rather than systematic.
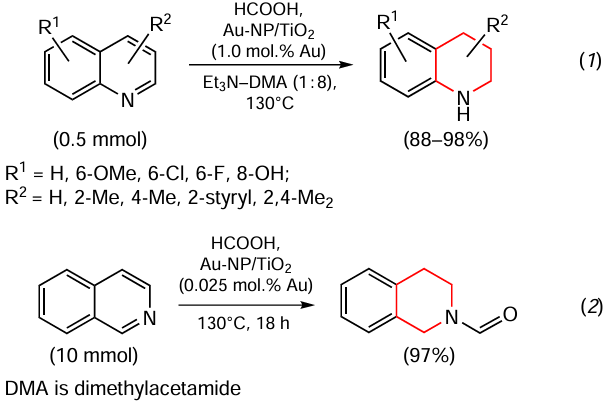
A method for the chemo- and regioselective transfer hydrogenation of quinoline derivatives to 1,2,3,4-tetrahydroquinolines under mild conditions (Scheme 1, reaction 1) is reported.[87] The liquid hydrogen carrier is formic acid and the catalyst is gold nanoparticles (NPs) supported on rutile, which is the most common polymorph of titania (Au-NP/TiO2). In addition, the authors of this study described a straightforward and selective method for the preparation of N-formylisoquinoline directly from isoquinoline via gold-catalyzed reductive one-pot N-formylation with formic acid (see Scheme 1, reaction 2). A similar reaction has also been carried out for quinoline derivatives.
The study [88] reports on gold nanoparticles deposited on a high-strength amine-functionalized mesoporous silicate SBA-15 (Santa Barbara Amorphous). This catalyst is highly active in transfer hydrogenation of structurally diverse N-heteroaromatic compounds, namely quinoline, isoquinoline, quinoxaline, acridine, phenanthroline, quinazoline and phenanthridine. The nitrogen-containing ring is selectively reduced to the corresponding amines in yields exceeding 90%. Formic acid is also used as a liquid hydrogen carrier, whereby gold nanoparticles with an initial size of 0.6 ± 0.2 nm aggregate to particles with a diameter (d ) of 4 – 5 nm and maintain this size in subsequent catalytic cycles (up to five cycles). At the same time, similar SBA-based catalysts with larger (d ≈ 5 – 35 nm) gold particles are inactive in transfer hydrogenation of hetarenes. Excess formic acid was also shown to furnish undesired hydrogenated formamide derivatives, which can be further quantitatively deformylated. The formic acid–triethylamine system can be used to suppress the side process of formylation.
The hydrogenation of the pyridine ring of quinolines with >99% conversion and remarkable selectivity in the presence of palladium nanoparticles (d < 1 nm) loaded onto nano-sized Fe3O4 particles (d = 8 – 10 nm) has been described.[89] In this case, the H2O-tetrahydroxydiboron system acts as the hydrogen source; in addition, water plays the role of a solvent. The authors proposed a plausible reaction mechanism. The said technique was used to synthesize a biologically active inhibitor of tubulin polymerization, which is a chemotherapeutic agent for cancer treatment.
Pi et al.[80] presented an example of efficient ligand-free transfer hydrogenation of various N-heteroaromatic quinoline compounds using Hantzsch esters as the hydrogen source (Scheme 2).
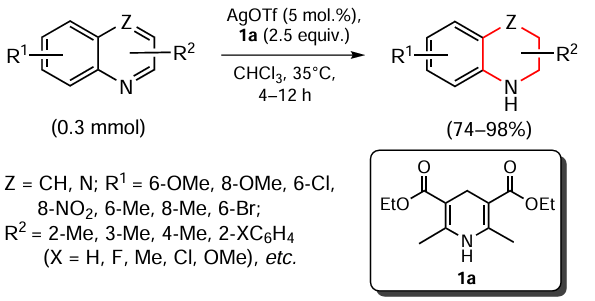
The process is carried out under mild conditions, silver salts (Ag2CO3 , AgOAc, AgSbF6 , AgOTs, AgNTf2 , AgNO3 , AgBF4 and AgOTf; Tf is trifluoromethanesulfonyl (triflyl)) have been tested as catalysts. Varying the nature of solvent revealed that the best yields of products were obtained in chloroform. The authors explain this fact by the activation of quinolines by halogen bonds formed as a result of the interaction of chlorine and nitrogen atoms. Diethyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (1a) was found to be optimal among the model Hantzsch esters, AgOTf was chosen as catalyst, and the hydrogenation proceeds at low heating to give the corresponding nitrogen-containing ring reduction products in yields up to 99%.
The authors proposed a possible reaction mechanism, which is shown in Scheme 3.
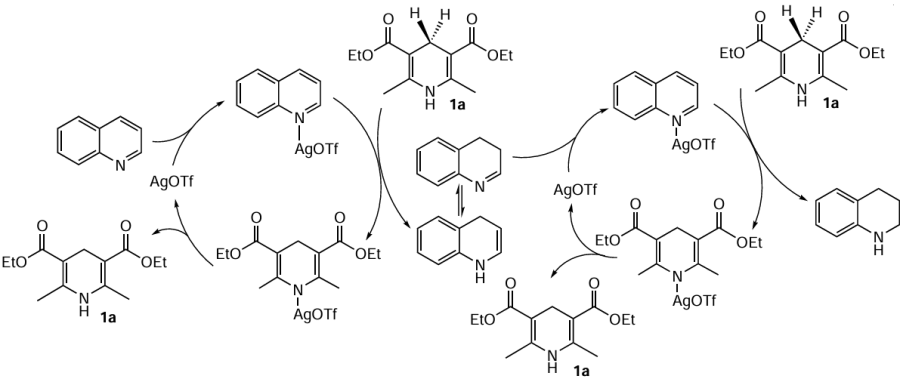
In this study, an asymmetric variant of this reaction was also realized in the presence of a number of model chiral phosphoric acids with an enantiomeric excess (ее) of up to 66%. In an attempt to expand the applicability of this method, other N-heteroaromatic compounds were investigated, with isoquinoline, N-methylindole and pyridine failing not be hydrogenated. This fact was not commented and cannot be explained within the proposed hydrogenation mechanism, which can be considered a significant drawback of this methodology.
Beller and co-workers [90] described a catalyst based on cobalt, i.e., a non-precious metal active in transfer hydrogenation of N-heterocycles such as quinolines, phenanthridine, phthalazine and 1,5-naphthyridine. This catalyst was obtained by pyrolysis of a mixture prepared from cobalt(II) acetate in an aqueous solution of melamine or waste melamine resins, the cobalt particle size not exceeding 20 nm. Formic acid was used as the hydrogen source. The authors have shown that in this case there is a selective reduction of the six-membered N-heterocycle, accompanied by the formation of N-formamide derivatives. This fact can be considered a significant drawback of the methodology, even taking into account the use of cheap cobalt catalysts instead of transition metal-based catalysts.
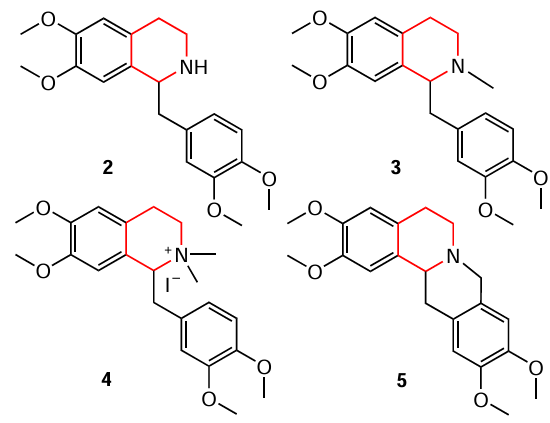
Du et al.[91] presented an efficient method for the transfer hydrogenation of isoquinolines carried out in the presence of copper salt Cu(OTf)2 as catalyst and boron-containing hydrogen source. Optimal hydrogenation conditions include 15 mol.% Cu(OTf)2 , 2 equiv. of oxazaborolidine–BH3 complex, 0.5 mmol of substrate, 2 ml of 1,2-dichloroethane, room temperature and argon atmosphere, and the reaction time of 24 h. A wide range of reduced products (1,2,3,4-tetrahydroisoquinolines, yields 61 – 85%) have been described such as unsubstituted and containing a 5- or 8-positioned bromine atom, 6-positioned chlorine atom or methoxy group, 8-positioned phenyl group 8, a number of 1-aryl derivatives in which the aryl substituent was p-tolyl, 4-methoxyphenyl, 1',4-biphenyl, 1-naphthyl(4-fluorophenyl)-1,2,3,4-tetrahydroisoquinoline and 4-(trifluoromethyl)phenyl, and 1-(3,4-dimethoxybenzyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline. The hydrogenation of isoquinolines bearing heterocyclic substituents at the 1-position was also carried out to afford 1-(2-furyl)-, 1-(2-thienyl)-, 1-(2-benzofuryl)-, 1-(2-benzofuryl)-, 1-(benzo[b]thiophen-2-yl)-1,2,3,4-tetrahydroisoquinolines. The authors proposed a plausible mechanism for the hydrogenation reaction. In addition, this method was used to synthesize the biologically active tetrahydroisoquinoline alkaloid (±)-norlaudanosine (2) in 62% yield, which is a key intermediate for the preparation of (±)-laudanosine (3), (±)-N-methyllaudanosine (4) and (±)-xylopinine (5) in one or two steps.
The transfer hydrogenation of N-hetarenes has been reported which gave rise to 1,2,3,4-tetrahydroquinoline derivatives in excellent yields (Scheme 4).[92] This process is carried out in the presence of a water-soluble and air-stable iridium complex 6 as the catalyst, formic acid as the hydrogen source and solvents (water, DMSO, toluene, THF, methanol, dichloromethane, acetonitrile) at room temperature for 12 h (reaction 1). This method is tolerant to various functional groups.
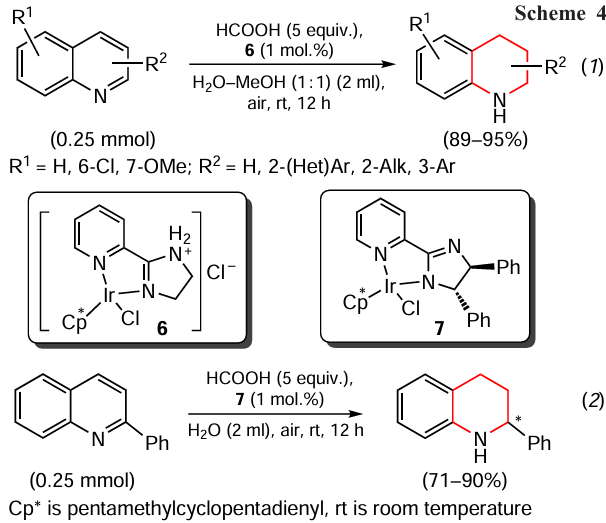
In addition to the optimized conditions, the authors also proposed the plausible pathway of the process and the possibility of enantioselective transfer hydrogenation in the presence of the chiral Ir catalyst 7 (see Scheme 4, reaction 2), which provides an access to chiral products in high yields and with good enantioselectivity.[92]
Mai and Nikonov [93] described transfer hydrogenation of a number of model N-heteroaromatic compounds in the presence of KOH and the ruthenium semi-sandwich complex [Cp(IPr)Ru(py)2][PF6]] (Cp is cyclopentadienyl, IPr is 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene, py is pyridine) based on N-heterocyclic carbene (NHC) in isopropyl alcohol, which acts as both hydrogen source and solvent. Only the heteroatom-containing ring was hydrogenated at 70°C; the maximum yield (72%) was obtained in the case of tetrahydroacridine, the yield for tetrahydroquinoline was 62% and 58% for 5,6-dihydrophenanthridine. Interestingly, when molecules containing two heterocycles, such as 1,5-naphthyridine, were hydrogenated, saturation occurred on only one of them, and the yield of the product was 67%. At the same time, 1,3,5-triazine could only be converted to 1,2-dihydro-1,3,5-triazine in 60% yield.
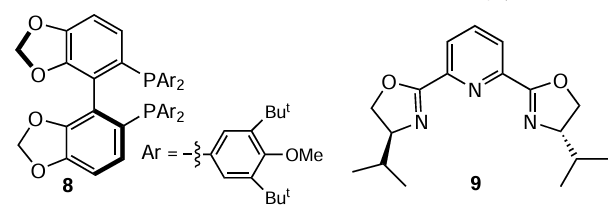
Another example of transfer hydrogenation of quinoline and its derivatives in the presence of a base metal catalyst, Fe(OTf)2 (1 mol.%), has been reported.[79] The hydrogen source in this case were Hantzsch esters, diethyl (1a) and di-tert-butyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (1b), and N 3,N5,2,6-tetramethyl-1,4-dihydropyridine-3,5-dicarboxamide (1c); the process was carried out at 40°C for 2 – 8 hrs. A number of substituted 1,2,3,4-tetrahydroquinolines were obtained in high yields; the reaction was tolerant to functional groups of different nature. The authors also tried to realize an asymmetric variant of this reaction using complexes of Fe(OTf)2 and chiral phosphine (8) or bis(oxazoline) ligands (9). However, only the racemic hydrogenation product of 2-methylquinoline, 2-methyl-1,2,3,3,4-tetrahydroquinoline, was detected in the resulting mixture.
Sundararaju and co-workers [94] described transfer hydrogenation of quinoline derivatives and the oxidative dehydrogenation of a cyclic amine, 1,2,3,4-tetrahydroquinoline. Both transformations took place in water in the presence of the air stable complex [Cp*Co(N͡ N)I]+I− (N͡ N is 8-aminoquinoline) (10) acting as the catalyst (Scheme 5). It is shown that the hydrogenation is promoted by formic acid as the hydrogen source, and the oxidation requires the presence of pressurized molecular oxygen.
Zhu et al.[95] carried out transfer hydrogenation of substituted quinolines in the presence of the similar air-stable N,N-bidentate cobalt complex bearing 2-[5-(tert-butyl)-1H-pyrazol-3-yl]pyridine instead of 8-aminoquinoline ligand, formic acid and special additives (Et3N and (or) PhMe). When implementing this procedure, the formylation of the target product occurs, which can be considered as its significant disadvantage. At the same time, the authors managed to show in several model examples the possibility of obtaining tetrahydroquinolines by adding an aqueous solution of sodium hydroxide and ethanol to the reaction mixture. However, no explanation was offered for this experimental fact, although the plausible mechanism of hydroformylation was presented.
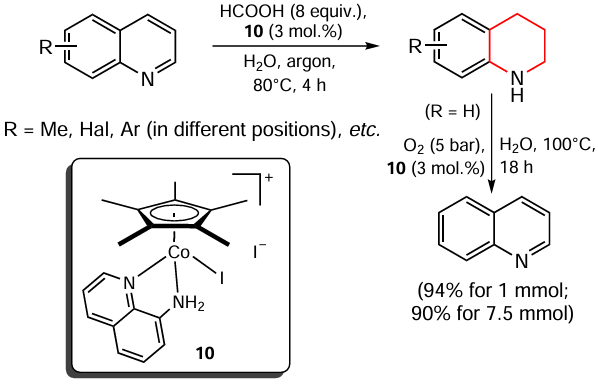
Kundu and co-workers [96] developed a transfer hydrogenation protocol for various N-heterocycles in the presence of a series of MnI complexes as catalysts and ammonia borane (BH3 · NH3) as the hydrogen source (Scheme 6). The manganese complex 11 was found to perform best. Based on the results of kinetic experiments, the authors proposed a putative reaction pathway. Although this protocol allows the hydrogenation of compounds such as quinolines, quinazolines, isoquinoline, acridine, benzothiazole, benzo[d]oxazole, it is more interesting from a fundamental point of view, since ammonia borane is a rather expensive reagent and the synthesis of the necessary manganese(I) complexes is rather laborious.

Guo et al.[97] described double axially chiral phosphoric acids, such as (R,R)-4,4'-bis(2-cyclohexyloxy-1-naphthyl)-2,2'-binaphtyl-3,3'-diylhydrophosphate (12a), as efficient organocatalysts for hydrogenation promoting hydrogen transfer to the heterocyclic skeleton of quinolines in the presence of Hantzsch ester 1b.
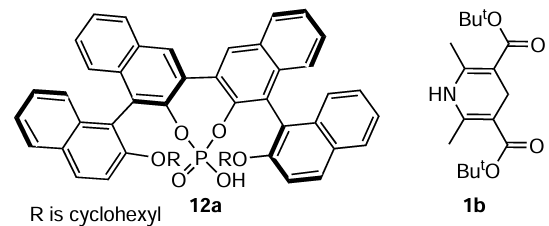
Small amounts of catalyst 12a (0.2 – 1 mol.%) were shown to be sufficient to afford tetrahydroquinoline derivatives with excellent enantioselectivities (up to 98%) and in high yields (Scheme 7, reaction 1). In addition, 2,3-disubstituted quinolines were hydrogenated with excellent diastereoselectivities and high enantioselectivities (up to 92%) (see Scheme 6, reaction 2) using Hantzsch esters 1a,b as the hydrogen source in solvents such as chloroform, dichloromethane, carbon tetrachloride, benzene, toluene, THF, ethanol, dioxane and diethyl ether. The hydrogenation was carried out in argon at 35°C for 12 – 20 h. Mild reaction conditions can be considered as an advantage of this procedure. However, the synthesis of axially chiral phosphoric acids is a complex multi-step process, which can be considered a significant drawback of the said method, despite the small amounts of these compounds required to carry out the transfer hydrogenation.
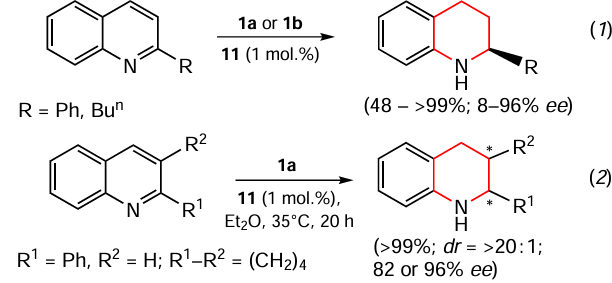
More and Bhanage [98] performed organocatalytic asymmetric transfer hydrogenation of 2-substituted quinolines using highly efficient phosphoric acid-based chiral catalysts 12b – e and Hantzsch ester 1a as the hydrogen source.
The authors have shown that enantiomerically pure 1,2,3,4-tetrahydroquinolines can be obtained in high yields with excellent enantioselectivity (up to 99%) in solution of diethyl carbonate. The effects of different chiral phosphoric acids, solvents, catalyst loading, temperature, and reaction time on the substrate conversion and enantioselectivity of the desired product are discussed. The authors note that diethyl carbonate, as a ‘green’ solvent, is an excellent substitute for organic solvents that are harmful to the environment. In addition, no transition metal catalysts are required in this case, which can be considered an advantage of this method.
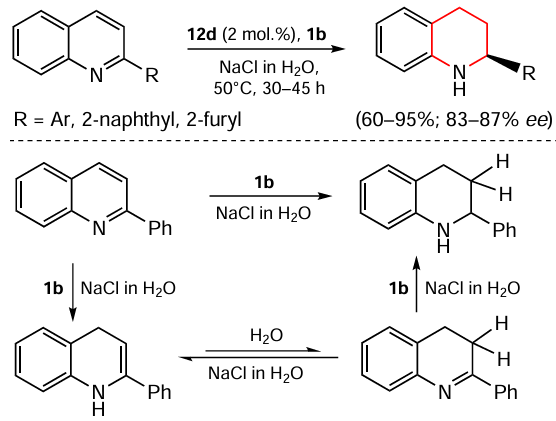
Rueping and Theissmann [99] described an example of an environmentally friendly, highly enantioselective transfer hydrogenation of 2-substituted quinolines. The process is catalyzed by Brønsted acid using a saturated aqueous solution of NaCl as the reaction medium (Scheme 8). It was found that the Hantzsch ester 1b acts exclusively as a hydride source rather than a proton source in this process, and that water plays an important role as a proton source in the hydrogenation.
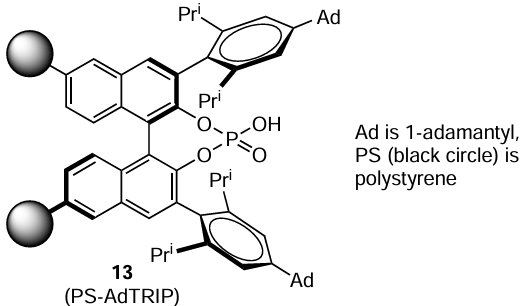
Nagorny and co-workers [100] used the chiral phosphoric acid PS-AdTRIP (13) immobilized on a polymeric support for transfer hydrogenation both in a batch reactor and in a continuous flow reactor. Such an acid served as a catalyst for the enantioselective reduction of various quinolines, 2H-1,4-benzoxazines, and 2H-1,4-benzoxazine-2-ones using Hantzsch ester 1a as the hydrogen source.Structure 13
It was found that a significant improvement in the enantioselectivity of hydrogenation was achieved by using a fluidized bed reactor packed with PS-AdTRIP catalyst and increasing the flow rate from 0.2 to 2.0 – 2.5 ml min–1. The authors pointed out that the optimal conditions of hydrogenation in such a reactor provided 4 – 6% higher selectivity than that for hydrogenation in a batch reactor with 2 mol.% of catalyst 13.
Hantzsch esters as the hydrogen source have also been used in the hydrogenation of 2-substituted quinolines,[101-103] benzoxazines,[101] quinoline-2-carboxylates [104] and other nitrogen-containing heterocycles.[103, 105]
The regioselective hydrogenation of the nitrogen-containing ring in quinoline derivatives, resulting in either 1,2-dihydroquinolines, which are inaccessible by direct hydrogenation (Scheme 9), or 1,2,3,4-tetrahydroquinolines, has been described.[83] This transformation is carried out by hydrosilylation followed by transfer hydrogenation.
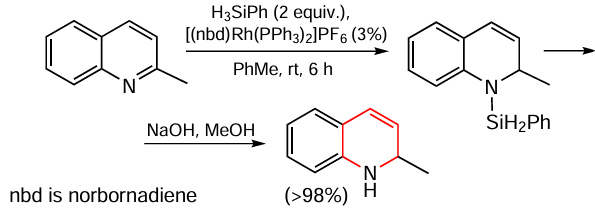
The initial in situ disproportionation of phenylsilane to H2SiPh2 and SiH4 , catalyzed by the [(nbd)Rh(PPh3)2]PF6 complex, is required for substrate reduction, as SiH4 was found to be the active reducing agent. The authors noted that the formation of N-silyl intermediates was never observed. Such a disproportionation reaction can be used to obtain SiH4 , which is usually difficult to obtain, which is one of the advantages of this technique. The model transfer hydrogenation reaction in isopropyl alcohol in the presence of the catalyst [Ir(cod)(NHC)PPh3]BF4 (cod is cycloocta-1,5-diene, NHC is 4-n-butyl-1-neopentyltriazole-5-ylidene) gives exclusively tetrahydrogenated products (yields up to 60%). In addition to quinoline and its derivatives, the authors applied this method to the hydrogenation of indole and acridine to indoline and 9,10-dihydroacridine, respectively, providing the yields of both products not exceeding 4%. These results indicate that this approach can be called effective only in the case of quinoline derivatives.
Barrios-Rivera et al.[106] described examples of asymmetric transfer hydrogenation of dihydroisoquinolines containing meta- or para-substituted aromatic rings at the 1-position (Scheme 10). The authors used an azeotropic mixture of formic acid and triethylamine (5 : 2) as the hydrogen source, dichloromethane as the solvent and 1 mol.% of ruthenium complex 14 as the catalyst. The corresponding tetrahydroisoquinolines were obtained in good yields and with high enantioselectivity. A probable pathway for the asymmetric reduction of model substrates is presented.
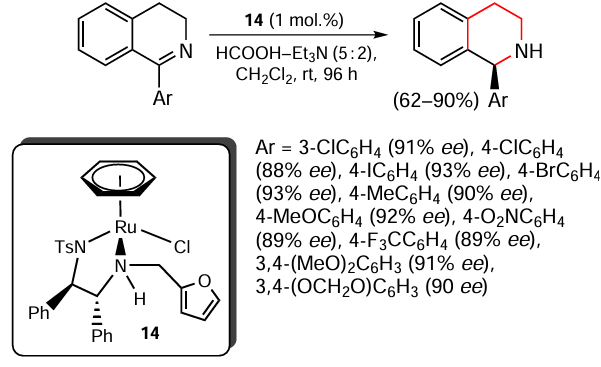
Transfer hydrogenation, including asymmetric hydrogenation, of substituted quinolines of different nature, in addition to the examples presented above, has also been carried out in the presence of ruthenium,[107, 108] rhodium [109] complexes, Noyori – Ikariya catalyst,[110] etc.[111, 112]
Indoline moieties, including chiral ones, are part of natural alkaloids and diverse biologically active compounds.[68] Although the demand for enantiomerically pure indolines is increasing over time, there are few examples of their preparation described in the literature. Even though indoles are quite stable, the pyrrole ring in their structure exhibits reduced aromaticity. In terms of atom-economic methods, the direct asymmetric hydrogenation of indoles is the most convenient approach to indolines.
N-protected indoles are successfully reduced in the presence of ruthenium, iridium, rhodium or palladium complexes.[68, 73, 113-115] However, deprotection requires additional steps and reduces the yield of the target product, thus increasing its cost. However, the hydrogenation of N-unsubstituted indoles is still quite challenging. Török and co-workers[116] presented a system for transfer hydrogenation of unprotected indoles and quinolines based on 10% Pd/C and formic acid. Depending on the reaction conditions (temperature and nature of the solvent), in addition to the target indolines and tetrahydroquinolines, N-formylated by-products were formed, which is considered to be a significant drawback of this procedure. In a follow-up study, Török and co-workers [117] obtained the corresponding indolines and tetrahydroquinolines under mild conditions in good yields in the presence of a system based on the presonicated Ni – Al alloy and water under mild conditions. In this case, water acted not only as a solvent but also as a hydrogen source. Consequently, the process of reducing heterocycles using such a catalytic system can be called transfer hydrogenation. However, the authors [117] provided no information on the enantioselectivity of these reactions.
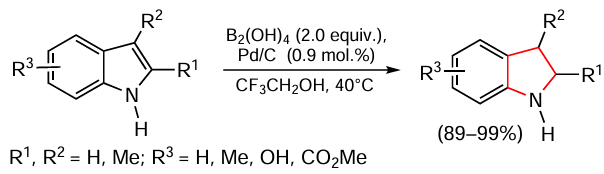
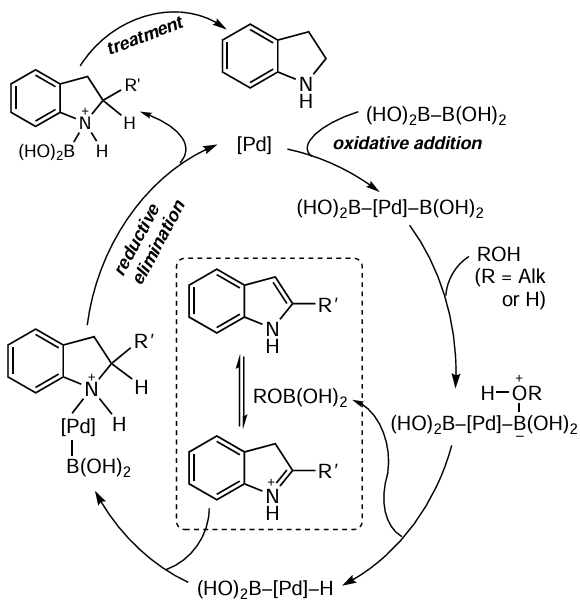
Zhou and Chen [118] carried out transfer hydrogenation in the presence of Pd/C catalyst (10 wt.%), trifluoroethanol as solvent and tetrahydroxydiborane (B2(OH)4) as the hydrogen source (Scheme 11). The yields of the products depended on the nature of the substituents and reached 99%, and in all cases the pyrrole ring was selectively hydrogenated, except for N-substituted indoles, which were not reduced under these conditions. The putative mechanism of transfer hydrogenation was also proposed (Scheme 12).
Bhatt and Natte [119] reported the use of ammonia borane as a hydrogen source for selective transfer hydrogenation. In the presence of a commercially available precatalyst RuCl3 · x H2O, the reduction of the nitrogen-containing ring in quinolines, quinoxalines, pyridines, pyrazines and indoles gave the corresponding N-heterocycles in moderate to high yields (Scheme 13).
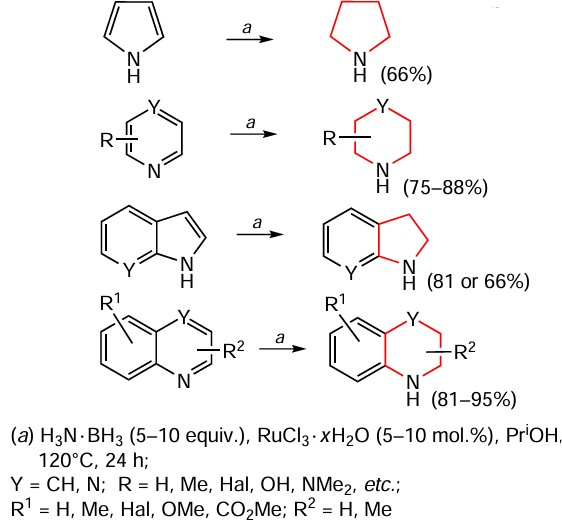
Isopropyl alcohol was used as the solvent and the process was carried out at elevated temperature for one day. The tolerance to various functional groups and the simplicity of the procedure provide access to a range of saturated N-heterocyclic compounds. In addition, it was noted that this procedure is easily scalable to gram quantities of substrate, which is important for drug development, and suggested a plausible mechanism of hydrogenation.
Cui et al.[120] described the transfer hydrogenation of quinoline and NH-indole derivatives in the presence of Cp2ZrH2 complex and ammonia borane (3.0 equiv.) in toluene providing the corresponding products in yields up to 94% (Scheme 14). The authors showed that this system is tolerant to the substituents of different nature in the substrate. Based on preliminary data obtained by NMR spectroscopy, study of the kinetic isotope effect and reaction stoichiometry, a concerted proton transfer mechanism is realized in this case.
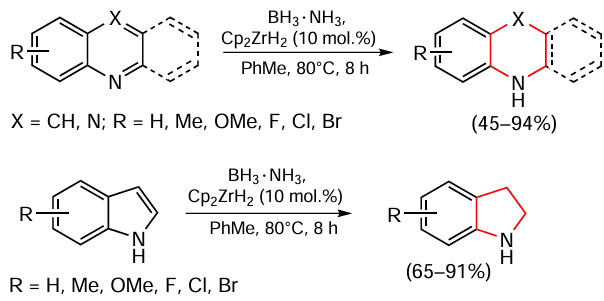
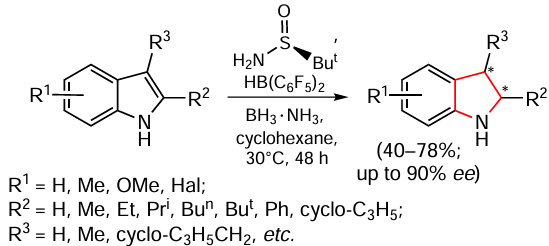
Zhao et al.[121] proposed an interesting method for the hydrogenation of N-unsubstituted indoles. Asymmetric transfer hydrogenation takes place in the absence of transition metal compounds but using a catalytic system consisting HB(C6F5)2 and (S)-tert-butylsulfinamide, with ammonia borane as the hydrogen source (Scheme 15). The authors proposed that the composition of HB(C6F5)2 and (S)-tert-butylsulfinamide acts on the principle of uncompensated sterically hindered frustrated Lewis acid – base pairs (FLP).[122-124] For various indoles with a free NH group, including 2-alkyl- or 2,3-dialkyl-substituted derivatives, the corresponding double bond hydrogenation products were obtained in moderate yields and high ее values up to 90%. At the same time, the authors have shown that in the case of hydrogenation of N-methyl-protected indoles, this reaction is characterized by low enantioselectivity, confirming the efficiency of using this method only with N-unsubstituted indoles.
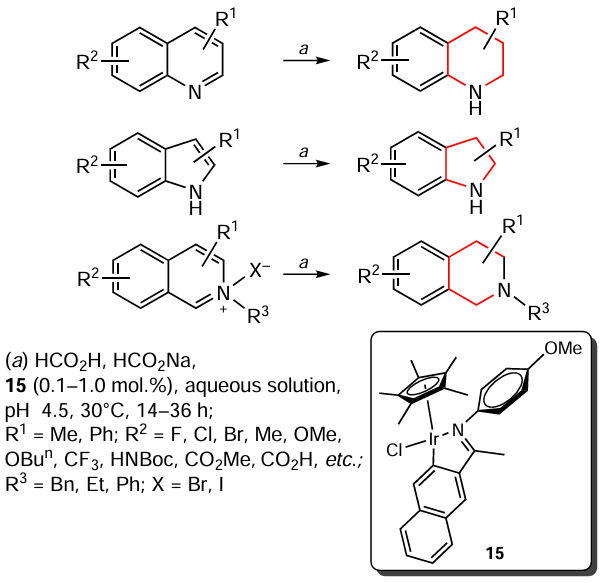
Talwar et al.[125] presented the transfer hydrogenation of various N-heterocycles, including quinolines, indoles, isoquinolines and their salts, in an aqueous solution of formic acid and sodium formate under mild conditions in the presence of iridium complex 15 (Scheme 16). Notably, this catalyst shows high values of the catalyst turnover number (TON), up to 7500, at its minimum loading of 0.01 – 1.0 mol.%. The authors attribute this fact to the homogeneous state of the catalytically active species. In addition, the hydrogenation process was carried out in a ‘green’ solvent,[126, 127] which, in combination with HCO2H, also acts as the hydrogen source, which is a significant advantage of this procedure.
The transfer hydrogenation reaction in an aqueous solution of HCO2H and HCO2Na and in the presence of a catalytic system based on [Cp*RhCl2]2 and 2,2'-bipyridine (bpy) was carried out a study.[128] This approach was shown to give the corresponding tetrahydroquinoxalines, dihydroquinoxalinones, tetrahydroquinolines and indolines in yields from good to quantitative, with the pH adjustment of the reaction solution being crucial to achieve high catalytic activity and acceptable selectivity. It was found that a medium pH < 7 is required to achieve optimum reduction conditions. In addition, the catalytic activity is determined by the quantitative ratio of substrate to catalyst and the nature of the substrate.
2.2. Oxygen-containing heterocycles
Chiral tetrahydrofuran and dihydrobenzofuran moieties are common structural elements of biologically active natural products. For this reason, hydrogenation, including asymmetric hydrogenation, can provide a straightforward route to enantioenriched tetrahydrofurans and dihydrobenzofurans from aromatic precursors that are generally readily available. For example, the first example of a total synthesis of thespesone and its synthetic enantiomer was described in 2010,[129] in which the number of steps was significantly reduced by using enantioselective hydrogenation to introduce a stereocentre into a bromodihydrobenzofuran precursor.
Despite considerable efforts in this direction over the last two decades, the methods for the enantioselective hydrogenation of furan or dihydrofuran derivatives remain fairly undeveloped. The choice of model substrates is mainly due to the specific tasks of the pharmaceutical and small-scale chemical industries, and the available publication lacks the putative reaction mechanisms and data on the nature of the catalytically active centres.
Nandy et al.[130] present a method for the efficient and versatile one-step reduction of a furan moiety to a tetrahydrofuran moiety (Scheme 17), leaving the labile functional group in the substrate intact and reducing the C = C double bond to a single bond.
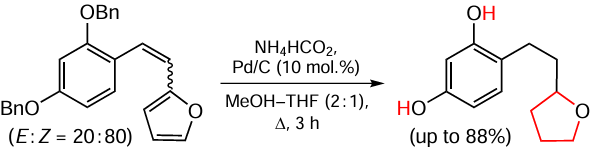
Transfer hydrogenation was carried out in a boiling mixture of methanol and THF in the presence of 10% Pd/C and an excess of ammonium formate, which acted as a hydrogen source. Similar substrates with substituted furan ring were also used in the reaction. Unfortunately, the possible transformation of the THF used as the solvent was not mentioned by the authors. However, the method per se has good synthetic potential, given that the reported yields of the products reach 88%.
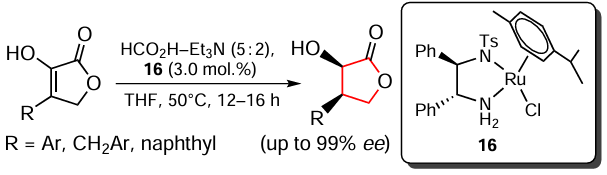
Hu et al.[131] developed an efficient method for the catalytic asymmetric transfer hydrogenation of β-substituted α-oxobutyrolactones, which gives rise to cis-β-substituted α-hydroxybutyrolactones in high yields, with high enantioselectivity and diastereoselectivity (Scheme 18). The RuCl[(R,R)-Tsdpen](p-cymene) complex (16) (Tsdpen is N-(p-tosyl)-1,2-diphenylethylene-1,2-diamine) was used as the catalyst but the authors noted that its use in the racemic form led to the racemates of the corresponding desired products. An azeotropic mixture of formic acid and triethylamine (in a ratio of 5 : 2) acts as the hydrogen source. In addition, it was found that this process can be carried out on a gram scale without loss of catalyst activity and enantioselectivity, and a possible reaction pathway is presented.
Zhang et al.[132] described the hydrogenation of furfurol to 2-methylfuran and(or) 2-methyltetrahydrofuran on a CuNi2Al bimetallic catalyst in the presence of isopropyl alcohol, which acts as both solvent and hydrogen source. Upon completion of the reaction, this catalyst can be used four times without significant loss of catalytic activity under the following conditions: loadings of furfurole and catalyst are 0.1 g and 0.025 g, respectively, 190 – 250°C, 4 h, 7 ml of methanol. Under optimum conditions, the highest yield of 2-methylfuran was 64.8% and 17.7% of 2-methyltetrahydrofuran (total yield 82.5%), obtained at 230°C for 4 h. The disadvantage of this method is the high hydrogenation temperature. In addition, the authors did not provide any information on mechanistic aspects of the reaction.
In addition to the transfer hydrogenation of N-heterocycles, similar reactions with benzofurans and furan derivatives were carried out in a study [119] to afford the corresponding alicyclic O-derivatives (Scheme 19).
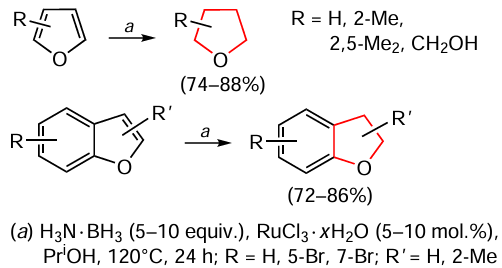
For example, unsubstituted benzofuran gives the desired dihydrobenzofuran in 86% yield. The introduction of a methyl group or a bromine atom in positions 2, 5 and 7 of this molecule, according to the data of work [119], has practically no effect on the hydrogenation process and gives partially reduced products in high yields. The relative amounts of the components of the reaction mixtures are similar to those for the protocol of hydrogenation of N-heterocycles, namely 0.5 mmol of hetarene, 5 equiv. of ammonia borane, 5 mol.% of RuCl3 · x H2O, 2 ml of isopropyl alcohol, 120°C, 24 h. However, the authors did not provide data on the possibility of scaling up the hydrogenation reaction of O-heterocycles, as in the case of the nitrogen analogues, which does not diminish the value of the proposed methodology and motivates its further improvement.
The reduction products of thiophenes, including dihydrothiophene and tetrahydrothiophene derivatives, play an important role in industrial, organic, biological and medicinal chemistries.[1, 3, 133-142] The hydrogenation of unsubstituted thiophene and its derivatives has been studied by many research groups (see, e.g.,[140, 141] and references cited therein), mainly because of the relevance of this process in the hydrodesulfurisation of oil and gas. At the same time, no examples of transfer hydrogenation of S-heterocycles could be found in the literature.
3. The Birch reduction
3.1. Nitrogen-containing heterocycles
Lei et al.[146] presented the results of the development of a highly selective ammonia-free Birch reduction in the presence of 3 to 6 equiv. of a model catalytic system based on the Na dispersion, 15-crown-5-ether and PriOH. It is suggested that the use of chelating 15-crown-5-ether favours the switching of the electron transfer induced by the disodium form of the crown ether (although a scheme of the putative mechanism is not presented in this work). From a practical point of view, this method requires only inexpensive, air- and moisture-stable reagents, small amounts of substrate (0.50 mmol, 1.0 equiv.) and solvent (3 ml THF), and very mild conditions (0°C). N-heterocycles such as acridine (affording 9,10-dihydroacridine in 99% yield), quinoline (1,2,3,4-tetrahydroquinoline, 83%), isoquinoline (1,2,3,4-tetrahydroisoquinoline, 80%) and N-methylindole (N-methylindoline, 72%) have been hydrogenated by this method. It was found that NH-free indole was not reduced by the Na–15-crown-5 – PriOH system. The high chemoselectivity of this method allows to expand the scope of the Birch reduction in the construction of complex molecules. In addition, the authors described the possibility of crown ether regeneration for subsequent transformations.
Gao et al.[147] developed a mechanochemical variant of the highly efficient and ammonia-free lithium-based flash Birch reduction. This method features operational simplicity and extremely short reaction time (not more than 1 min), due to the mechanical activation of lithium metal in situ and the absence of anhydrous solvents. The model catalytic system includes a mixture of 1.0 mmol substrate, 3.0 equiv. lithium (lithium wire), 6 equiv. ethylenediamine (as the hydrogen source) and 6 equiv. THF. This mixture was placed in a five ml stainless steel jar equipped with two stainless steel balls (d = 10 mm) and exposed to a Retsch MM400 mixer mill (30 Hz) for 1 min. The following N-heterocycles were used in the hydrogenation reaction: acridine (yield of 9,10-dihydroacridine is 97%), N-methylindole (yield of N-methylindoline is up to 60%), indole (yield of 4,7-dihydro-1H-indole is up to 48%). The authors found that the reduction of indole to the 4,7-dihydro derivative required the addition of ButOH as an additional hydrogen source. The advantages of this method include the fact that all the experimental manipulations can be carried out in air and that it can be scaled up to gram quantities of substrate, as demonstrated using acridine as an example.
Kondo et al.[148] presented a mechanochemical variant of ammonia-free sodium-based Birch reduction. In this case, D-(+)-glucose was used as the proton source (Scheme 20). Remarkably, all synthetic operations were carried out in the absence of inert gases and without prior activation of the metal.
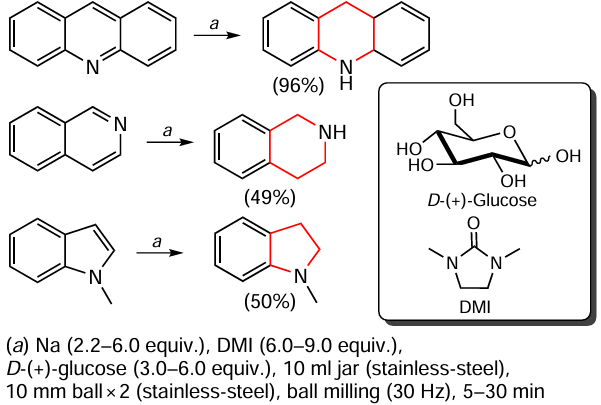
Acridine, isoquinoline and N-methylindole were hydrogenated in this way, using 1 mmol of substrate for the model reaction. Using acridine as an example, the authors increased the amount of substrate to 3 mmol and the yield of the 9,10-dihydro derivative remained virtually unchanged (95%). This clearly demonstrates the promise of this method, which allows the development of laboratory quantities of compounds containing N-saturated heterocycles. In addition, this study can be considered to be a further confirmation of the value of mechanochemistry as an important tool for enhancing the reactivity of metals, enabling transformations that are difficult or inefficient to achieve using classical chemical approaches.
An unusual method for the chemoselective Birch electroreduction of (het)arenes using rapid alternating polarity (rAP) is described (Scheme 21) *.[149] The authors note that this method is more suitable for (het)arenes with electron-withdrawing substituents giving products with a partially or fully hydrogenated N-heterocycle such as cyclohexyl 1-tert-butoxycarbonyl-3,4-dihydropyrrol-2-carboxylate (reaction 1) and ethyl 1-tert-butoxycarbonylindoline-2-carboxylate (reaction 2) as examples. Scheme 21 also shows the yields of the products obtained by the classical method of electrolysis under constant current conditions. It is believed that such a difference in yield is due to the fact that under rAP conditions only very rapid reactions occur at the cathode and anode, realized by rapid alternating polarity.
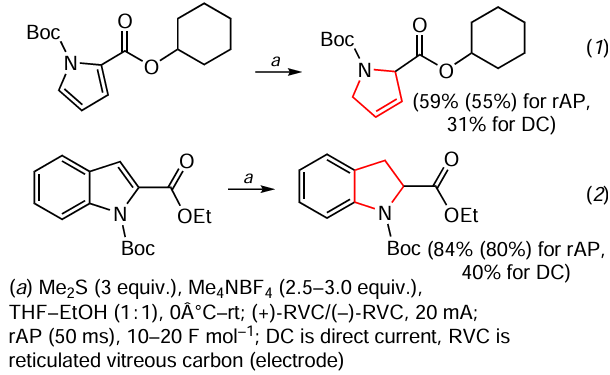
The experimental implementation of the method [149] requires the mixing of (hetero)aromatic substrate (0.1 mmol), tetramethylammonium tetrafluoroborate (0.25 mmol), THF and ethanol (1.5 ml each), followed by the addition of dimethylsulfide (0.3 mmol). After purging with argon for ~ 10 s, an anode and a cathode made of microporous reticulated vitreous carbon (RVC) are introduced into the resulting mixture. Electrolysis is carried out at 0°C or at room temperature for 2 h at a constant current of 20 mA with rapid alternating polarity (50 ms, frequency 10 Hz), the current passing through the solution being 10 – 20 F mol–1. The success of the reaction can be predicted from the reduction potential of the (het)arene substrate bearing the corresponding functional group, which can be easily measured by cyclic voltammetry (CV).** This method gave interesting results, but it requires special equipment and optimization of the experiment depending on the nature of the starting (het)arene.
Chatterjee and Kоnig [150] presented a straightforward method for the Birch reduction of N-heterocyclic compounds in moderate to good yields using photooxidation-reduction catalysis under visible (blue) light irradiation (Scheme 22). Diisopropyl ethylamine (DIPEA, 1 equiv.), Ir[dF(CF3)ppy]2-(dtbpy)PF6 complex (17) (dF(CF3)ppy is 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine and dtbpy — 4,4'-di-tert-butyl-2,2'-bipyridine) (1 mol.%) was used as photocatalyst, DMF (1 ml) as the solvent. The reduction was carried out in air at room temperature for 2 h. The authors presented a plausible reduction pathway, according to which the combination of energy and electron transfer processes leads to the anion-radical N-heterocycle, which is subsequently protonated to form a dearomatized product. Simplicity is undoubtedly a major advantage of this method, but the authors did not provide any information on its scalability.
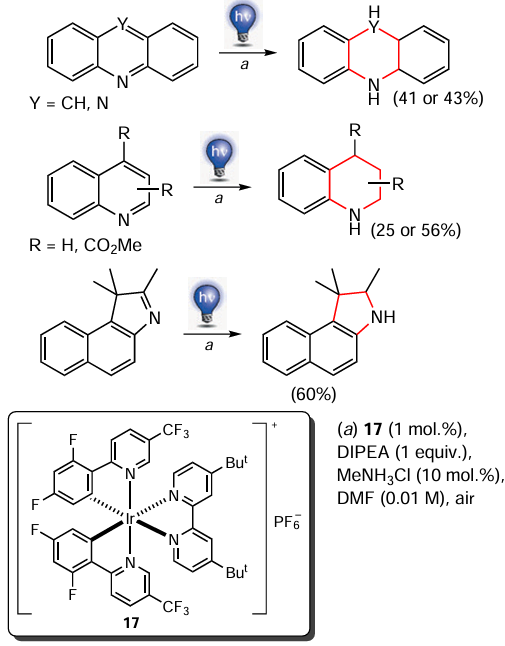
* In rAP reactions, the current is alternating, but with a square rather than the more common sinusoidal waveform. Reactions take place at different rates on the electrodes, and rapid polarity reversal allows only the fastest reactions (those that have time to be realized before the polarity is reversed) to occur, improving the selectivity of the process (see S.G.Davey. Nat. Rev. Chem., 5, 837 (2021); https://doi.org/10.1038/s41570-021-00344-8).
** Empirical guidance on the tolerance of this method to various functional groups is summarized in the [151].
3.2. Oxygen-containing heterocycles
Examples of electron-transfer Birch-type hydrogenations of O-heterocycles have been described in the literature. For example, benzofuran was reduced by such a reaction.[146] Using this substrate in an amount of 0.50 mmol (1.0 equiv.), the Na – PriOH– 15-crown-5 system (3.0 – 6.0 equiv.) and THF (3.0 mL) at 0°C, 2,3-dihydrofuran (39%) and 2-ethylphenol (42%), the product of the C – O bond cleavage of the heterocycle, were obtained. It should be noted that this study has already been cited above when discussing the hydrogenation of a large number of N-heterocycles (see Section 3.1). This provides further evidence that N-heterocycles are more susceptible to hydrogenation than their oxygen-containing counterparts.[3]
In addition to the reduction of N-hetarenes, the electrochemical Birch-type hydrogenation of five-membered O-heterocycles with different substituents has been carried out.[149] The authors also used two techniques, rapid alternating polarity (rAP) and classical direct current (DC) electrolysis. Compounds 18a – d were isolated as model products.
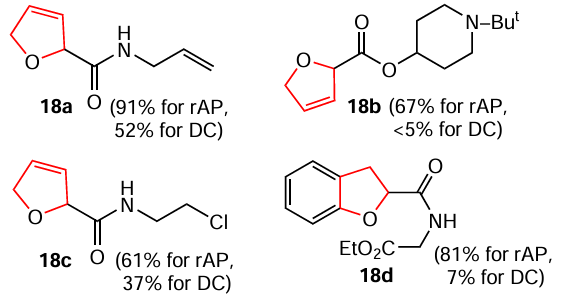
To conclude, this methodology is fairly versatile and needs further development.
3.3. Sulfur-containing heterocycles
The same study [149] describes the electrochemical reduction of five-membered S-heterocycles using the above methods for N- and O-containing heterocyclic substrates. The straightforward Birch hydrogenation of the corresponding furan derivatives afforded 2-phenyl-2,5-dihydrothiophene (19a) and 2-phenyl-2,3-dihydrobenzo[b]thiophene (19b). These compounds were previously obtained by complex four-step transformations.[151]
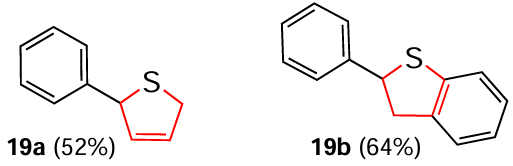
In addition, the authors [149] managed to isolate hydrogenation products such as cyclohexyl-2,5-dihydrothiophene-2-carboxylate (19c), (oxiran-2-ylmethyl)-2,5-dihydrothiophene-2-carboxylate (19d), ethyl (2,5-dihydrothiophene-2-carbonyl)glycinate (19e), (2-cyanoethyl) 2,5-dihydrothiophene-2-carboxylate (19f), tert-butyl(2,5-dihydrothiophene-2-carbonyl)-L-prolinate (19g) and L-mentyl ester of 2,3-dihydrobenzo[b]thiophene-2-carboxylic acid (19h). It should be noted that with the thiophene derivatives, the classical technique of reduction under direct electric current gave unsatisfactory results.
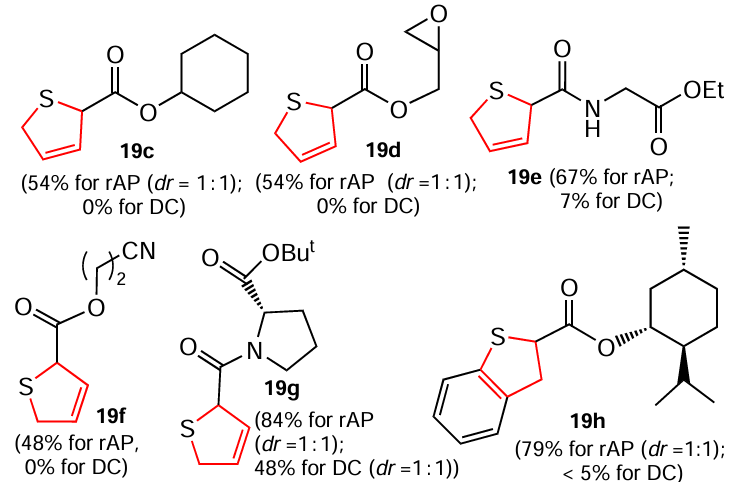
To summarize, the results reported [149] for the hydrogenation of (benzo)thiophenes not only extend the scope of the hydrogenation method, but also open up new possibilities for atom-economic transformations of S-hetarenes.
4. Photocatalytic reduction
Photochemical redox catalysis is one of the versatile tools designed to generate different radical and ion-radical centres by single electron transfer (SET). This process involves high-energy electronically excited states of redox catalysts that absorb visible light, allowing reactions to be carried out under milder conditions and diversifying modern synthetic protocols.[152-154] For example, Tan et al.[155] reported an unusual dearomatization of N-substituted indoles, which follows a radical-polar mechanism induced by a single electron transfer from photoexcited polysulfide anions in the presence of potassium formate and methanol (Scheme 23).
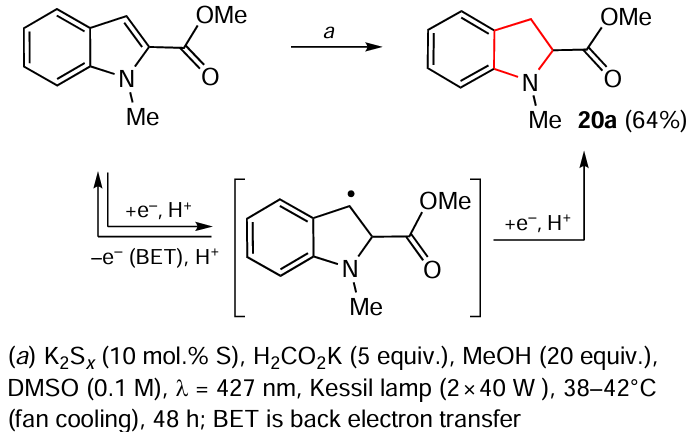
The authors carried out several model reactions using indole derivatives (0.5 mmol), K2Sx (10 mol.% sulfur), HCO2K (5 equiv.) and MeOH (20 equiv.) in DMSO at room temperature and irradiation with 427 nm light (two 40 W Kessil lamps). The process time varied from 6 to 48 h, depending on the nature of the substrate, and the yields of indolines 20a – e reached 78%. It should be noted that dearomatization of methyl N-Boc-indol-4-carboxylate afforded the corresponding indoline 20d in 69% yield and indoline-2-carboxylic acid 20e was formed as a by-product by hydrocarboxylation with concomitant formation of CO2 . A putative reaction mechanism was suggested. This photocatalytic protocol was found to be quite efficient for synthetic applications.
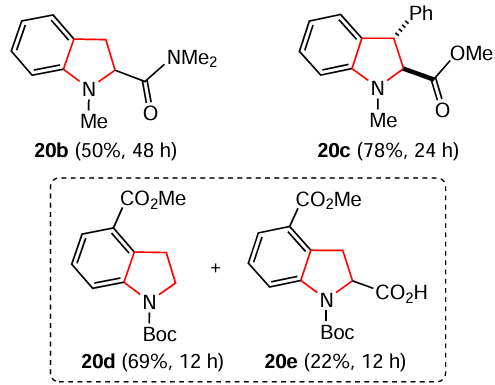
The authors [155] also carried out this reaction with O- and S-heterocycles. Benzofuran-2-carboxylate was efficiently dearomatized to dihydrobenzofuran 21 in high yield. In contrast, the dearomatization of benzothiophene-2-carboxylate to dihydrobenzothiophene 22a was accompanied by side formation of thiophenol 23 by a second SET process followed by the C – S bond cleavage. This reductive ring opening can be hampered by using carboxamide instead of ester to give compound 22b as a single product.
The results suggest that the methodology described in publication [155] requires further optimization and extension of a substrate range.
5. Electrochemical reduction
In recent decades, electrochemical synthesis has attracted increasing attention from researchers [156] due to its advantages such as simplicity of execution, good atom economy, substrate availability, high selectivity, low power consumption, reasonable reagent cost, environmental friendliness and low process temperatures. In addition, the electrons directly involved in these reactions can be considered one of the ‘green’ reactants,[157] and the electrodes can act as heterogeneous catalysts that can be easily separated from the reaction products.
In Scheme 21 (see Section 3.1) one of the possible variants of the electroreduction process was considered, but the authors of the study [149] showed that in this case the Birch reduction mechanism is realized.
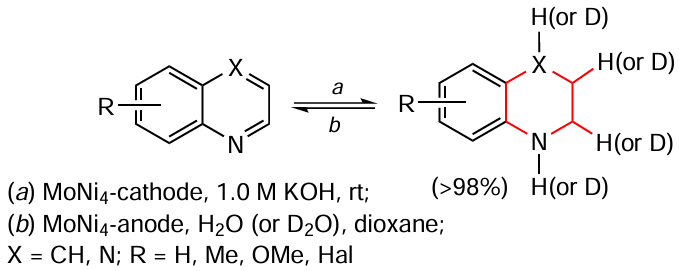
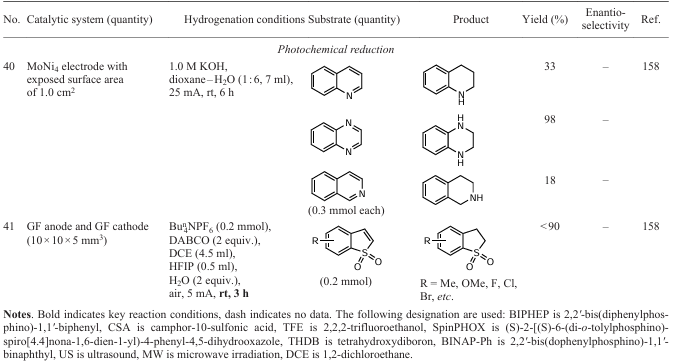
Li et al.[158] developed a procedure for electrochemical hydrogenation (deuteration) and reverse dehydrogenation of N-heterocycles on a bifunctional electrode consisting of MoNi4 at room temperature (Scheme 24). Water was used as the hydrogen source from which hydrogen was generated in situ, as confirmed by EPR spectroscopy. Formally, this process can also be called transfer hydrogenation. The yield of the hydrogenation product (deuteration) of, e.g., quinoxaline reaches 80% under optimum conditions, and that of the reverse dehydrogenation of 1,2,3,4-tetrahydroquinoxaline reaches 99%. The scaling of the process to gram quantities emphasizes the practical relevance of this procedure.
Nedeljkovic et al.[159] describes the electrochemical reduction of thiophenes using screen-printed electrodes (SPEs). The identified reaction products inluded compounds such as 4,5-dihydrothiophene, 2-ethylhexanthiol, diethyl sulfide, ethylethynyl sulfide, butenthiol, allyl methyl sulfide, 2-butylthiophene and thiophene-2-methanethiol. In addition, C5 – C7 hydrocarbons such as pentane, 2-methylbutane and heptane were found in the reaction mixture. Based on the composition of the products, it was assumed that the hydrogenation of thiophene at the combined SPE electrode can occur in two directions’--- the heterocycle ring opening followed by hydrogenation of the newly formed intermediates, or initial hydrogenation of the S-heterocycle with subsequent ring opening. The authors have found optimal desulfurisation conditions suitable for oil refining using an environmentally friendly, low-cost method in which a high degree of automation can be achieved.
Guo et al.[160] performed direct selective electrochemical reduction of benzo[b]thiophene-1,1-dioxides using 1,1,1,1,3,3,3,3-hexafluoropropan-2-ol (HFIP) as the hydrogen donor in a metal- and hydrogen-free mode (Scheme 25). Standard experimental conditions include the use of a substrate (0.2 mmol), Bun4NPF6 (0.2 mmol), 1,4-diazabicyclo[2.2.2]octane (DABCO, 2 equiv.), 1,2-dichloroethane (DCE, 4.5 mL), HFIP (0.5 mL), graphite felt (GF) anode and cathode, and a constant current of 5 mA. The reactions are carried out in air at room temperature for 3 h in an undivided cell and the quantity of electricity passed through the solution is 2.80 F mol–1.
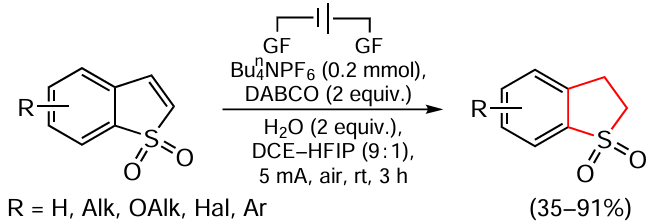
This method is characterized by its tolerance to various functional groups in the substrate, which has allowed the use of 43 compounds in the reaction. For example, benzo[b]thiophene-1,1-dioxides containing various electron-donating or electron-withdrawing substituents at the 5-position, including methyl, methoxy, phenyl groups or halogen atoms (fluorine, chlorine, bromine), give partial reduction products in 74 – 82% yields. In addition, 5-arylbenzo[b]thiophene-1,1-dioxides bearing substituents having different electronic properties are also suitable substrates and furnish the corresponding products in 35 – 87% yields. A substrate with a bulky naphthyl group also performed well in this process, giving a heterocyclic hydrogenated derivative in 72% yield. Moreover, benzo[b]thiophene-1,1-dioxides bearing electron-donating or electron-withdrawing substituents at the positions 4, 6 and 7 exhibit close reactivity, allowing the corresponding products to be obtained in 60 – 95% yields. The authors have also demonstrated the scalability of this process and proposed its putative mechanism.
6. Dearomatization
The following are examples of heterocyclic dearomatization carried out by various methods. Formally, chemical reactions of this type do not correspond to the stated subject of the review, but since the result is the transformation of hetarene into a compound with a partially or fully saturated heterocycle, it is appropriate to discuss them in a separate Section.
Chen et al.[161] described a process for the dearomatization of indoles, characterized by high regioselectivity and chemoselectivity and moderate to good yields. The reaction proceeds under mild conditions and is catalyzed by Co(OAc)2 (5 mol.%) in the presence of the phosphorus-containing ligand XantPhos (6 mol.%), pinacolborane (HBpin, 2.2 equiv.) and water (0.55 equiv.) in THF (Scheme 26). It has been shown that an inert atmosphere is required to implement the process, using HBpin reagent and H2O as the hydrogen source. The methodology was scaled up to gram quantities and the proposed mechanism of construction of the final hexahydropyrido[1,2-a]indole systems was presented.
Barnett et al.[162] carried out hydration of dihydropyran to 2-hydroxytetrahydropyran. The latter was identified as an intermediate formed in situ by mixing dihydropyran with water in an argon atmosphere at a pressure of 34.5 bar in the temperature range from room temperature to 200°C in the absence of a catalyst in a continuous flow or batch reactor. The quantity of this intermediate depends on the rate of mixing of the components and the temperature of the process. It should be noted that ‘acidic coke’ is formed as a by-product in the flow reactor and the yield of 2-hydroxytetrahydropyran is directly dependent on the reactor run time. Since the authors did not aim to obtain a saturated O-heterocycle, they simply identified 2-hydroxytetrahydropyran as one of the possible intermediates, the formation of which provided evidence for their plausible pathway. This transformation can be regarded as an unusual reaction that expands the boundaries of O-heterocyclic chemistry.
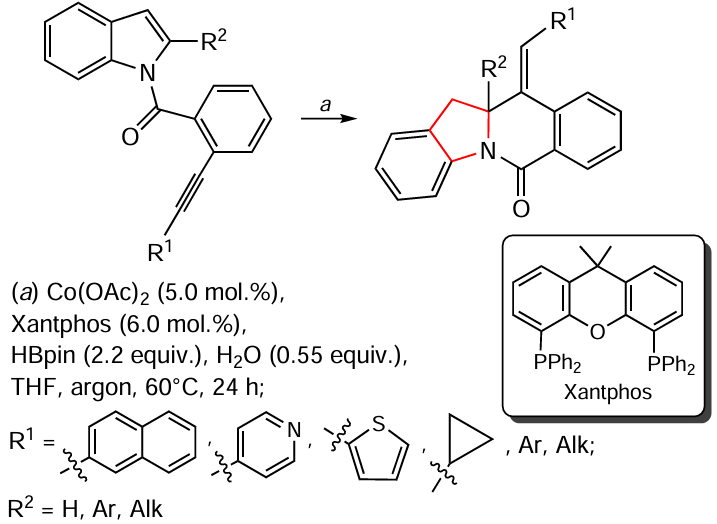
Ahn et al.[163] also noted the possibility of saturation of the C = C double bond of a six-membered O-heterocycle by hydration (Scheme 27). This process is realized in one of the steps of the total synthesis of (–)-hedycoropyran A, namely during the conversion of the substituted tetrahydropyran 24 to the diol 25. The catalyst is the OsO4 – 4-methylmorpholine-4-oxide (NMO) system and the reaction is carried out at room temperature for 12 h.
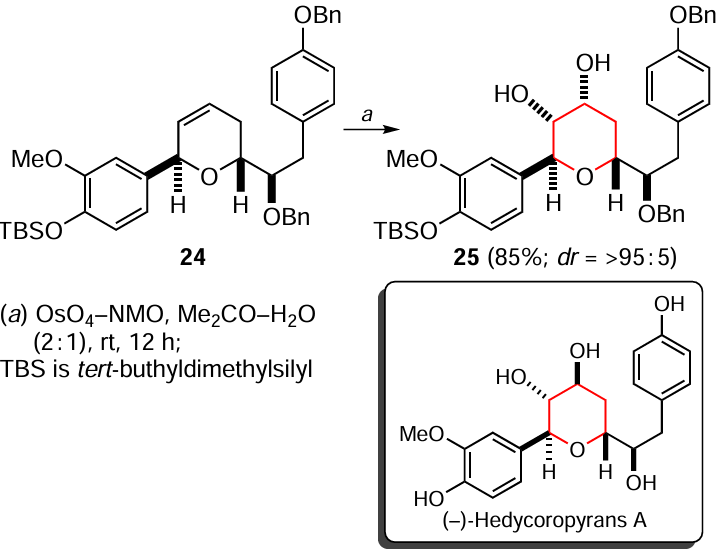
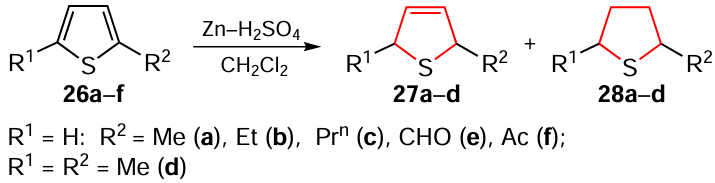
The reduction of alkyl-substituted thiophenes 26a – f in the presence of Zn – H2SO4 system to the corresponding 2,5-dihydro- (27a – d) and tetrahydrothiophenes (28a – d) has been described (Scheme 28).[164] It was found that 2-formyl and 2-acetyl derivatives 26e,f, in addition to the hydrogenation of the sulfur-containing aromatic ring, undergo a simultaneous reduction of the carbonyl substituent to an alkyl group to give the products 27a,b and 28a,b respectively. The optimum conditions of the process (variation of temperature, reaction time and reagent ratio) were chosed experimentally.
An unusual example of the dearomatization of 2-(chloromethyl)thiophenes 29 in the presence of a palladium-containing catalyst and allenyltributylstannane has been reported.[165] In this case, the dearomatization reaction proceeded selectively in a substrate-coordinating solvent (acetonitrile) to give only propargyl derivatives 30 in satisfactory or good yields, whereas in non-coordinating dichloromethane, allenes 31 were obtained as main products together with a small amount of propargyl compounds (Scheme 29). Palladium compounds such as Pd(PPh3)4 , Pd(OAc)2 , PdCl2 , Pd2(dba)3 (dba is dibenzylideneacetone) and the combination PdCl2 (5 mol.%) – PPh3 (10 mol.%) were tested as catalysts. The reactions were carried out under a nitrogen atmosphere at room temperature for 12 h. In addition to experimental data on the optimization of the reaction conditions, the authors present a putative pathway of such dearomatization.
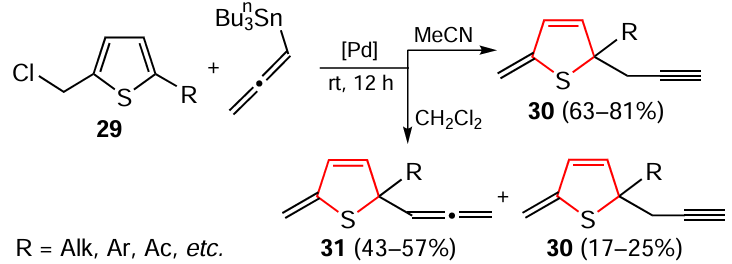
The important results of the study [165] include not only the methodology of dearomatization of five-membered S-heterocycles under mild conditions and the control of their selectivity using solvents of different nature. This approach opens a direct way to obtain reactive propargyl and allenyl saturated heterocycles from readily available hetarenes.
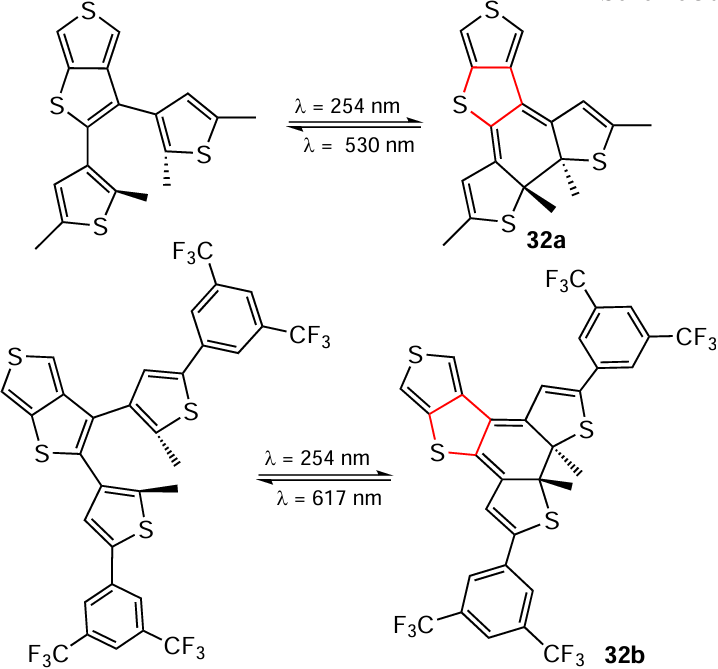
The study [166] gives examples of photochemical synthesis of π-conjugated polymers based on thieno[3,4-b]thiophene 32a,b containing saturated S-heteronuclei in acetonitrile (Scheme 30). The aim of this work was to obtain polymers with a specific architecture that would preserve the photochromic activity found in their low-molecular-weight analogues. For this reason, the authors did not study the dearomatization process in detail, but showed that processes of photochemical saturation of S-heterocycles under irradiation are possible in principle.
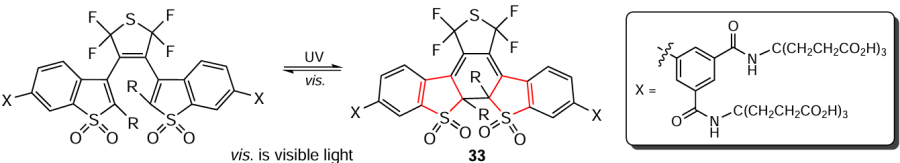
Similar systems 33 based on benzo[b]thiophene-1,1-dioxides have been synthesized in aqueous solutions (Scheme 31).[167]
Li et al.[168] describes the enantioselective partial dearomatization of tertiary alcohols 34 with 2-indole and 2-thienyl substituents by organocatalysis with the phosphoric acid derivative 12f. The authors demonstrated examples of such dearomatization in combination with asymmetric 1,10-conjugated addition, which afforded dihydrothiophene derivatives 35 α-substituted with indole and pyrrole moieties (Scheme 32). Two variations of the conditions are described: (a) thiophene 34 (0.20 mmol) and pyrrole (0.24 mmol), chlorobenzene (4 mL), –40°C, 12 h; (b) thiophene 34 (0.20 mmol) and pyrrole (2 mmol), toluene (4 mL), –20°C, 24 h.
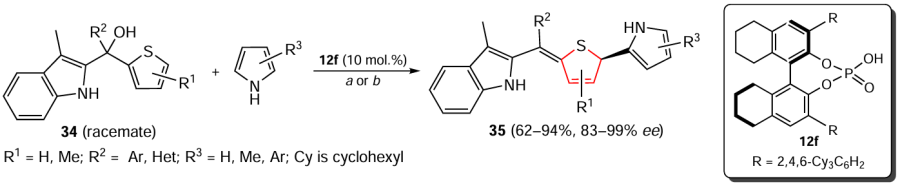
Under these conditions, dearomatization products are formed in good yields, characterized in most cases by acceptable ratios of E- and Z-isomers and high enantioselectivity. For example, the products 35 with aryl substituents R2, containing functional groups with different electronic properties and arrangement (ortho, meta and para), were obtained in 87 – 97% yields, with 92 – 99% ее and E : Z = >20 : 1. Furthermore, this process was found to be tolerant to polycyclic and heterocyclic aryl substituents. Notably, in substrates bearing two thienyl groups, one thiophene ring is dearomatized with high efficiency and chemoselectivity. It was shown that different pyrroles successfully act as nucleophiles, forming the corresponding enantioenriched S-heterocycles with rather good efficiency and selectivity. Other electron-rich arenes such as indole, naphthol, furan, thiophene and 1,3,5-trimethoxybenzene were also investigated as a second component. Indole gave the target product in good yield but with low Z,E selectivity and enantioselectivity, while other nucleophiles were generally inactive in similar reactions. In addition, the results of density functional theory (DFT) calculations were presented illustrating a possible reaction pathway in which multiple hydrogen bonds play a pivotal role in achieving excellent stereocontrol of the said process.[168] Using several examples, the authors showed that the method can be extended to the asymmetric dearomatization of selenophenes.
The selective hydrogenation of the C = C bond in the 2,5-dihydrothiophene ring to enantioenriched tetrahydrothiophene 36 was also described (Scheme 33).[168] In this case, NiCl2 was used as the catalyst, NaBH4 as the hydrogen source and a mixture of dimethoxyethane (DME) and methanol as the solvent. The reaction was carried out between 0°C and room temperature for 10 min.
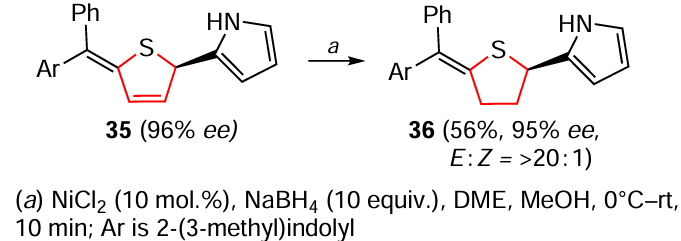
Given the variety of products that are accessible by the above techniques, it is clear that the results of the work[168] have good prospects for further development.
7. Conclusion
The presented material, although not exhaustive, shows that non-classical reduction methods (hydrogenation, saturation or dearomatization) allow the desired products to be obtained without the use of high temperatures and pressurized hydrogen gas. This means that there is no need in special apparatus for working with explosive reagents and, consequently, strict safety requirements, or complex equipment designed to carry out chemical processes under harsh conditions.[10, 68, 73, 169-171] For the sake of clarity, based on the results of the discussion of the reactions given in the present work, the Table 1has been compiled in which model examples of the classical methodology of hydrogenation in a hydrogen atmosphere and non-classical methods of saturation of hetarenes are compared. It should be noted that in 2018, a rather detailed review [14] was published devoted to the comparative analysis of classical methodologies for the hydrogenation of various heterocycles. Therefore, Table 1 only includes a few examples that were not included in the cited review because they appeared after 2018.
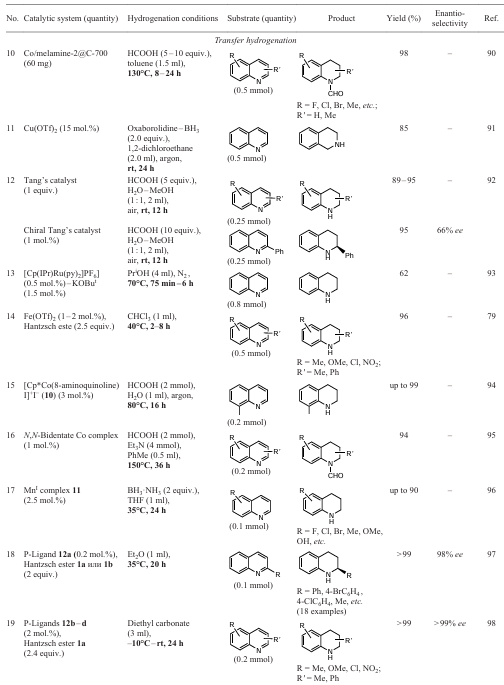
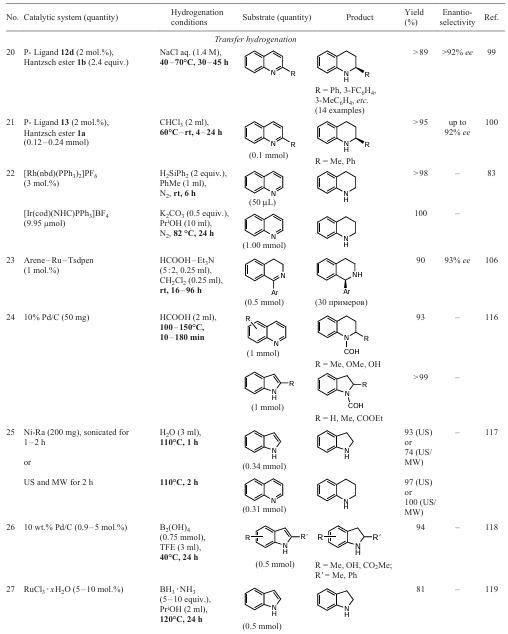
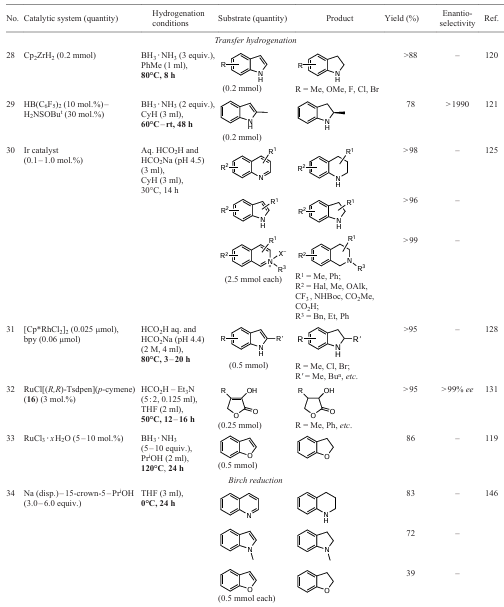

It should be noted that for all four general saturation methods (see Sections 2 – 5) and several individual examples of heterocyclic aromatization (see Section 6) presented in this work, there are actually no data on the possibility of their scalability to kilogram quantities of target products. However, some of the reactions appear at first glance to be promising basis for the further development of industrial technologies. In our opinion, the comparison of the above methods in terms of efficiency is not entirely correct, since the implementation of the Birch reduction methods in photocatalytic or electrochemical conditions depends primarily on the availability of suitable equipment, whereas transfer hydrogenation can be carried out in the presence of stirrers with thermostating of the reaction mixture and the necessary set of reagents. The nature of the substrate containing one or more heterocycles, the number and nature of its substituents, the thermostability of the starting compounds and target products and their solubility in appropriate media should also be considered.
We could find only one publication [117] describing the use of ultrasonic and/or microwave activation for the saturation of heterocycles. The findings of this work, namely the use of water as the hydrogen source and as the solvent, a readily available catalyst, Raney nickel (Ra – Ni), relatively mild conditions of catalyst activation and hydrogenation process, as well as the operational simplicity, make it possible to predict the direction of further development of such studies in the near future.
An analysis of the published data shows that most of the processes of hydrogenation and/or dearomatization of heterocyclic compounds are carried out by organic chemists in order to produce compounds substituted with a saturated heterocycle, without an in-depth study of the physicochemical features of the course of each reaction. The studies of catalytic chemists, on the other hand, aim to establish the physico-chemical regularities of a particular process, but the results of these few studies are not always taken into account. In addition, in most studies catalytically active centres are formed in situ during the reaction, and little is known about their nature and how they are formed, including under mild conditions. It is hoped that the combined efforts of and specialists in organic and catalytic chemistsries and in physicochemical analysis will make it possible to elucidate the fundamental features of saturation and dearomatization of heterocycles of different types.
8. List of abbreviations
Ad — 1-adamantyl,
BINAP-Ph — 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl,
BIPHEP — 2,2'-bis(diphenylphosphino)-1,1'-diphenyl,
Boc — tert-butoxycarbonyl,
bpy — 2,2'-bipyridine,
cod — cycloocta-1,5-diene,
Cp — cyclopentadienyl,
Cp* — pentamethylcyclopentadienyl,
CSA — camphor-10-sulfonic acid,
d — diameter,
DABCO — 1,4-diazabicyclo[2.2.2]octane,
dba — dibenzylydeneacetone,
DC — direct current,
DСE — 1,2-dichloroethane,
dF(CF3)ppy — 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine,
DIPEA — diisopropylethylamine,
DMA — dimethylacetamide,
DME — dimethoxyethane,
dtbpy — 4,4'-di-tert-butyl-2,2'-dipyridine,
ee — enantiomeric excess,
FLP — frustrated Lewis acid–base pairs,
GF — graphite felt,
HBpin — pinacolborane,
HFIP — 1,1,1,3,3,3‐ hexafluoropropan-2-ol,
IPr — 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene,
MW — microwave irradiation,
NADH — reduced nicotinamide adenine dinucleotide,
nbd — norbornadiene,
NHC — N-heterocyclic carbene,
Ni-Ra — Raney nickel,
NMO — 4-methylmorpholine 4-oxide,
NP — nanoparticle,
PS — polystyrene,
py — pyridine,
rAP — rapid alternating polarity,
RVC — reticulated vitreous carbon (electrode),
SBA — Santa Barbara Amorphous (highly stable mesoporous silica),
SET — single electron transfer,
SPE — screen-printed electrode,
SpinPHOX — (S)-2-[(S)-6-(di-о-tolylphosphino)spiro[4.4]nona-1,6-diene-1-yl)-4-phenyl-4,5-dihydrooxazole,
TBS — tert-butyldimethylsilyl,
Tf — trifluoromethanesulfonyl (triflyl),
TFE — 2,2,2-trifluoroethanol,
THDB — tetrahydroxydiboron,
TON — turnover number,
Ts — p-toluenesulfonyl (tosyl),
Tsdpen — N-(p-tosyl)-1,2-diphenylethylene-1,2-diamine,
US — ultrasonication.
References











