


Keywords
Abstract
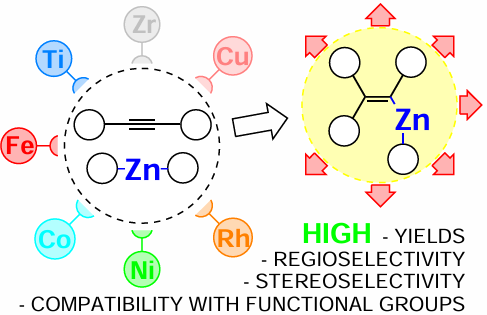
Over the last 10 years, conceptually new results have been obtained in the field of unsaturated organozinc reagents; these results need to be analyzed and integrated. This review systematically considers data on the catalytic carbozincation reactions of alkynes, which give alkenyl organozinc compounds, highly reactive intermediates for the synthesis of functionally substituted olefins. The reactions catalyzed by copper, iron, cobalt, nickel, and rhodium complexes are described. A separate part of the review addresses the catalytic reactions initiated by zirconium and titanium compounds. The reaction conditions are indicated; in some cases, putative reaction mechanisms are discussed.
The bibliography includes 135 references.
1. Introduction
Carbometallation reactions of acetylenes provide effective methods for the regio- and stereoselective synthesis of structurally diverse olefins. The most widely used transformations of this type include Zr-catalyzed Negishi methylamination,[1-3] cycloalumination,[4-7] Dzhemilev cyclomagnesiation,[8-13] carbocupration,[14] carbostannylation,[15] carboboration,[16, 17] and arylmagnesiation.[18]
The carbometallation of alkynes with organozinc reagents is one of the most popular approaches to the synthesis of various functionally substituted alkenes.[19-28] The considerable interest in the organozinc synthesis of these olefins is, first of all, due to the tolerance of Zn reagents to the presence of heterofuctional substituents in the substrates containing triple bonds.
This literature review is devoted to carbozincation reactions of structurally diverse acetylene derivatives induced by transition metal compounds. The mechanism of these reactions considerably depends on the nature of the transition metal, which is reflected in the structure of the review, which is divided into parts, each addressing the catalytic transformations induced by catalysts based on salts and complexes of a particular transition metal. It is noteworthy that carbozincation reactions are considered in quite a few reviews (e.g., Refs 20 – 28), which emphasizes high relevance of this subject for the development of organic chemistry. However, in the last 10 years, conceptually new results have been obtained in the field of unsaturated organozinc reagents, and these results are to be summarized and analyzed. First of all, this refers to recently developed methods for carbozincation of acetylenic compounds catalyzed by the Ti(OPri)4 – EtMgBr and Cp2ZrCl2 – EtMgBr systems (Cp is cyclopentadienyl). The reactions of terminal acetylenes with zinc catalyzed by iron(II) salts discovered at the same time also appear unusual. One more finding is related to the use of Rh catalysis, which allowed for decreasing the amount of the catalyst to 3 mol.%. These achievements can have a substantial impact on the subsequent trajectory of research in carbometallation of not only alkynes but also other unsaturated compounds. From this standpoint, the publication of this review is timely and useful for the development of organometallic chemistry.
By carbozincation of alkynes, we mean reactions in which the carbon and zinc atoms add to a triple carbon – carbon bond. A simple example is the reaction between an alkyne and an organozinc compound. Diarylzinc derivatives are known to be inert towards alkynes, while among dialkyl-containing organozinc compounds, only di(tert-butyl)zinc can be mentioned as being sufficiently nucleophilic to react with terminal acetylenes in refluxing tetrahydrofuran.[29-31] However, disubstiuted acetylenes cannot be carbozincated by this reagent. Allyl organozinc compounds have a higher reactivity, but in this case, the carbozincation reaction has limitations. For example, disubstituted acetylenes usually do not react. Meanwhile, 1-alkynylsilanes and 1-alkynyl sulfones with an activated triple bond readily undergo regioselective carbozincation under the action of allylzinc bromide in a THF solution.[32-34] There are data on successful carbozincation of N,N-diethylprop-2-yne-1-amine,[35, 36] but-3-yn-2-ol,[37-41] propargyl alcohol,[41, 42] and octa-1,3-diyne [43] under the action of allyl organozinc compounds. The reaction of phenyl- and butyl-substituted terminal acetylenes with allylzinc bromide gives a mixture of mono- and bis-addition products.
Thus, non-catalyzed carbozincation is characterized by the following features:
(1) the use of a narrow range of organozinc compounds with high nucleophilicity;
(2) the introduction of activated acetylenic substrates in the reaction;
(3) more drastic conditions of the reaction (refluxing or long reaction time) compared to those for analogous catalytic reactions.
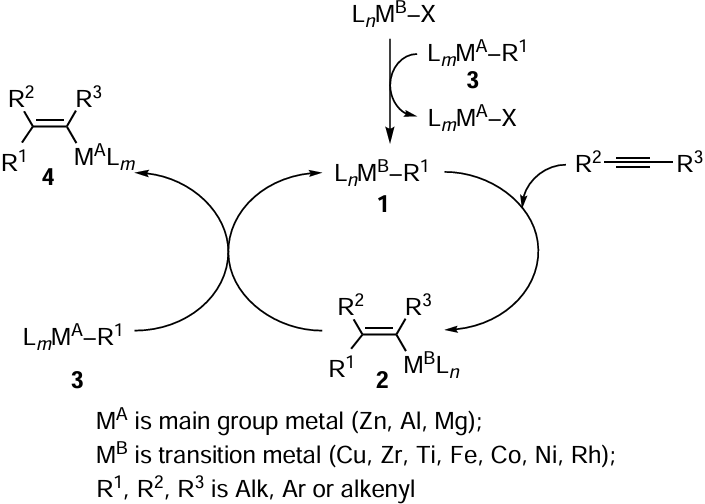
The use of transition metal catalysts based on transition metal salts and complexes is the most versatile and convenient approach to carbozincation of alkynes. The essence of catalytic carbometallation of alkynes is that organic compound of transition metal 1 is formed in situ and reacts with alkyne to give substituted vinyl derivative of the transition metal 2 (Scheme 1). The subsequent transmetallation of 2 with main group metal (Zn, Al, Mg) compound 3 results in carbometallation product 4 and regeneration of catalytically active species 1. There are various known approaches to initiation of the reaction, i.e., the initial generation of intermediate 1. The key method is the alkylation (arylation, alkenylation) of a transition metal salt with an organic compound of a main group metal. The direction of this reaction can be determined, at the qualitative level, from the electronegativity values of the transition and main group metals. The alkylation is expected to proceed towards the formation of the salt with more ionic bond. The Pauling electronegativity of zinc is 1.65, which is lower than the electronegativity of many transition d elements. For this reason, salts of most transition metals can theoretically catalyze carbozincation reactions. However, the alkylation of lanthanide and actinide salts or salts of d elements at the beginning of the period under the action of organozinc compounds is problematic.
This review addresses carbozincation reactions of acetylenic compounds catalyzed by copper, titanium, zirconium, iron, cobalt, nickel, and rhodium salts and complexes. It is noteworthy that all operations with organozinc compounds should be carried out in an inert gas atmosphere using standard Schlenk techniques. These reagents are exceptionally sensitive to the presence of water and other protic solvents, and volatile dialkylzinc derivatives can spontaneously ignite in air.
2. Catalysis by copper complexes
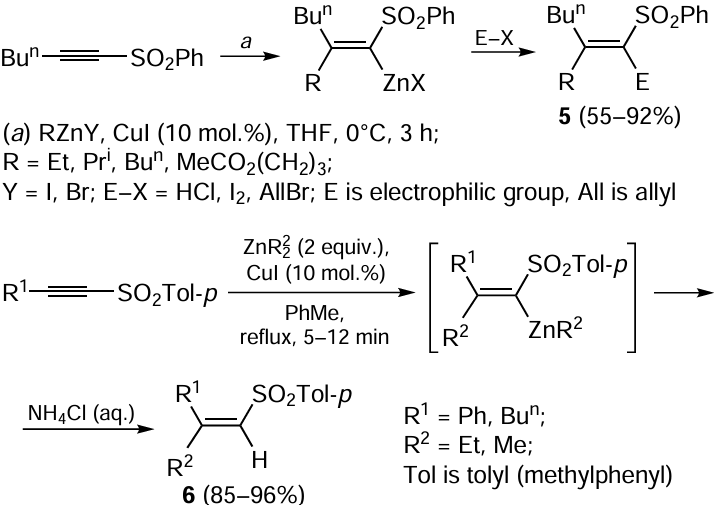
Organocopper compounds are reagents of choice for the carbometallation of alkynes, as they provide high stereo- and chemoselectivity of the reaction and are highly tolerant to the presence of functional groups in the acetylenic substrate molecule. Meanwhile, the carbometallation of functionally substituted acetylenes such as 1-alkynyl sulfones on treatment with organocopper reagents usually proceeds with low stereoselectivity.[44-49] This limitation of the carbocupration reaction can be circumvented by using organozinc reagents. Carbozincation of 1-alkynyl sulfones and sulfoxides catalyzed by copper salts has been comprehensively studied. CuI-Catalyzed carbozincation of the corresponding 1-alkynyl sulfones with alkylzinc halides and dialkylzinc reagents has a high regio- and stereoselectivity and gives exclusively (Z)-1-alkenyl sulfones 5 and 6 in good to high yields (Scheme 2). A similar carbocupration of alkynyl sulfones is non-selective, giving a mixture of two stereoisomers.[44] The Z- and E-isomer ratio for these products considerably depends on temperature and the nature of substituents at the triple bond and varies over a broad range from 42 : 58 to 0 : 100. Alkenyl- and arylzinc chlorides and bromides can be used as carbozincating reagents, apart from alkyl derivatives.[50] A minor decrease in the yield of the substituted alkenyl sulfone (down to 55%) takes place for carbozincation of (hex-1-yn-1-ylsulfonyl)benzene with carboxyl-containing reagent, MeOCO(CH2)3ZnI.[28, 51]
Cu-Catalyzed carbozincation of 1-alkynyl sulfoxides using organozinc reagents, including functionally substituted ones, is a stereoselective approach to the preparation of β,β-disubstituted vinyl sulfoxides. Since the sulfinyl group can be easily replaced by various functional groups, vinyl-substituted sulfoxides have found wide use in organic synthesis.
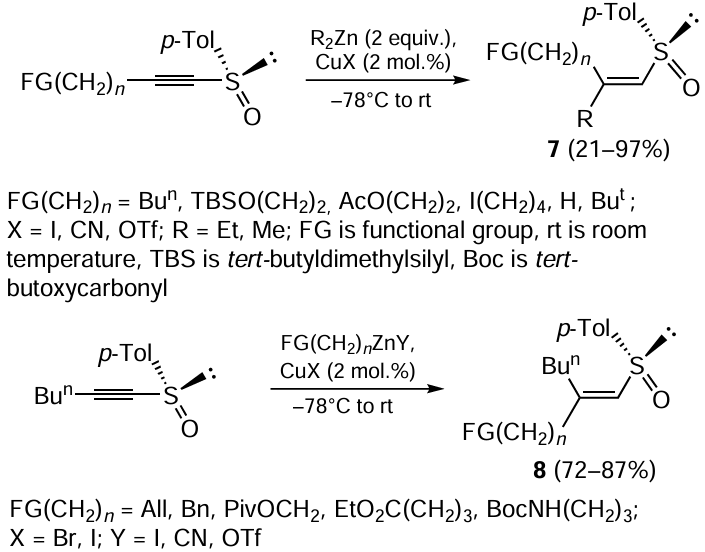
1-Alkynyl sulfoxides containing alkyl, acyl, tributylsilyloxy, or iodoalkyl group (Scheme 3) [52] were converted under the indicated conditions to functionally substituted 1-alkenyl sulfoxides 7, which formed as only Z-isomers in good yields. In the case of 1-alkynyl sulfoxides with a bulky tert-butyl substituent at the triple bond or a terminal triple bond, the yield of β,β-disubstituted 1-alkenyl sulfoxides 7 decreased to 21 – 24%. To obtain products 8, copper(I) iodide, cyanide, or trifluoromethanesulfonate (triflate, OTf) (2 mol.%) was allowed to react with 2 equiv. of an organozinc compound containing alkyl, allyl, benzyl, phenyl, pivaloyloxymethyl (PivOCH2), carboxyl, or amine group in the molecule.[52, 53]
Mention should be made of the advantages of using organozinc reagents over magnesium or lithium organic compounds for carbometallation of alkynyl sulfoxides. For example, the carbomagnesiation of 1-(hex-1-yn-1-ylsulfinyl)-4-methylbenzene catalyzed by Cu(OTf)2 proceeds non-selectively to give a mixture of two stereoisomers (E and Z) in 1 : 1.6 ratio.[54, 55] In addition, at room temperature, α-sulfinyl vinyl organozinc intermediates retain the stereoconfiguration at the double bond, which is confirmed by the products of their hydrolysis. Meanwhile, α-sulfinyl vinyl organolithium intermediates undergo stereoisomerization even at –78°C,[56, 57] which is attributable to higher ionicity of the C – Li bond and the energetic advantage of the configuration inversion.
CuI-Catalyzed carbozincation of 1-alkynyl sulfoximines results in the regio- and stereoselective formation of 1-alkenyl sulfoximines 9 exclusively as the syn-addition products (Scheme 4). This reaction was carried out for a broad range of organozinc compounds that contained alkyl, phenyl, and carboxyl groups.[58] The yields of the resulting alkenyl sulfoximines typically ranged from good to high. However, carbozincation on treatment with oct-1-ynylzinc bromide afforded the carbometallation product in a yield of only 18%.
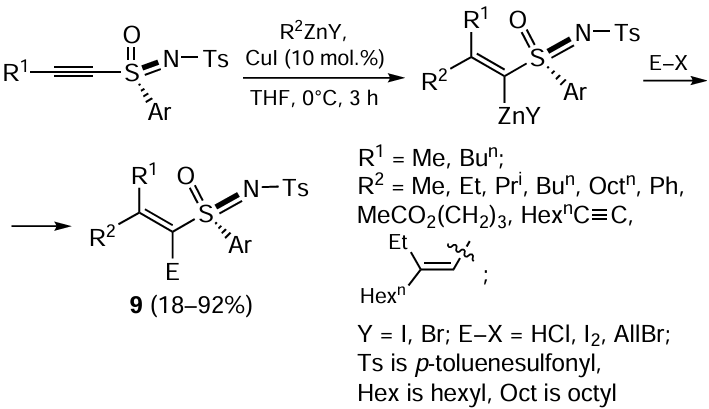
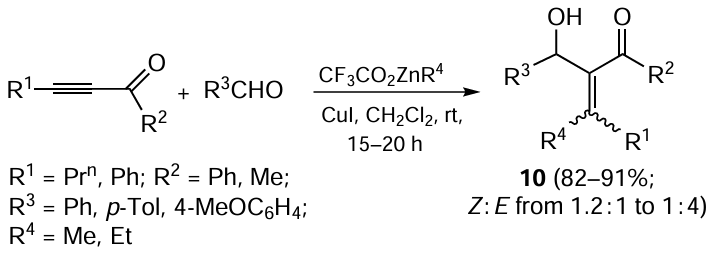
The reaction of 1-alkynyl ketones with organozinc reagents such as CF3CO2ZnR, where R = Et, Me, and Ph, in the presence of 5 mol.% CuI and aromatic aldehyde in dichloromethane at room temperature results in the regioselective formation of methylenehydroxyl-substituted 1-alkenyl ketones 10, mainly as Z-isomers, as shown in Scheme 5.[59] Although authors of the study call this reaction ‘CuI-catalyzed carbozincation of acetylenes’, it is more appropriate to consider it as carbozincation of 1-alkynyl ketones, because the transformation pathway proposed by the authors implies addition of the metal to the oxygen atom of the keto group in alkynyl ketone.[60] Presumably, this reactions proceeds via the step of formation of zinc-containing allenolate anion. A similar transformation of alkynylones to allenolates was reported by Noyori and co-workers [61] and by Japanese chemists.[62-64]
3. Catalysis and initiation of reactions by zirconium and titanium complexes
A number of publications [1, 65, 66] describe the carbozincation of non-functionalized terminal and dialkyl-substituted acetylenes with alkylzinc derivatives in combination with a stoichiometric amount of zirconocene dihalides (Scheme 6). This zirconocene-initiated reaction of alkynes with alkylzinc compounds (Et2Zn, Me2Zn, Bun2Zn, EtZnCl) was the first example of regio- and stereoselective (the syn- to anti-addition product ratio exceeded 98 : 2) synthesis of 1-alkenylzinc derivatives 11.[28, 66]
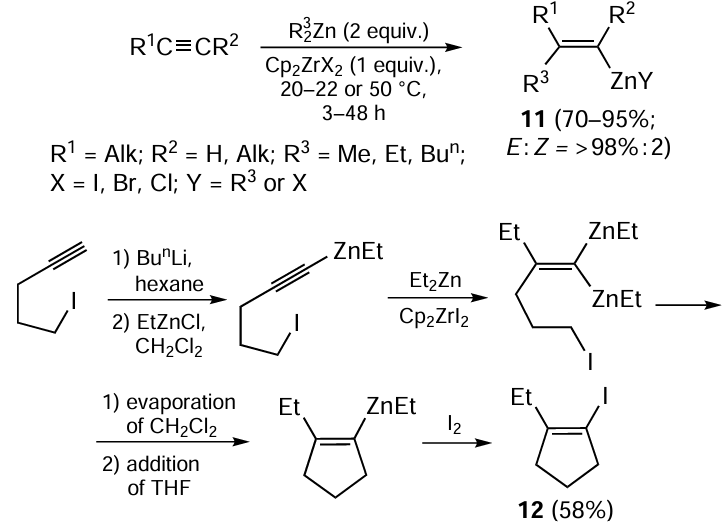
The carbozincation of terminal and dialkyl-substituted acetylenes with alkylzinc derivatives promoted by zirconocene diiodide (Cp2ZrI2) takes place at room temperature, while the use of Cp2ZrCl2 or Cp2ZrBr2 requires heating of the reaction mixture at 50°C for 48 h. The reaction proceeds stereoselectively as the syn-addition to the triple carbon–carbon bond. In the case of terminal acetylenes, the reaction mainly gives the regioisomer in which the zinc atom binds to the terminal carbon atom of the alkyne. The Zr-promoted carbozincation of 5-iodopent-1-yne with Et2Zn was utilized in a method for the synthesis of 1-ethyl-2-iodocyclopent-1-ene 12.[67] The allylzincation of dialkyl-substituted acetylenes (dec-5-yne and but-2-yne) with diallyl- or dicrotylzinc in the presence of a stoichiometric amount of Cp2ZrI2 proceeds mainly as the syn-addition to give carbometallation products in 84 – 92% yields.[33] An exceptionally high stereoselectivity was noted for the reaction involving dicrotylzinc.
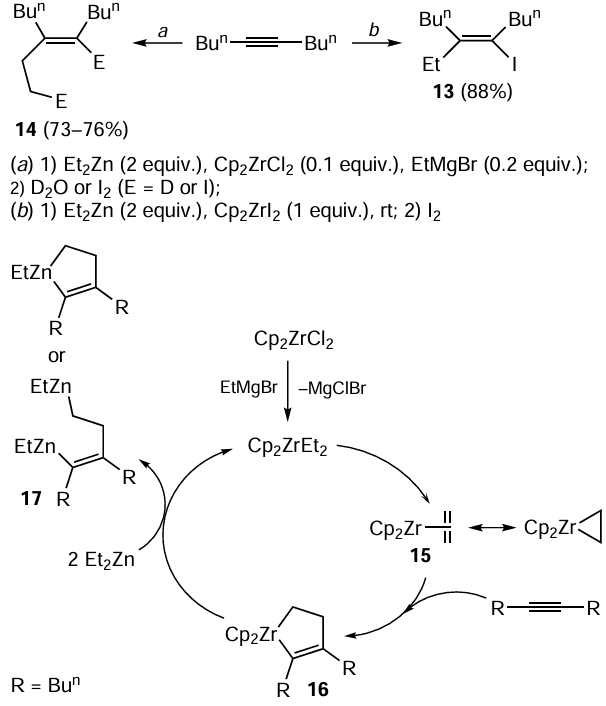
The reaction of dec-5-yne with Et2Zn can follow two pathways depending on the procedure. When 2 equiv. of Et2Zn and 1 equiv. of Cp2ZrI2 are used, the carbozincation followed by iodinolysis proceeds stereoselectively (> 98%) to give (E)-5-ethyl-6-iododec-5-ene 13 in 88% yield (Scheme 7).[66] However, analogous reaction of the same substrate with Et2Zn in the presence of catalytic amounts of Cp2ZrCl2 and EtMgBr follows the 2-zinco-ethylzincation pathway. The deuterolysis or iodinolysis give the corresponding dideuterated or diiodinated trisubstituted olefins 14 (see Scheme 7).[68] These pathways differ fundamentally by the fact that in the latter case, the key intermediate is zirconocene – ethylene complex 15, generated in situ upon the reaction of zirconocene dichloride with Grignard reagent (EtMgBr), rather than ethylzirconocene. The subsequent coordination of the acetylenic compound and coupling of the ethylene and acetylene moieties result in the generation of zirconacyclopentene intermediate 16, which is transmetallated with Et2Zn to give unsaturated organozinc compound 17.
Substituted propargylamines are successfully involved in the 2-zincоethylzincation reaction with Et2Zn catalyzed by the Cp2ZrCl2 – EtMgBr system. After hydrolysis (deuterolysis or iodinolysis), amino-substituted olefins 18 are obtained with high regio- and stereoselectivity (the ratio of the syn- and anti-addition products exceeds 95 : 5) (Scheme 8; the superscript ‘c’ stands for cyclo).[69, 70] The presence of nitrogen atom in the substrate does not induce destruction or deactivation of the catalytic complexes.
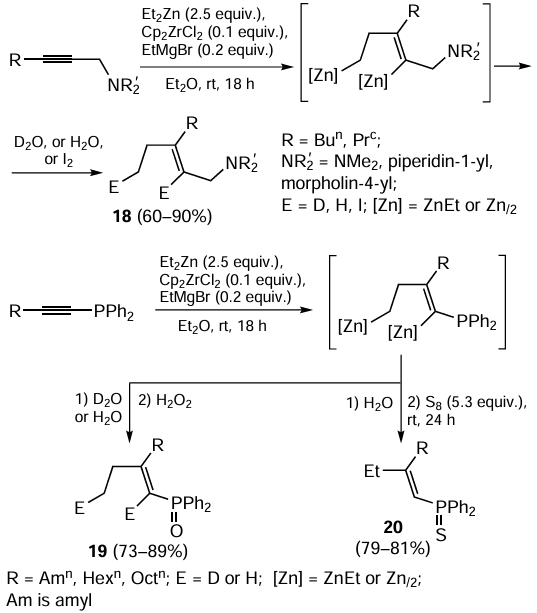
Under similar conditions, 1-alkynylphosphines are converted to phosphorus-containing organozinc compounds.[71] It is known that 1-alkenylphosphines are oxidized with air oxygen to 1-alkenylphosphine oxides.[72] Therefore, the obtained 1-alkenylphosphines were oxidized with an aqueous solution of H2O2 or elemental sulfur to 1-alkenylphosphine oxides 19 and 1-alkenylphosphine sulfides 20.
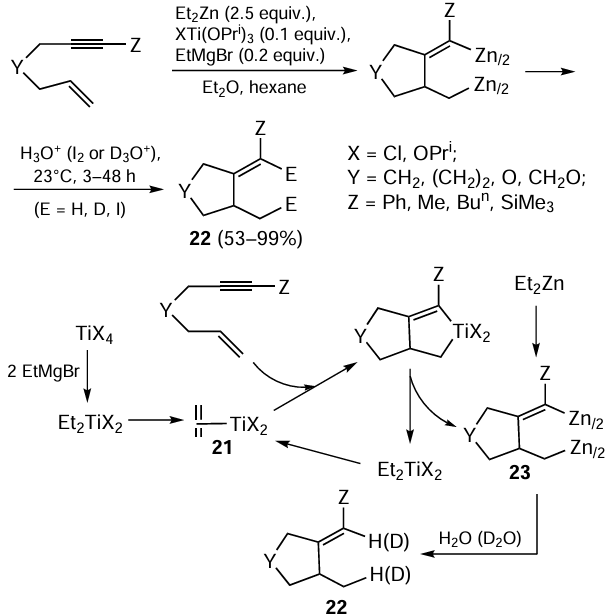
Thus, the formation of the key zirconocene–ethylene complex 15 in the reaction of substituted acetylenes with Et2Zn in the presence of catalytic amounts of Cp2ZrCl2 and EtMgBr promotes the formation of 2-zinco-ethylzincation products. Structurally similar titanium – ethylene complex 21 is formed upon the Kulinkovich reaction of Ti(OPri)4 with EtMgBr.[73] For this reason, it was obvious that the Ti(OPri)4 – EtMgBr system may be active in the carbozincation reaction, like the Cp2ZrCl2 – EtMgBr system noted above. Indeed, in 1998, Negishi and Montchamp reported [74] the first intramolecular cyclization of enynes with Et2Zn in the presence of catalytic amounts of Ti(OPri)4 and EtMgBr, which selectively gave alkylidenecyclanes 22 (Scheme 9). Formally, this reaction can also be considered as the carbozincation of the triple bond, because carbon and zinc atoms finally add to the triple bond. According to the proposed reaction mechanism,[74] coupling of the multiple bonds of the enyne molecule involving low-valent titanium – ethylene complex 21 leads to bicyclic titanacyclopentаdiene. The subsequent transmetallation of this product on treatment with Et2Zn, with the titanium atom being replaced by a zinc atom, yields carbozincation product 23. It is noteworthy that the presence of an oxygen atom in the enyne substrate molecule does not prevent its intramolecular cyclization.
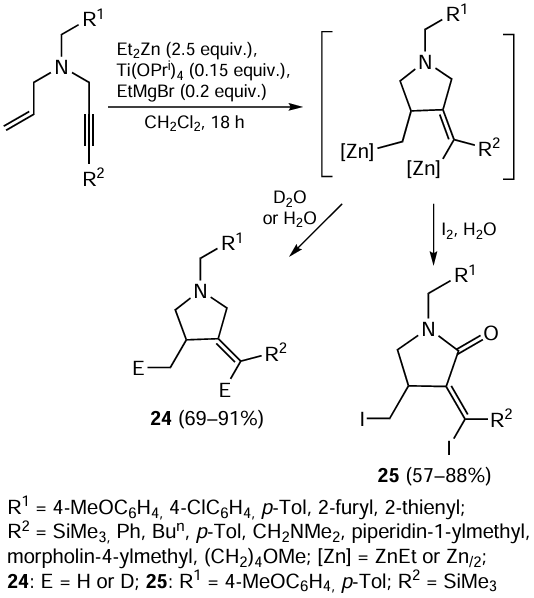
On treatment with Et2Zn and catalytic amounts of Ti(OPri)4 and EtMgBr in dichloromethane at room temperature followed by hydrolysis or deuterolysis, N-allyl-substituted propargylamines are converted to (Z)-methylenepyrrolidines 24 in good yields (Scheme 10).[75] If iodinolysis of the reaction mixture containing the organozinc intermediate is carried out instead of the hydrolysis, diiodinated pyrrolidin-2-ones 25 are formed as a result of oxidation involving the α-carbon atom of the pyrrolidine ring. The Ti–Mg-catalyzed heterocyclization of N-benzyl-N-(but-3-en-1-yl)hept-2-yn-1-amine with Et2Zn was used to obtain (Z)-1-benzyl-4-methyl-3-pentylidenepiperidine.[76]
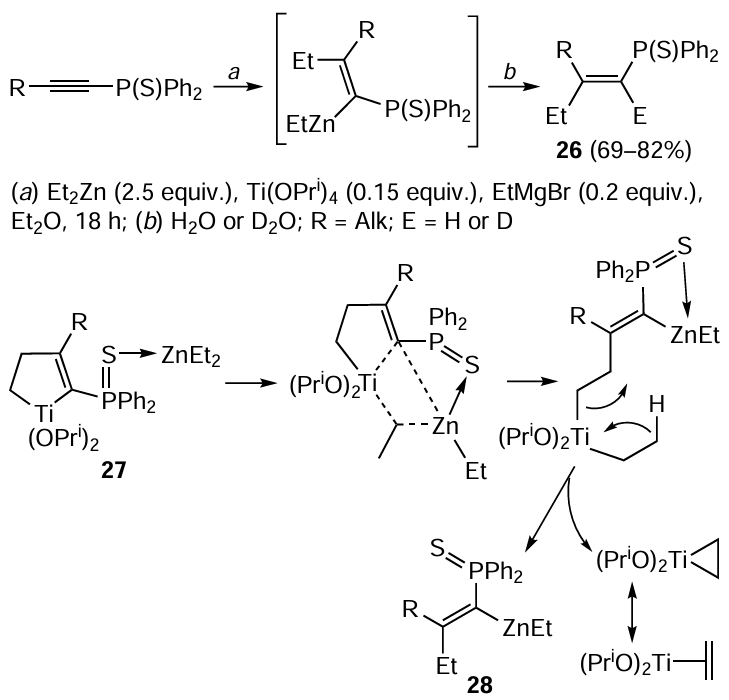
The Ti(OPri)4 – EtMgBr catalytic system is active towards the carbozincation of 2-alkynylamines and 1-alkynylphosphines on treatment with Et2Zn. As in the case of the Cp2ZrCl2 – EtMgBr system, this reaction leads to regio- and stereoselective (the ratio of the syn- and anti-addition products exceeds 95 : 5) formation of nitrogen- and phosphorus-containing olefins 18 – 20.[77, 78] The reaction presumably involves the formation of low-valent titanium – ethylene complex 21 by a mechanism similar to that depicted in Scheme 9. However, in the case of 1-alkynylphosphine sulfides, the conversion follows the ethylzincation pathway and gives, after hydrolysis or deutrolysis, ethyl-substituted olefin 26 (Scheme 11).[78] The authors believe that the presence of the sulfur atom at phosphorus facilitates the ligand exchange between Et2Zn and titanacyclopentene, which is formed upon the reaction of titanium – ethylene complex 21 with the acetylene derivative. The subsequent intramolecular disproportionation of alkenylethyl-titanium intermediate 27 is accompanied by regeneration of the titanium – ethylene complex and formation of ethylzincation product 28. The formation of a similar bimetallic complex was postulated in the Zr-catalyzed ethylmagnesiation reaction of non-activated olefins.[79]
4. Catalysis by iron complexes
The carbozincation of acetylenic compounds catalyzed by iron salts or complexes has been little studied, being a new area of metal complex catalysis. In 2021, regio- and stereoselective vinylzincation of terminal alkynes was performed, with the ratio of syn- and anti-addition products exceeding 95 : 5. As the reagent, divinyl organozinc complex 29 was used in the presence of iron-containing catalyst 30 stabilized by tridentate 1,10-phenanthrolinimine ligand; the reaction gave substituted dienes 31 in a yield of up to 98% (Scheme 12).[80]
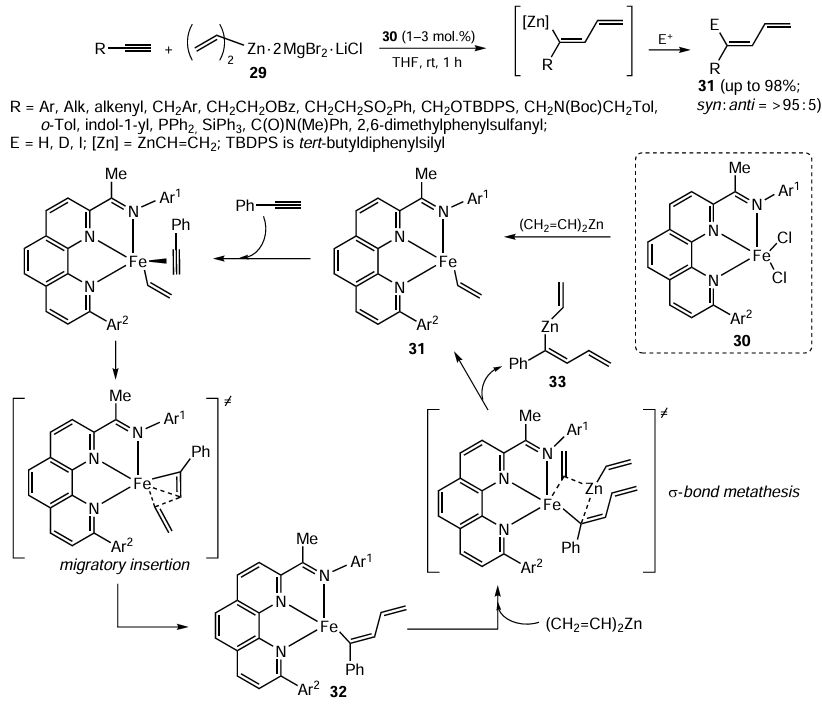
The developed method is characterized by high chemo-, regio-, and stereoselectivity of the formation of carbozincation products and high tolerance to the presence of N-, O-, S-, Si-, and P-containing groups in the acetylenic substrate. When FeCl2 is used as a catalyst in the absence of 1,10-phenanthrolinimine ligand, the reaction is non-selective and, together with various carbozincation products, gives by-products resulting from polymerization of acetylenic compounds and product mixtures difficult to analyze. The putative mechanism of the iron-catalyzed carbozincation includes reduction of the initial iron(II) complex 30 to the catalytically active vinylated iron(I) species 31. The coordination of the terminal acetylene molecule to complex 31 gives, after the migratory insertion step, organoiron intermediate 32. The subsequent transmetallation of this intermediate with divinylzinc via σ-bond metathesis step furnishes the alkyne carbozincation product, compound 33.
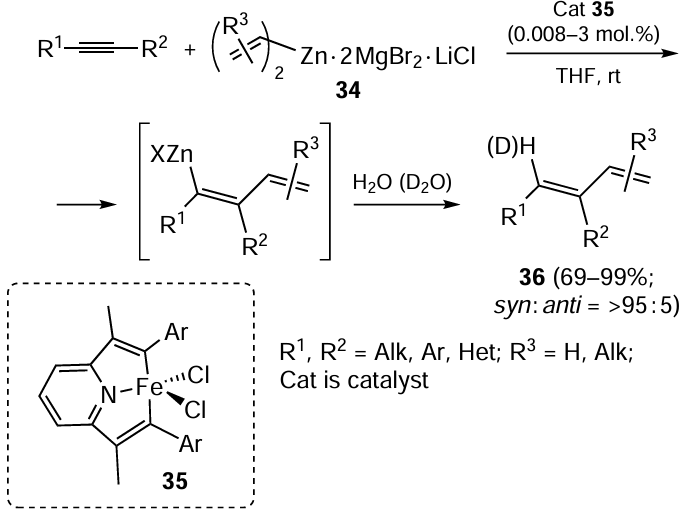
A similar strategy was implemented to perform regio- and stereoselective (with a content of syn-addition product of > 95%) Fe-catalyzed alkenylzincation of substituted acetylenes with complex 34, incorporating dialkenylzinc, MgBr2, and LiCl (Scheme 13).[81] Catalyst 35 represented a complex of FeCl2 with 2,6-bis[1-(phenylimino)ethyl]pyridine. The proposed approach is suitable for carbozincation of a broad range of terminal aryl-substituted acetylenes, unsymmetrical diaryl- and dialkylacetylenes and thus provides the synthesis of conjugated buta-1,3-diene derivatives 36.
An unusual version of carbozincation of terminal acetylenes was implemented with participation of alkyl iodides, bromides, and tosylates in the presence of zinc and catalytic amounts of FeBr2 and I2 (Scheme 14).[82-85] The reaction affords 1,2-disubstituted olefins 37 with Z-configuration of the carbon – carbon bond. The role of iodine is to activate zinc; it can be replaced by Me3SiI or CuBr2 (10 mol.%). This reaction was successfully performed for aryl-substituted terminal acetylenes containing various functional groups in the aryl substituent, e.g., dimethylamine, trifluoromethyl, thiomethyl, or thiophenyl group. The presence of a halogen atom (bromine, chlorine, fluorine) in the aryl moiety of arylacetylene does not prevent the reaction. However, when some functional group such as carbonyl, amide, ester, aldehyde, or cyanide group is present in the acetylenic substrate molecule, the yield of the carbozincation product decreases to 32 – 58% and the stereoselectivity is impaired.
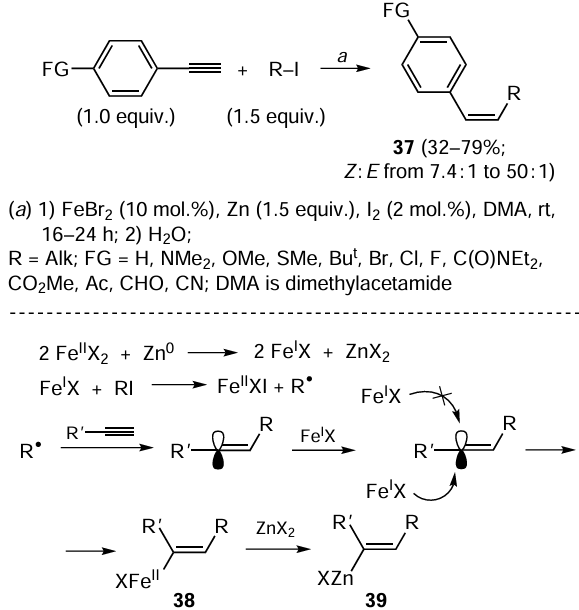
Presumably, the reaction is initiated by reduction of FeBr2 with zinc to give FeBr (see Scheme 14).[82, 86] The subsequent reaction of FeBr with alkyl iodide gives the alkyl radical, which reacts with terminal acetylene to give a substituted vinyl radical. The reaction of the formed radical with FeBr is controlled by steric factors and affords organoiron intermediate 38, which is transmetallated under the action of ZnX2 to give carbozincation product 39.
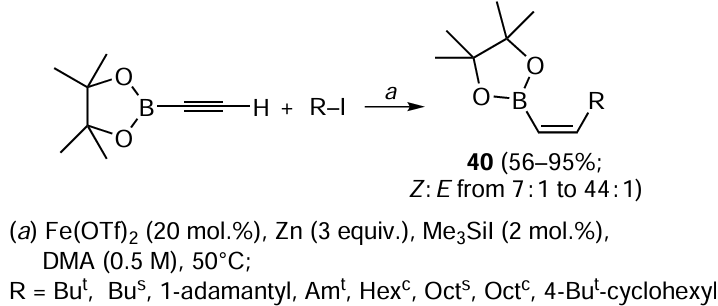
The developed strategy formed the basis for a new method of the synthesis of (Z)-1-alkenylboronates 40 by carbozincation of 1-ethynylboronate with various secondary and tertiary alkyl iodides and zinc in the presence of catalytic amounts of Fe(OTf)2 and Me3SiI (Scheme 15).[87] The model reaction of tert-butyl iodide with FeBr2 and I2 activator resulted in much poorer yields and stereoselectivities: 48% yield and Z : E = 12 : 1 vs. 95% yield and Z : E = 44 : 1 in the former case.
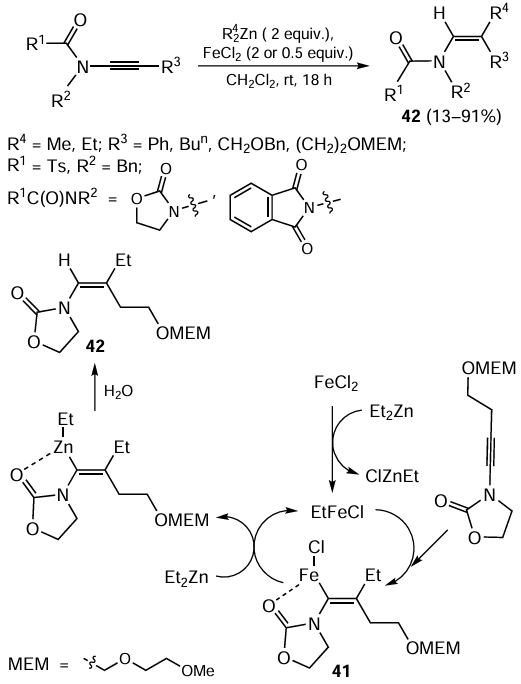
The carbometallation of ynamides is the most convenient and versatile method for the synthesis of enamides and selective synthesis of amino-functionalized olefins. Enamides are used in organic synthesis as useful building blocks for the introduction of nitrogen-containing functional groups into various aromatic or non-aromatic heterocycles. The stereoselective methyl- and ethylzincation of substituted ynamides was performed using dimethyl- or diethylzinc in the presence of a stoichiometric amount of FeCl2 (Scheme 16).[88] The authors detected the formation of only one stereoisomer. They assumed that the reaction proceeds via generation of the ethyliron derivative EtFeCl, which performs the carbometallation of ynamide. The subsequent transmetallation of the formed iron-containing intermediate 41 under the action of Et2Zn affords the carbozincation product, which is hydrolyzed to give substituted enamide 42. This mechanism is similar to that proposed for Cu-catalyzed carbomagnesiation of acetylenic compounds.[89]
5. Catalysis by cobalt complexes
The cobalt-catalyzed carbozincation of acetylenic compounds was performed for the first time by Oshima and co-workers.[90] The arylzincation by this procedure provides stereoselective (> 99%) carbometallation of disubstituted acetylenes by the ArZnI · LiCl complex in acetonitrile at 60°C to give the corresponding arylzincation products 43 in 80 to 91% yields (Scheme 17). The ArZnI · LiCl complex should be prepared in advance as a THF solution using the procedure reported by Krasovskiy et al.[91]
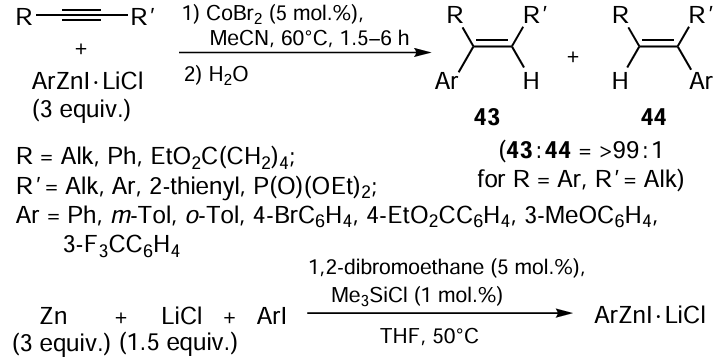
In the case of disubstituted acetylenes in which one substituent is an aryl ring, the carbozincation is not only stereoselective, but also regioselective, with products 43 and 44 being formed in 99 : 1 ratio. The zinc atom adds to the acetylene carbon atom bearing the aryl group. The carbozincation of unsymmetrical dialkyl-substituted and terminal acetylenes gives ~1 : 1 mixtures of regioisomers, which are formed in < 20% yields in the case of terminal acetylenes. Moderate yields of the carbometallation products (53 – 64%) are observed in the case of arylzincation of dodec-6-yne on treatment with the 3-CF3C6H4ZnI · LiCl and 4-BrC6H4ZnI · LiCl complexes containing electron-withdrawing substituents. The reaction is sensitive to steric factors and is hampered when bulky 2-methylphenyl organozinc reagent is used or if 2-methyldec-3-yne with the sterically hindered triple bond serves as the substrate.
This method of carbozincation of acetylenic compounds in the presence of cobalt(II) bromide catalyst has a number of drawbacks. The implementation of this method requires particularly aryl iodide, and the temperature of the synthesis of organozinc compound must be strictly controlled. Furthermore, different solvents are needed for different steps of the reaction, namely, tetrahydrofuran is used for the synthesis of arylzinc, while carbozincation of the acetylenic compound is performed in acetonitrile.
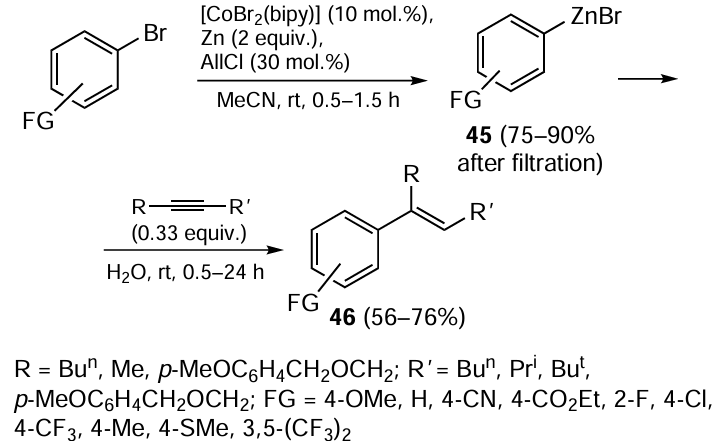
The above drawbacks were eliminated in the procedure proposed in two publications.[92, 93] The procedure includes the synthesis of arylzinc bromide by the reaction of aryl bromide with zinc powder in the presence of allyl chloride and a catalytic amount of the [CoBr2(bipy)] complex (bipy is 2,2'-bipyridine). The filtration of the reaction mixture gives a solution of arylzinc bromide 45 in acetonitrile, which is subsequently used for the carbozincation of alkynes. This procedure was successfully applied for the regio- and stereoselective synthesis (> 99% of the corresponding isomers) of trisubstituted olefins 46 using organozinc reagents containing either electron-donating or electron-withdrawing substituents (Scheme 18). As a result, alkenylaryl derivatives of ethers, esters, nitriles, chlorides, fluorides, and sulfides were obtained. Complex 45 is suitable for the arylzincation of acetylenic compounds with a sterically hindered triple bond. Meanwhile, the above method of Co-catalyzed carbozincation induced by ArZnI · LiCl (see Scheme 17) proved to be inapplicable for the carbometallation of sterically hindered alkynes.[90]
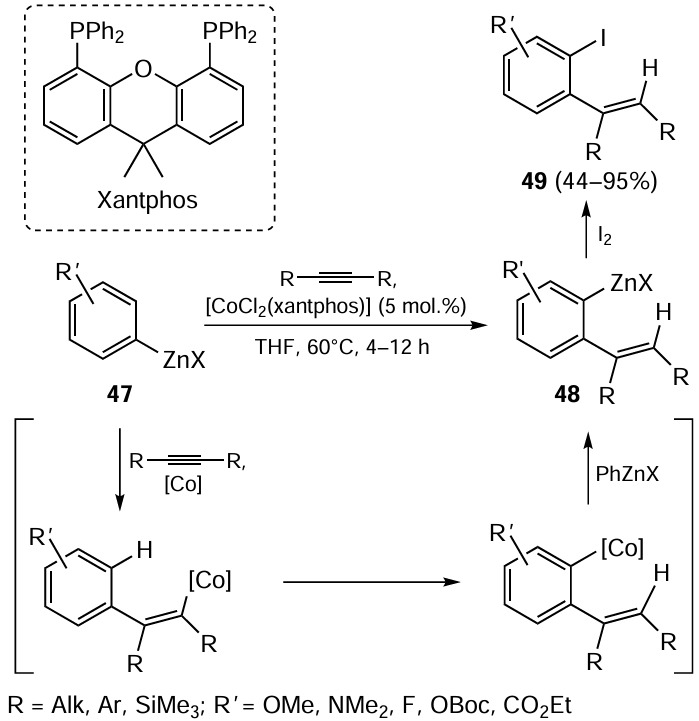
The arylzincation of disubstituted acetylenes with arylzinc halide 47 in the presence of 5 mol.% [CoCl2(xantphos)] in tetrahydrofuran at 60°C is accompanied by activation of the ortho-C – H bond in the benzene ring of the carbometallation product and formation of the organozinc compound 48 (Scheme 19).[94, 95] The subsequent iodinolysis gives arylalkene 49 with a iodine atom in the aryl substituent. For stereoselective transformation of arylalkylacetylenes into arylolefins 49 (isomer ratio E : Z = > 50 : 1), 10 mol.% P(OPh)3 is used as a co-ligand.[94] In the absence of the co-ligand, the ratio of E- to Z-isomer in the reaction product ranges from 6 : 4 to 7 : 3. In the presence of P(OPh)3 , the arylzincation of diphenylacetylene is also non-selective (E : Z = 6 : 4). The Co-catalyzed arylzincation cannot be performed for terminal acetylenes (phenylacetylenes, oct-1-yne) and sterically hindered bis(trimethylsilyl)acetylene.
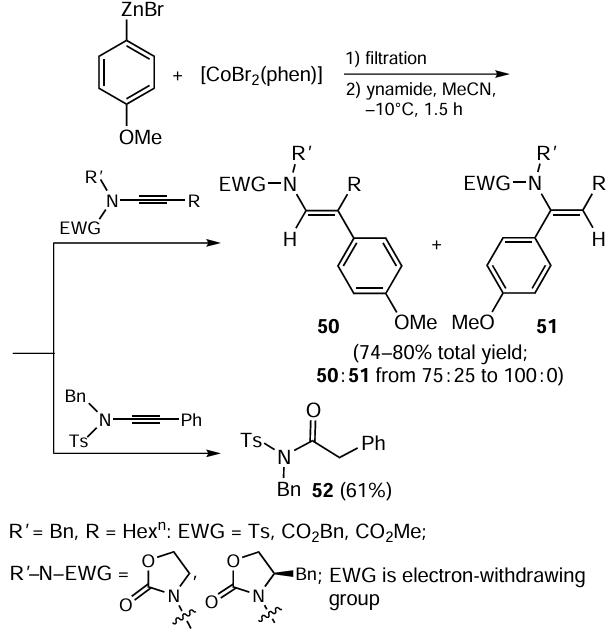
Ynamides react with arylzinc bromides (e.g., p-anisylzinc bromide) in the presence of 15 mol.% [CoBr2(phen)] (phen is phenanthroline), formed in situ upon the reaction of CoBr2 with phenanthroline in acetonitrile, to give enamides 50 and 51 (Scheme 20).[96] High yields and high selectivities are inherent in the carbozincation of 4-benzyl-3-(oct-1-yn-1-yl)oxazolidin-2-one, which may be attributable to the presence of the bulky benzyl substituent in the oxazolidine moiety of ynamide. Meanwhile, the presence of the sterically hindered phenyl substituent at the ynamide triple bond (see Scheme 20) prevents the triple bond carbometallation and results in the formation of amide 52 instead of enamide. This product is formed via the hydration under the action of trifluoroacetic acid. The presence of the trifluoroacetic acid in the reaction mixture is due to the conditions of preparation of arylzinc bromide by the [CoBr2(phen)]-catalyzed reaction of aryl bromide with zinc powder in the presence of 15 mol.% trifluoroacetic acid.
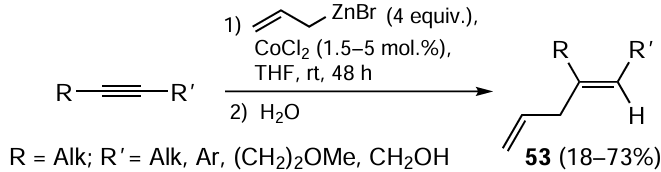
The allylmetallation reactions make an important tool for the synthesis of complex organic molecules with allyl groups.[97, 98] Allyl-substituted olefins 53 were obtained with high regio- and stereoselectivity by the reaction of disubstituted alkynes with 4 equiv. of allylzinc bromide in THF in the presence of catalytic amounts of CoCl2 (Scheme 21).[99, 100] Electron-withdrawing groups in the aryl substituent of the acetylenic substrate are favourable for higher yields of diene 53. This reaction with homopropargyl alcohols furnishes a mixture of two regioisomers in low yields.
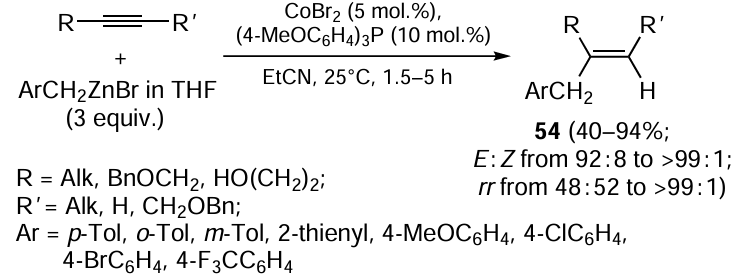
The benzylzincation of symmetrical acetylenes such as oct-4-yne and 1,4-dibenzyloxybut-2-yne with BnZnBr (3 equiv.) in the presence of catalytic amounts of CoBr2 and (4-MeOC6H4)3P results in the formation of trisubstituted olefins 54 in high yields (86 – 94%) and with high stereoselectivities (> 99%) (Scheme 22).[28, 101] High regioselectivity [regioisomeric ratio (rr) of more than 99 : 1] was also observed for the benzylzincation of terminal acetylenes. However, the carbozincation of unsymmetrical dialkyl-substituted acetylenes such as oct-2-yne gives 48 : 52 mixtures of regioisomers. The benzylzincation reaction is sensitive to steric and electronic effects. For instance, carbometallation of sterically hindered 2-methyldec-3-yne with benzylzinc bromide and carbometallation of oct-4-yne with electron-deficient 4-F3CC6H4CH2ZnBr barely take place.
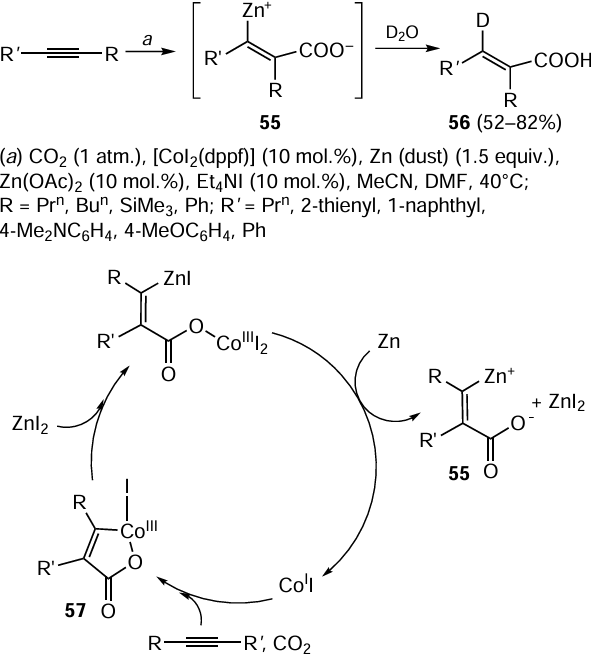
The regio- and stereoselective CoI2(dppf)-catalyzed [dppf is 1,1'-bis(diphenylphosphino)ferrocene] carbozincation of disubstituted acetylenes under the action of CO2 (1 atm) and zinc metal (1.5 equiv.) in the presence of catalytic amounts of Zn(OAc)2 and Et4NI (10 mol.%) can be exemplified by the synthesis of carboxylate-substituted alkenyl organozinc intermediates 55, which are converted to unsaturated carboxylic acids 56 after deuterolysis (Scheme 23).[102] Presumably, the reaction starts with the generation of low-valent CoI(dppf) upon the reaction of [CoI2(dppf)] with zinc.[103] The subsequent coupling of the acetylenic substrate with CO2 involving CoI complex gives cobaltacycle 57, the transmetallation of which via the reaction with ZnI2 and reduction by zinc lead to carbozincation product 55.
6. Catalysis by nickel complexes
Carbozincation of substituted acetylenes catalyzed by nickel complexes is a convenient selective method for the preparation of alkenyl organozinc compounds.[104] The alkylzincation of diphenylacetylene is conducted using 25 mol.% Ni(acac)2 (acac is acetylacetone) in a mixture of THF with N-methyl-2-pyrrolidone (NMP) in 3 : 1 ratio (v/v). The reaction is regio- and stereoselective (98% content of one isomer for both types of selectivity) at a temperature of –35°C within 3 – 18 h to give 1-alkenyl-substituted organozinc compound, which is hydrolyzed to yield substituted stilbenes 58 (Scheme 24).[105, 106]
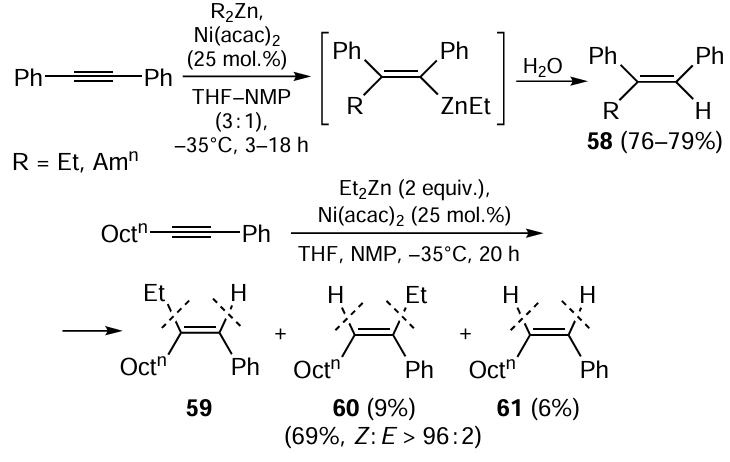
Regarding alkylarylacetylenes, the most regioselective (> 99% content of a single product) carbozincation is observed for methyl- and ethyl-substituted acetylenes. The syn-addition gives the regioisomer in which the zinc atom is attached to the acetylenic carbon atom bearing the aryl group. This attests to the crucial role of the electronic factor in the carbozincation of acetylenic compounds.
As the alkyl chain length increases, the selectivity of the reaction decreases. Indeed, the Ni(acac)2-catalyzed ethylzincation of octylphenylacetylene is non-regioselective. After the hydrolysis of the reaction mixture, a 7 : 1 mixture of two regioisomeric trisubstituted olefins 59 and 60 (70% total yield) and disubstituted alkene 61 (6% yield) are obtained (see Scheme 24).[105] The latter is formed upon the hydrolysis of the product of octylphenylacetylene hydrometallation. Hetarylacetylenes containing 2-thienyl, 5-pyrimidyl, and 2-pyridyl substituents were also introduced in the Ni-catalyzed ethylzincation reaction of disubstituted acetylenes on treatment with Et2Zn.
The addition of phenyl group to alkynes was successfully accomplished using Ni-catalyzed carbozincation with diphenylzinc.[105-107] In particular, a method for the synthesis of (Z)-tamoxifen, an antiestrogen antitumour agent effective for the treatment of metastatic breast cancer, was developed on the basis of the reaction of 1-phenylbut-1-yne with diphenylzinc.[104]
Phenyl-substituted propargyl ethers add organozinc compounds (Et2Zn, Pri2Zn, Ph2Zn) under mild conditions with excellent stereoselectivity (Z : E = 98 : 2) to give, after hydrolysis, (Z)-β-disubstituted allyl ethers.[106] The ratio of the resulting regioisomers is in the range from 90 : 10 to 100 : 0. The reaction carried out at –35°C is usually completed within 1 h. The ethyl- and methylzincation of (trimethylsilyl)phenylacetylene with Et2Zn or Me2Zn gives the regioisomer in which the zinc atom is attached to the acetylenic carbon atom bearing the trimethylsilyl group (Scheme 25).[106] It is noteworthy that the addition of methylcuprates to substituted alkynes is more difficult, requiring in some cases 120 h of the reaction.[108, 109]
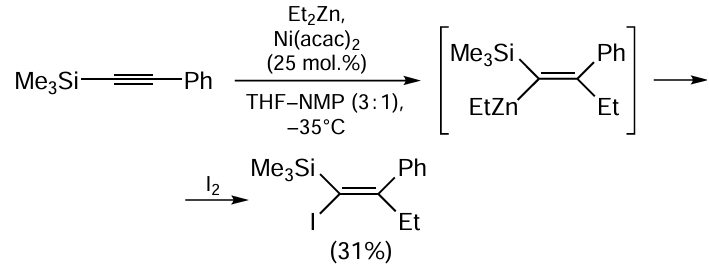
According to the putative mechanism, Ni(асас)2-catalyzed carbozincation of arylacetylenes starts with alkylation or arylation of Ni(acac)2 with dialkyl- or diarylzinc to give nickel-containing intermediate 62 (Scheme 26).[105, 106] The subsequent carbonickelation of arylacetylene with complex 62 results in the formation of alkenyl organonickel intermediate 63. Compound 62 is transmetallated with RZn(acac), which results in regeneration of the Ni(acac)2 catalyst and affords α-arylalkenyl organozinc compound 64.
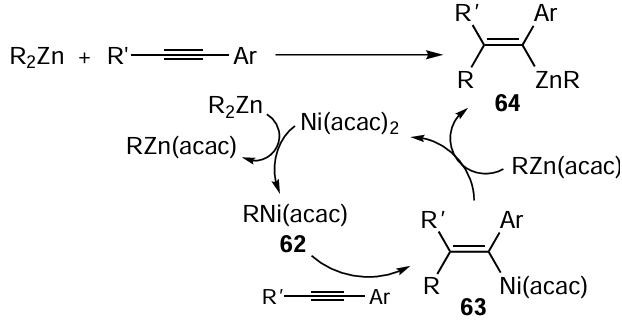
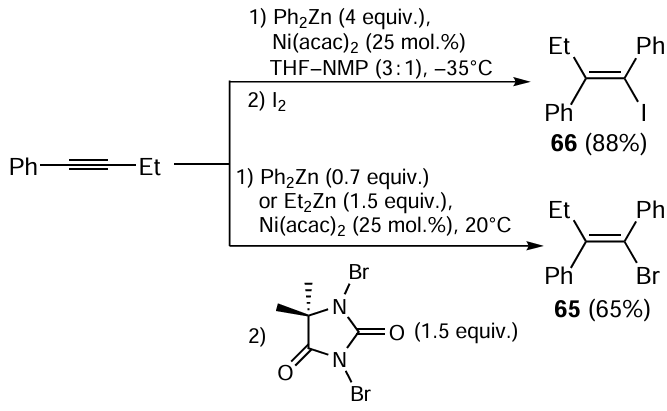
The carbozincation reaction was used for the stereoselective synthesis (97 – 100% content of a single isomer depending on the brominating agent) of (Z)-(1-bromobut-1-ene-1,2-diyl)dibenzene 65, the key intermediate for the preparation of a selective estrogen receptor modulator.[110] When a mixture of Ph2Zn and Et2Zn is used in the reaction in the presence of 25 mol.% Ni(acac)2 (Scheme 27) to obtain (Z)-(1-iodobut-1-ene-1,2-diyl)dibenzene 66, it is possible to decrease the amount of Ph2Zn to 0.7 equiv. instead of 4 equiv. needed in the absence of Et2Zn.[106] The proportion of the ethylzincation product is < 0.1% in this case. The authors attributed this result to the formation of mixed organozinc reagent PhZnEt and to the higher rate of transfer of the phenyl group to the nickel atom compared to the ethyl group.
A series of studies by Montgomery and Oblinger [111-113] demonstrated that Ni(cod)2 (cod is cycloocta-1,5-diene) initiates the intramolecular carbozincation of alkynyl enone with dialkylzinc or alkylzinc chloride (Scheme 28) giving rise to alkylidenecyclopentane compound 67. The key intermediate is the metallacyclopentane species formed as a result of coupling of double and triple carbon–carbon bonds with participation of nickel(0) π-complex.
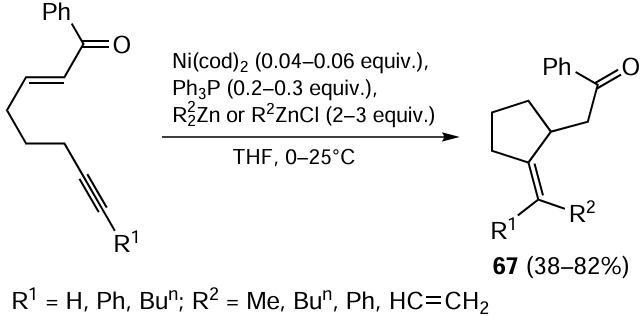
The reaction is exceptionally sensitive to the nature of the organozinc compound and to the presence of PPh3 . The reaction of (E)-1-phenyloct-2-en-7-yn-1-one with Bun2Zn in the absence of PPh3 gives a mixture of compounds 68 and 69 (Scheme 29).[114] In the presence of 25 mol.% PPh3, only 2-(2-methylienecyclopentyl)-1-phenylethan-1-one 69 is formed. In the case of carbocyclization of alkynylenone under the action of Me2Zn without PPh3 , the reaction is chemoselective, with target product 68 being formed in 82% yield.
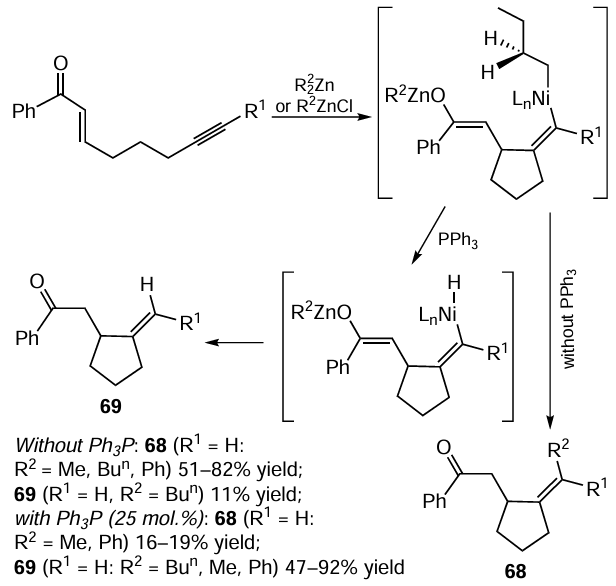
The mechanism of alkynylenone cyclization proposed by the authors (Scheme 30) is similar to the mechanism of enyne cyclization induced with the Ti(OPri)4 – EtMgBr system. In the case of pretreatment of Ni(cod)2 with triphenylphosphine, intermediate 70 is transmetallated with ZnR32 to give bis-zinc complex 71, which is hydrolyzed to give carbocycle 69. When no PPh3 is present in the reaction mixture, intermediate 70 undergoes reductive elimination, resulting in regeneration of LnNi0 and formation of carbocycle 72.
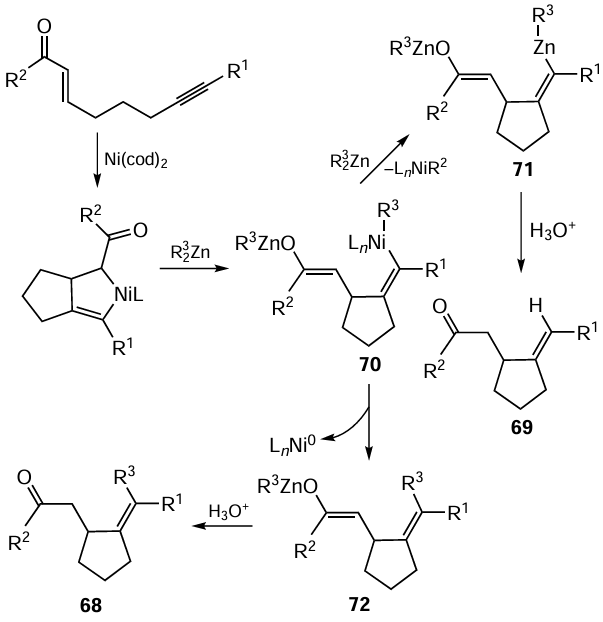
There is an example of intramolecular carbozincation involving the aldehyde group and the triple carbon – carbon bond in 5-ynals. This reaction is accompanied by the stereoselective formation of hydroxyl-substituted alkylidenecyclopentanes 73 (Scheme 31).[113] According to analysis of the product by NMR spectroscopy, the reaction gives only one stereoisomer.
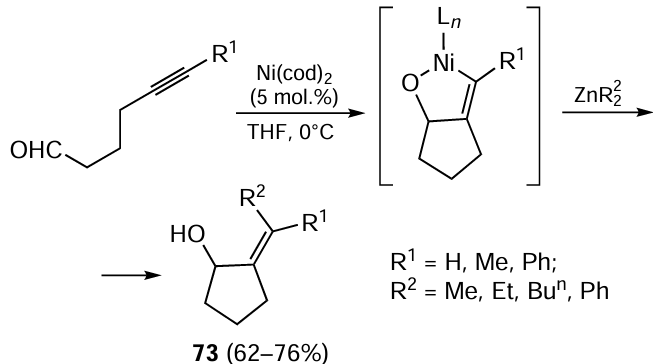
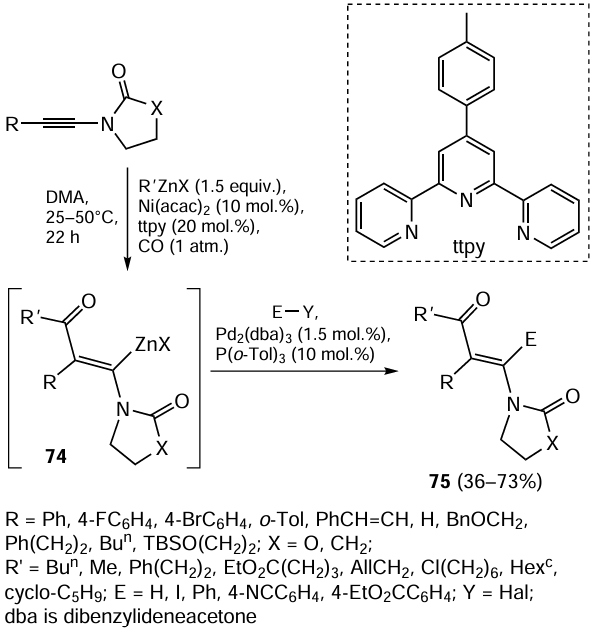
New synthetic potential of carbozincation is demonstrated by combination of Ni-catalyzed acylzincation of alkylzinc halides with ynamides induced by carbon(II) oxide (1 atm) and the subsequent cross-coupling of in situ formed alkenylzinc halides 74 with aryl halides, which affords substituted enones 75 (Scheme 32).[115] The Ni(acac)2 complex with the tridentate 4'-(p-tolyl)-2,2':6',2''-terpyridine (ttpy) ligand is used as the catalyst.
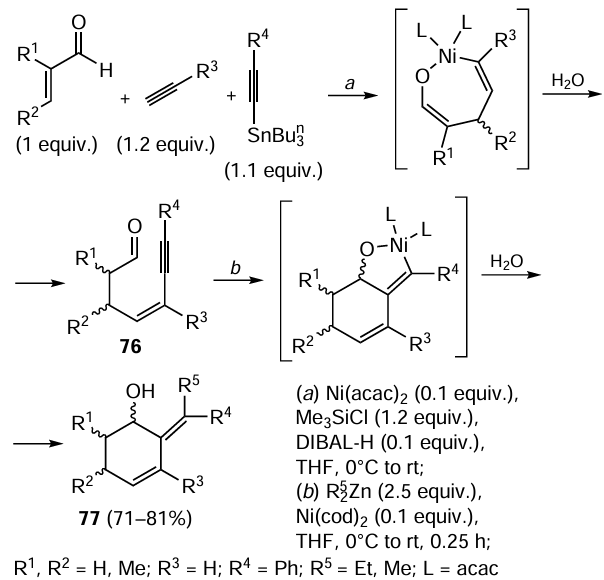
A series of studies [116-118] report coupling of a terminal acetylene, 1-alkynylstannane, and α,β-unsaturated aldehyde in the presence of trimethylchlorosilane induced by catalytically active nickel, generated in situ by the reaction of Ni(acac)2 with diisobutylaluminium hydride (DIBAL-H), which furnishes enynal 76 (Scheme 33). The subsequent reaction of enynal 76 in the presence of 0.1 equiv. of Ni(cod)2 with organozinc reagent, generated in situ by the reaction of 2.5 equiv. of ZnCl2 with organolithium or -magnesium compound, yields cyclohexenol 77.[116] When substituted acroleins (crotonaldehyde and methacrolein) are used, a mixture of diastereomers 77 in a ratio from 1.2 : 1 to 5.3 : 1 is formed. However, the exocyclic double bond formation in compound 77 is highly stereoselective in all cases. The authors did not detect the formation of a reaction product with a different stereoconfiguration of the double bond.
7. Catalysis by rhodium complexes
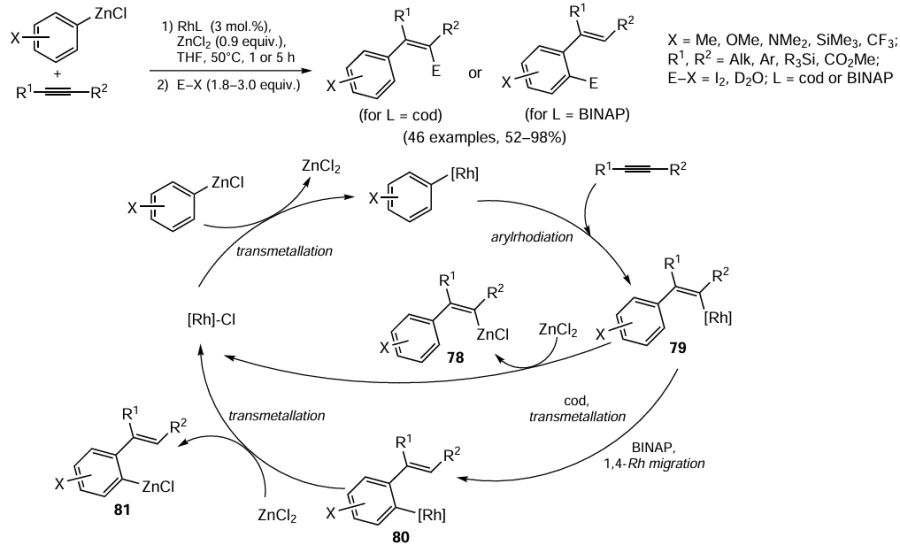
In the rhodium-catalyzed carbozincation of disubstituted acetylenes, an important factor is the ligand environment of the central atom in the catalyst [119, 120] (Scheme 34). When the ligand is cycloocta-1,5-diene, the carbozincation of non-functionalized alkynes affords 2-arylalkenyl-organozinc intermediates 78 via the formation of rhodium-containing intermediate 79 (see Scheme 34). However, when 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl (BINAP) is used as the ligand, Rh-intermediate 79 undergoes 1,4-migration of the rhodium-containing substituent to give intermediate 80, which is converted to ortho-alkenylaryl-organozinc compound 81 upon ZnCl2-induced transmetallation.
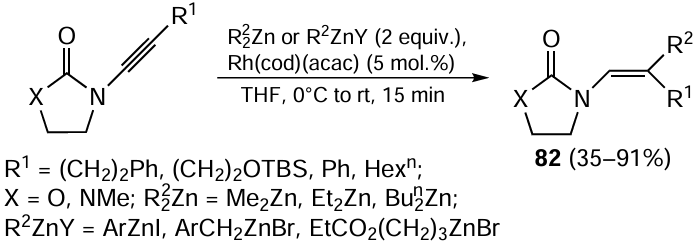
A regio- and stereoselective synthesis of substituted enamides 82 has been developed. The ratio of regioisomers formed in the synthesis is >19 : 1.[121, 122] The Rh(cod)(acac)-catalyzed carbozincation of ynamides containing oxazolidinone, pyrrolidinone, or imidaziolidinone moiety occurs on treatment with 2 equiv. of organozinc reagents in THF (Scheme 35). As organozinc reagents, dialkyl and diaryl zinc derivatives, including functionally substituted ones, were used.
8. Conclusion
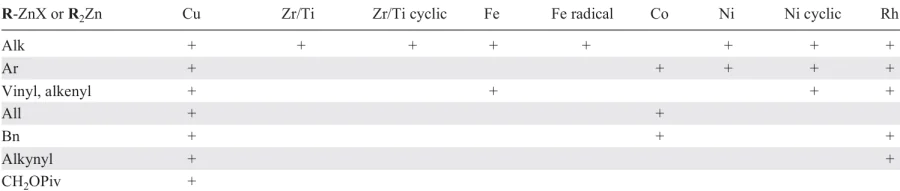
Three main types of mechanisms of catalytic carbozincation (designated as I – III) can be distinguished (Scheme 36). The first type is discussed in detail in the Introduction; the second one is associated with the generation of low-valent transition metal complexes and coupling reaction of the triple bond and another unsaturated bond. As the unsaturated bond, double carbon – carbon bond and aldehyde group were mentioned in the review. The third type mechanism includes generation of an alkyl radical, which reacts with terminal acetylene to give a substituted vinyl radical. All three mechanisms afford the carbometallation products. The review also presents reactions that do not completely fit into this pattern, but include similar carbometallation and transmetallation key steps.
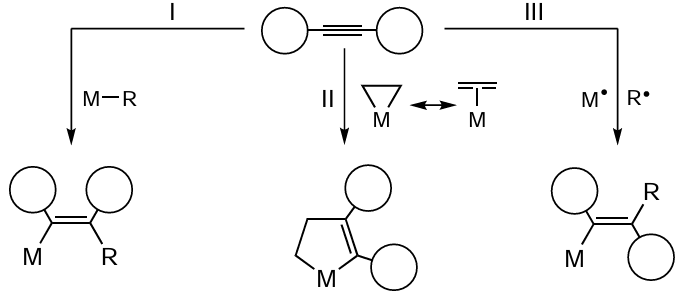
The effect of substituents at the acetylenic carbon atoms on the catalytic carbozincation reaction depending on the catalyst is illustrated in Table 1.* This Table analyzes the reactions that are described in this review. Note that due to the great variety of functionalized alkynes, all substituents in which the functional group is separated from the acetylenic carbon atom by three or more carbon atoms are indicated as alkyls. The columns that give examples of carbozincation by mechanisms II and III are marked by the words ‘cyclic’ and ‘radical’, respectively. The cells containing the ‘plus’ sign correspond to cases for which the corresponding reaction has been reported in the literature, i.e., the products formed as a result of carbozincation or further functionalization have been isolated and characterized. The empty cells correspond to combinations of reactants and catalysts that have not been reported to react to afford the target products. Actually, these are either unexplored reactions or reactions that follow a different pathway and do not give carbozincation products.
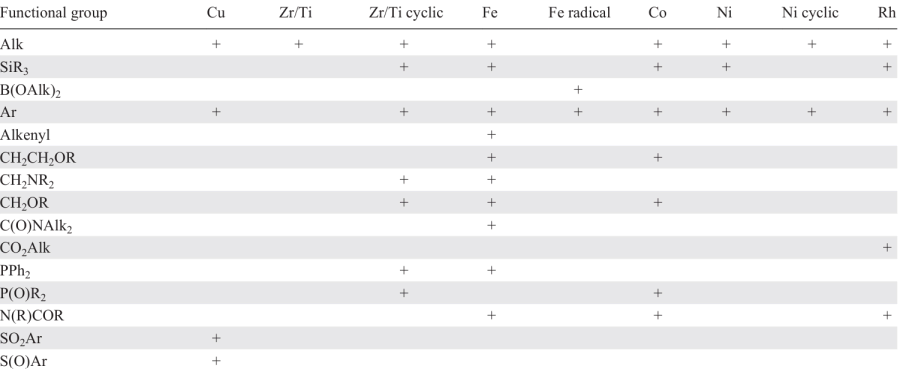
A similar Table was compiled to analyze the effect of substituents in the organozinc compound on the catalytic carbozincation reaction depending on the catalyst (Table 2). As can be seen from these Tables, there are still quite a few unexplored combinations of acetylenic substrates, organozinc compounds, and transition metal-based catalysts. It is worth noting that a systematic approach is applied to study the carbozincation involving iron- and copper-containing catalysts. In the former case, the influence of substituents at the triple bond has been thoroughly investigated; in the latter case, organozinc compounds of various natures have been used for the reaction.
Using Table 1 and Table 2, it is possible to select an appropriate combination of the reagents and the catalyst for a particular substrate. All cases marked by the ‘plus’ sign in these Tables mean that a synthetically available method has been developed. It is obvious that study of certain combinations of reagents and catalysts corresponding to empty cells in Table 1 and Table 2 may give rise to new catalytic approaches to the carbozincation of alkynes.
However, in our opinion, a more interesting and non-trivial task is to develop new methods for the carbozincation of acetylenic compounds coupled with the activation of aldehydes, ketones, nitriles, and small molecules (CO, CO2 , NO, SO2), This approach has good prospects for the synthesis of valuable organic compounds, including pharmaceuticals. This challenge is related to one more important question of why only particular transition metals were have been as carbometallation catalysts.
An expansion of the range of catalytic systems would open up new opportunities for the involvement of various substrates into this reaction. Therefore, one more promising trend in the studies of carbozincation is the search for new effective catalysts for this reaction based on other transition metals. Furthermore, rational design of catalytic systems could make it possible to decrease the catalyst amount to < 1%. The carboalumination and carbomagnesiation reactions alternative to carbozincation are highly potent tools for the formation of new carbon–carbon bonds. Using carbo- and cycloalumination reactions, effective methods for the synthesis of practically important classes of carbo- and metallacarbocycles, spirocarbocycles, and N-, O-, S-, and P-containing heterocycles have been developed,[72, 123-130] and new classes of biologically active terpenoid and steroid compounds have been prepared.[131-134] The carbomagnesiation of alkynes, like carbozincation, is carried out using a broad range of catalytic systems based on transition metals such as copper, silver, titanium, zirconium, chromium, manganese, iron, and nickel.[28] In this series, it is necessary to mention magnesium, a variety of organic derivatives of which are synthetically readily available.
Despite all advantages of carboalumination and carbomagnesiation reactions, the organozinc synthesis remains a widely used tool in the armoury of organic chemistry. This is caused by high tolerance of many functional groups to organozinc reagents, which are synthetically available almost to the same extent as organomagnesium compounds. Owing to the large number of solvents used in the catalytic carbozincation, ranging from dichloromethane to tetrahydrofuran, and to the broad range of catalytic systems considered in this review, this reaction is highly versatile. The subsequent development of efficient catalysts for the carbozincation of unsaturated compounds is of great interest as regards the design of efficient and facile methods for obtaining practically important classes of organic compounds.
* Here and in Table 2, the character R designates alkyl, aryl, benzoyl, alkoxyl, and other groups.
The review was written within the State Assignment of the Ministry of Science and Higher Education of the Russian Federation (subjects FMRS-2025-0029 and FFZZ-2025-0008).
9. List of abbreviations and symbols
The following abbreviations and symbols are used in the review:
acac — acetylacetone,
All — allyl,
Am — amyl (pentyl),
BINAP — 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl,
bipy — 2,2'-bipyridine,
Boc — tert-butoxycarbonyl,
Cat — catalyst,
cod — cycloocta-1,5-diene,
Cp — cyclopentadienyl,
dba — dibenzylideneacetone,
DIBAL-H — diisobutylaluminium hydride,
DMA — dimethylacetamide,
dppf — 1,1'-bis(diphenylphosphino)ferrocene,
E — electrophilic group,
EWG — electron-withdrawing group,
FG — functional group,
Hex — n-hexyl,
MEM — 2-methoxyethoxymethyl,
NMP — N-methyl-2-pyrrolidone,
Oct — octyl,
PivOCH2 — pivaloyloxymethyl,
phen — phenanthroline,
rr — regioisomeric ratio,
rt — room temperature,
TBS — tert-butyldimethylsilyl,
TBDPS — tert-butyldiphenylsilyl,
TfO — trifluoromethanesulfonate (triflate),
Tol — tolyl (methylphenyl),
ttpy — 4'-(p-topyl)-2,2':6',2''-terpyridine,
Ts — p-toluenesulfonyl (tosyl),
xantphos — 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene.
References










