


![13C NMR assignments of kraft and milled wood lignins.[27]](/storage/images/resized/bXULoBnafJZLmtYiJfh7tHtXXF7q01vXc7EIcB0n_xl.webp)
Keywords
Abstract
Present review focuses on the most recent results (the interim of 2019-2023) in a liquid-phase NMR of lignin and the lignin-derived products and related chemicals. Collected and discussed are the recent applications of 1H, 13C, and 31P NMR spectroscopy together with NMR of less popular nuclei to the structural studies and practical recommendations in this field. Owing to the complexity of lignin and the products of its transformation, their NMR spectra consist of a number of overlapping signals belonging to different structural types. In this regard, comprehensive studies of lignin by means of NMR over the past several decades revealed characteristic functional groups of lignin and lignin-related products together with spectral regions in which they resonate. Quantitative NMR spectra of 1H and 13C together with less popular nuclei like 31P are used to characterize different structural units of lignin (such as guacil, syringil, and p-hydroxyphenyl) providing aromatic and saturated carbons spread over many structural moieties.
The bibliography includes 54 references.
1. Introduction
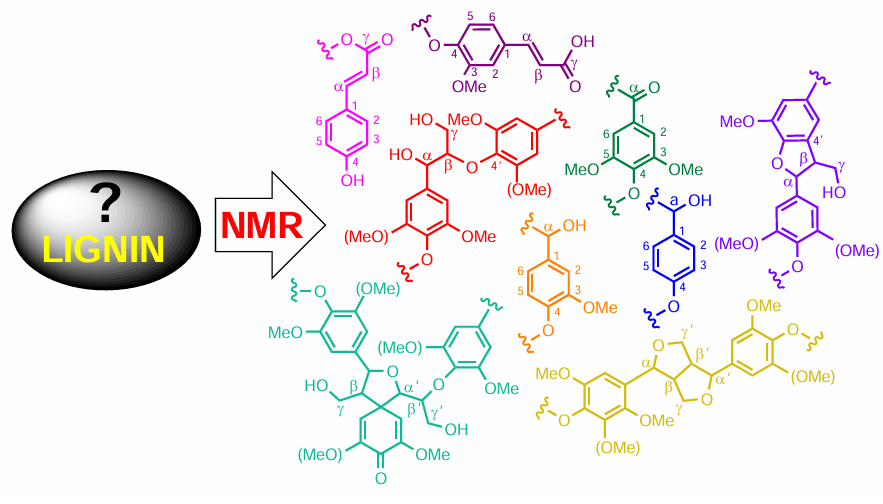
Lignin is a prospective biomass-derived source for the production of some crucial organic building blocks, typically derived from unsustainable and non-renewable petroleum feedstocks.[1] Lignin, a source of plant-based biomass, is a complex cross-linked phenolic polymer, as shown in Fig. 1.[2] As a complex polymer, lignin characterization is often challenging due to its random structure and multitudes of different repeating molecular substructures.
![[{"id":"z6KIGitfuY","type":"paragraph","data":{"text":"Identified structural units of lignin. Reproduced from Balakshin et al.[[ type=\"anchor\" referenceId=\"12325\" ]] under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/5Rvelweu59HaoD6M0CZPBpiUuseJxywQGMqOTUR5_xl.webp)
Identified structural units of lignin are shown in Fig. 1 and characteristic heteronuclear single quantum coherence (HSQC) NMR spectrum of spruce milled wood lignin is presented in FIg. 2. Aromatic structures present in lignin are as follows: syringyl unit, oxidized syringyl unit, guaiacyl unit, p-hydroxyphenyl unit, ferulic acid, cinnamyl alcohol end group, cinnamaldehyde end group, and p-coumaric acid, while the main linkages of lignin are β-aryl-ether β-O-4, resinol β-β, phenylcoumaran β-5 and spirodienone.
![[{"id":"3VBg21wtox","type":"paragraph","data":{"text":" Different lignin branching/cross-linking units: (<i>1</i>) etherified 4-O-5', (<i>2</i>) etherified 5-5', (<i>3</i>) dibenzodioxocin (DBDO), (<i>4</i>) α-O-Alk/β-O-4, and (<i>5</i>) γ-O-Alk/β-O-4. Given in the lower part is the HSQC NMR spectrum of spruce milled wood lignin in DMSO-<i>d</i><sub>6</sub>: (<i>a</i>) side chain region; (<i>b</i>) aromatic region. Reproduced from Balakshin et al.<sup>2</sup> under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/GqeMZRb7qOyCBfEtAqvYMl2WlHfykI66HTbfXhiV_xl.webp)
This polymer is crucial to the structural integrity of cell walls in wood and bark and has been extensively studied. As a by-product of paper production, lignin is used for fast energy generation. However, lignin proved to be an exceptional source of organic building blocks, many of which are currently obtained from petroleum feedstocks. Lignin, a natural biopolymer and an abundant by-product, is a particularly promising feedstock for carbon-based materials and a potentially sustainable alternative to phenolic resins, which are typically derived from crude oil. Among different types of biomass, lignin is the most abundant aromatic biopolymer and a potential source for industrial products.
The structure of lignin is highly dependent on its origin. While softwood lignins contain mainly guaiacyl units with negligible amounts of p-hydroxyphenyl groups, hardwood lignins are composed of guaiacyl and syringyl subunits. Grass lignins are constituted by guaiacyl (G), syringyl (S), and p-hydroxyphenyl (H) subunits.
In general, NMR provides direct information on lignin structure and its chemical composition. In particular, data from quantitative monodimensional 1H and 13C NMR spectra together with heteronuclear and multidimensional NMR data provide information on different types of lignin interunit bondings.[3] Unfortunately, monodimensional spectra suffer from significant signal overlap, which seriously hampers signal integration. Quantitative HSQC has been used to understand lignin framework, providing direct information on internal linkages and also giving general insights into lignin structure based on the early 1H and 13C NMR (see Refs [4-9]) together with 31P NMR (see Ref. [10]) data on modified lignin.
However, although a part of the lignin chemistry community has used HSQC as a quantitative or semi-quantitative tool in recent years, this is not scientifically rigorous and therefore, acceptable due to the intrinsic nature of the pulse sequence. Quantitative 1H – 13C HSQC measurements have recently greatly facilitated lignin analyses. In some cases, however, long acquisition times were needed for obtaining quantitative HSQC, which are incompatible with the chemical integrity of the (potentially functionalized) lignin sample.
Sette et al.[9] compared different methods that were developed for the more time-efficient quantitative HSQC measurements with respect to their usefulness in lignin analysis. As a result, reliable and reproducible results were obtained using both the QQ-HSQC and HSQC0 methods. Indeed, an obstacle to the development of a reliable relationship between lignin structure and upgradeability is the difficulty to quantitatively measure lignin structural features by means of classical HSQC. Amiri et al.[11] demonstrated that a modified HSQC method known as HSQC0, can accurately quantify lignin functionalities in extracted lignin. In 2024, Bourmaud et al.[12] developed a rapid elevated-temperature 1H – 13C heteronuclear single-quantum coherence zero (HSQC0) method that enables a precise quantification of the kraft lignin structural characteristics even with whole plant cell wall (known as ‘HSQC0 WPCW’). Such analysis illustrates how this analytical method can greatly facilitate the study of kraft lignin structure, which can then be used for the fundamental studies of lignin.
Recent applications of 1H, 13C, and 31P NMR spectroscopy together with NMR of less popular nuclei like 31P, 19F, and 15N to the structural studies of lignin provide a breakthrough and practical guide in this field. The structural NMR studies of lignin are developing on a backdrop of a marked progress in computational NMR of hydrogen [13-15] and carbon,[16-20] the elements which are the main constituents of lignin, together with NMR of phosphorous,[21][22] provided lignin is labelled with 31P isotope to offer lignin analysis by means of 31P NMR.
2. Most recent NMR studies of lignin and lignin-derived products
2.1. 1H and 13C one-dimensional NMR
A representative example of NMR studies of lignin carried out in the last five years is the paper by Levdansky et al.,[23] which deals with the NMR study of the soluble organosolv lignins extracted from fir and aspen wood. Fir and aspen lignins were found to be of the G and GS types, respectively. The one-dimensional 1H and 13C NMR spectra of these samples are illustrated in Fig. 3 and Fig. 4, respectively. The authors also found that the β-O-4' linkages in ethanol lignins extracted from fir and aspen were partially ethoxylated and contained the ethoxy groups in the α-position.
![[{"id":"PuOrpufs6s","type":"paragraph","data":{"text":" <sup>1</sup>H NMR spectra of ethanol lignins extracted from as pen (<i>a</i>) and fir (<i>b</i>). All <sup>1</sup>H NMR spectra were recorded on a Bruker Avance 600 NMR spectrometer operating at 600 MHz in DMSO-<i>d</i><sub>6</sub>. Reproduced with minor editing privileges from Levdansky et al.[[ type=\"anchor\" referenceId=\"12341\" ]] with the permission of the Siberian Federal University."}}]](/storage/images/resized/92VQKKztq7fPcBIhfwz9vuSA5GeJcvwLlxvEVL0v_xl.webp)
![[{"id":"hXebAuq6AB","type":"paragraph","data":{"text":"<sup>13</sup>C NMR spectra of ethanol lignins extracted from aspen (<i>a</i>) and fir (<i>b</i>). All <sup>13</sup>C NMR spectra were recorded on a Bruker Avance 600 NMR spectrometer operating at 150 MHz in DMSO-<i>d</i><sub>6</sub>. Reproduced with minor editing privileges from Levdansky et al.[[ type=\"anchor\" referenceId=\"12341\" ]] with the permission of the Siberian Federal University."}}]](/storage/images/resized/KoHT4JTbeKWU9v8r5uruaNHPlSlrAivDYEi1FKQ8_xl.webp)
Hopa and Fatehi [24] studied sulfoalkylated lignin derivatives obtained by sulfomethylation and sulfobutylation of kraft lignin. The 1H NMR spectra of kraft lignin and its sulfomethylated and sulfobutylated derivatives, shown in Fig. 5, were used to identify those structures. In the spectrum of kraft lignin, two broad peaks at 6.8 – 7.5 and 3.2 – 3.8 ppm were attributed to the protons of aromatic ring and methoxy groups, respectively. In the spectrum of sulfomethylated lignin, these peaks were found to be much weaker than those of kraft lignin due to the cleavage of aromatic ring and methoxy groups during sulfomethylation. The peak at 8.2 ppm (assigned by the authors to proton A) represented the hydrogen of the phenolic hydroxyls. In the spectrum of sulfomethylated lignin, the peak at 3.23 ppm (assigned to proton B) was attributed to the signal of the methylene groups. In the spectrum of sulfobutylated lignin, three peaks at 1.79, 1.58, and 2.63 ppm were assigned to C, D, and E protons, respectively, which were absent in the spectrum of the starting kraft lignin.
![[{"id":"Zz0LTmIwg_","type":"paragraph","data":{"text":"<sup>1</sup>H NMR spectra of kraft lignin (<b>KL</b>), sulfomethylated lignin (<b>SML</b>), and sulfobutylated lignin (<b>SBL</b>) recorded on the INOVA-500 NMR spectrometer (Varian) operating at 500 MHz in DMSO-<i>d</i><sub>6</sub>. TSP = Me<sub>3</sub>Si(CH<sub>2</sub>)<sub>2</sub>CO<sub>2</sub>H. Reproduced from Hopa and Fatehi[[ type=\"anchor\" referenceId=\"12342\" ]] under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/8SaEpmurHjPDMXqjmP89wI2ESKzLfUIokD7kOV4N_xl.webp)
In 2023, Zhao et al.[25] prepared a novel cationic tall oil lignin (TOL)-starch copolymer by the polymerization of TOL, starch and a cationic monomer of (3-acrylamidopropyl) trimethyl ammonium chloride (APTAC) through the free radical chain extension. Based on the 1H NMR and 1H – 1H Correlated Spectroscopy (COSY) experiments, it was found that TOL and starch were covalently linked by the APTAC to produce corresponding cationic copolymers. One-dimensional and 1H – 1H COSY spectra of the cationic copolymers prepared with a ratio of APTAC to TOL of 1 or 5 in the neutral or basic reaction medium (designated as CR1-pH7, CR1-pH11, CR5-pH7, and CR5-pH11, respectively) together with the pristine starch and TOL are shown in Fig. 6 and Fig. 7 as examples. For the origin and numbering of the fractions, see the original publication. In general, it was shown that the structure of cationic copolymers comprised anhydroglucose unit of starch, the phenolic structure of TOL, and the APTAC monomer. It was also found that the phenolic hydroxyl content of TOL was reduced by the reaction of the APTAC chain with the phenolic OH of TOL, significantly increasing the solubility, charge density, and molecular weight of the cationic copolymers.
![[{"id":"ZpNKsSIRss","type":"paragraph","data":{"text":"<sup>1</sup>H NMR spectra of cationic <b>TOL</b>–starch copolymers, starch and <b>TOL</b> recorded on the AVANCE NEO-1.2 GHz spectrometer (Bruker, USA) operating at 1.2 GHz in DMSO-<i>d</i><sub>6</sub>. Int. Std. is internal standard. Reproduced from Zhao et al.[[ type=\"anchor\" referenceId=\"12343\" ]] with the permission of Elsevier."}}]](/storage/images/resized/1oVxvLRUme3ABenb6SzqJ8v4kplxTRTMipYH3RAN_xl.webp)
![[{"id":"CWy31EEbDh","type":"paragraph","data":{"text":"2D <sup>1</sup>H – <sup>1</sup>H COSY spectra of cationic <b>TOL</b>–starch copolymers recorded on the AVANCE NEO-1.2 GHz spectrometer (Bruker, USA) operating at 1.2 GHz in DMSO-<i>d</i><sub>6</sub>. Reproduced from Zhao et al.[[ type=\"anchor\" referenceId=\"12343\" ]] with the permission of Elsevier."}}]](/storage/images/resized/ow8hOQBCxf2tytvi58jvzniZfpK4gGZPxVmT6LYC_xl.webp)
In 2023, Wang et al.[26] explored the free-radical in situ copolymerization study of lignin, acrylamide, and diallyldimethylammonium chloride by a combination NMR with other methods. It was shown that the presence of acrylamide in that reaction was essential for the polymerization of diallyldimethylammonium chloride with lignin to proceed due to the lower steric hindrance of the former (Fig. 8). The monomer conversion ratio and dynamic rheology of the reaction system indicated that lignin acted as an inhibitor in the corresponding copolymerization reaction.
![[{"id":"YTcrXpxzXo","type":"paragraph","data":{"text":"Copolymerization scheme of lignin, acrylamide, and diallyldimethylammonium chloride monitored by NMR. Reproduced from Wang et al.[[ type=\"anchor\" referenceId=\"12344\" ]] under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/Dj5fePSLvTHuAEhERAIzxO0sDRPEOQFBg9ztzTfC_xl.webp)
Mun et al.[27] studied the samples of kraft lignin from mixed hardwoods by 1H and 13C NMR. It is well known that the basic aromatic structure of hardwood lignin consists of guaiacyl (G) and syringyl (S) units, both of which can be seen in the 1H NMR spectra of acetylated kraft lignin (Ac-KL) and most clearly in the spectra of acetylated milled wood lignins (Ac-MWL) extracted from acacia (Ac-MWL-aca) and mixed hardwood (Ac-MWL-mhw) (see the marked yellow region in Fig. 9). The intensities of aromatic protons in Ac-KL were 0.52 for S and 0.37 for G units, while for Ac-MWL-mhw, the aromatic proton intensities were accordingly 0.84 for S and 0.57 for G units. Based on the 1H NMR spectral intensities of Ac-KL and Ac-MWLs, the authors detected the decreased number of aromatic protons, suggesting a highly condensed structure of KL. As shown in Table 1, the S/G molar ratio of Ac-KL was almost similar to that of Ac-MWL-mhw.
![<sup>1</sup>H NMR assignments and fractions of protons of acetylated kraft and milled wood lignins.[<a class="anchor" href="#reference-12345">27</a>]](/storage/images/resized/Srl9BsWDXdCQpQ7I7FiqLJeoWa5p5vTpfMkBt1j5_xl.webp)
![[{"id":"2maONk9K87","type":"paragraph","data":{"text":" <sup>1</sup>H NMR spectra of acetylated kraft (<b>Ac-KL</b>) and milled wood (<b>Ac-MWL</b>) lignins recorded on a JNM-ECZ500R NMR spectrometer (JEOL, Japan) operating at 500 MHz in CDCl<sub>3</sub>. Reproduced from Mun et al.[[ type=\"anchor\" referenceId=\"12345\" ]] under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/EQ2SF3NEatiaWeyVZwQivu0hUWWpNQsMoEGHPENu_xl.webp)
The 13C NMR spectra of KL and milled wood lignin (MWL) from acacia and mixed hardwood are presented in FIg. 10. The corresponding chemical shifts and signal intensities are listed inTable 2. The 13C NMR spectra of MWL-aca and MWL-mhw in aliphatic and aromatic regions were found to be essentially similar. However, the 13C NMR spectrum of KL showed decreased aliphatic and increased aromatic content. This phenomenon was typical for KL since significant structural changes occurred in the aliphatic and aromatic moieties of lignin during kraft pulping. In addition, demethylation of methoxyl groups and cleavage along Cγ leading to polycondensation resulted in a marked difference between KL and MWL spectra, as was claimed by the authors as one of the main results of their study.
![[{"id":"SE1_8jE5bu","type":"paragraph","data":{"text":" <sup>13</sup>C NMR spectra of kraft lignin and milled wood lignin recorded on a JNM-ECZ500R NMR spectrometer (JEOL, Japan) operating at 125 MHz in CDCl<sub>3</sub>. List of substructures <b>1 – 5</b> and side chains <b>6 – 15</b> of lignin and numbering of carbon atoms in the base fragment (highlighted with dotted lines). Reproduced from Mun et al.[[ type=\"anchor\" referenceId=\"12345\" ]] under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/oPGBdQraN77NlySy3fUpQLJopwHn1DQZ8KOmfASP_xl.webp)
Rönnols et al.[28] discussed the prospects for lignin analysis by the low-field benchtop NMR spectroscopy. It is well known that benchtop NMR spectroscopy is an emerging field with an appealing profile for analytical analysis of lignin. The instrumentation of the benchtop NMR spectroscopy offers the possibility to measure NMR spectra in situations where the use of the high-field NMR spectroscopy is too expensive or unavailable. In that paper, the ability to quantify different lignin samples using benchtop NMR was found to be rather promising. For example, the 1H NMR spectra of isolated lignin samples recorded at benchtop low and high magnetic fields showed three major peaks arising from aromatic and oxygenated aliphatic compounds together with that of the residual solvent with the corresponding spectral regions being clearly separated (Fig. 11).
![[{"id":"z9y5NSn1Nz","type":"paragraph","data":{"text":"<sup>1</sup>H NMR spectra of the isolated lignin samples recorded at low and high magnetic field strengths (43 and 60 MHz): the hardwood lignin sample at low field (<i>a</i>) and high field (<i>c</i>); softwood kraft lignin sample at low field (<i>b</i>) and high field (<i>d</i>). Spectra were recorded in DMSO-<i>d</i><sub>6</sub> using two benchtop NMR systems, Spinsolve 43 MHz Diffusion and Spinsolve 60 MHz Ultra (Magritek, Aachen, Germany), generating magnetic field strengths of 1 and 1.5 T, respectively. Reproduced from Rönnols et al.[[ type=\"anchor\" referenceId=\"12346\" ]] with the permission of Walter/De Gruyter GMBH & Co, KG."}}]](/storage/images/resized/iQuWwtt0vU7JLAQ80WxCMoqb9mwSvEUbWM2HPGv2_xl.webp)
2.2. 1H and 13C two-dimensional NMR
Two-dimensional NMR of lignin is currently used as either a qualitative or a semi-quantiative technique. An example of this is the fundamental study by Levdansky et al.,[23] who used HSQC spectra to confirm the presence of the β-O-4', β–β', and β-5' linkages in the organosolv lignin extracted from fir and aspen, as shown in Fig. 12 and Fig. 13. The main structural units of these ethanol lignins are also presented (see Fig. 12).
![[{"id":"kF6ls9PEf1","type":"paragraph","data":{"text":"Aliphatic region of the HSQC spectra of ethanol lignins extracted from fir (<i>a</i>) and aspen (<i>b</i>). All <sup>1</sup>H/<sup>13</sup>C NMR spectra were recorded on a Bruker Avance 600 NMR spectrometer operating at 600/150 MHz in DMSO-<i>d</i><sub>6</sub>. The main structural units of ethanol lignins extracted from fir and aspen wood: <b>A</b> is β-aryl ethers, <b>A'</b> is α-ethoxylated β-aryl ethers, <b>B</b> is pinoresinol, <b>C</b> is phenylcoumarans, <b>I</b> is terminal cinnamic alcohol groups, <b>J</b> is terminal cinnamic aldehyde groups, <b>pCA</b> is p-coumarates, <b>pBA</b> is p-hydroxybenzoates, <b>St</b> is stilbenes, <b>G</b> is guaiacyl units, <b>S</b> is syringyl units, <b>S'</b> and <b>S''</b> is oxidized syringyl units (<i>c</i>). Reproduced from Levdansky et al.[[ type=\"anchor\" referenceId=\"12346\" ]] with the permission of the Siberian Federal University."}}]](/storage/images/resized/xJimLRAtpGhs2IeqdtYc7KPphdVBWO6TB3NdRKt9_xl.webp)
![[{"id":"UIf5Eim3f4","type":"paragraph","data":{"text":"Aromatic region of the HSQC spectra of ethanol lignins extracted from fir (<i>a</i>) and aspen (<i>b</i>). All <sup>1</sup>H/<sup>13</sup>C NMR spectra were recorded on a Bruker Avance 600 NMR spectrometer operating at 600/150 MHz in DMSO-<i>d</i><sub>6</sub>. Reproduced from Levdansky et al.[[ type=\"anchor\" referenceId=\"12346\" ]] with the permission of the Siberian Federal University."}}]](/storage/images/resized/ogy3JjTi5cuF03vuZJz6N9PySjc0GehfwjrEpMGQ_xl.webp)
The same year, Li et al.[29] reported a kinetic study of the hydrogenolysis of lignin to monomers in a continuous flow reactor using 2D HSQC NMR spectra (Fig. 14). It was found that lignin end and internal units reacted essentially differently which provided insight into lignin hydrogenolysis pathways. As a result of that study, a continuous lignin upgrading process was developed using soluble lignin in a continuous flow reactor. The advantages of the flow-through system included the continuous production of lignin monomers monitored by 2D HSQC NMR by optimizing reaction parameters such as reaction temperature, pressure, and flow-rate in real time.
![[{"id":"2Rv6-fO_HH","type":"paragraph","data":{"text":"Partial 2D HSQC NMR spectra of copper-catalyzed alkaline hydrogen peroxide (Cu-AHP) lignin obtained from poplar — Cu-AHP lignin hydrogenolysis products after flow-tube reaction: lignin sidechain fingerprint region (<i>a</i>); aromatic region (<i>b</i>). NMR spectra were recorded in DMSO-<i>d</i><sub>6</sub>/pyridine-<i>d</i><sub>5</sub> on a Bruker Biospin AVANCE-III 700 MHz spectrometer fitted with a cryogenically-cooled 5 mm gradient probe. Reproduced with minor editing privileges from Li et al.[[ type=\"anchor\" referenceId=\"12347\" ]] with the permission of the Royal Society of Chemistry."}}]](/storage/images/resized/e4A8mITvUsIbgZPboo8CEUbTgTjp0RPt3hhlTjC3_xl.webp)
The resulting 2D HSQC NMR spectra showed that the major lignin sidechain units (β-O-4, β-5, and β-β) in the Cu-catalyzed alkaline hydrogen peroxide (Cu-AHP) lignin were well resolved in the sidechain fingerprint region. The aromatic signals were similar to those from kraft lignin, except for a small amount of the benzylic oxidation caused by the Cu-AHP pretreatment. The signals of the lignin side-chain units showed no obvious changes before and after the reaction.
In 2019, Liu et al.[30] reported a rapid lignocellulose fractionation method applied to common reed based on milling under mild alkaline conditions with using no organic solvents or other reagents. As a result, the lignin-containing cellulose nanofibre and native-like lignins were obtained. Films prepared from the suspension of the lignin-containing cellulose nanofibres showed very interesting properties such as transparency and hydrophobicity. Shown in Fig. 15 are characteristic partial 2D HSQC NMR spectra of reed straw enzyme and alkali-pretreated reed lignins. Overall, the isolated lignin preserved its native structure with high β-ether content. The resulting isolated high-quality lignin was found to be suitable for further upgrading by various lignin depolymerization processes.
![[{"id":"xDe_AwCcgo","type":"paragraph","data":{"text":"Partial 2D HSQC NMR spectra of reed straw enzyme lignins and alkali-pretreated reed lignin (1% NaOH). The retained polysaccharides are mainly xylan in the alkali-soluble lignin. For details, see original publication. NMR spectra were recorded in DMSO-<i>d</i><sub>6</sub>/pyridine-<i>d</i><sub>5</sub> on a Bruker Biospin AVANCE-III 700 MHz spectrometer fitted with a cryogenically-cooled 5 mm gradient probe. Reproduced from Liu et al.[[ type=\"anchor\" referenceId=\"12348\" ]] with minor editing privileges under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/Jy6Mz0OoIa1i7dqETHk9t9fZfGIK7Of1i8jeZajF_xl.webp)
Wang et al.[31] developed an integrated process based on hydrothermal pretreatment and Kraft delignification to deconstruct lignocellulosic biomass. Hydrothermal pretreatment was found not only to facilitate the production of xylo-oligosaccharides but also to reduce the chemicals dosage of the following delignification. The structural characteristics of lignin obtained from the integrated process were studied by 2D HSQC NMR (Fig. 16).
![[{"id":"Hj3anyyFM1","type":"paragraph","data":{"text":"Side-chain and aromatic regions in 2D HSQC NMR spectra of lignins obtained from raw materials and corresponding black liquor. For details, see original publication. NMR spectra were recorded on a Bruker AVIII 400 MHz spectrometer in DMSO-<i>d</i><sub>6</sub>. BE is benzyl ether, PB is <i>p</i>-hydroxybenzoate. Reproduced from Wang et al.[[ type=\"anchor\" referenceId=\"12349\" ]] with the permission of Wiley."}}]](/storage/images/resized/QE0Iwo9ixLWd7tAKQKN0X2Vsskx6RaYcExEKgqSx_xl.webp)
Zhao et al.[32] prepared alkaline and kraft lignins and carried out their comparative characterization by 2D HSQC NMR (Fig. 17 and Fig. 18). Using authenticated reference compounds, the phenylglycerol structures, characteristic of the cleavage of non-phenolic β-aryl ether by soda pulping, in the alkaline lignin were identified and quantified by 2D HSQC NMR. The content of phenylglycerol compounds in alkaline lignin was estimated to be 8 – 14%, which was much higher than that in kraft lignin.
![[{"id":"ueX-NrD2uM","type":"paragraph","data":{"text":" Partial 2D HSQC NMR spectra of nonacetylated/acetylated eucalyptus alkaline lignin (<b>AL</b>) and phenylglycerol compounds for peak assignment. The unknown peaks in (<b>A</b>) of eucalyptus <b>AL</b> were compared to the peaks in (<b>B</b>) from G-gly and (<b>C</b>) S-gly. The phenylglycerol structure in <b>AL</b> was also confirmed by matching the peaks from acetylated compounds and acetylated <b>AL</b> (<b>D</b>). For details, see original publication. NMR spectra were recorded on a Bruker AVANCE III HD 600 MHz spectrometer in DMSO-<i>d</i><sub>6</sub>. Reproduced from Zhao et al.[[ type=\"anchor\" referenceId=\"12350\" ]] with the permission of the American Chemical Society."}}]](/storage/images/resized/lSAoPuJcCzz08JNlpXKj1gxT87vLNJkTQUNjrEoX_xl.webp)
![[{"id":"4xav2wgwT8","type":"paragraph","data":{"text":"Partial 2D HSQC NMR spectra of enzyme lignin (<b>CEL</b>), alkaline lignin (<b>AL</b>), and kraft lignin (<b>KL</b>) of spruce and eucalyptus. For details, see original publication. NMR spectra were recorded on a Bruker AVANCE III HD 600 MHz spectrometer in DMSO-<i>d</i><sub>6</sub>. Reproduced from Zhao et al.[[ type=\"anchor\" referenceId=\"12350\" ]] with minor editing privileges with the permission of the American Chemical Society."}}]](/storage/images/resized/pa2levo7xwaAFbMoRlU5AODCDroSwISUBUNlR4Lh_xl.webp)
A large number of related 2D HSQC NMR studies have recently appeared devoted to the structural studies of lignin and lignin-related products, as exemplified below being among many other related papers.
Cao et al.[33] investigated the microwave-assisted depolymerization of various types of waste lignins elucidating how the structure of lignins recovered from various agricultural and industrial residues affects its downstream catalytic conversion. Three types of lignins, such as bio-enzymatic lignin, organosolv lignin, and Kraft lignin, were fully characterized by HSQC-NMR to obtain a detailed description of their structures, as shown in Fig. 19. The strong dependence of the conversion efficiency on the interunit linkages and functional groups of lignin structures was found. The strong metal – support interaction between CuO and sodium cyanoborohydride NaBH3CN not only facilitated lignin depolymerization via the promoted electron transfer, but also improved the stability of Cu catalysts under hydrothermal conditions.
![[{"id":"cvzY90K8Vu","type":"paragraph","data":{"text":"Side-chain and aromatic regions in 2D HSQC NMR of various lignin samples: bioenzymatic lignin (<b>BL</b>), organosolv lignin (<b>OL</b>) and Kraft lignin (<b>KL</b>). The main substructures identified by 2D NMR: <b>A</b> is β-O-4 linkage; <b>B</b> is β-5 linkage; <b>C</b> is β-β linkage; <b>S</b> is syringyl unit; <b>G</b> is guaiacyl unit; <b>H</b> is p-hydroxyphenyl unit; <b>F</b> is ferulic acid unit and <b>P</b> is p-coumaric acid unit. 2D HSQC NMR spectra of various types of lignins were recorded on a Bruker AVIII 400 MHz spectrometer. About 80 mg of lignin was dissolved in 0.6 mL of DMSO-<i>d</i><sub>6</sub>. Reproduced with minor editing privileges from Cao et al.[[ type=\"anchor\" referenceId=\"12351\" ]] with the permission of the Royal Society of Chemistry."}}]](/storage/images/resized/o9I5cj9WclNxX61pyQ9EYmeyQZUZnqIO46fY09Q1_xl.webp)
Chen et al.[34] developed an integrated biorefinery strategy towards phenolics, levulinic acid, and furfural during total utilization of lignin and carbohydrates in Eucalyptus grandis. It is well known that the depolymerization of lignin into the well-defined mono-aromatic chemicals suitable for downstream processing is recognized increasingly as an important starting point for the lignin valorization. In that study, the conversion of all three components of Eucalyptus grandis into the corresponding monomeric chemicals was investigated using solid and acidic catalysts in sequence. The 2D HSQC NMR spectra of the lignin oil shown in Fig. 20 confirmed that the main substructures, such as β-aryl ether (β-O-4), resinol (β-β), and phenyl coumaran (β-5) have disappeared after Pd/C treatment, indicating the C – O bonds dissociation in protolignin. New dominant cross peaks corresponding to a propanol moiety were observed in HSQC. The cross signals in the aromatic region corresponding to guaiacyl and syringyl units were also found.
![[{"id":"gknRgIueJv","type":"paragraph","data":{"text":"2D HSQC NMR spectra of the lignin oil obtained at 240°C, 4 h (DMSO-<i>d</i><sub>6</sub>). Aromatic region (<i>a</i>). The signals from the <b>G</b> and <b>S</b> units appeared at 112.2/6.71 (<b>G<sub>2</sub></b>), 115.0/6.64 (<b>G<sub>5</sub></b>), 120.0/6.55 (<b>G<sub>6</sub></b>) and 105.4/6.41 (<b>S<sub>2,6</sub></b>) ppm, respectively. Side chain region (<i>b</i>). The signals from the propanol moiety were found at 31.7/2.49, 34.5/1.68 and 60.1/3.40 ppm, respectively. 2D HSQC NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer in DMSO-<i>d</i><sub>6</sub>. Reproduced from Chen et al.[[ type=\"anchor\" referenceId=\"12352\" ]] under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/3NMbRjZUELSCHo5W6zzyEboirkdlMhEY3gf7Kf4j_xl.webp)
Kim et al.[35] carried out a 2D HSQC study of monolignol benzoates incorporation into the lignin of transgenic Populus trichocarpa (Fig. 21). It was found that monolignol benzoates incorporated into the lignin of triple-transgenic Populus trichocarpa, which is downregulated for the three endoplasmic reticulum cytochrome enzymes participating in oxidative radical coupling reactions during lignification in the cell wall. This study has demonstrated how the biosynthesis of monolignol benzoate and monolignol p-hydroxybenzoate in plant is involved in the lignin biosynthetic pathway by providing direct evidence for the monolignol benzoate and monolignol p-hydroxybenzoate structures in the transgenic Populus trichocarpa lignin. In addition, attention was drawn to the convoluted interactions of enzymes in the pathway to produce benzoates.
![[{"id":"l7SRYFjbQ-","type":"paragraph","data":{"text":"Aromatic region of 2D HSQC NMR spectra (DMSO-<i>d</i><sub>6</sub>/pyridine-<i>d</i><sub>5</sub>, 4 : 1, v/v) of enzyme lignins isolated from <i>Populus trichocarpa</i> and C4H/C3H-downregulated transgenics. 2D HSQC NMR spectra were recorded on a Bruker Biospin Avance 700 MHz spectrometer equipped with a 5 mm cryoprobe with inverse geometry in DMSO-<i>d</i><sub>6</sub>/pyridine-<i>d</i><sub>5</sub> (4 : 1, v/v). Reproduced from Kim et al.[[ type=\"anchor\" referenceId=\"12353\" ]] under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/yBOBKZMEL8SWQNuhHl5usBpNEGhquhIzkA8BuqTO_xl.webp)
Li et al.[36] used a number of methods to identify the biomass structural determinants defining the properties of the plant-derived renewable carbon fibre. The linkages and composition of lignin from eight sorghum feedstocks were analyzed by 2D HSQC NMR and the linkage profiles of uncondensed β-O-4, condensed β-5, and condensed β-β were calculated based on a number of aromatic rings of syringyl, guaiacyl and ρ-hydroxyphenyl units. At that, the frequencies of β-O-4, β-5, and β-β linkages were determined based on a series of 2D HSQC NMR experiments (Fig. 22 and Fig. 23). Considering the diverse range of lignin characteristics, lignin from the sorghum feedstock samples was found to be a perfect model precursor to elucidate the impact of biomass characteristics on the performance of the resultant carbon fibres. That 2D HSQC NMR study opened a new path to modify cell wall structures for manufacturing the high-quality carbon fibres.
![[{"id":"fyelQidnGo","type":"paragraph","data":{"text":"Internuitary linkages in the sorghum lignin as revealed by 2D HSQC NMR. Normalized lignin content in the samples: low (< 0.3, Sorghum 103, 205), moderate (0.5 – 0.9, Sorghum 101, 104, 201, 206), high (> 0.9, Sorghum 103, 205). Lignin (30 – 50 mg) was dissolved in 0.6 mL of DMSO-<i>d</i><sub>6</sub> and placed in an NMR tube. Adiabatic 2D <sup>1</sup>H/<sup>13</sup>C HSQC spectra were acquired on a Bruker Avance-III 400 MHz spectrometer equipped with a 5 mm broadband probe with a Z-gradient probe, Bruker). Reproduced from Li et al.[[ type=\"anchor\" referenceId=\"12354\" ]] under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/vBWeFeyjnQwhfyQc58D4MrZAxMIY9Hv5wWF04ceC_xl.webp)
![[{"id":"G6pPmL1jmW","type":"paragraph","data":{"text":"Aromatic regions of the 2D HSQC NMR spectra of lignin. <b>G<sub>2</sub></b>, <b>G<sub>5</sub></b> and <b>G<sub>6</sub></b> are carbon-2, carbon-5, and carbon-6 correlations from guaiacyl (<b>G</b>) units; <b>S<sub>2/6</sub></b> is carbon-2 and carbon-6 correlations from syringyl (<b>S</b>) units; <b>H<sub>2/6</sub></b> is carbon-2 and carbon-6 correlations from p-hydroxyphenyl (H) units; <b><i>p</i>CA<sub>2/6</sub></b> and <b><i>p</i>CA<sub>α</sub></b> are carbon-2 and carbon-6, and carbon-α correlations from p-coumarate (<b><i>p</i>CA</b>) units; <b>FA<sub>2</sub></b>, <b>FA<sub>5</sub></b> and <b>FA<sub>α</sub></b> are carbon-2, carbon-5, and carbon-α correlations from ferulate (<b>FA</b>) units. For more details, see original publication. Lignin (30 – 50 mg) was dissolved in 0.6 mL of DMSO-<i>d</i><sub>6</sub> and placed in an NMR tube. Adiabatic 2D <sup>1</sup>H/<sup>13</sup>C HSQC spectra were acquired on a Bruker Avance-III 400 MHz spectrometer equipped with a 5 mm broadband probe with a Z-gradient probe, Bruker). Reproduced from Li et al.[[ type=\"anchor\" referenceId=\"12354\" ]] under the Attribution NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/v7BicKB4J1bmtvHhFpUiBTqOT2hDChg0q8DKtMIe_xl.webp)
Liu et al.[37] reported the method for isolation and characterization of crude lignin of bark using the ethanol-water organosolv treatment. According to the two-dimensional 1H – 13C 2D HSQC NMR and monodimensional 13C NMR spectral data, crude lignin contained mixed polyphenolics, suberin compounds and carbohydrates (Fig. 24). Overall, this study demonstrated the potential to solubilize a high proportion of bark and provided insight into the structure–property relationships of crude lignin as a function of processing conditions.
![[{"id":"mgdC2xJgSv","type":"paragraph","data":{"text":" <sup>13</sup>C NMR spectra of organosolv crude lignin of pine bark (<i>a</i>), oak bark (<i>b</i>) and HSQC NMR spectra of organosolv crude lignin of pine bark (<i>c</i>), oak bark (<i>d</i>). The dimension of the axis in fig. <i>a</i> is similar to that of the axis in fig. <i>b</i>. <sup>13</sup>C NMR and <sup>1</sup>H NMR spectra were obtained on a Bruker Avance 300 MHz spectrometer at 298 K with a BBO probe. HSQC NMR spectra were obtained on a Bruker VANCE III 600 MHz spectrometer at 25°C equipped with a cryoprobe. <sup>13</sup>C NMR samples were prepared by dissolving 150 – 200 mg dried lignin powders or acetylated lignin powders in 450 μl DMSO-<i>d</i><sub>6</sub>, followed by the addition of 60 μl 50 mg μl<sup>–1</sup> chromium(III) acetylacetonate as a relaxation reagent. For <sup>1</sup>H NMR samples, 50 mg acetylated lignin samples were dissolved in 400 μl chloroform-d followed by the addition of 100 μl p-hydroxybenzaldehyde (5 mg ml<sup>–1</sup>) as an internal standard. For HSQC NMR, 30 mg dried lignin-rich compounds were dissolved in 500 μL DMSO-<i>d</i><sub>6</sub>. All these samples were transferred into 5 mm NMR tubes and caped. Reproduced with minor editing privileges from Liu et al.[[ type=\"anchor\" referenceId=\"12355\" ]] with the permission of the American Chemical Society."}}]](/storage/images/resized/cnfiUi4exzIuW2XgSa7sfSKBS67L6OdjysxOAdQr_xl.webp)
Rowlandson et al.[38] investigated the influence of aromatic structure of lignin on its thermal behaviour. The authors performed a systematic study of the chemical composition of lignins extracted using an identical organosolv isolation method but from different biomass feedstocks: hemp hurds, eucalyptus chips, flax straw, rice husk and pine. Basic results were obtained mainly from the 2D-NMR HSQC spectra shown in Fig. 25 for rice husk lignin, eucalyptus lignin and industrial lignin. It was found how the aromatic structure of lignin could affect the thermal behaviour of the polymer, which correlated with the structure of the resulting carbons. Carbons from lignins with a high content of syringyl units displayed a pronounced foaming behaviour which, upon activation, resulted in a high-surface area material with hierarchical porosity.
![[{"id":"1HpBYVauuj","type":"paragraph","data":{"text":"2D NMR HSQC spectra of the aromatic region (left) (δ<sub>C</sub>/δ<sub>H</sub> 95 – 140/6.0 – 7.8 ppm) and aliphatic side-chain region (right) (δ<sub>C</sub>/δ<sub>H</sub> 50 – 90/2.5 – 6.0 ppm) of rice husk lignin (<i>a</i>), eucalyptus lignin (<i>b</i>) and industrial lignin (<i>c</i>). All NMR spectra were recorded on a Bruker Avance III NMR spectrometer operating at 500.13 MHz (<sup>1</sup>H) and 125.77 MHz (<sup>13</sup>C). Lignin samples (~ 60 mg) were swollen in DMSO-<i>d</i><sub>6</sub>. 2D <sup>1</sup>H – <sup>13</sup>C HSQC spectra were acquired using a spectral width of 40‒180 ppm (<sup>13</sup>C) and 1.28‒10.68 ppm (<sup>1</sup>H). Reproduced with minor editing privileges from Rowlandson et al.[[ type=\"anchor\" referenceId=\"12356\" ]] with the permission of Springer Nature."}}]](/storage/images/resized/vxe3w5zjbWB00S26oRlDKIbLLGgfRfH8t3gtOBbU_xl.webp)
It was found that organosolv lignin samples contained syringyl unit, oxidized syringyl unit, guaiacyl unit, p-hydroxyphenyl unit, ferulic acid, cinnamyl alcohol end group, cinnamaldehyde end group, and p-coumaric acid with the following main linkages present: β-aryl-ether β-O-4, resinol β-β, phenylcoumaran β-5 and spirodienone (Fig. 26).
![[{"id":"V350bdp9Ue","type":"paragraph","data":{"text":"Aromatic structures present in lignin; syringyl unit (<b>S</b>), oxidized syringyl unit (<b>S'</b>), guaiacyl unit (<b>G</b>), p-hydroxyphenyl unit (<b>H</b>), ferulic acid (<b>FA</b>), cinnamyl alcohol end group (<b>I</b>), cinnamaldehyde end group (<b>J</b>), and p-coumaric acid (<b><i>p</i>CA</b>); the main linkages: β-aryl-ether β-O-4 (<b>A</b>), resinol β-β (<b>B</b>), phenylcoumaran β-5 (<b>C</b>) and spirodienone (<b>D</b>). Reproduced from Rowlandson et al.[[ type=\"anchor\" referenceId=\"12356\" ]] with the permission of Springer Nature."}}]](/storage/images/resized/FkxV4LJgHwx7uFCykht4rT7wzCsdojIYxl1NGbRW_xl.webp)
Tokunaga et al.[39] explored the nonproductive binding sites of lignin models with carbohydrate-binding module of cellobiohydrolase. In this study, three types of 13C-labeled β-O-4 lignin oligomer models were synthesized and characterized. According to the 2D 1H – 13C HSQC spectra of the 13C-labeled lignin models (Fig. 27), three types of the 13C labels were correctly incorporated in the aromatic rings and β-positions, α-positions and methoxy groups. The Trichoderma reesei carbohydrate-binding module 1 (TrCBM1) binding sites in lignin were analyzed by observing NMR chemical shift perturbations using the synthetic 13C-labeled β-O-4 lignin oligomer models. Obvious chemical shift perturbations were found in signals from the aromatic regions in oligomers bound to TrCBM1, whereas changes in the signals from aliphatic regions and methoxy groups were insignificant. These results suggested that hydrophobic interactions and π – π stacking were dominating factors in the nonproductive binding.
![[{"id":"8aZ3B_2dJV","type":"paragraph","data":{"text":"2D <sup>1</sup>H – <sup>13</sup>C HSQC spectra of the long-chain <sup>13</sup>C-labeled lignin oligomer models in the NMR experiments. Superimposed 2D <sup>1</sup>H – <sup>13</sup>C HSQC spectra of the aromatic region in the presence (red) and absence (blue) of 350 μM <i>Tr</i>CBM<sub>1</sub> (<i>a</i>). A magnified image of the HSQC signals from the C-5 positions (<i>b</i>). Superimposed 2D <sup>1</sup>H – <sup>13</sup>C HSQC spectra of aliphatic regions and methoxy groups in the presence (red) and absence (blue) of 350 μM <i>Tr</i>CBM<sub>1</sub> (<i>c</i>). For used designations and abbreviations, see original text. Each sample was dissolved in 50 mM acetic acid-<i>d</i><sub>4</sub> buffer, which was prepared using D<sub>2</sub>O (pD 5.0) and 10% (v/v) DMSO-<i>d</i><sub>6</sub>. Each 250 μL NMR sample was placed in a 5-mm Shigemi symmetrical microtube (Shigemi, Japan) and contained 20 μM 2,2-dimethyl-2-silapentane-5-sulfonic acid as an internal standard. All NMR spectra were recorded on a Bruker Avance III HD 600 spectrometer equipped with a cryogenic probe and Z gradient (Bruker BioSpin, USA). Reproduced with minor editing privileges from Tokunaga et al.[[ type=\"anchor\" referenceId=\"12357\" ]] under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/s3MlcIQvX9jzDANCjzBLPTtCFvxqDChBvBkKzUOX_xl.webp)
Ghavidel et al.[40] studied chemical reactivity and sulfofunctionalization of enzymatically produced lignin. The comparison between six different enzymatically hydrolyzed lignin samples revealed the fundamental role of β-aryl ether and residual carbohydrate on the physicochemical properties and chemical reactivity of the samples used to generate dispersants for the clay suspension. Based on the 2D HSQC spectra of six hydrolyzed lignin samples shown in Fig. 28, it was established that lignin samples with fewer β-O-4 and lignin-carbohydrate bonds (LCC) had a higher content of Ph – OH and carboxylate groups. The higher ratio of these bonds was associated by the authors with the superior contribution of the β-aryl ether and lignin-carbohydrate bonds. The samples with the lower sugar content appeared to be smaller and more porous, which increased the chemical reactivity of the hydrolyzed lignin by making it more accessible to the functionalization reactions.
![[{"id":"PsHFhdwHVI","type":"paragraph","data":{"text":"2D HSQC spectra of six hydrolyzed lignin samples (<b>HL1 – HL6</b>). The identified cross peaks are: β-O-4 arylether (β-O-4); β-O-4 aryl ether in guaiacyl β-O-4 (<b>G</b>); γ-ester GE (<b>LCC</b>); glycoside PG (<b>LCC</b>); methoxy group (OMe); syringyl (<b>S</b>); guaiacyl (<b>G</b>); Phenylcoumaran (β-5) (<b>C</b>); dibenzodioxcin (5-5-O-4) (<b>DBD</b>); resinol (<b>R</b>). For used designations and abbreviations, see original publication. The <sup>1</sup>H – <sup>13</sup>C HSQC spectra were recorded on a 500 MHz Bruker Avance NEO spectrometer equipped with a 5 mm Quattro Nucleus Probe and a field gradient. For the NMR experiments, 50 mg of the hydrolyzed lignin samples were added to 0.7 mL of DMSO-<i>d</i><sub>6</sub> and stirred at 45°C overnight. Reproduced with minor editing privileges from Ghavidel et al.[[ type=\"anchor\" referenceId=\"12358\" ]] with the permission of Elsevier."}}]](/storage/images/resized/1pkMxJHPbTBpAVFXzqw0hhB31UB2yIjbm8NjP39V_xl.webp)
Islam et al.[41] used N-methyl-2-pyrrolidone (NMP) pretreatment of lignocellulose to increase lignin yield and cellulose digestibility. The authors discovered a new N-methyl-2-pyrrolidone pretreatment solvent with excellent lignin solubility and preservation of β-O-4 linkages present in the pristine lignin structure. These results indicated that the formation of 4-(methylamino)butyric acid in the NMP pretreatment system increased the polarity of the solvent system. The increased polarity in turn increased the lignin removal and reduced the amount of lignin condensation. This result was attributed to the higher amount of the β-O-4 interunit linkages in the NMP lignin sample, which was mainly determined by HSQC analysis.
The 1H – 13C 2D HSQC NMR spectra shown in Fig. 29 demonstrated that the major substructures of the studied lignin were S and G units together with interunit β-O-4, β-β and β-5 linkages. The NMR signals were assigned by comparison with those of the benchmark ball mill lignin. The position and colour of the correlations shown represent the corresponding substructures, which are β-O-4 (green), β-5 (brown), and β-β (pink), S unit (grass green), G unit (light orange) and H unit (purple). The semi-quantitative values of major lignin moieties were determined using the aromatic units as an internal standard. For the ball milled lignin, α-position of β-O-4, β-β, and β-5 linkages appeared at δC/δH 72.56/4.82 ppm, δC/δH 85.66/4.60 ppm and δC/δH 86.1/4.1 ppm, respectively. With the NMP pretreatment, the corresponding peaks were observed at the same position with different intensities. The β-O-4 was found to be the predominant linkage of the studied lignin, which determined the integrity and potential of fractionated lignin for its effective valorization to produce aromatic chemicals.
![[{"id":"Hc4JnaRD6Q","type":"paragraph","data":{"text":" 2D HSQC spectra of the side-chain and aromatic regions of the ball milled lignin (<b>BM-L</b>) (<i>a</i>), NMP-pretreated lignin <i>(b</i>) and ethanol lignin from <i>Acacia confuse</i> wood (<i>c</i>). All 2D-HSQC <sup>1</sup>H – <sup>13</sup>C experiments were performed on a 500-MHz spectrometer JEOL ECZR equipped with a 5-mm probe. A standard pulse sequence was applied with the following parameters: spectral widths of 11 ppm in F2 (<sup>1</sup>H) with 1024 data points and 190 ppm in F1 (<sup>13</sup>C) with 256 data points, 64 scans, and 1.5 seconds interscan delay. For each test, 50 mg of purified lignin sample was dissolved in 0.5 ml of DMSO-<i>d</i><sub>6</sub>. Reproduced from Islam et al.[[ type=\"anchor\" referenceId=\"12359\" ]] with the permission of Springer Nature."}}]](/storage/images/resized/fPv9H9uPiHfZPfNSxocJFPdH95ih7NVenVCgeqZQ_xl.webp)
Diaz-Baca et al.[42] carried out the polymerization of tall oil lignin, starch and 2-methyl-2-propene-1-sulfonic acid sodium salt to produce flocculants for colloidal systems together with the sulfonated lignin – starch polymer, and described the use of the latter as a flocculant. Based on the advanced 1H, COSY, HSQC, HSQC-TOCSY, and HMBC NMR techniques (Fig. 30 and Fig. 31), it was confirmed that the phenolic substructures of tall oil lignin and the anhydroglucose unit of starch were covalently polymerized by the monomer to give the three-block copolymer.
![[{"id":"scf4RyfR94","type":"paragraph","data":{"text":"The <sup>1</sup>H – <sup>13</sup>C HSQC spectra of tall oil lignin (<b>TOL</b>), starch and anionic tall oil lignin-starch copolymers (<b>ALS-1</b> and <b>ALS-5</b>). Red circles indicate the corresponding cross-peaks of the aromatic region and methoxy groups. For details, see original publication. All <sup>1</sup>H – <sup>13</sup>C 2D-HSQC experiments were performed on a Varian UNITY INOVA™-500 MHz NMR spectrometer using 26 – 27 mg of previously dried samples in 500 μL of DMSO-<i>d</i><sub>6</sub>. Reproduced with minor editing privileges from Diaz-Baca et al.[[ type=\"anchor\" referenceId=\"12360\" ]] with the permission of the American Chemical Society."}}]](/storage/images/resized/JEZbYxpczUfbLrmG4MCAe4AkH2IYjzWitqfQtaja_xl.webp)
![[{"id":"oDn6hK44tp","type":"paragraph","data":{"text":"The <sup>1</sup>H – <sup>13</sup>C HSQC TOCSY spectra of tall oil lignin (<b>TOL</b>), starch, and anionic tall oil lignin-starch copolymers, <b>ALS-1</b> and <b>ALS-5</b>. Doted circles indicate TOCSY correlations of carbons (vertical) and protons (horizontal). For details, see original publication. All <sup>1</sup>H – <sup>13</sup>C 2D-HSQC experiments were performed on a Varian UNITY INOVA™-500 MHz NMR spectrometer using 26 – 27 mg of previously dried samples in 500 μL of DMSO-<i>d</i><sub>6</sub>. Reproduced with minor editing privileges from Diaz-Baca et al.[[ type=\"anchor\" referenceId=\"12360\" ]]<sup> </sup>with the permission of the American Chemical Society."}}]](/storage/images/resized/999jcPuoWyCCUNuM2AYZlW15jILpnrRBiKfWgYXh_xl.webp)
Karlsson et al.[43] studied lignin structure and reactivity in the organosolv process. The aim of this work was to study the reactivity of lignin during a cyclic organosolv extraction process while using physical protection strategies. Synthetic lignins obtained by mimicking the lignin polymerization process were used as reference compounds. The authors used state-of-the-art NMR analysis, which is a powerful tool for elucidating lignin interunit linkages and functionalities. This study revealed interesting fundamental aspects of lignin polymerization, such as identifications of molecular populations with high degrees of structural homogeneity and the emergence of branching points in the lignin structure. Analysis of the organosolv biorefinery lignin by HSQC NMR, as exemplified in Fig. 32, showed an abundance of aryl-ether linkages (β-O-4), confirming that physical protection did indeed occur due to performing the extraction in cyclic mode. A previously proposed intramolecular condensation reaction was substantiated, and new insights into the selectivity of this reaction were introduced, which was supported by the density functional theory (DFT) calculations (Fig. 33 and Fig. 34), where the important role of intramolecular π – π stacking was emphasized.
![[{"id":"shXb-Dsmvk","type":"paragraph","data":{"text":" 2D <sup>1</sup>H – <sup>13</sup>C HSQC NMR spectrum of the ethanol-soluble portion of the extracted lignin sample; f1 corresponds to the <sup>13</sup>C dimension and f2 — to the <sup>1</sup>H dimension. The NMR spectra were recorded in DMSO-<i>d</i><sub>6</sub> on a Bruker NMR spectrometer Avance III HD 400 MHz instrument (Bruker Corporation, USA) equipped with a 5 mm Z-gradient BBFO broadband smart probe. Reproduced from Karlsson et al.[[ type=\"anchor\" referenceId=\"12361\" ]]<sup> </sup>under the Attribution-Non Commercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/JzBzvYQ80tdlROQTU7wvNuK9XOTJC5PCVVTx30RW_xl.webp)
![[{"id":"q2xzvzqlim","type":"paragraph","data":{"text":"DFT models of three hexamers. Yellow dotted lines indicate distances between the π – π stacked aromatic rings for both sandwich and the T-shape type stacking. Reproduced with minor editing privileges from Karlsson et al.[[ type=\"anchor\" referenceId=\"12361\" ]] under the Attribution-Non Commercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/KqsOLAv73AQ4toId2H0BOIhEzSR2ZkQtRNvu882G_xl.webp)
![[{"id":"RWEu2XdTWc","type":"paragraph","data":{"text":"The hexamer used for the DFT simulation (<i>a</i>). Distances between the benzylic carbon and 5' atom on the next aromatic ring, marked with the yellow dotted lines on the simulations (<i>b</i>). Reproduced with minor editing privileges from Karlsson et al.[[ type=\"anchor\" referenceId=\"12361\" ]] under the Attribution NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/AaxtypKUryrzDT0S6cO46BAAnZOgVXTRpAmjFzvq_xl.webp)
Rinken et al.[44] investigated the role of solvent fractionation of lignin oil in structural profiling during lignin stabilization, and the carbohydrate nature in the H-transfer reductive catalytic fractionation was also evaluated. In particular, the solvent fractionation of the reductive catalytic fractionation lignin oil was studied as a facile method for producing lignin oil fractions for advanced characterization. Solvent fractionation is known to use small volumes of environmentally benign solvents like methanol, acetone, and ethyl acetate to produce multigram lignin fractions containing products in different molecular weight ranges. This feature allows the determination of structural heterogeneity across the entire molecular weight distribution of the reductive catalytic fractionation lignin oil by high-resolution HSQC NMR spectroscopy. Performed study provided a detailed insight into the role of the hydrogenation catalyst in stabilizing the lignin fragments and defining the structural features of the hemicellulose-derived carbohydrates in the lignin oil. The detailed characterization of the fractions by HSQC NMR measurements (Fig. 35) revealed the formation of the reduced β-O-4 linkages presenting a methylene group at the Cα position. It is generally accepted that stabilization processes primarily involve lignin monomer intermediates. The findings presented in this publication indicated that an additional route for the lignin stabilization has been established through the formation of the reduced β-O-4 linkages.
![[{"id":"E3JMk5TbSm","type":"paragraph","data":{"text":"2D <sup>1</sup>H – <sup>13</sup>C HSQC NMR spectrum of the EtOAc-insoluble fraction dissolved in DMSO-<i>d</i><sub>6</sub>. Zoomed spectral regions are given in insets. The <sup>13</sup>C−<sup>1</sup>H correlation signals attributed to the reduced β-O-4 linkages are highlighted in green. All samples were analyzed on a 800 MHz Bruker spectrometer equipped with inverse triple resonance cryoprobe with an ATM module. Reproduced from Rinken et al.[[ type=\"anchor\" referenceId=\"12362\" ]] under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/w5iZmyrO4hS4KaMsiIavsAe9CpRRvFB6oarUfXqY_xl.webp)
The next two short Sections 2.3 and 2.4 focus on the application of 31P, 19F, and 15N NMR to the structural studies of different types of lignin are by no means a comprehensive discussion of the subject.
2.3. 31P NMR
Due to the complexity of the repeating monomers in lignin, its 1H NMR spectra suffer from significant overlap when used for lignin analysis. However, 31P derivatization has been proven a useful technique for the NMR analysis of lignin. The structure of lignin can be effectively modified by the instantaneous incorporation of phosphorus into kraft lignin, so that 31P NMR could be applied to lignin analysis and structural studies. As an example, see paper by Puyadena et al.[45] on the phosphorus-containing lignin intermediates for the polyurethane and acrylic coatings for wood. The 100% natural abundance of the 31P isotope, the large chemical shift range, and the sharp signals provide perspectives of using 31P NMR for the lignin analysis.
One of the basic publications [46] of the last five years demonstrated that 31P NMR spectroscopy is a promising technique for the quantitative analysis of hydroxyl groups of lignin due to its unique characterization capability of the biorefinery process. The proposed protocol describes procedures for the preparation and analysis of phosphitylated lignin samples based on 31P NMR spectra together with the means of quantitative analysis of different types of hydroxyl groups. Compared to the traditional ‘wet-chemical techniques’, the above method of quantitative 31P NMR spectroscopy offered unique advantages in measuring phosphitylated hydroxyl groups from a single 31P NMR spectrum, as illustrated in Fig. 36 and Fig. 37. In addition, this method provided complete quantitative information about hydroxyl groups in small amounts of sample (~ 30 mg) within a relatively short experimental time (up to 2 h).
![[{"id":"AlK0CTanbi","type":"paragraph","data":{"text":"A quantitative <sup>31</sup>P NMR spectrum of a hardwood poplar lignin derivatized with 1-chloro-4,4',5,5'-tetramethyl-1,3-dioxa-2-phospholane (TMDP). All spectra were recorded on a Bruker Avance III HD 500-MHz spectrometer equipped with a 5-mm BBO probe in DMSO-<i>d</i><sub>6</sub>. Reproduced from Meng et al.[[ type=\"anchor\" referenceId=\"12364\" ]]<sup> </sup>under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)"}}]](/storage/images/resized/0kO6PKEGJGkMJXvPaLgwoLLvCCpURAT2HP8jUCsX_xl.webp)
![[{"id":"RDFcG6dzWj","type":"paragraph","data":{"text":"Quantitative <sup>31</sup>P NMR spectra of organosolv poplar, pine, and switchgrass lignins derivatized with TDMP. All spectra were recorded on a Bruker Avance III HD 500-MHz spectrometer equipped with a 5-mm BBO probe in DMSO-<i>d</i><sub>6</sub>. Reproduced from Meng et al.[[ type=\"anchor\" referenceId=\"12364\" ]] under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/LP815CQyfMZomCbuNx6AzvqhQd6YkL2z1GhbCwPR_xl.webp)
A similar study was carried out using quantitative 31P NMR of lignins and tannins from organosolv lignin, which was derivatized with TMDP.[47] Quantitative 31P NMR spectroscopy was shown to be a rapid and reliable method for the identification of unsubstituted, o-mono-substituted and o-disubstituted phenols, aliphatic hydroxyls and carboxylic acids in lignins and tannins. The methodology consisted of an in situ quantitative 31P labelling of lignin or tannin followed by the acquisition of a quantitative 31P NMR spectrum (Fig. 38). However, it was concluded that TMDP-based 31P NMR is unable to distinguish aryl-glycerol-β-aryl ethers, phenylcumarane and pinoresinols.
![[{"id":"dhgxf7ZBBx","type":"paragraph","data":{"text":"Quantitative <sup>31</sup>P NMR spectra of organosolv lignin derivatized with TMDP before (<i>a</i>) and after oxidation (<i>b</i>). The spectra were recorded on a 300 MHz NMR spectrometer in pyridine/deuterated chloroform. Reproduced from Argyropoulos et al.[[ type=\"anchor\" referenceId=\"12365\" ]] under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/nFC3PQISa9Avu0HLs1njax0U321GqitjjRQM7jhH_xl.webp)
Jia et al.[48] determined the role of lignin produced in the autohydrolysis process in the enzymatic hydrolysis of biomass. Based on the 31P NMR data, β-O-4 of lignin was reduced, while β-5 and β-β ratios increased with increasing the hydrolysis intensity. At that, the increase in the hydrolysis intensity significantly enhanced the content of condensed and non-condensed phenolic OH groups of lignin. It was also shown by 31P NMR that the cellulase enzyme adsorbs more readily onto lignin with higher phenolic content, and its association with lignin reduces its activity to hydrolyze cellulose microcrystals.
Mun et al.[49] carried out 31P NMR analysis of kraft lignin prepared from mixed hardwoods, as illustrated in Fig. 39, which shows the enlarged region (134 – 150 ppm) of phosphitylated hydroxyl groups in syringyl, guaiacyl and p-hydroxyphenyl moieties. A sharp peak at 174 ppm was attributed to the excess amount of unreacted TMDP indicating that all hydroxyls in kraft lignin were completely derivatized. The nature of the signal at 174 ppm was discussed in detail by Meng et al.[46]
![[{"id":"Wgo080NsXy","type":"paragraph","data":{"text":"<sup>31</sup>P NMR spectrum of kraft lignin prepared from mixed hardwoods showing phosphitylated hydroxyl groups in syringyl (<b>S</b>), guaiacyl (<b>G</b>) and p-hydroxyphenyl (<b>H</b>) moieties. Spectra were recorded on a 500 MHz spectrometer (Jeol, Japan) in pyridine/deuterated chloroform. Reproduced from Mun et al.[[ type=\"anchor\" referenceId=\"12367\" ]]<sup> </sup>under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/MVDjzPUMa5Xs7sl0GkCzmQ4MLsLWK3gHQQy9vAlw_xl.webp)
Purushothaman et al.[50] performed an extended NMR study of five technical lignins derived from different botanical origins (herbaceous, hardwood, and softwood) and covering three main industrial pulping methods (soda, kraft, and organosolv). The 31P NMR spectra (Fig. 40) confirmed that the base-catalyzed depolymerization residues were more condensed and had higher phenolic hydroxyl content, as compared to all the feed lignins. In general, a large decrease in aliphatic hydroxyl groups was observed in all lignin residues indicating on its depolymerization.
![[{"id":"W14HKr8Wta","type":"paragraph","data":{"text":"<sup>31</sup>P NMR spectrum of various lignin residues (<b>RL</b>) obtained after base-catalyzed depolymerization (after phosphitylation). All spectra were recorded on a Bruker AVANCE III 400 MHz instrument in DMSO-<i>d</i><sub>6</sub> using a standard phosphorous pulse programmer. Reproduced from Purushothaman et al.[[ type=\"anchor\" referenceId=\"12368\" ]] under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/PcDmw8BCXSGTQZZkMZJ7J6XMCYDk19Ln2nRJA65O_xl.webp)
It is well known that phenolic moieties strongly influence the reactivity and physical properties of lignin, and thus accurate quantification of phenolic groups in lignin is a critical analytical chemistry need. Kenny et al.[51] noted that 31P NMR is widely considered to be the standard method for lignin analysis, but this approach uses a hazardous and expensive derivatization agent, and the NMR spectroscopy experiments are time-consuming due to long relaxation times. The authors reported a complementary method that enables accurate identification and quantification of phenolic groups in lignin samples using pentafluoropyridine as a derivatizing reagent followed by the 19F NMR analysis. However, in this approach, the reactivity of the fluorinated compound towards aliphatic OH groups present in lignin is not quantitative, and signals arising from aliphatic hydroxy groups overlap with those from the phenolic groups, so that the analysis of the OH groups is in fact neither quantitative nor selective.
It should also be noted that an enhanced applicability and innovation of 31P NMR in the structural studies of lignins deals with the use of ionic liquids for the solvation of lignosulphonates. Indeed, 31P NMR spectroscopy is the most common and most accurate analytical method for the quantitative determination of the hydroxy group contents in technical lignins. However, for lignosulfonates, liquid-state NMR analysis is often limited due to solubility problems in commonly used solvent systems, which may arise from the wide range of lignosulfonates from different wood sources, pulping conditions, and purification procedures used in biorefineries. Finding a suitable solvent system is even more difficult for chemically modified or fractionated lignosulfonates. Wurzer et al.[52] proposed a novel and fast approach for the solubilization of genuine, modified, and fractionated lignosulfonates and subsequent quantitative analysis of hydroxy groups by 31P NMR after derivatization with TMDP.
Another application that deserves to be cited is the successful utilization of 31P NMR for studying the oxidation of kraft lignin in view of preparing lignin vitrimers, which are currently frontier materials in lignin chemistry. It is well known that the valorization of lignin into the commercial products by oxidative conversion is a widely studied strategy. However, in many cases, this approach has limited scope for integration into industrial processes.
In general, 31P NMR spectroscopy is used for either the qualitative or the quantitative determination of hydroxylated moieties as well as for determination of the condensation degree and the syringyl/guaiacyl ratio in lignins.
Of some practical interest is somehow also the benchtop NMR spectroscopy, which is used for the low-cost analytical analysis of lignin (Fig. 41). The benchtop NMR instrumentation offers the possibility to measure 31P NMR spectra in situations where the application of the high-field NMR spectroscopy is unavailable or too expensive. The former technique provides the ability to quantify different classes of compounds less accurately but at lower cost. Further examples of the benchtop NMR investigations of different lignins (like kraft softwood and hardwood lignins, organosolv hardwood lignin, and soda lignin) have been reported by Araneda et al.[53]
![[{"id":"-5WgKg8iuY","type":"paragraph","data":{"text":"Highfield and benchtop <sup>31</sup>P NMR spectra of phosphitylated lignin. All benchtop <sup>31</sup>P NMR spectra were obtained on a Nanalysis 60pro benchtop NMR spectrometer operating at 24.3 MHz <sup>31</sup>P frequency. All high-field <sup>31</sup>P NMR spectra were obtained on a Bruker 400 MHz Avance III spectrometer operating at 162 MHz <sup>31</sup>P frequency. Reproduced from https://cdnsciencepub.com/doi/full/10.1139/cjc-2022-0041 under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/wUqoGSm2oJuZtUlH8I1wlsD26tFc5kjpfLacTtbQ_xl.webp)
2.4. NMR of other nuclei
Examples of this type are very rare because NMR of other nuclei such as 15N or 19F requires isotopic labelling and/or chemical modification of lignin, which is rather expensive and/or time-consuming.
One of those rare examples appeared during the last 5 years, is the NMR study of the molecular interaction of lignin with amino acid residues of the 15N-labelled carbohydrate-binding module.[54] The authors reported NMR-based analyses of the binding sites of a carbohydrate-binding module of cellobiohydrolase 1 from a hypercellulase-producing fungus. A unique method was developed to obtain Trichoderma reesei carbohydrate-binding module 1 at the C-terminal domain (TrCBM1). Chemical shift analysis revealed that TrCBM1 was adsorbed onto cellohexaose in a highly specific manner via two subsites, the flat plane surface and the cleft, which were located on the opposite sides of the protein surface. Spectral assignments of 13C/15N-labelled TrCBM1 were performed by a standard sequential assignment procedure. As an illustrative example, the 1H – 15N HSQC spectrum of 15N-labelled TrCBM1 is shown in Fig. 42. The final backbone assignments of TrCBM1 were unambiguously made by the authors, however with the exception of eight residues.
![[{"id":"hSuKptnDUa","type":"paragraph","data":{"text":"Proposed structure of <i>Tr</i>CBM1 (<i>a</i>); 2D <sup>1</sup>H – <sup>15</sup>N HMQC spectra of NMR titration experiments using <sup>15</sup>N-labelled <i>Tr</i>CBM1 (<i>b</i>); (<i>c</i>) the close-up view of the region exhibited fairly large perturbations (<i>c</i>). For details, see original publication. NMR spectra were recorded on a Bruker Avance III 600 spectrometer equipped with a cryogenic probe and Z-gradient (Bruker BioSpin, USA). For NMR experiments, 150 μM of the <sup>13</sup>C/<sup>15</sup>N-labeled <i>Tr</i>CBM1 was dissolved in 45 mM sodium acetate buffer (pH 5.0), containing 10% D<sub>2</sub>O and 20 μM 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS). Reproduced with minor editing privileges from Tokunaga et al.[[ type=\"anchor\" referenceId=\"12372\" ]] under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/HJQEKSmUnSmn3Uinds8uwnXGl5jdDTwC2pTv2jBK_xl.webp)
The second example deals with the 19F NMR study of the phenolic hydroxyl groups in lignin modified by pentafluoropyridine, as was reported in 2023 by Kenny et al.[51] The authors proposed a complementary method based on 19F NMR spectroscopy that enabled accurate identification and quantification of phenolic groups in lignin samples by using pentafluoropyridine as a derivatizing reagent. The 19F NMR spectrum of the resulting tetrafluoropyridine ether (Fig. 43) showed signals of two sets of identical fluorine atoms, which gave rise to resonances in two distinct regions, namely a downfield (designated as ‘DF’ in Fig. 43) region, from –89 to –95 ppm (fluorine atoms and integration region are marked blue), and an upfield (designated as ‘UF’ in Fig. 43) region, from –155 to –162 ppm (fluorine atoms and integration region are marked red).
![[{"id":"HAqdb9puCb","type":"paragraph","data":{"text":"<sup>19</sup>F NMR spectrum of the reaction of pentafluoropyridine with a phenolic compound to form a tetrafluoropyridine ether. Resonances marked with a green asterisk belong to pentafluoropyridine, and the resonance marked with an orange asterisk relates for the internal standard, 4,4-difluorobenzophenone. Numbered resonances correspond to 4-propylphenol (<i>1</i>), 4-propylguaiacol (<i>2</i>) and 4-propylsyringol (<i>3</i>), respectively. Reproduced with minor editing privileges from Kenny et al.[[ type=\"anchor\" referenceId=\"12369\" ]] under the Attribution-NonCommercial 4.0 International Public License (CC BY-NC 4.0)."}}]](/storage/images/resized/5p53rxdbrWGaQKyLSOxkxE6HJQTgTGAXagc6LsT1_xl.webp)
However, the use of 19F NMR spectroscopy is still under revision and it is not at the level of 31P NMR for the quantitative and qualitative analyses of hydroxylated moieties of lignins. The former technique is especially suitable for the lignin-derived products such as pyrolysis oils.
3. Conclusion
Recent applications of the one- and two-dimensional 1H and 13C NMR spectroscopy together with NMR of less popular nuclei such as 31P, 19F and 15N to the structural studies of lignin and lignin-derived products in liquid phase have provided a solid breakthrough and a practical guide to their molecular organization. In this respect, 1H and 13C one-dimensional NMR spectra together with different modifications of two-dimensional 1H – 13C HSQC pulse sequences should first be mentioned. Also, NMR of less popular nuclei such as 31P, 19F, and 15N are of considerable interest and are a guiding thread in the structural studies of lignin.
Quantitative NMR spectra of 1H and 13C together with less popular nuclei such as 31P, 19F, and 15N are extensively used to characterize different structural units of lignin (syringyl, oxidized syringyl, guaiacyl, p-hydroxyphenyl, ferulic acid, cinnamyl alcohol end group, cinnamaldehyde end group, and p-coumaric acid, which provide the main lignin linkages — β-aryl-ether β-O-4, resinol β-β, phenylcoumaran β-5 and spirodienone) with aromatic and saturated carbons spread over many structural moieties. The structure of lignin is highly dependent on its origin. While softwood lignins contain mainly guaiacyl units with negligible amounts of p-hydroxyphenyl groups, hardwood lignins are composed of guaiacyl and syringyl subunits. In turn, grass lignins are composed of guaiacyl, syringyl and p-hydroxyphenyl subunits.
Due to the complexity of lignin and the products of its transformation, their NMR spectra consist of a number of overlapping signals belonging to different structural types. In this regard, comprehensive studies of lignin by NMR spectroscopy over the last few decades and especially the last five years have revealed characteristic functional groups of lignin and lignin-related products together with the spectral regions in which they resonate. These data provide a straightforward insight into the chemical structure and stereochemistry of different lignins.
4. List of abbreviations
Ac-KL — acetylated kraft lignin,
Ac-MWL — acetylated milled wood lignins,
Ac-MWL-aca — acetylated milled wood lignins from acacia,
Ac-MWL-mhw — acetylated milled wood lignins from mixed hardwood,
APTAC — (3-acrylamidopropyl) trimethyl ammonium chloride,
BL — bioenzymatic lignin,
COSY — COrrelated SpectroscopY,
Cu-AHP — Cu-catalyzed alkaline hydrogen peroxide lignin,
DF — downfield,
DFT — density functional theory,
DSS — 2,2-dimethyl-2-silapentane-5-sulfonic acid,
G — guaiacyl,
H — p-hydroxyphenyl,
HL — hydrolyzed lignin,
HMBC — heteronuclear multiple bond correlation,
HSQC — heteronuclear single quantum coherence,
HSQC-TOCSY — Heteronuclear Single Quantum Coherence in combination with TOtal Correlation SpectroscopY,
HSQC0 — heteronuclear single-quantum coherence zero,
Int. Std. — internal standard,
KL — kraft lignin,
MWL — milled wood lignin,
OL — organosolv lignin,
pBA — p-hydroxybenzoates,
pCA — p-coumarates,
S — syringyl,
SBL — sulfobutylated lignin,
SML — sulfomethylated lignin,
TMDP — 1-chloro-4,4',5,5'-tetramethyl-1,3-dioxa-2-phospholane,
TOL — Ttall Ooil Llignin,
TrCBM1 — Trichoderma reesei carbohydrate-binding module 1,
UF — upfield.
References









