


Keywords
Abstract
The stereochemistry of tautomeric functionalized azoles is a challenge for theoretical research, since the correct interpretation of the chemical behaviour and biological activity of these compounds depends on understanding the factors that determine the structural features and relative stability of their tautomers. Due to their physicochemical properties, 1,2,3- and 1,2,4-triazoles are the most promising research objects in various fields ranging from medicine, bioorganic chemistry and agrochemistry to materials science. This review is the first in-depth analysis of data on the structure and stereochemical features of functionalized 1,2,3-triazoles (their carbaldehydes, carbaldehydoximes, barbiturates and malononitriles), 1,2,4-triazoles and their model analogues, as well as related compounds, obtained by combined methods based on multipulse and multinuclear 1H, 13C, 15N and 29Si NMR spectroscopy and quantum chemistry calculations.
The bibliography includes 320 references.
1. Introduction
Azoles occupy a special place in heterocyclic chemistry because they are one of the most important classes of privileged heterocyclic scaffolds, which determines their drug-like and universal binding properties. In addition, their chemical structure is highly suitable to modification by some functional groups. Azole compounds are widely used in various fields of agriculture, materials science, industry, biochemistry, and organic and medicinal chemistry. Due to their intensive use in various branches of medicine, technology and agriculture, azoles have attracted the closest attention of researchers. Several monographs and reviews are devoted to the chemistry, properties and application of azoles, in particular, triazoles.[1-15] The results of our investigations of functionalized azoles and related compounds have been summarized in a monograph.[16] Several reviews have been published on the chemistry of azoles,[17-19] organosilicon azoles,[20] NMR spectroscopy and mass spectrometry of nitroazoles,[21][22] and structure and electronic effects of five-membered nitrogen-containing aromatic heterocycles.[23] In addition, the tautomerism and the stereochemical peculiarities of functional azoles have been extensively studied.[24-30] Problems of prototropy of NH-unsubstituted azoles [25] and silylotropy of their N-silylated analogues [27] studied by multinuclear and dynamic NMR spectroscopy, as well as tautomerism and structural features of functional azoles in the solid state studied by nuclear quadrupole resonance (NQR) [26] are discussed in detail. The results of stereochemical studies of biologically active azoles [28] and their organophosphorus derivatives [29][30] using NMR spectroscopy and quantum chemistry have been summarized and analyzed.
Despite the abundance of reviews devoted to the study of the structure of azoles, an overall systematization of data on structural studies of functionalized triazoles has not yet been undertaken.
Readily available triazoles are widely used as pharmaceuticals, high energy materials, agrochemicals, corrosion inhibitors, radiosensitizers, ionic liquids, plasticizers, dyes, precursors for nanocomposites, universal bases in peptide nucleic acids, optical brighteners, fluorescent whiteners, organocatalysts, and synthons for fine organic synthesis.[10-12][14-16][20][24][28] [31-44] Triazoles are a well-known and widely used scaffold in the pharmaceutical industry due to their broad spectrum of activity, high potency, good tolerability and oral availability. Triazoles have long been used to develop numerous drug scaffolds exhibiting anticancer, anti-HIV, antiviral, anti-inflammatory, antibacterial, antiproliferative, antidepressant, anticonvulsant, antioxidant, anticholinesterase, antifungal, analgesic activity, etc.[10-12] [14][15][36-42] [44-56] [57-68] The therapeutic effects of drugs containing 1,2,3- and 1,2,4-triazole moieties are also widely known. Triazole is now increasingly appearing in drug molecules, along with a growing understanding of its important role in drug structures.
Triazole, existing in one of two isomeric forms of 1,2,3- and 1,2,4-triazole, is a preferred object (scaffold) for research in various fields from medicinal and bioorganic chemistry, agrochemistry to materials science due to its physicochemical properties, e.g., large dipole moment, chemical stability (inert nature), tautomeric rearrangements, and the ability to mimic numerous functional groups, including amide ones.[4] [10][11] [32][33] [52][52][59] [69-73] This is a common motif in a variety of approved pharmaceuticals and experimental drugs: ‘There is more function to the 1,2,3-triazole unit than first meets the eye’.[74] In addition, the presence of three consecutive sp2-hybridized nitrogen atoms gives the 1,2,3-triazole ring a special electron-donating ability: compared to all-carbon aromatic non-functional rings, the triazole ring can thus act as a ligand for metal coordination or as a hydrogen bond acceptor and donor.[14] [22] [32][58][71] [73]
The triazole ring is a nitrogen-rich, highly electron-deficient heterocycle, and according to X-ray crystallography and molecular modelling, the triazole moiety is planar.[73] [75] It is also known that conjugation in triazoles does not extend through the triazole moiety (or is sometimes postulated to extend through the formally sp3-hybridized nitrogen atom (N-1) of the triazole).[73] [76][77] Triazole rings are widely used as rigid linkers because they provide a comparable dipole moment, which confines the substituents to an amide-like geometry.[11] [78][79] Triazoles have long attracted close attention of the scientific community due to their unique ability to be incorporated into other functional and bioactive molecules. The combination of two pharmacophores can successfully potentiate their specific biological activity. The identified compatibility of the triazole ring with the peptide moiety is of great interest in the development of new therapeutic targets.[11][70][80-83]
Hepatotoxicity induced by azole antifungals is a major problem in medical practice. Triazole-based drugs are known to have less toxicity and a broader range of antimicrobial properties than other azole analogues, particularly imidazoles. They have a higher affinity for fungal enzymes than for mammalian target enzymes, making them less toxic than imidazole analogues such as ketoconazole and miconazole. Currently available systemic triazoles are voriconazole, fluconazole, posaconazole, and itraconazole.[11][12][14] [84-95] Triazoles represent another group of azoles with a broader antifungal spectrum and a safer profile than imidazoles.
The presence of adverse effects in azole-containing drugs, such as hepatotoxicity and hormonal problems, requires a thorough review of the azole family in order to create drugs with low toxicity and minimal side effects.[12][14] [95] However, the prolonged use of triazole-based drugs at low concentrations provokes the development of drug resistance in some fungal species or even multidrug resistance to them and may cause serious health problems in the future. Therefore, there is a serious need to find new effective, non-toxic and cost-effective antibiotic triazole hybrids.[14] [94] [96]
The ability of triazole to mimic different functional groups, in particular amide moieties, justifies its widespread use as a bioisostere for the synthesis of novel promising molecules.[8] [10] [42] [49][70] [97-104] Triazoles have been successfully used as amide bond bioisosteres in peptidomimetics to modify the pharmacological properties of peptides or proteins. Amides are known to be present in many bioorganic molecules such as proteins, peptides and DNA, and are an important motif in various drugs and functional materials. Due to their unique structure, triazoles can also interact with numerous enzymes, proteins or receptors in the body through weak bonds and can therefore be considered as amide bioisosteres. Bioisosterism is widely used to rationally modify base compounds to increase selectivity, improve potency and pharmacokinetic properties, eliminate toxicity, and gain novel chemical space for intellectual property protection. The design of bioisosteres often introduces structural changes that can be either beneficial or detrimental to biological activity, with size, shape, electronic distribution, polarizability, dipole, polarity, lipophilicity, pKa and H-bond potentially playing key roles in ligand – receptor interactions in molecular recognition and mimicry.
In modern medicinal chemistry practice, the development and application of bioisosteres have been adopted as a fundamental tactical approach useful in addressing a number of aspects related to the design and development of drug candidates. This approach, combined with the optimization of physicochemical and pharmacokinetic properties, and the elimination or modification of toxicophores, opens up new opportunities in drug design and discovery, leading to the clinical development and market introduction of more effective drug candidates.[10] [42] [98] [104-107]
Recently, triazoles have been widely used in epigenetic therapy. Epigenetics is the study of heritable and stable changes in gene expression that occur through changes in the chromosome rather than in the DNA sequence. Epigenetic drugs are the future of cancer therapy. The triazole moiety is associated with compounds used in drug development that can potentially inhibit histone-modifying enzymes. Triazole isosteres have been successfully used in the therapy of epigenetic targets, including reader proteins.[11][70][100][106][108-125] Hybridization of the triazole moiety with different pharmacophores is an excellent strategy to create promising bioorganic molecules. The ease of preparation of triazole hybrids in good yields, high propensity to interact with multiple targets, anisosteric substitution of various functional groups and their high stability and better pharmacology encourage researchers to develop novel hybrids to study their medicinal properties.[108] [126-132]
The unusual properties of the triazole ring as an intriguing ring system for the development of a new medicinal chemistry strategy for the design and creation of triazole analogues as inhibitors of various biological targets were discussed in the review.[104] The versatility of triazole chemistry and the synthetic availability of this heterocycle make it an ideal building block for generating libraries of compounds for more comprehensive studies of structure – activity relationships. Triazole derivatives are readily accessible via click chemistry, giving rise to a diversity of compounds against numerous diseases, and their use in medicinal chemistry has recently attracted much attention from the global medical and chemical communities due to the development of their novel regioselective syntheses.[4] [32][133-141] In particular, the review [138] is devoted to the development of chemical catalytic synthesis of functionalized 1,2,3-trizoles and fills the gap in reports on the evolution of catalytic methods used in this field of organic chemistry in recent years (since 2015). The preparation of such compounds has mainly focused on new metal catalysts together with the implementation of green chemistry principles. The use of ionic liquids and other unconventional solvents reveals promising chemical routes, with special attention not only to the use of new catalysts, but also to the possibility of recycling the entire catalytic system. At the same time, the use of new metals alternative to the classical ones (Cu and Ru) allows for the implementation of numerous reactions, laying the foundation for a fruitful ‘green’ branch of chemical implementation of unusual molecules.
Halogenated triazole derivatives have found wide application in biology and medicinal chemistry as bioisosteres of nucleic bases in carbanucleosides, nucleosides, peptides and proteins. Coelho et al.[142] described the synthesis, functionalization and application of halotriazoles in biologically relevant molecules. Much attention is given to iodinated triazoles, which are capable of forming halogen bonds and providing efficient transport of anions across lipid bilayer membranes. The synthesis and modifications of halotriazole scaffolds in biologically active molecules, including nucleosides, carbohydrates, peptides and proteins, are discussed.
The triazole ring exhibits remarkable stability under reducing, oxidative, and hydrolytic conditions. In addition, the triazole ring has low multidrug resistance and toxicity, high bioavailability and stability under both basic and acidic conditions, which may be due to the presence of the nitrogen atom in the ring, for example, in enzyme-inhibitor interactions.[33] [46] [56][59] [70]
The importance of triazole compounds in medicinal chemistry is undeniable. Therefore, comprehensive comparative and targeted studies are needed to further understand the role of the triazole ring in molecular recognition. The unique properties and diverse biological activities of triazoles require a deeper understanding of the features of their electronic structure, spectral properties, stereochemical behaviour, reactivity and tautomeric transformations.
2. 1,2,3-Triazoles
1,2,3-Triazole is a five-membered, unsaturated aromatic heterocycle with π-excess nitrogen and a 6π electronic ring system, consisting of three consecutive nitrogen atoms and two carbon atoms with two double bonds. Of the three nitrogen atoms, one is of the pyrrole type, and the other two are of the pyridine type. All atoms in 1,2,3-triazoles are sp2 hybridized, and the available 6π electrons are delocalized around the ring, which is responsible for their aromatic character. The parent 1H-1,2,3-triazole is a colourless liquid, highly soluble in water, density 1.192, m.p. 23 – 25°С, b.p. 203°C. The dipole moment of the tautomeric mixture in benzene is 1.85 D. Depending on the substituents, 1,2,3-triazoles can have large dipole moments of up to 5 D. The pKa value of 1.17 for 1H-1,2,3-triazole indicates that it is a weak base, while the conjugate base has pKa = 9.4 indicating that it is a weak acid.[31] This means that 1,2,3-triazole is both a weak base and a weak acid, comparable in strength to phenol. In the solid state, triazole exists as a mixture of 1H and 2H tautomers in a 1 : 1 ratio.[143] Some of the basic properties of triazoles, such as dipole moment, planarity, and hydrogen bonding properties, are similar to those of amides. The two pyridine nitrogen atoms in the triazole have lone electron pairs (LEPs), which that act as hydrogen bond acceptors, similar to the amide oxygen. The CH bond in a triazole has a large dipole moment and can therefore function as a hydrogen bond donor, similarly to an NH amide. Because of these properties, 1,2,3-triazoles can form hydrogen bonds and participate in π-interactions.[7] [11] [31][32][104] [107] [143][144]
Tautomerism and reactivity of azoles, especially 1,2,3-triazoles, are discussed in detail in well-known studies by Elguero and co-workers.[2][3][145] [146] The reactivity of azoles with acetone was studied by 1H and 13C NMR at low temperatures in acetone-d6 .[146] The reaction mechanism theoretically calculated at the density functional theory (DFT) level was compared with experimental results. The calculated energy profiles of the reaction of various azole tautomers with acetone and a detailed analysis of the stability of these tautomers are discussed. The relative energies of the most stable tautomers and the equilibrium constants between azoles and adducts (α,α-dimethyl-azole – methanol) are given. The main results presented in the publication [146] allow the authors to correct some early literature data on the study of annular tautomerism of NH-azoles.
Although 1,2,3-triazoles do not occur in nature, this class of five-membered heterocyclic compounds is widely used in medicinal chemistry. The combination of such important properties of the triazole ring as chemical stability, high dipole moment, heteroaromatic nature of the cycle and the ability to donor – acceptor interaction during the formation of a hydrogen bond are productively used in organic synthesis to create new materials, biologically important molecules, new effective and safe drugs with antiviral anti-HIV, antimicrobial or antiepileptic activity. The most promising antibiotics are tazobactam and cefatrizine, anticancer and anti-HIV TSAO amidotriazoles (TSAO is tert-butyldimethylsilylspiroaminooxathioledioxide) (Fig. 1).[51] [137] [147]
![[{"id":"8rFFFIeuS4","type":"paragraph","data":{"text":" Promising triazole-based drugs"}}]](/storage/images/resized/1sGFgI9Yl7l6tfoBCidYXpelKpN525PpFdaZkJUn_xl.webp)
An active search for new inhibitors based on functionalized 1,2,3-triazoles using in silico approaches is currently underway. To date, several groups of potential 1,2,3-triazole-containing ligands have been found that show activity against a number of macromolecules (Table 1). The binding energies of the listed compounds with macromolecular structures were calculated using specialized software packages for docking modelling.
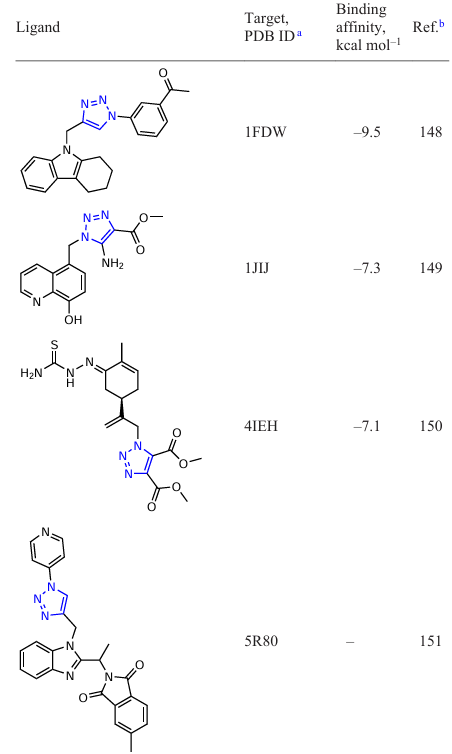
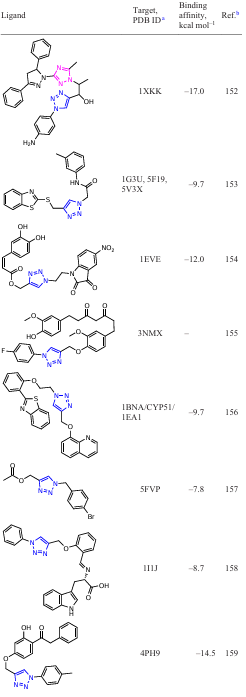
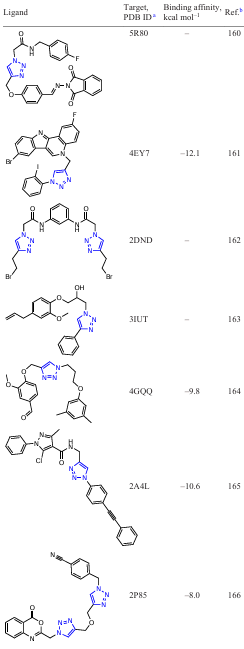
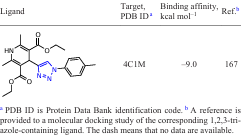
The results of such studies can be summarized in the form of a diagram of the distribution of target – ligand (1,2,3-triazole-based derivatives) binding energies (Fig. 2). As can be seen from these data, the strongest binding energies to target proteins occur with ligands including N1 – Ar derivatives of 1,2,3-triazoles. This may be due to the fact that in this case, a conjugation of the benzene ring with the triazole ring is formed via the ortho proton of the aryl moiety due to the formation of a hydrogen bond of the N∙∙∙H type.
![[{"id":"_s7aCUzTKx","type":"paragraph","data":{"text":"Summary bar chart of the distribution of target – ligand binding energies (1,2,3-triazole-based derivatives) according to studies.<sup>148 – 167</sup> Yellow bars represent average binding energies. Bn = CH<sub>2</sub>Ph."}}]](/storage/images/resized/v0W6Uny7JKyT3VumPiBnXrOKKlV6M8zl242z1AYc_xl.webp)
In addition to the above-mentioned 1,2,3-triazoles, 1,2,4-triazoles are also capable of acting as potential inhibitors of target proteins of some pathogenic microorganisms.[168-181]
Despite the availability of various synthetic approaches to 1,2,3-triazoles, the most practical and efficient method for obtaining 1,4-disubstituted 1,2,3-trizoles at present is the Cu(I)-catalyzed click reaction between azides and terminal alkynes. The limitations of this method are the inability to use disubstituted alkynes and to obtain N-unsubstituted triazoles. The disadvantages of the traditional thermal 1,3-dipolar cycloaddition of organic azides to alkynes (the Huisgen reaction) are the rather harsh conditions and the non-regiospecificity of the addition. In recent years, the interest of researchers in the use of water as a reaction medium has grown rapidly due to the increased rate and sometimes selectivity of reactions compared to conventional organic solvents, as well as the cost-effectiveness and environmental safety, in accordance with the requirements of green chemistry, and the importance of understanding the biochemical processes occurring in biological systems. The practical value of developing regiospecific methods for the synthesis of 1,2,3-triazoles in the absence of catalysts, including under physiological conditions, is due in particular to the limitations associated with the toxicity of Cu(I) in a living cell, despite the broad prospects for the application of bioorthogonal click reactions.
2.1. 1,2,3-Triazole-5-carbaldehyde oximes
Research on various types of azoles, including triazoles, has been going on for a long time. Recently, the high efficiency of using water as a solvent in the synthesis of functionalized 1,2,3-triazoles in almost quantitative yields at room temperature in the absence of a catalyst has been demonstrated. Functionalized 1,2,3-triazole-5-carbaldehyde oximes can be prepared in various ways as indicated in Scheme 1. It was found that the most practical, effective and convenient procedure includes the use of microwave irradiation (Scheme 2). For example, 4-substituted-1,2,3-triazole-5-carbaldehydeoximes (1 – 9) were obtained by reacting the corresponding derivatives of propynal, trimethylsilyl azide, and hydroxylamine in an aqueous methanol under microwave irradiation [182-186] (see Scheme 1 and Scheme 2).
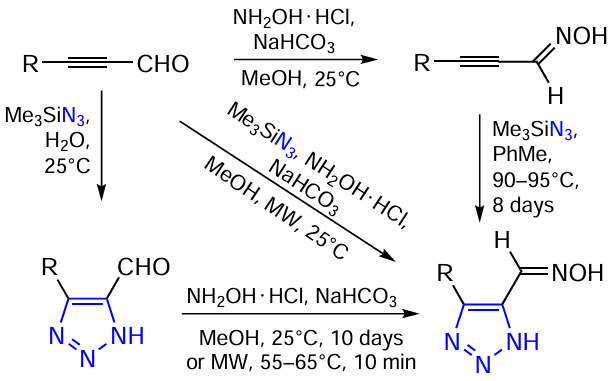
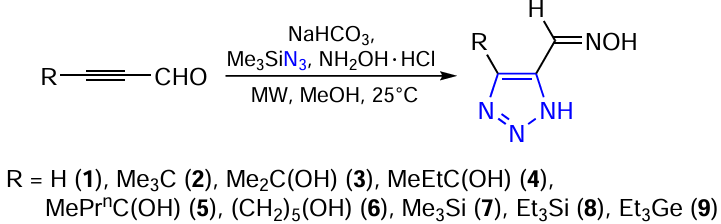
A more effective and convenient synthetic approach to 1,2,3-triazolecarbaldehyde scaffold is the direct use of the corresponding propynals. Propynals as bielectrophiles with a sterically unhindered aldehyde group occupy an important place among acetylenes with an activated triple bond. The presence of a silicon or germanium heteroatom at the triple bond of propynal stabilizes the aldehyde molecules and the resulting adducts, and subsequent demetallization under mild conditions can deliver heterocyclic analogues with a terminal triple bond.
Trialkylsilylpropynals are used in the synthesis of natural cytostatics, viz. forboxazoles, platelet aggregation inhibitor xemilofiban, as well as butadiynyl polyconjugated porphyrin assemblies, which are promising for obtaining materials used as sensors, reading devices, photochemical energy converters, magnetoactive materials (see Scheme 2).
The advantages of this synthesis are high yields, regioselectivity, absence of a catalyst and by-products, insensitivity of products to atmospheric moisture and oxygen, availability of starting reagents, cost-effectiveness corresponding to ideal synthesis and green chemistry requirements, and near physiological conditions.
The structure of oximes 1 – 9 was determined by multinuclear 1H, 13C, 15N, 29Si and two-dimensional (COSY-gp, NOESY-gp, HSQC-gp, HMBC-gp) NMR spectroscopy. It is well known, that 1,2,3-triazoles unsubstituted at the nitrogen atom exist in solution as an equilibrium mixture of three annular tautomers A, B, C, which causes significant experimental difficulties in determining their structure (Scheme 3).[24][25]
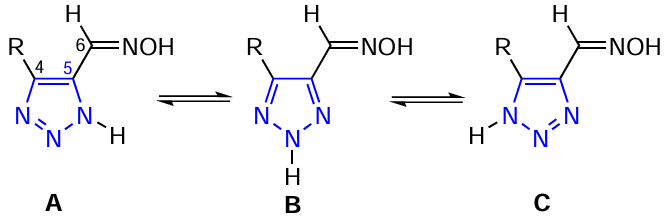
The prototropic transformations of the vast majority of azoles in solution are so fast on the NMR time scale that a decrease in the solution temperature does not lead to a transformation of the spectra and the acquisition of new information. In all cases, the spectra are averaged. Some slowing down of prototropy can only occur in structurally complicated tautomeric heterocycles. Since the tautomeric system of 1,2,3-triazole is also in a state of dynamic equilibrium, it is extremely difficult (or impossible) to detect NMR signals from the nitrogen atoms of triazoles of this type. The annular (ring) prototropic exchange in 1,2,3-triazole proceed also very rapidly.[2][3] [24][25] [28] In the 1H NMR spectra of the studied 1,2,3-triazoles, broad signals from the protons of NH and OH groups are observed in the region of 14 – 16 and 11 – 12 ppm, respectively. The broadening of the NH signals, as well as their downfield shift in the 1H NMR spectra indicates the involvement of the NH proton in the tautomeric process through the formation of hydrogen bonds. The signals from the carbon atoms in the 13C NMR spectra are also broadened, indicating the presence of an exchange prototropic process. Therefore, it is not possible to detect the signals from all the nitrogen atoms in the molecule in the 15N NMR spectra of an equilibrium tautomeric mixture of 1,2,3-triazoles (Table 5).
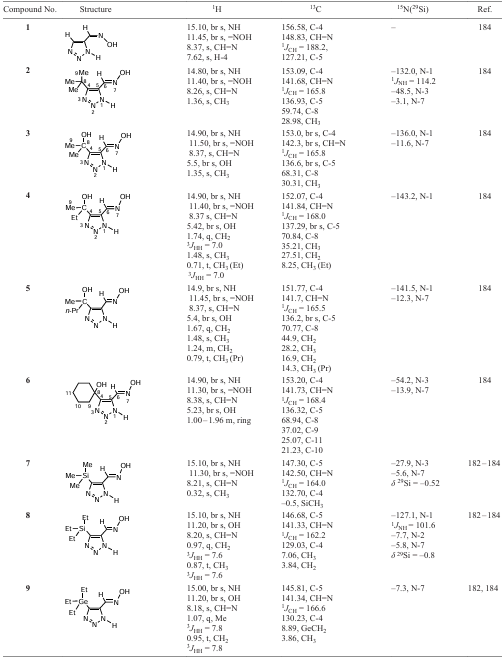
The orientation of the oxime group is determined from the 13C{H} NMR spectrum without proton decoupling and from the two-dimensional {1H – 13C} HMBC NMR spectra. The values of the 1JC6 – H coupling constants for most of the compounds studied are in the range of 162 – 168 Hz, indicating the existence of molecules 2 – 9 in the form of the E-isomer, while 1,2,3-triazolecarbaldehyde oxime (1) (1JC6 – H = 188.2 Hz) manifests itself as the Z-isomer (see Table 5).
The assignment of the C-4 and C-5 carbon signals of the triazolyl ring is based on two-dimensional spectra. In the {1H – 13C} HMBC NMR spectra of compounds 1 – 9, cross peaks of the protons of the methyl (or methylene) group of the substituent at position 4 with the C-4 carbon atom, as well as cross-peaks of the CH=N proton with the C-4 and C-5 carbon atoms are observed. The main spin-spin coupling constants (SSCCs) and NOESY correlation are given in Fig. 3 for compound 7 as an example.
![[{"id":"6ATL4dKWvw","type":"paragraph","data":{"text":"Main <i>J</i><sub>C – H</sub> and NOESY correlations for compound <b>7</b>. Reproduced from Semenov and Larina <sup>184</sup> with the permission of the American Chemical Society"}}]](/storage/images/resized/15oQCzWEr7l73G002eyPul1pfgwvNfc1ZCsTsX5x_xl.webp)
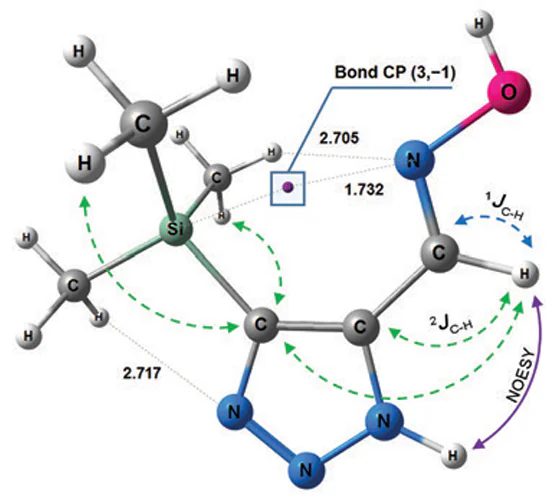
It was found that organosilicon derivatives of 1,2,3-triazole oximes (e.g. compound 7) can be stabilized by non-valent Si – N interactions (Fig. 4).[183] The electron density analysis carried out using the Localized Orbital Locator (LOL) descriptor [187][188] allowed the detection of the (3, –1) type bond critical point between the Si and N-7 atoms in all the main conformers. The energy of this tetrel bond is estimated to be ~ 1 kcal mol–1. Thus, the existence of a stable intramolecular interaction between the =NOH nitrogen and the silicon atoms, leading to the stabilization of the syn-periplanar form of silicon-containing oximes of 1,2,3-triazoles, cannot be denied.
![[{"id":"_7RzH2HQMF","type":"paragraph","data":{"text":"Equilibrium geometry of the <i>E-syn</i>-periplanar conformer of compound <b>7</b>, optimized at the MP2/aug-cc-pVTZ level. Interatomic distances are given in Å. Purple corresponds to the bond critical point (3, –1)."}}]](/storage/images/resized/RAJtzD8BxY8MlDqI3nWvU51E3JSvYDUYIscTUd1G_xl.webp)
Subsequently, stereochemical and computational NMR analyses were carried out on some representatives of functionalized 1,2,3-triazoles in search of the original tauto-conformers.[184] Of course, the search for each tauto-conformer for all 1,2,3-triazole derivatives is a very difficult task. In each case, at least 12 dynamic states should be taken into account: three prototropic tautomers for each of the four conformers. It is essential to consider the E- and Z-isomers relative to the double bond of the oxime moiety, as well as the syn- and anti-conformations of the oxime moiety relative to the triazole ring (rotation by the dihedral angle С-4–С-5–С-6 – N-7 (Fig. 5).
![[{"id":"ceW0nj9EUt","type":"paragraph","data":{"text":"Tautomerism (purple) and conformation isomerism (blue) in 1,2,3-triazoles. Reproduced from Semenov and Larina <sup>184 </sup>with the permission of the American Chemical Society"}}]](/storage/images/resized/AJR9ECr3jjeEySgQOXw2PzL7VAyqNeMpyS2AP7KT_xl.webp)
After performing the initial conformational search, the geometric parameters of all 108 putative tauto-conformers were optimized at the DFT level using the PBE0/cc-pVTZ//aug-cc-pVTZ computational scheme.[189][190] Basis sets extended by diffuse functions were used to account for non-valent interactions of nitrogen and oxygen LEPs. Non-specific solvation of the molecules was considered in the framework of Tomasi’s Polarizable Continuum Model (IEF-PCM).[191][192] Possible tauto-conformational states of 1,2,3-triazole oxime derivatives were studied and established using the correlation of experimental and theoretical NMR chemical shifts. Calculations of 1H and 13C NMR isotropic magnetic shielding constants and the corresponding chemical shifts have been carried out using the one-parameter hybrid functional mPW1PW91 [193] in combination with Pople’s triple-zeta basis set 6-311+G(d,p) [194] in the liquid phase of a particular solvent.
The studied NH-unsubstituted triazoles are in exchange dynamic equilibrium in solution due to annular prototropic tautomerism. This leads to a broadening of the signals in the NMR spectra, which makes it difficult to assign the signals reliably. The DP4+ approach [195] was used to identify the main tauto-conformational states that contribute to the actual spatial structure of these triazoles and determine the resulting NMR shielding constants (and their corresponding chemical shifts) (Fig. 6). For most of the 4-substituted 1,2,3-triazoles carbaldehyde oximes, experimentally unresolved NMR signals were assigned.
![[{"id":"fOmAVNa0N8","type":"paragraph","data":{"text":"DP4+ probability distribution of the main tauto-conformers of 2 – 9 (%). Reproduced from Semenov and Larina<sup>184</sup> with the permission of the American Chemical Society."}}]](/storage/images/resized/6K9CsMPih9x3rqrjSonhFL97vp04Ipekx9U7noQG_xl.webp)
2.2. 1,2,3-Triazole-5-carbaldehydes and related compounds
Functionalized 1,2,3-triazole-5-carbaldehydes, the precursors of 1,2,3-triazolecarbaldehyde oximes, were prepared by reacting the corresponding propynals with azides in water at room temperature (Scheme 4).[185][186]
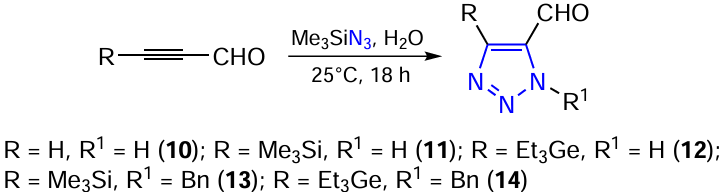
The resulting triazoles are biologically important compounds, building blocks for organic synthesis and ligands of metal complexes. The structure of the compounds has been determined by IR spectroscopy, X-ray diffraction analysis and multinuclear and two-dimensional NMR spectroscopy (Table 6).[185][186]
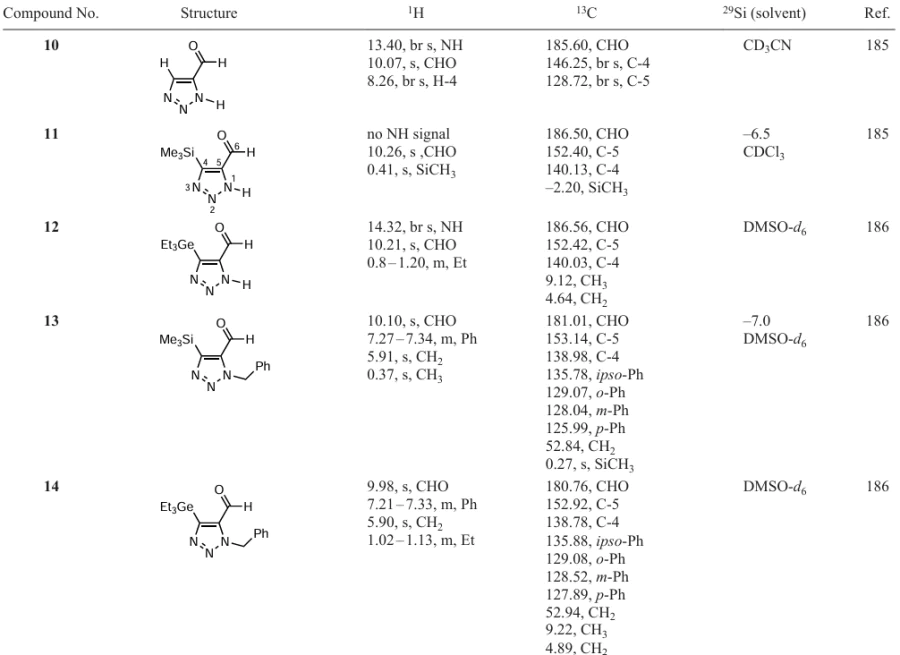
Unsubstituted formyl triazole 10 was isolated as a crystalline solid with m.p. 137 – 139°С (benzene – hexane) in 74% yield (see Scheme 4). The presence of a broadened signal at 8.26 ppm in the 1H NMR spectrum of a solution of triazole 10 in CD3CN, as well as signals with 128.72 and 146.25 ppm (C-4 and C-5) in the 13C NMR spectrum indicates migration of protons between nitrogen atoms in the triazole ring. The NH proton also appears as a broad singlet at 13.4 ppm. The 1H NMR spectrum of trimethylsilyl-substituted formyl triazole 11 in CDCl3 contains signals from the protons of the trimethylsilyl group and the aldehyde proton at 0.41 and 10.26 ppm, respectively. In CDCl3, the NH proton signal is not observed. In the spectra recorded in CD3CN and DMSO-d6 , the NH signal appears at 13.1 and 15.46 ppm, respectively. This is apparently due to the rapid prototropic exchange in chloroform and the associated strong broadening of the NH proton signal.[185][186]
Formyltriazoles 10 and 11 dimerize in solution to the corresponding tricyclic bis-hemiaminals 15 and 16 (Scheme 5).
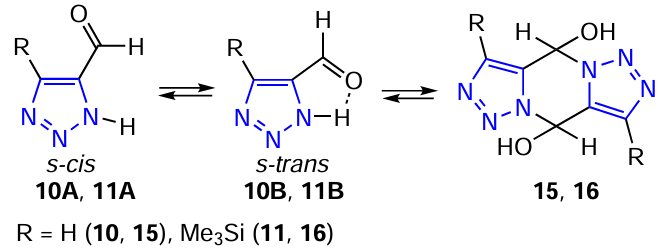
The 1H NMR spectrum (CD3CN) of the reaction mixture obtained from propynal and trimethylsilyl azide contains signals from the triazole proton 10, the methine proton in the pyrazine ring of tricyclic hemiaminal 15 at 6.46 ppm (2H, d) and methine proton in the triazole ring at 8.64 ppm (2H, d); the monomer-dimer ratio is 1 : 1. In CD3OD at room temperature, the monomer – dimer equilibrium (ratio is 1 : 3) is reached within 2 hours. However, the proton spectrum of the reaction mixture formed from trimethylsilylpropinal and trimethylsilyl azide contains signals belonging to triazole 11, as well as a signal at 7.33 ppm (2H, d), which is related to the CH proton in the pyrazine ring 16 (monomer : dimer ratio is 1 : 1).
According to the IR spectroscopy data, 1,2,3-triazole-5-carbaldehydes (10, 11) adopt the s-cis conformation of the carbonyl group relative to the C=C double bond of the triazole ring in the solid state. The X-ray diffraction analysis of the molecular structure of 4-trimethylsilyl-1,2,3-triazole-5-carbaldehyde (11) supports these data. The silicon atom in molecule 11 has a tetrahedral configuration. The distance between the silicon atom and the C4 atom in the triazole ring [1.858(7) Å] is slightly longer than the other bonds formed by the silicon atom. The triazole ring is planar: the N1, N2, N3, C4, and C5 atoms deviate from the mean-square plane by 1.13, 1.41, 0.54, 0.4, and 1.11°, respectively, i.e., not more than 1.5°. The aldehyde group is in the plane of the triazole ring.[185][186]
The results of quantum-chemical calculations (AM1) in the gas phase showed that structures 10B and 11B with intramolecular hydrogen bonds are more favourable. The energy of the hydrogen bond in 10B is 5.0 kcal mol–1, and the distance between NH hydrogen and the oxygen atom is 2.57 Å. The charges on the oxygen and hydrogen atoms are –0.28 and 0.29 e, respectively. The planar arrangement of the triazole ring and the aldehyde group favours stronger hydrogen bonding. The influence of medium polarity and temperature on the ability of 1,2,3-triazole carbaldehydes to dimerize to tricyclic bis-hemiaminals has been studied by IR and NMR spectroscopy.[185]
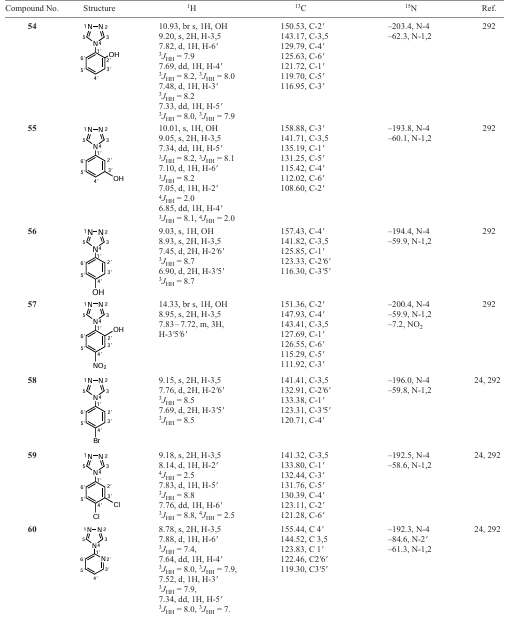
Polyfunctional 4-hydroxyalkyl-1H-1,2,3-triazole-5-carbaldehydes (A, B) were synthesized under optimal conditions for the preparation of 4-trialkylsilyl(germyl)-1H-1,2,3-triazole-5-carbaldehydes (11 – 14). It was found that, unlike element-containing propynals, g-hydroxyalkylpropynals add trimethylsilyl azide regioselectively under similar conditions: 4-hydroxyalkyl-1H-1,2,3-triazole-5-carbaldehydes (A) are formed in 70 – 95% yield, the content of minor isomers of 5-hydroxyalkyl-1H-1,2,3-triazole-4-carbaldehydes (B) is 10 – 20% (according to 1H NMR).[196] The non-catalytic addition of azides to the triple bond usually occurs non-regioselectively; the formation of 1,4-isomers (B) can be facilitated by the formation of an intramolecular hydrogen bond H – O∙∙∙H – N (Fig. 7).
![[{"id":"fJY9YxQ00L","type":"paragraph","data":{"text":"The formation of an intramolecular H-bond in 4(5)-hydroxyalkyl-1H-1,2,3-triazole-5(4)-carbaldehydes (<b>A, B</b>)."}}]](/storage/images/resized/BO9RpSfsMrbtnOuhvQ5YeNpZCnI9yERMVAq08UOF_xl.webp)
The stereochemical and configurational assignment of carbon-, silicon- and germanium-containing propynal oximes (17 – 21), the closest precursors of 1,2,3-triazole oximes, was performed using experimental measurements and ab initio high-level calculations of their 13C – 1H, 13C – 13C and 15N – 1H SSCCs (Fig. 8 and Table 7).[197]
![[{"id":"0GfBj6WFB2","type":"paragraph","data":{"text":"The effect of the nitrogen lone pair orientation on <sup>13</sup>C – <sup>1</sup>H, <sup>13</sup>C – <sup>13</sup>C and <sup>15</sup>N – <sup>1</sup>H coupling constants in the configurational isomers of propynal oximes <b>17 – 21</b>. Reproduced with minor editing privileges from Chernyshev et al.<sup>197</sup> with the permission of Wiley"}}]](/storage/images/resized/BESG9yUYxybWgz38N4MUMjePZa5k9xaCLv90fdHa_xl.webp)
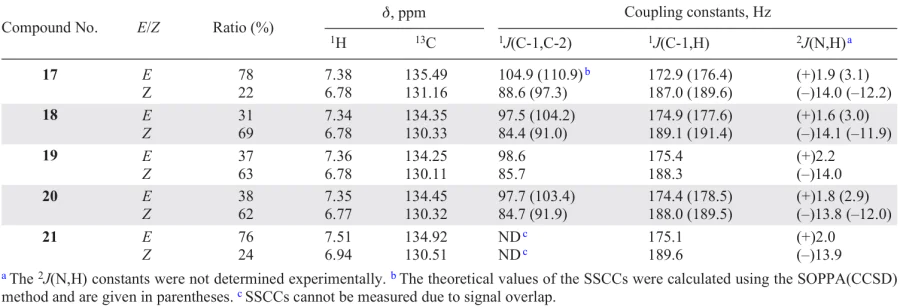
The results of the energy calculations of molecules 17 – 21 at the MP2/6-311G(d, p) level [194][198] showed that propynal oximes exist as non-equilibrium mixtures of E- and Z-isomers with an energy difference of less than 0.3 kcal mol–1. The configuration was determined experimentally from the 13C – 1H, 13C – 13C coupling constants obtained from proton-decoupled 13C NMR spectra and INADEQUATE spectra, respectively. The configurational assignment of acetylene oximes 17 – 21 was based on the known stereospecificity of the experimental 13C – 1H, 13C – 13C and 15N – 1H coupling constants towards the orientation of the nitrogen LEP in different isomers of the oximes, due to their second-order polarization propagator approach in combination with coupled cluster single and double excitation model SOPPA(CCSD) [199] calculations. The theoretical values of the 13C – 1H and 15N – 1H SSCCs are in good agreement with the experimental ones (see Table 7).
The Table 7 shows that the signals of the HC= proton and the C-1 carbon atom for all compounds 17 – 21 are shifted downfield and correspond to the E-isomer, and high-field signals correspond to the Z-isomer. Indeed, the 13C – 13C SSCCs are significantly higher, and the 13C – 1H SSCCs are significantly smaller for the low-field signal C-1 than for the high-field signal, which is due to the influence of the LEP of the azomethine nitrogen atom. In addition, calculations of the coupling constants, carried out at a high theoretical level, reproduce well the general tendency for the constants to differ depending on the orientation of the nitrogen atom LEP with respect to the neighbouring bonds and are in excellent agreement with their experimental values.
The essence of the method used to determine the configuration is that the LEP of the imine nitrogen in oximes makes a positive spatial contribution to the value of the coupling constant of the neighbouring Csp2 – Csp bond, which is in the cis-position to it. On the other hand, the transfer of the density of the unpaired electron from the azomethine nitrogen atom to the antibonding molecular orbital of the neighbouring bond, which is in the trans-position to the LEP (nσ – σ* interaction), leads to an elongation of this bond and to a decrease of the corresponding constant, i.e., the contribution LEP is negative at full value of this constant.
Although the calculated values of the 13C – 13C constants involving an sp-hybridized carbon of the triple bond are somewhat overestimated, the calculated differences between them in the E- and Z-isomers are very close to those observed experimentally.197 Thus, according to the data in Table 7, all constants show strong stereospecificity for the orientation of the nitrogen LEP in different isomers. A comparison of the theoretical and experimental values of the constants of the E and Z isomers of the said oximes clearly indicates that compounds 17 – 21 exist as a mixture of E and Z isomers in solution with a predominance of the E isomer of 4,4-dimethyl-2-pentynal oxime 17 and 3-triphenylgermyl-2-propynal oxime 15, while the Z-isomer predominated in the oximes of 3-trimethylsilyl-, 3-triethylsilyl- and 3-ethylgermyl-2-propynals 18, 19, 20, respectively.
Thus, the results obtained allow us to draw an unambiguous conclusion about the configurations of the entire series of oximes studied. The determining influence of the heteroatom at the triple bond on the isomeric composition of oximes has been demonstrated.
The structure of element-substituted NH-1,2,3-triazoloimines, 5-(1,3-thiazolan-2-yl)-1H-1,2,3-triazoles and 5-R-imino-[4-trialkylsilyl(germyl)]-1H-1,2,3-triazoles obtained from Si/Ge-substituted propynals, trimethylsilyl azide and functionalized primary amines by the microwave-assisted solvent- and catalyst-free synthesis has been studied.[200]
2.3. 1,2,3-Triazole barbiturates
Derivatives of barbituric acid are widely known in pharmacology as antimicrobial, antioxidant, fast-acting agents for emergency anesthesia and the prevention of epileptic seizures, relief of seizures, selective cell adhesion inhibitory and DNA-cleaving activity. Barbituric acid (pyrimidine-2,4,6-triones) is actively used in medicine as tranquilizer (phenobarbital, nembutal), anticancer (benzylbarbital) and hypnotic agents (Fig. 9).
![[{"id":"rpl3M5PZsK","type":"paragraph","data":{"text":"Known drugs based on barbituric acid"}}]](/storage/images/resized/1Vl2bIZVtgMnbCw3p9DLLsAO7XqbQf0VpaIXMYTV_xl.webp)
Even minor structural modifications, especially changes in the substituents on the C-5 atom, can have a decisive effect on the biological activity of these compounds. Important derivatives of barbituric acid are the 5-ylidene derivatives. Some of these have non-linear optical properties and are used as ligands in coordination chemistry. The combination of barbiturate heterocyclic rings and 1,2,3-triazole in a molecule affords new hybrid compounds with potential biological activity.[201][202]
Hydroxyalkyl-containing 5-(1H-1,2,3-triazol-4-ylmethylidene)barbiturates (22 – 29) were synthesized by a one-pot three-component reaction of 4-hydroxyalkynals, trimethylsilyl azide and barbituric acids in aqueous medium (Scheme 6). The reaction is catalyst-free and proceeds under mild eco-friendly conditions.
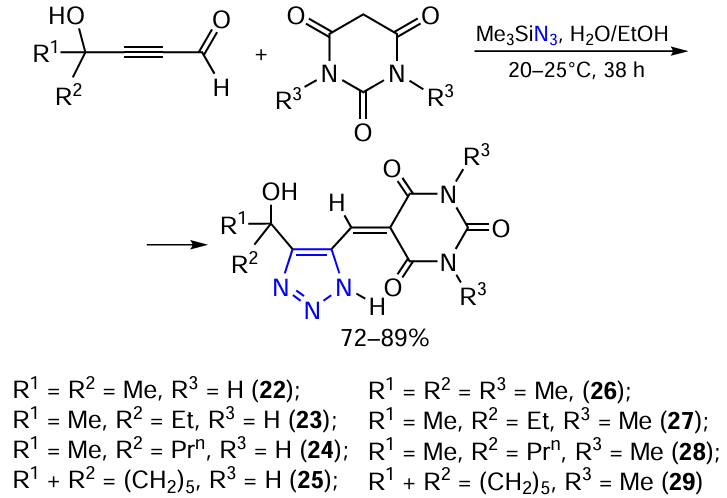
All products are easily isolated by successive washing of the crude precipitate with ethyl acetate and methanol. The isolated product yields are not quantitative because of possible losses during purification and minor polymerization of hydroxyalkynals. Steric hindrance has little effect on product yields. For example, hydroxycyclohexyl-substituted compounds 25 and 29 were isolated in 72 – 73% yields. Сompounds 22 – 29 are poorly soluble in most organic solvents. The structures of the 1,2,3-triazole barbiturates 22 – 29 have been confirmed by multinuclear 1H, 13C NMR and multipulse 2D NMR spectroscopy (Table 8).
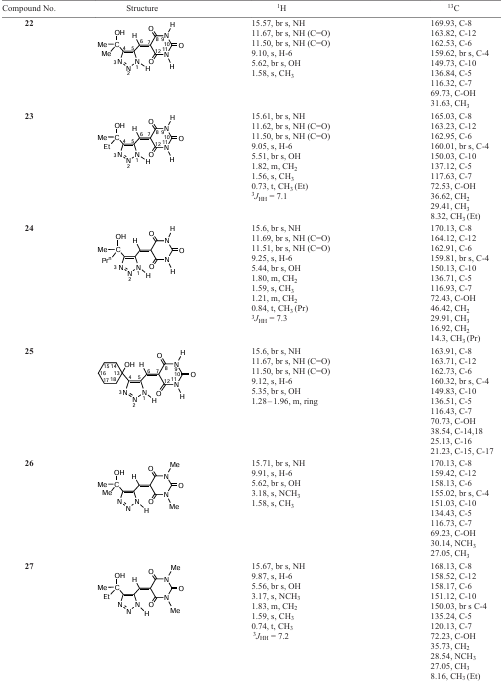
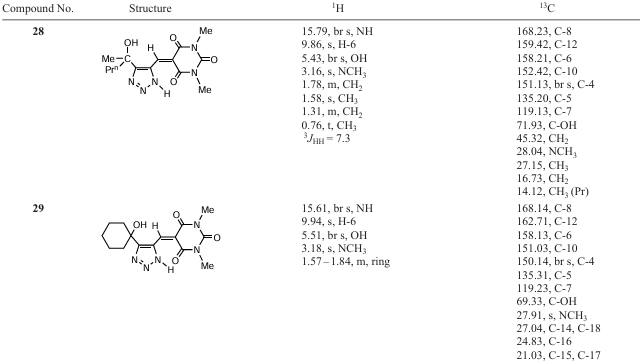
In the 1H NMR spectra, the olefin proton =CH appears at 9.00 – 9.95 ppm, and the NMe protons (26 – 29) are in the region of 3.1 – 3.18 ppm. The carbonyl carbon atoms in the 13C NMR spectra resonate at 165.0 – 170.1, 158.6 – 164.1 and 149.7 – 152.4 ppm, and the signals of the carbon atoms =CH and the triazole ring C-4 and C-5 appears at 158.1 – 162.9 ppm, 150.0 – 160.3 and 134.4 – 137.1 ppm, respectively.
The hybrid 1,2,3-triazole barbiturates, containing two important pharmacophores, are of interest as biologically active compounds and synthons for organic synthesis to produce new drugs and polydentate ligands for metal complexes and fluorophores for the creation of new materials.[201][202]
2.4. 1,2,3-Triazole malononitriles
Alkylidene- and arylidenemalononitriles are versatile synthons for the synthesis of structurally diverse 2,6-dicyanoanilines and heterocyclic compounds with good pharmaceutical profiles or fluorophores. 4-Substituted 1,2,3-triazole malononitriles 30 – 36 were obtained by a three-component reaction of the corresponding propynals, trimethylsilyl azide and malononitrile in water at room temperature using β-cyclodextrin as a catalyst (Scheme 7).[203] Supramolecular catalysis has attracted considerable attention from researchers in recent years for carrying out reactions under biomimetic conditions.
Of particular interest are cyclic oligosaccharides –cyclodextrins, which have a hydrophobic cavity. Among them, the most widely used is commercially available β-cyclodextrin (β-CD), the dimensions of the internal cavity of which allow the formation of inclusion complexes with a variety of guest molecules. Catalysis by cyclodextrins involves the reversible formation of host-guest complexes by non-covalent binding, similar to enzymes.[204] Cyclodextrins containing hydrophobic cavities selectively bind substrates and catalyze chemical reactions with high efficiency and selectivity to give, e.g., for example, heterocyclic [205-207] and heteroelement-containing [208] compounds. Since the catalytic action of cyclodextrins involves the encapsulation of a guest molecule in the host cavity, the authors studied the formation of the host – guest complex of β-CD with highly reactive hydrophobic trimethylsilyl- and triethylgermylpropynals RC≡CCH=O (R = Me3Si, Et3Ge) in an aqueous solution at room temperature. The complexation process depends on the size, shape and hydrophobicity of the guest molecule. 1H NMR spectroscopy has proved to be one of the most suitable and effective methods to study host – guest complex formation with cyclodextrin and since the inclusion of the guest molecule in the host cavity induces characteristic shifts of signals from the protons in the host molecule. Analysis of the 1H NMR spectrum in D2O in the region of 3.20 – 3.65 ppm revealed a significant effect of the nature of the element-containing group on the complexation process in the aqueous phase.[208]
Many compounds containing the NH-triazole moiety are used as antituberculosis, anti-HIV and anticancer agents, neurokinin-1 receptor antagonists, metallo-β-lactamase inhibitors and ligands to produce remarkable coordination materials. The toxicity of copper compounds, which induce metabolic disorders and oxidative damage in biological systems, limits their use in click chemistry, but also stimulates the important development of new metal-free methods for triazole synthesis.
The structure of 1H-1,2,3-triazole dicyanoalkylidenes 30 – 36 has been confirmed by 1H, 13C, 29Si and two-dimensional (COSY-gp, NOESY-gp, HSQC-gp, HMBC-gp) NMR spectroscopy (Table 10).[203]
The assignment of the C≡N carbon signals and the determination of the configurational structure of the molecule were made on the basis of 13C NMR spectra without proton decoupling. The value of the 3JCH constant (C≡N and HC=) indicates that one C≡N group is in cis-position (3JCH = 8.1 – 8.5 Hz), and the second is in trans-position (3JCH = 13.6 – 14.1 Hz) relative to the double bond HC=C (see Table 10 and Fig. 10).
![[{"id":"QvRySufFwY","type":"paragraph","data":{"text":"The structure of 4-substituted 1,2,3-triazole malononitriles <b>30 – 36</b>"}}]](/storage/images/resized/z6WpniP78RB7Y7C9CMKb5TCIIN8l0TWvNHhHt0i9_xl.webp)
The value of the chemical shift of the NH proton varies in the range of 15.8 – 15.9 ppm in the 1H NMR spectra of compounds 30 – 33, and in the range of 15.1 – 15.3 ppm for compounds 34 – 36 (see Fig. 10). In addition, the chemical shift of the =CH proton for compounds 30 – 33 appears at 8.54 – 8.58 ppm, and in compounds 34 – 36 it is varied in the range of 8.20 – 8.22 ppm. The downfield shift of the above proton signals in the 1H NMR spectra of 30 – 33 may indicate the presence of bifurcation hydrogen bonding in the molecule.[203]
The structure and fluorescent properties of ethynyl derivatives of 1,2,3-triazole have been reported.[210-212] The authors proposed two new approaches to the synthesis of triazoles containing 4- or 5-positioned ethynyl substituent, viz., copper-catalyzed azide-alkyne cycloaddition of organic azides to 1-iodobuta-1,3-diynes and a three-component reaction between 3-substituted propargyl aldehydes, amines and diazosulfonamides. Primary screening of biological properties revealed the low toxicity of the resulting 4,5-diethynyl-1H-1,2,3-triazoles, making them promising candidates for the further development of fluorescent labels for cytological studies of all molecules with fluorescent properties.
2.5. 2-Substituted 1,2,3-triazoles
The properties of 2-substituted 1,2,3-triazoles are very different from those of their N-1 and N-3 isomers, despite their structural similarity. Their different behaviour in biological systems may arise from the difference in the basicity between the N-1,3 and N-2 isomers. The 1,2,3-triazole ring is found in biologically active compounds such as fluorophores, optical coordination polymers, promising anticancer agents, dual orexin receptor antagonists used to treat insomnia, inhibitors of 2,3-oxidosqualene cyclase, α-glycosidase and serine hydrolase.[16] [213-225]
The nitrogen atoms in 1,2,3-triazole have different environments in tautomers A and B, and these tautomers have Cs and C2v symmetry, respectively, i.e. the 1H- and 3H-tautomers have Cs symmetry, and the 2H-tautomer has C2v symmetry (Scheme 8).
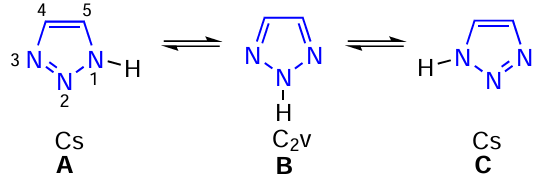
The tautomers A(C) and B have different physical, chemical and biological properties.[2][3] [16][25] [219] [224][226] The dipole moment of the N-1H tautomer is 4.38 D, while that of the N-2H tautomer is only 0.218 D. Based on the calculated charges for each carbon atom, Katritzky and Pozharskii [226] attributed an electron-rich character to the 1H-1,2,3-triazole π-system, while the N-2H tautomer was considered as π-electron deficient. Microwave and photoelectron spectroscopy data show that the 2H-tautomer predominates over the 1H-tautomer in the gas phase. The A(C) : B ratio calculated from the microwave spectrum of the triple 15N species is 1 : 1000. 1,2,3-Triazole exists as a 1 : 1 tautomeric mixture of A(C) and B in the solid state (X-ray crystallographic analysis). 4-Phenyltriazole is the 2H-tautomer and 4-nitrotriazole is the 1H-tautomer, stabilized by two intermolecular hydrogen bonds. Both tautomers are present in solution, while in polar media the 1H-form is predominantly stabilized. The low melting points of 1,2,3-triazole and 3(5)-methylpyrazole (they are liquids at ambient temperature) are explained by their existence as a mixture of tautomers.[24][25] [226]
There are conflicting reports on the structure of trimethylsilylated 1,2,3-triazole, N-trimethylsilyl-1,2,3-triazole. Previously, Birkofer and Wegner [227] suggested that the trimethylsilyl group in N-trimethylsilyl-1,2,3-triazole is rapidly exchanged between adjacent N-1 and N-2 atoms, i.e., is in dynamic equilibrium. According to other data,[228] N-trimethylsilyl-1,2,3-triazole exists as 2-trimethylsilyl-1,2,3-triazole (2H-tautomer). In order to clarify the dynamic behaviour of N-trimethylsilyl-1,2,3-triazole, a trimethylsilylation reaction of 1,2,3-triazole with hexamethyldisilazane was carried out (Scheme 9).
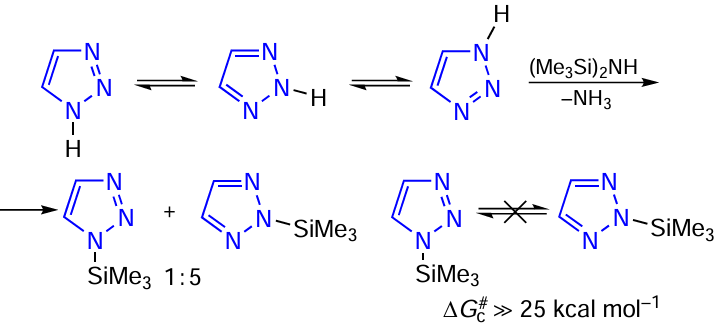
Multinuclear 1H, 13C, and 29Si NMR data showed that the reaction gives a mixture of 1-trimethylsilyl- (1-TMS) and 2-trimethylsilyl-1,2,3-triazoles (2-TMS) in a ratio of 1 : 5.[20][24][27] [229] Attempts to convert one form of TMS-1,2,3-triazole into another by heating the mixture (up to 65°C in chloroform and 200°C neat) have not change the spectra. Consequently, the free activation energies of mutual transitions 1-TMS 2-TMS are significantly greater than 25 kcal mol–1, and the molecules should not be attributed to tautomers, but to N-TMS-1,2,3-triazole isomers (see Scheme 9). NMR data indicate that the nature of the solvent (CDCl3) or neat liquid has no effect on the ratio of 1-trimethylsilyl- and 2-trimethylsilyl-1,2,3-triazoles in the mixture (Table 11).
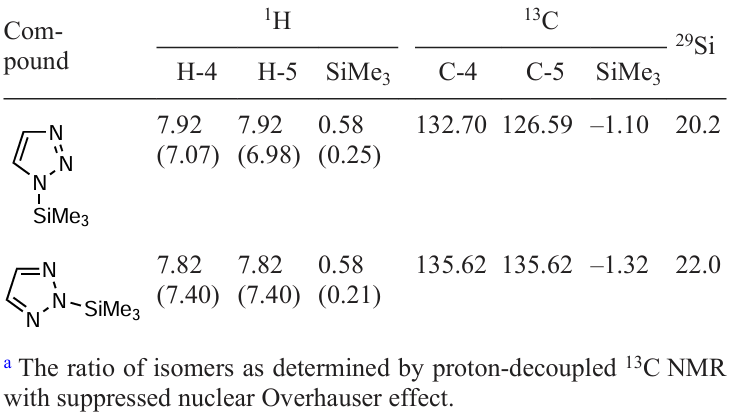
As shown in Table 11, H-4 and H-5 protons of 1-TMS-1,2,3-triazole are chemically equivalent in CDCl3. This indicates a high rate of the migration of trimethylsilyl groups between N-1 and N-3 atoms in chloroform at room temperature. The most probable reason for the silylotropy acceleration in 1-TMS-1,2,3-triazole may be the catalytic action of Me3SiCl formed during the reaction of 1-TMS-1,2,3-triazole with traces of HCl in CDCl3 .
These studies have shown that the authors [227] apparently did not observed the 1 2 exchange of the TMS group in N-trimethylsilyl-1,2,3-triazole but obtained a mixture of 1- and 2-TMS-1,2,3-triazoles, with a higher content of the latter. To further confirm this finding, the total energy values of 1-TMS- and 2-TMS-1,2,3-triazole were estimated by the ab initio method MP2/6-311++G(d ).[194][198] The resulting calculation data predict that in the gas phase, 1-trimethylsilyl-1,2,3-triazole should be more favourable (stable) by 4.8 kcal mol–1. At the same time, the calculated values of the dipole moments of the isomers 1-TMS and 2-TMS are 4.77 and 0.57 D, respectively. Based on the experimental values of the dipole moment of N-trimethylsilyl-1,2,3-triazole (1.54 D [227]) and the theoretical values obtained for each isomer, the ratio was estimated to be approximately 1 : 3 (0.23 : 0.77) for 1-TMS and 2-TMS, respectively. This is consistent with previous results obtained from NMR data.
Thus, it has been shown that trimethylsilylation of 1,2,3-triazole yields two isomers, 1-trimethylsilyl- and 2-trimethylsilyl-1,2,3-triazoles in a ratio of ~ 1 : 5, indicating the regioselectivity of the trimethylsilylation. Similarly, for 1-TMS-1,2,3-triazole, the 1 3 degenerate tautomeric rearrangement (migration) of the trimethylsilyl group is observed.
The unique properties of 2-substituted 1,2,3-triazoles have stimulated efforts to develop synthetic approach to these compounds in high yield and high selectivity. In this context, the chemistry and structural features of 2-substituted 1,2,3-triazoles are of increasing interest to the worldwide scientific community.[6] [216][218-220] [225][226][229-240]
2-Phenyl-1,2,3-triazole 37 was obtained by cyclization of phenylosazone glyoxal using copper triflate Cu(OSO2CF3) as a catalyst in high-boiling solvents such as o-xylene, toluene, ethylene glycol (Scheme 10).[230][231]
2-Phenyl-1,2,3-triazole is a promising precursor for the synthesis of nitro derivatives of 1,2,3-triazole — energetic materials in explosive compositions.[6] [16][17][25][241-247] Some examples of energetic compounds are presented in Table 12. In order to optimize the synthesis of 2-phenyl-1,2,3-triazole, i.e. to increase the yield and purity of the product and to simplify the procedure, the effect of the amount of catalyst on the yield of compound 37 was studied.[230][231] The optimum catalyst content in the reaction mixture was found to be 5%. The reaction kinetics was studied by 1H NMR spectroscopy. The structure of triazole 37 was confirmed by multinuclear 1H, 13C and 15N and 2D NMR spectroscopy (see Table 12). The 1H NMR spectra of compound 37 show resonance signals from the protons of the triazole ring at 8.11 ppm and signals of the phenyl protons: 7.41 for p-Ph, 7.55 for m-Ph and 8.03 ppm for o-Ph. The 13C NMR spectra contain signals from the C-4,5 atoms of the triazole ring (136.47 ppm) and signals from carbons of the phenyl ring. The two-dimensional 1H – 15N NMR spectrum of HMBC reveals two cross peaks at –58.6 and –120.2 ppm, assigned to the N-1,3 and N-2 atoms, respectively. The {1H – 15N} 2D spectrum shows a cross peak of the N-1,3 atoms with the protons of the triazole ring, as well as cross peaks of the N-2 atom with both protons of the triazole ring and the o-Ph and m-Ph protons. Thus, the use of copper triflate as a catalyst in high-boiling solvents (toluene, o-xylene, ethylene glycol) helped to optimize the synthesis and to obtain 2-phenyl-1,2,3-triazole of high purity (99.9%) in high yield (82 – 90%), and also to simplify the isolation step.[230][231]
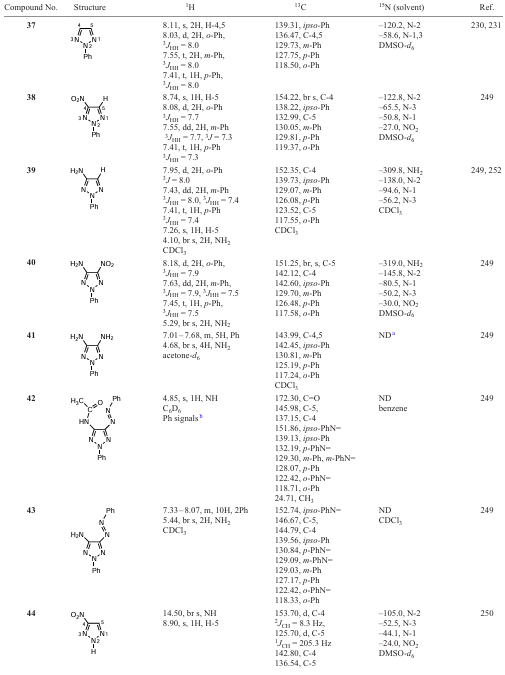
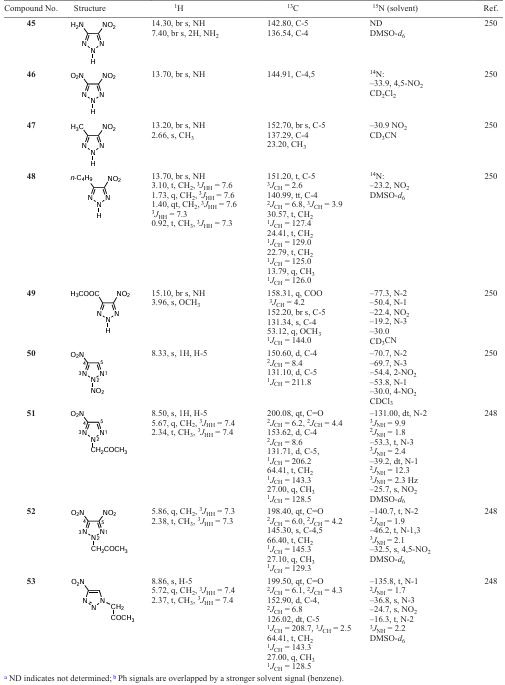
Amino and nitro derivatives of 1,2,3-triazoles are precursors to fluorescent fiber bleaching agents and compounds with chemosterilizing properties, and mainly explosives.[16][17] [24][25] [241] [248-252] 4-Nitro- and 4-amino-2-phenyl-1,2,3-triazoles were obtained by heterocyclization according to Scheme 11.[249] It is advisable to carry out the coupling of 2-nitroacetoxime (metazonic acid) with phenyldiazonium without isolating metazonic acid in its pure form, but using a solution of its sodium salt obtained by self-condensation of nitromethane. Cyclization of hydrazonooxime under the action of acetic anhydride in acetic acid leads in high yield to 4-nitro-2-phenyl-1,2,3-triazole 38, which can be reduced with hydrogen or over palladium and platinum catalysts under hydrogen pressure to 4-amino-2-phenyl-1,2,3-triazole 39 or non-catalytic reduction of 38 with nascent hydrogen (Scheme 11, Table 12).
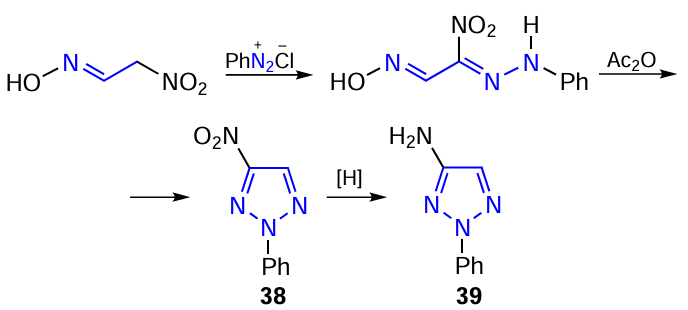
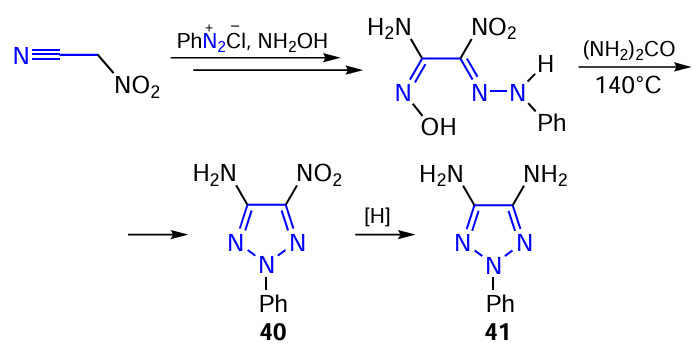
4-Amino-5-nitro-2-phenyl-1,2,3-triazole 40 is formed by cyclization of 2-nitro-2-phenylhydrazonoacetoxime with urea. Reduction of the nitro derivative 40 gives 4,5-diamino-2-phenyl-1,2,3-triazole 41 (Scheme 12).[249]
4-Acetylamino-5-phenylazo-2-phenyl-1,2,3-triazole 42 was also obtained by cyclization of the appropriate amidooxime in acetic anhydride. Treatment of compound 42 with an alkali solution afforded 4-amino-5-phenylazo-2-phenyl-1,2,3-triazole 43 (see Table 12).[249]
The 13C and 15N chemical shifts of the carbon and nitrogen atoms of the C-NO2 moiety are in the range of 151 – 158 and –(22 – 34) ppm, respectively, and have very small changes in both the carbon and nitrogen NMR scales depending on the substituents in adjacent position of the triazole ring (see Table 12). In fact, the 15N and 13C chemical shifts of the nitro group atoms in nitroazoles vary within a fairly narrow range, i.e., structural changes in the triazole ring do not significantly affect magnetic resonance factors of these atoms.[16][21] [24] The 13C (C – NO2) signal from the carbon atom in 13C NMR spectra of nitrotriazoles is shifted downfield by ~ 30 ppm compared to that of the unsubstituted triazole. The presence of the electron-withdrawing NO2 group in triazole reduces the shielding of the C – NO2 carbon atom. It should be noted that there are certain difficulties in recording (accumulating) signals from the carbon atom bonded to the nitro group. The C – NO2 carbon signal in nitroazoles usually exhibits a quadrupole broadening due to the 13C – 14N (NO2) spin-spin coupling. This broadening disappears in the 13C – {1H,14N} triple resonance experiment.[16][248] [250] [253] Therefore, the authors recorded the 13C spectra of nitroazoles under heteronuclear triple resonance conditions, i.e. observed the 13C nuclei with broadband decoupling from protons and selective decoupling from 14N nuclei of the NO2 group.[248] This produced a narrow 13C NMR signal of the NO2-bonded carbon atom. The known difference in the 15N chemical shifts of pyrrolic and pyridinic nitrogen atoms in azoles is ~ 100 ppm and is widely used in signal assignment.[16][21] [24][25][254] However, this difference is smaller for triazoles (see Table 12). The pyrrolic 15N chemical shifts for 1,2,3-triazoles, which are less sensitive to structural changes, are observed in the range of –(100 – 120) ppm, whereas the pyridinic nitrogen shifts vary in a wider range –(30 – 65 ppm), thus indicating a lower shielding.
It should be noted that the 15N chemical shifts of the nitrogen atom of the explosophoric nitro group N – NO2 , especially in N-nitrotriazoles, are in the range of –(50 – 60) ppm, i.e. the nitrogen signals are shifted upfield compared to those of the C – NO2 moiety.[16] [21] [24] [241] [250] see Table 12. Theoretical studies of the structure, detonation properties and stability of five-membered heterocycles, including 1,2,3-triazoles annulated with tetrazine-1,3-dioxides, have been carried out at the B3LYP/6-311++G(d,p) level.[255]
In addition, as can be seen from Table 12, it is not possible to detect 15N signals in the NMR spectra of the given compounds, and this mainly concerns the nitrogen atoms of the triazole ring. Firstly, and mainly this is due to the phenomenon of prototropic tautomerism, i.e., rapid proton exchange, the rate of which does not coincide with the NMR time scale. Secondly, lowering the temperature of the sample when recording spectra does not always provide the desired effect. It is not possible to observe individual tautomeric forms. Lower temperatures are required, which are beyond the capabilities of the solvents used. And thirdly, the molecules of some compounds contain no protons that would be located 2 – 3 bonds away from the nitrogen atom, which is a necessary condition for recording two-dimensional 1H – 15N HMBC spectra. Obviously, solid-state NMR would be useful in addressing these problems, but it should be noted that some of the compounds are oils.
However, Toppet et al.[256] [257] were able to observe individual tautomers on the example of 4(5)-vinyl-1,2,3-triazole and its polyvinyl derivatives using a low-frequency NMR spectrometer Varian XL 100-15 at 100 MHz (1H) and at 25.16 MHz (13C). 13C NMR studies of 4(5)-vinyl-1,2,3-triazole in DMF at –55°C allow the direct observation of three individual tautomers. The 13C chemical shift values of the three forms are given, together with their content in the tautomeric mixture. The exact tautomeric composition is best determined from a 1H spectrum recorded under identical conditions. The NH broad signal splits into three broad signals at 15.38 ppm (2-NH, 70%), 15.85 ppm (1-NH, 20%) and 15.98 ppm (3-NH, 10%).[257] The 13C NMR spectrum of poly-4(5)-vinyl-1,2,3-triazole in DMSO at room temperature shows three sets of two triazole carbon atoms corresponding to three tautomers: 1H (15 – 17%), 2H (66 – 70%) and 3Н (15 – 17%).[256] Thus, the 2H-tautomer predominates both in the 1,2,3-triazole itself and in 4(5)-substituted 1,2,3-triazoles.
The possibility of chlorotropy in 1,2,3-triazoles and benzotriazoles was considered,[258] as it had been reported that chlorine exchange took place in 1-chloro-4,5-diphenyl-1,2,3-triazole.[259] The intensive subsequent NMR study showed that the compounds were in fact symmetrical (2-chloro-4,5-diphenyl-1,2,3-triazole).[258]
Thus, generalizing the currently known experimental data, it is possible to systematize the data on chemical shifts of each type of carbon atom of the 1,2,3-triazole system (Fig. 11). The line charts show the characteristic ranges of the 13C NMR chemical shifts of the C-4, C-5, C-6 (C=O), C-6 (C=N) and C-8 atoms depending on the substituent at position 4. As can be seen, the most downfield regions obviously correspond to carbonyl and imine carbon nuclei at position 6. This is followed by the carbon nuclei at positions 4, 5, and 8 as the shielding increases. We believe that such systematized ranges will help in interpreting the spectral data of 1,2,3-triazoles.
![[{"id":"2BHsn1hEWk","type":"paragraph","data":{"text":"Line charts of the <sup>13</sup>C NMR chemical shift ranges of the carbon nuclei of 1,2,3-triazole systems as a function of the substituent at position 4 based on known experimental data (Refs 179 – 183, 194, 199, 200, 225 – 227, 245 – 249). For the atom numbering see Table 2."}}]](/storage/images/resized/qgzQYB6w34G7rGt517G0Azk7PriBR3JSPeP4p9N7_xl.webp)
2.6. Other substituted 1,2,3-triazoles
Continuing the discussion of theoretical research on 1,2,3-triazoles, it should be noted that approaches to studying the mechanisms of their formation within the framework of the DFT method are currently being actively developed. For example, Himo et al.[260] showed that the 1,3-dipolar Huisgen cycloaddition becomes non-concerted when copper(I) acetylides are reacted with azides and nitrile oxides, providing easy access to 1,4-disubstituted 1,2,3-triazoles. Notably, the process is very robust and has an unusually wide product coverage. Theoretical studies have revealed a stepwise mechanism involving unusual metallocyclic intermediates that appear to be common to several dipoles (Fig. 12).
![[{"id":"X8nMa901an","type":"paragraph","data":{"text":"Schematic representation of the energy profile of the reaction of copper(I) acetylides with organic azides to give 1,4-disubstituted 1,2,3-triazoles. Reproduced from Himo et al.<sup>260</sup> with the permission of the American Chemical Society."}}]](/storage/images/resized/c3j3sK3voXLQ8S2ElJ29SeSPT9Ew7OuSXMxipYXu_xl.webp)
The proposed mechanism explains key experimental observations. Firstly, the dramatic rate enhancement observed in the copper-catalyzed synthesis of 1,2,3-triazoles is in excellent agreement with the calculated activation barriers, which are up to 11 kcal mol–1 lower than those in the corresponding concerted cycloadditions. Secondly, the exceptional regioselectivity of the copper(I)-catalyzed processes is both predicted computationally and observed experimentally. Thirdly, the proposed mechanism suggests that dipoles other than the organic azide must be involved in a similar stepwise sequence with copper(I) acetylides.
Another study devoted to the quantum chemical investigation of the formation of 1,4-disubstituted 1,2,3-triazoles via the Cu(I)-catalyzed 1,3-dipolar cycloaddition reaction between aryl azides and monoterpene terminal alkynes showed that the [3 + 2] cycloaddition reaction proceeds efficiently, with high periselectivity and regioselectivity at the carbon – carbon triple bond, yielding the corresponding target products (Fig. 13).[261]
![[{"id":"799lNPD0kx","type":"paragraph","data":{"text":"Synthesis of heterocyclic systems by the CuAAC reaction of terminal alkynes with aromatic azides. Reproduced from Oubella et al.<sup>261</sup> under the CC BY license."}}]](/storage/images/resized/Hn0BMCZlz9RVho19p3QalbztZosRh1xjsEAAfrl3_xl.webp)
Mechanistic studies at the DFT level showed that the terminal alkyne is the most reactive of all dipolarophiles 7. In the absence of a catalyst, the said reaction proceeds via a concerted mechanism involving two energetically very close regioisomers. Meanwhile, the dinuclear Cu(I)-catalyzed reaction provides access to a single 1,4-substituted 1,2,3-triazole regioisomer via a stepwise pathway with a very low activation barrier, rationalizing the experimental observation.
Another example of the study of the [3 + 2] cycloaddition pathway of carvone azide acting as a 1,3-dipole with dimethyl acetylenedicarboxylate used as a dipolarophile was carried out at the B3LYP/6-311+G(d,p) level.[150]
Global reactivity indices using the global electron density transfer value were used to analyze this polar reaction.[262] The electron chemical potential value of carvone turned out to be significantly higher than that of dimethyl acetylenedicarboxylate. This indicates that there should be a global electron density transfer from carvone to dimethyl acetylenedicarboxylate during the above cycloaddition reaction. According to theoretical calculation, carvone acts as a nucleophile, and dimethyl acetylenedicarboxylate acts as a non-electrophile in this reaction. Other examples of detailed studies of the mechanisms for obtaining substituted 1,2,3-triazoles are also known,[263] including those using the DFT method.[264][265]
The introduction of triazole units into the structure of arabinogalactans (AG) was also investigated, which is of considerable interest for the development of various representatives of polyelectrolytes and ligands capable of binding to biological metals.[266] A promising area of application for triazole derivatives of AG is their quaternization to give cationic structures capable of conjugating with antitumour oligonucleotides, as is known for triazolochitosan; in addition, the ability of the conjugate to penetrate into target cells can be provided by the membrane tropism of AG. To develop a new family of AG-based materials, a copper-catalyzed reaction was carried out between AG with terminal alkyne groups (AG propargyl ethers), sodium azide and organyl halides. It is expected that the membrane-tropic properties of AG will impart enhanced biocompatibility to the metal complexes.
3. 1,2,4-Triazoles
1,2,4-Triazole derivatives have attracted much attention as an inexhaustible source for the research and development of compounds with various biological activities. 1,2,4-Triazoles have, as mentioned above, a wide range of therapeutically interesting potential drug candidates such as antimicrobial, antioxidant, analgesic, antiseptic, antiurease, diuretic, anticancer, anticonvulsant, anti-inflammatory, antidiabetic and antimigraine agents. The 1,2,4-triazole derivatives are used as pharmaceuticals, radiosensitizers, ionic liquids, dyes, precursors for nanomaterials, energetic materials (Refs [9-13] [15] [32][36] [45][49] [92][96][105] [111][112] [117] [267-274]). 1,2,4-Triazoles are also potential ligands for the synthesis of magnetoactive coordination compounds with electrophysical, optoelectronic, thermochemical, and electrochemical properties.[275-278] Interest in this class of compounds is constantly growing, as evidenced by the large number of publications devoted to the chemical, biological, and structural features of 1,2,4-triazoles.[146][279-288]
Regarding the tautomerism of 1,2,4-triazole, it can be generalized that in the solid state it is realized as an asymmetric form of the 1H(2H)-tautomer according to the results of X-ray structural analysis.[2][3][24][25] [146][289] Data from a large number of ab initio calculations of 1,2,4-triazole tautomers at different levels indicate the predominance of the 1H(2H)-tautomer over the 4H-tautomer in the gas phase, which is consistent with all experimental results.[2][3] [290][291] The 1H-tautomer also predominates in solution, with alkylation of 1,2,4-triazole yielding two isomers: 1-alkyl- and 4-alkyl-1,2,4-triazole in a ratio of ~ 10 : 1. A small amount of 2H-1,2,4-triazole (~ 5%) was detected in a highly polar solvent.[2][3][25][146]
The reaction mechanism of the 1,2,4-triazole with acetone, along with other azoles (pyrazole, imidazole, tetrazole), calculated at the B3LYP/6-311++G(d,p) level, was analyzed in detail in a study.[146] The calculated energy profiles of this reaction are shown in Fig. 14. These energy profiles clearly demonstrate that the reaction occurs exclusively at position 4 of the triazole ring (N-4), in contrast to other types of azoles.
![[{"id":"0_DSWlj8eG","type":"paragraph","data":{"text":"Theoretically calculated energy profiles of the reaction of the different tautomers with acetone. Relative energies are given in kJ mol relative to the most stable tautomer. The number 4 in 1<i>H</i> 1,2,4-triazole indicates that the triazole reacts by N(4). Reproduced with minor editing privileges from Claramunt et al.<sup>146</sup> under the CC BY license."}}]](/storage/images/resized/Pv8aC6WvW0xS81qnyBtZnHMVr17zjB84lMnbyDAQ_xl.webp)
The theoretical study of the formation mechanism of 1,2,4-triazoles is of particular interest for understanding the heterocyclization process. In turn, understanding the heterocyclization pathway helps to control the chemical process and to carry out targeted synthesis.
The mechanism of the heterocyclization of 4-substituted 1,2,4-triazoles and their structural features has been studied.[24] [292][293] The need to gain insight into the mechanism of this reaction is associated with the search for possible routes for the synthesis of 1,2,4-triazoles, promising precursors of thermochromic materials. 4-Substituted 1,2,4-triazoles were synthesized by fusion of diformylhydrazine with corresponding aminophenols or aminopyridines at 145 – 170°С in the yields of 46, 54 and 64% for the ortho-, meta- and para-isomers, respectively (Scheme 13).
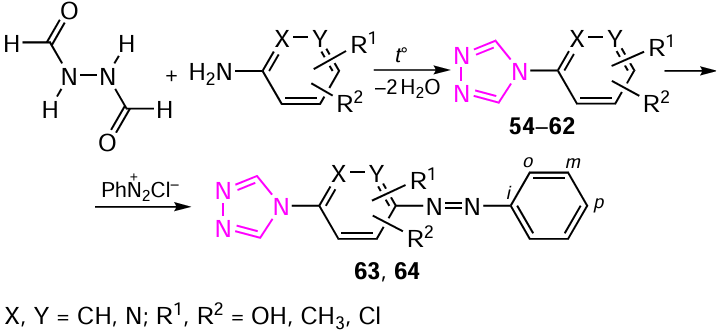
The resulting 1,2,4-triazoles 54 – 64 are stable crystalline compounds with high melting points, soluble in ethanol and DMSO. The compound 65, (1-methyl-4-nitropyrazol-5-yl)(1,2,4-triazol-4-yl)amine, was obtained by the vicarious nucleophilic substitution of 1-methyl-4-nitropyrazole with 4-amino-1,2,4-triazole 66 (Scheme 14 and Table 14).[28][294][295] Model compounds 66 and 67 are also presented in Table 14 for comparison. 4-Amino-1,2,4-triazole 66 is an efficient aminating agent in the vicarious C-amination of nitroazoles (and nitroarenes), which sometimes yields both aminonitro compounds and unexpected products as a result of addition or heterocyclization (see Scheme 14).[296-300]
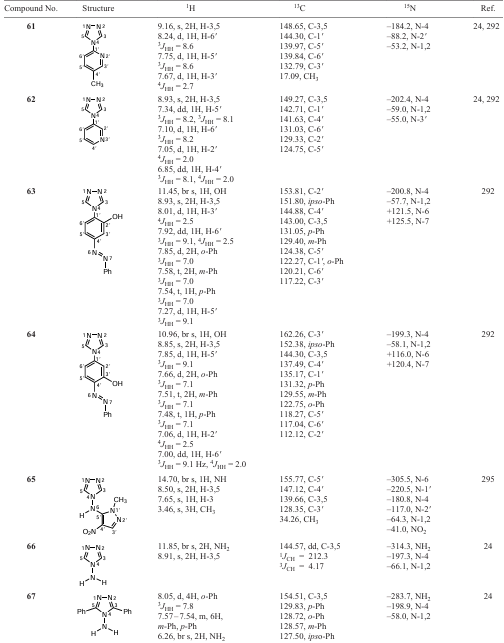
Together with the amination product (1-methyl-4-amino-5-nitrobenzimidazole 69), furazan derivative 68, 1-methyl-[4,5]-furazanobenzimidazole, is formed in a ratio of 2 : 1 (see Scheme 14).[28] [300] The reaction was monitored by 1Н NMR. It was shown that as reaction time increases (up to 14 days), furazanobenzimidazole predominates.
The structure of the investigated compounds was confirmed by multinuclear 1H, 13C, 15N and 2D NMR spectroscopy (COSY-gp, NOESY-gp, HSQC-gp, HMBC-gp) (54 – 69) (see Table 14). The two-dimensional {1H-15N} HMBC NMR spectra of compounds 54 – 67 show cross peaks of nitrogen atoms with the triazole ring protons H-3,5 and the corresponding protons of the phenyl (heteroaryl) moiety at –(53 – 66) ppm for the pyridine N-1,2 atoms, and in the range of –(184 – 203) ppm for the pyrrole N-4 atom. This indicates that the heterocyclization of compounds 54 – 64 delivers 1,2,4-triazoles.
Surprisingly, the chemical shift of the NO2 nitrogen in compound 65 is upfield shifted compared with those in other nitrotriazoles and nitropyrazoles.[16] [24][25] It is likely that the shielding of the nitrogen nuclear of the nitro group of 65 (–41.0 ppm) is increased due to the formation of the N – H∙∙∙O – N hydrogen bond (Fig. 15).
![[{"id":"Wa1qUYM-4h","type":"paragraph","data":{"text":"The hydrogen bonding in compound <b>65</b>"}}]](/storage/images/resized/AVsUBRxvETZkNcTacWHGvwKQGSLEq9YUkXE4soE5_xl.webp)
In addition, the resonance signal of the NH proton (14.7 ppm) in compound 65 is shifted downfield by ~ 3 – 4 ppm compared, e.g., with the NH signals in compounds 66 and 67, indicating the formation of a hydrogen bond.
The quantum-chemical simulation of the reaction pathway between diformylhydrazine with o- and p-aminophenols to afford 4-substituted 1,2,4-triazoles (54 and 56) has been carried out using the combined CCSD(T)/6-31+G(d)//B3LYP/ 6-311++G(d,p) approach (Scheme 15).[296]
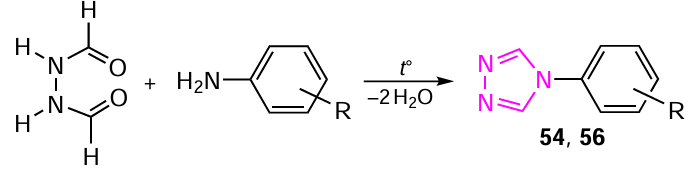
A mechanism for the reaction of diformylhydrazine with o- and p-aminophenols has been proposed, and the energy and geometric parameters of possible intermediates, transition states and final products were calculated (Fig. 16). For more detailed description of the reaction mechanism see Ref. [293].
![[{"id":"GtM0wuy3a_","type":"paragraph","data":{"text":"Energy profile of the reaction of diformylhydrazine (<b>1a, 1b</b>) with o-, p-aminophenols (<b>2a, 2b</b>) to give 1,2,4-triazoles <b>54, 56</b>. PRC is prereaction complex, TS is transition state, IC is intermediate compound. Reproduced from Chirkina and Larina<sup>293</sup> with the permission of Springer Nature."}}]](/storage/images/resized/YzNbJKgAFev1A3xWgxH0hbv584P41OAbKPIifJAZ_xl.webp)
The following elementary reaction steps have been identified, viz. the first is, nucleophilic addition of o- or p-aminophenol to one of the carbonyl groups of diformylhydrazine to form an unstable α-amino alcohol; the second is the dehydration of the geminal amino alcohol, which can proceed to furnish hydrazonamide or iminohydrazide intermediates. It has been shown that the formation of iminohydrazide gives rise to an additional process step, which involves the prototropic imino – amine tautomeric transformation, as a result of which iminohydrazide is converted into hydrazonamide. In this case, the water molecule formed during dehydration is involved in proton transport and contributes to this rearrangement. The third step is the heterocyclization of hydrazonamide via the nucleophilic attack of the nitrogen atom on the second carbonyl group to fumish a cyclic α-amino alcohol, see Scheme 16. Finally, the fourth and final step is the dehydration of the cyclic α-amino alcohol to give the final product, 1,2,4-triazole 54 or 56.
The formation of the target products, 4-(o-, p-hydroxyphenyl)-1,2,4-triazoles 54 and 56, decreases the free energy of the system by 54 – 55 kcal mol–1. Analysis of the geometric parameters of 4-(o-hydroxyphenyl)-1,2,4-triazole indicates the presence of a weak intramolecular hydrogen bond C – H∙∙∙O – H (2.618 Å) between the hydroxy group of the phenol moiety and the C – H bond of the triazole ring (Fig. 17).
![[{"id":"GFiLWGzYhx","type":"paragraph","data":{"text":"The hydrogen bonding in 4-(o-hydroxyphenyl)-1,2,4-triazole <b>54</b>"}}]](/storage/images/resized/FaAmcGCoYKsFRx6l4rF2LWEypZNB3kCOX7yo3y90_xl.webp)
Indeed, the experimental NMR data indicate the presence of the hydrogen bonding in ortho-hydroxyphenyl 1,2,4-triazole 54 (see Table 14). In the 1H NMR spectra of compound 54, a downfield shift of the broadened OH signal by about 2 ppm is observed compared with the para derivative. The position of the OH group in the phenyl moiety slightly affects the values of the 1H and 13C chemical shifts.
The reaction of 4-amino-1,2,4-triazole 66 with nitrobenzene under vicarious C-amination conditions delivers not only the expected amination product, nitroaniline 70, but also the condensation products bis(para-nitrophenyl)amine 71, (para-nitrophenyl)(1,2,4-triazol-4-yl)amine 72 and bis(para-nitrophenyl)(1,2,4-triazol-4-yl)amine 73 (Scheme 17).[28] [296] [299] [301]
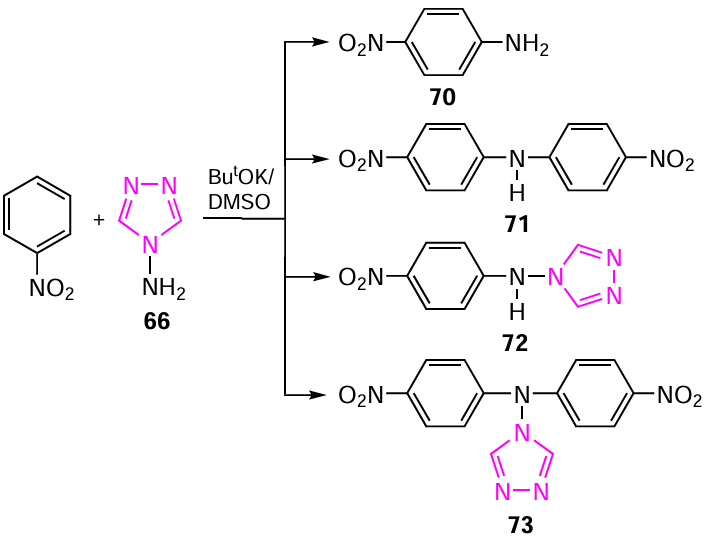
The ratio of the final products 70 – 73 depends significantly on the conditions of the vicarious nucleophilic substitution of hydrogen (temperature, reaction time and ratio of reagents). It was found that heating the reaction mixture for four hours gave two products: para-nitroaniline 70 and (para-nitrophenyl)(1,2,4-triazol-4-yl)amine 72. In addition, the reaction of nitrobenzene with 1,1,1-trimethylhydrazinium halides in absolute DMSO in the presence of ButOK at room temperature afforded both para-nitroaniline 70 and bis(para-nitrophenyl)amine 71. The nature of the halide anions (Cl, Br or I) had practically no effect on the yields of products 70 – 73. All compounds were identified and characterized by multinuclear and two-dimensional NMR spectroscopy (Table 16).
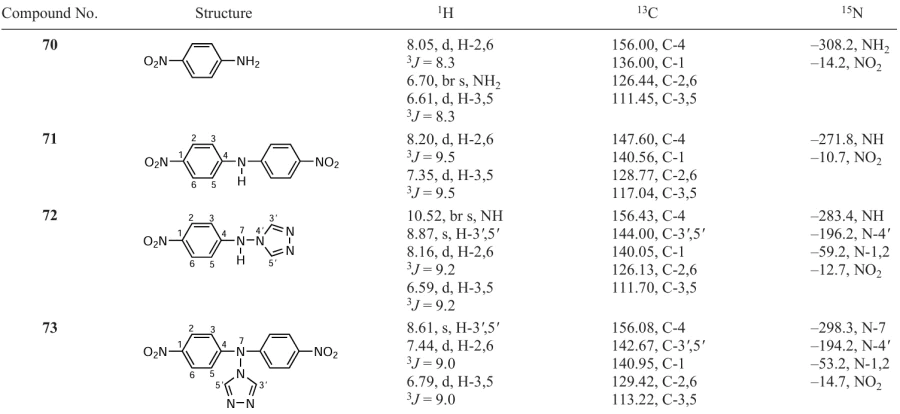
The 1H NMR spectrum of compound 71 showed a singlet signal of the NH proton (9.92 ppm) and two doublets of two pairs of equivalent protons of the benzene rings at 7.35 and 8.20 ppm with 3J = 9.5 Hz. In the 15N NMR spectrum of compound 71, signals characteristic of this type of compounds were detected, related to the nitrogen atom of two equivalent nitro groups (–10.7 ppm) and to the amino nitrogen atom (–271.8 ppm).[296]
4. Conclusion
The phenomena of tautomerism and isomerism in azoles, including functionalized triazoles, complicates the structural analysis of such compounds. One of the most important methods for studying nitrogen-rich compounds is 15N NMR spectroscopy, which allows both the study of the spatial and electronic structure and the prediction of the reactivity of nitrogen-containing heterocycles. The structural investigation of azoles using NMR spectroscopy, in particular, 15N NMR, in combination with quantum chemistry methods, represents a powerful and unique approach for studying the stereochemical behaviour and tautomeric properties of azoles.
Notably, the 15N nuclear shielding is highly sensitive to even minor variations in tautomeric equilibrium constants, molecular conformations, temperature and solvent properties. The solvent dependence can be explained both by the effects of non-specific solvation due to a change in the medium polarity and by the effects associated with the formation of hydrogen bonds and other non-valent interactions between the molecules of the solvent and the compounds under investigation — the so-called specific solvation.
The triazole ring plays an important role in medicinal chemistry due to its structural features, synthetic versatility and pharmacological potential. The ability of triazole to mimic various functional groups makes it widely used as a bioisostere for the synthesis of new promising molecules. The annular prototropic migration of the vast majority of azoles in solution is so fast on the NMR time scales that lowering the temperature of the solution often does not change the spectra and does not provide any new information. In all cases, the spectra are averaged, making it extremely difficult or impossible to detect NMR signals from individual nitrogen atoms of triazoles.
Therefore, when solving structural problems, quantum chemistry methods are often used to obtain theoretical values of 15N NMR chemical shifts. In the case of low-polarity aprotic solvents, the use of a continuum model is a sufficient condition for obtaining adequate theoretical 15N values. In the case of polar protic solvents, however, the continuum model is insufficient for a correct description of the solvation effects, and solvation must be taken into account explicitly, i.e. at least one solvent molecule must be added to the calculation space. Thus, the available experimental 15N NMR data will have high correlation parameters with the resulting theoretical values.
Theoretical study of stereochemical modifications of azoles in combination with modern experimental methods such as multipulse multinuclear 1H, 13C and 15N NMR spectroscopy is necessary for understanding the subtle structural features of compounds, allowing to predict their biological activities. Nitrogen-15 NMR spectroscopy is one of the most suitable, reliable and powerful express methods for determining the stereochemical structure and identifying the tautomeric forms of nitrogen-containing compounds.
Intermolecular hydrogen bonding in azoles has been shown to play a major role in the formation of a special type of compounds, viz., high-energy materials.[302-306] In recent years, there has been a growing interest in functionalized azoles and related nitrogen-rich compounds, as evidenced by the large number of publications on their chemical and structural studies.[307-320]
Acknowledgments. Authors are sincerely grateful to Dr. M.M.Demina for fruitful collaboration in the study of the structure of 1,2,3-triazoles.
5. List of abbreviation
AG — arabinogalactan,
AM1 — Austin Model 1 (a semi-empirical method),
B3LYP — Becke 3-parameter Lee-Yang – Parr exchange-correlation functional,
CCSD — second-order polarization propagator approximation with coupled cluster singles and doubles amplitudes,
COSY — COrrelated SpectroscopY,
CS — chemical shift,
DFT — density functional theory,
HIV — human immunodeficiency virus,
HMBC — heteronuclear multiple bond correlation,
HSQC — heteronuclear single-quantum correlation spectroscopy,
IC — intermediate compound,
IEF-PCM — equation formalism polarizable continuum model,
INADEQUATE — Incredible Natural-Abundance DoublE-QUAntum Transfer Experiment,
LEP — lone electron pair,
LOL — localized orbital locator,
MP2 — Møller – Plesset perturbation theory of the second order,
mPW1PW91 — Perdew – Wang exchange functional as modified by Adamo and Barone combined with PW91 correlation,
NOE — nuclear Overhauser effect,
NOESY — Nuclear Overhauser Effect SpectroscopY,
NQR — nuclear quadrupole resonance,
PBE — Perdew – Burke – Ernzerhof exchange-correlation functional,
PDB — protein data bank,
SOPPA — second-order polarization propagator approximation method,
SSCC — spin-spin coupling constant,
TS — transition state.
References
























