


Keywords
Abstract
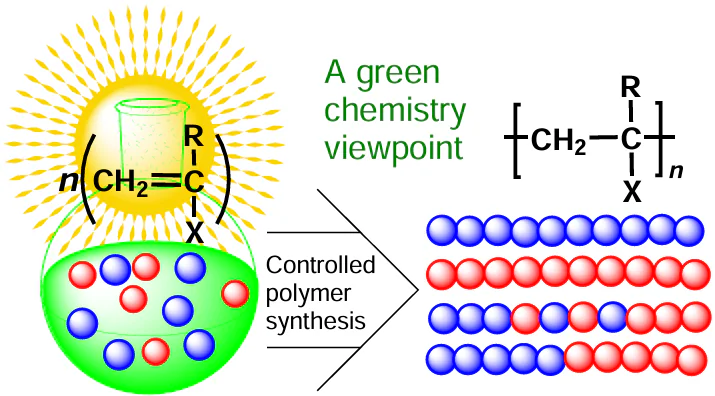
The discovery of reversible deactivation radical polymerization (RDRP), or controlled radical polymerization (CRP) has revolutionized the chemistry of synthetic polymers. This strategy opened up the way to polymer materials with controlled architecture, composition, and functions. Currently, owing to the use of novel approaches related to reversible chain deactivation, radical polymerization has gone beyond the polymer synthesis. It can be used to obtain not only macromolecular organic compounds, but also organic-inorganic hybrid materials, bioconjugates, promising polymers for electronics, energy production, medicine, and other high-tech fields. It is exceptionally important that some CRP methods have a clear-cut environmental component, since they are focused on compliance with the most important principles of green chemistry and the development of nature-like processes in the targeted synthesis of well-defined polymers with a specified set of properties and characteristics. This review considers particular examples and analyzes the possible prospects for the practical application of CRP methods and environmentally benign processes for the synthesis of high-tech functional polymers. A comparative analysis is performed for the classical methods of living radical polymerization of a wide range of monomers (reversible inhibition, reversible addition fragmentation chain transfer, and atom transfer radical polymerization involving transition metal complexes), the concept of organic photoredox catalysis, and green chemistry methodology as applied to the targeted synthesis of polymers with a specified set of properties and characteristics. The review gives analysis of the above CRP aspects, including the procedural details related to photoinitiation and organic photoredox catalysis, and their relationship with the key green chemistry principles. In our opinion, this will be of interest not only to specialists in the field of polymer chemistry, but also to a wide range of synthetic chemists and environmental scientists.
The bibliography includes 242 references.
1. Introduction
In recent years, the growth rate of polymer production has markedly exceeded the growth rate of global industrial production. It is noteworthy that polymers and materials based on them cover all areas of human activity and form the basis for manufacture of not only essential consumer goods such as garments and footwear, but also high-tech materials for electronics, medical equipment, pharmaceutical products, aircraft and automotive engineering, construction industry, etc. The wide application of polymer materials virtually in all spheres of human activity is caused by their functionality, exceptional durability, and relatively low production cost. It is noteworthy that polymer materials science is a relatively new area of materials chemistry: only a few years ago, in 2020, the polymer science celebrated the 100-year anniversary, because 1920 is the year of publication of H.Staudinger’s landmark work ‘Über Polymerisation’,[1] which laid the fundamentals of the modern physicochemistry of macromolecular compounds.
The materials formed by natural polymers, such as polysaccharides and proteins, have been used by mankind since the emergence of civilization. However, with the development of the world community, synthetic polymers have become more and more important; currently, approximately 90% of these polymers are obtained from fossil feedstock.[2] According to estimates of specialists,[3][4] while now about 6% of the global oil consumption is for the manufacture of plastics, with the current growth rate of the consumption of plastics, the percentage of oil spent for the manufacture of polymers is predicted to increase to 20% by 2050. The growth of the polymer production is accompanied by the increase in the environmental impact, including both consumption of energy and resources and contamination of land and water areas with polymer waste. Indeed, according to estimates of environmental scientists,[2][5] more than 90% of the manufactured polymers currently end up in landfills, thus contaminating the environment, in particular the global ocean. Therefore, considering the need to reduce the adverse environmental impact, an important challenge of modern synthetic polymer chemistry is to use principles and approaches of so-called green chemistry in the polymer production processes.[6][7] It is quite obvious that increasing the environmental friendliness of production processes is a significant aspect in the modern approaches to polymer synthesis. In particular, an effective and atom-economic synthesis of macromolecular compounds can be carried out using catalysts and/or water-based systems. The use of light, especially in the visible wavelength range, is more favourable regarding both the energy efficiency and environmental friendliness than the use of high temperatures and so on. It is clear that the second 100-year period that started for the polymer science poses great challenges and opens up great opportunities for polymer chemists as regards the development of new methods for the synthesis of polymer materials that would meet the varying demands of the society and simultaneously be safe for living nature.[8] In this respect, broad prospects are opened by original approaches to the controlled synthesis of macromolecules, or reversible deactivation radical polymerization (RDRP).[9-17] Today, this synthetic strategy is used most often to obtain polymer materials with controlled architecture, composition, and functions. It is exceptionally important that some CRP procedures have a clear-cut environmental component, because they are directed towards the compliance with the most important green chemistry principles in the targeted synthesis of polymers with a specified set of properties and characteristics. This review is first of all devoted to analysis of the modern trends in the controlled synthesis of macromolecules and, in particular, to organic photoredox catalysis, which is based on single-electron transfer processes involving metal-free catalysts,[18] which logically fits into the concept of green origin of polymeric materials.
2. Main aspects of the radical-initiated controlled synthesis of polymers and green chemistry principles
It is known[19] that the properties of polymers and polymer materials are determined, first of all, by their molecular weight characteristics and composition (in the case of copolymers) and also by the macromolecular architecture, including the polymer chain topology and functional groups present in the macromolecules. All these characteristics are specified directly during the polymer synthesis. It is not accidental that particular attention of polymer chemists has lately been devoted to the development of effective macromolecular engineering methods and precision methods for the synthesis of polymers of a specified structure, definite composition, and molecular weight (MW).[20] Controlled polymerization, or living polymerization is a tool of the targeted synthesis of macromolecular compounds, the main distinctive feature of which is the absence or minimization of irreversible chain termination, which leads to increasing dispersity and composition inhomogeneity of macromolecules and, hence, it is most often unfavourable for the properties of polymers.[19-21] The living polymerization methodology, first proposed by M. Szwarc for the synthesis of macromolecules in the presence of anions,[22][23] was later successfully extended to cationic and radical polymerization, and metathesis polymerization, including ring opening polymerization.[9-12][24]
It is no coincidence that particularly controlled/living polymerization is an important method for the synthesis of well-defined polymers with clearly specified properties and characteristics. This method combines the benefits of both cationic and anionic polymerization, ring opening polymerization, transition metal-catalyzed coordination polymerization, and free radical-initiated polymerization. As noted above, the term ‘living polymer’ was first proposed in 1956 by Szwarc.[22][23] Irrespective of the mechanism, polymerization is considered to be living if it is characterized by fast and efficient initiation, almost simultaneous growth of all chains, and a minor contribution of irreversible chain termination and transfer.[9-17]
Although living polymerization processes were first used for ionic polymerization, currently they are most widely used to prepare homo- and co-polymers under conditions of radical initiation or metal complex catalysis. Obviously, this is related to the fact that radical-initiated polymer synthesis is most widely used in industry to produce large-scale amounts of homo- and copolymers. In the last two decades, controlled radical polymerization has turned into a highly relevant trend of modern polymer chemistry. Apart from the above-mentioned absence (minor contribution) of irreversible chain termination or irreversible chain transfer, controlled processes are characterized by fast and efficient initiation, which promotes almost simultaneous growth of all polymer chains, together with linear increase in the average molecular weight of the polymer (Mn) with increasing conversion. Furthermore, polymerization products isolated at any monomer conversion can themselves act as macroinitiators. The addition of a fresh portion of the monomer results in the synthesis of post- or block-copolymers, including complex macromolecular structures. These features altogether ensure the formation of polymers with a narrow molecular weight distribution (MWD) characterized by low dispersity (Đ < 1.5) and controlled molecular weight, the parameters of which can be set directly during the preparation of the monomer composition. These methods open up wide opportunities for macromolecular design, including the synthesis of block copolymers, hybrid organic-inorganic polymer materials, and nano-sized polymer structures.[2][9-12] [21] It is no exaggeration to say that the implementation of CRP methods in the last two decades made a kind of revolution in the synthetic chemistry of polymers. It is not accidental that in 2019, this methodology was included in the list of the 10 most important new technologies of the 20th century, which, in the opinion of some scientists, can make the most significant contribution to the progress of human civilization.[25] In this regard, it is beyond doubt that taking account of the most important green chemistry principles in the controlled synthesis of polymers and nano-sized polymer structures will be favourable for further development of this area, in particular, considering the application in industry.
It is known [6] that the green chemistry concept was formulated as twelve principles more than 20 years ago by P.T.Anastas and J.C.Warner. Subsequently this concept was many times reconsidered, redefined, and corrected;[7] however, the key statements remained unchanged and, with certain assumptions, can be formulated in the following way:
(1) decrease (in the ideal case, elimination) of waste in the production (the Sheldon E-factor is close to zero);
(2) maximum incorporation of all substrates used in a chemical process into the final product (atom economy);
(3) development of the least hazardous routes of chemical synthesis, including low toxicity of both the reactants and final products;
(4) synthesis of safer chemicals (new products should not only possess specified properties, but also be less toxic than the reactants);
(5) the use of safer solvents and auxiliaries, e.g., aqueous solutions;
(6) energy efficiency principle (minimized energy expenditure);
(7) use of renewable raw materials;
(8) decrease in the number of intermediate steps and high selectivity of the synthesis;
(9) catalysis (it is better to use catalytic amounts of substances for the synthesis rather than stoichiometric ratios of components);
(10) synthesis of biodegradable products;
(11) real-time monitoring and control of the process in order to prevent the formation of hazardous substances able to cause environmental pollution;
(12) prevention of accidents.
Some of these principles have been already practically implemented in the living polymerization.[2] [26-29] Each of the three main types of controlled synthesis of macromolecules, that is, reversible inhibition, reversible chain transfer, and atom transfer, have their own specific features with regard to compliance with the green chemistry principles, depending on the mechanistic details of these reactions.
2.1. Reversible inhibition under thermal and photochemical initiation
Reversible inhibition radical polymerization is one of the most thoroughly investigated CRP techniques, which involves the use of stable radicals.[30-36] Most often, nitroxide spin adducts or their sources such as alkoxyamines, nitrones, nitroso compounds, etc., are used as stable radicals in reactions of this type. When sterically hindered nitroxides, characterized by high stability due to electron density delocalization, are introduced into the polymerization system, they can reversibly react with the growing macroradicals (~Pn•), thus preventing the irreversible chain termination (Scheme 1).
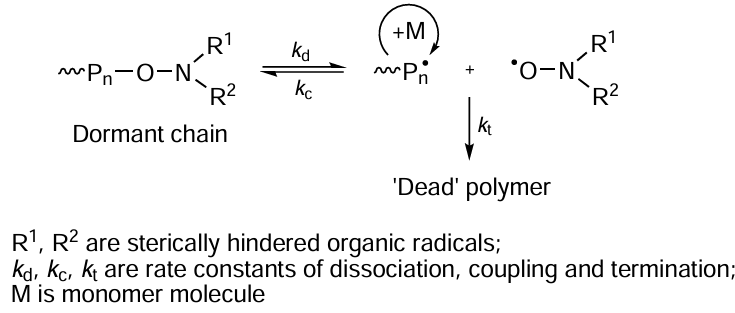
The key aspect of this mechanism is the formation of a relatively labile bond between the nitroxide radical and polymer macroradical; this bond can be reversibly cleaved upon either thermal activation (due to heating) or photoirradiation.
For the first time, nitroxide radicals, in particular 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO), were used as mediators by Georges et al.[37][38] and Hawker et al.[39] in the radical polymerization of styrene (ST), which gave polyST with a dispersity of 1.2. However, a considerable drawback of this process is high temperature of polymerization (130 – 140°C). Naturally, this imposes significant limitations on the practical use of this approach, and this is also at variance with one of the above key principles of green chemistry: the energy efficiency principle.[6][7]
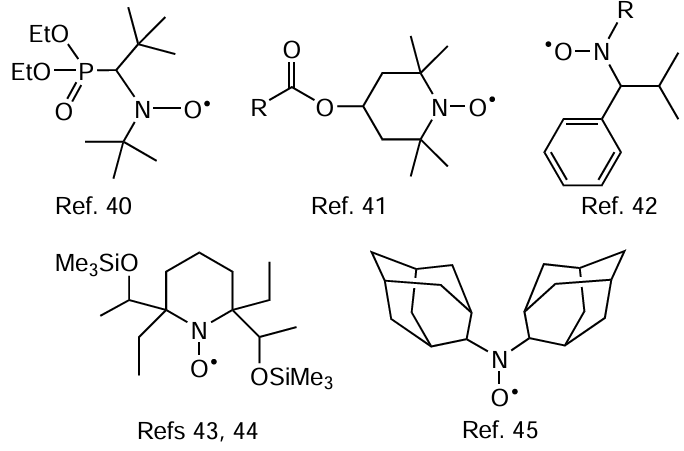
In a number of subsequent studies devoted to this type of CRP, a series of sterically hindered nitroxides of complex structures were proposed as polymerization mediators. The use of these compounds made it possible to somewhat decrease the temperature range of reversible inhibition (down to 70 – 100°C). The structures of some radicals of this type and references to the studies dealing with the polymer synthesis involving these radicals are shown below.
Another original approach to reversible inhibition CRP is related to the generation of sterically hindered macromolecular nitroxides directly in the polymerization system (in situ) using nitroso compounds, nitrones, and some other precursors of nitroxide spin adducts (Scheme 2).[21][46-50]
Considerable steric crowding around the reaction centre and the presence of macromolecular chain in the formed nitroxide spin adducts allow for the living-chain polymerization of a wide range of methacrylic monomers in a lower temperature range (60 – 70°C) [31] [51-54] than in the case of TEMPO and its analogues.
It is noteworthy that lowering the temperature of polymer synthesis is only one aspect of minimizing energy expenditure in the overall energy balance of an industrial process implied by the green chemistry principles mentioned above. Evidently, the use of photopolymerization approaches for the controlled synthesis of polymers would be much more efficient in this respect. Then it will be possible not only to reduce the polymerization temperature to room temperature, which significantly increases the energy efficiency of the process, but also to minimize the side disproportionation reaction between the growing macroradical and the nitroxide spin adduct. Most often, this disproportionation reaction decreases the number of reactive species and the yield of the polymer and also disrupts the control over the molecular weight characteristics of the polymer, which results in MWD broadening and increase in the dispersity.[55][56]
There are two options for conducting photopolymerization in the presence of nitroxides: the use of photosensitizers and the introduction of chromophore groups directly into the nitroxide molecules.
In 1996, Scaiano et al.[57] found that in the presence of xanthone as a photosensitizer, some alkoxyamines undergo homolytic cleavage under UV irradiation (λ ≈ 365 nm) to give simultaneously active carbon centred-radicals capable of initiating polymerization and stable nitroxide spin adducts providing reversible inhibition and control of the molecular weight characteristics.
Professor Yoshida,[58-63] who studied polymerization of methyl methacrylate (MMA) in the presence of 4-methoxy-TEMPO, a nitroxide that is used quite often for reversible inhibition CRP of styrene and its homologues, and some cationic photosensitizers, identified a number of features of this reaction typical of controlled processes. The number-average molecular weight of the polymer increased linearly with increasing conversion of the monomer, while the dispersity of the prepared samples decreased during the polymerization and did not exceed 1.5 – 1.7 (depending on the polymerization conditions and reactant ratio). The authors interpreted the observed facts by proposing an original radical-cation mechanism of photopolymerization in the presence of bis(alkylphenyl)iodonium hexafluorophosphate, which included a reinitiation step associated with the existence of a reversible equilibrium between a macromolecular nitroxide spin adduct, a growing macroradical, and the cation formed from 4-methoxy-TEMPO (Scheme 3).
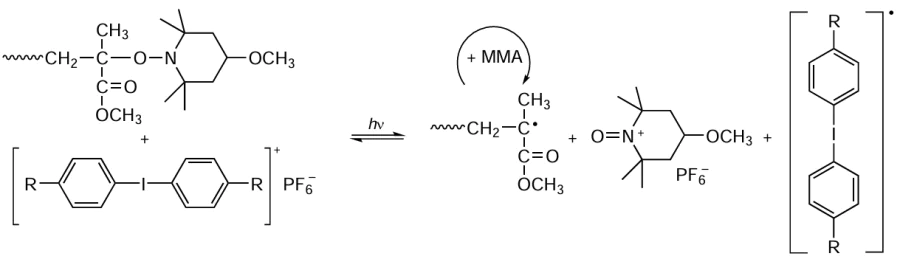
The approach considered above proved to be quite effective not only for the bulk photopolymerization of MMA, but also for solution photopolymerization [64] and for the dispersion polymerization of methacrylic monomers in aqueous media,[65] which smoothly fits into the green chemistry concept.
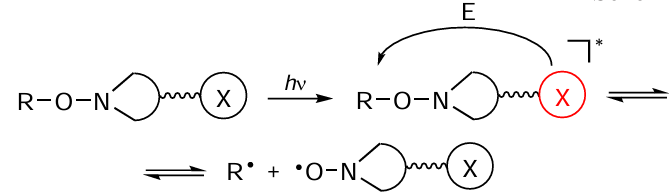
The polymerization in the presence of photosensitizers is provided by the intermolecular energy transfer from the photosensitizer to alkoxyamine followed by degradation of the photosensitizer.[57] Hence, for the nitroxide-mediated photopolymerization to proceed, the nitroxides should be coupled to an appropriate sensitizer. The irradiation of a chromophore with a light flux followed by the intramolecular energy transfer to alkoxyamine induces cleavage of the carbon – oxygen bond and furnishes a carbon-centred radical (R•) able to initiate polymerization and a stable nitroxide spin adduct according to Scheme 4.
This concept was further developed by Neckers and co-workers,[66] who synthesized a series of chromophore – alkoxyamine conjugates of various structures and tested them in photopolymerization reactions. The photopolymerization of MMA was implemented using PE-XTEMPO as a photoinitiator (PI); however, the resulting polymer was not photoreactive, i.e., it did not decompose into radicals on exposure to a light flux. This is likely to be responsible for the fact that polymerization did not follow the reversible inhibition mechanism.
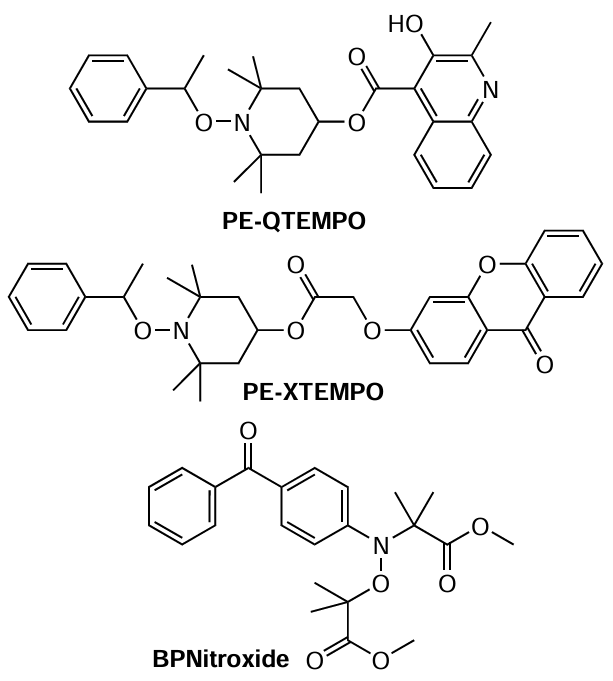
Later, Scaiano et al.[67] used structurally similar quinoline-containing alkoxyamine PE-QTEMPO, which, in the authors’ opinion, contained a more efficient molecular antenna, for polymerization of ST under UV irradiation. However, polymerization proceeded fairly slow: after 2 h, the conversion did not exceed 10%. The resulting polyST had a relatively broad MWD (Đ ~ 1.60), and Mn of the obtained samples did not exceed 2.7 kDa. Meanwhile, it should be noted that although this alkoxyamine did not show high activity in either ST polymerization or poly(methyl methacrylate) (PMMA) synthesis, it proved to be highly efficient for the control of the stereoregularity of macromolecules. The proportion of syndiotactic triads in the products of MMA polymerization involving this compound was more than 60%.
In order to improve the efficiency of energy transfer in these types of systems, several research groups have proposed the use of a new class of alkoxyamines in which the sensitizing chromophores were directly bound to the aminoxyl component.[68][69] In the authors’ opinion, in compounds of this type, the photochemical and photophysical properties of chromophores should have a more pronounced effect on the decay of alkoxyamines. With this assumption in mind, benzophenone-substituted nitroxide (BPNitroxide) was tested in the synthesis of polybutyl acrylate. This compound proved to be fairly effective regarding the monomer conversion; however, the molecular weight of the resulting polymer markedly deviated from the theoretical values, and the dispersity was more than 2.0,[70] which indicated a low control over the process.
Su et al.[71] synthesized a series of TEMPO-based photosensitive alkoxyamines with various chromophores including benzophenone, naphthalene, and quinoline. The results indicate that effective energy transfer sufficient for homolytic cleavage of the C – O bond and subsequent initiation of polymerization is possible in the indicated compounds. The authors noted that in this case, it was possible to synthesize PMMA by combining photosensitive nitroxide compounds as mediators with 2,2-dimethoxy-2-phenylacetophenone as a photoinitiator. The reaction was accompanied by linear growth of Mn with increasing degree of monomer conversion, with the dispersity being 1.3 – 1.4; in combination with other features of the process, this attests to controlled mechanism of polymerization.
Versace et al.[72] showed that the distance between the chromophore group and the aminoxyl bond is the main factor influencing the efficiency of photolysis. In particular, the location of chromophore in the α-position to the aminoxyl group is most favourable for influencing the polymerization process. Thus, the alkoxyamine BPNitroxide, which has already been mentioned above, can effectively initiate and control the butyl acrylate polymerization:[73][74] the monomer conversion reached 80% in less than 10 min. However, the dispersity of the synthesized samples proved to be fairly high. A low degree of control over the molecular weight characteristics was also observed in the case of alkoxyamines containing two benzophenone chromophore groups in the molecules.[75] An attempt to increase the regulating ability of alkoxyamines in the photopolymerization by replacing the benzophenone antenna with conjugated pyrene, anthraquinone, or naphthalene structures did not result in a pronounced increase in the degree of control over the molecular weight characteristics of polymers synthesized under irradiation with UV or visible light.[76] The use of nitrones containing benzoin, naphthalene, and pyrene chromophore antenna groups proved to be equally ineffective for conducting CRP under photoirradiation.[74][77] These nitrones are potentially capable of generating sterically hindered nitroxides and similar alkoxyamines in situ, but actually they do not provide for polymerization of ST and acrylic monomers by the reversible inhibition mechanism. According to Lin,[77] in this case, the low control over the photopolymerization may be caused, first, by the moderate rate of reinitiation and, second, a nitroxide with a sterically hindered antenna may have low efficiency in the reversible inhibition required for the control over the process of polymerization.
Exceptionally interesting results were obtained by Liu et al.,[78] who succeeded in performing the emulsion polymerization of MMA under irradiation in the presence of 4-hydroxy-TEMPO, an analogue of 4-methoxy-TEMPO considered above, and the commercial photoinitiator Irgacure 184 at room temperature. The polymerization was characterized by a linear increase in MW with increasing degree of conversion and the dispersity of the resulting polymers was 1.27 – 1.36. Unfortunately, the authors did not attempt to interpret the mechanism of polymerization, in particular, the most interesting step, рһotoreinitiation at room temperature (!?) in the absence of chromophore groups in the monomer or in the nitroxide. In our opinion, this seems to be problematic from the thermodynamic point of view.
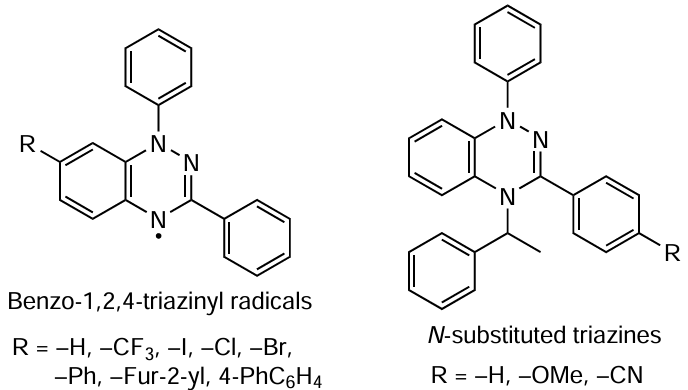
Naturally, the range of stable radicals that are able to act as effective radical mediators in CRP is not limited to nitroxides. Demetriou et al.[79] performed for the first time reversible inhibition polymerization of ST in the presence of benzo-1,2,4-triazinyl radicals. Using a number of stable radicals of this type, the authors prepared polyST with Ɖ = 1.11 – 1.17. The experimental results obtained in this work indicate that the structural characteristics of radicals, including steric hindrance, significantly affect their efficiency.
Areephong et al.[80] showed that targeted synthesis of these structures can provide quite a number of monomolecular photoinitiators, N-substituted triazine derivatives. Their ability to perform ST polymerization characterized by linear increase in MW with increasing monomer conversion was demonstrated. It was shown that ST and its homologues can undergo controlled homopolymerization and copolymerization with butyl acrylate and MMA in the presence of the above mediators.
Generally, while characterizing the CRP process involving nitroxides and other stable radicals under photoirradiation, it is necessary to note that although this approach allows polymerization of vinyl and (meth)acrylic monomers under milder temperature conditions, it is still inferior to the well-known high-temperature nitroxide-mediated radical polymerization of these monomers regarding the control over the molecular weight characteristics.[29-34] Meanwhile, some examples, including the examples given above, indicate that this trend may be of interest for both implementation of some of the above green chemistry principles and practical applications. It is not accidental that processes of this type are already practically used to fabricate luminescent coatings,[81] for laser printing,[82][83] and for other applications.[84][85]
The certain benefits of using particularly nitroxide spin adducts for the controlled radical initiated synthesis of macromolecules in the light of the basic principles of green chemistry are largely due to their low toxicity. Some representatives of nitroxides are used in medical practice, in particular as contrast agents in magnetic resonance imaging.[86][87] In addition, some nitroxide spin adducts possess antioxidant and radioprotective properties [88][89] and anticancer activity.[90-92] It was shown [93] that polymers obtained using nitroxides or sources of nitroxides do not have noticeable cytotoxicity and can be used as nanocontainers for targeted drug delivery. In the light of the above, the existing view that nitroxide-mediated radical polymerization as a type of controlled synthesis of macromolecules largely complies with some green chemistry principles appears quite substantiated.[28]
2.2. Reversible chain transfer
The reactions involved in the reversible addition fragmentation chain transfer (RAFT), or degenerative transfer (DT) radical polymerization as one of the three major types of CRP are shown in Scheme 5.[94-96]
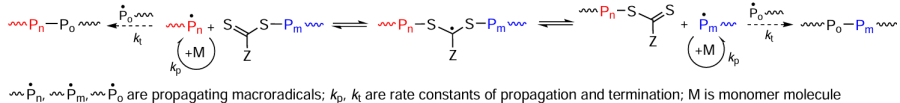
The reversible chain transfer (RCT) agents used most often in processes of this type induced by both heat treatment and photoirradiation are sulfur-containing compounds: dithioesters, dithiocarbamates, trithiocarbonates, and xanthates.[97-99] From the practical standpoint, a considerable drawback of this approach is the lack of versatility of RCT agents, i.e., it is necessary to select a particular regulator for a particular monomer or group of monomers in the case of homo- or copolymerization, respectively.
It is noteworthy that the first examples of photocontrolled radical polymerization with the use of thio compounds have been known since the 1980s.[99] However, in these reactions, the sulfur compounds were rather used as iniferters,[100] that is, agents that are simultaneously involved in the initiation, chain transfer, and chain termination steps. Typical photoiniferters are trithiocarbonates and disulfides with appropriate UV absorption ranges.
Thus, back in 1982 it was shown [101] that tetraethylthiuram disulfide (TTDS), dibenzoyl disulfide (DBDS), and S-benzyl-N,N-diethyldithiocarbamate (BDT) can be used to conduct reversible chain transfer polymerization of MMA and ST induced by UV irradiation. The synthesis of polymers is characterized by linear dependence of the number-average MW on the monomer conversion and some other features that attested to the controlled nature of polymerization.

Subsequently, the same authors investigated characteristic features of the photopolymerization of butyl acrylate in the presence of p-xylylene-bis-N,N-diethyldithiocarbamate (XDT).[102] This system was characterized by a substantial loss of activity of the terminal group of the growing polymer chain during the synthesis, which was due to the partial decomposition of XDT during polymerization. Better results were obtained by using dibenzyl trithiocarbonate (DBTTC) [103] for the synthesis of polyST, PMMA, and polybutyl acrylate under UV irradiation. The polymerization was accompanied by linear increase in the number-average MW of polymers with increasing degree of conversion of the monomer, and the resulting polymer had rather narrow MWD (Đ = 1.1 – 1.2).
Interesting results were reported by Wu et al.,[104] who proposed a new dithiobenzoate iniferter, 1-(ethoxycarbonyl)-1-propyl dithiobenzoate (EPDTB), for the controlled radical copolymerization of ST with maleic anhydride under irradiation (λ ≈ 310 nm). The study showed that the copolymer had a strictly alternating structure characteristic of the products of radical copolymerization of ST and maleic anhydride and, at the same time, it has low dispersity (Đ ~ 1.1 – 1.2). In combination with the data on polymerization kinetics, this confirmed the controlled nature of the process.
In recent years, reversible chain transfer agents have been used not only for thermal initiation of polymerization, but also for the synthesis of polymers in the presence of photoinitiators. This allowed the preparation of macromolecular compounds under mild temperature conditions, which complies with the energy efficiency principle of green chemistry.[6][7] Thus, Johnson and co-workers [105] used a system based on trithiocarbonate as an initiator and 10-phenylphenothazine (PPT) as a photocatalyst to prepare polyacrylates and polyacrylamides. This method also afforded a polymer with narrow MWD (Đ = 1.02 – 1.20), but polymerization proceeded at a low rate.[105]
A considerable drawback of the polymers produced using the above-mentioned RCT agents is the presence of sulfur-containing moieties in the macromolecular chain, the removal of which is an important and, in some cases, a challenging task. It was found that this process can be successfully performed with photocatalysis by eosin Y (EY). Modification of the polymer occurs in air in the presence of hexylamine and tributylphosphine,[106] which are needed as hydrogen atom donors for the formation of the HS terminal group (Scheme 6). In the absence of light, the classical nucleophilic degradation of the trithiocarbonate moiety takes place to give thiol. Then the thio group is substituted by a hydrogen atom under the action of radiation (see Scheme 6).
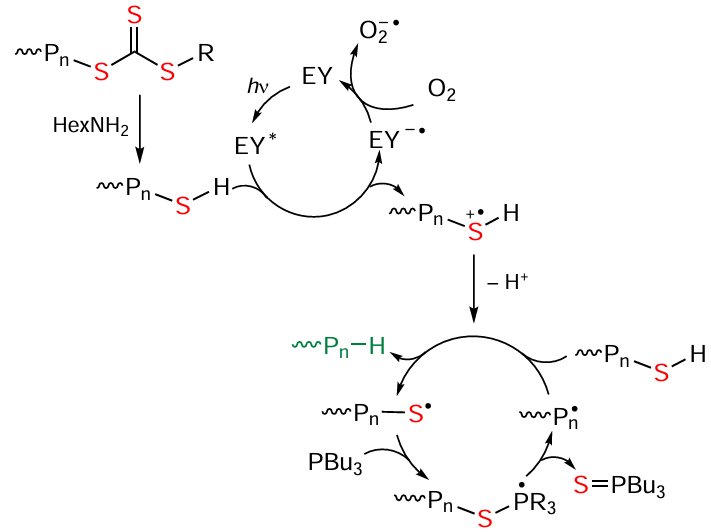
It is noteworthy that the introduction of amines to increase the control over the molecular weight characteristics in the reversible chain transfer photopolymerization of methacrylates under irradiation at λmax = 365 nm was also reported by Qiao and co-workers.[107][108]
As regards green chemistry principles and development of nature-like technologies, mention should be made of the results reported by Konkolewicz and co-workers,[109] who performed polymerization of MMA at room temperature on exposure to visible light or sunlight in the presence of the trithiocarbonate – tertiary amine system. The authors proposed an original two-step pathway for the reversible chain transfer polymerization in which tertiary amine does not act as a nucleophile, as shown in Scheme 6, but serves as a reducing agent that directly participates in the initiation of polymerization, making it possible to conduct the synthesis even without a classical radical initiator (Scheme 7).

Thus, it is obvious that promising methods based on the use of photoinduced electron transfer (PET RAFT process) [110-113] are a good alternative to the reversible chain transfer thermal polymerization involving sulfur-containing compounds regarding both the efficiency of controlled synthesis and keeping with green chemistry, since polymerization can proceed at room temperature and not only under UV irradiation, but in some cases, also on exposure to sunlight.[109] [114][115]
Another known type of chain transfer CRP, iodine transfer radical polymerization (ITRP), was proposed by Tatemoto and co-workers,[116] who tested this method for the preparation of fluorine-containing polymers with a relatively low dispersity.[116]
In the case of thermal ITRP, azobis(isobutyronitrile) (AIBN) and other sources of carbon-centred radicals are often used as initiators (In), and organoiodine compounds (alkyl iodides, phenyl iodides, mixed iodinated halogen-substituted alkanes, etc.) serve as polymerization mediators.[117] In this case, the initiator generates reactive radicals that cause polymerization, while the organoiodine compounds are involved in the chain transfer step, thus forming polymer molecules containing a labile bond with a iodine atom at the end (Scheme 8).
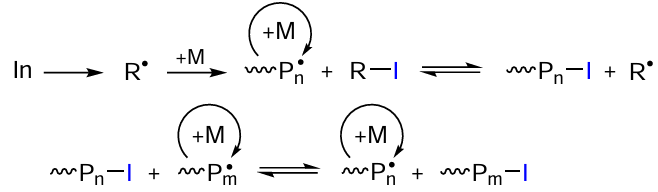
Polymer molecules with a terminal iodine atom can also be involved in the halogen atom transfer, i.e., living polymerization actually takes place, resulting in the formation of a polymer with rather narrow MWD (Mw/Mn < 1.5). Formally, the step by step growth of macromolecules during ITRP is provided by reversible transfer of an iodine atom between two growing radicals, which is characteristic of the degenerative chain transfer.[97] [118]
A considerable drawback of this method is the use of alkyl or aryl iodides, which are, as a rule, unstable due to the lability of the carbon – iodine bond, leading to the formation of molecular iodine.[119] The accumulation of molecular iodine in the system may often result in termination of the polymerization even at initial degrees of conversion. One more drawback of ITRP is related to the fact that the rate of the exchange reaction between dormant and active chains is fairly low. This drawback may be eliminated by adding compounds capable of actively detaching the iodine atom from the dormant polymer chain, e.g., germanium, tin, phosphorus and other derivatives. This leads to regeneration of the macroradical and transition of the chain from the dormant to active state.[120][121]
As in the case of atom transfer radical polymerization, this type of polymerization can be activated by introducing appropriate reducing agents into the system. For example, to increase the efficiency of processes of this type, Goto et al.[120][121] proposed adding amines as activators for polymerization involving organoiodine compounds. The authors [120][121] demonstrated that as a result of redox reactions, amines can reversibly detach the iodine atom located at the end of the polymer chain, thus bringing the chain to the active state (Scheme 9).
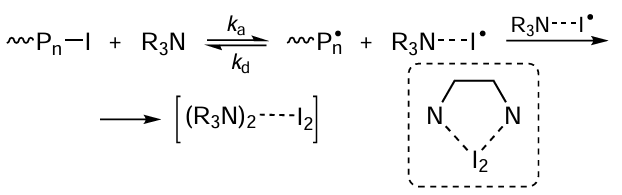
Goto et al.[122][123] pointed out that the effective control of the molecular weight characteristics in this type of reactions requires that amine should either actively react with the iodine atom and detach it from the polymer radical, thus activating the radical, or have chelating properties towards the iodine molecule. In other words, amine should be able to accept the released iodine, thus removing it from the reaction system, e.g., as an amine–iodine complex depicted in Scheme 9. In particular, high efficiency in MMA polymerization was found for N,N,N',N'-tetramethylethylenediamine and 1,4,8,11-tetraaza-1,4,8,11-cyclotetradecane; using these compounds, the authors were able to synthesize a polymer with quite narrow MWD (Ɖ ≈ 1.2 – 1.4) in high yield.[122] Further studies [123] markedly extended the list of amines that can act as catalysts for reactions of this type and the range of organoiodine initiators. It was found that the presence of iodine in the polymerization system promotes temporary transfer of the growing radicals into the dormant state; this increases the efficiency of the system in terms of synthesis of polymers with low dispersity. As a rule, tertiary amines are used as activators.
Apart from tertiary amines, onium salts such as ammonium, iodonium, phosphonium, and sulfonium iodides and bromides can serve as activators of radical polymerization using alkyl iodides.[123] For example, methyltributylphosphonium iodide (MeBu3P+I–) and tetrabutylammonium iodide (Bu4N+I–) showed a high catalytic activity in the polymerization of MMA initiated by 2-cyanopropyl iodide at temperatures above 70°C. In this case, polymerization proceeds up to high conversions, while dispersity values of the synthesized samples do not exceed 1.2, which attests in favour of the controlled nature of polymerization.
The authors of the above publication believe that the reaction of the added salt (A – I) with alkyl iodides (or polymers containing a iodine atom at the end of the chain) is stepwise and, in the general case, it can be described as a combination of three interrelated reactions (Scheme 10).
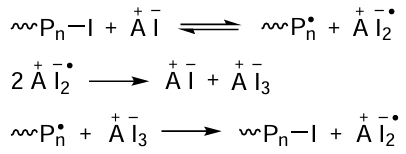
The formation of the derivatives Bu4N+I3– directly in the monomer medium was detected by UV spectroscopy, while the ability of polymer radicals to detach the iodine atom from Bu4N+I3– was unambiguously confirmed by investigation of the reaction products by NMR spectroscopy.
Sarkar et al.[124] utilized alkali and alkaline earth metal salts in combination with cyclic ethers as catalysts able to reversibly detach a iodine atom. 18-Crown ether and diglyme were used as complexing agents. On the one hand, ethers are needed in these reactions to increase the solubility of inorganic salts and, on the other hand, they promote elimination of the iodine atom. It was shown that a rational selection of an appropriate combination of an alkali metal iodide, an ether, and an organoiodine initiator may provide controlled polymerization of not only methacrylic esters (MMA, benzyl methacrylate, hydroxyethyl methacrylate, etc.), but also of acrylonitrile and ST.
Thus, in some cases, the ITRP strategy can be successfully used for thermally initiated polymerization of a broad range of monomers.
Apart from thermal initiation, iodine-containing compounds have proved to be no less effective in light-induced polymerization processes. For example, transition metal carbonyl complexes, in particular manganese carbonyl Mn2(CO)10 , were successfully used for polymerization of vinyl acetate, ST, and methyl acrylate in the presence of iodine derivatives.[125][126] It was established that the role of the metal complex was reduced to reversible detachment of the iodine atom, giving rise to reactive radicals in the system.
In addition to metal complexes, ITRP polymerization induced by photoirradiation, like thermally initiated polymerization, can be carried out in the presence of amines. Goto and Kaji[127] proposed using tributylamine (Bu3N), which, in combination with organoiodine compounds, leads to accumulation of carbon-centred radicals ~Pn• ) and iodine atoms (I•) in the polymerization system (Scheme 11). In the opinion of the authors cited above, the reaction can follow two mechanisms (a and b, Scheme 11). In the former case, light irradiation is used for direct photolysis of the carbon – iodine bond, and only after that, the iodine atom is accepted by the amine. In the latter case, complex formation between amine and the iodine-containing compound takes place first, and then the carbon–iodine bond photodissociates to form radicals (Scheme 11b).
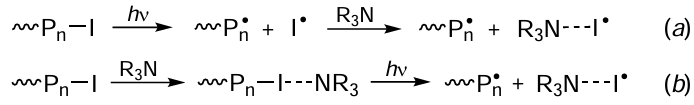
Zhu and co-workers[128] developed an original procedure for the polymerization of a number of methacrylic monomers initiated by 2-cyanopropyl iodide that occurs on exposure to blue light (l = 464 nm) without a photocatalyst. Polymerization effectively proceeded in polar solvents. In the authors’ opinion, polar solvents and functional monomers [glycidyl methacrylate, 2-hydroxyethyl methacrylate, and 2-(dimethylamino)ethyl methacrylate] can be used for direct complexation with the iodine-containing initiator, thus facilitating dissociation of the iodine – carbon bond.
Ohtsuki et al.[129] demonstrated that polymerization of methacrylic esters initiated by 2-cyanoisopropyl iodide in combination with Bu3N and some methine dyes (substituted carbocyanine iodides: 1,1'-diethyl-2,2'-cyanine iodide, 1,1'-diethyl-4,4'-quinocarbocyanine iodide, etc.) under UV or visible irradiation also proceeds in the controlled mode. It was found that the complexation of an organic catalyst with a iodine atom under irradiation is reversible. A necessary condition for the reversibility is the presence of diglyme in the reaction mixture as a polar solvent needed to dissolve the catalyst.
In general, it is noteworthy that the iodine atom transfer CRP induced by heating and/or photoirradiation is not only a fairly procedurally simple method for the preparation of homopolymers with specified molecular weight characteristics or copolymers of various architectures, but it is also commercially available from the practical standpoint.[130][131] However, a substantial drawback of processes of this type is the appearance of colour caused by the presence of iodine in the reaction system, which can also stain the polymer; this somewhat narrows down the range of possible practical applications of this approach.
2.3. Atom transfer polymerization involving metal-containing catalysts
The atom transfer radical polymerization (ATRP) is, on the one hand, a living radical polymerization method that has most rapidly progressed in recent years and, on the other hand, according to some researchers, particularly this method for polymer synthesis is most ‘dirty’ as regards compliance with green chemistry and environmental protection.[2] [28] [132-137] The classical pathway of this process (Scheme 12) is based on the catalytic Kharasch reaction [138] between alkyl halide (R – Hal) and transition metal complex (MtnLx), which can be easily and, what is important, reversibly switch from one oxidation state (n) to another one (n + 1).[10][21][139][140]
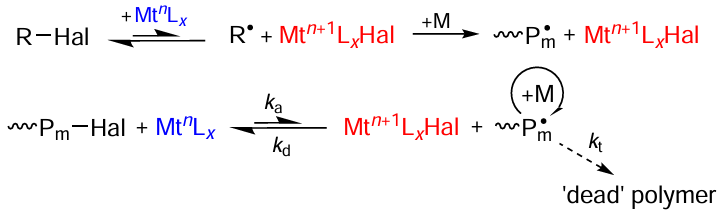
The above reaction leads to generation of radicals in the system, and the metal atom in the metal complex detaches the halogen atom from the initiator or from the growing polymer chain, thus switching to a higher oxidation state. Subsequently, the above oxidized metal complex acts as a deactivator and returns the polymer chain into the dormant state. Thus, during ATRP (see Scheme 12), the polymer chain passes through a series of activation (ka) and deactivation (kd) steps, which in the ideal case, ends with the exhaustion of the monomer and attainment of the limiting conversion. It should be noted that equilibrium in the ATRP process is generally shifted towards the dormant chains. Therefore, the current concentration of the growing macroradicals is moderate and the bimolecular chain termination reactions (kt) are minimized. The indicated regularities are favourable for the synthesis of polymers with narrow molecular weight distribution and provide broad opportunities for macromolecular design, including the synthesis of block copolymers and nano-sized macromolecular structures for numerous applications. The initially proposed ATRP procedure, the scheme of which is presented above (see Scheme 12) had some drawbacks and limitations, in particular those associated with the use of high concentrations of air-sensitive metal-containing catalysts. The remainder of the catalyst contaminates the polymer, which is especially undesirable for the macromolecular compounds intended for the use in microelectronics, medicine, and other fields sensitive to even trace amounts of a metal. The situation is aggravated by the fact that common catalysts of these reactions are complexes of copper, ruthenium, iron, and some other heavy metals, and some of them can have an adverse impact on the environment. In this regard, it is no surprise that this CRP method raised a number of questions regarding its compliance with the principles of green chemistry given in Section 1 of this review.[6] [7] [26-29] In view of the foregoing, development of ATRP methods based on the use of transition metal catalysts in low concentrations (at the ppm level relative to the monomer) or complete elimination of the use of metal complexes is important both for the prospects of practical applications and for the compliance with green chemistry principles.
A possible way to increase the efficiency of ATRP is to conduct the reaction shown in Scheme 12 in the opposite order: using a metal complex in a higher rather than lower oxidation state (so-called, reverse ATRP process). A minor amount of classical radical initiator with a short half-life at the polymerization temperature is required to generate radicals. This approach facilitates the polymerization from the methodological standpoint, since complexes of transition metals in higher oxidation states are more stable in air.[141] In some cases, reverse and normal initiation can proceed simultaneously in the atom transfer polymerization (simultaneous reverse and normal initiation ATRP, SR&NI ATRP). This can be achieved by using a conventional radical initiator, together with alkyl halide as a source of carbon-centred radicals and a metal complex in a higher oxidation state. This approach provides a significant increase in the catalyst activity and a decrease in the catalyst concentration in the system by several orders of magnitude.[142]
Among other approaches directed towards increasing efficiency of ATRP catalyzed by transition metal complexes, mention should be made of the methods involving the use of reducing agents for fast regeneration of the active form of the catalyst. As the reducing agents, amines, carbohydrates, ascorbic acid, and other compounds are often used.[143-145] These methods are called ‘activator generated by electron transfer ATRP’ and ‘activator regenerated by electron transfer ATRP’ (AGET ATRP and ARGET ATRP for the use of higher and lower oxidation states of the metal in the complex, respectively) and imply the use of activators for the electron transfer in ATRP processes.[4] The additional introduction of radical initiators such as AIBN into the polymerization system [initiators for continuous activator regeneration ATRP (ICAR ATRP)] is also often utilized to activate the atom transfer polymerization processes.[146]
Another interesting approach involves the use of zero-valent metals, in particular metallic copper and iron, as activators and reducing agents for metal complexes [so-called supplemental activator and reducing agent (SARA) ATRP].[147][148] The use of electrical current to activate the metal atom in the metal complex catalyst and to accelerate conversion of the inactive form to the active one (eATRP, that is, electrochemically mediated ATRP) has also been reported.[149] Furthermore, it is possible to use ultrasonic treatment to increase the efficiency of atom transfer polymerization processes, although the mechanism of this effect on the catalyst has not yet been ultimately clarified and raises certain questions.[150]
Evidently, the efficiency of ATRP processes depends not only on the structure of the metal complex catalyst, but also on the selection of the appropriate initiator, which usually contains a carbon – halogen bond. The concentration of the initiator largely determines the number of growing chains, provided that the initiation efficiency is high. As a rule, these initiators contain groups that are able to stabilize the generated radicals (e.g., benzyl, ester, or nitrile group), and their reactivity is inversely proportional to the energy of the carbon–halogen bond, which directly depends on the nature of the stabilizing group and the halogen atom.[151-153] It is not accidental that compounds used most often as initiators include benzyl halides, halogen-containing esters, carboxylic acid amides and nitriles, and alkyl halides. Derivatives containing bromine and chlorine atoms in the molecules are fairly efficient. Analogous iodine and fluorine derivatives are much less commonly used in processes of this type. If there are several halogen atoms in the initiator (including macroinitiator) molecule, it is possible to obtain polymers with a branched, star-like, or graft structure.[154]
The equilibrium between the active and dormant chains is largely determined by the metal complex catalyst. The metal atom should have a high affinity for halogen; this would facilitate fast elimination of the halogen atom to give a carbon-centred radical initiating polymerization. In addition, the metal in the catalyst must exist in two relatively stable oxidation states separated by one electron (Cu+ and Cu2+, Fe2+ and Fe3+, Ru2+ and Ru3+, etc.). Among transition metals, copper, iron, ruthenium, osmium, rhodium, titanium, rhenium, and cobalt are used most often in ATRP processes.
The ligand environment of the metal has a pronounced effect on the efficiency of the metal complex catalyst and its reversible transition from inactive to active form. The choice of the ligand is often a key issue, because this determines the catalytic activity of the complex in halogen atom transfer reactions and directly influences the solubility of the complex in the reaction medium.[2][10] [155] For copper complexes, the use of nitrogen-containing ligands is most preferable, while in the case of iron complexes, phosphine ligands are usually chosen.[156] For ruthenium, half-sandwich complexes with cyclopentadienyl or carborane ligands in combination with phosphines are usually employed.[157] The activity of copper-containing catalytic systems depends on the ligand denticity and the length of the carbon bridge between nitrogen atoms,[158] as well as on the steric and electronic effects.[159] Examples of nitrogen-containing ligands that are used most often for copper complexes are 2,2'-bipyridine, 4,4’-bis(5-nonyl)-2,2'-bipyridine, N,N,N',N'',N''-pentamethyldiethylenetriamine, 1,1,4,7,10,10-hexamethyltriethylenetetramine, tris[2-(N,N-dimethylamino)ethyl]amine, and tris(2-pyridylmethyl)amine.
Naturally, apart from the catalyst and initiator structures, the efficiency of ATRP can be markedly affected by other factors that determine the rate of establishment of the dynamic equilibrium in the system (see Scheme 12) such as the solvent, the temperature of the reaction mixture, the ratio between the components in the system, the presence of activators (promoters), etc.
As noted above, the major and most significant drawback of ATRP as the most effective type of CRP as regards compliance with green chemistry is the use of transition metal-based catalysts, including complexes of so-called heavy metals, which, in some cases, can have an adverse influence on humans and the environment as a whole.[160] The recently proposed original methods and approaches to conduct atom transfer polymerization considered above made it possible to substantially (down to ppm level) decrease the concentration of the catalysts.[2][161]Undoubtedly, these trends of development of this type of CRP logically fit into the green chemistry concept, in particular, they comply with the atom economy and catalysis principles noted above. It is noteworthy that even with exceptionally low concentrations of metal complex catalysts, it is possible to carry out polymerization up to conversions of nearly 100% and, in some cases, within time periods that are fairly short for controlled processes (5 – 6 h),[145][154] which also reasonably complies with the green chemistry principles.
The ATRP concept and its modified versions analyzed above were initially used to prepare homo- and copolymers in bulk and in organic solvents. It is quite obvious that bulk synthesis of polymers is in line with the green chemistry concept. Conversely, the use of organic solvents, in some cases, quite toxic ones (dimethylformamide, etc.), contradicts the principles of green chemistry. It is not accidental that in recent years, a number of catalytic systems and compositions have been developed to carry out CRP in aqueous media, which fully corresponds to the principles of green origin of polymer molecules. Of course, this concerns first of all (co)polymerization of hydrophilic monomers.[162][163] The main issue in conducting the polymerization in this case is the appropriate selection of metal complex catalysts that have an affinity for water and, as a consequence, good water solubility.
Most of the above ATRP processes effectively proceed in a temperature range of 60 – 80°C, which, as in the case of thermal initiation of classical radical polymerization, requires certain energy expenditures. Recently, a number of ATRP procedures that imply conduction of polymerization at room temperature (ambient temperature) under photoirradiation have been developed, which is in line with the green chemistry concept.[164][165]
In 2012, Hawker and Fors [166] describes an ATRP process that proceeded at room temperature in the presence of tris(2-phenylpyridine)iridium(III) [Ir(ppy)3] as a photocatalyst for MMA polymerization in combination with an organobromine initiator. The authors showed that by using 0.005 – 0.2 mol.% Ir(ppy)3 and visible light irradiation, it is possible to obtain PMMA with Đ = 1.22 and random copolymers of methacrylic acid with benzyl methacrylate with narrow MWD (Đ = 1.24). The authors suggested that chain propagation is regulated via the reversible detachment of a bromine atom from a dormant polymer chain by the metal complex. It was found that the intermediate compound that keeps bromine is tris(2-phenylpyridine)iridium(IV) bromide. The metal – bromine bond is formed as a result of photoinduced electron transfer from the iridium atom to the halogen atom upon light absorption.
It was shown that copper compounds can also act as catalysts for ATRP under irradiation.[165][167-170] Due to the external action on the redox process in the system, photo-ATRP makes it possible to control the polymerization over time by light switching on/off without any adverse influence on the molecular weight distribution of the final product.[169] The rate of polymerization depends on the radical generation rate, i.e., on the efficiency of Cu(II) reduction to Cu(I). Thus, the concentration of radicals in the system, and, hence, the polymerization rate can be controlled by changing the light intensity (brightness of the source and distance from the light source to the reaction vessel), radiation wavelength, and the concentration and Cu/ligand ratio. Photocontrolled ATRP can be performed using inexpensive light emitting diodes, UV lamps, or even sunlight as radiation sources,[170] which makes these processes similar to the synthesis of biopolymers in nature. A drawback of this type of processes is the small penetration depth of light, i.e., the intensity of light flux decreases as it moves towards the inner part of the reaction vessel, which in some cases reduces the degree of control over the process.
The subsequent studies in this area have raised the important question of what is the contribution of photochemical processes to other ATRP procedures as well.[171] In this connection, studies of the effect of light in various wavelength ranges on the oxidation/reduction of the metal atom in the metal complex during ATRP processes under photoirradiation are currently carried out by a number of research teams.[161] [167][168][170]
Meanwhile, optimization of ATRP processes by using photoirradiation as regards energy efficiency as one of the green chemistry principles has not eliminated the key drawbacks of this method related to the use of transition metals for polymerization. As has already been mentioned, the presence of even trace amounts of metals in the polymers used in medicine or microelectronics may be critical. The mere presence of heavy metals in the final materials, together with the necessity of regenerating expensive metal-containing catalysts, restrict the industrial applicability of both traditional (thermal) atom transfer radical polymerization and photocontrolled ATRP processes.
3. Organic photoredox catalysis
An alternative to metal complex catalysis based on the use of transition metal compounds as key reagents is provided by organic photoredox catalysis, which is based on redox transformations related to single electron transfer (SET).[172] In the case of atom transfer radical polymerization mechanism, processes of this type were called organocatalyzed ATRP (O-ATRP), or metal-free ATRP (MF ATRP).[131][173-175]
As regards keeping with green chemistry requirements, organic photoredox catalysis has two considerable advantages over organometallic catalysis, in particular ATRP:
(1) absence of metals, including heavy metals, in the reactions;
(2) energy efficiency: the possibility of performing polymerization at room temperature.
It is noteworthy that the possibility of using visible light brings these processes closer to the synthesis of polymers in nature under sunlight (nature-like technologies). Light-induced chemical reactions of molecules have been known for more than a hundred years. They underlie many biochemical processes that occur in nature, and in recent years, they have become an essential part of classical organic synthesis.[176][177] Recent years have witnessed a kind of renaissance of synthetic photochemistry related to the emergence of pioneering studies in the field of photoredox catalysis using visible light. Although it is well known that visible light is the cheapest source of energy, its enormous synthetic potential has been revealed only in recent years owing to the development and application of new catalysts and catalytic systems. Indeed, currently, photoredox catalysis using visible light has become a convenient and fairly promising method for the synthesis of a broad range of compounds, including polymers, which proceeds under mild conditions and at moderate temperatures.
3.1. Oxidative and reductive mechanisms
Metal-free ATRP, like the above-described atom transfer radical polymerization involving metal complexes, is based on the reversible transfer of a halogen atom between the dormant polymer chain and the catalyst (in this case, an organic compound), accompanied by switching of the polymer chain from the dormant state to the active state.[131] [178-181] Successful implementation of this mechanism requires that the organic catalyst effectively absorbs a light flux (electromagnetic radiation), being thus converted to the excited state. The excited state of the catalyst should have relatively long (several milliseconds) lifetime, because it must have time to undergo a redox reaction with the polymer chain containing a halogen atom at the end before returning to the ground state. Certainly, the oxidation or reduction of the photocatalyst must be reversible for the catalytic cycle to be closed.
In terms of the mechanism of activation of dormant chains, MF ATRP reactions can be conventionally subdivided into two main types. The first one is the so-called oxidative quenching cycle in which the organic photocatalyst (PC), when converted to the excited state (PC*), is oxidized to the corresponding radical cation (Scheme 13).
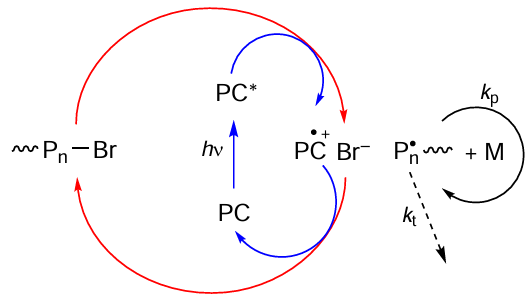
The putative mechanism of the oxidative quenching cycle [181] includes the initial excitation of the photocatalyst (PC) to the singlet or triplet state under irradiation. This is followed by single-electron transfer from the excited catalyst molecule (PC*) to the polymer chain existing in the dormant state, which finally results in the formation of an active radical. As this takes place, the photocatalyst is oxidized to give a radical cation, which forms an ion pair with the halide anion. According to quantum chemical calculations and kinetic measurements,[181] in this case, the reversible chain termination is due to the trimolecular reaction between the growing radical and the above-mentioned ion pair. Actually, this mechanism implies redox-active catalysis, that is, reversible electron transfer between the excited photocatalyst and the halogen atom of the initiator (see Scheme 13). As a result, a reactive radical inducing polymerization process is formed in the system.
The second type of MF ATRP mechanism is inherent in the processes that involve the reductive quenching cycle in which the catalyst in the excited state is reduced to a radical anion (Scheme 14). The implementation of this mechanism requires addition of reducing agents (RA) such as amines or other electron donors to the polymerization system.[182][183]
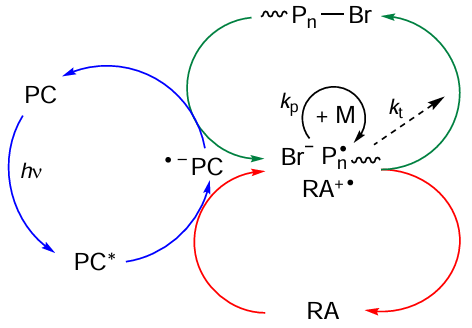
Actually, the second (reductive) mechanism of MF ATRP includes successive single-electron transfers between the electron donor (RA) and the photocatalyst, between the photocatalyst and bromine atom located at the end of the polymer chain, and between the bromine anion and the radical cation formed from the reducing agent (see Scheme 14).
Metal-free ATRP processes occurring by both mechanisms described above retain the key benefit of classical ATRP, that is, the possibility of preparing polymers with a definite MW and relatively low dispersity (Ɖ < 1.5) capable of subsequent transformations and modifications, including post-polymerization and block copolymerization, owing to the halogen atom located at the chain end.
3.2. Main types of catalysts
Since the MF ATRP mechanisms are based on reactions involving single-electron transfer (see Scheme 13 and Scheme 14), it is not accidental that polynuclear aromatic compounds prone to electron delocalization proved to be most efficient as catalysts for processes of this type. In particular, compositions based on perylene and its derivatives,[184][185] which were used for the synthesis of PMMA and MMA – n-butyl methacrylate copolymers, were among the first catalytic systems of this type to be proposed for the organophotocatalyzed polymerization of methacrylic monomers.
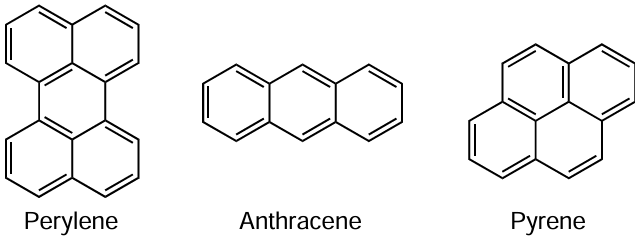
Miyake and Theriot [184][185] demonstrated that under visible light irradiation, perylene [184] and perylene-3,4,9,10-dicarboxydianhydride [185] detach a halogen atom from alkyl halide to give radicals, which can initiate polymerization of methacrylic monomers by the MF ATRP mechanism. Using perylene as a catalyst and ethyl 2-bromophenyl acetate as an initiator and irradiation at λ ≈ 380 nm, the authors synthesized polyMMA and poly(butyl methacrylate) with a relatively narrow MWD (Ð = 1.29 – 1.85). Study of the polymers by matrix-assisted laser desorption/ionization time-of-flight (MALDI TOF) mass spectrometry showed that the main array of the obtained macromolecules has a bromine atom at the end, which confirms that the polymerization follows the atom transfer mechanism. Meanwhile, the percentage of radical decay due to disproportionation or recombination reactions is rather high. This leads to a pronounced increase in the fraction of irreversible chain termination; this adversely affects the dispersity, which increases to 1.85 with increasing conversion.
Attempts to use other polynuclear aromatic hydrocarbons in the synthesis of polymers did not bring a significant increase in the degree of control of the polymerization process. For example, in the presence of anthracene as a catalyst and irradiation at λ ≈ 350 nm, MF ATRP proceeded at a relatively high rate, but it was impossible to obtain samples with a narrow MWD (Đ ³ 1.5).[186] According to the authors, this may be attributed to the occurrence of a side reaction of radical addition to anthracene during the process.
Pyrene, which contains four fused rings in the molecule, was a more effective photocatalyst of the polymerization of methacrylic monomers under similar conditions. In combination with tertiary alkyl bromides and benzyl bromides as initiators and sources of carbon-centred radicals under irradiation at λ ≈ 350 nm, the authors obtained PMMA with relatively low dispersity (Đ ≈ 1.3).[186] The proposed pyrene/ethyl 2-bromophenylacetate system also gave polystyrene and poly(tert-butyl acrylate) with relatively narrow MWD (Đ ≈ 1.3 – 1.4). The presence of bromine at the end of macromolecules was confirmed experimentally by post-polymerization of MMA and block-copolymerization of MMA with styrene.
The radical formation step and subsequent polymerization by the MF ATRP mechanism in the presence of perylene and pyrene as polynuclear aromatic hydrocarbons was considered by the authors in terms of the electron transfer from the excited photoactivator (or photosensitizer) to alkyl halide as an initiator within the oxidative quenching cycle (see Scheme 13), i.e., photoredox catalysis took place.
It should be noted that pyrene and its derivatives are widely used as photosensitizers and photoinitiators in cationic and free-radical polymerization.[187][188] In addition, perylene and pyrene as representatives of conjugated polycyclic hydrocarbons perfectly absorb light and show intense fluorescence.[189]
Polynuclear heterocyclic compounds containing nitrogen, oxygen, and sulfur atoms in the molecules also turned out to be quite effective catalysts of MF ATRP processes. For example, 10-phenylphenothiazine and its analogue, 10-methylphenothiazine, were tested in the synthesis of PMMA and polybenzyl methacrylate under photoirradiation.[190] It was found that 0.1 mol.% PPT and irradiation at λmax ≈ 380 nm causes polymerization of the above-mentioned methacrylic esters. The obtained PMMA and poly(benzyl methacrylate) samples showed a good relationship between the experimental and theoretically calculated Mn values and had low dispersity (Ð ≈ 1.2 – 1.3). Methylphenothiazine also allows for effective radical polymerization of MMA, but the dispersity of the resulting polymers proved to be somewhat higher (Ð ≈ 1.7).
It was shown that the photocontrolled polymerization induced by PPT, like the polymerization using the Ir(ppy)3 catalyst described above,[166] can be switched on and off by cyclic light irradiation of the reaction system. The polymerization in the dark (without irradiation) is attenuated, but starts all over again immediately after the repeated action of light. The PMMA samples obtained with participation of the PPT/halogenated organic compound system can be successfully used to prepare block copolymers, which confirms that polymerization proceeds in the living mode. The polymer containing a halogen atom at the chain end acts as the initiator. It was shown that PPT makes it possible to carry out polymerization of a wide range of monomers, including nitrogen-containing methacrylic monomers such as 2-(dimethylamino)ethyl methacrylate, furthermore, with a high degree of control of the molecular weight characteristics.[190]
Matyjaszewski and co-workers [191] used catalytic systems containing phenothiazines for polymerization of acrylonitrile under photoirradiation. Apart from PPT, analogous compounds, 4-methoxyphenylphenothiazine and naphthylphenothiazine, were tested in similar processes. After selection of the optimal conditions, controlled synthesis of polyacrylonitrile was performed. The dispersity of the products was ≈ 1.5, which, together with some features of the polymerization, attest to the MF ATRP mechanism.
Further modification of PPT-based photocatalysts by extending the conjugated system via introduction of additional aromatic moieties into the molecule expanded the range of thiazine photocatalysts suitable for the synthesis of methacrylic polymers by MF ATRP process.[192-194]
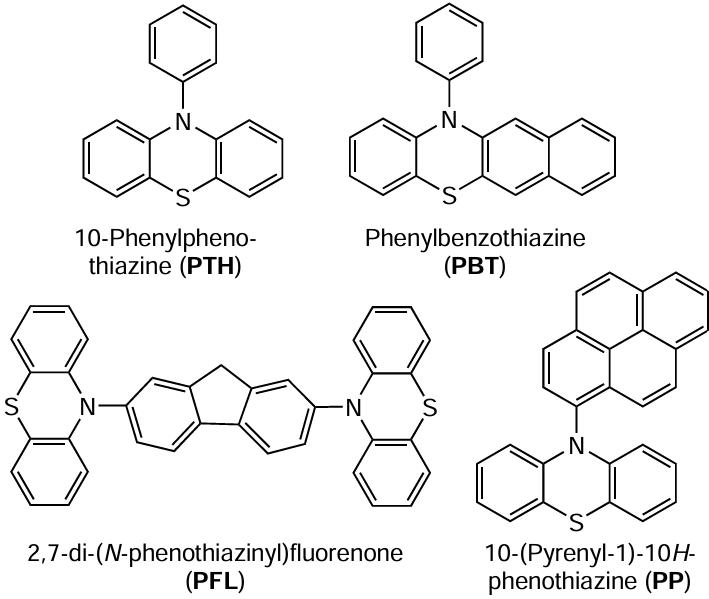
An increase in the size of the conjugated aromatic moiety shifts the absorption peaks of these compounds to the visible region. Hence, thiazine catalysts can be used not only under UV irradiation, but also under visible irradiation, which is important as regards green processes of polymer synthesis. It was shown that like PPT, phenylbenzothiazine (PBT) and 2,7-di(N-phenothiazinyl)fluorenone (PFL) combined with organobromine compounds can perform polymerization of methacrylic esters (MMA, benzyl methacrylate, and butyl methacrylate) as a controlled process. Depending on the nature of the monomer, the component ratio in the reaction medium, and time of synthesis, the dispersity values of polymers samples obtained in the presence of above-mentioned phenothiazines, PBT, PFL, and 10-(1-pyrenyl)-10H-phenothiazine (PP) were 1.3 – 1.7, 1.4, and 1.2 – 1.3, respectively. The living nature of the polymerization is confirmed by the possibility of obtaining a broad range of linear block copolymers based on methacrylic monomers with relatively narrow MWD (Ɖ ≈ 1.2 – 1.4).
In recent years, the ability of phenothiazines to behave as photoreducing agents has been often utilized in organic photocatalysis to perform dehalogenation reactions [195] and to form the carbon–carbon bonds with participation of aryl halide substrates.[196]
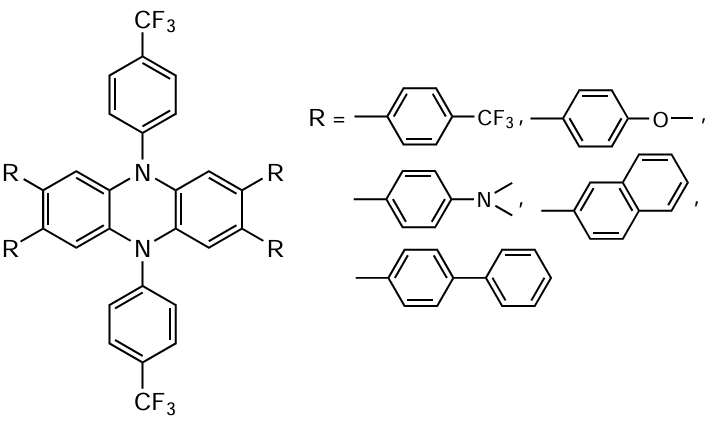
Some substituted phenothiazine analogues, in particular 9,10-(N,N-diaryl)dihydrophenazines,[197] the general formula of which is shown in Scheme 15, proved to be no less efficient catalysts of polymerization.
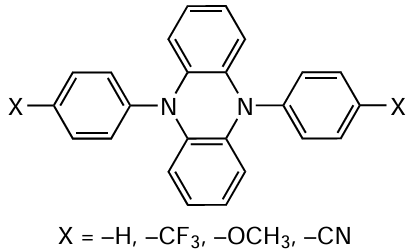
It is noteworthy that phenazine radical cations formed in the intermediate steps of the photoredox process are exceptionally stable and can be used as components in ferromagnetic organic materials.[198] In addition, phenazine and its cationic derivatives are successfully used as photocatalysts of some organic reactions,[199] and can also serve as parts of photoinitiating systems in the polymerization of a wide range of monomers,[200] including MF ATRP processes.[197] [201] Since 9,10-(N-aryl)dihydrophenazines are fairly sensitive to visible light irradiation, polymerization in the presence of these compounds can be carried out using long-wavelength radiation (400 – 700 nm). The introduction of electron-withdrawing substituents into the molecule of the indicated compound increases the catalyst efficiency. In particular, it was found [200] that 10-di(4-trifluoromethylphenyl)-9,10-dihydrophenazine makes it possible to obtain polymer samples with a very low dispersity (Đ = 1.10 – 1.18). Indeed, the synthesis of polymers in the presence of 9,10-(N-aryl)dihydrophenazines follows the MF ATRP mechanism, as indicated by the possibility of reinitiation of the reaction by conducting post-polymerization of MMA or block copolymerization of PMMA with butyl methacrylate and benzyl methacrylate. The extension of the system of conjugation in the catalyst also results in increasing catalyst activity in O-ATRP. By using 5,10-di(N-1-naphthyl)-5,10-dihydrophenazine, it was possible to synthesize PMMA with a rather narrow molecular weight distribution (Ɖ < 1.3), with the reaction proceeding up to high degrees of monomer conversion with excellent coincidence of the theoretical and experimental Mn values.[197][201]
In subsequent studies, Miyake and co-workers [201][202] found that the efficiency of the dihydrophenazine/organobromide compound catalytic system is influenced by not only the composition of the system, but also the nature of the solvent. Experimental studies and theoretical calculations showed that the solvent polarity is an important factor having a significant effect on the polymerization, including both the degree of charge transfer in the excited photocatalyst (PC*) and the ionic interaction between the catalyst radical cation and the bromide anion (РС+•Br–). As a result, the authors succeeded in selecting polar solvents the polymerization in which gave polymers with a very low dispersity (Ɖ < 1.10) at a high initiation efficiency (I* ≈ 100% and above).[202] A considerable drawback of these systems is the use of relatively high concentrations of the initiator (about 1000 ppm), which leads to the synthesis of polymers with a relatively low molecular weight (at 10 kDa level).
Further studies with the goal to modify dihydrophenazine-based catalytic systems by introducing various substituents have resulted in the design of a number of more effective catalysts (Scheme 16), which made it possible to carry out controlled polymerization of vinyl monomers using relatively low catalyst concentrations (at a level of 5 – 50 ppm).[203][204]
In addition to the phenazine-based catalytic systems considered above, Miyake and co-workers [205] tested another class of conjugated heterocyclic compounds, N-arylphenoxazines, as catalysts for MF ATRP.
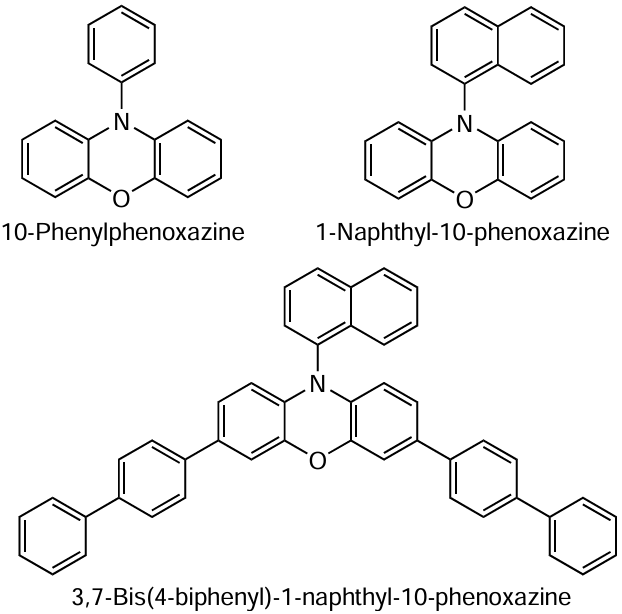
The efficiency of phenoxazine catalysts in polymerization processes was comparable to that of phenazines. Thus MMA polymerization under irradiation at 365 nm in the presence of 1-naphthyl-10-phenoxazine as a photocatalyst proceeded up to a high conversion (more than 95%) and was accompanied by a linear growth of the MW with increasing conversion, which is typical of the controlled synthesis of macromolecules, and the resulting polymer had narrow MWD (Ð = 1.11 – 1.21).
The modification of the phenoxazine ring by introducing biphenyl substituents into positions 3 and 7 gave rise to a new catalyst, 3,7-bis(4-biphenyl)-1-naphthyl-10-phenoxazine, with an extended conjugated system. Unlike 1-naphthyl-(10-phenoxazine), this catalyst induces a sharp increase in the molar extinction coefficient (ε) and red shift of the absorption band in the absorption spectrum (from 323 to 388 nm). The polymerization of MMA involving this catalyst under irradiation with white light gave a polymer with a low dispersity (Ɖ = 1.13 – 1.16), with the initiation efficiency being high (I* ≈ 98 – 100%).[206][207] The authors of these studies tested more than 20 catalysts with various substituents in the phenazine moiety and demonstrated that all of them initiate polymerization of MMA with high initiation efficiency (more than 95%) and provide the formation of polymers with rather narrow MWD (Ɖ < 1.3). The authors reasonably believe that high efficiency of the above compounds is due to their ability to efficiently absorb visible light and to their high reduction potentials.
From the practical point of view, it is important that these catalysts allow efficient synthesis of polymers not only in an inert atmosphere, but also in the presence of air oxygen.[208] A distinctive feature of the processes in this case is good correlation between the theoretically calculated and experimentally determined MW values, low dispersity of the synthesized samples (Ɖ ≈ 1.2 – 1.3), and high efficiency of initiation I* (84 – 99%). Note that in the case of living polymerization, by initiation efficiency is usually meant the fraction of initiator molecules that triggered the polymerization.
Apart from phenothazines, phenazines, and phenoxazines, catalytic systems based on other fused aromatic compounds,[209] including those containing selenium and tellurium,[210][211] have also found use in the catalysis of MF ATRP processes.
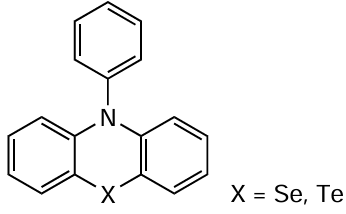
N-Phenylphenochalcogenazines, analogues of phenothazines containing selenium and tellurium atoms, were tested in the polymerization of MMA; however, they proved to be less effective photocatalysts than 10-N-phenylphenoxazine [211] regarding both the control over molecular weight characteristics of the synthesized polymers and efficiency of initiation.
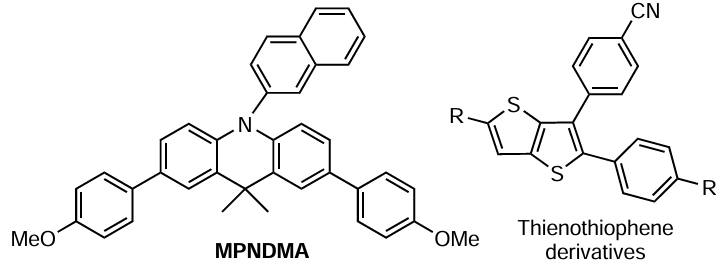
Yet another class of polynuclear heterocyclic compounds, dihydroacridines, were proposed as catalysts of MF ATRP for the polymerization of acrylic monomers.[212] It was found that, as in the case of phenoxazine and phenazine derivatives, the use of which in polymerization processes is described above, the activity of these catalysts is determined by the nature of substituents in positions 3, 7, and 10. An example of this type of compound is 3,7-bis(4-methoxyphenyl)-N-(2-naphthyl)-10,10-dimethylacridine (MPNDMA). Thus upon irradiation of butyl acrylate with light at λmax = 365 nm, the initiation efficiency depending on the composition of the dihydroacridine-based catalytic system varied from 35 to 100%. The dispersity of the samples ranged simultaneously from 1.35 to 1.93. The dispersity of the obtained polymers based on some acrylic monomers can be considerably decreased (down to Ɖ of approximately 1.2) by adding lithium bromide. Unfortunately, the mechanism of action of this activating agent is not proposed or discussed in the publication.[212]
It is noteworthy that acridine photocatalysts are actively used not only in polymerization but also in modern organic synthesis. In particular, they are used to carry out radical reactions involving thiols and disulfides and in dearylation reactions.[213][214] The authors of these studies proved the occurrence of so-called proton-coupled electron transfer in catalytic reactions involving acridine catalysts, which smoothly fits into the MF ATRP concept.
Yagci and co-workers.[215] reported the first application of a number of thienothiophenes, sulfur-containing heteroaromatic compounds, as catalysts for MMA polymerization by the MF ATRP mechanism. According to their study, in the presence of alkyl bromides as sources of carbon-centred radicals, these catalysts successfully initiate the polymerization of MMA under irradiation with light at λmax = 350 nm. The dependence of the polymer yield on time in semi-logarithmic coordinates is linear, which is characteristic of controlled radical polymerization reactions, and the dispersity of the synthesized samples is in the range from 1.3 to 1.8.
The initiation of MMA polymerization under irradiation with blue light was also observed using a number of other heterocyclic catalysts: 9-phenylcarbazole, 4,4'-bis(N-carbazolyl)biphenyl, and 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene in combination with ethyl 2-bromophenyl acetate as an initiator.[216] However, in the case of carbazole derivatives, polymers with a broad MWD (Ɖ ³ 1.80) were obtained. When 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene was used, the process was initiated at a catalyst concentration as low as approximately 5 ppm; however, the polymer dispersity was rather high (Ɖ ³ 1.50), irrespective of the ratio of components in the system. The presence of Br-containing groups at the end of the polymer chain, which confirms the ATRP mechanism of the polymerization, was proved by MALDI time-of-flight mass spectrometry and also by successful post-polymerization and synthesis of block copolymers.
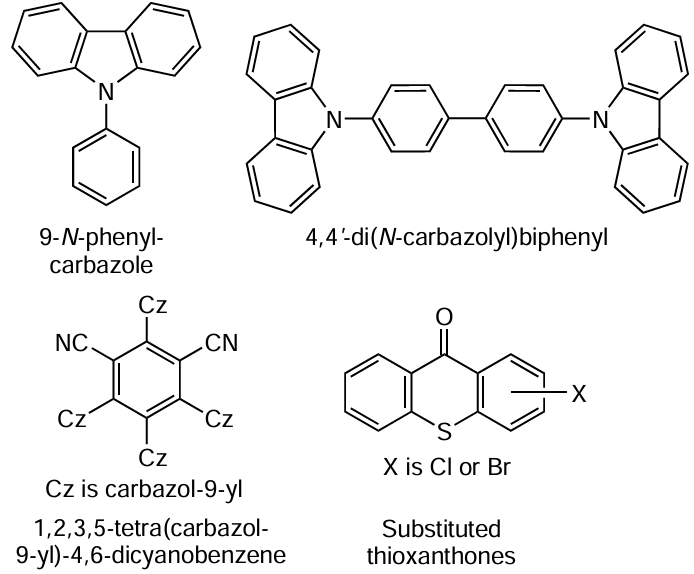
The above data on the use of polynuclear homo- and heterocyclic compounds unambiguously indicate that the presence of electron-donating substituents in the molecules has a beneficial effect on their reactivity towards MF ATRP processes, which logically fits into the above photopolymerization chart with oxidative quenching (see Scheme 13).
One more way of implementation of MF ATRP is the use of so-called type II photoinitiators [217][218] (benzoquinone, anthraquinone, thioxanthone, etc.) in combination with electron-donor compounds, e.g., amines, as sources of electrons, in the presence of alkyl halides as initiators and sources of carbon-centred radicals.[219][220]
Yagci and co-workers [219] carried out polymerization of MMA involving benzophenone and some thioxanthones under ultraviolet irradiation (λmax ≈ 350 nm) in the presence of pentamethyldiethylenetriamine (PMDETA) as electron donor and alkyl bromides as sources of carbon-centred radicals. The dispersity of the synthesized samples was at the level of 1.3 – 1.4. The presence of bromine-containing groups at the end of the polymer chain was confirmed by performing post-polymerization of MMA and block copolymerization of synthesized PMMA with butyl acrylate. Regarding the use of nature-like technologies for polymer synthesis, it is important that thioxanthones are effective as photocatalysts not only under UV irradiation, but also on exposure to visible light.
The polymerization mechanism proposed by the authors implies light-induced transition of thioxanthone to the excited state (Scheme 17) followed by electron transfer from amine to the catalyst. The resulting radical anion pair [thioxanthone + amine] reduces alkyl halide or a polymer molecule containing a terminal halogen atom to give radicals responsible for chain initiation and propagation. The back electron transfer from the halide anion to the amine radical cation actually completes the polymerization cycle by forming inactive macroalkyl halides, which return again to the polymerization system.
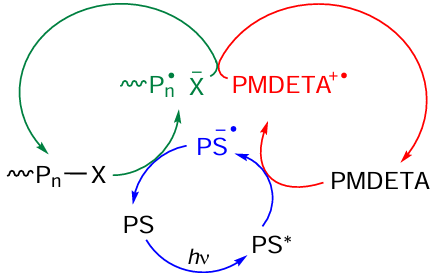
In addition to sulfur- and nitrogen-containing heterocyclic systems described above, oxygen-containing heterocyclic systems have been successfully applied for radical polymerization of vinyl and (meth)acrylic monomers. Fluorescein[220] and erythrosine B [221] proved to be fairly effective sensitizers in the presence of PMDETA. Using these compounds and irradiation at 400 – 500 nm in the presence of organobromine initiators, living polymerization of MMA, ST, 2-hydroxyhexyl methacrylate, and tert-butyl acrylate was performed. The presence of bromine-containing terminal groups in the polymer was experimentally confirmed by conducting post-polymerization and block copolymerization involving these groups.
Considering experimental results and quantum chemical calculation data,[220] a mechanism of quenching including the transfer of electrons from the donors (amines) to the photocatalyst, which is thus brought to the excited state, was proposed (similarly to Scheme 14). Fluorescein is converted from the neutral to radical anion form, while the amine, being an electron donor, is correspondingly converted to the radical cation. The photocatalyst radical anion further reacts with alkyl halide or the polymer chain containing a halogen atom and thus generates radicals that cause polymerization. The reversibility of the activation/deactivation process provides the necessary control over polymer MWD.
Thus, the above data indicate that conjugated systems based on polynuclear heterocyclic compounds are of obvious interest for the use in catalysis of MF ATRP processes. A certain drawback of the most efficient catalysts based on compounds of this type is their complex structure and, hence, most often complicated synthesis and, as a consequence, relatively high cost. In this regard, some natural and synthetic dyes with structures based on conjugated heterocyclic moieties that are prone to undergo single electron transfer reactions may be of certain interest for practical application in the controlled synthesis of polymers. These compounds are widely used in the chemical industry and analytical chemistry, as well as in medicine, and, therefore, they are quite affordable for practical application.
3.3. Synthetic and natural dyes as promising catalysts
As mentioned above, to achieve high efficiency, an organic catalyst should be characterized by
(1) high extinction coefficient (ε > 104) at wavelengths corresponding to the exciting radiation;
(2) relatively long (more than a microsecond) lifetime of the excited state.[131]
It should be noted that due to their nature, organic compounds are markedly inferior (by 1 to 3 orders of magnitudes) to transition metal complexes in characteristic (2). Meanwhile, by varying the structure of the organic catalyst, in particular by introducing chemically diverse substituents into the catalyst molecule, it is possible to influence to some extent the excited state lifetime. In addition, for the prospects of practical applications, it is important that the organic catalyst be cost affordable. In this regard, phenazine and phenoxazine dyes turned out to be very promising compounds for polymerization of a broad range of monomers by the MF ATRP mechanism.
Phenazine as a parent compound for the group of phenazine dyes (neutral red, safranin T, etc.) was successfully used to perform living polymerization of MMA and other methacrylic monomers under UV irradiation.[222][223] Ethyl 2-bromoisobutyrate (EBIB), tert-butyl bromide, and carbon tetrabromide were used as initiators and sources of carbon-centred radicals. The last-mentioned compound proved to be the compound of choice to attain high conversions; however, dependences of the polymer yield on the time of synthesis in the semi-logarithmic coordinates (ln[m0]/[m] vs. time) differ from linearity and show the presence of an induction period. When amine activators are added to the polymerization system, the induction period disappears, and the yield of polymer increases. The dependence of the number-average MW on the conversion is linear, which is characteristic of controlled radical polymerization processes. The resulting polymers were successfully used as macroinitiators for the synthesis of block copolymers and for post-polymerization. A distinctive feature of phenazine-based catalytic systems is the possibility of conducting polymerization under aerobic conditions (in the presence of air oxygen), which brings these processes closer to the synthesis of polymers in nature. It was shown experimentally that in the presence of amines, polymerization proceeds by the reductive MF ATRP mechanism (see Scheme 14). The dispersity of the synthesized samples varies in the range from 1.7 to 2.0, depending on the polymerization conditions. There is some discrepancy between the theoretically calculated and experimentally determined Mn values of PMMA samples, which is probably attributable to side reactions that take place in the system. In particular, the addition of the initiating radicals directly to phenazine rather than to the monomer is a possible reaction of this type.
As expected, phenazine derivatives containing electron-donating substituents in the aromatic rings, in particular, neutral red and safranin T, proved to be more efficient in the controlled synthesis of polymers.[224] In relation to MMA as a model monomer, it was found that in the presence of tert-butyl bromide and ethyl 2-bromoisobutyrate as sources of carbon-centred radicals, neutral red can initiate polymerization not only under UV irradiation, but also under irradiation with visible light, and both in a degassed reaction mixture and in the presence of air oxygen, which is very important regarding the use of nature-like technologies and green chemistry in the polymer synthesis.
In the presence of [neutral red – ethyl 2-bromoisobutyrate] composition, the yield of the polymer exceeded 90% within 4 hours, irrespective of the presence of oxygen in the reaction mixture. In this case, a light-emitting-diode strip with an emission spectrum similar to that of sunlight was used as a source of radiation. The addition of electron-donating activators, particularly secondary and tertiary amines, had a considerable effect on both the reaction kinetics and the molecular weight characteristics of polymers. In particular, tributylamine increases the polymerization rate in an early stage of the process; this results in decreasing number-average molecular weight of the polymer. The number-average MW followed a linear dependence on the conversion, which is typical of the controlled radical polymerization; the Đ values for the synthesized samples decreased during the process in the 2.2 – 1.6 range in the degassed reaction mixture and in the 2.8 – 1.5 range in the case of polymer synthesis in air (without degassing).
Similar patterns were observed in the polymerization of MMA involving the [safranin T – EBIB] composition.[224] In particular, irrespective of the presence of oxygen in the system, the reaction proceeded to a conversion of 90 – 95% in a fairly short period of time (within 4 h). In the case of degassed reaction mixture, after the addition of amines, longer times of synthesis were required to attain a high degree of conversion. It is of interest that the overall polymerization rate with the [safranin T – EBIB] and [safranin T – EBIB – tributylamine] initiating systems was higher in the presence of air oxygen than in the absence of oxygen. It should be noted that the numerical MW values of the polymers obtained using the [neutral red – EBIB] and [safranin T – EBIB] systems in the presence of air oxygen were virtually equal. The addition of isopropylamine may induce a decrease in MW of the PMMA samples in comparison with polymers obtained without the amine. Under anaerobic conditions, the dependence of Mn on the monomer conversion had two sections with different rates of increase in MW. During the process, the increase in the conversion was accompanied by decreasing Ɖ values of PMMA samples from 2.3 to 1.8. In the presence of oxygen, the [safranin T – EBIB – isopropylamine] composition promoted a linear increase in Mn with increasing degree of conversion of the monomer. In this case, the MWD curves were unimodal and shifted with time to greater MW values, while the dispersity of the synthesized polymer samples decreased from 2.8 to 1.7 during the polymerization.
It was shown that the [phenazine dye – EBIB – tributylamine] catalytic systems initiate polymerization of not only MMA, but of a number of other acrylic and methacrylic monomers such as butyl methacrylate, glycidyl methacrylate, methyl acrylate, and acrylonitrile.[224] In relation to polymerization of butyl methacrylate, it was shown that the catalytic system based on neutral red is more efficient than a similar composition based on safranin T. The overall polymerization rate and the limiting conversion of butyl methacrylate in the presence of [safranin T – EBIB – tributylamine] catalytic system are lower than in the case of neutral red under similar conditions. The dependence of Mn on the conversion is linear for both dyes, while the molecular weights of polymers obtained using safranin T are higher than those of polymers obtained with neutral red. The Đ value decreases with increasing monomer conversions in the presence of either of the dyes.
The polymerization of acrylonitrile in DMSO initiated by the [neutral red – EBIB – tributylamine] system occurs up to high degrees of conversion (up to 95%). The process was characterized by a linear increase in Mn with increasing conversion of the monomer and by unimodal MWD curves of the samples, which confirms the controlled nature of the polymerization. The Ɖ values decrease to 1.4 as conversion increases.
Spectroscopic and quantum chemical studies of the mechanism of polymerization in the presence of these dyes, together with published data on the reactivity of the dyes, suggest [225-227] the following chart of reactions between the components of this system (Scheme 18).
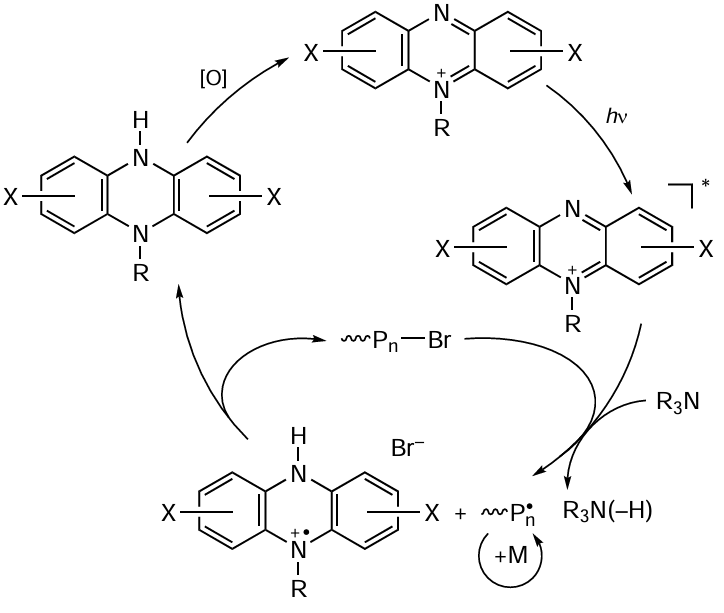
Thus, phenazine and especially phenazine-based dyes (neutral red and safranin T) are promising photocatalysts for the radical polymerization of a broad range of (meth)acrylic monomers, which can occur up to high monomer conversions over a short period of time at low concentrations of components of the catalytic system (0.01 mol.%). From the practical point of view, it is important that the proposed photocatalysts are fairly effective under the action of common white light, in particular in the presence of air oxygen in the reaction mixture.
Methylene blue (MB), the most well-known and widely practically used phenothiazine dye, the formula of which is depicted above, was also tested in controlled radical polymerization processes.[190][228][229] This dye is characterized by high ε values in the visible region and is widely used in biochemical studies.[230][231] The first attempts to use MB in MF ATRP processes were unsuccessful;[190] under white light irradiation in the presence of MB, the monomer conversion to the polymer did not exceed 1%.
Methylene blue proved to be a more effective catalyst when used as a part of a multicomponent photoinitiating system [iodonium salt + amine + dye],[232-234] which performs polymerization of (meth)acrylic monomers in high yield.
Methylene blue as a photocatalyst is of obvious interest as regards more persistent application of the green chemistry concept in polymerization processes. In particular, it was established that in combination with brominated initiators (2-bromoisobutyrate and other), MB provides effective polymerization of MMA and some other (meth)acrylic monomers under mild temperature conditions (room temperature) and under irradiation with visible light.[228][229] The polymer yield reaches 80% over a period of 7 h. The reaction is characterized by a linear growth of MW with increasing monomer conversion, which is characteristic of living polymerization. As expected, the introduction of activating reducing agents, e.g., glucose or ascorbic acid., into the polymerization system increases the total rate of polymerization and has a beneficial effect on both the process kinetics and the molecular weight characteristics of the synthesized polymers. In particular, it was shown that in the presence of ascorbic acid, MB is reduced to the leuco form. The reaction rate increases, and the monomer conversion actually reaches 100% in 2.5 h. As expected, MW of the polymer decreases compared to that of the samples obtained under the same conditions, but without addition of the activator. It is of interest that the combination of MB with ascorbic acid allows initiation of photocatalyzed polymerization of MMA in the absence of a halogen-containing initiator. Thus, ascorbic acid (or products of its oxidation during the reaction) is not only involved in the chain transfer, thus controlling MW of the polymer, but can also serve as an additional source of initiating radicals.
As activators and reducing agents, amines can also considerably influence the polymerization of MMA in the presence of MB. The type of this influence on the molecular weight characteristics of PMMA and on the polymerization rate directly depends on the structure of the amine. For example, the addition of primary and secondary amines, both aliphatic (PriNH2 , Pri2NH) and aromatic (PhNH2, Ph2NH) ones, causes a slight decrease in the conversion of MMA under both UV and white light irradiation. The average MW values of the samples are slightly lower than those of polymers synthesized without the use of amines. In the opinion of the authors,[228][229] the decrease in the monomer conversion and numerical values of MW is attributable to high reactivity of the above amines, in particular, to the presence of active hydrogen atoms in their molecules capable of being involved in chain transfer reactions.
Meanwhile, the type of influence of tertiary aliphatic amines, in particular tributylamine, on the polymerization depends on the wavelength of the light radiation used. If UV irradiation is used to initiate polymerization, the addition of tertiary amines containing no active hydrogen atoms into the polymerization system results, like in the case of ascorbic acid, in increasing monomer conversion and decreasing number-average MW, which indirectly indicates that polymerization initiation is additionally stimulated by the amine. When white light is used, tertiary amines have an even more pronounced effect on the monomer conversion; in the presence of Bu3N in the polymerization system, the conversion reaches 95% in 2.5 h.[228] The dependence of average MW on the conversion is linear, and the dispersity values remain at the level of 1.6 – 1.7 up to the limiting degrees of conversion. The results of studies indicate that by using MB as a representative of xanthene dyes, it is possible to carry out polymerization of methacrylic monomers by the MF ATRP method with a reductive quenching cycle of the catalyst excited state (see Scheme 14). In particular, detailed study of the polymerization mechanism, including the transformations of MB during the process, indicates that reduction of the cationic form of the photocatalyst (MB+) to the radical form (MB·), which finally makes it possible to observe a linear growth of MW with increasing conversion, characteristic of controlled radical polymerization, under irradiation at λmax ~ 450/510 nm (Scheme 19).
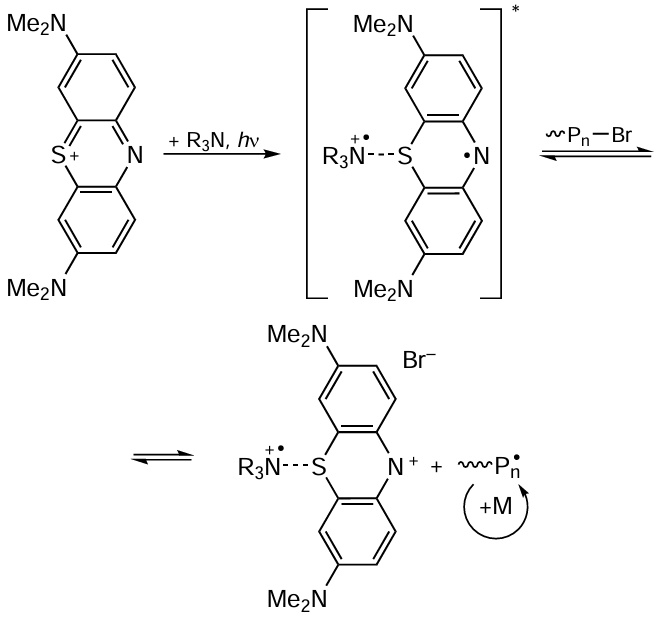
In terms of the economic cost and compliance with green chemistry, the use of organic catalysts is undoubtedly preferred over the use of metal complex ATRP regulators containing expensive organic ligands in the molecules. In addition, as mentioned above, the presence of even traces of transition metals in the synthesized polymer substantially restricts the scope of applicability of polymer materials in microelectronics, biomedicine, and some other high-tech fields. Therefore, the development of new high-performance metal-free (organic) catalysts for atom transfer radical polymerization is of obvious practical interest and is a highly relevant task.[235]
As regards the above-mentioned photocatalytic systems and compositions, an obvious advantage of dyes as catalysts of MF ATRP polymerization that meet all the basic requirements of green chemistry is not only their high efficiency combined with the possibility of controlled synthesis of polymers under visible light irradiation and under aerobic conditions, but also their ready availability. These compounds are widely used in analytical chemistry and medicine, they are produced on an industrial scale, and have relatively low cost. As a drawback associated with the use of these systems, note the presence of colouring in the initial stage of the synthesis, which, however, can be eliminated quite easily by purification of the polymer by standard repeated reprecipitation in appropriate solvents, usually hydrocarbons, which do not have a significant adverse effect on the environment.
4. Conclusion
It is clear that the development of human civilization cannot be conceived without polymers and macromolecular structures. The scope of their application in power engineering, microelectronics, construction industry, transport, agriculture, food industry, medicine, and pharmaceutical industry is rapidly increasing every year. Of particular importance are organic-inorganic composite materials, which have recently been actively used in regenerative medicine, wind energy, electric vehicles, aircraft and space technology, military-industrial complex, and other high-tech branches. Important parameters determining the properties and further applications of synthesized polymers and polymer materials are molecular weight characteristics and compositional uniformity. Evidently, the controlled radical polymerization strategy is the most effective tool for the targeted selection of the indicated characteristics. The procedures developed in recent years make it possible to synthesize polymers with both low and ultrahigh molecular weights (up to 2 × 106),[236-238] including multi-block copolymers and complex macromolecular structures,[237-239] and polymers for 3D printing.[240][241] Obviously, the growth of polymer production raises hot environmental issues [5][7] [242] regarding not only reuse and disposal of macromolecular compounds, but also development of effective methods for the synthesis of macromolecules that would have a minimum adverse effect on the environment. In this regard, controlled radical polymerization, especially using methods of organic photoredox catalysis that have been actively developed in recent years, appears to be the most effective and promising strategy for the synthesis of polymers with a specified set of properties and characteristics complying as fully as possible with green chemistry principles. The application of this strategy, including the use of organic compounds instead of metal complexes as catalysts under irradiation with visible light, as well as the possibility of polymerization under aerobic conditions at low temperatures corresponding to the ambient temperature, brings synthetic methods of polymer production closer to the processes of synthesis of macromolecules in nature.
It is known that Nature is, in some cases, a more ingenious and effective synthetic chemist than humans, if only because it can synthesize macromolecules with molecular weights of several tens and even hundreds of millions of daltons, with the synthesis proceeding under mild temperature conditions and using only the energy of the environment (solar light energy). Furthermore, polymers synthesized by Nature have often fairly complex molecular and supramolecular structures and, in some cases, also the required stereoisomerism. Unfortunately, the currently available techniques in the field of photoreox catalysis and their application in the synthesis of polymers, which are discussed in this review, are inferior in efficiency to not only nature-like technologies but, in some cases, also to classical methods using metal complex catalysts. Therefore, further research in the field of organic photoredox catalysis, including the development of new catalytic systems and compositions based on organic compounds, as well as the development of original methodological approaches in the photoinitiated controlled synthesis of polymers, seems to be a very relevant and significant task both for fundamental issues and for practical applications, including those keeping with principles of green chemistry and nature-like technologies.
In conclusion, it should be noted that controlled radical polymerization has turned in the last two decades into a highly relevant and actively developing trend of modern synthetic chemistry of macromolecular compounds. It is beyond doubt that a reasonable combination of the regularities of this process and green chemistry principles, including those related to organic photoredox catalysis, will promote effective development of the theoretical fundamentals and practical applications of polymer science in the coming years.
5. List of abbreviations and symbols
Mn — number-average molecular weight,
Ð — dispersity (Mw/Mn),
AIBN — azobis(isobutyronitrile),
ATRP — atom transfer radical polymerization,
A(R)GET — activator (re)generated by electron transfer,
CRP — controlled radical polymerization,
DT — degenerate chain transfer,
EBIB — ethyl 2-bromoisobutyrate.
ICAR — initiators for continuous activator regeneration,
ITRP — iodine transfer radical polymerization,
MF ATRP — metal-free atom transfer radical polymerization,
MMA — methyl methacrylate,
MW — molecular weight,
MWD — molecular weight distribution,
NMP — nitroxide-mediated polymerization,
O-ATRP — organocatalyzed atom transfer radical polymerization,
PBN — C-phenyl-N-tert-butyl nitrone,
PET — photoinitiated electron transfer,
PET-RAFT — photoinduced electron transfer – reversible addition-fragmentation transfer,
PMMA — polymethyl methacrylate,
PPT — 10-phenylphenothiazine,
RAFT — reversible addition-fragmentation transfer,
RCT — reversible chain transfer,
RDRP — reversible-deactivation radical polymerization,
SARA — supplemental activator and reducing agent,
SET-LRP — single electron transfer living radical polymerization,
SFRP — stable free radical polymerization,
SR&NI — simultaneous reverse and normal initiation,
St — styrene,
TEMPO — 2,2,6,6-tetramethylpiperidin-1-oxyl.
References


















