


Keywords
Abstract
The interest in hydrodechlorination (HDC) as a safe way to convert waste into high added value products is due to the significant amounts of chlorine-containing polymers in industrial and household waste plastics, a wide variety of liquid chlorine-containing wastes and their high environmental toxicity. The review analyzes the composition of chlorine-containing waste, including liquid products of pyrolysis, hydrocracking and hydrothermal treatment of polyvinylchloride and mixed waste plastics containing chlorinated polymers. The distinctive features of the HDC process are considered in comparison with hydrodesulfurization and hydrodeoxygenation reactions. The achievements of the last 15 years in the field of HDC catalysis are analyzed. Much attention is given to bimetallic noble metal catalysts and catalysts based on transition metal sulfides and phosphides. The prospects of sulfide and phosphide catalysts for processing complex mixtures of heteroatomic compounds, including products of pyrolysis of waste plastics mixtures, pyrolysis of polymer and biomass mixtures, hydroconversion of polymers in oil fractions are shown. The bibliography includes 294 references.
1. Introduction
The recycling and reuse of polymers, including the chlorinated polymers polyvinyl chloride (PVC) (or polyvinyl dichloride) and neoprene, is a global challenge, exacerbated by the accumulation of (micro)plastics in the environment and the toxicity of their degradation products.[1] [2] In 2019, 51.4 million tonnes of PVC were produced globally and 21.2 million tonnes of its waste were generated.[1] At present, there is no doubt about the importance of proper treatment of chlorine-containing organic wastes, which are characterised by toxicity and poor biodegradability, which implies their accumulation in the atmosphere, water, soil and living systems.
The developed technologies for processing chlorine-containing wastes should meet a number of requirements, including the absence of toxicity and commercial attractiveness of the resulting products, economic efficiency, versatility, flexibility of the technology to changes in the composition of the processed wastes, high degree of their transformation. Other requirements include the ability to use standard equipment that does not require high capital costs and special anti-corrosion coatings, as well as the use of readily available catalysts that can be easily regenerated or the ability to run the process in the absence of a catalyst.
It is not possible to identify a universal technology that is optimal for the processing of all types of chlorinated organic wastes due to significant differences in chlorine content, molecular weight, form of presence in the processed feedstock (chlorinated polymers, chlorinated solvents, chlorinated compounds in wastewater, etc.). The development of methods for the treatment of halogenated wastes is highlighted in a review,[3] technologies for the treatment of polychlorinated biphenyls are covered in reviews.[4-6] Reviews [7][8] consider trends in the chemical processing of PVC. Studies [3-8] have highlighted the prospects of hydrogen-based recycling processes for various halogenated wastes, including HDC and reductive dechlorination. The HDC process can be characterized as versatile, flexible, allowing, by varying the reaction parameters and the catalyst, to be involved in the treatment of different types of waste, including not only chlorine-containing waste, and characterized by the absence of highly toxic products. Hydrodechlorination can be integrated into the technological schemes of petrochemical plants for the production of hydrocarbons used as raw materials for petrochemicals and fuels.
Hydrodechlorination is a catalytic process, as are hydrodesulfurization, hydrodeoxygenation, etc. The use of various metals, both as reagents and as catalysts, in reductive dehalogenation processes is the subject of a review.[9] This review summarizes data on catalysts and technologies for the hydrodechlorination of various chlorinated wastes. During the last 10 – 15 years, the prospects for the use of some industrial catalysts in the treatment of such wastes have been demonstrated, new catalytic systems have been obtained and the characteristics of their deactivation have been described. Much knowledge has been accumulated on the use of palladium and platinum catalysts in HDC, including those unsupported. A new trend in HDC catalysis is associated with the use of transition metal phosphides. In the field of sulfide catalysts for HCD, there has been virtually no new work in recent years, but this direction seems promising from the point of view of its readiness for industrial implementation. We have tried to pay special attention to the catalytic HDC of complex recyclable wastes with high content of chlorine and other catalytic poisons, such as liquid chlorine-containing products of PVC processing, liquid chlorine-containing products of polymer blends, blends of PVC and oil fractions, etc. on sulfide catalysts. The treatment of these wastes is extremely challenging due to the simultaneous presence of various chlorine-containing compounds, polycyclic aromatic hydrocarbons, sulfur-, oxygen- and nitrogen-containing compounds, soot and mechanical impurities. In the context of the treatment of such wastes, a considerable amount of work has been devoted to sulfides as promising HDC catalysts. Due to the limited amount of new work in this field, we have tried to focus on the combined reactions of HDC and hydrogenolysis of other classes of compounds, as well as the deactivation of sulfides, which has not been analyzed in other review publications.
2. Brief characteristics of chlorine-containing organic wastes
In terms of reactivity and conditions for HDC, four conditional groups of chlorine-containing organic wastes can be identified (Fig. 1):
1. Wastes from production and use of chlorinated pharmaceuticals, chlorophenols, chlorinated pesticides;
2. Chlorinated organic solvents, chlorofluorocarbons;
3. Polychlorobiphenyls, polychlorobenzenes;
4. Chlorinated products from polymer treatment, including PVC and neoprene.
![[{"id":"GU48GK75cH","type":"paragraph","data":{"text":"Groups of chlorinated organic wastes in terms of reactivity and conditions for HDC"}}]](/storage/images/resized/wuh9omzDLw9rj3fYmzzRqbIYbgogSo1QtTzWVj6u_xl.webp)
The wastes in the first group have in common in that these compounds are usually found in wastewater and that the HDC process is carried out in the aqueous phase. For example, the following organochlorine pharmaceuticals have been found in the wastewaters from medical and pharmaceutical companies and in domestic wastewater: diclofenac (1), triclosan (2), chlorhexidine (3).[10] Terbuthylazine (4), clomazone (5) and other chlorinated pesticides have been found in the effluent of agricultural and pesticide companies. In this group of wastes, the most important by volume are chlorophenols (4-chlorophenol (6), 2,4-dichlorophenol (7), 2,4,5-trichlorophenol (8)),[11] which are intermediates in the production of herbicides, plant growth regulators, wood preservatives and dyes. The solubility of 4-chlorophenol in water at 25°C is 27 g L–1, of 2,4-dichlorophenol 4.5 g L–1 and of 2,4,5-trichlorophenol 0.28 g L–1.[11] Hydrodechlorination of chlorophenols, chlorine-containing pesticides and pharmaceuticals are usually carried out in aqueous phase under mild conditions (25 – 60°C, atmospheric pressure), which is favoured by thw low concentration of organochlorine compounds in water and rather high reactivity of chlorophenols compared to chloroalkanes, chlorobenzenes and polychlorobiphenyls.[10-14]
The next group of wastes are aliphatic organochlorine solvents (trichloromethane, dichloromethane, dichloroethane, tetrachloroethane, perchloroethylene) and chlorofluorocarbons. These compounds may either be present in wastewater or constitute a separate waste stream (chlorofluorocarbon oils and greases). The hydrodechlorination of aliphatic chlorinated compounds is carried out under more drastic conditions than the HDC of chlorophenols, due to the lower reactivity of the C – Cl bond in the molecules of aliphatic compounds.[15] The rate of hydrogenolysis of chloroaliphatic compounds is higher than that of chlorobenzenes. The reactivity of chloralkanes can be classified according to the position of the chlorine atom as follows: primary < secondary < tertiary; reactivity decreases with decreasing number of carbon atoms in the molecule.[1][9] The C – F bond has a higher dissociation energy (456 kJ mol–1) than the C – Cl bond (339 kJ mol–1),[9] allowing selective dechlorination of chlorofluorocarbons to produce fluorocarbons.[16][17] A special place in this group of compounds is occupied by the higher chlorofluorocarbons, i.e. oils, greases and hydraulic fluids, the treatment of which, due to the presence of additives and impurities from other oils, requires preliminary separation of mechanical impurities and water, careful selection of the catalyst and more drastic conditions due to the presence of a wide range of heteroatomic compounds.
Chlorinated wastes represented by aromatic compounds include chlorobenzenes (9) and polychlorobiphenyls (10). This group of wastes is characterized by a large number of isomers and the drastic conditions of the HDC process. The reactivity of the compound increases in the presence of electron-donating groups (OH, CH3) in the molecule, which weaken the C – Cl bond, and decreases in the presence of several chlorine atoms.[18] The reactivity of p- and m-dichlorobenzenes is comparable, whereas the reactivity of o-dichlorobenzene is significantly lower due to steric hindrance and deactivation of the o-position by the electron-withdrawing substituent. The same pattern is characteristic of polychlorobiphenyls.[5] Trichlorobenzenes are even less reactive than o-dichlorobenzene.[19]
Polychlorinated biphenyls are one of the most important chlorinated wastes that have been widely used as dielectrics in transformers, heat transfer fluids and hydraulic fluids. The composition, applications and toxicity of polychlorinated biphenyls are discussed in detail in reviews.[4][5] These wastes are always a mixture of compounds that differ in the number and position of the chlorine atoms and, consequently, in their reactivity, which makes their treatment quite challenging and increases the demands on the versatility of the dechlorination process. If the recycled waste consists of a mixture of lubricants with and without chlorine atoms, the requirements for the stability of the hydrogenolysis catalyst against catalytic poisons are even higher.
The processing of PVC (11), polyvinyl dichloride (12), neoprene (13) and chloroparaffins (14) by pyrolysis and hydroconversion methods produces liquid products together with gaseous and solid products.[20-22] The separation of these chlorinated wastes into a separate group is justified by the complexity and variability of their composition due to their origin from different types of plastics, the presence of plasticizers, dyes in polymer waste and the use of petroleum fractions as solvents for polymer wastes.[7][8][23][24] The composition of this group of wastes is discussed in more detail below, as it is these wastes that present the greatest challenge in selecting a catalyst and technology for the HDC.
The liquid product of PVC pyrolysis has a chlorine content of 100 to 3000 parts per million (ppm),[25][26] when PVC blends with polypropylene and polyethylene are processed using catalysts or HCl absorbers (e.g., MgO). The chlorine content can be as high as 13000 ppm in the case of thermal pyrolysis of waste plastic blends and a few per cent in the case of pyrolysis of pure PVC.[27] The concentration of organochlorine compounds can be up to 20 wt.%.[28]
The hydrocarbon composition of the liquid product of PVC pyrolysis depends on the pyrolysis temperature, the catalyst in choice and the composition of the polymer blend being treated. The product contains 10 – 25 wt.% of alkanes, 5 – 20 wt.% of olefins, and up to 90 wt.% of aromatic hydrocarbons, the content of which increases with increasing pyrolysis temperature.[29] Dehydrohalogenation leads to the formation of double bonds in the molecules, so that thermal polymer destruction yields oligomers containing polyunsaturated moieties, more often conjugated dienes, resulting in the Diels – Alder reaction to give polyaromatic compounds.[30][31] Monocyclic aromatic hydrocarbons are represented by benzene, toluene, ethylbenzene, styrene, xylenes, methylindene, methylphenylacetylene and others. Bicyclic aromatic hydrocarbons include naphthalene and its alkyl derivatives, polycyclic ones include anthracene, fluorene, acenaphthene, phenanthrene, pyrene, fluoranthene, chrysene and benzanthracene.[32-35] In addition, the products comprise biphenyl and its alkyl homologues. The content of tri- and tetracyclic aromatic hydrocarbons increases with increasing pyrolysis temperature. The total content of polycyclic aromatic hydrocarbons in the liquid product of PVC pyrolysis can reach 12 wt.%.[33] It should be noted that the content of aromatic hydrocarbons in the PVC pyrolysis product is significantly higher than in the pyrolysis product of polyethylene, polypropylene or polyethylene terephthalate.[35] The presence of polycyclic aromatic hydrocarbons complicates the treatment of the liquid product, not only because of their toxicity and the need to perform hydrogenation reactions simultaneously with HDC, but also because these hydrocarbons often contain chlorine atoms. Free radicals from polycyclic aromatic hydrocarbons and asphaltene-like compounds readily add chlorine atoms present in PVC pyrolysis products.[36]
Chlorinated compounds present in the PVC pyrolysis product are formed by two pathways:
1. Incomplete dechlorination during polymer chain scission followed by condensation of the resulting unsaturated compounds to aromatic compounds (Scheme 1, path I);[37]
2. Incomplete dechlorination during polymer chain scission (see Scheme 1, path II) to give chloroolefins;[37]
3. Addition of HCl or free chlorine radicals to tertiary carbon atoms in olefin molecules (see Scheme 1, path III).[38-41]
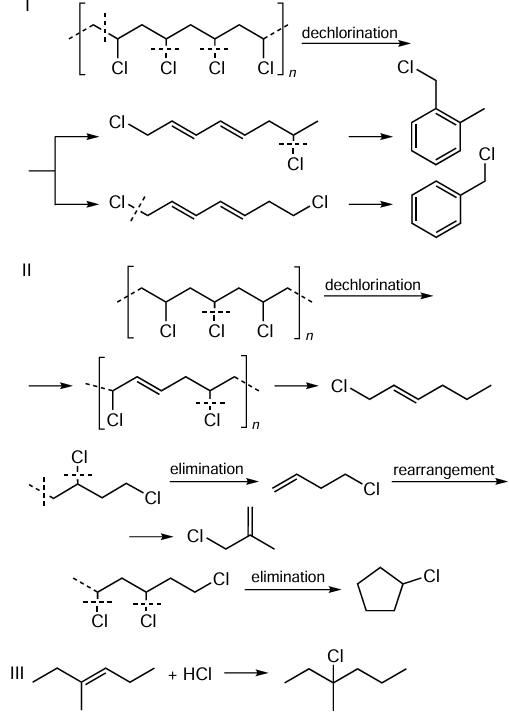
Under high-temperature pyrolysis conditions, paths II and III, characterized by the formation of the main chlorinated compounds of the liquid product, are more likely: a mixture of 2-chloro-2-methylpropane and 2-chloro-2-methylpentane (15) in the pyrolysis of pure PVP and its blends with polyethylene and polypropylene,[38] [41] a mixture of α-chloroethylbenzene and 2-chloro-2-phenylpropane (16) in the pyrolysis of a mixture of PVP and polystyrene (see Fig. 1).[39][40] In the pyrolysis of PVP/polyethylene blends, the maximum distribution of chlorinated compounds by number of carbon atoms falls on C8 – C11;[38] [40] in the pyrolysis of PVP/polystyrene blends, it falls on C6, C8 and C11.[39][40] α-Chloroethylbenzene derivatives with C8 and C9 alkyl chains, as well as 2-chloro-2,4-diphenylbutane and 2-chloro-2,4-diphenylpentane, are present in smaller amounts.[42]
It should be noted that PVC products often contain plasticizers such as dibutyl phthalate, diisobutyl phthalate, bis(2-ethylhexyl)phthalate and benzyl butyl phthalate.[43] The presence of phthalates in chlorinated polymer waste poses a risk of dioxin formation during pyrolysis, which complicates the hydrotreatment of the resulting pyrolysis liquid product.
Since polyvinyl chloride is often found in plastic household waste, a mixture of polymer waste containing polyethylene, polypropylene, polystyrene, polyethylene terephthalate, etc. is subjected to pyrolysis. When PVC and polyethylene terephthalate are pyrolyzed together, the composition of the liquid product changes: chlorinated esters of benzoic acid, chloroalkyl esters of terephthalic acid appear, and the total content of chlorine atoms in the product increases compared to the pyrolysis of other polymer blends.[44][45] The molecular mass distribution of chlorinated compounds also changes: compounds С15 – С16 and С18 – С21 appear. The maximum distribution of oxygenated compounds falls on С8, С11, С12.
Halogen atoms in polymer waste may also be present in halogenated flame retardants such as chlorinated paraffins, pentachlorocyclohexane, pentabromocyclohexane, hexabromocyclododecane, decabromodiphenyl ether, tetrabromobisphenol A, tris(1,3-dichloropropyl)phosphate, etc.[46-48] Halogenated flame retardants and their conversion products are highly toxic carcinogenic compounds. Although halogenated flame retardants are now used much less and are being replaced by other classes of flame retardants, polymers produced in the past and currently landfilled or recycled contain these compounds. Therefore, the liquid pyrolysis product of these wastes will contain chlorine and/or bromine compounds requiring hydrodehalogenation.
The liquid products of PVC pyrolysis differ significantly from hydroconversion products produced in solvent media (decalin, tetralin, vacuum gas oil, oil and oil residues [49-52]) or without solvent [53] that are characterized by a significantly lower chlorine content (up to 120 ppm)[50] compared to the pyrolysis product. The main chlorinated products include 1-chlorobutane, 2-chlorobutane, 1,3-dichlorobutane, 1,4-dichlorobutane and products of the interaction of chlorine free radicals with solvents, such as chlorotetralins and chlorodecalins.[54] Chlorobutanes are not primary products of polymer chain scission, but are formed by the reaction of HCl with olefins and dienes. When PVC undergoes hydrocracking in the absence of a solvent, the product contains more than 30 wt.% of bi- and polycyclic aromatic hydrocarbons,[55] whereas hydrocracking in tetralin produces mainly monocyclic aromatic hydrocarbons, viz., benzene, toluene, xylenes and butylbenzene.[56]
In general, the PVC hydroconversion/hydrocracking product differs from the pyrolysis product by a lower content of chlorine atoms and the presence of sulfur- and nitrogen-containing compounds, which are part of the oil fractions used as solvents. The pyrolysis product of polymer waste mixtures containing polyethylene terephthalate is characterized by a high content of esters, including chlorinated esters, which, together with the presence of polycyclic aromatic hydrocarbons, represents a limitation in the selection of a catalyst for the hydrocracking/hydrodehydrogenation of such products.[8]
3. Brief characteristics of hydrodechlorination and classification of catalysts
The hydrodechlorination process, as well as the hydrodesulfurization and hydrodeoxygenation processes widely used in industry, consists of the abstraction of a heteroatom from the substrate molecule to form a hydrocarbon. The main difference between hydrodechlorination and hydrodesulfurization is the rapid catalyst deactivation due to the formation of active metal chlorides, their leaching from the catalyst surface, changes in the dispersity of the active phase particles [57] and the deposition of carbon on the catalyst surface, which occurs more intensively than in hydrodesulfurization/hydrodeazotisation/hydrodeoxygenation.[58-63] The adsorption of HCl onto the surface of the catalyst leads to the formation of acidic sites on its surface, which promotes polymerization and the formation of active sites for the growth of coke deposits.[62] The resulting coke is fundamentally different from the carbon deposits formed on the surface of hydrodesulfurization catalysts because it contains chlorine atoms, which further increases the acidity of the catalyst surface and the rate of deposit formation reactions. At the same time, the acidic nature of the surface increases the adsorption of some basic compounds such as pyridine and quinoline.[64] Therefore, deactivation may be more pronounced when processing feedstock containing both chlorinated compounds and nitrogenous bases. Fig. 2 summarizes the main reasons for deactivation of HDC catalysts.
![[{"id":"ablRvllW2-","type":"paragraph","data":{"text":"Main reasons for HDC catalyst deactivation (Py is pyridine, М is the catalyst metal)"}}]](/storage/images/resized/aIHuIjrFYK3FZjzJLdYhKxTREukpsOGUMf8hfbUC_xl.webp)
It has been found [65] that microporous supports with a developed surface, such as activated carbon, deactivate faster than mesoporous and macroporous supports, such as Al2O3 or SiO2, precisely because of micropore clogging. However, Al2O3, MgO, TiO2 and ZrO2 react with HCl, resulting in support degradation, changes in pore volume and surface area, as shown by comparing the deactivation rate of palladium catalyst on different supports.[66] Therefore, it is recommended to use supports that do not react with HCl, such as activated carbon or AlF3, in the HDC process.[67]
Another feature of the HDC is that the HDC of chlorophenols and, in some cases, chlorobenzenes [68] is carried out in the aqueous phase when treating wastewater containing these compounds, also with the addition of inorganic and organic (e.g. amines) bases that bind the formed HCl with the removal of salts into the aqueous phase. In this case, the deactivation of the catalyst may be due to the formation of salts on its surface.[69-71]
In the HDC of complex mixtures, including PVC pyrolysis and hydroconversion products, chlorinated waste dielectrics, lubricants and technical fluids, there is competition between heteroatomic compounds and aromatic hydrocarbons for the active sites of the catalyst and deactivation of the catalyst by chlorine-, nitrogen- and, in some cases, sulfur- and oxygen-containing compounds. Therefore, when selecting a catalyst for HDC of such mixtures, it is necessary to be guided by the data on the activity of the catalyst in HDC, hydrodesulfurization, hydrodenitrogenation, hydrodeoxygenation and hydrogenation, as well as its stability over a long period of operation and the possibility of regeneration.
These characteristics of the HDC process largely determine the directions of current catalyst research. These include the selection of supports that are resistant to destruction in HCl-containing media and minimally adsorb HCl, or the non-use of supports in favour of unsupported catalysts, the optimization of pore size, the use of promoters that reduce the base metal deactivation, and the development of methods to restore catalyst activity. All HDC catalysts can be broadly classified into several groups according to the nature of the metal in choice (Fig. 3). It should be noted that this classification is very conditional and is proposed mainly to facilitate further consideration of HDC catalysts. For example, Fe-containing catalysts are not considered together with other elements of the iron triad, but are distinguished in a separate group due to their potential use as accessible and inexpensive protective layer catalysts. Catalysts containing Cu and Ag as the main components are also identified into a separate group as they are rarely used in HDC but can be considered promising if provided with promoters. Unsupported noble metal particles are considered separately as a very promising group of HDC catalysts for low-chlorine feedstock.
![[{"id":"kWOzyRR22N","type":"paragraph","data":{"text":" Classification of HDC catalysts"}}]](/storage/images/resized/3eIEqvbBMqyoiNPHV9fkPOpRnHgo8zsuZfkCd6EL_xl.webp)
4. Supported Fe-, Co- and Ni-based catalysts
Catalysts containing only Fe as the active component are rarely used in HDC due to their rapid deactivation and low activity compared to Ni and Co catalysts.[72-74] Ordóñez et al.[73][74] attempted HDC of tetrachloroethylele in the presence of red mud containing Fe, Ti, Al, Ca, Na, under rather drastic conditions (350°С, 10 MPa H2). A rapid deactivation of the catalyst was observed, slowing down during its sulfidation. The conversion of tetrachloroethylene on the sulfidized catalyst did not exceed 25%, whereas it was 18% on the non-sulfidized catalyst. Under the above conditions, such a low conversion of the aliphatic chlorinated compound indicates that Fe-containing catalysts can only be considered as protective layer catalysts prior to the main layer of an active catalyst, e.g. noble metal-based one. Nevertheless, bimetallic catalysts containing Fe as one of the components are being actively investigated, as will be discussed in Section 6.2.
Ni-containing catalysts are much more popular in the HDC reactions than Fe-containing catalysts, and Ni can be used either as the single active component or in combination with promoters. Suitable supports include Al2O3 (Ref. [75]), aluminosilicates,[76][77] activated carbon [78][79] and Nb2O5 (Ref. [80]). The activity and stability of the Ni catalyst is determined, on the one hand, by the dispersity and size of the metal particles, with smaller particles providing higher activity in HDC.[79] On the other hand, the reduction of the electron density on the Ni atom due to the interaction of Ni with the support or the introduction of promoters provides high activity in the HDC process.
Due to the above challenges, unsupported Ni catalysts have not found wide application in HDC. In the presence of Raney Ni in the aqueous phase in an alkaline medium at room temperature, an exhaustive HDC of chlorophenol has been observed, yielding phenol as the main product, but the high conversion is due to the fact that the organic and inorganic bases introduced into the reaction medium neutralize the released HCl, inhibit corrosion and leaching of Raney Ni.[81] At the same time, Raney Ni is less active in the HDC of chlorobenzene at H2 pressure of 1 MPa and a temperature of 70°C.[82] Similarly, low conversion of chlorobenzene was also observed using Al2O3 and SiO2 as supports, while Ni deposited on activated carbon showed very high activity. Since in this case the size of the Ni crystallites on all the supports tested was the same, the authors explained the differences in catalyst activity by the different ability of the supports to adsorb chlorobenzene.
The activity of the catalyst in the HDС reaction is affected not only by the particle size of the active phase, but also by the phase composition of the catalyst. For Al2O3-supported Ni catalysts, the degree of metal – carrier interaction is important.[83] Catalyst calcination at temperatures above 400°C results in chemical binding of Ni to Al2O3 in the form of spinel forms which are inactive in the HDС reaction. The Ni2+ species weakly bound to the support are more easily reduced to Ni0 and are active and stable in the HDC reaction.
During the HDC process, an increase in the size of Ni crystallites supported on aluminosilicate is observed due to their agglomeration upon reaction with HCl.[77] However, this does not significantly reduce the activity of the catalyst in the HDC of polychlorobiphenyls. The activity can be restored almost to the initial values by treating the catalyst with n-hexane to remove polymerization products from the surface, and then with alkali to remove chlorine-containing compounds, followed by reduction with Н2.
Doping the catalyst with boron and phosphorus helps to reduce the Ni crystallite size and increase H2 adsorption, as exemplified by NiP/SiO2 and NiB/SiO2 catalysts.[84] Boron and phosphorus dopants change the electron density of the Ni crystallites, and the electron density deficit favours the HDC reaction. In particular, the Ni/Nb2O5 catalyst (6 wt.% Ni) showed high activity in the HDC of 1,2,4-trichlorobenzene.[80][85] At a temperature of 250°C and atmospheric pressure of H2, the conversion of 1,2,4-trichlorobenzene was 63% with 18% selectivity to benzene. The optimum Ni content is important, where the adsorption of H2 and the surface area of the active phase species reach their maximum values; increasing the Ni content above 6% results in a decrease in the conversion of 1,2,4-trichlorobenzene.
Hydrodechlorination is much more efficient on Ni catalysts supported on activated carbon than on other commonly used supports (SiO2, Al2O3) due to the high particle dispersion and small crystallite size, the absence of interaction between the support and HCl and the destruction of the support.[65][67] [79][82] For example, the comparison of Ni/SiO2, Ni/Al2O3 and Ni/C catalysts in the HDC of chlorobenzene at a temperature of 70°C and H2 pressure of 1 MPa showed that the latter catalyst provides 100% conversion of chlorobenzene, whereas the first two catalysts do not allow more than 20% conversion, which is explained by the high dispersibility of the Ni species.[82] In addition, the surface of the activated carbon can be modified by various methods, such as treatment with HNO3 or oxidation, which increases the proportion of oxygen-containing functional groups on the surface and favours better binding of Ni or other metal to the support. This reduces the leaching of the active component from the catalyst surface under the action of HCl.[78]
Monometallic catalysts are characterized by rapid deactivation,[82] so the possibility of modifying catalysts by introducing a second component is being considered. The positive effect of doping Ni-based HDC catalysts with Mg has been described, resulting in an increase in catalyst activity and a slowing of deactivation.[86][87] The addition of MgO changes the electronic properties of Ni particles, promoting the desorption of H2 at lower temperatures and the adsorption of HCl, preventing the interaction of HCl with Ni species. In addition, the dispersity of Ni0 particles increases due to the formation of Ni/Mg solid solutions. Similarly, Ni/Pd bimetallic particles in carbon composites are resistant to agglomeration and oxidation to Ni2+.[88][89] The carbon coating of the nanoparticles (NPs) is also involved in the reaction. As suggested by Lokteva et al.,[88] it is involved in substrate activation and possibly in the H2 dissociation. Electron modification of Ni atoms at the expense of palladium and carbon atoms provides the reduction of Ni2+ to metallic Ni under reaction conditions and, consequently, more stable catalyst performance. The Sibunite-supported Pd/Ni catalyst shows more stable activity in the HDC of 1,2-dichloroethane than the monometallic Pd catalyst on the same support.[89] With increasing Pd content in the catalyst, the selectivity to ethylene decreases and the selectivity to ethane increases.
Monometallic Co catalysts are rarely used in the HDC because of their low specific catalytic activity and rapid deactivation compared to Ni and bimetallic catalysts.[90-92] Doping of Co catalysts with B and Pd improves the activity and stability of the resulting catalysts in the HDC.[92] When studying Co/carbon composites in the HDC of chlorobenzene, it was found that the presence of CoO provides higher catalyst activity than the presence of Co0 alone.[90] The activity of Co phases in the HDC reaction increases in the series CoO > Co3O4 > Co/C. In the Co-doped Pd catalysts, the formation of the PdCoO2 phase is observed.[91] In the course of HDC, Co chlorides and, to a lesser extent, Pd chlorides are formed, indicating that the deactivation of Pd-containing sites of the catalyst is prevented in the presence of Co atoms and the efficiency of Pd is increased.
To conclude the consideration of the iron triad elements in the HDC, we can note the feature characteristic of rapid deactivation for all three metals, which limits the use of monometallic catalysts in the above process. However, the introduction of the second component contributes to the modification of the electronic structure of the base metal, increases the stability and opens the possibility to regulate the selectivity of the hydrogenation reactions, which is important in the case of hydrotreatment of chloroalkanes. The possibility of using Fe-containing catalysts as protective layers in the processing of chlorine-rich raw materials can be noted.
5. Catalysts containing Cu and Ag as the main components
Monometallic catalysts containing Cu or Ag are very rarely used in the HDC due to their rapid and irreversible deactivation associated with the formation of Cu or Ag chlorides, which are not reduced by H2. Regeneration of the Cu surface is only possible at temperatures above 480°C, at which CuCl evaporates.[93] Nevertheless, it is technologically and economically impractical to carry out HDC at such high temperatures, so Cu and Ag are used in the composition of the HDC catalysts only in combination with other metals. However, these catalysts are discussed in a separate Section as they are promising for the selective dehydrochlorination of chloroalkanes to produce olefins.
The application of zeolite-supported catalysts Ag/Cu (Ref. 94), Cu/Ni (Ref. 95) in the HDC processes has been investigated. The use of β-zeolite-supported Ag/Cu catalyst (2 wt.% Ag, 2 wt.% Cu) in the HDC of 1,2-dichloroethane at 250°C gives a conversion of only 12%, which decreases about twofold with increasing catalyst time-on-stream (1000 h), with high selectivity to vinyl chloride (up to 100%), which is an undesirable product.[94] The Cu – Ni catalyst on the same support (2 wt.% Ni, 2 wt.% Cu) provides a conversion of 1,2-dichloroethane of ~7% with almost 100% selectivity to ethylene under the same conditions.[95] It is suggested that Cu atoms modify the β-zeolite acid sites, thereby increasing the selectivity to ethylene and decreasing the ethane yield. Similarly, Cu added to the Ni/SiO2 catalyst increases its selectivity to ethylene in the dehydrochlorination of 1,2-dichloroethane, with the conversion decreasing from 55% to 26 – 37% at 350°C, depending on the Cu content, compared to the nickel catalyst.[96] This effect was not explained by the dilution of Ni crystallites active in hydrogenation and HDC by inactive Cu crystallites rather than a decrease in the support acidity. The presence of Ni prevents surface deactivation by Cu and reduces the rate of catalyst deactivation by providing hydrogen dissociation and hydrogen atoms to the chlorinated Cu surface.[96][97]
Similarly, the addition of Ni to the Ag/SiBEA (dealuminated form of BEA zeolite, which is a high-silicon zeolite with large pores and a three-dimensional system of interconnected 12-membered ring channels) catalyst also increases its activity and reduces the deactivation rate.[98] Silver, in turn, increases the selectivity of the nickel catalyst to olefins in the HDC of chloroalkanes. However, in the presence of the Ag–Ni/SiBEA catalyst, the conversion of 1,2-dichloroethane does not exceed 10% even at 250°C.
In summary, Cu/Ag catalysts are characterized by low activity in the HDC reactions and cannot be regenerated. Bimetallic catalysts containing Cu or Ag can be promising in the HDC of dichloroalkanes to selectively produce olefins or monochloroalkanes.
6. Noble metal-based catalysts
6.1. Monometallic catalysts
6.1.1. Pd catalysts supported on metal oxides and zeolites
The use of noble metal-based catalysts in the HDC is due to their high activity. However, such catalysts have a disadvantage in that the chlorinated products formed during hydrogenolysis rapidly poison the metal sites. It is shown that the simultaneous presence of S-, N- and Cl-containing compounds in the raw materials reduces the activity of the Pd/Al2O3 catalyst due to palladium sulfidation, the formation of chloride salts of ammonium and nitrogenous bases, the blocking of active sites by nitrogenous bases and the destruction of the support.[99] The main work in the field of noble metal-based dechlorination catalysts is focused on optimising the composition of the support in order to avoid its destruction in contact with HCl, and on optimising the composition of the metal phase, in particular by introducing promoters.
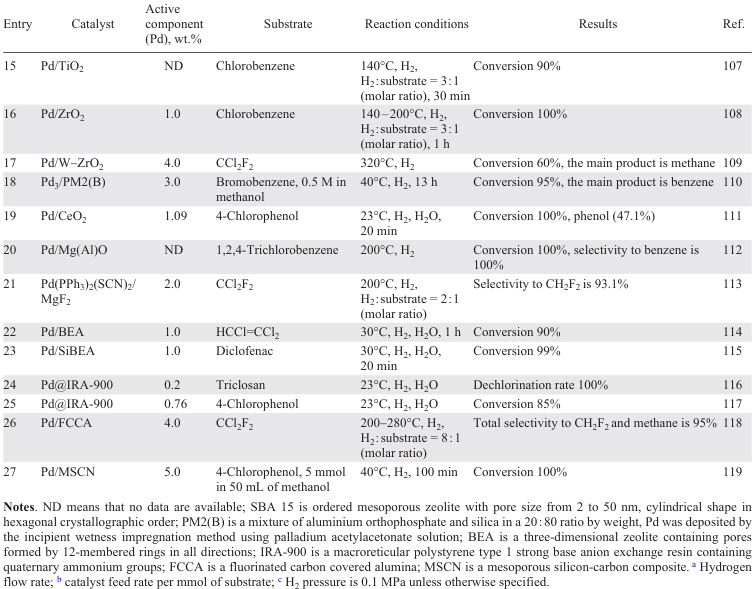
The diversity of supports [65] [99-119] applied is due to the search for the material that is most resistant to HCl, while at the same time providing maximum dispersion of the nanoparticles. Typically, supports for noble metal-based catalysts include various oxides such as Al2O3 (Refs [65], [99-103]), SiO2 (Refs [100], [104-110]), TiO2 (Ref. [107]), ZrO2 (Refs [100], [106-108]), CeO2 (Refs [100], [101]), Fe3O4 (Ref. [120]), MgO (Ref. [110]), Mg – AlO (Ref. [112]), salts (MgF2 (Ref. [113])), zeolites (SBA-15 (Ref. [100]), BEA (Refs[114][115])), ion-exchanging[116] and inert resins,[117] and also composites [105] [118][119] (Table 1).
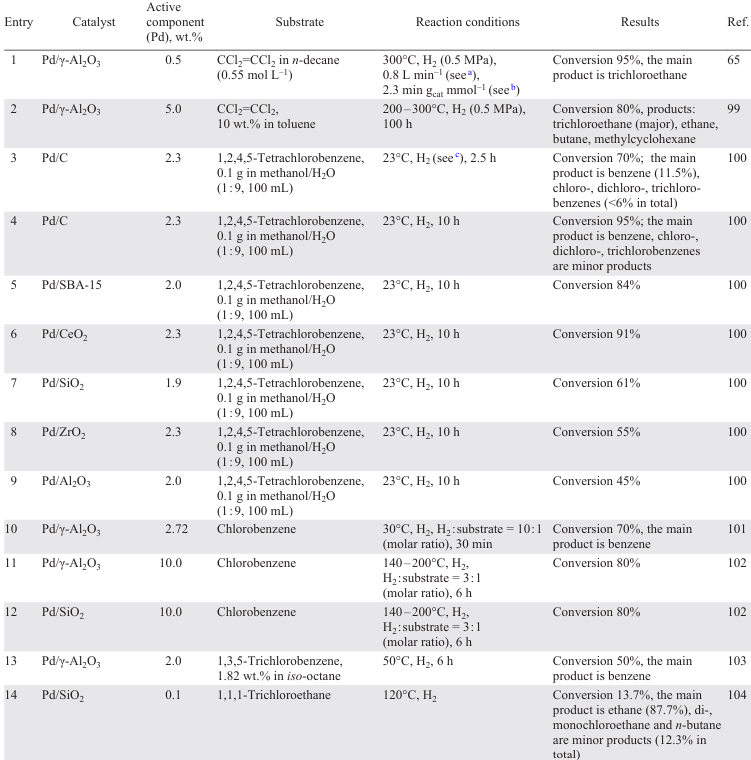
The study of the influence of the parameters of the HDC of tetrachloroethylene to ethane in the presence of 0.5 wt.% Pd/γ-Al2O3 showed that higher temperatures lead to a higher initial activity of the catalyst, determined by the presence of electron-deficient Pdn+ species and the size of Pd NPs on the support surface, and to faster deactivation, while increasing the H2 flow rate leads to higher activity and stability of Pd/γ-Al2O3. At the same time, changing the pressure has no significant effect.[65] Catalyst deactivation was faster when n-decane was used as a solvent (see Table 1, entry 1). In addition, the activity of Pd/Al2O3 is significantly affected by the metal content.[101] Catalysts with up to 2 wt.% Pd feature a higher dispersity, homogeneous distribution of active species interacting with the support without any formation of β-PdH, whereas higher metal loadings promote an agglomeration of PdO to give bulky clusters. The HDC of chlorobenzene affords benzene as the main product with a small admixture of cyclohexane (see Table 1, entry 10).[101] Exposure to microwave radiation [102] during the preparation of the supported catalysts promotes the formation of large PdO nanoparticles, the reduction of which improves the activity of the catalyst compared to those obtained by wet impregnation and calcination.
The modification of the Al2O3 support with Н8[Si(W2O7)6] reduces the size of the nanoparticles and the number of β-PdH active sites more firmly bound to the catalyst surface, and improves the stability of the catalyst by increasing the number of active sites (Pd+ and Pd2+ species) formed by the reaction of Pd with Н8[Si(W2O7)6] and the products of its thermal decomposition.[103] The Lewis acidity characteristics of the catalyst surface determine the possibility of adsorption and activation of 1,3,5-trichlorobenzene on the support, with a conversion reaching 50% with a selectivity to benzene of 30 – 40% in the presence of the modified support (see Table 1, entry 13) and hydrogen spillover from Pd0.
The interaction of the metallic particles of magnetite, used as a support, with the Pd active phase favours the electron transfer, leading to an even greater increase in the activity of the Pd/Fe3O4 catalyst, which reached 6100 L gPd–1 min–1 for the HDC of trichloroethylene and 3700 L gPd–1 min–1 for the HDC of chlorobenzene at minimum Pd loading, compared to the use of Pd NPs.[120] Higher loadings are accompanied by a decrease in the activity of Pd/Fe3O4 due to the formation of large three-dimensional Pd clusters with a smaller fraction of exposed metal. An additional advantage of this catalyst is that the use of magnetite as a support minimizes the loss of catalytic species. In addition, the ferromagnetism of the support makes it easy to separate the nanocatalyst from the spent water by magnetic separation.
The HDC of 1,2,4,5-tetrachlorobenzene in the presence of Pd/C, Pd/SBA-15, Pd/CeO2, Pd/SiO2, Pd/ZrO2 and Pd/Al2O3 (1.9 – 2.3 wt.% Pd; see Table 1, entries 4 – 9) has been investigated.[100] The high catalytic activity of Pd/C, Pd/CeO2 and Pd/SBA-15 compared to that of Pd/SiO2, Pd/Al2O3 and Pd/ZrO2 Pd/SiO2 is due to the high dispersity (44.9 – 59.7%) and high surface area (211.5 – 281.3 m2 g–1) of the catalysts of the first group, resulting in a greater availability of Pd sites and a significant resistance to deactivation than that of other catalysts. In general, the way in which the active component is deposited on the support affects the dispersity and electronic properties of the active phase.[107][108] The presence of electron-deficient palladium Pdn+ species on the surface is considered to be the main reason for the high activity of the catalyst in the HDC and its stability. While the activity depends almost entirely on the properties of the metal phase, the rate of catalyst deactivation is also determined by the nature of the support, mainly by its acid-base properties.[110] For example, the amount of coke on the surface of the Pd/Mg(Al)O catalyst decreases with increasing support basicity, while the conversion of 1,2,4-trichlorobenzene increases,[112] since the presence of basic sites on the surface favours the adsorption of chlorinated substrates (see Table 1, entry 20). Nevertheless, the problem of HCl interaction with the support and changes in the morphology of the latter has stimulated the search for ‘neutral’ supports that do not interact with HCl, which include carbon and its various modifications.
6.1.2. Pd catalysts on carbonaceous supports
Carbon-supported catalysts based on Pd NPs are highly active in HDC reactions (Table 3).[121-142] In dechlorination of various chlorinated substrates (mono-,[126] di-,[125][126] and trichloromethanes,[126] [141] chlorofluoromethanes,[132][133][142] trichloroethylene,[122] tetrachloroethylene,[121] [123][124][138] mono-,[136][137] di-,[136] tri- (Ref. [134]) and hexachlorobenzenes,[137] 4-chlorophenol[127-131] [135][139][140]), activated carbon, graphite, graphite nanofibres, carbon nanotubes, modified carbon, mesoporous carbon doped with boron or nitrogen, diamond particles with sizes smaller than 100 nm (UDD) are used as supports.
The structure of the carbonaceous supports significantly affects the catalytic performance of the Pd catalyst.[122] [136] Larger surface area, optimal size of micro- and mesopores of activated carbon, higher acidity of surface groups compared to limited porosity and low surface area of graphite and graphite nanofibres significantly improve the activity of Pd/C in the liquid-phase HDC of chlorobenzene to benzene (see Table 3, entry 22),[136] while in the liquid-phase HDC of trichloroethylene, 0.6 wt.% Pd/C showed the lowest activity — conversion after 50 min did not exceed 20% (see Table 3, entry 2).[122] However, in the gas phase process, the catalysts with a higher number of surface acid sites show low activity due to the interaction of chlorobenzene with them. In the gas phase HDC of 1,3-dichlorobenzene,[136] an opposite dependence was observed, and the activity of the catalysts decreased in the order Pd/graphite nanofibres > Pd/graphite > Pd/Cact, which may be due to the fact that surface oxygen-containing groups (especially carboxyl groups) inhibit the gas-phase HDC process (see Table 3, entries 23 – 25). The interaction of chlorine atoms in chlorinated substrates with the oxidized surface of the support further inhibits the process, so to improve the catalytic performance it is necessary to remove acidic oxygen-containing groups (carboxyl, anhydride) from the surface of the carbon support, which can be achieved by subjecting it to HNO3,[138] pyrolysis [138] and heat treatment.[142] In addition to removing oxygen-containing groups from the catalyst surface, its resistance to deactivation by chlorinated products can also be increased by doping with non-metals such as N [128][129][135] and B.[129] [138]
The high activity of the catalyst based on Pd NPs supported on high surface area graphite in the gas phase HDC of tetrachloroethylene (see Table 3, entry 3) [123] is due to the absence of sintering of metal particles and coke formation on the surface of 1 wt.% Pd/Cact and 1 wt.% Pd/nanofibre (Pd/CNF), together with the catalyst poisoning by chlorinated compounds.[121][123] Compared to 1 wt.% Pd/Cact, the carbon nanofibre-supported catalysts show poor activity in the gas phase HDC of tetrachloroethylene (60 and 40%, respectively, see Table 3, entries 4, 5), but a higher stability: the conversion of tetrachloroethylene reached 45% after 4 h of operation (see Table 3, entry 6), whereas 1 wt.% Pd/C was completely deactivated due to Pd oxidation and catalyst poisoning.[124]
Pd/C catalysts obtained by impregnation of sawdust with Pd2+ solution and subsequent pyrolysis showed different behaviour.[137] The gas phase dechlorination of chlorobenzene over catalysts with different Pd contents (0.6 and 0.9 wt.%) and nanoparticle sizes (2.6 and 3.7 nm) provided conversions of 85 – 90% and 65 – 90%, respectively. However, due to the significant adsorption of chlorinated benzenes on the surface of the catalyst with lower Pd content and larger particle size, the conversion in the liquid phase hydrogenation of hexachlorobenzene did not exceed 2%. This effect can be explained by the formation of a Pd surface carbide phase inhibiting the formation of PdH2, which is a hydrogen donor in the substrate hydrogenation process, during pyrolysis due to the interaction with the carbon support. The use of UDD [134] as a carbon support, the surface functional groups of which promote the formation of active Pd sites due to the metal–carrier interaction, made it possible to obtain exclusively benzene from 1,3,5-trichlorobenzene already after 30 min of the process (see Table 3, entry 18).
Calcination at higher temperature (1900°C) followed by steam gasification is accompanied by the formation of pure carbon materials characterized by a higher specific surface area and higher pore volume.[142] The use of the oil-soluble precursor (CH3COO)2Pd instead of PdCl2 to impregnate the resulting support affords highly dispersed Pd particles which exhibit high activity in the HDC of dichlorodifluoromethane [132][142] with selectivity to difluoromethane of up to 95% (Table 3, entry 33).
The activity of the catalyst is also influenced by the conditions of its thermal treatment.[140] The catalyst Pd/PBSAC, where PBSAC are polymer-based spherical activated carbon particles reduced at 80°C in H2, was catalytically inactive in the HDC of 4-chlorophenol. At the same time, the use of the catalyst calcined at 200ºC in air allowed an exhaustive dechlorination (1 h) to give only phenol without loss of activity during 100 h on stream (see Table 3, entry 30). The synergistic effect occurring between stabilized Pd NPs and the ordered mesoporous structure of carbon doped with N[128][129][135] and B [129][139] improves catalytic properties of Pd/C and increases stability of the catalyst in the HDC of 4-chlorophenol. The high dispersity of Pd and Pd2+, which contributes to stronger metal – carrier interactions, as well as the activation of the C – Cl bond cleavage by Pd2+ species [139] with the removal of the nucleophilic Cl– allow achieving 100% conversion of 4-chlorophenol at 30°C with a selectivity to cyclohexanone of 12% in 60 min and the use of the catalyst up to 6 cycles without loss of activity (see Table 3, entry 28). At the same time, in the presence of undoped 1 wt.% Pd/C in the second cycle, the conversion of 4-chlorophenol decreases to 40% (see Table 3, entry 29).[139] Exhaustive HDC of 4-chlorophenol over 2.5 wt.% Pd/C was observed for three cycles in a gas phase process at 200ºC (see Table 3, entry 14).[130] The comparison of Pd catalysts on different carbon supports (carbon nanotubes (CNTs), activated carbon, graphene) in the process of liquid phase HDC of 4-chlorophenol (40°С) showed that 5 wt.% Pd/CNTs performed best (see Table 3, entry 15).[131] This is due to the higher dispersity of smaller Pd particles, allowing more efficient diffusion of substrates or reaction products to the active sites on the CNT surface.
The impregnation of the activated carbon with different solutions makes it possible to vary the selectivity of the HDC process and to improve the stability of the catalyst.[141] The higher surface concentrations of electrodeficient Pd2+ species and acidic support groups obtained in catalysts based on Pd NPs deposited on carbons activated with KOH, NaOH and H3PO4 contribute to the enhanced adsorption of chlorinated compounds and, consequently, to the poisoning of the active sites. The catalysts obtained by impregnating carbon with solutions of ZnCl2 (65%) and FeCl3 (62%) showed high selectivity to olefins (see Table 3, entries 31, 32). However, the conversion in the presence of the FeCl3-impregnated catalyst decreased to 19% with increasing space time due to the sintering of the Pd particles resulting in loss of activity. The high stability of the ZnCl2-based catalyst is due to the influence of ZnCl2 on the process of redispersion of the Pd particles, resulting in the formation of uniformly distributed Pd particles of smaller size.
Reducing the size of the active site species is a key challenge in the preparation of supported catalysts. At the same time, it is not always possible to use supports with a highly developed surface, which provide high dispersity, due to intensive coke formation on their surface.[124] It is therefore necessary to find a balance between activity and stability of the catalyst. One solution is to use unsupported metal nanoparticles.
6.1.3. Pd nanoparticles
Catalysts based on nanoscale Pd particles stabilized by stabilizers [143-145] and embedded in a polymer[146-151] or SiO2 (Ref. [152]) are known for their high activity in the HDC of various chlorinated compounds (Table 5) and their convenient use (catalysts are easily separated from the reaction products and can be reused without reduction, purification and loss of activity).
The use of water-soluble polysaccharides,[143][144] e.g., carboxymethylcellulose, which contains carboxylate moieties in addition to hydroxy groups, allows for stronger binding of Pd particles, inhibiting particle growth and consequently their agglomeration. Carboxymethylcellulose-stabilized Pd NPs have smaller size and narrower size distribution than those obtained using D-glucose.[144] The small molecular volume of D-glucose allows the metal nanoparticles to bind more firmly to its surface, thereby limiting the access of H2 and substrate to the active sites in the HDC of trichloroethane, whereas the bulkier molecular structure of carboxymethylcellulose with a high degree of steric hindrance allows greater accessibility of the reactants to the active sites of the catalyst, thereby enhancing the catalytic activity of Pd.[144] The temperature of the catalyst synthesis is directly proportional to its activity. As the temperature decreases, the particles grow in size, become less dispersed and, consequently, the conversion of trichloroethylene degrades (see Table 5, entry 1).[143]
Encapsulation of Pd NPs in polymer matrix provides access to more active catalysts that are stable to deactivation. In particular, Pd NPs have been stabilized using sodium citrate and polydimethylsiloxane for use in the dechlorination of polychlorinated benzenes.[145] The activity of the NPs decreased as the degree of chlorination of the substrate increased. The use of poly(dimethylsiloxane) to stabilize the nanoparticles reduces deactivation and loss of nanoparticles, although the activity of the catalyst decreases. Poly(dimethylsiloxane) membranes are shown to effectively protect Pd catalysts in the HDC of trichloroethylene, preventing leaching of nanoparticles.[147] Highly dispersed Pd NPs have been obtained on the surface of poly(N-isopropylacrylamide), which are effective in the HDC of 4-chlorophenol (95% conversion at a substrate/Pd molar ratio of 100 : 1) and stable over three cycles without loss of activity (see Table 5, entry 6).[148] Such a catalyst is easy to use as it can be precipitated and separated from the reaction system by slightly raising the temperature of the aqueous solution. The use of polymeric hollow microspheres of hydrophilic polyacrylamide and a cross-linked hydrophobic shell of poly[styrene-co-2-(acetoxy)ethyl methacrylate-co-acrylamide] as microcapsules for the encapsulation of reagents and Pd NPs provided an access to a catalyst with high activity in the liquid-phase HDC of 4-chloro- and 2,4-dichlorophenols at 25ºC (see Table 5, entries 7, 8).[149] It has been shown that the HDC in water takes place in the microspheres cavities, which accumulate 2-chlorophenol molecules and release phenol. The catalyst for the 4-chlorophenol HDC retain its activity even after four cycles. Palladium nanoparticles stabilized in high-density polyethylene beads catalyze the sequential conversion of 4-chlorobiphenyl to bicyclohexenyl (see Table 5, entry 9),[150] 1,6-dichlorodibenzo-p-dioxin to dodecahydrodibenzo-p-dioxin (see Table 5, entry 3) [146] and 2,8-dichlorodibenzofuran to dodecahydrodibenzofuran (see Table 5, entry 4) [146] in supercritical CO2 resulting first in a stepwise removal of chlorine atoms, followed by a slower hydrogenation of benzene rings. The presence of supercritical CO2 promotes the swelling of high-density polyethylene, facilitating the access of the chlorinated substrate to the Pd NPs in the polymer structure.[146] [150]
Encapsulation of Pd nanoparticles in an ordered mesoporous SiO2 shell, which prevents nanoparticle aggregation,[152] opened an access to a catalyst for the dechlorination of 4-chlorophenol that is more efficient compared to the supported Pd catalysts, with the stability of the resulting catalyst decreasing only after five times of recycling (see Table 5, entry 11). The unique feature of this catalyst is that the nanoparticles are sandwiched between the SiO2 core and the mesoporous SiO2 shell, the pores of which allow molecules to pass through. Such a structure helps to reduce the loss of nanoparticles due to their leaching from the catalyst surface. It has also been proposed to use a magnetic porous carbon composite, synthesized from metal organic frameworks, to encapsulate Pd and Au nanoparticles.[151]The resulting nanocatalysts have magnetic properties and can be easily recovered from the reaction products. In this case, the catalyst was used for the HDC of 4-chlorophenol for several cycles without loss of activity (see Table 5, entry 10).
6.1.4. Pt nanoparticles
Compared to the numerous studies on catalysts based on Pd NPs, there are only a few known works dealing with the catalytic properties of Pt.[153-156]
In particular, the hydrogenation of chlorobenzene was carried out in the presence of colloidal Pt NPs stabilized with polyvinylpyrrolidone (PVP), the ratio of which had a significant effect on the selectivity and conversion of the substrate.[153] For example, a lower amount of stabilizing polymer led to agglomeration of the Pt NPs and consequently to a decrease in their catalytic activity, whereas a higher amount of PVP prevented the contact of the substrate with the catalyst surface and the desorption of the product. The introduction of additional amounts of polymer into the reaction mixture had an inhibitory effect on the reaction rate decreasing the selectivity to cyclohexane. In addition, the activity of the colloidal nanoparticles is also dependent on the preparation conditions, namely the presence of a break in stirring during the hydrogenation reaction, the increase in the duration of which favours agglomeration and possible precipitation of a small amount of Pt metal,[154] as well as the presence of different metal ions [155] in the reaction mixture: the addition of Mg2+ and K+ has a positive effect on the activity of the catalyst and the selectivity to cyclohexane, whereas Sn2+, Cu2+, Zn2+ and Mn2+ ions act as catalytic poisons, leading to a complete loss of activity of the colloidal Pt NPs.
The introduction of a ligand leads to the ‘ligand-acceleration phenomenon’, which can accelerate the initial reaction by a factor of 1 – 2. Bipyridyl and diethylenetriamine ligands decrease the selectivity of the HDC of chlorobenzene to cyclohexane, indicating that these ligands inhibit the hydrogenation of benzene, but accelerate the hydrogenation of chlorobenzene.[156] Acetylacetonate ligand enhances both the activity of the catalyst and the selectivity of the reaction. The activity of the Pt/PVP catalyst modified with polyamine ligands varies depending on the number of NH2 moieties: the conversion of chlorobenzene reaches 76.1% in the presence of the system containing 2 amino groups, and is 51.4% in the case of 3 amino groups, due to the change in the electron density of the Pt active site caused by the interaction of the metal with the polyamine ligands.
The main research directions in the field of the HDC catalysis by palladium and platinum nanoparticles include the search for ways to encapsulate nanoparticles in order to reduce their deactivation and mechanical losses,[145][149] the study of the possibility of separating and recycling nanoparticles,[148] including by magnetic separation,[151] and the synthesis of nanoparticles with a core–shell structure in order to reduce catalyst losses.[152]
6.1.5. Supported Pt, Ru and Rh catalysts
Catalysts based on platinum group metals (Pt, Ru, Rh) deposited on various supports have been underinvestigated compored to supported Pd catalysts. However, the former catalysts have shown high efficiency in the HDC of mono-, di-, tri- and tetrachloromethanes (Table 6).[157-170] The activity of metals in the HDC of dichloromethane decreased in the series: 0.80 wt.% Rh/C > 0.81 wt.% Ru/C > 0.83 wt.% Pt/C,[157] but the selectivity to non-chlorinated products in their presence reached 95% (see Table 6, entries 1 – 3). The use of 0.83 wt.% Pt/C allows to obtain methane (85 – 90%) and chloromethane (10 – 15%) as main products, while other catalysts afford C1+ hydrocarbons (methane (50 – 75%), ethane (15 – 20%), propane (9 – 11%) and up to 5% butane). The high stability of the Pt/C catalyst, which showed no signs of deactivation after 65 h on stream, is related to the re-dispersion of the particles during the reaction and, consequently, to the formation of much more finely dispersed, homogeneous particles, well distributed on the surface of the metal particles,[160] as well as to the predominance of Pt0 species, which are more resistant to poisoning during the adsorption of chloromethanes,[157][158] than the electrodeficient Ptn+ species. The deactivation of 0.81 wt.% Ru/C (45% decrease in conversion after 20 h on stream) and Rh/C (9% decrease in conversion after 65 h on stream) can be attributed to a decrease in the exposed surface area with active sites due to their poisoning by chlorinated hydrocarbons and metal sintering (in the case of 0.81 wt.% Ru/C).[157] Increasing the metal content of Pt/C from 0.5 to 2 wt.%[158][150] and consequently the number of Pt0 species on the catalyst surface allowed 100% trichloromethane conversion and 97.2% dichloromethane conversion in the HDC of the substrate mixture (see Table 6, entry 4). Raising the temperature to 400°C did not lead to poisoning of the Rh, Ru, Pt catalysts because of the large surface area of the activated carbon, which favours the dispersion of metal particles, and the acid-free nature of its surface, which prevents the formation of carbonaceous deposits (see Table 6, entries 5 – 10).[159] Increasing the temperature positively has a positive effect on the conversion and the yield of C1+ hydrocarbons in the HDC of di- and trichloromethane in the presence of 1 wt.% Rh/C and 1 wt.% Ru/C (see Table 6, cf. entries 2, 3 and 8, 9), whereas the use of 1 wt.% Pt/C has practically no effect on the quantitative composition of the products at different temperatures, indicating a different selectivity of the above catalysts (see Table 6, cf. entries 1 and 5). Contrary to the HDC of dichloroethane in the presence of 1 – 2 wt.% Pt/C,[158-160] the use of 2 wt.% Pt/SiO2 produces mainly ethane, the selectivity to which increases with increasing temperature.[165]
When Pt particles are supported on γ-Al2O3, they interact with Lewis sites on γ-Al2O3 to form electron-deficient Pt particles of smaller size (< 2 nm), which in turn undergo intense chloridation in the course of tetrachloromethane hydrogenation, resulting in catalyst deactivation (see Table 6, entry 13).[161] On SiO2 support, such metal–support interactions do not occur, so the deactivation of the HDC catalyst is less pronounced.
Platinum catalysts supported on Y-FAU and ZSM-5 zeolites obtained by wet impregnation and ion exchange method [163][164] performed well in the hydrogenation of tetrachloromethane and trichloromethane to methane and trichloroethane as well as in the hydrogenation of trichloroethylene to ethane (see Table 6, entry 17). However, the presence of Brønsted acid sites in zeolites also favours the side processes, viz., the formation of oxygenated compounds (COCl2) as a result of the reaction between intermediates and the zeolite framework oxygen. Phosgene formation can only be avoided by using a catalyst obtained by impregnation or reduction of ion-exchanged zeolites with NaBH4.
The study of the effect of the support (C, SBA-15, SiO2, Al2O3, CeO2, ZrO2) on the efficiency of the Ru catalyst in the HDC of 1,2,4-trichlorobenzene showed that higher catalytic activity and stability were observed when using 2 wt.% Ru/C (see Table 6, entry 19). This can be explained by the high dispersity and smaller Ru crystallite size due to the strong chemisorption of the chlorinated substrate leading to its complete hydrodechlorination.[166]
The comparison of the catalytic properties in the gas-phase HDC of chlorobenzenes showed that the catalyst based on 0.5 wt.% Pt/γ-Al2O3 (see Table 6, entry 20) is superior to that based on 0.5 wt.% Pt/AmLSA (AmLSA is alumina solid Lewis superacid). The high acidity of the Lewis centres in AmLSA favours the formation of a stable π-complex as a result of the interaction of adsorbed benzene-like intermediates with active sites, which reduces the catalyst surface area and hence its activity.[167] Conversely, in the gas-phase process, this effect led to an increase in the activity of the catalyst in the HDC of dichlorobenzene, where the resulting benzene was hydrogenated to cyclohexane, which was the only reaction product. In the HDC of 4-chlorophenol in the presence of Pt/NdC/SBA, in which Pt nanoparticles were supported on nitrogen-doped C/SBA (1 wt.%), exhaustive conversion and 100% selectivity to cyclohexanol were achieved (see Table 6, entry 28).[170] This effect can be explained by the influence of the N atoms on the size and electronic state of the Pt NPs on the catalyst surface and certainly by the electron-donating effect of the hydroxy group in the chlorophenol molecules, which favours the elimination of the chlorine atom.[10-14] However, the carbon-supported platinum group metals (Ru, Rh) showed no activity in the treatment of dielectric oil consisting of a mixture of chlorinated benzenes and polychlorinated biphenyls; the conversion after 48 h did not exceed 0.32% (see Table 6, entries 26, 27).[169] It is the insufficient activity of monometallic catalysts in the HDC of polychlorinated organic wastes and mixtures of heteroatomic compounds that determines the search for possibilities of modifying the active phase, including the introduction of a second component, in order to achieve more stable activity of the catalyst.
6.2. Bimetallic catalysts
6.2.1. Supported bimetallic catalysts
Catalytic HDC is considered to be a structure-sensitive process, the efficiency of which can be significantly affected by the size and structure of the supported metal particles.[171] Improving catalyst activity by modifying its geometric and electronic structure can be achieved by simultaneous double impregnation of the support followed by the reduction with H2/NaBH4 or by doping/promoting the active metal (Table 7).[172-207] The synthesis of bimetallic catalysts uses the solutions of Au,[172-174] Fe,[175][179] Ni,[96] [175-178] [180] [182] [197] Mo,[182] Cu,[176][183-188][191] Pt,[184][187][188][198][199] Ag,[98][189-196] Pd (Refs [172-174], [176][177][83] [185][189] [198][199][207]) Ga,[188] Ir,[200] Rh,[201] Co,[202] Yb,[203] Mg(O),[204] [205] alkali / alkaline earth metals,[205] and Bi.[207]
The addition of the second metal to the monometallic catalyst affects the HDC of trichloromethane and tetrachloromethane both in terms of both activity and product selectivity.[169-172] The addition of Au to Pd/Sibunite (2.8 – 7.9 wt.% metal/metal) significantly improves the stability of the catalyst, extending its lifetime in the HDC of CCl4 and increasing its selectivity to C1-2 hydrocarbons (see Table 7, entry 3).[172] In addition, the efficiency of the Au/Pd catalyst depends largely on the doping quality: well-mixed Au/Pd particles provide high catalytic performance, whereas the presence of fine unalloyed Pd particles results in rapid catalyst deactivation. In the case of 2.8 wt.% Pd/Sibunite, catalytic poisons are both carbonaceous (PdCx derived from Pd/Sibunite) and chlorinated deposits. It is shown [172] that the introduction of small amounts of Au (10 – 15 wt.%) into 2 wt.% Pd/C improves the selectivity to non-chlorinated C2+ hydrocarbons (see Table 7, entry 5), contrary to the data of the study.[172] When Pd (0.04 wt.%) is added to 1.2 wt.% Au/Al2O3, the rate of HDC of trichloromethane to methane increases (selectivity > 90%) due to the higher dispersion of Pd atoms on Au domains compared to 0.04 wt.% Pd/Al2O3, whereas 1.2 wt.% Au/Al2O3 shows no activity in this process (see Table 7, entry 4).[173]
The addition of Ni to catalytic systems based on Fe,[175] Ag [98] or Cu [96] has not only significantly increased the stability of the catalysts but also improved the selectivity to ethylene in the HDC of 1,2-dichloroethane. In this case, the monometallic Fe catalyst undergoes irreversible corrosion (see Table 7, entries 6, 7) [175] while the Ag (Ref. [98]) and Cu (Ref. [96]) catalysts irreversibly adsorb chlorine atoms to give metal chlorides, leading to rapid deactivation of the active sites of the catalysts (see Table 7, entries 1, 2). In the Fe – Ni system, where the metal particles are uniformly distributed on the polysulfone matrix, the HDC of a solution of a mixture of di- and trichloroethane takes place at the Fe – Ni interface, where Fe acts as a reducing agent and Ni as a catalyst.[175] The polymer matrix prevents the formation of an oxide layer on the nanoparticles, which affects the reduction rate of chlorinated hydrocarbons. Hydrogen produced from water, which promotes iron corrosion, is adsorbed on the Ni surface to generate a potent reducing agent Ni – H+, which reacts with the chlorine atom of the C – Cl bond to release HCl. Highly active AgNi/BEA catalysts (2 wt.% Ag, 2 wt.% Ni) obtained by calcination of zeolite impregnated with metal solutions, which yielded well-dispersed isolated mononuclear Ag(I) and Agnδ+ clusters and pseudo-tetrahedral Ni(II) incorporated into the BEA framework, provided almost quantitative selectivity to ethylene (see Table 7, entry 2).[98] The size of the metal particles did not exceed 3.1 nm. A monodisperse microporous distribution of particles was observed when Ni – Cu/SiO2 xerogel catalysts (0.83 wt.% Ni, 0.45 wt.% Cu, Ni : Cu molar ratio is 67 : 33) were obtained consisting of available Ni – Cu alloy crystallites of size 1.6 – 3.4 nm located inside silica particles (see Table 7, entry 1).[96] The addition of Cu to the system shifted the equilibrium towards the preferential formation of ethylene, whereas the addition of Ni resulted in ethane as the major product. The bimetallic Ni – Cu/SiO2 synergism is determined by the ability to activate H2 by dissociative chemisorption of hydrogen onto Ni, providing hydrogen atoms to regenerate chlorinated copper surfaces to metallic copper.
In the liquid-phase HDС of hexachlorobenzene, the dechlorination rate is proportional to the surface concentration of Pd in Pd – Ni/C (0.2 – 0.4 wt.% Pd, 0.19 – 2.08 wt.% Ni), and its increase is achieved by segregation of Pd on the catalyst surface (see Table 7, entries 8, 9).[176][177] Isolated Pd atoms on the surface of bimetallic particles show higher catalytic activity compared to atoms arranged in larger ensembles. The stability of the Pd – Ni/C catalyst (with a molar ratio of Pd : Ni = 2 : 98) increased by up to 6 h when tetramethylammonium chloride was added to the reaction mixture. The low stability of Pd–Ni/Al2O3 with a high Ni content (2 wt.%) is due to Ni particle aggregation on the Pd surface (0.5 wt.%).[178] The addition of 0.25 – 2 wt.% Fe to 0.5 wt.% Pd/Al2O3 in the HDC of chlorobenzene to benzene improves the catalyst’s activity and stability due to the formation of active Pdn+ species at the Pd – O – Fe interfaces.[179]
The hydrodechlorination of polychlorinated dibenzo- p-dioxins and polychlorinated dibenzofurans formed in fly ash from a solid waste incineration has been studied in the presence of mono-, bi- and trimetallic γ-Al2O3-supported catalysts containing 0.42 – 1.32 wt.% Mo, 0.31 – 1.07 wt.% Ni and 0.32 – 0.87 wt.% Pd.[182] The synergism between interacting metal particles leading to the formation of finely dispersed particles enables the use of smaller amounts of expensive Pd in bimetallic and trimetallic systems without reducing the catalyst’s activity. Monometallic 0.87 wt.% Pd/γ-Al2O3 shows quite high activity in the substrate dechlorination. The conversion of the substrates and the toxicity of the hydrogenation products decrease in the following order: MoPd (0.64 wt.% Mo, 0.32 wt.% Pd) > NiPd (0.32 wt.% Ni, 0.36 wt.% Pd) > MoNi (0.49 wt.% Mo, 0.31 wt.% Ni), which is consistent with the percentage of acid sites on the catalyst surface. The conversion of chlorinated compounds decreases in the series: NiPd (0.32 wt.% Ni, 0.36 wt.% Pd) > MoPd (0.64 wt.% Mo, 0.32 wt.% Pd) > MoNi (0.49 wt.% Mo, 0.31 wt.% Ni) > Pd (0.87 wt.% Pd) > MoNi. Pd) > MoNiPd (0.42 wt.% Mo, 0.35 wt.% Ni, 0.41 wt.% Pd) > Ni (1.07 wt.% Ni) > Mo (1.32 wt.% Mo) (see Table 7, entries 14 – 19).
An interesting solution to the problem of the HDC of trichloethylene has been the use of Pd-modified hydrotalcite (NiMgAl,[181] MgCuAl (Ref. [186])) as a support. The introduction of 0.1 – 0.5 wt.% Pd into NiMgAl, which has a sufficiently high surface concentration of Ni and high basicity of the support due to the presence of the Mg particles, at high temperatures promotes the interaction between Pd and Ni particles to give up to 80% ethylene (see Table 7, entry 13).[181] With higher Ni content in the catalyst, the ethylene fraction was 47% and the ethane fraction was 32%. The use of Mg- and Pd-enriched catalyst results mainly in ethane formation. Noble metals promoters, particularly Pt and Pd (0.5 wt.%), at times increase the stability of the hydrotalcite-based catalyst. This is because MgCuAl (Ref. [186]) is almost completely deactivated within 10 min due to the formation of CuCl2 on its surface (see Table 7, entry 24). The regeneration of the Cu chloride sites is due to the spillover of hydrogen from the noble metal, the nature of the metal being irrelevant.
The equilibrium in the HDC of 1,2-dichloroethane shifts towards the formation of ethylene when the Pd surface is modified with Ag [189-191][194] [195][198] or Cu [189][191] atoms or when the Ag surface is modified with Pd [193][198] atoms, due to the synergistic effect resulting from the interaction of the metal species. The Pd active sites mainly release ethane, while the Ag active centres mainly release chloroethylene (see Table 7, entries 27 – 29, 33, 35).[195] Active bimetallic species promote hydrogenolysis and the desorption of the evolving ethylene. Doping the catalyst with Cu increases the stability of the system due to the binding of adsorbed Cl to the catalyst surface. This effect is attributed to the weakening of the Cu – Cl bond upon the introduction of Cu or Pd atoms into the Pd–Cu bimetallic catalyst.[189][191]As with the addition of Au,[169-172] doping with Cu and Ag increases the dispersity of the Pd species on the surface. This increases the number and availability of active sites, thereby enhancing substrate adsorption. The study of the influence of the impregnation methods [189] (sequential and co-impregnation) of the Al2O3 support using Ag+ and Pd2+ solutions, followed by calcination and reduction with H2 at 400°С, revealed that the catalyst produced through sequential impregnation exhibits higher selectivity towards ethylene. In contrast, the catalyst produced through co-impregnation (sintering) displayed a disordered mixture of Ag and Pd particles. The Cu/Pd catalyst has lower selectivity than the Ag/Pd catalyst because there are fewer Cu atoms on the Pd surface for a similar amount of additive; in other words, Cu is less readily deposited on the Pd surface than Ag. This is due to the fact that Cu2+ is a divalent cation and its deposition requires two active H atoms generated by adjacent Pd active sites, as opposed to the monovalent Ag+ which requires only one active H atom. Palladium-modified Ag/support catalysts also showed selectivity to ethylene (see Table 7, entries 28, 34).[190][196] A study of the effect of the support material revealed that the use of PdAg/SiO2 and PdAg/γ-Al2O3 catalysts (0.47 – 0.78 wt.% Ag, 0.29 – 1.45 wt.% Pd) results in selectivities to ethylene of 97.3 and 94.6% respectively (see Table 7, entries 28, 34) due to a higher amount of isolated Pd species surrounded by Ag. In the presence of PdAg/SiO2, however, the selectivity to ethane was 91%. Impregnating the Ag/C catalyst with PdCl2 resulted in higher adsorption of trichloromethane onto the Ag centres and enhanced C–Cl bond cleavage to produce unsaturated hydrocarbons. The stability of AgPdCl/C was found to be 35 h (see Table 7, entry 32).[193] On the contrary, the use of Pd(NO3)2 impregnation solution provided a higher surface concentration of Pd with a larger particle size and the hydrogenation delivered ethane and propane.
The high acidity of the sulfated zirconium support favors the formation of smaller metal particles (< 5 nm) in the synthesis of the bimetallic Pd – Pt catalyst (total metal content is 0.5 wt.%, Pt : Pd = 1 : 3, 1 : 1, 3 : 1).[198] These species are well dispersed on the surface because the presence of acid groups enhances the hydrophilic character of the support and promotes the diffusion of the metal precursor. The higher activity and stability of the Pd–Pt catalyst (up to 80 h on stream) in the hydrogenation of dichloromethane (see Table 7, entry 35) compared to the monometallic Pd catalyst is due to the synergistic effect between Pd and Pt species (1 : 3). At the same time, the Pt catalyst is rapidly deactivated under these conditions due to HCl poisoning, deposition of coke and/or organochlorine compounds on the catalyst surface, metal sintering and changes in the degree of metal oxidation. The authors attribute the enhanced Pd – Pt/C [199] activity (see Table 7, entry 36) to the increased dispersity of smaller metal particles (0.7 nm at a Pd – Pt ratio of 1 : 1) compared to the study [198] and the optimal ratio of electrodeficient to zero-valent species.
Similar to the studies,[198][199] it was the addition of Pt to 10 wt.% Ir/SiO2 in a molar ratio of 1 : 4 (Ref. [200]) that provided practically 100% conversion in the HDC of CCl4 with the selectivity shifted towards methane (see Table 7, entry 37), whereas in the presence of 10 wt.% Ir/SiO2, only CHCl3 was formed. At the same time, the activities of Au/Ir and Pd/Ir catalysts drop sharply (conversion decreases from 90% (10 wt.% Ir/SiO2) to 4 – 11%, respectively) due to the rapid deactivation by the formation of dimeric products C2HxCly, indicators of coke formation on the catalyst surface, and Pd metal carbides, as reported in a study.[172] The microwave activation of the catalyst significantly reduced the degree of deactivation of Au/Ir (Au : Ir = 1 : 4 (molar ratio)) and Pd/Ir (Au : Ir = 1 : 4 (molar ratio)) catalysts (conversion increased to 38 and 65%, respectively). The increases selectivity to CHCl3, conversion of CCl4 and the stability of Ga – Pt/MgF2 (1 wt.% Pt, Pt : Ga atomic ratio = 1 : 0.28) were explained [188] by the Ga – Pt interaction, which improved the dispersibility of the Pt particles and altered the electronic environment of the Pt active sites. This resulted in faster desorption of Pt-modified adsorbed particles *CCl3, which is a precursor of chloroform. Modifying the surface structure of Cu-modified 0.5 wt.% Pt/SiO2 (Ref. [187]) increases selectivity to ethylene in the HDC of 1,2-dichloroethane due to decreased adsorption of the substrate and H2 and weaking binding of adsorbed ethylene (see Table 7, entry 25). In contrast, using 0.5 wt.% Pt/SiO2 produces ethane via the reaction of ethylene with surface hydrogen atoms. Varying the amounts of Pt (1.8 – 3.6 wt.%) and Cu (0.4 – 2 wt.%) during the catalyst synthesis, as well as the sequence of SiO2 support impregnation produced different outcomes in the HDC of 1,2-dichloroethane.[184] For example, the Pt16Cu84/SiO2 catalyst (Pt : Cu atomic ratio = 16 : 84) with a Cu-modified Pt surface is stable for up to 18 h on stream and has a selectivity to ethylene > 90% (see Table 7, entry 21). In the case of the Cu44Pt56/SiO2 catalyst being impregnated in reverse order (atomic ratio Pt : Cu = 56 : 44), in which the Cu shell was modified with Pt atoms, the selectivity changed towards the predominant formation of ethane (see Table 7, entry 22), in a manner similar to that observed with 3 wt.% Pt/SiO2.[187]
The presence of Rh in Rh – Pd sol – gel particles enabled the HDС of chlorinated derivatives of pyridine, indole, quinoline and isoquinoline to be carried out under mild conditions. The first step was the hydrogenolysis of the C – Cl bond to form heterocycles, which were then hydrogenated (see Table 7, entry 38).[201] Impregnation of the most active Pd/PVP catalyst with Co(OAc)2 solution reduced the time of HDC of chlorobenzene from 37 to 27 minutes with 100% conversion (see Table 7, entry 39).[202] At the same time, the Co/Pd catalyst exhibited high activity in the HDC of mono-, di- and polychlorosubstituted toluene, benzonitrile and biphenyls.
A more exotic choice of the second metal was the use of electron-donating Yb, which was present as a thin surface coating on Pd in the Yb – Pd/SiO2 catalyst (5 wt.% Pd, 5 wt.% Yb) and favoured hydrogen transfer via the YbH2 surface.[203] The 5 wt.% Yb/SiO2 catalyst was inactive in the HDC of 1,2-dichlorobenzene and chlorobenzene, and in the case of 5 wt.% Pd/SiO2, the conversion did not exceed 5%. At the same time, the activity of the bimetallic Pd – Yb increased sharply with doping, providing an eightfold increase in the yield of the hydrogenated dechlorinated product and enabling the selective formation of cyclohexane from 1,2-dichlorobenzene (see Table 7, entry 41). The stability of the catalyst is also noteworthy. After 18 hours on stream, the conversion rate decreased from 72% to 47%.
Activity of Pd- and MgO-based catalytic systems in the HDC of CCl2F2 is due to the synergistic effect of 5 wt.% Pd/C and 5 wt.% Pd/MgO components.[204] The reaction of MgO with the HF released during the hydrodefluorination of CCl2F2 produces MgF2 and also larger Pd particles in the Pd-MgO/C system (5 wt.% Pd, 10 wt.% Mg). This results in the formation of an electron-deficient Pd surface thereby increasing the desorption rate of the CF2* radical, which yields the predominant product CH2F2 (see Table 7, entry 42). Thus, the addition of MgO to the system shifts the equilibrium towards the selective formation of CH2F2, in contrast to the formation of CH4 over Pd/C. The conversion of 1,2,4-trichlorobenzene in the presence of 1 wt.% Pd/MgAlOx impregnated with solutions of alkali and Li, Na salts [205] increased to 77 – 85% in the dechlorination reaction, whereas in the presence of Pd/MgAlOx it was 65% (see Table 7, entry 43). Alkali modification of 1 wt.% Pd/MgAlOx significantly decreases the surface area and pore volume, reduced the crystallinity of the periclase phase of MgAlOx and decreased the dispersity of the Pd particles. This increased the activity of this catalyst. Interestingly, impregnating the catalyst with Cs+ salts led to an overlapping of the active sites and, consequently reduced the Pd metal’s ability to absorb H2, resulting in reduced conversion (down to 20%) and selectivity. It was found that 5 wt.% Pd/SiO2 catalysts modified with Ba2+ and Sr2+ salts provide specific rates of the HDC of mono- and dichlorobenzene that exceed those of unmodified 5 wt.% Pd/SiO2 by an order of magnitude.[206]
The influence of BiPd and Bi2Pd intermetallic species forming on the surface of the catalyst on its performance in the HDC of 2,4-dichlorophenol was studied by adding Bi as a second metal to catalysts containing 5 wt.% Pd/Al2O3 and 5 wt.% Pd/SiO2 (see Table 7, entries 44, 45).[207] Although the catalyst’s thermal stability increased with increasing Bi loading, its catalytic activity decreased sharply due to an increase in Bi2Pd particles forming on the Pd surface. However, substrate dechlorination selectivity reached 95%.
In summary, the analysis of the bimetallic supported HDC catalysts indicates that this research area is promising, as the introduction of a second (and occasionally a third) metal reduces the cost of catalysts compared to monometallic noble metal-based catalysts.
These catalytic systems have the main advantages of sufficiently high stability and the possibility of increasing selectivity to a more commercially valuable product by adding a second metal promoter. It would be interesting to study the possibility of using these catalysts in systems where several hydrogenolysis reactions (e.g. hydrogenolysis, hydrodesulfurisation, hydrodenitrogenation and hydrodeoxygenation) occur simultaneously. Such systems model the treatment of polymer waste mixtures. Supported catalysts are susceptible to deactivation due to three possible side processes: the chlorination of the metal surface due to the release of HCl, coke formation and metal sintering. Problems that may be expected in such catalytic systems are related to the condensation of polymer structures and subsequent coking of the support.
6.2.2. Bimetallic nanoparticles
The activity, stability and selectivity of catalysts in HDC and hydroskimming processes in general can be enhanced by the synergistic effect of interacting metallic particles occurred in bimetallic catalysts, which are prepared by doping/promoting monometallic catalysts or by in situ/ex situ synthesis of catalysts from two or more precursors. The HDC processes use the following nanoscale bimetallic catalysts: Pd/Co,[208] Pd/Cr,[208] Pd/Mn,[208] Pd/Ni,[208] Pd/Cu,[208] Pd/Fe,[208] Ni/Ru,[209], Ni/Fe,[208-212] Au@Pd,[213] Au/Pd,[120] [214-216] Pd/Fe [217] and Pd/Pt/Fe [218][219] (Table 9).
Doping the Pd/PVP catalyst with various transition metals (Co, Cr, Mn, Ni, Cu, Fe), significantly increased the dechlorination rate of perchlorobenzene to benzene (see Table 9, entries 1 – 6).[208] However, not all the metal promoters have improved the process. In particular, the Cu promoter reduced the dechlorination rate of perchlorobenzene to just 19% within 180 min (see Table 9, entry 2), whereas using the parent Pd/PVP in this reaction provided a quantitative yield within 270 min. Maximum dechlorination of the substrate (100%) in the presence of the bimetallic species Pd/Cr, Pd/Mn, Pd/Ni, Pd/Fe was achieved at longer reaction times (420 – 710 min), whereas the parent Pd/Co catalyst required only 240 min. Using NaBH4 as a reducing agent instead of H2 as in the previous reactions, a significant increase in the 100% dechlorination rate of perchlorobenzene over Pd/Co was achieved (from 240 min to 80 min at 55°C). This delivered smaller nanoparticles (4.4 nm).
The electron transfer that yields highly dispersed metallic Ru particles, which are uniformly distributed on the surface of the nanonickel catalyst results in the partial replacement of Ni0 particles by more reactive Ru3+ ions.[209] Formation of Ru3+ ions on the catalyst surface increases its activity and stability. Conversion of chlorobenzene increases from 43% (Ni) to 76% (Ni/Ru). However, stability of the catalysts decreases to 32% (Ni) after three cycles and to 71% (Ni/Ru) after four cycles (see Table 9, entry 7). This effect can be explained by the increased resistance of the electron-deficient Ni species to chlorine in the presence of Ru. Similarly [209] (by reductive desorption), highly active Ni/Fe catalysts were prepared and tested in the HDC of chlorophenols. The rate of this reaction increased linearly with the growth of the nickel content on the iron surface, with nickel favouring greater adsorption of H2.[210] Among the substrates, the rate decreased due to steric effects of substituents and an increase in adsorption of substrates on the catalyst surface in the following order: 2-chlorophenol > 4-chlorophenol > 2,4,4-dichloro- phenol > 2,4,6-trichlorophenol > 2,6-dichlorophenol (see Table 9, entries 8 – 12). Abstraction of chlorine atoms from the ortho-position occurs twice as easily as from the para-position. Replacing Ni with Pd in the Ni/Fe catalyst increases the rate of the HDC of 2,4-dichlorophenol [217] due to the substrate’s high chemisorption via formation of strong Pd – Cl bonds, as well as the adsorption of H2 and its subsequent dissociation into hydrogen atoms on the palladium surface (see Table 9, entry 22). A similar effect to that decribed in a study [210] was observed when using 22 mol.% Ni/Fe particles in the HDC of trichloroethylene, which adsorbed on the Ni surface, which also chemisorbed H2.[211] [212] In a study,[211] butane, hexane and octane were the main products (see Table 9, entry 13), whereas toxic vinyl chloride, 1,1-dichloroethylene and isomeric 1,2-dichloroethylene were formed only in trace amounts; in a study,[212] ethane predominated (see Table 9, entry 14). In addition, the HDC is promoted by the alkalization of the reaction solution,[212] [216] which reduces the poisoning of the catalyst by HCl.
High performance of 1 wt.% Pd@Au bimetallic catalysts, where Au is used as a support (see Table 9, entry 15), is due to the influence of Au atoms, which increase the activity of Pd sites [213] by increasing the occupancy of the Pd atom d-orbitals. The cleavage of C – Cl and C – Br bonds in the HDC of 4-halophenols (Hal = Cl, Br, I) mainly involves 4 – 7 adjacent Pd atoms forming the active site. In contrast, the cleavage of C – I bonds, which results in the formation of 4,4-dihydroxybiphenyl rather than phenol, is catalyzed by isolated Pd atoms. Palladium supported on Au NPs also performed well in the aqueous-phase HDC of trichloroethylene (see Table 9, entries 16, 17).[214][215] The activity of Pd/Au (172.8 – 943.4 L gPd–1 min–1 depending on the Pd content) was one order of magnitude higher than that of Pd, Pd/Al2O3 and Pd black nanoparticles (62, 12.2 and 0.42 L gPd–1 min–1).[215] Pd/PVP-supported Au and Cu nanoparticles were used as the catalyst for the HDC of the pesticide 4,4'-dichlorodiphenyltrichloroethane (DDT) and its derivatives, 4,4'-dichlorodiphenyldichloromethylmethane (DDD) and 1,1'-dichloro-2,2-bis(p-chlorophenyl)ethylene (DDE) to 1,1'-diphenylethane (see Table 9, entries 19 – 21).[216] The authors attribute the higher activity of Au/Pd compared to that of Cu/Pd and Pd/PVP nanoparticles to the ability of inert Au atoms to absorb chlorine atoms and weaken the C – Cl bond. This leads to dechlorination and prevents Pd poisoning. The fact that the Au and Pd metal species do not oxidize before or after reacting with a chlorinated substrate indicates that the catalyst is highly stable.
Summing up the review of bimetallic catalysts, we can conclude that they are effective in the HDC process. Firstly, they reduce the size of the resulting nanoparticles and, secondly, they lower the cost of the catalyst by adding the second metal (in rare cases, the third one [218][219]) (see Table 9, entries 25 – 28). However, research into the nanosized bimetallic catalysts is represented by a limited number of works; this area of catalysis will undoubtedly evolve.
7. Sulfide and phosphide catalysts
7.1. Mo and W sulfides
The low activity of Ni- and Co-containing catalysts has stipulated the interest to sulfide catalysts, which are traditionally used in hydrodesulfurization and hydrogenation reactions. A comparison of the activity of Raney Ni, Ni/SiO2 and sulfided Ni – Mo/Al2O3 catalysts in the HDC of di-, tri- and tetrachloromethane, trichloroethane, tri- and tetrachloroethylene has shown that the sulfided catalyst outperforms metallic Ni-containing catalysts in the HDC of all these compounds.[220] Another reason for the interest in sulfide catalysts is the rapid deactivation of noble metal-based catalysts, particularly when treating mixed feeds comprising chlorine, nitrogen and sulfur.[99] For example, in the hydrogenation of tetrachloroethylene using a Pd/Al2O3 catalyst, the conversion of tetrachloroethylene and the selectivity to ethane and ethylene decrease when thiophene and quinoline are added to the feedstock. In contrast, sulfide catalysts retain high activity even in the presence of N- and O-containing compounds.
Sulfide catalysts are used in petroleum refining and petrochemistry to process feedstocks containing a variety of heteroatomic compounds, polycyclic aromatic hydrocarbons, resins and asphaltenes. A vast experience of using such catalysts, including unsupported ones, in the hydroconversion and hydrocracking of heavy petroleum feedstock [221-223] enables us to recognize their promising potential in hydroskimming processes involving pyrolysis products from polymer waste mixtures containing various heteroatomic compounds. In these processes, there is competition between Cl-, S-, N- and O-containing compounds for active catalyst sites resulting in different selectivities for hydrodesulfurization, hydronitrogenation, hydrodeoxygenation and hydrogenation reactions.
All Ni – Mo sulfide catalysts outperform their non-sulfided analogues (i.e. the same catalysts in oxide form) in the HDC reactions.[224][225] The reaction rate in the presence of NiMoS is five times higher than that for the oxide form.[225] For non-promoted Al2O3-supported Mo, this difference is only two times higher.
Most publications focus on industrial Al2O3-supported NiMoS catalysts for hydrotreatment.[226-234] Thermobaric conditions include a temperature of 290 – 350°C and a hydrogen pressure of 3 – 6 MPa. These catalysts have been studied in the HDC of chloralkanes,[232][233] polychlorobiphenyls [230][231] [234][235] and chlorobenzenes.[228] Table 10 summarizes the activities of commercial sulfide catalysts in the hydrodechlorination reactions involving various substrates. MCM-41 aluminosilicates (see Table 10, entry 18) [236] and activated carbon (see Table 10, entries 7 – 9, 19, 11) [237-239] have also been proposed as supports. In the latter case, the catalyst was found to be more stable than those supported on Al2O3 due to the lack of support destruction in the HCl medium. It has been reported [239] that the ash content of this support has a small influence on the performance of the catalyst in the hydrogenation of chlorobenzene, chloronaphthalene and chloranthracene. With virtually exhaustive dechlorination of the above compounds, their hydrogenation was not observed under reaction conditions (260°C, 3 MPa H2, NiMo/C). The use of other sulfide catalysts (CoMoS and NiWS) in HDC processes has not been investigated.
As can be seen from Table 10, the concentration of chlorobiphenyls in the solvent plays a key role in the treatment process. The concentration of chlorobiphenyls or waste dielectric oil in the solvent is maintained within the 100 – 500 ppm range, at which concentration the exhaustive dechlorination is achieved already at 290 – 300°C. Increasing the concentration leads to decreased conversion and deactivation of the catalyst. There are no known technologies for the HDC of polychlorobiphenyls in a solvent-free mode or for using a substrate-rich mixture (10% or more) fed to the reactor. From an economic point of view, however, low concentrations of the substrate in the solvent are impractical. Therefore, the search for catalytic systems and technologies that can treat such raw materials in concentrated solution form seems promising. It is also relevant to search for available and easily recoverable protective layer catalysts.
The selectivity of dechlorination of feedstock containing various chlorinated compounds depends on the temperature. Using a NiMoS catalyst at low temperatures (~200°C, see Table 10, entries 6 – 9), rapid HDC of chloroalkanes (dichloroethane, trichloroethylene and perchloroethane) is observed, whereas the HDC rate of aromatic hydrocarbons is an order of magnitude lower. At 350°C, the reaction rates of chloroalkanes and chloroaromatics are equal.[15] Aromatic polychlorinated hydrocarbons undergo stepwise dechlorination involving the desorption of chlorine derivatives at an intermediate stage. In contrast, chloroalkanes are dechlorinated in one step without the intermediate desorption of chlorinated derivatives.
Frimmel and Zdrazil [240-242] obtained interesting data when examining the selectivity of various catalysts, including sulfide catalysts, in hydrodesulfurization and HDC reactions. The Ni catalyst in its reduced form performed best in the HDC and exhibited the lowest activity in the hydrodesulfurization reaction. Sulfide Ni – Mo catalysts displayed high activity in both HDC and hydrodesulfurization. Meanwhile, activated carbon-supported catalysts show higher activity in the HDC than those supported on Al2O3, enabling the selectivity of reactions to be regulated when treating mixed raw materials.
In general, NiMoS catalysts used in the o-dichlorobenzene/ 3-methylthiophene system were more active in the HDC reaction than in the hydrodesulfurization.[242] However, the analysis of a single substrate pair is insufficient to conclude that HDC does not inhibit hydrodesulfurisation (and vice versa). Such conclusions are quite appropriate for the competing reactions of hydrodesulfurization/hydrodenitrogenation, hydrodesulfurization/hydrogenation of aromatic hydrocarbons,[242] since considerable practical experience has been accumulated in studying them. However, there is very little experimental data for the competing reactions of HDC/hydrodesulfurization, HDC/hydrodenitrogenation, HDC/hydrogenation and HDC/hydrodeoxygenation,[240-244] therefore selecting a catalyst for refining mixed feedstock requires studying the inhibition of some reactions by others.
Deactivation of the sulfide catalyst in the HDC process can be due to the loss of sulfur from its surface to yield low-active oxide and oxysulfide phases; destruction of the support by interaction with HCl resulting in changes to its morphology,[232][238] formation of chlorinated deposits and coke.[232] As can be seen from Table 10 (entries 5, 10 and 16), sulfur-containing compounds are sometimes introduced into the Cl-containing feed stream entering the reactor in order to continuously maintain the catalyst in the sulfide form. However, none of the papers suggest that a decrease in the proportion of the sulphide phase could be the cause of catalyst deactivation. Sulfidation of the spent catalyst without first removing the deposits from its surface is ineffective in restoring its activity. Analysis of the surface of the spent NiMo/Al2O3 catalyst by energy dispersive X-ray spectroscopy reveals a significant accumulation of chlorine atoms. The catalyst’s surface area, volume and average pore diameter all decrease.[232] Deactivation is most significant when chlorine and nitrogen compounds are present in the feedstock simultaneously, resulting in the accumulation of NH4Cl and chloride salts of nitrogenous bases on the catalyst surface. This is demonstrated by the example of the co-hydroprocessing of chlorobenzene and pyridine.[64] The catalyst can be reactivated by removing deposits from its surface and sulfidating it. At the same time, a small positive effect of chlorobenzene has been reported [243] when carrying out hydrodenitrogenation of a mixture of chlorobenzene and quinoline over NiMo/Al2O3 sulfide catalyst. At least, the hydrodenitrogenation rate does not decrease in the presence of chlorobenzene, a phenomenon that can be attributed to the neutralizing effect of HCl on nitrogenous bases, suppressing their adsorption on acid sites.
Murena [244] highlighted the positive impact of thiophene on the HDC of chlorobenzene using a NiMo/Al2O3 sulfide catalyst. There was no mutual inhibition of the reactions, thiophene slightly accelerated the HDC reaction. This is likely due to the catalyst’s continuous regeneration through sulfidation — the replacement of chlorides by sulfides on the catalyst surface when a sulfur-containing compound is present. However, as previously mentioned, the introduction of quinoline [243] into this reaction system decreases the rates of both HDC and hydrodesulfurization, due to nitrogenous bases adsorbing onto the active sites and forming chloride salts that block the catalyst surface.
In conclusion, it should be noted that there is a very limited amount of new research in the field of sulfide catalysts for hydrodesulfurization. On the one hand, industrial sulfide catalysts for hydrodesulfurisation, as shown in Table 10, can be used to solve hydrodesulfurization problems and do not require detailed study. On the other hand, the search for HCl-insensitive supports is relevant, particularly for the hydroconversion of polymer waste in oil fractions. In this context, unsupported sulfide catalysts [245][246] with high carbonisation resistance may be of interest. Sulfide catalysts are promising for refining liquid polymer pyrolysis products due to their resistance to deactivation compared to Ni, Pd and Pt catalysts, and preliminary data suggests they have sufficient selectivity in both HDC and hydrogenation/hydrodesulfurization reactions.
It will be relevant to study the behaviour of sulfide catalysts that differ in terms of the active metal and promoter composition in competitive hydrogenolysis reactions involving different substrates. Determining the selectivity of each reaction, the inhibition of one reaction by another and the influence of each substrate on the rate of catalyst deactivation is promising.
7.2. Transition metal phosphides
Since the 2000s, transition metal phosphides have been actively investigated as hydrodesulfurization catalysts offering an alternative to conventional sulfide catalysts.[247-252] It has been reported [253] that phosphides of Mo and W are more active than sulfides of these metals. Ni2P shows the highest activity among the iron group phosphides.[249] [253] [254] One of the advantages of phosphide catalysts over sulfide ones is their high performance in hydrodesulfurization of sterically hindered heterocyclic compounds such as dibenzothiophene and its alkyl homologues.[250] [252] [254] Phosphides of Ni,[249] [253] Mo [251] and Ni – Mo phosphide catalysts have been most extensively studied in hydrodesulfurization reactions.[252] [255] Phosphides of these metals have also been found to be active in hydrodenitrogenation and hydrogenation of olefins and aromatic rings.[256][257] Several works are devoted to unsupported phosphide catalysts.[250] A significant block of research is focused on the hydroconversion of biomass products over phosphide catalysts, e.g., hydrodeoxygenation [258-262] and hydrodenitrogenation.[263]
Therefore, the interest in studying transition metal phosphides in HDC reactions is natural. On the one hand, these catalysts have all the advantages of sulfide catalysts, viz., high activity and resistance to HCl poisoning,[264][265] relatively low cost compared to platinum group metal-based catalysts. On the other hand, phosphide catalysts have received little attention in the context of the HDC process, particularly in the HDC of mixtures of chlorinated compounds, which are liquid products of chlorinated polymer waste. This section will consider Ni, Mo and Co phosphides, both supported and unsupported.(Ref.[266] see Table 8)
As mentioned above, the catalyst deactivates rapidly in the catalysis of HDC by Ni metal. This is due to the formation of NiCl2, which is then partially reduced by H2 to Ni0. However, at the same time, there is agglomeration and enlargement of the metal particles, which reduces the surface area of the active catalyst phase (see Fig. 2). Some of the chlorine ions are not desorbed, resulting in a loss of the active phase. In phosphide catalysts, Ni is present in the form of phosphides Ni2P, Ni3P or Ni12P5.[84] [267-270] In this case, there is a partial electron transfer from Ni to P atom, which decreases the electron density on the Ni atom, providing it with a small positive charge. This weakens the interaction between the Cl and Ni atoms, facilitating the desorption of Cl ions from the Ni active sites. Also, a steric effect occurs in which the P atom shields the Ni surface from Cl ions.
In addition, the interaction between Ni and P weakens the interaction of the adsorbed H2 with the catalyst’s active sites, reduces the energy barrier for hydrogen spillover, which favours the increase of the concentration of H atoms on the catalyst surface.[270] These hydrogen atoms, are then involved in the hydrogenolysis of the C – Cl bond and the desorption of Cl ions from the catalyst surface. Nickel phosphides are therefore superior to metallic Ni in terms of catalytic activity and stability. It is suggestes that the HDC process forms a phosphochloride phase [267] similar to the phosphosulfide phase in the hydrodesulfurisation process.[271] For example, a comparison of the activity of silica-supported Ni and NiP catalysts (5 wt.% Ni) in the HDC of chlorobenzene [267] showed that at 325°C, the activity of metallic Ni decreases after just 8 – 12 h from the reaction onset. Within 24 hours, the conversion of chlorobenzene decreases from 100% to 60%, whereas with Ni phosphide, it decreases from 100% to 80%. It is shown that the Ni3P/SiO2 catalyst is significantly superior in activity (the difference in conversion values is ~ 20%) and stability to Ni/SiO2 in the HDC of chlorobenzene at temperatures of 200 and 300°C.[269]
The presence of Lewis and Brønsted active sites on the surface of Ni phosphides favours the HDC due to the adsorption of polar Cl-containing molecules onto acid centres where the electron density is shifted away from the Cl atom.[266] Brønsted acid sites in phosphide catalysts are represented by P – OH groups,[268] which can not only promote the adsorption of organochlorine compounds and also act as sources of atomic hydrogen [272] due to the enhancement of hydrogen spillover. The presence of P – OH groups on the surfaces of the steam-treated Ni2P/SiO2 and Ni2P/MCM-41 catalysts was detected by diffuse reflection Fourier transform infrared spectroscopy.[272] Steam treatment was found to improve the catalysts’ activity in the HDC of chlorobenzene. This was attributed to the presence of P – OH groups and their role in forming atomic hydrogen on the catalyst surface. This atomic hydrogen is then involved in the hydrogenolysis of chlorobenzene molecules adsorbed on the active Ni sites. It has also been demonstrated that introducing 0.8 wt.% of steam into the H2 stream can significantly increase the conversion of chlorobenzene (from 8% to 68%). This is probably due not only to the formation of P – OH groups on the catalyst surface, but also to the reduction of the induction period inherent in phosphide catalysts, which will be discussed later. Additionally, the amount of coke deposits can be reduced due to their «washing out’ by water vapour.
The rate at which coke deposits are formed is determined by the support material, which also affects the catalyst activity and the formation of the active phase. For example, at the same Ni/P ratio during catalyst synthesis, the Ni2P phase is formed on SiO2 and TiO2, Ni and Ni3P phases are formed on γ-Al2O3, and Ni and Ni12P5 phases are formed on zeolite HY,[273] which is explained by the interaction of some of the phosphorus with γ-Al2O3 and zeolite to produce AlPO4. A comparison of the activity of Ni2P/TiO2, Ni2P/SiO2, Ni–Ni3P/γ-Al2O3 and Ni–Ni3P/HY [273] catalysts, all of which have the same nickel content, in the HDC of chlorobenzene shows that the interaction of TiOx with chlorinated compounds weakens the C – Cl bond and promotes hydrogenolysis. However, the catalyst deactivates rapidly (conversion decreases from 100 to 70% after 24 h at a temperature of 300°C and H2 pressure of 0.1 MPa) due to the formation of carbonaceous deposits. This has been confirmed by thermal gravimetric analysis. At the same time, no significant change in the Ni2P active phase was observed. Coke formation on HY zeolite (dealuminated Y zeolite) also proceeded very quickly due to its high micropore content, with chlorobenzene conversion decreasing from 100% to 30% after 10 hours. Furthermore, the presence of exposed Ni on the surface of this catalyst (Ni–Ni3P phase) facilitates the rapid formation of NiCl2, which also leads to a loss of activity. Therefore, silica with a small proportion of micropores is considered to be optimal support for phosphide hydrogenation catalysts. The conversion of chlorobenzene on Ni2P/SiO2 remains constant for at least 10 h.
A comparison of spent Ni2P/SiO2 and Ni/SiO2 catalysts [274] showed that the main reasons for deactivation of the Ni/SiO2 catalyst were the formation of NiCl2 and agglomeration of metal species due to cycles of NiCl2 formation and its subsequent reduction. No Cl bound to Ni was detected on the Ni2P/SiO2 catalyst surface; Cl atoms were only present in the coke deposits. The loss of activity is also due to the formation of coke deposits which block the active sites and thereby reduce the surface area. No changes in the size of the Ni2P crystallites were observed. It was found that the lower the Ni/P ratio, the greater the catalyst’s resistance to chlorine poisoning.[267][274] A different coke formation mechanism has been observed on the surface of Ni2P/SiO2 and Ni/SiO2 catalysts.[274] In the first case, coke on the Ni species arises from decomposition of the organochlorine substrate.[275] In the second case, coke is formed by polymerization of chlorinated compounds on the acidic catalyst sites (Lewis Niδ+ and Brønsted P – OH). Coke deposits can be more easily removed from the surface of a metal catalyst through hydrogen regeneration due to the hydrogenation of polycyclic aromatic moieties. Regeneration is not as easy in the case of the phosphide catalyst, probably due to the lower activity of nickel phosphide in hydrogenation reactions.
Table 11 summarizes the data on the catalysis of the HDC of various substrates using Ni phosphides. In contrast to Mo and W sulfides, Ni phosphides have not been investigated in the HDC of polychlorobiphenyls. Since the latter are less susceptible to hydrodechlorination than chloroalkanes, chlorobenzene and dichlorobenzenes, research in this area is relevant. In addition, studying Ni phosphide catalysts for the selective HDC of chloroolefins without hydrogenating double bonds is of great interest and seems quite feasible due to phosphides’ low activity in hydrogenation reactions.
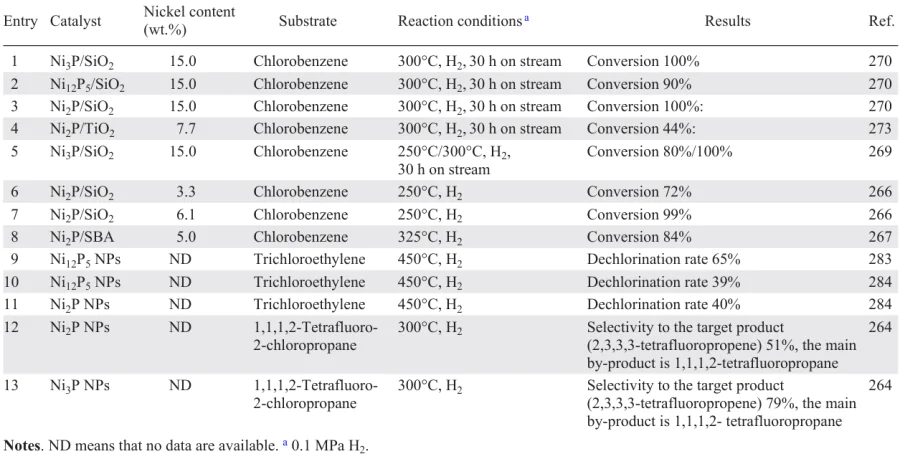
As mentioned earlier that phosphide catalysts are characterized by an induction period during which they demonstrate low activity, not only in HDC but also in hydrodesulfurization and hydrodenitrogenation processes.[253] This phenomenon is caused by excess phosphorus on the catalyst surface, which blocks its active sites. The presence or absence of the induction period depends on the Ni/P ratio during catalyst synthesis. During the catalyst reduction, excess P does not always desorb from the surface and its removal occurs already in the course of the reaction. The induction period decreases when the H2 flow rate, temperature and reduction time are increased during catalyst preparation.[270] [276] As an example, Fig. 4 shows the dependence of the chlorobenzene conversion on the catalyst time-on-stream for nickel phosphides synthesized at different initial Ni/P ratios is shown (see Table 11, entry 1).[270] Clearly, the induction time decreases as the increasing Ni/P molar ratio increases, being absent for Ni3P.
![[{"id":"DahGFjh7q0","type":"paragraph","data":{"text":"Chlorobenzene conversion as a function of catalyst time on stream for nickel phosphides synthesized at different initial Ni/P molar ratios (given in parentheses). Reaction conditions: 300°С, Н<sub>2</sub> 0.1 MPa. Reproduced from Liu et al.<sup>270</sup> with the permission of the American Chemical Society."}}]](/storage/images/resized/eU6hEs4CEizx3iA9Nmujn2DCfx5AudvG994pMoUg_xl.webp)
Since one way for processing polymer waste involves hydroconversion as part of oil fractionation,[52][245] the possibility of using phosphide catalysts in HDC has been investigated where hydrodesulfurization and hydrodenitrogenation reactions occur simultaneously. It has been reported [277][278] that the presence of H2S in the raw materials or hydrogen in the catalyst reduction process improves conversion in the HDC reaction. Sulfur atoms are embedded in the Ni2P crystallite structure to form a mixed phosphosulfide phase NiPxSy, which further reduce the electron density on the Ni atom. This favours hydrogen spillover and the regeneration of active sites.
In this context, it has been proposed that the catalyst should be passivated prior to reduction using a mixture of H2S (10 vol.%) and H2.[278] Passivation of phosphide catalysts is necessary because they oxidize rapidly when stored in air. Therefore, a stream of an inert gas containing 0.5 – 1 vol.% O2 is used to carry out mild oxidation of the catalyst surface, forming a protective oxide film. The catalyst is then reduced with H2 immediately prior to the hydrogenolysis reaction. Passivation using H2S provides higher catalytic activity than passivation using O2.[277] Surface SH moieties and P – OH groups may both be sources of hydrogen for the hydrogenolysis. However, it has been reported that passivation with NH3 negatively affects the activity of the catalyst, since nitrogen compounds bind strongly to the Ni2P active sites and are difficult to displace with sulfur.[278]
Returning to the treatment of S-, Cl-, O- and N-containing raw materials, it is important to note that, according to the data,[278] the substitution of sulfur by nitrogen on the surface of the NiP/MCM-41 catalyst (with a NiO and P2O5 content of 30 wt.% and a Ni/P ratio of 1.25) occurs much faster than the substitution of nitrogen by sulfur. In other words, after interacting with N-containing compounds, the catalyst is already inefficiently activated by H2S. Replacing sulfur with oxygen is slower than replacing oxygen with sulfur. Therefore, hydrodeoxygenation is not a problem when processing mixed feedstocks. However, when processing feedstocks containing organonitrogen compounds, it is particularly important to pre-passivate the phosphide catalyst by treating it with H2S. Introducing H2S (or organosulfur compounds) during the reaction process is ineffective because nitrogen compounds strongly bind to the catalyst’s active sites beforehand, resulting in irreversible loss of activity. The change in catalyst activity in the sequential hydrodesulfurization/hydrodeazotation/hydrodenitrogenation processes is shown. Following the hydrodenitrogenation cycle, the catalyst activity in the hydrodesulfurization recovers very slowly.[278] It can be concluded that the presence of nitrogenous compounds in the hydrodesulfurization feedstock is undesirable, as these compounds strongly adsorb onto the catalyst’s active sites and also form chloride salts that block these sites.[64]
The synergistic effect between Ni2P/Al2O3 and MoS2/Al2O3 catalysts has been described.[279] It was suggested that hydrogen atoms formed on the active sites of Ni2P migrate to the MoS2 phase, where they interact with sulfur to form H2S and crystal defects, which act as active sites in the hydrogenolysis reaction. Therefore, the presence of nickel phosphide on the surface of the MoS2 catalyst increases the concentration of sites responsible for the hydrogenation, resulting in a higher proportion of hydrogenation products among the reaction products. In contrast, mainly hydrogenolysis products are formed on the surface of the MoS2 catalyst. Introducting S atoms into the crystal lattice of the CoP catalyst [280] deactivates acid sites and alters the pathway of the hydrogenolysis or hydrogenation reactions, and also increases catalyst activity in these reactions by creating new active sites.
Changes in the morphology and composition of some supports when they interact with HCl [273] complicate the treatment of polymer waste pyrolysis products. Processing polymers containing Cl- and Br-containing flame retardants is also challenging. This raises the question of whether unsupported phosphide catalysts could be used in the HDC process. In recent years, such catalysts have been actively investigated for use in the hydroconversion of oxygenated compounds.[261][262][281-283] Ni phosphide species can be obtained from Ni(PH2O2)2 and NH4PH2O2 Ni using the hydrothermal method [282] or from NiCl2 and triphenylphosphine in the presence of hexadecylamine and octadecene to stabilize the resulting nanoparticles (average diameter 5.4 nm).[281] By varying the ratio of starting reagents, Ni2P, Ni5P4 and NiP2 species can be obtained.
Only two publications have been devoted to unsupported phosphide HDC catalysts.[283] [284] For example, Ni12P5 was obtained from Ni(NO3)2 and red phosphorus using the hydrothermal method in the presence of chelating agents (alkyl-substituted benzoic acids), which stabilize the resulting nanoparticles (diameter ~ 5 nm). These nanoparticles form clusters with a developed surface and internal pores.[283] The resulting catalyst was tested in the HDC of trichloroethylene. The dechlorination rate at 400 – 500°C reached 40 – 70% and increased with increasing P/Ni ratio (Table 11, entry 9). PVP was used to control the size of the Ni2P and Ni12P5 nanoparticles, which were obtained from Ni(NO3)2 and red phosphorus.[284] The nanoparticles were also obtained from Ni(NO3)2 and red phosphorus. The simplicity of this synthesis method and the non-toxicity of the reagents were noted. Polyvinylpyrrolidone favours the formation of the internal pore structure of the nanoparticle clusters, allowing greater accessibility of the active sites to the trichloroethylene substrate molecules, compared to samples synthesized in the absence of PVP. At a temperature of 500°C, the dechlorination rate of trichloroethylene reaches 70% (see Table 11, entries 10 and 11).
The possibility of fabricating magnetic particles of Ni phosphides and recovering them from reaction products by magnetic separation is being investigated.[285-287] For example, a method has been proposed for synthesizing ‘core – shell’ nanoparticles with an Ni core and Ni phosphide shell.[287] Firstly, Ni nanoparticles were obtained and then the surface was phosphatized by treating it with triphenylphosphine. The thickness of the shell was controlled by varying the phosphatization duration. The size ratio of the ferromagnetic core to the non-ferromagnetic shell is important for magnetic separation. Magnetic separation is impossible if the core size is insufficient.
Magnetic nanoparticles with a simple structure can be fabricated without a ferromagnetic core. These can be synthesized from inorganic salts of Ni and Na(PH2O2) [286] or triphenylphosphine.[287] Controlling the size of the resulting nanoparticles is crucial, either by using surfactants or by varying the synthesis conditions, as this determines their magnetic properties. Nickel is ferromagnetic, while nickel phosphide is paramagnetic. According to the data of Zheng et al.,[287] small nanoparticles (64 nm) are superparamagnetic, while large nanoparticles (85 – 95 nm) are ferromagnetic at room temperature. The saturation magnetization, residual magnetization and coercivity increase with the size of the Ni2P particles. In contrast, the magnetic properties of Ni phosphide nanoparticles were reported to weaken as the size increase.[286] The nanoparticles were synthesized using pulsed electrolysis. This weakening is due to P atoms penetrating the Ni crystal lattice and distorting it. Nanoparticles measuring 125 nm exhibited ferromagnetic properties, whereas those measuring 394 and 578 nm did not.
Clearly, the magnetic properties of nanoparticles are not determined by their size, but by their phase composition, which in turn depends on the method of their synthesis. The presence of ferromagnetic properties requires that parts of the Ni crystal lattice are not distorted by the incorporation of P atoms. The cited publications [286][287] did not explore the catalytic properties of these nanoparticles. To maintain the long-term activity of the HDC catalyst, there must be a sufficiently high proportion of the Ni phosphide phase on the surface of the clusters, along with a small proportion of metallic Ni. Such particles are unlikely to have ferromagnetic properties or be amenable to magnetic separation. This aspect, i.e. the balance between Ni and Ni phosphide phases, requires further investigation both for HDC processes, and other hydrogenolysis processes.
Compared to Ni phosphides, there is less research into Co phosphides as HDC catalysts.[19] [288] Silica-supported Co phosphides significantly outperformed metallic Co on the same support [288] in the HDC of chlorobenzene, exhibiting a similar activity pattern to Ni catalysts. Increasing the P/Co molar ratio to 2/1 favours hydrogen spillover and increases the proportion of weak acid sites, resulting in greater dispersion of the active phase and higher catalyst activity. A further increase in the above ratio leads to a decrease in the proportion of acid sites and metal active sites, as well as a reduction in the chlorobenzene conversion and the stability of catalyst operation. An increase in the P/Co ratio above 2/1 also leads to a shielding of the active sites by phosphorus. In catalyst samples with high Co content, the Co2P phase dominates, whereas in samples with high P content, the CoP phase prevails. Analysis of spent catalysts shows that Co2P catalysts are characterized by a higher chlorine content than CoP, i.e. the surface of catalysts with a high phosphorus content is more easily recovered from chlorides due to more intensive hydrogen spillover.[288]
Comparing the activity of Ni2P/SiO2 and CoP/SiO2, catalysts containing 5 wt.% of metal in the HDC of 1,2,4-trichlorobenzene (325°C, 0.1 MPa H2, catalyst time on steam 24 h) showed that the nickel catalyst is more active in the dechlorination reactions.[19] This catalyst enables the reaction to proceed to form benzene. In contrast, in the presence of cobalt phosphide the reaction only produces 1,2-dichlorobenzene and chlorobenzene, after which it practically stops (Scheme 2). Comparing the performance of these catalysts in the HDC of dichlorobenzenes shows that the activity of the cobalt catalyst is less active than the nickel catalyst. For example, the selectivity to benzene in the HDC of 1,4-dichlorobenzene is 90% for Ni2P/SiO2 and 80% for CoP/SiO2, whereas in the HDC of 1,2-dichlorobenzene these values are 72% and 55%, respectively.
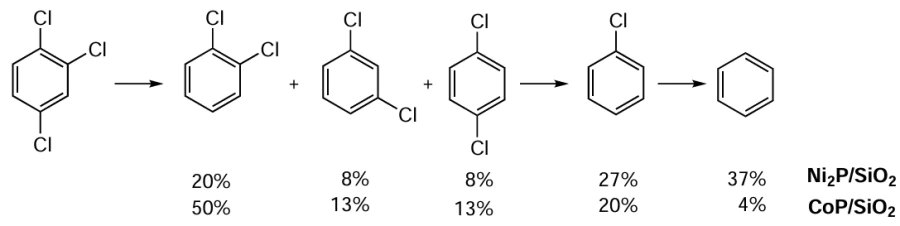
Molybdenum phosphide has been investigated as a catalyst for the HDC of trichloroethylene.[247] [289] The HDC of trichloroethylene was carried out at temperatures between 400 and 600°C in the presence of MoP/γ-Al2O3 catalyst (15 wt.% MoP).[247] At 400°C, a low degree of C – Cl bond conversion (~ 23%) was observed, which was only slightly higher than that observed for the Mo/γ-Al2O3 catalyst. The difference in catalytic activity becomes more pronounced at 500°C and disappears at 600°C. Temperatures below 400°C are insufficient for the active performance of MoP, whereas the catalyst is rapidly deactivated at temperatures above 500°C. In order to increase the activity and stability of unsupported MoP, Zhang et al.[289] suggested treating the catalyst with H2O2, which forms Mo – P – O units and creates unsaturated bonds on the surface, resulting in a defect-rich crystal lattice. This in turn creates a large number of active sites on the surface. The H2O2 treatment has also been proposed as a method of regenerating the spent catalysts. However, the narrow operating temperature range of MoP and the very high reaction temperature render this catalyst unsuitable for HDC.
In summary, the analysis of phosphide catalysts for HDC shows that this area of research is promising due to the low cost and availability of Ni, Co and Mo compared to noble metals. The stability of these catalysts is sufficiently high, and they can be regenerated, creating the conditions for their study in the hydrotreatment of chlorinated polymer waste. The synthesis of unsupported phosphide nanoparticles that can be used in the HDC of feedstocks prone to the formation of coke deposits, can be considered as an important and promising area of research. In turn, the possibility of obtaining magnetic nanoparticles and controlling their magnetic properties and catalytic activity could be a breakthrough trend in Ni phosphide catalysis research. It is of interest to study the activity and stability of phosphide catalysts in systems where several hydrogenolysis reactions occur simultaneously such as HDC, hydrodesulfurization, hydrodenitrogenation and hydrodeoxygenation. Such systems model the processing of polymer waste mixtures. Optimization of the phase composition of the catalysts and the initial metal/phosphorus ratio for different reaction systems might be considered relevant.
8. Conclusion
The hydrodechlorination of water-soluble chlorinated waste of a restricted composition, vis., chlorophenols, pesticides, pharmaceutical wastes characterized by low chlorine content and high reactivity, noble metal-based catalysts on various supports have been extensively studied. The main areas of focus in this field include studying the interaction between the active metal and the support in order to minimize particle agglomeration and protect the active component from chlorination and sintering; developing nanoparticles encapsulated in polymeric supports (including magnetic ones); and studying metal – promoter interactions and selecting dopants.
On the other hand, the pyrolysis products of polymers (not only polyvinyl chloride and neoprene, but also polyolefins, polystyrene, oxygenated polymers with additives of Cl- and Br-containing flame retardants [46-48] [290]), dielectric oils and chlorinated solvents, require catalysts that are resistant to deactivation, available and easy to recover. Analysis of the HDC catalysts shows that transition metal sulfides and phosphides best meet the above requirements. In this field, it seems relevant to study the hydroconversions of complex mixtures of Cl-, O-, S- and N-containing compounds, as well as developing supports that are resistant to HCl. It is also important to consider unsupported nanocatalysts, which are widely used in other hydrogenolysis processes.[222] [246][262][291][292] For nickel phosphides, creating magnetic nanoparticles that can be separated from the products by magnetic separation is promising.
A separate niche is occupied by the co-processing of chlorinated polymers and oil fractions,[51] coal distillates and biomass,[293][294] which can also be conditionally referred to as hydrodechlorination. Such raw materials place higher demands on the catalyst in terms of stability, availability, complete extraction of the active component, regeneration and reuse due to their extremely complex composition and the simultaneous presence of Cl-, O-, S- and N-containing compounds. There is little research in this field devoted to studying the competing runs of various hydrogenolysis, hydrogenation and hydrocracking reactions, one of which is the HDC reaction. Sulfide and phosphide catalysts can be considered the most promising for this group of HDC processes. However, questions remain regarding the optimization of support composition (or avoidance of support), methods of catalyst regeneration, and ways of slowing down catalyst deactivation, including preliminary moderate passivation.[278]
This review was financially supported by the Russian Science Foundation (Project No. 24-29-00401).
9. List of abbreviations
AmLSA — alumina solid Lewis superacid;
BEA — three-dimensional zeolite containing pores formed by 12-membered rings in all directions;
BOMC — B-doped ordered mesoporous carbon;
CNF — carbon nanofibres;
CNT — carbon nanotubes;
CoMoS — cobalt–molybdenum sulfide catalyst;
DDD — 4,4'-dichlorodiphenyldichloromethylmethane;
DDE — 1,1’-dichloro-2,2-bis(p-chlorophenyl)ethylene;
DDT — 4,4'-dichlorodiphenyltrichloroethane;
FAU(Y) — three-dimensional zeolite with large pores, having large cavities connected in the system by channels formed by 12-membered rings cycles of 12 elements (the ring comprises 12 cations (Si4+, Al3+) and 12 anions (O2–));
FCCA — fluorinated carbon covered alumina;
GNF — graphite nanofibres;
HDC — hydrodechlorination;
HDPE — high density polyethylene;
HSAG — high surface area graphite;
HY — dealuminated Y zeolite;
IRA-900 —macroreticular polystyrene type 1 strong base anion exchange resin containing quaternary ammonium groups;
MCM-41 — mesoporous material with a hierarchical structure from a family of silicate and alumosilicate solids;
MSCN — mesoporous silicon-carbon composite;
NaY — the sodium form of a Y-type zeolite;
NCN — nitrogen-doped carbon nanosheets;
NiMoS — nickel – molybdenum sulfide catalyst;
NiWS — nickel – tungsten sulfide catalyst;
NP — nanoparticles;
PBSAC — polymer-based spherical activated carbon;
Pd/C-S1 — Pd/C catalyst obtained by pyrolysis for 2.5 h (ramping time) and 3 h (isothermal stage);
Pd/C-S2 — Pd/C catalyst obtained by pyrolysis for 1.5 h (ramping time) and 3 h (isothermal stage);
PDMS — polydimethylsiloxane;
ppb — parts per billion;
ppm — parts per million;
PS-co-PAEMA-co-PAM — poly[styrene-со-2(acetoacetoxy)ethyl methyl acrylate-со-acrylamide];
PVC— polyvinyl chloride;
PVP — polyvinylpyrrolidone;
SAA — surface-active agent;
SBA-15 — ordered mesoporous zeolite with pore size from 2 to 50 nm, cylindrical shape in hexagonal crystallographic order;
SiBEA — de-aluminated form of BEA zeolite, which is a wide pored high-silicon zeolite with large pores and a three-dimensional system of interconnected 12-membered ring channels;
UDD — а diamond particles with sizes smaller than 100 nm;
ZSM-5 — quasi-tridimensional medium-pore aluminosilicate zeolite containing pores formed by 10-membered rings in one direction connected by zigzag channels formed by 10-membered rings.
References









