



Keywords
Abstract
The development of new catalysts is evolving in a few ways. One is to design complexes with increasingly complicated ligands. Although effective in terms of the selectivity achieved, this approach has the disadvantage of making catalytic systems progressively more expensive. Another path is to boost catalytic activity by modifying the common elements of the ligand environment. In particular, to date, there have been excellent reviews of the physical and chemical properties of various halogen-containing complexes but a systematic comparison of their catalytic performance is lacking. The present review partially fills this gap by comparing the activity and selectivity of catalysts containing Ru – I and Ru – Cl bonds. Though the influence of numerous parameters, viz., the leaving ability of a halogen, the stability of the complex, its redox potential, to name a few, does not allow for a reliable prognosis of their outcome for the catalyst efficiency in the general case, certain predictions and explanations can be made within some classes of reactions. Some of them are given in this review.
The bibliography includes 131 references.
1. Introduction
Ruthenium complexes are widely used in organic catalysis as catalysts for redox, C – H activation, metathesis, allylic activation, and other reactions.[1-10]
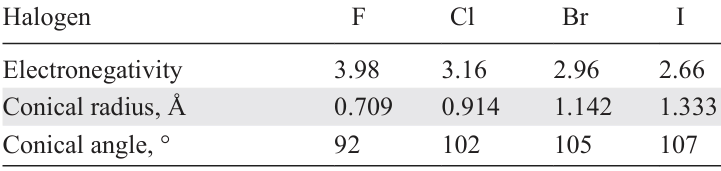
Often there is a halogen atom in the coordination sphere of the metal in a complex. Halogens differ in atomic radii, electronegativity and Pearson’s softness/hardness, which in turn is defined by polarizability, ionization energy and some other properties. A comparison of some steric and electronic features of halogen atoms is given in Table 1.[11] Due to these differences, the identity of a halogen atom coordinated to the metal can significantly influence catalytic activity of the complex. Fagnou and Lautens [11] presented a thorough investigation of such effects on the examples of various reactions. Chiusoli and co-workers [12] summarized the data on the addition of iodide to reaction mixtures and concluded that it can sometimes increase the activity of a catalyst due to the following factors:
1) Activation of an organic substrate. For example, a crucial step in the Monsanto process is the conversion of methanol into methyl iodide, which is more prone to oxidative addition;
2) The superior σ-donating ability of iodine. This increases the electron density on a metal centre, thus enhancing its nucleophilicity. As a result, oxidative addition proceeds more smoothly.
Redox potentials may also affect catalytic reaction rate. For example, a change in the halogen environment of the metal changes the potential of the transition by more than 0.2 V, as exemplified by the Ru(III)/Ru(II) transitions (Fig. 1).[13]
![[{"id":"6H_oU3fQ85","type":"paragraph","data":{"text":"Cyclic voltammetry data for Ru – Х complexes <i>cis</i>-(CO)-<i>trans</i>-(X)-[Ru(CO)<sub>2</sub>(HL)X<sub>2</sub>]"}}]](/storage/images/resized/AtC9SM0AQvrTeuKCKVjK48YjUGbT5Ir44HwZPqNJ_xl.webp)
This review considers the works devoted to catalysts based on Ru – Hal (Hal = I, Cl) complexes and compare the properties of these compounds depending on the halogen atom. In addition, it was of interest to find out in which cases the replacement of chlorine with iodine leads to an improvement (or deterioration) in the activity and/or selectivity of the catalyst. The reason for choosing the I – Cl pair is that it allows a representative comparison due to the amount of available data, which is sufficient, but not overwhelming. As for other halogens, fluorine also enables the design of high-performance catalysts. However, its specific electronic features and ability to form oligomers complicate matters, as these oligomers may serve as catalyst resting states.[14] Catalysts bearing Ru – Br bonds are represented in the literature rather sporadically and require further investigation. Therefore, we focused on studying the halogen effect by comparing the catalytic behaviour of iodine- and chlorine-containing complexes.
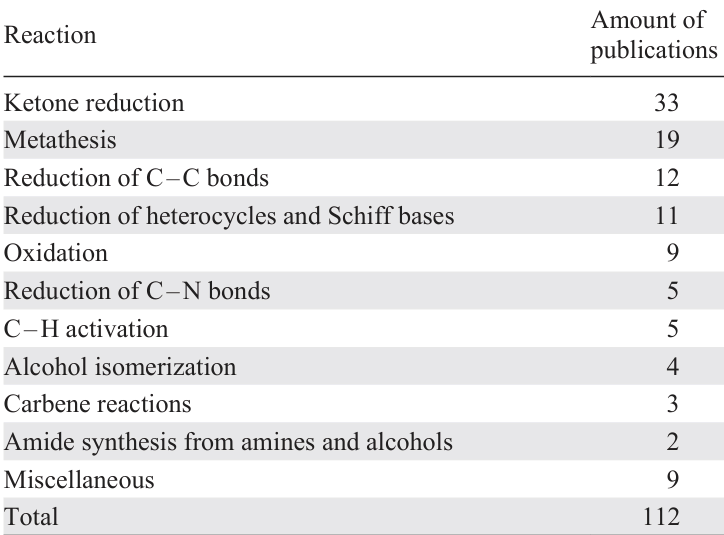
The literature survey revealed the following distribution of studies on Ru – I catalysts by reaction classes (Table 2). We categorized the available data on the application of Ru – I bond containing catalysts by reaction type from the most to the least frequently described in the literature.
2. Reduction of C=O bonds
The known reactions of ketones reduction can be divided in three groups:
1) Catalytical hydrogenation. Most of these works are aimed at finding ways to make these reactions enantioselective;
2) Hydrogen transfer, Meerwein – Ponndorf – Verley-type reactions;
3) Miscellaneous reactions.
2.1. Catalytic hydrogenation of ketones
The history of the development of methods for the catalytic hydrogenation of ketones goes back to Noyori’s works. In one of his early papers, a comparison of the neutral complexes RuX2(S-BINAP) for X = Cl, I is given (Scheme 1).[15]
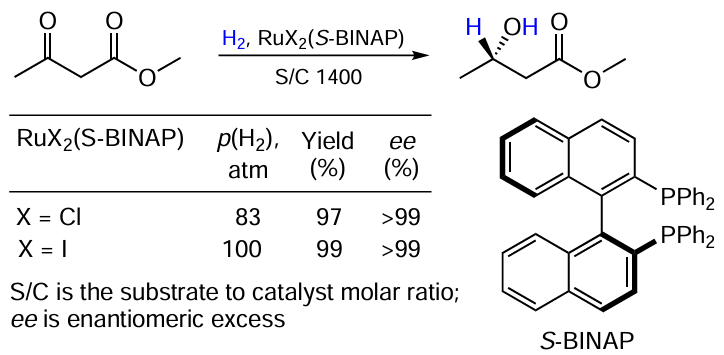
Both catalysts showed the same efficiency, though the chloride-substituted one operated at a slightly lower pressure (83 vs. 100 atm). Unfortunately, the data provided are insufficient to allow a reliable estimate of the difference in the catalytic activity to be made.
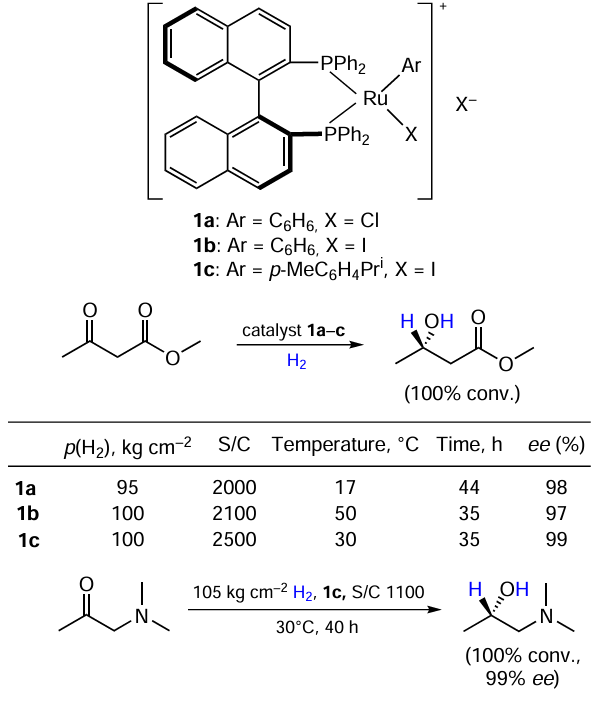
Cationic BINAP-ruthenium complexes were applied to the reduction of β-ketoesters (Scheme 2).[16][17] All catalysts 1a – с tested gave a 100% conversion of methyl acetoacetate, although the iodide complex 1b was used at a slightly higher temperature than its chloride counterpart 1a; on the other hand, the reaction with 1b ran faster. The variation in multiple reaction parameters does not allow claiming a significant difference in enantioselectivity between the catalysts. Сatalyst 1c also performed well in N,N-dimethyl aminoacetone reduction.
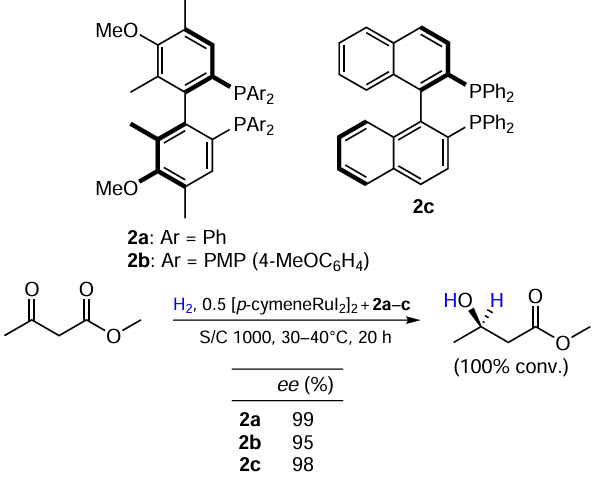
Yamamoto et al.[18] investigated the effect of a donor substituent in a phosphine ligand on the catalyst activity. Ligands 2a,b were synthesized and the effect of their in situ addition to [(p-cymene)RuI2]2-catalyzed reactions was studied (Scheme 3). As the reference ligand, (R)-BINAP was used. The catalysts 2a and 2b showed comparable performances in the reference system.
Interestingly, Chiba et al.[19] demonstrated the possibility of the similar conversion of α-ketoesters (Scheme 4). In this case, the halogen nature turned out to be crucial for both the activity and the selectivity of a catalytic system. The iodide complex delivered an enantiopure product in quantitative yield, whereas its chloride counterpart gave a 13% conversion with a low ee.
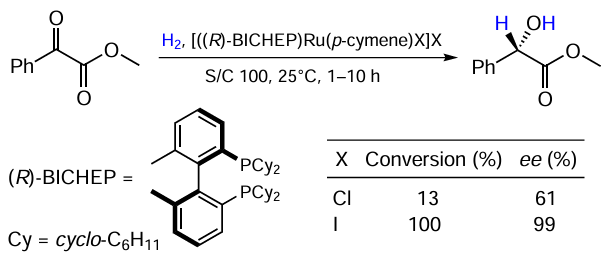
The authors also noted, referring to the unpublished data, that they had observed the reverse pattern when performing an analogous reaction with BINAP ligands with the chloride ligand proving superior to the iodide one. The chloride complex also showed slightly higher enantioselectivity in the reduction of α-ketoamides under the same conditions.
Complexes of the same type were successfully employed by Trost et al.[20] in a total synthesis of (–)-pseudolaric acid B to reduce a cyclic b-ketoester (yield 95%, ee > 90%).
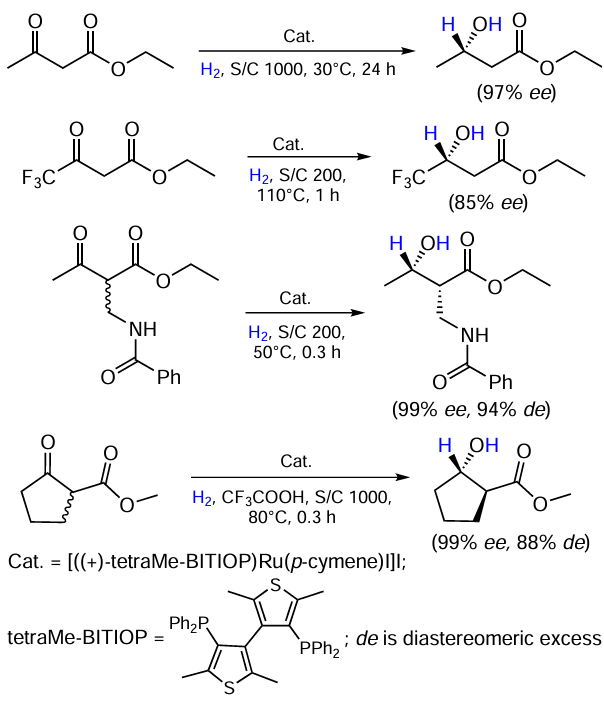
Benincori et al.[21] synthesized a series of Ru – I complexes with thiophene-substituted phosphine ligands (tetraMe – BITIOP) and tested them in the homogeneous hydrogenation of prostereogenic carbonyl functions in a- and b-ketoesters (Scheme 5). Various substrates underwent the reaction in high yields and with excellent enantioselectivities. Interestingly, a proton acid addition proved to be crucial for the cyclic substrate: ee did not exceed 62% in the absence of CF3COOH.
Weissensteiner and co-workers [22] synthesized biferrocene analogues of Ru(II)-BINAP complexes. It was found that the introduction of strong donor groups into aromatic rings of phosphine ligands decreased enantioselectivity of the hydrogenation of ethyl acetylacetate (Scheme 6). Further studies on this subject gave rise to a series of ferrocenylphosphine ligands, which, when added to [(p-cymene)RuI2]2, allow hydrogenation with excellent enantioselectivity.[23-30]
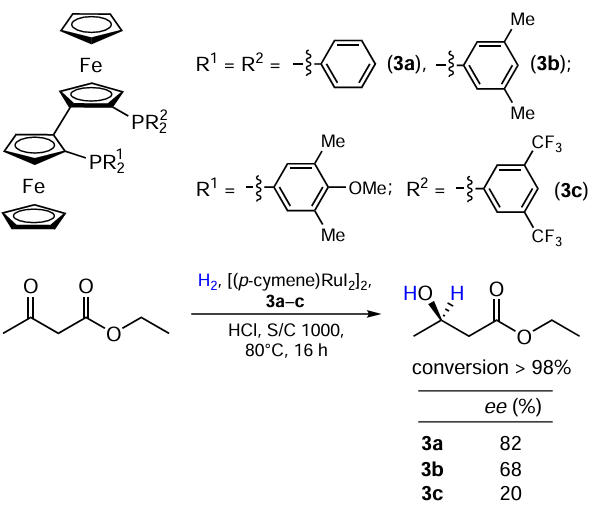
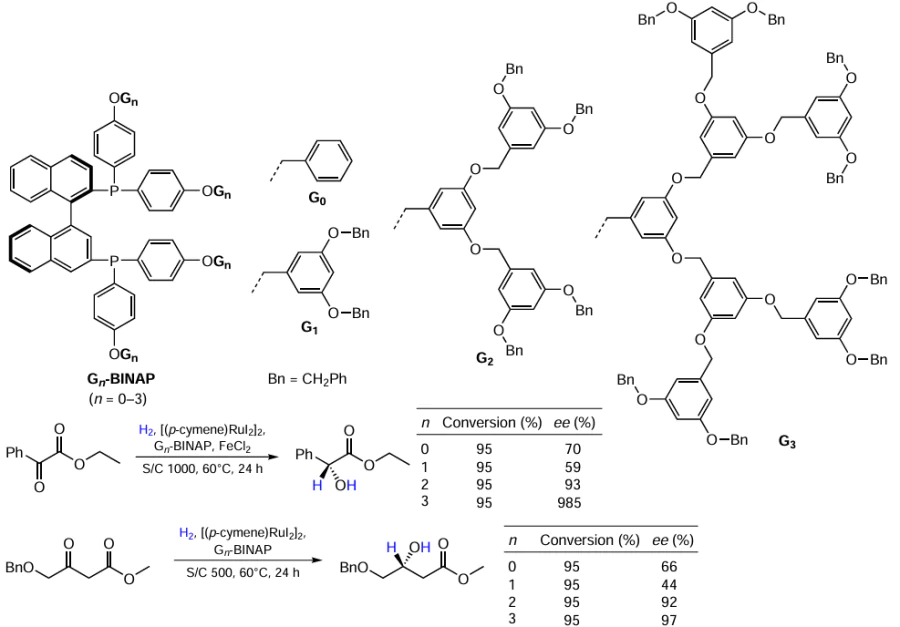
Ma et al.[31] developed a synthetic approach to dendritic BINAP ligands, Gn-BINAP. The corresponding [Ru(Gn-BINAP)] complexes obtained in situ from [(p-cymene)RuI2]2, provided highly enantioselective hydrogenation of α- and β-ketoesters, where the second- and third generation catalysts (G2, G3) performed best (Scheme 7).
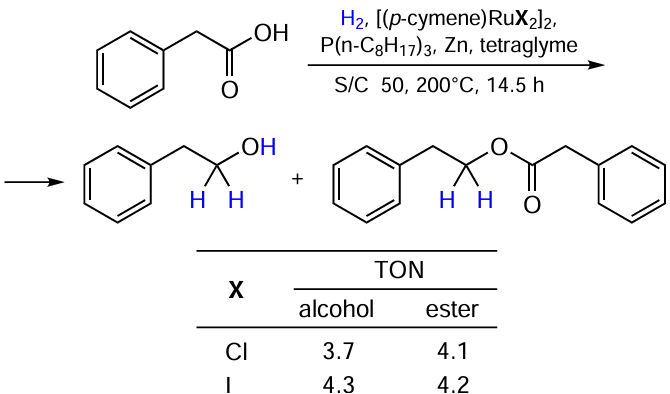
Ruthenium-halide complexes can also be applied to the hydrogenation of acids to alcohols (Scheme 8).[32] Chloride and iodide Ru complexes showed similar moderate activity.
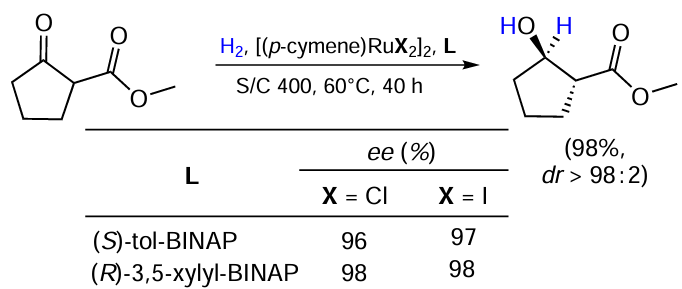
Madduri and Minnaard[29] compared the performance of catalytic systems based on [(p-cymene)RuHal2]2 complexes (Hal = Cl, I) with (S)-Tol-BINAP and (R)-3,5-xylyl-BINAP ligands in the hydrogenation of cyclic b-ketoester (Scheme 9). All the halogen–ligand combinations tested demonstrated similar efficiency. As compared to the above-mentioned method by Benincori et al.[21] (see Scheme 5), this protocol allows a slightly better diastereoselectivity, though at a higher catalyst loading.
In other reported examples of the С=О bond hydrogenation in the presence of the Ru – I catalysts bearing phosphine ligands, the yields and enantioselectivities were as high as in the examples considered above.[25] [33-36]
Slade et al.[37] obtained complexes with inositol-derived ligands and applied them to the ketone reduction. These catalysts provided rather poor enantioselectivities. The use of iodine complexes led to a slightly higher conversion (Scheme 10).
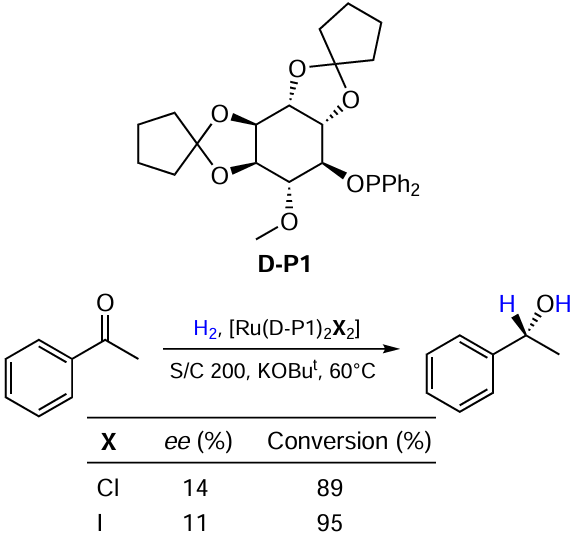
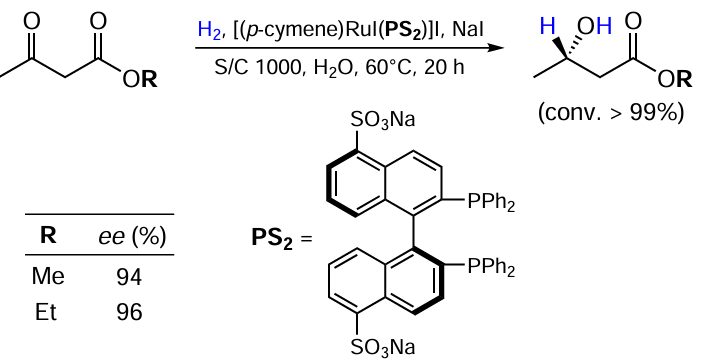
Carrying out reactions in aqueous medium is becoming increasingly important from a green chemistry perspective. Li and co-workers [38] successfully developed such a method for the reduction of ketoesters using sodium 5,5'-disulfonato (S)-BINAP ligand (Scheme 11). The process gives excellent yields and enantioselectivity. It should be noted that the water-soluble catalyst exhibited a very high catalytic stability in H2O. It was found that the reaction is promoted by the addition of sodium iodide in 100-fold excess with respect to Ru.
Thus, in most cases of the C=O bond hydrogenation there is little effect of halogen nature on catalyst activity and selectivity. In the one case [19] when such an effect is significant, is the reaction outcome strongly, up to complete reversion, is affected by the nature of other ligands. The choice between Ru – Cl and Ru – I complexes is largely due to their relative availability.
2.2. Transfer hydrogenation of ketones
Transfer hydrogenation is another approach to the reduction of ketones that is widely represented in the literature. Such reactions are usually carried out in isopropanol used as the hydrogen source in the presence of a strong base at 60 – 80°C.
In 2008, Zeng and Yu [39] developed NNC-pincer Ru complexes 4a – c with pyrazolyl N-heterocyclic carbene ligands and investigated their activity in transfer hydrogenation of ketones (Scheme 12). Complexes 4b,c were found to be superior to their analogue 4a. Catalysis with 4b afforded aromatic alcohols in 83 – 99% yields. The exception was o-bromoacetophenone, which give only 16% of the target product. Aliphatic ketones (cyclopentanone, cyclohexanone, n-hexyl methyl ketone) were also successfully reduced with excellent yields (95 – 100%) in 1.5 – 5 hours.
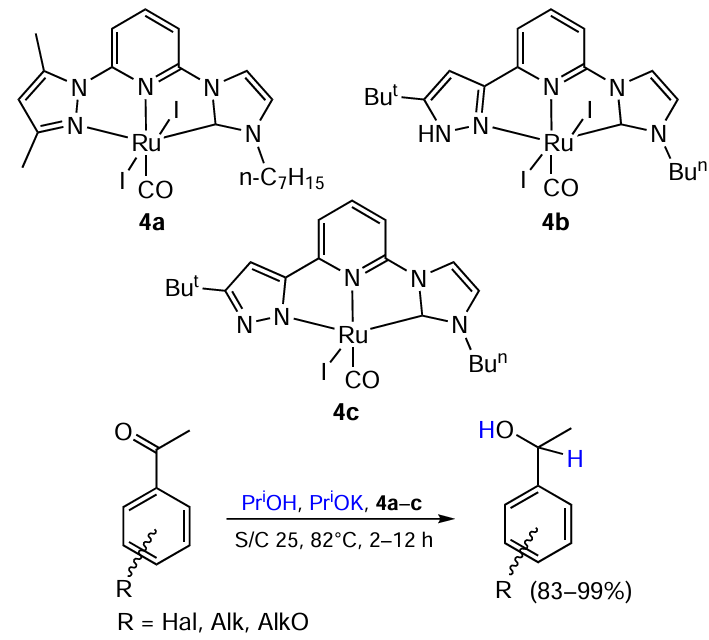
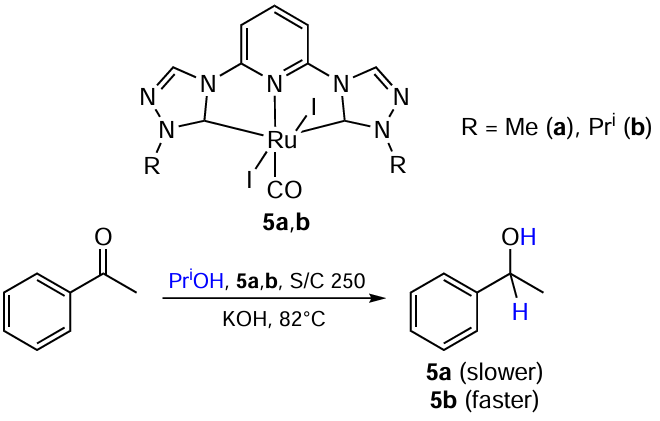
In 2011, Li et al.[40] explored CNC-pincer bis-carbene Ru complexes 5a,b with triazolyl ligands and compared their performance in the transfer hydrogenation of acetophenone (Scheme 13). It was found that the reaction runs faster when catalyzed by a complex with a bulkier ligand (5b). The authors suggested that this is due to the coordination saturation of the complexes, which is the reason for the reaction to proceed via a dissociative mechanism: the introduction of the bulky isopropyl group lengthens the metal–ligand distances (according to X-ray diffraction analysis), thus facilitating the ligand removal.
Remarkably, Naziruddin et al.,[41] who studied the catalytical properties of the closely related CCC-pincer bis-carbene Ru complexes 6a,b, observed the opposite tendency: the less hindered complex 6b proved to be more active than 6a (Scheme 14).
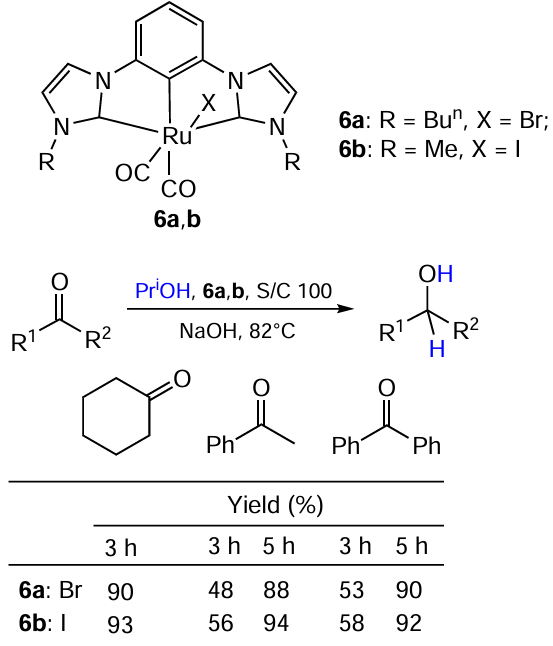
It was found that the reaction runs faster in the presence of the catalyst 6b, especially in the case of acetophenone. This can be related to the fact that there is a CO ligand in the trans position to the halogen: the strong trans-effect of iodine facilitates the CO dissociation from the complex better than the analogous effect of bromine despite the fact that the latter is enhanced by the steric effect of Bun group.
Samuelson and co-workers [42] used cyclometallated Ru half-sandwich complexes 7a – g in the transfer hydrogenation of aromatic ketones (Scheme 15). An iodine complex (7g) was less active than others, while there was no noticeable distinction between compounds 7a – f. This fact, along with results of the recyclability test, led the authors to conclusion that the complexes 7a – f are precatalysts, which are transformed into catalytically active nanoparticles, which in turn decompose in the course of the reaction. This was confirmed by the kinetic profile of the reaction (with an obvious induction period), the rapid colour change of the reaction mixture, the poisoning of the catalyst by phenanthroline, the reduction of aromatic nitro groups under the reaction conditions, and the drastic decrease in catalyst performance when reused. The presence of nanoparticles was confirmed by transmission electron microscopy (TEM) and dynamic light scattering (DLS). The lower activity of iodine complex 7g could be related to its low solubility.
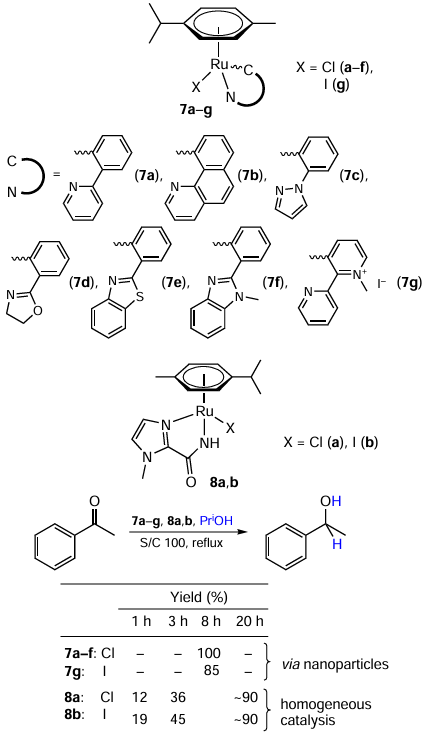
At the same time, similar complexes 8a – b studied by Citta et al.[43] most likely work in a homogeneous mode (see Scheme 15). Reasons for this idea are the retention of aromatic nitro group under the reaction conditions and a rather limited influence of phenanthroline addition on the catalyst’s performance (which would be poisoned if it were heterogeneous). It was found that the iodide catalyst performs better than the chloride one, especially at low reaction times. A plausible explanation is that it is due to the better leaving ability of iodide.[44][45]
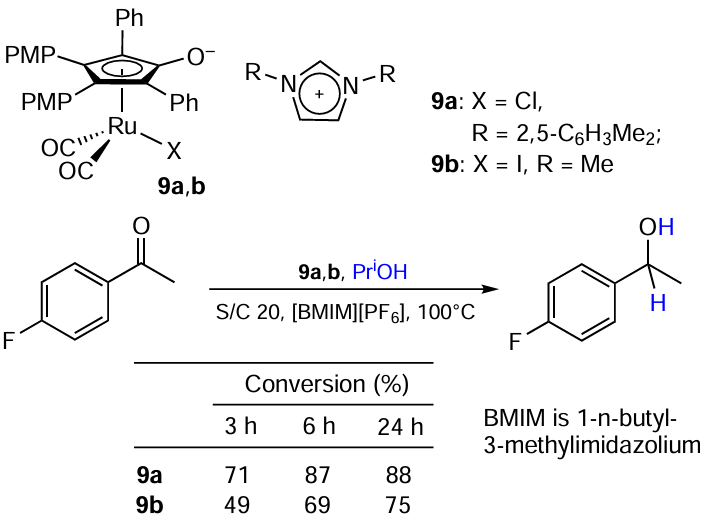
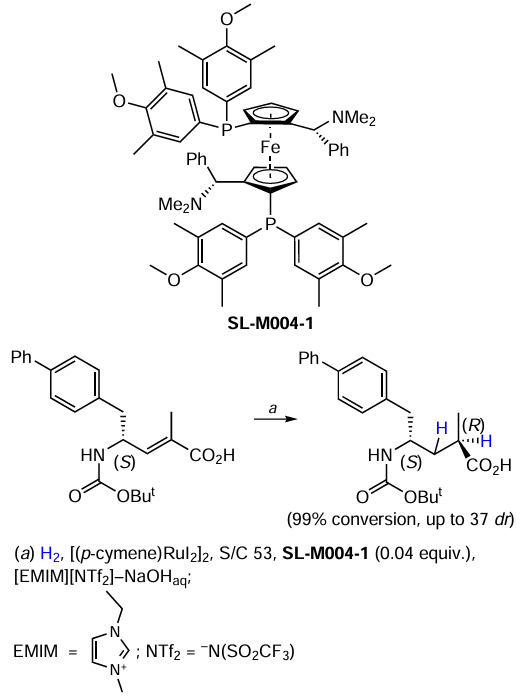
Cesari et al.[46] studied transfer hydrogenation of ketones in imidazolium ionic liquids (ILs) (Scheme 16). Interestingly, the neutral Ru complex shows no catalytic activity even in the IL media, whereas ion pairs 9a,b composed of the anionic Ru complex and imidazolium cation act as a pre-catalyst. The authors found that the steric bulk of a cation and the anion nature do not affect the performance of a catalyst. Nevertheless, it can be noted that the chloride catalyst performs slightly better.
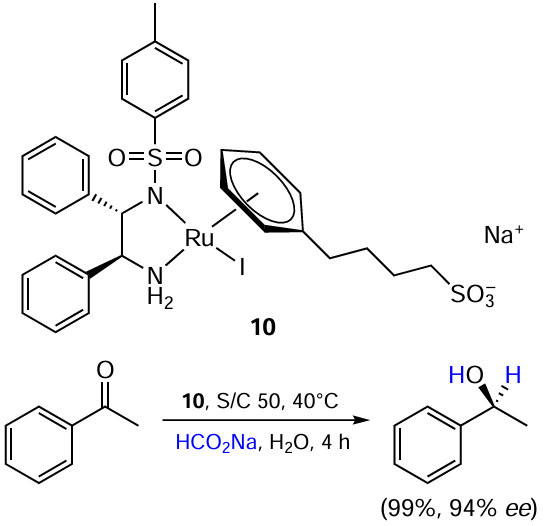
Virboul et al.[47] developed a synthetic approach to a water-soluble sulfonate-appended ruthenium(II) η6-arene complex 10 and studied its application in the asymmetric transfer hydrogenation of acetophenone in aqueous medium using sodium formate as the hydrogen donor (Scheme 17). Complex 10 proved to be an effective catalyst and afforded (S)-phenylethanol in nearly quantitative yield and 94% ee. The choice of the complex with an iodide ligand was dictated by its easier availability compared to its chloride analogue.
To conclude, the rate of dissociation of a ligand from the catalytic complex is important. Replacement of chloride with iodide improves the catalytic activity if it accelerates the elimination of a ligand.
2.3. Miscellaneous reactions of ketone reduction
Plietker and co-workers [48] reported a remarkable example of a selectivity switch in deuteration of polyfunctional organic molecules. Depending on the additive in choice (CuI, KOH, Zn powder), the precatalyst [RuCl2(PPh3)3] is converted into complexes 11a – c, which behave differently in the H – D exchange reactions: 11a catalyzes the reduction of alkynes, 11b catalyzes the reduction of carbonyl groups; 11a works in both С(sp3)- and С(sp2) – Н activation, whereas 11c was active in С(sp2) – Н activation only (Scheme 18).
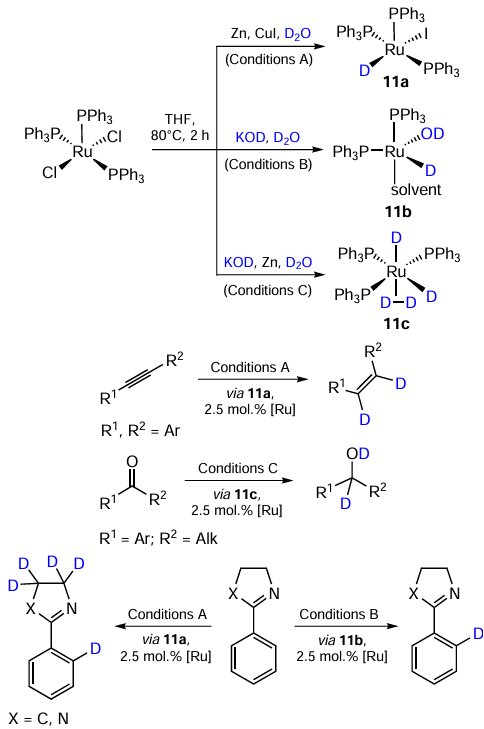
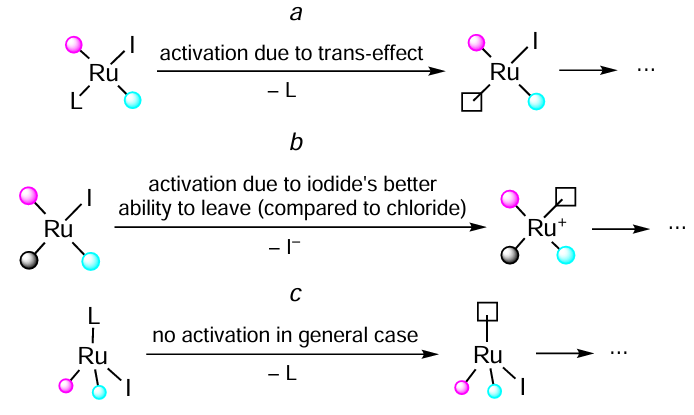
From the above data it can be seen that it is a ligand dissociation step that often determines the reaction rate. In such case, replacing chlorine with iodine can improve the catalyst activity by weakening the metal – ligand bond. Such a weakening can result from a stronger iodine trans-effect (Scheme 19, reaction a), or from the fact that iodide ion itself is a better leaving group than chloride one (see Scheme 19, reaction b). This effect is clearly observable for the transfer hydrogenation of carbonyl compounds. At the same time, for the catalytic hydrogenation of carbonyl compounds, the halogen effect appears to be less considerable, leading to insignificant and/or non-monotonous relationship between the halogen ligand nature and catalysts activity (see Scheme 19, reaction c).
3. Reduction of multiple carbon – carbon bonds
The reduction of multiple C – C bonds in the presence of Ru – I-containing catalysts are usually the reactions of hydrogenation. The majority of publications focus on achieving high stereoselectivity of the process. Activated (i.e. conjugated with electron-withdrawing groups (EWGs)) alkenes are the most common substrates for such reactions. In addition to alkene hydrogenation, alkyne hydrogenation to E-alkenes and alkenes reduction with formate have been reported.
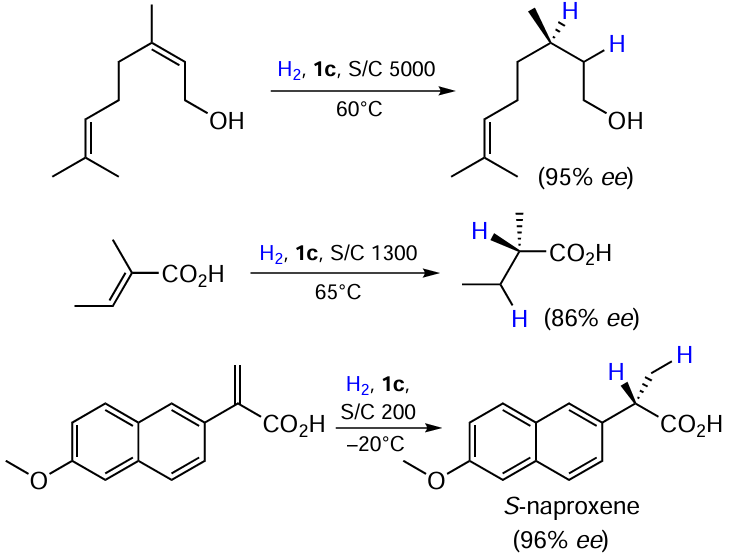
Sometimes the hydrogenation of C=C bonds has been investigated in parallel to C=O reduction under similar conditions, e.g., in the cited publications.[16-18] [25] [33-35] For example, geraniol and α,β-unsaturated acids were hydrogenated in high yields and with good selectivity using complex 1c (see Scheme 2, Scheme 20).
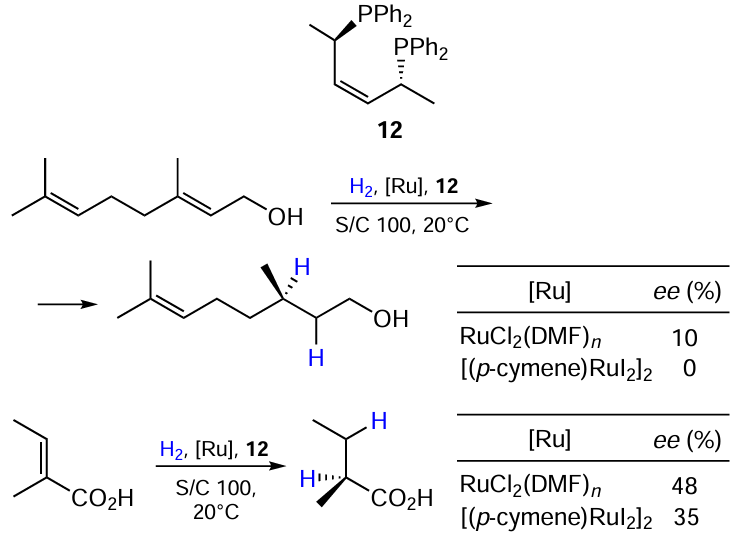
Cesarotti et al.[33] carried out asymmetric hydrogenation of C=C double bonds catalyzed by chloride- and iodide-containing Ru(II) catalysts with a chiral diphosphine ligand 12 and compared their activity (Scheme 21). The authors noted that the chloride-containing complex RuCl2(DMF)n clearly shows better enantioselectivity than the iodide-containing [(p-cymene)RuI2]2, although did not offer any explanation for this.
Yoshikawa et al.[49] showed that replacing the phosphine substituents in the ligand complex with more donor moieties (e.g., PPh2 to PCy2) in the asymmetric hydrogenation of a,b-unsaturated carboxylic acids significantly decreases the yield (Scheme 22). A similar trend was also observed in the reduction of the C=O bond.[34]
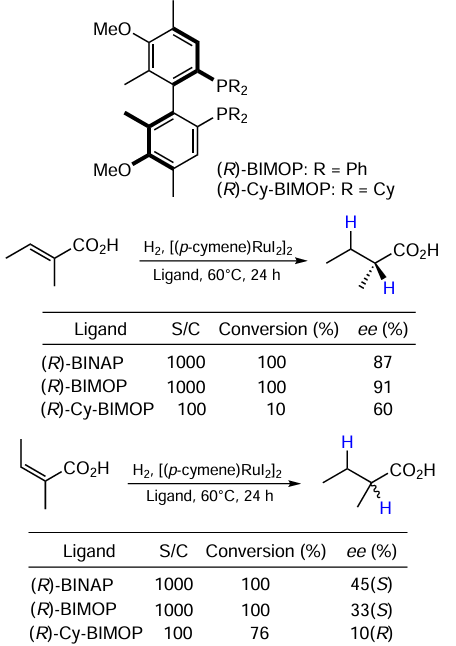
Other research in this direction has been performed by Benincori et al.[50][51] Their group developed bis(diphenylphosphino) heteroaryl ligands and successfully used them in the total synthesis of (–)-(S,S)-7-hydroxycalamenal.
Piscopo et al.[52] developed a protocol for the diastereoselective hydrogenation of an activated alkene under phase transfer catalysis conditions (Scheme 23). A convenient procedure for isolating the target product appears to be a considerable advantage of the method; the product enters the water phase (in the form of sodium salt) and can be precipitated on acidification, while the catalyst remains in the organic layer.
Ohta et al.[53] carried out the hydrogenation of a formally inactivated double bond in 1,1-disubstituted alkenes using BINAP-Ru(II) catalysts (Scheme 24). Under these conditions, two concurrent processes were observed: reduction and migration of the double bond to the ring.
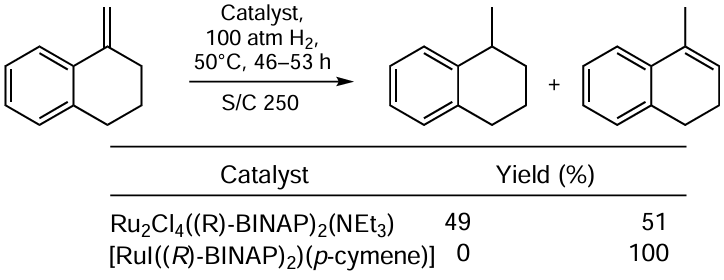
The Ru – I complex provided selective isomerization, whereas Ru – Cl complex gave rise to a mixture of the products in almost equal proportions. The authors note that the isomerization is not due to the protic species that could form in the process but involves metal hydride species.
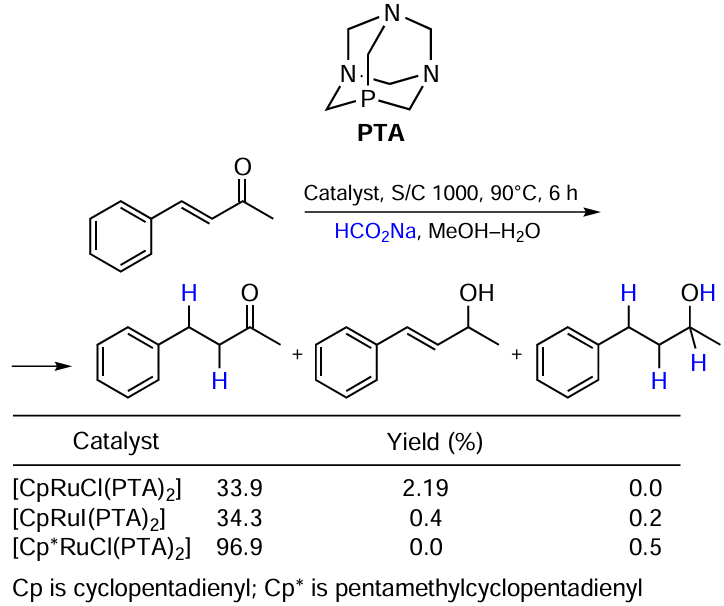
Bolaño et al.[54] carried out the transfer hydrogenation of activated alkenes using half-sandwich Ru-cyclopentadienyl complexes containing the cage-like water soluble monodentate phosphine 1,3,5-triaza-7-phosphaadamantane (PTA) (Scheme 25). The model reaction with benzylideneacetone showed that the replacement of chloride with iodide in Cp-based complexes [СpRuHal(PTA)2] had no significant effect on the hydrogenation outcome, although a slightly better selectivity to ketone was observed in the case of iodide-containing catalyst [СpRuI(PTA)2]. At the same time, the use of the [Сp*RuCl(PTA)2] catalyst changed the situation radically providing the highest selectivity to ketone. However, an attempt to use it for cinnamaldehyde hydrogenation resulted in the unselective reduction of C=O and C=C bonds. Unfortunately, the authors did not obtain and test a similar catalyst [Сp*RuI(PTA)2].
Yadav et al.[55] described the method for the transfer hydrogenation of both internal and terminal alkynes to alkenes using complex 13 containing an annulated p-conjugated imidazo-naphthyridine-based mesoionic carbene ligand (Scheme 26). A remarkable feature of this protocol is that it yields E-alkenes from internal alkynes, contrary to the usual stereoselectivity of catalytical hydrogenation methods and tolerates a number of reducible functional groups. The isomerization of Z-alkenes to their E-isomers was also observed under the reaction conditions. Based on this observation and the data on reaction kinetics, the authors supposed there are two catalytical cycles in the system: first, the starting material is reduced to Z-alkene. The latter isomerizes to the thermodynamically stable E-product via hydrogenation – rotation about the C – C bond — dehydrogenation sequence.
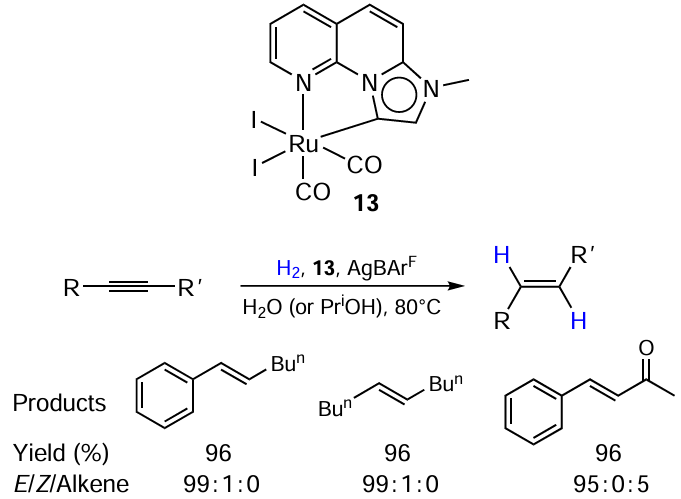
Thus, most reported applications of the catalysts with Ru – I bond to the reduction of carbon – carbon bonds are the protocols for alkene reduction. In addition, the pool of substrates is in most cases restricted to alkenes bearing strong electron-withdrawing groups. In the case of unactivated substrates, even a double bond as labile as exomethylene one is prone to simultaneous isomerization, and the catalyst bearing an iodine ligand shifts the selectivity completely towards the isomerization.
4. Reduction of heterocycles and Schiff bases
All the work mentioned in this Section is by the Zhou’s group. Technically, the reduction looks like hydrogenation in these works, but actually it is realized through the hydrogen borrowing from an organic reducing agent followed by its regeneration via hydrogenation.
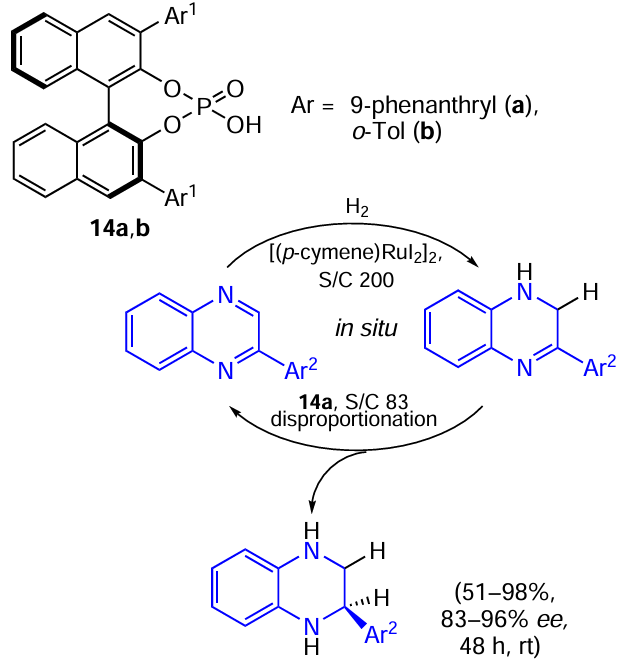
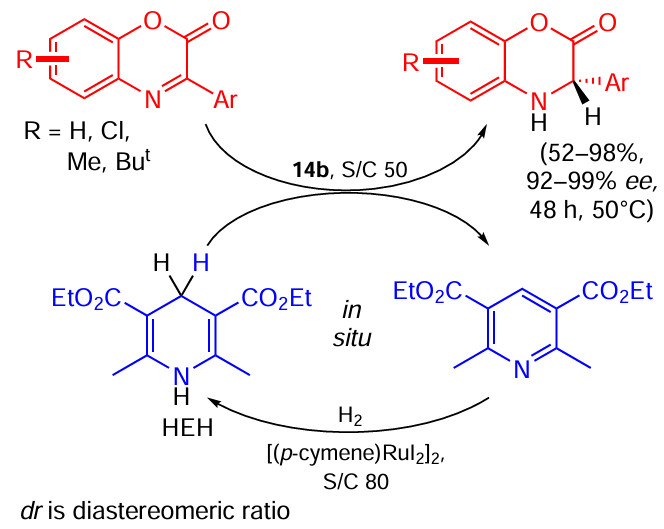
The first publication on this subject described the highly enantioselective reduction of quinoxalines using a metal/Brønsted acid relay catalysis system.[56] It was found that the process proceeds in two steps: at first, the starting material is hydrogenated to dihydroquinoxaline, which in turn disproportionates to the starting material and the target tetrahydroquinoxaline in the presence of a Ru catalyst with ligand 14a (a chiral Brønsted acid) (Scheme 27). The similar idea was then successfully applied to the reduction of benzoxazinones using the Hantzsch ester (HEH) as the hydrogen carrier and a chiral catalyst with ligand 14b (Scheme 28).[57]
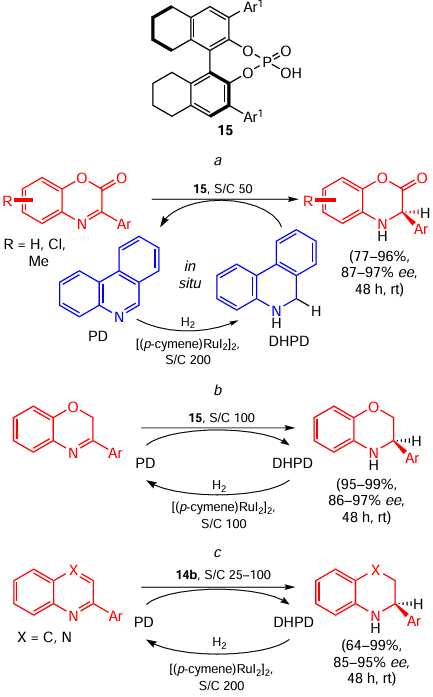
The next step in this work was to change the Hantzsch ester for phenanthridine (PD) as a hydrogen-transferring shuttle (Scheme 29, reactions a – c).[58] Such a replacement allowed for a decrease in the reaction temperature and broadened the set of suitable substrates improving functional group tolerance. Noteworthy, this also led to the inversion of the enantioselectivity of the process.
Other reported reactions implemented in the same nicotinamide adenine dinucleotidephosphate NADP(H)-mimetic fashion include the synthesis of tetrahydroquinolines [59] and the reduction of cyclic Schiff bases.[60]
The reactions described in this Section can be generalized by the Scheme 30. A hydrogen carrier reduces a substrate in the presence of a chiral acid catalyst leading to a chiral product (see Scheme 30, reaction a). The oxidized form of the carrier is then reduced by hydrogen. However, there is another way to get the similar product, that is, to use an achiral acid as a catalyst for hydrogenation, together with a chiral hydrogen carrier (see Scheme 30, reaction b). Implementations of this model are the subject of the rest of this Section.
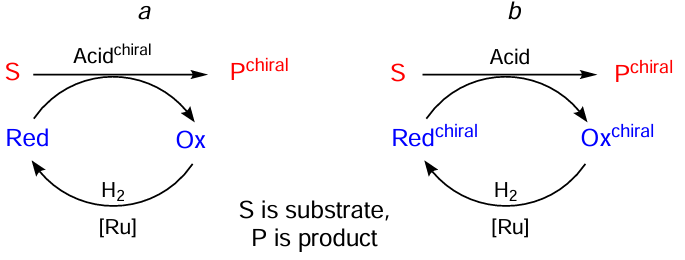
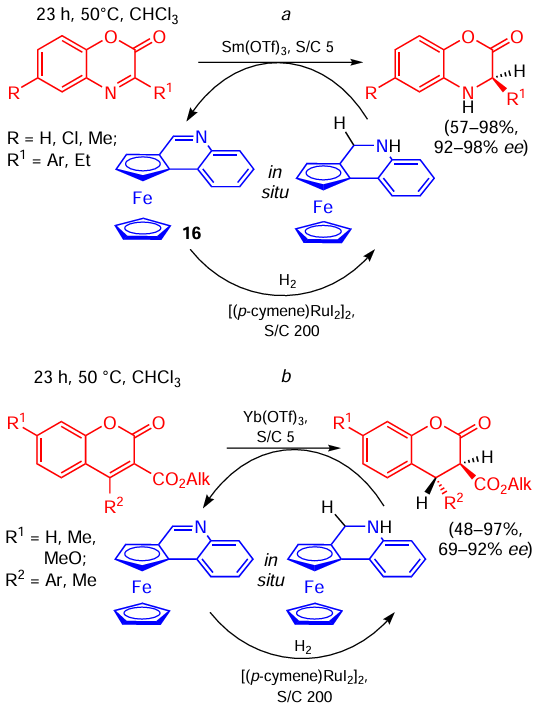
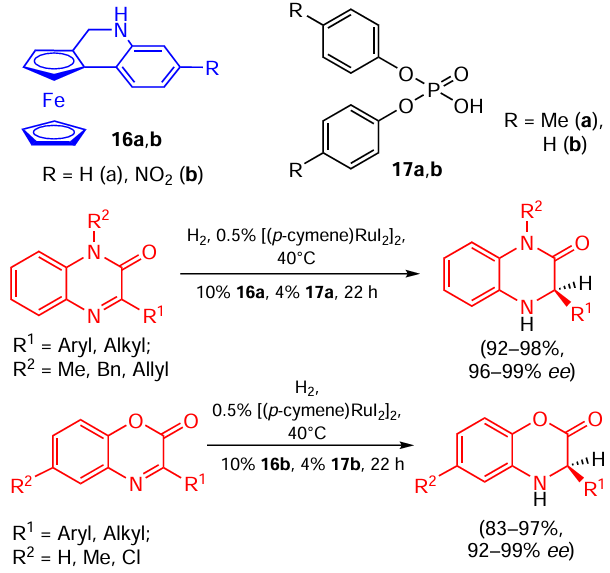
For example, the use of a chiral derivative of ferrocene 16 with a readily available Lewis acid as an achiral transfer catalyst allowed for the asymmetric reduction of benzoxazinones and coumarins under mild conditions in good yields and high enantioselectivity (Scheme 31).[61] The reaction performance, however, degraded when there was an aliphatic substituent (e.g., R1 = Et, reaction a, R2 = Me, reaction b, yields 57 and 48, respectively) in the heterocycle. For benzoxazinones this issue was later solved by using protic acids such as hydrophosphoryl compounds 17a,b instead of Lewis acids (Scheme 32).[62] Later, similar protocols were developed for the reduction of quinazolinones,[63] quinolines,[64] indenones [65] and for the reductive alkylation of flavonoids.[66]
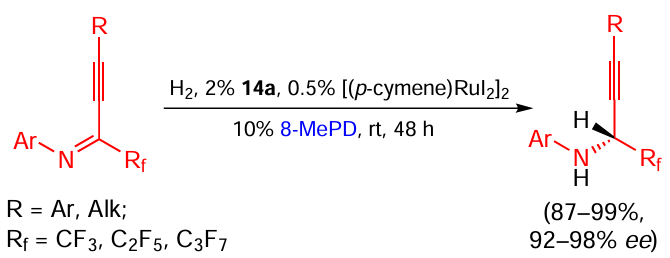
An example of the use of a protocol of this kind for the reduction of an acyclic compound is the asymmetric reduction of triple bond-conjugated fluorinated Schiff bases (Scheme 33).[67] The triple bond remains intact; however, the reaction is quite sensitive to the structure of a substrate: for example, the replacement of a perfluoroalkyl group with a non-fluorinated ester functionality leads to a drastic decrease both in the reaction yield and stereoselectivity (R = CO2Et, Ar = 4-MeOC6H4, yield 26%, 36% ee).
To sum up, there is a series of papers dedicated to the transfer hydrogenation of heterocycles, that is of great interest as an example of mimicking the biochemical processes (i.e., NAD- and FAD-mediated reactions). However, the role of ruthenium catalysts here is auxiliary and comes down just to the regeneration of the reduced form of a carrier molecule.
5. Reductive C – N coupling
The processes of this class described in the literature can be divided into several groups. The most straightforward one is represented by the Schiff base hydrogenation. The methylation of aldehydes using formaldehyde as both C1-synthone and reducing agent is the second group. Reductive amination without an external hydrogen source falls into the third one. In addition, the catalytic reactions of C – N amine-alcohol coupling, although not technically redox transformations, proceed via redox steps and are therefore also discussed in this Section.
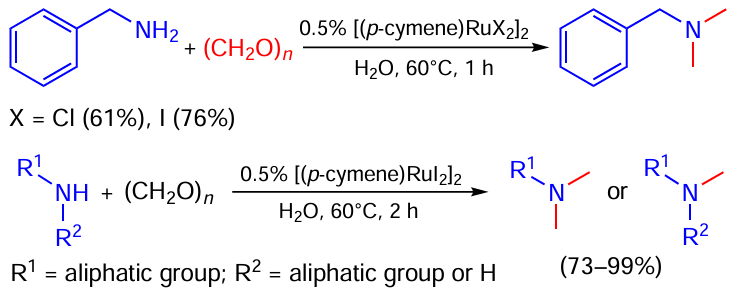
Van der Waals et al.[68] developed a method for amine methylation using paraform in aqueous media (Scheme 34). The iodide complex of ruthenium was found to provide slightly better yields than its chloride analogue. Both primary and secondary (including cyclic) amines perform well in the reaction.
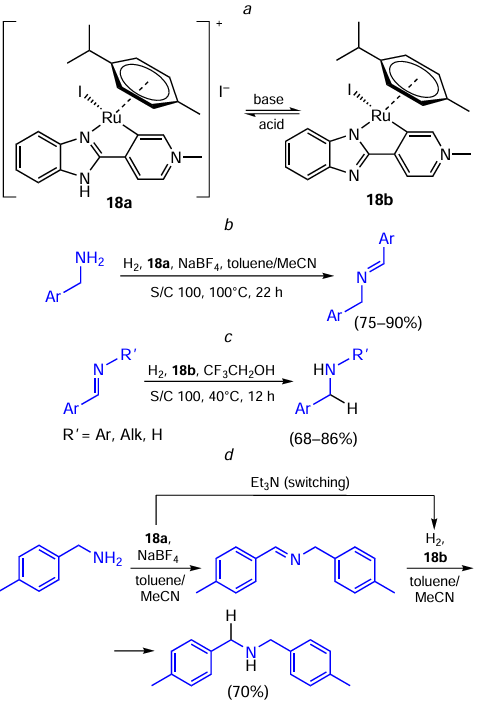
Semwal and Choudhury [69] found a catalyst that is a Ru(II)-based molecular switch existing in two forms 18a,b (Scheme 35, reaction a) and can catalyze either the dehydrogenative coupling of two molecules of amine to furnish the Schiff base (see Scheme 35, reaction b) or the Schiff base hydrogenation (see Scheme 35, reaction c). The reaction b proceeds much better in the presence of the Ru-imino state 18a, whereas the reaction c is catalyzed by the Ru-amido state 18b. Interestingly, the reaction с can be carried out equally well either with 18b or with the same loading of 18a by adding triethylamine in the reaction. Similarly, the catalytic hydrogenation of a Schiff base (see Scheme 35, reaction c) was catalyzed either by 18b, or by 18a in the presence of trifluoroacetic acid. The 18a ↔ 18b transformations upon base/acid addition were confirmed by UV-VIS-spectroscopy. The authors also showed an example of an assisted tandem catalysis and applied this system for a two-step one-pot synthesis of symmetric amines. First, benzylamine was converted to the corresponding Schiff base using 18a. Then, in the presence of 18b and when adding triethylamine, hydrogenation gave the desired amine product in good yield (see Scheme 35, reaction d ).
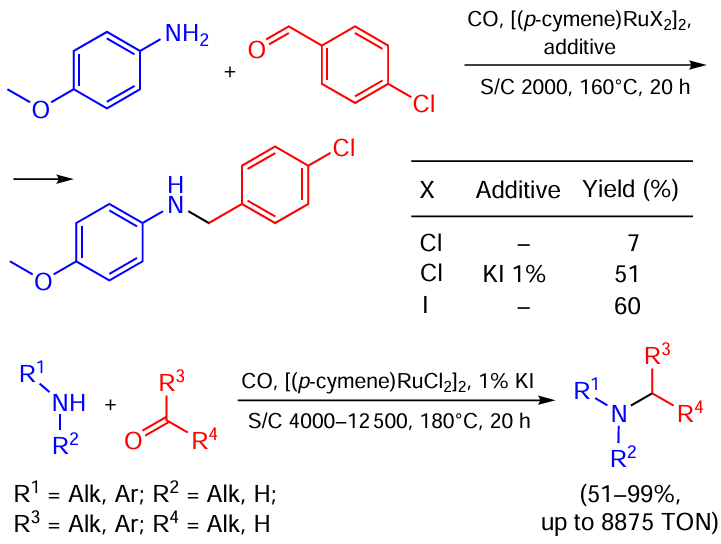
Chusov and co-workers [70] demonstrated the possibility to boost the catalytic activity by replacing chloride with iodide in the structure of a Ru catalyst for the reductive amination without an external hydrogen source (Scheme 36). The iodide catalyst turned out to be up to 15 times more active than its chloride counterpart. The corresponding increase in the yield (as compared to [(p-cymene)RuCl2]2) was observed for both [(p-cymene)RuI2]2 and [(p-cymene)RuCl2]2+KI systems (the latter implying the in situ generation of Ru – I species). However, for a few substrates [(p-cymene)RuI2]2 proved more efficient than [(p-cymene)RuCl2]2 + KI; this fact can be attributed to the possible side reactions induced by the excess of free iodide ions. The authors suppose [Ru(CO)3X3]– (X = Cl, I; the particle detected by ESI-MS) to be the real catalyst. The increased activity of the iodide complex can be explained by faster iodide substitution via a dissociative mechanism.
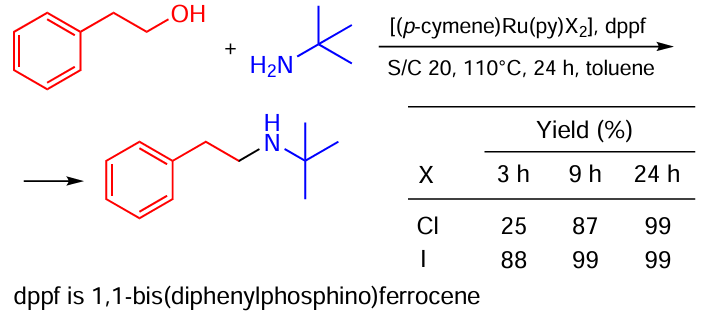
McGowan and co-workers,[71] studying hydrogen-borrowing alcohol – amine coupling, observed another example of the catalyst activation triggered by the swapping chlorine to iodine. A monomeric complex with iodine provided 88% reaction conversion in 3 h, while the same complex with chloride took 9 h to achieve the same result (Scheme 37).
Quite the opposite effect was reported for a fairly similar reaction by Rigo and co-workers [72] (Scheme 38). The Ru complex with X = Cl gave the desired product quantitatively, whereas its iodide analogue was almost completely inactive. We can propose the nature of a solvent as a reason for such an inversion: methanol solvates anions better than toluene, thus mitigating the leaving group effect and giving way to some other factors that determine the activity of a catalyst.

From the above data we conclude that replacing chloride with iodide in the catalyst molecule usually increases its activity in C – N coupling reactions. This conclusion is consistent to our view of dissociative mechanism for substitution in Ru complexes, although there are exceptions due to the variety of possible reaction pathways.
6. Oxidation reactions
The oxidation reactions carried out with Ru – I catalysts are the reactions of C – O bond dehydrogenation. They can be divided into two groups:
1) Dehydrogenation reactions in which hydrogen leaves as a molecule of H2;
2) Dehydrogenation reactions using a molecular oxidant to abstract hydrogen.
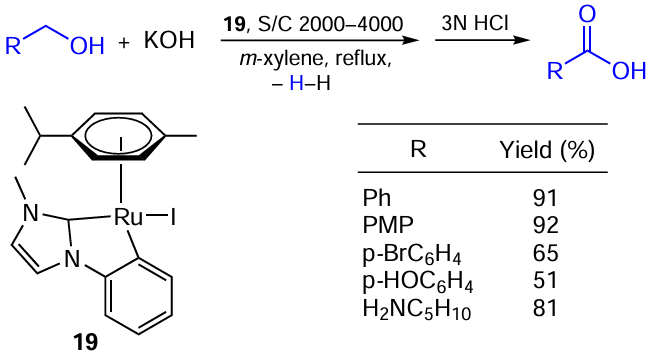
An example of a process of the first type has been reported by Verpoort and co-workers [73] [74]and concerned the dehydrogenative coupling of primary alcohols and hydroxide to afford carboxylic acids (Scheme 39). The NHC-based Ru – I catalyst 19 demonstrated remarkable efficiency with TON of up to 32800 together with the lowest loadings (250 ppm) ever reported for this reaction. Another advantage of this catalyst is that it allows carrying out dehydrogenation in air and under neat conditions.
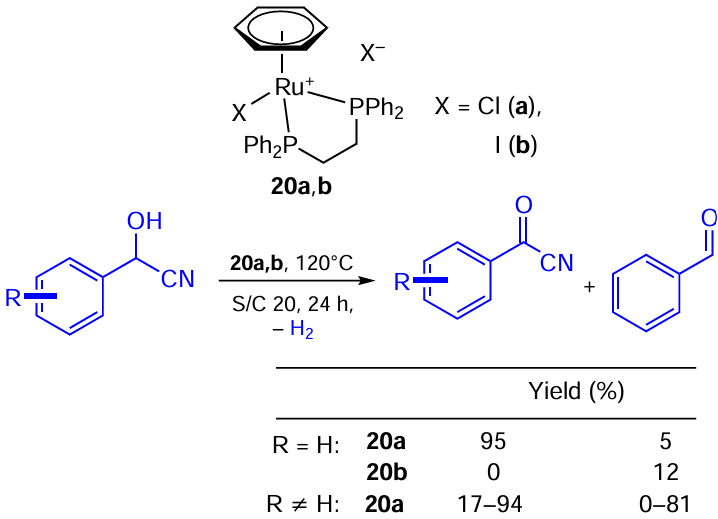
Another example of the C – O dehydrogenation is the acceptorless and base-free dehydrogenation of cyanohydrins developed by Hong and co-workers (Scheme 40).[75] Decyanation affording aldehydes was a notable side reaction in the process. A Ru – Cl bidentate phosphine complex performed better in this case, although the presumable mechanism includes halogenide substitution by alkoxide as the first step. It was found that the substrate scope is limited to aromatic compounds, and the performance of the catalytic system is very substrate-dependent.
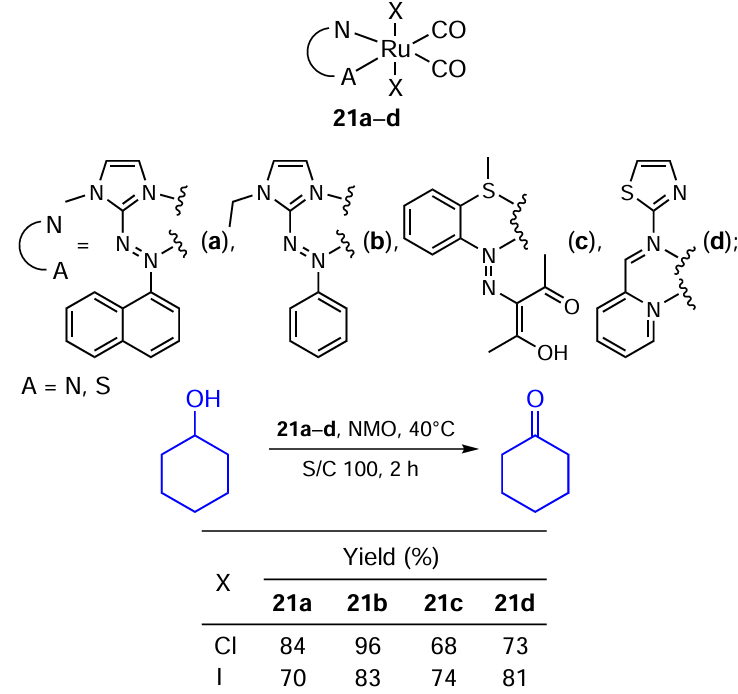
The catalytic oxidation of primary and secondary alcohols using N,N(S)-bidentate Ru(II)-halide-carbonyl complexes and N-methylmorpholine N-oxide (NMO) as the co-oxidant was explored by Sinha, Mondal and co-workers (Scheme 41).[13] [76-79] Electrochemical parameters of Ru complexes 21a – d, their frontier orbitals, and catalytic activity was studied. Among the imidazole-type complexes 21a,b, those containing the Ru – Cl bond show higher activity, while in the case of compounds 21c,d, the Ru – I complexes performed better. According to IR spectroscopy, the reaction proceeds via oxoruthenium species. The authors argue that the higher activity of the iodide complexes 21c,d may be due to their lower oxidation potential (measured by cyclic voltammetry (CVA)). The decrease in the activity for iodide complexes 21a,b is explained by a probable partial consumption of NMO for the oxidation of iodide.
An application of ruthenium iodide complexes for electrochemical oxidation has been developed by McElwee – White and co-workers.[80] Nafion films with Ru complexes were adsorbed onto glass carbon electrodes, which could then be used for oxidizing ethanol to acetaldehyde or acetic acid.
To sum up, the Ru – Hal-catalyzed alcohol dehydrogenation and intermolecular oxidation have been reported. For the latter, a comparison between Ru – Cl and Ru – I bond containing complexes led to the conclusion that the catalyst efficiency depends on the tradeoff between the ruthenium center redox lability (higher in the case of Ru – I) and the redox stability of the ligand environment (higher in the case of Ru – Cl).
7. Metathesis reactions
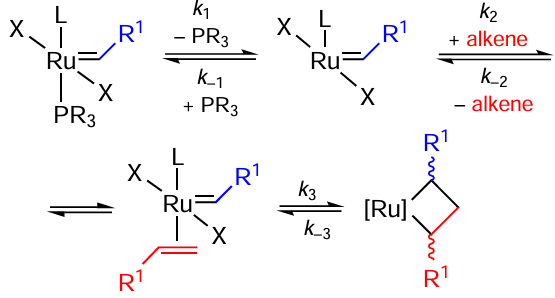
The reactions of metathesis draw thorough attention, especially in the context of the synthesis of polymers and biologically active compounds.[81-89] Ru – I bond containing catalysts have been used for metathesis reactions. The influence of halogen nature in general case is discussed by Grubbs and co-workers.[90] They proposed the metathesis pathway as depicted in Scheme 42 and argued that swapping X = Cl for X = I facilitates the reaction initiation as a phosphine ligand leaves the complex with iodide faster, perhaps due to the steric reasons. Nevertheless, the overall reaction is no faster or even slower with iodide complexes [91] as the k–1/k2 ratio increases 100 times when chlorine is replaced with iodine. The reason for this increase is unclear; however, the fact that cis-trans isomerization of halogens in the complex, required for alkene coordination, is hampered in Ru – I complexes due to the steric reason is cited as a feasible explanation.
The metathesis reactions described in the literature can be classified as follows:
1) Ring-opening cross-metathesis (ROCM) reactions;
2) Ring-opening metathesis polymerization (ROMP) reactions;
3) Ring closure metathesis (RCM) reactions.
The works dealing with these reactions are aimed at the improvement in reaction selectivity (e.g., RCM vs polymerization). Some publications on RCM report enantioselective protocols. A review of these research is given below.
7.1. Ring-opening cross-metathesis reactions
The activity of Ru – I bond containing complexes in this class of reactions has been studied by Hoveyda and co-workers.[92-95] The authors developed catalytic systems, provided an example of their application in total synthesis of Baconipyrone C and investigated the mechanism of the rearrangement of stereoisomers of Ru complexes with chirality on the metal atom. A comparison between analogous Ru – Cl and Ru – I complexes 22a,b in enantioselective ring-opening metathesis/cross-metathesis is given in Scheme 43. It is clear that although both 22a and 22b are suitable catalysts for the reaction but Ru – I complex 22b allows higher yields and enantioselectivities. The improvement in enantioselectivity can be explained by the higher steric hindrance of the complex with iodide; an increase in the yield may be due to the higher stability of the complex. The authors note that both Ru – Cl and Ru – I complexes generated in situ are as effective as pre-prepared ones in this reaction. Noteworthy, 22b is stable under air and column chromatography conditions, whereas 22a, its Ru – Cl counterpart, is not.
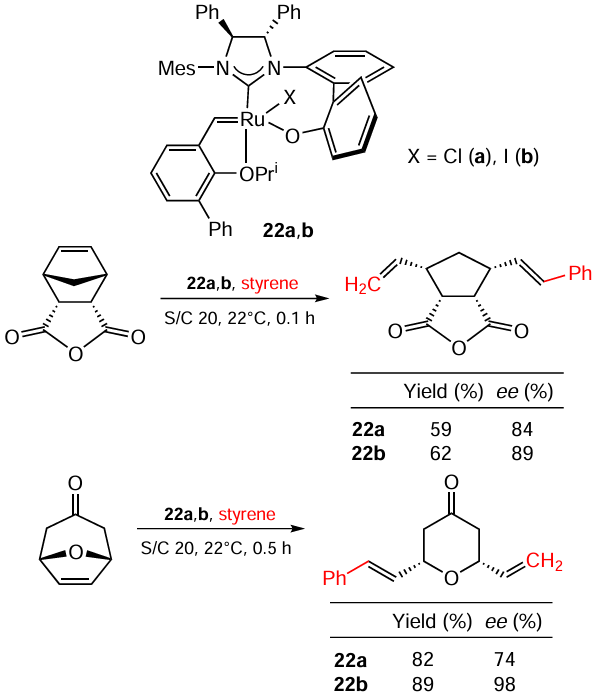
7.2. Ring-opening metathesis polymerization reactions
An early example of ROMP transformation was described by Slugovc and co-workers [96] (Scheme 44). It can be seen that the Ru – Cl catalyst initiates the polymerization of a cyclic alkene at room temperature while its Ru – I counterpart does not provide full conversion even when heated to 80°C for 19 h. The lower value of the number average molecular weight (Mn) implies the lower rate of the initiation in the case of the iodide complex.
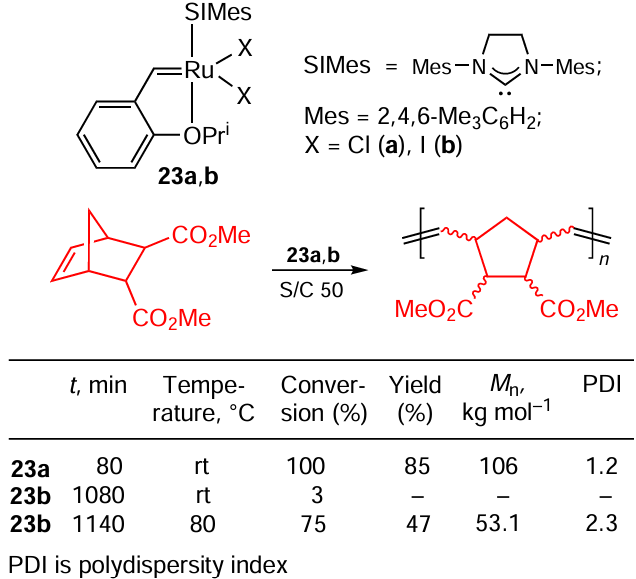
This is consistent with the idea of the higher activity of Ru – Cl complexes due to a higher k2/k–1 (see Scheme 42) ratio mentioned above (see the beginning of Section 7). In the case of polymerization, it is convenient to mix all the reactants and the catalyst under conditions where the reaction does not proceed, homogenize the mixture effectively, and then activate the catalyst by changing the conditions. Scheme 45 shows that this can be achieved using a Ru – I catalyst instead of its Ru – Cl analogue and thermal activation. This approach of latent metathesis has been successfully implemented by Grubbs and co-workers.[97] The authors designed a system based on catalyst 24 that is completely inert at rt but at 85°C gives high conversion of the substrate in only 2 h (see Scheme 45). Even better results were obtained when the corresponding imide was used instead of the anhydride: in this case the authors observed the full conversion of the monomer.
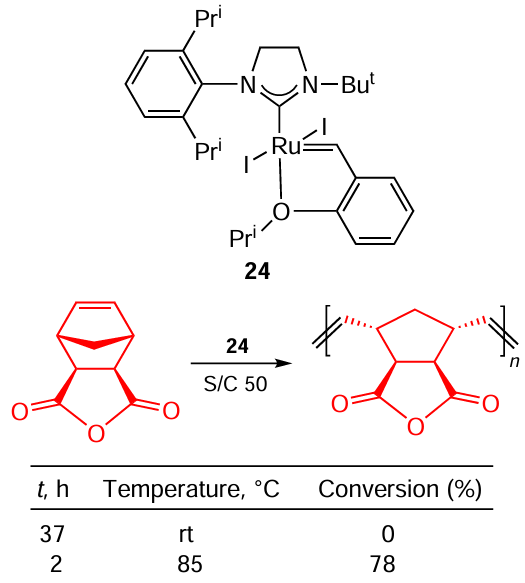
In the selective synthesis of a syndiotactic polymer, Ru – Cl and Ru – I catalysts did not show any significant difference in selectivity.[98]
Lemcoff and co-workers [99] developed a catalytic system for olefin metathesis reactions that is latent under standard conditions and becomes activated when illuminated with green (510 nm) light (Scheme 46). The irradiation sets off the in situ exchange of I in the precatalyst 25 for Cl (the latter comes from Cl-BODIPY) and subsequent trans-isomerization to give the resulting active cis-diiodo complex.
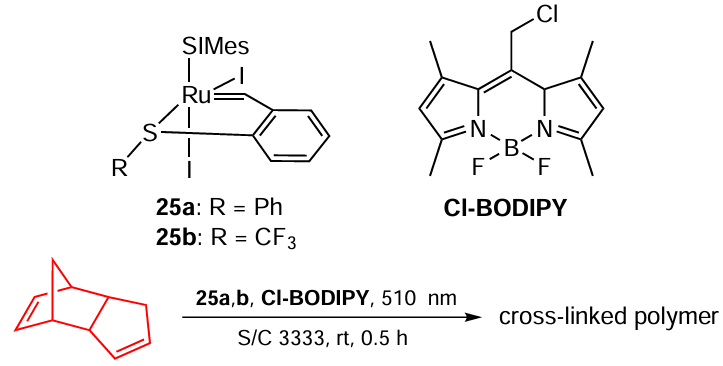
Thus, swapping Cl for I in the structures of Ru catalysts could be useful for designing of latent catalytic systems that are inert under standard conditions.
7.3. Ring closure metathesis reactions
As with catalysts for ROMP reactions, Ru – I complexes may also be used for RCM reactions as latent catalysts that can be conveniently stored.
For example, Gawin and Grela [100] described the carboxylate complex 26a (Scheme 47). This complex is inactive in metathesis reactions per se, but can be stored in solution for months and can be turned into an active catalyst 26b by mere addition of one equivalent of HCl in ether. Complex 26b was also compared to its ‘purely chloric’ counterpart 26c. As could be expected from previously discussed data and considerations, 26c performs slightly better.
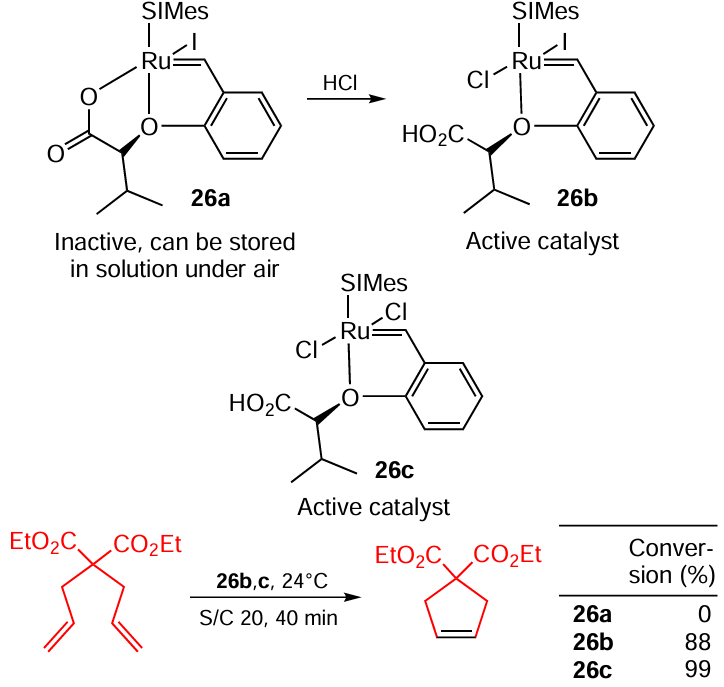
In the follow-up study,[101] a similar catalyst containing a quaternary benzimidazolium fragment was developed. This allowed a more convenient procedure for the separation of product from the catalyst.
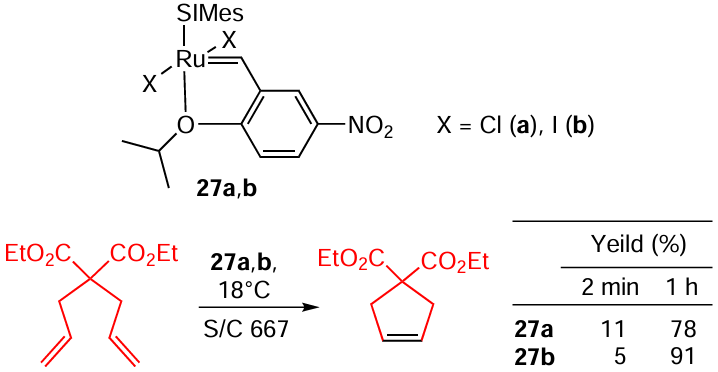
In some cases, a Ru – I complex can be more effective due to its higher stability in the reaction mixture as shown by Skowerski and co-workers.[102] Of the two nitro Grela-type catalysts, 27a,b it is actually the chloride complex 27a that is more active, providing a two times higher conversion at the third minute of the reaction. However, at long reaction times, more stable iodide complex 27b is superior, giving over 90% conversion (Scheme 48).
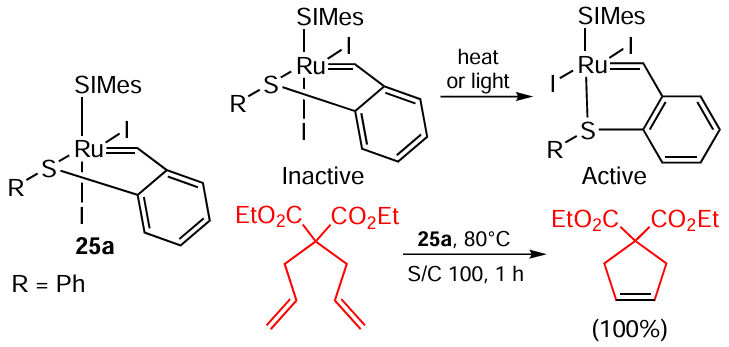
The group of Lemcoff [103-105] explored thioanalogues of Grubbs – Hoveyda catalysts. Iodide complexes require thermal or light activation to turn into their active forms (Scheme 49). It is noted that the catalysts of type 25 do not catalyze ROMP of cyclic alkenes under the reaction conditions. A protocol for the synthesis of macrocyclic lactones using such complexes via RCM of various acyclic dienes is also reported.
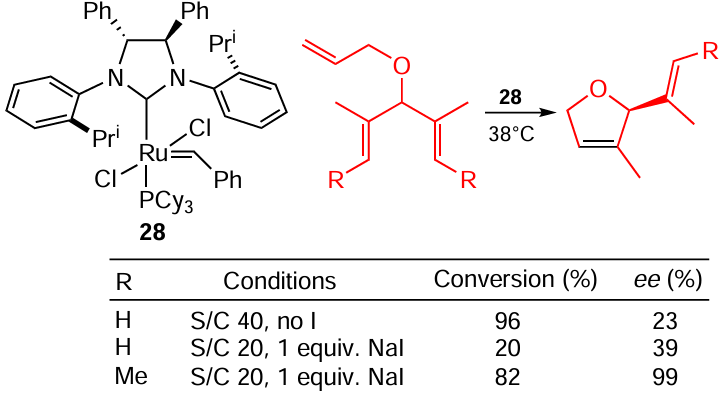
The first example of enantioselective Ru-catalyzed olefin RCM has been reported by Grubbs and co-workers.[106] In situ addition of an iodide ion deteriorates the conversion of the starting material, but improves enantioselectivity, probably for steric reasons (Scheme 50). The overall result is strongly dependent on the starting alkene identity.
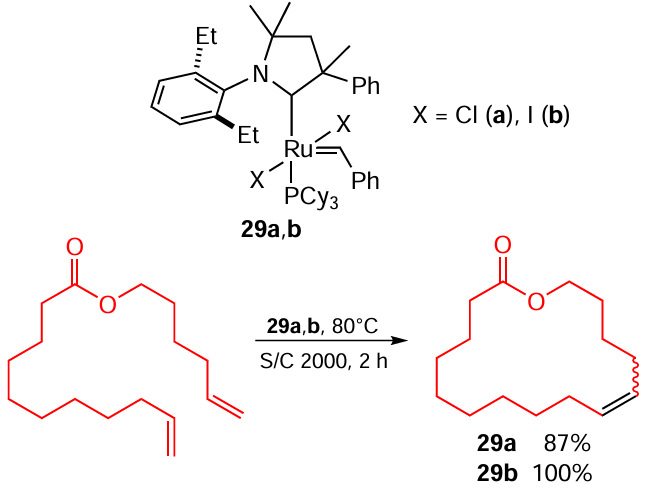
Fogg and co-workers [107] described the synthesis of macrocyclic esters via RCM The Ru – I catalyst 29b gives a better result than its Ru – Cl analogue 29a does due to its superior stability (Scheme 51). RCM reactions that deliver macrocyclic esters are of great practical interest, but their scale-up is hampered by the fact that ethylene is formed in too large quantities to be effectively removed from the reaction mixture during the course of the reaction. The presence of ethylene triggers side reactions of cross metathesis and destabilizes the catalyst forming a complex with a methylene unit coordinated to Ru. The problem of ethylene removal was solved by Bio and co-workers [108] who managed to achieve a continuous RCM process. As in the previous example, a more stable Ru – I complex proved to be more effective (Scheme 52).
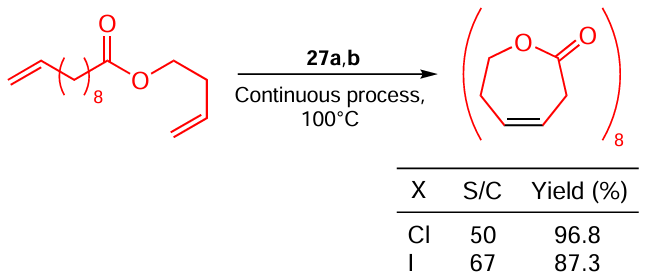
In summary, Ru – Cl metathesis catalysts are generally more active than their Ru – I counterparts for the reasons discussed at the beginning of this Section. However, there are examples where the catalyst’s stability is of primary importance (i.e. higher yields are obtained on Ru – I catalysts).
In those instances where stereoselectivity makes sense, Ru – I catalysts tend to be more stereoselective for steric reasons.
8. Alcohol isomerization reactions
This Section highlights the isomerization of allylic alcohols to ketones and the racemization of chiral alcohols.
8.1. Isomerization of allylic alcohols to ketones
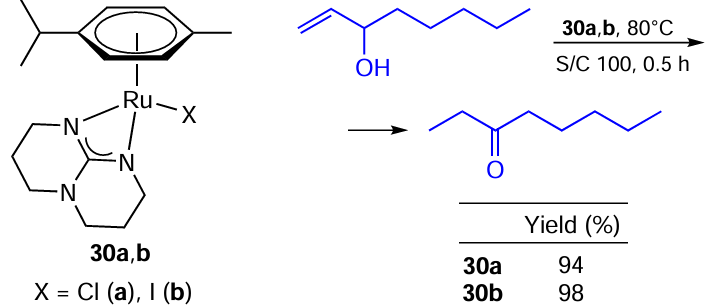
Gámez-Rivera et al.[109] described the rearrangement of allylic alcohols to ketones using mononuclear Ru(II) complexes 30a,b with the bicyclic guanidinate chelating ligand. The reaction runs well with both Ru – Cl and Ru – I catalysts providing excellent yields of the corresponding ketones (Scheme 53). The authors conclude that the halogen abstraction probably is not a vital stage of the reaction.
On the other hand, Gimeno and co-workers [110] carried out an analogous transformation in an aqueous medium using water-soluble Ru(II) complexes 31a,b, and found that the nature of the atoms coordinated to Ru had a significant impact on the catalytic activity (Scheme 54). The positively charged Ru – Cl complex 31b with an iodide counterion exhibits low activity due to the halogen exchange, which leads to a Ru – I complex that is latent in aqueous media. Under optimal conditions, only 15% yield was achieved in almost 3 h. The problem was solved by using non-coordinating SbF–6, which do not substitute halogens in the coordination sphere of the metal. The Ru – Cl complex 31a gives the desired product almost quantitatively in less than 2 h under the similar conditions.
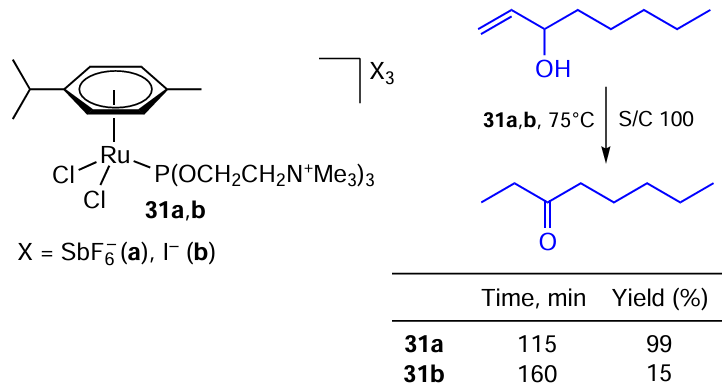
The observed difference in the influence of the nature of the halogen on the catalyst efficiency can be explained by the better solvation of the anion in water than in organic solvents, which allows the rupture of the ruthenium–chlorine bond to occur more easily compared to the ruthenium-iodine bond in aqueous media.
8.2. Racemization of chiral alcohols
The reactions of this class are useful for obtaining a single isomer from a racemate. Dynamic kinetical resolution (DKR) is an example of application of the reactions of this kind: if a catalyst allows reaching equilibrium between enantiomers fast, then, for example, an enzyme can be used to obtain its enantiomerically pure derivatives as shown in Scheme 55.
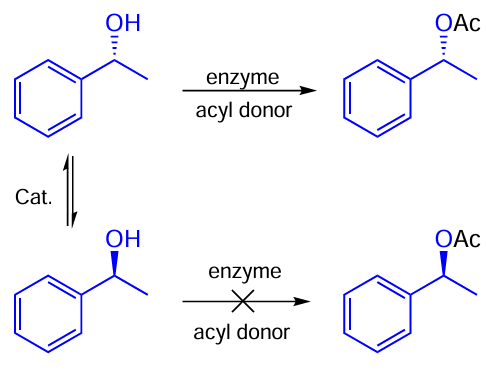
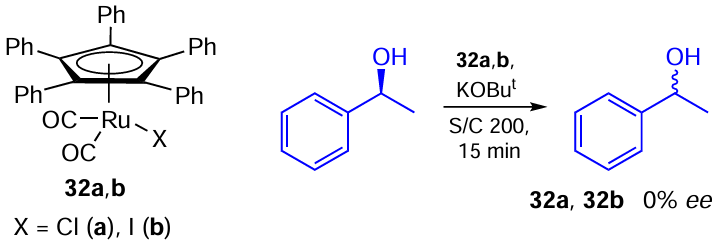
Two examples of such transformation on Ru – I catalysts have been reported in the literature. For example, Bäckvall and co-workers [111] described a racemization of (S)-1-phenylethanol using pentaphenylcyclopentadienyl carbonyl Ru – Hal complexes 32a,b (Scheme 56). The racemization rate was only slightly dependent on the halogen nature and for both catalysts 32a,b the complete racemization was achieved in 15 min. At the same time, the chloride complex 32a performed slightly better and provides full racemization in just ten minutes.
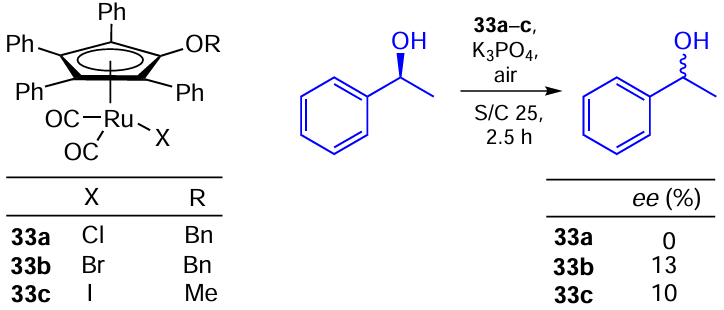
A similar reaction was carried out by Park and co-workers [112] using O-alkyl analogues 33a – c of the above Ru complexes 32a,b. Ru – Br and Ru – I catalysts 33b,c were slightly less active than their chloride analogue 33a (Scheme 57). Moreover, anchoring the above catalysts to a polymer support via the OR group gave rise to heterogeneous catalysts for this reaction, which showed comparable activity in the racemization of alcohols with the advantage of effective recovery and reusability.
To summarize, Ru – Cl complexes are usually more effective than their Ru – I analogues in the reactions of the isomerization of alcohols.
9. Synthesis of amides from alcohols and amines
Studies on using Ru – Hal complexes in the catalysis of the direct synthesis of amides from alcohols and amines are also very few. We found only two such examples. In both cases, Ru – I complexes do not perform better than Ru – Cl catalysts.
For example, Hong and co-workers [113] described Ru complexes 34, 35 with imidazolium-based NHC ligands (Scheme 58). A comparison of the catalytic activities of these complexes in the model reaction of amide synthesis revealed that replacing chlorine with iodine either slightly impairs the activity (cf. 34a,b) or has no effect (cf. 35a,b).
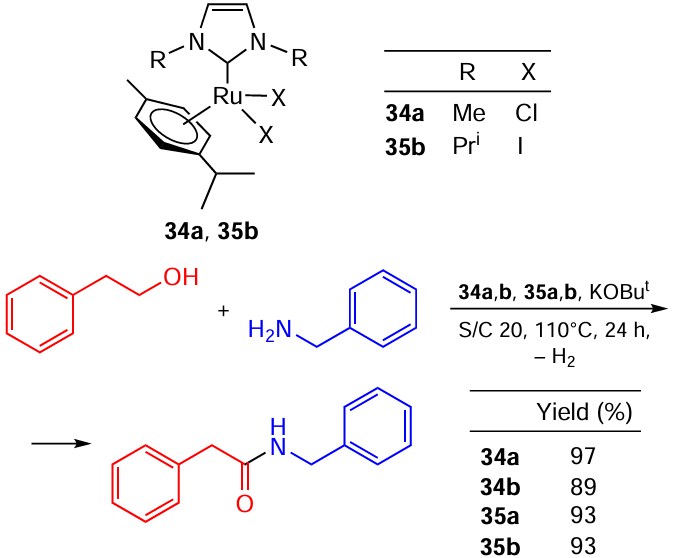
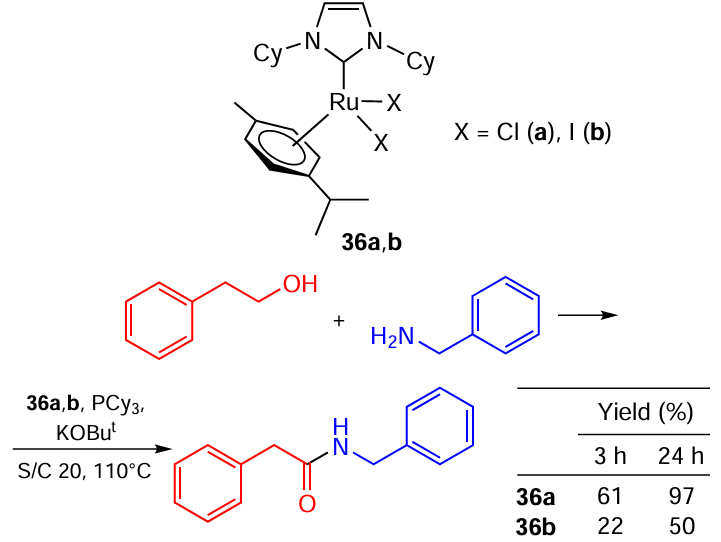
The situation changed dramatically when phosphines were added to the reaction mixture, as Madsen and co-workers [114] showed (Scheme 59). In the presence of PCy3, the Ru – Cl complex produced an amide in almost quantitative yield in 24 h, whereas the Ru – I catalyst gave less than 50% yield. At a reaction time of 3 h, the Ru – Cl catalyst was almost thrice more effective than the Ru – I one.
10. C – H activation reactions
C – H activation reactions carried out in the presence of catalysts containing the Ru – I bond can be divided into two groups:
1) C – H activation of gem-hydrogen of aliphatic alcohols towards addition reactions;
2) C – H activation of hydrogen in the aromatic ring.
These reactions will be discussed below.
10.1. C – H activation of aliphatic alcohols
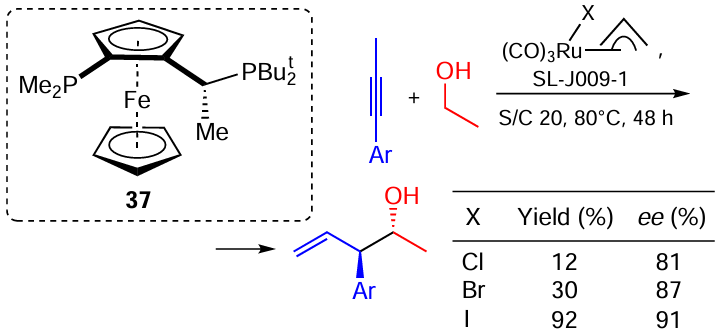
This type of reactions has been studied by the Krische’s group. In the context of our review, one of their papers [115] is of special interest as it describes complexes containing different halogens and provides an explanation for the observed halogen effect. In particular, the complex 37 catalyzes the coupling of allylic alcohols with triple bonds to afford homoallylic adducts (Scheme 60). Both the enantioselectivity and the yield of the product increases in a series Cl < Br < I. The authors managed to isolate the proposed catalytic species in all three cases and obtained X-ray crystallography data for them. It was found that the Ru – I complex has a strictly defined structure (the iodine atom occupies the ‘south’ position, allyl is oriented ‘upwards’, Scheme 61), whereas Ru – Br and Ru – Cl complexes are actually mixtures of stereoisomers.
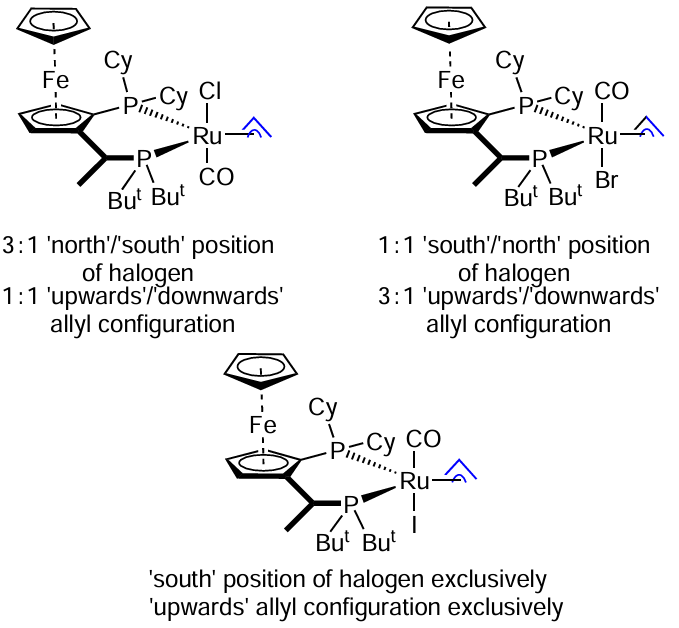
A well-determined structure may account for the better enantioselectivity. The authors argue that the higher yields can be explained by the steric reason and a non-classical hydrogen bond between the iodine and aldehyde hydrogen atoms stabilizing the transition state (Fig. 2). The existence of such a bond has been confirmed by quantum chemical calculations.
![[{"id":"TO5xb3s_ho","type":"paragraph","data":{"text":"Stabilization of the transition state by a non-classical hydrogen bond"}}]](/storage/images/resized/5vnSWLfilySawLTezlqPROdamcl09HClkjjCUZSD_xl.webp)
Other works of this research group describe similar reactions of the addition of alcohols to the multiple bonds of other compounds, vis., alkynes, dienes and cumulenes.[116-118] In all the cases catalysts with Ru – I bonds were superior to complexes with other halogens in terms of both activity and selectivity.
10.2. C – H activation in the aromatic ring
Clavier and co-workers [119] reports reactions of the C – H activation in the aromatic ring using [RuHal2(h6-p-cumene)] complexes 38a,b bearing phosphinous acid ligands. These reactions proved to be very sensitive to the nature of a halogen in a substrate. For example, the reaction with chlorobenzene is highly selective and gives the product in a good yield, while iodobenzene produces to a mixture of mono- and disubstituted products in less than 10% yield (Scheme 62). On the other hand, their results imply that the Ru – I complex 38b is outperforms its Cl-containing analogue 38a. Thus, the relationship between the reaction efficiency and the nature of halogens in a substrate and catalyst requires further investigation.
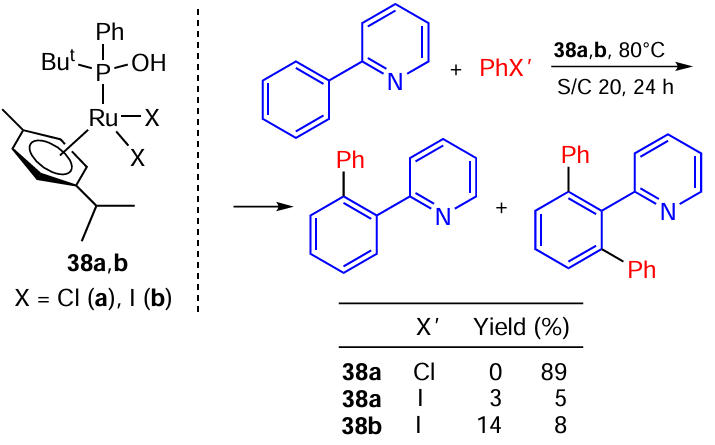
To sum up, in the case of halogen-containing carbonyl Ru complexes in the presence of JOSIPHOS ligands complexes with iodine are clearly superior to the Ru – Cl complexes in terms of both activity and selectivity due to their well-determined structures and hydrogen bonding. As for the aromatic C – H activation, a complex with iodine also shows better activity, although swapping chlorine for iodine in a substrate hampers the reaction.
11. Reactions proceeding via carbene-like intermediates
In this Section, we consider cyclopropanation reactions that proceed via the activation of a propargylic position, as well as the functionalization of 7-azaindoles at the pyridinium nitrogen on catalysts containing Ru – I bond.
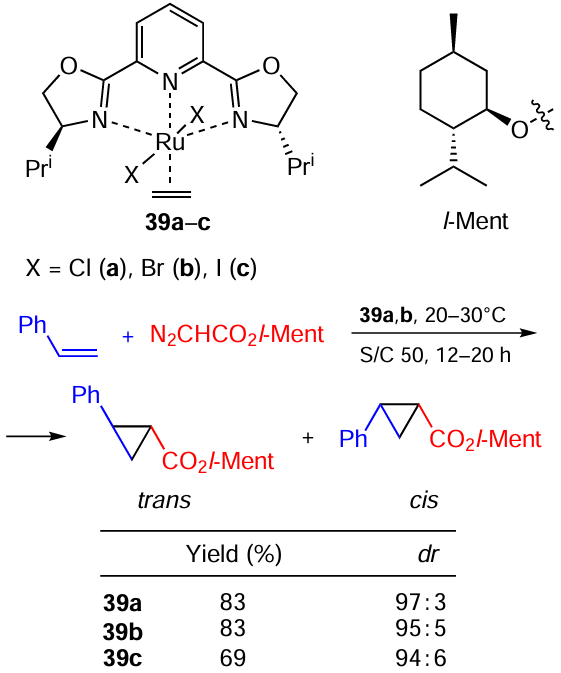
Nishiyama et al.[120] studied asymmetric catalytic [2 + 1] cycloaddition of diazoacetates to alkenes using chiral Ru(II)-bis-(2-oxazolin-2-yl)pyridine complexes 39a – c (Scheme 63). It was found that the transition from chlorine to bromine and especially iodine deteriorates both the activity and selectivity of the catalytic system.
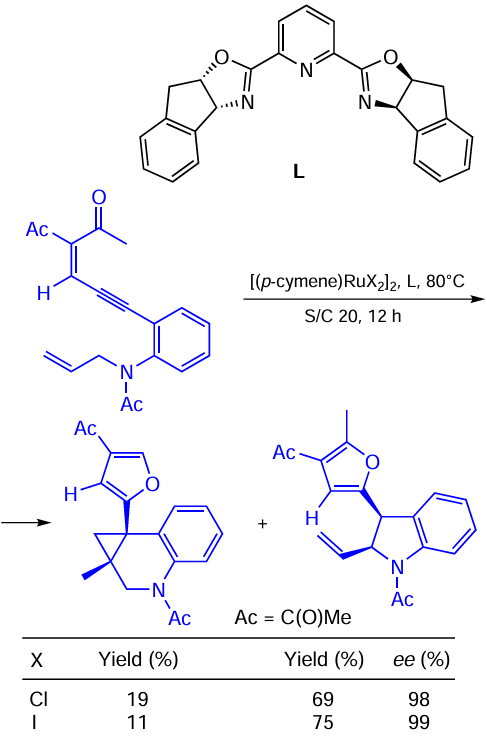
Zhu et al.[121] solved the issue of chemo-, enantio-and diastereoselectivity in the allylic C – H activation vs. cyclopropanation of donor – donor carbene precursors (Scheme 64). The catalytic system [(p-cymene)RuCl2]2/L selectively catalyzes the intramolecular C – H allylic insertion with N-allylic enynones to give vinyl-substituted dihydroindoles. The replacement of chlorine with iodine was shown to be among the modifications turning the selectivity in favour of the C – H activation product.
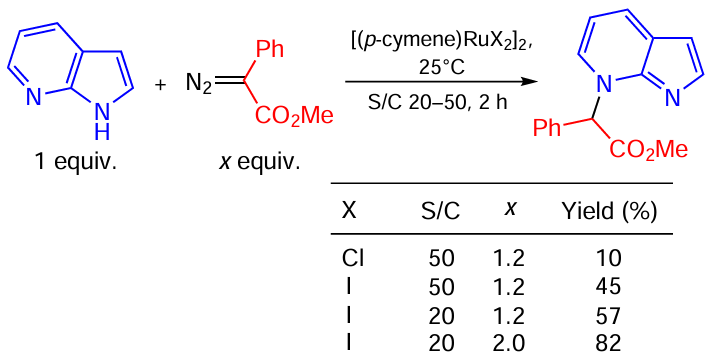
Sun and co-workers [122] carried out the site-selective functionalization of the pyridine nitrogen of 7-azaindoles with diazoacetate on in the presence of [(p-cymene)RuX2]2 (Scheme 65). It was found that the transition from chlorine to iodine under otherwise identical conditions leads to a more than fourfold increase in the product yield (from 10 to 45%). Further optimization of the reaction condition with the iodide catalyst increased the yield of the target product up to 82%.
Taking these data into account, it can be assumed that Ru – Cl catalysts perform better in cyclopropanation reactions than their Ru – I counterparts. However, this conclusion is rather doubtful due to the small number of examples.
12. Miscellaneous reactions
Reactions that we could not categorize into a general section are discussed here.
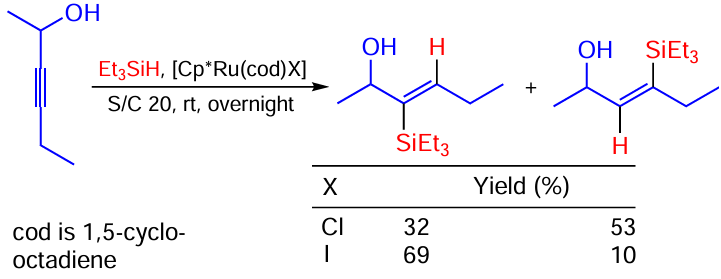
Furstner and co-workers [123] found that the hydrosilylation of unprotected propargylic alcohols in the presence of [Cp*Ru(cod)Hal] (Hal = Cl, I) follows an unconventional trans addition mode (Scheme 66). Comparing the efficiencies of the Cl- and I-containing catalysts revealed that Ru – Cl catalyst provides a slightly higher product yield (85 vs. 79%), albeit at the expense of low regioselectivity between proximal and distal products.
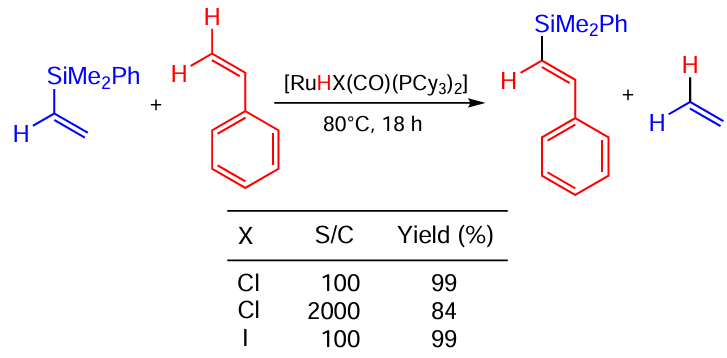
A remarkable highly stereo- and regioselective silylation of styrenes with vinylsilanes in the presence of RuHCl(CO)(PCy3)2 was reported by Kubicki and co-workers.[124] In this case, Cl- and I-containing catalysts showed equally high activity at an 1 mol.% loading, yielding the product quantitatively (Scheme 67). For Ru – Cl complex, the catalyst loading was decreased 20-fold with only a small decrease in the product yield.
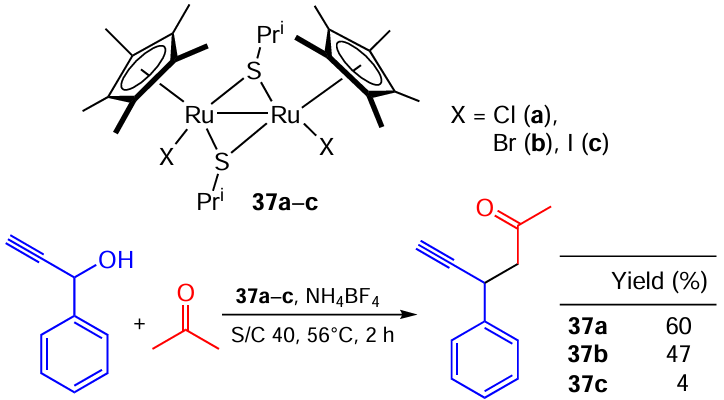
Nishibayashi and co-workers [125] studied the catalytic performance of thiolato-bridged dihaloruthenium complexes [{Cp*RuX(m-SR)}2] 37a – c (X = Cl, Br, I) in propargylic substitution reactions of 1-phenyl-2-propyn-1-ol depending on the nature of a ruthenium-coordinated halogen atom (Scheme 68). It is clear that switching from chlorine to bromine and especially iodine strongly reduces the catalytic activity of the system. The authors explained such a marked halide effect on the catalytic activity by the significant difference in redox behaviour observed for 37a – c by CVA. The reversible peaks are observed on the CVA curves of the complex 37a, whereas an irreversible oxidation is registered for bromide and iodide compounds 37b,c. This may be due to the instability of oxidized catalytic species, which is the cause of the decrease in the activity of the catalysts.
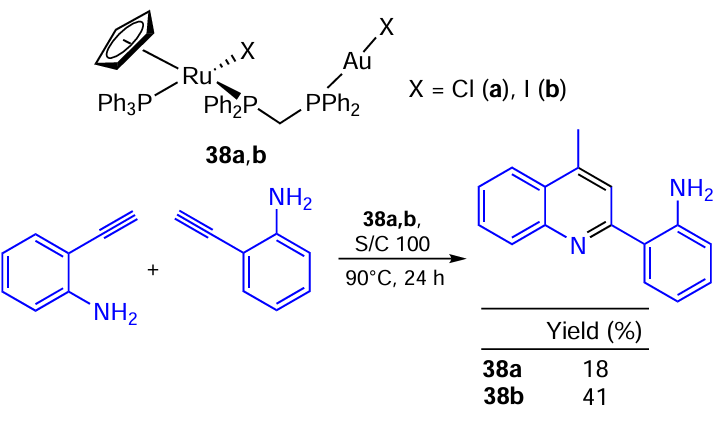
An interesting reaction of quinoline assembly from two o-ethynylaniline molecules on binuclear Ru – Au catalysts 38a,b was described by McElwhee-White and co-workers[126] (Scheme 69). Remarkably, the monometallic Au(I) catalyst produces an indole derivative, whereas the addition of a Ru – Hal centre to the catalyst molecule changes the reaction outcome. Catalyst 38b proved to be more active than its Ru – Cl analogue 38a, though only moderate yields were achieved.
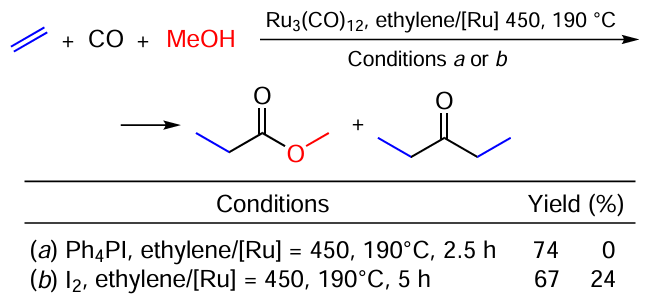
Reactions that allow the use of CO as a C1 synthon are of great importance. Hidai et al.[127] described an example of the direct synthesis of esters and ketones from carbon monoxide, alkenes and alcohols catalyzed by Ru – I systems (Scheme 70). The use of iodide promoters significantly increases the catalytic activity of the Ru precatalyst. Interestingly, ionic iodine salts, e.g., Ph4PI (see Scheme 70, conditions a) addition provided an ester selectively, while in the presence of covalent iodine compounds (I2, see Scheme 70, conditions b), a mixture of the ester and diethyl ketone is formed The authors suggested that the reaction pathway involves [HRu3(CO)11]– and [Ru(CO)3I3]– species, citing a paper by Dombek,[128] who identified these complexes by IR and NMR spectroscopy while studying the hydrogenation of CO to ethylene glycol using similar Ru – I catalytic system.
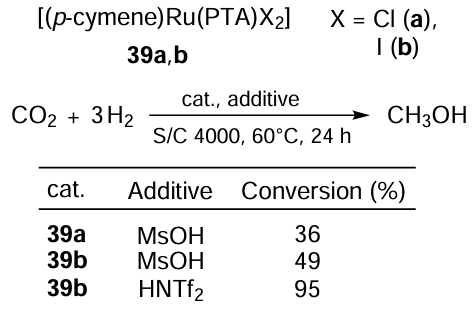
The group of Trivedi [129] developed a highly efficient homogeneous catalyst system for the acid-assisted hydrogenation of CO2 to methanol based on halide Ru complexes 39a,b with a PTA ligand (Scheme 71). Here, Ru – I complex 39b proved to be more active than its chlorine counterpart 39a. Using HNTf2 as a promoter resulted in 95% CO2 conversion to methanol.
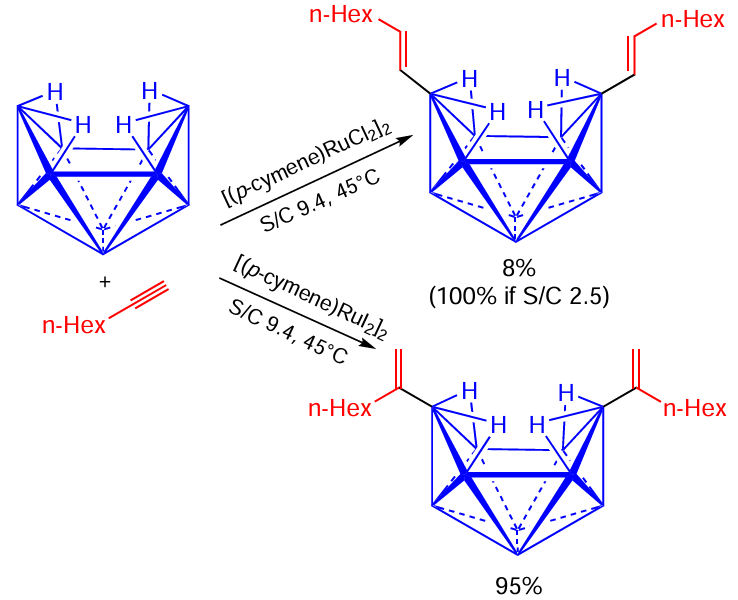
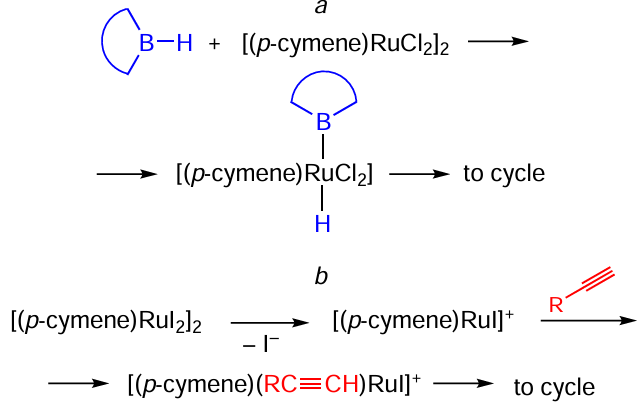
Sneddon and co-workers[130][131] used [(p-cymene)RuX2]2 (X = Cl, I) complexes in the hydroboration of acetylenes with decaborane (Scheme 72). In addition to the obvious gap between their activities, chlorine and iodine complexes demonstrated different regioselectivities: in the case of X = Cl decaborane adds to the terminal carbon of a triple bond, while with X = I it goes to the internal position. The authors suggested different reaction pathways for these two catalysts (Scheme 73). The reaction using Ru – Cl complex begins with the oxidative decaborane addition to form a metal – hydride Ru(IV) complex, which yields, after final reductive elimination, β-E-alkenyldecaborane (see Scheme 73, reaction a). As for the Ru – I complex, its reaction with decaborane involves first the dissociation of the iodide ion from the starting complex to give [(p-cymene)RuX]+, followed by the coordination of the alkyne to the resulting cationic species and subsequent reductive elimination to produce the α-isomeric product (see Scheme 73, reaction b). This explanation is in agreement with our earlier conclusion about the greater tendency of ruthenium iodide complexes to reactions proceeding by dissociative mechanism.
13. Conclusion
Summarizing the whole body of data provided above we must confess that there seem to be no single principle that would give a clue to predict the effect of replacing chlorine with iodine in the structure of a Ru complex on its catalytic performance. However, certain trends can be found and explained for some classes of reactions, and we can put forward reasons that may account for changes in the activity and selectivity of the catalytical system upon halogen replacement in many cases.
In many cases, reactions follow a dissociative pathway, in which the leaving ability of the ligand is highly important. This explains why the Ru – I catalyst can be more active, either due to the leaving ability of iodide or due to the activation of another ligand by trans-effect and steric hindrance (both are more prominent in the case of iodine than of chlorine, Scheme 74a). Examples of such behaviour are often observed in the transfer hydrogenation reactions. At the same time, these effects seem insignificant for some other reactions, e.g., multiple bond hydrogenation.
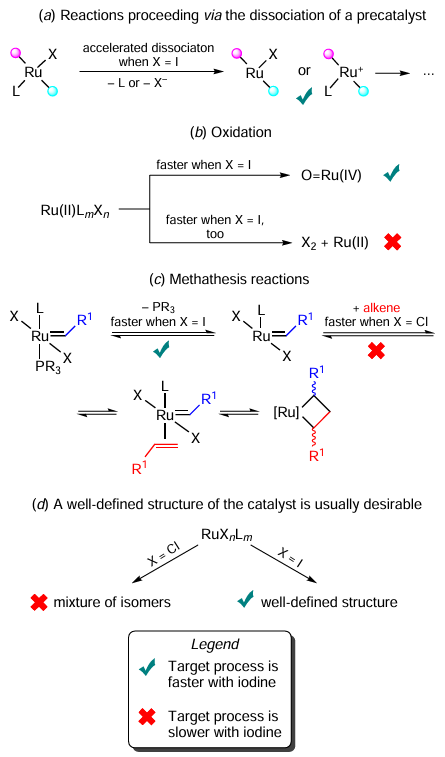
In oxidation reactions, the relative activity of Ru – Cl and Ru – I catalysts is likely determined, on the one hand, the redox lability of the Ru centre of Ru – I complexes, and on the other hand, the lower redox stability of the iodide ligands per se (see Scheme 74, reaction b). Depending on the structure of a complex, either factor may be decisive.
As for metathesis reactions, replacing Cl with I in the coordination sphere of a complex facilitates the reaction initiation (i.e., ligand abstraction from the complex). However, the subsequent alkene coordination step is slower, presumably due to the steric reason (see Scheme 74, reaction c). The latter effect usually prevails. This enables chemists to obtain latent catalytic systems that are activated by heating, irradiation, etc. Metathesis polymerization reactions can be modified to produce lower molecular weight polymers using Ru – I catalysts. However, there are also examples where Ru – I complexes give higher yields of products due to the higher catalyst stability under the reaction conditions. They also tend to provide higher stereoselectivity in metathesis reactions due to their higher steric crowding.
The C – H activation of allylic alcohols is an example that illustrates the high importance of determining the catalyst structure for the catalytic system to be efficient. The well-defined Ru – I complex performs better than its chloride counterpart, which is actually a mixture of stereoisomers (see Scheme 74, reaction d ). Non-classical interatomic interaction also contributes to the transition state stability in these reactions.
Nowadays, the highway of the development of catalytic systems is characterized by the progressive resort to more and more complex structures. Although this path can lead to impressive achievements in terms of obtained activity and selectivity, but often the complexity and expense of the resulting catalysts often renders them impractical to use. Simple structural modifications of the catalysts, or the use of simple additives in catalytic systems are more likely to be put into practice if they significantly improve the of the catalyst. One such modification is the halogen alternation in the structure of the complex, or the addition of a halide to the reaction mixture. We hope that patterns highlighted in this work will help to develop affordable high-performance catalysts with potential for practical use.
Acknowledgement
Literature search was carried out with the support from the Ministry of Science and Higher Education of the Russian Federation (Contract No. 075-00276-25-00). Analysis of data was supported by the Russian Science Foundation (Project No. 19-73-20212-П).
14. List of abbreviations
BICHEP — 6,6'-dimethylbiphenyl-2,2'-diyl)bis(dicyclohexylphosphine);
BIMOP — 6,6'-bis(dicyclohexylphosphino)-3,3'-dimethoxy-2,2',4,4'-tetramethyl-1,1'-biphenyl;
BINAP — 2,2'-bis(diphenylphosphino)-1,1»-binaphthyl;
BITIOP — 4,4'-bis(dicyclohexylphosphino)-3,3'-bitiophene;
BMIM — 1-n-butyl-3-methylimidazolium;
Cl-BODIPY — 8-(chloromethyl)-4,4-difluoro-1,3,5,7-tetramethyl-4-boro-3a,4a-diaza-s-indacene;
cod — 1,5-cyclooctadiene;
Cp — cyclopentadienyl;
Cp* — pentamethylcyclopentadienyl;
CVA — cyclic voltammetry;
Cy — cyclohexyl;
de — diastereomeric excess;
DHPD — dihydrophenantridine;
DKR — dynamic kinetical resolution;
DLS — dynamic light scattering;
dppf — 1,1-bis(diphenylphosphino)ferrocene;
dr — diastereomeric ratio;
ee — enantiomeric excess;
EWG — electron-withdrawing group;
FENAM — ferrocene-based NAD(P)H model;
HEH — Hantzsch ester;
IL — ionic liquid;
IMesH2 — 1,3-dimesityl-4,5-dihydroimidazol-2-ylidene;
8-MePD — 8-methylphenantridine;
NHC — N-heterocyclic carbene;
NMO — N-methylmorpholine N-oxide;
PD — phenanthridine;
PMP — p-methoxyphenyl;
PTA — 1,3,5-triaza-7-phosphaadamantane;
RCM — ring closure metathesis;
ROCM — ring-opening cross metathesis;
ROMP — ring-opening metathesis polymerization;
S/C — substrate/catalyst molar ratio;
SIMes — 1,3-bis(2,4,6-trimethylphenyl)-2-dihydroimidazol-2-ylidene;
TEM — transmission electron microscopy;
Tf — trifluoromethanesulfonyl (triflyl);
TOF — turnover frequency;
TON — turnover number.
References












