

Keywords
Abstract
The review integrates, for the first time, the results of studies carried out in the world over the last 15 – 20 years on the development and application of fundamental organometallic reactions catalyzed by transition metal complexes that give new carbon – carbon, metal – carbon, and heteroatom –carbon bonds and on the synthesis of small, medium, and large metallacarbocycles containing main group metals (Mg, Al, B). The review addresses data on the use of new organometallic reactions and metallacarbocycles for the preparation of practically important synthetic analogues of natural compounds, Z,Z-diene and Z,Z,Z-triene unsaturated carboxylic acids, acetogenins, lembehynes, regular isoprenoids, insect pheromones with a record high sterechemical purity, as well as toxins, heterocycles, and biaryls, the synthesis of which by other methods requires the use of expensive reagents or multistep procedures.
The bibliography includes 217 references.
1. Introduction
The discovery and practical use of novel organometallic reactions, reagents, and catalysts are considered to be important and fundamental achievements of organic and organometallic chemistry, which determined the prospects of development of modern chemistry and chemical engineering for future decades. Unfortunately, in the last 15 – 20 years, the rate of development of fundamental and applied studies in organoelement chemistry in Russia has markedly decreased, which resulted in the loss of Russia’s leadership in a number of research areas. Since the works in the field of organoelement chemistry have defined the original lines of research and the design of new chemical engineering processes for the production of a numerous valuable products and materials for decades to come, by writing this review, we intended was to draw attention of Russian researchers, especially young scientists, to this exceptionally promising and relevant area of organometallic chemistry. It is not an exaggeration to say that a great and crucial contribution to the development and formation of organometallic chemistry in Russia and in the world was made by Soviet and Russian scientists, whose achievements support the high prestige of Russian chemists on the Olympus of international science.
Following the historical justice and chronology of development of organometallic chemistry, first of all, it should be mentioned that dimethylarsine derivatives were first synthesized by pharmaceutical chemist C.L.Cadet. However, due to the difficulty of determining the structures of these compounds, R.W.Bunsen reproduced the Cadet synthesis, while scaling it up to several kilograms. Bunsen synthesized and fully characterized organoarsenic compounds, cacodyl (>As – As<) and cacodyl oxide (>As – O – As<), between 1799 and 1840.[1]
After these studies, in particular in 1849, E.Frankland obtained the first Zn-based organometallic compound.[2]
The S.N.Reformatsky’s discovery made in 1887 of the reaction providing the synthesis of β-hydroxy acids from zinc metal and α-haloacetic acid esters or aldehydes in a single preparative step is an important stage in the exploration of organozinc chemistry.[3]
In this series of brilliant discoveries, mention should be made of the Barbier – Grignard reaction reported in 1900, which can be used to prepare organomagnesium compounds from magnesium metal and organic halides in ether solvents. This reaction is among the most widely known and extensively used in organic chemistry.[4][5]
It is evident that the above works on the synthesis of the first organometallic compounds stimulated further research dealing with the preparation and investigation of properties of new classes of organometallic reagents.
In 1927, K.Ziegler reported the synthesis of organolithium compounds by reactions of aryl halides with lithium metal.[6]
The synthesis of the first organometallic compounds stimulated chemists to explore the applicability of these products for solving practical tasks.
For example, in 1881, M.G.Kucherov used organomercury compounds to perform acetylene hydration to acetaldehyde, while in 1884, H.Fenton performed hydroxylation of aromatic compounds with hydrogen peroxide using iron(II) compounds.[7][8]
The results of the above studies and the accumulated experience in the synthesis and application of organometallic compounds based on transition and main group metals allowed O.Roelen to accomplish the direct formylation and carbonylation of olefins in 1938, while W.Reppe developed unique catalytic transformations of acetylenes into cyclic polyenes.[9][10]
These fundamental reactions were destined to play a crucial role in the development of unique industrial processes for the production of a wide range of important monomers for the chemical, pharmaceutical, petrochemical, and petroleum refining industries.
The above-mentioned studies inspired vigorous development of the chemistry of organometallic compounds of transition metals in the early 1950s. For example, P.L.Pauson was the first to obtain bis(cyclopentadienyl)iron.[11] Somewhat later, in 1952, D.Burmingham, a gifted and lucky G.Wilkinson’s trainee, who was a post-graduate student at that time, prepared bis(cyclopentadienyl)zirconium and later obtained bis(cyclopentadienyl) Ti, Ni, V, Nb, and Ta complexes.[12][13]
Initially, the D.Birmingham’s fundamental results had no predictable practical value, but later, owing to the works of B.W.Kaminsky, H.H.Brintzinger, and D.Breslow, they were widely employed for the production of catalysts for olefin polymerization.[14][15]
The early 1950s witnessed one more discovery of K.Ziegler, who was able to demonstrate that mixing of Ti salts and compounds with alkylalanes gave complex catalysts soluble in organic solvents, which allowed the conduction of ethylene polymerization to polyethylene under mild conditions. Basing of these results, D.Natta and co-workers performed stereospecific polymerization of propylene to give syndiotactic polypropylene.[16]
It is not an exaggeration to say that both these discoveries, belonging to K.Ziegler and D.Natta, largely determined the prospects of development and establishment of the global production of high-molecular-weight polyolefins and polydienes, as well as materials based on these polymers.
In 1958, Günther Wilke, Director of the Institute for Coal Research (Germany), together with his trainees, made a landmark discovery that allowed the synthesis of nickel-containing complex catalysts soluble in organic solvents from nickel salts and trialkylalanes (Et3Al). These catalysts, like enzymes, are able to convert olefins and dienes into oligomers with high regio- and stereoselectivity under mild conditions.[17-19]
Subsequently, this discovery was called ‘nickel effect’ and was extensively studied by the German chemist’s school. For the first time in the world practice, they synthesized structurally diverse σ- and π-allyl nickel complexes and studied their structures and properties and also used these complexes for linear and cyclic oligomerization of olefins, conjugated dienes, acetylenes, and allenes.
These unusual and pioneering results of the German school of chemists initiated extensive research into metal complex catalysis at research centres, universities, and companies in various countries, primarily in Japan, the USA, France, Italy, UK, and the USSR (Russia).
Apart from nickel complexes, compounds and complexes of Co, Mn, Ti, Ir, Co, Fe, Pd, Rh, Ru, W, V, and later Au and Ag were practically implemented as homogeneous catalysts. This facilitated the development of promising processes such as homogeneous and asymmetric hydrogenation; oxidation of olefins and saturated hydrocarbons; metal complex-catalyzed polymerization of olefins, dienes, and acetylenes; telomerization of conjugated dienes with compounds containing active hydrogen atoms; linear and cyclic co-oligomerization of dienes with olefins; alkylation, hydroformylation, carbonylation, hydrocyanation, disproportionation, cyclopropanation, cross-coupling, and metathesis reactions.
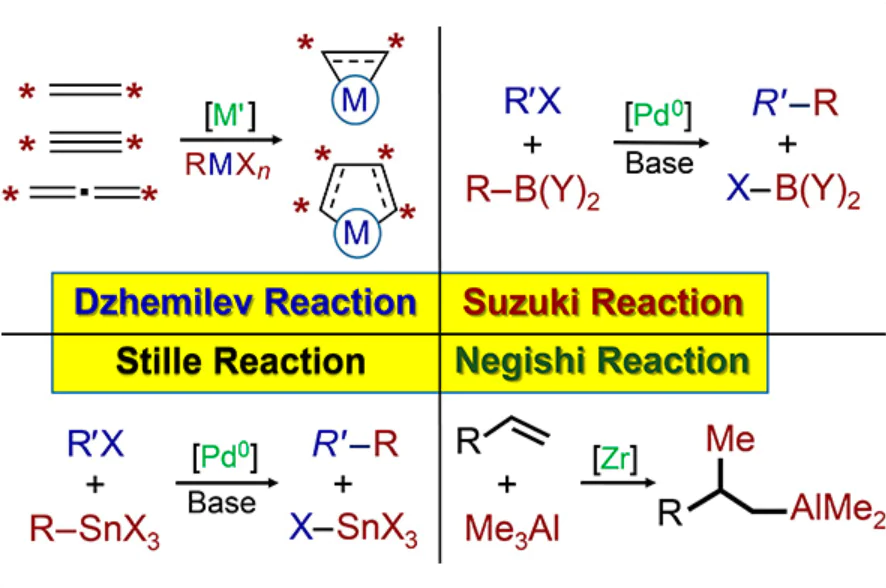
A special place in the above works belongs to the pioneering studies dealing with extension of the methods of transition metal catalysis into the chemistry of organometallic compounds based on main group metals (Mg, Zn, Al, Ga, In, B).
The fundamental studies carried out along this line in the last 15 – 20 years by U.M.Dzhemilev’s scientific school provided the emergence and fast development of the chemistry of small, medium, and large metallacarbocycles based on main group metals.[20-29]
It is important that the synthesized alumina- and magnesacyclic molecules contain simultaneously two metal–carbon bonds, which opens up extensive opportunities for targeted transformations into practically valuable small, medium, and large carbo-, heterocarbo-, and metallacarbocycles, analogues of natural compounds that were previously difficult to obtain.[30]
The original ideas on the development of a family of new organometallic reactions of catalytic cyclometallation of unsaturated compounds were based on a firm scientific foundation built by the studies of research groups headed by U.M.Dzhemilev and E.Negishi in the field of catalytic hydro- and carbometallation of non-activated alkenes with commercially available alkylalanes (Bui2AlH, Bui3Al, AlEt3, Et2AlCl) and simple organomagnesium compounds (RMgX, R2Mg) and also in the field of stereoselective carboalumination of alkynes with Me3Al.[31-38]
In particular, studies carried out by Dzhemilev’s school from the 1980s up to the 2000s resulted in the appearance of new catalytic organometallic reactions such as hydro- and carboalumination and β-alkylation of non-activated alkenes, which were characterized by high yields and high chemo-, regio,- and stereoselectivity (Scheme 1).[22] [25][26][39]
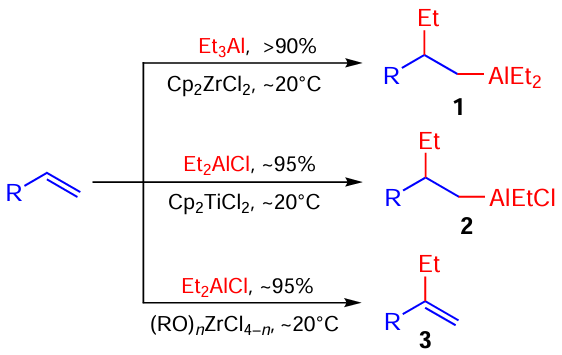
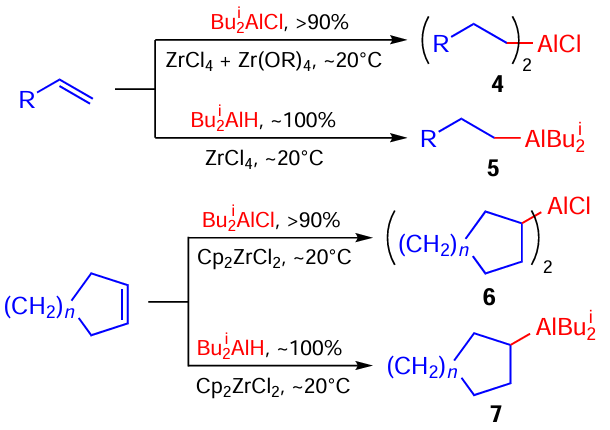
In addition to the above new fundamental reactions, our research group developed an industrially promising regioselective hydroalumination of cyclic and acyclic alkenes with commercially available Bui2AlH, Bui2AlCl, and Bui3Al to give practically important higher organoaluminium compounds (OAC) 4 – 7 (Scheme 2).[25]
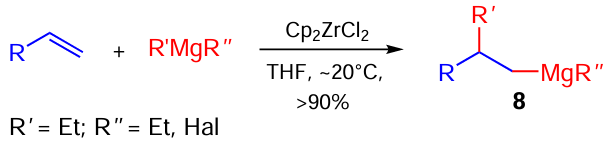
The subsequent fundamental studies of our research group [20-25] ensured implementation of the proposed concept and development of new catalytic 1,2-carbomagnesiation of α-olefins with Grignard reagents in the presence of Cp2ZrCl2 , which substantially expanded the limits of applicability of organomagnesium compounds in organic and organometallic synthesis.
Zirconium-catalyzed carbomagnesiation of non-activated alkenes, leading to carbomagnesiation products 8, has quickly become a part of synthetic organic and organometallic chemistry being called the Dzhemilev reaction (Scheme 3).[40]
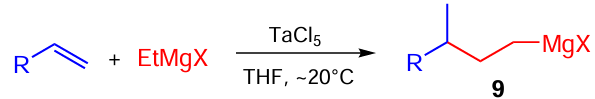
Later, it was shown that the use of TaCl5 , instead of Cp2ZrCl2, as a catalyst for this reaction changes the carbomagnesiation pathway, giving rise to the opposite regioisomer 9 (Scheme 4).[41]
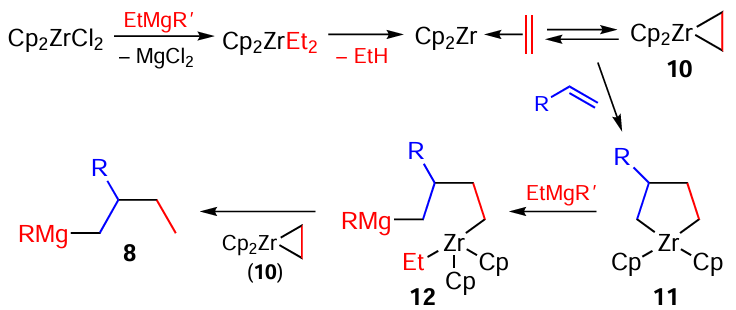
Professor F.J.Whitby from the University of Southampton performed a detailed study of the Dzhemilev reaction mechanism (Scheme 5).[40][42] According to the results, bis(cyclopenta- dienyl)zirconacyclopropane 10 and 3-substituted bis(cyclopentadienyl)zirconacyclopentane 11 are the key intermediates of this reaction. The latter reacts with excess Grignard reagent present in the reaction medium via the regioselective attack of the Zr – C bond,[2] resulting in the formation of intermediate acyclic organomagnesium compound 12. The final step of the reaction is the β-hydrogen transfer, which finally leads to the target organometallic compound (OMC) 8 and regeneration of the Zr cyclopropane complex.
A similar mechanism is involved when titanium and hafnium salts and complexes in combination with various alkyl and alkyl halide aluminium or magnesium derivatives are used as catalysts. The chemoselectivity of these reactions depends on the chosen unsaturated compound, the solvent, OMC, and the catalyst. It should be emphasized that in all cases, the reactions involve the transfer of a ligand from the catalyst central atom to a main group metal atom to give the target organometallic compound.[43-45]
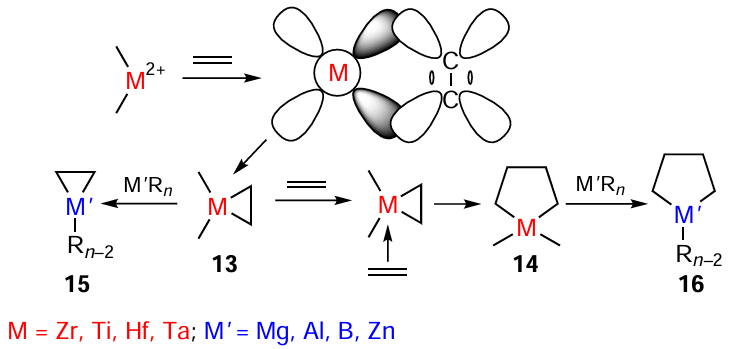
This effect formed the basis for the development of new catalytic organometallic reactions for the preparative-scale synthesis of Mg, Al, B, and Zn metallacycles using transition metal complexes the central atom of which can coordinate unsaturated compounds containing double, triple, or cumulative C – C bonds to give donor-acceptor complexes,[46-48] which results in the generation of three- 13 and five-membered 14 metallacycles based on Ti, Zr, Hf, or Ta (Scheme 6). According to the proposed idea, these products react with Mg, Al, B, and Zn alkyl or alkyl halide derivatives via ligand exchange to give the desired metallacarbocycles based on main group metals 15, 16.
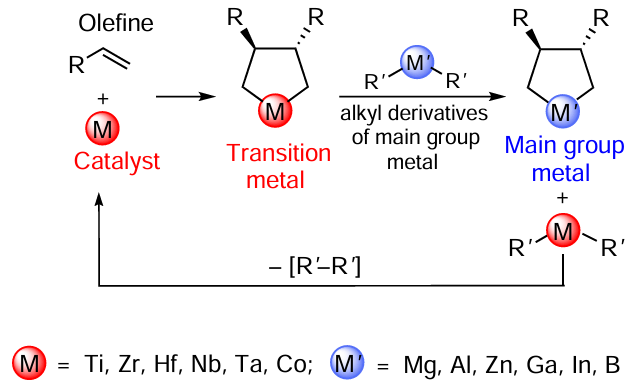
The long-term studies concerning reactions of structurally diverse unsaturated compounds with acyclic organometallic compounds based on main group metals in the presence of transition metal salts and complexes made it possible not only to develop a family of previously unknown catalytic organometallic carbo- and cyclometallation reactions of alkenes, alkynes, and 1,2-dienes and introduce them into the practical organic and organometallic synthesis, but also to formulate the main concept of the phenomenon that forms the basis for these reactions, that is, the catalytic replacement of transition metal atoms in the formed cyclic intermediates by main group metal atoms to give the target metallacarbocycles as shown below in Scheme 7.
The most important data on the preparative-scale synthesis, study of the properties, and applications of Al-, Mg-, B-, and Zn-based metallacarbocycles that are most interesting for practical and synthetic purposes are described below.
2. Catalytic cycloalumination of alkenes, alkynes, and allenes
The preparative-scale synthesis of 3-alkyl(aryl)-substituted aluminacyclopentanes by the Cp2ZrCl2-catalyzed reaction of α-olefins of various structures with Et3Al was first performed in 1985.[49][50]
Later, we studied the roles of various reaction characteristics such as solvent, temperature, the nature of starting OAC, ligand environment, and the central atom of the catalyst, which can crucially change the chemo-, stereo-, and regioselectivity of the synthesis of three- (17) and five-membered OAC (18, 19) and 1,4-dialuminium compounds (20) (Scheme 8).
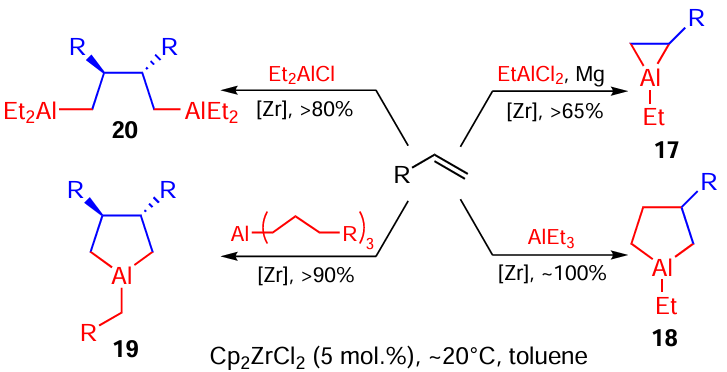
The developed reactions are versatile and provide the synthesis of the above classes of aluminacarbocycles with high regio- and stereoselectivity.[51-54]
The mechanism of this reaction was investigated by dynamic NMR spectroscopy in relation to cycloalumination of α-olefins with Et3Al in the presence of the Cp2ZrCl2 catalyst; the key reaction intermediates, that is, Zr – Al bimetallic complexes, responsible for the formation of target aluminacyclopentanes, were also identified (Scheme 9).[55][56]
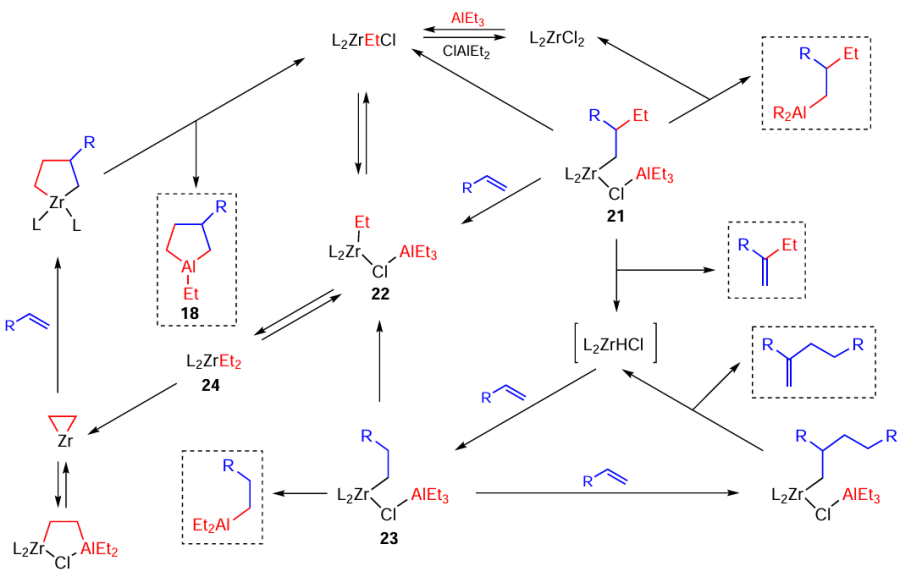
The rate constants for the formation of intermediate complexes and target aluminacarbocycles were measured and calculated by quantum chemical methods.
As shown in Scheme 9 depicting the mechanism of the catalytic cycloalumination of olefins, the replacement of the zirconium atoms by aluminium atoms occurs through the formation of Zr – Al bimetallic complexes 21 – 23 with simultaneous regeneration of catalytically active complex 24.
The presence of two reactive Al – C bonds in the resulting aluminacarbocycles inspired the authors to put effort into extensive studies of the targeted transformations of aluminacarbocycles in reactions with nucleophilic and electrophilic reagents and small molecules and carbocyclization, demetallation, and cross-coupling reactions.[57-60]
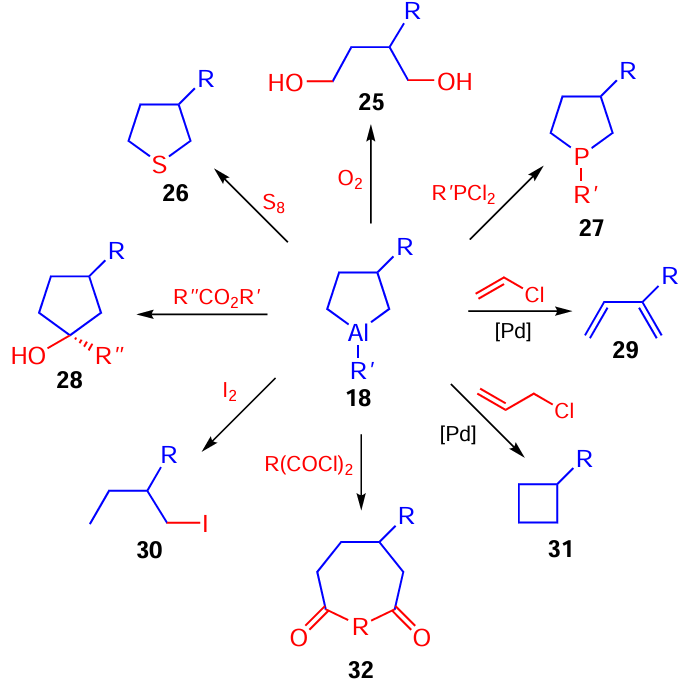
Out of numerous known functionalization reactions of aluminacyclopentanes 18, only some most interesting examples are depicted in Scheme 10; in our opinion, even these examples would allow the reader to conceive the huge potential of new developed organometallic reactions and reagents for the preparative-scale synthesis of structurally complex hetero- (26, 27) and carbocycles (28, 31, 32) and mono- (29, 30) and bis-functionalized (25) compounds, difficult to obtain by other methods, starting from readily available reactants in a single preparative step.[57-60]
In the next stage of research, we intended to find out whether the catalytic cycloalumination that we developed can be extended not only to alkenes, but also to other classes of unsaturated compounds such as substituted alkynes, allenes, and heteroolefins.
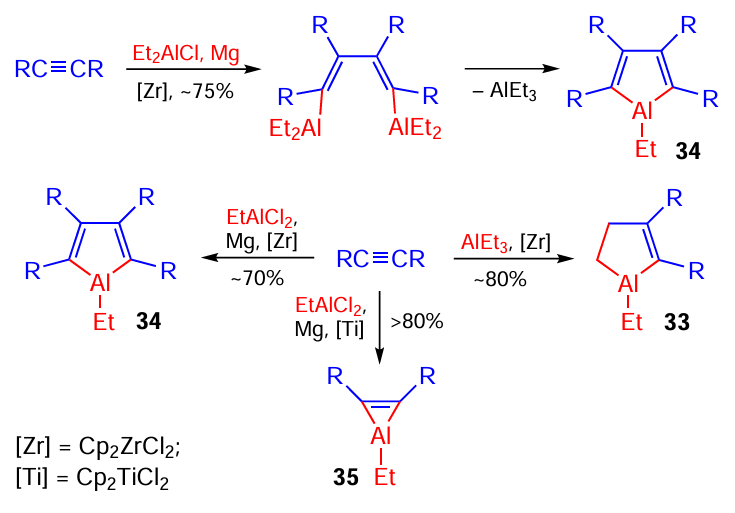
Scheme 11 illustrates the impressive achievements of our research group in the synthesis of previously unknown metallacarbocycles, that is, aluminacyclopent-2-enes 33, aluminacyclopentа-2,4-dienes 34, and aluminacyclopropenes 35 by cycloalumination of disubstituted acetylenes.[24][25] [61-65]
Equally interesting results were also obtained in the study of Zr- and Ti-catalyzed reactions involving various 1,2-dienes.[61][62] Unique 2-alkylidenealuminacyclopropanes 36, 2-alkylidenealuminacyclopentanes 37, 4-alkylidene-1-aluminacyclopent-2-enes 38, and bis-2,5-dialkylidenealuminacyclopentanes 39 were prepared for the first time and characterized (Scheme 12).
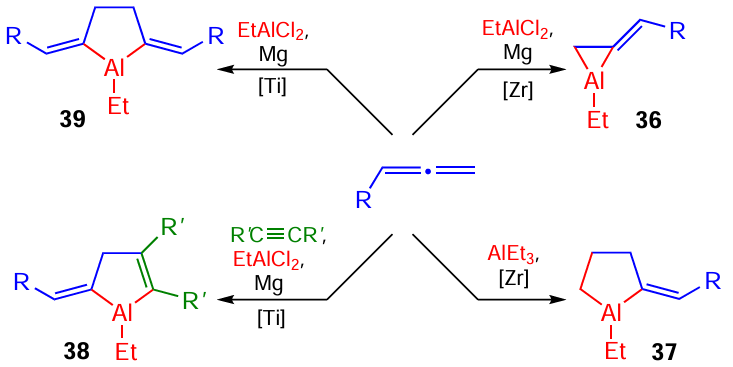
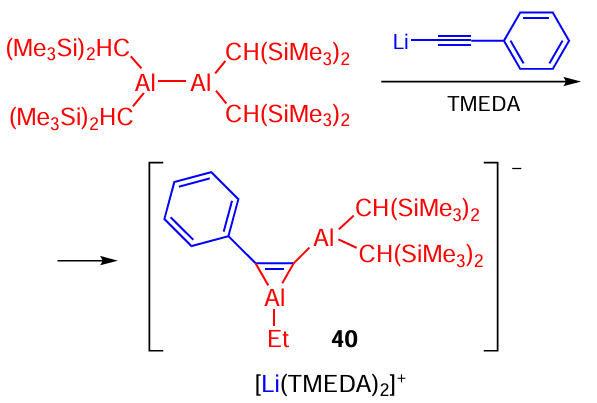
A few examples of the synthesis of aluminacyclopropanes, aluminacyclopropenes, and aluminacyclopentаdienes are presented below to compare the convenience, simplicity, and availability of the catalytic methods that we developed with alternative approaches to the synthesis of these products that appeared in the literature much later. For example, aluminacyclopropene was synthesized in 1999 by the reaction of tetrakis[bis(trimethylsilyl)methyl]dialane 40 with lithium phenylethynide involving the insertion of the anionic carbon atom into the Al – Al bond (Scheme 13).[66]
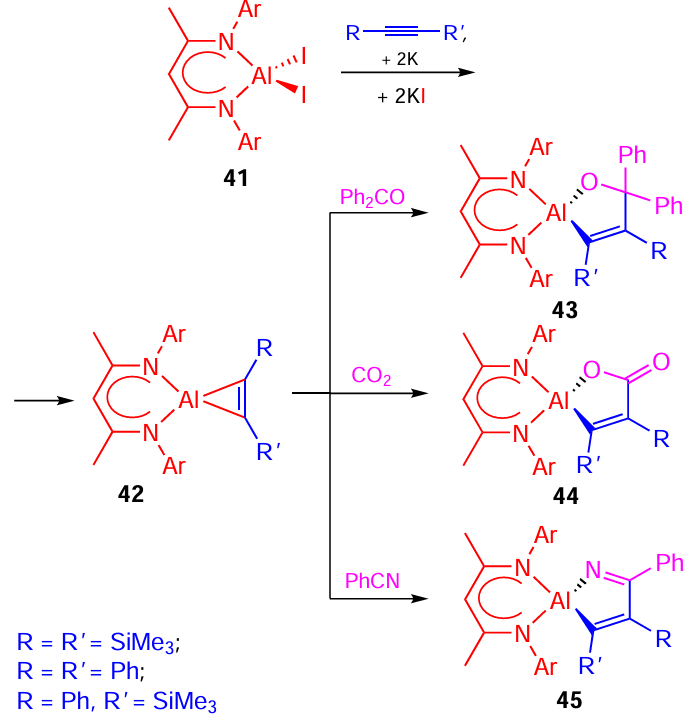
Later, the reduction of LAlI2 41 {L = HC[(CMe)(NAr)]2 , Ar = 2,6-Pri2C6H3} with potassium in the presence of alkynes C2(SiMe3)2 , C2Ph2 , and C2Ph(SiMe3) was carried out to synthesize aluminacyclopropenes 42 {LAl[η2-C2(SiMe3)2], LAl(η2-C2Ph2), and LAl[η2-C2Ph(SiMe3)]}; the chemical reactivity of products 42 towards nitriles, ketones, and CO2 was studied (Scheme 14).[67-72] A similar approach was used to prepare aluminacyclopropane.[73]
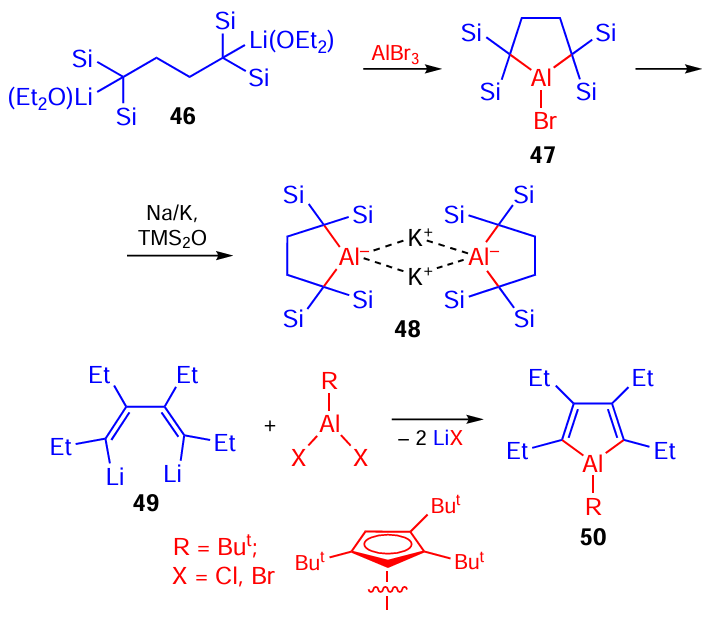
Aluminacyclopentanes 47 and 48 and aluminacyclopentа-2,4-dienes 50 were obtained at different times by the reaction of 1,4-dilithium butanes 46 and 1,4-dilithium 1,3-butadienes 49 with various aluminium halides (Scheme 15).[74][75] It was shown that aluminacyclopentadienes undergo ring expansion reactions accompanied by the insertion of alkynes and azides into reactive Al – C bonds.
Another considerable achievement of our research group is the development of an original one-pot method for the design of regular trans-isoprenoids using the discovered catalytic cycloalumination of α-olefins with Et3Al catalyzed by zirconium complexes (Scheme 16, the indicated yields are the overall yields per chain growth step).[62]
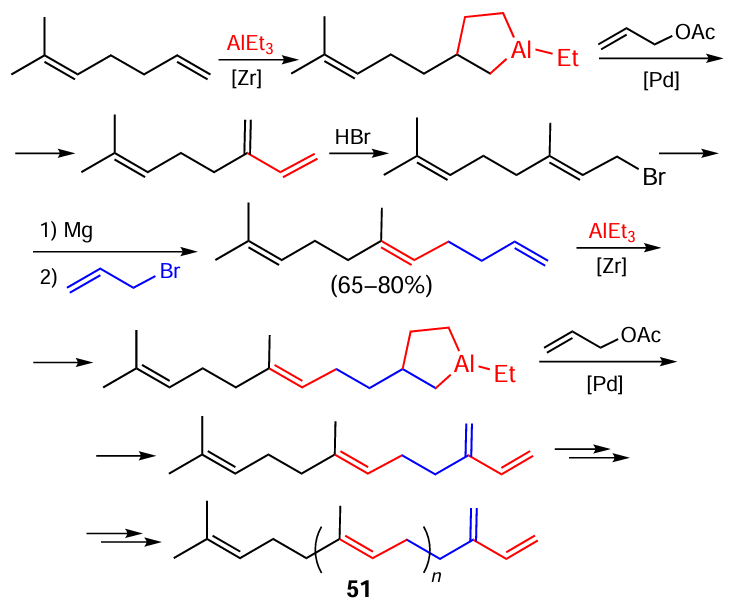
This method provides a route towards linear trans-isoprenoids 51 containing two to ten isoprene units in sufficiently high yields, which opens up prospects for scaling up of this process for industrial production of fragrances needed for perfumery, medicinal agents, and other valuable compounds.
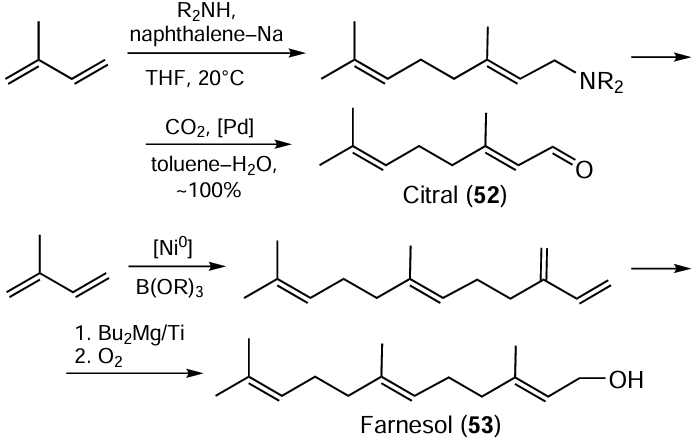
In view of the high value and good prospects of methods for the synthesis of useful linear regular isoprenoids, and as a continuation of the above works, we proposed one-pot methods for the preparation of industrially important citral (52) and farnesol (53) starting from isoprene according to the following sequence of reactions (Scheme 17).[62]
Successful development of studies along this line showed that the catalytic cycloalumination can be conducted for cyclic 1,2-dienes and acetylenes as well as for structurally diverse methylidene- and alkylidene-cycloalkanes, which were previously considered to be inactive in this reaction.
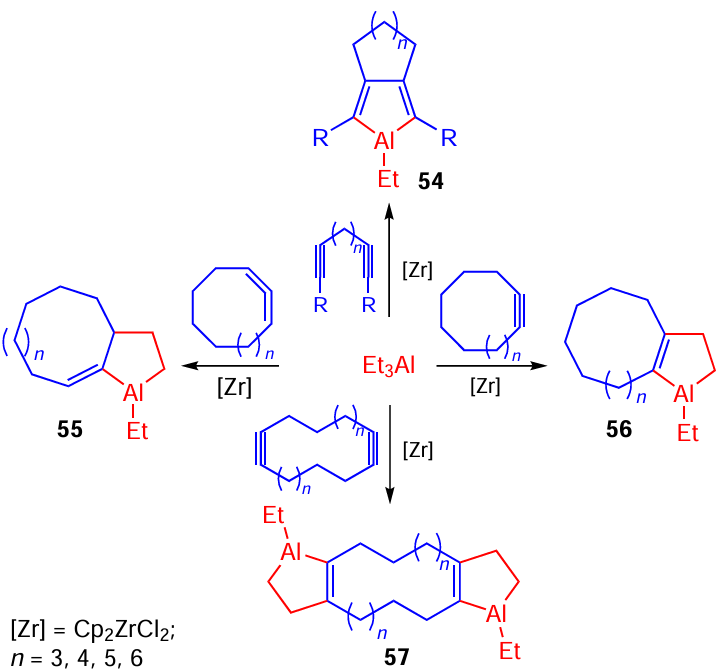
The Cp2ZrCl2-catalyzed cycloalumination of cyclic alkynes and allenes with Et3Al (5 mol.%, hexane, 6 h) gives rise to previously unknown bi- (54 – 56) and tricyclic (57) OAC in 65% to 95% yields (Scheme 18).[62-65] [76-78]
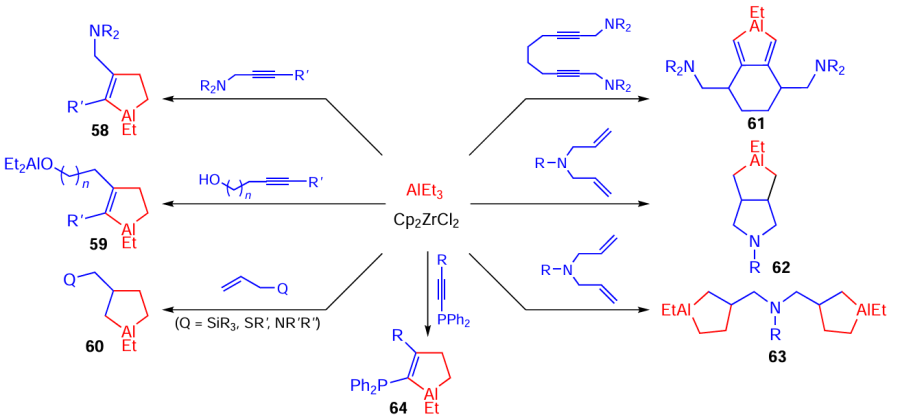
An important beneficial feature indicating the versatility of these reactions developed by our research group is the possibility of extending the reactions to N-, Si-, O-, S-, Se-, and P-containing alkenes and alkynes, which provides the synthesis of heteroatom-containing cyclic OAC 58 – 64 (Scheme 19).[79-81]
Non-trivial results were obtained in a study of the intermolecular cyclometallation of cyclic alkynes, in which we implemented an original method for the synthesis of macrocyclic C20 – C28 polyketones.[82][83]
The successive transformations of the obtained tricyclic alumina- (65) or magnesacyclopentаdienes (66) on treatment with α,ω-dicarboxylic acid α,ω-dihalides or chlorides followed by cleavage of double bonds by ozonolysis at the final stage furnished macrocyclic ketones 67 and 68 (Scheme 20).[84]
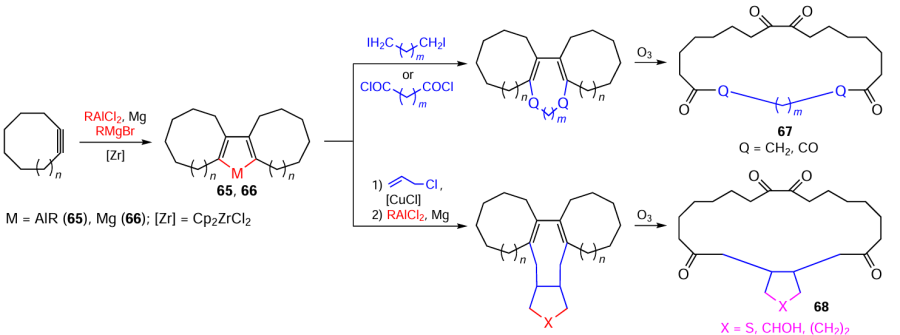
These studies open up prospects for the development of advanced processes for the production of effective fragrances for perfumery and antibacterial agents for medicine and original methods for the synthesis of macrocarbocycles of various structures.
The subsequent studies were aimed at expanding the scope of reactions and their applications. The catalytic cycloalumination of symmetrical and unsymmetrical cycloalkadiynes was carried out for the construction of macrocarbocycles with spirocyclopropane moieties (69 and 70). This was attained by carbocyclization of tricyclic OAC 57 containing annulated aluminacyclopentеne moieties on treatment with CH3OCH2Br, as shown in Scheme 21.[84-87]
It was found that Zr-catalyzed cycloalumination of methylidene- and alkylidene-cyclopropanes and methylidenecyclobutanes with triethylaluminium results in the formation of spiro-aluminacarbocycles 71 – 74 in high yields of ~ 95% (Scheme 22).[88][89]
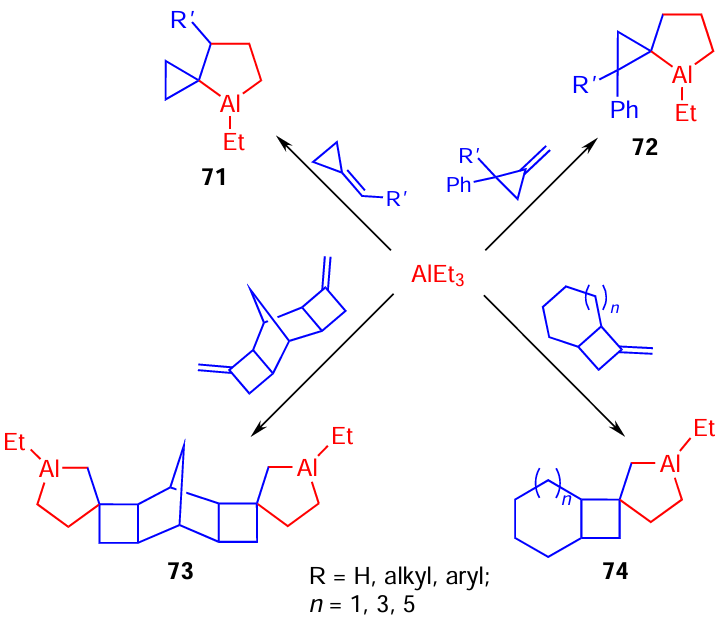
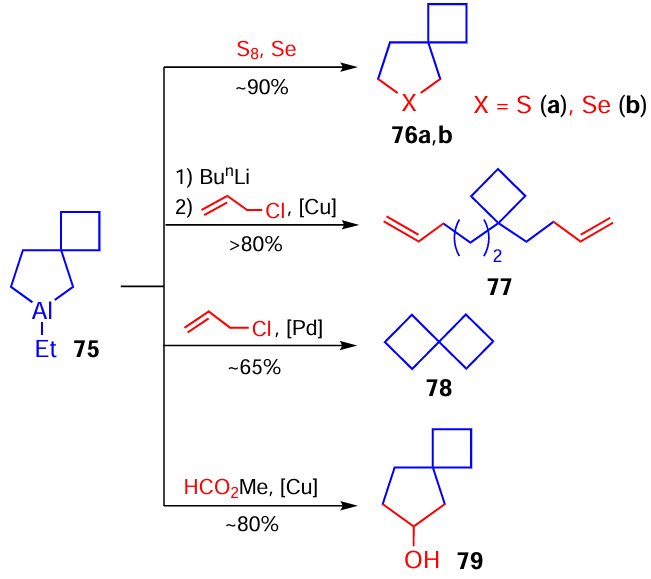
It was shown that the reactions of aluminaspiro[3.4]octane 75 prepared in situ with sulfur, selenium, methyl formate, and allyl chloride provide the synthesis of spiro[3.4]thia(selena)-heterocycles 76, dialkenyl cyclobutane derivative 77, spiro[3.3]heptane 78, and spiro[3.4]octanol 79, respectively (Scheme 23).[58] [89]
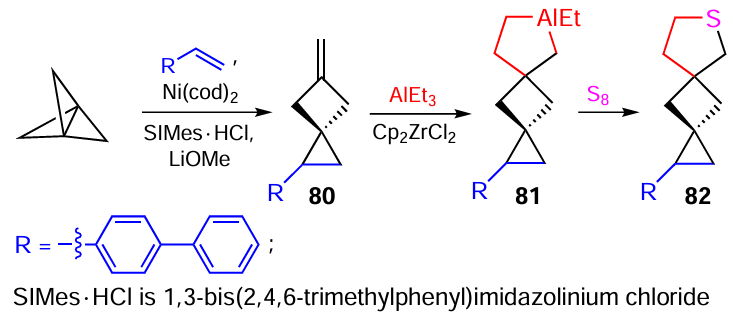
The reactions developed by our research group have become a part of practical organic synthesis and are often used in the total synthesis of carbo- and heterocycles difficult to obtain by other methods. For example, the one-pot preparation of 6-thiaspiro[3.4]octane from methylidenecyclobutane was successfully used in the total synthesis of difficult-to-obtain dispirothiophane 82 by successive transformations of 1-([1,1'-biphenyl]-4-yl)-5-methylenespiro[2.3]hexane (80) via the intermediate formation of aluminacyclopentane 81. In turn, spiro[2.3]hexane 80 was prepared by Ni-catalyzed cyclopropanation of 4-vinylbiphenyl with [1.1.1]propellane as a carbene precursor (Scheme 24).[90]
Another type of reactions that is gaining increasing popularity is the one-pot synthesis of cyclic organophosphorus compounds, namely, phospholanes 83, 84, phosphol-2-enes 85, and bis-phospholanes 86, with the key step being Zr-catalyzed cycloalumination of alkenes, alkynes, and α,ω-dienes of various structures with triethylaluminium (Scheme 25).[59][91-94]
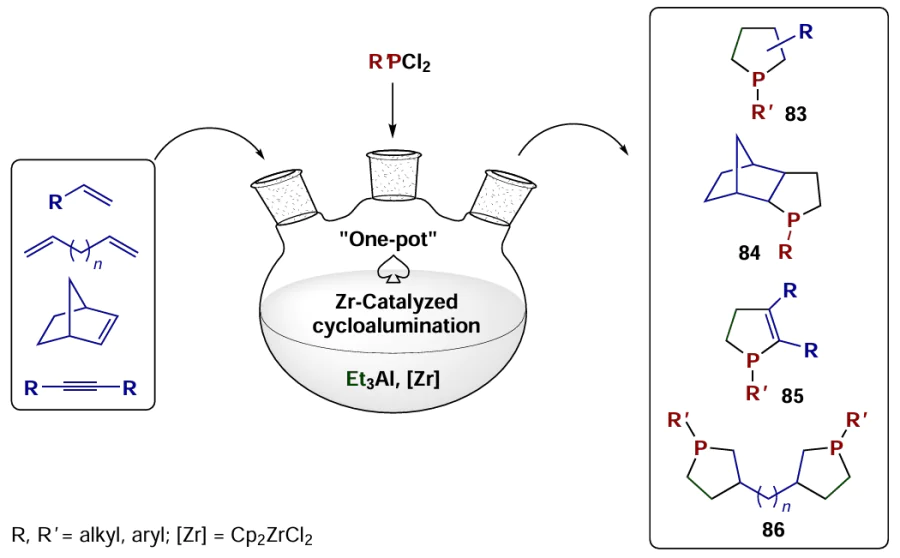
The above examples of the synthesis and targeted transformations of metallacarbocycles indicate the development of a new research area, that is, the chemistry of small, medium, and large (macrocyclic) aluminacarbocycles, which includes intensive exploration and spread of original catalytic organometallic reactions, which may crucially change the strategy of organic and organometallic synthesis in the future.
3. Catalytic cyclomagnesiation of alkenes, alkynes, and allenes
Studies of the catalytic hydro-, carbo-, and cycloalumination and 1,2-ethylmagnesiation of unactivated alkenes in the presence of transition metal complexes (Zr, Ti, Ta, Hf, Co) stimulated implementation of the idea of catalytic cyclomagnesiation of unsaturated compounds with magnesium alkyl derivatives or Grignard reagents, by analogy with the cycloalumination reactions, with the goal to develop original methods for the preparation of previously unreported substituted magnesacyclopentanes, magnesacyclopent-2-enes, and magnesacyclopentа-2,4-dienes, the synthesis of which by alternative methods appears intricate.
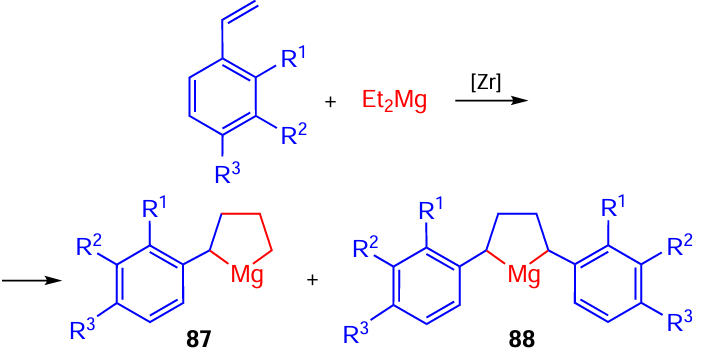
The possibility of synthesizing substituted magnesacyclopentanes by the reaction of alkenes with RMgX or R2Mg under the action of Zr complexes was first reported in 1989.[21] [52]
For example, styrene and its derivatives react with Et2Mg in the presence of Cp2ZrCl2 catalyst (conditions: 20°C, Et2O/THF) to give mono- (87) and diaryl-substituted (88) magnesacyclopentanes (Scheme 26).
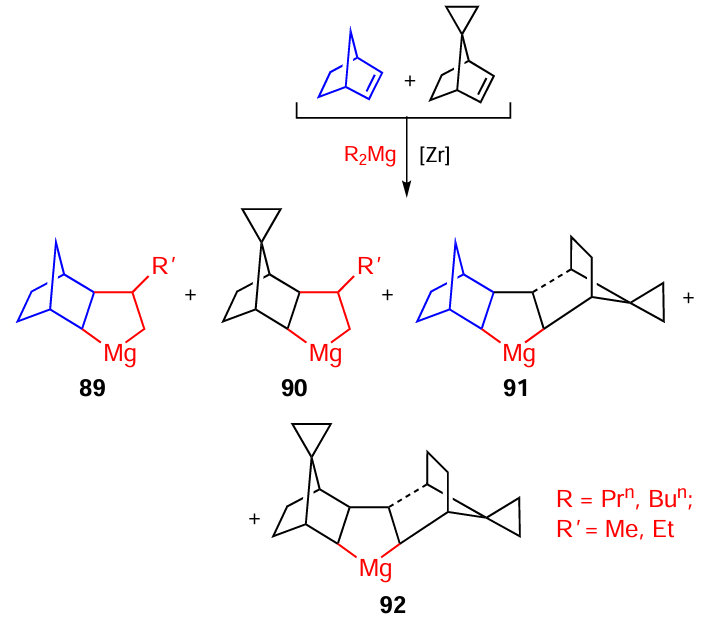
The cyclomagnesiation of norbornene and spirocyclopropylnorbornene, taken in an equimolar ratio, with dialkylmagnesium R2Mg results in the formation of tri- (89), tetra- (90), hexa- (91), and heptacyclic (92) magnesacyclopentanes in almost quantitative yields (Scheme 27).
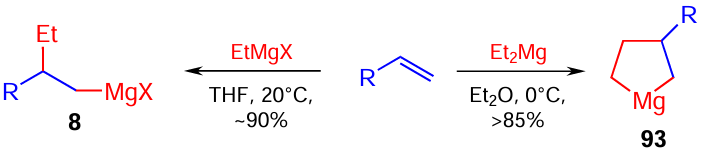
Conversely, EtMgX or Et2Mg react with α-olefins in the presence of the Cp2ZrCl2 catalyst (3 – 5 mol.%) to give either 1,2-ethylmagnesiation product 8 or cyclomagnesiation product 93, depending on the reaction conditions (Scheme 28).
The scope of applicability of the new cyclomagnesiation reactions developed by our research group is not limited to the above particular examples and has now been extended to a wide range of unsaturated compounds, which has been described in original papers,[95-99] reviews, and monographs.[24] [33][39]
Our attempts to extend the catalytic cyclomagnesiation reactions to other unsaturated compounds such as disubstituted alkynes were also a success. It was demonstrated for the first time that Zr-catalyzed cyclomagnesiation of disubstituted alkynes on treatment with RMgX or R2Mg (conditions: Et2O, ~20°C, 2 h) results in the formation of tetrasubstituted magnesacyclopentа-2,4-dienes 94 in up to 50% yields (Scheme 29).[100]
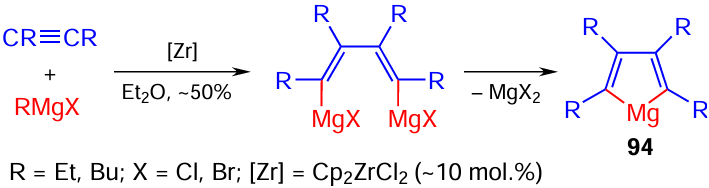
The reaction of an equimolar mixture of disubstituted alkynes and allenes under the above conditions furnishes unique cross-cyclomagnesiation products, 5-alkyl(aryl)idenemagnesa-2-cyclopentenes 95 in moderate yields. Apart from the major cross-cyclomagnesiation products, the reaction gives the corresponding tetrasubstituted magnesacyclopentа-2,4-dienes 94 and 2,5-bis-alkyl(aryl)idenemagnesacyclopentanes 96 (Scheme 30).[100]
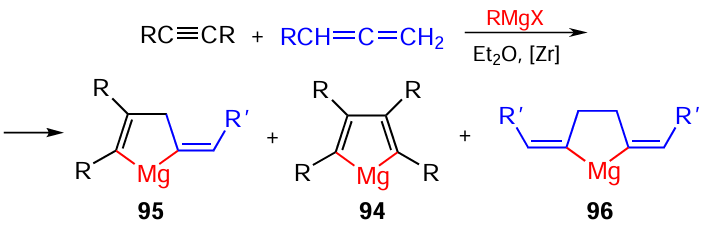
The in situ reaction of the resulting magnesacarbocycles with elemental sulfur (S8) and SOCl2 affords the corresponding thiophenes 97, dihydrothiophenes 98, and thiophanes 99 in > 60% yields (Scheme 31).[101]
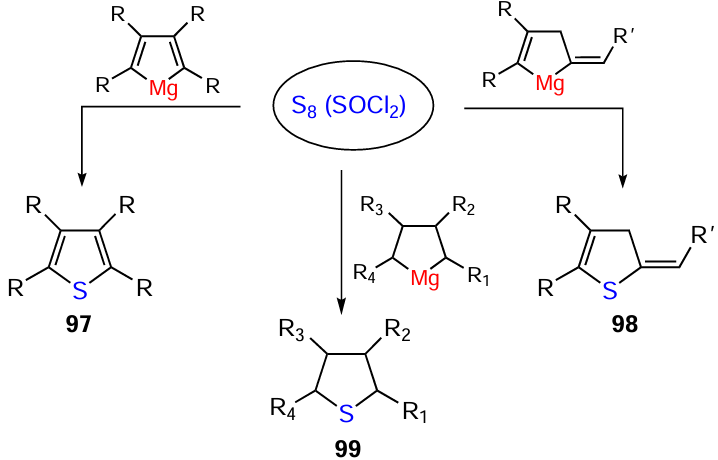
The simultaneous presence of two Mg – C bonds, in combination with the high reactivity of these bonds in magnesacyclopentаnes, magnesacyclopent-2-enes, and magnesacyclopentа-2,4-dienes, makes this class of organometallic compounds fairly promising for the development of one-pot processes for the synthesis of a broad range of practically valuable cyclic, acyclic, and heterocyclic compounds starting from available alkenes, alkynes, and 1,2-dienes.[24]
4. Catalytic cycloboration of alkenes and alkynes
The ever increasing interest in cyclic organoboron compounds is due to their high practical value.[102-104] Currently, effective fungicides and compounds with antiviral activity are known among these derivatives. Lately, increasing attention has been paid to organoboron compounds as monomers for non-linear optics. This stimulated our interest in this class of compounds, and we attempted to extend the developed cyclometallation reactions to the preparation of three- and five-membered boracarbocycles.
The first attempts to carry out direct catalytic cycloboration of alkenes with Et3B did not give the target boracarbocycles; therefore, the efforts were focused on investigation of the replacement of aluminium atoms in aluminacyclopentanes 18 prepared in situ by boron atoms on treatment with BF3 · Et2O. It was shown [105] that under certain conditions [hexane, –10°С, Cp2ZrCl2 (5 mol.%)], these reactions may lead to the target 1-fluoro-3-alkyl-substituted borolanes 100 as molecular complexes with EtBF2 in > 50% yields according to Scheme 32.
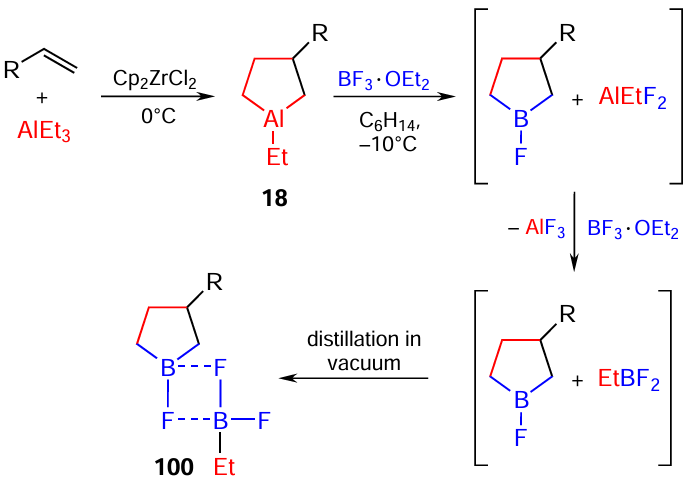
The idea of the possibility of catalytic synthesis of boracyclopentanes relied on the previous publications on the stoichiometric reactions of boron halides (RBX2) with cyclic or α,ω-dimetal organic compounds based on Li, Mg, Sn, Hg, Ir, and Ti.[106-110]
The structures of the obtained borolanes were proved using modern spectral methods and targeted chemical transformations.[111]
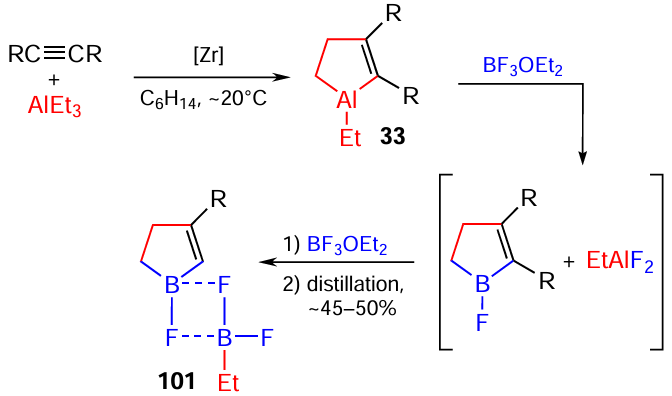
The replacement of alkenes by disubstituted alkynes in this reaction conducted under above-developed conditions [BF3 · Et2O (excess), hexane, –10°С, 0.5 h], which proceeds through the in situ formation of aluminacyclopent-2-enes 33, results in the formation of 4,5-dialkyl-substituted 1-fluoro-2,3-dihydro-1Н-boroles 101, which were isolated as molecular complexes with EtBF2 (Scheme 33).[111]
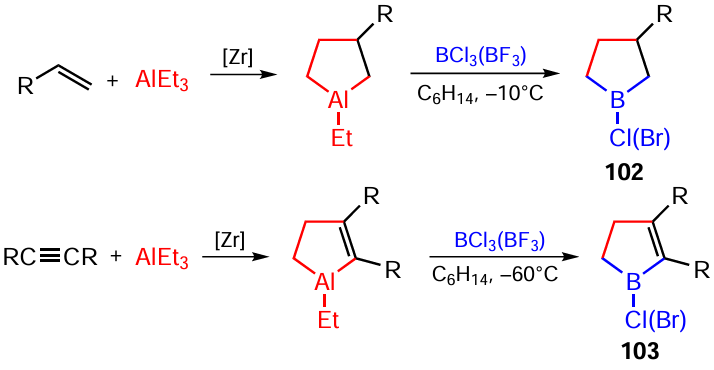
The use of BCl3 or BBr3 instead of BF3 · Et2O for transmetallation of aluminacarbocycles under the same conditions, with B : Al ratio being 2 : 1, provides an original route towards the synthesis of 1-chloro(bromo)-3-substituted borolanes 102 or 4,5-disubstituted 1-chloro(bromo)-2,3-dihydro-1H-boroles 103, difficult to prepare by other methods, which are formed in 70 – 80% yields (Scheme 34).[112]
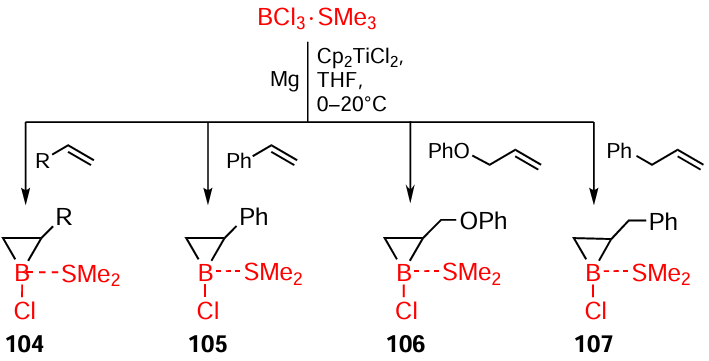
While continuing the attempts to implement direct catalytic cycloboration of alkenes, we accomplished the synthesis of three-membered boriranes 104 – 107, which are difficult to obtain by other methods, in 45 – 80% yields by the reaction of α-olefins with BF3 · Me2S or BF3 · THF and with zirconium complexes being replaced by titanium complexes (Scheme 35).[113][114]
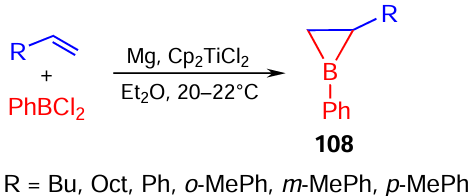
Single boriranes can be synthesized by direct cycloboration of α-olefins and styrenes on treatment with phenyldichloroboranes (PhBCl2), which affords target 1-phenyl-2-alkyl(aryl)boriranes 108 in a pure state, i.e., without being bound to electron-donor complex-forming agents (Me2S or THF), in 55 – 75% yields (Scheme 36).[115]
Apart from the above-described new organometallic reactions of catalytic cyclometallation of unsaturated compounds giving important aluma-, magnesa-, and boracarbocycles, significant progress has recently been made in the studies of catalytic carbo- and cyclozincation, which are widely addressed in original publications [116-120] and a recent exhaustive review.[121]
It is beyond doubt that the above original catalytic organometallic reactions will be further developed and will form a strong foundation for the development of modern industrial chemical processes and for the emergence of new lines of research.
5. Cross-cyclomagnesiation of allenes in the total synthesis of natural compounds and their synthetic analogues
It is known that most functionally substituted unsaturated cyclic and acyclic natural compounds containing Z-double bonds are important for the vital activity of living organisms including humans, animals, insects, and plants as intermediates in metabolic reactions such as biosynthesis of prostaglandins, leukotrienes, lembehynes, acetogenins, indolizidines, insect pheromones, peroxides, epoxides, carboxylic acids, steroids, terpenoids, and other important biologically active derivatives.
Of course, it impossible to describe the whole variety of natural unsaturated compounds, as well as their biological roles and properties within the scope of this review, since a huge array of research and discoveries in this area of chemical and biological science has been accumulated to date, the results being published in numerous papers, patents, monographs, and reviews.[122-128]
The above classes of natural compounds have a wide range of biological activity and are of exceptional interest for the development of advanced low-toxic, selective drugs with antimicrobial, antiviral, antiparasitic, fungicidal, or antitumor activity for practical medicine.
Since most of the mentioned classes of natural compounds are contained in living organisms in very small amounts, the isolation, purification, and practical use of these metabolites is faced with a number of difficulties.
Therefore, fundamental and applied research directed towards the development of new facile and regio- and stereoselective synthetic routes towards these classes of natural compounds and their synthetic analogues, which would make them practically available, is of high interest and applied value.
It is noteworthy that the formation of a single double bond of the specified Z-configuration in a molecule does not appear complicated, considering the accumulated world experience and currently existing synthetic methods. This can be done, for example, by using known reactions such as stereoselective hydrogenation, hydrofunctionalization, and carbometallation of acetylenes, olefin metathesis, and Wittig reaction. However, generation of two or more Z-configured double bonds in the molecules of target compounds with high stereochemical purity is a challenging task because of the poor accessibility of the starting reactants, and low stereoselectivity or multistep nature of the described methods.
Therefore, below we present examples of the total synthesis of natural insect pheromones, unsaturated Z,Z-dienoic acids, lembehynes, acetogenins, macrodiolides, isoprenoids, and their synthetic analogues containing 1Z,5Z-diene moieties characterized by very high stereochemical purity (> 99%) using new versatile reactions of catalytic cyclomagnesiation of various 1,2-dienes developed by our research group (Dzhemilev reaction).[24]
5.1. Organometallic reactions in the stereoselective synthesis of insect pheromones
According to published data, the molecules of a large number of pheromones of insects that threaten agricultural and ornamental plants contain a 1Z,5Z-diene moiety. As a rule, the known methods for the synthesis of pheromones consist of numerous steps (5 – 10 steps) and, most often, the target compounds are formed in very low yields (15 – 25%).[129-131]
Our research group addressed the above challenges related to the synthesis of insect pheromones by using the cross-cyclomagnesiation of aliphatic 1,2-dienes with О-containing allenes taken in 1.2 : 1 ratio under optimized conditions (Et2O, 20 – 22°С, 4 h). This resulted in an efficient strictly selective syntheses of hexadeca-7Z,11Z-dienal (109), a pheromone component of the citrus leaf miner moth (Phyllocnistis citrella); hexadeca-7Z,11Z-dien-1-yl acetate (110), an attractant for the pink cotton bollworm (Pestinophora gossypiella); pentacosa-7Z,11Z-diene (111) and nonacosa-7Z,11Z-diene (112), attractants for the fruit fly Drosophila melanogaster, which are highly demanded for agriculture and gardening and which are formed in high yields (60 – 75%).[132]
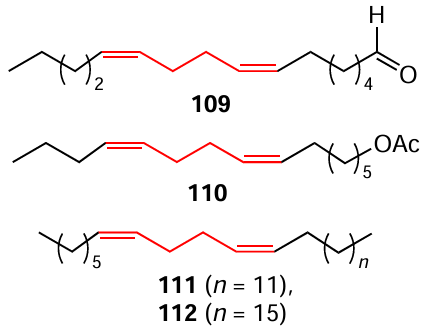
The above insect pheromones were synthesized by the Cp2TiCl2-catalyzed cyclomagnesiation of a mixture of 2-(7,8-nonadien-1-yloxy)tetrahydropyran (114) and 1,2-heptadiene (113) with EtMgBr (conditions: 113 : 114 : EtMgBr : Mg:[Ti] = 10 : 12 : 40 : 30 : 1, Et2O, 6 h, 20 – 22°С) to give magnesacyclopentane (115), which underwent acid hydrolysis to afford diene 116 in 94% yield. Refluxing of tetrahydropyran ether 116 in a МеОН – CHCl3 mixture (1 : 1) in the presence of p-TsOH resulted in the formation of hexadeca-7Z,11Z-dien-1-ol (117) in 89% yield. In the final step, the Swern oxidation of alcohol 117 with oxalyl chloride furnished target hexadeca-7Z,11Z-dienal (109), an attractant for the citrus leaf miner moth (Phyllocnistis citrella), in 92% yield.[132]
Compound 110, an attractant for the pink cotton bollworm Pestinophora gossypiella, was synthesized in an overall yield of 89% by acylation of the above unsaturated alcohol 117 with acetyl chloride (Scheme 37).[132]
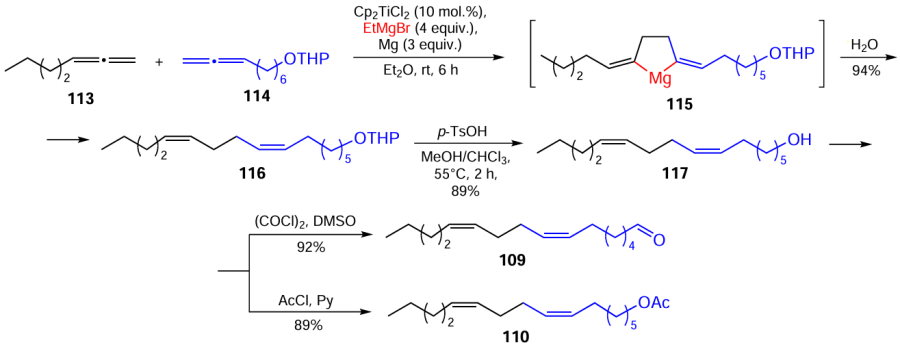
The use of catalytic cyclomagnesiation of 1,2-dienes enabled the development of a strictly stereoselective synthetic route to components of the fruit fly Drosophila melanogaster pheromone.[132]
Cross-cyclomagnesiation of a mixture of 1,2-hexadecadiene (118) or 1,2-eicosadiene (119) with 2-(7,8-nonadien-1-yloxy)tetrahydropyran (114) was performed by treatment of the mixture with EtMgBr in the presence of magnesium metal and the Cp2TiCl2 catalyst (10 mol.%) {conditions: 118 (119) : 114 : EtMgBr : Mg : [Ti] = 12 : 10 : 40 : 30 : 1, Et2O, 20 – 22°С, 6 h}, resulting in the intermediate formation of magnesacyclopentanes 120 and 121, the acid hydrolysis of which yielded dienes 122 and 123. These products were converted to pentacosa-7Z,11Z-dien-1-ol (124) and nonacosa-7Z,11Z-dien-1-ol (125) and the corresponding mesylates (126) and (127) (Scheme 38).[132]
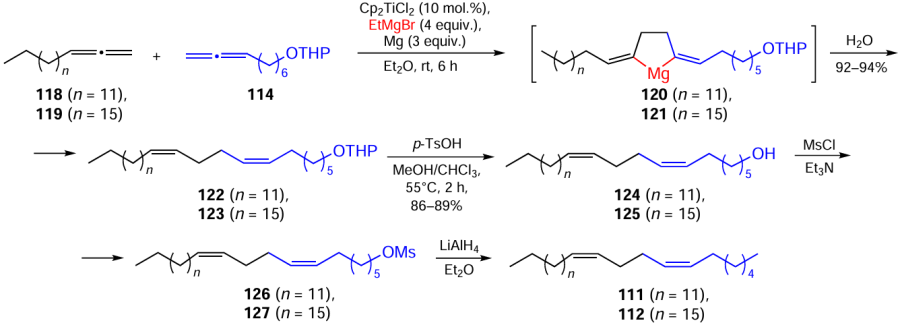
The subsequent reduction of mesylates (126) and (127) with LiAlH4 afforded the target pentacosa-7Z,11Z- (111) and nonacosa-7Z,11Z-dienes (112), which are sex attractants for Drosophilae insects.[132]
The total syntheses of natural insect pheromones demonstrated record high stereoselectivity (>99%) of the one-pot catalytic cyclomagnesiation of 1,2-dienes, which results in the formation of the 1Z,5Z-diene system in the target compounds.
It should be noted that none other known method or classical method described in the world literature for the construction of Z,Z-double bonds has equally high preparative efficiency or equally high stereoselectivity as the catalytic cyclomagnesiation of 1,2-dienes, in particular for the preparation of compounds containing 1Z,5Z-dimethylene-interrupted bonds.
5.2. Stereoselective synthesis of natural and synthetic analogues of Z,Z-diene and Z,Z,Z-triene carboxylic acids
The natural С16 – С34 unsaturated carboxylic acids containing bis-methylene-interrupted Z,Z-double bonds in the carbon chain, isolated previously from sea sponges and gymnosperm fruits, possess a wide spectrum of biological activities, including antimicrobial, antibacterial, antimalarial, antileishmanial, and antitumour activities.[133]
Several research groups established [134-140] that natural 5Z,9Z-dienoic acids are effective inhibitors of topoisomerases, cell cycle enzymes, which catalyze DNA relaxation reactions during replication, thus controlling processes important for the vital activity of cells.
In view of the important role of topoisomerases in the cell division, these enzymes are considered to be key molecular targets for the development of modern anticancer drugs.[141]
Unfortunately, in most cases, low content and difficulty of separation of mixtures of Z,Z-unsaturated carboxylic acids isolated from natural sources hamper evaluation of their biological activity, which is required to design modern medicinal agents. Therefore, active studies dealing with the development of total syntheses of natural 5Z,9Z-dienoic acids have been undertaken in top laboratories of various countries. Most often, the developed methods were not free from drawbacks as they consisted of numerous steps and gave the target acids in low yields; furthermore, these methods were not highly stereoselective and gave stereoisomeric mixtures of cis- and trans-carboxylic acids.
Meanwhile, the presence of trans-double bonds in the synthesized carboxylic acids reduces the efficiency of topoisomerase inhibition by two and even three orders of magnitude.
It should be emphasized that all problems listed above can be avoided if the versatile catalytic cyclomagnesiation of unsaturated compounds, namely 1,2-dienes, is used as the key method for the synthesis of Z,Z-diene carboxylic acids.[129]
In order to confirm this, we performed the intermolecular cyclomagnesiation of aliphatic and oxygen-containing 1,2-dienes with Grignard reagents (RMgX) in the presence of the Cp2TiCl2 catalyst (5 – 10 mol.%) under the developed conditions (Et2O, EtMgBr, Mg, ~20°C, 4 – 6 h). After hydrolysis, this gave Z,Z-diene compounds, which were then oxidized with the Jones reagent to give the target 5Z,9Z-diene carboxylic acids 128 (Scheme 39).[142]
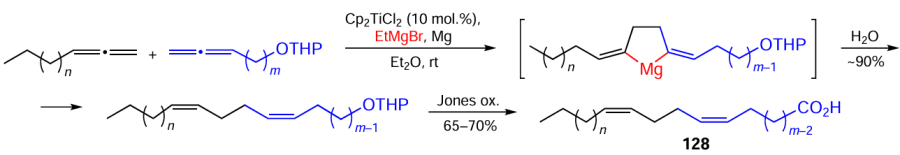
The above method was used to develop an effective preparative-scale stereoselective synthesis of a broad range of diene carboxylic acids containing a 1Z,5Z-diene group in various positions of the hydrocarbon chain relative to the carboxyl group.[143]
Thus, varying the position of the Z,Z-double bonds in unsaturated acids relative to the carboxyl group resulted in the preparation of compounds that had high inhibitory activity against topoisomerase I or topoisomerase II and acids with inhibitory activity against both topoisomerases simultaneously.[143]
As follows from the above, the catalytic cyclomagnesiation has a broad synthetic potential and allows the preparation of numerous natural and synthetic Z,Z-diene carboxylic acids with different positions of double bonds as single stereoisomers. This opens up prospects for practical application of this class of compounds for the development of modern and highly effective drugs.
The effective strategy for the synthesis of Z,Z-dienoic acids developed by our research group was successfully used to build higher unsaturated carboxylic acids 129 containing simultaneously three stereoisomerically pure Z,Z,Z-double bonds (Scheme 40).[144]
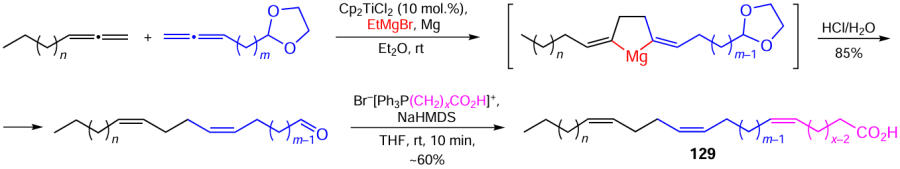
Using natural steroids, terpenoids, coordination compounds, and stereoisomerically pure natural and synthetic Z,Z-dienoic acids, we obtained hybrid molecules with various steroids, iodolactones, macrodiolides, and cyclophanes and extensively investigated the biological properties of the products.
The hybrid molecules possess high biological activity and are of exceptional interest as a base for the development of advanced anticancer drugs, in particular macrocyclic compounds.[145-150]
5.3. New organometallic reactions in the synthesis of natural acetogenins and lembehynes
Natural acetogenins were isolated from the leaves, roots, and bark of Annonaceae plants. These acetogenins are C32 – C34 carboxylic acids containing a hydroxylactone moiety.[151-153]
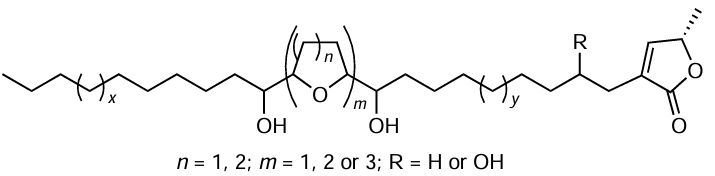
It is noteworthy that acetogenin molecules may contain additional hydroxy, keto, epoxy, tetrahydrofuran, and tetrahydropyran groups as well as double and triple bonds.
The enhanced interest of researchers in natural acetogenins and their derivatives is due to the high biological activity of these compounds, which includes insecticidal, fungicidal, antiprotozoal, immunosuppressive, and anticancer properties.[154]
In particular, natural acetogenins have a cytotoxic effect on multidrug-resistant tumours, being among the most potent currently known inhibitors of the mitochondrial complex.[155-157]
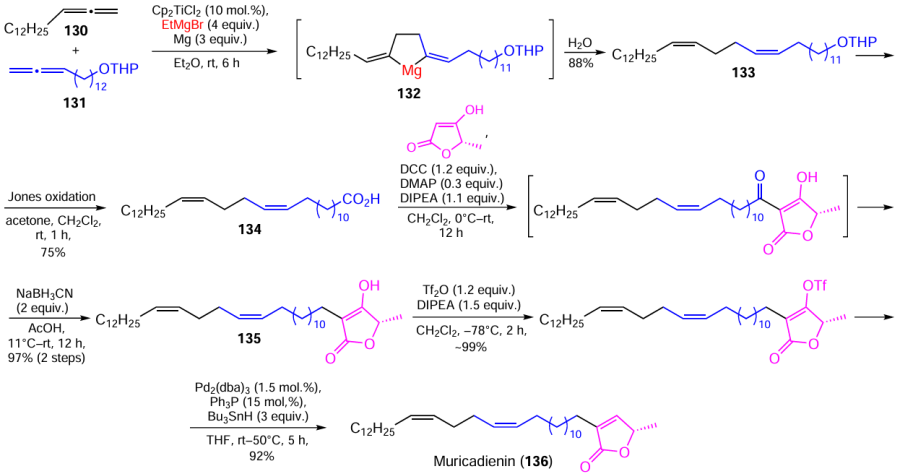
The practical value of natural acetogenins in the design of modern drugs stimulated us to develop an effective route for the total synthesis of muricadienin, a key precursor of this class of compounds. Muricadienin, first isolated in a minor amount from the roots of Annona muricata, exhibits pronounced anticancer, antiviral, and antibacterial properties.
13Z,17Z-Triaconta-13,17-dienoic acid (134), the key synthon for the preparation of muricadienin, was synthesized in two steps by the cross-cyclomagnesiation of 1,2-pentadecadiene (130) and 13,14-pentadecadien-1-ol tetrahydropyran (THP) ether (131) with EtMgBr catalyzed by Cp2TiCl2 (10 mol.%). This was followed by the Jones oxidation of the tetrahydropyran ether of triaconta-13Z,17Z-dien-1-ol (133), resulting from acid hydrolysis of dialkylidenemagnesacyclopentane (132). The final step of the synthesis was the Fries rearrangement giving the terminal α-substituted butenolide 135.[158][159] The overall yield of stereochemically pure muricadienin (136) was approximately 65% (Scheme 41).
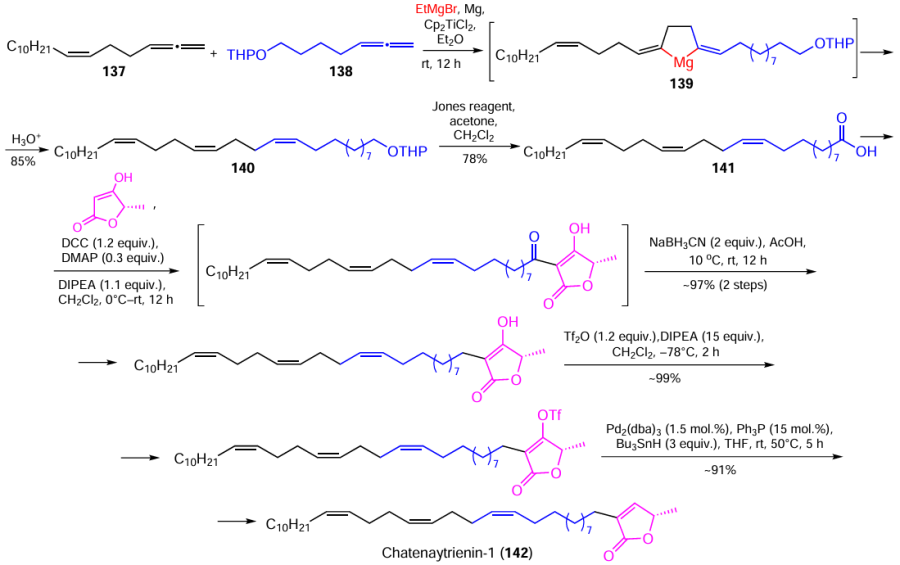
Using a similar procedure, chemists successfully synthesized a rare derivative of acetogenin, chatenaytrienin-1 (142), containing 13Z,17Z,21Z-double bonds in the hydrocarbon chain. 1-Dodecatriacontane-13Z,17Z,21Z-trienoic acid (141), the key synthon for the construction of the chatenaytrienin molecule, was synthesized in two steps by catalytic cross-cyclomagnesiation of heptadeca-1,2,6Z-triene (137) and 13,14-pentadecadien-1-ol pyran ether (138) (Scheme 42)[160]
The above examples of the synthesis of natural unsaturated carboxylic acids and acetogenins indicate that the catalytic cyclomagnesiation reaction of 1,2-dienes of various structures developed by our research group has a broad synthetic potential and is currently one of the most efficient synthetic tools for the stereoselective construction of 1Z,5Z-dienoic and 1Z,5Z,9Z-trienoic acids.
Currently, our efforts are directed towards the development of methods for the synthesis of a number of natural and synthetic long- and short-chain homologues of muricadienin and chatenaytrienins in order to produce them in reasonable amounts for large-scale biological assays for evaluation of their anticancer, antibacterial, and antiparasitic activities.
Lembehynes are structurally very similar to natural unsaturated carboxylic acids and acetogenins, as they contain 1Z,5Z-double bonds and an acetylenic alcohol in the terminal position. They were first isolated in minor amounts from the Indonesian sea sponge Haliclona sp.[161-163] and showed a high neuritogenic activity in pheochromocytoma (PC12) and neuroblastoma (2A) cells.
According to the predictions of the World Health Organization, due to the constant increase in the average life expectancy of people, neurodegenerative diseases will become by 2040 more common than cardiovascular diseases or cancer. The drugs currently used for the treatment of neurodegenerative diseases are aimed at mitigating the consequences of oxidative stress and neuronal death by reducing the effects of various neurotrophic factors and neuropeptides. Neurotrophic factors are unable to penetrate the blood – brain barrier (BBB) due to their large molecular size and hydrophilicity. This feature is a significant obstacle to the use of these drugs for the treatment of neurodegenerative diseases. Neuropeptides, on the other hand, are extremely expensive and often ineffective, and, in addition, they quickly degrade.
The lack of effective methods for the synthesis of natural lembehynes, along with their very low content in natural objects, were the main obstacles hampering comprehensive investigation of their biological properties and full disclosure of their biomedical potential in order to develop advanced drugs for the treatment of neurotrophic diseases.
Mention should be made of the pioneering study by Murakami and co-workers, who reported in 2001 the first 11-step total synthesis of lembehyne А, with the key synthon, 4Z,8Z-pentacosa-4,8-dien-1-ol, being prepared via a six-step route.[164]
Relying on the results we obtained previously in the synthesis of natural compounds containing a 1Z,5Z-diene moiety, we proposed a concept for using catalytic organometallic cyclomagnesiation of 1,2-dienes for the synthesis of neuritogenic acetylenic alcohols, including lembehynes A, B, and C.
Initially, we synthesized the key monomer for the preparation of lembehyne A (148),[165][166] particularly, cyclomagnesiation of a mixture of 1,2-nonadecadiene (143) and 4,5-hexadien-1-ol tetrahydropyran ether (144) with EtMgBr gave the corresponding 1,5-dialkylidenemagnesacyclopentane 145, which was hydrolyzed to give the tetrahydropyran ether of pentacosa-4Z,8Z-dien-1-ol (146) in ~ 89% yield. The subsequent refluxing of this product in a MeOH – CHCl3 mixture (1 : 1, v/v) in the presence of p-TsOH furnished the target pentacosa-4Z,8Z-dien-1-ol (147) in an overall yield of ~ 70% (Scheme 43).[165][166]
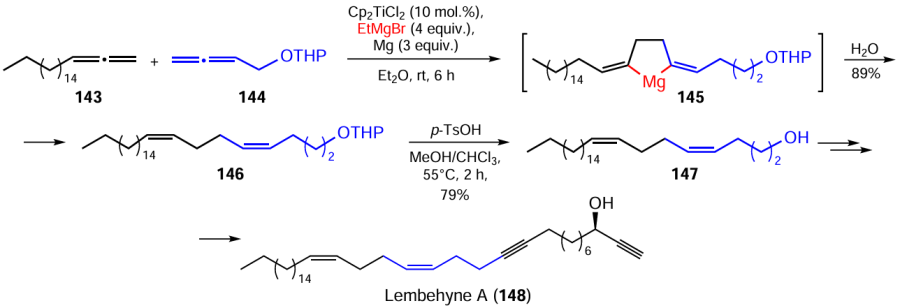
By analogy with the above pathway, the total synthesis of lembehyne B (152) was accomplished by cross-cyclomagnesiation of a mixture (1 : 1, mol./mol.) of 1,2-nonadecadiene (143) and dioxolane (149) under the following conditions: Et2O, ~20 – 22°C, 7 h. The hydrolysis of magnesacyclopentane 150 afforded tetraconta-13Z,17Z-dienal (151) in a yield of approximately 88%. The subsequent stereoselective transformations of aldehyde 151 furnished the desired lembehyne B (152) in an overall yield of 84% (Scheme 44).[165]
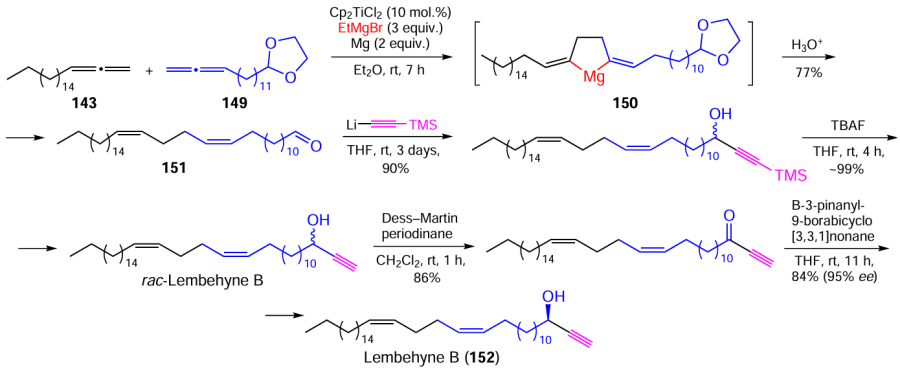
Using advanced cell technologies, it was shown for the first time [165] that lembehyne B functions as a selective inducer of early apoptosis in Jurkat, HL-60, and K562 cell cultures.
Basing on the experience in the stereoselective synthesis of natural pheromones, unsaturated carboxylic acids, acetogenins, and lembehynes containing 1Z,5Z-diene or 1Z,5Z,9Z-triene moieties in the molecules, we developed an effective method for the preparation of natural lembehyne С (153) (Scheme 45).[167]
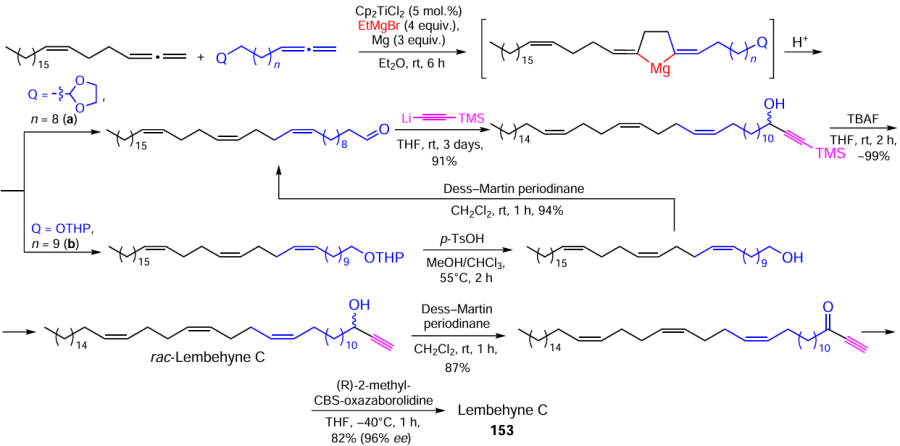
The range of biologically active compounds synthesized using the new catalytic homo- and cross-magnesiation of allenes as the key stage is not limited to the above examples; it should be noted that we have synthesized macrodiolides and cyclophanes, bis-hydroxyfurans, iodolactones, ionic liquids, and other 1Z,5Z-diene derivatives.[168-178]
6. Negishi, Suzuki – Miyaura, and Stille organometallic reactions
6.1. Negishi reaction
A considerable achievement in the field of organometallic synthesis is the Cp2ZrCl2-catalyzed acetylene carboalumination with Me3Al discovered in 1978 by Professor Negishi, an American chemist (Scheme 46).[179][180]
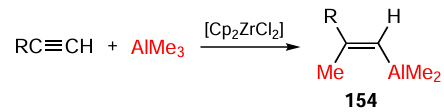
It was shown [180] that the mechanism of the Negishi reaction implies the initial formation of Zr–Al bimetallic complex 155, which is intramolecularly converted to organometallic compound 156 containing a Zr – CH3 bond.
The subsequent coordination and activation of the acetylene molecule affords π-complex 157. The next step is the anti-Markovnikov carbozirconation of activated acetylene to give syn-alkenylzirconocene 158. The transmetallation of the zirconium atom by aluminium under the chosen conditions results in the formation of the target alkenyldimethylaluminium 154 as shown in Scheme 47.
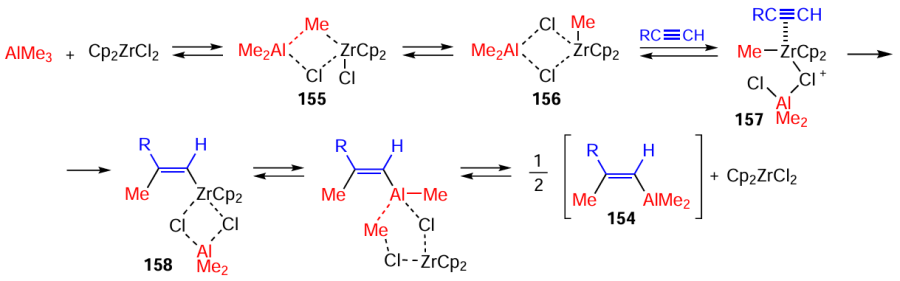
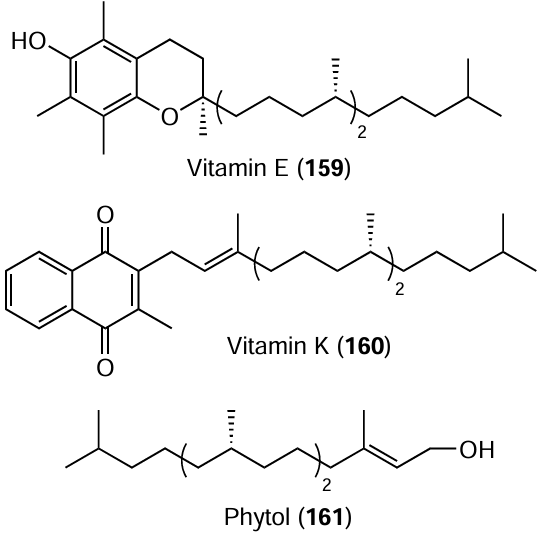
The discovered and thoroughly elaborated Negishi reaction, stereoselective caarboalumination of terminal and disubstituted acetylenes with Me3Al catalyzed by chiral Zr metallocene complexes, is an efficient synthetic tool for the construction of natural regular isoprenoids [vitamin Е (159), vitamin K (160), phytol (161)], medicinal agents, and toxins.[180-182]
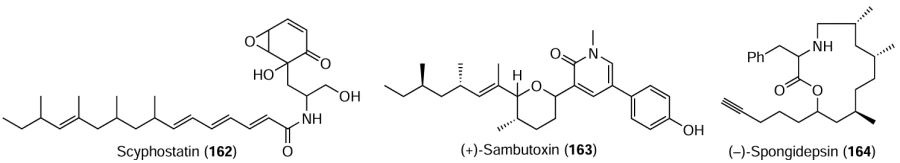
The synthesis of structurally complex natural compounds such as scyphostatin (162), (+)-sambutoxin (163), (–)-spongidepsin (164), and other important metabolites are of special interest and practical value.[183-186]
The above fundamental and applied studies conducted by Negishi and co-workers are widely addressed in the world literature, including relevant monographs, reviews, and original papers; therefore, it is inappropriate to discuss these results in detail in this review.[187-189]
6.2. Suzuki – Miyaura
The Suzuki – Miyaura reaction discovered in 1977 is one of important organometallic reactions widely used in the world practice.[190] This reaction is an effective tool for the construction of new carbon–carbon bonds. It represents the Pd-catalyzed reaction of aryl- and vinylboronic acids with aryl and vinyl halides (Scheme 48).[190-192]
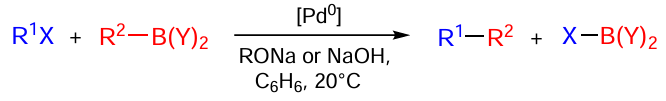
The organoboron nucleophiles used in the Suzuki–Miyaura reaction include boranes, boronic acids, and boronic acid esters.
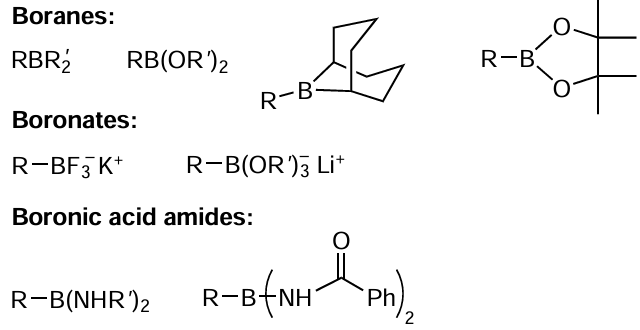
The ready availability of organoboron compounds and the simplicity of synthesis of the starting reactants make the Suzuki–Miyaura reaction quite attractive for the preparation of biaryls 165, bis-heterocycles 166, and natural metabolites 167, previously difficult to obtain, in 85 – 95% yields (Scheme 49).
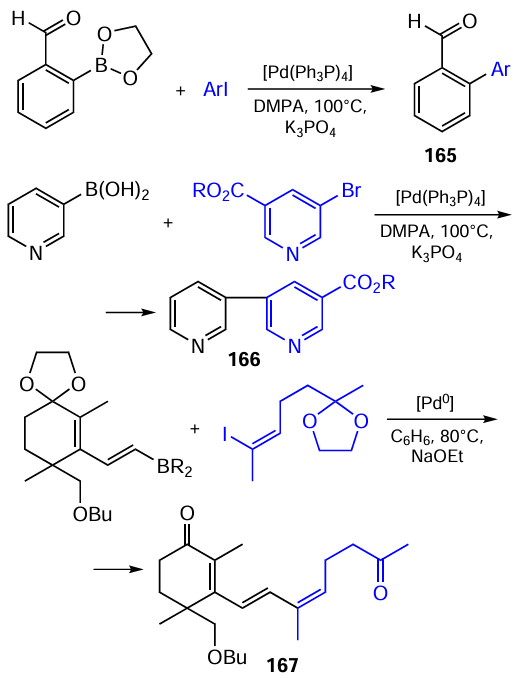
The mechanisms of the Suzuki – Miyaura reaction includes the oxidative addition of the appropriate aryl halides to the central Pd0 atom of the catalyst to give the R – Pd – X complex. Under the chosen reaction conditions, this complex reacts with a base (RONa) to give the compound R – Pd – OR, which reacts with an organoboron nucleophile giving the Pd2+ complex containing two covalently bound carbon moieties. The subsequent reductive elimination of the covalently bound carbon groups in the R 1 – Pd–R2 complex results in regeneration of the catalyst (Pd0) and gives the target compound, as depicted in the general Scheme 50.
Regarding the creativity and degree of originality of studies concerning the search for and implementation of new catalytic systems for the Suzuki–Miyaura reaction, apart from the study of conventional catalysts,[193] attention is attracted by recent works by Academician V.P.Ananikov’s school dealing with the potential use of aerobic bacterial cells Paracoccus yeei as a support for palladium nanoparticles,[194] which, as it was shown in the fundamental studies of this school, are responsible in most cases for the formation of target products.[195-203]
In addition to the high activity of the palladium/Paracoccus yeei catalyst comparable with that of the best commercial catalysts, one more appreciable benefit of this catalyst is that bacteria do not affect analysis of reaction mixtures by NMR spectroscopy and gas chromatography–mass spectrometry, which is extremely important for the study of catalytic reaction mechanisms.[204-206]
In the context of both fundamental and applied studies related to the use of the Suzuki–Miyaura reaction, there is one more line of research developed by Academician V.P.Ananikov’s school that, in our opinion, cannot be left aside, namely, the development of new criteria for safety assessment and degree of environmental impact of industrially promising reactions, that is, the Suzuki–Miyaura and Mizoroki – Heck reactions. The system uses bio-profiles derived from the cytotoxicity data of the starting reactants and reaction products.[207-212] The derived bio-profiles identify the compounds with the highest and lowest contributions to the ‘overall cytotoxicity’ of a particular chemical process. The bio-factors calculated from the bio-profiles reflect the variation of the ‘overall cytotoxicity’ during the reaction. The developed assessment criterion is characterized by simplicity, high information content, clarity, and versatility, which provides hope that it will be universally implemented in the near feature.
It should be noted that the popularity and the scope of application of the Suzuki reaction increase every year due to the efficiency of this method for the synthesis of a variety of natural compounds and drugs being implemented in industry.
6.3. Stille reaction
It is quite remarkable that the Stille reaction was discovered almost simultaneously with the Suzuki reaction and reported in 1978.[213-216]
The Stille reaction is the cross-coupling of aryl-, alkenyl-, alkynyl-, and alkylstannanes with aryl and alkenyl halides and pseudohalides (triflates and aryldiazonium or iodonium salts) catalyzed by low-valent palladium complexes.
Under the selected reaction conditions, the leaving halide or pseudohalide group is replaced by a group bound to stannane derivatives thus forming a new carbon–carbon bond (Scheme 51).
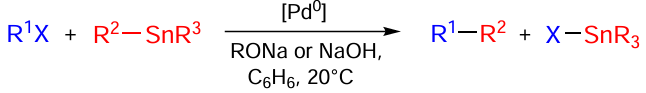
It should be noted that the reaction is carried out under inert atmosphere and in moisture- and oxygen-free solvents.
As regards the mechanism of the Stille reaction, it largely resembles the mechanism of the Suzuki reaction, including key steps such as the oxidative addition of the starting halides or pseudohalides to the central atom of the catalyst, ligand exchange, and reductive elimination to give a new carbon–carbon bond (Scheme 52).[217]
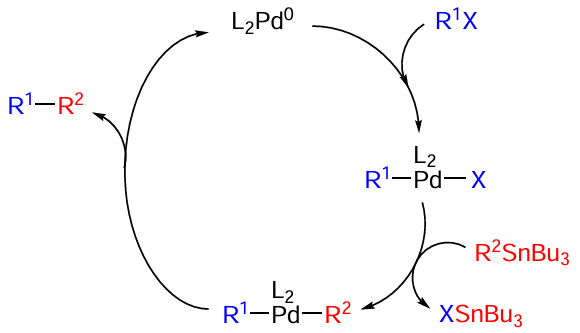
As a rule, these reactions occur at 20 – 100°С in aprotic and polar solvents and give the target products in fairly high yields (60 – 90%).
The cross-coupling method developed by Stille and co-workers is widely used not only in synthetic laboratory practice, but also in industry, especially for the production of drugs and natural metabolites.
7. Conclusion
An interesting and practically valuable trend of organic and organometallic chemistry is the discovery, development, and application of new effective organometallic reagents and fundamental reactions for the targeted transformation of saturated, unsaturated, and heteroatom-containing synthetic and natural organic compounds into target products, including the formation of new carbon – carbon, metal – carbon, and heteroatom – carbon bonds. This allows deliberate design of organic and organometallic compounds of a specified structure and configuration.
The present review integrates for the first time and gives a systematic account of the results of studies dealing with the development and application of new versatile fundamental organometallic reactions that are now widely used in synthetic practice and in industry.
The implementation of the versatile fundamental organometallic reactions described in this review has profoundly changed the field of organic and organometallic chemistry by giving rise to a new synthetic platform, which served as the basis for numerous efficient regio- and stereoselective methods. These methods provided the synthesis of various classes of metallacarbocycles, including small, medium, macrocyclic, and giant structures. A distinctive feature of these metallacarbocycles is their ability to be easily transformed into oxygen-, nitrogen-, sulfur-, and phosphorus-containing compounds over a single preparative step.
Particularly these studies led to the discovery of the catalytic replacement of transition metal atoms (Ti, Zr, Hf, Ta, Co) in metallacarbocycles by main group metal atoms (Mg, Zn, Al, Ga, In, B), which allowed the development of new fundamental reactions such as catalytic ethylmagnesiation, cyclomagnesiation, carbozincation, cycloalumination, and cycloboration of alkenes, alkynes, and 1,2-dienes. These reactions have gained worldwide recognition and have been given the general name Dzhemilev reaction.[24][40]
Mention should be made of the wide popularity and widespread use of organometallic reactions first proposed by Negishi, Suzuki – Miyaura, and Stille. These reactions have attracted considerable attention due to their efficiency for generation of new carbon – carbon bonds. Unlike these reactions, the Dzhemilev reaction is used to form not only carbon – carbon bonds, but also metal – carbon and heteroatom – carbon bonds in a single preparative step. This versatility allows the design of acyclic and cyclic metal- and heteroatom-containing organic compounds, which have high practical value. In view of the importance and practical value of the organometallic reactions and new synthetic methods discussed in this review, which allow the construction of functionally substituted compounds, carbocycles, metallacarbocycles, and heterocycles of a specified structure and configuration from readily available monomers and reactants over a single preparative step, we can predict with high probability that these results will be promising for the development of advanced processes for the production of useful compounds and materials for chemical, pharmaceutical, polymer, and defence industries and for the production of materials for microelectronics.
It is beyond doubt that all name organometallic reactions mentioned above are used in the world practice and are of exceptional interest for the elaboration and industrial use of modern chemical engineering processes as well as for the emergence of new research areas.
The review was written within the framework of the State Assignment of the Ministry of Science and Higher Education of the Russian Federation.
References
















