


Keywords
Abstract
N-(Het)aryl- and N-benzylamides are a practically relevant class of compounds that are widely used in medicinal chemistry, agrochemistry, catalysis, organic synthesis and materials chemistry. The amide group is one of the important structural moieties that form the basis of proteins, peptides, pharmaceuticals, polymeric functional materials, etc. To this day, the Ritter reaction, discovered in 1948, is often used in chemical synthesis to obtain amides, especially sterically crowded ones. Nitriles can act as a source of the amide function in the synthesis of N-(het)aryl- and N-benzylamides. The study of such transformations, which are carried out both under conditions of classical organic synthesis and using photochemistry, electrochemistry and photoelectrochemistry, is a relevant area of research, as evidenced by modern works on this topic. This review summarizes and analyzes the most significant achievements of recent years in the field of synthesis of N-(het)aryl- and N-benzylamides by reactions following the Ritter reaction pathway. Processes based on traditional methods of organic chemistry are presented, as well as modern of "green" chemistry approaches using photo- and electrochemical activation of reagents. In addition, known reactions of amidation of heteroaromatic substrates with nitriles, methods of amidation using three-component systems, and amidation of N-(hydroxymethyl)saccharin are considered in separate Sections. Proposed mechanisms of transformations that ensure the selectivity of the processes are discussed in detail.
The bibliography includes 122 references.
1. Introduction
Aromatic and heteroaromatic amide-substituted compounds are widely used in organic synthesis, medicinal chemistry, materials chemistry, agrochemistry, and organic catalysis.[1-3] The amide functionality is one of the most important moieties that form the basis of proteins, peptides, and many other biologically significant compounds.[4] Currently, about 25% of commercial pharmaceuticals and 75% of drug candidates contain at least one amide or amino group.[5] In addition, agrochemicals with herbicidal and antifungal activity (for example, propanil, boscalid), and many drugs used in practice (paracetamol, niclosamide, imatinib) contain an arylamide moiety (Fig. 1).[6-8]
![[{"id":"ZevCd7Kbxm","type":"paragraph","data":{"text":"Amide-substituted agrochemicals and pharmaceuticals"}}]](/storage/images/resized/QynIqISnSdSyc8h9txqXnpb58Y844wK7bpkI31OB_xl.webp)
Aromatic amides are essential precursors for the synthesis of many nitrogen-containing heterocyclic and carbocyclic systems, and also act as raw materials for modern materials used in various industries.[9-12] The practically important properties of nitrogen-containing compounds with an amide group stimulate the search for new reactions leading to compounds of this class, especially under environmentally friendly, so-called ‘green’, synthesis conditions.[13]
The reaction discovered by John Ritter[14][15] in 1948 and named after him involves the reaction of nitriles with alkenes or tertiary alcohols to give amides under acidic conditions (Scheme 1).[16] Under such conditions, alcohols 1 and alkenes 2 first form active carbocationic intermediates 3 and 4, which react with nitriles 5 to form key nitrilium ions 6 and 7 (their resonance structures are designated by numbers 8 and 9). After adding water, the latter furnish intermediates 10 and 11, which ultimately transform into the corresponding amides 12 and 13.
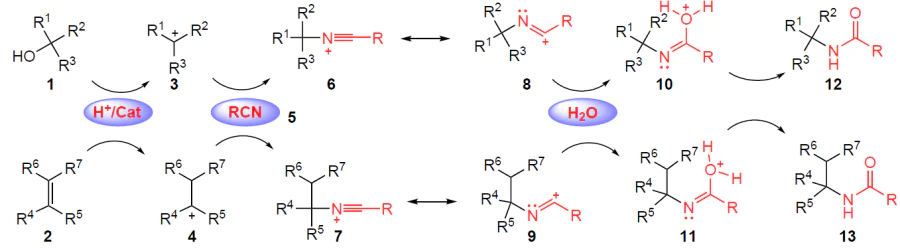
In recent decades, not only alcohols and alkenes but also alternative substrates have been used in the Ritter reaction. Thus, to date, variants of reactions proceeding according to the Ritter reaction type are known involving carboxylic acids,[17] haloalkynes,[18] aldehydes,[19] aryltriazenes,[20] aryldiazonium tetrafluoroborates,[21] cyclic diaryliodonium salts,[22] which open the way to various N-substituted amides, in particular N-benzylamides and N-arylamides. The Ritter reaction is used in the synthesis of biologically active molecules, namely antiallergic,[23] antibacterial,[24] anti-influenza [25] and antimicrobial drugs.[26] Reactions using nitriles as building blocks are one of the most attractive areas of organic synthesis, including electrochemical synthesis. Due to its good conductivity and relatively environmentally friendly properties, acetonitrile has become a sought-after reagent suitable for the production of nitrogenous compounds (e.g., amides and nitrogen heterocycles) or other cyano derivatives.[27]
The preparation of practically significant N-aryl- and N-benzylamides using nitriles as amidating reagents is considered a relevant area of research, as evidenced by new works that have appeared in the last decade, in which these transformations are carried out both under the conditions of classical organic synthesis and using electrochemical and photoelectrochemical approaches.[28][29] Nevertheless, in the available literature on this topic (see, e.g., Refs [30-35]), there is no systematic review devoted to the use of Ritter-type reactions for the synthesis of N-arylamides, N-benzylamides and N-hetarylamides, especially taking into account the latest advances in the field of photochemical and electrochemical methods. The present review aims to fill this gap: it summarizes and systematizes both traditional and modern approaches to the synthesis of N-substituted amides using nitriles as amidating reagents. Particular attention is paid to the possibilities of photo- and electrochemistry in the context of sustainable, highly selective and functionally directed organic synthesis.
The review begins with a discussion of Ritter-type reactions that yield N-aryl- and N-benzylamides under classical or photochemical conditions (Section 2) and with the use of electrochemistry (Section 3). It then presents known amidation reactions of some heteroaromatic substrates with nitriles (Section 4), and concludes with a discussion of amidation methods involving three-component systems and amidation of N-(hydroxymethyl)saccharin (Section 5).
2. Amidation of aromatic compounds with nitriles
2.1. Amidation of aryl- and benzylhalides
In 2013, Yu and co-workers[36] described Cu-catalyzed amidation of aryl halides 14 with acetonitrile (5a). Various N-arylamides 15 were obtained in yields from 39% to 75% using monohalogenated benzenes as substrates (Scheme 2). This reaction runs for 15 h on heating to 100°С in the presence of a mixture of bases, KOH (1.5 equiv.) and Cs2CO3 (0.5 equiv.).
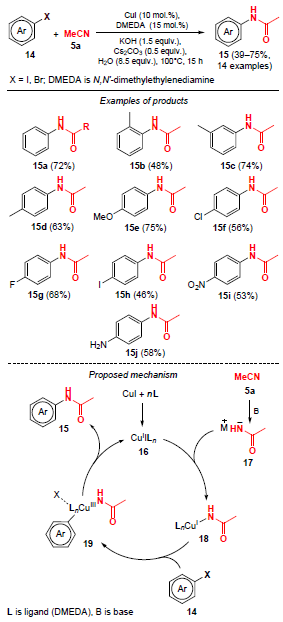
The plausible pathway of this catalytic process is shown in Scheme 2. The copper catalyst 16 reacts with amide salt 17, formed in the first step during the hydrolysis of acetonitrile under basic conditions, to give intermediate complex 18. Aryl halides 14 undergo an oxidative addition to complex 18 to afford intermediate 19. Next, reductive elimination gives the desired products 15 and catalyst 16 is released to complete the catalytic cycle.
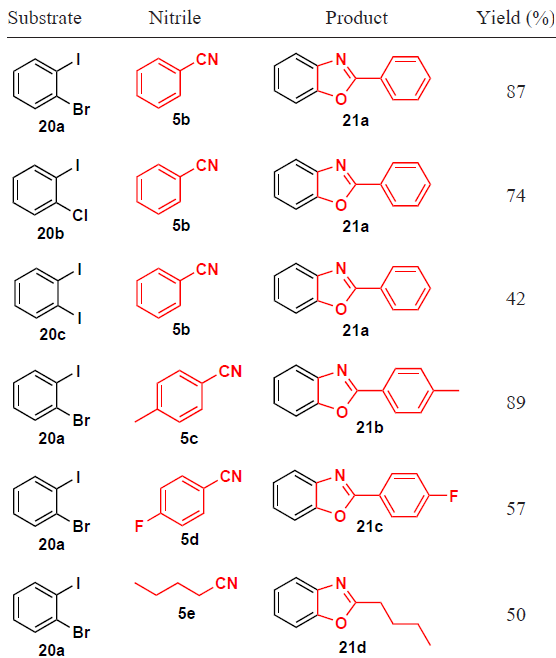
Using 1,2-dihalobenzenes (e.g., iodobenzenes 20) in this reaction, benzoxazoles 21 were isolated as reaction products rather than N-arylamides (Table 1).
A simple and efficient method for the synthesis of substituted N-benzylamides by coupling benzonitrile (5b) with benzyl halides 22 was proposed by Feng et al.[37] (Table 2). The reaction runs on heating to 80°C in a solvent-free fashion using FeCl2 · 4 H2O as a catalyst. This method provides an access to various N-benzylbenzamides 23 in high yields of up to 96% in 2 h. Acetonitrile turned out to be an unsuitable reagent for this transformation, since it gives no amidation products.
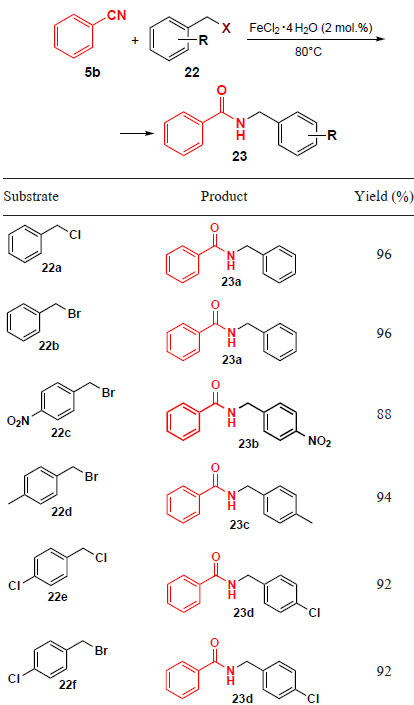
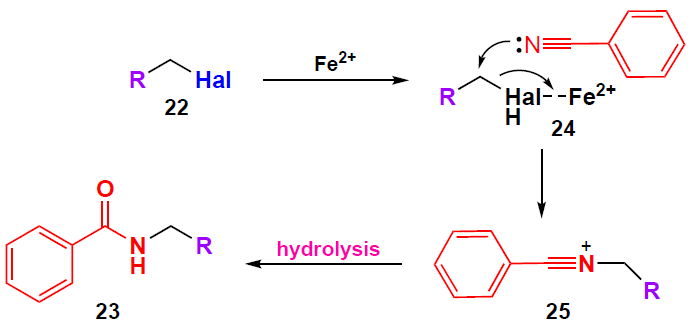
The proposed mechanism of the FeCl2 · 4 H2O-catalyzed coupling of compounds 5b and 22 is shown in Scheme 3. From a mechanistic point of view, the catalytic efficiency of Fe2+ is related to its properties as a Lewis acid, which promotes the amidation process by facilitating the cleavage of the C – Hal bond followed by nucleophilic substitution of the halogen atom in complex 24 with benzonitrile. In the final step, the hydrolysis of intermediate 25 gives N-benzylamide 23.
In 2018, Feng et al.[38] proposed a similar approach to N-benzylamides based on benzyl halides and nitriles using an low-cost and effective catalyst Zn(ClO4)2 · 6 H2O. The reaction was carried out by heating to 80°C for 3 – 5 h in the absence of solvent to give the corresponding amides in high yields (up to 98%).
A year later, Perera and Aakeröy[39] reported a Ritter-type reaction of acetonitrile-D3 (5f) with bromodiphenylmethane 26, which led to deuterated N-benzhydrylacetamide 27 in the presence of the Cat1 catalyst (Scheme 4).
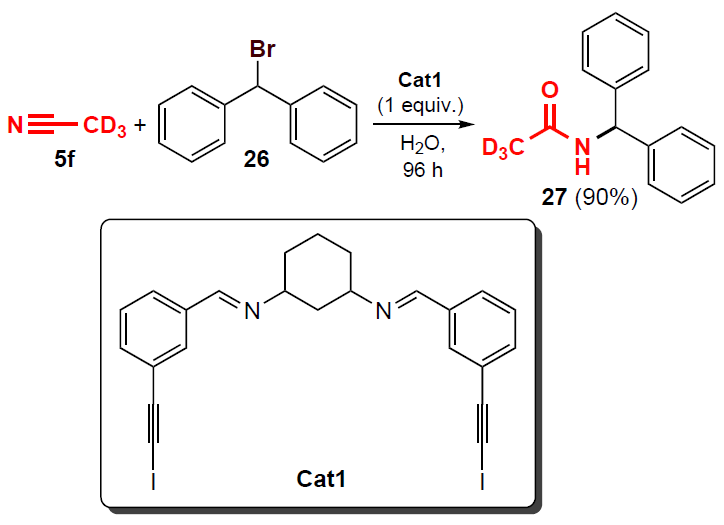
Deuteroacetonitrile acts both as a solvent and a nucleophile in this reaction. In the absence of a catalyst, the amidation product 27 is not formed even after > 96 h. According to 1H NMR data, the addition of 1 equiv. of Cat1 provides 90% conversion of substrate 26 to product after 96 h, with no by-products or decomposition of the catalyst per se being observed. When 10% of the same catalyst is added to the reaction mixture, the conversion of bromide 26 to amide 27 is 33%, and using 20% of the catalyst, the yield of the product increases to 63%.
2.2. Amidation of diaryliodonium salts
The synthesis of N-arylamides via copper-catalyzed amidation of diaryliodonium salts 28 with nitriles was proposed by Ji and co-workers [40] in 2021 (Scheme 5). Various substituted aromatic and aliphatic nitriles can be used in this reaction, with N-arylamides 15, 29 – 32 being obtained in moderate to high yields (57 – 90%).
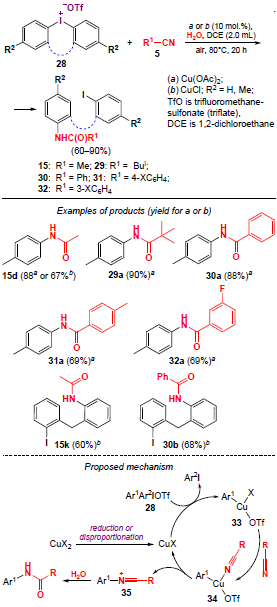
The plausible reaction pathway is shown in Scheme 5. Presumably, in the first step, the CuX2 catalyst disproportionates or is reduced to its active form CuX. Then, diaryliodonium salts 28 in the presence of a CuI-based catalyst form a highly active electrophilic copper(III) intermediate 33. The latter reacts with nitriles, and the reductive elimination of intermediate 34 produces aryl cationic species 35, which results in the release of the active form of the CuI catalyst and its regeneration in the catalytic cycle. In the final step, cations 35 are hydrolyzed to give the desired reaction products, N-arylamides.
When acyclic diaryliodonium salts are used in a similar reaction, N-arylamides are formed, and the coupling of cyclic diaryliodonium salts with nitriles leads to diarylmethanamides through the iodonium ring-opening with retention of the iodine atom in one of the aryl moieties. The catalyst Cu(OAc)2 turned out to be more suitable for the amidation of acyclic diaryliodonium salts, while the authors preferred CuCl for the formation of diarylmethanamides. A similar method for the preparation of diarylmethanamides (yields up to 82%) by CuCl-catalyzed reaction between cyclic diaryliodonium salts and nitriles was developed by Chen and co-workers.[22]
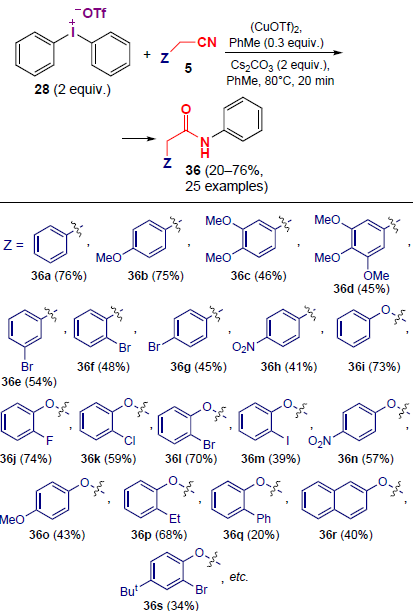
Another simple and practical synthetic approach to N-arylamides 36 using readily available benzyl nitriles and diaryliodonium salts 28 as electrophilic coupling partners was proposed in 2021 by Gillaizeau and co-workers[41] (Scheme 6). Benzyl nitriles with electron-donating and electron-withdrawing groups give the desired products in moderate and good yields (up to 76%) in this reaction. This method is characterized by high efficiency, the possibility of using a wide range of substrates and good tolerance to the functional groups therein.
2.3. Amidation of aryldiazonium tetrafluoroborates
In 2018, Tang and co-workers [42] proposed a direct synthesis of N-arylacetamides 15, which involved coupling of aryldiazonium tetrafluoroborates 37 with acetonitrile (5a) without the use of metal catalysts. The reaction proceeded under nitrogen atmosphere in the presence of a stoichiometric amount of K3PO4 base at 80°C for 12 h (Scheme 7).
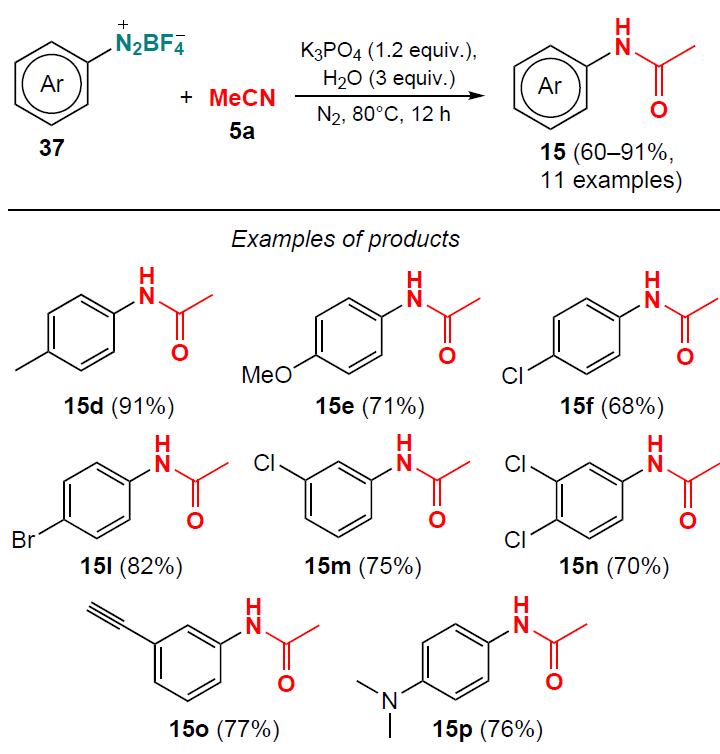
A number of N-arylacetamides 15 containing electron-donor or electron-withdrawing groups were obtained in high yields (up to 91%). The proposed approach allows avoiding the use of strong acids and air-sensitive reagents.
The authors believe that the amidation proceeds via the decomposition of aryldiazonium tetrafluoroborate to generate an aryl radical, as confirmed by experiments with a spin trap, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO).[42][43] The resulting radical then undergoes nucleophilic attack by nitrile and water to give the amidation product. However, another mechanism of the process is also possible, which involves the direct reaction of nitriles with aryldiazonium cations without the involvement of free radicals.[44][45]
2.4. Amidation of aryltriazenes
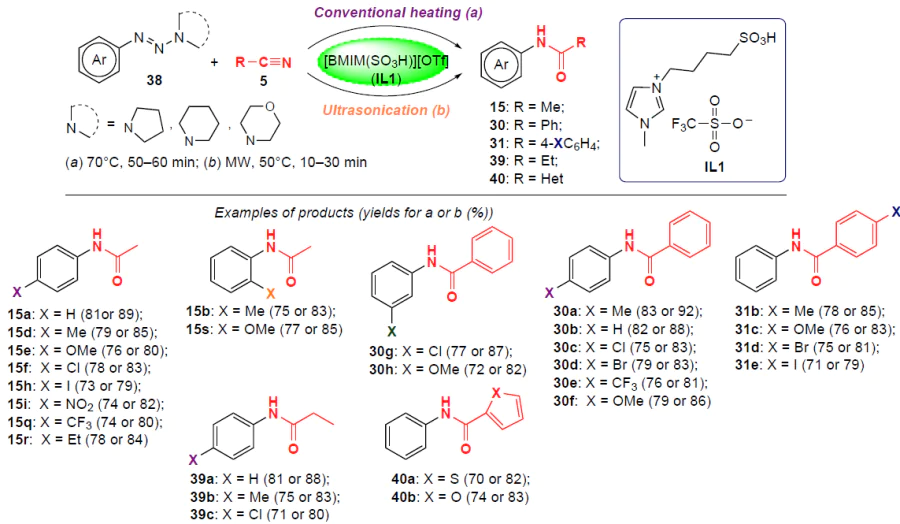
In 2021, Sutar and Kalkhambkar[46] proposed to obtain N-arylamides under mild conditions by coupling aliphatic, aromatic or heterocyclic nitriles with aryltriazenes 38 using the Brønsted ionic liquid [BMIM(SO3H)][OTf] (IL1) as a convenient promoter. The reaction was carried out in two ways (Scheme 8): on heating to 70°C for 1 h or under ultrasound (US) assistance at a lower temperature. The second method proved to be more efficient, providing the corresponding N-arylamides 15, 30, 31, 39 and 40 in high yields (up to 92%), while upon heating, the yields of amidation products did not exceed 83%.
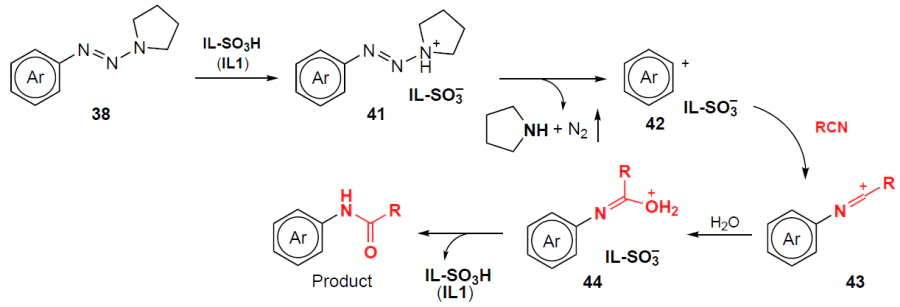
The probable mechanism of the process, presented in Scheme 9 on the example of pyrrolidine substrates 38, is as follows: in the first step, protonation of the aryltriazene at the pyrrolidine nitrogen atom occurs to give ammonium salt 41. Further elimination of pyrrolidine and molecular nitrogen leads to aryl cation 42, which in turn undergoes nucleophilic attack by the nitrile to afford cation 43. Subsequent hydrolysis and deprotonation of this cation yield the desired products, N-arylamides.
A method for the synthesis of N-arylacetamides 15 from aryltriazenes 38 and acetonitrile (5a) at room temperature was developed in 2021 by Liu and co-workers.[47] The catalyst-free reaction is carried out using the protic Brønsted ionic liquid IL2 as a potential promoter (Scheme 10). Triazenes with electron-donating and electron-withdrawing groups in the benzene ring proved to be suitable substrates for this reaction, affording the corresponding N-arylacetamides 15 in moderate yields (from 23% to 62%).
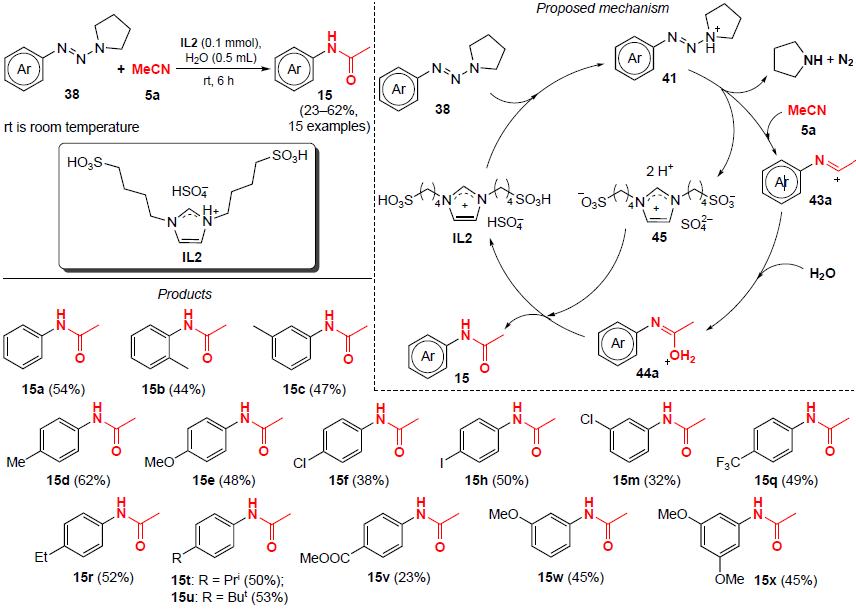
The plausible mechanism of the process is shown in Scheme 10. First, aryltriazene 38 is protonated to form ammonium salt 41. Next, the pyrrolidine molecule and molecular nitrogen (N2) are eliminated, and subsequent nucleophilic attack of nitrile 5a leads to cation 43a, which undergoes hydrolysis to generate intermediate 44a. In the final step, deprotonation and regeneration of IL1 from dication 45 yield product 15.
In 2019, Bräse and co-workers[20] obtained N-arylacetamides 15 (including pyrazolylbenzene derivatives), 30 and 47 from ortho-, meta- and para-substituted aryltriazenes 46 using acetonitrile as a solvent and reagent (Scheme 11; the black ball denotes a polymer support).
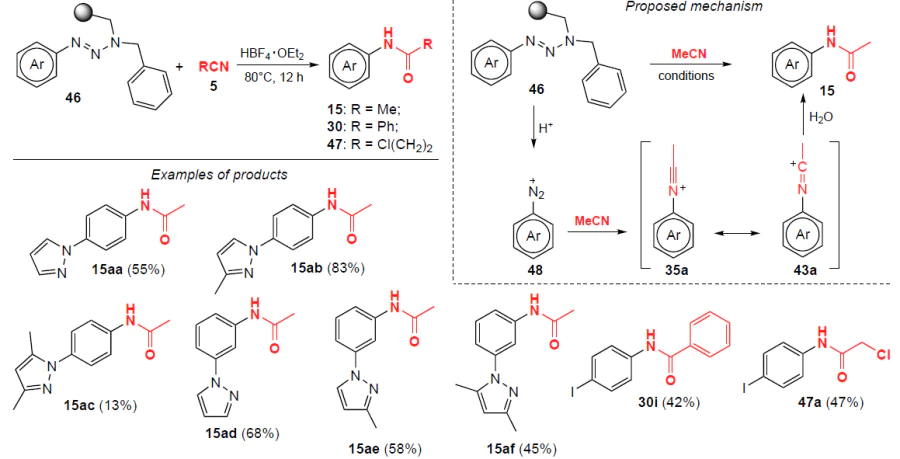
The corresponding arylamides are formed by heating the reagents to 80°C for 12 h (yields up to 83%) via cleavage of immobilized aryltriazenes in the presence of alkyl or aryl nitriles. Cations 48, 35а and 43а are the key intermediates of this transformation (see Scheme 11).
In 2020, Bugarin and co-workers[48] developed a photochemical approach to the synthesis of anilides by coupling π-conjugated aryltriazenes 49 with acetonitrile (Scheme 12). The reaction products are obtained in moderate to high yields (up to 99%) in the absence of metal catalysts under UV irradiation within 4 h.
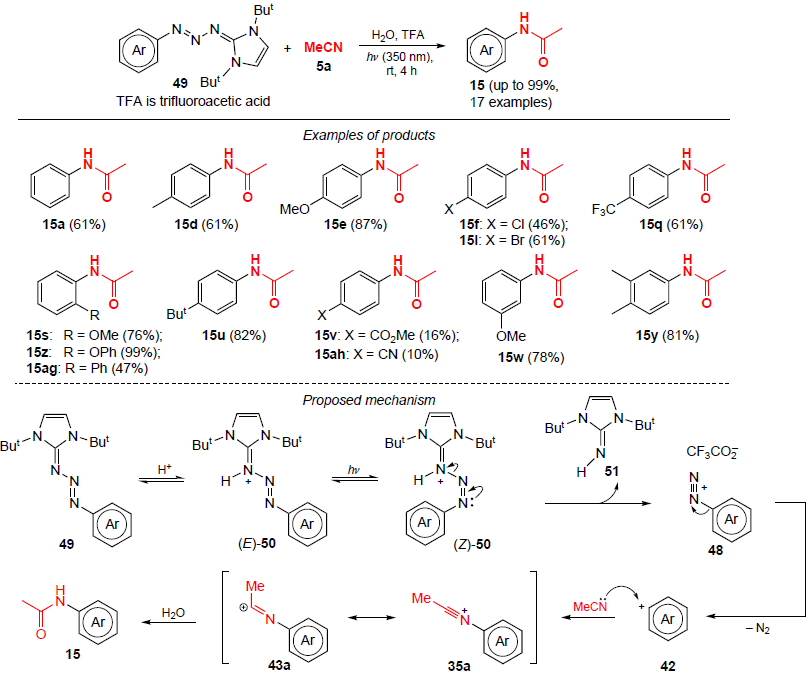
The proposed reaction pathway is shown in Scheme 12. In the initial step, triazene 49 is protonated with trifluoroacetic acid, then the protonated form of triazene undergoes photoisomerization from the E-isomer (E)-50 to the Z-form. Then, the protonated triazene (Z)-50 gives guanidine 51 and an unstable aryldiazonium salt 48, which decomposes to give the aryl cation 42 and molecular nitrogen. The subsequent nucleophilic attack of acetonitrile on carbocation 42 affords the intermediate nitrilium ion 35a, which tautomerizes to the iminium cation 43a. Subsequent hydrolysis of this cation leads to anilide 15.
2.5. Amidation of aryl triflates
In 2019, Li and co-workers [49] hypothesized that aryl triflates 52 can react with nitriles under the action of light in the presence of water to construct an amide bond (Scheme 13). The corresponding N-arylamides are obtained in moderate to good yields (35 – 70%). The reaction was monitored by gas chromatography with mass spectrometry (GC-MS). This reaction serves as an intermediate step for the further conversion of N-arylamides to anilines during alkaline hydrolysis.
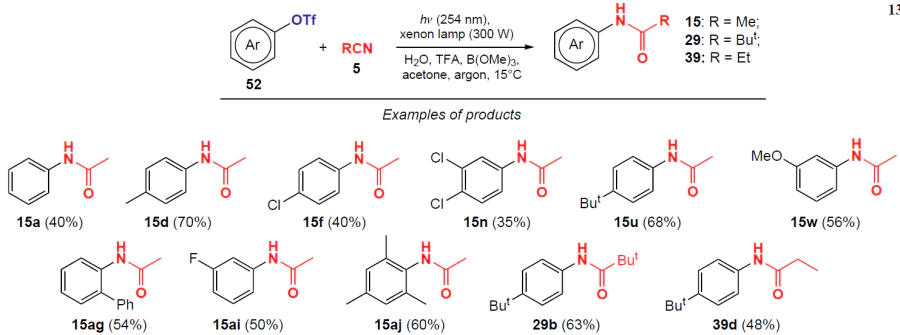
Aliphatic nitriles (MeCN, EtCN, ButCN) were found to be suitable reagents for this process, while the use of aromatic nitriles did not give amidation products under the above conditions. This is probably due to the fact that aromatic nitriles contain chromophore groups capable of absorbing energy, which, in turn, negatively affects the photoinitiation of the desired reaction.
2.6. Amidation of alkylbenzenes
In 2002, Ishii and co-workers [50] first reported a Ritter-type reaction of alkylbenzenes 53 with propionitrile (5g) using N-hydroxyphthalimide (NHPI) as the catalyst in combination with cerium(IV) ammonium nitrate (CAN) as an oxidizing agent. As a result, N-benzylpropionamides 54 were obtained from toluene derivatives with good selectivity in 69 – 93% yield (Scheme 14).
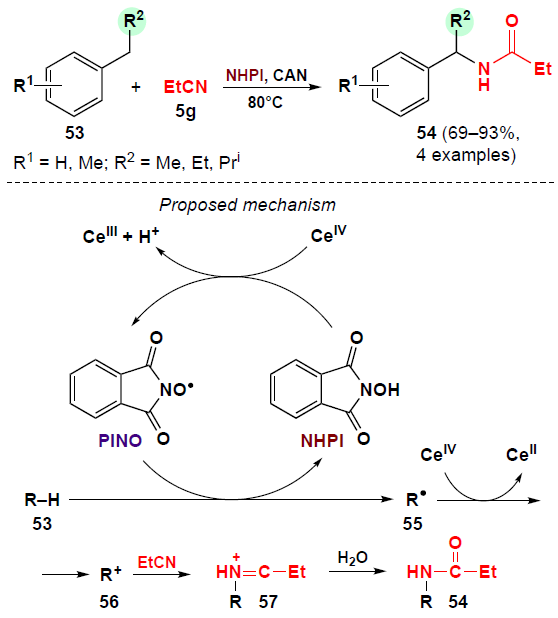
This reaction is presumably initiated by the reaction of NHPI with CAN to form phthalimide N-oxyl (PINO), which is considered a key intermediate for the generation of alkyl radicals (see Scheme 14).[51][52] The PINO radical abstracts a hydrogen atom from the substrate to generate a benzyl radical 55, which then undergoes single-electron oxidation in the presence of a cerium(IV) compound to form carbocation 56. The latter reacts with propionitrile to afford intermediate 57. Further nucleophilic attack by water contained in the solvent on intermediate 57 yields N-benzylpropionamide 54. If the reaction is carried out in an oxygen atmosphere, benzyl radicals 55 will react with O2 rather than with CeIV, giving rise to aromatic aldehydes.
A similar method for the oxidative amidation of alkylbenzenes 53 with acetonitrile (5a) mediated by sodium azide NaN3 was developed by Mohanan and co-workers[53] (Scheme 15). Most likely, the azide ion is first oxidized to an azide radical, which can activate the C – H bond by cleaving it to generate the benzyl radical 55. Further single-electron oxidation of the radical 55 affords cation 56. Subsequent reaction of the benzyl carbocation 56 with acetonitrile in the presence of water yields the corresponding N-benzylacetamides 58 (see Scheme 15).
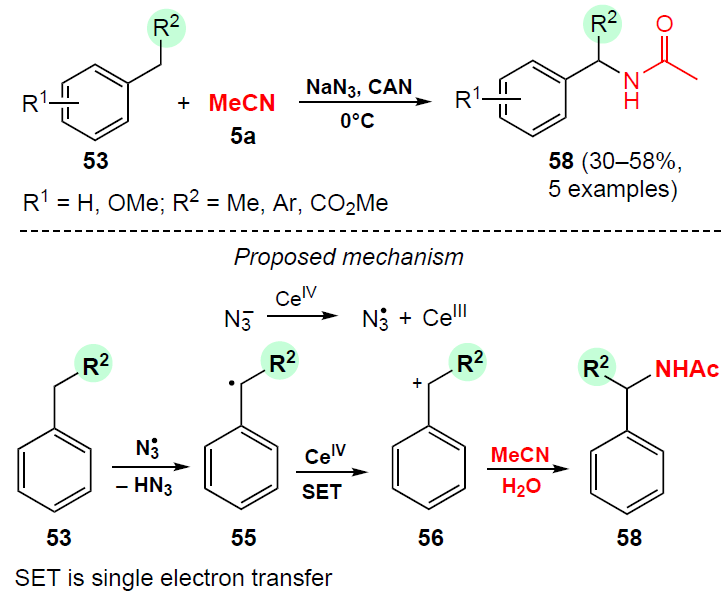
This approach is characterized by milder conditions compared to reactions carried out by Ishii and co-workers[50][51] and a broader range of substrates. However, N-benzylacetamides 58 are obtained in moderate yields (30 – 58%) due to the inevitable formation of benzaldehyde as a by-product.
In 2025, Huang, Xiao and co-workers[54] developed a photocatalytic strategy for the C – H amination of alkylarenes 53 with acetonitrile (Scheme 16). Under blue light irradiation and using CeI3 as a catalyst, bromination with tribromoacetic acid at the benzyl C(sp 3) – H bond first occurs. The resulting benzyl bromide then reacts with the nitrile to give the corresponding N-benzylamides in good yields (up to 84%).
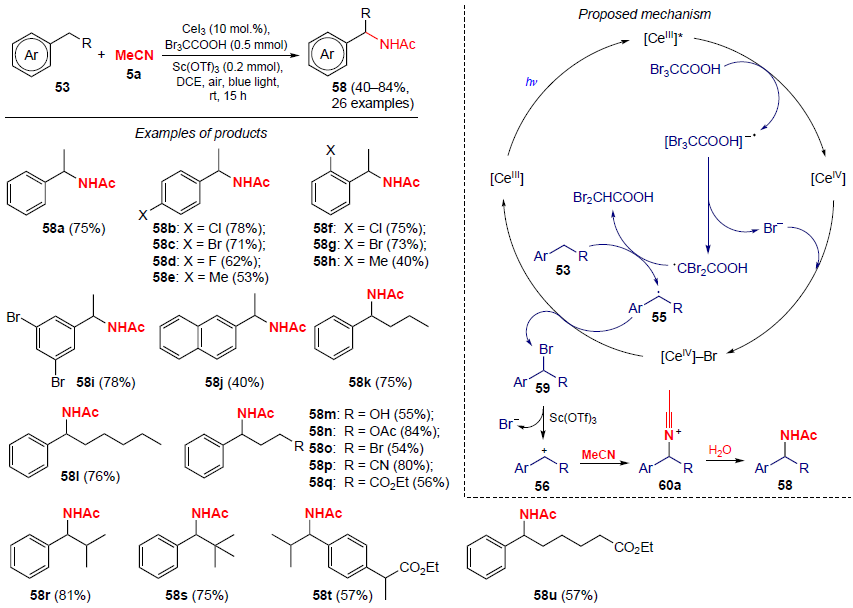
In the initial step of the process, the active form of the catalyst [CeIII]* is generated under photoinduction conditions. Then, the interaction of [CeIII]* with tribromoacetic acid initiates two redox transformations: the reduction of Br3CCOOH to the corresponding anion-radical [Br3CCOOH]– and the oxidation of [CeIII]* to [CeIV]. The unstable intermediate [Br3CCOOH] undergoes decomposition to the radical •CBr2COOH and the bromide anion (Br–). Then, the proton abstraction from alkylbenzene 53 takes place involving the unstable radical •CBr2COOH to give CHBr2COOH and benzyl radical 55. The reaction of the latter with [CeIV] – Br leads to intermediate 59 and the regeneration of cerium(III) in the catalytic cycle. In the presence of Lewis acid Sc(OTf)3 , intermediate 59 undergoes heterolysis to carbocation 56, which reacts with MeCN. The resulting nitrilium cation 60a is then hydrolyzed to the final product, N-benzylamide 58.
König and co-workers[55] proposed a method for the amidation of alkylbenzenes with acetonitrile under photochemical conditions in the presence of the iodonium complex iodine(III) – BF3 , obtained from iodobenzene, a carboxylate ligand, the oxidizing agent (Selectfluor) and Lewis acid (BF3). Such a catalytic system allows the isolation of N-benzylamide derivatives in a yield of up to 91% without the use of a metal catalyst or a photocatalyst.
Doyle and co-workers[56] developed a photocatalytic method for the selective amidation of primary and secondary benzyl C(sp3) – H bonds with nitriles and water using sulfonyloxyamide as a hydrogen atom transfer (HAT) reagent in the presence of the organic photocatalyst Cat2 (Scheme 17).
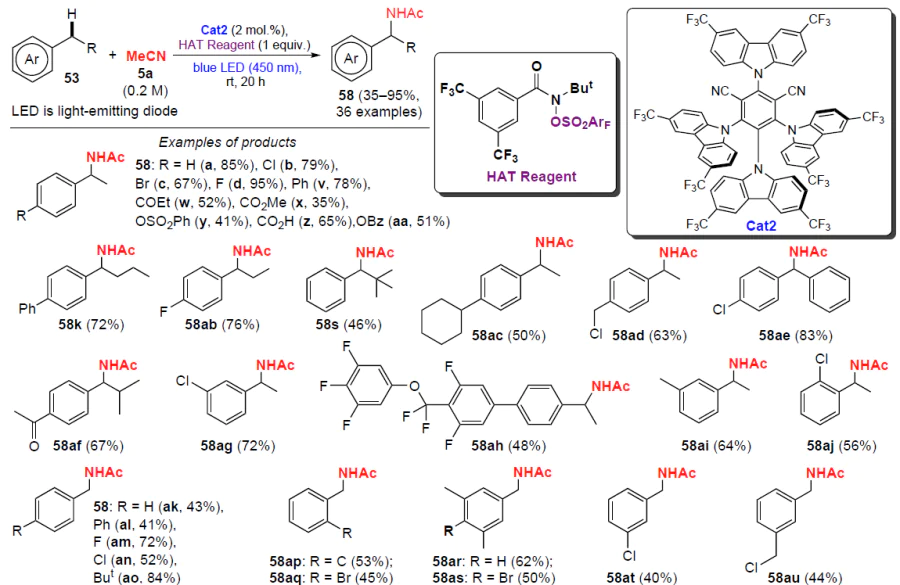
The amidation is carried out in several steps. At the initial step, homolytic cleavage of the N – O bond in the sulfonyloxyamide molecule occurs with the generation of an amidyl radical, which promotes the formation of a benzyl radical. When irradiated, the photocatalyst Cat2 undergoes conversion to the oxidized form, which reacts with the benzyl radical and gives a carbocation. The latter reacts with the nitrile to furnish the resulting N-benzylamides. The reduced form of the photocatalyst mediates the single-electron homolytic cleavage of the N – O bond in the sulfonyloxyamide molecule and is regenerated in the catalytic cycle. Other nucleophiles, such as amides, sulfonamides, carbamates and azoles, can also undergo this reaction.
Li et al.[57] developed a method for photocatalytic Ritter-type amidation of non-activated C(sp3) – H bonds in diarylmethanes without using metal catalysts and in the presence of molecular oxygen as a ‘green’ oxidizing agent (Scheme 18). Using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and tert-butyl nitrite (TBN) cocatalysts, amides were obtained in moderate to excellent yields (up to 86%) under mild conditions.
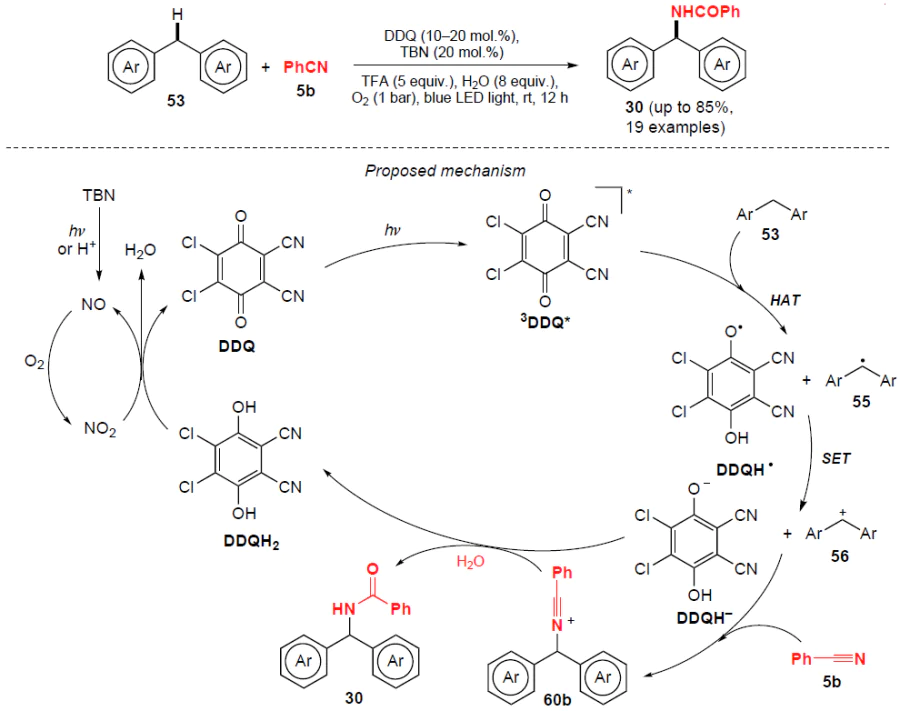
Most likely, the first step involves photolysis or acidolysis of TBN with the release of NO, which can be readily oxidized by oxygen (O2) to NO2 (see Scheme 18). Under visible light irradiation, DDQ is excited to the triplet state (3DDQ*). Hydrogen atom transfer between substrate 53 and 3DDQ* generates DDQH• and an alkyl radical 55, which undergoes subsequent electron transfer to afford the diphenylmethylium cation 56 and DDQH−. The carbocation 56 reacts with benzonitrile (5b) to form the nitrilium ion 60b, which is hydrolyzed to the amide 30. At the same time, the DDQH− anion is protonated to the corresponding hydroquinone (DDQH2), which can be reoxidized to DDQ with NO2.
Zhang et al.[58] proposed oxidative amidation of benzyl C(sp3) – H bonds with nitriles using Mn(OAc)3 as a catalyst and a Lewis acid. Primary, secondary, and tertiary alkyl, alkenyl, benzyl and aryl nitriles can be used in this reaction. N-Benzylamides are obtained in yields from 20 to 96% by heating to 90°C under nitrogen for 12 h in the presence of DDQ as an oxidizing agent and trifluoroacetic acid.
2.7. Amidation of arylketones
A large series of N-(2-oxo-1-arylcyclohexyl)amides 62 were synthesized via oxidative coupling of 2-arylcyclohexan-1-ones 61 with nitriles (Scheme 19).[59] The proposed approach features the use of 10 equiv. of nitrile, a wide range of substrates, and functional group tolerance. The reaction runs for 1.5 h in the presence of tetrafluoroboric acid and DDQ oxidizing agent to give the amidation products in yields of up to 85%.
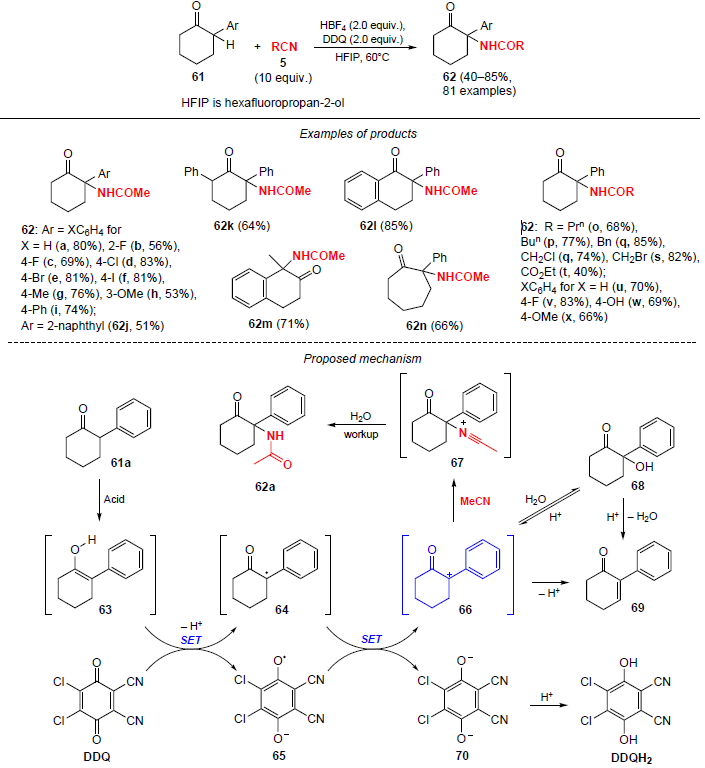
A plausible reaction pathway is shown inScheme 19. Under acidic conditions, substrate 61a is enolized to give intermediate 63, which, when treated with DDQ, undergoes a single-electron oxidation to radical 64, and DDQ is converted into the reduced radical form 65. In the next step, carbocation 66 is formed through a second sequential step of a single-electron oxidation of radical 64 by intermediate 65. In the next step, carbocation 66 is formed through the second successive step of a single-electron oxidation of radical 64 by intermediate 65. Then the process follows the classical route: carbocation 66 undergoes nucleophilic attack by acetonitrile to generate nitrilium cation 67, which is hydrolyzed to the desired amidation product 62a. If carbocation 66 has no time to interact with acetonitrile, then alcohol 68 is formed as a result of a side nucleophilic attack by water.
The second by-product is enone 69, which is formed by deprotonation of carbocation 66 or by elimination of water from 68. Enone 69 is obtained only in trace amounts, while alcohol 68 is generated in the initial reaction step and is completely converted to the desired amidation product 62a. It should be noted that SET from radical 64 is accompanied by the formation of dianion 70, which, upon acidification, yields the corresponding hydroquinone.
In 2023, the same research group[60] reported the synthesis of 3-acyl-3-pyrrolin-2-ones by coupling α-aryl ketones with substituted propionitriles using a similar oxidation system based on DDQ and HBF4. The process was carried out in two steps. In the initial step, the starting substrates were reacted to afford an intermediate under Ritter-type oxidative amidation conditions. The following intramolecular aldol condensation yielded the desired 3-acyl-3-pyrrolin-2-ones.
2.8. Amidation of benzyl alcohol derivatives
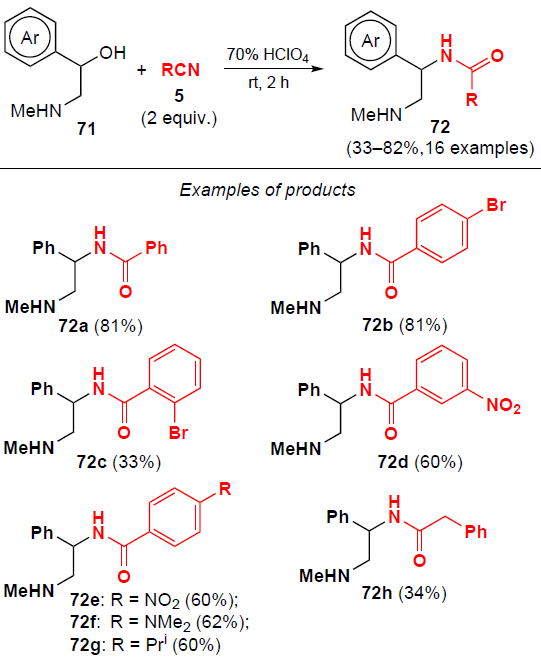
Sosnovskikh and co-workers[61] proposed the synthetic approach to 2-aminoethyl-1-benzamides from arylethanolamines 71. The reaction runs in the presence of excess perchloric acid and 2 equiv. of nitrile 5 at room temperature for 2 h (Scheme 20). Benzonitriles containing electron-withdrawing substituents (such as a nitro group or a halogen atom) in the para- and meta-positions readily reacted with substrates 71 at room temperature, affording products 72 in 60 – 81% yields. Benzonitriles with electron-donating groups (OMe, Alk) promoted competitive Friedel – Crafts alkylation to afford mixtures of products. 2-Aminoethyl-1-benzamides 72 were then used in a cyclization reaction to give imidazolines.
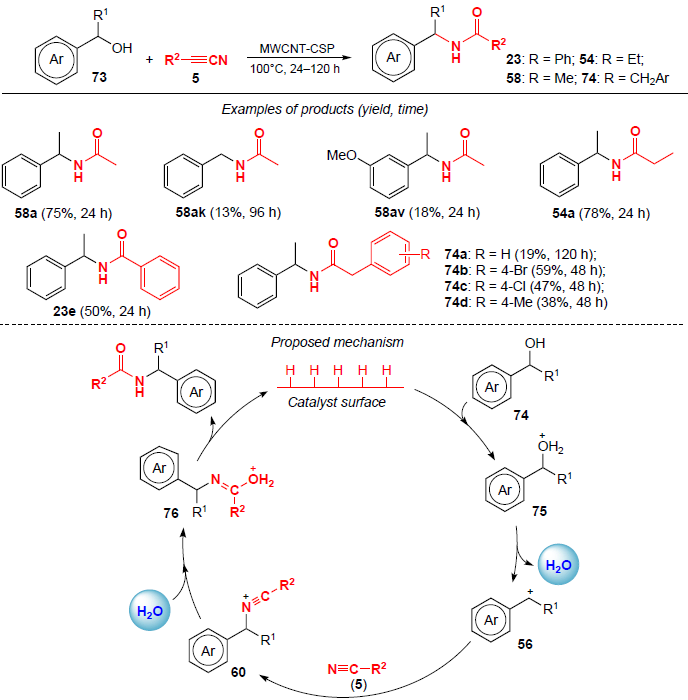
In 2023, Marques and co-workers[62] reported the synthesis of N-benzylamides using a heterogeneous MWCNT-CSP (multi-walled carbon nanotubes modified with p-toluenesulfonic acid) catalyst (Scheme 21). Heating benzyl alcohols 73 and nitriles to 100°C for 24 – 120 h yielded N-benzylamides 23, 54, 58 and 74 in moderate to good yields (up to 78%). According to the process pathway shown in Scheme 21, the authors suggest that substrate 73 is first protonated by acid, releasing a water molecule, followed by the formation of carbocation 56, which is captured by the nitrile and affords nitrilium cation 60. Subsequent hydrolysis of cation 60 leads to amidation products.
Wang and co-workers[63] developed a simple and efficient method for the synthesis of N-benzylamides and other amides by the reaction of secondary alcohols with nitriles mediated by Fe(NO3)3 · 9 H2O (Scheme 22, reaction (1)). Substrates with electron-donating groups gave the corresponding amides in good or high yields (up to 97%), while in the case of electron-withdrawing groups the yields of the products were significantly lower (up to 61%).
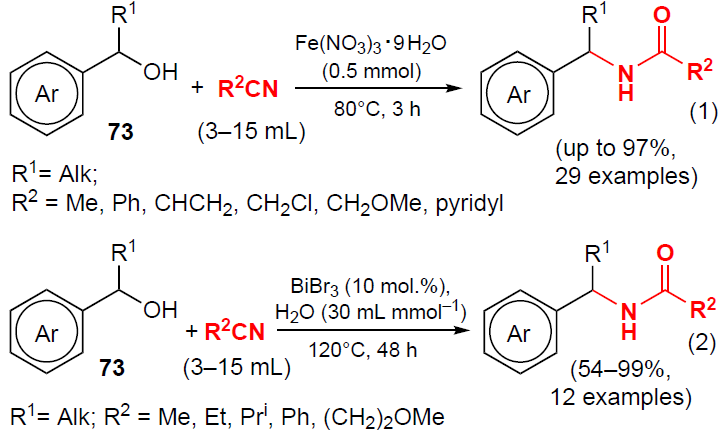
Ueno et al.[64] carried out an environmentally friendly reaction of benzyl alcohols with nitriles using commercially available bismuth salt BiBr3 as a catalyst (see Scheme 22, reaction (2)). The amidation products were formed in the presence of the bismuth salt at 120°C within 48 h. The yield of the corresponding desired N-benzylamides was improved by adding a small amount of water, and no deactivation of the bismuth salts was observed under these conditions.
Chen and co-workers[65] developed a method for the photocatalytic dehydration of benzyl alcohols suitable for their efficient direct conversion to the corresponding N-benzylamides using 2,4,6-triphenylpyrylium tetrafluoroborate (Cat3) as a photocatalyst (Scheme 23). The reaction of benzyl alcohols 73 and nitriles afforded the corresponding amides after 16 h of blue light irradiation in a nitrogen atmosphere. Benzyl alcohols containing electron-donating groups react quite readily (product yields range from 50% to 63%), while their analogues with electron-withdrawing groups give N-benzylamides in lower yields. The method can be applied to various alkyl nitriles, but with lengthening of the alkyl chain the activity of the nitriles decreases, which leads to a decrease in the yields of amidation products.
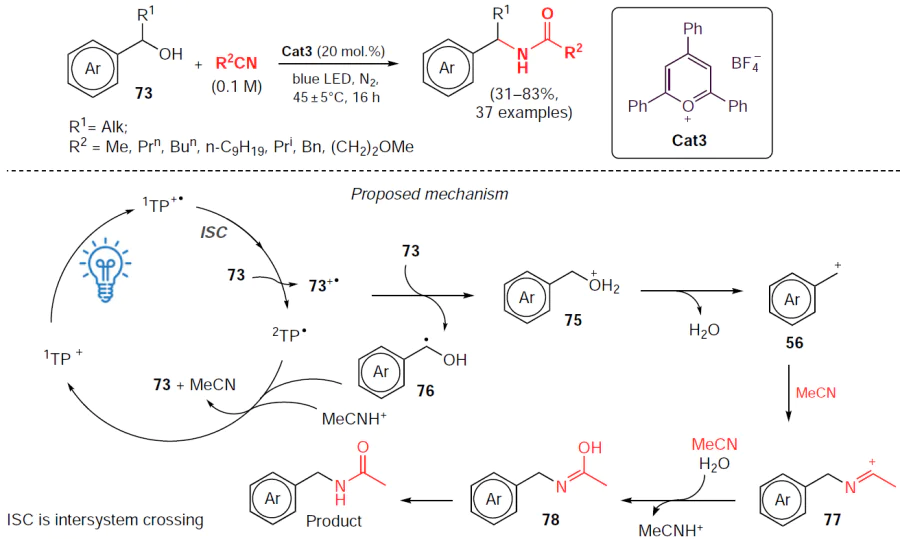
The probable mechanism of this process using the substituents R1 = H, R2 = Me as an example is shown in Scheme 23. At the initial step, under irradiation conditions, the photocatalyst (its cation is designated as TP) passes into an excited state to form a cation-radical. The interaction of benzyl alcohol with the active form of the photocatalyst and further transformation steps lead to the intermediate cationic form 75 with the release of radical 76. Then, cation 75 is dehydrated to the corresponding benzyl carbocation 56, which further reacts with acetonitrile and water to give successively cation 77, enol 78 and the amidation product.
2.9. Amidation of arylacetic acid derivatives
In 2022, König and co-workers[66] reported the decarboxylative amidation of carboxylic acids by a Ritter-type reaction using the iodine(III) – BF3 complex obtained from iodobenzene, Selectfluor as an oxidizing agent and BF3 as the Lewis acid. Carboxylic acids 79, irradiated with light in the presence of catalytic amounts of iodobenzene and stoichiometric amounts of Selectfluor in the presence of water and BF3, undergo an amidation to give N-benzylamides 58, 80 and 81 (Scheme 24). This reaction is applicable to both benzylic and aliphatic carboxylic acids.
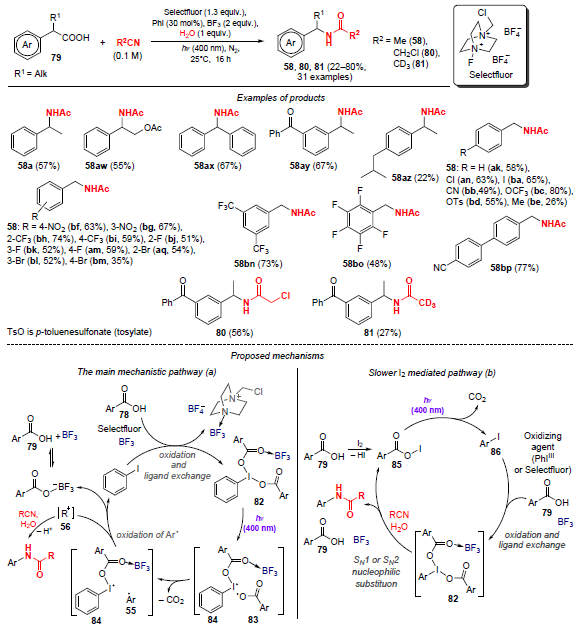
Based on the data obtained, the authors proposed two reaction pathways, shown in Scheme 24. Pathway a involves homolysis of the I – O bond of the initially formed iodonium complex 82, which gives rise to radicals 83 and 84. Next, decarboxylation of intermediate 83 occurs yielding benzyl radical 55 and further oxidation to carbocation 56. The next step is the nucleophilic addition of the nitrile to carbocation 56, followed by hydrolysis to the desired N-benzylamide.
The amidation process can be accomplished by an alternative mechanism when mediated by I2 (see Scheme 24, pathway b). In the first step, molecular iodine reacts with carboxylic acid 79 to give acyl hypoiodides RCO2I (85). Blue light irradiation of compounds 85 is accompanied by their decarboxylation to the corresponding alkyl iodides 86. Oxidation of the latter occurs either due to the action of Selectfluor or in a reaction with the fluoroborate complex PhIIII, in which case iodonium complex 82 is formed. Intermediate 82 undergoes rapid nucleophilic substitution in the presence of a nitrile with subsequent hydrolysis to the target amide.
In 2022, Yoon and co-workers[67] developed a method for oxidative decarboxylative coupling of carboxylic acids 79 with nitriles under visible light irradiation in the presence of Cu(OTf)2 and Na2CO3. The authors hypothesized that the photoactive copper(II) carboxylate chromophore, obtained in situ by the reaction of the carboxylic acid with Cu(OTf)2 in the presence of a base, leads to the dissociative formation of a carboxyl radical, which undergoes decarboxylation under blue light (427 nm) irradiation and generates a benzyl radical. The latter can be oxidized by copper(II) salts to form a carbocation, which then reacts with acetonitrile to give amidation products, N-benzylamides.
Ji and co-workers[68] proposed an effective and low-cost method for the synthesis of N-substituted amides based on the reaction of nitriles with (methoxymethyl)benzene derivatives, catalyzed by Fe(ClO4)3 · H2O. This catalyst proved to be quite effective for this reaction in the solvent-free protocol providing N-benzylamides in yields of up to 93%.
2.10. Functionalization of C(sp2) – H bonds in amidation of benzene derivatives
In 2015, Redshaw and co-workers[69] developed a copper-catalyzed amidation of benzene derivatives 87 with electron-donating groups using benzonitrile (5b) as an amide source. Interestingly, the reaction occurs selectively at the para-position (Scheme 25). The process runs in the presence of a catalyst, copper(II) trifluoromethanesulfonate, the iodine-centered electrophile 2-(diacetoxyiodo)mesitylene (MesI(OAc)2) and water (3 equiv.) in boiling dichloroethane for 15 h. The resulting N-arylbenzamides 30 are obtained in yields from 35% to 93%. The regioselectivity of this amidation is controlled by both steric and electronic factors, which leads to the selective formation of only para-amidation products. No other isomers were detected in the reaction mixture by 1H NMR spectroscopy. Aromatic compounds with electron-withdrawing groups, such as chlorobenzene and nitrobenzene, are unsuitable for this reaction, and no N-arylbenzamides were detected in this case.
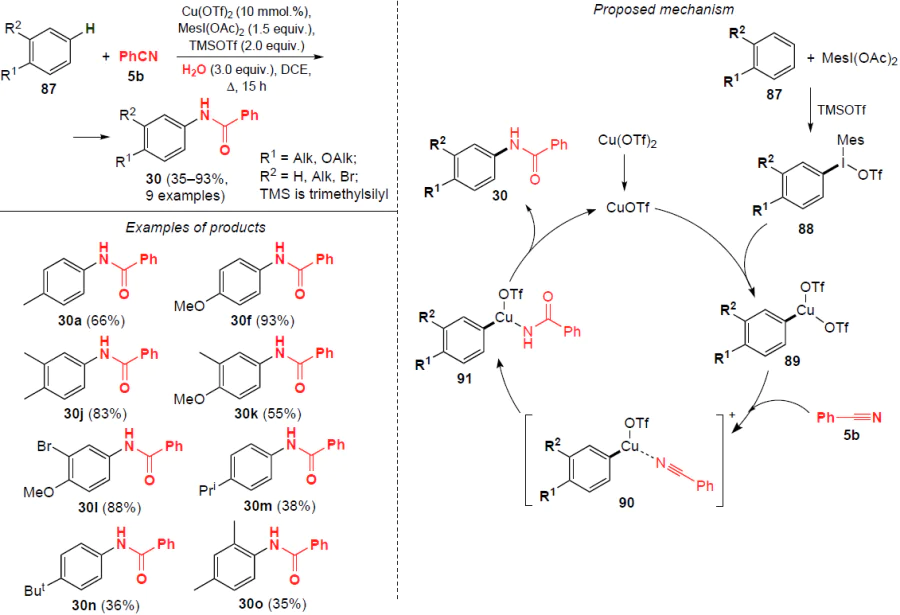
The probable mechanism of this catalytic transformation is shown in Scheme 25. In the first step, electrophilic substitution in the aromatic ring initiated by trimethylsilyl trifluoromethanesulfonate leads to diaryliodonium salts 88. The active form of the catalyst CuOTf, formed from Cu(OTf)2 via disproportionation or reduction, is oxidized by the diaryliodonium salt 88 to intermediate 89, which undergoes ligand exchange with benzonitrile (5b) to furnish complex 90. The proposed CuIII species promote the hydrolysis of the coordinated benzonitrile, forming intermediate 91, which then undergoes reductive elimination to N-arylbenzamides 30. In this case, the active form of the catalyst CuOTf is regenerated to participate in the catalytic cycle.
With an increase in the amount of aromatic substrate to 4 equiv., further arylation of the obtained N-arylamides occurs selectively in the meta position, which was shown by the example of the reaction between o-xylene 87a with various benzonitriles (Scheme 26). Benzonitriles containing both electron-donating and electron-withdrawing substituents undergo arylation to give the corresponding products 92 in moderate yields (36 – 51%).
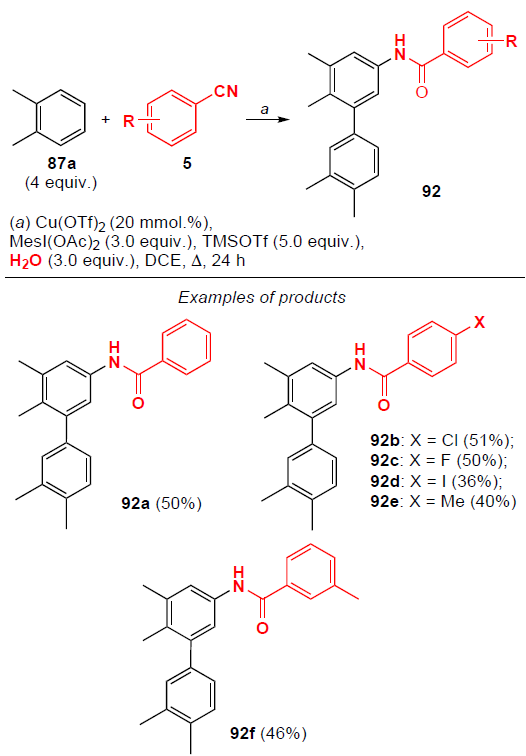
3. Electrooxidative amidation of aromatic compounds with nitriles
Organic electrosynthesis is an environmentally friendly alternative to traditional chemical reactions and allows valuable N-containing compounds to be obtained in an atom-efficient manner under mild conditions with a minimum number of steps.[70-74] Electrochemical approaches in which electrons are used as a ‘traceless’ reagent are also of interest for dehydrogenating C – H/N – H cross-coupling reactions.[75-77] Ritter-type reactions of electrochemical amidation of alkanes,[78][79] esters[80] and ketones[81] were described as early as the 1970s. Since then, the study of electrochemical processes following the Ritter-type reaction has slowed down somewhat. However, in the last few years, new electrochemical approaches have been proposed to the amidation of aromatic compounds by functionalizing the C(sp3) – H bonds at benzyl carbon atoms[82-91] and the C(sp2) – H bonds in aromatic centres.[89-95] Also worth mentioning are studies[96-102] on electrooxidative C – H/N – H coupling, implemented with the involvement of compounds of various classes.
3.1. Electrooxidative amidation of alkylbenzenes
In 2017, Ley and co-workers[82] reported the synthesis of N-benzylacetamides 58 from toluene derivatives by anodic oxidation in a flow reactor in the presence of methanesulfonic acid (MSA) as the Brønsted acid (Scheme 27).
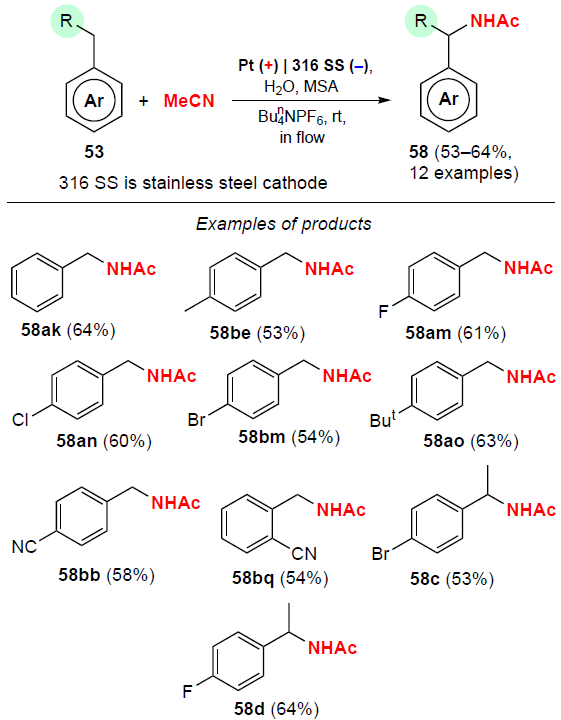
The amidation products of para-, meta- and ortho-substituted toluenes 53 were isolated in moderate yields (53 – 64%). In all cases, unreacted starting compounds were detected in the reaction mixture, indicating incomplete conversion of the substrates. By-products (< 2%) of this reaction are benzaldehydes and benzyl alcohols. The process does not proceed without the addition of acid.
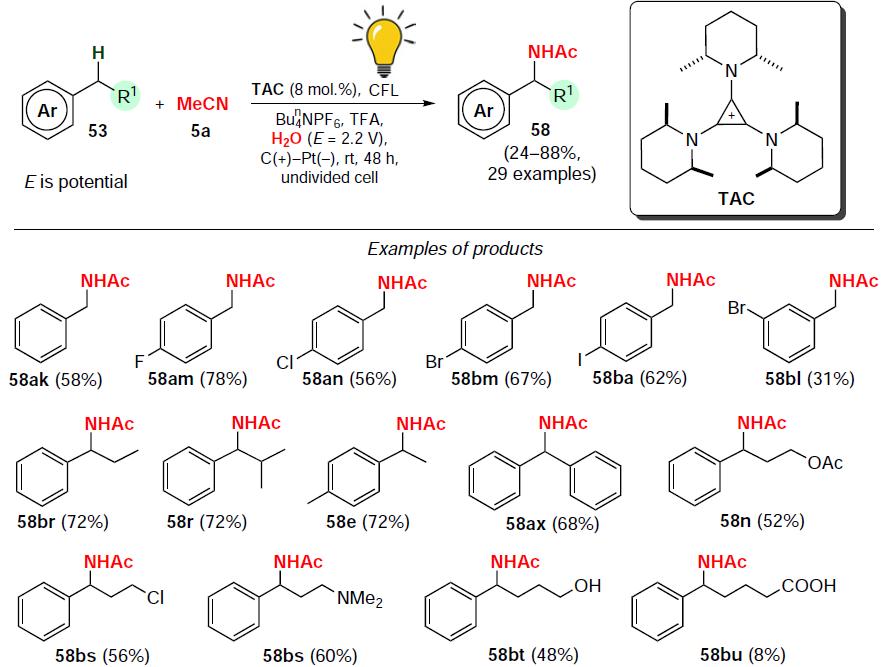
In 2021, Shen and Lambert[83] developed an electrophotocatalytic method for the synthesis of N-benzylacetamides 58 using tris(cis-2,6-dimethylpiperidin-1-yl)cyclopropenium (TAC) ion as a catalyst (Scheme 28). Aromatic compounds containing an ester, alcohol, chloroalkyl, or tosylate moiety were used in the amidation reaction. The target products 58 were obtained in moderate to good yields (31 – 78%), and the reaction did not proceed without application of the cell potential and in the absence of a catalyst. When the reaction was carried out in the absence of compact fluorescent lamp (CFL) irradiation, the amidation products were formed only in small quantities (yield of about 9%).
The mechanism of this electrophotocatalytic reaction is shown in Scheme 29. The TAC catalyst undergoes single-electron oxidation to generate a stable dication radical 93. Irradiation of the latter affords photoexcited intermediate 94, which is involved in the single-electron oxidation of substrate 53 to form cation radical 95.
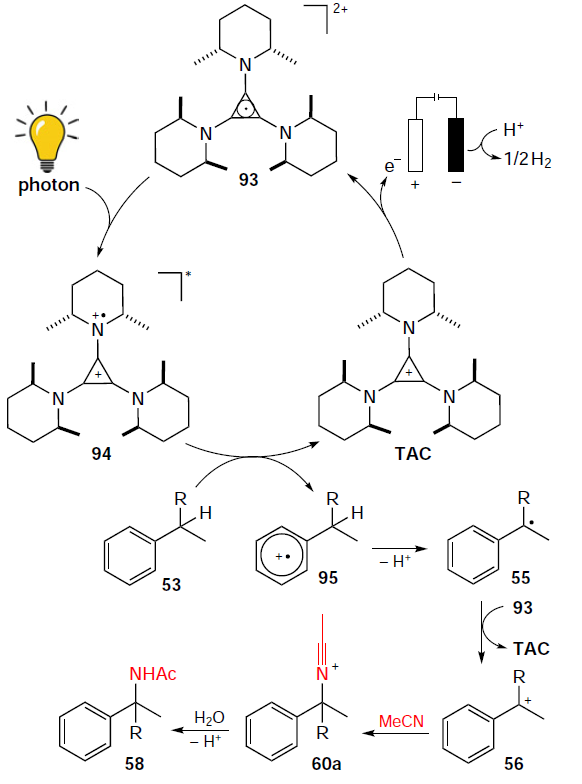
Deprotonation of this radical cation generates benzyl radical 55, which is then reoxidized either by TAC dication radical 93 or directly at the anode to yield carbocation 56. Nucleophilic addition of acetonitrile to cation 56 yields intermediate 60a. In the final step, cation 60a is hydrolyzed to the corresponding N-benzylacetamide 58. The key point of this approach is that the reaction occurs at the oxidation potential of the TAC electrophotocatalyst rather than the aromatic substrates, thereby minimizing the risk of side processes.
In 2022, Ye and co-workers [84] proposed another example of electrochemical amidation. The authors carried out the reaction using sulfuric acid in an undivided cell at room temperature (Scheme 30). The amidation products of toluene and ethylbenzene derivatives containing various functional groups (F, Cl, Br, I, CN, NO2, NH2, Alk, Ac, Ph, OMe) in the benzene ring are obtained in high yields (up to 85%). This method is characterized by high regioselectivity; substrates with several alkyl groups in the aromatic ring undergo amidation exclusively at one of them.
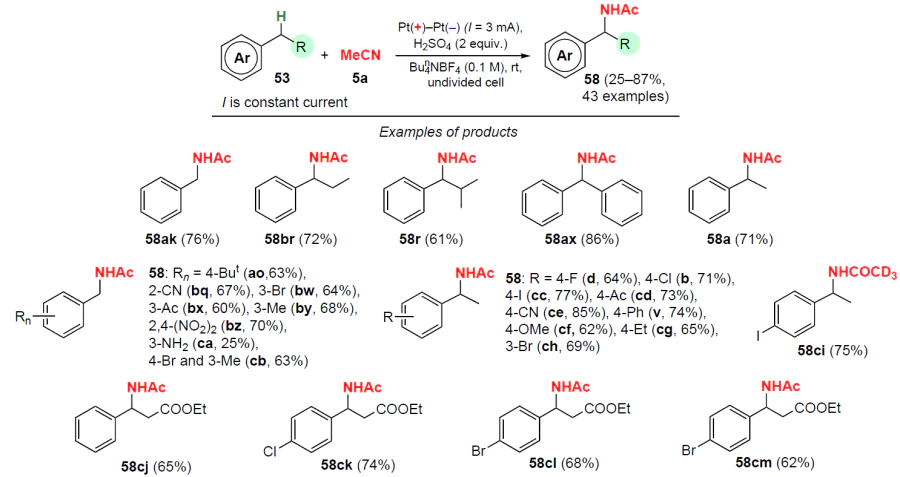
In the absence of sulfuric acid, there is virtually no reaction. Presumably, the sulfate anion SO42– undergoes single-electron oxidation at the anode to form SO4–. Being an active acceptor of the hydrogen atom, SO4– abstracts the hydrogen atom from the aromatic substrate to generate a benzyl radical, which is oxidized to a carbocation at the anode. The latter then undergoes a nucleophilic addition to the nitrile to give the corresponding N-benzylacetamides 58.
In 2023, Zhou and co-workers[85] reported a method for the amidation of alkylbenzenes under electrochemical conditions. The reaction was carried out under mild conditions at room temperature in an undivided cell in the presence of trifluoromethanesulfonic acid to furnish the corresponding N-benzylacetamides 58 in yields from 20 to 91% (Scheme 31). Substrates containing electron-donating substituents in the aromatic ring were more active in this reaction than their analogues with electron-withdrawing groups. This is apparently due to the higher electron density, which facilitates the activation of benzyl C – H bonds under direct current electrolysis conditions. The presence of several alkyl substituents in the ring has little effect on the yield of the target product. The yields of amidation products are significantly reduced when substrates with long alkyl chains or bulkier substituents are used.
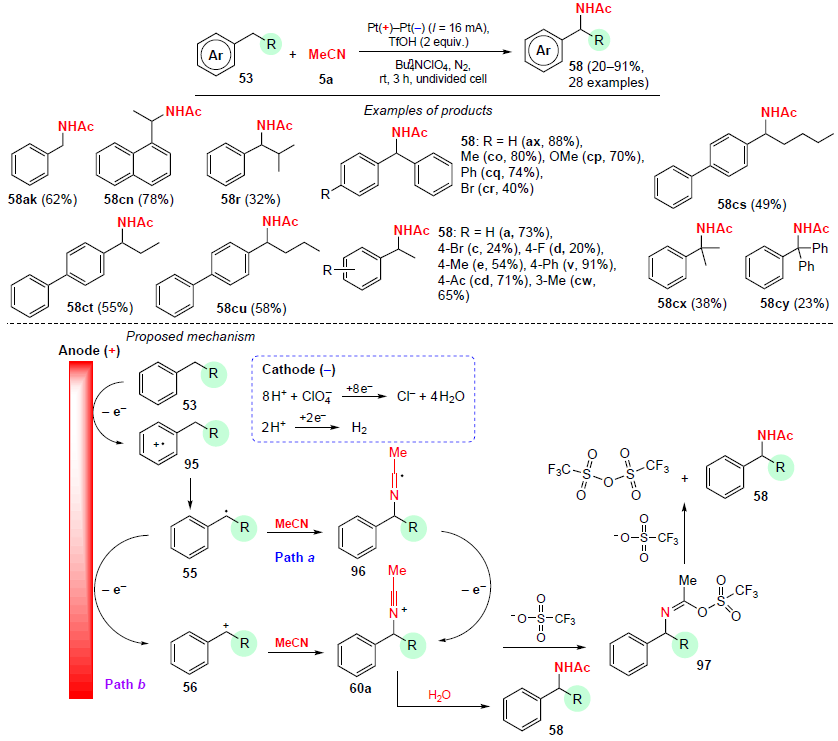
Based on the obtained experimental and literature data,[103-106] a mechanism for this process has been proposed (see Scheme 31). First, single-electron oxidation of substrate 53 occurs at the anode to form benzyl radical 55. Then, two reaction paths are possible. In path a, radical 55 reacts with MeCN to generate first radical 96 and then nitrilium cation 60a after another single-electron oxidation. In path b, radical 55 is oxidized to carbocation 56, which then reacts with MeCN to afford nitrilium cation 60a. The latter reacts with TfO– to form intermediate 97, which then undergoes detrifluoromethanesulfonylation to yield amidation product 58 and trifluoromethanesulfonic anhydride. The nitrilium cation 60a can be hydrolyzed directly to N-benzylamide 58. At the cathode, ClO4– is reduced to Cl– and H2O in the presence of H+.
Yin and co-workers[86] developed a new electrocatalytic protocol for the conversion of toluene to N-benzylacetamide using MeCN as an amidating reagent. The proposed approach involves the use of a hydrophobic electrode material based on polytetrafluoroethylene (PTFE)-coated carbon paper (CP) and a conductive membrane instead of the background electrolyte. Based on the experimental results and quantum chemical calculations, it was demonstrated that coating the electrode with such a polymer promotes the adsorption of toluene and its efficient dehydrogenation. In addition, the hydrophobic material prevents the adsorption of water onto the electrode, inhibiting the competitive reaction of water oxidation.
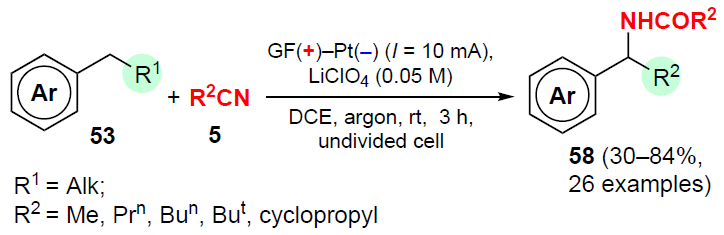
Sun and co-workers[87] described a method for the electrochemical amidation of alkylarenes with nitriles and water in the absence of an oxidizing agent and mediator, which provides an access to various N-benzylamides in moderate and good yields (Scheme 32).
In 2023, Budnikova and co-workers[90] carried out electrooxidative amidation of toluene derivatives in aceto- and propionitriles, leading to N-benzylamides in yields of up to 87% (Scheme 33). The reaction occurs under mild conditions (at room temperature) in the absence of metal catalysts, oxidizing agents and acid additives. For the substrates having several methyl groups in the aromatic ring (xylenes, mesitylene), the main product is amidation products with only one substituted methyl group, even when passing a larger amount of electricity and increasing the synthesis time. The presence of a bromine atom as a substituent in the ring reduces the yield of the target amidation product, since by-products, viz., aldehydes and dimers, are also formed.[91] The oxidation of ortho-, meta- and para-xylenes, in addition to N-benzylamides, produces by-products–aromatic aldehydes (yields of 8 – 12%). In all cases, products with only one amide group are formed during the reaction.
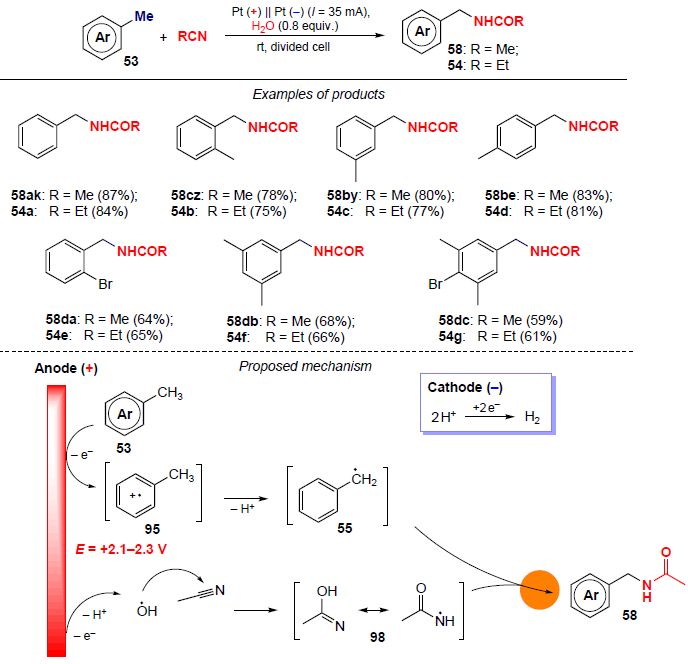
The mechanism of electrooxidative amidation of toluene derivatives with nitriles is shown in Scheme 33. It is assumed that the key step is the anodic oxidation of water generating hydroxyl radicals, which add to the nitrile to give spin adduct 98. Aromatic substrates with a methyl group (toluene, xylenes, mesitylene) are oxidized in the same potential range as water (+2.1 ÷ +2.3 V).[80][81] Probably, in this case, one electron is spent on the formation of the benzyl radical 55, and the second electron is spent on the generation of hydroxyl radicals followed by the formation of adduct 98. Further interaction of the two radicals affords amidation products 58. At the same time, the amidation can be realized by another route, where at the initial step, successive two-electron oxidation and deprotonation of methylarene to benzyl carbocation takes place.[83][86][87] Further nucleophilic addition of acetonitrile to the carbocation gives a nitrilium cation, which undergoes hydrolysis to the corresponding N-benzylamide.
In 2022, the same research group[88] described a one-pot protocol for the synthesis of N-benzylamides using amino acids 99 (2-phenylglycine, valine, alanine) as an amide source (Scheme 34).
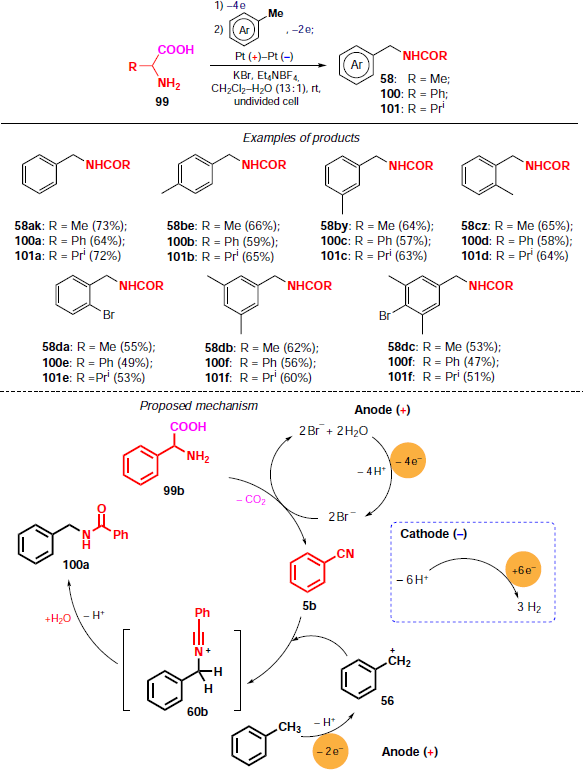
In the first step, amino acids undergo decarboxylation to nitriles in the presence of a mediator, potassium bromide, under anodic oxidation conditions. Subsequent addition of an aromatic substrate to the reaction mixture and passing electricity in the amount of 2 F leads to N-benzylamides 58, 100 and 101 in moderate yields (up to 73%).
The plausible mechanism of the process is shown in Scheme 34. Under conditions of anodic oxidation of the KBr mediator, hypobromite ions (BrO–) are generated, which selectively oxidize 2-phenylglycine (99b) to benzonitrile (5b). Addition of an aromatic substrate to the reaction mixture and subsequent two-electron oxidation in the presence of benzonitrile (5b) gives intermediate 60b. In the final step, hydrolysis of the nitrilium cation 60b affords N-benzylbenzamide 100a in 73% yield.
3.2. Electrooxidative functionalization of C(sp2) – H bonds in amidation of benzene derivatives
The first example of electrochemical amidation of trifluoromethylbenzene (87b) with acetonitrile was described in 2014 by Barba et al.[92] The reaction was carried out under anodic oxidation conditions in a potentiostatic mode at a potential of +2.8 V (vs. Ag/AgCl (sat.)) in absolute acetonitrile using tetra-n-butylammonium tetrafluoroborate as a background electrolyte (Scheme 35). The plausible mechanism of the process includes the formation of cation radicals 102 and 103.
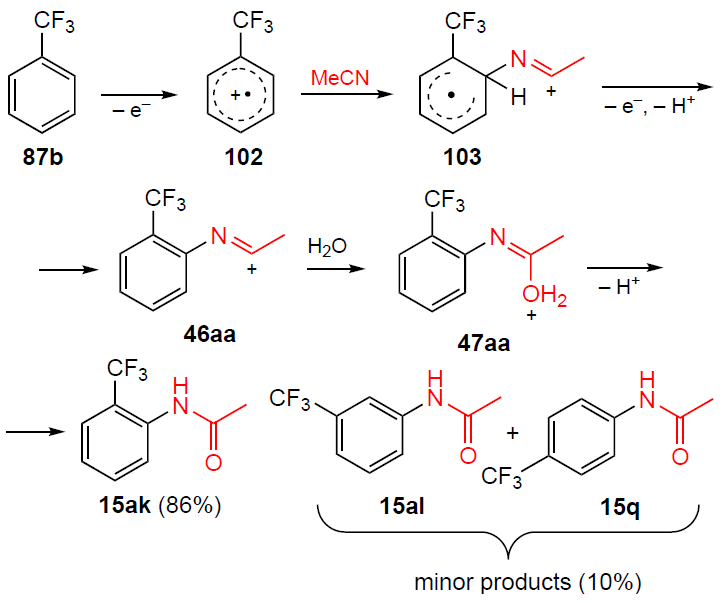
The reaction produces 2-(trifluoromethyl)acetanilide (15ak) as the major product (yield 86%), the by-product is a 10% mixture of 3-(trifluoromethyl)acetanilide (15al) and 4-(trifluoromethyl)acetanilide (15q) in a 3 : 1 ratio, as determined by gas chromatography (GC) and 1H NMR spectroscopy. No other substrates or nitriles were used in this reaction. The formation of 2-(trifluoromethyl)acetanilide (15ak) as the predominant isomer can be explained by the strong inductive effect of the trifluoromethyl group, which provokes a charge deficit in the adjacent positions, thereby facilitating the nucleophilic attack of the nitrile.
In 2022, Banerjee and co-workers[93] proposed an approach for the electrochemical amidation of phenols 104 with alkyl, aldehyde and ketone groups, allowing the preparation of various N-(4-hydroxyphenyl)acetamides 105 (Scheme 36).
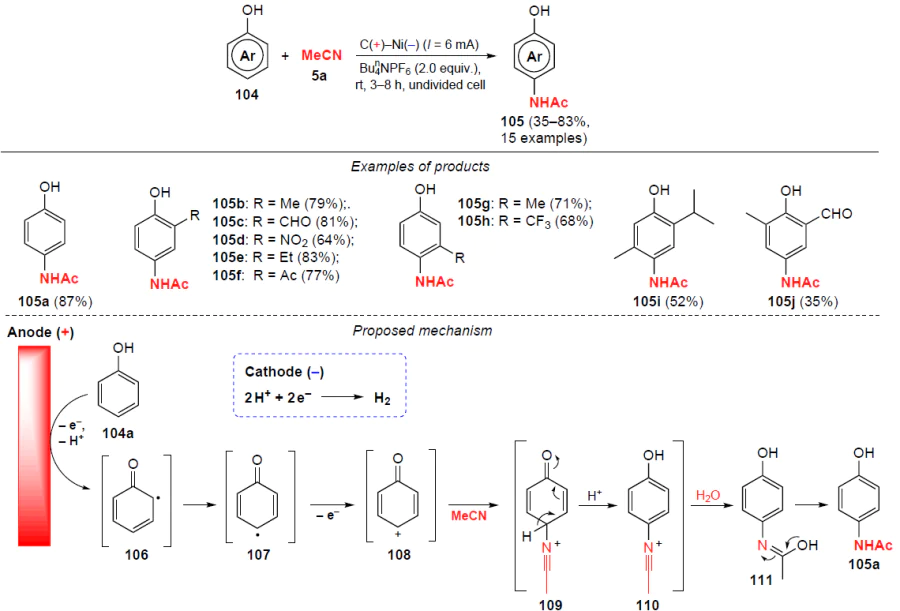
The reaction proceeds regioselectively in the para-position relative to the hydroxyl group in the absence of external oxidizing agents and catalysts, but this method is limited to phenolic substrates only. Substituents in the benzene ring affect the yield of the target products and the reaction time. The yields of N-arylacetamides 105 are lower for phenols with electron-withdrawing groups in the benzene ring (NO2, CF3, CHO, Ac), and more time is required for the reaction to be completed. Electrooxidation of phenols with electron-donating substituents (Me, Et) proceeds comparatively faster.
The probable mechanism of the process is shown in Scheme 36. The reaction is initiated by single-electron oxidation of phenol 104a to generate radicals 106 and 107, further single-electron oxidation of which gives carbocation 108. Carbocation 108 then undergoes classical Ritter reaction steps in the presence of acetonitrile to form nitrilium ions 109 and 110. The latter is then hydrolyzed and via the formation of hydroxyimine 111 yields the target product, acetaminophen 105a.
In 2023, Ye and co-workers[94] carried out amidation of benzene derivatives containing electron-withdrawing substituents in the aromatic ring in an undivided electrochemical cell (Scheme 37). Mono-, di-, and trisubstituted benzenes with CO2Me, SO3Me, CN, CF3 groups or Br, Cl, F atoms were used in this reaction, affording the corresponding N-arylacetamides 15 in moderate and good yields (from 39 to 75%). The process takes 10 h and requires the presence of 4 equiv. sulfuric acid. The method is atom-economical, since it utilizes cost-effective and readily available reagents.
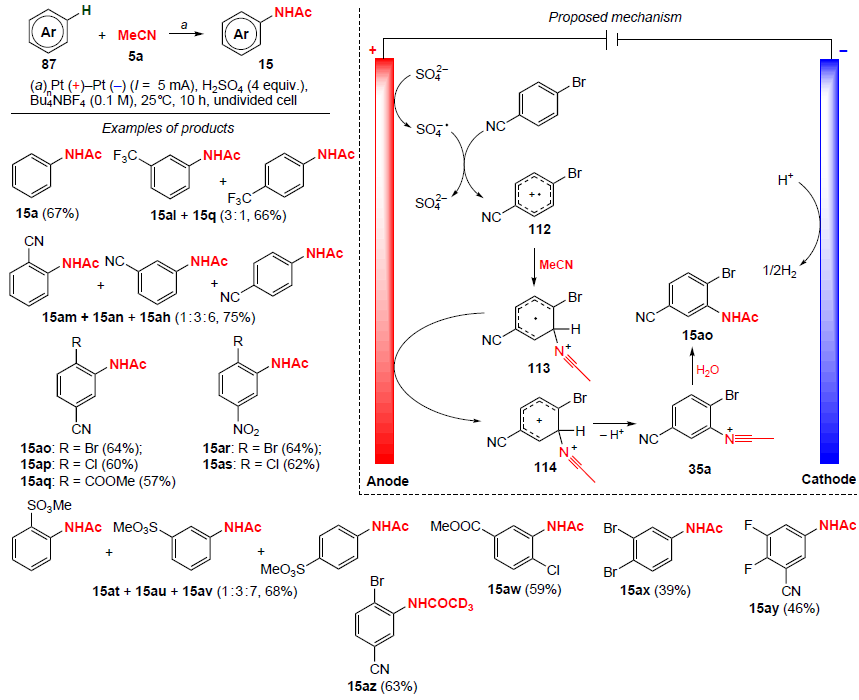
The proposed mechanism of amidation is shown in Scheme 37. In the first step, as a result of anodic oxidation of the sulfate ion, a sulfate anion radical is formed, which is involved in the single-electron oxidation of 4-bromobenzonitrile to generate cation radical 112. This cation radical then undergoes nucleophilic attack by acetonitrile to give radical 113, after which it undergoes further oxidation at the anode, affording cation 114. Deprotonation of the latter gives the nitrilium cation 35a, further hydrolysis of which leads to the target amidation product. Another possible reaction mechanism involves two-electron oxidation and deprotonation of the aromatic substrate 87 to furnish the corresponding cation, which then adds acetonitrile.
Yu.H.Budnikova and co-workers[90][95] developed an electrochemical protocol for the synthesis of anilides 15 and 42 in moderate to high yields (41 – 86%) using aceto- (5a) and propionitriles (5g) as amide sources (Scheme 38). This approach features mild conditions (room temperature, normal pressure); the reaction is carried out in an electrochemical cell with separation of the cathode and anodic spaces without the use of additives.
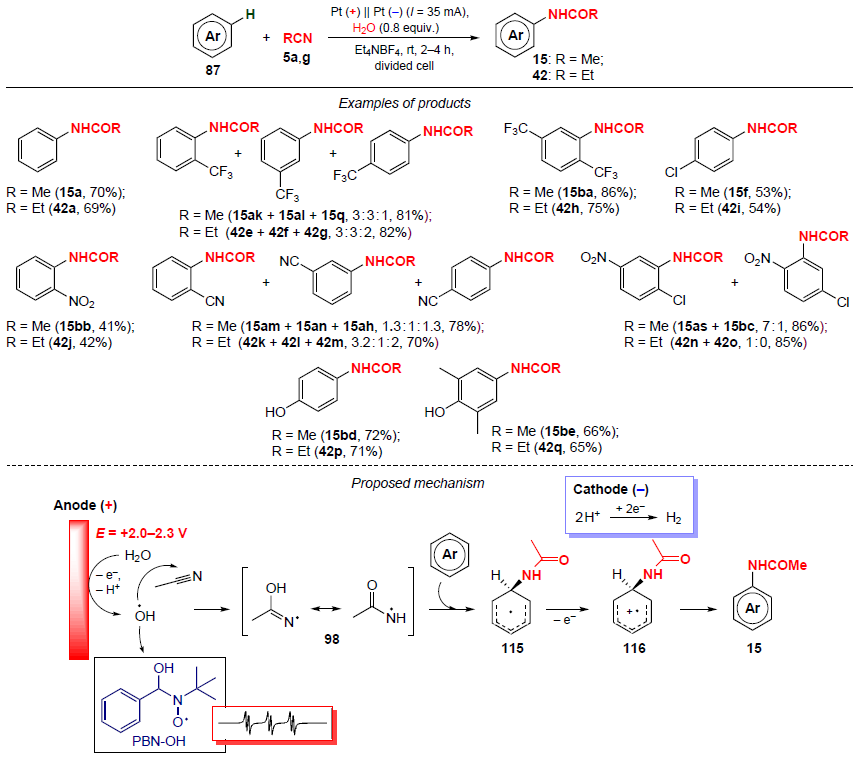
The proposed mechanism of the transformation is shown in Scheme 38. One of its key steps is the anodic oxidation of water to generate hydroxyl radicals (•OH), capable of adding to both the nitrile and the aromatic substrate. The formation of radicals during the electrochemical oxidation of aromatic substrates in acetonitrile was detected using electron paramagnetic resonance (EPR). When a benzene solution was oxidized in a MeCN – water (0.8 equiv.) mixture in the presence of N-tert-butyl-α-phenylnitrone (PBN) as a spin trap, the EPR spectrum of the spin adduct PBN-OH• was recorded. It is likely that the hydroxyl radical adds to the nitrile to afford the spin adduct 98, which then reacts with the aromatic substrate, which gives the amidation product 15 via intermediates 115, 116.
In 2022, Song and co-workers[107] reported electrophilic amidomethylation of aromatic compounds to give N-benzylacetamides 58. The authors used dimethyl sulfoxide and acetonitrile as sources of methyl (CH2) and amide (NHAc) units, respectively, and the process was carried out in a strongly acidic medium with prolonged heating to 60 – 80°C for 20 h (Scheme 39). The corresponding N-benzylacetamides 58 were isolated in moderate and good yields (31 – 82%). This method provides efficient practical synthesis of deuterated benzylamides from readily available deuterated solvents, DMSO-D6 and MeCN-D3 , and also allows functionalization of known drugs (gemfibrozil, naproxen, etc.).
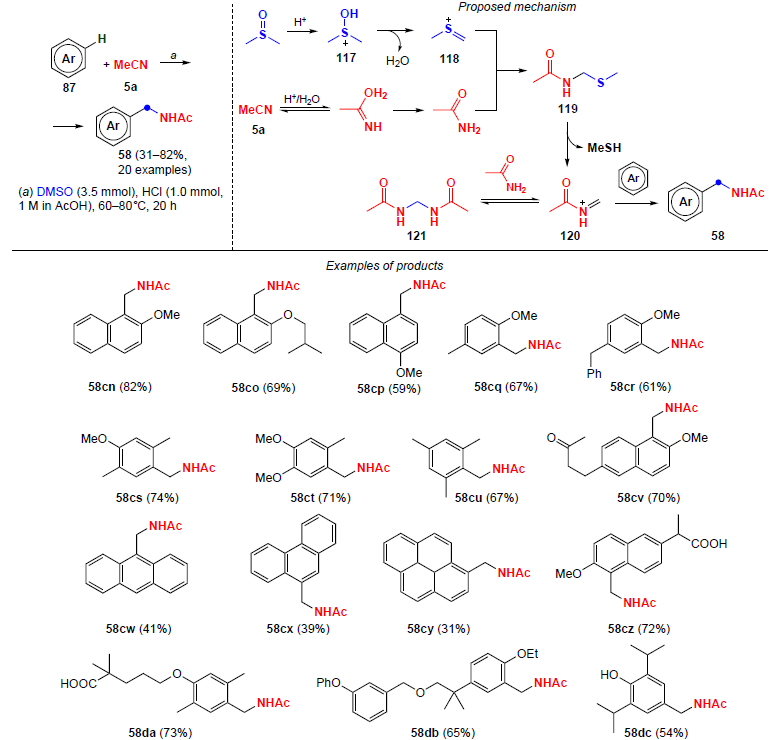
The proposed mechanism of the process is shown in Scheme 39. In the initial steps, dimethyl sulfoxide is protonated in an acidic medium to give an intermediate cationic form 117; its further dehydration leads to methyl(methylene)sulfonium 118. At the same time, acetonitrile undergoes acidic hydrolysis to acetamide, which reacts with the sulfonium cation 118 to afford intermediate 119. In an acidic medium, intermediate 119 decomposes to methanethiol and the ammonium cation N-methyleneacetamide 120. The latter reacts reversibly with acetamide and yields a by-product, N,N'-methylenediacetamide 121, which was isolated in an individual state.
Upon addition of an aromatic substrate to cation 120, the target amidomethylation occurs to afford N-benzylacetamide 58. The use of acetamide instead of acetonitrile also promotes the reaction, which could be evidence of the acidic hydrolysis of acetonitrile to acetamide as one of the key steps.
4. Amidation of heteroaromatic substrates
[]
4.1. Amidation of quinoxalin-2(1H)-ones
In 2019, Qu and co-workers[108] developed a method for direct oxidative palladium-catalyzed amidation of quinoxalin-2(1H)-ones 122 with acetonitrile (5a). The reaction proceeds at 100°C in the presence of an oxidizing agent, potassium persulfate (K2S2O8), for 12 h (Scheme 40). This method makes available various 3-acetaminoquinoxalin-2(1H)-ones 123 in good and high yields (up to 90%). Substrates containing electron-donating groups in the heterocycle (e.g. methyl) provide higher yields of products compared to quinoxalin-2(1H)-ones with electron-withdrawing substituents (F, Cl, Br). Nitroquinoxalin-2(1H)-ones were found to be unsuitable for this reaction, as was quinoxalin-2(1H)-one with a free nitrogen atom.
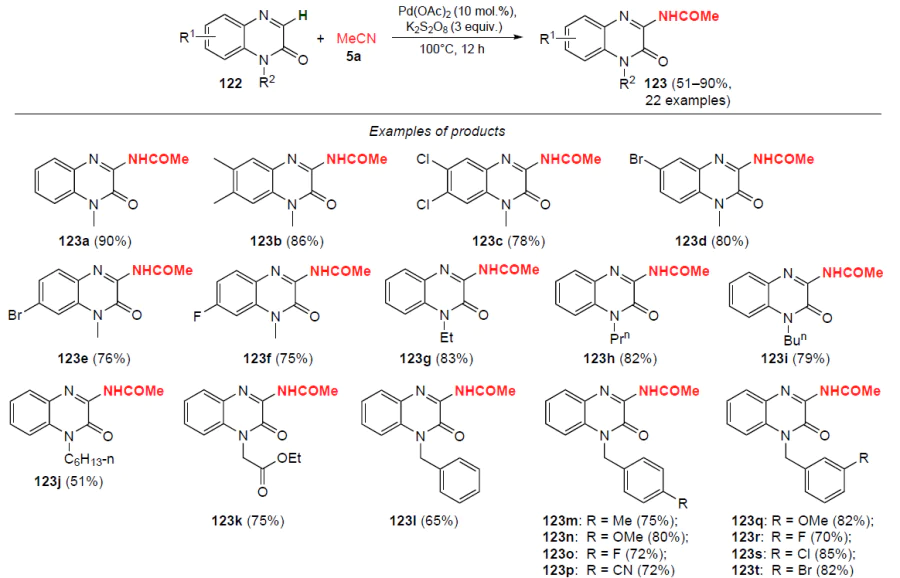
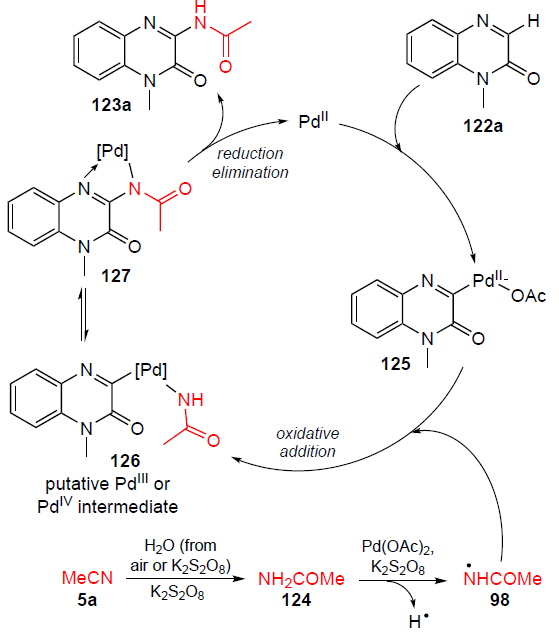
The proposed catalytic mechanism of the process using 1-methylquinoxalin-2(1H)-one (122a) as an example is shown in Scheme 41. Acetonitrile (5a) reacts with H2O under the action of K2S2O8 to give acetamide 124.[109] Promoted by K2S2O8 and the catalyst Pd(OAc)2 , homolytic cleavage of the N – H bond occurs in acetamide 124, forming radical 98. Metallation of 1-methylquinoxalin-2(1H)-one (122a) with palladium acetate affords intermediate 125, which undergoes oxidative addition to radical 98 to yield palladium(III) complex 126. Under dual palladium catalysis, PdIII intermediate 126 is oxidized to palladium(IV) compound 127 in the presence of a photoredox catalyst and/or an oxidizing agent.[110-112] Product 123a is formed by reductive elimination to release the PdII catalyst to be involved in the next catalytic cycle.
4.2. Amidation of xanthenes
In 2023, Li and co-workers[113] developed electrochemical amidation of xanthenes 128 with acetonitrile. The process takes place over 5 h in an undivided electrochemical cell equipped with graphite electrodes (Scheme 42). Xanthenes bearing Me, Ph, OMe and CF3 substituents were suitable substrates for this reaction, giving amidation products 129 in 56 – 75% yields.
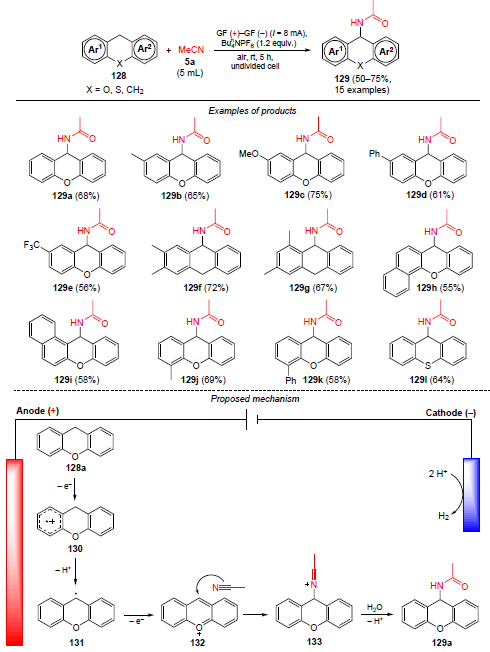
The plausible pathway of the electrochemical reaction with acetonitrile on the example of xanthene 128a is shown in Scheme 42. In the first step, single-electron oxidation of the substrate to cation-radical 130 takes place, followed by proton elimination and generation of radical 131. Next, anodic oxidation of radical 131 occurs to give cation 132, with reacts with acetonitrile via a nucleophilic addition mechanism to generate an intermediate nitrilium cation 133. At the final step, cation 133 is hydrolyzed to the desired amide 129a.
4.3. Amidation of quinoline N-oxides
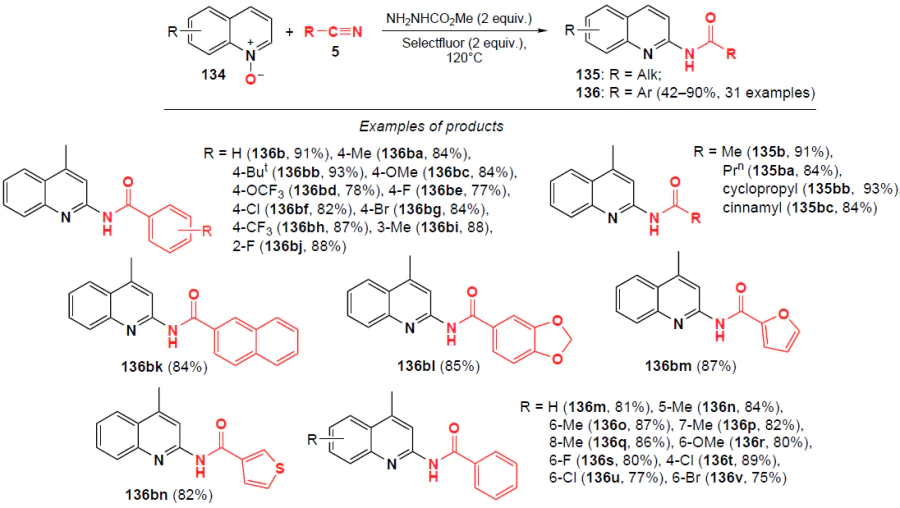
In 2018, He and co-workers[114] developed an approach to N-acylated 2-aminoquinolines 135, 136 based on quinoline N-oxides 134 using the [BMIM(SO3H)][OTf] (IL1) ionic liquid as a Brønsted acid (Scheme 43).* The reaction proceeds with 100% atom economy and high regioselectivity, and can involve aliphatic and aromatic nitriles, as well as diversely functionalized quinoline N-oxides. The advantage of this method is that the ionic liquid can be reused in subsequent reactions.
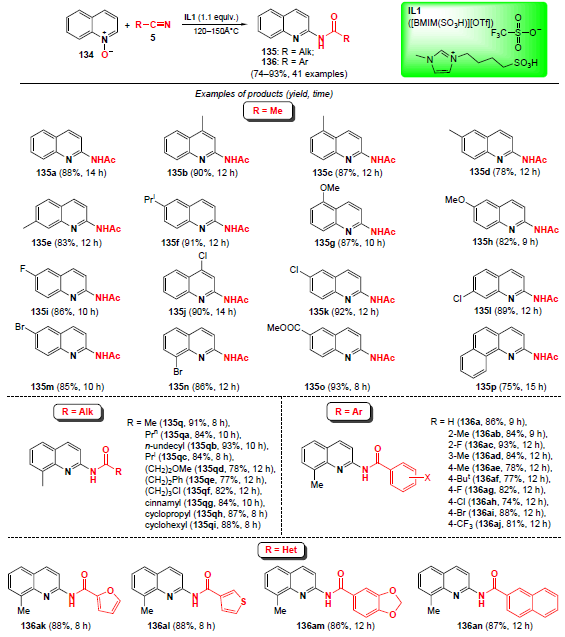
Based on these data and previous studies,[115][116] a probable mechanism for the process was proposed (Scheme 44). First, nitrile 5 is protonated by ionic liquid IL1 to generate cation 137. Then, the nucleophilic oxygen atom of quinoline N-oxide 134a attacks carbocation 137 to form intermediate 138, which undergoes intramolecular cyclization to yield cation 139. Finally, heterolytic cleavage of the N – O bond in this cation leads to intermediate 140, which undergoes deprotonation to afford the amidation product. The total amidation process involves cleavage of C – H and N – O bonds, formation of new C – N and C=O bonds, and a proton exchange between IL1 and quinoline N-oxide.
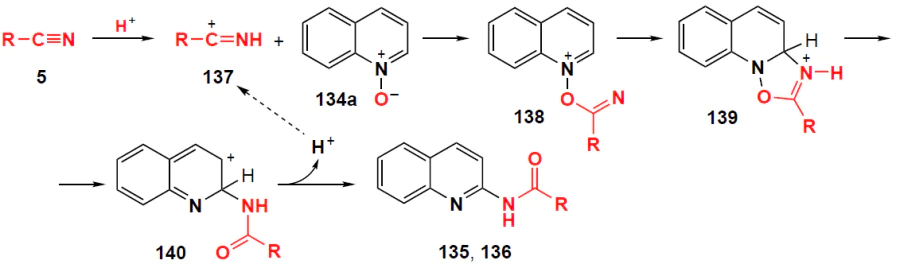
An alternative method for the high-temperature amidation of quinoline N-oxides 134 using p-toluenesulfonic acid monohydrate as a promoter and commercially available benzonitriles as amidating reagents was developed by Xiao and co-workers [117] (Scheme 45). This methodology does not require large amounts of organic solvent, is highly atom-economical (100% with respect to quinoline N-oxide), has simple reaction conditions, and yields of amidation products that range from good to excellent (75 – 94%).
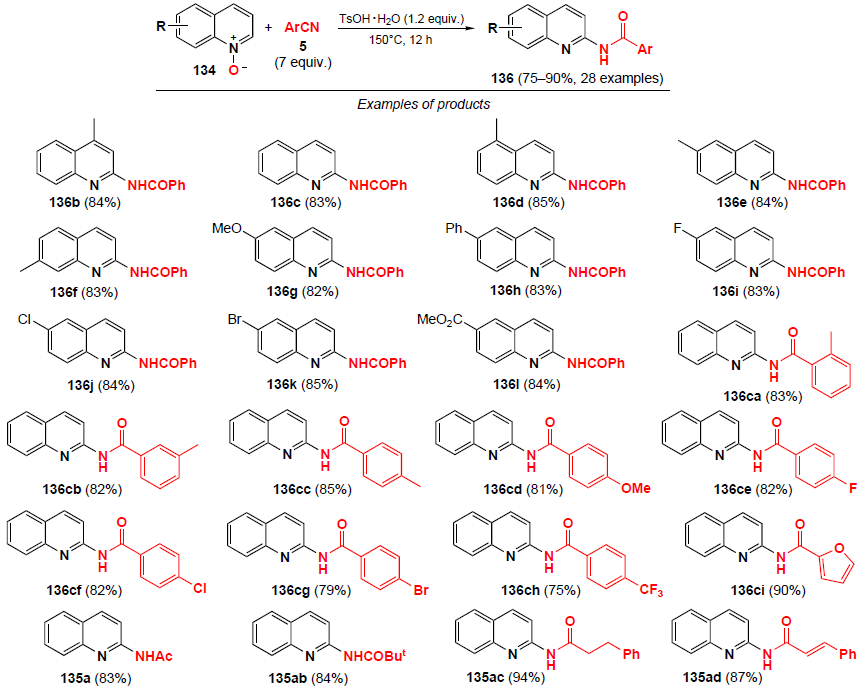
A probable mechanism of the process on the example of amidation of quinoline N-oxide 134a with benzonitrile (5b) is shown in Scheme 46. The amidation apparently occurs via 1,3-dipolar cycloaddition of quinoline N-oxide to cation 141, generated in situ under the conditions of the reaction of benzonitrile with TsOH · H2O,[118] affording a five-membered oxadiazolidine ring 139b. Then, intermediate 139b undergoes ring opening to give intermediate cationic form 140b, which is followed by a deprotonation/aromatization, which leads to the target product 136c and the regeneration of TsOH in the catalytic cycle.
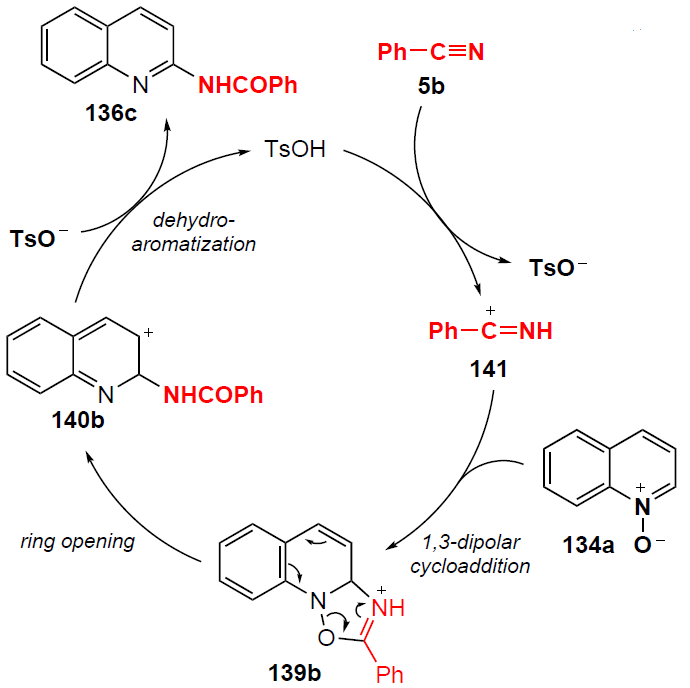
He and co-workers[119] proposed an effective alternative to amidation of quinoline N-oxides in the absence of metal catalysts, bases, and reducing agents using nitriles as the amide source and methyl carbazate as the radical activator and oxygen source (Scheme 47). Quinoline N-oxides and various functionalized nitriles readily react to furnish N-acylated 2-aminoquinolines in yields of up to 93%.
* In Schemes 43, 45, and 47, double letter numbering is used for some products: the first letter refers to substituents in the heterocycle, the second to those in the aroyl moiety.
5. Other examples of Ritter-type reactions
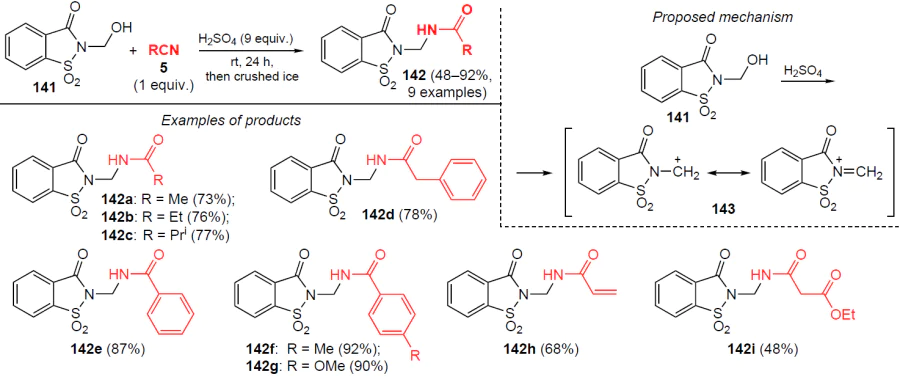
In 2024, de Oliveira and de Mattos[120] developed an approach to the amidation of N-(hydroxymethyl)saccharin (141) with nitriles, which allowed the synthesis of various N-[(saccharinyl)methyl]amides 142, which are of interest for medicinal chemistry. Amidation products 142 are obtained in yields of up to 92% within 24 h in the presence of 9 equiv. H2SO4 in the absence of solvent (Scheme 48). The rate-limiting step of this reaction is the formation of the relatively unstable intermediate carbocation 143, therefore, only a strongly acidic medium is effective for the amidation.
Malapaka and co-workers[121] described an acid-catalyzed three-component one-pot protocol to obtain 3-aryl-3-amidooxindoles directly from commercially available isatin derivatives 144, nitriles, and C-nucleophiles, tertiary arylamines 145 (Scheme 49).
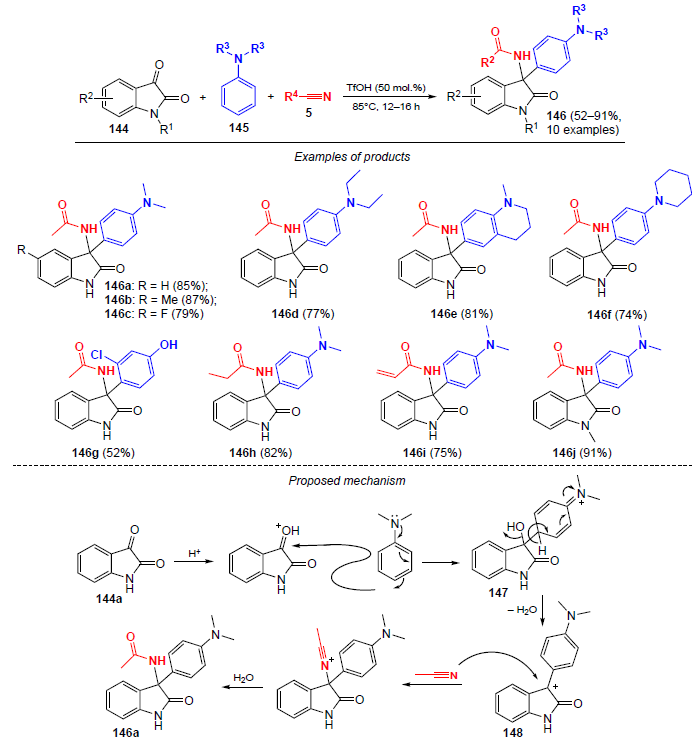
Various C-nucleophiles such as N,N-dimethylaniline (DMA), N,N-diethylaniline (DEA), 1-methyl-1,2,3,4-tetrahydroquinoline and 1-phenylpiperidine were used in this reaction. Tertiary amines reacted smoothly with isatin and acetonitrile upon heating to 85°C for 12 – 16 h in the presence of TfOH to give the amidation products 146 in high yields (up to 87%). The nucleophilic 3-chlorophenol was also subjected to a tandem Ritter reaction with isatin, furnishing the amide 146g in low yield (52%).
The use of propionitrile and acrylonitrile in the reaction with isatin and DMA led to the formation of amidation products in good yields (75 – 82%), but aromatic nitriles in this case were unreactive. N-Substituted isatins also undergo this reaction, as demonstrated by the example of the coupling of N-methylisatin with DMA and MeCN. As a result, N-{[3-(4-(dimethylamino)phenyl]-1-methyl-2-oxoindolin-3-yl}acetamide (146j) was obtained in 91% yield.
A probable mechanism of the process is illustrated by the example of the formation of N-{[3-(4-(dimethylamino)phenyl]-2-oxoindolin-3-yl}acetamide (146a) (see Scheme 49). In the first step, the carbonyl group of isatin 144a is protonated at position C(3). Next, the protonated form of isatin undergoes nucleophilic attack by DMA, which leads to intermediate 147. In turn, this intermediate undergoes rearomatization under dehydrating conditions to give intermediate carbocation 148. In the final step, nucleophilic attack of the nitrile at position C(3) followed by hydrolysis yields the amidation product.
The same research group[122] developed an acid-catalyzed multicomponent procedure for the synthesis of N-benzylamide derivatives from aldehydes 149, N,N-disubstituted arylamines 145 and nitriles (Scheme 50). Aromatic aldehydes with electron-withdrawing and electron-donating groups, aliphatic and aromatic nitriles, and C-nucleophiles (DMA, DEA, 1-methyl-1,2,3,4-tetrahydroquinoline, 1-methylindoline) were used in this reaction; the yields of the amidation products 150 reached 93%. The proposed method also allows the use of β-naphthols, 1,3-dicarbonyl compounds and 1,3,5-trimethoxybenzene as C-nucleophiles.
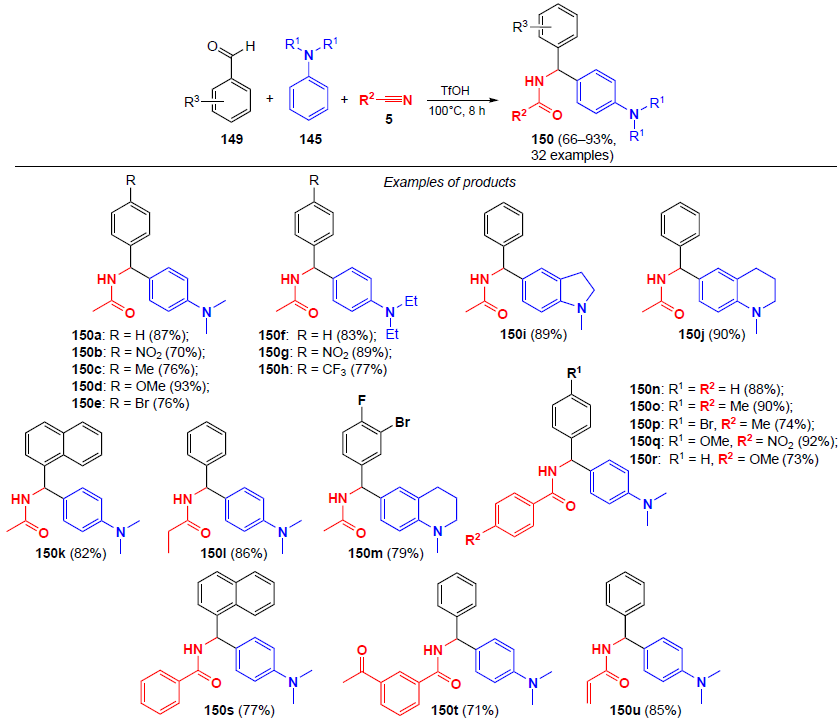
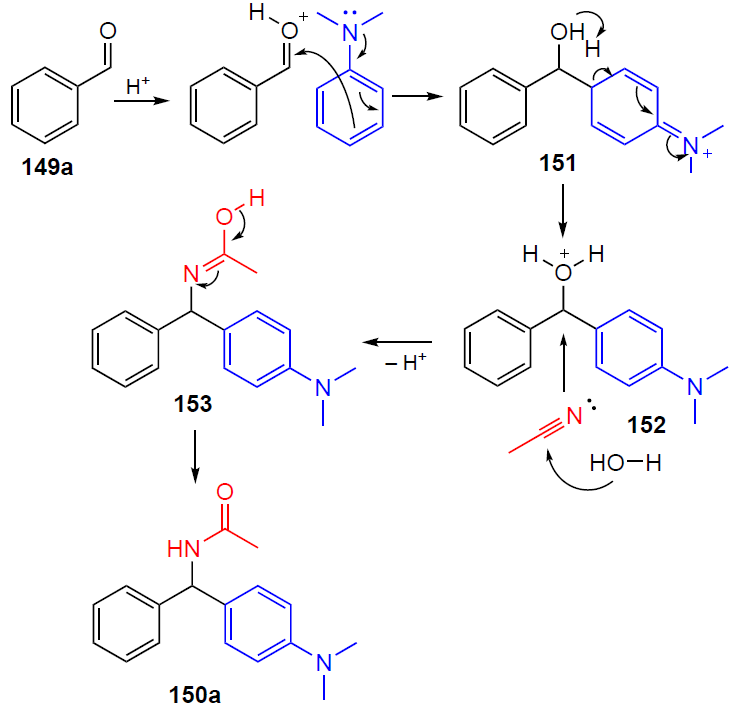
The plausible mechanism of the reaction is presented on the example of the preparation of N-{[4-(dimethylamino)phenyl](phenyl)methyl}acetamide (150a) (Scheme 51). In the first step, benzaldehyde undergoes protonation at the formyl oxygen atom. Next, nucleophilic addition of DMA to the protonated form of benzaldehyde occurs to give intermediate 151. The subsequent rearomatization of 151 furnishes intermediate 152. Intermediate 152 after dehydration undergoes nucleophilic attack by acetonitrile and water to the iminol form 153. The resulting iminol 153 ultimately tautomerizes to the target amide 150a.
6. Conclusion
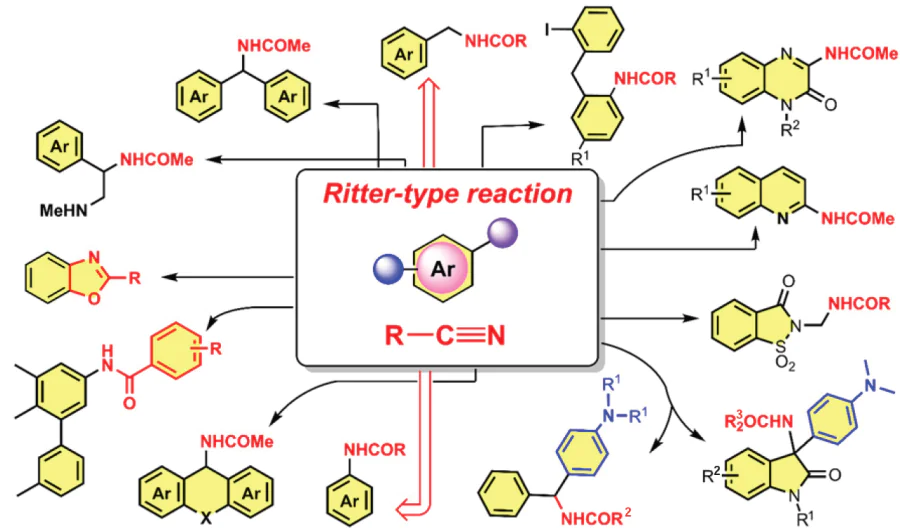
Organic compounds containing N-arylamide moieties are versatile components of pharmaceuticals, agrochemicals and organic functional materials. The search for new and improvement of known approaches to their synthesis have been developing rapidly in recent years. This review summarizes various transformations that follow the Ritter-type reaction pathway and are used in the synthesis of N-(het)arylamides and N-benzylamides both under the conditions of traditional organic synthesis and in their photo-, electro- and photoelectrochemical modifications (Scheme 52). Over the last 5 years, many significant works have appeared on the synthesis of N-arylamides and N-benzylamides involving nitriles, especially under electrochemical conditions. In this case, the latest publications have considered the functionalization of not only alkyl and benzyl C(sp3) – H bonds, but also aromatic C(sp2) – H bonds, which previously could not be involved in such transformations. This opens up new possibilities for improving this simple and atom-efficient method, which does not require preliminary functionalization of the substrates. There are very few known examples of obtaining N-arylamides by direct functionalization of aromatic C(sp2) – H bonds with nitriles, and some of them were developed only in 2023 – 2024.
The reactions considered in this review are used, inter alia, for the synthesis of biologically active compounds and compounds with potential activity. The authors not only developed new synthetic approaches to N-(het)arylamides and N-benzylamides, but also proposed mechanisms for these processes, which undoubtedly contributes to the understanding of the essence of known reactions and the discovery of new ones and approaches intended for obtaining nitrogenous compounds of other classes.
In recent years, there has been an increase in the number of studies in which the Ritter amidation reaction is a key step, indicating the expansion of the capabilities of this methodology. The involvement of more complex and multifunctional substrates, as well as the production of practically relevant biologically active molecules and natural products, emphasizes the promise of this synthetic approach. The authors hope that this review will inspire researchers to search for and develop new methods of Ritter amidation under classical conditions, as well as using electrochemistry and photoelectrochemistry. Such transformations should involve heteroaromatic substrates, examples of which are currently virtually absent, as well as complex molecules of practical value. Expanding the scope of application of the Ritter reaction and its modern versions can open new horizons in the field of organic synthesis and contribute to the development of effective pharmaceuticals and innovative materials.
7. List of abbreviations and designations
The following abbreviations and designations are used in the review:
B — base,
E — potential,
I — current,
L — ligand,
CAN — cerium(IV) – ammonium nitrate,
Cat — catalyst,
CFL — compact fluorescent lamp,
CP — carbon paper,
DCE — 1,2-dichloroethane,
DDQ — 2,3-dichloro-5,6-dicyano-1,4-benzoquinone,
DEA — N,N-diethylanilin,
DMA — N,N-dimethylanilin,
DMEDA — N,N'-dimethylethylenediamine,
EPR — electron paramagnetic resonance,
GC-MS — gas chromatography – mass spectrometry,
HAT — hydrogen atom transfer,
HFIP — hexafluoropropan-2-ol,
IL — ionic liquid,
LED — light-emitting diode,
Mes — mesityl,
MSA — methanesulfonic acid,
MWCNT-CSP — multi-walled carbon nanotubes modified with p-toluenesulfonic acid,
NHPI — N-hydroxyphthalimide,
PBN — N-tert-butyl-α-phenylnitrone,
PINO — phthalimide N-oxyl,
PTFE — polytetrafluoroethylene,
rt — room temperature,
SET — single-electron transfer,
SS — stainless steel (electrode),
ТАС — tris(cis-2,6-dimethylpiperidin-1-yl)cyclopropenium,
TBN — tert-butyl nitrite,
TEMPO — (2,2,6,6-tetramethylpiperidin-1-yl)oxyl,
TFA — trifluoroacetic acid,
TfO — trifluoromethanesulfonate (triflate),
TMS — trimethylsilyl,
TsO — p-toluenesulfonate (tosylate),
US — ultrasound.
References



![Designing Hypothesis of 2-Substituted-N-[4-(1-methyl-4,5-diphenyl-1H-imidazole-2-yl)phenyl] Acetamide Analogs as Anticancer Agents: QSAR Approach](/storage/images/resized/IYy5KbQWZ2uxYLvcvjXjlXRDR3xNOFyFK4Kl6043_small_thumb.webp)










