



Keywords
Abstract
Isocoumarins are isomers of a widely known class of aromatic organic lactones, coumarins, which possess unique photophysical properties. Despite the structural similarity of coumarins and isocoumarins, applications of the latter have long been limited to medicine and agriculture, in view of their fungicidal, antibacterial, and anti-inflammatory activities. Most organic compounds of this class have been isolated from living organisms. The effective method for the synthesis of isocoumarins discovered in the beginning of the 21st century, which is based on C – Н coupling of benzoic acids with acetylenes, made these compounds easily accessible. This gave impetus for their wide use in the design of materials for organic photonics and optoelectronics. The present review focuses on the photophysical properties of isocoumarins in comparison with coumarins and covers known methods for the design of the isocoumarin core published before March, 2025.
The bibliography includes 249 references.
1. Introduction
Over the last decades, organic luminophores have found wide use in various fields of science and human life, ranging from dyes and fluorescence markers to photonics and optoelectronics.[1-13] In view of the rapid development of technologies related to electroluminescent materials, the search for photoactive organic compounds with specified properties is a relevant task, which, in turn, motivates research in the molecular design and development of new methods for the synthesis of these systems.
Coumarin derivatives are among the most popular classes of organic luminophores;[14-26] however, the photophysical properties of related isocoumarin are still poorly studied, despite the structural similarity of these compounds. Isocoumarins differ from coumarins by the relative positions of the carbonyl group and the ether oxygen atom (Fig. 1).
![[{"id":"RzPcV9lfIB","type":"paragraph","data":{"text":"Structural formulas of coumarin and isocoumarin"}}]](/storage/images/resized/03PPrpQRmCNkv6Cv5cnQ48T9pz8LkyXAavecRK8L_xl.webp)
Although a few reviews on the methods for isocoumarin synthesis have been published in recent years, they mainly consider approaches to the synthesis of biologically active derivatives or natural products and cover the most advanced procedures.[27-33] The present review is focused on the photophysical properties of isocoumarins and applications associated with these properties. In particular, the review covers all currently available published examples of photoactive isocoumarins possessing promising properties such as solid-state luminescence, aggregation-induced emission (AIE), and thermally activated delayed fluorescence (TADF) for the subsequent use in organic light-emitting diodes (OLEDs). In addition, all methods for the design of isocoumarin core described in the literature up to March of 2025 are surveyed.
2. Luminophores based on isocoumarin derivatives
As noted above, isocoumarins are isomers of coumarins, well-known organic compounds, the photophysical properties of which have been studied in detail. Unlike isocoumarins, coumarins have already found practical use in various fields of optoelectronics, molecular probing, etc. However, the parent compound, unsubstituted coumarin, has a very modest fluorescence quantum yield (ϕF = 0.2%). If an electron-donating group is introduced in position 7 (and an electron-withdrawing group in position 3) of the coumarin core, the situation dramatically changes, and the luminescence quantum yield for this derivative increases by a large factor (up to 81%). [34] This is due to the fact that the electron-donating group activates the intramolecular charge transfer (ICT) as a result of conjugation, which can be represented as two resonance structures (Fig. 2).
![[{"id":"g3EWvdy7CY","type":"paragraph","data":{"text":"Carbon atom numbering in coumarin (on the left) and electronic effect of substituents in the particular positions of the coumarin core on the luminescent properties"}}]](/storage/images/resized/QCUmzHiziwa3EhuwEothl2KEkiZHVa44J57gGCpS_xl.webp)
Another way of coumarin modification that changes their photophysical parameters such as absorption and emission wavelengths and luminescence quantum yield is to extend the π-system by switching to annulated systems such as benzocoumarins. The photophysical properties of these derivatives, like those of coumarins, can be predicted from the nature and positions of electron-donating and/or electron-withdrawing substituents.[35]
A lot of coumarin-based dyes and molecular sensors have been prepared using these approaches.[14-18] In addition, fluorescent compounds were found among these derivatives, and they are actively used to produce materials with useful properties for various devices, including OLEDs based on the TADF effect.[19-25] Meanwhile, much less examples of studies of photophysical properties and applications in photonics have been reported for isocoumarins, which do not significantly differ from coumarins in the structure or practical potential. Indeed, no data on the photophysical characteristics of unsubstituted isocoumarin are available from the literature.
We assumed that a similar substituent effect could also be used in the case of isocoumarins, which structurally differ only in the position of the oxycarbonyl moiety. Thus, the introduction of a donor group in position 6 (or a donor group in position 7 and an acceptor group in position 3) should induce a similar fluorescence enhancement for isocoumarin derivatives (Fig. 3).
![[{"id":"2ERNBU_6YN","type":"paragraph","data":{"text":"Carbon atom numbering in isocoumarin (on the left) and anticipated effect of donor (D) and acceptor (A) groups at particular positions of the isocoumarin core on the luminescent properties"}}]](/storage/images/resized/khDliykRWF8OwLEI42malMyIDDr3YfWHRqRubnwy_xl.webp)
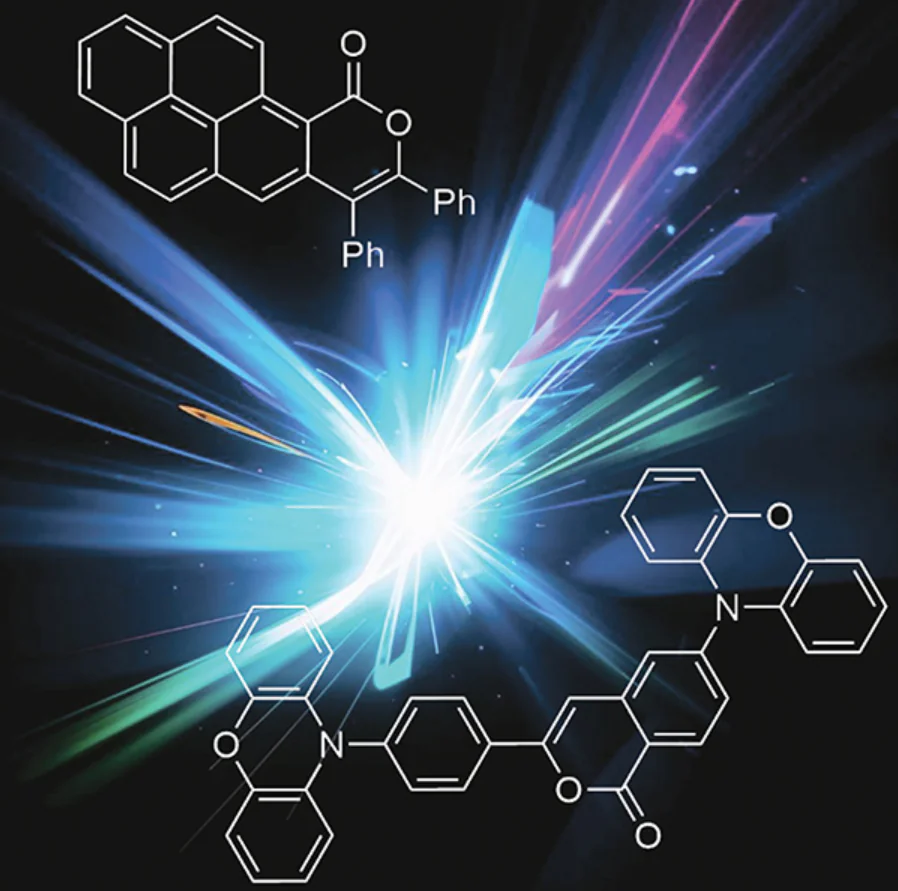
Indeed, although many isocoumarin derivatives (e.g., compound 1), like coumarins, exhibit very low fluorescence, it was shown in our study [36] that the presence of electron-donating substituents at position 6 leads to a substantial increase in the fluorescence quantum yield in solution (Fig. 4). For example, isocoumarin 2, containing diphenylamino group has a quantum yield of 95%. A major nonradiative relaxation pathway of isocoumarins is possibly photoinduced reversible lactone ring opening, as indicated by the deviation of the ether moiety from the plane of the molecule in the S1 excited state [according to the density functional theory (DFT) data]. Meanwhile, the presence of the Ph2N electron-donating group at position 6 stabilizes the S1 state of compound 2, and no distortion of the molecular geometry occurs.
![[{"id":"WIiiTilnl-","type":"paragraph","data":{"text":"Comparison of the ground state (S<sub>0</sub>) and the first excited state (S<sub>1</sub>) geometries for isocoumarins 1 (<i>a</i>) and 2 (<i>b</i>), possessing weak and strong fluorescence, respectively.<sup>36</sup> Red spheres designate the oxygen atoms; hydrogen atoms for compound 1 are omitted for simplicity; the distances between the C(1) and O(2) atoms in the six-membered ring are given according to DFT data."}}]](/storage/images/resized/u2QqaIN50CvSGpitNWwUwZdEUzGtEqv42yXlGuny_xl.webp)
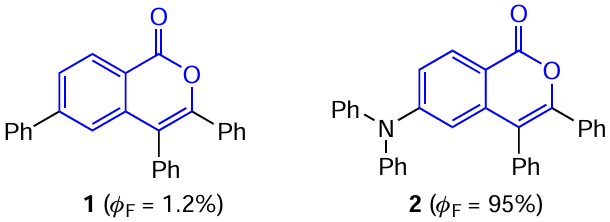
The attachment of isocoumarin at position 6 to other heterocyclic luminophores, e.g., isoquinolinium cations (structures 3 and 4) provides the possibility of fine tuning of the photophysical properties of compounds.[37] Thus, by varying the nature of substituents in the isoquinoline moiety, it is possible to switch the luminescent properties from high-efficiency π – π* type fluorescence with a quantum yield of 99% to intramolecular charge transfer, with the highest occupied molecular orbital (HOMO) being localized on the isocoumarin moiety, which has a low quantum efficiency (Fig. 5). The use of isocoumarin as the donor moiety in the donor – acceptor (D – A) systems makes it possible to achieve a low energy difference between the first singlet and triplet excited states (ΔEST), which may find use in the development of TADF emitters (see below, Section 2.2).
![[{"id":"rX5Gnxl8L2","type":"paragraph","data":{"text":"Effect of the nature of substituent in isocoumarin-substituted isoquinolinium cations <b>3</b> (<i>a</i>) and <b>4</b> (<i>b</i>) on the Δ<i>E</i><sub>ST</sub> value determining the photophysical properties of the compound.<sup>37</sup> Figure reproduced with permission from the American Chemical Society."}}]](/storage/images/resized/2S7Wezsm0Dgy8cDBHL5TjN80aJnKfV3r9GsjUT8W_xl.webp)
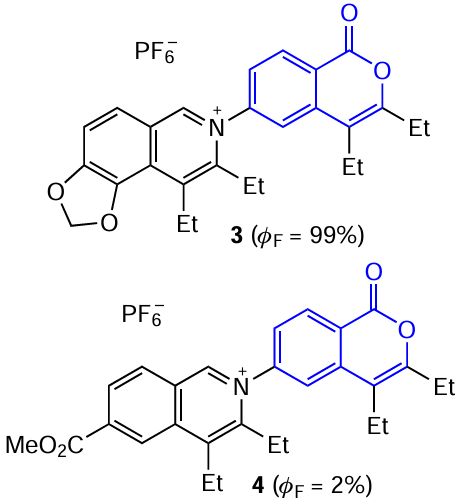
Due to the high electron deficiency and planar geometry of the isocoumarin core as well as to the π – π packing of molecules present in isocoumarin crystals, which provides for fast transport paths for electrons and holes, this structural moiety can be used to design light-emitting devices either as acceptor group in more complex polycyclic luminophores or as an electron-conductive linker.
2.1. AIE effect and solid-state luminescence of isocoumarins
It is known that most organic luminophores that exhibit pronounced luminescence in solution prove to have low efficiency as light-emitting materials because of the aggregation-caused quenching (ACQ) effect.[38-40] In this regard, of particular importance are compounds that exhibit the opposite effect, that is, aggregation-induced emission (AIE), which allows them to maintain high light-emission efficiency in the solid state.[41-43] The tremendous recent interest of researchers in AIE luminophores is caused particularly by their luminescence in the solid state, which is associated, among other factors, with the aggregation-induced delayed fluorescence (AIDF) mechanism caused by fixation of the perpendicular arrangement of the planar parts of molecules, thus providing a low degree of overlap of frontier orbitals.[44-46]
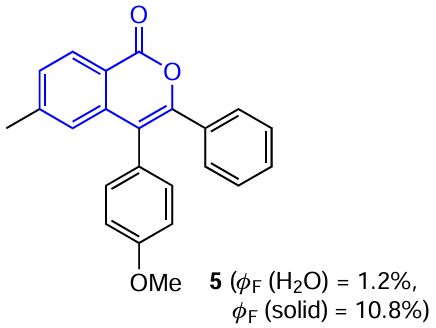
An important feature of isocoumarins, as well as coumarins, is the ability to form intermolecular contacts through lone pairs of electrons; this may potentially be useful for the production of materials exhibiting the AIE effect. In addition, it was found that the introduction of a MeO group in the para-position of one phenyl substituents in isocoumarin 5 leads to a pronounced fluorescence enhancement on going from solutions to the crystalline state (up to 11%).[47] Apparently, this fact is attributable to the formation of C – H⋯O, C – H⋯C, and O⋯O intermolecular contacts, which restrict the rotation of molecule and thus prevent the non-radiative energy loss (Fig. 6).
![[{"id":"9rGPJDgVKY","type":"paragraph","data":{"text":"Molecular structure (<i>a</i>) and crystal packing (<i>b</i>) of isocoumarin <b>5</b>, which demonstrates the AIE effect caused by the formation of intermolecular C – H⋯O, C – H⋯C, and O⋯O conacts.<sup>47</sup>"}}]](/storage/images/resized/fLWYuRdCuNaQUNSbcBSvFIB8hCieHcczDYlusJGb_xl.webp)
Tang and co-workers [47] were able to obtain polymer materials based on this isocoumarin moiety. Polymer 6 has a good thermal stability, optical transparency, and film-forming properties. In addition, this compound shows fluorescence enhancement upon the aggregation in THF – water mixtures with gradually increasing water content (Fig. 7).
![[{"id":"8_Ng80fk2J","type":"paragraph","data":{"text":"AIE effect of polymer 6 manifested as enhancement of the emission in THF – water mixtures with increasing water fraction (<i>f</i><sub>W</sub>) (<i>a</i>) and dependence of the relative fluorescence intensity (<i>I/I</i><sub>0</sub>) on the composition of the solvent (<i>b</i>).<sup>47</sup> Figure reproduced with permission from the Royal Society of Chemistry."}}]](/storage/images/resized/k6aR9RkfFVXcj7pVRW69mRhBXPEvt404OIRVizEX_xl.webp)
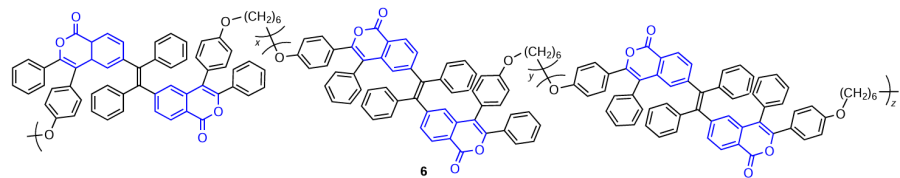
A similar effect of fluorescence enhancement in the blue spectral region on going to the crystalline state is inherent in 8-aminoisocoumarins, e.g., compound 7.[48] These compounds exhibit bright fluorescence in solutions and in thin films. In the emission spectra of these aminoisocoumarins in acetonitrile – water solutions with different water contents, the emission intensity gradually increases as the water fraction ( fw) increases from 10 to 80 vol.%. Since isocoumarins are readily soluble in most organic solvents (except for non-polar alkanes, benzene, etc.) and very poorly soluble in water, this system is used to detect the AIE effect. A solution of a test compound in an organic solvent is gradually diluted with water until aggregation of molecules occurs. For example, in the case of 8-aminoisocoumarin 7, further increase in the water content from 80 to 90 vol.% leads to a considerable increase in the fluorescence intensity caused by the change in the distribution order of the aggregates compared to the initial state (Fig. 8).
![[{"id":"lMHh6iKceG","type":"paragraph","data":{"text":"Dependences of the emission intensity of 8-aminoisocoumarin <b>7</b> in acetonitrile–water solutions on the content of water demonstrating the AIE effect.<sup>48</sup> Figure reproduced with permission from the Royal Society of Chemistry."}}]](/storage/images/resized/iyIWbcsxI5rTT5BSnawWsdioUYjYaQQdXay1kKNU_xl.webp)
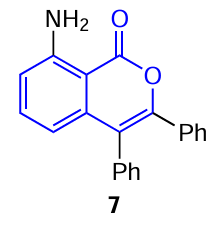
In 2009, Miura and co-workers [49] showed that 8-(arylamino)isocoumarins can also exhibit solid-state fluorescence in the 450 – 500 nm range of wavelengths. Meanwhile, 3,4-diphenylisocoumarin unsubstituted in the aromatic ring does not exhibit fluorescence, indicating that introduction of an amino group in position 8 of the isocoumarin system is essential for fluorescence properties. Notably, 8-(arylamino)isocoumarins exhibit emission with the intensity twice higher than the emission intensity of the classical coumarin-153 dye.
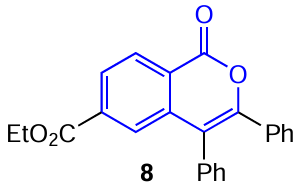
A similar behaviour in THF – H2O mixtures with various water contents was also observed for isocoumarin 8 with an electron-withdrawing ester (ethoxycarbonyl) group at position 6.[50] It was shown that increase in the water content from 90 to 99 vol.% induces a 2.5-fold enhancement of the emission in the fluorescence spectrum (Fig. 9); this may be attributed to the formation of dimers or more extended aggregates of this compound.
![[{"id":"Hev5Y_PJ1T","type":"paragraph","data":{"text":"Dependences of the emission intensity of 6-ethoxycarbonylisocoumarin <b>8</b> in acetonitrile – water solutions on the content of water demonstrating the AIE effect.<sup>50</sup> Figure reproduced with permission from Elsevier."}}]](/storage/images/resized/c8Jmh9nrztXzNPlIw6zY5NB4htmfhbPKGOMKZQ6H_xl.webp)
2.2. Isocoumarins as potential TADF materials for OLED technology
The significant breakthrough in the OLED technology made in recent years was due to the discovery of involvement of triplet states in emission through the TADF effect. Starting with the first study published in 2012 by Adachi co-workers,[51] quite a few TADF materials have been developed and successfully used in OLEDs. Nevertheless, studies aimed at increasing the stability, enhancing the performance, reducing the loss of efficiency during the operation of these devices, etc., are still relevant today.[52-55]
It should be borne in mind that the theoretical maximum external quantum efficiency (EQE) for OLEDs based on TADF effect is limited to 20% [provided that the internal quantum efficiency (IQE) of the device is 100% and the light scattering coefficient is 1/5]. In order to increase EQE, many researchers develop OLED displays based on exciplexes, which, however, show a considerable efficiency roll-off at a high excitation density and at a practical luminance of 1000 cd m–2. The research group headed by Professor Zhang [56] addressed this problem using an innovative strategy for the design of new type exciplex TADF emitters based on the introduction of a single-molecule TADF emitter as a component of the donor – acceptor (D – A) pair. This type of TADF emitters can utilize more triplet excitons and, hence, exhibit higher quantum efficiency in the devices. As the acceptor, the authors used triphenylphosphine oxide-substituted triazine 9 (code number PO-T2T), while dimethylacridine isocoumarin derivative 10 (code number MAC) served as the donor. The performance was additionally evaluated by comparing the results with those for a conventional donor, m-di(carbazol-9-yl)benzene (11, mCP).
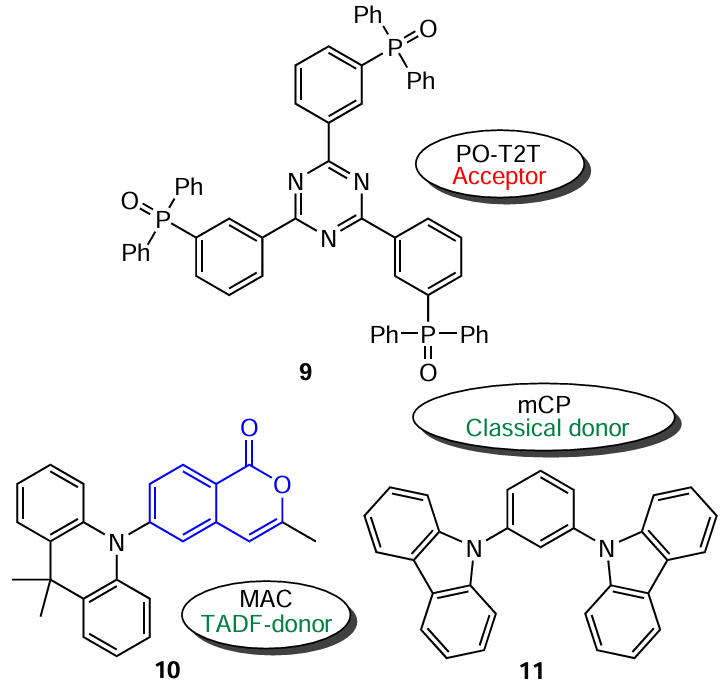
A study of electroluminescence (EL) using an optimized device based on the MAC (10) : PO-T2T (9) pair revealed a high EQE of 17.8%, which was the highest value of efficiency among the known exciplex OLED displays known up to 2016. This increase in efficiency is due to the additional reverse intersystem crossing (RISC) pathway inherent in isocoumarin molecules.[56]
Two more examples of the use of isocoumarin derivatives as dopants for the emitting films of exciplex TADF emitters were reported by the same research group.[57] Compounds 12 (PHzMCO) and 13 (PHzBCO) proposed by the authors were D – A structures with a phenoxazine ring as an electron donor and an isocoumarin core as an acceptor (Fig. 10).
![[{"id":"v3N9RUGipb","type":"paragraph","data":{"text":"EQE as a function of applied luminance of TADF-OLED cells based on mCP (<b>11</b>) films doped with 8 mass % isocoumarins <b>12</b> and <b>13</b>.<sup>57</sup> Figure reproduced with permission from the American Chemical Society."}}]](/storage/images/resized/OPQ6x02LM9yWbSy4WhxBLdGJyFnEbjnugHKjQvs5_xl.webp)
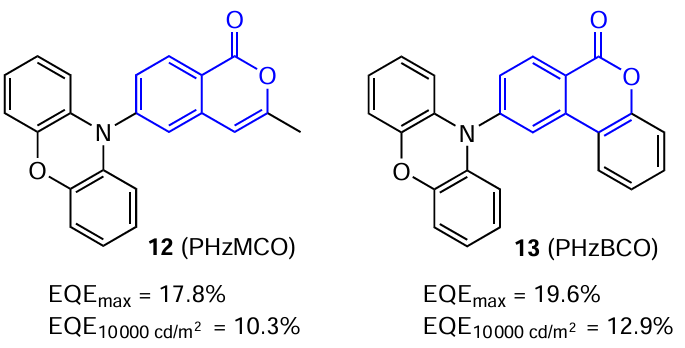
Both materials possess high photoluminescence quantum yields (PLQYs) of 0.47 and 0.52 as well as very low ΔEST values of 0.018 and 0.006 eV, respectively.[57] It is noteworthy that both films of compound 11 doped with isocoumarins PHzMCO and PHzBCO had the maximum PLQY values (0.47 and 0.52) when the doping level was 8 mass %. This can be attributed to the balance between insufficient exciton consumption at low dopant concentration and apparent exciton quenching at high dopant concentration. TADF devices based on these derivatives also showed high maximum external quantum efficiency (EQEmax) values, amounting to 17.8 and 19.6% for compounds 12 and 13, respectively. Moreover, both devices had a relatively low efficiency roll-off at high luminance, while maintaining the external quantum efficiency at 10 000 cd m–2 of 10.3 and 12.9%, respectively.[57] Thus, it was shown [57] that TADF emitters characterized by high PLQY values and low ΔEST values may effectively prevent the efficiency roll-off in TADF-OLED displays.
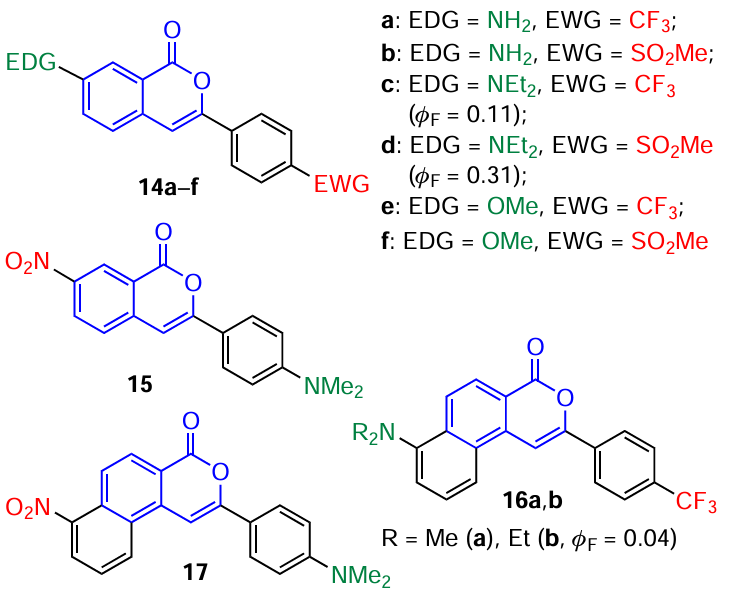
Abbiati and co-workers [58] were able to expand the library of D – π – A-type molecules in which isocoumarin acts as a core unit (π-linker between the donor and the acceptor). They prepared ten fluorescence dyes 14 – 17 sensitive to the polarity of the medium, which contained an isocoumarin-based bridge conjugated with electron-donating (EDG) and electron-withdrawing (EWG) groups. The authors used various combinations of D – A pairs, including those with reversed D – π – A system.
It is noteworthy that isocoumarins 14e,f, containing a methoxy group as EDG showed slight solvatochromism. Compounds 15 and 17 with reversed D – π – A system demonstrated interesting reversed solvatochromism in the visible region, but had low solubility and weak fluorescence. It was shown that compounds 14c,d containing an electron-withdrawing group at the para-position of the 3-phenyl substituent and the Et2N electron-donating group at position 7 of isocoumarin had the best photophysical and spectral characteristics for possible applications in biology and as advanced materials. In particular, they possessed a pronounced solvatochromic effect, good solubility in various solvents, constant absorption near the visible region, fairly intense fluorescence, and a large Stokes shift. However, the authors of the cited study did not investigate the potential TADF effect in these molecules.
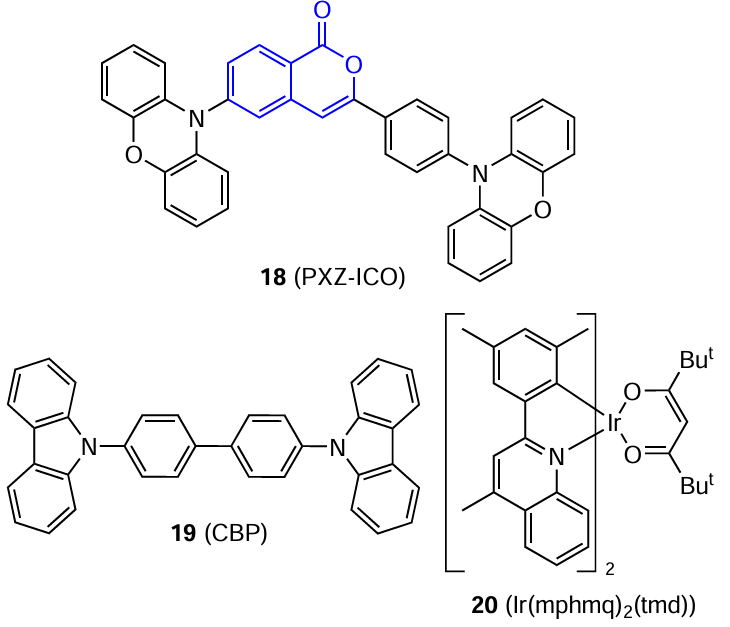
Recently, Bin and co-workers [59] showed that isocoumarins can act as structural units in donor – acceptor – donor (D – A – D) type photoactive compounds. The authors found that isocoumarin 18 (PXZ-ICO), containing two phenoxazine donor moieties, is characterized by a moderate energy difference between the singlet and triplet excited states, good transport properties, and high thermal stability. In addition, this compound exhibits good bipolar transport properties in the solid state at slightly higher hole current density compared to the electron current density, potentially making it a good candidate for use as a host material. The bis[2-(diphenylphosphino)phenyl] ether oxide (DPEPO) film doped with PXZ-ICO (10%) shows clear-cut delayed fluorescence with a lifetime of 343 μs, which attests to the TADF nature of derivative 18.
In addition, PXZ-ICO was used as the host material to fabricate a red phosphorescent OLED display using the iridium complex (Ir(mphmq)2(tmd) (20) [60] as an emitter in the emissive layer. The efficiency was compared with that of the reference sample based on 4,4-bis(N-carbazol-9-yl)-1,10-biphenyl (CBP, 19) (Fig. 11). Both devices exhibited electroluminescence with a maximum at 605 nm, indicating complete energy transfer from the host to the emitter. The device based on isocoumarin 18 surpassed analogue 19 in quantum efficiency (EQE = 18.6 and 15.3%, respectively) and also showed an exceptionally low efficiency roll-off (only 4%) upon an increase in the luminance up to 1000 cd m–2.[59]
![[{"id":"u4R2HRHzlh","type":"paragraph","data":{"text":"Electroluminescence spectrum at a luminance of 1000 cd m<sup>–2</sup> for OLED devices based on compounds <b>18</b> (PXZ-ICO) and <b>19</b> (CBP).<sup>59</sup>"}}]](/storage/images/resized/1ZmqtxpEQSwq7XnbwWvrGXWDFeGp6rkKLPuJ556D_xl.webp)
Molecular design is the most popular approach to the goal of decreasing the energy difference between the S1 and T1 states of the molecule. The overlap between HOMO and LUMO can be minimized by separating the donor and acceptor parts of the molecule using twisting. In relation to nitrogen analogues of isocoumarins 21 and 22, Lee and co-workers [61] showed not only the necessity of a twisted structure for the TADF effect to be manifested, but also the importance of taking into account the influence of all possible conformations of the molecule on the photophysical properties, which may account for the TADF effect in some slightly twisted molecules.
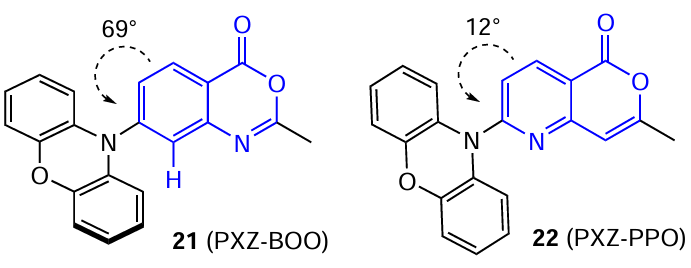
The authors compared the properties of two structurally similar emitter molecules, 21 (PXZ-BOO) and 22 (PXZ-PPO), containing phenoxazine moiety as the donor. The key difference between the structures is the position of the heterocyclic nitrogen atom. The nitrogen atom in the ortho-position relative to the electron-donating substituent in PXZ-PPO, which, unlike the C – H moiety, is unable to generate an effective steric hindrance to the intramolecular rotation, leads to lower twisting of molecule 22.
It was shown that planar conformation is most stable and most populated with electrons in the case of PXZ-PPO, whereas in the case of PXZ-BOO, it is the twisted conformation. However, the other metastable conformations also exist for both molecules. For compound 21, the planar conformer is not only less stable in the ground state, but it also has higher energy levels in the excited state than the highly twisted conformer, as evidenced by only one maximum present in the emission spectrum (Fig. 12a). This implies that this metastable conformation makes a negligibly low contribution to the photophysical properties of PXZ-BOO.
![[{"id":"RTYY67SzGL","type":"paragraph","data":{"text":"Absorption and emission spectra of compounds <b>21</b> (PXZ-BOO) and <b>22</b> (PXZ-PPO) in THF at room temperature (<i>a</i>) and energy levels of the excited states of PXZ-PPO found by DFT calculations (B3LYP/6-31G* basis set) (<i>b</i>).<sup>61</sup> Figure reproduced with permission from the Royal Society of Chemistry."}}]](/storage/images/resized/jNB0rpPbZCKzkBu9DWcEnZsGmBc4qwd9uwO8teyd_xl.webp)
Meanwhile, the emission spectrum of PXZ-PPO in solution exhibits two maxima (see Fig. 12a), one of which is absent from the emission spectrum in the crystalline state where the second long-wavelength maximum corresponds to the twisted metastable form. Despite the very low population of the twisted conformation in the ground state, this conformation is largely responsible for the photophysical properties of derivative 22, presumably, due to the fact that it has a lower excited state energy, which promotes its involvement into radiative processes during the energy transfer (see Fig. 12b).
It is noteworthy that OLEDs were fabricated and studied using compounds 21 and 22 as dopants (4.1 – 11.2 mass %) to 3,3'-di(9H-carbazol-9-yl)-1,1'-biphenyl (mCBP) in the emissive layer. Both devices showed maximum quantum efficiency values of 19.4 and 14.1% for PXZ-BOO and PXZ-PPO, respectively.[61]
An important issue is that excitation of the molecule can produce excitons in the locally excited (LE) and charge transfer (CT) states. In order to involve both types of excitons into radiative processes and thus to increase the efficiency of the device, Ma and co-workers [62] proposed for the first time a principle for the fabrication of photoemitting materials based on hybrid local charge transfer (HLCT). This approach is based on combining the two above types of excited states in one molecule. The HLCT materials harvest hot excitons by reverse intersystem crossing from the higher triplet state Tn (n > 1) to the S1 or S2 state; in this case, the exciton utilization efficiency (EUE) can theoretically reach 100%. The singlet locally excited state (1LE) in these molecules is characterized by a high degree of overlap of frontier orbitals, which results in a high luminescence efficiency. In addition, the charge transfer triplet state (3CT) provides RISC from Tn to S1 owing to the slight orbital overlap and, hence, small ΔEST value.
Li and co-workers [63] described materials based on isocoumarin as the acceptor unit in the HLCT molecule. To control the nature and the ratio of CT and LE contributions to the excited states, isocoumarins 23 and 24 with a methyl or phenyl group at position 3 (acceptor moiety) and various substituents at position 6 or 7 (donor moiety) were synthesized.
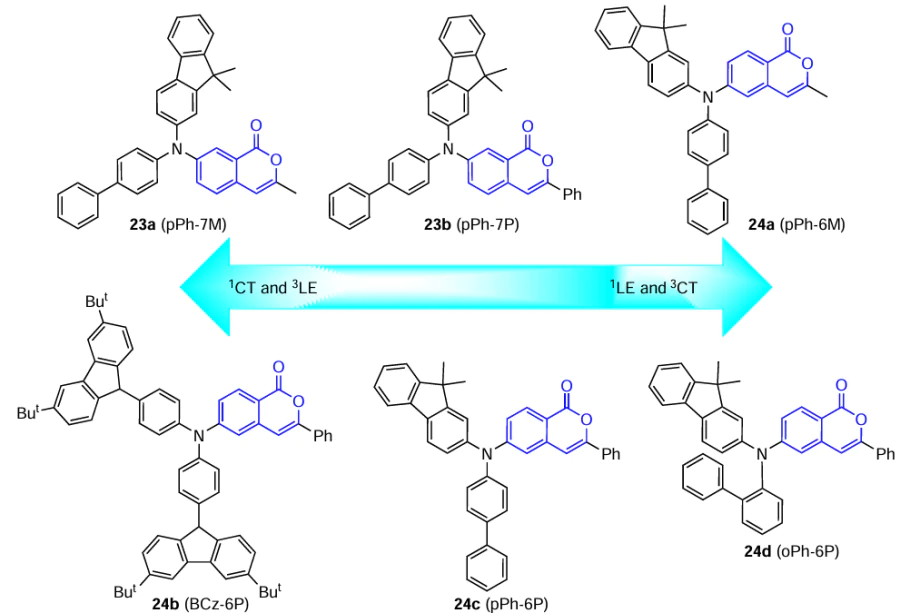
According to DFT calculations, the overlap of LUMO and HOMO in compound 24a (pPh-6M) is more pronounced than that in isomers with the same substituent at position 7. It is important that the energy differences between the S1 and T4 states (ΔES1T4) for compounds 23a,b (pPh-7P and pPh-7M) and between the S1 and T5 states (ΔES1T5) for pPh-6M are approximately 0.04 eV. This promotes harvesting of triplet excitons and their conversion to singlet excitons via reverse intersystem crossing from higher triplet states (hRISC).[63] Using natural transition orbital (NTO) analysis, it was shown that the S1 state in 24a has a hybrid structure combining the 1CT and 1LE characters, with the ratio of the 3CT and 3LE components for the T5 state being similar to that for the S1 state. It was also found that for isocoumarins pPh-7P and pPh-7M, the S1 excited state is mainly of the 1CT nature, while 3LE predominates in the T4 states.
Isocoumarins 23a,b and 24a were used as host materials doped with 10 mass % mCP (compounds 11) to fabricate films. These systems successfully suppressed the luminescence quenching upon aggregation of molecules in the film by restricting intermolecular interactions, which resulted in an increase in PLQY compared to that in undoped films. OLEDs based on these films showed EQEmax values exceeding the theoretical limit for conventional fluorescent materials (5%), which attests to effective utilization of hot excitons in radiative processes according to the HLCT mechanism.[63]
As a continuation of this study, Li and co-workers [64] compared the properties of compound 24c (oPh-6P) and two compounds 24b,d (BCz-6P and oPh-6P) containing different substituents at the nitrogen atom at position 6 of isocoumarin. According to DFT calculations, LUMO and HOMO in all three molecules markedly overlap, which leads to increasing contribution of the 1LE component to the S1 state, while the energy differences ΔES1T1 are 0.63, 0.70, and 0.78 eV for derivatives BCz-6P, pPh-6P, and oPh-6P, respectively. This restricts the RISC process from the T1 to S1 state and, hence, rules out the possibility of TADF mechanism in electroluminescence. In addition, the ΔES1T4 values of these compounds are 0.04, 0.14, and 0.14 eV, and the ΔES1T5 values are 0.12, 0.04, and 0.07 eV, respectively, which may be favourable for the hot exciton utilization mechanism. The contribution of the CT component in the S1 state is much higher for compound 24b than for pPh-6P or oPh-6P, mainly because of strong electron-donating properties of the carbazole ring. Meanwhile, the contribution of the LE component for the T4 and T5 states increases on going from BCz-6P to compounds with para- and ortho-biphenyl moieties at the nitrogen atom. Thus, the increase in the LE character for the first singlet excited state and the increase in the CT component for the high-lying T4 and T5 triplet states close in energy to S1 enable successful implementation of the HLCT mechanism in OLEDs based on these molecules.
2.3. Photoactive isocoumarins with extended π-system
An Indian research group headed by Gogoi [65] synthesized isocoumarins 25a – c annulated to a coumarin moiety to give an extended conjugated system. A study of the photoluminescent properties of these compounds showed that the unsubstituted molecules exhibit very low fluorescence, while the introduction of electron-donating substituents in position 6 of the isocoumarin ring and in position 7 of the coumarin moiety leads, as expected, to fluorescence enhancement due to appearance of intramolecular charge transfer (Fig. 13), according to assumptions depicted in Fig. 2 and Fig. 3.
![[{"id":"31lK5IlDDH","type":"paragraph","data":{"text":"Resonance structures of isocoumarins <b>25a – c</b> conjugated with the coumarin moiety and contribution of the structures to the intramolecular charge transfer."}}]](/storage/images/resized/YeNB4eGJuyB5KZG7BHILLFZ0bn2vEZ9VdnEMKjJP_xl.webp)
Unlike the compounds described above in which the intramolecular charge transfer takes place due to the presence of clear-cut donor and acceptor moieties in the molecules, an extended π-system is expected to favour that the π – π* type (LE) electron transition would be the major transition of the molecule in the excited state.
Tani and Ogawa [66] prepared polycyclic conjugated isocoumarins 26a – c.
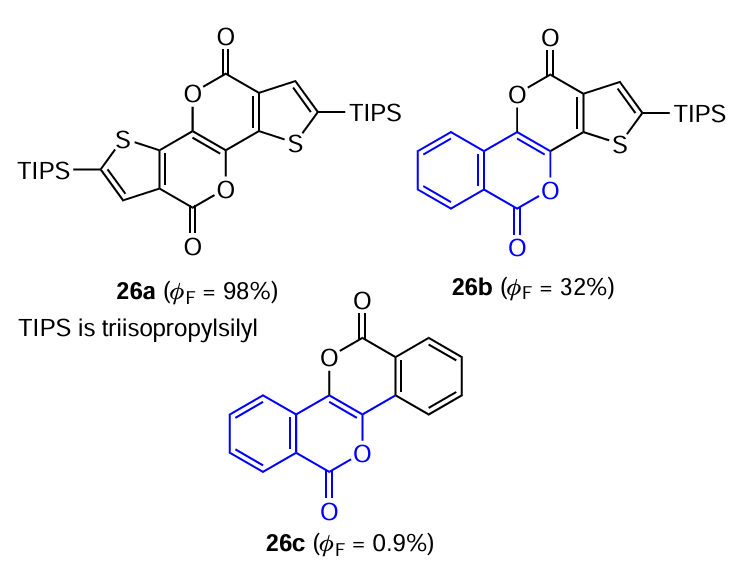
For all systems of this type, the absorption and fluorescence spectra are mirror images of each other, exhibiting vibrational fine structure and very small Stokes shifts, which is characteristic of molecules with π – π* type transitions (Fig. 14). These features may be attributable to the rigidity of the molecules and the structural similarity of their ground and first singlet excited states. The transition from benzo derivative 26c to thiophene derivatives 26a,b is accompanied by enhancement of the vibrational structure and decrease in the Stokes shifts in the spectra, being indicative of decreasing deformation of molecules in the S1 excited state. Thus, the luminescence quantum yields for solutions of these compounds in dichloromethane approach 100%.
![[{"id":"-xIAX14cmY","type":"paragraph","data":{"text":"Absorption (blue-coloured curves) and emission (red-coloured curves) of polycyclic conjugated isocoumarins <b>26a – c</b> (<i>a – c</i>, respectively).<sup>66</sup> Figure reproduced with permission from the American Chemical Society."}}]](/storage/images/resized/NlhnyArUb3AGCFKlOWivTemPsbUeGxdRhVUkNnVA_xl.webp)
Currently, the only example of the use of polyaromatic isocoumarin as a component of the OLED emissive layer is derivative 27, which contains a pyrene moiety in the molecule and represents a classical fluorescence emitter (Fig. 15). Using this compound, transparent films were manufactured, which were used as the emissive layer.[67] This OLED showed a luminance of 1740 cd m–2 at an applied voltage of 15 V.
![[{"id":"IQElMJx8NY","type":"paragraph","data":{"text":"Electroluminescence spectrum of 7,8-diphenyl-10<i>H</i>-phenaleno[1,9-gh]isocoumarin (<b>27</b>) (<i>a</i>) and current density <i>vs</i>. voltage (1) and luminance (2) curves for OLED with this compound as the emissive layer (<i>b</i>).<sup>67</sup> Figure reproduced with permission from Wiley."}}]](/storage/images/resized/UccuTr01sMCUmFpR8CCMhzTxSU3fAaksPef6r85X_xl.webp)
2.4. Molecular sensors based on isocoumarins
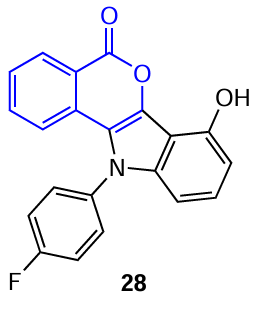
The first example of preparation of isocoumarin-based molecular sensor was reported in 2015 by Pramanik’s research group.[68] The authors proposed a new class of potentially biologically and fluorescence active isocoumarin systems (in particular, compound 28) annulated to 4-hydroxyindole, which were studied as sensors for the Cu2+ and Fe3+ ions.
The complex formation of molecular sensor 28 with the Cu2+ and Fe3+ ions was accompanied by pink colouring of the solution, which could be seen by a naked eye (Fig. 16a). The absorption spectra of solutions of this compound with increasing concentrations of copper ions recorded during titration also considerably changed, in particular, a long-wavelength absorption band appeared, probably corresponding to the metal-to-ligand charge transfer (MLCT) (see Fig. 16b). In addition, fluorometric titration in the presence of metal ions induces a noticeable fluorescence quenching for Fe3+ ions and especially Cu2+ ions (see Fig. 16c), which can also be observed visually in solutions of the samples under UV irradiation (see Fig. 16a).[68]
![[{"id":"crWRQYiMHK","type":"paragraph","data":{"text":"Change of the colour of solutions of compound <b>28</b> in the presence of metal ions under visible light (above) and after UV irradiation (below) (<i>a</i>), spectrophotometric titration of the solutions with Cu<sup>2+</sup> ions at different copper ion concentrations (<i>b</i>) and luminescence spectra of solutions of the sensor in the presence of various metal ions (<i>c</i>).<sup>68</sup> Figure reproduced with permission from the Royal Society of Chemistry."}}]](/storage/images/resized/qVGlpWLnrZiNdMpXQ1vKVfVIuMYnrN1qZ1sNBf85_xl.webp)
Isocoumarin-based chemosensor 29 (APICP) was reported by Yilmaz and co-workers.[69] In this case, fluorometric titration indicated the possibility of selective determination of Fe3+ and Hg2+ ions, the complexation of which leads to luminescence quenching (Fig. 17). The authors also successfully performed experiments on a human hepatocellular carcinoma cell line (HepG2) and concluded that this compound is applicable as an intracellular probe for fluorescent imaging to determine Fe3+ and Hg2+ ions in biological systems, with the limits of detection (LODs) being better than those of the probes developed earlier.
![[{"id":"srbunJ6rDx","type":"paragraph","data":{"text":"Complex formation reaction that underlies the operation of molecular sensor <b>29</b> designed for determination of Fe<sup>3+</sup> and Hg<sup>2+</sup> ions by intracellular imaging."}}]](/storage/images/resized/1HbdUESEZBVujh6OkztDlvOmqppg9EUvtUDD2UT5_xl.webp)
Korean research group headed by Son [70] prepared a functional MON material 30 (MON is a microporous organic network) based on isocoumarin.
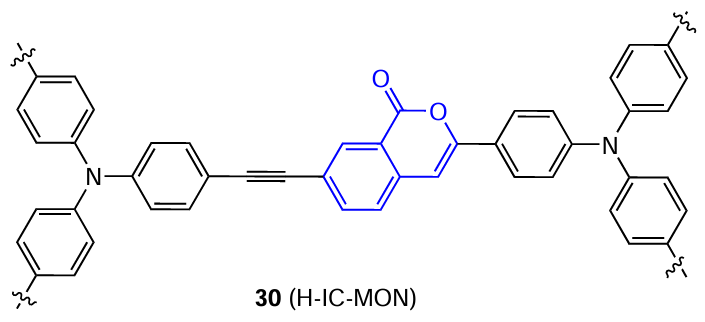
These compounds are synthesized by the Sonogashira reaction from arylalkynes and aryl halides. They are meant for detecting and scavenging environmental pollutants such as phenols and nitrophenols. The sensitivity of H-IC-MON to nitrophenols proved to be among the best up to the year 2016 compared to the results of determination of nitrophenols using other MON materials. Mention should be made of its high selectivity to nitrophenols in the presence of phenols, chlorophenol, and methylphenol. According to DFT calculations, this selectivity can be attributed to the fact that in this case, luminescence quenching occurs via electron transfer (ET) from LUMO of the sensor in the excited state to LUMO of the analyte, and this transfer is favourable only for nitrophenols.
3. Methods of synthesis
This Section gives a review of all currently available approaches (including unusual ones) to the creation of the isocoumarin skeleton, starting from early publications using multistep organic synthesis and ending with the most recent protocols, including metal-catalyzed single-step reactions, resulting in isocoumarins from reactants that are relatively easy to access. The Section includes two main parts, which describe methods of isocoumarin synthesis both catalyzed and not catalyzed by metal complexes.
3.1. Preparation of isocoumarins without the use of metal complex catalysis
The first synthetic routes to unsubstituted isocoumarin were proposed in the early and mid-20th century. In particular, Johnston’s research group [71] demonstrated the possibility of synthesis of isocoumarin from β-naphthol in five steps in an overall yield of 11.6% (Scheme 1).
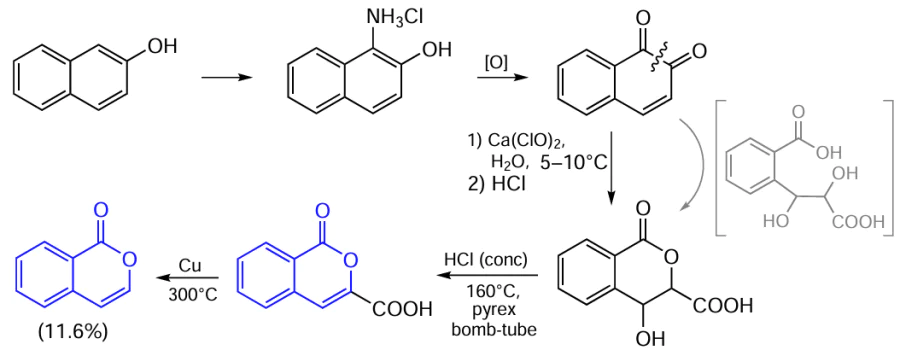
In this synthetic approach, the key step is the oxidation of 1,2-naphthoquinone with calcium hypochlorite followed by cyclization of the resulting intermediate (shown in brackets) to δ-lactone. The subsequent dehydration occurring on heating of the lactone with concentrated hydrochloric acid in a pyrex bomb-tube at 160°C and thermal decarboxylation of the resulting acid on treatment with a copper bronze powder at 300°C followed by instantaneous distillation under reduced pressure furnished the target isocoumarin.
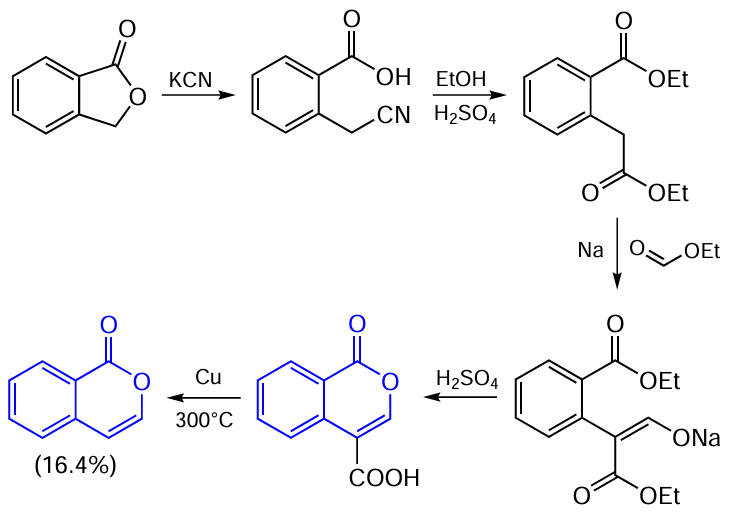
In another method for isocoumarin synthesis proposed in the same study,[71] phthalide, which can be prepared by the reduction of phthalic anhydride, was used as the starting compound. The overall yield of the product in this sequence of reactions was 16.4%, which is slightly higher than the yield in the previous method (Scheme 2).
A pronounced increase in the yield of unsubstituted isocoumarin was achieved by using the ozonolysis reaction. This approach allowed Warnell and Shriner [72] to synthesize isocoumarin in an overall yield of 63% in only two steps from indene (Scheme 3). The ozonolysis of indene in ethanol gave intermediate cyclic peroxy ether, which proved to be sufficiently stable to be isolated and, according to the authors, it did not decompose on exposure to various thermal and physical stimuli. Subsequently, this cyclic peroxy ether is cleaved on treatment with alkali to give aldehyde, which undergoes the acid-catalyzed cyclization, resulting in the expected unsubstituted isocoumarin.
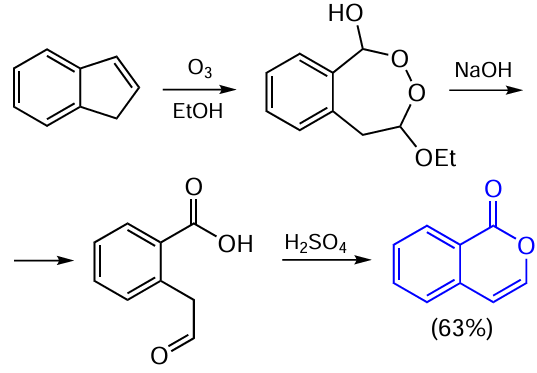
When starting indenes contain substituents, this ozonolysis-based protocol makes it possible to obtain isocoumarins containing various substituents in the aromatic ring. In addition, the use in the last step of reducing agents such as sodium borohydride or organoaluminium compounds, instead of acids, results in 3,4-dihydroisocoumarin, a highly practically important isocoumarin derivative.
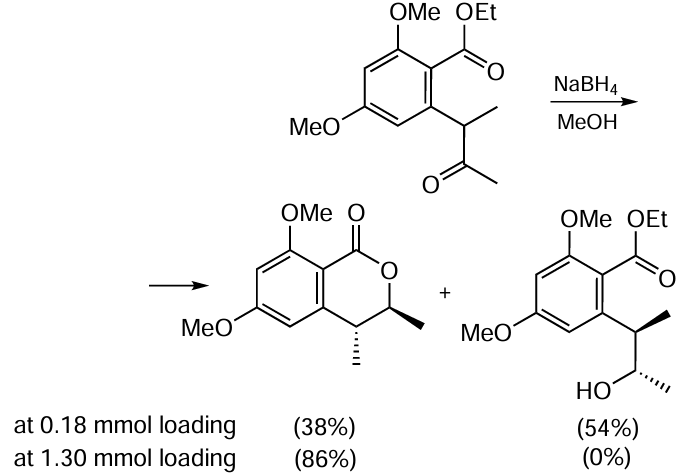
Staunton and co-workers [73] used the reduction of o-ethoxycarbonylbenzyl ketone with sodium borohydride to prepare a mixture of dihydroisocoumarin and hydroxy ester (Scheme 4). The latter product can be converted to the target lactone by treating the reaction mixture with sodium hydride. It is worth noting that dihydroisocoumarin is formed predominantly as the threo-isomer. If the amount of the starting benzoate is increased (from 0.18 to 1.30 mmol), the hydroxy ester is not formed and dihydroisocoumarin is produced as a mixture of threo- and cis-isomers in a ratio of 43 : 1.
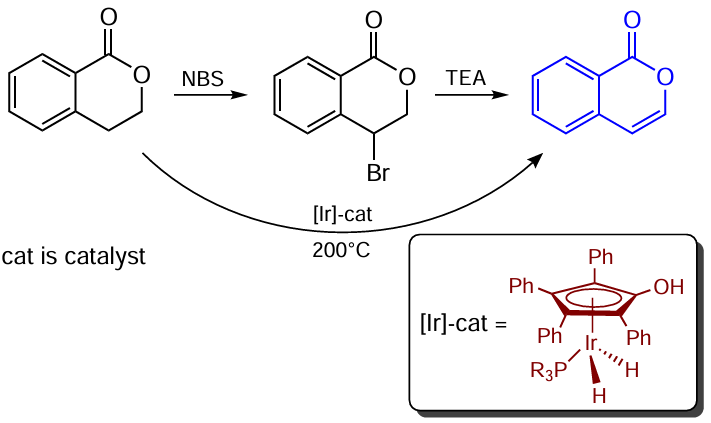
It is noteworthy that 3,4-dihydroisocoumarin can be converted to isocoumarin by bromination with N-bromosuccinimide (NBS) followed by dehydrohalogenation of the resulting bromo derivative in the presence of triethylamine (TEA) or pyridine [74][75] or by dehydrogenation catalyzed by hydroxycyclopentadienyl iridium complexes (Scheme 5).[76]
The principal ‘organic’ approach to the synthesis of substituted isocoumarins is based on various cyclizations of ortho-carboxybenzyl ketones and related compounds. The cyclization at elevated temperature can be effected by introducing good leaving groups as carboxy substituents.
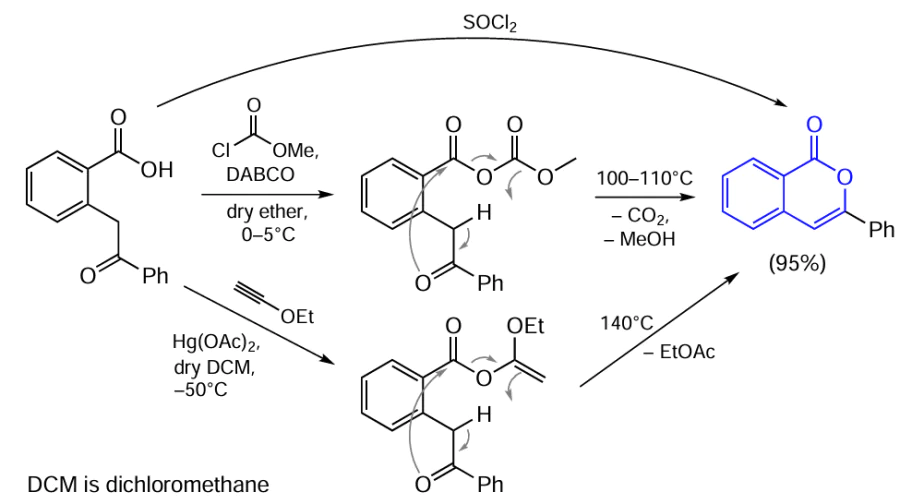
Treatment of ortho-carboxybenzyl ketone with methyl chlorocarbonate or its sodium salt in the presence of 1,4-diazabicyclo[2.2.2]octane (DABCO) resulted in the synthesis of acyclic mixed anhydride in a high yield (Scheme 6).[77] This anhydride is stable at temperatures of 70 – 80°C (both neat and in inert solvents), but on heating to 100 – 110°C, further transformations take place to give 3-phenylisocoumarin (95% yield). Another example of an applicable leaving group is the ethoxyvinyl group, which is generated by using ethoxyacetylene. Heating of the resulting compound to 140°C is accompanied by release of ethyl acetate and formation of isocoumarin. 3-Phenylisocoumarin can also be obtained directly by treating the starting ortho-carboxybenzyl ketone with thionyl chloride via the formation of reactive carboxylic acid chloride.
A simpler method for the preparation of isocoumarins from ortho-carboxybenzyl ketones and their esters is cyclization of appropriate substrates in the presence of acids under short-wavelength UV irradiation (UVC) in the 100 – 280 nm wavelength range. This approach can also be used to prepare annulated isocoumarins. In this case, acid-promoted cyclization of enolate takes place in the second step Scheme 7, reaction (1)].[78]
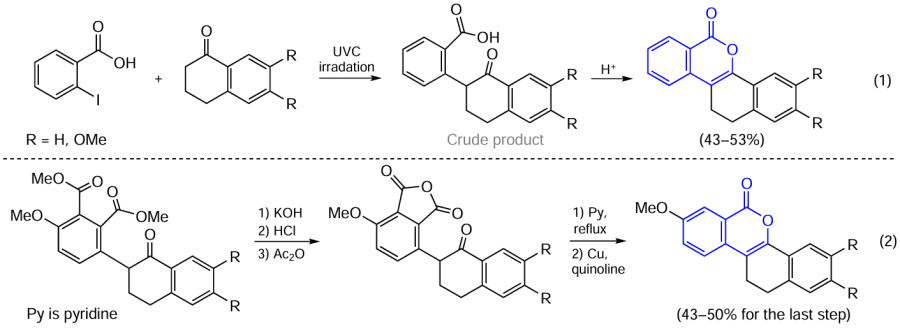
A similar method for the synthesis of such compounds is based on the intermediate formation of anhydrides, which are converted to isocoumarins via cyclization on refluxing in pyridine followed by decarboxylation [see Scheme 7, reaction (2)].[79]
8-Hydroxy-7-methoxycarbonyl-3,6-dimethylisocoumarin can also be formed from non-aromatic precursors. For example, in the first step of the synthesis of fredericamycin A (an antibiotic), Rao et al.[80] successfully formed the isocoumarin core by condensation of 1,3-diacetylacetone with acetone-1,3-dicarboxylate in the presence of alkali at room temperature (rt) (
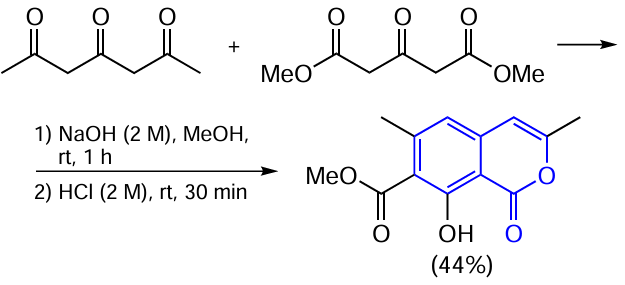
It should be noted that all of the cyclizations described above require the presence of a carbonyl group in the ortho-substituent of the benzoic acid derivative one carbon away from the aromatic ring. In some examples (see Scheme 1 , Scheme 2 and Scheme 3 ), this 2-oxoalkyl substituent is formed from some heterocyclic precursor via ring opening. In other cases, it is necessary to initially introduce 2-oxopropyl moiety into the substrate molecule.
Hegedus and co-workers [81] proposed a method for the synthesis of isocoumarins and dihydroisocoumarins based on the use of equimolar amounts of π-allyl nickel complexes to introduce an acetonyl substituent in the ortho-position of aromatic carboxylic acid esters and salts (Scheme 9).
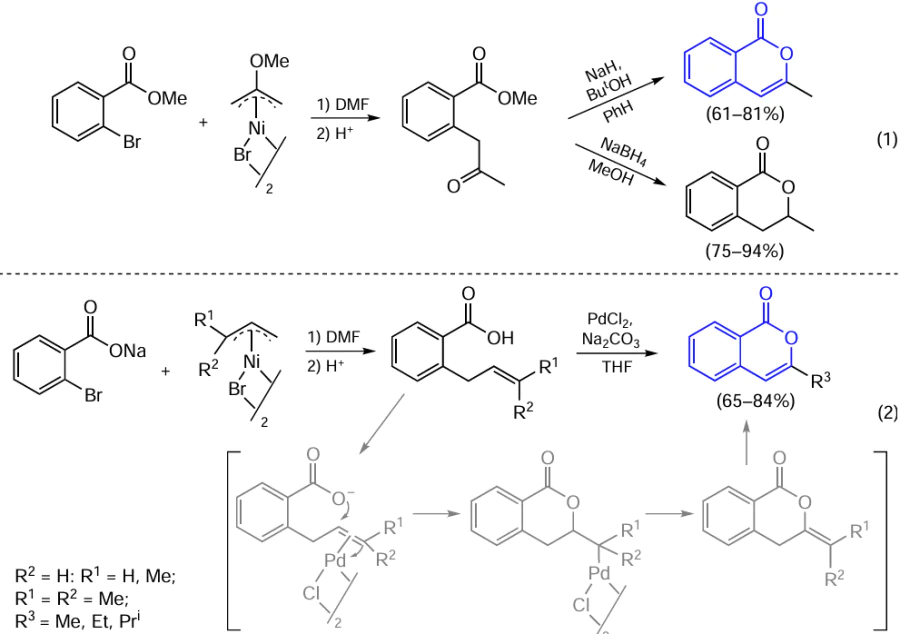
In the first step, aryl bromide reacts with 2-methoxyallyl nickel complex to form 2-methoxyallylbenzoate, which undergoes acid hydrolysis to give the corresponding ketone. Subsequently, this ketone is converted to isocoumarin on treatment with sodium hydride or to dihydroisocoumarin on treatment with a reducing agent [see Scheme 9, reaction (1)]. The reaction of sodium 2-bromobenzoate with 1-substituted π-allyl nickel complex affords acids containing a double bond and capable of forming olefin complexes with palladium chloride [see Scheme 9, reaction (2)]. The olefin coordination to the metal promotes nucleophilic cyclization. β-Elimination and subsequent isomerization afford the desired 3-methyl-substituted isocoumarins.
Walther’s research group [82] proposed a reverse approach based on the reaction of α-bromoketones with a stoichiometric amount of nickelacyclic carboxylate complex. In this case, the nickel complexes act as sources of the carboxylic acid moiety rather than of an allyl or 2-oxoalkyl moiety (Scheme 10).
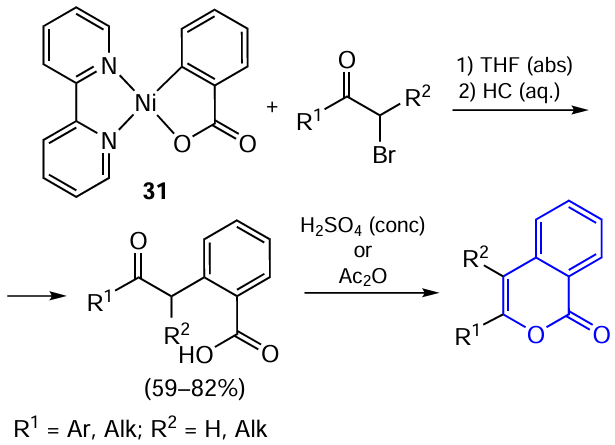
The staring nickel complex 31 was prepared by the oxidative addition/decarbonylation of phthalic anhydride in the presence of nickel(0) diene complex [(bipy)Ni0(cod)] (bipy is bipyridine, cod is cycloocta-1,5-diene). The subsequent reaction of complex 31 with α-bromoketones proceeds rather slowly and requires long-term stirring (from 24 to 96 h) of the reactants at room temperature. In some cases, a similar 2,9-dimethylphenanthroline nickel complex was used instead of the bipyridine complex to accelerate the reaction. α-Chloroketones did not react under ambient conditions; however, if 2,9-dimethylphenanthroline was used as an auxiliary ligand for the initial complex, the transformation did take place.
In one experiment, the authors were able to isolate crystalline nickel-containing intermediate 32 as a THF solvate ([(bipy)Ni+2(carboxylate)Br(thf)]). On the basis of X-ray diffraction data, assumptions about the reaction mechanism were made. As expected, the action of α-haloketone on the Ni – C bond induces bond cleavage to give a new nickel carboxylate complex with bidentate carboxylate group. The Ni – Br bond, the chelating bipyridine ligand, and additionally coordinated solvent molecule form the octahedral environment around the nickel centre.
In addition, the ortho-2-oxoalkyl derivatives of aromatic acids can be obtained by oxidation of the corresponding ortho-alkynyl derivatives catalyzed by platinum(ii) oxide.[83] The subsequent acid-induced cyclization of the intermediate affords 3-substituted isocoumarin.
The ortho-alkylation/cyclization of halogenated aromatic acids can also involve β-diketones. Liang and co-workers [84] proposed an approach to the synthesis of polysubstituted isocoumarins from ortho-iodo-substituted benzoic acids and β-diketones under the action of bases (Scheme 11). The applicable bases include Cs2CO3, K2CO3, Na2CO3, NaH, NaOH, etc. This reaction takes place in the absence of copper(I) salts, which are commonly used as catalysts in this type of reactions (see below, Scheme 25, Scheme 26 and Scheme 27).
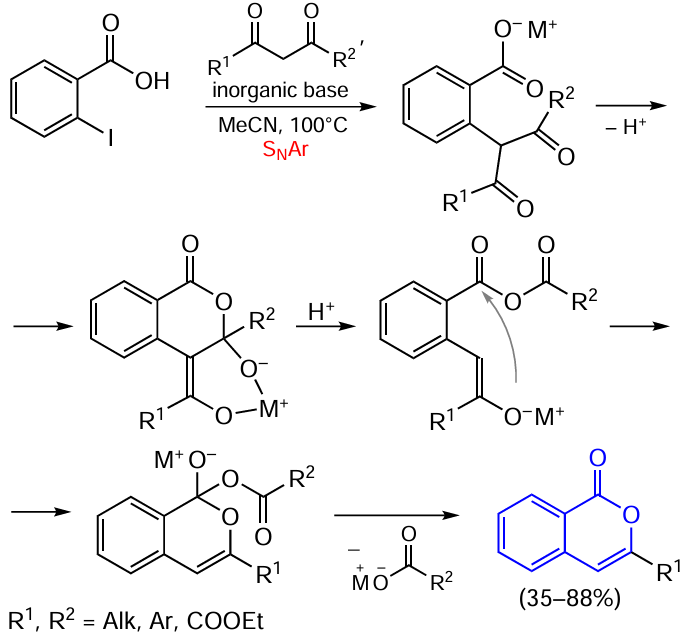
In the case of symmetrical β-diketones, this protocol allows the selective synthesis of the corresponding substituted isocoumarins in moderate to good yields. However, when unsymmetrical phenyl methyl β-diketone was taken, the authors isolated a mixture of 3-phenyl- and 3-methylisocoumarins in 14 : 3 ratio. The reactions of cyclic diketones and ketoester yield 3,4-substituted isocoumarins, since they are not accompanied by C – C bond cleavage.
As regards other approaches to the synthesis of isocoumarins, there is a method based on modification of the carboxy group to give an ester with a C – H bond located near the oxygen atom that is quite easily deprotonated.[85-87] This is followed by base-induced nucleophilic attack of the enol oxygen atom on the aldehyde or keto group directly bound to the aromatic ring at the ortho-position.
For example, alkali metal salts of 2-formylbenzoic acid and its derivatives are capable of condensation reactions with halogen-containing methyl ketones to give esters, which are converted to 3-acyl-substituted isocoumarins on treatment with organic bases (Scheme 12).[85]
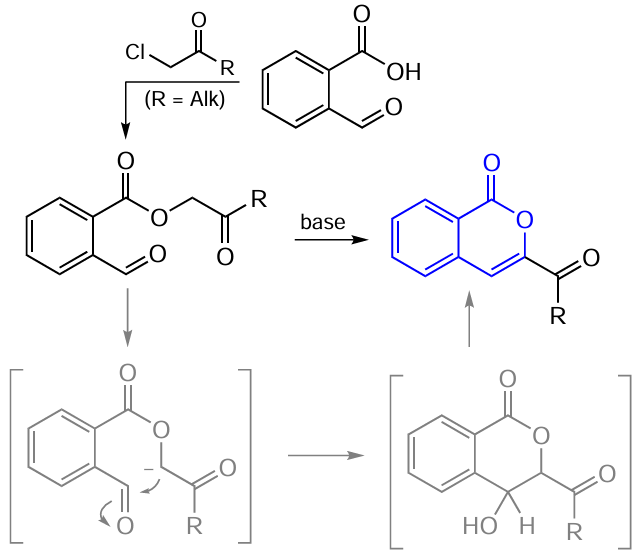
A somewhat different approach to the preparation of 3-substituted isocoumarins is the cyclization of ortho-alkynylbenzoic acids. Generally, ortho-alkynylbenzoic acids and related compounds are the major precursors for the formation of the isocoumarin skeleton without catalysis by metal complexes as well as for some metal-catalyzed transformations. Nevertheless, it should be borne in mind that the main method for the synthesis of these alkynyl derivatives from the corresponding halides is the palladium-catalyzed Sonogashira reaction.
The method based on the preparation of ortho-alkynylbenzoic acid derivatives was first reported in 1963 by Stephens and Castro.[88] The authors stated that 3-phenylisocoumarin can be obtained from o-iodobenzoic acid and copper(I) phenylacetylide (Scheme 13).
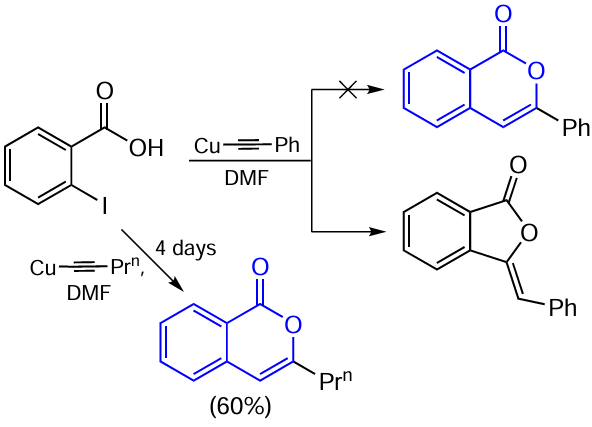
However, a few years later, the authors rejected their previous finding and reported that they had been wrong in determining the structure of the reaction product and that actually it was 3-benzylidenephthalide.[89] Moreover, this reaction can be used as a synthetic route particularly to phthalides. Only in the case of copper n-propylacetylide, the reaction gave 3-n-propylisocoumarin as a by-product. It should be noted that Batu and Stevenson [90] carried out a more detailed investigation of this reaction and found that increasing reaction time leads to a gradual increase in the product ratio in favour of 3-n-propylisocoumarin. After four days, the signal of phthalide was no longer observed in the NMR spectrum, and the target isocoumarin was isolated in a pure state in 60% yield.
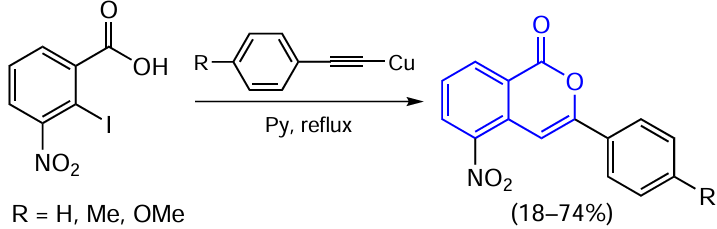
Forty years later, Threadgill and co-workers [91] reported a study in which they performed a similar reaction by the Castro – Stephens method and thus obtained 3-aryl-5-nitroisocoumarins (Scheme 14). The change in the cyclization pathway was attributed to the fact that the nitro group causes polarization of the intermediately formed alkyne, and the sp carbon atom remote from the aromatic ring becomes more electrophilic.
Electrophilic cyclization is the most popular method for the preparation of isocoumarin derivatives from ortho-alkynyl-substituted benzoic acids. For example, this approach was used by Rossi et al.,[92] who utilized molecular iodine as a source of electrophilic agent. In the first step, the triple bond is activated due to coordination of electrophilic I+ ion generated from molecular iodine. This is followed by the nucleophilic attack of the carboxyl oxygen atom on the triple bond carbon atom remote from the aromatic ring. After this, nucleophilic substitution reaction takes place according to the SN2 or SN1 mechanism with participation of the I– anion and elimination of methyl iodide (Scheme 15). It is noteworthy that the selectivities of reactions according to this approach are markedly different for the internal and terminal triple bonds.
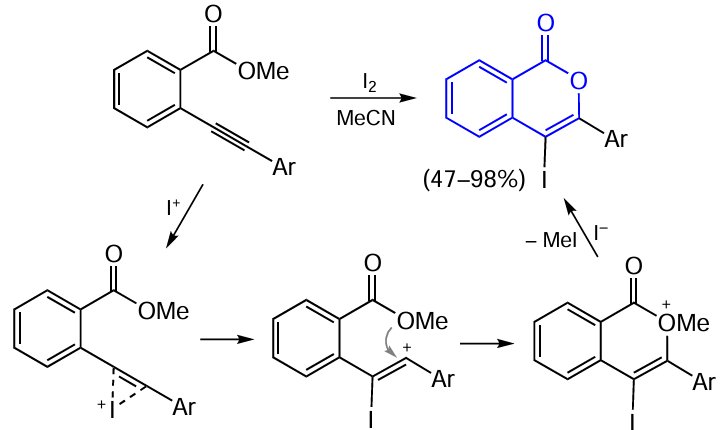
In the case of alkynylarene substrate with a terminal triple bond, it is assumed that the cyclization proceeds selectively via the formation of the intermediate benzyl cation in which the positive charge is better stabilized by the aromatic ring, despite the presence of an electron-withdrawing group in the ortho-position. This gives rise to a mixture of Z- and E-isomers of 3-iodomethylidenephthalide as the only product (Scheme 16).
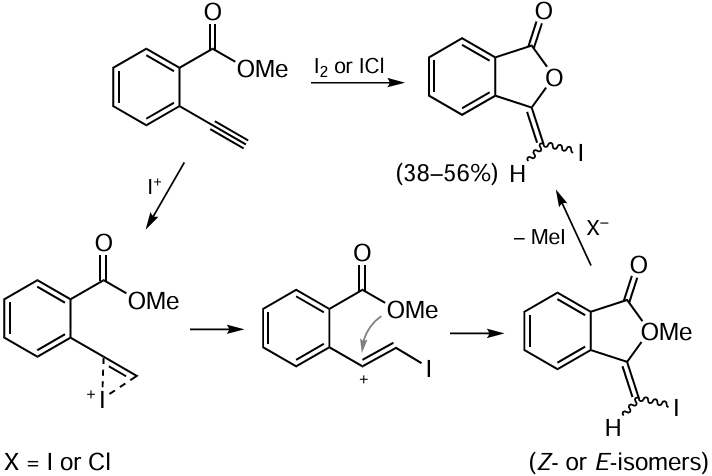
The iodine atom at position 4 of isocoumarin can be removed by palladium-catalyzed reduction with triethylammonium formate [92][93] or replaced via cross-coupling reaction.[92][94]
Apart from molecular iodine [92][95][96] and iodine monochloride,[92] other reagents or reagent mixtures can also serve as sources of electrophilic species, for example, Br2 – LiBr,[97] 4-O2NC6H4SCl,[98] PhSeCl,[98] HI,[98] HgSO4,[99] TMSCl – NCS (TMS is trimethylsilyl, NCS is N-chlorosuccinimide),[100] and B-chlorocatecholborane.[101] Other applicable electrophiles are hypervalent iodine compound — PhICl2,[102] or PhSCl and PhSeCl, which are formed in situ on treatment of disulfide RSSR or deselenide RSeSeR with oxidants PhICl2 or FeCl3, respectively,[103-105] and MeSCl, generated from dimethyl sulfoxide and SOCl2.[106] Oxone (K+[HSO5]–) is used as a green oxidant, which induces chalcogen–chalcogen bond cleavage in diselenides and ditellurides to give electrophilic species in situ under ultrasonic treatment.[107] o-Stilbenecarboxylic acids can also serve as substrates for electrophilic cyclization instead of o-alkynyl derivatives.[108]
The cyclization of o-alkynylbenzoates is promoted not only by electrophiles, but also by acids, which thus results in the synthesis of 3-substituted isocoumarins. Uchiyama’s research group [109] performed a detailed study of this reaction and found that isocoumarin formation upon 6-endo-cyclization occurs only in the presence of strong acids such as concentrated sulfuric acid, trifluoroacetic acid (TFA), or trifluoromethanesulfonic acid (TfOH). Conversely, under the action of weak bases, 5-exo-cyclization of substrates takes place to give phthalides (Scheme 17). Cyclizations of both types take place on refluxing the reactants in toluene in the presence of a catalytic amount of a strong acid or a weak base and give products with high selectivity and in high yields. Meanwhile, refluxing in toluene in a neutral medium or in a weak acid such as acetic acid or a strong inorganic base such as sodium hydride NaH does not induce the reaction by either of the above pathways.
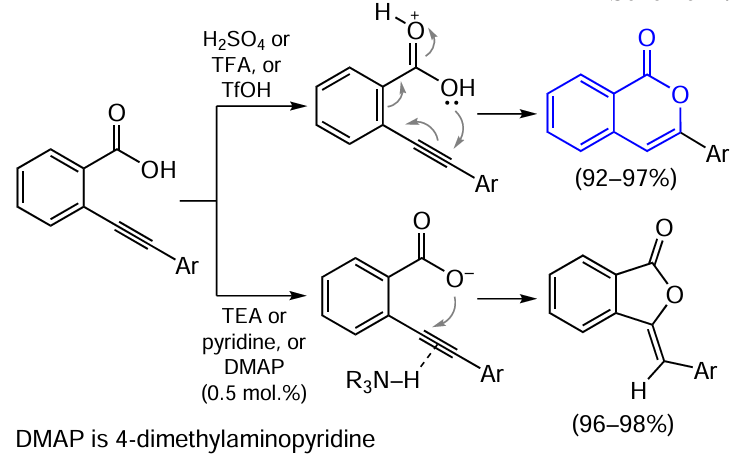
The authors explained this selectivity by the fact that the carbonyl group is protonated in the presence of strong acid catalysts, as should be expected in view of the basicity of the carbonyl oxygen atom in the carboxy group. Thus, the electron density shift on both carbon atoms of the triple bond promotes the Michael condensation (6-endo-type). In the presence of basic catalysts, the carboxylate anion can be obtained by deprotonation of the carboxylic acid, which favours 5-exo-cyclization of the intermediate.
By modification of this procedure, Alami and co-workers [110] were able to markedly reduce the time of the reaction. The authors used microwave irradiation and 20 mol.% p-toluenesulfonic acid (p-TsOH) as a catalyst; this decreased the reaction time from 24 h to 30 min. It is worth noting that not only aromatic acids, but also their esters, amides, and cyanides were used as the starting alkynyl compounds.
Aromatic aldehydes of this type can be involved in cyclization in the presence of acidic oxidants, e.g., the Jones reagent (a solution of CrO3 in aqueous H2SO4).[111] In order to avoid side cyclization reactions to give naphthol derivatives, 3 equiv. of the oxidant are used (Scheme 18).
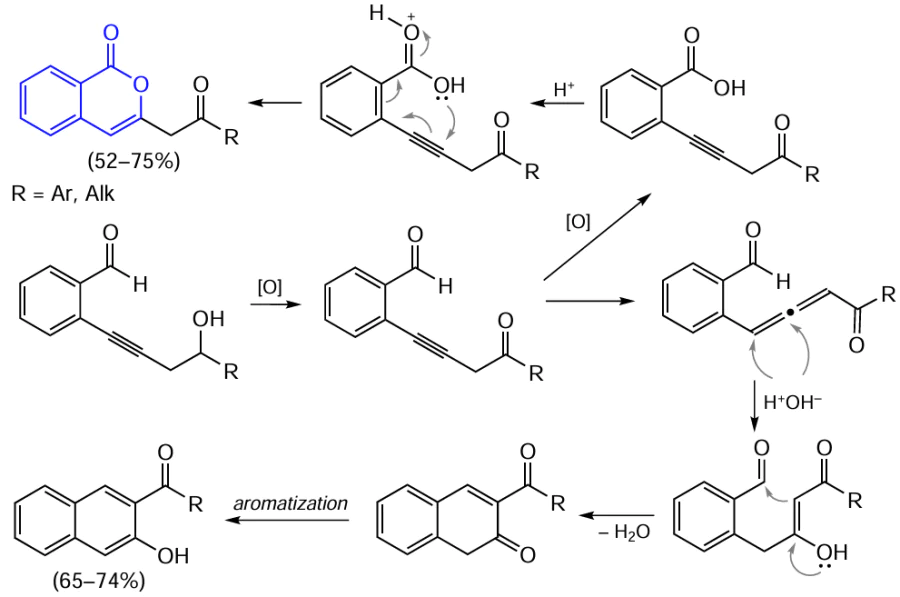
Note that in an earlier study, Tovar and Swager,[112] who performed acid-promoted cyclization of alkynyl-substituted benzoic acid esters, benzamides, benzaldehydes, and benzophenone derivatives, proposed a different cyclization mechanism resembling the mechanism of electrophilic cyclization (see Scheme 15 ), which starts with the triple bond activation. They assumed that the reaction proceeds via the formation of pyrylium salts, some of them were isolated and characterized by spectroscopic methods. The subsequent reaction in the presence of nitrogen bases gives substituted isoquinolines as the products, whereas hydrolysis of these pyrylium salts affords the corresponding isocoumarin derivatives (Scheme 19).
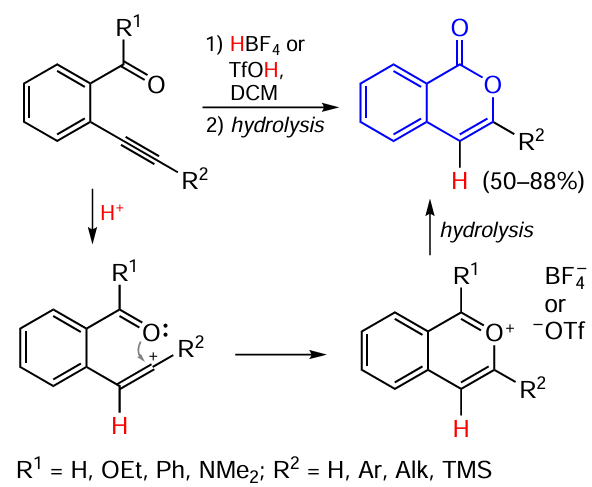
In this case, trifluoroacetic acid causes only partial cyclization and the reaction proceeds slowly. Meanwhile, the use of stronger acids, such as HBF4 and TfOH, provides instantaneous and virtually quantitative cyclization. In the absence of water traces to avoid further hydrolysis and at a relatively high dilution (0.005 M), which rules out significant intermolecular interaction, it is possible to observe cationic products, which either precipitate in a pure state or are detected by NMR in situ.
Mention should also be made of the cyclization of ortho-alkynyl-substituted benzoates performed by Lisowski and co-workers.[113] A specific feature of this study is that the authors synthesized isocoumarins by a two-step procedure consisting of the Sonogashira coupling of polymer-bound 2-bromobenzoate with terminal alkyne and subsequent cyclization of the intermediate o-alkynylbenzoate under the action of either electrophiles (E) or acid catalysts. The final step of the process is depicted in Scheme 20 (the black sphere implies the polymer support).
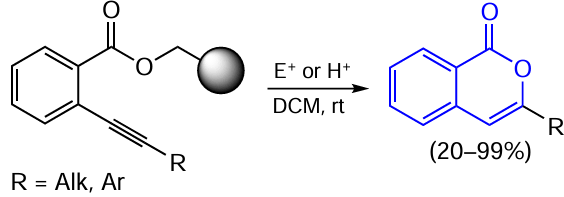
In this case, the polymer matrix performs several functions. The main advantages of using this matrix include protection of the carboxylic moiety and the ease of separation of the Sonogashira reaction by-products, diyne derivatives, which remain in solution and do not bind to the solid support in the first step. Therefore, it is easy to obtain only the desired cyclization product in the second step.
The reaction conducted in the presence of electrophilic reagents (I2 and ICl) resulted in the highly selective formation of 3-aryl- or alkyl-substituted 4-haloisocoumarins. In the case of R = Ar, no five-membered cyclic products, phthalides, were detected. The acid-catalyzed cyclization showed a lower efficiency of this approach; however, it allows selective formation of only 3-substituted isocoumarins without the halogen atom in position 4. Meanwhile, electrophilic cyclization using copper(II) halides mixed with Cy2NH · HX (Cy is cyclohexyl) gave at best a product mixture in 2.6 : 1 ratio (with a 4-halogen derivative impurity).
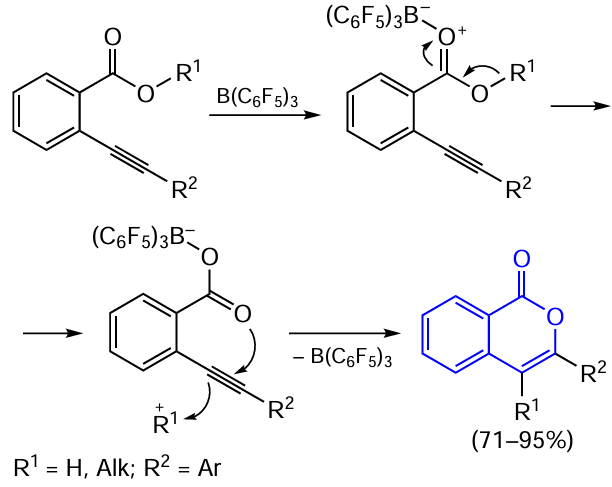
It should be noted that Lewis acids can be used to promote the cyclization instead of Brønsted acids.[114][115]An interesting example of cyclization of both o-alkynylarylcarboxylic acids and o-alkynylarylcarboxylates in the presence of Lewis acids was reported by Melen and co-workers [115] in 2017. The authors developed a new method for the synthesis of 3-substituted (R1 = H) and 3,4-disubstituted isocoumarins in high yields on treatment with B(C6F5)3 as a sterically crowded strong Lewis acid (Scheme 21). In this case, the ester group migrating to the triple bond of the alkynyl moiety apparently acts as the electrophile in the cyclization.
An attempt to use ortho-alkynyl-substituted benzaldehydes for the synthesis of isocoumarins was made by Youn and co-workers.[116] As a reagent promoting the cyclization, the authors used N-heterocyclic carbene (NHC) (10 – 20 mol.%) in combination with a base, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), taken in a 20 – 40 mol.% amount (Scheme 22).
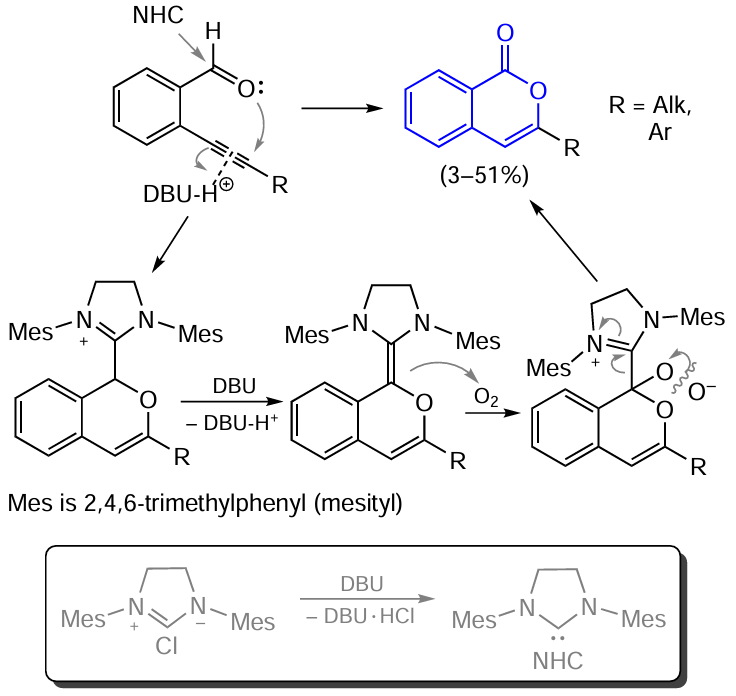
According to a mechanism proposed by the authors, deprotonation of NHC · HCl induced by DBU takes place in the first step. Then the released carbene attacks the carbonyl group of the aldehyde, while the protonated base acts simultaneously as an electrophile and transfers a proton to the triple bond, which results in the cyclization. The cationic intermediate thus formed loses a proton under the action of a base; this is followed by the addition of an oxygen molecule to the carbon atom bound to the heterocyclic carbene. The subsequent elimination of oxygen and release of NHC furnish the target product.
It should be noted that this reaction is not distinguished by high selectivity and, in most cases (except for R = Bun), isocoumarin is a minor product. The major product of this reaction is phthalide, which results from the nucleophilic attack on the triple bond carbon atom located more closely to the aromatic ring.
Another approach to the fabrication of the isocoumarin skeleton, differing from the above one, is based on the in situ generation of an aryne species.[117][118] This is done using o-(trimethylsilyl)phenyl triflate as the precursor and CsF as the source of fluoride ion, acceptor of the trimethylsilyl group. This method of benzyne generation is characterized by high rate even at room temperature and does not require strong bases. The synthesis of isocoumarin includes the insertion of aryne C≡C bond into the trifluoromethylated β-diketone, which is followed by cyclization (Scheme 23). The CF3 group acts as not only an acceptor, but also as a good leaving group, giving a fluoroform molecule in the presence of fluoride ions.
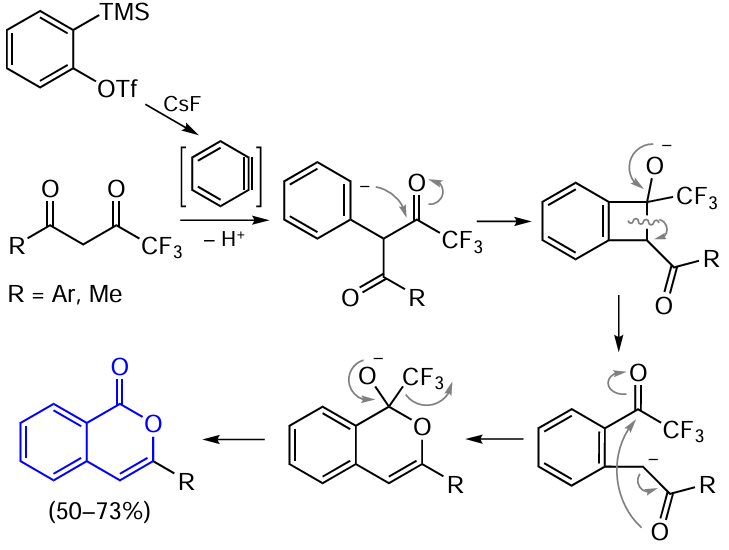
Instead of β-diketones, 4-hydroxycoumarin can act as the partner for the cyclization involving arynes;[119][120] the pyran ring in 4-hydroxycoumarin is opened on treatment with two benzyne molecules (Scheme 24).[118]
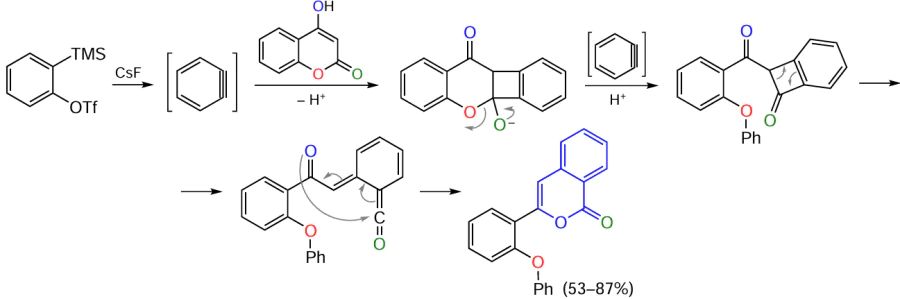
Thus, it can be concluded that the electrophilic cyclization of o-alkynylbenzoic acids and their derivatives and acid-catalyzed versions of the cyclization are today the key synthetic routes to isocoumarin derivatives free of metal complex catalysts. However, a number of drawbacks of this approach should be noted.
First, although the authors often emphasize the fact that their procedures do not involve organometallic catalysts for cyclization, the starting alkynyl compounds are poorly accessible and are commonly synthesized by the Pd-catalyzed Sonogashira reaction from ortho-iodo or ortho-bromo derivatives. Furthermore, the synthesis of ortho-halo derivatives most often requires conduction of additional steps. Nevertheless, this approach is used more often than the synthesis from ortho-2-oxoalkyl derivatives, since these reactants are even less accessible.
Second, an important issue is that the cyclization of alkynyl derivatives leads, in most cases, to 3-substituted isocoumarins. Meanwhile, the synthesis of 3,4-disubstituted products requires, except for a few examples (see Scheme 21),[115] an additional step [e.g., cross-coupling of isocoumarins that have a iodine or bromine atom or the B(OR)2 group in position 4 [101]].
The metal-catalyzed approaches to isocoumarin synthesis are considered below. It is shown that some of the above ‘organic’ reactions can also be implemented in the presence of metal salts or complexes as catalysts, which makes it possible to use milder conditions or to reduce the number of steps.
3.2. Metal complex catalysis in the synthesis of isocoumarins
The information on the metal-catalyzed approaches to the synthesis of isocoumarins is classified according to the nature of the metal and arranged starting from the late transition metals (Group 11) and ending with the middle transition series metals (Group 8). This is due not only to the relative chronological order of the development of addressed methods that employ particular metals. First of all, one should bear in mind that the late transition metals have a filled d-shell, which restricts their coordination capacity and often allows them to act only as Lewis acids. Meanwhile, on going from right to left along the row, the number of d-electrons in the metal atom decreases, which makes the catalytic processes involving these metals more diverse.
3.2.1. Catalysis by copper compounds
Non-catalyzed ortho-alkylation — cyclization reactions of ortho-halogen-substituted aromatic acids and β-diketones have already been described above (see Scheme 11). These reactions can also be promoted by copper(I) salts (Scheme 25). Despite the fact that the procedure of copper-catalyzed arylation of activated methylene compounds has been fairly well-developed,[121-124] there are only few examples of using this reaction for the synthesis of ortho-2-oxopropyl aromatic acid derivatives. For example, McKillop and co-workers,[125][126] who studied the alkylation of o-bromobenzoic acid, first mentioned the formation of isocoumarins in situ.
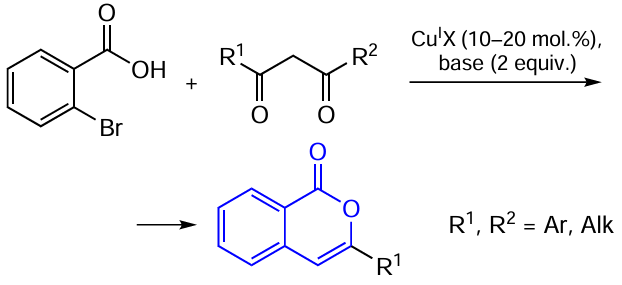
Subsequently, it was found that this α-arylation reaction of β-diketones proceeds for aromatic acids containing Cl, Br, or I atoms in the ortho-position.[127][128] According to control experiments, the formation of isocoumarins requires elevated temperatures and the cyclization step does not occur in the absence of copper(I) salts; in this case, the reaction stops after the formation of the ortho-alkylation product.
Subsequently, the use of copper-catalyzed cyclization of this type for the design of the isocoumarin core was extended to aromatic acid esters [129] or even amides,[130][131]which were initially used to prepare isoquinolone derivatives.[123] Moreover, in some cases, the acid is specially first converted to the amide and only after that, cyclization is carried out, with N-phenylacetamide functioning as the leaving group.[56] An approach using copper nanoparticles as a catalyst has also been developed.[132]
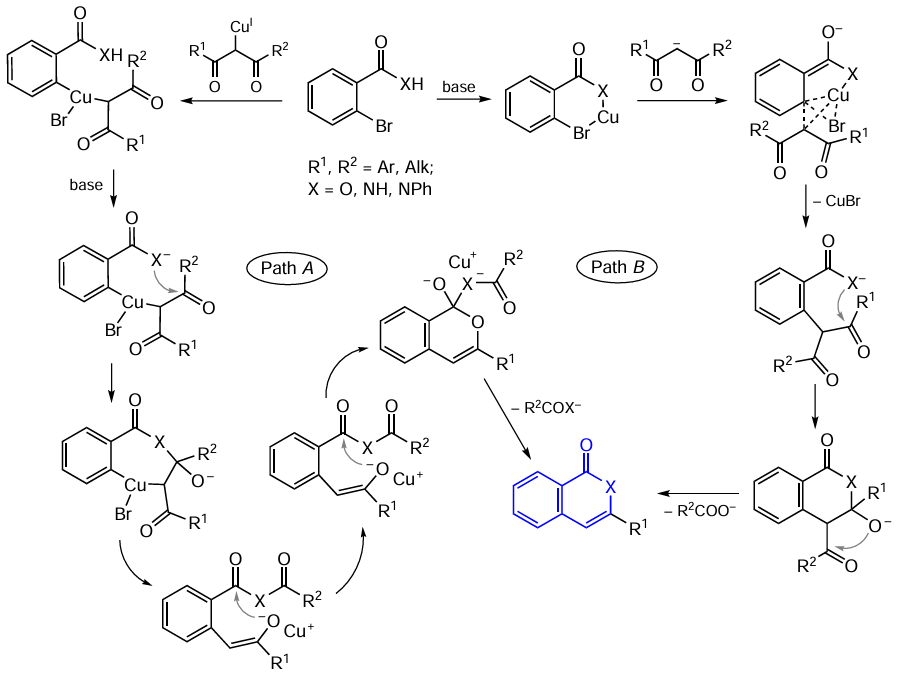
In view of the facts that the cross-coupling involving copper(I)-based catalytic systems has been less studied than the corresponding palladium-catalyzed reactions and several different reaction pathways have been established to date, the mechanisms of isocoumarin formation and, in particular, the mechanisms of cyclization to give the C – O bond, may be different. Indeed, a few possible reaction pathways have been postulated,[56] [123] [129-132] and they can be reduced to two major paths (Scheme 26). One of them (path A) is based on the sequence including oxidative addition/reductive elimination of organocopper intermediates (Ullman reaction type) followed by cyclization upon the attack of the enolate ion. The second one (path B) implies the formation of copper(I) benzoate under the action of a base and the subsequent copper assistance in the nucleophilic attack. Apparently, this reaction occurs via the insertion of copper into the C – Br bond, replacement of the halide ion as a result of the nucleophilic attack on the Cu centre, and reductive elimination, resulting in the ortho-alkylation product. Then the attack of the carboxylate ion on the carbonyl carbon atom induces cyclization to give the isocoumarin molecule.
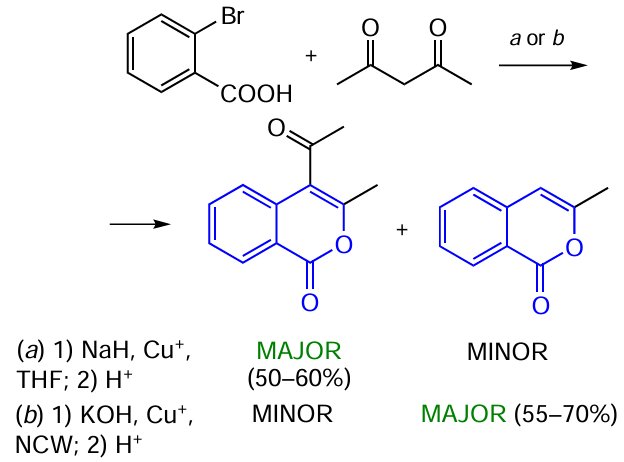
Bryson et al.[133] proposed an unusual method of Cu-catalyzed arylation of α-substituted β-dicarbonyl compounds for the synthesis of isocoumarins using near-critical water (NCW)* as the solvent. Reactions in near-critical water are considered to be environmentally safe; they are carried out in high-pressure stainless-steel reactors with heating or in Teflon or glass reactors under microwave irradiation.
While comparing the green approach with a similar reaction of o-bromobenzoic acid and acetylacetone in a standard organic solvent, one can notice the difference between their selectivities. 3,4-Disubstituted isocoumarin is the major product when the standard synthetic protocol is used starting from o-bromobenzoic acid in THF (Scheme 27, conditions a).[126] Meanwhile, the use of NCW and microwave irradiation results in the predominant formation of 3-methylisocoumarin.
A synthetic route to isocoumarins based on copper-catalyzed coupling reaction giving a C – C bond was reported by Shen and co-workers.[134] This process is a cascade copper-catalyzed intramolecular C-arylation and rearrangement process (Scheme 28).
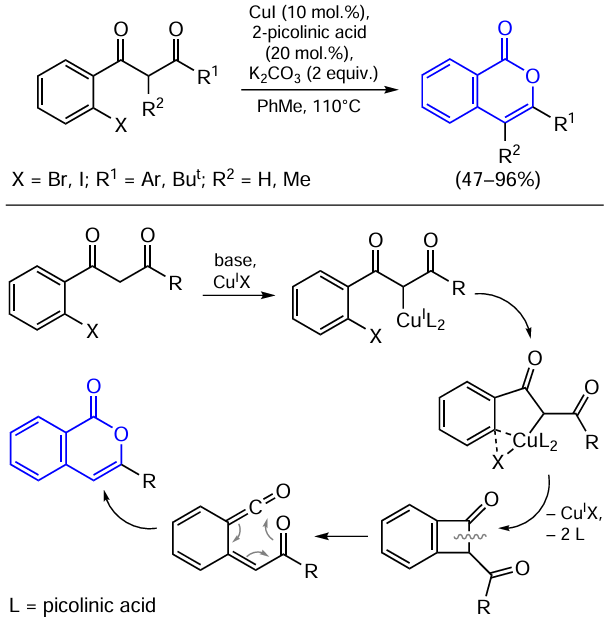
According to the mechanism proposed by the authors, first, a copper(I) organometallic complex is generated from CuI under the action of a base. The resulting complex undergoes oxidative addition/reductive elimination or another copper(I)-activated C – C coupling reaction to be converted to a four-membered cyclic intermediate. The subsequent rearrangement affords a ketene intermediate, which furnishes the desired isocoumarin product upon cyclization.
The cyclization of o-alkynylbenzoic acids described above (see Schemes 15 – 17, 20, 21) can also be promoted by divalent copper compounds. As has been mentioned by Lisowski and co-workers,[113] who synthesized isocoumarins with the use of a polymer matrix, the authors tested the procedure earlier developed by Li and co-workers.[135] This procedure gives mainly 4-chloro-substituted isocoumarins and a minor amount of 3-chlorinated derivatives. According to the proposed mechanism, first CuX2 is coordinated to the C≡C bond to give an intermediate alkyne complex. This is followed by the nucleophilic attack of the carboxy group on the activated triple bond and the subsequent cyclization. In the cyclization, CuX2 acts as a source of the halide ion at position 4 of isocoumarin, being thus reduced to copper(I) salt CuX. The authors believe that Cy2NH · HX performs two functions: (1) catalyzes the phase transfer in the CuX2/solvent/substrate/product system and (2) ensures the presence of free active X– anion to stimulate the reaction. However, this method cannot be classified as a copper-catalyzed process because the divalent copper salt is used in an amount of two equivalents. A catalytic version of this reaction was later proposed by Bihel’s research group.[136] The authors were able to prepare 3-substituted isocoumarins with high selectivity in the presence of copper(II) salts under microwave irradiation. The Cu0- and CuI-based catalysts provided lower yields of the target products.
Yet another CuI-catalyzed reaction is the synthesis of isocoumarins directly from ortho-halogenated benzoic acids and terminal alkynes. Initially, monovalent copper salts were used in the catalytic systems as mixtures with Pd0.[137] This resulted in the cascade process consisting of the palladium-catalyzed Sonogashira reaction and copper-catalyzed cyclization of ortho-alkynyl-substituted benzoic acids.
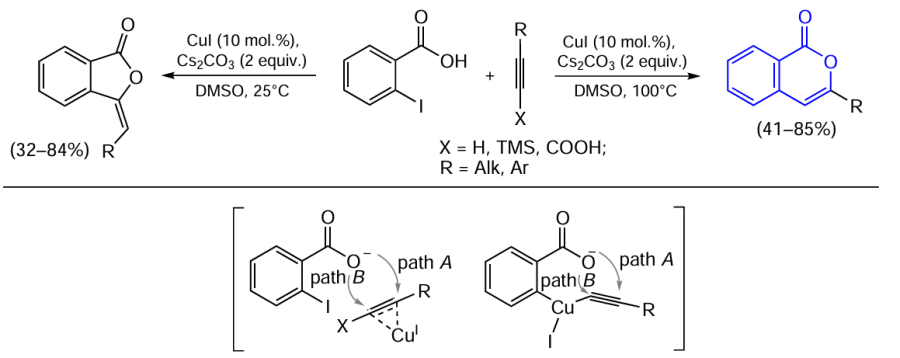
Later, it was found that the formation of isocoumarins from ortho-halobenzoic acids can also take place without a palladium co-catalyst. Although Inack-Ngi et al.[138] did not achieve high yields of products or reasonable selectivity (most reactions gave mixtures of isocoumarin and the corresponding phthalide in overall yields of 24 – 70%), further optimization of conditions enabled the synthesis of either isocoumarins or phthalides as the major products.[139]
In 2013, Lee and co-workers [139] found that the reaction selectivity can be controlled by varying the temperature. Thus, heating of the reaction mixture in DMSO to 100°C yielded mainly isocoumarins, while holding at 25°C for 12 h gave mainly the corresponding phthalides (Scheme 29). Additional studies of the reaction mechanism indicated that the ortho-alkynylbenzoic acid is not formed in this catalytic reaction. This also accounts for the fact that the reaction occurs not only with terminal alkynes, but also with TMS-protected analogues or alkynylcarboxylic acids. The authors suggest that the reaction involves an attack of the carboxylate ion on the alkyne triple bond, which occurs either before or simultaneously with the Sonogashira coupling reaction. In these reactions, path A may predominate at a temperature of 100°C, whereas path B predominates at 25°C.
In the same year, Pal and co-workers [140] proposed an improved green approach to the synthesis of isocoumarins by CuI-catalyzed reaction of o-iodobenzoic acids with terminal alkynes, but using polyethylene glycol (PEG-400) as a solvent under ultrasonic treatment. The authors were able to achieve good product yields (65 – 85%) and full selectivity of the process. In parallel with the above works, Guo[141] showed that internal alkynes can also participate in this reaction, which makes it possible to obtain 3,4-disubstituted isocoumarins from o-halobenzoic acids.
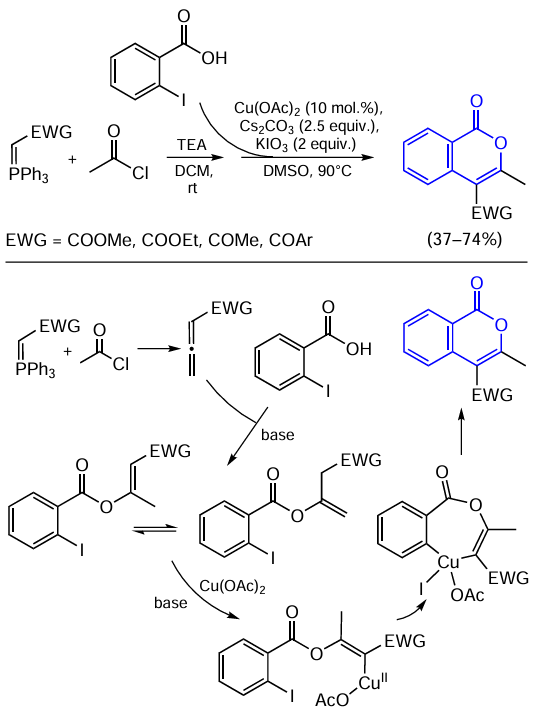
Chen and Liu [142] proposed an unusual method for the synthesis of isocoumarins by a copper-catalyzed reaction of acetyl chloride, o-halobenzoic acids, and Wittig reagents (Scheme 30). In the first step, acetyl chloride reacts with the Wittig reagents to give in situ the corresponding allene, which then undergoes the oxa-Michael reaction with o-iodobenzoate. The intermediate vinyl ether can exist as two tautomers. Under the action of the Cs2CO3 base, the nucleophilic carbon atom of the intermediate attacks Cu(OAc)2 according to the formal nucleophilic substitution of the acetate ligand to give copper(II) organometallic complex. The authors suggested that the subsequent oxidative addition of copper to the Ar – X bond gives a seven-membered intermediate containing copper in the unstable +4 oxidation state. Despite the fact that CuII-catalyzed cross-coupling reactions have been reported in the literature, the real mechanism of these reactions is, most often, more complicated and involves CuIII complexes.[143] The catalytic cycle is completed by reductive elimination to form the desired isocoumarin.
Mention should also be made of the synthesis of isocoumarins by a three-component coupling of the aryne precursors, terminal alkyne, and carbon dioxide catalyzed by the copper(I) complex with the NHC ligand (Scheme 31).[144]
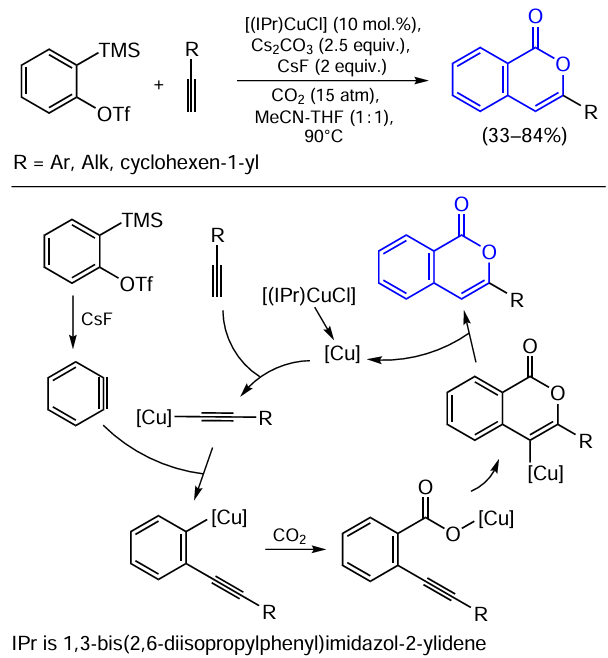
According to the mechanism of this reaction proposed by the authors, the NHC – copper hydroxide or carbonate, which is initially formed in situ, deprotonates the terminal alkyne to give copper acetylide. In parallel, aryne generation induced by the fluoride ion takes place, and the aryne reacts with the acetylide as an electrophile to give ortho-alkynyl copper complex. The formation of this intermediate is confirmed by the fact that no diphenylacetylene is formed in an additional experiment carried out under similar conditions in the absence of CO2 . This is followed by the insertion of CO2 into the Cu – C bond to give copper carboxylate, which undergoes cyclization to afford endocyclic organocopper intermediate. The catalytic cycle is completed by regeneration of the copper catalyst as a result of transmetallation of the endocyclic organocopper intermediate with cesium salts present in the reaction mixture and their subsequent protonation.
* NCW is liquid water in the temperature range from 200 to 350°C.
3.2.2. Catalysis by silver compounds
It is known that silver can also readily form alkyne complexes upon coordination to a triple bond and, therefore, this metal can catalyze the cyclization reactions involving ortho-alkynyl aromatic acids and their derivatives.[145-153] The first examples of these cyclizations catalyzed by silver compounds were reported in 1995 by Wakamatsu’s research group.[145][146] It was shown that the selectivity of the reaction was significantly dependent on the conditions and on the nature of the silver compound. For example, in the presence of Ag2CO3 (10 mol.%) as a catalyst in relatively non-polar solvents such as benzene, 1,4-dioxane, or dichloromethane, isocoumarin is formed as the major product, whereas more polar solvents such as acetonitrile, acetone, and dimethylformamide induce cyclization involving the triple bond carbon atom more proximate to the benzene ring to give the corresponding phthalide. Study of various silver compounds in DMF showed that silver(I) oxide produces a mixture of products; in the presence of silver(I) perchlorate, triflate, or nitrate, isocoumarins are formed predominantly; and, conversely, silver sulfate and halide, silver metal, and silver(I) carbonate provide the formation of phthalides.
Subsequently, it was shown that this reaction can also be used for the synthesis of 3-substituted isocoumarins from amides (in this case, isoquinolones or isoindolinones can be formed as by-products) [148][149] or esters.[150][151] The silver-catalyzed cyclization can also be carried out for aromatic aldehydes with an ortho-alkynyl substituent. However, in the presence of air oxygen, these substrates are converted in situ to the corresponding acids as a result of Ag-promoted generation of acyl radicals (the amount of AgBF4 is 10 mol.%) (Scheme 32).[152]
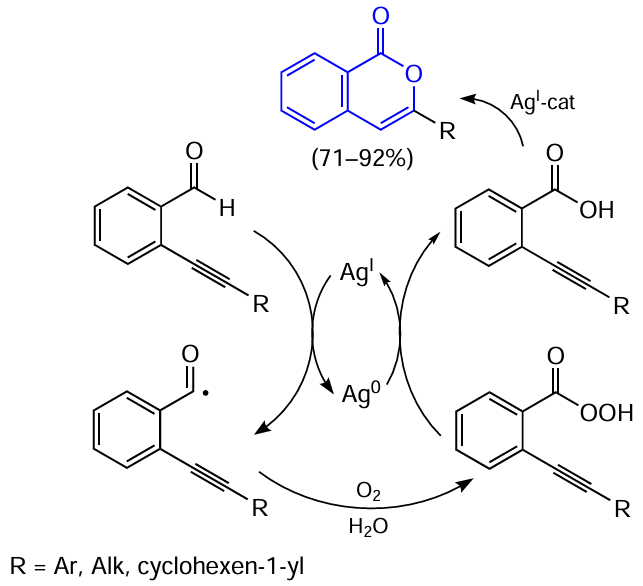
Finally, 2-(alkynonyl)alkynylbenzenes are the most exotic substrates that can participate in this type of cyclization. In this case, the alkynyl substituent at the carbonyl group acts as the leaving group at the last step of cyclization according to the retro-Favorskii reaction (Scheme 33).[153]
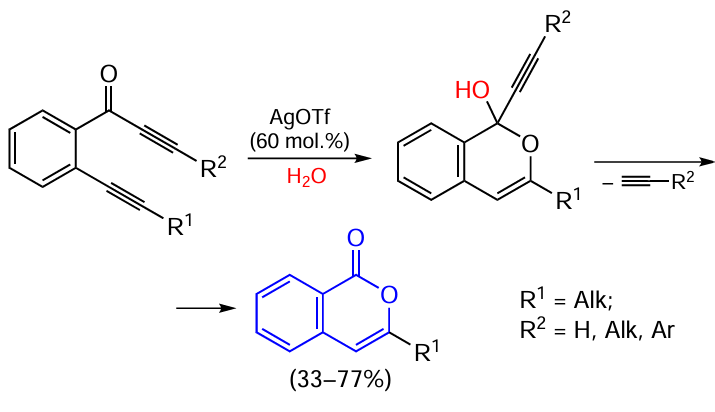
A somewhat different type of transformation induced by silver salts was reported by Panda et al.[154] The annulation reaction takes place for enol esters of ortho-iodinated aromatic acids or their mixtures with enol esters of unsubstituted analogues (Scheme 34).
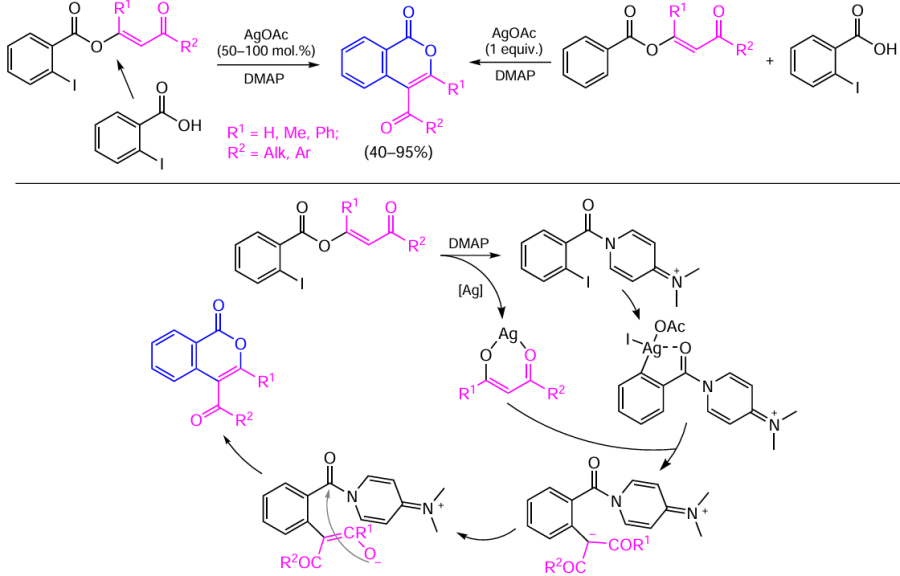
The authors assumed that the first step is the DMAP-induced attack of the enol ester carboxy group accompanied by enolate elimination; this is followed by insertion of silver into the C – I bond in the ortho-position of the resulting amide (or ortho-iodobenzoic acid in the case where it is used as a substrate in a mixture with vinyl benzoate). The subsequent replacement of the halide ligand with the enolate anion leads to enolate introduction in the ortho-position upon the reductive elimination of AgOAc. The reaction ends by the attack of the enolate on the carbonyl group of the amide accompanied by the elimination of DMAP and formation of the isocoumarin core. However, in view of the fact that the reaction yield drops to 66% as the amount of silver salt is reduced from one equivalent to 50 mol.%, this approach can hardly be considered to be a catalytic reaction as claimed by the authors.
3.2.3. Catalysis by gold compounds
Gold compounds, like silver compounds, are fairly often used in various cyclization reactions involving alkynes owing to their ability to coordinate acetylene derivatives, thus activating the multiple bond towards the addition of nucleophiles. In 2005, Michelet and co-workers[155] showed that gold(I) and gold(III) compounds catalyze the cyclizations of acetylenic acids resulting in γ-lactones. However, this selective procedure gave only five-membered lactone rings via exo-cyclization.
Several years later, van de Weghe’s research group [156] made an attempt to use this approach in the AuI or AuIII-catalyzed cyclization of o-alkynyl-substituted benzoic acids. As expected, most often, the reaction mainly gave five-membered phthalides, but in some cases, mixtures of products with significant proportions of isocoumarins were formed. The authors found an interesting fact: if an ester is taken as the starting reactant instead of an acid, cyclization selectively follows the 6-endo-pathway and gives exclusively the isocoumarin derivative. In this reaction, AuCl3 has a higher catalytic activity than AuCl. The proper acids can be used in this reaction if some phosphine ligand is added to gold(I) salt.[157] This catalytic approach was later successfully used in the total synthesis of citreoviranol[158] and exserolide F,[159] natural biologically active compounds.
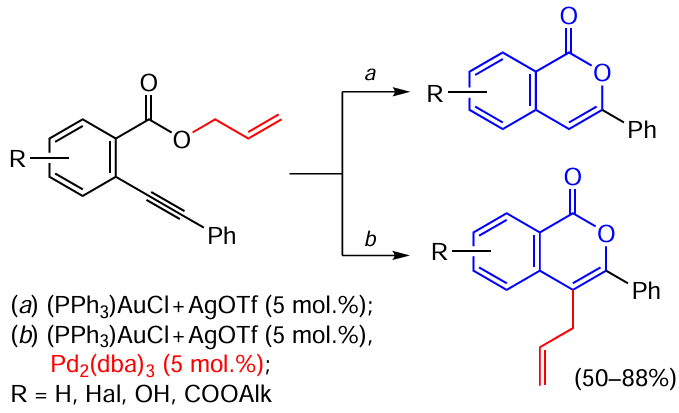
Blum’s research group[160][161] showed the applicability of gold compounds in combination with palladium complexes as catalysts. In the presence of a palladium co-catalyst, the Au-catalyzed cyclization of allyl o-alkynylbenzoates furnishes the isocoumarin core bound to the metal atom at position 4. This is followed by transmetallation (replacement of gold by palladium) and introduction of the allyl moiety, functioning as the leaving group, in position 4 of isocoumarin via Heck type reaction (Scheme 35).
Wang and co-workers [162] reported a study on the synthesis of indene-fused isocoumarins by AuI-catalyzed cyclization (Scheme 36). According to one of the putative reaction mechanisms proposed by the authors, the activation of a terminal alkyne by coordination of the carbophilic gold phosphine complex initiates the cyclization, which is followed by the intramolecular nucleophilic attack of the carboxy group (path A). Another route includes activation of an internal alkyne, while the carboxy group attacks the activated alkyne to give isocoumarin, which can be converted to the product (path B). However, the authors believe that in reality, the five-membered carbocycle and the six-membered heterocycle are apparently formed together. In the absence of a phosphine ligand, the catalytic reactions result in the formation of only isocoumarin moiety.
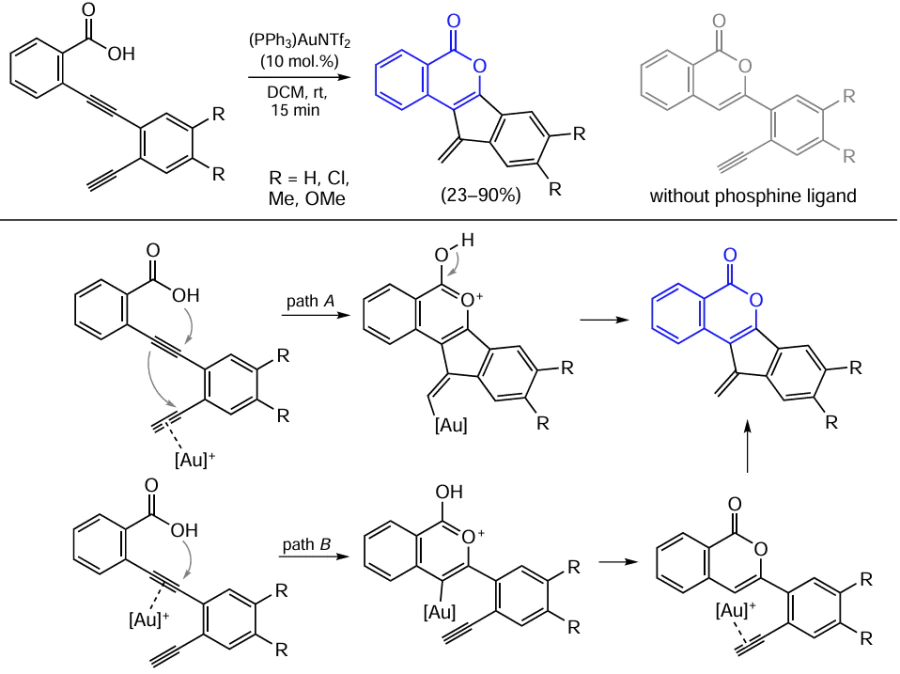
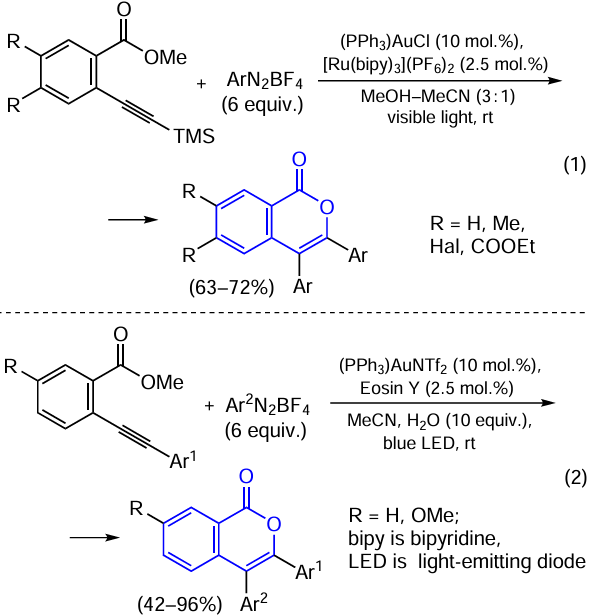
3,4-Diaryl-substituted isocoumarins can be synthesized from o-alkynylbenzoic acid esters under the action of the gold(I) phosphine complex in the presence of aromatic diazo compounds using photoredox catalysis. As photocatalysts, both ruthenium(II) bipyridine complexes Scheme 37, reaction (1)][163] and the eosin Y dye [see Scheme 37, reaction (2)][164] were used.
3.2.4. Catalysis by palladium compounds
The cyclizations of ortho-alkynylbenzoic acids to give isocoumarins described in detail in previous Sections can also be performed in the presence of palladium catalysts.[165][166] For example, Sashida and Kawamukai [165] proposed the first synthesis of isocoumarins based on this approach (Scheme 38). It is noteworthy that benzoic acids with bulky groups at the end of the triple bond show lower selectivity of ring closure due to steric hindrance and undergo a side cyclization reaction, which affords large amounts of phthalide. Moreover, Kundu’s [167] and Rossi’s [168] research groups found that even in the case where R is a linear alkyl group, in the presence of Pd(PPh3)4 or (PPh3)2PdCl2 complexes with bulky phosphine ligands, the reaction selectivity shifts towards the formation of phthalide.
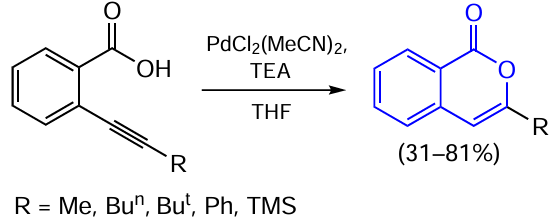
Currently, palladium catalysis of the cyclization of ortho-alkynylbenzoic acids and their esters is primarily used for both inter- [169-171] and intramolecular [172] tandem reactions in the presence of an olefin moiety.
The coordination of the triple bond to the palladium atom giving an alkyne complex activates the triple bond towards cyclization, which results in the formation of the isocoumarin skeleton bound to the palladium atom at position 4. The anionic ligand deprotonates the group R1, which results in the elimination of the corresponding unsaturated hydrocarbon. This is followed by the coordination of the activated olefinic moiety in the metal coordination sphere and its insertion into the M – C bond followed by β-elimination (Heck reaction). The regeneration of the catalytically active PdII species from the Pd0 compound occurs on treatment with an external oxidant (Scheme 39).
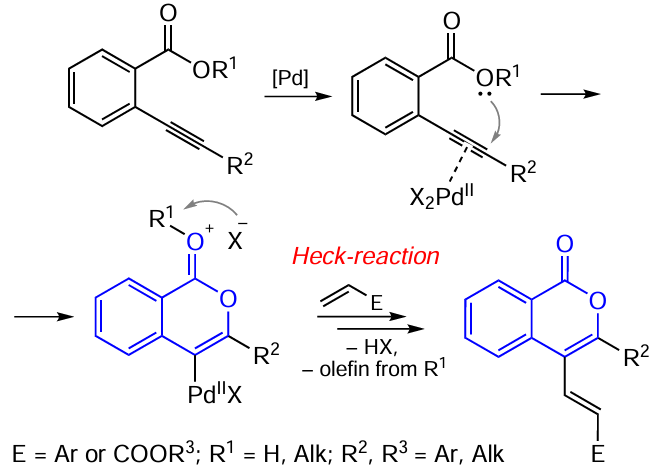
As mentioned above, the use of palladium catalysis in combination with agents that promote cyclization such as Lewis acids provides the synthesis of 3-substituted isocoumarins in a single step directly from o-halobenzoic acids (Scheme 40).[173]
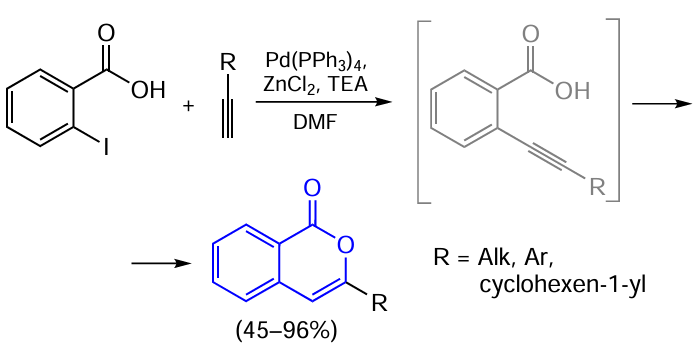
In the case of unsubstituted acetylene, the reaction, as expected, results in the formation of phthalide. In some cases, 3,4-disubstituted isocoumarins are formed upon the introduction of 1,4-disubstituted buta-1,3-diyne (arising as a Sonogashira reaction by-product upon the homocoupling of terminal alkynes) into o-iodobenzoic acid.
The formation of these by-products indicates the conceptual possibility of introducing internal alkynes into ortho-halobenzoic acids to obtain 3,4-disubstituted isocoumarins. Particularly this reaction was accomplished in 1989 in the studies of reactions of alkynes with arylpalladium complexes supervised by R.F.Heck, a Nobel Prize winner.[174] In relation to diarylacetylenes, it was found that they can react with methyl o-iodobenzoate in the presence of sodium acetate and triethylamine as a base (Scheme 41).
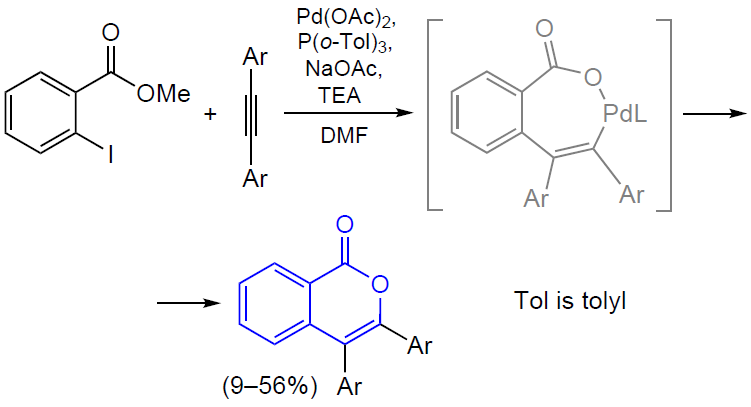
Subsequently these results were confirmed by Larock et al.[175] in relation to reactions of dialkylacetylenes with ortho-bromo and ortho-triflate derivatives.[176] This palladium-catalyzed approach to the synthesis of isocoumarins was further developed. The colloidal catalysis procedures that were elaborated in these studies use an aqueous solution of palladium nanoparticles stabilized by the micelles of polystyrene and polyethylene oxide (PS-PEO) copolymer with cetylpyridinium chloride (CPC) to perform reactions in dimethylacetamide (DMA)[177] and in water under microwave irradiation.[178]
Isocoumarins can also be obtained from ortho-alkenylbenzoic acid derivatives. In 1994, Hanaoka and co-workers[179] proposed their own version of the synthesis of 3-substituted isocoumarins by acyloxypalladation of o-alkenylbenzoic acids (Scheme 42). Benzoquinone was used as an oxidant, which regenerated the catalytically active species after β-elimination.
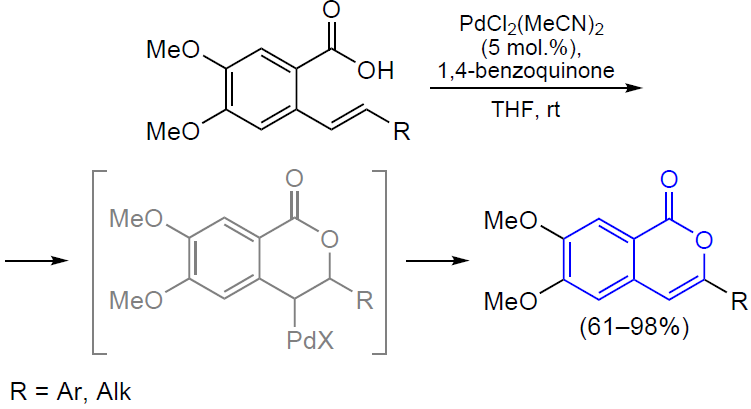
It is noteworthy that an allene group can play the role of the alkynyl substituent in the isocoumarin synthesis. For example, the tandem Stille reaction and heterocyclization of o-iodobenzoic acid involving various allenyltributyl tin derivatives in the presence of palladium acetate, phosphine ligand, and tetrabutylammonium bromide (TBAB) afforded 3-substituted isocoumarins (Scheme 43).[180] Despite the moderate yields of the reaction products (59 – 65%), this approach has high selectivity to particularly the isocoumarin skeleton.
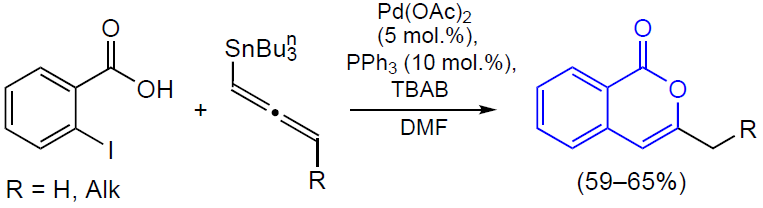
According to a probable reaction mechanism, the first step is the oxidative addition of o-iodobenzoic acid to the palladium complex (Scheme 44). Then the intermediate complex undergoes transmetallation followed by reductive elimination to give allenyl-substituted benzoic acid, which then undergoes PdII-promoted cyclization. Finally, this affords 3-substituted isocoumarin, with the alkyl chain of the substituent being increased by one methylene unit.
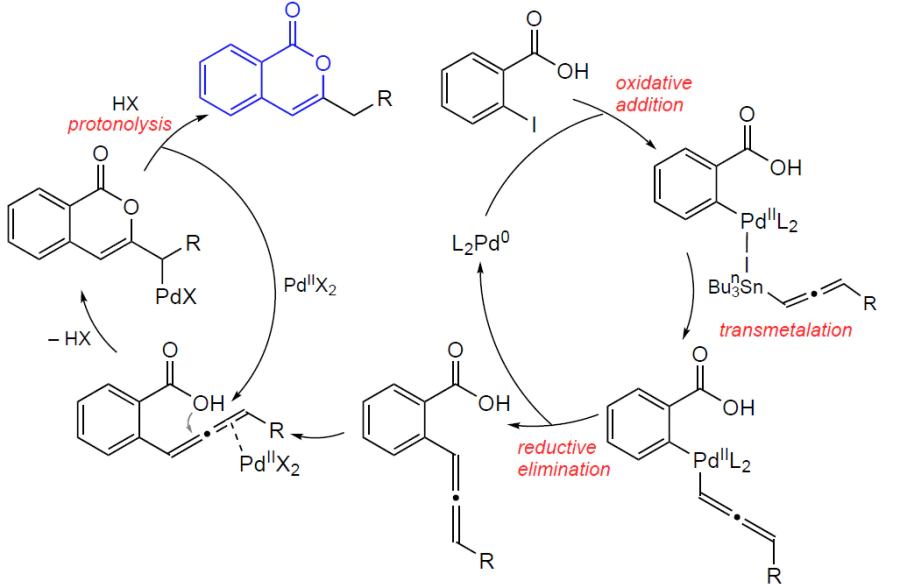
A year later, Chakravarty et al.[181] reported the synthesis of isocoumarin in which the authors used free allenes instead of organotin precursors. It is noteworthy that the reaction of unsubstituted terminal allenes afforded either 4-substituted isocoumarin as the only product (in the case of R = COOEt) or the product with an exocyclic double bond (when R = Ph) (Scheme 45).
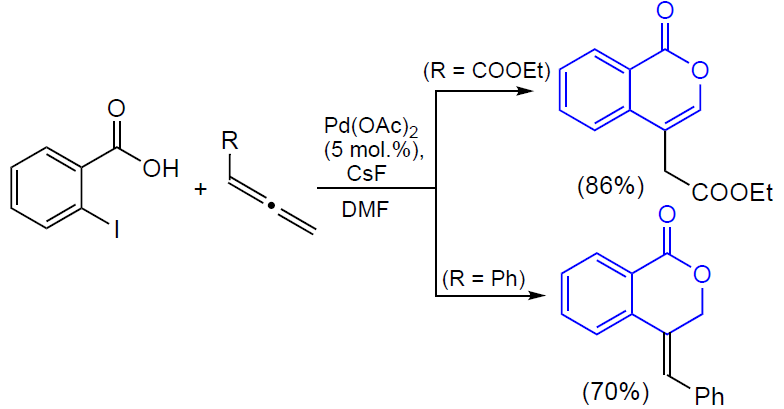
Several methods for the introduction of ortho-2-oxopropyl group into the substrate (see Scheme 9 and Scheme 10) in the presence of equimolar amounts of nickel complexes have been considered above. In recent years, these reactions were carried out in situ using palladium catalysts. For example, Zhang and Yu[182] reported an approach to the synthesis of isocoumarins by the retro-aldol condensation/α-arylation (Scheme 46).
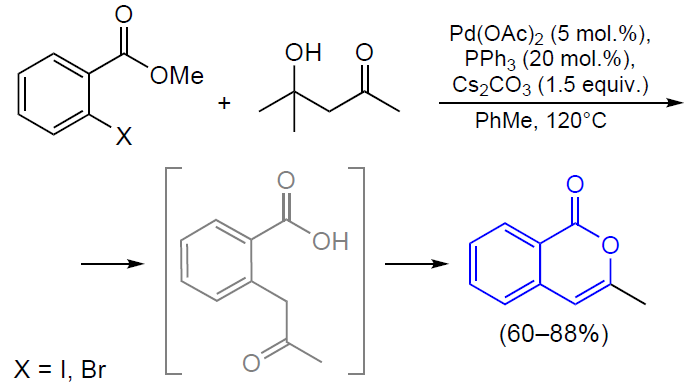
Presumably, the retro-aldol reaction of the β-hydroxycarbonyl compound coordinated to the palladium complex accompanied by the release of an acetone molecule affords a enolate species, the cross-coupling of which with the aryl moiety leads to the ortho-2-oxopropyl intermediate. The subsequent cyclization of the intermediate furnishes isocoumarin.
Somewhat later, the Motti’s research group[183] extended this procedure to usual ketones, first of all, those containing an acetyl group in the molecule (i.e., R1 = H). The use of α-substituted ketones (R1 ≠ H) gives 3,4-disubstituted isocoumarins in low yields of 10 – 40% (Scheme 47).
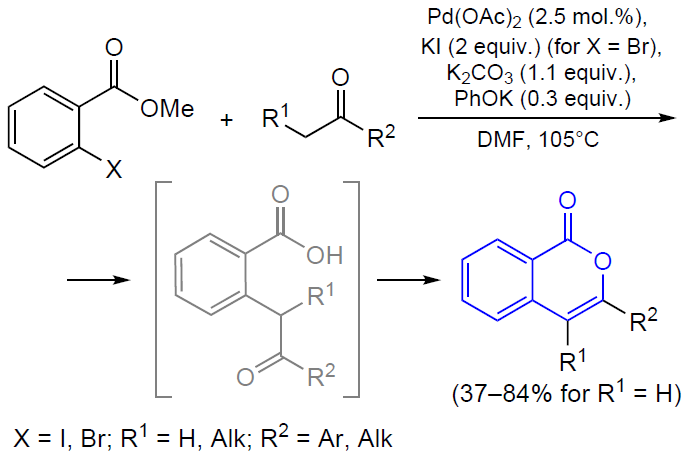
A fundamentally different type of the starting reactants meant for the synthesis of isocoumarins are ortho-2-oxopropyl-substituted aryl halides. A key distinction of this approach is the absence of the carboxy group in the substrate and, hence, in this case, it is necessary to add a carbonyl group from an external source. The simplest source of this group is carbon monoxide gas from a cylinder.[184][185] Thus isocoumarins were synthesized by the aryl halide carbonylation/intramolecular O-enolate acylation sequence (Scheme 48).
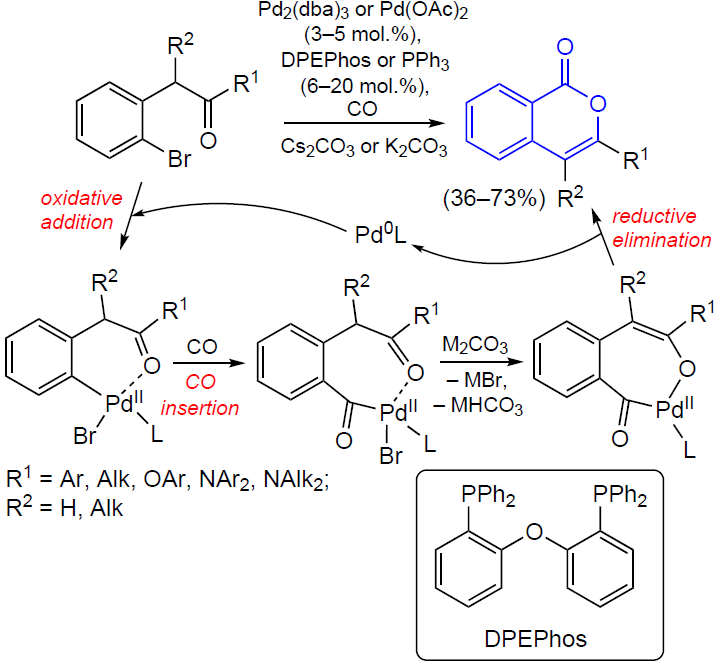
The first step is the oxidative addition of aryl halide to the palladium complex; this is followed by coordination of the CO molecule with the subsequent insertion into the Pd – C bond. After that, the α-position of the carbonyl compound is deprotonated under the action of the base to form the corresponding enolate, which replaces the halide ligand in the metal coordination sphere. In the final step of the catalytic cycle, the reductive elimination yields the expected isocoumarin.
The possible latent sources of CO are phenyl formate[186] or tert-butyl isocyanide, which give rise to the intermediate imine, being converted to the target isocoumarin after acid hydrolysis.[187]
The direct C – H activation on treatment with transition metals is the most efficient and atom-economic method for functionalization of organic compounds. There is an example of utilizing this approach to prepare isocoumarins, in particular using palladium complexes.
In 1998, Miura’s research group[188] proposed to use benzoic acids for the palladium-catalyzed oxidative cross-coupling with alkenes. Palladium(II) acetate (5 – 10%) mixed with the same amount of the Cu(OAc)2 oxidant in the presence of air oxygen served as the catalytic system. Unfortunately, this approach did not provide high yields of isocoumarins and had low selectivity, resulting in the formation of phthalide by-products. Later, Lee and co-workers[189] confirmed this result and revealed the following rule: isocoumarins are formed only provided that the second ortho-position in benzoic acid is unsubstituted. If a substituent is present at this position or if a substituent is absent due to an additional annulated ring, the benzylidenephthalide derivative is the only reaction product (Scheme 49). In addition, in 2022, Monopoli and co-workers [190] showed the possibility of performing this reaction in ionic liquids, which allowed a considerable decrease in the amount of the catalyst (down to 0.5 mol.%).
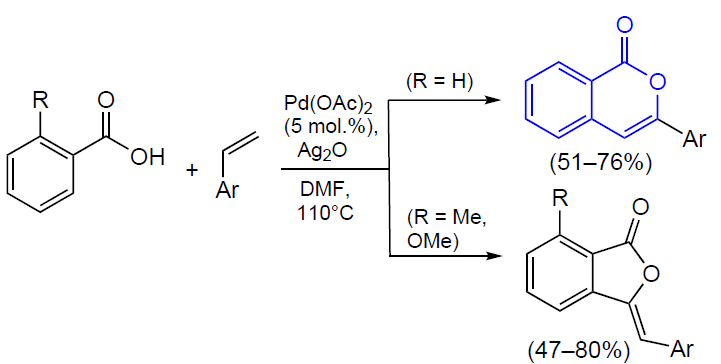
In the next example, the method using alkynes is not as atom-economic as in the case of Rh, Ir, or Ru (see Sections 4.2.6 and 4.2.7 below), because of the necessity to introduce a leaving bromine atom into the alkyne to implement the oxidative addition step. The research group headed by Jiang[191] developed an approach based on the nucleophilic addition/oxidative annulation sequence through C – H activation of the ortho-position in aromatic acids (Scheme 50).
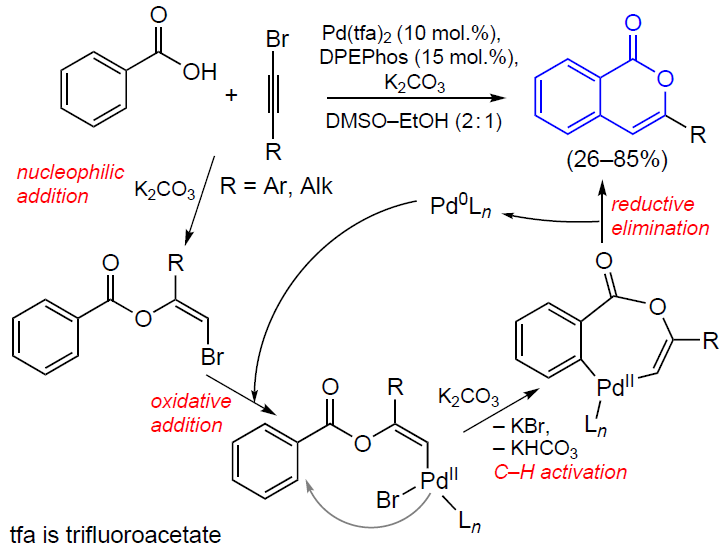
The reaction starts with deprotonation of benzoic acid under the action of a base, the subsequent nucleophilic attack of the resulting anion on the triple bond yields bromoalkene, which then undergoes oxidative addition to the palladium complex. The next stage of the catalytic cycle consists in the base-assisted activation of the C – H bond at the ortho-position of the aromatic ring. The seven-membered intermediate formed in this way undergoes reductive elimination to give 3-substituted isocoumarin.
An example of Pd-catalyzed annulation proceeding via the acyl C – H activation was described by Chen’s research group.[192] This approach is based on the palladium-catalyzed annulation of ortho-bromobenzaldehyde with aryldiazo esters. The authors suggest that the result is achieved via successive palladium carbene transfer and insertion into the Pd – C bond, migration of palladium to the oxygen atom, and activation of the acyl C – H bond followed by reductive elimination in a seven-membered palladacycle intermediate (Scheme 51).
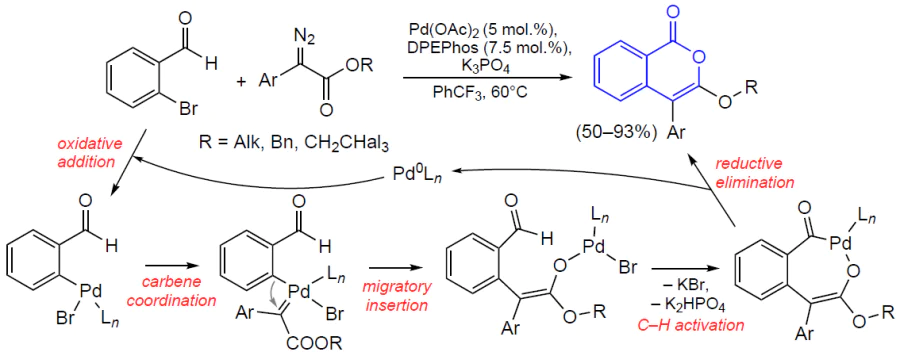
It may happen that the seven-membered intermediate is not actually formed, but the reaction includes the concerted attack of the enolate ion on the carbonyl carbon atom of the aldehyde group and hydride transfer to the metal followed by reductive elimination of an HBr molecule. In any case, this method is suitable only for the synthesis of isocoumarins containing an ether group at position 3.
In conclusion of the discussion of cyclizations of ortho-alkynylbenzoic acid derivatives, to avoid placing this information into separate Sections, it is necessary to mention that single examples of these reactions catalyzed by InBr3 (Ref. [193]) and Re(CO)5Cl (Ref. [194]) have also been reported.
3.2.5. Catalysis by nickel compounds
The isocoumarin synthesis using nickel catalysts is not a general approach, being described only in a few publications. The first example of Ni-catalyzed synthesis of isocoumarins was reported in 2008 by a group of Japanese chemists.[195] The authors proposed a procedure for the preparation of isocoumarins and α-pyrones by the addition of alkynes to anhydrides through the decarbonylation step (Scheme 52).
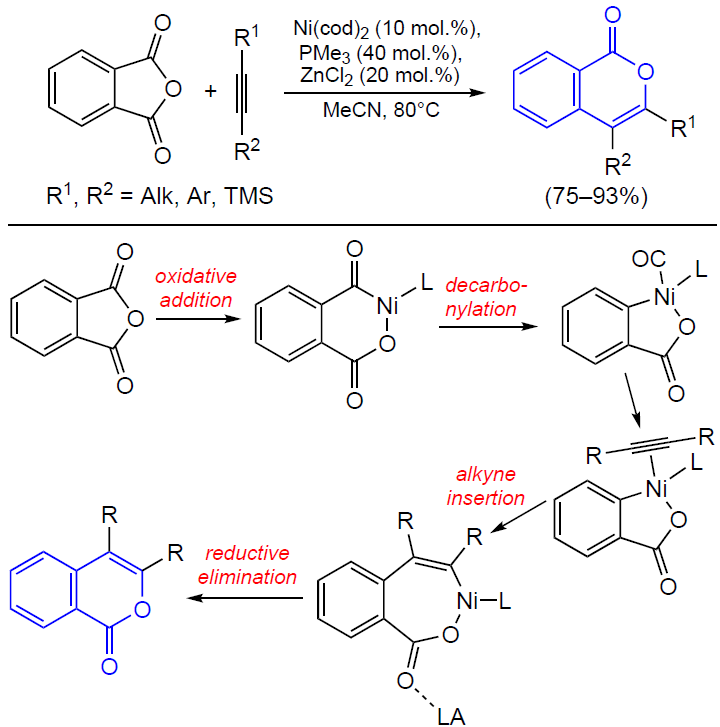
According to the proposed mechanism, the first step is nickel insertion into the C – O bond as a result of oxidative addition. The subsequent decarbonylation, replacement of the CO ligand by alkyne, and insertion of the alkyne into the Ni – C bond lead to seven-membered intermediate. In the authors’ opinion, the role of the Lewis acid, which can be represented by zinc(II) salts, BPh3, etc., is coordination to the carbonyl group in the seven-membered ring, which decreases the donor properties of the carboxyl ligand and thus facilitates the reductive elimination to give isocoumarin. A limitation of this procedure is that it is suitable only for internal alkynes.
The classical version of the synthesis of isocoumarins involving a nickel catalyst is based on the insertion of the metal atom into the C – Hal bond located at the ortho-position relative to the directing group. Thus, when aromatic acid amides are used as the starting compounds, the reaction gives isochromen-1-imines, the acid hydrolysis of which leads to isocoumarins.[196] The key issue for the formation of isocoumarin rather than isoquinolone skeleton is isomerization of the five-membered nickelacycle before the alkyne insertion controlled by the nature of the phosphine ligand (Scheme 53). A similar process based on the activation of the C – F bond in the ortho-fluorinated benzoic acids was reported in 2021 by Chatani and Matsuura.[197]
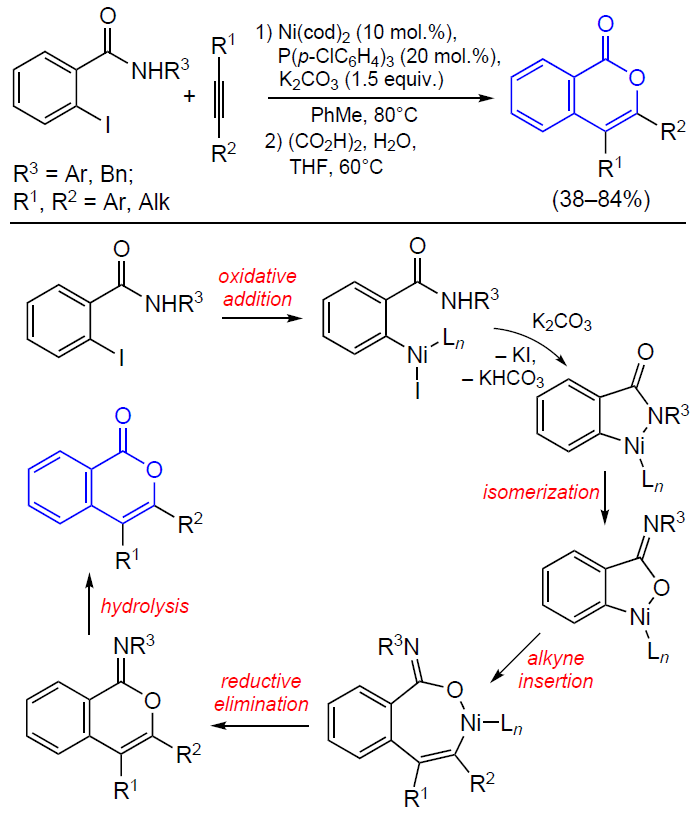
Fei et al.[198] reported the synthesis of isocoumarins from ortho-2-oxopropyl-substituted aryl halides catalyzed by the NiCl2 – dppe system [dppe is 1,2-bis(diphenylphosphino)ethane], similar to the Pd-catalyzed method (see Scheme 48). In this reaction, tert-butyl isocyanide is used as a latent source of CO.
3.2.6. Catalysis by Group 9 metal compounds
Although cobalt, rhodium, and iridium compounds are also capable of catalyzing the above reactions (e.g., the [(cod)IrCl]2 complex catalyzes the cyclization of ortho-alkynyl acids with a low selectivity),[199] this part of the review is devoted only to reactions proceeding through C – H bond activation at the ortho-position of aromatic substrates. Over the last two decades, this activation catalyzed by transition metals has become one of the most popular synthetic strategies owing to its efficiency, selectivity, and atom-economy [200-204] This procedure makes it possible to fabricate a variety of heterocyclic structures from simple and readily available reactants in only one step. The main benefit of this approach is that there is no need for additional modification of substrates by preliminary introduction of halogen atoms, multiple bonds, etc.
The rapid progress in the synthesis of isocoumarins was initiated by a study of Miura and co-workers [205] published in 2007. The authors found that rhodium(III) pentamethylcyclopentadienyl (Cp*) complex [Cp*RhCl2]2 taken in a relatively low amount (1 mol.%, which implies 2 mol.% Rh) performs annulation of acetylenes with benzoic acids in o-xylene at 120°C to give isocoumarins. This reaction proceeds through the direct C – H activation of the ortho-position of the acids without additional steps of substrate pre-functionalization (Scheme 54).
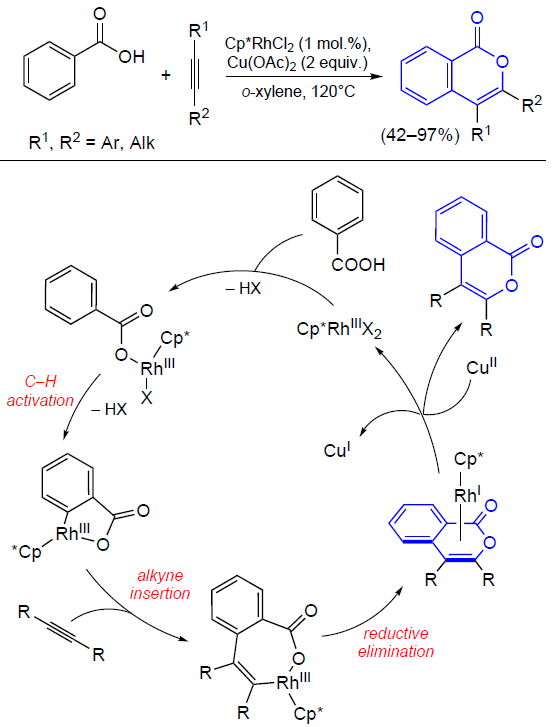
According to the hypothesized reaction mechanism, the carboxyl substituent acts as a directing group, determining the preferred C – H activation of the ortho-position of the benzene ring.[206][207] In the first step, rhodium is coordinated to the carboxy group to give the corresponding carboxylate. The subsequent C – H activation occurs with the assistance of acetate ion, which is coordinated to rhodium via exchange with the halide ligand, and results in the formation of a five-membered rhodacycle. The subsequent insertion of the alkyne molecule gives the key seven-membered cyclic intermediate, and the nature of substituents and ligands in the intermediate dictates the further reaction pathway. In particular, if the redox potential of the Cu2+/Cu1+ pair is sufficient to oxidize the intermediate rhodium(I) complex, isocoumarin is formed and the catalyst is simultaneously regenerated. The presence of five electron-donating methyl groups in the cyclopentadienyl ligand markedly facilitates this process. In recent years, we also demonstrated high catalytic efficiency of rhodium complexes with di- and triphenylcyclopentadienyl ligands in this reaction. [208-211]
It is noteworthy that in the presence of the iridium analogue [Cp*IrCl2]2, tetrasubstituted naphthalene derivative is formed as the major reaction product instead of isocoumarin (Scheme 55).[205] This product can be formed if the redox potential of the oxidant is too low. In this case, the more favourable process includes decarboxylation of the seven-membered iridacycle followed by the insertion of the second alkyne molecule into the intermediate complex, which is subsequently oxidized to give the naphthalene product. This redox process always proceeds more easily because the system loses the electron-deficient carboxy group upon decarboxylation. It is noteworthy that decarboxylation by itself requires elevated temperatures: the authors conducted this reaction at 160°C. In addition, our research group [212] found that the rhodium complex [CpRhI2]n with unsubstituted cyclopentadienyl (Cp) ligand used for the reaction of benzoic acid with tolane (diphenylacetylene) under similar conditions also provides the selective formation of tetraphenylnaphthalene in 91% yield. A similar chemoselectivity was found for the triple-decker rhodium complex (η-C4H4BMe)Rh(m-η:η-C4H4BMe)Rh(η-C4H4BMe) and for other rhodacarboranes.[213][214]
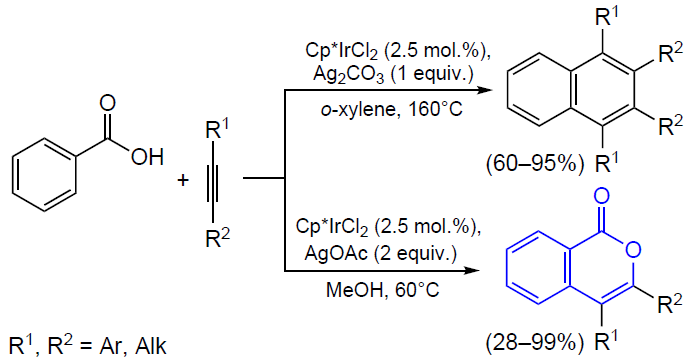
Nevertheless, at lower temperatures, the [Cp*IrCl2]2 complex can provide the formation of isocoumarins in high yields.[215][216] In this case, silver acetate is used as an oxidant, while methanol serves as a solvent, and the reaction proceeds even at 60°C (see Scheme 55). This procedure is best suited for dialkylacetylenes, while diarylacetylenes give the corresponding isocoumarins in lower yields.
An attractive technique is the use of similar cobalt compounds instead of expensive rhodium or iridium complexes. Cobalt(III) complexes are actually applicable as catalysts for the isocoumarin synthesis (Scheme 56),[217] but this requires high amounts of the catalyst (10 mol.%). The reaction mechanism is essentially the same as assumed for rhodium(III) and iridium(III) compounds. Silver carbonate and acetate used as oxidants as well as copper acetate show low efficiency compared to copper oxide. The NaOAc additive functioning as a source of acetate ions serves to facilitate the C – H activation step.
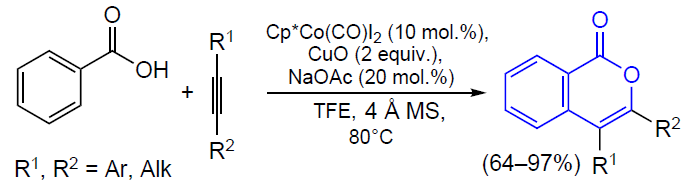
In 2016, Tanaka and co-workers [218] found that the introduction of two electron-withdrawing COOEt groups into the cyclopentadienyl ligand of the rhodium complex makes it possible to reduce the temperature of isocoumarin synthesis to room temperature, since the C – H activation step is facilitated due to increasing electrophilicity of the metal centre.
Despite all benefits of this approach, the main drawback is that this reaction cannot be carried out for terminal alkynes. This restriction is due to the fact that copper and silver salts used as oxidants are capable of forming acetylides; this leads to alkyne deactivation and possible dimerization to give diyne derivatives. Therefore, various research groups have explored the applicability of other external oxidants.
Air oxygen can be used as a more environmentally benign oxidant. The first use of oxygen in conjugation with a catalytic amount of copper acetate was also investigated by Miura’s group.[219] Despite the high efficiency of this approach to the synthesis of 3,4-disubstituted isocoumarins, an attempt to reduce the amount of copper(ii) acetate did not allow the involvement of terminal acetylenes in this reaction.
In 2020, Lu and co-workers[220] reported a study in which they used K2S2O8 as an oxidant to prepare isocoumarins from carboxylic acids and terminal alkynes. However, the yields of the target products were quite low (17 – 39%).
Cerium(iv) salts proved to be the most effective type of external oxidant. Nguyen et al.,[221] who studied the CoIII-catalyzed C – H activation, were able to prepare 3-substituted isocoumarins from terminal alkynes in fairly high yields (Scheme 57). It is worth noting that the product was still formed, although in a lower yield, when oxygen was the only oxidant used. The reaction proceeded more slowly in an inert atmosphere, which indicates that both CeIV salts and oxygen are needed to obtain products in high yields.
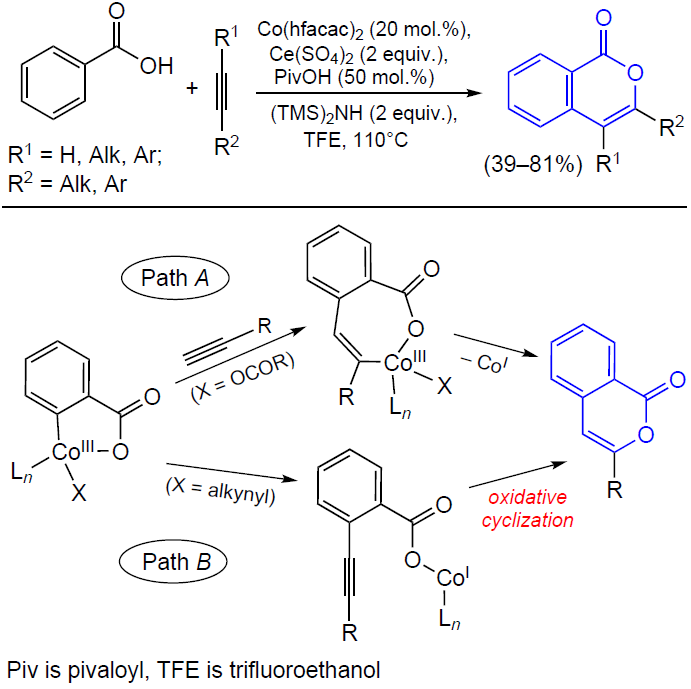
The authors assumed that the reaction proceeds via an intermediate CoIII complex, which is formed upon oxidation of Co(hfacac)2 (hfacac is hexafluoroacetylacetonate) in the presence of benzoic acid. After that, two reaction pathways are possible. One pathway implies the classical migratory insertion of alkyne followed by reductive elimination. The other pathway comprises the formation of an intermediate cobalt alkynyl complex followed by its reductive elimination to give ortho-alkynyl-substituted cobalt benzoate, which further undergoes the oxidative cyclization.
A fundamentally different approach to catalyst regeneration for the subsequent catalytic cycles in the isocoumarin synthesis via C – H activation is based on the use of electrical current as an oxidant.[222] This method was also successfully employed for iridium complexes.[223] However, the authors did not provide data on the applicability of terminal alkynes for this reaction. Nevertheless, it was shown that under the action of electrical current, terminal alkynes can be involved in the amide annulation reaction to afford the corresponding 3-substituted isoquinolones,[224] which may indicate the potential possibility of preparing also 3-substituted isocoumarins.
Apart from various external sources, electron pairs of the N – N, N – O, and O – O bonds, present directly in the substrate molecule, can also act as oxidants. The use of directing groups that contain an internal oxidant is a popular approach to C – H activation, in particular for the synthesis of isocoumarins (Scheme 58).[225-227]
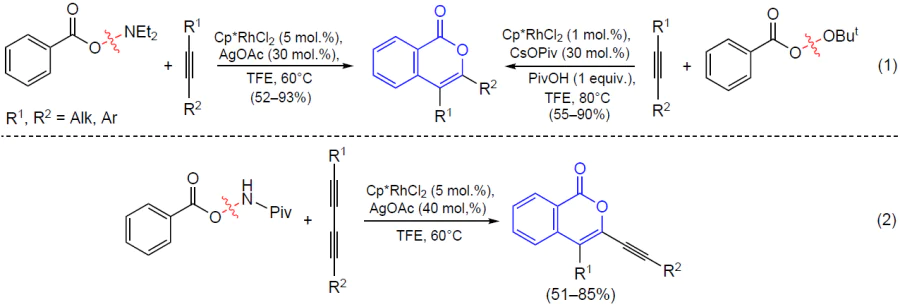
However, the authors of these studies also did not provide information on the possibility of using terminal alkynes under these conditions. In this case, the added silver [see the left-hand part of reaction (1) and reaction (2) in Scheme 58] apparently plays a dual role, acting as halide ion acceptor for activation of the catalyst and as a source of the acetate ion to facilitate the C – H activation and reoxidation of the metal centre. All steps of the reaction are identical to those of the classical oxidative coupling of aromatic acids and alkynes, except for the fact that the last step of the reaction is the O – O or O – N bond cleavage accompanied by Rh migration to the directing group either via reductive elimination/oxidative addition or by a concerted mechanism without a formal change in the oxidation state of the metal (Scheme 59).
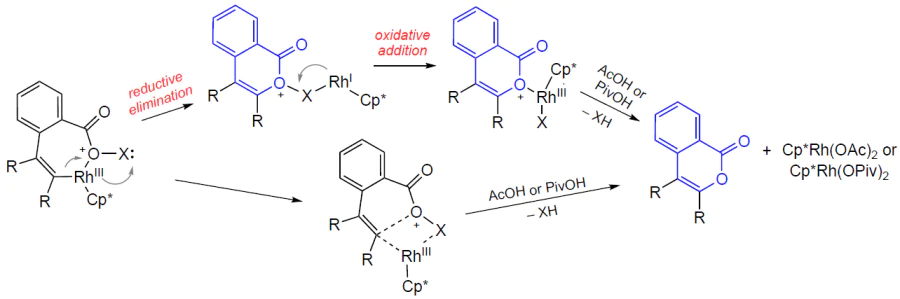
In addition, the N – O bond can serve as a common external oxidant in Cp*MIII-catalyzed (M is metal) oxidative coupling reactions proceeding via C – H activation. In relation to isocoumarin synthesis, Wang and co-workers [228] showed that organic compounds in which the carboxyl oxygen atom is bound to the NR2 group can be successfully used as external oxidants in the reactions catalyzed by Group 9 metal cyclopentadienyl complexes.
Instead of alkynes, other compounds can act as coupling partners, such as geminally substituted vinyl acetates,[229] diazo compounds,[230-235] sulfoxonium ylides,[236] and cyclic alkenyl carbonates.[237] Many of these compounds are also applicable for the synthesis of 3-substituted isocoumarins. For example, Cp*MIII-catalyzed reaction results in the introduction of 2-oxopropyl moiety in the ortho-position of the aromatic substrate (acid or amide), while the subsequent cyclization occurs in the presence of an added acid.[237]
Finally, in 2020, the same Miura’s group[238] proposed the use of vinylene carbonate as both a source of acetylene and an internal oxidant for the synthesis of 3- and 4-unsubstituted isocoumarins.
In turn, the use of dibasic terephthalic acid in the reaction can lead to the formation of more complex polycyclic products as a result of introduction of several alkyne molecules into the product molecule. For example, the reaction of terephthalic acid with oct-4-yne affords pyranoisocoumarins, which are symmetrical products resulting from the introduction of two alkyne molecules [49] Scheme 60, reaction (1)], whereas in the case of diphenylacetylene, introduction of three alkyne molecules to give benzoisocoumarins takes place [see Scheme 60, reaction (2)].[239] In addition, in our study submitted for publication,[240] it was shown that the use of the stepwise approach provides access to unsymmetrical pyranoisocoumarin derivatives [see Scheme 60, reaction (3)].
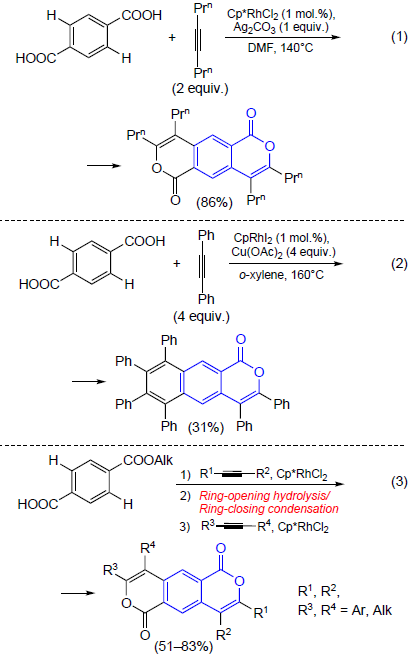
3.2.7. Catalysis by Group 8 metal compounds
While iron compounds act mainly as Lewis acids in the synthesis of isocoumarins,[104] ruthenium and osmium complexes can also catalyze reactions proceeding through C – H activation, like complexes of Group 9 metals. A large contribution to the development of ruthenium and osmium catalysis, in particulaar for the synthesis of isocoumarins, was made by Ackermann’s research group.[241-243] The authors showed that the known ruthenium catalyst [(p-Cym)RuCl2]2 (Cym is cymene) participates in both usual oxidative coupling reactions in the presence of a copper salt as an oxidant to give isocoumarins[241] and reactions involved in electrooxidation[242] or conducted in the presence of oxygen as the only oxidant.[243]
The [(p-Cym)RuCl2]2 complex was also used to prepare isocoumarin 18 (PXZ-ICO), which exhibits the TADF effect (see above), by unusual self-condensation of sulfoxonium ylide, which formally serves as a source of both carboxylic acid and alkyne in the C – H activation.[244] In addition, Ackermann and co-workers[245] published a study on the synthesis of isocoumarins catalyzed by osmium(II) complexes. The principle of this oxidative coupling based on C – H activation is similar to that of the reactions catalyzed by Group 9 metals; therefore, in this Section, we present only example of a synthesis of unsubstituted isocoumarin that follows a different strategy.
An unusual approach to isocoumarin synthesis is the metathesis reaction, which is commonly catalyzed by ruthenium carbene complexes widely known as the Grubbs catalysts. In a study of the cascade transvinylation of carboxylic acids/ring closing metathesis, Ostaszewski’s research group[246] mentioned, among other substrates, also the synthesis of unsubstituted isocoumarin by this method (Scheme 61).
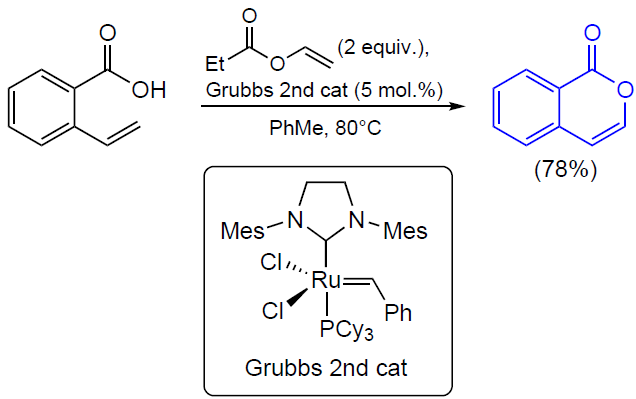
4. Conclusion
As it follows from the presented review, isocoumarins, like coumarins, their well-known isomers, have a great potential to be used as building blocks for the design of functional compounds for organic photonics, exhibiting useful photophysical properties depending on the nature of substituents. In particular, the introduction of electron-donating substituents in position 6 of the isocoumarin skeleton provides an almost 100% quantum efficiency of photoluminescence. Currently, there has been a pronounced increase in the research interest in isocoumarin derivatives as OLED components, in particular for high-efficiency devices. The development of this area is due to the ability of isocoumarins to exhibit the AIE effect and also to serve as building blocks for TADF emitters. In addition, review of the literature on the synthesis of isocoumarin derivatives provides the conclusion that the use of metal complex catalysis in the last decades has brought considerable progress in the development of synthetic approaches to the formation of the isocoumarin skeleton. Moreover, most advanced and effective metal-free organic methods are also based on the preliminary functionalization of available substrates by reactions catalyzed by transition metal complexes. The most atom-economic process is the oxidative coupling of aromatic acids with internal acetylenes or their analogues meant for the formation of the pyran ring, which proceeds via direct C – H activation of the ortho-position of the substrate. This statement suggests that a promising way for further use of 3,4-disubstituted isocoumarins is the design of photoswitchable compounds containing a diarylethene moiety, the synthetic equivalents of which are diarylacetylenes.[247-249]
Acknowledgements
This review was written with the financial support of the Russian Science Foundation (Project No. 19-73-20212). The authors are also grateful to the Ministry of Science and Higher Education of the Russian Federation (agreement No. 075-00276-25-00) and the National Research University Higher School of Economics for the access to electronic bases of scientific literature.
5. List of abbreviations and symbols
The following abbreviations and symbols are used in the review:
ϕF — fluorescence quantum yield,
ΔEST — energy difference between the first singlet and triplet excited states,
fw — fraction of water in the solvent mixture,
A — acceptor,
D — donor,
E — electrophile,
L — ligand,
M — metal,
S0 — ground state (of a molecule),
S1 — first singlet excited state (of a molecule),
T — triplet state (of a molecule),
ACQ — aggregation-caused quenching,
AIDF — aggregation-induced delayed fluorescence,
AIE — aggregation-induced emission,
bipy — bipyridine,
cat — catalyst,
cod — cycloocta-1,5-diene,
CPC — cetylpyridinium chloride,
Cp — cyclopentadienyl,
Cp* — pentamethylcyclopentadienyl,
CT — charge transfer,
Cy — cyclohexyl,
Cym — cymene,
DABCO — 1,4-diazabicyclo[2.2.2]octane,
dba — dibenzylideneacetone,
DBU — 1,8-diazabicyclo[5.4.0]undec-7-ene,
DCM — dichloromethane,
DFT — density functional theory,
DMAP — 4-dimethylaminopyridine,
DMA — dimethylacetamide,
dppe — 1,2-bis(diphenylphosphino)ethane,
EDG — electron-donating group,
EL — electroluminescence,
EQE — external quantum efficiency,
EQEmax — maximum external quantum efficiency,
ET — electron transfer,
EUE — exciton utilization efficiency,
EWG — electron-withdrawing group,
hfacac — hexafluoroacetylacetonate,
HLCT — hybrid local charge transfer,
HOMO — highest occupied molecular orbital,
hRISC — high-level reverse intersystem crossing,
ICT — intramolecular charge transfer,
IPr — 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ydene,
IQE — internal quantum efficiency,
LED — light emitting diode,
LE — locally excited state,
LOD — limit of detection,
LUMO — lowest unoccupied molecular orbital,
mCBP — 3,3'-di(9H-carbazol-9-yl)-1,1'-biphenyl,
Mes — 2,4,6-trimethylphenyl (mesityl),
MLCT — metal-to-ligand charge transfer,
NBS — N-bromosuccinimide,
NCS — N-chlorosuccinimide,
NCW — near-critical water,
NHC — N-heterocyclic carbene,
NTO — natural transition orbital,
OLED — organic light emitting diode,
PEG — polyethylene glycol,
Piv — pivaloyl,
PLQY — photoluminescence quantum yield,
PS-PEO — polystyrene – polyethylene oxide copolymer,
Py — pyridine,
RISC — reverse intersystem crossing,
rt — room temperature,
TADF — thermally activated delayed fluorescence,
TBAB — tetrabutylammonium bromide,
TEA — triethylamine,
TFA — trifluoroacetic acid,
tfa — trifluoroacetate,
Tf — trifluoromethanesulfonyl (triflyl),
TFE — trifluoroethanol,
TMS — trimethylsilyl,
Tol — tolyl,
TsOH — toluenesulfonic acid.
References














