


Keywords
Abstract
Catalytic C–N bond formation reactions play a crucial role in fine organic synthesis due to their wide application in the preparation of biologically active compounds and natural products. Recent advances in the modern theory of catalytic processes, particularly the development of the concept of dynamic catalysis, necessitate a re-evaluation and systematization of the vast experimental data accumulated in this field. This review provides a comprehensive analysis of amination reactions from the perspective of contemporary catalytic concepts. Special attention is given to the transformations of catalytic systems that lead to the formation of "cocktail"-type systems, as well as strategies for controlling these complex processes. Understanding these mechanisms is essential for the design of efficient catalytic systems applicable to cross-coupling reactions and other key synthetic transformations.
The bibliography includes 132 references.
1. Introduction
Transition metal catalysis of organic reactions is a key and vigorously developing area of modern chemistry.[1-5] The development or choice of a catalytic system is a cornerstone for the synthesis of compounds with high molecular complexity [6] and for the arrangement of industrial production.[7][8] This task is complicated by the high diversity of complexes containing transition metals and the absence of a direct relationship between the catalyst structure and key characteristics such as activity, selectivity, tolerance to various functional groups, etc.[9][10] The common views of a catalyst as a static site invariable during the reaction often do not reflect the real situation. Therefore, there is increasing interest in the formation of a comprehensive and consistent concept of catalysis that would explain and integrate the large body of empirical data accumulated over the past decades.
In the early stage, a considerable contribution to the development of the theory of heterogeneous catalysis was made by H.S.Taylor and A.A.Balandin. Taylor[11] put forward the hypothesis that atoms with lower coordination numbers located at the edges and corners of the crystal are the most active sites of the metal surface. Balandin[12][13] developed the multiplet theory of catalysis, in which catalytic processes were considered at the atomic level. According to this theory, for a chemical reaction to proceed, two conditions must be fulfilled: structural correspondence between the catalyst surface and the geometry of reacting substrates and energy correspondence between the sum of energies of the reacting bonds and the sum of energies of bonds between the reacting atoms and the catalyst surface.
The subsequent development of the theoretical views and accumulation of experimental data provided the understanding that active sites are not invariable during the reaction.[14][15] This gave rise to the concept of ‘cocktail’-type catalysts, which was first formulated by Ananikov et al.[16]
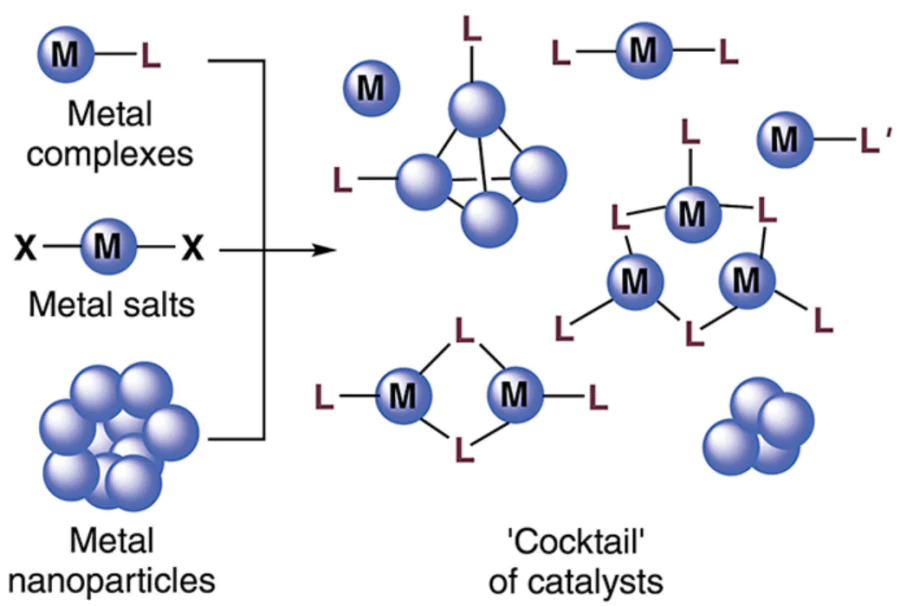
According to this concept, metal-containing compounds undergo various transformations to form a complex mixture of catalytically active sites.[17-20] In the reaction system, transition metal complex is capable of aggregation to increase the nuclearity and form species comprising 2, 3 ... n metal atoms (Fig. 1). Elimination of the auxiliary ligand from the metal coordination sphere gives rise to so-called ligandless complex, in which the metal is coordinated only by solvate molecules or reactants. Ligandless systems tend to aggregate to give metal clusters and nanoparticles (NPs), which can also act as active catalysts. The aggregation of ligandless systems with ligand-containing complexes or interaction of free ligands with metal aggregates leads to the formation of modified clusters and nanoparticles. In addition, ‘cocktail’-type systems are also formed upon mixing of two or more auxiliary ligands.
![[{"id":"0JlPFP4M_g","type":"paragraph","data":{"text":"Dynamic transformations of metal-containing systems in catalytic reactions"}}]](/storage/images/resized/iyRyu2bAOU8wdF8oq3O0RrNfEaB0X1wUnSO3iZa4_xl.webp)
The chemistry of ‘cocktail’-type catalysts is constantly expanding, which is facilitated by the development of instrumentation for the detection and investigation of a mixture of nano-sized and molecular components of the catalytic system. This is successfully done using electron microscopy,[21][22] solid-state NMR spectroscopy,[23, 24] mass-spectrometry,[25] and molecular modelling.[26] Currently, the dynamic catalysis was established for Mizoroki – Heck,[27][28] Suzuki – Miyaura,[29] hydrogenation,[30][31] hydrosilylation,[32][33] and many other[18] reactions.
The problem of transformation of catalytic systems in the C – C and C – X bond formation reactions receives a lot of attention in the scientific literature. There are two reviews published in 2004 and 2006 before the concept of dynamic catalysis appeared.[14][15] These publications consider the transformations of metal-containing compounds from the standpoint of the search for the ‘true catalyst’ responsible for the formation of the target product. The developed concept of the ‘cocktail of catalysts’ increased the relevance of studying the transformations of the catalyst precursor, and the problem formulation changed from determination of the type of catalytically active compound to determination of the composition of a mixture of active complexes and species in the reaction system.[17] The later reviews on the ‘cocktail’-type systems were devoted to C – C, C – S, and C – Se bond formation reactions[18] and particular examples of transformation of Pd complexes with N-heterocyclic carbenes (NHC),[34] and also to the history of development of the theoretical views in catalysis.[19] The reactions resulting in the C – N bond formation received much less attention.
The purpose of this review is to analyze the information gained over the last 25 years on the chemistry of C – N bond formation in the light of the concept of dynamic catalysis. Reactions of this type are of considerable interest as they are widely used in the synthesis of natural products and pharmaceuticals and in materials science.[35-41] Particular attention is paid to the evolution of catalytic systems and strategies for the control of these transformations.
2. Dynamic catalysis by transition metal complexes in the C – N bond formation reactions
2.1. Phosphine complexes of transition metals
Transition metal phosphine complexes are among the most common types of catalytic systems. A phosphine ligand may be a part of the catalyst precursor or may be added to the metal-containing compound. In the latter case, the phosphine complex is formed directly in the reaction system. The possibility of using these ligands as additives considerably simplifies selection of the optimal catalytic system by varying the nature and amount of the phosphine. The catalytic systems based on phosphine complexes often form a ‘cocktail’ of active sites. This process starts with the elimination of the phosphine ligand from the metal coordination sphere via dissociation, C – P coupling (formation of the phosphonium salt), or oxidation to form phosphine oxide.
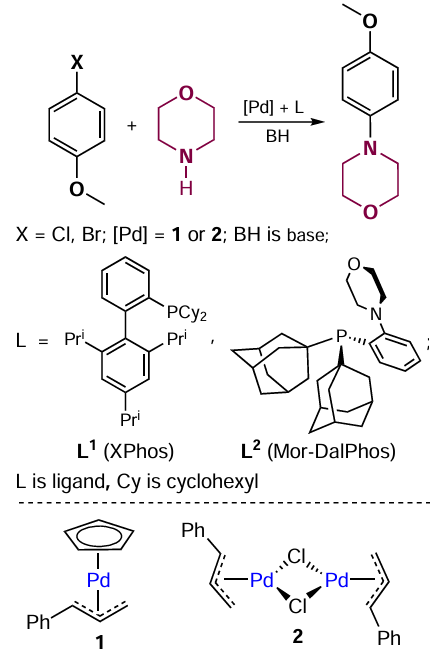
Catalytic systems based on palladium compounds, Pd2(dba)3 (dba is dibenzylideneacetone), Pd(OAc)2 , and allylic complexes 1 and 2 (Scheme 1) in the presence of added phosphine ligands such as PBut3, XPhos (dicyclohexyl[2',4',6'-tris(propan-2-yl)[1,1'-biphenyl]-2-yl]phosphine) and Mor-Dalphos (di(1-adamantyl)-2-morpholinophenylphosphine) have shown high activity in the Buchwald – Hartwig reaction involving 1-bromo-4-methoxybenzene and morpholine. The highest activity was found for complex 1 in combination with 2 equiv. of PBut3.[42] The addition of phosphines XPhos (L1) or Mor-DalPhos (L2) to solutions of compounds 1 and 2 resulted in the formation of [Pd(XPhos)] or [Pd(Mor-DalPhos)] complexes (see Scheme 1). The steric crowding prevented the arrangement of two phosphine ligands in the metal coordination sphere. The compounds [Pd(XPhos)] and [Pd(Mor-DalPhos)] are unstable and dissociate to release palladium metal and the free ligand.[43]
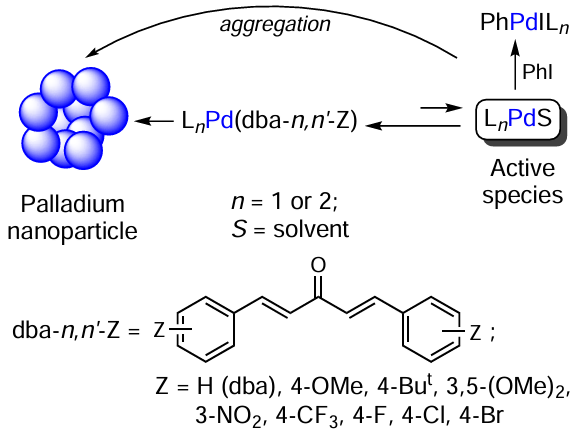
The [Pd2(dba-n,n'-Z)3] and [Pd(dba-n,n'-Z)2] complexes together with phosphines or N-heterocyclic carbenes were used as catalyst precursors in the Mizoroki – Heck, Suzuki–Miyaura, and Buchwald – Hartwig reactions (Scheme 2). Under these conditions, catalysts can undergo transformations involving dba cleavage and palladium aggregation. Palladium complexes with the dba-n,n'-Z ligands containing electron-donating Z-substituents showed higher catalytic activity in most reactions. The substituent effect was attributed to weakening of the metal–ligand bond, which increased the concentration of LnPd0S (S is the solvating ligand) actively reacting with aryl halides at the oxidative addition step. The additional role of the dba-n,n'-Z ligand was to retard the palladium aggregation. Thus, variation of Z-substituents enabled the fine tuning of the catalyst by changing the balance between various catalyst transformation processes.[44]
The commercially available [Pdx(dba)y] complexes can initially represent a ‘cocktail’-type catalyst and contain up to 40% palladium nanoparticles (Pd-NPs), which was demonstrated in relation to a number of samples derived from different sources.[45] The NP size varied in the 10 – 200 nm. A method for determining the purity of the original commercial complexes was proposed in the same study.[45] The [Pdx(dba)y] samples exhibited different catalytic activities in the Buchwald – Hartwig amination of 1-bromo-4-methoxybenzene. The yields of the product differed over very broad ranges from 12 to 96% (Table 1).[46]
![Yields of the products of amination of 1-bromo-4-methoxybenzene with aniline catalyzed by [Pd<sub>x</sub>(dba)<sub>y</sub>] samples received from various commercial sources](/storage/images/resized/IxfacZGprPujn6TBpdINgQe0CxKtKwsXjJnOE66L_xl.webp)
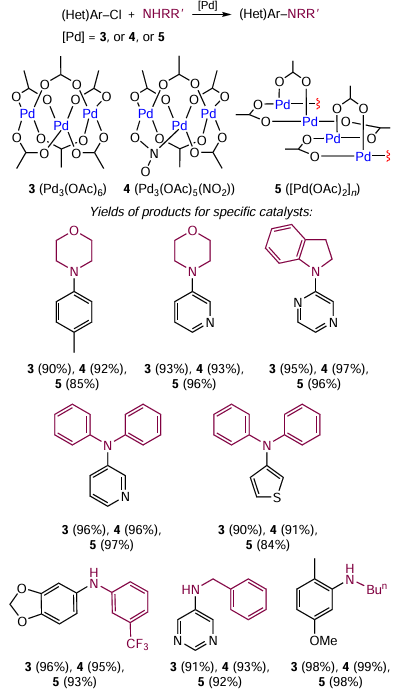
Similar studies were carried out for commercial palladium acetate, which exists as the trimer Pd3(OAc)6 (3) usually containing considerable amounts of the complex Pd3(OAc)5(NO2) (4) and polymer [Pd(OAc)2]n (5). Colacot and co-workers [47] prepared salt 3 in a pure state by a special procedure and compared its catalytic performance in the C – N bond formation with those of 4 and 5 (Scheme 3). All three compounds showed similar performances at 100°C, including polymer 5, which is insoluble in most organic solvents. Heating of 5 in toluene at 100°C did not induce changes in the polymer structure, whereas the addition of 1 equiv. of morpholine to the solution resulted in the deposition of palladium metal. Thus, commercially available palladium acetate samples are mixtures of compounds, three of which are capable of catalyzing C – N cross-coupling reactions.[48]
Another example of a dynamic catalytic system based on palladium complexes was described by Baird and co-workers.[49] As an efficient catalyst for the Buchwald – Hartwig reaction, the authors used complex 1, which was converted to complexes of higher nuclearity during the reaction in the presence of 1 equiv. of triphenylphosphine (Scheme 4). This gave rise to σ-allylic complex 6 and dimeric complex 7, which are presumably catalytically active intermediates (see Scheme 4).
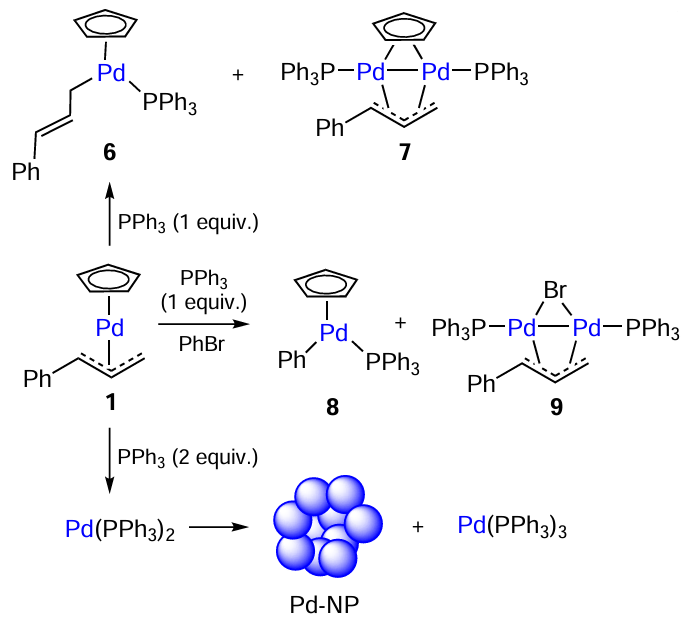
In the presence of excess bromobenzene, the reaction follows a different pathway giving two stable compounds 8 and 9, which show a moderate catalytic activity in the amination of 1-bromo-4-methoxybenzenemorpholine. In the presence of 2 equiv. of PPh3, complex 1 is reduced to [Pd(PPh3)2]. This compound is unstable and disproportionates to give [Pd(PPh3)3] and palladium metal.[50]
The three-coordinate complex can also be obtained by adding 3 equiv. of phosphine ligand to a solution of compound 1. However, this method is not applicable to all types of phosphines. In particular, it was possible to isolate complexes [Pd(PCy3)3] and [Pd(PMeBut2)3], whereas compound [Pd(PBut3)2] remained two-coordinate even in the presence of the corresponding free ligand.[51] Catalytic systems based on complex 1 in combination with phosphine ligands of various electronic and steric natures were also investigated for a number of other cross-coupling reactions.[52][53]
Phosphine complexes of palladium and nickel often tend to aggregate during the reactions of C – N bond formation. The nuclearity of metal-containing species can vary from small clusters such as dimers (M2) and trimers (M3) to large nanoparticles. The aggregation proceeding rapidly and in an uncontrolled manner may result in the formation of metal black and in the deactivation of the catalytic system. In some cases, clusters can be stabilized by ligands, which prevent their further aggregation. For example, a simple catalytic system, Pd(OAc)2 with two equivalents of PPh3, is converted to palladium(I) dimeric complex [Pd2(μ-PPh2)(μ2-OAc)(PPh3)2] containing acetate and phosphide bridging ligands. In the presence of halides, this dimer can be further converted to a trimer structure involving phosphide and halide bridging ligands.[54]
It is known that mono-, di-, and trimeric palladium complexes are catalytically active towards cross-coupling reactions.[55] The PdI dimeric systems can be formed both upon comproportionation between palladium(0) and palladium(II) complexes and upon leaching of active complexes from the nanoparticle surface.[56] Some palladium clusters are sufficiently stable to participate in catalytic cycles with their structure being preserved. For example, the Pd3 cage trimer-based catalyst exhibited high activity in the cycloisomerization of 2-phenylethynylaniline, with the trimer structure being preserved throughout the reaction, which was confirmed by NMR spectroscopy data and by extended X-ray absorption fine structure (EXAFS) spectrum.[57]
Generally, metal clusters represent an important component of ‘cocktail’-type catalytic systems, which is formed directly in the reaction medium and makes a considerable contribution to the substrate conversion.[58]
When the Pd + PR3 catalysts are used, the reaction system usually contains both monoligand and biligand complexes, which have different activity. The ratio of these compounds depends on the steric factor described by the percentage of the occupied volume of the metal coordination sphere (buried volume, Vbur). The activity of Pd complexes in the presence of phosphines in the reaction of bromobenzene with N-methylaniline was found to correlate with the Vbur value of the phosphine ligand. There is a threshold value of Vbur below which the reaction is critically retarded. This is probably due to the predominance of more active monoligand complexes in the case where bulky phosphines are used.[59] In the case of PBut3, the role of steric factor in the switching of the reaction mechanism to the monoligand one was additionally confirmed by quantum chemical simulation of the full catalytic cycle of the Buchwald – Hartwig reaction.[60]
The phosphine dissociation is not the only way to remove the ligand from the coordination sphere of the metal. Phosphines also tend to be oxidized to give phosphine oxides. Often, this process already takes place at the step of activation of the catalyst precursor, resulting in the formation of a mixture of metal-containing complexes. Triphenylphosphine reduces palladium chloride and acetate in the presence of various bases; most often, this requires 1 equiv. of the ligand.[54][61][62]
The phosphine oxidation takes place directly in the coordination sphere of the metal. Palladium acetate reacts with triphenylphosphine to give the [Pd(OAc)2(PPh3)2] complex (Scheme 5, part A). This product undergoes the P – O coupling reaction to give the [AcO – PPh3]+ cation and the Pd0(PPh3) complex.[63][64]
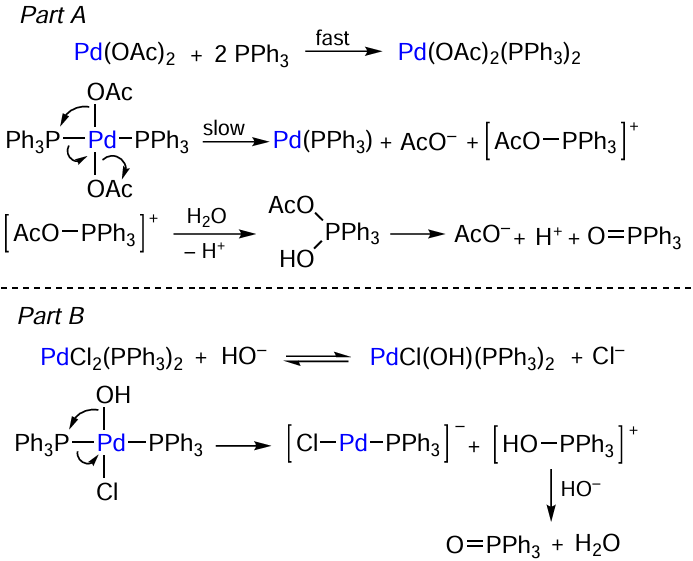
In the case of palladium halide, a base molecule serves as the reducing agent for the palladium complex.[65][66] The reaction starts with the ligand exchange between the halide anion and OH–; this is followed by P – O coupling yielding triphenylphosphine oxide via the formation of the cationic [HO – PPh3]+ intermediate (see Scheme 5, part B).
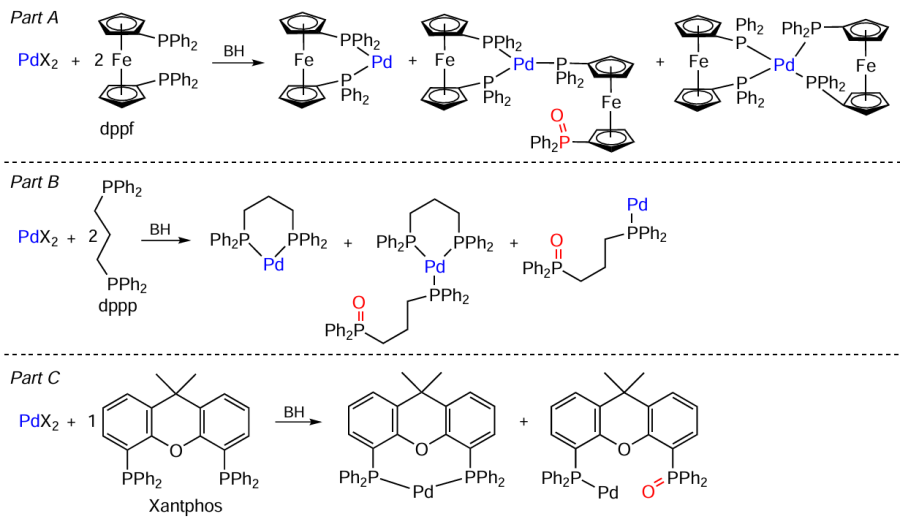
The transformation of phosphine ligands may be responsible for the formation of palladium nanoparticles. Bidentate phosphines are oxidized to monophosphine oxides, which retain the ability to coordination.[67][68] For example, in the presence of a base, the PdX2 + dppf (2 equiv.) catalytic system [where X = Cl, OAc; dppf is 1,1'-bis(diphenylphosphino)ferrocene] is converted to a mixture of palladium complexes (Scheme 6, part A), with the composition of this mixture being considerably dependent on the nature of the base. In the case of carbonates, no complexes containing phosphine oxide ligands are formed. The [PdCl2(MeCN)2] + dppp (2 equiv.) catalyst [dppp is 1,3-bis(diphenylphosphino)propane] is stable at 60°C in DMF; however, upon the addition of carbonate, it is reduced to a mixture of Pd0 complexes containing different numbers of phosphine and phosphine oxide ligands (see Scheme 6, part B). Similarly, in the presence of sodium acetate or caesium carbonate at 60°C, the Pd(OAc)2 + Xantphos catalytic system [Xantphos is 9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphine)] is converted to a mixture of Pd0 complexes (see Scheme 6, part C).[69]
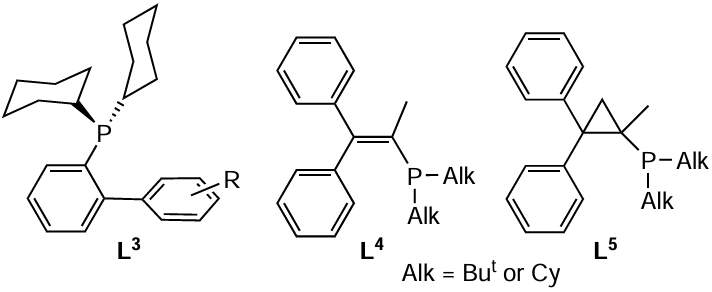
Owing to certain structural features, phosphine ligands are stable to oxidation. For example, dialkylbiarylphosphines L3 do not react with molecular oxygen. There are two hypotheses to explain this phenomenon: (1) interaction between the phosphorus lone pair (LP) of electrons and the π-electrons of the aromatic ring remote from phosphorus; (2) low stability of the pre-reaction complex of phosphine and oxygen due to steric repulsion. In any case, the oxidation of these phosphines should be preceded by the rotation of the dihedral angle around the P – CAr bond,* the potential barrier of which is estimated as 13.6 – 24.6 kcal mol–1.[70]
Other examples of oxidation-resistant phosphines are diphenylvinyl- and diphenylcyclopropylphosphines L4, L5. These ligands are air-stable and their palladium complexes effectively catalyze the Suzuki – Miyaura and Buchwald – Hartwig reactions.[71]
The phosphine ligand can be removed from the coordination sphere of the metal via reductive elimination giving the C – P-bond (or C – P coupling); this results in the corresponding phosphonium salt (Scheme 7). This reaction competes with the target reaction in the Buchwald – Hartwig catalytic cycle. Transition metals are known to catalyze the reaction between phosphines and aryl halides, thus promoting the formation of quaternized products.[72] The C – P coupling is reversible and, therefore, phosphonium salts can be used as precursors of phosphine ligands in the C – N cross-coupling.[73-76]
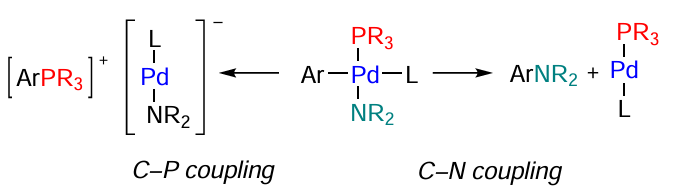
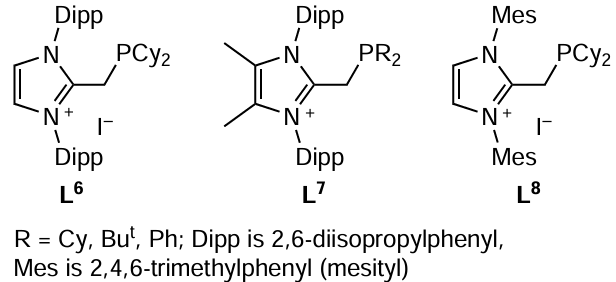
In the framework of dynamic catalysis, transformation of active sites takes place, which is favourable for the target reaction, because a required compound meant for the interaction with a particular substrate is present in the ‘cocktail’ of active species.[77] However, a drawback of these systems is the difficulty of their reuse (recycling). This problem was addressed by the development of imidazolium phosphines L6 – L8, water- and air-stable cationic ligands for metal complex catalysis. Owing to the ionic nature of these ligands, the stability of catalysts to aggregation increases, which opens up the possibility of catalyst recycling. Imidazolium oxides are stable to oxidation: after 8 cycles of the use of a palladium catalyst with this ligand, the yield of the formed phosphine oxide did not exceed 5%. Palladium complexes with imidazolium phosphines L6 – L8 effectively catalyzed the amination of 4-chloro-2-methylquinoline and 2-bromo-1,3,5-trimethylbenzene with 1-benzylpiperazine.[78]
*Here and below, the subscript denotes the moiety to which the carbon atom belongs.
2.2. Transition metal complexes with N-heterocyclic carbene ligands
Metal complex catalysts with N-heterocyclic carbene ligands (M + NHC) are widely used in cross-coupling reactions, in particular in C – N bond formation.[79-81]The binding energy between the NHC ligand and the metal is much higher than that in phosphine complexes.[82] Since dissociation of the M – NHC bond is difficult, previously it was believed that the [M – NHC] complexes are stable against degradation. However, later it was found that there are quite a few reactions in which NHC is eliminated from the metal coordination sphere and a ‘cocktail’-type catalytic system is formed.[34]
It was shown experimentally that the [M – NHC] complexes containing alkyl or aryl ligands are rather unstable and tend to undergo reductive elimination giving the CR – NHC bond.[83] Complexes of this type are formed as intermediates within the catalytic cycle of the Buchwald – Hartwig reaction and thus they can be transformed into ligandless compounds and palladium nanoparticles.
The CR – NHC coupling reaction was studied considering the model cationic palladium complexes 10 – 12 with imidazole (1,3-dimethylimidazol-2-idene) and cyclooctadiene (cod) ligands (Scheme 8). Cationic complex 10 has three types of Pd – C bonds, those with the σ-methyl group, π-alkene moiety, and carbene. Online monitoring of the decomposition of this complex in DMSO-d6 and in CDCl3 was performed using 1H NMR spectroscopy. In both solvents, the Pd – CMe signal disappeared, and a new signal corresponding to the azolium salt appeared at 2.56 (DMSO-d6) and 2.64 ppm (CDCl3). The formation of the CR – NHC coupling product was confirmed by mass spectrometry and elemental analysis.
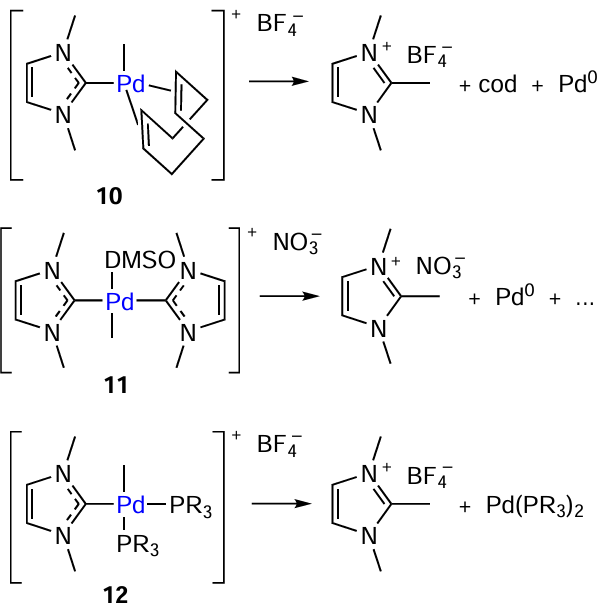
The decomposition of bis-imidazole complex 11 was studied in a similar way. This compound is more resistant to reductive elimination and slowly decomposes in DMSO-d6 at 120°C to give Pd0 complex and a mixture of unidentified organic products. Complex 12, containing NHC and phosphine ligands, underwent the CR – NHC reductive elimination, while the C – P coupling reaction does not take place. The CR – NHC coupling was also detected in the case of nickel and platinum complexes.[84][85]
The calculated CPh – NHC coupling reaction pathway for the compound [(NHC)Pd(Ph)(Br)(DMF)] was analyzed in terms of the Bader’s atoms in molecules theory,* which showed that the atomic rearrangement trajectory corresponds to the insertion of the phenyl ligand into the Pd – NHC bond rather than to the classical C – C coupling. This reaction is exergonic, with its potential barrier being low for most of catalytic processes. As a result of the CR – NHC coupling, the weakly bound solvating ligand leaves the coordination sphere of palladium, and polarization of the complex takes place with a gradual increase in the charge on the [CPh – NHC] structural moiety.[86] The solvent effect in the reaction was evaluated by comparing the molecular dynamic simulation data for this reaction in the gas phase and in the condensed state. According to the results, the solvent prevents the elimination of the solvating ligand and, hence, in the condensed state, the CR – NHC coupling proceeds in a four-coordinate complex. In the gas-phase version, a three-coordinate palladium complex reacts. As a consequence, the potential barrier of the reaction is higher in the condensed phase (17.7 vs. 21.8 kcal mol–1). In addition, a polar solvent stabilizes the CR – NHC coupling product by decreasing the Gibbs free energy of the reaction.[87]
In the presence of oxygen-containing bases, the catalyst precursors [(NHC)M(X)2L] (M = Pd or Pt) are converted to azolones O=NHC and ligandless M0 complexes. The oxidation of the NHC ligand by the O – NHC coupling mechanism is relevant for quite a few catalytic processes, particularly for the Buchwald – Hartwig reaction. Unlike the CR – NHC coupling, this reaction can take place in the catalyst precursors, resulting in their activation. The most probable mechanism of the O – NHC coupling is the reversible exchange of the anionic (X) or neutral (L) ligand with the RO– group followed by reductive elimination of the [RO – NHC]+ cation and formation of the M0 complex (Scheme 9). The NHC ligand acts as a two-electron intramolecular reducing agent, while the anion of the base serves as a source of the oxygen atom.
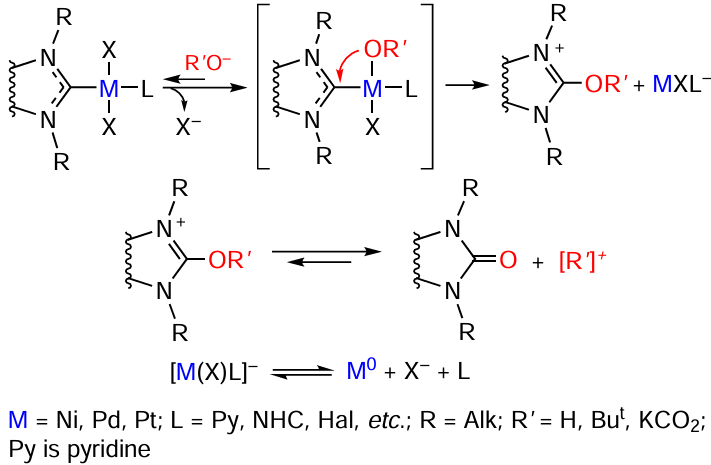
An alternative mechanism for the formation of azolones includes the nucleophilic attack of RO– on the carbene carbon atom of NHC. The second step is dissociation of the M – C bond accompanied by the release of M0 compound. The nucleophilic attack is hindered in the case of sterically bulky NHC ligands, thus reducing the probability that the reaction would proceed by this pathway. Among PdII, PtII, and NiII complexes for which the O – NHC coupling was found, palladium(II) compounds are most reactive.[88]
A detailed study of the transformation of the catalytic system based on monomeric and dimeric palladium complexes with the NHC ligand showed that in the Buchwald – Hartwig reaction, the Pd – NHC bond is cleaved without the loss of catalytic activity (Scheme 10). The ligand elimination occurs via H-NHC, CR – NHC, and O – NHC coupling reactions; the corresponding products were detected in the reaction mixture by high-resolution mass spectrometry. These processes trigger the formation of a ‘cocktail’ of active species, among which there are palladium clusters and nanoparticles. Metal aggregates detected in the reaction mixture by electron microscopy possessed catalytic activity. Leaching from the metal surface was confirmed by hot filtration experiments. Thus, dynamic catalysis is involved in the amination reaction. The [Pd – NHC] complexes are converted into mixtures of palladium compounds, including molecular complexes and nano-sized systems, which occur in dynamic equilibrium with one another (Scheme 11).[89]
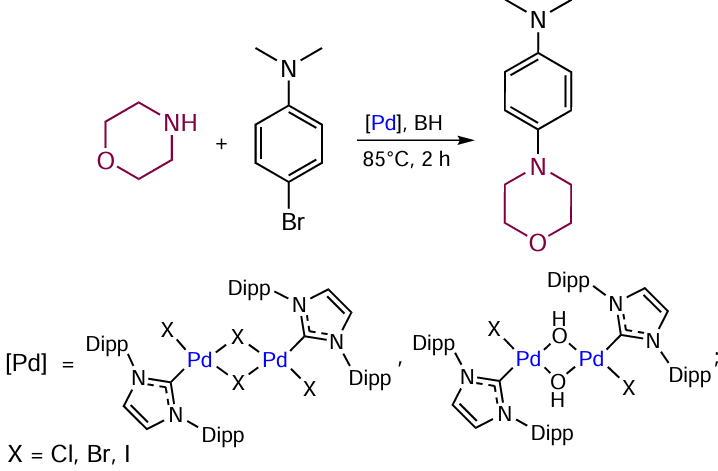
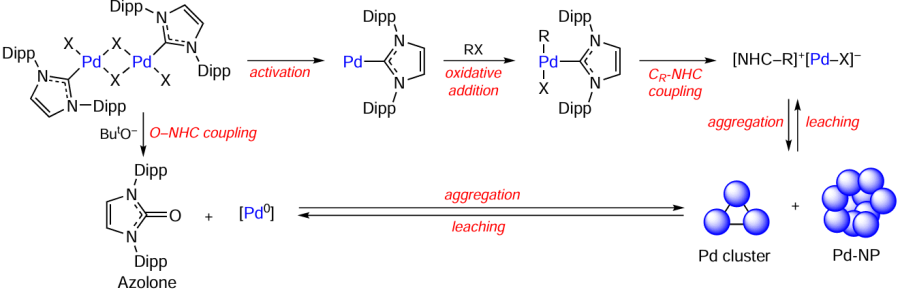
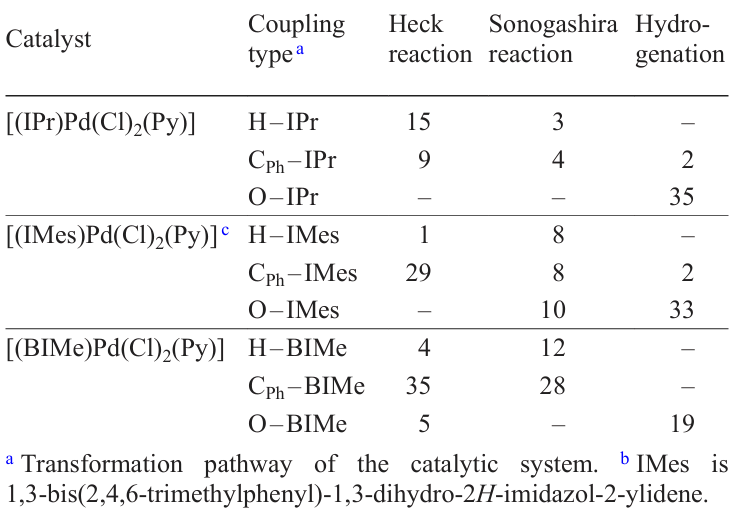
The contributions of the H – NHC, CR – NHC, and O – NHC couplings to the transformation of the Pd + NHC catalysts were quantitatively estimated by electrospray ionization mass spectrometry (Scheme 12). The essence of the method is in the use of deuterated standards for the organic coupling products, make it possible to plot the kinetic curves for each of these processes under conditions of cross-coupling and hydrogenation reactions. According to the results, 7 to 44% of the Pd + NHC catalyst are converted to the ligandless form in the Heck, Sonogashira, and transfer hydrogenation reactions (Table 2). The contributions of various processes of catalyst conversion depend on both the type of catalytic reaction and the nature of the NHC ligand. For example, transfer hydrogenation reactions give mainly azolones O=NHC. In the Heck and Sonogashira reactions, the Pd + IPr catalyst [IPr is 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene] is more susceptible to the H-IPr coupling, while the [Pd – BIMe] complex (BIMe is 1,3-dimethyl-1H-benzo[d]imidazol-2-ylidene) is converted to the Ph – BIMe product. The method is efficient for small catalyst loadings as low as 0.005 mol.%.[90]
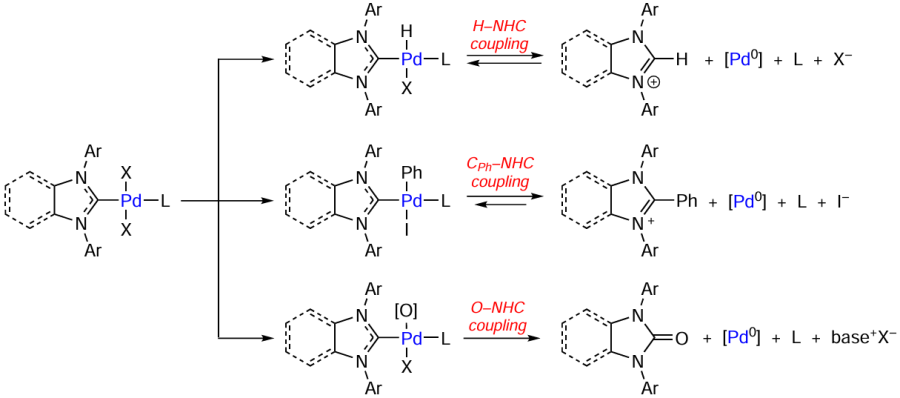
According to the results of quantum chemical calculations, the intermediates of cross-coupling reactions formed upon the oxidative addition of aryl halides to Pd0 complexes are easily involved in CPh – NHC coupling reactions.[91] The reaction product, the azolium salt [Ph – NHC]+X–, acts as a stabilizing agent for colloidal Pd nanoparticles, thus preventing precipitation of the palladium black and catalyst deactivation (Scheme 13). In systems of this type, the activity of the [Pd – NHC] complexes is mainly determined by their ability to decompose with metal–ligand bond cleavage and formation of clusters and nanoparticles. The formation of the ‘cocktail’-type catalysts is considerably affected by the nature of the coordinated halogen and type of NHC ligand.[27]
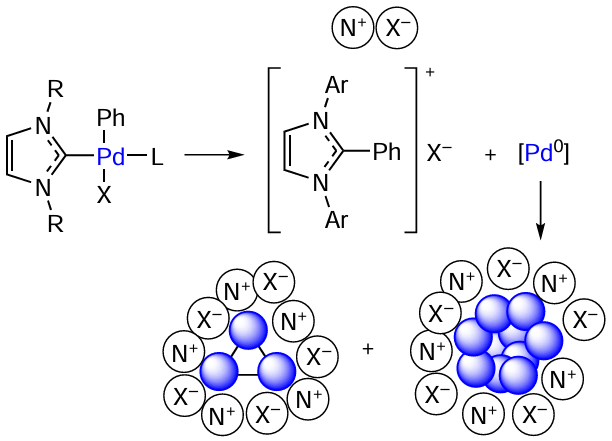
The effect of the NHC ligand on the CPh – NHC coupling was studied by molecular modelling. The [(NHC)Pd(Ph)(I)DMF] complexes undergo reductive elimination to give the products [Ph – NHC]+[Pd – I]–. These products are ion pairs in which the palladium complex anion is stabilized by an NHC-containing cation. Subsequently, the [Ph – NHC]+[Pd – I]– ion pairs decompose to azolium salts and low-coordinate Pd0 complexes, which, in turn, either aggregate or retain the molecular form and react with aryl halide according to the oxidative addition pathway. The calculated activation energies of the CPh – NHC coupling vary in the range of 17.9 – 25.1 kcal mmol–1 depending on the type of the ligand Scheme 14; the activation energies and reaction energies (in square brackets) in kcal mol–1 are given].
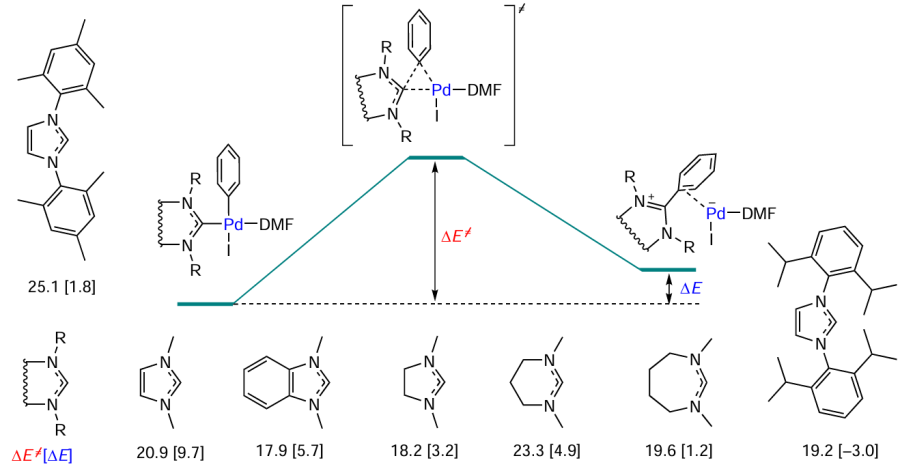
In most amination reactions, the temperature conditions make it possible to overcome these potential barrier values. The composition of the mixture of active compounds in the system substantially depends on the thermodynamic factor. The total energy of the CPh – NHC coupling ranges from –3.0 to 9.7 kcal mol–1. The reaction is more preferable for unsaturated NHC that contain bulky substituents. The obtained data reveal the mechanism of formation of ‘cocktail’-type systems. The use of the [Pd – NHC] complexes as catalyst precursors does not guarantee that the reaction would be homogeneous; therefore, thorough analysis of the nature of active species is needed in each particular case to determine the type of catalysis.[92]
Another way to cleave the bond between a transition metal atom and an NHC ligand is the N – NHC coupling. A necessary condition for this reaction is the presence of an amine in the reaction system. In the Buchwald – Hartwig reaction, amine is a starting substrate. In other cross-coupling reactions and the Mizoroki – Heck reaction, amine is sometimes used as a base.
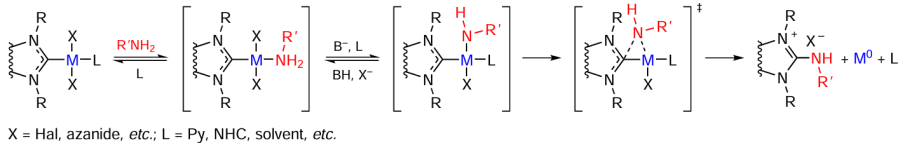
The Ni and Pd complexes [MII – NHC] react with primary aromatic and aliphatic amines in the presence of strong bases to give azole-2(5)-imines and M0 complexes. The reaction follows the mechanism of reductive elimination of NHC and azanide (N-deprotonated amine). In the first step, the amine is coordinated to the metal centre (Scheme 15). Deprotonation of the coordinated amine affords a key intermediate containing an azanide group as a ligand. The third step is reductive elimination to form a bond between the carbene carbon atom of NHC and the nitrogen atom. In the presence of potassium tert-butoxide, the N – NHC and O – NHC coupling reactions compete with each other. The predominant pathway is determined by the stoichiometric ratio between RNH2 and ButOK. The possible alternative mechanisms for this transformation are nucleophilic addition of amines to azalones and direct nucleophilic attack of the azanide on the carbene carbon atom. However, in the authors’ opinion, these reactions are unlikely.[93] In the case where the N – NHC coupling is undesirable, it can be suppressed by using sterically hindered NHC ligands.
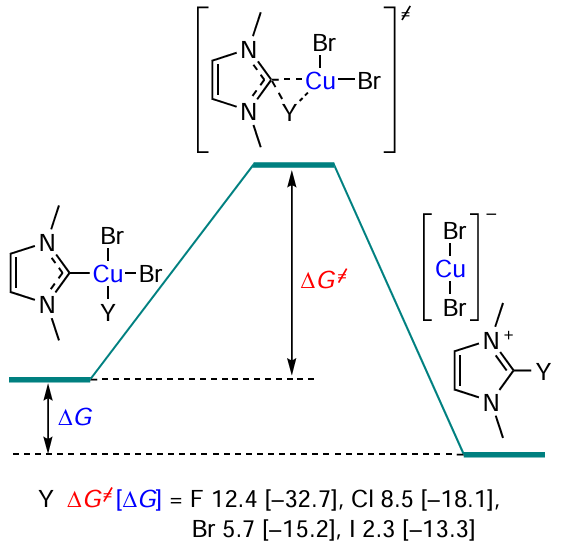
The C – N bond formation reactions are also carried out in the presence of copper complexes.[94-96] Compounds with the CuIII – NHC bond enable a specific Hal – NHC coupling reaction. The free activation energies of the reaction substantially depend on the halogen nature Scheme 16; the activation free energies and the reaction free energies (in brackets) in kcal mol–1 are given]. According to the results of quantum chemical calculations, stability of the [(NHC)Cu(X)(Br)2] complexes against the reductive elimination decreases in the series X = F > Cl > Br > I. It is noteworthy that the free energy of the reaction is inversely correlated with the potential barrier: the more drastic the conditions needed for the reaction to occur, the more thermodynamically favourable the reaction. The key factor determining the activation free energy of the Hal – NHC coupling is the strength of the Cu – Y bond. The correlation coefficient between the calculated values of activation free energies and the homolytic bond energy is 0.98.[97]
The synthetic protocol based on the self-activated catalyst generated from precursors, NiCl2Py2, IPr · HCl, and sodium tert-butoxide, makes it possible to carry out C – N coupling of low-reactive aryl chlorides with (hetero)aryl- and alkylamines. Under these conditions, the Ni + NHC catalytic systems are transformed by several competing mechanisms. According to mass-spectrometric study, the reaction mixture contained various NHC-containing organic products of catalyst transformation (Scheme 17).
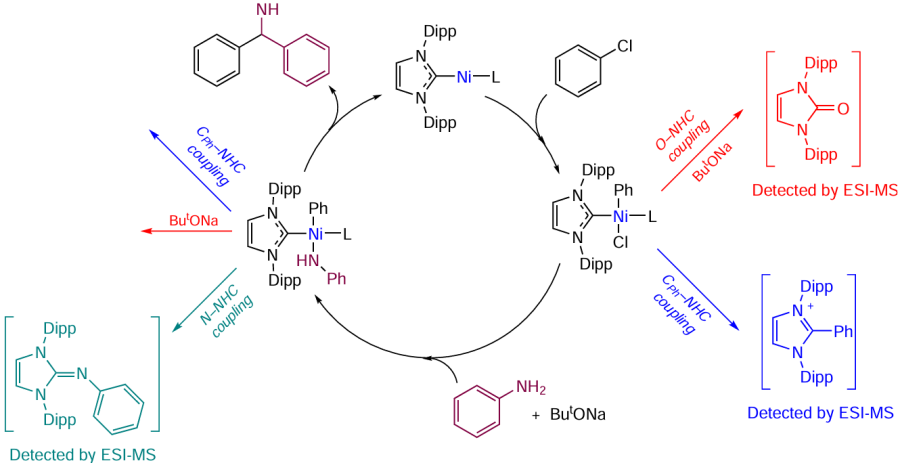
These transformations can occur at different steps of the amination catalytic cycle. Presumably, the first step of the oxidative addition of aryl chloride involves the [(NHC)Ni0L] complex and gives the intermediate [(NHC)Ni(Ph)(Cl)L]. This is followed by exchange of the halogen ligand for an amide group to give the intermediate [(NHC)Ni(Ph)(NHPh)L]. The C – N coupling product is formed at the final reductive elimination step. The side reactions accompanied by the removal of the NHC ligand from the Ni coordination sphere may take place in both of these intermediates, resulting in the gradual formation of ligandless nickel compounds. The latter are prone to aggregation to give inactive nickel nanoparticles, as was found by electron microscopy.[98] The intermediate [(NHC)Ni(Ph)(Cl)L] undergoes the CPh – NHC and O – NHC couplings to afford the corresponding azolium salt and azolone. The presence of both products in the reaction mixture formed by the Buchwald – Hartwig reaction was confirmed by mass spectrometry. The same transformations are inherent in the intermediate [(NHC)Ni(Ph)(NHPh)L], which, in addition, can undergo the N – NHC coupling reaction. The imine resulting from this reaction was also detected in the system. These results clearly demonstrate the dynamic nature of catalysis in C – N bond formation reactions.
An increase in the size of the NHC ligand in combination with the use of a mild reducing agent is a good method to suppress the O – NHC, N – NHC, and C – NHC couplings, which was demonstrated in relation to the Buchwald – Hartwig reaction involving various (hetero)arylamino-1,2,4-triazoles and aryl chlorides or bromides (Scheme 18). The [(NHC)Pd(X)2Py] complexes with various NHC ligands were used as catalysts. In the case of NHC with small-size substituents (methyl, butyl, and benzyl groups) at the heterocyclic nitrogen atom, metal complexes did not show catalytic activity towards the C – N coupling, and the initial complex decomposed with precipitation of palladium black. The transformation of the catalytic system proceeded via reductive elimination of NHC to give Pd0 ligandless systems unstable under the reaction conditions.
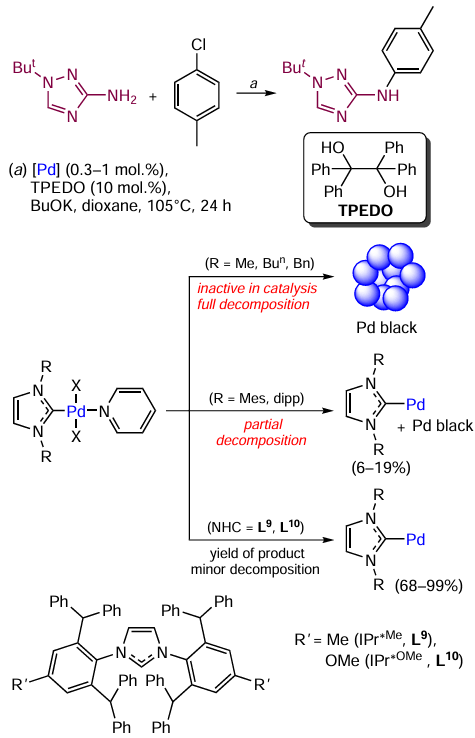
An increase in the steric bulk of the N-substituent in NHC was favourable for increasing stability of the catalyst. Complexes containing mesityl- and diisopropylphenyl-substituted ligands had a moderate catalytic activity. The highest yields of products were obtained when compounds with sterically hindered ligands L9 (IPr*Me) and L10 (IPr*OMe) were used. These catalytic systems had the highest stability, as the O – NHC, C – NHC, N – NHC, and H-NHC coupling reactions were suppressed. The key factor influencing the stability of complexes was the use of TPEDO (1,1,2,2-tetraphenylethane-1,2-diol), a mild reducing agent, which reduces the catalyst precursor, but does not participate in the O – NHC coupling.[99]
The addition of the electron-withdrawing substituent RSO2 to the carbon atom of the heterocycle also increases the steric bulk and modifies the electronic properties of NHC. The NHC-RSO2 ligand can be synthesized in a high yield by selective oxidation of accessible air-stable 4-RS-imidazolium salts with 30% hydrogen peroxide in the presence of ammonium heptamolybdate. The [Pd – IPr-RSO2] complexes obtained by C(2) – H-palladation of 4-RSO2-functionalized imidazolium salts showed higher activity in amination than the unsubstituted analogues [Pd – IPr]. The 4-aryl(alkyl)sulfonic group decreases the donor properties of NHC only to a minor extent; however, it substantially increases the π-acceptor properties and somewhat increases the buried volume of the ligand. The catalytic system based on modified NHC was also relatively stable in other cross-coupling reactions.[88]
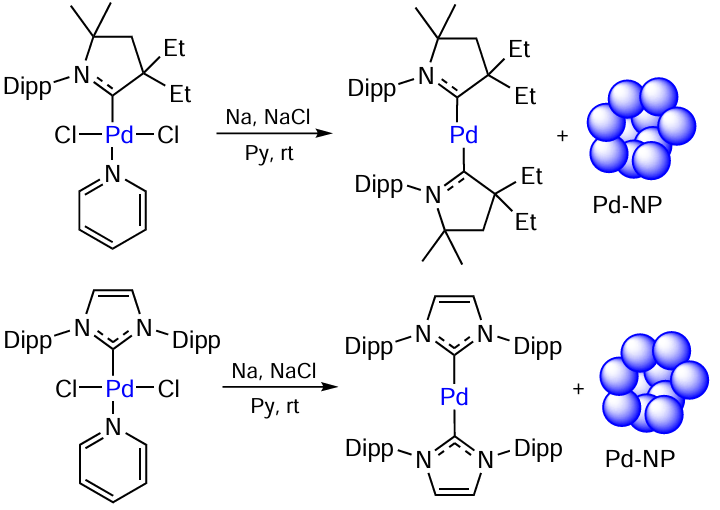
The transformations of the PdII + NHC catalyst precursor, resulting in the metal–ligand bond cleavage, were described above. The methods for retaining the ligand in the metal coordination sphere upon the activation of the catalytic system were also discussed. There is the third type of transformations of metal complexes that affords the compounds [(NHC)2Pd0] with two NHC ligands. The [(CAAC)Pd(Cl)2Py] complexes [CAAC is cyclic alkyl(amino)carbene] are reduced with the Na – NaCl system to be converted to the compounds [(CAAC)Pd(Py)], which disproportionate to give [(CAAC)2Pd] and Pd-NPs (Scheme 19). The [(CAAC)2Pd] complex is catalytically active in the Buchwald – Hartwig reaction involving aryl chlorides under mild conditions (60°C). Similar transformations are inherent in the [(IPr)Pd(Cl)2Py] complex. The product of reduction of this compound, [(IPr)2Pd], was isolated and used as a catalyst in the C – N bond formation reaction.[100] The susceptibility of some [Pd0 – NHC] complexes to disproportionation can also account for the formation of ‘cocktail’-type systems.
*See R.F.W.Bader. Atoms in Molecules: a Quantum Theory. (Oxford: Oxford University Press, 1990), 438 p.
2.3. Transition metal complexes with mixed ligand composition
Cocktail-type catalysts are formed in systems containing a mixture of various auxiliary ligands. This gives rise to an additional degree of freedom, which expands the variability of the catalytic systems. The reaction system comprises a mixture of active sites with ligands of a definite type, together with metal centres the coordination sphere of which includes simultaneously two different ligands. The variable composition is responsible for two benefits of multiligand systems. First, these catalysts are more versatile and are applicable to a wider range of substrates, which reduces the system optimization costs. Second, the performance of biligand catalysts may be enhanced due to the synergistic effect of the ligands. A disadvantage of these systems is, in some cases, a moderate selectivity.
The synergistic effect of the combination of NHC with phosphine ligands is manifested in the palladium catalysis of the Buchwald – Hartwig reaction (Scheme 20). The amination reactions of various substrates were carried out using three types of catalytic systems:
(1) [(IPr)PdCl2(Py)] complex containing only the NHC ligand (compound 13, 1 mol.%);
(2) RuPhos-[Pd(Cl)2(Py)] complex with a phosphine ligand (compound 14, 1 mol.%);
(3) catalytic system 15 comprising 1 mol.% [(IPr)PdCl2(Py)] and 1 mol.% RuPhos.
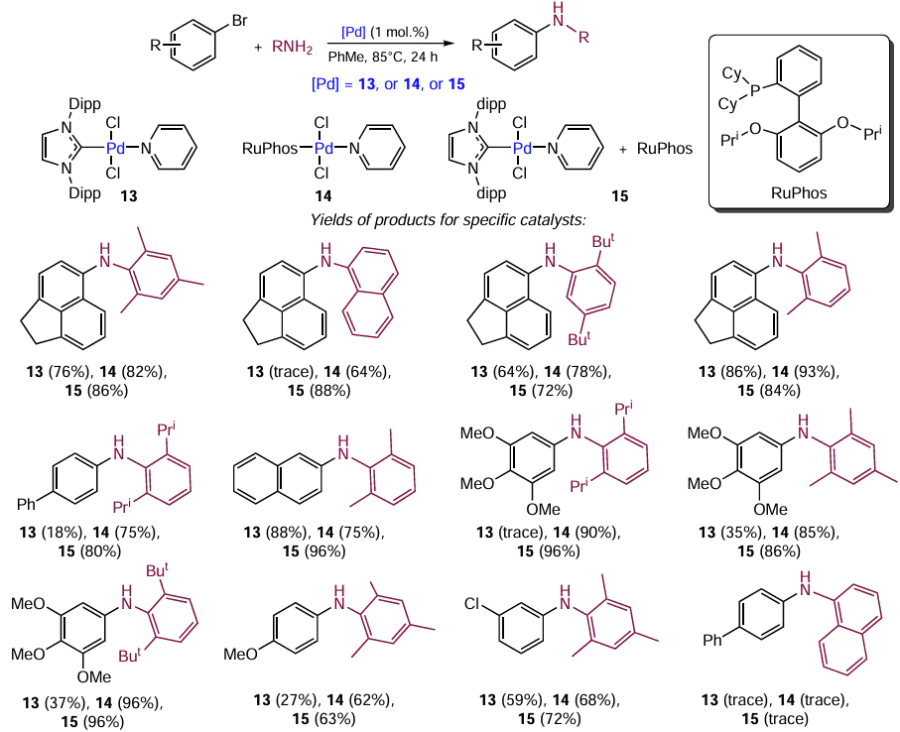
The reactions were conducted for 45 min and then the yields of the products were determined by 1H NMR spectroscopy. On average, the highest yields were observed when biligand system 15 was used. It is assumed that the catalytic cycle for this compound starts with the formation of the [NHC – Pd0] complex from the precursor [(IPr)Pd(Cl)2(Py)]. This is followed by the oxidative addition of aryl halide to the metal centre. The resulting intermediate [(NHC)PdII(X)(Ar)] is coordinated to the amine, and subsequently deprotonation, dehalogenation, and reductive elimination steps take place. The role of the phosphine ligand is apparently to accelerate the reductive elimination in the case where it is coordinated to the palladium intermediate containing an NHC ligand. In addition, the phosphine can stabilize ligand-free complexes resulting from the C – NHC or X – NHC coupling.[101] Mixed complexes containing the NHC and PR₃ ligands were also studied in other cross-coupling reactions.[102-107]
A good example of the synergistic effect is the use of mixed-ligand palladium complexes to catalyze the reactions of 1-bromonaphthalene with aniline (primary amine) and 1-bromo-4-methoxybenzene with diphenylamine (secondary amine) (Scheme 21). Generally, catalytic systems showed high performance in these reactions. In most cases, the target products were obtained in high yields and with high selectivity. Unlike the previous example in which the phosphine ligand was added separately, in this case, both ligands, NHC and phosphine, were parts of the catalyst precursor located in the trans-positions to each other. It is known that the 13C NMR chemical shift of the carbene carbon atom is sensitive to the electron-donating properties of the trans-located ligand in square-planar PdII complexes.[108] According to the 13C NMR spectroscopy, the electron-donating capacity of phosphine ligands decreases in the following order: RuPhos > SPhos ~ DavePhos > CyJohnPhos >> >> PPh3 > P(o-Tol)3 {SPhos is dicyclohexyl(2',6'-dimethoxy[1,1'-biphenyl]-2-yl)phosphine, DavePhos is 2-dicyclohexylphosphino-2'-(N,N-dimethylamino)biphenyl, CyJohnPhos is (2-biphenyl)dicyclohexylphosphine, Tol is tolyl}. A similar trend can be followed in the 31P NMR spectra: SPhos > RuPhos ~ DavePhos > CyJohnPhos >> PPh₃ > P(o-Tol)3. The highest catalytic activity in the Buchwald – Hartwig reaction with both primary and secondary amines was found for the (6-dipp)PdCl2 – SPhos complex. The yields of N-phenylnaphthalene-1-amine (16) and 4-methoxy-N,N-diphenylaniline (17) were 98 and 95%, respectively. The (6-Dipp)PdCl2 – SPhos catalyst with a donor phosphine ligand was also successfully used in some other amination reactions of aryl halides.[109]
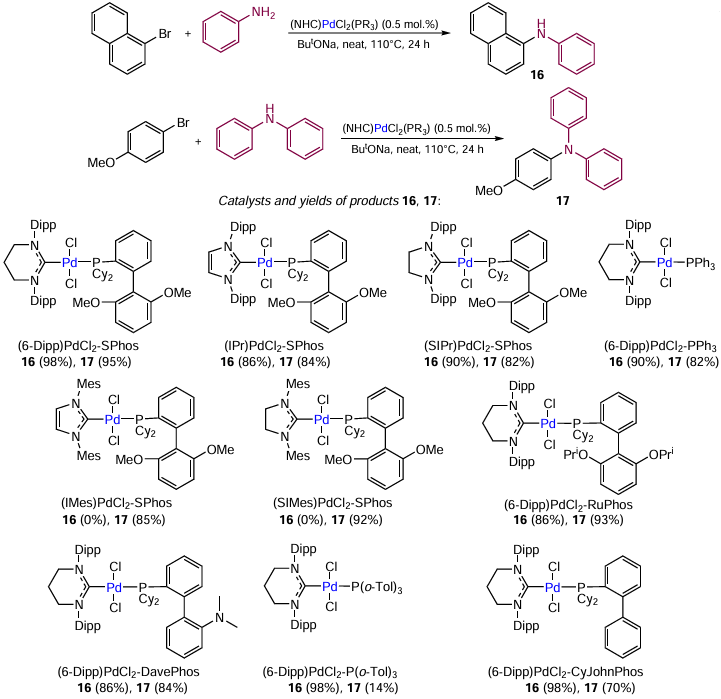
Complexes 18, containing simultaneously NHC and CAP ligands (CAP is 1,4,7-triaza-9-phosphatricyclo[5.3.2.1]tridecane), exhibited high activity in the reactions of secondary amines with substituted aryl chlorides. Characteristic features of the CAP ligand are strong donor properties and relatively small steric bulk. These mixed (NHC + CAP) compounds are cis-isomers, which are easily prepared by replacement of pyridine in the trans-[Pd(NHC)(Py)Cl2] complex or by mere addition of CAP to the [Pd(NHC)Cl2]2 dimer.[110]
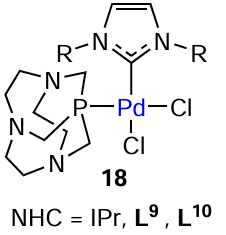
Many catalytic reactions are carried out in ionic liquids based on azolium salts as substitutes for environmentally harmful solvents. The success of these reactions is sometimes explained by the formation of complexes with NHC ligands in the system.[111] For example, the formation a mixed complex containing both NHC and phosphine ligands was detected at room temperature in the 1-n-butyl-3-methylimidazolium tetafluoroborate (bmim) ionic liquid (Scheme 22). Unlike above examples in which the formation of ‘cocktail’-type systems was initiated by the release of the NHC ligand from the metal coordination sphere, in this case, the diversity of active sites is provided by the fact that the ionic liquid itself can serve as a source of NHC ligand.[112]
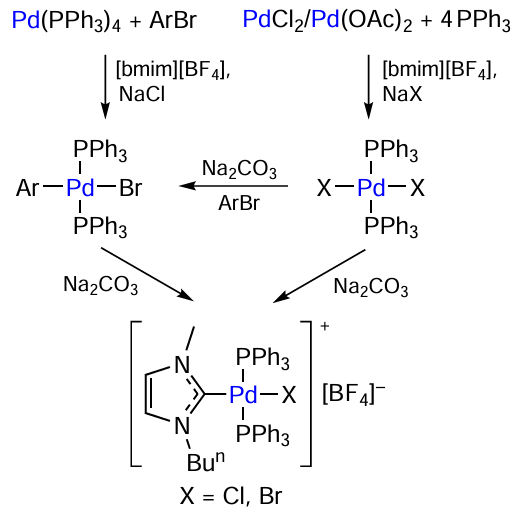
Catalytic systems based on two different biarylphosphine ligands, BrettPhos [2-(dicyclohexylphosphino)-3,6-dimethoxy-2',4',6'-triisopropyl-1,1'-biphenyl] and RuPhos, were used in the C – N cross-coupling (Scheme 23). One of the ligands was a part of the catalyst precursor, while the other one was used as an additive. Irrespective of which of the phosphines was added to the reaction system, ligand exchange took place to give a mixture of various palladium(0) complexes. This mixture of palladium-containing compounds reacted with aryl halides to afford the [BrPd(Ar)(BrettPhos)] and [BrPd(Ar)(RuPhos)] complexes. A number of key issues were identified by comparative analysis of catalysts based on P-ligands–BrettPhos, RuPhos, and their combination.
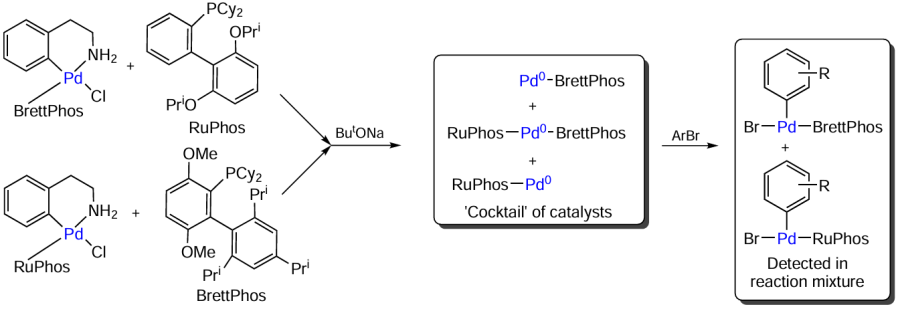
(1) BrettPhos-containing complexes effectively catalyze the monoarylation of primary amines, but show low activity in reactions with secondary amines.
(2) RuPhos-based catalysts effectively operate with secondary amines; however, in reactions with primary substrates, they often lead to the formation of double arylation products as impurities.
(3) Mixed BrettPhos+RuPhos system was highly effective in both types of reactions. The product yields in the arylation of secondary amines were comparable with the results obtained using the Pd-RuPhos catalyst. In the case of primary amines, optimization of temperature conditions made it possible to prepare secondary amines with a minor loss of selectivity compared to that for the Pd-BrettPhos catalyst. Moreover, in some examples of triarylamine synthesis, the multiligand system surpassed in efficiency both pure catalysts (Pd-RuPhos and Pd-BrettPhos).[113]
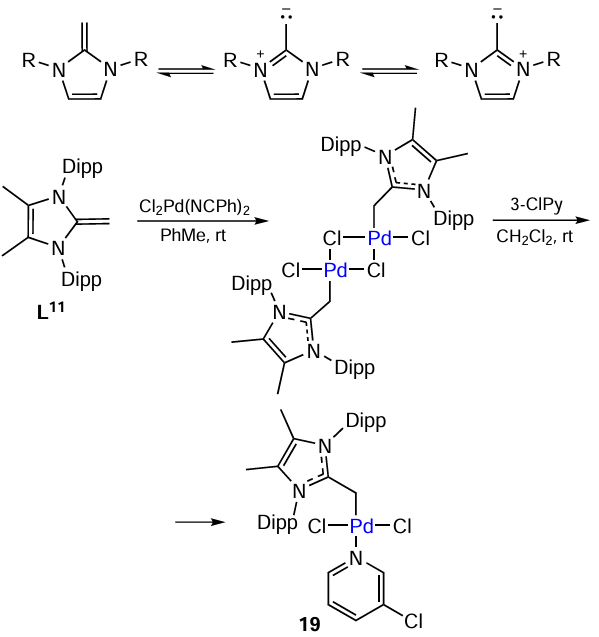
N-Heterocyclic olefins (NHO) were successfully used as ligand precursors in the Buchwald – Hartwig reaction involving various aryl halides and primary amines. These ligands (e.g., L11) were η1-coordinated to the palladium atom through the terminal carbon atom (Scheme 24). The compound [(NHO)PdCl2(3-ClPy)] (19) showed a moderate activity in the amination reaction. The best results were obtained when NHO was used as an additive to the palladium source, [Pd(cinnamyl)Cl]2 , in the presence of sodium tert-butoxide. The resulting catalytic system contained colloidal Pd nanoparticles and provided high yields of the target products. The reaction was highly selective, giving no products of double arylation of primary amines. The catalyst poisoning experiments attested to the heterogeneous nature of catalysis. The addition of mercury or small amounts of PMe3 stopped the reaction. After completion of the reaction, Pd-NPs were isolated from the reaction mixture and studied by transmission electron microscopy (TEM). Nanoparticles had a spherical shape, with their average size being 4.8 nm.[114]
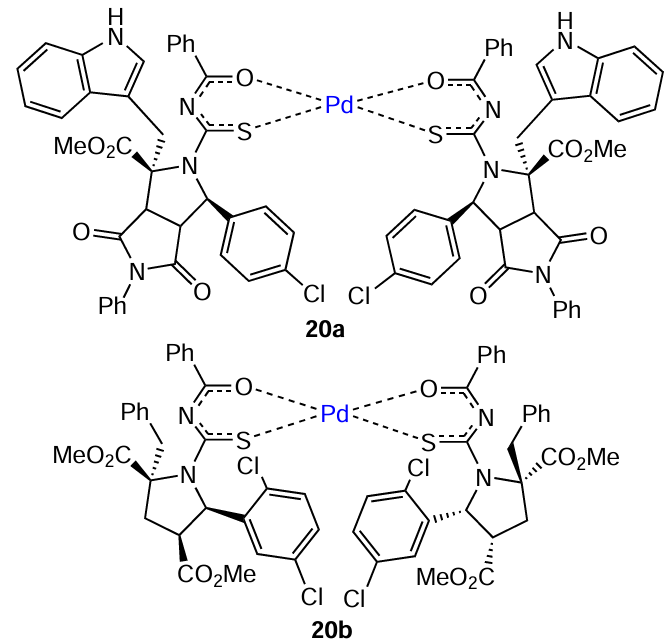
Stable PdII complexes 20 with two chelating O,S-ligands were used in the amination of primary arylamines in water. During the reaction, metal complexes were reduced and aggregated to give Pd-NPs, which also participated in the conversion of the starting substrates. The nanoparticle size varied from 3.5 to 5.0 nm, according to TEM data. According to elemental analysis data, NPs consisted of 98.5 – 98.9% Pd and also contained carbon (0.7 – 0.8%) and sulfur (0.2 – 0.3%). The catalytic activity of palladium aggregates was proved by mercury test and hot filtration. According to recycling experiments, NPs formed from palladium complexes 20a,b withstood six consecutive cycles without appreciable loss of the catalytic activity.[115]
Thus, the catalysis of C – N bond formation reactions, traditionally considered to be homogeneous, often has a more complex nature. A complex mixture of catalytically active sites of various natures can be formed from transition metal complexes in a reaction system. The formation of ‘cocktail’-type catalysts usually begins with the removal of the auxiliary ligand from the coordination sphere of the metal. This gives rise to labile ligandless compounds able to undergo reactions accompanied by increasing nuclearity of the system. The MII complexes tend to dimerize and trimerize, whereas the M0 complexes aggregate to give nanoparticles.
3. Dynamic catalysis by transition metals supported on a solid in the C – N bond formation reactions
Catalysis using metal nanoparticles immobilized on solid supports is a highly important area of modern organic synthesis. These catalytic systems differ considerably from the homogeneous ones by recyclability and lower amount of metal impurity in the target product.[116][117] The supported catalysts are characterized by high diversity caused by both the nanoparticle nature and size and the type of the support. The supports may have a porous structure or contain coordinating groups capable of binding to the metal centres. In heterogeneous systems, the active site structure can also change, which can lead to the formation of a ‘cocktail’ of metal-containing compounds. The transformations of supported catalysts include leaching of active species to the solution, migration of metal atoms over the support surface, redeposition of the metal from the solution, and modification of the metal surface by organic ligands. The dynamic nature of catalysis involving immobilized transition metal nanoparticles is also manifested in the C – N bond formation reactions.
The activity of carbon-supported palladium catalysts (Pd/C) received from various commercial sources was tested in the cross-coupling of iodobenzene with morpholine (Scheme 25). The yields of the target product (4-phenylmorpholine) varied over a broad range, from 0 to 63%, depending on the brand of the catalyst. This difference is presumably attributable to the size of palladium nanoparticles, the oxidation state of metal centres, and the surface morphology.[118] The choice of the solvent and the base had a minor effect on the catalyst performance, while the addition of 1 equiv. of phosphine ligand proved to be a necessary condition for the success of the reaction. In this case, the nature of the ligand played the crucial role.
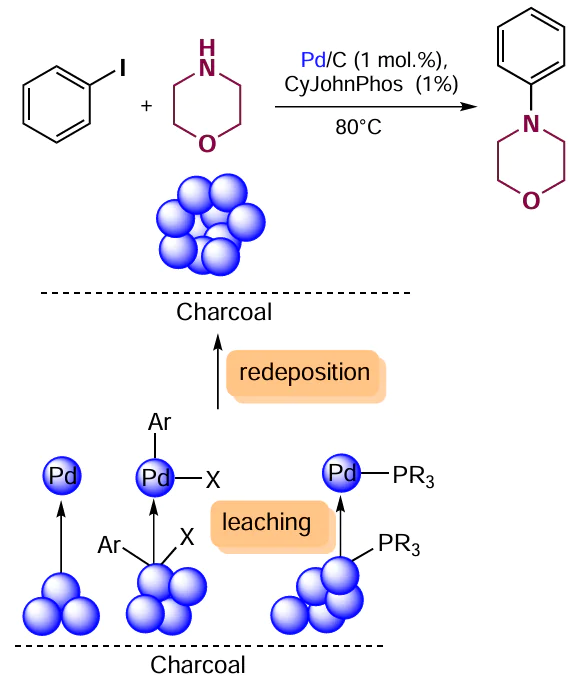
Several phosphines, that is, CyJohnPhos, XPhos, and dppf, were tested in this reaction. Reasonable yields of the amination product were achieved only with CyJohnPhos. Recycling of the Pd/C + CyJohnPhos system demonstrated that the catalyst can be reused for five successive cycles, provided that a fresh portion of the ligand is added in each cycle. In addition, the reaction was gradually retarded: the time required to achieve complete conversion was 1, 3, 5, 7, and 10 h, respectively. The decrease in the catalytic activity and the need to add the ligand indicate that palladium particles are leached from the support surface and may be redeposited.[119][120]It is known that leaching of the supported catalyst from the surface is promoted by oxidative addition of aryl halide[121] or coordination of phosphine ligands.[122] The leaching of active species was confirmed experimentally: a solution separated from the heterogeneous phase showed a catalytic activity towards the amination.
Similar experiments on the recycling of a catalytic system based on Pd/C and the dppf ligand in the N-phenylation of morpholine showed that 10% Pd/C can be reused for four cycles without significant loss of activity. According to inductively coupled plasma atomic emission spectrometry (ICP AES) data, only 1.1 mass % of palladium migrated to the solution over four cycles. Despite so small amount of leached metal, it was shown by experiments that the dissolved molecular fraction exhibits high catalytic activity. The Pd/C catalyst was first treated with the dppf ligand and sodium tert-butoxide and, after that, the mixture was filtered through a membrane filter. The resulting solution containing no solid phase successfully catalyzed the amination, providing a high yield of the product. This suggested that palladium dppf complexes are the active species. It is noteworthy that treatment of Pd/C with a base alone (without phosphine ligand) did not cause palladium leaching and did not yield a catalytically active filtrate. Additionally, it was found that treatment of the catalytically active solution with activated carbon eliminates the catalytic activity almost completely, which confirms the presence of palladium redeposition in the reaction medium.[123]
Palladium supported on multilayer carbon nanotubes (MWCNTs) or on phosphorus-doped carbon (PC) materials can be leached during the Buchwald – Hartwig, Sonogashira, and transfer hydrogenation reactions.[124]These processes are more intense during cross-coupling than during hydrogenation. Leaching also depends on the type of support: a larger amount of metal was leached from the Pd/PC catalyst than from Pd/MWCNTs. The key role in the metal transfer between the phases is played by the substrate molecules, because mere heating of the supported catalysts in a pure solvent leads to a markedly lower leaching of palladium. According to molecular modelling data, the oxidative addition of PhBr to palladium nanoparticles is thermodynamically favourable and can proceed at room temperature.[125] Subsequently, two reactions are possible: cross-coupling on the NP surface (the active sites remains in the solid phase) or leaching with a palladium atom transfer to a solution and catalysis by molecular complexes.
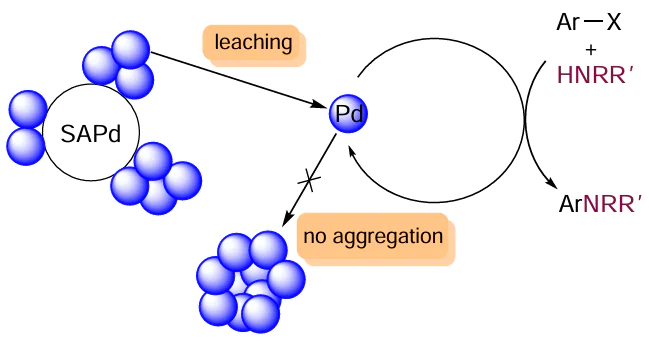
The support structure has a pronounced influence on the leaching and redeposition processes. Sulfur-modified gold-supported palladium catalyst (Pd/SA) provided high yields in the Buchwald – Hartwig reaction. As compared with the described catalysts based on carbon materials, these systems were much less susceptible to leaching. According to inductively coupled plasma mass spectrometry (ICP MS), the amounts of palladium immobilized in Pd/SA before and after the reaction were 79 ± 11 and 68 ± 18 μg, respectively.
The Pd/SA precatalyst presumably acts as a reservoir providing a low concentration of the active ligandless palladium species in the system (Scheme 26). On the one hand, this catalyst is characterized by a low level of leaching, and, on the other hand, the amount of leached active species is sufficient for the amination to proceed.[126] Due to low concentration of palladium in solution, aggregation does not occur in this system.
Owing to these properties, Pd/SA showed good results in recycling experiments for various substrates (Scheme 27). Each reaction was carried out 10 times with the same catalyst. The yields of products after the first cycle were high and amounted to 86 – 97%. In the tenth cycle, the catalyst did not lose activity, with the yields of the target products decreasing by only 1 – 2% (down to 85 – 95%).[127]
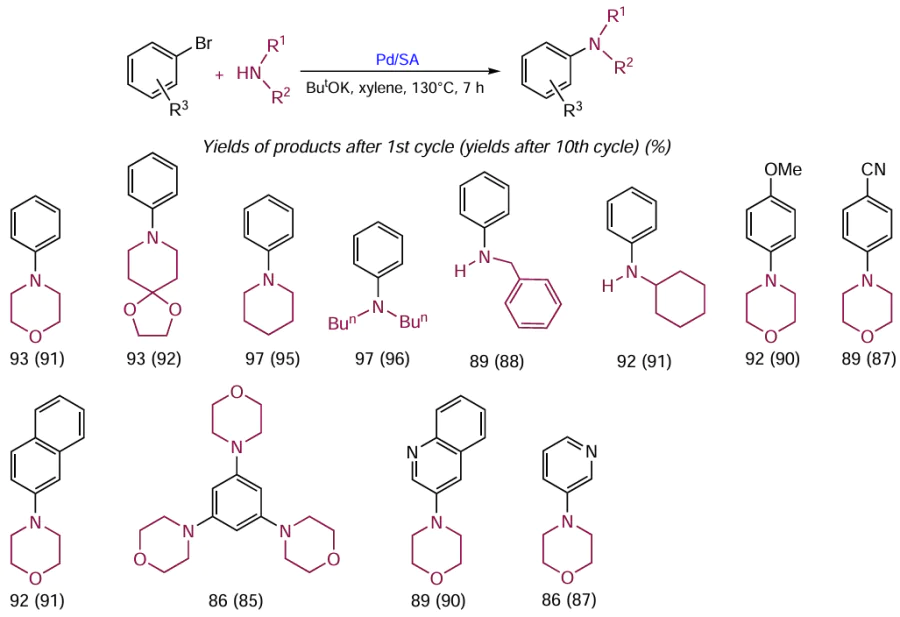
Further development of a new synthetic procedure resulted the appearance of the second-generation Pd/SA catalytic system. This system showed the same catalytic activity towards C – N bond formation as the first-generation catalyst, but was more resistant to leaching.[128]
Transition metal complexes immobilized on a solid support can form ‘cocktail’-type catalysts during cross-coupling reactions. Palladium compounds with NHC ligands (for example, IPr) bound to polystyrene support (Wang resin) through a silyl linker catalyze the amination of aryl chlorides (Scheme 28). The reaction proceeds within 10 min at low palladium loading (0.2 mol.%). This catalytic system exhibits dynamic properties consisting of Pd – NHC bond cleavage and formation of nanoparticles.
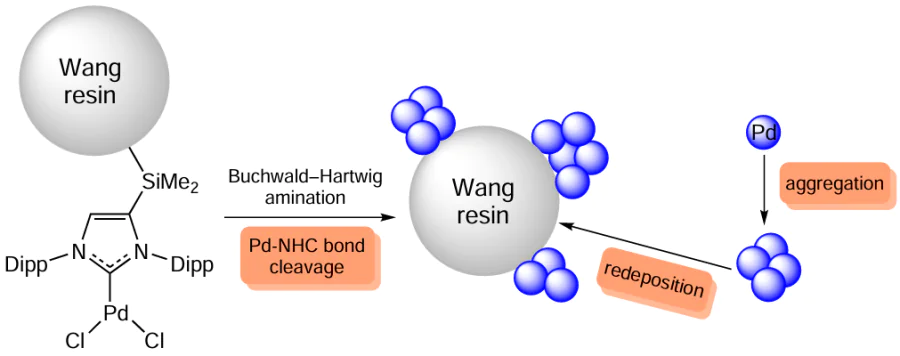
Analysis of the system after the reaction showed that the palladium NPs were mainly bound to the support, which adsorbed palladium compounds from the reaction mixture. Therefore, the amount of palladium compounds in the solution did not exceed 1 ppm. The authors did not specify whether the NP aggregation occurred on the support or in the solution. It is quite probable that both versions took place.
The catalyst reuse proved to be impossible: even in the second cycle, no catalytic activity was present.[129] It is noteworthy that the low palladium content in solution does not mean that there is no leaching of active species from the solid phase; instead, this may be due to the subsequent redeposition of nanoparticles on the support. Similar experiments were carried out for immobilized phosphine complexes. These systems also had a good activity in amination, with the level of solution contamination with palladium being low. However, in this case, the support surface was not analyzed after the reaction.[130]
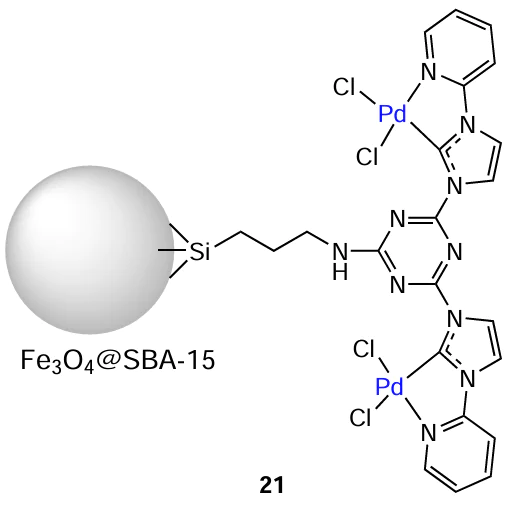
The heterogeneous precatalyst 21 based on the palladium complex with the NHC ligand immobilized on the magnetic silicate support Fe3O4@SBA-15 was leached during amination of p-bromotoluene. The amount of palladium that passed to the solution was 1.04 mass %. The authors assumed a heterogeneous nature of the catalysis without considering the possible contribution of leached Pd species to product conversion. The catalyst retained the activity in recycling experiments. The yields of the target product were 94 and 85% in the first and seventh cycles respectively.[131]
A single-atom palladium catalyst comprising palladium atoms coordinated to the heptazinic moieties of the graphitic carbon nitride C3N4 was tested in the arylation of primary and secondary amines involving a large number of substrates. The performance and stability of the Pd1@C3N4 catalytic system depended on the nature of the base used and the phosphine ligand added. Analysis of palladium distribution over the support surface after the reaction showed that in the presence of RuPhos as an additive, the catalyst structure was retained. In the case of PPh3 or PBut3 · HBF4, palladium nanoparticles were formed. The authors explained this phenomenon by leaching and subsequent redeposition of metal-containing compounds on the support or by migration of Pd atoms over the carbon nitride surface.[132]
During the amination reaction, supported catalysts can form ‘cocktail’-type systems owing to the leaching process, which results in the migration of metal atoms from the solid phase to the solution. If leaching proceeds actively, a fairly large amount of transition metal compounds are released to the solution, which leads to metal aggregation and redeposition on the support surface. When the concentration of metal atoms leached to the solution is moderate, aggregation does not occur, and the resulting mixture of metal complexes can make a crucial contribution to the formation of the target product. The metal leaching intensity is considerably influenced by the nature of the support, the use of additives (e.g., phosphine ligands or ammonium salts), temperature, and reaction medium.
4. Conclusion
Catalysts based on transition metal compounds undergo various transformations during C – N bond formation reactions. There are two conceptually different approaches to address this phenomenon in the literature. According to one approach, the search for the ‘true catalyst’ that makes the major contribution to the formation of the target product is carried out. This approach implies that the catalyst precursor is irreversibly converted to an active species that operates in a static mode, i.e., it remains invariable throughout the reaction. In the approach based on the concept of ‘cocktail’-type catalysts, a catalytic system is treated as a set of active sites that contribute to the formation of the product. In this case, the catalyst transformation can be reversible. For example, leaching of metal atoms from the solid phase to the solution and metal aggregation to give nanoparticles that can be redeposited on the solid support can proceed in one reaction system.
Dynamic catalysis is widely encountered in C – N bond formation reactions. ‘Cocktail’-type systems are formed in situ from various precatalysts (transition metal salts, metal complexes, supported metals, and single-atom catalysts). However, in most studies on the chemistry of amination, the authors do not take into account the diversity of transformations of the catalytic systems. A modern study of catalytic reactions requires the use of a set of physicochemical methods due to the large variety of active species in the reaction system and their low concentrations as well as short lifetimes of the formed intermediates. Further advancement of the concept of ‘cocktail’- type catalysts requires the development of new approaches to investigate complex reaction systems. The use of electron microscopy (nanofishing method) and solid-state NMR spectroscopy for catalytic processes was an important step forward in this direction.
The knowledge of the mechanisms of transformation of catalytic systems provides an opportunity for targeted control of the catalyst composition and stability. The retardation of the processes leading to elimination of auxiliary ligand from the metal coordination sphere is favourable for increasing the efficiency of the catalyst. For example, the oxidation of phosphine ligands can be retarded by using compounds such as diphenylvinyl- and diphenylcyclopropylphosphines or ligands with biaryl moieties. A similar effect is induced by increasing ligand to metal ratio using phosphine ligands as additives. The main processes resulting in NHC elimination are the H-NHC and CR – NHC coupling for the cross-coupling reactions and O – NHC coupling for transfer hydrogenation. These reactions are retarded when sterically bulky NHC and soft bases are used. An additional stabilization of the catalyst can be provided by using charged ligands, which reduce aggregation processes.
In the case of catalysts supported on solid materials, high performance was observed for systems that are characterized by slow leaching of metal atoms to the solution. These compounds provide a stable moderate concentration of active species in solution, which retards the aggregation and redeposition processes.
Acknowledgements
This study was prepared with the financial support of the Russian Science Foundation (Project No. 22-73-10109).
5. List of abbreviations and symbols
The following abbreviations and symbols are used in the review:
L — ligand,
M — metal,
S — solvating ligand,
Vbur — buried volume,
AAS — atomic absorption spectroscopy,
BIMe — 1,3-dimethyl-1H-benzo[d]imidazol-2-ylidene,
bmim — 1-n-butyl-3-methylimidazolium,
BrettPhos — 2-(dicyclohexylphosphino)-3,6-dimethoxy-2',4',6'-triisopropyl-1,1'-biphenyl,
CAAC — cyclic alkyl(amino)carbene,
CAP — 1,4,7-triza-9-phosphatricylo[5.3.2.1]tridecane,
cod — cycloocta-1,5-diene,
Cy — cyclohexyl,
CyJohnPhos — (2-biphenyl)dicyclohexylphosphine,
DavePhos — 2-dicyclohexylphosphino-2'-(N,N-dimethylamino)biphenyl,
dba — dibenzylideneacetone,
Dipp — 2,6-diisopropylphenyl,
dppf — 1,1'-bis(diphenylphosphino)ferrocene,
dppp — 1,3-bis(diphenylphosphino)propane,
EXAFS — extended X-ray absorption fine structure,
ICP AES — inductively coupled plasma atomic emission spectroscopy,
ICP MS — inductively coupled plasma mass spectrometry,
IMes — 1,3-bis(2,4,6-trimethylphenyl)-1,3-dihydro-2H-imidazol-2-ylidene,
Ipr — 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene,
LP — lone pair of electrons,
Mes — 2,4,6-trimethylphenyl (mesityl),
Mor-Dalphos — di(1-adamantyl)-2-morpholinophenylphosphine,
MWCNT — multi-walled carbon nanotube,
NHC — N-heterocyclic carbene,
NHO — N-heterocyclic olefin,
NP — nanoparticle,
PC — phosphorous-doped carbon material,
Py — pyridine,
RuPhos — 2-dicyclohexylphosphino-2',6'-diisopropoxybiphenyl,
SA — sulfur-modified gold support,
Sphos — dicyclohexyl(2',6'-dimethoxy[1,1'-biphenyl]-2-yl)phosphine,
TEM — transmission electron microscopy,
Tol — tolyl,
TPEDO — 1,1,2,2-tetraphenylethane-1,2-diol,
Xantphos — 9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphine),
XPhos — (dicyclohexyl[2',4',6'-tris(propan-2-yl)[1,1'-biphenyl]-2-yl]phosphine).
References














