



Keywords
Abstract
Nitrogen heterocycles hold leading positions in the field of medicinal chemistry and targeted search for next-generation pharmaceuticals. Among the enormous set of available heterocyclic structures, oxadiazole derivatives attract attention of scientists due to a wide range of pharmacological activity, including antimicrobial, antiinflammatory, antiproliferative, and other types of activity. This review summarizes the achievements in the field of synthesis and biological activity of oxadiazole derivatives over the past 10 years. Methods for the synthesis of various therapeutic agents based on 1,2,3- 1,2,4- and 1,2,5-oxadiazoles are considered, and new approaches to the fabrication of hybrid molecules (hybrids) consisting of two or more pharmacophore moieties are systematized. The review provides the first comprehensive consideration of the key structure–property relationships for each type of oxadiazole derivatives, which can serve as guidelines for specialists in various fields who develop strategies for the design of new pharmaceuticals.
The bibliography includes 173 references.
1. Introduction
The continual development of medicinal chemistry dictates the need to search for new potential drugs with optimized pharmacological profiles. Modern statistical analysis shows that approximately 60% of organic small-molecule drugs approved by the Food and Drug Administration (FDA) for the use in clinical practice contain at least one nitrogen heterocycle in the molecule.[1][2] Moreover, over the period from 2013 to 2023 alone, FDA approved 321 drugs, 82% of which were based on nitrogen-containing heterocycles.[3] For this reason, the search for next-generation drugs among nitrogen heterocycles is a highly relevant line of research in modern medicinal chemistry.
Heterocyclic chemistry is characterized by enormous structural diversity and the possibility of changing practically important properties of target structures at the atomic level.[4] In the series of privileged building blocks pertaining to heterocyclic compounds that have found use in medicinal chemistry, worthy of note are oxadiazoles, five-membered heterocycles containing two nitrogen atoms and one oxygen atom. Oxadiazoles may have four isomeric structures in which the oxygen atom and two nitrogen atoms occupy positions 1,2,3; 1,2,4; 1,3,4; or 1,2,5 (Fig. 1), with N – O bond being present in three of the four isomers. Although 1,2,3-oxadiazole derivatives exist only in the mesoionic form, their potential in medicinal chemistry is extensive, ranging from biorthogonal applications in the case of sydnones (1,2,3-oxadiazol-5-ones)[5] to the design of new exogenous nitric oxide donors in the case of sydnone imines (1,2,3-oxadiazole-5-imines).[6]
![[{"id":"FIKqkUnoGF","type":"paragraph","data":{"text":"Structures of oxadiazoles and heteroatom locants in particular isomers"}}]](/storage/images/resized/NTkpiDtocJLWouAbzBXu69x4T98mV5DnAVhvW40w_xl.webp)
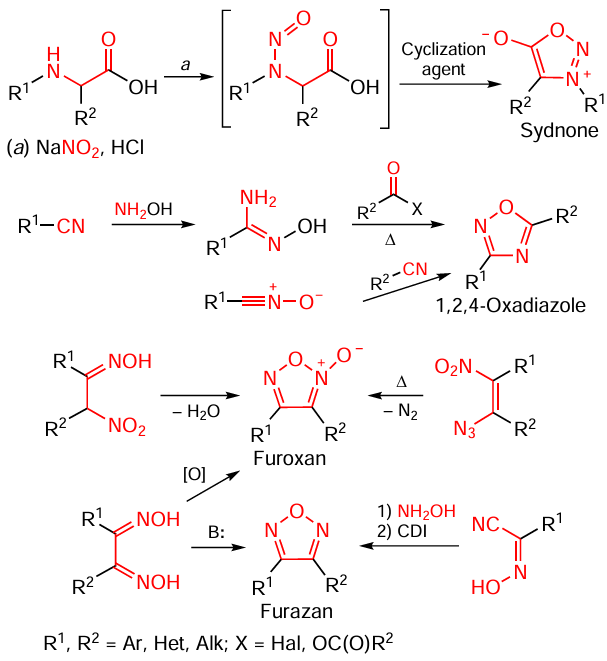
The major synthetic routes for the assembly of the heterocyclic cores for sydnones and sydnonimines,[7] 1,2,4-oxadiazoles,[8] 1,2,5-oxadiazoles (furazans), and their N-oxides (furoxans)[9] have been well studied. The heterocyclization reactions giving oxadiazoles used most commonly in synthetic practice are summarized in Scheme 1. The most popular method for the closure of the sydnone heterocyclic ring is the nitrosation of amino acid derivatives followed by cyclization. 1,2,4-Oxadiazoles are prepared by two standard synthetic routes, with nitriles being used as key substrates in both cases. The first method is based on the addition of hydroxylamine to the starting nitrile followed by cyclocondensation of the resulting amidoxime via the reaction with carboxylic acid anhydrides or chlorides or their synthetic equivalents. An alternative method consists in the use of nitriles as dipolarophiles in the 1,3-dipolar cycloaddition to nitrile oxides. The chemistry of 1,2,5-oxadiazoles is more diverse. There are three frequently used methods for the furoxan synthesis: oxidation of dioximes (glyoximes), dehydration of α-nitrooximes, and thermally induced elimination of molecular nitrogen in the cyclization of α-nitroazides. Under the action of bases, glyoximes are cyclodehydrated to give furazans; aminofurazans can also be prepared by treating cyanoxime with hydroxylamine followed by cyclodehydration in the presence of carbonyldiimidazole (CDI).
Among isomeric oxadiazole structures, pharmacologically oriented 1,3,4-oxadiazoles are among the longest known and best studied compounds. The recent works in this area were summarized in reviews published in 2018 and 2024.[10][11] However, the latest results on the synthesis and pharmacological studies of 1,2,3-, 1,2,4-, and 1,2,5-oxadiazoles have not been systematized, and data on these derivatives are fragmentary. Therefore, the present review summarizes the most recent achievements in the targeted synthesis of lead compounds based on oxadiazole derivatives with the N – O bond in the ring and discusses their pharmacologically relevant properties. Whenever possible, data on the possible mechanism of action of 1,2,3-, 1,2,4-, and 1,2,5-oxadiazoles as applied to a particular experimentally proven type of biological activity are given.
2. Synthesis and activity of 1,2,3-oxadiazoles and 1,2,3-oxadiazol-5-ones (sydnones)
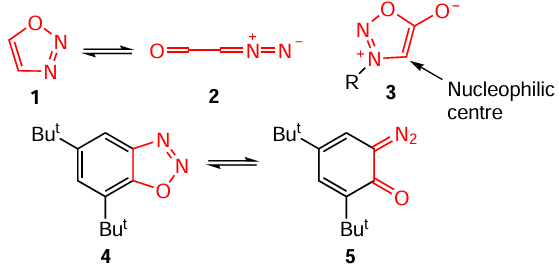
Unlike other isomers, unsubstituted 1,2,3-oxadiazole (1) is unstable and exists mainly as a tautomeric linear diazoketone, formyldiazomethane (2). A derivative with an exocyclic oxygen atom, 1,2,3-oxadiazol-5-one (sydnone) 3, which is a mesoionic heterocycle with spatially separated positive and negative charges, is used much more often (Scheme 2). However, the introduction of fused benzene rings or other electron-donating aromatic substituents may stabilize the 1,2,3-oxadiazole structure in the gas phase.[12] It was shown that some substituted benzannulated 1,2,3-oxadiazoles are in equilibrium with open-chain tautomers.[13] The relative equilibrium concentration of the components strongly depends on the solvent and degree of substitution: the diazoketone structure is stabilized by hydrogen bonds and polar interactions. Of the two compounds, 5,7-di-tert-butyl-1,2,3-benzoxadiazole (4) is more stable: it is 6.3 kJ mol–1 energetically more favourable in the gas phase than the valence isomer, diazocyclohexadienone 5. As regards the reactivity profile of sydnones, the C(4) atom of the heterocycle is the nucleophilic centre, providing the possibility of electrophilic functionalization. In the case of 3,5-disubstituted 1,2,3-oxadiazoles, the C(4) atom bears a relatively acidic hydrogen atom and can be deprotonated on treatment with strong organolithium bases.[7][14]
2.1. Compounds with antimicrobial activity
Studies of the antimicrobial activity of sydnones were initiated in 1968 by a publication by Naito et al.,[15] who obtained the first hybrids of penicillin with 3-arylsydnone that exhibited activity against penicillinase-producing bacterial strains. Since the discovery of pharmacophore properties of the sydnone moiety, many researchers have used it in the design of potential pharmaceutical agents by fabricating hybrids combining known pharmaceutical building blocks in their molecules.
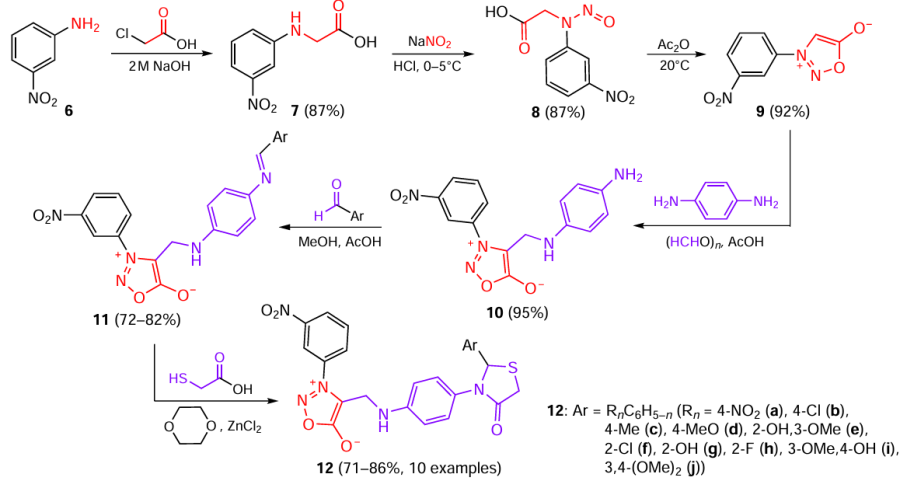
For example, Savant et al.[16] combined sydnone, Mannich base, and 4-thiazolidone moieties in one molecule with the goal to enhance the antimicrobial activity. In the first step, m-nitroaniline 6 was alkylated with chloroacetic acid in the presence of 2 M NaOH. Nitrosation of the resulting amino acid 7 followed by cyclization of intermediate nitrosocarboxylic acid 8 on treatment with acetic anhydride gave m-nitrophenylsydnone 9. Subsequently, compound 9 was aminomethylated by treatment with paraformaldehyde and p-phenylenediamine to afford sydnone 10, which was then condensed at the free amino group in the aromatic ring with substituted benzaldehydes in the presence of glacial acetic acid. The formed Schiff bases 11 were treated with thioglycolic acid in the presence of anhydrous zinc chloride as a Lewis acid, which resulted in the formation of 4-thiazolidones 12 in high yields (Scheme 3).
This series of compounds was studied for antibacterial activity in vitro against Gram-positive (Staphylococcus aureus and Streptococcus pyogenes) and Gram-negative (Escherichia coli and Pseudomonas aeruginosa) bacterial strains. Common antimicrobial agents including gentamicin, ampicillin, chloramphenicol, ciprofloxacin, and norfloxacin were used as the positive controls. The products were also investigated for activity against pathogenic yeast, including Candida albicans (the causative agent of candidiasis) and mould fungi such as black aspergillus (Aspergillus niger) and needle-shaped aspergillus (Aspergillus clavatus). In this case, antifungal drugs nystatin and griseofulvin served as the reference agents.
It was found that 4-nitro- (12a) and 4-chlorophenyl (12b) derivatives (Fig. 2) exhibited excellent activity that was superior to the activity of ampicillin against all of the bacterial strains, while in the case of Gram-negative strains, the activity was comparable with that of chloramphenicol. Similarly, the same sydnones demonstrated activity at the level of the reference drug nystatin against mould fungus A. niger [the minimum inhibitory concentrations (MICs) are presented in Table 1][16][17]. Compound 12c was most active against Gram-positive bacterium S. Pyogenes, 4-methoxyphenyl derivative 12d was active against Gram-positive S. aureus strain, while sydnone 12e had a high activity against Gram-negative E. coli. The activity of all other compounds ranged from moderate to good, while 2-hydroxy- and 2-chlorophenyl derivatives proved to be inactive against any of the listed strains.
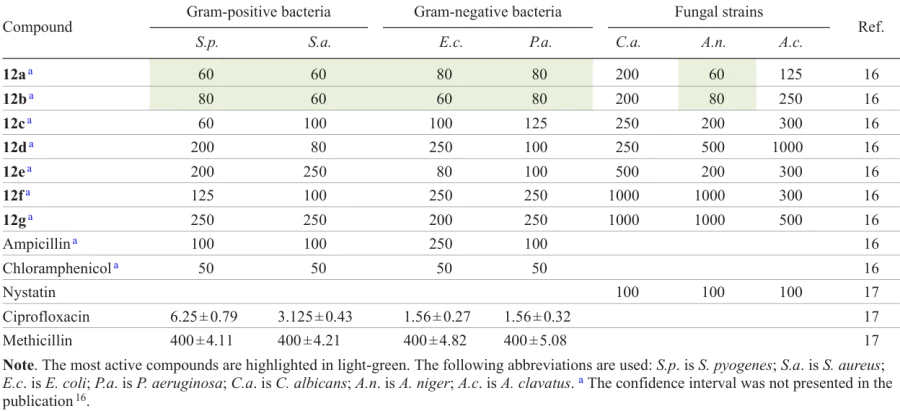
![[{"id":"PzIx1-3KmR","type":"paragraph","data":{"text":"Structures of thiazolidinone–sydnones <b>12 </b>with the most pronounced antimicrobial activities and active ingredients of commercial drugs (drawn in the box).<sup>16</sup>"}}]](/storage/images/resized/sA24kLOOMHqDAQQV2LRBSglFzkVMF4TVGqQXwqQO_xl.webp)
There are also other publications dealing with the antibacterial properties of sydnones.[18][19] The search for lead compounds was performed via the synthesis of various hybrid sydnones comprising effective and well-known pharmacological components possessing antibacterial properties such as aminocyanopyridine, and substituted hydrazines. The following trend was identified:[20] most of compounds that showed antimicrobial properties had a substituted phenyl group at the sydnone C(3) atom. Therefore, it was proposed [17] to synthesize conjugates of thiosemicarbazones with 3-arylsydnones and to assess their antibacterial activity against Gram-positive and Gram-negative bacteria, including methicillin-resistant Staphylococcus aureus (MRSA).
Substituted anilines 13 were converted to the corresponding aldehydes in five steps. First, substituted glycines 14 were obtained by the reaction of 13 with ethyl chloroacetate followed by alkaline hydrolysis. Nitrosation of compounds 14 and cyclization of the resulting N-nitroso derivatives 15 in the presence of acetic anhydride yielded 3-arylsydnones 16. The subsequent Vilsmeier – Haack formylation of sydnones 16 furnished 3-aryl-substituted 4-formylsydnones 17. These aldehydes were subjected to microwave (MW)-assisted condensation with N-(D-galactopyranosyl)thiosemicarbazide[21] (18) catalyzed by glacial acetic acid or 2-hydroxyethylammonium acetate (TEAA), resulting in the formation of conjugates 19 (Scheme 4). The use of ionic liquid provided advantages, including shorter reaction time and increased yield of the target product.[17]
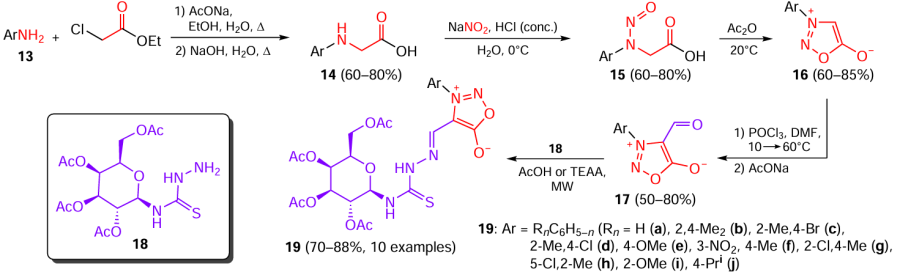
Conjugates 19a – f exhibited high inhibitory activity against both certain Gram-positive bacteria such as E. coli, P. aeruginosa, Klebsiella pneumoniae (Friedlander’s bacillus), and Salmonella typhimurium (causative agent of salmonellosis) and certain Gram-negative bacteria Bacillus subtilis (hay bacillus), S. aureus, S. epidermidis (epidermal staphylococcus), and Clostridioides difficile (causative agent of pseudomembranous colitis) (Table 2). The most active compounds of this series are depicted inFig. 3.
![[{"id":"q9EOb91rIH","type":"paragraph","data":{"text":"Structures of thiosemicarbazones <b>19 </b>with the most pronounced antibacterial action and active ingredients of commercial drugs (drawn in the box).<sup>17</sup>"}}]](/storage/images/resized/sHKw7h9boLH30Pk3vmB47YfMPubaAAeTTv37Wuwl_xl.webp)
Thiosemicarbazones 19b,d,f were the least effective inhibitors of Gram-positive bacteria, with MICs being in the range of 0.78 – 1.56 μg mL–1, which is lower than these values for the commercial antibacterial drug ciprofloxacin. Meanwhile, compounds 19b,c,f had the highest inhibitory effect against Gram-negative bacterial strains (MIC = 0.78 – 1.56 μg mL–1). Hence, compounds containing 2,4-dimethyl-, 4-bromo(or chloro)-2-methyl-, or 4-methyl-3-nitrophenyl group as a substituent in the heterocycle showed a more pronounced inhibitory effect against any of the tested bacterial strains, whereas the presence of methoxy group in the aromatic ring apparently decreased the inhibitory activity. Compounds 19b – d,f also exhibited high inhibitory activity against S. aureus DNA gyrase with half-maximal inhibitory concentration (IC50) in the 0.278 – 0.883 μM range and against DNA topoisomerase IV (IC50 = 1.316 – 2.315 μM). Furthermore, these four compounds had a low toxicity towards WI-38 normal cell line (IC50 > 73 μM).
Comparison of the activities of the prepared series of compounds 12 and 19 against two Gram-positive and Gram-negative bacterial strains (see Table 1 and Table 2) provides the conclusion that D-galactoso-conjugated thiosemicarbazones 19b,c,f were the most active antibacterial agents, although some compounds 12 showed the higher activity than ampicillin and methicillin.
2.2. Compounds with antifungal activity
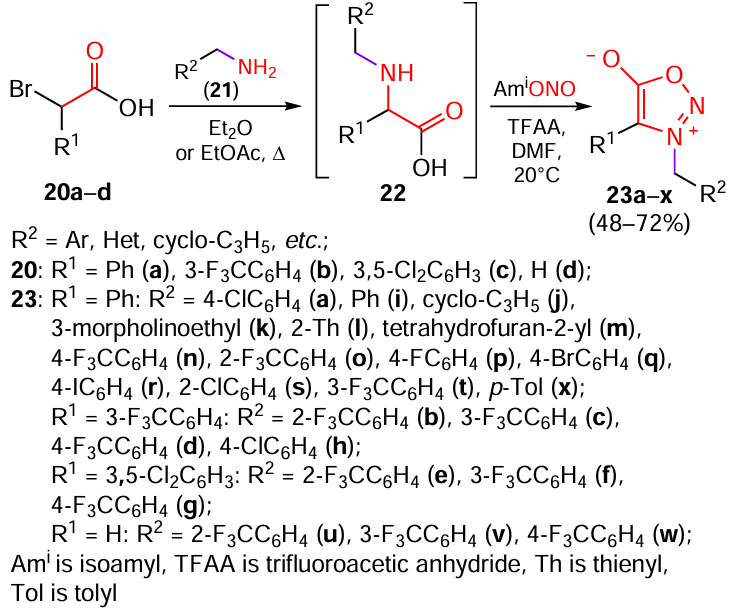
Owing to the discovery and commercialization of the insecticide triflumezopyrim,[22] the trend towards investigation of mesoionic compounds in the development of fungicides and insecticides has taken shape in the last decade.[23][24] Using a two-step one-pot procedure, Du et al.[25] prepared sydnonimines that had a good potential for the use in agriculture (Scheme 5). The first step of the synthesis consisted in the substitution of bromine atoms in acids 20a – d by amines 21, which readily proceeded on refluxing in diethyl ether or ethyl acetate. In the second step, nitrosation of the secondary amino group in compounds 22 was performed by treatment with isoamyl nitrite and trifluoroacetic anhydride, while the subsequent cyclization resulted in the target sydnones 23a – x.
Among the synthesized series of compounds, most trifluoromethylsydnone derivatives 23 (Fig. 4) exhibited a marked antifungal activity and inhibitory activity against tyrosinase. The best results in spore germination experiments were obtained for compound 23a, which showed an antifungal activity in vitro against Pyricularia grisea (rice blast pathogen) with half-maximal effective concentration (EC50) of 25 mg L−1 and an inhibition rate of 80%. Sydnones 23b – h proved to be active in vivo against the powdery mildew pathogen (Blumeria graminis) (EC50 = 400 mg L−1), with the inhibition rate being 80 – 95%. It should be noted that compound 23c (EC50 = 49 mg L−1) exhibited a considerable antifungal activity against the downy mildew pathogen (Pseudoperonospora cubensis), which was, however, somewhat lower than the results obtained for the kresoxim methyl used as a reference agent (EC50 = 44 mg L–1).
![[{"id":"GtyPD0khLI","type":"paragraph","data":{"text":"Structures of sydnones <b>23 </b>with the highest antifungal activity and the kresoxim-methyl fungicide (drawn in the box).<sup>25</sup>"}}]](/storage/images/resized/lN6ZUsHBA8bKYVoRXcT89eI8nyhu29ydL9IhXrWz_xl.webp)
The structure – activity relationships for a series of sydnones 23 indicate that trifluoromethyl groups present in the molecule (compounds 23b – h) increase the antifungal effect. Apparently, the presence of benzene rings in both substituents of sydnone is favourable for tyrosinase inhibition: the inhibition rate (IR) of compound 23i containing two phenyl groups (60%) was higher than that of compounds containing cyclopropane (23j, IR = 28%), morpholine (23k, IR = 33%), thiophene (23l, IR = 40%), or tetrahydrofuran (23m, IR = 14%) moieties. The introduction of electron-withdrawing substituents in the benzene ring leads to lower inhibition relative to compounds with unsubstituted phenyl groups: IR varies in the order 23i (R1 = R2 = Ph) > 23n (R1 = Ph, R2 = 4-F3CC6H4) ≈ 23o (R1 = R2 = 2-F3CC6H4) ≈ 23a (R1 = Ph, R2 = 4-ClC6H4) ≈ ≈ 23p – r (R1 = Ph, R2 = 4-HalC6H4) ≈ 23b – d,n,o,t–w (R1 or R2 = CF3C6H4). However, when an electron-donating group (e.g., 4-methyl) is present in the benzene ring, the tyrosinase inhibition rate by derivative 23x (R1 = Ph, R2 = p-Tol) increases to 61%. Meanwhile, no evident correlation was established between the fungicidal activity and the tyrosinase inhibition rate: hence, the antifungal activity of compounds 23 is not due to the enzyme inhibition alone.
2.3. Compounds with herbicide antidote and growth-regulating activity
A few publications of the past five years have been devoted to the development of ferrocene derivatives of sydnones and the study of their antidote activity against the metsulfuron-methyl herbicide (alternatively called Zinger WP). In particular, Shevaldina et al.[26] investigated the ferrocenylalkylation of 4-sulfanylsydnones (Scheme 6). The starting sydnones 24a – c were deprotonted on treatment with BunLi; then the resulting lithium intermediates 25a – c were allowed to react with elemental sulfur to give thiols 26a – c in moderate yields. Ferrocene was incorporated into the molecule via alkylation: ferrocene-containing secondary alcohols 27a,d were treated with n-butyllithium to give lithium alkoxides 28a,d. Upon acylation with ethyl chloroformate, these compounds were converted to intermediates 29a,d, which reacted with sulfanylsydnones 26 in the presence of triethylamine (TEA) to give 4-(ferrocenylalkylthio)sydnones 30a – f.
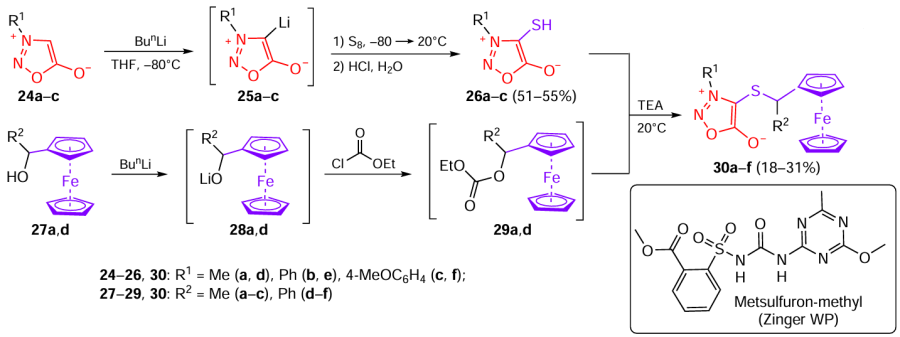
Compounds 30a–f did not show pronounced growth-regulating activity. A statistically significant herbicidal effect on the rooth growth was found only for derivatives 30a (R1 = R2 = Me), 30b (R1 = Ph, R2 = Me), and 30c (R1 = 4-MeOC6H4 , R2 = Me). However, compounds 30a,b and their analogue 30f (R1 = 4-MeOC6H4 , R2 = Ph) showed antidote activity against Zinger WP. Derivative 30b markedly suppressed the herbicidal action of this agent on the growth of plant roots, without affecting the sprouts. Meanwhile, compounds 30a,f had a significant antidote effect on the development of both roots and sprouts. Thus, the best antidote properties were found for sydnone derivative 30a that contains two methyl groups: for moderate herbicidal properties, it mitigated the herbicide action by 21% for roots and by 29% for sprouts. It is noteworthy that most of compounds exhibiting antidote activity contained a 1-ferrocenylethyl group, whereas compounds with R2 = Ph had a much lower activity.
Similarly, Kalganova et al.[27] investigated the properties of 4-[α-hydr(alk)oxy]ferrocenylmethyl sydnone derivatives (Scheme 7). As in the above study,[26] sydnones 24a – c were first converted to lithium intermediates 31a – c, which reacted with ferrocenecarbaldehyde to give intermediate lithium alkoxides 32a – c. Acidification of the reaction mixture furnished the target 4-(α-hydroxy)ferrocenylsydnones 33a – c, while treatment of ferrocene-containing alkoxides 32a – c with iodomethane gave methoxy derivatives 34a – c.
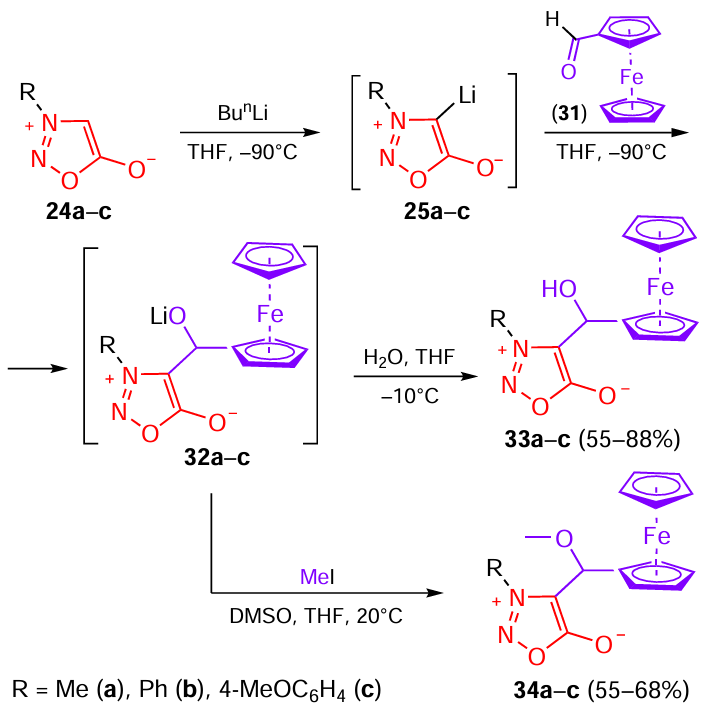
The activity of compounds 33a – c and 34a – c was evaluated by vegetation tests using cv. Krasnodarskaya 291 AMV corn seeds. It was found [27] that ferrocenyl sydnone derivatives (both hydroxymethyl 33a – c and methoxymethyl 34a – c compounds) possess low growth-regulating activity and have a minor effect on the growth of corn seeds. A statistically significant herbicidal effect manifested as 31% decrease in shoot length was observed only for compound 34b (R = Ph). Measurements of the root and shoot length indicate that the effects of the Zinger WP herbicide and ferrocene-containing mesoionic heterocycles 33a–c and 34a–c on the corn seed growth are often interrelated: derivative 34c (R = 4-MeOC6H4), which does not have significant growth-regulating activity, enhances the action of Zinger WP, i.e., it shows a negative effect. Derivatives 34a (R = Me) and 34b (R = Ph) considerably (by 18 – 25%) suppress the action of herbicides on the development of shoots. Thus, this series of compounds exhibits antidote activity against the metsulfuron-methyl herbicide rather than the growth-regulating activity.
Similar studies were carried out for α-thio-substituted acetylferrocenes containing a sydnone moiety at the sulfur atom (Scheme 8).[28] C(4)-unsubstituted sydnones 24a – d were first treated with n-butyllithium, and the lithium intermediates were treated with elemental sulfur to give lithium thiolates 35a – d. These thiolates are good nucleophiles and are easily alkylated with the corresponding chloroacetylferrocene derivative. This reaction is general and is successfully implemented for both 3-alkyl- and 3-aryl-substituted sydnones 36a – d.
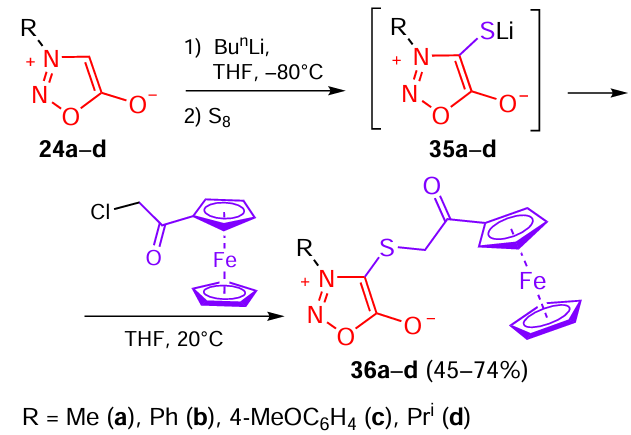
Ferrocenyl sydnone hybrids 36a – d were studied for the growth-stimulating activity and antidote activity against the metsulfuron-methyl herbicide in experiments on corn seedling growth. Treatment of corn seeds with derivatives 36d (R = Pri) and 36b (R = Ph) resulted in an increase in the fruit weight by 17 and 15%, respectively. However, acetylferrocenes did not show an antidote effect on Zinger WP: sydnone derivatives 36a,d, which had growth-stimulating properties, lost the stimulating effect in the presence of the herbicide and enhanced the inhibitory effect of metsulfuron-methyl.
Thus, the activity of sydnones is influenced by quite a few factors: the nature of the mesoionic moiety, the type of substituents in it, and the structure of the linker between ferrocene and sydnone. Compounds containing alkyl (in particular, methyl) and phenyl groups proved to be most active. However, currently, there are no sufficient data to definitively identify the relationship between the structure and properties of these sydnones.
2.4. Compounds with antioxidant activity
Due to the mesoionic nature of sydnones and unique distribution of charge density over the heterocycle, these compounds exhibit a substantial antioxidant activity.[29] Serrao et al.[30] reported the first study of the 1,6-addition and oxidative C – H functionalization reactions carried out between p-quinonemethylides 37 and sydnones 38 in the presence of catalytic amounts of copper(II) trifluoromethanesulfonate (triflate, TfO). When 20 mol.% Cu(OTf)2 was used and the reaction was carried out by refluxing in 1,2-dichloroethane (DCE), substituted pheno – sydnone hybrids 39a – m were obtained When an oxidant, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), was added to the system, oxidative C – C coupling of substrates 37 and 38 took place, resulting in the formation of products 40a – l in good yields (Scheme 9).
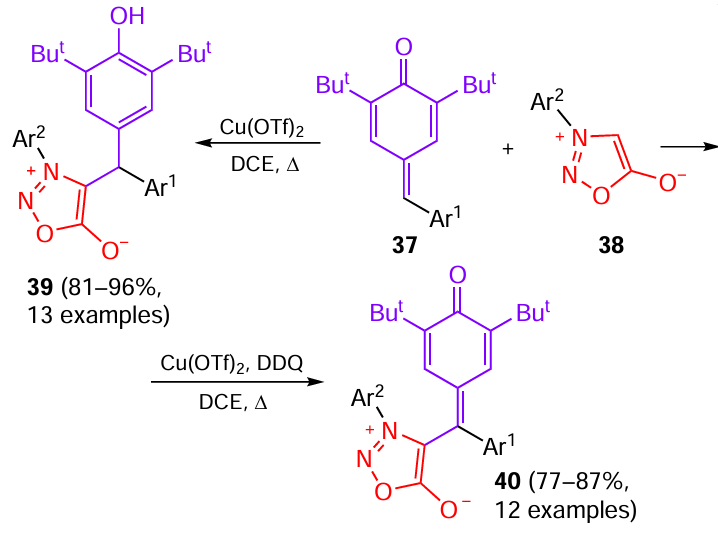
FIg. 5 shows derivatives that exhibited antioxidant activity exceeding that of ascorbic acid. It was found that high biological activity is inherent particularly in triaryl-substituted sydnones 39a – i; therefore, scavenging of 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical was studied for this series. Compounds 39a – c exhibited high activity owing to the presence of electron-donating methyl groups. The best DPPH scavenging ability was found for compound 39a, which contained methyl groups attached to the benzene rings both in the phenyl moiety and in sydnone. The antioxidant activity of products 39d – g decreased, due to the presence of electron-withdrawing groups in the aryl moiety, in the following order (the substituent is indicated in parentheses): 39d (Cl) > 39e (F) > 39f (NO2) > 39g (Br). Compounds 39d,e showed a moderate activity caused by the presence of p-tolyl groups. Derivative 39h, in which there are no additional substituents in any of the benzene rings, exhibited a moderate antioxidant activity, while thiophene-substituted compound 39i possessed clear-cut antioxidant behaviour. Thus, it was found [30] that all such compounds, except for bromo derivative 39g, have better free radical scavenging ability than ascorbic acid used as the reference.
![[{"id":"5hSrtKl1IU","type":"paragraph","data":{"text":"Structures of hybrid compounds <b>39 </b>consisting of phenol and sydnone moieties that showed pronounced antioxidant properties and ascorbic acid (drawn in the box) and IC<sub>50</sub> values for these compounds (in μg mL<sup>–1</sup>).<sup>30</sup>"}}]](/storage/images/resized/VnrgQOLMDxHEAO9J3rqGOYtoOc6QTbvTjsJI0isC_xl.webp)
2.5. Sydnone accumulation in mitochondria and fluorescence
Mitochondria are considered to be a promising target for cancer diagnosis and therapy. In recent years, mesoionic biorthogonal reagents, which include sydnones, have been used for cell labelling and drug delivery.[31] For example, Xu et al.[32] investigated the ability of fluorophore sydnones based on BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene) to act on mitochondria. These conjugates were synthesized from mesoionic compounds 41a,b in four steps (Scheme 10). First, esters 41a,b were hydrolyzed under mild conditions to acids 42a,b, which were then allowed to react with N-Boc-1,3‑diaminopropane (Boc is tert-butoxycarbonyl) to give compounds 43a,b. The Boc deprotection occurred on treatment with trifluoroacetic acid (TFA) through the formation of intermediates 44a,b. Finally, the reaction of compounds 44a,b with N-succinimide derivative BODIPY-NHS (45) in the presence of the Höing base (N,N-diisoropylethylamine) afforded the desired fluorophore conjugates 46a,b.
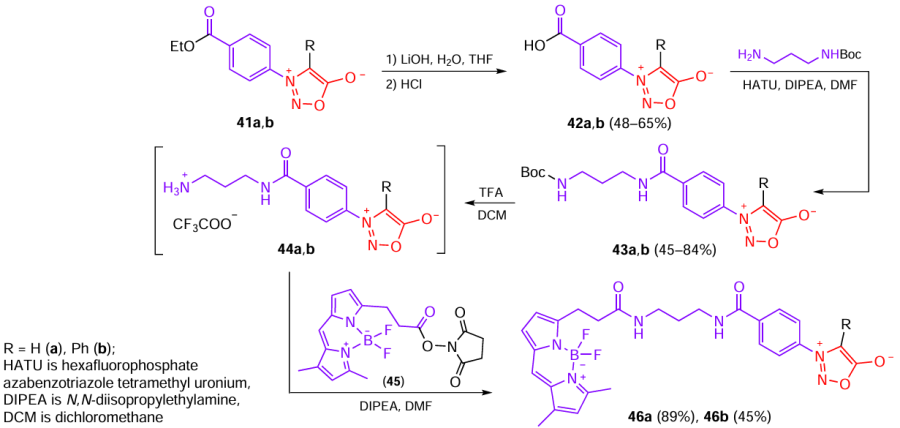
The results of biological experiments show that sydnone 46a has a high mitochondrial distribution with a Pearson correlation coefficient of 0.74. Contrary to expectations, the introduction of a phenyl group in the C(4) position in order to enhance the effect of this compound on mitochondria by increasing lipophilicity resulted in poor mitochondrial distribution with a Pearson correlation coefficient of 0.32. Since sydnones are mesoionic heterocycles, their affinity to mitochondria is presumably due to the delocalized positive charge. When sydnone contains a phenyl group at C(4), the conjugation can decrease the delocalized positive charge, which leads to decreasing accumulation of these derivatives in mitochondria.
2.6. Summary of the data on sydnone activity
Due to the lack of stability of 1,2,3-oxadiazole, the publications of the last five years virtually do not contain reliable data on their biological activity. A search of the Reaxys and Scifinder databases demonstrated that only eight papers containing new information on sydnone activity were published in the last five years, but in this review, we do not consider much more widespread sydnonimines. As regards the synthesis, all modifications mentioned above were directed towards combination of the sydnone core with a pharmacophore moiety such as ferrocene, BODIPY, D-galactose, or thiazolidinone. In most of considered studies, the selection of sydnone as a substituent to an active basis is not related to specific properties of sydnone, but rather to poor knowledge of this heterocycle and to synthetic interest. Therefore, it is difficult to assess the contribution of this moiety to various types of biological activity. However, even such a small sample of studies provides the conclusion that the highest activity is inherent in sydnones containing either aryl groups with electron-donating (see compounds 12c,d and 39a,b,c) or electron-withdrawing (12a, 23b – h) para-substituents or a small-size substituent at the C(4) atom of the heterocycle (30a, 33a, 46a).
3. Synthesis and activity of 1,2,4-oxadiazoles
From the medicinal chemistry standpoint, the 1,2,4-oxadiazole ring is considered to be a convenient and reliable bioisostere of the carboxyl group, and this finds use in the design of 1,2,4-oxadiazole-based therapeutic agents.[33] As a rule, monosubstituted 1,2,4-oxadiazoles are markedly less stable than disubstituted ones. The disubstituted oxadiazole ring is stable even to the action of strong acids such as concentrated sulfuric acid and strong bases. The C(3) and C(5) atoms in 1,2,4-oxadiazoles are electrophilic centres prone to form 3,5-disubstituted derivatives upon nucleophilic attack (Fig. 6).
![[{"id":"q1WLCqI58W","type":"paragraph","data":{"text":"Active centres of the 1,2,4-oxadiazole moiety (LG is leaving group)"}}]](/storage/images/resized/9BnIvZL74UO4WoB0OqLHIWzOD40MkuVX1eX03r4o_xl.webp)
Besides the fact that the 1,2,4-oxadiazole ring is a bioisostere of the carboxyl, ester, and amide groups characterized by hydrolytic instability,[34] it is also a hydrogen bond acceptor due to the electronegativity of nitrogen and oxygen atoms.[35] Nitrogen is a stronger hydrogen bond acceptor than oxygen.[36] 1,2,4-Oxadiazoles are actively involved in π – π stacking interactions.[37] It is noteworthy that compounds of this type to not tend to form toxic metabolites.[38] The set of the above properties makes 1,2,4-oxadiazole rings highly important structural moieties for drug design as regards interaction with receptors.
3.1. Compounds with antifungal activity
In an attempt to find new compounds with fungicidal activity that could become drug candidates, a series of various amides containing the 1,2,4-oxadiazole moiety was synthesized [39] (Scheme 11). 4-Cyanobenzoic acid 47 reacted with hydroxylamine in the presence of 8-hydroxyquinoline, used as a chelating agent to decrease the fraction of the undesirable benzamide by-product, to afford 4-carboxybenzamidoxime 48, which was then O-acylated and cyclized in the presence of the appropriate anhydride and pyridine (Py). Treatment of 4-(1,2,4-oxadiazolyl)benzoic acids 49a – c with thionyl chloride yielded chlorides 50a – c, which were subsequently allowed to react with secondary amines.
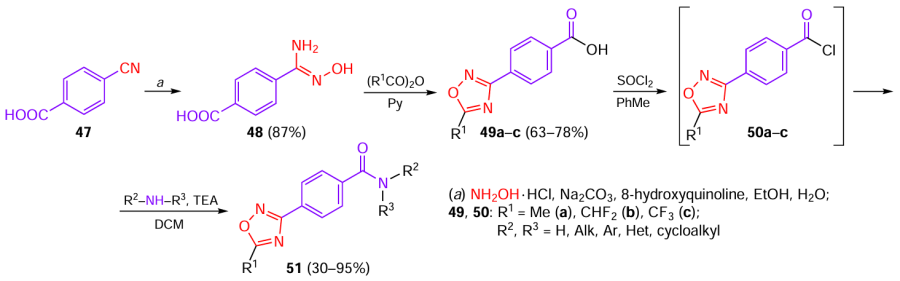
Compounds 51a – w (Fig. 7) at 50 μg mL–1 concentration exhibited moderate or high inhibitory effect against pathogens of rhizoctonia root rot of wheat (Rhizoctonia solani), apple Valsa canker (Valsa mali), white mould (Sclerotinia sclerotiorum), and botrytis gray mould (Botrytis cinerea), with the highest efficiency of inhibition being found for V. mali. The dependence of the inhibition rate on the substitutes present in the molecule was identified.
![[{"id":"mtZCrZRN2K","type":"paragraph","data":{"text":"Structures of 1,2,4-oxadiazoles <b>51 </b>exhibiting antifungal activity and boscalid (drawn in the box).<sup>39</sup>"}}]](/storage/images/resized/LJgvOa6sqZ4CnExx56CumDPkW4EhSWvwcRn4X7MC_xl.webp)
The fungicidal activity varied depending on substituents and differed for amides containing secondary and tertiary amino groups (Table 3). For secondary amides, the introduction of an aliphatic substituent (methyl, ethyl, or isopropyl) in the benzene ring resulted in increasing fungicidal effect (the activity changed in the order 51a < 51b < 51c); compound 51e containing an ortho-phenoxy group exhibited a satisfactory activity, while the presence of a para-phenoxy group (51f) decreased the inhibitory effect for all strains, except for S. sclerotiorum. In the presence of naphthalene moieity, the substituent position played a crucial role, with the most pronounced effect being observed for the unsubstituted naphthalene moiety (51g > 51h » 51i). An increase in the chain length between the amino group and the aromatic moiety increased the fungicidal activity (51j < 51k < 51l), while the chain branching was unfavourable for fungicidal properties (51j < 51k). Among tertiary amides, tetrahydroquinolines demonstrated satisfactory activity, with the best results being found for tetrahydroisoquinoline 51m; the inhibitory effect of this compound was comparable to that of compound 51g.
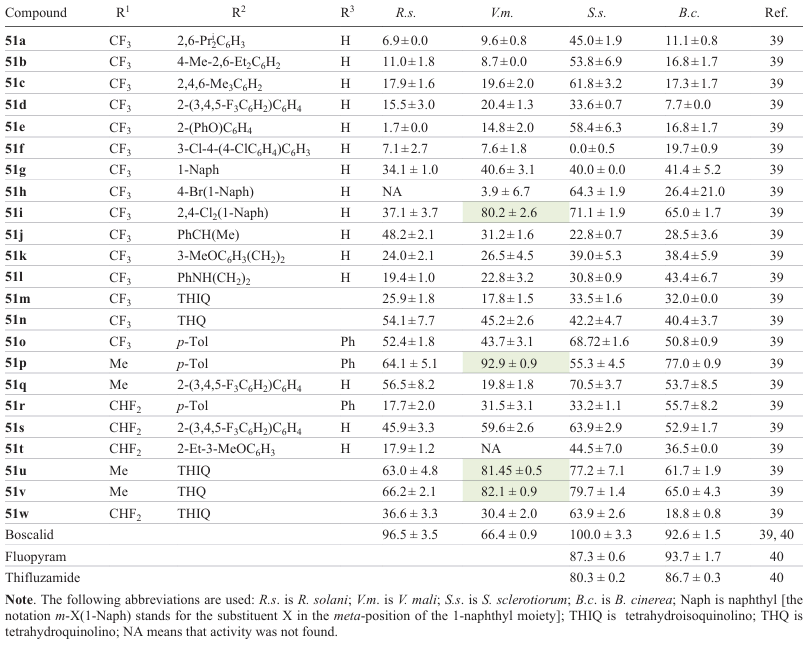
The substituent at the C(5) atom of 1,2,4-oxadiazole was found to affect the antifungal activity.[39] For this purpose, in the molecules of low-activity compound 51d, compounds 51k,o exhibiting moderate activity, and lead compounds 51m,n, which showed a pronounced activity, the CF3 group was formally replaced by CHF2 and CH3. The introduction of a methyl group into diphenylamine-substituted derivatives had a beneficial effect (51p > 51r ≈ 51o); a similar trend was followed for the ortho-(3,4,5-trifluoro-1,1'-biphenyl) derivatives: the introduction of a methyl group markedly improved the activity against the S. sclerotiorum strain (51q ≈ 51s > 51d). For the phenethyl substituent, the presence of a difluoromethyl group induced a considerable increase in the activity against V. mali and S. sclerotiorum (51t > 51k).
In view of the satisfactory activity of the indicated compounds against V. mali, the EC50 values were evaluated for compounds with IR > 80% at 50 μg mL–1; the general trend was in line with the primary screening results, and all test compounds had a better performance than the commercial agent boscalid. The IR values of compounds 51g,u were somewhat higher than 80%, and EC50 were about 10 μg mL–1. It is worth noting that although the inhibition rate for compound 51v was 81.45%, a decrease in the concentration did not induce an obvious decrease in activity, which provided a preferred EC50 value of 3.882 μg mL–1. The IR values for compounds 51m,w exceeded 90%, while the favourable EC50 values were below 5 μg mL–1.
Liu et al.,[40] who addressed 1,2,4-oxadiazole amides (Scheme 12) possessing antifungal properties, described the reaction of various (hetero)aromatic nitriles 52 with hydroxylamine. This gave amidoximes 53, which were then treated with ethyl oxalyl chloride in acetonitrile. Then esters 54 were hydrolyzed with LiOH in ethanol to give acids 55. Compounds 55 reacted with oxalyl chloride to give acid chlorides 56, which were converted to target amides 57a – w by the reaction with primary amines (Table 4).
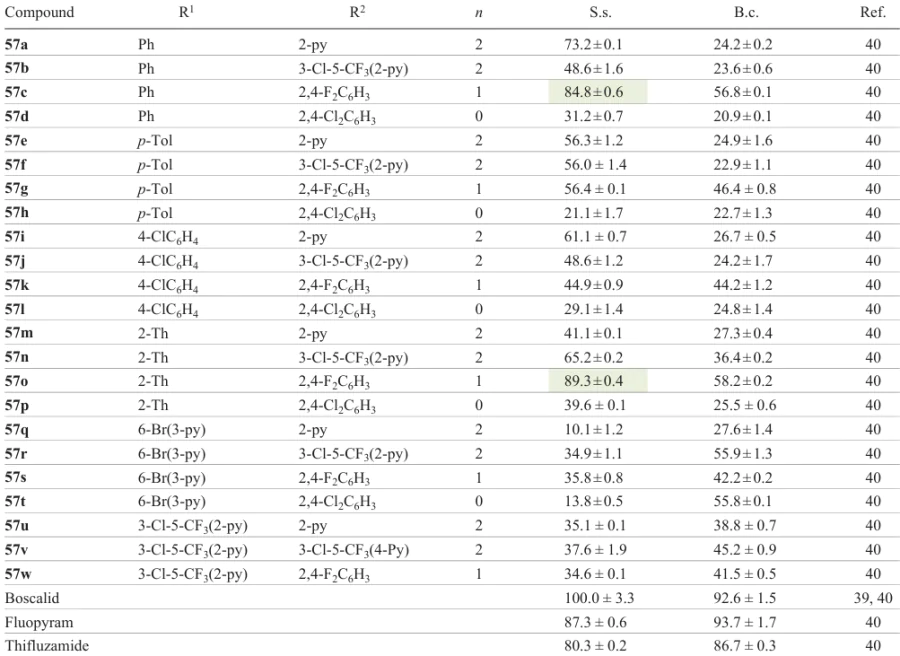
All compounds had moderate or high activity in the inhibition of mycelium growth at a concentration of 50 μg mL–1. In particular, compounds 57c,o,r,t showed a good antifungal activity against B. cinerea, with the inhibition rates being 56.8, 58.2, 55.9, and 55.8%, respectively. This, however, was lower than the inhibition rates of the reference agents fluopyram (87.3%) and thifluzamide (80.3%) (Fig. 8). Meanwhile, compounds 57a,c,i,n,o exhibited a high antifungal activity against S. sclerotiorum, with their inhibition rates being 73.2, 84.8, 61.1, 65.2, and 89.3%, respectively. Compound 57o had the best antifungal activity against S. sclerotiorum, which surpassed the activity of thifluzamide (4.3 μg mL–1) and was comparable with that of fluopyram (1.2 μg mL–1).
![[{"id":"gKj2WT8EpD","type":"paragraph","data":{"text":"Structures of 1,2,4-oxadiazoles-based amides <b>57 </b>with the highest antifungal activity and active ingredients of commercial products (drawn in the box).<sup>40</sup>"}}]](/storage/images/resized/RGfFGq8CUn9OkuANxzKQGdPORPt8kYOFCnJh7wd4_xl.webp)
Analysis of the structure–activity relationships for these 1,2,4-oxadiazoles showed that in the presence of an aryl substituent at C(3) and b-arylethylamine groups, the activity against B. cinerea and S. sclerotiorum was higher for compounds with unsubstituted pyridine (py) ring than for compounds containing a trifluoromethyl group and a chlorine atom in the pyridine ring (see Table 4). This can be illustrated by the following activity series of 1,2,4-oxadiazoles: 57a > 57b ≈ 57e > 57f ≈ 57i > 57j. However, when a heterocyclic substituent was introduced to C(3), the pattern was reversed, and the activity varied as follows: 57n > 57m ≈ 57r > 57q, 57v > 57u. The highest activity was found for compounds with n = 1 and 2,4-difluorophenyl group: the series were 57c > 57a ≈ 57b ≈ 57d and 57o > 57m ≈ 57n ≈ 57p, respectively. In addition, the antifungal effect of compounds with a thiophenyl group was substantially higher than that of compounds with other substituents (57o > 57c > 57g > 57k > 57w). A comparison of a series of compounds with identical substituents revealed the lowest activity for compounds with a 2,4-dichlorophenyl group and n = 0 (57o > 57n > 57m > 57p). Meanwhile, the presence of a 2,4-difluorophenyl group for n = 1 (57o) provided a higher antifungal activity against both B. cinerea and S. sclerotiorum. Thus, the large steric bulk of substituent R had an adverse effect on the fungicidal activity.
The results obtained in vitro were confirmed in vivo for lead compound 57o: it was able to combat the disease caused by S. sclerotiorum, which affected the cabbage leaves, and demonstrated curative and protective effects of 62.3% and 71.0% when used at a dose of 100 μg mL–1; in addition, the application of compound 57o can induce an obvious breakdown and shrinkage of the hyphal morphology of S. sclerotiorum.
Comparison of the inhibitory activity of test compounds 51 and 57 against four fungal strains with the activity of three commercial antifungal agents showed [39] [40] that 1,2,4-oxadiazole amides (in particular, compounds 51i,p,u,v) suppressed the growth of hypha of V. mali more actively than boscalid, while compounds 57c,o inhibited the growth of S. sclerotiorum more effectively than tifluzamide and comparably to fluopyram.
Liu et al.[41] prepared a series of pyrimidine ethers containing trifluoromethyl-1,2,4-oxadiazole, a structural moiety known for the ability to inhibit histone deacetylase and thus be useful for the treatment of rust on plants. Pyrimidinols and trifluoromethyl-1,2,4-oxadiazoles prepared via two- and three-step transformations, respectively, served as the key reactants (Scheme 13). In the first step, esters 58a – d were condensed with ethyl acetate in the presence of sodium hydride in THF. Cyclization of the resulting keto esters 59a – d with formamide afforded pyrimidinols 60. In some cases, the pyrimidine ring was additionally halogenated with N-bromo- (NBS) or N-chloro-succinimide (NCS). For the synthesis of trifluoromethyl-1,2,4-oxadiazoles, first, hydroxylamine was added to substituted aromatic nitriles 61a – c, then the resulting amidoximes 62a – c were cyclized on treatment with trifluoroacetic anhydride, and 1,2,4-oxadiazoles 63a – c and 67 thus formed were subjected to radical bromination of the methyl group with NBS in the presence of azobis(isobutyronitrile) (AIBN) as an initiator. At the final step, bromomethyl-1,2,4-oxadiazoles 64a–d underwent the nucleophilic substitution reaction with pyrimidinols 60 in the presence of potassium carbonate. A similar sequence of reactions was performed for 2-thienylnitrile 65, with amidoxime 66 and 1,2,4-oxadiazole 67 being isolated in intermediate steps. Finally, target products 68 and 69 were obtained.
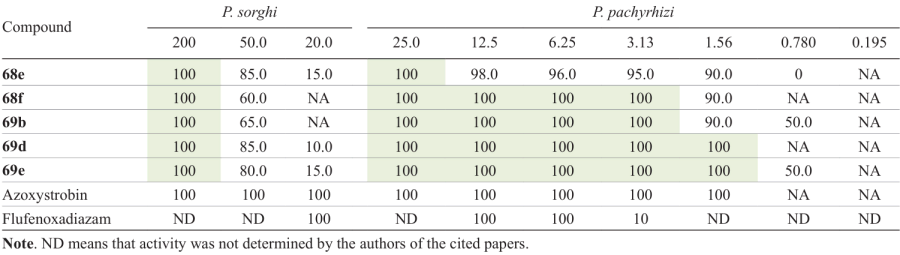
The results of biotesting of compound series 68 and 69 showed that some of them (Fig. 9) possess excellent activity against pathogens that cause rust disease of corn (Puccinia sorghi), soybean (Phakopsora pachyrhizi), and cereal crops (Puccinia rubigo). Most of 5-trifluoromethyl-1,2,4-oxadiazole derivatives at a dose of 200 mg L–1 showed a high antifungal activity against P. sorghi. For example, compounds 68a (R1 = H, R2 = CHFMe, R3 = Br), 68b (R1 = R3 = Cl, R2 = CHFMe), 68c (R1 = R3 = H, R2 = CHFMe), 68d (R1 = H, R2 = CHF2, R3 = Br), 68e (R1 = H, R2 = CHF2 , R3 = Cl), 68f (R1 = R3 = H, R2 = CHF2), 68g (R1 = R3 = H, R2 = Et), 69a (R2 = CHFCH3 , R3 = Cl), 69b, 69c (R2 = CHF2 , R3 = Cl), 69d, and 69e showed an excellent control effect * of 100% at a dose of 200 mg L–1 (see Table 3),[41] which was equal to that of azoxystrobin used as the positive control (100%). Compounds 68h (R1 = H, R2 = CHFCH3 , R3 = F) and 68i (R1 = F, R2 = CHF2 , R3 = H) at the same dose showed a good control effect against P. sorghi, 75% and 80%, respectively, which was, however, somewhat inferior to that of azoxystrobin (100%). When used at a dose of 50.0 mg L–1, compounds 68a,c – g and 69a – e were unable to completely inhibit the growth of P sorghi, and only 68e (85%), 69d (85%), and 69e (80%) demonstrated a good control effect, although it was somewhat lower than that of azoxystrobin (100%). When taken at a dose of 20.0 mg L–1, both series of compounds exhibited no significant control effect against P. sorghi, while azoxystrobin and flufenoxadiazam, a fungicide containing a 1,2,4-oxadiazole ring, showed 100% efficacy.
![[{"id":"mxFa3e_mUg","type":"paragraph","data":{"text":"Structures of pyrimidine ethers of trifluoromethyl-1,2,4-oxadiazoles <b>68 </b>and <b>69 </b>exhibiting the highest antifungal activity and active ingredients of commercial fungicides (drawn in the box).<sup>41</sup>"}}]](/storage/images/resized/FCa2MCpX7uwFfpAXhNd2QXtVzG5Y81hXaSkQ8zsk_xl.webp)
Compounds that were most active against P. sorghi (see Fig. 9) were also studied for the antifungal activity against P. pachyrhizi (Table 5). In the concentration range from 25.0 to 3.13 mg L–1, derivatives 68f and 69b,d,e showed an excellent activity against this strain, with the control effect being 100%. This value coincided with that of the commercial fungicide azoxystrobin. Meanwhile, 1,2,4-oxadiazole 68e provided a somewhat lower control (> 95%). As the dose was reduced to 1.56 mg L–1, the control effect of compounds 68e (90%), 68f (90%), and 69b (90%) decreased, while that of azoxystrobin was still 100%. It is noteworthy that compounds 69d,e at this dose showed an excellent activity against P. pachyrhizi, with the control effect being 100%. When the dose was halved (0.780 mg L–1), compounds 69b,e had a moderate control effect (50%), with that of azoxystrobin being 0%; as the concentration was further decreased down to 0.195 mg L–1, none of the five compounds provided any control of P. pachyrhizi. The above biological analysis indicates that these products have a higher activity against P. pachyrhizi than flufenoxadiazam (10% at a concentration of 3.13 mg L–1).
The structure – activity relationship was elucidated for compounds 68: when the benzene ring linked to trifluoromethyl-1,2,4-oxadiazole is unsubstituted (68a,d,c – g) or the trifluoromethyl-1,2,4-oxadiazole moiety is linked to a thiophene ring (69a – e), these derivatives provide a high protective effect against the rust disease. The activity also depends on the nature of substituents R2 (CHF2 > CHFMe > Pri > Et) and R3 (Cl > Br > H). Meanwhile, if the benzene ring at the trifluoromethyl-1,2,4-oxadiazole moiety contains F or Cl substituent, the activity disappears. The highest activity was found for thiophene derivative 69e (R2 = CHF2 , R3 = H), which provided 50% control of P. pachyrhizi at a concentration of 0.780 mg L–1, which is much better than the result obtained for the commercial fungicide azoxystrobin under similar conditions (0%). Field test results demonstrated that compound 69e has an excellent control effect (70.8%) against P. rubigo at a dose of 116 g per hectare. Analysis of acute toxicity showed that this derivative has low toxicity with a half-lethal dose (LD50) > 500 mg kg–1. In addition, according to the results of enzyme activity analysis, compound 69e is a strong non-selective inhibitor of histone deacetylases 4 and 6 with pIC50 = 5.4 and 8, respectively.41
* Control effect is defined by comparison of the frequency of fungal damage in the treated and control areas.
3.2. 1,2,4-Oxadiazoles with nematicidal activity
Plant-parasitic nematodes severely affect agricultural production worldwide, but there are few known effective non-toxic nematicides. Tioxazafen is a new nematicidal agent containing a 1,2,4-oxadiazole moiety for the treatment of seeds, which was developed by Monsanto and provides regular control of nematodes with a broad range of action in corn, soya, and cotton plant.[42] At the same time, field tests have shown that this product can also be effective against root knot nematodes in agricultural crops.[43] In recent years, several new products containing amide moieties and characterized by excellent broad-spectrum nematicidal activity (fluopyram, fluazaindolizine, and cyclobutrifluram) have been developed.[44][45] The introduction of amide moieties into 1,2,4-oxadiazole molecules became a promising strategy of the search for new nematocides. Apart from the antifungal activity, antihelmintic activity was evaluated for the series of compounds 57 (Fig. 10).
![[{"id":"i9gmXrKsm7","type":"paragraph","data":{"text":"Structures of 1,2,4-oxadiazole amides <b>57 </b>with nematicidal action and active substances of commercial products (drawn in the box).<sup>40</sup>"}}]](/storage/images/resized/H9JhOqsU0A1WtGugkfzHXLLTEHdbw9Uwa3Tyf25a_xl.webp)
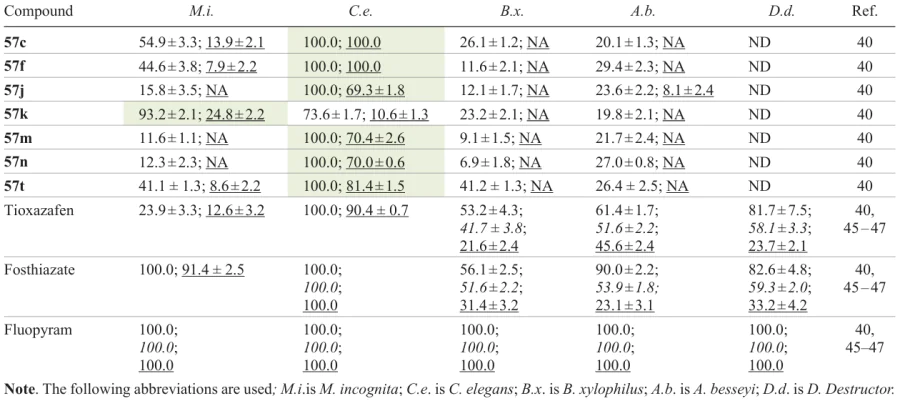
Compound 57k at a dose of 200 μg mL–1 showed an excellent nematicidal activity against southern root knot nematode (Meloidogyne incognita) with a corrected mortality rate 48 h after treatment being 93.2%, which exceeded this value for tioxazafen used as a positive control (23.9%) (Table 6). Meanwhile, compounds 57c,f,j,m,n,t showed a good anthelmintic activity against free-living soil nematode (Caenorhabditis elegans); the mortality rate was 100% at a dose of 200 μg mL–1. In particular, 1,2,4-oxadiazoles 57c,f exhibited 100% mortality rate even at a dose of 50 μg mL–1, which is superior to that of tioxazafen (90.4%) and similar to mortality characteristics of the commercial products fosthiazate and fluopyram. Also, the target compounds at high concentrations showed a certain nematicidal activity against rice leaf nematode (Aphelenchoides besseyi) and pine wood nematode (Bursaphelenchus xylophilus). The structure – activity relationship for various types of nematodes is variable, and no common structural patterns were identified. The most active derivative 57f, which shows both nematicidal and antifungal activities, was tested for the cytotoxicity against the human liver cell line L-02; the cytotoxicity proved to be lower than those of fluopyram and thifluzamide used as positive controls.
Another study[45] devoted to the search for new nematicidal agents was focused on the structural modification of fluopyram. The flexible ethyl group connecting the amide and pyridine moieties was replaced by structurally rigid 1,2,4-oxadiazole ring. For this purpose, a one-pot method was developed, which included the cyclization of amidoxime 70 with benzoyl chlorides followed by removal of the tert-butoxycarbonyl protecting group in intermediates 71. This gave intermediates 72 with a free amino group, which were acylated in situ to afford the target compounds 73 (Scheme 14).
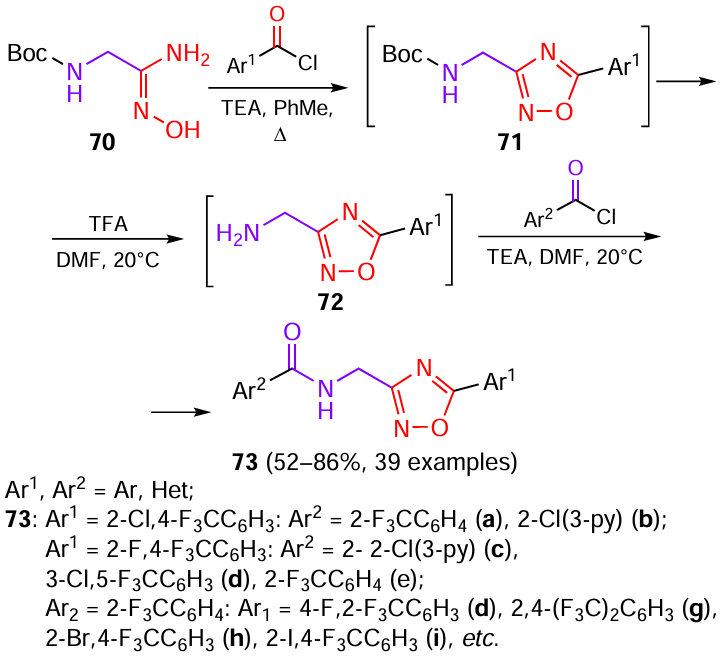
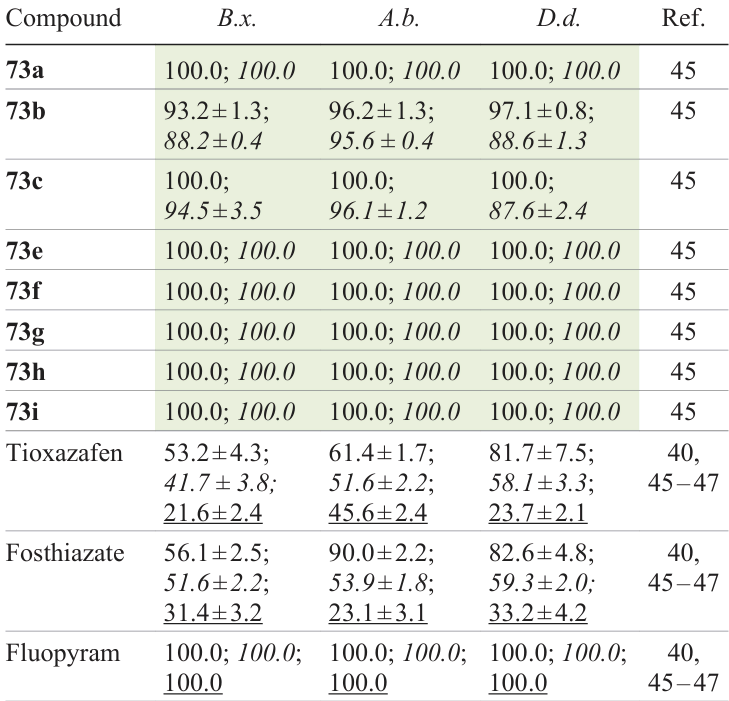
All 39 amide derivatives 73 were tested for nematicidal activity, and some of them showed outstanding results (Fig. 11). At a concentration of 200 mg L–1, the activity of compounds 73a – c,e – i against B. xylophilus, A. besseyi, and Ditylenchus destructor (potato rot nematode) exceeded 90%. The activity against the above nematodes for compounds 73a,e,f – i was retained at 100% level even when the concentration decreased down to 100 mg L–1, which was higher than the activity of the tioxazafen control (51.6, 81.7, and 82.6%, respectively) and comparable with the results for fluopyram (100%) (Table 7)[40][45-47].
![[{"id":"pMrL1UNXmZ","type":"paragraph","data":{"text":"Structures of 1,2,4-oxadiazole-based amides <b>73 </b>that showed a pronounced nematicidal activity.<sup>45</sup>"}}]](/storage/images/resized/fizvWgYNHzdQZLhcUOnUV0c1NE3orTpJRPG7eAdl_xl.webp)
For the most active compounds 73d – h, median lethal concentrations (LC50) against B. xylophilus, A. besseyi, and D. destructor were estimated to be 1.39 – 3.09 mg L–1. This means that these compounds were much more effective than the control nematicide, thioxazafen (106, 49.0, and 75.0 mg L–1 for the above nematodes, respectively). Compound 73d has a potent inhibitory effect on the nutrition, reproduction, and egg laying of nematodes and efficiently induced oxidative stress in nematode cells, thus damaging the intestine system. It is noteworthy that compound 73d considerably inhibited the succinate dehydrogenase (SDH) in nematodes, which resulted in blockage of the electron transfer in the respiratory chain and thus hampered the ATP synthesis, affecting the whole oxidative phosphorylation process in nematodes and finally resulting in their death.
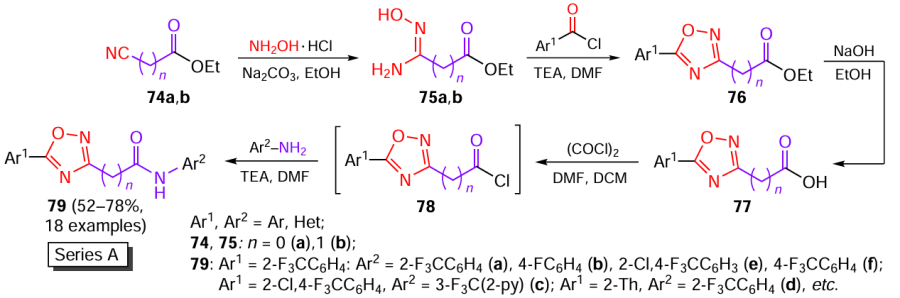
A similar synthetic approach was used to obtain three series (A – C) of 1,2,4-oxadiazole derivatives based on tioxazafen with high structural flexibility.[46] To this end, ethyl cyanoformates 74a,b were treated with hydroxylamine to give amidoximes 75a,b, which were then allowed to react with various aroyl chlorides in the presence of triethylamine. The resulting 1,2,4-oxadiazoles 76 were hydrolyzed to carboxylic acids 77, which were converted to acyl chlorides 78 on treatment with oxalyl chloride. Compounds 78 reacted in situ with primary amines to form compounds 79 belonging to series A (Scheme 15). Intermediate compounds 74 – 77 were isolated by procedures reported by Liu et al.[48] and Voronova et al.;[49] however, the yields of products were not indicated.
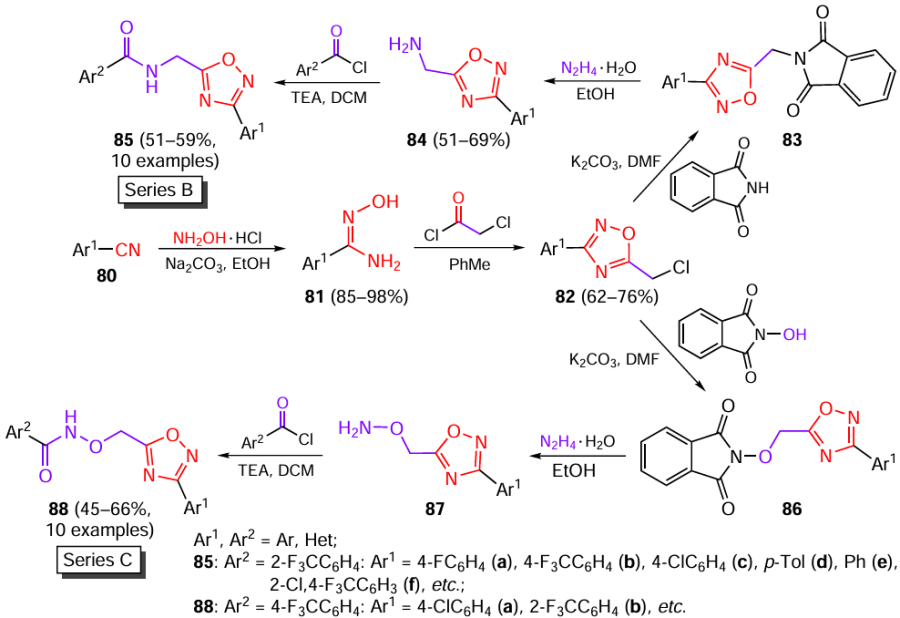
The pharmacologically oriented compounds of series B and C were synthesized starting from substituted benzonitriles 80, which were converted to amidoximes 81, and this was followed by the heterocyclization with chloroacetyl chloride[50] (Scheme 16). Using procedures reported by Ölmez and Waseer,[51] the resulting 5-chloromethyl-1,2,4-oxadiazoles 82 were treated with phthalimides. In the case of phthalimide with a free NH group, this gave derivatives 83, which were treated with hydrazine hydrate to obtain free primary amines 84, while the subsequent reaction with acyl chlorides furnished target compounds 85 (series B). A similar sequence of reactions for N-hydroxyphthalimide involved the intermediate formation of compounds 86 and 87 and resulted in the formation of 1,2,4-oxadiazoles 88 (series C). The yields of intermediates 82, 86, and 87 were not reported in the original publication.
The indicated series of amides A – C (Fig. 12, Table 8) were tested in vitro for the nematicidal activity against B. xylophilus, A. besseyi, and D. destructor. 1,2,4-Oxadiazoles 85b and 88a exhibited high activity against the B. xylophilus nematodes when incubated at concentrations of 200 μg mL–1 for 48 h, with the corrected mortality rates being 90.7 and 61.2%, respectively; this was higher than that for tioxazafen used as the control (41.7%). The results obtained for compounds 79a (45.3%), 79b (44.8%), 79c (47.1%), 79d (50.9%), 88a (37.1%), and 85a (38.8%) were compared with those of tioxazafen. Derivative 85b exhibited a high nematicidal activity against A. besseyi with a mortality rate of 93.6% at a dose of 200 μg mL–1, which surpassed the results for tioxazafen (75.6%) and fosthiazate (53.9%); also, this compound was active against D. destructor with corrected mortality rate of 88.7% (for 200 μg mL–1), which was better than that of tioxazafen (58.1%). For the most active compound 85b, median lethal concentrations were found; LC50 values were 37.2, 36.6, and 43.4 μg mL–1 for B. xylophilus, A. besseyi, and D. destructor, respectively; this was higher than LC50 of tioxazafen used as the positive control. According to tentative results of studies of the biological action mechanism, compound 85b not only suppressed the reproduction of B. xylophilus populations, but also affected the production of reactive oxygen species (ROS) and the accumulation of lipofuscin and lipids. In addition, this agent efficiently inhibited succinate dehydrogenase (IC50 = 45.5 μmol L–1).
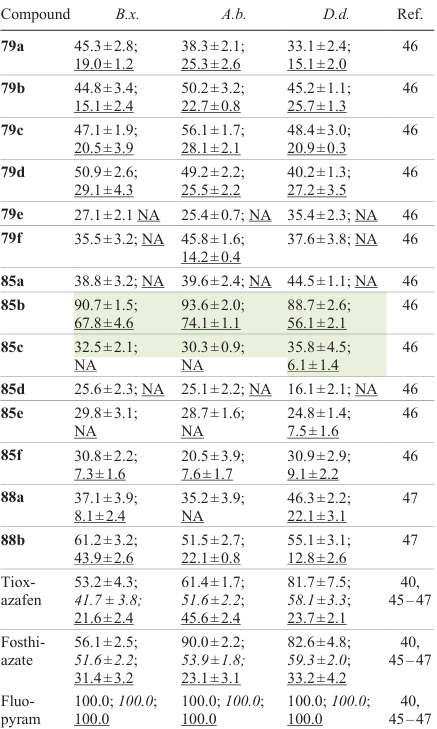
![[{"id":"_NPGwz9tnq","type":"paragraph","data":{"text":"Structures of 1,2,4-oxadiazole amides <b>79</b>, <b>85</b>, and <b>88 </b>possessing the highest nematicidal activity.<sup>46</sup>"}}]](/storage/images/resized/p2fOSqUEOiJg3l2YFkIXbxGwpNFbweM1rszgUL4K_xl.webp)
Using the obtained values of the nematicidal activity against B. xylophilus, Ou et al.[46] analyzed the structure – property relationships for the above series of compounds. It was found that upon the introduction of an amide moiety to the C(3) atom of 1,2,4-oxadiazole for Ar1 = 2-F3CC6H4 , the activity was higher for compounds with one substituent in the aromatic ring than for disubstituted compounds: 79f (Ar1 = 2-F3CC6H4, Ar2 = 4-F3CC6H4) > 79e (Ar1 = 2-F3CC6H4, Ar2 = 2-Cl,4-F3CC6H3). Meanwhile, the introduction of a trifluoromethyl group in the ortho-position of the aromatic ring in R2 led to increasing nematicidal activity against B. xylophilus: 79a > 79f. In addition, replacement of the amide moiety by a hydroxamate moiety decreased the nematicidal activity: series B compounds showed better results than analogous compounds of series C, e.g., 85b > 88b. In series B, the presence of an electron-donating group in the substituent R1 promoted an increase in the nematicidal activity against B. xylophilus: 85a > 85c (Ar1 = 4-ClC6H4 , Ar2 = 2-F3CC6H4) > 85e (Ar1 = Ph, Ar2 = 2-F3CC6H4) > 85d (Ar1 = p-Tol, Ar2 = 2-F3CC6H4). For Ar2 = 2-F3CC6H4 , the presence of a chlorine atom in the para-position of Ar1 decreased the activity: 85b > 85f (Ar1 = 4-FC6H4 , Ar2 = 2-Cl-4-F3CC6H3).
One more type of hybrid structures capable of acting as nematicidal agents combined 1,2,4-oxadiazole ring with a chalcone moiety.[47] According to early studies,[52] chalcone derivatives possess certain nematicidal activity. The chosen chalcones 89 and 90 were prepared by aldol condensation of benzaldehydes and acetophenones (Scheme 17). They were subjected to nucleophilic substitution reaction with chloroethyl 1,2,4-oxadiazole derivative 82 to give target compounds 91 and 92.
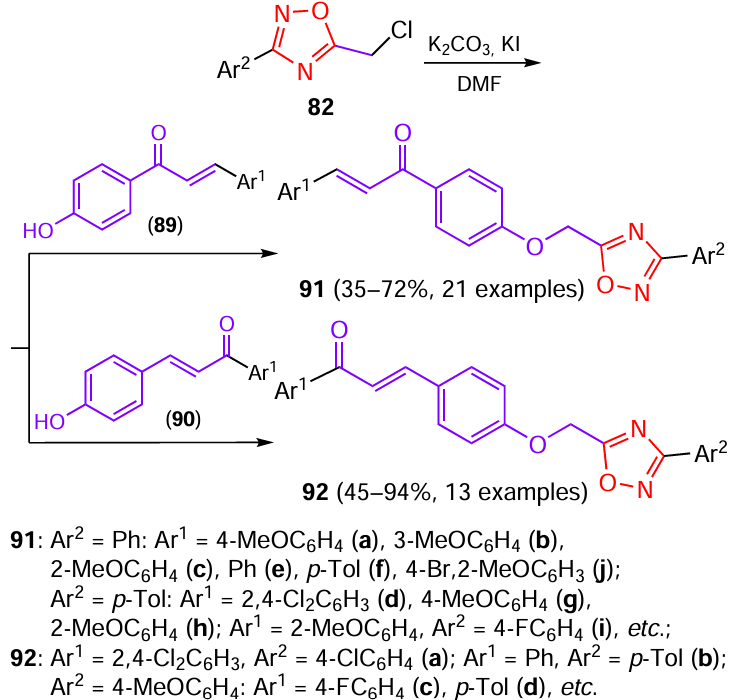
Compounds 91 and 92 were investigated for the activity against nematodes of three plants (Fig. 13, Table 9).
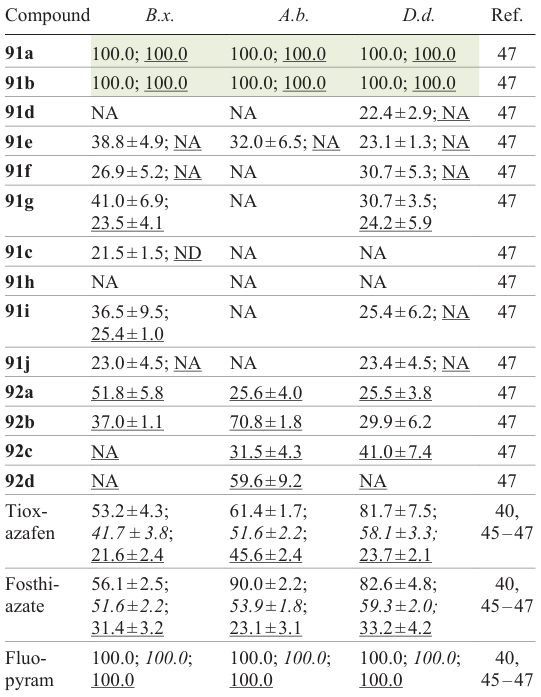
![[{"id":"yrkOG_Opvj","type":"paragraph","data":{"text":"Structures of hybrids <b>91 </b>and <b>92 </b>based on chalcone and 1,2,4-oxadiazole with high nematicidal activity and abamectin (drawn in the box).<sup>47</sup>"}}]](/storage/images/resized/O4XUQxazN3GcVQGTN9c7zMW4AVImaVxmtPfWdrlJ_xl.webp)
According to the results of biological assays, compounds 91a,b had a high nematicidal activity against B. xylophilus, A. besseyi, and D. destructor with LC50 values of 35.5, 44.7, and 30.2 μg mL–1 and 31.8, 47.4. and 36.5 μg mL–1 for these two compounds, respectively, which is superior to these values for known commercial products: tioxazefen, fosthiazate, and abamectin. Compounds 91a,b and 92a at a concentration of 50 μg mL–1 showed a higher nematicidal activity against B. xylophilus: the corrected mortality rates were 100, 100, and 51.8%, respectively; this surpasses the data for tioxazefen (34.3%), fosthiazate (43.9%), and abamectin (49.4%). Also, compounds 91a,b and 92b,d at a concentration of 50 μg mL–1 exhibited a good nematicidal activity against A. besseyi, with the corrected mortality rates being 100, 100, 70.8, and 59.6%, which is better than the results for tioxazefen (40.0%), and abamectin (42.3%). In addition, compounds 91a,b and 92c had a high activity against D. destructor characterized by corrected mortality rates of 100, 100, and 41.0%, respectively, which exceeded these characteristics for tioxazefen (29.0%), fosthiazate (33.3%), and abamectin (33.6%). However, the activity of any of the compounds taken in 10 μg mL–1 concentration against three types of nematodes was unsatisfactory.
Comparison of the nematicidal activity of the 1,2,4-oxadiazoles at three different concentrations (200, 100, and 50 μg mL–1) obtained in the above studies for five nematode species with the activity of three commercial agents (see Table 4 ) made it possible to identify the lead compounds. Thus amide 57k showed the highest activity against M. incognita, with the corrected mortality rate exceeding 90% at a concentration of 200 μg mL–1; this surpasses the results for tioxazafen by a large factor. Other active compounds of this series showed remarkable results against C. elegans: at a concentration of 200 μg mL–1, 1,2,4-oxadiazole amides 57c,f,j,m,n,t demonstrated 100% activity, and in the case of 57c,f, the maximum value was also retained upon a twofold decrease in the concentration. 1,2,4-Oxadiazole amides 73a,e – i provided the highest mortality rate for B. xylophilus, A. besseyi, and D. destructor at concentrations of 200 and 100 μg mL–1, while hybrids based on chalcones 91a,b showed 100% activity at concentrations of 100 and 50 μg mL–1, thus surpassing the tioxazafen and fosthiazate and being similar in activity to fluopyram. A high activity (corrected mortality rate > 85%) was also found for 1,2,4-oxadiazole amides 73b,c and 85b.
3.3. Compounds with anti-inflammatory activity
1,2,4-Oxadiazoles are well-known pharmacophores with a clear-cut therapeutic effect in inflammatory processes,[53] which allows them to be used for the treatment of Parkinson’s disease (PD).[54][55] The combination of a 1,2,4-oxadiazole ring and a flavonoid natural skeleton known to possess anti-inflammatory,[56] antioxidant, and neuroprotective properties[57] in a single molecule produced compounds with potential activity against PD.[58] The 1,2,4-oxadiazole ring was synthesized from nitrile derivatives using a standard two-step approach (Scheme 18). In the first step, nitriles 93a – o were converted to amidoximes 94a – o, which were introduced in one-pot cyclization with methyloxophenyl-4H-chromenecarboxylic acid using hexafluorophosphate benzotriazoletetramethyluronium (HBTU) as a coupling reagent and a Hünig’s base to give flavone derivatives 95a – o substituted at C(8).
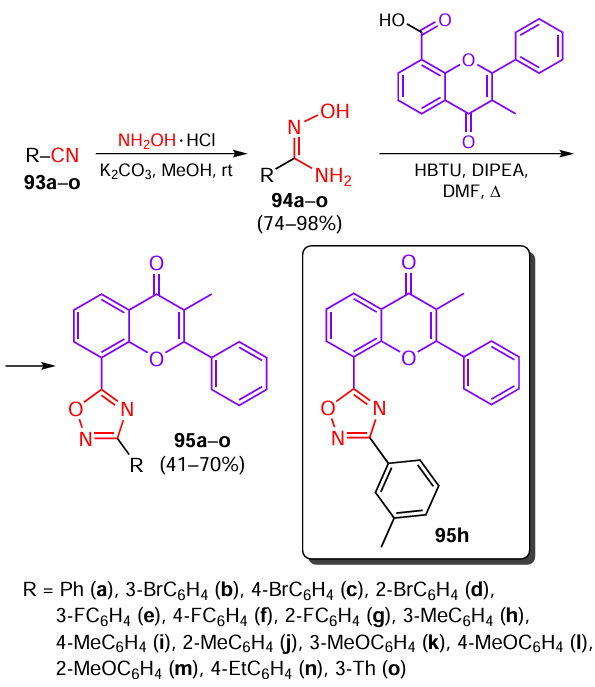
Compounds 95a – o were tested for the ability to inhibit the release of NO and ROS.[58] Derivatives 95a,e,i – k,m inhibited the NO formation in the range from 22.4 to 36.9%, while the inhibition rate for 1,2,4-oxadiazoles 95b,c,f – h,l,n was in the range from 41.6 to 64.2%. The inhibition of ROS production in LPS-induced BV2 nerve cells (LPS is lipopolysaccharide) was also studied: only compounds 95k,m showed a weak inhibitory effect at a concentration of 5 μM, whereas for other compounds of this series the activity at this concentration was relatively high. Thus, the substituents such as phenyl (95a), o-tolyl (95i), p-tolyl (95j), m-methoxyphenyl (95k), m-fluorophenyl (95e), or p-methoxyphenyl (95m) decreased the inhibitory activity of the compounds, while the best result was found for compound 95h. According to in vitro experiments, compound 95h exerted an anti-inflammatory effect by inhibiting MAPK and NF-κB signalling pathways, which was favourable for the balance between the M1 and M2 types of microglia. Meanwhile, using the BV2 cell model, it was found that compound 95h had an antineuronal apoptotic effect. In was found in in vivo study that 95h improved the motor capability and corrected the behavioural disorders and also increased the serum dopamine levels in PD mouse models.
Multiple inhibition of the biological targets involved in the biosynthesis of anti-inflammatory eicosanoids represents an innovative strategy for treating inflammatory diseases because of its higher efficacy and safety.[59] Potenza et al.[53] described 1,2,4-oxadiazole-based inhibitors of eicosanoid biosynthesis. The synthesis was performed according to a standard two-step protocol (Scheme 19): aromatic nitrile 96 was allowed to react with hydroxylamine, and the resulting amidoxime 97 was subjected to cyclization with various carboxylic acids in the presence of HBTU and DIPEA, which gave products 98a – k. Compounds 98h – k contained Boc-protected amino groups; the subsequent deprotection was performed by treatment with trifluoroacetic acid in dichloromethane.
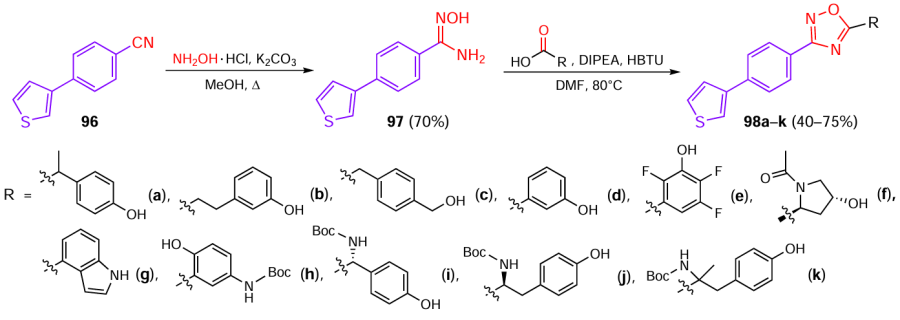
As a result of biological assays, three most active 1,2,4-oxadiazoles 98a,b,e were identified; each of them was characterized by IC50 in the micromolar range [a representative value is IC50 = 3.6 ± 0.7 μM for compound 98e and microsomal prostaglandin E2 synthase-1 (mPGES-1)]. In addition, these compounds acted as antagonists of the 5 lipoxygenase-activating protein (FLAP) (98a,b) and as multitarget inhibitors (98e) of arachidonic acid cascade enzymes including cyclooxygenase-1 (COX-1), 5 lipoxygenase (5-LO), and mPGES-1. According to results obtained in vivo, compound 98e can reduce leukocyte migration in a zymosan-induced peritonitis model and modulate the production of IL-1β and TNF-α.
Combining pyrazolo[3,4-d]pyrimidine moiety known for anti-inflammatory activity [60-62] with the 1,2,4-oxadiazole ring resulted in the formation of hybrids exhibiting synergistic properties.[63] The one-pot two-step preparation of intermediate pyrazolo[3,4-d]pyrimidines was the key to the synthesis of the target products (Scheme 20). 2-Ethoxymethylenemalononitrile was reacted with phenylhydrazines 99a,b, and the aminocyanopyrazoles 100a,b thus formed were heated in formic acid. This induced pyrimidine ring closure and afforded products 101a,b, which were then N-alkylated with ethyl chloroacetate in the presence of anhydrous potassium carbonate. Ethyl esters 102a,b were not isolated, but were subjected in situ to acid hydrolysis with concentrated hydrochloric acid. The carbonyl group of the resulting acids 103a,b was activated with CDI and subjected to heterocyclization with amidoximes to afford target 1,2,4-oxadiazoles 104a – h and 105a – h.
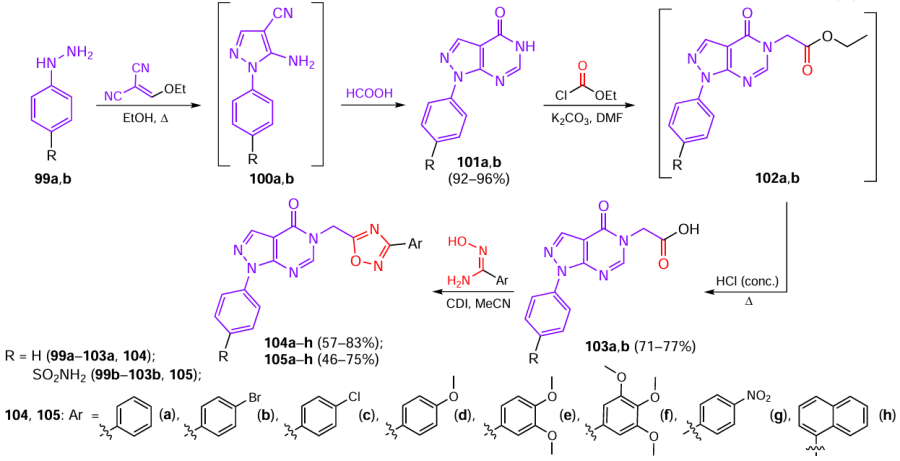
(1,2,4-Oxadiazolyl)pyrazolo[3,4-d]pyrimidine derivatives 104a – h and 105a – h were investigated for the inhibitory effect in vitro against COX-1, COX-2, and 5-LOX and for the ability to inhibit NO release, in order to evaluate the anti-inflammatory potential of the products. Most of the compounds showed the inhibitory activity at a micromolar level, while sulfamides 105a – h mainly exhibited selectivity to COX-2 with the following IC50 values (M):
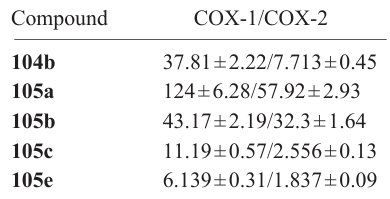
The highest activity was found for compound 105e, containing a dimethoxyphenyl substituent, which proved to be a good COX-2 and 5-LOX dual inhibitor and also demonstrated an acceptable inhibition of NO release (66.02%). Compounds 104e,f and 105e,f effectively inhibited 5-LOX, with the IC50 values being 2.833 ± 0.17, 1.952 ± 0.11, 2.662 ± 0.13, and 1.573 ± 0.08 μM, respectively), and NO release with inhibition rates of 73.85, 65.57, 66.02, and 72.28%. The anti-inflammatory analysis in vivo demonstrated that product 104e was most effective with minimal prevalence of gastric ulceration. Elucidation of the structure–activity relationship (SAR) for in compounds 105a – h showed that a combination of the pyrazolo[3,4-d]pyrimidine core and 1,2,4-oxadiazole with sulfonamide groups provides the highest inhibitory potential against COX-2. The presence of di- (104e, 105e) or trimethoxyphenyl (104h, 105h) groups in compounds increased the inhibitory activity against 5-LOX. When the molecule contained a sulfonamide group, halogen substituents in the benzene ring (104b, 105b,c) increased the COX-1/COX-2 selectivity of the inhibition, while the presence of a nitro group at the para-position (104g, 105g) provided the most effective inhibition of the inflammation mediator (NO) release.
3.4. Compounds with antiviral activity
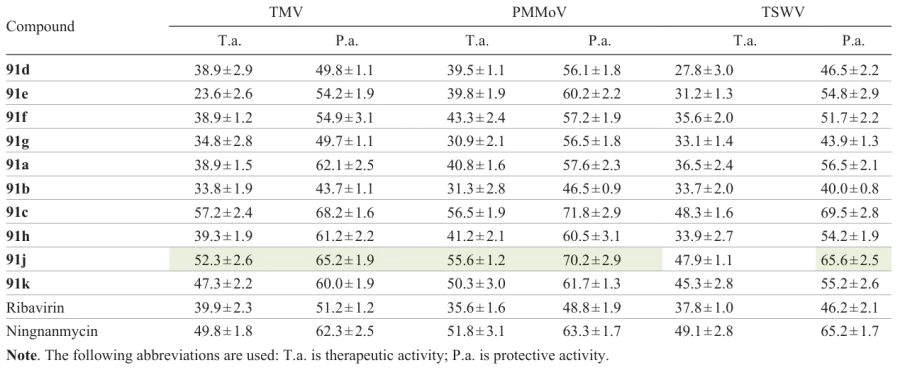
Compounds 91 and 92 described above were also investigated for the activity against the tobacco mosaic virus (TMV), pepper mild mottle virus (PMMoV), and the tomato spotted wilt virus (TSWV) (Fig. 14) by the half-leaf spot method.[51] In terms of the antihelminthic activity, lead compound 91j also demonstrated an excellent antiviral activity: the EC50 values against BTM, PMMoV, and TSWV were 210.4, 156.2, and 178.2 μg mL–1, respectively, which is superior to the values for ningnamycin (EC50 = 242.6, 218.4, and 180.5 μg mL–1).
![[{"id":"gxDa4AjXYp","type":"paragraph","data":{"text":"Structures of hybrids <b>91 </b>with high antiviral activity and active ingredients of commercial agents (drawn in the box).<sup>51</sup>"}}]](/storage/images/resized/753nfe83GdJqtg3jGp2Wr5ouimB7IMbwvscSpd5H_xl.webp)
Using the antiviral activity data, the structure – activity relationship was elucidated. Compounds 91 were found to possess a higher antiviral activity than isomeric compounds 92, which is in line with the correlation found for the nematicidal action. Derivatives containing a methoxy-substituted aryl moiety exhibited greater antiviral activity than compounds with other groups, with the activity varying in the order 91a > 91j ≈ ≈ 91f ≈ 91e. Furthermore, compound 91c (Table 10), possessing the highest activity in this series contains a methoxy group in the ortho-position of one benzene ring, with the antiviral properties of methoxy-substituted derivatives varying in the following order: 91c > 91a > 91b. An electron-withdrawing group (F atom) in Ar2 increases this type of activity compared to an electron-donating group (methyl), i.e., 91j > 91h > 91g > 91d.
In 2022, Kumar Kushwaha et al.[64] proposed a new synthetic approach to 3-isoxazolylmethyl(1,2,4-oxadiazole) derivative exhibiting activity against human immunodeficiency virus (HIV) (Scheme 21; the values in parentheses indicated under the structures are HIV-1 inhibition rates).[64] The starting pyran-3-carbonitriles 106a – e were obtained in one step by condensation of α-aroylketene dithioacetals with malononitrile, which was described in detail in earlier publications of this research group.[65][66] On treatment with hydroxylamine under mild basic conditions, pyrans 106a – e undergo heterocyclic ring opening followed by isoxazole ring closure, and the subsequent addition of the second hydroxylamine molecule affords amidoximes 107a – e. The intermediate amidoximes 107a – e were functionalized with a 3-trifluoromethylbenzoyl group in the presence of HATU, which was followed by microwave-assisted intramolecular cyclization to form final 1,2,4-oxadiazoles 109a – e. In the original publication,[64] the authors postulated acylation of the amino group in amidoximes 107; however, it seems more correct to consider intermediates 108 as O-acylated derivatives.
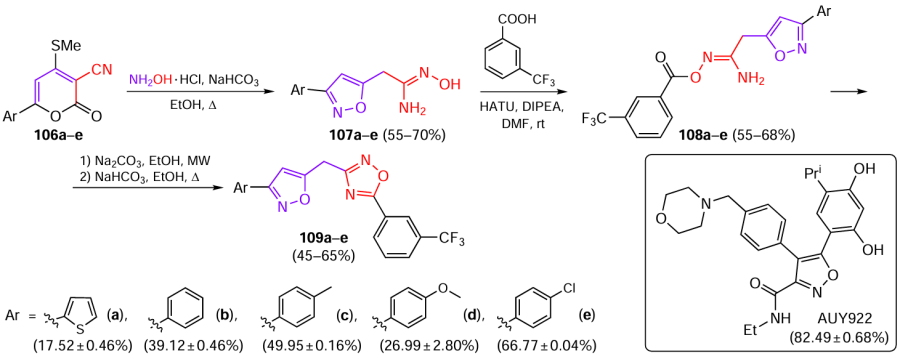
Derivatives 109a – e did not exhibit cytotoxic activity at a concentration of 100 nM. The potential of molecules 109a – e to inhibit HIV-1 replication was analyzed using enzyme-linked immunosorbent assay (ELISA) to detect p24 antigen in HIV-1 NL4.3 supernatants in CEM-GFP cells. Luminespib (commercial designation AUY922) was used as the positive control. Derivative 109e demonstrated the maximum anti-HIV activity, while 109c showed a moderate inhibition of HIV-1 replication. Therefore, compound 109e was used for determination of the therapeutic index (TI).* However, for 109e, TI was three times lower (154.96) that for AUY92 (469.58). Compound 109e did not inhibit any of the three major viral enzymes (reverse transcriptase, integrase, and protease), suggesting that the antiviral activity of 109e may be due to a target cell factor.
Qiu et al.[67] synthesized a series of benzimidazolyl-substituted 1,2,4-oxadiazoles that acted as potential immunomodulatory agents against hepatitis B virus (HBV). In the first step, 4-chloro-3-nitrobenzonitrile 110 reacted with hydroxylamine to form amidoxime 111, which cyclized with 3-substituted propionic acids yielding 1,2,4-oxadiazoles 112a – c (Scheme 22).
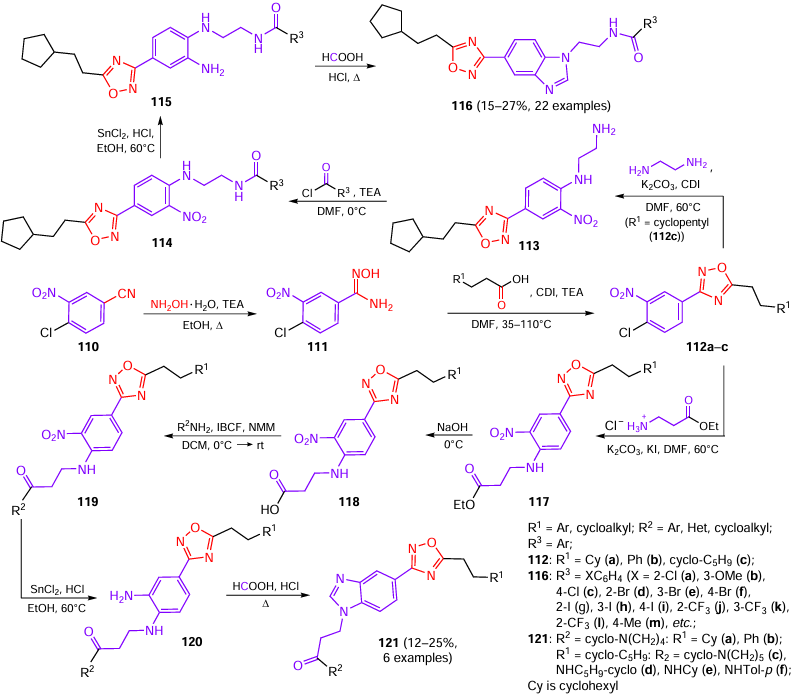
Next, two series of compounds were obtained. The synthetic protocol for the first series of target products began from intermediate compounds 112c. 1,2,4-Oxadiazole 112c reacted with ethylenediamine to give amine 113, which was then acylated to give amides 114. After reduction of the nitro group in these compounds with stannous chloride, the o-phenylenediamine derivatives 115 were subjected to cyclization with formic acid to obtain the target compounds.
The second series of target compounds was synthesized from 112a–c. The addition of β-alanine ethyl ester hydrochloride, potassium carbonate, and potassium iodide in DMF to solutions of 112a–c resulted in the formation of esters 117, which were converted to carboxylic acids 118 by alkaline hydrolysis. Then acids 118 and amines were converted to amides 119 in the presence of N-methylmorpholine (NMM) and isobutyl chloroformate (IBCF). The nitro group in amides 119 was reduced to amino group with stannous chloride to give substituted o-phenylenediamines 120. At the final step, refluxing of compounds 120 in formic acid resulted in the formation of the benzimidazole moiety. The second series of target products 121a – f was isolated in moderate yields (see Scheme 22). However, the authors did not present the yields of intermediate compounds.[67]
Relying on the obtained data on the degree of inhibition of HBV reproduction in vitro (Fig. 15), the authors described the influence of the structure on the activity for the compounds of both series in the following way: a cyclopentyl group considerably increases the activity of 1,2,4-oxadiazole derivatives against HBV. When the cyclopentyl group is replaced by a cyclohexyl or phenyl group, the anti-HBV activity sharply decreases (cf. activity of compounds 121a,b and L700), while the replacement of the pyrrolidine ring by a piperidine ring may increase the anti-HBV activity in vitro (121c > L700). Conversely, replacement of the pyrrolidine ring by a cyclopentylamine (121d), cyclohexylamine (121e), or p-toluidine (121f) moiety resulted in a substantial decrease in the activity against HBV. Interestingly, the amide bond isomerism is also important: the replacement of the C(O)NH group by the regioisomeric NHC(O) group increases the anti-HBV activity (121f > 116m). Furthermore, the introduction of substituents into the benzene ring increases the ability to kill the hepatitis virus. Compounds containing meta-substituents show a relatively higher activity than compounds substituted at other positions (116b > 116a,c; 116e > 116d,f; 116h > 116g,i; 116k > 116j,l).
![[{"id":"z-xT0R9g3T","type":"paragraph","data":{"text":"Structures of benzimidazolyl-1,2,4-oxadiazoles <b>116 </b>and <b>121 </b>possessing a strong anti-hepatitis activity and L700 and LAM agents (drawn in the box). The presented IR values were measured for human hepatoblastoma cell line (Hep-G2/2.2.15) at a concentration of 4 μM.<sup>67</sup>"}}]](/storage/images/resized/KGL6bno8hIZrZY4yrdi5ElhV3mnk4F449Rxifc5a_xl.webp)
Screening of the antiviral activity in vitro indicated that compound 116k can effectively inhibit HBV DNA replication for both drug-sensitive and lamivudine (LAM)-resistant HBV strains, with the IC50 values being 0.53 and 0.44 μM for these strains, respectively. A key characteristic for evaluation of promising anticancer agents is the selectivity index (SI), which is calculated as the ratio of IC50 values for normal cells and cancer cells. (According to literature recommendations,[68] compounds are considered to be selective when SI ≥ 3.) It is worth noting that SI for compound 116k was > 37, indicating a good safety profile. According to a preliminary study, depending on the dose, this compound can induce TLR8-dependent NF-κB activity, thus effectively activating TLR8. Enzyme-linked immunosorbent assay (ELISA) showed that 1,2.4-oxadiazole 116k stimulates secretion of the TNF-α and IL-12 in the supernatant of human peripheral blood mononuclear cells (PBMC).
Papain-like protease (PLpro) is a strategically important therapeutic target due to its key role in the life cycle of SARS-CoV-2.[69][70] A naphthylmethylbenzamide derivative with the commercial designation GRL0617 (Fig. 16) is characterized by high inhibitory activity against PLpro (IC50 = 2.1 μM), but has a slow metabolism. In this regard, being guided by the principles of bioisosterism, Qin et al. [71] prepared a few series of PLpro-inhibiting 1,2,4-oxadiazoles.
![[{"id":"UgA5MDaC_f","type":"paragraph","data":{"text":"Structures of 1,2,4-oxadiazoles <b>126 </b>and <b>129 </b>and GRL0617 (drawn in the box) with indicated anti-SARS-CoV-2 activity (in μM).<sup>71</sup>"}}]](/storage/images/resized/LJHR4fsQlI4zZEt3TLdpIXlz5Vt7hgiVFU4r9816_xl.webp)
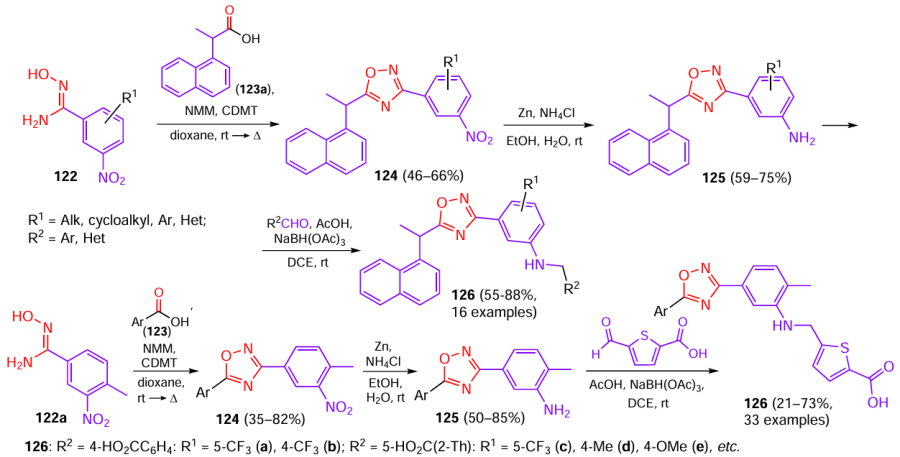
The 1,2,4-oxadiazole moiety was formed via the conventional cyclization of aromatic amidoximes 122 with carboxylic acids 123 (Scheme 23).[71] The carboxyl groups of acids 123 were preactivated by binding to 2-chloro-4,6-dimethyloxy-1,3,5-triazine (CDMT) in the presence of NMM in dioxane at room temperature. The intermediate esters were treated with nitroaromatic amidoximes 122 to form the corresponding 1,2,4-oxadiazoles 124, which were reduced with zinc and ammonium chloride to aminoaryl-1,2,4-oxadiazoles 125. The target compounds 126 were obtained by reductive amination of 125 with aldehydes in the presence of sodium triacetoxyborohydride in DCE at room temperature. Products 126 derived from amidoxime 122a (R1 = 4-Me) with participation of thiophene-2,5-dicarbaldehyde at the final step have a peripheral thiophenecarboxylic acid moiety complementary to a binding site of protease. 1,2,4-Oxadiazoles 126f,g, containing a biphenyl and carbazole instead of the naphthyl moiety, were synthesized using a similar sequence of transformations.
Aromatic amidoximes 94c,p,q and acids 123a,b were used as the starting reactants, being converted first to intermediates 127 and 128 and then to 1,2,4-oxadiazoles 129a – j (Scheme 24). The brominated intermediates 127b and 130 differ by the positions of substituents at the C(3) and C(5) atoms of the 1,2,4-oxadiazole ring: the former was prepared from p-bromophenyl amidoxime 94c, while in the synthetic pathway to compound 130, a para-bromine atom was present in carboxylic acid 123b. Subsequently, these aryl bromides were subjected to the Suzuki reaction with aromatic boronic acids, yielding compounds 129e–j, while the new C – N bond in product 129d was generated via a cross-coupling reaction with p-aminobenzoic acid.
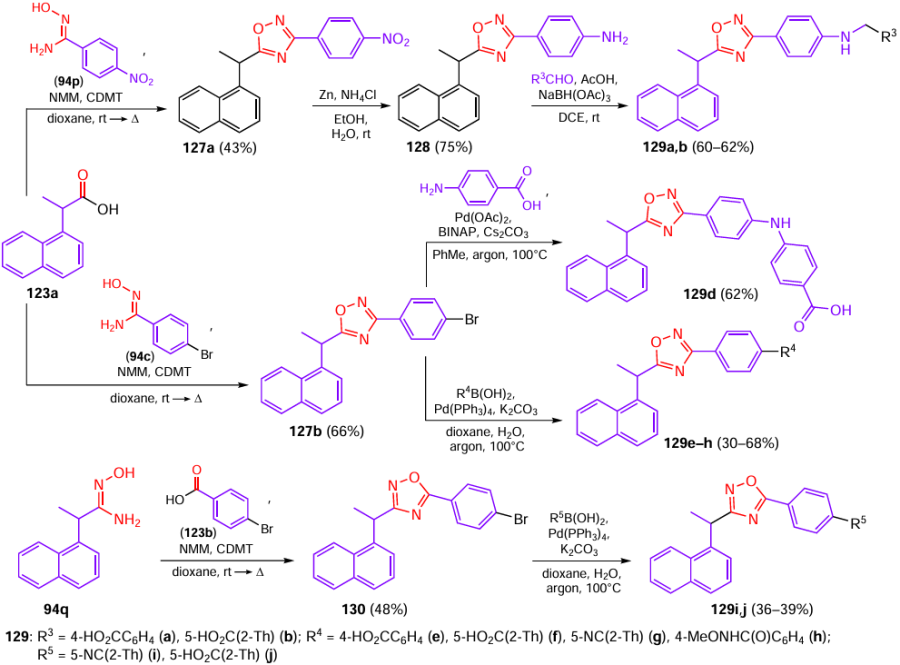
The targeted assembly of 1,2,4-oxadiazole structures furnished ten new compounds (see Fig. 16), which showed a comparable or higher inhibition rate of HIV protease, while possessing a more stable metabolism. Thus, most of derivatives containing benzoic acid moiety showed a potent inhibitory activity against PLpro. In particular, in comparison with compound GRL0617, derivatives 129f (R4 = 5-HO2C-2-Th) and 126g with enhanced metabolic stability exhibited a potent antiviral activity in cells infected with SARS-CoV-2 strains 2019-nCoV (EC50 = 5.4 and 4.3 μM vs. 44.1 μM for GRL0617) and omicron BA.1 (EC50 = 25.2 and 79.8 μM vs. 80.8 μM for GRL0617). Furthermore, these compounds had acceptable bioavailability and a favourable half-life (t1/2 > 93.2 min).
* TI = CC50/IC50, where CC50 is the half-maximal cytotoxic concentration.
3.5. Compounds with antioxidant activity
Studies of the antioxidant activity of heterocyclic compounds, especially with the aim of modulating the ROS generation processes, provide data for correlations with anticancer, anti-inflammatory, and even antimicrobial activity.[37][72][73] Mayer et al.[73] collected these data for (1,2,3-triazolyl)-1,2,4-oxadiazole systems. The condensation/cyclodehydration reaction sequence involving benzofuroxanyl- (131a) and dimethylbenzimidazolecarboxylic acid dioxide (131b) and substituted benzyl(1,2,3-triazolyl)carbamidoximes 132a – e resulted in the synthesis of target products 133a – e, 134a – e (Scheme 25).
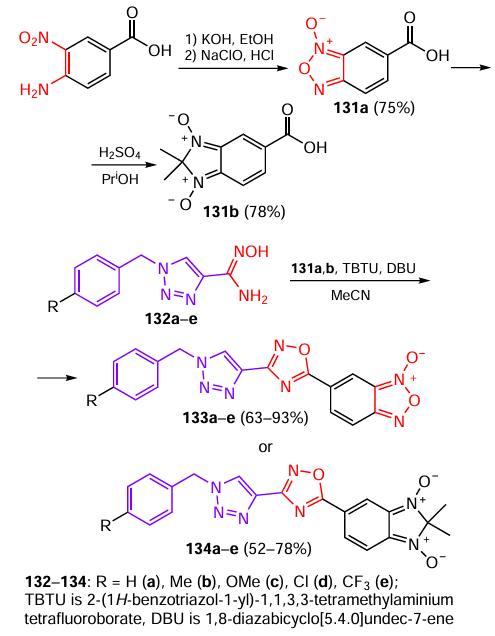
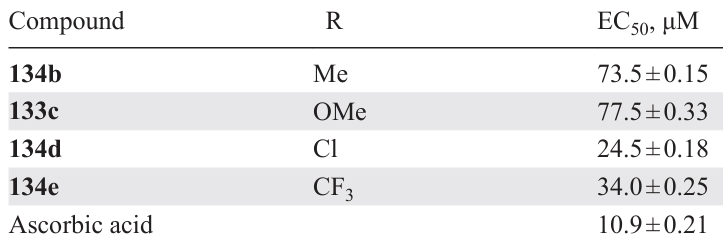
For compounds 133a – e and 134a – e, free radical scavenging experiments were carried out by UV absorption analysis using DPPH. A solution of ascorbic acid in ethanol served as a positive control. The best DPPH scavenging capability was found for derivatives 133c,b,d,e (Table 11); other compounds were inactive (EC50 > 200 μM). Thus, bis-N-oxides have higher antioxidant activity than similar mono-N-oxides.
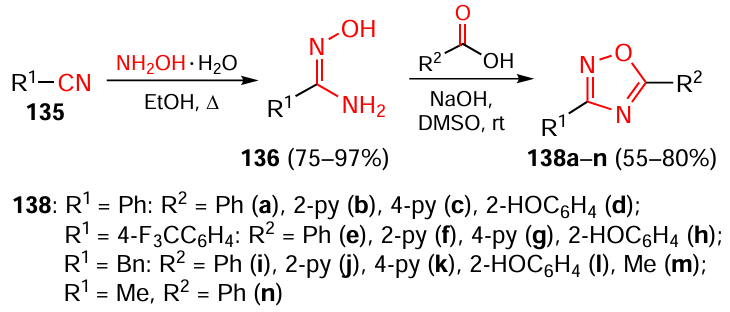
It is known that the key transcription factor Nrf2 is responsible for the antioxidant defence of many tissues and organs.[74] Ayoup[75] investigated the potential of 1,2,4-oxadiazole derivatives for the action on Nrf2 negative regulators, TrxR, IKK, and NF-κB, thus indirectly activating Nrf2 towards control of cancer diseases. The target products were obtained by standard methods from nitriles 135. Compounds 135 reacted with hydroxylamine to give amidoximes 136, which were subjected to cyclocondensation with carboxylic acids 137 in a basic medium (Scheme 26).
The antioxidant activity of 1,2,4-oxadiazoles 138a – n was studied using two methods: determination of free radical scavenging potential using DPPH and chelation of iron ions with 1,10-phenanthroline (Table 12). Phenol derivatives 138d,h,l showed the greatest free radical scavenging capacity among the compounds of this series. The hydroxyl groups of phenols are good hydrogen donors, which can react with ROS to give relatively long-lived radical species of antioxidant phenol derivatives, thus interrupting the cycle of formation of new ROS. The stability of this antioxidant compounds is due to stabilization of the phenoxyl radical through conjugation with aromatic π-electrons.[76]
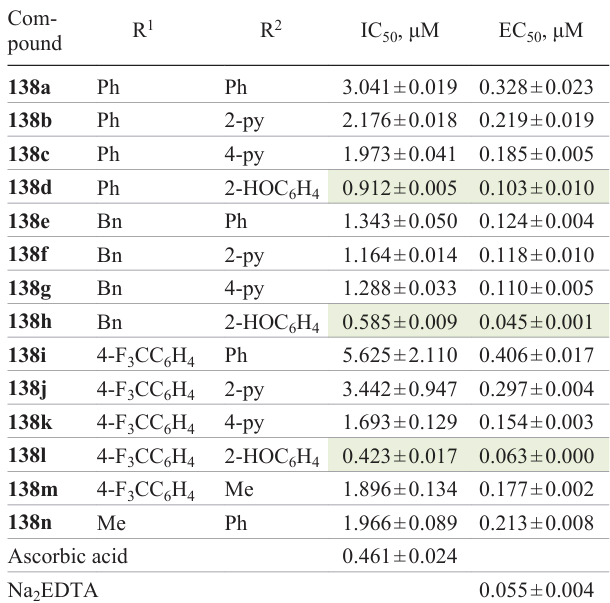
Among the tested phenolic derivatives, trifluoromethylphenyl-1,2,4-oxadiazole 138l was the most effective free radical scavenger, which may be attributed to the stabilizing effect of the electron-withdrawing trifluoromethyl substituent on the arising phenolic radical during the antioxidant process. The replacement of the phenol groups in compounds 138d,h,l by pyridin-2-yl (138b,f,j) or pyridin-4-yl (138c,g,k) moiety decreased the efficiency of ROS scavenging. The replacement of the pyridine ring at the C(3) position of oxadiazole by a benzene ring reduced the activity (138a,e,i). As regards the chelating activity, phenols 138d,h,l were also the most effective iron chelators among compounds of this series. Meanwhile, the EC50 values for 3-benzyl-substituted 1,2,4-oxadiazole 138h and 3-trifluoromethylphenyl analogue 138l were comparable to EC50 of a common chelator, ethylenediaminetetraacetate (EDTA), which may be due to iron chelating properties inherent in phenolic compounds.[77] Pyridine derivatives 138b,c,f,g,j,k chelated iron more efficiently than analogous phenyl-substituted 1,2,4-oxadiazoles 138a,e,i. This is consistent with earlier studies indicating the possible formation of heavy metal complexes with various nitrogen heterocycles, including 1,2,4-oxadiazoles.[78]
Additional biological assays were performed for the most active derivatives 138d,h,l. These 1,2,4-oxadiazoles decreased the generation of ROS in the HepG2 cells by a factor of three. In addition, compounds 138d,h,l in nanomolar concentrations inhibited thioredoxin reductase (TrxR1) (IC50 = 13.19 ± 0.55, 17.89 ± 1.11, 9.21 ± 1.45 nM), IKKα kinase (IC50 = 11.00 ± 0.31, 15.94 ± 0.45, 10.58 ± 0.30 nM), and NADPH oxidase (IC50 = 16.40 ± 0.46, 21.94 ± 0.62, 10.71 ± 0.30 nM) in vitro. As a result, the NF-κB level in HepG-2 cells decreased 7.6-, 1.4-, and 1.9-fold and Nrf2 was activated 2.36-, 1.78-, and 2.04-fold, respectively.
In recent years, some of the studies dealing with the development of drugs to treat Alzheimer’s disease have focused on antioxidant compounds, since the active generation of free radicals and hydrogen peroxide can lead to neuron degeneration.[79][80] A study by Ayoup et al.,[81] directed towards the search of compounds active against the Alzheimer’s disease, is devoted to the synthesis of 1,2,4-oxadiazole derivatives containing an arylalkyl or (hetero)aryl moiety at C(3) or C(5) (Scheme 27). Refluxing esters 139a– c with hydrazine hydrate in ethanol gave hydrazides 140a – c. The condensation of the hydrazides with o-nitrobenzaldehyde or 2-phenyl-1,2,3-triazole-4-carbaldehyde resulted in Schiff bases 141a – c and 142a – c, while refluxing of 140a – c with phthalic anhydride in acetic acid afforded N-phthalimide-substituted hydrazides 143a – c. Finally, the addition of quinaldic acid to hydrazides 140a – c in the presence of 1,3-dicyclohexylcarbodiimide (DCC) and 1-hydroxybenzotriazole (HOBt) furnished amide products 144a–c in high yields.
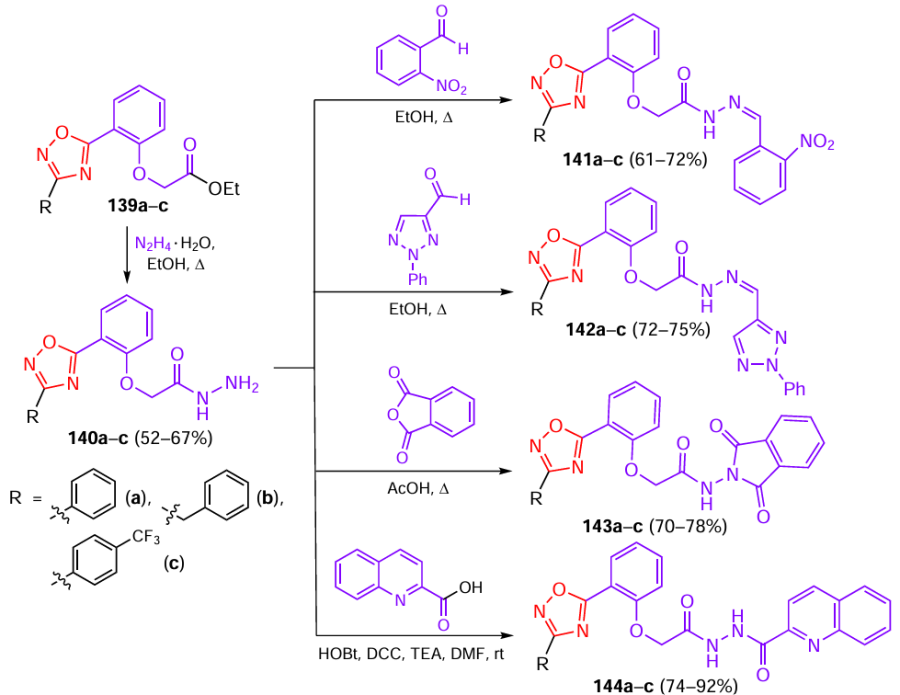
All of the synthesized 1,2,4-oxadiazoles were evaluated regarding their potential for the treatment of Alzheimer’s disease (Table 13). Eleven compounds showed a potent inhibitory effect (IC50 = 0.0010 – 0.0792 μM) against the acetylcholinesterase (AChE) enzyme, a promising target for the pharmaceutical agents used by Alzheimer’s patients. Their efficacy was 1.55 to 125.5 times higher than that of the commercial drug donepezil. However, these oxadiazole derivatives also exhibited selectivity and activity, although lower, against butyrylcholinesterase (BuChE). In addition, compound 141c had a higher antioxidant activity than quercetin.
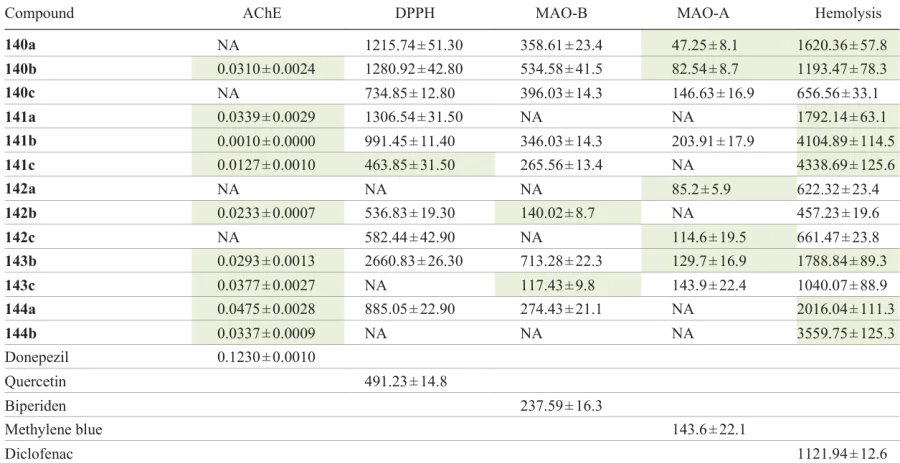
The authors [81] also evaluated the activity against a series of monoamine oxidases (MAO), because the inhibition of MAO-B is combined with a neuroprotective function [82] and decreases the risk of development of Alzheimer’s syndrome.[83] Compounds 142b,c provided a more pronounced inhibition of MAO-B than biperiden (IC50 = 237.59 μM), while derivatives 140a,b, 142a,c, and 143b showed excellent MAO-A inhibition potential with an efficacy 1.1 – 3.03 times higher than that of methylene blue. Most synthesized oxadiazole derivatives also demonstrated a strong protective effect against induced lysis of human red blood cells, indicating that these compounds could be safely used as potential therapeutic agents.
Study of structure–property relationships for the prepared series of compounds showed that the introduction of a benzyl group in the C(3) position of 1,2,4-oxadiazole (140b, 141b, 142b, 143b, and 144b) increased the AChE inhibition relative to the action of compounds containing phenyl or p-trifluoromethylphenyl substituent. Among the derivatives containing an ortho-hydrazide moiety in the benzene ring at the C(5) atom of 1,2,4-oxadiazole, only compound 140b with a benzyl substituent exhibited a high inhibitory activity against AChE. It is noteworthy that the presence of a polar electron-withdrawing o-nitrophenyl substituent (141a – c) led to an increase in the inhibition rate, whereas the introduction of a lipophilic electron-donating phenyltriazole (142a – c) had the opposite effect. Similarly, polar isoindoline derivatives (143a – c) had a higher inhibitory effect against AChE than lipophilic quinoline analogues (144a – c). As regards antioxidant potential, it can be concluded that 1,2,4-oxadiazole derivatives containing an N-acylhydrazone block (141a – c and 142a – c) are the most promising antioxidant drug candidates, while the introduction of a lipophilic electron-deficient p-trifluoromethylphenyl moiety to the C(3) atom of oxadiazole enhances the antioxidant potential of compounds 140c, 141c, and 142c. The lipophilic quinoline ring promoted a more effective antioxidant effect compared to polar isoindoline ring. Hydrazide derivatives 140a – c demonstrated a moderate inhibition of MAO-B, while N-acylhydrazone derivatives (142a, 142b, and 143b) showed a more pronounced effect. It is worth noting that MAO-B inhibition was enhanced to a greater extent when an isoindoline ring was introduced in the molecule (143a – c) than upon the introduction of a quinoline ring (144a – c). The introduction of an electron-deficient lipophilic p-trifluoromethylphenyl moiety in the 1,2,4-oxadiazole C(3) position (140c, 141c, and 143c) considerably increased the inhibitory potential against MAO-B. Considering the inhibition of MAO-A, a reasonable activity was found for both hydrazide and N-acylhydrazone derivatives; the phenyltriazole (142a – c) and isoindole (143a – c) moieties proved to be more favourable than the quinoline (144a – c) or o-nitrobenzene moiety (141a – c).
3.6. Compounds with antibacterial activity
Cinnamic acid and its derivatives are known for their antituberculosis activity.[84][85] The 1,2,4-oxadiazole ring is a known bioisostere of the carboxyl group possessing a low metabolic lability,[86] while the introduction of this moiety into cinnamic acid molecules may retard rotation around the double bond and lock the molecular conformation.[87] By replacing the carboxyl group by an oxadiazole ring, Upare et al.[37] prepared a series of new compounds with a potential activity against tuberculosis. The substituted cinnamic acids 145 were introduced into the condensation/cyclodehydration tandem reaction with amidoximes 146a,b in the presence of CDI, resulting in the formation of 3,5-disubstituted 1,2,4-oxadiazoles 147, 148 in good yields (Scheme 28).
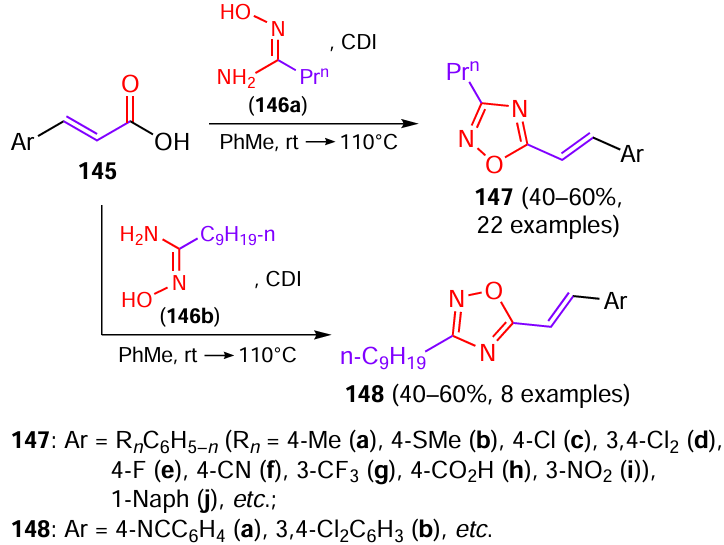
The antituberculosis activity of this series of styryl 1,2,4-oxadiazole derivatives was assessed in vitro against the mycobacterium tuberculosis strain H37Ra (Fig. 17). Compounds with electron-withdrawing substituents (147g – i) and halogen atoms (147c,e) proved to be most active, whereas the electron-donating substituents (147a,b) are least favourable for the activity. The elongation of the hydrocarbon chain, i.e., the increase in the lipophilicity of the C(3) position of 1,2,4-oxadiazole, is also favourable for the antituberculosis activity (compound 147d with a propyl substituent is less active than homologue 148b with a nonyl group; the same is true for IC50 values: 148a < 147f). Compound 147h, which had the lowest IC50 value in this series, proved to be most active against tuberculosis.
![[{"id":"vctEk6fyUP","type":"paragraph","data":{"text":"Structures of styryl-1,2,4-oxadiazoles <b>147</b>, <b>148</b>, possessing a high antituberculosis activity, with indicated IC<sub>50</sub> values (in μg mL<sup>–1</sup>).<sup>37</sup>"}}]](/storage/images/resized/3ydbTXJcqhwoRiRCTg6gGaYXiz4dcfjgnC7K3I4v_xl.webp)
Alsimaree et al.[88] combined a 1,2,4-oxadiazole ring and a benzoxazole moiety, often encountered in the antibacterial drugs, in the same molecule.[89] First, thiol 149 was treated with chloroacetic acid to be converted to thiocarboxylic acid 150, while benzonitriles 151a – h were converted to amidoximes 152a – h (Scheme 29). Next, the carboxyl group of compound 150 was activated by CDI, with the reaction pathway depending on the solvent and temperature: the reaction carried out in acetonitrile at room temperature afforded only O-acylated amidoximes 153a – h, while heating up to 90°C in DMF induces 1,2,4-oxadiazole ring closure. Thus, a series of desired benzoxazolyl-1,2,4-oxadiazoles 154a – h was synthesized.
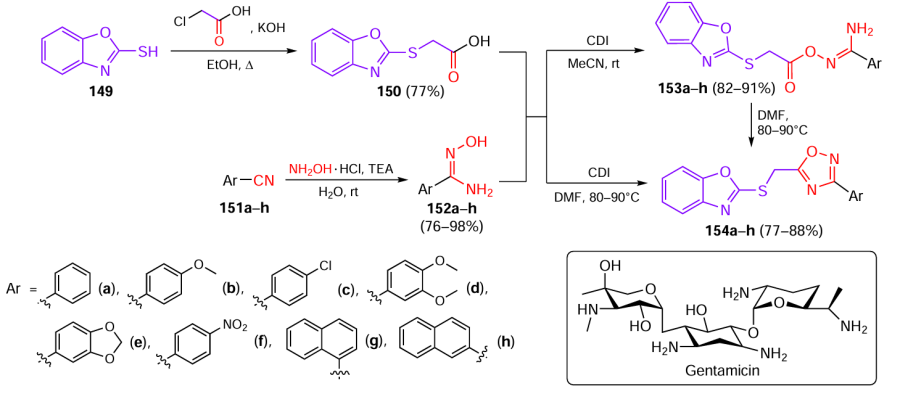
The antibacterial activity was assessed for all compounds 153a – h, 154a – h.[88] Derivatives 153a and 154c,e were most active: hybrid 153a with an unsubstituted phenyl group showed an exceptional activity against Gram-positive bacteria B. subtilis and S. aureus, with the zones of inhibition (ZI) determined by disk diffusion being 34 and 36 mm, respectively. In addition, this agent had a strong antibacterial effect against Gram-negative bacterium K. pneumoniae (ZI = 32 mm) and a moderate effect against Gram-negative E. coli (ZI = 15 mm). A high activity against B. subtilis, S. aureus, and K. pneumoniae was found for compounds 154e (p-methoxyphenyl substituent) and 154c (p-chlorophenyl substituent): ZI were 32, 36, and 34 mm and 31, 34, and 35 mm, respectively. For the most active derivatives, MICs were also evaluated (Table 14). In the case of S. aureus and K. pneumoniae, the minimum inhibitory concentration was lower than that for the positive control drug, gentamicin; for the B. subtilis strain, MIC of compounds 153a,e exceeded that of gentamicin and MIC of 154c was equal to that of gentamicin. However, even lead compounds inhibited E. coli more poorly than the reference agent.
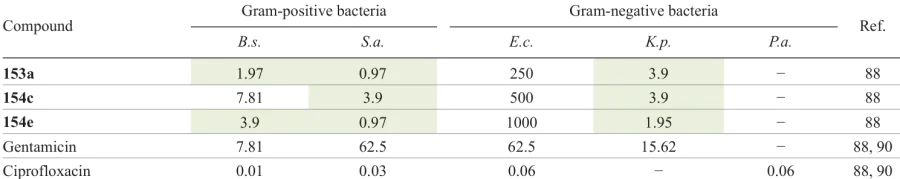
The DNA gyrase enzyme is a promising target for antibacterial drugs. Hybrid 1,2,4-oxadiazolyl(pyrrolidones) were synthesized to study the potential activity against DNA gyrase and topoisomerase IV (Scheme 30).[90] The compounds were prepared from carboxylic acids 155 and amidoximes 156 by acylation followed by cyclization to 1,2,4-oxadiazoles. This afforded target compounds 157 in moderate yields.
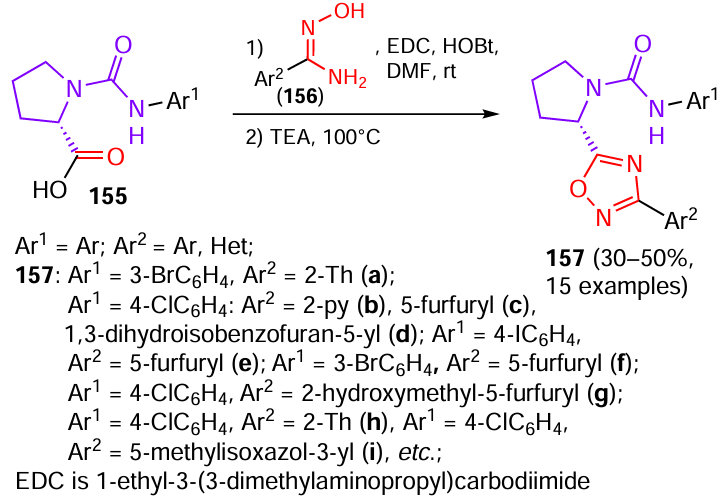
The prepared series of compounds was tested for inhibition of E. coli DNA gyrase; the most active compounds that showed an inhibitory activity level equal to or higher than the activity of novobiocin are depicted in Fig. 18. It is noteworthy that the p-chlorophenyl moiety was present in the structure of three of the five most active compounds (157b – d). The replacement of this substituent by m-bromophenyl (157f) or p-iodophenyl (157e) resulted in an at least 2.25-fold decrease in IC50, which attests to importance of the chlorophenyl moiety for the inhibitory activity of compounds. Furfuryl alcohol (5-hydroxymethylfuran) derivative 157c was most effective among the synthesized products and exhibited a stronger effect than novobiocin. In addition to the p-chlorophenyl substituent at nitrogen, 2-pyridyl (157b) or 1,3-benzodioxole moiety (157d) increased the efficiency of compounds against DNA gyrase. Meanwhile, compounds containing other heterocycles such as furan (157g), thiophene (157h), and isoxazole (157i) showed moderate activity, with residual enzyme activity values being 57, 84, and 89%, respectively.
![[{"id":"ERcNnyPSG9","type":"paragraph","data":{"text":" Structures of 1,2,4-oxadiazolyl(pyrrolidones) <b>157 </b>with a high antibacterial activity and novobiocin (drawn in the box) with indicated IC<sub>50</sub> value (in nM).<sup>90</sup>"}}]](/storage/images/resized/XIZuXjzxiTZUabfSShGtMehJsuY2qEnQWdf25DfI_xl.webp)
The antibacterial properties of the most active compounds were assessed using ciprofloxacin as a reference and the two-fold serial dilution procedure (Table 15). Generally, the tested 1,2,4-oxadiazole derivatives inhibited Gram-negative bacteria less effectively than Gram-positive ones. The most pronounced antibacterial activity was found for compound 157c, which had a lower minimum inhibitory concentration against S. aureus than ciprofloxacin. Activities similar to those of ciprofloxacin were found for compounds 157a,c against B. subtilis, 157d against S. aureus, and 157c also against E. coli and P. aeruginosa; the other products were less active than the reference agent.
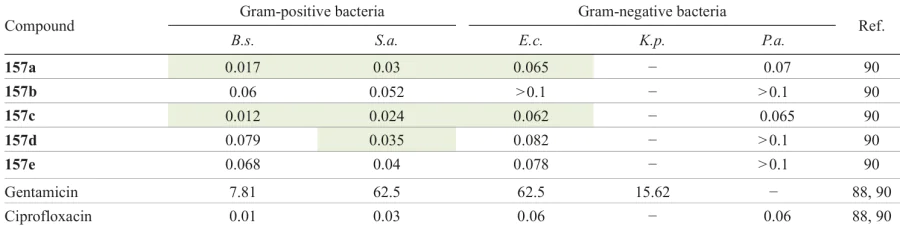
3.7. Compounds with anticancer activity
5-Fluorouracil (158) is a chemotherapeutic agent used to treat various types of cancer;[91] however, it is sparingly used due to low bioavailability and high toxicity.[92] The introduction of a 1,2,4-oxadiazole moiety exhibiting a pronounced antiproliferative activity against various human cancer cells into this molecule may increase the bioavailability of the resulting hydrides.
For this purpose, commercially available 5-fluorouracil was alkylated with 4-bromomethylbenzonitrile in the presence of DBU (Scheme 31). The resulting nitrile 159 was converted to amidoxime 160, which was then subjected to condensation with carboxylic acids 161 in the presence of EDC. The subsequent refluxing of the resulting intermediates in EtOH in the presence of AcONa afforded the target 1,2,4-oxadiazoles 162a – j.[93]
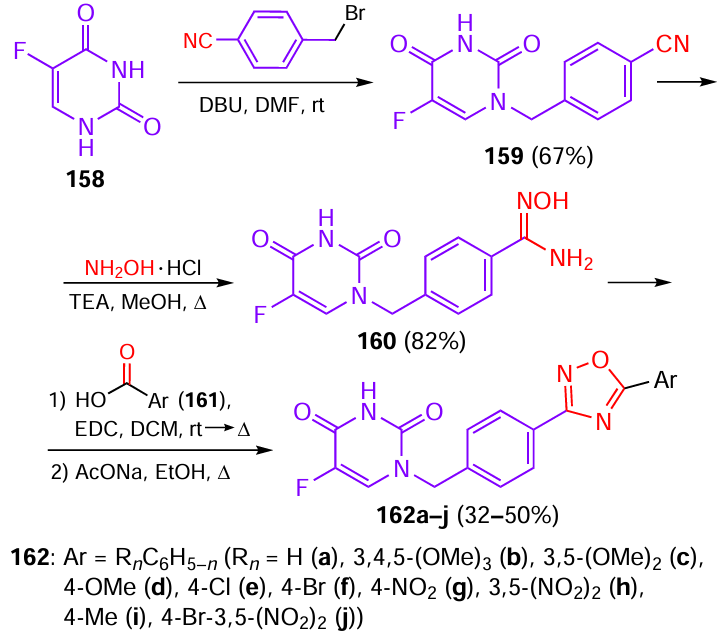
The anticancer activity of all compounds was studied against four human cancer cell lines, namely breast cancer (MCF-7), triple negative breast cancer (MDA-MB-231), non-small cell lung cancer (A-549), and prostate cancer (DU-145) cell lines (Table 16)[93-95]. Compounds 162a,b,c,d,i (Fig. 19) demonstrated promising anticancer activity: IC50 values were in the range from 0.011 ± 0.009 to 19.4 ± 8.11 μM, while IC50 for the reference compound (etoposide) varied from 1.91 ± 0.84 to 3.08 ± 0.135 μM. According to SAR studies, compound 162a with unsubstituted benzene ring attached to the 1,2,4-oxadiazole moiety possessed good anticancer effect against all four cell lines, while the introduction of three electron-donating methoxy groups into the benzene ring (162b) increased the activity. The anticancer activity of dimethoxy derivative 162c against the four cell lines was slightly lower than that of trimethoxy-substituted analogue 162b, while compound 162d with a p-methoxyphenyl group was even less active. Note that introduction of even a weak electron-donating methyl group in the para-position of the benzene ring (162i) led to an increase in the antiproliferative activity. In addition, compounds 162e (Ar = 4-ClC6H4), 162f (Ar = 4-BrC6H4), 162g (Ar = 4-NO2C6H4), 162h (Ar = 3,5-(NO2)2C6H3), and 162j (Ar = 4-Br-3,5-(NO2)2C6H2) containing electron-withdrawing substituents in the benzene ring had lower activities than above compounds 162a – d,i with electron-donating substituents.
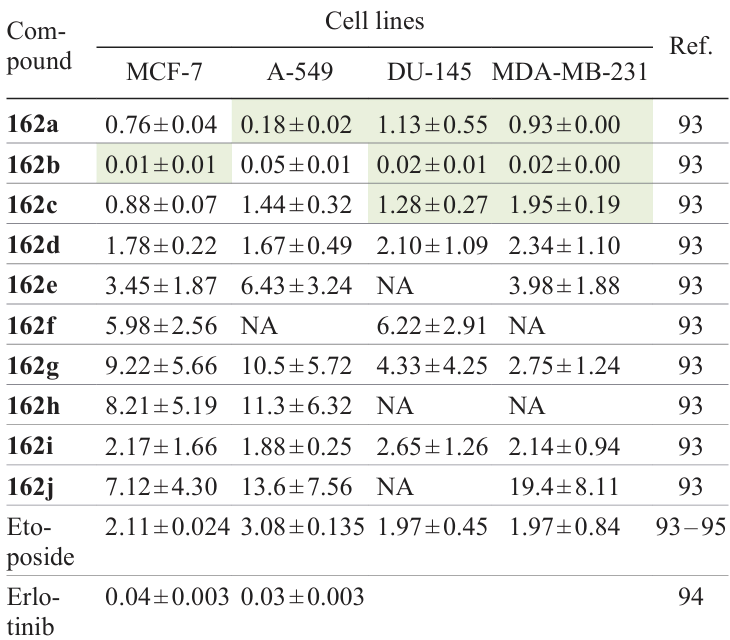
![[{"id":"HtXfzRdnI5","type":"paragraph","data":{"text":"Structures of hybrids <b>162 </b>based on 5-fluorouracil and 1,2,4-oxadiazole that have high antitumour activity and etoposide (drawn in the box).<sup>93</sup>"}}]](/storage/images/resized/mXUWnYiOk7AjE4aft8aamJB5g68vKt4HvxU2lPCB_xl.webp)
A combination of cytosolic serine/threonine protein kinase (BRAF) and tyrosine kinase (TK) inhibitors demonstrated high efficacy in suppressing tumour growth in clinical trials and can serve as an approach to overcome resistance to treatment.[96][97] Mohamed et al.[94] developed a new series of hybrid molecules containing 1,2,4-oxadiazole and quinazolinone moieties to be studied as multipurpose antiproliferative inhibitors (Scheme 32). First, amidoximes 53a – c were acylated with chloroacetyl chloride, and then intermediate compounds 163a – c were cyclized by refluxing in toluene to give 1,2,4-oxadiazoles 164a – c. The reaction of anthranilic acid (165) with isothiocyanates 166 yielded thiones 167, which were in tautomeric equilibrium with sulfanyl derivatives 168. The target products 169 were formed via nucleophilic substitution of the chlorine atom in 1,2,4-oxadiazoles 164a–c on treatment with 2-sulfanylquinazolinones 168.[98][99]
The antiproliferative activity of hybrids 169 was assessed using four human cancer cell lines: HT-29 (colon cancer), Panc-1 (pancreatic cancer), A-549, and MCF-7 with erlotinib as a positive control (Table 17).
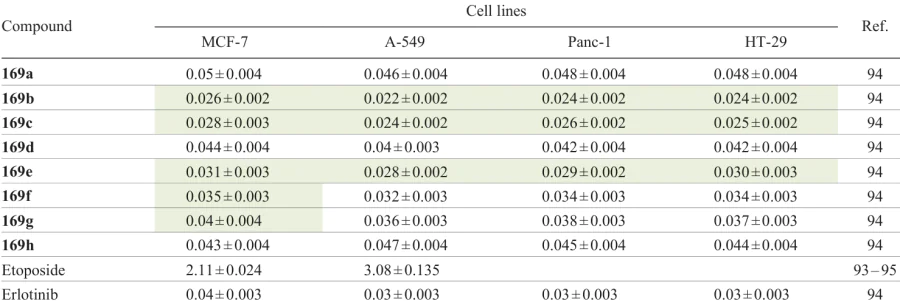
Generally, hybrid compounds 169 exhibit a substantial antiproliferative activity: the concentrations providing 50% inhibition of cell proliferation (GI50) were in the range from 24 to 70 nM. Compounds 169b,c,e – g (Fig. 20) were most effective among the whole series, with their GI50 values being 24, 26, 30, 34, and 38 nM, respectively, i.e., oxadiazoles 169b,c,e proved to be more effective than the reference drug erlotinib (GI50 = 33 nM). Of the whole series, derivative 169b exhibited the highest activity with the GI50 value of 24 nM, which is 1.4 times higher than that for the reference drug. Meanwhile, compounds 169d (Ar = p-Tol, R = 4-ClC6H4), 169f (Ar = 4-ClC6H4 , R = Et), and 169h (Ar = 4-ClC6H4 , R = All) showed GI50 = 42, 34, and 45 nM, respectively, being less effective than derivatives 169b,e and erlotinib. Thus, the antiproliferative activity of the compounds decreases depending on the substituent at the C(3) atom of the quinazoline moiety in the following order: phenyl > m-tolyl > p-tolyl > ethyl > allyl. The unsubstituted phenyl derivative 169a (Ar = R = Ph) was less effective than analogues 169b,c; hence, the para-substituents in the benzene ring attached to the C(3) atom of 1,2,4-oxadiazole influence the antiproliferative activity in the order Cl > OMe > H, irrespective of the nature of the substituent at the C(3) atom of quinazoline. The same order applies to other derivatives with this substitution pattern.
![[{"id":"sCfxGOzY3I","type":"paragraph","data":{"text":"Structures of hybrids <b>169 </b>based on 1,2,4-oxadiazole and quinazolinone that showed high anticancer activity and erlotinib (drawn in the box).<sup>94</sup>"}}]](/storage/images/resized/Bbut5J0iytK4f6h79mK25D0EaaLZZq1NQGkbhJed_xl.webp)
Hybrid structures 169 comprising 1,2,4-oxadiazole and quinazolinone were also investigated as inhibitors of epidermal growth factor receptors, EGFR, EGFRT790M and BRAFV600E. According to in vitro experiments, compounds 169b,c,e are potent antiproliferative agents able to act as EGFR/BRAFV600E dual inhibitors. In addition, these compounds have a pronounced antiproliferative effect on the EGFRT790M mutant of EGFR. Analysis of the cell cycle for the most active derivative 169b revealed cell cycle arrest in the G2/M phase, which may induce apoptosis.
Ashok et al.[95] combined three heterocyclic moieties possessing anticancer activity, pyrimidine, 1,2,4-oxadiazole, and 1,2,4-thiadiazole, in one molecule. Scheme 33 depicts the synthetic route to these structures. Thus, nitrile 170 was treated with hydroxylamine, and the resulting amidoxime was subjected to base-induced cyclization with carboxylic acids in the presence of 4-dimethylaminopyridine (DMAP). Finally, a series of compounds 171a – j was formed in moderate yields.
The anticancer activity of compounds 171a – j was assessed using MCF-7, A-549, Colo-205 (colon cancer), and A2780 (ovarian cancer) cell lines with etoposite as the positive control. Most derivatives exhibited high to moderate activity, with compounds 171b,c,d,i,j demonstrating the most promising anticancer activity in all cell lines (Table 18). According to a study of structure–activity relationships, compound 171b containing a 3,4,5-trimethoxyphenyl group at the C(3) position of oxadiazole showed an excellent anticancer activity, while the activity of analogues with 3,5-dimethoxy- (171c) or p-methoxyphenyl group (171d) was somewhat lower for all cell lines. When the p‑methoxyphenyl moiety was replaced by an electron-withdrawing substituent, namely p‑bromo (171e), p‑chloro (171f), or p-nitro (171g) group and 3,5-dinitrophenyl moiety (171h), the activity also decreased. Meanwhile, compound 171j with a 4-dimethylamino group in the benzene ring showed a high activity against all cell lines.
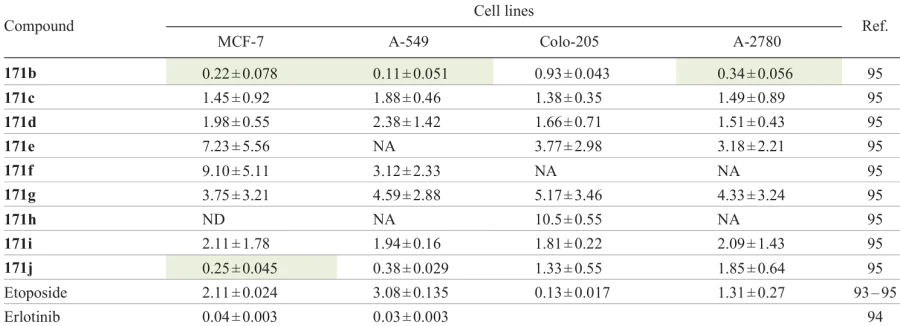
Thus, to summarize the data given in Tables 16 – 18,[93-95] it can be concluded that 1,2,4-oxadiazole — quinazolinone hybrids 169b,c,e containing p-chloro- or p-methoxyphenyl substituents at the C(3) position of quinazolinone 169b,c,e proved to be the most active compounds possessing antiproliferative action in the nanomolar concentration range. The highest activity against the MCF-7, DU-145, and MDA-MB-231 cell lines was found for 1,2,4-oxadiazolyl(fluorouracil) 162b with a dimethoxyphenyl substituent, while trimethoxyphenyl derivative 171b was the only compound composed of 1,2,4-oxadiazole, pyrimidine, and thiadiazole moieties that was more active than the reference etoposide against the A-2780 and A-549 cells. Generally, derivatives containing the electron-donating methoxy group were most active among the three above series of compounds.
3.8. Summary of the data on the activity of 1,2,4-oxadiazoles
1,2,4-Oxadiazoles, the most studied type of oxadiazoles, which have an N – O bond in the ring, exhibit a broad range of biological activity. Two main patterns of using active 1,2,4-oxadiazoles can be identified: cross-coupling with a known pharmacophore as a bioisostere linker (antiproliferative agents 162, 169, 171) or synthesis of arylcarboxamides that are active without the introduction of additional active groups (antifungal and nematicidal compounds 51, 57, 73, 79). Analysis of the structure–property relationships indicates that introduction of fluorine-containing substituents into the aromatic ring [fluorine atom (57c,o, 98e), difluoromethyl group (51w, 69e), or trifluoromethyl group (51g,m, 57f, 85b, 116k)] increases the antifungal, nematicidal, anti-inflammatory, and antiviral activity of compounds, while mono- (109e, 162d), di- (104e, 105e, 162c, 171c), and trimethoxyphenyl moieties (104f, 105f, 162b, 171b) are present in almost all lead compounds with high anti-inflammatory, antiviral, and antiproliferative activity.
4. Synthesis and activity of 1,2,5-oxadiazoles
1,2,5-Oxadiazole (furazan) and its N-oxide (furoxan) are π-excessive heterocycles with six electrons being distributed over five atoms. The electron-withdrawing N – O – N sequence of heteroatoms in the ring makes the carbon atoms in 1,2,5-oxadiazole active electrophilic centres, which has a pronounced effect on their reactivity.[100] The furoxan ring is much more labile towards nucleophiles than the furazan ring. In addition, the furoxan ring is capable of isomerization, which can lead to two regioisomers at the N-oxide group. This is an equilibium process involving the formation of a cis-1,2-dinitrosoethylene intermediate (Fig. 21).
![[{"id":"xdD8Avkbaj","type":"paragraph","data":{"text":"1,2,5-Oxadiazoles and their isomeric N-oxides"}}]](/storage/images/resized/PPDQT08NuoQzgEJFUZugTt3GfRJ9jMZlMugLq4v9_xl.webp)
The biological activity of furoxan derivatives is attributed to their ability to act as nitric oxide (NO) donors.[101] It has been established that numerous cardiovascular diseases and various immune and neurodegenerative disorders are directly related to impaired NO release in living organisms.[102] Furthermore, NO exhibits cytotoxic and cytostatic effects against various microorganisms, including protozoa such as Plasmodium falciparum, the causative agent of human malaria.[103] In addition, 1,2,5-oxadiazoles also exhibit cytotoxity,[104]immunosupression,[105]myorelaxant properties,[106]anticonvulsant activity,[107]and oxidase inhibition.[108]
4.1. Compounds with growth-regulating activity
The protection of plants from pests is crucial for growing agricultural crops. The pre-sowing seed treatment is the main preventive measure, since more than 70% of infective diseases are transmitted through seed material.[109]Currently, a wide range of growth stimulators and regulators (Phytolavin, Akpinol, Zircon) are used to improve the sowing quality of seeds:[110]they improve seed germination, enhance plant growth and development, and increase the plant resistance to diseases. The use of growth regulators increases the crop yield by up to 30% compared to the control.[111]However, the currently used plant growth regulators and stimulants are often toxic, harmful to the environment, and detrimental to the human health. A study by Chugunova et al.[112]is devoted to the development of low-toxic or non-toxic active compounds possessing above-indicated properties. Nucleophilic aromatic substitution reaction of 4,6-dichloro-5-nitrobenzofuroxan (172) with various amines followed by the neutralization of disubstituted amines 173 with hydrochloric acid resulted in the formation of a series of water-soluble benzofuroxan salts 174 (Scheme 34).
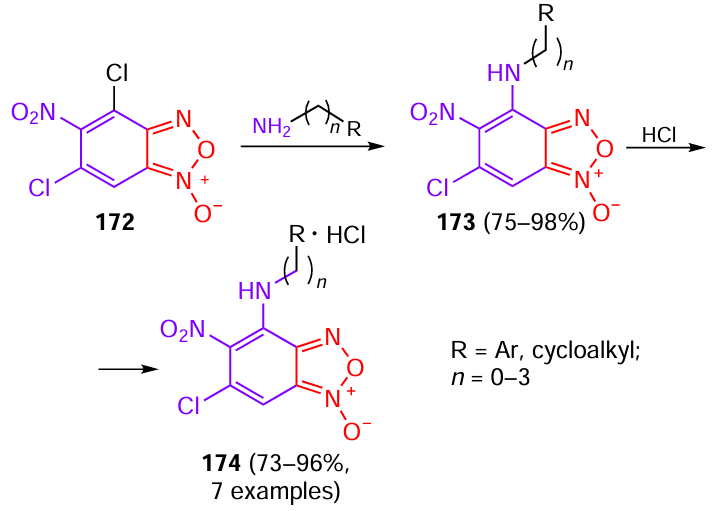
Benzofuroxan-based salts were evaluated as growth stimulators for various agricultural crops. To this end, the efficacy of pre-sowing treatment of agricultural seeds including wheat, alfalfa, barley, rice, and sorghum was assessed under laboratory conditions. The most active compounds are depicted in Fig. 22.
![[{"id":"keGmynNvrM","type":"paragraph","data":{"text":"Structures of benzofuroxan salts <b>174 </b>possessing growth-regulating activity.<sup>112</sup>"}}]](/storage/images/resized/pV47USMkuyscMo6uqtmBIHpr7hZoF0UEFyPVcDzZ_xl.webp)
The results of the analysis indicated that the laboratory germination of wheat seeds pretreated with salts 174 was higher than that in the control experiment (85.7%): for solutions of compound 174a at concentration of 10 or 50 mM, it was 96.5%; in the case of compound 174b at concentrations of 40 and 20 mM, it was 98.5 and 97.5%, while for compound 174c, the germination was 99% at a concentration of 40 mM. Similarly, the sowing qualities of alfalfa seeds pretreated with 10 mM (84.5%) and 50 mM (79.5%) solutions of salt 174a or a 40 mM solution of salt 174b (80%) were higher than those observed in the control experiment (76%). In addition, treatment with compounds 174a (10 and 50 mM doses), 174b (10 and 20 mM doses), and 174d (10 mM dose) had a beneficial effect on the suppression of fungal microflora. The treatment of rice seeds with solutions of salt 174a (10 and 50 mM), 174b (40 mM), and 174c (20 and 40 mM) resulted in 96% germination in the first-mentioned case and 99% germination in all other cases, which was considerably superior to the data for the control experiment (71.4%). The use of compounds 174b and 174c in low doses of 10 and 20 mM provided effective suppression of bacterial microflora. For the pretreatment of sorghum seeds, the experiment showed that laboratory germination after seed pretreatment with compounds 174b (20 mM) and 174d (20 and 40 mM) was 80.9, 71.4, and 80.9%; this was also higher than that in the control experiment (52.3%). Thus, these compounds at concentrations of 20 to 40 mM are effective for the pre-sowing treatment of agricultural crop seeds. The presence of arylamino group improves the growth-regulating properties, and compound 174a is active even at a concentration of 10 mM.
4.2. Compounds with anticancer activity
Furoxans are best known for their antiproliferative activity. Most of the used anticancer agents are poorly soluble in water, which has an adverse effect on their efficacy and safety upon oral, intramuscular, or intravenous administration.[113] Furthermore, this accounts for their low efficacy; therefore, auxiliary compounds are needed to use these agents; however, auxiliary components possess toxic side effects. Water-soluble aminobenzofuroxan salts 174 prepared by Chugunova et al.[112] were tested for the antiproliferative activity on cancer type cell cultures: SH-SY5Y (neuroblastoma), HepG, A-549, and HeLa (cell line derived from cervical cancer cells). To gain understanding how these compounds would affect normal cells, they were additionally tested for the effect on the viability of Hek-293 normal cell culture (Table 19). After 24 hours of incubation, the whole series exhibited cytotoxic properties, with the best results being against SH-SY5Y cells. The most pronounced effect for most cancer cell lines was observed in the case of salts 174c,d; however, compound 174d also exhibited high toxicity against normal cells, which was attributed to its good solubility in water and bioavailability. In turn, in the case of compound 174c, SI found as the ratio of IC50 values for the SH-SY5Y neuroblastoma cells and Hek-93 normal cells was > 4. In addition, comparative analysis of the half-maximal inhibition of cell culture growth was performed for two incubation times, 24 and 72 h. It was found that increase in the incubation time of the compounds with selected cell lines resulted in decreasing IC50. This may be due to both enhancement of the direct toxic effect of the test compounds over time and the presence of antiproliferative activity, which is manifested as an effect of the compound on the cell division, since a few cell cycles take place within 72 h. Compounds 174c,d also showed antileukaemic activity in vivo, which was manifested as increasing life span of mice with P388 murine leukaemia in comparison with the untreated control group.
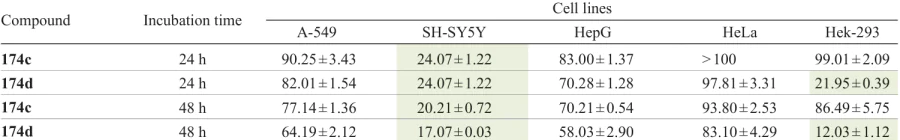
Nitric oxide is associated with the pathogenesis of a number of highly severe types of cancer such as malignant pleural mesothelioma (MPM). MPM cells are characterized by overexpression of NO synthases and increased production of NO compared to normal cells;[114]therefore, the use of NO donor compounds has the potential to saturate cancer cells with nitric oxide, thus causing their death.[115]Stebletsova et al.[116]described a series of NO donor (1,2,4-oxadiazolyl)furoxan derivatives (Scheme 35). Cyanoacetic acid 175 was subjected to an oxidation/dimerization cascade to give dicyanofuroxan 176, which was converted to amidoxime 177 by the reaction with 1 equiv. of hydroxylamine at low temperature. C(5)-Unsubstituted 1,2,4-oxadiazole 178a was prepared by acid-catalyzed cyclization of amidoxime 177 with trimethyl orthoformate in the presence of TsOH (Ts is p-toluenesulfonate). C(5)-substituted products 178b – l were obtained by a one-pot procedure: the carbonyl group of acids was activated with CDI, the resulting imidazole derivatives were introduced into acylation of amidoxime and then cyclized with 1,4-diazabicyclo[2.2.2]octane (DABCO).
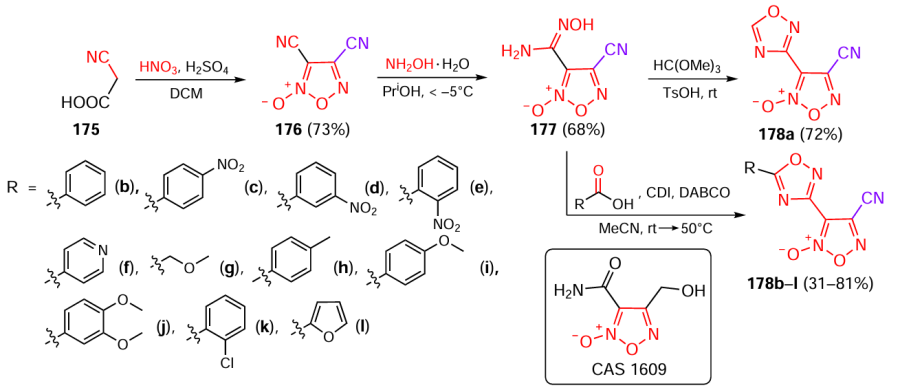
The anticancer properties of compounds 178a – l were tested on murine (AB1) and human (JU77) mesothelioma cell lines.[116]It is noteworthy that all compounds, except for 178g, showed a considerable cytotoxicity against both MPM cell lines at low micromolar concentrations (Table 20). The cytotoxicity of compounds 178a,f was similar to that of paclitaxel (PTX), while all other compounds surpassed this drug in activity against both cell lines. Nitrophenyl derivatives 178c,d,e had a higher cytotoxicity than unsubstituted phenyl derivative 178b, according to the pattern 178b ≈ 178c (para) ≤ 178d (meta) < < 178e (ortho). The highest activity was found for compounds 178e,l. In order to evaluate the selectivity of action of series 178a – l on cancer cells, the authors assessed the cytotoxicity against benign MRC-5 human lung fibroblasts. It is of interest that less cytotoxic compounds 178a,f had low selectivity to cancer cells, which is indicative of their limited therapeutic potential. Conversely, derivatives 178d,e showed 2.4 – 3.6 times higher cytotoxicity in human MPM cells compared to that in human lung fibroblasts. The anticancer activity is generally correlated with NO-donor properties: indeed, the most active anticancer agents 178e,l are also characterized by the highest percentage of NO release, and the whole series is characterized by higher NO-release properties than the reference agent, 4-hydroxymethyl-3-carbamoylfuroxan (CAS 1609) (see Scheme 35).
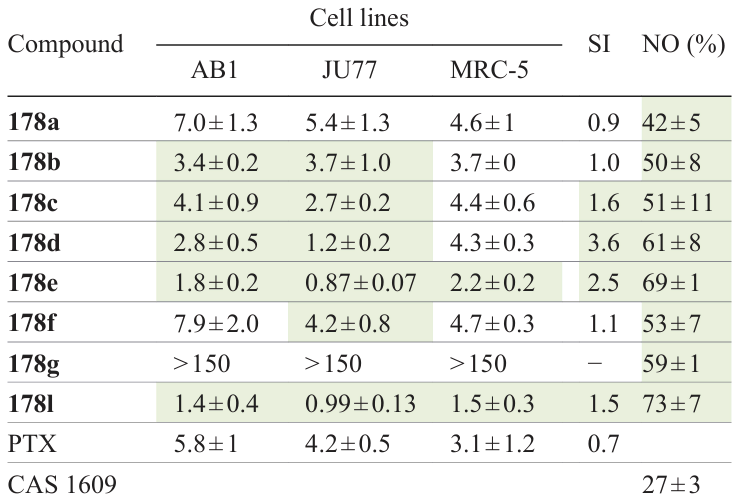
Another well-known approach to the preparation of new antiproliferative agents is the construction of heterocyclic moieties on a steroid skeleton. Vorontsova et al.[117]performed Riley oxidation of 2-acetyl-sibstituted steroid 179. The condensation of intermediate 1,2-dicarbonyl compound 180 with diaminofurazan 181 gave [1,2,5]oxadiazolo[3,4-b]pyrazine 182 with a steroid moiety (Scheme 36).
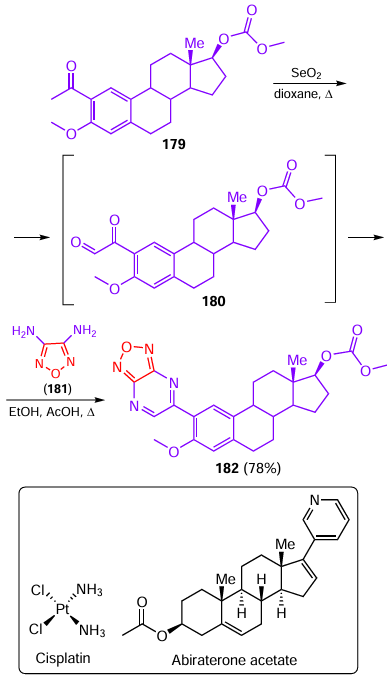
Compound 182 showed high activity against estrogen-dependent tumour cells: IC50 = 7.0 ± 0.6 and 9.3 ± 0.6 μM for MCF-7 and 22Rv1 (prostate cancer) cells, respectively. The results for this compound were comparable with those for cisplatin (Cys) (IC50 = 6.1 ± 0.6 and 9.7 ± 0.9 μM, respectively) and a hormonal drug, abiraterone acetate (IC50 = 6.1 ± 0.6 and 12.9 ± 1.0 μM, respectively).[117]
Currently, development of drugs inhibiting the formation of microtubules (microtubule-interfering agents, MIA) is a highly promising approach to the treatment of cancer; among these agents, phenstatin is widely used. Huang et al.[118]combined a furoxan moiety with the phenstatin scaffold and thus obtained a series of NO-donor benzophenone derivatives by the following synthetic protocol (Scheme 37). First, the hydroxyl group of the starting 2-methoxyphenol (183) was acylated with chloroacetyl chloride; this was followed by chemoselective acylation of compound 184 with substituted benzoic or naphthalenecarboxylic acids in the presence of Eaton’s reagent (10% solution of P2O5 in methanesulfonic acid) followed by hydrolysis. Intermediate benzophenones 185 were reacted with 2-bromoethanol or 2-(2-bromoethoxy)ethanol to give hydroxyethyl or hydroxyethoxyethyl derivatives 186, respectively. In the last step, the reaction of intermediates 186 with diphenylsulfonylfuroxan 187 in the presence of DBU afforded benzophenone and furoxan hybrids 188. In order to elucidate the effect of the linker length on the antiproliferative activity, one sulfonyl group of diphenylsulfonylfuroxan 187 was displaced by phenstatin (189), which gave additional derivative 190, in which the furoxan and phenstatin structural moieties were linked via an oxygen atom. The authors [118]did not report the yields of intermediate compounds, except for the yields of derivatives 185.
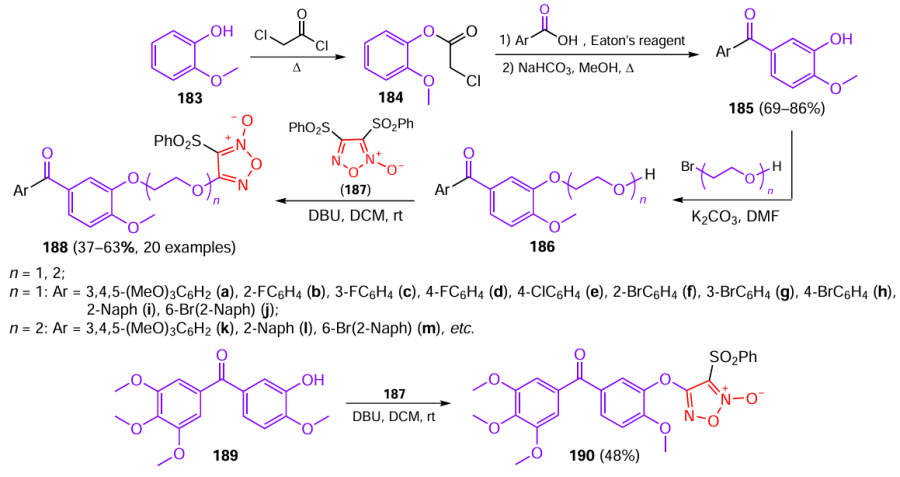
The target products 188a – m (Fig. 23) showed a moderate anticancer activity on the A-2780, MDA-MB-231, HCT-116, and A-549 cell lines, with fosbretabulin (CA-4) and paclitaxel being used as positive control (Table 21). It was found that hybrid compound 188a with an ethyl linker had a higher antiproliferative activity than phenstatin 189a, while the activity of analogues with an oxygen atom (190) or di(oxyethyl) linker (188k) had a lower activity; hence, the preferred length of the linker is two carbon atoms. As regards the substituent effect on the anticancer properties, the best results were achieved upon the introduction of the p-bromophenyl group (188h): the IC50 values for all four cell lines were in the range of 8 – 15 nM. The half-maximal inhibitory concentrations decreased in the presence of para-, meta-, and ortho-substituents in the order 188d > 188c > 188b and 188h > 188g > 188f, while the trend of inhibitory activity for the halogen atoms in the para-position was as follows: bromine > chlorine > fluorine (188h > 188e > 188d). Compound 188h showed the highest nitrite concentration (38.6 μM) among the whole series, thus supporting the correlation between the NO-donor properties and the antiproliferative activity.
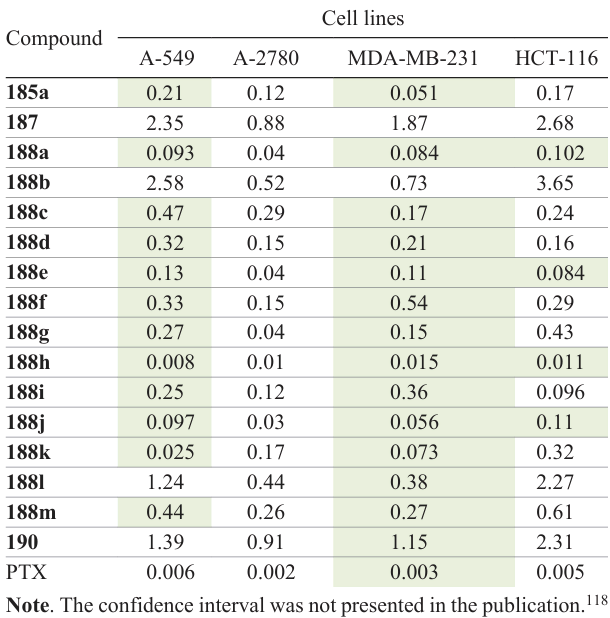
![[{"id":"N2pApUuirp","type":"paragraph","data":{"text":"Structures of hybrids <b>188 </b>based on benzophenone and furoxan possessing antiproliferative activity and active ingredients of commercial drugs (drawn in the box).<sup>118</sup>"}}]](/storage/images/resized/ZNWO0HsL92pbtbHSWoTmY1UzkAFb3O900VCYTV9J_xl.webp)
Mechanistic studies have shown that apart from NO-donor properties, compound 188h also tends to inhibit tubulin polymerization. This provides potent antiangiogenesis and more effective inhibition of colony formation by chemoresistant cancer cell lines, A2780/CDDP (0.015 μM) and A2780/PTX (0.021 μM), compared to that observed for paclitaxel (> 0.5 μM for A2780/CDDP) and cisplatin (> 50 μM for A2780/PTX), as well as cell cycle arrest and induction of apoptosis. In xenograft models in mice, compound 188h markedly inhibited the growth of paclitaxel-resistant tumours, while possessing low toxicity.
Combining NO-releasing furoxan with known anticancer drugs in a single molecule[118-122] substantially increased the efficacy and selectivity of the compounds. Wen et al.[119]prepared hybrids of esculetin, which has anti-inflammatory,[123]antioxidant,[124]and anticancer [125]activity, (phenylsulfonyl)furoxan (NO donor), and 7-(diethylamino)coumarin targeting mitochondria due to its lipophilic properties and depolarizing effect.[126]The starting compound 191 was oxidized with 30% hydrogen peroxide at room temperature (Scheme 38). Then, intermediate compound 192 was nitrated with fuming nitric acid in the presence of glacial acetic acid, then the furoxan ring was closed to give diphenylsulfonylfuroxan 187. Compounds 193a – d were prepared by the nucleophilic substitution of the sulfonyl group in furoxan 187 under the action of ω-bromoalkanols. The reactions of furoxan 193a – d and esculetin (194) in various ratios with potassium carbonate or sodium hydroxide as a base afforded mono- (195a – d) and difuroxan hybrids (196a – d) with one or two substituted hydroxyl groups, respectively.
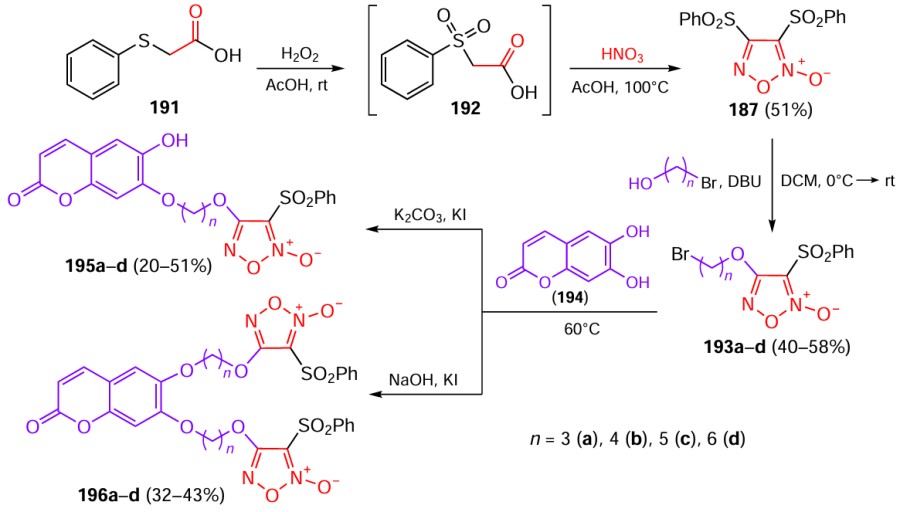
In view of the results of preliminary screening, a series of hybrids 198a – d was additionally synthesized from the most active substituted furoxan 195d with functional groups possessing high affinity to mitochondria (Scheme 39). The nucleophilic substitution reaction of 7-(diethylamino)coumarin-3-carboxylic acid (197) with dibromoalkanes afforded compounds 198a – d, which were then allowed to react with hybrid 195d at the hydroxyl group to give triheterocyclic structures 199a – d.
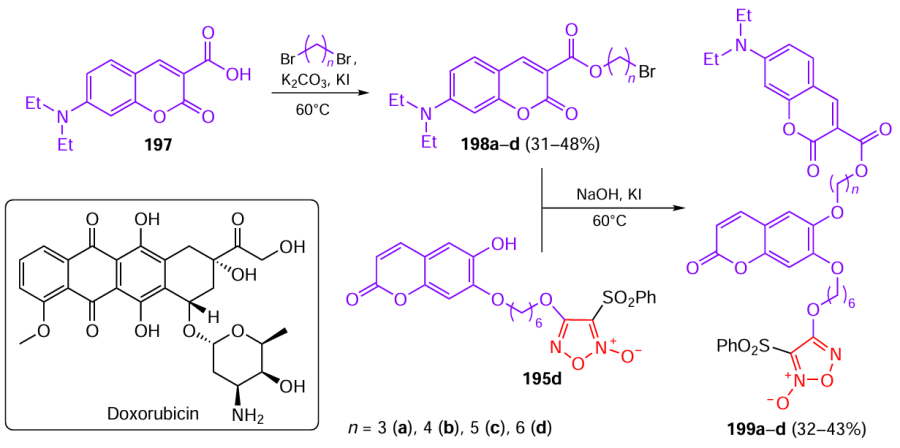
The antiproliferative activity of target products 195a – d, 196a – d, and 199a – d was evaluated against human cancer cells (MDA-MB-231, A-549, HCT-116, and MCF-7) and normal cells (L02 and MCF-10A). Doxorubicin (DOX) was used as a positive control. Table 22 summarizes the results for most active compounds.
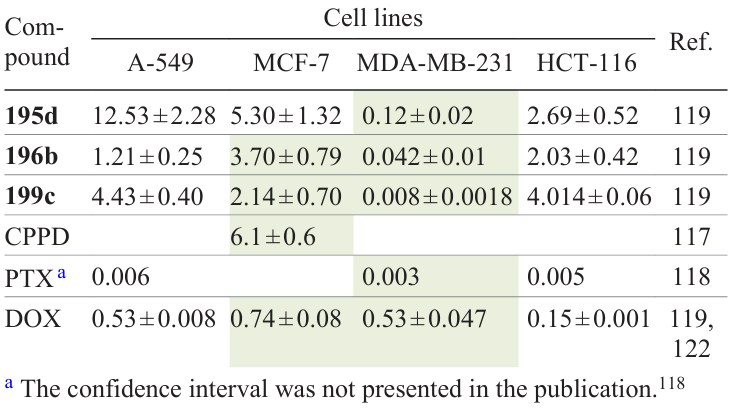
Among the compounds of this series, the best antiproliferative activity against MDA-MB-231 cell line (IC50 = 8 nM) with low toxicity to normal cells (L02, MCF-10A) was found for hybrid 199c, which comprises esculetin, (phenylsulfonyl)furoxan, and 7-(diethylamino)coumarin moieties. However, the IC50 values of this compound determined using other cancer cells were in the micromolar range. This fact was attributed [119]to the fact that MDA-MB-231 cells have a high mitochondrial membrane potential compared to other cancer and normal cells.[127]The action mechanism of compound 199c consists in the action on cell mitochondria, release of high NO concentration, and overexpression of cyclophilin D, which enhances the production of ROS and triggers the apoptosis of cancer cells. To achieve the anticancer effect, this hybrid can arrest the cell cycle in the G2/M phase. In addition, the tumour growth inhibition (TGI) in vivo by compound 199c at a 2.5 mg kg–1 dose exceeds that for DOX at the same concentration; the use of a higher dose (5 mg kg–1) provides TGI of up to 63.5%.
Gu et al.[120]prepared ternary molecular hybrids based on diphenylsulfonylfuroxan (187), piperidine, and pyrimidine (Scheme 40). Furoxan 187 was allowed to react with 4-hydroxypiperidine in alkaline medium to give derivative 199,[128] which then reacted with pyridine derivatives in a basic medium. The pyrimidine structural moieties were obtained in three steps. First, ethyl cyanoacetate 200, thiourea, and benzaldehyde were subjected to Biginelli condensation to give 5-cyano-6-phenylpyrimidine 201.[129] Then compound 201 was treated with aromatic, heteroaromatic, or aliphatic amines with various substituents to be converted to derivatives 202, the hydroxyl group of which was replaced by a chlorine atom on treatment with POCl3.[130] In the final step, furoxan 199 reacted with pyrimidine derivatives 203 in the presence of a base yielding the target products 204.
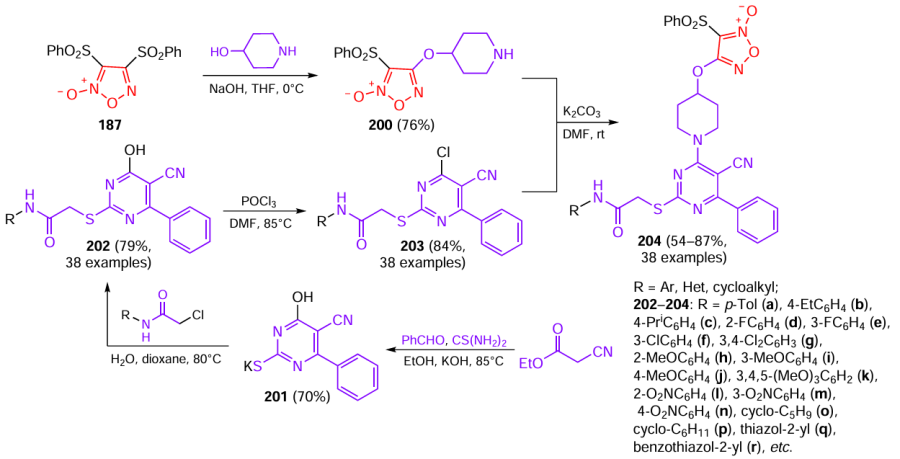
The antiproliferative activity of compounds 204 was evaluated [120]by incubating them with a number of cell lines, including MGC-803 (human gastric cancer), PC-3 (human prostate cancer), MDA-MB-231, and A-549, for 72 h; commercial 5-fluorouracil (5-FU) served as a positive control (Table 23). Most of the compounds showed moderate or good antiproliferative activity in vitro against all four cell lines, with MDA-MB-231 and PC-3 cells being more sensitive. For compounds with electron-donating alkyl groups such as Me, Et, and Pri, the activity decreased upon elongation of the aliphatic chain and upon increase in the substituent bulk, except for ortho-isomers. In the case of para-substituted phenyl group, the series of decreasing activity was as follows: 204a > 204b > 204c (Fig. 24). Ortho-fluorophenyl derivative 204d had a higher antiproliferative activity than meta-substituted product 204e against all cell lines except MDA-MB-231. The antiproliferative activity of the test compounds against A-549 cells generally decreased with decreasing polarity of the halogen atom (204e > 204f), while the antiproliferative activity of dichloro-substituted compounds was lower compared to that of analogous monosubstituted derivatives: 204f > 204g. It is noteworthy that compound 204k with a trimethoxyphenyl substituent was more active in in vitro experiments than monomethoxy derivatives 204h (R = 2-MeOC6H4), 204i (R = 3-MeOC6H4), and 204j (R = 4-MeOC6H4). The introduction of a strong electron-withdrawing nitro group in the para-position of the benzene ring (204n) resulted in a higher antiproliferative activity compared to those of ortho- (204l) and meta-isomers (204m). A bulky heterocyclic amine substituent reduced the antiproliferative activity of the derivatives, with the exception of MDA-MB-231 cell line, in which compounds 204o,p showed the best activity compared to those against the other three cancer cell lines.
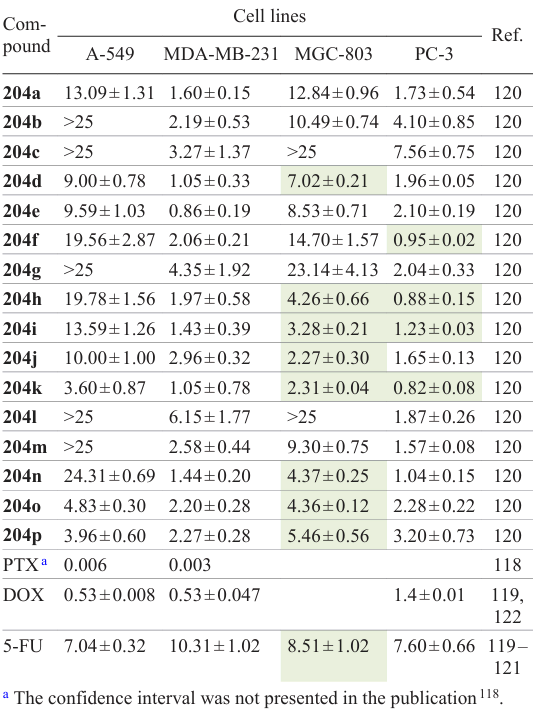
![[{"id":"weTJN5_G4q","type":"paragraph","data":{"text":"Structures of hybrids <b>204 </b>comprising piperidine, pyrimidine, and phenylsulfonylfuroxan moieties and possessing high anticancer activity and 5-fluorouracil (drawn in the box).<sup>120</sup>"}}]](/storage/images/resized/EGRi5jcYV4K3X0tPHpRbp6lsriO2mX1iqrSv5g2n_xl.webp)
Trimethoxyphenyl derivative 204k proved to be the most active compound of this series. Mechanistic studies have shown that this compound induces apoptosis by altering the expression of apoptosis-related proteins such as Bax, Bcl-2, and cytochrome C; it arrests the cell cycle in the G0/G1 phase and affects both mitochondrial and ROS-induced apoptosis pathways. In addition, analysis of NO-donor properties showed a positive correlation between antiproliferative activity and the nitric oxide release capacity for compounds 204. Indeed, compound 204k characterized by the most pronounced NO release showed activity in the nanomolar range of concentrations, which is markedly superior to 5-FU used as the reference; this attests to a synergism between the anticancer activity and intracellular formation of NO.
The preparation of coumarin derivatives of phenylsulfonylfuroxan with anticancer properties was reported by He et al.[121]In the first step, diphenylsulfonylfuroxan 187 was subjected to substitution reactions with various diols to give derivatives 205 (Scheme 41). The target compounds 206a – u were prepared by the reaction of coumarincarboxylic acids 207 with furoxans 205.
The in vitro anticancer activity of target products 206a – u was evaluated against four human cancer cell lines (A549, HT-29, MCF-7, and HepG2) using 5-FU as a positive control (Table 24). The inhibitory activity of compounds 206c,e – g,n,q – s,u in HepG2 cells (IC50 = 2.10 – 5.44 μM) was higher than that of the reference drug (5.49 μM), while derivatives 206g,k,l,n,o,q – s,u inhibited the growth of MCF-7 cells with IC50 < 10 μM. The most pronounced inhibitory effect of compound 206u against MCF-7 cells (4.40 μM) was close to that of 5-FU (4.59 μM). In HT-29 cells, the inhibitory activity of all target compounds (2.75 – 11.23 μM) was superior to the result for the positive control (14.12 μM), and in A549 cells, derivatives 206c,e – g,i – l,n,r,s,u were more active (2.34 – 6.87 μM) than 5-FU (7.04 μM), with the highest activity being inherent in compound 206g (2.34 μM). Thus, 206g proved to be the most active compound of this series against the four cancer cell lines.
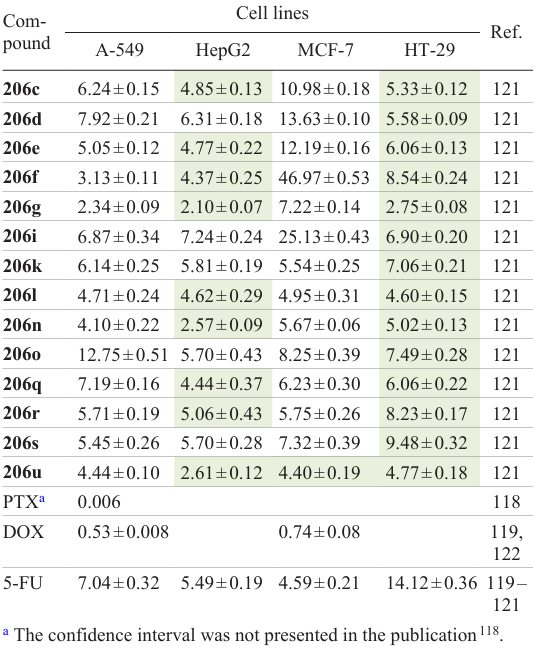
It should be noted that the antiproliferative properties of most coumarins are enhanced when a Mannich base is introduced in the C(8) position. The piperidine ring at the C(8) position of coumarins 206c,e,f,g enhances the inhibitory activity against HepG2 cells compared to that of unsubstituted coumarin, but in the case of piperidine derivatives and MCF-7 cells, the IC50 values markedly exceed the values for other lead compounds of this series. The presence of the N-methyl- or N-ethylpiperazine moiety at the same C(8) position enhances the proliferation-inhibiting effect of coumarins 206h – u in MCF-7 cells; this is especially pronounced for N-ethyl homologues 206o – u. A relationship between the activity and the linker structure was also detected: when butyne-1,4-diyl serves as the bridging R1 moiety, the target compounds have the most pronounced inhibitory effect, while an ester bridging group results in a moderate inhibition. If a diethylene glycol residue acts as the bridge, the inhibitory activity of these compounds against cancer cells decreases to even a greater extent. In addition, preliminary study of the inhibition mechanism showed that the most active compound 206g substantially enhances the apoptosis of HepG2 cells in a dose-dependent manner, but only slightly delay the cell cycle in the S phase.
The oncogenic metabolism in tumour cells generates an excess of acidic subproducts, including lactic acid and carbon dioxide.[131]The tumour cells counteract the intracellular acidosis by activating the concerted interaction between various transporters, ion exchangers, and carbonic anhydrases for adaptation and support of tumour growth.[132]Actually, human carbonic anhydrase (hCA) IX and XII inhibitors reduce the growth, proliferation, and metastatic potential of aggressive malignant tumours both in vitro and in vivo.[133]The goal of the study by Bua et al.[134]was to combine the NO-donor benzofuroxan structural motif and hCA inhibitor moiety (Scheme 42). Benzofuroxancarboxylic acid 208 reacted with amines 209, sulfonamides 210, or coumarins 211 by substituting the primary or secondary amino group or the hydroxyl group. The carboxyl group of the acid was activated by carbodiimide using azabenzotriazole, 4-dimethylaminopyridine, or N-hydroxysuccinimide. This gave target hybrids 212 – 214.
The prepared NO-donor hybrids 212 – 214 and the starting benzofuroxan 208 were evaluated for the inhibitory activity against cytosolic isoenzymes hCA I and II (which are not targets) and target membrane-bound isozymes hCA IX and XII and for NO-release properties. The most active compounds 213a,b showed a potent dual action: they had low nanomolar inhibitory concentrations for isozyme hCA IX (IC50 = 10.4 and 2.5 nM, respectively) and showed considerable NO release (66 ± 1.4% and 65 ± 1.5%, respectively). It is worth mentioning that compound 213a exhibited a pronounced antiproliferative effect against various cancer cell lines, in particular renal carcinoma cells (A-498). In these cells, the sulfonamide benzofuroxan derivative 213a substantially decreased the expression of hCA IX (IC50 was 3.0 – 6.9 μM after inhibition for 72 h) and iron regulatory proteins. The induction of apoptosis by activation of mitochondrial caspase and ferroptosis pathways was indicated by increasing concentrations of ROS and nitrites and decrease in the expression of ferritin-encoding genes. It is worth noting that in three-dimensional tumour models, compound 213a effectively reduced the spheroid size and viability. Experiments to assess in vivo toxicity in mice demonstrated that compounds 213a,b were well tolerated by the mice without significant impairment of renal function.
Chugunova et al.[122]investigated various types of activity (including anticancer and antimicrobial activities) of the hybrid systems containing phosphorylated sterically hindered phenols (SHP) and dinitrobenzofuroxans. The sterically hindered phenols with diaminopyridine or diaminophenyl moiety were used as the starting point due to high and selective cytotoxicity.[135]In the first step (Scheme 43), phenol 215 was phosphorylated, then compounds 216 were oxidized to quinonemethylides 217, which subsequently reacted with C-nucleophiles (2,6-diaminopyridine or 1,3-diaminobenzene).[136][137] This sequence of transformations furnished key intermediates 218a – g with two amino groups suitable for modification with a heterocyclic moiety, nitrochlorobenzofuroxan 219. This heterocycle is highly electrophilic and readily reacts with anilines. The phenolic OH group does not react under these conditions due to steric hindrance caused by the two tert-butyl groups. Depending on the used reactant ratio, the composition of the final products can be varied, which gave previously unknown phosphorylated sterically hindered phenol hybrids of 1 : 1 (220a – g and 220'e,g) or 2 : 1 (221a – g) composition. For some of the monosubstituted compounds (220e, 220g), isomerization of the N – O bond of the furoxan ring was observed, whereas for disubtituted analogues 221, no isomerization was detected.
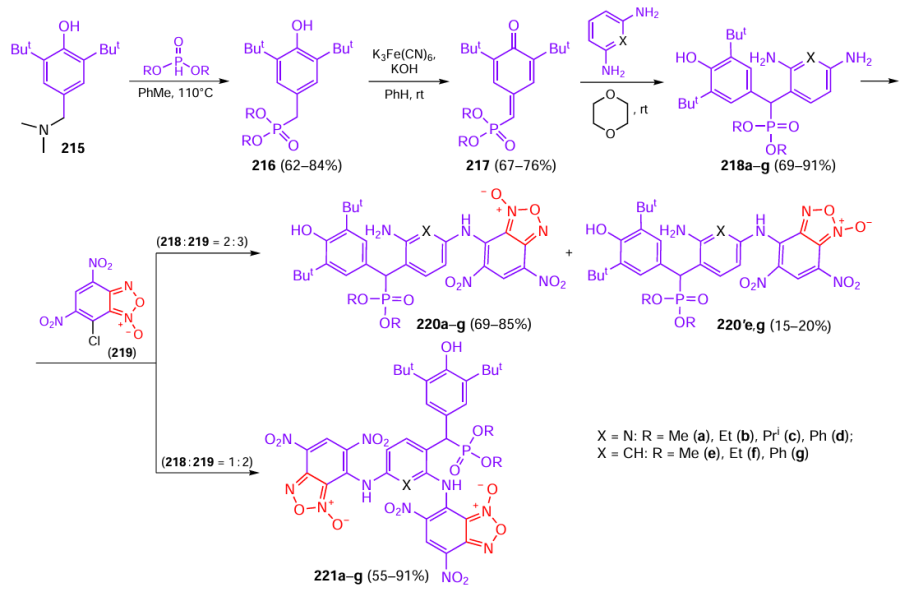
The antiproliferative activity of compounds 220, 221 was compared with the activity of the starting building blocks 218a – g and 219. Unlike the starting phosphates, nitrochlorobenzofuroxan 219 does not show cytotoxicity at millimolar concentrations (Table 25). The target products 220a – g, 221a – g had relatively high activity against all cancer cell lines used in experiments and moderate cytotoxicity against normal Chang-Liver cells (M-HeLa). As compared with the starting compounds, hybrid 220c comprising SHP and benzofuroxan moieties was 40 and 20 times more active than SHP 218c against M-HeLa and MCF-7 cells, respectively. The activity of hybrid 221d against M-HeLa and MCF-7 cells was 3.7 and 7.6 times higher than the activity of compounds 218d. However, it is noteworthy that the cytotoxicity of the lead hybrid compounds against normal cells also increased. These data indicate that finding the right balance between the efficacy and selectivity remains a challenging task. The IC50 values for compounds 220c and 221d against HuTu-80 (duodenal adenocarcinoma), MCF-7, and M-HeLa cell lines were either comparable with or higher than those for doxorubicin. Compound 221d with SI = 4 can be considered to be selective to MCF-7 and M-HeLa cells, with DOX being markedly inferior in the selectivity. Study of the cytotoxicity mechanisms suggests that it may be attributable to the induction of apoptosis via the intrinsic mitochondrial pathway and increased production of ROS. The tested compounds showed no haemolytic activity (IC50 > 100 μM) and were stable in the whole blood of mice for 1 h.
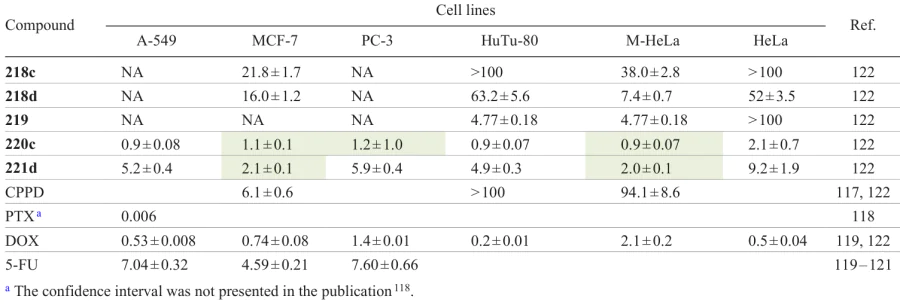
Thus, to summarize the data presented in Tables 21 – 25,[117-122] it can be concluded that phenstatin furoxan hybrids 188a – m are the most active compounds exhibiting antiproliferative activity in the nanomolar range against A-549, MDA-MB-231, and HCT-116 cell lines. The activity of most representatives of this series exceeded that of DOX, and lead compound 188h containing the p-bromophenyl substituent was similar in the characteristics to cisplatin. The lowest IC50 value against MDA-MB-231 cell line was found for esculetin furoxan derivative 199c. Piperidine pyrimidine phenylsulfonylfuroxan hybrids 204a – r proved to be the most effective inhibitors of MGC-803 and PC-3A. Among them, compound 204k with the trimethoxyphenyl substituent was most effective, with the half-maximal inhibitory concentration being lower than that for the reference etoposide. Meanwhile, coumarin furoxan derivatives 206a – u, among which piperidine derivative 206g was most active, were superior against HT-29 and HepG2 cells to 5-fluorouracil. Phosphorylated hybrid 220c based on SHP and benzofuroxan with an isopropyl substituent exhibited high antiproliferative activity against MCF-7, PC-3, and M-HeLa cell lines. However, none of the oxadiazoles tested against the HuTu80 and HeLa cells was superior to doxorubicin.
4.3. Compounds with antibacterial activity
Hybrids 220a – g and 221a – g based on phenols and benzofuroxan were also studied[122] for antimicrobial activity using Gram-positive (S. aureus, B. cereus, E. faecalis) and Gram-negative (E. coli, P. aeruginosa) bacteria and methicillin-resistant Staphylococcus aureus strains (MRSA-1, MRSA-2). The MRSA-1 strain is resistant to β-lactams and fluoroquinolones, and MRSA-2 is only β-lactam-resistant.
The antifungal activity of these compounds was assessed against C. albicans yeast. Neither benzofuroxan 219 nor 1 : 1 hybrids 220a – g showed activity against fungal or bacterial strains. Meanwhile, the introduction of a second benzofuroxan moiety enhanced the antimicrobial activity (see below):*[122][138][139] compounds 220a,c,d were found to exhibit selective antimicrobial activity against Gram-positive S. aureus, B. cereus, and E. faecalis comparable with the activity of the reference chloramphenicol and against the MRSA-1 strain; however, they were less active against MRSA-2. None of the compounds showed any effect against Gram-negative bacteria or C. albicans yeast. Thus, the antimicrobial effect is present only if at least two benzofuroxan moieties have been introduced into phenol. Overall, the combination of sterically hindered phenol moieties with benzofuroxan in a single molecule provides a number of benefits, including enhanced ROS production and greater selective cytotoxicity.
Imidazolethiones are cyclic analogues of thiourea with high pharmacological activity.[140]Hybrid molecules containing imidazolethiones are used to treat hyperthyroidism [141]and as antioxidants in vivo,[142]while the imidazolinethione moiety fused to cyclic compounds is used as a two-photon fluorescent probe for selective detection of hypochlorite anions in mitochondria.[143][144]However, furazan derivatives of imidazolethiones have not been obtained until recently; this was the goal of a study by Kurt and Sevgi.[138]In the first step Scheme 44), dichloroglyoxime (222) was treated with sodium carbonate to obtain highly reactive bis(nitrile oxide) 223, which was allowed to react with N,N'-diphenylthiourea (224) to give a mixture of syn- and anti-isomeric hydroxylamines 225. On refluxing in ethanol, stereoisomer mixture 225 was converted to pure syn-isomer 226. The target N,N'-diphenylimidazofurazanthione 227 was prepared by a microwave method in the presence of silica gel as a catalyst on a solid substrate.
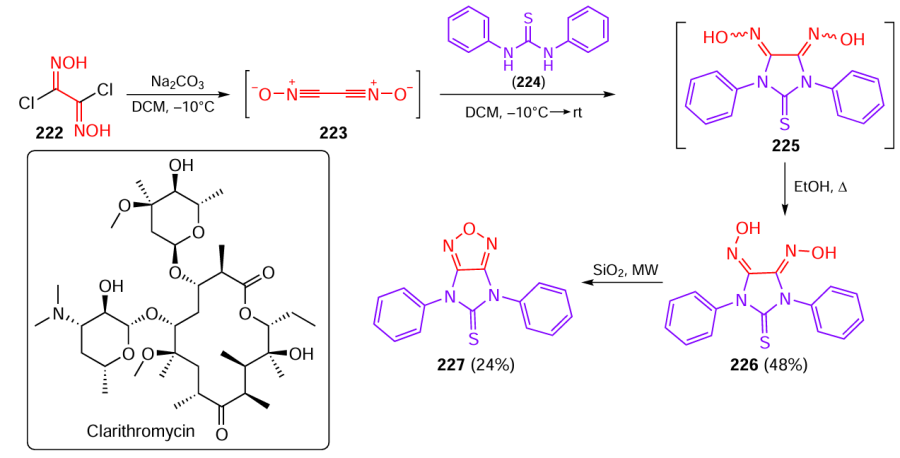
The biological activity of furazan 227 was demonstrated by assessing its antibacterial effect (see below) against a series of Gram-positive (S. aureus, MRSA, B. cereus, E. faecalis, S. epidermidis) and Gram-negative strains (E. coli, P. aeruginosa, K. pneumoniae). Gram-positive bacteria, especially Staphylococcus aureus, were more sensitive to furazan 227 (MIC = 64 – 128 μg mL–1). At the same time, compound 227 did not exhibit any antibacterial effect against Gram-negative strains. This paradox is often encountered in the literature: many antibacterial agents are less effective against Gram-negative bacteria than against Gram-positive strains, since the permeability of the outer membrane allows only slow penetration of the agent.[145]The cell wall compositions of Gram-positive and Gram-negative bacteria are markedly different, since Gram-negative bacteria have two cellular membranes and, furthermore, it is difficult for small molecules to cross the lipopolyssacharide-coated outer membrane.[146]
The antibiotic resistance of bacteria is a serious global health problem. Multidrug resistant (MDR) gram-negative bacteria cause particular concern due to reduced treatment options and high mortality.[147]Colistin, an antibiotic belonging to the polymyxin group, is an effective therapeutic agent against Gram-negative infections,[148]but wide use of this drug gave rise to drug resistance.[149] Harris et al.[139]prepared (1,2,5-oxadiazolyl)pyrazines, which increase the sensitivity of Gram-negative bacteria to colistin (Scheme 45). The first step was the reaction of diaminofurazan 181 with oxalic acid followed by treatment with a mixture of PCl5 with POCl3, which gave dichloride 228. Then compound 228 was treated with various amines to give disubstituted structures 229. The replacement of one chlorine atom in benzofurazan 228 by m-chloroaniline in the presence of triethylamine afforded unsymmetrical pyrazinylfurazan 230, which was then reacted with alkyl- or arylalkylamines to give products 231.
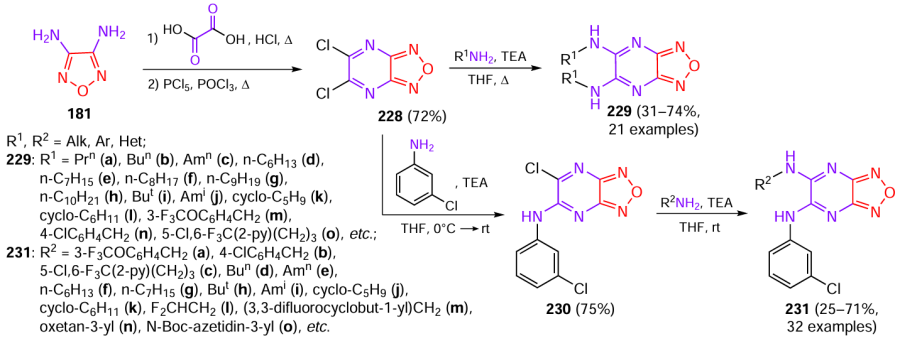
The colistin-potentiating activity of target compounds 229 and 231 was evaluated using minimum regrowth concentration (MRC) analysis (Table 26).[122][138][139] The MRC value was defined as the lowest concentration of compounds that inhibited bacterial growth in the presence of 2 μg mL–1 of colistin on an MDR E. coli strain, AR-0493. For compounds with MRC ≤ 0.5 μg mL–1, the minimum inhibitory concentration for E. coli cells was determined: all tested analogues had MIC > 64 μg mL–1, which is indicative of the absence of intrinsic antibacterial activity in compounds of this class.
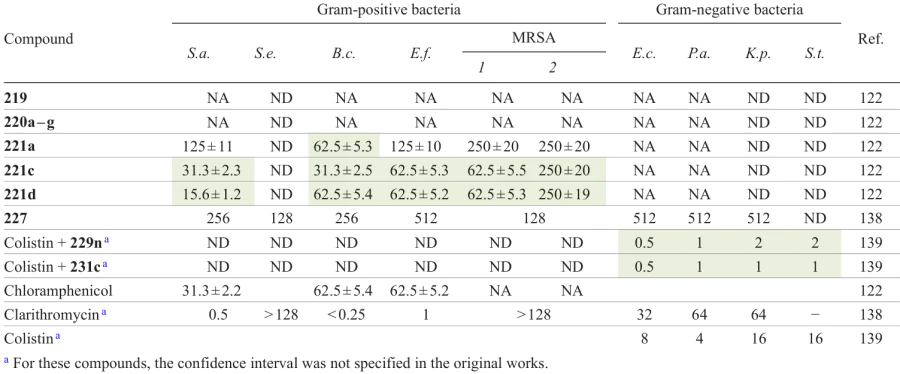
For a series of symmetrical diamines 229, SAR investigations were carried out. In the case of compounds 229a – h with hydrocarbon substituents, the antibacterial activity increased as the hydrocarbon chain increased up to seven carbon atoms and then decreased in the series 229a (C3; MRC > 16 mg mL–1) < 229d,e (C6 and C7; MRC = 0.5 mg mL–1) > 229g,h (C9 and C10; MRC > 16 mg mL–1). The optimal chain length for compounds in this series was determined to be 6 or 7 carbon atoms. Branched substituents, such as tert-butyl (229i, MRC = 8 mg mL–1) and isoamyl groups (229j, MRC = 2 mg mL–1), did not markedly alter the activity of derivatives compared to their linear analogues. The introduction of cyclic groups, e.g., cyclopentyl (229k, MRC = 8 mg mL–1) and cyclohexyl (229l, MRC = 4 mg mL–1), induced a decrease in the antibacterial action. Arylalkylamines 229m (MRC = 0.125 mg mL–1), 229n (MRC = 0.125 mg mL–1), and 229o (MRC = 0.25 mg mL–1) had the best activity among all compounds of this series; the structures of these compounds are depicted in Fig. 25.
![[{"id":"2Ph6zKimKD","type":"paragraph","data":{"text":"Structures of antibacterial pyrazinofurazans <b>229 </b>and <b>231</b>.<sup>139</sup>"}}]](/storage/images/resized/94st1YE1YBomskxKB2Q3xI7WlJaSIp7X8B1qCGYr_xl.webp)
Unsymmetrical amines 231 followed a trend similar to that of the symmetrical series: the activity also increased with increasing number of carbon atoms in the alkyl chain until the chain length reached seven, after which the activity decreased. The most pronounced antibacterial effect was found for compounds with C4 (231d), C5 (231e), and C7 (231g) chains as the substituent R2, which were characterized by MRC of 0.5 μg mL–1, while C6 derivative 231f had MRC = 1 μg mL–1. Branched isomers 231h,i (MRC = 2 μg mL–1) had a lower activity than linear analogues, whereas cyclic derivatives 231j,k always had MRC of 1 μg mL–1. The introduction of heteroatoms into substituents either had a minor effect on the activity (for difluorinated derivatives 231l,m, MRC = 1 – 2 μg mL–1) or, in the case of oxygen atom (231n; MRC > 16 μg mL–1) or NBoc group (231o; MRC = 4 μg mL–1), considerably decreased the activity. This tendency implies that hydrophobic substituents are generally preferred, which is consistent with the previously described membrane-penetrating ability of this class of compounds.[150][151] In the case of unsymmetrical pyrazinofurazans 231, the dependence of activity on the structure was also noted: compounds with chloro and trifluoromethyl substituents in benzene rings showed higher activity that the fluoro- and trifluoromethoxy-substituted compounds; the most active derivatives of series 231a – c had MRC = 0.25 μg mL–1.
Two compounds, 229n and 231c, were identified as possessing the highest activity that enhanced the action of colistin (see Table 26).[139]All investigated bacterial strains (E. coli, P. aeruginosa, K. Pneumoniae, and S. typhimurium) showed resistance to colistin with MIC being in the range from 4 to 16 μg mL–1. Derivatives 229n and 231c at concentrations of 1 μg mL–1 enhanced the action of colistin 32-fold and decreased MIC down to values equal to or lower than the clinical endpoint ** for all five strains.
* Numerical results on assessment of antibacterial activity are given at the end of the Section in Table 26.122,138–139
** The clinical endpoint is an event specified in advance that is used to evaluate the efficacy or safety of medical intervention in clinical trials.
4.4. Compounds with antiplatelet activity
Early studies of the inhibition of platelet aggregation by 1,2,5-oxadiazoles led to the discovery in the mid-1990s of a drug with the commercial code CAS 1609 (4-hydroxymethyl-3-carbamoylfuroxan), a therapeutic agent characterized by good vasodilatory properties [152][153] and antiplatelet activity.[154]
The search for new furoxan derivatives capable of NO release and, as a consequence, inhibition of thrombus formation was reported by Zhilin et al.[155]The starting 4-amino-1,2,5-oxadiazoles 232a – e and 233b were prepared by the procedure reported earlier.[156]Then these compounds were diazotized by treatment with NOBF4, and the intermediate diazonium salts 234a – e, 235b were subjected to azo coupling with potassium trinitroformate. Intermediates 236a – e, 237b were converted to water-soluble azasydnonylfuroxans (238a – e) and -furazans (239b) via a cascade of ZnCl2-catalyzed rearrangements (Scheme 46).
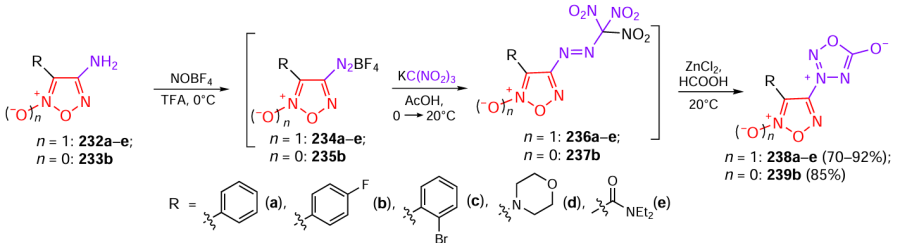
The prepared series of compounds was evaluated for antiplatelet (antiaggregant) activity in vitro using platelet-rich plasma (PRP). Three platelet aggregation inducers were used in the experiments, namely, adrenaline, adenosine diphosphate (ADP), and arachidonic acid. A sample was heated with PRP at a temperature of 37°C, then an inducer was added and light transmittance was recorded. None of the samples inhibited the arachidonic acid-induced platelet aggregation, but all compounds 238a – e and 239b showed excellent antiplatelet activities for ADP and adrenaline. In all experiments, the platelet aggregation remained at a negligible level, which is a unique feature. It should be noted that compounds 238b and 239b, distinguished by the presence of N-oxide group in 1,2,5-oxadiazole, showed almost identical antiplatelet activity, which attests to the crucial role of the azasydnone motif for this type of activity. It is worth noting that the addition of arachidonic acid at the seventh minute of the experiment immediately activated agglutination, indicating that after treatment with azasydnonyl(1,2,5-oxadiazoles), platelets are still able to interact with one another. A tenfold increase in the test concentration of compounds 238a – e and 239b led to a similar effect. These results indicate that compounds 238a – e and 239b inhibit the ADP- and adrenaline-induced platelet aggregation by a selective mechanism; It is important that ADP and adrenaline are considered to be the main agents that induce thrombus formation.[157][158]
4.5. Compounds with anti-inflammatory activity
While searching for new anti-inflammatory agents with improved pharmacological profiles, researchers are increasingly turning to plant extracts due to their high availability and low toxicity.[159]Caper spurge (Euphorbia lathyris) is used in traditional Chinese medicine for the treatment of terminal schistosomiasis, ascites, dropsy, and snake bites,[160]while Euphorbia factors L1, L2, L3 and some lathyrane diterpenoids isolated from Euphorbia lathyris inhibit LPS-induced NO production in RAW264.7 cells.[161]Meanwhile, NO can inhibit platelet aggregation, reduce neutrophil adhesion and activation, and suppress the overexpression of NO-synthase.[162][163] Thus, the synthesis of anti-inflammatory drugs based on furoxan hybrids is considered to be a promising strategy, because the introduction of an NO-donor moiety increases the anti-inflammatory activity.
Wang et al.[159]prepared three series of hybrids of the lathyrane diterpenoid and furoxan hybrids possessing anti-inflammatory activity (Scheme 47). Diphenylsulfonylfuroxan 187 reacted with various diols or Boc-protected ethanolamine in an alkaline medium; this was followed by deprotection to give compounds 240 and 241a – d, which were reacted with succinic anhydride in the presence of DMAP. Epoxylathyrol (242a) and lathyrol (242b) were used as the reagents for esterification of obtained acids 243 and 244a – d, which resulted in the formation of products 245a,b and 246a – h. A similar sequence of transformations of furoxan 248, derived from cinnamic alcohol (247), afforded, first, acid 249 and then hybrids 250a,b.[159]
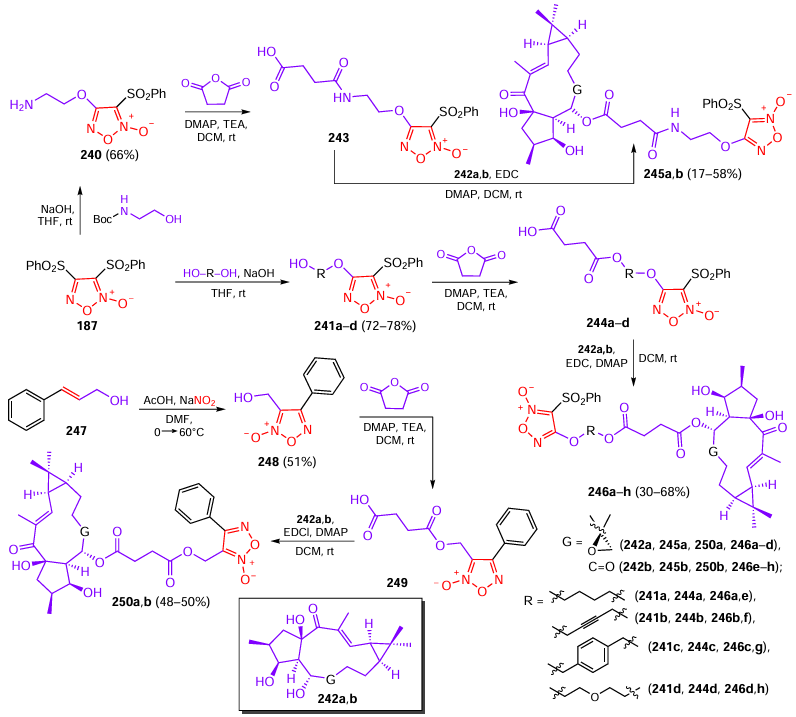
All synthesized phenylsulfonylfuroxan derivatives possess higher inhibitory activity than the starting diterpenoids, epoxylathyrol (242a) and lathyrol (242b) (Table 27). Hybrids 246f,h were the most active compounds of this series, and lathyrol derivatives were better than epoxylathyrol derivatves containing the same moieties (242b > 242a, 250b > 250a).
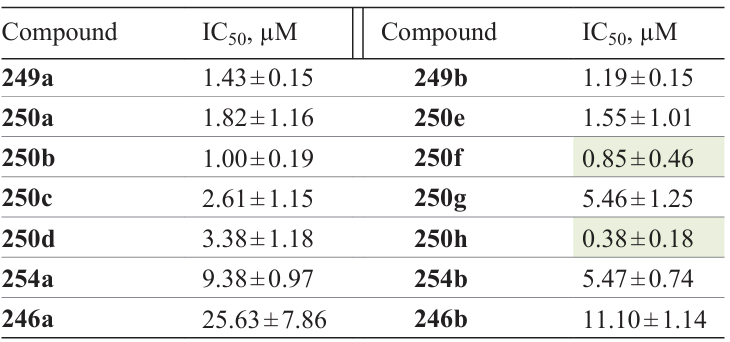
In order to investigate the relationship between the intrinsic ability to release NO and the activity of compounds, structures 246c,f,h were selected and used to determine the concentration of NO in RAW264.7 cells using Griess test. Compound 246h proved to be the most active NO donor with the highest nitrite concentration. The subsequent anti-inflammatory assay showed that 246h can substantially decrease ROS level, and its anti-inflammatory effect was associated with the inhibition of the transcroption activation of the Nrf2/HO-1 pathway.
4.6. Summary of the data on the 1,2,5-oxadiazole activity
1,2,5-Oxadiazoles are a class of compounds that was widely addressed in the literature of the last five years. Furoxan derivatives are of most interest due to their ability to release NO in the cell and antiproliferative activity. The optimal approach to study these compounds that has currently formed includes the construction of a furoxan ring on the basis of known anticancer therapeutic agents [e.g., phenstatin (hybrids 188) or esculetin (195, 196, 199)], which may provide IC50 values in the nanomolar range. Another promising trend is combination of two or more pharmacophore moieties in a single molecule: for example, piperidine, pyrimidine, and phenylsulfonylfuroxan in products 204 or SHP and benzofuroxan in 220 and 221; the synergistic action of the moieties makes it possible to achieve outstanding biological properties of the hybrids. Mention should also be made of studies dealing with the growth-regulating (for derivatives 178), antiplatelet (238), and anti-inflammatory (245, 246, and 250) activities of furoxan, although research along these lines is less popular. Meanwhile, furazans, which cannot release NO, but are much more hydrolytically stable, are promising building blocks for the production of antibacterial drugs (e.g., compounds 227 and 231).
Resorting to SAR studies, we concluded that furoxan derivatives with the following characteristics would be most active:
— a phenylsulfonyl group at the C(3) atom of the 1,2,5-oxadiazole ring (188h, 190, 199c, 206g),
— linkers with preferred chain length from two (188a, 206c,d) to seven carbon atoms (229d – e, 231b,c,d),
— more favourable substitution of the benzene ring is a para-bromine atom among halogens (188h) or a para-trifluoromethyl group (231a).
An increase in the antiproliferative activity of phenyl-substituted furoxan is also induced by the presence of electron-donating alkyl groups with a small-size short carbon chain (204a) or trimethoxy-substituted benzene rings (204k). Also, a direct correlation was found between the activity and the presence of fused aromatic amines (178a) or heterocyclic substituents, N-methyl or N-ethylpiperazine (206h – u).
5. Conclusion
Thus, analysis of the published data demonstrates a wide variety of biological activities found in different oxadiazole derivatives. In recent years, the search for lead compounds that possess better pharmacological properties than commercial drugs used in clinical practice among oxadiazoles containing an endocyclic N – O bond has been a highly relevant area of research at the intersection of organic and medicinal chemistry and clinical pharmacology.
1,2,4-Oxadiazoles are considered as the most common class of biologically active oxadiazoles with an endocyclic N – O bond. This is due not only to the highest hydrolytic stability of this ring among isomeric oxadiazoles, but also to the trend for using bioisosteres of therapeutic agents that persists in medicinal chemistry. Since 1,2,4-oxadiazole is a bioisostere of the fairly common ester and amide functional groups and in view of the high degree of elaboration and versatility of the methods for assembly of this ring via one- or two-step transformations of available nitriles, researchers often address these heterocyclic structures in order to enhance the pharmacological activity of known therapeutic agents. Another fairly important factor is high stability of the 1,2,4-oxadiazole ring to the action of various, sometimes quite harsh, reagents. This makes it relatively easy to perform post-functionalization of already assembled heterocyclic structures for finer tuning of pharmacological properties and for access to promising lead compounds. Apparently, this combination of facile synthetic approaches and predictable reactivity of 1,2,4-oxadiazoles made it possible to identify quite a few compounds of this class that have a wide range of pharmacological properties, including antibacterial, anticancer, nematicidal, antioxidant, anti-inflammatory, fungicidal, and antiviral activity.
The second most abundant class of pharmacologically active oxadiazole derivatives with endocyclic N – O bonds are 1,2,5-oxadiazoles; in recent years, studies have been focused on the derivatives of their oxides, furoxan. This is attributable to the wide range of NO-donor properties of furoxan derivatives, which is utilized in the targeted synthesis of anti-inflammatory and thrombolytic agents. In addition, in recent years, key trends have been identified towards NO-induced enhancement of antiproliferative and antibacterial properties; this increased the number of studies dealing with hybrid molecular structures that combine the NO-donor furoxan moiety with a known antibiotic or cytostatic drug. It is also important that particularly in the last five year, growth-regulating activity of furoxan derivatives wa found. This gives hope for achieving a new level of efficiency of the pre-sowing treatment of seeds of certain agricultural crops.
Finally, the least known subclass of pharmacologically active oxadiazoles with an endocyclic N – O bond are sydnones. This is due, to a certain extent, to the existence of only one admissible method for the assembly of their heterocyclic ring by nitrosation of substituted amino acids and to limited options for functionalization and post-modification of these derivatives. In addition, the sydnone moiety has a very limited hydrolytic stability. The pharmacological potential of sydnones is largely related to their possible applications in biorthogonal chemistry and in certain bioimaging methods. Nevertheless, in recent years, some sydnones have also been identified as lead compounds possessing pronounced antibacterial, antioxidant, and fungicidal activities. In addition, some sydnone derivatives have potent growth-regulating activity and herbicide antidote properties, which implies their potential practical use in agriculture.
In conclusion, it should be emphasized that the publications systematized in this review have mainly appeared over the last 5 to 10 years, which highlights the high relevance and demand for this scientific field on a global scale. Apart from the studies discussed in detail in this review, a number of other domestic publications of the past five years have been devoted to the biological activity[164-166] and organic materials science applications[167-173] of oxadiazoles and related nitrogen-and-oxygen systems. It is beyond doubt that the potential of development of this area is far from being exhausted, and efforts of many research groups in the coming years will be focused on more subtle optimization of the pharmacological properties of oxadiazole derivatives and hybrid structures based on them via targeted introduction of new pharmaceutical blocks and/or targeted post-modification of existing therapeutic agents. In addition, another relevant issue is more precise elucidation of the mechanisms of action of lead compounds of the oxadiazole series in in vivo models. We have no doubt that studies related to the targeted synthesis of pharmacologically active oxadiazoles will intensively develop in the coming years and will lead to the design of promising new-generation drugs based on them.
6. List of abbreviations
The following abbreviations and symbols are used in the review:
AChE — acetylcholinesterase,
ADP — adenosine diphosphate,
AIBN — azobis(isobutyronitrile),
All — allyl,
Am — amyl (pentyl),
Boc — tert-butoxycarbonyl,
BODIPY — 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene,
BRAF — serine/threonine protein kinase,
BuChE — butyrylcholinesterase,
CA-4 — fosbretabulin,
CAS 1609 – 4-hydroxymethyl-3-carbamoylfuroxan,
CC50 — half-maximal cytotoxic concentration,
CDI — 1,1'-carbonyldiimidazole,
CDMT — 2-chloro-4,6-dimethoxy-1,3,5-triazine,
COX — cyclooxygenase,
Cys — cisplatin,
DABCO — 1,4-diazabicyclo[2.2.2]octane,
DCC — 1,3-dicyclohexylcarbodiimide,
DCE — 1,2-dichloroethane,
DCM — dichloromethane,
DDQ — 2,3-dichloro-5,6-dicyano-1,4-benzoquinone,
DIPEA — N,N-diisopropylethylamine,
DOX — doxorubicin,
DPPH — 2,2-diphenyl-1-picrylhydrazyl,
EC50 — half-maximal effective concentration,
EDC — 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide,
EDTA — ethylenediaminetetraacetate,
ELISA — enzyme-linked immunosorbent assay,
FDA — Food and Drug Administration,
FLAP — 5 lipoxygenase-activating protein,
5-FU — 5-fluorouracil,
GI50 — concentration of 50% inhibition of cancer cell proliferation,
HATU — hexafluorophosphate azabenzotriazole tetramethyl uronium,
HBTU — hexafluorophosphate benzotriazoletetramethyluronoium,
HBV — hepatitis B virus,
hCA — human carbonic anhydrase,
HIV — human immunodeficiency virus,
HOBt — 1-hydroxybenzotriazole,
IBCF — isobutyl chloroformate,
IC50 — half-maximal inhibitory concentration,
IR — inhibition rate,
LC50 — median lethal concentration,
LD50 — median lethal dose,
LG — leaving group,
LO — lipoxygenase,
LPS — lipopolysaccharide,
MAO — monoamine oxidases,
MDR — multidrug resistance,
MIC — minimum inhibitory concentration,
mPGES-1 — microsomal prostaglandin E2 synthase-1,
MPM — malignant pleural mesothelioma,
MRC — minimum regrowth concentration,
MRSA — methicillin-resistant Staphylococcus aureus strain,
MW — microwave radiation,
NA — no activity,
Naph — naphthyl,
NBS — N- bromosuccinimide,
NCS — N-chlorosuccinimide,
ND — activity was not determined,
NHS — N-hydroxysuccinimide,
NMM — N-methylmorpholine,
PBMC — (human) peripheral blood mononuclear cells,
PD — Parkinson’s disease,
PLpro — papain-like protease,
PMMoV — pepper mild mottle virus,
PTX — paclitaxel,
PRP — platelet-rich plasma,
Py — pyridyne,
py — pyridyl,
ROS — reactive oxygen species,
rt — room temperature,
SAR — structure–activity relationship,
SDH — succinate dehydrogenase,
SHP — sterically hindered phenols,
SI — selectivity index,
TBAB — tetra-n-butylammonium bromide,
TBTU — 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate,
TEA — triethylamine,
TEAA — 2-hydroxyethylammonium acetate,
TFA — trifluoroacetic acid,
TFAA — trifluoroacetic anhydride,
TfO — trifluoromethanesulfonate (triflate),
TGI — tumour growth inhibition,
Th — thienyl,
THIQ — tetrahydroisoquinoline,
THQ — tetrahydroquinoline,
TI — therapeutic index,
TK — tyrosine kinase,
TMV — tobacco mosaic virus,
Tol — tolyl (methylphenyl),
TrxR1 — thioredoxin reductase,
Ts — p-toluenesulfonyl,
TSWV — tomato spotted wilt virus,
ZI — zones of inhibition.
Latin names of microorganisms:
Aphelenchoides besseyi — rice leaf nematode,
Aspergillus clavatus — needle-shaped aspergillus,
Aspergillus niger — black aspergillus,
Bacillus subtilis — hay bacillus,
Blumeria graminis — powdery mildew pathogen,
Botrytis cinerea — botrytis gray mould,
Bursaphelenchus xylophilus — pine wood nematode,
Caenorhabditis elegans — free-living soil nematode,
Candida albicans — diploid fungus,
Clostridioides difficile — causative agent of pseudomembranous colitis,
Ditylenchus destructor — potato rot nematode,
Escherichia coli — enteric bacterium,
Klebsiella pneumoniae — Friedlander’s bacillus,
Meloidogyne incognita — southern root-knot nematode,
Phakopsora pachyrhizi — soybean rust pathogen,
Plasmodium falciparum — causative agent of human malaria,
Pseudomonas aeruginosa — blue pus bacillus,
Pseudoperonospora cubensis — downy mildew pathogen,
Puccinia rubigo — cereal crop rust disease pathogen,
Puccinia sorghi — corn rust disease pathogen,
Rhizoctonia solani — pathogen of rhizoctonia root rot in wheat,
Salmonella typhimurium — causative agent of salmonellosis,
Sclerotinia sclerotiorum — white mould pathogen,
Staphylococcus aureus — golden staphylococcus,
Staphylococcus epidermidis — epidermal staphylococcus,
Streptococcus pyogenes — pyogenic streptococcus,
Valsa mali — apple Valsa canker pathogen.
References










![Metabolism of [14C]GSK977779 in Rats and Its Implication with the Observed Covalent Binding](/storage/images/resized/s6JtNKPXrzklKtbQZ4HrtzuJT0P7eHPX9U8x0nss_small_thumb.webp)

![Synthesis of multi ring-fused imidazo [1,2-a]isoquinoline-based fluorescent scaffold as anti-Herpetic agent](/storage/images/resized/ruydfaB80LDjlkYqsfOeUAZohOIODyq7bQzis5O7_small_thumb.webp)








