

Keywords
Abstract
Synthesis of new effective antitumour drugs is a topical issue in modern pharmacology. Cyclopalladated compounds are a very promising area of research. The review provides the first systematization of literature data on the results of studies of synthetic transformations of binuclear palladium cycles of various classes, occurring via the opening of the m-bridging fragment under the action of additional ligands (bridge-splitting reactions) to give new heteroleptic palladium complexes. Such atomeconomical transformations proceed under mild conditions and in high yields. The most significant results of the cytotoxic activity of heteroleptic palladium cycles are presented and leading drugs are identified. The presented information makes it possible to determine a universal strategy for the targeted formation of a ligand environment in palladium complexes, which are potential antitumour drugs.
The bibliography includes 80 references.
1. Introduction
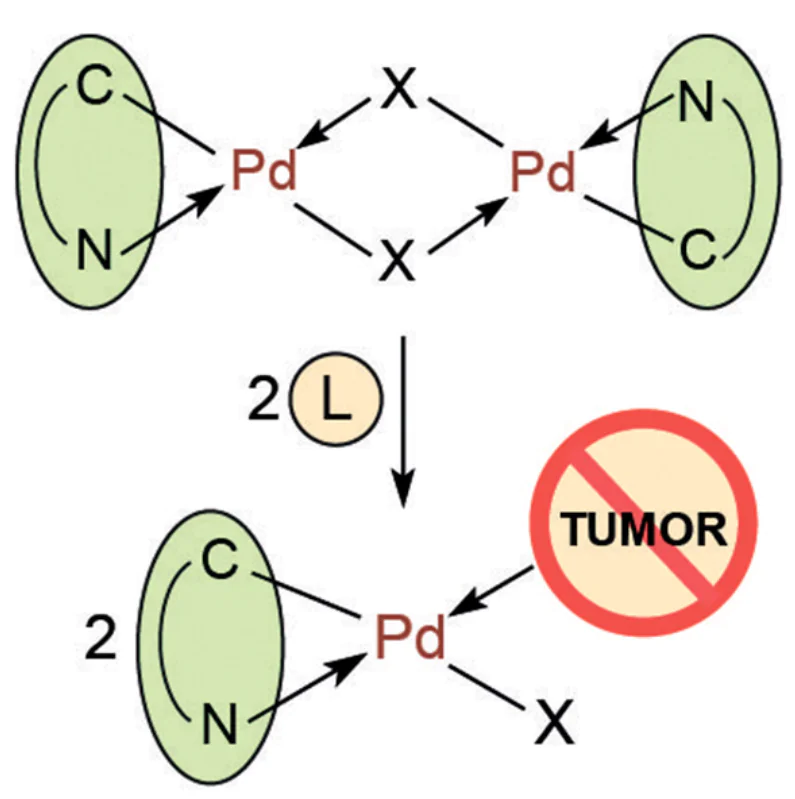
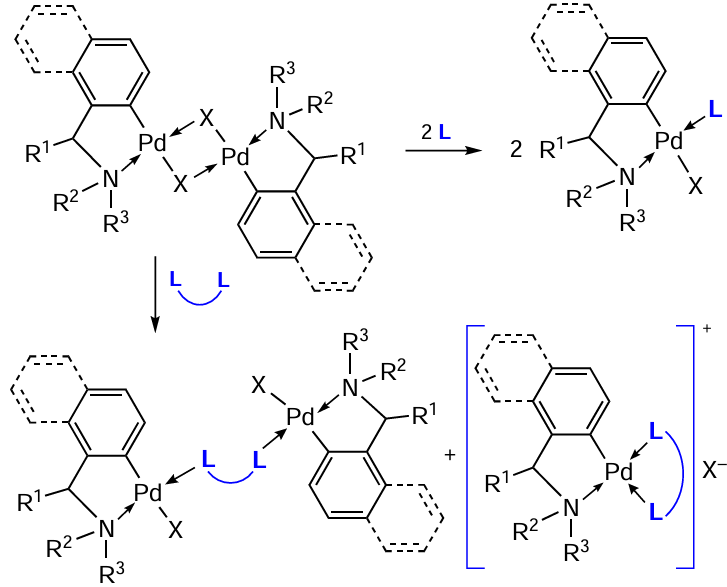
Binuclear palladacycles (PC) are versatile matrices for the synthesis of heteroleptic (mixed-ligand) palladium complexes. These complexes react readily with additional ligands (both mono and bidentate) due to the opening of m-bridging bonds (most ofter, halide bonds) to give the corresponding mononuclear complexes (Scheme 1 and Scheme 2). In this case, the choice of ligand (L) is virtually unlimited; it is possible to obtain neutral and ionic complexes depending on the structure of L. These can be well-proven pharmacophores, biologically active compounds, which allows obtaining hybrid structures.
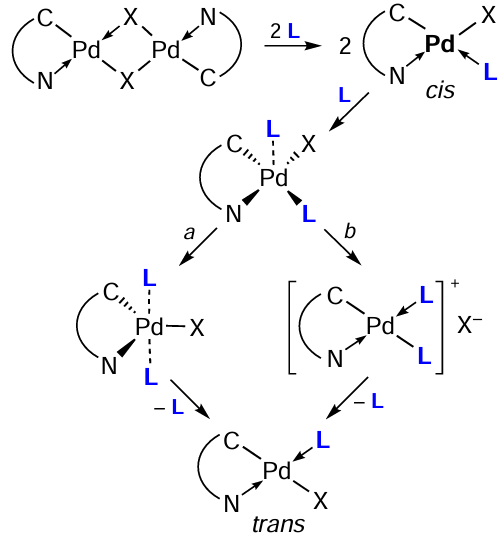
Transformations of this type have been studied in considerable detail, and publications from the seventies can be noted as pioneering works.[1][2] Theoretically, the reaction of a binuclear PC with an additional cleaving ligand can afford isomeric compounds (Scheme 1), those with the trans-position (trans-isomer) and with the cis-position (cis-isomer) of donor heteroatoms. With rare exceptions, palladium complexes were obtained as trans-isomers. A fairly logical mechanistic discussion and experimental evidence for this fact were given by Black et al.[3] The authors believe that in these reactions, a significant kinetic trans-effect of the σ-carbon ‘ligand’ determines, in the first step, the cleavage of the Pd – X (X = Hal) bond trans to the C-ligand. Therefore, the initial (kinetic) product of the bridge cleavage must be the cis-isomer. The subsequent rearrangement of the kinetic product can occur either via a five-coordinate transition state with concomitant ligand reorganization (pathway a) or the formal formation of an intermediate square-planar ion pair (pathway b). The subsequent conversion of the ionic intermediate to the neutral complex will be controlled by the significant kinetic trans effect of the σ-carbon ligand, leading to selective substitution of the C-trans ligand by the halide. In all cases, only the thermodynamically favourable trans isomers were formed.[3] Further studies confirm this pattern.
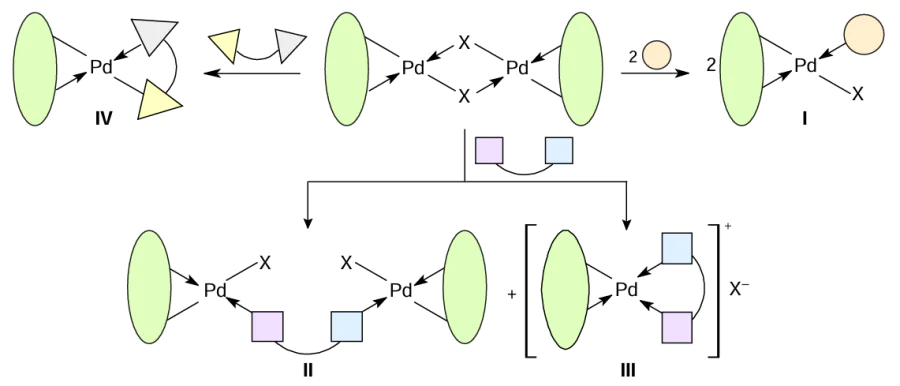
The main types of heteroleptic palladium complexes that can be obtained by bridge-splitting reactions of the parent binuclear PCs are shown in Scheme 2. When reacting with a monodentate ligand (pyridine and its derivatives, triarylphosphines, carbenes, amines), neutral complexes of type I are formed. Bidentate neutral ligands (biphosphines) may give both neutral (type II) and ionic (type III) heteroleptic PCs. Complexes of type III can also be obtained using 2,2'-bipyridyl (bipy), 1,10-phenanthroline (phen) and their derivatives as coligands. Neutral bis-chelate PCs of type IV are formed when the initial PCs react with anionic bidentate ligands (amino acids, β-diketones). For each type, this review considers specific examples of transformations occurring under mild conditions (organic solvent, room temperature (rt)) in high yields (70 – 90%). It should be especially noted that such bridge-splitting reactions are atom-economical, yielding a single product, which includes all the atoms of the reagents.
The synthesis of heteroleptic metal complexes is a very complex and relevant task of coordination chemistry. It is the coordination environment that determines both the biological and catalytic activities of metal complexes. Taking into account such factors as wide variability, stability of palladacycles, ease of opening of bridging bonds, the considered transformation of binuclear PCs (see Scheme 2) is an excellent effective strategy for obtaining new heteroleptic PCs.
The aim of almost all the works discussed herein was to find new effective antitumour drugs. An important stimulus for this was the success of using metallodrugs (e.g., cisplatin and its analogues) in clinical cancer therapy. Cyclopalladated compounds turned out to be very promising. The most significant results in their study were achieved by the research groups of A.C.F.Caires (Brazil), J.Albert (Spain) and K.Karami (Iran). The correctness of the comparison of the activity of the discussed PCs is ensured by the fact that the review analyzes the results within the framework of one study (protocol) or one research group following a certain methodology. In this case, the data for the reference drug (usually cisplatin) were taken into account to evaluate the potency of the compounds. More detailed information on the study of the plausible mechanism of cytotoxic action of the discussed PCs is available in many cited publications. This topic is quite controversial and is of independent interest.
2. Cyclopalladated amines
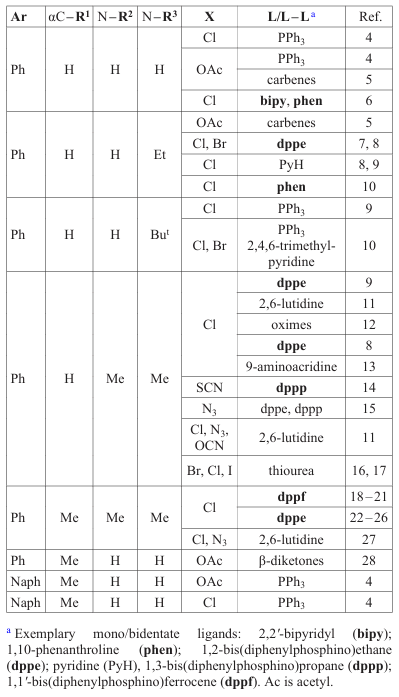
The most popular starting binuclear PCs are substituted benzylamines and their naphthyl analogues (Scheme 3, Table 1).[4-28] Coligands are represented by a variety of structural types. The most highly effective cytotoxic compounds are found among biphosphine derivatives.
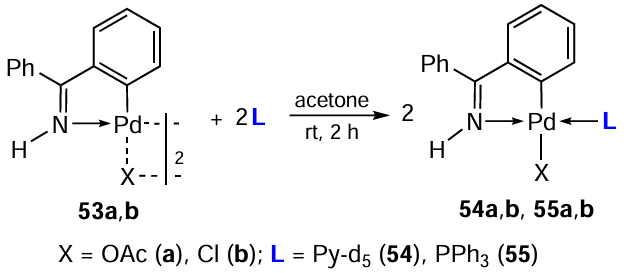
The study of the cytotoxic activity of biphosphine complexes was pioneered by Caires et al.[15]in 1999. In a follow-up study,[22]using binuclear PCs 1 – 3 as starting matrices, the authors synthesized heteroleptic palladium complexes 4 – 9 bearing the biphosphine coligand, 1,2-bis(diphenylphosphino)ethane (dppe) (Scheme 4, Scheme 5, Scheme 6). The structure of the resulting complexes, formed by the reaction of PC with dppe, was governed by the stoichiometry and nature of the solvents used. As a result, neutral bi- and mononuclear (4, 6, 8, 9) or ionic mononuclear (5, 7) complexes were obtained.
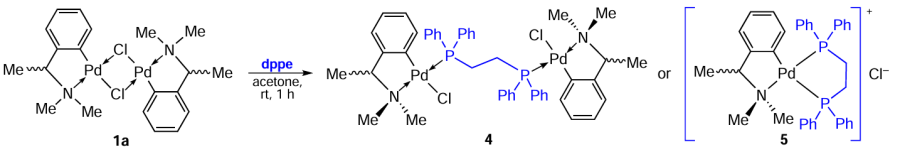
Ionic complexes 5 and 7 with chelating biphosphine ligands were obtained at a molar ratio of ligand : PC (1 or 2) = 2 : 1. In the case of the starting PC 3, a neutral mononuclear complex 9 is formed. Cyclopalladated complexes 4, 6 and 8 with a bridging biphosphine ligand were obtained at an equimolar ratio of the ligand to the starting PC (1 – 3 in dichloromethane). Complex 4 is formed as two enantiomers (S)-4 and (R)-4. In all above cases, the yields were at least 86%.
These drugs were tested in vitro for antitumour activity against a murine syngeneic melanoma model B16F10. The most potent were PC 4 and 5, which contain a benzyl aminate ring, and no difference in the activity of the enantiomeric forms (S)-4 and (R)-4 was observed. These compounds caused 100% death of tumour cells at concentrations below 1.25 μM. When using compounds 4, 5 as chemotherapeutic drugs in vivo, only the (S)-4 complex exhibited antitumour activity, significantly delaying the tumour evolution and death of treated mice. The authors hypothesize that both inactive complexes, 5 and (R)-4, do not have free access to tumour cells in vivo. Interestingly, PCs 7 and 9, which showed no activity in vitro, were active in vivo. Both preparations significantly delayed tumour development and death of treated animals, similar to compound (S)-4. All tested palladium complexes have low general toxicity.
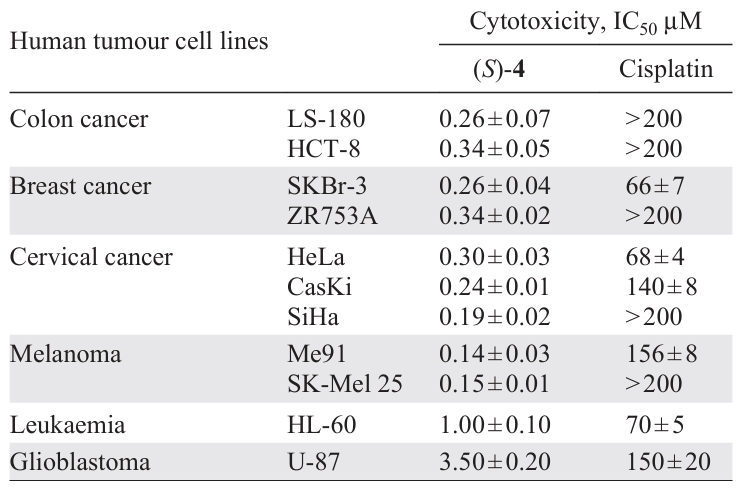
Extended studies [26]of the cytotoxic activity of compound (S)-4 have shown unique results — this PC demonstrates cytotoxicity significantly exceeding that of cisplatin and a broad spectrum of action (Table 2). In addition, the antiparasitic [29]and antifungal [30]activities of (S)-4 were studied in vitro and in vivo. The results obtained indicate its high multidisciplinary effectiveness. For the leading PC (S)-4, more in-depth studies of the mechanism of its cytotoxic activity were also carried out.[23][24] [26] [31][32]
The next, very promising group of PCs consists of chiral complexes 10 (Ref. 18) and 11 [19-21] with 1,10-bis(diphenylphosphine)ferrocene (dppf) ligand, based of N,N-dimethyl-1-phenethylamine. Compound 10 was obtained as two individual enantiomers (S)-10 and (R)-10. An equimolar mixture of reactants in dichloromethane was allowed to stand for 1 h in an inert atmosphere at room temperature. The yields were 97 and 95%. The use of a twofold excess of dppf leads to the ionic complex 11 with a chelating biphosphine ligand.[19]It is formed as the individual enantiomer (S)-11.
Caires et al.[18-21] investigated the mechanism of high cytotoxic activity (in vitro, in vivo) of complexes 10 and 11 and identified them as lead compounds for implementation in clinical practice.
The (R)-10 and (S)-10 complexes exhibited high cytotoxic activity in vitro and in vivo against Walker-256 carcinoma.[18]Histopathological analyses of the kidneys, spleen and liver of mice after administration of a high dose of the (S)-10 complex (100 mg kg–1) showed that the palladium complex did not cause tissue damage in these organs even when administered at a dose 50 times higher than the effective dose in rats bearing Walker-256 tumour (2 mg kg–1). The authors concluded that the antitumour activity of (R)-10 and (S)-10 PCs may be related to the ability of these complexes to inhibit the activity of cysteine proteases such as cathepsin B. Both complexes inhibited the enzyme activity in a similar manner (IC50 » 4 μM). In-depth kinetic studies were performed for the cyclopalladated (S)-10 complex. Interestingly, replacement of the dppf ligand with other biphosphines such as dppe resulted in the formation of PCs that were unable to inhibit cathepsin B activity.
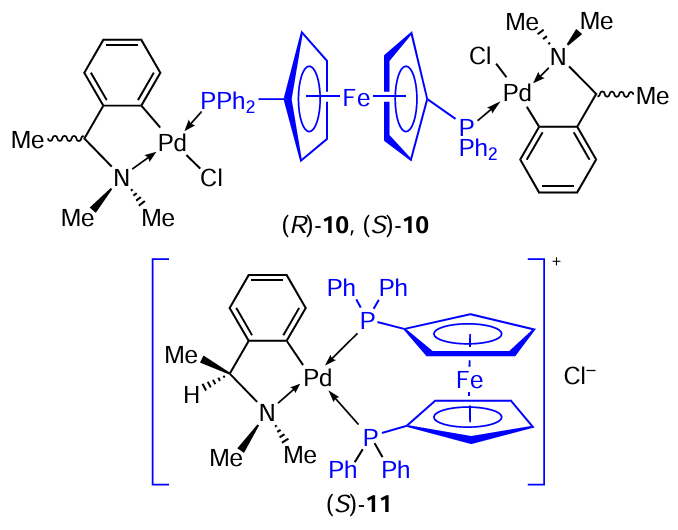
The biological activity of PC (S)-11 was explored.[19-21] In particular, the mechanism of K562 leukaemia cell death caused by the (S)-11 complex was studied.[19]The IC50 values were less than 5.0 μM. The complex was found to induce apoptosis in K562 cells by causing DNA fragmentation, as analyzed by electrophoresis. Toxicology studies showed that the (S)-11 complex induced no lesions for liver and kidney in mice fourteen days after administration of the drug (100 mg kg–1). These findings suggest a novel lysosomal pathway for (S)-11-induced apoptosis, in which lysosomes are the primary target and cathepsin B acts as a death mediator.
Further studies showed that the (S)-11 complex also induces apoptosis in leukaemia cells (HL60 and Jurkat) and is nontoxic to normal human lymphocytes.[20]The IC50 values obtained for both tumour cell lines were below 8.0 μM. Successful preclinical studies of (S)-11 have been carried out in an in vivo model in mice infected with B16F10-Nex2 melanoma, which were treated with (S)-11 intraperitoneally (8 mg kg–1 day–1) for ten consecutive days.[21]It should be noted that cyclopalladated compounds (S)-4, (S)-5, and (S)-10 have been patented by Caires et al.[25]as antitumour agents.
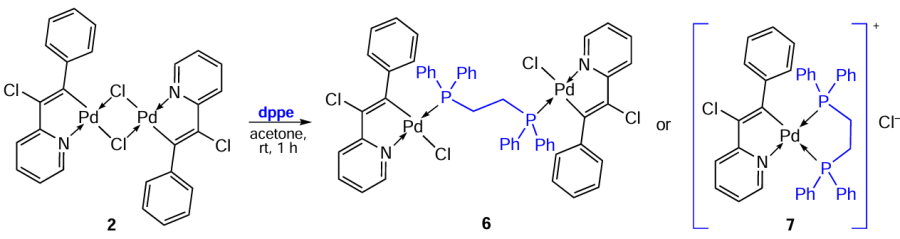
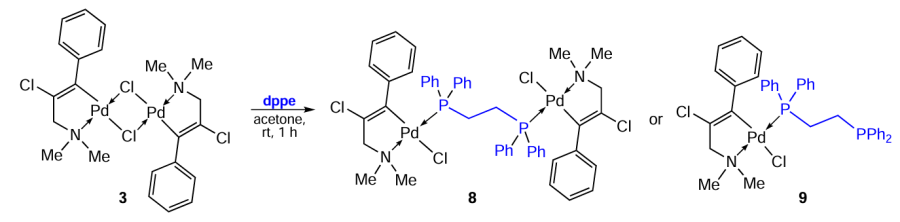
Karami et al.[7-9] studied palladium complexes of various types, including biphosphine ones (Scheme 7). Binuclear PCs 12a – c and their corresponding derivatives 13a, 14b, and 16c were synthesized on the basis of N-substituted benzylamines.[9] The methods for obtaining these compounds are practically identical and feature the simple conditions. Generally, the effective bridge-splitting reaction of the starting binuclear PC with the corresponding ligand occurs at room temperature in chloroform or dichloromethane. The target palladium complexes were isolated and purified by crystallization to give products with a preparative yield of 62% to 87%.
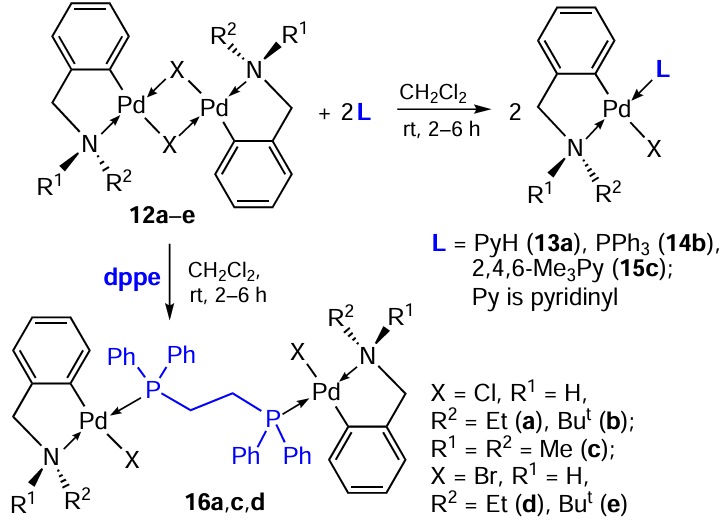
A comparative study of the cytotoxicity of the resulting compounds against four human tumour cell lines (HeLa, HT-29, K562 and breast cancer (MDA-MB-231)) (Table 3) showed that palladacycles 13a, 14b and 16c outperform (IC50 = 1.4 – 7.7 μM) cisplatin (IC50 = 4.8 – 51.3 μM). It should be noted that free ligands, benzylamines and dppe, are inactive. The authors suggest that the increased lipophilicity of 16c, associated with the presence of bulky phenyl groups of the biphosphine bridge, may facilitate the transport of this drug across cell membranes. In addition, the presence of the bridging dppe results in greater flexibility of the complex structure and ensures its more effective interaction with DNA. An additional study of complex 16c revealed high cytotoxicity of this compound against the cisplatin-resistant sub-line of K562R cells (IC50 = 0.25 ± 0.05 μM).[8] It has been experimentally confirmed that these compounds exert their cytotoxic effect via apoptosis.
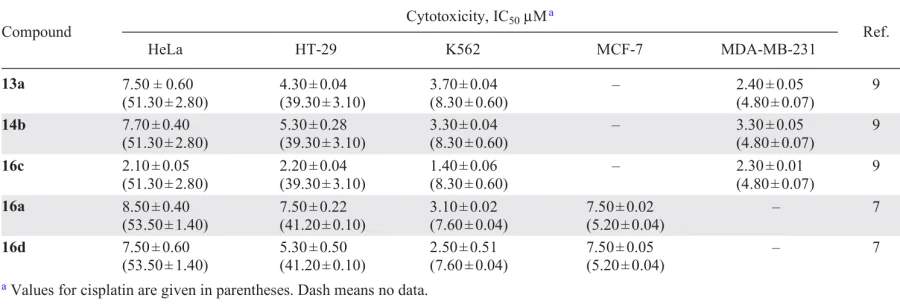
In order to assess the mechanism of cytotoxic activity, the DNA-binding properties of complexes 16a and 16d containing chloride or bromide ligands, respectively, were studied by electron absorption and fluorescence spectroscopy.[7] The authors concluded that both complexes can interact with DNA via intercalation and follow the order of binding affinity 16d > 16a, which correlates with their cytotoxic activity in vitro.
Monodentate ligands react with binuclear PC to give neutral mononuclear complexes 13 – 15 (see Scheme 7). The use of bidentate chelating ligands such as bipy or phen affords ionic complexes 17 – 19 (Scheme 8).[6][10] A special feature of the synthesis of these ionic complexes is that the parent binuclear PC is first treated with silver salts Ag(BF4) or Ag(CF3SO3) to remove the chloride ion, after which the appropriate ligand is added. The yields of complexes 17 – 19 after crystallization in hexane were 63 – 65%. Compounds 17 and 18 were tested for antitumour activity in vitro against two human cancer cell lines, Jurkat and MCF7 (breast adenocarcinoma), and normal peripheral blood mononuclear cells (PBMC).[6] Their activity (IC50 from 37 to 53 μM) was lower than that of cisplatin (IC50 = 8.9 and 12 μM). Both complexes exhibited low activity (IC50 > 100 μM) against PBMC cells (selectivity index (SI) is at least 1.5).
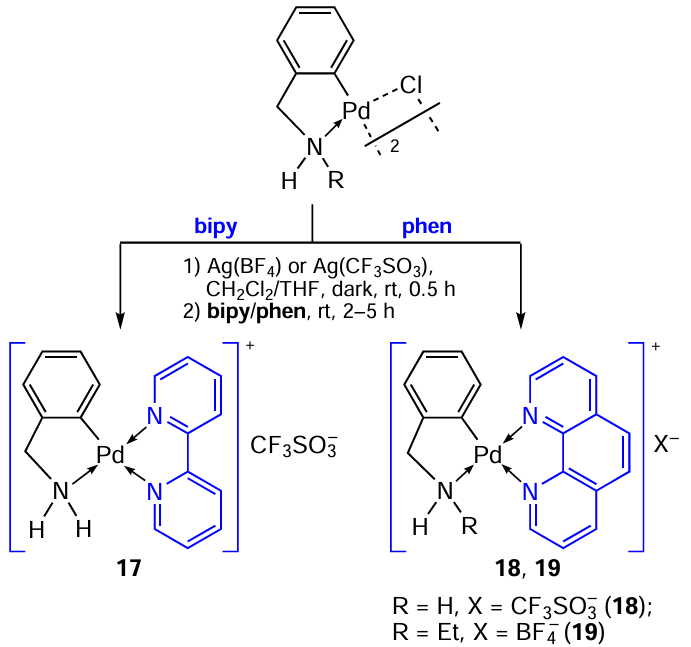
In continuation of their studies in the field of development and preparation of cyclopalladated compounds with antiproliferative activity, Karami et al.[5] synthesized for the first time palladium complexes 20a,b with two different cyclometallated aminobenzyl ligands, as well as N-heterocyclic carbene (NHC) as a coligand. NHC ligands are σ-donors that bind strongly to transition metals thus providing stability of the resulting complexes in the physiological environment. The antitumour properties of 20a,b were studied in vitro. Complexes 20a,b showed high activity and selectivity against HeLa and MCF-7 cells, while PC 20a with a free amino group showed superior antiproliferative activity. For example, the IC50 values as determined for complexes 20a and 20b and cisplatin with respect to MCF-7 are 0.01, 0.03 and 24.89 μM, respectively, and for HeLa, IC50 = 3.73, 5.98 and 26.19 μM.
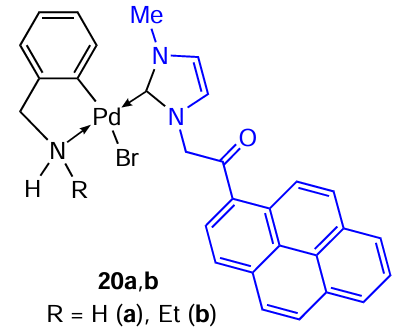
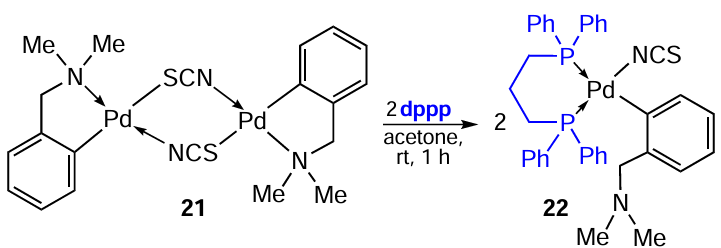
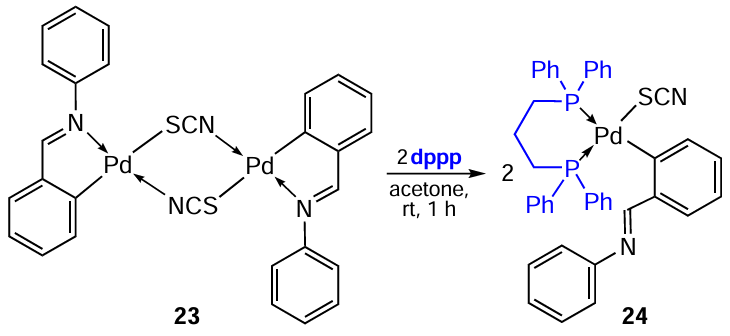
The C,N-palladacycle is generally very stable and is retained upon opening of the m-bridging bonds under the action of coligands. However, there are exceptions. In particular, in reaction with the chelating ligand 1,3-bis(diphenylphosphino)propane (dppp), thiocyanate-bridged PCs 21 and 23 are converted into mononuclear complexes 22 and 24, in which the Pd – C bond is retained and the Pd – N bond is opened (Scheme 9 and Scheme 10).[14][33]The reaction proceeds in acetone at room temperature in virtually quantitative yields. Benzyl aminate complexes 21 and 22 demonstrated satisfactory cytotoxic potential in vitro and in vivo, increasing the lifespan of mice bearing Ehrlich ascites tumour. At the same time, complex 21 demonstrated lower activity (IC50 = 47.86 ± 4.32 μM) compared to mononuclear derivative 22 (IC50 = 5.29 ± 3.89 μM) and cisplatin (IC50 = 33.77 ± 2.29 μM).
We would like to separately highlight a series of studies in which 2,6-dimethylpyridine (2,6-lutidine) is used as an coligand that destroys the bridge in binuclear PCs.[11] [27] [34][35] The choice of 2,6-lutidine was due to the fact that the study of the structure – activity relationship for a number of cyclopalladated compounds [Pd(C,N)(X)(L)] (C,N is ortho-metallated 1-phenylpyrazoles, 2-phenylpyridine, 1-(2'-pyridyl)indole); L is monodentate pyridines and phosphines; X = Cl, Br, I) revealed that 2,6-lutidine-containing complexes exhibit high cytotoxic activity.[3]
Binuclear PCs 1a,b and 12с,f – h react with 2,6-lutidine to give mononuclear heteroleptic complexes 25a – c,f – h in yields from 70 to 90% (Scheme 11).[11] [27]In this case, the outcome of the cleavage of the parent binuclear PC does not depend on the nature of the bridging group (X = Cl–, I–, N3–, NCO–).
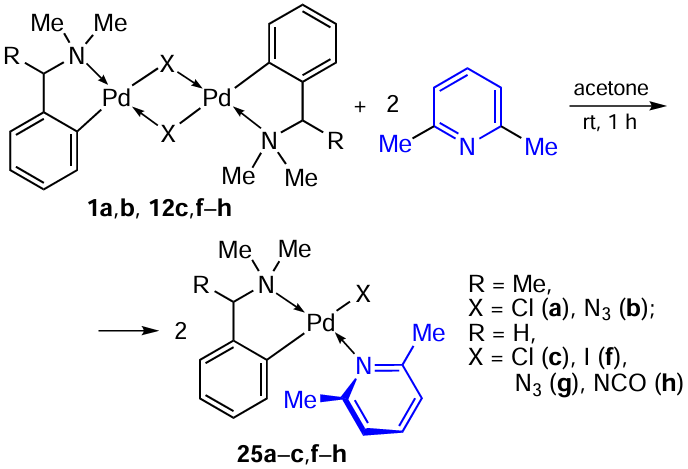
The inhibitory activity of complexes 25c,f – h and cisplatin in vitro was evaluated against a panel of murine {breast carcinoma (4T1) and melanoma (B16F10-Nex2)} and human melanoma (A2058, SK-MEL-110, and SK-MEL-5) tumour cell lines.[11] According to the obtained data, compounds 25c,f – h exhibited comparable levels of cytotoxic activity against all above tumour cell lines, indicating that the nature of the anionic ligand X did not play a significant role in the observed activity. The activity of 25c,f – h significantly exceeded that of cisplatin. For example, the IC50 values for compounds 25c,f – h in the A2058 cell line were 1.82 – 3.59 μM, while for cisplatin IC50 > 84 μM. The authors attribute the absence of dependence of the observed activity on the chemical nature of group X to the fact that hydrolytic ligand exchange occurs in solutions, ultimately leading to the formation of identical species in which the place of X is occupied by a solvent molecule. Compounds 25c,f – h showed activity against non-neoplastic cells (human lung fibroblasts, MRC-5) with IC50 values of 5 – 14 μM. The results of the cytotoxic activity of compounds 25c,f – h are further discussed below in comparison with their close PC analogues containing cyclopalladated oxime and 2,6-lutidine ligands.[35]
In a follow-up study,[27]similar PC 25a,b based on N,N-dimethyl-1-phenethylamine were synthesized and studied (see Scheme 11). The inhibitory effect of the resulting mononuclear derivatives and cisplatin was evaluated in vitro on human glioblastoma (U251 and T98G) and melanoma (HT144 and LB373) cell lines. Compounds 25a,b containing an α-methyl group at the benzyl position exhibited activity (IC50 = 0.3 – 6.7 μM) comparable to that of cisplatin (IC50 = 0.8 – 9.1 μM).
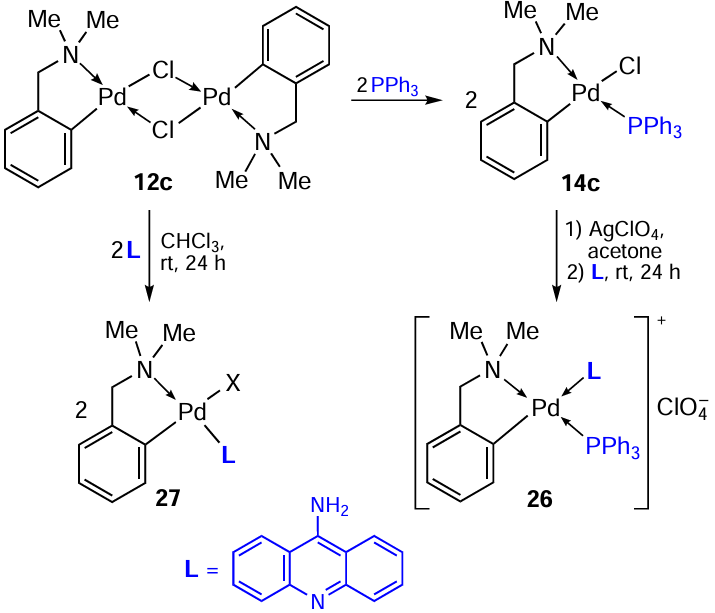
Further transformation of neutral PC 14c into ionic complex 26 has been proposed (Scheme 12).[13]The chloride ion is removed from the coordination sphere by treating compound 14c with Ag(ClO4) in acetone, AgCl is removed by filtration, and then a new ligand, 9-aminoacridine, is added, yielding ionic complex 26 in 65% yield. According to the classic route, neutral mononuclear complex 27 was also obtained in 90% yield. In addition, the cytotoxic activity of the resulting compounds against HL-60 tumour cells was studied. Both complexes 26 and 27 containing the aminoacridine coligand were significantly more active than cisplatin (IC50 = 15.6 μM). At the same time, ionic complex 26 was an order of magnitude more effective (IC50 = 0.52 μM) than neutral 27 (IC50 = 5.92 μM).
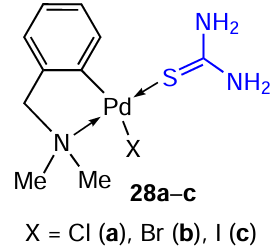
Thiourea has been studied as a coligand, which induces the cleavage of the halide bridges in benzyl aminate PC 12c and its analogues occurs.[16][17] The exchange of halide ligands in 12c occurs readily upon treatment of a solution of PC 12c in acetone with aqueous solutions of sodium bromide or iodide. The bridge-splitting reaction of the corresponding binuclear PCs with thiourea readily gives mononuclear chlorine-, bromine-, and iodine-containing palladium complexes 28a – c in yield of up to 80%. Evaluation of the cytotoxicity of these compounds showed that the iodine derivative 28c is the most potent (IC50 = 14.4 μM) against mouse mammary gland adenocarcinoma cells (LM3), being superior to cisplatin (IC50 = 30.3 μM) and the parent binuclear complex (IC50 = 36.3 μM). According to the results of the Salmonella typhimurium microsome test (Ames test), PC 28a – c do not cause genetic damage leading to gene mutations.
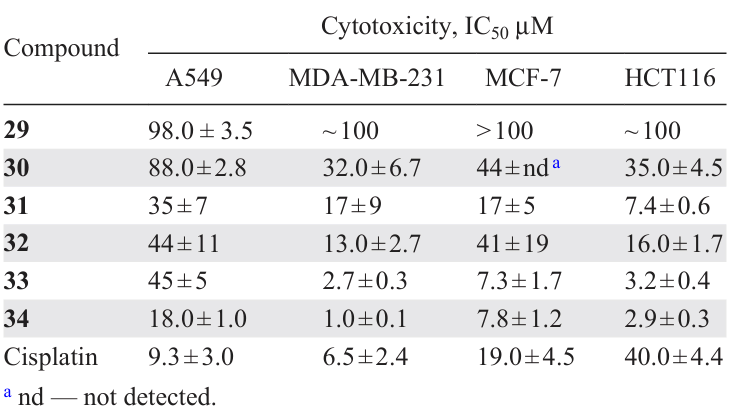
Albert et al.[4] carried out a systematic study of the methods of synthesis and properties of cyclopalladated primary amines. In particular, interesting data concerns the comparative evaluation of the cytotoxic activity of binuclear C,N – PCs 29 – 31 and their mononuclear triphenylphosphine derivatives 32 – 34 (Scheme 13, Table 4). The first group of compounds 29 – 31 has lower activity, and the transformation of binuclear C,N – PCs into mononuclear derivatives 32 – 34 significantly increases their cytotoxicity, especially against MCF-7 and MDA-MB-231 tumour cell lines. The structure of the cyclopalladated ligand also affects the said properties. Compounds 31 and 34, based on 2-aminobiphenyl, turned out to be slightly more active.
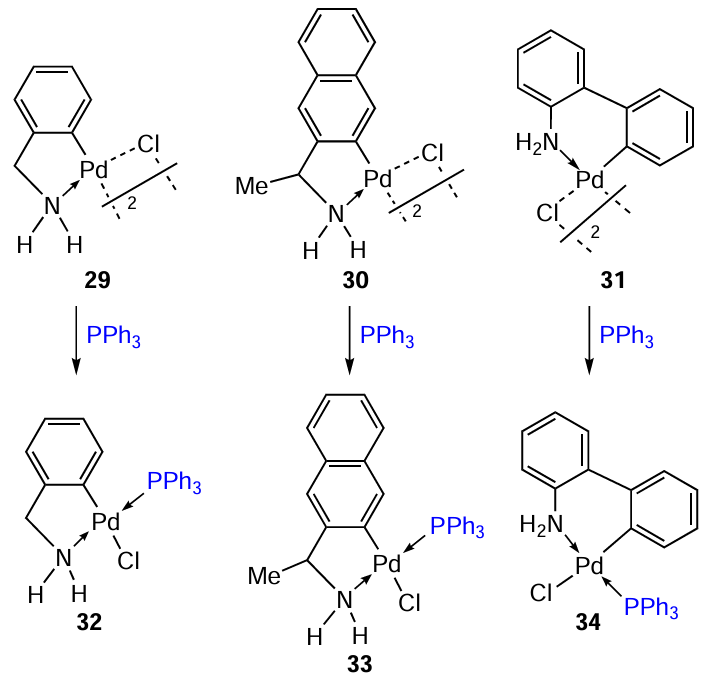
Cyclopalladated derivatives of 2-aminobiphenyl have been studied in a number of studies.[3][36-38] Bidentate ligands dppf and 1,3-bis(4-pyridine)propane (bpp) were used to transform cyclopalladated 2-aminobiphenyl 31.[36] Bridge-splitting reactions afforded binuclear palladium complexes 35 and 36 in yields of 68 and 76%, respectively (Scheme 14). Both complexes exhibited cytotoxic activity against human cancer cells Jurkat and SK-OV-3 (ovarian cancer) and were more active than cisplatin. Higher activity was observed when treating cells with complex 35 (IC50 = 5.2 μM and 2.3 μM, respectively). For 36, IC50 = 6.7 μM and 5.7 μM, respectively. Compounds 35 and 36 showed moderate toxicity against normal PBMC cells (IC50 = 39 and 31 μM, respectively).
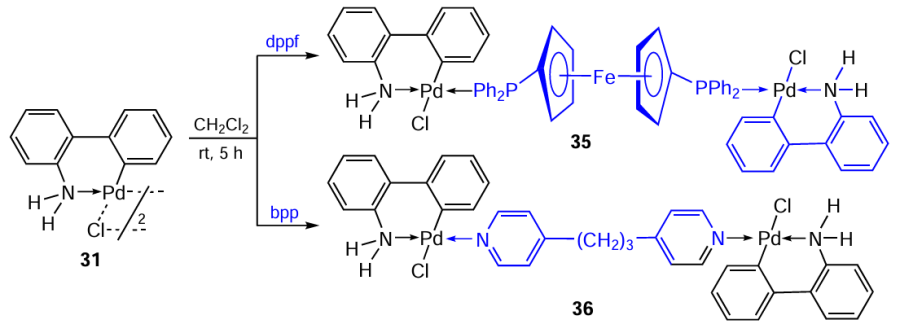
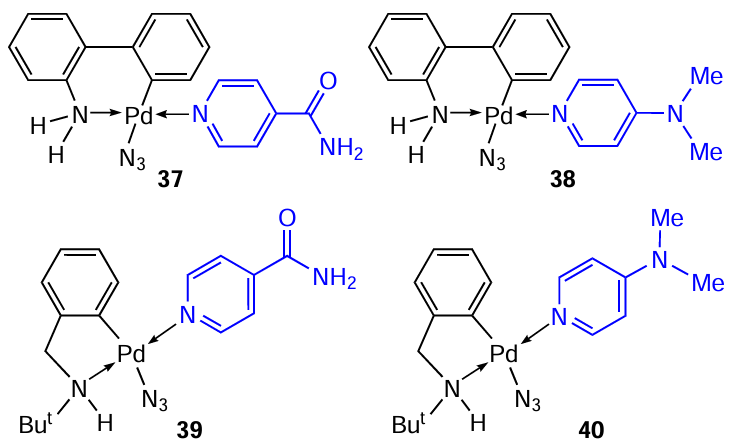
The follow-up study [37] describes the synthesis of new mononuclear derivatives of PC, compounds 37 – 40, containing 4-acetamido- and 4-dimethylaminopyridine as coligands. A feature of these structures is the presence of an azide ligand, which makes no substantive changes to the synthetic protocols for the corresponding complexes compared to their chloride analogues. A study of the cytotoxic activity of PC 37 – 40 against MCF-7 and HeLa tumour cells showed that their action does not exceed the activity of cisplatin. The best result was noted for complex 37 against HeLa cells (IC50 = 19.32 μM, IC50 = 26.19 μM for cisplatin).
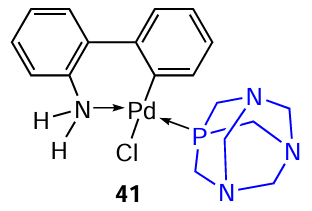
Based on biphenyl PC 31, complex 41 was obtained, containing 1,3,5-triaza-7-phosphaadamantane (PTA) as a coligand.[38]This is a rare example of a chloride bridge cleavage reaction that is carried out on heating (50°C). The isolated yield of the target PC 41 was 54%. Evaluation of the cytotoxic activity of compounds 31 and 41 showed that these complexes are capable of reducing the viability of human cancer cells (MCF-7 and Jurkat lines) in a dose-dependent manner, with a more pronounced reduction observed in the viability of normal fibroblast ASF-4 cells. The IC50 values of complexes 31 and 41 on Jurkat cells were 76 and 51 μM, respectively, indicating higher toxicity of the mononuclear derivative 41. Similarly, complex 41 also demonstrated greater toxicity on MCF-7 cells, with IC50 values of 49 and 35 μM for complexes 31 and 41, respectively. Compared to normal cells, both complexes showed selectivity indices SI < 2. The order of anticancer activity in vitro was found to correspond to the degree of binding of these complexes to DNA via intercalation.
3. Cyclopalladated oximes
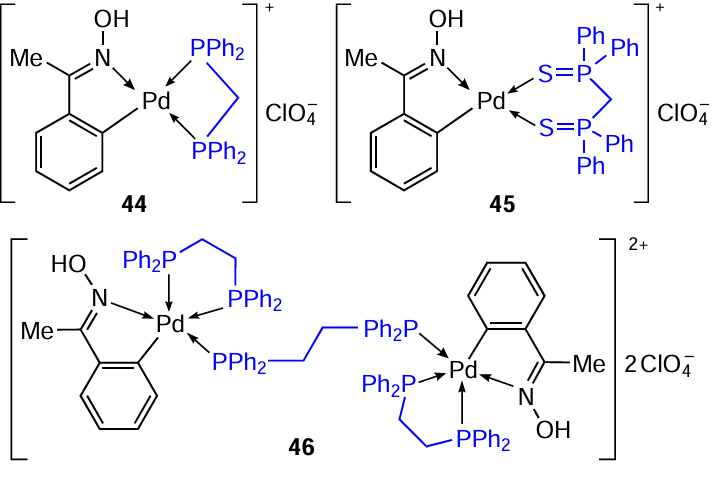
Oxime-derived palladacycles represent a large group of thermally stable palladium complexes that are insensitive to oxygen and atmospheric moisture. Direct ortho-palladation of benzophenone oxime was used to obtain binuclear PC 42, which was then used as a template for the synthesis of new heteroleptic palladium complexes with chelate phosphinylide 43a – d (Scheme 15) [39] and biphosphine 44 – 46 (Ref. 40) ligands. In order to obtain mononuclear complexes 43a–d, a mixture of PC 42 was reacted with NaClO4 and ylide (C,P – L) in CH2Cl2 at room temperature under a nitrogen atmosphere. For the synthesis of ionic complexes 44 – 46 by a similar method, bis(diphenylphosphine)methane (dppm), bis(diphenylthiophosphinoyl)methane (dppmS2), and dppe were used as bidentate cleaving ligands.[40]The structure of binuclear complex 46 was confirmed by X-ray diffraction analysis.
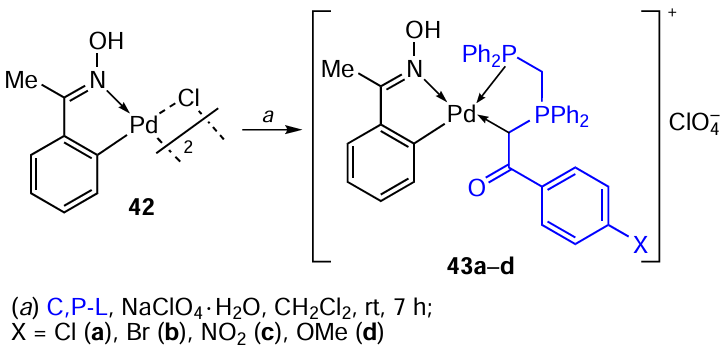
Studies of the antitumour properties of complexes 44 – 46 showed that all three complexes exhibit significant cytotoxic activity against A549, HT29, and HeLa cancer cells.[40]Complex 46 was the most potent against A549 (IC50 = 10 μM).
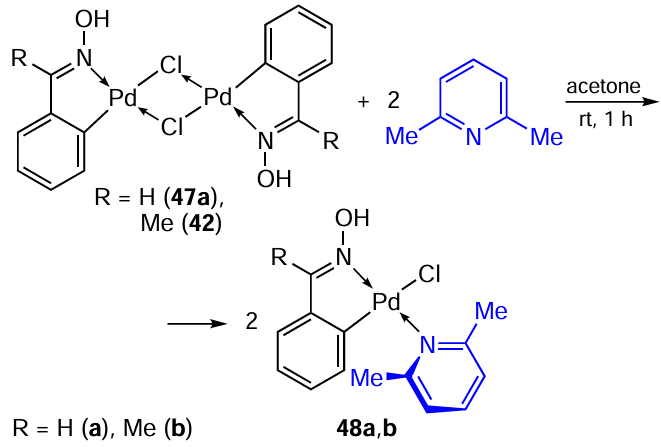
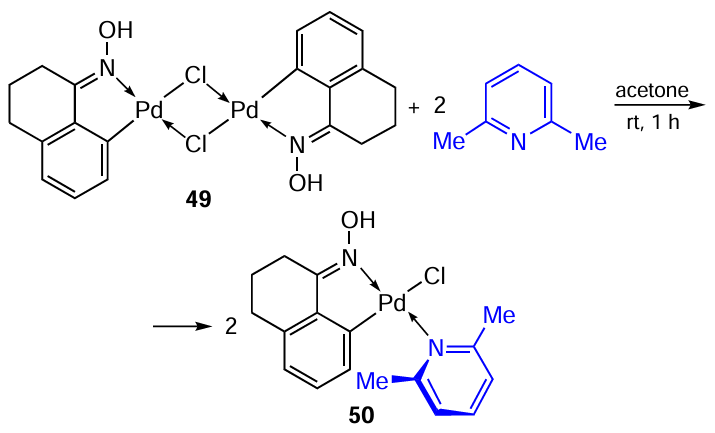
Mononuclear PCs 25a – c,f – h containing 2,6-lutidine as an additional ligand were discussed above.[11] [27]In 2025, Netto et al.[35]synthesized similar oxime-derived PCs 47 – 50 (Scheme 16, Scheme 17). Once again, we would like to emphasize the ease of transformations of this type, affording the target mononuclear complexes in high yields (61 – 94%). The cytotoxicity of compounds 48a,b, and 50 towards A549, MCF-7, and MDA-MB-231 tumour cells was revealed. For the MCF-7 cell line, a progressive increase in the cytotoxicity of the complex was observed depending on the type of the oxime ligand in the series 50 < 48b < 48a. The best result was obtained for 50 (IC50 = 14.78 μM), which is comparable to the effect of cisplatin (IC50 = 13.9 μM). In general, oxime-derived PCs 48a,b and 50 were less active against tumour cells than benzylamine-based PCs 25a – c,f – h,[11] [27]but less toxic against normal MRC-5 cells.
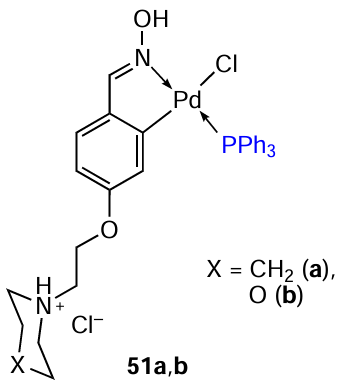
Ligand exchange was used to obtain PCs 51a,b, which contained substituted benzaldehyde oximes and PPh3 as ligands.[12]The cytotoxicity of compounds 51a,b was studied against cisplatin-resistant osteosarcoma SaOS-2 and U2OS cells and the non-tumour MC3T3-E1 cell line. It was found that the IC50 values for the two complexes in the three cell lines studied were close (IC50 = 24.55 – 43.55 μM). Although the IC50 values of complexes 51a,b to normal MC3T3-1 cells and tumour cells were similar, the authors note that palladium complexes do not show mutagenic properties. The low toxicity of complexes 51a,b in vivo to C. elegans confirms these findings.
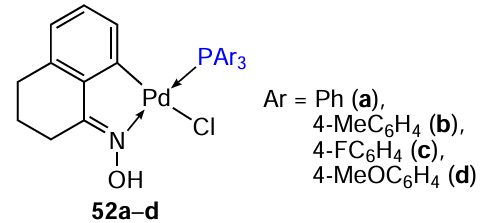
Mononuclear triarylphosphine derivatives 52a – d were obtained on the basis of the tetralone oxime-derived binuclear PC 49.[41] All cyclopalladated compounds were more active (IC50 = 19.22 – 26.33 μM) than cisplatin (IC50 > 50 μM) against MCF-7 tumour cells. The comparable activity level of 52a – d indicates that the substituents in the phosphine ligand do not significantly affect the activity of these compounds in vitro. Cytotoxicity of compounds 52a – c against normal MRC-5 cells is comparable (IC50 = 22.35 – 28.92 μM) to that of cisplatin (IC5 = 19.86 μM), while compound 52d is more active (IC50 = 8.12 μM).
4. Cyclopalladated imines
Albert et al.[42-45] reported the synthesis and studied the biological activity of binuclear cyclopalladated benzophenone imines 53a,b and their mononuclear derivatives 54a,b and 55a,b (Scheme 18, Table 5).[42-45] A review paper [45]summarized the results of the studies. The starting PCs 53a,b react quantitatively with deuteropyridine (Py-d5) in CDCl3 , allowing analysis of the corresponding reaction mixtures by NMR spectroscopy without the need to isolate complexes 54a,b. Synthesis of PPh3-containing сompounds 55a,b [42][43] was carried out in acetone (2 h, rt) and they were additionally purified by column chromatography. The yields were 43% and 79%, respectively. Palladacycles 55a,b show high antiproliferative activity against MDA-MB231 and MCF7 tumour cells with the IC50 values of 1 – 5 μM, which is lower than those of cisplatin (IC50 = 6.5 ± 2.4 μM and 19.0 ± 4.5 μM) in both cell lines. It should be noted that mononuclear phosphine derivatives 55a,b are significantly more active than the parent binuclear PCs 53a,b (IC50 = 10 – 18 μM). The authors explain the close IC50 values in 53a,b and 55a,b pairs against the MDA-MB231 and MCF7 cell lines by the fact that in the biological environment, these compounds can exist in the form of identical aqua cations formed as a result of the replacement of X ions by water molecules.
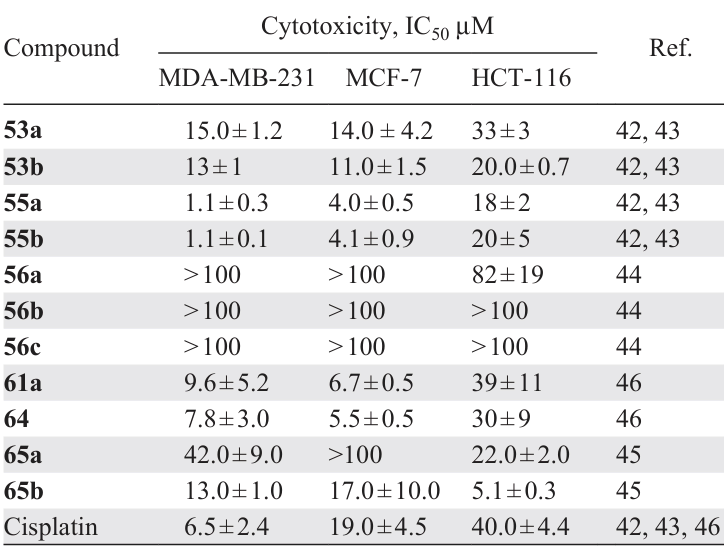
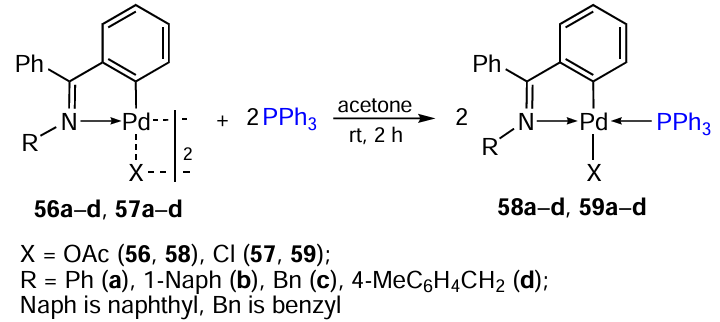
Based on N-substituted benzophenone imines, binuclear PCs with acetate (56a – d) and chloride (57a – d) bridges and the corresponding mononuclear triphenylphosphine derivatives 58a – d and 59a – d were obtained (Scheme 19).[44]The cytotoxicity of the synthesized palladium compounds against MDA-MB-231 and MCF-7 cell lines and cisplatin-resistant HCT-116 (colon adenocarcinoma) was studied. Surprisingly, almost all N-substituted derivatives proved to be inactive in all cases. The previously described unsubstituted nitrogen derivatives 53a,b and 55a,b[42][43] (see Table 5) showed a fairly high activity, being superior to cisplatin in activity against MDA-MB-231 and MCF-7. In this group, a clear tendency towards increased activity can be noted when moving from binuclear complexes (53a,b) to mononuclear triphenylphosphine derivatives (55a,b). The lowest activity (IC50 > 100 μM in all cell lines) is characteristic of naphthyl derivatives 58b, 59b. Another feature is that chlorine derivatives in all groups outperform the corresponding acetate complexes.
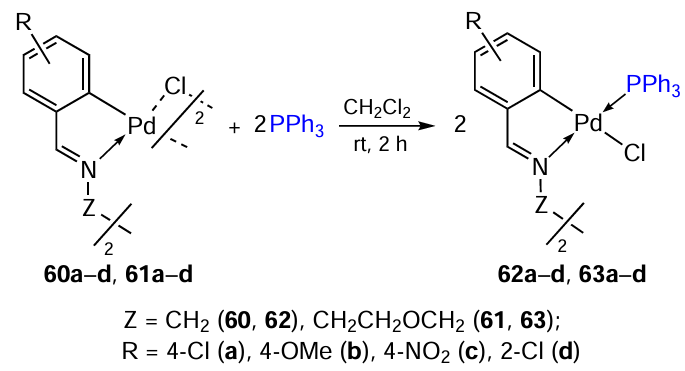
In order to obtain polynuclear palladium complexes, cyclopalladation of symmetrical diimines of salicylic aldehydes containing various bridges, viz., Z = CH2 (complexes 60a – d) or (CH2)2OCH2 (complexes 61a – d) and substituents in the aromatic ring was studied (Scheme 20).[46] These structural features make it possible to vary the conformational mobility, lipophilicity and hydrophilicity of the target molecule. The corresponding heteroleptic complexes 62a – d and 63a – d were obtained by cleaving the chloride bridges with triphenylphosphine. In addition, macrocyclic complex 64 (CHCl3, rt, 4 h, yield 91%) was obtained by reacting complex 61a with the ligand 2,2'-(ethylenedioxy)bis(ethylamine), which has high conformational flexibility.
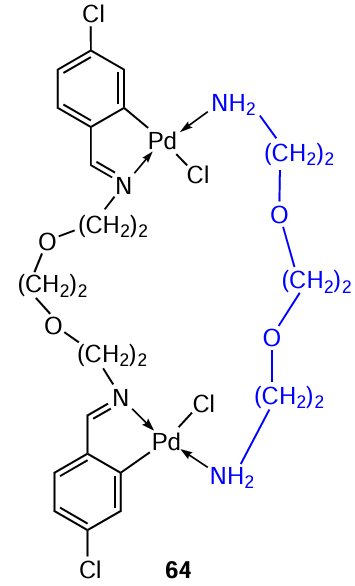
The cytotoxic activity of the resulting cyclopalladated compounds was evaluated using the HCT-116, MCF-7 and MDA-MB-231 cell lines.[46] Significant results (comparable or superior to cisplatin) were obtained for complexes 61a, 64, based on 4-chloro-substituted diimine with an ethylene glycol bridge (see Table 5). Complexes 60a, 62a, containing the NCH2CH2N unit, showed no activity (IC50 > 100 μM in all lines). The authors believe that the different cytotoxicity of the two series of described PCs is due to the different length and flexibility of the central saturated chain in the imine molecule, as well as its lipophilicity and hydrophilicity.
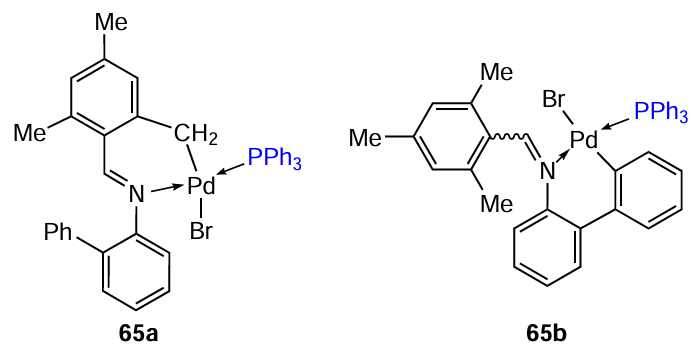
Regioselective cyclopalladation of (R)-N-([1,1'-biphenyl]-2-yl)-1-mesitylmethanimine was studied, resulting in endo- and exo-PCs (65a and 65b) containing a six-membered metallacycle.[45] Cyclopalladation of the starting imine on heating (90°C) occurs via the aliphatic methyl group (thermodynamic control), whereas at room temperature, it occurs via the aromatic ring (kinetic control). The cytotoxicity of compounds 65a,b was evaluated against MDA-MB-231, MCF-7 and HCT-116 cell lines (see Table 5). The ortho-palladated imine 65b showed significantly higher cytotoxicity compared to the endo-isomer 65a, although their overall activity did not exceed that of cisplatin. It should be noted that these complexes were nontoxic against normal BJ cells (65b: IC50 > 57 μM; 65a: IC50 > 100 μM).
Prince and co-workers[47-52] published a number of papers devoted to the study of PCs containing dppe as a coligand. The study [47] describes the synthesis of a new binuclear PC 66, which was formed in 88% yield via cleavage of the chloride bridge in the appropriate iminate PC with dppe under mild conditions (CH2Cl2 , rt, 6 h, inert atmosphere).
To evaluate the cytotoxic effect of 66, ME1402 melanoma cells and the metastatic cell lines WM1158 and 501 mel were used.[47] Three non-malignant fibroblast cell lines (DNB, FG-0, and CT-1) served as controls. In these cell lines, the IC50 was 0.19, 0.20, 0.25, 0.46, 0.40, and 0.43 μM, respectively. These results indicate that ME1402 cell lines were more sensitive to 66 than fibroblast cell lines. Complex 66 exhibited cytotoxicity at much lower concentrations than cisplatin. To assess the effect of 66 on tumour growth in vivo, six-week-old nude mice were injected subcutaneously with ME1402 cells. Test showed that 15 days of treatment with 66 resulted in a greater than 85% reduction in tumour weight, while treatment with cisplatin resulted in a 63% reduction.
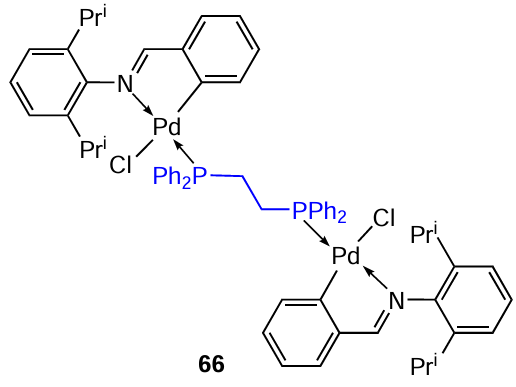
Subsequently, the antitumour activity of 66 was assessed in estrogen receptor-positive (MCF7) and estrogen receptor-negative (MDA-MB-231) breast cancer cells and in human breast cancer stem cells.[48] The IC50 values for MCF7 and MDA-MB-231 cells were 0.175 μM and 0.193 μM, respectively, while for non-malignant fibroblasts FG0 and DNB they exceeded 0.4 μM. Complex 66 was shown to induce DNA double-strand breaks leading to the intrinsic and extrinsic cell death pathways of autophagy and apoptosis, which are mediated by p38 MAP kinase.
The anticancer activity of 66 in alveolar rhabdomyosarcoma (aRMS, cell lines RH30 and AX-OH-1) and embryonic rhabdomyosarcoma (eRMS, cell lines RD and FL-OH-1) was studied in more detail.[49]For all lines, the IC50 values did not exceed 0.2 μM. To assess the selectivity of 66, its cytotoxicity against non-malignant mouse cells (FG0 and DMB fibroblasts and C2C12 myoblasts) was determined. The results showed that 66 is less cytotoxic in non-malignant cells with a selectivity index SI > 2 in all cases. According to clonogenic and migration assays, compound 66 inhibits the ability of RMS cells both to survive and migrate. Pharmacokinetic studies in mice showed that 66 has a satisfactory half-life, high volume of distribution, low clearance and good intraperitoneal absorption. Based on these data, the authors conclude that complex 66 may be a potent chemotherapeutic agent for the treatment of drug-resistant and advanced sarcomas with the desired mechanism of action.
In order to improve the pharmacological properties of compound 66, its water-soluble analogue 67 containing diethylene glycol substituents was obtained.[50]A dose-dependent decrease in the viability of MCF-7 and MDA-MB-231 cells was observed (IC5 = 0.49 and 0.58 μM). However, complex 66, which does not contain diethylene glycol units, had lower IC50 values of 0.180 and 0.190 μM in MCF-7 and MDA-MB-231 cells, respectively (Table 6). Based on the data obtained, the authors concluded that water solubility may not be the dominant factor affecting the antitumour activity of this class of metallodrugs.
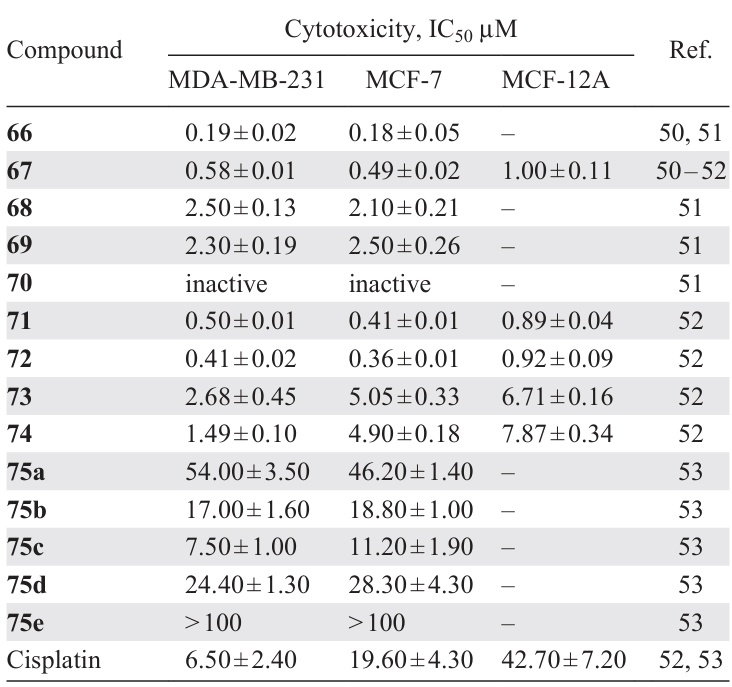
A number of binuclear benzylidene PCs 67 – 70 with biphosphine bridging ligands of various lengths were synthesized.[51] It was found that shortening (compound 68) or lengthening (compound 69) of the bridge negatively affects the cytotoxic activity of these compounds against MCF-7 and MDA-MB-231 tumour cells compared to compound 67 (see Table 6), and the introduction of a ferrocene moiety (70) completely deactivates the complex. These facts indicate that the conformational features of the metal complex are very important in terms of the efficiency of interaction with tumour cell DNA. The nature of the binding of complex 67 to DNA was studied by spectroscopy and electrophoresis, and it was found that 67 exhibits multimodal binding properties as a partial intercalator and DNA groove binder.
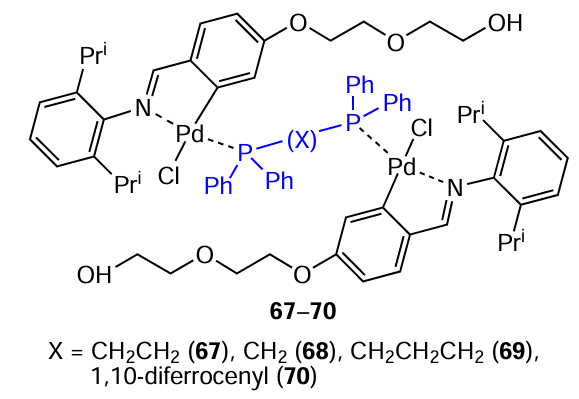
Compounds 71 and 72, analogues of PEGylated binuclear PC 67, were also obtained.[52] Two non-PEGylated palladacycles 73 and 74 were synthesized, bearing morpholine and fluorinated substituents, respectively. The antitumour activity of the resulting complexes was assessed against two tumour cell lines, MCF-7 and MDA-MB-231, and one non-neoplastic cell line MCF-12A (see Table 6). PEGylated palladacycles 67, 71 and 72 showed comparable potency against both cancer cell lines and relatively good selectivity for cancer cells compared to non-neoplastic cells. Compounds 73 and 74 were an order of magnitude less active than the PEGylated complexes but showed selectivity towards the more aggressive MDA-MB-231 cell line.
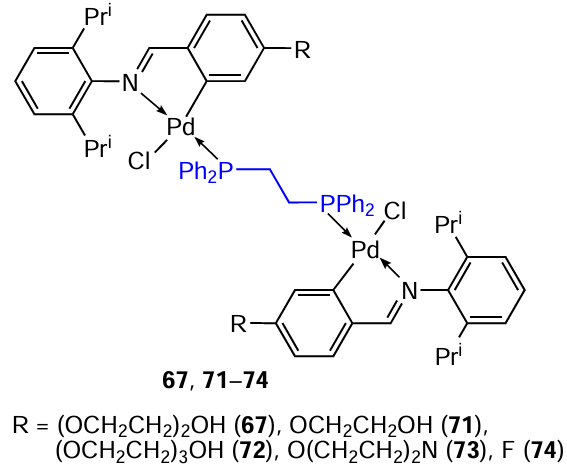
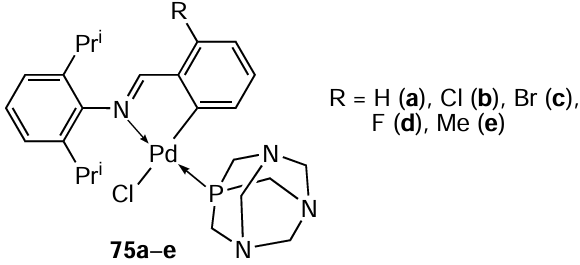
Water-soluble tertiary phosphine PTA was chosen as a ligand for the cleavage of chloride bridges of binuclear iminate PCs to obtain a series of aryl-substituted mononuclear derivatives 75a – e.[53] Under mild conditions (CH2Cl2, rt, 2 h), high yields (up to 90%) of the target PCs 75a – e were achieved. The potency of the resulting compounds as antitumour agents against MCF7, MDA-MB-231, and ME1402 cells was assessed in vitro. Complexes 75a – d were found to exhibit moderate cytotoxic activity (see Table 6). Activity against ME1402 melanoma was revealed only in complexes 75a – c. The nature of the different ortho substituents significantly affects the anticancer activity, which is most noticeable in the case of chloro- and bromo-substituted complexes 75b,c. In general, complexes 75b – d with substituents possessing an electron-withdrawing inductive effect in the aromatic ring (R = Cl, Br, F) are more active towards the tested cell lines compared to unsubstituted 75a and methyl-substituted 75e analogues. For compounds 75b – d, the IC50 values varied in the range from 11 to 32 μM when tested against the MCF7 cell line. The presence of a methyl substituent completely deactivated compound 75e (IC50 > 100 μM). The bromo complex 75c was unstable. The interaction of 75b with DNA was confirmed by various methods (gel electrophoresis, circular dichroism, UV-visible spectroscopy). These studies show that, unlike cisplatin, the binding mode of 75b to DNA does not involve intercalation. Initial results point to a possible electrostatic interaction with DNA. The authors note that the resulting PCs 75a – e are neutral complexes, so for electrostatic binding to occur, it is necessary to convert neutral complexes into their ionic forms. One of the possible transformations of these complexes is aquation or solvolysis of the Pd – C bond, which provides the transition to the cationic form.
Based on phosphaadamantane PTA, a water-soluble iminophosphorane ligand was obtained, which was further used in the synthesis of cyclopalladated compounds 76 – 80 (Scheme 21).[54] These results provide another example of the wide possibilities of converting the parent binuclear PCs into a variety of mononuclear derivatives. All reactions proceed in good yields (55 – 87%) in a suitable solvent for 1 h at room temperature. Only for the synthesis of complex 80b, an inert atmosphere is desirable. A distinctive feature of PCs 76 – 80 is their hydrophilicity. Compounds 79, 80a,b are soluble in water, and complexes 76 – 78 are soluble in a mixture of DMSO – H2O (1 : 99), which is an important characteristic for their biological studies.
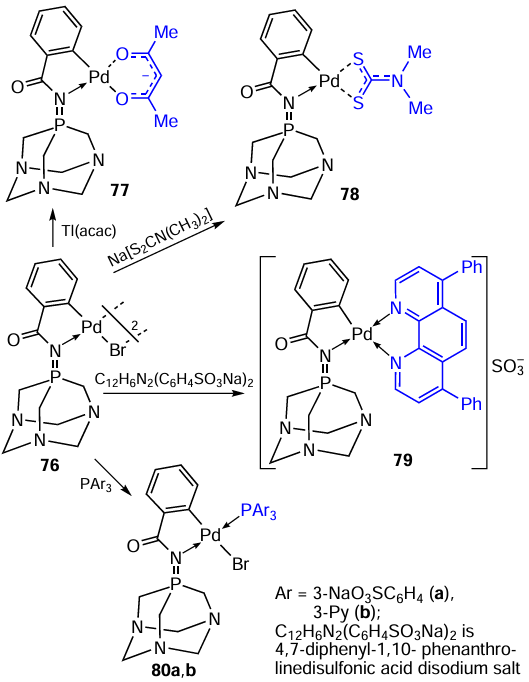
Compounds 76 – 80 were tested as anticancer agents in vitro against Jurkat-T and human prostate cancer DU-145 cells.[54]Complexes 76 and 77 can be distinguished from this group; their cytotoxic activity is similar to that of cisplatin. It should be noted that mononuclear derivatives 77 – 80 are significantly inferior in activity and selectivity to the parent PC 76. For example, IC50 values of compounds 76 – 79, 80a,b and cisplatin against DU-145 cells are 49.3, 96.2, 135, 187, 318, 299, 79 μM, respectively.
The promising results obtained for the biphosphine PC (S)-4 were developed by Spencer et al.,[55-58] whose objects of research were 1,4-benzodiazepine-based PCs. Using binuclear PCs 81a – g as starting compounds, the authors synthesized mononuclear compounds 82a – g and the biphosphine PC 83a (Scheme 22*, Table 7).[56][57] The resulting complexes were tested in vitro for antitumour activity against K562 cells.[57] All mononuclear complexes 82a – g showed weak activity (IC50 = 200 – 1000 μM). However, the in vitro activity of complex 83a was good (IC50 = 4.3 μM), significantly exceeding that for the previously described enantiomers of compound 4 (IC50 = 300 μM and 400 μM). Unfortunately, complex 83a showed high toxicity (IC50 = 0.30 ± 0.04 μM) towards normal Vero cells (cells of normal origin obtained from an adult African green monkey kidney epithelia).
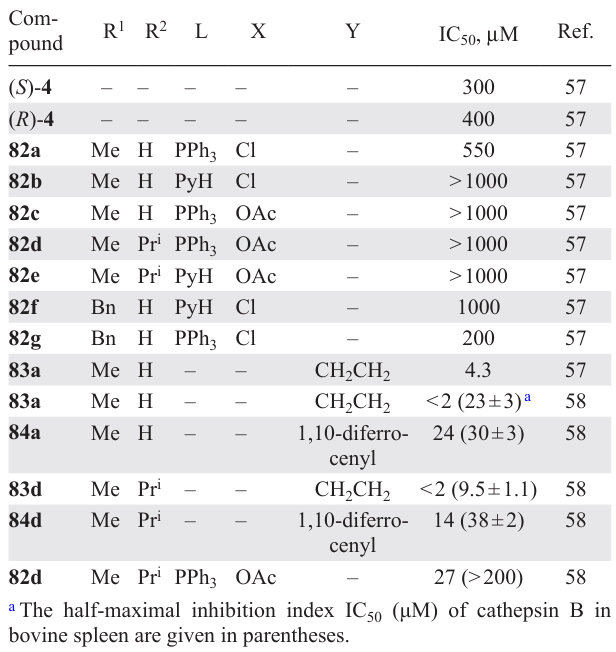
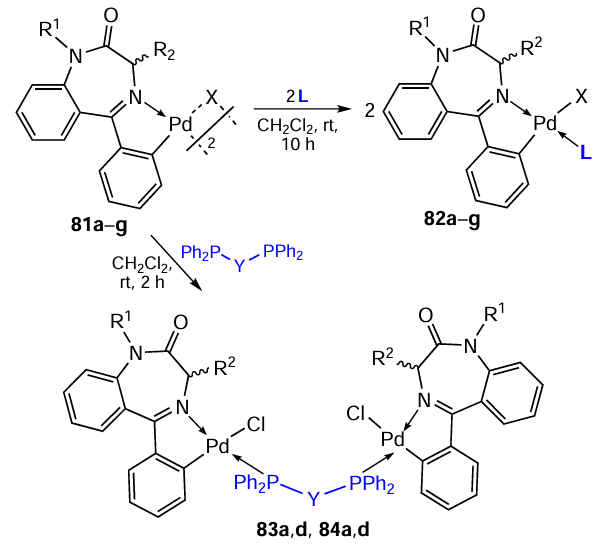
Biphosphine PCs 83a,d and 84a,d and mononuclear 82d were tested in vitro for cytotoxic activity against A2780/S cells and as inhibitors of the catalytic activity of cathepsin B, an enzyme involved in tumour progression (see Table 7).[58] Mononuclear PC 82d was found to exhibit minor cytotoxic and inhibitory activities against cathepsin B. Biphosphine PCs 83a,d and 84a,d are excellent inhibitors of cathepsin B and are cytotoxic against A2780/S. However, replacement of the ethylene bridge in 83a,d with a ferrocenyl moiety (84a,d) reduces the activity of the compounds.
* Information on the composition of the compounds (R, L, X, Y) is presented in Table 7.
5. Cyclopalladated N-heterocycles
Biaryl N-heterocyclic compounds can be considered as a structural analogue of conjugated arylimines, which are successfully used in the ortho-palladation reaction. This group of compounds is actively studied by Italian scientists.[59-62] The synthesis of PC 85 – 89 based on phenylpyridine and substituted phenylpyrimidine is described in publications.[59][60] Scheme 23 shows examples of the synthesis of mononuclear neutral (87, 88) and ionic (89) complexes. When 4-hydroxyacridine (4-OH-acrid) was used as a cleavage coligand, mononuclear bis-chelate PCs 87a and 88b were obtained in yields of 80% and 77%, respectively.[59] The low solubility of complex 88b reduced the capacity for studying its biological properties. Evaluation of the cytotoxicity of PC 87a against human ovarian carcinoma cells (lines A2780, OVACR 5 and OVACR 8) showed that its activity (IC50 = 2 – 7 μM) significantly exceeded that of cisplatin (IC50 = 7 – 53 μM). To obtain ionic complexes 89d,e, binuclear PC 86c was used as the starting material, which was preliminarily converted into the ionic intermediate [Pd(L)(MeCN)2][BF4] by treatment with AgBF4 in acetonitrile. The reaction of this intermediate with substituted bipyridines (4,4'-(R3)2-bipy) gives the target compounds 89d,e in yields of 97% and 60%, respectively.[60]
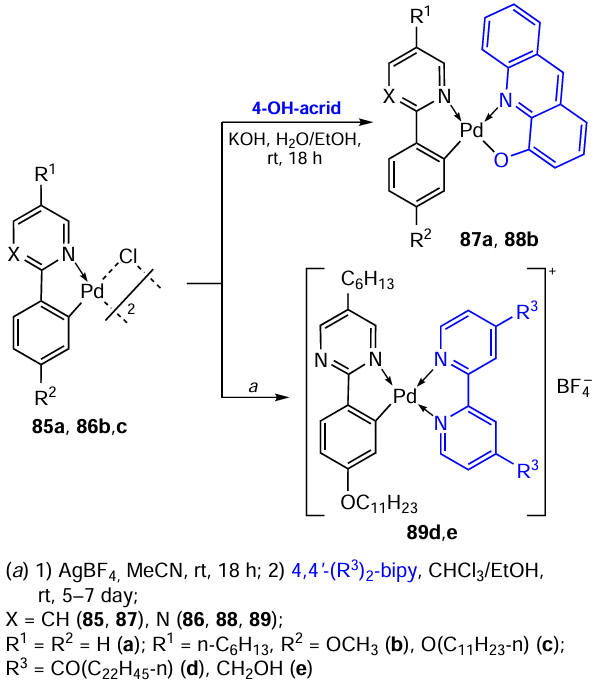
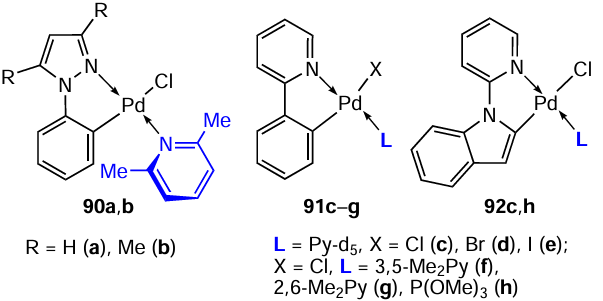
A large library of PCs based on N-donor heterocyclic ligands such as 1-phenylpyrazoles (90a,b), 2-phenylpyridine (91c – g) and 1-(2'-pyridyl)indoles (92c,h) was obtained.[34]Individual examples from more than 30 described structures are given. All mononuclear complexes were obtained by reacting the parent binuclear PCs with appropriate monodentate coligands. The effect of various structural factors, in particular, the nature of the halide, on the cytotoxic activity of the resulting compounds was studied. While the nature of the halide was of little importance for cationic metal intercalators where the halide serves as a counterion, the transition from the coordinated chloride to the bromide and iodide can be critical for enhancing activity in neutral complexes. Therefore, a number of corresponding chloride, bromide, and iodide derivatives 91c – e were obtained. However, cytotoxicity screening of the resulting complexes against mouse lymphoid leukaemia cells (L1210) showed that they exhibited identical cell growth inhibitory activity (IC50 = 11 – 12 μM). Obviously, halogen ions are exchanged for a water molecule (solvent) under experimental conditions to afford identical aqua complexes. One of the factors increasing the activity is steric hindrances around the metal centre, arising due to the presence of bulky ligands such as 2,6-dimethylpyridine. For example, replacing pyridine (91c) with 3,5-dimethylpyridine (91f) does not change the cytotoxic activity; the IC50 value is 11 μM for both complexes. The introduction of lutidine with two ortho-methyl substituents increases the cytotoxic activity tenfold. For 91g, IC50 = 1.2 μM, which is comparable with the activity of cisplatin (IC50 = 0.9 μM). In the case of a pair of complexes 90a and 90b, an increase in activity was also noted for methyl-substituted 1-phenylpyrazole 90b (IC50 = 6.4 μM and 3.0 μM, respectively). The most active compounds were selected for further in vivo studies, but none of the palladium complexes showed satisfactory activity against murine leukaemia P388 at doses below toxicity levels. The authors noted interesting features of the resulting complexes.34 Mononuclear palladium complexes 90 – 92 are stable at room temperature, but attempts to recrystallize them at elevated temperatures led to dissociation of ligand L to the parent μ-halodimers.
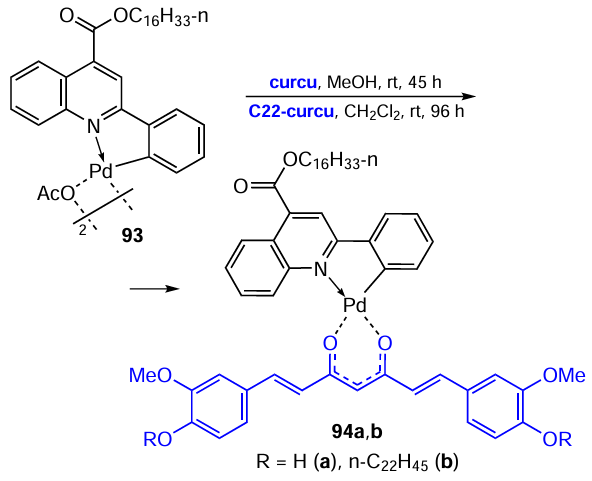
Pucci et al.[61] described an example of the successful use of β-diketones for the synthesis of heteroleptic palladium complexes (Scheme 24). The starting PC 93 was obtained by direct cyclopalladation of the acyl derivative of 2-phenylquinoline. When 93 reacts with curcumin (R = H) or its C22-ester (R = n-C22H45) under mild conditions, mononuclear complexes 94a,b are formed in 71% and 69% yields, respectively. The authors argued their choice of coligands by the fact that curcumin exhibits pronounced cytotoxic effects on various cell lines and, in particular, it suppresses the survival of human prostate cancer cells. In this regard, the cytotoxic effect of new mononuclear complexes 94a,b on the DU145 cell line was studied. Complex 94a was found to exhibit cytotoxic activity at a concentration of 0.1 μM at approximately 50% inhibition of cell proliferation, whereas pure curcumin was inactive at the same concentration. At a concentration of 0.1 μM, a similar percentage of proliferation inhibition (~ 55%) was observed for both the curcumin C22-ester ligand and complex 94b.
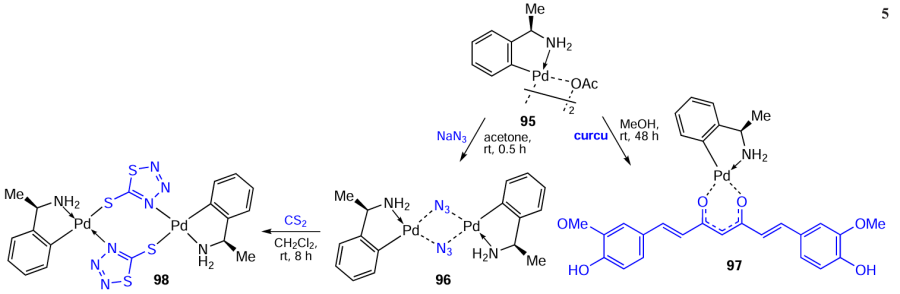
Karami et al.[28]also used curcumin as a bioactive coligand for the PC. Derivatives 96 – 98 were derived from binuclear cyclopalladated 2-phenethylamine 95 (Scheme 25). The authors demonstrate an unusual example of the transformation of the bridging fragment: in the first step, there is a direct exchange of acetate ion for azide ion (complex 96), followed by a cycloaddition of carbon disulfide to give a thiotriazole ring in the bridge of complex 98. Palladacycles 97 and 98 were isolated in good yields of 74% and 52%. The cytotoxic activity of complexes 97 and 98 in vitro against Jurkat, MCF-7 tumour cells and normal PBMC was studied. Good cytotoxicity against cancer cells was revealed for both complexes, while they both exhibited low toxicity (IC50 > 150 μM) against normal cells. Complex 97 showed higher toxicity against Jurkat than MCF-7 (IC50 = 45.16 μM and 75.5 μM, respectively). In contrast, complex 98 exhibited higher toxicity against MCF-7 cells (IC50 = 47.5 μM) than against Jurkat cells (IC50 = 72 μM).
6. Phosphorus-containing palladacycles
Iranian scientists [63-65] described the synthesis and results of the study of the biological activity of phosphorus-containing PCs (Scheme 26). Novel C,C-chelate phosphinylide complexes containing bridging (100) and terminal (101a,b) azide groups were obtained in 70 – 71% yields.[63]The DNA-binding properties of 101a,b were studied by electron absorption and fluorescence spectroscopy. It was found that both complexes can interact with DNA by intercalation, and the binding activity changes in the series 101a > 101b.
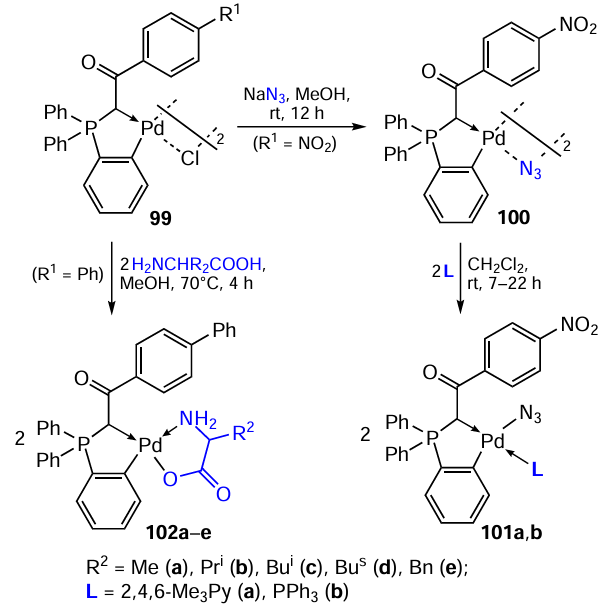
Using phenyl-substituted PC 99 (R1 = Ph) as the starting compound, mononuclear derivatives 102a – e were obtained, which contained amino acid coligands (see Scheme 26).[64] The bridge-splitting reaction proceeds in boiling methanol in yields of at least 80%. According to 1H and 31P-{1H} NMR spectroscopy, complexes 102 are formed as a mixture of geometric cis – trans isomers. The cytotoxic activity of 102a – e was assessed against K562 tumour cells, and a clear dependence on the nature of the amino acid moiety was established. Complexes containing phenylalanine (102e) and leucine (102c) showed IC50 values of 15.1 and 16.5 μM, respectively, which is comparable to that of cisplatin (19.4 μM). Unfortunately, the authors did not provide data for the parent binuclear PC 99 to assess the contribution of the amino acid moiety to the cytotoxicity of the resulting compounds.
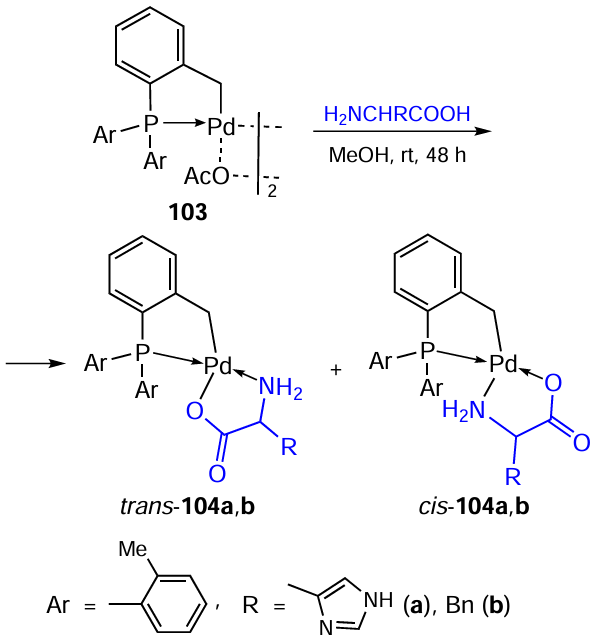
The synthesis of PCs 103 based on direct cyclopalladation of tri(ortho-tolyl)phosphine is described (Scheme 27).[65] In the reaction of compound 103 with amino acids (L-histidine or L-alanine), the acetate bridge opens to give the corresponding mononuclear bis-chelates 104a and 104b in yields of 73% and 76%, respectively. Based on NMR spectroscopy data, the authors concluded that the complexes were obtained as mixtures of geometric cis- and trans-isomers. The cytotoxic activity of complexes 104a and 104b was studied against B16F0 melanoma, C26 colon carcinoma, and normal fibroblast NIH cell lines. Although moderate cytotoxic activity was observed against cancer cells, both compounds demonstrated low toxicity against normal NIH cells (IC50 > 100 μM). The selectivity index SI > 2.4. Complexes 104a and 104b demonstrated higher cytotoxicity against B16F0 cells (IC50 = 30.5 and 31.5 μM) than against C26 cells (IC50 = 41.7 and 34.8 μM).
7. Terpene-based palladacycles
This Section presents the results obtained by the authors of this review, whose research focused on PCs with terpene ligands. The use of natural terpenoid derivatives as ligands opened the way to chiral metal complexes of high optical purity, which are of interest as potential pharmacological substances.[66]
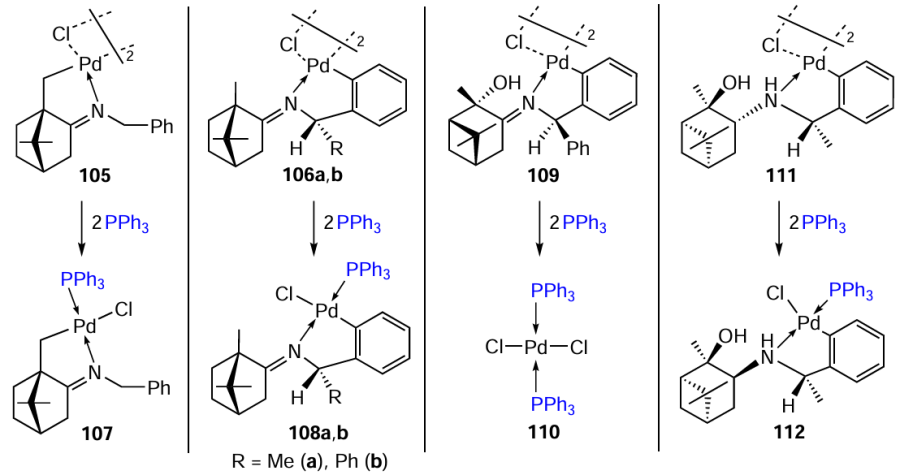
The bridge-splitting reaction of a number of PCs with triphenylphosphine was studied (Scheme 28). The stability of PCs with the bornane moiety should be noted first. In the reaction between compounds 105, 106a,b with triphenylphosphine (toluene, rt), the cyclopalladated motif is retained, the opening of chloride bridges takes place to furnish mononuclear mixed-ligand palladium complexes 107, 108a,b in 85 – 98%.[67][68] Unlike bornane structures, pinane iminate PC 109 is less stable, and when treated with triphenylphosphine under the said conditions, it decomposes to give dichloro(bis-triphenylphosphine)palladium 110.[68] However, its amine analogue 111 does not decompose under these conditions, and the corresponding mononuclear complex 112 is formed.[69]
Binuclear complex 111 was chosen as the starting material for the synthesis of mixed-ligand mononuclear derivatives 113a – d containing L-amino acids (proline (Pro), tyrosine (Tyr), serine (Ser), isoleucine (Ile), and cysteine (Cys)) as coligands (Scheme 29).[70]The isolated yields of complexes 113a – d ranged from 57% to 74%. It should be noted that in the case of Cys, complete degradation of PC 111 occurs to give palladium bis(cysteinate).
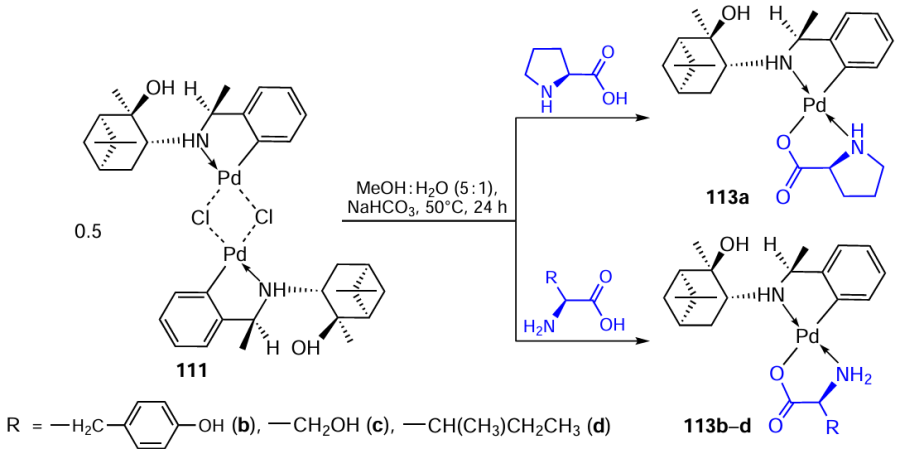
We proposed the trans-N,N geometry of complexes 113a – d, which is most characteristic of mononuclear palladium complexes of this type.[10] The high diastereoselectivity of the complexation reactions (> 95%, NMR) should be noted. It was previously found [71]that cyclopalladation of pinane benzylamine produces a new N-chiral centre of the S-configuration (PC 111). During proline chelation, the nitrogen configuration is also fixed. Analysis of 1H – 1H correlation interactions allows us to conclude that the nitrogen atom of the proline substituent in compound 113a has an S-configuration.
During the study the antitumour potential of novel C,N-PCs 113a – d containing L-amino acid ligands, we analyzed the effect of the studied compounds on the survival of A549, HeLa, neuroblastoma (SH-SY5Y), and laryngeal epidermoid carcinoma (Hep-2) tumour cells. Cisplatin was used as a reference substance, and the corresponding free ligands were used as comparative drugs. It was found that all mixed-ligand metal complexes 113a – d had a moderate effect on the viability of the cells of the above-mentioned lines. Compounds 113b,d exhibited the most pronounced cytotoxic effect on SH-SY5Y cells with IC50 = 28.65 ± 0.36 μM and 23.66 ± 0.41 μM, respectively. However, these values were higher than the IC50 of the starting binuclear complex 111 (6.27 ± 0.14 μM). Only for compound 113c, the toxic effect on Hep-2 cells was more than three times that of the parent compound 111. For the parent ligands (terpene amine and the amino acids Pro, Tyr, Ser, Ile and Cys), as well as for palladium biscysteinate, which does not contain terpene ligands, the IC50 values for all tumour lines exceeded 100 μM. These data allowed us to conclude that the cytotoxic activity of the tested compounds is mediated by coordination with the terpene ligand.
8. Conclusion
The data presented in this review irrefutable show that binuclear PCs are a versatile platform for the design of new cytotoxic palladium complexes. This versatility is due to two structural factors. First, the remarkable structural diversity of the starting ligands, the cyclopalladation of which gives rise to binuclear PCs with m-bridged fragments. First of all, the coligands include N-donor ligands such as amines, oximes, imines, heterocycles, as well as phosphorus-containing compounds, e.g., phosphines and phosphinylides. Secondly, the choice of a coligand is virtually unlimited, which facilitates the opening of bridge bonds and the formation of a new hybrid PC. Depending on the structure of the cleaving ligand, both mono- and bidentate, neutral and ionic PCs can be obtained. Therefore, there is a tool for the targeted formation of a ligand environment, including a macromolecular one, in metal complexes. This factor is crucial in terms of both the biological and catalytic activity of the target palladium complexes.
Particularly noteworthy is the ease of structural transformations, such as replacement of the bridging anion X, opening of m-bridging bonds, and the stability of palladacycles during such transformations. Only a few examples of opening of the metallacycle via the C – Pd bond[14][33] or its complete decomposition [68]upon reaction of the parent PC with a coligand have been described.
Palladium complexes are of undoubted interest as drugs with a wide pharmacological profile.[72-80] Based on the analysis of data on the cytotoxic activity of various types of PC, described in this review, it can be clearly concluded that cyclopalladated compounds are a very promising class of antitumour pharmaceuticals. Chiral biphosphine complexes (S)-4, (S)-5,26 (S)-10 (Ref. 18) and (S)-1[19-21] were identified as lead compounds, and their testing reached the preclinical stage. The results obtained allow enantiomeric purity to be considered a priority criterion for future therapeutics.
The presented PC transformation schemes (bridge-splitting reactions) enable fine-tuning (molecular editing) of the structure. These innovative approaches are highlighted in the American Chemical Society’s Analytical Digest as one of the eight key scientific trends for 2025: ‘Molecular editing allows chemists to create new molecules by precisely modifying existing large molecules; this allows them to create new compounds more efficiently and cost-effectively, and by reducing common synthetic steps, reduces the volume of toxic solvents and energy requirements for many transformations’.*
The information presented in the review will certainly be of interest to specialists working in the fields of coordination, bioinorganic, medicinal chemistry and metal complex catalysis.
This review was financially supported by the Ministry of Science and the Higher Education of the Russian Federation (State Assignment, Reg. No. 125020301261-5).
* https://www.cas.org/resources/cas-insights/scientific-breakthroughs-2025-emerging-trends-watch?tm_campaign=GLO_GEN_ANY_CIS_LDG&utm_medium=EML_CAS_ORG&utm_source=EM_subdrive 29.05.2025
9. List of abbreviations
The following abbreviations and designations were used in the review:
Ас — acetyl,
bipy — 2,2'-bipyridyl,
Bn — benzyl,
bpp — 1,3-bis(4-pyridyl)propane,
C. elegans — free-living soil nematode,
curcu — curcumin,
curcu22 — curcumin С22-ester,
dppe — 1,2-bis(diphenylphosphino)ethane,
dppf — 1,1'-bis(diphenylphosphino)ferrocene,
dppm — bis(diphenylphosphino)methane,
dppmS2 — bis(diphenylthiophosphinoyl)methane,
dppp — 1,3- bis(diphenylphosphino)propane,
IC50 — half-maximal inhibitory concentration,
MIC — minimum inhibitory concentration,
NHC — N-heterocyclic carbene,
4-OH-acrid — 4-oxyacridin,
PC — palladacycle,
phen — 1,10-phenanthroline,
PTA — 1,3,5-triaza-7-phosphaadamantane,
Py — pyridinyl,
SI — selectivity index.
Full names of cell lines:
4T1 — murine mammary carcinoma,
501mel — human melanoma,
A2058 — human melanoma,
A2780 — human ovarian carcinoma,
A549 — human lung carcinoma,
aARMS (RH30 and AX-OH-1 cell lines) — alveolar rhabdomyosarcoma,
B16F0 — human melanoma,
B16F10 — murine melanoma,
B16F10-Nex2 — murine melanoma,
BJ — human bland fibroblasts,
C26 — human colon carcinoma,
C2C12 — murine non-malignant myoblasts,
CasKi — human ovarian carcinoma,
CT-1 — human non-malignant fibroblasts,
CT26 — murine colon carcinoma,
DMB — murine non-malignant fibroblasts,
DNB — human non-malignant fibroblasts,
DU145 — human prostate cancer,
eRMS (RD and FL-OH-1 cell lines) — embryonic rhabdomyosarcoma,
FG0 — murine non-malignant fibroblasts,
FG-0 — human non-malignant fibroblasts,
HCT-8 — human colon carcinoma,
HCT-116 — human colorectal carcinoma,
HeLa — human cervical carcinoma,
Hep-2 — human laryngeal carcinoma,
HL-60 — human leukaemia,
HT-29 — human colorectal carcinoma,
HT144 — human melanoma,
Jurkat — human leukaemia,
K562 — chronic myeloid leukaemia,
L1210 — murine lymphoid leukaemia,
LB373 — human melanoma,
LM3 — murine mammary gland adenocarcinoma,
LS-180 — human colon cancer,
MC3T3-E1 — non-neoplastic cells,
MCF-7 — human breast duct adenocarcinoma,
MCF-12A — non-neoplastic cells of human breast cancer,
MDA-MB-231 — human breast adenocarcinoma,
ME1402 — human melanoma,
Me91 — human melanoma,
MRC-5 — human bland fibroblasts,
NIH — human fetal fibroblasts,
OVACR 5 — human ovarian cancer,
OVACR 8 — human ovarian cancer,
P388 — murine leukaemia,
PBMC — human peripheral blood mononuclear cells (T-lymphocytes),
Saos-2 — human osteosarcoma,
SH-SY5Y — human neuroblastoma,
SiHa — human cervical carcinoma,
SKBr-3 — human breast cancer,
SK-MEL-5 — human melanoma,
SK-MEL-25 — human melanoma,
SK-MEL-110 — human melanoma,
SK-OV-3 — human ovarian cancer,
T98G — human glioblastoma,
U251 — human glioblastoma,
U2OS — human osteosarcoma,
U-87 — human glioblastoma,
Vero — cells of normal origin obtained from an adult African green monkey kidney epithelia,
WM1158 — human melanoma,
ZR753A — human breast cancer.
References














