


Keywords
Abstract
The antitumour activity of curcumin has been studied in detail, but this natural compound has not found application in oncology due to its unacceptable pharmacological properties. Currently, monocarbonyl analogues of curcumin, particularly C5-curcuminoids, are considered the most promising for cancer therapy due to their satisfactory pharmacokinetic profile. This review provides a critical analysis of recent literature on the synthesis of monocarbonyl analogues of curcumin and the study of their antitumour activity. Unlike previously published reviews, it examines not only the synthesis methods and chemical properties of these compounds but also the mechanisms of their biological action. The key molecular targets that promote angiogenesis, proliferation, and growth of malignant tumours and regulate glycolysis in neoplastic cells are discussed. The information provided in this review may be of interest to specialists in organic and medicinal chemistry, as well as biochemistry and pharmacology.
The bibliography includes 168 references.
1. Introduction
Curcumin (1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione), a secondary metabolite of plants of the genus Curcuma of the ginger family (Zingiberaceae), began to be used as a physiologically active compound as early as the 19th century. The first mentions of the isolation and identification of this compound were found in a report by Vogel,[1] dated 1815, after which an era of improving the methods of its extraction from the plants Curcuma longa L. and Curcuma domestica L.[2] began. For a long time, curcumin was used in non-traditional Eastern (Ayurvedic) medicine for the prevention and treatment of various diseases, such as rheumatoid arthritis and osteoarthritis.[3] And only starting in 1913, when Lampe and Milobedzka[4] pioneered the synthesis of curcumin in its pure form by a five-step procedure and confirmed its structure, did a detailed study of the biological properties of this compound begin (Fig. 1). At present, curcumin has become an attractive object for a wide range of researchers, and on its basis, via various chemical transformations, a diversity of drugs has been obtained for the treatment of various diseases, including oncological[5][6] and neurodegenerative (such as Alzheimer’s and Parkinson’s diseases),[7-9] as well as bacterial[10][11] and fungal [12][13] infections and other types of pathologies.
![[{"id":"HLz76I7Z3F","type":"paragraph","data":{"text":"Key events in the history of curcumin research"}}]](/storage/images/resized/YVqTnSGkGReYKviOxCN2tl8zM3I5zlXElzEuXX7F_xl.webp)
Curcumin, in particular, has been found to enhance neuronal signalling and synaptic plasticity by modulating the pathways such as NF-κB transcription factor and PI3K/Akt (PI3K is a phosphoinositide 3-kinase, Akt is a kinase), and to reduce neuroinflammation and oxidative stress, thereby promoting synaptic integrity.[8] This compound also exhibits antibacterial activity against Mycobacteroides abscessus bacteria and can be used in photodynamic therapy for tuberculosis as a photosensitizer.[11] The antifungal activity of curcumin against Candida albicans fungi with a minimum inhibitory concentration of 250 μg mL–1 is due to changes in the membrane-associated properties of ATPase activity, ergosterol biosynthesis, and proteinase secretion.[13]
Currently, curcumin is most often used in medicine both as an independent dietary supplement to boost immunity and fight bacteria and viruses, and in combination with other drugs for cancer therapy. For example, curcumin has an inhibitory effect on the colorectal cancer cell (cell line CT26) growth. The IC50* values for curcumin are 23.52, 16.11, and 13.62 μM after 24, 48, and 72 h, respectively.[14] An in vivo study in mice showed a reduction in tumour volume when exposed to a curcumin dose of 15 mg kg–1. In turn, a combination of curcumin with paclitaxel demonstrated a synergistic antitumour effect on the MDA-MB 231 cell line (breast cancer),[15]which is reflected in an increase in the proportion of dead cancer cells from 34% (for curcumin) to 52% (for the combination of curcumin with paclitaxel).
The antitumour potential of curcumin is realized mainly due to the manifestation of antioxidant and anti-inflammatory properties. This leads to the modulation of various oncogenic signalling pathways due to the effect on certain intracellular molecular targets (Fig. 2), in particular many transcription factors (ERG-1,[16] ERE,[17] STAT-1,[18] STAT-3,[19] STAT-4,[20] STAT-5,[21] Notch-1,[22] NF-κB,[23] PPAR-γ,[24] WTG-1,[25] β-catenin [26]), protein kinases (MAPK,[27] EGFR,[28] ERK,[29] IL-1 RAK,[30] PKA(B,C),[31] JNK,[32] IKK [33]), proteins (Mcl-1,[34] Bcl family,[35] AP-1,[36] c-myc,[37] cyclin D1,[38] cytochrome С,[39] PARP,[40] Bax,[41] caspases,[42] FADD),[43] growth factors (FGF,[44] VEGF,[45] TGF-β1,[46] TF,[47] CTGF,[48] EGF [49]), cytokines (prostaglandins,[50] TNF,[51] IFN,[52] interkeukins,[53] COX-2,[54] MCP-1,[55] MIP[56]) and receptors (HER-2,[57] CXCR4,[58] EGFR,[59] H2R,[60] IL-8R,[61] LDL-R,[62] ITPR[63]). Another mechanism of curcumin’s antitumour action involves epigenetic regulation of cancer cell growth and proliferation. This is due to the fact that a fragment of curcumin can act as a ‘capping’ group in the structure of enzyme inhibitors involved in the deacetylation of proteins responsible for packaging cancer cell DNA.[64] Some of the most effective strategies for correcting the disadvantages of curcumin that hinder its use in oncology (e.g., poor water solubility and rapid metabolism)[65] include the production of its derivatives through chemical modification and the synthesis of its structural analogues. Structural modification, namely the introduction of physiologically active functionalities into the molecule,[66] conjugation with other pharmacophores,[67] and the development of curcumin-like compounds,[68] determines the improvement of pharmacological properties and enhanced biological activity. Currently, the most relevant trend is the chemical modification of curcumin. Its primary goal is to develop candidate drugs for the cancer therapy, which will undoubtedly pave the way for fully exploiting the therapeutic potential of this natural compound.
![[{"id":"QvIkddybte","type":"paragraph","data":{"text":"Molecular mechanisms of curcumin’s antitumour action. The following abbreviations are used: transcription factors: Bax is Bcl-2-associated X protein, COX-2 is cyclooxygenase 2, CTGF is connective tissue growth factor, CXCR4 is C – X – C – chemokine receptor type 4, EGF is epidermal growth factor, EGFR is epidermal growth factor receptor, ERE is estrogen response element, ERK is extracellular signal regulated kinase, FADD is Fas-associated protein with death domain, FGF is fibroblast growth factor, H2R is histamine H2 receptor, HER-2 is human epidermal growth factor receptor 2, IFN is interferon, IKK is IκB kinase, IL-1 RAK is interleukin-1 receptor-associated kinase, IL-8R is interleukin 8 receptor, ITPR is inositol 1,4,5-trisphosphate receptor, JNK is c-Jun N-terminal kinase, LDL-R is low-density lipoprotein receptor, MAPK is mitogen-activated protein kinase, MCP-1 is monocyte chemoattractant protein 1, MIP is macrophage inflammatory protein, PKA(B,C) is protein kinase A(B,C), PPAR-γ is Peroxisome proliferator-activated receptor gamma, STAT-1(3,4,5) is signal transducer and activator of transcription 1(3,4,5), TF is transcription factor, TGF-β1 is transforming growth factor beta 1, TNF is tumour necrosis factor, VEGF is vascular endothelial growth."}}]](/storage/images/resized/Vh0tE7M6LSLSUcoJHuyjCvwOG1kjr1qaPjYx860f_xl.webp)
This review summarizes and analyzes data on the synthesis of curcuminoids, which are structural analogues of curcumin with a single carbonyl group between the arylidene moieties, and their therapeutic potential. Carbo- and heterocyclic derivatives are considered, including 3,5-bis(arylidene)-4-piperidones as the most promising class. These compounds have the potential to be effective treatments for various cancers, as evidenced by studies of their specific molecular mechanisms of action in the occurrence and development of these pathologies. Unlike previously published review articles on similar topics,[69][70] we present the results of modern research on both the chemical synthesis and biological properties of such derivatives. The Section on chemistry considers methods for the synthesis and most typical transformations of monocarbonyl analogues of curcumin. In the biological part, special attention is paid to the antitumour effect of curcuminoids, in particular 3,5-bis(arylidene)-4-piperidones with pharmacophore substituents, against tumour cells of various morphologies. Systematization of material on the methods for the synthesis and biological properties of structurally diverse curcuminoids can form a basis for planning work carried out by specialists in organic and medicinal chemistry and pharmacology. Furthermore, the information presented in this review may be of interest to a wider audience.
* IC50 is the half-maximal inhibitory concentration. The lower its value, the more effective the substance is in terms of cytotoxic activity.
2. Chemistry of monocarbonyl analogues of curcumin
[]
2.1. Structure, properties and methods for the synthesis
Curcumin is a β-dicarbonyl compound with a seven-carbon chain (C7) between two benzene rings (shown in blue in Fig. 3), which exists in tautomeric equilibrium with its enol form in solution. Over time, all natural metabolites of curcumin (curcuminoids) were believed to contain the C7 chain.[71] However, in 2012, along with curcumin, its analogue, C5-curcumin, containing one carbonyl group between arylidene moieties,* was isolated from the plants Curcuma longa and Curcuma domestica.[72]
![[{"id":"wpisApHHUe","type":"paragraph","data":{"text":"Structures of natural curcumins and their monocarbonyl analogues — cyclic and heterocyclic C<sub>5</sub>-curcuminoids"}}]](/storage/images/resized/UvgORaI0XzZNcwuN5t9a5C4X8W7OimoPvxCUXSxL_xl.webp)
As noted in the Introduction, a significant drawback of natural C7-curcumin is its poor solubility in water (0.0004 mg mL–1 at pH 7.3) [65] because of its hydrophobicity and, consequently, low bioavailability (serum levels are < 60 nM).[73] Moreover, in aqueous solutions with acidic to neutral pH (gastric and intestinal conditions, respectively), the equilibrium is strongly shifted towards the keto form, which leads to poor solubility of curcumin at room temperature.[73]
There are two approaches to improving the water solubility and bioavailability of curcumin. The first involves encapsulation of curcumin into liposomes, micro- and nanoemulsions, as well as polymer complexes with poly(N-vinylpyrrolidone) and polymer nanoparticles. Such nanoparticles can be prepared using amphiphilic copolymers of N-vinylpyrrolidone and acrylic acid [74][75] or modified copolymers of lactic and glycolic acids.[76] Encapsulation of curcumin in nanosized carriers allows obtaining dosage forms that are stable under physiological conditions, capable of targeted delivery and highly potent. The mechanisms of interaction between drug-loaded nanoparticles and tumour cells are well studied and include endocytosis or membrane fusion,[75] which allows the active substance to be delivered to the endosomes and nucleus of the tumour cell. Despite the high efficiency of loading curcumin into nanoscale carriers and, consequently, significant improvement in curcumin biocompatibility, this approach has its drawbacks. For example, the size and shape of nanocarriers affect their stability, and the encapsulation method depends on the hydrophilicity (hydrophobicity) of the active molecule and affects its controlled release. Furthermore, mention should be made of the preparative difficulties and high production costs associated with the synthesis of nanocarriers, as well as the potential residual toxicity of solvents or surfactants (for nanoemulsions) and the high cost of such dosage forms.
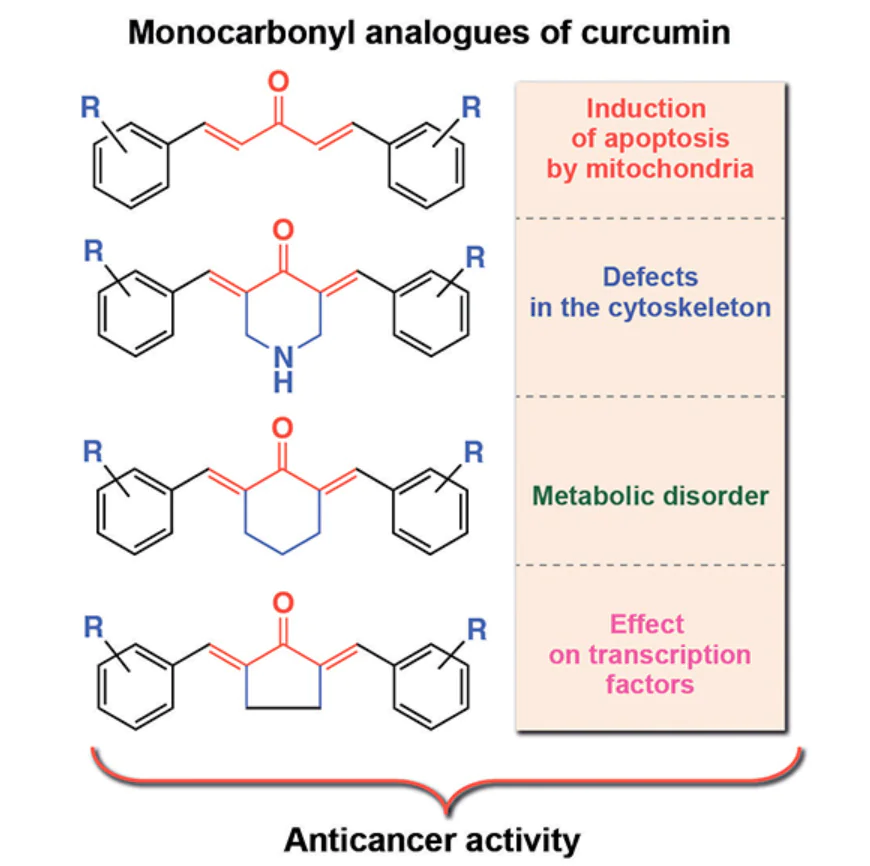
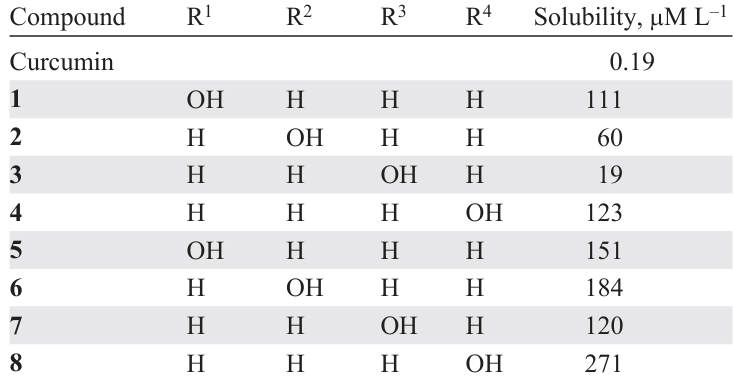
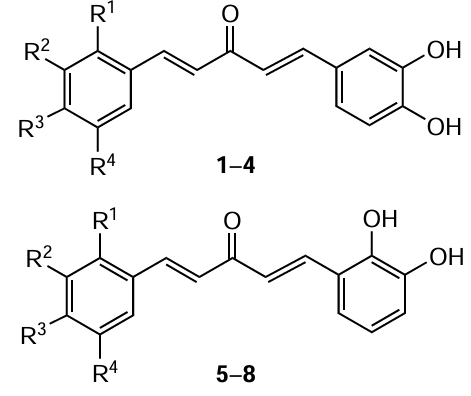
Another approach to circumventing the above-mentioned encapsulation issues is the use of synthetic curcumin analogues. In particular, biological studies of natural C5-curcumin and its monocarbonyl analogues (MACs) have shown that these compounds are highly soluble in water and possess cytostatic activity.[77] In particular, C5-curcuminoids 1 – 8 showed higher water solubility than natural curcumin (Table 1),[78] which is possibly due to the presence of polar hydroxy groups in the benzene rings. The cytostatic effect of MACs is associated with the presence of a conjugated dienone system in their structure, which acts as a pharmacophoric moiety. The plausible mechanism of the cytostatic action of MACs is their interaction with the sulfhydryl groups of thiols, in particular cysteine, in cancer cell receptors and in human topoisomerase IIα, an enzyme necessary for the proliferation and survival of cancer cells.[79] In turn, the amino groups in the nucleic acids of DNA of normal cells do not react with compounds of this class, which indicates the absence of genotoxicity.[80] This is a clear evidence of the advantage of MAC compared to commercial alkylating cytostatics, such as melphalan.
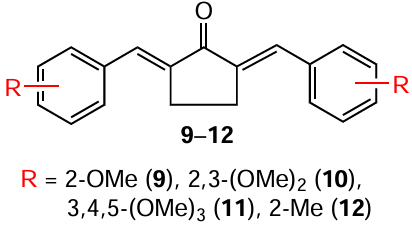
Another difference between MACs and curcumin is the higher stability of the former in the in vitro tests, which is associated with differences in their structure. The β-diketo group in the keto tautomer of natural C7-curcumin is a substrate for liver aldo-keto reductases, which likely contributes to its rapid metabolism in vivo. Due to the presence of a single carbonyl group, MAСs biodegrade much more slowly than C7-curcumin.[81] For example, the authors showed that after incubation in a 0.3% phosphate buffered sodium carboxymethylcellulose solution at pH 7.4 for 75 h, >4% of curcumin and < 10% of derivatives 9 – 12 with a five-membered ring degraded.**
Synthetic acyclic and carbocyclic C5-analogues of curcumin have been tested for various biological activities. Table 2[82-88] gives examples of both types of MACs that have demonstrated significant antioxidant, anti-inflammatory, antibacterial, or antitumour activity.
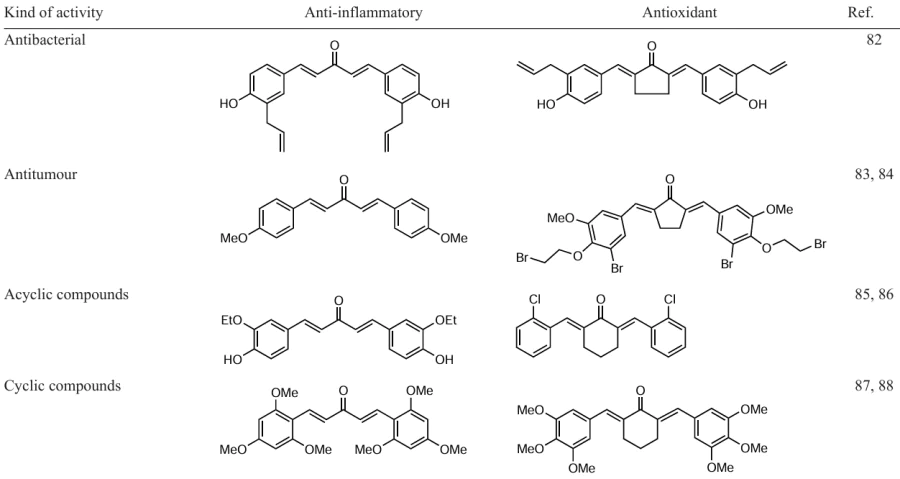
One of the first synthetic approaches to C5-curcuminoids, proposed[89] in 2009, was based on the oxidation of a secondary alcohol to a ketone followed by condensation with an aldehyde in toluene, catalyzed by the RhCl(PPh3)3 – BF3 · OEt2. As a result, dibenzylideneacetone (13) was obtained from isopropyl alcohol and benzaldehyde in 88% yield (Scheme 1). The authors showed that the reaction is extremely sensitive to the solvent used. For example, there is no reaction in tetrahydrofuran and acetonitrile, and in dichloromethane, the yield of product 13 drops to 52%.
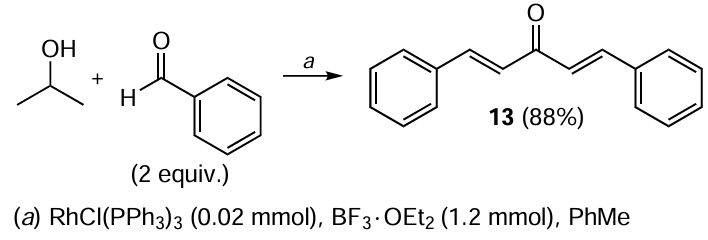
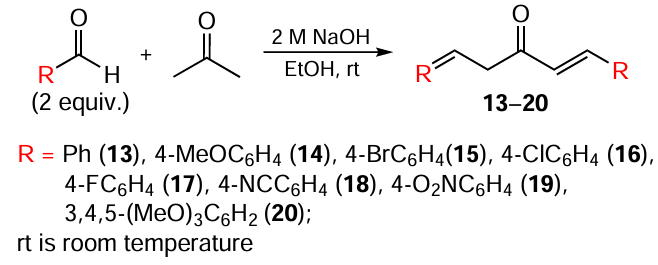
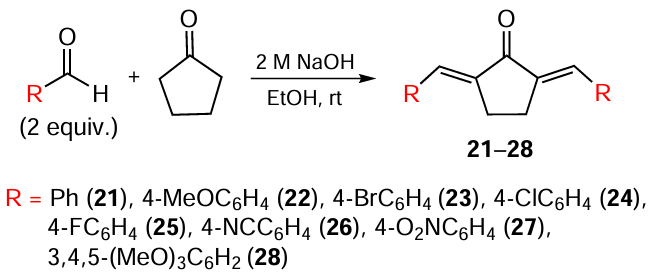
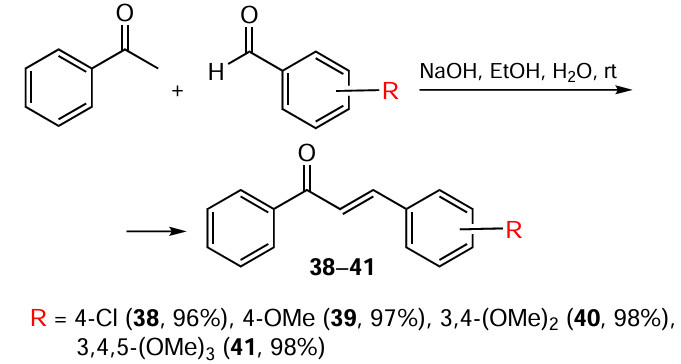
Despite the simplicity of the experiment and the high yield of the target product,[89] the expensive catalytic system significantly limits the laboratory and industrial application of this method. Acyclic and cyclic MACs are most often obtained through aldol/crotonic condensation involving aldehydes and ketones. Acetone serves as the ketone for the preparation of acyclic MACs 13 – 20 (Scheme 2), while cyclic analogues 21 – 36 are derived from cyclopentanone or cyclohexanone (Scheme 3, Scheme 4).[90-92] The catalyst in this reaction is NaOH, and the solvent is ethanol.
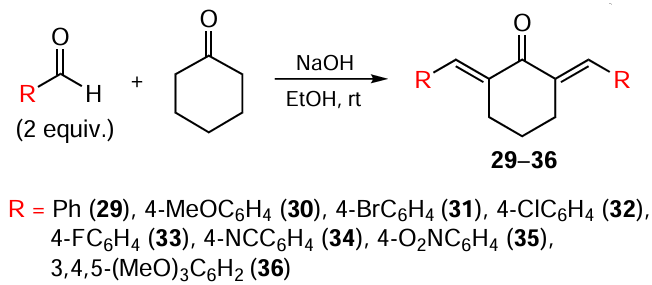
In the study,[93] C5-curcuminoids 15, 16, and 20 were synthesized by condensing acetone with a substituted aldehyde, taken in a 1 : 2 molar ratio, also in the presence of NaOH in EtOH (Scheme 5). The yields of the reaction products ranged from 93% to 96%, with the highest yield observed for compound 20.
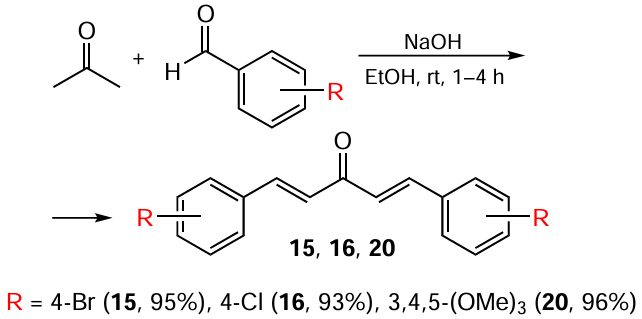
To obtain heterocyclic C5-curcuminoid 37, the authors [93] used thiophene-2-carboxaldehyde; the product yield was 92% (Scheme 6).
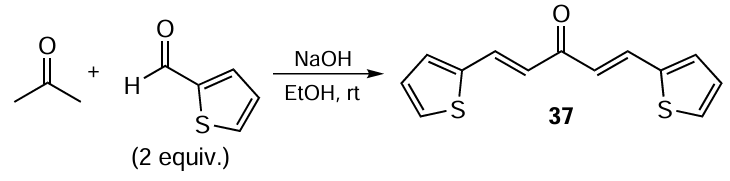
To compare the properties of chalcones, compounds 38 – 41 were prepared, which can be considered as unsymmetrical curcuminoids. In this case, an unsymmetrical ketone, such as acetophenone, was used in the reaction as a substrate in an equimolar ratio to the aldehyde (Scheme 7).[93]
In recent years, particular attention has been paid to heterocyclic MACs based on piperidone. These compounds, unlike C5-curcuminoids and cyclic MACs, are promising basis for the design of cytostatic agents used in the treatment of malignant tumours. This is primarily due to the extensive potential for their structural modification, which can improve their pharmacokinetic parameters and enhance antitumour activity. In particular, modification of the nitrogen atom of the piperidone ring allows the introduction of amide, sulfonamide, nitroxide, and phosphonate groups into the molecule, which are part of many pharmaceutical preparations and are capable of certain interactions with the corresponding receptor.[94-96] For example, in the 1990s, N-acyl-3,5-bis(benzylidene)-4-piperidone (42) was shown to exhibit > 8 times higher cytotoxic activity against P388 leukemia cells compared to 3,5-bis(benzylidene)-4-piperidone (43),[97] its precursor without an acetyl group.
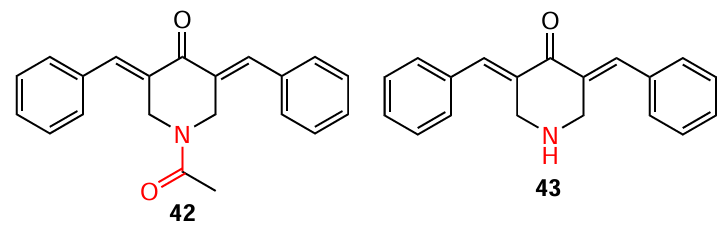
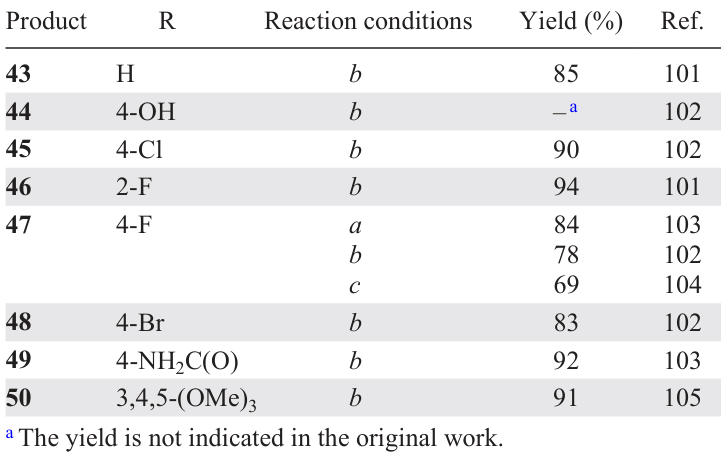
3,5-Bis(arylidene)-4-piperidones, which are cross-conjugated dienones, have found wide application in organic, organoelement and medicinal chemistry.[98] For example, compounds of this class have been used as precursors to new antiviral drugs, including those acting on drug targets of the SARS-CoV-2 coronavirus.[99] A promising trend is the study of the electrochemical behaviour of 3,5-bis(arylidene)-4-piperidones, since it contributes to the understanding of electron transfer processes and the mechanism of pharmacological action of compounds of this class, as well as their possible use in biochemical sensors for neurotransmitters.[100]The synthesis of 3,5-bis(arylidene)-4-piperidones is based on the classical aldol/crotonic condensation of cyclic ketones with two equivalents of aromatic aldehydes. This condensation can be accomplished using 4-piperidone hydrochloride monohydrate and the corresponding aryl aldehyde (1 : 2 ratio) in acetic acid by bubbling dry gaseous HCl through the reaction mixture or by using strong bases and Lewis acids (Scheme 8). To isolate 3,5-bis(arylidene)-4-piperidones 43 – 50 as free bases, the final step involves neutralizing the reaction mixture with a potassium carbonate solution in an acetone – water (5 : 1) mixture. The yields of these products and the conditions for their synthesis are presented in Table 3.[101-105]
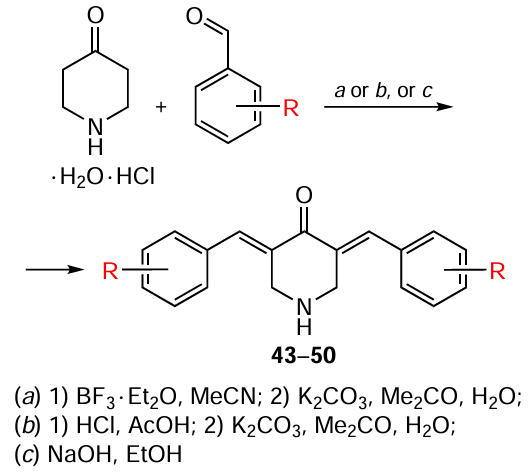
Although this method is quite in demand and widely used for the synthesis of 3,5-bis(arylidene)-4-piperidones, it has several limitations. First of all, it is not applicable to substrates containing labile groups, such as nitrile, phosphonate, and ester functionalities, or heterocyclic moieties (e.g., furanyl), in acidic or basic media. To address these limitations in the synthesis of compounds 43 – 50, catalytic systems are used such as boron trifluoride etherate, MgBr2 Et2O – MeOH – Et3N (see[103]), iodotrimethylsilane,[106] ytterbium trifluoromethanesulfonate (triflate, OTf),[107]lithium perchlorate in the presence of amines,[108]as well as SmI3,[109] RuCl3 (see [110]) and FeCl3 (see [111]) salts in ionic liquids.
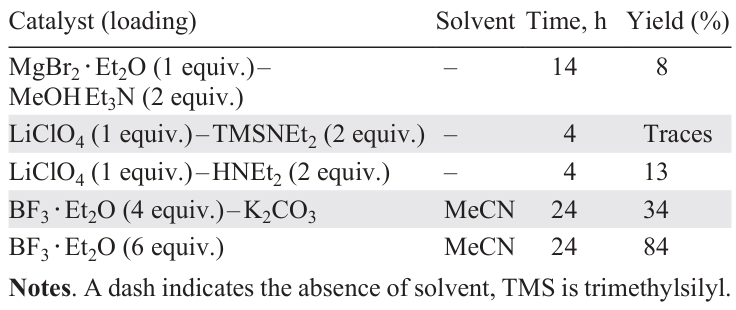
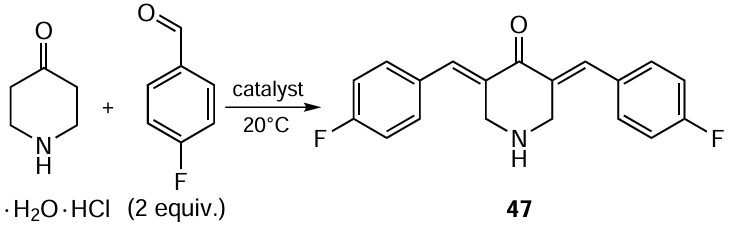
On the example of the condensation of 4-piperidone with p-fluorobenzaldehyde in the presence of Lewis acids and bases, Leonova et al.[103]showed that the use of Yb(OTf)3 or the MgBr2 · Et2O – MeOH – Et3N catalytic system affords compound 47 in low yield (Scheme 9, Table 4). A solvent-free system based on lithium perchlorate and diethylamine also proved ineffective in this reaction, while BF3 · Et2O in acetonitrile provided the target product 47 in high yield (84%).[103]
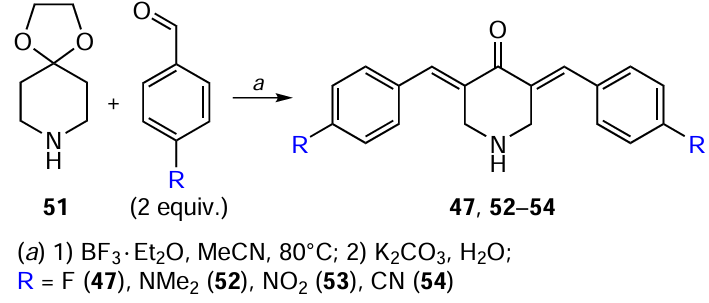
Not only 4-piperidone, but also its derivative with the dioxolane-protected carbonyl group, 2,3-dioxa-8-azaspiro[4.5]decane (51), can serve as a precursor for the synthesis of 3,5-bis(arylidene)-4-piperidones.[112]The reaction of this compound with 4-substituted benzaldehydes was carried out using BF3 · OEt2 (4 equiv.) as the catalyst in dichloromethane or acetonitrile (Scheme 10). To convert the resulting tetrafluoroborate salts into free bases, the reaction mixture was treated with an aqueous solution of potassium carbonate in the final step. The yield of products 47, 52 – 54 decreased in the following order: 52 > 53 > 47 > 54. This fact indicates that the presence of electron-donating groups in the aromatic ring positively affects the yield of the target bis(arylidene)piperidone, unlike electron-withdrawing groups.[103] [112]Thus, the yield of product 47 was as low as 55% after 14 h in boiling acetonitrile.
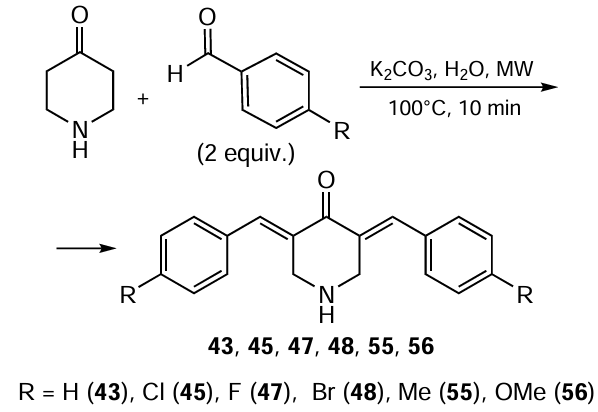
The condensation of 4-piperidone with benzaldehydes can be carried out under 200 W min–1 pulsed microwave irradiation (MW) in an aqueous solution of potassium carbonate as a catalyst or in a solvent-free fashion in the presence of alumina-supported potassium fluoride.[113]The advantages of this synthetic approach to 3,5-bis(arylidene)-4-piperidones are the reaction rate (at most 10 min), high yields of the target products (> 90%), ease of their isolation, and eco-friendly conditions (absence of a solvent and acidic medium).[114]For example, 3,5-bis(arylidene)-4-piperidones 43, 45, 46, 48, 55, 56 were synthesized by microwave-assisted condensation of 4-piperidone with 4-substituted benzaldehydes within 10 min in yields exceeding 90% (Scheme 11).[114]
It should be noted that the methods for producing 3,5-bis(arylidene)-4-piperidones using various catalytic systems are presented primarily in publications dated 2000 and earlier, while in studies from 2020 – 2025, such condensation processes are more often carried out in an acidic medium under microwave irradiation or ultrasonication. This is likely due to the unavailability and high cost of catalytic systems and low yields of the reaction products.
The next two Sections present the most popular chemical reactions for modifying 3,5-bis(arylidene)-4-piperidones. These are transformations involving the piperidine nitrogen atom, which can be roughly divided into two types: classical organic reactions and processes using click chemistry.
* Herein, this structural motif is designated as the C5 chain.
** Herein, the R group in an undefined position of the benzene ring can indicate a different number of substituents (from 1 to 5).
2.2. Classical organic reactions involving the 4-piperidone nitrogen atom
Conjugation of 3,5-bis(arylidene)-4-piperidones with two or more pharmacophoric moieties targeting multiple tumour cell signaling pathways can provide synergistic antitumour activity with a minimum number of side effects compared to single-target drugs. This Section discusses acylation and alkylation reactions that can be used to introduce a variety of substituents into the parent molecule.
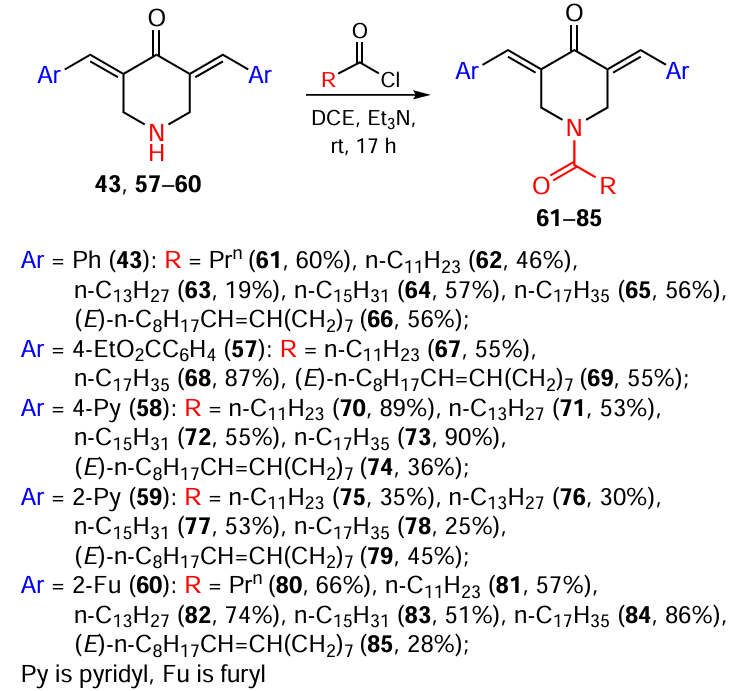
The simplest method for modifying the nitrogen atom of piperidones is their acylation with carboxylic acid chlorides. The reaction run in the presence of triethylamine at room temperature in 1,2-dichloroethane (DCE) (Scheme 12).[115]In this way, products containing residues of saturated (butyric, dodecanoic, tetradecanoic, palmitic, stearic) and unsaturated (oleic) fatty acids at the nitrogen atom were derived from 3,5-bis(arylidene)-4-piperidones 43, 57 – 60. The yields of compounds 61 – 85 varied in the range of 19 – 89%.
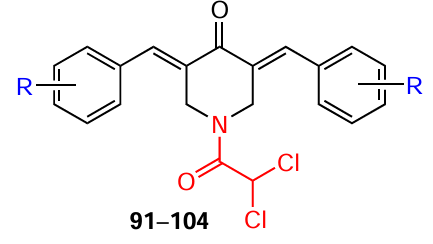
Dichloroacetyl chloride can be used as an acylating agent for NH-piperidone. In this case, the reaction is carried out in chloroform in the presence of triethylamine at room temperature (Scheme 13). As a result, bis(arylidene)-4-piperidones 43, 45 – 47, 50, 52, 53, 55, 56, 86 – 90 give rise to compounds 91 – 104 containing a 2,2-dichloroacetyl moiety at the piperidine nitrogen atom in yields from 60 to 80%, depending on the nature of the substituent on the benzene ring.[116][117] Such products are highly soluble in water and 0.1 M phosphate buffer (at a concentration of 0.4 mg mL–1), making possible studies of their biodegradation in a simulator of the intestinal environment.
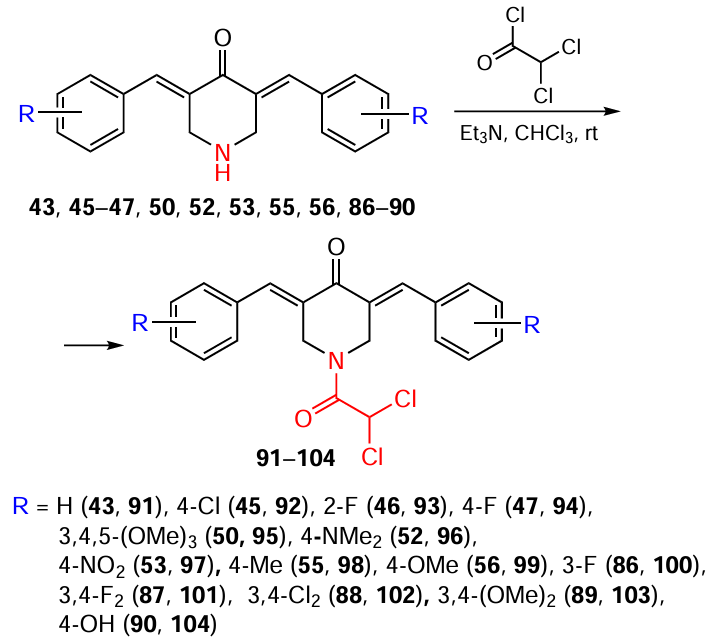
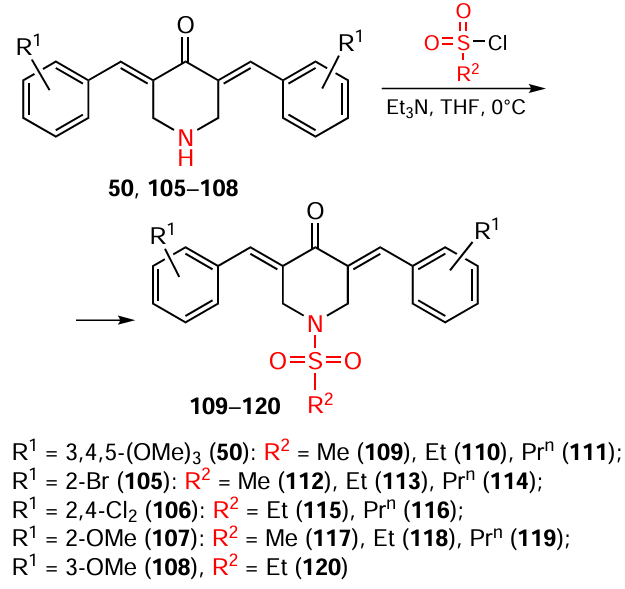
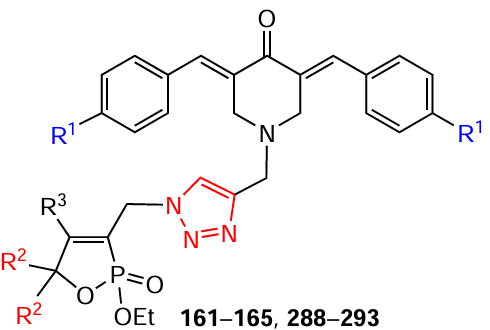
N-Sulfonation of piperidones 50, 105 – 108 with sulfonyl chloride in dry tetrahydrofuran in the presence of triethylamine at 0°C gives N-arenesulfonyl-3,5-bis(arylidene)-4-piperidones 109 – 120 in 70 – 91% yields (Scheme 14).[118]
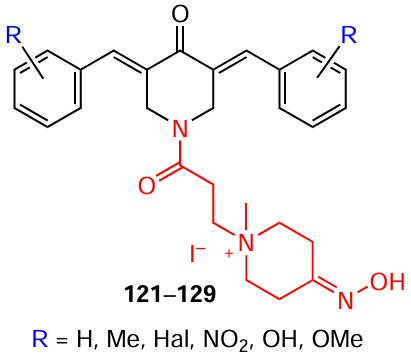
To obtain piperidones containing quaternary ammonium groups in the N-acyl moiety, in the first step, piperidones were acylated with acryloyl chloride, then the corresponding intermediate products were treated with 4-piperidone oxime, followed by quaternization with methyl iodide (Scheme 15; the yields of the compounds in the last step are indicated).[119]As a result, salts 121 – 129 were obtained in good yields.
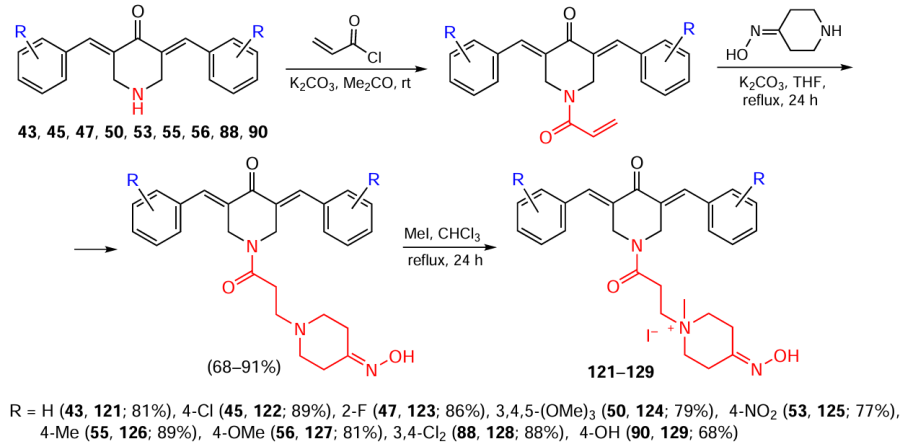
121 – 129 met four of the five criteria (except molecular weight) of Lipinski’s rule,* which specifies the physicochemical properties required for acceptable oral bioavailability of drugs.[119]
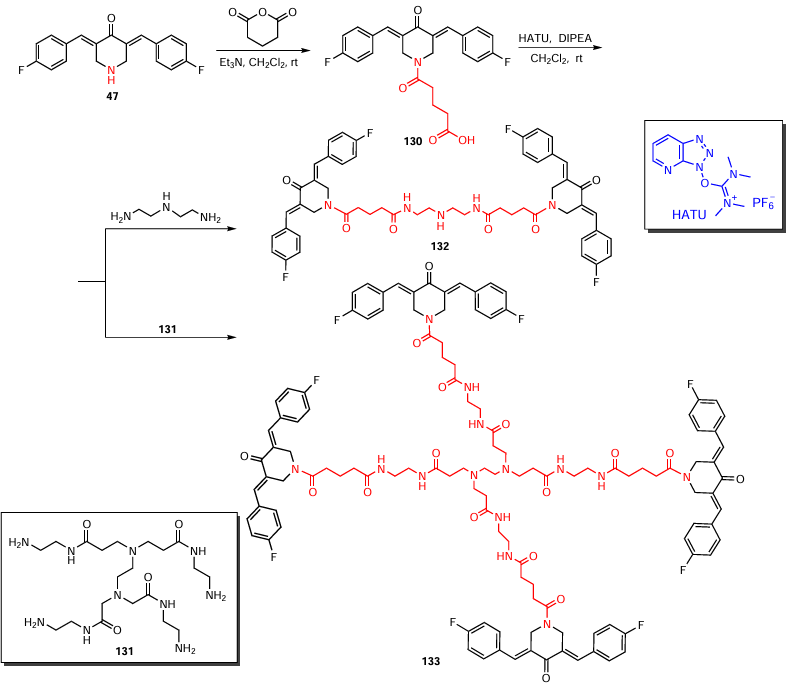
Acylation of piperidone 47 with glutaric anhydride was carried out in the presence of triethylamine in dichloromethane at room temperature, and amides were obtained from intermediate 130 by condensing it with polyamines (diethylenetriamine and oligoamine 131) in dichloromethane (Scheme 16). In the former case, product 132 was obtained, and the use of oligoamine gave dendrimer 133.[120] The authors additionally used an ionic liquid, 2-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate (HATU), as a condensation catalyst, and N,N-diisopropylethylamine (DIPEA) served as a base. The yields of products 132 and 133 were 40 and 46%, respectively. When assessing the hydrophilicity of the amide-containing compounds, unusual results were obtained. The authors of the study [120] found that piperidones 132 and 133 are more hydrophilic (log Po/w from 1 to 3) than the intermediate piperidone 130 (log Po/w > 3). Consequently, these compounds will be more soluble in water and characterized by higher bioavailability.
Piperidones bearing a hydrocarbon chain with terminal bisphosphonate groups at the nitrogen atom were obtained through a multistep transformation sequence. As the starting compound, the protected 4-piperidone 51 was used, which was first alkylated with 2-chloroethanol and then converted to the chloroethyl derivative 134. The key step was based on its reaction with tetraethyl ethylidenebisphosphonate, followed by crotonic condensation of intermediate 135 with aromatic aldehydes and pyridine-4-carbaldehyde (Scheme 17).[121]However, in this case, target products 136 – 139 were formed in low yields (£ 30%) due to hydrolysis of the ethoxy groups at the phosphorus atom.
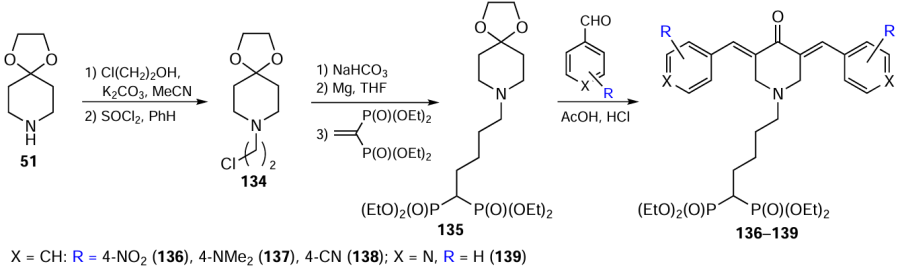
Despite the interest of researchers in classical methods of modifying piperidones, such methods are not without drawbacks, associated in some cases with multistep procedures, the need to remove excess reagent, or product loss due to its transition to the aqueous phase during the washing treatment, which reduces the yields.[120][121]
* The ‘rule of five’ formulated by Lipinski (see V.Ivanović, M.Rančić,B.Arsić, A.Pavlović. Pop. Sci. Art., 3 (1), 171 (2020)) includes the following parameters of the molecule: molecular weight ≤ 500, number of rotatable bonds ≤ 10, hydrogen bond acceptors ≤ 10, hydrogen bond donors ≤ 5 and partition coefficient in the octanol/water system (logPo/w) ≤ 4.15, which serves as a measure of the ratio of lipophilicity (solubility in fats) and hydrophilicity (solubility in water) of a substance.
2.3. Transformations based on ‘click’ chemistry methodology
In recent years, synthetic chemists have frequently used the ‘click’ methodology developed in 2001 by Sharpless.* This methodology is a set of highly efficient and selective reactions that rapidly connect molecular building blocks, usually carried out in an environmentally friendly solvent (or in the absence of one), and are characterized by stereospecificity and high product yield.[122]Click chemistry has become a powerful tool in organic and medicinal chemistry, enabling the easy, regio- and stereoselective production of potential medicinal agents, including cytostatics. The resulting 1,2,3-triazole ring is not simply a passive linker, but rather enhances the compound’s solubility in water and facilitates its binding to biological targets through hydrogen bonding and dipole-dipole interactions.[123]
The Huisgen reaction (a copper(I)-catalyzed 1,3-cycloaddition of azides to terminal alkynes) is widely used to synthesize MAC-based conjugates. This reaction produces 3,5-bis(arylidene)-4-piperidone derivatives in relatively high yields, with the isolation and purification of the desired products being simple.
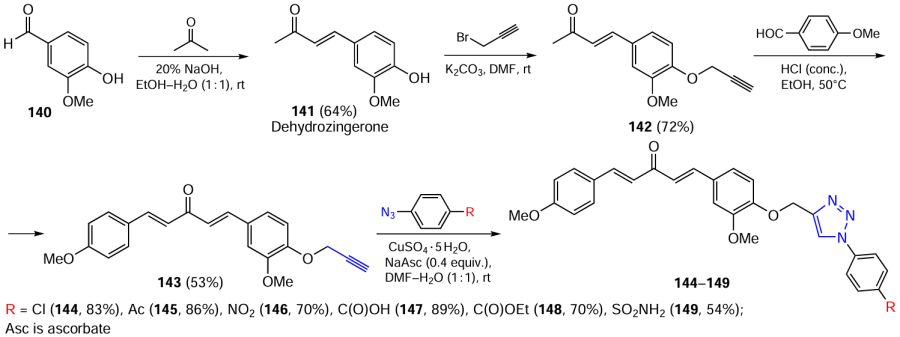
Unsymmetrical C5-curcuminoids with an aromatic moiety decorated with a triazole ring were synthesized via the reaction sequence shown in Scheme 18.[124]First, the aldol condensation of vanillin (140) with acetone was carried out at an equimolar ratio of reactants, then dehydrozingerone (141) was alkylated with propargyl bromide, and intermediate product 142 was condensed with anisaldehyde. The resulting unsymmetrical C5-curcuminoid 143 was then reacted with aryl azides. The yields of products 144 – 149 were 70 – 89%, depending on the nature of the substituent on the aromatic ring.
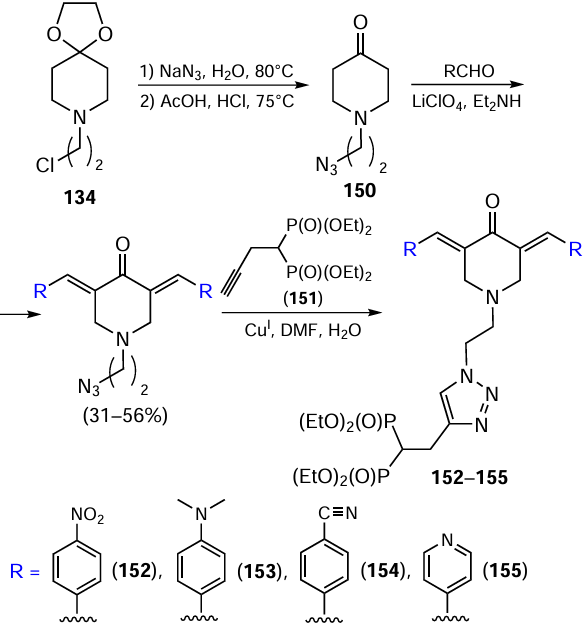
A large number of 3,5-bis(arylidene)-4-piperidone derivatives have been described, in which the phosphate group is attached to the heterocycle via a 1,2,3-triazole spacer. Three approaches to such compounds are used. For example, the authors of the study [121] introduced a bisphosphonate moiety into the piperidone molecule via a click reaction (Scheme 19). For this purpose, azide 150 was first obtained, which was condensed with various aldehydes to give the corresponding bis(arylidene) derivatives. The latter were used as components for 1,3-cycloaddition to bis(phosphonate)-substituted alkyne 151. At this step, the yields of compounds 152 – 155 were > 80%.
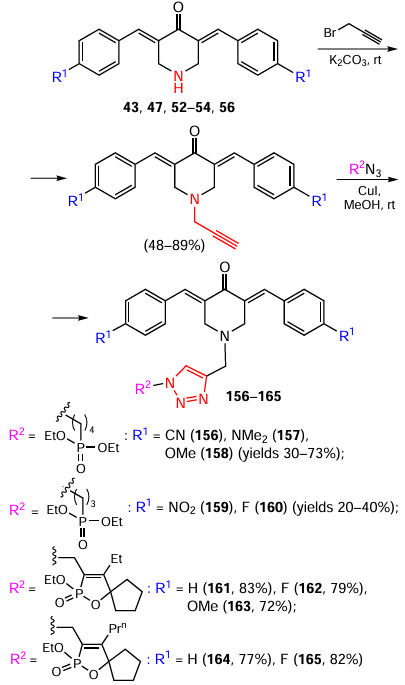
The second approach involves N-alkylation of the appropriate 3,5-bis(arylidene)-4-piperidones with propargyl bromide and their subsequent [2 + 3] cycloaddition with phosphonate-containing azides. Products 156 – 165 were obtained in yields ranging from 20 – 83%, depending on the starting azide (Scheme 20).[125][126]
The third approach is based on the initial alkylation of NH-piperidone with propargyl bromide. This is followed by cycloaddition of the corresponding azides to the terminal alkyne 166, and the final step is an aldol condensation involving 4-substituted benzaldehydes to afford products 156, 159, 160, 167 – 169 (Scheme 21).[125][127]
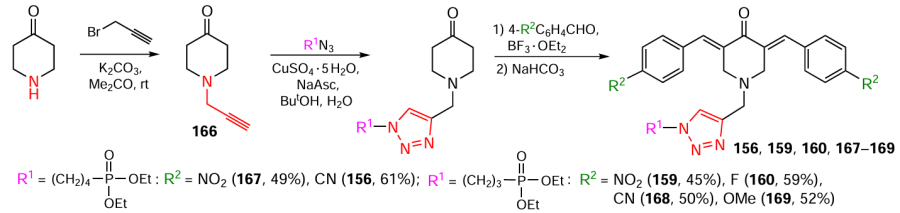
It is difficult to give preference to any one of the above methods for the synthesis of 1,2,3-triazole-substituted piperidones in terms of the product yield.
It should be noted that compounds 156 – 165, 167 – 169 (Refs 125 – 127) easily form stable complexes with inorganic salts (CaCl2, NaCl, Na2SO4, MgSO4) due to the presence of a phosphoryl group and a triazole ring in the molecule. Therefore, when isolating individual compounds, the use of drying and salting-out agents should be avoided. To remove copper(I) ions from the reaction mixture, treatment with an aqueous ammonia solution,[128]a 0.1 M ethylenediaminetetraacetate solution,[129]reversed-phase chromatography [130]or elution through neutral alumina [131]is usually used, although none of these methods provide their complete elimination.
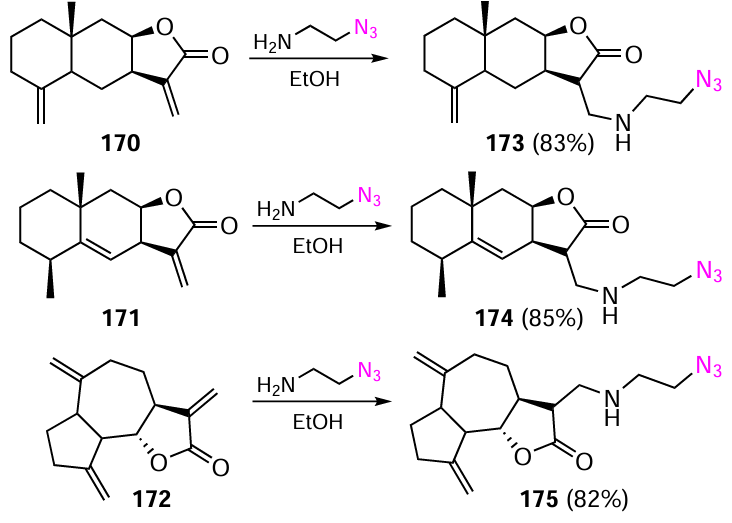
As a basis for the development of new antitumour drugs, the authors of the study [132]obtained a series of conjugates in which the pharmacophoric 3,5-bis(arylidene)-4-piperidone scaffold and a natural sesquiterpene lactone were linked via a triazole ring. For this purpose, an azide group was first introduced into the molecules of isoalantolactone (170), alantolactone (171), and dehydrocostuslactone (172). 2-Azidoethylamine was used as a reagent, which readily added to the activated exomethylene group of the lactones (Scheme 22).[132]The reaction proceeds at room temperature in ethanol to give azides 173 – 175 in high yields.
Next, bis(arylidene)piperidones 176 – 178, containing a N-acetylene moiety, were added to these azides using copper(I) bromide and DIPEA as a catalytic system (Scheme 23).[132]
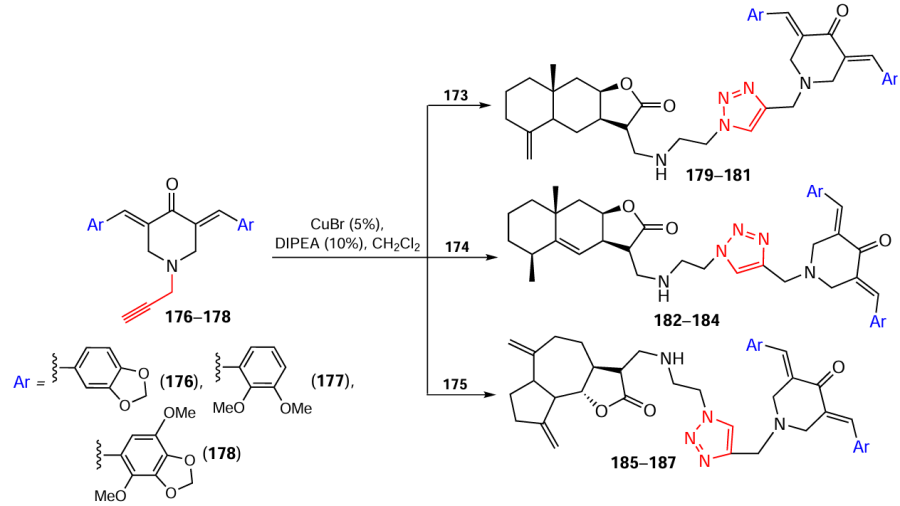
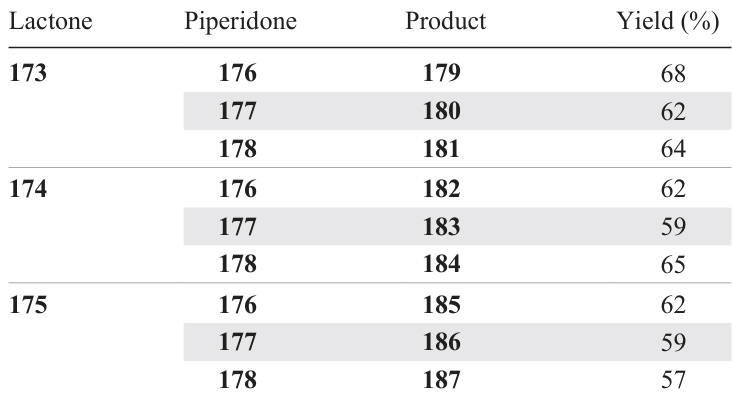
The target hybrid compounds 179 – 187 were isolated (Table 5) by column chromatography and characterized by spectral methods.
N-(1-Benzyltriazol-4-ylmethyl)-substituted piperidones were synthesized from piperidones 188 – 190 prepared in advance (Scheme 24).[133]The reaction with benzyl azides was carried out in the presence of copper(II) sulfate and sodium ascorbate. To remove excess benzyl azide, the product was washed several times with ethyl acetate and water. The yields of products 191 – 202 ranged from 70% to 85% and depended on the nature of the R1 and R2 substituents.
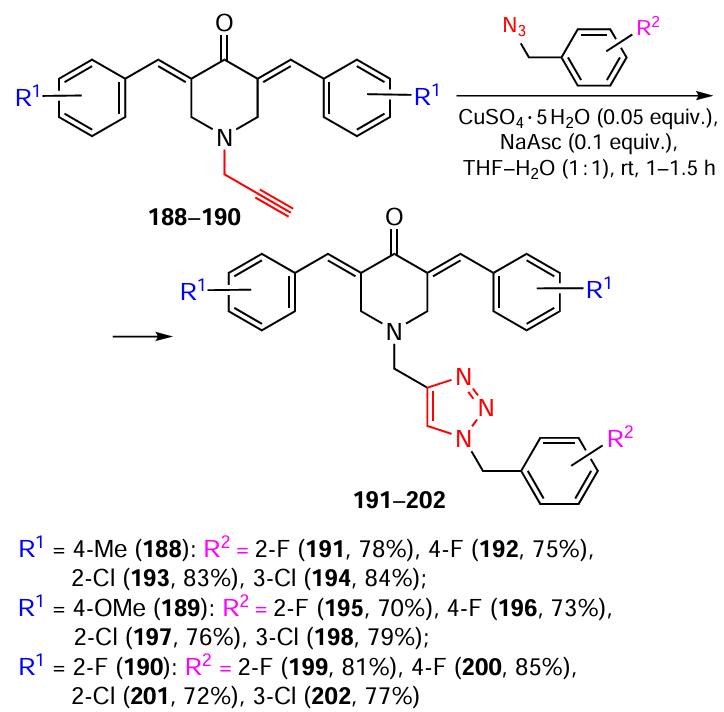
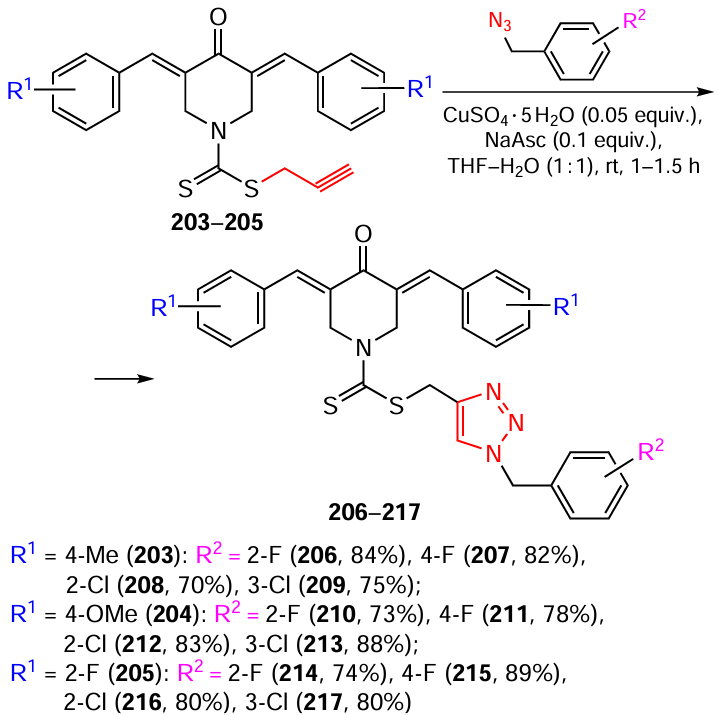
The synthesis of thiocarbamate-substituted N-(triazol-4-yl)bis(arylidene)piperidones from alkynes 203 – 205 by a similar method has also been reported.[133]The yields of products 206 – 217, which ranged from 70 – 89%, were affected by the nature of substituents on the benzene rings of both piperidone and azide (Scheme 25).
Therefore, the chemistry of monocarbonyl analogues of curcumin, including 3,5-bis(arylidene)-4-piperidones, is a promising area of organic chemistry. Developed procedures for structural modification of 3,5-bis(arylidene)-4-piperidones provide the high yields of the target compounds. Furthermore, parameters such as the number of reaction steps, solubility in water, and reagent cost are of great importance for the future use of such derivatives in pharmacy practice. Among the known methods for the synthesis of 3,5-bis(arylidene)-4-piperidones, choosing the most efficient one is extremely difficult. Since such compounds are typically obtained and structurally modified for a specific purpose, such as improving their pharmacological profile or enhancing a specific type of activity, it is no sense to compare different synthetic approaches. However, it should be noted that the use of click chemistry methodology in the synthesis of effective hybrid molecules has a number of advantages, primarily associated with the introduction of a triazole ring into the structure, which serves not only as a connecting unit, but also as an additional pharmacophoric group capable of affecting the pharmacokinetic and pharmacodynamic characteristics of the system as a whole.
* See H.C.Kolb, M.G.Finn, K.B.Sharpless. Angew. Chem., Int. Ed., 40 (11), 2004 (2001).
3. Mechanisms of antitumour action of synthetic analogues of curcumin
This Section presents an analysis of the literature on the biological effects of MACs. Particular attention is given to the classification of molecular targets that serve as potential therapeutic targets for compounds designed for use as antitumour agents. Taking into account the specific pathogenesis of malignant tumours, the influence of MACs on two key processes of carcinogenesis, viz., angiogenesis and the growth and proliferation of neoplastic cells, is discussed.
3.1. Effect on angiogenesis
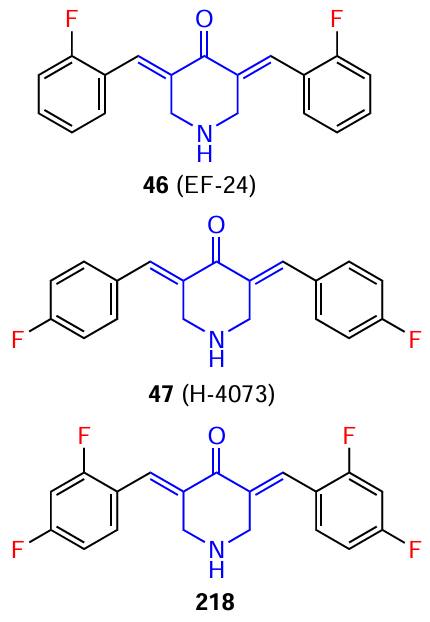
Studies investigating the effect of curcumin on angiogenesis were published as early as the first decade of the 21st century.[134][135] Like curcumin per se, its analogues also modulate key molecular pathways associated with the new blood vessel growth in tumours, limiting their blood supply.[136]For example, Subramaniam et al.[137]demonstrated the ability of 3,5-bis(2,4-difluorobenzylidene)-4-piperidone (218) to inhibit the formation of CD31-positive blood vessels in vivo in a mouse model of pancreatic cancer tumour xenografts. This effect is due to a decrease in the expression of VEGF and IL-8 proteins, which are potent promoters of the formation of new vessels that feed the tumour.[138]
Synthetic curcumin analogues 46 and 47 with the commercial codes EF-24 and H-4073, respectively, are among the curcuminoids with potential angiogenesis-inhibitory properties and the ability to suppress VEGF expression in cancer cells. For example, Kumar et al.[139]showed that piperidone 47 reduces VEGF levels by 36% in UM-SCC-74A head and neck tumour cells. Although there are no current data in the literature on the direct role of EF-24 and H-4073 in modulating tumour neovascularization, it can be assumed that, being effective inhibitors of VEGF,[140]as well as modulators of the NF-κB[141][142] and MAPK [143][144] signalling pathways, they are capable of inhibiting endothelial cell proliferation and migration. Consequently, this may lead to a decrease in tumour vascular density and a significant restriction of tumour blood supply, preventing their growth and metastasis.
Currently, the suppression of pathological angiogenesis using curcuminoids has received little attention, despite the potential of such research. A more in-depth understanding of the mechanisms of action and the active use of curcuminoids as inhibitors of pathological angiogenesis could play a significant role in boosting the effectiveness of treatment of cancer patients.
3.2. Effect on growth and proliferation of tumour cells
3.2.1. Nuclear molecular targets
In the search for new antitumour agents, some of the most frequently studied nuclear molecular targets are topoisomerases — enzymes that play an important role in processes associated with maintaining the correct structure of DNA in the cell.[145] In the last century, inhibition of topoisomerase II was established as the primary mechanism of action of the drugs amsacrine, etoposide, and doxorubicin, which are successfully used to treat malignant neoplasms.[146]In recent years, this trend has continued, and many new and already approved anticancer drugs target topoisomerases.[147-149] It has been found that inhibition of these enzymes is one of the mechanisms of cytotoxic action of a number of curcumin analogues.[115]
Fatty acids are known to be rapidly taken up by tumour cells and readily incorporated into the lipid bilayer of membranes, disrupting their fluidity and structure.[150]As demonstrated in Section 2, one promising avenue for modifying piperidones is their N-acylation. The introduction of essential fatty acid residues into the molecule increases its tumour-specificity compared to normal cells and enhances its ability to penetrate the extracellular membrane.[151]
Compounds 61 – 85 (Ref.[115]), conjugates of piperidones and fatty acids with different hydrocarbon chain lengths (see Scheme 12 and Table 5), turned out to be potent catalytic inhibitors of topoisomerase IIα in vitro, along with the reference antitumour drug sorafenib.
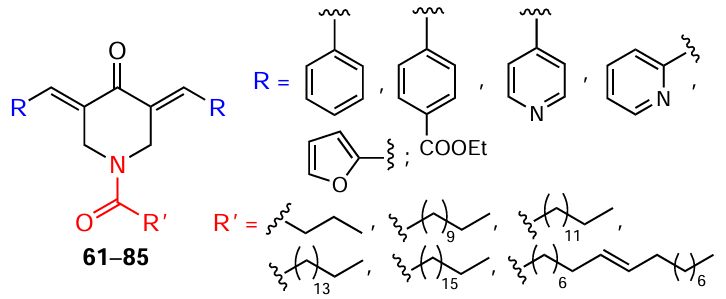
The findings reported in the cited study [115]on the inhibitory activity of these compounds directly correlated with the presence of cytotoxicity against a panel of following tumour cell cultures: CEM (human T-lymphocytes), HeLa (human cervical cancer), and L1210 (murine lymphocytic leukemia). Acylated piperidones 70 – 74 containing a 4-pyridyl substituent (IC50 in the range of 0.8 – 14 μM) demonstrated the highest efficiency. At the same time, compounds 80 – 85 with an electron-donating furan ring showed low cytotoxicity (IC50 = 47 – 250 μM). The authors attribute this fact to a decrease in the electrophilicity of the piperidone acceptor system, which adversely affects their thiolating ability. In turn, products 61 – 85 showed low toxicity against normal human fibroblast WI-38 cells, which is an advantage of the drug.[115]
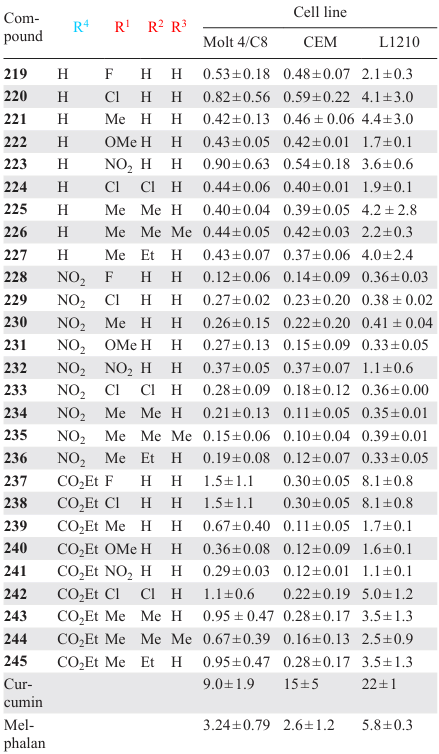
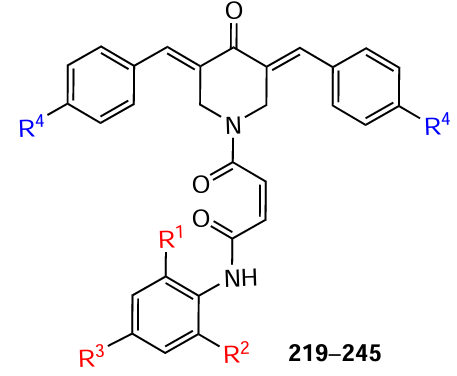
Piperidones 219 – 245, containing a maleic acid-based diamide moiety, were described.[152]These compounds demonstrated potent cytotoxicity against Molt 4/C8 and CEM human T lymphocytes, as well as against L1210 murine lymphocytic leukemia cells. The cytotoxic activity of these MACs exceeded that of curcumin and melphalan, as evidenced by lower IC50 values (Table 6). The authors confirmed that these compounds are effective inhibitors of topoisomerase IIα, which is considered to be the primary molecular target of their action.
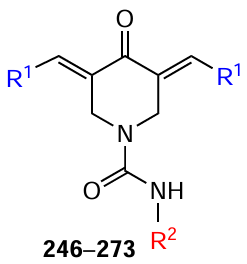
A significant inhibitory effect against DNA topoisomerase IIα was also demonstrated by carboxamide-substituted piperidones 246 – 273, obtained by Fawzy et al.[153]Most of the resulting compounds exhibited high cytotoxic activity against tumour cell lines such as HCT116 (human colon cancer), MCF-7 (breast ductal adenocarcinoma), and A431 (squamous cell carcinoma) (Table 7). The authors concluded that piperidones of this series containing electron-withdrawing groups (e.g., chlorine or fluorine atoms) in the benzene ring are more active than their analogues with electron-donating groups (methyl or methoxy).
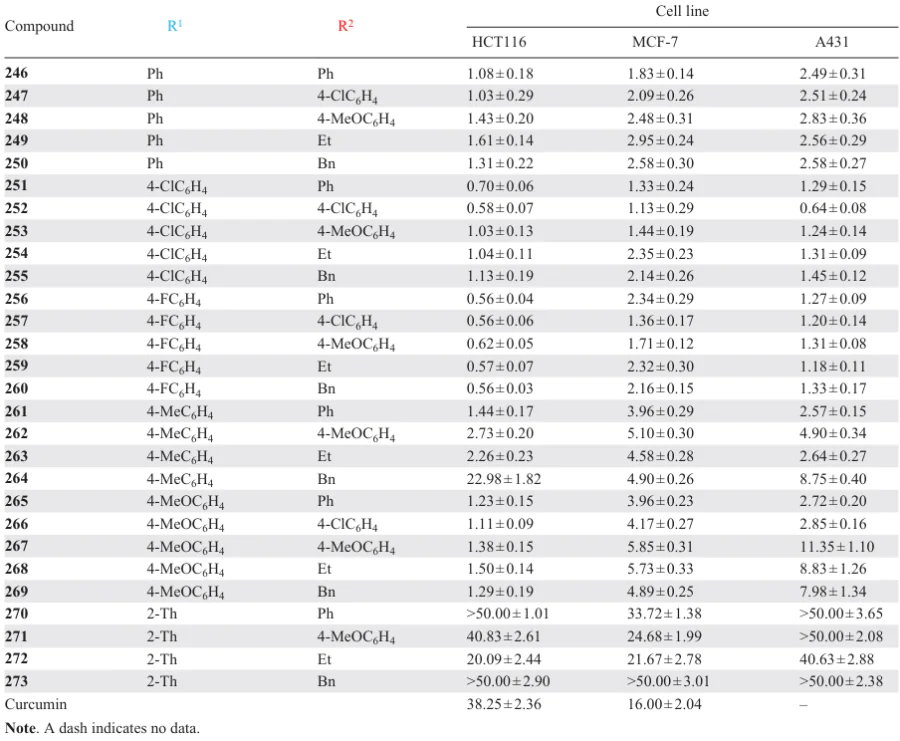
At the same time, the presence of a benzylidene moiety in the structure is preferable for activity compared to a heterocyclic (2-thienylidene) moiety, which should be taken into account when designing antitumour drugs. Furthermore, the cytotoxicity of piperidones 246 – 273 against the tumour lines studied was superior to that of curcumin.
Other important and no less popular targets for potential anticancer drugs include the following:
— poly(ADP-ribose) polymerase (PARP), an enzyme involved in various cellular processes, including DNA repair and cell cycle regulation;
— signal transducer and activator of transcription 3 (STAT3), a STAT family protein that mediates cellular responses to signals from interleukin receptors and tumour growth factors;
— the transcription factor NF-κB, which plays an important role in the cell cycle and apoptosis.
Modulators of these protein molecules are effectively used in combination cancer therapy for together with first-line drugs, such as cisplatin, since they have a different way of acting on the tumour cell, which allows for an enhanced positive effect of treatment.[154-156]
The enzymes listed above and the transcription factor NF-κB are also targets for synthetic analogues of curcumin. For example, it was shown[117] that bis(arylidene)piperidones 43 and 97 at concentrations < 1 μM cleave PARP, which contributed to the cytotoxic effect of these derivatives on Ca9-22, HSC-2, HSC-3, and HSC-4 oral squamous cell carcinoma cell lines. The IC50 values of these compounds were in the nanomolar range (Table 8) and were lower than those of the antitumour drug 5-fluorouracil, i.e., they are more potent.
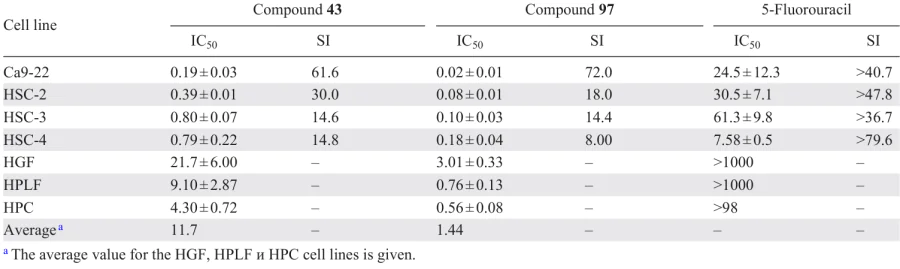
It should be noted that compounds 43, 97 were less toxic to normal HGF (human gingival fibroblasts), HPLF (human periodontal ligament fibroblasts) and HPC (human pulp cells) cells compared to malignant cells and showed high selectivity index* values, which is an important factor in the development of antitumour drugs.
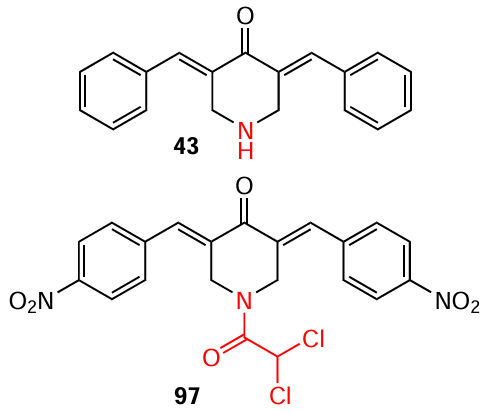
It was found that bis(arylidene)-4-piperidone 47 (H-4073) and its pyrroline-containing conjugate 274 (commercial code HO-3867) effectively inhibit phosphorylation of the activator STAT3.[157] Due to this, they exhibit targeted cytotoxicity against human pancreatic adenocarcinoma (AsPC-1) cells, causing cell cycle arrest in the G2/M phase and apoptosis. Compounds 47 and 274 at concentrations of 1.56 and 12.5 μM, respectively, led to > 60% cell death in the in vitro tests.
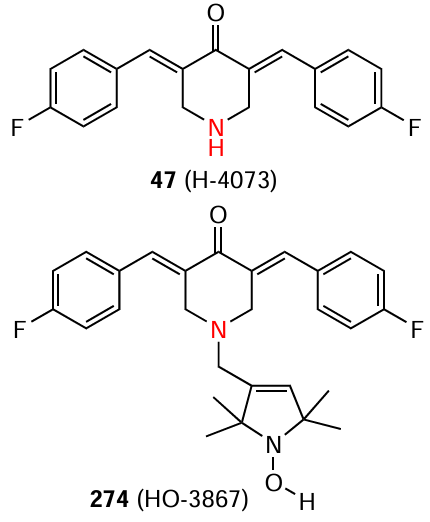
Unusual results on STAT3 inhibition were obtained by Mao et al.[120] The authors Dimer 132 and tetramer 133 in which bis(arylidene)-4-piperidone moieties were connected by oligoaminoamide bridges were synthesized and studied as potential antitumour agents.
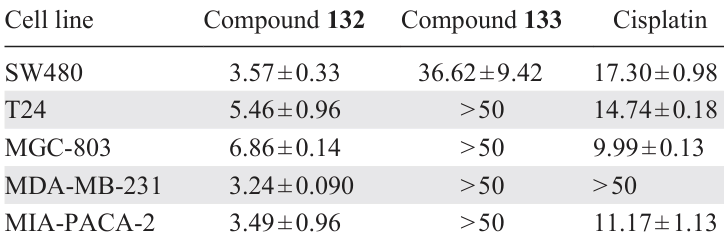
To study the STAT3 inhibition capacity, five cell lines (human colon adenocarcinoma SW480, bladder carcinoma T24, gastric cancer MGC-803, breast cancer MDA-MB-231, and human pancreatic adenocarcinoma MIA-PACA-2) were treated with compounds 132 and 133 at sequential gradient concentrations (1.5, 3, and 6 μM) for 24 h. The dimeric derivative 132 proved to be the most promising: it effectively inhibited STAT3 expression and thereby inhibited tumour cell growth in all five cell lines. Furthermore, the cytotoxic activity of compound 132 exceeded that of cisplatin (Table 9).
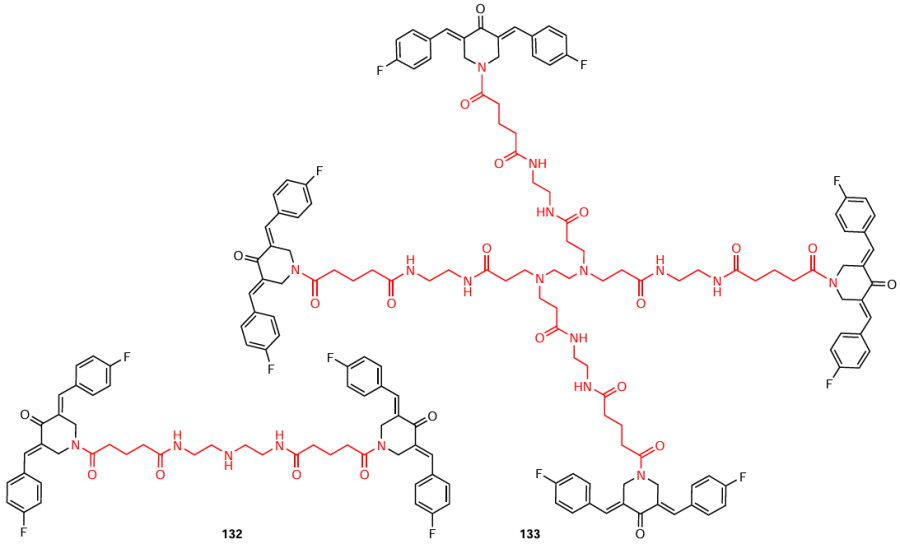
In vivo experiments using the SW480 xenograft model also confirmed the antitumour activity of dimer 132. Mao et al.[120]demonstrated a tumour mass reduction of more than 40% when animals were treated with this compound at a dose of 10 mg kg−1 and identified derivative 132 as an effective candidate for antitumour agents with a mechanism of action via STAT3 inhibition.
The same research group [158]synthesized an oxaliplatin derivative, a platinum(IV) complex 275 with a bis(arylidene)-4-piperidone moiety in one of the ligands. This system contains two pharmacophores and is capable of simultaneously acting on tumours by two different mechanisms, viz., STAT3 inhibition (piperidone) and DNA intercalation (platinum compound).
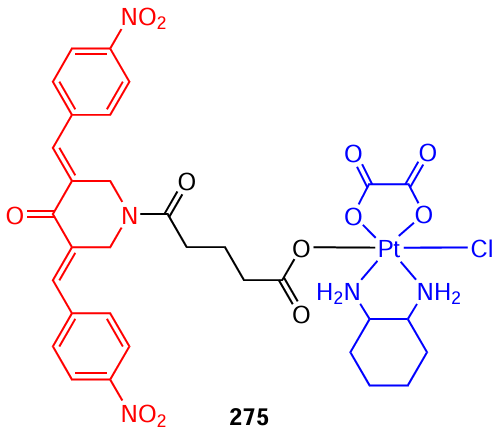
Complex 275 exhibited high antiproliferative activity against SW480 cells (human colon cancer) with IC50 = 12.79 μM.[158]The authors believe that it may have a chemoimmunotherapeutic effect by inhibiting the expression and phosphorylation of STAT3, thereby evoking CD4+ and CD8+ T lymphocyte immune responses, and inducing ferroptosis and apoptosis in tumour cells.
In conclusion of this Section, it is worth noting the aforementioned 3,5-bis(2-fluorobenzylidene)-4-piperidone (46, trade name EF-24). This compound has superior pharmacokinetic properties to curcumin, exhibits increased bioavailability, and displays antitumour and antiangiogenic activity while maintaining minimal toxicity to normal cells. Its high efficiency in inhibiting tumour cell growth is believed to be due to increased oxidative stress and inhibition of NF-kB, phosphoinositide 3-kinase, and the mitogen-activated protein kinase pathway, as well as regulation of HIF-1α expression (HIF-1α is a subunit of a heterodimeric transcription factor HIF-1).[159-161] Compound EF-24 has successfully passed preclinical trials and is planned for further studies as a new antitumour drug.[162]
* The selectivity index (SI) is the ratio of the IC50 value of a drug on normal cells to that on tumour cells.
3.2.2. Mitochondria-targeted molecular targets
For many years, mitochondria have firmly maintained their status as a key target for anticancer drugs.[163][164] This is due to the central role these organelles play in regulating cellular metabolism, energy synthesis, and initiating apoptosis[165] —processes that are essential for the survival of neoplastic cells. The properties of mitochondria provide unique opportunities for selectively targeting agents specifically to tumour cells. Therefore, mitochondria-targeted curcuminoids deserve special attention.
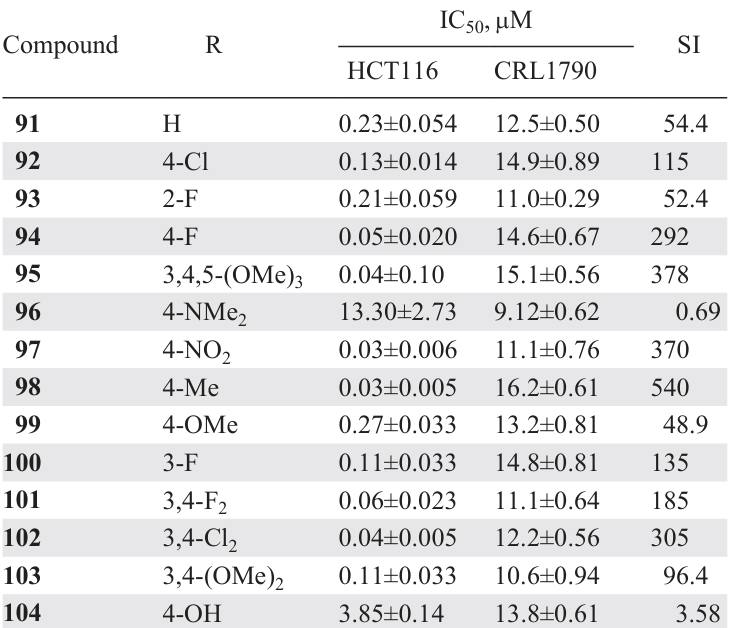
N-Dichloroacetyl-3,5-bis(arylidene)-4-piperidones 91 – 104 (see Scheme 13 ) are promising candidates for antitumour agents.[116]While highly toxic molecules toward the HCT116 tumour line with IC50 values in the nanomolar range (Table 10), most compounds (except for the dimethylamino derivative 96) were less toxic to the conditionally normal human colon cells CRL1790. When elucidating the mechanism of action of these compounds, the authors found that they are capable of reducing the potential of the mitochondrial membrane and leading to hyperproduction of reactive oxygen species in HCT116 cells, causing a therapeutic effect.
A similar mechanism of action has been described for unsymmetrical 3,5-bis(arylidene)-4-piperidones 276 – 287.[166]
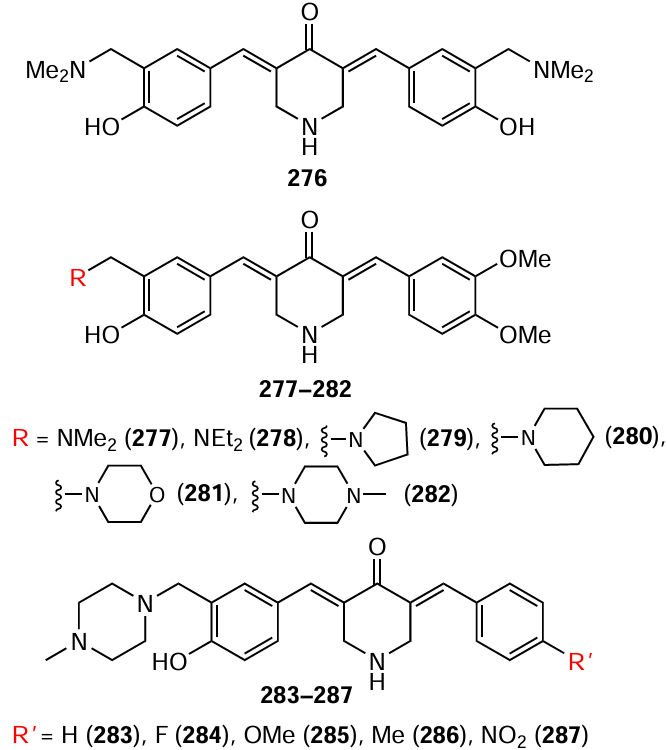
These compounds exhibited potent cytotoxic activity against HCT116 and HT29 colon cancer cell lines, as well as oral squamous cell carcinoma Ca9-22, HSC-2, HSC-3, and HSC-4 (Table 11), which was attributed to their ability to induce mitochondrial membrane depolarization and subsequent cell death via apoptosis.
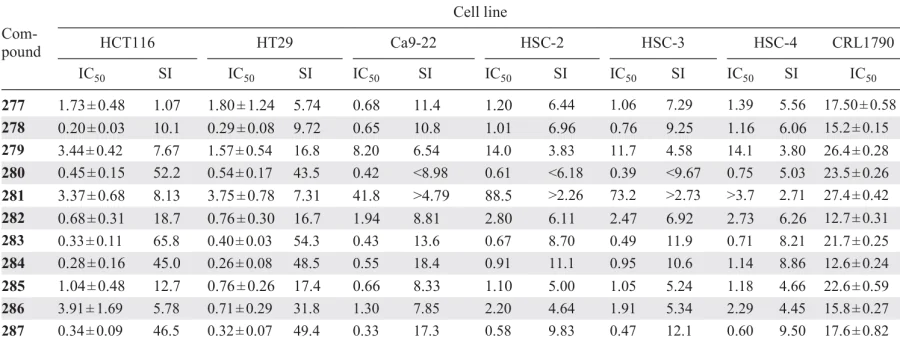
It is worth emphasizing that, similar to the aforementioned dichloroacetyl derivatives 91 – 104, compounds 277 – 287 showed reduced toxicity to normal CRL1790 cells (see Table 11). This selectivity is likely related to a mechanism of action based on modulation of mitochondrial function in cancer cells. Mitochondria are known* to be actively involved in tumour cell survival under stressful conditions, including hypoxia and nutrient deficiency, making them particularly vulnerable to various factors. Apparently, this is why mitochondria-targeting molecules preferentially affect neoplastic cells, minimizing damage to the healthy microenvironment.
In 2021, Roayapalley et al.[119]obtained and characterized a series of quaternary ammonium salts 121 – 129 with a similar mechanism of action (see Scheme 15).
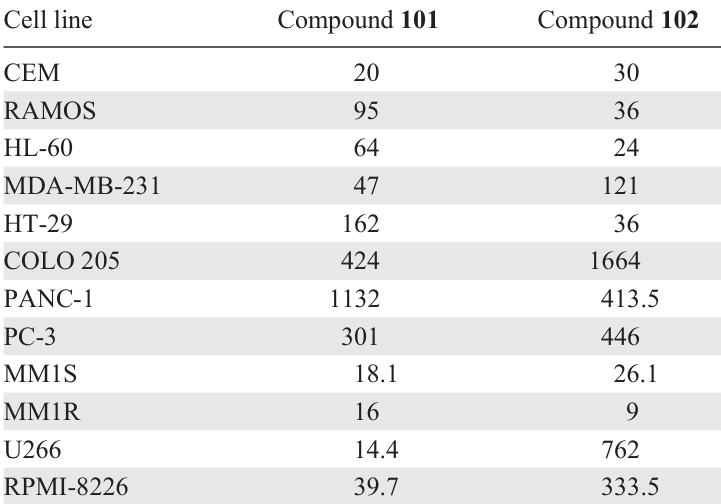
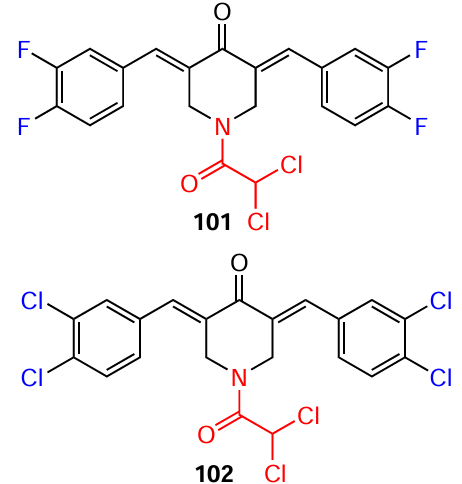
It has been established that apoptosis via the mitochondrial pathway is initiated primarily by various caspases. It was found[167] that N-dichloroacetyl-3,5-bis(3,4-dihalobenzylidene)-4-piperidones 101 and 102 exhibit cytotoxic activity in a wide panel of cell lines with IC50 values in the nanomolar range (Table 12). Tests were carried out on 12 cell lines: CEM T-lymphoblast cells, RAMOS B-lymphocytes, HL-60 promyelocytic leukemia, MDA-MB-231 breast adenocarcinoma, HT-29 and COLO 205 colorectal adenocarcinoma, PANC-1 pancreatic carcinoma, PC-3 prostate adenocarcinoma, and MM1S, MM1R, U266, and RPMI-8226 multiple myeloma. The authors found that the mechanism of toxic action of compounds 101 and 102 involves the activation of caspase 3 (or 7) and the induction of apoptosis.
Mandalapu et al.[133]showed that bis(arylidene)piperidones, in which the 1-benzyltriazole ring is linked to the nitrogen atom by methylene (191 – 202) and thiocarbamate (206 – 217) bridges (see Scheme 24, Scheme 25 ), exhibit high cytotoxicity against tumour cells, in particular breast (MCF-7, MDA-MB-231, and 4T1) and prostate (PC-3, DU-145) adenocarcinomas. The level of activity depended largely on the nature of the substituent on the benzene ring of the piperidone.
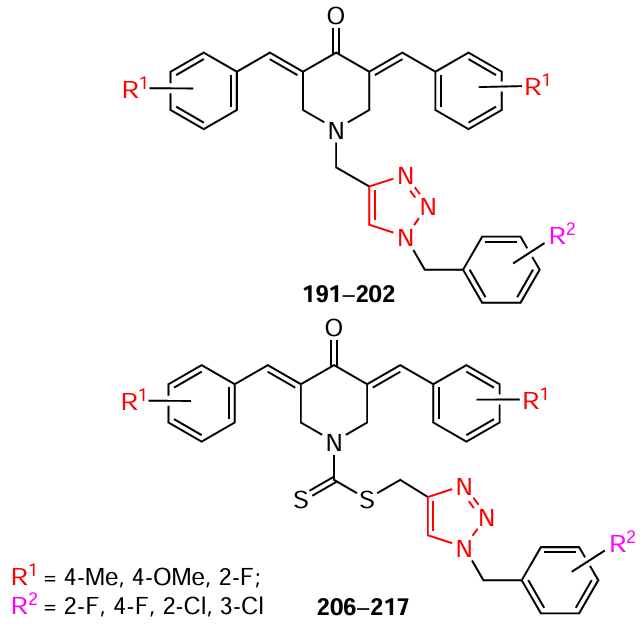
Compounds 191 and 206 (Table 13), containing methyl groups at the para-position of the benzene ring of piperidone and fluorine atoms at the ortho-position of the benzyl substituent, were shown to be the most effective against the studied cell lines. These compounds induced cell cycle arrest and mitochondrial apoptosis in these cell lines by modulating the expression levels of various signaling proteins, such as Akt, PCNA, Bax, and Bcl-2.[133]
These results indicate the great therapeutic potential of curcuminoids modified to selectively target cancer cell mitochondria, which may lead to the development of highly potent antitumour drugs based thereon.
* See C.L.Kuo, A.Ponneri Babuharisankar, Y.C.Lin, H.W.Lien, Y.K.Lo, H.Y.Chou, A.Y.L.Lee. J. Biomed. Sci., 29(1), 74 (2022).
3.2.3. Biochemical processes and molecular targets of the cytoplasmic space
This Section discusses the effects of synthetic curcumin analogues on processes associated with tumour cell metabolism and cytoskeletal health. For many years, the authors of this review have focused on the search for promising antitumour agents among conjugates of piperidones and various pharmacophores. In particular, good results in terms of cytotoxicity were obtained for 3,5-bis(arylidene)-4-piperidones 179 – 187, modified with sesquiterpene lactones (see Scheme 23 ).[132]
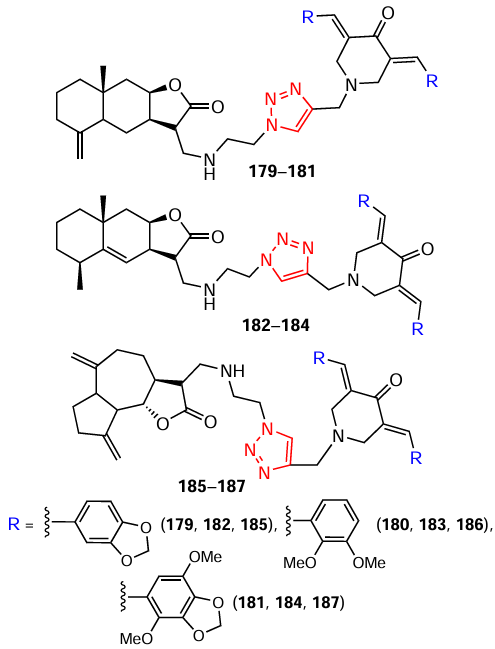
The study of biological properties of conjugates 179 – 187 revealed that they exhibit a selective cytotoxic effect against tumour cells of the epithelial adenocarcinoma of the mammary gland MCF-7, neuroblastoma SH-SY5Y and IMR-32, and human cervical adenocarcinoma HeLa (IC50 values range from 5 μM for the most potent substances) due to the inhibitory effect on the process of glycolysis, the main metabolic pathway of neoplastic cells.[132]Using in silico molecular screening, the ability of these conjugates to bind to the allosteric centre of the glycolytic enzyme pyruvate kinase M2, thereby reprogramming the tumour’s metabolic state, was demonstrated. This mechanism of action for piperidone derivatives was found for the first time in this study, expanding the antitumour potential of this class of compounds and laying the foundation for further exploration of new monocarbonyl analogues of curcumin that effectively target key glycolytic enzymes in cells.
A number of conjugates 161 – 165, 288 – 293 containing bis(arylidene)piperidone, 1,2,3-triazole, and 2-ethoxy-2,5-dihydro-5H-1,2-oxaphosphole-2-oxide motifs showed higher cytotoxic activity compared to curcumin against three cell lines: cervical carcinoma HeLa, lung adenocarcinoma A549, and neuroblastoma SH-SY5Y (Table 14).[126]
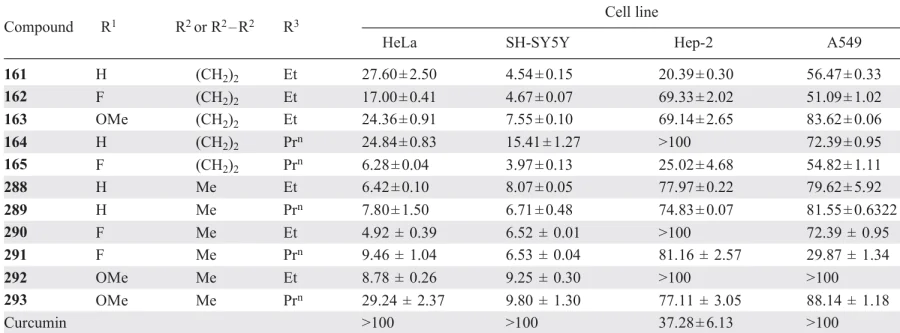
Compounds 162, 163, 290 and 291, which contain a fluorine atom in the benzene ring, demonstrated the most favourable cytotoxic profile, confirming the important role of substituents in biological activity. It is suggested that the mechanism of cytotoxic action of these conjugates was also related to their ability to inhibit glycolysis in cancer cells.
N-Alkanesulfonyl-3,5-bis(arylidene)-4-piperidones 109 – 120 (see Scheme 14 ) were shown to enhance the induction of the xenobiotic NAD(P)H-quinone dehydrogenase 1 (NQO1),* which metabolizes the cytosolic enzyme in murine hepatoma cells and stabilizes the apoptosis regulator p53.[118] Molecular docking studies, revealed hydrogen bonding between the sulfonyl oxygens of compounds 109 – 120 with the arginine hydrogen atoms in NQO1, along with π-cationic interactions between the benzene ring and the guanidine group of this amino acid.
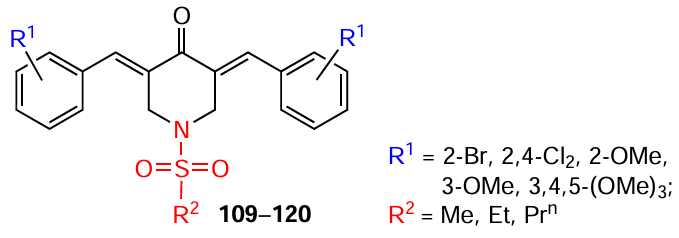
Another important cytoplasmic molecular target is tubulin, a building block protein for microtubules, which play a key role in the formation of the cytoskeleton and cell division. This makes tubulin a target for many anticancer drugs, as inhibiting its polymerization can stop tumour growth.[168]For example, Fawzy et al.[105] investigated the effect of bis(arylidene)piperidones 294, 295 at a concentration of 10 μM on tubulin polymerization in vitro. The authors found that diiodide 295 performed best in destabilizing microtubules compared to the positive control (vincristine). For compounds 294 and 295, the inhibition of tubulin polymerization in microtubules was ~25% and ~93%, respectively. Based on molecular docking data, the authors concluded that this is due to the presence of an iodine atom in the structure of compound 295, the size of which is ideal for binding to β-tubulin.
To illustrate the above, Fig. 4 summarizes the data demonstrating the broad spectrum of effects of MAСs on tumour cell signalling pathways, metabolism, and cellular structures. All of these mechanisms underlie the therapeutic potential of these compounds and make them promising candidates for the development of new, effective antitumour agents. It should be noted that this classification of MACs based on their effects on molecular targets is rather arbitrary, as even structurally similar compounds can disrupt the regulation of various signalling pathways involved in carcinogenesis.
![[{"id":"dEffZLvON8","type":"paragraph","data":{"text":"Molecular mechanisms of antitumour action of MACs and examples of chemical structures presented in the review"}}]](/storage/images/resized/1uNK1n645jOtdSZDoUpe0EHEvu4qtz1yhMETrl28_xl.webp)
* NAD(P)H-quinone dehydrogenase 1 is an enzyme that can reduce the level of reactive quinones responsible for oxidative stress and is located in the liquid part of the cell (cytosol).
4. Conclusion
To conclude, a review of the scientific literature shows that MACs are a promising class of compounds with diverse mechanisms of antitumour action that affect key signalling pathways, metabolic processes, and structural components of tumour cells (Fig. 5).
![[{"id":"QdDTJt6BD8","type":"paragraph","data":{"text":"Schematic representation of the diversity of monocarbonyl structural analogues of curcumin and their impact on the pathogenesis of cancer"}}]](/storage/images/resized/nQhJiiwK6KQrW2wfInG7FgpgeiIjQ71UWo5HZWXx_xl.webp)
The following nuclear targets acted upon by curcuminoids can be identified:
1) NF-κB, a key transcription factor regulating the expression of genes responsible for cell proliferation, survival, inflammation, and angiogenesis;
2) STAT3, a key regulator of cellular proliferation and immune response through signalling from cytokine and growth factor receptors;
3) topoisomerase IIα, an enzyme whose aberrant activity is an important determinant of tumour growth, metastasis, and resistance to therapy;
4) PARP is a receptor that plays a central role in DNA damage repair, initiating the repair of DNA breaks in tumour cells and promoting their survival.
Among the targets that promote angiogenesis, MACs influence HIF-1α, the key regulator of cellular adaptation to hypoxia, which stimulates angiogenesis by inducing VEGF, which determines the formation of new blood vessels. Furthermore, these compounds disrupt tumour cell energy metabolism by inhibiting key glycolytic enzymes and reducing anaerobic energy production. These processes lead to energy collapse in tumour cells and limit their ability to proliferate. At the same time, the targeted action of curcumin analogues on the mitochondria of transformed cells, through disruption of the transmembrane potential, provides tumour cell death via apoptosis. By disrupting the polymerization of tubulin within the microtubule structure, MACs cause destruction of the mitotic apparatus and block cell division, which facilitates the cessation of proliferation and the initiation of programmed cell death.
The active action of curcuminoids on the aforementioned targets ensures effective suppression of tumour cell proliferation, angiogenesis, and metastasis, as well as overcoming tumour resistance to treatment. Their unique selectivity for cancer cells and minimal toxicity to the normal microenvironment make these compounds attractive candidates for use in oncology pharmacology.
It should be noted that determining the most promising structural modification of MACs is difficult. Only two factors can be identified:
1. The presence of electron-withdrawing substituents on the benzene rings enhances the cytotoxic activity of 3,5-bis(arylidene)-4-piperidones against one cancer cell line compared to their analogues with electron-donating substituents. This effect is believed to be due to an increase in the partial positive charge on the carbon atom of the double bond, which, in turn, leads to increased reactivity of these derivatives when reacting with biogenic thiols involved in DNA repair in hypoxic tumour cells.
2. Piperidones containing amide, sulfonamide, phosphoryl and 1,2,3-triazole moieites at the nitrogen atom in some cases have higher cytotoxic activity compared to NH-piperidones due to additional centres for binding the substrate to the active centre of the enzymes.
Furthermore, a promising trend is the creation of hybrid molecular systems based on 3,5-bis(arylidene)-4-piperidones with two or more pharmacophoric moieties possessing cytotoxic properties. Such moieties could include, e.g., natural sesquiterpene lactones or metal complexes (platinum(IV), palladium(II), or copper(II)). In conclusion, it is important to emphasize that it is currently impossible to identify a single target for the antitumour action of curcuminoids, especially since it depends on the type of chemical modification aimed at a specific pathological process. Based on the data published in the modern literature, it can be concluded that the most frequently mentioned targets for which the effect of MACs has been proven are protein molecules involved in signalling pathways associated with apoptosis and proliferation of neoplastic cells. This is obviously explained by the fact that these two pathological processes have been most thoroughly studied in oncological diseases. However, further research will not only expand our understanding of the effects of these compounds on other tumorigenesis processes, such as angiogenesis and energy metabolism in cancer cells, but will also lead to the discovery of new promising therapeutic targets for the fight against cancer.
Acknowledgements
This review was written with the financial support of the Russian Science Foundation (Project No. 25-73-20033). The authors are also grateful to the Ministry of Science and Higher Education of the Russian Federation (State Task No. 075-00276-25-00) and Centre for Collective Use of INEOS RAS for the access to full-text versions of the cited publications.
5. List of abbreviations
The following abbreviations and designations are used in the review:
Asc — ascorbate,
Bax — Bcl-2-associated X protein,
COX-2 — cyclooxygenase 2,
CTGF — connective tissue growth factor,
CXCR4 — C – X – C – chemokine receptor type 4,
DCE — 1,2-dichloroethane,
DIPEA — N,N-diisopropylethylamine,
EGF — epidermal growth factor,
EGFR — epidermal growth factor receptor,
ERE — estrogen response element,
ERK — extracellular signal-regulated kinase,
FADD — Fas-associated protein with death domain,
FGF — fibroblast growth factor,
Fu — furyl,
H2R — histamine H2 receptor,
HATU — 2-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate,
HER-2 — human epidermal growth factor receptor 2,
IC50 — half-maximal inhibitory concentration,
IFN — interferon,
IKK — IκB кinase,
IL-1 RAK — interleukin-1 receptor-associated kinase,
IL-8R — interleukin 8 receptor 8,
ITPR — inositol 1,4,5-trisphosphate receptor,
JNK — c-Jun N-terminal kinase,
LDL-R — low-density lipoprotein receptor,
МАC — monocarbonyl analogues of curcumin,
MAPK — mitogen-activated protein kinase,
MCP-1 — monocyte chemoattractant protein 1,
MIP — macrophage inflammatory protein,
MW — microwave radiation,
NQO1 — NAD(P)H-quinone dehydrogenase 1,
PARP — poly(ADP-ribose) polymerase,
PI3K — phosphoinositide 3-kinase,
PKA(B,C) — protein kinase A(B,C),
PPAR-γ — peroxisome proliferator-activated receptor gamma,
Py — pyridyl,
rt — room temperature,
SI — selectivity index,
STAT-1(3,4,5) — signal transducer and activator of transcription 1(3,4,5),
TF — transcription factor,
TfO — trifluoromethanesulfonate (triflate),
TGF-β1 — transforming growth factor beta 1,
TMS — trimethylsilyl,
TNF — tumour necrosis factor,
VEGF — vascular endothelial growth.
References




















