


Keywords
Abstract
Most of the modern chemotherapeutic arsenal for the treatment of various types of cancer is based on pyrimidine nucleoside analogues. These include drugs that have been proven for decades (cytarabine, floxuridine, gemcitabine, capecitabine, azacitidine, and decitabine) as well as combinations of new antitumour agents (trifluorothymidine and tipiracil hydrochloride, decitabine and cedazuridine). New pyrimidine nucleoside analogues (doxyfluridine, tezacitabine, tiarabin, troxacitabine, etc.) and their depot forms (sapacitabine, MB-07133, etc.) are currently undergoing clinical trials for monotherapy or combination therapy for a wide range of oncological diseases. Over the past 15 years, publications have appeared describing various approaches to optimize the synthesis methods and structures of existing cancer drugs, as well as the design and synthesis of new pyrimidine nucleoside analogues with antitumour activity. This review summarizes new information and classical methods for synthesizing pyrimidine nucleoside analogues, as well as data on their antitumour activity, targets, and mechanisms of action. This review will be useful to a wide range of readers, including undergraduate and graduate students in chemistry and biology, and also specialists in chemistry, biology and medicine.
The bibliography includes 216 references.
1. Introduction
Natural nucleosides, in addition to their key role in the storage and transmission of genetic information, are involved in numerous cellular processes as substrates, regulators or cofactors. Nucleoside analogues with modified heterocyclic base and/or carbohydrate moiety, which have been acting as tools for affecting various pathogenic processes in the human body for more than half a century, still form the basis of antitumour and antiviral chemotherapy. Currently, 15 nucleoside analogues[1][2] are approved by the U.S. Food and Drug Administration (FDA) and are used for the treatment of cancer. The majority of the current chemotherapeutic arsenal against various types of cancer consists of drugs based on pyrimidine nucleoside analogues[3] (Fig. 1).
![[{"id":"v1oyfAKxr8","type":"paragraph","data":{"text":"Pyrimidine nucleoside analogues, fluorouracil and compounds <b>1 – 9</b>, are used in cancer therapy (listed under the name and the year of approval). Pyrimidine base modifications are highlighted in blue, carbohydrate moiety modifications are highlighted in green, and fragments of the depot forms are shown in pink."}}]](/storage/images/resized/0dPtCbUjUOWAabKYaWjt87zyUJMvqQX1XHyFQsRH_xl.webp)
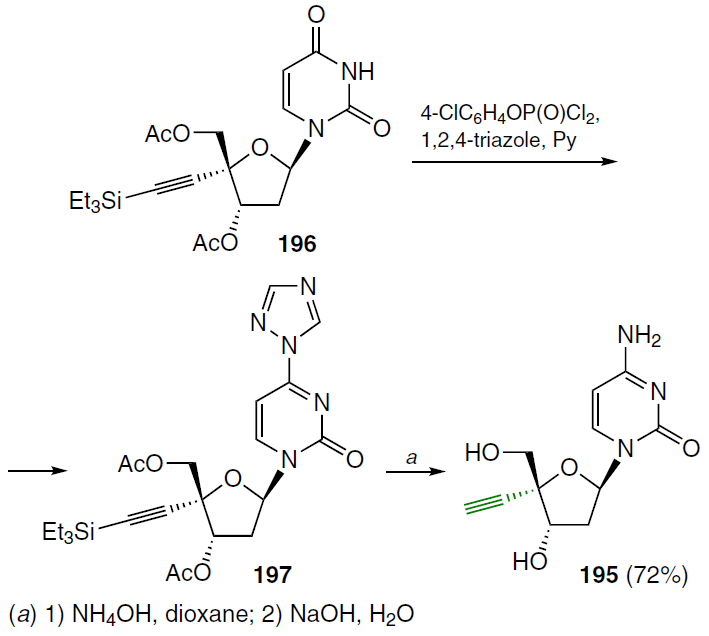
Although the first nucleoside drugs have been used in therapy for over fifty years, active research continues in the field of design and chemical synthesis, as well as an assessment of the anticancer potential of pyrimidine nucleoside analogues.[2][4][5] Novel pyrimidine nucleoside analogues (Fig. 2) and their depot forms* (Fig. 3) are currently undergoing clinical trials for use in monotherapy or combination therapy ([[ type="anchor" figureId="83241" ]]) of a wide range of oncological diseases.
![[{"id":"Qp4zQmKUGK","type":"paragraph","data":{"text":"Pyrimidine nucleoside analogues <b>10 – 16</b>, which have undergone clinical trials at various stages as potential cancer treatments (listed under the name). Modifications of the pyrimidine base are highlighted in blue, and those of the carbohydrate moiety are highlighted in green."}}]](/storage/images/resized/ZPsjmEuUYcACxBkEGrS36YsNqu9Wr2sjUCwqEdyS_xl.webp)
![[{"id":"gKfIUlFo2d","type":"paragraph","data":{"text":"Depot forms and prodrugs of pyrimidine nucleoside analogues <b>17 – 24</b>, which have undergone clinical trials at various stages as potential cancer treatments (listed under the title). Pyrimidine base modifications are highlighted in blue, carbohydrate moiety modifications are highlighted in green, and fragments of the depot forms and prodrugs are shown in pink."}}]](/storage/images/resized/bR3ubdSOBYHjMX7t5Wt00ywRVaSzFW4VIe8vv5QZ_xl.webp)
![[{"id":"wT7d39CbSn","type":"paragraph","data":{"text":"Combinations containing pyrimidine nucleoside analogues <b>25 – 27</b>, currently undergoing clinical trials at various stages as potential cancer treatments (listed under the title). Pyrimidine base modifications are highlighted in blue, and carbohydrate moiety modifications are highlighted in green."}}]](/storage/images/resized/ToyMuA3P05i9tM2il5vmEK8FtmbTrIvghNw4GK0L_xl.webp)
Nucleoside analogues are prodrugs,** and require cellular metabolism to become active. After transport across the plasma membrane, nucleoside analogues undergo first phosphorylation, while heterocyclic base analogues undergo ribosylation followed by phosphorylation to afford nucleoside 5'-monophosphate analogues. 2'-Deoxynucleosides are phosphorylated by 2'-deoxycytidine kinase, thymidine kinases 1 and 2, and 2'-deoxyguanosine kinase. The main rate-limiting enzyme for activation of most approved nucleoside analogues is 2'-deoxycytidine kinase (dCK).1 Although 2'-deoxycytidine is the preferred natural substrate for this enzyme, dCK also recognizes 2'-deoxyadenosine and 2'-deoxyguanosine as substrates. Cancer cells typically express 2'-deoxycytidine kinase at the protein level at 3 – 5 times higher levels than most normal cells, providing some selectivity for the action of nucleoside analogues. Thymidine kinases and 2'-deoxyguanosine kinase are base-selective, and certain modifications of the carbohydrate moiety can also interfere with the recognition of nucleosides by these kinases. Therefore, many modified nucleosides do not undergo first phosphorylation in cells. The first phosphorylation gives nucleoside 5'-monophosphates, which are converted into the corresponding nucleoside 5'-diphosphates and nucleoside 5'-triphosphates by various cellular kinases. To phosphorylate nucleoside 5'-monophosphates, the cell uses nucleoside monophosphate kinases or nucleotide kinases, which weakly distinguish between the ribose and 2'-deoxyribose fragments of the nucleotide but are specific to the nature of the base. The conversion of nucleoside 5'-diphosphates to nucleoside 5'-triphosphates is carried out by nucleoside diphosphate kinases, which are present in all cells and do not exhibit particular specificity to the types of nucleoside bases and are capable of recognizing 5'-diphosphates of ribonucleosides and 2'-deoxyribonucleosides as substrates.[6]
Nucleoside 5'-triphosphate analogues are substrates for DNA polymerases; the corresponding nucleotides are incorporated into DNA during replication or DNA synthesis during repair, leading to stalling of replication forks and chain termination.1 These events activate various DNA damage sensors that stimulate DNA repair, arrest cell growth, and often lead to apoptosis. Most cancer cells replicate their genome more frequently than normal adult cells, which are dormant and do not actively synthesize their DNA, providing a degree of selectivity for inhibiting cancer cell growth. Some nucleoside 5'-triphosphate analogues can act as substrates for RNA polymerases; the incorporation of the corresponding nucleotides into the growing RNA chain leads to transcription termination and instability of messenger RNA (mRNA) and ribosomal RNA (rRNA).
Nucleoside and nucleotide analogues (mono-, di-, or triphosphates) can also inhibit other key cellular enzymes, providing an alternative mechanism for cell growth suppression. Such enzymes include, for example, ribonucleotide reductase (RR), which removes the 2'-OH group from the carbohydrate moiety of ribonucleoside diphosphate to form 2'-deoxyribonucleoside diphosphate de novo; thymidylate synthase, which catalyzes the conversion of 2'-deoxyuridine-5'-monophosphate to thymidine-5'-monophosphate using 5,10-methylenetetrahydrofolate as a source of the methyl group; uridine-cytidine kinase 2, which converts uridine and cytidine to the corresponding 5'-monophosphates; and DNA methyltransferases, which catalyze the methylation of the 5-position of cytosine bases in newly synthesized DNA strands.[7] Some nucleoside analogues exhibit anticancer activity by inhibiting cellular enzymes not involved in nucleic acid synthesis.
Thus, pyrimidine and pyrimidine nucleoside analogues, mimicking natural compounds, act as substrates and/or inhibitors for numerous enzymes involved in cellular metabolism and catabolism. Unlike normal cells, the levels of many enzymes are elevated in actively dividing cancer cells. This ensures the selectivity of drug action and makes such proteins attractive targets for cancer chemotherapy. The targets and mechanisms of action of specific pyrimidine nucleoside analogues are described in the relevant sections of this review.
Over the years of research into pyrimidine nucleoside analogues, numerous compounds have been obtained and a large number of publications have emerged that analyze a diversity of aspects of this field, from the design and chemical synthesis of new molecules to clinical observations of the use of drugs in the treatment of oncological diseases. Reviews have been published on the mechanisms of action of pyrimidine nucleoside analogues and the pathways for the development of resistance to them,[2][8] as well as on the enzymatic targets to which inhibitors are directed: ribonucleoside reductase,[9][10] uridine-cytidine kinase 2 (UCK2),[11] thymidine phosphorylase,[12] DNA methyltransferase,[7] etc.
Over the past 15 years, a large number of publications have appeared devoted to various approaches to optimizing the structures of known drugs intended for the treatment of oncological diseases. The design and synthesis of new pyrimidine nucleoside analogues with antitumour activity have also been described. A number of reviews provide a fairly in-depth and detailed discussion of various aspects of the chemistry and/or research on the antitumour properties of individual groups of pyridine nucleoside analogues.[5][13-17] Reviews have been published that describe in detail individual targets of action[7][9-12][18] and/or mechanisms of antitumour activity and the development of drug resistance of compounds[2][8] approved for use in practice or undergoing clinical trials.[1][19-23] Works have also been presented that summarize data on optimizing the properties of existing drugs, in particular on the creation of targeted delivery methods and depot forms.[24-26] However, in our opinion, existing reviews do not provide a comprehensive understanding of the diversity of pyrimidine nucleoside analogues with antitumour activity, their role and potential in the treatment of cancer.
This review harmoniously combines all of these aspects in a brief summary. It summarizes information regarding classical and new methods for synthesizing pyrimidine nucleoside analogues, their antitumour activity against various cell lines, including both solid tumours and hematopoietic malignancies, as well as the targets and mechanisms of action of these compounds. Therefore, this review will be useful to a wide audience: students of chemistry and biology, researchers specializing in nucleosides, biologists, and oncologists. We hope that the information presented will serve as a basis for further research aimed at creating new therapeutic strategies in the fight against cancer.
* A depot form is a derivative of the active substance that ensures its slow release in the body over a long period of time.
** A prodrug is a molecule that possesses no intrinsic biological activity but can be converted into a biologically active drug through various metabolic steps.
2. Modification at the 2' position of the carbohydrate moiety
Two drugs used in cancer therapy,[4] cytarabine (1) and gemcitabine (3), are 2'-modified pyrimidine nucleoside analogues (Fig. 5).
Both drugs were created in the last century, and during their use, data was obtained not only on the effectiveness, targets, and mechanisms of targeted anticancer action, but also on the disadvantages, viz., side effects on the human body, the emergence of resistance, etc.[27][28] Numerous studies have been described aimed at optimizing the methods of synthesizing these drugs using modern approaches, and creating analogues and derivatives to improve therapeutic properties.[5] [14][17][29-33]
![[{"id":"MTIENIkHrM","type":"paragraph","data":{"text":"General structure of pyrimidine (Pyr) nucleosides (left, the arrow indicates the 2’-position of the carbohydrate moiety) and the structures of analogues <b>1</b> and <b>3</b> modified at this position."}}]](/storage/images/resized/Eu9RjoULsSrOHoWRxybVpqn0ODquRbepncbMqNJ3_xl.webp)
Gemcitabine (3)[14][33-37] is a 2',2'-difluorinated cytidine analogue marketed as the hydrochloride salt under the trade name Gemzar for the treatment of cancers such as non-small cell lung cancer, ovarian cancer, bladder cancer, pancreatic cancer, and breast cancer. In recent years, the need for effective methods for producing gemcitabine has increased significantly. The synthesis of gemcitabine (3) was patented[38] comprising the following sequence of transformations: complete protection of the NH2 and OH groups of cytidine with o-toluoyl chloride (o-TolC(O)Cl) followed by regioselective deprotection of position 2', oxidation of compound 28 with pyridinium dichromate (PDC), fluorination with diethylaminosulfur trifluoride (DAST) in the HF – pyridine (Py) system, and removal of the protecting groups with sodium methoxide (Scheme 1).
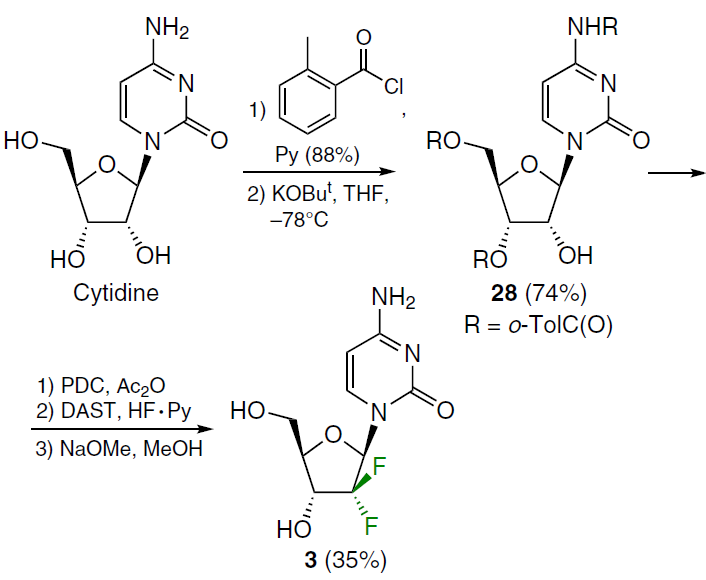
Enzymatic deamination of gemcitabine (3), rapid systemic clearance, and the emergence of chemoresistance limit the use of this drug. In recent years, data have been published on various strategies for creating gemcitabine (3) prodrugs with the aim of improving its pharmacokinetic properties, increasing efficiency and safety, as well as on the possibilities of using various strategies for its targeted delivery.[3][32][39][40] Three prodrugs of gemcitabine (3), known under the commercial codes NUC1031 (19), CP-4126 (20), and LY2334737 (21) (see Fig. 3), did not show sufficient benefits in clinical trials to warrant their recommendation for use.[3]
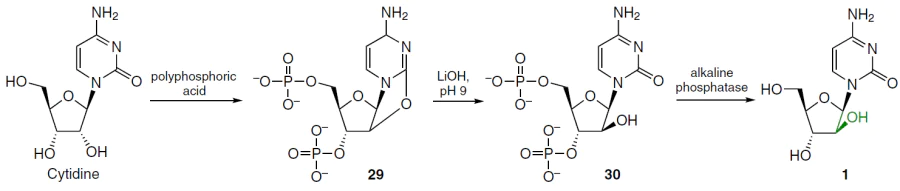
1-β-D-Arabinofuranosylcytosine (cytarabine, ara-C, 1)[41] (see Fig. 5) is one of the most potent drugs for the treatment of human acute myeloid leukemia, erythromyelosis, neuroleukemia and non-Hodgkin's lymphoma.[42-44] Cytarabine (1) was first synthesized by the Dekker's group[45] in 1959 (Scheme 2). Cytidine reacted with polyphosphoric acid to give 3',5'-O-diphosphorylated 2,2'-O-cyclocytidine 29, which was hydrolyzed with lithium hydroxide (pH 9) to afford cytarabine-3',5'-diphosphate 30. To obtain the target product 1, the phosphate groups were then removed by enzymatic hydrolysis with alkaline phosphatase.
Subsequently, other synthetic approaches to cytarabine (1) were proposed, some of which are described in Chapter 2 of the monograph Profiles of Drug Substances Excipients and Related Methodology,[41] dedicated to this drug. 1-β-D-Arabinofuranosyl-uracil (31) was the key intermediate in two other synthetic routes that allow the preparation of cytarabine (1) in kilogram quantities. It was obtained from uridine by a two-step protocol through the formation of 2,2'-anhydrouridine 32 (Scheme 3). Next, using one method, compound 31 was treated with hexamethyldisilazane (HMDS) under pressure at high temperature. In the final step, the reaction of intermediate 33 with methanol gave the target cytarabine (1) in a high yield of 50 g.[46]
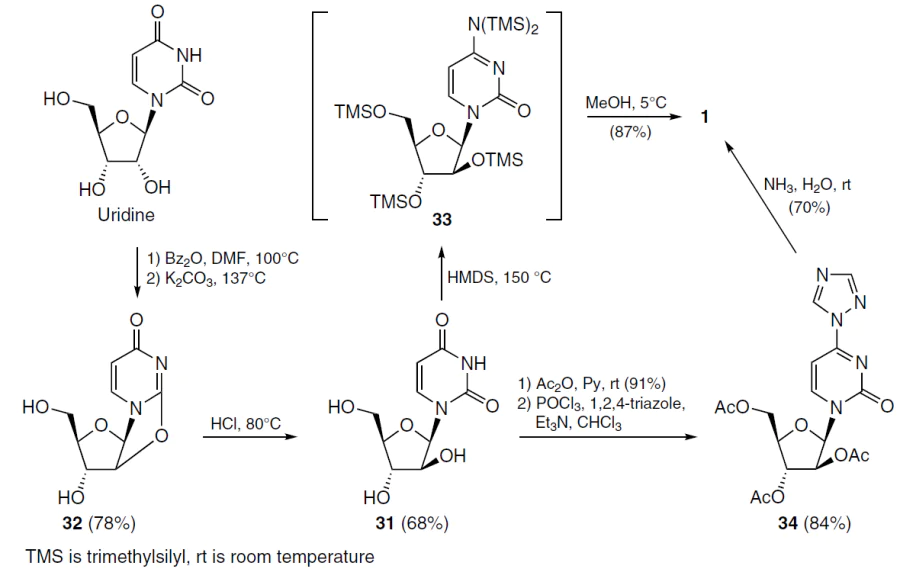
Another synthetic strategy[47] used the acyl-substituted 1,2,4-triazole derivative 34, which served as an intermediate, formed through sequential acylation and substitution of the oxygen atom at the 5-position of the uracil moiety with a triazole ring (see Scheme 3). Cytarabine (1) was obtained in kilograms after deprotection of compound 34 with ammonia.
The effectiveness of cytarabine (1) is limited by a number of disadvantages such as a short half-life in plasma (partly due to deamination to inactive 1-(arabinofuranosyl)uracil by cytidine deaminase[48]), low affinity for the key enzyme of the metabolic pathway — deoxycytidine kinase,[49] the development of resistance to this drug[50] and lack of activity against solid tumours. In an attempt to overcome these challenges, various types of depot forms and delivery systems of cytarabine (1) have been developed that can increase the half-life, improve stability, and provide targeted delivery of the drug to tumour cells.[1][30][51]
Proposed depot forms of cytarabine (1) include elacitarabine (CP-4055, 22),[52] which has entered phase III clinical trials as a drug for the treatment of leukemia, as well as the agent MB07133 (23)1 and aspacytarabine (24)[53] (see Fig. 3). A randomized phase III clinical trial in patients with acute myeloid leukemia (AML) found no significant differences in overall survival or adverse events between patients receiving elacitarabine (22) and the control group.[54] However, a lower dose of elacitarabine (22) in combination with idarubicin resulted in complete remission of the disease in 40% of cases.[55] Thus, despite the disappointing results of phase III studies of elacitarabine as a monotherapy for leukemia,[54] this drug may be useful in combination with other drugs or in patients with refractory disease.
Synergism has been revealed in combinations of elacitarabine with gemcitabine, irinotecan and topotecan, one of the targets of which is topoisomerase I, as well as the additive nature of taking this drug with idarubicin and cloretazine, which act by other mechanisms.[24][56] In addition to prodrugs, cytarabine (1) delivery systems are being actively developed for the treatment of various types of cancer. A liposomal form of cytarabine (DepoCyt, Pacira Pharmaceuticals, New Jersey, USA) is approved for the treatment of lymphomatous meningitis and is an example of the successful application of nanotechnology in oncology.[57]
Another trend for optimizing the anticancer properties of cytarabine (1) is the synthesis of 2'-alkyl analogues 35a,b, 36a,b.
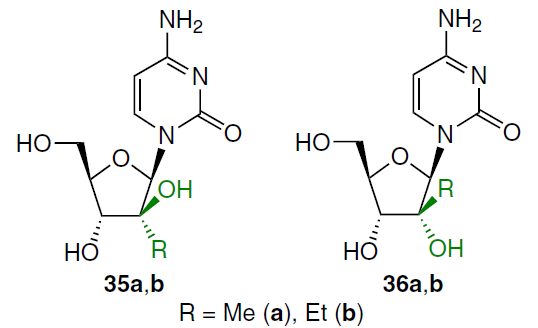
The synthesis of these compounds commenced with the treatment of 3',5'-protected 2'-keto-nucleoside 37 with alkyl magnesium bromide in diethyl ether while cooling under argon (Scheme 4).[58] The use of methyl magnesium bromide at ‒78°C led to a mixture of epimers 38a and 39a, while ethyl magnesium bromide at ‒50°C afforded a mixture of three nucleoside analogues (38b, 39b, and 40). Upon treatment of compound 37 with phenyl magnesium bromide at ‒78°C, the major product 38c was isolated in 84% yield, which is apparently due to steric hindrance when using a bulkier substituent.
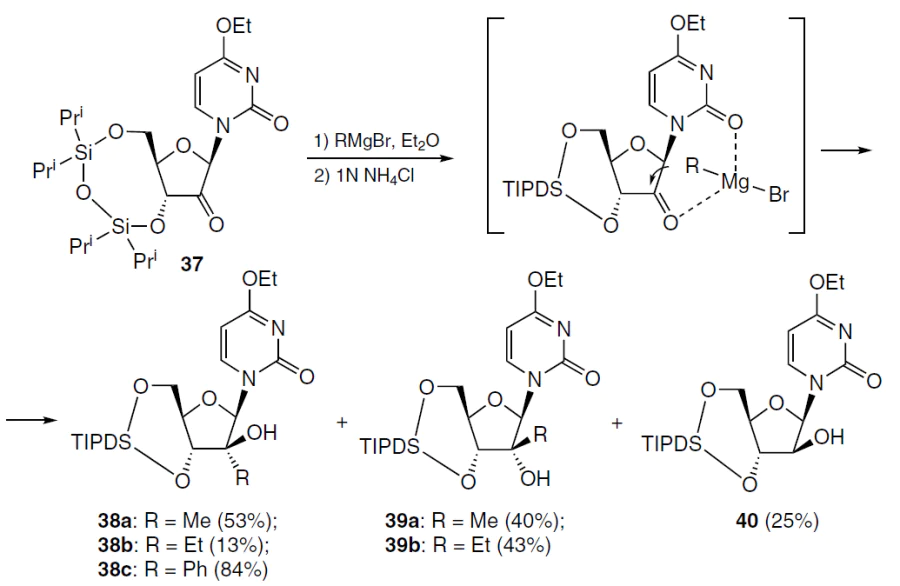
Removal of the tetraisopropyldisiloxanyl-1,3-diyl (TIPDS) protecting group in compounds 38a,b with tetra-n-butylammonium fluoride (TBAF) in THF followed by heating of intermediates 41a,b in methanol saturated with NH3 at 100°C in a sealed vial gave the corresponding 2'-alkyl-1-(arabinofuranosyl)cytosines 35a,b (Scheme 5).[58] Under similar conditions, compounds 39a,b were deprotected to afford 4-ethoxy-substituted 2'-methyl- (42a) and 2'-ethyl-β-D-ribofuranosylpyrimidin-2(1H)-ones (42b) in 94% and 79% yield, respectively. Ammonolysis of compounds 42a,b under the conditions described above furnished 2'-C-methyl- (36a) and 2'-C-ethyl derivatives (36b) in high yields.
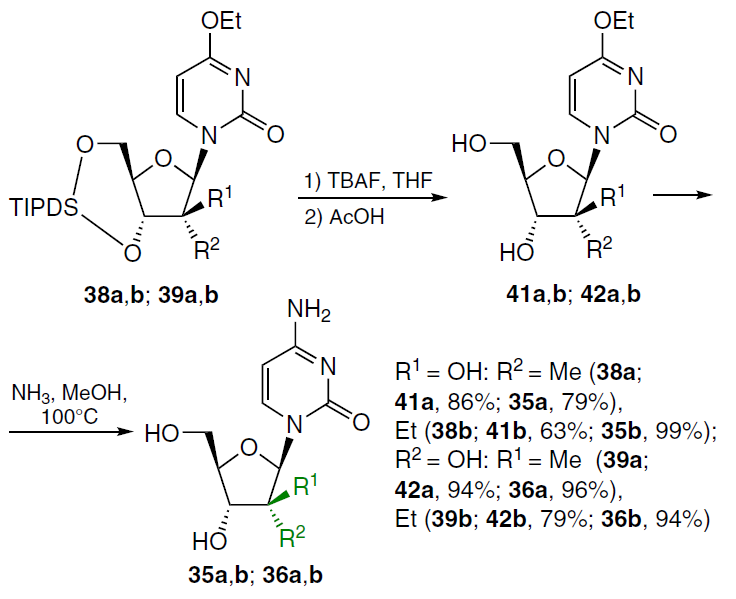
In addition to the above-mentioned 2'-alkylated analogues of cytarabine (1), derivatives with an ethynyl substituent at this position were synthesized. Two approaches to their preparation were proposed. Thus, the reaction of 1-[3',5'-O-[1,1,3,3-tetraisopropyldisiloxane-1,3-diyl]-β-D-erythrofuranosyl]uracil and LiC≡CR gave predominantly the α-adduct.[59] Treatment of the N4-benzoylcytosine derivative 43 with LiC≡CTMS in THF at ‒78°C led to a mixture of β- (44) and α-adducts (45), which was separated by column chromatography on silica gel, eluting with ethyl acetate in hexane with a concentration gradient (Scheme 6).[60] The silyl protection in the resulting intermediates was removed in the usual manner and the corresponding free nucleosides 46 and 47, respectively, were isolated.
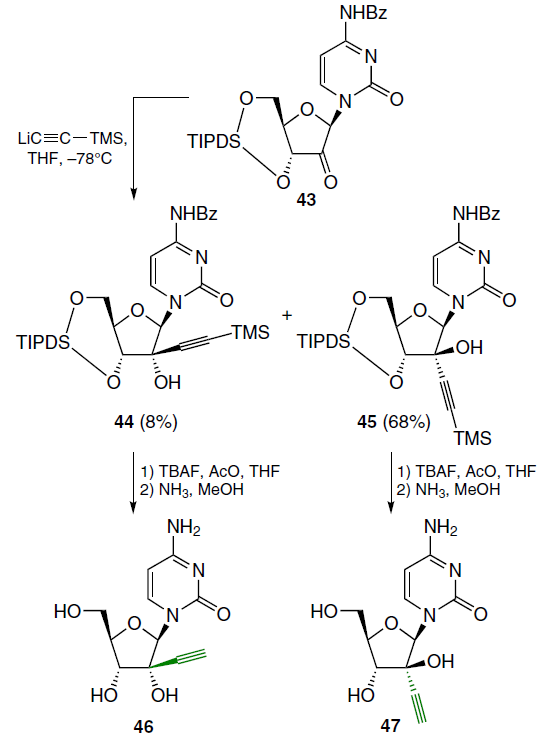
The synthesis of 2'-deoxy-2'-C-methyl- and 2'-deoxy-2'-C-ethylcytidines 48a,b is described. The hydroxyl group in the starting nucleosides 39a,b was deoxygenated with methyl oxalyl chloride in acetonitrile in the presence of two equivalents of 4-dimethylaminopyridine (DMAP). Thus obtained 2'-O-methyloxalyl esters 49a,b were heated without further purification in toluene at 100°C for 1 h in the presence of tri-n-butylstannane and azabis(isobutyronitrile) (AIBN) to give 2'-deoxy derivatives 50a,b in moderate yields. These compounds were then treated with TBAF in THF at room temperature to form nucleosides 51a,b in high yield. Further heating at 100°C of a solution of nucleosides 51a,b in methanol saturated with NH3 in sealed tubes for 2 days, followed by treatment of the residues dissolved in ethanol with a 2 M HCl solution gave the target products 48a,b in the form of hydrochlorides (Scheme 7).[61][62]
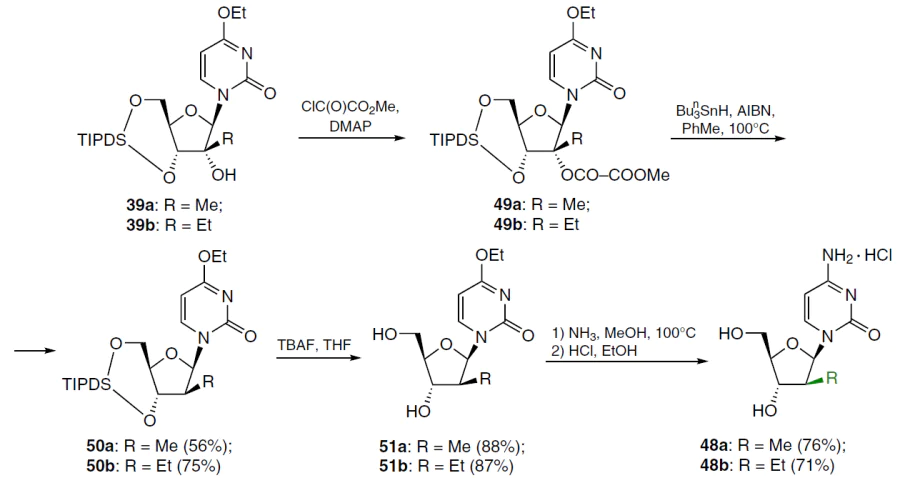
When nucleoside 38a was deoxygenated under the conditions shown in Scheme 7, the corresponding ester was isolated in very low yield, apparently due to steric hindrance during esterification. For this reason, 2'-deoxy-2'-C-methylcytidine (52) was obtained using a less bulky protecting group (Scheme 8).[62] Thus, nucleoside 38a at room temperature was treated with TBAF in THF, which furnished 4-ethoxy-1-(2'-C-methyl-β-D-arabinofuranosyl)-2(1H)-pyrimidinone (41a), which was acetylated with acetic anhydride in the presence of triethylamine and DMAP. In this case, 3',5'-di-O-acetyl derivative 53 was obtained in high yield, which was condensed with methyl oxalyl chloride in the presence of two equivalents of DMAP. The resulting 2'-O-methyloxalyl ester 54 was heated without further purification at 100°C in toluene for 1 h in the presence of tri-n-butylstannane and AIBN. Subsequent treatment of the deoxygenation product with sodium ethoxide in absolute ethanol gave a mixture of nucleosides 51a and 55, which were chromatographically separated to give the corresponding products in individual form. Ammonolysis of nucleoside 55 and subsequent treatment of the residue with a solution of HCl in ethanol afforded the hydrochloride of the target (2'R)-2'-deoxy-2'-C-methylcytidine (52) in high yield (see Scheme 8).[62]
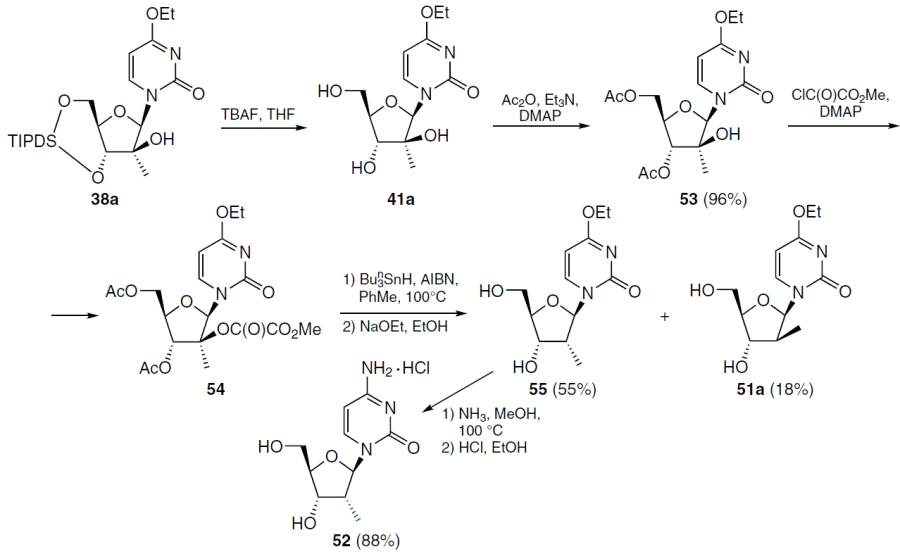
(2'S)-2'-C-Methylthymidine 56 was obtained by acid hydrolysis of 4-ethoxy-5-methyl-1-(2'-deoxy-2'-C-methyl-β-D-arabinofuranosyl)-2(1H)-pyrimidinone (57) in an aqueous–alcoholic medium (Scheme 9).[62]
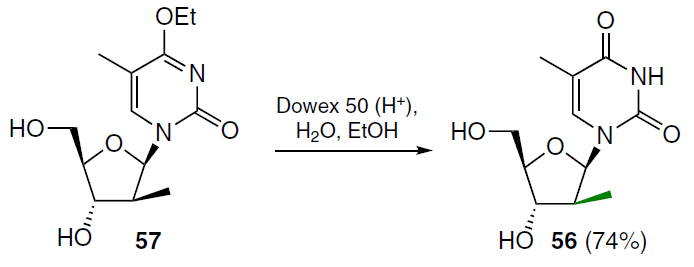
The synthetic approach to 1-(2'-deoxy-β-D-arabinofuranosyl)cytosines with azide (58) and amino groups (59) at position 2' involved the condensation of 5-O-benzoyl-3-O-acetyl-2-azido-3-deoxy-D-arabinofuranosyl chloride[63] with trimethylsilylated cytosine. This gave a mixture of α- and β-anomers, with the isolated yield after chromatographic separation of 9 and 39%, respectively. The protecting groups were removed by treatment with K2CO3 in methanol to afford the target 1-(2'-azido-2'-deoxy-β-D-arabinofuranosyl)cytosine 58.[63] Reduction of the latter on 5% Pd/C gave the 2'-amino-2'-deoxy derivative 59 in 83% yield.[64]
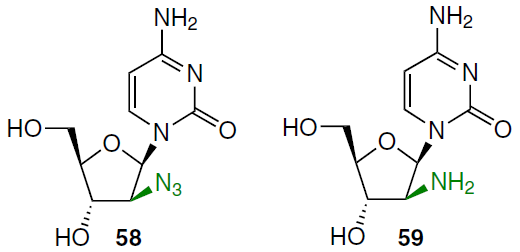
To introduce an azido group at the 2' position of uridine in an alternative method for the synthesis of azido derivative 58, the nucleophilicity of the carbonyl oxygen atom at position 2 was reduced in the first step. For this purpose, benzoyl protection of the N3 group was used.[65] Treatment of protected uridine 60 with diphenyl phosphorazidate (DPPA), triphenylphosphine, and diethyl azodicaboxylate (DEAD) afforded N3-benzoyl-1-(2'-azido-2'-deoxy-3',5'-O-(tetraisopropyl-disiloxane-1,3-diyl)-β-D-arabinofuranosyl)uracil (61) in moderate yield (Scheme 10).[66] After its debenzoylation with NH3 in methanol, 1-(2'-azido-2'-deoxy-3',5'-O-(tetraisopropyldisiloxane-1,3-diyl)-β-D-arabinofuranosyl)uracil (62) was obtained in high yield. Compound 62 was converted to the O4-triisopropylbenzenesulfonate derivative 63 by treatment with 2,4,6-triisopropylbenzenesulfonyl chloride (TPSCl) in dichloromethane and, without further purification, was treated with NH4OH in dioxane to give the cytosine derivative 64 (yield 83% based on compound 62). Deprotection of nucleoside 64 with TBAF in THF gave product 58 in quantitative yield as the hydrochloride.
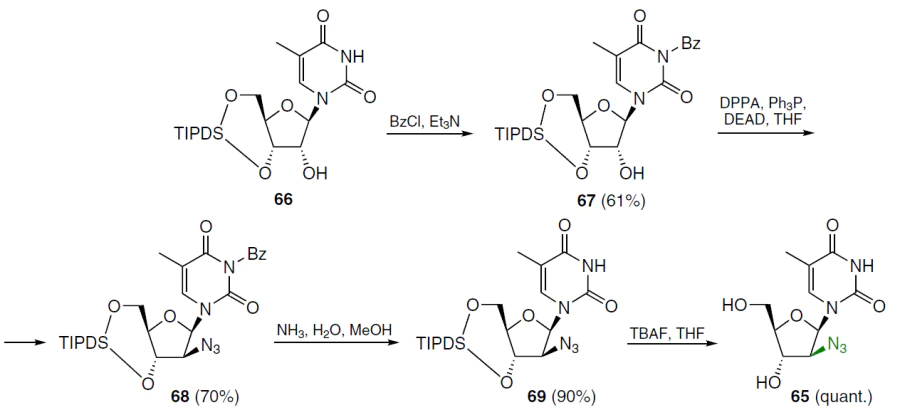
In the first step of the synthesis of 1-(2'-azido-2'-deoxy-β-D-arabinofuranosyl)thymine (65), N3-benzoyl derivative 67 was obtained from 1-[3',5'-O-(tetraisopropyldisiloxane-1,3-diyl)-β-D-ribofuranosyl]thymine (66) by treatment with benzoyl chloride in the presence of triethylamine in dichloromethane (Scheme 11).[66] Nucleoside 67 was treated with diphenyl phosphorazidate, triphenylphosphine, and diethyl azodicarboxylate in THF at room temperature to afford intermediate 68, which was purified by silica gel column chromatography in 70% isolated yield. Debenzoylation of compound 68 was achieved with a methanolic NH3 solution, and the reaction mixture was then treated with a THF solution of TBAF, affording the target product 65 in quantitative yield over two steps.[66]
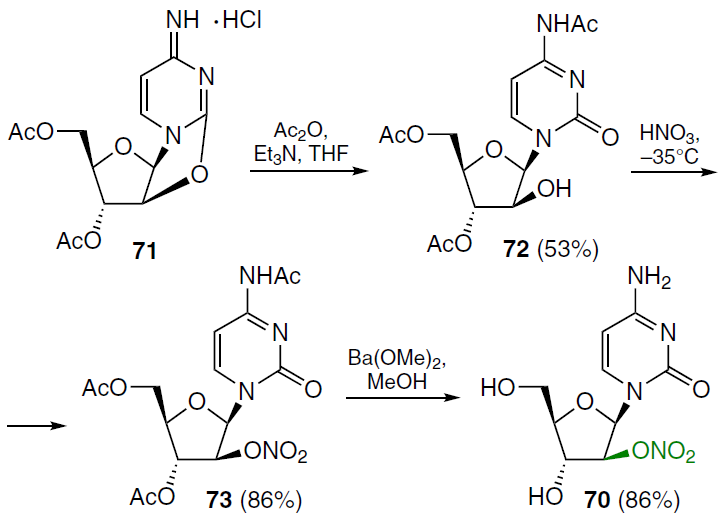
In order to overcome the susceptibility of cytarabine (1) to enzymatic deamination and thereby prevent its deactivation, 2'-O-nitro-β-D-arabinofuranosylcytosine (70) was prepared.[67] The first step of the reaction sequence shown in Scheme 12 comprised treating the starting 2,2'-anhydro-1-(3',5'-di-O-acetyl-β-D-arabinofuranosyl)cytosine hydrochloride (71) with acetic anhydride in THF in the presence of triethylamine, which led to 1-(3',5'-di-O-acetyl-β-D-arabinofuranosyl)-N4-acetylcytosine (72) in moderate yield. Esterification of compound 72 was accomplished with concentrated nitric acid at a temperature from –30 to –35°C, yielding 1-(3',5'-di-O-acetyl-2'-O-nitro-β-D-arabinofuranosyl)-N4-acetylcytosine (73). Deacetylation of compound 73 with barium methoxide in methanol gave the target nucleoside, 1-(2'-O-nitro-β-D-arabinofuranosyl)cytosine (70), in high yield.
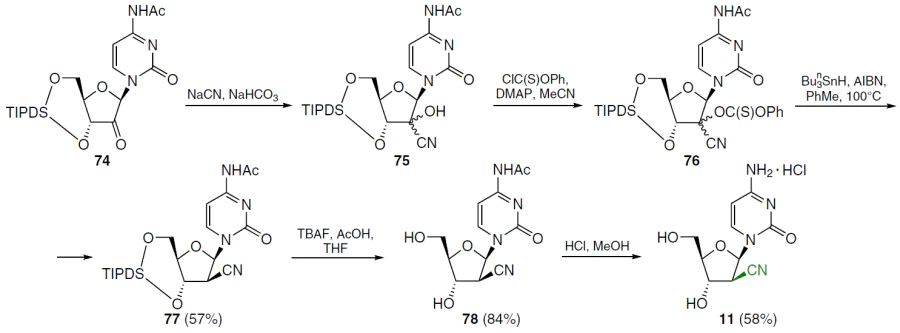
The synthesis of 1-(2'-C-cyano-2'-deoxy-β-D-arabinofuranosyl)cytosine (CNDAC, 11) was started by treating N4-acetyl-1-[3',5'-O-(tetraisopropyldisiloxane-1,3-diyl)-β-D-erythrofuran-2-ylosyl]cytosine (74) with sodium cyanide in the presence of NaHCO3 in a diethyl ether–water mixture (Scheme 13).[68] This afforded epimeric 2'-cyanohydrins 75, which were then reacted with phenyl chlorothiocarbonate in the presence of DMAP in acetonitrile. Deoxygenation of intermediate 76 was carried out by treatment with tri-n-butylstannane and 2,2'-azobis(isobutyronitrile) in toluene at 100°C. According to NMR spectroscopy data (nuclear Overhauser effect (NOE) experiments), radical deoxygenation proceeds stereospecifically to form arabinonucleoside 77, apparently due to the steric hindrance at the β-position. Desilylation of compound 77 with TBAF in the presence of acetic acid in THF afforded the N4-acetyl derivative 78 in high yield. Further treatment of the reaction mixture with HCl in MeOH at room temperature yielded 1-(2'-C-cyano-2'-deoxy-1-β-D-arabinofuranosyl)cytosine (11) as hydrochloride.[68]
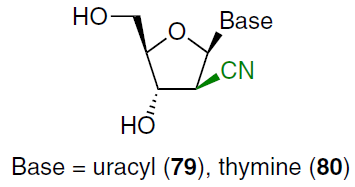
Under similar conditions (see Scheme 13), 1-(2'-C-cyano-2'-deoxy-β-D-arabinofuranosyl) derivatives of uracil (79) and thymine (80) were obtained.[69]
Condensation of 3-O-acetyl-5-O-benzoyl-2-deoxy-2-fluoro-D-arabinofuranosyl bromide (81)[70] with trimethylsilylcytosine or its 5-halo derivatives afforded protected nucleosides 82a – d, which were deprotected by ammonolysis to 2'-F-ara-C-analogues 83a – d (Scheme 14).[71]The 5-iodo derivative 83e was formed by iodination of compound 83a under the following conditions: AcOH, HIO3 , I2 , CCl4 , H2O, 40 – 50°C. Products 84a–d,f were obtained by condensation of bromide 81 with the corresponding trimethylsilyluracils and subsequent deprotection of compounds 85a–d,f. Iodination of nucleoside 85a (R = H) followed by ammonolysis afforded analogue 84e (see Scheme 14).71
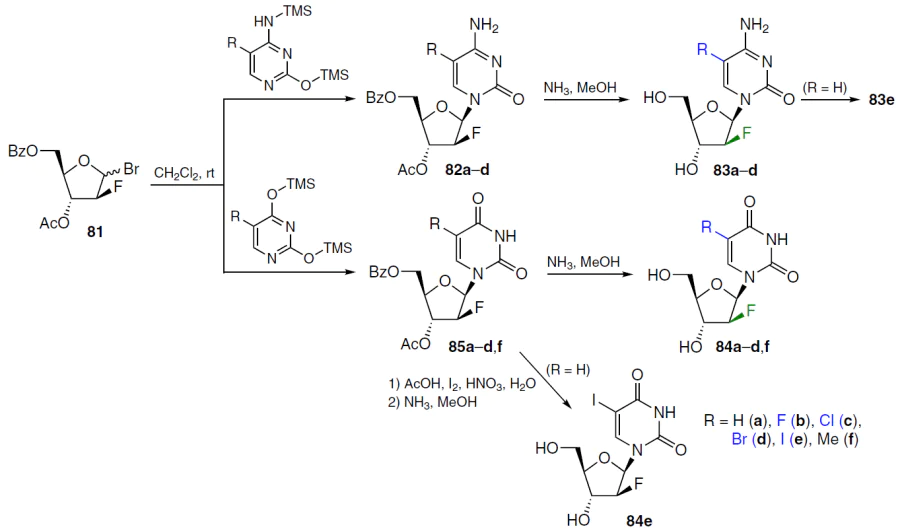
When a double bond is introduced into the 2' position of 2'-deoxycytidine, it forms, together with the secondary 3'-hydroxyl group, an allyl alcohol system, which is also found in a number of nucleoside antibiotics, including angustmycin A and neplanocin A. This structural feature may play an important role in the manifestation of biological activity due to increased reactivity and/or conformational fixation of the carbohydrate moiety. To test this hypothesis, 2'-deoxy-2'-methylidenecytidine (DMDC, 86a) was synthesized.[72] The starting 2'-ketonucleoside 37 was treated with an excess of methylenetriphenylphosphorane, previously obtained by the reaction of potassium hydride with methyltriphenylphosphonium bromide in dimethyl sulfoxide (Scheme 15). This resulted in the formation of 2'-methylidene nucleoside 87, desilylation of which by TBAF in THF afforded derivative 88 in high yield. Compound 88 was then converted by ammonolysis to the target nucleoside 86a, which was isolated as the hydrochloride.[72]
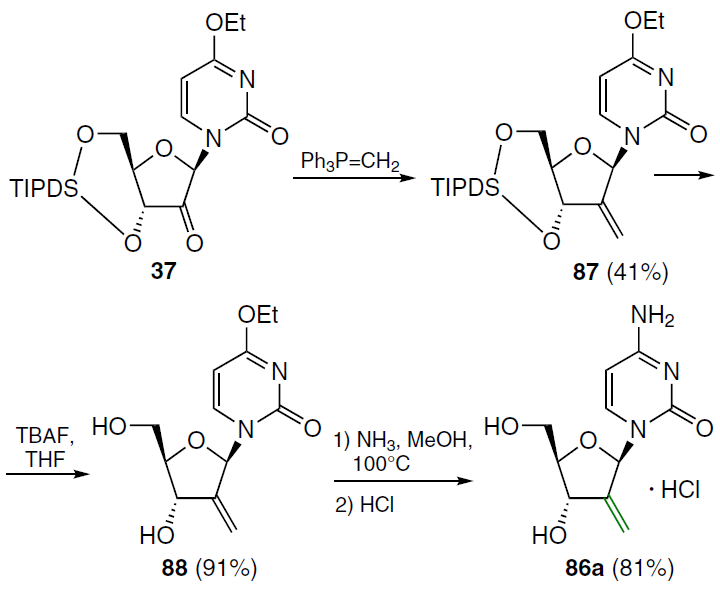
An alternative method for the synthesis of compound 86a is shown in Scheme 16. The starting N4-benzoyl-1-[3',5'-O-(tetraisopropyldisiloxane-1,3-diyl)-β-D-ribofuranosyl]cytosine (89) was oxidized[73] with the CrO3 – Py – Ac2O system (molar ratio 1 : 2 : 1) in dichloromethane, which led to the formation of the 2'-keto derivative 43, which was condensed with methylenetriphenylphosphorane in THF. In this case, compound 90 was isolated, and its deprotection with TBAF in THF followed by treatment with NH3 in MeOH gave the target nucleoside 86a.[74]
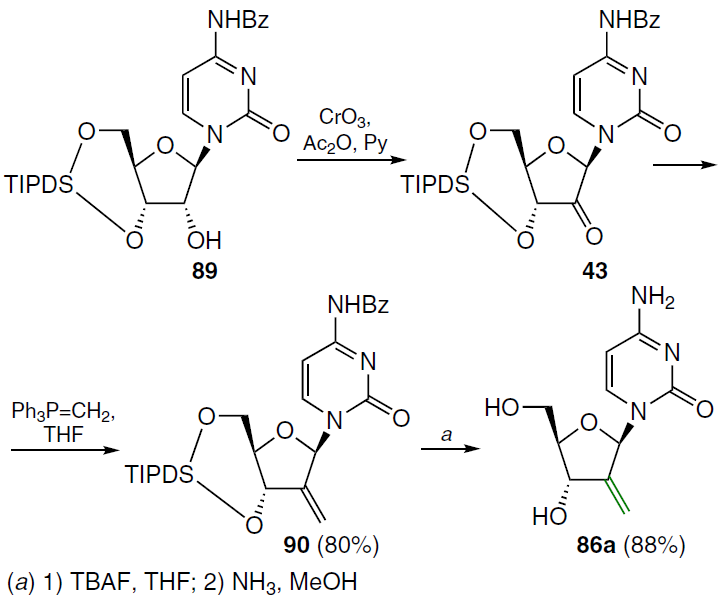
Derivatives of 2'-deoxy-2'-methylideneuridine and -cytidine containing various substituents at position 5 of the pyrimidine ring were also obtained. The synthesis of such products, as shown in Scheme 17, comprised an initial conversion of the starting nucleosides 91a – g under the action of 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane in pyridine to the corresponding 5-substituted 1-[3',5'-O-(tetraisopropyldisiloxane-1,3-diyl)-β-D-ribofuranosyl]uracils 92a – f and the above-described analogue 66 (R=Me). These compounds were then oxidized with the CrO3-Py–Ac2O system (molar ratio 1 : 2 : 1) in dichloromethane.[73] The resulting 2'-keto derivatives 93a – g were condensed with methylenetriphenylphosphorane in THF, leading to 2'-methylidene derivatives 94a – g, which were treated with TBAF in THF to afford the target uridines 95a – g.[74][75]
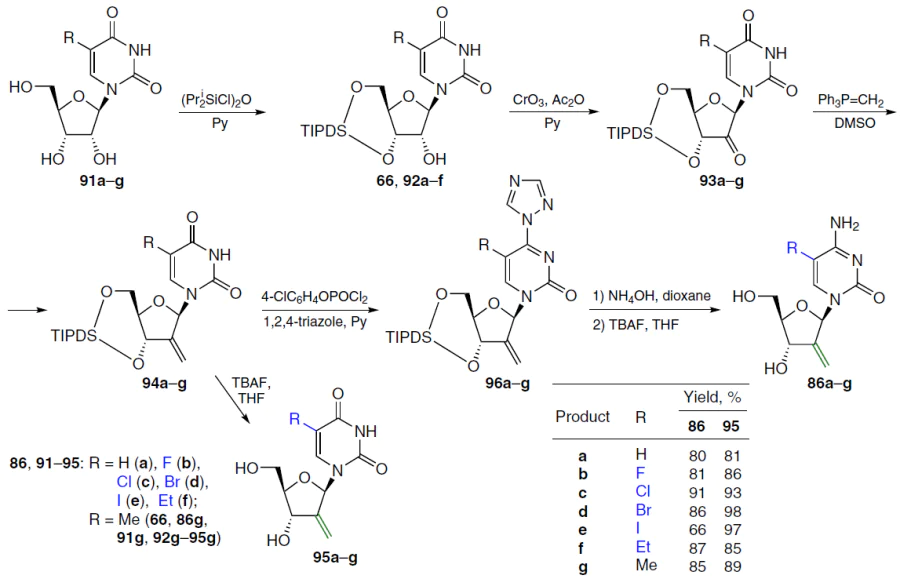
2'-Deoxy-2'-methylidenecytidine 86a and its analogues 86b – g (see Scheme 17) were synthesized from 2'-methylidene derivatives of uridine 94a – g. The latter were reacted with 4-chlorophenyl phosphorodichloridate and 1,2,4-triazole in pyridine, and then the resulting 4-triazolyl derivatives 96a – g were treated with NH4OH followed by deprotection with TBAF in THF to afford the target products 86a – g in high yields.
In an alternative approach to compounds 94 with a 2'-exomethylene group, 3',5'-silylene acetals 66 and 92a were first obtained in 76 – 84% yields, as described above; the 2'-OH groups in these compounds were then oxidized with 2-iodoxybenzoic acid (IBX). Subsequent Wittig methylenation afforded the desired olefins 94a,g (Scheme 18, reaction (1)). Compounds 94a,g were treated with the appropriate mercaptans in the presence of 2,2-dimethoxy-2-phenylacetophenone (DPAP) at room temperature under ultraviolet light irradiation. Derivatives 97a – c were obtained in moderate yields; in all cases, mixtures of diastereomers were observed, with the D-arabino form predominating.[76]
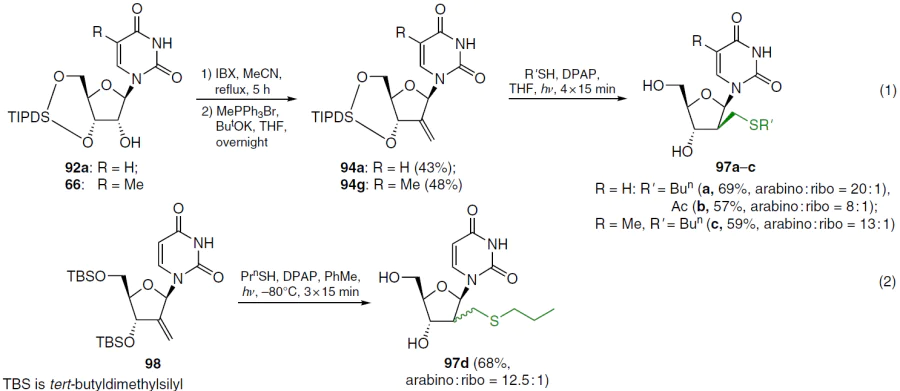
The use of tert-butyldimethylsilyl protection of the 5'- and 3'-hydroxy groups of uridine to obtain its 2'-alkylsulfanylmethyl derivatives is also described (see Scheme 18, reaction (2)). Carrying out the process at ‒80°C significantly increased the product yield and improved diastereoselectivity compared to a similar protocol at room temperature. The authors note that TIPDS protection is preferable, since the alternative method afforded the starting 3',5'-di-O-tert-butyldimethylsilyl nucleoside 98 in low yield, and the main reaction product was the regioisomeric 2',5'-di-O-tert-butyldimethylsilyl isomer.[77]
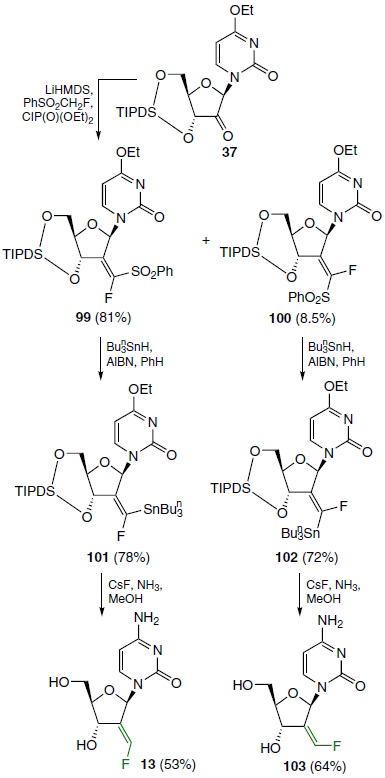
Using the Horner – Wittig reaction (or Julia – Lythgoe reaction), 2'-ketonucleoside 37 was converted into a mixture of easily separated fluorovinyl sulfones 99 (predominant product) and 100 (by-product). Next, two equivalents of tri-n-butylstannane were added to these compounds to obtain (fluorovinyl)stannanes 101 and 102 in good yields. Analysis of the crude reaction mixtures by 19F NMR spectroscopy confirmed the selectivity of the reaction, showing the absence of isomer 101 in product 102 and vice versa. Then (E)-2'-deoxy-2'-(fluoromethylidene)cytidine (13) was synthesized directly from compound 101 by treating it with a CsF – NH3 – MeOH mixture. (Z)-2'-Deoxy-2'-(fluoromethylidene)cytidine (103) was prepared from compound 102 under similar conditions (Scheme 19).[78]
Uridine was converted to 5'-O-TBDPS-2,2'-O-anhydrouridine 104 (TBDPS is tert-butyldiphenylsilyl), which was sequentially treated with 1,1'-carbonyldiimidazole and O-benzylhydroxylamine (Scheme 20).[79][80] The resulting 3'-O-(benzyloxyamino)carbonyl derivative without purification was converted into the 2'-N-benzyloxyamino-3'-O-carbonyl compound 105 in good yield. Treatment of this compound with cesium carbonate in methanol effectively cleaved the cyclic carbamate moiety to afford 2'-O-benzyloxyamino-2'-deoxyuridine 106. However, the latter did not give the desired 2'-hydroxylamino derivative 107 under hydrogenation conditions.
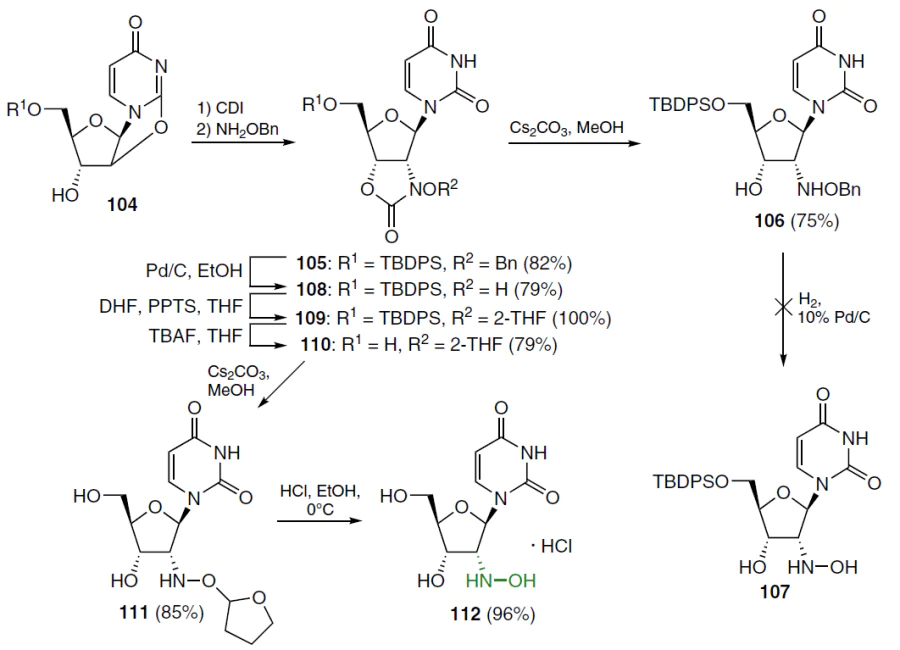
In an alternative method, hydrogenation of precursor 105 under the same conditions gave hydroxylamine 108. After protecting its N-hydroxy group with a tetrahydrofuran-2-yl (2-THF) moiety, compound 109 was obtained. The 5'-O-silyl group in this compound was removed with TBAF. Cleavage of the cyclic carbamate moiety in derivative 110 with CsCO3 in MeOH afforded analogue 111. Cleavage of tetrahydrofuranyl protecting group in the presence of 10% HCl in EtOH furnished the desired 2'-hydroxylamino-2'-deoxyuridine (2'-DHAU, 112) as the hydrochloride (see Scheme 20).[79][80]
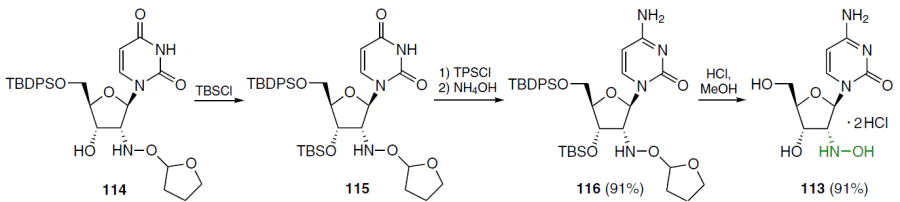
Cytidine analogue 113 was obtained from compound 114, in which the 3'-hydroxy group was first protected with TBS to give derivative 115 (Scheme 21).[79][80] From the latter, by a standard method, which involved the reaction with 2,4,6-triisopropylbenzenesulfonyl chloride in acetonitrile, O4-triisopropylbenzenesulfonate was obtained and converted without further purification into nucleoside 116 by treatment with NH4OH. The protecting groups of compound 116 were removed in a mixture of conc. HCl and MeOH to give 2'-hydroxylamino-2'-deoxycytidine (113) as dihydrochloride.
Among analogues of pyrimidine nucleosides modified at the 2'-position of the carbohydrate moiety, the synthesis of which is described in this Section, the compounds listed in Table 1 showed the most significant cytotoxicity against various tumour cell cultures.
The mechanisms of biological action of these drugs have been studied. For example, cytarabine (1) (see Fig. 1) enters cells primarily via the ENT1 transporter and is then sequentially phosphorylated by deoxycytidine kinase, deoxycytidylate kinase, and nucleoside diphosphate kinase (NDPK).[2] The effectiveness of this drug is directly related to the expression of these enzymes in the cell. The corresponding nucleotide is incorporated into the nascent DNA, leading to single-strand breaks, stalling the replication fork, and ultimately cell death.[2]
Gemcitabine (3) (see Fig. 1) enters cells primarily through the ENT1 and CNT3 transporters. Upon entering the cell, it is phosphorylated to the corresponding 5'-monophosphate (by dCK), 5'-diphosphate (by nucleotide monophosphate kinase (NMPK)) and 5'-triphosphate (by NDPK). Gemcitabine 5'-diphosphate inhibits ribonucleotide reductase, and its 5'-triphosphate terminates DNA synthesis, similar to cytarabine 5'-triphosphate. Furthermore, phosphorylated forms of gemcitabine can inhibit 3'-5' exonuclease, which removes some of the nucleotide analogues incorporated into DNA, enhancing their anticancer activity. These and other targets, as well as the mechanisms of action of gemcitabine (3), are described in detail.[2]
2'-C-cyano-2'-deoxy-1-β-D-arabinopentofuranosylcytosine (CNDAC, 11) (see Fig. 2) and its prodrug form sapacitabine (17) (see Fig. 3),[88] intended for oral administration,[20][89] have demonstrated clinical activity against AML.[90-92] The mechanism of action of CNDAC differs from that of other deoxycytidine analogues.[20] Following intracellular phosphorylation, the corresponding nucleotide is incorporated into the DNA chain; however, unlike cytarabine (1) and gemcitabine (3), this incorporation does not lead to inhibition of DNA replication, stalling of the replication fork, or arrest of the cell in S phase. Following incorporation of the next 2'-deoxynucleotide into DNA, instability of compound 11 causes β-elimination, resulting in conversion to 2',3'-didehydro-2',3'-dideoxy-2'-C-cyanocytidine (CNddC), which is virtually the 3'-terminal DNA chain terminator. The resulting single-strand breaks cannot be repaired by ligation and are converted to double-strand breaks during subsequent DNA replication.[93] Thus, after incubation of leukemic cells with cytostatic concentrations of drug 11, inhibition of the cell cycle in the S phase and its arrest in the G2 phase occur.[94] Acquired resistance to CNDAC is primarily mediated by a decrease in the activity of deoxycytidine kinase, which catalyzes the first step of phosphorylation of this compound.[95] The sensitivity of leukemia cells to CNDAC (11) is also critically dependent on the level of the SAMHD1 protein. This protein is a triphosphohydrolase that breaks down nucleoside triphosphates into deoxyribonucleosides and inorganic triphosphate.[95] Triphosphorylated forms of some anticancer nucleoside analogues, in particular cytarabine (1) and CNDAC (11), are substrates of this enzyme,[95][96] therefore high levels of SAMHD1* predetermine the low efficacy of therapy with these drugs in acute myeloid leukemia, acute lymphoblastic leukemia and Hodgkin's lymphoma.[97-99]
Tezacitabine (13) (see Fig. 2), being a structural analogue of deoxycytidine, like gemcitabine (3), is poorly susceptible to metabolism by cytidine deaminase.[100] Tezacitabine 13 is an antimetabolite inhibitor of human ribonucleotide reductase (hRR),[9] which consists of three subunits: hRRM1, hRRM2, and p53R2. The 5'-diphosphate of tezacitabine irreversibly inhibits the hRRM1 subunit, and its 5'-triphosphate is a terminator substrate for DNA polymerases.[100] Clinical evaluation of tezacitabine (13) included phase I and II studies, both as a monotherapy of refractory solid tumours and in combination with other agents, viz., cisplatin (for the treatment of gastrointestinal cancer) and 5-fluorouracil (for the treatment of advanced solid tumours). In all three cases, the trials did not progress beyond phase II due to lack of efficiency.
2'-Deoxy-2'-methylidenecytidine (DMDC, 86a) is also a structural analogue of deoxycytidine and is activated by intracellular phosphorylation to the corresponding 5'-diphosphate and 5'-triphosphate. The diphosphate inhibits the hRRM1 subunit, while the triphosphate inhibits DNA biosynthesis. These two nucleotide metabolites act in combination to cause DNA strand breaks, leading to apoptosis. Compound 86a is resistant to cytidine deaminase and has proven effective in xenograft cancer models where cytidine deaminase activity is high and the response to gemcitabine (3) is typically low. Phase I trials of DMDC as a solid tumour treatment revealed high levels of hematological toxicity, which suspended its development as a chemotherapeutic agent.[101] Subsequently, analogues 58, 86a and 86b demonstrated significant cytotoxic effects against T-cell leukemias in cell cultures and animal models.[87]
Thus, modification of the 2'-pyrimidine nucleoside position has proven its effectiveness and led to the development of several drugs. Currently, active research is underway to create depot forms and prodrugs to improve the pharmacokinetic properties, efficiency, and safety of existing drugs, as well as to explore various targeted delivery strategies and synthesize new analogues.
* SAMHD1 is a human protein 1 containing the SAM and HD domains.
3. Pyrimidine nucleoside analogues modified at the 3' position of the carbohydrate moiety
3'-Substituted pyrimidine nucleoside derivatives have attracted the interest of scientists for several decades. To date, many nucleoside analogues have been synthesized; however, unlike the compounds presented in the previous Section, none of them have been approved for cancer treatment (Fig. 6).
![[{"id":"PLFtPdGbdo","type":"paragraph","data":{"text":"General structure of pyrimidine nucleosides. The arrow indicates the position of the 3' carbohydrate moiety."}}]](/storage/images/resized/Prt11l46iBLtOFLempu8O3oWLCCuo0ttHEgA9GET_xl.webp)
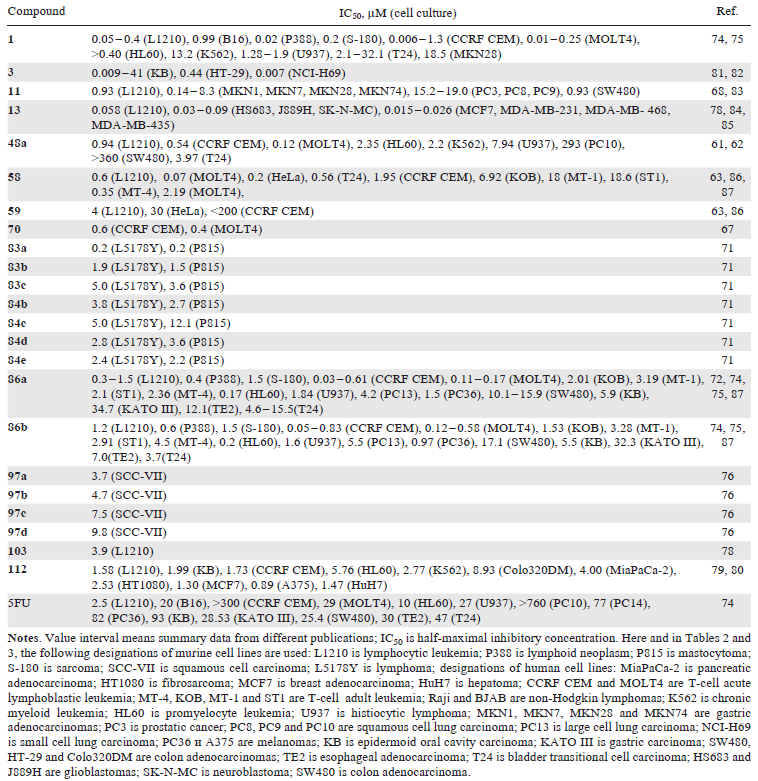
3'-Ethynyl-substituted nucleosides 14, 117 – 120 were synthesized by condensation of sugar 121, obtained in six steps from D-xylose,[102][103] with silylated nitrogenous bases under Vorbruggen reaction conditions.[104] Bis(trimethylsilyl)cytosine was reacted with compound 121 in the presence of tin tetrachloride in acetonitrile (Scheme 22). The target 2',3',5'-tri-O-benzoyl-3'-ethynylcytidine (122) was obtained as the major product in high yield.[103] Condensation of sugar 121 and the corresponding pertrimethylsilylated nucleobases (5-fluorocytosine, 5-fluorouracil, thymine) under the same conditions afforded the corresponding protected nucleoside precursors 118 – 120 (the structures are shown in the lower part of Scheme 22) in high yields. The reaction of bis(trimethylsilyl)uracil under the conditions described above yielded a mixture of N1- and N3-nucleosides in yields of 58% and 20%, respectively. However, if TMSOTf (TfO is trifluoromethanesulfonate (triflate)) was used as the Lewis acid instead of tin tetrachloride, the yield of product 123 reached 95%. To remove the protecting groups, nucleosides 122 and 123 were treated with ammonia in methanol at room temperature, and 3'-C-ethynyl nucleosides 14 (Ref. [105]) and 117 were isolated in crystalline form in high yields. Compounds 118 – 120 were prepared in a similar manner.
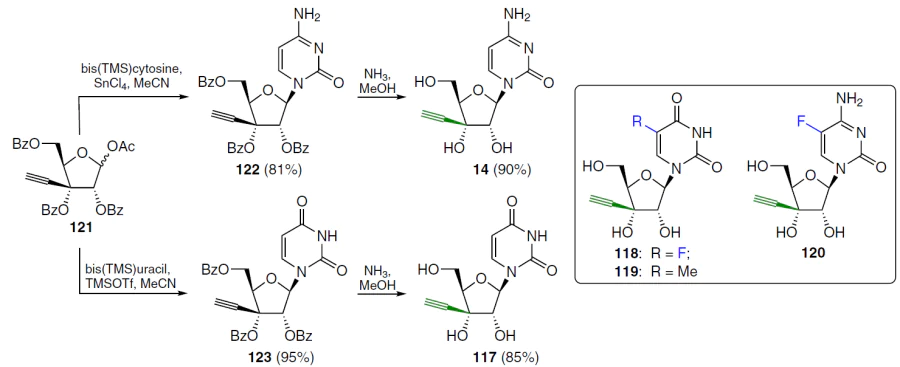
A further development of this area was the synthesis of uridine and cytidine derivatives containing propyn-1-yl, butyn-1-yl, vinyl, cyclopropyl, and ethyl groups at position 3'.[103] The synthesis of such compounds, shown in Scheme 23, involved the condensation of silylated uracil or cytosine with acylated sugar 124 in acetonitrile in the presence of TMSOTf (in the case of uracil) or SnCl4 (in the case of cytosine), followed by deprotection of intermediates 125 and 126 with NH3 in methanol. This gave 3'-C-substituted uridine derivatives 127a – e (yield 81 – 98%) and cytidine derivatives 128a – e (yield 68 – 94%).
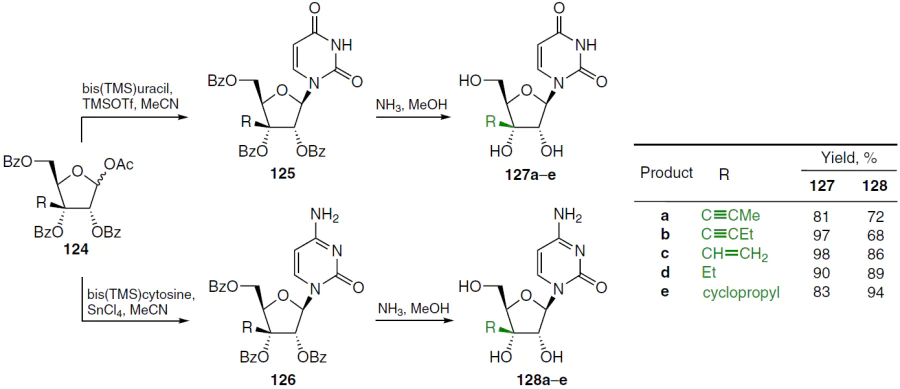
1-(3'-C-ethynyl-α-D-xylo-furanosyl)pyrimidines 129 and 130 (Scheme 24), which are 3'-epimers of compounds 14 and 117, were also synthesized. The reaction of the 2',5'-di-O-tert-butyldimethylsilyl-3'-ketouridine derivative (131) with HC≡CMgBr in THF at ‒78°C gave the xylo-adduct 132 with high stereoselectivity.[106][107] Similarly, the N4-acetyl-3'-ketocytidine derivative 133 was converted to xylofuranoside 134. Removal of the protecting groups in compounds 132 and 134 under standard conditions led to the corresponding 3'-C-ethynyl-β-D-xylofuranosyl derivatives of uracil (129) and cytosine (130).[103]
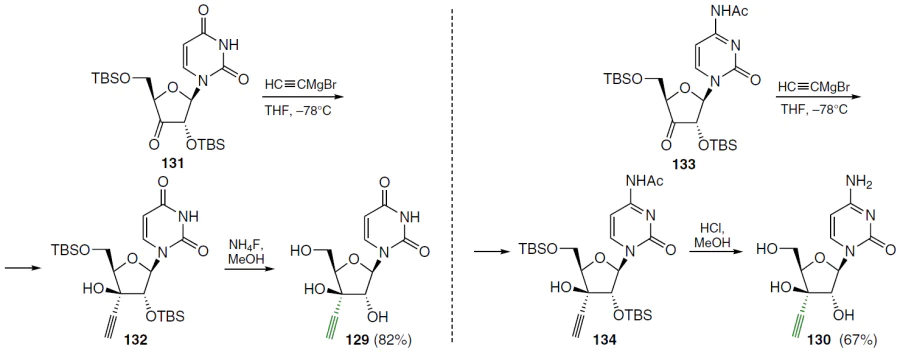
To determine the role of the 3'-hydroxyl group of compounds 14 and 117 in cytotoxicity, their 3'-deoxy analogues 136 – 138 were obtained (Scheme 25). In the starting compound 139 (Ref.[108]), the 5'-hydroxy group was protected by the action of benzoic anhydride in acetonitrile in the presence of DMAP, which gave derivative 140. Subsequent acid hydrolysis of the 2'-O-tert-butyldimethylsilyl group with a solution of HCl in MeOH afforded compound 141 (yield 56%). Treatment of this intermediate with 1,1'-thiocarbonyldiimidazole in acetonitrile afforded 2',3'-cyclic thiocarbonate 142, which was refluxed without further purification in the presence of tri-n-butylstannane and AIBN in toluene. This afforded an inseparable mixture of epimeric 3'-deoxy derivatives 143 in a ratio of ~ 4 : 1 in a high yield. The mixture was treated with tert-butylchlorodimethylsilane in the presence of imidazole in DMF and then separated by silica gel column chromatography. This furnished protected nucleosides 144 and 145, with the former predominating. Compound 144 was converted to the cytosine derivative 138 by reaction with 2,4,6-triisopropylbenzenesulfonyl chloride in acetonitrile in the presence of DMAP, followed by treatment with NH4OH. Removal of the protecting groups in epimers 144 and 145 afforded the target nucleosides 136 and 137 in quantitative yields.[60]
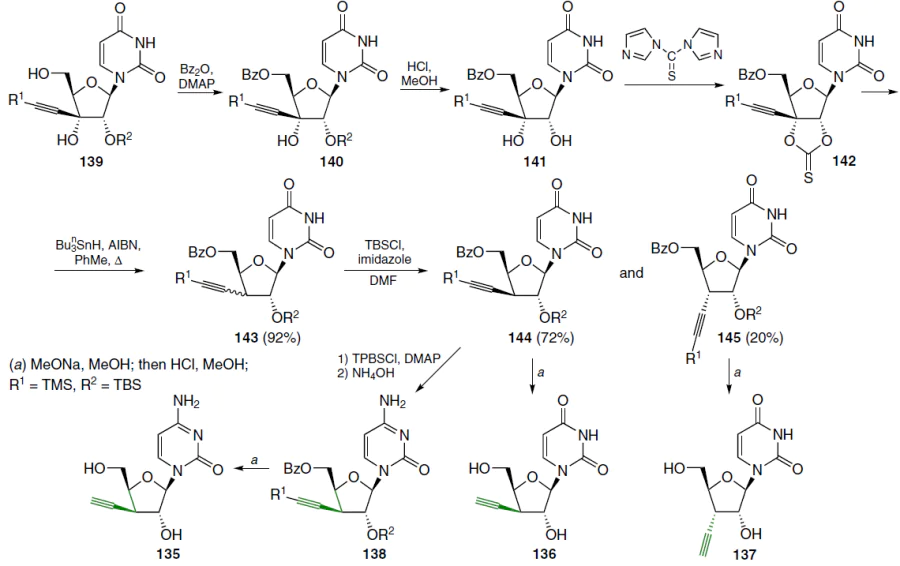
A study of the antitumour activity of compounds 14 and 117 – 120 in vitro showed compounds 14 and 117 to be the most potent inhibitors of the proliferation of L1210 and KB tumour cells (Table 2).[103]The activity of these derivatives 14 and 117 was also assessed against 36 human solid tumour cell lines. Cytidine analogue 14 demonstrated efficiency against 22 cell lines, with IC50 values ranging from nanomolar to subnanomolar, and was inactive against two cell lines. At the same time, the uridine analogue 117 had strong inhibitory activity against 12 cell lines with IC50 values in the subnanomolar range and was ineffective against six cell lines.[103]

In vitro studies of the antitumour activity of compounds 127a – e and 128a – e revealed that they either inhibited the growth of L1210 and KB cell lines 3 – 4 orders of magnitude worse than analogues 14 and 117, or were completely inactive. Evaluation of the efficiency of nucleoside analogues 136 – 138 in suppressing L1210 and KB cell growth showed that these compounds had no activity at concentrations below 350 μM.[60] Other analogues of ribo-, arabino- and deoxyribonucleosides containing various substituents at position 3' (methyl, fluoromethyl, hydroxymethyl, aminomethyl, azidomethyl and other groups) were also obtained;[109] however, they also did not exhibit noticeable cytotoxic properties in vitro even at the highest concentration (100 μM).
In vivo studies of compounds 14 (0.25 mg/kg dose) and 117 (2 mg/kg) in various human tumours as xenografts in nude mice demonstrated a more potent antitumour effect of these analogues compared to 5-fluorouracil (15 mg/kg).[103] Each of the doses for 14, 117, and FU used in this study represented the respective maximum nontoxic dose.[103][105]
The most interesting representative of the 3'-modified pyrimidine nucleoside analogues is 1-(3'-C-ethynyl-β-D-ribofuranosyl)cytosine (3'-ethynylcytidine (ECyd), TAS-106, 14) (see Fig. 2), which has demonstrated antiproliferative activity both in vitro and in vivo.[103][110-114] The cytotoxic mechanisms of compound 14 have been shown to be primarily related to the inhibition of RNA synthesis.[110][111][115] This nucleoside analogue is converted by uridine cytidine kinase (UCK, EC 2.7.1.48 enzyme classification) to 3'-ethynylcytidine-5'-monophosphate,[116] which is then phosphorylated to 3'-ethynylcytidine-5'-diphosphate (ECDP) and finally to 3'-ethynylcytidine-5'-triphosphate. The UCK family consists of two enzymes, UCK1 and UCK2 (Ref. [117]), and catalyzes the phosphorylation of uridine and cytidine to uridine 5'-monophosphate and cytidine 5'-monophosphate, respectively. These enzymes also catalyze the phosphorylation of several nucleoside analogues, such as 6-azauridine, 5-fluorouridine, and 5-methylcytidine.[118][119] UCK2 has been shown to be responsible for the phosphorylation of ECyd (14) under biological conditions.[120][121] 3'-Ethynylcytidine, which contains a covalently linked ethynyl group, exhibits pronounced cytotoxicity and radiosensitizing activity. As an analogue of cytidine, in the form of its 5'-triphosphate, it acts as a non-selective competitive inhibitor of the biosynthesis of all three RNA polymerases, thereby suppressing RNA synthesis.[21][122] 3'-C-Ethynylcytidine is an effective inhibitor of proliferation of > 40 types of cultured cancer cells, as well as human solid tumours xenografted into mice.[21][116] This drug has undergone phase I and phase II clinical trials under the commercial codes NCT00752011 and NCT00737360.[21] Phase I trials concluded that ECyd (14) can be administered either as an infusion or as a bolus injection, and that the main dose-limiting side effect is its neurotoxicity.[123-125] Phase II clinical trials showed no significant benefit from monotherapy with this drug. Further studies were ceased due to lack of efficiency and the occurrence of side effects.[126][127]
4. Pyrimidine nucleoside analogues modified at the 4'-position of the carbohydrate moiety
The literature describes the synthesis of pyrimidine nucleoside analogues modified at the 4' position of the carbohydrate moiety (Fig. 7).
![[{"id":"h1citm6w8t","type":"paragraph","data":{"text":"General structure of pyrimidine nucleosides. The arrow indicates the 4’ position of the carbohydrate moiety."}}]](/storage/images/resized/DxZIj3Z3ekSfyVTJ4p4VzYuOWwY9DuE4Jiz2Lbc2_xl.webp)
4.1. 4'-Methyl pyrimidine nucleosides
The synthesis of 4'-C-methyl derivatives consisted of the condensation of 1,2-di-O-acetyl-3,5-di-O-benzyl-4-C-methyl-β-D-ribofuranose (146) with silylated uracil under the conditions of the Vorbruggen method (TMSOTf, dichloromethane, room temperature, 2 h), leading to 2'-O-acetyl-3',5'-di-O-benzyl-4'-C-methyluridine (147), the yield of which was 93% after chromatographic purification (Scheme 26).[128] Deacetylation of compound 147 with saturated methanolic ammonia and subsequent debenzoylation by hydrogenolysis on Pd/C in methanol afforded 4'-C-methyluridine (148). To obtain 4'-C-methylcytidine (149), nucleoside 148 was converted to 2',3',5'-tri-O-acetyl-4'-C-methyluridine (150), which was treated with 4-chlorophenyl phosphorodichloridate in the presence of 1,2,4-triazole in pyridine. This gave 1-(2',3',5'-tri-O-acetyl-4'-C-methyl-β-D-ribofuranosyl)-4-(1,2,4-triazol-1-yl)-1H-pyrimidin-2-one (151) (yield 91%), which was dissolved in a mixture of NH4OH-dioxane and stirred for 3 h at room temperature to afford the target 4'-C-methylcytidine (149) in moderate yield.[128]
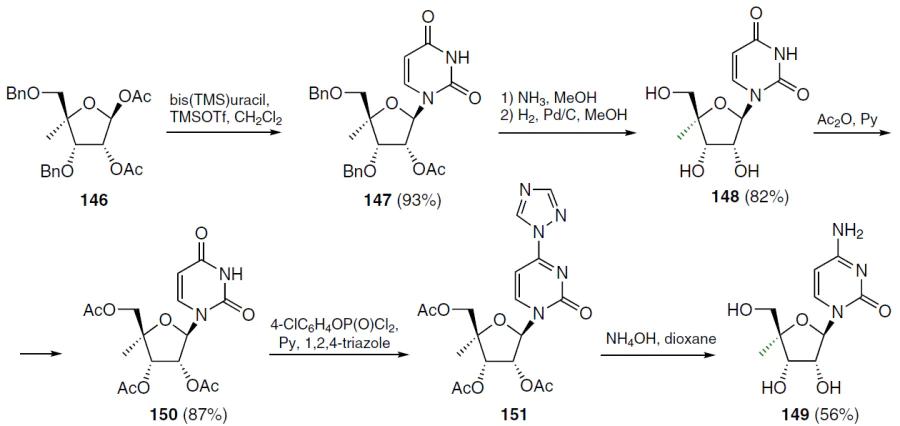
The synthesis of 2'-deoxy-4'-C-methylcytidine (152) commenced with the treatment of the starting 4'-C-methyluridine (148) with excess acetyl bromide in boiling acetonitrile, which furnished 2'-bromo-2'-deoxy derivative 153 (Scheme 27).[129] Heating a solution of compound 153 in toluene in the presence of tributylstannane and AIBN gave a mixture of 3',5'-di-O-acetyl-2'-deoxy-4'-C-methyluridine (154) and 3',5'-di-O-acetyl-4'-C-methyl-2,2'-anhydrouridine (155), with the former predominating. Using the methods described above, nucleoside 154 was converted first to triazolide 156 and then to the target 2'-deoxy-4'-C-methylcytidine (152).
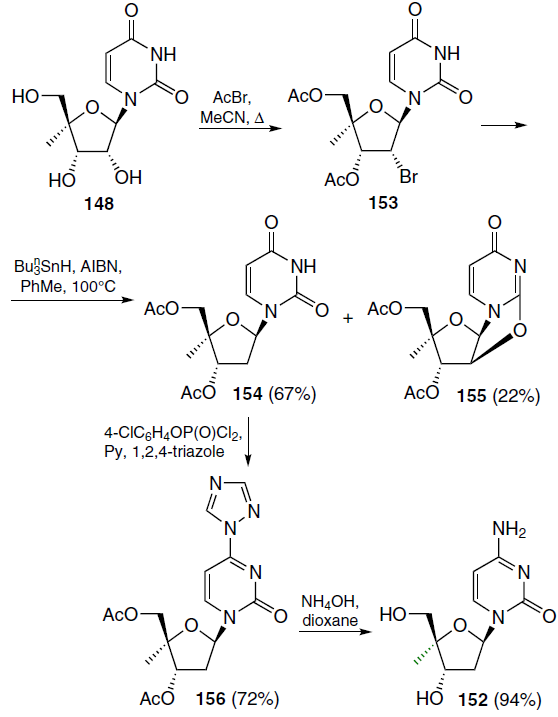
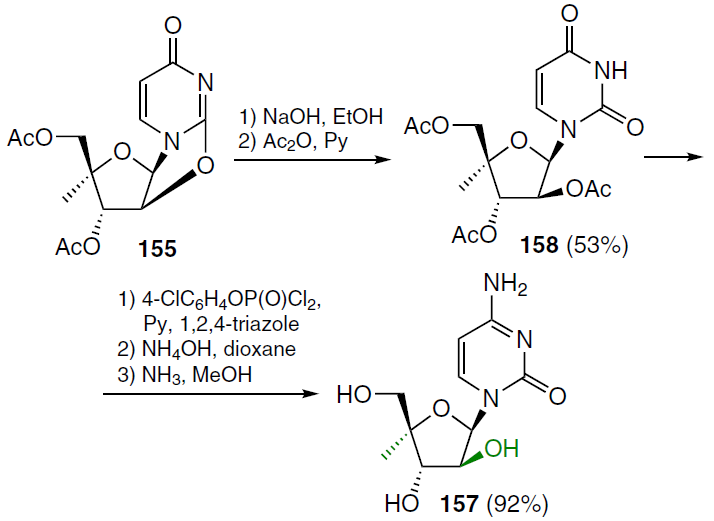
The reaction sequence used to prepare 1-(4'-C-methyl-β-D-arabinofuranosyl)cytosine (157) is shown in Scheme 28.[129] The starting 3',5'-di-O-acetyl-4'-C-methyl-2,2'-anhydrouridine (155) was treated sequentially with 1N NaOH in ethanol and acetic anhydride in pyridine to afford 1-(2',3',5'-tri-O-acetyl-4'-C-methyl-β-D-arabinofuranosyl)uracil (158) in moderate yield. The latter was treated with 4-chlorophenyl phosphorodichloridate in the presence of 1,2,4-triazole, followed by reaction with NH4OH-dioxane and then with NH3 in MeOH gave the desired nucleoside 157 in high yield.[129]
Iodination of the 5-position of the uracil residue in compound 154 with iodine in the presence of cerium(IV)-ammonium nitrate (CAN)[130] followed by the Heck reaction with methyl acrylate afforded the 5-methylacrylate derivative 159 in 56% yield. Сompound 159 was hydrolyzed under alkaline conditions; acidification of the reaction mixture with HCl afforded carboxylic acid 160, which was purified using reverse-phase octadecylsilyl (ODS) ion-exchange resin column chromatography. Final treatment of compound 160 with anhydrous KHCO3 and N-bromosuccinimide (NBS) in DMF[131] gave the target product 161 in high yield (Scheme 29).[132]
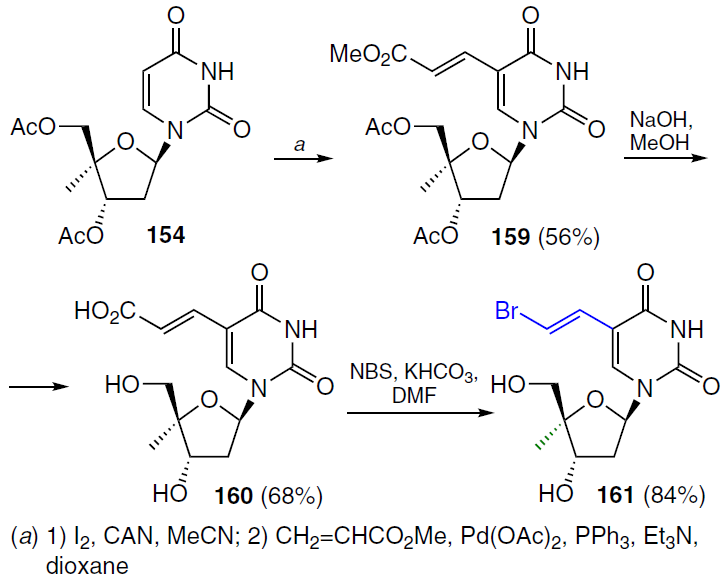
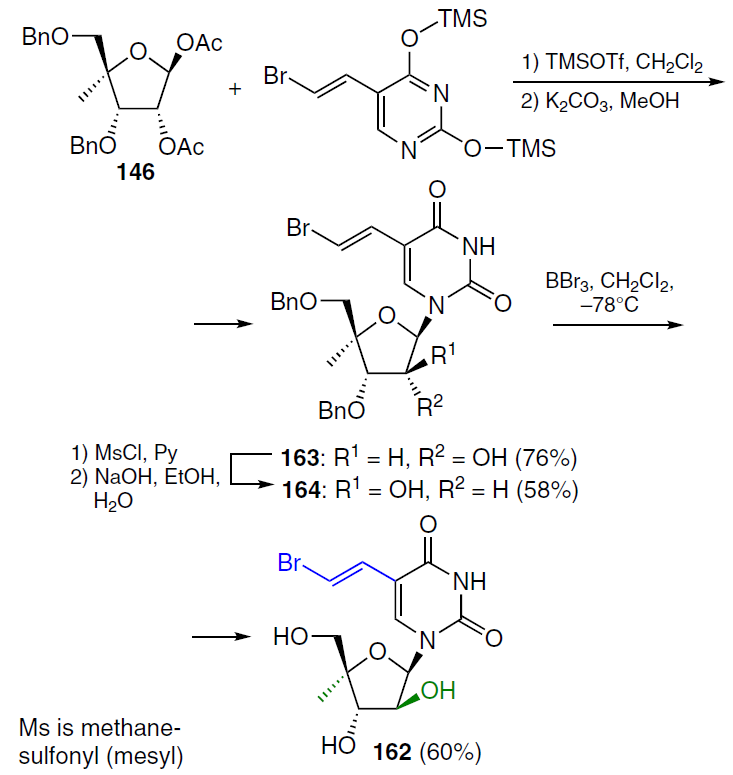
The synthetic route to 5-(2-bromovinyl)-1-(4'-C-methyl-β-D-arabinofuranosyl)uracil (162)[132] is shown in Scheme 30. Di-O-benzylated ribonucleoside 163 was prepared by condensation of protected 4-C-methyl-D-ribose 146 with silylated 5-bromovinyluracil in the presence of TMSOTf followed by treatment with potassium carbonate in methanol (yield 76% over two steps). Compound 163 was converted to an intermediate mesylate, which, when treated with an aqueous alcoholic NaOH solution, gave arabinonucleoside 164. Debenzoylation of the latter with BBr3 in dichloromethane at ‒78°C and subsequent quenching with a saturated Na2CO3 solution afforded the target analogue 162 in moderate yield.
4.2. 4'-Fluoromethyl pyrimidine nucleosides
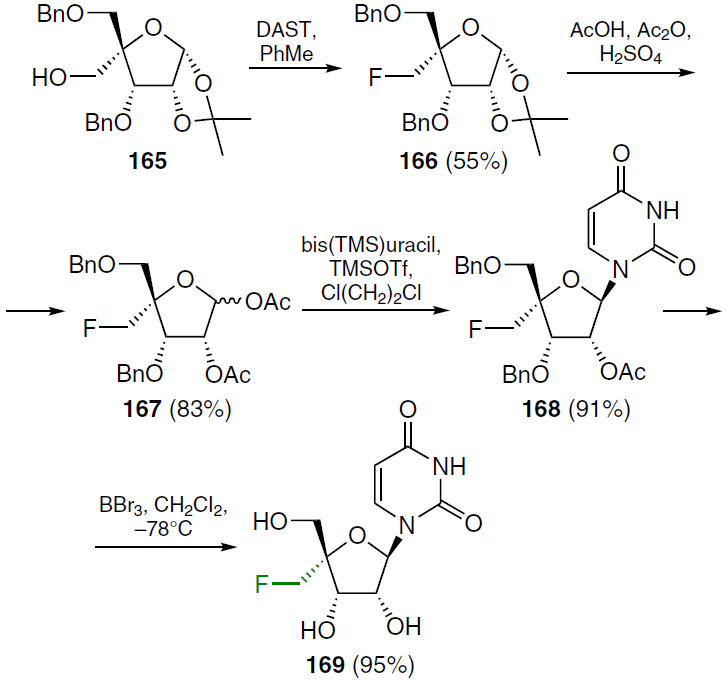
The preparation of nucleoside analogues with a fluoromethyl group at the 4'-position has been reported.[133] The starting protected 4-C-hydroxymethyl-D-ribofuranose (165) was treated with DAST in toluene at 60°C to give the corresponding 4'-fluoromethyl derivative 166 in moderate yield. Subsequent acetolysis of this compound gave a mixture of α- and β-anomers of 1,2-di-O-acetyl-3,5-di-O-benzyl-4-C-fluoromethylribofuranose (167), which was condensed in 1,2-dichloroethane with silylated uracil in the presence of TMSOTf. After chromatographic purification, 2'-O-acetyl-3',5'-di-O-benzyl-4'-C-fluoromethyluridine (168) was isolated in high yield. Compound 168 was treated with BBr3 in dichloromethane at ‒78°C to afford 4'-C-fluoromethyluridine (169) in 95% yield (Scheme 31).[133]
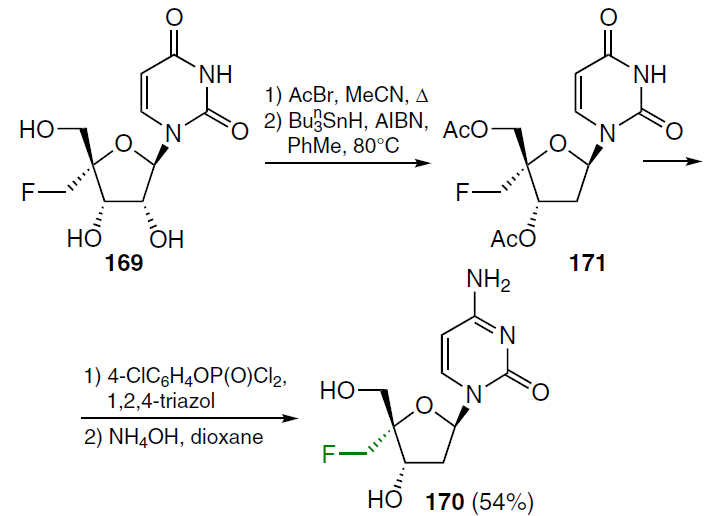
The first step in the synthesis of 2'-deoxy-4'-fluoromethylcytidine (170), shown in Scheme 32, involved treating ribonucleoside 169 with acetyl bromide in boiling acetonitrile and subsequent heating in the presence of tributylstannane and AIBN in toluene at 80°C to afford 2'-deoxy-3',5'-di-O-acetyl-4'-fluoromethyluridine (171). Compound 171 was treated sequentially with 4-chlorophenyl phosphorodichloridate, then with 1,2,4-triazole and NH4OH in dioxane. This gave 2'-deoxy-4'-fluoromethylcytidine (170) in 54% yield.[133]
4.3. 4'-Azidopyrimidine nucleosides
4'-C-Azido-β-D-ribofuranosyl nucleosides and their 2'-deoxy derivatives 172 – 176 were prepared in several steps (Scheme 33).[134] The synthesis of such analogues began with the conversion of the starting nucleosides 177 to 5'-deoxy-5'-iodo derivatives 178 using the PPh3 – I2 system. Subsequent treatment with sodium methoxide in methanol afforded compounds 179, which were reacted with ICl and sodium azide in DMF. This afforded the target 4'-C-azido derivatives 180 as a mixture with the by-product 4'-epimer in a ratio of ~ 20 : [134]
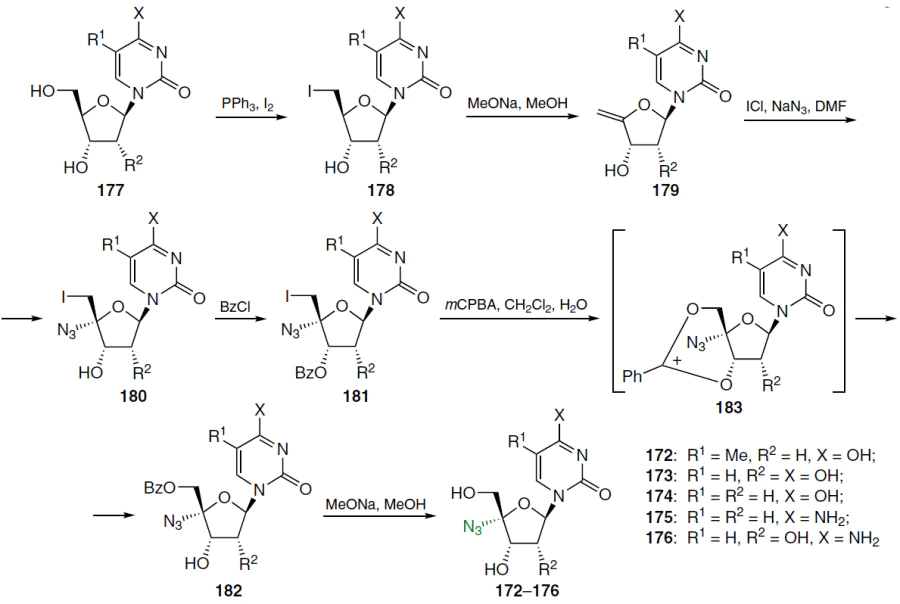
The conversion of the iodine atom at position 5' to a hydroxyl group is the greatest challenge. The reactivity of this iodine atom is reduced if the 4' carbon atom contains an electron-withdrawing substituent (e.g., an oxygen atom). To stabilize the nucleoside, it should be converted to 3'-O-benzoyl derivative 181 via esterification, after which the iodine atom can be activated by oxidation to the corresponding hypervalent state. Treatment of 4'-C-azido-3'-O-benzoyl-5'-deoxy-5'-iodine nucleoside 181 with m-chloroperoxybenzoic acid (mCPBA) in water-saturated dichloromethane was found to yield a number of products, with 5'-O-benzoyl-3'-hydroxy derivative 182 being isolated as the major component of the mixture (see Scheme 33). This compound is likely formed from the 3',5'-cyclic benzoxonium ion 183 by hydrolysis. The target nucleosides 172 – 176 were obtained in the individual state by hydrolysis of compounds 182 in the presence of a base followed by chromatographic purification.[134]
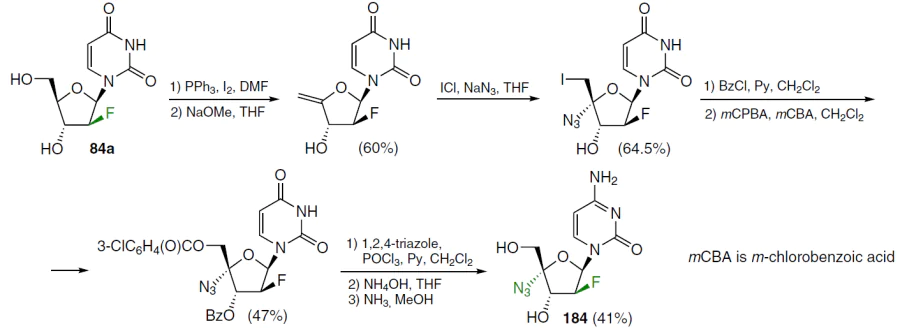
Similarly, azvudine[13] (184) was synthesized from 2'-deoxy-2'-fluoroarabinouridine (84a), obtained in three steps from 1,3,5-tri-O-benzoyl-2-deoxy-2-fluoro-D-arabinofuranoside in 78% yield (Scheme 34).[135]
4.4. Derivatives with unsaturated hydrocarbon substituents at the 4'-position
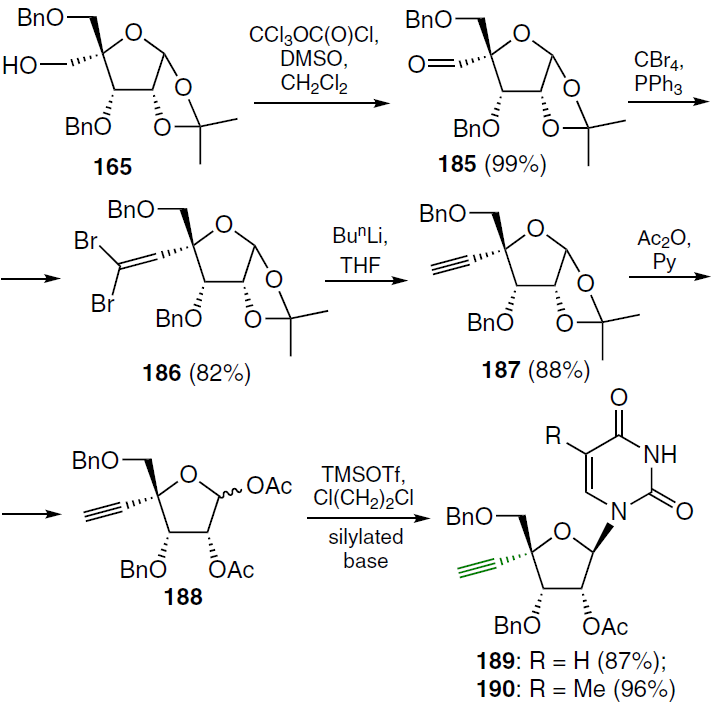
A series of nucleosides with an acetylene group at the 4' position of the carbohydrate moiety were obtained.[136] The synthesis of such compounds involved several steps (Scheme 35). In the first step, the starting riboside 165 (Ref. 128) was oxidized with a DMSO-diphosgene mixture in dichloromethane, which gave 4-aldehyde 185 in quantitative yield. It was treated with CBr4 and PPh3 under the Wittig reaction conditions to afford 4-(2,2-dibromovinyl) derivative 186. Treatment of the latter with n-butyllithium in THF gave 4-ethynyl-containing carbohydrate 187, which was hydrolyzed and then acetylated with the Ac2O-Py system to form the 1,2-di-O-acetyl derivative 188 (yield 88%). Further condensation with a silylated pyrimidine base (thymine or uracil) was carried out in the presence of TMSOTf in 1,2-dichloroethane. Uracil (189) and thymine (190) derivatives were isolated in high yields.[136]
The second step in the synthesis of the target nucleosides involved the transformation of the ribofuranosyl moiety of compound 189 into an arabinoside (Scheme 36).[136] For this purpose, nucleoside 189 was successively subjected to alkaline hydrolysis, treatment with MsCl in pyridine, and a NaOH solution in aqueous THF, which afforded 1-(4'-C-ethynyl-β-D-arabinofuranosyl)uracil (191) in good yield. Further reaction with BBr3 in dichloromethane and subsequent acetylation gave the 2',3',5'-tri-O-acetyl derivative 192, which was treated with 4-chlorophenyl phosphorodichloridate and 1,2,4-triazole in pyridine to form the corresponding 4-triazole derivative 193. The latter was converted to the target 1-(4'-C-ethynyl-β-D-arabinofuranosyl)cytosine (190) under basic conditions.
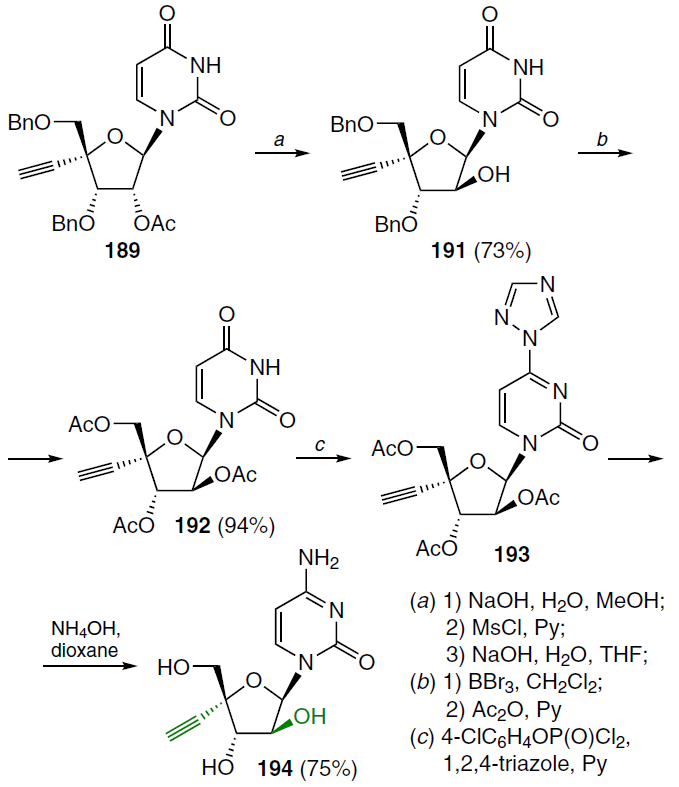
2'-Deoxy-4'-C-ethynylcytidine (195) was synthesized from 3',5'-di-O-acetyl-4'-C-triethylsilylethynyl-2'-deoxyuridine (196) (Scheme 37).[136] Starting compound 196 was treated with 4-chlorophenyl phosphorodichloridate and 1,2,4-triazole in pyridine, which delivered the 4-triazole derivative 197. Further steps of reaction with NH4OH in dioxane and alkaline hydrolysis gave rise to the target 2'-deoxy-4'-C-ethynylcytidine (195) in moderate yield.
Scheme 38 shows the synthetic route to 4'-substituted 2'-deoxycytidines. Aldehyde 198 was the starting compound, which was treated with an aqueous solution of formaldehyde and NaOH in dioxane for 10 min at room temperature. Subsequent reduction of the aldehyde function at position 4' with NaBH4 afforded compound 199a. Intermediate compound 199, obtained by manipulation with the protecting groups, was oxidized to the corresponding 4'-C-aldehyde 200 under the Swern reaction conditions.[137] Intermediate 200 was isolated as crystals and used for the synthesis of the target nucleosides 152, 195, 201 – 205.[138]
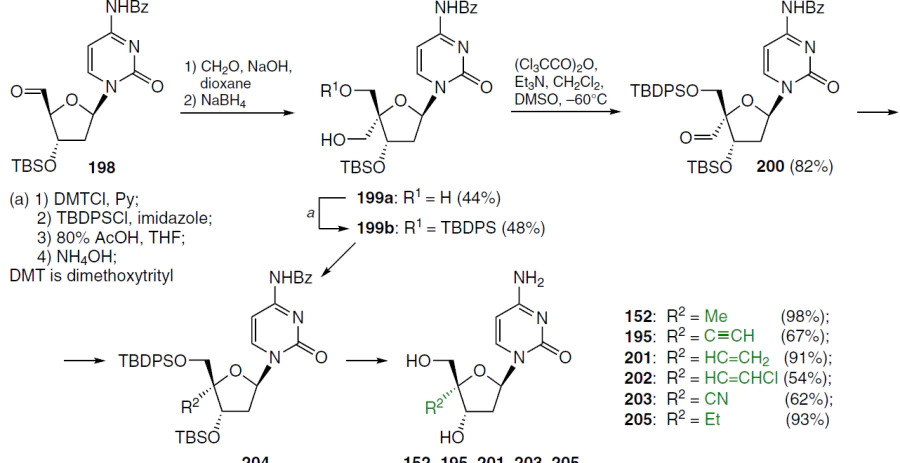
When subjected to the Wittig reaction with Ph3P=CH2 and Ph3P=CHCl, compound 200 gave 4'-C-ethenyl and 4'-C-(2-chloroethenyl) derivatives 204, (R2 = CH=CH2 and R2 = CH=CHCl respectively), with an isomer ratio of Z : E = 20 : 1; deprotection delivered the target nucleosides 201 and 202 in good yields. Compound 204 (R2 = CH=CHCl) was further treated with n-butyllithium in THF at ‒78°C to give debenzoylated 4'-C-ethynyl derivative deprotection of which led to 195.
Compound 200 was converted to the oxime 204 (R2 = CH=NOH), which was then dehydrated in a mixture of NaOAc and Ac2O and debenzoylated with 1,8-diazabicyclo- [5.4.0]undec-7-ene (DBU) in MeOH to form 4'-C-cyanide, which was deprotected to give product 203. 4'-C-Methyl derivative 152 was prepared from compound 199b using the I2 – imidazole – triphenylphosphine system via the 4'-C-iodomethyl analogue, which was hydrogenated on Pd/C in the presence of Et3N. Hydrogenation of compound 201 on Pd/C gave the 4'-C-ethyl derivative 205 (see Scheme 38).
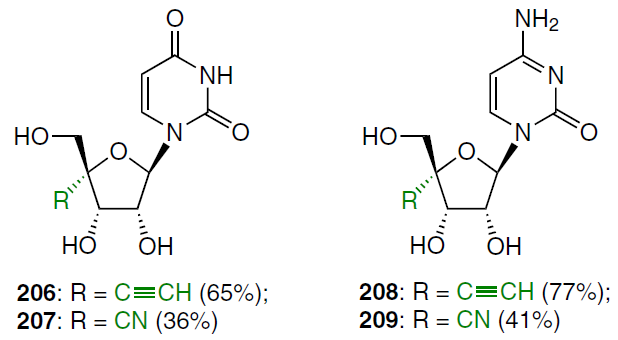
Ribonucleoside analogues were also synthesized: 4'-C-ethynyluridine (206), 4'-C-cyanouridine (207), 4'-C-ethynylcytidine (208) and 4'-C-cyanocytidine (209).
An in vitro study of the antitumour activity of 4'-modified nucleosides showed that compound 170 (see Scheme 32) exhibited significant activity against CCRF-HSB-2 lymphoblastic leukemia cells and had no significant effect on the growth of KB human oral carcinoma cells (Table 3).[133] Cytosine derivatives 175 and 176 (see Scheme 33) performed good in inhibiting the growth of the A3.01 cell line, a childhood acute lymphoblastic leukemia: complete inhibition of cell growth was recorded at IC100 concentrations of 1.9 and 74.1 μM, respectively.[134]
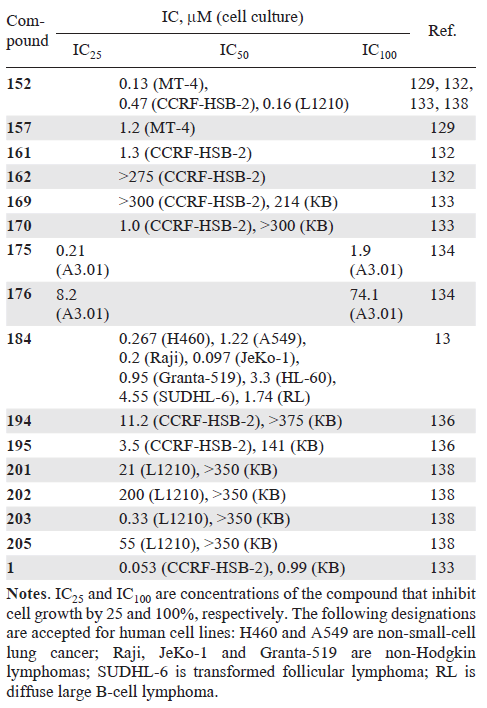
Data on the in vitro anticancer activity of azvudine (FNC, 184) (see Scheme 34)[13][19] are also presented in Table 3. A study of the antitumour activity of cytosine derivatives 194 and 195 against CCRF-HSB-2 and KB lymphoblastic leukemia cells showed that they exhibit significant activity in suppressing the growth of CCRF-HSB-2 cells.[133] The cytotoxicity of 4'-C-substituted nucleosides 152, 195, 201 – 203, and 205 – 209 was studied in vitro using L1210 mouse lymphocytic leukemia cells and KB cells (see Table 3). 4'-Methyl-substituted 2'-deoxycytidine 152 showed significant cytotoxicity against L1210 cells with an IC50 value of 0.16 μM. Analogues with cyano (203), ethynyl (195), ethenyl (201), and ethyl (205) groups at this position also exhibited cytotoxicity with IC50 values of 0.33, 0.80, 21, and 55 μM, respectively. At the same time, the 4'-C-[(2Z)-chloroethenyl] derivative 202 showed only insignificant antiproliferative activity.
As is evident from the data in Table 3, the 4'-substituents in 2'-deoxycytidines affect cytotoxicity in the following order: Me (152) > CN (203) > C≡CH (195) > CH=CH2 (201) > Et (205) > CH=CHCl (202). Activity is apparently related to the bulkiness of the substituents. Since 2'-deoxycytidine analogues can be phosphorylated by deoxycytidine kinase, and their cytotoxicity depends on the sensitivity of this enzyme to such compounds. The difference in activity against L1210 and KB cell lines may be related to the action of certain activation enzymes.[138] Ribonucleoside derivatives 206 – 209 did not exhibit cytotoxic properties.
Thus, the most promising representatives in terms of the presence of antitumour activity among 4'-modified analogues of pyrimidine nucleosides at the moment are 4'-methyl-2'-deoxycytidine (152) and azvudine (4'-azido-2'-deoxy-2'-fluoroarabinouridine, 184), which contains two substituents. Two more nucleoside analogues that combine a 4'-methyl substituent with other structural modifications — capecitabine (4) (see Fig. 1 ) and doxifluridine (10) (see Fig. 2 ) — are prodrugs of 5-fluorouracil and are mentioned in the next Section of this review. Based on the available literature data, it can be hypothesized that, unlike 2'-modified analogues of pyrimidine nucleosides, the introduction of one substituent at the 4'-position is not sufficient to exhibit cytotoxicity, and additional structural optimization is necessary to obtain highly active drugs of this class.
5. Modification of the heterocyclic backbone
5.1. 5-Fluorouracyl and fluoropyrimidines
The introduction of a fluorine atom into position 5 of the pyrimidine base has become one of the successful modifications of nucleosides in the development of drugs for the treatment of cancer (Fig. 8).[16]
![[{"id":"aZ_KL50qfM","type":"paragraph","data":{"text":" Structures of 5-fluoropyrimidines <b>2, 4, 7, 10, 25, 26, 210 – 213</b>. Modifications of the pyrimidine base are marked in blue, modifications of the carbohydrate moiety are marked in green, fragments of depot forms are marked in pink."}}]](/storage/images/resized/nsskE9ADjTu4G9OHttC6cF5VozIgX9bHP6peipyT_xl.webp)
Although the antitumour activity of 5-fluorouracil, the first and best-known drug in the fluoropyrimidine class, was discovered by Heidelberger et al.[139] in 1957, it remains the most commonly used chemotherapy drug today. It is effective against a wide range of solid tumours, is a central component of many combination treatment regimens for intractable cancers,[140] and is also used as monotherapy when more aggressive multidrug approaches are not feasible.[141][142]
The antitumour effect of 5-fluorouracil is due to its conversion into active intracellular metabolites: 5-fluoro-2'-deoxyuridine-5'-monophosphate (FdUMP), 5-fluoro-2'-deoxyuridine-5'-triphosphate (FdUTP), and 5-fluorouridine-5'-triphosphate (FUTP).[141][142] The metabolite FdUMP, having a high affinity for thymidylate synthase (TS), irreversibly inhibits this enzyme. Another mechanism of the cytotoxic effect of 5-fluorouracil is the interaction of FUTP with RNA polymerases and the incorporation of the corresponding nucleotide into RNA, leading to disruption of transcription, translation, and intracellular sorting of synthesized proteins.[141-143]
In 1957, a method for the synthesis of 5-fluorouracil was reported,[144] based on the interaction of ethyl pseudothiourea (214) with potassium 3-oxo-2-fluoro-3-ethoxypropenolate and subsequent hydrolysis (Scheme 39, Method A).
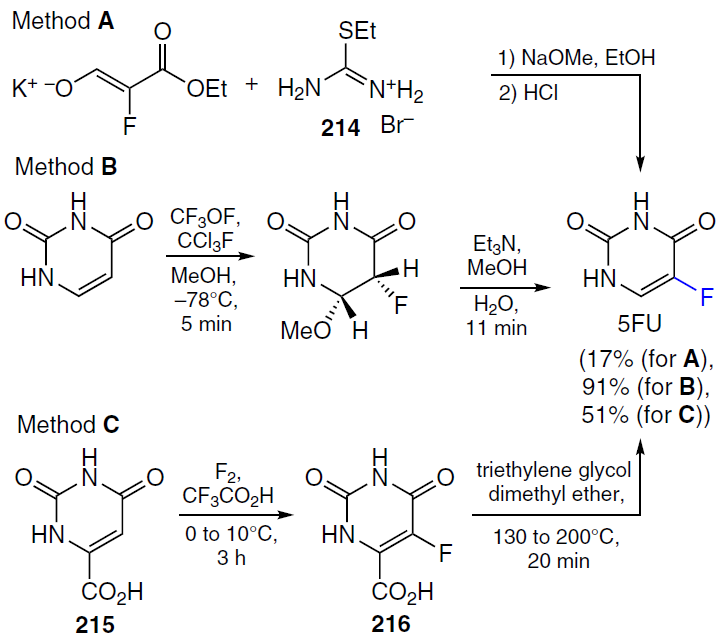
Two decades later, Robins et al.[145] presented a two-step protocol for the synthesis of 5-fluorouracil from uracil (see Scheme 39, Method B). In this procedure, trifluoromethyl hypofluorite was used to directly introduce a fluorine atom into the uracil ring, followed by an elimination reaction in methanol in the presence of a base to yield 5-fluorouracil. An alternative approach (method C) involved using inexpensive and readily available orotic acid (vitamin B13, 215) as the starting compound.[146] Its fluorination with gaseous fluorine in the presence of trifluoroacetic acid produced acid 216, which, after decarboxylation, delivered 5-fluorouracil in gram quantities.
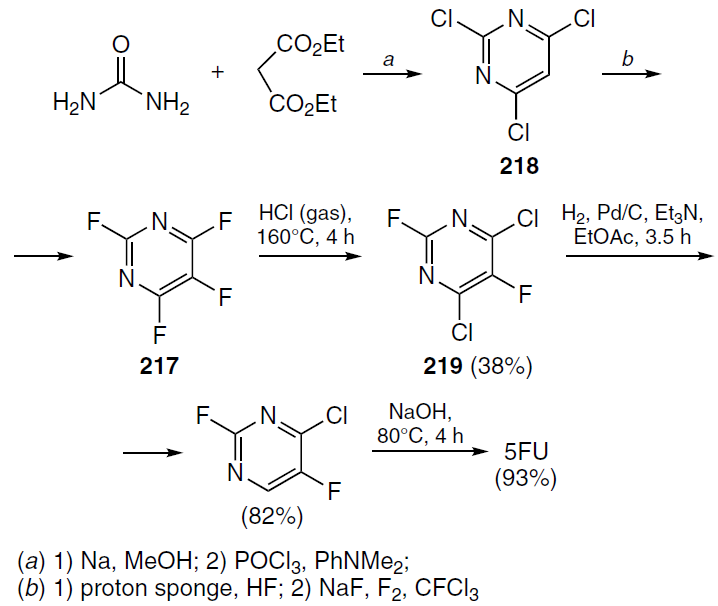
Another approach to 5-fluorouracil was proposed by Baasner and Klauke[147] (Scheme 40). This process involved the selective exchange of a fluorine atom for chlorine and subsequent hydrogenation. Tetrafluoropyrimidine (217) was synthesized in four steps from urea and diethyl malonate via the formation of 2,4,5-trichloropyrimidine (218). The substrate 217 was treated with gaseous hydrogen chloride, the dichloro derivative 219 was dechlorinated by palladium-catalyzed selective hydrogenation, and then treated with aqueous sodium hydroxide to yield 5-fluorouracil. These and other approaches to the synthesis of 5-fluorouracil and fluoropyrimidine-containing nucleoside analogs have been summarized and described in detail in review articles published in recent years.[1][14][16][148-150]
The bioavailability of unmodified 5-fluorouracil after oral administration can vary from 0% to 80% not only between patients but also within the same individual. This is primarily due to differences in the activity of dihydropyrimidine dehydrogenase, the enzyme responsible for the degradation of 5-fluorouracil. Due to the unpredictability and instability of the efficacy and toxicity of oral 5-fluorouracil, this drug is typically administered intravenously.
The development of effective fluoropyrimidine-based thymidylate synthase inhibitors continued with the advent of orally administered 5-fluorouracil-containing drugs such as capecitabine (4), tegafur, and the drug S-1, a combination of tegafur (25), gimeracil, and oteracil. These drugs are prodrugs, and their cytotoxicity is due to metabolic conversion to 5-fluorouracil.[25][151] Various methods of chemical synthesis and important aspects of the biological properties of 5-fluorouracil prodrugs, including floxuridine (2), capecitabine (4), doxifluridine (10), carmofur (210), NUC-3373 (211), etc., as well as combinations of fluoropyrimidines (trifluorothymidine (7) + tipiracil hydrochloride (8); tegafur (25) + uracil; tegafur (25) + gimeracil + oteracil; 5-fluoro-2'-deoxycytidine (26) + tetrahydrouridine (27)) are described in detail in a review by Shelton et al.1 published in 2016.
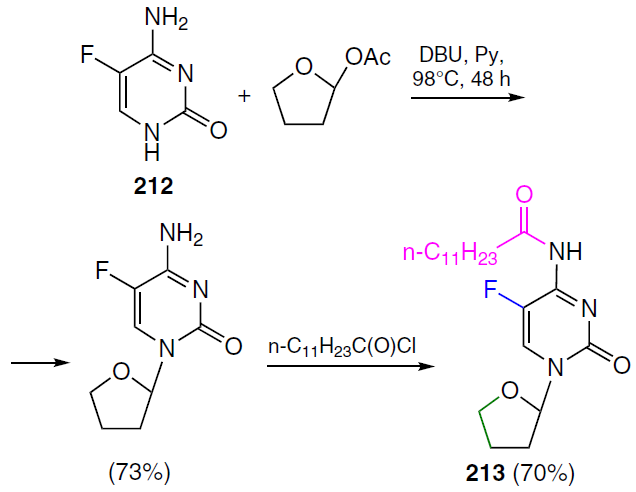
Frimpong et al.[152] proposed a 5-fluorouracil prodrug with the commercial code XYZ-I-73 (213), obtained by introducing a tetrahydrofuran ring into 5-fluorocytosine (212) and subsequent conjugation with lauroyl chloride (Scheme 41). The antiproliferative activity of this compound was demonstrated in MiaPaCa-2, PANC-1, and BxPC-3 pancreatic cancer cell lines. Prodrug XYZ-I-73 (213) demonstrated a more pronounced cytotoxic effect (IC50 = 3.6 ± 0.4 μM) compared to standard drugs gemcitabine (3) (IC50 = 24.2 ± 1.3 μM) and 5FU (IC50 = 13.2 ± 1.1 μM) and improved metabolic stability in vitro. It is of interest for further study as a potential drug for pancreatic cancer therapy.[152]
5.2. Azacitidine and decitabine
Azacitidine (5) and decitabine (6) are DNA methyltransferase (DNMT) inhibitors.[153] Both compounds are currently commercially available and approved for the treatment of myelodysplastic syndromes (MDS). Decitabine is also approved for use in patients with AML who have 20 – 30% blasts in the bone marrow.[154] These agents have shown excellent results in the treatment of elderly AML patients and those intolerant to more intensive therapy. Their relatively low toxicity allows them to be considered for maintenance therapy after initial treatment of AML.[155]
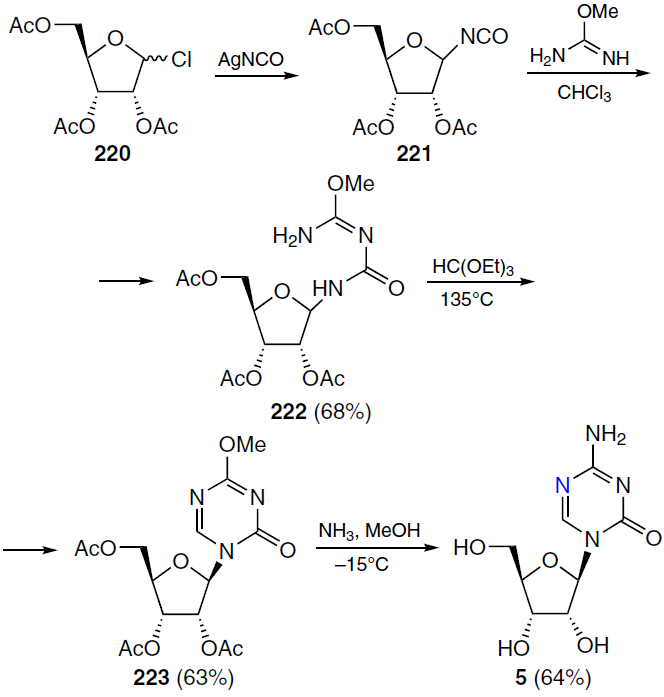
Azacitidine (5) (see Fig. 1), which has the international nonproprietary name 5-azacytidine, was isolated from the spore-forming bacteria Streptoverticillium ladakanus.[156] The synthesis of this compound was first described by Pískala and Šorm.[157] The authors described reaction of 1-chloro-2,3,5-tri-O-acetyl-D-ribofuranose (220) with silver cyanate, resulting in 2,3,5-tri-O-acetyl-β-D-ribofuranosyl isocyanate (221), which was treated without further purification with 2-methylisourea to obtain crystalline 1-(2,3,5-tri-O-acetyl-β-D-ribofuranosyl)-4-methylisobiuret (222). Condensation of this compound with ethyl orthoformate at 135°C afforded 1-(2,3,5-tri-O-acetyl-β-D-ribofuranosyl)-4-methoxy-2-oxo-1,2-dihydro-1,3,5-triazine (223), which, when treated with ammonia in methanol, yielded azacitidine (5) (Scheme 42). This method uses relatively readily available starting materials, and therefore, despite its complexity and multi-step nature, it remains relevant for the industrial synthesis of azacitidine (5).
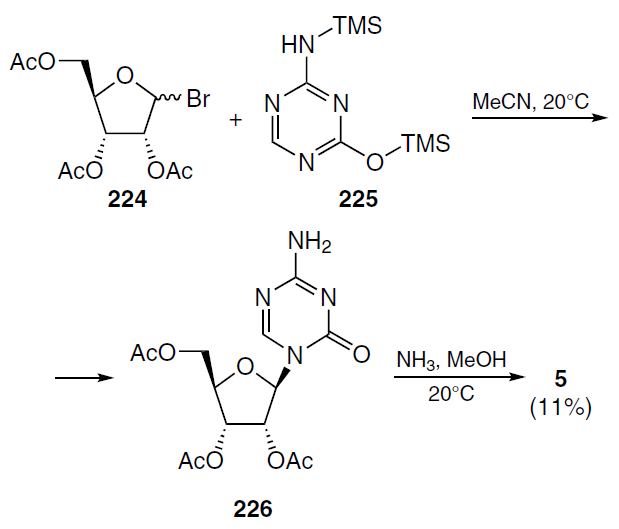
A method for the preparation of azacitidine based on the silyl version of the Gilbert – Johnson reaction was proposed by Winkley and Robins.[158] In this case, the condensation of 1-bromo-2,3,5-tri-O-acetyl-D-ribofuranose (224) with the trimethylsilyl derivative of 5-azacytosine 225 in anhydrous acetonitrile at room temperature gave compound 226. Removal of acetyl residues and subsequent purification allowed the preparation of azacitidine (5) in 11% yield (Scheme 43). The low yield of the target product limits the application of this method on an industrial scale.
The bioactivation of azacitidine begins with its phosphorylation by uridine cytidine kinase to 5-azacytidine-5'-monophosphate (5-AZA-CMP), which is converted to 5-azacytidine-5'-diphosphate (5-AZA-CDP) by cytosine nucleoside monophosphate kinase (CMPK). Nucleoside diphosphate kinase then phosphorylates 5-AZA-CDP to 5-azacytidine triphosphate (5-AZA-CTP), which is a substrate for RNA polymerases and disrupts protein synthesis. 5-AZA-CDP can be metabolized by ribonucleotide reductase to 2'-deoxyazacytidine-5'-diphosphate, the phosphorylated form of decitabine (6). After final phosphorylation, the decitabine-containing nucleotide acquires the ability to integrate into DNA and forms a covalent complex with DNMT, which leads to irreversible inhibition of the enzyme.
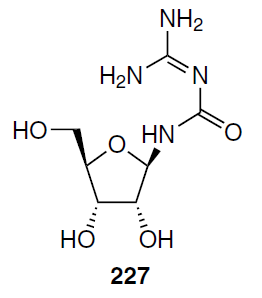
Azacitidine (5) has orphan drug status for the treatment of AML in Europe[159] and the United States.[160] It is also used to treat adult patients who are not candidates for hematopoietic stem cell transplantation and in the following diseases: high- or intermediate-risk MDS according to the International Prognostic Scoring System (IPSS), acute myeloid leukemia, or chronic myelomonocytic leukemia without features of MDS. Azacitidine was registered in Russia in 2010. The safety profile of this drug allows its effective use in elderly patients with comorbidities.[161][162] A disadvantage of azacitidine (5), which limits its scope of application, is its low stability in aqueous solutions. Azacitidine (5) hydrolyzes to give ribose guanylureide (227).[163][164]
Decitabine (AzadC, 6) (see Fig. 1),[165-167] or 5-aza-2'-deoxycytidine, 4-amino-1-(2-deoxy-β-D-erythrofuranosyl)-1,3,5-triazin-2(1H)-one, is an analogue of deoxycytidine. Early methods for preparing this compound gave very low yields of the β-stereoisomer. Decitabine (6) was first synthesized by Pliml and Šorm[168] in 1964 using a strategy similar to the synthesis of azacitidine, but the authors failed to isolate β-isomer from the mixture of anomers. Winkley and Robins[158] obtained pure β-decitabine by reacting a triacetyl-protected sugar with silylated 5-azacytosine 225, but the yield of the product was only 7%. In this case, the α-isomer was predominantly formed, for which the yield reached 52%.
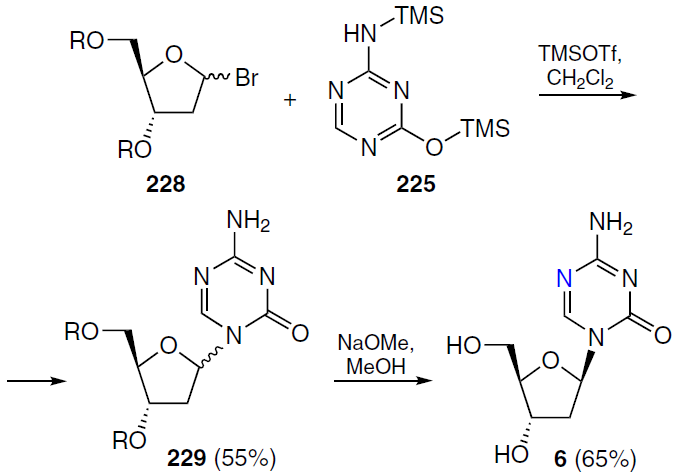
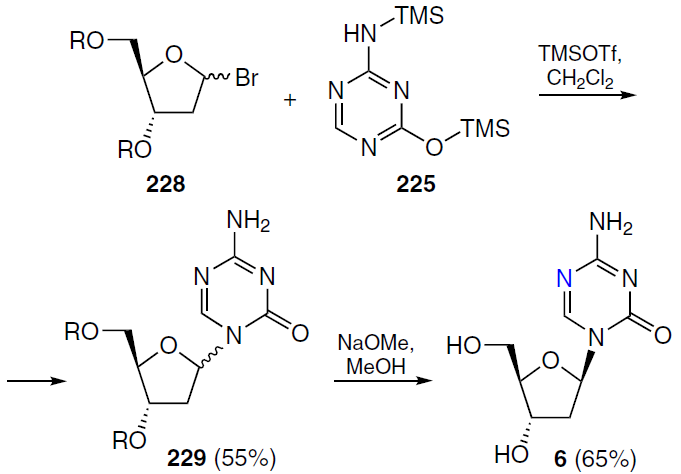
It was shown[169] that the selectivity of the glycosylation process depends on the solvent, reaction temperature, and the ratio of silylated triazine 225 to protected ribosaccharide 228 (Scheme 44). Varying these parameters leads to a change in the ratio of α- and β-glycosylated products from 1 : 1 to 1 : 3. Optimum conditions for large-scale synthesis involved the use of protected sugar 228 and silylated triazine 225 in a 1 : 1 ratio with the addition of 1.05 equiv. TMSOTf in cooled dichloromethane. It was found that in order to maintain the initial ratio of α- and β-forms and prevent isomerization of the target β-isomer into the undesirable α-anomer after completion of the reaction, immediate quenching of excess TMSOTf with an organic amine (preferably a primary one: MeNH2 or EtNH2) at a temperature not exceeding 0°C is required.[169] As a result, protected decitabine 229 was obtained as a mixture of α- and β-isomers in a ratio of 1 : 2.7. The final step in the synthesis of decitabine (6) involved removing the protecting groups of compound 229 with sodium methoxide in methanol. Subsequent filtration and recrystallization from a DMSO-methanol mixture gave decitabine (6) in kilogram quantities.
Another method for the industrial synthesis of decitabine has been patented (Scheme 45).[170] In this strategy, 2'-deoxy-D-ribose 230 was treated with acetyl chloride first in methanol and then in a mixture of pyridine and dichloromethane to form the bis(acetyl)-protected sugar 231. Subsequent condensation of this product with silylated 5-azacytosine in the presence of TMSOTf afforded a 1 : 1 mixture of α- and β-acylated compound 232. Acetyl groups were removed with a solution of ammonia in methanol, followed by crystallization to yield decitabine (6) with a purity of 90 – 99% according to HPLC. To improve purity, the resulting product was dissolved in DMSO, filtered, washed with a mixture of methanol and ethyl acetate, and dried in vacuo to give crystalline decitabine (6) with a purity of 99.8%.
The antileukemic activity of decitabine (6) was first demonstrated in 1968.[171] Its mechanism of action is dual: at high concentrations, a cytotoxic effect is observed, leading to cell death, and at lower concentrations, this drug affects cancer cells through DNA hypomethylation. This results in the reactivation of epigenetically suppressed genes, such as tumour suppressor genes, which may promote cell differentiation and restoration of normal cellular functions.[172][173] Clinical studies have shown the efficacy of low doses of decitabine (6), administrated by daily infusion for 5 days, in patients with MDS.[174]
A limiting factor in the use of decitabine is its low oral bioavailability due to rapid metabolism by cytidine deaminase (CDA) action.[175]To address this problem, another drug, cedazuridine (9) (see Fig. 1), was synthesized, which is an analogue of tetrahydrouridine with two fluorine atoms at the 2' position of the carbohydrate moiety. It is a competitive inhibitor of CDA and is part of the oral combination drug (cedazuridine + decitabine).[22][23][26] The combination of decitabine and cedazuridine is FDA-approved for the treatment of intermediate- or high-risk MDS and chronic myelomonocytic leukemia.[176] The European Medicines Agency also approved the use of the combination therapeutic agent cedazuridine–decitabine, trade name Inqovi®, in 2023.[159]
In an attempt to improve the bioavailability, metabolic stability, and cellular penetration of nucleoside analogues 5 and 6, a number of prodrugs have been obtained, some of which are shown in Fig. 9.
![[{"id":"tkBUNjppN9","type":"paragraph","data":{"text":"Examples of azacytidine and decitabine prodrugs"}}]](/storage/images/resized/LGE22CCd9JN84MAEF68mCi1o1qDRt2SQEPXNMgcN_xl.webp)
Structural modifications render these compounds poor substrates for cytidine deaminase. Guadecitabine (SGI-110, 18) (see Fig. 9), a prodrug of decitabine, has been clinically tested in combination with carboplatin for platinum-resistant ovarian cancer and hepatocellular carcinoma following sorafenib failure, and in combination with irinotecan for colorectal cancer.[177][178] Guadecitabine has been tested as a monotherapy for the treatment of high-risk myelodysplastic syndromes and chronic myelomonocytic leukemia, but it did not demonstrate a statistically significant improvement in overall survival compared with a control group of patients receiving alternative therapy.[179] Several acyl prodrugs of 5-azacytidine with increased lipophilicity and improved absorption have also been studied in clinical trials: 2'-deoxy-N4-[2-(4-nitrophenyl)ethoxycarbonyl]-5-azacytidine (NPEOC-DAC), 5-azacytidine-5'-elaidate (CP-4200), and 2',3',5'-triacetyl-5-azacytidine (TAC).[180]
5.3. 5-Alkynylpyrimidine nucleoside analogues
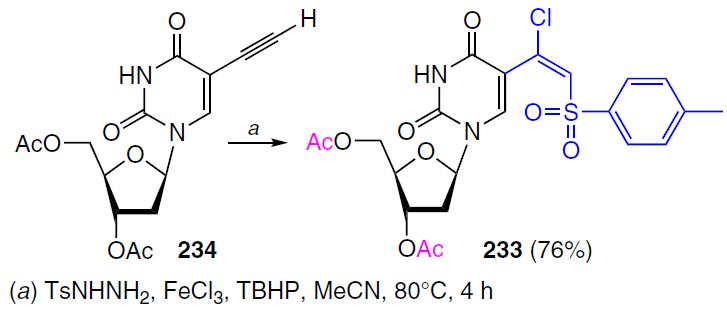
Several examples of reactions involving nucleosides with 5-alkynyl-substituent in the pyrimidine ring that yield products with antitumour activity have been reported. For example, compound 233, containing a 1-chloro-2-sulfonylvinyl fragment, was obtained by halovinylsulfonation of 2'-deoxy-5-ethynyluridine.[181] The reaction of 3',5'-di-O-acetyl-2'-deoxy-5-ethynyluridine (234) with TsNHNH2 (Ts is p-toluenesulfonyl (tosyl)) in the presence of FeCl3 · 6 H2O and tert-butyl hydroperoxide (TBHP) afforded the target product 233 in good yield (Scheme 46). Acetyl-protected (β-chloro)vinyl sulfone 233 inhibited the proliferation of murine leukemia (L1210), human T lymphocyte (CEM), and human cervical carcinoma (HeLa) cells with IC50 values of 5.6 ± 4.7, 11 ± 10, and 23 ± 8 μM, respectively.[182]
Ribonucleoside derivatives of furano[2,3-d]pyrimidine 235 (Ref. [183]) were synthesized by Pd-catalyzed cross-coupling of 5-iodouridines with the corresponding alkynes in two steps via the formation of intermediate 5-alkynepyrimidines 236 or in one step.[184][185] Treatment of substrates 235 with ammonia pyrrolo[2,3-d]pyrimidine ribonucleosides 237 (Scheme 47).[186][187] Similarly, 5'-norcarbocyclic analogues of furano- and pyrrolo[2,3-d]pyrimidine nucleosides were obtained from 1-(4'-hydroxy-2'-cyclopenten-1'-yl)-5-iodouracil.[188][189]
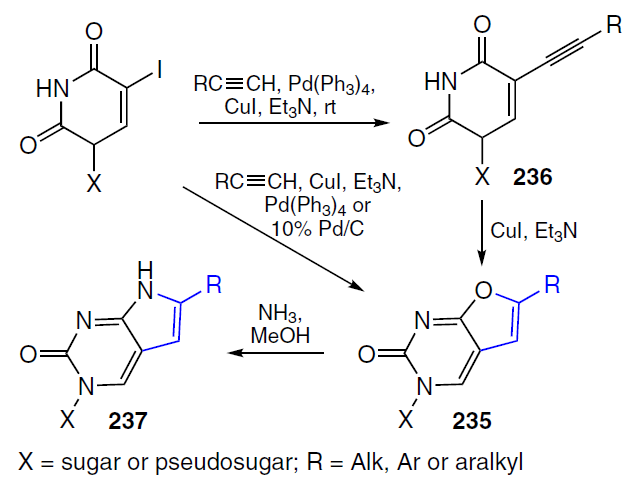
6-Octylpyrrolo[2,3-d]pyrimidine ribonucleoside 237a was shown to exhibit significant selective cytotoxicity against T98G glioblastoma (IC50 = 0.74 μM) compared to normal MRC-5 cells (IC50 > 130 μM) and the human cancer cell line HeLa (IC50 = 17.8 μM).[167] This compound is recommended for further study as a potential key structure in the development of an anti-glioma drug. Furano[2,3-d]pyrimidine 235a exhibited selective cytotoxicity against the following human cancer cells: HeLa (IC50 = 6.5 μM), HuTu80 (IC50 = 5.1 μM), KB-3-1 (IC50 = 8.2 μM).[189] Carbocyclic nucleoside 235a was found to induce cell death via the apoptotic pathway.[189]
6. Troxacitabine
The successful clinical use of cytarabine (1) and gemcitabine (3), as well as the need to overcome the side effects associated with these drugs, have led to the development of new cytidine nucleoside analogues. One such analogue is troxacitabine (β-L-dioxolanocytidine) (16) (see Fig. 2).[190]
In 1992, the first method for the diastereoselective synthesis of troxacitabine was presented, using L-ascorbic acid (238) as the starting material (Scheme 48).[191] The first step involved condensation of acid 238 and benzyloxyacetaldehyde dimethyl acetal in the presence of p-toluenesulfonic acid, affording a mixture of diastereomers 239. This mixture was then subjected to oxidative degradation and catalytic Wolfe oxidation in the presence of benzyltriethylammonium chloride to yield intermediate 240, which was isolated by flash chromatography. Subsequent steps of oxidative decarboxylation and N-glycosylation involving silylated N-acetylcytosine under Vorbruggen reaction conditions were completed by deprotection, resulting in the isolation of product 16 in an overall yield of 24%.[191] Other synthetic approaches to compound 16 have also been described.[1][192]
Unlike cytarabine (1) and gemcitabine (3), troxacitabine (16) has low affinity for cellular nucleoside transporters, and its membrane penetration occurs primarily by simple diffusion.[193] The cytotoxic metabolite of this drug is its 5'-triphosphate, which inhibits DNA biosynthesis by being a terminator for DNA polymerases. The first step in troxacitabine phosphorylation is realized by deoxycytidine kinase, and the conversion to 5'-di- and 5'-triphosphate is catalyzed by 3-phosphoglycerate kinase.[190] An important feature and advantage of compound 16 is its resistance to deamination by cytidine deaminase, resulting in a significantly longer half-life (approximately 80 h) compared to its predecessors. This is facilitated by the unique stereochemistry of troxacitabine (16). Natural nucleosides and all anticancer drugs based thereon have a β-D-configuration of the carbohydrate moiety; compound 16 is β-L-dioxolanylcytidine. Therefore, we are dealing with the first L-nucleoside analogue to exhibit antitumour activity.[194]
Phase II clinical trials showed that troxacitabine exhibits moderate activity against advanced and/or metastatic renal cell carcinoma. This drug has been used with varying degrees of success to treat leukemias and myelodysplastic syndromes, lymphoproliferative neoplasms and multiple myeloma, as well as pancreatic and lung cancer.[195] At maximum tolerated doses of 8 mg/m2/day administered intravenously every day for 5 days, it caused side effects such as mucositis, rash, and hand-foot syndrome.[196] Troxacitabine was granted orphan drug designation by the FDA and the European Medicines Agency (EMA) in 2005, but it was withdrawn in 2008 at the request of the sponsor and remains unlicensed.[195]
7. Carbocyclic analogues of pyrimidine nucleosides
Replacing the tetrahydrofuran ring in nucleosides with a cyclopentane moiety produces carbocyclic nucleoside analogues.[197] Compounds of this class lack a glycosidic bond, making them more resistant to cleavage by nucleoside phosphorylases and hydrolases. The conformational similarity between the cyclopentane and tetrahydrofuran moieties allows carbocyclic nucleoside analogues to act as substrates or inhibitors of enzymes catalyzing the synthesis and conversion of natural nucleosides and nucleotides.[197]
The cytotoxic drug cyclopentenylcytosine (CPEC, 241) was developed in 1979 as a cytosine-containing analogue of neplanocin A (242), a biologically active and toxic nucleoside isolated from natural sources. Among its purine and pyrimidine analogues, neplanocin A has proven to be the most potent in terms of antiviral activity and activity against murine leukemia and human tumour xenografts.[198]
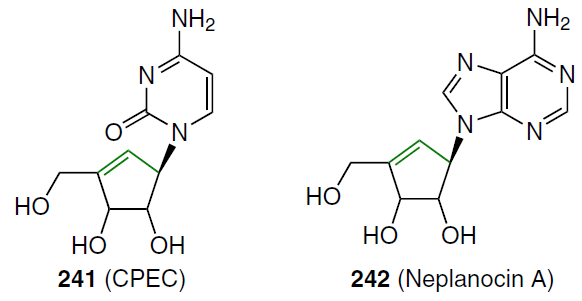
The synthesis of (1R,4R,5S)-1-[4,5-dihydroxy-3-(hydroxymethyl)-2-cyclopenten-1-yl]cytosine (241)* and a similar carbocyclic derivative of uracil was described by Lim et al.[199] Cyclopentenyl nucleosides were synthesized from optically pure 3-aminocyclopentene 243, obtained according to a previously described method[200] (Scheme 49). Condensation of the starting compound 243 with ethyl-(Z)-2-[(ethoxy- carbonyl)carbamoyl)-3-methoxyacrylate in the presence of triethylamine in ethanol gave intermediate 244, which was then cyclized to uracil derivative 245 in 80% yield. Further hydrolysis of compound 245 with LiOH in a water–methanol mixture followed by neutralization with Dowex 50W-X8 ion exchange resin in the H+ form yielded an intermediate carboxylic acid, which was decarboxylated without purification in the presence of copper powder in boiling quinoline to afford the protected uridine analogue 246a. Debenzylation of the latter with boron trichloride in dichloromethane was accompanied by removal of the isopropylidene group during subsequent workup and gave (1R,4R,5S)-1-[4,5-dihydroxy-3-(hydroxymethyl)-2-cyclopenten-1-yl]uracil (247) in moderate yield.[199]
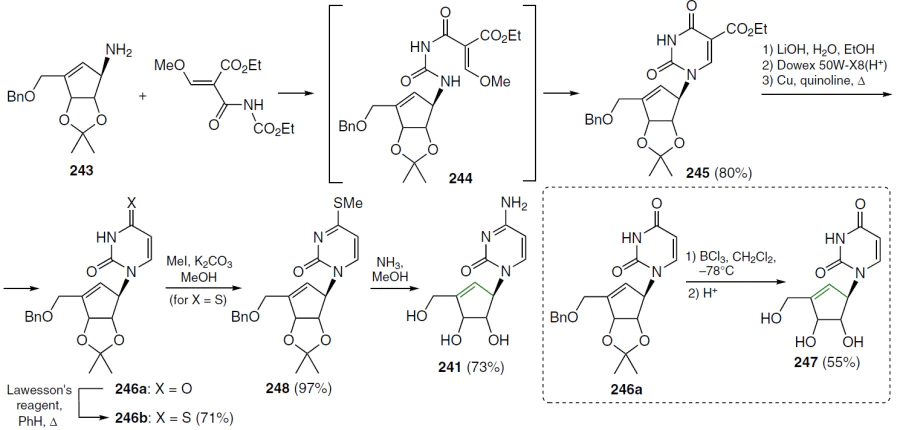
Conversion of compound 246a (X = O) to its thio analogue 246b (X = S) was accomplished by reacting the former with Lawesson's reagent in benzene. Methylthio derivative 248 was obtained in high yield from compound 246b (X = S) and methyl iodide in the presence of potassium carbonate in methanol. Finally, treatment of compound 248 with methanolic ammonia afforded the target product 241 (see Scheme 49).
The drug CPEC (241), in contrast to the inactive carbocyclic uracil derivative 247, showed significant cytotoxicity against the L1210 cell line, inhibiting cell growth by 58% at a concentration of 30 μM.[199] The efficiency of compound 241 was subsequently confirmed against various tumours, including neuroblastoma, but it did not find clinical use due to neurotoxicity.[201]
Song et al.[202] synthesized the optical isomer of compound 241, as well as 5-methyl- and 5-fluoro-substituted (1'S,2'R,3'S)-1-[2,3-dihydroxy-4-(hydroxymethyl)-4-cyclopenten-1-yl]pyrimidines shown in Scheme 50. The key step involved the condensation of cyclopentene 249 with N-benzoylated pyrimidine bases to form protected carbocyclic analogues 250a – c. The condensation was carried out using the Mitsunobu reaction: cyclopentenyl alcohol 249 was treated with N3-benzoyluracil,[203] N3-benzoylthymine,[203] or N3-benzoyl-5-fluorouracil[204] in the presence of DEAD and Ph3P in THF at room temperature. Debenzoylation of compounds 250a – c with saturated ammonia in MeOH delivered products 251a – c, and subsequent removal of the protecting groups (tert-butyl and isopropylidene) with aqueous trifluoroacetic acid at 50°C afforded carbocyclic analogues 252a – c. Protected uridine analogues 251a and 5-fluorouridine 251c were converted to the corresponding cytosine and 5-fluorocytosine derivatives using the described method,[205] followed by deprotection to isolate the cytosine (253a) and 5-fluorocytosine nucleosides (253c).
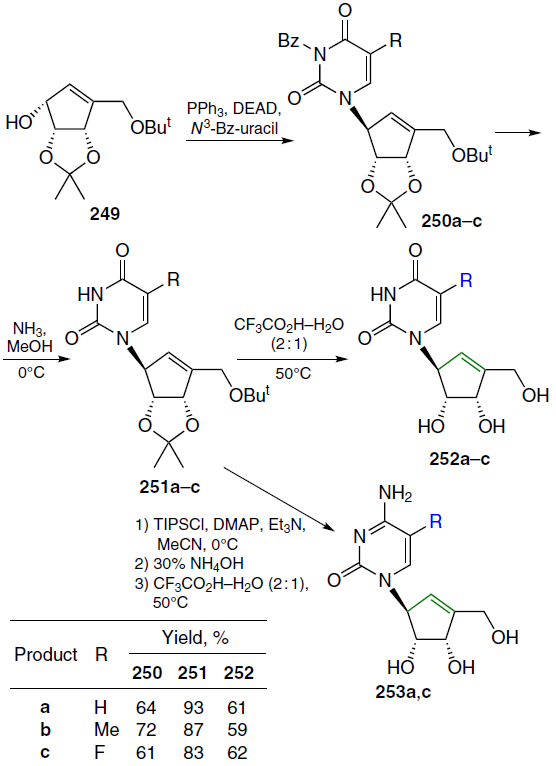
Nucleoside analogues 252 and 253 were shown to lack cytotoxicity against CEM cells. Under the same conditions, 1-[4,5-dihydroxy-3-(hydroxymethyl)-2-cyclopenten-1-yl]cytosine 241 exhibited high cytotoxicity (IC50 = 0.08 μM), while its 5-fluoro-containing analogue 254 was found to be inactive (IC50 = 51.8 μM).[202]
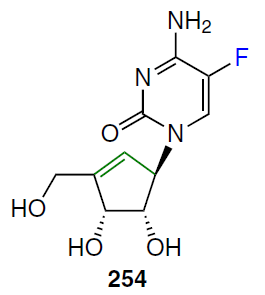
Choi et al.[206] described a method for the preparation of fluorocyclopentenyl nucleoside analogues of cytidine and uridine and studied their antitumour properties. The method involved converting the key compound, cyclopentenone 255, into a known glycosyl donor,[207] and then into the target cytosine derivative 12 (Scheme 51).
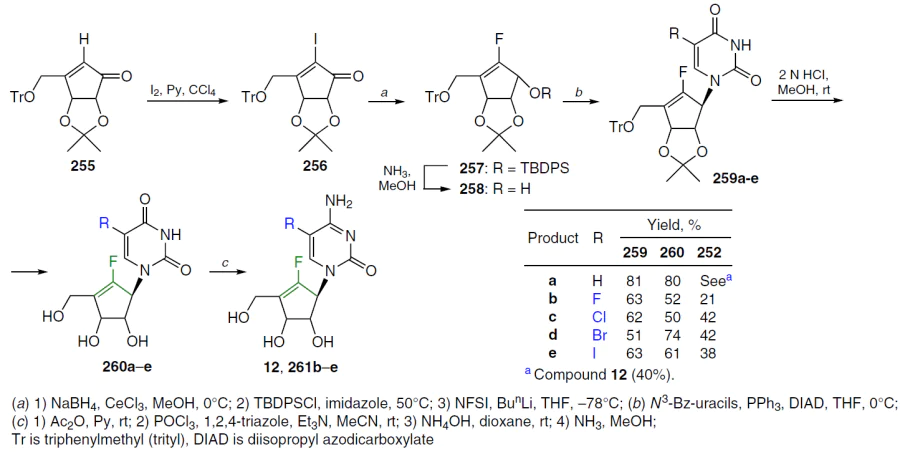
Iodination of cyclopentenone 255 with iodine in the presence of pyridine in CCl4 proceeded slowly, but when the solvent was replaced with THF, iodocyclopentenone 256 was obtained in fairly good yield. Reduction of this compound with sodium borohydride in the presence of cerium chloride, followed by protection of the hydroxyl group with a tert-butyldiphenylsilyl moiety, yielded a TBDPS ester, in which electrophilic substitution of the iodine atom by fluorine was achieved by the sequential addition of BunLi and N-fluorobenzenesulfonimide (NFSI) at –78°C. The resulting fluorocyclopentene 257 was then desilylated to form the key intermediate 258.[207] Condensation of the latter with N3-benzoyluracil under standard Mitsunobu reaction conditions[208] afforded uracil derivatives 259a – e. Treatment of these compounds with hydrochloric acid solution delivered uracil derivatives 260a – e.
The carbocyclic analogue of cytidine 12 was prepared in three steps: treatment of compound 260a (R = H) with acetic anhydride gave the triacetate, which was converted first to the triazole derivative by reaction with POCl3 and 1,2,4-triazole in the presence of triethylamine, and then to the corresponding cytosine derivative by reaction with ammonium hydroxide in 1,4-dioxane. Deacetylation of this compound with ammonia in methanol afforded the final product, 1-[(1R,2S,3R)-2,3-dihydroxy-4-(hydroxymethyl)-5-fluorocyclopent-4(5)-en-1-yl]cytosine (12) with the commercial code RX-3117. 5-Substituted in the pyrimidine ring 1-[(1R,2S,3R)-2,3-dihydroxy-4-(1-hydroxymethyl)-5-fluoro-cyclopent-4(5)-en-1-yl]cytosines 261b – e were obtained in a similar way (see Scheme 51).[209]
Compounds 261 exhibited no or 2 – 3 orders of magnitude lower antitumour activity against human cancer cells, including A549 (lung carcinoma), HCT-116 (colon carcinoma), SNU-638 (gastric adenocarcinoma), MDA-MB-231 (breast adenocarcinoma), SK-Hep-1 (epithelial tumour of the liver and intrahepatic bile ducts), and PC-3 (prostate adenocarcinoma) than cytosine 12 with a hydrogen atom at position 5 of the pyrimidine ring. The results of the antitumour activity study for compound 12 are presented in Table 4.[209]
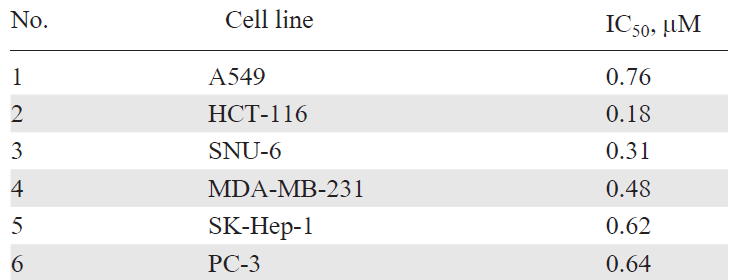
RX-3117 has an improved pharmacological profile compared to gemcitabine and other nucleoside analogues, high activity against various cancer cell lines and xenografts, including those resistant to gemcitabine (3), and excellent oral bioavailability.[206][210][211] This compound is not a substrate of cytidine deaminase. The drug's pharmacokinetic parameters were evaluated with daily oral administration of 700 mg (5 days a week for 3 weeks). Plasma levels were found to be micromolar, which is toxic to cancer cells. It demonstrated clinical activity in bladder and pancreatic cancers.[211]
Compound 12 is taken up by equilibrating nucleoside transporters and is selectively phosphorylated by uridine-cytidine kinase 2, which is expressed only in tumours. This drug has a dual mechanism of action: DNA damage and inhibition of DNA methyltransferase 1.[212][213]
The synthesis of a carbocyclic analogue of decitabine (cAzadC, 262) has been reported (Scheme 52).[214] The hydroxyl groups of the starting NHBoc-cyclopentane 263 (Boc is tert-butoxycarbonyl) were protected with benzyl groups, and then the Boc protection was removed from the amino group of intermediate 264 using trifluoroacetic acid. Compound 265 with the free amino group was reacted with methyl (1H-imidazole-1-carbonyl)carbaimidate (266), which was prepared from isomethylurea hydrochloride 267 via the formation of free base 268 with potassium hydroxide and its subsequent reaction with carbonyldiimidazole (CDI). The reaction of compounds 265 and 266 yielded the carbamoylurea-cyclopentane analogue of nucleoside 269, which was cyclized to triazine base 270 using triethyl orthoformate. The reaction of compound 270 with NH3 in methanol and deprotection with BCl3 in dichloromethane, followed by treatment with MeOH, column chromatography, and recrystallization from MeOH afforded the target product 262.
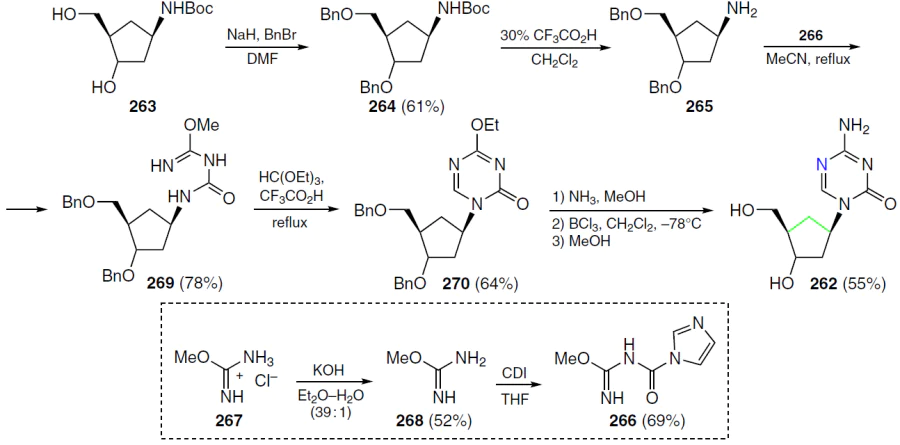
The carbocyclic analogue 262 is not only significantly more chemically stable than decitabine per se,[215] facilitating its administration, but also demonstrates high activity in inducing cancer cell death. Initial studies of the general toxicity of cAzadC indicate that this compound is less toxic and can be administered at significantly higher doses than decitabine.[216]
Methods for the chemical synthesis and study of the anticancer activity of C2',C4'-bridged bicyclic nucleosides derived from norbornane, as well as C3',C5'-bridged carbocyclic L-nucleosides and pyrimidine nucleoside analogues with various heteroatoms (nitrogen, sulfur, selenium, etc.) replacing the oxygen atom in the tetrahydrofuran ring, are described in detail by Guinan et al.[17]
* In this Section, the names of the compounds are given according to the IUPAC nomenclature, in contrast to the systematic and trivial names used above for nucleoside derivatives.
8. Conclusion
Pyrimidine nucleoside analogues remain an important and promising class of anticancer drugs. In the decades since the discovery of the first representatives of this class, significant advances have been made in understanding their mechanisms of action, optimizing their structure, and expanding their clinical applications. Both classical pyrimidine nucleoside analogue-based drugs — 5-fluorouracil and cytarabine — and modern drugs (e.g., gemcitabine, decitabine, and capecitabine) have made significant contributions to improving survival and quality of life in patients with various types of malignant neoplasms.
Literature data show that the effectiveness of pyrimidine nucleoside analogues stems from their ability to affect DNA replication and RNA biosynthesis, as well as nucleotide metabolism, leading to tumour cell growth arrest and death. However, despite their antitumour activity, these drugs also exhibit significant toxicity, limiting their clinical use. The development of drug resistance is also a serious problem, requiring a constant search for new ways to overcome it.
Current research in the field of pyrimidine nucleoside analogues aims to address a number of issues. Researchers are actively working to develop new drugs with increased selectivity, improved bioavailability, and the ability to overcome drug resistance mechanisms. Approaches to modifying the structure of the heterocyclic base, altering the carbohydrate moiety, and the synthesis of the prodrug forms allow the creation of compounds with a more favorable pharmacological profile and reduced toxicity. The development of targeted drug delivery technologies, including the use of nanoparticles and antibody conjugates, also opens up new opportunities for increasing the efficiency and selectivity of pyrimidine nucleoside analogues.
In conclusion, it can be noted that pyrimidine nucleoside analogues continue to play an important role in modern cancer therapy. Continued intensive research in this area should lead to the development of effective therapeutic methods for cancer control. Further efforts to develop new cytotoxic agents, optimize existing drugs, and understand the mechanisms of drug resistance will contribute to improved treatment outcomes and enhance patient quality of life. Data accumulated over years of development and clinical use of pyrimidine nucleoside analogues can be used as a solid foundation for future research in anticancer drug design.
This review was financially supported by the Russian Science Foundation (Project No. 23-64-10018).
9. List of abbreviations and designations
The following abbreviations and designation are used in the review:
AIBN — azobis(isobutyronitril),
AML — acute myeloid leukemia,
ara-C — 1-β-D-arabinofuranosylcytosine (cytarabine),
5-AZA-CDP — 5-azacytidine-5'-diphosphate,
5-AZA-CMP — 5-azacytidine -5'-monophosphate,
5-AZA-CTP — 5-azacytidinetriphosphate,
AzadC — 5-aza-2'-deoxycytidine (decytabine),
Boc — tert-butoxycarbonyl,
CAN — cerium(IV)-ammonium nitrate,
сAzadC — carbocyclic analogue of decytabine,
CDA — cytidindesaminase,
CDI — carbonyldiimidazole,
CMPK — cytosine nucleoside monophosphate kinase,
CNDAC — 2'-deoxy-2'-С- cyanocytidine,
CNddC — 2',3'-didehydro-2',3'-dideoxy-2'-C-cyanocytidine,
CPEC — cyclopentenylcytosine,
DAST — diethylaminosulful trifluoride,
DBU — 1,8-diazabicyclo[5.4.0]undec-7-ene,
dCK — deoxycytidine kinase,
DEAD — diethylazodicarboxylate,
2'-DHAU — 2'-hydroxylamino-2'-deoxyuridine,
DIAD — diisopropylacetylenedicarboxylate,
DMAP — 4-dimethylaminopyridine,
DMDC — 2'-deoxy-2'-methylydenecytidine,
DNMT — DNA-methyltransferase,
DPPA — diphenylphosphoryl azide,
DPAP — 2,2-dimethoxy-2-phenylacetophenone,
ECyd — 3'-ethynylcytidine,
ECDP — 3'-ethynylcytidine-5'-diphosphate,
EMA — European Medicines Agency,
FDA — U.S. Food and Drug Administration,
FNC — azvudine,
FdUMP — 5-fluoro-2'-deoxyuridine-5'-monophosphate,
FdUTP — 5-fluoro-2'-deoxyuridine-5'-triphosphate,
5-FU — 5-fluorouracyl,
FUTP — 5-fluorouridine-5'-triphosphate,
IBX — 2-iodoxybenzoic acid,
IC50 — half-maximal inhibitory concentration,
IPSS — international prognostic scoring system,
HMDS — hexamethyldisilazane,
hRR — human ribonucleotide reductase
mCBA — m-chlorobenzoic acid,
mCPBA — m-chloroperoxybenzoic acid,
MDS — myelo-dysplastic syndrome,
Ms — methanesulfonyl (mesyl),
NBS — N-bromosuccinimide,
NDPK — nucleotide diphosphate kinase,
NFSI — N-fluorobenzenesulfonimide,
NMPK — nucleotide monophosphate kinase,
NPEOC-DAC — 2'-deoxy-N4-[2-(4-nitrophenyl)ethoxycarbonyl]-5-azacytidine,
PDC — pyridinium dichromate,
Py — pyridine,
Pyr — pyrimidin-1-yl,
RR — ribonucleotide reductase,
rt — room temperature,
SAMHD1 — SAM domain and HD domain-containing human protein 1,
TAC — 2',3',5'-triacetyl-5-azacytidine,
TBAF — tetra-n-butylammonium fluoride,
TBDPS — tert-butyldiphenylsilyl,
TBHP — tert-butylhydroperoxide,
TBS — tert-butyldimethylsilyl,
2-THF — tetrahydrofuran-2-yl,
TIPDS — tetraisopropyldisiloxane-1,3-diyl,
TMS — trimethylsilyl,
TfO — trifluoromethanesulfonate,
TPSCl — triisopropylbenzenesulfonyl chloride,
Ts — p-toluenesulfonyl (tosyl),
Tol — tolyl,
Tr — triphenylmethyl (trityl),
TS — thymidilate synthase,
UCK — uridine-cytidine kinase.
References





















