

Keywords
Abstract
Post-translational modifications of histones, the protein components of nucleosomes, regulate the local state of chromatin for optimal gene expression. Among these modifications, a balance of methylation and demethylation of particular lysine residues in histones is important, as under physiological conditions this balance provides the permissiveness of chromatin for template processes. An imbalance of methylation and demethylation leads to transcriptional deregulation, a characteristic feature of malignant cells. A specific viewpoint of the authors is a multidisciplinary approach to the analysis of complex and contradictory data on epigenetic regulators. This review presents general information about human histone lysine demethylases (KDM). For the KDM2 subgroup, structural features, mechanism of catalysis (histone demethylation), and the role in tumour biology are considered. Since KDM2 functions are not limited to enzymatic activity, the pharmacological approach for anticancer therapy implies both the design of demethylation blockers, with KDM2 being retained in the cells, and the search for possible tools for total elimination of these proteins.
The bibliography includes 174 references.
1. Introduction
Epigenetic (occurring without changes of the DNA nucleotide sequence) regulation of gene expression is an important mechanism for maintaining homeostasis.[1-3] In eukaryotic cells, genomic DNA forms complexes with histone proteins (chromatin). The intricate organization of chromatin ensures compact storage of genetic information and allows for the regulation of access (permissiveness) for proteins to carry out DNA-dependent processes such as transcription, damage repair, and replication.[4-6] The chromatin state is regulated epigenetically, with histone methylation and acetylation being among the most important modifications.[1][7-10] These modifications are accomplished owing to the covalent binding of amino acid residues to methyl or acetyl groups transported by ‘writer’ enzymes from donor metabolites.[7][10][11] The modified histone sites are recognized by ‘reader’ proteins and removed by ‘eraser’ enzymes. The coordinated functioning of these mechanisms provides ‘the histone code’, which determines the local and genome-wide organization of chromatin.[7][11][12] Epigenetic modifications of chromatin are inherited in cell generations.
Methylation occurs for lysine and arginine residues. The present review considers methylation of lysine residues in histones that are commonly designated as H2A, H2B, H3, and H4. The best studied methylation sites are H3K4, H3K9, H3K27, H3K36, H3K79, and H4K20 where K is the lysine residue in the corresponding position of histone. The lysine residue may be non-methylated or may contain one (me1), two (me2), or three (me3) CH3 groups, that is, it can be mono-, di-, or trimethylated.[7][13][14] The site and the degree of methylation of lysine residues influence gene expression. The methylated H3K4, H3K36, and H3K79 sites are usually associated with transcriptional activation, whereas methylated H3K9, H3K27, and H4K20 are usually related to transcriptional restriction or suppression.[15-17] The methylation-demethylation balance of histone lysines is supported by lysine methyltransferases (‘writers’) and lysine demethylases (K-DeMethylases (KDM)) (‘erasers’).[7][14][18][19] Disruptions of this balance occur during pathological processes, mainly in tumour and ageing cells.[7][8][19][20]
This review addresses the subgroup 2 lysine demethylases (KDM2), focusing on their structural features, biological functions, and mechanism of histone demethylation. These fundamental issues form the basis for analysis of the role of KDM2 as targets for anticancer therapy. The complexity of epigenetic regulation determines the concept of development of small-molecule compounds that inhibit the catalytic function of KDM2 while preserving these proteins in the cell. An alternative approach is to design chemical tools for elimination of KDM2. We consider this problem to be multifaceted, and, therefore, we address the intricate aspects of epigenetic regulation as parts of a general biological phenomenon and emphasize the contradictory nature of this mechanism in cancer cells.
2. General overview of lysine demethylases
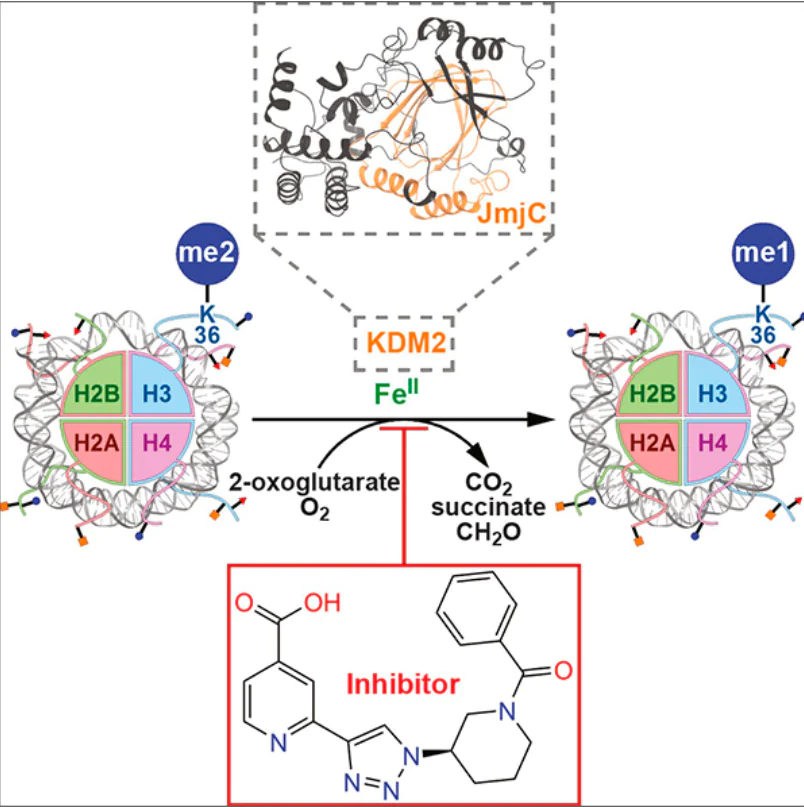
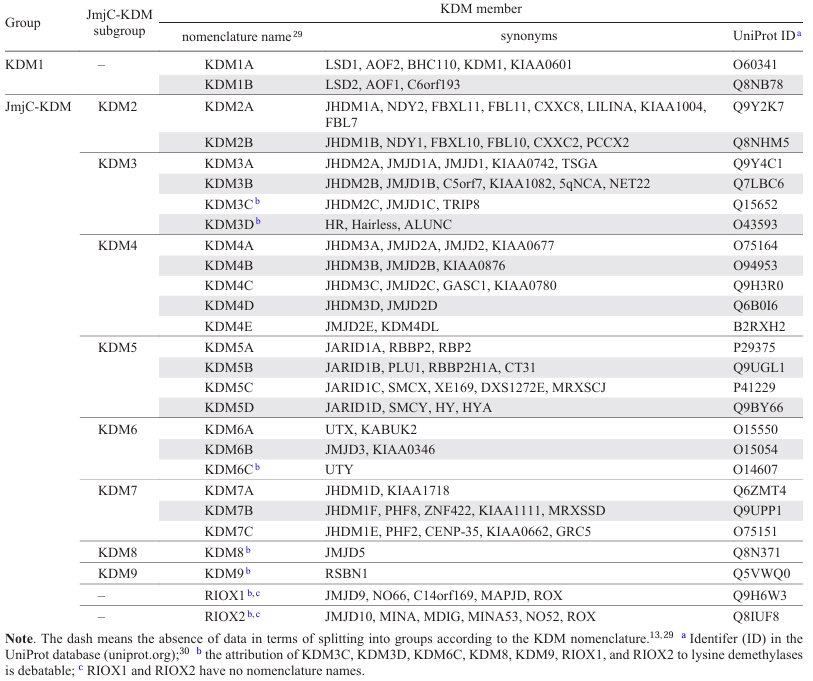
Lysine demethylases are a subclass of oxidoreductases.[21] A key function of these proteins is to remove the methyl groups from lysine residues in histones.[13][14] This role determines the importance of KDM for ontogenesis, for numerous physiological processes, and also for the pathogenesis of socially significant diseases.[8][19][20] The family of human KDM comprises approximately 30 members classified according to their structure and the enzymatic mechanism. The KDM1 group includes FAD-dependent (FAD is flavin adenine dinucleotide) aminooxidases KDM1A and KDM1B. These enzymes remove methyl groups from mono- and dimethylated lysine residues.[13][14] Another group consists of KDM2-9, which are 2-OG-dependent (2-OG is 2-oxoglutarate) oxygenases containing the Jumonji C (JmjC) domain. This group designated as JmjC-KDM uses FeII ions and 2-OG to remove methyl groups from mono-, di, and trimethylated lysine residues.[13][14] The activity of particular JmjC-KDM members (e.g., KDM3B[22] and KDM5C[23][24]) toward demethylation of histone arginine residues has been reported.
The relationship between particular members of JmjC-KDM can be identified by generating the phylogenetic tree (Fig. 1) based on the homology of the JmjC domain amino acid sequences. Analysis of the phylogenetic tree makes it possible to identify paralogues, that is, proteins of the same organism that are similar in structure and function. For example, the closest paralogues for KDM2A are KDM2B and KDM7A-C. KDM can exist as a few isoforms, i.e., the products of mRNAs transcribed from a single gene. However, not all of the proteins that have the JmjC domain refer to KDM family. Relevance of demethylases KDM3C, KDM3D, KDM6C, KDM8, and KDM9 and ribosomal oxygenases RIOX1 and RIOX2 to KDM is being debated.[3][13][14][25] The nomenclature of human KDM is given in Table 1[26-30].
![[{"id":"A9nYlXuTCz","type":"paragraph","data":{"text":"Phylogenetic tree of human KDM.<sup>13, 25 – 28</sup>"}}]](/storage/images/resized/wvZdpwKZj4pGLQ9JcJdLYW9efldjS6qwKdrlWycL_xl.webp)
3. Structure of KDM2 proteins
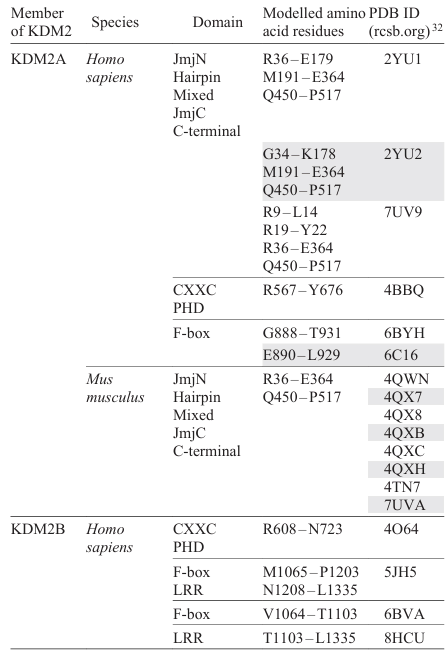
The KDM2A and KDM2B proteins have a similar set of domains, but different numbers of amino acid residues. The identity and similarity of their canonical sequences do not exceed 51% and 65%, respectively. The domain structures for canonical isoforms of human KDM2A and KDM2B are depicted in Fig. 2a,b according to the UniProt database[30] and published data.[13][14][25] The tertiary structures of these proteins predicted using the AlphaFold3 artificial intelligence tool[31] are shown in Fig. 2c,d; the models of protein domains are available in the Protein Data Bank database (PDB, rcsb.org).[32]
![[{"id":"epbrviijgt","type":"paragraph","data":{"text":"Structural features of KDM2: domain architectures of KDM2A (<i>a</i>) and KDM2B (<i>b</i>) and predicted (AlphaFold3) tertiary structures of full-length KDM2A (<i>c</i>) and KDM2B (<i>d</i>) proteins. The amino acid sequences were taken from the UniProt database (protein IDs are Q9Y2K7 and Q8NHM5). The coloured written words indicate domains designated by the following abbreviations: JmjN is Jumonji N; JmjC is Jumonji C; CXXC is the zinc finger domain CXXC; PHD is the plant homeodomain; LRR is the leucine-rich repeat domain containing six [LRR(1-6)] or seven [LRR(1-7)] repeats.<sup>14, 30, 31</sup>"}}]](/storage/images/resized/K30LDoapLZQmWyPB0kEwRpexAN7Wx0MvfGQd2rK7_xl.webp)
The JmjC[33] catalytic domain in KDM2A and KDM2B is a double-stranded β-helical fold. Two four-stranded antiparallel β-sheets form a β-sandwich structure of JmjC in KDM2A (Fig. 3a).[14][34][35] The cavity of the barrel-like JmjC structure accommodates a FeII cofactor with octahedral coordination held by interaction with conserved amino acid residues, H212, D214, and H284 for KDM2A[34][36] or H242, D244, and H314 for KDM2B based on sequence alignment. Fig. 3a,b shows the coordination of amino acid residues in the crystal structure of KDM2A (PDB ID: 4QX7) relative to the NiII ion, which replaces the endogenous iron(II) to stabilize the crystal structure.[36] The co-substrate 2-OG (see Fig. 3a,b), which provides additional coordination of the ion, and the substrate, i.e., the methylated lysine residue of histone peptide (K36me2), are located in the vicinity of the NiII ion.[36]
![[{"id":"6MlVyPgxsQ","type":"paragraph","data":{"text":"3D visualization of the crystal structures of KDM2 fragments: KDM2A complex with the histone peptide (PDB ID: 4QX7) in accordance with the protein domain architecture (<i>a</i>), arrangement of amino acid residues and 2-OG near NiII, which replaces endogenous FeII in the protein (PDB ID: 4QX7) (<i>b</i>), the amino acid sequence of the histone peptide (PDB ID: 4QX7) (<i>c</i>), and domain architectures and structures of KDM2A (PDB ID: 4BBQ, 6BYH) (<i>d, e</i>) and KDM2B (PDB ID: 4O64, 5JH5) (<i>f, g</i>). Here and in Figs. 4 – 7, 9: the digits 1 – 5 in the upper left corners of the boxes designate the same images with different magnifications.<sup>32</sup>"}}]](/storage/images/resized/zecYCdd97rdupY4Y401UlaIRXWLXEYKGAL5RVjhG_xl.webp)
In the KDM2A protein, the JmjN[33] domain (see Fig. 3a) ensures the structural integrity of JmjC. The amino acid residues of both domains are involved in intermolecular interactions; the JmjN residues do not participate in the formation of the active site located in JmjC.[14][35] The JmjN domain is required for catalysis.[37] The Hairpin, Mixed, and C-terminal domains in KDM2A also support the structural integrity of JmjC and interact with the histone peptide (see Fig. 3a,c) for its demethylation.[34][36] The KDM2A and KDM2B proteins contain zinc finger domains: CXXC and PHD (see Fig. 3d, f ). The CXXC domain identify the non-methylated CpG* dinucleotides in DNA;[38] the PHD domain contributes to CXXC stabilization[38] and can probably recognize histone modifications.[39-41] The F-box and LRR domains (the latter contains repeats enriched with leucine residues[42]) (see Fig. 3e,g) promote the formation of multiprotein complexes.[43-45]
The PDB contains geometric parameters of KDM2A and KDM2B fragments of mammals (Table 2) determined by experimental methods. No data of the JmjC domain of KDM2B are available in this database.
Han et al.[34] investigated the crystal structures of individual KDM2A fragments (PDB ID: 2YU1, 2YU2). The M1 – W516 fragment has a demethylase activity toward the H3K36me2 peptide. The same fragment devoid of the mobile R384 – R449 region is also able to demethylate H3K36me2.[34] Presumably, the Q450 – W516 region may play a role in intramolecular or intermolecular interactions and is important for acquiring an active conformation and maintaining the structural integrity through interaction with the M1 – R383 region containing JmjC.[34] The interaction between these regions occurs via amino acid residues of one α-helix (W334, L337, Y340, V341, I344) and hydrophobic residues of two other α-helices (L453, L461, L464, L468; L488, I489, V492, L495, L499) (Fig. 4a), and also the residues V64 and P512, R175 and W516, Y335 and P479, L350 and I482.[34]
![[{"id":"GTiw422ymj","type":"paragraph","data":{"text":"Arrangement of amino acid residues in the crystal structure of KDM2A (PDB ID: 4QX7): interacting residues of α-helices in the C-terminal domain (<i>a</i>), residues surrounding the histone peptide, and the surface section showing the U-shaped folding structure of the peptide (<i>b</i>).<sup>32</sup>"}}]](/storage/images/resized/jnVsx6VI8dr5Al8zeoe138Llccf7Jj18vO7vr0Yt_xl.webp)
Interactions of the KDM2A N-terminal region, including the R36 – E364 and Q450 – P517 residues, with the H3(A29 – Y41)K36me1/2/3 peptide were found in the structures PDB ID: 4QWN, 4QX7, 4QX8, 4QXB, 4QXC, 4QXH, 4TN7.[36] The histone peptide binds to KDM2A in such a way that the A29 – P30 – A31 – T32 – G33 – G34 – V35 residues enter the narrow region of the KDM2A channel; K36me1/2/3 falls into the JmjC domain cavity; and the K37 – P38 – H39 – R40 – Y41 residues are incorporated into a wider part of the KDM2A channel.[36] The histone peptide binds to KDM2A, thus forming a U-shaped conformation (see Fig. 4b). Apparently, this occurs through the G34 and P38 residues, which change the initial conformation of the peptide chain.[36]
Upon binding of the H3(A29 – Y41)K36me1/2/3 peptide, the Q181–M191 loop segment, which is disordered in the free state, becomes ordered as a result of interaction with the A31 – G34 segment.[36] This loop segment is present only in KDM2A and KDM2B, according to amino acid sequence alignment. Upon binding of H3(A29 – Y41)K36me1/2/3, the K323 and F324 residues of KDM2A undergo a conformational change: a flap covering the T32 – G34 residues of the histone peptide. In addition, histone peptide residues (A31; G33; G34; V35; K36me1/2/3; K37; Y41) were found to interact (through hydrogen bonds) with KDM2A residues (I188; N186; R325, Y199; K323; S145; Q116; T115, V113, R36), respectively (see Fig. 4b).[36]
It should be emphasized that G34A and P38A mutations in the H3(A29 – Y41)K36me2 peptide reduce the demethylase activity of KDM2A three- and sevenfold, respectively, while mutation Y41A reduces the demethylase activity by 40%. The Y41 residue of the histone peptide is bound in a separate pocket of KDM2A (see Fig. 4b); the possible phosphorylation of Y41 apparently affects the peptide demethylation.[36] Moreover, combined mutations in the G33A – G34A – V35R – K36me2 peptide segment make KDM2A inactive. The point mutation G34V found in paediatric glioblastoma causes a similar effect.[36] Thus, the presence of glycine residues in positions 33 and 34 of the histone peptide is optimal; other residues are likely to cause steric constraints being located in the narrow part of the KDM2A channel.[36]
The N186A mutation in KDM2A decreases the demethylation of the H3(A29 – Y41)K36me2 peptide by 60%; the double K323A – F324A mutation results in threefold decrease in the activity, while replacement of S145A precludes demethylation.[34][36] These data confirm the importance of Q181 – M191 and K323 – F324 segments of KDM2A for peptide stabilization and demethylation. The S145 residue is crucially important for the demethylase activity of KDM2A, assumedly due to the hydrogen bonding between the oxygen atom of the S145 side chain and the NH group in the backbone of K36me2.[34][36]
The interaction details of KDM2A or KDM2B with the nucleosome were investigated using biochemical methods, electron microscopy (PDB ID: 7UV9), and X-ray diffraction (PDB ID: 7UVA).[46] Upon binding of the E2 – Q685 (KDM2A) or A2 – G734 (KDM2B) fragment to the N-terminal moiety of nucleosome H3, the nucleosomal terminal DNA rotates by ~ 45° from its initial coiled state (Fig. 5).[46] In the unfolded state of the nucleosome, KDM2A residues (K99, R105, R128, R152, R319) (see Fig. 5) or KDM2B residues (K129, R135, R158, K182) can interact with DNA regions, thus influencing the demethylase activity.[46] Single mutations K99E, R105E, R128E, R152E, R319E and combined mutations R128E + R152E, K99E + R105E + R128E + R152E in KDM2A increase the average relative levels of H3K36me2 up to 0.60, 0.67, 0.63, 0.74, 0.73 and 0.82, 0.81, respectively, vs. 0.34 for intact KDM2A, i.e., the demethylase activity of KDM2A decreases upon mutations.[46] This is probably caused by the fact that R152E and K99E + R105E + R128E + R152E mutations decrease the KDM2A affinity for the nucleosome ~ 4- and ~ 20-fold, respectively.[46]
![[{"id":"bJNqVntrun","type":"paragraph","data":{"text":"Images of the KDM2A interaction with the nucleosome in the structure (PDB ID: 7UV9) obtained with electron microscopy.<sup>32, 46</sup>"}}]](/storage/images/resized/Cwk4VcNDjlzxHZuIlw8rQ8dj5CFtNbHCjyTYCZSd_xl.webp)
The interaction of KDM2A with the nucleosome depends on the DNA length: the E2 – Q685 fragment more efficiently binds to the nucleosomes that have a linker [the DNA length is 185 base pairs (bp)] than to linker-free nucleosomes (147 bp).[46] This dependence may be due to the interaction of the CXXC domain with the linker DNA containing CpG dinucleotides, since, according to electron microscopy data, the JmjC domain does not interact with linker DNA.[46] The KDM2A protein binds to short (119 bp) nucleosomes more efficiently than to elongated (147 bp) linker-free ones, but less efficiently than to linker-containing counterparts (apparently, due to activation of the H3 N-terminal region).[46]
In addition, it is important to detect the paralogue-specific interaction of KDM2A with the nucleosome acidic patch formed by the Q24, E56, E61, E64, N68, D72, N89, D90, E91, and E92 residues in H2A histone and the Q47, E113 residues in H2В histone.[46] The N-terminus of KDM2A (M1 – K35) can bind to the nucleosome acidic patch through R9, R13, and R19 residues located between H2AE56, H2BQ47, H2BE113; H2AE61, H2AD90, H2AE92; H2AN68 and H2AD72 residues (see Fig. 5), whereas the KDM2B N-terminus (M1 – V65) does not bind to the nucleosome acidic patch. Mutations R9A, R13A, and R19A in KDM2A increase the average relative levels of H3K36me2 up to 0.54, 0.66, and 0.55, respectively, while fragments with the truncated N-terminus (G16 – Q685 and R36 – Q685) increase the respective values to 0.85 and 0.93 vs. 0.36 for intact KDM2A (E2 – Q685), implying a decreased demethylase activity of mutant and truncated KDM2A forms.[46] Since the E2 – Q685 fragment of KDM2A showed a higher demethylase activity than the A2 – G734 fragment of KDM2B (average relative levels of H3K36me2 were 0.24 and 0.67, respectively), the chimeric protein KDM2A(E2 – K35) – KDM2B(R66 – G734) demonstrated a higher activity (0.27) than KDM2B (0.67), while KDM2B(A2 – V65) – KDM2A(R36 – Q685) exhibited a lower activity (0.84) than KDM2A (0.24).[46] It is assumed that the acidic residues in the N-terminal segments of KDM2A (E23 – E33) (see Fig. 5) and KDM2B (D51 – E61) have a stabilizing effect owing to electrostatic interactions with the histone surface; in the case of KDM2A, this is confirmed by the increased average relative level of H3K36me2 (0.78) upon replacement of E23 – E33 acidic residues to neutral residues (0.36 for intact KDM2A).[46] These data indicate the high significance of the N-terminus of KDM2A (M1 – K35) for H3K36me2 demethylation and a weak influence on the nucleosome binding. It is possible that binding of the N-terminus to the nucleosome acidic patch promotes the orientation of KDM2A for effective demethylation of H3K36me2.[46] Since the N-terminus of KDM2B (M1 – V65) does not bind to the nucleosome acidic patch, effective H3K36me2 demethylation through KDM2B does not occur.[46] Apparently, KDM2B uses other proteins for orientation on the nucleosome and optimization of demethylation.[46]
The protein fragments containing the CXXC domain interact with synthetic double-stranded DNA.[38] These domains in KDM2A and KDM2B preferentially bind to non-methylated CpG dinucleotides.[38] The crescent-like CXXC domain (Fig. 6) interacts with DNA through the positively charged surface and specifically recognizes CpG via the M600 – K601 – Q602 residues (in KDM2A) or M642 – K643 – Q644 residues (in KDM2B), which are hydrogen-bonded to the heterocyclic bases of nucleotides (models based on the crystal structures [PDB ID: 4BBQ and 4O64]).[38] The CXXC domain in intact KDM2A is responsible for binding to a double-stranded DNA [the dissociation constant (Kd) is 1.2 μM]; binding is weaker in the M600A mutant (Kd = 2.9 μM) and is absent for Q602A mutation.[38] The D590 side chain in KDM2A or D632 side chain in KDM2B apparently stabilize the recognition of CpG through hydrogen bonding with the M600, K601 or M642, K643 residues (see Fig. 6), thus providing the binding of CXXC to CpG.[38] Hence, owing to the complex structure of KDM2, the action of separate domains is cooperative: CХХC recognizes non-methylated CpG dinucleotides, thus directing the protein toward demethylation of lysine residues by JmjC. In other words, the CХХC domain may be favourable for positioning the demethylase on the template, providing an effective catalysis, without directly participation in it.
![[{"id":"UkLkgd5iX2","type":"paragraph","data":{"text":"Overlay of 3D structures of KDM2A (PDB ID: 4BBQ) and KDM2B (PDB ID: 4O64) containing CXXC and PHD domains.<sup>32, 38</sup>"}}]](/storage/images/resized/DzONfzvZz2GNCTy6b9AsUylsIAN1JGi0Ka2Zs17c_xl.webp)
The CXXC domain in KDM2B can be stabilized by the PHD domain (see Fig. 6) through hydrogen bonds between the R610 and E687, P714, N715 amino acid residues and through hydrophobic interactions between L656, A660, L685, I694 residues.[38] Similar stabilization through interactions between R568 and E641, P667, N668, as well as between L614, V618, L639, I648 is likely, for KDM2A based on tertiary structure alignment (PDB ID: 4BBQ, 4O64). Comparison of amino acid sequences suggests that the PHD domains in KDM2A and KDM2B do not bind to histone terminal regions,[38] although binding of the PHD domain of KDM2B to H3K36me2 and H3K4me3 has been reported.[39] The PHD domain of KDM2A does not bind to histone peptides.[47] Phosphorylation of T632 in this domain weakens the binding to chromatin.[48] The role of the PHD domain in KDM2A and KDM2B requires further investigation.
Interactions of F-box and LRR domains of KDM2A and KDM2B in complexes with other proteins have been investigated.[43-45] Gorelik et al.[43] developed ubiquitin variants (UbV) for inhibiting the activity of SKP1 – CUL1 – F-BOX complexes (SCF)** of E3 ligases through binding of UbV to the SKP1 – F-BOX interface in competition with CUL1.[43] The crystal structures of SKP1 – UbV – F-BOX complexes (PDB ID: 6BYH, 6C16, 6BVA) demonstrate the interaction of the SKP1 protein with the F-box domain of KDM2A and KDM2B, thus confirming the importance of this domain for the formation of multiprotein complexes. The interaction of SKP1 with KDM2A or KDM2B F-box domain can occur through L105, N108, D117, V123, E133, I135, F139, E149 (in SKP1) and V897, E896, Y904, V901, R915, V916, W898, K918 (in KDM2A) or V1069, E1068, Y1076, V1073, R1087, V1088, W1070, R1090 (in KDM2B) (Fig. 7a).[43]
![[{"id":"wZINZ99glp","type":"paragraph","data":{"text":"3D visualization of crystal structure fragments of multiprotein complexes with KDM2: overlay of the KDM2A F-box domain structure (PDB ID: 6BYH) on KDM2B F-box domain in the complex with SKP1 (PDB ID: 6BVA) (<i>a</i>), and structures of PCGF1 complex with LRR(1-7) domain of KDM2B, BCORL1 (PDB ID: 5JH5) (<i>b</i>) and PCGF1 complex with LRR(1-7) domain of KDM2B, BCOR (PDB ID: 8HCU) (<i>c</i>).32, 43 – 45 The following designations are used: PCGF1 is the Polycomb group RING finger protein 1; BCOR is BCL-6 corepressor; BCORL1 is BCL-6 corepressor-like protein 1."}}]](/storage/images/resized/iyW8EhRzJMnILZiwE5eUtCtl6CcBuRpQgfwKcPZx_xl.webp)
The LRR(1-7) domain of KDM2B is also important for the formation of multiprotein complexes. Wong et al.[44] showed the possibility of assembling a four-component non-canonical Polycomb repressive complex 1 (PRC1.1) by combining the SKP1 – KDM2B and BCORL1 – PCGF1 heterodimers. This assembly can be accomplished using the full-length SKP1 protein and KDM2B (D1059 – S1336 region), BCORL1 (D1594 – S1711), and PCGF1 (L150 – K255) fragments.[44] The crystal structure of the complex (PDB ID: 5JH5) shows binding of the BCORL1 – PCGF1 dimer to the KDM2B LRR(1-7) domain.[44] The BCORL1 – PCGF1 dimer interacts with KDM2B LRR(1-7) domain through D1594, Q1664 (in BCORL1); R197, K234 (in PCGF1); and N1135, D1224 (in KDM2B) residues, respectively (see Fig. 7b).[44] Interestingly, the BCOR – PCGF1 dimer with the BCOR protein homologous to BCORL1 shows a different binding pattern to the LRR(1-7) domain of KDM2B.[45] The crystal structure of the complex (PDB ID: 8HCU) also reveals the interaction of the N1619, P1620, P1621 residues (in BCOR) with W1143, P1115, L1118 residues (in KDM2B) (see Fig. 7c). The residues of two acidic regions of BCOR (E1607 – E1611 and E1624 – D1631) are also important for BCOR – PCGF1 heterodimer binding to the LRR(1-7) domain of KDM2B; this is confirmed by biochemical experiments and simulation of the SKP1 – KDM2B – BCOR – PCGF1 complex based on the crystal structure (PDB ID: 8HCU).[45]
* CpG is a dinucleotide consisting of cytosine (C) and guanine (G) bases arranged in the 5'-C – G-3' sequence.
** SKP1 is the S-phase kinase-associated protein 1, CULl is cullin-1, F-BOX is the F-box domain-containing protein.
4. Functions, catalytic mechanism, and biological significance of KDM2
This Section addresses the action of KDM2 proteins for demethylation of lysine residues and the mechanism of this process. Note that the demethylation is not the only function of these enzymes; recent studies have expanded the views on KDM2: apart from catalysis, these proteins may also perform non-enzymatic functions. In this case, the significance of KDM2 increases; however, deeper insight in the fundamental problem complicates the practical aspect, that is, the development of chemical tools to modulate epigenetic events in cells for therapeutic purposes.
4.1. Histone demethylase activity of KDM2A
The KDM2A protein was established to act as a demethylase for H3K36me. Analysis of interactions of KDM2A with methylated histone peptides, core histones, and mono- and oligonucleosomes as substrates indicated that this enzyme demethylates mainly H3K36me2 and, to a lesser extent, H3K36me1.[49] The demethylation of H3K36me2 by KDM2A in cell culture was confirmed by immunocytochemistry.[49-51] A decrease in the KDM2A content in cervical carcinoma cells did not induce global changes in the H3K36me2 content, but substantially increased H3K36me2 in CpG islands (according to chromatin immunoprecipitation with quantitative polymerase chain reaction).[52] The demethylation was found in mouse hepatocytes as an increased amount of H3K36me2 in the C/EBPα locus* upon the knockdown of the kdm2A gene.[53] The gene specificity of the effect is important: in the C/EBPβ locus, the knockdown of kdm2A did not increase the H3K36me2 level; moreover, the knockdown of kdm2A virtually did not affect the amount of H3K36me3 either.[53] Furthermore, an increased H3K4me3 in p15INK4B and p27Kip1 promoter regions** upon knockdown of kdm2A in stem cells from apical papilla was not accompanied by significant changes in the H3K36me2 level.[54]
The geometric parameters of crystal structures containing complexes of mouse KDM2A fragments and H3K36me reveal the details of interaction between KDM2A and H3K36me; as a result, the absence of demethylase activity of KDM2A toward H3K36me3 (Ref. 49) can be attributed to steric constraints in the catalytic site preventing demethylation of this substrate.[36] A study of the KDM2A demethylation mechanism using quantum mechanics/molecular mechanics (QM/MM) approach confirmed the role of steric constraints in KDM2A-mediated demethylation of H3K36me3.[55] Thus, H3K36me2 is recognized as the main histone substrate of KDM2A.[36][49-53] In some cases, KDM2A was found to exhibit demethylase activity toward H3K36me1[49][51] and H3K4me3.[54] The necessity of the JmjC domain of KDM2A for demethylation has been mentioned.[36][49][53]
* C/EBPα is the gene that encodes the CCAAT/enhancer-binding protein alpha; C/EBPβ is the gene encoding the CCAAT/enhancer-binding protein beta.
** p15INK4B gene encodes the inhibitor of cyclin-dependent kinase 4B; p27Kip1 gene encodes the inhibitor of cyclin-dependent kinase 1B.
4.2. Histone demethylase activity of KDM2B
Like KDM2A, the KDM2B protein exhibits demethylase activity toward histone lysine residues with various degrees of methylation; the key role belongs to the JmjC domain.[50][56-58] A study of the effect of this enzyme on demethylation of H3K4me3 and H3K36me2 in mouse embryonic fibroblasts revealed no global histone modification, which suggests a locus-specific function of KDM2B.[39] This hypothesis was confirmed by chromatin immunoprecipitation: KDM2B binds to Ccl7 and Xist gene promoters,* thereby considerably decreasing the amount of H3K4me3 in these promoter regions, whereas the level of H3K36me2 remains unchanged or increases.[39] A decrease in the H3K4me3 level, with the content of H3K36me2 being invariable, was also found[39] in the Crabp2 and RipK3 promoters.** These data confirm the demethylase activity of KDM2B toward H3K4me3 and the absence of this activity toward H3K36me2, but demethylation of H3K36me2 by KDM2B under other conditions cannot be ruled out.[39]
Furthermore, KDM2B demethylates H3K36me2 to a higher extent than H3K36me1, while demethylase activity toward H3K4me3 is virtually absent.[56] Kang et al.[57] established the demethylating action of this enzyme toward H3K79me2 and H3K79me3, which is confirmed by experiments on incubation of GST-labelled (GST is glutathione S-transferase) KDM2B containing M1–G734 amino acid residues with core histones and nucleosomes from 293Т cells. In addition, KDM2B was shown to demethylate H3K4me and H3K36me.[57] A variety of histone substrates of KDM2B have been reported, including H3K4me3,[39][50][58] H3K9me3,[59] H3K36me1,[56] H3K36me2,[56][58][60] H3K36me3,[60] H3K79me2,[57] and H3K79me3.[57]
The substrate specificity of the KDM2A[52][54] and KDM2B[39] [56] enzymes in particular cases is determined by the functional conditions: the chromatin ‘landscape’ that either promotes or hinders demethylation, as well as other features of catalysis in multiprotein complexes.[39] In this context, functioning of KDM2 implies non-enzymatic effects. This important aspect introduces a new level of complexity in interpreting the biological role and significance of KDM2 as therapeutic targets (see Sections 4.4, 5, and 6).
* Ccl7 gene encodes the C – C motif chemokine 7; Xist gene encodes the X-inactive specific transcript.
** Crabp2 gene encodes the cellular retinoic acid-binding protein 2; RipK3 gene encodes the receptor-interacting serine/threonine-protein kinase 3.
4.3. Mechanism of KDM2A-mediated demethylation
For demethylation of the Nɛ-methylated lysine residue, the KDM2A and KDM2B proteins use the FeII cofactor, the 2-OG co-substrate, and an oxygen molecule. As a result of demethylation, the lysine residue loses a methyl group; succinate, CO2 , and formaldehyde molecules are released.[13][14][61]The mechanism of demethylation by KDM2A has been studied in detail.[34][36][55] The putative mechanism of demethylation of Nε-methylated lysine residue by KDM2A is shown in Fig. 8. In the case of KDM2B, study of the demethylation mechanism is hampered, because no JmjC-containing structure is available in PDB.
![[{"id":"zgqXZdN8E1","type":"paragraph","data":{"text":"Mechanism of KDM2A-dependent demethylation of lysine residues (from Refs 34, 36, 55, and 61). The shown Fe-involving bonds should be considered as coordination bonds. E is the energy value between the current and transition (≠) states on the way to the next state. The energy values were found for H3K36me2 using quantum chemical calculations at the UB3LYP/def2-TZVP + ZPE level.<sup>55</sup> See text for the legend (<i>a – k</i>)."}}]](/storage/images/resized/16qpKSPjdLsAnCjIm2ynGfRAYkW8vA2abRTG0lQR_xl.webp)
The demethylation of the Nɛ-methylated lysine residue of histone is preceded by a series of interrelated events. Presumably,[34][36] FeII, 2-OG, and the methylated lysine residue successively bind to the JmjC domain of KDM2A. First, FeII is positioned in the JmjC cavity and coordinates H212, D214 and H284 amino acid residues, forming a catalytic site. This complex acquires an octahedral geometry in which the FeII ion is positioned approximately at the octahedron centre, the H212 and D214 residues are located in a square plane on one edge of the octahedron, while H284 occupies an apical position relative to the square plane (see Fig. 8a and Fig. 9a). In such a complex, FeII can coordinate another three water molecules (see Fig. 8a), two of which would be located in the square plane opposite to the H212 and D214 residues, while the third one occupies the apical position opposite to H284.[34][36] Mutations H212A[49][51][53] and D214A[36] eliminate the demethylase activity of KDM2A. In the KDM2B protein, H242A mutation (similar to the H212A in KDM2A) also renders the enzyme inactive.[57]
![[{"id":"FQ9v7tgEUP","type":"paragraph","data":{"text":"Explanation to the demethylation mechanism based on 3D visualization of the crystal structure (PDB ID: 4QX7): KDM2A complex with the histone peptide according to the protein domain architecture and amino acid sequence of the peptide; amino acid residues coordinated by NiII, which replaces the endogenous FeII (<i>a</i>), and arrangement of amino acid residues and 2-OG near the NiII ion (<i>b, c</i>).<sup>32</sup>"}}]](/storage/images/resized/pR0q0anyroxaMaqdGTz4wVca6aZJEiWwfqZSlulX_xl.webp)
Second, the 2-OG co-substrate enters the KDM2A JmjC cavity and is held in the site formed by the N142, I144, L201, T209, Y222, K229, and V286 amino acid residues (see Fig. 9b). It is assumed[34][36][55] that 2-OG has initially an ‘off-line’ orientation[62] (see Fig. 8b, Fig. 9b), i.e., carboxylate oxygen at the C(1) atom of 2-OG occupies the apical position of the octahedron opposite to the H284 residue, while carbonyl oxygen at the C(2) atom is located in the square plane of the octahedron opposite to D214. The 2-OG carboxylate oxygen atom at C(1), which is not coordinated to FeII, can be hydrogen-bonded to the hydroxyl group of Y222 where the side chain points ‘toward the metal’ (see Fig. 8b).[34] The 2-OG carboxylate oxygen atoms at C(5) can form intermolecular hydrogen bonds with T209, K229, and N142 residues.[34][55] The aliphatic part of 2-OG is involved in hydrophobic interactions with I144 and V286.[34] Binding of 2-OG in the JmjC domain cavity is favourable for stabilization of the I144 – H150 region through co-substrate interaction with N142 and I144.[34] This is confirmed by X-ray diffraction data, namely, a clearer electron density of this region in KDM2A bound to 2-OG compared to the structure in which 2-OG is absent.[34] Furthermore, mutations in the I144 – H150 and V108 – V110 interacting segments reveal the importance of 2-OG-induced stabilization for recognition of H3K36me2 substrate and for the catalytic activity of KDM2A.[34] Mutations that eliminate the demethylase activity toward H3K36me2 include V108D, D109A, V110D, E147A, and F148A.[34]
Third, 2-OG-induced stabilization is favourable for binding of the histone substrate. The H3 oligopeptide (A29 – K36me – Y41 residues) binds via interactions described in Section 3, while K36me enters in the KDM2A JmjC cavity formed by the I144, S145, Y199, L201, D214, F215, V220, G297, and N298 residues (see Fig. 8c, Fig. 9c).[36][55] Hydrophobic interactions arise between the side chain of Ne-methylated lysine and the I144, Y199, L201, and F215 residues.[36][55] The Y199 and F215 residues promote binding of the K36me side chain through cation – π interactions.[34] Mutations Y199A and F215A reduce the demethylase function of KDM2A (the residual activity is 30 – 40%).[36] The Nɛ-protons of mono- and dimethylated K36 can form hydrogen bonds with the carbonyl oxygen atom of the N298 side chain; such interaction is more common for K36me2.[36][55] The N298 orientation is determined by hydrogen bonds between the amino group in its side chain and the D214 carboxylate oxygen atom not coordinated to FeII.[36][55] The mutation N298A leads to the loss of KDM2A demethylase activity, thus indicating the importance of the interactions described above.[34][36] The Nɛ-protons of K36me1 and K36me2 form hydrogen bonds with the hydroxyl group of Y222, its side chain being directed ‘toward the metal’ (see Fig. 8c).[36] The same protons can form hydrogen bonds with the 2-OG carboxylate oxygen atom bound to C(1), which is not coordinated to FeII (see Fig. 8c).[36]
Finally, demethylation of the Nε-methylated lysine residue requires an oxygen molecule. Simulation of O2 transport toward the enzyme catalytic site for the complex of KDM2A with the H3 peptide (A29 – K36me2 – Y41) showed that, apart from the I144, L201, T209, V213, V220, and Y222 residues, oxygen diffusion is affected by histone residues G34, V35, K37, and P38.[55] In this complex, the oxygen molecule preferentially approaches the apical coordination position of the octahedron and is located opposite to H284 (see Fig. 8c).[55] Molecular dynamic simulation also showed that O2 approaches a coordination position in the square plane of the octahedron opposite to the H212 residue, which leads to its capture by a hydrophobic cage consisting of Y222, F231, F276, I278, and V286 and to a long residence time.[55]
Binding of the oxygen molecule in the apical coordination position of the octahedron (opposite to H284) and the formation of FeIII-superoxo complex imply 2-OG reorientation from the ‘off-line’ to ‘in-line’ position,[62] since O2 binding in the apical coordination position is hampered due to the ‘off-line’ orientation of 2-OG. The reorientation consists in the shift of the 2-OG C(1) carboxylate group, in which the oxygen atom located opposite to H284 ends up opposite to H212.[55] The reorientation of 2-OG is hampered if the side chain of Y222 in KDM2A points ‘toward the metal’.[55] For the 2-OG reorientation to be possible, the Y222 side chain must rearrange from ‘toward the metal’ to ‘away from the metal’ position.[55] Thus, first, Y222 moves to the ‘away from the metal’ orientation (see Fig. 8d) and, after that, 2-OG is rotated to the ‘in-line’ coordination mode and the oxygen molecule binds at the apical coordination position opposite to H284 (see Fig. 8e).[55] The energies for Y222 transition from the ‘toward the metal’ to ‘away from the metal’ orientation in the absence and in the presence of O2 are similar: 22.9 and 25.8 kcal mol–1, respectively (according to the UB3LYP/def2-TZVP + ZPE quantum chemical calculations).[55] The E134, S203, D291, and R325 amino acid residues destabilize the transition state on the way of Y222 side chain from ‘toward the metal’ to ‘away from the metal’ position. Conversely, the R137, C207, K229, V286, and E315 residues stabilize this transition state, with K229 making the greatest contribution.[55]
If Y222 points ‘away from the metal’, the 2-OG rotation from the ‘off-line’ to ‘in-line’ mode in the absence of O2 may occur almost without energy consumption (~ 1 kcal mol–1), although free optimization of the product shows partial ‘in-line’ orientation of 2-OG.[55] The 2-OG rotation from the ‘off-line’ to ‘in-line’ position in the presence of O2, together with the formation of FeIII-superoxo complex, occurs at an energy of 4.4 kcal mol–1 (see Fig. 8e).[55] Furthermore, when the Y222 residue points ‘away from the metal’, its hydroxyl group can form a hydrogen bond with the C(5) carboxylate oxygen atom of 2-OG.[36][55] This interaction is apparently important for the demethylase activity of KDM2A. The results of QM/MM simulation indicate that in the case of mutant Y222A, the 2-OG rotation from the ‘off-line’ to ‘in-line’ position requires no energy.[55] However, according to experiments, mutation Y222F reduces the demethylase activity, while Y222A eliminates this activity.[34] These data attest to the importance of the conformation of the Y222 side chain for maintaining the demethylase activity of KDM2A, namely, control of 2-OG orientation, stable interactions with the C(1) and C(5) carboxylate oxygen atoms of 2-OG and with the Nɛ-protons of the methylated lysine. Importantly, 2-OG transition from the ‘off-line’ to ‘in-line’ position is possible only when KDM2A interacts with H3K36me1 and H3K36me2. In the case of H3K36me3, the transition is hindered due to steric constraints arising between the ζ-methyl group of K36 and the FeII-non-coordinated C(1) carboxylate oxygen atom of 2-OG (see Fig. 8d ).[36][55]
The successive binding of FeII, 2-OG, and the histone substrate (H3K36me1 or H3K36me2) in the cavity of the KDM2A JmjC domain and oxygen diffusion to the catalytic site lead to the formation of the FeIII-superoxo complex (see Fig. 8e).[55] According to molecular dynamic simulation data, interactions between D214 and N298 in this superoxo complex are enhanced compared to those in the enzyme – substrate complex, which is manifested for H3K36me1 and H3K36me2.[55] The simulation of the FeIII-superoxo complex indicates that, as a result of interaction with T209, the C(5) carboxylate oxygen atoms of 2-OG are less flexible in H3K36me1 than in H3K36me2.[55]
In the FeIII-superoxo complex, the distal oxygen atom (Od) attacks the 2-OG C(2) carbon atom (see Fig. 8e), which results in an intermediate peroxide compound and CO2 release (see Fig. 8g).[55] The energies required for this process, taking into account the mono- and dimethylated states of the lysine substrate, are 10.8 and 14.9 kcal mol–1, respectively, suggesting a lower energy consumption at this step for the monomethylated substrate.[55]
It is noteworthy that the D109, I144, K229, and E315 residues stabilize the transition state (see Fig. 8f ) for the formation of the intermediate peroxide compound (see Fig. 8g), while D74, K323, R325, and H3K37 destabilize this transition state.[55] The intermediate peroxide compound is decomposed without energy consumption via the homolytic cleavage of the bond between distal (Od) and proximal (Op) oxygen atoms followed by compound rearrangement to yield a ferryl complex with succinate (see Fig. 8h).[55] The complex derived from dimethylated lysine is more stable than the complex containing the monomethylated substrate.[55] Molecular dynamics for ferryl complexes showed that D214 and N298 residues of KDM2A play an important role in the stabilization and orientation of H3K36me1 and H3K36me2 side chains via the formation of hydrogen bonds with Nɛ-protons, with N298 interactions being more frequent.[55] In the case of H3K36me1, there are two relatively stable orientations (the ζ-methyl group is directed either toward or away from the ferryl oxygen atom); however, for H3K36me2, there is only one relatively stable orientation in which one ζ-methyl group points toward the ferryl oxygen atom (see Fig. 8h).[55]
Interactions in the ferryl complex induce the homolytic cleavage of the C – H bond in the ζ-methyl group of H3K36me2 to produce the corresponding radical, while the proton forms a bond with the ferryl oxygen atom (see Fig. 8i).[55] This provides σ transfer of an electron from the C – H bond to the iron cation to give intermediate FeIII – OH complex (see Fig. 8i).[55] The energy required to form this complex is ~ 22 kcal mol–1 for H3K36me2.[55] The N142, D167, K229, and E315 residues stabilize the transition state of the FeIII – OH complex, while D109, I144, and R325 destabilize this state.[55]
The hydroxyl group of the intermediate FeIII – OH complex adds to the substrate radical almost without an energy barrier (1.4 kcal mol–1), with the FeIII – OH bond being cleaved and iron being reduced to FeII (see Fig. 8j).[55] The transition state of this process is stabilized by N142, D167, and E315 and destabilized by I144 and R325.[55] The resulting hydroxylated substrate decomposes to generate formaldehyde and the demethylated substrate (see Fig. 8k).[14][61] After removal of demethylation products from the JmjC cavity, the enzyme can again participate in the catalytic cycle. An important factor is the correlated motion of the amino acid residues of KDM2A (which stabilizes or destabilizes various steps of the demethylation mechanism) with the secondary structure elements of the protein.[55] The coordinated motion was detected for H45 – R105 and S145 – F165; R36 – F165 and K195 – P235; and R36 – P235, C265 – F295, Y330 – Q355, and K475 – L495 regions, while R36 – F295 and E375 – N435 regions were shown to exhibit an uncorrelated motion.[55] The relationship between the moving regions is determined by catalytic steps and the degree of methylation of the lysine residue.[55]
Cheng et al.[36] and Thomas et al.[55] reported the demethylation mechanism for H3K36me1 and H3K36me2 by KDM2A and proposed an explanation to the experimentally observed absence of the activity toward H3K36me3,[49] although crystal structures demonstrating H3K36me3 binding to the KDM2A JmjC domain were obtained.[36] Presumably, this domain acts as a ‘reader’ rather than ‘eraser’ of the H3K36me3 mark. Interestingly, Gao et al.[54] suggested the ability of KDM2A to demethylate H3K4me3. Since the degrees of methylation of H3K36me3 and H3K4me3 are identical, there is a different binding mode of H3K4me3 to KDM2A JmjC that provides the demethylation of this residue. Structural differences between KDM2A and KDM2B determine their substrate specificity: recognition of different sites in histones and dependence of the function on the degree of methylation of the lysine residue. Resolving the crystal structures of KDM2B complexes with histone substrates would help to clarify the mechanism of KDM2 substrate specificity.
4.4. KDM2 proteins as not only histone lysine demethylases
Studies of intracellular localization showed that KDM2 are nuclear proteins.[49-52][56][63] It is reasonable to assume that, apart from the demethylation of lysine residues in histones, KDM2 perform other functions, i.e., non-histone and non-enzymatic (other than catalytic) ones. Indeed, KDM2 can demethylate lysine residues in non-histone proteins.[64][65] KDM2A can inhibit the NF-κB transcription factor via demethylation of p65 subunit at K218me1 and K221me2.[64] The human KDM2A and clawed frog (Xenopus laevis) kdm2A/B proteins demethylate the lysine residues in the L275 – A360 amino acid sequence of β-catenin, a protein important for cell contact with the microenvironment and for regulation of gene expression. The demethylation promotes degradation of β-catenin.[65]
The KDM2A protein, unlike KDM2B, binds to heterochromatin, which is important for organization of the chromatin structure.[63][66] The KDM2A isoform interacts with PRC2 (Polycomb repressive complex 2), which methylates H3K27.[67] In addition, KDM2A can interact with the PFKFB3 protein,* thus mediating its degradation,[68] with the transcription factor 7-like 2 (TCF7L2), thus causing its destabilization irrespective of the catalytic activity of JmjC,[69] and with the DNA repair protein, p53-binding protein 1 (53BP1), thereby promoting genomic instability.[70]
Furthermore, KDM2B interacts with c-Jun transcription factor and with Sin3A transcriptional repressor, a paired amphipathic helix protein.[71] The KDM2B isoform is incorporated in the SCF complex of ubiquitin ligase E3 [44][72] and in the PRC1.1 complex, which regulates transcription and monoubiquitinates the H2AK119 mark.[44][73] Interaction with KDM2B stabilizes the estrogen receptor; apparently, this interaction is independent of the JmjC domain.[74]
Thus, KDM2 enzymes, being components of multiprotein complexes that vary in composition and function, may perform other roles besides the direct demethylation of lysine residues in histones. The functional importance of these complexes is generally similar to that of demethylation: heterochromatin for local transcriptional repression is formed in one way or another.
The non-enzymatic functions of KDM2 are linked to structural specialization of domains (see Section 3) and diversity of isoforms. Detection of truncated KDM2A[51] and KDM2B[39][75] isoforms lacking the N-terminal region and JmjC domain confirms functions other than demethylation. An interaction between the long and short KDM2B isoforms with chromatin-associated Polycomb repressive proteins, independent of demethylase activity, was detected.[76] The phenotypic differences arising upon the loss of long or short KDM2B isoforms suggest that the isoforms have different functions.[77] Interestingly, the short (but not long) isoform of KDM2A may mediate the decrease in the H4K20me3 level, thus activating the transcription.[78] The short isoform, unlike the long one, can form heterochromatin bodies by interacting with heterochromatin protein 1 homolog alpha (HP1a). This isoform is devoid of demethylase activity. Therefore, its binding to pericentromeric regions of chromosomes results in local accumulation of H3K36me, chromatin compaction, and reduced gene expression.[79] In addition, short KDM2 isoforms may restrict the Wnt signalling pathway and bind to transcription factor 7-like 1 (TCF7L1).[80] According to other data,[81] KDM2 proteins act as repressors of genes whose promoters are enriched with CpG islands. Attenuated expression is not limited to KDM2 demethylase activity and may be attributed to the regulation of RNA polymerase II.[81] In addition, the demethylase activity of KDM2 makes a minor contribution to the depletion of H3K36me2 from the promoters carrying CpG islands, without marked disruption of gene expression.[81]
The biological role of KDM2A was established for various processes such as maintenance of genomic stability,[36][63] [70] differentiation,[82] glucogenesis,[53] circadian rhythm regulation,[83] and proliferation.[54][68][78][84] The role of KDM2A in various types of cancer is considered, including breast,[78][85-89] gastric,[90-92] lung,[93][94] colorectal,[64][95] hepatic,[96][97] bladder,[98] and ovarian cancers[99] and myeloma.[68] As a rule, KDM2A emerges as a factor of disease progression; however, some studies[64][68][86][89] suggest an opposite effect of KDM2A.
Processes mediated by KDM2B include normal nervous system development,[75-77] control of cell proliferation and size,[39][50][56][72] senescence,[56][58][60][100] metabolic reprogramming,[60][101] apoptosis,[102] and glycolysis.[103] The role of KDM2B has been studied in leukaemia[104-107] and other cancers, including breast,[74][102] [108][109] ovarian,[110-112] cervical,[50][72] [103]prostate,[102][113][114] bladder,[115][116] gastric,[117][118] and pancreatic.[119] This role may be anti-oncogenic,[50][72][105][109] although KDM2B is mainly considered to be a factor of tumour progression. The outcome is determined by the combination of conditions. Thus, the KDM2B function can be manifested in cooperation with other transcriptional proteins. In Ewing sarcoma cells, KDM2B cooperates with DHX9 helicase: this interaction optimizes chromatin permissiveness for RNA polymerase II in the region of the YAP1 gene, thereby inducing the pro-oncogenic Yes-associated protein 1.[59]
The above results make it possible to consider KDM2 as therapeutic targets. The situation is complicated by the fact that the effects of these proteins may be linked to the demethylase activity[93][94][98][104][111][112] or be unrelated to the catalytic function.[72][78][85][106] Thus, KDM2 proteins participate in the regulation of numerous processes.[120-123] The relationship of their effects is determined by particular conditions and requires detailed investigation: in some cases, certain functions prove to be decisive. A deeper understanding of the role of KDM2 could be facilitated by the development of small-molecule compounds as pharmacological modulators of these proteins.
* PFKFB3 is 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3.
5. Small-molecule KDM inhibitors
This Section presents structures of low molecular weight organic compounds that target lysine demethylases. Since the medicinal chemistry of these compounds is relatively a new field, their number is moderate. It is important to identify the benefits and drawbacks of modulators of various demethylases, not only KDM2. Detailed analysis of chemical modulators of particular KDM subgroups can be found elsewhere.[124-126]
X-Ray crystallography and NMR spectroscopy enable a detailed investigation of the domain architecture and spatial structure of KDM by visualizing the 3D organization of proteins, thus facilitating the development of inhibitors for these enzymes. The known KDM blockers vary in efficiency and selectivity.[124-126] Currently, studies of KDM1 inhibitors are in progress: nine KDM1A antagonists are in various stages of clinical trials.[125][127] Among them, seven compounds are covalent inhibitors based on tranylcypromine (TCP), while two compounds encoded as CC-90011 and SP-2577 are reversible inhibitors (Fig. 10). Pulrodemstat (CC-90011), a selective reversible KDM1A inhibitor (with half-maximal inhibitory concentration, IC50 , of 0.3 nM), induces differentiation of acute myeloid leukaemia cells and small cell lung cancer cells.[128][129] Seclidemstat (SP-2577), a reversible KDM1A inhibitor (IC50 = 1.3 μM), is investigated for efficacy, tolerability, and safety.[129-131]
![[{"id":"KkM7bh7z_N","type":"paragraph","data":{"text":"KDM1A inhibitors in clinical trials"}}]](/storage/images/resized/wmtHGtJyX7zw9PxWQd6wsFPObRckunwam0e71Xzv_xl.webp)
For JmjC-KDM inhibitors, the isoform specificity and bioavailability are relevant. The known classes include 2-OG analogues [N-oxalylglycine (NOG),[132] dimethyloxalylglycine (DMOG),[132] and 2-hydroxyglutaric acid (2HG)[133]], caffeic acid;[127] pyridine derivatives [pyridine-2,4-dicarboxylic acid (2,4-PDCA),[134] compounds encoded as JIB-04,[135] KDM5-C49,[136] and KDM5-C70[136]], 8-hydroxyquinoline derivatives (IOX1,[137] B3,[138] and CBA-1 [139]), pyrimidine derivatives (GSK-J1[140] and GSK-J4[140]), and pyrido[3,4-d]pyrimidin-4(3H)-one derivatives (GSK467 [136] and Compound-54k[141]); and inhibitors based on 3-aminoisonicotinic acid (QC6352 [142] and TACH101 [143]), pyrazolo[1,5-a]pyrimidin-7(4H)-one (CPI-455),[144] and 1,3-benzodioxole (JDM-7)[145] (Fig. 11). Virtually all of the listed inhibitors compete with 2-OG for binding to the enzyme active site.
![[{"id":"gziH1_sXKF","type":"paragraph","data":{"text":"JmjC-KDM inhibitors"}}]](/storage/images/resized/qKSHYVFhToMozmHMoOOcnTqYB3IQT1JP9aplYD8G_xl.webp)
Peptide inhibitors that disrupt the protein–protein interactions are also being investigated. These compounds, developed using the peptidomimetic approach, demonstrated higher selectivity than metal chelators.[146] The catalytic action of KDM4A and KDM4C was retained in truncated peptides, H3(A7 – K14)K9me3 and H3(A7 – T11)K9me3, respectively.[146] Binding of uracil, a known iron chelator, to the lysine amino group at position 9 of similar peptides, resulted in the formation of KDM4-Tat peptides (see Fig. 11) with inhibitory activity against KDM4A and KDM4C.[146] Kawamura et al.[147] developed the cyclic peptide CP2 (see Fig. 11), a histone substrate-competitive inhibitor of KDM4A-C with high selectivity compared to other JmjC-KDM proteins.
Results of crystal structure investigation of the KDM4A complex with CP2 (PDB ID: 5LY1) showed that the ligand is located in the histone-binding groove of KDM4A, and the R6 amino acid residue is bound in the sub-pocket occupied by the H3K9me3 substrate.[147] The positively charged residues at position 6 of the cyclic peptide are necessary for enhancing the inhibitory activity.[147] Optimization of the CP2 peptide resulted in the CP2f-7 cyclic peptide (see Fig. 11), which proved to be more effective in inhibiting KDM4A/C according to the AlphaScreen method (IC50 = 6.0 and 2.2 nM for KDM4A and KDM4C, respectively).[148] Although high selectivity and efficacy have been achieved for peptide inhibitors, their low cellular uptake and instability pose a challenge in the development of practically useful products.
Of note is the search for new agents targeting auxiliary domains of JmjC-KDM with high-throughput screening of chemical libraries. The benzofuran derivative B2 found in such screening (see Fig. 11) is a ligand that disrupts the interaction between the PHD1 domain of KDM5A and histone.[149] The binding of B2 to the PHD1 domain was confirmed with NMR spectroscopy.[149]
Efforts have been made to develop small-molecule inhibitors of KDM4, most of which are close analogues of 2-OG targeting the JmjC catalytic pocket.[150][151] However, the high polarity of this pocket complicates the development of this chemotype.[142] The 2,4-PDCA inhibitor showed a potent activity against KDM4, but low selectivity.[134][137] Other broad spectrum inhibitors such as IOX1 proved to have low activity in cell culture experiments, despite the high activity in cell-free assays.[134][137] This discrepancy is apparently attributable to low cellular uptake of inhibitors and competition with the endogenous 2-OG. Some inhibitors are in various phases of clinical trials. For example, caffeic acid is being studied as a KDM4C inhibitor.[127][152] The compound TACH101 showed high enzyme-inhibitory activity against KDM4 (IC50 = 41 – 270 nM) as well as selectivity and antiproliferative activity in cultured tumour cells and in in vivo models.[143]This compound is in Phase I clinical trials,[153] which confirms the potential of KDM4 as a therapeutic target.
Compounds possessing selectivity to KDM2 have been identified (Fig. 12). Daminozide (1), a plant growth regulator, selectively inhibits KDM2/7 at submicromolar concentrations. The selectivity of inhibition by 1 is two orders of magnitude higher for KDM2/7 than for other demethylases. Daminozide interacts with the 2-OG binding site and chelates the metal in the catalytic site through hydrazide carbonyl and hydrazide dimethylamino groups.[154]
![[{"id":"JQnDg_xjhe","type":"paragraph","data":{"text":"KDM2 inhibitors with differential selectivity to other JmjC-KDM proteins"}}]](/storage/images/resized/c4DnyZOsEVAda4x6X9VuO0BIYaHtzfnC3Nws8SKJ_xl.webp)
The TC-E5002 (2) selective inhibitor based on hydroxamic acid has been obtained. This compound proved to effectively inhibit KDM7A and KDM7B (IC50 = 0.2 – 1.2 μM), being 1 – 2 orders of magnitude less selective to KDM2A and demethylases 5A, 4A, 4C, and 6A. The inhibitory activity of 2 is affected by the 9-cyclopropylnonanoic acid moiety. Compound 2 was able to exhibit an antiproliferative activity.[155]
An important step in the design of selective blockers was the development of 1,2,3-triazole derivative 3.[156] This compound proved to be highly effective against KDM2A with IC50 of 63 nM, which was several orders of magnitude lower than the corresponding values for lysine demethylases 3A, 4A, 4C, 4E, 5C, and 6B. The X-ray diffraction data for the co-crystal of a close analogue of 3, 2-(1H-1,2,3-triazol-4-yl)isonicotinic acid, with KDM4A confirmed binding to the metal ion in the JmjC domain. The synthesis of other analogues yielded selective inhibitors of various JmjC-KDM. In particular, azetidine-substituted triazoles exhibited selectivity to KDM5C, while piperidine derivatives were selective for KDM2A. In view of the high degree of homology between the catalytic domains of KDM2 and KDM7, the authors suggested that compound 3 is a potent inhibitor for both subgroups.[156] It is noteworthy that the effect of 3 markedly exceeds that of other KDM2/7 inhibitors such as 1 and 2. This emphasizes the potential of 3 as a drug candidate.
High-throughput screening identified N,N-dimethylaniline (4, 5) and toluene (6) derivatives, which bind to KDM2A and KDM4A with micromolar Kd values. These compounds cause histone hypermethylation in pancreatic ductal carcinoma cells.[157] However, authors did not report the consequences of this effect: whether the hypermethylation resulted in deregulation of gene expression or affected cell viability?
McAllister et al.[124] presented a number of patented structures of lysine demethylase inhibitors. Quanticel Pharmaceuticals patented the inhibitory action of 2-(1-methyl-1H-imidazol-4-yl)isonicotinic acid (7) against KDM2B, -4A, -4C, -5A, and -5B (in cell-free systems, IC50 < 0.1 μM). 1,2-Diazole derivative 8 inhibited KDM2B, -4C, -5A, and -5B at comparable concentrations and exhibited activity in breast cancer cells (ZR-75-1 line, IC50 < 0.1 μM for demethylation of H3K4me3). 2-Aminomethylpyridine derivative 9, which inhibits KDM2B and KDM5B (IC50 < 0.25 μM) and, to a lesser extent, KDM6A (IC50 > 2.5 μM), is mentioned in EpiTherapeutics patents.
McAllister et al.[124] and Wang et al.[157] did not report the activities for 4 – 9 against KDM7. However, the structural similarity of KDM2A and KDM7A (see Fig. 1) made it possible to develop a common inhibitor, the substituted indoline 10 (IC50 ~ 0.2 μM).[126][158] This compound proved to have very high selectivity to the indicated isoforms (> 75-fold relative to other JmjC-KDM). At 1 – 10 μM, 10 inhibited demethylation of H3K36me2, while in HAP1 cells (1 μM), it caused considerable changes in the expression of ~ 30 genes. According to kinetic studies, this derivative did not compete with either 2-OG or the peptide substrate, which distinguished it from most KDM2 inhibitors.[158] Hence, compound 10 was a breakthrough in the development of selective KDM2 inhibitors. The relevance of development of this agent consists in its use as a unique chemical tool for studying the properties of lysine demethylases in cell-free and, more importantly, in cell-based systems.
Despite the achievements of medicinal chemistry of JmjC-KDM inhibitors, their optimization requires further biological studies, as well as the synthesis of focused chemical libraries, and elucidation of detailed structure–activity relationships.[14][125][126][159] The implementation of candidate compounds into medical practice remains critical. The difficulties consist in not only the structural similarity of particular demethylases, low bioavailability, and other undesirable properties. Along with these important, but potentially surmountable obstacles, it is necessary to mention a fundamental problem of non-enzymatic functions of KDM. To solve this issue, strategies beyond the scope of classical enzyme inhibition are required.
One such innovative strategy is based on the use of targeted degraders obtained by the PROTAC (PROteolysis Targeting Chimera) technique, that is, chimeric molecules for targeted protein proteolysis. These are bifunctional molecules capable of recruiting a target protein (in this case, KDM) to E3 ubiquitin ligase for poly-ubiquitination and degradation in the proteasome. A benefit of the PROTAC approach for targeting KDM is the possibility to remove the whole protein.[160] The first preclinical trials of PROTAC agents targeting KDM demonstrated effective and selective elimination of particular isoforms (e.g., KDM4A – C, with KDM4D remaining intact), which emphasizes high specificity with respect to the target.[161] These degraders cause the expected effects, namely, accumulation of methylated lysine residues in histones, suppression of tumour cell proliferation, delayed cell cycle, and apoptosis.[161-163] A few compounds targeting various KDM have been described: RDN8011,[161] GT-653,[164] YTHu78,[165] and Compound-4[166] (Fig. 13).
![[{"id":"f6fBmMfpnm","type":"paragraph","data":{"text":"KDM inhibitors based on PROTAC technique; proteins targeted for degradation are indicated under the compound codes"}}]](/storage/images/resized/1ZWnRGapSTiBYCEQZ3fZpsX7SN1dfYbQsaIe5ceI_xl.webp)
Thus, the development of KDM degraders using PROTAC is an innovative strategy to overcome key limitations of classical inhibitors. This strategy not only provides new potent chemical tools for in-depth study of the biological functions of KDM in vitro and in vivo, but also opens up prospects for the design of highly effective and selective agents. No examples of ‘molecular glues’ (see Section 6) specific to KDM have been reported in the literature. In the case of molecular glues, success was achieved for other proteins: IKZF1 transcription factor (IKAROS family zinc finger protein 1), cyclin-dependent kinase 12 (CDK12), and GSPT1 translation factor (G1 to S phase transition protein 1).[167][168] Development of molecular glues for KDM is a subject for future research.
6. Conclusion
Epigenetic labelling of the genetic apparatus carried out by few classes of specialized enzymes is an elegant method conserved in evolution for biological regulation. Small chemical groups such as acetyl, methyl, and phosphate, prove to be potent modifiers of vital essential processes such as maintenance of genome structure and regulation of gene transcription. It is just to ‘mark’ particular (not any!) amino acid residues in DNA-binding proteins (histones) with these groups to ultimately influence the fate of a cell via changing the gene expression profile at the local and genome-wide levels. The efficiency of this regulation is in the fact that this influence does not require modification of gene structure, a long-term process that is reliably controlled in eukaryotes. Epigenetic regulation is especially important for cell responses to external stimuli.
This review considers mainly the chemical aspects of epigenetic chromatin labelling in relation to methylation of lysine residues in histones regulated by KDM2 subgroup demethylases. The complex mechanism of catalysis, depending on the multidomain structure of the enzyme, the degree of methylation, and the location of the lysine residue in the histone substrate, was demonstrated. Demethylation is a multistage event including a series of sophisticated stages unveiling as a dynamic interaction between the enzyme and the substrate.
Evaluation of biological consequences of chemical modification of chromatin is faced with certain difficulties. Fig. 14 shows that one and the same amino acid residue in the histone may be modified by both acetylases and methyltransferases. However, the results of acetylation and methylation are usually opposite: activation or inhibition of gene expression. This way of controlling histone conformation allows for fine-tuned regulation of transcription, which is optimal for the expression of genes at a specific locus in a given situation. However, due to the redundancy of amino acid residues as targets for epigenetic labelling, it is difficult to reliably predict the action of lysine demethylases. This uncertainty applies to a greater extent to cell-based than to cell-free systems.
![[{"id":"v3lrLzSZya","type":"paragraph","data":{"text":"Multiplicity of epigenetic modifications of histones: lack of strict selectivity of amino acid residues for particular modifications: methylation (me), acetylation (ac), and phosphorylation (p); H2A, H2В, H3, H4 are histone variants; for example, in H3 histone, the same lysine residues can be methylated as well as acetylated.<sup>10</sup>"}}]](/storage/images/resized/3SXnxM9ULRtKxyLrPQ6Nbh8cE0NaptYpggu676hb_xl.webp)
The solution of this problem may be facilitated by development of small-molecule tools for modulation of KDM2 proteins in cells. Currently, the main focus of this research is the design of KDM2 inhibitors. This is justified by increasing number of works supporting the role of KDM2 in various tumour types (see Section 4.4). It is reasonable to assume that KDM2 can act as intracellular targets for anticancer therapy and that a decrease in the demethylase activity could become a promising component of treatment protocols, given that the KDM2 structure makes these proteins druggable.
Analysis of the structure and properties of compounds developed for KDM2 modulation provides the following considerations. First, it is difficult to obtain inhibitors with high preference for a specific isoform due to the structural similarity of proteins. Second, high selectivity may not be a mandatory requirement for KDM2 inhibitors as anticancer drugs. The site specificity of demethylation implies that the result of action of an inhibitor in the cell is determined by the significance of methylation of particular sites for viability, that is, by the role of genes whose promoters are methylated in a given cell line. In these situations, the use of an isoform-selective inhibitor is insufficient to induce cell death: an inhibitor is effective if the targeted KDM is critical for viability. Since this situation is seldom encountered, combinations of inhibitors of single isoforms of one or several subgroups seem reasonable. In addition, the introduction of functionally opposite chemical groups (acetyl or methyl) into adjacent or closely spaced amino acid residues (see Fig. 14) may counterbalance the effect of removal of one mark. For example, a lysine demethylase inhibitor would preserve the methyl group, thus locking the chromatin, but acetylation in a nearby site would cause local permissiveness. A number of questions remain, namely, what will be the effect of surrounding residues that are phosphorylated or modified in another way? How is gene expression changed in this case, and how can the effect of KDM2 inhibitor be evaluated?
Third, the situation is complicated by a general biological feature of these enzymes. The action of KDM is not limited to enzymatic function; inhibition of the demethylating activity (while maintaining protein integrity) is not equivalent to inhibition of gene expression or protein degradation. Apart from the catalysis, functions important for the design of inhibitors have been established for methyltransferases, the KDM antagonists.[169] Apparently, KDM are not an exception, although the role of KDM2 beyond the demethylation has not been adequately studied. The data provided in Section 4.4 and Refs [170-174] indicate that these proteins are not only enzymes. If KDM incorporated in heterochromatin complexes do not necessarily exhibit a demethylating activity but the transcription repression is achieved, then the use of catalytic inhibitors is insufficient.
Therefore, in addition to catalytic cycle inhibitors that do not alter the presence of protein in the cell, other strategies are the development of PROTAC and the comparison of the effects of two fundamentally different tools. It is necessary to get more detailed views on the PROTAC structure: the optimal linker length, finding an appropriate flexibility – rigidity balance for the linker, conjugation with a KDM-recognizing chemical group, and so on. The results of these studies (in silico calculations, chemical synthesis, and cell culture experiments) performed by our research team are patentable. It is also of interest to design ‘molecular glues’ that fix protein–protein complexes to hinder conformational mobility and prevent substrate interaction dynamics. In these situations, there is no need to remove the target protein from the cell.
Finally, the question of significance of KDM2 inhibition in chemotherapeutic combinations remains unsolved. Whatever the mechanism of inactivation, the biological role of these proteins is determined by specific chemical processes in local chromatin regions and beyond. It is necessary to identify clinical situations in which regulation with either a single agent or with drug combinations, would be important for achieving the notoriously difficult goal of killing tumour cells. Can one hope for a therapeutic ‘window’, that is, higher cytotoxicity of KDM2 antagonists against tumour cells compared to normal cells? What are the mechanisms of emergence of tumour cell resistance to these compounds? These issues are beyond the chemical aspects and markedly expand the importance of epigenetic mechanisms of biological regulation, both as fundamental processes in eukaryotic cells and as therapeutic targets.
The authors are grateful to M.M. Erokhin (Institute of Gene Biology, Russian Academy of Sciences) for discussing the non-catalytic functions of lysine demethylases. The review was prepared with financial support of the National Research Nuclear University MEPhI (Priority 2030 program).
7. List of abbreviations and symbols
The following abbreviations and symbols are used in the review:
2,4-PDCA — pyridine-2,4-dicarboxylic acid,
2HG — 2-hydroxyglutaric acid,
2-OG — 2-oxoglutarate,
53BP1 — p53-binding protein 1,
BCOR — BCL-6 corepressor,
BCORL1 — BCL-6 corepressor-like protein 1,
C/EBPα — gene encoding the CCAAT/enhancer-binding protein alpha,
C/EBPβ — gene encoding the CCAAT/enhancer-binding protein beta,
Ccl7 — gene encoding the C – C motif chemokine 7,
CDK12 — cyclin-dependent kinase 12,
CpG — a dinucleotide consisting of cytosine (C) and guanine (G) bases arranged in the 5'-C – G-3' sequence,
Crabp2 — gene encoding the cellular retinoic acid-binding protein 2,
CUL1 — cullin-1,
CXXC — CXXC zinc finger domain,
DHX9 — DExH-box helicase 9,
DMOG — dimethyloxalylglycine,
Е — energy,
FAD — flavin adenine dinucleotide,
F-BOX — F-box domain-containing protein,
GSPT1 — G1 to S phase transition protein 1,
GST — glutathione S-transferase,
H2A, H2B, H3, H4 — histones,
H3K36me2 — histone H3 lysine 36 dimethylated,
HP1a — heterochromatin protein 1 homolog alpha,
IC50 — half-maximal inhibitory concentration,
ID — identifier,
IKZF1 — IKAROS family zinc finger protein 1,
JmjC — Jumonji C domain,
JmjN — Jumonji N domain,
Kd — dissociation constant,
KDM — lysine demethylases (K-DeMethylases),
KDM2 — subgroup 2 lysine demethylases,
KDM2A — lysine demethylase 2A,
LRR — leucine-rich repeat domain,
NF-κB — nuclear factor kappa-light-chain-enhancer of activated B cells,
NOG — N-oxalylglycine,
Od — distal oxygen atom,
Op — proximal oxygen atom,
p15INK4B — gene encoding the inhibitor of cyclin-dependent kinase 4B,
p27Kip1 — gene encoding the inhibitor of cyclin-dependent kinase 1B,
PCGF1 — Polycomb group RING finger protein 1,
PDB — protein data bank,
PFKFB3 — 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3,
PHD — plant homeodomain,
PRC1.1 — non-canonical Polycomb repressive complex 1,
PRC2 — Polycomb repressive complex 2,
PROTAC — proteolysis targeting chimera,
QM/MM — quantum mechanics/molecular mechanics,
RIOX — ribosomal oxygenases,
RipK3 — gene encoding the receptor-interacting serine/threonine-protein kinase 3,
SCF — SKP1 – CUL1 – F-BOX complex,
SKP1 — S-phase kinase-associated protein 1,
TCF7L1 — transcription factor 7-like 1,
TCF7L2 — transcription factor 7-like 2,
TCP — tranylcypromine,
UbV — ubiquitin variant,
Xist — gene encoding the X-inactive specific transcript,
YAP1 — gene encoding the Yes-associated protein 1,
bp — base pair.
References



























