

Keywords
Abstract
This review summarizes the modern views on the mechanism, active site structure, and thermodynamics of the electrochemical oxygen reduction reaction catalyzed by heteroatom-doped carbon nanostructures for the subsequent rational design of catalytic sites and development of promising metal-free cathode materials for fuel cells for next-generation energy systems. The effect of both classic (B, N, S, P, O, Hal) and advanced "hybrid" (Si, Se) dopants in various states and in two- or threecomponent forms on the activity modulation of oxygen adsorption sites and on the reaction mechanism is considered. Particular attention is paid to the evolution of the views on the role of silicon, which switched from considering silicon to be inert due to its high oxophilicity to the discovery of unique catalytic properties of silicon in SiNx type catalytic sites. The conclusions made in the review open up new opportunities for the targeted design of hierarchical materials with adjustable properties via precise control of the composition and structure of heteroatom defects.
The bibliography includes 252 references.
1. Introduction
The growing global demand for environmentally clean energy with low carbon dioxide emissions is one of the modern challenges for the development of energy sources and sustainable growth of the energy and transport sectors of the economy. According to the Paris agreement on prevention of the climate change,[1] the emission level of greenhouse gases by 2030 in Russia should not exceed 70% of that in 1990. In 2018, the emission level was 52% of that in 1990 and, hence, the reserve for its increase is very small.[2]
One source of green energy is hydrogen, the oxidation of which in fuel cells [3][4] makes it possible to convert the chemical energy of a fuel and an oxidant into electrical energy (Fig. 1).[5] The only by-product formed in this process is water, which enables energy generation with the lowest carbon footprint in the case of environmentally friendly hydrogen production, for example, in solar–water electrolyzers.[6][7]
![[{"id":"cFBffBUdWX","type":"paragraph","data":{"text":"Schematic diagram of a proton exchange membrane hydrogen fuel cell (on the left) and alkaline fuel cell (on the right). The acronyms are expanded in the text."}}]](/storage/images/resized/uaUFn9IvFD9xgpr0sQ3iFPLAvMgff4AAqNXuHjxv_xl.webp)
The fuel cell technology has a long history. A fuel cell prototype, gas battery, was designed back in 1839 by Grove,[8] who used hydrogen, oxygen, and sulfuric acid as the fuel, oxidant, and electrolyte, respectively. In a modern fuel cell, the cathode and the anode with deposited catalysts needed to accelerate the hydrogen oxidation reaction (HOR) at the anode and the oxygen reduction reaction (ORR) at the cathode can be separated by either liquid or solid polymer electrolyte.[9][10] It should be noted that the efficiency of the fuel cell is not limited by the Carnot cycle efficiency,[11][12] since the fuel oxidation, unlike that in internal combustion engine, is isothermal and is determined by the overpotential (hOx/Red , where Ox is oxidant, Red is reductant), that is, the difference between the thermodynamic (U 0) and experimental (U ) half-reaction potential at each electrode. Meanwhile, the maximum energy conversion efficiency of a fuel cell, which includes both the energy input into the fuel cell system (DHf) and the maximum amount of this energy (DGf) available for performing external work, can be found as the ratio of these two quantities (htherm = DGf /DHf) and reaches ~ 0.83 under the standard conditions. In the general case, htherm is in the range from 0 to 1 only for exothermic reactions (ΔH < 0, ΔG < 0), which are actually used in chemical batteries, including fuel cells. The fundamental thermodynamic and electrochemical principles governing the operation of hydrogen fuel cells are considered in detail in reviews[12][13] devoted to fuel cell technology.
Currently, platinum is used most often in fuel cells as the catalyst.[14][15] Molecular hydrogen is easily oxidized at the anode by the Volmer – Tafel mechanism, with the overpotential not exceeding 50 mV.[16] Conversely, the oxygen reduction proceeds slowly at the cathode due to a multistep mechanism (Fig. 2), with the experimental overpotential being at best 300 – 350 mV when immobilized platinum nanoparticles are used.[17] This high value, aggravated by active site poisoning,[17][18] substantially decreases the fuel cell efficiency and requires large amounts of expensive metal (20 – 40%); therefore, the development of new inexpensive, highly efficient catalysts for ORR and understanding of the mechanism of their action are exceptionally important, being at the junction of experimental and theoretical chemistry of materials.
![[{"id":"E5kQgJSRlr","type":"paragraph","data":{"text":"Catalytic cycles of molecular oxygen electroreduction in acidic medium via two-electron (on the left) and four-electron (on the right) pathways. Proton and electron charges are omitted for clarity."}}]](/storage/images/resized/3bZGLEyGyouG7NDvYvjSPoiDAiWgNVSZAa3aGCYR_xl.webp)
In order to reduce the cost of cathode materials and decrease the ORR overpotential, carbon nanomaterials (CNMs), including graphene, nanotubes, etc., are doped with metals (Fe, Co, Cu)[19-22] or heteroatoms (N, S).[23-26] The resulting materials possess excellent mechanical properties and high electrocatalytic activity at low metal content (hORR = 390 mV at Fe content of about 0.05 at.%);[22] however, the metal active sites are prone to poisoning by carbon monoxide resulting from degradation of the carbon component[27] or to demetallation under harsh conditions of the reaction carried out at low pH values.[28] The number of studies devoted to the search for new catalysts for this reaction is growing rapidly in the world, whereas in Russia, this problem has attracted attention only quite recently.[29-48]
Metal-free heteroatom-doped CNMs are largely devoid of these drawbacks. Nevertheless, they are less active than metal (or heteroatom)-containing CNMs, and their overpotential is at least 200 – 400 mV higher.[49][50] However, doping with two heteroatoms with higher and lower electronegativity (N, S or N, Si) gives rise to synergistic effect of dopants in accelerating ORR.[41][48][51][52] As a result, these materials have a low ORR overpotential and may prove to be stable, cheap, and environmentally friendly catalysts,while the diversity of active sites in these catalysts dictates the need for detailed investigation of the mechanism of the target reaction in order to select the most promising representatives and methods for the subsequent targeted synthesis. A useful tool is provided by quantum chemical computational methods, which can identify the preferable pathway, determine the ORR free energy profile and overpotential, and thus evaluate a broad range of structurally and compositionally variable heteroatom-doped CNMs before real experiments.
This review presents a detailed consideration of the mechanisms, structures of key intermediates, and thermodynamic features of the oxygen electroreduction reaction at various active sites of heteroatom-doped carbon nanostructures for the subsequent rational design of active sites and development of promising metal-free catalysts for the cathode half-reaction of fuel cells and generation of environmentally friendly energy. The review addresses the effect of classic (B, N, S, P, O, Hal) and advanced ‘hybrid’ (Si, Se) dopant elements taken separately in various states or as two- or three-component species on the modulation of the adsorption activity of substrate carbon atoms in the initial oxygen adsorption and the mechanism and overpotential of the target reaction. The causes for the high efficiency and prospects of Si,N-doped carbon structures in accelerating ORR are identified. The need for this review is dictated by recent achievements, as well as the fact that the latest review on this topic by Ma et al.[24] appeared in 2019.
Before considering particular representatives of ORR catalysts, it is necessary to briefly get acquainted with the mechanism of this reaction. As a result of diffusion of molecular oxygen to the fuel cell cathode, the O2 molecule is adsorbed at the active site to form the O2* adsorbate.# The O2* adsorbate structures modelled by the Pauling type adsorption, in which oxygen is attached by one atom of the molecule to one substrate atom, (OO*), Yeager type adsorption, which implies [2 + 2]-cycloaddition (*OO*), and Griffith type adsorption, which is based on [1 + 2]-cycloaddition (O*O*), are shown in Fig. 3. The O – O bond dissociation in the O2* adsorbate can be accompanied by adsorbate isomerization to dioxo adsorbate (2 O*) by the dissociative pathway (see Fig. 2). The subsequent reduction of O2* under the action of [H+ + e–] (acid medium) or [H2O + e–] (alkaline medium) follows the associative four-electron (4e) pathway through intermediate peroxy (HOO*), oxo (O*), and hydroxy (HO*) intermediates and ends in the regeneration of the active site and formation of two H2O molecules. This ORR pathway competes with the two-electron (2e) reduction of HOO* to hydrogen peroxide (H2O2); depending on the particular goal, H2O2 can be considered as either a target[53] or undesirable product that decreases the selectivity of the catalyst.[54] An alternative and a more attractive dissociative 4e pathway of ORR involves the formation of 2 O* intermediate, which is more stable than O2*, and/or the formation of oxo-hydroxy (O*HO*) and dihydroxy (HO*HO*) intermediates. The last-mentioned species is then isomerized to the O* adsorbate or reduced to the HO* adsobate (see Fig. 2).
![[{"id":"7B1DLv4KDG","type":"paragraph","data":{"text":"Possible structures of O<sub>2</sub>* adsorbates."}}]](/storage/images/resized/wEombDTA9uK8Xhw1fvbTDeNzntbplRTDFvq3yLqL_xl.webp)
# The asterisk indicates the catalyst active site, and the adsorbed species are shown in bold.
2. Nørskov equation and computational hydrogen electrode model
The main principles and notions that are used in the text below were considered in our previous paper.[36] Studies of electrochemical and photoelectrochemical processes have greatly advanced with the emergence of the computational hydrogen electrode (CHE) model developed and implemented into the practice of quantum chemical calculations in 2004 by Nørskov et al.[55] (see also publication by Oberhofer[56]). The Nørskov equation (1) relates the Gibbs free energy of intermediates of chemical reactions involving an electron, needed to construct the Gibbs free energy profile of an electrochemical reaction, to the electrode potential, i.e., DG = f (U).[55] Owing to the conceptual simplicity and computational efficiency, the CHE model enables both in-depth investigations of reaction pathways and large-scale screening calculations to identify the optimal materials and active sites of catalysts. It is important to note that construction of free energy profiles of an electrochemical reaction using the Nørskov equation makes it possible to determine the minimum overpotential associated with the high thermodynamic stability of a certain i-th intermediate at U = U0, with the activation barrier for the transformation of this intermediate being equal, in the minimum estimate, to the free energy change of the electron transfer step (i → i + 1). Among the set of contributions to overpotential (activation, ohmic, diffusion, etc.), this approach formally takes into account the thermodynamic contribution (ηtherm) to the activation overpotential: ηact = ηtherm + ηkinet , whereas inclusion of the additional overpotential caused by kinetic barriers (ηkinet) requires identification of transition states and is fairly time-consuming. The Nørskov equation has the form
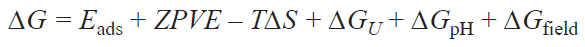
where Eads is the difference between the total electronic energies of a molecule adsorbed on the catalyst active site (asterisked), the adsorbate, and the catalyst (the negative or positive sign of Eads is indicative of exo- or endothermic adsorption), which can be calculated by equation (2)
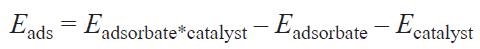
ZPVE and DS are the zero-point vibration energy of molecules and the entropy change in the system calculated in a similar way; T is absolute temperature (298 K); DGU is the Gibbs free energy of an electron calculated by equation (3):
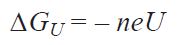
where n is the number of the transferred electrons, e is the electron charge, and U is the electrode potential; DGpH is the correction factor to the free energy of H+ ions, which takes into account the concentration dependence of the entropy and can be found from the equation
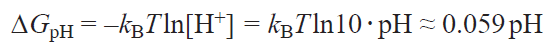
where kB is the Boltzmann constant, pH is the measure of acidity. Usually, pH values of 0 (DGpH = 0, acid medium) and 13 (DGpH ≈ –0.77 eV, alkaline medium) are used. Finally, the last term in equation (1), DGfield , represents a correction term to the free energy arising from the presence of the electrical double layer. According to published data,[55][57] this value for ORR intermediates on carbon surfaces can be neglected. A negative or positive sign of ΔG attests to exergonic or endergonic nature of the formation of individual intermediates in the ORR catalytic cycle.
A change in the charge distribution near the electrode surface has a considerable influence on the interaction of reactants with the surface by altering the orientation of water molecules and other polar compounds in the interfacial layer and, hence, on the free energy of intermediates and transition states of the electrochemical reaction. The application of the microkinetic model of ORR, which combines the hydrogen electrode model with the electric field effect, showed that the latter has a pronounced effect on the stability of HOO*, O2*, and H2O2* adsobates on the Pt(111), Au(111), and Au(100) surfaces depending on the medium acidity, i.e., DGfield = f(pH).[58] This model successfully reproduces polarization curves and makes it possible to predict the onset potentials and reproduce the experimental Tafel slopes, which opens up new opportunities for the search for advanced catalysts.
If the standard hydrogen electrode potential (U0 = 0) is taken as the reference, the free energy of the proton H+ in the [H+ + e–] system is equal to that for 1/2 H2 at pH = 0, hydrogen pressure of 1 bar, and a temperature of 298 K, which corresponds to the thermodynamic equilibrium in the H2 D 2 [H+ + e–] system. Thus, the free energy of the hydroxide anion (HO–) under similar conditions can be calculated according to the equilibrium reaction H2O D H+ + HO– in the aqueous solution.
The change in free energy ΔG in each elementary step of the ORR catalytic cycle (see Fig. 2) for the first four terms of the Nørskov equation (1) can, in the general case, be calculated using equations (5) – (10).
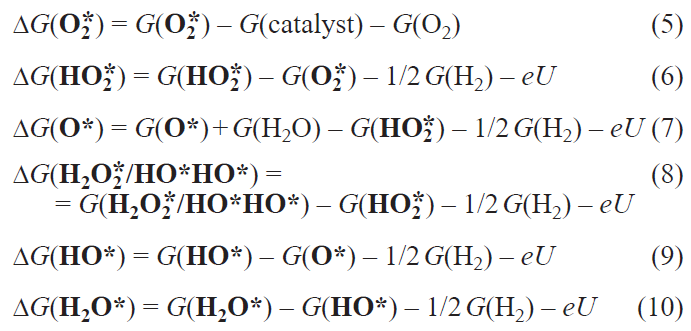
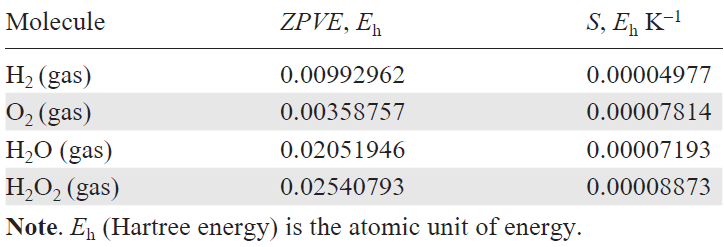
To increase the accuracy of calculations, simplify the comparison, and enable the reproducibility of the results between various research teams, it was recommended[55][59] to use ZPVE and S corrections to the free energy of H2 and O2 in the gas phase and ZPVE value for H2O and H2O2 taken from US National Institute of Standards and Technology (NIST) databases[60] (Table 1), while entropy S for H2O and H2O2 can be derived from calculations at a pressure of 0.035 bar (saturated vapour pressure of H2O at 298 K). For other structures (intermediates of the ORR catalytic cycle), the ZPVE and S values are calculated. The free energies of O2 and H2O2 were calculated from the free energy changes in the reactions O2 + 2 H2 → 2 H2O and O2 + H2 → H2O2 to be –4.92 eV (4e × 1.23 V) and –1.39 eV (2e × 0.695 V), respectively, for the forward reactions under standard conditions. The overpotential hORR (V) is found using relation (11), where DDGmin is the smallest decrease in the free energy in one step of the ORR catalytic cycle involving an electron at U = 0 in an acidic medium (DGpH = 0):
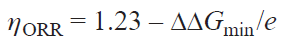
The H+ and e– transfer steps in the ORR catalytic cycle are usually considered to be synchronous. Nevertheless, they can be separated in time and be affected by various factors. According to Oberhofer,[56] the order of these steps does not matter from the thermodynamic standpoint, because free energy is a thermodynamic function of state. This means that the free energy difference does not depend on the pathway by which transition between two states (intermediates) takes place. In the case of electrochemical reactions, the free energy calculated by equation (1) does not depend on the mechanism (synchronous or stepwise) of proton and electron transfer.
The computational hydrogen electrode model has initiated quite a number of theoretical electrochemical studies described below; however, CHE model has some significant drawbacks due to its conceptual simplicity. The major one is simulation of the processes at a constant (most often, zero) charge; meanwhile, the reaction proceeds at a constant potential, i.e., the surface charge is unknown in advance. The subsequent iterations to improve the CHE model led to the emergence of the grand canonical ensemble method within density functional theory (GC-DFT) resulting from combining the gas cluster procedure for modelling small molecular ensembles with DFT for accurate calculation of the energy characteristics of these ensembles. This approach provides an optimal balance between calculation accuracy and computational efficiency.
Kim et al.[61] showed that the charge of the system affects more strongly ORR on 2D materials, in particular on NPy-doped# graphene than on 3D metals, and the energy of the reaction can change by more than 1 eV. This is due to the fact that a change in the charge of 2D materials considerably alters the population of electronic states because of their low density. Study of the thermodynamics of ORR on NGr-doped graphene (NGr is the graphitic nitrogen atom) using DFT and electrochenical model with an fixed potential (Uconst) also revealed a dependence of the adsorption energy of the intermediates in the 4e pathway (HOO*, O*, HO*) on the electrode potential U, which is at variance with the results of DFT calculations for constant charge (QConst), in which this dependence is neglected.[29][62] However, to the surprise of the authors,[62] the energy difference between HOO* and HO*, equal to ~ 3.24 eV, is also retained at a constant potential. As U decreases, the synchronous electron – proton transfer switches to successive electron and proton transfer, with the U value for this transfer depending on the type of nitrogen dopant.[29] The results of DFT calculations for a constant potential indicate the need to revise the previously proposed mechanisms of electrocatalysis, taking into account the influence of charge.
The second drawback is related to the widespread use of the approach based on investigation of the relative stability of intermediates and equilibrium thermodynamic profiles of ORR, which provides theoretical values of overpotentials, but does not give information about the reaction rate. Activation energy is traditionally considered to be the cornerstone of classical electrochemistry.[63] The solely thermodynamic approach, which prevailed until recently in DFT calculations owing to the CHE model, often neglects the fact that the kinetic barrier cannot always be overcome even for a thermodynamically allowed reaction (DG < 0). An evolutionary leap is the transition from the calculated hydrogen electrode model to microkinetic model,[58] which implies inclusion of electric field effects into the CHE model. Using this approach, it is possible to take into account the influence of electrode potential U on the adsorption energies of intermediates, calculate the currents, plot polarization curves that can be directly compared with experimental results, and explain the anomalous behaviour of surfaces [e.g., Au(100)[58] where pH change affects the 4e and 2e ORR pathways in different ways]. Without calculation of the activation barriers and taking account of the process kinetics, it is impossible to accurately predict the Tafel slopes and catalyst selectivity in the presence of competing reactions. Thus, modern computational electrochemistry moves from ‘volcano’ thermodynamic plots to comprehensive microkinetic description involving the electrical double layer dynamics and energy barriers of particular steps.
The selection of computational protocols to study thermodynamics and mechanism of ORR is determined by the investigation object that can be either periodic model of the catalyst or an idealized finite structure in which dangling C – C bonds are replaced by the C – H bond (quantum dot model). Typically, a model of the catalyst consists of a relatively large number of heavy atoms (> 50), including transition metal atoms, which imposes restrictions on the choice of the calculation protocol (method or basis set) that would satisfy the trade-off between the accuracy and time consumption. In this regard, DFT methods are currently used most often for theoretical investigation of electrocatalytic reactions at the interface.[36][64][65]
For computational evaluation of the performance and selectivity of ORR electrocatalysts, generalized gradient approximation (GGA) functionals are used most often due to their good correlation with experimental values for bulk and surface properties,[64] as well as the computational speed. Among GGA functionals, Perdew – Burke – Ernzerhof (PBE) exchange correlation functional in combination with a pseudopotential calculated by the projector augmented-wave (PAW) method is most popular.[66-68] However, it is well known that in some cases, gas-phase energy calculations at the GGA level do not coincide with experimental results, in particular, when molecules have multiple bonds (e.g., O2) or contain transition metals.[64] [69]This is caused by the fact that functionals of this group do not adequately describe the exchange contribution to the energy of molecules with strong static correlation effects. The errors are partly compensated and decrease on going from the local density approximation (LDA) to GGA functionals and further to meta-GGA and hybrid functionals.
The so-called meta-GGA functionals are based on a more advanced approach taking into account the electron density, electron density gradient, and kinetic energy density approximation; they provide better results in describing covalent and metallic bonds.[64] For example, hybrid functionals include accurate non-local Hartree – Fock exchange energy and require enormous computational cost, especially when plane-wave basis sets are used; however, they show a better energy accuracy for molecular systems.[65] Data on the use of high-level ab initio methods such as CCSD(T)## to study the ORR mechanisms are scarce; however, the results are in good agreement with the results obtained using PBE0-D3(BJ) and HSE06-D3(BJ) hybrid functionals[70] and Becke-3 hybrid functional with the Lee – Yang – Parr correlation (B3LYP).[71]Currently, there are more than 200 DFT functionals; the comparative statistical results of their testing are presented in a number of publications.[65][72-78]
It is important to take into account the effect of solvation on ORR thermodynamics. In an original publication, Nørskov et al.[55] demonstrated increase in the stability for some intermediates of ORR catalytic cycle on the surface of the Pt(111) model platinum catalysts upon taking into account the solvation of, in particular, hydroxy (HO*) and peroxy (HOO*) adsorbates. Nevertheless, the results of theoretical studies[79-81] devoted to ORR thermodynamics in relation to the FeN4-doped periodic graphene in the gas phase, considered in explicit and implicit solvation models, attest to weak dependence of overpotential on the inclusion of solvation: the error of hORR prediction is ± 40 mV. In terms of kinetics, the inclusion of solvation in the calculations of the free energy of transition states for hydrogen transfer from a carbon substrate to an oxygen-containing adsorbate is exceptionally important for the prediction of the heights of activation barriers;[82] nevertheless, the Bell – Evans – Polanyi principle is widely used to study ORR pathways due to the similarity of the elementary steps of the reaction.[83]
The electronic structure of model catalysts and intermediates of the ORR catalytic cycle can be supplemented by using information on the dynamic aspects of charge transfer and electron correlation in the systems and by considering the dynamics of surface oscillations and adsorbate — catalyst interactions solved within the framework of the Green’s function method.[84][85]
# NPy stands for pyridinic nitrogen atom.
## CCSD(T) is the coupled cluster method that includes single-, double-, and perturbative triple excitations.
3. Undoped carbon nanomaterials
Nanomaterials consisting of sp2-hybridized carbon atoms (graphene, nanotubes, fullerenes, quantum dots, etc.) possess a set of unique properties that include high chemical stability, mechanical strength, and thermal and electrical conductivity. The combination of these properties determines the role of these materials not only in ORR catalysis and electro-Fenton process,# but also in other fields such as microelectronics, design of molecular sensors, energy storage devices, and so on.[86] However, reports on the use of pristine carbon nanomaterials in the catalysis of the 4e ORR pathway are few, due to low catalytic activity of these materials, which requires the presence of topological or electronic structure defects caused by the presence of heteroatoms.[69][87-96]
# The electro-Fenton process is the in situ electrocatalytic generation of hydroxyl (HO·) radicals from H2O2 in the presence of Fe2+ ions in O2-saturated aqueous solutions. It is widely used for the decomposition of toxic organic compounds in sewage water.
3.1. Graphene and graphene-like quantum dots
Low-temperature and quantum chemical studies of molecular oxygen adsorption on graphene and single-walled carbon nanotubes are indicative of predominantly physical nature of the interaction between graphene and oxygen, since O2 adsorption is slightly exothermic, with Eads being only –0.1 eV. Conversely, in the case of carbon nanotubes, Eads = –0.2 eV, which indicates weak chemisorption of the oxygen molecule.[97-99] Сhemisorbed O2* is higher in energy than the isolated O2 molecules (in the triplet ground state), and, hence, is metastable.[100] Due to weak antibonding nature of the p-bond in O2*, the dissociation of oxygen to give the diepoxy adsorbate (2*O*) on the graphene surface has a very high activation barrier, E ≠ ≈ 2.8 eV,[99][101] which is higher than the barrier for the destruction of the carbon substrate.[30]
Analysis of the free energy profile of the associative 4e-pathway of ORR on the basal carbon atoms of graphene (Fig. 4) indicates that the formation of the peroxy HOO* adsorbate is markedly endergonic (DG = 0.88 eV);[69] therefore, the adsorbate desorption into the near-cathode space as a peroxide anion or radical-anion is more likely than the subsequent reduction via the 4e-pathway. Note that these results should be treated critically since exergonicity (–DG0 = 4eU0) of the reaction 2 H2 + O2 → 2 H2O is 4.92 eV rather than ~ 1.4 eV indicated in the profile. However, it is well known that pristine graphene is a poor catalyst for 4e ORR due to the very high overpotential (hORR ~ 0.8 V).[102] Nevertheless, pristine graphene-based materials are used to accelerate 2e ORR to give H2O2 . The mechanism, thermodynamics, and kinetics of hydrogen peroxide formation at the peripheral carbon atoms of the zigzag and armchair edges have been described in detail by Ly et al.,[103] who demonstrated the possibility of changing ORR selectivity toward the 2e pathway at the electrode potential U < 0.55 V in acidic medium and –0.22 V in alkaline medium. The ways for increasing the catalytic activity of graphene in 2e ORR are discussed in detail in a recent review.[86]
![[{"id":"QMAfs-pi9X","type":"paragraph","data":{"text":"Free energy profiles of the associative 4e pathway of ORR on the periodic models of graphene and N-doped graphene (<i>a</i>), and on the (585) defect structure (<i>b</i>) and graphitic N-doped graphene (<i>c</i>).<sup>69</sup>"}}]](/storage/images/resized/pjXp4kb0NlJkFV1qsym2sApTpIJT96sZ57BpOItS_xl.webp)
One way for enhancing the graphene activity in the 4e ORR pathway is to disrupt the p-electron system by generating structural defects (Stone – Wales defects, vacancies, holes, etc.)[69][91] and also to increase the number of zigzag edge carbon atoms (graphene ribbon).[89] Structural defects induce changes in the charge and/or spin density, giving rise to the appearance of oxygen adsorption sites and ORR catalysis.[102] Defects can be experimentally generated by annealing nitrogen-doped carbon nanomaterials at temperatures above 1000°C in an inert atmosphere, which leads to partial elimination of heteroatoms to generate vacancies or by removal of particular carbon atoms by argon plasma.[89][92] Defects in the graphene structure can accelerate not only ORR, but also the electrochemical water decomposition into hydrogen and oxygen.[104]
Using high-resolution transmission electron microscopy (HRTEM), Girit et al.[105] showed experimentally that structural defects in graphene represent a dynamic system, with armchair edge carbon atoms being more mobile and more prone to be removed by irradiation. Theoretical analysis of the dynamics and stability of carbon vacancies were performed by Kim et al.[106]
As a result of removal of two neighbouring basal carbon atoms from graphene and transformation of four coupled hexagons into two pentagons and one octagon to give so-called (585)-defect (see Fig. 4b), Zhao et al.[69] theoretically demonstrated changes in the thermodynamic profile of the 4e pathway of ORR. As compared with basal carbon atoms as active sites of graphene or nitrogen-doped analogue (see Fig. 4a,c), the carbon atoms that form a (585)-defect are more active in the target reaction. The free energy values of oxy O* and hydroxy HO* intermediates are similar to those for an ‘ideal’ ORR catalyst, whereas the elementary step of formation of the peroxy HOO* intermediate by the reaction * + O2 + [H+ + e–] is weakly endergonic. The authors experimentally showed that pyrolysis of the PAF-40 organic framework containing phenyl and triazine groups at 700 – 800°C gives rise to nitrogen-doped graphite-like material, while further holding at 900 – 1000°C decreases the nitrogen content (mainly graphitic type nitrogen) to 0.2 at.%. Comparison of these materials in 4e ORR revealed higher catalytic activity of the last-mentioned one caused by the presence of structural defects in the carbon matrix.[69] Jiang et al.[89] showed that the activity of carbon atoms in the 4e pathway of ORR increases in the following order: basal << armchair edge < hole << defect (basal pentagon) < zigzag edge. In the last two structures, all elementary steps of O2 reduction are exergonic (Fig. 5), and particularly these structures appear to make the major contribution to the ORR catalysis by pristine carbon materials.
![[{"id":"k8QNDsBY_e","type":"paragraph","data":{"text":"Free energy profile for ORR on various carbon atoms of graphene (<i>a</i>) with structure defects as a basal pentagon (<i>b</i>), hole (<i>c</i>), zigzag edge (<i>d</i>) and armchair edge (<i>e</i>).<sup>89</sup>"}}]](/storage/images/resized/eF4yUHBDAMGQb3nBrq3jUuGFShCNqJbJSdazn7VG_xl.webp)
Zhang et al.[107] used graphene quantum dot C100H26 to design a number of structural defects, including Stone – Wales defects, C-terminated single and double vacancy, pentagon ring defect at the zigzag edge (after removal of CH), and various combinations of pentagon and octagon rings resulting from linking of two quantum dots with armchair or zigzag edges. The authors calculated the free energies of intermediates along the 2e and 4e pathways of ORR in acidic medium by the B3LYP/6-31G(d,p) method and determined the activation barriers. It was shown that for structures containing Stone – Wales defects or vacancies in the basal position, the charge distribution is identical to that in the original defect-free structure, and such sites are ineffective in ORR catalysis. The most pronounced change in the charge (up to ± 0.2e) was noted for the carbon atoms that form the zigzag edge and a pentagon ring defect at the zigzag edge with various combinations of links. The two last-mentioned defects proved to be most active in the four-electron ORR. The reduction of hydroxy adsorbate (HO*) to H2O and active site regeneration was found to be the rate-determining step at the zigzag edge defect [the activation energy (E≠) was 0.48 eV, hORR ≈ 0.7 V], while in the case of carbon atoms forming a junction between different-type edges, the rate is limited by the reduction of the peroxy HOO* adsorbate to the O* intermediate (+H2O) (E≠ = 0.35 eV, hORR ≈ 0.3 V).
3.2. Carbon nanotubes
Like graphene, pristine carbon nanotubes are not very effective in the 4e pathway of ORR, although they experience deformation stresses. The transition from a planar structure to a closed cylindrical structure slightly increases the catalytic activity.[108] Nevertheless, data on the use of undoped carbon nanotubes in ORR are few because it is necessary to generate defects.[92-96] Combination of internal stress, which increases with decreasing diameter, with the Stone – Wales defects may give rise to nanoscale plastic deformation effect (tensile strain) in carbon nanotubes.[109]
According to the BLYP/DNP calculation data, the adsorption energy of molecular oxygen depends not only on the chirality of nanotubes, but also on the distance of the active site from basal carbon.[110] In the case of (4,4)-armchair nanotubes, Eads of molecular oxygen on the basal carbon atoms is only –0.01 eV, and on moving toward the nanotube open end, it reaches –0.12 eV. These atoms also exhibit better adsorption properties for ORR intermediates such as HOO*, O*, and HO*; therefore, the catalytic activity in the 4е pathway of ORR is mainly inherent in the peripheral carbon atoms of nanotubes,[87] which is in line with experimental results.[90] In the case of (8,0)-zigzag nanotube, the adsorption energy of O2 is –0.15 eV, irrespective of the active site position on the nanotube surface.[110] The free energy profiles for the 4e pathway of ORR on the peripheral carbon atoms of model (4,4)-armchair and (8,0)-zigzag nanotubes are shown in Fig. 6.
![[{"id":"SpErGhKA_W","type":"paragraph","data":{"text":"Structures of (4,4)-armchair- and (8,0)-zigzag carbon nanotubes, fused-linear I(4,4)-(8,0) and bent L(4,4)-(8,0) structures based on them (above); free energy profiles of the 4e associative pathway for ORR (below). The following designations are used: B is basal, Z is zigzag, A is armchair.<sup>110</sup> Copyright Elsevier."}}]](/storage/images/resized/pfwQLVKbfM1sSQbmS5Rm92riXi5y25wcTicjsfn7_xl.webp)
The zigzag nanotubes are characterized by equilibrium between the initial state of the system (* + O2) and the peroxy HOO* adsorbate (DDG = –0.08 eV, BLYP/DNP calculation), resulting from transfer of the first electron; this makes this reaction the rate-determining step; the calculated overpotential hORR amounts to 1.15 V and decreases with increasing nanotube diameter; for example, for the (15,0)-zigzag nanotubes, hORR = 0.98 V.[111] Further electroreduction of HOO* via the associative 4e pathway is accompanied by a decrease in DG in each step. Conversely, the active site of the armchair nanotube tends to form the quasi-stable epoxy *O* intermediate (an oxygen atom is adsorbed simultaneously on two adjacent carbon atoms) via the transfer of two electrons. The subsequent reduction of this adsorbate proceeds slowly compared to its accumulation over a wide range of electrode potentials, 0 < U < 1.23 V, while the step *O* + [H+ + e–] → HO* is rate-determining. The combined thermodynamic picture of ORR on basal carbon atoms of (15,0)-zigzag nanotubes was reported by Chen et al.;[111] in this case, * + O2 + [H+ + e–] D HOO* is an equilibrium step (DDG = 0); and the endergonicity of *O* + [H+ + e–] → HO* amounts to 0.17 eV at U = 0.
By using Green’s functions, it was shown theoretically that connection of (5,5)-armchair- and (10,0)-zigzag carbon nanotubes with open ends (see Fig. 6) leads to charge transfer from the armchair segment to the zigzag one, emergence of new electronic states near the Fermi level (EF), and averaging of the conduction band compared to the constituent parts;[112] this is beneficial for the catalytic activity.[110] The increase in the number of junctions in the synthesis of single-walled carbon nanotubes can be both random and temperature-controlled.[113] Comparison of the total energies of joint carbon nanotubes depending on the angle between the axes of symmetry of the segments indicates increased stability of bent structures.[114] As a result, the basal carbon atoms in a junction are more efficient as catalysts for the 4e pathway of ORR (see Fig. 6b) than the terminal atoms of the constituent parts (see Fig. 6а). The calculated overpotentials (hORR) for straight I(4,4)-(8,0) and bent L(4,4)-(8,0) joint carbon nanotubes are rather high and amount to 0.88 V, while the reactions HOO* + [H+ + e–] → O* (+H2O) and O* + [H+ + e–] → HO* are rate-determining steps, respectively.
3.3. Fullerenes
Fullerenes, unlike other carbon allotropes, have a strictly defined molecular formula,[115] while the absence of defects prevents the appearance of any satisfactory catalytic activity.[116] In addition, poor wettability in aqueous solutions and low electrical conductivity hinder electron transfer, which markedly limits their use for accelerating electrocatalytic processes.[117]
Nevertheless, Wang et al.[59] investigated the mechanism of associative 4e ORR pathway on C60 fullerene and on heteroatom-doped fullerene-based C59X structures (X = B, N, P, S, and Si) in an alkaline medium by the B3LYP/6-31G(d,p) method. The adsorption of molecular oxygen on the active sites of the above structures is exergonic, with that on C59Si being highly exergonic because of high oxophilicity of the silicon atom. The free energy profile for ORR in the C60 active site at high negative potentials U is characterized by decreasing DG in each step, i.e., the reaction proceeds spontaneously. As U increases to –0.6 V, the rate-determining step of transfer of the third electron, O* + H2O + e– → HO* + HO–, first passes through the equilibrium and then becomes endergonic.
Gao et al.[118] experimentally studied C60 fullerene as the ORR catalyst and demonstrated that the onset potential (Uonset) and the half-wave potential (U1/2) amount to 0.62 and 0.53 V, respectively, while the limiting current density (jL) is very low (–0.24 mA cm–2). The addition of carbon nanotubes (Uonset = 0.84 V, U1/2 = 0.80 V, jL = –0.32 mA cm–2) during the catalyst preparation stage markedly increased the catalytic activity (Uonset = 0.91 V, U1/2 = 0.84 V, jL = –1.73 mA cm–2). According to Raman spectroscopy data, carbon nanotubes in the composite were disordered due to the adsorption-induced charge transfer to C60 .
Later, the model of the above composite in ORR was studied using the BLYP/DNP method in relation to C60 fullerene complex with (15.0)-zigzag C300H30 nanotube[111] in which the pairs of C60 carbon atoms and nanotube basal atoms located most closely to each other are considered as active sites (AS-2 and AS-3) (Fig. 7). This complex has a total electronic energy 0.4 eV lower than that of separate components at a distance of 3.70 Å between them. Analysis of the free energy profile for the associative 4е pathway of ORR in acidic medium in the carbon nanotube active site (AS-3) in the complex attests to the presence of an endergonic step of formation of the peroxy intermediate (* + O2 + [H+ + e–] → HOO*). As this takes place, DG of the system increases by 0.39 eV at U = 0, which implies deterioration of the catalytic activity of the basal carbon atoms of nanotubes. Conversely, the C60 fullerene active site (AS-2) in the complex demonstrates a satisfactory thermodynamic picture of ORR as a result of a decrease in the free energy of the peroxy HOO* intermediate. The calculated overpotential hORR is 0.58 V, which does not agree well with the experimental data of Gao et al.;[118] this may point to drawbacks of the Mulliken charge system and the need for using alternative approaches for charge estimation. Mulliken charge analysis of the active sites in the complex attests to a fivefold charge increase to +0.01e on the AS-2 carbon atom of the C60 component, while the charges on the terminal carbon atoms of nanotubes (AS-1) decrease. Chen et al.[111] also showed that the reaction * + O2 + [H+ + e–] → HOO* is the rate-determining step for the C60 active site in an acidic medium, with the overpotential hORR being 1.19 V.
![[{"id":"hS1MfMS_RP","type":"paragraph","data":{"text":"Structure of the complex of C<sub>60</sub> fullerene with (15,0)-zigzag carbon nanotube (<i>a</i>) and free energy profiles for 4e ORR pathway on some carbon atoms (<i>b</i>).<sup>111</sup> Copyright Elsevier."}}]](/storage/images/resized/uRqkhf2bikQyaeQr0kAQWhlmF2d2tbPncuzQS2u3_xl.webp)
4. Heteroatom-doped carbon nanomaterials
As shown in Section 3, pristine carbon nanomaterials can accelerate ORR mainly owing to the active sites located at zigzag edges and carbon atoms that form structural defects, in particular at the junctions of different-type edges. Nevertheless, their experimental current – voltage characteristics are unsatisfactory for commercial use in fuel cells (high overpotentials, low absolute values of limiting current density, etc.). A good alternative to structural defects are electronic defects formed upon doping of carbon nanomaterials with heteroatoms such as N, S, F, P, B, O, Br, I, Se, etc.[91][119-123] In practice, both types of defects are usually present, giving rise to synergistic effect on the catalytic activity.[91]
The introduction of a heteroatom into an unsaturated carbon matrix disrupts its π-electron structure due to differences in the atomic radius and electronegativity compared to those of substrate carbon atoms, thus leading to a change in the physical and chemical properties of the material.[24] The location and configuration of dopants play a key role in the formation of local sites with decreased (or increased) charge and/or spin density that are more effective for oxygen adsorption, decrease the activation barriers, and exhibit the catalytic activity in ORR.[24] [91][120][124-126] Heteroatom-doped carbon nanomaterials may demonstrate high efficiency and selectivity in the 4е pathway of ORR; they are considered to be most promising metal-free catalysts for fuel cells.[127]
4.1. Nitrogen doping
The heteroatom-doped carbon structures were first reported in 1999 when Huang et al.[128] described the synthesis of arrays of vertically oriented carbon nanotubes doped with nitrogen by pyrolysis of iron(II) phthalocyanine (FePc) in an Ar/H2 atmosphere at 800 – 1100°C as a nanocomposite for LED displays. For a decade, nitrogen was considered an undesirable impurity in carbon nanotubes, before Gong et al.[23] showed that nitrogen-doped nanotubes have a high catalytic activity in ORR, resistance to carbon monoxide poisoning in alkaline medium, large surface area, good electrical conductivity, controlled morphology, and economic attractiveness.[129] They showed better performance in ORR and longer service life in alkaline fuel cells compared to commercial Pt/C catalysts.[23] Subsequently, a wide range of materials based on nitrogen-doped graphene and graphite were synthesized, most of which were also effective toward the 4e pathway of ORR, had excellent current–voltage characteristics, and, unlike Pt/C, they were resistant to catalytic poisons (methanol and carbon monoxide).[130-137]
Synthetic approaches to nitrogen-doped carbon materials are highly diverse and are not limited to a single standard procedure. The classic preparation method consists in thermal or, less commonly, mechanochemical post-treatment of carbon materials with reactive nitrogen-containing compounds such as urea, nitric acid, and especially ammonia.[126] A drawback of this method is the predominant modification of faces and cavities of materials.
The second approach is based on in situ pyrolysis or chemical vapour deposition of nitrogen- and carbon-containing compounds such as heterocycles, melamine, or amino sugars; as a result, nitrogen atoms may be incorporated into the arising carbon framework.[133] Currently, the hydrothermal carbonization of cheap and readily available biomass rich in nitrogen and carbon is a more attractive approach,[137] which makes it possible to obtain nitrogen-containing carbon intermediates for further treatment and application.[126] An alternative technique is based on the pyrolysis of ionic liquids containing nitrile (C≡N) groups in an inert atmosphere.[138-140] The number of existing examples of various nitrogen-doped carbon materials (graphene, graphite, nanotubes, porous carbon, etc.) is enormous.[24][134][137-146] Transition metal complexes and metal-organic frameworks (MOFs) can also be used to produce nitrogen-containing carbon materials that are suitable, in particular, for accelerating ORR, as well as for use in supercapacitors or as cathode materials for lithium and sodium batteries.[147-149] In the latter case, more selective insertion of nitrogen atoms of a certain nature is possible, and the metal is considered only as their carrier.[50] Since a substantial contribution of more active metal centres to the acceleration of ORR may be achieved when the metal concentration is < 0.05 at.%[150] or if trace amounts of metals (e.g., Fe, Co, Mn) are present during the high-temperature annealing of doped carbon materials,[151] it is impossible to experimentally evaluate the activity of non-metal sites in the presence of metal sites, while removal of the latter requires thorough post-synthetic treatment[152-154] and is economically unfeasible.
The nitrogen atom is a unique dopant for modulating ORR activity since it has an atomic radius close to that of carbon (rN = 65 pm, rC = 70 pm) and higher electronegativity (Pauling electronegativities: χN = 3.04, χC = 2.55).[155] The mechanism of the electrocatalytic action of dopants including nitrogen comprises increase in the oxygen chemisorption energy, stabilization of the O2* adsorbate, and promoting ORR on carbon atoms adjacent to the heteroatom.[29][156] In doped carbon materials, the nitrogen atom can exist in four main forms: pyrrolic (NPyr), pyridinic (NPy), pyridinic N-oxide, and graphitic (quarternary, NGr) nitrogen; depending on the form, nitrogen is located in the basal plane or the substrate cavity or at the edge (Fig. 8). The relative content and chemical nature of dopants on the surface of heteroatom-doped materials can be determined using high-resolution X-ray photoelectron spectroscopy (HR-XPS).[24] After a quarter of century since the first synthesis of nitrogen-doped carbon materials, the nature of their active sites in ORR still remains a matter of debate.[24][26] [91][120][122]
![[{"id":"OAuCxFtZGx","type":"paragraph","data":{"text":"Schematic drawing of nitrogen-doped graphene and N1s electron binding energy according to HR-XPS.<sup>129</sup> Copyright American Assiciation for the Advancement of Science."}}]](/storage/images/resized/R3dU9vPKIDP51unDsYwLOdowXuDF4kJjY3onzpYx_xl.webp)
If pyridinic and pyrrolic nitrogen atoms are located at the edges or participate in the formation of hole defects of carbon material, the number of electrons in the π-system of the material does not change. Conversely, replacement of a carbon atom at some distance from the edge by a graphitic nitrogen atom does not distort the structure, but the number of electrons in the π-system increases in proportion to the number of nitrogen atoms.[126][129][141] The diversity of carbon materials dictates the need to analyze the influence of various nitrogen species on the reactivity of active sites to gain a deeper understanding of the ORR mechanism and optimize experimental studies for the targeted synthesis of the most active catalysts.
Li et al.[157] theoretically evaluated the basic properties of the heteroatom in graphene and (5,5)-armchair, (9,0)-zigzag, and (11,0)-zigzag nanotubes doped with pyrrolic, pyridinic, and graphitic nitrogen atoms using the Fukui function. The calculations showed that pyridinic nitrogen has the greatest capacity, while graphitic nitrogen has the lowest capacity for donating electrons and that the Lewis basicity of various nitrogen forms depends not only on the nature of the material (graphene, nanotubes) and the location of the dopant in the material (edge, basal defect), but also on the chirality and diameter of the nanotube.
Until recently, there were two points of view regarding the nature of catalytic sites in nitrogen-doped carbon materials. According to one of them, high activity in ORR may be related to the increased number of electrons in delocalized π-orbitals of the carbon framework due to the lone electron pair (LEP) of pyridinic nitrogen; this enhances the nucleophilicity of π-electrons and, hence, causes more effective attack on the oxygen molecule.[91]This hypothesis is erroneous, since LEP of the pyridine nitrogen atom lies in the substrate plane. The pyridinic N-oxide and pyrrolic nitrogens are apparently inactive in ORR.[91][141] It is more realistic that both pyridinic and graphitic nitrogen atoms exhibit electron-withdrawing properties caused by strong –I-effect with respect to neighbouring carbon atoms giving rise to the partial positive charge d+ on carbon (Fig. 9)[158]; this enhances the catalytic activity[24][119][126][143][159] and may indicate the charge control of ORR, as was shown for metal centres.[40] This assumption is supported by the nature of the ground state of oxygen molecule, which is a triplet biradical.
![[{"id":"0uon6XD8FD","type":"paragraph","data":{"text":"Charge distribution in undoped (CNT) and graphitic N-doped (N-CNT) (5,5)-armchair carbon nanotubes.<sup>158</sup> Copyright American Chemical Society."}}]](/storage/images/resized/Big3gTIqWW8ioQvsEi1iA6QpmMhG9GFVficOUE3a_xl.webp)
According to another version, graphitic nitrogen atoms are responsible for acceleration of ORR in the doped carbon material, since the quaternary nitrogen atom introduces one additional electron into the π-system. As a result, an oxygen molecule is adsorbed on the active site of this catalyst via both oxygen atoms and neighbouring substrate atoms (*OO*, Yeager model) rather than via one oxygen atom (OO*, Pauling model) (see Fig. 3).[48] The increase in the reactivity of nitrogen-doped carbon structures is due to the decrease in the energy gap (Egap) between the frontier molecular orbitals, the highest occupied (HOMO) and lower unoccupied (LUMO) orbitals, caused by increasing EHOMO . This facilitates the electron transfer from the catalyst to the adsorbed oxygen molecule.[158]Recent studies pointed out a synergistic effect on ORR acceleration in the presence of pyrrolic and graphitic nitrogen;[48] [160][161] the former is responsible for the onset potential Uonset due to low hORR , while the latter enhances the electrical conductivity and increases the limiting current density,[141] as the O2 adsorption becomes less endergonic; however, it provides higher hORR .[48]
Zheng et al.[101] considered the adsorption and dissociation of molecular oxygen on the periodic model of graphene doped with graphitic nitrogen using the PBE/PAW method. As the active sites, the authors considered two pairs of neighbouring atoms of the >N – Ca=Cb< structural moiety (Fig. 10). The energy of adsorpion of molecular oxygen (Eads) is ~ 0.25 eV higher on these active sites than on the basal carbon atoms of undoped graphene and amounts to –0.34 eV; this indicates activation of carbon atoms covalently bound to the dopant in the initial step of ORR. The activation energy barriers (E≠) for O2* dissociation at the active sites of the >N – Ca=Cb< moiety are 1.85 and 1.70 eV by pathways A and B, respectively, with the free energy change for the formation of diepoxy (2*O*) and oxo-epoxy intermediates (O**O*) being 0.88 and 1.06 eV. From these results, the authors concluded that the adsorption and dissociation of molecular oxygen on nitrogen-doped graphene proceed more easily than on the pristine material, but less efficiently than on platinum.
![[{"id":"sKWtIER3z2","type":"paragraph","data":{"text":"Dissociation mechanism of <b>O</b><sub>2</sub>* adsorbate on the graphitic nitrogen-doped graphene model.<sup>101</sup> TS is transition state. Copyright Elsevier."}}]](/storage/images/resized/N0L5nLgRHc6XCOq9Hrt9IV4Zo467suaOam1rly3h_xl.webp)
While studying the 4е mechanism of ORR on graphene with a vacancy resulting from removal of two neighbouring basal carbon atoms (585 defect), Zhao et al.[69] plotted a free energy profile for nitrogen-doped graphene (see Fig. 4), which indicated that the active site tends to undergo irreversible oxidation to give stable oxo adsorbate (O*), while its subsequent electroreduction to the hydroxy HO* intermediate is highly endergonic: DDG = ~ 1 eV. This result contradicts experimental data on the high activity of nitrogen-doped carbon materials in ORR.
The same year, Saidi[162] calculated the overpotentials, hORR , for the 4е associative and dissociative pathways of ORR using a number of model structures [C46H20 , graphene quantum dot (GQD) model] doped with pyrrolic, pyridinic, and graphitic type nitrogen (N-GQD-x) (Fig. 11). Among the large number of potential catalytic sites, the highest activity was inherent in vinyl carbon atoms at pyridine nitrogen (C1=C2, N-GQD-2) incorporated in an armchair edge: hORR = 0.55 V. Model structures doped with graphitic nitrogen proved to be poor ORR catalysts, with their hORR slightly increasing as the dopant moves from the edge (0.79 V for N-GQD-3) one atom inside the structure (0.86 V for N-GQD-4). The authors found a linear correlation between the free energies of HO* and HOO* intermediates, apparently due to the same type of their binding to the active site, which allowed them to identify the DG(O*) value as the only descriptor of catalytic activity. For the active site of the N-GQD-2 catalyst, ORR is rate-limited by the formation of HOO*, while in the case of N-GQD-3 and N-GQD-4, the reduction of the HO* intermediate is the rate-determining step.
![[{"id":"63_lddKpvH","type":"paragraph","data":{"text":"Model structures of nitrogen-doped graphene quantum dots (the values in parentheses are the calculated overpotentials, η<sub>ORR</sub> , V).<sup>162</sup> Copyright American Chemical Society."}}]](/storage/images/resized/AhTLbIN4bjXVDNr4zIJ636rRykwDjtNzBNVkNcAx_xl.webp)
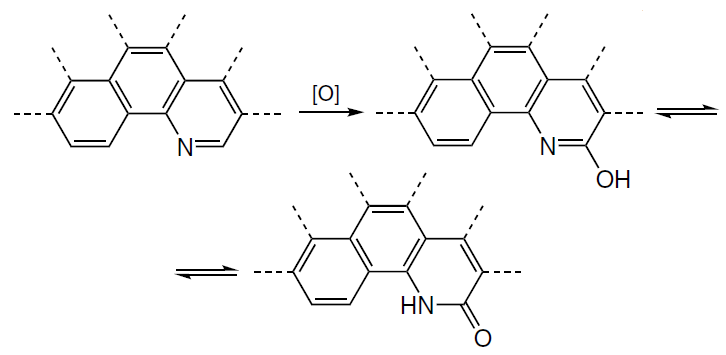
Later, theoretical notions about the influence of the pyridine nitrogen atom on the high catalytic activity in ORR for nitrogen-doped carbon materials were confirmed experimentally. For ORR catalyzed by nitrogen-doped graphite in an acidic medium (0.1 M H2SO4), Guo et al.[49] found, using HR-XPS, the formation of a stable 2-pyridone type intermediate (400.2 eV, see Fig. 8) upon the oxidation of the Ca carbon atom in the pyridine ring (Scheme 1). This confirms the hypothesis that the active sites for ORR are formed near the dopant pyridinic nitrogen atom located on the armchair edge. Experiments on CO2 adsorption demonstrated that the dopant also gives rise to active sites with the Lewis basicity at which the reaction takes place. The minimum experimental overpotential value hORR for the composition-optimized catalyst was 0.5 – 0.6 V, which is consistent with the theoretically predicted value.[162] Theoretical investigation of N-terminated N-doped carbon nanotubes with pyridinic nitrogen and a monovacancy indicates that the appearance of the 2-pyridone intermediate in experiments[49] may be attributable to the fact that 2-pyridone is more stable than tautomeric 2-hydroxypyridine in a wide range of electrode potentials U and corresponds to a local minimum on the potential energy surface (PES) of ORR, i.e., it is a reaction product (Fig. 12).[39]
![[{"id":"v4xS5ntuQM","type":"paragraph","data":{"text":"Structures of model N-doped nanotubes: N<sub>3</sub>/(6,6)N<sub>3</sub>CNT (<i>a</i>) and H – N<sub>3</sub>/(6,6)N<sub>3</sub>CNT (<i>b</i>) in which C<sub>2</sub>-vinyl groups studied in ORR are shown in pale-olive colour; structures of <b>O</b><sub>2</sub>* adsorbates on N<sub>3</sub>/(6,6)N<sub>3</sub>CNTs (<i>c</i>); and free energy profile for ORR on the C<sup>5</sup>=C<sup>6</sup> vinyl site (<i>d</i>).<sup>39</sup>"}}]](/storage/images/resized/BdsDt9uuQfVNI67aGRQHegBShluuqFETKR9UlWwn_xl.webp)
Duan et al.[163] plotted the thermodynamic profile of the 4е pathway of ORR using the graphene model doped with the pyridinic nitrogen atom at the zigzag edge; the rate-determining step was similar to that in the active site of the model N-GQD-2 structure,[162] and hORR was 0.53 V.
Zhang et al.[119][164] investigated the mechanism of ORR on coronene and circum- and dicircumcoronene doped with graphitic nitrogen at the zigzag edge and theoretically showed that the calculated overpotential hORR (V) increases in the series: C23H12N (0.79) < C53H18N (0.88) < C95H24N (1.05). This may indicate a decrease in the catalytic performance with increasing number of carbon atoms in the substrate and, hence, this accounts for the higher performance of small quantum dots in ORR.
A year later, Matsuyama et al.[165] investigated the ORR mechanisms for dicircumcoronene doped with graphitic nitrogen at various positions and demonstrated that on doping at the central benzene ring, the minimum hORR is 0.55 V, which is similar to the value found for the active site at the zigzag edge when pyridinic nitrogen is the dopant.[162] For the above catalysts, the rate-determining step of ORR is the reduction of the HO* intermediate to H2O, which leads to active site regeneration.
Calculations using periodic models of graphene doped with graphitic nitrogen indicate that the doping reaction is endothermic and that the structure with one NGr type nitrogen atom in the basal plane is most stable.[166] The overpotential in the 4e pathway of ORR on this catalyst amounts to 0.84 V, with the formation of the peroxy HOO* intermediate being the rate-determining step.[167]
Chai et al.[168] performed a theoretical study of the joint effect of vacancies, Stone–Wales defects in the graphene matrix, and doping with graphitic nitrogen on the ORR thermodynamics (Fig. 13). As a result, only one structure, N-SW-G-3, proved to be potentially suitable for ORR: the calculated hORR value was 0.43 V.
![[{"id":"uqzfToELB0","type":"paragraph","data":{"text":"Nitrogen-doped models of graphene containing Stone-Wales defect (N-SW-G)<sup>168</sup> and monovacancy (MV-c-PN).<sup>163</sup> Copyright American Chemical Society."}}]](/storage/images/resized/fxACcUJY2pfFh9LfDfTmGpoFQtXZ0iuzjBD1Y3IY_xl.webp)
A group of authors[163] developed and successfully synthesized a highly effective porous graphite-like catalyst for ORR. It was prepared by impregnation of silica gel with dicyandiamide and thermal polymerization followed by modification with (3-aminopropyl)triethoxysilane, dimethylimidazole, and zinc nitrate, which are necessary for the growth of the ZIF-8 metal-organic framework, and subsequent pyrolysis. It was shown that the experimental onset potential Uonset (0.855 V) and the half-wave potential U1/2 (0.95 V) of the synthesized material do not differ much from these characteristics for commercial 20% Pt/C (0.846 and 0.96 V, respectively). Having performed the physicochemical studies of the material using 13C NMR spectroscopy, the authors proposed a model for graphene defect comprising a monovacancy and pyridinic nitrogen (MV-c-PN) (see Fig. 13) as the active site. Analysis of the free energy profile for ORR (Fig. 14) points to the reduction of the HO* intermediate as the rate-determining step, with calculated hORR being 0.44 V.
![[{"id":"-apfISwRPO","type":"paragraph","data":{"text":"Free energy profiles for ORR on the models of N<sub>Py</sub>-doped graphene at the zigzag edge (<i>a</i>) and monovacancy in the basal position (<i>b</i>)."}}]](/storage/images/resized/mUN2L8cxc6LVprM38V786GQMW3jEjaN2DprQLT5S_xl.webp)
Tian et al.[169] synthesized a pyrrolic nitrogen-doped (up to 4.22 at.%) carbon nanomaterial by pyrolysis of a resin based on 3-halo-3-aminophenol (Hal = F, Cl, Br) and formaldehyde followed by treatment with alkali. The resulting material showed good capacitance characteristics for use in capacitors the pseudo-capacitance of which increased with increasing number of pyrrole nitrogen atoms (up to 354 F g–1) as a result of a redox reaction (Scheme 2). This clearly demonstrates that pyrrolic nitrogen atoms in the carbon substrate can participate in the redox reaction when U 0 is 0.3 – 0.4 V, and this has an adverse effect on the catalytic activity of the material in ORR. The mechanism of the target reaction was investigated by Xia et al.,[170] who used a model of graphene quantum dot doped with pyrrolic nitrogen in the substrate cavity and demonstrated the possibility of irreversible oxidation of active site carbon atoms covalently bonded to the dopant (the Ca atom) in the stage of adsorption and dissociation of molecular oxygen. The Eads value is approximately –7 eV, while the O* or HO* adsorbates are the most stable species regarding the free energy, depending on the electrode potential U.
Finally, a large-scale theoretical study of the ORR mechanism on nitrogen-doped graphene (53 model structures) containing one or two heteroatoms was performed by Ganyecz and Kállay.[171] It was shown that doping of graphene with two nitrogen atoms in the meta-position relative to each other reduces the adsorption energy of ORR intermediates, while two ortho-nitrogen atoms, conversely, increase the adsorption energy. The external location of the dopant at the zigzag edge of the graphene sheet also promotes the adsorption of reduced oxygen species, while the basal position hinders the adsorption. The dissociative pathway of ORR was also investigated, but it proved to be thermodynamically favourable only for one of numerous considered structures. By correlation analysis of the energy relationship between the energies of HOO* and HO* intermediates, the authors estimated the minimum possible overpotential, which was 0.33 V for model structures doped with two ortho-nitrogen atoms. At high temperatures required for the preparation of doped carbon materials, the N−N bond proves to be unstable, which results in the destruction of catalyst active sites of this type.[172]
The mechanism of ORR on nitrogen-doped carbon nanotubes (N-CNTs) is less studied than that for graphene and is more contradictory. Wang et al.[173] reported a systematic B3LYP/6-31G(d) study of a series of single-walled (5,5)-armchair nanotubes of ~ 8 Å length (for N-CNTs) doped with heteroatoms (N, P, Si, B or S) in the basal plane. The minimum calculated overpotential of N-CNTs in ORR was found to be 0.74 V. The activity of the catalyst toward oxygen adsorption is determined by the charge distribution in the structure and increases with increasing positive charge on carbon. The authors calculated the dissociation energy of O2* adsorbate on the model N-CNT structure (E≠ = 0.35 eV) and the energy for first reduction step, O2* + H* → HOO* (E≠ = 0.09 eV). The results indicate that ORR follows the associative pathway, with catalysis as a whole being determined by adsorption of O2 . The rate-determining step is the transfer of the last, fourth electron and reduction of the HO* intermediate of ORR, respectively. Among descriptors determining the activity of doped carbon nanotubes, the authors listed the ionization potential, energy gap between the frontier orbitals (Egap), and the dipole moment of the catalyst. Free energy analysis for oxygen-containing intermediates made it possible to predict the optimal DG value for the hydroxy HO* intermediate (0.16 eV), which can be achieved by designing an active site through the formation of defects, introduction of heteroatoms, and the variation of the surface chirality and curvature of nanotubes.
Using the PBE/PAW method, Xu et al.[66] studied the effect of the position of the nitrogen atom in a similar single-walled (5,5)-armchair nanotube in which the rate-determining step of ORR in the case of periodic N-CNT-1 model doped with nitrogen in the basal position is the formation of the HOO* intermediate upon the transfer of the first electron (Fig. 15), hORR = 0.58 V. The introduction of a second nitrogen atom in the para-position to the first one (N-CNT-2 model) increases the ORR overpotential by 0.2 V.
![[{"id":"x4Mv0l0XRY","type":"paragraph","data":{"text":"Free energy profiles for ORR on model nitrogen-doped (5,5)-armchair carbon nanotubes.<sup>66</sup> Copyright American Chemical Society."}}]](/storage/images/resized/rgbgo3fg0qGsaAL9wVtshLQLPfCsFrDLX3yIfL6n_xl.webp)
The authors also considered the finite models for nitrogen-doped carbon nanotubes in which the dopant atom is located at the armchair edge (NPy) (N-CNT-3 mode) or separated by one atom (N-CNT-4) and some other structures less active in ORR that contain a monovacancy and a pyridone moiety. When the electrode potential U is zero, there is equilibrium between the initial state of the system (O2 + *) and the HOO* adsorbate formed upon the first electron transfer in the N-CNT-3 active site, which has an adverse effect on the overpotential, hORR = 1.09 V. The highest activity in ORR was found for the N-CNT-4 model structure in which the rate-determining step is reduction of the HO* intermediate, and the calculated hORR is 0.47 V. Note that the adsorption of O2 on N-CNT-2 is weakly exothermic (Eads = –0.22 eV), while in other cases, it is weakly endothermic (Eads = 0.36 – 0.49 eV).
Gíslason and Skúlason[67] investigated the free energy profiles for the associative 4e pathway of ORR using the PBE/PAW method in relation to 72 periodic models of carbon nanotubes of various diameters and chiralities doped with graphitic nitrogen into basal position (13 armchair, 17 zigzag, and 44 chiral nanotubes). The highest activity was found for the carbon atoms at the heteroatom of (14.7)-, (12.6)-, and (8.8)-nanotubes, with the minimum overpotential hORR = ~ 0.35 V, which is 0.1 V lower than the value predicted for Pt(111) at the same level of theory. The first step of O2 electroreduction to the peroxy HOO* adsorbate is the rate-determining step.
The mechanism of ORR on nitrogen-doped fullerene C59N in alkaline medium was theoretically studied using the B3LYP/6-31G(d,p) method by Wang et al.[59] Analysis of frontier orbitals shows that nitrogen atom decreases both the ionization potential of fullerene and Egap more appreciably than other dopants such as S, B, Si, or P. The energy released upon the Pauling type (OO*) adsorption of oxygen molecule on the active site of N-doped fullerene C59N is 0.37 eV. The calculated overpotential is fairly large (0.73 V) and corresponds to the reduction of HO* intermediate to H2O and regeneration of the active site. On the basis of analysis of the thermodynamic profiles of ORR on heteroatom-doped fullerenes, the authors concluded that only C59N can accelerate the target reaction.
By pericyclic replacement of the acetylene carbon atom by a nitrogen atom during co-pyrolysis of graphdiyne-2 and melamine, Zhao et al.[174] and Shang et al.[175] prepared nitrogen-doped graphdiyne (N-GDY) containing sp-hybridized nitrogen atoms and tested the resulting material in ORR. The material had a good catalytic activity (hORR = 0.36 V) comparable to that of commercial Pt/C catalyst in an alkaline medium, while in acidic medium, it was more active than metal-free heteroatom-doped carbon-based catalysts. Bader charge analysis allowed the authors to confirm the assumption about participation of sp-hybridized nitrogen atoms in the formation of active sites for the adsorption of molecular oxygen.
It was only in 2023 that the mechanism of oxygen electroreduction on the N-GDY model catalyst (Fig. 16) was theoretically considered using the PBE-D2/PAW and RPBE-D2/PAW methods (taking into account the solvent nature in an explicit form as a cluster with H2O).[68] It was shown that the dissociative pathway of ORR is preferable for the N-GDY-1 catalyst, while the reduction of the HO* adsorbate of the N(OH)-GDY-1 type is weakly endergonic (DDG = 0.13 eV). The presence of a stable intermediate in ORR prompted the authors to study the catalytic activity of the N(OH)-GDY-1 structure, on which ORR preferably proceeds via associative pathway because of catalyst poisoning. A similar ORR pathway was also found for the N-GDY-2 active site in which the dopant is pyridinic nitrogen, and a weakly exergonic step of formation of the HOO* intermediate (DDG = –0.22 eV) is involved in O2 reduction. The authors demonstrated the crucial role of the solvent in stabilization of ORR intermediates; and analysis of free energy diagrams taking into account the solvent confirms a more adequate assessment of overpotential compared to that in the gas phase. The poisoned N(OH)-GDY-1 catalyst has a relatively low overpotential (0.46 V) close to the experimental value (0.36 V),[174] whereas the N-GDY-2 model, containing a pyridine nitrogen atom has a low catalytic activity (hORR = 0.75 V). The N-GDY-3 model structure proved to be prone to poisoning to give stable HO* adsorbate.
![[{"id":"9pcnpN6aAo","type":"paragraph","data":{"text":"Free energy profiles for ORR on active sites of model nitrogen-doped graphdiynes.<sup>68</sup>"}}]](/storage/images/resized/qOiRXT2GiiS4LwUNByKxBSNCmaqpFZzjoNlZN9s6_xl.webp)
It is noteworthy that nitrogen-doped carbon materials are good catalysts for many electrochemical reactions, including ORR. Quantum chemical representations of the role of various forms of nitrogen indicate that oxygen adsorption is most effective on carbon atoms covalently bonded to graphitic nitrogen atoms in CNMs; however, hORR is high (~ 0.8 V). The opposite situation is observed for a pyrrolic nitrogen atom: hORR is low (~ 0.3 V), while the adsorption of oxygen is more endergonic. Thus, NPy and NPyr complement each other in ORR catalysis, the former being responsible for the limiting current and the latter being responsible for the onset potential. Along with graphitic and pyrrolic forms, the pyridinic form of nitrogen also gives rise to active sites of ORR. On the other hand, the presence of free LEP accounts for complex-forming properties toward metal ions and easy metal chelation to give stable complexes. For example, common metals such as Co, Mn, Ni, Cu, and especially Fe in metal- and nitrogen-doped carbon materials are excellent catalytic sites of ORR exhibiting higher activity than carbon sites in both the adsorption and hydrogen reduction steps.[40] Therefore, metal ions inevitably accompany the pyridinic nitrogen atom in heteroatom-doped carbon materials, and experimental assignment of activity to a particular catalytic site is difficult, while the removal of trace metal impurities is economically unprofitable.
4.2. Silicon doping
In recent years, there has been growing interest in silicon as a dopant for carbon materials, since it is the second most abundant element in the Earth’s crust and a key material in microelectronics, while being safe for humans and inexpensive. The introduction of silicon atoms into the carbon matrix enhances semiconductor properties[176] by increasing the energy gap between the conduction band and the valence band.[177] Recent studies demonstrate the high potential of silicon-doped carbon nanostructures as anode materials for lithium-ion batteries;[178] among these structures are carbon nanotubes with silicon content of ~ 0.3 at.%.[179] Various applications of silicon-containing materials in fuel cells are discussed in a recent review.[180]
Low electronegativity of silicon (Pauling electronegativity χSi = 1.9,[155] χC = 2.55) accounts for its electron-donating properties. Doping carbon structures with silicon leads to accumulation of negative charge on carbon atoms adjacent to the heteroatom; as indicated by González et al.,[176] ‘silicon-doped carbon electrons are more easily transferred to O2 than undoped ones, which favours ORR activity’. This statement is faulty, since silicon is one of the most oxophilic elements, with the standard enthalpy of formation of the Si – O bond (DHf298) being ≈ 800 kJ/mol;[181] therefore, it will be the first to be irreversibly oxidized with oxygen.[38][45-47] The possibility of formation of stable oxygen-containing adsorbates with participation of silicon was predicted theoretically[39][41][45-47][59][173][182-187] and confirmed experimentally by HR-XPS, and silicon-doped catalysts have a higher oxygen content than nitrogen-doped analogues.[41][176][187] Note that the effect of irreversible silicon centre poisoning on the catalytic activity was described for the first time in our publications,[41][186] and this is the key issue for high catalytic activity of silicon-doped carbon materials.[38]
Another problem arising when carbon materials are doped with silicon is related to the large atomic radius of the heteroatom (rSi = 110 pm, rC = 70 pm), which induces an increase in the internal stress in the structure and decreases the stability of the material; in the case of carbon nanotubes, the tubular structure is broken.[176][188] For this reason, silicon doping is often performed in conjunction with O- or N-doping to preserve the structure of the material.
The main methods for the synthesis of silicon,heteroatom-doped carbon materials include chemical vapour deposition using tetraethoxysilane[176] and triphenylsilane;[189] pyrolysis of silicon-containing precursors [(3-aminopropyl)triethoxysilane and silicone oil,[187][190] tetraethoxysilane and lysine[191]] in the presence of iron catalysts; and doping by high-temperature annealing in the presence of silicon-containing macrocycles, e.g., silicon phthalocyanine dichloride (PcSiCl2).[41] Simultaneous doping of carbon materials with silicon and nitrogen provides a synergistic effect in accelerating ORR,[187] and intercalated silicon nanoparticles (as oxide or carbide) may generate edge sites that enhance the catalytic activity.[176]
Due to large atomic radius, silicon can replace either one or two carbon atoms in the substrate structure to form three- (Si – C3) and four-coordinate (Si – C4) environment (Fig. 17).[187] Three-coordinate silicon is characterized by a smaller sum of the angles at the heteroatom, resulting in deviation of the dopant from the substrate plane.[192] An increase in the degree of pyramidality of the silicon atom increases the adsorption and catalytic activity, in particular, in the deprotonation reactions of H2O and NH3 .[193] Silicon-doped carbon nanotubes have a lower thermodynamic stability than pristine nanotubes regarding both the cohesive energy and Gibbs free energy and have large energy gaps Egap .[192] The calculated formation energy of silicon-doped fullerenes and carbon nanotubes (Si – C3) is two to three times smaller in magnitude than that of nitrogen-doped analogues.[59][173]
![[{"id":"JISWUYt7IU","type":"paragraph","data":{"text":"Graphene structural fragment doped with three- and four-coordinate silicon."}}]](/storage/images/resized/MwZ3q79p1L2oxr5jnwTiKHHbkyxagqYJPdS1fkGS_xl.webp)
Yuan et al.[194] simulated a number of silicon-doped (n,n)-armchair carbon nanotubes of various diameters (Si – C3/CNTs) and theoretically showed that the charge on the carbon atoms adjacent to the dopant increases in magnitude as the nanotube diameter decreases (n < 7). Meanwhile, in the case of larger-diameter nanotubes (8 < n < 14), the charge is invariable; therefore, the energy released upon the adsorption of O2 molecule on a silicon centre increases with decreasing diameter of the nanotubes.[47][182]
The Griffith type adsorption of oxygen molecules on three- and four-coordinate silicon atoms in graphene (see Fig. 3) is highly exothermic compared to the nitrogen-doped analogue: Eads for O2* is –2.60 and –1.67 eV, respectively. Note that the O∙∙∙O distance in O2* adsorbate substantially increases from 1.23 Å in an isolated oxygen molecule (1.26 and 1.28 Å for O2* on graphene and nitrogen-doped graphene) to 1.53 and 1.47 Å for O2* on four- and three-coordinate silicon,[187] indicating considerable activation of the oxygen molecule and the single-bond character of the O – O bond. The exothermic effect of the adsorption of molecular oxygen on the heteroatom in a silicon-doped (5,5)-armchair carbon nanotube (Si – C3/CNT) is 1.5 eV.[173]
In 2023, Cao et al.[195] proposed a mechanism for O2 activation on silicon-doped graphene-like g-SiC6 material based on the selective introduction of electrons with antiparallel spins into the oxygen molecule during its adsorption on two silicon centres to give highly active bridging *OO* adsorbate, while silicon hybridization changes from sp2 to sp3. This is followed by O – O bond cleavage in the adsorbed O2 molecule with a very low activation barrier (E≠ = 0.09 eV). The authors also showed that a carbon monoxide molecule (CO) can be adsorbed on activated *OO* or O* adsorbate and is easily oxidized to CO2 . In contrast, Fang et al.[178] investigated the mechanism of CO2 electroreduction to CH4 , in which silicon-doped carbon materials exhibited a good catalytic activity.
The mechanism of oxygen electroreduction on silicon-doped graphene and (10,0)-zigzag carbon nanotube was studied by Zhang et al.[182] As can be seen from the thermodynamic free energy profile for the associative 4e pathway of ORR (Fig. 18), the three-coordinate silicon atom in the Si – C3/Gr catalyst is irreversibly oxidized to stable HO* adsorbate, and the subsequent reduction is highly endergonic: DDG = 1.54 eV. A similar pattern is observed for the active site of the Si – C4/Gr catalyst; however, the ORR intermediates are less stable compared to those on Si – C3/Gr. The authors also studied the more favourable dissociative channel of ORR on Si – C4/Gr [see Fig. 18b, which shows the possibility of dissociation of the O – O bond in the O2* adsorbate (DDG≠ = 0.95 eV) to give more stable dioxy 2 O* and oxo hydroxy O*HO* adsorbates]. When the graphene substrate is replaced with a zigzag carbon nanotube, the adsorption of molecular oxygen on the silicon centre of the Si – C4/CNT catalyst becomes weakly endergonic, whereas the subsequent electroreduction via the associative pathway proceeds spontaneously: DDG < 0. The calculated overpotential on the silicon centre of the Si – C4/CNT catalyst is hORR = 0.81 V, which is 0.3 – 0.4 V greater than the experimental value.[176][187][196] It is noteworthy that analysis of the free energy profile of ORR on silicon-doped (5,5)-armchair carbon nanotubes indicates the formation of stable HO* intermediate,[173] which is expected to slow down the target reaction on the heteroatom active site, while the difference between the experimental and calculated overpotential values may indicate the existence of catalytic sites more active in ORR than silicon, probably carbon atoms adjacent to the heteroatom.
![[{"id":"ZHGHxvXNp5","type":"paragraph","data":{"text":"Free energy profiles for associative (<i>a</i>) and dissociative (<i>b</i>) pathways of ORR on some silicon-doped carbon structures.<sup>182</sup>"}}]](/storage/images/resized/9osA1FLKISaceMi2eIN5GqFvF5XZELhvoaeb4csf_xl.webp)
Zhang et al.[182] also considered the free energy profile of ORR on an active site inside (10,0)-zigzag nanotube, which, similarly to Si – C4/Gr, proved to be prone to irreversible oxidation to give the HO* adsorbate. Thus, the curvature of the substrate surface on going from planar graphene to curved surface of a carbon nanotube considerably affects both the adsorption and catalytic activity of the silicon centre in ORR.
We studied the effect of surface curvature of carbon materials on the thermodynamic and kinetics of ORR using the B3LYP/6-31 1G(d,p) and wB97XD/6-311G(d,p) methods (wB97XD is the Chai and Head-Gordon hybrid functional with dispersion corrections) in the gas phase by considering Buckminsterfullerene, circumcoronene, and (3,3)- and (6,6)-armchair carbon nanotubes doped with three-coordinate silicon atom (Si – C3/NCM) (NCM is nanocarbon material).[45-47] In addition to the commonly accepted associative ORR pathway on a heteroatom, a dissociative reaction channel was considered and proved to be thermodynamically more favourable in the initial stage of ORR. The reaction proceeds via insertion of one oxygen atom of O2* adsorbate into the Si – C bond without participation of a hydrogen atom and the subsequent single reduction of the adduct to form oxysilanol *O*HO* as a local minimum on the ORR PES (Fig. 19). Further reduction of the *O*HO* adsorbate is unlikely, because it is highly endergonic. For structures with low curvature of the carbon sheet [circumcoronene, (6,6)-armchair nanotube], the energy gap DDG between the *O*HO* oxysilanol and the global minimum in the ORR PES corresponding to silanol HO* is ~ 1 eV, whereas for structures with greater curvature [Buckminsterfullerene, (3,3)-armchair nanotube], it is only ~ 0.5 eV.
![[{"id":"yBz1qlDiI7","type":"paragraph","data":{"text":"Optimized structure of model Si-doped circumcoronene (<i>a</i>), charge density distribution map (<i>b</i>). and free energy profiles for associative (<i>c</i>) and dissociative (<i>d</i>) pathways of ORR on a silicon site in acidic medium at U = 0.<sup>46</sup>"}}]](/storage/images/resized/X6TkvY2e9DSyYgO6pHMc0j2tt8nnoYRBiwggjDbn_xl.webp)
Thus, according to the set of calculated energy parameters, ORR on three-coordinate silicon atoms in the Si – C3/GQD type structures mainly proceeds by the dissociative 4e pathway. In addition, O2 can be reduced not only to silanol HO* as a result of transfer of three out of the four possible electrons, but also to oxysilanol *O*HO*, resulting from a cascade of O2 adsorption/isomerization elementary steps followed by synchronous protonation/transfer of only one electron. The formation of an adsorbate with a similar composition was recently confirmed by HR-XPS in relation to multi-walled carbon nanotubes doped with triphenylsilane a 800 – 900°C.[189] In this case, in addition to a major peak corresponding to SiO3C and a minor peak for SiO2, a medium-intensity peak was observed at 102.5 – 102.9 eV and was attributed by the authors to a silicon atom covalently bonded to two oxygen atoms and two carbon atoms simultaneously (SiO2C2).
The predominant formation of the last-mentioned species, apart from the SiO4 group (peak at 103.7 eV), is a result of pyrolysis of glucose and graphitic nitride (g-C3N4) in the presence of SiCl4 at 800°C, which was also described by Yang et al.[197] The calculations of the associative pathway using a graphene quantum dot, in which a four-coordinate silicon atom was simultaneously bonded to two oxygen atoms and two carbon atoms with a planar bond configuration, showed a high overpotential hORR = ~ 1 V (Fig. 20), which was in poor agreement with the experimental data, indicating this value to be 0.43 V. Despite the high nitrogen content in the material (13.86%), the possibility of formation and participation of silicon sites containing Si – N bond was not considered for ORR; however, exactly the presence of these sites may be responsible for the high activity of Si,N-doped carbon nanomaterials in the target reaction. This was shown by Kruusenberg et al.[41] using multi-walled carbon nanotubes doped with silicon phthalocyanine dichloride.
![[{"id":"TtSrIrxZ8G","type":"paragraph","data":{"text":"Structure of Si-doped graphene quantum dot Si-GLC (<i>a</i>), and free energy profiles of the associative pathway of ORR on the Si-GLC silicon site and carbon site of Si-free GLC structure (<i>b</i>).<sup>197</sup>"}}]](/storage/images/resized/PETvy7oNAcEhp4TYMY3FrWnpejq6tWqwYrRouZ3I_xl.webp)
As a result of irreversible oxidation of the silicon atom in the Si – C3/NCM catalyst to stable (oxy)silanols (*O*HO*, and HO*),[46] the carbon atoms that adjacent to a structural defect (Fig. 21) prove to be inactive in ORR, since adsorption of O2 to give 1,2-dioxetanes *OO* is highly endergonic on these sites. Thus, it can be concluded that silicon-poisoned Si(O)(OH) – C3/GQD structures cannot accelerate the target reaction.
![[{"id":"I6c8l8etta","type":"paragraph","data":{"text":"Structures of the silicon-poisoned quantum dot Si(O)(OH) – C<sub>3</sub>/GQD and 1,2-dioxetanes *<b>OO</b>* based on it. The corresponding E<sub>ads</sub> and ΔG (in parentheses) values (eV) are given under the structures."}}]](/storage/images/resized/mgpsFiolEsxMoHpGn8b6HO8RzYVfMhhXqWxKvYgh_xl.webp)
Bai et al.[198] performed a more comprehensive study of the mechanism, thermodynamics, and kinetics of ORR on silicon-doped graphene (Si – C4/Gr) and showed that all possible elemental steps of oxygen electroreduction occur in a small active region of the substrate around the dopant and adjacent four carbon atoms. The O2* dissociation and HOO* reduction to O* + H2O proved to be the fastest steps, while the reduction of O* to HO* was, conversely, the slowest and, hence, the rate-determining step of ORR: E≠ = 1.13 eV. The authors noted the good resistance of the silicon active site to carbon monoxide poisoning compared to Pt(111).
The thermodynamics of the associative ORR pathway was systematically studied for model graphenes X – C3/Gr and X – C4/Gr, where X = N, Al, Si, P, S, Ga, Ge, As, In, Sn, Te, and Tl.[185] Among the large number of considered structures, silicon and aluminium atoms were found to be dopants most susceptible to oxidation.[199] Wang et al.[59] studied the mechanism of ORR on silicon-doped C59Si fullerene in an alkaline medium and showed the irreversible formation of HO* adsorbate upon oxidation of the heteroatom over a wide potential range.
Golzani et al.[200] investigated the adsorption of O2 and CO2 molecules on silicon-decorated graphdiyne as a potential material for gas sensors; the adsorption energies were –2.2 and –0.63 eV, respectively.
Thus, easy oxidation of three- and four-coordinate silicon in CNMs to give stable, i.e., catalytically inactive, silicon-oxidized species prevents the occurrence of ORR catalytic cycle, while the role of catalytic sites of the reaction is performed by carbon atoms at the periphery and within topological defects of the carbon material.
4.3. Silicon and nitrogen doping
The influence of the silicon decoration with nitrogen in graphene, Si – N4/Gr (Fig. 22), on the adsorption of molecular oxygen and kinetics of particular steps of ORR on a silicon site was studied by Chowdhury and Datta.[184] The replacement of four carbon atoms with more electron-withdrawing nitrogen atoms makes the formation of O2* adsorbate 2.70 eV more exothermic. The O – O bond length in O2*, which is 1.55 Å long, attests to considerable substrate activation, which is needed for subsequent electroreduction. Comparison of the dissociation energy (E≠ = 0.53 eV) and the energy of the first step of O2* electroreduction to give the peroxy HOO* intermediate by the associative pathway (E≠ = 0.61 eV) evidences in favour of the dissociative pathway. The reduction of 2 O* to oxo hydroxy O*HO* adsorbate is also fast (E≠ = 0.47 eV); however, the next step of reduction of O*HO* to O* + H2O requires overcoming a relatively high barrier, E≠ = 1.13 eV. The possibility of electroreduction of O*HO* to the dihydroxy HO*HO* intermediate was not considered. Along the associative pathway of ORR, the above step of formation of the HOO* intermediate is rate-determining, while the steps HOO* → HO*HO* and O* → HO* are fast (Fig. 22). The Eads value for an H2O molecule on silicon is lower than that for an oxygen molecule, which attests to easy desorption of the product of ORR in the first turnover of the catalytic cycle and substrate adsorption in the beginning of the second turnover. The authors also demonstrated the impossibility for ORR to proceed along the less favourable 2e pathway, since the adsorption of H2O2 molecules on silicon induces spontaneous dissociation to give the dihydroxy HO*HO* intermediate. Unfortunately, the free energy profile of ORR was not considered in the study.
![[{"id":"Ckgh6jMwCl","type":"paragraph","data":{"text":"Mechanism of electroreduction of molecular oxygen on silicon,nitrogen-doped graphene, Si – N<sub>4</sub>/Gr.<sup>184</sup> Copyright American Chemical Society."}}]](/storage/images/resized/UBIf9estWgCmwKuomB9PpNTLZhdfCV0MZWtO2Irj_xl.webp)
It is noteworthy that no activation energy analysis is usually carried out for elementary steps of ORR, since determination of the activity of a particular catalytic site requires calculation of free energies, i.e., solving a rather laborious vibration task and search for transition states.[162] This purely thermodynamic approach establishes a minimum set of requirements for analysis of the target reaction based on binding of intermediates and implies the absence of additional barriers, e.g., O2 and H2O adsorption/dissociation or proton/electron transfer reactions. The theoretical substantiation for this approach is based on the similarity of the elementary steps and the Bell – Evans – Polanyi principle,[83] which states that the lower the free energy of the product of elementary step relative to the prereaction complex, the lower the activation energy and that the reaction accompanied by the smallest decrease in DG is the rate-determining step.
In an experimental and theoretical study, Kruusenberg et al.[41] showed that doping of multi-walled carbon nanotubes with silicon phthalocyanine dichloride provides synergistic effect in acceleration of ORR due to the simultaneous presence of groups in which silicon is covalently bound to nitrogen (SiNx) and to oxygen (SiOy) atoms in the material. The ORR overpotential (~ 0.3 V) for this material is 14 mV lower than that for commercial Pt/C catalyst, indicating the high potential of silicon in accelerating the target reaction. According to theoretical study of the ORR mechanism on SiNx-containing (6,6)-armchair carbon nanotubes, where x = 3 and 4, the replacement of carbon atoms in the silicon local coordination sphere with nitrogen atoms increases the charge on silicon from +0.1e (SiC3) to +0.2e (SiN3) and +0.35e (SiN4), which in turn inevitably increases the oxophilicity and finally leads to poisoning of the silicon site by the products of partial oxygen reduction (Fig. 23). These products may be represented by the silanediol 2 HO* and hydroxysilanone O*HO* pair (for x = 3)[39] or silanol HO* and silanone O* pair (x = 4)[38] at low and high electrode potentials U, respectively, which does not agree with experimental results.
![[{"id":"wSrvNMaTBU","type":"paragraph","data":{"text":"Structure of model SiNx-doped nanotubes and free energy profiles on the Si sites of the Si – N<sub>3</sub>/(6,6)CNT (<i>a</i>),<sup>39</sup> Si – N<sub>3</sub>/N<sub>3</sub>(6,6)CNT (<i>b</i>),<sup>39</sup> and Si – N<sub>4</sub>/(6,6)CNT (<i>c</i>)<sup>38</sup> catalysts at some electrode potentials <i>U</i> in acidic medium. The bond lengths in Å are indicated at the structures."}}]](/storage/images/resized/YLyJgE0z5Gq9KAMjLxe60BEJ8MTuQpnUN4XmzaF7_xl.webp)
The subsequent search for the active sites on the surface of Si-poisoned carbon nanotubes revealed the potential possibility of oxygen adsorption on carbon atoms bound to the SiOnHmNx defect. In the case of x = 3, the O2* adsorbates formed upon [2 + 3]-cycloaddition of the oxygen molecule to the C=N – Si moiety to increase the silicon coordination number to six proved to be most stable (Fig. 24).[39] Meanwhile, in the case of х = 4, the calculations predicted a moderately endergonic (0.43 to 0.72 eV) formation of O2* 1,2-dioxetane involving the C1=C2 vinyl moiety (C2 site) at the SiOnHmN4 defect (see Figs 23 c and 25).[38] Theoretical study of ORR thermodynamics showed that for SiOnHmN3-containing structures, the reduction of O2* adsorbates can lead to elimination of silicon atom in the form of orthosilicic acid as the most preferred pathway. Conversely, the reduction of 1,2-dioxetane at the C2 site of the Si(O)(OH)N4-doped structure follows the dissociative pathway in which O*HO* + [H+ + e–] D HO*HO* is the rate-determining step (see Fig. 25).[38] The calculated overpotentials hORR are 0.61 [Si(OH)N4] and 0.31 V [Si(O)N4]; the latter value is in line with the experimental Uonset value,[41] while Tafel analysis attests to the presence of two linear segments, which may correspond to two different oxidized catalyst species differing in activity: Si(O)(OH)N4 and Si(OH)2N4.
![[{"id":"GgX_-_LoJB","type":"paragraph","data":{"text":"Structure of <b>O</b><sub>2</sub>* adsorbates formed at the C<sub>2</sub> sites of Si-poisoned structures: Si(O)OH – N<sub>3</sub>/(6,6)CNT (<i>a</i>), Si(OH)<sub>2</sub> – N<sub>3</sub>/(6,6)CNT (<i>b</i>), Si(O)OH – N<sub>3</sub>/(6,6)N<sub>3</sub>CNT (<i>c</i>), and Si(OH)<sub>2</sub> – N<sub>3</sub>/(6,6)N<sub>3</sub>CNT (<i>d</i>). Free oxygen adsorption energies are indicated below the structures (eV).<sup>39</sup>"}}]](/storage/images/resized/AU1e8KZFatrlIqCFYsPJisNnK4WN150OdlloOf9n_xl.webp)
![[{"id":"XlDQdep4Ol","type":"paragraph","data":{"text":"Free energy profiles for intermediates of ORR at the C<sub>2</sub> vinyl sites of Si-poisoned structures: Si(O) – N<sub>4</sub>/(6,6)CNT (<i>a</i>) and SiOH – N<sub>4</sub>/(6,6)CNT (<i>b</i>) at some electrode potentials <i>U</i> in acid medium.<sup>38</sup>"}}]](/storage/images/resized/Ir8I5tGmqCkRURnotTGqjLzGWMXDU5p1zLrVN0aZ_xl.webp)
Saeidi et al.[201] theoretically predicted the prospects for the use of silicon,nitrogen-doped graphene (Si – N4/Gr) in the electrocatalytic reduction of NO to N2 owing to high activation of the substrate molecule, with the limiting potential of the reaction (Ulim) being 0.56 V.
Thus, the replacement of carbon atoms in the local coordination sphere of silicon with nitrogen atoms enhances the oxophilicity of silicon in CNMs and promotes the shift of catalysis to neighbouring carbon centres. Among the large number of silicon-poisoned forms, structural defects such as SiOnHmN4 have the best promoting effect on the neighbouring C2 dicarbon site toward 4e dissociative pathway of ORR.
4.4. Sulfur doping
Doping of carbon nanomaterials with sulfur atoms also increases the electrocatalytic activity.[24][91][124][202-204] However, the nature of this effect differs from that in the case of nitrogen- and silicon-doped structures considered above in which modulation of the ORR activity is due to the charge generation on the neighbouring carbon atoms. Unlike nitrogen or silicon atoms, the electronegativity of sulfur is close to that of carbon (Pauling electronegativity χS = 2.58,[155] χC = 2.55); therefore, no charge redistribution takes place, and the catalytic activity of sulfur-doped carbon nanomaterials in ORR is attributable to an increase in spin electron density on carbon atoms adjacent to the dopant.[205][206] In view of the larger atomic radius of sulfur (rS = 100 pm, rC = 70 pm), the introduction of sulfur into the basal position of graphene and carbon nanotubes is a more complex process than doping with nitrogen atoms.[205]
Sun et al.[207] synthesized sulfur-doped carbon nanospheres by solvatothermal treatment of saccharose and diphenyl sulfide and subsequent pyrolysis. Analysis by the HR-XPS method demonstrated that sulfur occurs in the prepared material mainly in the aromatic thiophene or thiopyran rings and, to a minor extent, in the sulfone C – SO2 – C and sulfate C – SO3H groups. The obtained S-doped carbon nanospheres showed a low productivity for HO2– and enhanced activity in the 4е pathway of ORR compared to that of sulfur-undoped carbon analogue. The experimental half-wave potential (U1/2) and onset potential (Uonset) were 0.725 and 0.862 V for the most active catalyst sample, which corresponds to overpotential (hORR) of ~ 0.5 V.
By mechanochemical treatment of graphite in the presence of elemental sulfur, Jeon et al.[205] obtained graphene doped with sulfur at the periphery of the carbon sheet. The authors noted the increased electrocatalytic activity and resistance of the material to methanol poisoning. Similar results were obtained by Maiti et al.,[208] who performed chemical vapour deposition of thiophene and toluene on the substrate. Macias et al.[209] described sulfur-doped hollow carbon nanocylinders for which the measured half-wave potential (U1/2) was as low as ~ 0.6 V. Other methods for the synthesis of sulfur-doped carbon nanomaterials as catalysts for ORR are discussed in a recent review.[123]
Jeon et al.[205] used the C100H26 graphene quantum dot model to theoretically study the spin density and Mulliken charge density distributions after the adsorption of a sulfur atom on the basal plane, i.e., sulfur (including oxidized sulfur) doping at the armchair and zigzag edges; they also considered two-graphene clusters connected by two disulfide bridges (Fig. 26). In all of these structures, the introduction of a sulfur atom does not change the charge density, whereas the replacement of a carbon atom in one face substantially increases the spin density on the C atoms adjacent to the dopant.
![[{"id":"DdynUSbO_S","type":"paragraph","data":{"text":"Structural fragments of sulfur-doped graphene.<sup>205</sup> The values in parentheses are the maximum values of spin and charge densities (in <i>e</i> units) on carbon atoms adjacent to sulfur. Copyright Wiley-VCH Verlag GmbH & Co. KGaA."}}]](/storage/images/resized/RRxnk9liytltevBCpWvrlcQWMDksY852jaSYCF67_xl.webp)
Banerjee et al.[206] carried out a large-scale theoretical study of ORR mechanism for a model graphene quantum dot (S-GQD-1) doped with sulfur (in particular, in the oxidized state) at the basal position and at opposing zigzag edges and for the model dimer resulting from doping with two sulfur atoms at the basal plane of one quantum dot and at the zigzag edge of the other quantum dot (S-GQD-2) attached as shown in Fig. 27.
![[{"id":"GkGnWdKm24","type":"paragraph","data":{"text":"Model S-GQD structures (<i>a, b</i>) and free energy profiles for ORR on S-GQD structures (<i>c – f</i>).<sup>206</sup> See the text for explanations of Figs <i>a–f</i>. Copyright Elsevier."}}]](/storage/images/resized/4kgZT3VqxrBEzE1roaBnp9HjEjXY8tCAOdvy4ftD_xl.webp)
The authors addressed only the adsorption energy and free energy for the intermediates along the associative pathway of ORR, that is, HOO*, O*, and HO*, at various active sites, while the adsorption of oxygen molecule was not considered. The first reduction step yielding the peroxy HOO* intermediate at C0 – C2 sites, where a sulfur atom replaces a zigzag edge carbon atom (C0) and two adjacent carbon atoms (C1 and C2) (group A in S-GQD-1) is endergonic; on going from C0 to C2 , DDG increases from 0.5 to 1.8 eV (see Fig. 27c), which implies weak adsorption activity of the C1 and C2 sites that form group A. The replacement of a carbon atom with sulfur in the basal position of S-GQD-1 (group B) leads to irreversible oxidation of sulfur to give O* (see Fig. 27d, C0 site, black curve).
The C1 site of group B is active in ORR; however the reduction of O* to HO* on this site is endergonic; conversely, the C2 site is inactive. The most satisfactory thermodynamic picture of the 4e ORR pathway is provided by the C1 carbon atom adjacent to two thiopyran sulfur atoms at the zigzag edge (see Fig. 27a, group C). The HO* electroreduction to give water molecule, which results in regeneration of the active site, is the rate-determining step (see Fig. 27e); the calculated hORR of 0.5 V is in good agreement with the experimental value.[207] Similar results were obtained in an earlier study by Maiti et al.[208] The second most active ORR catalytic site is the C1 carbon atom covalently bonded to the sulfonic sulfur atom (see Fig. 27а, f, group D). The formation of the HOO* intermediate at this site is the rate-determining step, hORR = 0.52 V. The other potential catalytic sites for ORR in the S-GQD- structure and the C2 site of the S-GQD-2 dimer are less effective in ORR.
Thus, activity modulation in S-doped carbon structures can be achieved by introduction of one sulfone sulfur atom or by replacement of two neighbouring carbon atoms of the zigzag edge (C1 site, see Fig. 27a, group C). Maiti et al.[208] theoretically showed that the catalytic activity can be further increased if both sulfur atoms forming group C are oxidized to sulfones; in this case, the overpotential decreases down to 0.4 V.
The kinetics of 2e and 4e pathways of ORR on the periodic graphene model doped with sulfur via replacement of one carbon atom in the basal position was studied by Lu et al.[210] It was shown that the most likely mechanism of ORR on heteroatoms involves the initial formation of molecular hydrogen peroxide (E≠ = 0.53 eV for HOO* + H* → H2O2 + *), because migration of hydroxyl group from the sulfur atom to the neighbouring carbon atom to give the O*HO* intermediate and the subsequent electroreduction to O* + H2O involved in the alternative dissociative 4e pathway are kinetically less preferable (E≠ = 0.75 and 0.68 eV, respectively). The slowest step of the catalytic cycle of ORR is the final electroreduction of the HO* intermediate to H2O and active site regeneration (E≠ = 0.62 eV). The proposed kinetics of ORR on S-doped graphene is in line with experimental results[207] according to which the yield of H2O2 varied from 10 to 40% depending on the conditions of synthesis of the catalyst and electrode potential U.
The mechanism and kinetics of the 4е ORR pathway on the model (5,5)-armchair carbon nanotube (S-CNT) doped with sulfur (as sulfonium cation) at the basal position were studied by Tavakol and Keshavarzipour.[211] Analysis of the total electronic energy profile of the oxygen electroreduction in the S-CNT active site (a carbon atom at the heteroatom) shows high catalyst performance, and calculation of the activation barriers indicates that electroreduction of dihydroxy adsorbate (2 HO*) to hydroxy adsorbate (HO*) is the rate-determining step, E≠ = 1.65 eV. The free energies of the ORR intermediates are not indicated in the publication.
4.5. Sulfur and nitrogen doping
The above-discussed carbon nanomaterials doped with sulfur atoms alone may possess a satisfactory or good catalytic activity in 4e ORR; the activity can be markedly increased by simultaneous doping with sulfur and nitrogen.[212] For example, the replacement of the C4 carbon atom in S-GQD-2 with graphitic nitrogen increases the catalytic activity of the C1 carbon atom (group F in Fig. 27b), while the ORR overpotential decreases to 0.51 V.[206] Note that without nitrogen doping the HOO* formation on the S-GQD-2catalyst is endergonic.
Using the method of template formation of the catalyst, Liu et al.[213] prepared N,S-doped three-dimensional carbon foams from saccharose and thiourea in various ratios as precursors. These materials varied in morphology, surface area, and N : S ratio. The sample obtained at an initial N : S ratio of 1 : 4 exhibited activity in ORR comparable to that of commercial Pt/C catalyst. The authors attributed the high activity to the synergistic effect of the co-doping with these heteroatoms and to high contents of the dopants in the material (N, 6.53%, S, 2.88%).
Villemson et al.[48] synthesized S,N-doped carbon composite nanomaterial by pyrolysis of reduced graphene oxide, multi-walled carbon nanotubes, and O-methylisourea bisulfate containing 93.5 at.% C, 2.4 at.% O (as C – OH, C – O – C, and N – O), 3.1 at.% N (the ratio of pyrrolic, pyridinic, and graphitic nitrogen was ~ 4 : 4 : 2), and 1 at.% S (the C – S and C – SOx ratio was ~ 65 : 35). Circumcoronene with isolated nitrogen and sulfur atoms (N,S,O-GQD) was chosen as the model structure for theoretical investigation of the ORR mechanism [B3LYP/6-311G(d) basis set, gas phase] (Fig. 28a). In view of the identical nature of the vinyl C2 sites, the authors elucidated the linear dependence of the free energy of Yeager type O2* adsorbates, 1,2-dioxetanes, on the adsorption energy (see Fig. 28b); this dependence was used to classify the active sites into potentially active (AS 1 – 3), weakly active (AS 4 – 6), and inactive ones.
![[{"id":"5wnbhOKLMz","type":"paragraph","data":{"text":"Model structure N,S,O-GQD (<i>a</i>) and ΔG(<b>O</b><sub>2</sub>*) as a function of adsorption energy (<i>b</i>). The coloured ovals in Fig. <i>a</i> indicate the studied C<sub>2</sub> sites; R<sup>2</sup> in Fig. <i>b</i> is the coefficient of determination."}}]](/storage/images/resized/26J9omCvvy7OvZVkadfV66GZ0Ox9AUIR08T2g3LR_xl.webp)
Analysis of free energy profiles of ORR at AS 1 – 3/4 active sites showed that O2 adsorption is weakly or moderately endergonic, which corresponds to diffusion-controlled ORR in agreement with the experiment, with the dissociative 4e pathway being preferred, because DG(O*HO*) < DG(HOO*). In the case of low electrode potentials U, all steps of O2* reduction are exergonic, i.e., they proceed spontaneously (Fig. 29a). For AS-1 (C2 site at the NGr atom), an increase in the electrode potential to 0.48 V in an acidic medium gives rise to equilibrium between epoxy (*O*) and hydroxy (HO*) intermediates. Thus, this is the rate-determining step, and the ORR overpotential is 0.75 V, which is in agreement with the value of 0.79 V predicted by Saidi.[162] The AS-2 active site at the thiopyran sulfur atom, which is the second most efficient in O2 adsorption, differs from AS-1 by a lower overpotential, hORR = 0.61 V, while the nature of the rate-determining step is identical to that for AS-1 (see Fig. 29b). The vinyl moiety at zigzag edge separated by one carbon atom from NPyr and forming the C2 active site (AS-3), showed improved thermodynamics of elementary steps of O2* reduction, similar to that of the ideal ORR catalyst (hORR = 0.34 V); however, it has a reduced adsorption activity towards O2 (see Fig. 29с). The free energy profile of ORR for AS-4 formed by the Ca and Cb pyrrolic carbon atoms is identical to that for AS-3, while the target reaction is highly restricted by O2 adsorption on this site. The calculated hORR value equal to 0.36 V is quite low compared to the previously predicted one (0.81 V),[162] which may be attributed to the difference in the type of considered edges and curvature of surface structures. The obtained theoretical results provide the conclusion that the Cb=Cg vinyl moiety at pyrrolic nitrogen of the zigzag edge (AS-3) is the most likely effective catalytic site and that the increase in the current found experimentally for low potentials U may be due to the involvement of C2 vinyl sites adjoining graphitic nitrogen in ORR; these sites are more active in the O2 adsorption step, but less effective than AS-3, AS-1, or thiopyran sulfur atom (AS-2).
![[{"id":"qtzTemEa_-","type":"paragraph","data":{"text":"Free energy profiles for intermediates of the ORR catalytic cycle at AS-1 (<i>a</i>), AS-2 (<i>b</i>), AS-3 (<i>c</i>), and AS-4 (<i>d</i>) active sites of N,S,O-GQD structures at various potentials <i>U </i>in acidic medium."}}]](/storage/images/resized/pDNkeT3A3AfnJBc23A2Q5RZyPbZ1T4ZrViIDSAV1_xl.webp)
High activity of S,N-doped carbon nanomaterials in ORR was also demonstrated by Song et al.,[52] who prepared a catalyst by pyrolysis of ZIF-8 metal-organic framework, followed by impregnation with thiourea and annealing at 1000°C. It was shown that the relative configuration of heteroatoms in the material has a crucial effect on the catalytic activity; HR-XPS analysis of the catalyst optimized in composition attests to the presence of pyridinic, graphitic, and other nitrogen forms and thiophene/thiopyran (C – S – C) and thiazine/thiazole (C – S – N) sulfur atoms. The material containing a covalently bound pair of heteroatoms exhibited catalytic activity superior to that of commercial Pt/C catalyst in an alkaline medium, while the overpotential hORR was close to zero (–0.004 V). Meanwhile, for an isolated heteroatom pair and sulfur-free nitrogen-doped materials, hORR was 0.270 and 0.403 V, respectively.
The mechanism and free energy profiles of the associative 4e pathway of ORR on periodic N- and S,N-doped graphene models are depicted in Fig. 30. However, the obtained theoretical data are at variance with the high catalytic activity of the material found in the experimental part of the study, since the free energy profiles of ORR (see Fig. 30d – f ) and the corresponding active sites of N- and S,N-doped graphenes indicate the presence of endergonic electroreduction of HO* to HO–. Conversely, a similar N-doped structure considered by Saidi[162] not only has no steps accompanied by increase in DG, but also has a calculated overpotential hORR of 0.86 V. Therefore, the nature of highly active catalytic sites of S,N-doped carbon nanomaterials in ORR has not yet been ultimately established.
![[{"id":"d9T9aeLz4o","type":"paragraph","data":{"text":"Structure of the Pauling type <b>O</b><sub>2</sub>* adsorbates (<i>a – c</i>) and free energy profiles for ORR (<i>d – f</i>) at active sites of N- and S,N-doped graphenes.<sup>52</sup> Copyright Royal Society of Chemistry."}}]](/storage/images/resized/ba7rDRqs6IbnTATp6Ax83TyYhXp6cuXOzszHJDDO_xl.webp)
4.6. Boron doping
Like nitrogen, boron has an atomic radius close to that of carbon (rB = 85 pm, rC = 70 pm); therefore, replacement of carbon atoms in carbon nanomaterials with boron atoms induces minor distortions of the original structure.[122] Boron has one valence electron less than carbon and its vacant 2pz orbital can be conjugated with the delocalized π-system of the carbon material, thus enhancing the sp2 hybridization of the structure and, therefore, improving the catalytic activity of boron-doped carbon materials in ORR.[214][215] Unlike nitrogen, boron is an electron donor with respect to carbon (Pauling electronegativity χB = 2.04, χC = 2.55, χN = 3.04),[155] with the electronegativity difference being somewhat greater for the C and B pair than for the N and C pair. This is indicative of preferred adsorption of molecular O2 on the positively charged boron atom as the ORR active site, and boron is considered to be a promising dopant for accelerating the target reaction.[216][217]
Therefore, numerous efforts have been made toward the targeted synthesis of boron-doped carbon materials and investigation of their catalytic activity.[122][123] Boron-doped carbon nanotubes have been obtained by chemical vapour deposition of organoboron compounds, for example, triphenylborane and benzene, in the presence of ferrocene as a catalyst; the boron content in the material was 2.24 at.%.[216] A similar heteroatom content (2.37 at.%) was achieved by the post-synthetic treatment of carbon nanotubes with boric acid followed by annealing at 1000°C under inert atmosphere. The use of HR-XPS method to characterize the catalyst demonstrated that boron can occur in a carbon matrix as B3C, B4C, B(O)2C, and B(O)C2 .[217] It was found that the activity of boron-doped carbon nanotubes in ORR in an alkaline medium increases in direct proportion to the heteroatom content, being, however, lower not only than the activity of a commercial Pt/C sample, but also than that of nitrogen-doped analogues,[218][219] and the average number of transferred electrons is less than three, which attests to the predominance of the 2e pathway for the formation of H2O2 .[216] The main drawback of boron-doped carbon materials is stronger binding of boron atoms to oxygen atoms than to carbon atoms, resulting in the formation of B – O – C moieties, deterioration of the microstructure, and a decrease in the catalytic activity.[24] The ways to increase the selectivity to the 2e mechanism of ORR on boron doped carbon materials are addressed in recent studies (see, e.g., Ref. [220] and Refs [221],[222] on B,N co-doping).
Fazio et al.[215] reported a theoretical study of the associative and dissociative mechanisms and kinetics of ORR on the model structures of BC53H18 circumcoronene (Fig. 31) and periodic graphene (B-Gr) doped with three-coordinate boron. It was shown that the adsorption of molecular oxygen on boron atoms is endergonic (DG = 0.54 eV, B3LYP(SMD,H2O)/6-31G* calculations)# and gives Pauling type O2* adsorbate (OO*) (see Fig. 3). The subsequent electroreduction of O2* via the associative ORR pathway is kinetically more favourable in the first step of hydrogen transfer (Langmuir – Hinshelwood model). Oxygen in the intermediate O* adsorbate can be incorporated into the B – C bond to form more stable oxy *O* intermediate. The calculated overpotential for the 4e pathway of ORR in acidic medium amounts to 0.29 V, and the reduction of OO* to the peroxy HOO* intermediate is the rate-determining step. Note that the calculated onset potential (Uonset = 0.05 V) for boron-doped graphene in an alkaline medium are in line with the experimental value (0.035 V).[223]
![[{"id":"UbmF4cECcu","type":"paragraph","data":{"text":"Associative mechanism for ORR on boron-doped circumcoronene BC<sub>53</sub>H<sub>18</sub>. The solid black line denotes the Langmuir – Hinshelwood (LH) mechanism, dotted red line corresponds to the Eley–Rideal (ER) mechanism.<sup>215</sup> Copyright Elsevier."}}]](/storage/images/resized/q0uFYIYgvYKfhzFBMMVzP578BWqtc1OqjWXwv3Mo_xl.webp)
The number of electrons transferred in ORR on boron-doped carbon nanostructures (n < 3) indicated above implies that the electroreduction of HOO* is the point of bifurcation of the 2e and 4e pathways of ORR. Indeed, the calculations of the activation barriers for the reactions HOO* + H* → H2O2 (DG≠ = 0.76 eV) and HOO* + H* → O* + H2O (DG≠ = 0.67 eV)[215] reveal a minor difference between the rates of these two reactions, although the latter reaction is highly exergonic (see Fig. 31); this accounts for decreasing selectivity to the 4e pathway of ORR. The authors also showed that arising hydrogen peroxide can be isomerized on the boron atom and the adjacent carbon atom (BC active site) to give the dihydroxy 2 HO* adsorbate with an activation barrier DG≠ of 0.92 eV. However, the initial step of H2O2 adsorption/isomerization on this active site was not considered; therefore, it cannot be ruled out that the formation of H2O2* may be more endergonic than that resulting from adsorption of molecular oxygen (DG = 0.54 eV).
Using M06-2X(CPCM,H2O)/6-311+G(2d,2p) calculations (CPCM is the polarizable continuum model), Ashraf et al.[224] found the adsorption energies for intermediates and activation barriers for the elementary steps of the associative 4e pathway of ORR in relation to boron-doped (6.0)-zigzag carbon nanotube BC134H12 . The adsorption of O2 on the heteroatom was shown to be barrierless and be accompanied by a decrease in the energy of the system by 0.72 eV (Pauling type OO* adsorbate). The reduction of this adsorbate to HOO* is also barrierless, with the reduction of O* to HO* being the slowest step: E≠ = 0.34 eV. The same publication describes the possible mechanism of ORR on a carbon nanotube doped with two boron atoms in the para-positions relative to each other in the benzene ring. In this case, Griffith type *OO* adsorbate characterized by increased stability is formed initially (Eads = –0.92 eV). The authors believe that the next step, which is rate-determimimg, is the O – O bond cleavage giving the dioxo 2 O* adsorbate, with the activation barrier (E≠) being 0.91 eV. An alternative option of protonation of one of the oxygen atoms in the *OO* adsorbate was not studied.
Studies devoted to the catalytic activity of B,N-doped carbon nanomaterials in ORR deserve special consideration. It was shown theoretically that chemical bonding between the electron-rich nitrogen atom and the electron-deficient boron atom results in charge neutralization, which adversely affects catalyst activity in the initial stage of O2 adsorption.[212][225] Zhao et al.[225] prepared two types of B,N-doped carbon nanotubes by simultaneous or successive doping with heteroatoms. As was to be expected, successive doping leads to uniform distribution of dopants in the carbon matrix, whereas co-doping promotes the formation of B – N bonds. According to experimental results, the catalyst with isolated heteroatoms makes the major contribution to the 4e ORR pathway, unlike the catalyst containing the C=N – B=C moiety. Nevertheless, it was shown theoretically and experimentally that the presence of covalent bonds between heteroatoms of boron nitride (BN)x groups, including those adjoining Stone–Wales defects, in the carbon material plays a crucial role in the targeted synthesis of H2O2 (the selectivity in the experiment exceeded 94%).[222]
Thus, the structure of the most promising active sites has not yet been established by quantum chemical methods, and the development of applications of B- and B,N-doped carbon nanomaterials in the 4e pathway of ORR is associated with optimization of synthesis methods, control of heteroatom distribution, and the development of new approaches to increasing the selectivity of the target reaction.
# SMD is universal solvation model based on electron density.
4.7. Miscellaneous non-metal dopants
4.7.1. Phosphorus
Phosphorus is an abundant element in the Earth’s crust and an attractive dopant for modulating the catalytic activity of carbon nanostructures. The electronic properties of phosphorus are similar to those of nitrogen, but the electronegativity is much lower (Pauling electronegativity χN = 3.04, χР = 2.06) and the atomic radius is much greater (rB = 100 pm, rC = 70 pm), which hampers the replacement of carbon atoms in the carbon matrix by phosphorus.[122] For this reason, phosphorus doping remained poorly studied until recently.[126][226]
According to experiments, phosphorus in carbon nanomaterials exhibits electron-donating properties,[227] and these materials possess excellent thermal and electron conductivity.[228] In the carbon matrix, phosphorus forms covalent bonds not only with carbon (P – C), but also with oxygen (P – O and P=O).[229]Various methods for the synthesis of phosphorus-doped carbon materials are discussed in detail in reviews.[123][226] Note that P-doped carbon nanotubes have increased catalytic activity and selectivity compared to undoped ones (the average number of transferred electrons reaches 3.9), but they are inferior to other heteroatom dopants (e.g., N and S).
Few theoretical studies note a considerable distortion of the flat graphene structure (P – C3/Gr)[230] and the tubular carbon nanotube structure[173] upon doping with three-coordinate phosphorus and also the ease of formation of stable oxygen adsorbates, O* and HO*, in Р sites during ORR (Fig. 32). The four-coordinate phosphorus atom in the P – C4/Gr structure is less prone to oxidation, since the first electroreduction step of the adsorbed O2 molecule affords unstable HOO* peroxy adsorbate (DDG > 0). The P(O) – C4/Gr catalyst containing oxidized heteroatom and carbon atoms covalently bound to phosphorus proved to be inactive in ORR as the formation of the HOO* intermediate is highly endergonic. The last step of the catalytic cycle of ORR, the HO* reduction to H2O and regeneration of the active heteroatom site in graphene doped with three-coordinate phosphorus (P – C3/Gr), is endergonic. Analogous results were obtained for fullerene where the final step of the catalytic cycle of ORR is equilibrium at U = 0 in acidic medium.[59]
![[{"id":"4qhBvL1cbN","type":"paragraph","data":{"text":"Free energy profiles for associative pathway of ORR on carbon atoms at the heteroatom (<i>a</i>) and phosphorus atom (<i>b</i>) of various P-doped graphenes at the electrode potential <i>U </i>of 0.455 V in alkaline medium.<sup>230 </sup>Copyright American Chemical Society."}}]](/storage/images/resized/7tgQ3CUAAJ60CCzQL5ijDcWoxaXzq5ADEZdpQ7Dm_xl.webp)
4.7.2. Oxygen
Oxygen attracts considerable attention as a dopant that modifies the electronic structure of carbon materials owing to the predominant acceleration of the two-electron pathway of ORR and formation of hydrogen peroxide.[231][232] The origin of the catalytic activity of O-doped carbon nanomaterials has not yet been adequately addressed, and the data on this issue are contradictory. The oxygen atom is the most electronegative element (Pauling electronegativity χO = 3.44,[155] χC = 2.55) capable of forming various stable functional groups with carbon in carbon materials, such as C – OH, C – O – C, C=O, and C(O)OH.[233]
Lu et al.[234] synthesized oxidized carbon nanotubes that were highly active in the synthesis of H2O2 and theoretically considered O-doped graphene model in the 2e pathway of ORR. An ether carbon atom in the basal position and a zigzag edge carbon atom covalently bound to the carboxyl group were proposed as active sites. Wang et al.[235] designed a zinc air battery in which an oxidized porous carbon nanomaterial served as the cathode. In addition to generation of electricity, this battery produced hydrogen peroxide with a selectivity of ~ 54%. By varying the ratio of oxygen-containing functional groups in the catalyst, it was experimentally found that C – O – C and CH=O groups make the largest contribution to acceleration of the 2e pathway of ORR. However, in 2024, Song et al.[236] theoretically studied the kinetics of 2е ORR pathway and demonstrated the crucial role of the carboxy groups (COOH) and water molecules (medium) in this process. As a result of HOO* reduction to H2O2 , the H* atom adsorbed on the carbon substrate is first desorbed from the catalyst surface as a cation and then attacks the water molecule to give the oxonium cation H3O+ and synchronously protonates the a-oxygen atom of the HOO* adsorbate. Thus, the presence of an oxygen atom in the carbon matrix deteriorates the selectivity, and the 4e mechanism of ORR is accompanied by two-electron O2 electroreduction.
4.7.3. Selenium
Selenium-doped carbon materials are much less common than the S-doped materials, which is due to larger atomic radius of selenium (rSe = 115 pm, rC = 70 pm), high selenium polarizability compared to that of sulfur, and electronegativity identical to that of carbon (Pauling electronegativities χSe = χC = 2.55, Ref. 155). In terms of abundance in the Earth’s crust, selenium is a trace element with an average content of about 500 mg per ton. However, it is more abundant than noble metals such as Pt, Pd, and Au. The latest achievements in the synthesis of Se-doped carbon materials and remarkable examples of application in heterogeneous (electro)catalysis, energy storage/conversion, and neutralization of pollutants were described in a recent review by Dyjak et al.[237]
Jin et al.[238] pioneered the synthesis of Se-doped carbon electrocatalysts for ORR. The authors used reduced graphene oxide (rGO) and mixtures of rGO with carbon nanotubes (CNTs) as starting materials, while diphenyl diselenide served as a source of selenium. By annealing at 900°C, the authors obtained composites containing selenophene moieties according to HR-XPS data. They noted that the predominant factor responsible for the activity of Se-doped carbon catalysts is the modified spin density of the material. In alkaline medium, the optimized catalyst based on rGO/CNT containing 1 mass% Se showed high activity and stability comparable to that of a commercial Pt/C sample, and ORR followed the 4e pathway (n = 3.95). Therefore, it was hypothesized that, the large size of the dopant induces pronounced deformation of the material structure and, as a result, causes charge density changes, which promotes chemisorption of oxygen molecules. Choi et al.51 prepared S- and Se-decorated N-doped rGO/CNT as highly active ORR catalysts in acidic medium, while selenium co-doping provided a higher catalytic activity than sulfur doping alone. As noted by Dyjak et al.,[237] the Se-doped catalyst was highly contaminated with cobalt and iron, as the catalyst synthesis involved chlorides of these metals. The content of selenium was comparable to that of Co, but markedly lower than the Fe content (0.05 and 0.22 at.%, respectively), although the doped catalyst was treated with acid. Thus, the presence of traces of Co and Fe could account for the high activity of the material in ORR.[239] A similar statement is true for Se-doped rGO-catalyst for ORR contaminated with copper.[240] Nevertheless, the catalyst demonstrated high activity and selectivity for the 4e pathway compared to those of selenium-free material; it was assumed that a selenium atom linking two rGO sheets (C – Se – C moiety) acts as the catalytically active site.[240]
A vivid example of the high catalytic activity towards ORR of selenium as a dopant in carbon materials free from metal impurities is the use of the catalyst described in 2022 by Hu et al.[241] The sample was prepared using the template method (ZIF-8 framework, pyrolysis at 1000°C); single Se atoms (~ 2 mass %) were uniformly distributed in the nitrogen-containing carbon material, with Se atom acting as an active site for the 4e pathway of ORR. The results of experiments showed higher activity and stability of the Se,N-doped catalyst compared to the commercial Pt/C sample in an alkaline medium. This catalyst is promising as a cathode material in zinc air batteries: the output power density exceeds that of Pt/C (176.9 and 115.4 mW cm–2, respectively) and the open-circuit voltage is 1.45 V.
In the same study, the authors theoretically considered the associative 4e ORR mechanism on N- and Se,N-doped periodic graphene and Se6 cluster models (Fig. 33). As shown in Fig. 33a, the replacement of the CH group at the zigzag edge of N-doped graphene by a Se atom induced charge density redistribution near the dopant and a change in the adsorption energy of ORR intermediates (such as HOO*, O*, and HO*) relative to the Se-undoped analogue. Analysis of the Gibbs free energy diagram (see Fig. 33b) shows that the peroxy HOO* adsorbate is in equilibrium with the initial state of the catalytic system (* + O2), and the electroreduction of O* to HO* on the selenium atom is accompanied by an increase in the Gibbs energy: DDG = 0.23 eV. Conversely, the formation of the HOO* adsorbate at the Se6 cluster is more endergonic: DDG = 0.70 eV. Unfortunately, the results of the only theoretical work[241] devoted to the catalytic activity of selenium-containing metal-free carbon nanomaterials in ORR are at variance with the high catalytic activity of both Se- and Se,N-doped carbon materials observed in experiments. This attests to inconsistency of the chosen structural model of the catalytic site or the possibility of ORR proceeding via a more favourable dissociative 4e pathway.
![[{"id":"TH692QV_SC","type":"paragraph","data":{"text":"Charge density redistribution in the Se,N-doped graphene model (<i>a</i>) and free energy profiles for the 4e associative mechanism if ORR on the N- and Se,N-doped graphene and Se<sub>6</sub> cluster models (<i>b</i>).<sup>241 </sup>Copyright Wiley-VCH Verlag."}}]](/storage/images/resized/FzMGcwaLjuCX6FUp9rQWhynbG2L43O83MquDj3HQ_xl.webp)
4.7.4. Halogens
Halogens (F, Cl, Br, and I) have a higher electronegativity than the carbon atom (Pauling electronegativity χF = 3.98, χCl = 3.16, χBr = 2.96, χI = 2.66, χC = 2.55);[155] therefore, doping can generate a partial positive charge on carbon atoms adjacent to the dopant and improve the catalytic properties of the resulting materials.[102] It was shown experimentally that the χHal value does not correlate with the catalytic activity: Br- and I-doped carbon nanomaterials are most active in ORR,[242] whereas Cl-doped analogues are least active.[243] This is due to the high strength of the CAr – Cl bond, which is a covalent bond, and hence, the catalyst structure is more rigid and is less prone to transformations. Unlike chlorine, bromine and iodine form weaker and partially ionic bonds with carbon atoms; this leads to positive charge on the carbon atom on which ORR catalysis takes place.
The progress in the synthesis of halogen- and (metal)halogen-,heteroatom-doped carbon materials for use in various electrocatalytic processes is discussed in detail in recent reviews.[242][244] The key methods for halogenation of carbon materials include high-temperature annealing[245] and grinding of graphite[243] in a ball mill in the presence of Hal2 , and hydrothermal synthesis in the presence of metal halides such as ZnF2 , etc.[246]
Fluorine doping endows carbon materials with unique properties, including increased ORR activity (hexp = 0.319 V),[243] durability, high corrosive resistance (due to doping at the periphery) in highly acidic and alkaline media, in particular at high electrode potentials U.[244] Sun et al.[247] showed experimentally that the presence of ionic, and especially partially ionic C+ – F– bonds increases the activity of carbon nanomaterials toward ORR compared to materials containing only covalent C – F bonds. It was noted that halogen doping of graphene results in the formation of both structural defects that can be beneficial for the catalytic activity and oxygen chemisorption and defects related to the change in the type of carbon hybridization from sp2 (>C=CF–) to sp3 (–CF3) as a result of attachment of fluorine atoms.[248] The presence of fluorine atoms induces considerable changes in the geometric and electronic structures of carbon materials and has an adverse effect on the electrical conductivity. For example, an increase in the fluorine content in graphene induces transformation of the metal-like conductive state into an insulator,[246] due to increase in the band gap (Egap) from ~ 0 to 3 eV. Note that chlorine and bromine doping does not change the band gap, which is 0.9 and ~ 0 eV, respectively.[249]
The optimal content of fluorine in graphene for catalysis of 4e ORR is ~ 2.6 at.%,[250] while an increase in the content to 3.4 at.% shifts ORR towards the 2e pathway, with the selectivity to H2O2 increasing to 97.5% and to 792.6 mmol h–1 g–1 for U = 0.2 – 0.3 V and pH = 1.[246] According to quantum chemical calculations, the high activity and selectivity of F-doped graphene in the synthesis of H2O2 are due to the increased fluorine content as >CF2 and CF3 groups, the carbon atoms in which interact more weakly (than that in the =CF group) with the a-oxygen atom in the peroxy HOO* intermediate, which promotes its reduction and easy desorption into the cathode space as HOO–.
Of particular interest is the study by Jeon et al.,[243] who showed that ball milling of graphite in the presence of halogens (Cl2 , Br2 , and I2) results in simultaneous doping at the periphery and delamination due to electrostatic repulsion of halogen atoms from neighbouring sheets. The resulting materials had a good electrocatalytic activity and selectivity in the 4е pathway of ORR, stability to carbon monoxide poisoning, and durability. The quantum chemical calculations performed in this study made it possible to determine the adsorption activity of electron-deficient carbon atoms (C – X) of the zigzag edge toward molecular O2; Eads was –0.71 (X = Cl), –0.75 (Br), and –1.04 eV (I), respectively, and the catalytic activity was inversely proportional to Eads(O2). The calculated overpotentials hORR were 0.38 – 0.5 (X = F, Fig. 34),[251] 0.61 (Cl), 0.57 (Br), and 0.48 V (I), with the reduction of the HO* adsorbate to H2O and regeneration of the catalyst active site being the rate-determining step.[252]
![[{"id":"GxJMGskb9Z","type":"paragraph","data":{"text":"Model of fluorine-doped graphene (<i>a</i>) and dependence of the calculated onset potential U<sub>onset</sub> on the free energy of the hydroxyl <b>HO</b>* adsorbate on carbon atoms as catalytic sites for ORR (<i>b</i>).<sup>251</sup> Copyright Elsevier."}}]](/storage/images/resized/8Uu6qFc122PIgOsRsKxUgp4RoIDFvPk0J4vdpdav_xl.webp)
Thus, despite the difficulties associated with the phosphorus atom size and susceptibility to oxidation, phosphorus remains a promising dopant for CNMs to improve the characteristics of catalysts. The unique electronic properties of phosphorus enable the design of new functional materials with controllable activity and selectivity in catalytic processes, which necessitates further research to optimize the synthesis of effective catalysts and research into the ORR mechanisms on carbon atoms with various phosphorus-oxidized defects. In addition to heteroatoms that can be oxidized, peripheral carbon atoms of the substrate can also be oxidized by molecular oxygen. The introduction of oxygen into the carbon matrix reduces the ORR selectivity by increasing the probability of the 2e pathway, which allows the use of these materials in specific energy generation devices in which H2O2 is formed as a by-product. Study of the application of chalcogens such as selenium as dopants to accelerate ORR is still in its infancy, and theoretical ideas about the structure of active sites are fairly ambiguous and require further comprehensive investigation.
5. Conclusion
The development of views about the structure of active sites and the mechanism of electrocatalytic processes, in particular, oxygen reduction reaction, is of great practical importance for modern society on the way to develop highly active, durable, and inexpensive catalysts for the target process, to introduce them into practice of fuel cells, and to generate the trend toward improving the environmental situation by reducing carbon dioxide emissions. In view of the accumulated knowledge in this field, the application of quantum chemical methods for determining the nature of active sites not only makes it possible to evaluate the activity of a particular potential catalytic site, the microstructure of which is derived from experimental results, but also serves as an independent tool for catalyst design. Thus, theoretical approach supplements experimental studies, being often independent and, in some cases, the only one available approach. Theoretical calculations provide quantitative comparison of the activities of ORR sites different in nature by analysis of adsorption energies of catalytic cycle intermediates and the calculated overpotentials as a measure of efficiency, and predict the nature of the rate-determining step, which may serve as the basis for the design of highly active catalysts. In addition, computational methods in the study of ORR and other (electro)chemical processes involving heterogeneous catalysts are the only source of information about functioning of active sites depending on their composition, structure, and architecture, which often cannot be gained in experiments.
Despite the fact that the computational hydrogen electrode method and more advanced approach, the microkinetic model, can be used to successfully solve problems related to oxygen reduction reaction mechanisms by quantum chemical calculations, there are a number of unresolved issues and challenges that require close attention and further working through. First, computational models often simplify the actual reaction conditions by simulating ideal two-dimensional and one-dimensional catalyst surfaces; meanwhile, real carbon materials are characterized by a complex structure with numerous defects, non-uniform distribution of heteroatoms, and a porous surface. Amorphous carbon structures, which are difficult to simulate due to poor ordering of surface structures, occupy a special place. Second, the presence of an electrical double layer affects the structural and energy parameters of ORR reactants and intermediates, induces electric fields in solution and spatial charge near the electrode thus forming a complex picture that is difficult to reproduce using computational methods. After the introduction of the Nørskov microkinetic model into computational practice, it became possible to take into account the effect of the electrical double layer. Third, the calculation of states and trajectories of transition states and intermediates presents a considerable challenge, because many intermediates exist as a few conformations in dynamic equilibrium, which requires computation of a multitude of options and state ensembles. To overcome these difficulties, it is necessary to develop new methods and approaches, including the use of machine learning, to take into account real reaction conditions, increase the simulation accuracy, and expand the capabilities of considering integrated phenomena.
The data presented in this review provide the conclusion that carbon nanomaterials (graphene, nanotubes, etc.) co-doped with two heteroatoms with higher and lower electronegativity, where the first heteroatom is nitrogen and the second one is silicon or sulfur (selenium), tend to show high activity in ORR, compared with the activity of the commercial Pt/C sample in alkaline media because of synergistic effect of dopants and prove to be promising metal-free catalysts for ORR. The nature of this phenomenon is related to the presence of a covalent N – Y bond (Y = S, Si, Se) and a different radius and/or different electronegativity of the heteroatom compared to those of the carbon atom, which distorts the electronic and geometric structure and, hence, promotes ORR. In some cases (e.g., for chalcogens), the structure of the active site is not entirely clear. The views on the applicability of certain elements as dopants have reversed from complete rejection to unconditional acceptance. For example, it was found that silicon, which was previously considered to be unsuitable for modulating ORR activity when incorporated into carbon nanomaterials because of high oxophilicity, is a unique element widely occurring in the Earth’s crust, capable of sharply increasing the catalytic activity of carbon materials, especially in combination with other heteroatoms (first of all, nitrogen atoms of various nature). Catalytic sites can be represented by vinyl carbon atoms covalently bonded to nitrogen atoms at oxidized silicon atom in SiN4-doped carbon nanomaterials.
The advantage of carbon nanomaterials containing two heteroatoms simultaneously, one of which is nitrogen, is the synergism of different types of dopants, which provides an optimal combination of electronic and geometric influence on active sites. In addition, wide diversity of catalytic sites, both heteroatom- and carbon-based ones, forming N – Y and N – Cx–Y bonds at edges, holes, and basal planes of the carbon substrate opens up broad opportunities for the targeted design of highly efficient, new-generation catalytic materials with controlled properties.
6. List of abbreviations and symbols
The following abbreviations and symbols are used in the review:
h — overpotential,
χ — electronegativity,
wB97XD — Chai and Head-Gordon hybrid functional with dispersion corrections,
E≠ — activation energy,
Eads — adsorption energy,
EF — Fermi energy,
Egap — energy gap between frontier orbitals,
DHf — enthalpy of formation,
DHf298 — standard enthalpy of formation,
DG — free energy change (relative to the reactants),
DDG — change in the free energy difference,
DG≠ — free activation energy,
jL — limiting current density,
NGr — graphitic nitrogen atom,
NPy — pyridinic nitrogen atom,
NPyr — pyrrolic nitrogen atom,
r — atomic radius,
S — entropy,
U — electrode potential (relative to the standard hydrogen electrode),
U 0 — standard electrode potential,
U1/2 — half-wave potential,
Uonset — onset potential,
AS — active site,
B3LYP — Becke-3 hybrid functional with the Lee –Yang – Parr correlation,
CCSD(T) — coupled cluster method with single-, double-, and perturbative triple excitations,
CHE — computational hydrogen electrode,
CNM — carbon nanomaterial.
CNT — carbon nanotubes,
CPCM — polarizable continuum model,
DFT — density functional theory,
GC-DFT — grand canonical ensemble density functional theory,
GDY — graphdiyne,
GGA — generalized gradient approximation,
GLC — graphene-like carbon,
GQD — graphene quantum dot,
Gr — graphene,
HOMO — highest occupied molecular orbital,
HOR — hydrogen oxidation reaction,
HRTEM — high-resolution transmission electron microscopy,
HR-XPS — high-resolution X-ray photoelectron spectroscopy;
LUMO — lowest unoccupied molecular orbital,
LDA — local density approximation,
LEP — lone electron pair,
MOF — metal-organic framework,
ORR — oxygen reduction reaction,
Ox — oxidant,
PAW — projector augmented-wave method,
PBE — Perdew – Burke – Ernzerhof exchange correlation functional
PES — potential energy surface,
Pc — phthalocyanine,
Red — reducing agent,
rGO — reduced graphene oxide,
SMD — solvation model based on density,
TS — transition state,
ZPVE — zero-point vibrational energy.
References















