

Keywords
Abstract
Improving hydrocarbon feedstock processing efficiency is an important objective for modern chemical science. To address this issue, technologies for catalytic conversion of light alkanes and alkenes should be developed. Metal-modified zeolites demonstrate the required catalytic performance for selective dehydrogenation, aromatization, and oxidation of light hydrocarbons. Understanding how metal-containing zeolites work on the molecular level can help in the successful design of industrial catalysts. This review summarizes the fundamental studies on the mechanisms of light alkane and alkene activation and transformation on zeolites modified with copper, zinc, silver, and indium. The available data on the structure and composition of active metal-containing sites, the nature of the key surface intermediates, and possible transformation pathways for C1 – C4 alkanes and C2 – C4 alkenes are systematized and critically analyzed. Conclusions regarding the basic principles and characteristics of metal-modified zeolite catalysis are provided.
The bibliography includes 393 references.
1. Introduction
Increasing the efficiency of hydrocarbon processing is a major challenge facing modern science. This challenge involves fundamental research and applied development of methods for the catalytic conversion of light alkanes and alkenes. Being components of natural and associated petroleum gases, these alkanes and alkenes are readily available, relatively inexpensive, but underutilized feedstocks for producing valuable chemicals. One of the promising areas in this field is the development of catalysts for the selective dehydrogenation, aromatization, and oxidation of light hydrocarbons based on metal-modified zeolites that exhibit the necessary catalytic properties.[1-10] Understanding how these catalysts operate at the molecular level can contribute to the successful solution of this practical challenge.
Zeolites, also known as molecular sieves, are crystalline microporous aluminosilicates with a regular micropore structure. Zeolites were first described by the Swedish chemist and mineralogist Axel Cronstedt in 1756. He noted that stilbite heating produced a large amount of steam, the source of which was the water contained in the material’s pores. Therefore, Cronstedt named this type of mineral zeolites, meaning ‘boiling stones’ (from the Greek ζέω — to boil, λίθος — stone). The structural elements of the zeolite framework are TO4 tetrahedra, where the T atom is either silicon or aluminum, and each oxygen atom belongs to two tetrahedra.[11][12] Moreover, each AlO4 tetrahedron has one negative charge, which is compensated by extra-framework cations. If the countercation is H+, then a Brønsted acid site (BAS) is formed in the zeolite structure, which is a bridged Si – O(H) – Al group (Fig. 1). The protonating power of such BASs is comparable to that of concentrated sulfuric acid.[13] Zeolite properties can be controlled at the synthesis stage by varying the aluminum content, which is characterized by such an important parameter as the Si/Al or SiO2/Al2O3 ratio. By changing the Si/Al ratio, one can affect both the strength and concentration of the BASs.[14]
![[{"id":"T1D_cuMn4R","type":"paragraph","data":{"text":"Structure of the Brønsted acid site, Si–О(Н) – Al group (<i>а</i>), and its position at the intersection of channels in H-ZSM-5 zeolite (<i>b</i>)."}}]](/storage/images/resized/JxeXdmanE3gGdN1MgV4rDFa4eKIuBbHe3r3XglH1_xl.webp)
To date, more than 250 structural types of zeolites,[15] both natural and synthetic, are known. Each type has a unique micropore structure, also known as channels, whose size varies from 1.5 to 15 Å for different zeolites and is comparable to the kinetic diameters of the molecules. For example, Fig. 2 shows the framework structure of the five most commonly used zeolites.[16] The structure of zeolite channels, due to their limited internal space and geometry, affects the diffusion of reactants and products and largely determines the direction of a reaction. This is called the molecular sieve effect,[17-19] which, inter alia, hinders or even prohibits the reaction to occur whose transition state does not correspond to the geometry and size of the pores.
![[{"id":"OC4Su5exh0","type":"paragraph","data":{"text":"Structures of zeolites of various topologies: MFI (ZSM-5), FER (ferrierite), FAU (Y), MOR (mordenite), and BEA (beta). The sizes of the channel windows and cavities formed by intersecting channels are indicated."}}]](/storage/images/resized/K3PQWV477iNpH8RIRjpS7pUAvSqm9PBLhLcXcv3E_xl.webp)
The data accumulated to date[20-27] indicate that zeolites without modifying additives are capable of converting light alkanes and alkenes into valuable aromatic compounds, benzene, toluene, and xylene (BTX), due to the presence of BASs in their structure and the effect of limited pore volume.[28] However, the parallel process of hydrocarbon cracking involving BASs notably reduces the yield of aromatic compounds, and a significant proportion of the reagent is spent on the formation of by-products, mainly methane.
The properties of metal-containing zeolites have been studied for 30 years, starting with the works.[23][24][29-34] It has been found[3][4][20][22][26][27][35-41] that loading zeolites with various metals largely alters their catalytic performance compared to unmodified zeolites: the conversion via aromatization begins to prevail over cracking, and in the case of methane, its oxidation to methanol becomes possible.[42-52] This is most pronounced in the case of group 11 – 13 metals, of which zinc, gallium, indium, copper, and silver are of interest for the conversion of light alkanes and alkenes.[2]
The change in the catalytic properties of metal-containing zeolites clearly results from a change in the mechanism of their action compared to unmodified zeolites. Therefore, the attention of researchers[1][5][6][8][9][35][50][53-55] is focused on studying the mechanisms of activation and transformation of light alkanes and alkenes on metal-modified zeolites, as well as the effect of the nature and composition of metal-containing sites on their properties. However, in studies devoted to this topic, the nature of the active metal-containing sites and the mechanisms of activation and transformation of light alkanes and alkenes with their participation are described differently, and the reported results are often mutually exclusive. This complicates the development of industrial processes for the catalytic conversion of light alkanes and alkenes into chemically useful products on metal-modified zeolites. It is therefore necessary to critically analyze the available data and draw general conclusions about possible directions for further research in the field of application of metal-modified zeolites, which is the purpose of this review.
In the literature, there are review articles on the following related topics: preparation of metal-modified zeolites,[2][6][9][53][56] study of the composition and structure of metal-containing sites and species introduced into zeolites,[4][6][50][53][54] study of the catalytic properties of metal-modified zeolites.[3][5][7-9] Also noteworthy there are few reviews that consider the mechanisms of activation and transformation of alkanes and alkenes.[1][6][9][53][55][57][58] Publications[6][9][53][58][59] are devoted to the studies of the mechanisms of ethane and propane conversion over Ga-modified zeolites. Reviews[1][59] describe the findings of methane activation mechanisms on Mo- and Cu-modified zeolites and discuss the types of active sites. The main topic of the review[55] is the surface hydrocarbon intermediates found on zeolites during the transformation or adsorption of various substrates, which also include alkanes and alkenes. Plausible mechanisms for the oligomerization of ethylene and propylene on metal-containing zeolites are also discussed,[57] with particular attention to the spectroscopic characteristics of the key intermediates.
The purpose of this review is to analyze the results of the studies for several hydrocarbons, namely C1 – C4 alkanes and C2 – C4 alkenes, and zeolites containing Cu, Zn, Ag, or In as modifying additives. The review provides a summary of the state-of-the-art understanding of the activation and transformation mechanisms for light hydrocarbons on metal-containing zeolites.
2. How do Brønsted acid sites work?
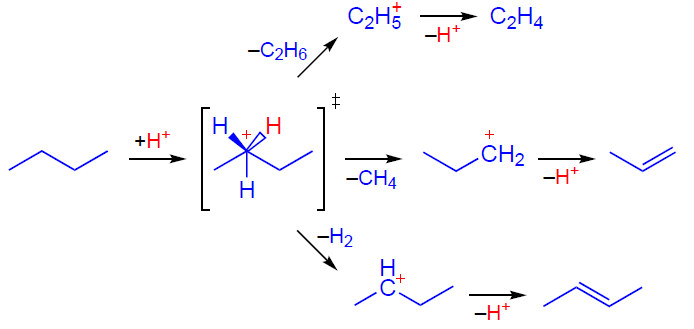
The activation and transformation of alkanes involving zeolite BASs can occur via two competing pathways. The mechanism involving acyclic carbenium ions[22][27][35][60-65] suggests that activation occurs by a bimolecular hydride transfer from an alkane molecule, such as butane, to a carbenium ion R+ (Scheme 1), which can be formed by the protonation of impurity alkenes, thermal cracking of the starting alkane, or with the participation of Lewis acid sites (LASs), such as extra-framework aluminum species. The resulting carbenium ion undergoes deprotonation, hydride transfer, isomerization, β-cleavage, and oligomerization, which explains the observed transformations and products at temperatures below 550 – 600 K, high partial pressure, high conversion of the starting alkane, and the presence of micropores of 7 – 11 Å in size. At temperatures of 623 – 823 K, low alkane concentrations, low conversion, the presence of strong BASs, and smaller micropores (< 7 Å), a monomolecular carbonium-ion or protolytic mechanism is realized.[21][66-76] According to this mechanism (Scheme 2), the alkane molecule is protonated by a BAS to generate a pentacoordinate carbonium ion as a transition state. This ion is unstable and readily converts into various carbenium ions, methane, ethane, and molecular hydrogen. Thus, attack by the BAS on the C – H or C – C bond in the alkane molecule results in dehydrogenation or cracking, respectively. As can be seen, both alkane transformation mechanisms result in the formation of alkenes, key intermediates on the pathway to aromatic hydrocarbons, and C1 – C2 alkanes.
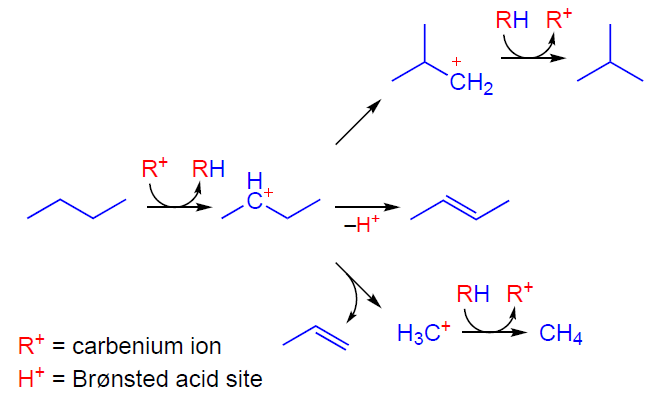
The activation and transformation of light alkenes on the zeolite BAS can be represented as follows. At low temperatures (≤373 K), the transformation mainly follows the oligomerization pathway, which occurs at a high rate and delivers a large variety of oligomeric alkenes.[28][77-85] At higher temperatures, the alkene additionally undergoes isomerization, cracking, and aromatization.[28][77] Based on the available data,[79][86-89] the mechanism of activation and subsequent oligomerization of an alkene, e.g., butene-2, can be described as follows. First, the alkene molecule is adsorbed on the zeolite BAS forming a π-complex (Scheme 3), which is observed by IR spectroscopy.[78][79][90] Next, the C=C double bond is protonated, and the resulting intermediate is assumed to exist on the zeolite surface in the form of either a carbenium ion[87][88][91-93] or an alkoxide species.[78][79] The subsequent reaction between the carbenium ion and the second alkene molecule gives a dimeric product (Scheme 4), which can undergo further transformations according to the above mechanism.[89]
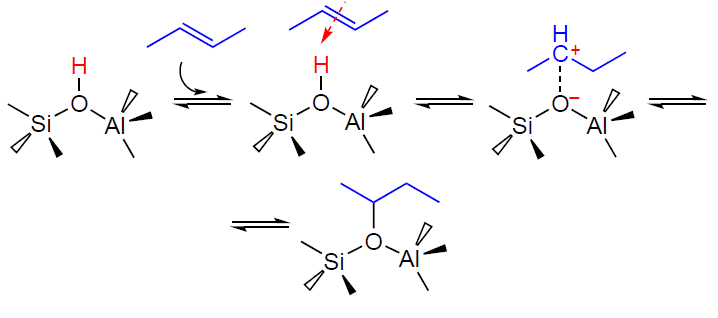
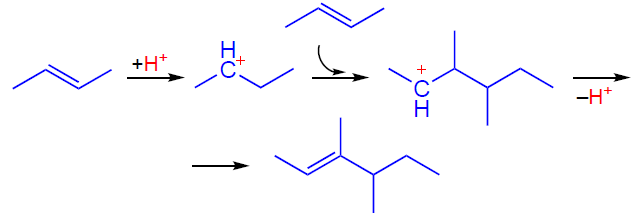
It should be noted that the nature of the intermediates formed from alkenes on the surface of solid acid catalysts remains unclear. Based on quantum chemical calculations, it is concluded[94-100] that acyclic carbenium ions are not stabilized on the zeolite surface and represent transition states formed in reactions involving light alkenes. Indeed, the formation of acyclic carbenium ions on zeolites (or other solid acid catalysts) has not yet been directly observed by spectroscopic methods,[55][90][101] and alkoxide species have been detected spectroscopically during the transformation of alkenes and alcohols on zeolites.[89][102-108] However, the formation of carbenium ions on zeolites was demonstrated by NMR using trap molecules, acetonitrile and ammonia.[109-112] Further evidence for the existence of carbenium ions as intermediates was provided by the 13C label transfer in alkenes[113] and the reaction of alkenes and alcohols with CO to generate carboxonium cations.[114][115] Thus, it can be concluded that the two types of species, carbenium ions and alkoxides, are in equilibrium, as described by Scheme 3, with the equilibrium shifted toward the alkoxides.
Further transformation of oligomeric alkenes involving zeolite BASs follows the conjunct polymerization mechanism,[116-119] which suggests the formation of both alkanes and aromatic compounds as products.[88] This mechanism includes a hydride transfer step (Scheme 5), responsible for the simultaneous formation of saturated hydrocarbons and unsaturated carbenium ions. Cyclopentenyl cations (CPCs), such as 1,2,3-trimethylcyclopentenyl cation, are detected as characteristic intermediates by various spectroscopic methods.[87-89][93][120-123]
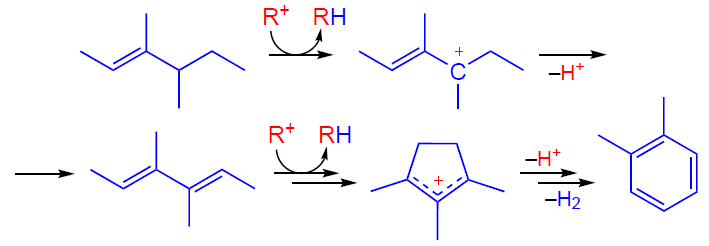
It should be noted that a fairly large number of publications are devoted to the study of the alkene transformation on unmodified zeolites (H-form) using spectroscopic methods.[78-80][87][89] [92][93][98][101][102][104][124-126] In some cases (see, e.g., Ref. [125]), the interpretation of spectral data is ambiguous, which is separately analyzed in the works.[121][122] In this Section, the mechanisms of the transformation of alkanes and alkenes involving zeolite BAS are described in general terms, indicating the most characteristic features. Additional information on this topic can be found in review articles.[22][35]
3. Metal-containing sites in zeolites
3.1. Methods for the introduction of metal sites and their structure
As a result of modification, the following types of metal-containing species can be introduced into zeolites: ions Mn+, [M=O](n – 2)+, MHx(n – x)+ or M(OH)(n – 1)+ (M = Cu, Zn, Ga, Ag, In, etc., n = 1 – 3), which are stabilized in the cation-exchange positions of the zeolite framework (Si – O– – Al), as well as MxOy or [MxOy]n+ oxo-clusters (x, y = 2 – 5) of variable composition and structure. The method of introducing metal-containing species determines their composition and structure,[127] which are studied by X-ray absorption methods (extended X-ray absorption fine structure (EXAFS), X-ray absorption near-edge structure (XANES)), X-ray photoelectron spectroscopy (XPS), electron paramagnetic resonance (EPR), electron spectroscopy, and IR spectroscopy (FTIR) of adsorbed probe molecules. This issue is covered in a number of review articles.[4][50][53][128] Fig. 3 shows the structures of some metal-containing ions and oxo-clusters optimized by density functional theory (DFT) methods.[49][50][129-132]129 – 132
![[{"id":"FlL8_zyqbC","type":"paragraph","data":{"text":"Structures of metal-containing sites and species in ZSM-5 zeolite, optimized by the DFT methods: Ag<sup>+</sup> (<i>a</i>) and Zn<sup>2+</sup> (<i>b</i>) ions, [Cu<sub>3</sub>(μ-O)<sub>3</sub>]<sup>2+</sup> (c) and Zn<sub>3</sub>(μ-O)<sub>3</sub> (<i>d</i>) species."}}]](/storage/images/resized/IB7uuzReiMXyKJr17ZBi737kNIuodXNKm0fVB76h_xl.webp)
Ion exchange and incipient wetness impregnation are traditional and widely used methods for zeolite modification.[2][4][30][36][127][133][134] The use of traditional methods generally leads to a mixture of different metal-containing species in zeolites.[36][135][136] In the case of ion exchange, Mn+ or M(OH)(n – 1)+ ions are predominantly introduced into the zeolite, and their amount is limited by the number of cation-exchange sites (Si – O– – Al sites). Incipient wetness impregnation produces not only metal ions but also oxo-clusters inside the zeolite channels and oxide species on its outer surface. It should be noted that the presence of metal-containing species of varying nature in a zeolite makes it impossible to draw definitive conclusions about the activity and role of different types of sites in the observed reactions, complicating the study of hydrocarbon transformation mechanisms on zeolite catalysts. This can be demonstrated using examples, which are considered hereinbelow in this Section.
The pioneering studies of the catalytic properties of Zn-modified zeolites in the conversion of light alkanes were carried out for samples in which zinc was introduced by ion exchange and incipient wetness impregnation methods.[23][24][27][29] It was shown that the conversion of light alkanes and alkenes did not depend on the method of zeolite modification used.[27] Such samples contain both Zn2+ ions and zinc oxo-clusters (of ZnO composition) inside the zeolite channels,[135][137-143] which does not allow one to reliably determine which type of species is responsible for the activity of the catalysts. In particular, ZnO/ZSM-5 zeolite containing only zinc oxo-clusters demonstrated good performance in the conversion of propane to aromatic hydrocarbons.[144] Zeolites in which zinc was present only in the form of Zn2+ ions also demonstrated the ability to activate light alkanes,[145-147] including methane.[146-148] Almutairi et al.[127] suggest that ZnO oxo-clusters are more active than Zn2+ ions. Results of other studies[136][149-153] indicate the key role of Zn2+ ions in the activation and transformation of alkanes and alkenes.
In studies,[51][52][154-158] various methods for loading indium into zeolites were used, viz., ion exchange and incipient wetness impregnation with an In(NO3)3 solution, mechanical mixing with In2O3 or InCl3, and solid-state exchange with In2O3 in an oxidizing or reducing atmosphere. These methods lead to different states of indium in the zeolite and, as a consequence, to different catalytic properties of the resulting In-modified zeolite. It was shown that the zeolite prepared by mechanical mixing with In2O3 oxide had comparatively high BTX selectivity and high alkene yield.[154][158] The authors suggested that the active sites are the In3+ sites, which increase the dehydrogenation capacity of the catalyst. However, it is found[156][157] that reductive treatment of a mixture of zeolite and indium(III) oxide improves the BTX selectivity and the alkene yield. Accordingly, it has been suggested that In2O oxide is also capable of effectively dehydrogenating alkanes. Studies[159-165] have established that the ground state of indium in zeolites obtained by mixing with In2O3 followed by reductive treatment is In+ ions. It has also been shown that indium(I) cations are easily oxidized by oxygen to form [In=O]+ oxo-ions.[159][160][162] Furthermore, in the presence of paired cation-exchange Si – O– – Al sites, for example, in CHA zeolite, the formation of [In2(m-O)2]2+ oxo-clusters is possible.[166]
The above examples demonstrate that the available data contains certain contradictions and inconsistencies, which can be found not only in the studies devoted to Zn- and In-containing zeolites. This is because many authors failed to take into account the presence of different types of metal-containing species in the tested samples. Therefore, the question about the types of species present in a zeolite is very important, since, as described in the following Sections, the nature of the metal-containing sites affects their ability to activate and convert light alkanes and alkenes. This issue must be considered when analyzing literature data and planning fundamental and applied research, particularly when choosing a method for introducing metals into zeolites. The following can be recommended: experiments should be conducted using zeolite samples selectively modified with one specific type of metal-containing species. Only then can unambiguous results be obtained, allowing the properties of the sites of different nature to be accurately determined.
To selectively introduce one type of species, special techniques are required, such as vapor deposition of organometallic or inorganic volatile compounds.[127][131][167-171] Let us consider several examples.
The method of selective introduction of Zn2+ ions is based on the reaction of metallic zinc vapor with BASs,[167] which can be described by the equation
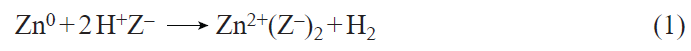
where Z– is the Si – O– – Al cation-exchange site. Deng and co-workers[148][172] believe that this method of introducing zinc affords Zn+ – O – Zn2+ bridging structures with an extra-framework oxygen atom, the origin of which is unclear, since the reaction between the BASs and Zn atoms occurs after the removal of all adsorbed molecules (O2, H2O, etc.) from the zeolite. In addition, the amount of H2 released, as shown in a study,[173] strictly corresponds to the amount of zinc introduced and the stoichiometry of equation (1).
For the selective introduction of zinc oxo-clusters into zeolite, a method has been developed[131] which is based on the adsorption of dimethyl zinc vapor into the pores of the Li-form of zeolite, followed by its hydrolysis with water vapor and thermal treatment of the sample. This procedure results in the formation of small (ZnO)x oxo-clusters within the zeolite pores, where x = 2 – 5 according to EXAFS data. Li+ ions are then replaced by NH4+, the decomposition of which at elevated temperatures (723 – 773 K) yields BASs. This method does not result in the exchange of protons from BASs for Zn2+ ions, which cannot be avoided using traditional methods.
The selective modification of zeolite with ions of the In+ or [In=O]+ type can be carried out using the known techniques.[159-165] The H-form zeolite powder is mixed with indium(III) oxide, after which the resulting mixture is treated in a stream of H2 at an elevated temperature. As a result, indium is stabilized in the form of In+ ions in the cation-exchange positions of zeolite. This is assumed[159-162] to occur via a redox reaction between BASs and In2O (the involvement of metallic In0 is also considered), as a two-step reaction (reactions 2, 3; Z– = Si – O––Al)
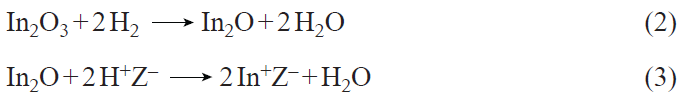
The resulting In+ ions are stable to reduction up to 973 K,[164] but are readily oxidized by oxygen to [In=O]+ oxo-ions according to reactions 4, 5.[159][160][162]
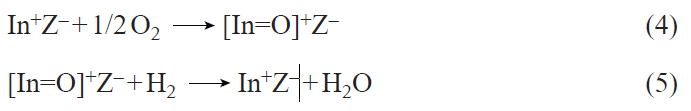
3.2. Nature of metal-containing site catalytic activity
As will be shown in the following Sections of the review, catalytically active sites in metal-modified zeolites can be considered within the framework of the paired Lewis site M – O model (by analogy with the Mn+ – O2– site on the surface of metal oxides[174][175]), where M is an extra-framework metal cation and O is an oxygen atom located either in the Si – O––Al fragment of the zeolite framework or as part of the MxOy and [MxOy]n+ oxo-clusters. The concept of frustrated Lewis acid–base pairs (FLPs) is used in homogeneous catalysis[176][177] and, due to the limited pore volume effect, is also applicable to zeolite systems. Examples of the properties of Zn2+ ions[145-147][178] found in zeolites, as well as Ga-, In-, and Ag-containing sites[179-181] as applied to the dissociation of the C – H bond in methane indicate that the paired M – O site model can be used to describe the observed alkane transformations, as discussed in detail in the next Section.
Possible correlations between the properties of the Zn2+ ion site and its reactivity were analyzed using quantum chemistry methods (Fig. 4).[182] The adsorption energy ΔEads of the probe molecule on the Zn2+ ion in beta zeolite and the intrinsic activation energy ΔE ≠int for the C – H bond cleavage in methane, performed by the paired Zn2+ – O– site, where O– is the oxygen atom from the Si – O––Al site (Fig. 4a), were chosen as parameters for comparison. The obtained results show (see Fig. 4b) that the ΔE ≠int value correlates with the ΔEads parameter (R2 = 0.80) only using a specific probe, e.g., pyrrole, which is capable of simultaneously interacting with both atoms of the Zn2+ – O– site (see Fig. 4c). Obviously, this is a consequence of the fact that the structure of the pyrrole adsorption complex reproduces the structure of the transition state for the methane dissociation reaction (see Fig. 4a,c). For other probes (CO, acetonitrile, pyridine), which can be adsorbed only on the Zn2+ ion, a weak correlation of the parameters was observed (R2 = 0.44 – 0.57). This result confirms the need to consider the active Zn2+ sites in zeolites as Zn2+ – O– Lewis pairs to adequately describe their reactivity. As a result, it can be concluded that the Lewis acid-base pair concept is applicable to describing the properties of Zn2+ ions in zeolites. Unfortunately, similar studies for other metals incorporated into zeolites are not yet available. However, this concept can be extended to zeolite systems containing Cu2+, [In=O]+, and Ag+ ions. This is discussed in the following Sections of the review.
![[{"id":"AeqL9B-GLb","type":"paragraph","data":{"text":"The main steps of methane dissociation on the paired Zn<sup>2+</sup> – O<sup>–</sup> site (<i>а</i>). The following energy parameters are indicated: adsorption energy ΔE<sub>ads</sub>, intrinsic ΔE<sup>≠</sup><sub>int</sub> and apparent ΔE<sup>≠</sup><sub>app</sub> activation energies, a change in the reaction energy ΔE<sub>r</sub>. The correlation between the activation barrier ΔE<sup>≠</sup><sub>int</sub> for methane dissociation and adsorption energy ΔE<sub>ads</sub> of pyrrol (<i>b</i>). Structure of the adsorption complex of pyrrole on Zn<sup>2+</sup> – O– site in beta zeolite (<i>с</i>). Reproduced with minor changes from Kolganov et al.<sup>182</sup>"}}]](/storage/images/resized/FGw2NaCCy7Vcpys8DerBEINNK3pSBQW4z1ndBB9P_xl.webp)
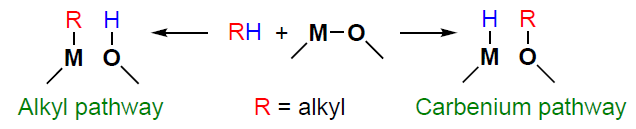
In the studies,[20][39][136][145-147][149][183-191] it is concluded that metal-containing sites are directly involved in the cleavage of C – H bonds in alkanes and in the dehydrogenation step. There are various assumptions about how exactly the activation of the C – H bond occurs.[189][192-194] According to some reports, heterolytic cleavage of the C – H bond is possible, but two reaction pathways are proposed, viz., alkyl[136][145-147][149][184-189][195-197] or carbenium pathways,[39] [198-202] which lead to the formation of metal-alkyl or methoxide intermediates, respectively, on the zeolite surface. Taking into account the model of the Lewis paired M – O site, the principle of metal-containing site action can be described by the general Scheme 6. It should be separately mentioned that the possibility of homolytic cleavage of the C – H bond in alkanes is also not excluded,[188] but is mainly considered for copper-containing zeolites.[49][129][203-207] In the following Sections of the review, we analyze the available data for methane, C3 – C4 alkanes, and C2 – C4 alkenes.
For the sake of completeness, it should be noted that there is an alternative point of view. In particular, it has been suggested that the activation of the alkane molecule occurs with the participation of the zeolite BASs via carbenium-ion[27][30] (see Scheme 1) or carbonium-ion[26][36] [208][209] (see Scheme 2) pathways. In this case, the role of the metal-containing sites consists either in the rapid removal of hydrogen formed during the cleavage of the C – H bond from the BASs via recombination and the release of H2 ,[29][36][210-213] or in increasing the BAS strength,[209] which should reduce the energy barrier of the protonation step. However, this view on how metal-modified zeolites work is refuted by the existing data. For example, it was demonstrated that the protonating strength of BASs did not change when Zn2+ ions were introduced into the zeolite.[214] Further, we also show that, during the light alkane and alkene transformation, the intermediates typical for the mechanisms involving BASs are generally not detected.
4. Activation and transformation of methane
Methane, being an accessible natural resource, has enormous potential as an inexpensive feedstock for the chemical industry. Developing a technology that would ensure the direct catalytic conversion of methane into valuable chemical products is an important task for modern science.[201][215-222] Selective low-temperature conversion of methane to methanol,[223][224] acetic acid,[225-227] and aromatic compounds[228-230] over metal-modified zeolite catalysts is considered a promising alternative to high-temperature and energy-intensive processes such as syngas production.[231] Therefore, the catalytic properties of metal-modified zeolites for methane activation and conversion are the subject of many modern fundamental and applied research. This Section examines the results of studies of methane activation and transformation mechanisms on the zeolites modified with Cu, Zn, Ag, and In.
4.1. Zn-modified zeolites
The ability of Zn-modified zeolites to convert light alkanes has been the subject of intensive research.[2][3][5][6][53][209][232-244] This is due not least to the fact that such materials exhibit activity in the conversion of methane into higher molecular weight hydrocarbons.[200][222][245-252] However, one of the main problems in understanding the activity of zinc-containing zeolites is still the methane activation step, which is believed[145-148][172][200][225][245][253][254] to be accomplished by cleavage of the C – H bond on paired Zn – O surface sites.
Several possible pathways for methane dissociation on Zn-containing sites in zeolites are discussed. Using 13C MAS NMR (MAS is magic angle spinning), Kolyagin et al.[146][147] found, and this was later confirmed by other researchers,[233] that methane activation on Zn2+ ions in zeolites occurs already at room temperature under non-oxidative conditions, and heterolytic cleavage of the C – H bond leads to the formation of Zn – CH3 species (Scheme 7a). In other words, the alkyl activation pathway is realized. This conclusion is confirmed by the IR spectroscopy data.[145] In the study,[148] during the room-temperature activation of methane on Zn-modified ZSM-5 zeolite, mainly methoxy species were detected (13C MAS NMR). The authors suggested that the mechanism presented in Scheme 7b is realized. Homolytic cleavage of the C – H bond in the methane molecule occurs on O– sites of the Zn+ – O– – Zn2+ bridged structure, and the resulting methyl radical is stabilized on the zeolite framework as a methoxy O – CH3 species. Interestingly, in both cases, the same zinc introduction procedure and similar ZSM-5 zeolite samples were used, and methane conversion was carried out under identical non-oxidative conditions. However, the authors obtained contradictory results. The formation of methoxy species after methane activation on zinc-containing beta zeolite was also observed by Luzgin et al.,[200][245] who concluded that the C – H bond is heterolytically cleaved on (ZnO)x oxo-clusters to form Zn – O – CH3 surface species (see Scheme 7c). This result was later reproduced by Wu et al.,[253] and a similar methane activation pathway was proposed. However, it was subsequently established using 13C MAS NMR and IR spectroscopy that (ZnO)x oxo-clusters are fundamentally incapable of dissociative methane adsorption,[131] meaning that in the cited studies,[200][245][253] where zeolites were modified with zinc by impregnation, C – H bond activation occurred on Zn2+ ions.
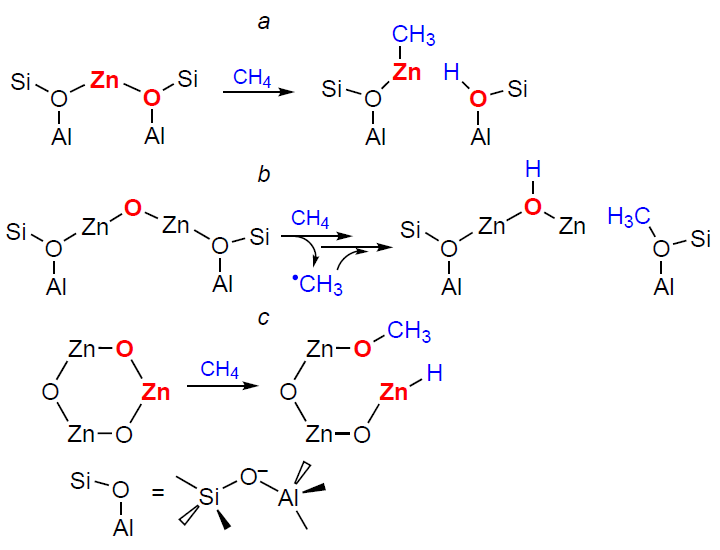
An explanation for the existing contradiction regarding the implementation of either the alkyl or carbenium pathway of methane activation was proposed (Fig. 5).[178]Based on the 13C and 1H MAS NMR spectra (see Fig. 5a,b), it is shown that the dissociation of the C – H bond on the Zn2+ sites gives only Zn – CH3 species and BAS, while the addition of oxygen to the studied samples leads to the generation of methoxide intermediates and other oxygen-containing surface species (see Fig. 5c). These data confirm the conclusions[145-147] about the activation of methane via the alkyl pathway. One can believe that the reported detection of oxygen-containing species, in particular surface methoxides,[148][172][200][245][253] results from the presence of oxygen impurities in the samples. Thus, the conclusions[148][200][253] about the mechanism of methane activation on Zn-modified zeolites should be revised. The transformations of methane observed in the presence of oxygen can be explained by the oxidation of Zn – CH3 species (Scheme 8), the properties of which are obviously similar to those of various organometallic compounds.[255-257] This is also supported by the results of a study on the reactivity of Zn – CH3 species on the surface of ZSM-5 zeolite toward various substrates (О2, Н2О, СО2, etc.).[253]
![[{"id":"0Pnl1y0Uuq","type":"paragraph","data":{"text":"<sup>13</sup>С (<i>a</i>) and <sup>1</sup>Н (<i>b</i>) MAS NMR spectra of methane-<sup>13</sup>С and Zn – CH<sub>3</sub> species on Zn<sup>2+</sup>/H-ZSM-5 zeolite after transformation at 298 – 523 K. <sup>1</sup>Н MAS NMR spectrum of a pure Zn<sup>2+</sup>/H-ZSM-5 zeolite is shown as a grey line in (<i>b</i>). <sup>13</sup>С MAS NMR spectrum of the surface intermediates generated from methane-<sup>13</sup>С in the presence of oxygen (СН<sub>4</sub> : О<sub>2 </sub>= 4 : 1) on Zn<sup>2+</sup>/H-ZSM-5 zeolite after transformation at 298 – 473 K (<i>c</i>). The symbol * denotes a spinning side band."}}]](/storage/images/resized/TfRgpmlh5ppToJ2S0mYbhOAkDBbayZiCrf10V91B_xl.webp)
4.2. Cu-modified zeolites
It is known that the methane monooxygenase enzyme, which contains copper – oxygen sites, is capable of oxidizing methane to methanol with a selectivity of 100% at room temperature.[258][259] This fact prompted researchers to study the properties of Cu-containing zeolites,[42] the ability of which to activate and convert methane to methanol at relatively low temperatures (< 250°C) was first demonstrated by Groothaert et al.[260] Post-synthetic modification allows introducing Cu-containing species of various nature, viz., copper ions,[261] polynuclear oxo-clusters,[262][263] linear CuO-like chains[264] into the zeolite pores. It has now been shown that methane can be activated by copper species of the following types: [Cu2+ – OH]+, [Cu2+O]+ and Cu2+ ions,[204][205][265][266] binuclear[203] [Cu2(m-O)]2+ and trinuclear[48][49][59][129][205][267-269] [Cu3(m-O)3]2+ oxo-clusters.
It is believed that the presence of an extra-framework (i.e., not belonging to the zeolite framework) oxygen-containing ligand is a key factor determining the properties of copper species.[49][129][214][270]Quantum-chemical analysis of the electronic structure of Cu – O – Cu moieties in bi- and trinuclear copper oxo-clusters shows[129] that the oxygen site is an О– • anion-radical, i.e., the fragment has a Cu2+–О– • – Cu+ structure. The putative mechanism of methane activation was studied using DFT methods.[49][129] [203][204][270] Let us describe the obtained results using the example of the [Cu2(m-O)]2+ binuclear oxo-cluster (Scheme 9a). The activation of the C – H bond involves its homolytic cleavage during the adsorption of methane on the О– • oxygen site in the [Cu2(m-O)]2+ oxo-clusters. In this case, the [Cu2(m-OH)]2+ • species, the structure of which is described as a delocalized radical,[129][203] and a methyl radical are formed. The methyl radical then rebounds to a [Cu2(m-OH)]2+ • species or a Si – O– – Al site of the zeolite framework. In the first case, a [Cu2(m-CH3OH)]2+ species is formed. In the second case, a Si – O(CH3) – Al methoxy species and a [Cu2(m-OH)]+ species are formed. In both cases, two Cu2+ sites are reduced to the Cu+ state. It was noted,[129] that the recombination pathway of the methyl radical is not strictly defined, as it is related to the spatial separation of the radical pair. Therefore, in addition to those indicated, other variants are possible. Furthermore, the formation of a Si – O(CH3) – Al species is thermodynamically most favorable, but the species per se is the least reactive.
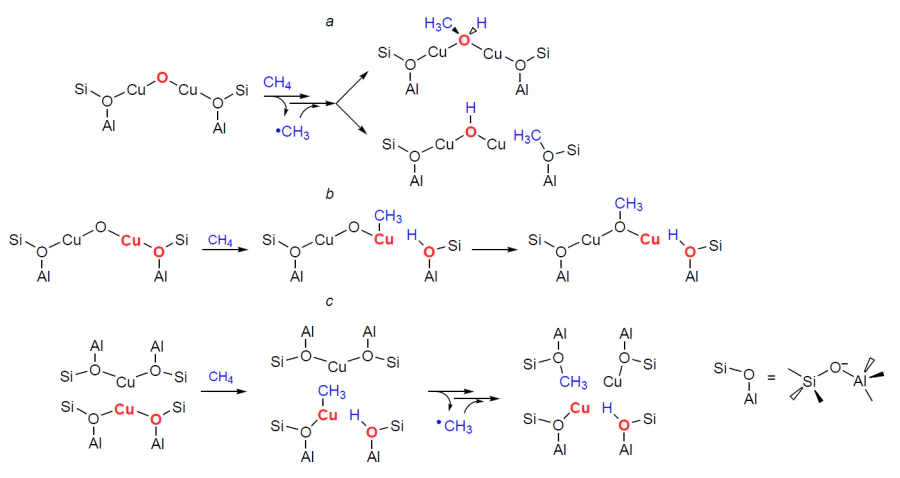
Another point of view is that the activation of methane occurs at the paired Cu – O sites, where O originates from the Si – O– – Al site, by the heterolytic cleavage of the C – H bond.[48][266] Sushkevich et al.[48] suggest that such a mechanism is mediated by [Cu2(m-O)]2+ oxo-clusters (see Scheme 9b). The resulting intermediate [Н3С – Cu(m-O)Cu]+ is metastable and quickly transforms into a [Cu(m-OCН3)Cu]+ species, in which both Cu2+ sites are reduced to the Cu+ state. According to the DFT calculations, the methane activation energy is 90 kJ mol–1. Interestingly, for the previously described homolytic cleavage of the C – H bond, a value of about 90 kJ mol–1 was also obtained.[129] As a result, it is impossible to draw an unambiguous conclusion about which mechanism is realized. Importantly, in the case of methane transformation involving [Cu3(m-O)3]2+ oxo-clusters, the activation barrier is significantly lower and amounts to 51 – 54 kJ mol–1 for homolytic cleavage.[129]
It is found[265][271][272] that methane is also activated by Cu2+ ions bearing no oxygen-containing ligands. The mechanism of such site action was studied using the DFT method (see Scheme 9c).[267]It was shown that two adjacent Cu2+ ions were required for methane activation. One of the sites acts as a part of a Cu – O pair, where heterolytic dissociation of the C – H bond occurs. The primary intermediate is Cu – CH3 species, which is then converted to a methoxy species, Si – O(CH3) – Al, with the intermediate formation of a methyl radical. In this process, both Cu2+ sites are reduced to the Cu+ state.
As can be seen, various methane activation pathways involving copper-containing sites in zeolites are discussed in the literature. A common feature of the described mechanisms (see Scheme 9) is the formation of methoxide surface species from methane. Indeed, the formation of different types of methoxy species has been detected using 13C NMR and IR spectroscopy.[48][207][227][267][271-275] The nature of the surface intermediates was studied (Fig. 6).[265] [272][276] It was found that, in addition to methanol, Si – O(CH3) – Al-type species were generated from methane with the participation of Cu2+ ions, while the involvement of the Cu – O – Cu-type species, which are part of [Cu3(m-O)3]2+ oxo-clusters, led to the formation of Cu – O(CH3) – Cu and Cu – (CH3OH) – Cu species. Thus, it can be concluded that the mechanisms discovered by theoretical methods of quantum chemistry were spectroscopically confirmed, although not all studies have reported experimental data for the zeolite systems studied.
![[{"id":"kqDZ_NmZbL","type":"paragraph","data":{"text":"<sup>13</sup>С MAS NMR spectra of methoxy species generated from methane-<sup>13</sup>С at 523 K on the surface of Cu<sup>2+</sup>/H-ZSM-5 and CuO/H-ZSM-5 zeolites containing either Cu<sup>2+</sup> ions or [Cu<sub>3</sub>(m-O)<sub>3</sub>]<sup>2+</sup> oxo-clusters, respectively (<i>a</i>). Structures of methoxy species and chemical shifts of their constituent carbon atoms obtained by DFT calculations (<i>b</i>). The Figure contains a fragment of a Figure from a previously published article by the authors.<sup>272</sup>"}}]](/storage/images/resized/haVzqU27nZmfamP2ShtqawAtlhaJlsCAVnHhE9cp_xl.webp)
The transformation of methane to methanol on copper-containing zeolites has the following features. First, in the course of the reaction with methane, CuII sites are reduced to the CuI state,[277-280] which is inactive.[266] [281] As a result, a separate step of active site regeneration is required, which can be accomplished by treating the zeolite with oxygen[282][283] or water,[48] although the ability of water to oxidize copper sites in zeolites is a focus of active debate.[266][284-286] Secondly, activation of methane leads to the formation of methoxide surface intermediates,[48][207][227][265][272][274][275] which are converted to methanol upon treatment of the zeolite with water at room or elevated temperature.[43][49][282][287]Thus, the product is a mixture of water and methanol, which can be separated, e.g., by distillation, which, in turn, requires additional energy costs.[288][289] Therefore, the possibility of a reaction between methoxy species and other substrates, carbon monoxide and benzene, was studied.[227][273] The 13C MAS NMR method showed (Fig. 7) that the reaction between these substrates and methoxy species was possible, and therefore copper-containing zeolites are capable of catalyzing the reactions of benzene methylation with methane and methane carbonylation with carbon monoxide.[273] Interestingly, in one of the cases, benzaldehyde is formed as a product, which indicates the ability of copper sites to oxidize not only methane, but also toluene, which opens up additional prospects for the use of these catalysts.
![[{"id":"3bW0StMKJ0","type":"paragraph","data":{"text":"<sup>13</sup>С MAS NMR spectra of methoxy species with the addition of СО (<i>a</i>) or benzene (<i>b</i>) adsorbed on the surface of copper-modified ZSM-5 zeolites, before and after transformation at temperatures of 523 or 573 K. The symbol * denotes a spinning side band."}}]](/storage/images/resized/IWwlZiQTNpQOA6SkpUMMnj5tzdgmN0FFP61YQkvP_xl.webp)
4.3. Ag-modified zeolites
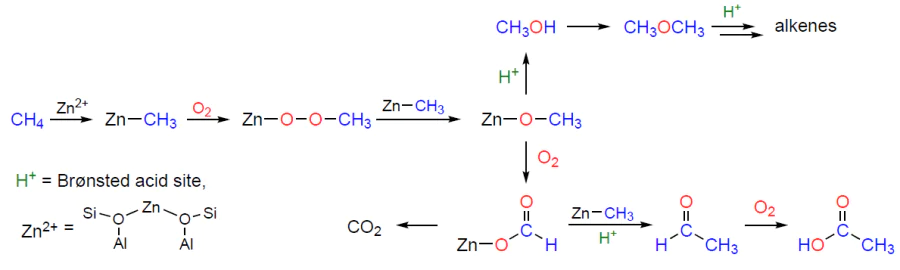
Another example of metal-modified zeolites capable of activating methane for subsequent transformations is Ag-containing zeolites. Since the discovery of their activating properties, several attempts have been made[290-295] to determine how methane activation occurs with the participation of Ag sites, which can take the form of the Ag+ species[295] or Ag3+ species.[296] These species are believed to promote the dissociation of the C – H bond to form Ag – H hydrides and surface methoxy groups.[290][291][293][294] However, this activation pathway was initially supported only by indirect data. In particular, the formation of Ag – H species was observed only for methane conversion on Ag – Y (Ag-FAU) zeolite, but not on Ag-ZSM-5 zeolite.[294] Methoxy species were not detected in any of the studies. Furthermore, according to the results of quantum chemical calculations,[197] the alkyl pathway for methane activation is energetically more favorable than the carbenium pathway (see Scheme 6). In other words, the dissociation of the C – H bond should afford Ag – CH3 species and an O – H group, but this, too, was not confirmed experimentally. Later, the formation of methoxy species was detected by 13C MAS NMR during the activation of methane on Ag+/H-ZSM-5 zeolite (Fig. 8),[180] while Ag – CH3 species (dC from –5 to –15 ppm)[297] were not detected. Thus, the accumulated experimental data support the carbenium pathway of methane activation on Ag+-containing zeolites (Scheme 10a).
![[{"id":"UbKFNUSR4T","type":"paragraph","data":{"text":"<sup>13</sup>С MAS NMR spectra of methane-<sup>13</sup>С on Ag/H-ZSM-5 zeolite after its transformation at 298 – 823 K. The symbol * denotes a spinning side band. The Figure contains a fragment of a Figure from a previously published article by the authors.<sup>180</sup>"}}]](/storage/images/resized/tpofKuLgPK7bxhtxdqUbQDyGFSFXq4omHOnNhziG_xl.webp)
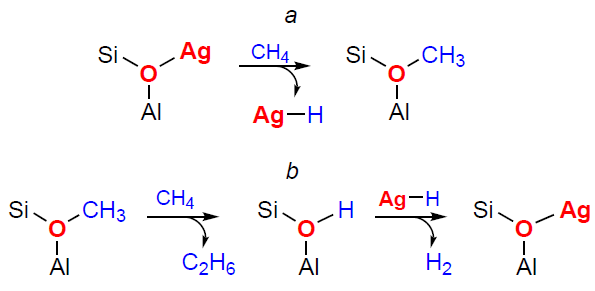
Interestingly, methane activation and the formation of surface methoxy species are followed by further methane transformation.[180] Ethane, ethylene, and benzene/toluene are formed sequentially as intermediates and transformation products. The data obtained indicate that methoxy species, which react with methane molecules, are responsible for the formation of the first C – C bond (ethane molecules) (see Scheme 10b). Thus, Ag-modified zeolites can be considered promising catalysts for the direct conversion of methane to ethylene and aromatic hydrocarbons.
4.4. In-modified zeolites
The first studies of indium-modified zeolites were conducted in parallel with those of Ag-containing zeolites. In particular, it was shown that methane reacted with ethylene or benzene on the In/H-ZSM-5 zeolite to give propylene or toluene, respectively.[292][298] Similar transformations were observed on the Ag/H-ZSM-5 zeolite.[290][291][293] Based on these data, it was suggested that the mechanisms of indium- or silver-containing zeolite action were similar, and that methane activation on In sites should proceed via the carbenium pathway (Scheme 11a) to give surface methoxy species, the properties of which determine all the observed alkane transformations. As described above, the implementation of such a mechanism for Ag+-containing zeolites has been experimentally confirmed (see Scheme 10a). A study of the methane activation mechanism on In-CHA zeolite using DFT calculations has shown[166] that C – H bond dissociation should occur via the alkyl pathway, with the active site including an extra-framework oxygen atom (Scheme 11b). However, the authors have not performed calculations for the carbenium pathway of methane activation, which does not allow us to compare the energy profiles of the two reactions and, accordingly, to make a definitive conclusion about which C – H bond dissociation pathway is realized under these conditions.
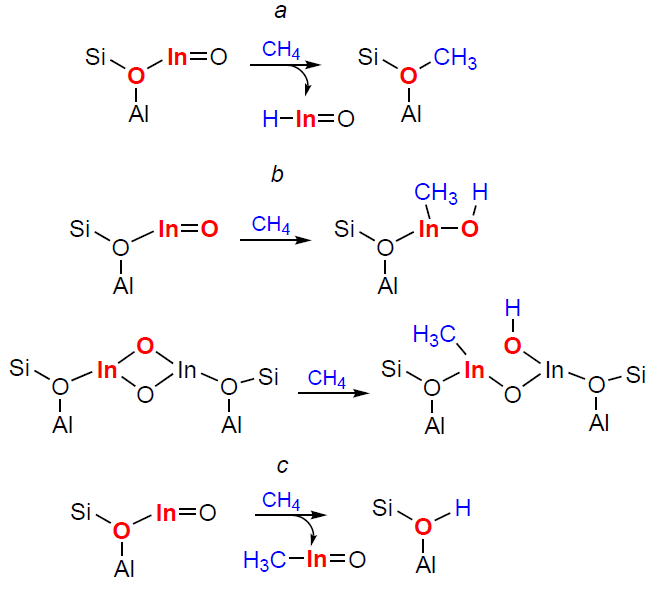
Gabrienko et al.[181] studied methane transformation on In/H-ZSM-5 zeolites by MAS NMR and XPS techniques (Fig. 9). It was found that In+ ions did not activate methane molecules. On the contrary, [In=O]+ ions do activate them, but both In – CH3 species and O – CH3 methoxy species were detected as surface intermediates. In addition, the formation of new BASs was noted (see Fig. 9a,b). At the same time, the reduction of [In=O]+ ions to In+ was detected by XPS (see Fig. 9c). These results were interpreted as follows: dissociation of the C – H bond in methane occurs at the paired In – O sites (see Scheme 11c), but the oxygen atom belongs to the Si – O––Al sites of the zeolite framework. The intermediate is the H3C – In=O species, which is then oxidized to the H3C – O – In=O species by the [In=O]+ sites. Thus, [In=O]+ ions in zeolites activate methane via the alkyl pathway. The In – CH3 species is the primary intermediate formed from methane. Methoxy species are the oxidation product of indium-methyl. Thus, the carbenium pathway for methane activation proposed in studies[290][291][293] is not supported experimentally. The activation pathway of methane involving an extra-framework oxygen atom in the indium-containing species is also not realized (see Scheme 11b), since in the 1H MAS NMR spectra, only the resonance of the Si – O(H) – Al groups was observed, and not the In – OH ones.
![[{"id":"tclo6ipz7Z","type":"paragraph","data":{"text":"<sup>13</sup>С (<i>a</i>) and <sup>1</sup>Н (<i>b</i>) MAS NMR spectra of methane- <sup>13</sup>С on InO<sup>+</sup>/H-ZSM-5 zeolite after transformation at 298 and 523 K. The symbol * denotes the spinning side bands. In 3d<sub>5/2</sub> core-level X-ray photoelectron spectra (<i>c</i>) of In<sup>+</sup>/H-ZSM-5 (1), InO<sup>+</sup>/H-ZSM-5 (2), InO<sup>+</sup>/H-ZSM-5 zeolites after interaction with methane at 523 K (3), difference (3) – (2) spectrum is shown as spectrum (4)."}}]](/storage/images/resized/0BNEvVrf0ck80hOVNIznaRPXGb1mtGLaOdq6vHQT_xl.webp)
4.5. General patterns of methane activation
This Section presents and analyzes the research findings on the properties of zeolites modified with Cu, Zn, Ag, and In, as they relate to methane activation and transformation. Based on the accumulated experimental data, it is concluded that methane activation on metal-containing sites occurs primarily via heterolytic cleavage of the C – H bond. Section 3, using the example of Zn2+ ions in zeolite beta, discusses how the properties of such sites should be described in terms of the Lewis acid–base pair concept, or, in other words, the paired Zn2+ – O– site. A comparison of the properties of zeolites modified with various Group 11 – 13 metals suggests that this approach can also be applied to the Cu2+, Ag+ and [In=O]+ ionic sites. Dissociation of the C – H bond in methane can occur via either the alkyl or carbenium pathways, depending on the nature of the metal M in the paired M – O site. The primary intermediates formed on the surface of metal-modified zeolites are either metal – methyl or methoxide species. Metal–methyl species are capable of oxidation to methoxides by molecular oxygen or, for example, with the assistance of [In=O]+ sites. These methoxide species can then be converted to methanol, formate, acetate, and other oxygen-containing products. An important property of methoxy species is their ability to methylate the benzene ring, which opens an alternative pathway for converting methane into aromatic hydrocarbons.
5. Transformation of light alkanes
Light alkanes, particularly propane, butane, and isobutane, are the main components of liquefied petroleum gas, a crude oil refinery product, and natural gas. Primary uses of light alkanes include steam cracking[299] to produce ethylene and propylene, and catalytic cracking[300] to produce С2 – С4 alkenes and aromatic hydrocarbons, benzene, toluene, ethylbenzene, and xylenes (BTEX). Alkenes and aromatic hydrocarbons are indispensable in the chemical industry as basic components to produce synthetic fibers, plastics, dyes, rubber, and other valuable products. Zeolites are used as catalysts or catalyst components in hydrocarbon processing,[301][302] because they possess a range of important properties, such as thermal and chemical stability, variable Brønsted acidity, and a molecular sieve effect. However, the presence of strong BASs in zeolites promotes the formation of carbon deposits or coke[303] and undesirable methane,[21][66][67] leading to catalyst deactivation and low selectivity for target products.[22][304][305] To improve the catalytic properties, zeolites are loaded with various promoting additives, viz., alkaline earth metals (Mg, Ca, Sr, Ba),[306] transition metals (Fe, Co, Zn, Ga, etc.),[243][307-314] rare earth elements (La, Ce, etc.),[315-317] as well as phosphorus.[315][318] Zeolites modified with zinc and indium are among the most active and selective (for BTX) catalysts for the aromatization of light alkanes.[23][27][137][154][319] However, much about the mechanism of their action is still unclear. Therefore, fundamental research aimed on the mechanisms of light alkane transformation on Zn- and In-modified zeolites is extremely relevant, as it is necessary for further applied work on the development of industrial catalysts. This Section discusses the results of the studies into the mechanisms of light alkane transformation on zinc- and indium-modified zeolites.
5.1. Zn-modified zeolites
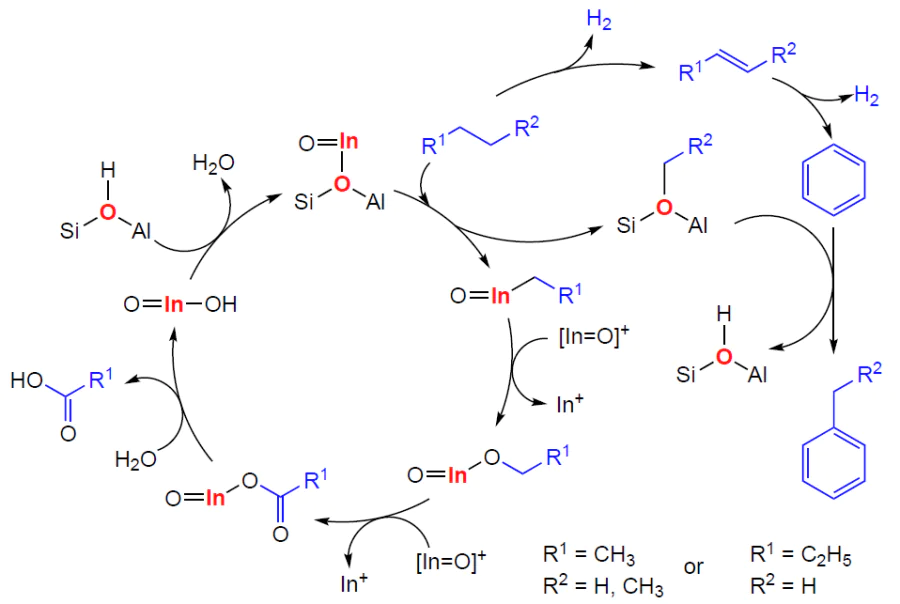
Zn-modified zeolites were among the first zeolites to be studied, and continue to be actively researched, for the development of the light alkane aromatization catalysts. Examples include publications[27][36][134][312][313][320][321], which pioneered research in this area, as well as recent review articles,[3-6][9][35] which summarize the data accumulated to date on catalytic processes, including the most topical ones. It has been shown that efficient aromatization of light alkanes requires a bifunctional catalyst containing BASs and dehydrogenating component,[27][36] namely, metal cations or oxo-clusters. The metal atoms in these species, being coordinatively unsaturated, represent sites capable of donor-acceptor interactions and, therefore, are capable of catalyzing the dehydrogenation of alkanes. Loading zeolites with zinc species as a dehydrogenating component significantly alters the alkane conversion pathway[20][26][27][35][36] compared to unmodified zeolites. The general scheme of alkane conversion on Zn-modified zeolites is shown in Fig. 10. The key reactions are dehydrogenation and hydrogenolysis of the starting alkane, oligomerization of the resulting alkenes, and aromatization of the oligomeric alkenes. However, there is no consensus regarding the mechanism of alkane activation and transformation and the role of BASs and zinc-containing sites at each step of the conversion.
![[{"id":"-b4k1QCfSl","type":"paragraph","data":{"text":"Scheme of С<sub>3</sub> – С<sub>4</sub> alkane conversion on Zn-containing zeolites"}}]](/storage/images/resized/2IE66KUyVNafsmyWIY48NghzNq3meIuLNF7Utrnf_xl.webp)
The studies[26][27][30][36][208] suggest that the activation and transformation of alkanes occur via carbenium-ion (see Scheme 1) or carbonium-ion (see Scheme 2) pathway, with zinc sites catalyzing the recombinational desorption of hydrogen atoms formed during the cleavage of the C – H bond involving BASs.[208] It is assumed that zinc oxide species are the active species. However, the nature of the migrating species (Н+, Н– or Н0) and the mechanism of their desorption from the metal oxide surface remain unclear. In this context, it is also important to note the following. The transformation of alkanes on Zn-containing zeolites begins in some cases already at 296 – 298 K,[147][322-324] whereas mechanisms involving BASs require temperatures ≥550 K. Dehydrogenation of C3 – C4 alkanes also occurs over ZnO-modified silicalite-1,[325][326] which has the structure of ZSM-5 zeolite but does not contain BASs. Furthermore, a comparison of the activity of Zn2+ ions and ZnO oxo-clusters has shown that the former type of zinc species is more active in the dehydrogenation reactions of С3 – С4 alkanes.[322][323] [327] Thus, the hypothesis that light alkanes are activated only with the participation of BASs is not supported experimentally.
According to another hypothesis, the activation of light alkanes occurs through the dissociation of the C – H bond on zinc sites followed by dehydrogenation.[20][146][145-147][149][184][186][187][189] One of the first hypotheses about how this might occur was put forward by van Santen and co-workers[184][186] when studying ethane dehydrogenation on Zn-ZSM-5 zeolite using the DFT method: the zinc sites in the Zn2+ and [Zn – O – Zn]2+ ionic species acts as acceptors of the ethyl C2H5 fragment during the heterolytic cleavage of the C – H bond. The nature of the surface hydrocarbon intermediates generated from alkanes was studied experimentally by 13C MAS NMR using propane and Zn-ZSM-5 zeolite as examples.[146] Later, the process of C2 – C4 alkane transformation on Zn-modified zeolites containing Zn2+ ions or ZnO oxo-clusters was investigated.[147][149] [322-324] The results obtained show that the only surface intermediates formed from alkanes are zinc–alkyl fragments (Fig. 11a).[322] It is important to note that, according to 13C MAS NMR data, n-propyl and n-butyl fragments are formed from propane and n-butane. This indicates the activation of only the methyl groups of alkanes with the participation of zinc sites. This conclusion is confirmed by the results of a study of the hydrogen–deuterium (H/D) exchange reaction between C3 – C4 alkanes and BASs of Zn/H-BEA and Zn/H-ZSM-5 zeolites:[328-331] the exchange is regioselective, and only the methyl groups are involved (see Fig. 11b).[328]
![[{"id":"Z1_lKiK_9k","type":"paragraph","data":{"text":"<sup>13</sup>С MAS NMR spectra of propane-1-<sup>13</sup>С and propane-2-<sup>13</sup>С on Zn<sup>2+</sup>/H-BEA zeolite after transformation at 473 K (<i>а</i>). <sup>1</sup>Н MAS NMR spectrum of propane-d<sub>8</sub> on Zn<sup>2+</sup>/H-BEA zeolite in situ at 423 K (<i>b</i>). The symbol • denotes the position of the <sup>13</sup>C atom in the hydrocarbon moiety. The Figure contains a fragment of a Figure from a previously published article by the authors.<sup>328</sup>"}}]](/storage/images/resized/dTJDXkplA1zkwHhRv4VsSuzdzuKnvZ44H73XYTFM_xl.webp)
Thus, it can be concluded that the activation of light alkanes on Zn-modified zeolites is indeed accomplished with the participation of Zn-containing sites through the dissociation of the C – H bond via the alkyl pathway, and only the methyl groups are involved. In the case of Zn-modified zeolites, the active species are ions Zn2+ and ZnO oxo-clusters,[322-324] and n-alkyl fragments and hydroxyl Si – O(H) – Al (BAS) or Zn – OH groups are formed (Scheme 12a,b). A study of the properties of [Zn – O – Zn]2+ species by the DFT method has shown[189] that the cleavage of the C – H bond in ethane with the participation of such sites (see Scheme 12c) occurs with a lower activation barrier, but the further transformation of the zinc–ethyl intermediate, on the contrary, is thermodynamically less favorable compared to the reactions involving Zn2+ ions. In addition, according to DFT calculations, the structures of [Zn – O – Zn]2+ type are the least stable form of zinc-containing species in ZSM-5 zeolite.[332][333]
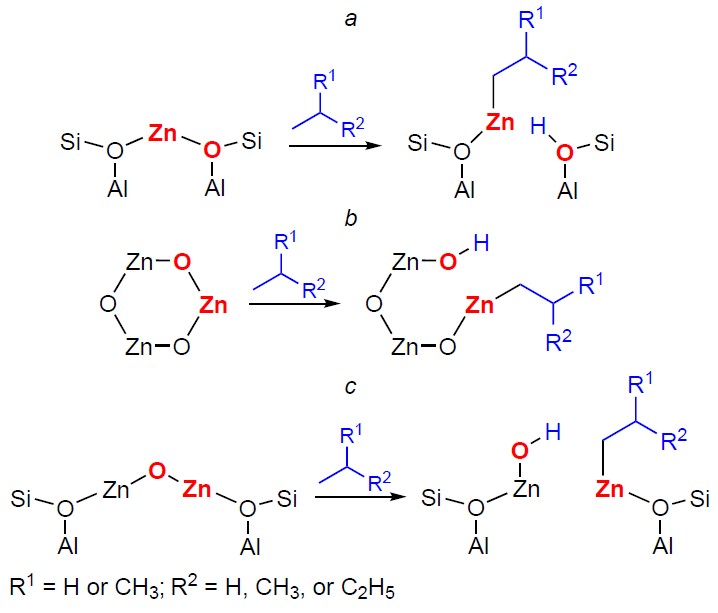
It is important to note that when analyzing the results of the H/D exchange kinetics study for C2 – C4 alkanes with BASs, it has been found[151][328-330] that loading the zeolite with Zn2+ ions and ZnO oxo-clusters significantly increases the reaction rate and decreases the activation barrier (Fig. 12).[328] Since H/D exchange occurs as a result of the reversible cleavage and formation of the C – H (C – D) bond in the alkane, this finding indicates that the bond activation involves not only zinc sites but also BASs, which also play a role in this process. Unfortunately, there is still no clear explanation of the mechanism of hydrogen – deuterium exchange between an alkane and BASs on Zn-modified zeolites. The promoting effect of zinc is attributed to the formation of adsorption molecular complexes of alkanes with Zn2+ sites, in which the C – H bond is polarized, resulting in the alkane molecule being more easily protonated by the BAS.[131][328][330][334] In other words, the activation of the C – H bond is the result of the joint action of the paired Zn – O site and the BAS. Alternatively, the exchange reaction is preceded by the dissociation of the C – H bond in the adsorption complex on the paired Zn – O site, forming zinc–alkyl intermediates that interact with the BASs (Scheme 13).[151][322][329] In 2025, Lashchinskaya et al.[335] attempted to clarify this issue using quantum chemical calculations (by the example of methane) and concluded that the second option is being realized.
![[{"id":"EH6V8ZdpxY","type":"paragraph","data":{"text":"Arrhenius plot for the H/D exchange reaction of the methyl (○) and methylene (□) groups of propane-d<sub>8</sub> with BASs on H-BEA and Zn<sup>2+</sup>/H-BEA zeolites. The Figure contains a fragment of a Figure from a previously published article by the authors.<sup>328</sup>"}}]](/storage/images/resized/IFbEWEM62UxfjaZG3diUx8Esbjo956gsU4uF0dfi_xl.webp)
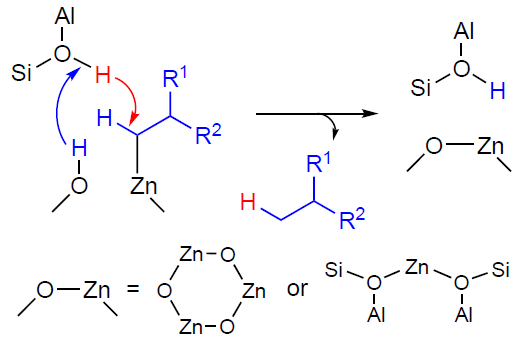
Further transformation of light alkanes, namely, the zinc – alkyl intermediates formed therefrom, occurs via dehydrogenation into alkenes. Based on 13C MAS NMR and IR data, it was suggested that the dehydrogenation step occurred sequentially (Scheme 14a): 1) elimination of the hydride ion from the β-carbon atom (position C-2) in the zinc – alkyl fragment to form an alkene molecule; 2) recombination of H+ and H– ions from the surface – OH and –Zn – H groups to form H2.[147][322][336][337] An alternative pathway is a one-step dehydrogenation via recombination of the H atom at the β-carbon atom of the zinc–alkyl moiety with H+ from the adjacent –OH group (see Scheme 14b). Note that in both cases, the –OH group involved in the transformation is initially formed as a result of the dissociative adsorption of the alkane on the paired Zn – O site (see Scheme 12a,b). A comparison of the two pathways by the DFT method has shown[189] [338] that single-step dehydrogenation is thermodynamically and kinetically more favorable, and the Zn – H species detected by the FTIR method are[189] a product of the dissociative adsorption of H2 molecules on the paired Zn – O sites, which is observed already at a relatively low temperature (≥373 K).[141][339-341]
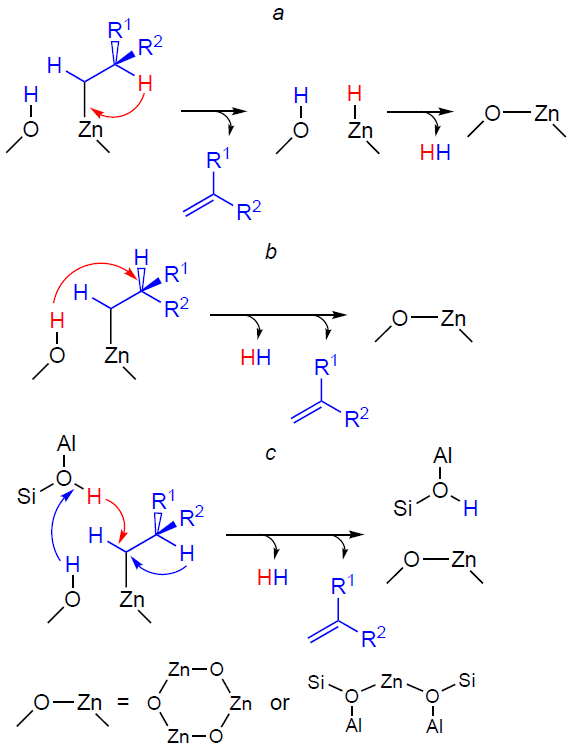
The results discussed above, obtained for the H/D exchange involving alkanes,[151][328-330] indicate that BASs, which protonate zinc – alkyl intermediates, are located near Zn2+ ions and ZnO oxo-clusters in zeolites. Furthermore, the exchange reaction is regioselective for the methyl groups of alkanes; in other words, BASs selectively protonate zinc – alkyl fragments at the α-carbon atom (position C-1) (see Scheme 13). Taking these data into account, it was proposed that the observed transformations of alkanes, in particular their dehydrogenation, involved both zinc sites and BASs.[152] [324][329] Therefore, the dehydrogenation reaction pathway can be described as follows: H+ in the BAS (Si – O(H) – Al group) attacks the C – H bond at the α-carbon atom, and the hydride transfer from the β-carbon atom occurs simultaneously (see Scheme 14c).
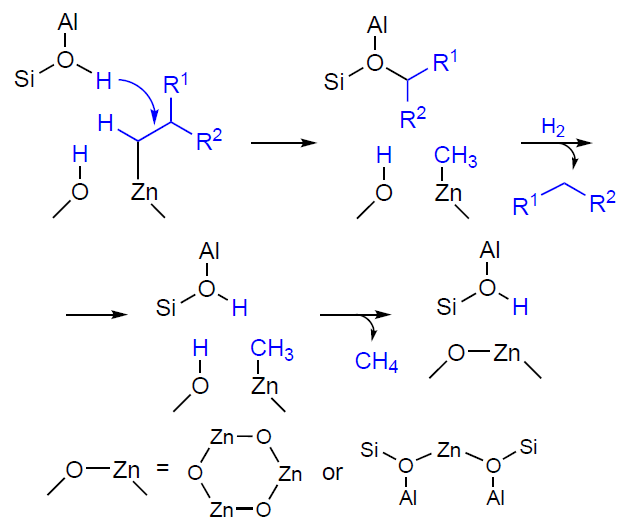
It is interesting to note that loading zeolites with zinc, according to the available data,[23][25][27][29][146][147] promotes not only the dehydrogenation of the starting alkane, but also the hydrogenolysis, which occurs yielding by-products, methane and ethane. Some authors[27][35] have suggested that the main source of C1 – C2 alkanes is protolytic cracking. However, the analysis of the products and kinetics of the transformation of C2 – C4 alkanes by 13C and 1H MAS NMR in situ shows[146][152][153][322][323][327] that the nature of the observed transformations indicates the occurrence of the direct hydrogenolysis as the main pathway for methane formation. The hydrogen sources required for hydrogenolysis are the dehydrogenation and aromatization steps. If the alternative pathway, protolytic cracking (see Scheme 2), were to occur, the 13C MAS NMR spectra would show a characteristic 13C label transfer in the starting alkane molecule,[93][126][342-344] and the products of the transformation would be an alkane and an alkene, e.g., methane and ethylene in the case of propane, or methane and propylene in the case of butane. For Zn-modified zeolites, the 13C label transfer was not detected,[322-324] and C3 – C4 alkenes and C1 – C2 alkanes were identified as the primary products of the transformation of C3 – C4 alkanes, simultaneously. Thus, the conversion of C3 – C4 alkanes on Zn-modified zeolites proceeds in two parallel routes: dehydrogenation and direct hydrogenolysis (see Fig. 10). The mechanism of the hydrogenolysis of C3 – C4 alkanes on the paired Zn – O sites with the participation of BASs was proposed (Scheme 15).[152] [322] The key step involves the protonation of the C – C bond in the zinc – alkyl intermediate to generate Zn – CH3 and an alkoxy species, the desorption of which from the zeolite surface with the participation of hydrogen formed during the dehydrogenation step affords methane.
Data on the kinetics of C3 – C4 alkane transformation on Zn-modified zeolites beta[322][323][327] suggest the existence of a reaction pathway that enables methane transformation under non-oxidative conditions in the presence of higher alkanes. A characteristic feature of alkane conversion is that, at the initial moment, the amounts of products formed do not correspond to the stoichiometry of the hydrogenolysis reaction. For example, butane hydrogenolysis produces propane and methane, according to reaction (6):
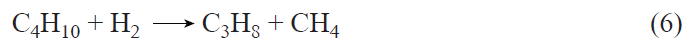
However, it was found experimentally that significantly more propane was formed than methane (Fig. 13a).[323] This observation was explained by the presence of a pathway for methane consumption, i.e., its conversion, according to the gross reaction
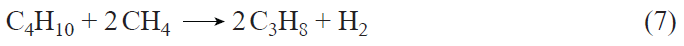
![[{"id":"c7hiJ5wcvI","type":"paragraph","data":{"text":"Kinetics of butane conversion on ZnO/H-BEA zeolite at 553 K <i>in situ</i> (<i>a</i>): kinetic curves for butane, propane, ethane, and methane. Experimental curves are indicated by dots, and simulated curves are indicated by solid lines. <sup>13</sup>C MAS NMR spectra of methane and butane or methane-<sup>13</sup>C and butane on Zn<sup>2+</sup>/H-ВЕА zeolite after conversion at 523 K (<i>b</i>). The Figure contains a fragment of a Figure from a previously published article by the authors.<sup>323</sup>"}}]](/storage/images/resized/WDGVMQqDAmsaNRhgi8LT29roSavVoZAACRrBpJwI_xl.webp)
which formally represents a complex process of co-conversion of butane and methane, yielding two propane molecules. Gabrienko et al.[323] also provided additional evidence in favor of this suggestion. A comparison of the 13C MAS NMR spectra of the mixtures of methane and butane or 13CH4 and butane after their adsorption and reaction on Zn2+/H-BEA zeolite (see Fig. 13b) showed[323] that the products of 13C-enriched methane and butane contained a greater number of 13C atoms, including alkanes, propane (16.4 and 17.0 ppm) and ethane (5.9 ppm). This directly indicates that methane is involved in the conversion with butane to give C2 – C3 alkanes.
The mechanism of the co-conversion of methane with alkanes on Zn-modified zeolites was studied.[200][245] It was established that the reaction involved surface methoxy species, which were formed from methane and further methylate the aromatic products of alkane conversion (see Section 4). For the formation of methoxy species from methane, as described above (see Fig. 5, Scheme 8), the presence of oxygen (oxidative conditions) is necessary. According to the studies,[322][323][327] co-conversion is also possible under non-oxidative conditions; however, it delivers different products. Arzumanov et al.[322] suggested that the methane conversion pathways under oxidative and non-oxidative conditions differ. Under non-oxidative conditions, the reaction between methane and alkane involves surface Zn – CH3 species (Scheme 16), which are formed either directly from methane (see Scheme 7) or during the hydrogenolysis of C3 – C4 alkanes (see Scheme 15). Activation of the C – C bond in the alkane can occur on BAS and afford a methoxy species, which quickly recombines with Zn – CH3 . It should be noted that, given its practical importance, the mechanism for the co-conversion of methane with higher alkanes under non-oxidative conditions requires further research, particularly using quantum chemistry methods. Understanding the pathway of this process could facilitate the development of effective methane conversion catalysts based on Zn-modified zeolites.
5.2. In-modified zeolites
According to available data,[154] In-modified zeolites exhibit the highest activity and selectivity for the conversion of light alkanes to BTX compared to zeolites containing other metals. However, in contrast to, e.g., Zn-containing zeolites, the catalytic properties of In-containing zeolites for the conversion of С2 – С6 alkanes have been studied significantly less.[51][52][154-158][345-347] A limited number of studies[52][345-355] have also been devoted to investigating the mechanisms of the activation and transformation of С2 – С4 alkanes on In-modified zeolites. These studies utilized various methods for loading indium into zeolites: ion exchange and incipient wetness impregnation with an In(NO3)3 solution, mechanical mixing with In2O3 or InCl3, and solid-state exchange with In(NO3)3 in an oxidative or reductive atmosphere. These methods lead to the different states of indium in zeolite and, consequently, different catalytic properties of the resulting In-modified zeolite. As a result, the literature contains conflicting conclusions regarding the activity of various types of In-containing species and the alkane conversion pathway.
The properties of In – Y zeolite loaded with indium by the ion exchange method were studied.[345] Based on the results of quantum-chemical calculations and analysis of the state of indium sites by the FTIR method, the [InOH]2+ site, stabilized on the zeolite framework by two Si – O––Al sites, was selected as the active one. The authors concluded that the activation and transformation of ethane occurs via heterolytic dissociation of the C – H bond on the In – O paired site, the oxygen atom of which is a part of either the extra-framework O – H ligand (Scheme 17a) or the Si – O––Al bridged fragment (see Scheme 17b). The initial step of C – H bond cleavage is characterized by an activation energy of 96 and 59 kJ mol–1 for the first and second variants, respectively, whereas the subsequent steps of ethylene and H2 formation have activation barriers of the order of 120 – 170 kJ mol–1. Moreover, the catalytic experiments on ethane conversion on In – Y were conducted at a temperature of 973 K, which appears excessively high given the relatively low activation energies. It should also be noted that, prior to the catalytic tests, the In – Y zeolite was calcined in air at 993 K, which could have caused the dealumination of the zeolite framework and removal of surface hydroxyl groups. In this regard, one can express doubts about the correctness of the choice of the active site model for carrying out DFT calculations, which probably does not correspond to the real state of indium species in the zeolite modified by the ion exchange method.
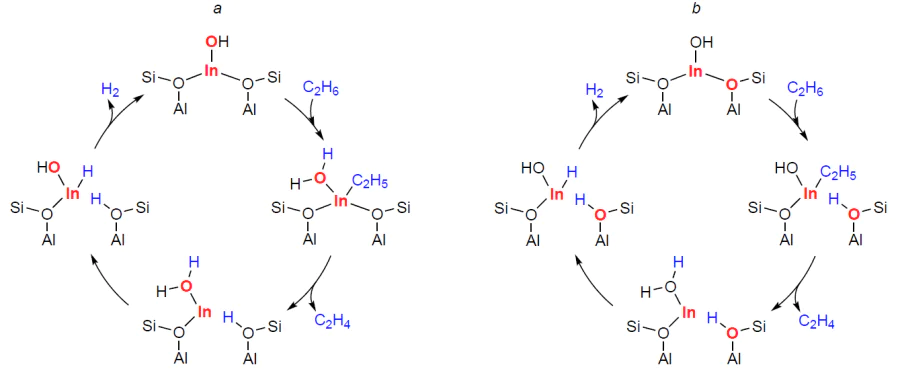
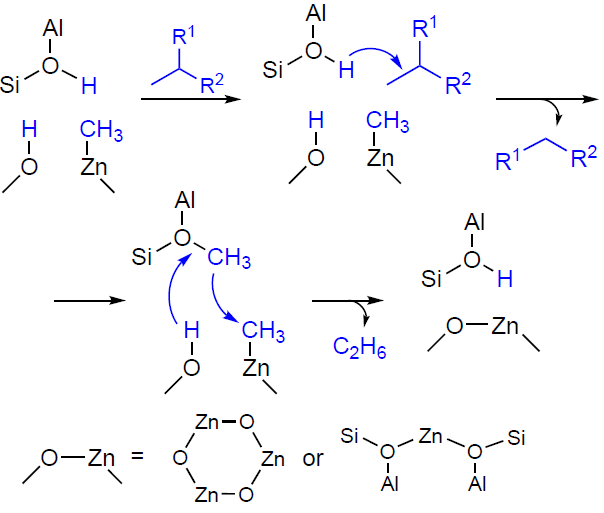
It has been shown that the reductive treatment of a mixture of ZSM-5 zeolite and indium(III) oxide increases BTX selectivity and alkene yield in the conversion of С3 – С6 alkanes.[156][157] Accordingly, it has been suggested that In2O species, In+ ions, and [InH2]+ ions are also capable of effectively dehydrogenating alkane.[52][156][157] Based on the results of studying the conversion of ethane on In-CHA after the reductive treatment with H2 , Maeno et al.[52] came to the conclusion that the active species are the [InH2]+ ions, and not In+ or [InH]2+ ones, and that the dehydrogenation of ethane occurs via a concerted (Scheme 18a) or alkyl mechanism (Scheme 18b). However, the activation enthalpies calculated by the DFT method for these two processes were 230 and 280 kJ mol–1, respectively, indicating that such reactions are kinetically prohibited. In addition, ethane conversion on In-CHA was observed at temperatures of 933 – 973 K, while FTIR and XANES techniques provided only indirect evidence of ethane interaction with In-containing sites, which also casts doubt on the authors’ conclusions about the activity of the reduced or hydride forms of indium. It can be assumed that, at such a high temperature, the conversion of ethane occurs with the participation of residual BASs, as demonstrated in a study.[349] The transformation of ethane and propane on In-CHA after treatment with H2 has also been studied.[346, 347] The authors proposed In+ ions as the active sites, which activate alkanes by oxidative addition to give the intermediate [alkyl – InH]+ (see Scheme 18c), while the In+ site changes the oxidation state to In3+. The high activation energy of such a process (250 kJ mol–1), as well as the fact that the conversion of alkanes occurs at a temperature of 873 K, do not allow, as in the previous case, to agree with the authors’ hypothesis about the possibility of such a pathway for activating alkanes.
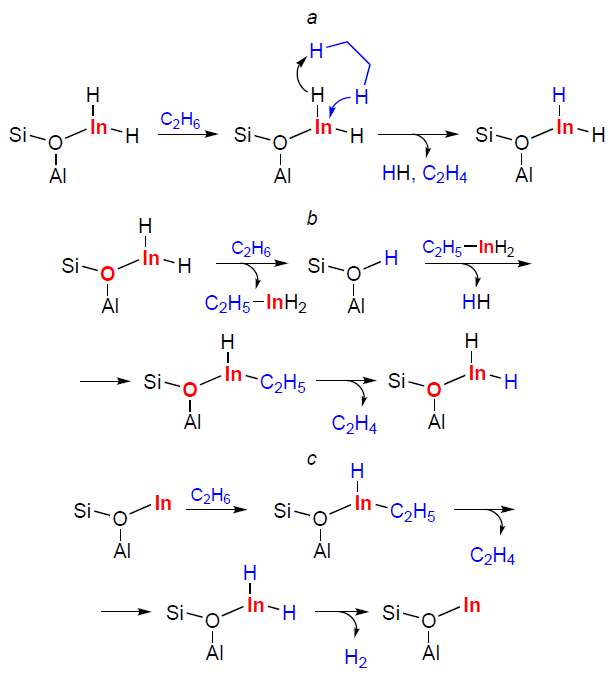
It has been established that the ground state of indium in zeolites obtained by mixing them with In2O3 followed by the reductive treatment are In+ ions.[159-165] In addition, it has been shown [159][160][162] that indium(I) cations are easily oxidized by oxygen to oxo-ions [In=O]+ or [In2(m-O)2]2+ oxo-clusters in the presence of paired cation-exchanged Si – O– – Al sites, for example, in CHA zeolite.[166] A comparison of the properties of In+ and [In=O]+ ions with respect to propane transformation has shown[349] that the first type of sites is incapable of activating alkane at a temperature of 298 – 723 K (a conversion involving BASs was observed), whereas, in the presence of the second type sites, propane conversion occurs at 423 K (in the case of butane and isobutane, at 296 K[351][353]). This result clearly demonstrates the significantly higher activity of oxidized forms of indium and refutes the conclusions made in the works[52][346][347] regarding the activity of the reduced or hydride forms of indium.
The mechanisms of the activation and transformation of С2 – С4 alkanes on ZSM-5 and beta zeolites modified with [In=O]+ ions were studied by FTIR, 13C MAS NMR, and DFT methods.[349][351][353][355] It is shown that the activation of alkanes results in the formation of n-alkyl species, for example, n-propylindium, which has characteristic signals at 19.1 (–СН3), 20.2 (–СН2–), and 25 (In – СН2–) ppm (Fig. 14a).[349] Thus, as in the case of alkane transformation on Zn-containing zeolites, only methyl groups are activated. In this case, dissociation of the C – H bond can be accomplished in two ways, which are described by reactions (8) and (9) (Z– = Si – O––Al, n = 2 – 4):
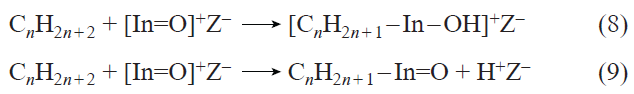
![[{"id":"AkuZFjYfjo","type":"paragraph","data":{"text":"<sup>13</sup>С MAS NMR spectra of propane-1-<sup>13</sup>С and propane-2-<sup>13</sup>С on InО<sup>+</sup>/H-ВЕА zeolite after transformations at 423 (<i>а</i>) and 573 K (<i>b</i>). The symbol • denotes the position of the <sup>13</sup>C atom in the hydrocarbon moiety. The symbol * denotes a spinning side band."}}]](/storage/images/resized/GK9j8KyqzpFVcAzbLxgXqIg6kYSy3Orn2hKKUc6G_xl.webp)
According to the FTIR data,[351][353] In – OH hydroxyl groups are formed simultaneously with n-alkyls. For example, during the conversion of butane, a νOH band at 3450 cm–1 is observed in the IR spectra (Fig. 15a).[351] This indicates that the dissociation of the C – H bond in alkanes corresponds to equation (8). Quantum-chemical calculations for isobutane transformation, performed using the DFT method, confirm this conclusion (see Fig. 15b):[353] path A, corresponding to equation (8), is kinetically and thermodynamically more favorable compared to path B (equation (9)). Along with alkyl species, alkenes are detected in the IR spectra (see Fig. 14a and Fig. 15a),[349][351] and in the case of butane and isobutane already at a temperature of 296 K. Based on these data, a mechanism for the activation and dehydrogenation of alkanes was proposed, which for С2 – С4 alkanes can be represented by Scheme 19.[349][351][353][355]
![[{"id":"QYJ6gEFcxu","type":"paragraph","data":{"text":"FTIR spectrum of butane after transformation on InО<sup>+</sup>/H-ZSM-5 zeolite at 296 K (<i>а</i>). Possible pathways for the dissociation of the C – H bond in the methyl group of isobutane on [In=O]<sup>+</sup> oxo-ions in ZSM-5 zeolite (<i>b</i>): pathway A corresponds to equation (8), and pathway B corresponds to equation (9)."}}]](/storage/images/resized/7ip6TQGqEg60Kgzs3kzhTAFZyqpVJ7A3h8H1ZuXF_xl.webp)
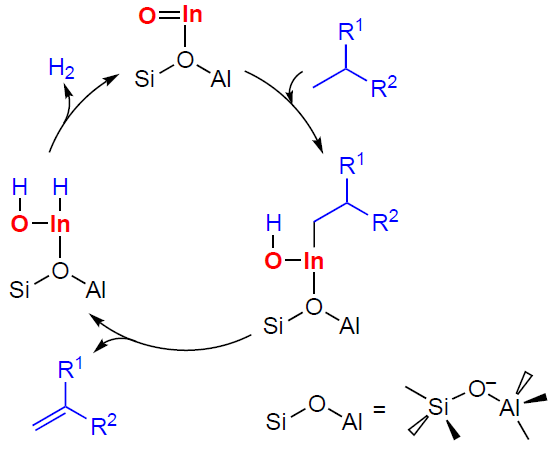
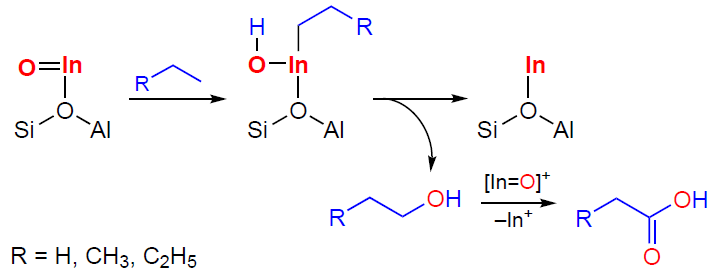
It has also been shown[349][351][353][355] that In-containing zeolites are capable of oxidizing С2 – С4 alkanes to carboxylic acids: the 13C MAS NMR spectra show signals of surface С2 – С4 carboxylate ions, e.g., at 186 ppm (COO–) and 31 ppm ( – CH3 group of the acetate ion CH3COOIn=O) (see Fig. 14b).[349] These ions are converted into the corresponding acids upon water addition. It is interesting to note the following results, which suggest a mechanism for the oxidation of alkanes. Using 13C MAS NMR, it was shown that during the transformation of propane-1-13C, the 13C label is selectively transferred to the methyl group of the acetate ion and simultaneously to the methyl group of toluene (see Fig. 14b),[349] and, according to GC-MS data, acetic acid and toluene are formed in equal quantities. Propionic acid is also formed, but to a lesser extent. The butane oxidation affords acetic acid (or surface acetate) as the main product. Propionic acid is also formed, with butanoic acid being formed in significantly smaller quantities than С2 – С3 acids. All this indicates that the reaction proceeds in two pathways: direct oxidation of the alkane, associated with the activation of the C – H bond in the methyl group (Scheme 20), and indirect oxidation, which results from the activation of the C – C bonds (Scheme 21). It should be noted that, based on the ratio of the amounts of acetic and propionic acids formed from butane, the pathway via the dissociation of the internal C – C bond predominates. The terminal C – C bond is activated by [In=O]+ sites less effectively. Noteworthy, isobutane or the surface intermediates formed during its dissociation are not directly oxidized (the main oxidation products are butanoic and С2 – С3 acids),[353] apparently due to steric hindrance experienced by the corresponding hydrocarbon species in the limited pore volume of the zeolite.
5.3. General patterns of alkane transformation
This Section discusses the results of the studies devoted to the properties of zeolites modified with Zn and In, as applied to the activation and transformation of С2 – С4 alkanes. The conversion of light alkanes over zinc-modified zeolites containing Zn2+ ions or ZnO oxo-clusters involves several key steps, viz., dehydrogenation to alkenes, parallel hydrogenolysis to form С1 – С2 alkanes, oligomerization of alkenes, and aromatization of oligomeric unsaturated hydrocarbons. The dehydrogenation and hydrogenolysis steps are carried out due to the combined action of the paired Zn – O sites and zeolite BASs, while the key role is played by the surface zinc-n-alkyl species formed as a result of the activation of the C – H bond in the methyl groups of alkanes. It has been found that Zn2+ ions exhibit greater activity compared to ZnO oxo-clusters.[322][323][327] The results of studies of In-modified zeolites indicate that In+, [InH2]+, and [InH]2+ ions are not active. On the other hand, [In=O]+ oxo-ions are capable of effectively activating alkanes for their subsequent transformations, which include parallel reactions of dehydrogenation to alkenes and partial oxidation to carboxylic acids. Furthermore, it has been found[349][351][353][355] that the transformation of light alkanes does not afford С1 – С2 alkanes. This is a distinctive feature of such catalysts compared to unmodified and Zn-modified zeolites. Dehydrogenation occurs through the activation of the C – H bond in the methyl groups of alkanes, forming [CnH(2n + 1) – In – OH]+ (n = 2 – 4) surface species as intermediates. It is noteworthy that dehydrogenation of alkanes is observed even at low temperatures (296 – 298 K). This points to high activity of the zeolites containing [In=O]+ sites. Oxidation of alkanes involves the C – H and C – C bond activation and can proceed in two pathways. Direct oxidation of alkanes is possible, resulting in the corresponding carboxylic acids (propane to propionic acid, butane to butanoic acid), as well as indirect oxidation, which involves the cleavage of C – C bonds in alkanes and gives acetic acid as the main product. This reaction pathway involves various ways of alkanes dissociation, followed by oxidation of the resulting hydrocarbon fragments.
6. Oligomerization and aromatization of alkenes
Light С2 – С4 alkenes, by-products of liquefied natural gas and naphtha processing, are readily available and inexpensive feedstocks for the chemical industry. Furthermore, as discussed in Sections 3 and 5, alkenes are the intermediates in the conversion of light alkanes over zeolite catalysts to oligomeric alkenes and aromatic hydrocarbons. The incorporation of metals such as zinc, gallium, nickel, etc. into zeolites improves the conversion of alkenes and selectivity for dimeric products and BTX, and decreases the yield of С1 – С2 alkanes.[91][356-363] This indicates a change in the mechanism of alkene transformation compared to that on unmodified zeolites. However, the role of metal-containing sites in the transformation of alkenes remains unclear, since in most cases there are no experimental (e.g., spectroscopic) data on the key surface intermediates formed during the oligomerization and aromatization of alkenes on metal-modified zeolites.[57] Conclusions regarding the mechanisms of these reactions are primarily based on quantum chemical calculations, which, as noted above, do not always provide adequate predictions of possible hydrocarbon transformation pathways. This Section discusses current experimental and theoretical results obtained in the studies of the mechanisms of С2 – С4 alkene transformation on zeolites modified with Cu, Zn, Ag, and In.
6.1. Formation of alkene π-complexes
The promoting effect of metal-containing sites can be explained by their ability to specifically interact with alkene molecules. The presence of such interaction is confirmed by FTIR data,[130][351][364-375] according to which a significant shift in the ν(C=C) vibration frequency is observed during the adsorption of С2–С4 alkenes on Cu-, Zn-, Ag-, and In-modified zeolites. As an example, we can cite the results for propylene adsorbed on ZSM-5 zeolites (Fig. 16),[90][366][368] which contain cationic sites of various nature: BASs, copper, zinc, and silver ions. These results indicate the formation of π-complexes between propylene and cationic sites: ν(C=C) = 1653 cm–1 for gaseous alkene, whereas ν(C=C) = 1545 – 1595 cm–1 in the case of adsorption on metal-containing zeolites. The mechanisms of such complex formation were studied[372][374][376] using FTIR and DFT methods, in which a correlation was found between the value of the shift in the vibration frequency ν(C=C) and the heat of adsorption of the alkene on the cationic site. However, it has also been shown[376] that there is no universal mechanism for the formation of π-complexes, since it is determined not only by the contributions of σ- and π-bonding, but also by the structure of the cation orbital shell, as well as the properties of individual cation orbitals. The observed π-complexes of alkenes are stable: if alkenes intensively oligomerize on H-form zeolites already at low temperatures (≤296 K),[28][77][124][377] then the transformation of alkenes on Zn-, Cu-modified zeolites is observed starting from a temperature in the range of 423 – 573 K,[366][368] and for Ag-containing samples the initiation of the reaction occurs only at 623 – 673 K and above.[90][180] Thus, the adsorption of alkenes on metal-containing cationic sites is thermodynamically more favorable compared to BASs,[130][353] which leads to the stabilization of alkene molecules and the prevention of rapid and non-selective oligomerization involving carbenium ions (see Scheme 4).
![[{"id":"C3qNGLYkDf","type":"paragraph","data":{"text":"FTIR spectra of propene adsorbed on H-ZSM-5 (<i>a</i>), Cu<sup>2+</sup>/H-ZSM-5 (<i>b</i>), Zn<sup>2+</sup>/H-ZSM-5 (<i>c</i>), Ag<sup>+</sup>/H-ZSM-5 (<i>d</i>) zeolites at 296 K."}}]](/storage/images/resized/SwghuX2RTrepZDPPOt4iqpWloCwBoxmYMnRo8nAh_xl.webp)
6.2. Mechanism of alkene oligomerization
In early studies[358][361] it was assumed that alkene oligomerization on Zn- and Cu-modified zeolites occured at metal-containing sites yielding allylic intermediates, which were later detected by 13C MAS NMR and IR spectroscopy in the case of С3 – С4 alkenes.[130][149][152][366][368][371] Allyl species were also observed during the transformation of С3 – С4 alkenes on Ag- and In-containing zeolites.[90][351][353][378] The 13C MAS NMR spectra of the H-ZSM-5 zeolite presented in Fig. 17,[90][366] clearly demonstrate that the nature of the alkene (propene) transformation changes, e.g., upon the incorporation of Zn2+ ions into the H-ZSM-5 zeolite. This observation indicates a change in the transformation pathway. Propene actively oligomerizes on H-ZSM-5 zeolite already at 296 K, and with increasing temperature it is converted into aromatic hydrocarbons and alkanes with the intermediate formation of CPCs (see Fig. 17a), which corresponds to the conjunct polymerization mechanism (seeScheme 5).[116-119] Zn-modified zeolites (see Fig. 17b,c),[130][366][371] as well as zeolites containing Ag+ or Cu2+ ions,[90][368] are characterized by relatively slow oligomerization of alkenes at 298 – 523 K, with metal–allyl species and delocalized carbanion species formed as intermediates. Thus, alkene molecules prefer to interact with metal-containing sites rather than BASs, forming π-complexes and allyl structures.
![[{"id":"_yVPKg_5Pf","type":"paragraph","data":{"text":"<sup>13</sup>С MAS NMR spectra of propene-3-<sup>13</sup>С (<i>a, b</i>) and propene-2-<sup>13</sup>С (c) on H-ZSM-5 (<i>a</i>) and Zn<sup>2+</sup>/H-ZSM-5 (<i>b, c</i>) zeolites after transformation at 296 – 673 K. The symbol • denotes the position of the <sup>13</sup>C atom in the hydrocarbon moiety. The symbol * denotes a spinning side band."}}]](/storage/images/resized/aZSHXgpS2aWddiOIwN3POXDr9voSzkomD64JAy6o_xl.webp)
The results of the studies[90][130][366][368][371] allow us to describe the mechanism of С3 – С4 alkene oligomerization by the general Scheme 22. The key steps of the transformation are the alkene adsorption on the cationic site or metal (Zn, Cu, Ag) oxo-cluster to give a π-complex, the dissociation of the C – H bond in the allyl position on the paired M – O site resulting in a metal-allyl intermediate, which can have the structure of either σ-allyl or π-allyl, the growth of the hydrocarbon chain due to the coordination and incorporation of a second alkene molecule into the M – C bond in the metal – allyl with the double bond opening. This mechanism was first proposed[366] for Zn-containing beta and ZSM-5 zeolites by analogy with the properties of organozinc compounds[379-381] and the generally accepted mechanism of polymerization on various organometallic catalysts.[382]
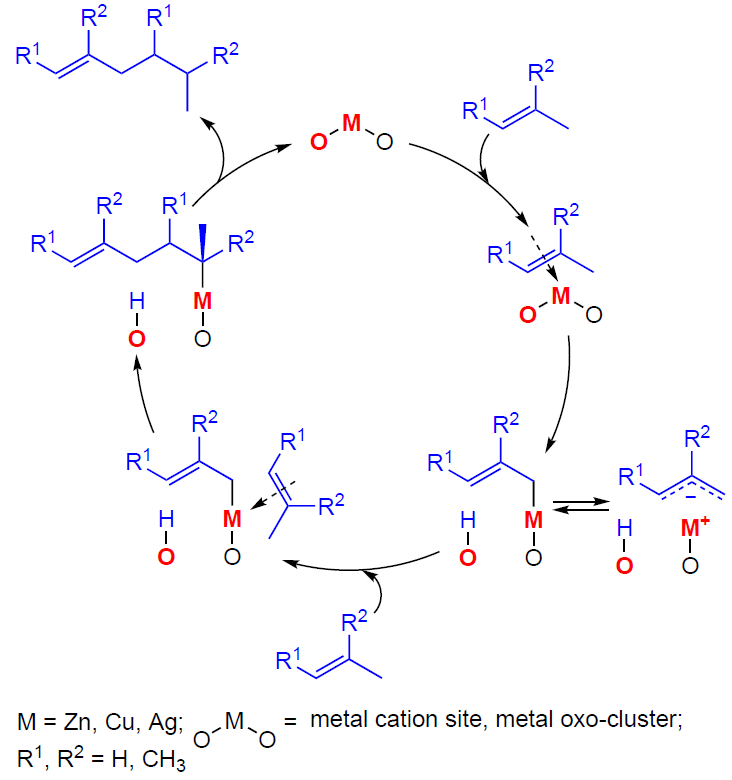
The transformation of С3 – С4 alkenes on In-modified zeolites is of special attention. According to the results of spectroscopic studies,[349][351][353] oligomerization occurs involving both [In=O]+ and BAS. This follows from the nature of the observed intermediates, which include alkoxy species, CPCs, π-complexes of alkenes with [In=O]+, as well as indium–allyl species. DFT calculations for isobutene[353] indicate that the adsorption of alkenes on [In=O]+ oxo-ions is, on average, slightly more favorable than the adsorption on BASs. In this case, alkenes, being adsorbed on In-containing zeolites, can interact simultaneously with both [In=O]+ and BAS. A mechanism of the oligomerization involving [In=O]+ sites has been proposed, similar to that of zeolites containing Zn, Cu, and Ag, in which the stage of the C – H bond dissociation in the allyl position is described by Scheme 23.[351][353]
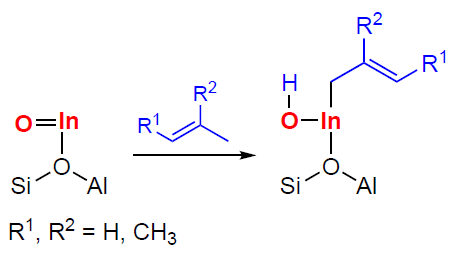
The oligomerization of ethylene on metal-modified zeolites should be considered separately. There is no allyl position in the ethylene molecule, so the oligomerization pathway described for С3 – С4 alkenes (see Scheme 22) cannot be realized for it. The transformation of ethylene on Ni-, Ga-, and Zn-modified zeolites has been studied in the works,[237][370][383-391] but conclusions about the possible mechanism were based primarily on the results of quantum chemical calculations. It is assumed that ethylene oligomerization involves metal-containing sites, which, upon interaction with the alkene, form surface alkyl,[383-386] vinyl,[387][388] or metallacyclic[237][389] species, although none of these intermediates has yet been reliably confirmed experimentally. This issue is discussed in detail in our review article.[57]
One example of the detection of surface hydrocarbon species during ethylene transformation is given in the study[370] devoted to NMR and IR spectroscopic studies of Zn2+/ZSM-5 zeolite. It has been found that, firstly, such a catalyst selectively converts ethylene to butene at a temperature of 296 – 473 K and, secondly, the dimerization intermediate is but-3-en-1-ylzinc (Fig. 18). According to quantum chemical calculations, this hydrocarbon fragment can be formed in two ways. The first is the dissociative adsorption of an ethylene molecule on a paired Zn – O site, forming a vinylzinc species (Scheme 24a), followed by the insertion of a second ethylene molecule into the Zn – C bond of the vinylzinc. The second pathway involves the adsorption of two ethylene molecules on a single Zn2+ ion, followed by the synchronous opening of C=C bonds and the formation of a bridged –O – (CH2)4–Zn– intermediate, the deprotonation of which leads to but-3-en-1-ylzinc (see Scheme 24b). Desorption of but-3-en-1-ylzinc delivers butene. The proposed mechanism is the only example that has both spectroscopic and quantum-chemical confirmation.
![[{"id":"9mu8lbFf4l","type":"paragraph","data":{"text":"<sup>13</sup>С MAS NMR spectra of ethylene-<sup>13</sup>С<sub>1</sub> on Zn<sup>2+</sup>/ZSM-5 zeolite after transformation at 296, 423 K (<i>a</i>). The symbol • denotes the position of the <sup>13</sup>C atom in the hydrocarbon moiety. The symbol * denotes a spinning side band. FTIR spectra of ethylene adsorbed on Zn<sup>2+</sup>/ZSM-5 zeolite (<i>b</i>) at 296, 373 K. Structure and spectral characteristics of the ethylene dimerization intermediate (<i>c</i>)."}}]](/storage/images/resized/NIGX7LMXRpgoW3PPODldFjTSgfUg8PbDzuciC8yh_xl.webp)
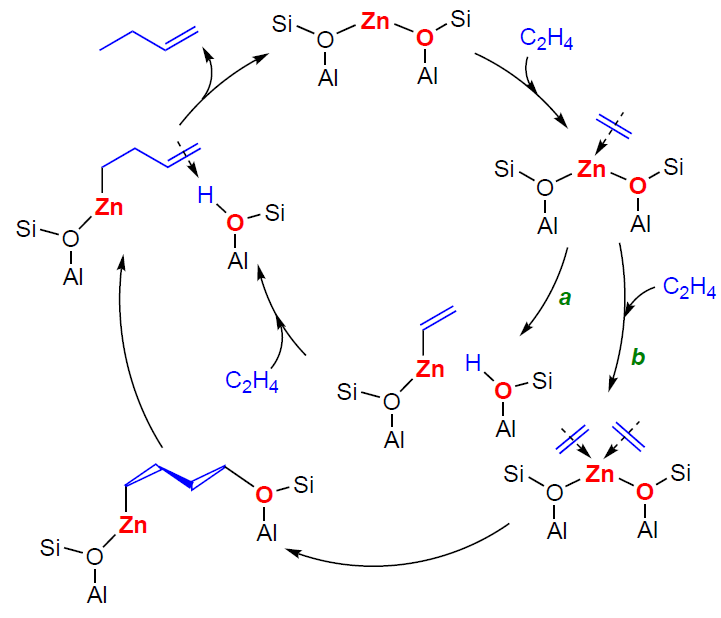
6.3. Mechanism of alkene aromatization
Oligomeric alkenes formed from С2 – С4 alkenes are further converted into aromatic hydrocarbons, mainly benzene and toluene. One possible variant of such a transformation is the aromatization of oligomeric alkenes with the participation of zeolite BASs (see Scheme 5) as discussed in a study.[363]However, in this case, the intermediates in this transformation will be various carbenium ions, including CPCs, and alkanes will inevitably be formed as products (Fig. 17a),[90] which is not observed when using zeolites modified with Cu, Zn, Ag (Fig. 17b,c).[90][130][366][368][371] The spectroscopic data are fully consistent with the results of a study of the catalytic properties of H-ZSM-5 and Zn-ZSM-5 zeolites for butene conversion.[392] It has been found that the introduction of zinc into the zeolite leads to a significant increase in the yield of BTX and, simultaneously, a decrease in the amount of the resulting С1 – С2 alkanes. Ono et al.[392] explained these results by the involvement of zinc sites in the aromatization reaction and suggested that zinc – allyl intermediates should form on the catalyst surface. The latter were subsequently detected by 13C MAS NMR and FTIR techniques,[130][149][152][322-324][366][371] not only at the early stages of the transformation but also in the final steps, when they exist in the form of delocalized carbanionic (polyene) species. The key role of allyl species is also emphasized in publications[356][361] [393] devoted to other metal-modified zeolites.
Taking into account the fact that delocalized carbanionic (polyene) species were detected by spectroscopic methods on the surface of Ag- and Cu-modified zeolites,[90][368][378] it is logical to propose a general mechanism for the aromatization of alkenes with the participation of metal-containing sites (Zn, Ag, Cu) and the detected intermediates (Scheme 25), the details of which are discussed in the works.[90][130][324][371] The main steps of this mechanism are the sequential dehydrogenation reactions of oligomeric alkene on paired M – O sites, forming allylic and delocalized carbanionic (polyene) species as surface intermediates. Dehydrogenation of hydrocarbon intermediates can occur via hydride ion elimination, according to the mechanisms discussed in Section 5 (see Scheme 14a,b).
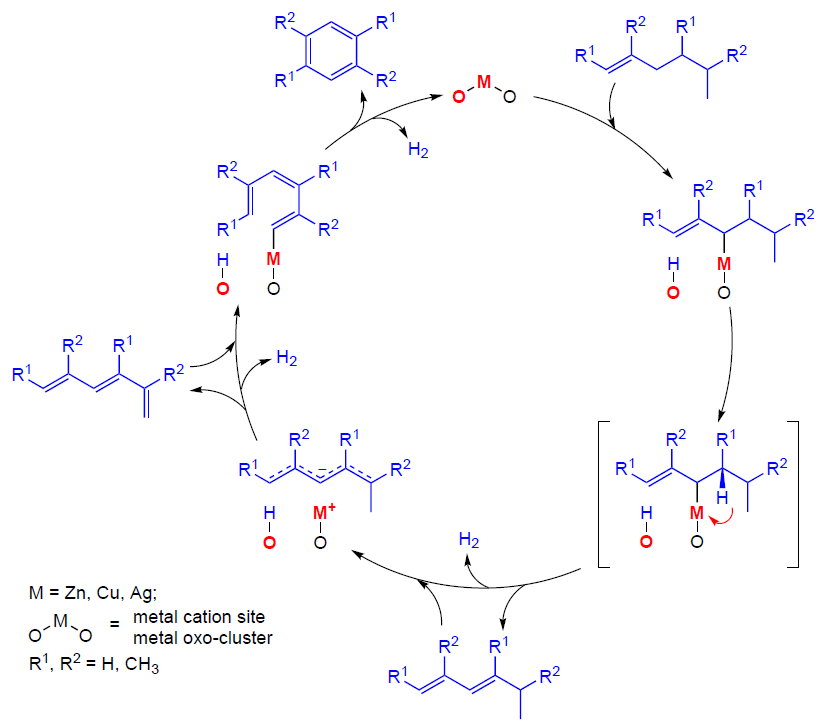
Studies [349][351][353][355] describe the formation of allyl and delocalized carbanion species, as well as alkoxy species and CPCs, during the conversion of С2 – С4 alkenes over In-modified zeolites. Interestingly, C1 – C2 alkanes are not formed in this process. While the detection of CPCs and alkoxy species supports mechanisms involving BASs (see Scheme 3,Scheme 4,Scheme 5), this contradicts the fact that alkanes are not formed in this process. Apparently, the aromatization of oligomeric alkenes occurs via the pathway described for Cu-, Zn-, and Ag-containing zeolites (see Scheme 25), but the resulting intermediate polyene hydrocarbons can desorb from the [In=O]+ sites and interact with BASs, leading to their protonation followed by cyclization, resulting in the formation of CPCs. However, the contribution of BASs to aromatization is minimal. Accordingly, the dehydrogenation steps of oligomeric hydrocarbons involve the dissociation of C – H bonds on the [In=O]+ sites (see Scheme 23).
6.4. General patterns of alkene transformation
This Section presents and analyzes the results of the studies on the properties of zeolites modified with Cu, Zn, Ag, and In, as applied to the activation and conversion of С2 – С4 alkenes. It has been convincingly demonstrated that the introduction of metal-containing species into the zeolite alters the mechanism of alkene oligomerization and aromatization compared to the transformation on unmodified zeolites (H-form). In the case of modified zeolites, the key intermediates were shown to be π-complexes of alkenes with Mn+ ionic sites, σ- and π-allyl species formed by the dissociation of the C – H bond in the methyl group of alkenes on paired M – O sites, as well as delocalized carbanionic (polyene) species. It was found that the adsorption of alkenes on metal-containing sites is thermodynamically more favorable than on BASs (except for In sites), and the resulting π-complexes with cationic species are stable at elevated temperatures. This leads to their transformation involving the metal-containing sites rather than BASs, which, inter alia, explains the absence of undesirable C1 – C2 alkanes in the conversion products. It was found[130][366][368][371] that the properties of cationic sites (Cu2+, Zn2+) and oxide-like species (ZnO, [Cu3(m-O3)]2+) were similar in terms of the alkene transformation mechanism, with the exception that π-complexes with oxide-like species may be less stable, which is expressed in the greater involvement of BASs in the transformations.
Oligomerization of С3 – С4 alkenes involving metal-containing species occurs through the dissociation of the C – H bond at the allyl position on the paired M – O site to generate a metal – allyl intermediate. The next step is the subsequent insertion of a second alkene molecule into the M – C bond in the allyl species with the opening of the double bond. Desorption of the resulting hydrocarbon fragment yields an oligomeric alkene with a single C=C bond. Ethylene oligomerization also involves metal-containing sites, although this has only been experimentally demonstrated for Zn-modified zeolites. Two possible pathways for this reaction are the formation of a zinc-vinyl intermediate via dissociation of the C – H bond on the paired Zn – O site, followed by the insertion of a second alkene molecule into the Zn – C bond, or the formation of a bridged intermediate, Zn – (CH2)4 – O, from two ethylene molecules.
The aromatization of oligomeric alkenes is also mediated by metal-containing sites. The pathway of this process involves the dissociation of the oligomeric alkene on the paired M – O site to form an allyl moiety. Subsequently, this hydrocarbon intermediate undergoes sequential dehydrogenation by eliminating hydride ions, forming diene and triene structures and molecular hydrogen. The surface form of the unsaturated hydrocarbon fragments is delocalized carbanionic (polyene) species, whose negative charge is distributed over five carbon atoms. Dissociation and dehydrogenation of the triene intermediate produce aromatic hydrocarbons (BTX).
7. Conclusion
This review demonstrates that important fundamental results have been obtained to date describing the mechanisms of metal-containing zeolite action for the activation and conversion of light alkanes and alkenes, including methane. The analysis of the available data demonstrates both similarities and differences in the properties of species containing Group 11 – 13 metals, namely Cu, Zn, Ag, and In, incorporated into zeolites. The obtained information is highly relevant for the development of approaches to the rational design and control of the catalytic properties of metal-modified zeolites for the efficient processing of hydrocarbon feedstock.
To the question of whether metal-containing species are involved in the activation and transformation of light alkanes and alkenes, one can confidently give a positive answer. A pool of experimental, spectroscopic, and catalytic data reliably demonstrates that the hydrocarbon transformation mechanism changes when zeolites are modified with Cu, Zn, Ag, and In. The observation of various metal – hydrocarbon surface intermediates as well as calculations of possible transformation pathways using quantum chemistry methods provide a direct indication of how the interaction of alkane and alkene molecules with metal-containing sites occurs, leading to their activation and transformation. A summary of the available data allows us to state that the active species are paired M – O sites in the composition of cationic sites or metal oxo-clusters, which perform the dissociation of the C – H bonds in methyl, allyl, and vinyl groups. Furthermore, paired M – O sites are capable of dehydrogenating alkanes and alkenes, oligomerizing and aromatizing alkenes. However, it should be noted that some of the results described in the review (e.g., the H/D exchange data) indicate the possibility of the joint action of M – O sites and BASs in activating and dehydrogenating alkanes. Therefore, it remains unclear whether the so-called synergistic effect occurs in this case and what its nature is.
An important conclusion from the presented analysis of the literature data is that unambiguously interpretable results can only be obtained using a combination of methods (FTIR, 13C and 1H NMR, DFT). Authors of future studies should be cautioned against relying solely on the results of quantum chemical calculations when establishing the mechanisms of zeolite catalyst action. As shown in this review, theoretical methods often provide only possible scenarios for a catalytic reaction, but not all of these will be realized under experimental conditions. Therefore, it is important to support such results with spectroscopic data.
In the field of zeolite catalysis, the authors believe the following areas of further research are promising. As shown in Section 3, methane activation involving Cu-, Zn-, Ag-, and In-containing sites delivers metal-methyl and methoxide species on the zeolite surface. Therefore, it is of interest to study the properties of zeolites modified simultaneously with various metals, which may exhibit activity in the C – C coupling reaction, opening a new avenue for efficient methane processing. Fundamental research in this area will allow us to test this hypothesis. From the point of view of applied research, it is necessary to study the properties of metal-modified zeolites in relation to the joint conversion of methane with higher hydrocarbons, benzene and alkanes, as well as СО2, СО, О2, H2O, etc. The results presented in Section 3 on the reactivity of the surface intermediates formed from methane indicate the promise of this research area.
To improve the efficiency of С2 – С4 alkanes and alkenes conversion, i.e., to reduce methane yield, further fundamental research is needed using modern methods utilizing in situ and operando modes (NMR, XPS, FTIR, etc.). Specifically, the kinetic parameters and mechanisms of the dehydrogenation and hydrogenolysis steps, which, according to available data, occur in parallel with the participation of metal-containing and Brønsted acid sites, require clarification. Is it possible to direct alkane conversion primarily through the dehydrogenation pathway? Under what reaction conditions and catalyst composition will the contribution of the methane-alkane co-conversion reaction be maximized? Answering these questions will enable a rational approach to the development of a catalyst for the conversion of С2 – С4 alkanes and alkenes to BTX.
In conclusion, it should be noted that Cu, Zn, Ag, and In-modified zeolites are undoubtedly promising catalysts for the conversion of light alkanes and alkenes into valuable chemicals. However, further applied research in this area is required to develop industrial catalysts. The results of fundamental research, already published and to be obtained in the future, could provide a reliable foundation for rapidly achieving this practical goal.
This work was supported by Russian Science Foundation (Grant No.22-13-00029-П). In part of data collecting on alkane and alkene transformation on Cu-, Zn-, Ag-modified zeolites, the work was performed in the framework of the governmental assignment for Boreskov Institute of Catalysis (Project FWUR 2024-0032).
8. List of abbreviations and designations
BAS — Brønsted acid site,
BTEX — benzene, toluene, ethylbenzene, and xylenes,
BTX — benzene, toluene, and xylenes,
CPC — cyclopentenyl cation,
DFT — Density functional theory,
EXAFS — Extended X-ray absorption fine structure,
FTIR — Fourier transform infrared spectroscopy,
GC-MS — gas chromatography–mass spectrometry,
LAS — Lewis acid site,
MAS — magic angle spinning,
NMR — nuclear magnetic resonance,
XANES — X-ray absorption near edge structure,
XPS — X-ray photoelectron spectroscopy.
References











![A [Cu2O]2+ core in Cu-ZSM-5, the active site in the oxidation of methane to methanol](/storage/images/resized/mxFdPe9qujsfvfYfcN0QOclAiYORFb0xrRlwV8gs_small_thumb.webp)




