

Keywords
Abstract
Since its discovery in 1995, the Soai reaction remains the only experimental example of a chemical reaction capable of amplifying enantiomeric excess to a virtually enantiopure state. This review consolidates the results of three decades of research, including data from recent years that have broadened the understanding of the reaction mechanism. Competing catalytic models (dimeric, tetrameric, hemiacetal, and others) are critically reviewed; a broad spectrum of chiral triggers, ranging from circularly polarized light and chiral minerals to cryptochiral isotopomers, is systematized; and the phenomenon of absolute asymmetric synthesis is analyzed. The position of the Soai reaction among other chirality amplification mechanisms and its significance as a model for the origin of biological homochirality are discussed.
The bibliography includes 159 references.
1. Introduction
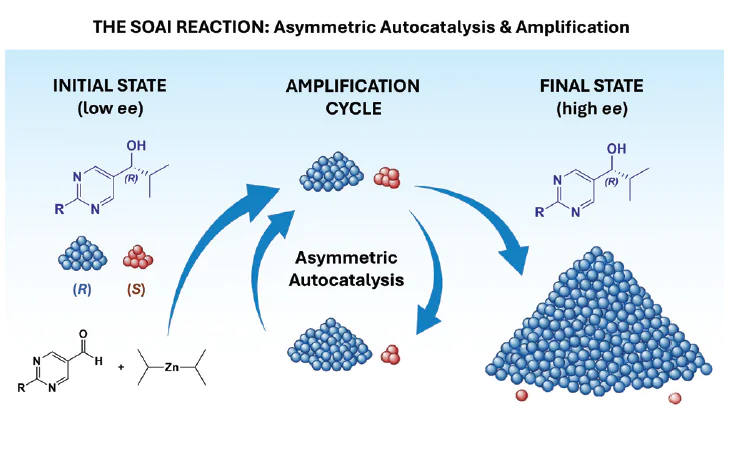
The Soai reaction, a highly efficient autoamplifying asymmetric autocatalysis (AAA) reaction, is observed during the alkylation of heterocyclic aldehydes of a certain structure with diisopropylzinc. The uniqueness of the process lies in the fact that the chiral reaction product catalyzes its own formation, with its enantiomeric excess being higher than that of the original catalyst.
Despite the significance of this discovery, there are only a limited number of comprehensive reviews in the literature. Most of these were prepared by Professor Soai’s research group and are regularly updated with new data. The most comprehensive presentation of current concepts is outlined in the monograph ‘Asymmetric Autocatalysis: The Soai Reaction’,[1] published in 2022 in a limited edition. The first chapter of the book is open access and provides a basic understanding of the AAA phenomenon and the characteristics of the Soai reaction. Publications by other authors generally have a narrower focus and cover individual aspects, viz., the study of the mechanism,[2][3] spontaneous generation of chirality,[4] kinetics of the process[5] or the connection of AAA with the origin of biological homochirality.[6][7]
The uniqueness and elegance of the Soai reaction encourages the systematization of accumulated data in a new format that is broader than existing specialized reviews and more accessible than a fundamental monograph.
The purpose of this review is to achieve this goal. In addition to the AAA phenomenon per se and its applicability (Section 2), and the reaction pathway (Section 3), chiral initiators (Section 4), unusual aspects of asymmetric induction (Section 5), and spontaneous generation of chirality (Section 6) are discussed in detail.
2. Soai reaction and autoamplifying asymmetric autocatalysis (AAA) mechanism
2.1. Scope of application of the Soai reaction
The Soai reaction is feasible only for a narrow range of aldehydes (Scheme 1).[8][9] Pyrimidines 1a,b are the most effective and thoroughly studied substrates. Their diversely substituted analogs 1с – k,[10-15] and a number of pyridine[16][17] and quinoline[18-20] derivatives demonstrate similar reactivity and are also suitable for this reaction. Diisopropylzinc is invariably used as the alkylating reagent,[5][21-23] and toluene is the most suitable solvent.
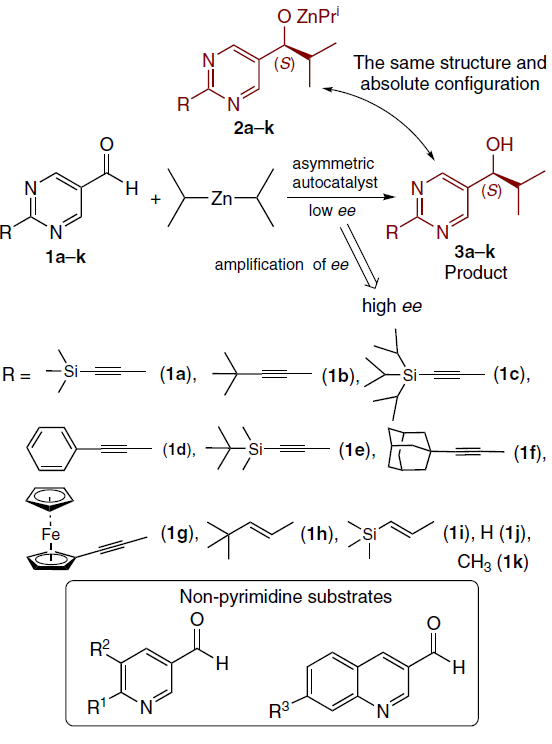
Products with very high ee (≥ 99%) are accessible via a reaction sequence using the product obtained in the previous step as a catalyst (Scheme 2).[10][11][14][15] [24-29]
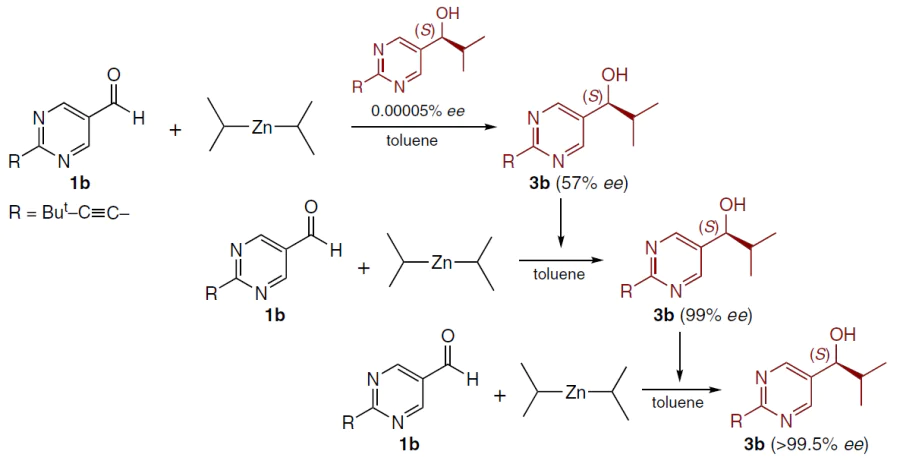
Fig. 1 shows the efficiency of chiral amplification in the Soai reaction. Three successive cycles of asymmetric autocatalysis result in a 630 000-fold increase in the amount of the major enantiomer, yielding a virtually enantiomerically pure product.[24]
![[{"id":"-v82Ymf9Tp","type":"paragraph","data":{"text":"Amplification of enantiomeric excess in asymmetric autocatalysis of compound <b>3b</b>.<sup>24</sup> Copyright Wiley 2003."}}]](/storage/images/resized/3Cnn7gcLRGZOcuKqXMIYgPDfd0FxpNw1MSSNkTh1_xl.webp)
2.2. History of the discovery of the Soai reaction
2.2.1. Theoretical basis and predictions. The Frank’s model
In 1953, Frank[30] proposed a kinetic model that could lead to the emergence of asymmetric autoamplification. The main principles of the Frank’s model are self-replication and mutual inhibition of competing chiral forms.[6] It is assumed that each enantiomer of a chiral molecule is capable of catalyzing its own formation (Scheme 3):
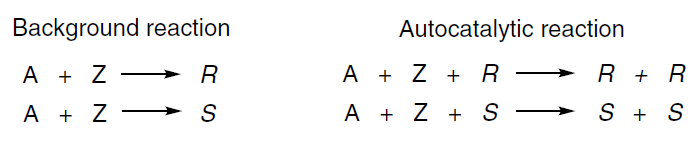
The Frank’s concept of ‘mutual antagonism’ [30] means that each enantiomer can also inhibit the formation of the opposite one. This is achieved by the interaction of the enantiomers to form a kinetically inactive, or less reactive, heterochiral complex (Scheme 4):[31]
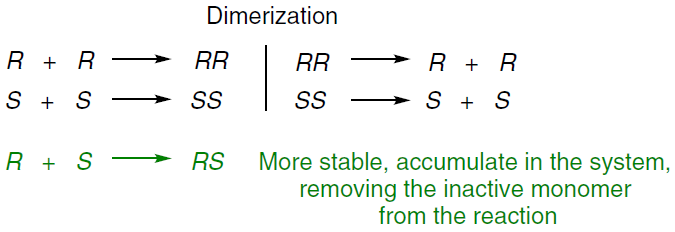
This process effectively excludes the minor enantiomer from the catalytic cycle, allowing the major enantiomer to be self-replicated.
The idea of forming chiral asymmetry in molecules was further developed by Decker,[32] and also Goldanskii and Kuzmin,[33] who demonstrated the importance of oligomerization processes for the possibility of implementing autocatalysis.
2.2.2. Non-linear effects in catalysis
Another important step of the AAA concept development was the discovery by Wynberg and Feringa[34] of the so-called ‘antipodal interaction effect,’ which Kagan and co-workers[35][36] later characterized as ‘nonlinear’. With a positive nonlinear effect, even a small enantiomeric excess of the catalyst yields a product with a higher ee than would be expected from a linear relationship, i.e., amplification occurs. With a negative nonlinear effect, on the contrary, even the use of a nearly enantiomerically pure catalyst leads to a product with a lower enantiomeric excess (Fig. 2).[36-38]
![[{"id":"peRIi4V75w","type":"paragraph","data":{"text":"Nonlinear effects in chemical reactions"}}]](/storage/images/resized/SCVacDR5NSvPSdTL18SUgHeWmgKudEZeV2SJ8BDp_xl.webp)
The combination of the principles of the Frank’s kinetic model with the concept of the nonlinear effect of Kagan gave rise to a more general term to describe the observed phenomena, which was called the ‘reservoir effect’.[36]
2.2.3. Discovery of a real chemical system
At the end of the last century, asymmetric catalytic alkylation of aldehydes with organozinc reagents catalyzed by chiral amino alcohols was actively studied.[22][39][40] Diethyl zinc was most commonly used,[22][41-46] however, in a number of works, other organometallic zinc derivatives containing methyl, n-propyl, isopropyl, n-butyl, n-pentyl, 2-furyl and vinyl groups were also studied.[22]
In 1989, the Soai’s group found that the presence of a nitrogen atom in the aldehyde molecule significantly accelerates the reaction. For example, 3-pyridinecarbaldehyde[47] reacted with diethyl zinc 16 times faster than benzaldehyde (Scheme 5).[45][46]
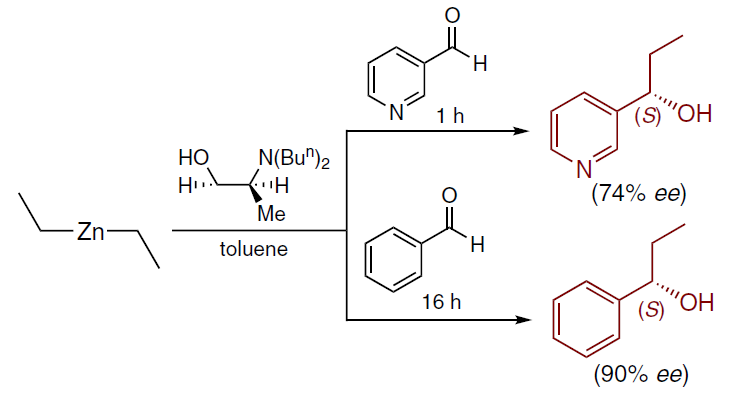
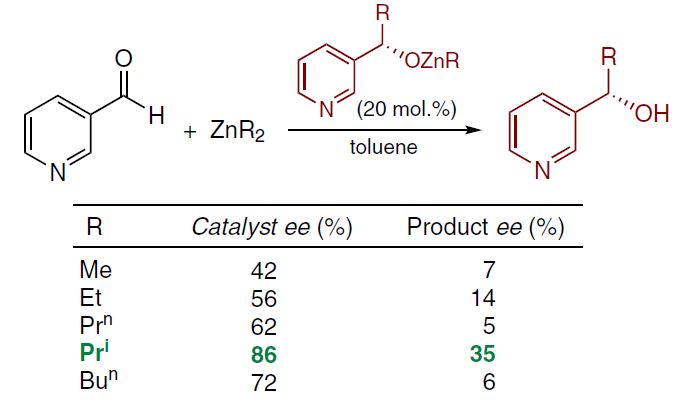
This fact indicated the possibility of autocatalysis, which led to a more in-depth study of the catalytic alkylation of pyridine-3-carbaldehyde[21] with various dialkyl zinc derivatives in the presence of chiral alcohols — the products of enantioselective alkylation. Asymmetric autocatalysis was observed using Me2Zn, Et2Zn, Prn2Zn, Pri2Zn and Bun2Zn as alkylating reagents. The highest enantiomeric excess in the resulting product was observed in the reaction with Pri2Zn (Scheme 6).[21][22]
The next step was to use a substrate with a second nitrogen atom at position 3 relative to the aldehyde group, and in 1995, the AAA in the reaction of diisopropylzinc with pyrimidine-5-carbaldehyde 1j was discovered.[10] Autoamplification, an increase in the optical purity of the newly formed product compared to that of the catalyst, was observed in this reaction (Scheme 7).
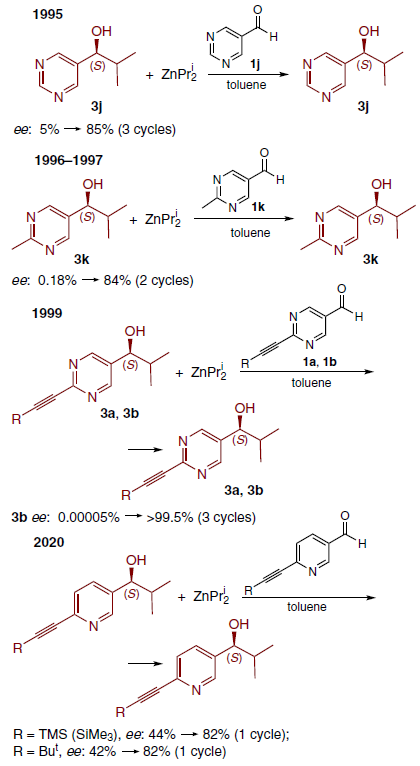
It was subsequently found that in the case of methyl-substituted pyrimidine carbaldehyde 1k, AAA is much more pronounced.[15][48] In 1999, the synthesis of aldehydes 1a,b was described, which still remain the most effective substrates for studying AAA in the Soai reaction.[11] Finally, in 2020, it was shown that pyridine carbaldehydes with similar acetylene substituents also exhibit a high degree of amplification, comparable to that observed for 1a and 1b.[5]
The Soai reaction serves as a benchmark for amplifying asymmetric autocatalysis, the efficiency of which is concentrated around a limited number of substrates and an organozinc reagent. This narrow specificity of the system confirms the fact that the mechanisms of self-replication and mutual inhibition require a strict match between the steric and electronic parameters of the reactants, a feature characteristic of only certain heteroaromatic aldehydes and diisopropylzinc.
3. Studies of the Soai reaction mechanism
Elucidating the mechanism of autocatalytic asymmetric amplification observed in the Soai reaction is an important issue. Numerous publications, which will be discussed in this Section, have been devoted to various approaches to its solution.
However, due to the technical difficulties inherent in these studies (high sensitivity of organozinc compounds to air moisture and oxygen, as well as light, complexity of equilibria between catalyst oligomers, ambiguity of kinetic data), it has rarely been possible to propose a complete catalytic cycle leading to autoamplification and reliably confirm it with experimental and calculated data.
3.1. Experimental and calculated data on the composition of the reaction mixture of the Soai reaction
In general, asymmetric autoamplifying autocatalysis can be described as a combination of a positive nonlinear effect and autocatalysis. As mentioned above, the nonlinear effects are due to the participation of oligomeric species in catalysis. Thus, it could be assumed that the autocatalyst in the Soai reaction is an oligomeric alkoxide formed in the reaction mixture prior to its hydrolytic decomposition.
Accordingly, the very first publication,[49] in which the possible structure of the catalyst using (S)-3k was discussed, suggested that it had the form of a macrocyclic dimer 4k.
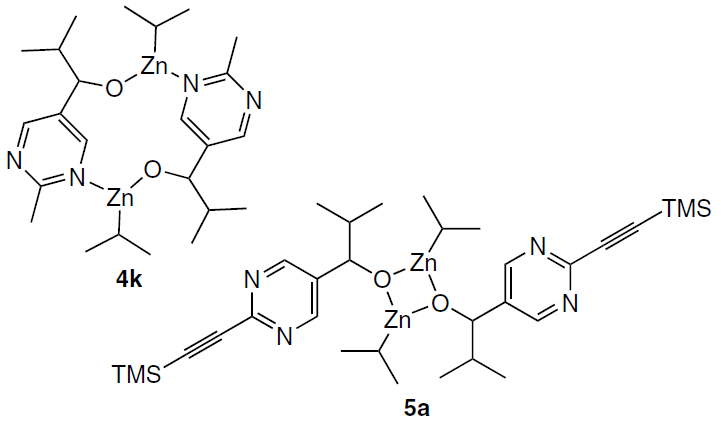
However, experimental and calculated data for (R)-3a reported in follow-up publications[50][51] indicated that the square dimer 5a is the major component of the reaction mixture containing the alkoxides, the products of the Soai reaction, whereas the analogous macrocyclic dimer 4a is approximately 10 kcal mol–1 less stable, so that its concentration in the reaction mixture is negligible.
3.1.1. Kinetic studies of the Soai reaction pathway
A significant number of studies have been devoted to investigating the kinetics of the Soai reaction using microcalorimetry, mass spectrometry, IR, and NMR spectroscopy. It was assumed that determining the reaction order in different reactants would yield useful data on the degree of catalyst oligomerization. However, an analysis of the results presented in Table 1[52-56] reveals that the conclusions regarding the reaction order with respect to different reactants vary significantly. This is likely due to the complexity of the equilibria (or quasi-equilibria) between various oligomers, whose concentrations change in the course of the reaction, as well as the use of simplified kinetic analysis schemes.
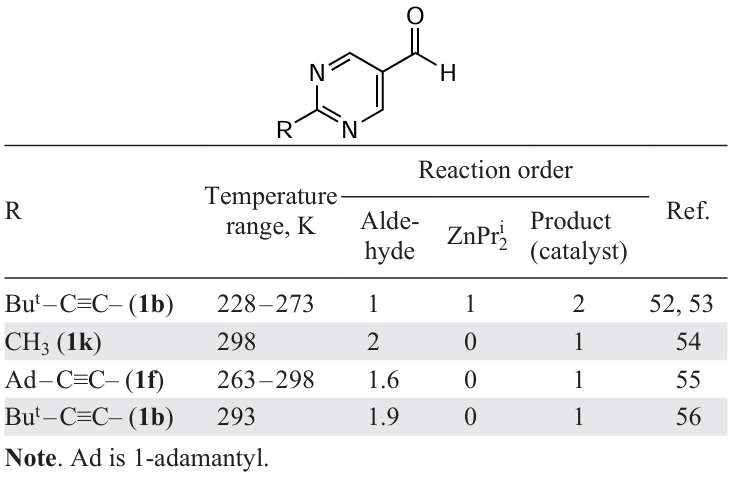
Thus, the zero order of the reaction in diisopropylzinc, which is most frequently encountered in the results of kinetic measurements, is not consistent with the experimental and calculated data on the association constants of diisopropylzinc with aldehyde and oligomers of various structures (see below). At the same time, zero orders in both the aldehyde and diisopropylzinc may indicate that the measurements were carried out under conditions of saturation of the reactive complex with both reagents. Experimental and methodological difficulties in studying the kinetics of the Soai reaction are discussed in detail in the reference.[57]
A more productive approach appears to be modeling the catalytic curves and enantiomeric excess accumulation curves during the reaction based on calculated data. Examples of this approach are given later in this Section.
3.1.2. Study of the Soai reaction mixtures by NMR spectroscopy
The catalyst structure in solution was studied using 1H NMR spectroscopy. Broadened signals at 298 K were observed in the spectra recorded in toluene-d8 (Fig. 3), indicating the presence of dynamic equilibria in the system.[50]
![[{"id":"Dtjd3Zs75H","type":"paragraph","data":{"text":"<sup>1</sup>Н NMR spectra (500 MHz, toluene-d<sub>8</sub>) of homochiral (<i>a</i>) and heterochiral (<i>b</i>) dimeric alkoxide <b>5a</b> at 298 K.<sup>50</sup> Copyright Wiley 2004."}}]](/storage/images/resized/dmcv76NhJbMxdKKpiMw4MimudIIrlwYnlM6oF86D_xl.webp)
Based on these spectral data, some conclusions about the steady-state composition of the Soai reaction can be drawn. For example, the spectra of the chiral and racemic alkoxides differ significantly, clearly ruling out a monomeric structure for the alkoxide. Furthermore, the shape of the pyrimidine proton signals in the spectrum of the racemic alkoxide (d 8.70 – 8.80 ppm) indicates that the concentrations of the homochiral (RR + SS) and heterochiral (RS) dimers are approximately equal. This indicates their similar stability and rules out the possibility of a ‘reservoir effect’ (see Section 2.2.2) as a source of nonlinear effects in the Soai reaction.
More clear 1H NMR spectra of the reaction mixture were obtained in THF-d8 (Fig. 4). Comparison of Fig. 3 and Fig. 4 shows that the only significant difference is the signal broadening in deuterotoluene. In addition, the statistical distribution between the homo-5a(RR) and heterochiral 5a(RS) dimers is conserved. Thus, although the Soai reaction does not take place in tetrahydrofuran, the spectra recorded in this solvent can be used to determine the structure of the major components of the reaction mixture.
![[{"id":"R5ZYfLlrz5","type":"paragraph","data":{"text":"<sup>1</sup>Н NMR spectra (500 MHz, THF-d<sub>8</sub>) of homochiral (<i>a</i>) and heterochiral (<i>b</i>) dimeric alkoxide <b>5a</b> at 298 K.<sup>50</sup> Copyright Wiley 2004."}}]](/storage/images/resized/qMCE36dPmOenntalJSouUtJfEPU5ozARil7HUkHP_xl.webp)
This was achieved by exploiting the symmetry features of 5a(RR) and 5a(RS), which are evident from their spectra. The 1H NMR spectrum of enantiomerically pure 5a(RR) shows four doublets arising from methyls in ZnOCHCHMe2 and ZnCHMe2, indicating that both pairs of methyl groups are diastereotopic. In the spectrum of racemic dimer 5a, containing virtually equal amounts of 5a(RR) and 5a(RS), three additional doublets of methyl groups appear, that corresponds to isochronism of the ZnCHMe2 pair in this case. These data are fully consistent with the structures of 5a(RR) and 5a(RS).
Interestingly, the absence of autocatalysis in tetrahydrofuran (well-resolved signals) and its presence in toluene (broadened signals) indirectly indicate that it is the active catalyst, which is in exchange with 5a(RR) and 5a(RS), that can contribute to the broadening of the lines in the spectra recorded in toluene.
At 353 K, the signals of 5a(RR + SS) and 5a(RS) coalesce (Fig. 5), indicating reversible dissociation of the dimers into a racemic mixture of monomers 2a(R) and 2a(S). The free energy of activation of this process was determined to be 19.1 kcal mol–1. At temperatures of 298 K and above, two dynamic processes leading to the line broadening in the spectra were identified, viz., dissociative exchange between homo- and heterochiral dimers and exchange between the isopropyl groups of the alkoxide and diisopropyl zinc (see Fig. 5, Fig. 6a).[50][58]
![[{"id":"3wlaVdAapx","type":"paragraph","data":{"text":"Determination of the activation barrier for the dissociation of dimer <b>5a</b> (toluene-d<sub>8</sub>) from the temperature dependence of the signal shape of pyrimidine protons.<sup>58</sup> Copyright Springer 2008."}}]](/storage/images/resized/6jFCnY3gIlclBTaQ5Sglcproh8VDS2QlyAQhRbk7_xl.webp)
![[{"id":"9KoslA_0Fx","type":"paragraph","data":{"text":"The <sup>1</sup>H – <sup>1</sup>H EXSY NMR spectrum (500 MHz, toluene-d<sub>8</sub>, 298 K) of a mixture of product <b>3a</b>, (catalyst) of the Soai reactions and Pr<sup>i</sup><sub>2</sub>Zn in a ratio of 1 : 3 (<i>а</i>).<sup>58</sup> Copyright Springer 2008. Cluster intermediate <b>7k</b> (calculated by DFT (B3LYP/6-31G*) for alkoxides <b>3k</b> and Me<sub>2</sub>Zn, used as a simpler model) (<i>b</i>).<sup>50</sup> Copyright Wiley 2004."}}]](/storage/images/resized/DyO32rWye32fxBNhtodmxJ6GwlSAdTLlxMVnYSk1_xl.webp)
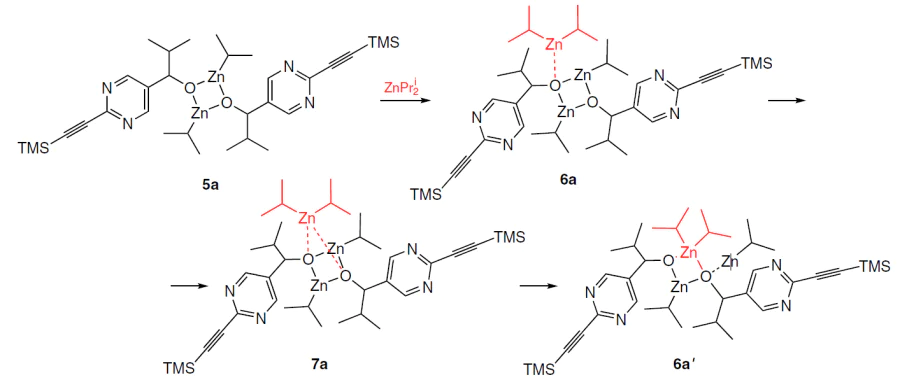
The exchange of isopropyl groups occurs between the alkoxide and diisopropyl zinc in solution (Scheme 8), was evidenced by the EXSY 1H – 1H NMR spectrum (Fig. 6a). According to the calculation results, this process is mediated by a symmetrical trinuclear cluster 7a, in which a small change in the bond lengths in the R2Zn – ZnR unit leads to the observed rearrangement (see Fig. 6b).[50]
To obtain data on zinc binding by the alkoxide and the reagent, corresponding NMR measurements were performed. The binding constant for diisopropylzinc by the alkoxide was determined from the dependence of 13C chemical shifts on the diisopropylzinc concentration at 298 K. Although similar data from 15N heteronuclear multiply bound correlation spectroscopy (HMBC) spectra were obtained at a different temperature and were not subjected to quantitative analysis, the obvious similarity of the chemical shift dependences on the diisopropylzinc concentration indicates that binding occurs almost exclusively at the nitrogen atoms of the alkoxide, and this coordination affects the closely positioned triple bond.
Interestingly, the binding constant of diisopropylzinc measured in tetrahydrofuran (0.4 M–1) is approximately 10 times lower than the value obtained in toluene, which indicates that the inhibitory effect of tetrahydrofuran is associated with the blocking of the coordination centers by this solvent.
The binding constants of diisopropylzinc with aldehyde 1a, measured at 213 K (Fig. 7b), are close to those for alkoxide 2a at 298 K. Consequently, at room temperature, the aldehyde does not compete with the alkoxide for binding the organometallic reagent; the reaction occurs between the aldehyde and associates of the alkoxide with diisopropylzinc.
![[{"id":"esrIvuJm6k","type":"paragraph","data":{"text":"Determination of the binding constants of Pr<sup>i</sup><sub>2</sub>Zn with dimer <b>5a</b> (<i>a</i>) and aldehyde <b>1a</b> (<i>b</i>) in toluene-d<sub>8</sub> by NMR spectroscopy. Copyright Springer 2008."}}]](/storage/images/resized/blz8GNsM8T4AuJI1z2wKhtjdzIhN4NgANiwemPkH_xl.webp)
Also of interest is a fact that can be interpreted as an effect similar to the action of shift reagents. Thus, the signal from the methyl groups of pure Pri2Zn in toluene-d8 is observed at d 1.20 ppm (doublet) with minimal temperature dependence. At the same time, for excess Pri2Zn in the presence of an alkoxide, two signals appear with a pronounced temperature dependence of the chemical shifts, different for each signal (Fig. 8). Given the rapid exchange between coordinated and uncoordinated Pri2Zn, these effects are apparently significantly more pronounced for coordinated molecules.
![[{"id":"LhQMIgk_Dv","type":"paragraph","data":{"text":"A fragment of the <sup>1</sup>H NMR spectrum of alkoxide <b>2a</b> (500 MHz, toluene-d<sub>8</sub> , 0.08 M) with signals from the methyl groups of excess Pr<sup>i</sup><sub>2</sub>Zn (0.04 M) shifted relative to their characteristic position at δ 1.20.<sup>50</sup> Copyright Wiley 2004."}}]](/storage/images/resized/9bPOgUzkJHzs0qQJmFhpJeyJ39leSbbt7AiCgfRh_xl.webp)
Thus, the set of spectral data indicated that dimers 7a are the main component of the Soai reaction mixture, whereas at decreased temperatures they associate to give a tetramer, which, apparently, is the true catalyst.
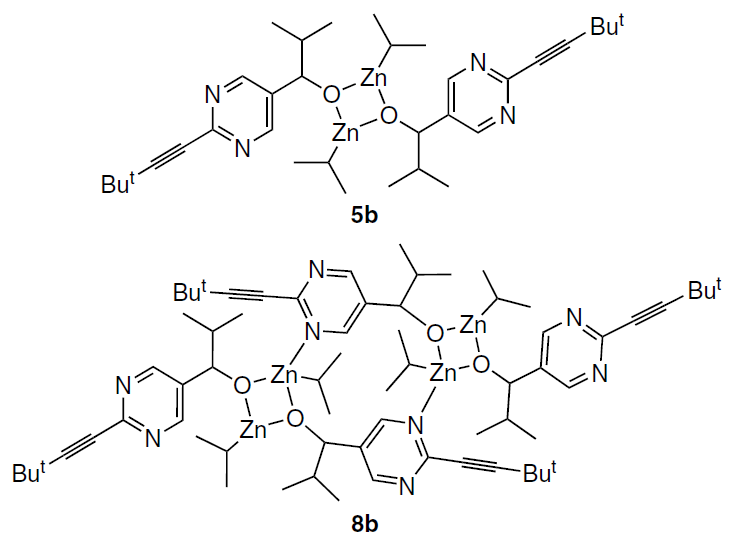
This conclusion was confirmed by a non-obvious nuclear Overhauser effect (NOE) detected for alkoxides 2b in the phase-sensitive 1Н – 1Н NOESY NMR spectrum, which, together with the calculated data, allowed the SMS (Square – Macro – Square) structure of tetramer 8b to be assigned to the species in equilibrium with the square dimer 5b.[59]
In addition, this conclusion was confirmed by diffusion-ordered spectroscopy (DOSY) by comparing the diffusion coefficients measured for the signals observed in the spectrum of the Soai reaction mixture with the values for porphyrin molecules with a molecular mass close to that of 8b.[23][60]
Important information about the catalyst structure was obtained by studying the pronounced temperature dependence of the pyrimidine proton signal in the 1H NMR spectrum of alkoxide 2b (Fig. 9).[2][59] As the temperature decreased to 263 K, only significant signal broadening was observed. At 253 K, the second low-intensity signal appeared, noticeably downfield shifted relative to signal 2b.
![[{"id":"F-U4CjXK-H","type":"paragraph","data":{"text":"Reversible temperature dependence of the signal shape from two pyrimidine protons in the <sup>1</sup>H NMR spectrum (500 MHz, toluene-d<sub>8</sub>) of enantiomerically pure alkoxide <b>2b</b>.<sup>2, 59</sup> Copyright American Chemical Society 2012."}}]](/storage/images/resized/AOVabat0Gr1meg8BqKS37VZQRgOgBLJam3p4QqiN_xl.webp)
With a further decrease in temperature, the shape of both signals becomes more complex; in the range of 193 – 213 K, the ratio of the integrated intensities for the two signals is 1 : 3 (see Fig. 9).[2][59]
These results were interpreted as strong evidence for the formation of tetrameric catalyst 8b in a defined conformation with orthogonally arranged pyrimidine rings (Scheme 9).
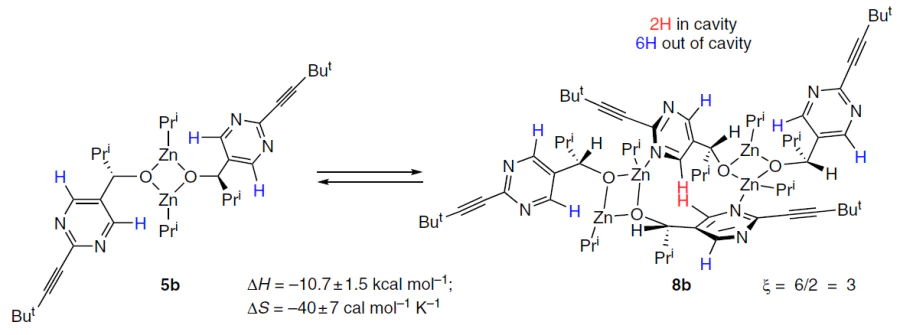
Calculations showed that in this configuration, two of the eight pyrimidine protons are located in a specific cavity, resulting in a significant downfield shift of their signals. A simple observation (Fig. 10) leads to a ratio of 1 : 3 for protons with d 9.5 and d 8.5, which is in good agreement with the experimental data obtained from 1H NMR spectra (see Fig. 9).[2][59]
![[{"id":"7qNqvf4JPu","type":"paragraph","data":{"text":"Calculated spectra of dimer <b>5b</b>, tetramer <b>8b</b> and their complexes with one and two Pr<sup>i</sup><sub>2</sub>Zn molecules.<sup>2, 59</sup> Copyright American Chemical Society 2012."}}]](/storage/images/resized/ba50ixnT5mRGeC0dIGPuRvULDJPgk908uiFVcG9p_xl.webp)
The two-dimensional EXSY (chemical exchange) spectrum shown in Fig. 11 demonstrates the diversity and complexity of exchange processes observed in the Soai reaction mixture at low temperatures. These processes apparently include dimer – tetramer equilibria, equilibria of alkoxides with their Pri2Zn complexes, and (possibly) conformational equilibria.[23]
![[{"id":"8B9J8rQxBQ","type":"paragraph","data":{"text":"A fragment of <sup>1</sup>Н – <sup>1</sup>Н EXSY NMR spectrum (500 MHz, toluene-d<sub>8</sub> , 298 K) of alkoxide <b>2b</b> (98% <i>ее</i>); mixing time 250 ms.<sup>23</sup> Copyright Royal Society of Chemistry 2007."}}]](/storage/images/resized/aljGwjTxBVRg65ulfYoA8p4El3yyPqJG94O9kp5i_xl.webp)
3.2. Identification of minor components of the Soai reaction mixtures
3.2.1. Intermediate acetal formation in the Soai reaction
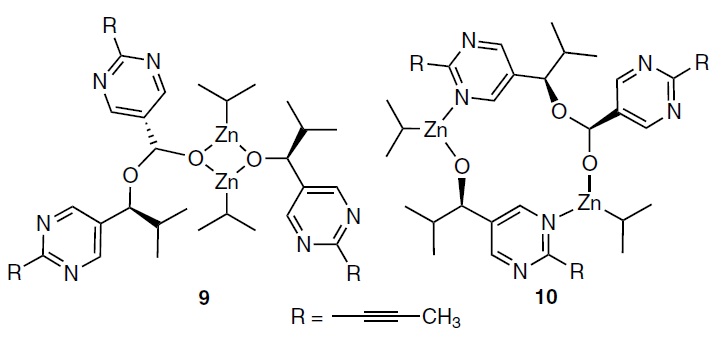
When conducting low-temperature NMR studies, an intermediate was detected, which accumulated during the reaction and was completely decayed by its end at the optimal temperature of 273 K. At the same time, at 253 K, detectable amounts of this intermediate remained in the reaction mixture, which made it possible, based on the data of two-dimensional NMR spectroscopy, to assume the formation of an acetal moiety.[61]
The structure of the observed intermediate could not be fully established. The authors propose several possible structures (9 and 10) that are determined based on calculations and are in line with experimental observations.[61]
3.2.2. The role of the acetal intermediate in the catalytic cycle of the Soai reaction
The possible role of the observed intermediate in the catalytic cycle was subsequently explored.[62] Three questions were addressed:
1. Is the acetal an important intermediate in the Soai reaction? Should any proposed catalytic cycle contain an acetal intermediate?
2. If this is not the case, how can the kinetics of the acetal formation and decay be explained?
3. What are the structure and origin of the acetal?
To answer these questions, an approach that included formal kinetic analysis in combination with DFT calculations of possible intermediates and transition states was used. After modeling of kinetic curves (using thermodynamic parameters obtained by calculation) they were compared with the experimental ones (Fig. 12).[62]
![[{"id":"l1pGaUqDXi","type":"paragraph","data":{"text":"Kinetic curves for aldehyde decay (circles) and acetal formation (squares) at 273 K (<i>a</i>) and 253 K (<i>b</i>),<sup>61 </sup>as well as the results of their simulation (<i>c</i>) and (<i>d</i>). Сopyright Wiley 2012. Conditions: [Ald] = 0.022 M, Pr<sup>i</sup><sub>2</sub>Zn = 0.044 M, starting concentration [P]<sub>0</sub> = 0.0011 M, toluene-d<sub>8</sub>. The red curve shows kinetics of a monomolecular decay with k<sub>d,Ac</sub> = 6.7.<sup>62</sup> Сopyright Chemical Society of Japan 2015."}}]](/storage/images/resized/4PPoDZYwnRndOA5di5y5iN41zWlmA7X3nPPAL3t7_xl.webp)
This approach was applied to reactions involving aldehyde 1b (Scheme 10).[62]
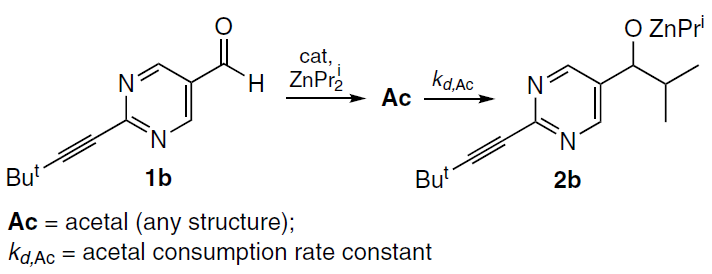
Kinetically, reactions from Scheme 10 can be described by Equations (1) – (5). For the first case, a reaction scheme was used in which the acetal is the precursor to the reaction product.
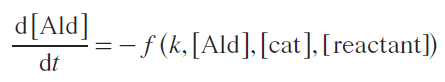
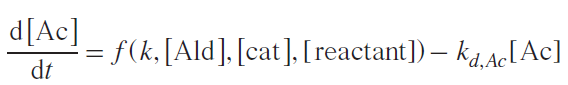
Replacing the first term in the second equation with the left-hand side of the first equation, the Equation (3) was obtained:
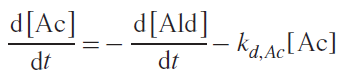
Integration of equation (3) (details are available in the original publication) results in the Equation (4):
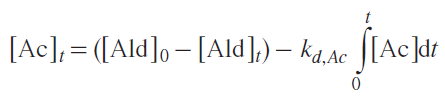
Replacing the integral with a sum leads to Equation (5), which can be used to compare calculated and experimental aldehyde concentrations:
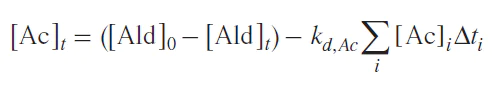
where f (k, [Ald], [cat], [reactant]) is a function in general form describing the formation of acetal from the starting aldehyde, kd,Ac is the rate constant of the acetal decay, [Ald]0, [Ald]t, [Ac]t are concentrations of aldehyde and acetal in the beginning of the reaction and at time t.
Table 2 shows that the experimentally measured running aldehyde concentrations are several orders of magnitude lower than the values calculated by the Equation (4). The low value of the acetal decay rate constant from the consecutive scheme indicates that all the reacted aldehyde must remain in the acetal form for a relatively long time, which is inconsistent with the experimental data. Therefore, it can safely be concluded that the acetal is not formed via a consecutive reaction pathway and is not an intermediate in the catalytic cycle of the Soai reaction. This conclusion holds true for a wide range of acetal decay rate constants, as well as for the bimolecular mechanisms of this process.
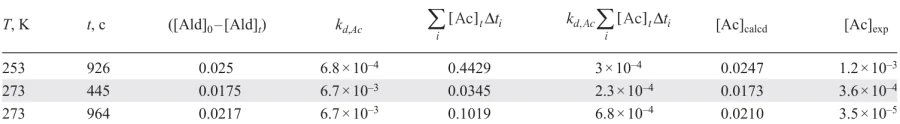
In two other possible schemes for the acetal formation, it is considered a byproduct that is not involved in the catalytic cycle of the Soai reaction. It can be assumed that the acetal (Ac) is formed from the reaction product (P) and the unreacted aldehyde (Ald) (Scheme 11).
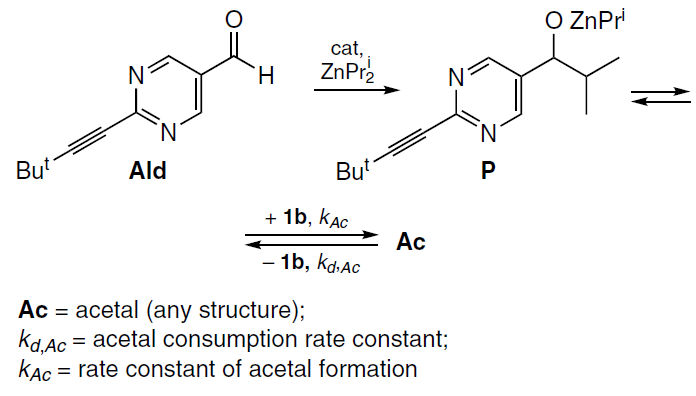
Within this model, the kinetic equation for the bimolecular formation of an acetal can be written as follows:
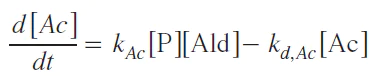
In this case, the decrease in acetal concentration at the end of the reaction occurs due to a decrease in aldehyde concentration.
When the aldehyde concentration reaches its maximum, the right-hand side of Equation (6) becomes zero, leading to an equation for the rate constant of acetal formation, where all concentrations are taken at the point of maximum acetal concentration:
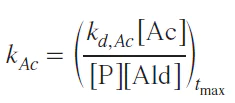
Equation (7) allowed to calculate the rate constants for acetal formation and compare them with experimental data (see Fig. 12). It should be noted that the calculated time of maximum acetal concentration does not correspond to the experimental data.
To further verify these conclusions, calculations were performed for the most probable acetal formation pathway, viz., the reaction of a square dimer with an aldehyde, mediated by the corresponding adduct (Scheme 12). The calculated thermodynamic parameters of the equilibria are summarized in Table 3.
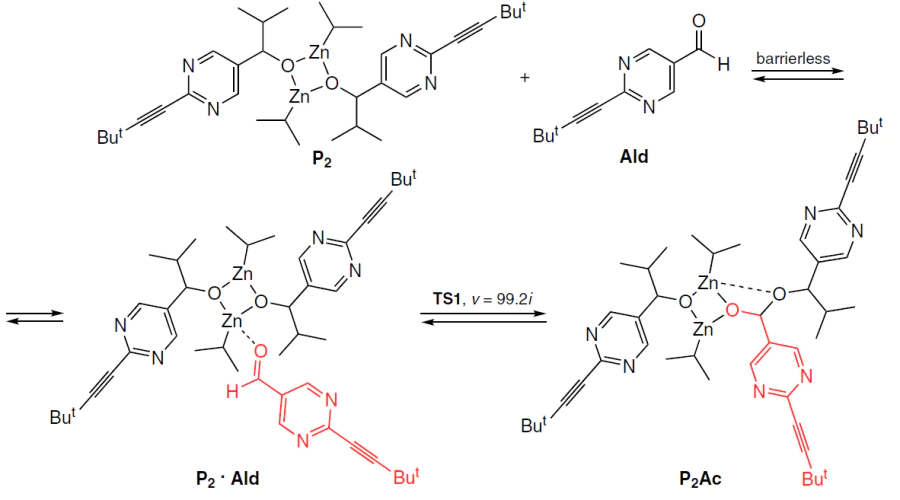

From the data presented in Table 3, it is clear that the formation of acetal from aldehyde and square dimer is thermodynamically forbidden. The rate constants for the corresponding reactions indicate slow formation and rapid decay of acetal. Similarly, the reaction product of the alkoxide monomer with aldehyde exhibits a rapid reverse reaction with regeneration of the aldehyde.
Therefore, the combination of thermodynamic and kinetic data contradicts the proposed acetal formation pathway.
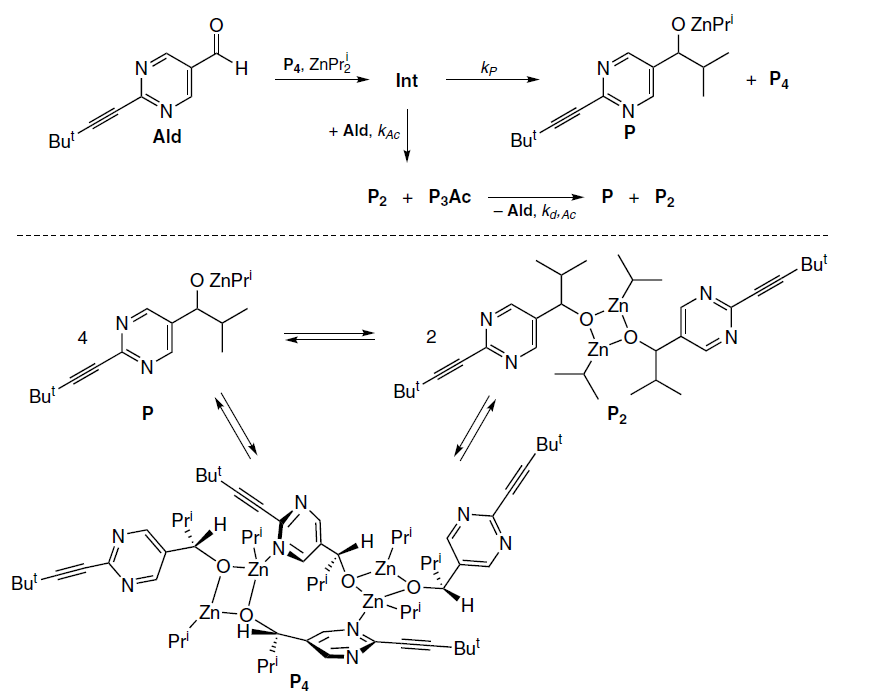
A third possible mechanism was the formation of acetal from an intermediate Int preceding the reaction product P, with the acetal decay affording the same product (Scheme 13).
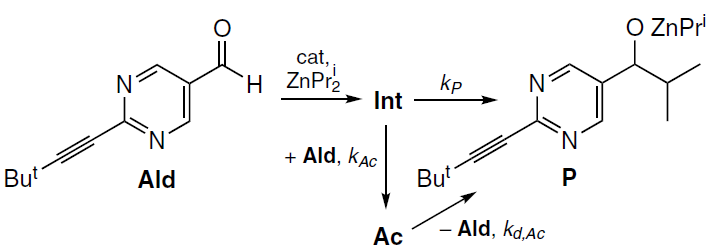
In this case, the rate of acetal formation should be maximum at the moment of maximum rate of aldehyde decay, which is in good agreement with the experimental kinetic data (see Fig. 12). The acetal concentration is determined by the relative values of kP and kAc rate constants.[62]
The acetal behavior in this case must meet the following requirements: it must originate from a catalytic cycle intermediate, yield a reaction product after decay, be thermodynamically unstable, but kinetically stable enough to decompose slowly with an activation energy of ~16 kcal mol–1. In addition, according to experimental data, it must be able to reversibly regenerate the aldehyde.[61] It was assumed that these strict requirements for the possible structure of the acetal would help to determine the most realistic scenario for the formation of the acetal from a chemical point of view.
According to previously published DFT calculations,[23][50][58][59] the aldehyde alkylation product has the structure of a pentameric alkoxide Р4 · Р. Under thermodynamic equilibrium conditions, square dimers Р2 and their associates are the most stable species in the reaction mixture of the Soai reaction, while all ‘odd’ oligomers (trimers, pentamers, etc.) are destabilized. Therefore, to achieve thermodynamic equilibrium, the adduct Р4 · Р formed in the catalytic cycle must react with another molecule of the monomeric alkoxide to afford a stable dimer Р2.
The possibility of a reaction of excessive starting aldehyde with a P4 · P molecule to afford the P5 · Ac adduct, was considered (Scheme 14).
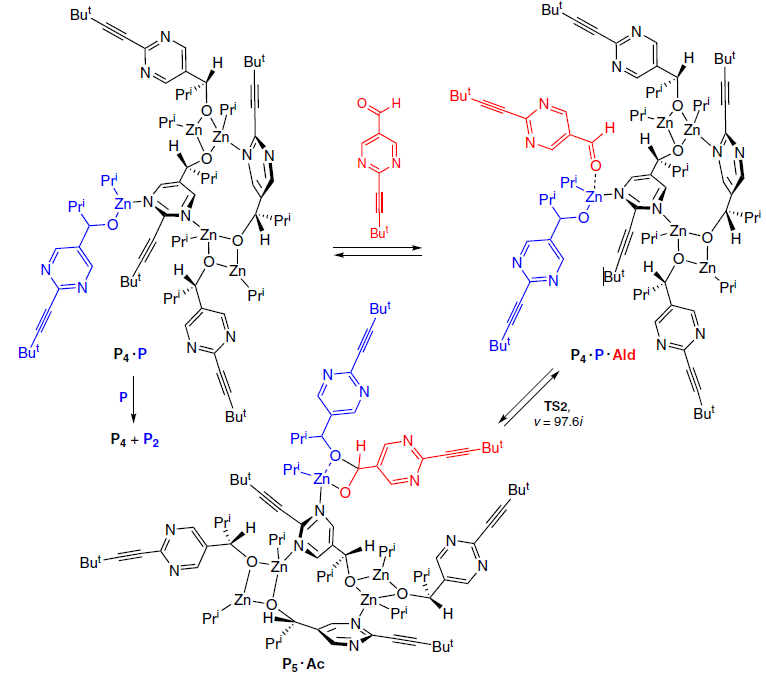
Indeed, calculations showed that such a process can occur via the TS2 transition state with a low activation barrier (∆G≠298 = 6.4 kcal mol−1), delivering an adduct of the pentamer and P5 · Ac acetal. However, it turned out that this reaction is reversible, and it is impossible to obtain the acetal in detectable concentrations in this way.
On the other hand, the P3 · Ac acetal is quite stable due to the formation of a Zn3 cluster with close Zn – Zn bond lengths (Fig. 13).[62] On this basis, it was concluded that once the P5 · Ac adduct is formed, it converts into a more stable combination P2 + P3Ac (Scheme 15).
![[{"id":"L5vGJEgbOu","type":"paragraph","data":{"text":"Optimized structure of the acetal <b>P3 · Ac</b>. The Zn – Zn distances are 3.02, 3.09, and 3.19 Å.<sup>62</sup> Copyright Chemical Society of Japan 2015."}}]](/storage/images/resized/ymPTVN5qaMpDtFkokAvmBxVSJ5cwSEUn26KJuqNi_xl.webp)
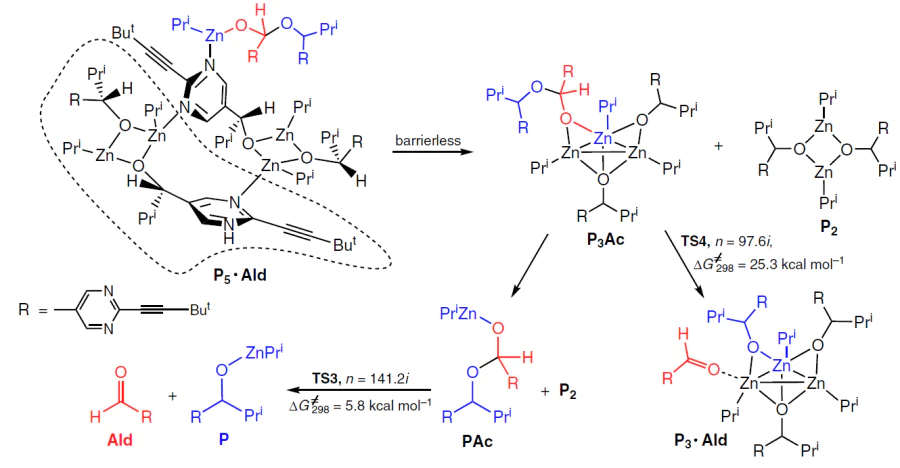
The activation barrier for regeneration of Ald from P3Ac (∆G≠298 = 25.3 kcal mol–1) was found to be high enough to explain the kinetic trapping leading to P3Ac but too high to explain the kinetics of the acetal decay.
On the other hand, P3Ac is capable of dissociating into PAc and P2 with an effective activation barrier of ~ 18 kcal mol−1, followed by rapid regeneration of the aldehyde from PAc, which is consistent with the experimentally observed rate of the acetal decay.
Therefore, the calculation results are in line with the modified kinetic scheme (Scheme 16).
3.2.3. Detection of acetal intermediates by mass spectrometry
The composition of the Soai reaction mixture was studied in detail using high-resolution mass spectrometry.[50][63] Based on the analysis of over 800 mass spectra, various intermediates containing an acetal moiety were identified. This enabled the authors to propose a catalytic cycle leading to the enantioselective formation of an alkoxide; however, quantum chemical calculations for this cycle were not performed. This catalytic cycle is not discussed in this review, since the measured concentrations of the observed acetal intermediates were extremely low.[50]
A critical examination of the mechanism described above can be found in the study.[58]
3.2.4. X-ray diffraction studies of crystals obtained from the Soai reaction mixtures
In 2015, the first direct evidence of the existence of tetrameric aggregates in the solid phase was reported.[64] Isolating single crystals of the Soai reaction intermediates presented a complex experimental challenge, because the alkyl zinc alkoxides are extremely sensitive to air moisture and oxygen, and exist in solution as a dynamic mixture of rapidly exchanging oligomers. Soai and co-workers[64] overcame these difficulties by varying the crystallization conditions. It was found that the crystal structure critically depends on the presence of the excess of diisopropyl zinc (Pri2Zn) and the enantiomeric purity of the starting alcohol (Fig. 14).
![[{"id":"k_Q5tDlzw2","type":"paragraph","data":{"text":"Crystallization of zinc alkoxides.<sup>64</sup> Сopyright Wiley 2015."}}]](/storage/images/resized/AqmR5JEI5EUPa9sG4DXtOiY86ZMm0kmz1sBvdeMm_xl.webp)
In the cited and a follow-up study,[64][65] six key crystal types were isolated and characterized:
— crystals A and В — tetramers obtained from the enantiomerically pure (S)-alcohol (А) and racemic (В) alcohol in the presence of a large excess of Pri2Zn,
— crystals C and D — oligomers obtained from the enantiomerically pure (R)- alcohol (С) and a racemate using a small amount of Pri2Zn (1 – 2 equiv.),
— crystals E and F — enantiomerically pure (E) and racemic (F) complexes crystallized with tetrahydrofuran and excess Pri2Zn.
3.2.4.1. Tetramers (crystals A and В)
Crystal A structure (space group P212121) is a tetrameric aggregate [(S)-2b]4 · [Pri2Zn]6 . The molecule core includes a 12-membered macrocycle linking two dimeric subunits, each containing a square Zn2O2 moiety. The macrocycle is formed by the coordination of zinc atoms of one dimer with the pyrimidine nitrogen atoms of the other dimer (Fig. 15).
![[{"id":"CumgwJh9gR","type":"paragraph","data":{"text":"Molecular structures and a simplified structure of the tetramer crystallized with excess Pr<sup>i</sup><sub>2</sub>Zn. Crystal of enantiomerically pure tetrameric alkoxide <b>A</b> (<i>а</i>). Crystal of racemic tetrameric alkoxide <b>B </b>(<i>b</i>).<sup>64</sup> Сopyright Wiley 2015."}}]](/storage/images/resized/0RSSefJi5gHsUnbKW9c4qnMdQloLHr7Zk2mHWuka_xl.webp)
The lattice cell contains six Pri2Zn molecules coordinated to the pyrimidine nitrogen atoms. The Zn2O2 moieties contain coordinatively unsaturated zinc atoms, are located within a chiral cavity, and are well positioned to bind the aldehyde carbonyl oxygen atom.[64][65]
Crystal B belongs to the space group C2/c. Its structure differs significantly from that of its homochiral analog, despite the similar chemical composition, ([(S)-2b]2 · [(R)-2b]2 · [Pri2Zn]4). The SMS structure motif is retained, but the outer pyrimidine rings are oriented in opposite directions relative to the plane of the macrocycle, whereas in the homochiral compound, the Zn2O2 squares are located on the same side of the macrocycle (see Fig. 15).[64]
The Zn – N distances in the 12-membered macrocycle are 2.20 Å, compared to 2.26 Å in the homochiral analog, indicating the greater stability of the former compound. At the same time, the opposite orientation of the pyrimidine fragments reduces the accessibility of the Zn centers and hinders the approach of the aldehyde.[64][65] DFT studies of the tetramers confirm these findings.[65]
3.2.4.2. Oligomeric chains (crystals C and D)
Reducing the diisopropylzinc content in the reaction mixture leads to the formation of higher oligomeric structures (crystals C and D) due to the alternation of Zn2O2 squares and 12-membered macrocycles. Unlike tetramers, in oligomers (both enantiomerically pure and racemic), the Zn2O2 squares are always located on opposite sides of the macrocycle, and their structure is virtually random, which contrasts with the significant difference in the structure of tetramers.[65] The results obtained (Fig. 16) indicate that the reaction mixture may contain higher-order oligomers. Besides, a possibility of the existence of the heterochiral structures resembling the crystal A or homochiral tetramers similar to crystal B cannot be excluded.
![[{"id":"2HgJPE0TB5","type":"paragraph","data":{"text":"Molecular structure and simplified structure of an oligomer co-crystallized with 1 – 2 equiv. Pr<sup>i</sup><sub>2</sub>Zn. Crystal of enantiomerically pure oligomer <b>C</b> (<i>а</i>). Crystal of racemic oligomer <b>D</b> (<i>b</i>).<sup>64</sup> Сopyright Wiley 2015."}}]](/storage/images/resized/RXnVSn0N8YH0vGyd5QDdpLT7h8NQrRPb5im6AWGC_xl.webp)
3.2.4.3. Crystals E and F in the presence of THF
Crystallization of enantiomerically pure or racemic alkoxides in THF yielded crystals of tetramers E and F containing THF. Both tetramers form a Zn2O2 square and a 12-membered macrocycle. For the enantiomerically pure tetramer (E), the square dimer is located on the same side of the 12-membered ring as in the THF-free crystal (A). In the case of the racemic tetramer (F), the square dimer is located on the opposite side, similar to the THF-free tetramer (B). However, instead of oligomerization via Zn – N bonds, the zinc coordination sites were occupied by THF molecules (Fig. 17).[65]
![[{"id":"pP-NcshZcA","type":"paragraph","data":{"text":"Crystal structure of tetramers crystallized with excess Pr<sup>i</sup><sub>2</sub>Zn (crystals <b>E</b> and <b>F</b>).Copyright Chemical Society of Japan 2016."}}]](/storage/images/resized/dmlv0zK2JnPi3DhVeq5PtRLSNK2lztofS1PrS1TE_xl.webp)
This observation explains the well-known fact of inhibition of the Soai reaction by donor solvents: THF destroys the supramolecular architecture of the catalyst, acting as a competitive inhibitor.[64-66]
Thus, the tetrameric structure contains two THF molecules and is free of coordinatively unsaturated zinc atoms, which are believed to be highly reactive. This feature explains the low reaction rate of asymmetric autocatalysis of 5-pyrimidylalkanol in the presence of THF.[65]
3.2.4.4. Change in crystal structure when replacing the alkyl group at the zinc atom
The preparation of a single crystal of an alkoxide containing a diethylzinc (Et2Zn) molecule instead of diisopropylzinc was described.[65] In this case, the oligomeric structure is formed even in the presence of excess diethylzinc. In this oligomer, the Zn2O2 square is located on the side of the 12-membered macrocyclic structure. An additional Et2Zn is coordinated to each alkoxide unit (Fig. 18), which differs significantly from the structure with isopropyl zinc alkoxides. This result clearly demonstrates that the steric effect of the isopropyl group plays an important role in the formation of the tetrameric structure.
![[{"id":"mutvIge4nQ","type":"paragraph","data":{"text":"Oligomeric structure of Et<sub>2</sub>Zn-alkoxide.<sup>65</sup> Сopyright Chemical Society of Japan 2016."}}]](/storage/images/resized/6bw4rDy4X7M2IIHfun1mQ1nBb3iCmITxdH73EDKC_xl.webp)
3.3. Study of catalytic cycles using quantum chemical calculations
3.3.1. Early results
The first results of quantum chemical calculations related to the Soai reaction were published in 2004.[51] Furthermore, the structures of the main components capable of coexisting in the reaction mixture were determined for the first time.
In particular, the structures of the square 5k and macrocyclic 4k dimers, as well as their complexes with Me2Zn (which was used in the calculations instead of Pri2Zn), were optimized. Later, more accurate calculations sometimes took conformational and entropic effects into account, but the topology of these molecules and their relative stability were generally in line with these initial calculations.
Thus, the formation of a trinuclear cluster was revealed upon coordination of dialkyl zinc with an oxygen atom, and it was found that dimer 5k was significantly more stable than 4k (Fig. 19).[51] All these conclusions were subsequently confirmed experimentally (see Section 3.1.2).
![[{"id":"OrKrTcvkVn","type":"paragraph","data":{"text":"Calculated structures and relative energies of formation (kJ mol<sup>–1</sup>) of square (<i>a</i>) and macrocyclic (<i>b</i>) dimers, as well as their complexes with Me<sub>2</sub>Zn. Energies are quoted relative to the summed energies of two aldehydes and two (or three) molecules of Me<sub>2</sub>Zn.<sup>51</sup> Сopyright National Academy of Sciences 2004."}}]](/storage/images/resized/qjPSy0LQ8KNxhLvqcARtQCEb4KroNZbACJ6xZAFC_xl.webp)
Although, according to calculations, trimeric alkoxides are thermodynamically less favorable than dimeric or tetrameric ones, some of the structures described in this work were discussed in later studies as intermediates in the proposed catalytic cycles (Fig. 20).[51]
![[{"id":"xEOf70yoC0","type":"paragraph","data":{"text":"Calculated structures and relative energies of formation (kJ mo<sup>l–1</sup>) of trimeric alkoxides.<sup>51</sup> Сopyright National Academy of Sciences 2004."}}]](/storage/images/resized/t1xkkw9is93DufjVN6sBPCILgGyAuiN82jIH7TPz_xl.webp)
For example, the macrotricyclic trimer (see Fig. 20d ) is mentioned in the catalytic cycle starting from the macrocyclic dimer 4k (see Section 3.3.2), and the bicyclic trimer analog (see Fig. 20b) was later proposed as an intermediate formed in the side process of acetal decay (see Section 3.2.2).
Finally, each of the simplified tetrameric structures shown in Fig. 21 was used as a model in one or another of the proposed catalytic cycles (see Sections 3.3.2 – 3.3.7). This does not mean that other associates cannot be modeled from four alkoxide molecules. However, the strict structural and stability requirements for potential catalysts for the Soai reaction made possible to confidently exclude them from consideration (e.g., a polymer chain or macrocyclic ring are extremely unstable).
![[{"id":"mImdfvj3wH","type":"paragraph","data":{"text":"Calculated structures and relative energies of formation (kJ mol<sup>–1</sup>) of tetrameric alkoxides.<sup>51</sup> Сopyright National Academy of Sciences 2004."}}]](/storage/images/resized/MBCrrbzE9f8Jw8FSgTxl3a4GFKekeTvjR1sBS91c_xl.webp)
3.3.2. Macrocyclic dimer as a catalyst (or template), tetramer-‘barrel’ as a product
As noted above, the macrocyclic dimer 4k was considered as the first candidate for the role of catalyst in the Soai reaction.[49] However, its absence in the reaction mixture (see Section 2) along with its low thermodynamic stability (see Fig. 19)[51] had not confirmed such assumptions.
However, in a series of papers,[67-70] published in 2008 – 2011, this structure was used as a starting point in suggested catalytic cycles (Scheme 17).
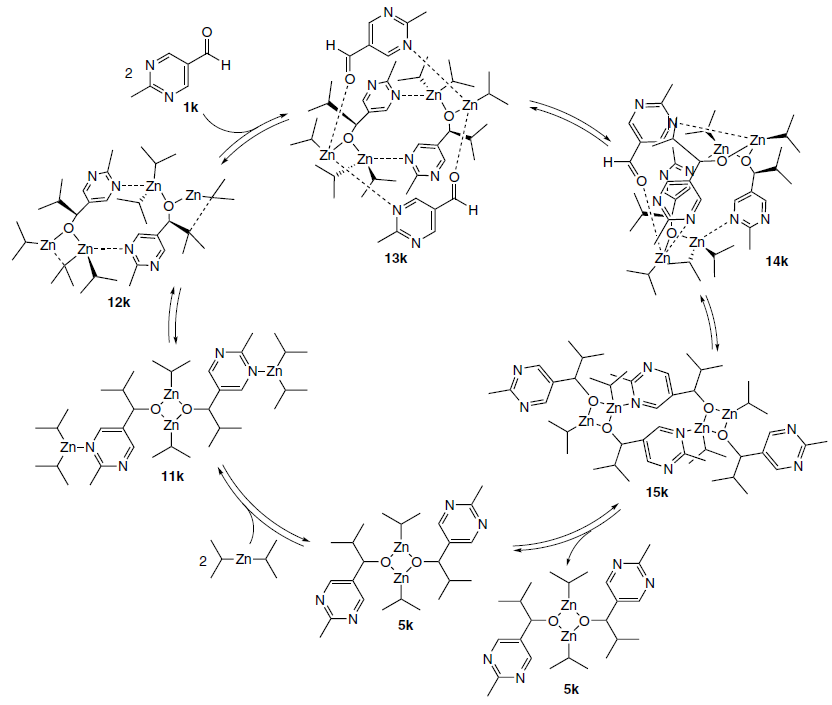
It was assumed that coordination of two diisopropylzinc molecules with the square dimer 11k promotes stabilization of the macrocyclic isomer, forming adduct 12k. Then, association of two molecules of substrate 1k occurs, followed by stereoselective migration of the isopropyl group. This delivers intermediate 14k, which is an alkoxide trimer with a coordinated aldehyde molecule. A second transfer of the isopropyl group completes the formation of the alkoxide tetramer 15k with a ‘barrel’-like configuration, which dissociates into two square dimers, completing the catalytic cycle (see Scheme 17).[67-70]
In this mechanism, the catalyst is formally the macrocyclic dimer 4k. However, kinetic analysis showed that the experimentally observed level of amplification could not be achieved with a dimeric catalyst.[68] This allowed the authors to suggest that, in addition to two sequential stereoselective alkylation reactions, the amplification of the enantiomeric excess of the product also involves a ‘reservoir effect’, viz., the removal of the minor enantiomer from the catalytic cycle by forming a more stable heterochiral oligomer.
A more detailed stereochemical analysis of the enantioselective steps of the proposed catalytic cycle revealed a complex picture caused by the conformational lability of key intermediates. Eight competing transition states and three catalytic cycles were analyzed, significantly reducing the clarity of the authors’ proposed amplification scheme.[69] A serious problem is also the systematic neglect of the entropic contribution in the calculations, which can be particularly critical given the large number of steps that involve the formation of ordered structures from small molecules.
3.3.3. SMS tetramer as a catalyst
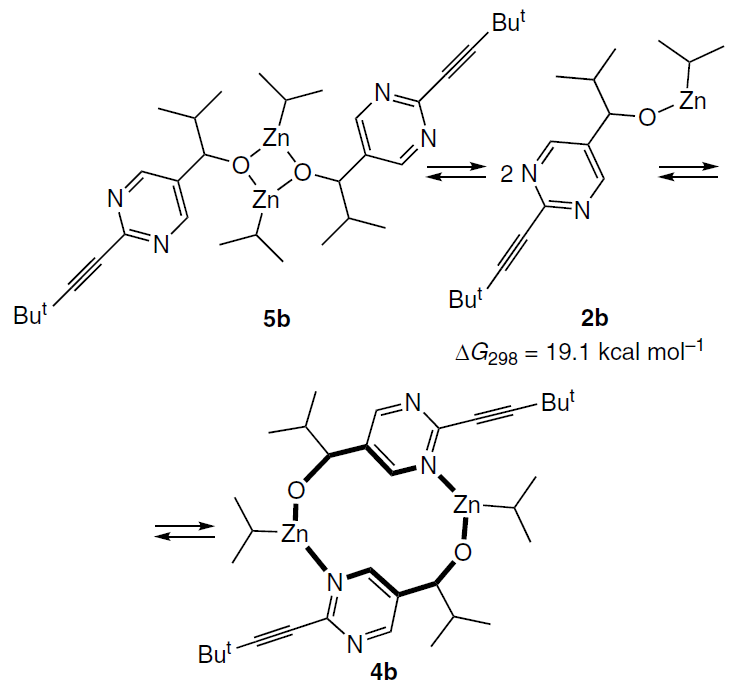
In Section 3.1.2, it was shown that data from numerous NMR experiments are consistent with the square dimer 5b being the main component of the Soai reaction mixture at room temperature. At the same time, the structure of the macrocyclic dimer 8b seemed more promising in terms of its structural potential for the formation of stereoselective transition states.
The nature of the signal shape changes in the 1H NMR spectra with decreasing temperature indicated further oligomerization of the alkoxides to form tetrameric (and even hexameric) associates (see Section 3.1.2).
Interestingly, the formation of the dimeric macrocycle from the more stable square dimer 5b requires its dissociation into monomers 2b, associated with overcoming a high activation barrier (see Section 3.1) (Scheme 18).
On the other hand, the formation of a similar macrocyclic unit within the tetramer can occur through the association of two molecules of 5b. This process is virtually barrierless and affords an SMS tetramer with a structure matching the experimental data (Scheme 19).
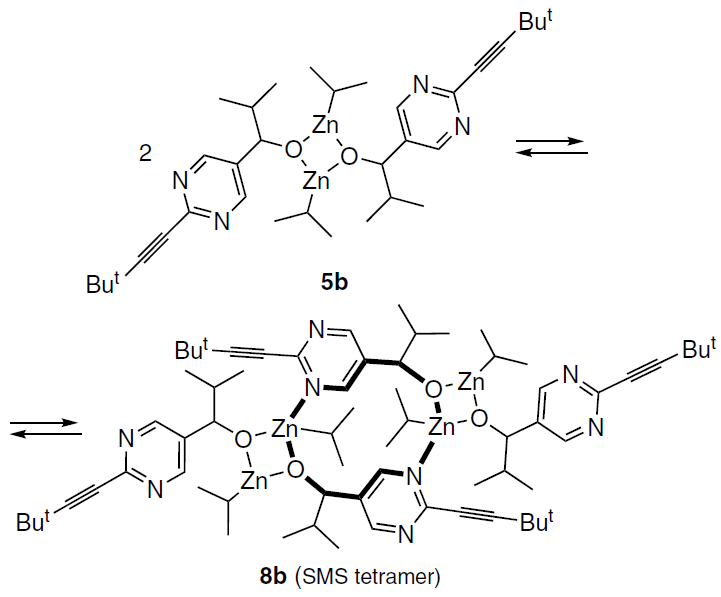
Moreover, the free energy values of this process calculated using the B3LYP functional were in good qualitative agreement with the experimental data (see Fig. 9). At room temperature, two dimers are significantly more stable than the tetramer, whereas at 193 K, the formation of the tetramer becomes exergonic (Table 4).[59] At the same time, the M05-2X functional significantly overestimates the free energy of tetramerization.
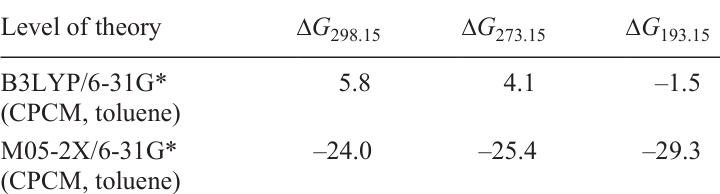
Despite the presence of two four-membered Zn – O – Zn – O rings in the tetramer structure, the macrocycle itself is conformationally labile. Therefore, further analysis required studying the conformational equilibria of homo- and heterochiral alkoxides for various structures.
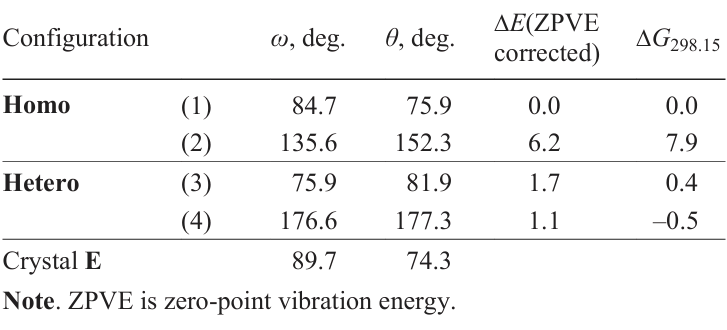
To characterize the macrocycle conformations, the parameters ω (the angle between the planes of pyrimidine rings) and θ (a dihedral angle between two Zn – O bonds of the macrocycle) were introduced (Fig. 22).[59] By scanning these two parameters, conformational minima were localized for homo- and heterochiral tetramers.
![[{"id":"fcHTR2fgeC","type":"paragraph","data":{"text":"Characteristic conformational parameters of a macrocycle: ω (an angle between the planes of pyrimidinyl rings) and θ (a dihedral angle between two Zn – O bonds of the macrocycle).<sup>59</sup> Copyright American Chemical Society 2012."}}]](/storage/images/resized/fV9TTBal0d3PT3TS0odWi8D6Z5NdjbloXN4WiJWy_xl.webp)
In each case, four conformational minima were found, two of which were significantly (by 10 – 15 kcal mol−1) more stable than the other two. Coordination of one or two diisopropylzinc molecules did not alter these conformational preferences.
For the homochiral tetramer, the most stable conformation contained virtually orthogonal pyrimidine rings; the dihedral angle θ between the two Zn – O bonds was also close to 90°. For the heterochiral tetramer, a similar conformation (with a smaller angle between the pyrimidine rings) was only slightly less stable. At the same time, the conformation with practically coplanar pyrimidine rings and Zn – O bonds was stable only in the case of the heterochiral dimer (Table 5, Fig. 23b).[2][59] Thus, structural diversification was observed between homo- and heterochiral tetramers.
![[{"id":"sbUTd2377M","type":"paragraph","data":{"text":"Homochiral tetramer framework (with opposite chirality sign) found in the crystal structure of <b>E</b> (<i>а</i>).<sup>65</sup> Сopyright Chemical Society of Japan 2016. Macrocycle structures calculated using the B3LYP/6-31G* method (<i>b</i>).<sup>2, 59</sup> Сopyright American Chemical Society 2012."}}]](/storage/images/resized/6lwFG3Fs8ULZ6eLkktmJLh7vDzCY6xpVijiCOnCU_xl.webp)
The Homo(1) structure is particularly intriguing as a potential homochiral catalyst, as the nearly orthogonal arrangement of the pyrimidine rings creates a cavity that is suitable for substrate coordination (see Section 3.3.5).[2][59] In the closely related Hetero(3) structure, the ω angle is 10° smaller (see Table 5).
It is interesting to note that the structure of the homochiral alkoxide in the crystal structure of E (Fig. 23a)[65] is similar to that of the Homo(1) tetramer (see Table 5), whereas in heterochiral crystals only conformations with coplanar pyrimidine rings were observed (see Section 3.2.4.3).
3.3.4. Effect of diisopropylzinc coordination on tetramer conformations
The first step in the catalytic cycle of the Soai reaction must be the coordination of the alkylating agent (diisopropylzinc) with the catalyst. The thermodynamic parameters of this process and the geometries of the resulting complexes were calculated.[59]
The main results are presented in Fig. 24.[57] In the case of a homochiral tetramer (see Fig. 24a), after coordination of Pri2Zn, the pyrimidine rings remain practically orthogonal, leaving the cavity formed by the macrocycle unoccupied permitting the aldehyde coordination.
![[{"id":"hiWqHNBo9u","type":"paragraph","data":{"text":"Calculated structures of complexes of homo- (<i>a</i>) and heterochiral (<i>b</i>) tetramers with Pr<sup>i</sup><sub>2</sub>Zn.<sup>57</sup> Сopyright Royal Society of Chemistry 2023."}}]](/storage/images/resized/R1f2sUxO12S4Mv1kAMyTlYZ2srt3vgd5TqZq8Ghf_xl.webp)
At the same time, in both possible isomers of the heterochiral tetramer complex, when diisopropylzinc is coordinated, the cavity closes, that prevents the effective coordination of the aldehyde (Fig. 24b).[57]
This effect was interpreted as an important stereodiscriminating factor, allowing to consider only the homochiral tetramer as an effective catalyst for the Soai reaction. This fully accounts for the observed autocatalysis and autoamplification phenomena.
3.3.5. Transition state for the SMS tetramer as a catalyst
In this model, it is proposed that the cavity formed by the orthogonal pyrimidine rings in the adduct of С2-symmetric homochiral tetramer with diisopropylzinc coordinates the substrate to form the transition state TS1, computed using the DFT method (Fig. 25а).[59]
![[{"id":"Ud17zC3Anu","type":"paragraph","data":{"text":"Transition state for the enantioselective transfer of the isopropyl group computed using the B3LYP/6-31G* method (CPCM, toluene) (<i>а</i>). Spatial structure <b>TS1</b> (<i>b</i>).<sup>59</sup> Сopyright American Chemical Society 2012."}}]](/storage/images/resized/M3yeawBybFoHAZzpYV55PMssedWuq1GTbB5bbS3k_xl.webp)
An important feature of this process is the complete impossibility of forming an adduct leading to the alternative enantiomer, since the coordination of the aldehyde using another prochiral plane is excluded for geometric reasons.
Fig. 25b[59] shows the role of the specific substrate structure in the formation of the TS1 transition state. The planar pyrimidine ring and acetylene moiety perfectly fit the cavity size, with the hydrogen atom and nitrogen atom of the substrate pyrimidine ring being involved in non-covalent stabilizing interactions, and the tert-butyl group acting as an ‘anchor’ holding the substrate molecule in an orientation favorable for the transfer of the isopropyl group from zinc to carbon via CH∙∙∙НС and CH∙∙∙N interactions.[57]
3.3.6. Calculation of the catalytic cycle and its kinetic simulation
The catalytic cycle involving direct alkylation of a coordinated aldehyde with diisopropylzinc is shown in Fig. 26, and the calculated data for the key steps are presented in Table 6.[59]
![[{"id":"8nHhIv7Bwa","type":"paragraph","data":{"text":"Reaction energy profile and catalytic cycle for the direct alkylation of an aldehyde coordinated to diisopropylzinc.<sup>59</sup> Сopyright American Chemical Society 2012."}}]](/storage/images/resized/7EBK3T4lsq9cgFyDSLHdNFjUviyNSdJ4OtuLVMg4_xl.webp)
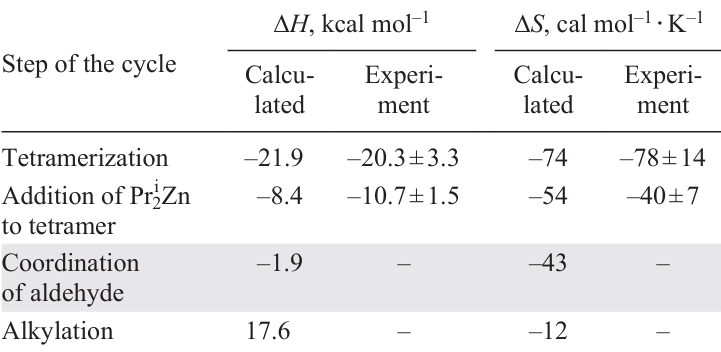
After the formation of a tetramer (in a specific conformation containing orthogonal pyrimidine rings) from two molecules of square dimer 4b, coordination of diisopropylzinc occurs yielding adduct 8b (see Fig. 26).[59] A molecule of substrate 1b then coordinates to the zinc atom in the three-dimensional cavity of the catalyst to give the ternary complex 8b · Pri2Zn · 1b. After enantioselective alkylation via transition state TS1, the catalyst – product complex 8b · 2b is formed, which dissociates, returning the catalyst to the cycle and amplifying the chirality of the system due to the newly formed chiral alkoxide 2b.
Next, based on the calculated catalytic cycle and a number of logical assumptions about the nature of equilibria in the system, a modeling of the kinetic curves was carried out (see Fig. 27a).[59] This made possible reproducing the experimental data (Fig. 27b)[55] for the inverse Arrhenius dependence observed in the Soai reaction.
![[{"id":"_89KFTgsQq","type":"paragraph","data":{"text":"Computed (<i>а</i>) and experimental (<i>b</i>) kinetic curves for the reaction of diisopropylzinc with aldehyde <b>1b</b> at different temperatures (0.015 M <b>1b</b>, Pr<sup>i</sup><sub>2</sub>Zn (1.5 equiv.), enantiomerically pure <b>3b</b>(1 mol.%)).<sup>55, 59</sup> Сopyright American Chemical Society 2010 & American Chemical Society 2012."}}]](/storage/images/resized/noKWWz69SvS4LDASUDT8kwDIohzc6ayR6jFZAN7k_xl.webp)
3.3.7. ‘Floor-to-floor’ model
Based on crystallographic data of Soai et al.,[64][65] a transition state called ‘Floor-to-floor’ was proposed.[71]
In this model, the ‘floor’ of the catalyst is the plane of the homochiral tetramer macrocycle (Fig. 28a)[71] with protruding Zn2O2 nodes. The ‘floor’ of the substrate is the plane of the pyrimidine ring of the aldehyde.
![[{"id":"imcacx_Ufb","type":"paragraph","data":{"text":"Homo- (<i>а</i>) and heterochiral (<i>b</i>) tetramers, their intermediates with aldehyde <b>1а</b> and transition states.<sup>71</sup> Сopyright Springer Nature 2020."}}]](/storage/images/resized/H5CqQkgTgwiGr0OBcrHWhHhcgsapMicq7bESnE33_xl.webp)
The aldehyde coordinates to the tetramer via two points, viz., the carbonyl oxygen coordinates to the coordinatively unsaturated zinc in the Zn2O2 square and the pyrimidine nitrogen coordinates to another zinc atom in the macrocycle. This bidentate bonding rigidly fixes the orientation of the aldehyde. The alkyl group is transferred from the pre-activated Pri2Zn molecule via coordination to the tetramer ‘arm’.
In the heterochiral tetramer (see Fig. 28b)[71] such orientation of the aldehyde relative to the catalyst is impossible because of steric hindrances created by the different arrangement of the pyrimidine rings.
Such a significant difference in the structures (see Fig. 28) and energies of formation of reactive complexes (Fig. 29)[71] is quite sufficient to explain the AAA in the Soai reaction.
![[{"id":"0iSinv92hC","type":"paragraph","data":{"text":"Computed energy profiles of catalytic reactions.<sup>71</sup> Сopyright Springer Nature 2020."}}]](/storage/images/resized/5AiSDqluZK1l2UKk8vjRH46SHxwpcR7zFWO2EEuJ_xl.webp)
At the same time, it should be noted that the full catalytic cycle was not calculated in these studies, and the possible involvement of alkoxides with different structures was not considered. Therefore, the obtained results should be considered an interesting finding, useful for discussing possible mechanisms, rather than a ‘demystification’ of the Soai reaction, as the authors claimed.
Besides, the high substrate specificity of the Soai reaction has been studied in considerable detail. Specifically, it was concluded that the role of the isopropyl group in the effectiveness of AAA lies in its steric demands, preventingthe formation of cubic alkoxides. The latter, due to their closed structure and high stability, prevent the catalytically active alkoxides from being involved in reversible equilibria.[5]
3.3.8. General conclusions on the current view on the AAA mechanism in the Soai reaction
A consensus that AAA in Soai reaction stems from the structural diversification of the oligomeric intermediates, with only the homochiral species being able to act as active catalysts is provisionally achieved.
Given the rapid conformational equilibria in solution, the question of the specific structure of the active catalyst is less important if the first condition is met. In other words, the simultaneous occurrence of several mechanisms is possible.
Furthermore, the different stabilities of homo- and heterochiral reaction products can enhance the amplifying effect of the first factor.
4. Chiral initiators. Sources of symmetry breaking
The most remarkable property of the Soai reaction is its sensitivity to a broad spectrum of chiral triggers. Due to its powerful amplification mechanism, the formation of a specific enantiomer can be initiated by factors considered insignificant in classical chemistry. The reaction links the macroscopic chirality of organic products with the microscopic or physical chirality of the initiators.
4.1. Circularly polarized light
Circularly polarized light (CPL) is a truly chiral physical field and is considered one of the most likely primary causes of the emergence of homochirality of organic matter on prebiotic Earth.[72-74] In the context of the Soai reaction, CPL acts as a primary trigger, initiating the process through absolute asymmetric synthesis or asymmetric photolysis of racemic mixtures.[8][74-79] The fundamental basis of this process is circular dichroism–the difference in the molar extinction coefficients of enantiomers with respect to left- and right-handed polarized light.
Irradiation of racemic leucine with r-CPL results in the preferential destruction of one of the enantiomers, creating a small enantiomeric excess (< 2%) due to the difference in extinction coefficients. This value alone is chemically insignificant, but using leucine as an additive in the Soai reaction can significantly amplify the enantiomeric excess (Scheme 20).[80]
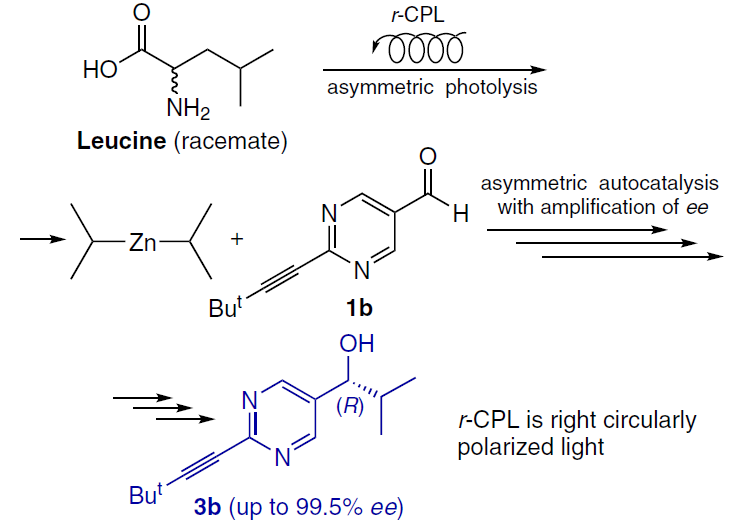
Moreover, the Soai reaction can be initiated without the involvement of chiral molecules, for example, by direct irradiation of a racemic pyrimidine alcohol with CPL. Exposure to r-CPL or l-CPL (usually in the UV range, e.g., at 313 nm, which corresponds to the alcohol absorption band) creates a microscopic initial ee of the product (~ 10–5 – 10–4), followed by autocatalytic alkylation of pyrimidine-5-carbaldehyde with diisopropylzinc.[76]
The use of tunable CPL sources, such as synchrotron radiation, demonstrated a strict dependence of the sign of the resulting enantiomer not only on the polarization direction but also on the irradiation wavelength, which is completely consistent with circular dichroism spectra.
This made it possible to establish for the first time a direct correlation between photon chirality and the chirality of an organic compound with high optical purity, demonstrating a complete mechanism for the transition from physical chirality to chemical homochirality (Scheme 21).[47][81]
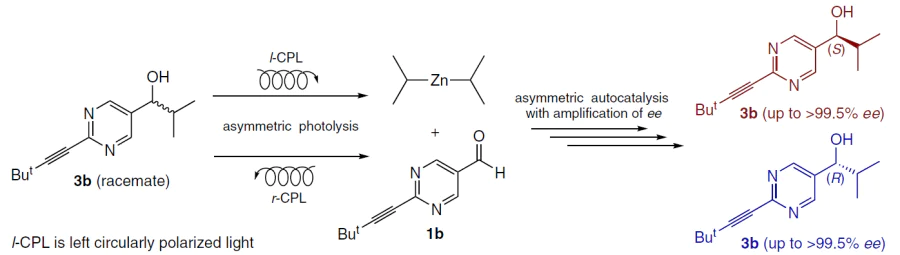
4.2. Chiral crystals of inorganic compounds
The crystal lattices of many abundant minerals, such as quartz and cinnabar, belong to chiral space groups.
4.2.1. Quartz (SiO2)
Quartz, one of the most abundant minerals in the Earth’s crust, exists in two enantiomorphic forms: dextrorotatory (d-quartz, space group P3121) and levorotatory (l-quartz, P3221). Soai et al.[8][47][82][83] showed that quartz powder can act as a heterogeneous chiral inducer. The following correlation was established (Scheme 22):[84]
— d-quartz induces the formation of (S)-pyrimidine alcohol (up to 97% ee);
— l-quartz induces the formation of (R)-pyrimidine alcohol.
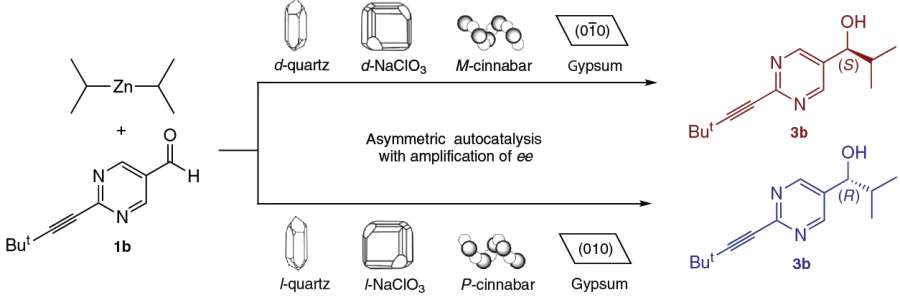
The observed S-path in d-quartz and R-path in l-quartz are stabilized by interactions with the surface, while their inverses are destabilized by the spatial repulsion of the isopropyl groups from the lattice (Fig. 30).[84] Calculations showed that enantioselectivity is due to the helical chirality of the quartz crystal structure.
![[{"id":"H1F4YQ6025","type":"paragraph","data":{"text":"Optimized transition state structures. The purple dashed lines indicate bond formation and cleavage, and the red dashed lines indicate stabilizing Zn – O contacts. Numerical values represent interatomic distances in Å.<sup>84</sup> Сopyright Royal Society of Chemistry 2025."}}]](/storage/images/resized/BpBDqV8krXOhQIavYnmce5kwv2tPymGl5hOwIY3v_xl.webp)
This discovery supports the Bernal’s hypothesis that chiral mineral surfaces on the early Earth could have served as templates for the first enantioselective synthesis.[85] Adsorption of reagents on a chiral surface creates an asymmetric environment sufficient to initiate an autocatalytic process.[84][86]
4.2.2. Cinnabar (HgS), gypsum, and other minerals
Cinnabar (mercury(II) sulfide) crystallizes in helical structures designated as P-helix (right-handed) and M-helix (left-handed).
— P-Cinnabar initiates the formation of (R)-pyrimidine alcohol;
— M-Cinnabar initiates the formation of (S)-pyrimidine alcohol.[8][47][81][83][87]
Other minerals, such as sodium chlorate (NaClO3) and bromate (NaBrO3)[8][47][81][88][89] and retgersite (nickel sulfate, NiSO4 · 6 H2O),[90] are also effective as chiral initiators (see Scheme 22). The ability of such diverse inorganic lattices to initiate organic stereochemistry underscores the universal nature of the principles of chiral recognition at interfaces.
The most unexpected result is the possibility of using gypsum (CaSO4 · 2H2O) as an initiator. The crystal structure of gypsum is centrosymmetric (achiral). However, when the crystal is cleaved, specific faces, (010) and (0 – 10) are formed, which are enantiotopic. This means that the two-dimensional surfaces of the faces themselves are mirror images of each other, despite the achirality of the bulk crystal.
— The reaction on the (010) face affords an (R)-alcohol;
— The reaction on the (0 – 10) face gives an (S)-alcohol (see Scheme 22).[47][81][83][91]
— This discovery is of fundamental importance, as it demonstrates that even achiral minerals can act as local sources of chirality if their surfaces interact with organic substrates in a specific way.
4.3. Chiral crystals of achiral organic compounds
Crystallization of achiral compounds to form enantiomorphic crystals (spontaneous resolution) is a fundamental mechanism for the transition from the achiral state to macroscopic chirality. It is known that approximately 5 – 10% of all achiral organic molecules crystallize in chiral space groups, forming crystalline conglomerates. The Soai’s group systematically explored the ability of such crystal surfaces to act as heterogeneous chiral initiators of primary asymmetric alkylation (Scheme 23).
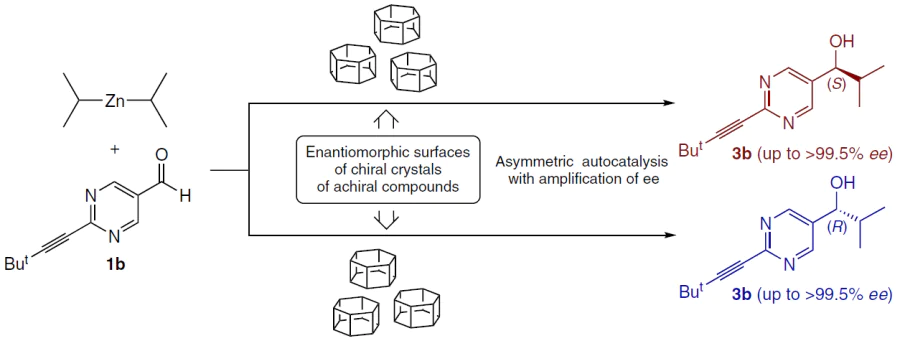
Fundamental prebiotic molecules, such as the nucleic acid bases cytosine and adenine, are strictly achiral when isolated. However, when precipitated, they can form enantiomorphic crystal lattices (particularly as salts such as adenine dinitrate). Using suspensions of such crystals as a heterogeneous matrix in the Soai reaction ensures high final enantioselectivity of the process due to subsequent autocatalysis. A clear correlation is observed between the absolute crystal structure and the sign of the enantiomeric excess of the resulting pyrimidine alcohol.[47][79][81][83][92][93]
Of particular importance in the context of prebiotic chemistry is the behavior of the simplest amino acid, glycine. Being achiral in solution, glycine is capable of forming polymorphic modifications that crystallize in chiral space groups: the β-form (group P21) and the γ-form (enantiomorphic groups P31 and P32).[94] It has been experimentally proven that heterogeneous induction on the surface of crystals of space group P31 selectively initiates the formation of the (R)-isomer of alcohol, while crystals of group P32 direct the reaction towards the formation of the (S)-enantiomer.[95]
Glycine derivatives such as hippuric acid (N-benzoylglycine),[47] and triglycine sulfate[96] also form enantiomorphic crystals capable of initiating the reaction.
Furthermore, chiral crystal structures can be found in some hydrocarbons and their derivatives,[97-102] as well as a number of metal complexes (Fig. 31).[103][104]
![[{"id":"JfvPbjVEf2","type":"paragraph","data":{"text":"Examples of chiral crystal structures suitable for initiating the Soai reaction."}}]](/storage/images/resized/FOXtNvyWT6HpXyfJ5IBUcvuT6yNRUdfO1P5OjZYC_xl.webp)
In all these cases, the surface of the chiral crystal acts as a template capable of inducing the Soai reaction, resulting in a product with a high enantiomeric excess.[76][97][105-109]
These data indicate that spontaneous crystallization of simple achiral molecules can create a local chiral environment that influences the synthesis of more complex chiral structures.
4.4. Isotopic chirality
One of the most impressive evidence of the sensitivity of the Soai reaction is its ability to distinguish chirality arising solely from isotopic substitution.[8][47][81][110-115] Electronic and steric differences between isotopes (e.g., 12C and 13C) are negligible, making such chirality virtually invisible to conventional methods.
4.4.1. Carbon isotopic chirality
In the study,[116] the chiral isotopomer of dimethylphenylcarbinol was used, the molecule of which is achiral if both methyl groups contain 12C. However, if one methyl group is 13C-enriched, a chiral center emerges (Scheme 24).
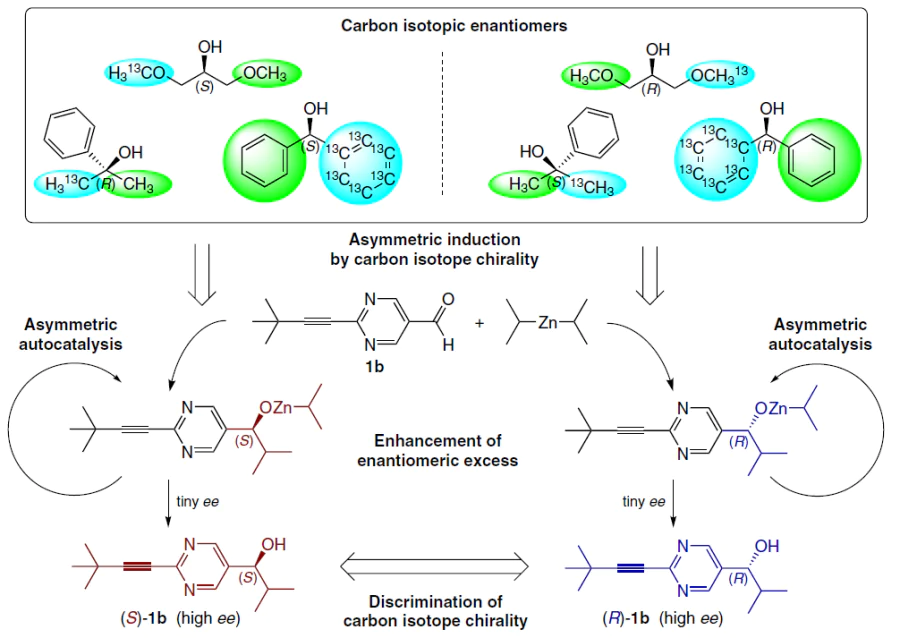
It was found that:
— (R)-[13C]-dimethylphenylcarbinol induces the formation of (S)-pyrimidine alcohol;
— (S)-[13C]-dimethylphenylcarbinol induces the formation of (R)-pyrimidine alcohol.[116]
Discrimination is based on microscopic differences in C – C bond distances and vibrational energies, which the autocatalytic system amplifies to a macroscopic chemical outcome.
4.4.2. Other isotopes
Similar discrimination was achieved for other isotopes (Scheme 25):[81]
— Hydrogen (H/D), using chiral glycine-α-d and deuterated primary alcohols;[114][115]
— Nitrogen (15N/14N), a chiral 15N-diamine, directs stereoselectivity;[117]
— Oxygen (18O/16O), isotopomers of diols also act as initiators.[118][119]
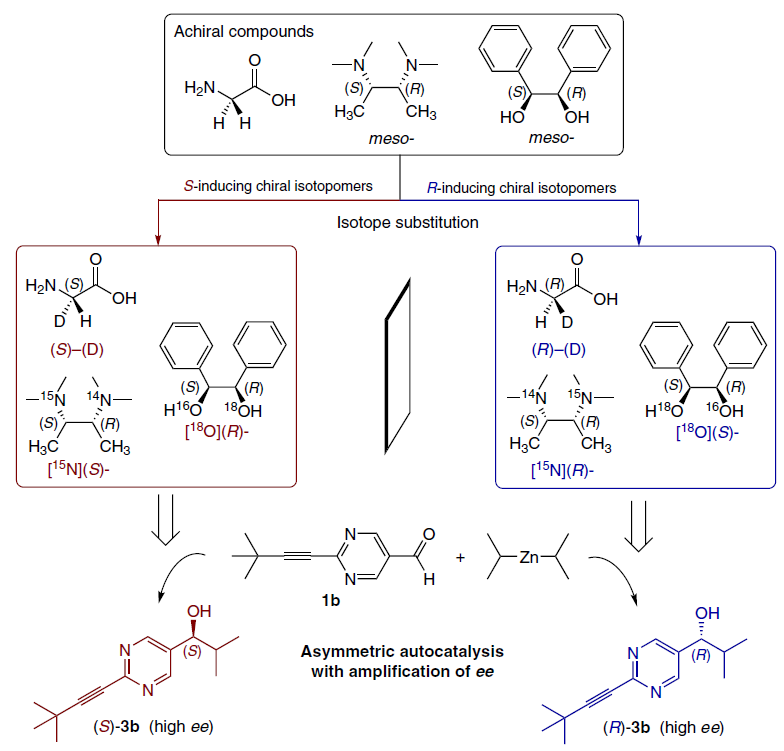
These results prove that information about symmetry breaking, stored at the level of an atomic nucleus, can be transmitted to the molecular level through chemical kinetics.
4.5. Cryptochirality and hydrocarbons
The reaction can be initiated by chiral hydrocarbons that do not contain functional groups capable of coordinating with zinc (Fig. 32).[71][118][120-126]
![[{"id":"gzJBCJ_3yr","type":"paragraph","data":{"text":"Chiral hydrocarbons suitable for initiating the Soai reaction."}}]](/storage/images/resized/YIbnNDk6Ri5l0kxtndT37lib8DVDQxynN9MF1Z3u_xl.webp)
[2.2] Paracyclophanes — planar chiral hydrocarbons — often induce reverse stereoselectivity compared to their heteroatomic analogs (containing amino or hydroxy groups).[124] It was shown that monosubstituted [2.2] paracyclophanes initiate the formation of alcohol with ee’s up to 97% after amplification.[122]
Chiral quaternary hydrocarbons (e.g. 5-ethyl-5-propylundecane), which have immeasurably small optical rotation, successfully initiate the reaction, probably due to weak van der Waals dispersion interactions with the π-system of the aldehyde.[71][120][121]
Alkenes, allenes, and polycyclic aromatic compounds can also be successfully used as chiral initiators of the Soai reaction.[118][123][125-128]
These data demonstrate the remarkable sensitivity of the Soai reaction to various sources of symmetry breaking, making it a powerful tool for investigating the origin of chirality using asymmetric autocatalysis.
5. Unusual aspects of asymmetric induction in the Soai reaction
Further confirmation of the uniqueness of the Soai reaction is provided by the anomalous fact that a single chiral source can yield both enantiomers depending on external conditions (temperature changes, the presence of achiral additives), as well as the phenomenon of ultra-remote intramolecular asymmetric induction. These data demonstrate the potential flexibility of this stereoselective synthesis and indicate that the initial chiral trigger does not always uniquely determine the final product configuration.
5.1. Asymmetric autocatalysis induced by a mixture of chiral and achiral compounds
The use of achiral additives in asymmetric catalysis can lead to changes in reactivity and selectivity. [129-132] Furthermore, it was found that achiral additives can work in combination with a chiral initiator, causing an inversion of enantioselectivity (Fig. 33).[133-135]
![[{"id":"FevWKhbtoE","type":"paragraph","data":{"text":"Chiral and achiral β-amino alcohols inducing inversion of enantioselectivity in asymmetric autocatalysis."}}]](/storage/images/resized/BrmR276EvKlfGiIYATpJqNYHx7CNaSDK0vgVagLo_xl.webp)
5.1.1. Reversal of enantioselectivity in the presence of achiral β-amino alcohols
(1S,2R)-N,N-dimethylnorephedrine (DMNE) is known to initiate the formation of (S)-5-pyrimidine alcohol, and the opposite enantiomer initiates the formation of (R)-alcohol.[45] However, the use of a mixture of (1S,2R)-DMNE with achiral amino alcohols (e.g., N,N-dibutylaminoethanol (DBAE)) affords the (R)-enantiomer (Scheme 26).[133][134]
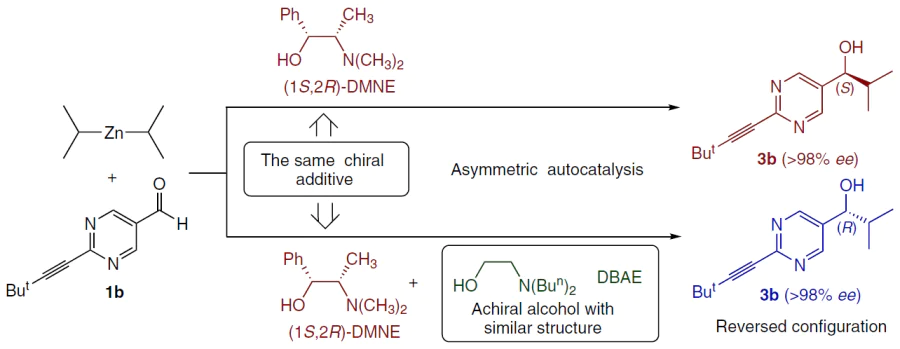
At a ratio of N,N-dibutylaminoethanol (DBAE) : DMNE = 65 : 35, the expected (S)-product was formed, but with a slight change in the ratio (to 70 : 30), reversal occured to furnish the (R)-alcohol with a high ee. In hexane, 5 mol.% of DBAE is sufficient for the reversal. According to the results of kinetic studies, this is due to the formation of catalytically active heteroaggregates of zinc alkoxides (Fig. 34).[134]
![[{"id":"5WC6Ga_Qj-","type":"paragraph","data":{"text":"Reversal of enantioselectivity using a mixture of chiral DBAE and achiral DMNE.<sup>134</sup> Сopyright American Chemical Society 2008."}}]](/storage/images/resized/zgnUYat22VxcNHkAjQd1XzYa5AQ2oSVZyw6ZujHV_xl.webp)
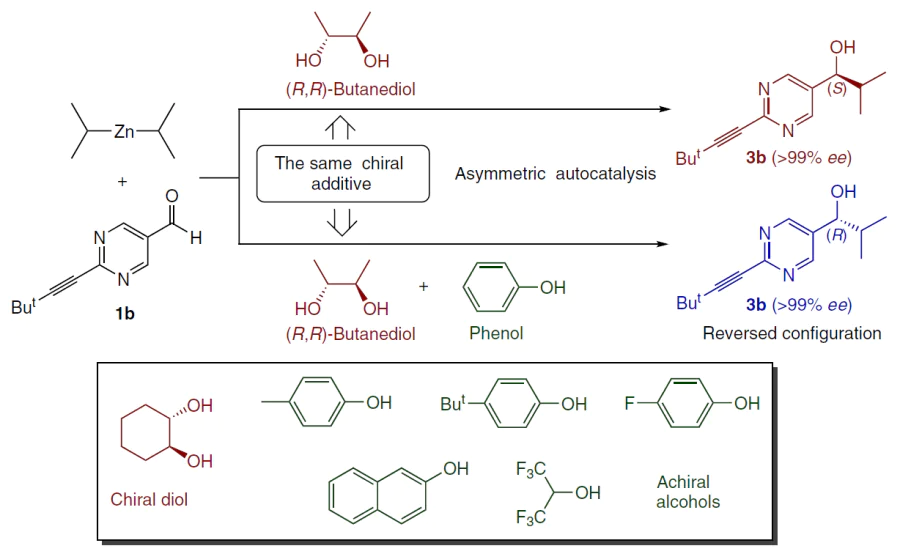
5.1.2. Inversion of enantioselectivity of chiral diols by achiral alcohols
A similar effect was observed with chiral diols. (2R,3R)-butane-2,3-diol alone induced the formation of (S)-alcohol. The addition of phenol (diol : phenol < 20 : 80) leads to reversal and the formation of (R)-alcohol (Scheme 27). The critical transition point is between ratios of 25 : 75 and 20 : 80. A similar result is reproduced with other para-substituted phenols and naphthols, allowing the synthesis of both enantiomers from a single chiral source.[136]
5.1.3. Cooperative action of two chiral β-amino alcohols
Combining two chiral catalysts can also lead to unexpected results (Scheme 28).
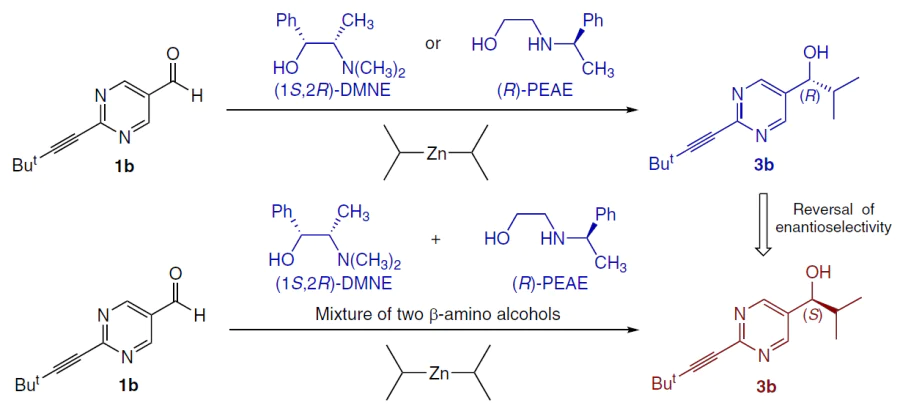
For example, a mixture of (1R,2S)-DMNE and (R)-1-phenyl-2-(1-pyrrolidinyl)ethanol ((R)-PEAE) (both individually afford (R)-product) at a certain ratio induced the formation of (S)-product (Fig. 35).[137][138]
![[{"id":"_8v1zIfLPV","type":"paragraph","data":{"text":"A mixture of two catalysts both yielding (R)-product may afford (S)-product depending on their relative quantities.<sup>138</sup> Сopyright Royal Society of Chemistry 2023."}}]](/storage/images/resized/wDz6sx63Ogf58aVh9mt496MtTK1n0CeutdvNi1SZ_xl.webp)
Furthermore, the competitive autocatalysis method allows to compare the catalysts ‘powers’. For example, using a mixture of (1S,2R)-DPNE (delivering S-product) and (1R,2S)-DBNE (delivering an R-product), an (R)-alcohol is formed, indicating the greater asymmetric power of DBNE. Quantitative evaluation showed that DBNE is approximately 2.8 times more effective than DMNE.[139]
5.2. Abnormal effect of the reaction temperature
In reactions with adamantyl-ethynyl derivatives,[12] an inverse dependence of reaction rate on the temperature was observed. At 263 K, the reaction rate is more than 20 times higher, and the incubation period is shorter than at 298 K, due to increase of the concentration of catalytically active zinc alkoxide tetramers with decreasing temperature.[55][138]
A rare phenomenon of a temperature-dependent selectivity switch has been observed with certain aromatic alcohols and amines (Scheme 29).
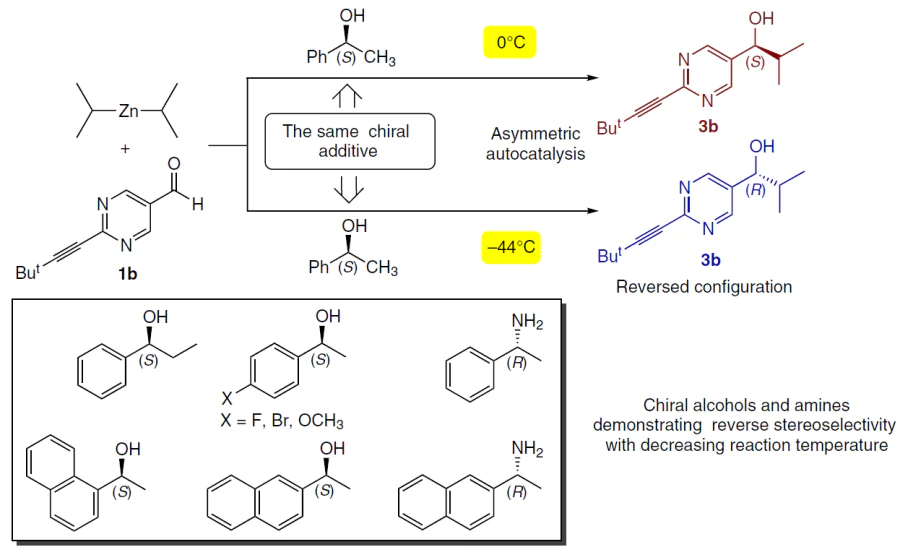
In particular, when initiating the reaction with (S)-1-phenethyl alcohol at 0°C, the (S)-product is formed, whereas at –44°C, the same initiator affords the (R)-product.[140]
5.3. Effect of the position of the nitrogen atom in aza[6]helicenes
Azahelicenes are effective chirality inducers, but their behavior depends on the position of the heteroatom in the molecule (Scheme 30):[141][142]
— 1-aza[6]helicene: the (P)-isomer induces the formation of the (S)-product, and the (M)-isomer induces the formation of the (R)-product (this corresponds to the behavior of ordinary carbo- and thiohelicenes).
— 2-aza[6]helicene: reverse stereochemistry is observed — the use of the (P)-isomer delivers (R)-alcohol, and the (M)-isomer gives (S)-alcohol.
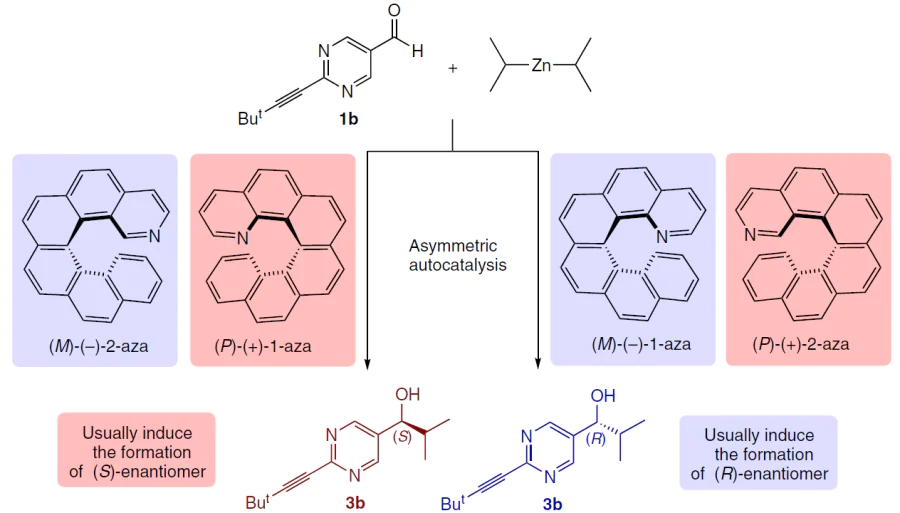
The position of the nitrogen atom in aza[6]helicenes determines the structure of the reactive precursor, and weak intramolecular disperse interactions create asymmetry in the transition states (Scheme 31).[142]
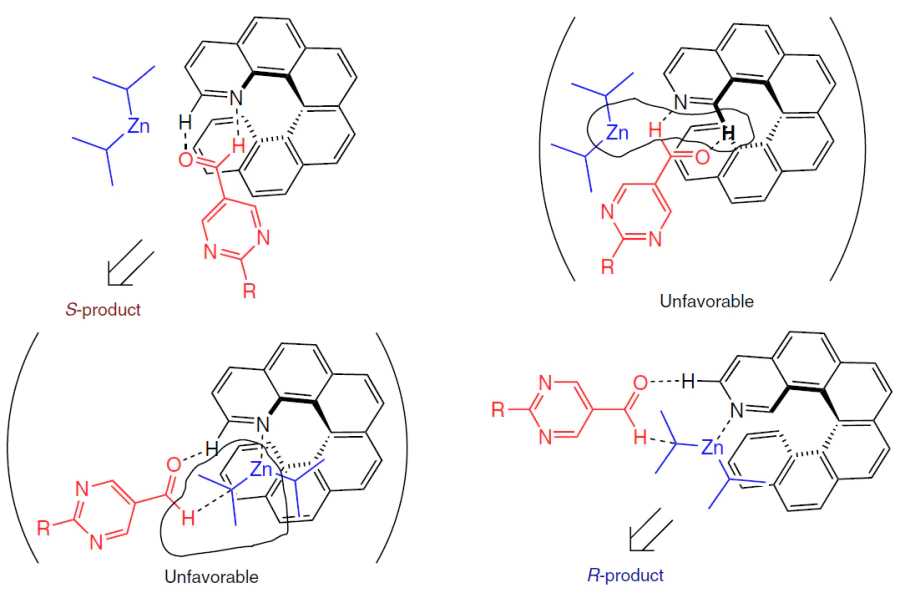
Thus, the correlation between the helicity of azahelicene and enantioselectivity may depend not only on the helicity per se, but also on the position of the nitrogen atom in its molecule.
5.4. Ultra-remote asymmetric control
The reaction of (S)-hydroxyaldehydes tethered via a disilylalkyl chain with Pri2Zn gives an (S,S)-product with high enantiomeric and diastereomeric excess. Subsequent amplification yields products with ee > 99.5% and DL : meso = 96 : 4 (Scheme 32).[26]
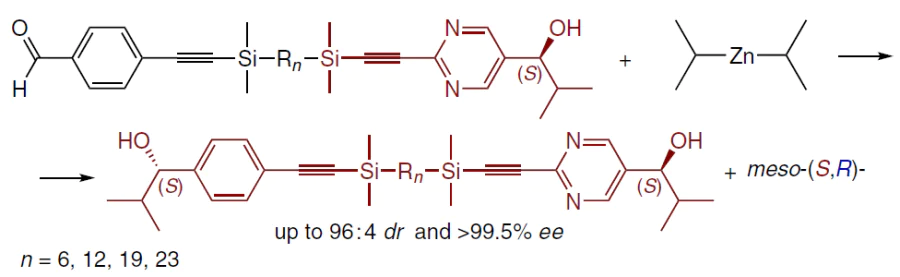
The addition of Pri2Zn in such systems occurs stepwise to afford a mono-zinc alkoxide of hydroxyaldehyde, followed by the formation of a bis(zinc alkoxide) with intramolecular stereocontrol (up to ultra-remote 1,39-induction through 39 bonds at n = 23). Thus, the configuration of one end of the molecule determines the stereochemical outcome of the reaction at the other end (Scheme 33).[26]
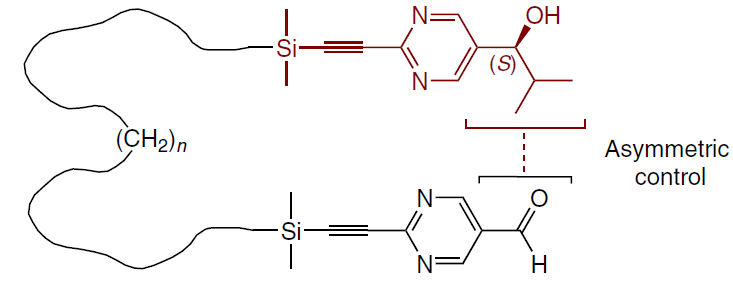
The presence of other chiral additives (e.g., (R)-pyrimidylalkanol 2b of opposite chirality) during the reaction did not affect its stereoselectivity. This indicates the significant power of intramolecular asymmetric induction and its predominance over other intermolecular factors.[26]
The reaction of bis(pyrimidine-5-carbaldehyde) linked by a 1,12-disilyldodecane chain with Pri2Zn in the presence of the corresponding (S,S)-diol with a diastereomeric ratio DL : meso = 62 : 38 and 7% ee as the initial autocatalyst followed by amplification resulted in the formation of the (S,S)-product with DL : meso = 96 : 4 and ee >99.5% (Scheme 34), which corresponds to a 2000-fold increase in the amount of the (S,S)-diol, while the amounts of the meso form and the (R,R)-diol increased by only 76 and 6 times, respectively (Fig. 36).[138]
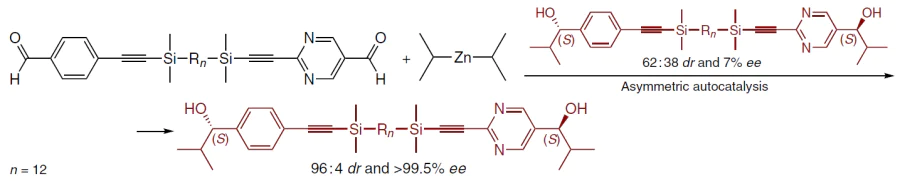
![[{"id":"zZBleTdZ1R","type":"paragraph","data":{"text":"Asymmetric autocatalysis and amplification of bis(pyrimidyl-5-alkanol).<sup>138</sup> Сopyright Royal Society of Chemistry 2023."}}]](/storage/images/resized/greswZSkbOOjU0QRdtmGBpCNogIIe1AomOuq1ldj_xl.webp)
By using hexakis(2-ethynyl-5-pyrimidylalkanol)hexaalkylsilane with 59% ee in the reaction with Pri2Zn and the corresponding hexaaldehyde and subsequent amplification, the amount of the (S6)-form of hexanol increased by incredible 92000-folds over 5 catalytic cycles. Other isomers containing the R-configuration virtually disappeared, with the exception of 2% of the (S5R)-form (Fig. 37).[25]
![[{"id":"iZYukPcnRs","type":"paragraph","data":{"text":"Asymmetric autocatalysis with the use of hexaaldehyde (<i>а</i>). Rates of amplification of the enantiomeric excess of hexanol (<i>b</i>). Visualization of the change in the enantiomeric excess of the products at each reaction cycle (<i>c</i>).<sup>25</sup> Сopyright Wiley 2014."}}]](/storage/images/resized/UYu4wMrxTcTch8kfTPXFqBBU3Lf2STfrPZXsG2pe_xl.webp)
The above results expand the understanding of the mechanisms of chiral amplification and provide new opportunities for controlling stereoselective syntheses.
6. Spontaneous generation of chirality
The unique ability of the Soai reaction to amplify the enantiomeric excess of a product leads to the phenomenon of spontaneous generation of chirality in the absence of any chemical or physical inducers. This phenomenon was first described by Professor Soai’s group in 1997 in a Japanese patent.[143]
The main difference between spontaneous chirality generation and AAA in the presence of triggers or inducers described in the previous Section, is the stochastic distribution of the product chirality sign in a series of experiments (Scheme 35). In other words, in a sufficiently large pool of reactions conducted under strictly achiral conditions, approximately the same number of reactions affording R and S products should be observed.[77]
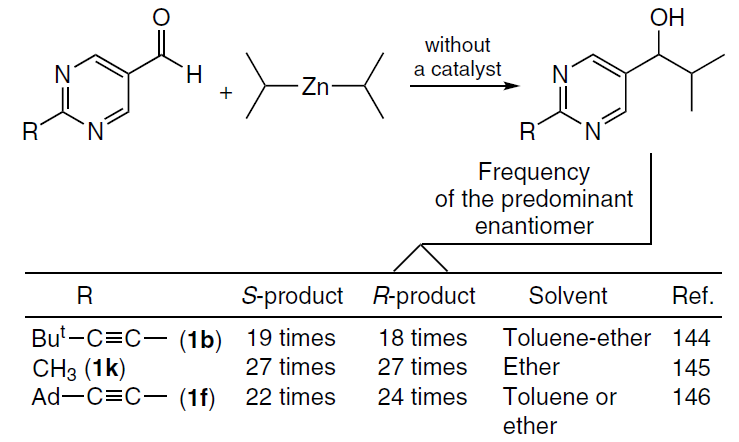
Based on this criterion and three independent reports[144-147] published in 2003, a consensus was reached on the reality of spontaneous generation of chirality in the Soai reaction.
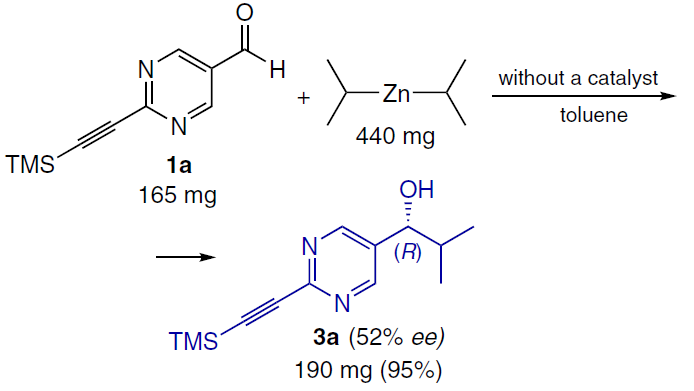
Typically, such experiments yield low ee values (from 1 to 12 – 15%). Moreover, the enantiomeric excess values are also random, as confirmed by the results of a preparative experiment aimed at synthesizing a racemic mixture (Scheme 36). In practice, this means that to obtain a racemic product, the reaction should be carried out using an achiral amino alcohol with different structure.[145]
As noted earlier, the spontaneous generation of chirality in the Soai reaction is due to an efficient mechanism for amplifying vanishingly small enantiomeric excesses.
Obviously, no real racemate is a mixture of equal numbers of molecules with opposite chirality. There is always a statistical fluctuation leading to the predominance of one of the enantiomers. Both the magnitude of this excess and its sign are random.
In the vast majority of chemical reactions, this microscopic imbalance (statistically equal to 0.21% ee, according to one estimate[144]) persists until the end of the process, but does not affect the macroscopically measured value of the enantiomeric excess of the product.
Interestingly, spontaneous generation of chirality can be detected by NMR spectroscopy (Fig. 38).[145] As shown above, the protons of the pyrimidine ring in the heterochiral dimer appear as two broadened singlets of equal intensity. Meanwhile, only one singlet is observed in the spectrum of the homochiral dimer. This makes possible the detection of the spontaneous generation of chirality (predominance of homochiral dimers) immediately after mixing the reactants.
![[{"id":"sWHmaJ0F-x","type":"paragraph","data":{"text":"<sup>1</sup>H NMR spectra (500 MHz) of reaction mixtures obtained in six experiments on the reaction of aldehyde <b>1a</b> with a large excess of diisopropylzinc in a solvent-free fashion. In experiments B, D, and E, spontaneous generation of chirality was observed with enantiomeric excesses of 84% (R), 69% (R), and 32% (S), respectively.<sup>145</sup> Сopyright Royal Society of Chemistry 2003."}}]](/storage/images/resized/CF5rsJi6mODcU7rGbt7q2DJD9OBOEBwEFGJSh27I_xl.webp)
Fig. 39[145] shows the kinetic curves obtained in two experiments monitoring the reaction involving aldehyde 1a. In the first case (see Fig. 39a), the reaction proceeded slowly, at a roughly constant rate, and did not result in an enantiomeric excess in the product. In the second case (see Fig. 39b), the curve acquired a sigmoidal shape, characteristic of an autocatalytic process. The second half of the substrate was consumed twice as fast as the first, and the product was optically active (6% ee).[145]
![[{"id":"v0ysSr0UhO","type":"paragraph","data":{"text":"Experimental curves of aldehyde 1a decay in the reaction with diisopropylzinc in toluene-d<sub>8</sub> at 273 K. Starting amounts: (<i>а</i>) <b>1а</b> (0.01 mol), Pr<sup>i</sup><sub>2</sub>Zn (0.02 mol) (<i>ее</i> of the product is below the detection limit of HPLC); (<i>b</i>) <b>1а</b> (0.02 mol), Pr<sup>i</sup><sub>2</sub>Zn (0.04 mol) (product ее 6%).<sup>145</sup> Сopyright Royal Society of Chemistry 2003."}}]](/storage/images/resized/zpMpEx3nOprGTAjMrrVPVobzs9Y0oO7qIgtapINH_xl.webp)
In the case of a classical enantioselective reaction, one might conclude that increasing the reactant concentration results in the improvement of the enantiomeric excess. However, the difference in the results of the two experiments merely indicates the stochastic nature of the spontaneous generation of chirality.
The difference between amplifying autocatalysis and spontaneous chirality generation is clearly illustrated inFig. 40, which compares the results of reactions with decreasing catalyst concentration and in its absence.[145]
![[{"id":"xGN_lDbR7v","type":"paragraph","data":{"text":"Dependence of the enantiomeric excess of the product on the concentration of the catalyst for the reaction of aldehyde <b>1a</b> (0.025 mol) with Pr<sup>i</sup><sub>2</sub>Zn (0.0425 mol) in toluene at 273 K in the presence of different amounts of catalyst (from 0.002 to 20 mol.%). The value ‘0’ on the x-axis corresponds to the catalyst-free reaction.<sup>145</sup> Сopyright Royal Society of Chemistry 2003."}}]](/storage/images/resized/Fi6fvMeS6pI7JQOC7b0fXrcz1jpnZYF9K9iLUbk9_xl.webp)
At relatively high catalyst concentrations, reproducible enantiomeric excess values close to 100% ee were observed. In the catalyst concentration range of 5 – 10 μmol L–1, the results were also reproducible, but the ee values decreased to 70 – 80%.
At a concentration of 0.5 μmol L–1, a large spread of enantiomeric excess values of the product was observed, ranging from 5 to 75%. However, all products d the same chirality as the original catalyst.
A similar distribution of enantiomeric excesses is observed for reactions in the absence of a catalyst, but in this case, products of the R or S configuration are formed with approximately equal frequency.
The existing consensus regarding the spontaneous generation of chirality in the Soai reaction allows this transformation to be classified as an example of absolute asymmetric synthesis. A detailed discussion of the evolution of ideas about absolute asymmetric synthesis and the modern interpretation of this phenomenon is provided in the Mislow’s review.[77]
As the first reliably established example of absolute asymmetric synthesis, the Soai reaction is frequently mentioned in the literature in connection to the origin of chiral life on Earth. Although it is difficult to imagine life existing under the conditions of the Soai reaction, the very fact of the existence of such a transformation suggests the possibility of similar processes occurring under conditions closer to prebiotic ones. Such reactions could serve as the impetus for the emergence of homochiral biological systems.
Recently, attempts have been made to search for systems similar to the Soai reaction (involving organometallic reagents and catalysts prone to oligomerization) that might exhibit similar effects.[146][148] In a number of cases, macroscopic (although usually low) enantiomeric excess values have been recorded for products obtained in achiral media.
However, no ee amplification was observed in these reactions. Apparently, the spontaneous emergence of chirality at individual stages is a fairly common phenomenon. However, the lack of a highly efficient autoamplification mechanism ultimately leads to the dissipation of chirality on a macroscopic scale.[149]
7. Conclusion
The Soai reaction is an unique phenomenon in organic chemistry, remaining to date the only proven example of highly efficient asymmetric autoamplifying autocatalysis consistent with the Frank’s kinetic model.
Based on the data reviewed herein, the following conclusions can be drawn:
— Exceptional sensitivity: The Soai reaction demonstrates the ability to detect and amplify vanishingly small symmetry violations that are undetectable by other methods. The spectrum of reaction initiators covers:
Physical fields (circularly polarized light);
Chiral surfaces of inorganic minerals (quartz, cinnabar) and achiral minerals with enantiotopic faces (gypsum);
Isotopic chirality (substitution H/D, 12C/13C, 14N/15N, 16O/18O);
Chiral crystals of achiral organic compounds;
Cryptochiral hydrocarbons and other surprising observations.
— Spontaneous generation of chirality: The Soai reaction provides the first well-established example of absolute asymmetric synthesis. In the absence of external chiral inducers, the reaction can enhance microscopic statistical fluctuations in the composition of a racemic mixture, delivering products with high enantiomeric excess, the sign of which is stochastically distributed.
— Amplification mechanism: The nature of the giant chirality amplification in the Soai reaction remains a subject of active research and scientific debate. Current concepts link the reaction mechanism to supramolecular interactions and the formation of complex oligomeric associates of zinc alkoxides. The system achieves a dynamic equilibrium in which the key role is played by differences in stability and reactivity between homochiral and heterochiral aggregates. The effects of mutual inhibition and nonlinear dependence of the reaction rate on the enantiomeric excess of the catalyst make it possible to theoretically substantiate the phenomenon of autocatalysis, but the exact structure of the catalytically active species and the details of the transition states structures are still a subject of discussion.
— Significance for the problem of the origin of life: despite the fact that the chemical conditions of the Soai reaction (anhydrous medium, organozinc reagents) are far from biological, it serves as fundamental evidence of the fundamental possibility of the emergence of homochirality from achiral matter through the mechanisms of autocatalysis and competitive inhibition.
— Significance for the development of the theory of amplifying autocatalysis and the mechanism of spontaneous generation of chirality: there is a significant number of studies on kinetic and stochastic modeling of AAA, which were not considered in this review, as they are beyond the competence of the authors. The interested reader can access the state of research in this area in the relevant works.[150-159]
Thus, the Soai reaction links microscopic chirality at the nuclear level and the structure of the smallest species with the macroscopic chirality of organic matter, demonstrating the impressive power of symmetry breaking laws.
This review demonstrates the diverse manifestations of highly effective AAA, leading to the formulation of new chemical and physicochemical problems worthy of in-depth consideration.
The uniqueness and value of the Soai reaction extend beyond the mere alkylation of specific substrates with diisopropylzinc, especially given the narrow range of compounds capable of effective autoamplification. On the contrary, the Soai reaction has proven itself as a sensitive indicator for identifying chemical systems capable of initiating asymmetric autocatalysis. This provides unique insight into the nature of the chiral organization of effective inducers.
More broadly, the discovery of the Soai reaction has stimulated a significant amount of interdisciplinary research that would unlikely be initiated in any other context. This gives reason for optimism about the future development of this field.
This review was financially supported by the Russian Science Foundation (Project No. 22-13-00275-P).
8. List of abbreviations, designations and terms
ААА — autoamplifying asymmetric autocatalysis (a phenomenon in which a chiral product is a catalyst for its own formation, and its enantiomeric excess is higher than that of the starting catalyst),
B3LYP, M05-2X — functionals used in quantum chemical calculations (DFT),
CPL — circularly polarized light (a physical factor used as an initiator of chirality),
DBAE — N,N-dibutylaminoethanol,
DBNE — N,N-dibutylnorephedrine,
DFT — density functional theory,
DL : meso — the ratio of diastereomers (racemic pair to meso form),
DMNE — N,N-dimethylnorephedrine,
DOSY — diffusion ordered spectroscopy,
dr — diastereomeric ratio,
ee — enantiomeric excess,
EXSY — exchange spectroscopy,
NOE — nuclear Overhauser effect (an effect used in NMR to determine the three-dimensional structure of molecules),
NOESY — nuclear Overhauser effect spectroscopy,
PEAE — (S)-1-phenyl-2-(1-pyrrolidinyl)ethanol,
P/M — helical chirality notations (plus/minus) applied to helicenes and cinnabar crystals,
SMS — the structural type of tetramer (‘square-macrocycle-square’), which is the active form of the catalyst.
The used terms:
Absolute asymmetric synthesis — synthesis of chiral compounds from achiral ones in the absence of chiral reagents or catalysts;
antipodal interaction effect — the first name of the nonlinear effect;
conglomerate — a mixture of crystals of individual enantiomers formed by crystallization of a racemate;
cryptochirality — chirality that does not result in measurable optical rotation but can initiate a reaction;
enantiotopic faces–faces of an achiral crystal (e.g., gypsum) that are mirror images of each other;
floor-to-floor — a transition state model in which the plane of the catalyst macrocycle and the plane of the substrate are oriented in a specific way;
Frank’s model — a kinetic model describing the emergence of chirality through self-replication and mutual inhibition of enantiomers;
isotopomers — molecules that differ only in isotopic composition and are capable of exhibiting isotopic chirality;
macrocyclic dimer — alternative structure of a large ring dimer;
mutual antagonism (mutual inhibition)–the suppression of the formation of one enantiomer by another, often through the formation of an inactive heterochiral complex;
nonlinearly dependent on the enantiomeric excess of the catalyst (a positive NLE leads to amplification),
square dimer (Zn2O2) — structural element of zinc alkoxide oligomers;
nonlinear effect (NLE) — a phenomenon in which the enantiomeric excess of the product is nonlinearly dependent on the enantiomeric excess of the catalyst (a positive NLE leads to amplification);
reservoir effect — removal of the minor enantiomer from the catalytic cycle by forming more stable inactive aggregates;
space group — symmetry characteristic of a crystal;
spontaneous generation of chirality–the formation of enantiomeric excess from achiral starting materials without involving chiral inducers;
tetramer-barrel — one of the theoretically calculated conformations of the tetrameric catalyst;
ultra-remote asymmetric control — transfer of chiral information over a long distance through a molecular chain (e.g. 1.39-induction).
References












